frÉquence et rÉparation de dommages À l'adn associÉs …
TRANSCRIPT

SANDRINE LACOSTE FRÉQUENCE ET RÉPARATION DE DOMMAGES À
L'ADN ASSOCIÉS À LA 4-(MÉTHYLNITROSAMINO)-1-(3-PYRIDYL)-1-
BUTANONE (NNK), UNE NITROSAMINE SPÉCIFIQUE DU TABAC, ÉVALUÉS À L'AIDE DU
TEST DES COMÈTES.
Thèse présentée à la Faculté des études supérieures de l’Université Laval
dans le cadre du programme de doctorat en biologie cellulaire et moléculaire pour l’obtention du grade de Philosophiae Doctor (Ph. D.)
FACULTÉ DE MÉDECINE
UNIVERSITÉ LAVAL QUÉBEC
JANVIER 2007 © Sandrine Lacoste, 2007

ii
Résumé La fumée de tabac contient plusieurs substances carcinogènes qui mènent à la formation
constante de petites quantités de dommages à l'ADN dans les poumons des fumeurs ainsi
que des non-fumeurs exposés à la fumée environnementale de tabac. La 4-
(méthylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) est l'une de ces substances et elle
semble plus particulièrement associée avec le développement des adénocarcinomes, la
forme de cancer pulmonaire dont l'incidence progresse le plus rapidement ces dernières
années. Dans les cellules pulmonaires, la NNK est bioactivée via des cytochromes P450 en
intermédiaires réactifs capables de méthyler ou de pyridyloxobutyler l'ADN. Les
dommages résultant de ces deux modes d'activation de la NNK peuvent être investigués
séparément en utilisant des analogues qui génèrent sélectivement l'un ou l'autre type
d'intermédiaires réactifs. Le test des comètes est une technique simple, très sensible et
couramment utilisée pour étudier au niveau cellulaire les dommages à l'ADN qui sont peu
fréquents. Les travaux présentés dans cette thèse montrent que certains des dommages
résultant de l'activation de la NNK peuvent être investigués de manière spécifique à l'aide
de cette technique, et ce à des fréquences de dommages qui se rapprochent de celles
correspondant à une exposition réelle à la fumée de tabac. Parmi ces dommages, un type
d'adduits encore inconnu associé à la pyridyloxobutylation de l'ADN a pu être mis
indirectement en évidence. Il s'agit vraisemblablement de la forme formamidopyrimidine
(fapy) d'une lésion primaire formée dans les cellules. La vitesse de réparation d'un type de
dommage influe sur le risque qu'il a d'être impliqué dans la transformation maligne des
cellules. La disparition des dommages dans le temps a pu être suivie avec le test des
comètes afin d'investiguer la réparation dans des cellules capables ou pas de bioactiver la
NNK. Le suivi post-traitement des dérivés fapy associés à la pyridyloxobutylation de
l'ADN, a montré un phénotype ne dépendant pas du type cellulaire mais plutôt du statut de
p53 dans les cellules. En effet, au lieu de diminuer après la fin du traitement, la fréquence
des adduits fapy dans les fibroblastes augmente dans un premier temps et ce, seulement
dans les cellules ayant une protéine p53 fonctionnelle. La nature de ce phénotype particulier
n'est pas clairement identifiée, mais elle est vraisemblablement liée à la réparation des
dommages à l'ADN.

iii
Abstract Tobacco smoke contains several carcinogens that lead to the frequent formation of rare
DNA damage in lungs of smokers. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)
is one of these substances that seems more particularly associated with the development of
adenocarcinoma. During the last 30 years, the frequency of this lung cancer type has
increased significantly. In lung cells, cytochromes P450 can bioactivate NNK into reactive
species capable of either methylating or pyridyloxobutylating DNA. The use of analogs
capable of generating only one type of NNK-associated reactive species allows to
investigate methylation and pyridyloxobutylation separately. The comet assay is a simple
and sensitive technique that is commonly used to investigate low frequency DNA damage
at the cellular level. The work presented here show how some of the NNK-related DNA
damage can be investigated specifically with this technique at damage frequencies that are
relevant to a real exposure to cigarette smoke. One of the adduct type resulting of DNA
pyridyloxobutylation that we studied here had never been demonstrated before. It
corresponds likely to the formamidopyrimidine (fapy) form of a lesion primarily formed in
cells. The repair rate of a damage type influences the probability that it has to be implicated
in mutagenesis. The time course of different damage types was documented with the comet
assay in order to investigate the repair of NNK-related damage in different cell types that
can either bioactivate NNK or not. When the fapy adducts associated with
pyridyloxobutylation were investigated post-treatment, their time course did not depend on
the cell type but showed a p53-dependant phenotype. In fact, instead of decreasing because
of repair, the frequency of these fapy adducts in fibroblasts first increased post-treatment
and this increase seemed associated with p53 proficiency. The cause of this phenotype is
not clearly elucidated but it should be related to DNA damage repair.

iv
Remerciements Mon parcours pour arriver à cette étape finale de la préparation de ma thèse de doctorat a
été plutôt long et difficile. Mais loin de me décourager de la recherche, ce cheminement
m’a donné le genre de force que l'on gagne à avoir su surmonter les difficultés. J'ai
maintenant acquis la conviction profonde d'avoir choisi la bonne voie et de faire ce pour
quoi je suis faite.
Evidemment, je n’en suis pas arrivée là toute seule et je tiens ici à remercier tous ceux qui
m’ont aidée et soutenue, de près ou de loin, dans ce long travail.
Je tiens avant tout à remercier mon directeur de recherche, le Docteur Régen Drouin, pour
m'avoir accueillie dans son laboratoire. Il m'a offert la chance de travailler dans le domaine
qui m'intéresse et je lui en serai toujours reconnaissante. Sa générosité, sa ténacité, et
surtout sa capacité à gérer le stress forcent mon admiration plus que je ne saurais
l'exprimer. Merci également à André Castonguay pour avoir accepté la co-direction de ce
travail de thèse, me donnant ainsi l'occasion d'entrer dans le monde complexe et fascinant
de la carcinogenèse associée à la NNK.
Les personnes que j’ai cotoyées quotidiennement dans le laboratoire m'ont apporté
énormément de support, qu'il soit d'ordre technique ou moral. Merci, Nathalie, pour ta
gentillesse, ton énergie et pour ton rire fabuleux que l’on a souvent l’occasion d’entendre.
Merci aussi d’avoir été là pour répondre à mes questions et pour écouter mes complaintes
de toutes natures. Merci, Isabelle et Marc, d'être des piliers de ce laboratoire et de
contribuer à en faire une machine qui avance. Merci, Mélissa, pour ta gentillesse et ta
vision résolument positive du monde. Merci à tous les étudiants , en particulier à Patrick
Rochette, avec qui j’ai beaucoup discuté et argumenté, et à Jean-François Cloutier, mon
prédécesseur dans l’étude de la NNK, et qui a toujours pris le temps de répondre à mes
questions, même après son départ. Merci à tous les autres qui participent à faire de ce
laboratoire un endroit plein de vie : Marie-Chantal, Denis, Macoura, Kada, Oumar, Walid.
Merci enfin, à Christiane qui prend toujours le temps de nous écouter, même quand elle est
débordée de travail.

v
Il me faut aussi remercier tous ceux qui font partie de ma vie ou qui l’on traversée au fil des
années. Comme ils ne sont pas tous susceptibles de lire ces pages un jour, je ferai en sorte
de les remercier en personne. Mais je veux ici souligner l'influence de personnes qui me
sont chères. Merci à mes parents, Paulette et Bernard, de m’avoir donné tout ce qu’ils
pouvaient et probablement même un peu plus. Merci à Maryse et Nanou pour tout ce
qu’elles m'ont apporté d’écoute et de réconfort depuis ma plus tendre enfance. Merci aussi
à Géraldine pour son amitié durable et qui ne se dément pas au fil des années.
Sans vous, je ne serais pas la personne que je suis aujourd’hui.

vi
"Le début de la science moderne date du moment où aux questions générales se sont
substituées des questions limitées ; où au lieu de demander : "Comment l'univers a-t-il été créé ? De quoi est faite la matière ? Quelle
est l'essence de la vie ?", on a commencé à se demander : "Comment tombe une pierre ? Comment l'eau coule-t-elle dans un tube ? Quel est le cours du sang dans le corps ?".
Ce changement a eu un résultat surprenant. Alors que les questions générales ne
recevaient que des réponses limitées, les questions limitées se trouvèrent conduire à
des réponses de plus en plus générales." François Jacob, Le jeu des possibles

vii
Contribution aux travaux publiés Les paragraphes suivants décrivent ma contribution aux articles qui sont présentés dans
cette thèse.
Chapitre 2 : Ce manuscrit a été soumis pour publication à la revue
« Médecine/Sciences ». Il est le résultat de mon travail et de celui du Docteur Patrick
Rochette qui était un étudiant au doctorat dans le laboratoire du Docteur Drouin à ce
moment-là. Ma contribution a été de concevoir le plan de l'article et d'en écrire le texte.
Patrick Rochette a conçu et réalisé les figures et a révisé le texte. Ma contribution relative à
la rédaction de cet article est de 85 %.
Chapitre 3 : Ces travaux ont été publiés dans la revue « Mutation Research. Fundamental
and molecular mechanisms of mutagenesis ». Ma contribution pour cet article concerne la
détermination du modèle expérimental, la préparation des protocoles, la réalisation des
manipulations ainsi que la rédaction de l'article. Les Docteurs Drouin et Castonguay ont
supervisé l'ensemble du projet et ont corrigé la qualité de l'écriture de l'article. Dans
l'ensemble des travaux incluant la rédaction, ma contribution relative est de 100 %.
Chapitre 4 : Ces travaux ont été soumis pour publication à la revue « Mutation Research.
Fundamental and molecular mechanisms of mutagenesis ». Ma contribution pour cet article
concerne la détermination du modèle expérimental, la préparation des protocoles, la
réalisation des manipulations ainsi que la rédaction de l'article. Les Docteurs Drouin et
Castonguay ont supervisé l'ensemble du projet et ont corrigé la qualité de l'écriture de
l'article. Dans l'ensemble des travaux incluant la rédaction, ma contribution relative est de
100 %.

viii
Table des matières Résumé....................................................................................................................................ii Abstract..................................................................................................................................iii Remerciements.......................................................................................................................iv Contribution aux travaux publiés..........................................................................................vii Table des matières............................................................................................................... viii Liste des tableaux et figures....................................................................................................x Liste des abréviations.............................................................................................................xi Chapitre 1 Introduction.....................................................................................................1
1.1 Le cancer du poumon et ses origines ......................................................................1 1.1.1 Incidence et mortalité......................................................................................1 1.1.2 Association avec la consommation de cigarettes............................................2 1.1.3 Histologie des cancers du poumon .................................................................3 1.1.4 Évolution de la cigarette en relation avec l'évolution du cancer du poumon .7
1.2 La théorie de la mutagenèse somatique ..................................................................8 1.2.1 Comment la cigarette mène au cancer du poumon ? ......................................8 1.2.2 La théorie de la mutagenèse somatique ..........................................................9
1.3 Les substances carcinogènes de la fumée de tabac...............................................10 1.4 La NNK dans les cellules pulmonaires.................................................................12
1.4.1 Détoxication et/ou activation métabolique ...................................................12 1.4.2 Les dommages à l'ADN générés par la NNK ...............................................14
1.5 Les altérations génétiques dans les cancers pulmonaires .....................................21 1.5.1 Région chromosomique 3p ...........................................................................24 1.5.2 Régions chromosomiques 9p21 ....................................................................26 1.5.3 Le gène TP53 ................................................................................................26 1.5.4 Les gènes KRAS et EGFR .............................................................................27
1.6 Quels mutagènes du tabac peuvent être associés à quelles mutations ? ...............28 1.6.1 Les points chauds de mutation de TP53 et KRAS .........................................29 1.6.2 La nature des mutations générées .................................................................34 1.6.3 Bilan..............................................................................................................35
1.7 Les systèmes de réparation des dommages à l'ADN ............................................36 1.7.1 Quel système de réparation pour quel type de dommage ? ..........................36 1.7.2 La réparation par réversion directe des dommages ......................................39 1.7.3 La réparation par excision de bases (REB)...................................................41 1.7.4 La réparation par excision de nucléotides (REN).........................................46 1.7.5 Les autres systèmes de réparation.................................................................51
1.8 Les méthodes d'analyse des dommages à l'ADN et de leur réparation.................52 1.8.1 La "ligation-mediated PCR" .........................................................................52 1.8.2 Le test des comètes .......................................................................................55
1.9 Les objectifs de l'étude..........................................................................................58 1.9.1 Les objectifs de l'article 1 (chapitre 2)..........................................................58 1.9.2 Les objectifs de l'article 2 (chapitre 3)..........................................................59 1.9.3 Les objectifs de l'article 3 (chapitre 4)..........................................................60
Chapitre 2 Comprendre la mutagenèse somatique via la cartographie des dommages à l’ADN............................................................................................................61

ix
2.1 Chapô ....................................................................................................................62 2.2 Article ...................................................................................................................63
2.2.1 Résumé (anglais)...........................................................................................64 2.2.2 Introduction...................................................................................................65 2.2.3 Le cancer de la peau et les différentes longueurs d'ondes d'UV...................67 2.2.4 Rôle de la méthylation des cytosines sur la mutagenèse par les UV............68 2.2.5 Etude du taux de réparation au niveau nucléotidique ...................................68 2.2.6 Identification de carcinogènes impliqués dans le cancer du poumon...........69 2.2.7 Lien étiologique entre l'aflatoxine et le carcinome hépatocellulaire ............71 2.2.8 Conclusion ....................................................................................................72 2.2.9 Remerciements..............................................................................................73 2.2.10 Références.....................................................................................................74 2.2.11 Légendes des figures.....................................................................................78
Chapitre 3 Formamidopyrimidine adducts are detected using the comet assay in human cells treated with reactive metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) ..........................................................................84
3.1 Résumé en français ...............................................................................................85 3.2 Article ...................................................................................................................86
3.2.1 Abstract.........................................................................................................87 3.2.2 Introduction...................................................................................................88 3.2.3 Materials and Methods..................................................................................91 3.2.4 Results...........................................................................................................95 3.2.5 Discussion...................................................................................................100 3.2.6 Acknowledgements.....................................................................................104 3.2.7 References...................................................................................................105 3.2.8 Table ...........................................................................................................110 3.2.9 Legends to Figures......................................................................................111
Chapitre 4 Repair kinetics of specific types of nitroso-induced DNA damage using the comet assay in human cells. ........................................................................117
4.1 Résumé en français .............................................................................................118 4.2 Article .................................................................................................................119
4.2.1 Abstract.......................................................................................................120 4.2.2 Introduction.................................................................................................121 4.2.3 Materials and Methods................................................................................124 4.2.4 Results.........................................................................................................127 4.2.5 Discussion...................................................................................................132 4.2.6 Acknowledgements.....................................................................................137 4.2.7 References...................................................................................................138 4.2.8 Legends to figures.......................................................................................144
Chapitre 5 Discussion...................................................................................................152 Chapitre 6 Conclusions générales.................................................................................157 Références bibliographiques (Chapitres 1 et 5) ..................................................................159

x
Liste des tableaux et figures Figure 1 : Lieu de formation dans le poumon des carcinomes épidermoïdes et des
adénocarcinomes.....................................................................................................4 Figure 2 : Image au microscope électronique représentant une bronchiole terminale qui
débouche dans les sacs alvéolaires. ........................................................................6 Figure 3 : Schéma montrant le lien entre les cigarettes, les substances carcinogènes qu'elles
contiennent et la survenue du cancer du poumon. ..................................................9 Tableau 1 : Évaluation de la participation de carcinogènes spécifiques dans le cancer du
poumon humain induit par la cigarette. ................................................................11 Figure 4 : Voies de bioactivation de la NNK et de la NNAL. .............................................13 Figure 5 : Les sites nucléophiles sensibles à l'alkylation dans l'ADN. ................................15 Figure 6 : Formation de HPB suite à l'hydrolyse acide ou thermique neutre d'un ADN
pyridyloxobutylé...................................................................................................17 Tableau 2 : La formation des adduits et leur importance dans la tumorigenèse chez le rat et
la souris. ................................................................................................................18 Tableau 3 : Les adduits de la pyridyloxobutylation de l'ADN et leur capacité à relâcher le
HPB.......................................................................................................................20 Figure 7 : Accumulation des altérations dans la progression de la carcinogenèse
pulmonaire. ...........................................................................................................22 Figure 8 : Les voies de suppresseurs de tumeurs impliquées dans le cancer du poumon....24 Figure 9 : Spectre des codons mutés dans TP53 pour les tumeurs pulmonaires de fumeurs
(tous niveaux de consommation confondus) et de non-fumeurs. .........................30 Tableau 4 : Bilan des études de formation préférentielle d'adduits de la NNK ou du BPDE
dans des séquences contenant le codon 12 de KRAS ou les différents points chauds de mutation de TP53. ................................................................................32
Tableau 5 : Bilan des associations que l'on retrouve le plus fréquemment entre la quantité et le type de consommation de tabac, les carcinogènes du tabac, les mutations présentes dans les tumeurs pulmonaires et les deux principaux types histologiques de CPNPC.......................................................................................36
Figure 10 : Les dommages à l'ADN les plus communs et leur mode de réparation. ...........37 Figure 11 : Les dommages d'alkylation dans les cellules, leurs conséquences possibles et
les systèmes de réparation mis en jeu pour les réparer. ........................................38 Figure 12 : La réversion de l'alkylation réalisée par l'AGT. ................................................40 Figure 13 : La réparation par excision de bases...................................................................43 Figure 14 : Les différentes étapes de voie globale de la réparation par excision de
nucléotides. ...........................................................................................................47 Figure 15 : Influence de la relaxation de la chromatine induite par p53 sur la réparation des
lésions induites par les UV. ..................................................................................50 Figure 16 : Schéma des différentes étapes de la technologie LMPCR. ...............................54 Figure 17 : Evolution de l'aspect des comètes en fonction de la fréquence de dommages
dans les cellules. ...................................................................................................56 Figure 18 : Les différentes étapes du test des comètes. .......................................................56 Figure 19 : Analyse des images de comètes avec le logiciel CASP (Comet Assay Software
Project)..................................................................................................................57

xi
Liste des abréviations 1-mA 1-méthyladénine
3-mA 3-méthyladénine
3-mC 3-méthylcytosine
7-m-fapy ou N7-m-fapy 7-méthylformamidopyrimine
7-mG ou N7-mG 7-méthylguanine
7-pob-fapy ou N7-pob-fapy 7-pyridyloxobutylformamidopyrimine
7-pobG ou N7-pobG 7-pyridyloxobutylguanine
AAF acétylaminofluorène
AAG (ou MPG) alkyladénine glycosylase
Alk alkylation
AlkB 3-alkyladénine-ADN glycosylase
AP apurinique/apyrimidique
APE1 (ou HAP1) AP endonucléase 1
AGT O6-alkylguanine-ADN alkyltransférase
BPDE (+/-)-anti-benzo[a]pyrène-7,8-diol-9,10-époxyde
BRCA1 "breast cancer 1"
CASP Comet Assay Software Project
CBC cassure bicaténaire
CBP "CREB Binding Protein"
CDKN2A "cyclin-dependent kinase inhibitor 2A"
CMC cassure monocaténaire
CPNPC cancer pulmonaire non à petites cellules
CPPC cancer pulmonaire à petites cellules
CSA, B "Cockayne syndrome A, B"
CYP cytochrome P450
DCP ou "CPD" dimère cyclobutylique de pyrimidines
DDB protéine "Damaged DNA-Binding"
DMS diméthylsulfate
dRP 5'-désoxyribose 5-phosphate
dRPase 5'-désoxyribose 5-phosphatase
EGFR "epidermal growth factor receptor"

xii
ERCC1 "excision repair cross-complementing, group 1"
fapy formamidopyrimidine
fapy A, G formamidopyrimidine adénine, guanine
Fpg formamidopyrimidine glycosylase
G REN réparation par excision de nucléotides globale
GADD45 "growth arrest and DNA damage-inducible protein 45"
HAP hydrocarbure aromatique polynucléé
HAP1 (ou APE1) "human AP endonuclease 1"
HPB 4-hydroxy-1-(3-pyridyl)1-butanone
HR23B "human RAD23 homolog B"
KRAS Kirsten rat sarcoma virus
LigI, III Ligase I, III
LMPCR "ligation-mediated PCR"
MPG (ou AAG) méthyl-purine ADN glycosylase
NDMAOAc N-acétoxynitrosométhylméthylamine
NNAL 4-(méthylnitrosamino)-1-(3-pyridyl)-1-butan-1-ol
NNAL-Gluc NNAL-glucuronide
NNK 4-(méthylnitrosamino)-1-(3-pyridyl)-1-butanone
NNKOAc 4-(acétoxyméthylnitrosamino)-1-(3-pyridyl)-1-butanone
O4-mT O4-méthylthymine
O4-alkT O4-alkylthymine
O6-alkG O6-alkylguanine
O6-mG O6-methylguanine
O6-mG:T appariement entre une O6mG et une T
O6-pobG O6-pyridyloxobutylguanine
P phosphate
PCNA proliferating cell nuclear antigen
Polβ,δ,ε polymérase β,δ,ε
PP(6-4) photoproduit (6-4)
RASSF1A "Ras-association domain family 1, isoform A"
RB rétinoblastome
REB ou BER réparation par excision de bases
REN ou NER réparation par excision de nucléotides

xiii
RENH raboutage des extrémités non homologues
RFC "replication factor C"
RH recombinaison homologue
RM réparation des mésappariements
RPA protéine de réplication A
TC REN réparation par excision de nucléotides couplée à la transcription
TFIIH general transcription factor IIH
UVA, B, C Ultraviolets de type A, B ou C
XPA, B, C, D, F, G Xeroderma Pigmentosum A, B, C, D, F, G
XRCC1 "X-ray repair cross-complementing, group 1"

Chapitre 1 Introduction
1.1 Le cancer du poumon et ses origines
1.1.1 Incidence et mortalité Il y a 100 ans, le cancer du poumon était une maladie excessivement rare. Lorsque Adler
publia en 1912 un livre recensant les cas de cancer du poumon relatés dans la littérature
mondiale, il n'en comptabilisa que 374 (Adler, 1912 ; Spiro et Silvestri, 2005). Ce n'est qu'à
partir de la fin des années 20 que l'on commença à parler d'épidémie et l'on remarqua dès
lors que les patients atteints de cancer du poumon étaient aussi de gros fumeurs (Ochsner,
1973). Au cours du XXème siècle, l'incidence du cancer du poumon n'a cessé d'augmenter.
De nos jours, il s'agit du type de cancer le plus fréquent dans le monde, avec environ 1,2
million de nouveaux cas par an, ce qui représente 12,3 % des cancers diagnostiqués pour
l'année 2000 au niveau mondial (Parkin et al., 2001).
Dans plusieurs régions du monde, les taux de cancer du poumon ont atteint un pic et
commencent à diminuer, en tout cas pour les hommes. Ils continuent cependant à
augmenter pour les femmes (Devesa et al., 2005). Ce décalage dans le temps s'explique
également par la consommation de tabac qui, chez les femmes, a commencé plus
tardivement et a continué d'augmenter jusque dans les années 70. On estime que cette
tendance à l'augmentation chez les femmes ne devrait pas s'inverser avant au moins 2010
(Thomas et al., 2005). Il a également été suggéré qu'à consommation de tabac équivalente,
les femmes seraient plus susceptibles de développer un cancer du poumon que les hommes,
avec un risque de 1,5 à 2,7 fois plus élevé, mais ces données sont contestées par d'autres
études qui n'ont pas observé de différence significative (Patel, 2005 ; Thomas et al., 2005).
Malgré certains progrès thérapeutiques, environ 90 % des patients meurent de leur maladie
et l'on estime à 1,1 million, dont ≈ 75 % d'hommes, le nombre de personnes décédées de
cancer du poumon en 2000, ce qui en fait la première des causes de mort par cancer dans le
monde, avec 17,8 % des décès (Parkin et al., 2001).
On estime qu'au Canada, 22 700 nouveaux cas de cancer du poumon seront diagnostiqués
en 2006, et que 19 300 personnes en décèderont. De manière comparable à ce que l'on

2
observe au niveau mondial, l'incidence et la mortalité du cancer du poumon au Canada ont
commencé à décroître pour les hommes depuis 1988, alors que les deux paramètres
continuent à augmenter pour les femmes (NCIC, 2006).
1.1.2 Association avec la consommation de cigarettes Alors que le tabac est consommé depuis très longtemps, il ne s'est popularisé qu'à partir de
la première guerre mondiale chez les hommes (Spiro et Silvestri, 2005). Les femmes, elles,
n'ont commencé à fumer de manière importante qu'à partir des années 40. Quelque soit le
pays dont on analyse les statistiques, on peut systématiquement faire un parallèle entre
l'incidence de cancers du poumon et la consommation de tabac, mais avec environ 20 à 30
ans de décalage (Bray et al., 2004 ; Spiro et Silvestri, 2005).
La relation entre la dose (le nombre de cigarettes fumées par jour, le degré d'inhalation,
l'âge du début de la consommation) et le risque de cancer du poumon est également très
claire. Ainsi, une personne qui a fumé toute sa vie a 20 à 30 fois plus de risques de
développer un cancer du poumon qu'un non-fumeur (Parkin et al., 2001).
On estime globalement que 86 % des cancers du poumon chez l'homme et 46 % chez la
femme sont dus à la consommation de cigarettes, ce qui en fait de loin, la première cause de
cancer du poumon au niveau mondial (Parkin et al., 2001). Il existe de fortes variations
régionales dans la proportion des cancers associés à la cigarette chez les femmes, qui sont
essentiellement liées à des différences dans l'ancienneté des habitudes de consommation de
cigarettes. En effet, dans les régions où la consommation de tabac est ancienne et forte chez
les femmes, comme par exemple le Royaume-Uni, c'est près de 80 % des cancers
pulmonaires qui sont liés à la cigarette, tandis que dans certains autres pays où les femmes
fument peu ou depuis moins longtemps, tels l'Espagne et le Portugal, l'incidence des
cancers du poumon féminins est proche de celle des femmes non-fumeuses des autres pays
et ces cancers ne sont pas du tout associés à la consommation de tabac (Parkin et al., 2001).

3
1.1.3 Histologie des cancers du poumon
1.1.3.1 Classification des tumeurs du poumon Les différentes classes de cancers pulmonaires recencées par l'Organisation Mondiale de la
Santé sont les suivantes (Travis et al., 2004) :
• Carcinome épidermoïde • Carcinome à petites cellules • Adénocarcinome • Carcinome à grandes cellules • Carcinome adénosquameux • Carcinomes avec éléments pléomorphes, sarcomatoïdes ou sarcomateux • Tumeur carcinoïde • Tumeurs des glandes bronchiques Les quatre premières catégories sont de loin les plus fréquentes et à eux seuls, les
carcinomes épidermoïdes et les adénocarcinomes totalisent 80 à 90 % des cancers du
poumon recensés, quel que soit le lieu dans le monde (Devesa et al., 2005). Par définition,
chaque catégorie histologique à une origine tissulaire différente dans le poumon. Les
carcinomes épidermoïdes dérivent de l'épithélium bronchique tandis que les
adénocarcinomes se développent dans des régions situées plus à l'extrémité de l'arbre
bronchique (Figure 1).
On classe tous ces types histologiques en deux catégories distinctes qui sont considérées
comme fondamentales du point de vue clinique, biologique ou génétique : (1) les
carcinomes pulmonaires à petites cellules (CPPC) et (2) tous les autres types qu'on désigne
par le terme de carcinomes pulmonaires non à petites cellules (CPNPC).

4
Figure 1 : Lieu de formation dans le poumon des carcinomes épidermoïdes (1) et des adénocarcinomes (2).
Images tirées de http://www.spiral.univ-lyon1.fr.

5
1.1.3.2 Fréquence relative des différents types histologiques L'incidence relative des différents types de cancer du poumon est différente pour les
femmes et pour les hommes. Les carcinomes épidermoïdes sont plus communs chez les
hommes tandis que les femmes développent plus d'adénocarcinomes (Patel, 2005 ; Thomas
et al., 2005). Les adénocarcinomes sont également le type de cancer le plus fréquent chez
les personnes n'ayant jamais fumé de tabac, quel que soit leur sexe.
La fréquence relative des différents types histologiques de cancer du poumon a évolué avec
le temps. Chez les hommes, les carcinomes épidermoïdes, qui étaient beaucoup plus
fréquents il y a 25 ans, tendent à diminuer au profit des adénocarcinomes, ces derniers
devenant même la forme prédominante de cancer du poumon chez les hommes dans
certains pays comme l'Islande ou les États-Unis. Chez les femmes, tous les types
histologiques progressent mais la prédominance des adénocarcinomes tend à s'accentuer
avec le temps (Devesa et al., 2005).
1.1.3.3 Les cellules à l'origine de l'adénocarcinome Déterminer le type cellulaire à l'origine d'un cancer est toujours difficile mais ça l'est
particulièrement dans le cas des cancers du poumon qui sont le plus souvent diagnostiqués
à des stades avancés (Minna et al., 2002). Pour la plupart des types de CPNPC, les cellules
cancéreuses présentent des caractéristiques correspondant à des cellules différenciées de la
région où la tumeur s'est développée. Par exemple, les carcinomes épidermoïdes présentent
la kératinisation caractéristique des cellules épithéliales de la trachée et des voies aériennes
proximales.
Les adénocarcinomes, quant à eux, présentent des marqueurs cellulaires caractéristiques des
cellules de Clara ou des pneumocytes de type II (Kim et al., 2005). Les cellules de Clara
constituent la majorité de l'épithélium des bronchioles et des bronchioles terminales alors
que les pneumocytes de type I et de type II constituent l'épithélium alvéolaire (Figure 2).
Jusqu'à très récemment, on pensait que la présence de tels marqueurs caractéristiques dans
les tumeurs indiquait nécessairement que les adénocarcinomes pulmonaires dérivent de la
transformation maligne de cellules de Clara ou de pneumocytes de type II. En réalité, il
semblerait que ce sont des cellules souches bronchioalvéolaires se trouvant à la jonction
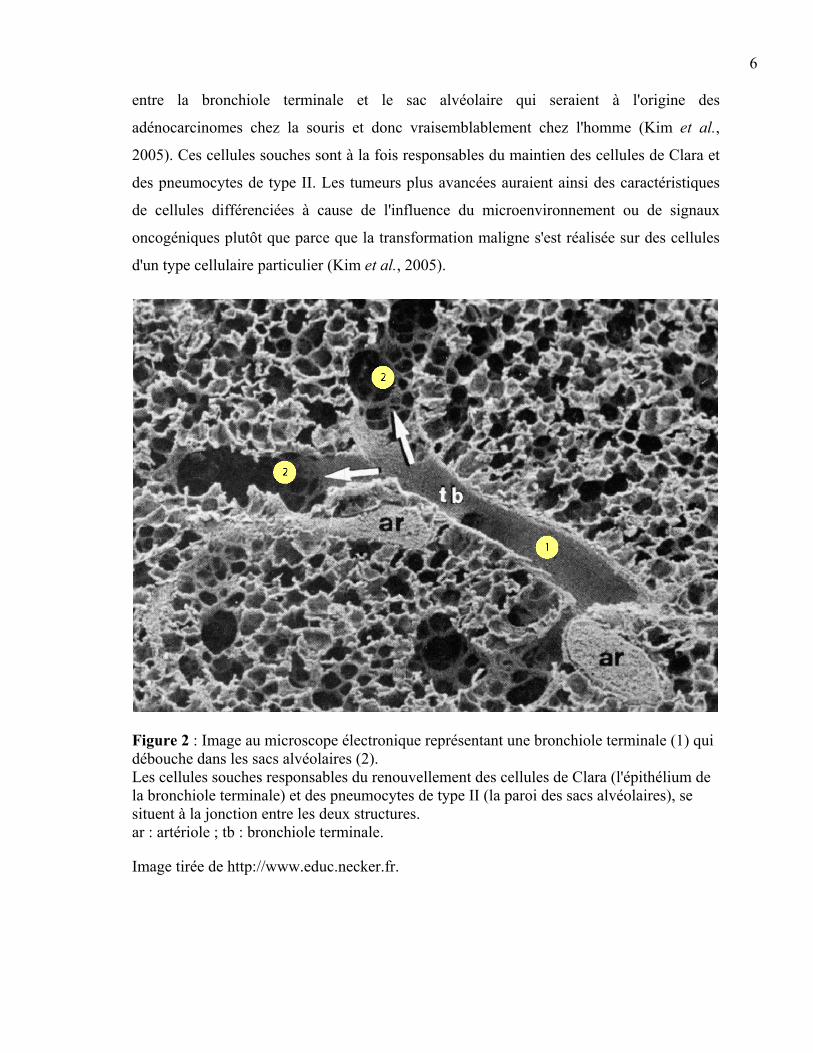
6
entre la bronchiole terminale et le sac alvéolaire qui seraient à l'origine des
adénocarcinomes chez la souris et donc vraisemblablement chez l'homme (Kim et al.,
2005). Ces cellules souches sont à la fois responsables du maintien des cellules de Clara et
des pneumocytes de type II. Les tumeurs plus avancées auraient ainsi des caractéristiques
de cellules différenciées à cause de l'influence du microenvironnement ou de signaux
oncogéniques plutôt que parce que la transformation maligne s'est réalisée sur des cellules
d'un type cellulaire particulier (Kim et al., 2005).
Figure 2 : Image au microscope électronique représentant une bronchiole terminale (1) qui débouche dans les sacs alvéolaires (2). Les cellules souches responsables du renouvellement des cellules de Clara (l'épithélium de la bronchiole terminale) et des pneumocytes de type II (la paroi des sacs alvéolaires), se situent à la jonction entre les deux structures. ar : artériole ; tb : bronchiole terminale.
Image tirée de http://www.educ.necker.fr.

7
1.1.4 Évolution de la cigarette en relation avec l'évolution du cancer du poumon
Après que les relations entre les cigarettes et le cancer du poumon aient été clairement
établies par plusieurs grandes études épidémiologiques dans les années 50, la fabrication et
la composition des cigarettes ont évolué pour tenter de rendre les cigarettes moins nocives
(Hoffmann et Hoffmann, 1997 ; Hoffmann et al., 2001). La quantité de nicotine a été
abaissée et des filtres ont été ajoutés pour diminuer la quantité de goudron inhalée. En
parallèle, l'évolution des procédés de fabrication a mené à des cigarettes contenant plus de
nitrates afin de favoriser la combustion du tabac. Une meilleure combustion défavorise la
formation de certaines substances carcinogènes du goudron tels que les hydrocarbures
aromatiques polynucléés (HAP). Cependant ces nitrates favorisent aussi, de manière
indépendante de la combustion, la formation d'autres composés carcinogènes. En effet,
l’oxyde d’azote, le nitrate et le nitrite de méthyle du tabac provoquent, lors de la
fermentation du tabac et sa combustion, la N-nitrosation de la nicotine pour former des N-
nitrosamines (Hoffmann et Hoffmann, 1997 ; Hoffmann et al., 2001). Les N-nitrosamines,
contrairement aux HAP, constituent donc une famille de composés spécifiques du tabac et
il a été montré que chez l'animal, plusieurs d'entre elles, dont la NNK, ont une action
carcinogénique dirigée de manière quasiment exclusive contre le poumon et ce,
indépendamment de leur mode d'administration (Hoffmann et Hoffmann, 1997 ; Hoffmann
et al., 2001).
Toutes ces modifications dans la composition des cigarettes ont eu des conséquences sur la
nature et la quantité de substances carcinogènes auxquelles les fumeurs sont exposés. En
particulier, cela a eu une influence sur l'intensité de l'inhalation. La fumée des cigarettes
fortes n'est qu'inhalée superficiellement, menant à un dépôt des carcinogènes chimiques
essentiellement dans la région bronchique, celle où se développent les carcinomes
épidermoïdes. Par contre, la présence d'un filtre donne une fumée plus légère qui permet
des inhalations plus profondes et donc l'exposition de régions pulmonaires plus
périphériques, celles où se développent les adénocarcinomes. Il a également été suggéré
que la réduction de la quantité de nicotine dans les cigarettes pousserait à une inhalation
plus profonde, le fumeur voulant "compenser" afin d'obtenir la quantité de nicotine dont il a

8
besoin (Hoffmann et Hoffmann, 1997 ; Hoffmann et al., 2001). Des résultats récents ont
montré une association entre la consommation de cigarettes à faible taux de goudron et la
formation préférentielle de tumeurs au niveau périphérique, c'est à dire dans le parenchyme
pulmonaire (Brooks et al., 2005). Ainsi, les changements de composition des cigarettes
n'auraient pas pour effet de diminuer la dangerosité de la cigarette, mais simplement de
déplacer le problème : les carcinomes épidermoïdes ont diminué mais les adénocarcinomes
ont progressé (Wynder et Muscat, 1995).
1.2 La théorie de la mutagenèse somatique
1.2.1 Comment la cigarette mène au cancer du poumon ? Le lien entre la consommation et la formation ultime d'un cancer du poumon s'établit de la
façon suivante (Figure 3). Une personne fume des cigarettes à cause de sa dépendance à la
nicotine. En même temps que la nicotine, la fumée apporte dans les poumons un certain
nombre de substances carcinogènes, dont des HAP et des N-nitrosamines telle que la NNK.
Ces substances sont activées métaboliquement dans les cellules pulmonaires en
intermédiaires réactifs qui peuvent interagir avec l'ADN et ainsi former des dommages. La
cellule possède différents systèmes de détoxication (excrétion), de réparation des
dommages, d'élimination des cellules anormales (apoptose) qui lui permettent de prévenir
la transformation maligne. Mais si ceux-ci persistent et que les cellules répliquent un ADN
endommagé, il y a risque de mutations, c'est-à-dire de changements dans la séquence
d'ADN de gènes impliqués notamment dans le maintien de l'intégrité du génome et le
contrôle du cycle cellulaire.

9
Figure 3 : Schéma montrant le lien entre les cigarettes, les substances carcinogènes qu'elles contiennent et la survenue du cancer du poumon.
Adapté et traduit de (Hecht, 1999b).
1.2.2 La théorie de la mutagenèse somatique La théorie de la mutagenèse somatique a été énoncée pour la première fois par Thilly en
1983 (Thilly, 1983) puis modifiée par Holmquist et Gao en 1997 (Holmquist et Gao, 1997).
Elle indique le lien existant entre les substances génotoxiques et la transformation maligne
en établissant que la probabilité qu'une mutation se retrouve dans une tumeur est le résultat
des étapes séquentielles de la mutagenèse. Elle se formule de la manière suivante :
Cela signifie que la probabilité Pi que la ième position nucléotidique soit mutée dans une
tumeur [Pi (mutation dans la tumeur)] dépend de la probabilité de plusieurs événements qui
interviennent successivement. Il faut que cette position nucléotidique soit effectivement
endommagée par l'agent mutagène [Pi (endommagée par mutagène)], que ce dommage ne
soit pas réparé [Pi (non réparée)] et que la base endommagée soit mal répliquée par une
polymérase [Pi (base endommagée lue par la polymérase)]. Enfin, pour que la mutation se
retrouve dans une grande quantité de cellules de la tumeur, il faut qu'il y ait eu une étape de
sélection de la mutation [Pi (sélection)]. La probabilité Pi (mutation dans la tumeur),
lorsqu'elle est déterminée pour chaque position nucléotidique le long d'un gène, donne le
spectre mutationnel d'un mutagène donné (Holmquist et Gao, 1997).

10
1.3 Les substances carcinogènes de la fumée de tabac Le courant principal de fumée dégagé par une cigarette, donc la fumée à laquelle les
poumons d'un fumeur sont exposés, contient deux phases : une phase gazeuse et une phase
particulaire. La phase particulaire contient au moins 3 500 composés parmi lesquels se
trouvent l'essentiel des composés carcinogènes. Le Centre International de Recherche sur le
Cancer (IARC) a évalué et reconnu 55 de ces composés comme ayant "suffisamment de
preuves de carcinogénicité" chez l'homme ou les animaux de laboratoire (Hoffmann et
Hoffmann, 1997). Parmi eux, 20 ont été montrés comme capable de générer des tumeurs
chez au moins une espèce animale (Hecht, 1999b).
Déterminer la contribution relative de ces nombreuses substances présentes dans la fumée
de cigarettes dans le développement du cancer pulmonaire est très difficile. Hecht (Hecht,
1999b) a évalué les rôles des différents carcinogènes présents dans la fumée de cigarettes
en considérant les critères suivant (Tableau 1) :
• La quantité présente dans la cigarette • La carcinogénicité chez les animaux de laboratoire • L'absorption de la substance chez l'homme • La métabolisme et la formation d'adduits chez l'homme • La possibilité de jouer un rôle dans les changements moléculaires observés dans les
gènes des tumeurs chez l'homme Pour chaque substance, ces critères ont été évalués sur une échelle de 1 à 4 selon le barème
suivant :
1. les données sont inadéquates 2. les preuves sont faibles ou équivoques 3. il existe certaines preuves mais les études sont limitées 4. les preuves sont claires ; les études sont robustes et reproductibles Un score final a ainsi été attribué pour plusieurs composés de la fumée de cigarette
(Tableau 1).

