les néoplasies myéloprolifératives (nmp) · abl1, de diagnostic de la lmc, de la pv, de la mf...
TRANSCRIPT

Les néoplasies myéloprolifératives (NMP)
Survol clinique, traitement, et soins infirmiers.
Catherine de Ravinel Inf BscCentre d’oncologie Hôpital Notre-Dame Séminaire de l’AQIO 2017

Conflits d’intérêts et remerciements
• Je n’ai reçu aucun honoraire pour la présentation de cette conférence
•Remerciements à la compagnie Novartis, pour la mise à disposition de matériel pédagogique
•Remerciements à Dr Harold Olney, hémato-oncologue à l’hôpital Notre-Dame du CHUM pour la révision du contenu.

Plan de la présentation • Présentation générale des néoplasies myéloprolifératives
(NMP) /mise en contexte.
• Survol de chacune des maladies, symptômes, traitements, et soins infirmiers reliés.
• Histoire de cas
• Outils de suivi pour les infirmières
• Outils pour les patients.
• Questions

Mise en contexte • Au centre d’oncologie de l’hôpital Notre-Dame du CHUM suivi des patients qui sont sous
chimio PO. Ce suivi se fait à l’aide de différents outils qui ont été développés au fil du temps.
• Les 2/3 de ces patients sont atteints de NMP (PV, TE, MF et LMC, ainsi que d’autres types d’hémopathies plus rares)
• Cette présentation met l’accent sur les PV, TE et MF
• Maladies relativement peu connues, même par les infirmières en oncologie. (Pas de chimio IV, pts rarement hospitalisés sauf dans le cas de la MF (médecine de jour et hospitalisation)
• Suivi global
• Ca fait plus de 5 ans que je fais le suivi des patients chimio PO. Intérêt pour partager cette activité clinique en oncologie, y ayant découvert un univers complexe, mais passionnant.

Quelques faits sur les NMP• Les néoplasies myéloprolifératives (NMP) sont un groupe
d’affections malignes hématologiques de faible incidence caractérisé par des cellules souches hématopoïétiques clonales de lignées myéloïdes. 1 (à l’inverse des Leucémies aigues, ces cellules conservent leur capacité de différenciation)
• Les NMP sont classés en sous-types par la détection du chromosome Philadelphie (Ph +; soit la présence gène de fusion BCR-ABL1) pour confirmer le diagnostic de la leucémie myéloïde chronique (LMC) ou les NMP Ph négative. 1
• Les trois NMP Ph- classiques (et ce sont celles qui nous intéressent aujourd’hui) sont : • La polcythemie vraie (PV) ou maladie de Vaquez• La thrombocytémie essentielle (TE)• La myélofibrose (MF)
1. Site WEB du Groupe québécois de recherche en LMC-NMP

Précédemment désignées « maladies myéloprolifératives » les NMP sont
considérées depuis 2008 comme des cancers par l’OMS, ce qui peut causer de la confusion et de l’anxiété chez les patients,
qui entendent parfois de la part du MD qu’il ne s’agit pas d’un cancer.

Extrait d’une note d’évolution de Mme V, 75 ans, qui vient d’être diagnostiquée avec une TE
Des explications lui ont été données sur la nature de la maladie qui est une néoplasie de la moelle osseuse, mais avec un très faible risque de transformation vers une leucémie, et un excellent pronostic.

Évolution naturelle des néoplasies myéloprolifératives
8 % - 30 %
2 % - 7 %
2 % - 7 %
Passamonti F, et coll. Oncotarget. 2011;2(6):485-490.
PV
TE
MF LMA
MED/JAKm/0001

Néoplasies myéloprolifératives (NMP) : Incidence estimative
D’après : 1. Moulard O, et coll. Eur J Haematol 2014; 92(4):289-97; 2. Titmarsh GJ, et coll. Am J Hematol 2014; 89(6):581-7; 3. Mehta J, et coll. Leuk Lymphoma 2014; 55(3):595-600.
Sous-type Incidence estimée par 100 000/année1-3
Polycythémie vraie (PV) 0,01 – 2,8
Thrombocythémie essentielle (TE)
0,21 – 2,27
Myélofibrose (MF) primaire 0,1 – 1,3
Myélofibrose consécutive à une PV 0,12 – 0,42
Myélofibrose consécutive à une TE 0,22 – 0,36
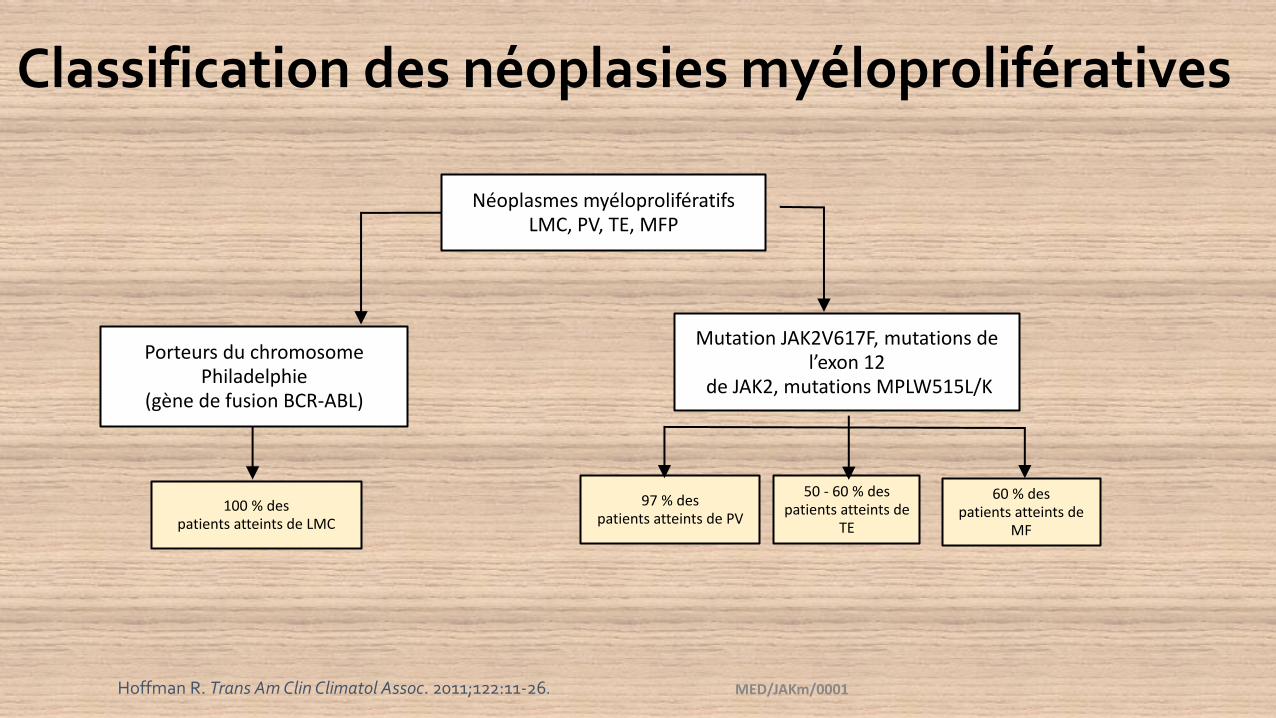
Classification des néoplasies myéloprolifératives
Hoffman R. Trans Am Clin Climatol Assoc. 2011;122:11-26.
Néoplasmes myéloprolifératifsLMC, PV, TE, MFP
Porteurs du chromosome Philadelphie
(gène de fusion BCR-ABL)
Mutation JAK2V617F, mutations de l’exon 12
de JAK2, mutations MPLW515L/K
100 % despatients atteints de LMC
97 % despatients atteints de PV
50 - 60 % despatients atteints de
TE
60 % despatients atteints de
MF
MED/JAKm/0001
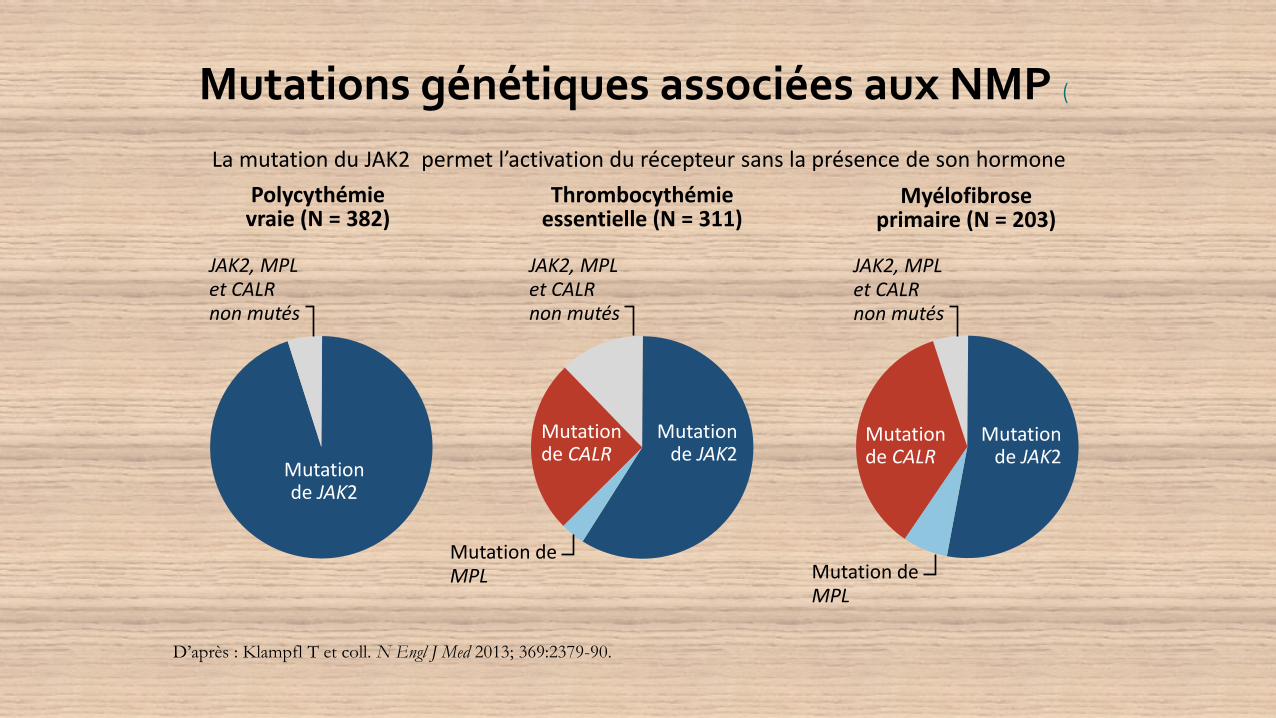
Mutations génétiques associées aux NMP (
D’après : Klampfl T et coll. N Engl J Med 2013; 369:2379-90.
Polycythémievraie (N = 382)
Myélofibroseprimaire (N = 203)
Thrombocythémieessentielle (N = 311)
JAK2, MPLet CALRnon mutés
Mutation de JAK2
JAK2, MPLet CALRnon mutés
Mutation deMPL
Mutation de JAK2
Mutation de CALR
JAK2, MPLet CALRnon mutés
Mutation de JAK2
Mutation de CALR
Mutation deMPL
La mutation du JAK2 permet l’activation du récepteur sans la présence de son hormone

La plupart des patients atteints de NMP présentent donc une mutation
génétique, la plus courante étant JAK2 (75 % des patients)
JAK2 est une protéine (enzyme)de type tyrosine kinase codée par un gène situé sur le chromosome 9. Une fois la mutation présente, les cellules myéloides sont plus réactives aux cytokines, et vont se multiplier. Une fois mutée, la cellule est protégée de l’apoptose.

