de la crise au syndrome - des ped auvergne rhône … · - environ 0,5% de la population, - 1...
TRANSCRIPT

De la crise au
Syndrome
Dr Dorothée Ville, Service
Neuropédiatrie, HFME Lyon

INTRODUCTION Pathologie fréquente:
- environ 0,5% de la population,
- 1 nouveau cas/40 000 dans la 1ere année.
- Bien équilibrée dans 70% des cas
particularités de l’enfant:
- sa présentation et ses syndromes particuliers
- leur potentiel retentissement neurologique plus sévère que chez les adultes
- L’évolution ave l’âge (maturation cérébrale)
- leur spécificité par rapport au traitement
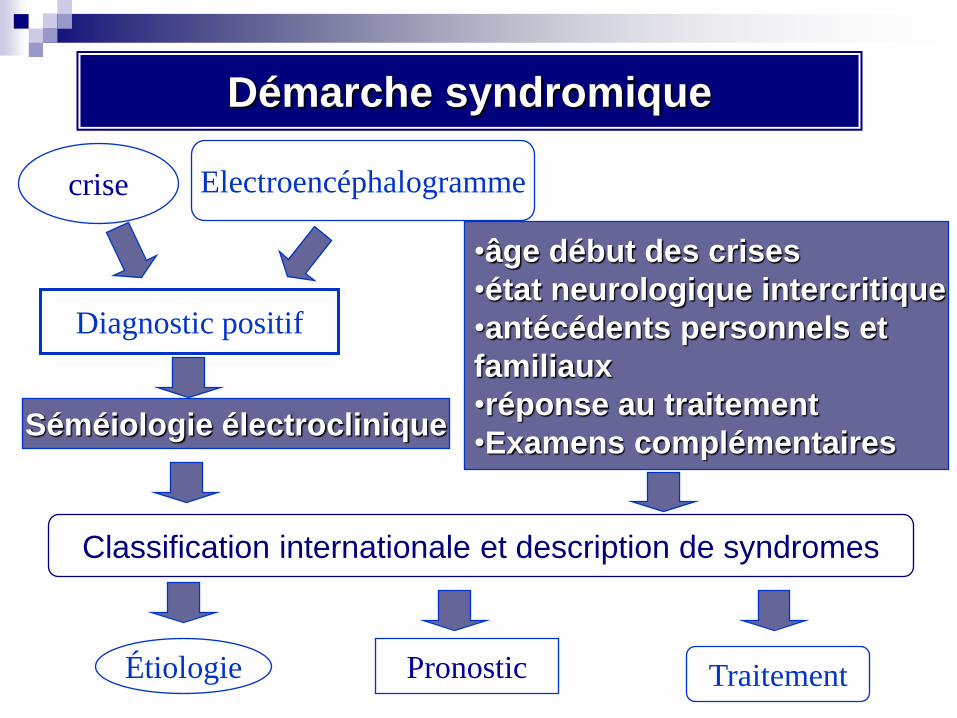
Démarche syndromique
crise Electroencéphalogramme
Diagnostic positif
Classification internationale et description de syndromes
Étiologie Pronostic Traitement
Séméiologie électroclinique
•âge début des crises
•état neurologique intercritique
•antécédents personnels et
familiaux
•réponse au traitement
•Examens complémentaires

Classification Internationale des
épilepsies
Définie en 1981, revue en 1989, en cours de
révision
Repose à la fois sur:
- Le caractère généralisé ou partiel
- La notion d’épilepsie symptomatique,
idiopathique, cryptogénique (introduit un
aspect étiologique, et physiopathologique)

classification internationale de l’épilepsie (d’après la Ligue Internationale Contre l’Epilepsie
1989)
- Epilepsies et syndromes épileptiques focaux
- Epilepsies et syndromes épileptiques
généralisés
- Epilepsies dont le caractère focal ou généralisé
n’est pas déterminé
- Syndrome spéciaux
Cette classification est importante,
car a des implications sur le pronostic, et le
traitement.
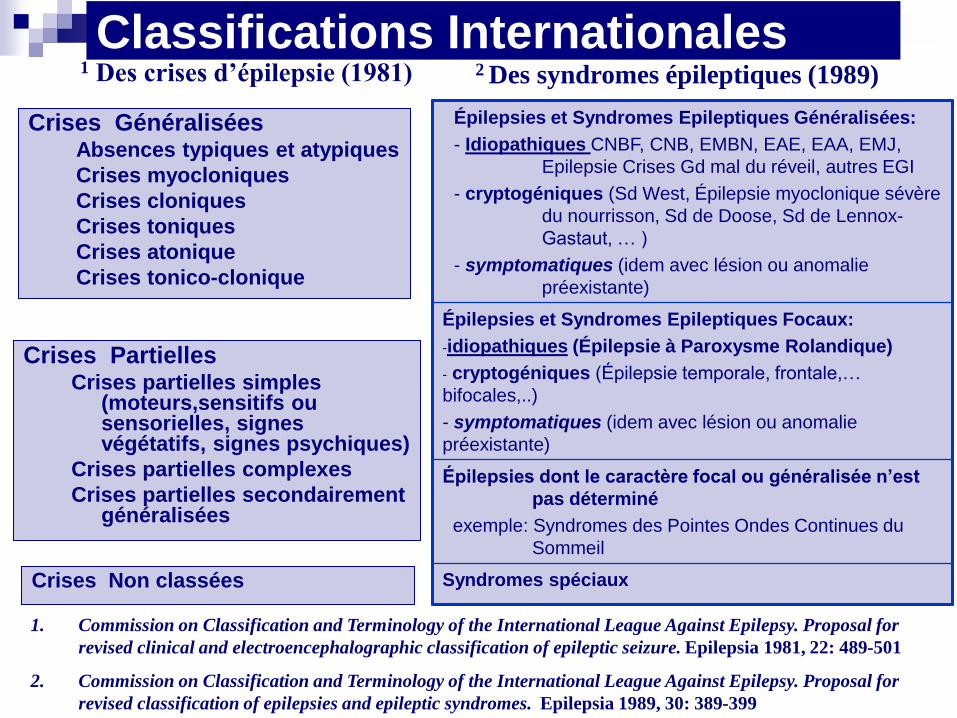
Classifications Internationales
Crises Généralisées Absences typiques et atypiques
Crises myocloniques
Crises cloniques
Crises toniques
Crises atonique
Crises tonico-clonique
1 Des crises d’épilepsie (1981) 2 Des syndromes épileptiques (1989)
Crises Partielles Crises partielles simples
(moteurs,sensitifs ou sensorielles, signes végétatifs, signes psychiques)
Crises partielles complexes
Crises partielles secondairement généralisées
Crises Non classées
Épilepsies et Syndromes Epileptiques Généralisées:
- Idiopathiques CNBF, CNB, EMBN, EAE, EAA, EMJ,
Epilepsie Crises Gd mal du réveil, autres EGI
- cryptogéniques (Sd West, Épilepsie myoclonique sévère
du nourrisson, Sd de Doose, Sd de Lennox-
Gastaut, … )
- symptomatiques (idem avec lésion ou anomalie
préexistante)
Épilepsies et Syndromes Epileptiques Focaux:
-idiopathiques (Épilepsie à Paroxysme Rolandique)
- cryptogéniques (Épilepsie temporale, frontale,…
bifocales,..)
- symptomatiques (idem avec lésion ou anomalie
préexistante)
Épilepsies dont le caractère focal ou généralisée n’est
pas déterminé
exemple: Syndromes des Pointes Ondes Continues du
Sommeil
Syndromes spéciaux
1. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for
revised clinical and electroencephalographic classification of epileptic seizure. Epilepsia 1981, 22: 489-501
2. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for
revised classification of epilepsies and epileptic syndromes. Epilepsia 1989, 30: 389-399

Les épilepsies symptomatiques
- congénitales: malformations cérébrales,
anoxie et accident vasculaire anténatal,
anomalies chromosomiques, maladies
métaboliques.
- acquises: séquelles de souffrance
néonatale, d’accident vasculaire cérébral,
de traumatisme crânien, d’infection du
système nerveux central, d’hémorragie
cérébro-méningée.

Les épilepsies idiopathiques
liées à l’âge et à la maturation cérébrale,
généralement bénignes, sans maladie
neurologique sous-jacente;
parfois familiales
Définies par un syndrome électroclinique
précis
dans certains cas, des mutations sur un
canal ionique ont été trouvées.

