monoxyde d’azote et homéostasie cardiovasculaire v3.pdf · no- monoxyde d’azote et...
TRANSCRIPT

Monoxyde d’azote et homéostasie cardiovasculaire

2 NO - Monoxyde d’azote et homéostasie cardiovasculaire
Copyright © 2005Menarini France
1, Rue du Jura - Silic 52894633 Rungis Cedex
Tél. : 01 45 60 77 20Fax : 01 46 87 94 31
Conception graphique et réalisation :Samoa - Versailles
Monoxyde d’azoteet homéostasie cardiovasculaire
Jean-Baptiste MichelXxxxxxxYyyyy
&
Christophe HeymesXxxxxxxYyyyy

3NO - Monoxyde d’azote et homéostasie cardiovasculaire
SommaireIntroduction et historique 4 - 5
Structure fonction du système cardiovasculaire 6Stucture-fonction du cœur 6
• Mécanique des cardiomyocytes : origine moléculaire de la contraction 8• Rôle du calcium dans la contraction/couplage excitation-contraction 9• Le cœur en tant que pompe 10• La Loi de Starling et la courbe pression/volume 11• La mesure des vitesses de contraction et de relaxation chez l’homme 11• Le rôle des vaisseaux 11
Structure-fonction artérielle 12• Compartimentation structurale et fonctionnelle de la paroi artérielle 12• Rôle des grandeurs hémodynamiques 13
Bases tissulaires et moléculaires del’endothélium-dépendance de la vasorelaxation 15
Tonus vasomoteur et définition de la pression artérielle 15• Signalisation intracellulaire du tonus vasomoteur 15
Les NO synthases 16
Régulation de la NO synthase endothéliale 18• Calcium-dépendance 18• Phosphorylations 18
Cibles moléculaires du NO 19• La guanylate cyclase soluble 19• Localisation et action des phosphodiestérases 19• Second messager : GMP cyclique 20• Cible du GMP cyclique : les G-kinases 21• NO au-delà de la réponse vasomotrice 24• Autres interactions de NO 25
Pharmacologie du NO 27Agonistes du système NO 27
• Les dérivés nitrés 27• Molécules activant la sécrétion de NO 28
- Système peptidomimimétique et endothélium 28- Système adrénergique et endothélium 29
• Potentialisateurs de NO 29
Antagonistes du système NO 30• Les arginines substituées 30• Antagonistes de NO 32
Rôle physiologie du monoxyde d’azote 33NO et vaisseaux : effet vasodilatateur 33
• Adaptation à l’effort 33• NO comme neuromédiateur périphérique : exemple de l’érection 34• NO synthases et rein 35
NO synthases et cœur 36• Rôle physiologique des donneurs de NO dans le cœur sain 36• Rôle physiologique du NO endogène dans le cœur sain 38• Les NOS du myocyte et le rôle autocrine du NO 38• NOS3 et le rôle paracrine du NO 39
Rôle du monoxyde d’azote en pathologie cardiovasculaire 40Pathologie par excès de NO : le choc septique 40
Pathologies dans lesquelles un défaut de NO est impliqué 40• L’hypertension artérielle 40• NO et insuffisance cardiaque 43
- Insuffisance cardiaque 43- NO vasculaire et insuffisance cardiaque 43- NO myocardique et insuffisance cardiaque 44
Le remodelage vasculaire 45
Monoxyde d’azote et athérosclérose 46
Conclusion 47
Bibliographie 47

4 NO - Monoxyde d’azote et homéostasie cardiovasculaire
Monoxyde d’azote et homéostasie cardiovasculaire
Le monoxyde d’azote ou NO a envahi le domaine designalisation cellulaire et des communicationsintercellulaires dans la biologie de nombreux sys-
tèmes. Le nombre de publications consacrées au sujeten témoigne : 55 articles étaient consacrés à NO en1985, toutes disciplines confondues, 5.000 en 1994.L’intérêt croissant porté à cette molécule simple par lesbiologistes de tous les horizons, l’a fait désigner molé-cule de l’année par la revue internationale “Science”en 1992. Enfin, le NO a valu à ses découvreurs, RobertF. Furchgott, Ferid Murad et Luis J. Ignarro, le prixNobel en 1998. Robert Furchgott a démontré l’endothé-lium-dépendance de la vasodilatation artérielle en1980 ; Ferid Murad a démontré à partir de 1977 que lesdérivés nitrés et en particulier la nitroglycérine étaientvasodilatateurs en relarguant du monoxyde d’azote eten activant la guanylate cyclase dans l’organisme invivo ; enfin Louis Ignarro démontre que le facteur deFurchgott relargué par l’endothélium n’est autre que lemonoxyde d’azote mis en évidence par Murad. A cestrois prix Nobel, il faut ajouter les travaux de SalvatorMoncada qui a identifié la NO synthase endothélialecomme l’enzyme clée productrice de monoxyded’azote dans l’endothélium.
NO est un agent médiateur dans le système immuno-inflammatoire, le système nerveux central et le sys-tème cardiovasculaire. NO est une molécule trèsancienne, présente dans la nature depuis plusieurscentaines de millions d’années et parfaitement conser-vée à travers les millions d’années de l’évolution desespèces biologiques. C’est une molécule très labiledont la demi-vie est de quelques secondes. Dans cesystème cardiovasculaire NO joue un rôle primordialde communication entre les différents types cellulairesconstituant et régulant l’homéostasie cardiovasculaire,et de signalisation spécifique dans certaines des cellu-les le constituant. En physiologie, NO est constammentimpliquée dans l’adaptation du système vasculaire àl’augmentation de besoins métaboliques périphériquesque représentent l’exercice physique pour les vais-seaux à destinée musculaire, la digestion pour les vais-seaux mésentériques, l’activité intellectuelle pour lesvaisseaux cérébraux… En pathologie, un excès de NOest responsable de l’état de choc vasoplégique en rap-port avec une vasodilatation périphérique excessive.Un défaut de NO pourrait participer à de nombreuxphénomènes tels que l’hypertension artérielle, le vieil-lissement, l’athérosclérose…
De l’implication de NO en physiologie et en pathologiecardiovasculaire naît l’intérêt thérapeutique qu’il pour-rait y avoir à moduler l’expression et/ou l’activité decette molécule par des agents pharmacologiques.
Introduction

5NO - Monoxyde d’azote et homéostasie cardiovasculaire
HistoriqueComme souvent en sciences, l’historique de ladécouverte des rôles de NO en biologie s’est faitprogressivement à travers l’association fortuited’informations provenant de domaines différents.C’est à partir de 1980 que vont converger cesdifférentes informations vers la découverte deNO et des différentes enzymes capables de leproduire dans les organismes vivants. A partir delà, les applications de NO à la physiologie et lapathologie des différents systèmes va se dévelop-per exponentiellement dans les années 90.
Cette convergence viendra des trois grandes dis-ciplines où à posteriori, NO est majoritairementimpliquée :
1) de la corrélation entre la production endogènede nitrate et l’activité cytotoxique du systèmeinflammatoire ;
2) de la mise en évidence du mode d’action vasodilatateur des dérivés nitrés et de la découverte de la fonction vasodilatatrice de l’endothélium ;
3) des recherches sur la nature non-adrénergiqueet non-cholinergique de certaines neurotransmissions.
Au début du 20ème siècle, une production endo-gène de nitrate avait été montrée dans l’orga-nisme humain. L’excrétion urinaire de nitrate estsupérieure à l’apport alimentaire chez l’homme.En l’absence de nitrate dans les apports alimen-taires, il en persiste une excrétion urinaire nonnégligeable. L’observation fut également faited’une augmentation d’excrétion de nitrate en casd’infection. Expérimentalement, l’injectiond’endotoxine bactérienne s’accompagne d’uneaugmentation d’excrétion urinaire de nitrate ;des souris génétiquement déficientes en activitémacrophagique ont un taux extrêmement basd’excrétion urinaire de nitrate. In vitro, la stimula-tion des macrophages par des lippolysaccharides(LPS), fragment membrane de bactéries, produitun efflux de nitrate dans le milieu de culture, etcette production est dépendante de l’ajoûtd’arginine au milieu. Enfin, fût faite l’observationque le pouvoir toxique des macrophages sur lescellules tumorales, les bactéries, les champignons,dépend de la présence d’arginine, s’accompagned’une production de citrulline et peut être blo-qué par des dérivés substitués de l’arginine telque la L-Nitro-arginine.
Dès la deuxième partie du 19ème siècle, un effetbénéfique de la nitroglycérine avait été observédans l’angine de poitrine. C’est en 1867, qu’unmédecin écossais Thomas Brunton rapportaitl’efficacité de dérivés nitrés à traiter l’angine depoitrine (sédation des douleurs et baisse de lapression artérielle). Alfred Nobel, inventeur dela dynamite et fondateur du prix du même nom,utilisant le pouvoir anthropique de la nitroglycé-rine dans la mise au point d’explosif, souffrantlui-même d’angine de poitrine, écrivait à un deces amis : “Je perçois comme une ironie au faitque mon médecin me prescrive de la nitroglycé-rine à usage interne”. Un siècle plus tard, lepouvoir vasodilatateur de la nitroglycérine étaitreconnu. Dans les années 80, l’équipe deNeedleman et d’Ignarro montrait que la nitrogly-cérine n’était pas directement active, que lanitroglycérine produisait des nitrates qui eux-mêmesn’étaient pas directement vasodilatateurs maisqu’une molécule intermédiaire, très labile, quine fut pas identifiée comme NO à cette date,était responsable de l’effet vasodilatateur.
En parallèle, c’est en 1980 que fut découvert parFurchgott et Zawadzski la fonction vasodilatatricede l’endothélium. Jusqu’à cette époque, ondécrivait un tonus vasoconstricteur principalementlié à la production de catécholamines par les ter-minaisons du système nerveux sympathique,interagissant avec des récepteurs sur les cellulesmusculaires lisses vasculaires. L’angiotensine IIavait les mêmes effets. La vasodilatation s’expli-quait par une diminution de ce tonus constricteur.D’autres molécules telles que l’acétylcholine,injectées par voie intraveineuse chez l’animal, se
Avant abrasion
NE - 8
-7,5
-7,4
ACh - 8
- 7
- 6
- 5- 4
Après abrasion
NE- 7,5
ACh - 7- 6 - 5
- 4
- 6
Figure 1

6 NO - Monoxyde d’azote et homéostasie cardiovasculaire
comportaient comme de puissants vasodilatateurssans que l’on en comprenne le mécanisme phar-macologique d’action.Furchgott et Zawadzki montrèrent le rôle obliga-toire de l’endothélium dans cette réponse et dece fait montrèrent une communication entre cel-lules endothéliales et cellules musculaires lissesautres que la production de prostaglandines.Leur expérience fut simple (figure 1) : un anneauaortique est précontracté par la norépinéphrine(catécholamine contractant directement la cellulemusculaire lisse via un récepteur α-1) ; en présenced’un endothélium intact, l’acétylcholine induit unerelaxation dose-dépendante de l’anneau ; quandl’endothélium est abrasé, l’acétylcholine induitune contraction supplémentaire. Là encorel’agent intermédiaire entre l’endothélium et lescellules musculaires lisses ne put être immédia-tement identifié et il fut appelé EDRF (pourEndothelium Dependent Relaxing Factor).Il fut ensuite montré que l’action de l’EDRF étaitpharmacologiquement bloquée par l’hémoglobine,et potentialisée par la superoxyde dismutase(SOD).
Il fut rapidement montré que l’EDRF agissait viala génération de GMP cyclique dans la cellulemusculaire lisse et que ce phénomène médiaitles effets vasodilatateurs de diverses molécules :bradykinine, histamine, nucléotides adényliques,thrombine, substance P, 5-hydroxytryptamine,sérotonine…
Ce n’est qu’en 1987-1988 que Palmer etMoncada (figure 2) montrèrent que l’EDRF et NOétaient une seule et même molécule, produitepar l’endothélium à partir de l’arginine. Ilsidentifièrent la production de NO par chémolu-ninescence. Ils montrèrent que le monoxyded'azote modulait puissamment les grandeurshémodynamiques de pression et de flux sanguin.Les différentes enzymes capables de produire NOà partir de l’arginine furent ensuite rapidementidentifiées dans les différents systèmes nerveuxcentraux, macrophage, endothélium. NO fut alorségalement reconnu comme un neurotransmetteur.
Nitro-argininearginine…..
…
.
NOPression
Nitro- arg Arginine
Flux
Nitro- arg Arginine
100
50
Figure 2

7NO - Monoxyde d’azote et homéostasie cardiovasculaire
Stucture-fonctiondu cœur
Le cœur est composé de deux parties fonction-nellement et anatomiquement distinctes, le cœurdroit et le cœur gauche (figure 3). Chacune deces deux parties est elle-même subdivisée enoreillette et ventricule. Les deux parties du cœurfonctionnent bien entendu de façon synchrone,mais le ventricule droit est moins musclé que legauche car les résistances pulmonaires sont plusfaibles que les résistances périphériques. La
circulation sanguine a un sens et va du coeurgauche au coeur droit et il existe entre les diffé-rentes parties de la circulation un système devalves qui empêche le sang de refluer là d'où ilvient. Les oreillettes et les ventricules sont séparésles uns des autres par des valves qui s'ouvrentvers la cavité ventriculaire et empêche le refluxdans les oreillettes. Ces valves sont fibreuses,
non musculaires, mais sont rattachées à despiliers musculaires qui en assurent l'ouverturesynchrone. Les valves en nid-de-pigeon des orificespulmonaires et aortiques empêchent le reflux dusang artériel dans les ventricules. Le cœur est un organe comme les autres et possèdecomme tout organe une vascularisation propreavec des artères et des veines dites coronaires,mais les artères du cœur, les artères coronairessont les premières de l'arbre vasculaire artériel,et les veines, collectées par le sinus coronaireles dernières du système veineux. Les artèrescoronaires prennent naissance dans le creux desvalves sigmoïdes aortiques antérieures, circulent
à la surface du myocarde(sur l'épicarde) et plon-gent dans l'épaisseur dumuscle qu'elles irriguent,donc de l'extérieur versl'intérieur. Les veinescoronaires sont collectéesdans une sorte de vastepoche, appellée sinuscoronaire, qui s'ouvredans l'oreillette droite. Les cavités cardiaquessont tapissées par unendothélium appelléendocarde qui joue unrôle fonctionnel impor-tant. Les cavités ventricu-laires proprement ditessont faites d'anfractuositéscomplexes qui forment unréseau musculaire trabécu-laire ; les cavités desoreillettes sont sillonnéesde fins muscles appellésmuscles pectinés. La portionsuperficielle du myocarde,ou épicarde, est recou-verte d'un sac étanche
appellé péricarde, fait de tissu conjonctif. A lacoupe le myocarde ventriculaire apparaît forméde travées fibreuses en spirales, ou en “corde demontagne”, orientées selon un axe dirigé en baset à gauche, cet axe est le véritable axe de forcedu cœur.Le cœur ne possède ni innervation motrice, niinnervation sensitive, mais il possède une inner-
Structure-fonctiondu système cardio-vasculaire
Veine cave supérieure
Artère pulmonaire
Veine pulmonairedroite
Oreillette droite
Oreillette gauche
Veine pulmonaire gauche
Tronc pulmonaire
Artère pulmonaire
Crosse aortique
Veine cave inférieurAorte
Ventricule gauche
Ventriculedroit
Figure 3
8 NO - Monoxyde d’azote et homéostasie cardiovasculaire
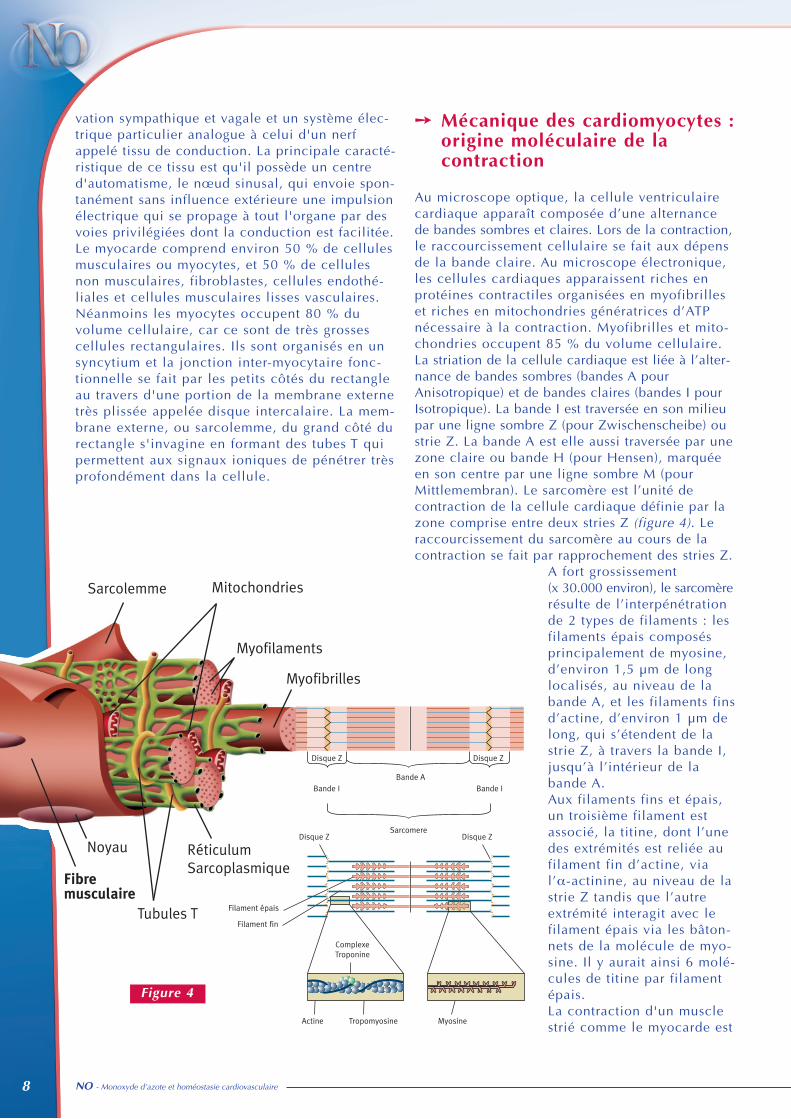
vation sympathique et vagale et un système élec-trique particulier analogue à celui d'un nerfappelé tissu de conduction. La principale caracté-ristique de ce tissu est qu'il possède un centred'automatisme, le nœud sinusal, qui envoie spon-tanément sans influence extérieure une impulsionélectrique qui se propage à tout l'organe par desvoies privilégiées dont la conduction est facilitée.Le myocarde comprend environ 50 % de cellulesmusculaires ou myocytes, et 50 % de cellulesnon musculaires, fibroblastes, cellules endothé-liales et cellules musculaires lisses vasculaires.Néanmoins les myocytes occupent 80 % duvolume cellulaire, car ce sont de très grossescellules rectangulaires. Ils sont organisés en unsyncytium et la jonction inter-myocytaire fonc-tionnelle se fait par les petits côtés du rectangleau travers d'une portion de la membrane externetrès plissée appelée disque intercalaire. La mem-brane externe, ou sarcolemme, du grand côté durectangle s'invagine en formant des tubes T quipermettent aux signaux ioniques de pénétrer trèsprofondément dans la cellule.
➙ Mécanique des cardiomyocytes :origine moléculaire de lacontraction
Au microscope optique, la cellule ventriculairecardiaque apparaît composée d’une alternancede bandes sombres et claires. Lors de la contraction,le raccourcissement cellulaire se fait aux dépensde la bande claire. Au microscope électronique,les cellules cardiaques apparaissent riches enprotéines contractiles organisées en myofibrilleset riches en mitochondries génératrices d’ATPnécessaire à la contraction. Myofibrilles et mito-chondries occupent 85 % du volume cellulaire. La striation de la cellule cardiaque est liée à l’alter-nance de bandes sombres (bandes A pourAnisotropique) et de bandes claires (bandes I pourIsotropique). La bande I est traversée en son milieupar une ligne sombre Z (pour Zwischenscheibe) oustrie Z. La bande A est elle aussi traversée par unezone claire ou bande H (pour Hensen), marquéeen son centre par une ligne sombre M (pourMittlemembran). Le sarcomère est l’unité decontraction de la cellule cardiaque définie par lazone comprise entre deux stries Z (figure 4). Leraccourcissement du sarcomère au cours de lacontraction se fait par rapprochement des stries Z.
A fort grossissement(x 30.000 environ), le sarcomèrerésulte de l’interpénétrationde 2 types de filaments : lesfilaments épais composésprincipalement de myosine,d’environ 1,5 µm de longlocalisés, au niveau de labande A, et les filaments finsd’actine, d’environ 1 µm delong, qui s’étendent de lastrie Z, à travers la bande I,jusqu’à l’intérieur de labande A. Aux filaments fins et épais,un troisième filament estassocié, la titine, dont l’unedes extrémités est reliée aufilament fin d’actine, vial’α-actinine, au niveau de lastrie Z tandis que l’autreextrémité interagit avec lefilament épais via les bâton-nets de la molécule de myo-sine. Il y aurait ainsi 6 molé-cules de titine par filamentépais.La contraction d'un musclestrié comme le myocarde est
Figure 4
Sarcolemme Mitochondries
Myofilaments
Noyau
Tubules T
RéticulumSarcoplasmique
Myofibrilles
Disque Z
Disque Z
Filament épais
Filament fin
ComplexeTroponine
TropomyosineActine Myosine
Disque Z
Bande ABande I Bande I
Sarcomere
Fibremusculaire
Disque Z