Traitement• À ce jour le seul traitement curatif pour les NMP est la greffe de
cellules souches hématopoïétiques. (risque élevé de morbidité et de mortalité) Les autres traitements sont largement palliatifs et jouent un rôle limité dans l’évolution de la maladie. Ces traitements visent à améliorer la qualité de vie des patients par le traitement des symptômes liés aux NMP et empêcher la mortalité précoce de thrombose, toutefois leur utilisation est mal standardisée et peut être associée à des effets secondaires significatifs.
• Le traitement palliatif (mais offrant une espérance de vie comparable à celle de la population en général pour au moins deux décennies dans le cas de la TE et de la PV) dépend de plusieurs facteurs comme par exemple l’âge, les antécédents de thrombose, ou la présence ou non de mutation génétique.
• Source: Groupe québécois de recherche en LMC-NMP
La thrombocytémieessentielle (TE)

La thrombocytémie essentielle (TE) est caractérisé par une thrombocytosepersistante, à la suite d’une hyperplasie mégacaryocytaire 1 et de l’absence d’une fibrose significative dans la moelle osseuse.2
1. Mégacaryocyte: Cellule géante de la moelle osseuse, responsable de la production des plaquettes
2. Site WEB du groupe québécois de recherche en LMC -NMP

Thrombocytémie essentielle (TE): Aperçu (1/2)
• La TE représente environ 24 % des cas de NMP1
• Environ 60 % des patients ont la mutation JAK 2
• Depuis l’adoption des critères diagnostics révisés de l’OMS (2008), l’incidence de la TE a augmenté, passant à 1-2,5/100 0002,3
• Le seuil plaquettaire a été abaissé, passant de 600 x 109 plaquettes/L à ≥ 450 x 109/L en 2008
• Il se peut que l’incidence réelle soit plus élevée, car plusieurs patients ne présentent aucun symptôme
D’après : 1. Rollison DE, et coll. Blood 2008; 112(1):45-52; 2. Birgegård G. Ann Hematol 2009; 88:1-10; 3. Brière JB. Orphanet J Rare Dis 2007; 2:3.

Thrombocytémie essentielle (TE)Aperçu (2/2)
• Âge médian au diagnostic :65-70 ans1
• Survie médiane > 18-20 années2-3
• Surreprésentation des femmes et des jeunes gens2-3
D’après : 1 Brière JB. Orphanet J Rare Dis 2007; 2:3; 2 Wolanskyj AP et coll. Mayo Clin Proc 2006; 81(2):159-66; 3. Radaelli F et coll. Hematology 2008; 13(4):195-202.

Critères de l’OMS 2016 en matière de diagnostic de la TE
D’après : Alter DA, et coll. Blood 2016; 127(20):2391-2405.
Exigences relatives au diagnostic de la TE :
Satisfaire aux 4 critères majeurs OU aux 3 premiers critères majeurs ainsi qu’au
critère mineur
Critères majeurs
1. Nombre de plaquettes ≥ 450 × 109/L
2. Biopsie de la moelle osseuse indiquant la prolifération prédominante de la lignée mégacaryocytaire ainsi qu’une augmentation du nombre de gros mégacaryocytes matures hyperlobés. (La BMO est nécessaire, entre autres pour éliminer la MF)
3. Ne pas satisfaire aux critères de l’OMS en matière d’anomalie moléculaire BCR-ABL1, de diagnostic de la LMC, de la PV, de la MF primaire, des SMD et d’autres néoplasies myéloïdes
4. Présence d’une mutation JAK2, CALR ou MPL
Critère mineur
Présence d’un marqueur clonal ou absence de données probantes indiquant unethrombocytose réactive (tx par certains médicaments, pathologies inflammatoires)

Manifestations cliniques de la TE1,2,3
• Généralement asymptomatique avant et après le diagnostic• Prédisposition aux occlusions vasculaires (artérielles et veineuses) souvent à
des sites inhabituel (veine sagittale dans les méninges, intraabdominale)• Hémorragie (accumulation du facteur de coagulation Von Willebrand autour
des plaquettes et quand très élevé il y a déplétion fonctionnelle avec coagulopathie secondaire)
• Potentiel de splénomégalie légère• Principaux symptômes: faiblesses, céphalées, neuropathies périphérique• Érythromélalgie, érythème et sensation de brûlure distale (main, pied)
probablement par microthrombose dans les capillaires
• D’après: 1. Brière JB . Orphanet J Rare Dis 2007;2;3 2.Michiels JJ Clin ApplThromb Hemost 1999;5:147-51 3.LiesveldJ et coll. Tiré de : Merck Manual of Diagnosis and Therapy. Édition en ligne consultée en juin 2016.

Facteurs pronostiques relatifs à la TE
Principaux facteurs de risque de thrombose1
• Âge > 60 ans
• Antécédents de thrombose
• Facteurs de risque cardiovasculaire
• Mutation JAK2V617F
Ces facteurs composent le International Prognostic Score of thrombosis in World Health-essential thromocythemia (IPSET-thrombosis) [cotation pronostique de thrombose en vertu des critères de l’Organisation mondiale de la santé en matière de thrombocythémie]
Autres facteurs de risque possibles
• Nombre de plaquettes > 1500 × 109/L2
• Antécédents d’hémorragie grave2
− Ou saignements mineursavec le nombre de plaquettes > 1000 × 109/L
• Diabète3
• Hypertension3
D’après : 1. Barbui T, et coll. Blood 2012; 120(26):5128-33. 2. Brière JB. Orphanet J Rare Dis 2007; 2(1); 3. Wolanskyj AP et coll. Mayo Clin Proc 2006; 81(2):159-66.

Objectifs du traitement de la TE
• Prévenir les complications thrombotiques et hémorragiques sans augmenter le risque de saignement1
• Maîtriser et traiter les symptômes associés à la TE, dont les symptômes microcirculatoires ou vasomoteurs (p. ex., céphalées, étourdissements, paresthésies, rougeur et sensation de brûlure aux extrémités, douleurs inhabituelles à la poitrine)1,2
• Améliorer la qualité de vie2
• Minimiser le risque de transformation maligne ou de MFconsécutive à une TE2
D’après : 1. Tefferi A, et coll. Am J Hematol 2015; 90(2):162-73; 2. Haznedaroglu IC. Expert Opin. Pharmacother 2013; 14(11):1431-6

Traitement de la TE (1/2)
• •
Traitement Notes concernant l’efficacité, l’innocuité et la tolérabilité
Agents antiplaquettaires(p. ex., AAS 80 mg/jour par voie orale)
• À ce jour, l’utilisation de l’aspirine seule n’a jamais fait l’objet d’étude, mais parfois donnée seule en TE et on observe une diminution du risque de thrombose.
• L’association d’un agent antiplaquettaire et d’un agent cytoréducteur a permis la diminution de la fréquence de la thrombose associée à la TE1
Hydroxyurée(HU)
• Dose initiale de 500 mg/jour par voie orale et ajustée pour des plaquettes < 400 x 109/L2
• Maîtrise relativement efficace du nombre de plaquettes1
• L’anémie, la neutropénie, la fièvre, les ulcères et les lésions cutanées comptent parmi les quelques effets secondaires associés à l’HU1,3,4
D’après : 1. Brière JB. Orphanet J Rare Dis 2007; 2:3; 2. Tefferi A, et coll. Am J Hematol. 2015; 90(2):162-73; 3. Finazzi G, et coll. Blood 2007; 109(12):5104-11; 4. NajeanY. Ann Med Interne (Paris) 1998; 149(2):94-100; 5. Begna K, et coll. Blood Cancer J 2016; 6:e427.

Traitement de la TE (2/2)Traitement Notes concernant l’efficacité, l’innocuité et la tolérabilité
Interféron
• Réduit efficacement le nombre de plaquettes1
• Effets secondaires parfois significatifs, dont des symptômes pseudogrippaux1
• À utiliser en tant que traitement de 2e ou de 3e intention (ou de première intention chez les femmes enceintes où l’HU est contre-indiquée)
• Dose habituelle : 3 millions d’unités par voie sous-cutanée trois fois par semaine et chez les patients avec assurance privé, forme Pégylé peut être donner 1 fois semaine (90 mg)
Busulfan
• Peut convenir comme traitement de deuxième intention; réservé chez des patients âgés de plus de 80 ans car augmente risque de transformation leucémique5
• Il pourrait être nécessaire de diminuer ou arrêter la dose si le nombre de plaquettes diminue en deçà de 300 x 109/L
• Le traitement doit être interrompu si le nombre de plaquettes diminue en deçà de 150 à 200 x 109/L
• C’est un traitement de choix de 2ème intention
D’après : 1. Brière JB. Orphanet J Rare Dis 2007; 2:3; 2. Tefferi A, et coll. Am J Hematol. 2015; 90(2):162-73; 3. Finazzi G, et coll. Blood 2007; 109(12):5104-11; 4. Najean Y. Ann Med Interne (Paris) 1998; 149(2):94-100; 5. Begna K, et coll. Blood Cancer J 2016; 6:e427.

Hydroxyurée (HU) 1/2•Classe: antinéoplasique •Mode d’action: inhibiteur de la synthèse de l’ADN 1
•Médicament cytotoxique, donc les précautions s’imposent (séance d’information chimio po)•Sur la liste du NIOSH•Dose de départ:de 500 mg/jour par voie orale et
ajustée pour des plaquettes < 400 x 109/L 2
• 1. Monographie de produit, Bristol-Myers-Squibb, Canada 2. Tefferi A, et coll. Am J Hematol. 2015; 90(2):162-73;

Principaux effets secondaires❖Diarrhées et constipation❖Ulcères❖Nausées et vomissements❖Rash, rougeur et ulcération au niveau de la peau❖Anémie, neutropénie ❖Fièvre
Hydroxyurée (HU) 2/2

Rôle de l’infirmière dans le suivi des patients avec TE
Gestion des symptômes de la maladieGestion des effets secondaires des traitementsSuivi des résultats des prises de sang, et communiquer résultats aux patientsSupport psycho-socialApproche interdisciplinaire est essentielle, on suit le patient dans sa globalité.