Les épilepsies cryptogéniques
aucune cause n’a pu être mise en évidence,
mais dont l’évolution, et le retentissement
sur le développement font suspecter une
étiologie sous-jacente
Population en voie de régression en raison
de l’augmentation des étiologies retrouvées
Concept remis en cause (« épilepsie
probablement symptomatique »)
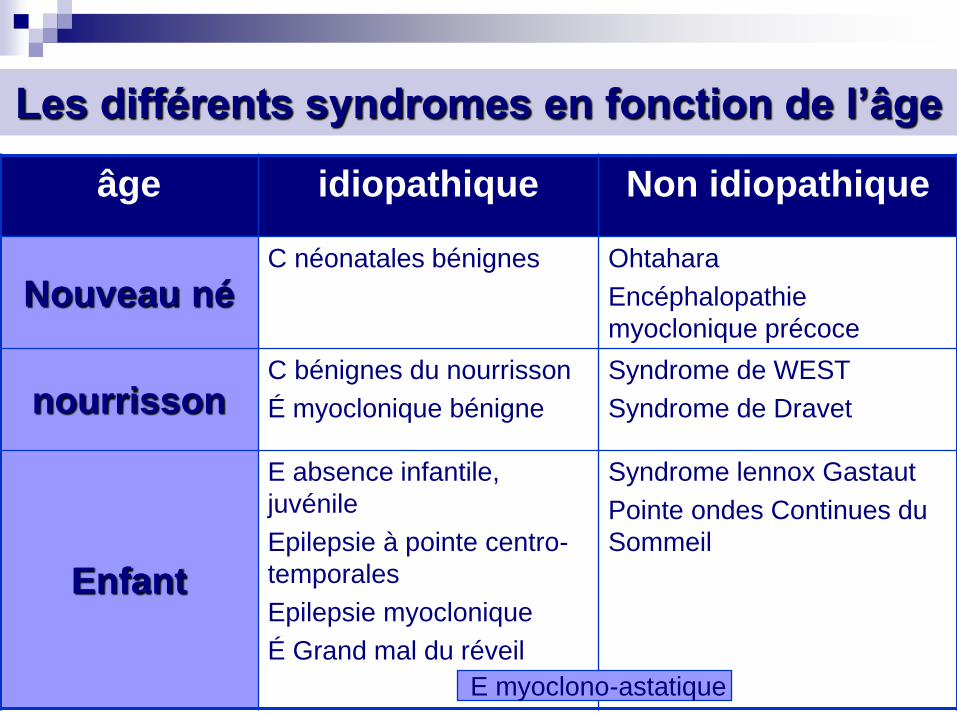
Les différents syndromes en fonction de l’âge
âge idiopathique Non idiopathique
Nouveau né C néonatales bénignes Ohtahara
Encéphalopathie
myoclonique précoce
nourrisson C bénignes du nourrisson
É myoclonique bénigne
Syndrome de WEST
Syndrome de Dravet
Enfant
E absence infantile,
juvénile
Epilepsie à pointe centro-
temporales
Epilepsie myoclonique
É Grand mal du réveil
Syndrome lennox Gastaut
Pointe ondes Continues du
Sommeil
E myoclono-astatique

Les épilepsies par tranche d’âge
Les différents
syndromes
Le nouveau né
Le nourrisson
L’enfant
Les étiologies

Séméiologie des
crises

Modes de présentation particuliers
chez l’enfant
Mon enfant a fait un malaise
Mon enfant tombe (chute tonique, chute atonique, spasme, myoclonie)
Mon enfant a des secousses
Mon enfant a les yeux qui tournent (déviation de l’œil, secousses oculaires, secousses des paupières)
Mon enfant fait une « absence »
traduction en termes médicaux

I- CRISES GENERALISEES
1. Crises toniques
2. Myoclonies
3. Crises Tonico-cloniques généralisées
4. Absences typiques et atypiques
5. Crises atoniques

II-CRISES PARTIELLES (1)
- manifestations motrices: cloniques (cr bravais-
jacksonnienne), toniques, versives, posturales, +/-
phonatoires,
- crises avec manifestations neurovégétatives: (vomissements, pâleur, rubéfaction, sueur, mydriase,
sensation épigastrique),
crises avec signes somesthésiques, sensoriels (paresthesies, troubles proprioceptifs, signes visuels, auditifs,
gustatives, vertiges)
Crises somato-psychiques (dream state, illusions,
hallucinations…)
Ces crises peuvent être avec ou sans rupture de contact

II-CRISES PARTIELLES (2)
Attention aux crises faussement
généralisées (notamment nouveau-né,
nourrisson)
Attention au début et à la fin des crises:
éléments importants pour la localisation
Propagation, généralisation des crises

II-CRISES PARTIELLES (3)
Localisation temporale: rupture de contact,
manifestations neurovégétatives, automatismes, Cr
versives, confusion post-critique
Localisation frontale: hypertonie, agitation, crises
posturales, souvent nocturnes
Localisation occipitale: signes visuels, déviation
des yeux,secousses oculaires…
Localisation pariétale: paresthésies, sensation
vertigineuse, rotation lente.
Localisation centrale: secousses cloniques,
crises Bravais-jacksonniennes

Exemples de
syndrome

Syndromes particuliers
du nouveau né
Ex convulsions du 5eme jour
Familiales ou non
Orage de crises partielles; bilan négatif
Examen neurologique et EEG intercritique
normal
Evolution favorable sur le plan épilepto et
neuro (TRT par VPA)
Gènes codant pour des canaux potassiques
Les convulsions néonatales bénignes
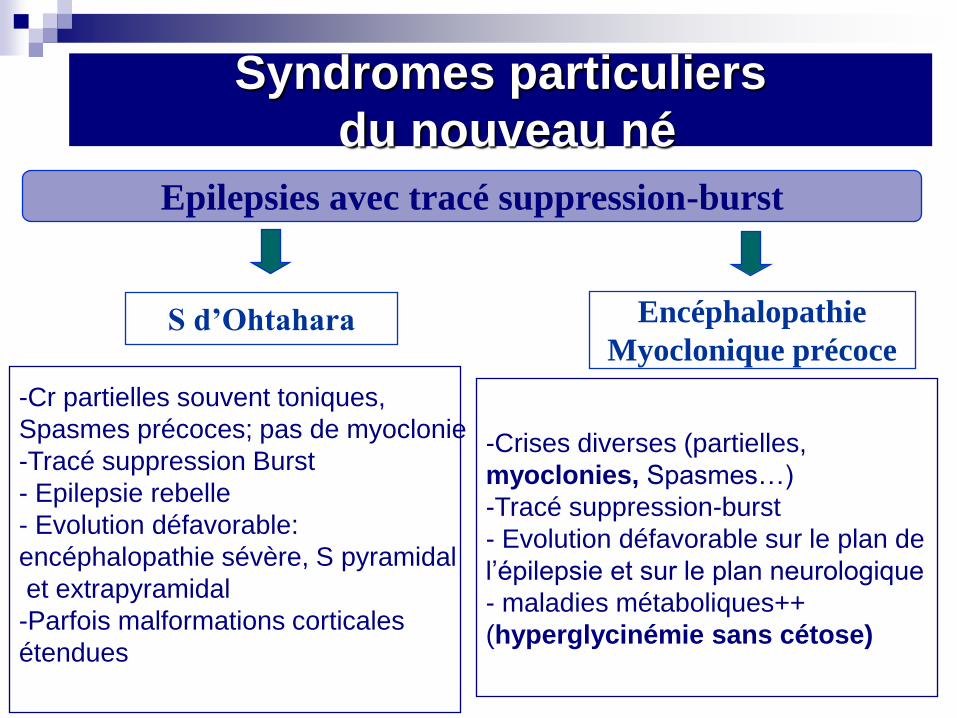
Epilepsies avec tracé suppression-burst
S d’Ohtahara Encéphalopathie
Myoclonique précoce
-Cr partielles souvent toniques,
Spasmes précoces; pas de myoclonie
-Tracé suppression Burst
- Epilepsie rebelle
- Evolution défavorable:
encéphalopathie sévère, S pyramidal
et extrapyramidal
-Parfois malformations corticales
étendues
-Crises diverses (partielles,
myoclonies, Spasmes…)
-Tracé suppression-burst
- Evolution défavorable sur le plan de
l’épilepsie et sur le plan neurologique
- maladies métaboliques++
(hyperglycinémie sans cétose)
Syndromes particuliers
du nouveau né

LE NOURRISSON
Le syndrome de West
Le syndrome de Dravet
Les convulsions infantiles bénignes

LES ETIOLOGIES
- Crises occasionnelles en particulier les crises en contexte fébrile; âge du syndrome H.H, de l’encéphalite herpétique, post traumatiques (HSD)
- malformations cérébrales, maladies métaboliques, anomalies chromosomiques
- syndromes neurocutanés: Bourneville, Sturge-Weber...

