51710181 1 13 usercom 15 ta - mettler toledo · 1/2002 informations pour les utilisateurs des...
TRANSCRIPT

1/2002
Informations pour les utilisateurs dessystèmes l’analyse thermique METTLER TOLEDO
15Cher clientCette année est pour nous, en analyse thermique, une année toute particulière. En effet,pour la première fois dans l’histoire de la société, nous lançons sur le marché un appa-reil d’analyse mécanique dynamique (DMA). Nous avons, malgré la conjoncture, pris letemps nécessaire au développement d’un appareil DMA qui se différencie nettement desappareils actuellement sur le marché et qui ouvre de toutes nouvelles possibilités.Vous disposez grâce à la méthode IsoStep et à la méthode étendue de la cinétique sansmodèle, de nouveaux moyens intéressants, également dans le domaine DSC et TGA.
Interprétation des courbes DMA, 1ère partieGeorg Widmann, Dr. Jürgen Schawe, Dr. Rudolf Riesen
Cette première partie introduit la technique de mesure, elle ne traite pas des mesures DMAisothermes. La deuxième partie (UserCom16) décrira plus en détail les différents aspectsdes mesures isothermes, principalement le comportement élastique en fonction de la fré-quence.
IntroductionPar élasticité, nous désignons le mode de déformation des matériaux sous l’action deforces extérieures. Le module d’élasticité d’un matériau est le rapport de la contraintemécanique sur la déformation relative.En analyse mécanique dynamique DMA, des déformations mécaniques sinusoïda-les, d’une fréquence f, sont appliquées à un échantillon et les forces engendrées sont me-surées.Inversement, il est également possible d’appliquer une force d’amplitude définie sur unéchantillon et de mesurer la déformation correspondante.
Conseils TASommaire
Conseils TA- Interprétation des courbes DMA,
1ère partie
Nouveautés- STARe V7.00- Manuels d’application
Applications- Analyse thermique de toners- Caractérisation des résines dans les
procédés lithographiques- Analyse quantitative de mélanges de
polyoléfines- Etude de réactions de durcissement
à l’aide de la méthode IsoStep- Décomposition thermique du sulfate
de cuivre pentahydraté- Etude du délaminage et de la forma-
tion de mousse à l’aide du couplageTMA-MS
Dates

UserCom 1/20022
Dispositifs de mesureIl existe différents dispositifs de mesure:- Cisaillement pour les échantillons dont
la valeur du module de cisaillements’étend sur une plage très étendue, de1 kPa à 2 GPa environ. Les liquides vis-queux ainsi que les échantillons solides,tels que les polymères à l’état vitreux,peuvent donc être mesurés.
- Flexion en trois points pour les échan-tillons rigides, ayant un module d’élas-ticité jusqu’à 1000 GPa.
- Single et Dual Cantilever (simple oudouble levier) pour les échantillonsayant une déformation trop importantedans l’essai de flexion en trois points.
- Mesure en traction pour les barres fines,les films et les fibres.
- Mesure en compression pour les échan-tillons dont le module est inférieur à1 GPa environ.
Grandeurs mesurées
Les données brutes, c’est-à-dire, les ampli-tudes mesurées de la force et de l’allonge-ment, Fa, La, et leur décalage de phase δpermet de calculer les propriétés du maté-riau :• module complexe (M*), module d’élas-
ticité E* pour une tension normale oumodule de cisaillement G* pour unecontrainte en cisaillement.
où g est un facteur géométrique calculé àpartir des dimensions de l’échantillon.
E et G sont reliés par le coefficient de Pois-son, µ :
Pour la majorité des matériaux isotropes,µ est compris entre 0,2 et 0,5. Pour ces ma-tériaux, E est de 2,4 à 3 fois au maximumplus grand que G. Dans la zone élastiquedes matériaux non chargés, E ≈ 3 G, et àl’état vitreux, E = 2.7 G.Pour les matériaux anisotropes, comme lesmatériaux synthétiques renforcés de fibresunidirectionnellement, E peut être plus decent fois plus grand que G.Lorsque le matériau est chauffé, le moduled’élasticité diminue de plusieurs ordres degrandeur par paliers. Le palier correspondà un pic dans le module de perte. Si lestransitions dépendent de la fréquence, ils’agit de transitions de relaxation décaléesvers les températures plus élevées lorsquela fréquence augmente.
Technique de mesure• La mesure DMA est généralement effec-
tuée à amplitude de déplacementimposée, avec une force maximaleprédéfinie, qui ne doit pas être dépasséequelle que soit la rigidité de l’échan-tillon.Un choix non judicieux des amplitudesde déplacement ou de la force peutnuire à la précision de mesure. Les am-plitudes optimales sont supérieures àrespectivement 1 µm et 10 mN, dans lamesure où l’amplitude de déplacementne dépasse pas 1% de la dimension cor-respondante de l’éprouvette. Le modulepourrait varier pour des amplitudesplus importantes (non-linéarité despropriétés de l’échantillon).
• module d’élasticité dynamique, M’(proportionnel à l’énergie emmagasi-née, réversible par élasticité).
• module de perte, M” (proportionnel àl’énergie transformée en chaleur de fa-çon irréversible).
• Facteur de perte, tan δ. Les matériauxpurement élastiques ne présentent pasde décalage de phase, δ, les matériauxpurement visqueux présentent un déca-lage de phase de 90°. Le facteur de pertedes matériaux viscoélastiques varie de 0à l’infini (δ = 90°).
La tan δ correspond au rapport M” surM’ .
Les modules sont calculés selon les formu-les suivantes :
Figure 2: Force et déplacement à une fréquence, f, de 1 Hz. Le décalage de phase δ est calculé à partir du dé-calage de temps ∆ : δ = 2πf∆.
-1.2
-1
-0.8
-0.6
-0.4
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 0.2 0.4 0.6 0.8 1 1.2 1.4
Time in s
Force in N
Displacement in µm
∆
Figure 1: Dispositifs de mesure DMA/SDTA861e (schémas). 1: Cisaillement, 2: Flexion trois points, 3: DualCantilever (analogue à la flexion mais l’éprouvette est à double levier), 4: Single Cantilever (éprouvette àsimple levier), 5: Traction de barres minces, de films et de fibres, 6: Compression.Le dispositif de serrage est représenté en noir, l’échantillon en rouge. Les parties fixes sont hachurées.
4 5 6321
3UserCom 1/2002
• En raison de la faible conductivité ther-mique des matériaux synthétiques etdes dimensions relativement importan-tes des échantillons, les vitesses dechauffe employées – exceptées pourles mesures préliminaires – sont≤ 3 K/min. Ceci s’applique égalementà la vitesse de refroidissement.
• Afin de déterminer l’influence de lafréquence, les mesures sont souventeffectuées à plusieurs fréquences soit enmode multifréquence soit en série defréquences fixées séparément.
Outre les mesures avec un programme detempérature dynamique, le DMA/SDTA861e
permet d’effectuer des mesures isothermesavec des valeurs, sélectionnées par l’utili-sateur, croissantes ou décroissantes par pa-lier, de :• la fréquence,• l’amplitude de déplacement,• l’amplitude de force.
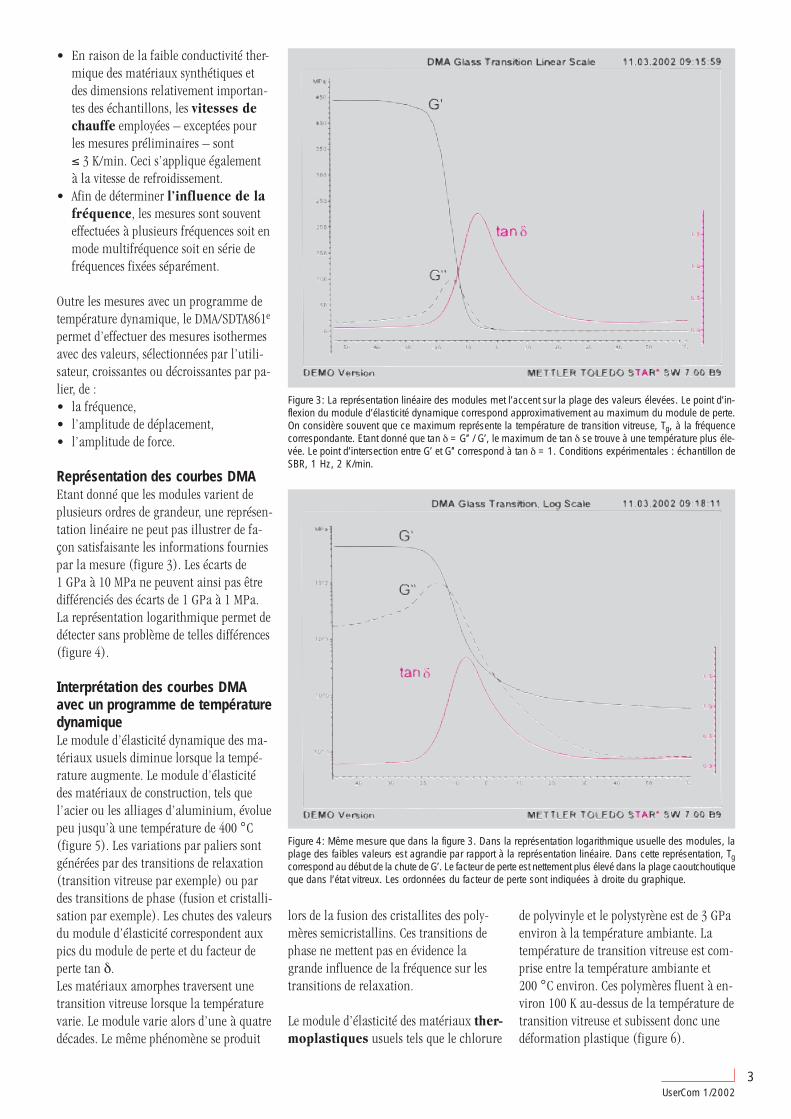
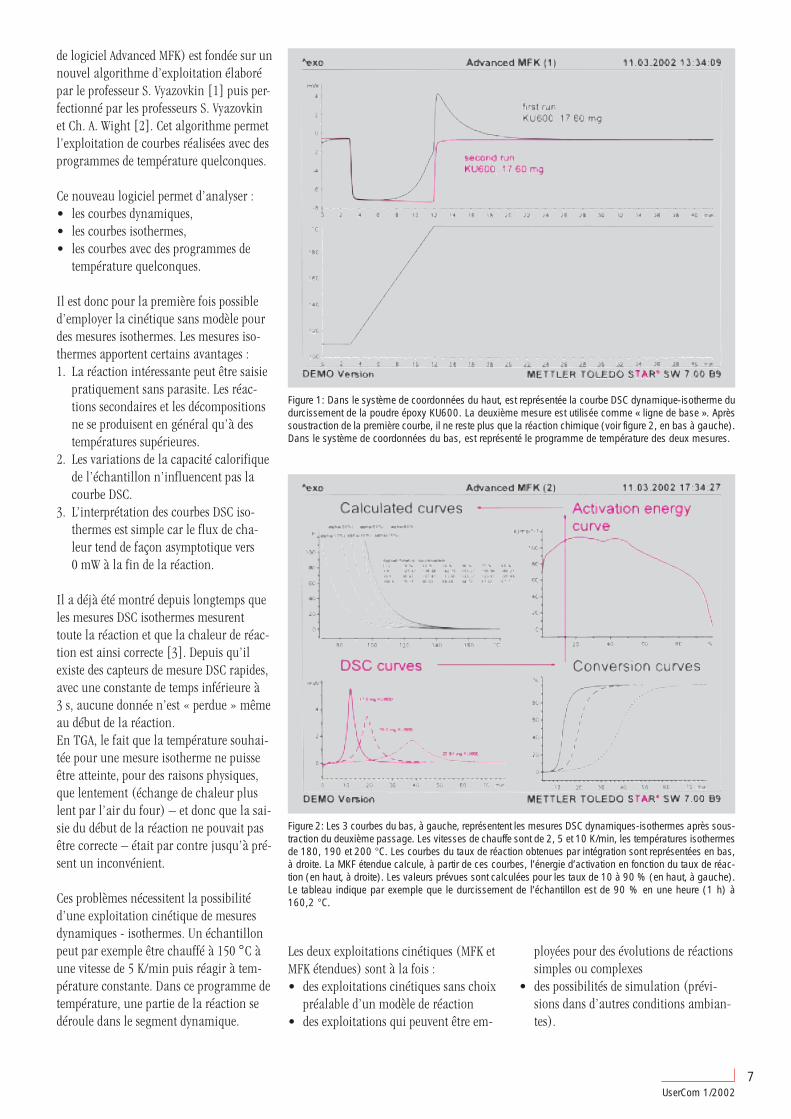
Représentation des courbes DMAEtant donné que les modules varient deplusieurs ordres de grandeur, une représen-tation linéaire ne peut pas illustrer de fa-çon satisfaisante les informations fourniespar la mesure (figure 3). Les écarts de1 GPa à 10 MPa ne peuvent ainsi pas êtredifférenciés des écarts de 1 GPa à 1 MPa.La représentation logarithmique permet dedétecter sans problème de telles différences(figure 4).
Interprétation des courbes DMAavec un programme de températuredynamiqueLe module d’élasticité dynamique des ma-tériaux usuels diminue lorsque la tempé-rature augmente. Le module d’élasticitédes matériaux de construction, tels quel’acier ou les alliages d’aluminium, évoluepeu jusqu’à une température de 400 °C(figure 5). Les variations par paliers sontgénérées par des transitions de relaxation(transition vitreuse par exemple) ou pardes transitions de phase (fusion et cristalli-sation par exemple). Les chutes des valeursdu module d’élasticité correspondent auxpics du module de perte et du facteur deperte tan δ.Les matériaux amorphes traversent unetransition vitreuse lorsque la températurevarie. Le module varie alors d’une à quatredécades. Le même phénomène se produit
lors de la fusion des cristallites des poly-mères semicristallins. Ces transitions dephase ne mettent pas en évidence lagrande influence de la fréquence sur lestransitions de relaxation.
Le module d’élasticité des matériaux ther-moplastiques usuels tels que le chlorure
de polyvinyle et le polystyrène est de 3 GPaenviron à la température ambiante. Latempérature de transition vitreuse est com-prise entre la température ambiante et200 °C environ. Ces polymères fluent à en-viron 100 K au-dessus de la température detransition vitreuse et subissent donc unedéformation plastique (figure 6).
Figure 3: La représentation linéaire des modules met l’accent sur la plage des valeurs élevées. Le point d’in-flexion du module d’élasticité dynamique correspond approximativement au maximum du module de perte.On considère souvent que ce maximum représente la température de transition vitreuse, Tg, à la fréquencecorrespondante. Etant donné que tan δ = G’’ / G’, le maximum de tan δ se trouve à une température plus éle-vée. Le point d’intersection entre G’ et G’’ correspond à tan δ = 1. Conditions expérimentales : échantillon deSBR, 1 Hz, 2 K/min.
Figure 4: Même mesure que dans la figure 3. Dans la représentation logarithmique usuelle des modules, laplage des faibles valeurs est agrandie par rapport à la représentation linéaire. Dans cette représentation, Tgcorrespond au début de la chute de G’. Le facteur de perte est nettement plus élevé dans la plage caoutchoutiqueque dans l’état vitreux. Les ordonnées du facteur de perte sont indiquées à droite du graphique.