Outils (infirmière et patients)
•Séance d’information chimio PO• Fiches éducatives CHUM• Le GEOQ (Groupe étude en oncologie
du Québec) • Les plans de transfert

La polycythémie vraie (PV)

Polycythémie vraie (PV)
La PV est principalement caractérisé par une augmentation de la masse des globules rouges, du nombre de plaquettes et du nombre de globules blancs qui sont les résultats de la prolifération clonale des cellules souches des lignées érythroïdes, mégacaryocytaires et myéloïdes. 1
1. Site WEB du groupe québécois de recherche en LMC-NMP

Polycythémie vraie (PV) : Aperçu
• La PV représente environ 45 % des NMP1
• Également appelée maladie de Vaquez
• Incidence : ~ 1-2/100 000 par année2-4
• Âge médian au moment du diagnostic :60 ans5
• Survie après 15 ans : 65 %6
• Presque tous les patients (plus de 97%) atteints de PV présentent la mutationJAK2V617F 5,7
D’après : 1. Rollison DE, et coll. Blood 2008; 112(1):45-52; 2. Girodon F, et coll. Haematologica 2009; 94(6):865-9; 3. Johansson P, et coll. Semin Thromb Hemost 2006; 32(3):171-3; 4. McNally RJ, et coll. Hematol Oncol 1997; 15:173-89; 5. Stuart BJ, et coll. Am Fam Physician 2004; 69(9):2139-44; 6. Passamonti F, et coll. Am J Med 2004; 117(10):755-61; 7. Barosi G et coll.
Blood 2009; 113(20):4829-33

Exigences relatives au diagnostic de la PV :Les 3 critères majeurs OU les 2 premiers critères majeurs et le critère mineur
Critères majeurs de l’OMS (2016) en matière de diagnostic de la PV
*Plus de 25 % supérieure à la valeur théorique normale moyenneD’après : Alter DA, et coll. Blood 2016; 127(20):2391-2405
Critères majeurs
Hb > 165 g/L (hommes), > 160 g/L (femmes) OU
Hématocrite > 49 % (hommes), > 48 % (femmes) OU
augmentation de la masse érythrocytaire*
Biopsie de la moelle osseuse (pas faite d’emblée au Québec en PV) indiquant une
hypercellularité anormale selon l’âge touchant les 3 lignées (panmyélose), y
compris une prolifération érythroïde, granulocytaire et mégalocytaire nette
présentant des mégacaryocytes polymorphes matures (différences de taille)
Présence de la mutation JAK2V617F ou de celle de l’exon 12 de JAK2
Critère mineur
Taux d’érythropoïétine (une cytokine/hormone pour les précurseurs des érythrocytes dans
la moelle osseuse (c'est un facteur de croissance))sérique sous la normale (compensation rénale en produisant moins d’EPO)

PV : Manifestations cliniques
D’après : 1. Schafer AI. Blood 2006; 107(11):4214-22; 2. Finazzi G, et coll. Blood 2007; 109(12):5104-11; 3. Stuart BJ, et coll. Am Fam Physician 2004; 69(9):2139-4;4. Spivak JL. Blood 2002; 100(13):4272-90; 5. Hoffman R. Semin Oncol 2002; 29(3S10):3-9; 6. Liesveld J et coll. Tiré de : Merck Manual of Diagnosis and Therapy. Édition en ligne, consultée le 7 juin 2016. Rege-Cambrin G et coll. Cancer Genet Cytogenet 1987;25(2):233-45; 8. Tefferi A. Am J Hematol 2008; 83:491-7; 9. Langabeer SE, et coll. Eur J Haematol 2015; 95(4):270-9.
Manifestations cliniques1,4-6
• Souvent asymptomatique• Augmentation du taux d’hémoglobine et de l’hématocrite (sup à 0,49
ou 49%)• Thrombose• Hyperviscosité sanguine accrue • Symptômes constitutionnels (PV de stade avancé) : prurit, (la
dégranulation des basophiles à la chaleur libère des histamines qui provoquent des démangeaisons) perte de poids, faiblesse, sueurs nocturnes, douleur osseuse, splénomégalie

Facteurs associés à un mauvais pronostic 1-4
• Antécédents de thrombose veineuse• Âge > 60 ans• Leucocytose (toute la lignée myéloide est
affectée)
PV : Facteurs pronostiques
D’après : 1. Passamonti F, et coll. Am J Med 2004; 117(10):755-61; 2. Barosi G et coll. Curr Opin Hematol. 2009; 16:129-34; 3. Tefferi A. Am J Hematol 2008;83:491-7;4. Testa JR et coll. Am J Hematol 1981; 11(1):29-45.

Objectifs du traitement de la PV
• Maîtrise de l’hématocrite (diminution de l’hématocrite et du taux d’hémoglobine, à l’aide de différents traitements ou agents)
• Prévention des événements thromboemboliques
• Élimination ou atténuation des symptômes associés à la PV
• Réduire la splénomégalie pour contrôler les symptômes.
MED/JAKp/0001

Options thérapeutiques contre la PV(1/2)
Traitement Notes concernant l’efficacité, l’innocuité et la tolérabilité
Phlébotomie • Objectif : maintien de l’hématocrite à < 45 %1
• Permet une maîtrise efficace de l’érythrocytose2
AAS (80 mg/jour par voie orale)
• Diminution du risque d’événements cardiovasculaires1
• Donné en association avec HU• Associé à un risque accru d’hémorragie grave
Hydroxyurée
• Empêche la thrombose de manière efficace en cas de PV1
• Dose initiale de 500 mg/jour par voie orale, ajustement selon la réponse1
• Associée à certaines toxicités, dont la neutropénie, l’anémie, les ulcères buccaux et des jambes ainsi que les lésions cutanées2
D’après : 1. Sirhan S, et coll. Clin Lymphoma Myeloma Leuk 2015; 15(12):715-27; 2. Finazzi G, Barbui T. Blood. 2007;109(12):5104-5111;3. Silver RT. Cancer. 2006;107(3):451-458; 4. Quintás-Cardama A. J Clin Oncol. 2009;32(27):5418-5424; 5. Vannucchi AM, et coll. N Engl J Med. 29 janv. 2015;372(5):426-35.6. Novartis Pharma Canada inc. Monographie de Jakavi. 2015; 7. Begna K, et coll. Blood Cancer J 2016; 6:e427.

Options thérapeutiques contre la PV(2/2)
Traitement Notes concernant l’efficacité, l’innocuité et la tolérabilité
Interféron-α
• Associé à la survenue fréquente d’effets secondaires, dont la neutropénie, les symptômes pseudogrippaux, la dépression, les infections, la diarrhée, la vision trouble et l’asthénie3,4
• Dose habituelle : 3 millions d’unités par voie sous-cutanée trois fois par semaine mais forme pégylé disponible si couverture d’assurance
Ruxolitinib
• Le seul médicament approuvé par Santé Canada pour le traitement de la PV
• Pour les patients présentant une réponse inadéquate ou des effets indésirables inacceptables à l’hydroxyurée5,6
• Permet une maîtrise efficace de l’hématocrite et du volume de la rate ainsi qu’une diminution efficace des symptômes associés à la PV5,6
• Dose selon le nombre de plaquettes6 (10 mg BID)
Busulfan
• Emploi possiblement approprié comme traitement de deuxième intention chez les patients plus âgés7
• Il pourrait être nécessaire de diminuer ou cesser la dose si le nombre de plaquettes diminue en deçà de 300 x 109/L
• Le traitement doit être interrompu si le nombre de plaquettes diminue en deçà de 150 à 200 x 109/L
D’après : 1. Sirhan S, et coll. Clin Lymphoma Myeloma Leuk 2015; 15(12):715-27; 2. Finazzi G, Barbui T. Blood. 2007;109(12):5104-5111;3. Silver RT. Cancer. 2006;107(3):451-458; 4. Quintás-Cardama A. J Clin Oncol. 2009;32(27):5418-5424; 5. Vannucchi AM, et coll. N Engl J Med. 29 janv. 2015;372(5):426-35.6. Novartis Pharma Canada inc. Monographie de Jakavi. 2015; 7. Begna K, et coll. Blood Cancer J 2016; 6:e427.
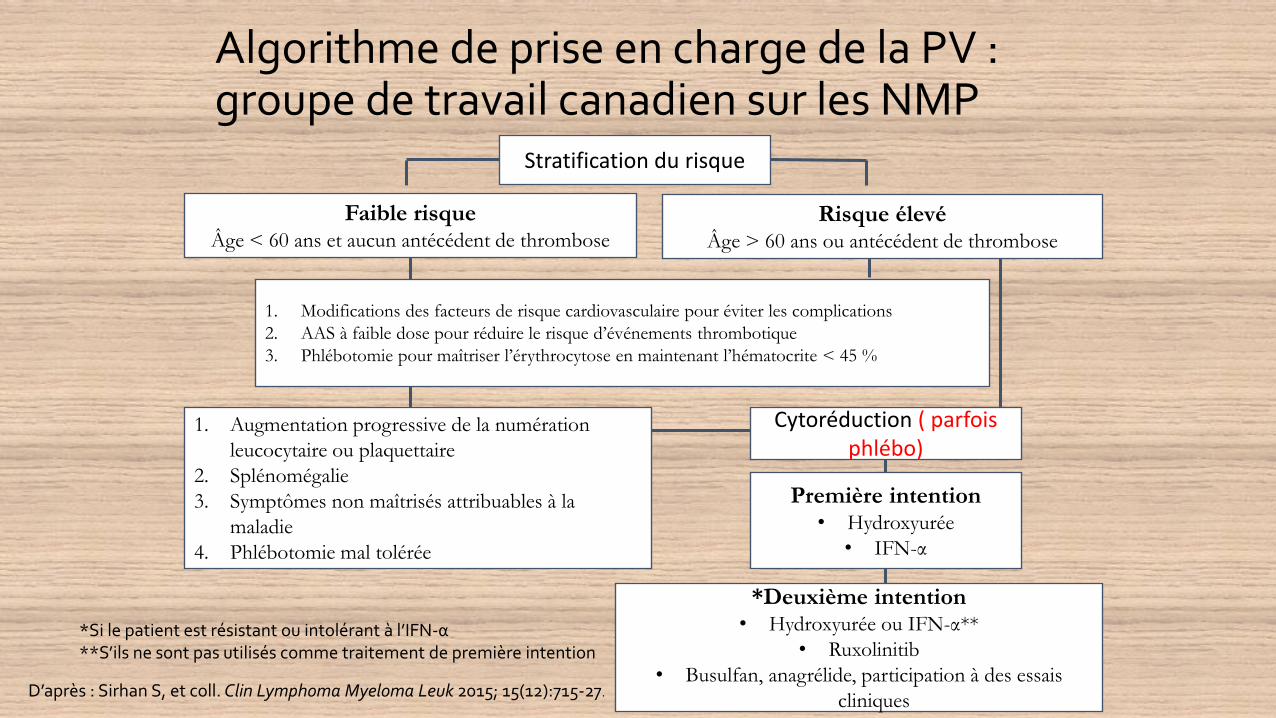
Algorithme de prise en charge de la PV : groupe de travail canadien sur les NMP
D’après : Sirhan S, et coll. Clin Lymphoma Myeloma Leuk 2015; 15(12):715-27.
Stratification du risque
Faible risqueÂge < 60 ans et aucun antécédent de thrombose
*Deuxième intention• Hydroxyurée ou IFN-α**
• Ruxolinitib
• Busulfan, anagrélide, participation à des essais
cliniques
*Si le patient est résistant ou intolérant à l’IFN-α**S’ils ne sont pas utilisés comme traitement de première intention
1. Augmentation progressive de la numération
leucocytaire ou plaquettaire
2. Splénomégalie
3. Symptômes non maîtrisés attribuables à la
maladie
4. Phlébotomie mal tolérée
Première intention• Hydroxyurée
• IFN-α
Cytoréduction ( parfois phlébo)
Risque élevéÂge > 60 ans ou antécédent de thrombose
1. Modifications des facteurs de risque cardiovasculaire pour éviter les complications
2. AAS à faible dose pour réduire le risque d’événements thrombotique
3. Phlébotomie pour maîtriser l’érythrocytose en maintenant l’hématocrite < 45 %