LES SPASMES INFANTILES (0,3 à 0,5/1000)
Concept d’encéphalopathie
épileptique
Urgence diagnostique et thérapeutique+++
Triade classique Spasmes infantiles
Hypsarythmie
Régression psycho-motrice
Association Spasmes- retard mental
Rôle des spasmes et de l’hypsarythmie
Rôle de l’étiologie sous-jacente
75 à 90% de RM
50 à 60% font encore des
crises à long terme
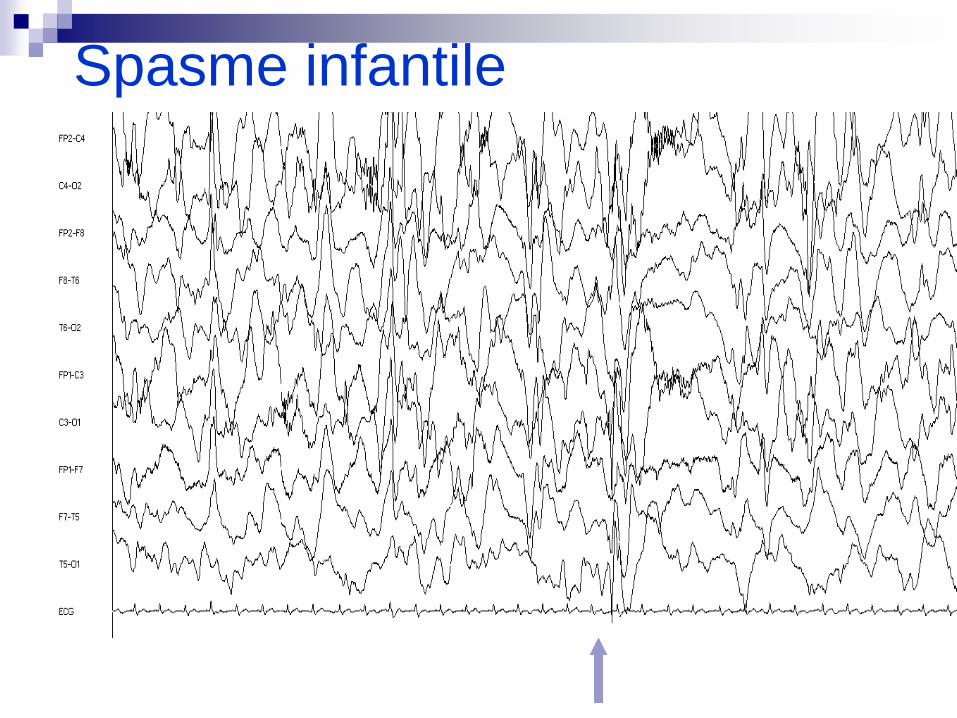
Spasme infantile
Sal… Thibault NEM
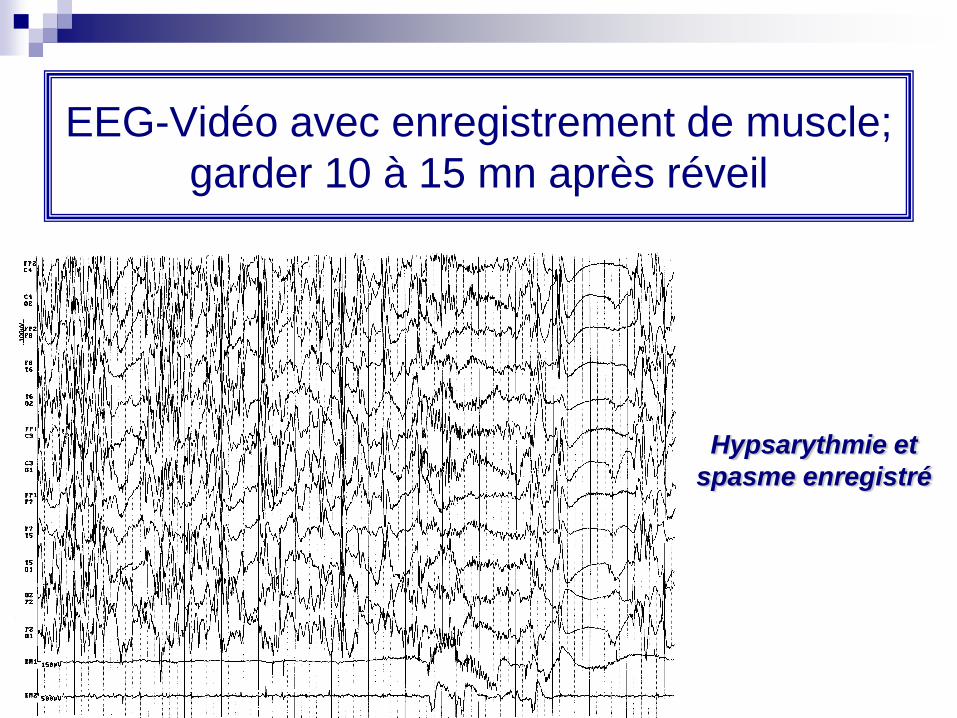
EEG-Vidéo avec enregistrement de muscle;
garder 10 à 15 mn après réveil
Hypsarythmie et
spasme enregistré

