neutropénies isolées de l’adulte orientations...
TRANSCRIPT

Neutropénies isolées de l’adulte Orientations diagnostiques
Journées de l’AIH
-
Strasbourg 2015
-
Flore Sicre
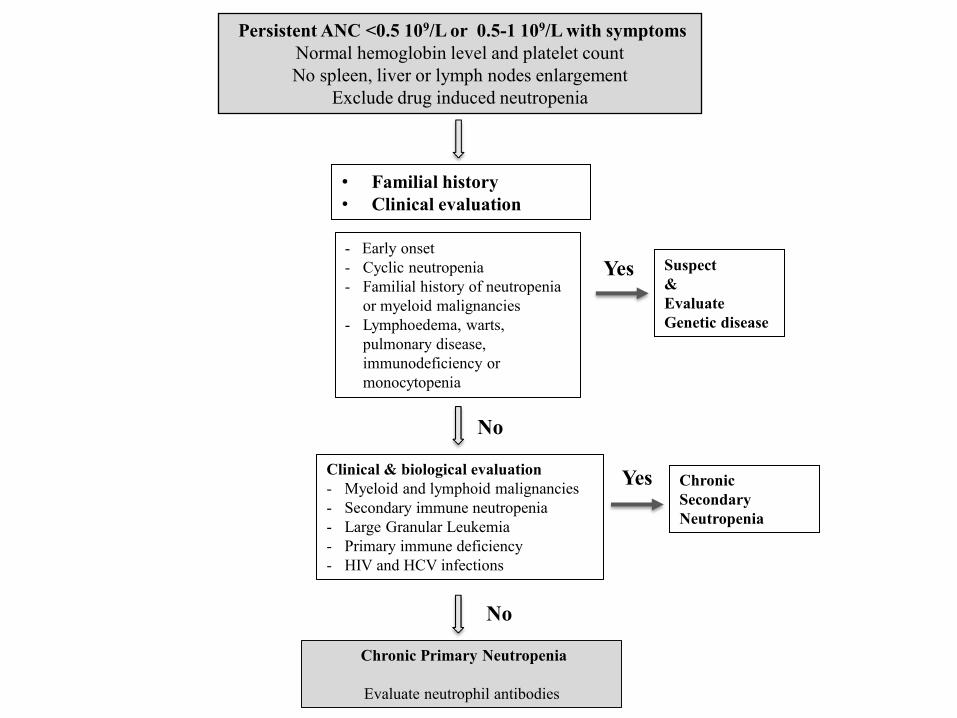
Persistent ANC <0.5 109/L or 0.5-1 109/L with symptoms
Normal hemoglobin level and platelet count
No spleen, liver or lymph nodes enlargement
Exclude drug induced neutropenia
• Familial history
• Clinical evaluation
Clinical & biological evaluation
- Myeloid and lymphoid malignancies
- Secondary immune neutropenia
- Large Granular Leukemia
- Primary immune deficiency
- HIV and HCV infections
Chronic Primary Neutropenia
Evaluate neutrophil antibodies
- Early onset
- Cyclic neutropenia
- Familial history of neutropenia
or myeloid malignancies
- Lymphoedema, warts,
pulmonary disease,
immunodeficiency or
monocytopenia
Suspect
&
Evaluate
Genetic disease
Chronic
Secondary
Neutropenia
Yes
No
Yes
No

Cas n°1
Mademoiselle A. 26 ans – avril 2015
– Neutrophiles : 1 G/L
– Hb 13 g/dl VGM 84 fl
– Lymphocytes 2 G/L, Monocytes 0,5 G/L
– Plaquettes 250 G/L
MT pour explorations complémentaires

Interrogatoire
Origine congolaise
Atcd familiaux = 0
Hémogramme depuis 2001 : PNN 0,6-1,2 G/L
Infections, aphtose, arthralgies, sd sec = 0
Examen clinique normal
Explorations à Nice en 2001
• Myélogramme : moelle de richesse normale, maturation granuleuse normale
• Caryotype 46, XX

Examens complémentaires ?
Pas obligatoires !
- NFS, Frottis
- EPP
- FAN, Anti-DNA
- Sérologie HIV, HCV

Diagnostic ?
Neutropénie ethnique
• Isolée
• Origine ethnique compatible
• Asymptomatique
• Ancienne
• Fluctuante

Neutropénie ethnique ou bénigne
• Décrit en 1941 (Forbes)
• Variation normale du chiffre de PNN
• Normales définies / ethnies caucasiennes
• Non pathologique
• Ethnies principalement concernées – Africaines
– Afro-américaine
– Moyen-orient (juives yemenite, bedouins..)
– ….

Neutropénie ethnique : mécanisme ?
• Richesse médullaire normale
• CFU-GM similaire
• Margination : capacité de démargination moindre après stimuli physico-chimiques
• Durée de vie neutrophiles similaire
• Diminution de capacité de migration des neutrophiles moelle vs sang périphérique



Neutropénies ethniques : Autres populations ?
« Juifs » yéménites : 11.7%
« Arabes » bedouins : 20% « Ougandais » : 30%