Rôle de l’infirmière dans le suivi des patients avec PV
Gestion des symptômesGestion des effets secondaires des traitementsTraitement (ex: phlébotomies)Suivi des résultats des prises de sang, et communiquer résultats aux patientsSupport psycho-socialApproche interdisciplinaire est essentielle, on suit le patient dans sa globalité.
La myélofibrose (MF)

La myélofibrose est un cancer rare de la moelleosseuse qui fait partie des cancers du sang regroupéssous le nom de néoplasies myéloprolifératives (NMP),caractérisée par un développement anormal des cellulessanguines produites dans la moelle osseuse. Laformation de tissu cicatriciel fibreux qui en résulteentraîne une anémie grave, de la fatigue et unehypertrophie de la rate et du foie.(hépatosplénomégalie)
Source: Site Web de la société de Leucémie et lymphome du Canada

Quelques faits sur la myélofibrose
• Hyperplasie mégacaryocytaire atypique (trop et anormale en anatomie)1-2
• Myéloprolifération chronique 1-2 ( surproduction des trois lignées myéloïdes, (en raison de la mutation JAK 2 ou CALR ) ce qui entraine la production de cytokines, qui à leur tour stimulent le développement de tissus cicatriciel dans la moelle )
• Fibrose (cicatrise) de la moelle osseuse 1-2
• La production de fibrose supplante la capacité de la moelle osseuse de produire suffisamment de cellules sanguines normales. 3
• On se souvient que presque tous les patients avec MF présentent une mutation génétique (JAK 2 ou CALR )
• D’après : 1. Tefferi A. Tiré de : Ansell SM, éd. Rare Hematological Malignancies. 2008;29-49; 2. Abdel-Wahab OI, et coll.Annu Rev Med 2009; 60:233-45 3. Site Web de la société de Leucémie et lymphome du Canada

Myélofibrose: statistiques (1/2)
• Fréquence: 0,4 -1,5 cas /100 000 1 (c’est donc la moins fréquente des NMP mais aussi la survie est la plus courte, donc on accumule moins de patients avec le temps)
• Âge médian au moment du diagnostic: 67 ans
• La MF peut être primaire ou consécutive à la TE ou la PV 2
➢Risque de MF consécutive à la TE ou la PV après 10 à 15 ans: 10-20% 3
• Risque à 10 ans de transformation en LMA : 6-18% 4
• D’après : 1. Tefferi A. Tiré de : Ansell SM, éd. Rare Hematological Malignancies. 2008;29-49; 2 Mesa RA, et coll. Blood 2005; 105(3):973-7 3. . Passamonti F, et coll. Oncotarget 2011; 2(6):485-90 4.Geyer HL, et coll. Blood 2014; 124(24):3529-37;

Myélofibrose: statistiques (2/2)
• Survie médiane: 5,7 ans selon les facteurs de risque1
➢Survie médiane de 2 ans chez les patients atteints de MF présentant un risque élevé
➢La cause de décès de 50% des patients atteints de MF primaire est attribuable à la maladie (ex:LMA, complication de cytopénie en maladie avancée)2
➢ D’après 1 Tefferi A. Am J Hematol 2008; 83:491-7: 2. Cervantes F, et coll. Blood 2009; 113(13):2895-901;

Myélofibrose (MF) : Manifestations cliniques
Anémie grave, érythropoïèse inefficace• Hématopoïèse extramédullaire ± hépatosplénomégalie
(c’est la rate qui prend le relais pour l’hématopoièse)
D’après : Tefferi A. Am J Hematol 2013; 88:142-50; Tefferi A. Blood 2011; 117(13):3494-504; et Tabarroki A, et coll. Expert Opin Investig Drugs 2012; 21(8):1141-54.
Prurit avec basophilie
Symptômes constitutionnels (dus aux cytokines, produits par les précurseurs de la lignée myéloide)• Fatigue, sueurs nocturnes, fièvre et douleur, rash
Autres complications possibles • Infarctus splénique, cachexie, thrombose/hémorragie,
ascite, hypertension portale symptomatique, hypertension pulmonaire, épanchement pleural, douleur diffuse
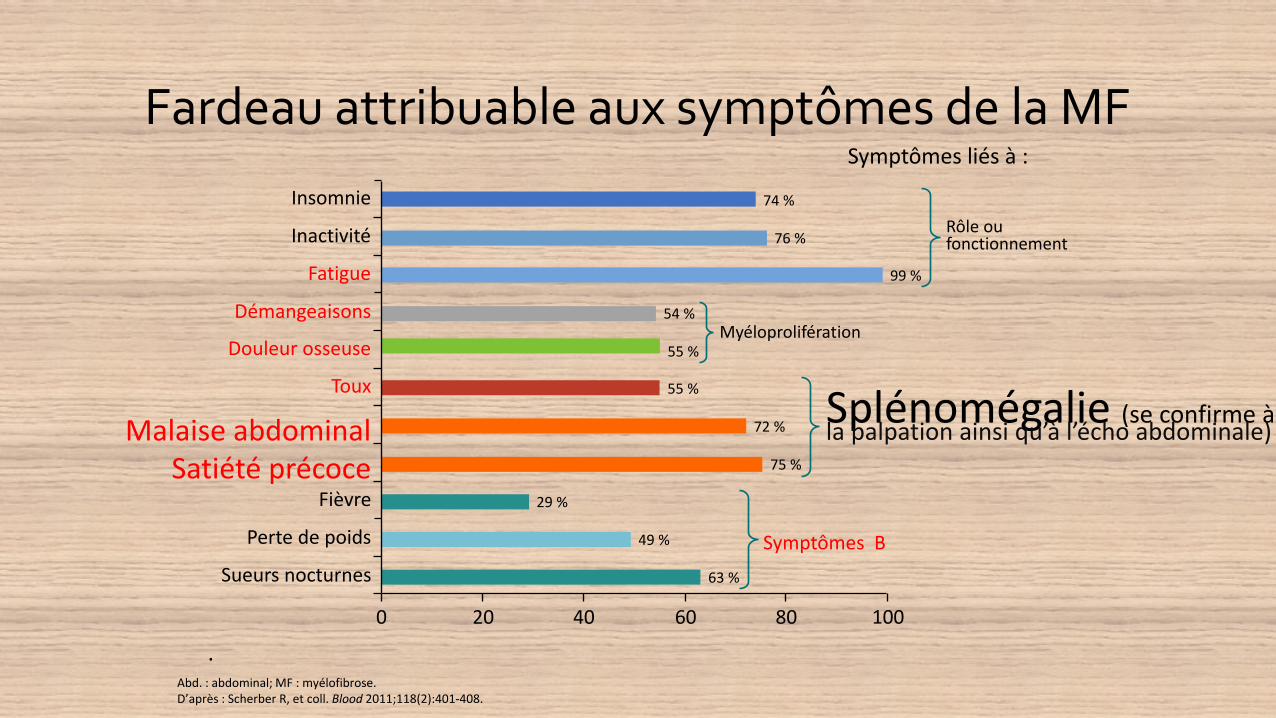
Fardeau attribuable aux symptômes de la MF
Abd. : abdominal; MF : myélofibrose.D’après : Scherber R, et coll. Blood 2011;118(2):401-408.
Symptômes liés à :
Symptômes B
Rôle ou fonctionnement
Myéloprolifération
Splénomégalie (se confirme à la palpation ainsi qu’à l’écho abdominale)
Insomnie
Inactivité
Fatigue
Démangeaisons
Douleur osseuse
Toux
Malaise abdominalSatiété précoce
Fièvre
Perte de poids
Sueurs nocturnes
0 20 40 60 80 100
74 %
76 %
99 %
54 %
55 %
55 %
72 %
75 %
29 %
49 %
63 %
.

Résultats des analyses de laboratoireet anomalies moléculaires associés
à la MFRésultats d’analyses de laboratoire anormaux1,2
• Anémie : le plus fréquent; Hb < 10 g/dL chez ~ 50 % des patients(on voit beaucoup de pts qui sont dépendants aux transfusions)
• Leucoérythroblastose
• Leucocytose (en début de maladie) ou leucopénie (en maladie plus avancée)
• Thrombocytose (en début de maladie)ou thrombocytopénie (en maladie plus avancée)
• Augmentation du taux de LDH sérique
• Augmentation des cellules progénitrices hématopoïétiques circulantes exprimant CD34
Anomalies moléculaires3,4
• Mutation JAK2V617F présente chez ~ 50 % des patients
• Mutation CALR chez 30 % des patients.
• Mutation MPLW515/K chez ~ 11 % des patients
• Plusieurs autres mutations possibles
D’après : 1. Tefferi A, et coll. Cancer 2007; 109(10):2083-8; 2. Abdel-Wahab OI, et coll. Annu Rev Med 2009; 60:233-45; 3. Tefferi A, et coll. Nat Rev Clin Oncol 2009; 6:627-37;4. Cervantes F, et coll. Blood 2009; 113(13):2895-901.