UserCom 1/20024
La température de transition vitreuse desélastomères tels que le caoutchouc na-turel NR est inférieure à la températureambiante, aucun fluage ne se produit en
raison de la réticulation chimique (figure5). Cette faible réticulation apparaît lorsde la vulcanisation du caoutchouc initiale-ment thermoplastique.
Les matériaux thermodurcissables telsque les résines époxy sont constitués demacromolécules réticulées tridimension-nellement. La plage de la température detransition vitreuse se trouve nettement au-dessus de la température ambiante. En rai-son de leur réticulation stœchiométrique,ces matériaux ne fluent pas lorsque latempérature continue à augmenter. Lesmatériaux initiaux des thermodurcissablessont constitués de plusieurs composants,souvent appelés «résine» et «durcisseur».Le durcissement des matériaux thermo-plastiques initiaux évolue avec la réticula-tion tridimensionnelle et la température detransition vitreuse augmente de 50 à 300 K(figure 10).
Lorsque les macromolécules ont été ali-gnées par un traitement adapté, on parlede polymères orientés. Ceux-ci sont ani-sotropes, c’est-à-dire, leurs propriétésdépendent fortement de la direction. Cecis’applique également aux polymères ren-forcés de fibres.
Plusieurs transitions de relaxation sont ob-servées dans les matériaux amorphes etsemicristallins, la transition qui se produità la plus haute température est appeléepour des raisons historiques relaxationααααα ou transition vitreuse. Elle est attri-buée au mouvement moléculaire interactifdans une zone de quelques nanomètresalors que la relaxation secondaire, ourelaxation βββββ concerne les mouvementsde segments courts.Les processus de relaxation dépendent,contrairement aux processus de fusion, decristallisation et de réactions chimiques,de la fréquence et peuvent donc être fa-cilement détectés. La transition vitreusevarie (est décalée) de 5 à 10 K par décadede fréquence. L’influence de la fréquenceest encore plus importante sur la relaxa-tion β décalage de 10 K/décade au mini-mum (figure 7).
Les mélanges incompatibles de polymè-res amorphes ou les copolymères en blocsprésentent les deux transitions vitreusesdes deux composants alors que les mélan-ges compatibles et les copolymères statisti-
Figure 5: Partie élastique, E’, du module E* de quelques matériaux en fonction de la température. E’ de l’acierdiminue peu (210 GPa à la température ambiante, 177 GPa à 400 °C). Par contre, la chute du module d’é-lasticité (proportionnel à la rigidité) des matériaux organiques, en un ou plusieurs paliers, est pratiquementtotale. Tg est la température de transition vitreuse, Tm la température de fusion. CFE: résine époxy renforcée defibres de verre, PP: polypropylène, PS: polystyrène, NR: caoutchouc naturel.
Temperature
E'
0 100 200 300 400 °C-100
10 GPa
1 GPa
10 MPa
1 MPa
Steel
Aluminum alloys
CFE
PP
NR Tg
Tg
Tg
Tm
RT
Tg
PS
Figure 6: Comportement type d’un thermoplastique amorphe : à l’état vitreux, le polymère a un comportementélastique pratiquement idéal. Au niveau de la transition vitreuse le comportement devient de type « leathery »puis « caoutchoutiquex, souple ». La largeur de la zone d’élasticité caoutchoutique augmente avec la massemolaire moyenne. Un écoulement plastique suit si la température de décomposition du thermoplastique estsuffisamment élevée. La zone viscoélastique comprend la partie du type « leathery », le plateau coutchoutiqueet la zone de fluage.
10 GPa
1 GPa
100 MPa
10 MPa
1 MPa
Temperature
G'
Glassy Leathery Rubbery Plateau
Elas
tic F
low
Liqu
id F
low
0
1.5
1
0.5
tan δ
G'
tan δ

5UserCom 1/2002
ques ne présentent qu’une transition vi-treuse située entre celles de chacun descomposants (figure 8).Les fractions peuvent être estimées à l’aidedes courbes des polymères purs.
Les propriétés des thermoplastiques semi-cristallins dépendent du taux de cristal-linité. Certains matériaux synthétiques,tels que le polyéthylène-térephtalate, res-tent amorphes par trempe à partir du
fondu et cristallisent à une température su-périeure à Tg lorsque la température aug-mente (figure 9).
Dans le cas des thermodurcissables,on s’intéresse au comportement des maté-riaux thermoplastiques initiaux, à l’aug-mentation du module lors de la gélifica-tion et à la transition vitreuse du matériaudurci et fini (figure 10). De telles mesures,pour lesquelles le module s’étend sur plusde 4 décades, ne sont possibles qu’en ci-saillement.
ConclusionLes mesures DMA donnent d’une part unaperçu de la mobilité moléculaire en fonc-tion de la température et de la fréquence,elles permettent d’autre part de déterminerdes propriétés de matériaux intéressantespour l’ingénieur : rigidité, amortissementet température d’utilisation des matériauxpar exemple. Les mesures DMA mettent enparticulier en évidence l’influence de l’hu-midité ou du degré de durcissement parexemple sur la transition vitreuse (voirUserCom11, p. 8...13).
Figure 9:Courbes du haut : ramollissement du polyéthylène-térephtalate à la transition vitreuse et cristallisationau niveau de C. Le module augmente ensuite encorelégèrement en raison de l’apparition de cristallitesplus parfaits. Les cristallites fondent enfin. Seule latransition vitreuse présente un glissement lorsque lafréquence augmente.Courbes du bas : le module d’un thermoplastiquesemicristallin tel que le polypropylène, augmente avecle taux de cristallinité. L’évolution de la transition vi-treuse des matériaux très cristallins est moins impor-tante.
Tg
Tm
Increasingcrystallinity
E'
1 Decade
PPTg
C
Tm
PET1,10, 100 Hz
Temperature
0 100 200 300 °C-100
Figure 7: Module de cisaillement et facteur de perte, mesurés pour trois fréquences variant par décades suc-cessives. Le décalage de relaxation β est plus important que celui de la transition vitreuse. Le facteur de pertene dépasse normalement la valeur 1 qu’à la transition vitreuse.
Temperature
tan δ
G'
Glass Transitionβ-Relaxation
1, 10, 100 Hz
10 GPa
1 GPa
100 MPa
10 MPa
1 MPa
G'
0
1
tan δ
Figure 8: Mélanges de polymères ou copolymères compatibles et incompatibles comparés aux homopolymèresd’origine.
Temperature
G'
Blends or copolymers
1 GPa
100 MPa
10 MPa
1 MPa
Pure polymerwith low Tg
Pure polymerwith high T g
Incompatible
Compatible