Reduced Neutrophil Count in People of African DescentIs Due To a Regulatory Variant in the Duffy AntigenReceptor for Chemokines Gene
David Reich1,2*, Michael A. Nalls3,4, W. H. Linda Kao5, Ermeg L. Akylbekova6, Art i Tandon1,2, Nick
Patterson2, James Mullikin7, Wen-Chi Hsueh8, Ching-Yu Cheng5,9, Josef Coresh5, Eric Boerwinkle10, Man
Li5, Alicja Waliszewska2,11, Julie Neubauer2, Rongling Li12, Tennille S. Leak13, Lynet te Ekunwe6, Joe C.
Files14, Cheryl L. Hardy14, Joseph M. Zmuda13, Herman A. Taylor15,16,17, Elad Ziv18,19,20, Tamara B.
Harris4, James G. Wilson21,22*
1 Department of Genetics, Harvard Medical School, Boston, Massachusetts, United States of America, 2 Broad Institute of Harvard and MIT, Cambridge, Massachusetts,
United States of America, 3 Laboratory of Neurogenetics, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
4 Laboratory of Epidemiology, Demography and Biometry, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
5 Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, United States of America, 6 Jackson Heart Study Analysis Group,
Jackson State University, Jackson, Mississippi, United States of America, 7 Comparative Genomics Unit, Genome Technology Branch, National Human Genome Research
Institute, Rockville, Maryland, United States of America, 8 Division of Medical Genetics, Department of Medicine, Department of Epidemiology and Biostatistics, Institute
for Human Genetics, University of California San Francisco, San Francisco, California, United States of America, 9 Inherited Disease Research Branch, National Human
Genome Research Institute, Baltimore, Maryland, United States of America, 10 Human Genetics Center, University of Texas Health Science Center at Houston, Houston,
Texas, United States of America, 11 Laboratory of Molecular Immunology, Center for Neurologic Disease, Brigham and Women’s Hospital, Boston, Massachusetts, United
States of America, 12 Department of Preventive Medicine, Center for Genomics and Bioinformatics, University of Tennessee Health Science Center, Memphis, Tennessee,
United States of America, 13 Department of Epidemiology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America,
14 Department of Medicine, Division of Hematology, University of Mississippi Medical Center, Jackson, Mississippi, United States of America, 15 Jackson State University,
Jackson, Mississippi, United States of America, 16 Tougaloo College, Jackson, Mississippi, United States of America, 17 University of Mississippi Medical Center, Jackson,
Mississippi, United States of America, 18 Division of General Internal Medicine, Department of Medicine, University of California San Francisco, San Francisco, California,
United States of America, 19 Department of Epidemiology and Biostatistics, Institute for Human Genetics, University of California San Francisco, San Francisco, California,
United States of America, 20 Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, California, United States of America,
21 V.A. Medical Center, Jackson, Mississippi, United States of America, 22 University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Persistently low white blood cell count (WBC) and neutrophil count is a well-described phenomenon in persons of Africanancestry, whose etiology remains unknown. We recently used admixture mapping to identify an approximately 1-megabaseregion on chromosome 1, where ancestry status (African or European) almost entirely accounted for the difference in WBCbetween African Americans and European Americans. To identify the specific genetic change responsible for thisassociation, we analyzed genotype and phenotype data from 6,005 African Americans from the Jackson Heart Study (JHS),the Health, Aging and Body Composition (Health ABC) Study, and the Atherosclerosis Risk in Communities (ARIC) Study. Wedemonstrate that the causal variant must be at least 91% different in frequency between West Africans and EuropeanAmericans. An excellent candidate is the Duffy Null polymorphism (SNP rs2814778 at chromosome 1q23.2), which is theonly polymorphism in the region known to be so differentiated in frequency and is already known to protect againstPlasmodium vivax malaria. We confirm that rs2814778 ispredictive of WBCand neutrophil count in African Americans abovebeyond the previously described admixture association (P= 3.86 102 5), establishing a novel phenotype for this geneticvariant.
Citat ion: Reich D, Nalls MA, Kao WHL, Akylbekova EL, Tandon A, et al. (2009) Reduced Neutrophil Count in People of African Descent Is Due To a RegulatoryVariant in the Duffy Antigen Receptor for Chemokines Gene. PLoS Genet 5(1): e1000360. doi:10.1371/journal.pgen.1000360
Editor: Peter M. Visscher, Queensland Institute of Medical Research, Australia
Received September 3, 2008; Accepted December 30, 2008; Published January 30, 2009
This is an open-access article distributed under the terms of the Creative Commons Public Domain declaration which stipulates that, once placed in the publicdomain, this work may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose.
Funding: Research support for JHSwas provided by R01-HL-084107 (JGW) from the National Heart, Lung, and Blood Institute and contracts N01-HC-95170, N01-HC-95171, and N01-HC-95172 from the National Heart, Lung, and Blood Institute and the National Center on Minority Health and Health Disparities. Researchsupport for Health ABC was provided by the Intramural Research Program of the National Institute on Aging, and contracts N01-AG-6-2101, N01-AG-6-2103, andN01-AG-6-2106. The Atherosclerosis Risk in Communities Study is a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, and N01-HC-55022. Support for the ARIC admixture mapping studies wasprovided by R21DK073482 and K01DK067207 (WHLK). Genotyping for both the JHS and Health ABC was supported by grant U54 RR020278 from the NationalCenter for Research Resources to the Broad Institute of Harvard and MIT; a subsidy from this grant covered half the cost of Health ABC genotyping. DR wassupported by a Burroughs Wellcome Career Development Award in the Biomedical Sciences, and methodological and statistical analysis was supported by grantU01-HG004168.
Compet ing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected] (DR); [email protected] (JGW)
PLoS Genetics | www.plosgenetics.org 1 January 2009 | Volume 5 | Issue 1 | e1000360
Reduced Neutrophil Count in People of African DescentIs Due To a Regulatory Variant in the Duffy AntigenReceptor for Chemokines Gene
David Reich1,2*, Michael A. Nalls3,4, W. H. Linda Kao5, Ermeg L. Akylbekova6, Art i Tandon1,2, Nick
Patterson2, James Mullikin7, Wen-Chi Hsueh8, Ching-Yu Cheng5,9, Josef Coresh5, Eric Boerwinkle10, Man
Li5, Alicja Waliszewska2,11, Julie Neubauer2, Rongling Li12, Tennille S. Leak13, Lynette Ekunwe6, Joe C.
Files14, Cheryl L. Hardy14, Joseph M. Zmuda13, Herman A. Taylor15,16,17, Elad Ziv18,19,20, Tamara B.
Harris4, James G. Wilson21,22*
1 Department of Genetics, Harvard Medical School, Boston, Massachusetts, United States of America, 2 Broad Institute of Harvard and MIT, Cambridge, Massachusetts,
United States of America, 3 Laboratory of Neurogenetics, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
4 Laboratory of Epidemiology, Demography and Biometry, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
5 Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, United States of America, 6 Jackson Heart Study Analysis Group,
Jackson State University, Jackson, Mississippi, United States of America, 7 Comparative Genomics Unit, Genome Technology Branch, National Human Genome Research
Institute, Rockville, Maryland, United States of America, 8 Division of Medical Genetics, Department of Medicine, Department of Epidemiology and Biostatistics, Institute
for Human Genetics, University of California San Francisco, San Francisco, California, United States of America, 9 Inherited Disease Research Branch, National Human
Genome Research Institute, Baltimore, Maryland, United States of America, 10 Human Genetics Center, University of Texas Health Science Center at Houston, Houston,
Texas, United States of America, 11 Laboratory of Molecular Immunology, Center for Neurologic Disease, Brigham and Women’s Hospital, Boston, Massachusetts, United
States of America, 12 Department of Preventive Medicine, Center for Genomics and Bioinformatics, University of Tennessee Health Science Center, Memphis, Tennessee,
United States of America, 13 Department of Epidemiology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America,
14 Department of Medicine, Division of Hematology, University of Mississippi Medical Center, Jackson, Mississippi, United States of America, 15 Jackson State University,
Jackson, Mississippi, United States of America, 16 Tougaloo College, Jackson, Mississippi, United States of America, 17 University of Mississippi Medical Center, Jackson,
Mississippi, United States of America, 18 Division of General Internal Medicine, Department of Medicine, University of California San Francisco, San Francisco, California,
United States of America, 19 Department of Epidemiology and Biostatistics, Institute for Human Genetics, University of California San Francisco, San Francisco, California,
United States of America, 20 Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, California, United States of America,
21 V.A. Medical Center, Jackson, Mississippi, United States of America, 22 University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abstract
Persistently low white blood cell count (WBC) and neutrophil count is a well-described phenomenon in persons of Africanancestry, whose etiology remains unknown. We recently used admixture mapping to identify an approximately 1-megabaseregion on chromosome 1, where ancestry status (African or European) almost entirely accounted for the difference in WBCbetween African Americans and European Americans. To identify the specific genetic change responsible for thisassociation, we analyzed genotype and phenotype data from 6,005 African Americans from the Jackson Heart Study (JHS),the Health, Aging and Body Composition (Health ABC) Study, and the Atherosclerosis Risk in Communities (ARIC) Study. Wedemonstrate that the causal variant must be at least 91% different in frequency between West Africans and EuropeanAmericans. An excellent candidate is the Duffy Null polymorphism (SNP rs2814778 at chromosome 1q23.2), which is theonly polymorphism in the region known to be so differentiated in frequency and is already known to protect againstPlasmodium vivax malaria. We confirm that rs2814778 ispredictive of WBCand neutrophil count in African Americans abovebeyond the previously described admixture association (P= 3.86 102 5), establishing a novel phenotype for this geneticvariant.
Citation: Reich D, Nalls MA, Kao WHL, Akylbekova EL, Tandon A, et al. (2009) Reduced Neutrophil Count in People of African Descent Is Due To a RegulatoryVariant in the Duffy Antigen Receptor for Chemokines Gene. PLoS Genet 5(1): e1000360. doi:10.1371/journal.pgen.1000360
Editor: Peter M. Visscher, Queensland Institute of Medical Research, Australia
Received September 3, 2008; Accepted December 30, 2008; Published January 30, 2009
This is an open-access article distributed under the terms of the Creative Commons Public Domain declaration which stipulates that, once placed in the publicdomain, this work may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose.
Funding: Research support for JHSwas provided by R01-HL-084107 (JGW) from the National Heart, Lung, and Blood Institute and contracts N01-HC-95170, N01-HC-95171, and N01-HC-95172 from the National Heart, Lung, and Blood Institute and the National Center on Minority Health and Health Disparities. Researchsupport for Health ABCwas provided by the Intramural Research Program of the National Institute on Aging, and contracts N01-AG-6-2101, N01-AG-6-2103, andN01-AG-6-2106. The Atherosclerosis Risk in Communities Study is a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, and N01-HC-55022. Support for the ARIC admixture mapping studies wasprovided by R21DK073482 and K01DK067207 (WHLK). Genotyping for both the JHS and Health ABC was supported by grant U54 RR020278 from the NationalCenter for Research Resources to the Broad Institute of Harvard and MIT; a subsidy from this grant covered half the cost of Health ABC genotyping. DR wassupported by a Burroughs Wellcome Career Development Award in the Biomedical Sciences, and methodological and statistical analysis was supported by grantU01-HG004168.
Compet ing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected] (DR); [email protected] (JGW)
PLoS Genetics | www.plosgenetics.org 1 January 2009 | Volume 5 | Issue 1 | e1000360
Reduced Neutrophil Count in People of African DescentIs Due To a Regulatory Variant in the Duffy AntigenReceptor for Chemokines Gene
David Reich1,2*, Michael A. Nalls3,4, W. H. Linda Kao5, Ermeg L. Akylbekova6, Art i Tandon1,2, Nick
Patterson2, James Mullikin7, Wen-Chi Hsueh8, Ching-Yu Cheng5,9, Josef Coresh5, Eric Boerwinkle10, Man
Li5, Alicja Waliszewska2,11, Julie Neubauer2, Rongling Li12, Tennille S. Leak13, Lynette Ekunwe6, Joe C.
Files14, Cheryl L. Hardy14, Joseph M. Zmuda13, Herman A. Taylor15,16,17, Elad Ziv18,19,20, Tamara B.
Harris4, James G. Wilson21,22*
1 Department of Genetics, Harvard Medical School, Boston, Massachusetts, United States of America, 2 Broad Institute of Harvard and MIT, Cambridge, Massachusetts,
United States of America, 3 Laboratory of Neurogenetics, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
4 Laboratory of Epidemiology, Demography and Biometry, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
5 Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, United States of America, 6 Jackson Heart Study Analysis Group,
Jackson State University, Jackson, Mississippi, United States of America, 7 Comparative Genomics Unit, Genome Technology Branch, National Human Genome Research
Institute, Rockville, Maryland, United States of America, 8 Division of Medical Genetics, Department of Medicine, Department of Epidemiology and Biostatistics, Institute
for Human Genetics, University of California San Francisco, San Francisco, California, United States of America, 9 Inherited Disease Research Branch, National Human
Genome Research Institute, Baltimore, Maryland, United States of America, 10 Human Genetics Center, University of Texas Health Science Center at Houston, Houston,
Texas, United States of America, 11 Laboratory of Molecular Immunology, Center for Neurologic Disease, Brigham and Women’s Hospital, Boston, Massachusetts, United
States of America, 12 Department of Preventive Medicine, Center for Genomics and Bioinformatics, University of Tennessee Health Science Center, Memphis, Tennessee,
United States of America, 13 Department of Epidemiology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America,
14 Department of Medicine, Division of Hematology, University of Mississippi Medical Center, Jackson, Mississippi, United States of America, 15 Jackson State University,
Jackson, Mississippi, United States of America, 16 Tougaloo College, Jackson, Mississippi, United States of America, 17 University of Mississippi Medical Center, Jackson,
Mississippi, United States of America, 18 Division of General Internal Medicine, Department of Medicine, University of California San Francisco, San Francisco, California,
United States of America, 19 Department of Epidemiology and Biostatistics, Institute for Human Genetics, University of California San Francisco, San Francisco, California,
United States of America, 20 Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, California, United States of America,
21 V.A. Medical Center, Jackson, Mississippi, United States of America, 22 University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Persistently low white blood cell count (WBC) and neutrophil count is a well-described phenomenon in persons of Africanancestry, whose etiology remains unknown. We recently used admixture mapping to identify an approximately 1-megabaseregion on chromosome 1, where ancestry status (African or European) almost entirely accounted for the difference in WBCbetween African Americans and European Americans. To identify the specific genetic change responsible for thisassociation, we analyzed genotype and phenotype data from 6,005 African Americans from the Jackson Heart Study (JHS),the Health, Aging and Body Composition (Health ABC) Study, and the Atherosclerosis Risk in Communities (ARIC) Study. Wedemonstrate that the causal variant must be at least 91% different in frequency between West Africans and EuropeanAmericans. An excellent candidate is the Duffy Null polymorphism (SNP rs2814778 at chromosome 1q23.2), which is theonly polymorphism in the region known to be so differentiated in frequency and is already known to protect againstPlasmodium vivax malaria. We confirm that rs2814778 ispredictive of WBCand neutrophil count in African Americans abovebeyond the previously described admixture association (P= 3.86 102 5), establishing a novel phenotype for this geneticvariant.
Citation: Reich D, Nalls MA, Kao WHL, Akylbekova EL, Tandon A, et al. (2009) Reduced Neutrophil Count in People of African Descent Is Due To a RegulatoryVariant in the Duffy Antigen Receptor for Chemokines Gene. PLoS Genet 5(1): e1000360. doi:10.1371/journal.pgen.1000360
Editor: Peter M. Visscher, Queensland Institute of Medical Research, Australia
Received September 3, 2008; Accepted December 30, 2008; Published January 30, 2009
This is an open-access article distributed under the terms of the Creative Commons Public Domain declaration which stipulates that, once placed in the publicdomain, this work may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose.
Funding: Research support for JHSwas provided by R01-HL-084107 (JGW) from the National Heart, Lung, and Blood Institute and contracts N01-HC-95170, N01-HC-95171, and N01-HC-95172 from the National Heart, Lung, and Blood Institute and the National Center on Minority Health and Health Disparities. Researchsupport for Health ABCwas provided by the Intramural Research Program of the National Institute on Aging, and contracts N01-AG-6-2101, N01-AG-6-2103, andN01-AG-6-2106. The Atherosclerosis Risk in CommunitiesStudy is a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, and N01-HC-55022. Support for the ARIC admixture mapping studies wasprovided by R21DK073482 and K01DK067207 (WHLK). Genotyping for both the JHSand Health ABC was supported by grant U54 RR020278 from the NationalCenter for Research Resources to the Broad Institute of Harvard and MIT; a subsidy from this grant covered half the cost of Health ABC genotyping. DR wassupported by a Burroughs Wellcome Career Development Award in the Biomedical Sciences, and methodological and statistical analysis was supported by grantU01-HG004168.
Compet ing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected] (DR); [email protected] (JGW)
PLoS Genetics | www.plosgenetics.org 1 January 2009 | Volume 5 | Issue 1 | e1000360
Phénotype duffy null/null = absence d’expression DARC sur GR (≠ ¢ endotheliales) Récepteur chemokine (CXC et CC) : CXCL8 et RANTES (CCL5) Recrutement des neutrophiles Mécanisme ? Absence de transduction du signal Régulateur des chemokines circulantes
Autres arguments - Phénotype duffy null/null associé à la R Plasmodium vivax - Zones géographiques neutropénies ethniques / paludisme - Phénotype fréquent au sein de la pop juive yéménite
Genome-Wide Association Study of White Blood CellCount in 16,388 African Americans: the ContinentalOrigins and Genetic Epidemiology Network (COGENT)
Alexander P. Reiner1,2. * , Guillaume Lett re3,4. , Michael A. Nalls5. , Santhi K. Ganesh6. , Rasika Mathias7. ,
Melissa A. Aust in2,8. , Eric Dean9. , Sampath Arepalli5, Angela Brit ton5, Zhao Chen10, David Couper11, J.
David Curb12, Charles B. Eaton13, Myriam Fornage14, Struan F. A. Grant15, Tamara B. Harris16, Dena
Hernandez5, Naoyuki Kamat ini17, Brendan J. Keat ing15, Michiak i Kubo18, Andrea LaCroix1,2, Leslie A.
Lange19, Simin Liu20, Kurt Lohman21, Yan Meng22, Emile R. Mohler III23, Solomon Musani24, Yusuke
Nakamura25, Christopher J. O’Donnell26,27, Yukinor i Okada17, Cameron D. Palmer22, George J.
Papanicolaou26, Kushang V. Patel16, Andrew B. Singleton5, Atsushi Takahashi17, Hua Tang28, Herman A.
Taylor Jr.29,30, Kent Taylor31, Cynthia Thomson32, Lisa R. Yanek7, Lingyao Yang33, Elad Ziv9, Alan B.
Zonderman34, Aaron R. Folsom35" , Michele K. Evans36" , Yongmei Liu21" , Diane M. Becker7" , Beverly M.
Snively33" , James G. Wilson37" *
1 Department of Epidemiology, University of Washington, Seattle, Washington, United States of America, 2 Division of Public Health Sciences, Fred Hutchinson Cancer
Research Center, Seattle, Washington, United States of America, 3 Montreal Heart Institute, Montreal, Canada, 4 Departement de Medecine, Universite de Montreal,
Montreal, Canada, 5 Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, United States of America, 6 Division of
Cardiovascular Medicine, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, United States of America, 7 Department of Medicine, The Johns
Hopkins University School of Medicine, Baltimore, Maryland, United States of America, 8 Department of Epidemiology and Institute for Public Health Genetics, School of
Public Health, University of Washington, Seattle, Washington, United States of America, 9 Department of Medicine, University of California San Francisco, San Francisco,
California, United States of America, 10 Division of Epidemiology and Biostatistics, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson,
Arizona, United States of America, 11 Department of Epidemiology, University of North Carolina School of Public Health, Chapel Hill, North Carolina, United States of
America, 12 Department of Geriatric Medicine, John A. Burns School of Medicine, University of Hawaii, Honolulu, Hawaii, United States of America, 13 Center for Primary
Care and Prevention, Alpert Medical School of Brown University, Providence, Rhode Island, United States of America, 14 Houston Institute of Molecular Medicine,
University of Texas, Houston, Texas, United States of America, 15 Center for Applied Genomics, Division of Human Genetics, Children’s Hospital of Philadelphia Research
Institute, Philadelphia, Pennsylvania, United States of America, 16 Laboratory for Epidemiology, Demography, and Biometry, National Institute on Aging, National
Institutes of Health, Baltimore, Maryland, United States of America, 17 Laboratory for Statistical Analysis, Center for Genomic Medicine (CGM), Institute of Physical and
Chemical Research (RIKEN), Yokohama, Japan, 18 Laboratory for Genotyping Development, CGM, RIKEN, Yokohama, Japan, 19 Department of Genetics, University of
North Carolina, Chapel Hill, North Carolina, United States of America, 20 Departments of Epidemiology and Medicine, University of California Los Angeles, Los Angeles,
California, United States of America, 21 Center for Human Genomics, Department of Epidemiology and Prevention, Division of Public Health Sciences, Wake Forest
University School of Medicine, Winston-Salem, North Carolina, United States of America, 22 Program in Medical and Population Genetics, Broad Institute, Cambridge,
Massachusetts, United States of America, 23 Cardiovascular Division, Vascular Medicine Section, Department of Medicine, University of Pennsylvania School of Medicine,
Philadelphia, Pennsylvania, United States of America, 24 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States of America,
25 Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science, University of Tokyo, Tokyo, Japan, 26 National Heart, Lung, and Blood Institute
(NHLBI), Division of Cardiovascular Sciences, Bethesda, Maryland, United States of America, 27 NHLBI’s Framingham Heart Study, Framingham, Massachusetts, United
States of America, 28 Department of Genetics, Stanford University School of Medicine, Stanford, California, United States of America, 29 Jackson State University,
Tougaloo College, Jackson, Mississippi, United States of America, 30 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States
of America, 31 Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, California, United States of America, 32 Nutritional Sciences, Arizona Cancer Center,
University of Arizona, Tucson, Arizona, United States of America, 33 Department of Biostatistical Sciences, Division of Public Health Sciences, Wake Forest School of
Medicine, Winston-Salem, North Carolina, United States of America, 34 Laboratory of Personality and Cognition, National Institute on Aging, National Institutes of Health,
Baltimore, Maryland, United States of America, 35 Division of Epidemiology and Community Health, University of Minnesota, Minneapolis, Minnesota, United States of
America, 36 Health Disparities Research Section, Clinical Research Branch, National Institute on Aging, National Institutes of Health, Baltimore, Maryland, United States of
America, 37 Department of Physiology and Biophysics, University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Total white blood cell (WBC) and neutrophil counts are lower among individuals of African descent due to the commonAfrican-derived ‘‘null’’ variant of the Duffy Antigen Receptor for Chemokines (DARC) gene. Additional common geneticpolymorphisms were recently associated with total WBC and WBC sub-type levels in European and Japanese populations.No additional loci that account for WBC variability have been identified in African Americans. In order to address this, weperformed a large genome-wide association study (GWAS) of total WBCand cell subtype counts in 16,388 African-Americanparticipants from 7 population-based cohorts available in the Continental Origins and Genetic Epidemiology Network.In addition to the DARC locus on chromosome 1q23, we identified two other regions (chromosomes 4q13 and 16q22)
PLoS Genetics | www.plosgenetics.org 1 June 2011 | Volume 7 | Issue 6 | e1002108
Genome-Wide Association Study of White Blood CellCount in 16,388 African Americans: the ContinentalOrigins and Genetic Epidemiology Network (COGENT)
Alexander P. Reiner1,2. * , Guillaume Lettre3,4. , Michael A. Nalls5. , Santhi K. Ganesh6. , Rasika Mathias7. ,
Melissa A. Aust in2,8. , Eric Dean9. , Sampath Arepalli5, Angela Brit ton5, Zhao Chen10, David Couper11, J.
David Curb12, Charles B. Eaton13, Myriam Fornage14, Struan F. A. Grant15, Tamara B. Harris16, Dena
Hernandez5, Naoyuki Kamatini17, Brendan J. Keat ing15, Michiak i Kubo18, Andrea LaCroix1,2, Leslie A.
Lange19, Simin Liu20, Kurt Lohman21, Yan Meng22, Emile R. Mohler III23, Solomon Musani24, Yusuke
Nakamura25, Christopher J. O’Donnell26,27, Yukinori Okada17, Cameron D. Palmer22, George J.
Papanicolaou26, Kushang V. Patel16, Andrew B. Singleton5, Atsushi Takahashi17, Hua Tang28, Herman A.
Taylor Jr.29,30, Kent Taylor31, Cynthia Thomson32, Lisa R. Yanek7, Lingyao Yang33, Elad Ziv9, Alan B.
Zonderman34, Aaron R. Folsom35" , Michele K. Evans36" , Yongmei Liu21" , Diane M. Becker7" , Beverly M.
Snively33" , James G. Wilson37" *
1 Department of Epidemiology, University of Washington, Seattle, Washington, United States of America, 2 Division of Public Health Sciences, Fred Hutchinson Cancer
Research Center, Seattle, Washington, United States of America, 3 Montreal Heart Institute, Montreal, Canada, 4 Departement de Medecine, Universite de Montreal,
Montreal, Canada, 5 Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, United States of America, 6 Division of
Cardiovascular Medicine, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, United States of America, 7 Department of Medicine, The Johns
Hopkins University School of Medicine, Baltimore, Maryland, United States of America, 8 Department of Epidemiology and Institute for Public Health Genetics, School of
Public Health, University of Washington, Seattle, Washington, United States of America, 9 Department of Medicine, University of California San Francisco, San Francisco,
California, United States of America, 10 Division of Epidemiology and Biostatistics, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson,
Arizona, United States of America, 11 Department of Epidemiology, University of North Carolina School of Public Health, Chapel Hill, North Carolina, United States of
America, 12 Department of Geriatric Medicine, John A. Burns School of Medicine, University of Hawaii, Honolulu, Hawaii, United States of America, 13 Center for Primary
Care and Prevention, Alpert Medical School of Brown University, Providence, Rhode Island, United States of America, 14 Houston Institute of Molecular Medicine,
University of Texas, Houston, Texas, United States of America, 15 Center for Applied Genomics, Division of Human Genetics, Children’s Hospital of Philadelphia Research
Institute, Philadelphia, Pennsylvania, United States of America, 16 Laboratory for Epidemiology, Demography, and Biometry, National Institute on Aging, National
Institutes of Health, Baltimore, Maryland, United States of America, 17 Laboratory for Statistical Analysis, Center for Genomic Medicine (CGM), Institute of Physical and
Chemical Research (RIKEN), Yokohama, Japan, 18 Laboratory for Genotyping Development, CGM, RIKEN, Yokohama, Japan, 19 Department of Genetics, University of
North Carolina, Chapel Hill, North Carolina, United States of America, 20 Departments of Epidemiology and Medicine, University of California Los Angeles, Los Angeles,
California, United States of America, 21 Center for Human Genomics, Department of Epidemiology and Prevention, Division of Public Health Sciences, Wake Forest
University School of Medicine, Winston-Salem, North Carolina, United States of America, 22 Program in Medical and Population Genetics, Broad Institute, Cambridge,
Massachusetts, United States of America, 23 Cardiovascular Division, Vascular Medicine Section, Department of Medicine, University of Pennsylvania School of Medicine,
Philadelphia, Pennsylvania, United States of America, 24 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States of America,
25 Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science, University of Tokyo, Tokyo, Japan, 26 National Heart, Lung, and Blood Institute
(NHLBI), Division of Cardiovascular Sciences, Bethesda, Maryland, United States of America, 27 NHLBI’s Framingham Heart Study, Framingham, Massachusetts, United
States of America, 28 Department of Genetics, Stanford University School of Medicine, Stanford, California, United States of America, 29 Jackson State University,
Tougaloo College, Jackson, Mississippi, United States of America, 30 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States
of America, 31 Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, California, United States of America, 32 Nutritional Sciences, Arizona Cancer Center,
University of Arizona, Tucson, Arizona, United States of America, 33 Department of Biostatistical Sciences, Division of Public Health Sciences, Wake Forest School of
Medicine, Winston-Salem, North Carolina, United States of America, 34 Laboratory of Personality and Cognition, National Institute on Aging, National Institutes of Health,
Baltimore, Maryland, United States of America, 35 Division of Epidemiology and Community Health, University of Minnesota, Minneapolis, Minnesota, United States of
America, 36 Health Disparities Research Section, Clinical Research Branch, National Institute on Aging, National Institutes of Health, Baltimore, Maryland, United States of
America, 37 Department of Physiology and Biophysics, University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Total white blood cell (WBC) and neutrophil counts are lower among individuals of African descent due to the commonAfrican-derived ‘‘null’’ variant of the Duffy Antigen Receptor for Chemokines (DARC) gene. Additional common geneticpolymorphisms were recently associated with total WBC and WBC sub-type levels in European and Japanese populations.No additional loci that account for WBC variability have been identified in African Americans. In order to address this, weperformed a large genome-wide association study (GWAS) of total WBCand cell subtype counts in 16,388 African-Americanparticipants from 7 population-based cohorts available in the Continental Origins and Genetic Epidemiology Network.In addition to the DARC locus on chromosome 1q23, we identified two other regions (chromosomes 4q13 and 16q22)
PLoS Genetics | www.plosgenetics.org 1 June 2011 | Volume 7 | Issue 6 | e1002108
Genome-Wide Association Study of White Blood CellCount in 16,388 African Americans: the ContinentalOrigins and Genetic Epidemiology Network (COGENT)
Alexander P. Reiner1,2. * , Guillaume Lettre3,4. , Michael A. Nalls5. , Santhi K. Ganesh6. , Rasika Mathias7. ,
Melissa A. Aust in2,8. , Eric Dean9. , Sampath Arepalli5, Angela Brit ton5, Zhao Chen10, David Couper11, J.
David Curb12, Charles B. Eaton13, Myriam Fornage14, Struan F. A. Grant15, Tamara B. Harris16, Dena
Hernandez5, Naoyuki Kamat ini17, Brendan J. Keat ing15, Michiak i Kubo18, Andrea LaCroix1,2, Leslie A.
Lange19, Simin Liu20, Kurt Lohman21, Yan Meng22, Emile R. Mohler III23, Solomon Musani24, Yusuke
Nakamura25, Christopher J. O’Donnell26,27, Yukinor i Okada17, Cameron D. Palmer22, George J.
Papanicolaou26, Kushang V. Patel16, Andrew B. Singleton5, Atsushi Takahashi17, Hua Tang28, Herman A.
Taylor Jr.29,30, Kent Taylor31, Cynthia Thomson32, Lisa R. Yanek7, Lingyao Yang33, Elad Ziv9, Alan B.
Zonderman34, Aaron R. Folsom35" , Michele K. Evans36" , Yongmei Liu21" , Diane M. Becker7" , Beverly M.
Snively33" , James G. Wilson37" *
1 Department of Epidemiology, University of Washington, Seattle, Washington, United States of America, 2 Division of Public Health Sciences, Fred Hutchinson Cancer
Research Center, Seattle, Washington, United States of America, 3 Montreal Heart Institute, Montreal, Canada, 4 Departement de Medecine, Universite de Montreal,
Montreal, Canada, 5 Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, United States of America, 6 Division of
Cardiovascular Medicine, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, United States of America, 7 Department of Medicine, The Johns
Hopkins University School of Medicine, Baltimore, Maryland, United States of America, 8 Department of Epidemiology and Institute for Public Health Genetics, School of
Public Health, University of Washington, Seattle, Washington, United States of America, 9 Department of Medicine, University of California San Francisco, San Francisco,
California, United States of America, 10 Division of Epidemiology and Biostatistics, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson,
Arizona, United States of America, 11 Department of Epidemiology, University of North Carolina School of Public Health, Chapel Hill, North Carolina, United States of
America, 12 Department of Geriatric Medicine, John A. Burns School of Medicine, University of Hawaii, Honolulu, Hawaii, United States of America, 13 Center for Primary
Care and Prevention, Alpert Medical School of Brown University, Providence, Rhode Island, United States of America, 14 Houston Institute of Molecular Medicine,
University of Texas, Houston, Texas, United States of America, 15 Center for Applied Genomics, Division of Human Genetics, Children’s Hospital of Philadelphia Research
Institute, Philadelphia, Pennsylvania, United States of America, 16 Laboratory for Epidemiology, Demography, and Biometry, National Institute on Aging, National
Institutes of Health, Baltimore, Maryland, United States of America, 17 Laboratory for Statistical Analysis, Center for Genomic Medicine (CGM), Institute of Physical and
Chemical Research (RIKEN), Yokohama, Japan, 18 Laboratory for Genotyping Development, CGM, RIKEN, Yokohama, Japan, 19 Department of Genetics, University of
North Carolina, Chapel Hill, North Carolina, United States of America, 20 Departments of Epidemiology and Medicine, University of California Los Angeles, Los Angeles,
California, United States of America, 21 Center for Human Genomics, Department of Epidemiology and Prevention, Division of Public Health Sciences, Wake Forest
University School of Medicine, Winston-Salem, North Carolina, United States of America, 22 Program in Medical and Population Genetics, Broad Institute, Cambridge,
Massachusetts, United States of America, 23 Cardiovascular Division, Vascular Medicine Section, Department of Medicine, University of Pennsylvania School of Medicine,
Philadelphia, Pennsylvania, United States of America, 24 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States of America,
25 Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science, University of Tokyo, Tokyo, Japan, 26 National Heart, Lung, and Blood Institute
(NHLBI), Division of Cardiovascular Sciences, Bethesda, Maryland, United States of America, 27 NHLBI’s Framingham Heart Study, Framingham, Massachusetts, United
States of America, 28 Department of Genetics, Stanford University School of Medicine, Stanford, California, United States of America, 29 Jackson State University,
Tougaloo College, Jackson, Mississippi, United States of America, 30 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States
of America, 31 Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, California, United States of America, 32 Nutritional Sciences, Arizona Cancer Center,
University of Arizona, Tucson, Arizona, United States of America, 33 Department of Biostatistical Sciences, Division of Public Health Sciences, Wake Forest School of
Medicine, Winston-Salem, North Carolina, United States of America, 34 Laboratory of Personality and Cognition, National Institute on Aging, National Institutes of Health,
Baltimore, Maryland, United States of America, 35 Division of Epidemiology and Community Health, University of Minnesota, Minneapolis, Minnesota, United States of
America, 36 Health Disparities Research Section, Clinical Research Branch, National Institute on Aging, National Institutes of Health, Baltimore, Maryland, United States of
America, 37 Department of Physiology and Biophysics, University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Total white blood cell (WBC) and neutrophil counts are lower among individuals of African descent due to the commonAfrican-derived ‘‘null’’ variant of the Duffy Antigen Receptor for Chemokines (DARC) gene. Additional common geneticpolymorphisms were recently associated with total WBC and WBC sub-type levels in European and Japanese populations.No additional loci that account for WBC variability have been identified in African Americans. In order to address this, weperformed a large genome-wide association study (GWAS) of total WBCand cell subtype counts in 16,388 African-Americanparticipants from 7 population-based cohorts available in the Continental Origins and Genetic Epidemiology Network.In addition to the DARC locus on chromosome 1q23, we identified two other regions (chromosomes 4q13 and 16q22)
PLoS Genetics | www.plosgenetics.org 1 June 2011 | Volume 7 | Issue 6 | e1002108

