homéostasie du fer, biosynthèse...
TRANSCRIPT

Homéostasie du Fer, Biosynthèse de
l’hème et Porphyries
Pr Jean-Charles Deybach
Faculté Médecine - Université D. Diderot – Paris
P2 - 2012
1

I. Le Fer
2

• Fe++, ferreux (réduit)
• Fe+++, ferrique (oxydé)
Le fer biologiquement actif est sous forme Fe++(ferreux) seul à
passer les membranes cellulaires
Le Fer: Indispensable à toute forme de vie sur Terre
• Transport d’oxygène (Hème)
• Facteur limitant de l’erythropoïèse (formation des globules rouges) • Réactions de transfert d’électrons
• Synthèse d’ADN, d’ARN, des protéines
• Respiration cellulaire
Fe3+ Fe2+ - e-
+ e-
3
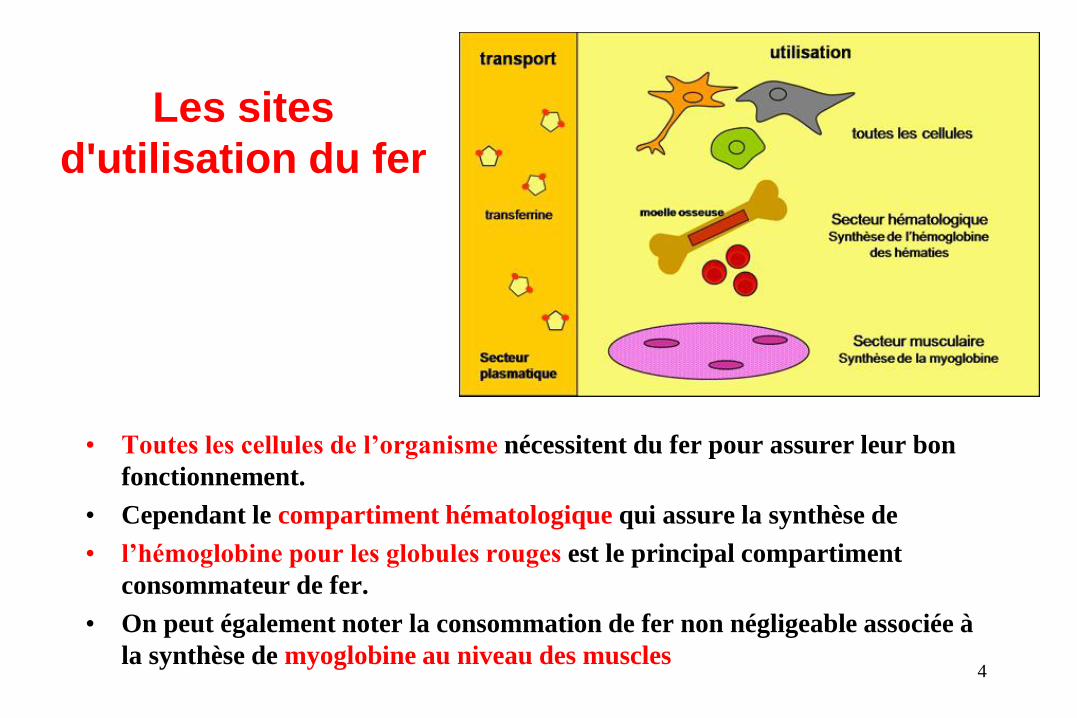
Les sites
d'utilisation du fer
• Toutes les cellules de l’organisme nécessitent du fer pour assurer leur bon
fonctionnement.
• Cependant le compartiment hématologique qui assure la synthèse de
• l’hémoglobine pour les globules rouges est le principal compartiment
consommateur de fer.
• On peut également noter la consommation de fer non négligeable associée à
la synthèse de myoglobine au niveau des muscles
4

Catalyse la production de formes radicalaires de l’oxygène (ROS)
H2O2+ Fe2+ OH + OH + Fe3+
O2 + Fe3+ + Fe2+ . - O 2
2 + 2 H+ O2 + H2O2 - . O 2
•Cassures de l’ADN
•Inactivation des enzymes
•Peroxydation lipidique
Le FER : Indispensable à toute forme de vie…
…éventuellement nocif !
5

Fer et pathologie
• Hémochromatoses héréditaires
• Acéruloplasminémie
• Maladie de Wilson
• Maladie de Menkes
• Anémies sidéroblastiques
• Ataxie de Friedreich
• Vieillissement
• Maladies inflammatoires
• Infections chroniques
• Cancers
• Anémies ferriprives
• Surcharges en fer
secondaires
Défauts « quantitatifs » Défauts génétiques
Pathologies associées
• Maladies neurodégénératives :
Parkinson, Alzheimer, Huntington
• Porphyrie cutanée tardive
• Porphyrie érythropoïétique
6
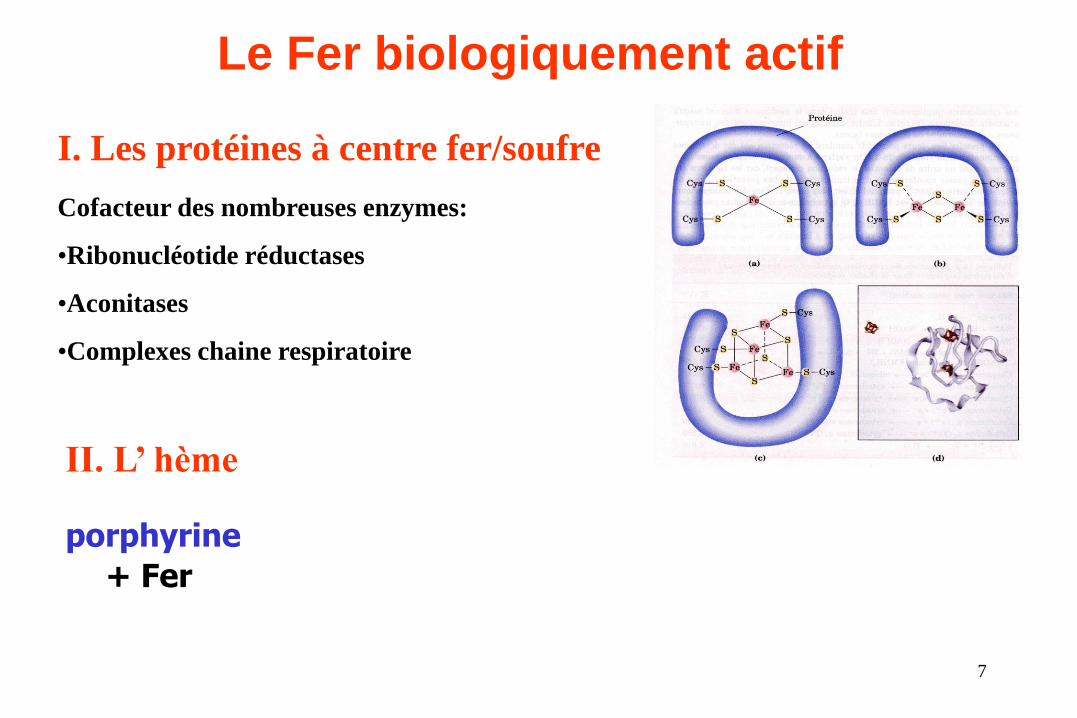
Le Fer biologiquement actif
I. Les protéines à centre fer/soufre
II. L’ hème
porphyrine
+ Fer
Cofacteur des nombreuses enzymes:
•Ribonucléotide réductases
•Aconitases
•Complexes chaine respiratoire
7
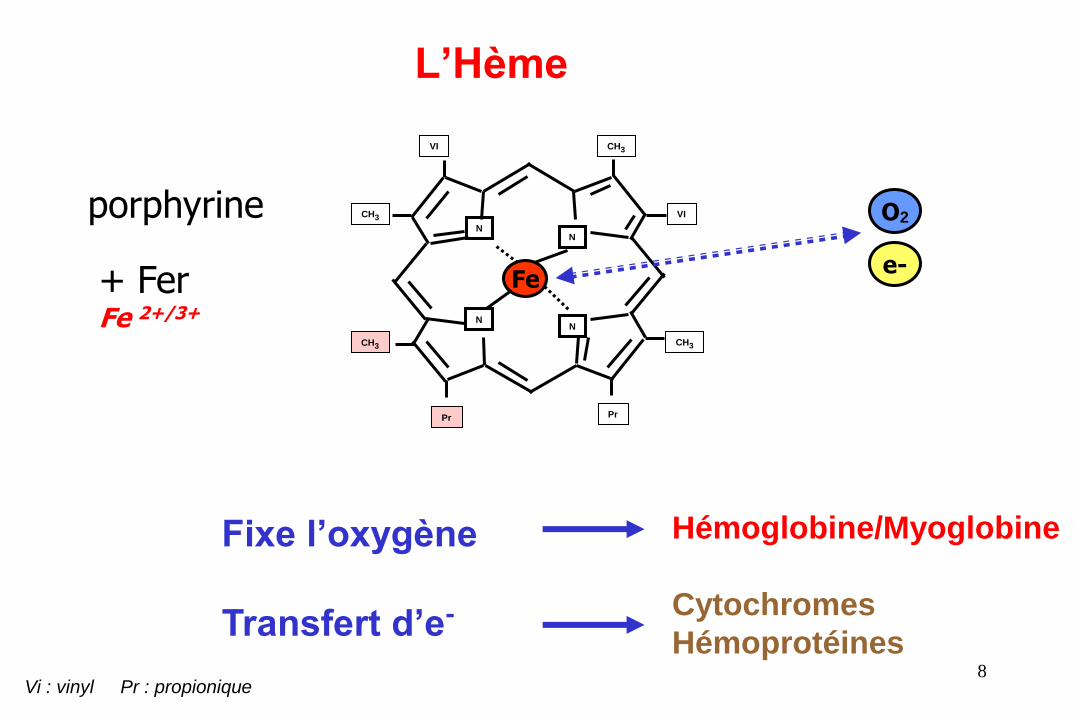
L’Hème
Fe
N N
N N
CH3
VI
VI
CH3
CH3
CH3
Pr Pr
porphyrine
+ Fer Fe 2+/3+
Fixe l’oxygène
Transfert d’e-
Hémoglobine/Myoglobine
Cytochromes
Hémoprotéines
Vi : vinyl Pr : propionique
O2
e-
8

Cytochromes Cytochromes P450
mitochondriaux microsomaux
Hémoglobine Myoglobine
NO synthases Guanylate
cyclase soluble
Catalase Peroxidase
Cyclooxygénase Tryptophane
dioxygénase
Quelques Hémoprotéines
9