UserCom 1/20026
Nouveautés
STARe V7.00
La nouvelle version de logiciel STARe
V7.00, compatible Windows2000 etWindowsNT, est la version qui succède àtoutes les versions STARe ≤ V6.20 (Unixou WindowsNT).La compatibilité avec les anciennes ver-sions est assurée. Vous pouvez ainsi char-ger sans problème les anciennes donnéesdans la nouvelle version de logiciel et leséditer si nécessaire.
Cette nouvelle version ouvre les possibilitéssuivantes :• Commande du DMA/SDTA861e de
METTLER TOLEDO• Représentation et exploitation des cour-
bes DMA• Nouvelle cinétique sans modèle étendue
(option de logiciel : Advanced ModelFree Kinetics)
• Nouvelle analyse IsoStep (séparationdes effets cinétiques des variations de lacapacité calorifique) (option de logiciel :IsoStep)
Outre ces 4 possibilités importantes supplé-mentaires, le logiciel a également été per-fectionné dans de nombreux petits détails :• Possibilités de formatage (choix des
couleurs, police d’écriture, taille de lapolice et style de ligne)
• Différents systèmes de coordonnées avecnouvelles possibilités, améliorées,d’échelles : linéaire, semi-logarithmi-que (linéaire-logarithmique ou loga-rithmique-linéaire), logarithmique
• Nouvelles exploitations des courbes,également dans les systèmes de coor-données non linéaires
• Fonction d’approximation polynomialeavec possibilités étendues
• Méthode Cp-Saphir améliorée, lorsquele programme de température est com-posé de plusieurs segments dynamiques,ce que nous recommandons pour lesmesures de cp sur une large plage detempérature.
Cinétique sans modèle étendueLa cinétique sans modèle (option de logi-ciel MFK) permettait jusqu’à présent d’ex-ploiter des courbes de mesure dynamique.La nouvelle cinétique sans modèle (option
Lorsque les mesures DMA sont effectuéessur des échantillons inconnus, il est tou-jours préférable d’effectuer auparavantune mesure DSC à 20 K/min sur une largeplage de température. La courbe DSC obte-nue permet de déduire une plage de tempé-rature judicieuse pour les mesures DMAafin d’éviter une fusion ou une décomposi-tion totale de l’échantillon par exemple.Ainsi une deuxième mesure sur le mêmeéchantillon est possible, tout au plus lagéométrie doit être redéfinie.
Les mesures DSC peuvent en général facili-ter l’interprétation des courbes DMA (et in-versement !). Les mesures DSC et DMAfournissent différentes informations et nepeuvent pas mutuellement se remplacer,elles se complètent par contre très bien.
Figure 10: Module de cisaillement et facteur de perte du système résine époxy liquide à la température am-biante DGEBA (éther diglycidylique du bisphénol A) et DDM (diaminodiphénylméthane). Vitesse de chauffe etde refroidissement : 3 K/min, fréquence :10 Hz, géométrie de l’échantillon : 11 mm de diamètre, 0,2 mmd’épaisseur. Le mélange des 2 composants est solidifié à l’état vitreux à la température initiale de -50 °C, ildevient liquide au-dessus de 0 °C. Le module d’élasticité diminue alors de 7 décades et le facteur de perteaugmente de 5 décades. Au cours du durcissement, la résine gélifie à 140 °C. Après que la températurefinale, de 210 °C, a été atteinte, le thermodurcissable est refroidi au-dessous de la température de transitionvitreuse, de 145 °C, le module augmente de nouveau.
7UserCom 1/2002
de logiciel Advanced MFK) est fondée sur unnouvel algorithme d’exploitation élaborépar le professeur S. Vyazovkin [1] puis per-fectionné par les professeurs S. Vyazovkinet Ch. A. Wight [2]. Cet algorithme permetl’exploitation de courbes réalisées avec desprogrammes de température quelconques.
Ce nouveau logiciel permet d’analyser :• les courbes dynamiques,• les courbes isothermes,• les courbes avec des programmes de
température quelconques.
Il est donc pour la première fois possibled’employer la cinétique sans modèle pourdes mesures isothermes. Les mesures iso-thermes apportent certains avantages :1. La réaction intéressante peut être saisie
pratiquement sans parasite. Les réac-tions secondaires et les décompositionsne se produisent en général qu’à destempératures supérieures.
2. Les variations de la capacité calorifiquede l’échantillon n’influencent pas lacourbe DSC.
3. L’interprétation des courbes DSC iso-thermes est simple car le flux de cha-leur tend de façon asymptotique vers0 mW à la fin de la réaction.
Il a déjà été montré depuis longtemps queles mesures DSC isothermes mesurenttoute la réaction et que la chaleur de réac-tion est ainsi correcte [3]. Depuis qu’ilexiste des capteurs de mesure DSC rapides,avec une constante de temps inférieure à3 s, aucune donnée n’est « perdue » mêmeau début de la réaction.En TGA, le fait que la température souhai-tée pour une mesure isotherme ne puisseêtre atteinte, pour des raisons physiques,que lentement (échange de chaleur pluslent par l’air du four) – et donc que la sai-sie du début de la réaction ne pouvait pasêtre correcte – était par contre jusqu’à pré-sent un inconvénient.
Ces problèmes nécessitent la possibilitéd’une exploitation cinétique de mesuresdynamiques - isothermes. Un échantillonpeut par exemple être chauffé à 150 °C àune vitesse de 5 K/min puis réagir à tem-pérature constante. Dans ce programme detempérature, une partie de la réaction sedéroule dans le segment dynamique.
Figure 1: Dans le système de coordonnées du haut, est représentée la courbe DSC dynamique-isotherme dudurcissement de la poudre époxy KU600. La deuxième mesure est utilisée comme « ligne de base ». Aprèssoustraction de la première courbe, il ne reste plus que la réaction chimique (voir figure 2, en bas à gauche).Dans le système de coordonnées du bas, est représenté le programme de température des deux mesures.
Figure 2: Les 3 courbes du bas, à gauche, représentent les mesures DSC dynamiques-isothermes après sous-traction du deuxième passage. Les vitesses de chauffe sont de 2, 5 et 10 K/min, les températures isothermesde 180, 190 et 200 °C. Les courbes du taux de réaction obtenues par intégration sont représentées en bas,à droite. La MKF étendue calcule, à partir de ces courbes, l’énergie d’activation en fonction du taux de réac-tion (en haut, à droite). Les valeurs prévues sont calculées pour les taux de 10 à 90 % (en haut, à gauche).Le tableau indique par exemple que le durcissement de l’échantillon est de 90 % en une heure (1 h) à160,2 °C.
Les deux exploitations cinétiques (MFK etMFK étendues) sont à la fois :• des exploitations cinétiques sans choix
préalable d’un modèle de réaction• des exploitations qui peuvent être em-
ployées pour des évolutions de réactionssimples ou complexes
• des possibilités de simulation (prévi-sions dans d’autres conditions ambian-tes).

UserCom 1/20028
Ces exploitations cinétiques conviennentparticulièrement bien aux études sur la sé-curité et aux optimisations de procédés.
Bibliographie[1] S. Vyazovkin, Evaluation of the activation
energy of thermally stimulated solid-statereactions under an arbitrary variation ofthe temperature, J. Comput. Chem., 1997,v. 18, N3, 393-402.
[2] S. Vyazovkin, C.A. Wight, Estimatingrealistic confidence intervals for theactivation energy determined fromthermoanalytical measurements, Anal.Chem., 2000, v. 72, N14, 3171-3175 andto S. Vyazovkin, Modification of the inte-gral isoconversional method to accountfor variation in the activation energy,J. Comput. Chem., 2001, v. 22, N2, 178-183
[3] G. Widmann, Thermochimica Acta, 11(1975) 331-333
IsoStepLe programme de température de cette mé-thode est composé de nombreux segmentsdynamiques intercalés de segments isother-mes.
L’exploitation est fondée sur les idées de S.C. Mraw et H. Dörr. Les segments isother-mes permettent de corriger la dérive iso-therme des segments dynamiques, ce quiconduit à une meilleure précision de la dé-termination de la cp. Le palier isothermepeut contenir également des informationscinétiques d’une réaction chimique parexemple.
Cette nouvelle technique permet ainsi• de déterminer la cp avec une meilleure
précision à l’aide de la référence saphiret
• de séparer les effets cinétiques des varia-tions de la capacité calorifique.
A titre d’exemple, une mesure du PET a été exploitée à l’aide de la méthode IsoStep.
Une application IsoStep est illustrée à la page 18.
Figure 3: Programme de température IsoStep cons-titué de différents segments dynamiques et isother-mes.
Temperature
Time
Figure 4: Dans le système de coordonnées du haut sont représentées les mesures du PET et du saphir corri-gées par la valeur à blanc. Elles permettent de calculer les 3 autres courbes :1. Courbe Cp (montre une augmentation de cp à 75 °C (transition vitreuse) et une diminution de cp à
170 °C (cristallisation à froid))2. Courbe réversible (courbe du flux de chaleur calculée à partir de la courbe de la capacité calorifique)3. Courbe non réversible (montre une relaxation d’enthalpie à 75 °C environ (125 min) et une cristallisa-
tion à froid à 120 °C environ (170 min))
On trouve des applications intéressantes dans les domaines suivants :
Industrie Effets analysés avec IsoStep
Automobile et Réactions de durcissement, influence de l’humidité,aéronautique transition vitreuse, vitrification
Chimie Réactions exothermiques (études sur la sécurité), tran-sition vitreuse, cinétique, cristallisation, polymorphie,séchage, capacité calorifique
Electronique Réactions de durcissement, transition vitreuse, vitrification
Peintures et vernis Réactions de durcissement, influence de l’humidité,transition vitreuse, séchage, vitrification
Caoutchoucs Transition vitreuse, séparation de phase, fusion,(élastomères) vulcanisation
Matériaux synthétiques Réactions de durcissement, influence de l’humidité, relaxa-(thermoplastiques, ther- tion d’enthalpie, transition vitreuse, cristallisation à froid,modurcissables, fibres, séparation de phase, fusion, fusion et cristallisation,films, textiles, colles, vitrification, capacité calorifiqueemballages et câbles)
Produits alimentaires Influence de l’humidité, gélatinisation, transition vitreuse,viscosité, polymorphie, séchage
Pharmacie Influence de l’humidité, transition vitreuse, fusion (paliersisothermes -point de fusion), cristallisation, polymorphie,séchage, capacité calorifique, décomposition

9UserCom 1/2002
Titre Langue / No. de référence Résumé
Manuel Manuel : Le manuel d’applications, adapté aux substances de test, convient plus particu-didacticiel Allemand: 51 709 919 lièrement à l’étude autodidacte de l’analyse thermique.
Anglais: 51 709 920 22 exemples illustrent tout que l’on peut faire en analyse thermique.Français: 51 709 921
Manuel av. substances de test :Allemand: 51 140 877Anglais: 51 140 878Français: 51 140 879
Manuel sur Allemand: 51 725 001 Les comportements thermiques de plus de 20 thermoplastiques sont discutés àles thermo- Anglais: 51 725 002 partir de plus de 100 courbes DSC. 16 mesures TMA et 9 TGA complètent l’ensem-plastiques ble. Les effets étudiés sont : fusion, cristallisation, transition vitreuse, ramollisse-
ment, séchage, décomposition thermique, stabilité à l’oxydation, dilatation etretrait. Des questions pratiques telles que la détermination thermogravimétriquedes teneurs de constituants, ou comment éviter une erreur de matériau, sontégalement traitées.
Manuel Anglais: 51 725 058 Après une introduction sur l’analyse thermique de même que sur la structure etsur les les propriétés des élastomères, plus de 50 exemples d’analyses d’élastomères sontélastomères présentés, ils prennent en compte non seulement les mesures DSC, TGA et TMA(NOUVEAU) mais aussi des méthodes couplées de l’analyse des gaz et de l’analyse mécanique
dynamique (DMA). La partie pratique du manuel est divisée en deux chapitres.Le premier traite les fondements des effets thermiques et leur exploitation. L’ana-lyse de la composition des substances à l’aide de la TGA ainsi que les mesurespour la détermination de la vulcanisation, de la cristallisation et de la transitionvitreuse y sont également discutées. Il comprend de plus des remarques sur l’opti-misation des mesures et des exploitations des résultats. Il est suivi d’un chapitred’exemples pratiques d’analyse d’élastomères, en allant d’exemples relativementsimples jusqu’à des analyses complexes de systèmes compliqués.
Manuel phar- Allemand: 51 725 005 47 exemples sélectionnés illustrent les possibilités d’application de l’analyse ther-maceutique Anglais: 51 725 006 A mique dans l’industrie pharmaceutique. Le comportement à la fusion, la poly-
morphie, la pureté, le taux d’humidité de même que la stabilité des substancesactives et auxiliaires entre autres sont analysés à l’aide des mesures DSC, TGA,EGA et TOA. L’influence des conditions expérimentales et l’étalonnage des appa-reils sont de plus discutés.
Manuel sur Allemand: 51 725 003 Les applications de l’analyse thermique de protéines, d’hydrates de carbone, deles produits Anglais: 51 725 004 graisses et d’huiles sont illustrée par 53 courbes DSC, 2 TGA et une TMA. Lesalimentaires principaux effets étudiés sont : la dénaturation des protéines, le gonflement de
l’amidon dans l’eau, la fusion du sucre, la décomposition thermique du sucre etde l’amidon, la fusion et la cristallisation des graisses, des huiles et du chocolat.
Manuel Anglais: 51 725 056 Le manuel a pour objet le couplage de la thermogravimétrie (TGA) et de l’ana-TGA-EGA lyse des gaz. La première partie traite des bases de la spectroscopie de masse(NOUVEAU) (MS), de la spectroscopie IRTF et de leur interprétation. La deuxième partie,
pratique, présentent 17 exemples d’application de ces techniques de couplage.Des échantillons organiques et inorganiques de même que des systèmes de poly-mères sont étudiés. Outre les couplages conventionnels TGA-MS et TGA-FTIR, unexemple de couplage TMA-MS est également présenté.
Manuels d’application
C’est avec plaisir que nous vous proposons deux nouveaux manuels d’application, qui peuvent vous apporter une aide précieuse dansvos tâches journalières.
Le manuel sur les élastomères et celui sur les techniques de mesure couplées (TGA-FTIR et TGA-MS) étendent encore le nombre de vo-lumes d’applications.