Reduced Neutrophil Count in People of African DescentIs Due To a Regulatory Variant in the Duffy AntigenReceptor for Chemokines Gene
David Reich1,2*, Michael A. Nalls3,4, W. H. Linda Kao5, Ermeg L. Akylbekova6, Art i Tandon1,2, Nick
Patterson2, James Mullikin7, Wen-Chi Hsueh8, Ching-Yu Cheng5,9, Josef Coresh5, Eric Boerwinkle10, Man
Li5, Alicja Waliszewska2,11, Julie Neubauer2, Rongling Li12, Tennille S. Leak13, Lynet te Ekunwe6, Joe C.
Files14, Cheryl L. Hardy14, Joseph M. Zmuda13, Herman A. Taylor15,16,17, Elad Ziv18,19,20, Tamara B.
Harris4, James G. Wilson21,22*
1 Department of Genetics, Harvard Medical School, Boston, Massachusetts, United States of America, 2 Broad Institute of Harvard and MIT, Cambridge, Massachusetts,
United States of America, 3 Laboratory of Neurogenetics, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
4 Laboratory of Epidemiology, Demography and Biometry, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
5 Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, United States of America, 6 Jackson Heart Study Analysis Group,
Jackson State University, Jackson, Mississippi, United States of America, 7 Comparative Genomics Unit, Genome Technology Branch, National Human Genome Research
Institute, Rockville, Maryland, United States of America, 8 Division of Medical Genetics, Department of Medicine, Department of Epidemiology and Biostatistics, Institute
for Human Genetics, University of California San Francisco, San Francisco, California, United States of America, 9 Inherited Disease Research Branch, National Human
Genome Research Institute, Baltimore, Maryland, United States of America, 10 Human Genetics Center, University of Texas Health Science Center at Houston, Houston,
Texas, United States of America, 11 Laboratory of Molecular Immunology, Center for Neurologic Disease, Brigham and Women’s Hospital, Boston, Massachusetts, United
States of America, 12 Department of Preventive Medicine, Center for Genomics and Bioinformatics, University of Tennessee Health Science Center, Memphis, Tennessee,
United States of America, 13 Department of Epidemiology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America,
14 Department of Medicine, Division of Hematology, University of Mississippi Medical Center, Jackson, Mississippi, United States of America, 15 Jackson State University,
Jackson, Mississippi, United States of America, 16 Tougaloo College, Jackson, Mississippi, United States of America, 17 University of Mississippi Medical Center, Jackson,
Mississippi, United States of America, 18 Division of General Internal Medicine, Department of Medicine, University of California San Francisco, San Francisco, California,
United States of America, 19 Department of Epidemiology and Biostatistics, Institute for Human Genetics, University of California San Francisco, San Francisco, California,
United States of America, 20 Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, California, United States of America,
21 V.A. Medical Center, Jackson, Mississippi, United States of America, 22 University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Persistently low white blood cell count (WBC) and neutrophil count is a well-described phenomenon in persons of Africanancestry, whose etiology remains unknown. We recently used admixture mapping to identify an approximately 1-megabaseregion on chromosome 1, where ancestry status (African or European) almost entirely accounted for the difference in WBCbetween African Americans and European Americans. To identify the specific genetic change responsible for thisassociation, we analyzed genotype and phenotype data from 6,005 African Americans from the Jackson Heart Study (JHS),the Health, Aging and Body Composition (Health ABC) Study, and the Atherosclerosis Risk in Communities (ARIC) Study. Wedemonstrate that the causal variant must be at least 91% different in frequency between West Africans and EuropeanAmericans. An excellent candidate is the Duffy Null polymorphism (SNP rs2814778 at chromosome 1q23.2), which is theonly polymorphism in the region known to be so differentiated in frequency and is already known to protect againstPlasmodium vivax malaria. We confirm that rs2814778 ispredictive of WBCand neutrophil count in African Americans abovebeyond the previously described admixture association (P= 3.86 102 5), establishing a novel phenotype for this geneticvariant.
Citat ion: Reich D, Nalls MA, Kao WHL, Akylbekova EL, Tandon A, et al. (2009) Reduced Neutrophil Count in People of African Descent Is Due To a RegulatoryVariant in the Duffy Antigen Receptor for Chemokines Gene. PLoS Genet 5(1): e1000360. doi:10.1371/journal.pgen.1000360
Editor: Peter M. Visscher, Queensland Institute of Medical Research, Australia
Received September 3, 2008; Accepted December 30, 2008; Published January 30, 2009
This is an open-access article distributed under the terms of the Creative Commons Public Domain declaration which stipulates that, once placed in the publicdomain, this work may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose.
Funding: Research support for JHSwas provided by R01-HL-084107 (JGW) from the National Heart, Lung, and Blood Institute and contracts N01-HC-95170, N01-HC-95171, and N01-HC-95172 from the National Heart, Lung, and Blood Institute and the National Center on Minority Health and Health Disparities. Researchsupport for Health ABC was provided by the Intramural Research Program of the National Institute on Aging, and contracts N01-AG-6-2101, N01-AG-6-2103, andN01-AG-6-2106. The Atherosclerosis Risk in Communities Study is a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, and N01-HC-55022. Support for the ARIC admixture mapping studies wasprovided by R21DK073482 and K01DK067207 (WHLK). Genotyping for both the JHS and Health ABC was supported by grant U54 RR020278 from the NationalCenter for Research Resources to the Broad Institute of Harvard and MIT; a subsidy from this grant covered half the cost of Health ABC genotyping. DR wassupported by a Burroughs Wellcome Career Development Award in the Biomedical Sciences, and methodological and statistical analysis was supported by grantU01-HG004168.
Compet ing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected] (DR); [email protected] (JGW)
PLoS Genetics | www.plosgenetics.org 1 January 2009 | Volume 5 | Issue 1 | e1000360
Reduced Neutrophil Count in People of African DescentIs Due To a Regulatory Variant in the Duffy AntigenReceptor for Chemokines Gene
David Reich1,2*, Michael A. Nalls3,4, W. H. Linda Kao5, Ermeg L. Akylbekova6, Art i Tandon1,2, Nick
Patterson2, James Mullikin7, Wen-Chi Hsueh8, Ching-Yu Cheng5,9, Josef Coresh5, Eric Boerwinkle10, Man
Li5, Alicja Waliszewska2,11, Julie Neubauer2, Rongling Li12, Tennille S. Leak13, Lynette Ekunwe6, Joe C.
Files14, Cheryl L. Hardy14, Joseph M. Zmuda13, Herman A. Taylor15,16,17, Elad Ziv18,19,20, Tamara B.
Harris4, James G. Wilson21,22*
1 Department of Genetics, Harvard Medical School, Boston, Massachusetts, United States of America, 2 Broad Institute of Harvard and MIT, Cambridge, Massachusetts,
United States of America, 3 Laboratory of Neurogenetics, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
4 Laboratory of Epidemiology, Demography and Biometry, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
5 Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, United States of America, 6 Jackson Heart Study Analysis Group,
Jackson State University, Jackson, Mississippi, United States of America, 7 Comparative Genomics Unit, Genome Technology Branch, National Human Genome Research
Institute, Rockville, Maryland, United States of America, 8 Division of Medical Genetics, Department of Medicine, Department of Epidemiology and Biostatistics, Institute
for Human Genetics, University of California San Francisco, San Francisco, California, United States of America, 9 Inherited Disease Research Branch, National Human
Genome Research Institute, Baltimore, Maryland, United States of America, 10 Human Genetics Center, University of Texas Health Science Center at Houston, Houston,
Texas, United States of America, 11 Laboratory of Molecular Immunology, Center for Neurologic Disease, Brigham and Women’s Hospital, Boston, Massachusetts, United
States of America, 12 Department of Preventive Medicine, Center for Genomics and Bioinformatics, University of Tennessee Health Science Center, Memphis, Tennessee,
United States of America, 13 Department of Epidemiology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America,
14 Department of Medicine, Division of Hematology, University of Mississippi Medical Center, Jackson, Mississippi, United States of America, 15 Jackson State University,
Jackson, Mississippi, United States of America, 16 Tougaloo College, Jackson, Mississippi, United States of America, 17 University of Mississippi Medical Center, Jackson,
Mississippi, United States of America, 18 Division of General Internal Medicine, Department of Medicine, University of California San Francisco, San Francisco, California,
United States of America, 19 Department of Epidemiology and Biostatistics, Institute for Human Genetics, University of California San Francisco, San Francisco, California,
United States of America, 20 Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, California, United States of America,
21 V.A. Medical Center, Jackson, Mississippi, United States of America, 22 University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abstract
Persistently low white blood cell count (WBC) and neutrophil count is a well-described phenomenon in persons of Africanancestry, whose etiology remains unknown. We recently used admixture mapping to identify an approximately 1-megabaseregion on chromosome 1, where ancestry status (African or European) almost entirely accounted for the difference in WBCbetween African Americans and European Americans. To identify the specific genetic change responsible for thisassociation, we analyzed genotype and phenotype data from 6,005 African Americans from the Jackson Heart Study (JHS),the Health, Aging and Body Composition (Health ABC) Study, and the Atherosclerosis Risk in Communities (ARIC) Study. Wedemonstrate that the causal variant must be at least 91% different in frequency between West Africans and EuropeanAmericans. An excellent candidate is the Duffy Null polymorphism (SNP rs2814778 at chromosome 1q23.2), which is theonly polymorphism in the region known to be so differentiated in frequency and is already known to protect againstPlasmodium vivax malaria. We confirm that rs2814778 ispredictive of WBCand neutrophil count in African Americans abovebeyond the previously described admixture association (P= 3.86 102 5), establishing a novel phenotype for this geneticvariant.
Citation: Reich D, Nalls MA, Kao WHL, Akylbekova EL, Tandon A, et al. (2009) Reduced Neutrophil Count in People of African Descent Is Due To a RegulatoryVariant in the Duffy Antigen Receptor for Chemokines Gene. PLoS Genet 5(1): e1000360. doi:10.1371/journal.pgen.1000360
Editor: Peter M. Visscher, Queensland Institute of Medical Research, Australia
Received September 3, 2008; Accepted December 30, 2008; Published January 30, 2009
This is an open-access article distributed under the terms of the Creative Commons Public Domain declaration which stipulates that, once placed in the publicdomain, this work may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose.
Funding: Research support for JHSwas provided by R01-HL-084107 (JGW) from the National Heart, Lung, and Blood Institute and contracts N01-HC-95170, N01-HC-95171, and N01-HC-95172 from the National Heart, Lung, and Blood Institute and the National Center on Minority Health and Health Disparities. Researchsupport for Health ABCwas provided by the Intramural Research Program of the National Institute on Aging, and contracts N01-AG-6-2101, N01-AG-6-2103, andN01-AG-6-2106. The Atherosclerosis Risk in Communities Study is a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, and N01-HC-55022. Support for the ARIC admixture mapping studies wasprovided by R21DK073482 and K01DK067207 (WHLK). Genotyping for both the JHS and Health ABC was supported by grant U54 RR020278 from the NationalCenter for Research Resources to the Broad Institute of Harvard and MIT; a subsidy from this grant covered half the cost of Health ABC genotyping. DR wassupported by a Burroughs Wellcome Career Development Award in the Biomedical Sciences, and methodological and statistical analysis was supported by grantU01-HG004168.
Compet ing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected] (DR); [email protected] (JGW)
PLoS Genetics | www.plosgenetics.org 1 January 2009 | Volume 5 | Issue 1 | e1000360
Reduced Neutrophil Count in People of African DescentIs Due To a Regulatory Variant in the Duffy AntigenReceptor for Chemokines Gene
David Reich1,2*, Michael A. Nalls3,4, W. H. Linda Kao5, Ermeg L. Akylbekova6, Art i Tandon1,2, Nick
Patterson2, James Mullikin7, Wen-Chi Hsueh8, Ching-Yu Cheng5,9, Josef Coresh5, Eric Boerwinkle10, Man
Li5, Alicja Waliszewska2,11, Julie Neubauer2, Rongling Li12, Tennille S. Leak13, Lynette Ekunwe6, Joe C.
Files14, Cheryl L. Hardy14, Joseph M. Zmuda13, Herman A. Taylor15,16,17, Elad Ziv18,19,20, Tamara B.
Harris4, James G. Wilson21,22*
1 Department of Genetics, Harvard Medical School, Boston, Massachusetts, United States of America, 2 Broad Institute of Harvard and MIT, Cambridge, Massachusetts,
United States of America, 3 Laboratory of Neurogenetics, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
4 Laboratory of Epidemiology, Demography and Biometry, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
5 Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, United States of America, 6 Jackson Heart Study Analysis Group,
Jackson State University, Jackson, Mississippi, United States of America, 7 Comparative Genomics Unit, Genome Technology Branch, National Human Genome Research
Institute, Rockville, Maryland, United States of America, 8 Division of Medical Genetics, Department of Medicine, Department of Epidemiology and Biostatistics, Institute
for Human Genetics, University of California San Francisco, San Francisco, California, United States of America, 9 Inherited Disease Research Branch, National Human
Genome Research Institute, Baltimore, Maryland, United States of America, 10 Human Genetics Center, University of Texas Health Science Center at Houston, Houston,
Texas, United States of America, 11 Laboratory of Molecular Immunology, Center for Neurologic Disease, Brigham and Women’s Hospital, Boston, Massachusetts, United
States of America, 12 Department of Preventive Medicine, Center for Genomics and Bioinformatics, University of Tennessee Health Science Center, Memphis, Tennessee,
United States of America, 13 Department of Epidemiology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America,
14 Department of Medicine, Division of Hematology, University of Mississippi Medical Center, Jackson, Mississippi, United States of America, 15 Jackson State University,
Jackson, Mississippi, United States of America, 16 Tougaloo College, Jackson, Mississippi, United States of America, 17 University of Mississippi Medical Center, Jackson,
Mississippi, United States of America, 18 Division of General Internal Medicine, Department of Medicine, University of California San Francisco, San Francisco, California,
United States of America, 19 Department of Epidemiology and Biostatistics, Institute for Human Genetics, University of California San Francisco, San Francisco, California,
United States of America, 20 Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, California, United States of America,
21 V.A. Medical Center, Jackson, Mississippi, United States of America, 22 University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Persistently low white blood cell count (WBC) and neutrophil count is a well-described phenomenon in persons of Africanancestry, whose etiology remains unknown. We recently used admixture mapping to identify an approximately 1-megabaseregion on chromosome 1, where ancestry status (African or European) almost entirely accounted for the difference in WBCbetween African Americans and European Americans. To identify the specific genetic change responsible for thisassociation, we analyzed genotype and phenotype data from 6,005 African Americans from the Jackson Heart Study (JHS),the Health, Aging and Body Composition (Health ABC) Study, and the Atherosclerosis Risk in Communities (ARIC) Study. Wedemonstrate that the causal variant must be at least 91% different in frequency between West Africans and EuropeanAmericans. An excellent candidate is the Duffy Null polymorphism (SNP rs2814778 at chromosome 1q23.2), which is theonly polymorphism in the region known to be so differentiated in frequency and is already known to protect againstPlasmodium vivax malaria. We confirm that rs2814778 ispredictive of WBCand neutrophil count in African Americans abovebeyond the previously described admixture association (P= 3.86 102 5), establishing a novel phenotype for this geneticvariant.
Citation: Reich D, Nalls MA, Kao WHL, Akylbekova EL, Tandon A, et al. (2009) Reduced Neutrophil Count in People of African Descent Is Due To a RegulatoryVariant in the Duffy Antigen Receptor for Chemokines Gene. PLoS Genet 5(1): e1000360. doi:10.1371/journal.pgen.1000360
Editor: Peter M. Visscher, Queensland Institute of Medical Research, Australia
Received September 3, 2008; Accepted December 30, 2008; Published January 30, 2009
This is an open-access article distributed under the terms of the Creative Commons Public Domain declaration which stipulates that, once placed in the publicdomain, this work may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose.
Funding: Research support for JHSwas provided by R01-HL-084107 (JGW) from the National Heart, Lung, and Blood Institute and contracts N01-HC-95170, N01-HC-95171, and N01-HC-95172 from the National Heart, Lung, and Blood Institute and the National Center on Minority Health and Health Disparities. Researchsupport for Health ABCwas provided by the Intramural Research Program of the National Institute on Aging, and contracts N01-AG-6-2101, N01-AG-6-2103, andN01-AG-6-2106. The Atherosclerosis Risk in CommunitiesStudy is a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, and N01-HC-55022. Support for the ARIC admixture mapping studies wasprovided by R21DK073482 and K01DK067207 (WHLK). Genotyping for both the JHSand Health ABC was supported by grant U54 RR020278 from the NationalCenter for Research Resources to the Broad Institute of Harvard and MIT; a subsidy from this grant covered half the cost of Health ABC genotyping. DR wassupported by a Burroughs Wellcome Career Development Award in the Biomedical Sciences, and methodological and statistical analysis was supported by grantU01-HG004168.
Compet ing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected] (DR); [email protected] (JGW)
PLoS Genetics | www.plosgenetics.org 1 January 2009 | Volume 5 | Issue 1 | e1000360
Phénotype duffy null/null associé à la R Plasmodium vivax ? - Zones géographiques neutropénies ethniques / paludisme

