bilan d’hémostase pré-opératoire dépistage des … fv bilan...• faible vpn •...
TRANSCRIPT

Bilan d’hémostase pré-opératoire et
dépistage des anomalies
DIU Maintien des compétences en anesthésie pédiatrique
Janvier 2011Dr François VOILLET

Bilan d’hémostase préopératoire
Détecter une anomalie congénitale de l’hémostase
Prévenir les complication hémorragiques
Épargner le capital veineux
Limiter la spoliation sanguine
Éviter tout examen douloureux ou invasif inutile
Limiter les examens coûteux

Bilan d’hémostase préopératoire
• Particularités physiologiques de l’enfant
• Pathologies congénitales
• Données épidémiologiques
• Évaluation clinique
• Interprétation du bilan biologique
• Hémostase et anesthésie loco-régionale

Particularités physiologiques de l’enfant
• Déficit en Vitamine K1– Hypovitaminose profonde rare (supplémentation
systématique à la naissance)– Carence modérée non exceptionnelle (allaitement maternel,
sténose du pylore)
• Immaturité hépatique– Défaut de synthèse du facteur IX (≥30%)– Valeurs adultes atteintes entre 6 et 12 mois– Les facteurs I et V se normalisent à 3 jours de vie– Les taux de fg, de facteur VIII et de facteur Willebrand sont
identiques à ceux de l’adulte

– Les plaquettes néonatales sont hyporéactives à la
thrombine, l’ADP, l’adrénaline et au thromboxane A2.
– Le temps de saignement est plus court (↑ taille des GR, ↑Ht, ↑
de l’activité et des formes multimériques du facteur de Willebrand)
– État d’hypercoagulabilité du nouveau né (↓AT3, protéines C et S, cofacteur 2 de l’héparine)
Interprétation d’un allongement du TQ ou du TCA délicate chez le nouveau né et dans les 6ers mois de vie
Particularités physiologiques de l’enfant

Pathologies congénitalesde l’hémostase

Maladie de Willebrand
• La + fréquente des anomalies constitutionnelles de l’hémostase (1 à 3% de la population)
• Transmission autosomique dominante
• Facteur de Willebrand (vWF) glycoprotéine synthétisée par les cellules épithéliales et mégacaryocytes
• Rôles du facteur de Willebrand:– Interaction plaquette-paroi vasculaire lésée et entre les plaquettes
– Transport et protection du Facteur VIII
• Taux plus faible de vWF chez les sujets de gpe sanguin O