Rappel bref sur
le métabolisme
du fer
Protéines essentielles Mutations dans les
gènes humains
codant pour les
protéines du fer :
• les anémies
• les surcharges
Quelques exemples
de maladies héréditaires
Le Fer dans l’histoire humaine : trop ou pas assez…
10
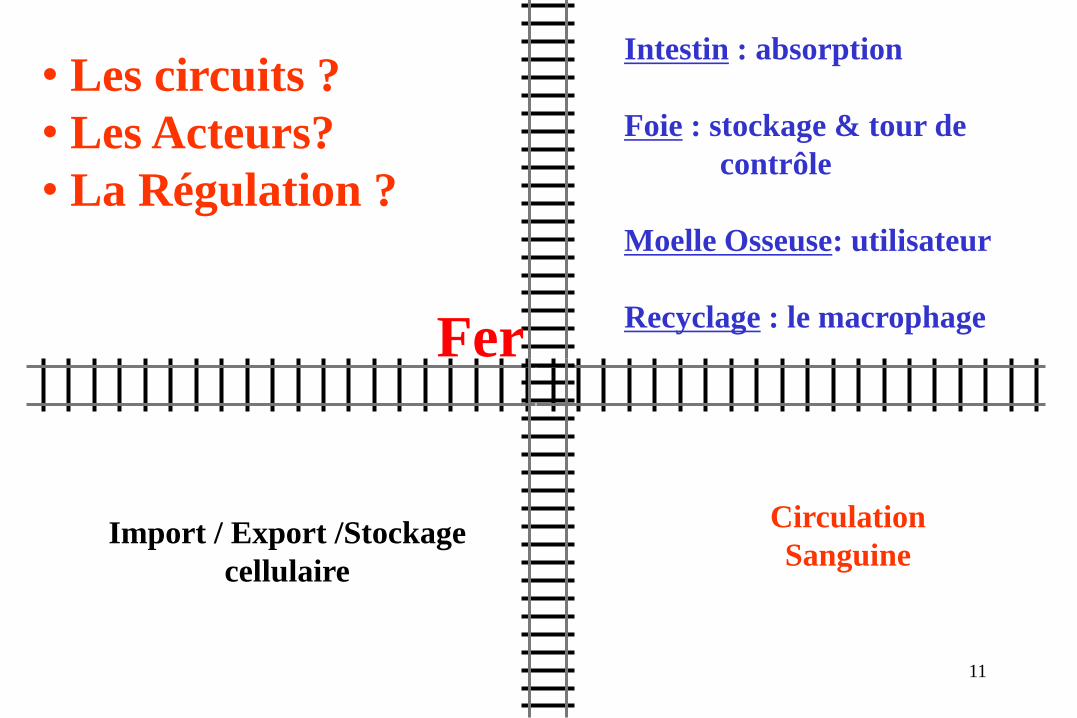
• Les circuits ?
• Les Acteurs?
• La Régulation ?
Intestin : absorption
Foie : stockage & tour de
contrôle
Moelle Osseuse: utilisateur
Recyclage : le macrophage
Circulation
Sanguine Import / Export /Stockage
cellulaire
Fer
11
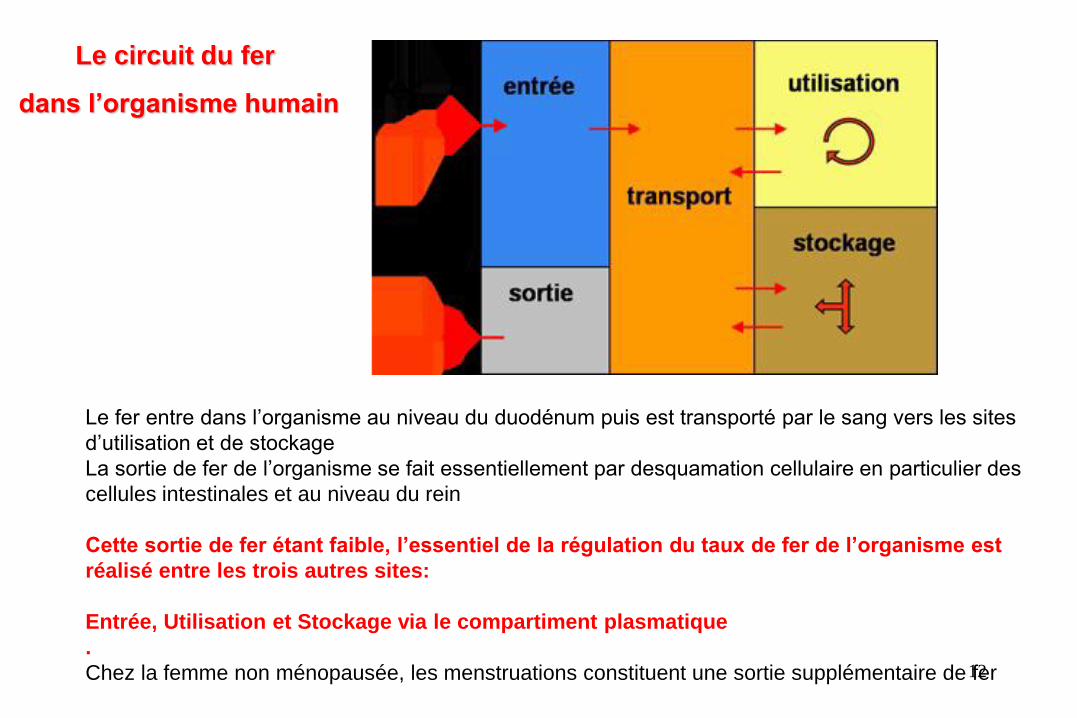
Le fer entre dans l’organisme au niveau du duodénum puis est transporté par le sang vers les sites
d’utilisation et de stockage
La sortie de fer de l’organisme se fait essentiellement par desquamation cellulaire en particulier des
cellules intestinales et au niveau du rein
Cette sortie de fer étant faible, l’essentiel de la régulation du taux de fer de l’organisme est
réalisé entre les trois autres sites:
Entrée, Utilisation et Stockage via le compartiment plasmatique
.
Chez la femme non ménopausée, les menstruations constituent une sortie supplémentaire de fer
Le circuit du fer
dans l’organisme humain
12
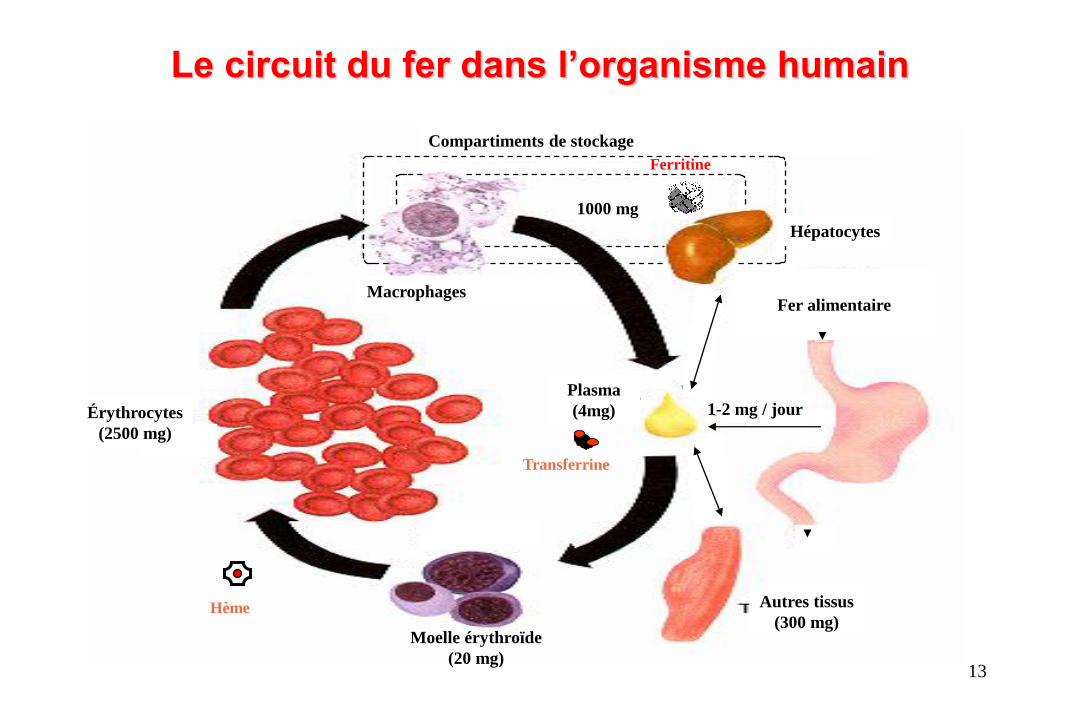
Le circuit du fer dans l’organisme humain
Macrophages
Compartiments de stockage
Fer alimentaire
Plasma
(4mg)
Moelle érythroïde
(20 mg)
Autres tissus
(300 mg)
Érythrocytes
(2500 mg)
1000 mg
1-2 mg / jour
Hépatocytes
Transferrine
Ferritine
Hème
13
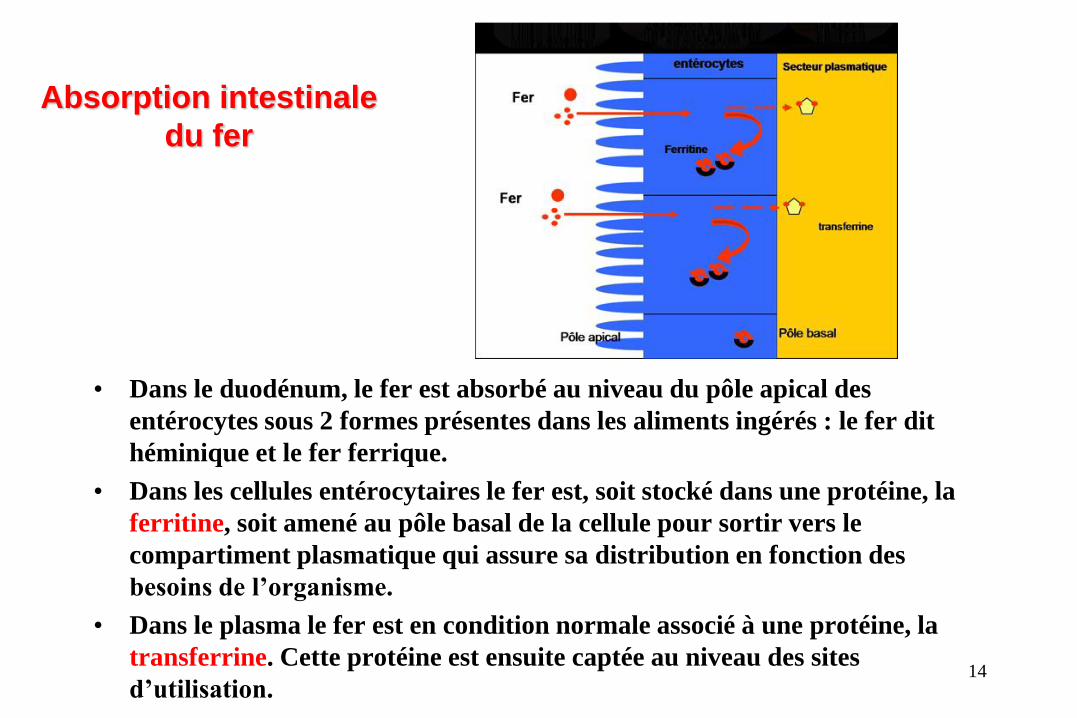
• Dans le duodénum, le fer est absorbé au niveau du pôle apical des
entérocytes sous 2 formes présentes dans les aliments ingérés : le fer dit
héminique et le fer ferrique.
• Dans les cellules entérocytaires le fer est, soit stocké dans une protéine, la
ferritine, soit amené au pôle basal de la cellule pour sortir vers le
compartiment plasmatique qui assure sa distribution en fonction des
besoins de l’organisme.
• Dans le plasma le fer est en condition normale associé à une protéine, la
transferrine. Cette protéine est ensuite captée au niveau des sites
d’utilisation.
Absorption intestinale
du fer
14
Absorption intestinale du fer
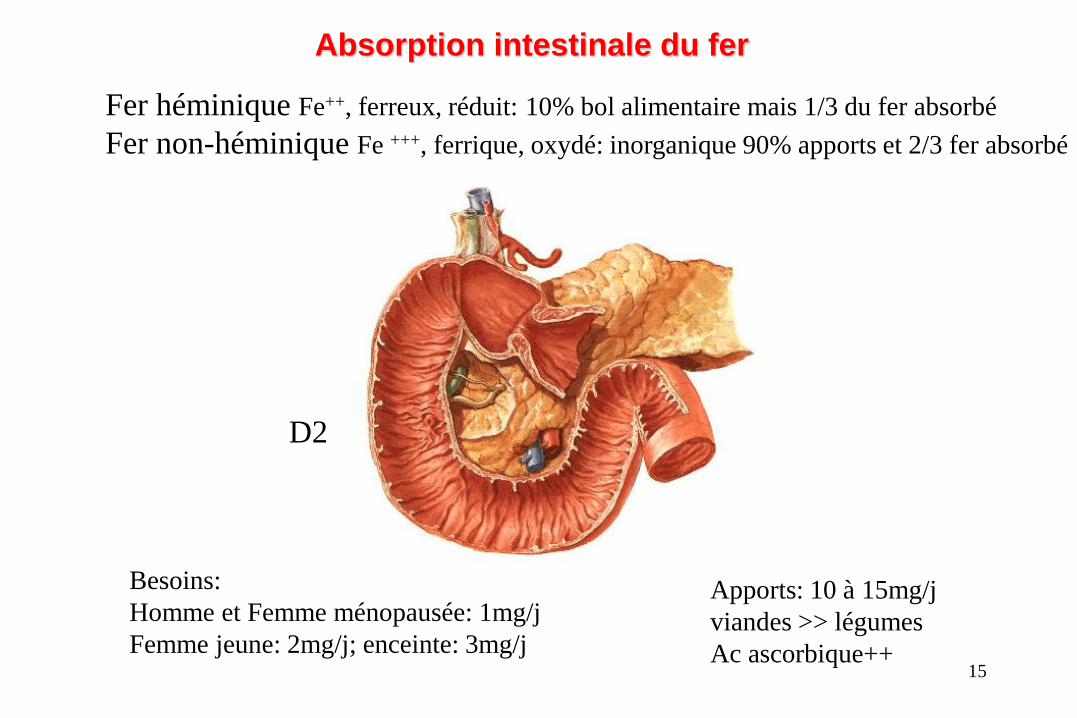
Fer héminique Fe++, ferreux, réduit: 10% bol alimentaire mais 1/3 du fer absorbé
Fer non-héminique Fe +++, ferrique, oxydé: inorganique 90% apports et 2/3 fer absorbé
Besoins:
Homme et Femme ménopausée: 1mg/j
Femme jeune: 2mg/j; enceinte: 3mg/j
Apports: 10 à 15mg/j
viandes >> légumes
Ac ascorbique++
D2
15

L’absorption
enterocytaire du Fer
• Au pôle apical des entérocytes l’hème se lie à son récepteur HCP (Heme Carrier Protein)
puis va être dégradé par une enzyme l’hème oxygénase, libérant ainsi le fer qui s’associe
alors à la ferritine ou est utilisé pour les besoins cellulaires.
• Le fer ferrique (Fe3+) présent dans l’alimentation d’origine végétale est réduit en fer
ferreux (Fe2+) par une enzyme (Dcytb) et entre dans la cellule en utilisant un transporteur
d’ion divalent (DMT1).
• Le fer sera alors soit stocké fixé à la ferritine soit envoyé à la membrane basale de
l’entérocyte et pris en charge par une autre protéine de transport, la ferroportine.
• C’est la ferroportine qui permet la sortie du fer de la cellule vers le compartiment
plasmatique.
• Le fer ferreux étant très réactif il sera oxydé en fer ferrique par l’héphaéstine et pris en
charge par la transferrine. 16
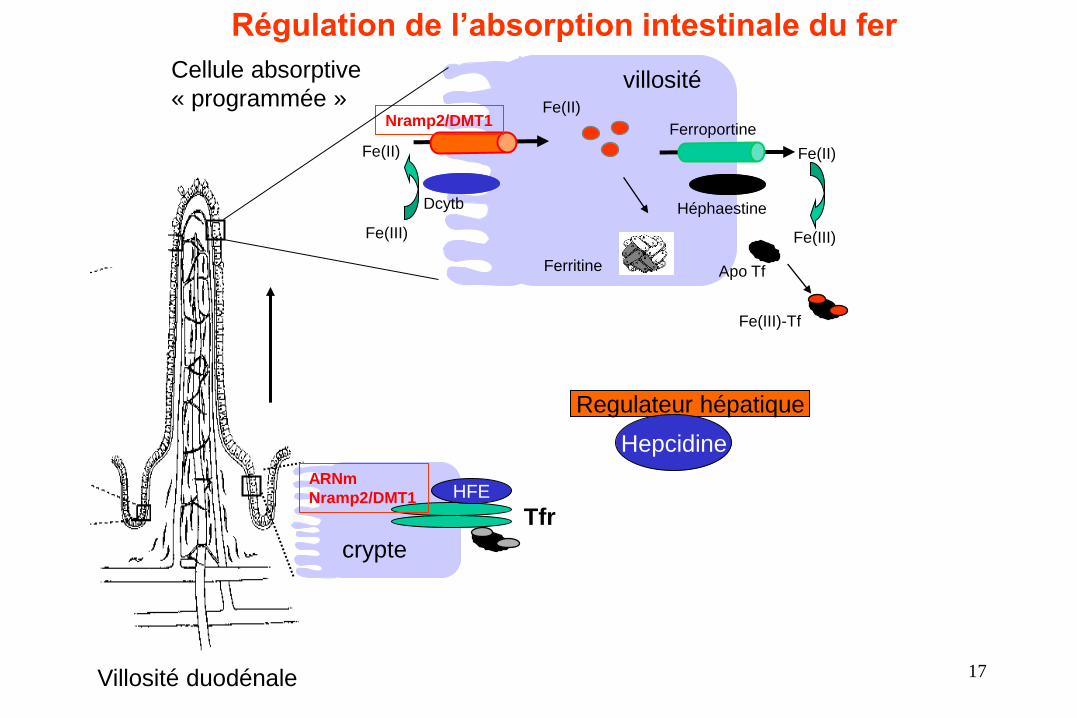
Regulateur hépatique
Villosité duodénale
Hepcidine
crypte
HFE
Fe(II)
Fe(II)
Fe(III)
Dcytb
Nramp2/DMT1 Ferroportine
Héphaestine
Fe(II)
Fe(III)
Apo Tf
Fe(III)-Tf
Ferritine
Régulation de l’absorption intestinale du fer
Tfr
Cellule absorptive
« programmée »
ARNm
Nramp2/DMT1
villosité
17