Reduced Neutrophil Count in People of African DescentIs Due To a Regulatory Variant in the Duffy AntigenReceptor for Chemokines Gene
David Reich1,2*, Michael A. Nalls3,4, W. H. Linda Kao5, Ermeg L. Akylbekova6, Art i Tandon1,2, Nick
Patterson2, James Mullikin7, Wen-Chi Hsueh8, Ching-Yu Cheng5,9, Josef Coresh5, Eric Boerwinkle10, Man
Li5, Alicja Waliszewska2,11, Julie Neubauer2, Rongling Li12, Tennille S. Leak13, Lynet te Ekunwe6, Joe C.
Files14, Cheryl L. Hardy14, Joseph M. Zmuda13, Herman A. Taylor15,16,17, Elad Ziv18,19,20, Tamara B.
Harris4, James G. Wilson21,22*
1 Department of Genetics, Harvard Medical School, Boston, Massachusetts, United States of America, 2 Broad Institute of Harvard and MIT, Cambridge, Massachusetts,
United States of America, 3 Laboratory of Neurogenetics, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
4 Laboratory of Epidemiology, Demography and Biometry, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
5 Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, United States of America, 6 Jackson Heart Study Analysis Group,
Jackson State University, Jackson, Mississippi, United States of America, 7 Comparative Genomics Unit, Genome Technology Branch, National Human Genome Research
Institute, Rockville, Maryland, United States of America, 8 Division of Medical Genetics, Department of Medicine, Department of Epidemiology and Biostatistics, Institute
for Human Genetics, University of California San Francisco, San Francisco, California, United States of America, 9 Inherited Disease Research Branch, National Human
Genome Research Institute, Baltimore, Maryland, United States of America, 10 Human Genetics Center, University of Texas Health Science Center at Houston, Houston,
Texas, United States of America, 11 Laboratory of Molecular Immunology, Center for Neurologic Disease, Brigham and Women’s Hospital, Boston, Massachusetts, United
States of America, 12 Department of Preventive Medicine, Center for Genomics and Bioinformatics, University of Tennessee Health Science Center, Memphis, Tennessee,
United States of America, 13 Department of Epidemiology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America,
14 Department of Medicine, Division of Hematology, University of Mississippi Medical Center, Jackson, Mississippi, United States of America, 15 Jackson State University,
Jackson, Mississippi, United States of America, 16 Tougaloo College, Jackson, Mississippi, United States of America, 17 University of Mississippi Medical Center, Jackson,
Mississippi, United States of America, 18 Division of General Internal Medicine, Department of Medicine, University of California San Francisco, San Francisco, California,
United States of America, 19 Department of Epidemiology and Biostatistics, Institute for Human Genetics, University of California San Francisco, San Francisco, California,
United States of America, 20 Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, California, United States of America,
21 V.A. Medical Center, Jackson, Mississippi, United States of America, 22 University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Persistently low white blood cell count (WBC) and neutrophil count is a well-described phenomenon in persons of Africanancestry, whose etiology remains unknown. We recently used admixture mapping to identify an approximately 1-megabaseregion on chromosome 1, where ancestry status (African or European) almost entirely accounted for the difference in WBCbetween African Americans and European Americans. To identify the specific genetic change responsible for thisassociation, we analyzed genotype and phenotype data from 6,005 African Americans from the Jackson Heart Study (JHS),the Health, Aging and Body Composition (Health ABC) Study, and the Atherosclerosis Risk in Communities (ARIC) Study. Wedemonstrate that the causal variant must be at least 91% different in frequency between West Africans and EuropeanAmericans. An excellent candidate is the Duffy Null polymorphism (SNP rs2814778 at chromosome 1q23.2), which is theonly polymorphism in the region known to be so differentiated in frequency and is already known to protect againstPlasmodium vivax malaria. We confirm that rs2814778 ispredictive of WBCand neutrophil count in African Americans abovebeyond the previously described admixture association (P= 3.86 102 5), establishing a novel phenotype for this geneticvariant.
Citat ion: Reich D, Nalls MA, Kao WHL, Akylbekova EL, Tandon A, et al. (2009) Reduced Neutrophil Count in People of African Descent Is Due To a RegulatoryVariant in the Duffy Antigen Receptor for Chemokines Gene. PLoS Genet 5(1): e1000360. doi:10.1371/journal.pgen.1000360
Editor: Peter M. Visscher, Queensland Institute of Medical Research, Australia
Received September 3, 2008; Accepted December 30, 2008; Published January 30, 2009
This is an open-access article distributed under the terms of the Creative Commons Public Domain declaration which stipulates that, once placed in the publicdomain, this work may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose.
Funding: Research support for JHSwas provided by R01-HL-084107 (JGW) from the National Heart, Lung, and Blood Institute and contracts N01-HC-95170, N01-HC-95171, and N01-HC-95172 from the National Heart, Lung, and Blood Institute and the National Center on Minority Health and Health Disparities. Researchsupport for Health ABC was provided by the Intramural Research Program of the National Institute on Aging, and contracts N01-AG-6-2101, N01-AG-6-2103, andN01-AG-6-2106. The Atherosclerosis Risk in Communities Study is a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, and N01-HC-55022. Support for the ARIC admixture mapping studies wasprovided by R21DK073482 and K01DK067207 (WHLK). Genotyping for both the JHS and Health ABC was supported by grant U54 RR020278 from the NationalCenter for Research Resources to the Broad Institute of Harvard and MIT; a subsidy from this grant covered half the cost of Health ABC genotyping. DR wassupported by a Burroughs Wellcome Career Development Award in the Biomedical Sciences, and methodological and statistical analysis was supported by grantU01-HG004168.
Compet ing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected] (DR); [email protected] (JGW)
PLoS Genetics | www.plosgenetics.org 1 January 2009 | Volume 5 | Issue 1 | e1000360
Reduced Neutrophil Count in People of African DescentIs Due To a Regulatory Variant in the Duffy AntigenReceptor for Chemokines Gene
David Reich1,2*, Michael A. Nalls3,4, W. H. Linda Kao5, Ermeg L. Akylbekova6, Art i Tandon1,2, Nick
Patterson2, James Mullikin7, Wen-Chi Hsueh8, Ching-Yu Cheng5,9, Josef Coresh5, Eric Boerwinkle10, Man
Li5, Alicja Waliszewska2,11, Julie Neubauer2, Rongling Li12, Tennille S. Leak13, Lynette Ekunwe6, Joe C.
Files14, Cheryl L. Hardy14, Joseph M. Zmuda13, Herman A. Taylor15,16,17, Elad Ziv18,19,20, Tamara B.
Harris4, James G. Wilson21,22*
1 Department of Genetics, Harvard Medical School, Boston, Massachusetts, United States of America, 2 Broad Institute of Harvard and MIT, Cambridge, Massachusetts,
United States of America, 3 Laboratory of Neurogenetics, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
4 Laboratory of Epidemiology, Demography and Biometry, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
5 Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, United States of America, 6 Jackson Heart Study Analysis Group,
Jackson State University, Jackson, Mississippi, United States of America, 7 Comparative Genomics Unit, Genome Technology Branch, National Human Genome Research
Institute, Rockville, Maryland, United States of America, 8 Division of Medical Genetics, Department of Medicine, Department of Epidemiology and Biostatistics, Institute
for Human Genetics, University of California San Francisco, San Francisco, California, United States of America, 9 Inherited Disease Research Branch, National Human
Genome Research Institute, Baltimore, Maryland, United States of America, 10 Human Genetics Center, University of Texas Health Science Center at Houston, Houston,
Texas, United States of America, 11 Laboratory of Molecular Immunology, Center for Neurologic Disease, Brigham and Women’s Hospital, Boston, Massachusetts, United
States of America, 12 Department of Preventive Medicine, Center for Genomics and Bioinformatics, University of Tennessee Health Science Center, Memphis, Tennessee,
United States of America, 13 Department of Epidemiology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America,
14 Department of Medicine, Division of Hematology, University of Mississippi Medical Center, Jackson, Mississippi, United States of America, 15 Jackson State University,
Jackson, Mississippi, United States of America, 16 Tougaloo College, Jackson, Mississippi, United States of America, 17 University of Mississippi Medical Center, Jackson,
Mississippi, United States of America, 18 Division of General Internal Medicine, Department of Medicine, University of California San Francisco, San Francisco, California,
United States of America, 19 Department of Epidemiology and Biostatistics, Institute for Human Genetics, University of California San Francisco, San Francisco, California,
United States of America, 20 Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, California, United States of America,
21 V.A. Medical Center, Jackson, Mississippi, United States of America, 22 University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abstract
Persistently low white blood cell count (WBC) and neutrophil count is a well-described phenomenon in persons of Africanancestry, whose etiology remains unknown. We recently used admixture mapping to identify an approximately 1-megabaseregion on chromosome 1, where ancestry status (African or European) almost entirely accounted for the difference in WBCbetween African Americans and European Americans. To identify the specific genetic change responsible for thisassociation, we analyzed genotype and phenotype data from 6,005 African Americans from the Jackson Heart Study (JHS),the Health, Aging and Body Composition (Health ABC) Study, and the Atherosclerosis Risk in Communities (ARIC) Study. Wedemonstrate that the causal variant must be at least 91% different in frequency between West Africans and EuropeanAmericans. An excellent candidate is the Duffy Null polymorphism (SNP rs2814778 at chromosome 1q23.2), which is theonly polymorphism in the region known to be so differentiated in frequency and is already known to protect againstPlasmodium vivax malaria. We confirm that rs2814778 ispredictive of WBCand neutrophil count in African Americans abovebeyond the previously described admixture association (P= 3.86 102 5), establishing a novel phenotype for this geneticvariant.
Citation: Reich D, Nalls MA, Kao WHL, Akylbekova EL, Tandon A, et al. (2009) Reduced Neutrophil Count in People of African Descent Is Due To a RegulatoryVariant in the Duffy Antigen Receptor for Chemokines Gene. PLoS Genet 5(1): e1000360. doi:10.1371/journal.pgen.1000360
Editor: Peter M. Visscher, Queensland Institute of Medical Research, Australia
Received September 3, 2008; Accepted December 30, 2008; Published January 30, 2009
This is an open-access article distributed under the terms of the Creative Commons Public Domain declaration which stipulates that, once placed in the publicdomain, this work may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose.
Funding: Research support for JHSwas provided by R01-HL-084107 (JGW) from the National Heart, Lung, and Blood Institute and contracts N01-HC-95170, N01-HC-95171, and N01-HC-95172 from the National Heart, Lung, and Blood Institute and the National Center on Minority Health and Health Disparities. Researchsupport for Health ABCwas provided by the Intramural Research Program of the National Institute on Aging, and contracts N01-AG-6-2101, N01-AG-6-2103, andN01-AG-6-2106. The Atherosclerosis Risk in Communities Study is a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, and N01-HC-55022. Support for the ARIC admixture mapping studies wasprovided by R21DK073482 and K01DK067207 (WHLK). Genotyping for both the JHS and Health ABC was supported by grant U54 RR020278 from the NationalCenter for Research Resources to the Broad Institute of Harvard and MIT; a subsidy from this grant covered half the cost of Health ABC genotyping. DR wassupported by a Burroughs Wellcome Career Development Award in the Biomedical Sciences, and methodological and statistical analysis was supported by grantU01-HG004168.
Compet ing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected] (DR); [email protected] (JGW)
PLoS Genetics | www.plosgenetics.org 1 January 2009 | Volume 5 | Issue 1 | e1000360
Reduced Neutrophil Count in People of African DescentIs Due To a Regulatory Variant in the Duffy AntigenReceptor for Chemokines Gene
David Reich1,2*, Michael A. Nalls3,4, W. H. Linda Kao5, Ermeg L. Akylbekova6, Art i Tandon1,2, Nick
Patterson2, James Mullikin7, Wen-Chi Hsueh8, Ching-Yu Cheng5,9, Josef Coresh5, Eric Boerwinkle10, Man
Li5, Alicja Waliszewska2,11, Julie Neubauer2, Rongling Li12, Tennille S. Leak13, Lynette Ekunwe6, Joe C.
Files14, Cheryl L. Hardy14, Joseph M. Zmuda13, Herman A. Taylor15,16,17, Elad Ziv18,19,20, Tamara B.
Harris4, James G. Wilson21,22*
1 Department of Genetics, Harvard Medical School, Boston, Massachusetts, United States of America, 2 Broad Institute of Harvard and MIT, Cambridge, Massachusetts,
United States of America, 3 Laboratory of Neurogenetics, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
4 Laboratory of Epidemiology, Demography and Biometry, Intramural Research Program, National Institute on Aging, Bethesda, Maryland, United States of America,
5 Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland, United States of America, 6 Jackson Heart Study Analysis Group,
Jackson State University, Jackson, Mississippi, United States of America, 7 Comparative Genomics Unit, Genome Technology Branch, National Human Genome Research
Institute, Rockville, Maryland, United States of America, 8 Division of Medical Genetics, Department of Medicine, Department of Epidemiology and Biostatistics, Institute
for Human Genetics, University of California San Francisco, San Francisco, California, United States of America, 9 Inherited Disease Research Branch, National Human
Genome Research Institute, Baltimore, Maryland, United States of America, 10 Human Genetics Center, University of Texas Health Science Center at Houston, Houston,
Texas, United States of America, 11 Laboratory of Molecular Immunology, Center for Neurologic Disease, Brigham and Women’s Hospital, Boston, Massachusetts, United
States of America, 12 Department of Preventive Medicine, Center for Genomics and Bioinformatics, University of Tennessee Health Science Center, Memphis, Tennessee,
United States of America, 13 Department of Epidemiology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania, United States of America,
14 Department of Medicine, Division of Hematology, University of Mississippi Medical Center, Jackson, Mississippi, United States of America, 15 Jackson State University,
Jackson, Mississippi, United States of America, 16 Tougaloo College, Jackson, Mississippi, United States of America, 17 University of Mississippi Medical Center, Jackson,
Mississippi, United States of America, 18 Division of General Internal Medicine, Department of Medicine, University of California San Francisco, San Francisco, California,
United States of America, 19 Department of Epidemiology and Biostatistics, Institute for Human Genetics, University of California San Francisco, San Francisco, California,
United States of America, 20 Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, California, United States of America,
21 V.A. Medical Center, Jackson, Mississippi, United States of America, 22 University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Persistently low white blood cell count (WBC) and neutrophil count is a well-described phenomenon in persons of Africanancestry, whose etiology remains unknown. We recently used admixture mapping to identify an approximately 1-megabaseregion on chromosome 1, where ancestry status (African or European) almost entirely accounted for the difference in WBCbetween African Americans and European Americans. To identify the specific genetic change responsible for thisassociation, we analyzed genotype and phenotype data from 6,005 African Americans from the Jackson Heart Study (JHS),the Health, Aging and Body Composition (Health ABC) Study, and the Atherosclerosis Risk in Communities (ARIC) Study. Wedemonstrate that the causal variant must be at least 91% different in frequency between West Africans and EuropeanAmericans. An excellent candidate is the Duffy Null polymorphism (SNP rs2814778 at chromosome 1q23.2), which is theonly polymorphism in the region known to be so differentiated in frequency and is already known to protect againstPlasmodium vivax malaria. We confirm that rs2814778 ispredictive of WBCand neutrophil count in African Americans abovebeyond the previously described admixture association (P= 3.86 102 5), establishing a novel phenotype for this geneticvariant.
Citation: Reich D, Nalls MA, Kao WHL, Akylbekova EL, Tandon A, et al. (2009) Reduced Neutrophil Count in People of African Descent Is Due To a RegulatoryVariant in the Duffy Antigen Receptor for Chemokines Gene. PLoS Genet 5(1): e1000360. doi:10.1371/journal.pgen.1000360
Editor: Peter M. Visscher, Queensland Institute of Medical Research, Australia
Received September 3, 2008; Accepted December 30, 2008; Published January 30, 2009
This is an open-access article distributed under the terms of the Creative Commons Public Domain declaration which stipulates that, once placed in the publicdomain, this work may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose.
Funding: Research support for JHSwas provided by R01-HL-084107 (JGW) from the National Heart, Lung, and Blood Institute and contracts N01-HC-95170, N01-HC-95171, and N01-HC-95172 from the National Heart, Lung, and Blood Institute and the National Center on Minority Health and Health Disparities. Researchsupport for Health ABCwas provided by the Intramural Research Program of the National Institute on Aging, and contracts N01-AG-6-2101, N01-AG-6-2103, andN01-AG-6-2106. The Atherosclerosis Risk in CommunitiesStudy is a collaborative study supported by National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, and N01-HC-55022. Support for the ARIC admixture mapping studies wasprovided by R21DK073482 and K01DK067207 (WHLK). Genotyping for both the JHSand Health ABC was supported by grant U54 RR020278 from the NationalCenter for Research Resources to the Broad Institute of Harvard and MIT; a subsidy from this grant covered half the cost of Health ABC genotyping. DR wassupported by a Burroughs Wellcome Career Development Award in the Biomedical Sciences, and methodological and statistical analysis was supported by grantU01-HG004168.
Compet ing Interests: The authors have declared that no competing interests exist.
* E-mail: [email protected] (DR); [email protected] (JGW)
PLoS Genetics | www.plosgenetics.org 1 January 2009 | Volume 5 | Issue 1 | e1000360
Phénotype duffy null/null associé à la R Plasmodium vivax ? - Zones géographiques neutropénies ethniques / paludisme
The effect of Duffy ant igen receptor for chemokineson severity in sickle cell disease
Despite the identical genotypes, the clinical course ofsickle cell disease (SCD) is extremely variable, promptingthe search for genetic and biological predictors of diseaseseverity.1,2 Raised white blood counts (WBC) have beenknown to be a marker of disease severity in SCD since the1990s.1-3 “Benign ethnic neutropenia” has long beenobserved in hematology clinics in peoples of Africandescent. Recent work has shown that this relates to a sin-gle nucleotide polymorphism (SNP) rs2814778 T/C posi-tion -33 in the FY gene promoter resulting in lack of Duffyexpression on red blood cells, and also a lower WBC.4-7
Afenyi-Annan et al. also found that the association with FYstatus held with and without hydroxycarbamide therapy.5
Relationships between the Duffy phenotype and markersof disease severity or the postulated disease phenotypes inSCD have been investigated but no clear consensus hasbeen reached.5,8-10 However, using a multi-organ chronicdisease score, Afenyi-Annan et al. reported an overallincrease in the number of organs damaged and also anassociation with macroalbuminuria as shown by 1+ urinedip positivity in the Duffy negative group.5
For this study, DNA was extracted from buffy coatsobtained via the sickle cell gene bank based at King’sCollege Hospital, London, UK (REC 07/H0606/165). Allpatients were of African origin; they were genotyped forSNP rs2814778 in the DARC (Duffy Antigen Receptor forChemokines) promoter region using a TaqMan allelic dis-crimination assay and proprietary PCR primers from ABIbiosystems.
Biological data including hemoglobin (Hb), lactate dehy-drogenase (LDH), WBC, neutrophil count, HbF, reticulo-cyte count, ferritin, creatinine, urine albumin creatinineratio (ACR), cystatin C and erythropoietin levels were col-lected from routine blood results in patients attendingsteady state clinic during a 2-year period from January 1st
2009 to December 31st 2010. Estimated glomerular filtra-tion rates (eGFRs) were calculated using the 4-pointModification of Diet in Renal Disease (MDRD) formula.Clinical data, including the presence of specific complica-tions (stroke, priapism, leg ulcers, acute chest syndrome,avascular necrosis, retinopathy, tricuspid regurgitant jetvelocity ³2.5 m/s and gallstones) were collected from theelectronic patient record system and sickle cell database.Definitions of clinical complications were made using localdiagnostic guidelines which are based on published litera-ture as reviewed by Ballas et al.11 Diagnosis of chronic sick-le lung disease was made on chest CT scans as reviewedby a consultant chest radiologist. Admission data werealso collected for the 2-year study period, including lengthof stay, time to readmission and number of admissions.Only admissions lasting over 24 h requiring inpatient staywere included; visits to the Accident and EmergencyDepartment were excluded. Patient age at the end of thestudy period was recorded. Variables were log transformedwhere appropriate to obtain a normal distribution.
A total of 272 patients were genotyped for SNPrs2814778(T/C) in the DARCpromoter. The Duffy pheno-type was predicted to be negative based on the absence ofthe DARC -33T allele. The genotype was DARC 33C/C in243 (89%) of patients. The Duffy phenotype was predict-ed to be positive based on the presence of the DARC 33Tallele, with DARC -33T/C and -33T/T in 26 (10%) and 3(1%) patients, respectively (Table 1). We observed the per-centage of Duffy negative subjects to be much higher than
the 63% and 73% reported in African Americans by Nallset al. and Afenyi-Annan et al., respectively. The differenceis likely to be related to the different degrees of Europeanadmixture. The UK cohort is predominantly West Africanwith a smaller number of Afro-Caribbeans (Table 1).
The clinical characteristics and Duffy genotypes (andphenotypes) of the study group (and subgroups) are out-lined in Tables 1 and 2. There was no significant differencebetween the frequencies of rs2814778 T or C allelesbetween the different sickle genotypes. The predictedDuffy phenotypes were used to look for associations withmarkers of disease severity.
Laboratory data were available for the entire studygroup. Multiple regression analysis using a random effectsmodel was used to analyze the data, enabling the pooling
haematologica 2013; 98:e87
LETTERS TO THE EDITOR
Table 1. Summary of demographic data and Duffy genotypes/ phe-notypes for study group and admission cohort. Laboratory data wereavailable on the whole cohort.
Whole cohort Admitted patients(% of whole cohort) (% of subcategory)
N. of patients 272 112 (41)
Male: Female 107:165 (39:61) 48:64 (43:57)
Mean age (range) 36 (17 to 74) 34 (18 to 74)
Sickle genotypesHb SS 174 (64) 89/174 (51)Hb SC 80 (30) 17/80 (21)Hb Sβ+ 12 (4) 1/12 (8)Hb Sβ0 6 (2) 5/6 (83)
Alpha globin genotypes 245 (90) 101/245 (41)a a/ a a 148 (58) 61/148 (41)a a/ a- and a-/ a- 95 (37) 38/95 (40)a a/ a a a 2 (<1) 2/2 (100)
Duffy genotypesC/C 243 (89) 97/243 (40)C/T 26 (10) 14/26 (54)T/T 3 (1) 1/3 (33)
Duffy phenotypes Duffy negative 243 (89) 97/243 (40)Duffy positive 29 (11) 15/29 (52)
Table 2. Summary of demographic data and Duffy genotypes/ phe-notypes for sickle cell anemia patients with clinical and end-organdamage data available.
SCA patients only (n = 180) Patients with clinical data available
Male: Female 70:110 (39:61)
Mean age (range) 34 (17 to 68)
Sickle genotypesHb SS 174 Hb Sb0 6
Alpha globin genotypes 162/180 (90)a a/ a a 92/162 (57)a a/ a- and a-/ a- 68/162 (42)a a/ a a a 2/162 (1)
Duffy genotypes-46 C/C 163/180 (91)-46 C/T 14/180 (8)-46 T/T 2/180 (1)
Duffy phenotypes Duffy negative 163/180 (91)Duffy positive 17/180 (9)
The effect of Duffy ant igen receptor for chemokineson severity in sickle cell disease
Despite the identical genotypes, the clinical course ofsickle cell disease (SCD) is extremely variable, promptingthe search for genetic and biological predictors of diseaseseverity.1,2 Raised white blood counts (WBC) have beenknown to be a marker of disease severity in SCD since the1990s.1-3 “Benign ethnic neutropenia” has long beenobserved in hematology clinics in peoples of Africandescent. Recent work has shown that this relates to a sin-gle nucleotide polymorphism (SNP) rs2814778 T/C posi-tion -33 in the FY gene promoter resulting in lack of Duffyexpression on red blood cells, and also a lower WBC.4-7
Afenyi-Annan et al. also found that the association with FYstatus held with and without hydroxycarbamide therapy.5
Relationships between the Duffy phenotype and markersof disease severity or the postulated disease phenotypes inSCD have been investigated but no clear consensus hasbeen reached.5,8-10 However, using a multi-organ chronicdisease score, Afenyi-Annan et al. reported an overallincrease in the number of organs damaged and also anassociation with macroalbuminuria as shown by 1+ urinedip positivity in the Duffy negative group.5
For this study, DNA was extracted from buffy coatsobtained via the sickle cell gene bank based at King’sCollege Hospital, London, UK (REC 07/H0606/165). Allpatients were of African origin; they were genotyped forSNP rs2814778 in the DARC (Duffy Antigen Receptor forChemokines) promoter region using a TaqMan allelic dis-crimination assay and proprietary PCR primers from ABIbiosystems.
Biological data including hemoglobin (Hb), lactate dehy-drogenase (LDH), WBC, neutrophil count, HbF, reticulo-cyte count, ferritin, creatinine, urine albumin creatinineratio (ACR), cystatin C and erythropoietin levels were col-lected from routine blood results in patients attendingsteady state clinic during a 2-year period from January 1st
2009 to December 31st 2010. Estimated glomerular filtra-tion rates (eGFRs) were calculated using the 4-pointModification of Diet in Renal Disease (MDRD) formula.Clinical data, including the presence of specific complica-tions (stroke, priapism, leg ulcers, acute chest syndrome,avascular necrosis, retinopathy, tricuspid regurgitant jetvelocity ³2.5 m/s and gallstones) were collected from theelectronic patient record system and sickle cell database.Definitions of clinical complications were made using localdiagnostic guidelines which are based on published litera-ture as reviewed by Ballas et al.11 Diagnosis of chronic sick-le lung disease was made on chest CT scans as reviewedby a consultant chest radiologist. Admission data werealso collected for the 2-year study period, including lengthof stay, time to readmission and number of admissions.Only admissions lasting over 24 h requiring inpatient staywere included; visits to the Accident and EmergencyDepartment were excluded. Patient age at the end of thestudy period was recorded. Variables were log transformedwhere appropriate to obtain a normal distribution.
A total of 272 patients were genotyped for SNPrs2814778(T/C) in the DARCpromoter. The Duffy pheno-type was predicted to be negative based on the absence ofthe DARC -33T allele. The genotype was DARC 33C/C in243 (89%) of patients. The Duffy phenotype was predict-ed to be positive based on the presence of the DARC 33Tallele, with DARC -33T/C and -33T/T in 26 (10%) and 3(1%) patients, respectively (Table 1). We observed the per-centage of Duffy negative subjects to be much higher than
the 63% and 73% reported in African Americans by Nallset al. and Afenyi-Annan et al., respectively. The differenceis likely to be related to the different degrees of Europeanadmixture. The UK cohort is predominantly West Africanwith a smaller number of Afro-Caribbeans (Table 1).
The clinical characteristics and Duffy genotypes (andphenotypes) of the study group (and subgroups) are out-lined in Tables 1 and 2. There was no significant differencebetween the frequencies of rs2814778 T or C allelesbetween the different sickle genotypes. The predictedDuffy phenotypes were used to look for associations withmarkers of disease severity.
Laboratory data were available for the entire studygroup. Multiple regression analysis using a random effectsmodel was used to analyze the data, enabling the pooling
haematologica 2013; 98:e87
LETTERS TO THE EDITOR
Table 1. Summary of demographic data and Duffy genotypes/ phe-notypes for study group and admission cohort. Laboratory data wereavailable on the whole cohort.
Whole cohort Admitted patients(% of whole cohort) (% of subcategory)
N. of patients 272 112 (41)
Male: Female 107:165 (39:61) 48:64 (43:57)
Mean age (range) 36 (17 to 74) 34 (18 to 74)
Sickle genotypesHb SS 174 (64) 89/174 (51)Hb SC 80 (30) 17/80 (21)Hb Sβ+ 12 (4) 1/12 (8)Hb Sβ0 6 (2) 5/6 (83)
Alpha globin genotypes 245 (90) 101/245 (41)a a/ a a 148 (58) 61/148 (41)a a/ a- and a-/ a- 95 (37) 38/95 (40)a a/ a a a 2 (<1) 2/2 (100)
Duffy genotypesC/C 243 (89) 97/243 (40)C/T 26 (10) 14/26 (54)T/T 3 (1) 1/3 (33)
Duffy phenotypes Duffy negative 243 (89) 97/243 (40)Duffy positive 29 (11) 15/29 (52)
Table 2. Summary of demographic data and Duffy genotypes/ phe-notypes for sickle cell anemia patients with clinical and end-organdamage data available.
SCA patients only (n = 180) Patients with clinical data available
Male: Female 70:110 (39:61)
Mean age (range) 34 (17 to 68)
Sickle genotypesHb SS 174 Hb Sb0 6
Alpha globin genotypes 162/180 (90)a a/ a a 92/162 (57)a a/ a- and a-/ a- 68/162 (42)a a/ a a a 2/162 (1)
Duffy genotypes-46 C/C 163/180 (91)-46 C/T 14/180 (8)-46 T/T 2/180 (1)
Duffy phenotypes Duffy negative 163/180 (91)Duffy positive 17/180 (9)
©Fer
rata
Sto
rti F
ound
ation
No
com
mer
cial u
se
of multiple observations for the same individual. Thesedata included both acute and steady-state results. Analysisshowed significantly lower WBC and neutrophil countsfor Duffy negative patients (P=0.001). Interestingly, raisedCystatin C and lower estimated glomerular filtrationmeasurement using the Hoek formula12 were significantlyassociated with the DARC positive genotype (P=0.002 and0.001, respectively), although no association was foundwith macroalbuminuria as defined by a urine albumin:cre-atinine ratio over 30, in line with National Institute forHealth and Clinical Excellence (NICE) guidelines.13 TheWBC and neutrophil rise in the acute phase was propor-tional to the starting value. Duffy status had no additionaleffect on hematologic parameters in the acute phase.These results are summarized in Table 2.
Admission data were available for the whole cohort.Among the 272 patients, 112 (41%) had at least oneadmission during the 2-year study period. There was nodifference in the distribution of Duffy phenotypesbetween the admitted and non-admitted groups; 89% ofthe total cohort were Duffy negative compared to 87% ofthe admitted group. The effect of Duffy status was ana-lyzed using logistical regression. There were a total of 313
admissions ranging from 1 to 19 per patient (mean 3).Length of stay ranged from 1 to 74 days (mean 7). Sixty-nine of the 112 patients (61%) were readmitted within thestudy period (time to readmission 3-627 days, mean 158).There were no significant effects of Duffy status on lengthof stay or time to readmission.
Analysis of clinical complications was limited to the 180patients with Hb SS and Hb Sb0 as patients with Hb SCand Hb Sb+ tend to have a milder disease with fewer com-plications (Table 2). The groups were compared usingFisher’s exact test. Notably, the incidence of leg ulcers wassignificantly higher in Duffy positive compared to Duffynegative patients (P=0.04). Age, gender and alpha globingenotype had no effect on the frequency of leg ulcers. Wenoted a difference in leg ulcers between the Fy -ve and Fy+ve patients in the study by Afenyi-Annan et al. whencompared to our present study: the population studied byAfenyi-Annan et al. had 25% Fy +ve patients indicating agreater degree of European admixture. The number ofsickle-related complications also appeared to be signifi-cantly associated with Duffy status, but this effect disap-peared when corrected for age. All other associations werenon-significant.
haematologica 2013; 98:e88
LETTERS TO THE EDITOR
Table 3. Laboratory values in Duffy negative and Duffy positive patients. Difference between the groups is expressed as a percentage orabsolute difference except for Hb where absolute difference was used as the parameter.
Laboratory variable Mean value (range) Difference between P value(all genotypes n=272) Fy –ve Fy +ve groups (95%CI)
(n=243) (n=29)
WBC (x109/L)* 8.95 (3.31–18.86) 10.08 (4.99–20.91) 14% (4–26) 0.008
Neutrophils (x109/L)* 4.12 (1.75–9.64) 5.75 (2.69–13.17) 23% (9–38) 0.001
LDH (IU/L)* 385.74 (169.40–985.89) 435.47 (144.67–938.00) 7% (-4–19) 0.2
Reticulocyte count (x109/L)* 287.14 (67.30–592.48) 272.86 (110.48–477.43) -3% (-11–7) 0.6
Hb F (%)* 9.02 (0.2–31.1) 4.96 (1.5–12.9) -10% (-39–32) 0.6
Ferritin (ng/mL)* 505.89 (9.17–8163.67) 628.13 (43.67–3911.40) 49% (-15–125) 0.06
MDRD eGFR(ml/min/1.73m2)* 133.40 (2.93–376.09) 119.42 (65.03–264.16) -11.82% (-30.07–6.42) 0.2
Cystatin C* 0.91 (0.53–5.21) 1.08 (0.74–3.25) 20% (7–35) 0.002
Hoek formula eGFR* 97.20 (11.56–150.92) 83.42 (55.46–110.47) -16% (-25–-7) 0.001
Erythropoetin* 84.98 (12.02–401.00) 80.75 (26.56–249.08) -8% (-25–9) 0.3
Macroalbuminuria 54 (22%) 9 (30%) 1.57 (0.68–3.66 ) 0.06
Hb (g/dL) 9.41 (4.34–14.98) 9.40 (5.64–13.60) -0.20 (-0.67–0.28) N/S
Analysis of laboratory variables was corrected for age, sex, sickle genotype and alpha globin genotype, and for whether acute or steady-state. Fy +ve: Duffy positive; Fy –ve:
Duffy negative. *Variables log transformed prior to inclusion in the analysis. Macroalbuminuria is defined as an ACR >30.
Table 4. Clinical complications and end-organ damage in Duffy positive and Duffy negative patients.
Clinical complication Complications/ Data available PComplication/ Data available (% with complications)
Fy –ve (n = 163) Fy +ve (n = 17)
Chest crisis n = 40/157 36/142 (25) 4/15 (27) N/S
TRJet >2.5 m/s n = 28/134 26/123 (21) 2/11 (12) N/S
Sickle cell lung disease$n =23/127 21/115 (18) 2/12 (12) N/S
All respiratory complications n = 38/177 34/161 (21) 4/16 (25) N/S
Stroke n = 20/157 16/142 (11) 4/15 (27) N/S
All cerebral complications n = 30/177 25/161 (16) 5/16 (31) N/S
Leg ulcers n = 22/158 17/143 (12) 5/15 (33) 0.04
Avascular necrosis n = 31/149 25/133 (19) 5/16 (31) N/S
End-stage renal failure n = 6/159 5/144 (4) 1/15 (7) N/S
At least 1 complication n = 89/177 76/161 (47) 13/16 (81) 0.02
Limited to SCA n = 180. $Sickle cell lung disease diagnosed on CT findings.
of multiple observations for the same individual. Thesedata included both acute and steady-state results. Analysisshowed significantly lower WBC and neutrophil countsfor Duffy negative patients (P=0.001). Interestingly, raisedCystatin C and lower estimated glomerular filtrationmeasurement using the Hoek formula12 were significantlyassociated with the DARC positive genotype (P=0.002 and0.001, respectively), although no association was foundwith macroalbuminuria as defined by a urine albumin:cre-atinine ratio over 30, in line with National Institute forHealth and Clinical Excellence (NICE) guidelines.13 TheWBC and neutrophil rise in the acute phase was propor-tional to the starting value. Duffy status had no additionaleffect on hematologic parameters in the acute phase.These results are summarized in Table 2.
Admission data were available for the whole cohort.Among the 272 patients, 112 (41%) had at least oneadmission during the 2-year study period. There was nodifference in the distribution of Duffy phenotypesbetween the admitted and non-admitted groups; 89% ofthe total cohort were Duffy negative compared to 87% ofthe admitted group. The effect of Duffy status was ana-lyzed using logistical regression. There were a total of 313
admissions ranging from 1 to 19 per patient (mean 3).Length of stay ranged from 1 to 74 days (mean 7). Sixty-nine of the 112 patients (61%) were readmitted within thestudy period (time to readmission 3-627 days, mean 158).There were no significant effects of Duffy status on lengthof stay or time to readmission.
Analysis of clinical complications was limited to the 180patients with Hb SS and Hb Sb0 as patients with Hb SCand Hb Sb+ tend to have a milder disease with fewer com-plications (Table 2). The groups were compared usingFisher’s exact test. Notably, the incidence of leg ulcers wassignificantly higher in Duffy positive compared to Duffynegative patients (P=0.04). Age, gender and alpha globingenotype had no effect on the frequency of leg ulcers. Wenoted a difference in leg ulcers between the Fy -ve and Fy+ve patients in the study by Afenyi-Annan et al. whencompared to our present study: the population studied byAfenyi-Annan et al. had 25% Fy +ve patients indicating agreater degree of European admixture. The number ofsickle-related complications also appeared to be signifi-cantly associated with Duffy status, but this effect disap-peared when corrected for age. All other associations werenon-significant.
haematologica 2013; 98:e88
LETTERS TO THE EDITOR
Table 3. Laboratory values in Duffy negative and Duffy positive patients. Difference between the groups is expressed as a percentage orabsolute difference except for Hb where absolute difference was used as the parameter.
Laboratory variable Mean value (range) Difference between P value(all genotypes n=272) Fy –ve Fy +ve groups (95%CI)
(n=243) (n=29)
WBC (x109/L)* 8.95 (3.31–18.86) 10.08 (4.99–20.91) 14% (4–26) 0.008
Neutrophils (x109/L)* 4.12 (1.75–9.64) 5.75 (2.69–13.17) 23% (9–38) 0.001
LDH (IU/L)* 385.74 (169.40–985.89) 435.47 (144.67–938.00) 7% (-4–19) 0.2
Reticulocyte count (x109/L)* 287.14 (67.30–592.48) 272.86 (110.48–477.43) -3% (-11–7) 0.6
Hb F (%)* 9.02 (0.2–31.1) 4.96 (1.5–12.9) -10% (-39–32) 0.6
Ferritin (ng/mL)* 505.89 (9.17–8163.67) 628.13 (43.67–3911.40) 49% (-15–125) 0.06
MDRD eGFR(ml/min/1.73m2)* 133.40 (2.93–376.09) 119.42 (65.03–264.16) -11.82% (-30.07–6.42) 0.2
Cystatin C* 0.91 (0.53–5.21) 1.08 (0.74–3.25) 20% (7–35) 0.002
Hoek formula eGFR* 97.20 (11.56–150.92) 83.42 (55.46–110.47) -16% (-25–-7) 0.001
Erythropoetin* 84.98 (12.02–401.00) 80.75 (26.56–249.08) -8% (-25–9) 0.3
Macroalbuminuria 54 (22%) 9 (30%) 1.57 (0.68–3.66 ) 0.06
Hb (g/dL) 9.41 (4.34–14.98) 9.40 (5.64–13.60) -0.20 (-0.67–0.28) N/S
Analysis of laboratory variables was corrected for age, sex, sickle genotype and alpha globin genotype, and for whether acute or steady-state. Fy +ve: Duffy positive; Fy –ve:
Duffy negative. *Variables log transformed prior to inclusion in the analysis. Macroalbuminuria is defined as an ACR >30.
Table 4. Clinical complications and end-organ damage in Duffy positive and Duffy negative patients.
Clinical complication Complications/ Data available PComplication/ Data available (% with complications)
Fy –ve (n = 163) Fy +ve (n = 17)
Chest crisis n = 40/157 36/142 (25) 4/15 (27) N/S
TRJet >2.5 m/s n = 28/134 26/123 (21) 2/11 (12) N/S
Sickle cell lung disease$n =23/127 21/115 (18) 2/12 (12) N/S
All respiratory complications n = 38/177 34/161 (21) 4/16 (25) N/S
Stroke n = 20/157 16/142 (11) 4/15 (27) N/S
All cerebral complications n = 30/177 25/161 (16) 5/16 (31) N/S
Leg ulcers n = 22/158 17/143 (12) 5/15 (33) 0.04
Avascular necrosis n = 31/149 25/133 (19) 5/16 (31) N/S
End-stage renal failure n = 6/159 5/144 (4) 1/15 (7) N/S
At least 1 complication n = 89/177 76/161 (47) 13/16 (81) 0.02
Limited to SCA n = 180. $Sickle cell lung disease diagnosed on CT findings.

