cinétique électrochimique - f-legrand.fr · frédériclegrandlicencecreativecommons1 cinétique...
TRANSCRIPT

Frédéric Legrand Licence Creative Commons 1
Cinétique électrochimique
1. Phénomènes électrochimiques1.a. Conducteurs électroniques et ioniques
Dans un conducteur électronique, le courant électrique est dû au déplacement desélectrons. Il s’agit le plus souvent d’un solide. Les métaux (Fe, Pt, etc.) et les semi-conducteurs (C, Si) sont des conducteurs électroniques. Un conducteur électronique estcaractérisé par sa conductivité électrique γ. La loi d’Ohm permet d’exprimer la densitéde courant volumique en fonction du champ électrique :
−→j = γ
−→E (1)
Dans un conducteur ionique, le courant électrique est dû au déplacement d’ions positifs etnégatifs. Un milieu conducteur ionique est aussi appelé électrolyte. Une solution aqueusecontenant des ions est un exemple d’électrolyte. Les ions d’éléments chimiques différentsont en général des mobilités différentes. Comme la conductivité est proportionnelle à laconcentration des ions (pour une solution très diluée), on définit pour chaque ion uneconductivité molaire en divisant sa conductivité par sa concentration :
λi =γiCi
(2)
Les conductivités molaires des ions usuels sont données dans des tables. La page Conduc-tivité électrique donne des valeurs de conductivités molaires.
Par exemple, la conductivité d’une solution aqueuse de NaOH (soude caustique) deconcentration c = 0, 1 mol/L = 100 mol/m3 peut être estimée en considérant que seulsles ions sodium et hydroxyle contribuent à la conductivité :
γ = c(λNa+ + λOH−) = 100(5 + 20)10−3 = 2, 5 S/m (3)
On voit sur cet exemple que la conductivité d’une solution est très faible comparée àcelle des métaux.
Il peut être nécessaire de séparer deux solutions aqueuses tout en permettant lepassage d’un courant ionique entre les deux, au moyen d’une jonction. On peut utiliserpour cela un matériau poreux, comme un papier filtre ou un verre fritté, qui laisse passerles ions pour assurer le passage du courant entre les deux solutions, tout en ralentissantfortement le mélange des réactifs. Les jonctions doivent être les plus fines possibles, pourne pas trop abaisser la résistance globale du circuit.
1.b. Réactions électrochimiquesUne réaction électrochimique se déroule à l’interface entre un conducteur électronique
et un électrolyte. Le conducteur électronique est appelé électrode.Une réaction électrochimique d’oxydation est une perte d’électron d’une espèce de
l’électrolyte, de la forme :
νr Red→ νoOx+ n e− (4)

Frédéric Legrand Licence Creative Commons 2
Les électrons sont directement captés par l’électrode. Le réducteur doit donc entrer direc-tement en contact avec l’électrode pour subir cette oxydation. Dans certains cas, l’espèceoxydée est le métal de l’électrode, ou un solide accroché à sa surface.
Une réaction électrochimique de réduction est un gain d’électron de la forme :
νr Red← νoOx+ n e− (5)
Les électrons sont prélevés directement à la surface de l’électrode.Lorsque la réaction globale se déroulant sur une électrode est une oxydation, l’élec-
trode est appelée anode. Lorsque la réaction globale est une réduction, on parle de ca-thode. Sur une anode, il se déroule en général des réactions d’oxydation et de réduction,mais le bilan de ces réactions conduit à un prélèvement d’électrons à l’électrolyte. Autre-ment dit, le courant allant du circuit électronique vers l’anode est positif. Il y a donc uncourant ionique positif de l’anode vers la cathode. Les cations se dirigent vers la cathode.
Électrolyte
Anode Cathode
Circuit électronique
e- e-
I > 0
I
I
Oxydationprépondérante
Réductionprépondérante
1.c. Transports de matière dans l’électrolyteLe transport des ions dans l’électrolyte est nécessaire pour assurer le passage du cou-
rant avec le minimum de résistance électrique. Les espèces électroactives, c’est-à-direcelles qui contribuent aux réactions électrochimiques, ne sont pas toujours en quantitésuffisante pour assurer une bonne conductivité de l’électrolyte. On doit alors ajouter unsel de fond à la solution pour augmenter sa conductivité. En principe, aucun des ions dece sel de fond ne doit être électroactif. Par ailleurs, les espèces électroactives, qui sont sou-vent des ions, doivent parvenir aux électrodes pour subir les réactions électrochimiques.Il y a trois types de transport :
. Le transport par migration (ou par conduction), causé par le champ électrique. Ilest caractérisé par la loi d’Ohm.
. Le transport par diffusion, causé par les différences de concentration entre lesdifférentes parties de l’électrolyte.

Frédéric Legrand Licence Creative Commons 3
. Le transport par convection, causé par les mouvements macroscopiques de fluide.
Les transports par diffusion et par convection concernent aussi bien les ions que lesespèces non chargées.
La diffusion d’une espèce est un phénomène se déroulant à l’échelle moléculaire. Elleprovient de l’agitation thermique des molécules, qui a tendance à uniformiser la concen-tration. La diffusion se fait toujours des régions de plus forte concentration vers celles deplus faible concentration. Elle intervient principalement au voisinage des électrodes, dansune couche de diffusion dont l’épaisseur est notée δ. Elle est de quelques micromètres.
La convection peut être naturelle ou forcée. Dans les expériences de laboratoire,on établit une convection forcée en agitant la solution, avec un agitateur motorisé. Laconvection intervient principalement en dehors de la couche de diffusion. Dans les pileset accumulateurs commerciaux, il n’y a pas de convection.
La migration intervient partout dans l’électrolyte.La figure suivante montre les différents types de transport et les zones où ils inter-
viennent :
DiffusionMigration
DiffusionMigration
ConvectionMigration
δ
Agitation
AnodeCathode
E
Il arrive qu’une réaction électrochimique nécessite un ion qui tend à s’éloigner de l’élec-trode par son sens de migration. Par exemple, considérons l’oxydation de l’ion Fe2+
en ion Fe3+ sur une anode. Comme le champ électrique est dirigé de l’électrode versl’électrolyte, il tend à éloigner l’ion de la surface de l’électrode. Dans ce cas, seule ladiffusion et la convection permettent à l’ion de parvenir à la surface, et doivent contrerl’effet de la migration. À proximité immédiate de la surface, seule la diffusion intervient.Néanmoins, la convection a une grande importance car elle modifie l’épaisseur δ de lacouche de diffusion. Au voisinage de la surface, le courant de migration et le courant dediffusion sont en sens inverse : il faut que le courant de diffusion soit prépondérant. Enpratique, cela nécessite une forte agitation de manière à réduire l’épaisseur de la couchede diffusion.

Frédéric Legrand Licence Creative Commons 4
Anode
Fe2+e-
E
Fe2+
Courant de migration
Courant de diffusion
Gradient de concentration
Fe2+
Fe2+
Fe2+
Fe2+
Fe2+
1.d. Vitesses de réaction et intensité du courantConsidérons une électrode, sur laquelle se déroule a priori les deux réactions électro-
chimiques suivantes :
νr Red→ νoOx+ n e− (6)
νr Red← νoOx+ n e− (7)
Pour simplifier, on ne considère qu’une réaction d’oxydation et une réaction de réduction.En réalité, il peut y en avoir plusieurs.