9NO - Monoxyde d’azote et homéostasie cardiovasculaire
due au glissement les uns sur les autres des fila-ments qui composent le sarcomère. Ce glisse-ment n'est possible que si l'ATPase de la myosineest activée, ce qui permet l'hydrolyse de l'ATP etce faisant (i) libère l'énergie nécessaire à lacontraction et (ii) diminue la concentration intra-cellulaire en ATP au dessous d'un certain seuil,car l'ATP empêche les relations entre ces fila-ments. Cette activation est déclenchée par uneaugmentation de la concentration intracellulaireen calcium, elle-même sous la dépendance d'uneexcitation électrique. Le couplage excitation-contraction est un phénomène complexe quimet en jeu de très nombreuses protéines mem-branaires.La contraction est un glissement des deux typesde filaments (fins et épais) les uns par rapportaux autres. Le glissement est obtenu par le mou-vement des ponts qui se replient sur eux mêmes,les extrémités distales des filaments épais se rap-prochent ainsi des lignes Z, les extrémités dista-les des filaments minces se rapprochent les unesdes autres et peuvent même, dans les contractu-res extrèmes, se chevaucher. Le glissement n'estpas strictement linéaire car les ponts sont émispar le filament épais par paires et d'une manièrehélicoïdale, de plus le filament épais est dans unberceau de filaments minces. Les filaments épais sont composés de 300 à 400molécules de myosine, molécules en forme decanne de hockey, d'un poids moléculaire élevé(500.000), dont la crosse (c'est la tête de lamolécule) fait issue hors du filament en formantles ponts et dont le manche forme le corps dufilament épais. La myosine est à la fois une protéinede structure et un enzyme. La tête de la moléculenon seulement peut se lier à l'actine en formantun pont, mais aussi posséder une activité enzy-matique ATPasique. L'hydrolyse de l'ATP permetin situ la transduction de l'énergie chimiqueprésente dans la molécule d'ATP en énergiemécanique. Par ailleurs des concentrations élevéesd'ATP dissocient les filaments minces et les filamentsépais et relaxent le muscle. En activant l'ATPasede la myosine, on diminue la concentrationintracellulaire en ATP, ce qui permet aux filamentsde se réassocier. La régulation de la contraction,battement par battement, sera donc celle del'hydrolyse de l'ATP.Le filament mince est composé de moléculesd'actine, protéines globulaires (Poids Moléculaire60.000), arrangées de façon hélicoïdale en deuxlongs polymères autour d'un dimère triplementhélicoïdal, la tropomyosine. L'ensemble ressem-ble à un collier de perles, les perles étant lesmolécules d'actine et le cordon étant la tropo-
myosine. C'est sur ces perles que s'articulent lesponts. Le filament mince supporte également lesystème de régulation qui met en œuvre lacontraction ou la relaxation. Ce système est uncomplexe composé de trois protéines, les tropo-nines I, C et T, ce complexe se trouve disposé surle collier de perles à intervalles réguliers, tousles 40 nm. La troponine T lie le complexe à latropomyosine, cette dernière amplifiera le signalémis par le complexe régulateur et le transmettraà l'ensemble du filament mince. La troponine Ien l'absence de calcium inhibe la contraction etrelaxe le muscle en empêchant la formation desponts. La troponine C fixe le calcium et lèvecette inhibition.
➙ Rôle du calcium dans lacontraction/couplageexcitation-contraction
L'ion calcium joue un rôle central dans le couplageexcitation-contraction. La contraction cardiaquerésulte de la dépolarisation membranaire et del’entrée de Ca2+ par les canaux calciques de type L.La quantité de Ca2+ qui entre dans la cellule n’estpas suffisante pour déclencher directement lacontraction, mais elle entraîne une libération desstocks intracellulaires (“Calcium induced-cal-cium release”) par des canaux de relargage(récepteurs de la ryanodine, RyR2) présents surla membrane du réticulum sarcoplasmique (RS).Ces canaux sont situés dans une zone appeléetriades, en face des canaux L, dans des invagina-tions de la membrane plasmique (tubules T).C'est dans ces zones que naissent les “étincelles”de Ca2+ “calcium sparks”, éléments élémentairesde la sortie de Ca2+ des réservoirs du RS. L'augmentation du Ca2+ libre dans le cytoplasmedéclenche la contraction. L'ion Ca2+ libre se fixesur la troponine C. En l'absence de Ca2+, la tro-ponine I interagit avec la tropomyosine pourempêcher l'actine de s'associer à la myosine.Une fois le Ca2+ lié à la troponine C, un change-ment conformationnel des protéines du complexetroponine I - tropomyosine apparaît. Des ponts seforment entre l'actine et la myosine, permettantle glissement des filaments épais sur les filamentsfins, ce qui engendre la contraction. L'énergie dela contraction est produite par le clivage del'ATP par les sites ATPasiques de la myosine enprésence de Mg2+. La relaxation se produit lorsque le Ca2+ est libérépar la troponine C, puis repompé par la pompecalcique du réticulum (SERCA), dont l'activité estcontrôlée par le phospholamban. L’inhibition

10 NO - Monoxyde d’azote et homéostasie cardiovasculaire
exercée par le phospholamban sur l’activité deSERCA est levée lorsque celui-ci est phosphorylé.Le Ca2+ repompé au niveau des tubules longitudi-naux circule dans le réticulum pour atteindre lesciternes terminales et permettre une nouvellecontraction. En diastole, c'est essentiellementl'échangeur Na+/Ca2+ qui est responsable de lasortie des ions Na+ (3 ions Na+ pour 1 ion Ca2+),et les ions Na+ sont extrudés par la Na+/K+ ATPase(2 ions K+).
➙ Le cœur en tant que pompe
La principale fonction du cœur est d’assurer undébit sanguin et d’engendrer des pressions deperfusion permettant de subvenir aux besoinspériphériques mais aussi à ses propres besoins.La fonction ventriculaire gauche est la plus étu-diée car c’est de son niveau de performance quedépend l’éjection du sang dans l’aorte et donc laperfusion systémique. De plus, au cours des car-diopathies, c’est principalement son atteinte quiest responsable de la morbi-mortalité. On peutschématiquement définir quatre phases dans lecycle cardiaque, à savoir deux lors de la systoleet deux lors de la diastole. D’un point de vueexpérimental et clinique, l’établissement de cou-bes pression-volume permet de bien les illustrer(figure 5).
Phase 1 : la systole commence à la fermeture dela valve mitrale. La valve aortique n’est pasencore ouverte. Le cœur est en fin de diastole(télédiastole) et le remplissage est terminé.L’arrivée de l’onde de dépolarisation déclencheà ce moment la contraction ventriculaire avecaugmentation de la pression dans la cavité sansvariation de son volume : c’est la systole isovolu-mique.
Phase 2 : la contraction se poursuivant, la pressiondans le ventricule devient supérieure à cellerégnant dans l’aorte. Cette différence de pressionprovoque l’ouverture passive mais rapide dessigmoïdes aortiques, marquant le début de laphase d’éjection. A la fin de la contraction, lapression ventriculaire gauche diminue rapidementet la valve aortique se referme lorsque la pressionaortique devient supérieure à celle du ventriculegauche : la diastole commence.
Phase 3 : la valve aortique est fermée et la valvemitrale n’est pas encore ouverte alors que lasystole est terminée. C’est la phase de relaxationisovolumique pendant laquelle la pression baissesans variation de volume. Cette chute de pressionest d’autant plus rapide que le ventricule gaucheest capable de se relâcher facilement, en d’autrestermes que sa distensibilité ou compliance estgrande. La différence de pression qui tend alorsà s’établir de part et d’autre de la valve mitraleprovoque son ouverture, caractérisant la dernièrephase du cycle cardiaque : le remplissage.
Figure 5
Pres
sion
ven
tric
ulai
re g
auch
e (m
mH
g)
Volume ventriculaire gauche (ml)
DiastoleSystole
b : ouverture de la valve aortique
d : ouverture de la valve mitrale
a : télédiastole
c : télésystole
b bc bc
a a
d
1. Contraction isovolumique
2. Ejection 3. Relaxation isovolumique
4. Remplissage
Illustration des quatre grandes phases du cycle cardiaque à partir d’une courbe pression-volume

11NO - Monoxyde d’azote et homéostasie cardiovasculaire
Phase 4 : la phase de remplissage comprend troisétapes successives qui permettent de ramener leventricule à son état télédiastolique initial. Lamajeure partie du remplissage est rapide etsecondaire à un appel de sang provoqué parl’ouverture de la valve mitrale : le sang pénètrealors brusquement dans le ventricule qui setrouve dans un état de relaxation complète et lavalve mitrale se ferme partiellement. Il s’ensuitune étape de remplissage lent qui est raccourcie,voire absente lorsque la fréquence cardiaques’élève. C’est à la fin de cette période, alors quele remplissage du ventricule est presque terminé,que survient la contraction de l’oreillette provoquantune augmentation transitoire et faible de lapression ventriculaire.
➙ La Loi de Starling et la courbe Pression/Volume
Cette relation Pression/Volume représente le travailexterne du myocarde, lequel peut schématiquementaugmenter soit lorsque la quantité de sang àéjecter augmente, c'est le cas lorsque le cœur estdilaté, soit lorsque les résistances à l'éjection dusang sont augmentées, c'est la cas dans les surchargesde pression.La courbe Pression/Volume peut être répétée,chez un même sujet, en variant les conditions decharge. Cette relation porte un nom particulieren physiologie cardiaque et s'appelle loi deStarling. Cette loi est basée sur des expériencestrès simples faites sur cœur isolé, elle dit simple-ment qu'en dehors de toute innervation par lesystème nerveux autonome le cœur va spontané-ment éjecter autant de sang qu'il en reçoit. La loipermet de prédire le volume de sang éjecté àchaque systole pour différentes pressions deremplissage, c'est-à-dire pour différentes pressionsveineuses. Lorsqu'on augmente la pression veineuse,on augmente le volume de sang qui reste à la finde la diastole et l'on dilate la cavité ventriculaire.Le volume qui sera ensuite éjecté à chaquebattement augmentera proportionnellement à lapression veineuse et donc au volume télédiastolique.Les expériences de Starling (et Frank) ont aussiconsisté à faire l'expérience inverse c'est-à-dire àmontrer que le volume télédiastolique augmenteen réponse à une élévation de la pression d'éjectionou du volume éjecté.
➙ La mesure des vitesses decontraction et de relaxation chez l'homme
La vitesse maximale à laquelle s'élève la pressionintraventriculaire pendant la période de contractionisovolumétrique qui sépare la fermeture des valvesmitrales et l'ouverture des valves sigmoïdes aortiquesse mesure couramment en dérivant la pressionpar rapport au temps, le dP/dt max est un bonindex de contractilité mais d'utilité assez limitéecar il dépend de très nombreux paramètres et enparticulier il est directement proportionnel à lapression du VG. Un meilleur indice de contractilité(car indépendant des conditions de post-chargeet pré-charge) réside dans le calcul de l’élastanceventriculaire, égale à la pente de la droite quipasse par tous les points télésystoliques issus descourbes pression-volume. La vitesse de relaxationpeut aussi être évaluée par le dP/dt min, avec lesmêmes réserves que pour le dP/dt max, pendantla phase de relaxation isovolumétrique quisépare la fermeture des valves aortiques etl'ouverture des mitrales.
➙ Le rôle des vaisseaux
Le cœur ne travaille pas isolé, le VG éjecte dans unsystème à haute pression et à faible capacitance, lesystème artériel périphérique, l'oreillette droitepar contre se remplit à partir d'un système à faiblepression et à forte capacitance, le système veineuxqui représente plus de la moitié du volume vas-culaire. Entre les deux se trouve le lit capillairequi représente 1.000 fois le diamètre artériel. Lesystème pulmonaire est un peu particulier puisquela pression de l'artère pulmonaire est relativementfaible.Les grosses artères, et en particulier l'aorte quiest branchée directement sur le VG, opposentpeu de résistance à l'éjection systolique, la zonede résistance se situe beaucoup plus en aval auniveau des artérioles et du sphincter précapillaire.Ceci est très particulier au VG. A droite l'artèrepulmonaire n'oppose pas non plus beaucoup derésistance, mais sa pression est faible car lesartérioles pulmonaires n'ont pas de zone derésistance importante comme leur contrepartiepériphérique. Il ne faut pas confondre ici résistance avec charge.La véritable charge opposée à l'éjection du VGn'est pas constituée par les seules résistancespériphérique mais par l'impédance caractéristi-que de l'aorte qui est un paramètre complexe qui

12 NO - Monoxyde d’azote et homéostasie cardiovasculaire
inclut les résistances, mais aussi l'inertance dusang, la compliance du vaisseau (ou son inversela rigidité) et enfin les ondes en retour. L'éjectionsystolique propulse une onde qui se heurte aufront opposé par les résistances artériolaireslesquelles renvoie une onde en retour qui s'opposeà l'éjection systolique. Lors du vieillissement enl'absence de toute hypertension artérielle (quiaugmente les seules résistances artériolaires)l'impédance aortique, et donc la charge du VG,augmente parce que l'âge rend les vaisseaux, eten particulier l'aorte, plus rigides.La structure des artères et des veines est différenteet ces différences rendent compte des propriétésde ces deux types de vaisseaux : les grosses artèressont beaucoup plus riches en tissu élastique queles artérioles et que les veines. Les capillaires sontdépourvus de tissu élastique. Le muscle lisse estplus abondant au niveau des grosses artères, saufl'aorte, qu'à celui du sphincter précapillaire etdes artérioles. Le tissu fibreux est plus abondantdans les grosses artères et les grosses veines quedans les vaisseaux de petit calibre. La compliance de l'aorte joue un rôle mécaniqueimportant. Lors de l'éjection le sang éjecté vadistendre l'aorte, dès l'occlusion des valves aorti-ques les artères distendues vont restituer l'énergiecinétique qu'elles ont emmagasinées pendant lasystole et revenir progressivement à leur positioninitiale. Ce phénomène va à la fois prolongerl'action de la pompe cardiaque, en amortir lecaractère pulsatile et maintenir la pression arté-rielle diastolique. Le débit dans les vaisseauxrestera certes encore pulsatile, mais les pulsationsseront amorties et d'autant moins marquées quel'on sera plus loin du cœur.La circulation coronaire n'est qu'un exemple descirculations régionales, le premier dans l'ordrepuisque les artères coronaires sont les premièresartères à quitter l'aorte dans la concavité mêmedes valves sigmoïdes aortiques. La circulation dusystème coronaire se produit uniquement aucours de la diastole, au moment où les valvesaortiques se referment, elle est liée à la relaxationdu tissu myocardique. La circulation coronaire,comme la plupart des circulations régionalesest soumise au phénomène d'autorégulation,c'est-à-dire que dans des limites de pression deperfusion du réseau coronaire assez larges ledébit coronaire est inchangé, ce qui évite aumyocarde les à coups.
Stucture-fonction artérielle
➙ Compartimentation structurale et fonctionnelle de la paroiartérielle
En physiologie, la structure de la paroi artérielleest compartimentée en trois couches : - une couche endoluminale, l'endothélium, composée
d'une monocouche de cellules endothéliales reposant sur une membrane basale ;
- une couche médiale se décomposant en un compartiment cellulaire musculaire lisse et un compartiment matriciel composé essentiellementd'élastine, de collagène et de protéoglycans ;
- une couche externe de tissu conjonctif plus lâche, composée de fibroblastes périvasculaireset de collagène.
L'endothélium a une fonction essentielle de bar-rière physique, métabolique et pharmacologiqueliée aux jonctions serrées qui relient les cellulesendothéliales entres-elles. Ces jonctions sont lefait de protéines intercellulaires : connexines,cadherines, exprimées sur la membrane cellulaire.Cette fonction de barrière est physiologiquementmodulée en fonction des sites vasculaires entredeux extrêmes : l'endothélium vasculaire desvaisseaux cérébraux, extrêmement imperméable(barrière hémato-encéphalique) et l'endothéliumdes capillaires glomérulaires assurant la fonctionde filtration rénale des composantes plasmatiques(genèse de l'urine initiale). L'endothélium desartères de conductance et de résistance est essen-tiellement imperméable. L'endothélium capillaireest plus fenestré. Cette fonction de barrière, enrapport avec l'imperméabilité endothéliale,assure la compartimentation des secteurs dans lesystème artériel. La couche endothéliale fait l'in-terface avec le sang circulant, dont elle reçoitdes signaux qu'elle intègre et transmet soit versle sang circulant, soit vers les cellules musculai-res lisses sous-jacentes. L'ensemble, élémentsfigurés du sang-plasma-endothélium, constitue uncompartiment fonctionnel physiologique et phar-macologique qui communique avec le compartimentsous-jacent musculaire lisse. En physiologie lescommunications directes compartiment sanguin-média artérielle sont peu nombreuses. La pathologievasculaire est souvent caractérisée par un phénomènede perte partielle ou totale de l'imperméabilitéendothéliale : phénomène de décompartimentation.

13NO - Monoxyde d’azote et homéostasie cardiovasculaire
La média artérielle est la couche centrale, structu-rante de la paroi artérielle, responsable de lafonction vasomotrice, de la fonction de contentionet de la fonction d'amortissement. La fonctionvasomotrice est en rapport avec les capacitéscontractiles toniques de la cellule musculairelisse. La matrice extracellulaire assure la fonctionde contention du sang dans l'espace vasculaire.La fonction d'amortissement du caractère phasiquedes contraintes, est en rapport avec les propriétésélastiques de la matrice extracellulaire et avecl'état de contraction ou de relaxation de la cellulemusculaire lisse des vaisseaux de conductance.Les cellules musculaires lisses, outre leur fonctionvasomotrice, biosynthétisent et maturent lamatrice extracellulaire collagène et élastique.Elles ont par ailleurs, un potentiel hypertrophique,prolifératif et migratoire en réponse à des phéno-mènes d'activation. Plus les artères sont proches ducœur plus elles sont riches en matrice, plus elless'en éloignent, plus elles se cellularisent.La média baigne dans le liquide interstitiel pro-venant essentiellement du plasma sanguin. Lesprotéines plasmatiques et les nutriments filtrentfaiblement à travers la paroi sous l'effet des gra-dients de concentration (diffusion) et du fluxd'eau généré par la pression artérielle ; c'est ceque l'on appelle la convection. Ces protéinespeuvent s'adsorber au cours de leur transportinterstitiel sur des éléments de la matrice extra-cellulaire, en particulier sur les protéoglycans.Cette fonction est très dépendante de l'intégrité etde l'endothélium. Un endothélium lésé augmenteconsidérablement le transport des protéines plas-matiques.L'ensemble, cellules musculaires lisses-matriceextracellulaire-liquide interstitiel, constitue uncompartiment fonctionnel, séparé du compartimentéléments figurés du sang-plasma-endothélium,mais communiquant indirectement avec lui enphysiologie. L'adventice est la couche la plus externe. Outre lecollagène et les fibroblastes, elle véhicule des capillairesnourriciers et les terminaisons du système nerveuxautonome. Dans les organes pleins, ce tissu fibreuxpérivasculaire, fait l'interface entre la paroi artérielleet le tissu plein environnant spécifique de chaquefonction (muscle cardiaque, épithélium rénal)…Elle véhicule donc des signaux venant du versantexterne des vaisseaux, signaux neurogènes, ou signauxvenant du tissu environnant, vers la média artérielle.Du fait de la présence de capillaires permettant lamigration et l'accumulation des monocytes-macrophageset de lymphocytes, elle est le principal site desréponses immuno-inflammatoires en pathologievasculaire.
En pathologie, cette compartimentation physiologiquepeut disparaître du fait, par exemple, de la disparitiondes cellules endothéliales, comme dans l'athéromecompliqué. Il y a alors un phénomène de décomparti-mentation auquel la paroi artérielle répond en induisantune migration endoluminale des cellules musculaireslisses, leur prolifération intimale, puis la réendothé-lialisation sur le versant luminal et une réactioninflammatoire sur le versant adventitiel. L'ensemblede la réponse ou remodelage tend à cicatriser ourecompartimenter la paroi.
➙ Rôle des grandeurshémodynamiques
Dans le système nerveux central ou endocrinien,les signalisations et communications cellulairessont uniquement d'ordre biochimique : une cellulereçoit un signal moléculaire, l'intègre, génère unenouvelle molécule qui transmet un nouveau signalà une autre cellule. Dans le système cardiovas-culaire, ce type de signalisation existe égalementet des molécules endocrines, présentes dansl'ensemble de l'organisme, ou des moléculesparacrines agissant localement ou régionalementont un rôle tout à fait prépondérant. Néanmoins,ces signaux d'ordre biochimique ne résument pastoutes les communications et signalisations dansle système cardiovasculaire. La phylogenèse asélectionné des cellules et un système capabled'intégrer des signaux d'ordre hémodynamique,c’est à dire d'ordre de la mécanique des fluides.En effet le système cardiovasculaire est un tissusolide, visco-élastique, contenant le sang, unfluide. Les battements cardiaques animent defaçon phasique ce fluide en générant une énergiecinétique (flux) transmises à la colonne sanguine,transformée en partie en énergie potentielle(pression) du fait des résistances périphériques àl'écoulement. La contraction cardiaque est doncun générateur phasique de pression (énergiepotentielle) et de débit (énergie cinétique). Laparoi vasculaire va percevoir ces énergies sousforme de contraintes tensionnelles (potentielles)et de cisaillement (cinétiques). Le cisaillementcorrespondant au coefficient de frottement de laphase liquide en mouvement (le sang) sur laphase solide immobile. Quatre grandeurs hémo-dynamiques permettent de définir ces contraintes :la pression, la vitesse, la dimension vasculaire etla fréquence cardiaque.La pression est la grandeur d'énergie potentiellegénérée par l'éjection systolique dans le systèmevasculaire résistif. Ce sont les résistances péri-phériques à l'écoulement du sang qui génère la