Neutropénies ethniques : autres polymorphismes ?
rs4065321 : 17q12, gène CSF3, R-GCSF • 1 étude européenne • 2 études japonaise GB/PNN plus bas
210 VOLUME 42 | NUMBER 3 | MARCH 2010 NATURE GENETICS
ARTI CLES
The recent progress in genome-wide association studies (GWAS) has
led to the identification of many loci associated with common diseases
as well as with quantitative traits. We report here a GWAS for a range
of hematological and biochemical traits. We used genome-wide SNP
data from ten cohorts including a total of ~14,700 Japanese individuals.
The genotypes were originally obtained as part of the BioBank Japan
project for ongoing GWAS. An advantage of our sample is that the
structure of the Japanese population has been extensively studied1.
Furthermore, individual data for factors that may confound the results
of the association studies were available, and we were able to adjust for
these factors.
RESULTS
The GWAS results are summarized in Table 1 (hematological traits)
and Table 2 (biochemical traits). Quantile-quantile (Q-Q) plots
are shown in Supplementary Figure 1 and Manhattan plots are
shown in Supplementary Figure 2. Regional plots are shown in
Supplementary Figure 3.
White blood cell count
GWAS for white blood cell count (WBC) revealed four newly associ-
ated loci, including rs4895441 in the HBS1L-MYB locus (P = 1.67 ×
10−9), rs3094212 in CDSN-PSORS1C1 in the human MHC region
(P = 6.76 × 10−9), rs445 in CDK6 (P = 2.44 × 10−8) and rs12313946
in the RAP1B-NUP107-SLC35E3-MDM2 locus (Table 1). We also
confirmed the previously reported association of WBC with rs4065321
in the GSDM1-PSMD3-CSF3-MED24-THRA locus2 (P = 2.94 ×
10−14), which includes the CSF3 gene, encoding granulocyte colony-
stimulating factor (Table 1).
Variants in the HBS1L-MYB region were initially reported to be
associated with fetal hemoglobin (HbF) levels in adults3. Subsequently,
variants in the HBS1L-MYB locus were reported to be associated with
red blood cell, platelet and monocyte counts4. In our study, we repli-
cated this association with WBC in a larger data set.
The WBC-associated SNP with the third-lowest P-value, rs3094212,
is located in the human MHC region in 6p21. The fourth-lowest
P-value was observed for rs445 in CDK6 (Table 1). CDK6 encodes
cyclin-dependent kinase-6, which is a regulator of cell cycle progres-
sion. The SNP with the fifth-lowest P-value was rs12313946, which is
located in a linkage disequilibrium (LD) block that includes RAP1B,
NUP107, SLC35E3 and MDM2 (Supplementary Fig. 3a).
Red blood cell traits
We performed GWAS for the following six erythrocyte-related traits:
red blood cell count (RBC), hemoglobin concentration (Hb), hemato-
crit (Ht), mean corpuscular volume (MCV), mean corpuscular
hemoglobin (MCH) and mean corpuscular hemoglobin concentra-
tion (MCHC). In total, we found 8 RBC loci, 2 Hb loci, 2 Ht loci,
15 MCV loci, 15 MCH loci and 7 MCHC loci (Table 1). We confirmed
the previously reported associations of erythrocyte-related traits with
the following ten loci2,4–7: HBS1L-MYB, TMPRSS6, PDGFRA-HK1,
CCND3, RCL1, MARCH8, CITED2, TFRC-ZDHHC19, CD164 and
HBA2-HBA1-LUC7L-ITFG3-RGS11 (Table 1). We also found some
associations between these loci and erythrocyte-related traits that, to
our knowledge, have not been reported previously, including PDGFRA-
HK1 with RBC and MCH, CCND3 with RBC, CD164 with RBC and
MCH, PRKCE with RBC, MARCH8 with MCH, and TYMP with MCH.
Regional plots for these loci are shown in Supplementary Figure 3b.
Genome-wide association study of hematological and biochemical traits in a Japanese population
Yoichiro Kamatani1,2, Koichi Matsuda1, Yukinori Okada3, Michiaki Kubo4, Naoya Hosono4, Yataro Daigo1,2,
Yusuke Nakamura1,5 & Naoyuki Kamatani3
We report genome-wide association studies for hematological and biochemical traits from ~14,700 Japanese individuals. We
identified 60 associations for 8 hematological traits and 29 associations for 12 biochemical traits at genome-wide significance
levels (P < 5 × 10–8). Of these, 46 associations were new to this study and 43 replicated previous reports. We compared these
associated loci with those reported in similar GWAS in European populations. When the minor allele frequency was >10% in
the Japanese population, 32 (94.1%) and 31 (91.2%) of the 34 hematological loci previously reported to be associated in a
European population were replicated with P-values less than 0.05 and 0.01, respectively, and 31 (73.8%) and 27 (64.3%) of the
42 European biochemical loci were replicated.
1Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science and 2Department of Medical Genome Sciences, Graduate School of Frontier
Sciences; the University of Tokyo, Tokyo, Japan. 3Laboratory for Statistical Analysis, 4Laboratory for Genotyping Development and 5Center for Genomic Medicine,
RIKEN, Kanagawa, Japan. Correspondence should be addressed to N.K. ([email protected]).
Received 25 August 2009; accepted 22 December 2009; published online 7 February 2010; doi:10.1038/ng.53 1
© 2
010
Na
ture
Am
eri
ca
, In
c. A
ll r
igh
ts r
es
erv
ed
.
210 VOLUME 42 | NUMBER 3 | MARCH 2010 NATURE GENETICS
ARTI CLES
The recent progress in genome-wide association studies (GWAS) has
led to the identification of many loci associated with common diseases
as well as with quantitative traits. We report here a GWAS for a range
of hematological and biochemical traits. We used genome-wide SNP
data from ten cohorts including a total of ~14,700 Japanese individuals.
The genotypes were originally obtained as part of the BioBank Japan
project for ongoing GWAS. An advantage of our sample is that the
structure of the Japanese population has been extensively studied1.
Furthermore, individual data for factors that may confound the results
of the association studies were available, and we were able to adjust for
these factors.
RESULTS
The GWAS results are summarized in Table 1 (hematological traits)
and Table 2 (biochemical traits). Quantile-quantile (Q-Q) plots
are shown in Supplementary Figure 1 and Manhattan plots are
shown in Supplementary Figure 2. Regional plots are shown in
Supplementary Figure 3.
White blood cell count
GWAS for white blood cell count (WBC) revealed four newly associ-
ated loci, including rs4895441 in the HBS1L-MYB locus (P = 1.67 ×
10−9), rs3094212 in CDSN-PSORS1C1 in the human MHC region
(P = 6.76 × 10−9), rs445 in CDK6 (P = 2.44 × 10−8) and rs12313946
in the RAP1B-NUP107-SLC35E3-MDM2 locus (Table 1). We also
confirmed the previously reported association of WBC with rs4065321
in the GSDM1-PSMD3-CSF3-MED24-THRA locus2 (P = 2.94 ×
10−14), which includes the CSF3 gene, encoding granulocyte colony-
stimulating factor (Table 1).
Variants in the HBS1L-MYB region were initially reported to be
associated with fetal hemoglobin (HbF) levels in adults3. Subsequently,
variants in the HBS1L-MYB locus were reported to be associated with
red blood cell, platelet and monocyte counts4. In our study, we repli-
cated this association with WBC in a larger data set.
The WBC-associated SNP with the third-lowest P-value, rs3094212,
is located in the human MHC region in 6p21. The fourth-lowest
P-value was observed for rs445 in CDK6 (Table 1). CDK6 encodes
cyclin-dependent kinase-6, which is a regulator of cell cycle progres-
sion. The SNP with the fifth-lowest P-value was rs12313946, which is
located in a linkage disequilibrium (LD) block that includes RAP1B,
NUP107, SLC35E3 and MDM2 (Supplementary Fig. 3a).
Red blood cell traits
We performed GWAS for the following six erythrocyte-related traits:
red blood cell count (RBC), hemoglobin concentration (Hb), hemato-
crit (Ht), mean corpuscular volume (MCV), mean corpuscular
hemoglobin (MCH) and mean corpuscular hemoglobin concentra-
tion (MCHC). In total, we found 8 RBC loci, 2 Hb loci, 2 Ht loci,
15 MCV loci, 15 MCH loci and 7 MCHC loci (Table 1). We confirmed
the previously reported associations of erythrocyte-related traits with
the following ten loci2,4–7: HBS1L-MYB, TMPRSS6, PDGFRA-HK1,
CCND3, RCL1, MARCH8, CITED2, TFRC-ZDHHC19, CD164 and
HBA2-HBA1-LUC7L-ITFG3-RGS11 (Table 1). We also found some
associations between these loci and erythrocyte-related traits that, to
our knowledge, have not been reported previously, including PDGFRA-
HK1 with RBC and MCH, CCND3 with RBC, CD164 with RBC and
MCH, PRKCE with RBC, MARCH8 with MCH, and TYMP with MCH.
Regional plots for these loci are shown in Supplementary Figure 3b.
Genome-wide association study of hematological and biochemical traits in a Japanese population
Yoichiro Kamatani1,2, Koichi Matsuda1, Yukinori Okada3, Michiaki Kubo4, Naoya Hosono4, Yataro Daigo1,2,
Yusuke Nakamura1,5 & Naoyuki Kamatani3
We report genome-wide association studies for hematological and biochemical traits from ~14,700 Japanese individuals. We
identified 60 associations for 8 hematological traits and 29 associations for 12 biochemical traits at genome-wide significance
levels (P < 5 × 10–8). Of these, 46 associations were new to this study and 43 replicated previous reports. We compared these
associated loci with those reported in similar GWAS in European populations. When the minor allele frequency was >10% in
the Japanese population, 32 (94.1%) and 31 (91.2%) of the 34 hematological loci previously reported to be associated in a
European population were replicated with P-values less than 0.05 and 0.01, respectively, and 31 (73.8%) and 27 (64.3%) of the
42 European biochemical loci were replicated.
1Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science and 2Department of Medical Genome Sciences, Graduate School of Frontier
Sciences; the University of Tokyo, Tokyo, Japan. 3Laboratory for Statistical Analysis, 4Laboratory for Genotyping Development and 5Center for Genomic Medicine,
RIKEN, Kanagawa, Japan. Correspondence should be addressed to N.K. ([email protected]).
Received 25 August 2009; accepted 22 December 2009; published online 7 February 2010; doi:10.1038/ng.531©
2010
Na
ture
Am
eri
ca
, In
c. A
ll r
igh
ts r
es
erv
ed
.
210 VOLUME 42 | NUMBER 3 | MARCH 2010 NATURE GENETICS
ARTI CLES
The recent progress in genome-wide association studies (GWAS) has
led to the identification of many loci associated with common diseases
as well as with quantitative traits. We report here a GWAS for a range
of hematological and biochemical traits. We used genome-wide SNP
data from ten cohorts including a total of ~14,700 Japanese individuals.
The genotypes were originally obtained as part of the BioBank Japan
project for ongoing GWAS. An advantage of our sample is that the
structure of the Japanese population has been extensively studied1.
Furthermore, individual data for factors that may confound the results
of the association studies were available, and we were able to adjust for
these factors.
RESULTS
The GWAS results are summarized in Table 1 (hematological traits)
and Table 2 (biochemical traits). Quantile-quantile (Q-Q) plots
are shown in Supplementary Figure 1 and Manhattan plots are
shown in Supplementary Figure 2. Regional plots are shown in
Supplementary Figure 3.
White blood cell count
GWAS for white blood cell count (WBC) revealed four newly associ-
ated loci, including rs4895441 in the HBS1L-MYB locus (P = 1.67 ×
10−9), rs3094212 in CDSN-PSORS1C1 in the human MHC region
(P = 6.76 × 10−9), rs445 in CDK6 (P = 2.44 × 10−8) and rs12313946
in the RAP1B-NUP107-SLC35E3-MDM2 locus (Table 1). We also
confirmed the previously reported association of WBC with rs4065321
in the GSDM1-PSMD3-CSF3-MED24-THRA locus2 (P = 2.94 ×
10−14), which includes the CSF3 gene, encoding granulocyte colony-
stimulating factor (Table 1).
Variants in the HBS1L-MYB region were initially reported to be
associated with fetal hemoglobin (HbF) levels in adults3. Subsequently,
variants in the HBS1L-MYB locus were reported to be associated with
red blood cell, platelet and monocyte counts4. In our study, we repli-
cated this association with WBC in a larger data set.
The WBC-associated SNP with the third-lowest P-value, rs3094212,
is located in the human MHC region in 6p21. The fourth-lowest
P-value was observed for rs445 in CDK6 (Table 1). CDK6 encodes
cyclin-dependent kinase-6, which is a regulator of cell cycle progres-
sion. The SNP with the fifth-lowest P-value was rs12313946, which is
located in a linkage disequilibrium (LD) block that includes RAP1B,
NUP107, SLC35E3 and MDM2 (Supplementary Fig. 3a).
Red blood cell traits
We performed GWAS for the following six erythrocyte-related traits:
red blood cell count (RBC), hemoglobin concentration (Hb), hemato-
crit (Ht), mean corpuscular volume (MCV), mean corpuscular
hemoglobin (MCH) and mean corpuscular hemoglobin concentra-
tion (MCHC). In total, we found 8 RBC loci, 2 Hb loci, 2 Ht loci,
15 MCV loci, 15 MCH loci and 7 MCHC loci (Table 1). We confirmed
the previously reported associations of erythrocyte-related traits with
the following ten loci2,4–7: HBS1L-MYB, TMPRSS6, PDGFRA-HK1,
CCND3, RCL1, MARCH8, CITED2, TFRC-ZDHHC19, CD164 and
HBA2-HBA1-LUC7L-ITFG3-RGS11 (Table 1). We also found some
associations between these loci and erythrocyte-related traits that, to
our knowledge, have not been reported previously, including PDGFRA-
HK1 with RBC and MCH, CCND3 with RBC, CD164 with RBC and
MCH, PRKCE with RBC, MARCH8 with MCH, and TYMP with MCH.
Regional plots for these loci are shown in Supplementary Figure 3b.
Genome-wide association study of hematological and biochemical traits in a Japanese population
Yoichiro Kamatani1,2, Koichi Matsuda1, Yukinori Okada3, Michiaki Kubo4, Naoya Hosono4, Yataro Daigo1,2,
Yusuke Nakamura1,5 & Naoyuki Kamatani3
We report genome-wide association studies for hematological and biochemical traits from ~14,700 Japanese individuals. We
identified 60 associations for 8 hematological traits and 29 associations for 12 biochemical traits at genome-wide significance
levels (P < 5 × 10–8). Of these, 46 associations were new to this study and 43 replicated previous reports. We compared these
associated loci with those reported in similar GWAS in European populations. When the minor allele frequency was >10% in
the Japanese population, 32 (94.1%) and 31 (91.2%) of the 34 hematological loci previously reported to be associated in a
European population were replicated with P-values less than 0.05 and 0.01, respectively, and 31 (73.8%) and 27 (64.3%) of the
42 European biochemical loci were replicated.
1Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science and 2Department of Medical Genome Sciences, Graduate School of Frontier
Sciences; the University of Tokyo, Tokyo, Japan. 3Laboratory for Statistical Analysis, 4Laboratory for Genotyping Development and 5Center for Genomic Medicine,
RIKEN, Kanagawa, Japan. Correspondence should be addressed to N.K. ([email protected]).
Received 25 August 2009; accepted 22 December 2009; published online 7 February 2010; doi:10.1038/ng.531
© 2
010
Na
ture
Am
eri
ca, In
c. A
ll r
igh
ts r
es
erv
ed
.