LES DIFFERENTES ETIOLOGIES
Spasmes symptomatiques: (60% Dg sur IRM)
malformations cérébrales
Séquelles d’anoxo-ischémie, d’AVC anté ou périnatal,
foetopathie CMV, méningite, post traumatique
•Maladies chromosomiques (T21, mais aussi 1P3-6, gARX)
•Maladies métaboliques (toutes les causes d’épilepsie
précoce, Attention à la mitochondrie)
Les Spasmes idiopatiques: (10% cas): Tracé
hypsarythmique, et spasmes typiques, développement
normal avant sp, peu de régression, bonne évolution
Spasmes cryptogéniques
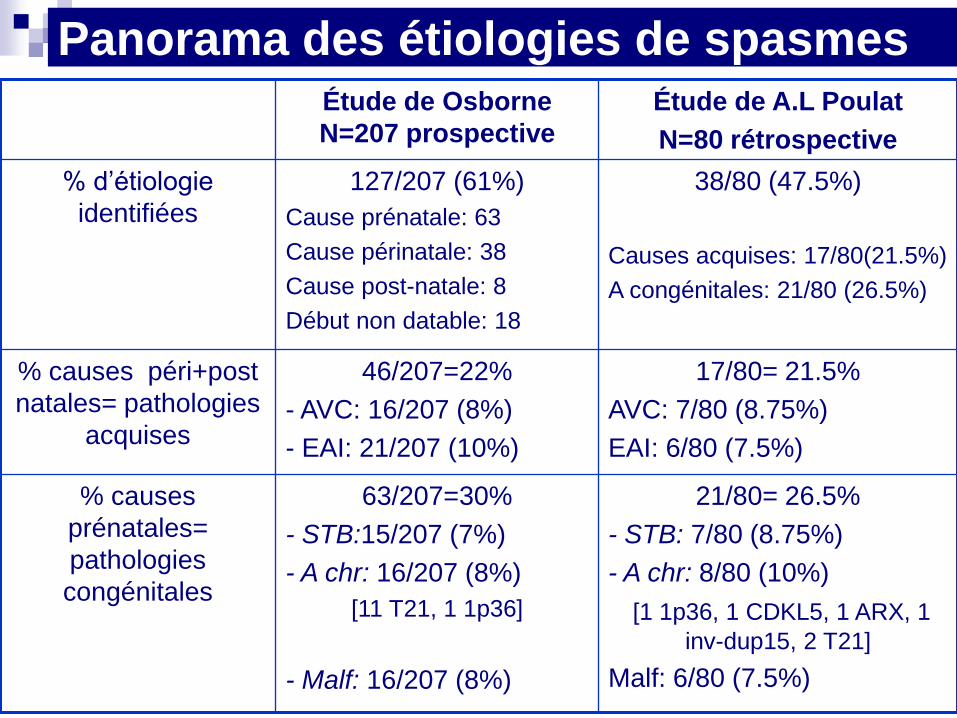
Panorama des étiologies de spasmes Étude de Osborne
N=207 prospective
Étude de A.L Poulat
N=80 rétrospective
% d’étiologie
identifiées
127/207 (61%)
Cause prénatale: 63
Cause périnatale: 38
Cause post-natale: 8
Début non datable: 18
38/80 (47.5%)
Causes acquises: 17/80(21.5%)
A congénitales: 21/80 (26.5%)
% causes péri+post
natales= pathologies
acquises
46/207=22%
- AVC: 16/207 (8%)
- EAI: 21/207 (10%)
17/80= 21.5%
AVC: 7/80 (8.75%)
EAI: 6/80 (7.5%)
% causes
prénatales=
pathologies
congénitales
63/207=30%
- STB:15/207 (7%)
- A chr: 16/207 (8%)
[11 T21, 1 1p36]
- Malf: 16/207 (8%)
21/80= 26.5%
- STB: 7/80 (8.75%)
- A chr: 8/80 (10%)
[1 1p36, 1 CDKL5, 1 ARX, 1
inv-dup15, 2 T21]
Malf: 6/80 (7.5%)
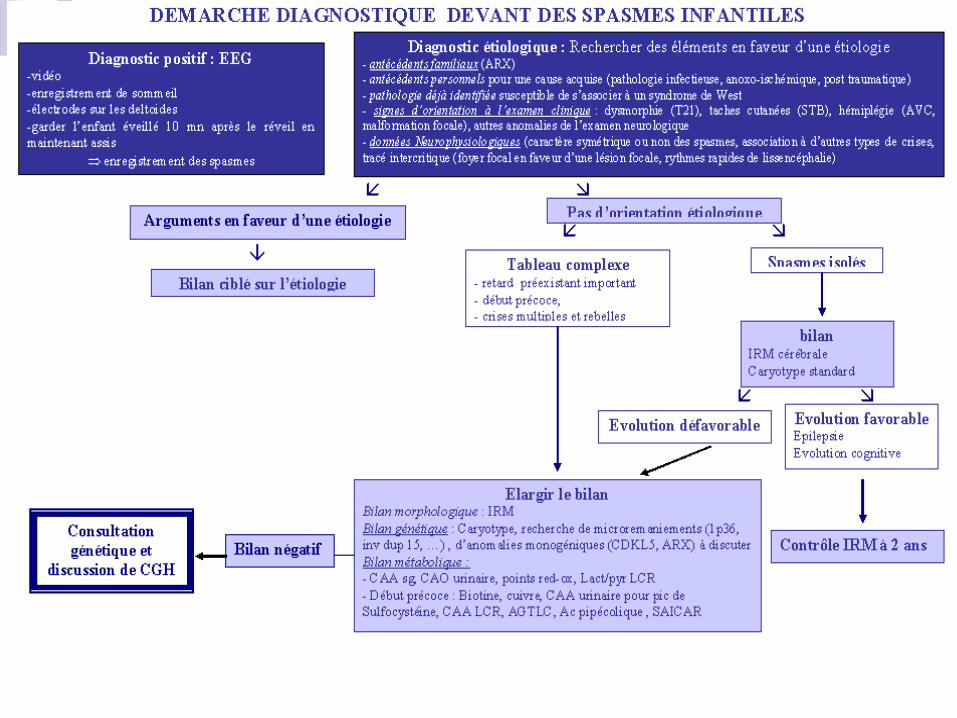
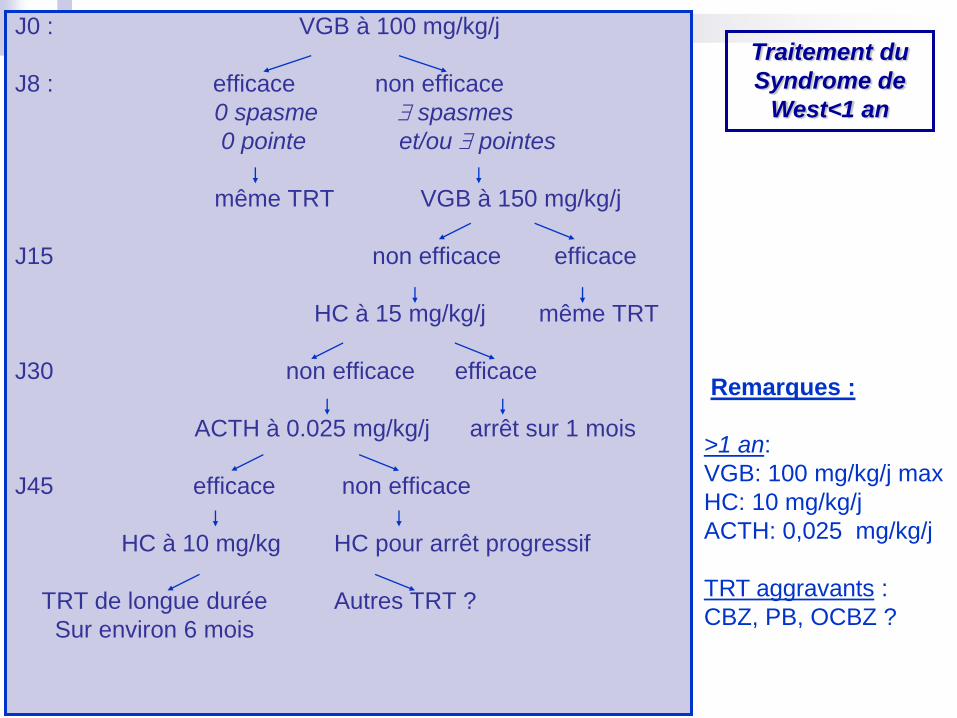
J0 : VGB à 100 mg/kg/j
J8 : efficace non efficace
0 spasme spasmes
0 pointe et/ou pointes
même TRT VGB à 150 mg/kg/j
J15 non efficace efficace
HC à 15 mg/kg/j même TRT
J30 non efficace efficace
ACTH à 0.025 mg/kg/j arrêt sur 1 mois
J45 efficace non efficace
HC à 10 mg/kg HC pour arrêt progressif
TRT de longue durée Autres TRT ?
Sur environ 6 mois
Remarques :
>1 an:
VGB: 100 mg/kg/j max
HC: 10 mg/kg/j
ACTH: 0,025 mg/kg/j
TRT aggravants :
CBZ, PB, OCBZ ?
Traitement du
Syndrome de
West<1 an


Béatrice
Tess1
Tess 2
Théo
Gabriel

Syndrome de Dravet
Diagnostic sur l’histoire clinique: - Crises fébriles particulières: précoces, longues,
parfois avec un caractère partiel
- développement initial normal (jusqu’à 18, 24 mois)
- EEG et imageries normaux; photosensibilité précoce
Evolution: - Épilepsie sévère: avec apparition de crises
fréquentes, plus courtes toujours favorisée par la fièvre (abs, myoclonies, TCG, cr toniques, cr part)
- Stagnation des acquisitions, peu de langage, pas acquisition scolaire, ataxie

Le syndrome de Dravet
Étiologie:
mutation du gène SCNA1 dans 60 à 80% des cas
Le plus souvent sporadique
Causes du retard mental dans le syndrome de
dravet
Rôle des états de mal sur le développement
psychomoteur
Autres explications??