Maladie de Willebrand
• Manifestations cliniques:– Hémorragies cutanéo-muqueuses
• Ecchymoses
• Epistaxis
• Gingivorragies
• Hémorragies sur plaies superficielles
– Maladie très hétérogènes dans son expression clinique
– La caractérisation du type et sous type est importante pour guider le choix thérapeutique
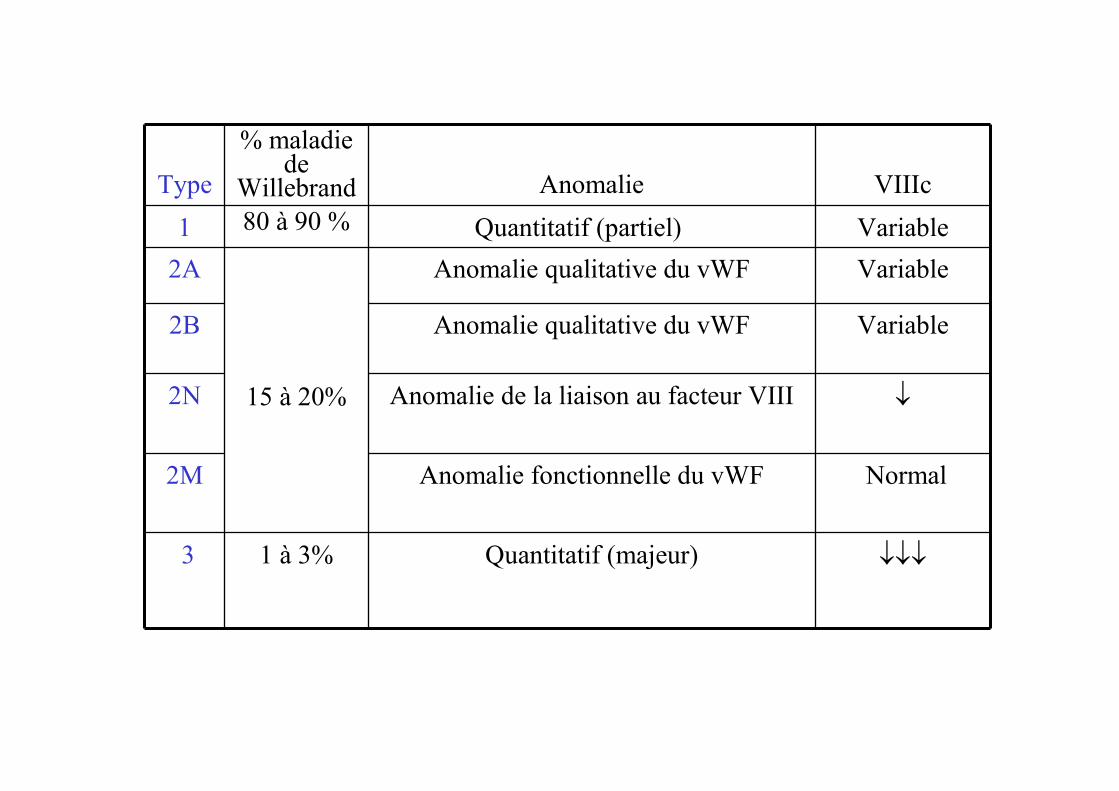
Type
% maladie de
Willebrand Anomalie VIIIc
1 80 à 90 % Quantitatif (partiel) Variable
2A
15 à 20%
Anomalie qualitative du vWF Variable
2B Anomalie qualitative du vWF Variable
2N Anomalie de la liaison au facteur VIII ↓
2M Anomalie fonctionnelle du vWF Normal
3 1 à 3% Quantitatif (majeur) ↓↓↓

Maladie de Willebrand
• Tests de dépistage– TS augmenté, TO augmenté
– TCA souvent allongé (peut être Nle)
– Plaquettes souvent Nle (sauf IIb)
• Tests spécifiques de 1° intention - Buts– Diagnostic de Willebrand
– Typage : intérêt pronostique et thérapeutique
– Activité cofacteur de la ristocétine: vWf RCO
– Dosage du vWf antigène : vWf Ag

Maladie de WillebrandTests spécifiques
ActivitActivitéé cofacteur de la cofacteur de la ristocristocéétinetine: : vWfvWf RCORCO
• Agglutination de plaquettes Nle en présence du plasma du
patient (vWf patient) et ristocétine (agrégamétrie)
• Ristocétine permet l’interaction vWf/plaquette (GPIb IX)
• Défaut d’agglutination = défaut quantitatif OU qualitatif de
vWf

Maladie de WillebrandTests spécifiques
Dosage du Dosage du vWfvWf antigantigèène : ne : vWfvWf Ag Ag
• Quantité de vWf circulant
• Par méthode ELISA
• Interprété en fonction du groupe sanguin
� Nle 50 – 200%
� Nle > 40% (groupe O)

Maladie de WillebrandTests spécifiques
Calcul du ratio Calcul du ratio vWfvWf RCO/ RCO/ vWfvWf AgAg
– Ratio proche de 1 = anomalie quantitative
– Ratio < 0.7 = anomalie qualitative
(activité du vWf trop basse par rapport au taux de vWf)

Maladie de Willebrandet Desmopressine
• DDAVP multiplie par 2 ou 3 les concentrations de vWF et FVIII chez les patients répondeurs
• Indiquée dans les Willebrand type 1
• Utile pour préparer un patient répondeur avant la chirurgie contre-indiquée chez l’enfant de moins de 2 ans.
• L’enfant doit être testé avant le jour de la chirurgie (dose test 0,2 mcg/kg en 30 min)
• Si le patient est répondeur, perfusion de DDAVP (0,3 mcg/kg), 1 h avant la chirurgie, éventuellement répétée 12 h après
• Restriction hydrique (risque d’hyponatrémie)