11
Composé(s)
Présence dans la fumée de cigarette
Carcinogénicité pulmonaire chez
les rongeurs
Absorption chez l'homme
Métabolisme et adduits chez
l'homme
Changements moléculaires chez l'homme
Score final
HAP spécifiques 4 4 4 3 3 18
Aza-arènes 3 3 1 1 2 10
NNK, N-nitrosodiéthylamine 4 4 4 3 3 18
Métaux 4 4 1 1 1 11
Composés organiques divers 4 3 1 1 1 10
Radicaux libres/dommages oxydatifs 3 1 3 3 1 11
Tableau 1 : Évaluation de la participation de carcinogènes spécifiques dans le cancer du poumon humain induit par la cigarette. HAP : Hydrocarbures aromatiques polynucléés incluant benzo[a]pyrène, benzofluoranthènes, dibenzo[a,i]pyrène, dibenz[a,h]anthracène et 5-méthylchrysène ; NNK : 4-(méthylnitrosamino)-1-(3-pyridyl)-1-butanone ; Métaux : nickel, chrome, cadmium, polonium-210 et arsenic ; Composés organiques divers : inclut 1,3-butadiène, éthyl carbamate et aldéhydes.
Extrait et adapté de (Hecht, 1999b). Les preuves les plus importantes d'implication dans le cancer pulmonaire chez l'homme
sont donc pour les HAP et la NNK. En effet, ces composés sont présents dans la fumée de
cigarettes, ils sont des carcinogènes efficaces chez les rongeurs, leur absorption et leur
activation métabolique a clairement été démontrée chez l'homme et enfin, des dommages et
des mutations pouvant être reliés avec l'exposition à ces substances ont pu être observés
dans des tissus humains.
Comme nous l'avons vu précédemment, l'évolution des cigarettes dans le temps s'est
accompagnée d'une diminution des HAP et d'une augmentation en parallèle de toutes les N-
nitrosamines spécifiques du tabac, y compris la NNK (Hoffmann et Hoffmann, 1997 ;
Hoffmann et al., 2001). Ainsi, la diminution des carcinomes épidermoïdes et
l'augmentation généralisée des adénocarcinomes est corrélée avec la diminution des HAP et
l'augmentation de la NNK et l'on suspecte que la NNK serait, au moins en partie,
responsable de l'augmentation récente de la fréquence relative des adénocarcinomes
(Wynder et Muscat, 1995). L'association entre NNK et adénocarcinomes est également
indiquée par le fait que les animaux de laboratoires exposés à la NNK développent

12
essentiellement des adénomes et des adénocarcinomes pulmonaires (Hoffmann et al.,
1996 ; Hoffmann et Hoffmann, 1997).
1.4 La NNK dans les cellules pulmonaires Comme la NNK est le composé de la fumée de tabac qui a été plus particulièrement étudié
dans ce travail de thèse, il est utile de se concentrer sur le devenir de ce composé dans
l'organisme.
La NNK est la plus abondante et la plus carcinogène des N-nitrosamines spécifiques du
tabac. Une de ses principales caractéristiques est sa très grande spécificité pour le poumon.
En effet, elle génère des tumeurs pulmonaires chez les rongeurs quel que soit son mode
d'administration : la nourriture, l'eau de boisson, de manière sous-cutanée, intrapéritonéale
ou autre (Hecht, 1998 ; Hecht, 1999b). Toutefois, d'autres types de tumeurs peuvent être
retrouvés, notamment de la cavité orale et nasale, du pancréas, du foie et de la trachée
(Hecht, 1998).
1.4.1 Détoxication et/ou activation métabolique Le devenir de la NNK dans l'organisme dépend de sa biotransformation par des enzymes
spécifiques. Il est donc susceptible d'être différent d'une espèce à l'autre, d'un tissu à l'autre
et même d'un individu à l'autre au sein de la même espèce. La présence de ces enzymes est
responsable de la spécificité pulmonaire de la carcinogénicité de la NNK. La
biotransformation de la NNK a été extensivement étudiée (Hecht, 1998), y compris dans
des cellules pulmonaires fraîchement isolées (Smith et al., 1999) et avec des microsomes
constitués à partir de cellules pulmonaires de différents individus (Smith et al., 2003). Dans
les cellules pulmonaires humaines comme celles de rongeurs, la réaction la plus fréquente
est la réduction (réaction a de la Figure 4) de la NNK en 4-(méthylnitrosamino)-1-(3-
pyridyl)-1-butanol (NNAL). Cette transformation est réversible, mais elle se fait
majoritairement dans le sens de la formation de NNAL.

13
Figure 4 : Voies de bioactivation de la NNK et de la NNAL.
Adapté de (Hecht, 1998). La NNK et la NNAL peuvent subir les mêmes types de biotransformations. Leur N-
oxydation génère des métabolites chargés faciles à excréter. Il s'agit donc d'une voie de
détoxication, mais qui reste mineure quantitativement. La voie de détoxication majeure de
la NNK passe par la formation de NNAL qui a la particularité de pouvoir se conjuguer à
l'acide glucuronique pour former le NNAL-glucuronide (NNAL-Gluc). La NNAL et sa
forme glucuronidée sont détectées dans les urines de personnes qui ont été exposées à la
fumée de cigarettes, même de manière passive comme c'est le cas lors d'une exposition à la
fumée environnementale de tabac (Hecht et al., 1993 ; Hecht et al., 1994 ; Anderson et al.,
2001 ; Hecht, 2002 ; Anderson et al., 2003 ; Hecht, 2004). Ils constituent des biomarqueurs
intéressants de l'exposition à la fumée de tabac parce qu'ils sont spécifiques de la NNK, un
composé qui n'existe que dans les cigarettes (Hecht, 2002). La NNK n'est pas détectée dans
les urines et la NNAL n'est pas présente dans la fumée de tabac, si bien que la quantité
urinaire de NNAL et de NNAL-Gluc constituent un marqueur de l'absorption humaine de la
NNK (cf. Tableau 1). Une plus forte concentration de NNAL urinaire chez la population

14
noire américaine après chaque cigarette fumée (en comparaison avec la population
caucasienne) a d'ailleurs été associée avec leur risque plus élevé de développer un cancer
du poumon (Muscat et al., 2005). La NNAL-Gluc n'étant pas carcinogène en elle-même, il
a été suggéré que le rapport NNAL-Gluc:NNAL dans l'urine pourrait être un indicateur de
la capacité de détoxication d'un individu et donc un possible indicateur de risque individuel
de cancer (Hecht, 2002).
La NNK et la NNAL peuvent également être bioactivées dans certaines cellules par α-
hydroxylation, ce qui mène à la formation d'intermédiaires réactifs capables d'alkyler
l'ADN. Dépendamment de la position de l'α-hydroxylation, l'intermédiaire réactif généré
mène à la méthylation (voie c de la Figure 4) ou à la pyridyloxobutylation de l'ADN (voie b
de la Figure 4). Cette α-hydroxylation pulmonaire est majoritairement réalisée par des
cytochromes P450 (CYP) chez les rongeurs (Hecht, 1998) mais semble plus complexe chez
l'homme où la bioactivation de la NNK est quantitativement moins importante et implique
d'autres voies d'activation impliquant des lipooxygénases, et des cyclooxygénases (Hecht,
1998 ; Smith et al., 1999 ; Rioux et Castonguay, 2000 ; Smith et al., 2003). Il a cependant
été clairement démontré que les CYP contribuent à la bioactivation de la NNK chez
l'homme (Smith et al., 2003). Plusieurs CYP interviennent, et ce de manière variable en
fonction des individus, mais les CYP2A6 et/ou CYP2A13 ainsi que le CYP2B6 semblent
être particulièrement déterminants (Smith et al., 2003). La contribution relative de l'un ou
l'autre des différents CYP semble pouvoir varier d'une personne à l'autre (Smith et al.,
2003). Enfin, l'α-hydroxylation de la NNK dans une préparation enrichie en pneumocytes
de type II humains s'est avérée plus importante que dans des préparation de cellules
pulmonaires en général ou de macrophages alvéolaires, indiquant que les capacités
d'activation dépendent aussi du type cellulaire à l'intérieur d'un même tissu (Smith et al.,
1999).
1.4.2 Les dommages à l'ADN générés par la NNK
1.4.2.1 Les sites réactifs de l'ADN L'alkylation peut avoir lieu sur tous les hétéroatomes (N et O) disponibles de la molécule
d'ADN. Ces sites susceptibles d'être alkylés sont indiqués à la Figure 5. Parmi eux, certains

15
ont une plus grande réactivité due au fait qu'ils sont plus nucléophiles : (1) la N7 des
guanines, (2) la N3 des adénines, (3) la N1 des adénines, la N3 des guanines et la O6 des
guanines (Friedberg et al., 2006).
Figure 5 : Les sites nucléophiles sensibles à l'alkylation dans l'ADN.
Différents agents alkylants peuvent avoir des sites nucléophiles préférentiels différents et
donc générer des types de dommages différents (Friedberg et al., 2006). Il est donc difficile
de définir a priori quels dommages sont générés par un agent alkylant donné car plusieurs
paramètres interviennent :
1. Plus l'agent alkylant est un électrophile fortement réactif, plus il va avoir de cibles
différentes. Les électrophiles fortement réactifs peuvent alkyler les sites nucléophiles
faibles et forts tandis que les électrophiles plus faiblement réactifs ne vont alkyler que
les sites nucléophiles les plus forts.
2. L'environnement nucléaire, notamment les interactions avec les histones et
l'organisation en chromatine d'une manière générale, affectent la répartition des charges
dans la molécule d'ADN.
3. La structure tridimensionnelle de l'ADN influence la capacité des agents alkylants les
plus volumineux à pouvoir endommager certaines positions nucléotidiques. Par

16
exemple, dans la conformation hélicale classique de type B, les atomes N7 et O6 de la
guanine se trouvent dans le sillon majeur de l'ADN, tandis que la position N3 des
adénines se trouve dans le sillon mineur de la double hélice, ce qui la rend relativement
moins accessible.
En fait, les agents alkylants sont regroupés en deux catégories dépendamment de leur
mécanisme d'action. L'étape déterminante pour les réactions de type SN1 est la formation du
carbocation (ex : CH3+) qui, une fois formé, va réagir rapidement avec n'importe quel site
nucléophile de la molécule d'ADN. Les intermédiaires réactifs qui résultent de la
bioactivation de la NNK (Figure 4) agissent comme les carbocations des agents de type
SN1. Ces agents alkylants tendent à générer une grande variété de lésions. Pour les réactions
de type SN2, un état de transition complexe est formé entre l'agent alkylant électrophile et le
site nucléophile cible. L'étape limitante pour ce deuxième type d'agent alkylant est la
capacité pour le nucléophile d'interagir avec la molécule alkylante électrophile. Ce sont ces
réactions qui sont particulièrement dépendantes du niveau de nucléophilie de l'atome ciblé
dans l'ADN. Les agents alkylants SN2 tendent donc à favoriser les réactions avec les centres
les plus nucléophiles, telle que la position N7 des guanines.
La séquence primaire de l'ADN a aussi de l'importance dans la réactivité des bases, ce qui
mène à une formation préférentielle des adduits à certaines positions nucléotidiques. Ainsi,
la position en N7 d'une guanine sera plus facilement sujette à une attaque électrophile si
celle-ci est flanquée d'autres guanines (Richardson et Richardson, 1990). Au delà de cette
constatation, les agents de type SN1 vont alkyler plus volontiers au centre d'une série de G
tandis que les agents de type SN2 le feront plus souvent en 5' d'une série du même genre
(Cloutier et al., 1999 ; Cloutier et al., 2001a).
1.4.2.2 Les dommages de la NNK générés in vivo
1.4.2.2.1 Chez les rongeurs Différents adduits ont pu être détectés in vivo chez des animaux traités avec la NNK. La
détection des adduits résultant de la méthylation de l'ADN n'a pas posé de problèmes mais
la pyridyloxobutylation de l'ADN n'a été pendant longtemps déduite qu'indirectement via la
détection du céto alcool 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB). En effet, la libération

17
de HPB lors de l'hydrolyse acide ou thermique neutre d'un ADN indique que cet ADN a été
pyridyloxobutylé, mais sans que l'on sache nécessairement quels types de dommages ont
été générés. Ces dommages instables lors de l'hydrolyse de l'ADN sont restés longtemps
non identifiés et ont donc été qualifiés de "dommages relâchant le HPB" (Hecht, 1998).
Figure 6 : Formation de HPB suite à l'hydrolyse acide ou thermique neutre d'un ADN pyridyloxobutylé.
S. Hecht (Hecht, 1998) a fait un bilan des adduits retrouvés in vivo pour les différentes
études réalisées chez les rongeurs. Il a ainsi déterminé leur quantité relative en fonction des
animaux et des tissus à l'intérieur de la même espèce.

18
Animal Tissu Quantité relative d'adduits Autres dommages
poumon 7-mG > adduits relâchant HPB > O6-mG > O4-mT 8-oxoG muqueuse nasale 7-mG > O6-mG > adduits relâchant HPB CMC Rat F-344
foie 7-mG > adduits relâchant HPB > O6-mG > O4-mT CMC, 8-oxoG
Souris A/J poumon 7-mG > O6-mG > adduits relâchant HPB > O4-mT 8-oxoG
Quantité relative dans les différents tissus Adduits Rat F-344 Souris A/J 7-mG muqueuse nasale > poumon* > foie* foie > poumon
O6-mG muqueuse nasale > poumon* > foie* foie > poumon O4-mT foie > poumon > muqueuse nasale n.d.
Adduits relâchant HPB poumon* > foie* > muqueuse nasale foie > poumon Tableau 2 : La formation des adduits et leur importance dans la tumorigenèse chez le rat et la souris. Ces valeurs peuvent être différentes selon le type cellulaire et le temps écoulé avant le dosage. Les adduits considérés comme importants dans la mutagenèse chez chaque animal sont indiqués en gras et en italique. 7-mG : 7-méthylguanine ; O6-mG : O6-méthylguanine ; O4-mT : O4-méthylthymine ; 8-oxoG : 8-oxoguanine ; CMC : cassures monocaténaires de l'ADN * à faibles doses de NNK ; c'est l'inverse à hautes doses
Traduit et adapté de (Hecht, 1998). Comme le prédit la théorie de la mutagenèse somatique, pour que des adduits participent à
des mutations présentes dans les tumeurs, il faut qu'ils soient produits dans les tissus cibles,
qu'ils ne soient pas réparés, de sorte qu'ils persistent suffisamment pour être mal répliqués
et enfin qu'il y ait une sélection des cellules portant la mutation. Le Tableau 2 donne une
idée de la formation et de la persistance des dommages associés avec la NNK dans les
tissus. Ces résultats sont importants pour estimer la contribution d'un type de dommages
dans la mutagenèse mais ils ne sont pas suffisants. Ainsi, les rats F-344 traités avec la NNK
présentent des tumeurs dans tous les organes qui accumulent les dommages (poumon,
cavité nasale et foie) mais les souris A/J ne présentent que des tumeurs du poumon, alors
même que tous les types d'adduits sont en plus grande quantité dans le foie (Tableau 2).
Donc même si l'on peut faire des corrélations entre accumulation de dommages et la
transformation maligne, la correspondance n'est pas complète et très insuffisante pour
prédire la survenue des mutations et des tumeurs.
D'autre part, les grandes différences constatées d'une espèce animale à l'autre (même aussi
proches que la souris et le rat), d'un tissu à l'autre voire d'un type cellulaire à l'autre à

19
l'intérieur d'un même tissu rendent très difficiles les tentatives de faire un schéma général
des dommages fait par la NNK dans un organisme. Ainsi, même si ces données sont
essentielles pour comprendre la mutagenèse liée à la NNK, il est très difficile de prédire ce
qui se passe chez l'homme d'après les résultats obtenus chez les rongeurs.
1.4.2.2.2 Chez l'homme Plusieurs études ont mesuré les dommages d'alkylation dans les poumons chez l'homme
(Hecht, 1998). Les 7-mG se sont avérées 7,5 – 25 fois plus fréquentes que les adduits
relâchant le HPB, ce qui est comparable à ce qui a été trouvé chez des rats traités à la NNK.
Les dommages relâchant le HPB sont nécessairement associés avec la bioactivation de la
NNK mais ce n'est pas nécessairement le cas des 7-mG qui peuvent être générés par
plusieurs autres agents alkylants. Des études ont rapporté des niveaux de 7-mG plus élevés
chez les fumeurs que chez les non-fumeurs (Hecht, 1998).
1.4.2.3 Importance relative des deux voies d'activation de la NNK dans la mutagenèse
La NNK peut être activée selon deux modes d'α-hydroxylation différents (Figure 4) chez
tous les animaux testés (Hecht, 1998 ; Hecht, 1999a). Il y a donc à la fois production
d'adduits résultant de la méthylation et de la pyridyloxobutylation dans les tissus. Leur
quantité relative (Tableau 2) reflète la capacité pour les cellules (1) de les produire en
bioactivant la NNK et (2) de les laisser s'accumuler. Leur implication dans la mutagenèse a
été en partie estimée à partir de ces informations, mais aussi en testant la tumorigénicité
d'analogues de la NNK qui ne font que méthyler ou que pyridyloxobutyler l'ADN.
Ainsi, les deux voies d'activation de la NNK semblent nécessaires pour la transformation
maligne dans le poumon chez le rat. En effet, ni la N-nitrosodiméthylamine (qui ne peut
que méthyler l'ADN) ni la N'-nitrosonornicotine (qui ne peut que pyridyloxobutyler l'ADN)
ne sont des carcinogènes pulmonaires efficaces. Cela suggère que seule la NNK fournit le
mélange approprié de dommages de la méthylation et de la pyridyloxobutylation nécessaire
pour la tumorigenèse pulmonaire chez le rat (Hecht, 1998). Au contraire, chez la souris A/J,
les analogues qui ne peuvent que méthyler l'ADN sont très tumorigènes tandis que les
molécules qui ne font que pyridyloxobutyler l'ADN ne le sont que très faiblement, d'où la

20
plus grande importance relative attribuée aux adduits de la méthylation chez cet animal (en
gras dans le Tableau 2).
Encore une fois, les différences observées entre le rat et la souris rendent difficile une
extrapolation sur l'implication de chaque mode d'activation dans la transformation maligne
chez l'homme. Cependant, le fait que les cellules humaines exposées à la fumée de
cigarettes présentent les deux types de dommages (7-mG et adduits relâchant le HPB)
suggère que les deux voies d'activation pourraient être importantes (Hecht, 1998).
1.4.2.4 Identification des dommages de la pyridyloxobutylation Des études récentes réalisées in vitro avec un analogue de la NNK générant uniquement le
métabolite pyridyloxobutylant de la NNK : la 4-(acétoxyméthylnitrosamino)-1-(3-pyridyl)-
1-butanone (NNKOAc) ont permis d'identifier plusieurs adduits résultant de cette voie
d'activation. Le Tableau 3 résume les types de dommages identifiés et leur capacité à
relâcher le HPB lors d'une hydrolyse acide ou thermique neutre de l'ADN pyridyloxobutylé
(Hecht, 1998 ; Haglund et al., 2002 ; Wang et al., 2003 ; Hecht et al., 2004 ; Sturla et al.,
2005).
Position des adduits Relâchant le HPB
Guanine N7 Oui O6 Oui N2 Non
Thymine O2 Non
Cytosine
O2 Oui
Squelette ADN Phosphate Non
Tableau 3 : Les adduits de la pyridyloxobutylation de l'ADN et leur capacité à relâcher le HPB (Hecht, 1998 ; Haglund et al., 2002 ; Wang et al., 2003 ; Hecht et al., 2004 ; Sturla et al., 2005).

21
La formation de O6-pyridyloxobutylguanine (O6-pobG) a d'abord été montrée sur de l'ADN
traité in vitro avec la NNKOAc (Hecht, 1999a). Il est resté longtemps le seul adduit bien
caractérisé associé avec la pyridyloxobutylation de l'ADN. La présence de petites quantités
de O6-pobG a été détectée dans le foie mais pas dans les poumons de souris A/J traitées
avec la NNK. Chez ces mêmes souris traitées avec la NNKOAc, des O6-pobG ont été
détectées dans le foie et dans les poumons (Thomson et al., 2003).
Cloutier et al. (Cloutier et al., 2001b) ont montré la formation d'une quantité importante de
CMC dans l'ADN traité à la NNKOAc. Ces cassures ont été associées à la formation
d'adduits au niveau des phosphates (Figure 5). La présence de tels
pyridyloxobutylphosphotriesters a été confirmée plus récemment dans le foie de souris
traitées avec la NNK (Haglund et al., 2002).
L’utilisation de conditions plus douces d’hydrolyse de l’ADN a permis d'identifier les
autres dommages aux bases générés in vitro sur de l'ADN ou des désoxyguanosines traités
avec la NNKOAc (Wang et al., 2003 ; Hecht et al., 2004 ; Sturla et al., 2005). La formation
de dommages aux positions N7 et N2 des guanines ainsi qu'à la position O2 des cytosines et
des thymines a ainsi été montrée (Tableau 3). Parmi ceux-ci, les 7-pyridyloxobutylguanines
(7-pobG) sont de loin les plus fréquentes et seules les 7-pobG et les O2-
pyridyloxobutylcytosines sont des adduits relâchant le HPB (Wang et al., 2003 ; Hecht et
al., 2004 ; Sturla et al., 2005). Ces adduits de la pyridyloxobutylation ont été quantifiés très
récemment dans les tissus (foie et poumon) de rats traités à la NNK (Lao et al., 2006). Une
quantité importante de 7-pobG a été mesurée dans tous les tissus mais la quantité relative
de O6-pobG s'est avérée beaucoup plus faible que ce qui avait été observé in vitro, en
particulier dans le foie mais aussi dans les poumons. Cela est vraisemblablement lié à une
réparation plus efficace des O6-pobG par rapport aux 7-pobG dans les deux types de tissus.
Des O2-pobT en quantité importante ont également été détectées dans ces tissus, ce qui
indique probablement une difficulté à les réparer pour les cellules (Lao et al., 2006).
1.5 Les altérations génétiques dans les cancers pulmonaires Les principales anomalies moléculaires qui ont été associées avec le développement du
cancer du poumon se rangent dans les catégories suivantes (Osada et Takahashi, 2002) :

22
• Méthylation aberrante de promoteurs • Activation de proto-oncogènes, de facteurs de croissances et de récepteurs • Expression de l'activité télomérase • Perte de fonction de gènes suppresseurs de tumeurs • Perte de composantes des voies menant à l'apoptose • Perte potentielle des mécanismes de réparation de l'ADN • Angiogenèse tumorale Beaucoup de gènes ont pu être associés à ces différents types d'anomalies moléculaires et
présentent un fonctionnement déficient dans les tumeurs pulmonaires. Certains gènes plus
fréquemment altérés ont pu être mis en évidence et la Figure 7 montre les différentes étapes
de la transformation maligne dans le poumon, telle qu'elle peut être comprise à ce jour. La
séquence des événements peut être reconstituée en tenant compte de la fréquence de ces
modifications et du stade de la transformation maligne auquel elles peuvent être détectées
(Osada et Takahashi, 2002).
Figure 7 : Accumulation des altérations dans la progression de la carcinogenèse pulmonaire.
Extrait et traduit de (Osada et Takahashi, 2002).

23
Certaines particularités ont pu être distinguées dans la séquence des événements associés
avec le développement des CPPC par rapport aux CPNPC. En particulier, les mutations de
TP53 sont plus fréquentes dans les CPPC alors que les mutations de KRAS sont plus
fréquentes dans les CPNPC. En effet, TP53 est muté dans 80 % des CPPC mais seulement
dans 50 % des CPNPC (Agarwal et al., 1998). De la même manière, RB est muté dans
quasiment tous les CPPC (> 90 %) mais plutôt rarement dans les CPNPC (15 – 30 %)
(Hensel et al., 1990 ; Xu et al., 1991). Des études cytogénétiques ont également permit
d'identifier des régions subissant des pertes d'hétérozygotie qui sont plus spécifiques d'un
type ou l'autre de cancer du poumon (Girard et al., 2000). Cependant, beaucoup de gènes
semblent communs aux deux grandes catégories de cancer pulmonaire et l'on peut ainsi
considérer les grandes étapes de la transformation maligne indiquées à la Figure 7 comme
étant généralisable à tous les cancers du poumon.
Les voies cellulaires les plus fréquemment affectées dans le cancer du poumon sont
indiquées dans la figure 8. Les deux voies majeures de suppresseurs de tumeurs impliquées
dans le cancer du poumon (p14ARF-p53 et p16INK4a-RB) sont liées fonctionnellement et
sont des composantes des points de contrôle et des inhibiteurs de la croissance cellulaire.

24
Figure 8 : Les voies de suppresseurs de tumeurs impliquées dans le cancer du poumon. Rouge : molécules fréquemment altérées dans le cancer du poumon ; Bleu foncé et clair : molécules qui sont moins fréquemment ou rarement altérées, respectivement.
Extrait de (Osada et Takahashi, 2002).
1.5.1 Région chromosomique 3p Des études de délétion à haute résolution ont permis de détecter des délétions dans la région
chromosomique 3p dans 91 % des CPPC, 95 % des carcinomes épidermoïdes et 71 % des
adénocarcinomes, ce qui en fait un marqueur quasi universel de la cancérogenèse
pulmonaire (Wistuba et al., 2000). La perte de cette région chromosomique a même été
identifiée dans l'épithélium normal et dans des lésions préinvasives chez des fumeurs et elle
a été associée avec un début de la consommation de tabac à un âge précoce. La prévalence
de la perte d'hétérozygotie en 3p21 est significativement plus importante chez les fumeurs
et les ex-fumeurs par rapport aux non fumeurs (Sozzi et al., 1997 ; Hirao et al., 2001).
Ainsi, la région chromosomique 3p a pu être associée avec des événements précoces dans
la transformation maligne au niveau pulmonaire.

25
Une influence de p53 sur la perte d'hétérozygotie en 3p21 a été démontrée. En effet, si la
délétion initiale dans la région 3p a bien été associée avec une exposition aux carcinogènes,
une perte subséquente plus extensive a été associée avec le statut de p53. En effet, les
tumeurs avec une p53 mutante ont significativement plus de risque de perte d'hétérozygotie
à un ou plusieurs locus de 3p21 que les tumeurs avec une p53 sauvage (Marsit et al., 2004).
RASSF1A (Ras-association domain family 1, isoform A) est l'un des gènes suppresseur de
tumeurs candidat présent dans la région mais il n'est vraisemblablement pas le seul. Une
perte d'hétérozygotie à ce locus a été associée à une méthylation aberrante de ce gène sur
l'autre allèle (Agathanggelou et al., 2001). RASSF1A est un inhibiteur de la croissance
cellulaire mais son mécanisme d'action n'est pas clairement élucidé (Agathanggelou et al.,
2005). De nombreux gènes sont régulés de manière dépendante de RASSF1A
(Agathanggelou et al., 2003) et plusieurs de ces gènes sont également régulés par RAS
activé, mais de manière opposée. RASSF1A et RAS activé ont donc un effet réciproque sur
l'expression de certains gènes, ce qui suggère leur implication dans la même voie mais via
des effets opposés (Agathanggelou et al., 2005).
Le gène XPC est un autre candidat présent dans la région 3p qui pourrait participer à la
carcinogenèse pulmonaire. XPC est l'une des protéines impliquées dans la détection des
dommages devant être réparés par la voie de réparation par excision de nucléotides (REN)
(cf. section 1.7.4). Des polymorphismes dans ce gène ont été associés avec un plus grand
risque de développer un cancer du poumon (Marin et al., 2004 ; Hu et al., 2005 ; Lee et al.,
2005) et il a été montré que 100 % des souris déficientes pour le gène Xpc développent
spontanément des tumeurs du poumon, essentiellement des adénomes (Hollander et al.,
2005). De plus, une perte allélique au locus de XPC a été montrée dans une grande
proportion (75 %) des CPNPC chez l'homme (Miyakis et al., 2003 ; Hollander et al., 2005).
Ainsi, la perte de XPC pourrait être un événement précoce dans la transformation maligne
pulmonaire qui favoriserait la formation subséquente de mutation induites par les
carcinogènes pulmonaires (Hollander et al., 2005).

26
1.5.2 Régions chromosomiques 9p21 Des pertes alléliques en 9p21 ont également été rapportées dans les lésions épithéliales
prémalignes et dans les cellules bronchiques normales (Sanz-Ortega et al., 2001).
La région chromosomique 9p21 contient le locus p16INK4a/p14ARF/p15INK4b (gène
CDKN2A). Or, les protéines de la famille INK4 telles que p15 et p16 sont des inhibiteurs
des kinases 4 et 6 cyclines - dépendentes (Figure 8) et sont donc des suppresseurs de
tumeurs qui inhibent la croissance cellulaire (Caputi et al., 2005). p16 est inactivé dans plus
de 40 % des CPNPC, soit par perte d'hétérozygotie, soit par hyperméthylation du gène
(Belinsky, 1998). Les inactivations de p16 ne sont pas significativement plus fréquentes
dans les CPNPC chez les fumeurs que chez les non-fumeurs mais les inactivations par
mutations ponctuelles semblent plus fréquentes chez les fumeurs (Sanchez-Cespedes et al.,
2001).
1.5.3 Le gène TP53 Le gène TP53 est fréquemment muté dans tous les types de cancer y compris le cancer du
poumon. Les mutations de TP53 sont associées à des étapes assez tardives de la progression
maligne et de l'invasion (Figure 7). La protéine p53 est impliquée dans de nombreuses
fonctions cellulaires en relation avec le maintien de l'intégrité du génome, tels que la
régulation du cycle cellulaire, le déclenchement de l'apoptose ou l'induction de certains
systèmes de réparation des dommages à l'ADN (Agarwal et al., 1998).
Des banques de données existent où sont recensées les mutations de TP53 trouvées dans les
tumeurs (http://www-p53.iarc.fr/AdvancedCriteria.asp) (Olivier et al., 2002). Dans les cas
où l'information est disponible, des corrélations peuvent être recherchées entre le type
histologique de tumeurs pulmonaires, le niveau de consommation de tabac et la nature ainsi
que la fréquence de mutations trouvées dans le gène TP53 (Greenblatt et al., 1994).
Cependant, ce type d'étude est souvent limité par des incertitudes concernant une éventuelle
consommation passée de tabac chez les patients et/ou l'exposition passive à la fumée. Une
étude récente a pu surmonter ces problèmes en exploitant les structures déjà mises en place
pour une étude épidémiologique du type cas-contrôle concernant le cancer du poumon.

27
Cette étude était déjà en cours en Russie et les informations concernant la consommation de
tabac des patients et d'autres facteurs de risque étaient très bien documentées (Le Calvez et
al., 2005). En investiguant les tumeurs de ces patients, ils ont trouvé 64 % des tumeurs
mutées pour TP53, s'étendant de 47 % des tumeurs pour les gens n'ayant jamais fumé à
77 % des tumeurs pour les fumeurs actuels. Le risque relatif d'avoir une mutation dans
TP53 a été estimé à 1,7 pour les ex-fumeurs et 5,2 pour les fumeurs actuels par rapport aux
non fumeurs. Parmi les fumeurs, il existe une évolution linéaire du risque relatif en fonction
de la quantité de tabac consommée. Ainsi, le risque relatif d'avoir une mutation dans TP53
atteint la valeur de 13 pour les plus gros consommateurs (plus de 50 paquets - années). Des
différences entre les types histologiques ont également pu être constatées. À consommation
de tabac égale, les mutations de TP53 sont plus fréquentes dans les carcinomes
épidermoïdes et les autres types histologiques que dans les adénocarcinomes. En fait, il n'y
a aucune augmentation significative du risque d'avoir une mutation dans TP53 en fonction
de la quantité de cigarette pour les adénocarcinomes (Le Calvez et al., 2005).
1.5.4 Les gènes KRAS et EGFR L'oncogène KRAS est fréquemment muté dans les tumeurs du poumon et ce principalement
au niveau du codon 12. L'EGFR (epidermal growth factor receptor) est un récepteur
tyrosine kinase qui joue un rôle crucial dans la prolifération cellulaire, la survie et la
différentiation des cellules (Castillo et al., 2004). Lorsqu'elles sont mutées, les protéines
KRAS et EGFR sont activées constitutivement et favorisent la croissance cellulaire (Figure
9).
Environ 30 % des adénocarcinomes ont des mutations dans le codon 12 de KRAS ou le
domaine tyrosine kinase de EGFR, ces mutations étant mutuellement exclusives. Par
contre, de telles mutations s'avèrent rares dans les autres types histologiques (Rodenhuis et
Slebos, 1992 ; Cooper et al., 1997 ; Shigematsu et al., 2005). Une étude plus récente,
réalisée sur 215 adénocarcinomes, indique des résultats allant dans le même sens avec
53,5 % de tumeurs présentant une mutation dans EGFR et 10 % présentant une mutation
dans KRAS, ces deux types d'anomalies étant toujours mutuellement exclusives (Tam et al.,
2006).

28
La même étude que citée précédemment (cf. section 1.5.3) à propos de la fréquence de
mutations dans TP53 en fonction de la consommation de tabac (Le Calvez et al., 2005) s'est
intéressée aussi aux mutations dans le codon 12 de KRAS. De telles mutations existent dans
15 % des tumeurs mais ne sont pas plus fréquentes chez les gros fumeurs que chez les non-
fumeurs (Le Calvez et al., 2005). La seule catégorie présentant une fréquence plus élevée
de mutations dans KRAS correspond aux tumeurs s'étant développées chez les ex-fumeurs
(ayant arrêté il y a plus de 10 ans). D'une manière générale, les mutations dans KRAS
semblent associées aux adénocarcinomes chez les ex-fumeurs (65 % des tumeurs mutées)
(Le Calvez et al., 2005). Cependant, une association entre le fait de fumer et la fréquence
de mutation de KRAS a été montrée dans certaines études (Ahrendt et al., 2001 ; Tam et al.,
2006) et des mutations du codon 12 de Kras ont été retrouvées chez des souris traitées à la
NNK ou avec des analogues générant l'un ou l'autre de ses métabolites réactifs (Ronai et
al., 1993).
Il n'y pas de points chauds de mutations dans le gène EGFR même si la majorité des
mutations ont été trouvées sur les exons 19 et 21 (Tam et al., 2006). En effet, sur 131
mutations différentes analysées, pas moins de 40 types de mutations différentes ont été
recensées, essentiellement des mutations ponctuelles et des délétions (Tam et al., 2006). Il
est important de noter que la fréquence des mutations dans EGFR est associée à la
consommation de tabac, mais de manière inversement proportionnelle à l'exposition. Ainsi,
ce sont les non-fumeurs qui sont le plus susceptibles d'avoir une mutation dans EGFR :
74 % d'entre eux contre seulement 19 % des fumeurs (Tam et al., 2006).
1.6 Quels mutagènes du tabac peuvent être associés à quelles mutations ?
Les mutations dans TP53 et KRAS (dans le codon 12) ont la particularité d'être en grande
majorité des mutations ponctuelles survenant à des endroits précis qui constituent des
points chauds de mutation. Ainsi, il devient possible de faire des associations entre les
profils de mutations trouvées dans les tumeurs et les substances potentiellement à l'origine
de ces tumeurs. Ces analyses se font à deux niveaux : (1) le lieu de formation des mutations
(la position des codons mutés) et (2) la nature des mutations (transition, transversion...).

29
1.6.1 Les points chauds de mutation de TP53 et KRAS Comme les mutations dans le gène KRAS se concentrent au niveau d'un nombre limité de
codons (surtout le codon 12), c'est essentiellement au niveau du gène TP53 que s'est faite
l'étude des spectres de codons mutés. On a ainsi pu constater que, dans TP53, les codons
mutés ne sont pas nécessairement les mêmes dans les tumeurs de fumeurs et dans les
tumeurs de non-fumeurs. La Figure 9 montre le nombre de tumeurs recensées dans la base
de donnée du Centre international de Recherche sur le Cancer (Olivier et al., 2002) qui
présentent une mutation à chaque codon particulier de TP53 (des codons 120 à 300 où sont
concentrées l'immense majorité des mutations de TP53) chez les fumeurs et chez les non-
fumeurs.

30
Figure 9 : Spectre des codons mutés dans TP53 pour les tumeurs pulmonaires de fumeurs (tous niveaux de consommation confondus) et de non-fumeurs.
Les données sont extraites de la banque des mutations de TP53 du Centre International de Recherche sur le Cancer (Olivier et al., 2002) version R10, Juillet 2005 (http://www-p53.iarc.fr/AdvancedCriteria.asp). Certains des codons de TP53, comme les codons 248 et 273, sont fréquemment mutés dans
tous les types de cancers. Mais d'autres, comme les codons 157 et 245 semblent être des
points chauds de mutations particuliers aux cancers du poumon chez les fumeurs. Selon la
théorie de la mutagenèse somatique (cf. section 1.2.2.), l'influence de la consommation de

31
cigarette sur la probabilité de transformation maligne peut se faire à plusieurs niveaux, mais
l'explication la plus évidente est qu'il doit y avoir des dommages spécifiques aux
carcinogènes de la fumée de tabac qui sont générés aux positions nucléotidiques
correspondantes de TP53 et que ces dommages sont peu ou pas réparés.
1.6.1.1 Fréquence de formation de dommages à des sites fréquemment mutés Denissenko et al. (Denissenko et al., 1996) ont montré que le (+/-)-anti-benzo[a]pyrène-
7,8-diol-9,10-époxyde (BPDE), la forme activée du benzo[a]pyrène, génère
préférentiellement des dommages au niveau de points chauds de mutations de TP53 (aux
codons 157, 248 et 273) à l'intérieur des cellules. Les dommages sont également réparés
plus lentement à ces positions nucléotidiques particulières (Denissenko et al., 1998).
Plus récemment, un système in vitro a été mis au point pour étudier la fréquence de
formation d'adduits particulier au niveau de séquences fréquemment mutées dans les
cancers du poumon (Tretyakova et al., 2002). Ainsi, plusieurs études dont les résultats sont
résumés dans le Tableau 4, ont été réalisées pour déterminer si les adduits du BPDE ou de
la NNK sont plus fréquemment formés sur les guanines situées dans les séquences
correspondant au codon 12 de KRAS ou aux différents points chauds de mutation de TP53.

32
Codon 12 de KRAS (GGT) Points chauds de mutation dans TP53
Lieu de formation ? Effet de Me des C ? Lieu de formation ? Effet de Me des C ?
NNK (O6-mG, O6-pobG, N7-mG)
Formation préférentielle des 3 types d'adduits à la
2ème G (3)
Inhibition de la formation des 3 types
d'adduits aux G (4)
Pas de formation préférentielle des
adduits (6)
Inhibition de la formation des 3 types
d'adduits aux G (6)
BPDE (N2-BPDE-G)
Formation préférentielle des
adduits à la 1ère G (1)
Ne favorise pas la formation d'adduits
au codon 12 (2)
Formation préférentielle des adduits à tous les
points chauds sauf ceux de l'exon 5 (1)
Favorise la formation des adduits
aux G (1,5)
Tableau 4 : Bilan des études de formation préférentielle d'adduits de la NNK ou du BPDE dans des séquences contenant le codon 12 de KRAS ou les différents points chauds de mutation de TP53. L'influence éventuelle d'une méthylation (Me) en position 5 de la cytosine sur la formation préférentielle d'adduits sur la guanine adjacente a aussi été investiguée. (1) (Tretyakova et al., 2002) ; (2) (Hu et al., 2003) ; (3) (Ziegel et al., 2003) ; (4) (Ziegel et al., 2004) ; (5) (Matter et al., 2004) ; (6) (Rajesh et al., 2005). N.B. Dans cette dernière étude, la formation de N7-pobG a également été investiguée mais a donné le même résultat que pour les autres adduits de la NNK.
Ces études ont déterminé que les adduits du BPDE se forment préférentiellement à tous les
points chauds de mutation de TP53 excepté ceux de l'exon 5 (exon qui contient les codons
157 et 158). Cette formation préférentielle est encore favorisée lorsque la cytosine
adjacente de la guanine affectée est méthylée en position 5, comme c'est le cas dans les
CpG de TP53 correspondant aux points chauds de mutation. Par contre, aucun des adduits
de la NNK qui ont été testés ne semblent être formés de façon préférentielle à ces mêmes
positions. En revanche, on observe une formation préférentielle de tous les types d'adduits
au niveau du codon 12 de KRAS, simplement c'est plutôt au niveau de la 1ère guanine du
codon pour ceux formés par le BPDE et de la 2ème guanine pour ceux résultant de la NNK.
Il est intéressant de noter que la méthylation des cytosines ne favorise pas mais au contraire
inhibe la formation des adduits de la NNK au niveau des guanines du codon 12 de KRAS,
position qui n'est de toute façon pas méthylée dans les cellules. Ainsi, ces résultats
semblent impliquer les adduits du BPDE dans les mutations observées dans KRAS comme
dans TP53. Il faut cependant noter que tous les types de dommages connus de la NNK (cf.
section 1.4.2.4) n'ont pas été investigués et certains autres adduits pourraient participer dans

33
les mutations observées de TP53, en particulier au niveau de l'exon 5. D'autres facteurs
épigénétiques que la méthylation des cytosines adjacentes pourraient aussi influencer la
formation des adduits in vivo. En effet, de tels facteurs ont été suggérés pour expliquer que
le codon 12 de KRAS puisse être plus fréquemment endommagé (notamment par le BPDE)
dans l'ADN génomique qu'il ne l'est dans la même séquence amplifiée par PCR (Hu et al.,
2003). De la même manière, certains éléments spécifiques à l'ADN génomique doivent
intervenir pour expliquer la formation préférentielle d'adduits du BPDE au codon 157 de
l'exon 5 in vivo (Denissenko et al., 1996) alors que celle-ci ne semble pas exister sur la
même séquence in vitro (Tretyakova et al., 2002).
1.6.1.2 Un effet du tabac sur les autres étapes de la mutagenèse somatique ? En dehors de la fréquence de formation et de la vitesse de réparation des dommages, il n'est
pas impossible que les étapes de la réplication et de la sélection des mutations favorables
soient également affectées différemment chez les fumeurs et les non-fumeurs. En effet, la
mortalité cellulaire associée à l'exposition aux substances toxiques de la fumée de tabac a
deux conséquences majeures. La première, c'est que ces cellules mortes doivent être
remplacées afin que l'intégrité du tissu pulmonaire soit maintenue. Cela nécessite une
division cellulaire (cf. section 1.1.3.3) qui n'aurait pas eu lieu autrement, donnant ainsi des
chances supplémentaires de répliquer un ADN endommagé et donc de fixer des mutations
au niveau de cellules souches responsables du maintien du tissu (Kim et al., 2005). La
seconde conséquence, c'est que les cellules mortes à cause de la fumée de tabac laissent un
vide qui pourrait favoriser l'expansion de cellules adjacentes qui seraient déjà porteuses de
mutations favorables.
Une hypothèse comparable impliquant l'importance de la mortalité des cellules adjacentes
pour favoriser la croissance des cellules mutées a déjà été proposée sous le terme de théorie
du "double punch solaire" (Brash et al., 1996). Cette théorie indique que le développement
d'un cancer de la peau nécessite au moins deux expositions solaires : une pour générer des
mutations, et une seconde pour tuer des cellules afin de laisser de la place pour l'expansion
clonale des cellules mutées. Dans le cas de la consommation de tabac, l'exposition aux
carcinogènes est évidemment chronique.