14 NO - Monoxyde d’azote et homéostasie cardiovasculaire
pression artérielle. Ces résistancespériphériques sont le fait du tonusvasomoteur exercé par les cellulesmusculaires lisses artérielles. Le tonusest sous le contrôle de médiateursextracellulaires : catécholamines desterminaisons nerveuses sympathiques,angiotensine, etc ; et du couplagepharmaco-mécanique entre cesmédiateurs extracellulaires, et lasignalisation intracellulaire respon-sable de la contraction. Cette énergiepotentielle (pression) va permettrela répartition des flux en fonctiondes régulations locco-régionales desrésistances. Elle va en particulierpermettre la perfusion coronaire endiastole. La pression génère unecontrainte sur la paroi artérielle qui selon la loide Laplace est proportionnelle également à ladimension si le vaisseau est assimilé à un cylindre :T = P x r où T est la tension, P la pression, r lerayon du cylindre. Cette tension génère descontraintes au sein de la paroi qui se répartissent
sur son épaisseur (figure 6).La vitesse du sang va générer un frottement
sur la surface vasculaire qui se définitcomme la contrainte de cisaillement.
Elle est proportionnelle à lavitesse et à la viscosité san-
guine et inversement pro-portionnelle à la dimension :
t = µ v/r ; où t est lecisaillement, µ laviscosité du sang, vla vitesse, r le rayon(figure 7).La contraction car-diaque est phasique,c’est à dire que cescontraintes poten-tielles et cinétiques,
générées par lecœur, passent par un
pic à chaque systole.La fréquence et l'inten-
sité du pic de contraintedéfinit la fatigue du bioma-
tériau visco-élastique qu'est laparoi vasculaire.
Comme structuralement la paroi artérielle estcompartimentée, la perception des contraintespar cette même paroi est également comparti-mentée en physiologie : la média artérielle, dontla cellule musculaire lisse est le seul constituantcellulaire, perçoit et répond essentiellement à lacontrainte tensionnelle ; l'endothélium, faisantl'interface entre la phase liquide en mouvementet la paroi, perçoit et répond à la contrainte decisaillement. La fatigue est essentiellement perçuepar les constituants de la matrice extracellulaire.Comme nous le verrons plus loin, la contraintephysiologique de cisaillement module par sonintensité la production endothéliale de monoxyded'azote qui joue ainsi un rôle majeur dans larégulation du tonus vasculaire basal et dans sonadaptation à l'augmentation périphérique desbesoins métaboliques.Les médiateurs moléculaires régulant la vasomo-tricité sont également compartimentés. Soit lesmolécules agissent de façon centrifuge : généréesau niveau du compartiment sanguin elles interfè-rent principalement avec l'endothélium ; soitelles agissent de façon centripète, générées parle métabolisme du tissu environnant le vaisseauou au niveau de l'adventice (neuromédiateurs),elles communiquent directement à la média.
Figure 6
Contrainte decisaillement
r
cisaillement = fct.µ V
r
endothéliumµ = viscositéV = vitesser = rayon
Figure 7
r media
P h

15NO - Monoxyde d’azote et homéostasie cardiovasculaire
Tonus vasomoteuret définition de lapression artérielle
On ne peut aborder les questions touchant aurôle du monoxyde d'azote dans la circulation,sans aborder au préalable, ce qu'est le tonusvasomoteur diffus et les résistances périphériquesà l'écoulement sanguin, définissant la pressionartérielle. La pompe cardiaque est un générateur deflux (énergie cinétique), dont une partie, du fait del'existence de résistances périphériques à l'écoulement,est transformée en pression (énergie potentielle). Lesrésistances périphériques ou tonus vasomoteur,définissant la pression artérielle, sont le fait d'unniveau permanent de contraction partielle de lacellule musculaire lisse artériolaire en rapportavec une certaine quantité de pontages entrel'actine et la myosine dans ces cellules. Cettecontraction partielle et tonique est le fait durelarguage permanent de médiateurs extracellu-laires tel que les catécholamines relarguées parles terminaisons du système nerveux sympathique, oude la production d'angiotensine II ou d'autrespeptides vasoconstricteurs.
➙ Signalisation intracellulaire du tonus vasomoteur
A l'état désafférenté de tous médiateurs extracellulaires,la cellule musculaire lisse est spontanément relaxée,sans aucun pontage entre l'actine et la myosine.L'apport de médiateurs extracellulaires (norépinéphrine),liant des récepteurs à 7 domaines transmembranaires(récepteurs α1– adrénergiques), couplés à la mobilisationdu calcium et à l'activation de la protéine kinase-C(PKC) va provoquer la contraction de la cellule mus-culaire lisse. C'est la voie dite de la phospholipase-C.Cette voie est composée de différents éléments : desrécepteurs à sept domaines transmembranairesspécifiques des amines pressives (α1- adrénergiques)
et des peptides vasoconstricteurs comme l'angio-tensine II, la vasopressine, l'endothéline… Cesrécepteurs sont couplés à une activité de phos-pholipase membranaire par l'intermédiaired'une protéine G de type Gq. L'activité de laphospholipase C produit un messager solubledans le cytosol, les inositols phosphates (IP), àpartir des phospho-inositides membranaires, etdu dyacylglycérol (DAG), hydrophobe et lipophile,qui reste à la membrane où il active la protéinekinase C transloquée. Les inositols phosphatesvont être directement responsables de la libérationdu calcium à partir du réticulum sarcoplasmique etindirectement responsables de la dépolarisation de lamembrane (entrée de sodium, changement du potentielde membrane) et de l'ouverture des canaux calciquesvoltage-dépendants faisant entrer du calcium dumilieu extracellulaire vers le milieu intracellulaire(figure 8).
Cette mobilisation du calcium après liaison à lacalmoduline va activer une cascade de phospho-rylation aboutissant au pontage de l'actine et dela myosine et à la contraction tonique de lacellule musculaire lisse. L'activation de la PKCpar le dyacylglycérol va entraîner la phosphory-lation de différentes cibles protéiques intracellulairesimpliquées dans la contraction (caldesmone, calponine),mais également dans l'activation cellulaire(modifications phénotypiques, facilitation de lamitose).
Les cellules musculaires lisses vasculaires ont dece fait un tonus contractile (ni complètementcontractées, ni relaxées). Ce tonus est entretenuen permanence par le relarguage de médiateursextracellulaires, contrôlé au niveau du systèmenerveux central au périphérique régulant l'ho-méostasie du milieu intérieur (SRA), couplé à lavoie de la phospholipase-C dans la cellule mus-culaire lisse.
C'est l'ensemble de cette chaîne physiologique :contrôle central, médiateurs extracellulaires,
Bases tissulaires etmoléculaires del'endothélium-dépendancede la vasorelaxation

16 NO - Monoxyde d’azote et homéostasie cardiovasculaire
signaux intracellulaires, dépendant des phospholipases,qui définit les résistances périphériques diffuses àl'écoulement et donc la pression artérielle. Le rôle del'endothélium, et en particulier de la production dumonoxyde d'azote, va être de moduler localement lasignalisation cellulaire entretenue par cette voie et deréguler ainsi la répartition des flux (énergie cinétique).
Les NO synthasesLa synthèse de monoxyde d'azote impliquel 'oxydat ion du groupement guanidine de laL-arginine en présence d'oxygène. Ce processusconsomme des électrons et induit la formationde monoxyde d'azote et de L-citrulline. Cette réactionest sous le contrôle d'enzymes appelées NO synthases(NOS). On connaît trois isoformes correspondantaux trois grands sites de production de NO : laNO synthase neuronale (NOS I), la NO synthaseinductible macrophagique (NOS II), la NO synthaseendothéliale (NOS III).
Les NO synthases neuronales et endothélialessont dites constitutives et calcium-dépendantes.Elles sont fortement exprimées dans les cellulesendothéliales et certains neurones. Elles sontrégulées essentiellement au niveau de leur activité etpeu au niveau transcriptionnel.
La NO synthase macrophagique est dite inductible etcalcium-indépendante. Les monocytes-macrophagesquiescents ne possèdent pas l'enzyme, l'activation dela cellule s'accompagne de l'apparition irréversible del'enzyme. L'expression de la NOS II est doncessentiellement régulée à un niveau transcriptionnel
(présence ou non dela molécule). La NOSinductible peut êtreexprimée dans denombreux types cellu-laires, incluant lescellules musculaireslisses en réponse àdes stimuli pro-inflammatoires.
Mais ce schéma derégulation n'est pasexclusif, il est seule-ment prépondérant. Ilexiste de faibles tauxde NO synthasesinductibles, constituti-vement expriméesdans les cellules mus-
culaires lisses. L'activation chronique de la NOsynthase endothél ia le s 'accompagne d'uneaugmentation d'expression des ARN messagerscodant pour la protéine (induction d'expression).Un point fondamental dans le fonctionnementdes NO synthases est la nécessité de co-facteurs,indispensables à leur action. Ces co-facteurs,régulent le flux d'électrons entre le domaineréductase et le domaine oxydase de l'enzyme.Les NO synthases possèdent des sites de liaisonpour la NADPH (Nicotinamide AdénineDinucléotide Phosphate réduit), pour la FlavineAdénine Dinucléotide (FAD) et la FlavineMonoNucléotide (FMN), pour l'hème, pour lecomplexe Ca-calmoduline. L'activité des NOsynthases est également augmentée par la tétra-hydrobioptérine (H4B) et la protéine de chocthermique 90 (HSP90). Le transfert séquentield'électrons entre ces différents co-facteurs facilitentl ’ac t iv i té ca ta ly t ique des NO synthases . Cesélectrons, générés par le domaine réductase,transitent par le fer héminique des NO synthasesqui les donnent directement au groupement guanidinede l'arginine (figure 9). En fait les gènes codantspour les NO synthases correspondent à desfusions entre des gènes codants pour des fonctions deNADPH réductase (donneur d'électrons libres) etdes NO synthases, oxydases à proprement parlé.De ce fait on décrit un fonctionnement articuléde l'enzyme à partir de ces deux domainesNADPH reductase/NO synthase. L'arginine est unacide aminé essentiel capté par la cellule àl'aide d'un système de transport spécifique.Parmi les isoformes de NO synthases, seule laforme endothéliale possède un site de myristilation(accrochage d'une chaîne d'acide gras). Cette
Ca ++
α-adrenergiqueangiotensine IIendothélinekinines
PLC
Ca ++calmoduline phosphorylations
IPPKC
contractionCellules musculaires lisses
DAG
Figure 8

17NO - Monoxyde d’azote et homéostasie cardiovasculaire
addition d'une chaîne d'acide gras permet son associationconstante avec les conches ; lipidiques des membranescellulaires alors que les deux autres NO synthases quine possèdent pas ce bras lipidique, sont hydrosolubleset présentes dans le cytosol des cellules. Cette association de la NO synthase endothéliale avecles membranes est réputée faciliter par proximité ladiffusion du NO vers le sang circulant et vers les cellulesmusculaires lisses sous-jacentes. L'association à lamembrane se fait préférentiellement au niveau descavéoles. Les cavéoles sont des replis ultrastructurauxde la membrane plasmique enrichie d'une part encholestérol et d'autre part d'une protéine spécifiqueappelée cavéoline. La cavéoline joue un rôle importantdans la régulation de l'activité de la NO synthaseendothéliale.
Un autre point d'intérêt, est la capacité des NO synthasesde produire des anions superoxydes. Cet anion super-oxyde est généré en l'absence d'arginine, alors quel'addition d'arginine réduit la formation de formesradicalaires. Ce point est important et introduit les rapportscomplexes, mais absolument permanents entre lemonoxyde d'azote et les phénomènes d'oxydationdans le système cardiovasculaire. Par exemple, lors del'intoxication chronique à la Nitro-Arginine chez l'animal,la NOS II est induite dans les cellules musculaires lissesartérielles et la paroi produit plus d'oxygène radicalaire.Les trois isoformes de NO synthases ont une homologiede séquence d'environ 50 %. Les NOS II et III ont unpoids moléculaire de 130 kD et sont légèrement pluspetites que la NOS I qui fait 150 Kd. Ces trois protéinesont conservé des homologies substantielles avec la
réductase du cytochrome P-450. Cette enzyme (laréductase) se comporte comme un donneur d'électronspour le cytochrome. La phylogénèse des gènes codantspour les NO synthases vient d'une fusion d'un segmentd'ADN codant pour la réductase et d'un segmentd'ADN codant pour un équivalent de P-450 rendantainsi compte des similitudes de fonctions entre lesdeux types d'enzymes. Le NO exerce un rétro-contrôlenégatif sur l'activité des NO synthases par le biais del'hème. La citrulline est recyclée en L-arginine moyennantl'incorporation d'un atome d'azote. L'arginine est àl'interface entre différentes voies métaboliques et designalisation dans les cellules production de NO, cyclede l'urée, production de créatine.
Le monoxyde d'azote est le premier exemple d'unsystème de signalisation complètement nouveau, trèsdifférent du concept devenu classique de médiateurextracellulaire, synthétisé et sécrété par une cellule etse liant à un récepteur sur la membrane d'une autrecellule.
Le monoxyde d'azote est un radical gazeux simple quipeut se lier facilement de façon covalente à de nom-breuses cibles moléculaires, alors que les médiateursclassiques ont une structure définie, leur donnant leurspécificité de liaison à un récepteur ayant une structurecomplémentaire. NO n'est pas stocké dans des granuleset il diffuse librement à partir de son site de synthèsedans toutes les directions. NO est soluble dans l'eau etdans les lipides, ce qui permet sa diffusion d'une celluleà l'autre.
NADP+
NADPH
Fe3+ H B4
2
+
NO
e-
L-arginine+O
L-citrulline
Domaine C-terminalréductase
Domaine N-terminalhème
NN
NN
FADFMN
Figure 9

18 NO - Monoxyde d’azote et homéostasie cardiovasculaire
Régulation de laNO synthaseendothéliale
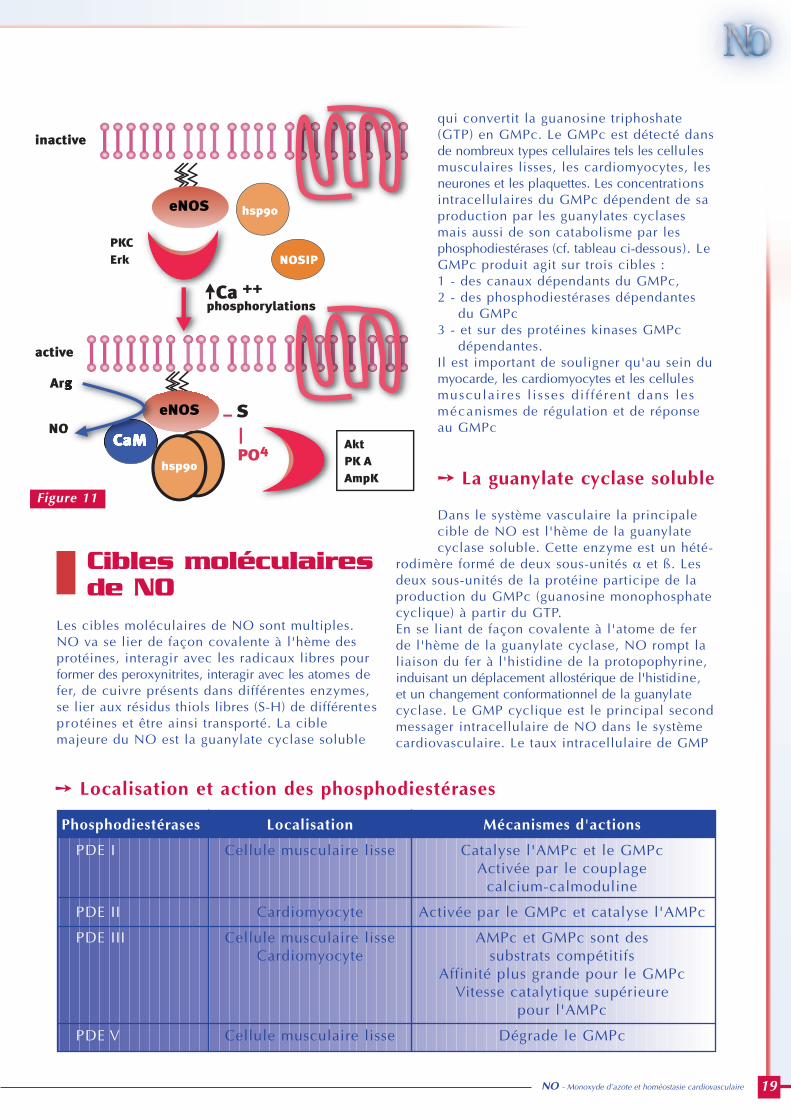
➙ Calcium-dépendance
La régulation de l'activité de la NO synthaseendothéliale (e-NOS) fait appel à des mécanismes debiochimie intracellulaire complexes, ne touchantpas seulement directement l'enzyme elle-même,mais également tout un assemblage de protéinesrégulatrices, interférant avec l'enzyme, cibles dephosphorylations potentielles : Heat ShockProtein 90 (HSP 90), NOSIP (NO synthase inter-acting protéine), Erk (extracellular signal relatedkinase), cavéoline. C'est cet assemblage protéique quiprobablement régule au long cours la productionde NO par l'endothélium.Deux voies régulent principalement l'activité dela NO synthase endothéliale : la voie du calciumet les phosphorylations possibles par les protéines-kinases. La première voie (calcium) est le principaleffecteur de l'activation pharmacologique de lae-NOS par les peptides vaso-actifs (acétylécholine,bradykinine, angiotensine II), la seconde est leprincipal effecteur de l'activation par les forcesde cisaillement. Cette répartition des rôles n'estpas univoque, le cisaillement mobilise égalementle calcium, certains agonistes de la NO synthaseagissent via l'activation de protéines-kinases.A l'état non-stimulé la e-NOS est associée à la
membrane plasmique au niveau des cavéoles, oùelle est réprimée par la cavéoline-1. Lorsqu'unligand comme l'acétylcholine ou la bradykininelie un récepteur à 7 domaines transmembranairessur la membrane, l'interaction ligand-récepteurmembranaire va activer la phospholipase-C,générer des inositols phosphates, et mobiliser lecalcium dans la cellule endothéliale.Le calc ium ains i mobi l i sé va se f ixer sur lacalmoduline et l'ensemble, 4 molécules de Ca-une molécule de calmoduline, va déplacer lacavéoline de son site de liaison répresseur sur lae-NOS et permettre ainsi l'activation de l'enzyme.C'est le mode d'activation aiguë (burst phase) dela NO synthase (figure 10).
➙ Phosphorylations
Le second système de régulation agit à plus longterme via la phosphorylation de l'enzyme par lesprotéines-kinases. Deux systèmes de protéines-kinases semblent jouer un rôle prépondérantdans l'activation au long cours de la e-NOS, lavoie PI-3 kinase/Akt et la protéine-kinase A (PKA)activée par l'AMP cyclique. Ces deux enzymesphosphorylent probablement les sérines en position1179 et 635 de la e-NOS provoquant son activation etsa translocation. D'autres protéines kinases peuventégalement moduler l'activité de la e-NOS : laPKC semble avoir un rôle répresseur, alors que laCaM-kinase et l'AMP-kinase sont activatrices(figure 11).
Ces systèmes de phosphory-lation interfèrent avec lalocalisation subcellulaire del'enzyme aux cavéoles, àl'appareil de Golgi et dansle cytosol. La phosphoryla-tion de l'enzyme dépend desa localisation et inverse-ment sa phosphorylationpermet sa translocationd'un compartiment (cavéo-les où elle est facilementréprimable par la cavéoline)à un autre (cytosol où ellen'est plus réprimable par lacavéoline).
........ ... .... .Ca++
cavéole
cavéoline eNOS
calmoduline
eNOS
arginine
NO.
NO synthase endothéliale inactive NO synthase activée par Ca++
Figure 10
19NO - Monoxyde d’azote et homéostasie cardiovasculaire
Cibles moléculairesde NO
Les cibles moléculaires de NO sont multiples.NO va se lier de façon covalente à l'hème desprotéines, interagir avec les radicaux libres pourformer des peroxynitrites, interagir avec les atomes defer, de cuivre présents dans différentes enzymes,se lier aux résidus thiols libres (S-H) de différentesprotéines et être ainsi transporté. La ciblemajeure du NO est la guanylate cyclase soluble
qui convertit la guanosine triphoshate(GTP) en GMPc. Le GMPc est détecté dansde nombreux types cellulaires tels les cellulesmusculaires lisses, les cardiomyocytes, lesneurones et les plaquettes. Les concentrationsintracellulaires du GMPc dépendent de saproduction par les guanylates cyclasesmais aussi de son catabolisme par lesphosphodiestérases (cf. tableau ci-dessous). LeGMPc produit agit sur trois cibles :1 - des canaux dépendants du GMPc,2 - des phosphodiestérases dépendantes
du GMPc3 - et sur des protéines kinases GMPc
dépendantes.Il est important de souligner qu'au sein dumyocarde, les cardiomyocytes et les cellulesmuscula i res l i s ses d i f fé rent dans lesmécanismes de régulation et de réponseau GMPc
➙ La guanylate cyclase soluble
Dans le système vasculaire la principalecible de NO est l'hème de la guanylatecyclase soluble. Cette enzyme est un hété-
rodimère formé de deux sous-unités α et ß. Lesdeux sous-unités de la protéine participe de laproduction du GMPc (guanosine monophosphatecyclique) à partir du GTP.En se liant de façon covalente à l'atome de ferde l'hème de la guanylate cyclase, NO rompt laliaison du fer à l'histidine de la protopophyrine,induisant un déplacement allostérique de l'histidine,et un changement conformationnel de la guanylatecyclase. Le GMP cyclique est le principal secondmessager intracellulaire de NO dans le systèmecardiovasculaire. Le taux intracellulaire de GMP
eNOS hsp90
NOSIPPKCErk
Ca ++phosphorylations
inactive
active
hsp90
AktPK AAmpK
PO4CaM
Arg
SNO
eNOS
Figure 11
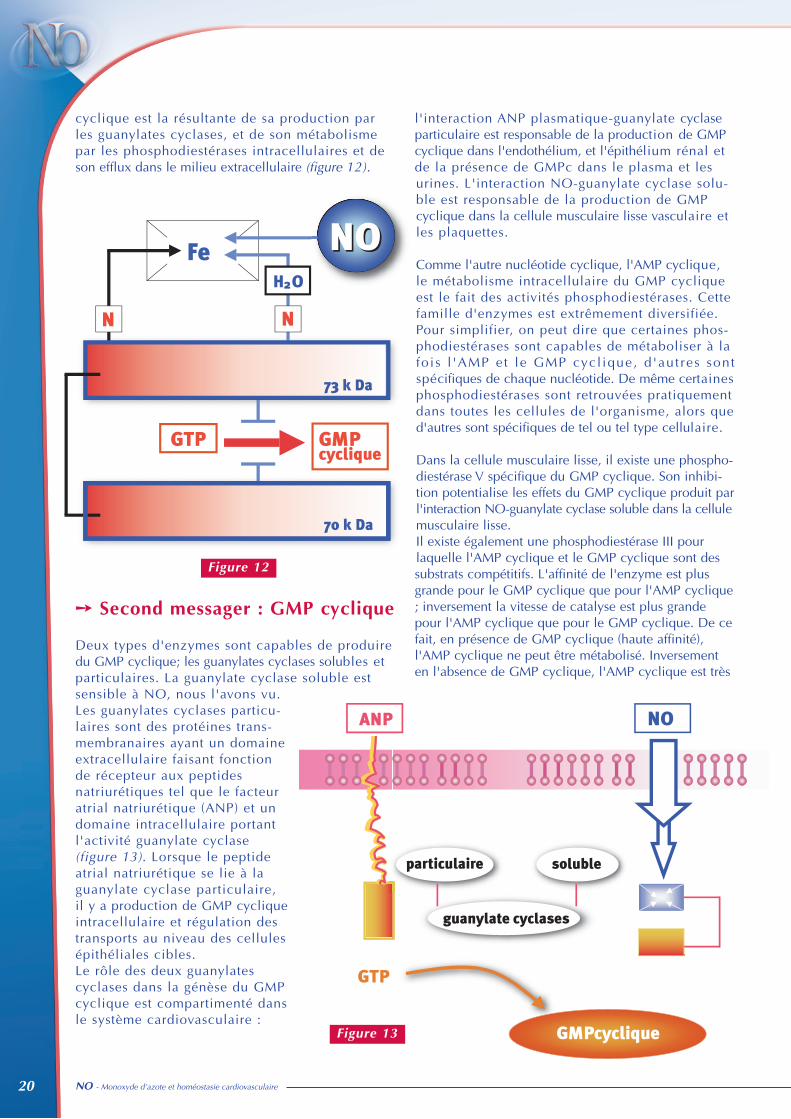
➙ Localisation et action des phosphodiestérases
Phosphodiestérases Localisation Mécanismes d'actions
PDE I Cellule musculaire lisse Catalyse l'AMPc et le GMPcActivée par le couplage
calcium-calmoduline
PDE II Cardiomyocyte Activée par le GMPc et catalyse l'AMPc
PDE III Cellule musculaire lisse AMPc et GMPc sont desCardiomyocyte substrats compétitifs
Affinité plus grande pour le GMPcVitesse catalytique supérieure
pour l'AMPc
PDE V Cellule musculaire lisse Dégrade le GMPc
20 NO - Monoxyde d’azote et homéostasie cardiovasculaire
cyclique est la résultante de sa production parles guanylates cyclases, et de son métabolismepar les phosphodiestérases intracellulaires et deson efflux dans le milieu extracellulaire (figure 12).
➙ Second messager : GMP cyclique
Deux types d'enzymes sont capables de produiredu GMP cyclique; les guanylates cyclases solubles etparticulaires. La guanylate cyclase soluble estsensible à NO, nous l'avons vu.Les guanylates cyclases particu-laires sont des protéines trans-membranaires ayant un domaineextracellulaire faisant fonctionde récepteur aux peptidesnatriurétiques tel que le facteuratrial natriurétique (ANP) et undomaine intracellulaire portantl'activité guanylate cyclase(figure 13). Lorsque le peptideatrial natriurétique se lie à laguanylate cyclase particulaire,il y a production de GMP cycliqueintracellulaire et régulation destransports au niveau des cellulesépithéliales cibles. Le rôle des deux guanylatescyclases dans la génèse du GMPcyclique est compartimenté dansle système cardiovasculaire :
l'interaction ANP plasmatique-guanylate cyclaseparticulaire est responsable de la production de GMPcyclique dans l'endothélium, et l'épithélium rénal etde la présence de GMPc dans le plasma et lesurines. L'interaction NO-guanylate cyclase solu-ble est responsable de la production de GMPcyclique dans la cellule musculaire lisse vasculaire etles plaquettes.
Comme l'autre nucléotide cyclique, l'AMP cyclique,le métabolisme intracellulaire du GMP cycliqueest le fait des activités phosphodiestérases. Cettefamille d'enzymes est extrêmement diversifiée.Pour simplifier, on peut dire que certaines phos-phodiestérases sont capables de métaboliser à lafo is l 'AMP et le GMP cycl ique, d 'aut res sontspécifiques de chaque nucléotide. De même certainesphosphodiestérases sont retrouvées pratiquementdans toutes les cellules de l'organisme, alors qued'autres sont spécifiques de tel ou tel type cellulaire.
Dans la cellule musculaire lisse, il existe une phospho-diestérase V spécifique du GMP cyclique. Son inhibi-tion potentialise les effets du GMP cyclique produit parl'interaction NO-guanylate cyclase soluble dans la cellulemusculaire lisse. Il existe également une phosphodiestérase III pourlaquelle l'AMP cyclique et le GMP cyclique sont dessubstrats compétitifs. L'affinité de l'enzyme est plusgrande pour le GMP cyclique que pour l'AMP cyclique; inversement la vitesse de catalyse est plus grandepour l'AMP cyclique que pour le GMP cyclique. De cefait, en présence de GMP cyclique (haute affinité),l'AMP cyclique ne peut être métabolisé. Inversementen l'absence de GMP cyclique, l'AMP cyclique est très
ANP NO
GTP
GMPcyclique
particulaire soluble
guanylate cyclases
Figure 13
Fe
N
H O2
N
GTP GMPcyclique
70 k Da
73 k Da
NONO
Figure 12