rs9131 : 4 q13, CXL2 Genome-Wide Association Study of White Blood CellCount in 16,388 African Americans: the ContinentalOrigins and Genetic Epidemiology Network (COGENT)
Alexander P. Reiner1,2. * , Guillaume Lett re3,4. , Michael A. Nalls5. , Santhi K. Ganesh6. , Rasika Mathias7. ,
Melissa A. Aust in2,8. , Eric Dean9. , Sampath Arepalli5, Angela Brit ton5, Zhao Chen10, David Couper11, J.
David Curb12, Charles B. Eaton13, Myriam Fornage14, Struan F. A. Grant15, Tamara B. Harris16, Dena
Hernandez5, Naoyuki Kamat ini17, Brendan J. Keat ing15, Michiak i Kubo18, Andrea LaCroix1,2, Leslie A.
Lange19, Simin Liu20, Kurt Lohman21, Yan Meng22, Emile R. Mohler III23, Solomon Musani24, Yusuke
Nakamura25, Christopher J. O’Donnell26,27, Yukinor i Okada17, Cameron D. Palmer22, George J.
Papanicolaou26, Kushang V. Patel16, Andrew B. Singleton5, Atsushi Takahashi17, Hua Tang28, Herman A.
Taylor Jr.29,30, Kent Taylor31, Cynthia Thomson32, Lisa R. Yanek7, Lingyao Yang33, Elad Ziv9, Alan B.
Zonderman34, Aaron R. Folsom35" , Michele K. Evans36" , Yongmei Liu21" , Diane M. Becker7" , Beverly M.
Snively33" , James G. Wilson37" *
1 Department of Epidemiology, University of Washington, Seattle, Washington, United States of America, 2 Division of Public Health Sciences, Fred Hutchinson Cancer
Research Center, Seattle, Washington, United States of America, 3 Montreal Heart Institute, Montreal, Canada, 4 Departement de Medecine, Universite de Montreal,
Montreal, Canada, 5 Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, United States of America, 6 Division of
Cardiovascular Medicine, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, United States of America, 7 Department of Medicine, The Johns
Hopkins University School of Medicine, Baltimore, Maryland, United States of America, 8 Department of Epidemiology and Institute for Public Health Genetics, School of
Public Health, University of Washington, Seattle, Washington, United States of America, 9 Department of Medicine, University of California San Francisco, San Francisco,
California, United States of America, 10 Division of Epidemiology and Biostatistics, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson,
Arizona, United States of America, 11 Department of Epidemiology, University of North Carolina School of Public Health, Chapel Hill, North Carolina, United States of
America, 12 Department of Geriatric Medicine, John A. Burns School of Medicine, University of Hawaii, Honolulu, Hawaii, United States of America, 13 Center for Primary
Care and Prevention, Alpert Medical School of Brown University, Providence, Rhode Island, United States of America, 14 Houston Institute of Molecular Medicine,
University of Texas, Houston, Texas, United States of America, 15 Center for Applied Genomics, Division of Human Genetics, Children’s Hospital of Philadelphia Research
Institute, Philadelphia, Pennsylvania, United States of America, 16 Laboratory for Epidemiology, Demography, and Biometry, National Institute on Aging, National
Institutes of Health, Baltimore, Maryland, United States of America, 17 Laboratory for Statistical Analysis, Center for Genomic Medicine (CGM), Institute of Physical and
Chemical Research (RIKEN), Yokohama, Japan, 18 Laboratory for Genotyping Development, CGM, RIKEN, Yokohama, Japan, 19 Department of Genetics, University of
North Carolina, Chapel Hill, North Carolina, United States of America, 20 Departments of Epidemiology and Medicine, University of California Los Angeles, Los Angeles,
California, United States of America, 21 Center for Human Genomics, Department of Epidemiology and Prevention, Division of Public Health Sciences, Wake Forest
University School of Medicine, Winston-Salem, North Carolina, United States of America, 22 Program in Medical and Population Genetics, Broad Institute, Cambridge,
Massachusetts, United States of America, 23 Cardiovascular Division, Vascular Medicine Section, Department of Medicine, University of Pennsylvania School of Medicine,
Philadelphia, Pennsylvania, United States of America, 24 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States of America,
25 Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science, University of Tokyo, Tokyo, Japan, 26 National Heart, Lung, and Blood Institute
(NHLBI), Division of Cardiovascular Sciences, Bethesda, Maryland, United States of America, 27 NHLBI’s Framingham Heart Study, Framingham, Massachusetts, United
States of America, 28 Department of Genetics, Stanford University School of Medicine, Stanford, California, United States of America, 29 Jackson State University,
Tougaloo College, Jackson, Mississippi, United States of America, 30 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States
of America, 31 Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, California, United States of America, 32 Nutritional Sciences, Arizona Cancer Center,
University of Arizona, Tucson, Arizona, United States of America, 33 Department of Biostatistical Sciences, Division of Public Health Sciences, Wake Forest School of
Medicine, Winston-Salem, North Carolina, United States of America, 34 Laboratory of Personality and Cognition, National Institute on Aging, National Institutes of Health,
Baltimore, Maryland, United States of America, 35 Division of Epidemiology and Community Health, University of Minnesota, Minneapolis, Minnesota, United States of
America, 36 Health Disparities Research Section, Clinical Research Branch, National Institute on Aging, National Institutes of Health, Baltimore, Maryland, United States of
America, 37 Department of Physiology and Biophysics, University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Total white blood cell (WBC) and neutrophil counts are lower among individuals of African descent due to the commonAfrican-derived ‘‘null’’ variant of the Duffy Antigen Receptor for Chemokines (DARC) gene. Additional common geneticpolymorphisms were recently associated with total WBC and WBC sub-type levels in European and Japanese populations.No additional loci that account for WBC variability have been identified in African Americans. In order to address this, weperformed a large genome-wide association study (GWAS) of total WBCand cell subtype counts in 16,388 African-Americanparticipants from 7 population-based cohorts available in the Continental Origins and Genetic Epidemiology Network.In addition to the DARC locus on chromosome 1q23, we identified two other regions (chromosomes 4q13 and 16q22)
PLoS Genetics | www.plosgenetics.org 1 June 2011 | Volume 7 | Issue 6 | e1002108
Genome-Wide Association Study of White Blood CellCount in 16,388 African Americans: the ContinentalOrigins and Genetic Epidemiology Network (COGENT)
Alexander P. Reiner1,2. * , Guillaume Lettre3,4. , Michael A. Nalls5. , Santhi K. Ganesh6. , Rasika Mathias7. ,
Melissa A. Aust in2,8. , Eric Dean9. , Sampath Arepalli5, Angela Brit ton5, Zhao Chen10, David Couper11, J.
David Curb12, Charles B. Eaton13, Myriam Fornage14, Struan F. A. Grant15, Tamara B. Harris16, Dena
Hernandez5, Naoyuki Kamatini17, Brendan J. Keat ing15, Michiak i Kubo18, Andrea LaCroix1,2, Leslie A.
Lange19, Simin Liu20, Kurt Lohman21, Yan Meng22, Emile R. Mohler III23, Solomon Musani24, Yusuke
Nakamura25, Christopher J. O’Donnell26,27, Yukinori Okada17, Cameron D. Palmer22, George J.
Papanicolaou26, Kushang V. Patel16, Andrew B. Singleton5, Atsushi Takahashi17, Hua Tang28, Herman A.
Taylor Jr.29,30, Kent Taylor31, Cynthia Thomson32, Lisa R. Yanek7, Lingyao Yang33, Elad Ziv9, Alan B.
Zonderman34, Aaron R. Folsom35" , Michele K. Evans36" , Yongmei Liu21" , Diane M. Becker7" , Beverly M.
Snively33" , James G. Wilson37" *
1 Department of Epidemiology, University of Washington, Seattle, Washington, United States of America, 2 Division of Public Health Sciences, Fred Hutchinson Cancer
Research Center, Seattle, Washington, United States of America, 3 Montreal Heart Institute, Montreal, Canada, 4 Departement de Medecine, Universite de Montreal,
Montreal, Canada, 5 Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, United States of America, 6 Division of
Cardiovascular Medicine, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, United States of America, 7 Department of Medicine, The Johns
Hopkins University School of Medicine, Baltimore, Maryland, United States of America, 8 Department of Epidemiology and Institute for Public Health Genetics, School of
Public Health, University of Washington, Seattle, Washington, United States of America, 9 Department of Medicine, University of California San Francisco, San Francisco,
California, United States of America, 10 Division of Epidemiology and Biostatistics, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson,
Arizona, United States of America, 11 Department of Epidemiology, University of North Carolina School of Public Health, Chapel Hill, North Carolina, United States of
America, 12 Department of Geriatric Medicine, John A. Burns School of Medicine, University of Hawaii, Honolulu, Hawaii, United States of America, 13 Center for Primary
Care and Prevention, Alpert Medical School of Brown University, Providence, Rhode Island, United States of America, 14 Houston Institute of Molecular Medicine,
University of Texas, Houston, Texas, United States of America, 15 Center for Applied Genomics, Division of Human Genetics, Children’s Hospital of Philadelphia Research
Institute, Philadelphia, Pennsylvania, United States of America, 16 Laboratory for Epidemiology, Demography, and Biometry, National Institute on Aging, National
Institutes of Health, Baltimore, Maryland, United States of America, 17 Laboratory for Statistical Analysis, Center for Genomic Medicine (CGM), Institute of Physical and
Chemical Research (RIKEN), Yokohama, Japan, 18 Laboratory for Genotyping Development, CGM, RIKEN, Yokohama, Japan, 19 Department of Genetics, University of
North Carolina, Chapel Hill, North Carolina, United States of America, 20 Departments of Epidemiology and Medicine, University of California Los Angeles, Los Angeles,
California, United States of America, 21 Center for Human Genomics, Department of Epidemiology and Prevention, Division of Public Health Sciences, Wake Forest
University School of Medicine, Winston-Salem, North Carolina, United States of America, 22 Program in Medical and Population Genetics, Broad Institute, Cambridge,
Massachusetts, United States of America, 23 Cardiovascular Division, Vascular Medicine Section, Department of Medicine, University of Pennsylvania School of Medicine,
Philadelphia, Pennsylvania, United States of America, 24 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States of America,
25 Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science, University of Tokyo, Tokyo, Japan, 26 National Heart, Lung, and Blood Institute
(NHLBI), Division of Cardiovascular Sciences, Bethesda, Maryland, United States of America, 27 NHLBI’s Framingham Heart Study, Framingham, Massachusetts, United
States of America, 28 Department of Genetics, Stanford University School of Medicine, Stanford, California, United States of America, 29 Jackson State University,
Tougaloo College, Jackson, Mississippi, United States of America, 30 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States
of America, 31 Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, California, United States of America, 32 Nutritional Sciences, Arizona Cancer Center,
University of Arizona, Tucson, Arizona, United States of America, 33 Department of Biostatistical Sciences, Division of Public Health Sciences, Wake Forest School of
Medicine, Winston-Salem, North Carolina, United States of America, 34 Laboratory of Personality and Cognition, National Institute on Aging, National Institutes of Health,
Baltimore, Maryland, United States of America, 35 Division of Epidemiology and Community Health, University of Minnesota, Minneapolis, Minnesota, United States of
America, 36 Health Disparities Research Section, Clinical Research Branch, National Institute on Aging, National Institutes of Health, Baltimore, Maryland, United States of
America, 37 Department of Physiology and Biophysics, University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Total white blood cell (WBC) and neutrophil counts are lower among individuals of African descent due to the commonAfrican-derived ‘‘null’’ variant of the Duffy Antigen Receptor for Chemokines (DARC) gene. Additional common geneticpolymorphisms were recently associated with total WBC and WBC sub-type levels in European and Japanese populations.No additional loci that account for WBC variability have been identified in African Americans. In order to address this, weperformed a large genome-wide association study (GWAS) of total WBCand cell subtype counts in 16,388 African-Americanparticipants from 7 population-based cohorts available in the Continental Origins and Genetic Epidemiology Network.In addition to the DARC locus on chromosome 1q23, we identified two other regions (chromosomes 4q13 and 16q22)
PLoS Genetics | www.plosgenetics.org 1 June 2011 | Volume 7 | Issue 6 | e1002108
Genome-Wide Association Study of White Blood CellCount in 16,388 African Americans: the ContinentalOrigins and Genetic Epidemiology Network (COGENT)
Alexander P. Reiner1,2. * , Guillaume Lettre3,4. , Michael A. Nalls5. , Santhi K. Ganesh6. , Rasika Mathias7. ,
Melissa A. Aust in2,8. , Eric Dean9. , Sampath Arepalli5, Angela Brit ton5, Zhao Chen10, David Couper11, J.
David Curb12, Charles B. Eaton13, Myriam Fornage14, Struan F. A. Grant15, Tamara B. Harris16, Dena
Hernandez5, Naoyuki Kamat ini17, Brendan J. Keat ing15, Michiak i Kubo18, Andrea LaCroix1,2, Leslie A.
Lange19, Simin Liu20, Kurt Lohman21, Yan Meng22, Emile R. Mohler III23, Solomon Musani24, Yusuke
Nakamura25, Christopher J. O’Donnell26,27, Yukinor i Okada17, Cameron D. Palmer22, George J.
Papanicolaou26, Kushang V. Patel16, Andrew B. Singleton5, Atsushi Takahashi17, Hua Tang28, Herman A.
Taylor Jr.29,30, Kent Taylor31, Cynthia Thomson32, Lisa R. Yanek7, Lingyao Yang33, Elad Ziv9, Alan B.
Zonderman34, Aaron R. Folsom35" , Michele K. Evans36" , Yongmei Liu21" , Diane M. Becker7" , Beverly M.
Snively33" , James G. Wilson37" *
1 Department of Epidemiology, University of Washington, Seattle, Washington, United States of America, 2 Division of Public Health Sciences, Fred Hutchinson Cancer
Research Center, Seattle, Washington, United States of America, 3 Montreal Heart Institute, Montreal, Canada, 4 Departement de Medecine, Universite de Montreal,
Montreal, Canada, 5 Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland, United States of America, 6 Division of
Cardiovascular Medicine, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, United States of America, 7 Department of Medicine, The Johns
Hopkins University School of Medicine, Baltimore, Maryland, United States of America, 8 Department of Epidemiology and Institute for Public Health Genetics, School of
Public Health, University of Washington, Seattle, Washington, United States of America, 9 Department of Medicine, University of California San Francisco, San Francisco,
California, United States of America, 10 Division of Epidemiology and Biostatistics, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson,
Arizona, United States of America, 11 Department of Epidemiology, University of North Carolina School of Public Health, Chapel Hill, North Carolina, United States of
America, 12 Department of Geriatric Medicine, John A. Burns School of Medicine, University of Hawaii, Honolulu, Hawaii, United States of America, 13 Center for Primary
Care and Prevention, Alpert Medical School of Brown University, Providence, Rhode Island, United States of America, 14 Houston Institute of Molecular Medicine,
University of Texas, Houston, Texas, United States of America, 15 Center for Applied Genomics, Division of Human Genetics, Children’s Hospital of Philadelphia Research
Institute, Philadelphia, Pennsylvania, United States of America, 16 Laboratory for Epidemiology, Demography, and Biometry, National Institute on Aging, National
Institutes of Health, Baltimore, Maryland, United States of America, 17 Laboratory for Statistical Analysis, Center for Genomic Medicine (CGM), Institute of Physical and
Chemical Research (RIKEN), Yokohama, Japan, 18 Laboratory for Genotyping Development, CGM, RIKEN, Yokohama, Japan, 19 Department of Genetics, University of
North Carolina, Chapel Hill, North Carolina, United States of America, 20 Departments of Epidemiology and Medicine, University of California Los Angeles, Los Angeles,
California, United States of America, 21 Center for Human Genomics, Department of Epidemiology and Prevention, Division of Public Health Sciences, Wake Forest
University School of Medicine, Winston-Salem, North Carolina, United States of America, 22 Program in Medical and Population Genetics, Broad Institute, Cambridge,
Massachusetts, United States of America, 23 Cardiovascular Division, Vascular Medicine Section, Department of Medicine, University of Pennsylvania School of Medicine,
Philadelphia, Pennsylvania, United States of America, 24 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States of America,
25 Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science, University of Tokyo, Tokyo, Japan, 26 National Heart, Lung, and Blood Institute
(NHLBI), Division of Cardiovascular Sciences, Bethesda, Maryland, United States of America, 27 NHLBI’s Framingham Heart Study, Framingham, Massachusetts, United
States of America, 28 Department of Genetics, Stanford University School of Medicine, Stanford, California, United States of America, 29 Jackson State University,
Tougaloo College, Jackson, Mississippi, United States of America, 30 Department of Medicine, University of Mississippi Medical Center, Jackson, Mississippi, United States
of America, 31 Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, California, United States of America, 32 Nutritional Sciences, Arizona Cancer Center,
University of Arizona, Tucson, Arizona, United States of America, 33 Department of Biostatistical Sciences, Division of Public Health Sciences, Wake Forest School of
Medicine, Winston-Salem, North Carolina, United States of America, 34 Laboratory of Personality and Cognition, National Institute on Aging, National Institutes of Health,
Baltimore, Maryland, United States of America, 35 Division of Epidemiology and Community Health, University of Minnesota, Minneapolis, Minnesota, United States of
America, 36 Health Disparities Research Section, Clinical Research Branch, National Institute on Aging, National Institutes of Health, Baltimore, Maryland, United States of
America, 37 Department of Physiology and Biophysics, University of Mississippi Medical Center, Jackson, Mississippi, United States of America
Abst ract
Total white blood cell (WBC) and neutrophil counts are lower among individuals of African descent due to the commonAfrican-derived ‘‘null’’ variant of the Duffy Antigen Receptor for Chemokines (DARC) gene. Additional common geneticpolymorphisms were recently associated with total WBC and WBC sub-type levels in European and Japanese populations.No additional loci that account for WBC variability have been identified in African Americans. In order to address this, weperformed a large genome-wide association study (GWAS) of total WBCand cell subtype counts in 16,388 African-Americanparticipants from 7 population-based cohorts available in the Continental Origins and Genetic Epidemiology Network.In addition to the DARC locus on chromosome 1q23, we identified two other regions (chromosomes 4q13 and 16q22)
PLoS Genetics | www.plosgenetics.org 1 June 2011 | Volume 7 | Issue 6 | e1002108
rs12149261 and every other typed SNP in the genome. There
was reduced local LD within the chromosome 16 duplicated
region, relative to the surrounding chromosome 16 SNP (FigureS5). Three SNPshad r-squared valuesof . 0.20 with rs12149261:
one located 20 kb away on chromosome 16 in the HYDIN gene
(rs1774524; r2= 0.27), and two located on chromosome 1 at, 120 Mb near the HYDIN paralogue (rs12087334 and
rs4659245; r2= 0.25 and 0.22, respectively). Moreover, com-bined analysis of the 4 cohorts typed on the Affymetrix GWA
platform showed that the chromosome 16q22 association signalat rs12149261 (P= 2.126 102 18) was completely abolished after
conditioning on chromosome 1 DARC rs2814778 (P= 0.36).
While defects in theHYDIN gene result in hydrocephalus[19,20],this genomic region has not previously been associated with
WBC. Together, these results demonstrate that the chromosome
16 HYDIN association finding is most likely a probe cross-hybridization artifact due to inter-chromosomal sequence simi-
larity with the duplicated segment on chromosome 1q21 near the
DARC region and that the polymorphisms associated with WBC
in the studies using the Affymetrix arrays actually map to the
chromosome 1 region.
Discovery of a novel CXCL2 association finding on
chromosome 4q13 and replication in other ethnic
populations
A novel SNP association on chromosome 4q13 was identified
in our African-American WBC discovery GWAS. The lead SNP
rs9131 is located in the 39 UTR of the CXCL2 gene, which
encodes a macrophage-derived chemotactic cytokine for
polymorphonuclear leukocytes. In African Americans, the
minor T allele (MAF= 23%) was associated with lower WBC.
Several additional SNPs in the chromosome 4 chemokine gene
cluster had P-values ranging from 102 5 to 102 7, including
rs2367291 located upstream of CXCL1 (Figure 3A) Further
adjustment for rs9131, however, abolished these associations
(data not shown). Based on HapMap phase 2 and 1000 genomes
data, rs9131 is in perfect LD with 7 other inter-genic SNPs in
Figure 2. Manhattan plot of meta-analysis P-values in GWAS for total WBC count. Horizontal axis indicates chromosomal position. Verticalaxis indicates 2 log10 P-values from inverse variance-weighted fixed effects meta-analysis. The red horizontal line indicates the genome-widesignificance threshold of P= 2.56 102 8. Association signals are present at 1q23, 4q13, and 16q22. The P-values for the broad chromosome 1 signal aretruncated at 102 20. This region spans nearly 90 Mb on both arms of chromosome 1 and results artifactually in two apparently distinct peaks becauseof the lack of genotyped or imputed SNPs around the centromere.doi:10.1371/journal.pgen.1002108.g002
Table 3. Meta-analysis results of genome-wide significant SNPs for white blood cell count subtypes.
Cell type Chromosome 1q23 DARC rs2814778 T allele Chromosome 4q13 CXCL2 rs9131 T allele
N Effect size (Standard Error) P-value N Effect size (Standard Error) P-value
Neutrophils 5609 +0.305 (0.009) 1.06 102 237 7353 2 0.038 (0.008) 1.56 102 6
Lymphocytes 5642 +0.020 (0.007) 3.86 102 3 7390 +0.010 (0.005) 0.06
Monocytes 5593 +0.048 (0.004) 6.06 102 27 7330 2 0.004 (0.004) 0.23
Eosinophils 5411 +0.012 (0.003) 8.66 102 5 6402 2 0.0005 (0.003) 0.85
Basophils 5104 +0.002 (0.0008) 3.56 102 3 6052 2 0.0007 (0.0007) 0.27
Effect size represents the effect of a minor allele on natural log-transformed white blood cell count.
doi:10.1371/journal.pgen.1002108.t003
GWAS of WBC in African Americans
PLoS Genetics | www.plosgenetics.org 5 June 2011 | Volume 7 | Issue 6 | e1002108
CXCL2=MIP2α
• Sécrétée ds les tissus peripheriques (infection, inflammation) : chimiotactisme
• Souris : IV -> polynucléose (idem GCSF)
• Migration PNN : moelle -> sang peripherique
Neutropénies ethniques : autres polymorphismes ?