34
1.6.2 La nature des mutations générées Au delà du lieu de formation des mutations, leur nature peut également renseigner sur les
dommages qui en sont à l'origine. En effet, le type de mutation généré par une polymérase
via l'incorporation du mauvais nucléotide lors de la réplication va dépendre de la nature de
la base endommagée présente sur le brin répliqué. Certains types de mutations peuvent
ainsi être associés avec le(s) mutagène(s) responsable(s).
Les transversions G → T dans le gène TP53 sont très courantes dans les tumeurs
pulmonaires de fumeurs actuels, moins fréquentes chez les ex-fumeurs et rares chez les
gens n'ayant jamais fumé (Le Calvez et al., 2005). Ce type de transversion est très
spécifique du cancer du poumon associé avec la consommation de tabac (Ruggeri et al.,
1993 ; Greenblatt et al., 1994). Les transversions G → T ont été associées à l'exposition aux
HAP, et en particulier au benzo[a]pyrène (Hainaut et Pfeifer, 2001 ; Pfeifer et al., 2002 ;
Pfeifer et Hainaut, 2003 ; Lewis et Parry, 2004), pour les raisons suivantes : (1) elles sont
situées à des positions nucléotidiques fréquemment endommagées par le BPDE
(Denissenko et al., 1996) ; (2) les dommages du BPDE sont réparés de façon plus lente sur
le brin non transcrit (Denissenko et al., 1998) et (3) la réplication de ces adduits génère des
transversions G → T (Maher et al., 1990 ; Ruggeri et al., 1993 ; Seo et al., 2000 ; Yoon et
al., 2001 ; Jackson et al., 2006). Cependant, il a été suggéré que d'autres facteurs plus
spécifiques au tissu pulmonaire, comme par exemple la formation de 8-oxoG, pourraient
aussi intervenir pour générer ce type de transversion (Rodin et Rodin, 2000 ; Rodin et
Rodin, 2002 ; Rodin et Rodin, 2004).
Les transitions G → A dans le gène TP53 sont, quant à elles, beaucoup plus associées avec
le fait de n'avoir jamais fumé. Ce type de mutation pourrait d'ailleurs subvenir
spontanément dans les cellules suite à la désamination d'une guanine en xanthine, qui serait
ensuite mal appariée lors de la réplication de l'ADN (Eritja et al., 1986). En fait, la
fréquence des G → A dans TP53 diminue de manière progressive avec l'exposition au tabac
(Le Calvez et al., 2005). C'est le type de mutation que l'on retrouve de manière largement
majoritaire (96 %) dans le codon 12 de Kras chez les souris exposées à la NNK (Jackson et
al., 2006). Dans le cancer pulmonaire chez l'homme, le codon 12 de KRAS (GGT) peut

35
subir à la fois des transitions G → A (GAT ou AGT) et des transversions G → T (TGT ou
GTT), mais ces dernières se trouvent exclusivement chez les fumeurs, anciens ou actuels
(Le Calvez et al., 2005). Ainsi, les transitions G → A peuvent être associées, dans une
certaines mesure, avec une exposition à la NNK, d'autant plus que l'on sait que la NNK
mène à la formation de O6-alkylG qui génèrent quasi spécifiquement ce type de transition
lors d'une réplication (Loechler et al., 1984 ; Foiles et al., 1991 ; Ronai et al., 1993).
1.6.3 Bilan L'ensemble des données dont on dispose actuellement concernant (1) la composition de la
fumée de tabac et son évolution, (2) le type et le lieu de formation des dommages à l'ADN,
(3) la nature des mutations associées et (4) les types histologiques des tumeurs pulmonaires
ainsi que leur évolution dans le temps, tendent à indiquer que la tumorigenèse pulmonaire
pourrait survenir par des voies différentes en fonction des circonstances (Tableau 5).
Ainsi, la tumorigenèse chez les fumeurs actuels ou ayant arrêté récemment serait plus
associée à une mutation de TP53 et liée à la quantité de tabac consommée, tandis que
transformation maligne menant aux adénocarcinomes et/ou chez des fumeurs ayant arrêté il
y a longtemps serait plus liée à KRAS (Le Calvez et al., 2005). A l'appui de cette hypothèse
indiquant deux types de tumorigenèse pulmonaire se trouve aussi le fait que les tumeurs
mutantes pour KRAS ont tendance à être TP53 sauvage et vice-versa (Le Calvez et al.,
2005).

36
Gros fumeur Non fumeur (fumeur passif ?) Cigarettes fortes Cigarettes légères Fumeur actuel Tabac sans fumée
Ex-fumeur HAP (benzo[a]pyrène) N-nitrosamines (NNK)
Dommages aux points chauds de mutation de TP53 Dommages au codon 12 de
KRAS
Transversions G → T Transitions G → A Mutation dans TP53 Mutation dans KRAS
Carcinome épidermoïde Adénocarcinome
Tableau 5 : Bilan des associations que l'on retrouve le plus fréquemment entre la quantité et le type de consommation de tabac, les carcinogènes du tabac, les mutations présentes dans les tumeurs pulmonaires et les deux principaux types histologiques de CPNPC.
1.7 Les systèmes de réparation des dommages à l'ADN
1.7.1 Quel système de réparation pour quel type de dommage ? La molécule d'ADN subit en permanence des modifications, que ce soit de manière
spontanée ou à cause de l'action d'agents environnementaux internes ou externes à la
cellule. Il existe plusieurs systèmes de réparation plus ou moins spécialisés et qui sont
destinés à maintenir l'intégrité de l'information génétique. La Figure 10 fait un bilan des
types de dommages existant et des stratégies mises en place pour les réparer dans les
cellules de mammifères.

37
Figure 10 : Les dommages à l'ADN les plus communs et leur mode de réparation. HAP : hydrocarbures aromatiques polynucléés ; cis-Pt : cisplatine ; MMC : mitomycine C ; 8-oxoG : 8-oxoguanine ; 7-mG : 7-méthylguanine ; PP(6-4) : photoproduits (6-4) ; DCP : dimères cyclobutyliques de pyrimidines.
Extrait, traduit et adapté de (Hoeijmakers, 2001). Les dommages de l'alkylation ne sont pas seulement pris en charge par le système de
réparation par excision de bases (REB), contrairement à ce qui semble suggéré sur la Figure
10. En fait, la diversité de ces dommages et de leurs conséquences possibles fait que tous
les types de systèmes de réparation participent à contrer les effets néfastes de l'alkylation.
La Figure 11 montre les alkylations possibles dans l'ADN et l'ARN, leurs modes de
réparation et les conséquences possibles en termes de toxicité, de mutation et d'instabilité
génomique.
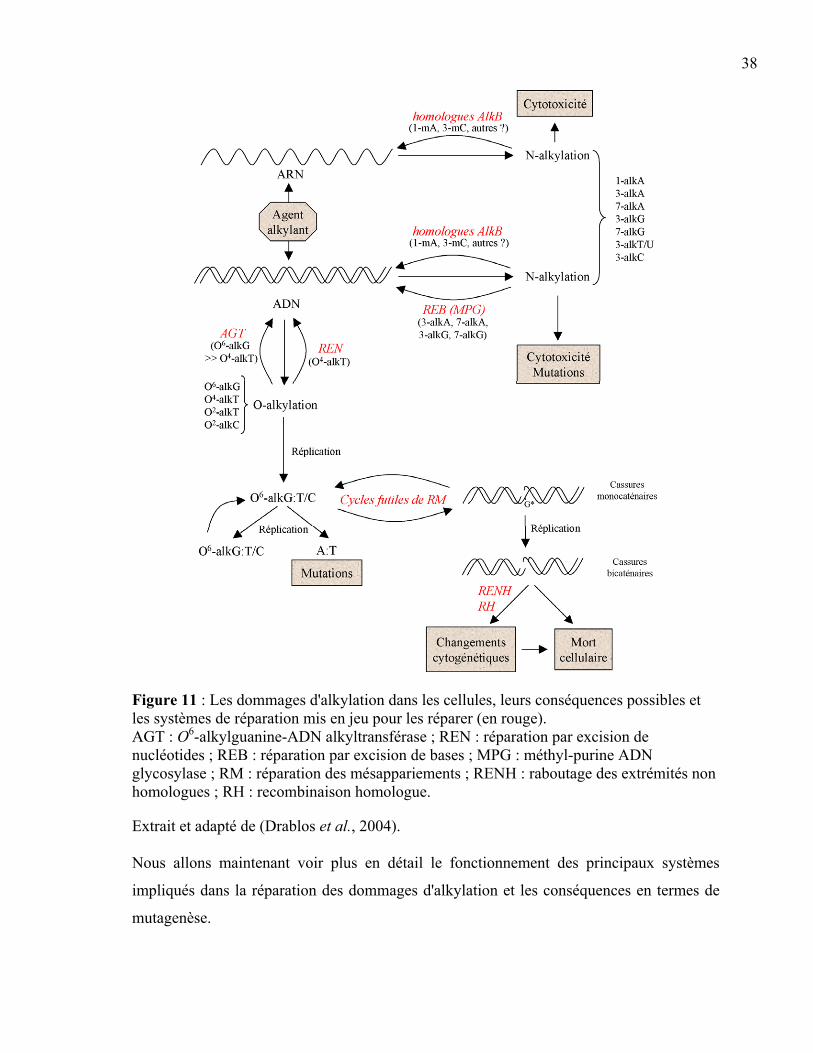
38
Figure 11 : Les dommages d'alkylation dans les cellules, leurs conséquences possibles et les systèmes de réparation mis en jeu pour les réparer (en rouge). AGT : O6-alkylguanine-ADN alkyltransférase ; REN : réparation par excision de nucléotides ; REB : réparation par excision de bases ; MPG : méthyl-purine ADN glycosylase ; RM : réparation des mésappariements ; RENH : raboutage des extrémités non homologues ; RH : recombinaison homologue.
Extrait et adapté de (Drablos et al., 2004). Nous allons maintenant voir plus en détail le fonctionnement des principaux systèmes
impliqués dans la réparation des dommages d'alkylation et les conséquences en termes de
mutagenèse.

39
1.7.2 La réparation par réversion directe des dommages La réversion directe des dommages est en général réalisée par des protéines très
spécialisées dans la réparation d'un dommage particulier. La proportion des dommages à
l'ADN qui sont réparés ainsi est très faible mais ce type de réparation est déterminant quand
il s'agit des dommages d'alkylation. En effet, les quelques enzymes connues chez les
mammifères qui sont capables de réaliser une réversion directe sont des enzymes révertant
des alkylations. Elles se répartissent en deux catégories : la O6-alkylguanine-ADN
alkyltransférase et les analogues d'AlkB.
1.7.2.1 La O6-alkylguanine-ADN alkyltransférase (AGT) La O6-alkylguanine-ADN alkyltransférase (AGT) est une enzyme capable de générer la
réversion directe des O6-alkylguanines (O6-alkG) et des O4-alkylthymines (O4-alkT) en G
et T normales. Cela se fait par le transfert irréversible du groupement alkyl sur un des
résidus cystéine de la protéine (Figure 12), ce qui la rend inactive. Celle-ci est ensuite
dégradée via une ubiquitinylation (Srivenugopal et al., 1996 ; Xu-Welliver et Pegg, 2002).
Ainsi, les niveaux constitutifs d'AGT vont déterminer la capacité initiale, pour une cellule,
de réparer ce type de dommages (Pegg, 2000).

40
Figure 12 : La réversion de l'alkylation réalisée par l'AGT. R : résidu d'alkylation
Extrait de (Duguid et al., 2003). L'AGT est capable d'enlever plusieurs groupements alkyl différents, y compris des
groupements très encombrants (Pegg, 2000). Ainsi, elle peut réparer plusieurs des
dommages associés à la NNK : les O6-mG et les O4-mT résultant de la méthylation, mais
aussi les O6-pobG résultant de la pyridyloxobutylation de l'ADN (Thomson et al., 2003).
La réparation des O6-pobG serait capable d'interférer avec celle des O6-mG et c'est l'un des
mécanismes fréquemment proposé pour expliquer l'action synergétique de la
pyridyloxobutylation et de la méthylation dans la mutagenèse associée à la NNK (Peterson
et al., 1993). La capacité pour l'AGT de réparer un type d'alkylation ou un autre dépend de
la taille du groupement à éliminer puisque l'activité de l'enzyme est dépendante de l'espace
disponible dans la poche enzymatique. En effet, des différences dans l'efficacité de l'AGT
de différents organismes ont pu être expliquées de cette manière (Mijal et al., 2004),
notamment le fait que l'AGT humaine répare plus efficacement les O6-mG que les O6-pobG
alors que l'AGT de rongeurs a la même efficacité pour les deux types d'adduits (Mijal et al.,
2004). De la même manière, certaines variations dans la séquence en acides aminés de la
protéine ont une influence sur la taille de la poche enzymatique et donc sur l'efficacité de
l'enzyme face à certains types de dommages. De telles variations interindividuelles de
l'enzyme AGT ont été associées avec des variations dans les capacités de réparation des O6-
pobG à l'intérieur de la population humaine (Mijal et al., 2004). Bien que cela n'ait pas été
démontré, l'importance relative de chacune des voies d'activation de la NNK dans la
cancérogenèse du poumon chez les rongeurs vs. les êtres humains mais aussi entre
différents individus à l'intérieur de l'espèce humaine pourraient en être affectés.

41
1.7.2.2 Les analogues de AlkB Il existe un autre mode de réversion directe des alkylations qui est réalisée par des
déméthylases oxydatives qui fonctionnent de manière analogue à la protéine AlkB
bactérienne. Elles réparent les 1-méthyladénines (1-mA) et les 3-méthylcytosines (3-mC)
en oxydant directement le groupement méthyl, ce qui mène à la libération de formaldéhyde
(Drablos et al., 2004). Huit homologues à AlkB ont été identifiés dans le génome humain
mais la fonction particulière de chacun est en grande partie inconnue, même si on a pu les
associer avec des localisations intracellulaires différentes. La méthylation en 1 des adénines
ou en 3 des cytosines altère les ponts hydrogènes interbrins et survient plus facilement sur
des substrats monocaténaires. Ainsi, ces analogues de l'AlkB pourraient avoir un rôle
particulièrement important dans la réversion de l'alkylation sur les acides nucléiques
monocaténaires, c'est-à-dire les ARN et l'ADN en cours de réplication ou de transcription
(Drablos et al., 2004). Un des avantages de la réversion directe dans ces circonstances est
qu'elle ne génère pas de cassures monocaténaires, contrairement aux autres systèmes de
réparation comme la réparation par excision de bases.
1.7.3 La réparation par excision de bases (REB)
1.7.3.1 Les différents modes de REB La réparation par excision de bases (REB) est le mode privilégié de réparation des
dommages aux bases. Ce type de réparation fonctionne en deux étapes : une étape
spécifique d'un type de dommages qui est réalisée par une glycosylase et une étape plus
générale impliquant l'action séquentielle de plusieurs protéines et menant à l'insertion de un
ou plusieurs nucléotides commençant au niveau de la base endommagée (Figure 13). La
REB est donc initiée par des glycosylases. La reconnaissance des dommages et la liaison de
l'enzyme peuvent varier d'une glycosylase à l'autre mais elles ont toutes en commun de
fonctionner avec un phénomène de "base flipping" c'est-à-dire que la base anormale se
retrouve expulsée à l'extérieur de la double hélice afin de se retrouver dans le site actif de
l'enzyme (Nilsen et Krokan, 2001).
Les glycosylases bifonctionnelles (Figure 13A) enlèvent la base anormale et forment une
base de Schiff avec l'atome C1' du désoxyribose. Le clivage du brin d'ADN en 3' du

42
désoxyribose se fait ensuite par β-élimination (activité 3' β-lyase de l'enzyme). L'enzyme
HAP1 (aussi appelée APE1) est l'endonucléase majeure pour les sites abasiques (sites AP)
et se trouve à l'intersection de toutes les voies du REB. HAP1 coupe en 5' du site AP
(Figure 13A), générant ainsi une extrémité 3'-OH (Parikh et al., 1999) pouvant servir de
substrat pour une néosynthèse par la polymérase β (Polβ) (Lindahl et Wood, 1999). Puis la
ligase III (LigIII) qui est dans un complexe avec XRCC1, vient sceller la cassure
monocaténaire résiduelle (Cappelli et al., 1997 ; Nash et al., 1997 ; Tomkinson et Mackey,
1998).

43
Figure 13 : La réparation par excision de bases. La réparation est initiée par des ADN glycosylases et peut être suivi d'une néosynthèse courte (1 nucléotide) ou plus longue (1 à 4 nucléotides), dépendamment du type de glycosylase. La réparation de la 8-oxoguanine (8-oxoG) ou de l'uracile (U) sont représentées comme exemple de réparation avec une glycosylase bifonctionnelle (A) ou monofonctionnelle (B et C). La protéine catalytique de chaque étape est soulignée.
Extrait et traduit de (Nilsen et Krokan, 2001). Les enzymes monofonctionnelles (Figure 13B et C) hydrolysent le lien glycosidique et
libèrent la base anormale. HAP1 vient alors couper en 5' du site AP (Kubota et al., 1996 ;

44
Klungland et Lindahl, 1997 ; Parikh et al., 1999) et il y a alors 2 possibilités : la voie de
réparation "courte" (Figure 13B) et la voie "longue" (Figure 13C). Pour la voie "courte", la
Polβ est recrutée directement par l'endonucléase HAP1 et vient insérer un seul nucléotide
(Bennett et al., 1997 ; Fortini et al., 1998). Le groupement 5'-désoxyribose 5-phosphate
(dRP) est enlevé grâce à l'activité 5'-désoxyribose 5-phosphatase (dRPase) de la Polβ
(Matsumoto et Kim, 1995). Cette voie "courte" dépendante de la Polβ semble être
prédominante et survenir dans 75 à 90 % des réparations par REB dans les cellules
humaines (Nealon et al., 1996 ; Sobol et al., 1996 ; Bennett et al., 1997). La voie "longue"
consiste en l'addition de 2 à 8 nucléotides (le plus souvent 2) et se trouve très utilisée si le
groupement dRP est réfractaire à la coupure par la Polβ (Gary et al., 1999). Une synthèse
est alors réalisée qui implique le déplacement du brin déjà en place et le groupement dRP
est alors éliminé dans l'oligonucléotide ainsi écarté (Figure 13C). Ce déplacement de brin
peut être réalisée par la Polβ (Klungland et Lindahl, 1997 ; Dianov et al., 1999) mais
surtout par les Polδ et Polε (Matsumoto et al., 1999 ; Pascucci et al., 1999). L'antigène
nucléaire des cellules en prolifération (PCNA) et le facteur de réplication C (RFC) sont
indispensables à cette étape lorsque la synthèse de réparation est faite par Polδ ou Polε, ce
qui semble être le cas la plupart du temps (Stucki et al., 1998). Les ligases I (LigI) et III
(LigIII) sont toutes les deux capables de sceller la cassure monocaténaire résiduelle
(Klungland et Lindahl, 1997), mais comme la LigI interagit avec PCNA (Levin et al.,
1997), on suppose que c'est plutôt elle qui intervient lors d'une réparation par la voie
"longue".
1.7.3.2 La méthyl-purine ADN glycosylase (MPG) La méthyl-purine ADN glycosylase (MPG) aussi appelée alkyladénine glycosylase (AAG)
est une glycosylase monofonctionnelle qui excise une grande variété de purines altérées
telles que les 3-mA, 7-mG, hypoxanthine, éthènoadenine et éthènocytosine (O'Connor et
Laval, 1991 ; Samson et al., 1991 ; Saparbaev et Laval, 1994 ; Izumi et al., 1997).
L'efficacité de l'excision par la MPG dépend du type de dommages, les 3-mA étant excisées
plus efficacement que les 7-mG (Plosky et al., 2002).

45
En ce qui concerne les dommages associées à la NNK, la MPG devrait intervenir pour
réparer les 7-mG. Cependant, il a été montré que cette voie n'est pas la seule capable
d'éliminer les 7-mG puisque ces adduits sont également réparés dans des cellules murines
déficiente en cette glycosylase (Plosky et al., 2002). Il est possible que la MPG excise aussi
les 7-pobG de la NNK, mais cette enzyme semble moins efficace que son homologue
bactérien (AlkA) pour exciser les dommages aux bases correspondant à des lésions
encombrantes (Mattes et al., 1996). Les adduits encombrants tendent plutôt à être réparés
par le système de réparation par excision de nucléotides (Levy et al., 1992 ; Hoare et al.,
2000 ; Alekseyev et al., 2004 ; Bedard et Massey, 2006).
1.7.3.3 La formamidopyrimidine glycosylase (Fpg) La formamidopymidine glycosylase (Fpg) est une glycosylase bifonctionnelle bactérienne
qui excise les purines oxydées tels que les 8-oxoG et les dérivés formamidopyrimidines
(fapy) de purines (fapyA ou fapyG) et de nombreuses autres purines modifiées. Les
substrats de la Fpg bactérienne sont plus variés que ses homologues chez les mammifères.
Notamment, la Fpg est capable d'exciser des dommages aux bases correspondant à des
lésions encombrantes, y compris des dommages d'alkylation, du moment qu'ils sont sous
forme fapy (Chetsanga et Frenette, 1983 ; Li et al., 1997 ; Tudek, 2003). Ainsi, les
bactéries peuvent utiliser la REB pour réparer plusieurs adduits, tels que les adduits fapy de
l'aflatoxine B1, qui doivent nécessairement être réparés par REN chez les mammifères
(Tudek, 2003 ; Alekseyev et al., 2004).
1.7.3.4 La régulation de la REB Tous les types de lésions aux bases ne sont pas réparés avec la même efficacité dans une
cellule donnée (Cappelli et al., 2000). La quantité disponible d'enzyme initiatrice de la
réparation, à savoir la glycosylases spécifique, est un facteur limitant mais ça n'est pas le
seul paramètre déterminant l'efficacité de la REB d'un type de dommage donné (Cappelli et
al., 2001). En effet, une estimation de la quantité présente par cellule de certaines protéines
participant à la REB a permis de constater qu'un dommage n'est pas nécessairement réparé
plus rapidement parce qu'il y a plus de molécules participant à sa réparation dans la cellule
(Cappelli et al., 2001). La régulation de la REB se ferait donc aussi à travers des

46
modifications post-traductionnelles et des variations dans les interactions entre protéines
(Izumi et al., 2003). Ainsi, même si l'excision par la MPG est une étape limitante dans la
réparation des 7-mG (Ye et al., 1998), il a été montré que la surexpression de cette
glycosylase ne protège pas les cellules de l'action des agents alkylants (Ibeanu et al., 1992),
ce qui indique que la quantité de glycosylase n'est pas le seul facteur déterminant la vitesse
de réparation des dommages d'alkylation par la REB. En fait, l'endonucléase HAP1 serait
une protéine clé impliquée dans la coordination de la REB (Izumi et al., 2003).
La protéine p53 est capable de stimuler la REB dans les cellules en interagissant
directement avec HAP1 et la Polβ (Zhou et al., 2001). En effet, p53 stabilise l'interaction
entre la Polβ et l'ADN et cette fonction est très importante vu qu'il y a diminution très
importante de la REB si p53 est enlevée des cellules (Zhou et al., 2001). De plus, suite à
différents stress génotoxiques, il a été montré que p53 induit la MPG mais pas HAP1
(Zurer et al., 2004). Cependant dans certains cas, comme lorsque les cellules sont traitées
avec de l'oxyde nitrique, un autre mécanisme a été démontré où p53 induit la MPG mais a
secondairement un effet de répression en trans de l'ARNm de cette même enzyme,
vraisemblablement pour limiter les risques d'accumulation de sites AP dans les cellules au
delà des capacités de prise en charge par HAP1 (Zurer et al., 2004).
1.7.4 La réparation par excision de nucléotides (REN)
1.7.4.1 Les différents modes de la REN
Le système de réparation par excision de nucléotides (REN) est plus particulièrement
spécialisé dans les dommages encombrants qui provoquent une distorsion importante de la
double hélice d'ADN (Gillet et Scharer, 2006). Mais comme nous l'avons vu
précédemment, la REN peut aussi prendre en charge d'autres types de dommages, servant
ainsi de système de soutien à la REB. C'est au niveau de l'efficacité de la réparation que le
type de dommages va avoir le plus d'influence, les dommages distordant le plus la double
hélice étant ceux qui sont le plus facilement excisés par REN (Branum et al., 2001). Les
dommages de la NNK susceptibles d'être réparés par la REN sont donc très variés mais elle
devrait être impliquée plus particulièrement dans la réparation des dommages dérivant de la
pyridyloxobutylation étant donné que les adduits générés sont plus volumineux que ceux

47
résultant de la méthylation. En particulier, la REN pourrait intervenir dans la réparation des
pyridyloxobutylphosphotriesters. En effet, bien que la réparation de ces adduits n'ait jamais
été étudiée spécifiquement, on sait que la machinerie du REN est capable d'exciser des
dommages aux phosphates plus petits tels que les méthylphosphotriesters (Branum et al.,
2001).
Il existe deux voies d'initiation de la REN : une voie globale capable de réparer les
dommages partout dans le génome (G REN) et une voie couplée à la transcription (TC
REN) où la REN est initiée par l'arrêt de la polymérase au niveau des dommages. La Figure
14 indique les différentes étapes de la G REN.
Figure 14 : Les différentes étapes de voie globale de la réparation par excision de nucléotides. N.B. : Dans la partie B, il faut lire HR23B et non R23B.
Extrait et traduit de (Gillet et Scharer, 2006).

48
La séquence des événements associés à la REN a été reconstituée comme suit (Riedl et al.,
2003 ; Tapias et al., 2004 ; Gillet et Scharer, 2006). Les lésions induisant une distorsion de
l'ADN sont détectées par le complexe XPC-HR23B qui stabilise la courbure de l'ADN.
XPC-HR23B recrute le complexe TFIIH au site de la lésion. TFIIH déroule la double hélice
jusqu'à ce qu'une de ses sous-unités hélicase (XPD dans la Figure 14) rencontre une base
modifiée chimiquement. La seconde sous-unité hélicase (XPB dans la Figure 14) continue à
dérouler l'ADN pour créer une structure ouverte en "bulle" de 20 pb. RPA, XPA et XPG
sont alors recrutées pour assembler le complexe de "préincision". ERCC1 et XPF
rejoignent le complexe et une double incision est réalisée (en 5' par ERCC1-XPF et en 3'
par XPG). RPA reste liée à l'ADN monocaténaire et facilite la synthèse d'ADN liée à la
réparation par la Polδ (ou Polε) avec l'aide de RFC et PCNA. La LigI vient alors sceller la
cassure résiduelle.
La réparation couplée à la transcription se déroule de la même manière, si ce n'est que la
détection du dommage, et donc l'initiation de la réparation, se fait par la polymérase qui est
bloquée au niveau du dommage sur le brin en cours de transcription. Les protéines CSA et
CSB sont alors nécessaires mais le fonctionnement exact de ces deux protéines n'est encore
complètement élucidé (Costa et al., 2003).
L'efficacité différentielle dans la capacité pour la REN de réparer un type de dommages
plutôt qu'un autre a une conséquence très concrète lorsque les cellules ont à faire face à
plusieurs types de dommages simultanément, ce qui est évidemment le cas de celles
soumises à la fumée de cigarette. En effet, il a été montré que certains adduits tels que ceux
résultant de l'action de l'acétylaminofluorène (AAF) qui sont très efficacement reconnus par
les protéines de la REN, sont capables d'en monopoliser une large fraction et ce aux
dépends de la réparation des autres types d'adduits présents (Buterin et al., 2002). On peut
anticiper, à partir de ce mécanisme, l'existence d'un effet génotoxique "hyper additif"
lorsque les cellules sont simultanément exposées à plusieurs agents génotoxiques (Buterin
et al., 2002). Cette compétition des adduits pour être réparés par REN pourrait participer au
fait que les deux voies d'activation de la NNK sont nécessaires à la transformation maligne
in vivo (cf. section 1.4.2.3). En effet, s'il y a simultanément méthylation et

49
pyridyloxobutylation, il y a une plus grande diversité de dommages produits en même
temps dans les cellules qui sont en concurrence pour les mêmes systèmes de réparation.
1.7.4.2 Les remaniements de la chromatine associés à la REN Dans les cellules vivantes, l'ADN est organisé en chromatine et pour qu'une réparation par
REN puisse avoir lieu, il doit y avoir remodelage de cette chromatine afin que la
machinerie de réparation ait accès aux dommages (Hara et al., 2000 ; Ura et al., 2001 ;
Gillet et Scharer, 2006). Suite à une irradiation UV, il a été montré que l'acétylation des
histones est augmentée et que la stabilisation des histones hyperacétylés, par l'action des
inhibiteurs d'histone désacétylases, favorise la réparation des dommages (Figure 15). Cela
suggère qu'en réponse à une irradiation UV, l'acétylation des histones participe à la réponse
cellulaire globale afin de favoriser la réparation des dommages (Ramanathan et Smerdon,
1989 ; Gillet et Scharer, 2006). La protéine p53 est nécessaire à la relaxation globale de la
chromatine suite à une irradiation UV et cela de manière indépendante de son action
comme facteur de transcription (Allison et Milner, 2004).
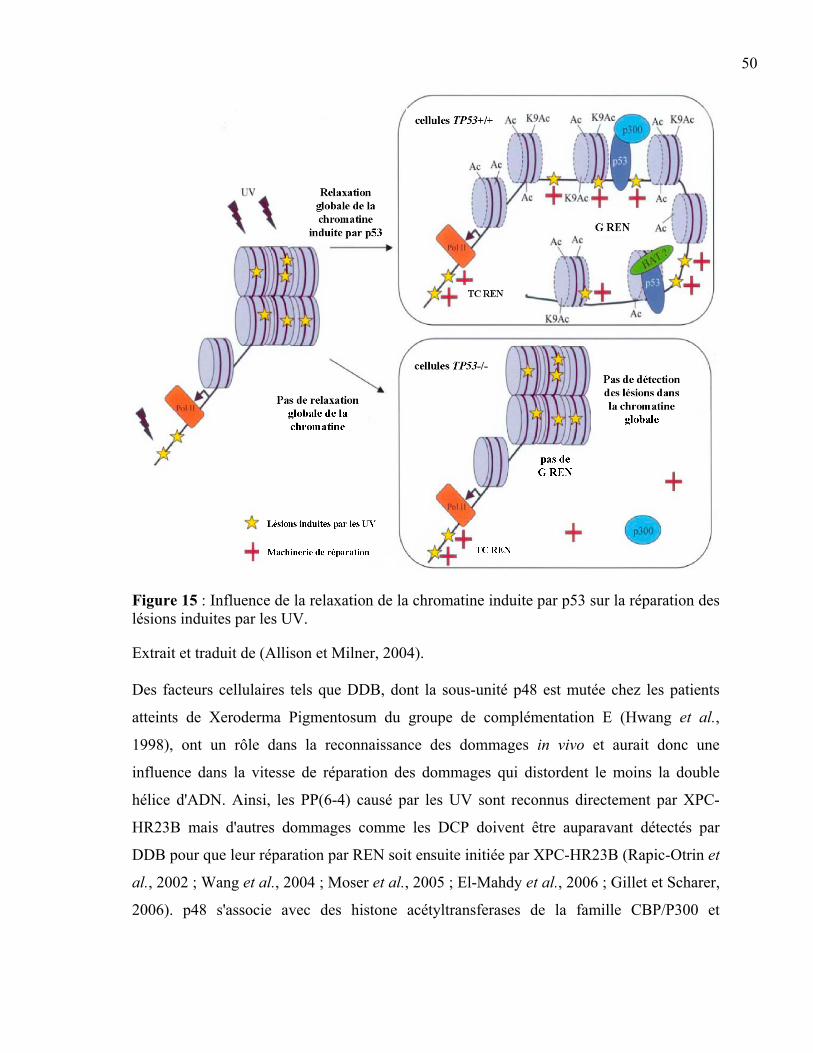
50
Figure 15 : Influence de la relaxation de la chromatine induite par p53 sur la réparation des lésions induites par les UV.
Extrait et traduit de (Allison et Milner, 2004). Des facteurs cellulaires tels que DDB, dont la sous-unité p48 est mutée chez les patients
atteints de Xeroderma Pigmentosum du groupe de complémentation E (Hwang et al.,
1998), ont un rôle dans la reconnaissance des dommages in vivo et aurait donc une
influence dans la vitesse de réparation des dommages qui distordent le moins la double
hélice d'ADN. Ainsi, les PP(6-4) causé par les UV sont reconnus directement par XPC-
HR23B mais d'autres dommages comme les DCP doivent être auparavant détectés par
DDB pour que leur réparation par REN soit ensuite initiée par XPC-HR23B (Rapic-Otrin et
al., 2002 ; Wang et al., 2004 ; Moser et al., 2005 ; El-Mahdy et al., 2006 ; Gillet et Scharer,
2006). p48 s'associe avec des histone acétyltransferases de la famille CBP/P300 et

51
favoriserait aussi la REN par l'induction d'un remodelage local de la chromatine au niveau
du site endommagé (Datta et al., 2001).
1.7.4.3 La régulation de la REN Il a clairement été montré par plusieurs équipes que la perte de p53 mène à une déficience
de la REN suite à une irradiation UV (Ford et Hanawalt, 1995 ; Ford et Hanawalt, 1997 ;
Therrien et al., 1999 ; Zhu et al., 2000). La régulation de la REN par p53 se fait à plusieurs
niveaux. En effet, une p53 fonctionnelle est nécessaire pour avoir un niveau basal des
protéines XPC et p48 mais elle intervient aussi pour induire une réponse adaptative plus
tardive en activant la transcription de ces mêmes gènes suite à une irradiation UV (Hwang
et al., 1999 ; Adimoolam et Ford, 2002). BRCA1 favorise aussi la transcription de XPC,
p48 et GADD45 d'une manière indépendante de p53, favorisant ainsi la REN (Hartman et
Ford, 2002). L'exposition chronique à des dommages réparés par REN mène ainsi à une
adaptation des cellules qui sont alors capables de réparer plus rapidement des dommages
subséquents (Ye et al., 1999). Cette augmentation de la quantité de facteurs impliqués dans
la reconnaissance des dommages favorise la réparation de dommages moins bien reconnus
et donc qui ne sont pris en charge que secondairement par XPC-HR23B, comme c'est le cas
pour les DCP.
1.7.5 Les autres systèmes de réparation Comme nous l'avons vu dans la Figure 11, d'autres systèmes de réparation interviennent
dans la réparation des dommages d'alkylation. Comme leur implication est plus marginale,
quantitativement parlant, ils ne seront pas détaillés ici mais il semble tout de même
important de dire à quel niveau ils interviennent et les conséquences que cela peut avoir en
terme de cytotoxicité et de mutagenèse.
La réplication des O6-mG résulte en l'intégration d'une T en face de la G alkylée, d'où la
génération de transversions G → A à la réplication suivante, comme nous l'avons vu
précédemment (cf. section 1.6.2). Ces mésappariements O6-mG:T sont reconnus par le
système de réparation des mésappariements (RM) qui excise le brin qui vient d'être répliqué
afin de corriger le problème (Figure 11). Mais lors de la néosynthèse d'ADN subséquente,

52
le brin répliqué comporte toujours la O6-mG et il y a de nouveau réplication anormale avec
intégration d'une T. Il s'en suit toute une série de cycles futiles de tentative de RM
(Margison et Santibanez-Koref, 2002 ; Drablos et al., 2004). Ces cycles futiles de RM sont
responsables de la cytoxicité des agents méthylants. En effet, alors que les agents
méthylants sont toujours mutagènes, ils ne sont pas cytotoxiques dans les cellules
déficientes en RM. Cette induction de la mort cellulaire serait liée à la formation secondaire
de cassures bicaténaires (CBC) suite à la réplication d'un ADN présentant des CMC
générées lors des cycles futiles de RM (Margison et Santibanez-Koref, 2002 ; Drablos et
al., 2004).
Les CBC, en ce qui concerne les dommages d'alkylation, résultent en général de la
réplication d'un ADN présentant des CMC, comme celles générées lors d'événements de
réparation non complétés ou résultant directement de l'action de l'agent alkylant, comme
c'est le cas suite à un traitement à la NNK (cf. section 1.4.2.4). Dépendamment du moment
dans le cycle cellulaire, les CBC sont réparées par RENH ou par RH (Figure 10).
Lorsqu'elles ne sont pas réparées, elles sont susceptibles de mener à la perte de matériel
génétique (délétions) ou à des réarrangements chromosomiques et provoquer ultimement la
mort cellulaire ou la transformation néoplasique (Mills et al., 2003).
1.8 Les méthodes d'analyse des dommages à l'ADN et de leur réparation
Les techniques pour étudier les dommages à l'ADN sont assez nombreuses et le but n'est
pas ici de les recenser de manière exhaustive mais plutôt de décrire celles qui seront plus
particulièrement mentionnées ou utilisées dans le corps de cette thèse : la "Ligation-
mediated PCR" (LMPCR) et le test des comètes.
1.8.1 La "ligation-mediated PCR" La "ligation-mediated PCR" (LMPCR) est une technique de séquençage génomique qui
permet de détecter au niveau nucléotidique le lieu et la fréquence de formation des
dommages dans une section prédéterminée du génome. Les dommages générés peuvent être
détectés s'il s'agit de CMC ou de dommages pouvant être convertis en CMC par un

53
traitement chimique ou enzymatique. Ces cassures doivent en outre posséder une extrémité
5' avec un phosphate et être d'une fréquence minimale approximative de 1 cassure par
5 x 105 bases (Drouin et al., 1996). Les différentes étapes de la technique sont indiquées
dans la Figure 16.
Une extension est réalisée à partir d'une amorce spécifique du gène d'intérêt et s'arrête au
lieu de la cassure. Un adaptateur asymétrique est alors lié à toutes les extrémités ainsi
générées disposant d'un phosphate (P) en 5' et une amplification par PCR est réalisée entre
une deuxième amorce spécifique du gène d'intérêt choisie en 3' de la première amorce et
une amorce spécifique de l'adaptateur. Enfin, les produits PCR ainsi obtenus sont mis à
migrer dans un gel d'acrylamide, transférés sur une membrane et détectés à l'aide d'une
sonde spécifique de la région d'intérêt. Les produits d'amplification ont une taille et une
quantité qui dépendent respectivement du lieu et de la fréquence des cassures à chaque
position nucléotidique, donc de la fréquence des dommages générés dans l'ADN à l'origine.
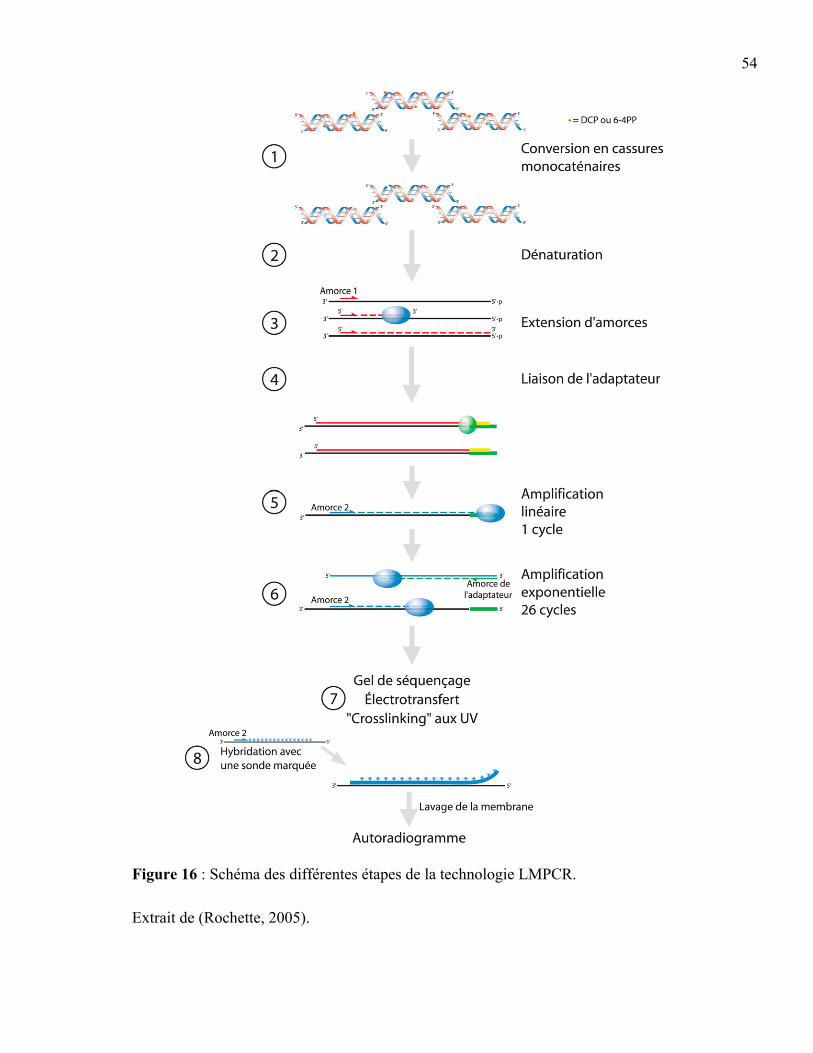
54
Figure 16 : Schéma des différentes étapes de la technologie LMPCR.
Extrait de (Rochette, 2005).