DMT1/Nramp2
• Cible de la Régulation par l’Hepcidine :
dégradation des ARNm DMT1 dans les c\ cryptiques
Inhibition de l’absorption du Fer dans les c\ villositaires
par le blocage de la Ferroportine
• Transport du fer à travers la membrane apicale des c\ duodénales
• Transport intra c\ du fer de l’endosome vers le cytoplasme
18

Ferroportine (IREG1) : exporteur basal du fer,
Dcytb (« duodenal cytochrome b ») : ferriréductase apicale Fe3+ Fe2+
Hephaestine : ferroxydase membranaire Fe2+ Fe3+
Autres protéines de l'absorption du fer
19

Le passage plasmatique
du fer
• La transferrine Trf est en condition normale le transporteur du
fer dans le plasma.
• C’est une protéine synthétisée par le foie. On parle
d’apotransferrine lorsque la transferrine n’est pas chargée en fer
et d’holotransferrine lorsqu’elle transporte du fer.
• Cette dernière est captée par des récepteurs à la transferrine
(TFR1 et TFR2) en fonction des besoins des différents organes et
tissus. 20
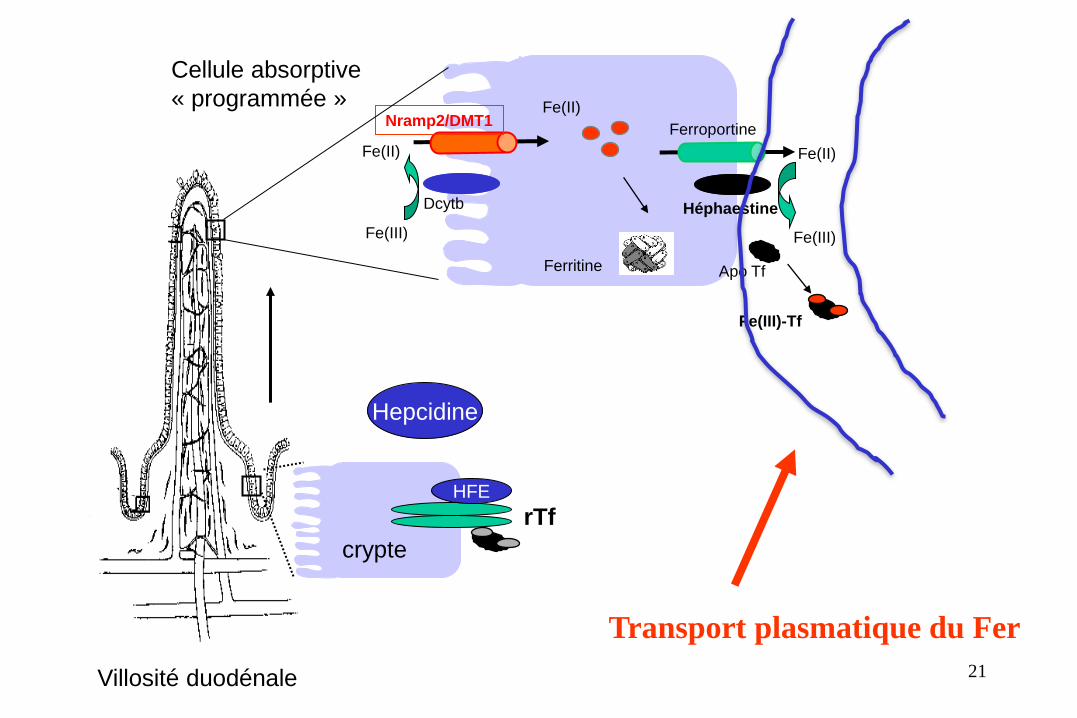
Villosité duodénale
Hepcidine
crypte
HFE
Fe(II)
Fe(II)
Fe(III)
Dcytb
Nramp2/DMT1 Ferroportine
Héphaestine
Fe(II)
Fe(III)
Apo Tf
Fe(III)-Tf
Ferritine
rTf
Cellule absorptive
« programmée »
Transport plasmatique du Fer
21

Le transport plasmatique du fer
• La Transferrine (Tf) : Glycoprotéine de 80 kDa
• Synthétisée par le foie
• Deux sites de liaison du Fe3+
• Taux plasmatiques soumis à de nombreuses variations :
– ▲ si Fe ▼et inversement
– ▲avec ▲des estrogènes (grossesse, pilule, THS ménopause)
– ▼infections, inflammations, cancers
22
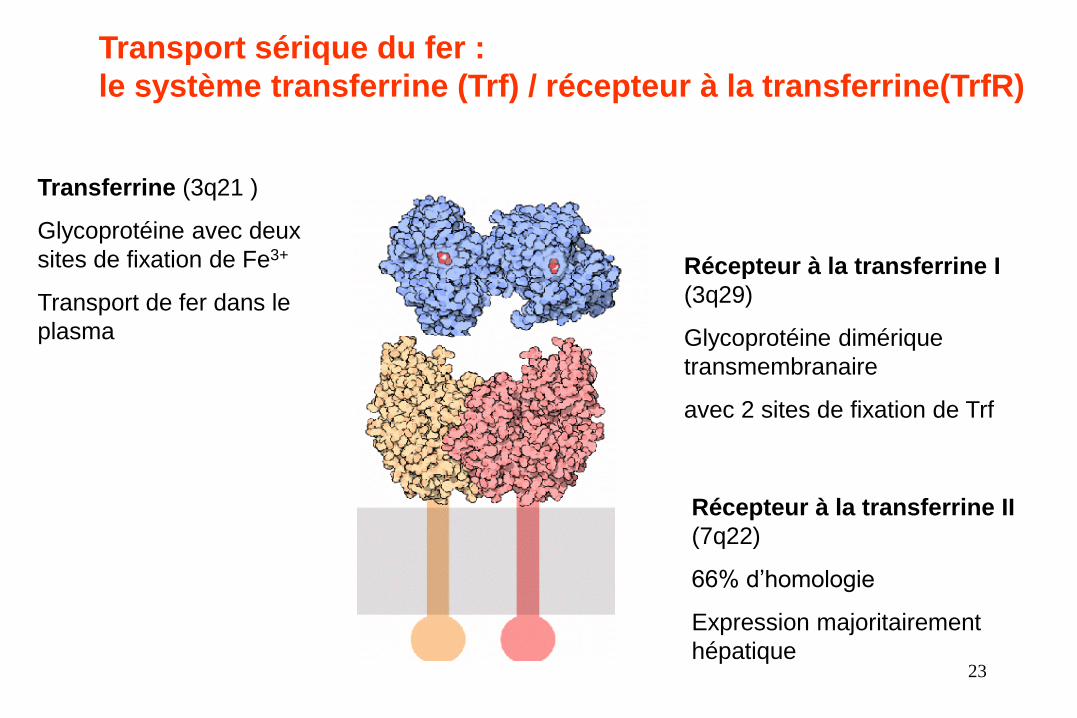
Transferrine (3q21 )
Glycoprotéine avec deux
sites de fixation de Fe3+
Transport de fer dans le
plasma
Récepteur à la transferrine I
(3q29)
Glycoprotéine dimérique
transmembranaire
avec 2 sites de fixation de Trf
Récepteur à la transferrine II
(7q22)
66% d’homologie
Expression majoritairement
hépatique
Transport sérique du fer :
le système transferrine (Trf) / récepteur à la transferrine(TrfR)
23

Captation du fer lors de
l’érythropoïèse
• Les globules rouges (GR) ou hématies ou érythrocytes sont issus de cellules précurseurs, les
érythroblastes, et fabriqués lors de l’érythropoïèse dans la moelle osseuse. C’est le site
majeur d’utilisation du fer par l’organisme pour la synthèse de l’hémoglobine
• Au niveau de la membrane de l’érythroblaste, l’holotransferrine est captée par le récepteur
à la transferrine 1 (TFR1). Un processus d’endocytose permet l’entrée du complexe
fer/transferrine/récepteur dans la cellule
• Dans l’endosome le fer se dissocie de la transferrine. Il est réduit par l’enzyme STEAP3 et
transporté par DMT1 afin d’être utilisé pour la synthèse de l’hème.
24

• Les macrophages en particulier ceux présents au niveau du foie et de la rate assurent stockage
et recyclage du fer à partir des hématies sénescentes.
• Cette dégradation aboutit à un recyclage soit de l’hème soit du fer après dégradation de l’hème.
• Le fer stocké dans la ferritine sera ensuite redistribué à l’organisme en fonction de la demande
en fer. La sortie du fer du macrophage est assurée par la ferroportine. La ceruloplasmine oxyde
le fer ferreux en fer ferrique.
• Le fer ferrique s’associe ensuite à la transferrine.
Recyclage du fer par
les macrophages
25

Globules rouges
Foie
Macrophage
(rate, moelle, foie)
Hème oxygénase
Fe(II)
Céruloplasmine Fe(II)
Fe(III)
Fe(III)-Tf
Ferroportine
Apo-Tf
Hepcidine
Recyclage du Fer par les macrophages
26
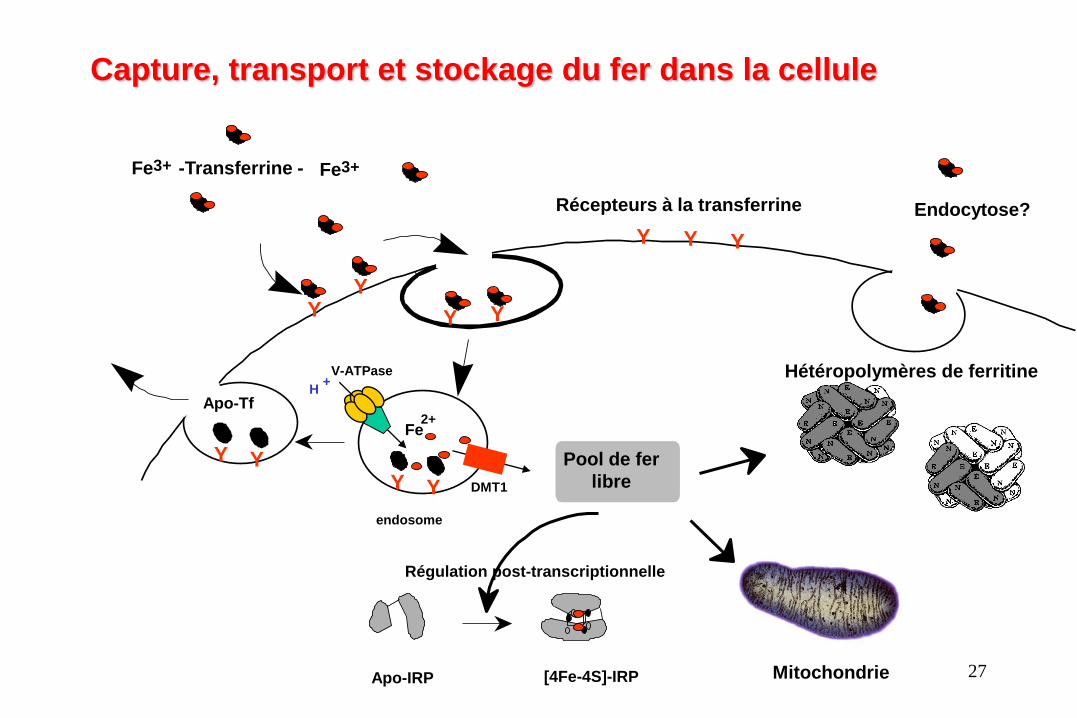
Y
Y
Pool de fer
libre
Y
Y Y
Y
Y Y
Récepteurs à la transferrine
Y Y
Fe 2+
Apo-IRP [4Fe-4S]-IRP
Hétéropolymères de ferritine
Y Y
endosome
Apo-Tf
DMT1
H +
V-ATPase
3+ Fe -Transferrine - 3+ Fe
Capture, transport et stockage du fer dans la cellule
Mitochondrie
Endocytose?
Régulation post-transcriptionnelle
27

Rôle du foie dans la
régulation du
métabolisme du fer
• L’hépatocyte est capable de stocker le fer via le processus d’endocytose du
complexe fer/transferrine/TFR1 suivi de la liaison du fer à la ferritine
- Il peut en fonction des besoins de l’organisme restituer du fer via la protéine
d’export du fer cellulaire (ferroportine) vers le compartiment plasmatique. Puis la
ceruloplasmine oxyde le fer Fe2+ en Fe3+ lequel peut alors être pris en charge par
la transferrine.
- Il synthétise et secrète des molécules clés de la régulation du métabolisme du fer :
la transferrine, la ceruloplasmine et l’hepcidine décrite comme « l’hormone du
métabolisme du fer». 28

Hepcidine, un régulateur de l’homéostasie du fer
Synthétisée par le foie comme un peptide de 85 AA
peptide mature (20-25 AA) dans le sérum et l’urine
23 AA Peptide signal Pro-région
Site de coupure de propeptide convertase
peptides matures (20 or 25 AA) 36 AA
25 AA 20 AA
29

Action de
l’hepcidine
• L’hepcidine en s’associant avec la ferroportine entraîne la dégradation de cette
protéine, stoppant ainsi la sortie de fer des cellules vers le plasma. De cette façon
l’hepcidine contrôle l’absorption intestinale et le relargage de fer par les
macrophages.
• L’hepcidine à un rôle de frein sur l’entrée du fer dans le plasma. Ainsi une
augmentation de la production d’hepcidine va entraîner une diminution de l’entrée
du fer tandis qu’une inhibition de la production d’hepcidine va au contraire aboutir à
une augmentation de l’entrée du fer. . 30