Enjeu d’1 diagnostic précoce et du
traitement dans le syndrome de Dravet
Stratégie thérapeutique: association VPA,CLB,STP
Étude de Chiron and al: essai contrôle versus placebo en association avec Urbanyl et Valproate :
à 2 mois:
- 71% une diminution de plus de 50% du nombre de crises sous Stiripentol contre 5% sous placebo
- 9/21 étaient sans crises sous Stiripentol contre 0/20 sous placebo.
Effet du Topiramate; rôle du Keppra?
TRT à éviter: CBZ, LTG, VGB, PB

Convulsions infantile bénignes S de Watanabe
Formes familiales(AD) ou non
Tableau d’orage de crises partielles (temporales)
souvent stéréotypées, chez un enfant normal
auparavant, sans cause évidente.
Ex neuro et EEG intercritique normaux.
Evolution favorable
Liaison avec le chromosome 9, 16.
En cas de cas sporadique, diagnostic de présomption et
d’élimination, confirmé par l’évolution

LES EPILEPSIES CHEZ
L’ENFANT ET
L’ADOLESCENT
toutes les causes déjà citées chez le nouveau-né et le nourrisson (malformations, métaboliques, chromosomiques)
Les causes occasionnelles existent mais sont plus rares
Il existe également des syndromes propres à l’enfant et l’adolescent

LES EPILEPSIES IDIOPATHIQUES
Les épilepsies absences: brefs épisodes de rupture de contact pluriquotidiens, Abs typique avec P.O à 3 Hz
Les épilepsies à paroxysme rolandique: crises nocturnes avec clonies hémiface, ou généralisées; EEG: pointes amples centro-temporales, activé ds le sommeil
Les épilepsies Grand Mal du réveil de l’adolescent: début à l’adolescence, crises TCG après dette de sommeil, stress, alcool.
Les épilepsies myocloniques juvéniles: à l’adolescence; myoclonies matinales (lâchage d’objets), +/- TCG; EEG: bien organisé, bouffées généralisées

L’épilepsie
Absence de
l’enfance

L’épilepsie absence de l’enfance Fréquence: environ 12% des épilepsies de l’enfant (chiffres variables)
avec 60 à 70% de filles
Âge de début 4 à 10 ans (pic 5-7 ans)
Étiologie: facteurs génétiques?, facteurs acquis? Microdysgénésies?
Sémiologie: Durée 4 à 20 sec
Début et fin brusque
Symptomatologie: altération marquée de la conscience, arrêt de l’activité en cours, automatismes (persévérations), quelques manifestations toniques, cloniques
Répétition: de 10 à > 100/j
Facteurs favorisants: HPN, +/- SLI, stress, émotion, relâchement
Formes cliniques: EA infantile/juvénile, EA myoclonique, EA avec myoclonies palpébrales, épilepsie myoclonique juvénile, épilepsie absencess précoces…

EEG: tracé correctement organisé, bouffées de PO intercritiques, absences typiques enregistrées, sensibilité à l’HPN
Traitement: Dépakine, Zarontin, Lamotrigine en 1ère intention
Évolution: - Disparition des absences dans 90% des cas (délai moyen de 6.6
ans)
- Crises TCG dans 36 à 60% des cas (facteurs prédictifs de TCG: début tardif, états de mal absence, traitement tardif
- Rémission complète: chiffres variables (70% en cas de traitement adapté, 18% en cas de traitement incorrect
- Neuropsychologique: favorable; place de troubles attentionnels et exécutifs? 1/3 auraient des difficultés d’insertion socioprofessionnelle à l’âge adulte (surtout formes atypiques)
L’épilepsie absence de l’enfance

Marie Brune I… née le 02/07/2000
Ou la moitié d’absence
•Développement psychomoteur normal
•Épilepsie partielle débutant en 2005, suivie
d‘une épilepsie partielle continue
•IRM: hypersignal centropariétal gauche puis
apparition d’une atrophie hémisphérique
diagnostic d’encéphalite de Rasmussen
Échec du traitement par corticoide

Décision chirurgicale
WISC IV
Quotient Intellectuel Global Non calculé
• Indice de Compréhension Verbale 120
• Indice de Raisonnement Perceptif 109
• Indice de Mémoire de Travail 85
• Indice de Vitesse de Traitement 81
IRM fonctionnelle du langage
Dominance hémishérique droite

Hémisphérotomie le 18 juin 2007
• Pas de complication post-opératoire immédiate
• Anatomopathologie compatible
• Récupération rapide de la marche; persistance d’une hémiparésie, prédominante au membre sup; pas de difficulté de langage
• Évaluation neuropsy à 6 mois post-op:
ICV à 138(120),
IRP à 102 (109)
avec qq difficultés en mémoire de travail
• Pas de crise post-opératoire permettant un arrêt progressif du TRT AE
• Hyperphagie et prise de poids conduisant à poser le diagnostic de syndrome hypothalamique

Évolution en juillet 2010
• Neuropsycho: scolarité normale sans
aménagement; persistance d’une petite plainte en
mémoire de travail
• Stabilisation du poids (mesures diététiques, TRT
par hormone de croissance)
• Apparition depuis Mai 2010 d’épisodes brefs de
rupture de contact, avec secousses des paupières
Enregistrement EEG de 24 heures
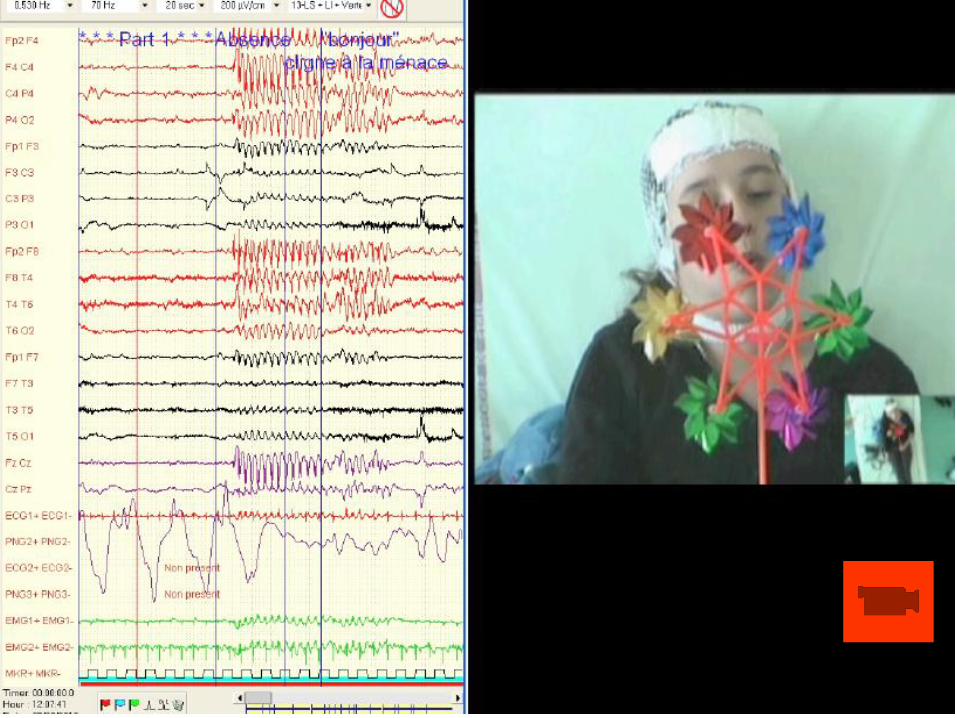