Lorsque ne moles d’électrons sont reçues par l’électrode (grandeur algébrique), laquantité de charge transférée est donnée par la loi de Faraday (inventeur de l’électrochi-mie) :
Q = neF (8)
La constante de faraday F est la quantité de charge d’une mole d’électrons :
F = Nae = 96485 C ·mol−1 (9)
Si l’on considère la réaction (6) seule, on définit sa vitesse (vitesse d’oxydation) par :
Vox =1
νo
1
S
dnoxdt
=1
n
1
S
dnedt
= − 1
νr
1
S
dnreddt
(10)
où S est l’aire de la surface de l’électrode en contact avec l’électrolyte. Dans une ré-action se faisant en volume, la vitesse est une grandeur intensive définie en divisant letaux de variation des nombres de mole par le volume de la solution. Pour une réactionélectrochimique, on divise par la surface car le taux de production est proportionnelle àla surface. Une électrode deux fois plus grande donne deux fois plus de produits pour lamême durée.
En considérant la réaction (7) seule, on définit la vitesse de réduction :

Frédéric Legrand Licence Creative Commons 5
Vred = −1
νo
1
S
dnoxdt
= − 1
n
1
S
dnedt
=1
νr
1
S
dnreddt
(11)
Considérons la réaction globale :
νr Red = νoOx+ n e− (12)
Sa vitesse est :
V = Vox − Vred (13)
Lorsque cette vitesse est positive, il y a globalement une oxydation : l’électrode est uneanode. Lorsque cette vitesse est négative, il y a globalement une réduction : l’électrodeest une cathode. Lorsque cette vitesse est nulle, l’électrode est à l’équilibre.
La loi de Faraday permet d’exprimer le flux de quantité de charge qui parvient àl’électrode (compté de l’électrolyte vers l’électrode) :
dQ
dt= F
dnedt
= FnS(Vox − Vred) (14)
On est alors amené à définir la densité de courant en divisant par l’aire :
La densité de courant de l’électrode vers l’électrolyte est :
j = nF (Vox − Vred) (15)
Pour une anode, cette densité de courant est positive. La densité de courant est utilepour les calculs théoriques, car elle ne dépend pas de la taille des électrodes. Expéri-mentalement, on utilise plutôt l’intensité du courant I = Sj. En régime stationnaire,l’intensité du courant sur l’anode est l’opposée de l’intensité sur la cathode. Comme lesaires peuvent être différentes, on écrira :
Saja = −Scjc (16)
Le courant anodique est positif, le courant cathodique est négatif.

Frédéric Legrand Licence Creative Commons 6
2. Courbes courant-potentiel2.a. Montage à trois électrodes
Ce montage permet de mesurer les potentiels des deux électrodes et le courant élec-trique. La troisième électrode est une électrode de référence, c’est-à-dire une demi-piledont le couple a un potentiel qui reste pratiquement constant. L’extrémité d’une élec-trode de référence est une jonction en verre fritté, qui permet le passage du courantionique entre l’intérieur de l’électrode et l’electrolyte étudié. Ce courant doit néanmoinsrester extrêmement faible (de l’ordre du nano-ampère), car la résistance électrique duverre fritté est grande.
V
A
U
ER
CE ET
ii
i
Les deux électrodes sont appelées électrode de travail et contre électrode. On utilise sou-vent des électrodes chimiquement inertes, par exemple des électrodes en platine. L’élec-trode de travail est celle qu’on étudie. Par convention, l’intensité du courant est mesuréede l’électrode de travail vers l’électrolyte. Lorsqu’elle est positive, l’électrode de travailest le siège d’une réaction globale d’oxydation (mais il peut y avoir aussi une réduction).
Une électrode de référence couramment utilisée est l’électrode au calomel saturée(ECS), composée de mercure au contact d’une solution saturée de calomel (Hg2Cl2).L’électrode à proprement parler est en mercure liquide. La réaction électrochimique sedéroulant sur cette électrode (dans un sens ou dans l’autre) est :
Hg2Cl2 + 2e− = 2Hg(l) + 2Cl− (17)
L’électrode de référence doit être parcourue par un courant pratiquement nul (de l’ordredu nano ampère). Dans ces conditions, son potentiel est pratiquement égal à son potentielde Nernst :
EECS = E0Hg2Cl2/Hg
− 0, 056 log(aCl−) (18)

Frédéric Legrand Licence Creative Commons 7
La solution contenue dans l’électrode ECS est une solution saturée enKCl, ce qui permetde maintenir constante l’activité en chlolure. Le potentiel de l’électrode ECS est E =0,246V à température ambiante.
Dans le montage à trois électrodes, le potentiel de l’électrode de travail est fourni parla tension V :
EET = EER + V (19)
Le générateur de force électromotrice U permet de faire circuler un courant entre l’élec-trode de travail et la contre-électrode, dans un sens ou dans l’autre. L’objectif est demesurer l’intensité I pour différentes valeurs de EET . Le fait de fixer la force électromo-trice U ne permet pas toujours de maintenir le potentiel de l’électrode de travail fixe,car le passage du courant s’accompagne de modifications importantes dans l’électrolyte.On est alors amené à utiliser un circuit de commande spécifique appelé potentiostat, quiasservit la tension U en fonction d’une valeur de commande de EET . La méthode demesure est appelée ampérométrie, car la grandeur mesurée est l’intensité du courant. Ilest aussi possible d’imposer le courant et de mesurer le potentiel (potentiométrie).
2.b. Courbe courant-potentielUne courbe courant-porentiel permet d’étudier, pour un système constitué d’une élec-
trode de travail et d’un électrolyte donnés, la cinétique globale du système. Celle-ci ré-sulte à la fois de la cinétique de la réaction électrochimique (appelée aussi cinétique dutransfert de charge) et de la cinétique du transport de matière dans l’électrolyte.
On s’intéresse dans un premier temps au cas où un seul couple redox intervient dansles réactions électrochimiques, aussi bien sur la branche anodique que sur la branchecathodique :
νr Red = νoOx+ n e− (20)
Pour une réaction rapide, l’aspect général de la courbe courant-potentiel est le suivant(au voisinage de l’équilibre) :

Frédéric Legrand Licence Creative Commons 8
E
I
Red Ox
Red Ox
Potentiel d'équilibre
Surtension
Le potentiel est celui de l’électrode de travail. L’intensité est celle du courant allant ducircuit électronique de commande vers l’électrode de travail, donc de cette électrode versl’électrolyte.
Le potentiel à courant nul est le potentiel d’équilibre. Lorsque l’électrode a ce poten-tiel, la réaction (20) est à l’équilibre sur l’électrode : la vitesse de la réduction compenseexactement la vitesse de l’oxydation. À l’échelle moléculaire, les deux réactions inversesse produisent sans cesse mais le bilan macroscopique est nul. Lorsqu’un seul couple in-tervient, et s’il existe un seul point de courant nul, le potentiel d’équilibre est égal aupotentiel de Nernst :
E(I = 0) = EOx/Red = E0Ox/Red +
RT
nFln
(aνooaνrr
)(21)
Le quotient de réaction argument du logarithme est celui de la réaction (20), qui comporteéventuellement d’autres espèces que l’oxydant et le réducteur.