Régulation de la
synthèse d’hepcidine
La synthèse de l’hepcidine est finement régulée par différents signaux émanant de l’organisme en
fonction de ses besoins en fer. Ainsi on distingue:
- Le signal inflammatoire qui stimule la production d’hepcidine afin d’empêcher l’accès au fer pour des
agents pathogènes
- Le signal érythropoïétique qui inhibe la production d’hepcidine afin d’assurer une entrée de fer
plasmatique nécessaire à la fabrication de nouvelles globules rouges
- Le signal hypoxique (manque d’oxygène ) qui inhibe la production d'hepcidine.
- Le signal lié au statut en fer de l’organisme qui met en jeu de nombreuses molécules nouvellement
identifiées. Pour certaines, des mutations de leur gène sont responsables de maladies de surcharges en fer. 31
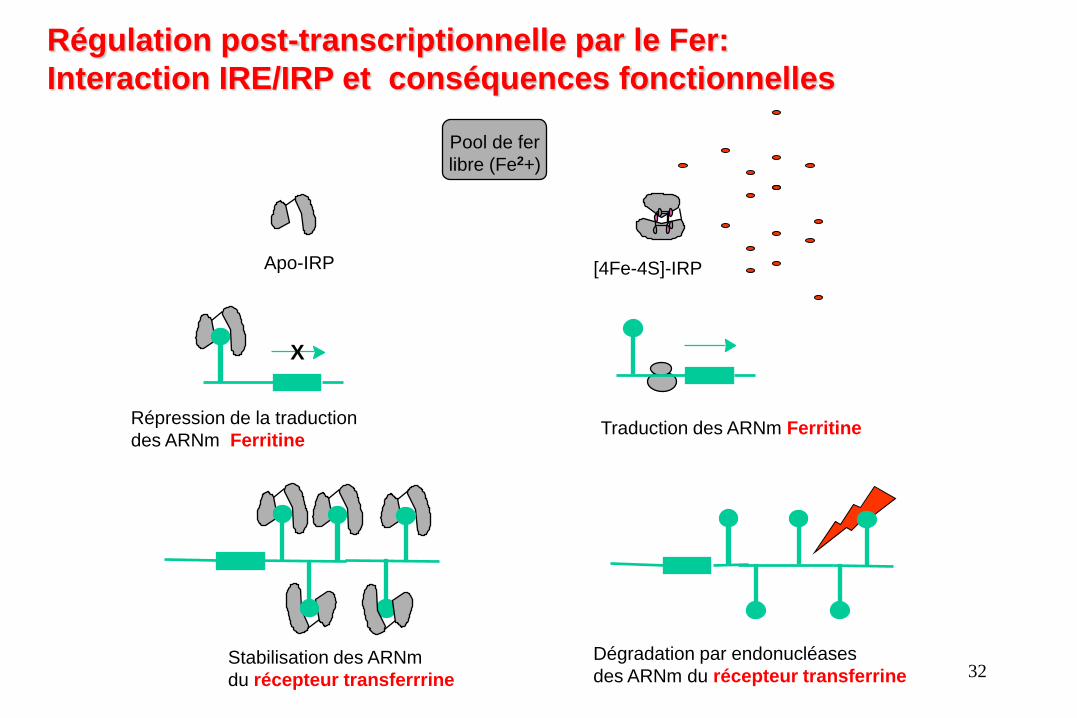
Apo-IRP [4Fe-4S]-IRP
Pool de fer
libre (Fe2+)
Répression de la traduction
des ARNm Ferritine
X
Traduction des ARNm Ferritine
Dégradation par endonucléases
des ARNm du récepteur transferrine Stabilisation des ARNm
du récepteur transferrrine
Régulation post-transcriptionnelle par le Fer:
Interaction IRE/IRP et conséquences fonctionnelles
32
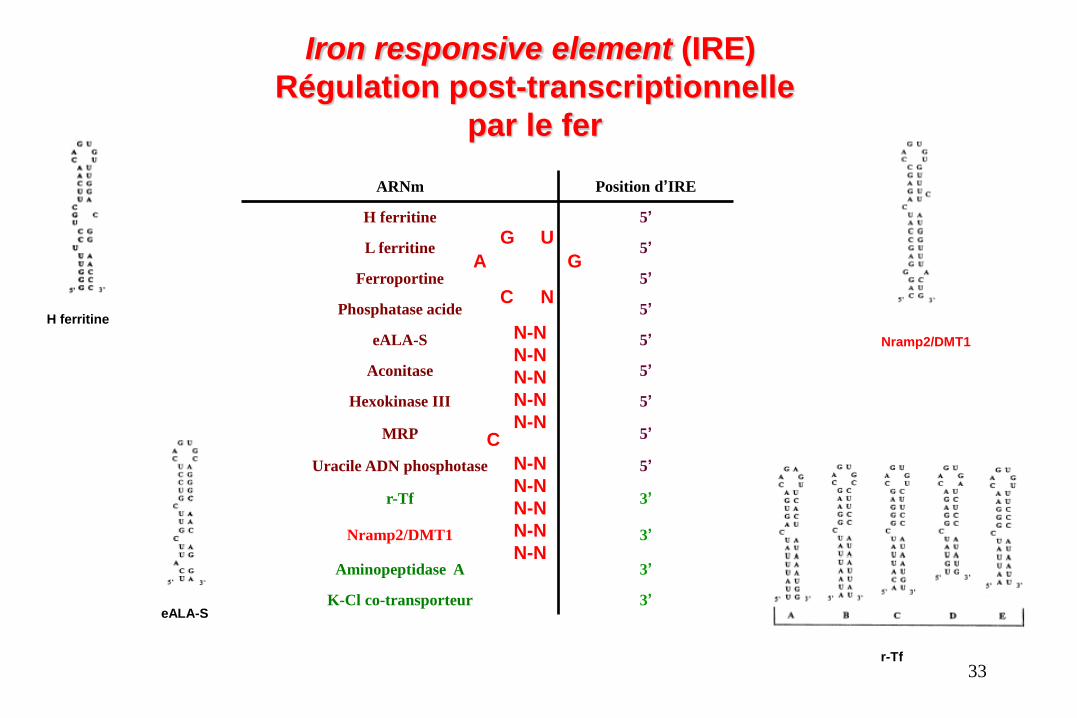
ARNm Position d’IRE
H ferritine 5’
L ferritine 5’
Ferroportine 5’
Phosphatase acide 5’
eALA-S 5’
Aconitase 5’
Hexokinase III 5’
MRP 5’
Uracile ADN phosphotase 5’
r-Tf 3’
Nramp2/DMT1 3’
Aminopeptidase A 3’
K-Cl co-transporteur 3’
G
A
U
N C
C
G
N-N
N-N
N-N
N-N
N-N
N-N
N-N
N-N
N-N
N-N
H ferritine
eALA-S
r-Tf
Nramp2/DMT1
Iron responsive element (IRE)
Régulation post-transcriptionnelle
par le fer
33

Le stockage du fer
• Le fer peut aussi être stocké, principalement au niveau du foie et de la rate. Les
globules rouges ont une durée de vie limitée au terme de laquelle ils sont phagocytés
par les macrophages du système réticuloendothélial en particulier au niveau de la
rate.
• Le fer ainsi recyclé pourra être remis en circulation dans le plasma et réutilisé par
l’organisme ou stocké dans les hépatocytes lorsqu’il est en excès.
• Dans ce cas un système de régulation dit de rétro-contrôle est mis en place par les
hépatocytes dans le foie, et va intervenir afin de diminuer la concentration de fer
dans le plasma ceci en agissant sur l’absorption intestinale et la sortie de fer du
système réticuloendothélial.
• En absence de ce rétro-contrôle des problèmes de surcharge en fer de l’organisme
vont apparaître.
34

noyau ferrique : jusqu’à 4500 atomes de fer par molécule
H (11q23) L (19q13)
hépatocytes : fer absorbé au niveau duodénal
macrophages de la rate, du foie, de la moelle osseuse :
fer recyclé héminique
ferritine mitochondriale (5q23.1)
coquille protéique : hétéropolymère de 24 su (H et L)
Stockage du fer : la ferritine
35
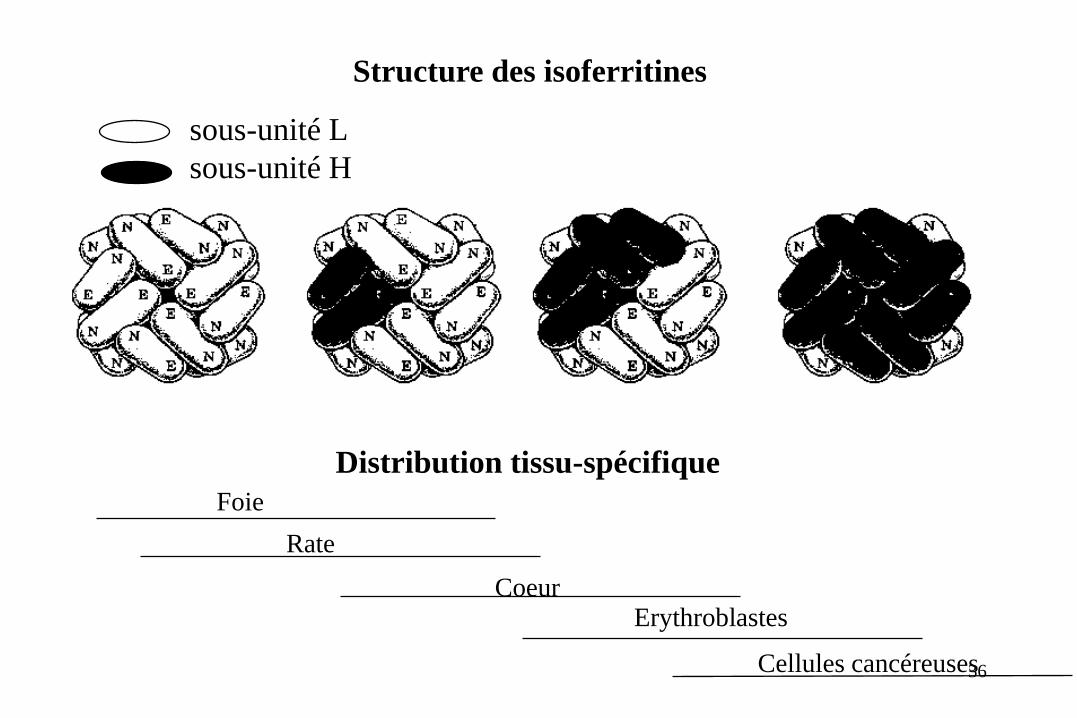
Structure des isoferritines
sous-unité L
sous-unité H
Distribution tissu-spécifique
Foie
Rate
Coeur Erythroblastes
Cellules cancéreuses 36

Activité ferroxydase - Catalyse Fe2+ Fe3+ Contrôle le pool de fer libre et joue un rôle dans la
défense contre le stress oxydatif
-
-
La sous-unité H ferritine :
La sous-unité L ferritine facilite la formation du noyau
ferrique à l’intérieur de la molécule de ferritine
Pas de redondance fonctionnelle entre les deux sous-unités
L’inactivation des deux allèles H ferritine chez la souris
(HFt -/-) est létale entre 3.5 et 9.5 jours de développement
embryonnaire
Structure et fonction de la ferritine
37

La ferritine sérique
• Valeurs normales: 20 à 250 µg/l
• Propriétes: Contient peu de fer
Partiellement glycosylée
Composée principalement de sous-unité L
• Les causes d’élévation de la ferritine sérique sont multiples:
Surcharge en fer génétique ou secondaire
Etats inflammatoires et infectieux
Syndrome cataracte-hyperferritinémie
Autres: cancer…….
38

EXPLORATION DU METABOLISME DU FER
• (Fer Sérique ±)
• Ferritine
• Transferrine
• CTF (Capacité Totale de Fixation)
• sTrfR (Récepteur soluble à la transferrine)
• IRM (contenu hépatique en fer +++)
• Histologie moëlle : coloration de Perls, sidéroblastes, ME
39

Fer et pathologie
• Hémochromatoses héréditaires
• Acéruloplasminémie
• Maladie de Wilson
• Maladie de Menkes
• Anémies sidéroblastiques
• Ataxie de Friedreich
• Vieillissement
• Maladies inflammatoires
• Infections chroniques
• Cancers
• Anémies ferriprives
• Surcharges en fer
secondaires
Défauts « quantitatifs » Défauts génétiques
Pathologies associées
• Maladies neurodégénératives :
Parkinson, Alzheimer, Huntington
• Porphyrie cutanée tardive
• Porphyrie érythropoïétique
40

Les carences en Fer
• carences d’absorption: gastrectomie,
malabsorption
• carences d’apport: nourisson, malnutrition
• augmentation des besoins: grossesse
• augmentation des pertes: hémorragies
• inflammations subaiguës ou chroniques
• Cancers
• médicaments: cortisone, aspirine, allopurinol, …
41

Les carences martiales
ETIOLOGIES
• Mal absorption ……..
• Fuite (saignements TD, reins, métrorragies…….)
• Inflammatoire (hepcidine)
BIOLOGIE
• Décompensée: anémie ferriprive microcytaire
• Compensée :
ferritine basse, CTF bas, rTfs élevé
=> intérêt rTfs en cas d’ inflammation 42

Les surcharges en Fer
• Hémochromatose génétique;
• Hemochromatose secondaire (Transfusion, alcool)
• Dysérythropoïèse: insuf. médullaire, anémie de
Biermer, thalassémie, anémies sidéroblastiques,
myélodysplasies,..
• Cytolyse: hépatique, musculaire, anémie
hémolytique, fibrose, Carcinome Hépato Cellulaire
43

Fer et pathologie
• Hémochromatoses héréditaires
• Acéruloplasminémie
• Maladie de Wilson
• Maladie de Menkes
• Anémies sidéroblastiques
• Ataxie de Friedreich
• Vieillissement
• Maladies inflammatoires
• Infections chroniques
• Cancers
• Anémies ferriprives
• Surcharges en fer
secondaires
Défauts « quantitatifs » Défauts génétiques
Pathologies associées
• Maladies neurodégénératives :
Parkinson, Alzheimer, Huntington
• Porphyrie cutanée tardive
• Porphyrie érythropoïétique
44