CXCL2=MIP2α
• Sécrétée ds les tissus périphériques (infection, inflammation) : chimiotactisme
• R= CXCR2
• Souris : IV -> polynucléose (idem GCSF)
• Migration PNN : moelle -> sang peripherique
The Journal of Clinical Investigation http:/ /www.jci.org Volume 120 Number 7 July 2010
of neutrophils in the blood were from Cxcr2–/– cells (P < 0.0001).
Of note, the number of neutrophils in the spleen, another reser-
voir for neutrophils, was comparable between Cxcr2–/– and WT
cells (Figure 1C). Neutrophil trafficking from the bone marrow
was estimated by calculating the percentage of neutrophils in the
blood relative to the total number of neutrophils in the blood,
bone marrow, and spleen (neutrophil distribution index; NDI;
ref. 5). Consistent with previous studies (22, 38), under basal
conditions, 1.84% ± 0.32% of WT neutrophils were present in the
blood (Figure 1D). In contrast, the percentage of Cxcr2–/– neutro-
phils in the blood was 0.57% ± 0.18% (P = 0.02). No perturbation
in other hematopoietic lineages was observed (Figure 1E), which
indicates that the observed differences in neutrophil chimerism
are not caused by altered engraftment of Cxcr2–/– hematopoietic
stem cells. Consistent with this observation, the number and
cytokine responsiveness of myeloid progenitors in the bone mar-
row were comparable between WT and Cxcr2–/– cells (Figure 1F).
Myelokathexis is characterized by the accumulation of mature,
often hypersegmented or dysplastic, neutrophils in the bone mar-
row (10). Consistent with this phenotype, we observed that the
percentage of Gr-1hiSSChi cells — representing the most mature
neutrophils (39) — relative to the total Gr-1+ myeloid cell popula-
tion was higher for Cxcr2–/– than WT cells (Figure 2, A and B). To
confirm this finding, Cxcr2–/– and WT Gr-1+ myeloid cells were
m
m
m
Downloaded from http://www.jci.org on March 16, 2015. http://dx.doi.org/10.1172/JCI41649
The Journal of Clinical Investigation http:/ /www.jci.org Volume 120 Number 7 July 2010
of neutrophils in the blood were from Cxcr2–/– cells (P < 0.0001).
Of note, the number of neutrophils in the spleen, another reser-
voir for neutrophils, was comparable between Cxcr2–/– and WT
cells (Figure 1C). Neutrophil trafficking from the bone marrow
was estimated by calculating the percentage of neutrophils in the
blood relative to the total number of neutrophils in the blood,
bone marrow, and spleen (neutrophil distribution index; NDI;
ref. 5). Consistent with previous studies (22, 38), under basal
conditions, 1.84% ± 0.32% of WT neutrophils were present in the
blood (Figure 1D). In contrast, the percentage of Cxcr2–/– neutro-
phils in the blood was 0.57% ± 0.18% (P = 0.02). No perturbation
in other hematopoietic lineages was observed (Figure 1E), which
indicates that the observed differences in neutrophil chimerism
are not caused by altered engraftment of Cxcr2–/– hematopoietic
stem cells. Consistent with this observation, the number and
cytokine responsiveness of myeloid progenitors in the bone mar-
row were comparable between WT and Cxcr2–/– cells (Figure 1F).
Myelokathexis is characterized by the accumulation of mature,
often hypersegmented or dysplastic, neutrophils in the bone mar-
row (10). Consistent with this phenotype, we observed that the
percentage of Gr-1hiSSChi cells — representing the most mature
neutrophils (39) — relative to the total Gr-1+ myeloid cell popula-
tion was higher for Cxcr2–/– than WT cells (Figure 2, A and B). To
confirm this finding, Cxcr2–/– and WT Gr-1+ myeloid cells were
m
m
m
Downloaded from http://www.jci.org on March 16, 2015. http://dx.doi.org/10.1172/JCI41649
Neutropénies ethniques : autres polymorphismes ?

Diagnostic d’élimination
=
Faisceau d’arguments clinico-biologiques
Polymorphisme duffy null/null
≠
VPP-VPN
Neutropénie ethnique : critères diagnostiques ?

Symptomatique ?
Ancienneté ?
Ethnie ?
Isolée : clinique et biologique
> 0.5 – 0.8 G/L
Bilan de base : frottis, EPP
FAN, anti DNA, anti nucléaires solubes, LGL
Contrôle NFS
Myélogramme ?
Neutropénie ethnique : critères diagnostiques ?

Symptomatique ?
Ancienneté ?
Ethnie ?
Isolée : clinique et biologique
> 0.5 – 0.8 G/L
Bilan de base : frottis, EPP
FAN, anti DNA, anti nucléaires solubes, LGL
Contrôle NFS
Myélogramme ?
Neutropénie ethnique : critères diagnostiques ?

• 2012, Melle J, 25 ans • Origine turque, juive ashkénaze • Neutropénie isolée évoluant depuis 2003, isolée
PNN 0.7 et 1.6 G/L • Antécédents personnels :
– Episode anorexie sévère 2003-2004 résolu définitivement depuis 2008
– Raynaud depuis 2004 capillaroscopie anormale, « bilans » auto-immunité répétés (médecine interne) négatif depuis 2004 incluant des anti-granuleux
• Pas d’atcd familiaux notables
Cas n°2

• Asymptomatique : – pas d’infections, – pas d’aphtose, pas de photosensibilité, – pas d’arthralgies, pas d’arthrites – Pas de lymphoedeme – Raynaud – BMI=17
• Lymphopénie modérée, gamma 7,9 g/dl • Monocytes normaux 0,45 G/L
Cas n°2

• Neutropénie auto-immune ?
• Neutropénie ethnique ?
• Neutropénie carentielle (dosage du cuivre et
du fer normal)
Cas n°2 : conclusion ??