Hémophilie A ou B
FVIII Sévérité Traitement
< 2% Sévère FVIII
2-5 % Modérée FVIII
6- 30 % Mineure FVIII ou DDAVP
• Incidence estimée de 1/10 000 et 1/ 65 000
• Récessif lié à l’X
• La sévérité des manifestations cliniques est corrélée au taux de facteur VIII ou IX
- formes sévères (50% des cas): hématomes volumineux dès les 1ers traumatismes et/ou hémarthroses spontranées
- formes modérées et mineurs: hémorragies provoquées

Autres pathologies
• Déficit en facteur II,V et X: rares
• Déficit en facteur VII:– 1/ 500 000
– Transmission autosomique récessive
– Expression clinique variable et sévérité non corrélée aux taux de VIII
– Formes graves: hémorragies intracérébrales ou hémarthroses
– Formes modérées: hémorragies cutanéo-muqueuses ou provoquées

Autres pathologies
• Déficit en facteur XI (Hémophilie C)– 1/ 100 000– Transmission autosomique récessive– Plus fréquent chez les juifs Ashkénases– Saignements spontanés très rares– Hémorragies essentiellement post traumatiques ou post chirurgicales
• Déficit en facteur XIII– Hémorragie à la chute du cordon ombilical (90%)– Hémorragie post traumatiques retardée et progressive– Anomalie de cicatrisation
• Thrombopénie et thrombopathie

Données épidémiologiques
• Prévalence des formes asymptomatiques d’anomalies congénitales de l’hémostase avec un saignement faible: 1/ 40 000
• Prévalence des désordres acquis asymptomatiques: 1/ 2 000
• Prévalence globale des anomalies congénitales àpotentiel hémorragique: 1/ 6 500

Evaluation clinique• ATCD familiaux de saignements• ATCD personnels de saignements
• Hémorragie importante après une chirurgie mineure• Caractère retardé et touchant app locomoteur• Saignements lors d’une circoncision• Hémarthrose• Caractère immédiat et cutanéomuqueux• Epistaxis• Ecchymoses• Pétéchies• Saignement au cordon ombilical
• Prise médicamenteuse (Aspirine, AINS +++)• Notion de consanguinité
Déficit en facteur XIII
Hémophilies
Maladie de Willebrand

• Intérêt de l’utilisation de questionnaires standardisés
Evaluation clinique
Pour savoir s’il existe un risque hémorragique particulier avant une intervention chirurgicale chez votre enfant répondez par OUI ou par NON aux 10 questions suivantes:
1. Il a été transfusé ou il a sagné plus de 24h après un petit traumatisme ou une intervention mineure telle q’une circoncision, ou une suture simple
2. A la naissance, son ombilic a saigné plus de 12h après la chute du cordon
3. Il a saigné la nuit ou le lendemain après une extraction dentaire
4. Il a eu du sang dans les urines
5. Les frères ou sœurs ou vous même avaient déjà présenté l’une de ces hémorragies
6. Il se fait des bleus facilement
7. Il a été opéré pour des saignements de nez
8. Le chirurgien a dû faire une suture pour une extraction dentaire
9. Il a saigné plus d’un quart d’heure après une prise de sang
10. Vous savez qu’il saigne facilement
Pour savoir s’il existe un risque hémorragique particulier avant une intervention chirurgicale chez votre enfant répondez par OUI ou par NON aux 10 questions suivantes:
1. Il a été transfusé ou il a sagné plus de 24h après un petit traumatisme ou une intervention mineure telle q’une circoncision, ou une suture simple
2. A la naissance, son ombilic a saigné plus de 12h après la chute du cordon
3. Il a saigné la nuit ou le lendemain après une extraction dentaire
4. Il a eu du sang dans les urines
5. Les frères ou sœurs ou vous même avaient déjà présenté l’une de ces hémorragies
6. Il se fait des bleus facilement
7. Il a été opéré pour des saignements de nez
8. Le chirurgien a dû faire une suture pour une extraction dentaire
9. Il a saigné plus d’un quart d’heure après une prise de sang
10. Vous savez qu’il saigne facilement