21NO - Monoxyde d’azote et homéostasie cardiovasculaire
activement métabolisé (vitesse de catalyse élevée) parcette enzyme. Dans la cellule musculaire la présencede GMP cyclique dépendante de NO potentialise doncles effets de l'AMP cyclique (figure 14).
Au contraire dans la cellule cardiaque il existeune phosphodiestérase II, dont l'activité catalytique estspécifique de l'AMPc, mais qui est activée par leGMP cyclique. Dans le cardiomyocyte, l'augmentation deGMP cyclique active la dégradation de l'AMPcyclique.
C'est par ce mode d'action que l'on explique enpartie les effets inotropes négatifs du NO-GMPcyclique dans le cœur. Dans la cellule musculaire ilexiste une phosphodiestérase I non-spécifique,capable d'hydrolyser le GMP cyclique et l'AMPcyclique, activée par le couplage calcium-calmo-duline.La seconde voie permettant à la cellule de sedébarrasser du GMP cyclique est de l'égresseractivement dans le milieu extracellulaire. Dansle milieu extracellulaire les nucléotides cycliquessont plus stables du fait de l'absence de phos-phodiestérases extracellulaires. Le GMP cycliquedu milieu extracellulaire ne peux pas réentrerdans la cellule. Du fait de la compartimentationdu rôle des guanylates cyclases, le taux de GMPcyclique dans le plasma et les urines est essen-tiellement le fait des peptides natriurétiques.
➙ Cible du GMPcyclique : les G kinases
Dans la cellule musculaire lisse,le principal effecteur du GMPcyclique est la protéine kinaseGMP cyclique-dépendante (GK-I)ou kinase G. Cette kinase va elle-même phosphoryler plusieurs ciblesmoléculaires à l'intérieur de lacellule et aboutir à la relaxationde la cellule musculaire lisse. Lakinase-G-1 est relativement spéci-fique de la cellule musculairelisse qui en est très riche. Parexemple si l'on utilise des anti-corps anti kinase-G sur un tissu(le myocarde), ces anticorps nemarqueront que la média des artèrescoronaires (figure 15).Les cellules musculaires lisses en cultu-res primaires sont également très richesen kinase-G et il faut de très nombreuxpassages (une quinzaine) pour que lacellule musculaire lisse se dédifféren-cie au point de perdre l'expression dela kinase-G.
Dans la cellule épithéliale, il existe une autre isoformede la G-kinase dite GK-II également sensible au GMP-cyclique. Sa cible préférentielle est la régulation de laconductance chlore (CFTR). Si le CFTR est phosphorylépar la GK-II, le chlore s'accumule dans le milieu extra-cellulaire, retenant le sodium et l'eau. Comme nousl'avons vu, la cellule épithéliale, en particulier rénaleest surtout sensible aux peptides extracellulaires activantla guanylate cyclase particulaire. Le GMP cyclique ainsi formé va activer la G-kinase IIqui en phosphorylant le CFTR, enrichira le milieuextracellulaire (urine initiale) en chlore, sodium et eauprovoquant natriurèse et diurèse. On voit que ces deuxsystèmes : NO, guanylate cyclase soluble, G-kinase-I,cellules musculaires lisses d'une part ; peptides natriu-rétiques, guanylate cyclase particulaire, G-kinase-II,cellules épithéliales rénales d'autre part sont indépendants,avec des fonctions physiologiquement bien comparti-mentées. Il y a une exception à cette règle, qui sont lescellules myoépithélioïdes sécrétrices de rénines. Cescellules musculaires lisses de l'artériole afférente auglomérule, transdifférentiées en cellules sécrétrices derénine, expriment préferentiellement la G-kinase II(épithéliales). L'activation de la G-kinase II via le NOet le GMP cyclique dans la cellule myoépithélioïdeaugmente la sécrétion de rénine probablement par uneffet d'hyperpolarisation membranaire.
Domaine régulateur Site catalytique
PDE II
PDE II
AMPc
AMPc
AMP
GMPc
GMPcGMP
Figure 14

22 NO - Monoxyde d’azote et homéostasie cardiovasculaire
En l'absence de GMP cyclique, la kinase-G estprésente dans le cytosol des cellules mais estinactive. C'est une protéine bicaténaire, les deuxchaînes étant reliées par un pont disulfure. Unedes chaînes porte l'activité enzymatique et est enpermanence réprimée par un des domaines del'autre chaîne. Ce domaine est appelé pseudo-substrat car il contient un enchaînement d'acidesaminés proche de celui reconnu comme ciblepar l'activité enzymatique de l'autre chaîne. Cedomaine “pseudo-substrat” lie de façon non-covalente le site actif de la kinase située surl'autre chaîne. La chaîne contenant le domainepseudo-substrat possède également un domainede liaison pour le GMP cyclique. Lorsque leGMP cyclique interagit avec son site de liaison,le pseudo-substrat se décroche du site actif etl'enzyme peut fonctionner. On peut comparer lemode d'action de cette enzyme à une épingle ànourrice fermée en l'absence du second messager etne pouvant pas agir, ouverte par l'interaction duGMP cyclique et phosphorylant alors ses cibles.Cette activation par le GMP cyclique est réversible.Les cibles de la G-kinase dans la cellule musculairelisse, aboutissant à la relaxation, sont nombreuses etprobablement pas toutes ident i f iées . Lesphospholambans, protéines associées à lal'ATPase calcique du réticulum sarcoplasmique,responsable du repompage du calcium, en sontune. La phosphorylation des phospholambanslibère l'activité de la calcium-ATPase augmentantle repompage du calcium et diminuant ainsi le
taux de calcium libreintracytosolique. Surtoutla kinase-G interfère avecla voie de phospholipase-Cmajoritairement respon-sable de la contractionde la cellule musculairelisse.
L'activité probablementprédominante de laG-kinase est de découpléle système récepteur àsept domaines transmem-branaires de leurs signalisa-tion intracellulaire. Enl'absence de kinase-Gactive, les récepteurs activéssont couplés à la signalisa-tion intracellulaire.Après phosphorylationpar la kinase-G le cou-plage n'est plus possibleet le calcium ne peutplus être mobilisé dans
la cellule en réponse à la liaison ligand (catécho-lamine par exemple) - récepteur. Ceci est le faitde la phosphorylation de l'IRAG (protéine associéeaux récepteurs aux IPs du réticulum sarcoplasmique).La GKI induit également un phénomène d'hyper-polarisation cellulaire en augmentant la sortie depotassium. Enfin la GKI phosphoryle laMBS/PP1M, un complexe protéique associé àl'actine et à la myosine, inhibant leur interaction(voir tableau ci-contre).
Dans l'expérience initiale de Furchgott, l'effet vasodi-latateur de l'endothélium, par le fait de NO, nécessitaitla contraction préalable des cellules musculaires lissespar des agents activant la voie de la phospholipase-Ctelle la norépinéphrine ou l'angiotensine II. En présenced'une voie de la phospholipase-C ainsi activée dans lacellule musculaire lisse, le NO libéré par l'endothéliumétait capable de relaxer le vaisseau. Inversement, si levaisseau n'est pas préalablement contracté par un activateurde la PLC, il n'y a pas d'effet direct de NO. Il a étérécemment montré dans les cellules transfectées que lakinase-G et le second messager (GMP cyclique) étaientnécessaires à cet effet (épingle à nourrice ouverte).En l'absence de GMP cyclique (épingle à nourricefermée), la kinase-G seule n'a pas d'effet dedécouplage entre l'activation des récepteurs et lacontraction. En l'absence de kinase-G, le GMPcyclique seul n'a également pas d'effet.
domaine d’inhibition(pseudo substrat)
domaine de liaison du GMPc
Kinase G-I
domaine d’activité enzymatique
PM 75 000PM 75 000
PM 82 000PM 82 000
NH2
COOH
SS
Figure 15

23NO - Monoxyde d’azote et homéostasie cardiovasculaire
L'effet vasodilatateur de NO apparaît donccomme une modulation du tonus constricteur liéà l'activation de la voie de la phopholypase Cdans la cellule musculaire lisse : en l'absence deNO l'activité de la voie de la constriction (PLC)est maximum ; en présence de NO la voie de laconstriction est inhibée proportionnellement à laquantité de GMP cyclique produit dans la cellulecible musculaire lisse.
La kinase-G est essentiellement la cible du GMPcyclique comme son nom l'indique, et le GMPcyclique à une forte affinité pour la kinase-G.Mais le taux d'AMP cyclique est plus élevé quecelui de GMP cyclique dans la cellule musculairelisse. L'AMP cyclique est capable de lier lakinase-G avec une plus faible affinité que leGMPc, et de l'activer. Il apparaît donc que l'AMPcyclique a un effet relaxant de la cellule musculairel i sse médiée par la même s ignal i sa t ion intra-cellulaire que le GMP cyclique, la kinase-G. C'estcomme cela que l'on explique les effets vasodila-tateurs de la stimulation béta-adrénergique, desprostaglandines vasodilatatrices d'origine endothélialeet de l'adénosine.
Dans la cellule musculaire, les activateurs del'adénylate cyclase diminuent la mobilisation ducalcium et sont donc relaxants, alors que dans lemyocyte cardiaque, les mêmes agonistes adréner-giques augmentent la mobilisation du calcium etsont inotropes et chronotropes positifs. La différenceentre les deux spécificités cellulaires de réponsevient probablement de la richesse des cellulesmusculaires lisses en kinase-G, sensibles auxnucléotides cycliques. Dans la cellule endothélialel'activité ß-3-adrénergique stimule la productiond'AMP cyclique et l'activation de la PKA quielle-même phosphoryle la e-NOS, augmentantson activité.
Au sein du cardiomyocyte, les trois effecteurs lesplus importants du GMP cyclique sont les phos-phodiestérases de type II et III, et la protéinekinase G (PKG). L’activation de la PKG provoquede nombreux effets cellulaires :1 - une inhibition des canaux calciques potentiel-
dépendant pour de fortes concentrations en GMPc (>10 µM),
2 - une diminution de la sensibilité de l’appareil contractile au calcium, effet qui pourrait s‘expliquer par la phosphorylation de la troponin I,
3 - une phosphorylation du récepteur de laryanodine (RyR2) qui correspond au canal calcique du réticulum sarcoplasmique,
4 - une stimulation de la synthèse d’ADP-ribose cyclique, un activateur endogène de RyR2.
Il existe également au sein du cardiomyocyte desvoies de signalisation indépendantes du GMPc.En effet, le NO et les peroxynitrites (associationdu NO et des anions superoxydes) peuvent modifier lacontraction cardiaque par oxydation de résidusthiols sur des protéines régulatrices myocardiques.RyR2 et le canal calcique de type L sont ainsirégulés par poly-S-nitrosylation. De plus, le NOet/ou les peroxynitrites peuvent aussi contrôlerl’activité d’enzymes régulant la consommationd’oxygène et la génération d’ATP dans le cœur.Ainsi, les donneurs de NO inhibent directementle créatine kinase et réduisent l’activité de lachaîne respiratoire mitochondriale. En entrant encompétition avec l’oxygène sur la cytochromeoxydase, le NO pourrait moduler la sensibilitéapparente des mitochondries pour l’oxygène.
Le NO et/ou le GMPc apparaissent donc commedes régulateurs de plusieurs grandes fonctions dumyocyte cardiaque (figure 16).
Cibles potentielles de phosphorylationpar la G-kinase de type I dans la cellule
musculaire lisse vasculaire
• IRAG (protéine régulatrice associéeau récepteur canal à l'Inositol Phosphate, sa phosphorylation inhibe la sortie de calcium)
• Canal potassique (efflux depotassium et hyperpolarisation membranaire)
• Protéine Gq de couplage entre les récepteurs à 7 domainestransmembranaires et laphospholipase-C
• Phospholambans associés à laCa-ATPase du réticulum, leurphosphorylation augmentel'activité de recaptage du calcium par le réticulum
• MBS/PP1M – complexe protéique associé aux protéines contractiles dont la phosphorylation inhibe l'interaction actine-myosine

24 NO - Monoxyde d’azote et homéostasie cardiovasculaire
➙ NO au delà de la réponsevasomotrice
Au-delà de ses effets fonctionnels sur la vasomo-tricité, NO a également des effets sur la trophicitévasculaire. En particulier il a été montré que NOa un effet antiprolifératif sur les cellules musculaireslisses dépendant de la production de GMPc.Nous avons vu que l'activation de la voie de laphospholipase C, non seulement augmentait letonus constricteur, mais augmentait également lavoie de la protéine kinase-C impliquée dans lesmodifications phénotypiques associées à l'activationdes cellules musculaires lisses. En découplant lesrécepteurs de la phospholipase C, le NO inhibecette voie. De ce fait, la production de NO participe àla désactivation des cellules musculaires lissesen présence d'endothélium.
Au delà de ses effets sur les cellules musculaireslisses, NO diffuse également vers le secteur sanguinoù il a un effet anti-agrégant plaquettaire. En casd'agrégation plaquettaire libérant des moléculesvasoactives en face d'un endothélium intact,l'augmentation de production de NO, représenteune contre-régulation majeures désintégrantl'agrégat plaquettaire, dilatant le vaisseau en
regard, prévenant la thrombose. En l'absenced'endothélium, cette contre-régulation ne peutavoir lieu.De même le monoxyde d'azote endothélialeinhibe l'adhésion et la transmigration des monocytescirculants et de ce fait leur activation en macro-phages.En résumé, une activation permanente de la voiede la contraction (tonus vasomoteur), de lasignalisation dépendante de l'activité de la phos-pholipase-C, est nécessaire pour révéler l'effetrelaxant de la signalisation NO-GMP cyclique-kinase-G. En l'absence de NO, le taux de GMP cycliquesest bas et le niveau d'activation de la signalisationrécepteurs -proté ines G-phosphol ipase C es trenforcé dans les cellules musculaires lisses. Lesystème de signalisation intracellulaire de lacontraction et de la relaxation de la cellule musculairelisse fonctionne comme une chaîne son-micro-phone- amplificateur, où les sons initiaux sontles l igands des récepteurs à sept domainestransmembranaires (micro). Le NO module l'amplifi-cateur : beaucoup de NO, l'amplificateur estcomplètement b loqué, en l 'absence de NOl'amplificateur fonctionne au maximum de sescapacités (figure 17).
eNOSmyocyte
nNOSmyocyte
Cytochromeoxydase
Créatinekinase
Canal calciquedu réticulum
Modulation de lacontractibilité cardiaque
Modulation del'énergétique cardiaque
Guanylylcyclase
GMPc
PDE2
PDE3
PK-GMPc
NO
Figure 16