55
La technologie LMPCR permet d'identifier quelles positions nucléotidiques sont
endommagées par un agent génotoxique donné et avec quelle fréquence. La réparation peut
également être suivie en quantifiant les produits PCR obtenus avec l'ADN de cellules
récoltées à différents intervalles de temps après le traitement. Cette technique a été utilisée
pour montrer la corrélation entre les sites de formation préférentielle des dommages par un
agent génotoxique donné et les sites fréquemment mutés dans les tumeurs associées à cet
agent endommageant, comme par exemple pour les dommages du benzo[a]pyrène dans le
gène TP53 en relation avec les sites mutés dans les tumeurs du poumon (cf. section
1.6.1.1).
1.8.2 Le test des comètes Le test des comètes permet d'étudier la fréquence globale de dommages générés dans des
cellules individuelles. Comme pour la LMPCR, les dommages sont détectés sous forme de
cassures mais la technique est beaucoup plus sensible puisque que des dommages d'une
fréquence de l'ordre de 1 cassure pour 108 bases peuvent être détectés (Singh et al., 1990).
Une première version de la technique a d'abord été mise au point par Östling et Johanson
(Ostling et Johanson, 1984) comme une technique microélectrophorétique permettant de
visualiser des cassures radioinduites dans des cellules individuelles. Pour réaliser cette
technique, un faible nombre de cellules irradiées avaient été suspendues dans de l'agarose et
placées en fine couche sur une lame puis lysées, soumises à une électrophorèse et colorées
avec un colorant à ADN fluorescent. Les images ainsi obtenues ressemblaient à des
"comètes", ce qui est à l'origine du nom le plus fréquemment donné à la technique. Östling
et Johanson observèrent que la quantité d'ADN libérée de la tête de la comète durant
l'électrophorèse est fonction de la dose d'irradiation (Figure 17).

56
.
Figure 17 : Evolution de l'aspect des comètes en fonction de la fréquence de dommages dans les cellules.
Une version alcaline du test a permis d'augmenter sa sensibilité et de détecter les sites alcali
labiles en plus des cassures directement générées par l'agent génotoxique (Singh et al.,
1988). De nombreuses variations de ce test sont aujourd'hui utilisées, incluant certaines qui
présentent une étape de digestion avec des glycosylases spécifiques permettant d'augmenter
simultanément la sensibilité et la spécificité du test (Collins, 2004). Mais les principes de la
technique sont toujours les mêmes et sont indiqués à la Figure 18.
Figure 18 : Les différentes étapes du test des comètes.

57
La fréquence de dommages générés peut être estimée par différents paramètres mais le
pourcentage d'ADN dans la queue semble le plus intéressant étant donné qu'il évolue de
manière linéaire avec la dose de radiations ionisantes pour des doses allant de 0 à 10 Gy
(Collins, 2004). Le pourcentage d'ADN dans la queue est déterminé en mesurant, à l'aide
d'un logiciel spécialisé, le nombre de pixels présents dans la queue et dans la tête de chaque
comète individuelle (Figure 19). En résumé, le test des comètes permet d’étudier la
fréquence globale des cassures au niveau de chaque cellule individuelle, alors que la
technologie LMPCR permet une étude de la fréquence des dommages au niveau de la
séquence nucléotidique pour la moyenne d’une population cellulaire.
Figure 19 : Analyse des images de comètes avec le logiciel CASP (Comet Assay Software Project).
Image extraite de (Konca et al., 2003). Étant donné sa grande sensibilité, les applications les plus fréquentes du test des comètes
sont en rapport avec la recherche et l'estimation de la fréquence de dommages
correspondant à des expositions réelles à des agents génotoxiques connus ou soupçonnés. Il
peut aussi servir à estimer la génotoxicité de substances qui sont encore mal connues (Tice

58
et al., 2000). Certaines études se sont intéressé à la détection de dommages à l'ADN
associés à l'exposition à la fumée de cigarette mais avec des résultats contradictoires : la
plupart n'ont pas réussi à mettre en évidence un plus grand nombre de dommages dans les
cellules de fumeurs par rapport à celles de non fumeurs (Speit et al., 2003).
1.9 Les objectifs de l'étude La NNK est un carcinogène présent dans la fumée de tabac et il ne fait aucun doute que
cette substance est capable à elle seule de mener à la formation de tumeurs pulmonaires
chez les rongeurs (Hecht, 1998). Mais il est particulièrement difficile de tirer des
conclusions quant à l'implication de la NNK dans les cancers du poumon chez l'homme à
partir des données obtenues chez l'animal à cause des différences que nous avons décrites
précédemment. Il est donc important d'étudier la fréquence et la réparation des dommages à
l'ADN associés à la NNK dans des cellules humaines si l'on veut mieux comprendre
l'implication de la NNK dans la transformation maligne associée à la cigarette.
1.9.1 Les objectifs de l'article 1 (chapitre 2) La technologie LMPCR permet d'étudier le lieu et la fréquence de formation de dommages
à l'ADN générés par un agent génotoxique donné et de suivre leur réparation dans le temps.
Ainsi, des corrélations peuvent être faites entre les lieux préférentiels de formation des
dommages et les sites fréquemment mutés dans les tumeurs. Ce court article de synthèse
avait pour objectif de faire le point sur les informations qui ont pu être obtenues à ce jour
grâce à cette technique dans la perspective de mieux comprendre les deux premières étapes
intervenant dans la mutagenèse somatique que sont la formation des dommages et leur
réparation.
La technologie LMPCR a permis de fournir, dans un certain nombre de cas, une
confirmation au niveau moléculaire de liens étiologiques qui étaient déjà établis suite à des
études épidémiologiques. Ainsi, l'étude au niveau nucléotidique de la position et de la
fréquence de formation des dommages générés par différentes longueurs d'ondes d'UV ont
permis de confirmer que les DCP générés par les UVB et possiblement aussi les UVA sont
la cause des mutations associées avec les cancers cutanés non mélanocytiques. De la même

59
manière, la formation préférentielle des adduits du BPDE, la forme activée du
benzo[a]pyrène de la fumée de cigarette, a pu être montrée à des positions nucléotidiques
fréquemment mutées dans les tumeurs pulmonaires, confirmant la participation de cet agent
mutagène dans la transformation maligne associée à la cigarette.
Si la NNK participe également à la mutagenèse associée avec la consommation de tabac, il
devrait y avoir une formation de dommages associés dans les cellules à des endroits qui
sont mutés dans les tumeurs. Et comme pour le BPDE, cela doit pouvoir être investigué
avec la technologie LMPCR. Cette technique a effectivement été utilisée pour étudier
certains dommages associés à la NNK, notamment ceux correspondant à la méthylation de
l'ADN que ce soit ceux formés sur de l'ADN purifié ou dans les cellules. Les dommages
résultant de la pyridyloxobutylation de l'ADN, et plus particulièrement les CMC, ont aussi
pu être cartographiés sur de l'ADN nu et mis en rapport avec des positions fréquemment
mutées dans les tumeurs pulmonaires.
1.9.2 Les objectifs de l'article 2 (chapitre 3) La formation in vivo et la réparation des dommages associés à la NNK et résultant de la
pyridyloxobutylation de l'ADN sont très mal connus, à l'exception notable des O6-
alkylguanines. S'il est probable que ces derniers participent à la mutagenèse associée à
l'exposition à la NNK chez l'homme, il est possible que d'autres types de dommages soient
impliqués. Cependant on ignore à peu près tout de la fréquence de formation et de la
réparation de ces dommages dans les cellules humaines. Bien que cette technique apporte
beaucoup d'informations pertinentes, il s'est avéré très difficile de générer une fréquence de
pyridyloxobutylation de l'ADN suffisamment élevée pour que les dommages puissent être
étudiés par la technologie LMPCR. Nous avons alors décidé d'utiliser une autre technique
capable de détecter des dommages beaucoup moins fréquents : le test des comètes. Les
fréquences de dommages nécessaires à l'étude sont alors plus faciles à obtenir et se
rapprochent de celles générées suite à une exposition réelle à la fumée de cigarette.
Cependant, le test des comètes souffre de son manque de spécificité étant donné que seules
des cassures peuvent être détectées. Les objectifs principaux de ce travail étaient :

60
1. Tester différentes stratégies qui permettent d'améliorer la sensibilité et la spécificité du
test des comètes.
2. Identifier la nature des dommages à l'ADN observés avec le test des comètes en se
référant aux dommages associés à la NNK qui ont déjà été identifiés.
3. Estimer la fréquence relative de formation de ces différents types de dommages
associés à la NNK dans des cellules humaines.
1.9.3 Les objectifs de l'article 3 (chapitre 4) L'article précédent nous a permis de mettre en évidence que certains dommages associés
avec la NNK peuvent être étudiés assez spécifiquement par le test des comètes. Nous
souhaitions pouvoir investiguer la réparation globale de ces dommages dans différents
types cellulaires. Le test des comètes n'est que très rarement utilisé pour déterminer des
cinétiques de réparation de dommages et lorsque c'est le cas, il ne s'agit pas nécessairement
de dommages dont la nature est clairement identifiée. Ainsi, les principaux objectifs de cet
article étaient de :
1. Déterminer si le test des comètes peut être utilisé pour obtenir des cinétiques de
réparation d'adduits spécifiques comme les 7-mG générés par le diméthylsulfate ou les
dimères cyclobutyliques de pyrimidines résultant d'une irradiation aux UV.
2. Investiguer les cinétiques de réparation dans différents types cellulaires de tous les
types de dommages associés à la NNK que l'on est capable d'étudier spécifiquement
avec le test des comètes.
3. Déterminer pour chaque type de dommages s'il existe des différences notables dans les
vitesses de réparation d'un type cellulaire à l'autre.
4. Si les cinétiques de réparation diffèrent entre les cellules testées, déterminer si ces
différences pourraient être liées au type cellulaire, notamment selon que ces cellules
sont compétentes ou non pour activer la NNK.

Chapitre 2 Comprendre la mutagenèse somatique via la cartographie des dommages à l’ADN
(soumis à la revue « Médecine Sciences »)

62
2.1 Chapô La théorie de la mutagenèse somatique indique que la transformation maligne des cellules
est intimement liée à la formation de dommages à l'ADN et au risque que ces derniers
puissent mener à des mutations favorables à la croissance cellulaire. Elle énonce donc le
lien logique qui associe l'exposition à des agents génotoxiques et le cancer. Les spectres de
mutations, notamment dans le gène TP53, recensent les mutations dans un type de cancer
donné en fonction de leur position nucléotidique. Les positions les plus fréquemment
mutées sont différentes selon les cancers et reflètent donc la "signature" du ou des agents
mutagènes qui en sont à l'origine. La technologie LMPCR permet de cartographier au
niveau nucléotidique les sites les plus fréquemment endommagés par un agent génotoxique
donné. Ainsi, on a la possibilité de chercher une corrélation entre ces sites fréquemment
endommagés et les sites fréquemment mutés dans un type de cancer. Cela a été notamment
appliqué pour confirmer au niveau moléculaire le lien étiologique reliant une exposition
aux UVB et le cancer de la peau ainsi que celui qui s'établit entre plusieurs substances de la
fumée de tabac et le cancer du poumon.

63
2.2 Article
Comprendre la mutagenèse somatique via la
cartographie des dommages à l’ADN Titre en anglais : Mapping DNA damage to understand somatic mutagenesis.
Sandrine Lacoste1, Patrick J. Rochette1* & Régen Drouin1
1 Service de Génétique, Département de Pédiatrie, Faculté de Médecine et des Sciences de la Santé, Université de Sherbrooke, 3001, 12ème Avenue Nord, Sherbrooke, Qc, Canada J1H 5N4
b Adresse actuelle: Department of Therapeutic Radiology, Yale School of Medicine, 333 Cedar St. / HRT 210, New Haven, CT, USA 06520-8040
___________________________ Adresse pour la correspondance et tirés à part : Dr Régen Drouin
Service de génétique Département de pédiatrie, local 1428 Faculté de Médecine et des Sciences de la Santé 3001, 12e Avenue Nord Sherbrooke (Québec) J1H 5N4 Canada Tél : (819) 820 6827 Fax : (819) 564 5217 Courriel : [email protected]

64
2.2.1 Résumé (anglais) Somatic mutation theory explains how DNA damage can lead to the malignant
transformation of cells. It therefore elucidates the connection between genotoxic agents and
cancers. Mutational spectra, which tend to be characteristic of a cancer type, are available
for certain genes like TP53 which is frequently mutated in tumors. A mutational spectrum
could therefore be the signature of the genotoxic agent(s) at the origin of the malignant
transformation. Ligation-mediated PCR (LMPCR) is a genomic sequencing method that
can be used for the mapping of DNA damage at nucleotide resolution. Such a mapping can
then be compared to a mutational spectrum to test the hypothesis that implies one agent can
cause mutations into one cancer type. LMPCR has been used this way to map DNA
damage generated by different UV wavelengths. The frequently damaged sites following
UVB irradiation correlate with the mutational spectrum of TP53 in skin cancer. Similarly,
BPDE, the activated form of the benzo[a]pyrene present in tobacco smoke, generates
frequent adducts at sites corresponding to mutation hotspots of TP53 in lung cancers. Still,
the correlation between BPDE damage sites and TP53 mutations is not perfect and this
suggests a role of other genotoxic substances that are also present in tobacco smoke, such
as the nitrosamine NNK. LMPCR has also been used to investigate the role of Aflatoxin B1
(AFB1) in hepatocellular carcinoma. AFB1 can actually damage DNA at codon 249 of
TP53, which is frequently mutated in hepatocellular carcinoma, but also at many other
sites. This led to the assumption that another mechanism unrelated to AFB1, such as the
synergetic effect the hepatitis B virus infection, could favorably select this peculiar
mutation. Finally, and beyond this objective of better understanding somatic mutagenesis,
LMPCR is commonly used whenever DNA damage frequency and/or repair is to be
investigated.

65
2.2.2 Introduction L'ADN contenu dans chacune de nos cellules est constamment endommagé par des stress
endogènes, comme les espèces oxygénées réactives produites par le métabolisme cellulaire,
et par des stress exogènes, tels que les ultraviolets (UV) [1]. Selon les cas, ces
modifications peuvent perturber la double hélice d'ADN, empêcher sa transcription aussi
bien que sa réplication et bien sûr, mener à des mutations, c'est-à-dire à des modifications
de l'information génétique codée par la molécule. Pour favoriser la survie de la cellule et
maintenir la qualité de l'information codée, plusieurs systèmes de réparation existent qui
sont plus ou moins spécialisés selon le type de dommages ou d'altérations.
Certains changements dans l'information génétique des cellules somatiques (mutations
ponctuelles et/ou réarrangements chromosomiques) sont une cause primaire de
transformation maligne de ces cellules. L'accumulation de ces altérations au niveau des
gènes importants dans le maintien de l’intégrité cellulaire, tel le contrôle du cycle cellulaire,
la réparation et l'apoptose, peut amener la multiplication anarchique de cellules anormales
et la génération d'une tumeur.
Afin d'expliquer la relation existant entre les dommages à l'ADN, les mutations et la
formation des tumeurs, la théorie de la mutagenèse somatique a été énoncée par Thilly en
1983 [2] puis améliorée par Holmquist et Gao en 1997 [3]. Elle tente de prédire la
probabilité pour qu'un dommage généré par un agent mutagène puisse mener à la
transformation maligne d'une cellule. Cette probabilité dépend de plusieurs facteurs
intervenant successivement : (1) la fréquence des dommages à l'ADN, (2) la vitesse de leur
réparation, (3) le risque d'une erreur lors de la réplication de l'ADN endommagé et enfin (4)
la sélection des mutations « avantageuses » (Figure 1).
Le fait de mieux connaître où sont générés les dommages (à quelles positions
nucléotidiques) et la vitesse à laquelle ils sont réparés est donc fondamental pour
comprendre ce qui relie les dommages à l'ADN et la transformation maligne des cellules.
La technologie LMPCR (ligation-mediated PCR ou réaction de polymérisation en chaîne
impliquant la liaison d’un adaptateur) permet d'investiguer ces deux paramètres. En effet,
cette technique permet de déterminer la fréquence des dommages générés par un agent

66
génotoxique donné à chaque position nucléotidique d'une séquence d'ADN. Cependant,
l’étude des dommages par la technologie LMPCR est restreinte aux cassures
monocaténaires de l'ADN ou aux lésions pouvant être converties en de telles cassures par
une réaction chimique ou enzymatique. Le principe de la technique est illustré en Figure 2.
Depuis quelques années, des bases de données existent qui recensent les mutations
retrouvées dans les tumeurs humaines (http://p53.free.fr ; http://www-p53.iarc.fr). Il s'agit
essentiellement de mutations dans le gène TP53 puisqu'il a le double avantage d'être très
fréquemment muté (dans plus de 50 % des tumeurs humaines) et de subir essentiellement
des mutations ponctuelles (m/s 2000, nº12, p.1387). Le gène KRAS est également
investigué, même si le nombre de mutations concernées est beaucoup plus restreint et que
cela apporte donc moins d'informations. Ces bases de données permettent de reconstituer
des spectres de mutations indiquant les sites mutés dans un type de tumeur donné en
fonction de la position nucléotidique. L'étude des spectres de mutations de TP53 dans
différents types de tumeurs a permis de faire apparaître des "points chauds" de mutation
(hotspots) spécifiques à certains types de cancers, amenant l'idée que ces spectres
mutationnels sont en quelque sorte la signature du ou des agents mutagènes à l'origine de la
transformation maligne [4]. La technologie LMPCR s'est avérée l'un des outils essentiels
pour vérifier si ces points chauds de mutations correspondaient effectivement à des
positions nucléotidiques fréquemment endommagées et/ou réparées lentement. Ainsi, des
liens étiologiques entre certains agents génotoxiques et certains types de cancer ont pu être
établis au niveau moléculaire.

67
2.2.3 Le cancer de la peau et les différentes longueurs d'ondes d'UV Le cancer de la peau a depuis longtemps été associé à l’exposition aux UV du soleil et plus
particulièrement aux UVB (280 - 320 nm) qui, contrairement aux UVC (100 – 280 nm), ne
sont pas arrêtés par la couche d’ozone. Les mutations de TP53 les plus fréquentes dans ce
type de cancer sont des transitions C → T (une cytosine est remplacée par une thymine) y
compris quelques transitions en tandem CC → TT. Cela est cohérent avec l'hypothèse que
ces mutations sont la conséquence de la formation de dimères de pyrimidine (DCP),
correspondant à des liaisons covalentes entre deux pyrimidines adjacentes, suite à une
irradiation UV. Huit points chauds de mutation ont été mis en évidence dans les cancers de
la peau, situés aux codons 151/152, 177, 196, 245, 248, 278, 286 et 294 de TP53.
Une première étude réalisée avec des cellules irradiées aux UVC soit à une longueur d'onde
proche de celle qui est la mieux absorbée par l'ADN (260 nm), a montré que certains sites
fréquemment endommagés par les UVC correspondent à des codons de TP53 fréquemment
mutés dans les tumeurs non-mélanocytiques de la peau: particulièrement le codon 286 mais
aussi les codons 151, 278 et 294 [5]. Une expérience comparable réalisée dans notre
laboratoire mais cette fois avec des UVB [6], a révélé que les 8 points chauds de mutation
sont alors fréquemment endommagés, et non plus simplement 4 d'entre eux. Ainsi, le
spectre mutationnel des cancers de la peau correspond mieux aux dommages générés par
une exposition aux UVB qu'à ceux dus aux UVC. D'autre part, il semblerait que les
couches supérieures de l'épiderme soient suffisantes pour protéger de l'effet génotoxique
des UVC mais pas de celui des UVB [7]. Ainsi, bien que les UVC, de par leur longueur
d’onde, soient plus efficaces pour générer des dommages à l’ADN, ils ne sont pas
nécessairement le meilleur modèle expérimental pour mimer une exposition solaire de la
peau.
Les UVA, quant à eux, représentent 5,1 % de l’énergie solaire terrestre (contre 0,3 % pour
les UVB). Cependant, ils sont 105 fois moins énergétiques que les UVB et l'on a longtemps
considéré que leur nocivité était négligeable par rapport autres longueurs d'onde d'UV.
Jusqu'à récemment, les UVA étaient reconnus uniquement pour leur capacité à oxyder
indirectement l’ADN via la production d’espèces réactives oxygénées. Or le spectre des

68
mutations obtenues suite à une irradiation aux UVA montre une prédominance de
transversions T → G [8], ce qui s'explique difficilement par les seuls dommages oxydatifs,
qui ne génèrent habituellement pas ce genre de mutation. Depuis longtemps, la formation
de DCP par les UVA avait été soupçonnée puis détectée tout en restant un sujet de
controverse [9]. La cartographie des DCP générés suite à une irradiation aux UVA a été
réalisée et a confirmé leur formation préférentielle aux dipyrimidines TT (Figure 3). La
formation de ces TT est cohérente avec le spectre mutationnel des UVA qui avait été
investigué chez les rongeurs [8, 10]. Ainsi, s'il y a encore à faire pour bien comprendre le
lien entre les DCP générés sur les TT et les transversions T → G, la LMPCR a permis
d'élucider l'origine d'une fraction des mutations induites par les UVA.
2.2.4 Rôle de la méthylation des cytosines sur la mutagenèse par les UV La détermination de la fréquence des dommages au niveau nucléotidique a aussi permis de
mettre en évidence l'influence de la méthylation des cytosines (Cm) sur la mutagenèse par
les UV. Plusieurs phénomènes concourent à une surreprésentation des Cm parmi les sites
fréquemment mutés. Le premier est que la formation des DCP est plus fréquente aux sites
5'-CCG et 5'-TCG où les cytosines sont méthylées [11, 12]. Ceci serait lié au fait que la
longueur d’onde maximale d’absorption des cytosines méthylées (273 nm) est différente de
celle des cytosines non methylées (267 nm), ce qui les rend plus susceptibles d'être
impliquées dans la formation d'un DCP aux longueurs d'onde présentes dans la lumière
solaire. D'autre part, la probabilité de désamination spontanée d'une cytosine méthylée, ce
qui génère une thymine (T), serait plus grande lorsqu'elle se retrouve impliquée dans un
DCP [13, 14]. La principale conséquence est un risque élevé qu'une ADN polymérase ait à
répliquer une molécule d'ADN avec un DCP du type T^T, T^U ou U^U (qui résulterait
respectivement de la désamination de DCP Cm^Cm, Cm^C ou C^C). Celle-ci tend alors à
incorporer des adénines en face des DCP, générant ainsi les transitions C → T ou
CC → TT caractéristiques des UV [14-16].
2.2.5 Etude du taux de réparation au niveau nucléotidique La correspondance entre sites fréquemment endommagés et sites mutés n'est jamais
parfaite. Certains sites, comme par exemple le codon 290 de TP53, sont fréquemment

69
endommagés par les UV, mais ne constituent pas pour autant un point chaud de mutation
dans les cancers de la peau. Au contraire, certains sites comme les codons 248 et 278 de
TP53 sont fréquemment mutés alors qu'ils sont peu susceptibles de générer des DCP. Ceci
reflète évidemment l'influence des autres étapes intervenant dans la mutagenèse somatique
(Figure 1). La vitesse de réparation à chaque site a l'avantage de pouvoir être investiguée
avec la LMPCR et des variations de 1 à 15 dans la vitesse de réparation (quantifiée en
"temps nécessaire pour 50 % de réparation") ont effectivement été mesurées entre les sites
réparés les plus rapidement et les plus lentement [17]. Le contexte de séquence aurait donc
une influence énorme sur la vitesse de réparation d'un dommage. On a ainsi déterminé que
certains sites fréquemment mutés correspondaient à des sites réparés lentement, notamment
les codons 177, 196, 245, 248 et 278 de TP53 [18]. Cependant, certains contestent que la
vitesse de réparation puisse être une étape déterminante pour expliquer le spectre
mutationnel des tumeurs de la peau [19].
2.2.6 Identification de carcinogènes impliqués dans le cancer du poumon Le cancer du poumon continue d'être la principale cause de mortalité par cancer [20]. Il a
été associé depuis très longtemps à l'exposition au tabac et en particulier à la fumée de
cigarette. Celle-ci comprend pas moins de 20 carcinogènes différents capables d'induire des
tumeurs pulmonaires chez des rongeurs [21]. Ces substances carcinogènes sont réparties
dans plusieurs classes de composés dont les principales sont les hydrocarbures aromatiques
polynucléés (HAP), contenus dans les "goudrons" de la cigarette, et les nitrosamines qui
sont des dérivés de la nicotine. La technologie LMPCR a été utilisée pour cartographier les
dommages générés par plusieurs carcinogènes présents dans la fumée de tabac.
Le gène TP53 est muté dans 70 % des cancers du poumon chez l'homme (http//p53.free.fr).
Dans 40 % des cas, il s'agit de transversions G → T et dans 24 % de transitions G → A [4].
Les codons les plus fréquemment mutés dans les cancers du poumon associés à la cigarette
ont été identifiés aux codons 157, 158, 175, 245, 248, 249 et 273 de TP53 (Figure 4).
Certains sont aussi fréquemment mutés dans les cancers du poumon chez les non-fumeurs
(codons 158, 175, 249 et 273) tandis que d'autres sont plus spécifiques des cancers associés
à la consommation de cigarettes: les codons 157 et 245. Le point chaud de mutation au

70
codon 157 est particulièrement intéressant vu qu'il est spécifique au cancer du poumon. En
effet, il n'existe pas dans les autres types de tumeurs. Le gène KRAS est un gène très
important dans les cancers du poumon. C'est l'un des premiers gènes altérés et il est activé
par des mutations ponctuelles dans 30 % des adénocarcinomes pulmonaires et des
carcinomes à larges cellules [22]. Les mutations de KRAS semblent particulièrement
spécifiques des adénocarcinomes pulmonaires et la plupart se retrouvent dans le codon 12.
Elles sont composées à 80 % de transversions G → T et à 20 % de transitions G → A.
Une étude par LMPCR en 1996 [23] a montré une formation préférentielle dans les cellules
traitées au BPDE (la forme activée du benzo[a]pyrène, un des HAP présent dans la fumée
de cigarette) de dommages sur des guanines au niveau des points chauds de mutation de
TP53, en particulier les codons 157, 248 et 273. Les codons 248 et 273 sont fréquemment
mutés dans tous les types de cancer, mais le codon 157 est, lui, très spécifique du cancer du
poumon chez les fumeurs. Cette étude correspond à la première démonstration moléculaire
d'un lien étiologique entre un carcinogène donné et le cancer pulmonaire. D'autres HAP de
la fumée de cigarette ont montré des profils comparables de dommages, bien qu'avec des
différences concernant leur fréquence relative à une position donnée. En effet, plusieurs
autres HAP ont montré une formation préférentielle de dommages à des sites correspondant
aux points chauds de mutation des codons 157, 158, 245, 248 et 273 de TP53 [24]. Ainsi,
les HAP en général semblent contribuer significativement au spectre mutationnel du cancer
du poumon chez l'homme. Comme pour les DCP, il a été montré une possible influence de
la vitesse de réparation des dommages du BPDE dans la probabilité de formation des
mutations. En particulier, la réparation aux points chauds de mutations des codons 157, 248
et 273 s'est avérée 2 à 4 fois plus lente qu'au niveau d'autres sites endommagés [25].
La NNK, une nitrosamine du tabac qui est un carcinogène pulmonaire prouvé chez l'animal,
a été également étudiée dans notre laboratoire. Des dérivés réactifs de la NNK créent
également beaucoup de dommages à des codons fréquemment mutés dans le cancer du
poumon, notamment le codon 12 de KRAS ainsi que les codons 241, 245 et plus
modérément le codon 248 de TP53 [26]. La NNK pourrait donc également participer aux
mutations retrouvées dans les cancers pulmonaires.

71
2.2.7 Lien étiologique entre l'aflatoxine et le carcinome hépatocellulaire D'autres agents génotoxiques ont aussi été investigués par LMPCR pour vérifier leur
éventuelle implication en relation avec un type de cancer. C'est le cas l'aflatoxine B1
(AFB1) qu'on suppose être impliquée dans la formation du carcinome hépatocellulaire
(CHC). Cette mycotoxine est considérée comme la cause majeure de CHC dans les régions
où la nourriture est fortement contaminée par le champignon qui la produit. Un point chaud
de mutation unique, une transversion G → T, a été observé dans ces tumeurs au codon 249
de TP53. Denissenko et al. [27] ont constaté par LMPCR que si l'AFB1 crée effectivement
des dommages sur de nombreuses guanines de TP53, ça n'est pas particulièrement le cas au
niveau du codon 249, même si des dommages sont observés à cette position nucléotidique.
Ainsi, l'exposition à l'AFB1 ne suffirait pas à expliquer à elle seule le fait que ce codon de
TP53 est un point chaud de mutation dans le CHC. Le rôle synergique d'une infection
parallèle avec le virus de l'hépatite B a été suggéré qui participerait à sélectionner les
mutants au codon 249.

72
2.2.8 Conclusion La technologie LMPCR apporte donc des informations importantes qui permettent de faire
le lien entre les dommages à l'ADN causés par certaines substances génotoxiques et des
mutations caractéristiques trouvées dans certaines tumeurs. Cependant, la mutagenèse
somatique n'a pas été le seul domaine de connaissance à bénéficier de la possibilité de
déterminer des fréquences de dommages au niveau nucléotidique. Notamment, de
nombreuses investigations ont été réalisées pour étudier l'influence de différents paramètres
sur la vitesse de réparation. Notamment, le fait qu'un brin soit transcrit ou pas (réparation
couplée à la transcription) [4], l'éloignement d'une région par rapport au promoteur [28], ou
encore l'implication de différentes protéines sur tel ou tel aspect de la réparation [29, 30].

73
2.2.9 Remerciements Les travaux présentés dans cette synthèse ont été financés par le l’Institut national du
cancer du Canada et les Instituts de recherche en santé du Canada. R. Drouin détient une
chaire de recherche du Canada en « Génétique, mutagenèse et cancer ».

74
2.2.10 Références 1 Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature
2001; 411: 366-74.
2 Thilly WG. Analysis of chemically induced mutation in single cell populations.
Basic Life Sci 1983; 23: 337-78.
3 Holmquist GP, Gao S. Somatic mutation theory, DNA repair rates, and the
molecular epidemiology of p53 mutations. Mutat Res 1997; 386: 69-101.
4 Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor
suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 1994;
54: 4855-78.
5 Tornaletti S, Rozek D, Pfeifer GP. The distribution of UV photoproducts along the
human p53 gene and its relation to mutations in skin cancer. Oncogene 1993; 8: 2051-7.
6 Drouin R, Therrien JP. UVB-induced cyclobutane pyrimidine dimer frequency
correlates with skin cancer mutational hotspots in p53. Photochem Photobiol 1997; 66:
719-26.
7 Therrien JP, Rouabhia M, Drobetsky EA, Drouin R. The multilayered organization
of engineered human skin does not influence the formation of sunlight-induced cyclobutane
pyrimidine dimers in cellular DNA. Cancer Res 1999; 59: 285-9.
8 Drobetsky EA, Turcotte J, Chateauneuf A. A role for ultraviolet A in solar
mutagenesis. Proc Natl Acad Sci USA 1995; 92: 2350-4.
9 Sage E, Perdiz D, Grof P, et al. DNA damage induced by UVA radiation: role in
solar mutagenesis. In: E. Sage, R. Drouin and M. Rouabhia (Eds.), From DNA photolesions
to mutations, skin cancer and cell death. The Royal Society of Chemistry, 2005, pp. 33-47

75
10 Rochette PJ, Therrien JP, Drouin R, et al. UVA-induced cyclobutane pyrimidine
dimers form predominantly at thymine-thymine dipyrimidines and correlate with the
mutation spectrum in rodent cells. Nucleic Acids Res 2003; 31: 2786-94.
11 Tommasi S, Denissenko MF, Pfeifer GP. Sunlight induces pyrimidine dimers
preferentially at 5-methylcytosine bases. Cancer Res 1997; 57: 4727-30.
12 You YH, Pfeifer GP. Similarities in sunlight-induced mutational spectra of CpG-
methylated transgenes and the p53 gene in skin cancer point to an important role of 5-
methylcytosine residues in solar UV mutagenesis. J Mol Biol 2001; 305: 389-99.
13 Lemaire DG, Ruzsicska BP. Kinetic analysis of the deamination reactions of
cyclobutane dimers of thymidylyl-3',5'-2'-deoxycytidine and 2'-deoxycytidylyl-3',5'-
thymidine. Biochemistry 1993; 32: 2525-33.
14 Tu Y, Dammann R, Pfeifer GP. Sequence and time-dependent deamination of
cytosine bases in UVB-induced cyclobutane pyrimidine dimers in vivo. J Mol Biol 1998;
284: 297-311.
15 Taylor JS. New structural and mechanistic insight into the A-rule and the
instructional and non-instructional behavior of DNA photoproducts and other lesions.
Mutat Res 2002; 510: 55-70.
16 Lee DH, Pfeifer GP. Deamination of 5-methylcytosines within cyclobutane
pyrimidine dimers is an important component of UVB mutagenesis. J Biol Chem 2003;
278: 10314-21.
17 Gao S, Drouin R, Holmquist GP. DNA repair rates mapped along the human PGK1
gene at nucleotide resolution. Science 1994; 263: 1438-40.
18 Tornaletti S, Pfeifer GP. Slow repair of pyrimidine dimers at p53 mutation hotspots
in skin cancer. Science 1994; 263: 1436-8.

76
19 Rodin SN, Rodin AS, Juhasz A, Holmquist GP. Cancerous hyper-mutagenesis in
p53 genes is possibly associated with transcriptional bypass of DNA lesions. Mutat Res
2002; 510: 153-68.
20 Remontet L. Estimations nationales : tendance de l'incidence et de la mortalité par
cancer en France entre 1978 et 2000. BEH 2003; 41-42: 190-93.
21 Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst 1999;
91: 1194-210.
22 Devereux TR, Taylor JA, Barrett JC. Molecular mechanisms of lung cancer.
Interaction of environmental and genetic factors. Giles F. Filley Lecture. Chest 1996; 109:
14S-19S.
23 Denissenko MF, Pao A, Tang M, Pfeifer GP. Preferential formation of
benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science 1996; 274: 430-
2.
24 Smith LE, Denissenko MF, Bennett WP, et al. Targeting of lung cancer mutational
hotspots by polycyclic aromatic hydrocarbons. J Natl Cancer Inst 2000; 92: 803-11.
25 Denissenko MF, Pao A, Pfeifer GP, Tang M. Slow repair of bulky DNA adducts
along the nontranscribed strand of the human p53 gene may explain the strand bias of
transversion mutations in cancers. Oncogene 1998; 16: 1241-7.
26 Cloutier JF, Drouin R, Weinfeld M, O'Connor TR, Castonguay A. Characterization
and mapping of DNA damage induced by reactive metabolites of 4-(methylnitrosamino)-1-
(3-pyridyl)-1-butanone (NNK) at nucleotide resolution in human genomic DNA. J Mol Biol
2001; 313: 539-57.
27 Denissenko MF, Koudriakova TB, Smith L, O'Connor TR, Riggs AD, Pfeifer GP.
The p53 codon 249 mutational hotspot in hepatocellular carcinoma is not related to
selective formation or persistence of aflatoxin B1 adducts. Oncogene 1998; 17: 3007-14.

77
28 Tu Y, Tornaletti S, Pfeifer GP. DNA repair domains within a human gene: selective
repair of sequences near the transcription initiation site. EMBO J 1996; 15: 675-83.
29 Tu Y, Bates S, Pfeifer GP. Sequence-specific and domain-specific DNA repair in
xeroderma pigmentosum and Cockayne syndrome cells. J Biol Chem 1997; 272: 20747-55.
30 Therrien JP, Drouin R, Baril C, Drobetsky EA. Human cells compromised for p53
function exhibit defective global and transcription-coupled nucleotide excision repair,
whereas cells compromised for pRb function are defective only in global repair. Proc Natl
Acad Sci USA 1999; 96: 15038-43.

78
2.2.11 Légendes des figures Figure 1 Représentation schématique de la théorie de la mutagenèse somatique telle
qu’élaborée par Thilly en 1983 et améliorée par Holmquist et Gao en 1997. Cette théorie
tente de prédire la probabilité pour qu'un dommage généré par un agent mutagène (ici les
dimères cyclobutyliques de pyrimidines générés par les UV) puisse participer à la
transformation maligne d'une cellule. La probabilité qu'une mutation donnée soit associée à
une transformation maligne donnée suppose (1) que des dommages en quantité suffisante
surviennent à cette endroit, (2) qu'ils ne soient pas réparés ou réparés suffisamment
lentement pour que (3) la polymérase de réplication de l'ADN ait le temps de venir
répliquer cet ADN endommagé, qu’elle interprète mal cette base endommagée et ainsi
qu’elle "fixe" la mutation (en incorporant un mauvais nucléotide en face de la base
endommagée) et finalement que (4) cette mutation influe sur le phénotype cellulaire dans
une fonction impliquée dans la transformation maligne.
Figure 2 Représentation schématique de la technique LMPCR. On présente ici
l'exemple de dommages à l'ADN qui doivent être convertis en cassures pour être étudiés
par cette technique. Cependant, certains agents endommageant induisent des cassures
monocaténaires directement, ce qui élimine l’étape de conversion. Les étapes de la
technique sont les suivantes. Une extension est réalisée à partir d'une amorce spécifique du
gène d'intérêt et s'arrête au lieu de la première cassure rencontrée qui correspond au lieu du
dommage initial. L'amorce spécifique amène un premier niveau de spécificité à la
technique. Cette première extension d’amorce génère un bout franc sur lequel on vient lier
un adaptateur. Il en résulte des fragments d’ADN dont les deux extrémités sont connues
qu'on peut alors amplifier par PCR avec une amorce correspondant à l’adaptateur et une
seconde amorce spécifique du gène d’intérêt. Cette seconde amorce est choisie légèrement
en aval de la première afin d’amener un deuxième niveau de spécificité à la technique. Les
produits PCR ainsi obtenus ont une taille et une quantité qui dépendent respectivement de
la position nucléotidique des dommages de départ et de leur fréquence initiale à chaque
position donnée. Une migration dans un gel d'acrylamide suivie d'un électro-transfert sur
une membrane de nylon puis d'une hybridation avec une sonde spécifique radioactive
permettent de visualiser l'ensemble des produits PCR.

79
Figure 3 Exemple d’application de la technique LMPCR extrait de [10] . Ces
autoradiogrammes montrent la cartographie des DPC obtenus sur le gène aprt (adénine
phosphoribosyltransférase) suite à une irradiation aux UVA, UVB, UVC ou à la lumière
solaire simulée (SSL). Les quatre premières colonnes de chaque autoradiogramme
correspondent à la LMPCR faite sur de l’ADN spécifiquement cassé au niveau des G, des
A, des T et C ou sur des C seuls. Elles permettent de suivre la séquence sur le gène et donc
de situer le lieu des dommages. Chacune des flèches à droite représente un site
dipyrimidinique, c’est-à-dire un site potentiel de DCP. Les bandes sur l’autoradiogramme
correspondent à des sites effectivement endommagés. Leur intensité dépend du nombre de
molécules d'ADN au départ qui étaient cassées à cette position nucléotidique. La colonne
identifiée « NoUV » correspond à la LMPCR faite sur de l’ADN non irradié. Elle sert de
contrôle négatif et constitue un indicateur du bruit de fond généré par la technique. On peut
remarquer que les UVA induisent presque exclusivement leur DCP sur des sites TT alors
que les autres longueurs d’ondes sont moins exclusives et endommagent tous les types de
dipyrimidines. Le tableau recense les dommages quantifiés à partir de ces deux
autoradiogrammes et des autoradiogrammes correspondants au brin opposé (non montrés).
La localisation de ces dommages aux TT corrèle très bien avec le spectre de mutations aux
UVA précédemment publié [8].
Figure 4 Spectre des codons mutés de TP53 dans les tumeurs pulmonaires. Les
graphiques représentent le pourcentage de mutations ponctuelles recensées dans TP53 à un
codon donné dans les tumeurs pulmonaires chez des (A) non fumeurs ou (B) des fumeurs.
Ainsi, on peut déterminer les mutations de TP53 qui sont spécifiquement associées à
l'exposition à la fumée de tabac (http://www-p53.iarc.fr).