UserCom 1/200210
Applications
Analyse thermique de tonersDr. Markus Schubnell
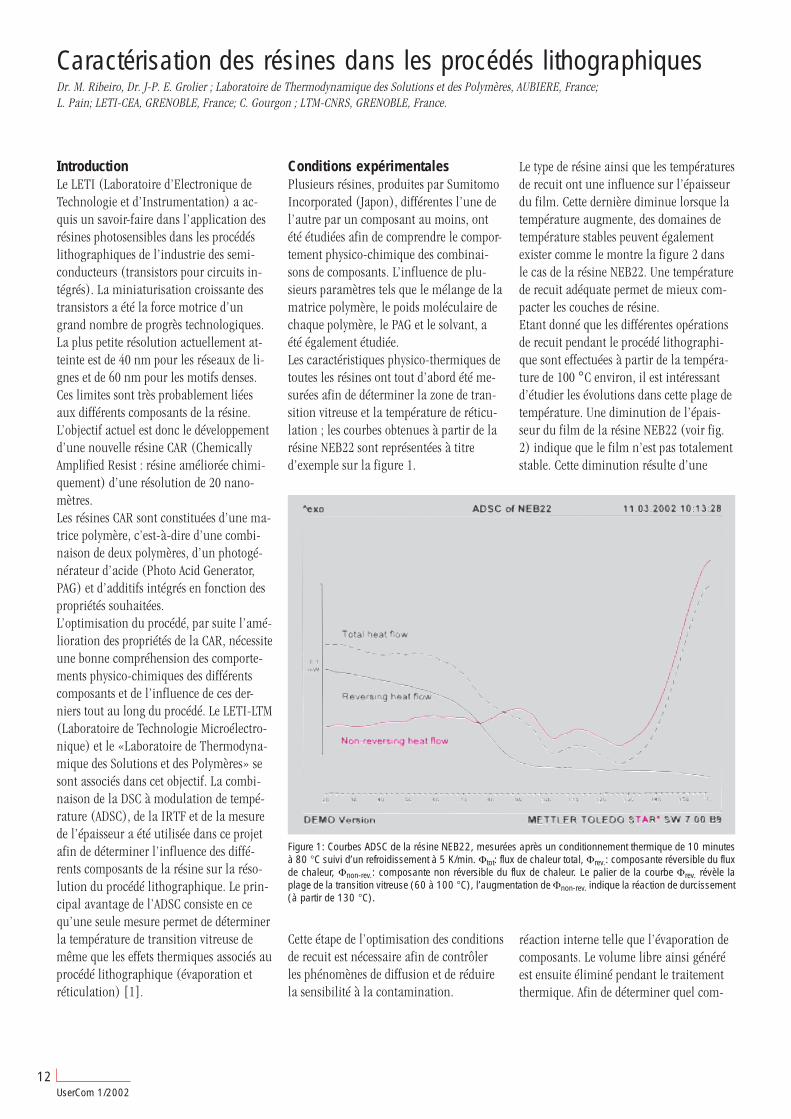
IntroductionLes toners, tels qu’ils sont employés dansles imprimantes laser actuelles et dans lesphotocopieurs, sont en règle générale desmélanges complexes de matériaux de basethermoplastiques auxquels différents addi-tifs tels que solvants, colorants, stabilisa-teurs UV et autres sont mélangés.Les toners sont caractérisés entre autrespar la température de transition vitreusedu matériau de base et par la températureet l’enthalpie de fusion des additifs. Cesparamètres peuvent être déterminés de fa-çon simple et fiable par l’analyse thermi-que. Les méthodes plus particulièrementemployées pour cela sont l’analyse calori-métrique différentielle (DSC) et l’analysemécanique dynamique (DMA).Cet article compare les mesures de ces deuxtechniques à l’aide d’un exemple de toner.Les appareils utilisés sont le DSC821e et leDMA/SDTA861e.
Mesures DSCL’échantillon est tout d’abord chauffé à10 K/min puis refroidi également à10 K/min et une nouvelle fois chauffé à10 K/min. La première montée en tempé-rature présentent 2 pics endothermiquesincomplets, séparés l’un de l’autre. Un lé-ger décalage de la ligne de base a, enoutre, été constaté. La courbe du refroidis-sement présente plusieurs pics exothermi-ques décalés à des températures inférieuresà celle des pics de la courbe de montée entempérature. Un palier dans la ligne debase est de plus également révélé de 70 °Cà 45 °C, approximativement à la mêmetempérature pour la montée en tempéra-ture. Ceci signifie une superposition d’unefusion et d’une transition vitreuse pendantla première montée en température. A ladeuxième montée en température, seul unpic très marqué apparaît. Le deuxième picendothermique de la première montée en
température peut donc être attribué à unerelaxation d’enthalpie, il se produit lors dela montée en température à la suite d’unerelaxation d’enthalpie à l’état vitreux (voirUserCom 10, page 13 ff).
Mesures DMAEn analyse mécanique dynamique, unéchantillon est soumis à une force pério-dique, sinusoïdale. Dans la zone linéaire(Hooke), les déformations générées sont de
Cette interprétation peut être vérifiée si lesnouveaux échantillons sont chauffés avecdifférentes vitesses de chauffe. Le pic de fu-sion devrait rester dépendant de la vitessede chauffe alors que la relaxation d’enthal-pie devrait se décaler vers les températuressupérieures lorsque la vitesse de chauffeaugmente. Les résultats expérimentauxcorrespondants sont montrés sur la figure2 sur laquelle les courbes DSC de la pre-mière montée en température avec unevitesse de chauffe de 0.5, 10 et 150 K/minsont tracées. La figure montre effectivementque la relaxation d’enthalpie se décale versles températures plus élevées tandis que lepoint de fusion, à 60 °C, reste pratique-ment inchangé.
forme sinusoïdale mais décalées dans letemps par rapport à la force. Des informa-tions sur la dynamique moléculaire del’échantillon peuvent être déduites du rap-port des amplitudes de la force et de la dé-formation ainsi que du décalage de phase.Les résultats quantitatifs fournis par l’ana-lyse mécanique dynamique sont le moduled’élasticité et le module de perte de mêmeque le facteur mécanique de perte (amor-tissement).Un paramètre important dans les mesuresDMA est la période, c.-à-d. la fréquence, àlaquelle l’échantillon est soumis. L’in-fluence de la fréquence sur les courbesexpérimentales permet de déterminer cer-tains effets. Ainsi, la cristallisation et la fu-
Figure 1: Courbes DSC de la première montée en température, du refroidissement et de la deuxième montéeen température d’un échantillon de toner.

11UserCom 1/2002
sion par exemple sont des processus prati-quement indépendants de la fréquence. Lesphénomènes de relaxation tels que la tran-sition vitreuse dépendent par contre tou-jours de la fréquence. Si nous partons duprincipe de la superposition d’une fusionet d’une transition vitreuse dans le cas del’échantillon de toner, les deux processusdoivent pouvoir être différenciés par la dif-férence de leur comportement en fonctionde la fréquence.L’échantillon est constitué de fine poudre.Une possibilité de mesurer les matériauxsous forme de poudre en DMA consiste àcompacter la poudre en une éprouvette cy-lindrique comprimée. Ces éprouvettes sontensuite serrées dans les supports de cisail-lement de l’appareil DMA. Des éprouvettesde 12 mm de diamètre et de 1,5 mm dehauteur ont été réalisées de cette manièreavec le matériau considéré. Une pressionde 100 N/cm2 a été appliquée.La figure 3 montre l’évolution de la com-posante élastique du module de cisaille-ment de l’échantillon en fonction de latempérature, aux fréquences 1, 10, 100 et800 Hz. La vitesse de chauffe employée estde 2 K/min. L’essai a été effectué à dépla-cement imposé, la valeur de consigne del’amplitude ayant été fixé à 1 µm. Uneamplitude maximale de la force 3 N a de
le processus de fusion, indépendant de lafréquence. Le deuxième palier nettementvisible aux fréquences plus élevées corres-pond à la température de transition vi-treuse des composants thermoplastiquesde l’échantillon.
ConclusionUne fusion et une transition vitreuse super-posées peuvent être facilement séparées àl’aide de la DSC et de la DMA. En DSC, lesdeux effets peuvent être séparés en faisantintervenir l’influence de la vitesse dechauffe sur la transition vitreuse. En DMA,la séparation est obtenue par les différen-ces dans l’influence de la fréquence. Lesdeux méthodes fournissent en principe desrésultats équivalents. La sensibilité de laDSC aux transitions vitreuses est, par na-ture, nettement plus faible que la sensibi-lité aux processus de fusion. La DMA est
plus été définie. La courbe du module ob-tenue à une fréquence de 1 Hz présente unlarge palier. Un deuxième palier de plus enplus décalé est révélé lorsque la fréquenceaugmente. La température initiale de lapremière chute de température reste prati-quement toujours la même, indépendam-ment de la fréquence. Ce palier décrit donc
Figure 2: Première montée en température du toner avec différentes vitesses de chauffe.
Figure 3: Composante élastique du module de cisaillement d’une éprouvette de toner à différentes fréquen-ces. Les 4 courbes ont été obtenues avec une vitesse de chauffe de 2 K/min.
par contre nettement plus sensible aux va-riations de la mobilité moléculaire tellesqu’elles se produisent de manière équiva-lente lors de la fusion et des transitionsvitreuses. La grande plage de fréquencesdu DMA/SDTA861e apportent de nouvellespossibilités en analyse mécanique dynami-que.
UserCom 1/200212
Caractérisation des résines dans les procédés lithographiquesDr. M. Ribeiro, Dr. J-P. E. Grolier ; Laboratoire de Thermodynamique des Solutions et des Polymères, AUBIERE, France;L. Pain; LETI-CEA, GRENOBLE, France; C. Gourgon ; LTM-CNRS, GRENOBLE, France.
Le type de résine ainsi que les températuresde recuit ont une influence sur l’épaisseurdu film. Cette dernière diminue lorsque latempérature augmente, des domaines detempérature stables peuvent égalementexister comme le montre la figure 2 dansle cas de la résine NEB22. Une températurede recuit adéquate permet de mieux com-pacter les couches de résine.Etant donné que les différentes opérationsde recuit pendant le procédé lithographi-que sont effectuées à partir de la tempéra-ture de 100 °C environ, il est intéressantd’étudier les évolutions dans cette plage detempérature. Une diminution de l’épais-seur du film de la résine NEB22 (voir fig.2) indique que le film n’est pas totalementstable. Cette diminution résulte d’une
IntroductionLe LETI (Laboratoire d’Electronique deTechnologie et d’Instrumentation) a ac-quis un savoir-faire dans l’application desrésines photosensibles dans les procédéslithographiques de l’industrie des semi-conducteurs (transistors pour circuits in-tégrés). La miniaturisation croissante destransistors a été la force motrice d’ungrand nombre de progrès technologiques.La plus petite résolution actuellement at-teinte est de 40 nm pour les réseaux de li-gnes et de 60 nm pour les motifs denses.Ces limites sont très probablement liéesaux différents composants de la résine.L’objectif actuel est donc le développementd’une nouvelle résine CAR (ChemicallyAmplified Resist : résine améliorée chimi-quement) d’une résolution de 20 nano-mètres.Les résines CAR sont constituées d’une ma-trice polymère, c’est-à-dire d’une combi-naison de deux polymères, d’un photogé-nérateur d’acide (Photo Acid Generator,PAG) et d’additifs intégrés en fonction despropriétés souhaitées.L’optimisation du procédé, par suite l’amé-lioration des propriétés de la CAR, nécessiteune bonne compréhension des comporte-ments physico-chimiques des différentscomposants et de l’influence de ces der-niers tout au long du procédé. Le LETI-LTM(Laboratoire de Technologie Microélectro-nique) et le «Laboratoire de Thermodyna-mique des Solutions et des Polymères» sesont associés dans cet objectif. La combi-naison de la DSC à modulation de tempé-rature (ADSC), de la IRTF et de la mesurede l’épaisseur a été utilisée dans ce projetafin de déterminer l’influence des diffé-rents composants de la résine sur la réso-lution du procédé lithographique. Le prin-cipal avantage de l’ADSC consiste en cequ’une seule mesure permet de déterminerla température de transition vitreuse demême que les effets thermiques associés auprocédé lithographique (évaporation etréticulation) [1].
Conditions expérimentalesPlusieurs résines, produites par SumitomoIncorporated (Japon), différentes l’une del’autre par un composant au moins, ontété étudiées afin de comprendre le compor-tement physico-chimique des combinai-sons de composants. L’influence de plu-sieurs paramètres tels que le mélange de lamatrice polymère, le poids moléculaire dechaque polymère, le PAG et le solvant, aété également étudiée.Les caractéristiques physico-thermiques detoutes les résines ont tout d’abord été me-surées afin de déterminer la zone de tran-sition vitreuse et la température de réticu-lation ; les courbes obtenues à partir de larésine NEB22 sont représentées à titred’exemple sur la figure 1.
Cette étape de l’optimisation des conditionsde recuit est nécessaire afin de contrôlerles phénomènes de diffusion et de réduirela sensibilité à la contamination.
réaction interne telle que l’évaporation decomposants. Le volume libre ainsi généréest ensuite éliminé pendant le traitementthermique. Afin de déterminer quel com-
Figure 1: Courbes ADSC de la résine NEB22, mesurées après un conditionnement thermique de 10 minutesà 80 °C suivi d’un refroidissement à 5 K/min. Φtot: flux de chaleur total, Φrev.: composante réversible du fluxde chaleur, Φnon-rev.: composante non réversible du flux de chaleur. Le palier de la courbe Φrev. révèle laplage de la transition vitreuse (60 à 100 °C), l’augmentation de Φnon-rev. indique la réaction de durcissement(à partir de 130 °C).