25NO - Monoxyde d’azote et homéostasie cardiovasculaire
Beaucoup de NO correspond au choc septique ;dans ce cas les agents presseurs usuels (catécho-lamines, angiotensines II…) sont inefficaces àrétablir une pression artérielle normale (les sonsinitiaux sont là, les micros présents mais l'amplinon branché). Inversement en l'absence de NOcorrespondant à l'intoxication chronique par desantagonistes de l'arginine sur les NO synthases,les sons initiaux ne sont pas plus forts : mêmestaux de catécholamine, d'angiotensine II, mais latransduction du signal fonctionne au maximum,provoquant une augmentation intense de la pressionartérielle.
➙ Autres interactions de NO
NO est un radical, donneur d'électron dont lescibles sont potentiellement multiples. NO peutinteragir comme ligand de toutes les hèmes-protéines.Il interagit avec l'anion superoxyde pour formerdes péroxynitrites. Il interagit avec les atomes deFer et de Cu présents au site actif de nombreusesenzymes. Il se lie aux radicaux sulfhydril (SH)des cystéines pour former des nitrosothiols quipeuvent servir de réservoir intermédiaire pour
NO et le véhiculer dans le plasma et surtout lestissus. Il peut être capté par les protéines glycéesréalisant un phénomène de “quentching” oud'absorbance.
Certainement que les interactions entre NO et lestress oxydatif sont la seconde cible d'intérêt decette molécule dans le système cardiovasculaire,du fait de l'importance des phénomènes d'oxydationdans le développement de l'athérosclérose. Lestress oxydatif existe dans tous les systèmesvivants aérobies dont les métabolismes dépendent del'apport en oxygène. Les cellules vivantes sontextrêmement bien équipées pour résister austress oxydatif des espèces radicalaires.Le terme de stress oxydant est usuellement utilisé,mais il n'est pas très clair au plan de la chimie.Certaines espèces radicalaires comme l'anionsuperoxyde fonctionnent généralement commedes agents réducteurs (donneur d'électron). Enfait, on définit le stress oxydant comme la productiond'espèces radicalaires, très réactives, dans lessystèmes vivants. Néanmoins certaines espècesradicalaires tel le NO, ne sont pas forcément délétères. Al'inverse la réaction de Fenton qui produit l'eauoxygénée (H2 O2) à partir de l'eau, et de formes
Gprot
signalisationde la contraction
ligandrécepteur
son
micro
réponse
NOamplificateur
Figure 17

26 NO - Monoxyde d’azote et homéostasie cardiovasculaire
radicalaires de l'oxygène en présence de fer est en soibeaucoup plus toxique. L'eau oxygénée est un produitstable dont les capacités oxydantes sont pérennes.
Le stress oxydant prend habituellement une connotationdélétère de réactions biochimiques potentiellementtoxiques pour le vivant. Dans le système cardiovasculaire,le stress oxydant est réputé jouer un rôle importantdans le développement de l'athérosclérose. Une descibles majeures du stress oxydant vasculaire sont lesLDLs. Les LDL oxydées, et non les LDL natives, peuventêtre internalisées par les macrophages tissulaires et lescellules musculaires lisses induisant un dépôt massif decholestérol dans la paroi artérielle (génération de“foam cell”).
Les cellules endothéliales, musculaires lisses, lesmacrophages et les polymorphonucléaires produisentdes espèces radicalaires capables d'oxyder les LDL. LesLDL oxydées activent les cellules musculaires lissesconduisant au recrutement local de cellules inflamma-toires. La mesure usuelle des effets délétères des espècesradicalaires est leur capacité à initier une péroxydationlipidique.
L'anion superoxyde réagit avec NO pour former del'acide péroxynitreux. L'acide péroxynitreux est unpuissant oxydant qui transfert des atomes d'oxygène,oxyde les résidus tyrosines et les thiols des protéines,initie la peroxydation lipidique et sert de donneur deradicaux hydroxyls.
Après son interaction avec l'anion superoxyde, NOperd ses fonctions vasodilatatrices pour s'intégrer dansles cycles auto-entretenus d'oxydation des systèmesvivants. En présence d'agents antioxydants, la fonctionvasodilatatrice de l'endothélium est potentialisée. Lescellules endothéliales produisent les deux espèces : leNO et l'anion superoxyde. Les cellules musculaires lissesproduisent également l'anion superoxyde. C'est lasuperoxyde dismutase qui est l'enzyme principalementimpliquée dans l'inactivation de l'anion superoxyde.Lorsque la SOD est inhibée par le diéthyldithiocarbamateou par un déficit en cuivre (le cuivre est un co-facteurnécessaire à l'activité de la SOD) NO est beaucoupmoins actif. Une autre interaction possible entre NO etla production de radicaux libres, passe par le fait qu'enl'absence d'arginine, son substrat, la NO synthasepourrait générer de l'anion superoxyde.
Le phénomène de glycation des protéines est un phé-nomène général, passif non enzymatique (de ce faitqu'il s'oppose au terme de glycosylation qui impliquel'adjonction de sucre à une protéine par une réactionenzymatique), lié au temps (âge) et à la concentrationen glucose (diabète).Le glucose ou d'autres sucres (ribose) vont se déposersur les fonctions amines des protéines du vivant, enparticulier sur les lysines pour former différents adduitsintermédiaires conduisant à de véritables phénomènesde pontages non-enzymatiques entre protéines.
Ces phénomènes de glycation touchent les protéinesplasmatiques, en particulier l'albumine, mais égalementles protéines de la matrice extracellulaire, en particulierles collagènes. Les sucres plasmatiques convectés parle flux d'eau transpariétal ou les riboses libérés par lamort cellulaire (les riboses rentrent dans la constitutionde l'ADN) vont se complexer aux fonctions aminespour former des pontages interfibrillaires appelés AGE(pour Advanced Glycation End-products).
Il a été montré que ces produits de glycation, en parti-culier les produits intermédiaires, étaient capablesd'inactiver NO et de ce fait d'en limiter la diffusiondans des situations comme le vieillissement ou le diabète,où les produits de glycation s'accummulent sur lesconstituants de la matrice extracellulaire.L'accumulation des produits de glycation a d'autresconséquences comme celle d'augmenter la perméabilitéendothéliale aux protéines plasmatiques.
Ces phénomènes inactivation de NO, augmentation dela perméabilité endothéliale, pontage et rigidificationdes protéines fibrillaires de la matrice extracellulairefont probablement le lien entre l'accumulation de produitsde glycation et le développement de pathologie vasculaireavec l'âge et le diabète.

27NO - Monoxyde d’azote et homéostasie cardiovasculaire
Comme toute approche pharmacologique, on peutdiviser les outils disponibles en deux grandes classes :les agonistes du système mîmant ses effets et lesantagonistes du système bloquant ces mêmes effets.L'approche pharmacologique est en règle utilisée àdeux fins différentes : soit d'ordre thérapeutique,l'agoniste ou l'antagoniste sont utilisés comme agentsthérapeutiques dans une situation donnée, soit d'ordrephysiopathologique, l'agoniste ou l'antagoniste sontutilisés comme un outil montrant l'implication dusystème endogène dans telle ou telle situation patho-logique. Cette deuxième approche est souvent difficiled'interprétation, en particulier en pharmacologie clinique,où les possibilités d'interventions multiples sont limitées.
Agonistes dusystème NO
Ils sont de deux grands types : les substances quirelarguent directement NO et les substances quiactivent la sécrétion de NO à partir de l'endothélium.On peut y ajouter les substances qui potentialisent leseffets de NO.
➙ Les dérivés nitrés
Par définition, comme leur nom l’indique, lesdérivés nitrés ou nitrodilatateurs sont des moléculespossédant un atome d’azote couplé à un ou plusieursatomes d’oxygène.Les dérivés nitrés sont des molécules anciennes.Nous l’avons vu, les effets thérapeutiques desdérivés nitrés furent rapportés dès 1867 ; et sonmédecin prescrivait déjà de la nitroglycérine àAlfred Nobel, à la fin du siècle dernier pour soulagerson angine de poitrine. Leurs effets biologiqueset thérapeutiques sont en rapport avec leurscapacités de se comporter comme des donneursdirects de NO. Ils incluent la nitroglycérine, lenitroprussiate de sodium, l’isosorbide dinitrate,la linsidomine, le nitrite d’amyl. Néanmoins il ya de nombreuses différences entre le mode defonctionnement du NO endogène et des dérivésnitrés exogènes. Ces différences incluent desvariations du métabolisme de ces substances enNO et comment ce métabolisme a lieu dans différentstissus.
La chimie des dérivés nitrés concerne les capacitésoxydatives de l’atome d’azote qui les constitue.La majorité des substances naturelles contiennentdes atomes d’azote dans leur forme réduite. Ceciinclue tous les amino acides, les peptides, lesacides nucléiques… 99 % de toute l’azote présentedans l'organisme est réduite. En particulier l’activitébactérienne à métaboliser les produits azotés nechange pas cet état réduit.
Le monoxyde d’azote représente une exception àcette règle dans la biochimie du vivant. Puisquede l’atome d’azote de l’arginine (appartenant au99 %) passe d’un état réduit à un état oxydé sousl’effet des NO synthases. Le point commun desdérivés nitrés est qu’ils contiennent de l’azotedans un état d’oxydation différent, ce qui permetla libération de NO indépendamment de l’activité desNO synthases. Pour certains dérivés nitrés lerelarguage de NO vient d’une simple réductionpar l’échange d’un électron. C’est le cas dunitroprussiate de sodium, où l’exposition à lalumière, où la rencontre d’agents réducteurscomme le déthiothreitol, le glutathion, ou despréparations de microsomes hépatiques, libèrentNO du nitroprussiate. Les membranes cellulaireselles-mêmes peuvent servir d’agents réducteurspour le nitroprussiate (y compris les membranesdes globules rouges, le NO alors produit, pouvant selier “in situ” à l’hémoglobine et induire la toxi-cité hématologique de ce type de molécule). Lecyanure libéré par la réaction peut égalementêtre toxique.
Les autres dérivés nitrés sont plus stables etnécessitent une activité enzymatique, différentede celle des NO-synthases pour relarguer NO.L’exemple le plus classique est la nitroglycérinequi, exposée au tissu vivant, relargue NO. Lesrésidus sulfhydrils (S-H) présents dans les protéinescellulaires servent de co-facteurs, d’agentsréducteurs, à la réaction. Le glutathion est lasource intracellulaire majeure de thiols. Mais sides donneurs de thiols augmentent la sensibilitéà la nitroglycérine, leurs déplétions ne semblentpas être la cause du phénomène de toléranceaux dérivés nitrés. L’enzyme responsable de laréaction est probablement la glutathion S-transférase.Cette enzyme est présente dans la cellule musculairelisse. Le bromosulfopthaléine qui l’inhibe, inhibela réponse aux dérivés nitrés. In vivo, la nitrogly-cérine se comporte essentiellement comme un
Pharmacologie du système NO

28 NO - Monoxyde d’azote et homéostasie cardiovasculaire
vasodilatateur des gros troncs coronaires et unvasodilatateur veineux.
La nitroglycérine a peu d’effet sur les vaisseauxrésistifs au contraire d’autres vasodilatateurscomme les inhibiteurs de l’adénosine déaminase(dipyridamole) ou certains inhibiteurs de phos-phodiestérases (papavérine).
Cette différence dans la cible, vaisseaux deconductance-vaisseaux de résistance, est en rapportavec la capacité des gros vaisseaux et l’incapacitédes petits vaisseaux (diamètre < 100 mm) demétaboliser la nitroglycérine en NO. Ce phénomèneest probablement en rapport avec l’abondanceou l’insuffisance de radicaux thiols intracellulaires,essentiellement le glutathion. Dans des expériences invitro, la réponse à la nitroglycérine est dépendante dela taille des vaisseaux, l’adjonction de cystéine(acide aminé porteur de thiol) rétablit uneréponse homogène des vaisseaux de toutes taillesà la nitroglycérine. La nitro-cystéine dilate defaçon identique les vaisseaux de grandes et depetites tailles.
Au total, le nitroprussiate est le plus actif maiségalement le plus toxique des dérivés nitrés.L’activité de la nitroglycérine est très dépendantede la capacité des tissus à la biotransformer enNO. Les dérivés nitrés se comportent donccomme des donneurs de NO, mais non sélectifs(nitroprussiate) pouvant aboutir à une vasodilatationextrêmement intense (proche de celle observéedans le choc septique) ; ou bien sélectifs (trinitrine)mais sur des bases non physiologiques, ne tenantpas compte des phénomènes de compartimentation.Des molécules pharmacologiquement actives,stimulant ou potentialisant spécifiquement le NOd’origine endothéliale, prenant en compte lacompartimentation du système sont très intéressantes.
➙ Molécules activant la sécrétion de NO
Système peptidomimétique etendothélium
Comme nous l’avons vu, dans son expérienceinitiale Furchgott employait l’acétylcholine pourrévéler la relaxation dépendante de l’endothélium. Enprésence de l’endothélium, l’acétylcholine parl’intermédiaire de ses récepteurs muscariniquesest vasodilatatrice. En l’absence d’endothélium,l’acétylcholine par l’intermédiaire de ces mêmes
récepteurs muscariniques est directementconstrictrice de la cellule musculaire lisse. Ceparadoxe n’est pas propre à l’acétylcholine.
La majorité des peptides et des amines vasoactifssont ambigus : ils possèdent à la fois des récepteurs à7 domaines transmembranaires couplés à lamobilisation du calcium dans la cellule endothélialeet dans la cellule musculaire lisse. L’interactionligand-récepteur sur la cellule endothéliale aboutit àl’augmentation de libération de NO et induit unevasodilatation indirecte ; alors que l’interactiondu même ligand avec le même récepteur sur lacellule musculaire lisse conduit à une constrictiondirecte. Ceci est dû au fait que la voie de signalisationintra cellulaire activée par le peptide ou l’amineest la même dans les deux types cellulaires. Cessubstances vasoactives se lient à un récepteur àsept domaines transmembranaires couplé à l’activitéde la phospholipase C par l’intermédiaire d’uneprotéine G et provoquent la mobilisation du calciumdans la cellule cible. Dans la cellule endothé-liale l’activation de cette voie de signalisation vaaboutir à l’augmentation d’activité de la NO synthasecalcium-calmoduline dépendante. Dans la cellulemusculaire lisse l’activation de cette voie vaaboutir à la contraction calcium-dépendante.Finalement un peptide, une amine, une substancevasoactive peut être à la fois vasodilatatriceendothélium-dépendante ou directement vaso-constrictrice.Tout dépend de son site d’administration exogène(pharmacologie ) , de la dens i té de récepteursdisponibles sur les deux cellules cibles et deleur richesse respective en enzymes capables dedégrader le principe actif.
De nombreuses substances se comportent commedes agonistes aigus de la sécrétion de NO parl’endothélium, l’agrégation plaquettaire, l’ATP,ADP, la sérotonine, la substance P, la stimula-tion α2 adrénergique, la bradykinine, l’angioten-sine II, l’endothéline 1, l’histamine…
Néanmoins, l'administration ou la potentialisationchronique de ces substances aboutit rapidementà des phénomènes de désensibilisation. En particulier,la potentialisation chronique de ces peptidesendogènes (quand i l s ex is tent ! ) peut abou-t i r, au-delà de la stimulation du calcium, à uneactivation de la PKC (inhibitrice de la NO synthase) etde la voie de production des formes radicalairesde l'oxygène via l'activation de la NADPH oxy-dase membranaire. L'effet vasodilatateur initial,se transforme rapidement en effet pro-oxydant.

29NO - Monoxyde d’azote et homéostasie cardiovasculaire
La major i té de ces subs tances peuvent avoi régalement une action constrictrice directe. Toutdépend de leur site de production prédominant,plasma versus milieu interstitiel en physiologie,et du taux d’occupation préalable des récepteursendothéliaux lorsqu’ils sont injectés par voieintraveineuse dans le compartiment plasma-endothélium.
Système adrénergique etendothélium
En général, les ß-bloqueurs, par leur effet dediminution du débit cardiaque et donc probablementleur tendance à diminuer le cisaillement endothélialsont considérés habituellement comme délétèrespour la fonction endothéliale. Par exemple, ilssont contre-indiqués de ce fait dans l’artérite desmembres inférieurs.
Néanmoins la pharmacologie d'interaction desagonistes et antagonistes adrénergiques avecl'endothélium est souvent complexe du fait : 1er - de la diversité des récepteurs
α et ß-adrénergiques sur l'endothélium ; 2ème - des effets agonistes partiels possibles des
inhibiteurs du fait de leurs analogiesstructurelles avec les antagonistes.
Seuls les récepteurs α2 adrénergiques semblent pré-sents sur l'endothélium. Leur stimulation active la voiede la phosphodipase C dans l'endothélium, mobilise lecalcium et provoque une sécrétion accrue de monoxyded'azote. Par contre, plusieurs types de récepteursß-adrénergiques sont présents et fonctionnels surl'endothélium humain, probablement les récepteursß1-adrénergique ; sûrement les récepteurs ß2 et ß3-adrénergiques. Leur stimulation active la sécrétion demonoxyde d'azote en interagissant avec la protéinekinase A (PKA), à laquelle ils sont couplés via l'AMPcyclique. La PKA phosphoryle la NO synthase. Ceteffet pharmacologique ß-adrénergique (phosphorylation)est plus pérenne que celui de la stimulationα-adrénergique (mobilisation du calcium).Les antagonistes ß-adrénergiques peuvent-être peusélectifs, touchant les différents récepteurs ß. Dans cecas, ils peuvent avoir un effet bronchoconstricteur liéà leur antagonisme des récepteurs ß2 prédominantsur la cellule musculaire bronchique. La stimulationß2-adrénergique provoque une bronchorelaxation pareffet direct de génération d'AMP cyclique dans la cellulemusculaire lisse bronchique (stimulation de la PKGcomme dans la cellule musculaire lisse vasculaire).Les ß-bloqueurs les plus récents sont sélectifs desrécepteurs ß1-adrénergiques présents dans le cœur.On les dit alors “cardiosélectifs” car dénués d'effet
bronchoconstricteur. Par contre du fait de leur analogiestructurale avec l'Isuprel, béta stimulant de référence,ils peuvent avoir des effets agonistes sur les récepteursß2 et ß3.
C'est le cas en particulier du Nebivolol, extrêmement sélec-tif des récepteurs ß1 cardiaque en terme d'antagonisme etpossédant probablement des propriétés stimulantes agonistesdes récepteurs ß3, provoquant une vasorelaxation péri-phérique en rapport avec une augmentation pérennede sécrétion de monoxyde d'azote par l'endothélium.
➙ Potentialisateurs de NO
Une autre manière pharmacologique de manipuler lesystème NO est d’en faciliter l’action, soit en protégeantNO lui-même, soit en potentialisant les effets du GMPcyclique, second messager de NO dans la cellule muscu-laire lisse. Les anti-oxydants potentialisent les effets deNO. Ceci fut montré par l’équipe de P. Van Houtteinitialement dans une expérience sur anneau artériel,où l’adjonction de super-oxyde dismutase (chélateurdes formes réactives de l’oxygène) potentialisait les effetsrelaxants endothélium dépendant de l’acétylcholine.
Comme nous l’avons vu, NO peut interagir avecl’anion superoxyde pour former du peroxynitrite. Lasuperoxyde dismutase est capable de dismuter l’oxygèneradicalaire. La simple exposition de cellules endothélialesà la superoxyde dismutase induit une relaxation descellules musculaires lisses indépendamment de toutestimulation. Le blocage de la SOD par le diéthyldithio-carbamate diminue les effets de NO montrant quel’activité de la SOD est dépendante de la présenced’un atome de zinc à son site actif. Des animaux adultesrendus déficients en zinc, deviennent hypertendus etprésentent une relaxation endothélium-dépendante trèsperturbée. De ce fait, l’ensemble des molécules ayantà long terme une activité anti-oxydante : vitamine E,probucol, statines… devrait faciliter les effets de NO.
NO agit comme relaxant de la cellule musculairelisse en utilisant le GMP cyclique comme secondmessager. Dans la cellule musculaire lisse, lemétabolisme du GMP cyclique est le fait d’enzy-mes intracellulaires appelées phosphodiestérases.Il existe plusieurs formes de ces enzymes capablesde dégrader le GMPc, mais la phosphodiestérase-V(PDE-V) est spécifique du GMPc et de la cellulemusculaire lisse. De ce fait, cette PDE-V est unecible moléculaire intéressante à bloquer afin depotentialiser les effets de NO. Les inhibiteurs spécifiquesde PDE-V facilitent et prolongent les relaxationsdépendantes du monoxyde d'azote. Ils ont un effethypotenseur généralisé chez l’animal hypertendu.