80
Figure 1

81
Figure 2

82
Figure 3
UVA UVB SSL UVC T-T 57% 27% 29% 26% C-C 14% 30% 26% 35% C-T 11% 17% 17% 15% T-C 18% 26% 28% 24%

83
Figure 4

Chapitre 3 Formamidopyrimidine adducts are detected using the comet assay in human cells treated with
reactive metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)
(Mutation Research. 2006 Aug 30;600(1-2):138-149)

85
3.1 Résumé en français La 4-(méthylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) est un carcinogène pulmonaire
puissant qui est spécifique de la fumée de cigarette. Sa bioactivation dans les cellules mène
à une petite quantité de méthylation et de pyridyloxobutylation de l'ADN. Considérant sa
grande sensibilité, le test des comètes semble une technique de choix pour investiguer les
dommages associés à la NNK. Plusieurs stratégies ont été utilisées pour augmenter la
spécificité de cette technique: (1) utiliser des analogues qui produisent une variété limité de
dommages, en ne générant que la méthylation ou que la pyridyloxobutylation de l'ADN ;
(2) utiliser des cellules ayant différentes capacités de bioactivation ; (3) utiliser la
conversion alcaline et/ou des enzymes spécifiques pour cliver au niveau de certaines classes
particulières de dommages ; (4) utiliser différentes conditions de lyse pour convertir une
classe spécifique de lésions à l'ADN en lésions reconnues par une enzyme. Nous avons
déterminé que plusieurs dommages associés à la NNK peuvent être détectés avec une
certaine spécificité avec le test des comètes. En ce qui concerne la méthylation de l'ADN, il
s'agit de sites AP et d'adduits formamidopyrimidine (fapy) beaucoup plus fréquents. Ces
fapy correspondent à des N7-mG formées dans les cellules et dont le cycle s'est ouvert
pendant le test sous l'effet de la lyse à pH 10. La pyridyloxobutylation de l'ADN mène à la
formation d'alkylphosphotriesters et d'une quantité à peu près égale d'adduits fapy. Par
analogie avec les dommages de la méthylation, nous pensons que ces adduits correspondent
à la forme fapy des N7-pyridyloxobutylguanines (N7-pobG). Ces N7-pobG sont instables
et ceci constitue la première démonstration indirecte qu'ils sont formés dans des cellules
humaines. Mais contrairement aux N7-m-fapy, la longueur ou le pH de la lyse n'ont aucune
influence sur la fréquence de N7-pob-fapy détectés. Ainsi, ces adduits seraient
effectivement formés dans les cellules et ne résulteraient pas des conditions expérimentales.
Ces N7-pob-fapy ont un fort potentiel mutagènique et nous pensons que le test des comètes,
malgré ses limitations, est une bonne technique pour les étudier, considérant l'instabilité des
lésions primaires qui en sont à l'origine.

86
3.2 Article
Formamidopyrimidine adducts are detected using the
comet assay in human cells treated with reactive
metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-
butanone (NNK)
Sandrine Lacostea, André Castonguayb, Régen Drouina,*
a Service of Genetics, Department of Paediatrics, Faculty of Medicine and Health Sciences, University of Sherbrooke, 3001, 12th Avenue North, Sherbrooke, Que., Canada J1H 5N4
b Faculty of Pharmacy, Laval University, Quebec City, Que., Canada G1K 7P4
Keywords: 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK); Formamidopyrimidine adducts; Formamidopyrimidine glycosylase; Comet assay; Tobacco; DNA damage
Corresponding author:
* Régen Drouin Service of Genetics Department of Paediatrics Faculty of Medicine and Health Sciences University of Sherbrooke 3001, 12th Avenue North Sherbrooke, Qc, Canada J1H 5N4
Tel: (819) 820-6827 Fax: (819) 564-5217 E-mail: [email protected]

87
3.2.1 Abstract 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is the most lung-specific of the
carcinogens present in tobacco smoke. Its bioactivation in cells leads to a small amount of
methylation or pyridyloxobutylation DNA damage. Considering its great sensitivity, the
comet assay seems a technique of choice to investigate NNK-related damage. Several
strategies were used to impart some specificity to the assay: (1) using analogs that produce
a limited variety of DNA lesions, as they mimic either the methylation or the
pyridyloxobutylation pathway; (2) using cells with different bioactivation abilities; (3)
using alkali conversion and/or enzymes specific for cleaving particular classes of damage;
(4) using different lysis conditions to convert a specific class of DNA lesions into enzyme-
sensitive lesions. We determined that several NNK-associated lesions can be detected with
some specificity with the comet assay. For the methylation pathway, they are AP sites and
the more frequent formamidopyrimidine (fapy) adducts. These fapy adducts correspond to
N7-methylguanines generated in the cells that were ring-opened during the assay by the
lysis solution at pH 10. For the pyridyloxobutylation pathway, alkylphosphotriesters and a
roughly equal frequency of fapy sites were detected. By analogy to the methylation
damage, these fapy adducts are thought to be the ring-opened form of N7-
pyridyloxobutylguanines (N7-pobG). N7-pobG are unstable and this constitutes the first
indirect demonstration of their formation in cells. But contrary to N7-m-fapy, the lysis time
or pH did not influence the frequency of N7-pob-fapy adducts detected, suggesting that
they already exist in the cells and are not related to the experimental conditions. These N7-
pob-fapy have a strong mutagenic potential and we think that the comet assay, in spite of its
limitations, is a good way to study them considering their low frequency and the inherent
instability of the adduct from which they originate.

88
3.2.2 Introduction N-Nitrosamines are thought to play a major role in carcinogenesis related to tobacco
consumption. Among the tobacco specific nitrosamines, 4-(methylnitrosamino)-1-(3-
pyridyl)-1-butanone (NNK) is the most abundant and the strongest carcinogen [1,2]. It
exhibits lung-selective toxicity and induces primarily adenocarcinoma of the lung on
laboratory animals [3]. This led to the assumption that NNK accounts for recent increases
in the relative incidence of human lung adenocarcinoma in smokers [4,5].
Bioactivation to reactive intermediates is necessary for NNK to be genotoxic and its
activation has been extensively studied [6] in freshly isolated human lung cells [7] and in
peripheral human lung microsomes [8]. The two α carbons of the nitrosamino group of
NNK can be hydroxylated, yielding unstable intermediates that can lead to DNA alkylation
(Fig. 1). The α-methylene hydroxylation of NNK leads to the formation of
methyldiazohydroxides, which can methylate DNA and the α-methyl hydroxylation of
NNK results in the formation of oxobutyldiazohydroxides, which can alkylate DNA
through a reaction of pyridyloxobutylation. Both types of α-carbon hydroxylation clearly
exist in human lung cells [3,7] and both pathways seem to participate in NNK-related lung
carcinogenesis in humans [3,9]. But the extent to which they do, remains to be determined.
These pathways can be investigated separately through the use of analogs that generate
either one type of reactive species or the other. For instance, N-
nitroso(acetoxymethyl)methylamine (NDMAOAc) and 4-(acetoxymethylnitrosamino)-1-
(3-pyridyl)-1-butanone (NNKOAc) mimic respectively the methylation or the
pyridyloxobutylation pathway (Fig. 1). NNK bioactivation, that is the biotransformation in
alkylating reactive species, is not as frequent as other possible biotransformations
corresponding for example to detoxification pathways, but it is thought to be responsible
for the carcinogenetic effect of NNK. In fact, the relative occurrence of bioactivation in
reactive species seems significantly higher in alveolar type II cells and macrophages than in
other pulmonary cell types, making them potential target for NNK toxicity [7].
DNA adducts generated by NNK through the methylation or the pyridyloxobutylation
pathway (Table 1) have been extensively studied to determinate which of them could be

89
relevant to carcinogenesis [3]. Laboratory animals treated with NNK show the following
adducts in their lung: N7-methylguanine (N7-mG), O6-methylguanine (O6-mG) and to a
lesser extend of O4-methylthymine (O4-mT), all these are due to the methylation pathway.
Some pyridyloxobutyl adducts have also been detected such as O6-pyridyloxobutylguanine
(O6-pobG) or phosphate adducts (pyridyloxobutylphosphotriesters) [6,10,11]. NNK
generates single-strand breaks [6] but exactly how they are produced remains
undetermined. It has been suggested that they derived from the methylation pathway by
spontaneous or enzymatic depurination of N7-mG or N3-mA [3] but they more likely result
from the pyridyloxobutylation pathway by the generation of
pyridyloxobutylphosphotriesters [10,11]. The O6-alkyl adducts (O6-mG and O6-pobG) are
considered as major players in NNK-related carcinogenicity because of their mutagenetic
potential and the fact that they are repaired both by the same O6-methylguanine
methyltransferase which is easily overwhelmed by low adduct levels [3]. Still,
carcinogenicity of the pyridyloxobutylation pathway has been also associated with the
presence of another type of damage. This adduct type, that accounts for at least 50% of the
DNA binding obtained through this pathway, is the most prevalent adduct from the
pyridyloxobutylation pathway and remained uncharacterized until recently. It was therefore
only qualified according to its ability to release 4-hydroxy-1-(3-pyridyloxobutyl)-1-
butanone (HPB) (Fig. 1) upon acid or neutral thermal hydrolysis. Recently, several HPB-
releasing adducts have been obtained and identified in DNA and/or deoxyguanosine treated
with NNKOAc in vitro: N7-pyridyloxobutylguanine (N7-pobG) [12] and O2-
pyridyloxobutylcytosine (O2-pobC) [13], the former being by far the most abundant [14].
Other adducts have also been characterized that do not release HPB. They derive from the
pyridyloxobutylation of the N2 position of guanine [12] and more frequently of the O2
position of thymine [13]. None of these newly characterized adduct (HPB-releasing or not)
have yet been demonstrated to occur in animals or cells, probably because of their low
frequency of occurrence and/or their instability, which makes them difficult to observe
[12,14].
The comet assay is a commonly used, sensitive method to investigate DNA damage in
individual cells [15]. It has already been used to detect NNK-associated damage in the
metabolically competent lymphoblastoid cell line MCL-5 [16] and in white blood cells

90
where NNK seems to act synergistically with UVA to generate DNA damage [17]. In both
studies, the type of damage detected could not be identified. In fact, this technique
completely lacks specificity in that it can determine only one parameter: the average break
frequency. On the contrary, a technique like ligation-mediated PCR (LMPCR), which is
commonly used in DNA damage studies, gives several information concerning the breaks:
the exact genomic base position along with some chemical information such as the
existence of a 5' phosphate termination. Nevertheless, the comet assay is extremely
sensitive, detecting breaks as infrequent as 1 break/108 bases [18] versus 1 break/5 x 104
bases for LMPCR [19] and can therefore be useful to detect DNA damage that is
infrequent. In fact, specificity must be sacrificed for sensitivity in cases like NNK-
associated damage where damage frequency can be limited by factors such as the ability of
the cell to biotransform the exogenous pre-mutagen into intracellular DNA-damaging
species. Moreover, some specificity can be imparted to the assay by the following
treatments: (1) using analogs that produce a limited variety of DNA lesions such as
NDMAOAc or NNKOAc; (2) using a variety of cells with different activation abilities:
U937, a histiocytic lymphoma cell line that bioactivates NNK [20], NCI-H23, a lung
adenocarcinoma cell line originally obtained from a smoker and normal lymphocytes; (3)
generating the single-strand breaks from DNA using chemical treatment (comet assay at
different pH) and/or enzymes specific for cleaving particular classes of DNA lesions; (4)
using chemical treatments (different lysis conditions) that convert a specific class of DNA
lesions into enzyme-sensitive lesions. We used all four specificity enhancement methods to
distinguish various types of NNK-associated damage that can be specifically studied using
the comet assay.

91
3.2.3 Materials and Methods
3.2.3.1 Caution All work involving NNK and its derivatives should be performed with protective clothing
and in a well-ventilated fume hood.
3.2.3.2 Cell culture U937 (human histiocytic lymphoma CRL-1593.2) and NCI-H23 (human lung
adenocarcinoma CRL-5800) cell lines were obtained from the ATCC. Both were grown in
RPMI 1640 with 2 mM L-glutamine modified to contain 10 mM HEPES, 1 mM sodium
pyruvate, 4.5 g/L glucose, 1.5 g/L bicarbonate and supplemented with 10% FBS (v/v) at
37°C in humidified atmosphere of 5% CO2. U937 cells were maintained at low density
(below 4 x 105 cells/mL). Lymphocytes were isolated from the blood of a healthy young
non-smoker with Histopaque-1077 (Sigma-Aldrich Canada, Oakville, ON) as described by
the manufacturer. They were kept overnight or for a maximum of 48 h (until the comet
assay) in RPMI 1640 with 2 mM L-glutamine modified to contain 10 mM HEPES, 1 mM
sodium pyruvate, 4.5 g/L glucose, 1.5 g/L bicarbonate and supplemented with 10% FBS
(v/v) at 37°C in humidified atmosphere of 5% CO2.
3.2.3.3 Chemicals NDMAOAc (purity 99.8%) was purchased from LKT laboratory (St. Paul, MN) and
NNKOAc (purity > 94%) synthesised as described previously [10]. They were both
dissolved in DMSO to a stock solution of 0.1 M (kept at –80°C) and diluted before use in
DMSO to an optimal concentration so that the percentage of DMSO would be below 10%
in the final treatment solution. Any solution or material that has been in contact with either
NDMAOAc or NNKOAc was treated with 5 M NaOH to allow complete inactivation prior
to disposal. Dimethyl sulfate (DMS) (purity ≥ 99.0%) was purchased as a 10.5 M solution
from Fluka (Neu-Ulm, Switzerland). It was diluted in PBS prior to use.

92
3.2.3.4 Treatment of cells
Cells were trypsinized for 5 min at 37°C (NCI-H23) or directly taken from the culture
medium (U937 and lymphocytes) and 6.25 x 104 cells for each treatment condition
transferred in an 1.5 mL tube. Cells were centrifuged at 3000 x g for 4 min and the pellet
was resuspended in 125 μL of complete culture medium (0.5 x 106 cells/mL). Cells were
then treated with increasing doses of NDMAOAc or NNKOAc for 2 h at 37°C in a cell
incubator or 6 min with 10 μM DMS at room temperature. After the alkylation treatment, 1
mL of cold PBS was added and cells were kept on ice as much as possible for the
subsequent steps. They were centrifuged at 3000 x g for 4 min.
To standardize the measured percent tail DNA, UVB-irradiated U937 cells were used. For
these, an equivalent number of cells in PBS was irradiated with a UV tube
(FS20T12/UVB/BP, Philips) filtered with a screen of cellulose acetate (Kodacel TA-407,
clear 0.015-inch, Eastman Kodak) before centrifugation at 3000 x g for 4 min. An
estimation of the frequency of cyclobutane pyrimidine dimers (CPD) generated by UVB
irradiation is available [21]: 32.7 CPD/(J m-2 10-9 bases).
3.2.3.5 Comet assay The procedure of Singh and al. [22] was followed with some modifications. Prior to the
experiment, precleaned slides (Surgipath, Winnipeg, MB) were precoated with UltraPure
agarose (Invitrogen, Carlsbad, CA) 1% in H2O and dried. The cell pellet was resuspended
in low melting point (LMP) agarose (Bio-Rad Laboratories, Hercules, CA) 1% in PBS kept
at 37°C. The agarose/cells mix (50 μL / slide) was spread on the precoated slides. Before
the LMP agarose solidified, a cover slip 22 mm x 22 mm was added, then the slides were
kept at 4°C for 5 min. They were then placed in a lysis solution (2.5 mM NaCl; 0.1 mM
EDTA; 10 mM Tris pH 10, Triton X-100 1%) at 4°C for 1 h or the indicated time. When
specified, the lysis solution was the same but set at pH 7.5. After lysis, slides were quickly
rinsed with cold PBS and equilibrated with 3 x 5 min washes of the cold enzyme buffer
prior to the digestion per se. After digestion, the slides were quickly rinsed with cold PBS,
then transferred to an electrophoresis tank in a cold chamber containing 1200 mL of an
electrophoresis solution (300 mM NaOH; 1 mM EDTA) either at pH > 13 or adjusted to pH

93
12.2. If not indicated, the pH of the electrophoresis solution was pH > 13. The slides
remained in this solution for 40 min to allow DNA unwinding and an eventual alkali
conversion of some lesions, before a current of 0.3 V/cm was applied for 15 min. The slides
were then neutralized with 3 x 5 min washing steps with Tris-HCl pH 7.5, and then stained
with 50 μL SYBR Gold (Molecular Probes, Eugene, OR). Twenty-five consecutive
nucleoids in the centre of each slide were photographed using an Olympus BX 61
microscope.
3.2.3.6 Enzymatic digestions When indicated, an enzymatic digestion was performed during the comet assay after the
lysis and prior to the transfer in the electrophoresis solution. The enzyme buffer was either
50 mM NaCl; 1 mM EDTA; 100 μg/mL BSA; Tris 10 mM pH 7.6 for T4 endonuclease V
(T4 endo V) digestion or 0.1 M KCl; 0.5 mM EDTA; 0.2 mg/mL BSA; 40 mM HEPES-
KOH pH 8 for endonuclease III (endo III) or formamidopyrimidine glycosylase (fpg)
digestion. After equilibration in the cold enzyme buffer, the excess liquid was dabbed off
with tissue. Slides were then treated with 50 μL of enzyme buffer alone or buffer
containing the enzyme under a cover glass in a moist box at 37°C for 45 min (endo III or
fpg) or 1 h (T4 endo V).
Endo III and fpg were obtained from Dr. Serge Boiteux and T4 endo V from Dr. R.
Stephen Lloyd. They were used here at concentrations established as optimal for detection
of their main substrate: oxidized pyrimidines, 8-oxoguanine and CPD, respectively.
3.2.3.7 Analysis of the comets and statistics TIFF format images were exported to be analysed with the CASP freeware [23] to
determine the percent tail DNA of each nucleoid. For each slide, the median percent tail
DNA of 25 nucleoids was determined and the average and standard deviation of 2/3
independent experiments (2/3 slides) calculated. The correlation (r2) of the average percent
DNA with dose applied to the cells could then be determined. To verify if the difference in
the average percent tail DNA between pH > 13 and pH 12.2 obtained with UVB-irradiated

94
cells was significant, a Student’s t-test was performed to obtain the probability (p) of the
null hypothesis.

95
3.2.4 Results NNK-associated damage has been characterized by others (Table 1). We report here that
the comet assay, when used with different strategies to increase its specificity, can be used
to detect several types of damage quite specifically with the great sensitivity permitted by
this technique.
3.2.4.1 Direct breaks and alkali-labile damage With the classical comet assay, single-strand breaks are revealed thanks to an alkaline
unwinding at pH > 13 followed by an electrophoretic migration. More breaks result in more
DNA in the tail of the nucleus after migration. Therefore this assay reveals typically both
direct breaks and alkali-labile damage. Performing the assay at a lower pH such as 12.2
makes it possible to discriminate these two types of damage as most of the alkali-labile
damage are stable until the pH is above 12.5 [24]. We used this to detect direct breaks
and/or alkali-labile damage generated through the methylation and/or the
pyridyloxobutylation pathway.
U937 cells were treated with increasing doses of NDMAOAc (0–15 μM) (Fig. 2A) or
NNKOAc (0–100 μM) (Fig. 2B) before being submitted to the comet assay. In both cases,
the number of DNA breaks, as measured by the average median percent tail DNA,
increased with dose when the assay was performed at pH > 13. The correlation of the
percent tail DNA with the dose was good for both NDMAOAc (r2 = 0.965) and NNKOAc
(r2 = 0.991) treatments. When the assay was performed at a lower pH (pH 12.2) that does
not convert alkali-labile site in breaks, some breaks were still detected after NNKOAc but
not after NDMAOAc treatment (Fig. 2A and B). This indicates that NNKOAc, unlike
NDMAOAc, generates direct breaks and not just alkali-labile sites. The measured percent
tail DNA seemed lower at pH 12.2 than at pH > 13 (Fig. 2B), suggesting that the breaks
measured at the higher pH could correspond to a mix of direct breaks and alkali-labile sites.
Still, the correlation with the dose of the percent tail DNA for NNKOAc-treated cells at pH
12.2 (r2 = 0.845) was not as good as the one obtained at pH > 13, making it difficult to be
sure that the pH 12.2 was as good as the higher pH to estimate the number of breaks.

96
To determine if the difference observed between the assays at different pH was related to a
difference in the quantification of the breaks or an actual difference in their frequency, the
two parallel pH regimens were tried on U937 irradiated with increasing doses of UVB (0–
8 J/m2). UV irradiation generates cyclobutane pyrimidine dimers (CPD) that are converted
to breaks by the T4 endonuclease V (T4 endo V) digestion. No breaks were observed,
whatever the pH, when the slides were not submitted to the T4 endo V digestion prior to
the alkaline treatment (data not shown). This system permits therefore a direct comparison
of the two variations of the assay when just breaks and no alkali-labile sites are present.
Fig. 2C shows that both pH give a linear dose response but that the correlation with the
dose is actually better at pH > 13 (r2 = 0.990) than at pH 12.2 (r2 = 0.961) and that the
higher pH should therefore be preferred. Still, the difference does not seem sufficient to
explain the lower percent tail DNA observed at pH 12.2 for the higher doses of NNKOAc
(Fig. 2B) and it should be envisioned than both direct breaks and alkali-labile sites are
present at the higher pH. More importantly, these results allowed us to calibrate our assay,
as UVB have been measured to generate an estimation of 32.7 CPD/(J m-2 10-9 bases) [21].
The percent tail DNA measured with our comet assay at pH > 13 can therefore be
associated with a certain dose of UVB and converted into a number of lesions per 109 bases
(Fig. 2C).
3.2.4.2 Lesion specific enzymes on U937, NCI-H23 and lymphocyte cells The use of enzymes specific for cleaving particular classes of DNA lesions can increase
greatly the sensitivity and specificity of the comet assay, as it can reveal DNA damage that
are not or are only weakly alkali-labile. Considering the enzymes available, their specificity
and the DNA damage identified for both activation pathways (Table 1), we determined that
two major classes of damage could be researched for both NNKOAc and NDMAOAc
treatments: AP sites or formamidopyrimidine (fapy) lesions. Both correspond to secondary
lesions of an N7 alkylation, which is common with both activation pathways of NNK (Fig.
1 and Table 1). AP sites can be detected as alkali-labile and/or endonuclease III (endo III)
sensitive sites whereas fapy lesions can be detected as formamidopyrimidine glycosylase
(fpg) sensitive sites. Both enzymes have already been tried in comet assays studying
alkylation damage [25-27]. Comet assays at pH > 13 with an additional digestion step prior

97
to the DNA unwinding step in the alkaline solution were therefore performed on U937,
NCI-H23 and normal lymphocytes treated with NDMAOAc or NNKOAc.
The results, as shown in Figs. 3 and 4A and B, are the same for U937 and NCI-H23 cells.
The dose of NDMAOAc applied to the cells had to be reduced compared to previous assays
to stay in the detection limits of the technique. At these doses (<1 μM), no alkali-labile sites
were detected from the background in NDMAOAc treated cells. But a significant frequency
of endo III-sensitive sites and more frequent fpg-sensitive sites were measured: 11.0 and
16.7 times more fpg-sensitive sites than endo III-sensitive sites for U937 and NCI-H23,
respectively. The NNKOAc treatments could be performed at the same doses as previously.
Endo III-sensitive sites were present in U937 and NCI-H23 cells (Fig. 4A and B) but their
number did not increase with the dose and therefore seemed non specific to the alkylating
agent. Direct breaks/alkali-labile sites were detected and a roughly equal frequency of fpg-
sensitive sites was added by the fpg digestion: fpg-sensitive sites were 0.81 and 0.72 time
the frequency of direct breaks/alkali-labile sites in U937 and NCI-H23 cells, respectively.
The lymphocytes showed a pattern slightly different (Figs. 3C and 4C). Endo III digestion
following the NDMAOAc treatment did not increase the number of breaks detected (Fig.
3C). On the contrary, the frequency of fpg-sensitive sites was so high that the comet assay
could not measure them at a dose as low as 1 μM. But the median percent tail DNA was
actually proportional to all the doses ≤0.5 μM. The pattern obtained for lymphocytes
treated with NNKOAc was also different (Fig. 4C). The direct breaks/alkali-labile sites as
well as the fpg-sensitive sites ceased to be proportional to the mutagen dose when it was
>20 μM. But the relative frequency of fpg-sensitive sites versus direct breaks/alkali-labile
sites for doses ≤20 μM was comparable to that observed for the other cell types: 0.73.
Endo III-sensitive sites were observed at doses >10 μM but in a manner that was function
of but not proportional to the dose applied.
3.2.4.3 Influence of the lysis on fpg-sensitive sites It was suggested in previous reports that the fpg-sensitive sites observed with the comet
assay following an alkylation treatment [26,27] corresponded to N7-mG, from which the
ring-opening had been triggered by the lysis solution at pH 10. Actually, fpg cannot detect

98
N7-mG unless they have been modified into formamidopyrimidines (fapy). The efficiency
of a strong base to stimulate the fapy transformation of a N7-mG is well documented
[28,29] but it remained uncertain whether our lysis solution could have the same kind of
effect. To investigate this question, we decided to used DMS as a source of N7-mG as 70%
of DNA damage generated by this alkylating agent corresponds to N7-mG.
U937 cells were first treated with increasing doses of DMS and submitted to a comet assay.
They presented a high frequency of fpg-sensitive sites at doses where no alkali-labile sites
could be detected (data not shown). We therefore decided to investigate if the lysis
conditions had an influence on the frequency of these DMS-related fpg-sensitive sites.
U937 cells were treated with 10 μM DMS for 6 min at room temperature and slides were
prepared as previously for the comet assay. They were kept in the lysis solution for 0.5–
24 h. At the end of each lysis time, slides were transferred in Tris-HCl pH 7.4 to neutralize
and stop an eventual effect of the lysis pH. Fig. 5A shows that the number of fpg-sensitive
sites detected after 10 μM DMS increased gradually with the time of lysis when it was
performed at pH 10 but not at pH 7.5. This means that the lysis at pH 10 was both
necessary and sufficient to generate the fpg-sensitive lesions observed in cells treated with
DMS. These results are consistent with the hypothesis that N7-mG can be a source of fpg-
sensitive sites measured with the comet assay. It is therefore likely that the fpg-sensitive
sites observed following a NDMAOAc treatment have the same origin (Fig. 5A).
By analogy, NNKOAc-related fpg-sensitive sites could correspond to ring-opened N7-
pobG. In order to investigate if they follow the same kind of pattern as N7-mG, different
lysis time at pH 10 were tried on U937 cells treated with 50 μM NNKOAc. The frequency
of fpg-sensitive sites remained grossly the same whatever the lysis time at pH 10 (Fig. 5B).
If our hypothesis is correct, this could either mean that 30 min (the shortest time of lysis
tried) was sufficient to obtain a ring-opening of all the N7-pobG and/or that the fapy lesions
already existed in cells prior to the lysis. We therefore decided to investigate the effect of a
lysis time < 30 min. In order to eliminate any effect on the results of a possible lower
efficiency of the lysis by these smaller lysis time, cells were lysed 5–60 min at pH 10 (Fig.
5C) or pH 7.5 (Fig. 5D) then submitted to the fpg-digestion, then lysed another 12 h at pH
10 in order to complete the lysis before the following steps of the comet assay. Fig. 5C and

99
D show that the fpg-sensitive sites are detected after as little as 5 min of lysis whatever the
pH and that the percent tail DNA obtained was comparable to the one obtained for 1 or
24 h of lysis.

100
3.2.5 Discussion The results of our present study show that the comet assay is sufficiently sensitive and
specific to measure NNK-related DNA damage. In peculiar, it permits an investigation of
potentially important damage of the pyridyloxobutylation pathway which is far less
documented than that of the methylation pathway. A major damage of this pathway
correspond to N7-pobG, an HPB-releasing adduct, that had never been shown in cells to
this day. Our result indicate that this adduct could exist in its fapy form in human cells
treated with NNKOAc.
The DNA damage observed with these comet assays can be identified by taking three
factors into account: (i) the adduct types already known from the methylation and the
pyridyloxobutylation pathways (ii) their chemical properties notably their alkali-lability at
different pH and (iii) the specificity of the lesion specific enzymes that recognized them.
Following a NDMAOAc treatment, the following categories of damage were detected with
our comet assays: alkali-labile sites, endo III-sensitive sites and fpg-sensitive sites. AP sites
are in part alkali-labile at pH > 13 [25] but not at pH 12.2 and should therefore account for
the breaks observed at high dose (alkali-labile sites) in cells treated with NDMAOAc
without enzymatic digestion (Fig. 2A). Actually, only a 4 hours treatment at pH 12.8 and
24°C can induce breakage at >98% of AP sites [30], which means that even at pH > 13 for
40 min at 4°C, the alkali conversion into breaks was not complete. That is why alkali-labile
sites could not be detected from the background at the lower doses tested (Fig. 3) whereas
some AP sites, as endo III-sensitive sites, could still be seen. It should be noticed that fpg is
known to have also an enzymatic activity on AP sites [31] and that part of the fpg-sensitive
sites could therefore correspond to abasic sites as well. Still the endo III treatment, which is
much more specific for that type of adduct, gives only a low frequency of breaks (Fig. 3),
excluding the hypothesis that AP sites account for the high frequency of fpg-sensitive sites.
Moreover, these fpg-sensitive sites detected following the NDMAOAc treatment resemble
those that can be seen after a DMS treatment (Fig. 5A). They correspond therefore more
likely to N7-mG produced in the cells that underwent a ring-opening in the lysis solution at
pH 10, a transformation that makes them detectable by fpg [31,32]. The 1 h lysis time that

101
we used here (Fig. 2) is not sufficient to observe a complete opening as the maximum
frequency of DMS-related fpg-sensitive sites is obtained only by a 12 h lysis (Fig. 5A).
This means that only a fraction of N7-mG (≈25%) are detected in our experiments.
The DNA damage detected following the NNKOAc treatment can be divided into two
categories: direct breaks/alkali-labile sites and fpg-sensitive sites. The phosphate adducts
(pyridyloxobutylphosphotriesters) that are produced by the pyridyloxobutylation pathway
(Table 1) can potentially generate direct breaks in cells as they affect the sugar phosphate
backbone of the DNA double helix. As a matter of fact, direct breaks are seen in DNA
treated in vitro with NNKOAc [10]. Alkylphosphotriesters are alkali-labile adducts and
could therefore explain both the direct breaks and the alkali-labile sites, detected at pH > 13
but not at pH 12.2 (Fig. 2B). This would mean that not all alkylphosphotriesters generate
direct breaks but rather that more and more of them can be seen with the comet assay as pH
increases. Unidentified alkali-labile damage, shown in hepatocytes of NNK-treated rats,
had such a pH dependency [33]. Their estimated frequency in U937 cells treated with
50 μM NNKOAc for 2 h is 75 lesions per 109 bases. Overall, these results confirm the
hypothesis that the DNA single-strand breaks generated by a NNK treatment are due to the
pyridyloxobutylation pathway and not to the methylation pathway.
The second type of damage detected with these assays following the NNKOAc treatment
correspond to fpg-sensitive sites. Their estimated frequency in U937 cells treated with
50 μM NNKOAc for 2 h is 82 lesions per 109 bases. By analogy to those observed after the
NDMAOAc treatment and considering the ability of fpg to detect adducts that present a
bulky chemical lesion at the guanine N7 atom [29,32], these adducts could be the
secondary fapy form resulting from the formation of N7-pobG (Fig. 1). The existence of
such adducts would therefore be an indirect evidence that the N7-pobG, which is an HPB-
releasing adduct, is actually formed in cells treated with NNKOAc, as expected from
analysis of adducts generated to DNA in vitro [13]. Still, we failed to see an effect of the
lysis conditions on these fpg-sensitive sites. This is not sufficient to infirm our hypothesis
as the ring-opening of these adducts could be much easier than for N7-mG. If so, the fact
that 5 min lysis at neutral pH is sufficient to see them (Fig. 5D) suggests that these lesions
already exist in the cell prior to the lysis.

102
The slightly different profile of the normal lymphocytes treated with NDMAOAc or
NNKOAc versus the other cell types could result from specific features of this cell type.
The fact that the number of adducts at higher dose ceased to be proportional to the dose
could be related to differences in the ability of lymphocytes to convert NDMAOAc and
NNKOAc in their reactive species. Esterases are necessary to this activation and such
enzymes are not in an equal amount in all cell types. Type II pneumocytes and to a lesser
extend lung histiocytes, that correspond to the cell types from which originated NCI-H23
and U937, respectively, show some esterase activity [34]. A smaller esterase activity in
lymphocytes could limit the proportion of NNKOAc molecules that is activated in reactive
species at higher doses, generating a smaller frequency of damage than expected from the
results at smaller doses. Moreover, lymphocytes are known to be extremely sensitive to UV
irradiation, this hypersensitivity being associated with a deficiency in the rejoining of
excision breaks due to a limited amount of deoxyribonucleosides available in this cell type
that is not actively proliferating [35,36]. In lymphocytes treated with NNKOAc at a high
dose, this lack of deoxyribonucleosides could account for the observed accumulation of
non-filled AP sites (generated spontaneously or by glycosylase excision).
More important than the frequency of the formed adducts is their biological significance,
their impact in the cells, notably towards the DNA polymerases and the possibility of
miscoding during translesional synthesis. The pyridyloxobutylphosphotriesters that
generate direct breaks could be a major problem if they persist a long time in cells, as it is
the case for the single-strand breaks observed in rat liver after an NNK treatment [37],
possibly due to the persistence of a blocking residue in 3’OH [10]. The alkylation of a
guanine at the N7 position, as it happens through both pathways of activation of NNK,
gives a non stable adduct that can form secondary lesions by rupture of (i) the glycosidic
bond, which leads to an AP site or (ii) the C8-N9 bond of the imidazole ring, which leads to
a fapy residue [38,39]. N7-mG are very common adducts generated by methylating agents
but they do not represent a major disturbance for the cells. But as they are unstable, they
can lead to AP sites or N7-m-fapy (Fig. 1) which both block polymerase elongation and can
be mutagenetic in case of translesional synthesis [28,38,39]. Still, neither AP sites nor fapy
adducts seem to be very frequent in cells treated with NDMAOAc, suggesting that N7-
alkylation of guanines is not major in mutagenesis associated with the methylation pathway

103
of NNK activation. On the contrary, N7-alkylation of the pyridyloxobutylation pathway
could be a major mutagenic pathway. Little is known about the N7-pobG and its possible
fapy derivative (Fig. 1) but N7-pobG adducts are unstable and could therefore be part of the
HPB-releasing adducts which have been associated with the carcinogenicity of the
pyridyloxobutylation pathway of NNK [6,12]. It should be noticed that these adducts are
somewhat similar to the N7-aflatoxin B1-G and its fapy form, the later being particularly
relevant as it is both genotoxic (it blocks the polymerase) and miscoding, resulting in the
mutations observed in hepatocellular carcinoma [40]. But only mutagenesis analysis with
these adducts would determine if they could actually participate in NNK-related
carcinogenesis. However, their great instability [12,14] could make that kind of study very
difficult. The comet assay is therefore interesting to get at least some information on how
cells deal with the fapy form, as they can be followed as fpg-sensitive sites following a
NNKOAc treatment.

104
3.2.6 Acknowledgements The authors are grateful to Dr. Jean-François Cloutier for the synthesis of NNKOAc, Dr.
Serge Boiteux for kindly supplying the endonuclease III and the formamidopyrimidine
glycosylase, and Dr. R. Stephen Lloyd for the T4 endonuclease V. The authors sincerely
thank Dr. Gerald P. Holmquist for important discussion and carefully editing the
manuscript. The research carried out in the laboratory of R.D. was funded by the Institute
of Genetics of the CIHR and the Canada Research Chairs Program. R.D. holds the Canada
Research Chair in « Genetics, Mutagenesis and Cancer ».

105
3.2.7 References [1] S.S. Hecht, Tobacco smoke carcinogens and lung cancer, J. Natl. Cancer Inst. 91
(1999) 1194-1210.
[2] G.N. Wogan, S.S. Hecht, J.S. Felton, A.H. Conney, L.A. Loeb, Environmental and
chemical carcinogenesis, Semin. Cancer Biol. 14 (2004) 473-486.
[3] S.S. Hecht, DNA adduct formation from tobacco-specific N-nitrosamines, Mutat.
Res. 424 (1999) 127-142.
[4] D. Hoffmann, A. Rivenson, S.S. Hecht, The biological significance of tobacco-
specific N-nitrosamines: smoking and adenocarcinoma of the lung, Crit. Rev. Toxicol. 26
(1996) 199-211.
[5] S.S. Devesa, G.L. Shaw, W.J. Blot, Changing patterns of lung cancer incidence by
histological type, Cancer Epidemiol. Biomarkers Prev. 1 (1991) 29-34.
[6] S.S. Hecht, Biochemistry, biology, and carcinogenicity of tobacco-specific N-
nitrosamines, Chem. Res. Toxicol. 11 (1998) 559-603.
[7] G.B. Smith, A. Castonguay, P.J. Donnelly, K.R. Reid, D. Petsikas, T.E. Massey,
Biotransformation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-
1-butanone (NNK) in freshly isolated human lung cells, Carcinogenesis 20 (1999) 1809-
1818.
[8] G.B. Smith, J.R. Bend, L.L. Bedard, K.R. Reid, D. Petsikas, T.E. Massey,
Biotransformation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in peripheral
human lung microsomes, Drug Metab. Dispos. 31 (2003) 1134-1141.
[9] D.H. Phillips, Smoking-related DNA and protein adducts in human tissues,
Carcinogenesis 23 (2002) 1979-2004.
[10] J.F. Cloutier, R. Drouin, M. Weinfeld, T.R. O'Connor, A. Castonguay,
Characterization and mapping of DNA damage induced by reactive metabolites of 4-

106
(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) at nucleotide resolution in human
genomic DNA, J. Mol. Biol. 313 (2001) 539-557.
[11] J. Haglund, A.P. Henderson, B.T. Golding, M. Tornqvist, Evidence for phosphate
adducts in DNA from mice treated with 4-(N-Methyl-N-nitrosamino)-1-(3-pyridyl)-1-
butanone (NNK), Chem. Res. Toxicol. 15 (2002) 773-779.
[12] M. Wang, G. Cheng, S.J. Sturla, Y. Shi, E.J. McIntee, P.W. Villalta, P. Upadhyaya,
S.S. Hecht, Identification of adducts formed by pyridyloxobutylation of deoxyguanosine
and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically
activated form of tobacco specific carcinogens, Chem. Res. Toxicol. 16 (2003) 616-626.
[13] S.S. Hecht, P.W. Villalta, S.J. Sturla, G. Cheng, N. Yu, P. Upadhyaya, M. Wang,
Identification of O2-substituted pyrimidine adducts formed in reactions of 4-
(acetoxymethylnitrosamino)- 1-(3-pyridyl)-1-butanone and 4-(acetoxymethylnitros-
amino)-1-(3-pyridyl)-1-butanol with DNA, Chem. Res. Toxicol. 17 (2004) 588-597.
[14] S.J. Sturla, J. Scott, Y. Lao, S.S. Hecht, P.W. Villalta, Mass spectrometric analysis
of relative levels of pyridyloxobutylation adducts formed in the reaction of DNA with a
chemically activated form of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-
pyridyl)-1-butanone, Chem. Res. Toxicol. 18 (2005) 1048-1055.
[15] R.R. Tice, E. Agurell, D. Anderson, B. Burlinson, A. Hartmann, H. Kobayashi, Y.
Miyamae, E. Rojas, J.C. Ryu, Y.F. Sasaki, Single cell gel/comet assay: guidelines for in
vitro and in vivo genetic toxicology testing, Environ. Mol. Mutagen. 35 (2000) 206-221.
[16] L. Wolz, G. Krause, G. Scherer, The comet assay with MCL-5 cells as an indicator
of genotoxic treatment with chemicals and cigarette smoke condensates, Altern. Lab. Anim.
30 (2002) 331-339.
[17] C.H. Chuang, M.L. Hu, Synergistic DNA damage and lipid peroxidation in cultured
human white blood cells exposed to 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone and
ultraviolet A, Environ. Mol. Mutagen. 47 (2006) 73-81.