Pour placer le système hors d’équilibre, il faut appliquer un potentiel différent dupotentiel d’équilibre. La différence entre le potentiel appliqué et le potentiel d’équilibreest la surtension :
η = E − E(I = 0) (22)
Lorsque le potentiel est supérieur au potentiel d’équilibre, c’est-à-dire lorsque la surten-sion est positive, la réaction prépondérante est l’oxydation. Le courant est donc positif.On reporte la réaction prépondérante sur la branche d’oxydation anodique comme indi-qué sur la figure. Si la surtension est négative, la réduction est prépondérante donc lecourant est négatif.
Théoriquement, la surtension représente la différence de potentiel entre l’électrodeet l’électrolyte (à une constante additive près). En pratique, l’électrode de référence està une certaine distance de l’électrode de travail et la chute de tension ohmique entreces deux électrodes intervient. Celle-ci augmente proportionnellement à l’intensité. Pour
Frédéric Legrand Licence Creative Commons 9
minimiser cet effet, l’électrode de référence doit être placée au plus près de l’électrodede travail et l’électrolyte doit contenir une forte concentration d’ions pour minimiser sarésistance.
2.c. Système rapideLe système étudié est constitué d’un couple redox et d’une électrode donnés. La
vitesse de la réaction électrochimique dépend du couple considéré, mais aussi du matériauconstituant l’électrode.
Un système est dit rapide lorsque la vitesse est limitée par le transport de matière etpas par le transfert de charges entre les espèces électroactives et les électrodes.
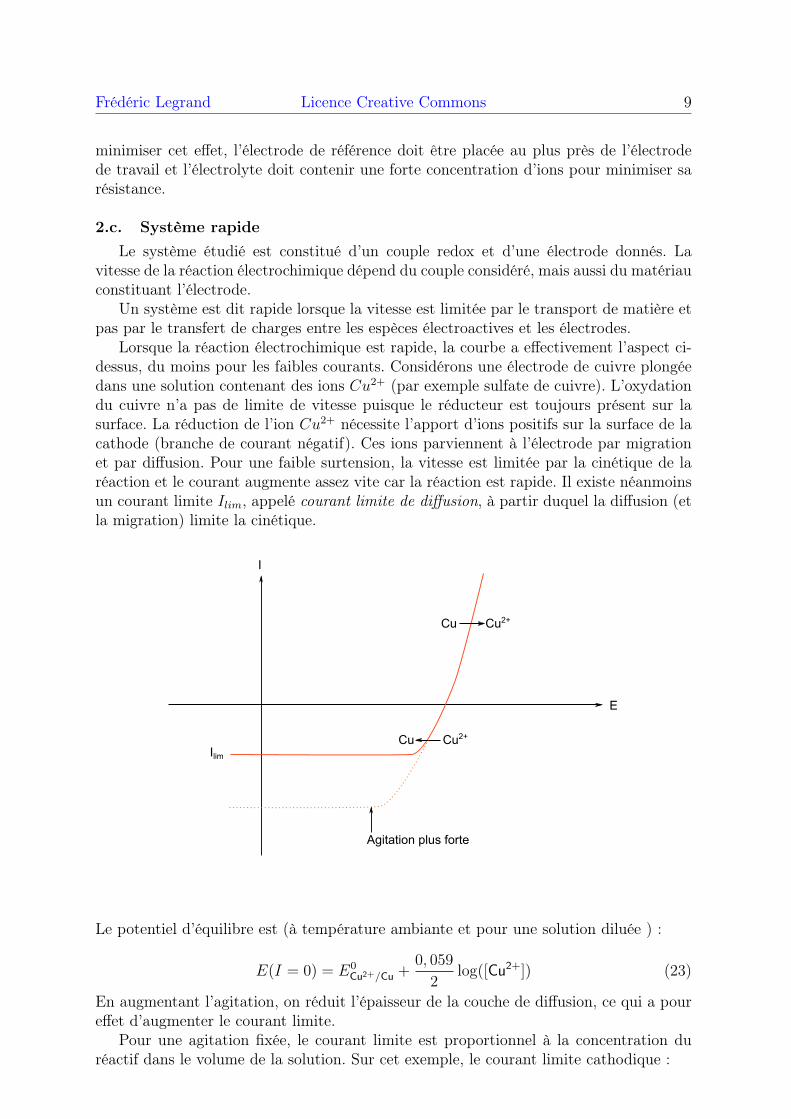
Lorsque la réaction électrochimique est rapide, la courbe a effectivement l’aspect ci-dessus, du moins pour les faibles courants. Considérons une électrode de cuivre plongéedans une solution contenant des ions Cu2+ (par exemple sulfate de cuivre). L’oxydationdu cuivre n’a pas de limite de vitesse puisque le réducteur est toujours présent sur lasurface. La réduction de l’ion Cu2+ nécessite l’apport d’ions positifs sur la surface de lacathode (branche de courant négatif). Ces ions parviennent à l’électrode par migrationet par diffusion. Pour une faible surtension, la vitesse est limitée par la cinétique de laréaction et le courant augmente assez vite car la réaction est rapide. Il existe néanmoinsun courant limite Ilim, appelé courant limite de diffusion, à partir duquel la diffusion (etla migration) limite la cinétique.
E
I
Cu Cu2+
IlimCu Cu2+
Agitation plus forte
Le potentiel d’équilibre est (à température ambiante et pour une solution diluée ) :
E(I = 0) = E0Cu2+/Cu +
0, 059
2log([Cu2+]) (23)
En augmentant l’agitation, on réduit l’épaisseur de la couche de diffusion, ce qui a poureffet d’augmenter le courant limite.