13UserCom 1/2002
posant est responsable de cette instabilité,des résines de différentes formulations,dans lesquelles un des composants a étéretiré, ont été préparées et leurs caractéris-tiques thermiques déterminées. Les résul-tats suivants ont été obtenus [2]:• La matrice polymère seule présente une
limite thermique à 115 °C environ.
• L’addition d’agents réticulants ne changepas de manière significative les courbesADSC. Nous avons en outre étudié uneautre résine, la NEB33, qui diffère dela NEB22 par le type d’agent réticulant.Les deux résines présentent le mêmecomportement thermique.
• L’introduction d’un photogénérateur
(PAG) modifie les signaux thermiques.La figure 3 montre que l’instabilité ob-servée aux environs de 100 °C est due àl’évaporation du PAG. Cette tendance àl’évaporation est confirmée par les me-sures IRTF.
• La part de solvant dans la matrice poly-mère, non négligeable, a une influencesur l’épaisseur pendant le recuit. Unegrande partie du solvant devrait êtreéliminée au début du traitement, vers80°C . La mesure DSC montre néan-moins que du solvant résiduel, proba-blement bloqué par la formation d’unepellicule, s’évapore à des températuresplus élevées, (figure 4). Une tempéra-ture d’évaporation du solvant de 140 °Ca été déterminée avec la DSC. Le pic en-dothermique apparent de la courbe dela figure 4, vers 130 °C, montre claire-ment qu’une vitesse d’application troplente retient très probablement plus desolvant, conduisant ainsi à une résinemoins «stable». La présence de solvanta été confirmée par les mesures IRTF,montrant un pic vers 1740 cm-1, attri-bué à la liaison C=O du solvant.L’intensité de ce pic décroît rapidementlorsque la température de mesure at-teint 150 °C [3].
L’influence d’autres paramètres de la ma-trice polymère tels que la polydispersité dumélange ou et la masse molaire, a été étu-diée à l’aide des mêmes techniques. Il aété montré qu’une diminution de la massemolaire permet d’obtenir une meilleure ré-solution optique. Une résolution de 30 nma été finalement obtenue pour les réseauxde lignes.
L’optimisation du procédé à l’aide del’analyse thermique de différents maté-riaux et d’autres techniques est encore encours. Différentes résines sont étudiées enfonction des paramètres du procédé.
ConclusionLa combinaison de méthodes d’analyse(DSC, ADSC, IRTF) présentée dans cet arti-cle permet d’obtenir des informations inté-ressantes pour l’amélioration de la formu-lation de la CAR. L’étude du comportementthermique permet d’optimiser le procédé et
Figure 2: Evolution de l’épaisseur du film de la résine NEB22 après application en fonction de la températurede traitement.
Thickness / nm
Bake Temperature / °C
Stable Temperature Domain 1
160
165
170
175
180
185
190
50 100 150
Stable Temperature Domain 2
Figure 3: Courbes ADSC de la matrice polymère avec PAG (légende des courbes : voir fig. 1). L’influence duPAG et l’évaporation du solvant résiduel (pic à 35°C) fournit une toute autre évolution de Φnon-rev. que cellemontrée sur la figure 1.

UserCom 1/200214
de comprendre le comportement des diffé-rents composants de la résine. La techniqueADSC est donc une technique intéressante
Figure 4: Composante non-réversible du flux de chaleur (Φnon-rev.) de la NEB22 non conditionnée, avec re-vêtement appliqué à différentes vitesses de rotation (1000, 2000, 4000 et 6000 t min-1). L’évaporation dusolvant est plus importante lorsque le revêtement est effectué à une vitesse plus élevée. L’intensité du picendothermique à 130 °C diminue donc, ce qui indique une diminution de l’évaporation.
pour mesurer des effets thermiques prati-quement simultanés : transition vitreuse etévaporation/durcissement.
Bibliographie[1] Resolution limit of negative tone chem-
ically amplified resist used for hybridlithography. Influence of the molecularweight. L. Pain, C. Higgins, B. Scarfoglière,S. Tedesco, B. Dal’Zotto, C. Gourgon, M.Ribeiro, T. Kusumoto, M. Suetsugu andR. Hanawa. Journal of Vacuum Sciencesand Technology. B., 2000, 18(6), 3388.
[2] High Resolution Negative ChemicallyAmplified Resists (CARs) for E-Beamlithography: the impact of CAR com-pounds on lithographic properties.C. Gourgon, L. Pain, C. Higgins,B. Scarfoglière, L. Mollard, B. Dal’zotto,S. Tedesco, M. Ribeiro, J-P. E. Grolier andY. Quere. Proc. 12th International Confer-ence on Photopolymers, 16.-18. Oktober2000, McAfee, New Jersey, USA.
[3] Properties of High Resolution NegativeChemically Amplified Resists (CARs) forE-Beam lithography. (Affiche) M. Ribeiro,L. Pison, C. Gourgon, L. Pain, Y. Quereand J.-P. E. Grolier. 16th IUPAC Confer-ence on Chemical Thermodynamics,(ICCT 2000) , 6.-11. August 2000, Hali-fax, Canada.

15UserCom 1/2002
Analyse quantitative de mélanges de polyoléfinesD. Seifert, Institut für Nichtmetallische Werkstoffe, Fachgebiet Polymertechnik, Technische Universität Berlin, Fasanenstr. 90, 10623 Berlin
IntroductionLes matériaux synthétiques recyclés à par-tir des rebuts d’emballage sont en généraldes mélanges de polymère. Ces mélangessont majeure partie constitués de polyo-léfines, c’est-à-dire de polyéthylène et depolypropylène. Ces derniers peuvent êtrerecouvrés par dissolution pour une réutili-sation des matériaux. Le recyclage par dis-solution est un procédé qui permet d’obte-nir de meilleures propriétés du matériauainsi obtenu que par les procédés de fusionusuels [1].Afin de fabriquer des granulats avec despropriétés élevées et constantes, d’autresétapes de procédé ont été développées pourséparer le recyclé polyoléfine en fonctionde ses composants, c’est-à-dire en fonctiondu polyéthylène basse densité (LDPE), dupolyéthylène haute densité (HDPE) et dupolypropylène (PP) [2].La qualité de la séparation peut être éva-luée par la composition du recyclé. Cet ar-ticle décrit une nouvelle méthode qui per-met de déterminer la composition d’unéchantillon polyoléfine à l’aide de mesuresDSC. Cette méthode repose sur des considé-rations physiques et se distingue par le faitqu’elle différencie non seulement les poly-éthylènes du polypropylène mais aussi lespolyéthylènes basse et haute densité.Cette méthode consiste à décrire les cha-leurs de fusion observées sur la courbe defusion d’un échantillon polyoléfine par lesfractions de masse (celles-ci dépendent dela composition de l’échantillon) et les en-thalpies normales de fusion des différentscomposants.La courbe de fusion d’un mélange ne peutpas être immédiatement décrite commesomme des courbes de fusion des compo-sants purs, il faut donc introduire des fac-teurs correctifs, déterminés par itération àpartir des résultats de mesure d’une séried’échantillons polyoléfines ayant une com-position connue.
Conditions expérimentalesUne série d’échantillons de compositionconnue, c’est-à-dire d’échantillons d’éta-lonnage, a tout d’abord été réalisée. Lescompositions de ces échantillons ont étéchoisies de sorte que les trois répartitions(représentées en pourcentages de poids)5:5:90, 10:10:80 et 20:20:60 soient réaliséespour chacun des trois polymères commecomposant principal. Ces neuf mélangessont représentés graphiquement sur la fi-gure 3. Les échantillons ont été mélangésdans la solution, séparés et séchés. Afind’obtenir des résultats représentatifs statis-tiquement, trois creusets de mesure ont étéréalisés par mélange.Les échantillons ont été mesurés avec unDSC822e de METTLER TOLEDO, selon lesprogrammes de température suivants :1. Mise en température de l’échantillon,
de 20 °C à 200 °C à 10 K/min (sup-pression/équlibrage de l’histoire ther-mique de tous les échantillons).
2. Refroidissement de 200 °C à 20 °C à10 K/min.
3. Température constante pendant 10 mi-nutes.
4. Mise en température de 20 °C à 200 °Cà 10 K/min. (mesure proprement dite).
5. Refroidissement de 200 °C à 20 °C à10 K/min.
différentes histoires thermiques de l’échan-tillon, seule la deuxième courbe a été étu-diée.L’exploitation doit être reproductible. Laligne de base est extrapolée en ligne droiteà partir du domaine liquide. L’ensemble dela courbe est tout d’abord intégrée pour ladétermination des enthalpies de fusion.Dans une prochaine étape, les chaleurs defusion des polyéthylènes et du polypropy-lène sont séparées, ce qui est relativementsimple car le PE et le PP ne sont pratique-ment pas miscible au sens thermodynami-que d’une part et les deux signaux ne sesuperposent que très peu dans la courbe defusion d’autre part. Il est ainsi possible defaire une séparation verticale au niveau duminimum entre les pics de fusion du PE etdu PP.Il faut de plus séparer les chaleurs de fu-sion du LDPE et du HDPE. Cette séparationest plus difficile car ces deux polyoléfinessont en partie miscibles d’une part et leurstempératures de fusion sont proches l’unede l’autre d’autre part. Les signaux se su-perposent en conséquence dans la courbede fusion. Il faut donc séparer trois cas(voir figure 2):1. Le LDPE et le HDPE présentent chacun
un maximum distinct, nettement dé-fini : il existe un minimum clairementdéfini entre les deux maxima. Ce cas seprésente lors que le rapport des massesdu LDPE / HDPE est suffisammentgrand, c’est-à-dire lorsqu’il y a plus deLDPE que de HDPE dans l’échantillon.
2. Le LDPE ne présente qu’un « épaule-ment » dans le pic du HDPE. Ce cas seprésente lorsque le LDPE et le HDPE setrouvent approximativement dans lesmêmes quantités dans le mélange.
3. Le LDPE n’est plus détecté dans le picdu HDPE. Ce cas se présente lorsque lerapport de masse LDPE / HDPE est troppetit, c’est-à-dire lorsqu’il y a moins deLDPE que de HDPE dans l’échantillon.
Exploitation des courbes de fusionEtant donné que les courbes de fusion desthermoplastiques diffèrent en fonction des
Figure 1: Profil de température du programme de mesure
0
50
100
150
200
0 0.5 1 1.5
Time [h]
Tem
pera
ture
[°C
]