Évolution entre 2010 et 2012
• Discussion syndromique? Épilepsie Absence idiopathique?
• Échec VPA mais arrêt des absences sous Zarontin
Dernière consultation le 28 mars 2012:
aucune récidive de crise malgré un traitement par
Zarontin à petites doses (6 ml/jour)
la 6ème se passe bien, même si elle nécessite plus
d’efforts de travail. pas d’AVS, ni d’autre rééducation que
les 3 séances de kinésithérapie motrice/semaine.
Hémiparésie persistante
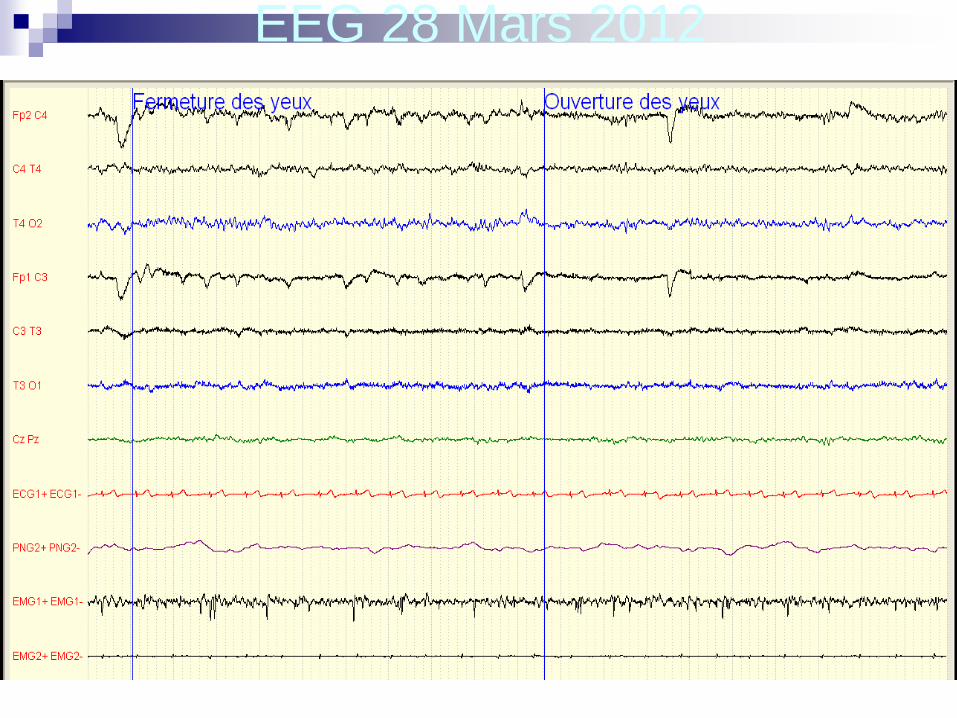
EEG 28 Mars 2012
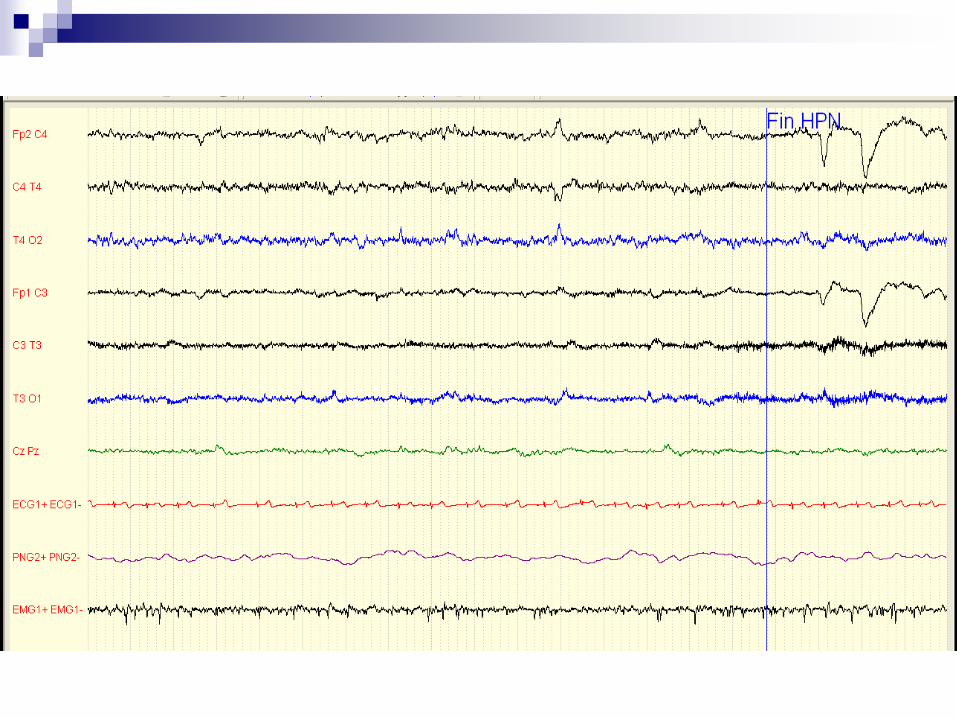
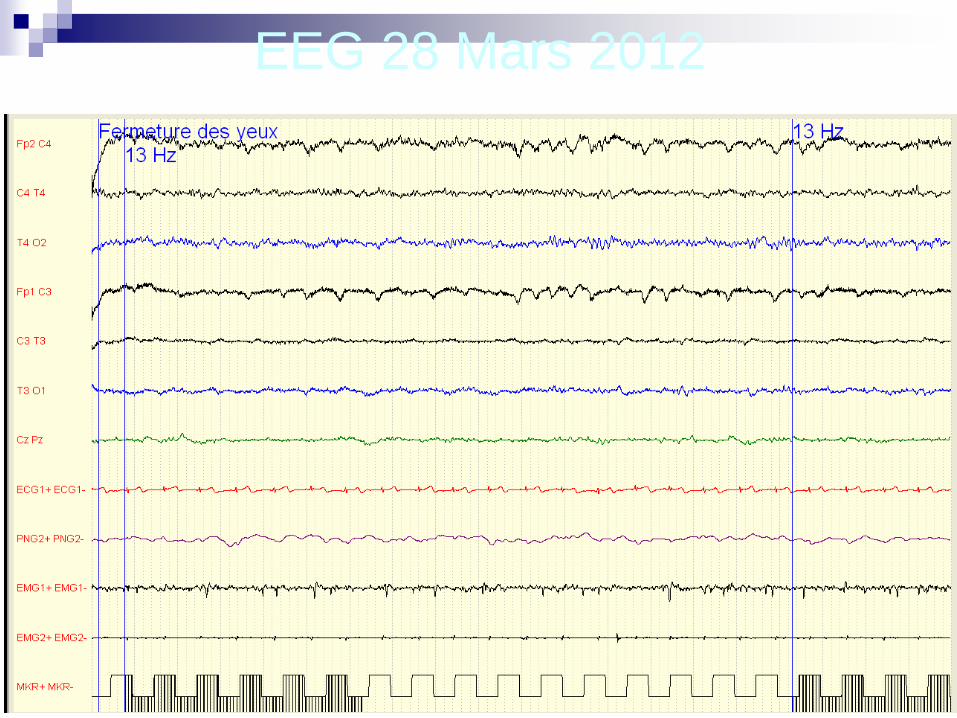
EEG 28 Mars 2012

épilepsies généralisées non idiopathiques
Lennox-Gastaut -Crises: abs atypiques, crises
toniques, TCG; début 3-5 ans
-EEG: P.O.L bifrontales 2,5 Hz
polypointes, rythmes rapides
Dans le sommeil.
-Causes diverses, parfois
symptomatique, peut succéder
aux spasmes.
-Epi rebelle, retard psycho
-Moteur, S frontal
-TRT: VPA+LTG, PTH, Taloxa
E myoclono-astatique
ou S de Doose -Crises: myoclono-astatiques,
toniques, TCG, status
Myoclonique; début 2-4 ans.
-EEG: lent, bouffées diffuses
- Pas d’étiologie mais ATCD
de CF , EGI ds la famille
-Pronostic variable ( Doose
Bénin, Doose sévère)
-TRT: VPA+ LTG, ESM, TPM,
Keppra