Type 1: la plus fréquente mutations gène HFE (C282Y, H63D)
(pénétrance variable, contribution du déficit en hepcidine)
Hémochromatoses Héréditaires
Autosomique récessive
Autosomique dominante
Type 4: mutations des gènes Ferroportine, H Ferritine
Type 3: mutations du gène TfR2
Type 2: hémochromatoses juvéniles
Déficit en Hepcidine
Déficit en Hémojuvéline
45

Hemochromatose génétique liée à HFE
(HFE1)
• Maladie autosomique et dominante
• Développement d’une surcharge en fer progressive
(foie, pancréas), devenant secondairement macrophagique
• Atteintes viscérales multiples
hépatomégalie (+/- cirrhose)
myocardiopathie
douleurs osteo-articulaires, mélanodermie
• Due à des mutations du gène HFE (C282Y)
• Traitement par saignées
46

Gène Chromosome Défaut moléculaire Pathologie associée
Transferrine 3q21 M, D Atransferrinémie congénitale
HFE 6p21 M Hémochromatose (HFE-1)
Hemojuveline 1q21 M Hémochromatose juvénile (HFE-2)
Hepcidine 19q13 M Hémochromatose juvénile (HFE-2)
r-Tf2 7q22 M Hémochromatose méditerranéenne (HFE-3)
Ferroportine 2q32 M Hémochromatose dominante (HFE-4)
H ferritine 11q12 M dans l’IRE Hémochromatose dominante
L ferritine 19q13 M dans l’IRE Cataracte-hyperferritinémie
URO-D 1p34 M, I, D Porphyrie cutanée tardive
Ferrochélatase 18q21.3 M Protoporphyrie érythropoïétique
Céruloplasmine 3q21-q24 M, I, D Acéruloplasminémie
ALAS2 Xp11.21 M Anémie sidéroblastique
ABC7 Xq13 M Anémie sidéroblastique et ataxie
ATPase 7A Xq13-q21.1 M Maladie de Menkes
ATPase 7B 13q14.3 M Maladie de Wilson
FRDA 9q13 Expansion GAA Ataxie de Friedreich
Inconnu 15q5.1-15.3 Inconnu Anémie dysérythropoïétique congénitale de type I
Inconnu 20q11.2 Inconnu Anémie dysérythropoïétique congénitale de type II
Inconnu 15q21-25 Inconnu Anémie dysérythropoïétique congénitale de type III
47

48

II. Biosynthède de l’hème et Porphyries
Les pigments de la vie :
La chlorophylle
La vitamine B12
Le coenzyme F430
L’hème
Molécule “mère” : l’uroporphyrinogène III
49
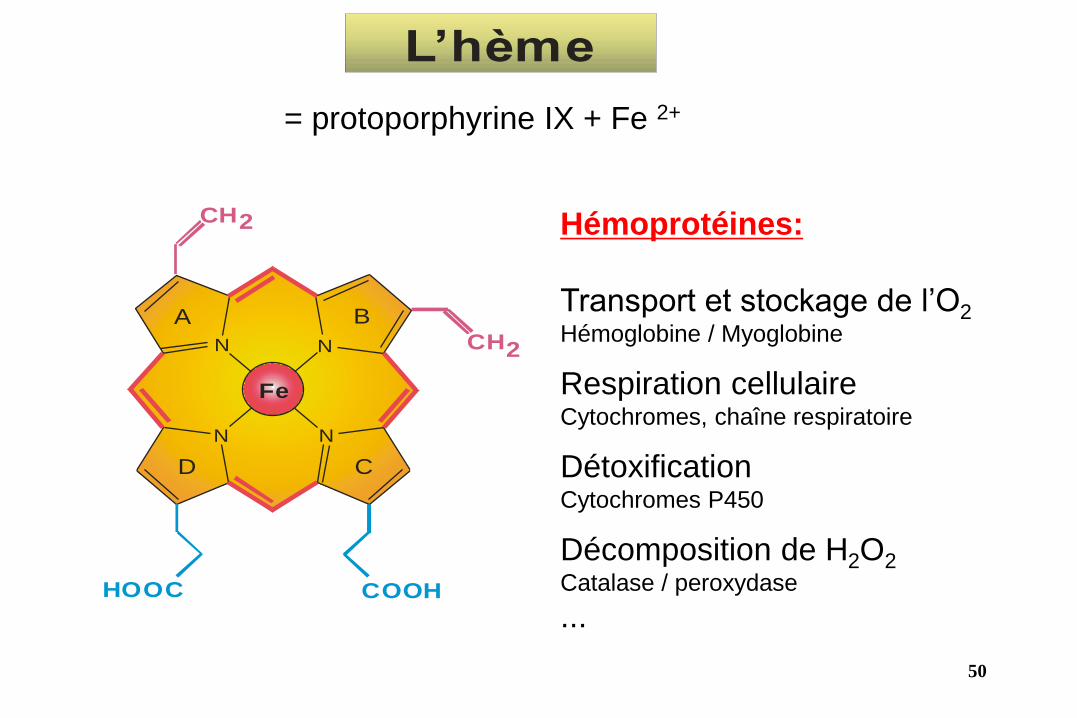
N
Fe
N
NN
CH2CH3
CH3
H3C
H3C
CH2
HOOC COOH
A B
CD
Hémoprotéines:
Transport et stockage de l’O2 Hémoglobine / Myoglobine
Respiration cellulaire Cytochromes, chaîne respiratoire
Détoxification Cytochromes P450
Décomposition de H2O2 Catalase / peroxydase
...
L’hème
= protoporphyrine IX + Fe 2+
50
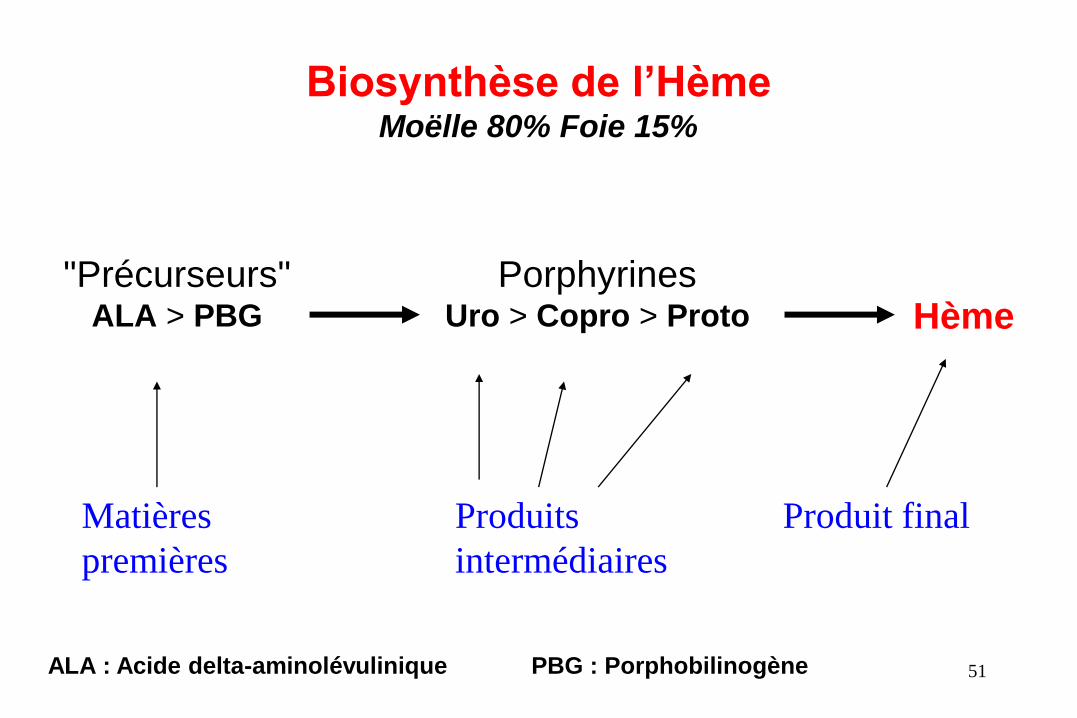
Biosynthèse de l’Hème Moëlle 80% Foie 15%
"Précurseurs" ALA > PBG
Porphyrines Uro > Copro > Proto Hème
Matières
premières
Produit final Produits
intermédiaires
ALA : Acide delta-aminolévulinique PBG : Porphobilinogène 51
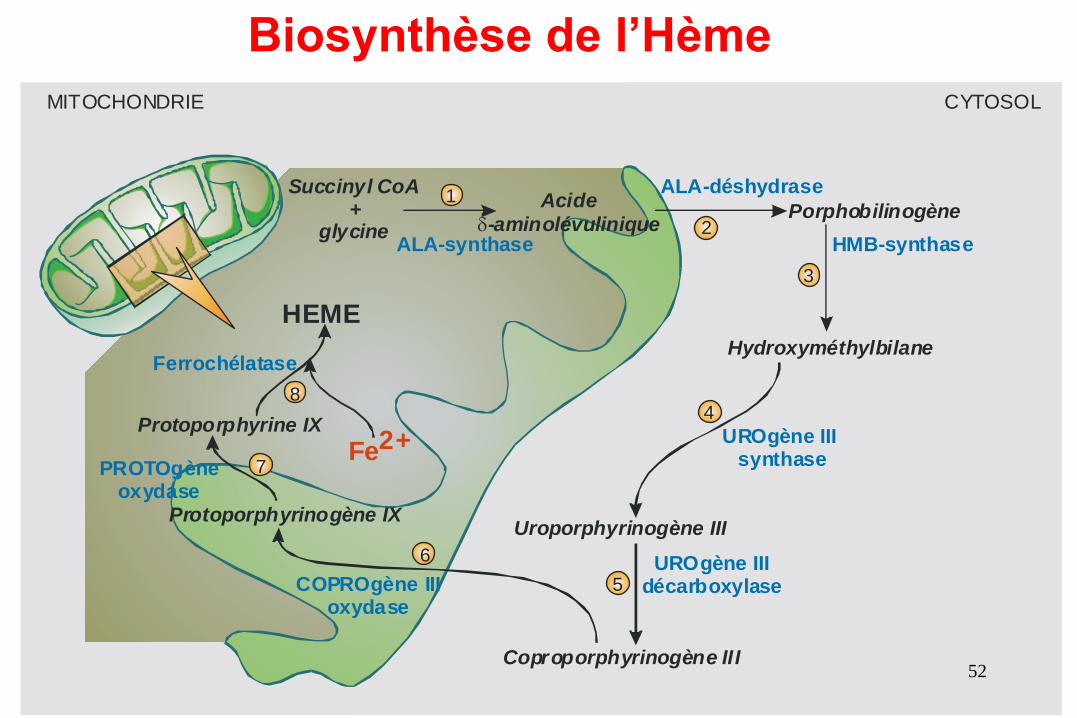
ALA-déshydrase
ALA-synthase HMB-synthase
UROgène III synthase
UROgène III décarboxylaseCOPROgène III
oxydase
PROTOgèneoxydase
Ferrochélatase
Porphobilinogène Acide
-aminolévulinique
Hydroxyméthylbilane
Succinyl CoA+
glycine
Uroporphyrinogène III
Coproporphyrinogène III
Protoporphyrinogène IX
Protoporphyrine IX
MITOCHONDRIE CYTOSOL
Fe2+
HEME
8
7
6
4
3
2
1
5
Biosynthèse de l’Hème
52

Régulation de la synthèse d’Hème
• 2 sites majeurs de production :
– La moëlle : 80%
– Le foie et autres tisus : 20%
Flux métabolique adapté et donc différent
• 2 systèmes de régulation « tissus spécifiques »
– Erythroïde (E)
– Non érythroïde (NE) = ubiquitaire
– Spécificité 1ère étape : ALA Synthase, 2 gènes différents, même
activité enzymatique
– Etapes 2 et 3 : 2 promoteurs alternatifs (E, NE)
53

Gènes de la biosynthèse de l’Hème
• ALAS 2
• ALAS 1
• ALAD
• PBGD
• UROS
• UROD
• CPO
• PPOX
• FECH
X E
3 NE
9 NE/E
11 NE/E
10 U
1 U
3 U
1 U
18 NE/E
E : Erythroïde NE : Non Erythroïde U : Ubiquitaire 54
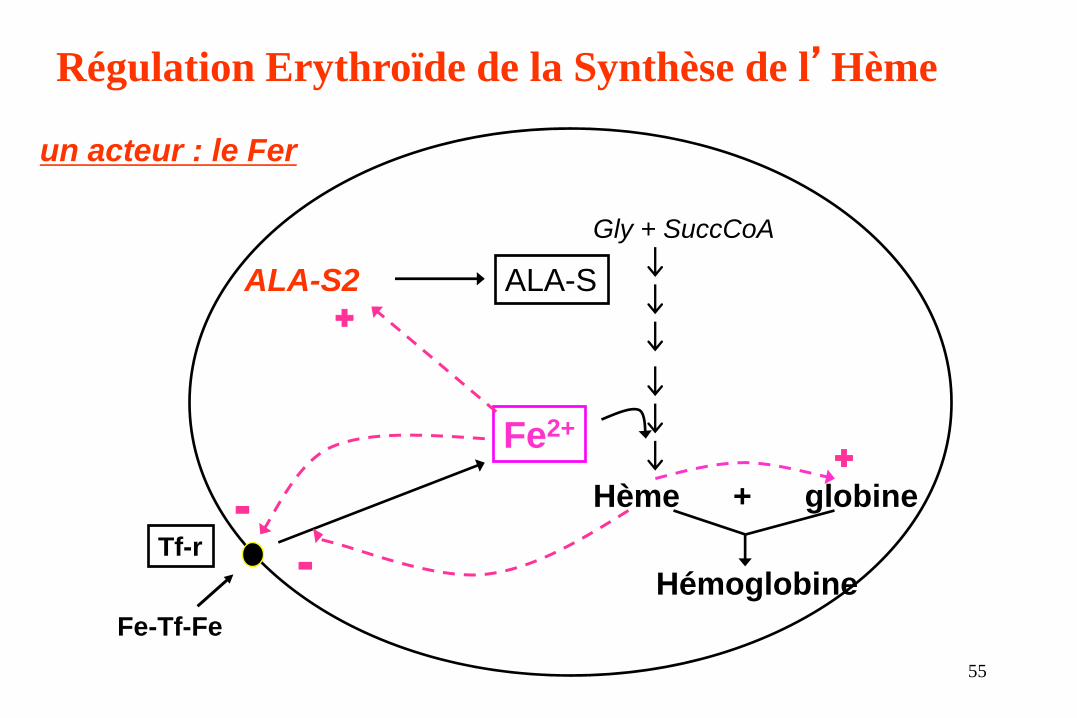
Fe-Tf-Fe
Fe2+
Hème + globine
Hémoglobine
Gly + SuccCoA
Tf-r
ALA-S
Régulation Erythroïde de la Synthèse de l’Hème
ALA-S2
+
+
-
-
un acteur : le Fer
55