UserCom 1/200216
Dans le premier cas, la séparation est rela-tivement simple. La limite est tracée au ni-veau du minimum entre les domaines defusion du LDPE et du HDPE .Dans le deuxième cas, la première dérivéede la courbe de fusion doit être tracée. Laséparation verticale entre le LDPE et leHDPE est alors tracée au niveau de l’épau-lement du pic PE, c’est-à-dire au niveau dela plus faible pente.Dans le troisième cas, les surfaces partiel-les du LDPE et du HDPE ne peuvent pasêtre séparées. La chaleur de fusion duLDPE est nulle dans cette exploitation decourbe.
Etalonnage du système à trois com-posantsLes enthalpies de fusion standards (∆H0)des trois polyoléfines à l’état pur, c’est-à-dire non mélangé, sont tout d’abord déter-minées. Les trois polyoléfines sont désignéspar un indice correspondant dans toutesles formules ci-dessous, soit : L pour LDPE,H pour HDPE et P pour PP.∆H0
L = 91 J/g∆H0
H = 129 J/g∆H0
P = 84 J/gSi les trois polymères n’avaient pas d’in-
fluence mutuelle lors de la cristallisationet de la fusion, les chaleurs de fusion, ∆H,déterminées lors de l’exploitation des cour-bes de fusion, devraient être égales au pro-duit de la fraction de masse x et de l’en-thalpie standard de fusion ∆H0 du compo-sant. La chaleur de fusion du PP seraitdonc par exemple :
Cette formule simple ne peut pas être em-ployée pour les mélanges de polyoléfinespour les raisons suivantes :1. La ligne de base comme extrapolation
linéaire à partir du fondu ne corres-pond pas à la réalité thermique dans cedomaine de température. Elle devraitprésenter une courbure convexe au ni-veau des températures plus basses [3].Cette construction de la ligne de baseest néanmoins conservée pour la repro-ductibilité.
2. Le polyéthylène et le polypropylène nesont pratiquement pas miscibles ther-modynamiquement mais les deuxpolyéthylènes LDPE et HDPE le sontpartiellement. Ceci signifie que les sur-faces des pics des différents composantssont influencées par l’enthalpie de mé-lange.
3. Les verticales coupent les intégrales defaçon reproductible, mais coupent –arbitrairement au sens physique – ledébut et la fin des courbes de fusion.
Il faut donc introduire pour le calcul del’enthalpie de fusion prévue un facteurcorrectif. Ce facteur doit être déterminé sé-parément pour chaque composant. Ce fac-teur (appelé cP pour le PP) doit de mêmeêtre déterminé pour chaque système DSC etaprès chaque ajustage de l’appareil. La for-mule (1) doit donc être étendue à :
Etant donné qu’il n’est pas possible de me-surer avec une fiabilité suffisante la cha-leur de fusion absolue des recyclés, lescalculs seront faits par la suite avec lesfractions de surface. La fraction de surfaced’un composant est la chaleur de fusiondu composant, déterminée sur la courbe defusion, divisée par la somme des chaleursde fusion ; on aura donc pour le PP parexemple :
En remplaçant par les enthalpies dans l’é-quation (3) par celles de l’équation (2) onobtient :
Pour les deux polyéthylènes on aura :
En calculant les fractions de masse xL, xHet xP à partir de ces trois formules, on ob-tient :
Figure 2: Courbes de fusion d’un mélange à trois composants avec différentes caractéristiques du pic LDPE-HDPE. De haut en bas : cas 1, cas 2 et cas 3.

17UserCom 1/2002
Afin de déterminer les facteurs correctifscL, cH et cP, les mélanges de compositionconnue ont tout d’abord été mesurés et lescourbes exploitées. Les fractions de massexL, xH et xP ont été calculées avec les va-leurs initiales cL=cH=cP= 1,0. Ces frac-tions de masse déterminées expérimentale-ment ont été comparées avec les fractionsde masse de l’échantillon initial, la diffé-rence ∆xP a alors été déterminée pourchaque mélange d’étalonnage. Les troisfacteurs correctifs ont été modifiés (par ité-ration) jusqu’à ce que la somme des diffé-rences ∆xPP soit minimale. Les résultats demesure du composant PP sont alors pris encompte car ce composant est le seul à pou-voir être exploité sur toute la plage descompositions.Un exemple de résultat d’une telle itérationest montré sur la figure 3. Cette itérationfournit les facteurs correctifs cL = 1,76,cH = 1,46 et cP = 1,31. Les trois facteurscorrectifs sont supérieurs à 1 car les surfa-ces déterminées pour le mélange sont plusgrandes que celles des différents compo-sants en raison du choix de la ligne debase.La figure 3 montre que les données expéri-mentales des échantillons riches en LDPEcorrespondent bien aux fractions calculéesà partir des pesées initiales alors que la
méthode n’est plus applicable lorsque lerapport de masse HDPE / LDPE est trop im-portant. Pour les autres compositions, l’er-reur maximale est de 3 % en poids (pour lePP) et de 6 % en poids (pour les LDPE etHDPE).
Détermination de la compositiondes trois composantsTrois creusets ont été préparés pour chaqueéchantillon de composition inconnue,d’une part pour obtenir une certaine sécu-rité statistique et d’autre part pour vérifierl’homogénéité des échantillons. Ceux-ciont été mesurés avec le programme de tem-pérature décrit ci-dessus. L’exploitation descourbes doit être effectuée selon les mêmescritères de reproductibilité que ceux em-ployés lors de l’exploitation des échantil-lons d’étalonnage. Les formules (7), (8) et(9), les valeurs connues des enthalpiesstandards de fusion et les facteurs correc-tifs permettent de déterminer la composi-tion de l’échantillon à partir des fractionsde surface des courbes exploitées.Lorsque aucun LDPE ne peut être détectédans la courbe de fusion, on ne peut dé-duire aucune donnée quantitative de cecomposant mais seulement une informa-tion du type « xL est inférieure à 20 % ».La position exacte de la limite dépend du
système de mesure employé et des condi-tions générales de la mesure.
Si des mélanges d’autres polyoléfines doi-vent être étudiés, un nouvel étalonnagedoit être effectué pour ces types de maté-riaux, la détermination des enthalpiesstandards de fusion et des facteurs correc-tifs doit donc être effectuée. Etant donnéque les recyclés représentent un mélangede nombreux différents types de matériauxsynthétiques, aucun étalonnage adapté nepeut être effectué. Il faut plutôt employerdes types les plus proches du recyclé.Cette méthode permet de fournir des résul-tats comparables, même si l’étalonnagen’a pas été effectué avec les types de polyo-léfines adaptés.
ConclusionCet article décrit une méthode qui permetde déterminer la composition d’un échan-tillon de polyoléfine à l’aide de la DSC.Cette méthode est fondée sur le fait que lesenthalpies des trois composants LDPE,HDPE et PP sont obtenues à partir des en-thalpies standards de fusion et de la con-centration de ces trois composants dans lemélange. Etant donné que le comporte-ment des composants n’est pas idéal dansun mélange, la relation décrite ci-dessusdoit être corrigée par un facteur. Ces fac-teurs sont déterminés à l’aide de mélangesdont les compositions sont connues. L’ex-ploitation des courbes des mélanges d’éta-lonnage ainsi que celle des échantillons àétudier doivent être effectuées de façon re-productible.
Bibliographie:[1] Klein, F., „Verfahrensentwicklung,
Werkstoffeigenschaften und Wirt-schaftlichkeitsbetrachtung für dasKunststoffrecycling über Lösen vonMischthermoplasten.“, SchriftenreiheKunststoff-Forschung Band 48,Technische Universität Berlin.
[2] N.N., DKR im Blick 03/2000, DeutscheGesellschaft für Kunststoffrecycling mbH
[3] Alsleben, M., Schick, C., Mischok, W.,Thermochimica Acta, 187 (1991),261-268Figure 3: Diagramme ternaire pour les trois composants LDPE, HDPE et PP.
0.00 0.25 0.50 0.75 1.00
0.00
0.25
0.50
0.75
1.00 0.0
0.2
0.4
0.6
0.8
1.0
Mass f
racti
on P
P
Sample weight Experimental
Mass fraction HDPE
Mass fraction LDPE

UserCom 1/200218
Etude de réactions de durcissement à l’aide de la méthodeIsoStepDr. Uwe Hess
IntroductionLes réactions de durcissement jouent unrôle important dans la fabrication des ma-tériaux synthétiques. On peut citer outreles préimprégnés (prepregs) et les vernis,les matériaux composites. Ceux-ci sont, depar leurs propriétés mécaniques et leur baspoids moléculaire, des matériaux impor-tants, ils sont souvent constitués de fibresde verre ou de carbone associées par unerésine durcie. La résine et le matériau fi-breux déterminent les propriétés du maté-riau composite. Etant donné que ces ma-tériaux sont employés dans des domainesdans lesquels la sécurité a une grande im-portance, par exemple dans la constructionaéronautique et la construction automo-bile, le comportement au durcissement desrésines doit être caractérisé avec précision.
Un matériau totalement durci est relative-ment rigide et peut être inadapté dans cer-taines applications en raison de sa fragi-lité. On préfère dans ce cas un matériaunon totalement durci. Un durcissement in-complet peut par contre avoir pour effetune modification à long terme des proprié-tés. Un critère important pour la détermi-nation du degré de durcissement d’unmatériau est la température de transitionvitreuse. La température de transition vi-treuse d’un matériau augmente avec sondegré de durcissement. Une étude DSCexacte est alors toujours rendue difficile,voire impossible, en présence d’une réac-tion de durcissement simultanée, dont lepic exothermique est superposé à la transi-tion vitreuse. Dans le cas des résines dur-cissant à chaud en particulier, il peut seproduire une vitrification pendant la réac-tion. La résine passe alors d’un état liquideà un état vitreux qui ralentit considérable-ment la réaction.
En règle générale, on souhaite caractériserle matériau et connaître les conditions res-trictives du processus de durcissement etdu stockage pour l’utilisation finale. Lesquestions classiques sont donc :
• Quelle relation existe-t-il entre la tem-pérature de transition vitreuse et lespropriétés du matériau ?
• Dans quelles conditions une vitrifica-tion du matériau se produit-elle ?
• Dans quelles conditions un durcissementincomplet d’un matériau se poursuit-ilpendant le stockage ou l’utilisation ?
Justement lors d’applications dont la sécu-rité est importante, le procédé de fabrica-tion doit garantir un matériau uniformé-ment durci et assurer une stabilité à longterme du matériau dans des conditionsdéfinies.Dans l’exemple suivant, le comportementau durcissement d’une résine constituéede deux composants, DGEBA (Diglycidyl-ether de bisphénol A) et DDM (Diamino-Diphenylméthane) a été étudié.Une réaction très lente, donc une duréeplus longue, induit une meilleure réticula-tion du matériau, les températures de tran-sition vitreuse sont toujours décalées versles valeurs plus élevées. Si la températurede transition vitreuse est plus élevée que latempérature de réaction actuelle, il se pro-duit soudain une vitrification. Dans cetétat à présent solide, la réaction de durcis-sement est nettement ralentie [1]. Cecipeut par exemple conduire à des problèmespour des formes d’outil présentant des dif-férences de température marquées lorsquela montée en température de certaines zo-nes est trop lente et par suite lorsque leprocessus de durcissement n’est pas com-plet.
Afin de déterminer en DSC si une vitrifica-tion du matériau a eu lieu, la courbe de lacapacité calorifique doit être séparée dupic de réaction. Les mesures présentéesdans cet article ont été effectuées à l’aidede la nouvelle méthode IsoStep et lesrésultats comparés avec ceux obtenus avecla DSC conventionnelle.
Mesure et résultatsLes échantillons ont été chauffés alternati-vement lentement et légèrement plus rapi-dement : les longueurs des segments desphases isothermes et de montée en tempé-rature sont respectivement de 60 et 30 s, lesvariations incrémentales de la températuresont respectivement de 0,5 et 1 K. La courbede DSC conventionnelle a été mesurée avecla même vitesse moyenne de chauffe.L’échantillon chauffé relativement rapide-ment présente le comportement montré surla figure 1 : la courbe DSC révèle une tran-sition vitreuse à -20 °C et un pic de durcis-sement avec un maximum à 115 °C envi-ron. La capacité calorifique augmente avecune vitesse croissante pendant la réaction,la plus grande augmentation étant à laplus grande vitesse (pic DSC). Ceci résultedes mouvements moléculaires supplémen-taires aux nœuds de réticulation [2].Unevitrification est révélée par une baissebrusque de la capacité calorifique. Mais ilne se produit apparemment pas de vitrifi-cation dans ces conditions expérimentaleset la réaction reste contrôlée chimiquementpendant tout le processus de durcissement.La courbe cinétique montre la vitesse deréaction.
Une courbe DSC ainsi que les courbes de lacapacité calorifique et cinétique d’unéchantillon chauffé lentement sont repré-sentées sur la figure 2. Dans ces condi-