Mohamed né le 20 05 2008
Qui devient muet après une agression…
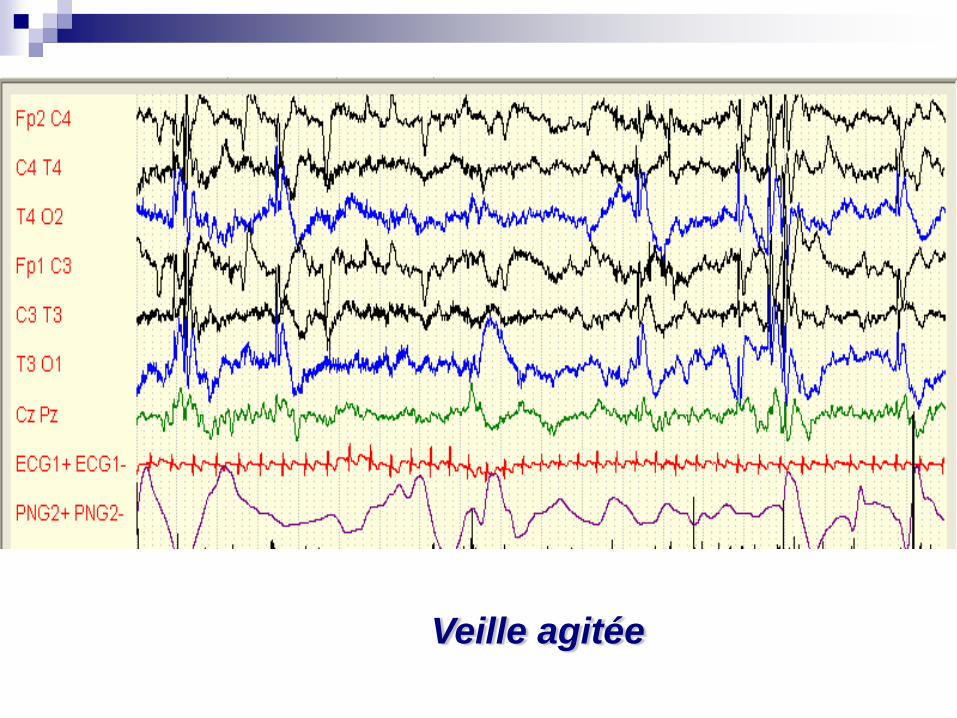
Veille agitée
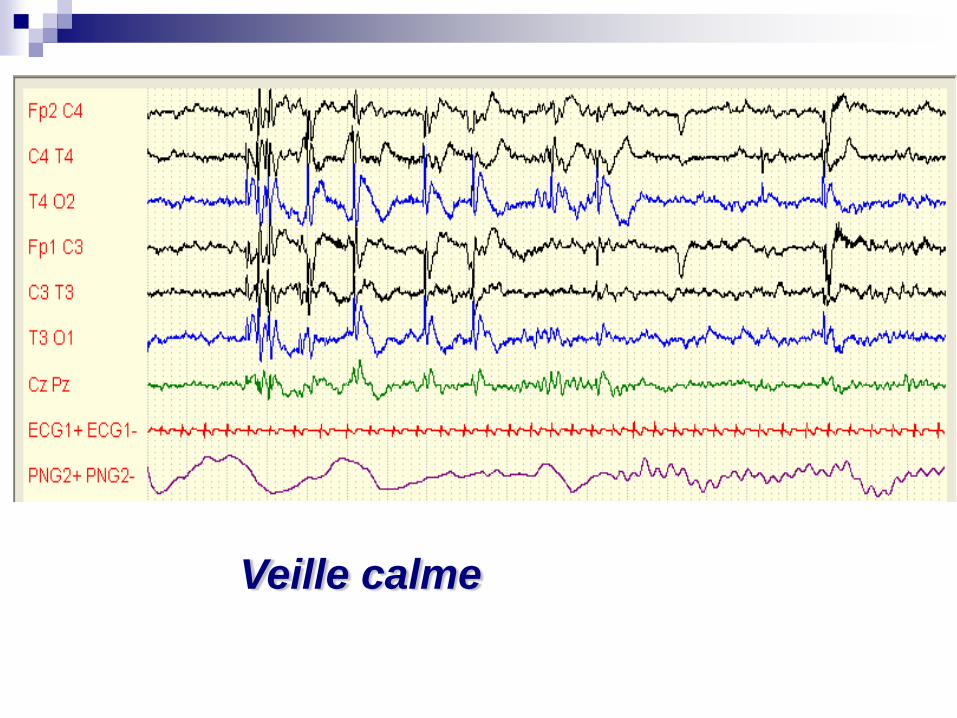
Veille calme
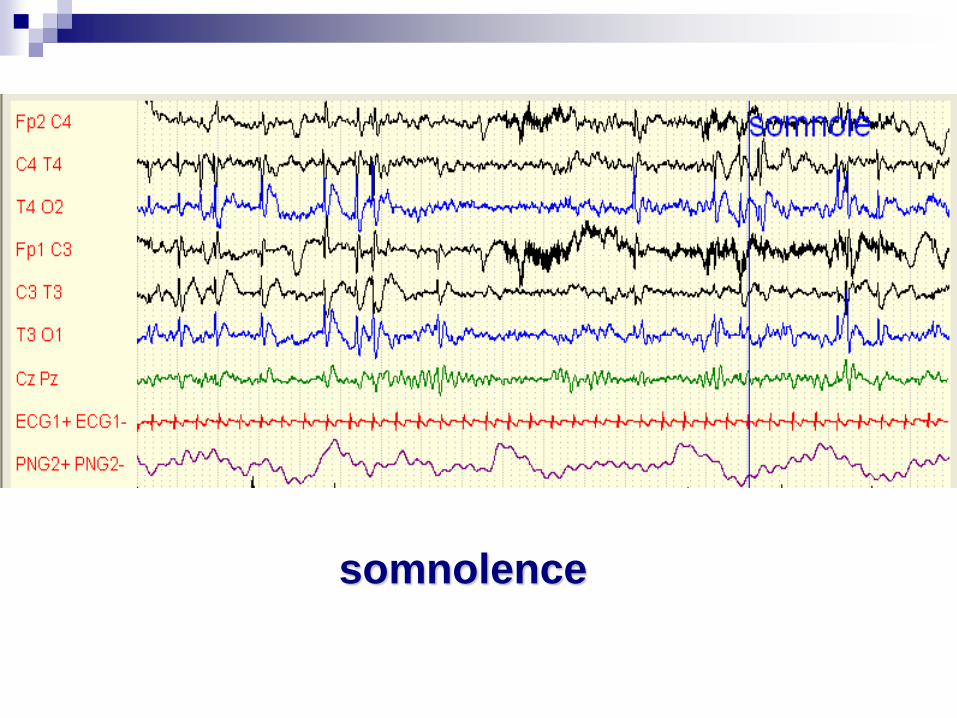
somnolence
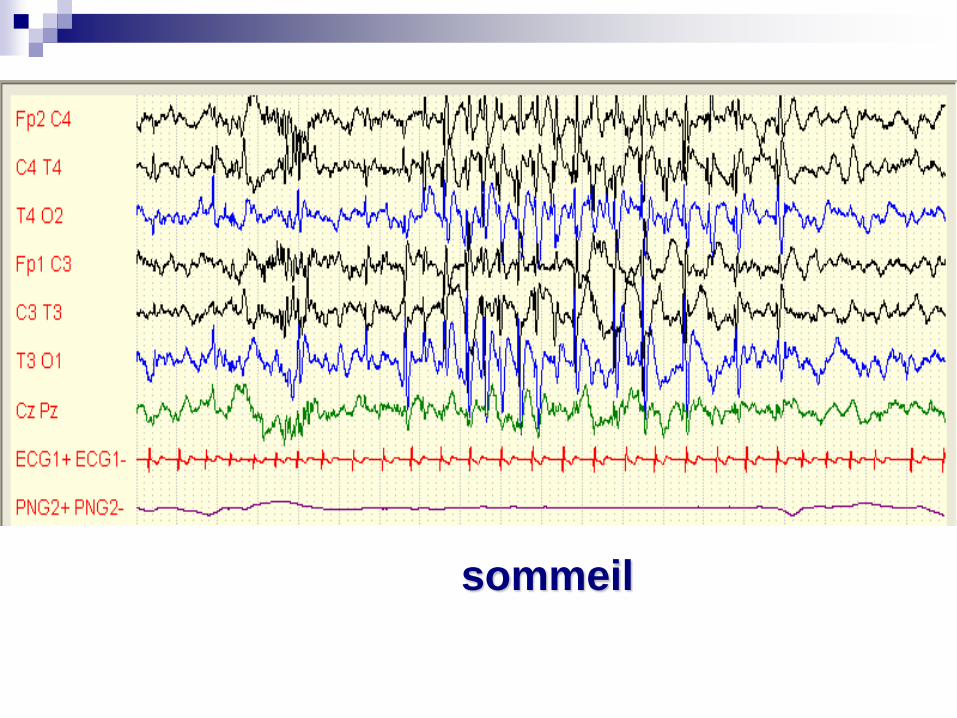
sommeil
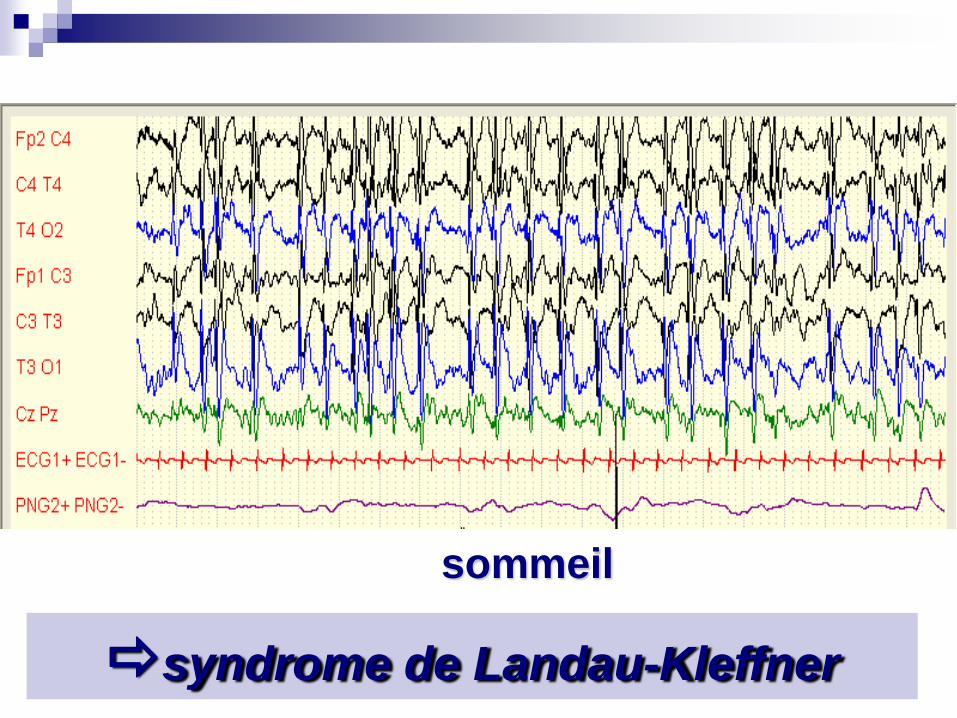
sommeil
syndrome de Landau-Kleffner