• Mme A, 1950
• 1992 -2008 : neutropénie de margination car test adrénaline + ( PNN 0.3-0.8 G/L)
• 2008 : nouvelles explorations (déménagement) - Sd sec clinique, schilsom grade III BGSA - FAN 1/400, anti DNA natif + IgG - Atteinte pulmonaire : micronodules + alvéolite
lymphocytaire (sarcoïdose)
• 2009 : gingivostomatite rétractile mixte -> GCSF
Cas n°3

Répéter l’interrogatoire et les explorations
GS >> LES (neutropénies sévères)
anti-SSA/SSB +++
autres ANCA
BGSA au moindre doute clinique/ biologique
Thyroïdite auto-immune ?
Neutropénies immunologique secondaires

Neutropénies & GS
Brito Zeron, Semin Arthritis Rheum 2009
PNN < 1 G/L (10%) : 50% hospitalisation pour infection (vs 9%, p=0.002)

Mme D. 1960
2006 : Entéropathie auto-immune non coeliaque (pas
d’atrophie villositaire, infiltrat T CD4+, auto anticorps
négatif, HLA non DQ2/DQ8) non peu sévère, traitée par
entocort à partir de 2008
2009 : hospitalisation pour réévaluation de l’entéropathie
• NFS :
– Hb 13 g/dL VGM 86
– Leucocytes 3 G/L
– PNN 0.4 G/L, Ly 2 G/L, mono 0.4G/L, PNE 0.18 G/L
– Plaquettes 218 G/L
Cas n°4

Interrogatoire :
• Aucun nouveau traitement,
• Pas de notion de virose
• Pas d’arthralgie, arthrite, syndrome sec, aphtose
Examen clinique :
• Apyrétique, pas de sd infectieux
• Pas d’hépato-splénomégalie
• Pas d’adénopathies
Cas n°4

Examens complémentaires :
• Myélogramme : moelle de richesse normale sans
blocage de maturation granuleuse, sans dysplasie.
Discret excès de lymphocytes (25%)
• Caryotype : 46 XX
• Dosage vitaminique (folates/B12/fer): normaux
• EPP : discrète hypergamma polyclonale 15 g/L
Cs d’hématologie pour évaluation diagnostique
Cas n°4

Examens complémentaires :
• Frottis sanguin : LGL ?
• Phénotypage lymphocytes sang
• FAN, anti-DNA, anti-ENA, FR, complément
• Coombs
• Sérologie VIH, VHC
• Dosage du cuivre
Cas n°4 : examens complémentaires

• Frottis sanguin : neutrophiles 0.5 G/L, présence de rares
LGL
• Phénotypage lymphocytes sang :
– présence d’un excès de LGL+ CD3+ CD57+ CD95+ (40%
des ly CD3+)
• FAN -, anti-DNA -, anti-ENA-, FR –
• complément normal
• Coombs négatif
• Sérologie VIH, VHC négatives
• Dosage du cuivre normal
Cas n°4 : examens complémentaires

• Lymphoprolifération LGL T CD8+ associée à une
neutropénie
– Clonalité T : présence d’un clone T majoritaire sur une
population oligoclonale
– Clonalité T sur la biopsie digestive : présence d’un clone T
différent
Cas n°4 : conclusion

• Persistance d’une neutropénie fluctuante asymptomatique
0.3 – 1 G/L : surveillance simpe
• Novembre 2011 : hospitalisation pour une fièvre à 40°C avec
point d’appel dentaire
– Antibiothérapie intraveineuse
– Début d’un traitement par GCSF : réponse rapide, PNN 6
G/L à h48
• Poursuite du GCSF en prophylaxie secondaire
– 1 injection / semaine 5 μg/kg : PNN > 1 G/L
– Mauvaise tolerance : céphalées, douleurs osseuses
– Echec de modulation des doses
Cas n°4 : évolution

• M3 : décision de débuter un traitement par MTX 20
mg/semaine en SC
– Traitement de l’entéropathie auto-immune
– Traitement de la neutropénie associée au LGL
Cas n°4 : évolution

• M3 : décision de débuter un traitement par MTX 20
mg/semaine en SC
– Traitement de l’entéropathie auto-immune
– Traitement de la neutropénie associée au LGL
• M3 MTX :
– PNN > 1 G/L
– Entéropathie : régression de l’infiltrat T CD4
– Pas de nouvel épisode infectieux bactérien mais un zona
– Asthénie importante depuis le début du MTX rythmé par
les injections
– Persistance des LGL T CD8+ clonaux (50%)
Cas n°4 : évolution

• M6 MTX : arrêt par la patiente
– PNN 0.5 et 1.5 G/L à l’arrêt
– Troubles digestifs stables
– Pas de nouvel épisode infectieux bactérien
Surveillance seule depuis 2012 : pas de
complications
Cas n°4 : évolution

• Lymphocytes à grands grains ou LGL, sujet sain :
• 10 à 15% lymphocytes normaux (0,25 G/L)
• 95% phénotype NK
• 5% T cytotoxiques
• Brouet et al.(1975) : excès de LGL > 2 G/L, > 6 mois
• Entité clinique :
• 55 – 60 ans
• Splénomégalie, manifestations articulaires (PR atypique)
• Neutropénie (asymptomatique), érythroblastopénie
• Auto-immunité biologique
• Evolution chronique, indolente / hétérogène
LGL

LGL
• Caractérisation phénotypique (CMF) :
LGL T : TCRαβ, CD3+ CD8+ CD4- CD57+.
LGL NK : CD3-CD8+CD57+CD16+
➛ Phénotype clinique identique
• Clone T : LGL T, syndrome de Felty
OMS 2008
leucémies à LGL T
lymphoproliférations chroniques NK
Excès de LGL, > 6 mois, sans autre étiologie
Critères diagnostiques n’incluent ni clonalité, ni seuil

LGL
Somatic STAT3 Mutations in Large Granular Lymphocytic
Leukemia
Hanna L.M. Koskela, M.D., Samuli Eldfors, M.Sc., Pekka Ellonen, B.Sc., Arjan J. van
Adrichem, M.Sc., Heikki Kuusanmäki, B.Sc., Emma I. Andersson, B.Sc., Sonja Lagström,
M.Sc., Michael J. Clemente, M.Sc., Thomas Olson, B.Sc., Sari E. Jalkanen, M.Sc., Muntasir
Mamun Majumder, M.Sc., Henrikki Almusa, M.Sc., Henrik Edgren, M.Sc., Maija Lepistö,
M.Sc., Pirkko Mattila, Ph.D., Kathryn Guinta, B.Sc., Pirjo Koistinen, M.D., Ph.D., Taru
Kuittinen, M.D., Ph.D., Kati Penttinen, B.M., Alun Parsons, M.Sc., Jonathan Knowles, Ph.D.,
Janna Saarela, M.D. Ph.D., Krister Wennerberg, Ph.D., Olli Kallioniemi, M.D., Ph.D., Kimmo
Porkka, M.D., Ph.D., Thomas P. Loughran Jr., M.D., Caroline A. Heckman, Ph.D., Jaroslaw
P. Maciejewski, M.D., Ph.D., and Satu Mustjoki, M.D., Ph.D.
Hematology Research Unit Helsinki, Department of Medicine, University of Helsinki and Helsinki
University Central Hospital (H.L.M.K., E.I.A., S.E.J., K. Penttinen, K. Porkka, S.M.), the Institute
for Molecular Medicine Finland (S.E., P.E., A.J.A., H.K., S.L., M.M.M., H.A., H.E., M.L., P.M.,
A.P., J.K., J.S., K.W., O.K., C.A.H.), and the Haartman Institute (P.M.), University of Helsinki,
Helsinki; the Department of Medicine, Oulu University Hospital and University of Oulu, Oulu
(P.K.); and the Department of Medicine, Kuopio University Hospital, Kuopio (T.K.) — all in
Finland; the Department of Translational Hematology and Oncology Research, Taussig Cancer
Institute, Cleveland Clinic, Cleveland (M.J.C., K.G., J.P.M.); and Penn State Hershey Cancer
Institute, Pennsylvania State College of Medicine, Hershey (T.O., T.P.L.).
Abstract
BACKGROUND—T-cell large granular lymphocytic leukemia is a rare lymphoproliferative
disorder characterized by the expansion of clonal CD3+CD8+ cytotoxic T lymphocytes (CTLs)
and often associated with autoimmune disorders and immune-mediated cytopenias.
METHODS—We used next-generation exome sequencing to identify somatic mutations in CTLs
from an index patient with large granular lymphocytic leukemia. Targeted resequencing was
performed in a well-characterized cohort of 76 patients with this disorder, characterized by clonal
T-cell–receptor rearrangements and increased numbers of large granular lymphocytes.
RESULTS—Mutations in the signal transducer and activator of transcription 3 gene ( STAT3)
were found in 31 of 77 patients (40%) with large granular lymphocytic leukemia. Among these 31
patients, recurrent mutational hot spots included Y640F in 13 (17%), D661V in 7 (9%), D661Y in
7 (9%), and N647I in 3 (4%). All mutations were located in exon 21, encoding the Src homology 2
(SH2) domain, which mediates the dimerization and activation of STAT protein. The amino acid
changes resulted in a more hydrophobic protein surface and were associated with phosphorylation
of STAT3 and its localization in the nucleus. In vitro functional studies showed that the Y640F
and D661V mutations increased the transcriptional activity of STAT3. In the affected patients,
downstream target genes of the STAT3 pathway (IFNGR2, BCL2L1, and JAK2) were up-
Copyright © 2012 Massachusetts Medical Society.
Address reprint requests to Dr. Mustjoki at the Hematology Research Unit Helsinki, University of Helsinki and Helsinki UniversityCentral Hospital, P.O. Box 700, Haartmaninkatu 8, FIN-00029 Helsinki, Finland, or at [email protected]. Koskela, Loughran, Heckman, and Maciejewski and Mr. Eldfors contributed equally to this article.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
NIH Public AccessAuthor ManuscriptN Engl J Med. Author manuscript; available in PMC 2013 June 26.
Published in final edited form as:
N Engl J Med. 2012 May 17; 366(20): 1905–1913. doi:10.1056/NEJMoa1114885.
NIH
-PA
Auth
or M
anu
scrip
tN
IH-P
A A
uth
or M
anu
scrip
tN
IH-P
A A
uth
or M
an
uscrip
t
Mutations gain de fonction STAT3 exon 21 40% LGL T Associée neutropénie et RA
NEJM 2012
Coexistence de plusieurs clones STAT3 mutés mutations différentes

LGL
Infections virales chroniques : HIV, HCV, HBV, CMV, HTLV1
Auto immunité
Immunodépression : transplantation d’organes solides, post allogreffe de moelle
Déficits immunitaires : DICV
Hémopathies : MDS (AA?), myelome, MGUS, lymphomes bas grade et haut grade
Cancers solides
Splénectomie (clonales +)
ITK
T >> NK

LGL
Phénotype biologique :
• Neutropénies, érythroblastopénie
• FR
• Hypergamma polyclonale
Pronostic : bon
• Formes clonales / tumorales
• Infections sévères rares
Traitements ? Edx, Mtx
Bareau B Haematologica 2010

Loughran, Leukemia 2015
Prospective (1999-2009) : 59 pts, FU médian 76 mois Methotrexate 10 mg/m2/sem PO + PDN 2 mois Cyclophosphamide 100 mg/j PO + PDN 2 mois
MTX
CY
LGL +
- PNN < 0.5/infection - Anémie tf

Cas n°5
Mme L. 1975 Atcd personnels : - myopie sévère (sœur idem) - G1P1, accouchement par césarienne (grossesse RAS) (NFS normale 06/2013) - Appendicectomie - Pas d’infections fréquente Atcd familiaux (fratrie de 7) RAS – fils de 2 ans en bonne santé 12/05/15 : asthénie, NFS GB = 0.03 G/L 15/05/15 : H pour sepsis sévère sans point d’appel clinique • GB 0,160 • Hb 9.2 VGM 79 retic 27 G/L • Plaquettes 218 G/L Myélogramme : richesse diminuée, agranulocytose, lignée erythroblastique et mégacaryocytaire bien représentée Antibiothérapie à large spectre et Remplissage vasculaire + GCSF (16/05)

Etiologie ? - Interrogatoire : aucune prise médicamenteuse récente en dehors de doliprane (stimulation ovarienne car désir de 2nde grossesse) - Pas d’infection virale, pas de vaccin - Pas d’arthralgies, d’arthrite, de rash cutanée, pas d’apthose Examens para-cliniques : - BOM (moelle de richesse diminuée) : moelle hypoplasique avec absence
presque complète de la lignée granuleuse - Caryotype normal - Clone HPN : négatif - PCR CMV, EBV, parvo B19 négatives - Scanner : doute sur une lésion de la tete du pancréas, comblement des
cellules du sinus maxillaire - EPP : hypergamma polyclonale 16 g/L
03/06/15 (j7 GCSF) absence de modification de l’hémogramme GB = 0,1 G/L

Autres explorations ?

Autres explorations ?
Agranulocytose symptomatique persistante à j7 de GCSF
Hypothèses ? 1. Agranulocytose médicamenteuse 2. Infection virale 3. Immunologique
• LGL • Auto-immune IIr • Auto-immune Ive • Idiopathique
4. Constitutionnelle

Autres explorations ?
1. Agranulocytose médicamenteuse
• Attendre : 21 jours parfois 2. Infection virale 3. Immunologique
• LGL : phenotypage lymphocytaire • Auto-immune Iir : FAN, anti-DNA, anti-ENA, FR,
compléments, thyroidite • Auto-immune Ive : anticorps antigranuleux • Idiopathique (clone T ?)
4. Constitutionnelle : pas à ce stade (recherche d’un involution graisseuse du pancréas)

Autres explorations ?
1. Agranulocytose médicamenteuse
• Attendre : 21 jours parfois 2. Infection virale 3. Immunologique
• LGL : phenotypage lymphocytaire • Auto-immune Iir : FAN, anti-DNA, anti-ENA, FR,
compléments, thyroidite • Auto-immune Ive : anticorps antigranuleux • Idiopathique (clone T ?)
4. Constitutionnelle : pas à ce stade (recherche d’un involution graisseuse du pancréas)

Autres explorations ?
• LGL : équilibre population B/T/NK • FAN, anti-DNA, anti-ENA, FR, TRAK négatif, anti
TPO négatifs • C3, C4, CH50 augmentés • Anticorps antigranuleux (CMF) : négatifs • IRM pancréas : normale
23/06/15 : arrêt du GCSF (échec), sortie à domicile sous vfend (ECBC + pyo) et antibiothérapie

Autres explorations ?
Evolution : • Stabilité des GB < 0.2 G/L sf 1 fois reascencion
transitoire à PNN 1.2 G/L 20/07/15 puis rechute GB < 0.2 G/L à partir du 11/08/15
• Pas de nouvelle épisode infectieux

Neutropénie chronique sévère acquise de l’adulte symptomatique
2 hypothèses diagnostiques : • Constitutionnelle
- NFS normale en 2013 - Sd GATA2 ? Teloméropathie ?
• Immunologique : neutropénie sévère idiopathique de l’adulte
Traitement ?

Neutropénie chronique sévère acquise de l’adulte symptomatique
Traitement ? • Absence d’argument clinique / biologique forme
congenitale • Tableau compatible avec neutropénie
« idiopathique » • Echec vrai du GCSF (> 21 jours)
• Ciclosporine 3 mg/kg x2/J j1 (28/08/15)
PNN > 1 G/L depuis le 14/09 stable Bonne tolérance de la ciclosporine

Neutropénie chronique sévère acquise de l’adulte symptomatique
= Neutropénies « auto-immunes » primitives
+ Neutropénies chroniques idiopathiques

Neutropénie chronique sévère acquise de l’adulte symptomatique
Sicre Blood 2015, pre published online
Etude rétrospective (cohorte prospective) : 108 pts PNN < 0,5 ou < 1 G/L + spt FU médian 8.3 ans

Neutropénie chronique sévère acquise de l’adulte symptomatique
Sicre Blood 2015, pre published online

Neutropénie chronique sévère acquise de l’adulte symptomatique
Sicre Blood 2015, pre published online

Neutropénie chronique sévère acquise de l’adulte symptomatique
Sicre Blood 2015, pre published online
• Femme d’âge moyen • Marqueurs cliniques / biologiques d’auto-immunité >50% • Pas de « sous entité » clinique/pronostique identifiable Aspect médullaire Anticorps anti granuleux Auto immunité biologique Clone T
• Le plus souvent peu asymptomatique • Infections « neutropénique » peu fréquentes • Pas de symptômes = pas de traitement • 1ère ligne traitement = GCSF dose minimale efficace • Pas d’évolution vers SMD/LAM

Neutropénies sévères congénitales à l’âge adulte ?
• Neutropénie congénitales sévères «classiques » - Exceptionnel - Patient symptomatique avant l’âge adulte - Forme cyclique le plus souvent - A discuter avec les généticiens : C Bellané-Chantelot (Pitié Salpetrière)

Neutropénies sévères congénitales à l’âge adulte ?
• Syndrome MonoMAC - Mutation AD GATA2 (atcd familiaux inconstant) - Polymorphe, expressivité variable : lymphoedeme, verrues
et condylomes, infection à mycobactéries atypiques, infections bactériennes (pneumopathies), surdité
- Neutropénie parfois isolée mais le plus souvent monocytopenie associée, pancytopénie à moelle pauvre
- IC de SMD/LAM >70% à 60 ans (chimioréfractaire & infection fongique invasive)
Probablement le plus fréquent des sd de prédisposition génétique aux hémopathies myéloïdes
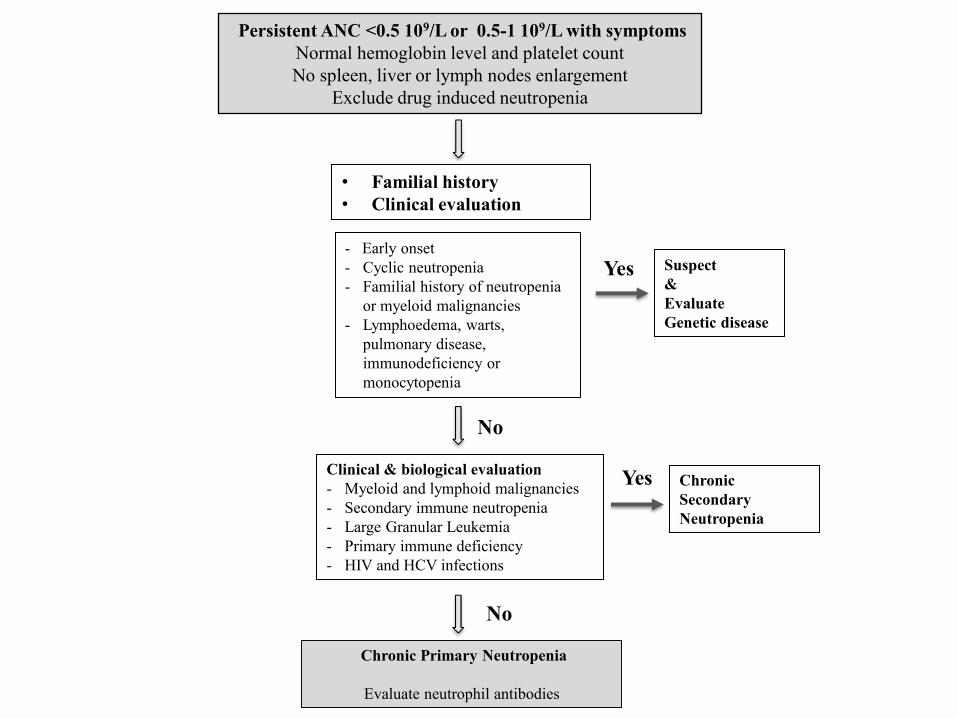
Persistent ANC <0.5 109/L or 0.5-1 109/L with symptoms
Normal hemoglobin level and platelet count
No spleen, liver or lymph nodes enlargement
Exclude drug induced neutropenia
• Familial history
• Clinical evaluation
Clinical & biological evaluation
- Myeloid and lymphoid malignancies
- Secondary immune neutropenia
- Large Granular Leukemia
- Primary immune deficiency
- HIV and HCV infections
Chronic Primary Neutropenia
Evaluate neutrophil antibodies
- Early onset
- Cyclic neutropenia
- Familial history of neutropenia
or myeloid malignancies
- Lymphoedema, warts,
pulmonary disease,
immunodeficiency or
monocytopenia
Suspect
&
Evaluate
Genetic disease
Chronic
Secondary
Neutropenia
Yes
No
Yes
No