30 NO - Monoxyde d’azote et homéostasie cardiovasculaire
Antagonistes dusystème NO
Là également ils se divisent en deux grandesclasses : les molécules capables de bloquer laproduction de NO par les NO synthases et lesmolécules capables d’inactiver NO.
➙ Les arginines substituées
C’es t à l ’équipe de Moncada que l ’on doi tla première utilisation pharmacologique d’unanalogue substitué sur la fonction amino-guanidine del’arginine comme antagoniste de la productionde NO dans le système cardiovasculaire. Ce premierantagoniste compétitif de la L-arginine sur lesNO synthases est le N-monométhyl-L-arginine(L-MMA) (figure 18).Comme dans d’autres systèmes enzymatiques, lamodification chimique du substrat naturel (L-arginine)produit un inhibiteur compétitif. D’autres moléculesdu même type se sont également avérées trèsefficaces :- la L-N-nitro arginine + méthyl ester (L-NNA,
L-NAME), - la N-N-dimethyl L-arginine (L-ADMA),- la L-canavanine,- la N-iminoethyl-L ornithine (L-NIO),
Ces différentes molécules ont en commun d’êtreau départ une molécule d’arginine substituée surla fonction amino-guanidine. Si les fonctionsNH2 de la L-arginine sont substituées, l’analogueen résultant ne peut pas servir de substrat. Unallongement trop important de la chaîne diminuele pouvoir antagoniste.
Le problème posé par l’utilisation de ces analoguessubstitués est qu’ils ne sont pas spécifiques detelle ou telle isoforme de NO synthase. Même sila L-NIO, la N-amino-L-arginine et la L-canavaninesont réputées plus actives sur la NOS inductible,alors que la N-nitro-arginine est plus inhibitricede NO synthase endothéliale, que sur l'isoformeinductible, il n’en demeure pas moins que l’arginineest le substrat commun aux différentes isoformeset que les analogues substitués ont un pouvoirinhibiteur sur les différentes NO synthases.
L’intérêt de ces molécules est d’ordre pharmaco-logique. Ce sont des molécules puissammentactives par voie orale, bien absorbées, rentrantdans les cellules cibles par le même mécanismeactif que l’arginine elle-même.
Dès les premières expériences avec la L-NMMA,l’équipe de Moncada démontrait que l’adjonction deces antagonistes dans un système vasculaire réactifinduisait une contraction tonique des cellules muscu-laires lisses vasculaires, et que in vivo cette contraction
L- arginine
Analoguessubstitués surle groupementaminoguanidine= antagonistes
NH2
NH2HN
NH
N R
NH2HN2
HN2
COOH
COOH
Figure 18

31NO - Monoxyde d’azote et homéostasie cardiovasculaire
se traduisait par une augmentation de pression et unediminution de débit. Ces expériences réalisées audépart en aiguë sur organes isolés furent étendues àl’administration chronique de ces antagonistes dans desorganismes entiers vivants. Du fait de sa solubilité,c’est le L-NAME qui est le plus utilisé par voie oraledans différentes espèces animales. Ces molécules sontfréquemment utilisées en pharmacologie aiguë pourmontrer l’endothélium-NO-dépendance d’un phénomène.Par exemple, les effets vasodilatateurs du nébivolol sontbloqués par les arginines substituées ce qui suggèrefortement l’endothélium-dépendance du phénomène.
Chez le rat de laboratoire, l'administration chronique deL-NAME s'accompagne d'une hypertension artérielledont le niveau est proportionnel à la dose administrée,d'une diminution globale des débits: débit cardiaque,débit rénal… et d'une diminution des dimensions deslumières vasculaires et des cavités cardiaques. Cesrésultats, utilisant un antagoniste du système NO,montrent qu'il existe en permanence un tonus endogènevasodilatateur dépendant de NO. La suppression partielleou complète de ce tonus augmente de façon diffuse leniveau de contraction des cellules musculaires lisses
vasculaires ayant pour conséquences : une augmentationdes pressions, une diminution des débits et un remodelagevasculaire caractérisé par une diminution du diamètre dela lumière artérielle.
En l'absence de NO (L-NAME), le taux de GMPcyclique est très bas et la G-kinase ne peut découplerle système membranaire de transduction du signal devasoconstriction : pour la même quantité d'angiotensine IIet de récepteur, la transduction du signal est maximum,conduisant à une vasoconstriction accrue. En présenced'un inhibiteur du système rénine-angiotensine, malgrél'absence de remontée du taux de GMP cyclique, undes principaux systèmes vasoconstricteurs ne peut pastransduire le signal. De ce fait le signal de vasocons-triction revient à la normale. En l'absence de NO(L-NAME) il existe une hypersensibilité du systèmevasculaire à l'angiotensine II et au catécholaminesendogènes, une hypersensibilité aux donneurs de NOexogènes (dérivés nitrés) et une désensibilisation partielleà l'angiotensine II exogène.
Ces arginines substituées peuvent être égalementutilisées par voie locale afin de bloquer in situ
L-arginine
L-citrulline
L-NA
hémoglobineanion superoxyde
NO synthase
NO
1er messagerGTP
bleu de méthylène LY 83 583
GMP cyclique2nd messager effets physiologiques
Guanylate Cyclase soluble
Figure 19

32 NO - Monoxyde d’azote et homéostasie cardiovasculaire
un phénomène d'hyperhémie. Par exemplele L-NAME administré in situ dans un modèled'inflammation locale cutanée et sous-cutanéeprovoquée par injection locale de bradykinine,d'histamine… supprime l'œdème, l'accumulationdes éléments figurés du sang, d'hyperhémie…
Les guanidines sont une autre classe de substancesamino-aromatiques capables d'inhiber les NOsynthases , mais e l les sont peu spéci f iques(inhibition du métabolisme de l'histamine, inhibitionde la glycation des protéines...). Certains dérivésdes guanidines comme la 7-nitro-indazole semblentinhiber re la t ivement spéci f iquement la NOSneuronale plutôt que la NOS endothéliale.L 'aminoguanidine, peu spéci f ique, b loquel 'ac t ivité de la NO synthase inductible.
➙ Antagonistes de NO
Comme nous l'avons vu, NO est une espèce radicalairequi se lie de façon covalente à ses cibles. Uneautre approche possible du blocage de NO est dele mettre en présence d'autres hèmes protéines,auxquelles ils se lient. Ces hèmes protéines rentranten compétition avec la guanylate cyclase. C'estle cas de l'hémoglobine. Comme l'oxygène, NOlie l'atome de fer de l'hème. L'affinité de NOpour l'hémoglobine est 100.000 fois supérieure àcelle de l'oxygène, la présence d'oxygène augmentela liaison de NO à l'hémoglobine (figure 19).
En présence d'hémoglobine extracellulaire, leNO libéré par l'endothélium, est capté et ne peutactiver la guanylate cyclase soluble intracellulairedes cellules musculaires lisses. Par exemple, dessubstituts de synthèses transporteurs d'oxygène,mimant les effets de l'hémoglobine, ont un effethypertenseur quand ils sont injectés par voiegénérale par captage du monoxyde d'azote endo-théliale.
En corollaire, une molécule comme le bleu deméthylène, qui lie l'atome de fer de l'hème,inhibe la liaison de NO à la guanylate cyclase età l'hémoglobine (le bleu de méthylène peut êtreutilisé pour reverser l'intoxication de l'hémoglobinepar le nitroprussiate). Ces antagonistes sontessentiellement utilisés in vitro en pharmacologie.

33NO - Monoxyde d’azote et homéostasie cardiovasculaire
NO et vaisseaux :effet vasodilatateur
Le monoxyde d'azote d'origine endothéliale estessentiellement impliqué dans la régulation desénergies cinétiques dans le système circulatoire : - Le cisaillement endothélial lié au flux sanguinentretient une activité basale de sécrétion deNO par l'endothélium. Ce phénomène est diffusà tout l'organisme, générant un “tonus” vasodi-latateur qui s'oppose au “tonus” vasoconstric-teur et participe ainsi au maintien de la pressionartérielle dans les limites physiologiques.- L’endothélium-dépendance est le mécanismemajeur d'adaptation du système vasculaire deconductance aux besoins énergétiques périphéri-ques. Ce mécanisme permet l'adaptation spécifi-que des vaisseaux de conductance concernés parle territoire périphérique métaboliquement actif.Par exemple c'est le mécanisme par lequel lesartères coronaires s'adaptent à l'augmentationde travail cardiaque, les artères des membres àl'exercice physique, l'artère mésentérique à ladigestion, etc.
➙ Adaptation à l’effort
L'augmentation du métabolisme musculaire(exercice) va libérer localement des substancesdérivées de l'ATP comme l'adénosine.L'adenosine a une demi-vie très courte, elle estmétabolisée in situ et a une action directe sur lacellule musculaire lisse vasculaire au contact dutissu. L'adonosine se lie à un récepteur et activela production d'AMP cyclique dans la cellulemusculaire lisse artérielle. Ce phénomène restestrictement limité aux artérioles terminales intra-musculaires. C'est un phénomène fonctionnelle-ment centripète. Par exemple, des moléoculescomme le dipyridamole (persantine) ou le carbo-chromène qui inhibent la dégradation de l'adé-nosine, ont un effet de vasodilatateur strictementartériolaire, sans effet sur les vaisseaux deconductance. Du fait du métabolisme immédiatde ces substances, il n'y a pas de possibilité dediffusion du processus de vasodilatation métabo-lique aux vaisseaux de conductance(figure 20).
Rôle physiologiquedu monoxyde d'azote
résistanceséchangescapillaires
vasodilatationendothélium-dépendante
conductance
communicationscentrifuges communications
centripètes
vasodilatation métabollique Figure 20

34 NO - Monoxyde d’azote et homéostasie cardiovasculaire
L'adaptation des vaisseaux de conductance à lavasodilatation métabolique périphérique passe parl'augmentation de vitesse d'écoulement du sang dansce territoire (augmentation du gradient de pression pardiminution des résistances les plus périphériques),conduisant à l'augmentation du cisaillement endothélial,à la mobilisation du calcium et à la phosphorylation dela NO synthase dans la cellule endothéliale, à l'aug-mentation d'activité de la NO synthase constitutivecalcium-calmoduline dépendante, et à la productionde NO (figure 21).
Au repos, il existe un flux sanguin cisaillant l'endothéliumet générant NO, à l'exercice, le cisaillement augmente et laproduction de NO aussi, relaxant plus les cellules muscu-laires lisses sous-jacentes et adaptant le calibre du vaisseaude conductance à l'augmentation de débit. Ce phénomèneest un phénomène de proche-amont, centrifuge, localisé
aux seuls vaisseaux de conduc-tance concernés par le territoireen vasodilatation métabolique, oùles médiateurs viennent du com-partiment sang-endothélium etvont vers la cellule musculairelisse. Ce mécanisme permet larépartition harmonieuse du débitcardiaque en fonction desbesoins métaboliques. Cettedébit-dépendance de la vasodila-tation peut s'explorer chezl'homme en mesurant les varia-tions de diamètre de vaisseaux deconductance en réponse à l'exer-cice ou à une épreuve d'ischémietransitoire reproduisant une vaso-dilatation artériolaire périphérique(figure 22).
Cette description ne tientcompte que d'un phénomèned'adaptation immédiate portantsur l'activité de la NOS IIIendothéliale, dépendante dutaux de calcium libre intracel-lulaire et du niveau de phos-phorylation de l'enzyme.Lorsque la stimulation par lecisaillement est pérenne (exer-cices répétés, entraînement) larégulation ne porte plus seule-ment sur l'activité de l'enzymemais également sur une induc-tion d'expression de la protéine(augmentation du nombre demolécules versus augmentationde l'activité de chaque molé-
cule) dans les cellules endothéliales. Inversement enl'absence de stimulation chronique par le cisaillement,non seulement l'activité mais également l'expressionde la NOS endothéliale diminue.
➙ NO comme neuromédiateurpériphérique : exemple de l'érection
A l'inverse de la communication centrifuge duNO endothélial: flux sanguin-cisaillement endothélial-cellule musculaire lisse, la signalisation du NOneurogène est centripète : terminaisons nerveusesadventitielles-cellules musculaires lisses.
C'est dans le tractus intestinal que le NO neurogène aété le mieux analysé. La présence de NO synthaseneuronale a été mise en évidence dans les plexus
Guanylate cyclase solubleGTP
GMP cyclique
relaxationCa++
Kinase G
Cellule musculaire lisse
Contraintes hémodynamiquesde cisaillement
Ca++
récepteurs
bradykinineacétylcholinedopamineangiotensineendothéline
arginine citrulline
NO
kinases
NO
Cellule endothéliale
synthase constitutive
Figure 21

35NO - Monoxyde d’azote et homéostasie cardiovasculaire
mésentériques. Des sourisinvalidées pour le gènede la NO synthase neuro-nale présentent unehypertrophie circulaire dela musculeuse intestinaleavec sténose hypertrophi-que du pylore.
C'est dans l'érectionpénienne que le rôle vascu-laire de la NO synthase neu-ronale a été le mieux identi-fié. Les plexus pelviens, lesnerfs caverneux et les termi-naisons nerveuses de l'ad-ventice des artères et dessinusoïdes de la périphériedes corps caverneux sontparticulièrement riches enNO synthase neuronales.A l'état flacide, le flux san-guin dans les corps caver-neux est limité par le tonusvasoconstricteur imposé parle système nerveux sympathique α-adrénergique sur lessphincters artériolaires pré-caverneux. L'érection est laconséquence d'une activation centrale des voiesnitroxydergiques à destinée pelvienne. Le NO sécrétépar les terminaisons nerveuses va activer la voie duGMP cyclique et de la G-kinase dans la cellule muscu-laire lisse des sphincters pré-capillaires, et empêcher letonus vasoconstricteur provoquant une augmentationimmédiate du remplissage sanguin des corps caverneux.Ces phénomènes sont neurogènes, la stimulation desécrétion de NO neuronal par des neuropeptides estresponsable de la vasodilatation des entrées vasculaires.Les inhibiteurs de phosphodiesterase V, potentialisant letaux intracellulaire de GMP cyclique, prolongentl'érection. C'est comme cela qu'agit le viagra.Les plexus pelviens ne sont pas les seules terminaisonsdu système nerveux autonome à posséder la NOsynthase neuronale. En particulier, il existe une inner-vation autonome du système vasculaire coronarien,non-adrénergique, non-cholinergique, impliquant laNO synthase neuronale (nitroxydergique) et participantdu contrôle de la vasorelaxation coronaire en associationavec le contrôle métabolique et la flux-dépendance.
➙ NO synthases et rein
Dans le rein les trois isoformes de NO synthasessont présentes en physiologie.- La NO synthase endothéliale (NOS III) est présentedans l'endothélium des vaisseaux corticaux, desartérioles pré et post glomérulaires, des capillairesglomérulaires.- La NO synthase macrophagique inductible(NOS II) est constitutivement exprimée dans lerein et il y a de nombreuses évidences que cetteisoforme soit impliquée dans l'homéostasie dusodium. La NOS II semble être exprimée dans lescellules musculaires lisses des artérioles corticales etdans différents segments du néphron.- La NO synthase neuronale (NOS I) est essen-tiellement exprimée dans les cellules épithélialesde la macula densa de l’appareil juxtaglomeru-laire (cellules assurant les communications entre letubule distal et les artérioles du système porteglomérulaire, contenant les cellules myoépithé-loïdes sécrétrices de rénine, et contrôlant lapression de perfusion dans le glomérule : retro-contrôle tubulo-glomérulaire).
La production de NO par le rein participe de la régula-tion d'excrétion du sodium et de la sécrétion de rénine.Néanmoins, du fait de la présence des trois isoformes dansdes compartiments différents du rein : compartimentvasculaire, compartiment glomérulaire, compartiment tubulaireépithélial, il est difficile actuellement de faire une analyse fine
Ca ++
Flux-AgonistesBradykinineVasopressine (V1)EndothélineATPThrombine
PlaquettesMacrophages
PLC
Ca ++
calmoduline
NO synthase (III)
L- Arginine NO
Cellules musculaires lisses
…………….
phosphorylations
Cellules endothéliales
Figure 22

36 NO - Monoxyde d’azote et homéostasie cardiovasculaire
de la participation spécifique de telle ou telle isoforme à lasécrétion de rénine ou à la fonction tubulaire, voire à lafonction glomérulaire.
Dans la cellule myoépithéloïde de l'artériole afférente auglomérule, la sécrétion de rénine est sous le contrôle de lavoie des inositols phosphates : plus le calcium est mobilisé,moins la rénine est sécrétée. La cellule myoépithéloïdefonctionne comme une cellule musculaire lisse. Tous lespeptides vasoactifs comme l'angiotensine II en mobilisantle calcium dans la cellule myoépithéloïde, diminuent lasécrétion de rénine, In vivo, il semble que l'augmentationde production de NO augmente la sécrétion de rénine. Enfait la NO synthase neuronale des cellules épithéliales dela macula densa participe au rétro-contrôle tubulo-glomé-rulaire et à la régulation de la sécrétion de rénine. En casde charge sodée, la réabsorption du chlore, du sodium etdu potassium (Na.K.2Cl) est augmentée au niveau de lamacula densa et la sécrétion de NO par ces cellules estdiminuée, provoquant une vasoconstriction de l'artérioleafférente au glomérule. Inversement en cas de régime sanssel ou de prescription de natriurétiques thiazidiques(Lasilix), qui inhibe la réabsorption Na.K.2Cl, l'activité dela NO synthase neuronale épithéliale de la macula densaest augmentée provoquant une vasodilatation de l'artérioleafférente au glomérule, associée à une augmentation desécrétion de rénine et de production d'angiotensine II,provoquant une vasoconstriction de l'artériole efférente auglomérule. Ces deux phénomènes associés, vasodilatationpréglomérulaire, NO-dépendante, vasoconstriction post-glomérulaire angiotensine II dépendante participe dumaintien de la filtration glomérulaire.
Lors de leur transdifférentiation en cellules myoépithélioïdesà rénine, les cellules musculaires lisses changent leurpatron d’expression génique et en particulier expriment defaçon constitutive la G-kinase II épithéliale. Dans les cellulesmyoépithélioïdes, la GKII est associée aux granules derénine. Cette isoforme des GKs joue le rôle prédominantdans l’effet inhibiteur du NO/GMPc sur la sécrétion derénine.
En physiologie, la réponse à l'administration aiguë d'unantagoniste de l'arginine sur les NO synthases se caractérisepar une diminution du flux sanguin rénal, une diminutionde la filtration glomérulaire et du coefficient de filtration,une diminution de sécrétion de rénine, une diminution del'excrétion du sodium et une augmentation importante dela protéinurie.
En ce qui concerne l'excrétion sodée, un défaut de NO estassocié à une rétention hydrosodée sans que l'on sacheclairement définir le lien de cause à effet. Il est possibleque le rôle pathologique d'un défaut de NO dans l'hyper-tension artérielle passe par un défaut rénal d'excrétion dusodium, nous y reviendrons.
NO synthases etcœur
Les trois isoformes de NOS sont exprimées dansle myocarde mais leur expression est spécifiquede la cellule considérée. NOS3 (eNOS) est présente àla fois au niveau des cellules endothéliales situéesau niveau des coronaires intramyocardiques et del’endocarde mais aussi dans les cardiomyocytes auri-culaires, ventriculaires et les cellules du nœudauriculo-ventriculaire. Dans les cellules endothélialescomme dans les cardiomyocytes, cette isoforme estlocalisée au niveau des cavéoles de la membraneplasmique. A l’état basal, l’activité de NOS3 descellules endothéliales est inhibée du fait de sonassociation avec la cavéoline-1, protéine structuraledes cavéoles. Les cardiomyocytes expriment constituti-vement eNOS également associée aux cavéolines(cavéoline-3), et nNOS (NOS1) au niveau de leurreticulum sarcoplasmique. En revanche, dans lesconditions physiologiques, ils n’expriment pas iNOS(NOS2). Cette isoforme est uniquement exprimée dansdes situations de souffrance cardiaque. L’ensemblereprésente une source de production cardiaque de NOimportante et variée.Les études relatives aux effets du NO sur la fonctionmyocardique ont principalement porté sur lesmodulations par le NO de la contractilité, de larelaxation, et de la distensibilité.
➙ Rôle physiologique des donneursde NO dans le cœur sain
Les donneurs de NO (exogène ou endogène) produisentdes effets biphasiques sur la force de contractionmyocardique en fonction des concentrations utilisées.Ainsi, de faibles concentrations de donneurs de NOstimulent le courant calcique (myocyte auriculairehumain), potentialisent la stimulation ß-adrénergique,et amplifient l’inotropisme de myocytes isolés.Ces effets sont consécutifs à l’inhibition de PDE IIIet donc à une faible augmentation de la concentrationen AMPc intracellulaire. A plus fortes concentrations,les donneurs de NO inhibent la stimulation ß-adré-nergique du courant calcique et réduisent l’effetinotrope positif de ces agonistes sur des cardio-myocytes isolés. Ces effets font intervenir la PDE IIou la PKG (figure 23).In vivo (i.e. sur des préparations multicellulaires),les effets inotropes directs du NO sur la contractionmyocardique sont généralement masqués par seseffets hémodynamiques vasculaires qui, par la miseen jeu du baroreflexe et la loi de Frank-Starling,vont modifier la contractilité. Afin d’éviter les