107
[18] N.P. Singh, D.B. Danner, R.R. Tice, L. Brant, E.L. Schneider, DNA damage and
repair with age in individual human lymphocytes, Mutat. Res. 237 (1990) 123-130.
[19] R. Drouin, H. Rodriguez, G.P. Holmquist, S.A. Akman, Ligation-mediated PCR for
analysis of oxidative DNA damage, in: G.P. Pfeifer (Ed.), Technologies for Detection of
DNA Damage and Mutations, Plenum Press, New York, 1996, pp. 209-223.
[20] N. Rioux, A. Castonguay, The induction of cyclooxygenase-1 by a tobacco
carcinogen in U937 human macrophages is correlated to the activation of NF-kappaB,
Carcinogenesis 21 (2000) 1745-1751.
[21] J. Cadet, E. Sage, T. Douki, Ultraviolet radiation-mediated damage to cellular DNA,
Mutat. Res. 571 (2005) 3-17.
[22] N.P. Singh, M.T. McCoy, R.R. Tice, E.L. Schneider, A simple technique for
quantitation of low levels of DNA damage in individual cells, Exp. Cell Res. 175 (1988)
184-191.
[23] K. Konca, A. Lankoff, A. Banasik, H. Lisowska, T. Kuszewski, S. Gozdz, Z. Koza,
A. Wojcik, A cross-platform public domain PC image-analysis program for the comet
assay, Mutat. Res. 534 (2003) 15-20.
[24] A. Eastman, M.A. Barry, The origins of DNA breaks: a consequence of DNA
damage, DNA repair, or apoptosis? Cancer Invest. 10 (1992) 229-240.
[25] P. Fortini, G. Raspaglio, M. Falchi, E. Dogliotti, Analysis of DNA alkylation
damage and repair in mammalian cells by the comet assay, Mutagenesis 11 (1996) 169-
175.
[26] K.J. Angelis, M. Dusinska, A.R. Collins, Single cell gel electrophoresis: detection
of DNA damage at different levels of sensitivity, Electrophoresis 20 (1999) 2133-2138.
[27] G. Speit, P. Schutz, I. Bonzheim, K. Trenz, H. Hoffmann, Sensitivity of the FPG
protein towards alkylation damage in the comet assay, Toxicol. Lett. 146 (2004) 151-158.

108
[28] T.R. O'Connor, S. Boiteux, J. Laval, Ring-opened 7-methylguanine residues in
DNA are a block to in vitro DNA synthesis, Nucleic Acids Res. 16 (1988) 5879-5894.
[29] Q. Li, J. Laval, D.B. Ludlum, Fpg protein releases a ring-opened N-7 guanine
adduct from DNA that has been modified by sulfur mustard, Carcinogenesis 18 (1997)
1035-1038.
[30] T. Lindahl, A. Andersson, Rate of chain breakage at apurinic sites in double-
stranded deoxyribonucleic acid, Biochemistry 11 (1972) 3618-3623.
[31] S. Boiteux, T.R. O'Connor, F. Lederer, A. Gouyette, J. Laval, Homogeneous
Escherichia coli FPG protein. A DNA glycosylase which excises imidazole ring-opened
purines and nicks DNA at apurinic/apyrimidinic sites, J. Biol. Chem. 265 (1990) 3916-
3922.
[32] C.J. Chetsanga, M. Lozon, C. Makaroff, L. Savage, Purification and
characterization of Escherichia coli formamidopyrimidine-DNA glycosylase that excises
damaged 7-methylguanine from deoxyribonucleic acid, Biochemistry 20 (1981) 5201-
5207.
[33] K. Demkowicz-Dobrzanski, A. Castonguay, Comparison of DNA alkali-labile sites
induced by 4-(methylnitrosamino)-1(3-pyridyl)-1-butanone and 4-oxo-4-(3-pyridyl)butanal
in rat hepatocytes, Carcinogenesis 12 (1991) 2135-2140.
[34] S. Kudo, Differentiation of Clara cells and pneumocytes of the rat by means of
enzyme- and immunohistochemistry, Anat. Rec. 238 (1994) 49-56.
[35] M.H. Lankinen, L.M. Vilpo, J.A. Vilpo. UV- and gamma-irradiation-induced DNA
single-strand breaks and their repair in human blood granulocytes and lymphocytes, Mutat.
Res. 352 (1996) 31-38.
[36] M.H. Green, A.P. Waugh, J.E. Lowe, S.A. Harcourt, P.H. Clingen, J. Cole, C.F.
Arlett, Protective effect of deoxyribonucleosides on UV-irradiated human peripheral blood
T-lymphocytes: possibilities for the selective killing of either cycling or non-cycling cells,
Mutat. Res. 350 (1996) 239-246.

109
[37] R. Jorquera, A. Castonguay, H.M. Schuller, DNA single-strand breaks and toxicity
induced by 4-(methyl-nitrosamino)- 1-(3- pyridyl)-1-butanone or N-nitrosodimethylamine
in hamster and rat liver, Carcinogenesis 15 (1994) 389-394.
[38] J. Laval, S. Boiteux, T.R. O'Connor, Physiological properties and repair of
apurinic/apyrimidinic sites and imidazole ring-opened guanines in DNA, Mutat. Res. 233
(1990) 73-79.
[39] B. Tudek, Imidazole ring-opened DNA purines and their biological significance, J.
Biochem. Mol. Biol. 36 (2003) 12-19.
[40] M.E. Smela, M.L. Hamm, P.T. Henderson, C.M. Harris, T.M. Harris, J.M.
Essigmann, The aflatoxin B(1) formamidopyrimidine adduct plays a major role in causing
the types of mutations observed in human hepatocellular carcinoma, Proc. Natl. Acad. Sci.
USA 99 (2002) 6655-6660.

110
3.2.8 Table Table 1 DNA damage generated through the methylation and the pyridyloxobutylation pathways
and their stability
Adducts position Activation pathway
Pyridyloxobutylation
GuanineN7 Yes Stable Yes HPB-releasingO 6 Yes Stable Yes StableN 2 No Yes Stable
ThymineO 4 Yes Stable NoO 2 No Yes Stable
CytosineO 2 No Yes HPB-releasing
DNA backbonePhosphate No Yes Stable
Methylation

111
3.2.9 Legends to Figures Figure 1 Activation of NNK, NDMAOAc and NNKOAc in reactive species. The
DNA damage types that are studied with the comet assay in this work are presented as well
as the way they can be detected.
Figure 2 Comparison of the comet assay at pH > 13 and pH 12.2 in U937 cells treated
with NDMAOAc (A), NNKOAc (B) or UVB followed by T4 endonuclease V enzymatic
digestion (C). Each value is the average median percent tail DNA ± S.D. of three
independent experiments. * p < 0.02
Figure 3 Comet assay at pH > 13 of U937 (A), NCI-H23 (B) and normal lymphocytes
(C) treated with increasing doses of NDMAOAc. The assay included a treatment with the
enzyme buffer, endo III or fpg. Each value is the average median percent tail DNA ± S.D.
of three independent experiments. For each type of enzymatic treatment, the calculated
slope is indicated as well as the ratio of fpg-sensitive sites vs. endo III-sensitive sites.
Figure 4 Comet assay at pH > 13 of U937 (A), NCI-H23 (B) and normal lymphocytes
(C) treated with increasing doses of NNKOAc. The assay included a treatment with the
enzyme buffer, endo III or fpg. Each value is the average median percent tail DNA ± S.D.
of three independent experiments. For each type of enzymatic treatment, the calculated
slope is indicated as well as the ratio of fpg-sensitive sites vs. direct breaks/alkali-labile
sites.
Figure 5 Influence of the lysis time on the fpg-sensitive sites detected with the comet
assay. U937 cells were treated with (A) 10 μM DMS then lysed for 0.5–24 h at pH 10
followed by fpg digestion or not or lysed at pH 7.5 followed by fpg digestion; (B) 50 μM
NNKOAc then lysed for 0.5–24 h at pH 10 followed by fpg digestion or not; (C and D)
50 μM NNKOAc then lysed for 5–60 min at pH 10 (C) or 7.5 (D) followed by a fpg
digestion or not then a new 12 h lysis at pH 10. Each value is the average median percent
tail DNA ± S.D. of two independent experiments.

112
Figure 1
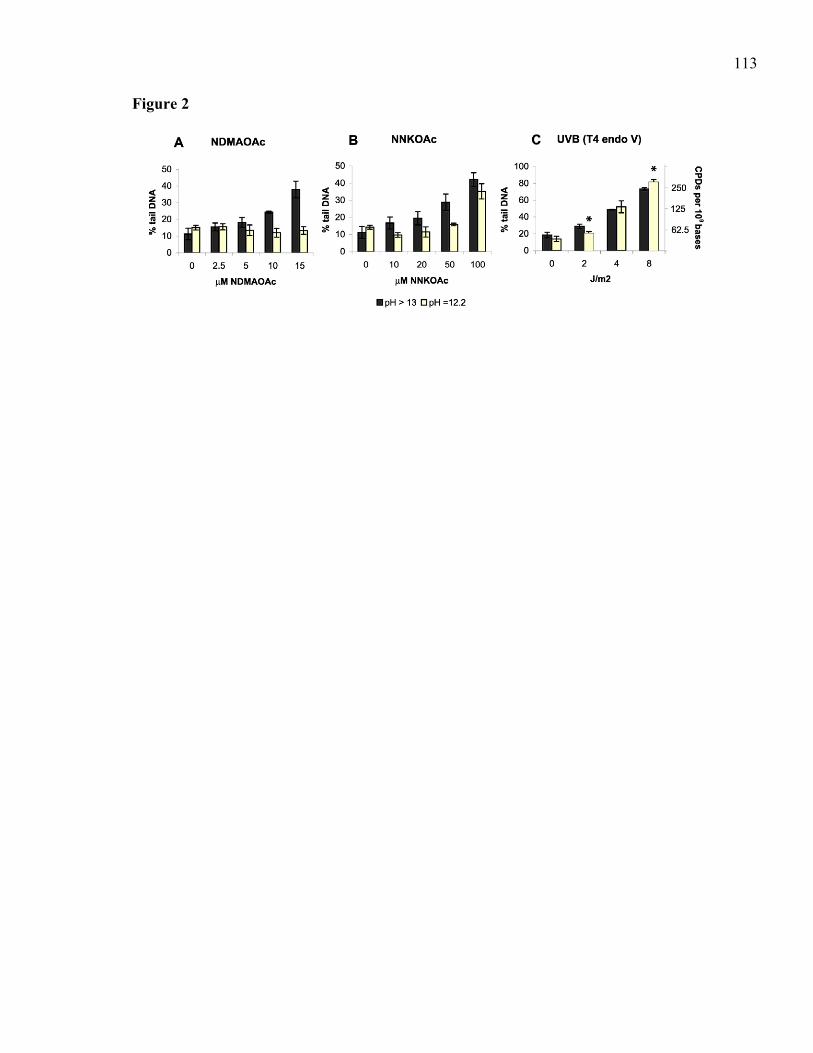
113
Figure 2

114
Figure 3
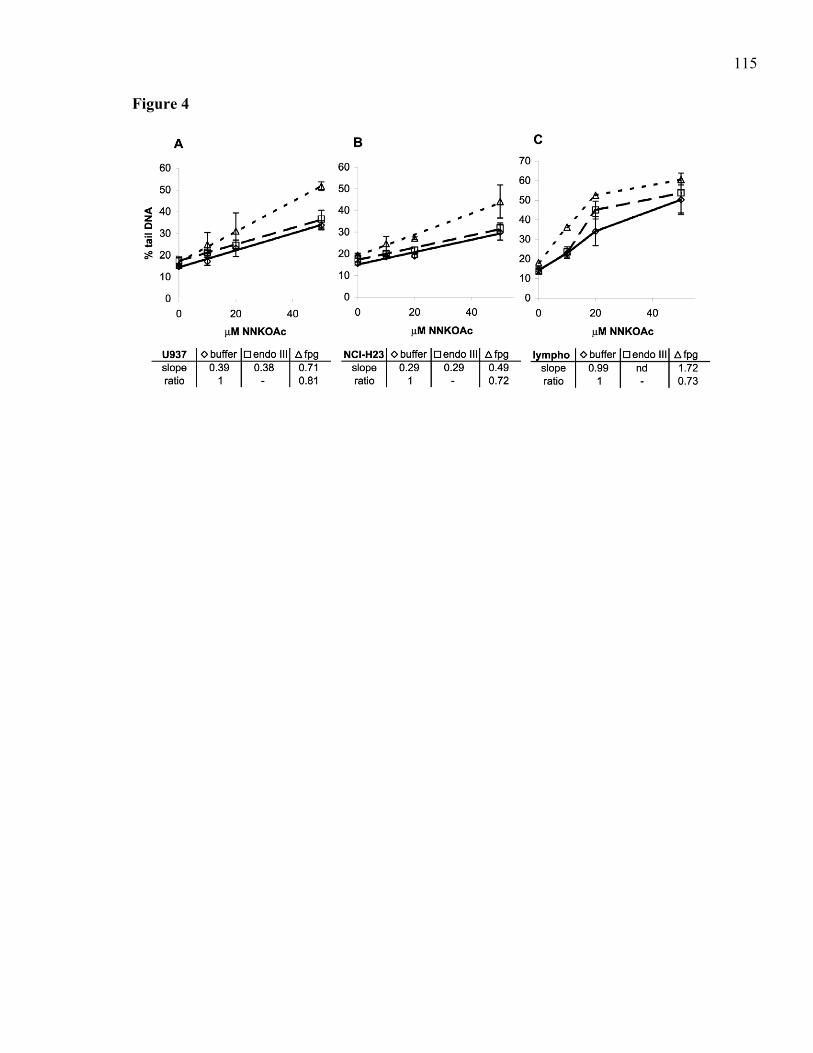
115
Figure 4
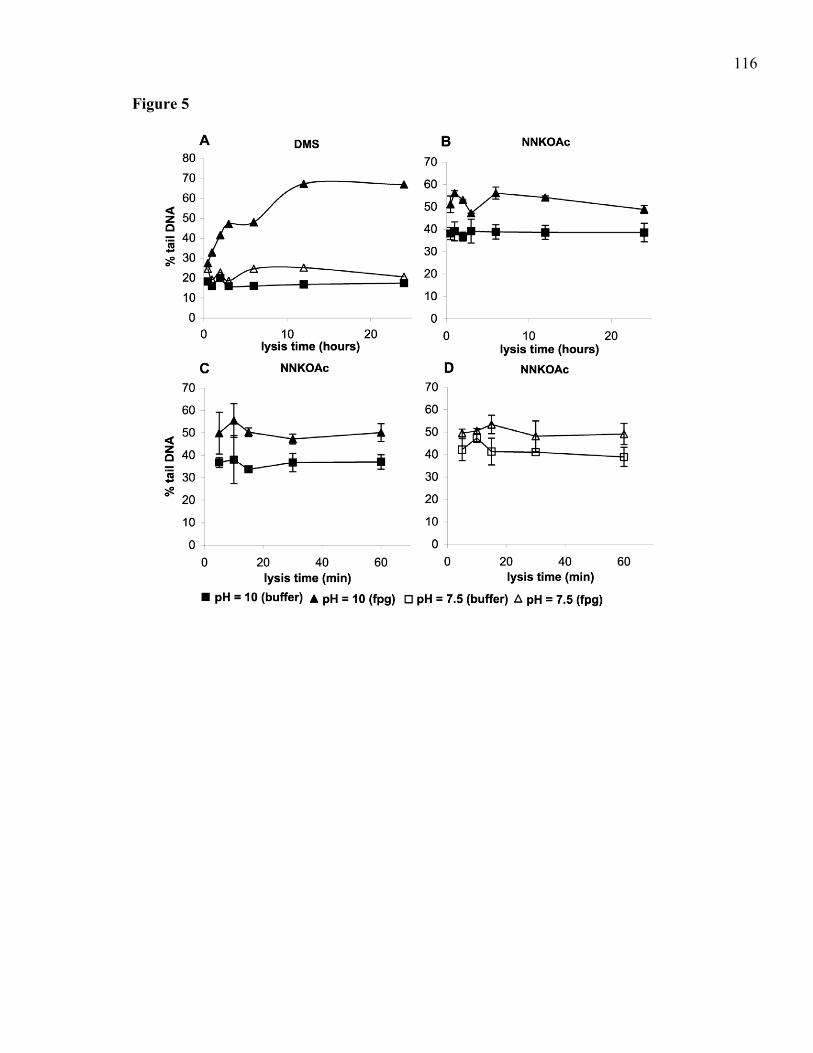
116
Figure 5

Chapitre 4 Repair kinetics of specific types of nitroso-induced DNA damage using the comet assay in human
cells. (accepté pour publication par la revue « Mutation Research, Fundamental and molecular
mechanisms of mutagenesis »)

118
4.1 Résumé en français Le test des comètes est sensible et permet de détecter des fréquences de dommages à l'ADN
inférieures à 1 pour 107 bases. Nous avons montré précédemment que plusieurs types de
dommages associés à la 4-(méthylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), un pro-
mutagène spécifique du tabac, peuvent être étudiés spécifiquement avec cette technique. On
connait peu de chose sur la réparation de ces dommages. Nous avons d'abord vérifié que le
test des comètes permettait de quantifier la réparation dans les fibroblastes de certains
dommages spécifiques tels que les 7-méthylguanines (7-mG) générés par le diméthylsulfate
ou les dimères cyclobutyliques de pyrimidines résultant d'une irradiation aux UVB. Puis
nous avons suivi l'évolution, formation et réparation, après des doses aiguës de métabolites
activés de la NNK, de la fréquence des dommages dans des cellules normales (fibroblastes
et lymphocytes) et dans des cellules capables d'activer la NNK (U937 et NCI-H23). La
NNK peut être activée dans les cellules en métabolites réactifs qui vont soit méthyler soit
pyridyloxobutyler l'ADN. La fréquence des 7-mG résultant de la méthylation évolue après
le traitement de manière suffisamment différente pour déduire que les U937 les réparent
rapidement, les lymphocytes les réparent lentement et que leur taux de réparation dans les
NCI-H23 et les U937 est intermédiaire. La pyridyloxobutylation génère des dommages
sensibles à la formamidopyrimidine (fapy) glycosylase (Fpg), que l'on pense être la forme
secondaire fapy de 7-pyridyloxobutylguanines produites dans les cellules. L'évolution post-
traitement de la fréquence de ces dommages semble dépendre encore plus clairement du
type cellulaire : les fibroblastes et les NCI-H23 présentent une augmentation initiale rapide
de la fréquence des sites sensibles à la Fpg qui n'existe pas chez les lymphocytes et les
U937. Cette augmentation semble associée avec la présence d'une protéine p53
fonctionnelle dans les fibroblastes. Ainsi, nos résultats montrent que des cinétiques de
réparation peuvent être investiguées avec le test des comètes mais que l'activation du pro-
mutagène et/ou la manière dont un adduit est formé peuvent affecter la quantification de la
réparation.

119
4.2 Article
Repair kinetics of specific types of nitroso-induced DNA
damage using the comet assay in human cells.
Sandrine Lacostea, André Castonguayb and Régen Drouina,*
a Service of Genetics, Department of Paediatrics, Faculty of Medicine and Health Sciences, University of Sherbrooke, 3001, 12th Avenue North, Sherbrooke, Que., Canada J1H 5N4
b Faculty of Pharmacy, Laval University, Quebec City, Que., Canada G1K 7P4 Key words: 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK);
Formamidopyrimidine adducts; p53; Comet assay; DNA repair; Chromatin. Corresponding author : * Régen Drouin
Service of Genetics Department of Pediatrics Faculty of Medicine and Health Sciences University of Sherbrooke 3001, 12th Avenue North Sherbrooke, Qc, Canada J1H 5N4 Tel: (819) 820-6827 Fax: (819) 564-5217 E-mail: [email protected]

120
4.2.1 Abstract The comet assay is sensitive and can detect DNA damage frequencies less than one in 107
bases. We have previously shown that several types of DNA damage associated with 4-
(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a tobacco-specific pro-mutagen, can
be investigated with some specificity using this technique. Little is known about their
repair. We verified the ability of the comet assay to quantify the repair kinetics of specific
types of damage in normal fibroblasts, e.g., dimethylsulfate-induced 7-methylguanines (7-
mG) and UVB-induced cyclobutane pyrimidine dimers. The time course, formation and
repair, of DNA damage after acute doses of NNK reactive metabolites, were then compared
in normal human cells (fibroblasts and lymphocytes) and in cells proficient for activating
NNK (U937 and NCI-H23). NNK can be activated in cells into reactive metabolites that
can either methylate or pyridyloxobutylate DNA. The 7-mG generated by methylation gave
post-treatment patterns that were sufficiently different between cell types to conclude that
repair of 7-mG in U937 cells was fast, repair in lymphocytes was slow, and repair in NCI-
H23 cells and fibroblasts displayed intermediate rates. Pyridyloxobutylation generated
formamidopyrimidine (fapy) glycosylase (fpg)-sensitive sites that could be the fapy form of
7-pyridyloxobutylguanines produced in cells. For this type of adducts, the post-treatment
patterns of adduct frequency as a function of time depended even more clearly on the cell
type: fibroblasts and NCI-H23 cells showed an initial rapid increase in fpg-sensitive
damage frequency that did not occur in lymphocytes and U937 cells. This increase seemed
associated with p53 proficiency in fibroblasts. Our results show that repair kinetics can be
investigated with the comet assay and that differences between cell types can be observed
with that technique. But it seems that pro-mutagen activation and/or the way a type of
adducts is formed can affect the quantification of the repair.

121
4.2.2 Introduction 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), is the most abundant and the most
potent carcinogen among the N-nitrosamines present in tobacco smoke [1,2]. NNK is
specific to tobacco and has been associated with recent increases in the relative incidence of
human lung adenocarcinoma versus squamous cell carcinoma in smokers [3,4].
In metabolically competent cells, especially lung, nasal mucosa and liver, NNK can be
metabolically activated into two different reactive species capable of either methylating or
pyridyloxobutylating DNA (Fig. 1). Both activation pathways can be mimicked by analogs
that generate either one of the two reactive metabolites. Such analogs were used to
determine which pathway is important to tumorigenesis in animals; the results suggest that
both are important to carcinogenesis [5-8]. The detection of NNK-associated damage
corresponding to both pathways of activation in human smokers and in human cells is
consistent with results from animal studies [9-13]. The nature of the DNA damage that is
actually responsible for NNK-associated carcinogenesis was postulated to involve O6-
alkylguanines (O6-methylguanines and O6-pyridyloxobutylguanines) [14-17] but other
types of damage could participate as well. Little is known about the repair of NNK-
associated damage except for O6-alkylguanines that are known to be repaired by direct
reversion by the alkylguanine transferase [14,15].
Like other carcinogens present in tobacco smoke, NNK is present in low quantity in the
mainstream smoke of each cigarette: 80 – 770 ng or less that 4 nmol [1,13]. Lung cells of
smokers deal with the chronic maintenance of a low frequency of alkylation damage, due to
low level of carcinogens per cigarette and their necessity for bioactivation. Still, most
tobacco mutagenicity studies investigate repair in cells that were submitted to high acute
doses of damaging agent and it is not clear whether the repair rates measured under those
conditions reflect the repair in cells having a lower frequency of DNA damage. Exploring
this issue requires the use of a technique capable of detecting very rare damage frequencies
and following the damage frequency through time. The 32P-post-labeling technique [18]
and an immunoblot assay [19] have already been used to study global repair of low
frequencies (10-50 adducts per 108 nucleotides) of damage generated by another tobacco-

122
related carcinogen: benzo[a]pyrene-diol-epoxide (BPDE). Other types of polycyclic
aromatic hydrocarbon adducts have also been studied using the 32P-post-labeling technique
[20].
The comet assay is another technique to study rare damage and we have already shown that
it can be used to detect quite specifically some of the NNK-associated damage at
frequencies as low as 60 lesions per 109 bases [21]. The 7-mG generated by the methylation
pathway of NNK activation can be investigated as formamidopyrimidine (fapy) glycosylase
(fpg)-sensitive sites (Fig. 1) by the comet assay. We have shown that the lysis step
transforms 7-mG into a fapy fpg-sensitive form and that a 12 h lysis during the comet assay
is sufficient for a complete conversion of all adducts. On the other hand, the
pyridyloxobutylation pathway of NNK generated direct breaks/alkali-labile sites (ALS) and
a roughly equal frequency of fpg-sensitive damage. The nature of the detected damage has
been discussed previously [21] and based on (1) the damage already known to be generated
by DNA pyridyloxobutylation, (2) enzyme specificity and (3) alkali-lability of the damage,
we suggested that direct-breaks/ALS were alkyl-phosphotriesters, whereas the fpg-sensitive
damage could be fapy secondary lesions deriving from 7-pyridyloxobutylguanines (7-pob)
in the cells (Fig. 1). Contrary to what was shown for 7-mG, the fpg-sensitive sites obtained
by pyridyloxobutylation corresponded to damage that are actually produced within the cells
and did not simply result from the lysis step of the comet assay [21]. If such 7-pob-fapy
adducts are actually formed, they should be significant in terms of mutagenesis as they
resemble several other adducts, such as the 7-Aflatoxin B1 fapy adduct, that is known to
participate in the mutational load associated with an exposure to Aflatoxin B1 [22].
Following the frequency of each type of damage with time in cells that were treated with
NNK analogs that generate one active metabolite or the other should provide insights on
how the damage is repaired in cells, and more particularly on whether they persist longer in
some cell types than others.
The types of DNA damage most frequently studied with the comet assay are of oxidative
origin and UV damage because of the availability of specific glycosylases that cleave the
DNA at adducted bases [23-30]. Alkylation damage has also been investigated using
several approaches: (1) as abasic (AP) sites secondary to DNA alkylation [31]; (2) as 3-

123
methyladenine glycosylase (AlkA)-sensitive damage, which can correspond to several sorts
of alkylation damage [27-29,32]; (3) through the ability of the fpg glycosylase to detect 7-
mG after a ring-opening [33]. Some of these studies questioned the repair capacities of the
cells [28,29,31,34] but did not truly show repair kinetics.
In this work, we use the comet assay as a technique to study repair kinetics of specific types
of low frequency DNA damage. It was first verified that the repair kinetics of
dimethylsulfate-induced 7-mG and UVB-induced cyclobutane pyrimidine dimers (CPD)
could be determined in normal human fibroblasts. Then, the post-treatment time courses of
damage frequencies were obtained for all NNK-associated types of damage that can be
investigated with some specificity using the comet assay, in normal human cells
(fibroblasts and lymphocytes) and in human cell types proficient for activating NNK (U937
and NCI-H23).

124
4.2.3 Materials and Methods
4.2.3.1 Cell Culture U937 (human histiocytic lymphoma CRL-1593.2) and NCI-H23 (human lung
adenocarcinoma CRL-5800) cell lines were obtained from the ATCC. U937 cells are p53-
deficient because of a point mutation affecting a splice donor site [35] and NCI-H23 cells
express a full length mutant p53 protein (M246I) [36]. Both cell types were grown in RPMI
1640 with 2 mM L-glutamine modified to contain 10 mM HEPES, 1 mM sodium pyruvate,
4.5 g/L glucose, 1.5 g/L bicarbonate and supplemented with 10% fetal bovine serum (FBS)
(v/v). U937 cells were maintained at low density (below 4 × 105 cells/mL). Lymphocytes
were isolated from the blood of a healthy young non-smoker with Histopaque-1077
(Sigma-Aldrich Canada, Oakville, ON) as described by the manufacturer. They were kept
overnight or for a maximum of 48 h (until the comet assay) in RPMI 1640 with 2 mM L-
glutamine modified to contain 10 mM HEPES, 1 mM sodium pyruvate, 4.5 g/L glucose,
1.5 g/L bicarbonate and supplemented with 10% FBS (v/v). Normal skin fibroblasts were
grown in DMEM with 2 mM L-glutamine and supplemented with 10% FBS (v/v). Li-
Fraumeni p53-deficient skin fibroblasts (LF041) as well as LXSN or E6-expressing lung
fibroblasts were obtained and cultured as described previously [37]. Briefly, LF041 are a
spontaneously immortalized (i.e., post-crisis; approximately 200 population doublings) skin
fibroblast strain (a gift of M. Tainsky, University of Texas M. D. Anderson Cancer Center,
Houston, TX) that have lost one p53 allele and carry a frameshift mutation at codon 184 in
the remaining copy. They were grown in DMEM with 2 mM L-glutamine and
supplemented with 10% FBS (v/v). LXSN and E6-expressing fibroblasts correspond to
low-passage lung fibroblasts infected with a replication-defective retrovirus (LXSN)
expressing G418 resistance, and the HPV E6 oncoprotein derived from the high-risk HPV
type 16. They were grown in Ham's F-10 medium containing 15% FBS (v/v). All cells were
kept at 37°C in humidified atmosphere of 5% CO2.
4.2.3.2 Chemicals N-Nitroso(acetoxymethyl)methylamine (NDMAOAc) (CAS # 56856-83-8) (purity 99.8%)
was purchased from LKT laboratory (St. Paul, MN) and 4-(acetoxymethylnitrosamino)-1-

125
(3-pyridyl)-1-butanone (NNKOAc) (purity >94%) synthesised as described previously [38].
They were both dissolved in DMSO as a stock solution of 0.1 M (kept at –80°C).
Treatments were made with ready-to-use aliquots of 10 μM (NDMAOAc) or 1 mM
(NNKOAc) dilutions in DMSO kept at -80°C until the last minute. Any solution or material
that has been in contact with either NDMAOAc or NNKOAc was treated with 5 M NaOH
to allow complete inactivation prior to disposal. Dimethylsulfate (DMS) (CAS # 77-78-1)
(purity ≥99.0%) was purchased as a 10.5 M solution from Fluka (Neu Ulm, Switzerland). It
was diluted 1/10,000 in PBS prior to use. Trichostatin A (TSA) (CAS # 58880-19-6)
(purity ≥98%) was obtained from Sigma-Aldrich (St. Louis, MO) and dissolved in DMSO
as a stock solution of 0.5 mg/mL. Aliquots of a 10 μg/mL dilution in DMSO of TSA were
prepared and kept at -20ºC until use.
4.2.3.3 Treatment of cells
Cells were trypsinized for 5 min at 37°C (NCI-H23 or fibroblasts) or directly taken from
the culture medium (U937 and lymphocytes). Approximately 6.25 × 104 cells for each
treatment condition were centrifuged 4 min at 3000 × g then suspended in 125 μL of
complete culture medium (0.5 × 106 cells/mL). The treatment doses were chosen based on
results published previously showing doses dependent increases of the percent tail DNA for
the same DNA damaging agents [21]. The DNA damage frequency had to be high enough
to allow a repair study, but in the range where the percent tail DNA measured is
proportional to the dose applied. It corresponded to (1) either 0.25 μM NDMAOAc (U937),
0.1 μM NDMAOAc (NCI-H23, lymphocytes and fibroblasts), 50 μM NNKOAc (all cell
types) for 2 h at 37°C in a cell incubator or (2) 6 min with 10 μM DMS (fibroblasts) at
room temperature or (3) 4 J/m2 UVB (fibroblasts) with a UV tube (FS20T12/UVB/BP,
Philips) filtered with a screen of cellulose acetate (Kodacel TA-407, clear 0.015-inch,
Eastman Kodak). After the treatment, cells were centrifuged at 3000 × g for 4 min, rinsed
with warm PBS then suspended in complete culture medium for 0, 0.5, 1, 2, 3, 6, 12 or
24 h. For each experiment, cells that were not treated but kept for 0 or 24 h in culture
medium were used as control in order to determine the level of background percent tail
DNA that was not related to the DNA damaging agent. When indicated, 10 ng/mL TSA

126
was added to the repair culture medium for 0–6 h. At each time post-treatment, 800 μL cold
PBS was added to the cells in culture medium. Cells were then centrifuged at 3000 × g for
4 min and suspended in Low Melting Point (LMP) agarose (Bio-Rad Laboratories,
Hercules, CA) then put on a slide for the comet assay.
4.2.3.4 Comet assay The procedure was performed as previously described [21]. Briefly, the cell pellet was
suspended in LMP agarose (1% in PBS kept at 37°C) and the agarose/cells mix
(50 μL / slide) was spread on slides with a 22 mm × 22 mm coverslip above it. After 5 min
at 4°C for solidification of the agarose, the coverslip was removed and the slides were
placed in a lysis solution (2.5 mM NaCl; 0.1 mM EDTA; 10 mM Tris-HCl pH 10, Triton
X-100 1%) at 4°C. The following steps were performed when all the slides corresponding
to all post-treatment times were collected in the lysis buffer. Lysis time was therefore >13 h
for all slides except for the 24 h post-treatment slides where it was only >1 h. After lysis,
slides were quickly rinsed with cold PBS and equilibrated with 3 × 5 min washes of
enzyme buffer (0.1 M KCl; 0.5 mM EDTA; 0.2 mg/mL bovine serum albumin (BSA);
40 mM HEPES-KOH pH 8). Each slide was then treated with 50 μL of enzyme buffer
alone or buffer containing endonuclease III (endo III) or formamidopyrimidine glycosylase
(fpg) under a coverslip in a humidified box at 37°C for 45 min. The same procedure was
used for the T4 endonuclease V (T4 endo V) digestion except that the enzyme buffer was
50 mM NaCl; 1 mM EDTA; 100 μg/mL BSA; Tris-HCl 10 mM pH 7.6 and the digestion
lasted 1 h. Following digestion, the slides were quickly rinsed with cold PBS and then
transferred for 40 min to an electrophoresis tank in a cold chamber containing the
electrophoresis solution (300 mM NaOH; 1 mM EDTA, pH >13) to allow DNA unwinding
and alkali-labile conversion of some lesions. Then a current of 0.3 V/cm was applied for
15 min. The slides were then neutralized with 3 × 5 min washing steps with Tris-HCl
pH 7.5, and stained with 50 μL SYBR Gold (Molecular Probes, Eugene, OR). Twenty-five
consecutive nucleoids in the centre of each slide were photographed using an Olympus
BX 61 microscope.

127
Endo III and fpg were kindly provided by Dr. Serge Boiteux and the T4 endo V by Dr. R.
Stephen Lloyd. They were all used at the same concentrations as previously reported [21].
4.2.3.5 Comets and statistical analyses TIFF format images were exported to be analyzed with the CASP software [39] to
determine the percent tail DNA of each nucleoid. For each slide, the median percent tail
DNA of 25 nucleoids was determined and the average and standard deviation of three
independent experiments (three slides) calculated. Two independent experiments were
performed for fibroblasts treated with DMS and lymphocytes treated with NNKOAc. When
indicated, a Student t-test was performed to determine the probability (p) of the null
hypothesis.
For each type of damage, the initial damage frequency (after the acute pro-mutagen dose) is
measured by the proportion of DNA in the tail. It corresponded to the average median
percent tail DNA measured for treated cells at 0 h minus the average median percent tail
DNA measured for non treated cells at 0 h (background); all other parameters, including
enzymatic digestion during the comet assay, being equal. The time to 50% repair for of
each type of damage corresponds to the time post-treatment when the percent tail DNA
associated with this type of damage has decreased of half of its initial value. In the case of
the fpg-sensitive damage associated with NNKOAc, the damage frequency at each time
post-treatment corresponded to the value of the percent tail DNA for fpg-digested slides
minus percent tail DNA for non digested slides (buffer). To follow more easily their repair,
the fpg-sensitive damage frequency was represented specifically on another graphic and the
initial damage frequency was brought to 100%.
4.2.4 Results
4.2.4.1 Repair kinetics of glycosylase-specific adducts in normal fibroblasts
Normal human skin fibroblasts were treated with 10 μM of DMS for 6 min at room
temperature (Fig. 2A) or irradiated with 4 J/m2 UVB (Fig. 2B), then allowed to repair in
complete culture medium for 0–24 h. At each time post-treatment, cells were harvested and

128
analyzed with a comet assay that included or not a digestion step with a specific
glycosylase.
Fig. 2A shows that the percent tail DNA in fpg-digested slides decreased regularly with
time after fibroblasts were treated with DMS. Neither breaks nor ALS or endo III-sensitive
sites could be detected from the background during the time course post-treatment (slides
treated with endo III in Fig. 2A), indicating that all the percent tail DNA obtained above the
background in the fpg-digested slides corresponded specifically to fpg-sensitive damage
that is not AP sites. Time to 50% repair was 12.5 h and all damage had disappeared after 24
h. Similarly, Fig. 2B shows the regular decrease of the percent tail DNA in UVB-irradiated
fibroblasts when slides were digested with T4 endo V. Time to 50% repair was 20 h and
≈30% of the damage remained after 24 h.
4.2.4.2 Post-treatment time courses of the DNA damage generated by NDMAOAc U937 and NCI-H23 correspond to cell types that proficiently activate NNK. U937 cells
were treated with 0.25 μM NDMAOAc, an NNK analog that generates specifically the
methylating reactive metabolite (Fig. 1), for 2 h then allowed to repair for 0–24 h in
complete culture medium before a comet assay including an endo III or an fpg digestion
was performed. As seen previously for DMS damage (Fig 2A), the absence of breaks or
ALS or endo III-sensitive sites (endo III-digested slides in Fig. 3A) indicates that the
percent tail DNA observed in fpg-digested slides corresponds specifically to fpg-sensitive
damage that is not AP sites. In U937 cells, the fpg-digested damage was half of its initial
frequency after 5.2 h and was at the background level after 24 h (Fig. 3A). NCI-H23 cells
had to be treated with a lower dose of NDMAOAc (0.1 μM) for the percent tail DNA to be
in a range analysable with the comet assay. At 6 h post-treatment and later, the fpg-
sensitive damage frequency in NCI-H23 cells (Fig. 3B) decreased as it did in U937 cells
(Fig. 3A), but the time course was rather complex within the first 5 h post-treatment in
NCI-H23 cells (Fig. 3B). The time to 50% repair was therefore difficult to determine
precisely in NCI-H23 cells but the fpg-sensitive damage frequency diminished clearly more
slowly than in U937 cells, as it was half of its initial value only after 9.8 h.

129
The post-treatment time courses of NDMAOAc-associated fpg-sensitive damage were also
realized for normal human skin fibroblasts and lymphocytes. Cells were treated for 2 h with
0.1 μM NDMAOAc and allowed to repair for 0–24 h. The fpg-sensitive damage frequency
in fibroblasts (Fig. 3C) and in lymphocytes (Fig. 3D) decreased with time similarly to what
was observed for NCI-H23 cells (Fig. 3B). The fpg-sensitive damage frequency was half of
its initial value at 10.2 h and 14.6 h post-treatment for fibroblasts and lymphocytes,
respectively.
4.2.4.3 Post-treatment time courses of the DNA damage generated by NNKOAc U937 cells, NCI-H23 cells, normal human fibroblasts and lymphocytes were treated for 2 h
with 50 μM NNKOAc, an NNK analog that generates specifically the pyridyloxobutylating
reactive metabolite (Fig. 1), and then allowed to repair for 0–24 h in complete culture
medium before a comet assay was performed; the assay included or not an fpg digestion.
NNKOAc generates direct breaks/ALS and fpg-sensitive damage (Fig. 1). A parallel
digestion with endo III was also tried for the assays in fibroblasts (data not shown). This
endo III digestion did not significantly increase the percent tail DNA at any time post-
treatment, indicating that AP sites did not contribute to the fpg-sensitive damage frequency.
Fig. 4 shows the evolution with time of the percent tail DNA in non-digested slides (buffer)
and in fpg-digested slides for each cell type tested. The direct breaks/ALS frequencies can
be followed directly in non-digested slides (buffer). They decreased quickly in U937 cells
(Fig. 4A) and in NCI-H23 cells (Fig. 4B) but not as much in fibroblasts (Fig. 4C). Their
time to 50% repair in these cells was 0.8 h, 0.9 h and 8.7 h, respectively.
The fpg-sensitive damage corresponded to the percent tail DNA that was added by the fpg
digestion and their repair is presented more specifically in Fig. 4. The fpg-sensitive damage
frequency induced by NNKOAc decreased continuously with time in U937 cells (Fig. 4A)
with a pattern that is very similar to that observed for the fpg-sensitive damage induced by
NDMAOAc in the same cell type (Fig. 3A). Their time to 50% repair was 8.5 h and the
percent tail DNA was at the background level after 24 h. Surprisingly, the fpg-sensitive
damage frequencies seemed to evolve differently in NCI-H23 (Fig. 4B) and in fibroblasts
(Fig. 4C) during the first 6 h post-treatment, making it difficult to quantify the repair in

130
these cells. In NCI-H23 and fibroblasts, the damage frequencies seemed to increase to
150% and 161% of their initial value at 2 h or 1 h post-treatment, respectively. Although
these increases were not statistically significant, they were totally reproducible, as they
could be observed for both cell types for each independent experiment at a similar intensity
and at the same time post-treatment. After this peak, the fpg-sensitive damage frequencies
decreased rapidly and became very similar to what could be observed for U937 cells at 6 h
post-treatment and later. In lymphocytes (Fig. 4D), the time course of the fpg-sensitive
damage frequency resembled more the one observed for U937, as it never increased above
the initial frequency. Still, the fpg-sensitive damage stopped decreasing after 6 h post-
treatment and were still at this level (≈6% tail DNA above the background) after 24 h.
4.2.4.4 Influence of the p53 status on the post-treatment time course of NNKOAc-associated fpg-sensitive damage in fibroblasts
The complex time courses observed for the fpg-sensitive damage frequencies during the
first 6 h post-treatment in NCI-H23 cells and fibroblasts (Fig. 4B and C) makes it unclear
what happens during the repair in these cells. Considering the implication of p53 in
numerous mechanisms associated with the cell response to genotoxic stress, we
investigated a possible influence of the p53 status on the post-treatment evolution of the
fpg-sensitive damage frequency. Lung fibroblasts transfected with an empty LXSN vector
or expressing the human papillomavirus (HPV) E6 oncoprotein (that functionally
inactivates p53) were used, as well as Li-Fraumeni p53 deficient skin fibroblasts (LF041
cells). All these cells have already been used to investigate the implication of p53 on DNA
repair [37].
Fig. 5 shows the post-treatment time courses of NNKOAc-related damage in LXSN-
transfected fibroblasts (Fig. 5A), E6-transfected fibroblasts (Fig. 5B) and LF041 fibroblasts
(Fig. 5C). Fibroblasts proficient for p53 (Fig. 5A) showed an initial increase in fpg-
sensitive damage frequency similar to what was observed previously for NCI-H23 and
normal fibroblasts (Fig. 4B and C), whereas fibroblasts deficient for p53 (Fig. 5B and C)
showed no such initial increase. Once again, the increase in fpg-sensitive damage frequency
observed in LXSN-transfected fibroblasts was not statistically significant but was totally
reproducible.