Fe2+
Hème
Cytochromes
Hémoprotéines
Gly + SuccCoA
ALA-S
Régulation Hépatique de la Synthèse de l’Hème
ALA-S1
Apo-protéines
? -
-
un acteur : l’Hème
56
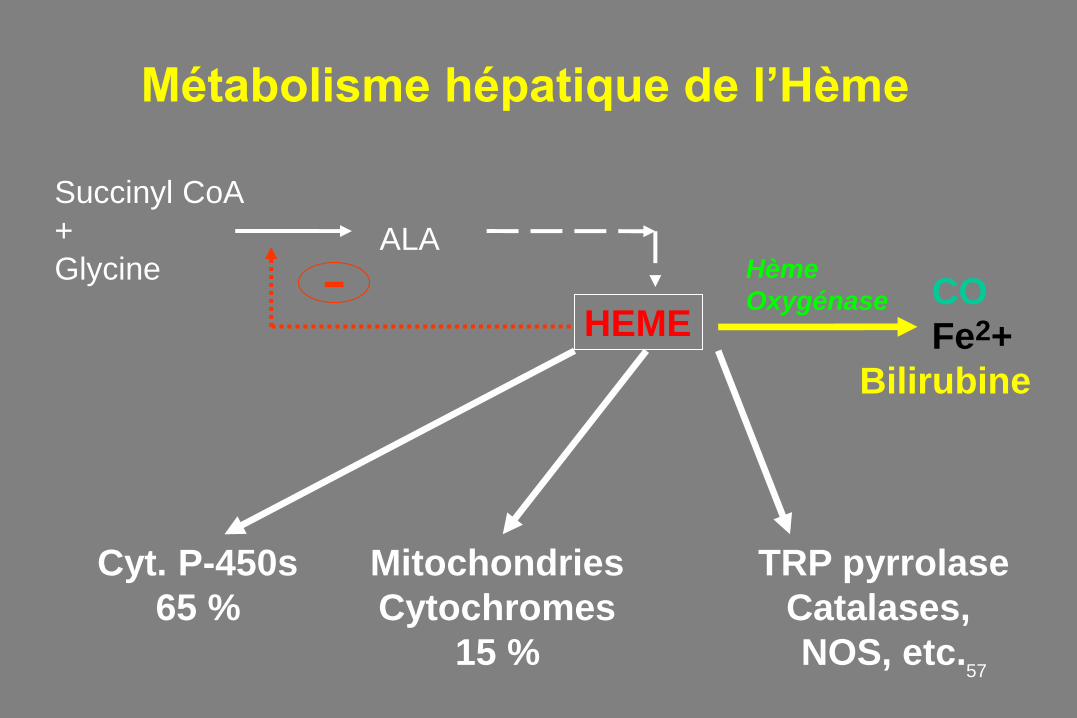
Métabolisme hépatique de l’Hème
Succinyl CoA
+
Glycine
TRP pyrrolase
Catalases,
NOS, etc.
Mitochondries
Cytochromes
15 %
Cyt. P-450s
65 %
HEME CO
Fe2+
Bilirubine
Hème
Oxygénase
ALA
-
57

Maladies héréditaires rares de la biosynthèse de l’hème
Centre Français des Porphyries
CHU Louis Mourier –– Université D. Diderot Paris 7
Les Porphyries
www.porphyrie.net / www.porphyria-europe.org 58

Porphyries
Urines « porto »
Du Grec Porphyre : pigment rouge
59

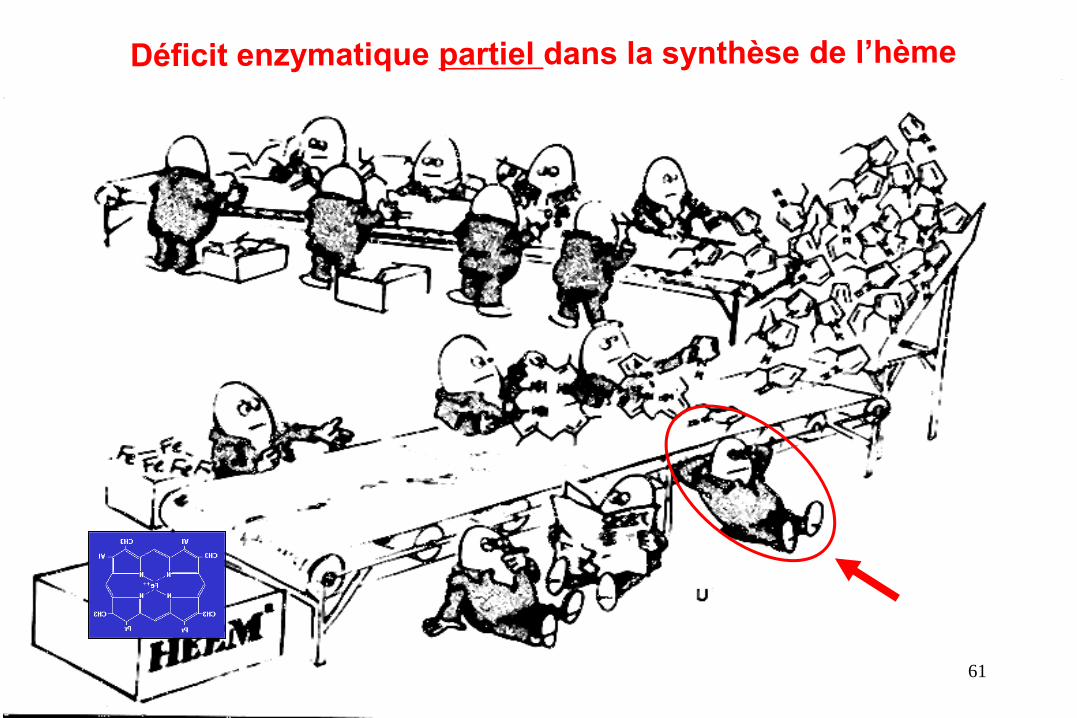
Les Porphyries héréditaires
• 8 Maladies génétiques rares (1/50 000)
• Dues à un défaut partiel des enzymes de la synthèse de l’hème
• Production anormale de porphyrines et/ou des précurseurs
dans le foie et /ou la moëlle (►urines, selles, sang)
• Crises aiguës neuroviscérales intermittentes et/ou signes
cutanés photo induits (dermatoses bulleuses ou algiques)
• Pénétrance faible et expression clinique variable
• Facteurs déclenchants exogènes et endogènes
…du grec « porphyre » : pigment rouge 60
61
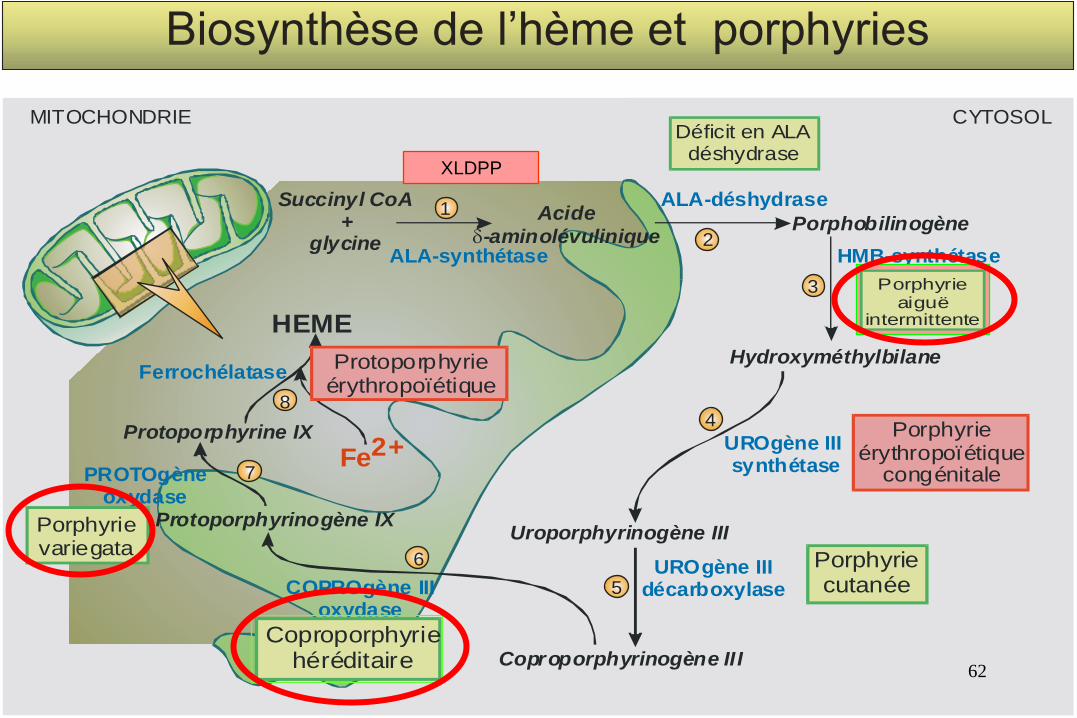
ALA-déshydrase
ALA-synthétase HMB-synthétase
UROgène III synthétase
UROgène III décarboxylaseCOPROgène III
oxydase
PROTOgèneoxydase
Ferrochélatase
Porphobilinogène Acide
-aminolévulinique
Hydroxyméthylbilane
Succinyl CoA+
glycine
Uroporphyrinogène III
Coproporphyrinogène III
Protoporphyrinogène IX
Protoporphyrine IX
MITOCHONDRIE CYTOSOL
Fe2+
HEME
8
7
6
4
3
2
1
5
Déficit en ALAdéshydrase
Porphyrieérythropoïétique
congénitale
Porphyriecutanée
Coproporphyriehéréditaire
Porphyrievariegata
Protoporphyrieérythropoïétique
Biosynthèse de l’hème et porphyries
Porphyrieaiguë
intermittente
XLDPP
62

Classification des principales Porphyries
Porphyries hépatiques (A. dominantes)
Porphyrie aiguë intermittente PAI
Coproporphyrie héréditaire CH
Porphyrie variegata PV
Porphyrie cutanée tardive PCT
Porphyries érythropoïétiques
Porphyrie érythropoïétique congénitale PEC
Protoporphyrie érythropoïétique PPE
Protoporphyrie dominante liée à l’X
Aiguës
63

Porphyries : les symptômes
Crises aiguës « neurologiques », liées à l'accumulation des précurseurs (ALA,PBG)
et/ou au manque d’hème
Seulement les Porphyries hépatiques aiguës :
PAI, CH, PV
Lésions cutanées, induites par le rayonnement solaire
(photosensibilité), dues à l'accumulation de
porphyrines dans la peau
Toutes les porphyries, sauf la PAI
64

Melle C…, 19 ans, (Mère: Infirmière)
Janv 1998 : Hôpital Louis Mourier,
Service des Urgences Chirurgie :
- Douleurs abdominales +++, nausées ++, vomissements ++
- Examen clinique négatif sauf tachychardie isolée
- Radio abdomen négative, Échographie négative
- Biologie normale…Na : 133
- Aucun ATCD familial notable
- Pas de réel diagnostic de sortie : pancréatite médic.(paracetamol)? / kyste ovarien? /
«agitation nerveuse»
- hospitalisation « écourtée » en chirurgie après tt intempestif par Haldol
Pendant les 3 mois suivants : plusieurs autres hôpitaux de la région N-O
- Perte de 10 kilos
- Douleurs abdominales récurrentes, intenses, sur plusieurs jours, en
période lutéale +++, uniquement soulagées par les opiacés
- Pas de diagnostic sinon histrionisme et/ou toxicomanie,
Porphyrie hépatique aiguë
65