Pour une agitation fixée, le courant limite est proportionnel à la concentration duréactif dans le volume de la solution. Sur cet exemple, le courant limite cathodique :
Frédéric Legrand Licence Creative Commons 10
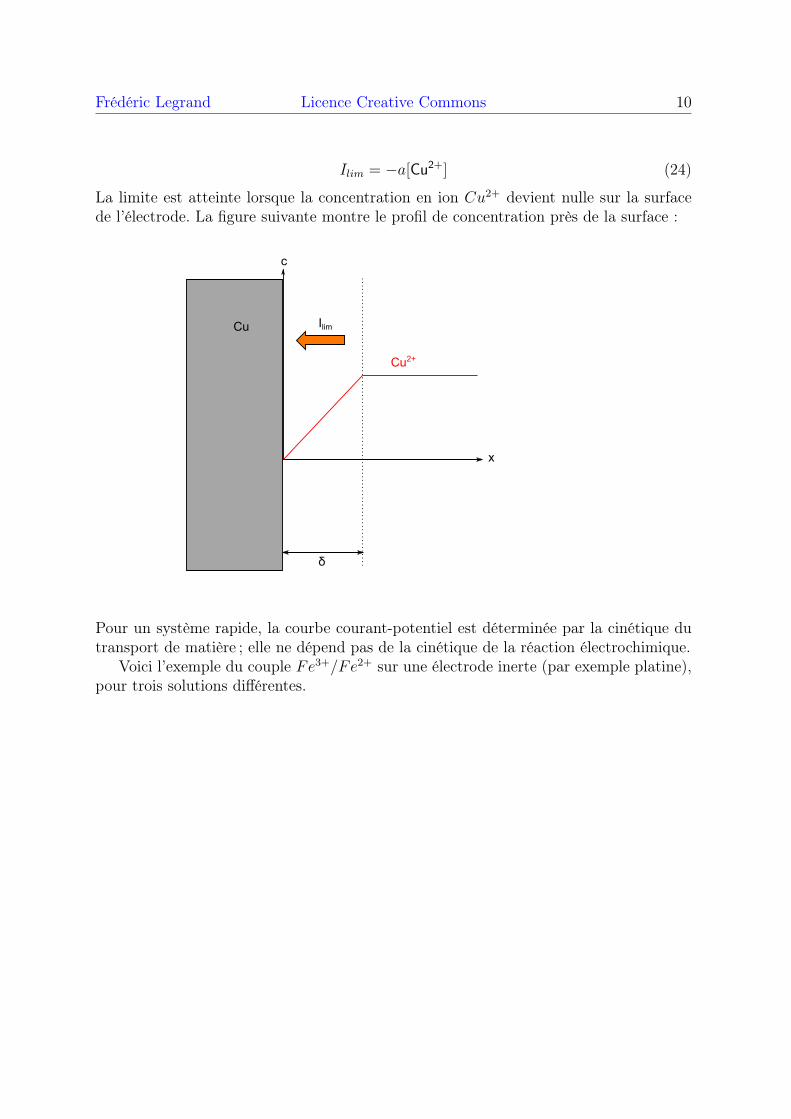
Ilim = −a[Cu2+] (24)
La limite est atteinte lorsque la concentration en ion Cu2+ devient nulle sur la surfacede l’électrode. La figure suivante montre le profil de concentration près de la surface :
x
c
Cu
δ
Ilim
Cu2+
Pour un système rapide, la courbe courant-potentiel est déterminée par la cinétique dutransport de matière ; elle ne dépend pas de la cinétique de la réaction électrochimique.
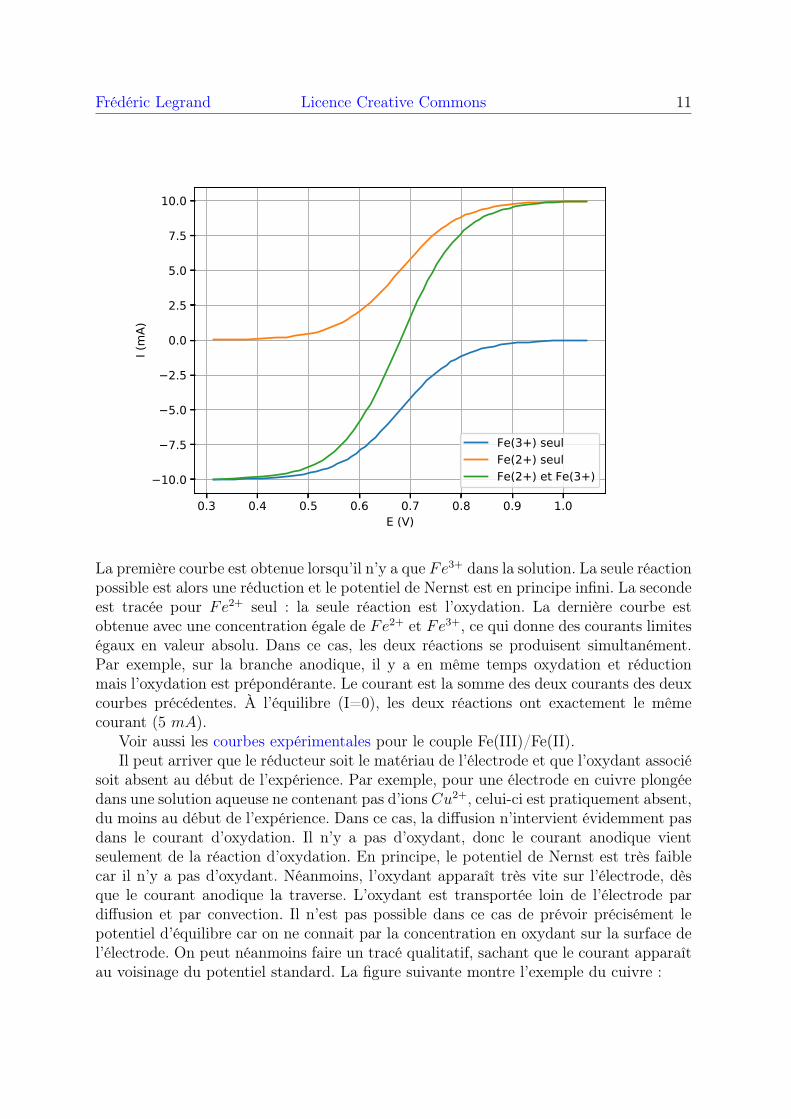
Voici l’exemple du couple Fe3+/Fe2+ sur une électrode inerte (par exemple platine),pour trois solutions différentes.
Frédéric Legrand Licence Creative Commons 11
0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0E (V)
10.0
7.5
5.0
2.5
0.0
2.5
5.0
7.5
10.0
I (m
A)
Fe(3+) seulFe(2+) seulFe(2+) et Fe(3+)
La première courbe est obtenue lorsqu’il n’y a que Fe3+ dans la solution. La seule réactionpossible est alors une réduction et le potentiel de Nernst est en principe infini. La secondeest tracée pour Fe2+ seul : la seule réaction est l’oxydation. La dernière courbe estobtenue avec une concentration égale de Fe2+ et Fe3+, ce qui donne des courants limiteségaux en valeur absolu. Dans ce cas, les deux réactions se produisent simultanément.Par exemple, sur la branche anodique, il y a en même temps oxydation et réductionmais l’oxydation est prépondérante. Le courant est la somme des deux courants des deuxcourbes précédentes. À l’équilibre (I=0), les deux réactions ont exactement le mêmecourant (5 mA).