131
4.2.4.5 Influence of chromatin relaxation on the post-treatment time course of NNKOAc-associated fpg-sensitive damage in fibroblasts
Trichostatin A (TSA), an histone deacetylase inhibitor, can mimic the type of global
chromatin relaxation that happens following a genotoxic stress [40]. To investigate whether
chromatin relaxation is an event that can influence the post-treatment time course of
NNKOAc-associated fpg-sensitive damage, both normal and LF041 fibroblasts were
treated for 2 h with 50 μM NNKOAc, then allowed to repair for 0–6 h in complete culture
medium containing 10 ng/mL TSA (Fig. 6A and B). Fig. 6C and D shows the time course
of the calculated fpg-sensitive damage frequencies for these experiments plus those
previously obtained (already shown in Figs. 4C and 5C) for the same cell types without
TSA. In normal fibroblasts (Fig. 6C), the increase in fpg-sensitive damage frequency post-
treatment seemed at first attenuated by the presence of TSA, as the peak in damage
frequency still existed at 1 h post-treatment but reached only 118% of the initial value
versus 160% in non-treated cells. At 2 h post-treatment, the fpg-sensitive damage
frequency started to increase regularly, so that at 6 h post-treatment, it was higher in TSA
treated cells (180% of the original damage frequency) than in non-treated cells (95%). The
effect was similar in LF041 cells (Fig. 6D) as TSA made, at first, the fpg-sensitive damage
frequency decreased faster (until 1 h post-treatment) but increased later with a peak at 3 h
post-treatment, when it was 150% of the original damage frequency versus 74% for non-
treated cells. Overall, the presence of TSA during the repair seemed to diminish at first the
fpg-sensitive damage frequency while it had a rather opposite effect later during the time
course, when the damage frequency is increased in TSA treated cells versus non-treated
cells. Interestingly, this effect was specific to the fpg-sensitive damage as in the same cells,
the time courses of the direct breaks/ALS frequencies were not affected by either the p53
status or the presence of TSA (Figs. 4C, 5C, 6A and B).

132
4.2.5 Discussion We demonstrate in this report that the comet assay can be used to quantify the repair
kinetics of very low damage frequencies of specific types of DNA damage such as 7-mG or
CPD. When this technique was applied to investigate the repair kinetics of specific types of
NNK-associated damage, differences in repair between cell types could be observed for
each type of adducts investigated individually and for several of them, the post-treatment
time course of damage frequency seemed to be affected by formation of new lesions and
therefore did not represent simply repair kinetics.
The repair in normal fibroblasts of DNA damage commonly studied (generated by DMS
and UVB) was first investigated. DMS quickly produces a high frequency of 7-mG adducts
that are converted by the lysis step into fpg-sensitive sites [21,33], whereas UVB generates
CPD that can be detected as T4 endo V-sensitive sites using the comet assay. The time
courses obtained for these glysosylase-sensitive sites corresponded therefore to repair
kinetics for 7-mG and CPD in fibroblasts (Fig. 2A and B). The repair of the damage
generated by ≈250 times the dose of DMS we used in the present experiment has already
been studied [41] as well as the repair of higher frequencies of CPD. The later have been
shown to be repaired more quickly when the adduct frequency is lower because of the
possibility to saturate the repair capacity of the cells [42]. In the present experiments, we
show repair kinetics for adduct frequencies that are far below the saturation of the repair
capacities of the cells. These kinetics should therefore represent what happens in cells
exposed to low doses of DNA damaging agent. Moreover, the repair kinetics were clearly
different for 7-mG and CPD, and could be used as references for archetypical DNA repair
by base excision repair (7-mG) or nucleotide excision repair (CPD) at very low damage
frequency in fibroblasts.
We applied the comet assay to study repair kinetics of NNK-associated damage in different
cell types, including in cells that activate NNK (U937 and NCI-H23 cells) and in normal
cells frequently used to study DNA repair (fibroblasts and lymphocytes). The most
common adduct generated by methylation in cells that bioactivate NNK is 7-mG.
NDMAOAc, an NNK analog that generates selectively the reactive methylating metabolite

133
(Fig. 1), was used to treat all cell types and the frequency of 7-mG (as fpg-sensitive sites)
was then followed for 24 h. Surprisingly, the time courses observed for these NDMAOAc-
produced 7-mG adducts did not correspond to simple repair kinetics for most cell types
investigated (Fig. 3). It is especially intriguing for fibroblasts as the post-treatment time
course of these NDMAOAc-induced 7-mG adducts would be expected to be exactly the
same as for the DMS-produced 7-mG (Fig. 2A). These results are consistent with the fact
that NDMAOAc is a pro-mutagen that requires hydrolysis in cells to generate reactive
alkylating SN1 metabolites (Fig. 1), whereas alkylating SN2 DMS reacts directly with
DNA. So what was seen within the first hours post-treatment in NDMAOAc-treated cells
likely results of a balance between repair and formation of new 7-mG because of an
intracellular pro-mutagen pool continuously being converted to reactive metabolites. This
complicates comparison of repair kinetics between cell types, but considering the initial
adduct frequency in each cell type as well as the time necessary for the damage frequency
to decrease by 50 %, it is still possible to conclude that differences exist in the repair
capacities of these cells. U937 cells seemed to repair 7-mG quite fast. On the other hand,
lymphocytes repaired 7-mG quite slowly and the damage frequency kept increasing post-
treatment: it was significantly higher (p <0.02) at 3 h post-treatment than at 0 h (Fig. 3D),
which indicates that the formation of new damage was faster than the repair in
lymphocytes, even after the damaging agent was removed from the culture medium. This is
consistent with our previous results indicating that lymphocytes accumulate more
NDMAOAc-produced 7-mG than the other cell types [21]. Finally, NCI-H23 cells and
normal fibroblasts seemed to have an intermediate repair capacity for 7-mG, that is to say
they repaired faster than lymphocytes but more slowly than U937 cells. Overall, these
differences observed between cell types indicate that the rate at which the damage is
formed can influence the time course of the damage post-treatment, even after an acute
dose.
NNKOAc, a NNK analog that generates a reactive pyridyloxobutylating metabolite, was
also used to generate damage in the same cell types. The types of damage that can be
investigated using the comet assay are direct breaks/ALS and fpg-sensitive damage. [21].
As seen previously for 7-mG, the cell types had an influence on the repair that could be
observed. In lymphocytes, breaks accumulated post-treatment (Fig. 4D) in a way that

134
resembles what happens during the repair of UV irradiated lymphocytes [43]. This has been
associated with a shortage of deoxynucleotides available in these non-dividing cells, that
results in a deficiency in DNA repair synthesis that make excision breaks persist longer.
This accumulation of breaks should therefore reflects events in relation with repair more
than the formation of new damage post-treatment. In the other cell types, the time courses
post-treatment for direct breaks/ALS (Fig. 4) corresponded to classical repair kinetics,
although the repair seemed clearly faster in U937 and NCI-H23 cells (half-life <1 h) than in
fibroblasts (half-life ≈9 h). Simple repair kinetics are consistent with our hypothesis that
direct breaks/ALS could correspond to one single type of damage and also suggest that,
even if NNKOAc requires hydrolysis in the cells in order to generate DNA damage (Fig.
1), pro-mutagen activation did not participate in what was observed after termination of the
NNKOAc treatments. A rapid repair of direct breaks/ALS is not necessarily consistent with
data in the literature concerning repair of NNK-associated breaks. Notably, breaks in the
liver DNA of rodents treated with NNK were shown to persist up to 2-3 weeks [44].
Moreover, DNA treated in vitro with NNKOAc showed breaks presenting a blocking group
in 3' that could prevent repair [38]. Still, the differences in NNK-associated damage
formation and repair that have been shown between species (mice versus rat versus
humans) [13] and between cell types within the same species, as we showed here, indicate
that it is difficult to imply conclusions from results obtained with a different animal model.
The time course post-treatment of the fpg-sensitive damage generated by NNKOAc was
clearly more complex than what was observed for the direct-breaks/ALS followed
simultaneously in the same cells. For U937 and lymphocytes, the fpg-sensitive damage
frequency decreased more or less regularly after the end of the treatment, whereas NCI-H23
and fibroblasts seemed to show first a rapid increase in the fpg-sensitive damage frequency
before the decrease. The initial increase in fpg-sensitive damage frequency post-treatment
can be associated with p53 proficiency in fibroblasts, whether they originate from skin
(normal or LF041 fibroblasts) or lung (LXSN and E6-transfected fibroblasts). But this
association cannot be easily generalized to other cell types tested, as normal lymphocytes
(p53 proficient) did not show it, whereas NCI-H23 cells (p53 deficient because of a
missense mutation in codon 246 [36]) did present the increase in fpg-sensitive site
frequency. Regardless of its cause, a complex evolution of the fpg-sensitive damage

135
frequency during the 6 h post-treatment reminds of what was observed for the time course
of the 7-mG generated with NDMAOAc in the same cells. But in that case, another type of
damage followed simultaneously (namely direct breaks/alkali-labile sites) had rather simple
repair kinetics, making it unlikely that formation of new damage, due to activation of new
NNKOAc pro-mutagen after the end of the treatment, is responsible for the observed
phenomenon. An alternative explanation could be that the fpg-sensitive damage that is
detected correspond to a secondary lesions. Therefore, the time course post-treatment that is
observed could measure several events happening simultaneously such as the conversion of
primary lesions (not recognized by the fpg enzyme) into secondary lesions (fpg-sensitive)
and the repair of both primary and secondary lesions. The hypothesis of the fpg-sensitive
damage being secondary damage is consistent with our previous suggestion that they
correspond to the secondary formamidopyrimidine form of 7-pobG generated in cells (Fig.
1). But this needs to be confirmed through the use of alternative methods, such as liquid
chromatography with tandem mass spectrometry, that will identify unambiguously the
nature of the damage that is observed.
Our results also suggest that in fibroblasts, p53 proficiency or chromatin relaxation
(provoked by an histone deacetylase inhibitor during the repair) could influence the time
course post-treatment of the fpg-sensitive damage associated with pyridyloxobutylation.
Although the mechanism is not clear on how this could influence what was measured in
cells, they could both be connected to an influence on the repair rate of some of the
damage. If the fpg-sensitive damage is actually secondary lesions, the influence on repair
could happen on the primary adduct or the secondary or both. The use of analytic methods
that clearly identify the nature of the damage should allow a better identification and a
better understanding of the observed phenotype. But it is conceivable that the influence of
p53 and chromatin relaxation could be connected to the same mechanism. In fact, global
chromatin relaxation following a genotoxic stress (such as a UV irradiation) happens in
order to favor the access of the repair machinery to the damaged DNA present in condensed
chromatin [45]. This mechanism was shown to be p53-dependent and to occur at UV doses
that are insufficient to activate p53 as a transcription factor [45], indicating that the
mechanism is transcription independent and that it can be triggered by very low damage
frequencies.

136
In summary, we have shown in this work that the comet assay can be used to specifically
assess the repair kinetics of some rare damage. The repair kinetics of DMS-induced 7-mG
and UV-induced CPD can be easily obtained and could be used as archetypical repair in
fibroblasts by base excision repair or nucleotide excision repair, respectively. The
investigation of the repair of NNK-associated DNA damage also showed that differences
between cell types for the repair of the same type of DNA damage can be shown with this
technique. But it seems that for some types of damage, the quantification of the repair can
be made difficult due to the formation of new lesions after the end of the treatment, and
therefore that what is measured do not simply represent repair kinetics.

137
4.2.6 Acknowledgements The authors are grateful to Dr. Jean-François Cloutier for the synthesis of NNKOAc, Dr.
Serge Boiteux for kindly supplying the endonuclease III and the formamidopyrimidine
glycosylase, and Dr. R. Stephen Lloyd for the T4 endonuclease V. The authors sincerely
thank Drs. Gerald P. Holmquist, Marc-Edouard Mirault and Timothy R. O’Connor for
important discussion and carefully editing the manuscript. The research project was funded
by the Institute of Genetics of the CIHR and the Canada Research Chairs Program. R.D.
holds the Canada Research Chair in « Genetics, Mutagenesis and Cancer ».

138
4.2.7 References [1] S.S. Hecht. Tobacco smoke carcinogens and lung cancer, J. Natl. Cancer Inst. 91
(1999) 1194-1210.
[2] G.N. Wogan, S.S. Hecht, J.S. Felton, A.H. Conney, L.A. Loeb. Environmental and
chemical carcinogenesis, Semin. Cancer Biol. 14 (2004) 473-486.
[3] S.S. Devesa, G.L. Shaw, W.J. Blot. Changing patterns of lung cancer incidence by
histological type, Cancer Epidemiol. Biomarkers Prev. 1 (1991) 29-34.
[4] D. Hoffmann, A. Rivenson, S.S. Hecht. The biological significance of tobacco-
specific N-nitrosamines: smoking and adenocarcinoma of the lung, Crit. Rev. Toxicol. 26
(1996) 199-211.
[5] S.S. Hecht, C.B. Chen, T. Ohmori, D. Hoffmann. Comparative carcinogenicity in
F344 rats of the tobacco-specific nitrosamines, N'-nitrosonornicotine and 4-(N-methyl-N-
nitrosamino)-1-(3-pyridyl)-1-butanone, Cancer Res. 40 (1980) 298-302.
[6] S.S. Hecht, N. Trushin, A. Castonguay, A. Rivenson. Comparative tumorigenicity
and DNA methylation in F344 rats by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and
N-nitrosodimethylamine, Cancer Res. 46 (1986) 498-502.
[7] D. Hoffmann, A. Rivenson, S. Amin, S.S. Hecht. Dose-response study of the
carcinogenicity of tobacco-specific N-nitrosamines in F344 rats, J. Cancer Res. Clin.
Oncol. 108 (1984) 81-86.
[8] L.A. Peterson, S.S. Hecht. O6-methylguanine is a critical determinant of 4-
(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung, Cancer
Res. 51 (1991) 5557-5564.
[9] G.B. Smith, A. Castonguay, P.J. Donnelly, K.R. Reid, D. Petsikas, T.E. Massey.
Biotransformation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-
1-butanone (NNK) in freshly isolated human lung cells, Carcinogenesis 20 (1999) 1809-
1818.

139
[10] G.B. Smith, J.R. Bend, L.L. Bedard, K.R. Reid, D. Petsikas, T.E. Massey.
Biotransformation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in peripheral
human lung microsomes, Drug Metab. Dispos. 31 (2003) 1134-1141.
[11] P.G. Foiles, S.A. Akerkar, S.G. Carmella, M. Kagan, G.D. Stoner, J.H. Resau, S.S.
Hecht. Mass spectrometric analysis of tobacco-specific nitrosamine-DNA adducts in
smokers and nonsmokers, Chem. Res. Toxicol. 4 (1991) 364-368.
[12] S. Kato, E.D. Bowman, A.M. Harrington, B. Blomeke, P.G. Shields. Human lung
carcinogen-DNA adduct levels mediated by genetic polymorphisms in vivo, J. Natl. Cancer
Inst. 87 (1995) 902-907.
[13] S.S. Hecht. Biochemistry, biology, and carcinogenicity of tobacco-specific N-
nitrosamines, Chem. Res. Toxicol. 11 (1998) 559-603.
[14] L. Liu, X. Qin, S.L. Gerson. Reduced lung tumorigenesis in human methylguanine
DNA--methyltransferase transgenic mice achieved by expression of transgene within the
target cell, Carcinogenesis 20 (1999) 279-284.
[15] J. Engelbergs, J. Thomale, M.F. Rajewsky. Role of DNA repair in carcinogen-
induced ras mutation, Mutat. Res. 450 (2000) 139-153.
[16] G.T. Pauly, L.A. Peterson, R.C. Moschel. Mutagenesis by O(6)-[4-oxo-4-(3-
pyridyl)butyl]guanine in Escherichia coli and human cells, Chem. Res. Toxicol. 15 (2002)
165-169.
[17] R.S. Mijal, N.A. Loktionova, C.C. Vu, A.E. Pegg, L.A. Peterson. O6-
pyridyloxobutylguanine adducts contribute to the mutagenic properties of
pyridyloxobutylating agents, Chem. Res. Toxicol. 18 (2005) 1619-1625.
[18] D.R. Lloyd, P.C. Hanawalt. p53-dependent global genomic repair of
benzo[a]pyrene-7,8-diol-9,10-epoxide adducts in human cells, Cancer Res. 60 (2000) 517-
521.

140
[19] M.A. Wani, Q. Zhu, M. El-Mahdy, S. Venkatachalam, A.A. Wani. Enhanced
sensitivity to anti-benzo(a)pyrene-diol-epoxide DNA damage correlates with decreased
global genomic repair attributable to abrogated p53 function in human cells, Cancer Res. 60
(2000) 2273-2280.
[20] D.R. Lloyd, P.C. Hanawalt. p53 controls global nucleotide excision repair of low
levels of structurally diverse benzo(g)chrysene-DNA adducts in human fibroblasts, Cancer
Res. 62 (2002) 5288-5294.
[21] S. Lacoste, A. Castonguay, R. Drouin. Formamidopyrimidine adducts are detected
using the comet assay in human cells treated with reactive metabolites of 4-
(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), Mutat. Res. 600 (2006) 138-149.
[22] M.E. Smela, M.L. Hamm, P.T. Henderson, C.M. Harris, T.M. Harris, J.M.
Essigmann. The aflatoxin B(1) formamidopyrimidine adduct plays a major role in causing
the types of mutations observed in human hepatocellular carcinoma, Proc. Natl. Acad. Sci.
USA 99 (2002) 6655-6660.
[23] A.R. Collins, S.J. Duthie, V.L. Dobson. Direct enzymic detection of endogenous
oxidative base damage in human lymphocyte DNA, Carcinogenesis 14 (1993) 1733-1735.
[24] A.R. Collins, D.L. Mitchell, A. Zunino, J. de Wit, D. Busch. UV-sensitive rodent
mutant cell lines of complementation groups 6 and 8 differ phenotypically from their
human counterparts, Environ. Mol. Mutagen. 29 (1997) 152-160.
[25] K.J. Angelis, M. Dusinska, A.R. Collins. Single cell gel electrophoresis: detection
of DNA damage at different levels of sensitivity, Electrophoresis 20 (1999) 2133-2138.
[26] M.P. Sastre, M. Vernet, S. Steinert. Single-cell gel/comet assay applied to the
analysis of UV radiation-induced DNA damage in Rhodomonas sp. (Cryptophyta),
Photochem. Photobiol. 74 (2001) 55-60.
[27] M. Dusinska, A. Collins, A. Kazimirova, M. Barancokova, V. Harrington, K.
Volkovova, M. Staruchova, A. Horska, L. Wsolova, A. Kocan, J. Petrik, M. Machata, B.

141
Ratcliffe, S. Kyrtopoulos. Genotoxic effects of asbestos in humans, Mutat. Res. 553 (2004)
91-102.
[28] J. Blasiak, M. Arabski, R. Krupa, K. Wozniak, M. Zadrozny, J. Kasznicki, M.
Zurawska, J. Drzewoski. DNA damage and repair in type 2 diabetes mellitus, Mutat. Res.
554 (2004) 297-304.
[29] M. Arabski, G. Klupinska, J. Chojnacki, P. Kazmierczak, M. Wisniewska-
Jarosinska, J. Drzewoski, J. Blasiak. DNA damage and repair in Helicobacter pylori-
infected gastric mucosa cells, Mutat. Res. 570 (2005) 129-135.
[30] C.C. Smith, R. O'Donovan M, E.A. Martin. hOGG1 recognizes oxidative damage
using the comet assay with greater specificity than FPG or ENDOIII, Mutagenesis (2006).
[31] P. Fortini, G. Raspaglio, M. Falchi, E. Dogliotti. Analysis of DNA alkylation
damage and repair in mammalian cells by the comet assay, Mutagenesis 11 (1996) 169-
175.
[32] A.R. Collins, M. Dusinska, A. Horska. Detection of alkylation damage in human
lymphocyte DNA with the comet assay, Acta Biochim. Pol. 48 (2001) 611-614.
[33] G. Speit, P. Schutz, I. Bonzheim, K. Trenz, H. Hoffmann. Sensitivity of the FPG
protein towards alkylation damage in the comet assay, Toxicol. Lett. 146 (2004) 151-158.
[34] A.R. Collins, A.G. Ma, S.J. Duthie. The kinetics of repair of oxidative DNA
damage (strand breaks and oxidised pyrimidines) in human cells, Mutat. Res. 336 (1995)
69-77.
[35] K. Sugimoto, H. Toyoshima, R. Sakai, K. Miyagawa, K. Hagiwara, F. Ishikawa, F.
Takaku, Y. Yazaki, H. Hirai. Frequent mutations in the p53 gene in human myeloid
leukemia cell lines, Blood 79 (1992) 2378-2383.
[36] P.M. O'Connor, J. Jackman, I. Bae, T.G. Myers, S. Fan, M. Mutoh, D.A. Scudiero,
A. Monks, E.A. Sausville, J.N. Weinstein, S. Friend, A.J. Fornace, Jr., K.W. Kohn.
Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer

142
Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123
anticancer agents, Cancer Res. 57 (1997) 4285-4300.
[37] J.P. Therrien, R. Drouin, C. Baril, E.A. Drobetsky. Human cells compromised for
p53 function exhibit defective global and transcription-coupled nucleotide excision repair,
whereas cells compromised for pRb function are defective only in global repair, Proc. Natl.
Acad. Sci. USA 96 (1999) 15038-15043.
[38] J.F. Cloutier, R. Drouin, M. Weinfeld, T.R. O'Connor, A. Castonguay.
Characterization and mapping of DNA damage induced by reactive metabolites of 4-
(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) at nucleotide resolution in human
genomic DNA, J. Mol. Biol. 313 (2001) 539-557.
[39] K. Konca, A. Lankoff, A. Banasik, H. Lisowska, T. Kuszewski, S. Gozdz, Z. Koza,
A. Wojcik. A cross-platform public domain PC image-analysis program for the comet
assay, Mutat. Res. 534 (2003) 15-20.
[40] C.P. Rubbi, J. Milner. p53 is a chromatin accessibility factor for nucleotide excision
repair of DNA damage, EMBO J. 22 (2003) 975-986.
[41] N. Ye, G.P. Holmquist, T.R. O'Connor. Heterogeneous repair of N-methylpurines at
the nucleotide level in normal human cells, J. Mol. Biol. 284 (1998) 269-285.
[42] S. Courdavault, C. Baudouin, S. Sauvaigo, S. Mouret, S. Candeias, M. Charveron,
A. Favier, J. Cadet, T. Douki. Unrepaired cyclobutane pyrimidine dimers do not prevent
proliferation of UV-B-irradiated cultured human fibroblasts, Photochem. Photobiol. 79
(2004) 145-151.
[43] M.H. Green, A.P. Waugh, J.E. Lowe, S.A. Harcourt, P.H. Clingen, J. Cole, C.F.
Arlett. Protective effect of deoxyribonucleosides on UV-irradiated human peripheral blood
T-lymphocytes: possibilities for the selective killing of either cycling or non-cycling cells,
Mutat. Res. 350 (1996) 239-246.

143
[44] R. Jorquera, A. Castonguay, H.M. Schuller. DNA single-strand breaks and toxicity
induced by 4-(methyl-nitrosamino)- 1-(3- pyridyl)-1-butanone or N-nitrosodimethylamine
in hamster and rat liver, Carcinogenesis 15 (1994) 389-394.
[45] S.J. Allison, J. Milner. Remodelling chromatin on a global scale: a novel protective
function of p53, Carcinogenesis 25 (2004) 1551-1557.

144
4.2.8 Legends to figures Figure 1 Bioactivation of NNK and NNK analogs in cells. NDMAOAc leads to the
formation of 7-mG that are detected with the comet assay as fpg-sensitive sites after the
lysis step at pH 10. NNKOAc leads to the formation of alkylphophotriesters and fpg-
sensitive damage. The latter could be 7-pob-fapy adducts but their formation is hypothetical
and remains to be proven.
Figure 2 Time course of the percent tail DNA measured using the comet assay in
normal fibroblasts treated with (A) 10 μM DMS for 6 min at room temperature or (B)
4 J/m2 UVB, then allowed to repair for 0–24 h in complete culture medium. To generate
breaks at specific adducts, the slides were treated with (A) endo III or fpg or (B) T4 endo V
enzyme. Data represent the averages (± S.D.) for (A) two or (B) three independent
experiments.
Figure 3 Time course of the percent tail DNA measured using the comet assay in cells
treated with NDMAOAc. (A) U937 cells were treated with 0.25 μM NDMAOAc for 2 h.
(B) NCI-H23 cells, (C) fibroblasts and (D) lymphocytes were treated with 0.1 μM
NDMAOAc for 2 h. All cells were allowed to repair for 0–24 h after the treatment in
complete culture medium. During the comet assay, slides were either digested with endo III
or fpg. Data represent the averages (± S.D.) for three independent experiments.
Figure 4 Time course of the percent tail DNA measured using the comet assay in cells
treated with 50 μM NNKOAc for 2 h then allowed to repair for 0–24 h in complete culture
medium. (A) U937 cells, (B) NCI-H23 cells, (C) fibroblasts and (D) lymphocytes. During
the comet assay, slides were either non-digested (buffer) or digested with fpg. Data
represent the averages (± S.D.) for three independent experiments. The fpg-sensitive
damage corresponds specifically at each time post-treatment to the percent tail DNA for
fpg-digested slides minus the percent tail DNA for buffer-treated slides.
Figure 5 Time course of the percent tail DNA measured using the comet assay in
fibroblasts of known p53 status. Normal lung fibroblasts transfected with (A) LXSN alone

145
or (B) LXSN expressing E6 are therefore p53 proficient and p53 deficient, respectively. (C)
LF041 fibroblasts are p53 deficient cells. All cells were treated with 50 μM NNKOAc for
2 h then allowed to repair for 0–24 h in complete culture medium. During the comet assay,
slides were either non-digested (buffer) or digested with fpg. Data represent the averages
(± S.D.) for three independent experiments.
Figure 6 Time course of the percent tail DNA measured using the comet assay in (A)
normal or (B) LF041 fibroblasts that were treated with 50 μM NNKOAc for 2 h then
allowed to repair for 0–6 h in complete culture medium containing 10 ng/mL of TSA.
During the comet assay, slides were either non-digested (buffer) or digested with fpg. Data
represent the averages (± S.D.) for three independent experiments. The time course for fpg-
sensitive damage specifically, in presence of TSA or not, is presented for (C) normal and
(D) LF041 fibroblasts with the percent tail DNA normalized to be 100% at 0 h post-
treatment.

146
Figure 1
NNK
α-methyl hydroxylation
α-methylene hydroxylation
NNKOAc NDMAOAc
pyridyloxobutyldiazohydroxide methyldiazohydroxide REACTIVE METABOLITES
esterase esterase
CYP450
7-pobG
alkylphosphotriesters other types of
pyridyloxobutylation damage
secondary transformation ?
fpg-sensitive
7-pobfapy
7-mG
O6-mG O4-mT
comet assay lysis step pH 10
7-mfapy
?

147
Figure 2

148
Figure 3
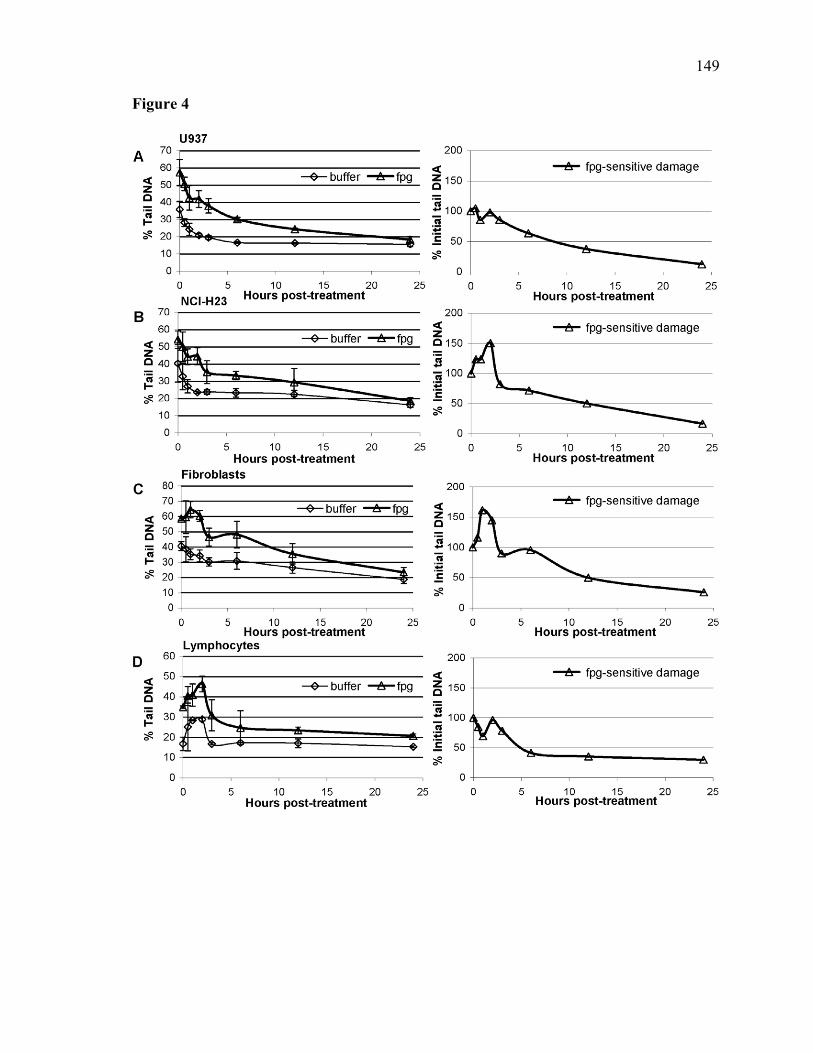
149
Figure 4

150
Figure 5

151
Figure 6

152
Chapitre 5 Discussion
La NNK participe au développement des cancers pulmonaires associés à la consommation
de tabac et en particulier des adénocarcinomes (Hecht, 1998 ; Hecht, 1999b). Cependant,
comme la fumée de cigarettes contient de nombreuses substances génotoxiques, il est très
complexe de distinguer les effets carcinogènes de la NNK de ceux de ses autres
composantes. Pour déterminer en quoi cette substance particulière participe à la
transformation maligne chez l'homme, il faut connaître le type et la fréquence des
dommages associés à la NNK générés dans les cellules humaines.
On peut estimer la probabilité pour un type de dommages donné de participer à générer les
mutations présentes dans les tumeurs si l'on connait : (1) la fréquence et le lieu de
formation de ces dommages, (2) leur vitesse de réparation, (3) le potentiel pour les
polymérases de placer la mauvaise base lors de la réplication d'un brin d'ADN présentant ce
type de dommages et enfin (4) les altérations génétiques retrouvées dans les tumeurs pour
déterminer si elles peuvent être générées par le type de dommages en question. Ces
investigations ont été réalisées pour les O6-akylguanines (Rodenhuis et Slebos, 1992 ;
Cooper et al., 1997) mais restent à faire pour la plupart des autres adduits associés à la
NNK, en particulier ceux liés à la pyridyloxobutylation les plus récemment identifiés.
Avec le travail présenté dans cette thèse, plusieurs types de dommages associés à la NNK
ont été investigués à faible fréquence et de manière spécifique à l'aide du test des comètes :
(1) les 7-mG résultant de la méthylation de l'ADN, ainsi que (2) les cassures directes / sites
alcali labiles et (3) les 7-pob-fapy dérivant de la pyridyloxobutylation de l'ADN. Des
résultats concernant la fréquence de formation de ces dommages et leur vitesse de
réparation ont été obtenus qui peuvent servir de base pour discuter de leur éventuelle
participation dans la mutagenèse associée à la NNK.
Les 7-mG sont des dommages d'alkylation souvent étudiés, mais qui ne sont pas
nécessairement pertinents en terme de mutagenèse étant donné qu'ils ne sont ni
cytotoxiques, ni mutagènes. Cependant, ils peuvent subir une dépurination spontanée, et
mener ainsi à la formation de sites AP qui sont eux, cytotoxiques et mutagènes. Nous avons

153
montré qu'à des fréquences de dommages faibles, il n'y a pas accumulation de sites AP
après un traitement méthylant, mais plutôt une disparition progressive des 7-mG due à leur
réparation. La présence de ce type d'adduits a été montrée dans les poumons de fumeurs
(Hecht, 1998). Cependant, comme ils ne sont pas mutagènes en tant que tels et que les
cellules semblent capables de les réparer sans qu'il y ait accumulation de sites AP, il est peu
probable que les 7-mG participent significativement à la mutagenèse associée à la NNK.
Les cassures directes / sites alcali labiles détectés par le test des comètes sont fréquents
dans les cellules traitées avec un analogue pyridyloxobutylant de la NNK. La réparation de
ces dommages s'est avérée très rapide (demi-vie < 1 h) dans les cellules d'une lignée
histiocytique (U937) et dans les cellules d'une lignée dérivée d'un adénocarcinome
pulmonaire (NCI-H23) alors qu'elle est plus lente dans des fibroblastes de peau normaux
(demi-vie ≈ 9 h). Ces dommages dérivent très vraisemblablement de la formation de
pyridyloxobutylphosphotriesters, c'est-à-dire de l'alkylation des phosphates du squelette
sucre / phosphate de l'ADN. La proportion de ces dommages détectés par le test des
comètes correspondant à des CMC versus de simples adduits aux phosphates n'est pas
clairement établie, vu que ce test n'est pas capable de les distinguer. Mais les deux sont
susceptibles d'avoir des conséquences dans les cellules. Les CMC sont habituellement
réparées très rapidement à cause des risques de perte de matériel génétique associés avec la
réplication d'un ADN cassé (Caldecott, 2001 ; Khanna et Jackson, 2001). La formation de
ces cassures pourrait impliquer la NNK dans les pertes d'hétérozygoties associées avec le
développement du cancer du poumon (cf. section 1.5). En accord avec cette hypothèse, une
étude récente a montré une quantité plus importante d'aberrations chromosomiques dans les
adénocarcinomes de souris induits par la NNK par rapport aux tumeurs spontanées (Herzog
et al., 2006). Il a également été montré qu'un analogue pyridyloxobutylant de la NNK,
utilisé à des doses très faibles, mène à une quantité importante de changements dans le
caryotype de cellules pulmonaires humaines en culture (Herzog et al., 2006). En ce qui
concerne les pyridyloxobutylphosphotriesters qui ne mèneraient pas à des CMC, la manière
dont ils sont réparés dans les cellules n'est pas encore clairement connue. Contrairement à
ce qui se passe chez les bactéries, il n'existe pas dans les cellules de mammifères une
protéine alkylguanine transférase capable de réaliser la réversion des méthylations aux
phosphates (Margison et Santibanez-Koref, 2002). Ces derniers doivent donc être pris en

154
charge par un autre système de réparation. Il a été montré que les méthylphosphotriesters
peuvent être détectés et excisés in vitro par la machinerie du système de REN (Branum et
al., 2001) mais il reste à prouver que ce type de réparation intervient effectivement dans les
cellules. Un adduit sur un phosphate n'est pas a priori un problème pour le maintien de
l'intégrité du génome, puisque les phosphates ne portent pas l'information génétique.
Cependant, les conséquences peuvent être importantes si le groupement provoque une
distorsion de la double hélice d'ADN. En effet, cela s'accompagne généralement d'une
perturbation locale dans l'appariement entre bases azotées. Il est alors concevable, même si
cela reste à démontrer, que de tels dommages puissent participer à la formation de
mutations ponctuelles lors de la réplication d'un brin endommagé.
Les 7-pobG sont des adduits fréquents associés à la pyridyloxobutylation de l'ADN et qui
relâchent le HPB. Leur formation a été montrée sur de l'ADN pyridyloxobutylé in vitro
(Wang et al., 2003 ; Sturla et al., 2005) et dans les tissus de rat traités à la NNK (Lao et al.,
2006). On s'attend à ce que de tels dommages soient générés dans n’importe quel type
cellulaire traité avec un agent pyridyloxobutylant. Faute d’avoir une méthode spécifique,
nous n’avons pas pu les détecter avec nos tests des comètes. Cependant, nous avons
observé des dommages sensibles à la Fpg, qui sont vraisemblablement la forme secondaire
fapy de ces 7-pobG. Comme il s’agit d’une démonstration indirecte, la nature des
dommages sensibles à la Fpg devra être confirmé par des méthodes d’analyse plus
spécifiques, telle que la chromatographie alliée à la spectrométrie de masse. On sait très peu
de choses sur les 7-pobG et sur leur forme fapy, si ce n'est qu'il s'agit d'adduits encombrants
qui ressemblent aux adduits générés en N7 par l'aflatoxine B1. Ces derniers sont mutagènes
en générant des transversions G → T et il pourrait donc en être de même pour ces adduits
de la NNK, même si cela reste à démontrer. Les transversions G → T dans TP53 sont les
mutations caractéristiques retrouvées dans les tumeurs pulmonaires associées à la
consommation de tabac. Elles sont habituellement attribuées à la formation de dommages
liés au benzo[a]pyrène aux positions nucléotidiques correspondantes de TP53. Cependant,
il est concevable que les 7-pobG et/ou leur forme fapy puissent contribuer à la formation de
telles mutations. Il ne semble pas y avoir une formation préférentielle de 7-pobG au niveau
des séquences correspondant aux points chauds de mutation de TP53 (Rajesh et al., 2005)

155
mais il reste à vérifier ce qui se passe effectivement dans les cellules, étant donné
l'importance que peut revêtir le contexte chromatinien et/ou épigénétique dans la
probabilité de formation des dommages. On ne dispose pas d'un mode spécifique de
détection des 7-pobG et il serait donc complexe d'étudier le lieu et la fréquence de
formation de ces dommages avec la technologie LMPCR (cf. section 1.8.1), par exemple
dans TP53. Cependant, ce type d’analyse pourrait être réalisé pour investiguer la forme 7-
pob-fapy qui est reconnue spécifiquement par la glycosylase Fpg.
En suivant l'évolution avec le temps de la fréquence des adduits fapy (sensibles à la Fpg)
après un traitement pyridyloxobutylant, nous avons observé un phénotype lié à p53 dans les
cellules. En effet, si ces adduits fapy semblent se former à une fréquence comparable dans
toutes les cellules après une pyridyloxobutylation, certaines cellules présentent une
augmentation importante de la fréquence de ces dommages juste après la fin du traitement.
L’observation de ce phénomène dans plusieurs types cellulaires ayant différents statuts
pour p53 nous a permis de questionner la nature de ce qui se passe dans les cellules. Dans
les fibroblastes, qu'ils soient d'origine cutanée ou pulmonaire, l'augmentation de la
fréquence des sites fapy semble clairement associée avec la présence d'une p53
fonctionnelle, mais cette association n'est pas systématique dans toutes les cellules testées.
En effet, les lymphocytes ne présentent pas d'augmentation de la fréquence des sites fapy
suite au traitement, alors que ce sont des cellules normales (p53 sauvage). Cependant, on
sait que les lymphocytes sont déficients pour la voie ATR-p53 suite à une irradiation UV
(Jones et al., 2004). Il est donc possible que la différence observée entre lymphocytes et
fibroblastes soit liée à cette différence dans la capacité d'induire p53 suite à certains stress
génotoxiques. Ainsi, la différence observée pourrait être en fait une indication de la
manière dont le phénotype associé à p53 est déclenché dans les cellules. D'autre part, on a
observé dans les cellules NCI-H23 que l'évolution de la fréquence des sites fapy post-
traitement était comparable à celle dans les fibroblastes normaux. Or, ces cellules sont
déficientes pour p53. En effet, les NCI-H23 expriment une protéine p53 mutante (Met →
Ile au codon 246) de pleine longueur mais qui a perdu ses capacités de transactivation.
Encore une fois, cette observation pourrait constituer une indication sur la nature du
phénomène associé à p53. En effet, si le rôle de p53 impliqué dans ce phénotype est
indépendant de ses capacités de transactivation, c'est-à-dire qu'il implique plutôt des

156
intéractions protéine / protéine, il est possible qu'une protéine mutante telle que celle des
cellules NCI-H23 soit capable d'agir comme une protéine sauvage. Toutes ces hypothèses
restent à explorer.
La manière dont p53 est capable d'influencer la fréquence des adduits fapy mesurée après la
fin du traitement reste à déterminer. Nous avons montré avec le test des comètes que cette
fréquence pouvait également être affectée par la relaxation globale de la chromatine, et ce
quel que soit le statut de p53 dans les cellules. p53 et la relaxation de la chromatine
influencent la vitesse de disparition des dommages réparés par REN (Allison et Milner,
2004) et l'on peut poser l'hypothèse que la fréquence des sites fapy détectée à un moment
donné pourrait être affectée par la vitesse de la réparation des 7-pobG (lésions primaires
non détectées) et / ou des lésions secondaires fapy qui en dérivent.
Il serait important de mieux comprendre ce phénotype lié à p53, et en particulier, en quoi il
pourrait être influencé différemment en fonction des mutations de p53. En effet, dans des
cellules comme les NCI-H23 (p53 mutante), on observe une forte augmentation de la
fréquence de sites fapy qui sont potentiellement très mutagènes, alors que la cellule est
déficiente dans la plupart de ses fonctions lui permettant de maintenir l’intégrité de son
génome (arrêt du cycle cellulaire, induction de gènes impliqués dans la réparation des
dommages, etc.). Les cellules NCI-H23 sont des cellules d’adénocarcinome pulmonaire et
il serait particulièrement intéressant de déterminer s’il existe une corrélation entre ce
phénotype lié à p53 et les mutations de TP53 plus spécialement associées au cancer du
poumon.