Critères de l’OMS en matière de diagnostic de la MF primaire manifeste
D’après : Alter DA, et coll. Blood 2016; 127(20):2391-2405.
Critères majeurs
Présence d’une prolifération mégacaryocytaire et d’atypie, accompagnées d’une fibrose réticulinique et/ou d’une fibrose collagène de grade 2 ou 3 (MO essentielle pour déterminer l’étendue de la fibrose)
Ne pas satisfaire aux critères de l’OMS en matière de : TE, PV, LMC BCR-ABL1, syndromes myélodysplasiques ou d’autres néoplasies myéloïdes
Présence d’une mutation des gènes JAK2, CALR ou MPL, ou présence d’un autre marqueur clonal ou absence de MF réactive en l’absence de ces mutations.
Critère mineur
Présence d’au moins un des symptômes suivants confirmée à deux reprises consécutives :a. Anémie non attribuable à une comorbiditéb. Leucocytose ≥ 11 x 109/Lc. Splénomégalie palpabled. Augmentation du taux de LDH à une valeur supérieure aux limites normales de
l’intervalle de référence institutionnele. LeucoérythroblastoseExigences relatives au diagnostic de la MF primaire :
Les trois critères majeurs et au moins 1 critère mineur
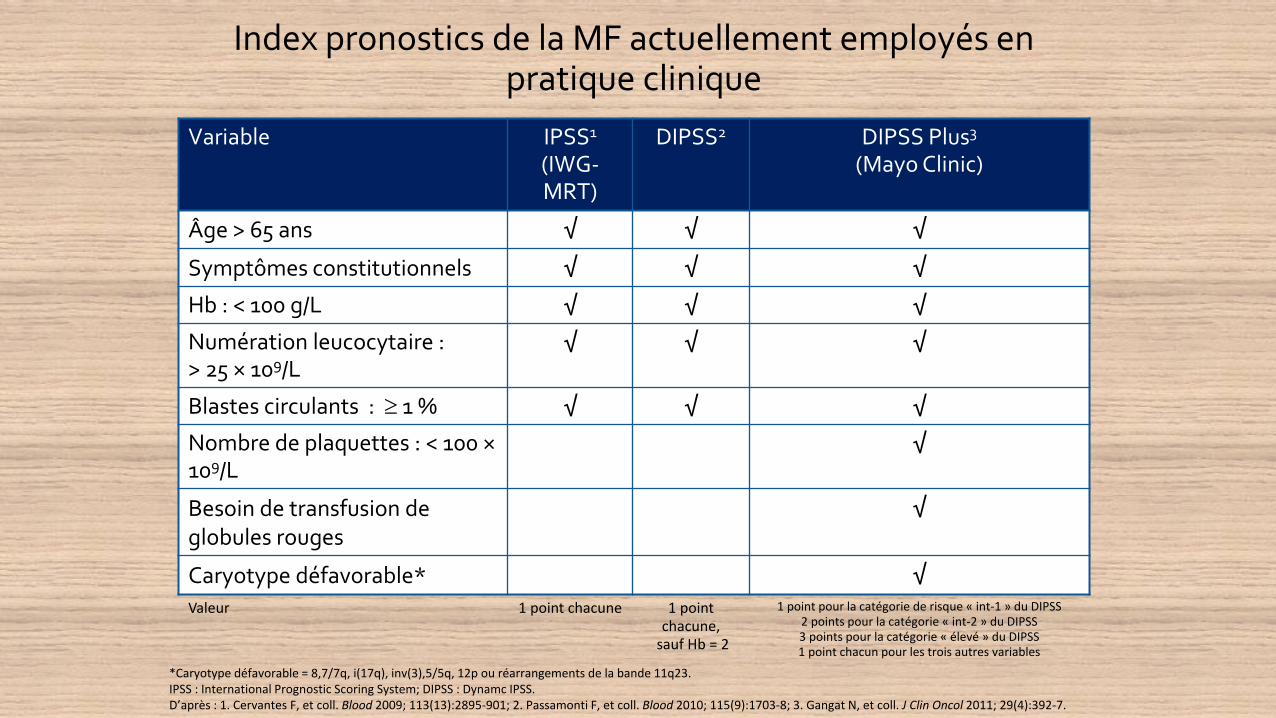
Index pronostics de la MF actuellement employés en pratique clinique
Variable IPSS1
(IWG-MRT)
DIPSS2 DIPSS Plus3
(Mayo Clinic)
Âge > 65 ans √ √ √
Symptômes constitutionnels √ √ √
Hb : < 100 g/L √ √ √
Numération leucocytaire : > 25 × 109/L
√ √ √
Blastes circulants : 1 % √ √ √
Nombre de plaquettes : < 100 ×109/L
√
Besoin de transfusion de globules rouges
√
Caryotype défavorable* √
Valeur 1 point chacune 1 point chacune,
sauf Hb = 2
1 point pour la catégorie de risque « int-1 » du DIPSS2 points pour la catégorie « int-2 » du DIPSS3 points pour la catégorie « élevé » du DIPSS1 point chacun pour les trois autres variables
*Caryotype défavorable = 8,7/7q, i(17q), inv(3),5/5q, 12p ou réarrangements de la bande 11q23.IPSS : International Prognostic Scoring System; DIPSS : Dynamc IPSS.D’après : 1. Cervantes F, et coll. Blood 2009; 113(13):2895-901; 2. Passamonti F, et coll. Blood 2010; 115(9):1703-8; 3. Gangat N, et coll. J Clin Oncol 2011; 29(4):392-7.
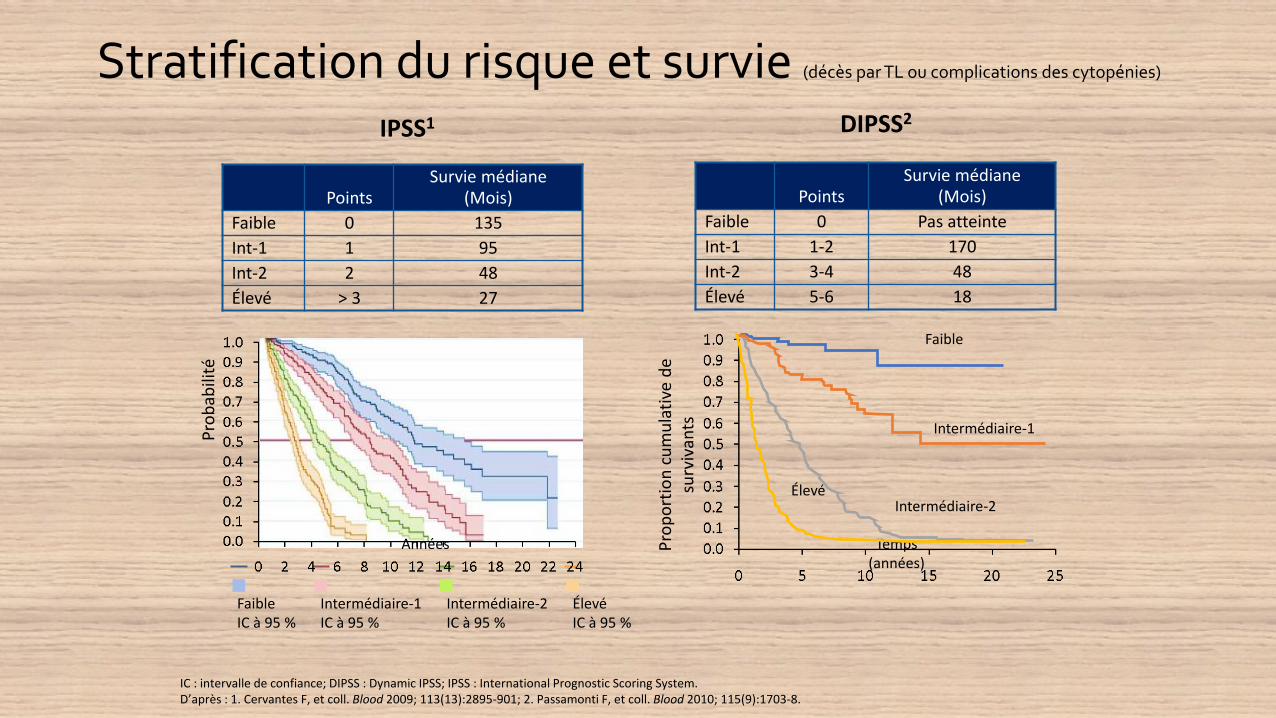
Stratification du risque et survie (décès par TL ou complications des cytopénies)
IPSS1 DIPSS2
PointsSurvie médiane
(Mois)
Faible 0 Pas atteinte
Int-1 1-2 170
Int-2 3-4 48
Élevé 5-6 18
PointsSurvie médiane
(Mois)
Faible 0 135
Int-1 1 95
Int-2 2 48
Élevé > 3 27
IC : intervalle de confiance; DIPSS : Dynamic IPSS; IPSS : International Prognostic Scoring System.D’après : 1. Cervantes F, et coll. Blood 2009; 113(13):2895-901; 2. Passamonti F, et coll. Blood 2010; 115(9):1703-8.
Faible Intermédiaire-1 Intermédiaire-2 ÉlevéIC à 95 % IC à 95 % IC à 95 % IC à 95 %
Années
Pro
bab
ilité
Temps (années)
Pro
po
rtio
n c
um
ula
tive
de
surv
ivan
ts
Faible
Intermédiaire-1
Intermédiaire-2Élevé

Objectifs de traitement de la MF
• Diminuer la prolifération excessive de tissu myéloide
• Diminuer la splénomégalie ainsi que les symptômes constitutionnels
• Diminuer les cytopénies• Minimiser le risque de transformation en
leucémie.