Melle C., Suite
Avril 1998 : Hôpital Beaujon service de gynéco-obstétrique
- douleurs abdominales intenses +++
- examen clinique et imagerie négatifs
- Cœlioscopie exploratrice : RAS, petit kyste ovaire dte
- Porphyrie aiguë enfin évoqué par un jeune interne
- Envoi d’urines au CFP : ALA, PBG
diagnostic de Porphyrie Aiguë confirmé
• Traitement par Arginate d’hémine : Normosang®
• résolution de la crise en 4 jours,
• diagnostic biologique de Porphyrie Aiguë Intermittente (PAI)
• évolution : crises récurrentes chroniques
66
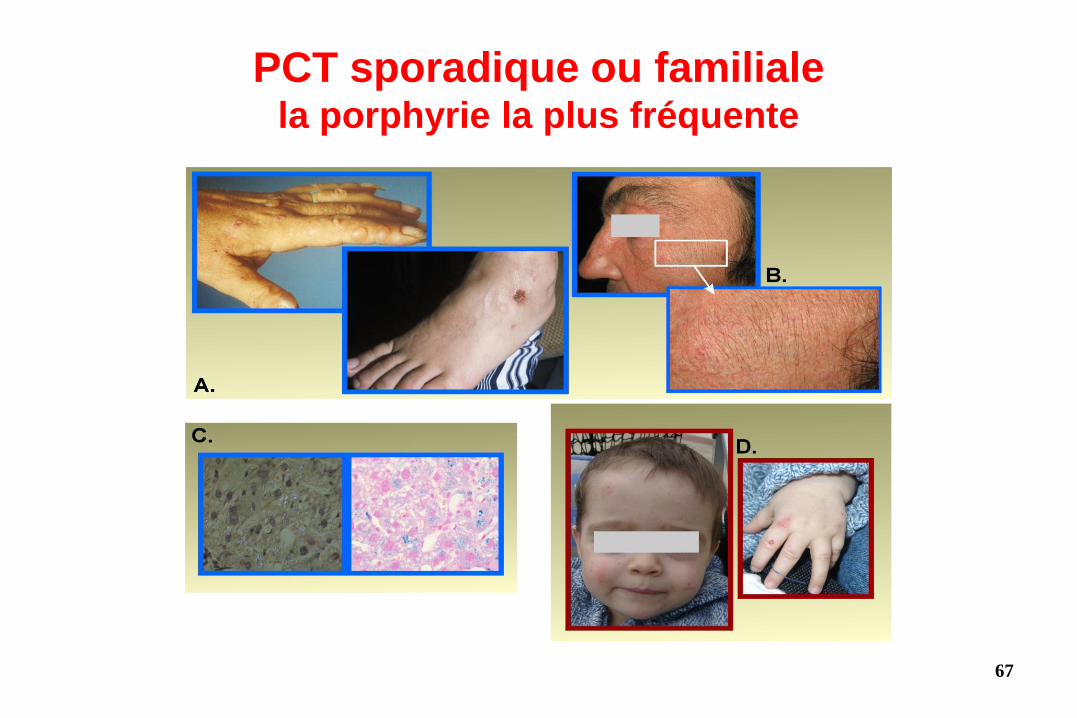
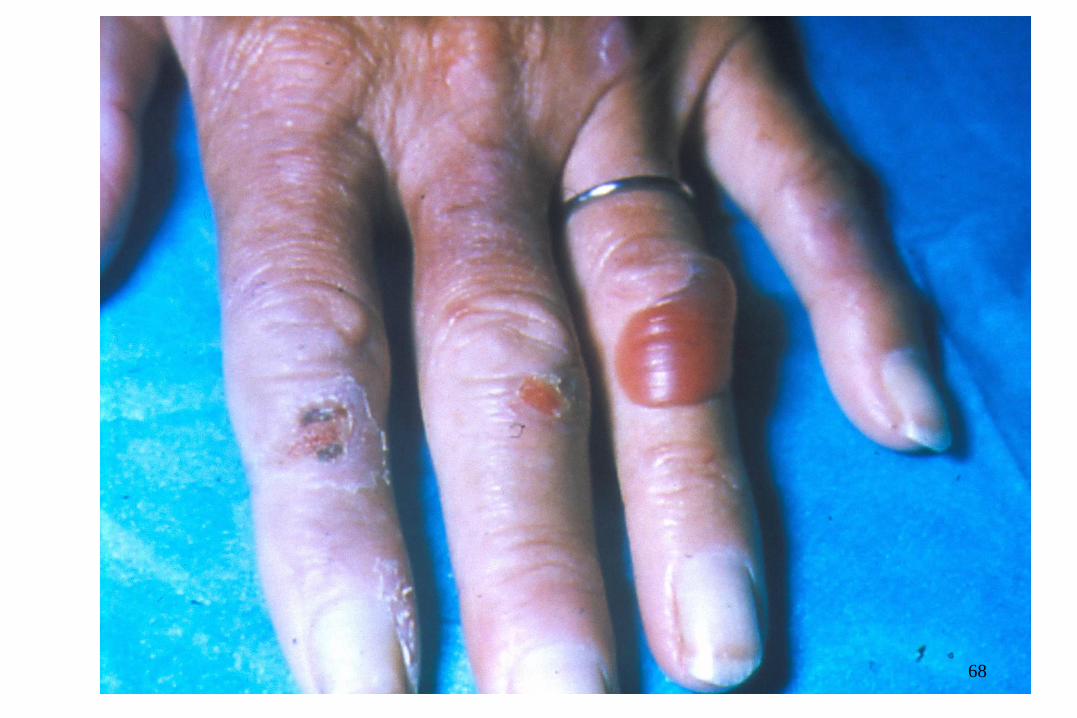
PCT sporadique ou familiale la porphyrie la plus fréquente
67
68
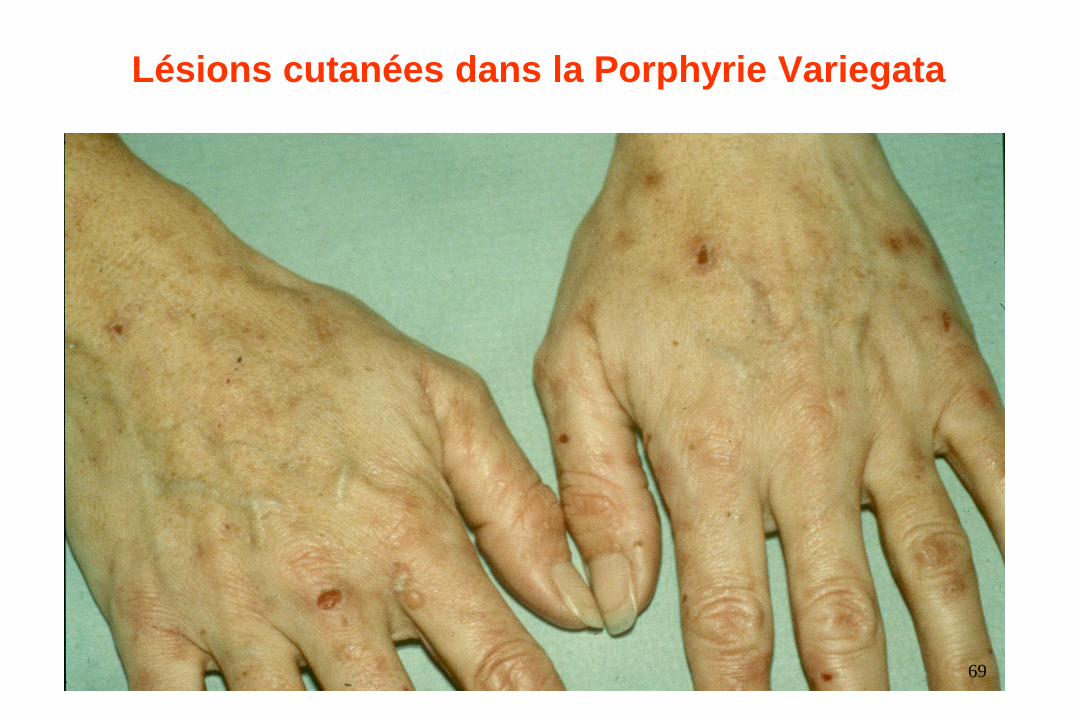
Lésions cutanées dans la Porphyrie Variegata
69

70

Les Porphyries Erythropoïétiques
• Porphyrie Erythropoïétique Congénitale (Maladie de Günther) – Autosomique et récessive : 2 allèles UROS mutés
• Protoporphyrie Erythropoïétique – Pseudo dominante : Un allèle FECH muté + un allèle FECH
hypomorphe
• Protoporphyrie erythropoïétique dominante liée à l’X
71
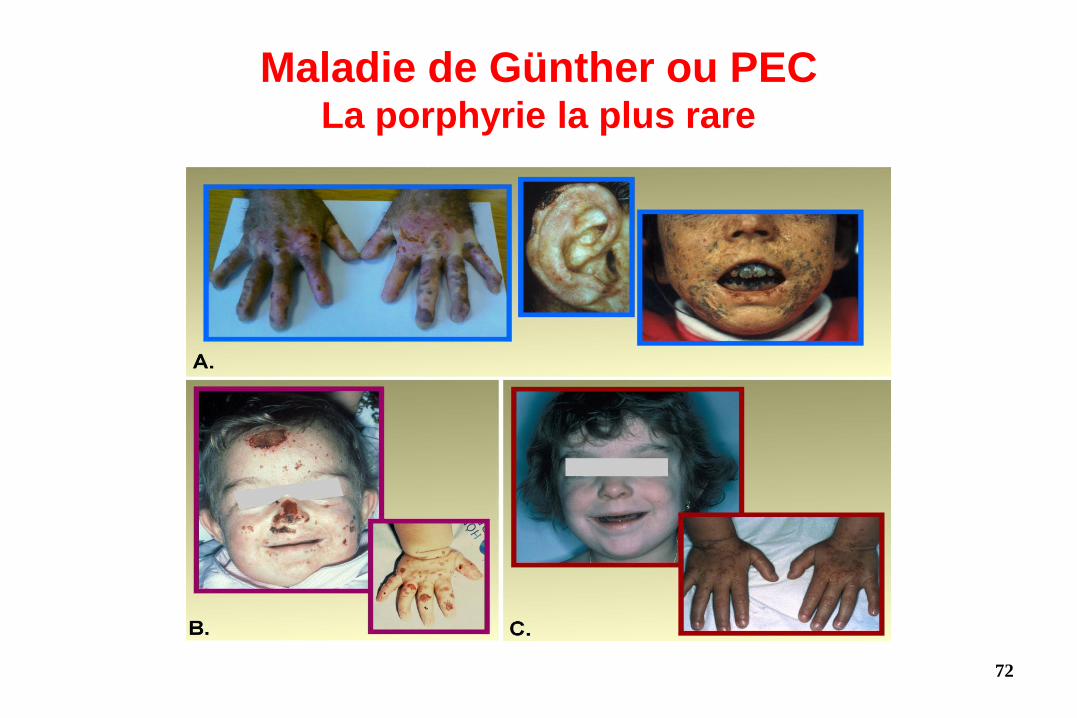
Maladie de Günther ou PEC La porphyrie la plus rare
72
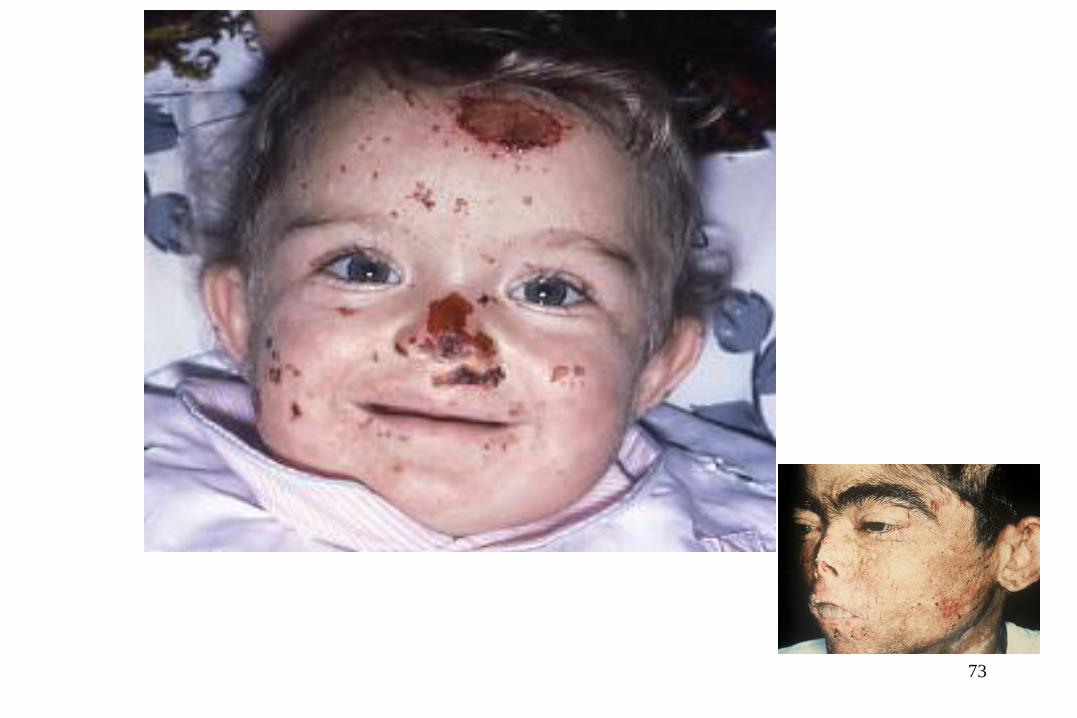
73

Après greffe de moelle
74
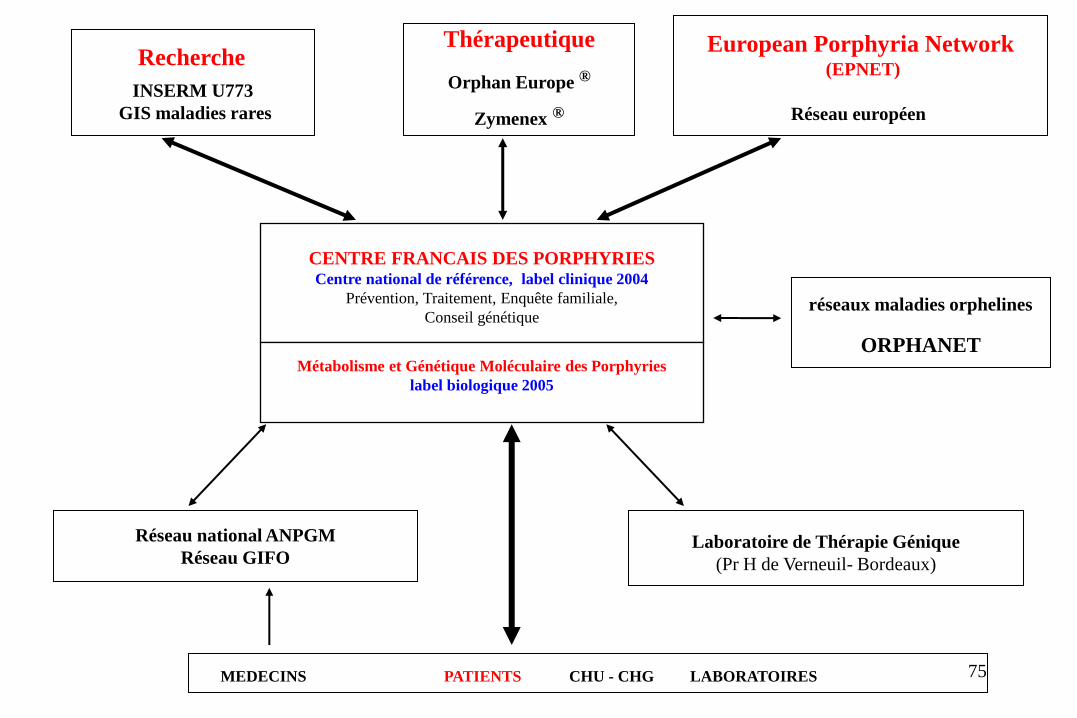
Réseau national ANPGM
Réseau GIFO
Laboratoire de Thérapie Génique
(Pr H de Verneuil- Bordeaux)
CENTRE FRANCAIS DES PORPHYRIES Centre national de référence, label clinique 2004
Prévention, Traitement, Enquête familiale,
Conseil génétique
Métabolisme et Génétique Moléculaire des Porphyries label biologique 2005
réseaux maladies orphelines
ORPHANET
Recherche
INSERM U773
GIS maladies rares
Thérapeutique
Orphan Europe ®
Zymenex ®
European Porphyria Network (EPNET)
Réseau européen
MEDECINS PATIENTS CHU - CHG LABORATOIRES
75