37NO - Monoxyde d’azote et homéostasie cardiovasculaire
artefacts dus à ces effets systémiques, la fonctioncardiaque est évaluée après application locale(intracoronaire) des donneurs de NO. Ainsi, chezdes sujets sains, l’injection intracoronaire d’undonneur de NO (nitroprussiate de sodium) modifieles deux principaux déterminants de la fonctiondiastolique du ventricule gauche : la relaxation etla compliance. Après 5 minutes d’administrationde nitroprussiate, la relaxation plus pécoce duventricule gauche entraîne une baisse des pressionsmaximales engendrées par le ventricule gauche.De plus, le nitroprussiate entraîne une augmentationdu volume télédiastolique et une baisse de la pressiontélédiastolique du ventricule gauche caractériséepar le déplacement vers le bas et la droite de larelation pression-volume diastolique du ventriculegauche. Ces effets sont alors en faveur d’une meilleuredistensibilité du ventricule gauche. En revanche,les paramètres de la fonction systolique ne sontpas ou peu modifiés.Pour certains auteurs, ces effets du NO sont attribuésaux modifications de la sensibilité au calcium des myo-filaments. La sensibilité au calcium des myofilamentsest connue pour intervenir dans la réponse à l’étirementdu muscle cardiaque, ce qui laisse supposer que le NOpeut moduler la réponse de Frank-Starling. Autrementdit le NO, en modifiant les propriétés diastoliques ducœur, pourrait améliorer la réponse du myocarde à uneélévation de la précharge. Il a été ainsi démontré quel’inhibition non sélective des NOS par le L-NMMA oudu NO par l’hémoglobine diminuait significativementl’augmentation du débit cardiaque induit par une
augmentation de laprécharge. L’effet duL-NMMA, opposé àcelui du nitroprus-siate, est égalementinhibé en présence deL-arginine. Cet effetdu L-NMMA est posi-tivement corrélé auniveau de préchargeventriculaire, ce quisuggère que la libéra-tion et/ou l’action duNO sont plus impor-tantes lors d’augmen-tation du volume ven-triculaire télésystoli-que. Ceci suggèreque l’étirement de lachambre ventricu-laire gauche induitune libération localeaccrue de NO quiaugmente la distensi-
bilité et le volume de remplissage ventriculaire, etdonc le débit cardiaque. L’influence de l’étirement surla libération du NO myocardique semble un méca-nisme de régulation majeur. Le processus de mécano-transduction mis en jeu n’est pas complètement établi,mais ferait intervenir le cytosquelette d’actine qui estconnecté aux cavéoles, site majeur de localisation desNOS au niveau des cellules endothéliales. Pour résumé, l’isoforme de NOS impliquée dans cesmodifications de la fonction diastolique est sujet àdébat. Une majorité d’auteurs tiennent le NO d’origineendothéliale (NOS3) pour responsable de l’améliorationde la relaxation et de la distenibilité NO-dépendantes,la majeure partie de ces effets étant observés aprèsstimulation par la substance P qui libère le NO d’origineendothéliale.Ces travaux montrent que le NO exogène modifieraitprincipalement la fonction diastolique, en particulierla relaxation avec peu ou pas d’effet direct sur la fonctionsystolique. Cependant, les effets des donneurs de NOdoivent être interprétés avec prudence, car leurspropriétés chimiques ne sont pas celles du NO endogène.De plus, le site (membrane versus reticulum sarcoplas-mique) et la vitesse de production du NO endogène(NOS2>NOS3≥NOS1) sont apparus comme des para-mètres essentiels pour déterminer l’implication de cemessager.
p+
Ca ++
phosphorylation
adenylatecyclase
PK A
AMP
Cardiomyocyte
NO Guanylate cyclaseGMPc
GTP
PDE II+
ATP
isuprel
récepteur ß
AMP cyclique
Figure 23

38 NO - Monoxyde d’azote et homéostasie cardiovasculaire
➙ Rôle physiologique du NOendogène dans le cœur sain
Contrairement aux effets des donneurs de NO, le rôledu NO endogène (produit par les NO synthases) sur lacontractilité myocardique basale est variable selon lesétudes et surtout plus controversé. Les mécanismesdépendants du NO endogène impliqués dans la régulationdes propriétés contractiles sont complexes d’autant quela plupart des études sont réalisées sur des préparationsmulticellulaires et ne permettent donc pas de distinguerl’origine cellulaire du NO (cellules endothéliales, cellulesmusculaires…). Par ailleurs, il est maintenant reconnuque certains types cellulaires, tels les cardiomyocytes,possèdent une ou plusieurs isoformes de NOS (NOS3et NOS1), pouvant en outre être situées dans des loca-lisations sub-cellulaires différentes (figure 24). Cettevariabilité de localisation intracellulaire va conférer auxdifférentes isoformes de NOS présentes dans unemême cellule des fonctions différentes voire opposées,et ce bien qu’elles produisent toutes du NO.
➙ Les NOS du myocyte et le rôle autocrine du NO
Jusqu'à récemment, NOS3 semblait être la seule isoformeconstitituve de NOS exprimée dans le cardiomyocyte etdonc la seule isoforme impliquée dans les régulations dela fonction myocardique à l'état basal. Il existait en effet
des arguments fonctionnels en faveur d’un rôle autocrinede la NOS3. Des myocytes ventriculaires isolés et stimulésélectriquement sont capables d’engendrer du NO, pro-duction associée à une atténuation de la relation positivecontraction-fréquence. Ces deux phénomènes sont abolispar un inhibiteur non-sélectif des NOS. Les premièresétudes rapportent également une réduction de l’effetinotrope positif induit par les catécholamines.Cependant, les résultats concernant l’implication de cetteisoforme sont en partie contradictoires. Ainsi, les effetsinotropes observés lors d'une inhibition non sélective desNOS n'ont pas été retrouvés lors de plusieurs étudesréalisées chez des souris déficientes en NOS3 (NOS3-/-).Certains auteurs observent chez ces souris une augmenta-tion de la réponse ß-adrénergique qui est concomitanted'une augmentation de la transitoire calcique. D’autreséquipes, au contraire, montrent une diminution voireune absence de la réponse ß-adrénergique, voire décriventune diminution de cette dernière. Chez l’homme, l’inhi-bition non-sélective des NOS n’entraîne pas de modificationsde la force de contraction et de la relaxation myocardique,aussi bien à l’état basal que sous stimulation ß-adrénergique,suggérant que le NO endogène n’interviendrait pas demanière significative dans la régulation de la fonctioncardiaque (figure 25).L’ensemble de ces travaux suggèrent que le NO produitpar la NOS3 myocytaire à l’état de base ou après stimula-tion béta-adrénergique n’a pas ou peu d’effet modulateurde la fonction contractile.
acetylcholine
vaisseau
contraction
noradrenaline
NO
NO
NO
NO
SR
nNOS
eNOS
eNOS
nNOS
nNOS
CaCa
Signalisation AMPc
Signalisation NO/GMPc
NO/GMPc independant
Ca
ICa
ß1
ß3
RYR PLB
ATPase
SERCA
? ?
?
Gi
Gi
M2
Na
K
Myocyte cardiaque
Figure 24

39NO - Monoxyde d’azote et homéostasie cardiovasculaire
Les divergences actuelles s’expliquent, en partie, par lesdifférences de préparations utilisées et de protocoles expéri-mentaux. Ces dernières années, deux explications sontvenues éclairer ces divergences. D’une part, le fait que lescatécholamines puissent induire des effets inotropes opposéspeut s’expliquer par l’activation de plusieurs sous-types derécepteurs ß-adrénergiques couplés à des voies de signalisationdifférentes en fonction des concentrations de ß-agonisteset/ou de la sélectivité de ces derniers. En effet, dans le myo-carde humain, l’activation des récepteurs ß3-adrénergiquesstimule la voie du NO conduisant à une diminution de lacontractilité, alors que la stimulation des récepteurs ß1 etß2-adrénergiques induisait un effet inotrope positif.La seconde explication réside dans la découverte récente del'expression de NOS1, au niveau du réticulum sarcoplasmiqueà proximité des récepteurs à la ryanodine. Ces travaux sug-gèrent ainsi que cette isoforme pourrait être un des élémentsmodulateurs du couplage excitation-contraction, et ainsiresponsable des modifications de la fonction systoliqueNO-dépendante en particulier par la régulation des flux cal-ciques. Afin de mieux comprendre le rôle de cette isoformesur la fonction cardiaque, des études ont alors été réaliséessur des souris dont le gène NOS1 a été invalidé. Cependant,ces travaux mettent en évidence deux phénotypes cardia-ques différents. Certains auteurs rapportent l'absence de dif-férence in vivo de la pression artérielle, de la relaxation, dela fréquence cardiaque et une élévation isolée du paramètrede contractilité dP/dt/P entre les souris déficientes en NOS1et les souris contrôles. A l'opposé, d’autres auteurs décriventune augmentation de la contraction basale et une améliora-tion de la relaxation de myocytes isolés chez les souris défi-cientes en NOS1, comme chez les souris contrôles traitéespar un inhibiteur sélectif de la NOS1 (LVNIO). Ces résultatsont été confirmés par l'analyse de la fonction cardiaque paréchocardiographie. La fraction d'éjection des souris déficien-tes en NOS1 était significativement plus élevée que celle des
contrôles. De plus, ils démontrent que chez la souris déficienteen NOS1 la transitoire calcique était potentialisée suite àl’augmentation du courant calcique via les canaux calciquesde type L et par une augmentation de la charge calcique duréticulum sarcoplasmique.La stimulation ß-adrénergique des souris déficientes enNOS1 est également modifiée. Mais une fois de plus lesrésultats sont opposés d’un groupe de recherche à l’autre.Ainsi, les souris déficientes en NOS1 présentent une diminu-tion de la réponse à la stimulation ß-adrénergique par rapportaux souris contrôles (résultat opposé pour NOS3-/-) aussi bienpar la technique de courbe pression-volume in vivo que parla technique de myocyte isolé. Ainsi NOS1 participerait à laréserve contractile myocardique et induirait des effets opposésà ceux de NOS3 sur les flux calciques. Les auteurs suggèrentque NOS1 régulerait l'activité des récepteurs à la ryanodine(RyR2) via des interactions entre NOS1-RyR2. Ces résultats,concernant NOS1, n'ont pas été confirmés par le secondgroupe. A l’inverse, les myocytes isolés de souris déficientesen NOS1, comme ceux des souris traitées par LVNIO, sontcaractérisés par une augmentation de leur réponse contractileà l'isoprotérénol. L’ensemble de ses données suggèrent fortement que NOS1interviendrait dans la régulation des flux calciques et doncde la contractilité.
➙ NOS3 endothéliate et le rôle paracrine du NO
L’effet paracrine de la NOS3 a été étudié in vitro dansdes co-cultures de cellules endothéliales et de myocytescardiaques, où l’effet inotrope négatif de la bradykininesur les myocytes est bloqué par un inhibiteur de NOS.Cette diminution de contractilité semble due auxcellules endothéliales, ce peptide vasorelaxant n’exerçantpas d’effet inotrope sur les cardiomyocytes en mono-culture. Cette observation a été confirmée ex vivo surdes muscles papillaires ou des cœurs isolés, où lastimulation de l’activité de NOS3 (vasculaire et endo-cardique), par la bradykinine ou la substance P, accélèrela relaxation diastolique, sans modification de lafonction systolique. L’utilisation d’électrodes à por-phyrines, sur ces cœurs isolés, a en parallèle permisde détecter un authentique flux sortant de NO lors dela diastole cardiaque. Ce flux sortant est accru lorsd’élévation de la précharge et est stimulé par desfacteurs mécaniques qui augmentent la compressiondu myocarde, tels que l’augmentation du volume télé-diastolique ou de la pression de transmurale. Chezl’homme, l’injection intracoronaire de substance Pinduit également l’accélération de la relaxationcardiaque. Ces études montrent donc que l’activitéde NOS3 myocardique (vasculaire et endocardique)règle des paramètres fondamentaux de la contrac-tilité (cardiomyocytes) cardiaque.
[NO]
Cœur normalForce
Stimulation ß adrenergique
État Basal
Effet inotrope positif après inhibition des NOS
Figure 25

40 NO - Monoxyde d’azote et homéostasie cardiovasculaire
De l'implication physiologique de NO découleson rôle physiopathologique dans un certainnombre de pathologies cardiovasculaires. Unexcès de NO peut être pathologique. Un défautd'activité de NO peut participer de nombreusespathologies : hypertension, diabète, athérosclérose,insuffisance cardiaque. Néanmoins dans cespathologies une insuffisance de productionbasale de NO n'est pas forcément en cause. Ilpeut s'agir d'un défaut d'efficacité du NO endo-thélial ; d'une non-adaptation à l'augmentationde cisaillement (exercice) ; et surtout de phénomènede décompartimentation plus qualitatif que quan-titatif, correspondant en règle à un déplacementd'expression de la NO III endothéliale, vers laNOS II inductible dans les cellules musculaireslisses, et à un défaut immédiat d'adaptation auxconditions de flux.
Pathologie par excès de NO :le choc septique
Le choc septique se caractérise par un dysfonc-tionnement cardiovasculaire et des défaillancesmultiviscérales. Les facteurs pro- et anti-inflam-matoires, dont les niveaux sont modifiés au coursde cette maladie, régulent l’expression et/ouactivité de NOS2. En effet, les médiateurs pro-inflammatoires (tumor necrosis factor-α, interleu-kine-1ß, interleukine-6, interféron-γ), toutcomme le lipolysaccharide (LPS), peuvent provo-quer l’induction de NOS2 dans les cellules car-diaques (cardiomyocyte, cellules endothélialesmicrovasculaires et endocardiques, fibroblastes,cellules musculaires lisses), et dans les cellulesinflammatoires (macrophages, neutrophiles).Dans la paroi artérielle, l’induction de NOS2joue un rôle fonctionnel dans la vasodilatationassociée au choc septique. Dans le cœur, le NOproduit par NOS2 peut être soit sans effet, soitinhiber la contractilité basale, ou encore atté-nuer la réponse contractile aux agonistes ß-adré-nergiques. Bien que variés, ces résultats permet-tent de conclure que, dans certaines situations,une production mesurable de NO par NOS2 car-
diaque entraîne un dysfonctionnement contrac-tile, dont le mécanisme le plus étudié impliquele GMPc. L’activité de NOS2 peut égalementinhiber la respiration cellulaire et l’utilisation del’oxygène, un aspect métabolique rencontré dansle choc septique.
Pathologies dans lesquelles un défaut de NOest impliqué
➙ L'hypertension artérielle
Comme nous l'avons vu l'injection aiguë d'antago-niste de la NO synthase ou l'inhibition chroniquede la production de NO sont associées à une dimi-nution des flux et à une augmentation de la pres-sion artérielle. L'inhibition chronique de l'activitédes NO synthases s'accompagne rapidement de tou-tes les conséquences organiques d'une hypertensionartérielle sévère au long cours, en particulier uneartériolosclérose extrêmement intense. Elle se tra-duit par des oblitérations vasculaires dans le sys-tème nerveux central (AVC), dans le rein, condui-sant à une néphro-angiosclérose aboutissant, enconjonction avec l'atteinte glomérulaire, à l'insuffi-sance rénale. Dans le cœur se développent desmicro-infarctus conduisant à des cicatrices fibreusesmultiples. Dans ce modèle d'intoxication chroniquepar des dérivés substitués de l'arginine, le rôle res-pectif des différentes NO synthases ne peut êtredéfini précisément du fait de la non-spécificité par-tielle du blocage. Néanmoins ces effets délètèressont essentiellement médiés par la NOS endothé-liale. Le second point est qu'il existe dans cemodèle une diminution chronique des dimensionscardiovasculaires aboutissant à une diminutionglobale du volume du contenant et probablement àune diminution globale du volume contenu (volumesanguin total). Enfin, si la suppression chronique deproduction de NO contracte fonctionnellement lesartérioles, et les remodèle chroniquement, le rôlerespectif de ce remodelage constrictif fonctionnel
Rôle du monoxyde d'azote enpathologie cardiovasculaire

41NO - Monoxyde d’azote et homéostasie cardiovasculaire
et structural, versus l'augmentation d'agrégabilitéplaquettaire due au défaut de NO, dans la survenuedes oblitérations artériolaires, reste à préciser.
Ces résultats expérimentaux laissent clairement sup-poser que NO est impliqué dans la régulation de lapression artérielle et donc qu'un défaut de NOpourrait être en cause dans l'hypertension.L'invalidation spécifique du gène de la NOS endothé-liale s'accompagne d'une augmentation de la pres-sion artérielle moyenne de l'ordre de 15 à 20 mmHg.
Par rapport aux autres modèles d'hypertensionartérielle expérimentale, le modèle d'intoxicationchronique à la L-NAME se caractérise donc par :- la survenue précoce d'une atteinte structurale
du rein avec protéinurie abondante, conduisant rapidement à une insuffisance rénale chronique caractérisée par, à la fois, une atteinteglomérulaire et tubulaire.
- la survenue d'accidents vasculaires du système nerveux central, se traduisant par des monoplégies,en rapport avec des infarctus de la mœllecervicale haute et du cervelet ;
Par contre un point commun avec la majorité desautres modèles d'hypertension, en particulier avecl'hypertension génétique du SHR, est la sensibilitédu modèle aux inhibiteurs du système rénine angio-tensine malgré l'absence d'activation détectable decelui-ci. L’intoxication chronique au L-NAME nes'accompagne pas d'une augmentation de la sécré-tion de rénine par le rein à un stade initial ; enrevanche dès ce stade, les inhibiteurs de l'enzymede conversion ou les antagonistes de l'angiotensineII sont capables de prévenir ou de réverser la montéeen pression. Cette sensibilité aux antagonistes dusystème rénine-angiotensine s'explique par la signa-lisation du GMP cyclique/GKI dans la cellule mus-culaire lisse. En l’absence de GMPc, le couplage durécepteur à l’angiotensine II à sa signalisation effec-trice intracellulaire est maximal. La suppressionmême partielle de l’interaction ligand (AII)-récep-teur diminue cette signalisation intracellulaire, et dece fait, la pression artérielle.
Au-delà de ses effets vasomoteurs le blocage de laNO synthase s'accompagne d'un phénotype pro-inflammatoire et procoagulant de la paroi artérielle.En physiologie, le monoxyde d'azote prévientl'apoptose des cellules endothéliales et l'adhésiondes leucocytes et des plaquettes à l'endothélium.Lors d'une intoxication chronique au L-NAME,l’adhésion des monocytes à l'endothélium estaugmenté ; le nombre de cellules inflammatoiresadventitielles périvasculaires, augmente autour des
vaisseaux dans le cerveau, le rein, le cœur. Cette activation se traduit par une augmentation duniveau de stress oxydant dans la paroi artérielle etune augmentation d'expression des gènes pro-inflammatoires dépendant de NF-κB, comme certainesinterleukines (IL-6) ou le Facteur Tissulaire.
L'augmentation d'expression du Facteur Tissulaire etd'inhibiteurs de la fibrinolyse, participe du phéno-type procoagulant de la paroi artérielle et de l'appa-rition de phénomènes thrombotiques touchant prin-cipalement les artérioles du système nerveux cen-tral. Cette physiopathologie n'est probablement pastrès éloignée de celle qui relie les accidents vascu-laires cérébraux thrombotiques à l'hypertensionartérielle chez l'homme.
L'étape suivante fût de s'intéresser aux autres modèlesd'hypertension expérimentale, en particulier aumodèle du rat spontanément hypertendu, représentantle modèle de l'hypertension artérielle primitive,génétiquement déterminée. Dans ce modèle, il estrapporté de nombreux résultats divergents d'étudespharmacologiques utilisant des agonistes (acétyl-choline) stimulant la sécrétion de NO par l'endo-thélium. Certains auteurs rapportent une diminutionde réponse à l'acétylcholine, d'autres une réponsenon différente de celle des témoins normotendus.
Les études utilisant des antagonistes montrent quele blocage des NO synthases a des effets similaireschez l'animal normotendu et chez l'animal hyper-tendu, suggérant qu'il n'éxiste pas de défaut majeurquantitatif de production de NO endogène dans cemodèle. Il en est de même dans les autres modèlesd'hypertension artérielle secondaire à un excèsd'apport sodé ou à un excès de rénine, où le tauxbasal de GMP cyclique est rapporté normal, nondifférent du groupe témoin normotendu. Dans lemodèle de surcharge corticoïde, la vasodilatationendothélium dépendante semble même exacerbée.Dans ce modèle la fonction endothéliale à produiredu NO semble jouer un rôle de contre-régulation,limitant l'augmentation de pression.
Le rôle de l'activation de la voie arginine-NO-GMPcyclique rénale dans la régulation de l'homéostasiesodée est suggéré par les expériences de rein isoléperfusé. L'administration aiguë de L-NAME danscette préparation diminue le flux plasmatique rénalet augmente d'autant plus la pression de perfusionque les animaux ont été pré-traités de façon chroni-que par un régime riche en sel. Il a été montré qu'àpression de perfusion constante, la nitro-argininediminuait l'excrétion urinaire de sodium in vivo etqu'un prétraitement des animaux par un régime

42 NO - Monoxyde d’azote et homéostasie cardiovasculaire
riche en sel augmentait la réponse aux antagonistesde la NO synthase.
Le rôle de la voie arginine-NO-GMPcyclique rénaledans la contre-régulation à un apport excessif en selest également fortement suggéré par la réponse durat de Dahl à l'administration de L-arginine. Le ratde Dahl sensible au sel est un modèle génétiqued'hypertension en interaction avec l'environnementsodé. La souche de rat, dite sensible au sel, est prati-quement normotendue lorsque le régime sodé estnormal, mais la pression artérielle augmente signifi-cativement lorsque ces animaux sont soumis à unrégime riche en sel. Dans la souche résistante au sel,l'apport excessif de sel ne s'accompagne pas d'uneaugmentation détectable de la pression artérielle, etl'administration de fortes doses de L-arginine n'a pasd'effet sur la pression artérielle. En revanche,l'administration aiguë d'un antagoniste de l'arginineaugmente la pression et diminue la filtration glomé-rulaire, plus dans la souche résistante soumise à lasurcharge sodée que dans la souche sensible. Al'inverse, l'administration aiguë de fortes doses deL-arginine diminue la pression artérielle dans lasouche sensible au sel soumise à une intoxicationchronique par le sel, et l'administration chroniquede L-arginine prévient le développement de l'hyper-tension. Dans ce modèle un certain nombre d'argumentsmilite en faveur de l'implication de la NOS IIinductible plus que de la NOS III endothéliale.Dans nos propres expériences, il existe une aug-mentation d'expression de la NOS II inductible dansle modèle DOCA-sel, au niveau du rein.
L'ensemble de ces données physiopathologiques esten faveur d'un rôle important de cette voie dansl'adaptation à la surcharge sodée. Dans le modèlephysiologique de surcharge en sel, l'activation de lavoie du NO-GMPc semble importante pour le maintiende la pression artérielle à un niveau normal. Dansle modèle DOCA sel, l'élévation de pression arté-rielle a lieu malgré l'activation de cette voie. Chezle rat de Dahl sensible au sel, un déficit génétiquementdéterminé de cette voie pourrait rendre compte del'élévation de pression artérielle en présence d'unexcès de sel.
Chez l'homme, le rôle de la fonction vasodilatatriceendothéliale dans l'hypertension n'a pu être abordéque par l'étude de l'action loco-régionale d'agentspharmacologiques ayant pour cible l'endothélium.Il a été rapporté, chez des patients ayant une hyper-tension essentielle, une diminution de la réponsevasodilatatrice à l'injection intra-artérielle d'acétyl-choline, comparée à celle observée chez un groupetémoin normotendu. Cette diminution de sensibilité
à l'acétylcholine s'accompagne, chez les malades,d'une diminution de la réponse vasoconstrictriceaux antagonistes de l'arginine.
Inversement, la perfusion de L-arginine, si elle n'apas d'effet sur le tonus vasodilatateur de base chezle témoin normotendu ni chez l'hypertendu, aug-mente la sensibilité à l'acétylcholine chez le témoinnormotendu et ne le modifie pas chez l'hypertendu.Cependant, il apparaît plus intéressant de tester laréponse vasodilatatrice à l'augmentation de fluxinduite par une ischémie transitoire de l'avant-braspar exemple, qu'à une stimulation pharmacologiquepar l'acétylcholine.
Ces études ne permettent pas de conclure sur unéventuel déficit quantitatif global de cette voie dansl'hypertension artérielle essentielle humaine.Néanmoins, elles démontrent le rôle limitant dusubstrat (L-arginine) dans des conditions de fortestimulation chez l'individu normal sain, et des dif-férences notables de réponse aux agents pharmaco-logiques de cette voie chez l'hypertendu : diminutionde sensibilité à l'acétylcholine, aux antagonistes del'arginine et à la L-arginine elle-même. Un effetbénéfique de la perfusion de substrat exogène (L-argi-nine) a été également rapporté dans l'hypercholesté-rolémie expérimentale et humaine. Ces travauxd'investigation clinique ont jusqu'à maintenantutilisé essentiellement une approche de stimulationlocale de l'endothélium-dépendance soit par desagents pharmacologiques (acétylcholine), soit pardes variations aiguës de flux (manœuvre d'ischémies-reperfusion).
L'hypertension artérielle est un phénomène d'aug-mentation diffuse du tonus constricteur (et nonlocal), et l'endothélium-dépendance un phénomèned'adaptation constant et immédiat loco-régional àl'exercice. Vouloir cliniquement définir la dysfonctionendothéliale de l'hypertendu nécessite probablementde s'intéresser à l'adaptation à l'effort de cespatients en terme de montée de pression, de variationsdes dimensions vasculaires, de flux… en trouvantdes marqueurs périphériques de cette dysfonction.
NO est impliqué dans la régulation de la pressionartérielle. Un défaut de NO donne une hypertensionartérielle. Un médicament qui stimule la production deNO par l'endothélium diminue la pression artérielle.