Objectifs thérapeutiques et options de traitement contre la myélofibrose (MF)
D’après : Barbui T, et coll. J Clin Oncol 2011;29(6):761-770, Vannucchi AM. Hematology Am Soc Hematol Educ Program 2011; 2011:222-30; et Novartis Pharma Canada inc. Monographie de Jakavi. Date de révision : 23 novembre 2015.
Options Éléments à prendre en compte
Approche curative
• Seulement permise par la greffe allogénique de cellules souches• Patients relativement jeunes et en bonne santé présentant un INT-2 ou élevé• Précédée d’un conditionnement myéloablatif ou de réduction des doses
Traitement de la maladie
Ruxolitinib : (traitement de 1ère ligne pour les patients à risque int 2 ou élevé)• Approuvé pour le traitement de la splénomégalie et/ou des symptômes qui y sont associés
chez les patients adultes atteints de myélofibrose primaire (aussi connue sous le nom de myélofibrose idiopathique chronique), de myélofibrose consécutive à une polycythémievraie ou de myélofibrose consécutive à une thrombocythémie essentielle
• Dans le cadre d’essais cliniques, il a entraîné une diminution du volume de la rate, une diminution des symptômes constitutionnels et une amélioration de la qualité de vie en plus de présenter un avantage sur le plan de la survie
Traitement des symptômes
Santé Canada n’a approuvé aucun traitement conventionnel contre la MF (tous empiriques)
• Androgènes, corticostéroïdes, antinéoplasiques, agents érythropoïétiques, immunomodulateurs, interféron
• Destiné aux patients qui ne sont pas admissibles à une transplantation• Maîtrise des symptômes et amélioration de la qualité de vie
Étude clinique /traitement par un
inhibiteur de la JAK†
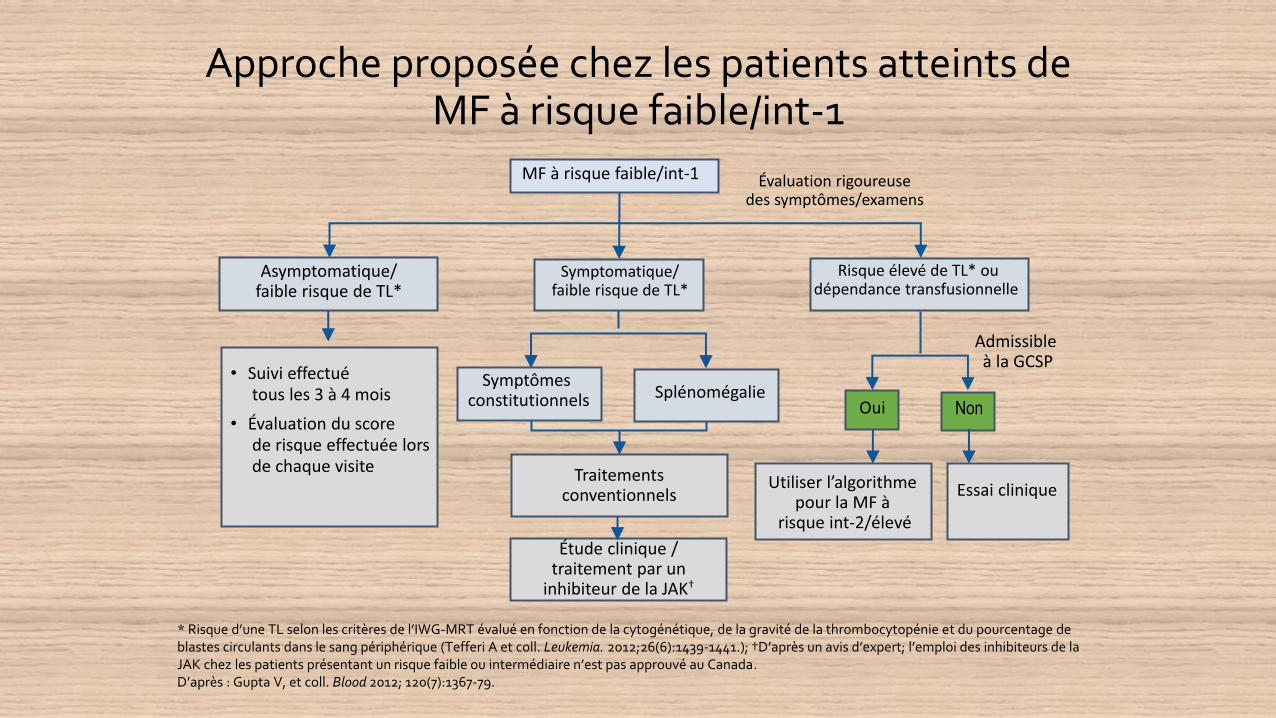
Approche proposée chez les patients atteints de MF à risque faible/int-1
* Risque d’une TL selon les critères de l’IWG-MRT évalué en fonction de la cytogénétique, de la gravité de la thrombocytopénie et du pourcentage de blastes circulants dans le sang périphérique (Tefferi A et coll. Leukemia. 2012;26(6):1439-1441.); †D’après un avis d’expert; l’emploi des inhibiteurs de la JAK chez les patients présentant un risque faible ou intermédiaire n’est pas approuvé au Canada.D’après : Gupta V, et coll. Blood 2012; 120(7):1367-79.
Évaluation rigoureusedes symptômes/examens
• Suivi effectuétous les 3 à 4 mois
• Évaluation du scorede risque effectuée lorsde chaque visite
Admissible à la GCSP
Utiliser l’algorithme pour la MF à
risque int-2/élevé
Essai clinique
MF à risque faible/int-1
Symptomatique/faible risque de TL*
Asymptomatique/faible risque de TL*
Risque élevé de TL* oudépendance transfusionnelle
Symptômes constitutionnels Splénomégalie
Oui Non
Traitements conventionnels
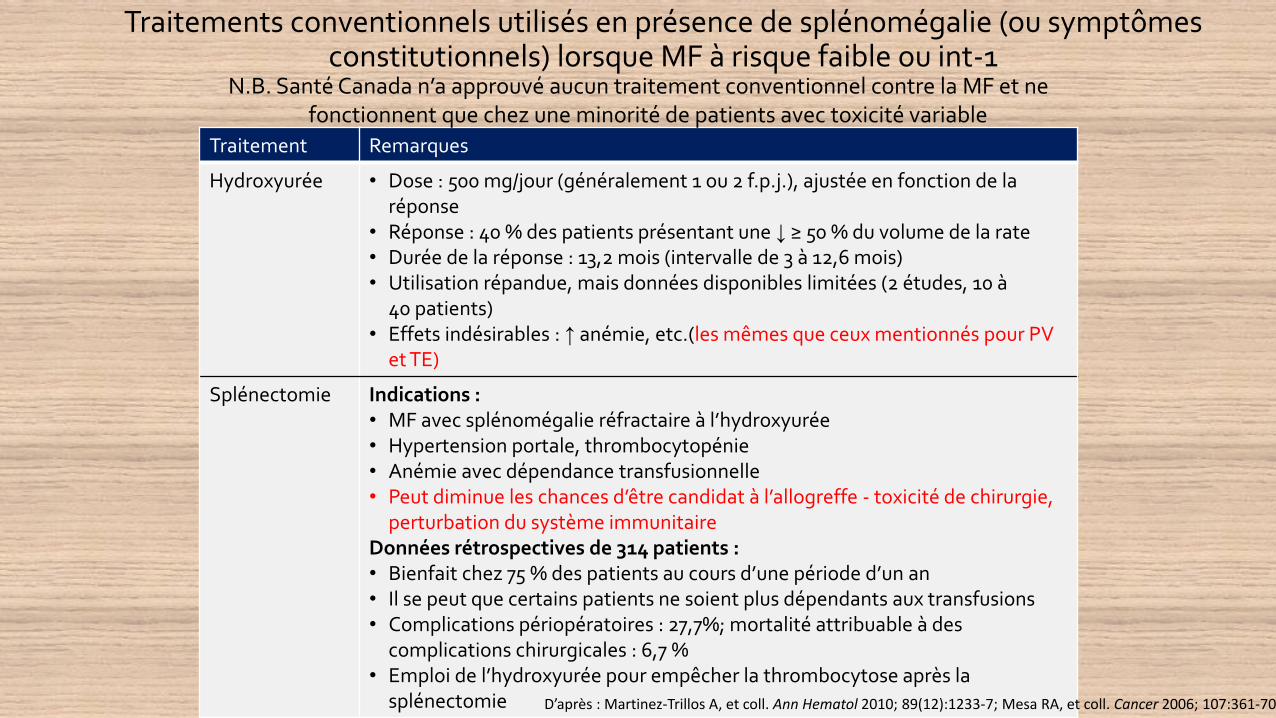
Traitements conventionnels utilisés en présence de splénomégalie (ou symptômes constitutionnels) lorsque MF à risque faible ou int-1
Traitement Remarques
Hydroxyurée • Dose : 500 mg/jour (généralement 1 ou 2 f.p.j.), ajustée en fonction de la réponse
• Réponse : 40 % des patients présentant une ↓ ≥ 50 % du volume de la rate• Durée de la réponse : 13,2 mois (intervalle de 3 à 12,6 mois)• Utilisation répandue, mais données disponibles limitées (2 études, 10 à
40 patients)• Effets indésirables : ↑ anémie, etc.(les mêmes que ceux mentionnés pour PV
et TE)
Splénectomie Indications : • MF avec splénomégalie réfractaire à l’hydroxyurée• Hypertension portale, thrombocytopénie• Anémie avec dépendance transfusionnelle• Peut diminue les chances d’être candidat à l’allogreffe - toxicité de chirurgie,
perturbation du système immunitaireDonnées rétrospectives de 314 patients :• Bienfait chez 75 % des patients au cours d’une période d’un an• Il se peut que certains patients ne soient plus dépendants aux transfusions• Complications périopératoires : 27,7%; mortalité attribuable à des
complications chirurgicales : 6,7 %• Emploi de l’hydroxyurée pour empêcher la thrombocytose après la
splénectomie D’après : Martinez-Trillos A, et coll. Ann Hematol 2010; 89(12):1233-7; Mesa RA, et coll. Cancer 2006; 107:361-70.
N.B. Santé Canada n’a approuvé aucun traitement conventionnel contre la MF et ne fonctionnent que chez une minorité de patients avec toxicité variable

Traitements conventionnels utilisés en MF à risque faible/int-1 (patients anémiques seulement)(1/2)
Traitement Remarques
Darbépoïétine alfa
• Dose : 150-300 mcg par voie sous-cutanée une fois par semaine• Cible : Patients n’ayant subi aucune transfusion et dont le taux
d’Hb < 100 g/L• Réponse : 25 à 50 % des patients (durée médiane d’un an)• Peut aggraver la splénomégalie
Érythropoïétine • Dose : 20 000-40 000 U par voie sous-cutanée une fois par semaine (ne
fonctionne pas toujours. Les transfusions font partie de la panoplie de traitements, avec la chélation qui va avec.)
Prednisone • Dose : 0,5 mg/kg par voie orale une fois par jour• Réponse : 15 à 25 % des patients pendant une période d’un à deux ans• Effets indésirables à long terme
Androgènes
• Dose : Énanthate de testostérone, 400 à 600 mg par voie intramusculaire une fois par semaine
• Réponse : 30 % des patients pendant une période d’un an• Nécessite une surveillance hépatique et un dépistage du cancer de la
prostate
D’après : Tefferi A. Am J Hematol 2013; 88:142-50; Tefferi A. Blood 2011; 117(13):3494-504; Cervantes F, et coll. Cancer J 2007; 13:377-83; Cervantes F, et coll. Brit J Haematol 2005; 129(6):771-5; Tefferi A, et coll. J Clin Oncol 2009; 27:4563-9; Tabarroki A, et coll. Expert Opin Inv Drugs 2012; 21(8):1141-54; et Cervantes F. Blood 2014; 124(17):2635-42.