19UserCom 1/2002
tions, une diminution brusque de la capa-cité calorifique à 100 °C, correspondant àune vitrification, est mise en évidence pen-dant le processus de durcissement sur lacourbe de la capacité calorifique. Le trèslent processus laisse à l’échantillon letemps de former un réseau polymère rela-tivement dense, dont la température detransition vitreuse se trouve au-dessus dela température de réaction. La réaction estcontrôlée en raison de la vitrification pardiffusion et ralentit. Lors d’une nouvellemontée en température, la transition vi-treuse du matériau partiellement durci estatteinte à 150 °C. A partir de cette tempé-rature, le matériau est de nouveau liquideet la vitesse de la réaction, de nouveaucontrôlée chimiquement, augmente. Uneexploitation cinétique du pic de durcisse-ment à l’aide de la courbe cinétique seraitjudicieuse car la capacité calorifique n’in-tervient pas.
ConclusionIsoStep est une méthode très bien adap-tée pour séparer les transitions vitreusesdes processus cinétiques superposés telsque les réactions de durcissement, la cris-tallisation et l’évaporation. L’étude desréactions de durcissement de systèmes derésine en particulier est très importante caril est possible d’obtenir des informationspermettant la caractérisation des maté-riaux synthétiques, sur les conditions defabrication et sur le comportement à longterme, c.-à-d. le vieillissement. Les résinesde polymères sont employées dans des ma-tériaux aussi importants que les matériauxcomposites, les vernis et les préimprégnés(Prepregs).
Bibliographie[1] S. Montserrat, Y. Calventus and
P. Colomer, UserCom 11, 2000[2] J. E. K. Schawe, Therochim. Acta, 2002,
submitted
Figure 1: La courbe DSC conventionnelle a été enregistrée pour une vitesse de chauffe constante de 1 K/min.La courbe de la cp et la courbe cinétique ont été enregistrées pour des segments isothermes et de montée entempérature de 30 s avec des variations de température de 1 K.
Figure 2: La courbe DSC a été enregistrée pour une vitesse de chauffe de 0,5 K/min. La courbe de la cp et lacourbe cinétique de la méthode IsoStep ont été obtenues à partir de mesures effectuées avec des segmentsisothermes et de montée en température de 60 s, l’augmentation de température étant de 0,5 K. La réactionde durcissement relativement lente entraîne une vitrification de l’échantillon suivie d’une transition vitreuse.

UserCom 1/200220
Décomposition thermique du sulfate de cuivre pentahydratéDr. Jean-Nicolas Aebischer, Dozent für Physikalische Chemie, Hochschule für Technik und Architektur Freiburg,Bd. Pérolles 80, CH-1705 Fribourg, e-mail: [email protected]
IntroductionLors de l’apprentissage d’une nouvelle mé-thode il est toujours conseillé d’étudierd’abord un système dont le comportementà des conditions données est connu. La dé-composition thermique du CuSO4 ⋅ 5 H2Oest très bien connue et l’interprétation dela courbe TGA ne pose pas de problème,même à un débutant.
Conditions expérimentalesLes mesures ont été effectuées avec un TGA/SDTA851e (Plage : 5 g; Résolution 1 µg),couplé à un spectromètre de masse InficonThermostar QMS (Masse : 1-300).
Exploitation des résultatsLes trois premières étapes révèlent une di-minution totale relative du poids de35,58 % (Figure 1). Ceci correspond à laperte des 5 molécules d’eau. Une nouvelleperte de 32,87 % se produit lors de la der-nière étape de sorte qu’il ne reste que duCuO.Le tableau 1 récapitule les pertes de poidsrelatives et les réactions nettes à chaqueétape.
Confirmation des étapes de réac-tion proposées par la mesure deflux importants d’ionsLa justesse des réactions proposées peutêtre vérifiée par la mesure en continu desflux d’ions pour les rapports m/z de l’eauet du SO3.Comme le montre la figure 2, l’eau est ef-fectivement séparée dans les 3 premièresétapes. Par contre, aucune formation deSO3 n’est présente dans la dernière étape,qui correspond après le calcul de la dérivéeà une double perte de masse.
Interprétation des observations ex-périmentalesLe fait que le SO3 ne soit pas observé peutavoir deux origines. Premièrement, il sepeut que ce ne soit pas du SO3 mais de l’O2et du SO2 qui se séparent du réseau cristal-
Tableau 1: Réactions nettes et pertes de masse relatives correspondantes lors de la décomposition thermiquedu CuSO4 ⋅ 5 H2O dans le domaine de température de 40 °C à 840 °C; (calc) se réfère à la perte de massecalculée, (exp) à la perte de masse déterminée expérimentalement, toutes les valeurs sont rapportées à lamasse initiale de l’échantillon.
Etape Réaction nette Perte de masse relativeen %
1-3 CuSO4 ⋅ 5 H2O(s) → CuSO4(s) + 5 H2O(g) 35.89 (calc)35.58 (exp)
4 CuSO4(s) → CuO(s) + SO3(g) 32.07 (calc)32.87 (exp)
Tableau 2: Caractéristiques thermochimiques de l’oxygène, du dioxyde de soufre et du trioxyde de soufre.
SO3 (g) SO2 (g) O2 (g)
∆fH° / kJmol-1 -395.77 -296.81 0
S° / JK-1mol-1 256.77 248.223 205.07
cp / JK-1mol-1 50.644 39.874 28.915
lin CuSO4. Deuxièmement, du SO3 peut ef-fectivement s’être formé mais il n’est passtable aux conditions présentes de tempé-rature et de pression.La réponse à la question de la stabilité duSO3 peut être fournie à l’aide de calculsthermochimiques.Pour la réaction de décomposition, on
trouve pour 25 °C les caractéristiques ther-mochimiques suivantes :
SO3(g) → SO2(g) + 0.5 O2(g)
A la température ambiante, la plus grandestabilité se trouve totalement du côté duSO3.
Figure 1: Courbe TGA de la décomposition thermique du CuSO4 ⋅ 5 H2O dans le domaine de température de40 °C à 840 °C.

21UserCom 1/2002
Les 4 équations ci-dessous permettent dedéterminer la température à partir de la-quelle la plus grande stabilité est plutôt ducôté du dioxyde de soufre ou de l’oxygène.
Aux températures inférieures à 780 °C,l’enthalpie libre de réaction de Gibbs, de ladécomposition du SO3 est positive puischange de signe, la plus grande stabilité setrouve alors du côté du SO2 et du O2.Ce fait permet d’expliquer le fait observéque le flux d’ions du SO3 reste constant enfonction du temps. On ne peut pas répon-dre ainsi à la question des produits primai-res de décomposition. Le fait que du O2 etdu SO2 soient observés dans les deux der-nières étapes contredit toutefois une sépa-ration par étape de l’oxygène et du dioxydede soufre.
Qu’apporte cet exemple pour uneformation pratique ?La réaction de décomposition du CuSO4 ⋅5 H2O est un exemple idéal à partir duquelle principe du couplage TGA-MS peut êtreexpliqué aux étudiants. L’observation toutd’abord décevante que du SO3 ne se formepas dans les présentes conditions, permetde mettre en évidence l’intérêt de l’effetsynergique du couplage.Cette expérience est intéressant pour lesétudiants dans la mesure où les bases dethermodynamique chimique acquises dansles cours peuvent être employées pour ex-pliquer ces observations.
Etude du délaminage et de la formation de mousse à l’aide ducouplage TMA-MSCyril Darribère
IntroductionL’analyse thermomécanique (TMA) permetde mesurer la variation de la dimensiond’un échantillon lors d’une variation detempérature. Le couplage de la TMA avecla spectroscopie de masse (MS) permet lamesure simultanée des gaz combustibles etdes gaz de décomposition en présence et deleurs influences sur la dimension del’échantillon. On peut ainsi étudier parexemple à quelles températures des pro-duits de décomposition sont générés etquels sont-ils lors du délaminage de pla-ques à circuit imprimés ou comment laformation de mousse d’un matériau syn-thétique a lieu ? Le couplage de la TMAavec l’analyse des gaz (MS ou IRTF) per-met une rapide explication des variations
des dimensions générées par les processusde décomposition ou une optimisation duprocédé de transformation en mousse.Le TMA/SDTA840 du système STARe deMETTLER TOLEDO a été relié à un spec-tromètre de masse Inficon ThermostarQMS (masse 1-300) par l’intermédiaired’un capillaire en quartz chauffé, de ma-nière analogue à celle pratiquée habituel-lement pour le couplage TGA-MS [1].
Délaminage et décomposition d’uneplaque à circuits imprimésLes plaques à circuits imprimés (PCB,Printed Circuit Board), constituées d’unematrice en résine époxy renforcée d’untissu de fibres de verre, sont souvent em-ployées comme supports de composants
électroniques.L’évolution de la dilation d’échantillonsdécoupés à l’emporte-pièce (diamètre :4 mm, épaisseur : 1.683 mm) a été mesu-rée à l’aide d’une sonde de mesure sphéri-que, la force appliquée étant de 0,05 N.Après un prétraitement jusqu’à 100 °C, leséchantillons sont chauffés de 30 °C à650 °C, à une vitesse de 20 K/min sous unbalayage d’azote de 10 ml par minute.Comme le révèle la courbure de la courbeTMA de la figure 1, la plaque à circuits im-primés présente une transition vitreuse à92 °C. Le soudain changement dans la di-mension de l’échantillon selon l’axe z àdes températures au-dessus de 320 °C mon-tre le délaminage de l’échantillon.Le processus est simultanément suivi avec
Figure 2: Courbes TGA-MS de la décomposition thermique du CuSO4 ⋅ 5 H2O dans la plage de température de40 °C à 840 °C. Les flux d’ions ont été parfois multipliés et décalés pour une meilleure représentation.