Voir aussi les courbes expérimentales pour le couple Fe(III)/Fe(II).Il peut arriver que le réducteur soit le matériau de l’électrode et que l’oxydant associé
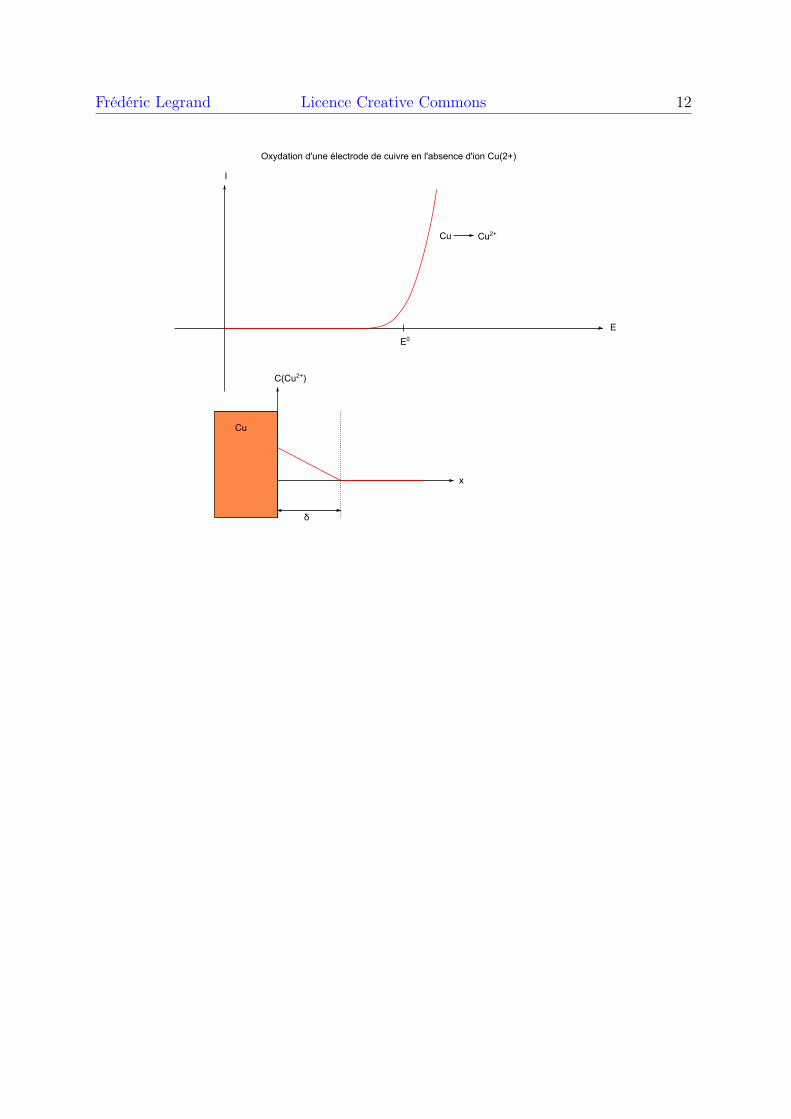
soit absent au début de l’expérience. Par exemple, pour une électrode en cuivre plongéedans une solution aqueuse ne contenant pas d’ions Cu2+, celui-ci est pratiquement absent,du moins au début de l’expérience. Dans ce cas, la diffusion n’intervient évidemment pasdans le courant d’oxydation. Il n’y a pas d’oxydant, donc le courant anodique vientseulement de la réaction d’oxydation. En principe, le potentiel de Nernst est très faiblecar il n’y a pas d’oxydant. Néanmoins, l’oxydant apparaît très vite sur l’électrode, dèsque le courant anodique la traverse. L’oxydant est transportée loin de l’électrode pardiffusion et par convection. Il n’est pas possible dans ce cas de prévoir précisément lepotentiel d’équilibre car on ne connait par la concentration en oxydant sur la surface del’électrode. On peut néanmoins faire un tracé qualitatif, sachant que le courant apparaîtau voisinage du potentiel standard. La figure suivante montre l’exemple du cuivre :
Frédéric Legrand Licence Creative Commons 12
E
I
Cu Cu2+
E0
Cu
C(Cu2+)
δ
Oxydation d'une électrode de cuivre en l'absence d'ion Cu(2+)
x

Frédéric Legrand Licence Creative Commons 13
2.d. Système lentDans un système lent, la cinétique des réactions électrochimiques est prépondérante
par rapport à la cinétique du transfert de matière, du moins au voisinage de l’équilibre.Le transfert d’électrons entre les espèces électroactives et l’électrode limite la vitesse.Lorsque la réaction est très lente, on observe qu’un seuil de surtension est nécessairepour déclencher le courant.
La théorie donne l’expression du courant en fonction du potentiel (avec des hypothèsessur le mécanisme réactionnel). Elle est donnée ici à titre indicatif, pour montrer commentles courbes suivantes sont tracées :
ε =nF
RT(E − E0) (25)
j = nFCrede
(1−α)ε − Coxe−αε1k0
+ 1kred
e(1−α)ε + 1koxe−αε
(26)
Voir courbe courant potentiel pour la définition des constantes et le tracé de la courbe.La figure suivante montre la courbe pour un système très lent, aussi lent pour la réduc-
tion que pour l’oxydation. Par convention, le potentiel standard est nul. Les constanteskox et kred contrôlent les vitesses des échanges de matière et k0 la vitesse de la réaction.Ces trois constantes sont en m/s. Les concentrations sont en mol/m3.
1.00 0.75 0.50 0.25 0.00 0.25 0.50 0.75 1.00E (V)
1000
750
500
250
0
250
500
750
1000
j(A
m2 )
Surtension seuil d'oxydation
Surtension seuil de reduction
E Nernst
Ox+Red
La courbe courant-potentiel d’un système très lent est caractérisée par un intervalle depotentiel où le courant est pratiquement nul. Pour cette raison, le potentiel de l’électroden’est pas défini lorsque le courant est nul. Il faut appliquer une surtension supérieure àun seuil pour déclencher l’oxydation (de même pour la réduction).
La figure suivante montre les tracés lorsque seul l’oxydant et seul le réducteur sontprésents :
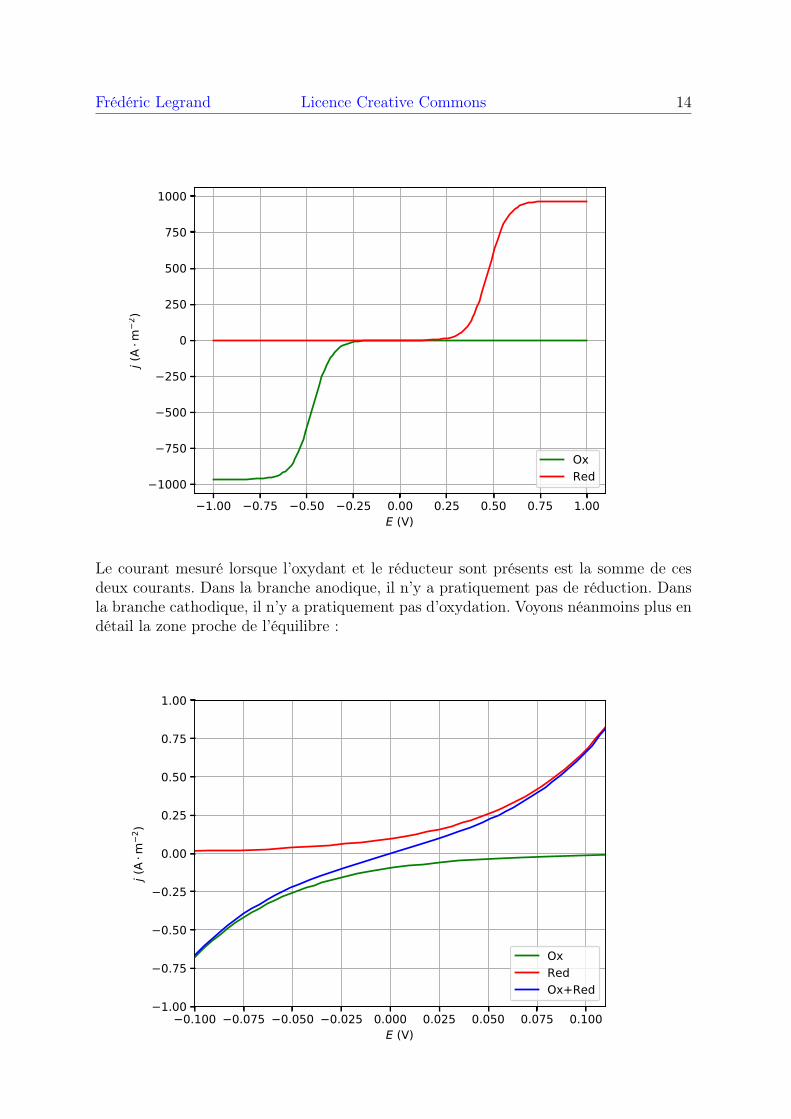
Frédéric Legrand Licence Creative Commons 14
1.00 0.75 0.50 0.25 0.00 0.25 0.50 0.75 1.00E (V)
1000
750
500
250
0
250
500
750
1000
j(A
m2 )
OxRed
Le courant mesuré lorsque l’oxydant et le réducteur sont présents est la somme de cesdeux courants. Dans la branche anodique, il n’y a pratiquement pas de réduction. Dansla branche cathodique, il n’y a pratiquement pas d’oxydation. Voyons néanmoins plus endétail la zone proche de l’équilibre :
0.100 0.075 0.050 0.025 0.000 0.025 0.050 0.075 0.100E (V)
1.00
0.75
0.50
0.25
0.00
0.25
0.50
0.75
1.00
j(A
m2 )
OxRedOx+Red

Frédéric Legrand Licence Creative Commons 15
Lorsque E est proche du potentiel d’équilibre, il existe un très faible courant d’oxydationet un très faible courant de réduction. Lorsque la surtension dépasse le seuil, le courantaugmente de manière exponentielle. En pratique, les appareils de mesure ordinaires nedétectent aucun courant en dessous du seuil.