Traitements conventionnels utilisés en MF à risque faible/int-1 (patients anémiques seulement)(2/2)
Traitement Remarques
Danazol • Dose : 600 mg par voie orale une fois par jour• 37% de réponses sur une période de 6 à 24 mois• Toxicité modérée
Immunomodulateurs • Doses :− Thalidomide 50 à 200 mg par voie orale/jour (augmenter de 50 à
200 mg toutes les semaines jusqu’à une dose de 400 à 800 mg/jour) − Lénalidomide 10 mg par voie orale/jour (5 mg/jour si le nombre de
plaquettes < 100 x 109/L)− Pomalidomide 2 mg par voie orale une fois par jour
• ↓ de 20 à 40 % de l’anémie; ↓ de 20 à 60 % de la thrombocytopénie; ↓ de 1 à 40 % de la splénomégalie
• Lénalidomide : Efficacité ++ en présence de l’anomalie del(5q31)• Pomalidomide : aucun effet sur la splénomégalie, effet modeste sur
l’anémie• Ajout de corticostéroïdes :↑ de la cytotoxicité et ↑ de la tolérance aux
immunomodulateurs
N.B. Santé Canada n’a approuvé aucun traitement conventionnel contre la MF
D’après : Tefferi A. Am J Hematol 2013; 88:142-50; Tefferi A. Blood 2011; 117(13):3494-504; Cervantes F, et coll. Cancer J 2007; 13:377-83; Cervantes F, et coll. Brit J Haematol 2005; 129(6):771-5; Tefferi A, et coll. J Clin Oncol 2009; 27:4563-9; Tabarroki A, et coll. Expert Opin Inv Drugs 2012; 21(8):1141-54; et Daver N et coll. Leuk Res. nov. 2013; 37(11): 1440–1444.
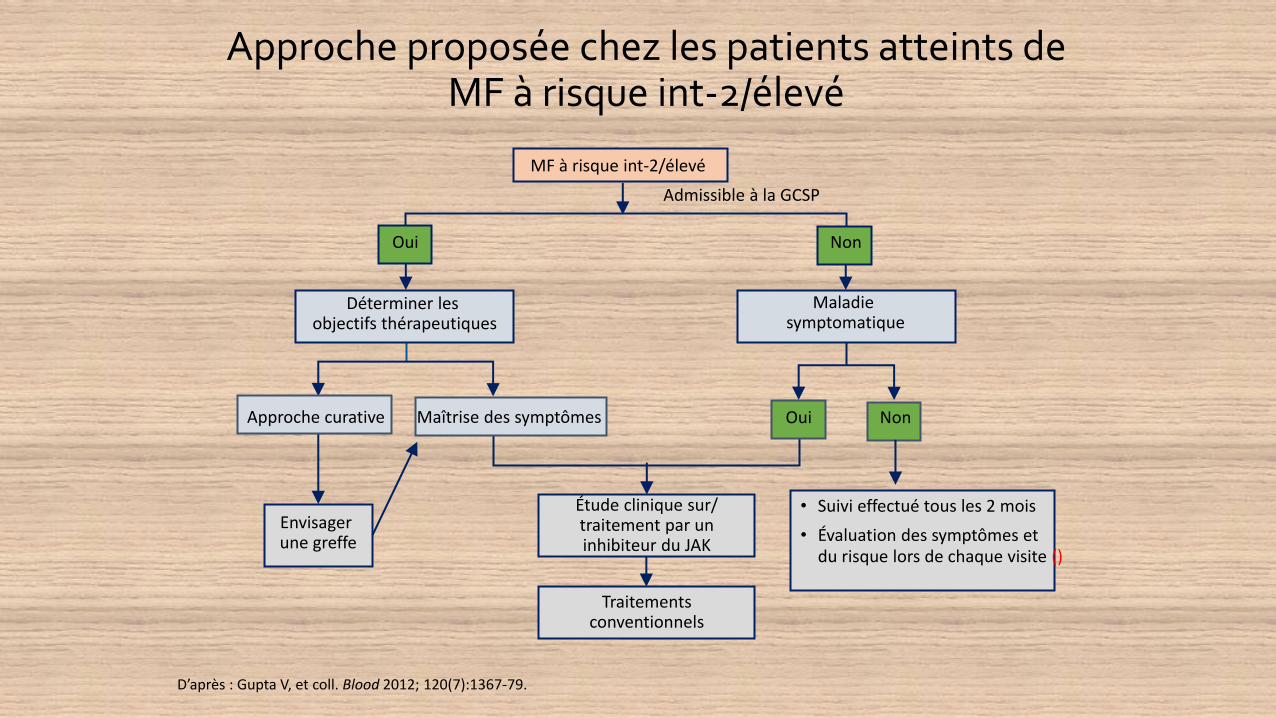
Approche proposée chez les patients atteints de MF à risque int-2/élevé
D’après : Gupta V, et coll. Blood 2012; 120(7):1367-79.
Étude clinique sur/traitement par un inhibiteur du JAK
Admissible à la GCSP
• Suivi effectué tous les 2 mois
• Évaluation des symptômes etdu risque lors de chaque visite ()
Envisager une greffe
MF à risque int-2/élevé
Déterminer les objectifs thérapeutiques
Maladie symptomatique
Approche curative Maîtrise des symptômes Oui Non
Oui Non
Traitements conventionnels

• C’est un agent antinéoplasique, inhibiteur de la tyrosine-kinase JAK 21 (thérapie ciblée)
• Approuvé par Santé Canada en juin 2012.
• N’est pas sur la liste du NIOSH
• Médicament d’exception, utilisé dans le traitement de la MF à risque int 2 ou élevé (remboursé par la RAMQ en médicament d’exception)
• 1. Monographie de produit, produite par Novartis Canada Inc
Ruxolitinib 1/2

Ruxolitinib (2/2)Mode d’action
La signalisation tributaire des enzymes JAK est • déréglée chez les patients atteints de MF indépendamment de la présence ou de l’absence de la mutation du gène porteur de JAK2V617F. 1
Quel que soit leur état mutationnel, les patients atteints de MF •répondent à l’inhibition de la voie JAK3
L’inhibition de JAK• 2 entraîne une diminution rapide du taux de cytokines inflammatoires (et donc diminution de la fibrose) associées à la MF4
Mode d’action: Le • ruxolitinib inhibe la voie de signalisation JAK-STAT.(Autrement dit, inhibe l’activité enzymatique de JAK 2 (muté ou non) et limite la myéloprolifération (mégacaryocytes↓cytokines ↓ tissus cicatriciel ↓)Agit autant au niveau de la • rate que de la moelle osseuse
1. 1.Monographie de produit, produite par Novartis Canada Inc

Ruxolitinib pour le traitement de la MF : Raisons justifiant son emploi
Les essais cliniques démontrent que le ruxolitinib procure des bienfaits significatifs comparativement au placebo et au meilleur traitement existant sur les plans suivants :
• Réduction du volume de la rate• Diminution des symptômes constitutionnels• Amélioration de la qualité de vie• Diminution du taux de cytokines inflammatoires sérique• Stabilisation ou diminution de la fibrose • Avantage potentiel sur le plan de la survie
D’après : Verstovsek S, et coll. N Engl J Med 2012; 366(9):799-807; 2. Harrison CN, et coll. N Engl J Med 2012; 366(9):787-98.3. Verstovsek S, et coll. Blood 2011; 118(21) : Résumé 278.

Effets secondaires principaux du Ruxolitinib 1
Hématologiques❖
An• émie (82,4 %), (besoins transfusionnels qui augmentent au début (pendant environ 8 à 12 semaines) et qui se stabilisent par la suite) Thrombocytop• énie (69,8 %) Ecchymoses (• 21,3%)Neutrop• énie (15,6 %),
Non hématologiques❖
Hausse des concentrations d’ALT (• 26,9 %)Hausse des concentrations d’AST (• 19,3%)Hypercholestérolémie• ( 16,6%)Étourdissements (• 15,0 %)
• Céphalées (13,6 %).
• 1. Monographie de produit, produite par Novartis Pharma Canada inc.

Ruxolitinib : Examens de départ et surveillance pendant le suiviExamens ou surveillance
Recommandations
Formule sanguine• Avant le début du traitement et toutes les 2 à 4 semaines jusqu’à
stabilisation de la dose utilisée, puis au besoin selon l’état clinique du patient1
Bilans des fonctions hépatiques et rénales
• Il faut effectuer des bilans des fonctions hépatiques et rénales avant de commencer un traitement et périodiquement par la suite1
Surveillance de l’innocuité du traitement sur le plan cardiaque
• Surveillance du pouls et de la pression artérielle1
• ECG avant l’amorce du traitement et périodiquement par la suite afin de déceler un ralentissement de la fréquence cardiaque et un allongement de l’intervalle PR1
Infections
• On doit effectuer un test cutané à la tuberculine et/ou un test de libération de l’interféron gamma avant d’amorcer le traitement afin de déceler la présence d’une infection tuberculeuse chez les patients à risque. Les résultats de ces épreuves doivent cependant être interprétés avec prudence chez les patients gravement malades ou immunodéprimés vu l’éventualité d’un résultat faussement négatif 1
• Sérologie afin de dépister une hépatite1
• Envisager de vacciner contre le zona1
D’après : 1. Novartis Pharma Canada inc. Monographie de Jakavi. Date de révision : 23 novembre 2015; 2. Avis du comité de professionnels.