76

77
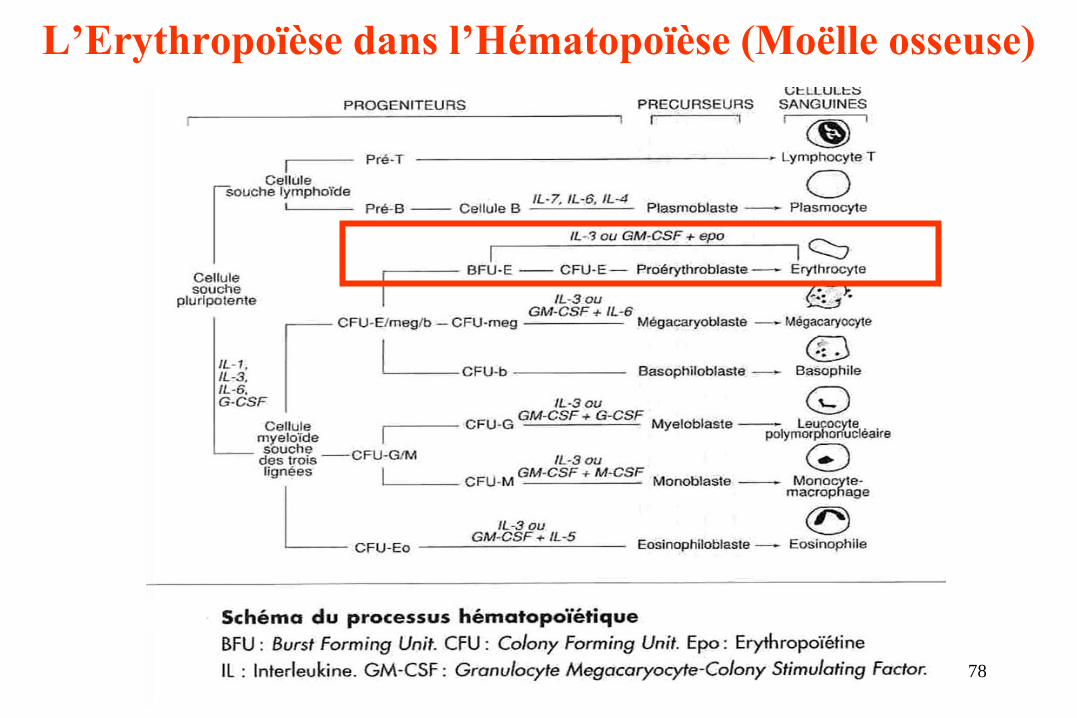
L’Erythropoïèse dans l’Hématopoïèse (Moëlle osseuse)
78

79

Facteurs limitants de l’érythropoïèse
• Erythropoïétine (Rein)
• Synthèse d’ADN ► dTTP ► Thymidilate Kinase ►Vit B12 et
Ac Folique
• Synthèse d’Hémoglobine ► Globine + Hème
• Synthèse d’Hème = protoporphyrine + Fe2+ ► Fer
80

Prolongements cliniques : Les anémies
• Anémies carencielles:
Carence en Fer (anémie microcytaire)
carence en Vit B12 / Folates; déficit en EPO
• Anémies hémolytiques: • Anomalies corpusculaires congénitales: :
- de la membrane GR : sphérocytose, elliptocytose…
- de l’Hémoglobine : drépanocytose, thalassémies…
- déficit enzymatique énergétique: G6PDH, pyruvate kinase, GSH-
synthétase…
• Anomalies extracorpusculaires acquises::: infections parasitaires
(paludisme)…
81
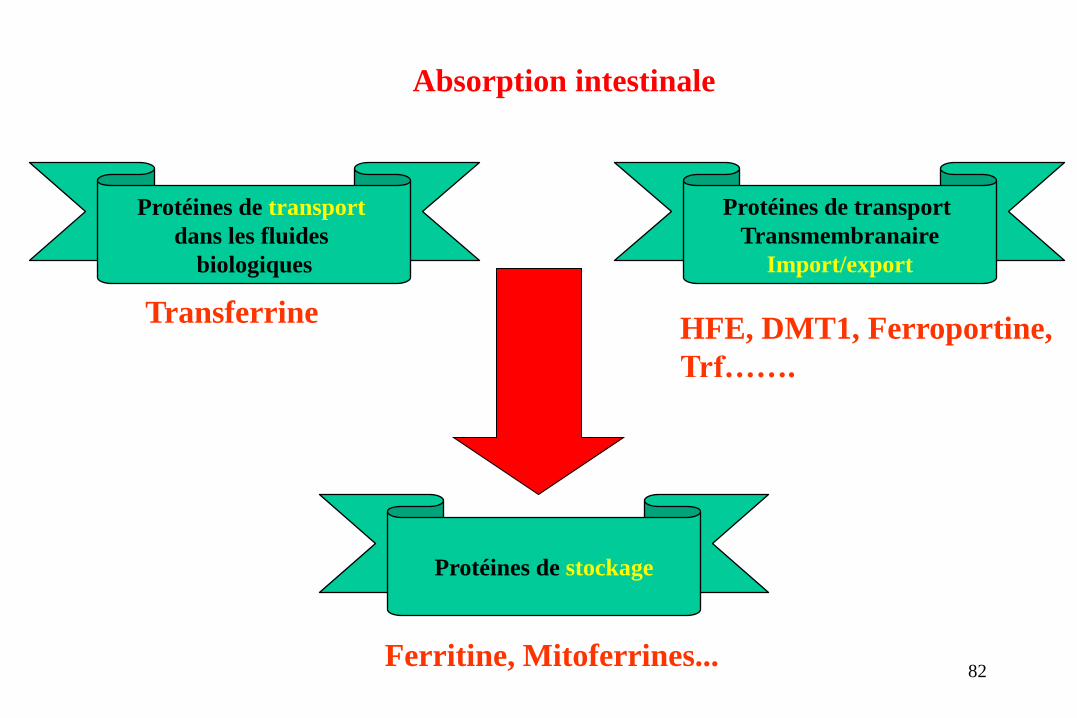
Protéines de transport
dans les fluides
biologiques
Protéines de transport
Transmembranaire
Import/export
Protéines de stockage
Transferrine HFE, DMT1, Ferroportine,
Trf…….
Ferritine, Mitoferrines...
Absorption intestinale
82
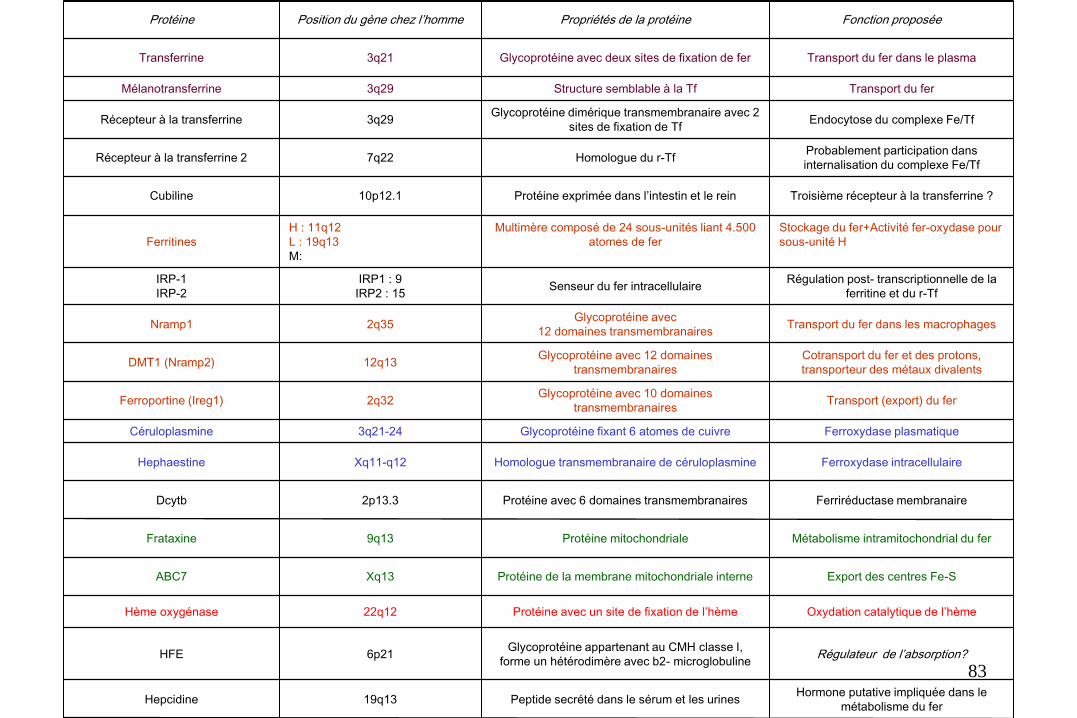
Régulateur de l’absorption? Glycoprotéine appartenant au CMH classe I,
forme un hétérodimère avec b2- microglobuline 6p21 HFE
Transport du fer dans les macrophages Glycoprotéine avec
12 domaines transmembranaires 2q35 Nramp1
Hormone putative impliquée dans le
métabolisme du fer Peptide secrété dans le sérum et les urines 19q13 Hepcidine
Oxydation catalytique de l’hème Protéine avec un site de fixation de l’hème 22q12 Hème oxygénase
Export des centres Fe-S Protéine de la membrane mitochondriale interne Xq13 ABC7
Métabolisme intramitochondrial du fer Protéine mitochondriale 9q13 Frataxine
Ferriréductase membranaire Protéine avec 6 domaines transmembranaires 2p13.3 Dcytb
Ferroxydase intracellulaire Homologue transmembranaire de céruloplasmine Xq11-q12 Hephaestine
Ferroxydase plasmatique Glycoprotéine fixant 6 atomes de cuivre 3q21-24 Céruloplasmine
Transport (export) du fer Glycoprotéine avec 10 domaines
transmembranaires 2q32 Ferroportine (Ireg1)
Cotransport du fer et des protons,
transporteur des métaux divalents
Glycoprotéine avec 12 domaines
transmembranaires 12q13 DMT1 (Nramp2)
Régulation post- transcriptionnelle de la
ferritine et du r-Tf Senseur du fer intracellulaire
IRP1 : 9
IRP2 : 15
IRP-1
IRP-2
Stockage du fer+Activité fer-oxydase pour
sous-unité H
Multimère composé de 24 sous-unités liant 4.500
atomes de fer
H : 11q12
L : 19q13
M:
Ferritines
Troisième récepteur à la transferrine ? Protéine exprimée dans l’intestin et le rein 10p12.1 Cubiline
Probablement participation dans
internalisation du complexe Fe/Tf Homologue du r-Tf 7q22 Récepteur à la transferrine 2
Endocytose du complexe Fe/Tf Glycoprotéine dimérique transmembranaire avec 2
sites de fixation de Tf 3q29 Récepteur à la transferrine
Transport du fer Structure semblable à la Tf 3q29 Mélanotransferrine
Transport du fer dans le plasma Glycoprotéine avec deux sites de fixation de fer 3q21 Transferrine
Fonction proposée Propriétés de la protéine Position du gène chez l’homme Protéine
83
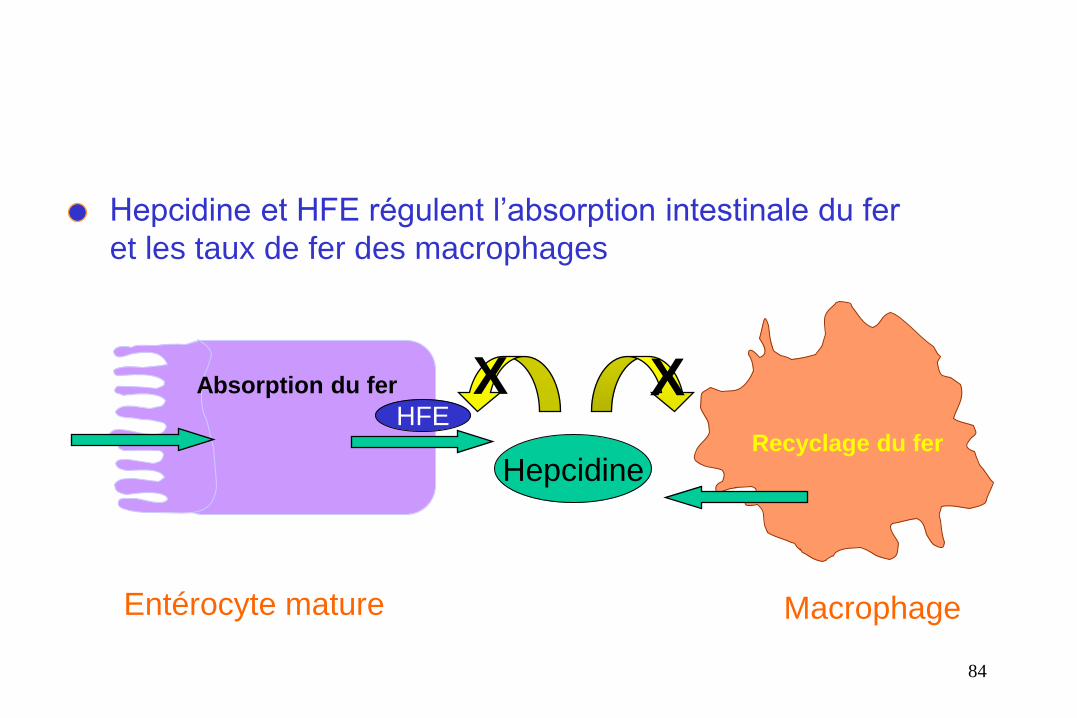
Hepcidine et HFE régulent l’absorption intestinale du fer
et les taux de fer des macrophages
Absorption du fer
Recyclage du fer
Entérocyte mature Macrophage
Hepcidine
X X HFE
84