Pour bien voir à la fois les forts et les faibles courants, on peut tracer le logarithmede la valeur absolue du courant :
1.00 0.75 0.50 0.25 0.00 0.25 0.50 0.75 1.00E (V)
2
1
0
1
2
3
log(
j) (A
/m^2
)
Ox+Red
La branche cathodique est à gauche, la branche anodique à droite. Lorsque la réduc-tion est négligeable sur la branche anodique, celle-ci est pratiquement une droite (avantla limite de courant), ce qui montre le caractère exponentiel de la courbe. De même,lorsque l’oxydation est négligeable sur la branche cathodique, celle-ci est pratiquementrectiligne. L’intérêt de cette représentation est de permettre la détermination du courantà l’équilibre par intersection de ces deux branches rectilignes. Ce courant est ici égal à0,1A ·m−2.
2.e. Oxydation et réduction du solvantOn considère le cas d’un électrolyte liquide dont le solvant est l’eau. L’eau peut subir
aussi bien une oxydation qu’une réduction.La réaction d’oxydation de l’eau est :
H2O→1
2O2 + 2e− + 2H+ (27)
Le potentiel de Nernst s’écrit :
Enernst = E0 − 0, 06 pH +0, 06
2log
(P
1/2O2
)(28)

Frédéric Legrand Licence Creative Commons 16
En réalité, la réaction se fait avec le dioxygène dissout dans l’eau. La présence de lapression partielle de dioxygène dans la loi de Nernst vient de l’équilibre entre le dioxygèneatmosphérique et celui dissout dans la solution. Dans les conditions ordinaires, la pressionpartielle d’oxygène est de l’ordre de 1 bar. Dans ce cas, la quantité de dioxygène dissoutest trop faible pour donner lieu à une réduction visible sur les courbes courant-potentiel.
Le potentiel standard est E0 = 1,23V. Le potentiel standard apparent est E0 −0, 06pH pour une pression partielle d’oxygène de 1 bar. Pour l’eau, le courant limite dediffusion est en principe infini car elle est toujours présente en abondance sur l’électrode.Voyons ce que devrait donner la courbe courant-potentiel si le couple était rapide, à pHnul.
0.0 0.5 1.0 1.5 2.0 2.5E (V)
0
250
500
750
1000
1250
1500
1750
2000
j(A
m2 )
H2O (pH=0)
Le courant d’oxydation apparaît pour un potentiel de l’ordre du potentiel standard ap-parent.
Voyons les résultats expérimentaux. Pour tracer une courbe courant-potentiel del’eau, il faut ajouter des ions pour augmenter la conductivité de l’eau. La courbe ci-dessous est obtenue avec une solution d’acide sulfurique de concentration 0,5mol · L−1.L’électrode de travail est en platine.

Frédéric Legrand Licence Creative Commons 17
0.0 0.5 1.0 1.5 2.0 2.5E (V)
0
250
500
750
1000
1250
1500
1750
2000
j(A
m2 )
H2O + H2SO4 0,5MPt
On voit que le courant d’oxydation apparaît pour un potentiel d’environ 1,5V, bienau dessus du potentiel standard apparent. Cela montre que l’oxydation de l’eau (sur leplatine) est très lente.
L’eau est aussi réduite selon la réaction :
1
2H2 + OH− ← H2O+ e− (29)
Le potentiel standard apparent à pH nul est 0V. Voyons la courbe courant-potentielcomplète obtenue avec une électrode de travail en platine dans la solution d’acide sulfu-rique :
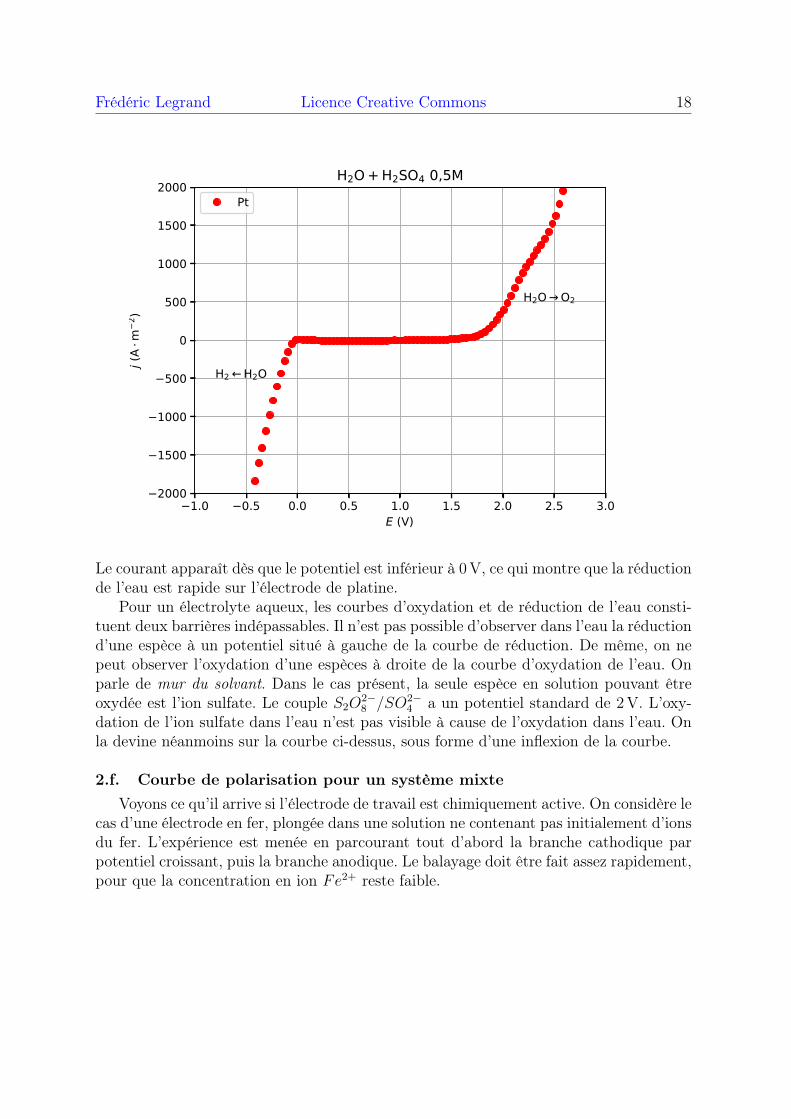
Frédéric Legrand Licence Creative Commons 18
1.0 0.5 0.0 0.5 1.0 1.5 2.0 2.5 3.0E (V)
2000
1500
1000
500
0
500
1000
1500
2000
j(A
m2 )
H2O O2
H2 H2O
H2O + H2SO4 0,5MPt
Le courant apparaît dès que le potentiel est inférieur à 0V, ce qui montre que la réductionde l’eau est rapide sur l’électrode de platine.