157
Chapitre 6 Conclusions générales
L'objectif principal du travail présenté dans cette thèse était d'investiguer les dommages
associés à la NNK, formation et réparation, à des fréquences se rapprochant de celles d'une
exposition réelle à la fumée de tabac. Le test des comètes qui mesure les dommages
présents au niveau de cellules individuelles, s'est avéré une technique de choix pour aborder
les questions posées. En effet, elle permet d'investiguer les dommages à des fréquences très
faibles et elle peut être réalisée sur n'importe quel type de cellules humaines. D'autre part,
elle ne nécessite pas de manipuler l'ADN à l'extérieur des cellules, ce qui est un avantage
lorsque l'on veut investiguer des dommages instables. Par contre, ce test ne peut détecter les
dommages que sous forme de cassures et il ne permet pas de savoir où ces derniers sont
formés dans le génome. En utilisant différentes stratégies, incluant l'utilisation d'analogues
de la NNK et différentes variantes du test des comètes (variation du pH de la lyse et de la
conversion alcaline, utilisation de glycosylases pour détecter spécifiquement certains types
de dommages...), nous avons pu surmonter les problèmes de spécificités de la technique
pour un certain nombre de dommages et ainsi les investiguer dans plusieurs types
cellulaires. D'autre part, nous avons pu mettre indirectement en évidence la formation d'un
type d'adduits associé à la pyridyloxobutylation qui était jusqu'alors inconnu et qui a les
caractéristiques d'un dommage à protentiel mutagénique élevé : les 7-pob-fapy.
En se basant sur la fréquence connue de DCP générée par une dose particulière d'UV, nous
avons pu associer des valeurs de pourcentage d'ADN dans la queue, telles que mesurées par
le test des comètes, avec des valeurs en fréquence de dommages générés dans les cellules.
Ainsi, une estimation de la fréquence de formation de certains dommages par les analogues
de la NNK a pu être réalisée dans différents types cellulaires. Les variations observées dans
les valeurs mesurées ont systématiquement pu être associées avec des variations dans la
capacité des cellules à activer les analogues en intermédiaires réactifs. En effet, les
dommages ont été plus facilement formés (à des doses plus faibles) par l'analogue de la
NNK méthylant l'ADN que par celui pyridyloxobutylant l'ADN, ce qui reflète la capacité
pour les estérases cellulaires d'activer chacun de ces composés. D'autre part, de petites
différences ont pu être observées entre types cellulaires : les cellules NCI-H23 et U937,

158
correspondant à des types cellulaires bien pourvus en estérases, ont semblé mieux activer
l'analogue pyridyloxobutylant l'ADN que les lymphocytes. Toutes ces données renforcent
l'idée que la formation des intermédiaires réactifs, c'est à dire la capacité de bioactiver la
NNK, est l'un des facteurs fondamentaux déterminant la quantité de dommages subis par
une cellule à un moment donné.
Les cinétiques de réparation de chacun de ces différents types de dommages ont également
été réalisées avec le test des comètes. Certaines des différences observées pour ces
cinétiques ont pu, encore une fois, être associées avec l'activation des analogues via la
formation de nouveaux dommages. Ainsi, l'analogue méthylant l'ADN, qui est le plus
facilement activé dans les cellules, a semblé capable de générer des nouveaux dommages
(des 7mG) dans les cellules, même après la fin du traitement. Combiné avec des capacités
différentes pour les cellules de réparer ces mêmes dommages, les cinétiques observées se
sont avérées très différentes d'un type cellulaire à l'autre. Les cellules U937, qui activent
bien l'analogue méthylant mais réparent bien les 7-mG, ont vu leur fréquence de dommages
diminuer rapidement après la fin du traitement. Tandis que les lymphocytes, qui activent
moins bien mais réparent mal les 7-mG, ont continué à accumuler des dommages jusqu'à
3 h après la fin du traitement. En ce qui concerne les dommages associés à la
pyridyloxobutylation de l'ADN, les différences dans les cinétiques ont semblé refléter plus
spécifiquement des différences dans les capacités de réparation des cellules. Par exemple,
les cellules U937 et NCI-H23 ont semblé réparer beaucoup plus rapidement les
cassures / sites alcali labiles générés que les fibroblastes soumis à la même fréquence de
dommages. D'autre part, la réparation des sites fapy associés à la pyridyloxobutylation, a
permis de mettre en évidence un nouveau phénotype associé à p53 qui reste à caractériser,
mais qui semble lui aussi pouvoir être associé à des différences intercellulaires dans les
capacités de réparation des dommages.
Pour conclure, ce travail a permis d'améliorer les connaissances concernant les
conséquences cellulaires d'une exposition à la NNK, et les résultats obtenus vont dans le
sens des données épidémiologiques et des données obtenues avec des modèles animaux, à
savoir que la NNK a probablement une participation significative dans la transformation
maligne chez les fumeurs et les personnes exposées à la fumée environnementale de tabac.

159
Références bibliographiques (Chapitres 1 et 5) Adimoolam S et Ford JM (2002) p53 and DNA damage-inducible expression of the
xeroderma pigmentosum group C gene. Proc Natl Acad Sci USA 99:12985-12990. Adler I (1912) Primary malignant growths of the lungs and bronchi. Longsmans, Green
and Company, New York. Agarwal ML, Taylor WR, Chernov MV, Chernova OB et Stark GR (1998) The p53
network. J Biol Chem 273:1-4. Agathanggelou A, Bieche I, Ahmed-Choudhury J, Nicke B, Dammann R, Baksh S, Gao B,
Minna JD, Downward J, Maher ER et Latif F (2003) Identification of novel gene expression targets for the Ras association domain family 1 (RASSF1A) tumor suppressor gene in non-small cell lung cancer and neuroblastoma. Cancer Res 63:5344-5351.
Agathanggelou A, Cooper WN et Latif F (2005) Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res 65:3497-3508.
Agathanggelou A, Honorio S, Macartney DP, Martinez A, Dallol A, Rader J, Fullwood P, Chauhan A, Walker R, Shaw JA, Hosoe S, Lerman MI, Minna JD, Maher ER et Latif F (2001) Methylation associated inactivation of RASSF1A from region 3p21.3 in lung, breast and ovarian tumours. Oncogene 20:1509-1518.
Ahrendt SA, Decker PA, Alawi EA, Zhu Yr YR, Sanchez-Cespedes M, Yang SC, Haasler GB, Kajdacsy-Balla A, Demeure MJ et Sidransky D (2001) Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer 92:1525-1530.
Alekseyev YO, Hamm ML et Essigmann JM (2004) Aflatoxin B1 formamidopyrimidine adducts are preferentially repaired by the nucleotide excision repair pathway in vivo. Carcinogenesis 25:1045-1051.
Allison SJ et Milner J (2004) Remodelling chromatin on a global scale: a novel protective function of p53. Carcinogenesis 25:1551-1557.
Anderson KE, Carmella SG, Ye M, Bliss RL, Le C, Murphy L et Hecht SS (2001) Metabolites of a tobacco-specific lung carcinogen in nonsmoking women exposed to environmental tobacco smoke. J Natl Cancer Inst 93:378-381.
Anderson KE, Kliris J, Murphy L, Carmella SG, Han S, Link C, Bliss RL, Puumala S, Murphy SE et Hecht SS (2003) Metabolites of a tobacco-specific lung carcinogen in nonsmoking casino patrons. Cancer Epidemiol Biomarkers Prev 12:1544-1546.
Bedard LL et Massey TE (2006) Aflatoxin B(1)-induced DNA damage and its repair. Cancer Lett 241:174-183.
Belinsky SA (1998) Role of the cytosine DNA-methyltransferase and p16INK4a genes in the development of mouse lung tumors. Exp Lung Res 24:463-479.
Bennett RA, Wilson DM, 3rd, Wong D et Demple B (1997) Interaction of human apurinic endonuclease and DNA polymerase beta in the base excision repair pathway. Proc Natl Acad Sci USA 94:7166-7169.
Branum ME, Reardon JT et Sancar A (2001) DNA repair excision nuclease attacks undamaged DNA. A potential source of spontaneous mutations. J Biol Chem 276:25421-25426.

160
Brash DE, Ziegler A, Jonason AS, Simon JA, Kunala S et Leffell DJ (1996) Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J Investig Dermatol Symp Proc 1:136-142.
Bray F, Tyczynski JE et Parkin DM (2004) Going up or coming down? The changing phases of the lung cancer epidemic from 1967 to 1999 in the 15 European Union countries. Eur J Cancer 40:96-125.
Brooks DR, Austin JH, Heelan RT, Ginsberg MS, Shin V, Olson SH, Muscat JE et Stellman SD (2005) Influence of type of cigarette on peripheral versus central lung cancer. Cancer Epidemiol Biomarkers Prev 14:576-581.
Buterin T, Hess MT, Gunz D, Geacintov NE, Mullenders LH et Naegeli H (2002) Trapping of DNA nucleotide excision repair factors by nonrepairable carcinogen adducts. Cancer Res 62:4229-4235.
Caldecott KW (2001) Mammalian DNA single-strand break repair: an X-ra(y)ted affair. Bioessays 23:447-455.
Cappelli E, Degan P et Frosina G (2000) Comparative repair of the endogenous lesions 8-oxo-7, 8-dihydroguanine (8-oxoG), uracil and abasic site by mammalian cell extracts: 8-oxoG is poorly repaired by human cell extracts. Carcinogenesis 21:1135-1141.
Cappelli E, Hazra T, Hill JW, Slupphaug G, Bogliolo M et Frosina G (2001) Rates of base excision repair are not solely dependent on levels of initiating enzymes. Carcinogenesis 22:387-393.
Cappelli E, Taylor R, Cevasco M, Abbondandolo A, Caldecott K et Frosina G (1997) Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J Biol Chem 272:23970-23975.
Caputi M, Russo G, Esposito V, Mancini A et Giordano A (2005) Role of cell-cycle regulators in lung cancer. J Cell Physiol 205:319-327.
Castillo L, Etienne-Grimaldi MC, Fischel JL, Formento P, Magne N et Milano G (2004) Pharmacological background of EGFR targeting. Ann Oncol 15:1007-1012.
Chetsanga CJ et Frenette GP (1983) Excision of aflatoxin B1-imidazole ring opened guanine adducts from DNA by formamidopyrimidine-DNA glycosylase. Carcinogenesis 4:997-1000.
Cloutier JF, Drouin R et Castonguay A (1999) Treatment of human cells with N-Nitroso(acetoxymethyl)methylamine: distribution patterns of piperidine-sensitive DNA damage at the nucleotide level of resolution are related to the sequence context. Chem Res Toxicol 12:840-849.
Cloutier JF, Castonguay A, O'Connor TR et Drouin R (2001a) Alkylating agent and chromatin structure determine sequence context-dependent formation of alkylpurines. J Mol Biol 306:169-188.
Cloutier JF, Drouin R, Weinfeld M, O'Connor TR et Castonguay A (2001b) Characterization and mapping of DNA damage induced by reactive metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) at nucleotide resolution in human genomic DNA. J Mol Biol 313:539-557.
Collins AR (2004) The comet assay for DNA damage and repair: principles, applications, and limitations. Mol Biotechnol 26:249-261.
Cooper CA, Carby FA, Bubb VJ, Lamb D, Kerr KM et Wyllie AH (1997) The pattern of K-ras mutation in pulmonary adenocarcinoma defines a new pathway of tumour development in the human lung. J Pathol 181:401-404.

161
Costa RM, Chigancas V, Galhardo Rda S, Carvalho H et Menck CF (2003) The eukaryotic nucleotide excision repair pathway. Biochimie 85:1083-1099.
Datta A, Bagchi S, Nag A, Shiyanov P, Adami GR, Yoon T et Raychaudhuri P (2001) The p48 subunit of the damaged-DNA binding protein DDB associates with the CBP/p300 family of histone acetyltransferase. Mutat Res 486:89-97.
Denissenko MF, Pao A, Pfeifer GP et Tang M (1998) Slow repair of bulky DNA adducts along the nontranscribed strand of the human p53 gene may explain the strand bias of transversion mutations in cancers. Oncogene 16:1241-1247.
Denissenko MF, Pao A, Tang M et Pfeifer GP (1996) Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science 274:430-432.
Devesa SS, Bray F, Vizcaino AP et Parkin DM (2005) International lung cancer trends by histologic type: male:female differences diminishing and adenocarcinoma rates rising. Int J Cancer 117:294-299.
Dianov GL, Prasad R, Wilson SH et Bohr VA (1999) Role of DNA polymerase beta in the excision step of long patch mammalian base excision repair. J Biol Chem 274:13741-13743.
Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, Pena-Diaz J, Otterlei M, Slupphaug G et Krokan HE (2004) Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair (Amst) 3:1389-1407.
Drouin R, Rodriguez H, Holmquist GP et Akman SA (1996) Ligation-mediated PCR for analysis of oxidative DNA damage, in: Technologies for detection of DNA damage and mutations (Pfeifer GP ed), pp 209-223, Plenum Press, New York.
Duguid EM, Mishina Y et He C (2003) How do DNA repair proteins locate potential base lesions? a chemical crosslinking method to investigate O6-alkylguanine-DNA alkyltransferases. Chem Biol 10:827-835.
El-Mahdy MA, Zhu Q, Wang QE, Wani G, Praetorius-Ibba M et Wani AA (2006) Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J Biol Chem 281:13404-13411.
Eritja R, Horowitz DM, Walker PA, Ziehler-Martin JP, Boosalis MS, Goodman MF, Itakura K et Kaplan BE (1986) Synthesis and properties of oligonucleotides containing 2'-deoxynebularine and 2'-deoxyxanthosine. Nucleic Acids Res 14:8135-8153.
Foiles PG, Akerkar SA, Carmella SG, Kagan M, Stoner GD, Resau JH et Hecht SS (1991) Mass spectrometric analysis of tobacco-specific nitrosamine-DNA adducts in smokers and nonsmokers. Chem Res Toxicol 4:364-368.
Ford JM et Hanawalt PC (1995) Li-Fraumeni syndrome fibroblasts homozygous for p53 mutations are deficient in global DNA repair but exhibit normal transcription-coupled repair and enhanced UV resistance. Proc Natl Acad Sci USA 92:8876-8880.
Ford JM et Hanawalt PC (1997) Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J Biol Chem 272:28073-28080.
Fortini P, Pascucci B, Parlanti E, Sobol RW, Wilson SH et Dogliotti E (1998) Different DNA polymerases are involved in the short- and long-patch base excision repair in mammalian cells. Biochemistry 37:3575-3580.

162
Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA et Ellenberger T (2006) DNA repair and mutagenesis - 2nd ed. American Society for Microbiology, Washington, DC.
Gary R, Kim K, Cornelius HL, Park MS et Matsumoto Y (1999) Proliferating cell nuclear antigen facilitates excision in long-patch base excision repair. J Biol Chem 274:4354-4363.
Gillet LC et Scharer OD (2006) Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev 106:253-276.
Girard L, Zochbauer-Muller S, Virmani AK, Gazdar AF et Minna JD (2000) Genome-wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non-small cell lung cancer, and loci clustering. Cancer Res 60:4894-4906.
Greenblatt MS, Bennett WP, Hollstein M et Harris CC (1994) Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 54:4855-4878.
Haglund J, Henderson AP, Golding BT et Tornqvist M (2002) Evidence for phosphate adducts in DNA from mice treated with 4-(N-Methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Chem Res Toxicol 15:773-779.
Hainaut P et Pfeifer GP (2001) Patterns of p53 G-->T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis 22:367-374.
Hara R, Mo J et Sancar A (2000) DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol Cell Biol 20:9173-9181.
Hartman AR et Ford JM (2002) BRCA1 induces DNA damage recognition factors and enhances nucleotide excision repair. Nat Genet 32:180-184.
Hecht SS (1998) Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol 11:559-603.
Hecht SS (1999a) DNA adduct formation from tobacco-specific N-nitrosamines. Mutat Res 424:127-142.
Hecht SS (1999b) Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst 91:1194-1210.
Hecht SS (2002) Human urinary carcinogen metabolites: biomarkers for investigating tobacco and cancer. Carcinogenesis 23:907-922.
Hecht SS (2004) Carcinogen derived biomarkers: applications in studies of human exposure to secondhand tobacco smoke. Tob Control 13 Suppl 1:i48-56.
Hecht SS, Carmella SG, Foiles PG et Murphy SE (1994) Biomarkers for human uptake and metabolic activation of tobacco-specific nitrosamines. Cancer Res 54:1912s-1917s.
Hecht SS, Carmella SG, Murphy SE, Akerkar S, Brunnemann KD et Hoffmann D (1993) A tobacco-specific lung carcinogen in the urine of men exposed to cigarette smoke. N Engl J Med 329:1543-1546.
Hecht SS, Villalta PW, Sturla SJ, Cheng G, Yu N, Upadhyaya P et Wang M (2004) Identification of O2-substituted pyrimidine adducts formed in reactions of 4-(acetoxymethylnitrosamino)- 1-(3-pyridyl)-1-butanone and 4-(acetoxymethylnitros- amino)-1-(3-pyridyl)-1-butanol with DNA. Chem Res Toxicol 17:588-597.
Hensel CH, Hsieh CL, Gazdar AF, Johnson BE, Sakaguchi AY, Naylor SL, Lee WH et Lee EY (1990) Altered structure and expression of the human retinoblastoma susceptibility gene in small cell lung cancer. Cancer Res 50:3067-3072.

163
Herzog CR, Desai D et Amin S (2006) Array CGH analysis reveals chromosomal aberrations in mouse lung adenocarcinomas induced by the human lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Biochem Biophys Res Commun 341:856-863.
Hirao T, Nelson HH, Ashok TD, Wain JC, Mark EJ, Christiani DC, Wiencke JK et Kelsey KT (2001) Tobacco smoke-induced DNA damage and an early age of smoking initiation induce chromosome loss at 3p21 in lung cancer. Cancer Res 61:612-615.
Hoare S, Zou Y, Purohit V, Krishnasamy R, Skorvaga M, Van Houten B, Geacintov NE et Basu AK (2000) Differential incision of bulky carcinogen-DNA adducts by the UvrABC nuclease: comparison of incision rates and the interactions of Uvr subunits with lesions of different structures. Biochemistry 39:12252-12261.
Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411:366-374.
Hoffmann D et Hoffmann I (1997) The changing cigarette, 1950-1995. J Toxicol Environ Health 50:307-364.
Hoffmann D, Hoffmann I et El-Bayoumy K (2001) The less harmful cigarette: a controversial issue. a tribute to Ernst L. Wynder. Chem Res Toxicol 14:767-790.
Hoffmann D, Rivenson A et Hecht SS (1996) The biological significance of tobacco-specific N-nitrosamines: smoking and adenocarcinoma of the lung. Crit Rev Toxicol 26:199-211.
Hollander MC, Philburn RT, Patterson AD, Velasco-Miguel S, Friedberg EC, Linnoila RI et Fornace AJ, Jr. (2005) Deletion of XPC leads to lung tumors in mice and is associated with early events in human lung carcinogenesis. Proc Natl Acad Sci USA 102:13200-13205.
Holmquist GP et Gao S (1997) Somatic mutation theory, DNA repair rates, and the molecular epidemiology of p53 mutations. Mutat Res 386:69-101.
Hu W, Feng Z et Tang MS (2003) Preferential carcinogen-DNA adduct formation at codons 12 and 14 in the human K-ras gene and their possible mechanisms. Biochemistry 42:10012-10023.
Hu Z, Wang Y, Wang X, Liang G, Miao X, Xu Y, Tan W, Wei Q, Lin D et Shen H (2005) DNA repair gene XPC genotypes/haplotypes and risk of lung cancer in a Chinese population. Int J Cancer 115:478-483.
Hwang BJ, Ford JM, Hanawalt PC et Chu G (1999) Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci USA 96:424-428.
Hwang BJ, Toering S, Francke U et Chu G (1998) p48 Activates a UV-damaged-DNA binding factor and is defective in xeroderma pigmentosum group E cells that lack binding activity. Mol Cell Biol 18:4391-4399.
Ibeanu G, Hartenstein B, Dunn WC, Chang LY, Hofmann E, Coquerelle T, Mitra S et Kaina B (1992) Overexpression of human DNA repair protein N-methylpurine-DNA glycosylase results in the increased removal of N-methylpurines in DNA without a concomitant increase in resistance to alkylating agents in Chinese hamster ovary cells. Carcinogenesis 13:1989-1995.
Izumi T, Tatsuka M, Tano K, Asano M et Mitra S (1997) Molecular cloning and characterization of the promoter of the human N-methylpurine-DNA glycosylase (MPG) gene. Carcinogenesis 18:1837-1839.

164
Izumi T, Wiederhold LR, Roy G, Roy R, Jaiswal A, Bhakat KK, Mitra S et Hazra TK (2003) Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology 193:43-65.
Jackson MA, Lea I, Rashid A, Peddada SD et Dunnick JK (2006) Genetic alterations in cancer knowledge system: analysis of gene mutations in mouse and human liver and lung tumors. Toxicol Sci 90:400-418.
Jones GG, Reaper PM, Pettitt AR et Sherrington PD (2004) The ATR-p53 pathway is suppressed in noncycling normal and malignant lymphocytes. Oncogene 23:1911-1921.
Khanna KK et Jackson SP (2001) DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet 27:247-254.
Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT et Jacks T (2005) Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121:823-835.
Klungland A et Lindahl T (1997) Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J 16:3341-3348.
Konca K, Lankoff A, Banasik A, Lisowska H, Kuszewski T, Gozdz S, Koza Z et Wojcik A (2003) A cross-platform public domain PC image-analysis program for the comet assay. Mutat Res 534:15-20.
Kubota Y, Nash RA, Klungland A, Schar P, Barnes DE et Lindahl T (1996) Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J 15:6662-6670.
Lao Y, Villalta PW, Sturla SJ, Wang M et Hecht SS (2006) Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem Res Toxicol 19:674-682.
Le Calvez F, Mukeria A, Hunt JD, Kelm O, Hung RJ, Taniere P, Brennan P, Boffetta P, Zaridze DG et Hainaut P (2005) TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Res 65:5076-5083.
Lee GY, Jang JS, Lee SY, Jeon HS, Kim KM, Choi JE, Park JM, Chae MH, Lee WK, Kam S, Kim IS, Lee JT, Jung TH et Park JY (2005) XPC polymorphisms and lung cancer risk. Int J Cancer 115:807-813.
Levin DS, Bai W, Yao N, O'Donnell M et Tomkinson AE (1997) An interaction between DNA ligase I and proliferating cell nuclear antigen: implications for Okazaki fragment synthesis and joining. Proc Natl Acad Sci USA 94:12863-12868.
Levy DD, Groopman JD, Lim SE, Seidman MM et Kraemer KH (1992) Sequence specificity of aflatoxin B1-induced mutations in a plasmid replicated in xeroderma pigmentosum and DNA repair proficient human cells. Cancer Res 52:5668-5673.
Lewis PD et Parry JM (2004) In silico p53 mutation hotspots in lung cancer. Carcinogenesis 25:1099-1107.
Li Q, Laval J et Ludlum DB (1997) Fpg protein releases a ring-opened N-7 guanine adduct from DNA that has been modified by sulfur mustard. Carcinogenesis 18:1035-1038.
Lindahl T et Wood RD (1999) Quality control by DNA repair. Science 286:1897-1905.

165
Loechler EL, Green CL et Essigmann JM (1984) In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc Natl Acad Sci USA 81:6271-6275.
Maher VM, Yang JL et McCormick JJ (1990) Ability of adducts formed in a shuttle vector by reactive metabolites of 1-nitropyrene and benzo(a)pyrene to induce mutations when the plasmid replicates in human cells. Acta Biol Hung 41:173-186.
Margison GP et Santibanez-Koref MF (2002) O6-alkylguanine-DNA alkyltransferase: role in carcinogenesis and chemotherapy. Bioessays 24:255-266.
Marin MS, Lopez-Cima MF, Garcia-Castro L, Pascual T, Marron MG et Tardon A (2004) Poly (AT) polymorphism in intron 11 of the XPC DNA repair gene enhances the risk of lung cancer. Cancer Epidemiol Biomarkers Prev 13:1788-1793.
Marsit CJ, Hasegawa M, Hirao T, Kim DH, Aldape K, Hinds PW, Wiencke JK, Nelson HH et Kelsey KT (2004) Loss of heterozygosity of chromosome 3p21 is associated with mutant TP53 and better patient survival in non-small-cell lung cancer. Cancer Res 64:8702-8707.
Matsumoto Y et Kim K (1995) Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 269:699-702.
Matsumoto Y, Kim K, Hurwitz J, Gary R, Levin DS, Tomkinson AE et Park MS (1999) Reconstitution of proliferating cell nuclear antigen-dependent repair of apurinic/apyrimidinic sites with purified human proteins. J Biol Chem 274:33703-33708.
Matter B, Wang G, Jones R et Tretyakova N (2004) Formation of diastereomeric benzo[a]pyrene diol epoxide-guanine adducts in p53 gene-derived DNA sequences. Chem Res Toxicol 17:731-741.
Mattes WB, Lee CS, Laval J et O'Connor TR (1996) Excision of DNA adducts of nitrogen mustards by bacterial and mammalian 3-methyladenine-DNA glycosylases. Carcinogenesis 17:643-648.
Mijal RS, Thomson NM, Fleischer NL, Pauly GT, Moschel RC, Kanugula S, Fang Q, Pegg AE et Peterson LA (2004) The repair of the tobacco specific nitrosamine derived adduct O6-[4-Oxo-4-(3-pyridyl)butyl]guanine by O6-alkylguanine-DNA alkyltransferase variants. Chem Res Toxicol 17:424-434.
Mills KD, Ferguson DO et Alt FW (2003) The role of DNA breaks in genomic instability and tumorigenesis. Immunol Rev 194:77-95.
Minna JD, Roth JA et Gazdar AF (2002) Focus on lung cancer. Cancer Cell 1:49-52. Miyakis S, Liloglou T, Kearney S, Xinarianos G, Spandidos DA et Field JK (2003)
Absence of mutations in the VHL gene but frequent loss of heterozygosity at 3p25-26 in non-small cell lung carcinomas. Lung Cancer 39:273-277.
Moser J, Volker M, Kool H, Alekseev S, Vrieling H, Yasui A, van Zeeland AA et Mullenders LH (2005) The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions. DNA Repair (Amst) 4:571-582.
Muscat JE, Djordjevic MV, Colosimo S, Stellman SD et Richie JP, Jr. (2005) Racial differences in exposure and glucuronidation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Cancer 103:1420-1426.
Nash RA, Caldecott KW, Barnes DE et Lindahl T (1997) XRCC1 protein interacts with one of two distinct forms of DNA ligase III. Biochemistry 36:5207-5211.

166
NCIC (2006) Statistiques canadiennes sur le cancer 2006, Société canadienne du cancer et Institut national du cancer du Canada, Toronto.
Nealon K, Nicholl ID et Kenny MK (1996) Characterization of the DNA polymerase requirement of human base excision repair. Nucleic Acids Res 24:3763-3770.
Nilsen H et Krokan HE (2001) Base excision repair in a network of defence and tolerance. Carcinogenesis 22:987-998.
Ochsner A (1973) Corner of history. My first recognition of the relationship of smoking and lung cancer. Prev Med 2:611-614.
O'Connor TR et Laval J (1991) Human cDNA expressing a functional DNA glycosylase excising 3-methyladenine and 7-methylguanine. Biochem Biophys Res Commun 176:1170-1177.
Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC et Hainaut P (2002) The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat 19:607-614.
Osada H et Takahashi T (2002) Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene 21:7421-7434.
Ostling O et Johanson KJ (1984) Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem Biophys Res Commun 123:291-298.
Parikh SS, Mol CD, Hosfield DJ et Tainer JA (1999) Envisioning the molecular choreography of DNA base excision repair. Curr Opin Struct Biol 9:37-47.
Parkin DM, Bray FI et Devesa SS (2001) Cancer burden in the year 2000. The global picture. Eur J Cancer 37 Suppl 8:S4-66.
Pascucci B, Stucki M, Jonsson ZO, Dogliotti E et Hubscher U (1999) Long patch base excision repair with purified human proteins. DNA ligase I as patch size mediator for DNA polymerases delta and epsilon. J Biol Chem 274:33696-33702.
Patel JD (2005) Lung cancer in women. J Clin Oncol 23:3212-3218. Pegg AE (2000) Repair of O(6)-alkylguanine by alkyltransferases. Mutat Res 462:83-100. Peterson LA, Liu XK et Hecht SS (1993) Pyridyloxobutyl DNA adducts inhibit the repair
of O6-methylguanine. Cancer Res 53:2780-2785. Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS et Hainaut P (2002)
Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 21:7435-7451.
Pfeifer GP et Hainaut P (2003) On the origin of G --> T transversions in lung cancer. Mutat Res 526:39-43.
Plosky B, Samson L, Engelward BP, Gold B, Schlaen B, Millas T, Magnotti M, Schor J et Scicchitano DA (2002) Base excision repair and nucleotide excision repair contribute to the removal of N-methylpurines from active genes. DNA Repair (Amst) 1:683-696.
Rajesh M, Wang G, Jones R et Tretyakova N (2005) Stable isotope labeling-mass spectrometry analysis of methyl- and pyridyloxobutyl-guanine adducts of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in p53-derived DNA sequences. Biochemistry 44:2197-2207.
Ramanathan B et Smerdon MJ (1989) Enhanced DNA repair synthesis in hyperacetylated nucleosomes. J Biol Chem 264:11026-11034.

167
Rapic-Otrin V, McLenigan MP, Bisi DC, Gonzalez M et Levine AS (2002) Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic Acids Res 30:2588-2598.
Richardson FC et Richardson KK (1990) Sequence-dependent formation of alkyl DNA adducts: a review of methods, results, and biological correlates. Mutat Res 233:127-138.
Riedl T, Hanaoka F et Egly JM (2003) The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J 22:5293-5303.
Rioux N et Castonguay A (2000) The induction of cyclooxygenase-1 by a tobacco carcinogen in U937 human macrophages is correlated to the activation of NF-kappaB. Carcinogenesis 21:1745-1751.
Rochette PJ (2005) Cartographie des dimères cyclobutyliques de pyrimidines (DCP) induits par les UVA et étude des effets de certains gènes de réparation des mésappariements et du gène p53 muté sur la réparation par excision de nucléotides des DCP. Faculté de médecine, Université Laval, Québec.
Rodenhuis S et Slebos RJ (1992) Clinical significance of ras oncogene activation in human lung cancer. Cancer Res 52:2665s-2669s.
Rodin SN et Rodin AS (2000) Human lung cancer and p53: the interplay between mutagenesis and selection. Proc Natl Acad Sci USA 97:12244-12249.
Rodin SN et Rodin AS (2002) On the origin of p53 G:C --> T:A transversions in lung cancers. Mutat Res 508:1-19.
Rodin SN et Rodin AS (2004) On the excess of G --> T transversions in the p53 gene in lung cancer cell lines. Reply to Pfeifer and Hainaut. Mutat Res 545:141-144; author reply 145-146; discussion 147.
Ronai ZA, Gradia S, Peterson LA et Hecht SS (1993) G to A transitions and G to T transversions in codon 12 of the Ki-ras oncogene isolated from mouse lung tumors induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and related DNA methylating and pyridyloxobutylating agents. Carcinogenesis 14:2419-2422.
Ruggeri B, DiRado M, Zhang SY, Bauer B, Goodrow T et Klein-Szanto AJ (1993) Benzo[a]pyrene-induced murine skin tumors exhibit frequent and characteristic G to T mutations in the p53 gene. Proc Natl Acad Sci USA 90:1013-1017.
Samson L, Derfler B, Boosalis M et Call K (1991) Cloning and characterization of a 3-methyladenine DNA glycosylase cDNA from human cells whose gene maps to chromosome 16. Proc Natl Acad Sci USA 88:9127-9131.
Sanchez-Cespedes M, Decker PA, Doffek KM, Esteller M, Westra WH, Alawi EA, Herman JG, Demeure MJ, Sidransky D et Ahrendt SA (2001) Increased loss of chromosome 9p21 but not p16 inactivation in primary non-small cell lung cancer from smokers. Cancer Res 61:2092-2096.
Sanz-Ortega J, Saez MC, Sierra E, Torres A, Balibrea JL, Hernando F, Sanz-Esponera J et Merino MJ (2001) 3p21, 5q21, and 9p21 allelic deletions are frequently found in normal bronchial cells adjacent to non-small-cell lung cancer, while they are unusual in patients with no evidence of malignancy. J Pathol 195:429-434.
Saparbaev M et Laval J (1994) Excision of hypoxanthine from DNA containing dIMP residues by the Escherichia coli, yeast, rat, and human alkylpurine DNA glycosylases. Proc Natl Acad Sci USA 91:5873-5877.
Seo KY, Jelinsky SA et Loechler EL (2000) Factors that influence the mutagenic patterns of DNA adducts from chemical carcinogens. Mutat Res 463:215-246.

168
Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba, II, Fong KM, Lee H, Toyooka S, Shimizu N, Fujisawa T, Feng Z, Roth JA, Herz J, Minna JD et Gazdar AF (2005) Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 97:339-346.
Singh NP, Danner DB, Tice RR, Brant L et Schneider EL (1990) DNA damage and repair with age in individual human lymphocytes. Mutat Res 237:123-130.
Singh NP, McCoy MT, Tice RR et Schneider EL (1988) A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 175:184-191.
Smith GB, Bend JR, Bedard LL, Reid KR, Petsikas D et Massey TE (2003) Biotransformation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in peripheral human lung microsomes. Drug Metab Dispos 31:1134-1141.
Smith GB, Castonguay A, Donnelly PJ, Reid KR, Petsikas D et Massey TE (1999) Biotransformation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in freshly isolated human lung cells. Carcinogenesis 20:1809-1818.
Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K et Wilson SH (1996) Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature 379:183-186.
Sozzi G, Sard L, De Gregorio L, Marchetti A, Musso K, Buttitta F, Tornielli S, Pellegrini S, Veronese ML, Manenti G, Incarbone M, Chella A, Angeletti CA, Pastorino U, Huebner K, Bevilaqua G, Pilotti S, Croce CM et Pierotti MA (1997) Association between cigarette smoking and FHIT gene alterations in lung cancer. Cancer Res 57:2121-2123.
Speit G, Witton-Davies T, Heepchantree W, Trenz K et Hoffmann H (2003) Investigations on the effect of cigarette smoking in the comet assay. Mutat Res 542:33-42.
Spiro GS et Silvestri GA (2005) One hundred years of lung cancer. Am J Respir Crit Care Med 172:523-529.
Srivenugopal KS, Yuan XH, Friedman HS et Ali-Osman F (1996) Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemistry 35:1328-1334.
Stucki M, Pascucci B, Parlanti E, Fortini P, Wilson SH, Hubscher U et Dogliotti E (1998) Mammalian base excision repair by DNA polymerases delta and epsilon. Oncogene 17:835-843.
Sturla SJ, Scott J, Lao Y, Hecht SS et Villalta PW (2005) Mass spectrometric analysis of relative levels of pyridyloxobutylation adducts formed in the reaction of DNA with a chemically activated form of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem Res Toxicol 18:1048-1055.
Tam IY, Chung LP, Suen WS, Wang E, Wong MC, Ho KK, Lam WK, Chiu SW, Girard L, Minna JD, Gazdar AF et Wong MP (2006) Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin Cancer Res 12:1647-1653.
Tapias A, Auriol J, Forget D, Enzlin JH, Scharer OD, Coin F, Coulombe B et Egly JM (2004) Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J Biol Chem 279:19074-19083.

169
Therrien JP, Drouin R, Baril C et Drobetsky EA (1999) Human cells compromised for p53 function exhibit defective global and transcription-coupled nucleotide excision repair, whereas cells compromised for pRb function are defective only in global repair. Proc Natl Acad Sci USA 96:15038-15043.
Thilly WG (1983) Analysis of chemically induced mutation in single cell populations. Basic Life Sci 23:337-378.
Thomas L, Doyle LA et Edelman MJ (2005) Lung cancer in women: emerging differences in epidemiology, biology, and therapy. Chest 128:370-381.
Thomson NM, Kenney PM et Peterson LA (2003) The pyridyloxobutyl DNA adduct, O6-[4-oxo-4-(3-pyridyl)butyl]guanine, is detected in tissues from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-treated A/J mice. Chem Res Toxicol 16:1-6.
Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC et Sasaki YF (2000) Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen 35:206-221.
Tomkinson AE et Mackey ZB (1998) Structure and function of mammalian DNA ligases. Mutat Res 407:1-9.
Travis WD, Brambilla E, Muller-Hermelink HK et Harris CC (2004) World Health Organization classification of tumours, pathology and genetics: tumours of the lung, pleura, thymus and heart, IARC, Lyon.
Tretyakova N, Matter B, Jones R et Shallop A (2002) Formation of benzo[a]pyrene diol epoxide-DNA adducts at specific guanines within K-ras and p53 gene sequences: stable isotope-labeling mass spectrometry approach. Biochemistry 41:9535-9544.
Tudek B (2003) Imidazole ring-opened DNA purines and their biological significance. J Biochem Mol Biol 36:12-19.
Ura K, Araki M, Saeki H, Masutani C, Ito T, Iwai S, Mizukoshi T, Kaneda Y et Hanaoka F (2001) ATP-dependent chromatin remodeling facilitates nucleotide excision repair of UV-induced DNA lesions in synthetic dinucleosomes. EMBO J 20:2004-2014.
Wang M, Cheng G, Sturla SJ, Shi Y, McIntee EJ, Villalta PW, Upadhyaya P et Hecht SS (2003) Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chem Res Toxicol 16:616-626.
Wang QE, Zhu Q, Wani G, Chen J et Wani AA (2004) UV radiation-induced XPC translocation within chromatin is mediated by damaged-DNA binding protein, DDB2. Carcinogenesis 25:1033-1043.
Wistuba, II, Behrens C, Virmani AK, Mele G, Milchgrub S, Girard L, Fondon JW, 3rd, Garner HR, McKay B, Latif F, Lerman MI, Lam S, Gazdar AF et Minna JD (2000) High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res 60:1949-1960.
Wynder EL et Muscat JE (1995) The changing epidemiology of smoking and lung cancer histology. Environ Health Perspect 103 Suppl 8:143-148.
Xu HJ, Hu SX, Cagle PT, Moore GE et Benedict WF (1991) Absence of retinoblastoma protein expression in primary non-small cell lung carcinomas. Cancer Res 51:2735-2739.

170
Xu-Welliver M et Pegg AE (2002) Degradation of the alkylated form of the DNA repair protein, O(6)-alkylguanine-DNA alkyltransferase. Carcinogenesis 23:823-830.
Ye N, Bianchi MS, Bianchi NO et Holmquist GP (1999) Adaptive enhancement and kinetics of nucleotide excision repair in humans. Mutat Res 435:43-61.
Ye N, Holmquist GP et O'Connor TR (1998) Heterogeneous repair of N-methylpurines at the nucleotide level in normal human cells. J Mol Biol 284:269-285.
Yoon JH, Smith LE, Feng Z, Tang M, Lee CS et Pfeifer GP (2001) Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer Res 61:7110-7117.
Zhou J, Ahn J, Wilson SH et Prives C (2001) A role for p53 in base excision repair. EMBO J 20:914-923.
Zhu Q, Wani MA, El-Mahdy M et Wani AA (2000) Decreased DNA repair efficiency by loss or disruption of p53 function preferentially affects removal of cyclobutane pyrimidine dimers from non-transcribed strand and slow repair sites in transcribed strand. J Biol Chem 275:11492-11497.
Ziegel R, Shallop A, Jones R et Tretyakova N (2003) K-ras gene sequence effects on the formation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-DNA adducts. Chem Res Toxicol 16:541-550.
Ziegel R, Shallop A, Upadhyaya P, Jones R et Tretyakova N (2004) Endogenous 5-methylcytosine protects neighboring guanines from N7 and O6-methylation and O6-pyridyloxobutylation by the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Biochemistry 43:540-549.
Zurer I, Hofseth LJ, Cohen Y, Xu-Welliver M, Hussain SP, Harris CC et Rotter V (2004) The role of p53 in base excision repair following genotoxic stress. Carcinogenesis 25:11-19.