UserCom 1/200222
la spectroscopie de masse. La plaque à cir-cuits imprimés émet des ions de fragmen-tation à m/z 79 et 94. Ces valeurs peuventêtre facilement attribuées au brome et aubromure de méthyle, c’est-à-dire aux pro-duits de décomposition du tétrabromobis-phénol A (TBBA, agent ignifuge). Les pro-duits de réaction contenant du brome sontdéjà détectés après la transition vitreuse.La forte augmentation des gaz de décom-position contenant du brome à partir de330 °C montre également l’origine du dé-but du délaminage.
Transformation en mousse d’unepoudre de polymèreLes granulés synthétiques contenant dugaz dissout ou un combustible chimiqueprésentent des propriétés particulières lors-qu’ils sont soumis à la chaleur. L’augmen-tation de la pression de vaporisation ducombustible et le ramollissement du poly-mère engendrent une importante augmen-tation du volume dans la plage de tempé-rature entre 80 et 190 °C. L’augmentationdu volume peut être facilement mesuréeavec la TMA.Un creuset en oxyde d’aluminium de 150 µlest rempli de 3 à 4 mg de poudre couverted’un disque en verre quartzeux de 6 mmde diamètre. Une sonde de mesure sphéri-que, de 3 mm de diamètre, applique sur ledisque une charge de 0,01 N. Les échan-tillons sont chauffés de 50 à 400 °C à unevitesse de 15 K/min.La mesure TMA est représentée sur la figure2. La transformation en mousse est mesu-rée par la soudaine augmentation du si-gnal qui atteint sont maximum à 142 °C.L’augmentation maximale de volume estde 6000 % environ (avec le dispositif demesure choisi et la force appliquée par lasonde de mesure, de 0,01 N). La mousse« s’écroule » aux températures supérieuresà 150 °C, en raison de la poursuite du ra-mollissement. Comme le montre les cour-bes MS, la décomposition du polymère seproduit à une température supérieure à240 °C.Le type de combustible a été identifié par lesignal MS m/z 43, qui est simultané au dé-but de la dilatation mesurée en TMA. Cetion de fragmentation indique que le com-bustible en présence est le isopentane.Des informations sur le polymère ont étéobtenues par l’exploitation des modèles defragmentation à températures élevées. Onpeut déduire de l’évolution de la décompo-
sition qu’il s’agit d’un copolymère thermo-plastique, d’un mélange de chlorure devinyle (HCl m/z 36) et de styrène (benzènem/z 78) par exemple.
ConclusionLes possibilités d’application du couplagede l’analyse thermomécanique et de laspectroscopie de masse ont été démontréesà l’aide de l’étude d’une plaque à circuitsimprimés et d’un copolymère transforma-ble en mousse. L’évolution des dimensionsdes échantillons a été corrélée avec le dé-
gagement des substances gazeuses. Des in-formations sur la composition des échan-tillons ont été obtenues par la spectroscopiede masse. La combinaison de la TMA et dela MS est particulièrement bien adaptée àl’étude de l’influence de la température surles variations soudaines de volume et àl’étude de leur origine.
Bibliographie[1] Evolved Gas Analysis, Collected Applica-
tions for Thermal Analysis,Mettler-Toledo GmbH (2001) Seite 5 ff.
Figure 1: Courbes TMA et MS d’une plaque à circuits imprimés jusqu’à la température de 650 °C. L’évolutiondes ions de fragmentation m/z 79 et 94, caractéristiques du brome et du bromure de méthyle est égalementtracée.
Figure 2: Courbes TMA et MS d’une poudre de polymère transformable en mousse. Les ions de fragmentationm/z 36, 43 et 78 correspondent respectivement au HCl, isopentane et benzène. m/z 78 a été multiplié par500 pour une meilleure visualisation.

23UserCom 1/2002
Dates
Exhibitions, Conferences and Seminars - Veranstaltungen, Konferenzen und Seminare6 th International Conference on Pharmacy andApplied Physical Chemistry May 26-30, 2002 Ascona, SwitzerlandAFCAT (Association de Calorimétrie et d’Analyse Thermique) May 27-29, 2002 Paris, France7 th LAEHNWITZSEMINAR on CALORIMETRY and17th IUPAC Conference on Chemical Thermodynamics 28.07-02.08, 2002 Rostock, GermanyESTAC 8 August 25-29, 2002 Barcelona, SpainMATERIAUX 2002 October 21-25, 2002 Tours, FranceEXPOQUIMIA November 26-30, 2002 Barcelona, Spain
TA Customer Courses and Seminars in Switzerland - Information and Course Registration:TA-Kundenkurse und Seminare in der Schweiz - Auskunft und Anmeldung bei:Frau Esther Andreato, METTLER TOLEDO GmbH, Schwerzenbach, Tel.: ++41 1 806 73 57, Fax: ++41 1 806 72 40, e-mail: [email protected]/Kurse:TMA-DMA/SW Basic (Deutsch) 16. September 2002 TMA-DMA/SW Basic (English) September 23, 2002TGA (Deutsch) 17. September 2002 TGA (English) September 24, 2002DSC Basic (Deutsch) 18. September 2002 DSC Basic (English) September 25, 2002DSC Advanced (Deutsch) 19. September 2002 DSC Advanced (English) September 26, 2002SW Advanced (Deutsch) 20. September 2002 SW Advanced (English) September 27, 2002Seminare:Einführung in die Thermische Analyse 25. November 2002 GreifenseeDer Glasübergang – die Kerngrösse amorpher Stoffe 26. November 2002 GreifenseeValidierung und CFR21 Part11 27. November 2002 GreifenseeInfoseminar:SETA2002 Infoday TA: Thermische Analyse in Forschung und Qualitätssicherung 04. Juni 2002 Greifensee
TA-Kundenkurse und Seminare (Deutschland)Für nähere Informationen wenden Sie sich bitte an: Frau Ina Wolf, METTLER TOLEDO GmbH, Giessen, Tel.: ++49 641 507 404Kundenkurse und Workshops:Branchenworkshop: “TA & spektroskopische Methoden bei der Entwicklung undQualitätssicherung von Kunststoffen” 22/23.10.2002 Giessen (DE)Workshop: “Interpretation thermoanalytischer Kurven” 24.10.2002 Giessen (DE)DSC-Workshop 29/30.10.2002 Giessen (DE) TG-Workshop 31.10/01.11.2002 Giessen (DE)Fachseminare:Thermische Analyse in der pharmazeutischen Forschung und Produktion 06. Juni 2002 München (DE)Thermoanalytische und spektroskopische Methoden an Kunststoffen,Veranstaltung von METTLER TOLEDO und Thermo Nicolet 20. Juni 2002 Frankfurt/M. (DE)
Cours et séminaires d’Analyse Thermique en FranceRenseignements et inscriptions par Christine Fauvarque, METTLER TOLEDO S.A., Viroflay, Tél.: ++33 1 3097 1439, Fax: ++33 1 3097 1660Cours clients :TG et logiciel STARe 15 octobre 2002 Viroflay (France) DSC avancé et logiciel STARe 17 octobre 2002 Viroflay (France)DSC et logiciel STARe 16 octobre 2002 Viroflay (France) TMA et logiciel STARe 18 octobre 2002 Viroflay (France)Journées d’information :Journée d’information 11 juin 2002 Lyon (France) Journée d’information 11 septembre 2002 Paris Nord (France)Séminaires :L’analyse thermique et les techniques de couplage TGA-IRTF,TGA-SM, TGA-GCMS, UVDSC, 1 journée complète 14 octobre 2002 Paris
Cours et séminaires d’Analyse Thermique en BelgiumRenseignements et inscriptions par Pat Hoogeras, N.V. METTLER TOLEDO S.A., Zaventem, Tél.: ++32 2 334 02 09, Fax: ++32 2 334 03 34Workshops:Combined techniques - basics and applications of TGA-MS and TGA-FTIR June 3, 2002Kinetics - basics and the application of kinetics to crystallization and reactions October 24, 2002ADSC - basics and the application of temperature modulated techniques such as ADSC, IsoStep, SSA, ... October 25, 2002Infoday:DMA and Elastomers - introduction of the new METTLER TOLEDO DMA/SDTA861e and applications on Elastomers October 28, 2002

UserCom 1/200224
TA Customer Courses and Seminars in the NetherlandsVoor verdere informatie kunt U kontakt opnemen met: Hay Berden, Mettler-Toledo B.V., Tiel, Tel.: ++31 344 63 83 63
Corsi e Seminari di Analisi Termica per Clienti in ItaliaPer ulteriori informazioni prego contattare:Simona Ferrari, Mettler-Toledo S.p.A., Novate Milanese, Tel.: ++39 02 333 321, Fax: ++39 02 356 2973, E-mail: [email protected] per Clienti:DSC base 4 Giugno, 17 Settembre 2002 Novate MilaneseDSC avanzato 5 Giugno, 18 Settembre 2002 Novate MilaneseTGA 6 Giugno, 19 Settembre 2002 Novate MilaneseTMA 7 Giugno, 20 Settembre 2002 Novate Milanese
Cursos y Seminarios de TA en EspañaPara detalles acerca de los cursos y seminarios, por favor, contacte con:Francesc Catala, Mettler-Toledo S.A.E., Tel: ++34 93 223 76 00, E-Mail: [email protected]
Seminario de aplicaciones TA: Madrid, Mayo 22 Seminario de aplicaciones TA: Barcelona, Mayo 29Seminario para usuarios STARe: Madrid, Mayo 23 Seminario para usuarios STARe: Barcelona, Mayo 30
TA Customer Courses and Seminars for Sweden and the Nordic countriesFor details of training courses and seminars please contact:Catharina Hasselgren, Mettler-Toledo AB, Tel: ++46 8 702 50 24, Fax: ++46 8 642 45 62, E-mail: [email protected]
TA Customer Courses and Seminars in the UKFor details of training courses and seminars please contact:Rod Bottom, METTLER TOLEDO Ltd Leicester, Tel: ++44 116 234 5025, Fax: ++44 116 236 5500
TA Customer Courses and Seminars in the USA and CanadaBasic Thermal Analysis Training based on the STARe System Versions 6 and 7 is being offered in California and at the Columbus, Ohio Headquarters.The training includes lectures and hands-on workshops.For information contact: Jon Foreman at +1 614 438 4687, fax: +1 614 438 4693 or by e-mail: [email protected]
TA Customer Training October 9-10, 2002 Columbus (OH)
TA Customer Courses in the South East Asia Regional Office, Kuala Lumpur.For information on dates please contact:Malaysia: Ricky Tanat at ++603 78455773, fax: 603 78458773 Thailand: W. Techakasembundit at ++662 7230336, fax: 662 7196479Singapore: Edwin Ho at ++65 8900011, fax: 65 8900013 Or SEA regional office: Soosay P. at ++603 78455373, fax: 603 78453478
TA Customer Courses and Seminars in JapanFor details of training courses and seminars please contact:Yasushi Ikeda at METTLER TOLEDO Japan, Tel.: ++81 3 5762 0606; Fax: ++81 3 5762 0971
TA Customer Training August 27-29, 2002 Tokyo & Osaka Service CenterInfoday Seminar October 29, 2002 Tokyo Technical CenterInfoday Seminar November 1, 2002 Osaka Branch
For further information regarding meetings, products or applications, please contact your local METTLER TOLEDO representative.Bei Fragen zu weiteren Tagungen, den Produkten oder Applikationen wenden Sie sich bitte an Ihre lokale METTLER TOLEDO Vertretung.Internet: http:/www.mt.com
Editorial teamMETTLER TOLEDO GmbH, Analytical, Sonnenbergstrasse 74, CH-8603 Schwerzenbach, Switzerland
Dr. J. Schawe, Dr. R. Riesen, J. Widmann, Dr. M. Schubnell, C. Darribère, Dr. M. Wagner, Dr. D. P. May Ni Jing Urs JörimannPhysicist Chem. Engineer Chem. Engineer Physicist Chem. Engineer Chemist Chemist Chemist Electr. Engineere-mail: [email protected], Tel.: ++41 1 806 73 87, Fax: ++41 1 806 72 60
Layout and ProductionPromotion & Dokumentation Schwerzenbach, Walter Hanselmann ME-51710181
Printed on 100% chlorine-free paper, for the sake of the environement.