Pour un électrolyte aqueux, les courbes d’oxydation et de réduction de l’eau consti-tuent deux barrières indépassables. Il n’est pas possible d’observer dans l’eau la réductiond’une espèce à un potentiel situé à gauche de la courbe de réduction. De même, on nepeut observer l’oxydation d’une espèces à droite de la courbe d’oxydation de l’eau. Onparle de mur du solvant. Dans le cas présent, la seule espèce en solution pouvant êtreoxydée est l’ion sulfate. Le couple S2O
2−8 /SO2−
4 a un potentiel standard de 2V. L’oxy-dation de l’ion sulfate dans l’eau n’est pas visible à cause de l’oxydation dans l’eau. Onla devine néanmoins sur la courbe ci-dessus, sous forme d’une inflexion de la courbe.
2.f. Courbe de polarisation pour un système mixteVoyons ce qu’il arrive si l’électrode de travail est chimiquement active. On considère le
cas d’une électrode en fer, plongée dans une solution ne contenant pas initialement d’ionsdu fer. L’expérience est menée en parcourant tout d’abord la branche cathodique parpotentiel croissant, puis la branche anodique. Le balayage doit être fait assez rapidement,pour que la concentration en ion Fe2+ reste faible.
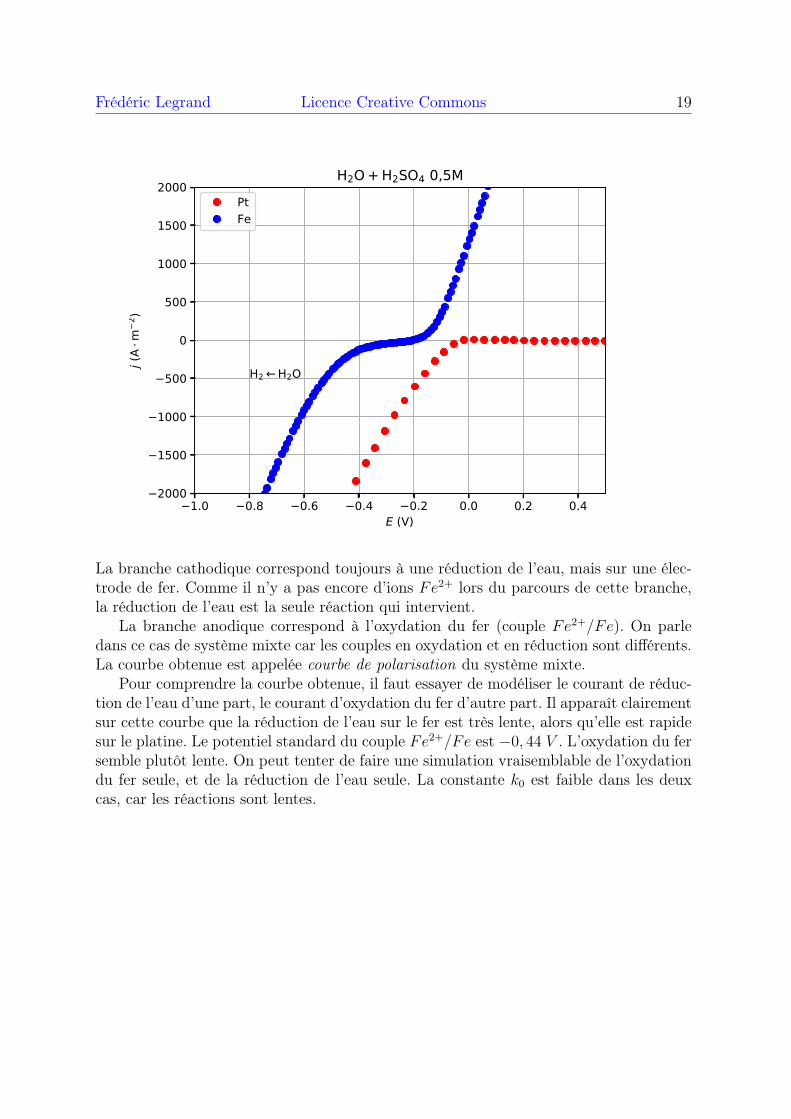
Frédéric Legrand Licence Creative Commons 19
1.0 0.8 0.6 0.4 0.2 0.0 0.2 0.4E (V)
2000
1500
1000
500
0
500
1000
1500
2000
j(A
m2 )
H2O O2
H2 H2O
H2O + H2SO4 0,5MPtFe
La branche cathodique correspond toujours à une réduction de l’eau, mais sur une élec-trode de fer. Comme il n’y a pas encore d’ions Fe2+ lors du parcours de cette branche,la réduction de l’eau est la seule réaction qui intervient.
La branche anodique correspond à l’oxydation du fer (couple Fe2+/Fe). On parledans ce cas de système mixte car les couples en oxydation et en réduction sont différents.La courbe obtenue est appelée courbe de polarisation du système mixte.
Pour comprendre la courbe obtenue, il faut essayer de modéliser le courant de réduc-tion de l’eau d’une part, le courant d’oxydation du fer d’autre part. Il apparaît clairementsur cette courbe que la réduction de l’eau sur le fer est très lente, alors qu’elle est rapidesur le platine. Le potentiel standard du couple Fe2+/Fe est −0, 44 V . L’oxydation du fersemble plutôt lente. On peut tenter de faire une simulation vraisemblable de l’oxydationdu fer seule, et de la réduction de l’eau seule. La constante k0 est faible dans les deuxcas, car les réactions sont lentes.