43NO - Monoxyde d’azote et homéostasie cardiovasculaire
➙ NO et insuffisance cardiaque
Insuffisance cardiaqueLa fonction de la pompe cardiaque se définitcomme génératrice de débit. L’insuffisance car-diaque se définit donc comme l’incapacité de lapompe à assurer des débits adaptés aux besoinspériphériques. Quelque soit son étiologie initiale,l’insuffisance cardiaque se caractérise par uneaugmentation de la résistance à l’écoulementsanguin, le site de cette résistance étant variable :augmentation des résistances périphériques dansl’hypertension artérielle, obstacle à l’éjectionventriculaire, augmentation de l’impédance proprede la géométrie ventriculaire dans les cardiomyo-pathies primitives ou ischémiques (infarctus).Quelque soit son étiologie, l’insuffisance cardiaques’exprime sur le plan hémodynamique par uneaugmentation de la pression télédiastolique duventricule gauche, traduisant cette augmentationd’impédance à l’écoulement sanguin, cette reré-partition des énergies circulatoires de moins decinétique vers plus d’energie potentielle.
La complexité des mécanismes de régulation dela fonction cardiaque par le NO s'accroît dans lecadre des pathologies, telles l’insuffisance cardiaque.Ceci est lié aux modifications :
1 - d'expression et d'activité des NO synthases (secondaire aux altérations de la biodisponibilitédes substrats, des cofacteurs et des agents régulateurs),
2 - de localisation des NOS,3 - de la biodisponibilité du NO (augmentation
du stress oxydatif),4 - de la sensibilité des vaisseaux et du cœur au NO.Tous ces paramètres varient en fonction de lapathologie et de son stade d'évolution. Par ailleursil faut distinguer l’action du NO vasculaire et cardiaque.
NO vasculaire et insuffisance cardiaqueL'augmentation des résistances périphériques aucours de l'insuffisance cardiaque est seulementpartiellement explicable par l'activation des systèmespresseurs, système catécholaminergique et systèmerénine-angiotensine. Il semble donc qu'il existe desperturbations locales de la débit-dépendance donttémoigne l'absence d'adaptation correcte des vascu-larisations périphériques à l'augmentation de besoinmétabolique. Cette désadaptation à l'effort est enrapport avec des phénomènes physio-pathologiquespériphériques parmi lesquels le NO joue un rôledéterminant.La dysfonction de pompe cardiaque retentit de façondifférenciée en amont et en aval du ventricule gauchedéfaillant (figure 26). En aval, la diminution de débit
Amont : circulation pulmonaire
Vitesse
Pression
Dimension
Vitesse
Pression
Dimension
Aval: circulation artérielle systémique
Cisaillement Cisaillement ~Figure 26

44 NO - Monoxyde d’azote et homéostasie cardiovasculaire
cardiaque s'accompagne d'une tendance à la diminu-tion de la pression artérielle. La diminution de pressiontend à diminuer le rayon endoluminal en faisant régresserle système sur la courbe de la relation pression volume.De ce fait, malgré la baisse possible de débit, la diminutionassociée de dimension tend à maintenir constante lescontraintes de cisaillement principales déterminantes dela sécrétion de NO par l'endothélium. Cet effet périphé-rique chronique de baisse d'énergie cinétique est essen-tiellement médié par une diminution de phosphorylationde la NO synthase endothéliale, dépendante d'une dimi-nution d'activité de la voie PI-3 Kinase/Akt.Au contraire, dans la petite circulation pulmonaire, enamont du ventricule gauche défaillant, la même diminutionde débit s'accompagne d'une augmentation de pression.Les propriétés visco élastiques (déformation) de la paroiartérielle périphérique sont différentes de celles de lacirculation pulmonaire.
Dans le système artériel pulmonaire proche du systèmeveineux, une faible variation de pression s'accompagned'une forte variation de dimension dont témoignent lesvariations de forme de l'arc moyen gauche cardiaque àla radio de thorax de face. L'arc moyen gauche concave,lorsque la pression artérielle pulmonaire est physiologique,devient convexe dans l'insuffisance cardiaque, traduisantl'augmentation de dimension vasculaire en rapport avecl'augmentation de pression. De ce fait au cours del'insuffisance cardiaque le débit tend à diminuer et lerayon à augmenter dans la circulation pulmonaire induisantune forte diminution des contraintes de cisaillement surl’endothélium. De ce fait la sécrétion de NO par l'endo-thélium pulmonaire diminue alors que la sécrétiond'endothéline augmente. Des travaux expérimentauxrécents mettent bien en évidence des différences deréponse entre la petite circulation pulmonaire et lacirculation artérielle systémique. L'augmentation du tauxd'endothéline circulante dans l'insuffisance cardiaquetraduit donc la dysfonction endothéliale pulmonairesecondaire à la dysfonction diastolique du ventriculegauche. Il est probablement inversement proportionnel àl'activité de la NO synthase endothéliale pulmonaire.
NO myocardique et insuffisance cardiaqueLa diminution d'activité vasculaire de NOS3 est bienétablie et plus communément appelée dysfonction endothé-liale. Cette diminution de production endothéliale de NOserait liée à une diminution des flux sanguins secondaireà l’effondrement du débit cardiaque. Les mécanismes decette dysfonction associent une diminution de la productiondu NO (diminution de l'expression de NOS3, déficienceen substrat et cofacteurs (L-arginine et/ou THB4), diminutiondu nombre de transporteurs de la L-arginine) et/ou uneaugmentation de sa dégradation (accroissement du stressoxydatif). Il faut cependant noter que bien que la pro-duction de NO par NOS3 soit diminuée au cours de
l’insuffisance cardiaque, cette isoforme est toujoursfonctionnelle et capable d’avoir un effet sur la fonctioncardiaque. Ainsi, certains auteurs ont démontré, chezdes patients en insuffisance cardiaque terminale, que lastimulation de cette NOS3 endothéliale par la substanceP se traduisait par une amélioration significative du travailcardiaque.Plusieurs études ont mis en évidence une augmentationde la concentration de NO cardiomyocytaire au coursde l’insuffisance cardiaque à la fois expérimentale maiségalement chez l’homme. Jusqu’à ces dernières années,le dogme était que cette production accrue de NO semblaitavant tout le fait de NOS2 et/ou NOS3 cardiomyocytaires.L’hypothèse de l'implication de NOS2 dans l'insuffisancecardiaque reposait sur des études qui démontraient laprésence d'une activité NO synthase indépendante ducalcium, et une expression d'ARNm et de protéine deNOS2 dans différents modèles expérimentaux d’insuffi-sance cardiaque mais aussi dans le myocarde de patientsen insuffisance cardiaque. De manière identique, plusieursétudes chez l’animal et l’homme avaient montré uneaugmentation de l’expression de NOS3 dans les cardio-myocytes au cours de l’insuffisance cardiaque. Du pointde vue fonctionnel et à l’état basal, le NO produit parces isoformes n’a aucune influence sur la contractilitécardiaque aussi bien au niveau cellulaire (cardiomyocyteisolé) qu’au niveau de l’organe entier (cœur isolé perfusé,perfusion intracoronaire d’inhibiteur de NOS). De plusla sur-expression cardiaque et/ou l’invalidation des gènesde NOS2 et NOS3 chez des souris n'a entraîné que desmodifications mineures de la fonction cardiaque.A l’opposé, au cours de l’insuffisance cardiaque, le NOendogène du cardiomyocyte semblerait avoir un impactmajeur sur les réponses chronotrope et inotrope suite àune stimulation ß-adrénergique. Ces dernières sont eneffet diminuées chez les patients en insuffisance cardiaque.Ces phénomènes sont en partie liés :- à un changement de la densité des récepteurs ß-adré-nergiques : diminution des récepteurs inotropes positifsß1 associée ou non à une augmentation du sous-typeinotrope négatif ß3. En fait, ces récepteurs ß3 exerçe-raient leur effet inotrope via la production de NO.Toutefois la stimulation par un agoniste ß3-adrénergiquede cardiomyocytes ventriculaires issus de patients eninsuffisance cardiaque n'engendre qu'un effet inotropenégatif très faible.- à l’augmentation de production de NO endogène desmyocytes cardiaques. En effet, les études réalisées chezl’animal ou chez l'Homme démontrent que l'inhibitionnon sélective des NO synthases lors de l'insuffisancecardiaque augmente la réponse contractile à une stimu-lation ß-adrénergique (figure 27). Toutefois, les isoformesde NO synthases impliquées sont une fois encore discutées.Il semblerait quand même que le NO produit parl’isoforme NOS1 soit un des éléments majeurs de ladiminution de la réponse ß-adrénergique associée à

45NO - Monoxyde d’azote et homéostasie cardiovasculaire
l’insuffisance cardiaque. En effet, des données récentesexpérimentales et cliniques, ont montré que la majoritédu NO produit lors de l’IC l’était au niveau du myocytecardiaque et était le fait de l’isoforme NOS1. De plus,les auteurs ont montré que l’inhibition sélective et aiguede cette isoforme avait pour conséquence l’améliorationde la réponse contractile ß-adrénergique. Ceci pourraitsuggérer que la stimulation ß-adrénergique de la pro-duction de NO, via la NOS1, pourrait agir commecontre-régulateur physiologique pour limiter l’effetinotrope positif et/ou délétère des catécholamines dansle cœur.
Le remodelagevasculaire
L'hypertension artérielle s'accompagne d'un remodelagevasculaire, associant une hypertrophie des cellulesmusculaires lisses de la média et une accumulation decollagène.
Si ce remodelage pariétal en rapport avec l'augmentationde contrainte tensionnelle à la paroi est connu depuislongtemps, la description de celui associé aux variationsde contraintes de cisaillement est plus récente. Lemodèle le plus classique est celui de la fistule artério-veineuse chez l’animal comme chez l’homme enhémodialyse ou porteur d’une malformation artérioveineuse.Une fistule artérioveineuse crée un court-circuit entrele vaisseau artériel afférent et le vaisseau veineux
efférent, augmentant le débit dans ces vaisseauxet le cisaillement à la paroi.
Cette augmentation chronique de cisaillements’accompagne d’un remodelage structurelexpansif de la paroi vasculaire associant unélargissement de la lumière artérielle, unehypertrophie en série des cellules musculaireslisses conduisant à une diminution d’épaisseurmalgré l’augmentation globale de la massepariétale. Cette augmentation de calibrevasculaire s’accompagne d’une augmentationd’activité de la NO synthase qui se traduitpar une augmentation de GMP cyclique dansla paroi, et si le stimulus dure, par une aug-mentation d’expression de la NO synthase IIIendothéliale (figure 28).
L’exercice physique répété, ou l’augmentationchronique d’activité métabolique s’accompagneégalement, mais à un degré moindre d’unélargissement du calibre artériel et d’uneaugmentation de la NO synthase.Inversement une diminution chronique du
flux chez l’animal comme chez l’homme, en rapportavec une diminution chronique d’activité métabolique(atrophie d’un membre par exemple, ou un rein fonc-tionnel), ou un blocage chronique de la NO synthasepar administration d’une arginine substituée, s’accom-pagne d’une diminution chronique du calibre vascu-laire.
Ces phénomènes montrent qu’au-delà de leur fonctionvasodilatatrice, les NO synthases sont également impliquéesdans la structuration tubulaire, normale et pathologique,de la paroi artérielle. Dans les modèles sus-décrits, lesvariations de cisaillement sont constantes sur l’ensemblede la circonférence de l’artère et les variations deproduction de NO sont également circonférentielles.
NO est impliqué dans la structuration de paroi vasculaire.Il inhibe la prolifération des cellules musculaires lisses etaugmente le diamètre luminal vasculaire. Un médicamentqui stimule la sécrétion de NO pourrait jouer un rôledans la prévention des sténoses.
[NO]
Cœur pathologiqueForce
Stimulation ß adrenergique
État Basal
Effet inotrope positif après inhibition des NOS
Réduction de la force de contraction associée à la pathologie
Augmentation de la production de NO associée à la pathologie
Figure 27

46 NO - Monoxyde d’azote et homéostasie cardiovasculaire
Monoxyde d’azote etathérosclérose
L’implication du monoxyde d’azote dans la pathologieathéromateuse est complexe, dépendant du maintien dela fonction de barrière de l’endothélium vasculaire(compartimentation) et probablement de l’expressionpréférentielle de la NO synthase endothéliale versus laNO synthase inductible (inflammatoire).
Il faut différencier les stades évolutifs de l’athérosclérose :stade précoce de dépôts lipidiques sous-intimaux danslesquels l’augmentation de perméabilité endothéliale auxprotéines plasmatiques et aux éléments figurés du sangjoue un rôle déterminant ; stades tardifs de plaques pluscomplexes et pathogènes, constituées d’un centre lipido-fibrino-cruorique encapsulé dans une chape fibreuseluminale, en général réendothélialisée, et la médiaartérielle externe.
Dans la phase précoce, la perte partielle de fonction debarrière endothéliale joue un rôle majeur, comme cela aété récemment montré en microscopie intravitale, dansl’adhésion des plaquettes et l’adhésion et la transmigrationdes monocytes à travers l’endothélium. Ces deux phénomè-nes, adhésion plaquettaire et transmigration monocytairejouent un rôle initiateur des dépôts lipidiques intimaux.Ces deux phénomènes sont prévenus par une sécrétionadaptée et compartimentée de monoxyde d’azote parl’endothélium fonctionnellement intact. De ce fait, unedes questions posées est de savoir le lien entre facteursde risque et dysfonction endothéliale.
Ce lien a été étudié à travers dif-férents facteurs de risque :hypercholestérolémie, diabète,hypertension, tabagisme ; maistoujours de façon indirecte chezl’homme, en testant l’hypothèseque la dysfonction endothélialevasomotrice (perturbation de laréponse vasodilatatrice à unagent pharmacologique ou àune augmentation de flux) étaitreprésentative de la perte defonction de barrière. La majoritédes études prospectives vontdans ce sens donnant à la per-turbation de la fonction endo-théliale vasomotrice une bonnevaleur pronostique de dévelop-pement de complications clini-ques. A partir de là, plusieurshypothèses sont possibles :
- Les facteurs de risque perturbent le transport de l’arginine,substrat de la NO synthase. En effet des étudesexpérimentales et quelques unes cliniques chez l’homme montrent qu’une supplémentation importante en arginine peut reverser la dysfonction endothéliale vasomotrice, en particulier dans l’hypercholesterolémie.
- Les facteurs de risque augmentent le stress oxydant dans la paroi artérielle et perturbent de ce fait la fonctionvasomotrice dépendante de l’endothélium via le monoxyde d’azote.
- L’hypercholestérolémie perturbe l’ultrastructure et le fonctionnement des cavéoles des cellules endothéliales et de ce fait les régulations physiologiques de productionde monoxyde d’azote.
Dans les phases plus tardives de l’athérothrombose (plaquesvulnérables), le rôle du monoxyde d’azote est certainementplus complexe en grande partie lié aux phénomènes dedécompartimentation de sa production secondaire àl’expression de la NO synthase inductible dans les cellulesmusculaires lises et les macrophages.
Dans le contexte clinique des différentes formes d’angor,cette dysfonction de la production de NO par l’endothélium,joue probablement un rôle majeur dans le déclenchementdes symptômes douloureux à l’effort, voire au repos, parfoismême en l’absence de lésions morphologiquement décela-bles des coronaires. Cette dysfonction pourrait égalementjouer un rôle dans la survenue des accidents aigus, oblitérants,des vaisseaux coronaires. En l’absence de NO, l’agrégationplaquettaire devient vite irréversible.
artère
témoin fistule artério-veineuse
GMPc
diamètre
veine artèreveine
remodelageexpansif
Figure 28

47NO - Monoxyde d’azote et homéostasie cardiovasculaire
ConclusionL’implication de NO dans la physiopathologie cardiovasculaire est certainement à la fois prédominante etcomplexe, et un schéma trop univoque d’identification entre diminution de production de NO et pathologiescardiovasclaires comme l’hypertension, l’athérosclérose, ou l’insuffisance cardiaque est certainement tropsimpliste. Il pourrait s’agir, soit plus souvent de phénomènes de décompartimentation dans la production deNO, soit d’un véritable déficit global, en particulier à l’effort. L’approche pharmacologique, chez l’homme estdifficile, et une diminution de la réponse endothéliale à l’acéthylcholine exogène ne peut pas s’interprétersimplement comme un déficit de NO endogène.
La NO synthase endothéliale, si elle est la principale isoforme du système cardiovasculaire n’est pas la seule.Les deux autres isoformes existent de façon physiologique dans le rein, le cœur et la paroi des artères. Enphysiologie, NO fonctionne plus comme un système d’adaptation loco-régional, entre autres aux besoinsmétaboliques de tel ou tel organe, que comme un système régulé à un niveau général. Une dysrégulationgénérale de la production de NO par l’endothélium serait plus à rechercher dans le cadre d’épreuves d’effortque dans des conditions basales de repos.
Par contre, il apparaît très clairement, du fait de l’implication permanente de NO en physiologie cardiovasculaire,qu’il pourrait exister un réel et important bénéfice thérapeutique à stimuler de façon chronique la productionde NO par l’endothélium, rétablissant une compartimentation physiologique de production de NO, relaxant lescellules musculaires lisses sous-jacentes, inhibant leur activation pathologique dans la paroi artérielle,prévenant l’agrégation plaquettaire dans le compartiment sanguin.
Bibliographie• Arnal JF, Dinh-Xuan AT, Pueyo M, Darblade B, Rami J. Endothelium-derived nitric oxide and
vascular physiology and pathology. Cell Mol Life Sci. 1999;55:1078-87.
• Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Physiol Cell Physiol. 2003;285:C499-508.
• Fulton D, Gratton JP, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn't calcium/calmodulin enough? J Pharmacol Exp Ther. 2001;299:818-24.
• Gauthier C, Langin D, Balligand JL. Beta3-adrenoceptors in the cardiovascular system. Trends Pharmacol Sci. 2000;21:426-31.
• Massion PB, Feron O, Dessy C, Balligand JL. Nitric oxide and cardiac function: ten years after, and continuing. Circ Res. 2003;93:388-98.
• Nadar S, Blann AD, Lip GY. Endothelial dysfunction: methods of assessment and application tohypertension. Curr Pharm Des. 2004;10:3591-605.
• Paulus WJ, Shah AM. NO and cardiac diastolic function. Cardiovasc Res. 1999;43:595-606.
• Shah AM, MacCarthy PA. Paracrine and autocrine effects of nitric oxide on myocardial function.Pharmacol Ther. 2000;86:49-86.
• Tong BC, Barbul A. Cellular and physiological effects of arginine. Mini Rev Med Chem. 2004;4:823-32.

1, Rue du Jura - Silic 52894633 Rungis CedexTél. : 01 45 60 77 20Fax : 01 46 87 94 31
1 TEM
036 0
4/0
5 -
Docum
ent
éta
bli e
n a
vri
l 2005 -
Concepti
on g
raphiq
ue e
t ré
alisati
on :
Sam
oa -
Vers
aille
s