Pointes Ondes Continues du Sommeil
Crises: pas toujours au premiers plan: chutes, absences atypiques, crises partielles…. Début vers 3-4 ans
Régression psycho-motrice (tableau dissocié)
EEG: Pointes-ondes continues dans le sommeil
Faire un EEG + sieste devant une régression des acquisitions
Etiologie: cryptogénique ou symptomatique (anoxo-ischémie anté ou périnatale)
TRT:Benzo, ESM, Hydrocortisone (corticodépendance), et prise en charge éducative, suivi neuropsy, penser à l’hémisphérotomie
KEPPRA SABRIL
REGIME CETOGENE EPITOMAX
CALLOSOTOMIE
CHIRURGIE DE L’EPILEPSIE
???
inovelon
lyrica
vimpat
dépakine
dihydan
gardénal
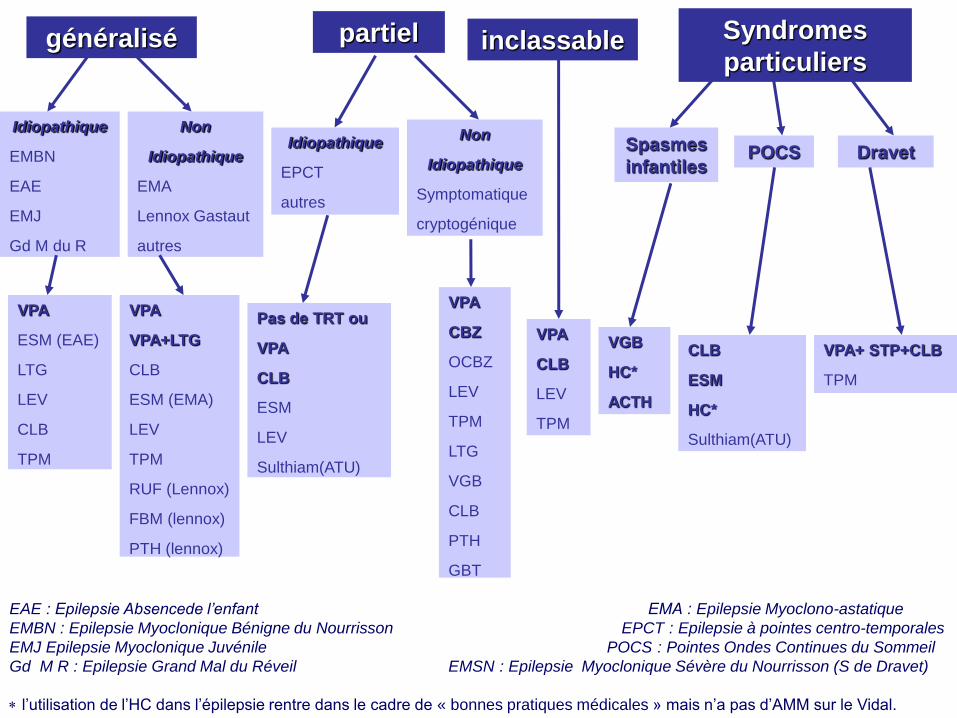
généralisé partiel inclassable Syndromes
particuliers
Idiopathique
EMBN
EAE
EMJ
Gd M du R
Non
Idiopathique
EMA
Lennox Gastaut
autres
Idiopathique
EPCT
autres
Non
Idiopathique
Symptomatique
cryptogénique
Spasmes
infantiles POCS Dravet
VPA
ESM (EAE)
LTG
LEV
CLB
TPM
VPA
VPA+LTG
CLB
ESM (EMA)
LEV
TPM
RUF (Lennox)
FBM (lennox)
PTH (lennox)
Pas de TRT ou
VPA
CLB
ESM
LEV
Sulthiam(ATU)
VPA
CBZ
OCBZ
LEV
TPM
LTG
VGB
CLB
PTH
GBT
VGB
HC*
ACTH
CLB
ESM
HC*
Sulthiam(ATU)
VPA+ STP+CLB
TPM
VPA
CLB
LEV
TPM
EAE : Epilepsie Absencede l’enfant EMA : Epilepsie Myoclono-astatique
EMBN : Epilepsie Myoclonique Bénigne du Nourrisson EPCT : Epilepsie à pointes centro-temporales
EMJ Epilepsie Myoclonique Juvénile POCS : Pointes Ondes Continues du Sommeil
Gd M R : Epilepsie Grand Mal du Réveil EMSN : Epilepsie Myoclonique Sévère du Nourrisson (S de Dravet)
l’utilisation de l’HC dans l’épilepsie rentre dans le cadre de « bonnes pratiques médicales » mais n’a pas d’AMM sur le Vidal.
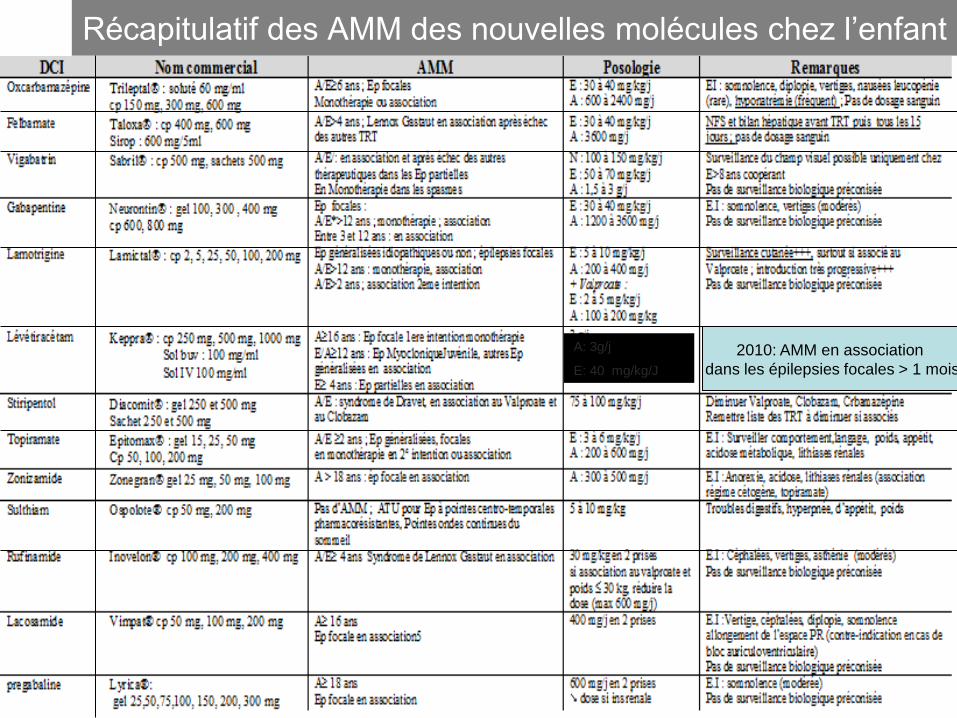
Récapitulatif des AMM des nouvelles molécules chez l’enfant
2010: AMM en association
dans les épilepsies focales > 1 mois
A: 3g/j
E: 40 mg/kg/J