Considérations posologiques sur le ruxolitinib (1/2)La dose d’attaque• recommandée est fondée sur le nombre de plaquettes au départ.Réalisation d’une formule sanguine• : avant d’entreprendre le traitement, puis toutes les 2 à 4 semaines jusqu’à stabilisation de la dose utilisée et au besoin selon l’état clinique du patient par la suite.
Nombre de plaquettes > 200 000/mm³ → 20 mg 2 f.p.j. par voie orale
Nombre de plaquettes entre 100 000 et 200 000/mm3 → 15 mg 2 f.p.j. par voie orale
Nombre de plaquettes entre 50 000 et 100 000/mm3 → 5 mg 2 f.p.j par voie orale
• Il faut envisager de réduire la dose si le nombre de plaquettes diminue pour éviter toute interruption du traitement en raison d’une thrombocytopénie.
• Il faut interrompre le traitement si le nombre de plaquettes chute sous le seuil de 50 000/mm3. Lorsque le nombre de plaquettesfranchit de nouveau cette valeur minimale, il est possible de reprendre le traitement. Même en deça du seuil de 50 000/mm3 lletraitement peut être donné, si pas d’autres alternatives.
D’après : Novartis Pharma Canada inc. Monographie de Jakavi. Date de révision : 23 novembre 2015.
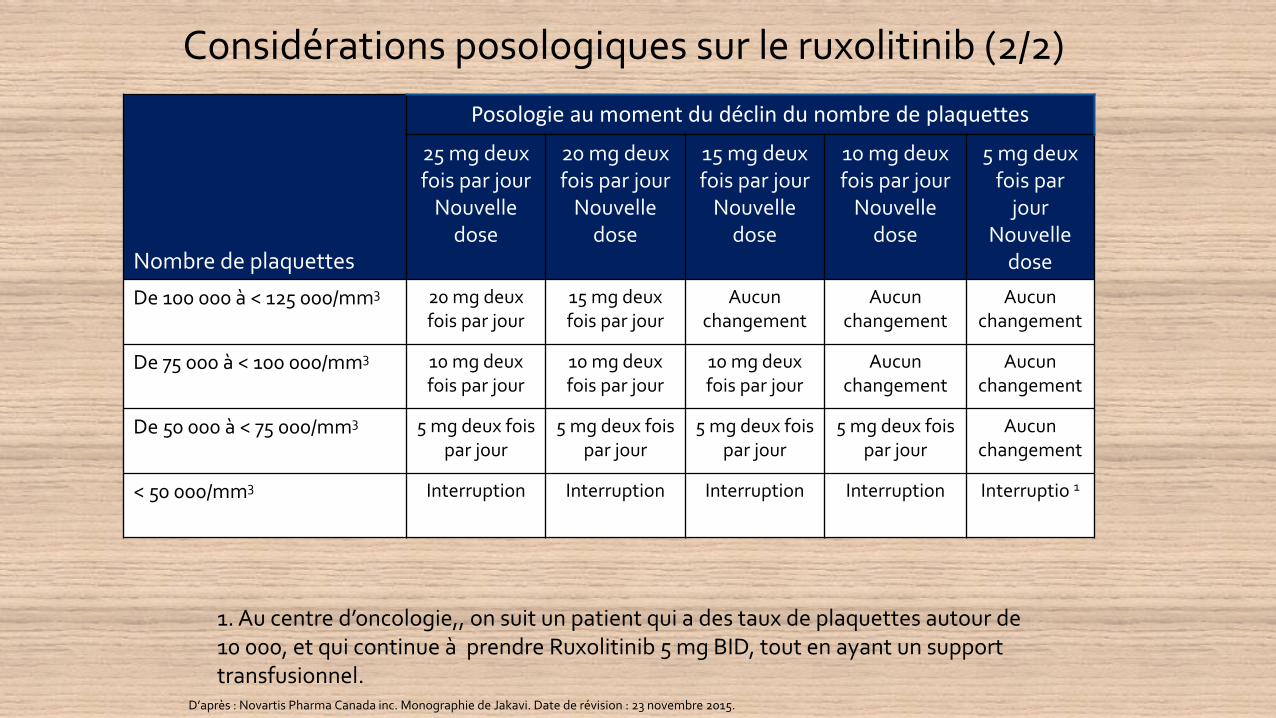
Considérations posologiques sur le ruxolitinib (2/2)
Nombre de plaquettes
Posologie au moment du déclin du nombre de plaquettes
25 mg deux fois par jour
Nouvelle dose
20 mg deux fois par jour
Nouvelle dose
15 mg deux fois par jour
Nouvelle dose
10 mg deux fois par jour
Nouvelle dose
5 mg deux fois par
jourNouvelle
dose
De 100 000 à < 125 000/mm3 20 mg deux fois par jour
15 mg deux fois par jour
Aucun changement
Aucun changement
Aucun changement
De 75 000 à < 100 000/mm3 10 mg deux fois par jour
10 mg deux fois par jour
10 mg deux fois par jour
Aucun changement
Aucun changement
De 50 000 à < 75 000/mm3 5 mg deux fois par jour
5 mg deux fois par jour
5 mg deux fois par jour
5 mg deux fois par jour
Aucun changement
< 50 000/mm3 Interruption Interruption Interruption Interruption Interruptio 1
D’après : Novartis Pharma Canada inc. Monographie de Jakavi. Date de révision : 23 novembre 2015.
1. Au centre d’oncologie,, on suit un patient qui a des taux de plaquettes autour de 10 000, et qui continue à prendre Ruxolitinib 5 mg BID, tout en ayant un support transfusionnel.

Conseils pratiques relatifs au ruxolitinibAnémie• Patients n’ayant pas de
dépendance transfusionnelle :Poursuivre le traitement à la même dose; il est possible que le taux d’hémoglobine s’améliore
• Patients ayant une dépendance transfusionnelle : réduire la dose de ruxolitinib ou envisager d’ajouter ou de remplacer ce médicament par des agents stimulant l’érythropoïétine
Interruption du traitement Diminution progressive de la •dose; n’arrêter le traitement de façon brusque car risque de tempête de cytokine
Éducation des patients :Les informer de la réapparition •des symptômes en cas d’interruption1
Stratégies permettant •d’accroître l’adhésion au traitement
S’assurer que les patients disposent •d’une quantité suffisante de médicament (p. ex., lorsqu’ils partent en vacances)
D’après : 1. Verstovsek S, et coll. N Engl J Med 2012; 366(9):799-807. Annexe supplémentaire.

Ruxolitinib (Jakavimd) NOM DU PATIENT : ___________________________________
DATE DE NAISSANCE : ____________________________________
Rédaction : mai 2015 ; mise à jour : septembre 2016. Préparé par le département de pharmacie du CHUM en collaboration avec le comité sur l’évolution
de la pratique en soins pharmaceutiques de la Direction québécoise de cancérologie du ministère de la Santé et des Services sociaux.
Mise en garde
Les meilleurs soins ont été apportés à ces informations qui sont basées sur des données disponibles au moment de leur rédaction.
Pour en savoir plus, veuillez-vous référer à la monographie du produit.
INFORMATIONS À L’INTENTION DES PHARMACIENS COMMUNAUTAIRES
Afin d’optimiser les soins au patient, voici des informations supplémentaires concernant le nouveau traitement
Oui Non Notes
Formulaire de médicament/patient d’exception faxé ce jour à la
RAMQ
OU complété et remis au patient pour ses assurances privées
Patient inscrit à un programme d’accès de la compagnie
Enseignement effectué par le pharmacien d’oncologie SVP valider la compréhension
Interactions médicamenteuses vérifiées par le pharmacien d’oncologie À revérifier selon votre dossier-patient
Commentaires : ______________________________________________________________________
______________________________________________________________________
Coordonnées
Pharmacie oncologie Tél. :
Pharmacien d’oncologie Nom : Tél. :
Infirmière pivot Nom : Tél. :
Principaux effets indésirables à surveiller (liste non-exhaustive)
Myélosuppression
- Il s’agit de l’effet secondaire le plus commun de ce médicament et est généralement réversible en
réduisant la dose ou en interrompant temporairement le traitement
- Peut survenir de quelques jours à quelques semaines après le début du traitement
- La thrombocytopénie (70%) et l’anémie (environ 80%) sont plus fréquentes que la neutropénie
(environ 20%). - Recommander les mesures de prévention des infections.
Référer si : - fièvre ≥38°C x 1h ou >38,3°C ou symptômes d’infection (frisson, toux, maux de gorge, douleur
urinaire, etc.) (urgence) - signes et symptômes d’infections, de saignements ou d’anémie (fatigue importante, essoufflement,
etc.).
Saignements (environ 30 %)
- Les hémorragies rapportées durant le traitement étaient majoritairement des ecchymoses
- Suivre les signes et symptômes de saignement.
Référer si : - Saignement digestif, hématurie ou hémorragie intracrânienne suspectée
- Saignement mineur >15 minutes ou accompagné d’étourdissement ou ecchymoses spontanées.
Diarrhées (environ 14 %)
- Favoriser une hydratation suffisante.
- Légères à modérées (jusqu’à 6 selles molles par jour) : lopéramide 2 cos STAT puis 1 co après chaque
selle liquide (ad 8 cos/j).
Référer si : - Toutes diarrhées qui ne s’atténuent pas après 24 heures de traitement adéquat.
- Diarrhées sévères (> 7 selles molles/j), sang dans les selles, fièvre, altération de l’état mental,
déshydratation.
Suggestions de paramètres à surveiller en milieu communautaire : -Ecchymose, étourdissement, céphalée, tension artérielle, fréquence cardiaque
Références utiles à consulter
- Site du Groupe d’étude en oncologie du Québec (inscription en ligne gratuite): http://www.geoq.info/ - Document «OnCible» disponible sur le GEOQ et sur Vigilance Santé
- Cancer Care Ontario (anglais et français): http://cancercare.on.ca/toolbox/patientdruginfo/ - BC Cancer Agency (anglais): http://www.bccancer.bc.ca/HPI/DrugDatabase/DrugIndexPro/default.htm

Rôle de l’infirmière dans le suivi des patients avec MF
Gestion des symptômesGestion des effets secondaires des traitements Traitement (Ex: transfusions)Suivi des résultats des prises de sang, et communiquer résultats aux patientsSupport psycho-social (aspect particulièrement important chez les patients avec MF) Approche interdisciplinaire est essentielle, on suit le patient dans sa globalité.

Histoire de cas

• 2012 Anémie macrocytaires c hémolyse, suivi d’abord à l’hôpital de Ste-Eustache. (Entre décembre 2008 et novembre 2012, hb 130 67 )
• Entre novembre 2012 et février 2013, 10 culots sanguins reçus.
• Plusieurs examens effectués non-concluants.• MO ne met pas en évidence de dx clair, par ailleurs
hépatomégalie avec splénomégalie à 16 cm• Dans ce contexte, référé à HND
Histoire de Monsieur L.

• Tests effectués à nouveau dont , JAK 2, hep, VIH, doppler (hépatosplénomégalie) et BMO
• MO montre mégacaryocytes pléomorphes avecmyélofibrose suggestifs d’une MF primaire.
• Doppler montre une rate à 18 cm (taille normale d’une rate < 11-12 cm)
• MF soupçonnée, même si recherche de mutation JAK 2 est toujours en cours.

Entre février • 2013 et mars 2013, GB sont passés de 47,7 à 26,5 donc cytoréductionn’est pas indiquée. (dx de MF pas encore confirmé)
Eprex• néanmoins prescrit pour diminuer besoins transfusionnels (moelle fibrosée)
Avril • 2013: Rate à 5 cm sous le rebord costal, JAK 2 positif, MF

• Pt à risque élevé avec un IPSS à 3 (Hb , leucocytes et rate) donc Roxolitinib prescrit en avril 2014 à 20 mg BID (plaquettes à 376 000) en plus de l’Eprex et du support transfusionnel (qui était déjà en place)
• Suivi étroit de sa formule sanguine ainsi que de sa fonction rénale et hépatique.

• Mai 2013: Amélioration de l’état global, pas de symptômes B. dlr rebord costal
• Août 2013:État stable, toujours transfusion dépendant, ferritine à 2500 Echo montre une régression de la taille de la rate. LDH (enzyme) à 1600.
• Pt répond bien au Ruxolitinib, et augmenté a 25mg BID. Par ailleurs Eprex cessé. (échec)

• Oct 2013: Réponse toujours adéquate au point de vue clinique et radiologique. Rate à 1cm sous le rebord costal. (donc par rapport à avril. )Par ailleurs, Exjade débuté, contrôle de la créatinine + serré.
• Février 2014 : Hospitalisé pour arthralgie et infection virale. rate 4 cm sous le rebord costal.
• Pendant l’année suivante, jusqu’en février 2015, état demeure stable, même si au mois d’octobre de la rate à 8cm sous le rebord costal.
• Décès le 28 avril 2015 des complications d’une pneumomie.

Questions ?