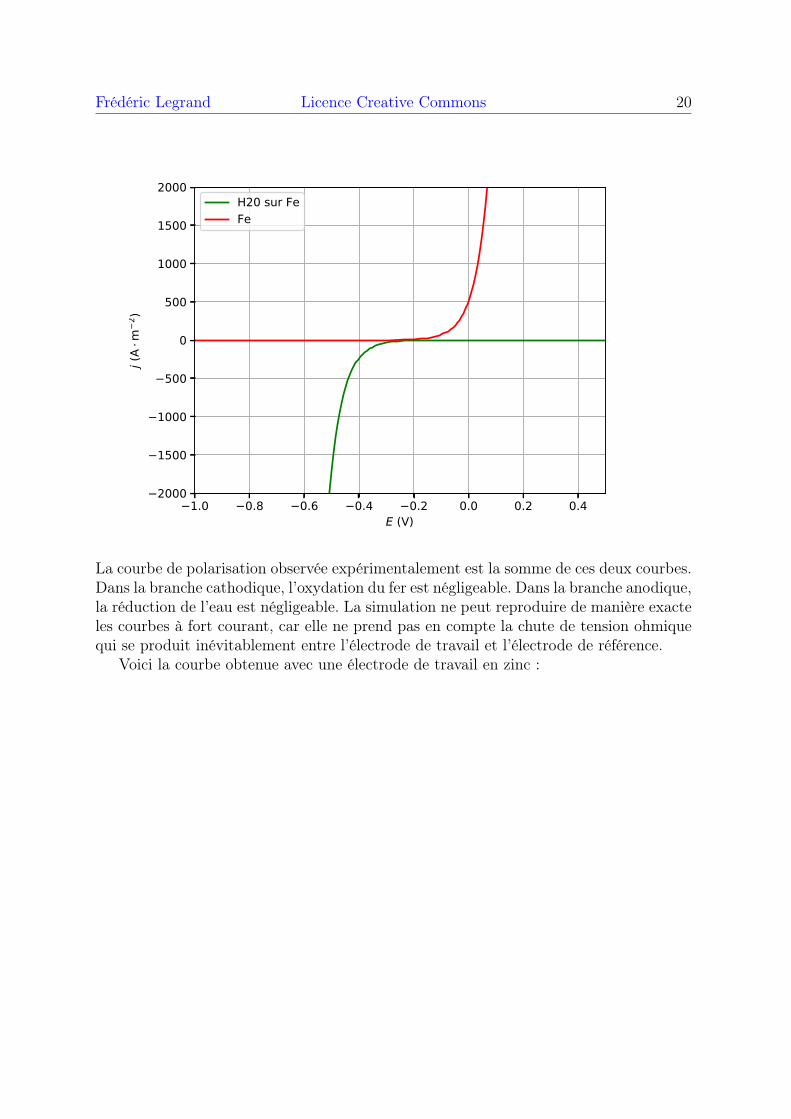
Frédéric Legrand Licence Creative Commons 20
1.0 0.8 0.6 0.4 0.2 0.0 0.2 0.4E (V)
2000
1500
1000
500
0
500
1000
1500
2000
j(A
m2 )
H20 sur FeFe
La courbe de polarisation observée expérimentalement est la somme de ces deux courbes.Dans la branche cathodique, l’oxydation du fer est négligeable. Dans la branche anodique,la réduction de l’eau est négligeable. La simulation ne peut reproduire de manière exacteles courbes à fort courant, car elle ne prend pas en compte la chute de tension ohmiquequi se produit inévitablement entre l’électrode de travail et l’électrode de référence.
Voici la courbe obtenue avec une électrode de travail en zinc :
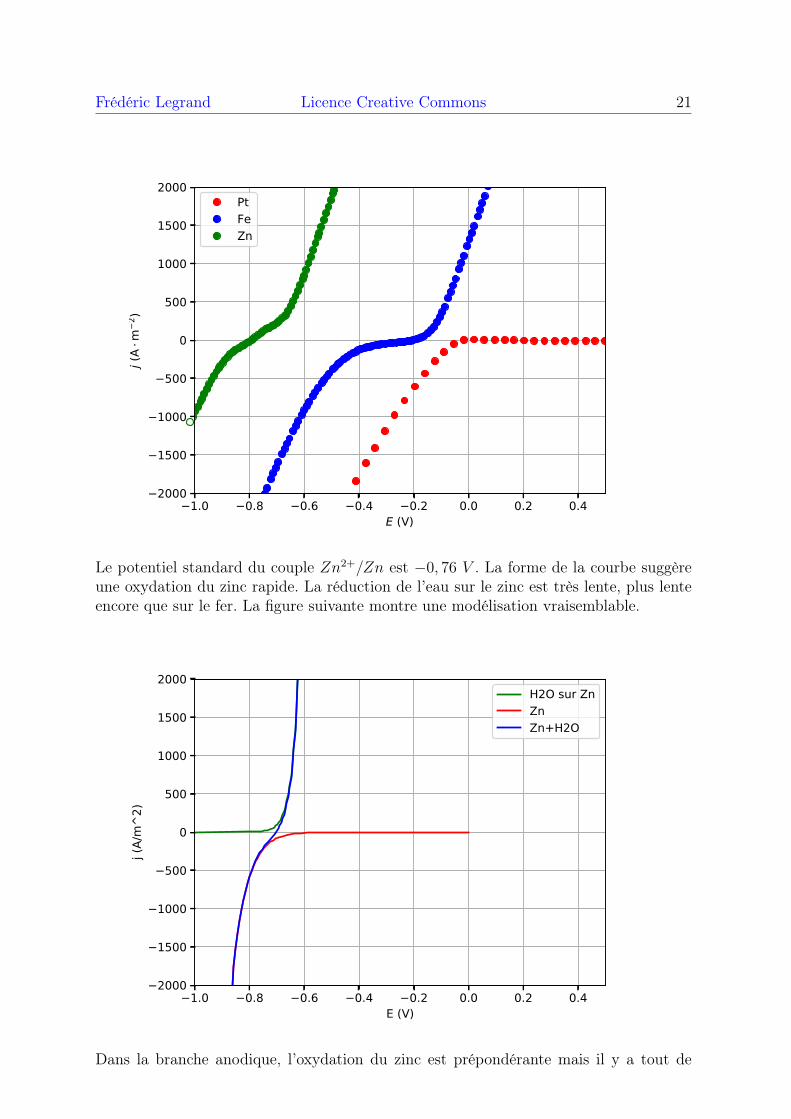
Frédéric Legrand Licence Creative Commons 21
1.0 0.8 0.6 0.4 0.2 0.0 0.2 0.4E (V)
2000
1500
1000
500
0
500
1000
1500
2000
j(A
m2 )
PtFeZn
Le potentiel standard du couple Zn2+/Zn est −0, 76 V . La forme de la courbe suggèreune oxydation du zinc rapide. La réduction de l’eau sur le zinc est très lente, plus lenteencore que sur le fer. La figure suivante montre une modélisation vraisemblable.
1.0 0.8 0.6 0.4 0.2 0.0 0.2 0.4E (V)
2000
1500
1000
500
0
500
1000
1500
2000
j (A/
m^2
)
H2O sur ZnZnZn+H2O
Dans la branche anodique, l’oxydation du zinc est prépondérante mais il y a tout de

Frédéric Legrand Licence Creative Commons 22
même réduction de l’eau au voisinage de l’équilibre. Dans la branche cathodique, laréduction de l’eau est prépondérante mais il y a aussi oxydation du zinc.
2.g. Vagues successivesLorsque deux couples d’oxydoréduction sont présents dans l’électrolyte, ayant des po-
tentiels standard bien distincts, on observe deux vagues d’oxydation successives. Utiliserla simulation courbe courant potentiel pour obtenir ce type de courbes.