Ecchymoses et Tb de l’hémostase
• Questionnaire standardisé
• 228 enfants sans ATCD de saignement
• Comparaison à 31 patients avec tb de l’hémostase– Bleus « faciles »: 24% vs 67%
– Bleus au mois 1 fois/sem: 36% vs 68%
– Epistaxis: 39% vs 69%
– Bleus sur plusieurs parties du corps: 4,9% vs 38,5%
– Bleus étendus: 3,5% vs 29,6%
– Hématome étendu: 2,7% vs 21,7%
Bleeding/bruising symptomatology in children with and without bleeding disorders.Nosek-Cenkowska et al. Thromb Haemost 1991; 65: 237-41

Épistaxis et Tb de l’hémostase
• Étude rétrospective– Screening 3681 enfants, 178 présentant des épistaxis récurrentes– 103 garçons/75 filles– Âge médian: 84 mois [15-219]– 33% (n=59) porteur d’une coagulopathie:
• Maladie de Willebrand: 56%• Défaut d’agrégation plaquettaire: 17%• Thrombopénie: 12%• Déficit facteur VIII modérée: 5%• Syndrome de Bernard-Soulier: 3%• Déficit en facteur VII: 1,5%• Déficit en facteur IX: 1,5%• Déficit en facteur XI: 1,5%• ACC: 1,5%
Clinical and laboratory features of 178 children with recurrent epistaxis.Sandoval et al. J Pediatr Hematol Oncol; 24: 47-9

• Examen clinique– En général normal– Signes cliniques orientant vers une pathologie de l’hémostase:• Ecchymoses, hématomes• Pâleur (anémie congénitale ou acquise)• Ictère, hépatomégalie, splénomégalie• Pétéchies• Hémarthrose• Douleur, déformation osseuse (leucémie)• Epaississement de la peau et de la langue (amylose)
Évaluation clinique

Bilan biologique préopératoire
• Enfant ayant déjà eu un contrôle de l’hémostase normal: (au-delà de l’âge de 6 mois)
⇒ pas de bilan
• Enfant ayant acquis l’âge de la marche et intervention mineure: ⇒ pas de bilan
• Dans les autres cas, bilan systématique:
⇒ NFS plaquettes
⇒ TP, TCA
• Si interrogatoire positif, exploration de l’hémostase primaire:
TS, PFA100

Problèmes pratiques de prélèvement
• Difficulté technique
• Prélèvements sur microtubes souvent coagulé
• Interprétation difficile des test globaux chez le nouveau-né (rapport citrate/ volume plasmatique)
• Possibilité de recours aux techniques de prélèvements capillaires par microméthode chez le nouveau né et le prématuré

Interprétation du bilan biologique
• Bilan initial anormal à recontrôler
Burk et al, Pediatrics, 1992
- 1603 (3 à 16 ans) avant amygdalectomie
- Bilan préopératoire: plaquette,TP, TCA,TS
- 31/ 1 603 soit 2% résultats anormaux
- 15/ 1 603 soit 1% résultats anormaux confirmés
(14 TCA allongé et 1 TS allongé)
11 ACC antiprothrombinase
1 Hémophilie A atténuée
1 maladie de Willebrand

Anomalie de l’hémostase primaire

Le temps de saignement
• Exploration in vivo des fonctions plaquettaires et vasculaire
• Techniques:– Duke
• Incision lobe de l’oreille
• Abandonnée (peu reproductible)
– Ivy:• Tensiomètre à 40 mm Hg
• Incision face antérieure de l’avant bras au mieux standardisée (Surgicutt junior)
• Valeur normale < 8 mn

Intérêt du temps de saignement
• A abandonner en systématique• Faible VPN• Difficulté technique à assurer une reproductibilitédu test
• Garde un intérêt en cas de doute sur l’hémostase primaire (histoire clinique positive, TCA et num plaquettaire Nx)
• Tend à être remplacer par les études des fonction plaquettaires in vitro: Platelet Function Analyser (PFA)
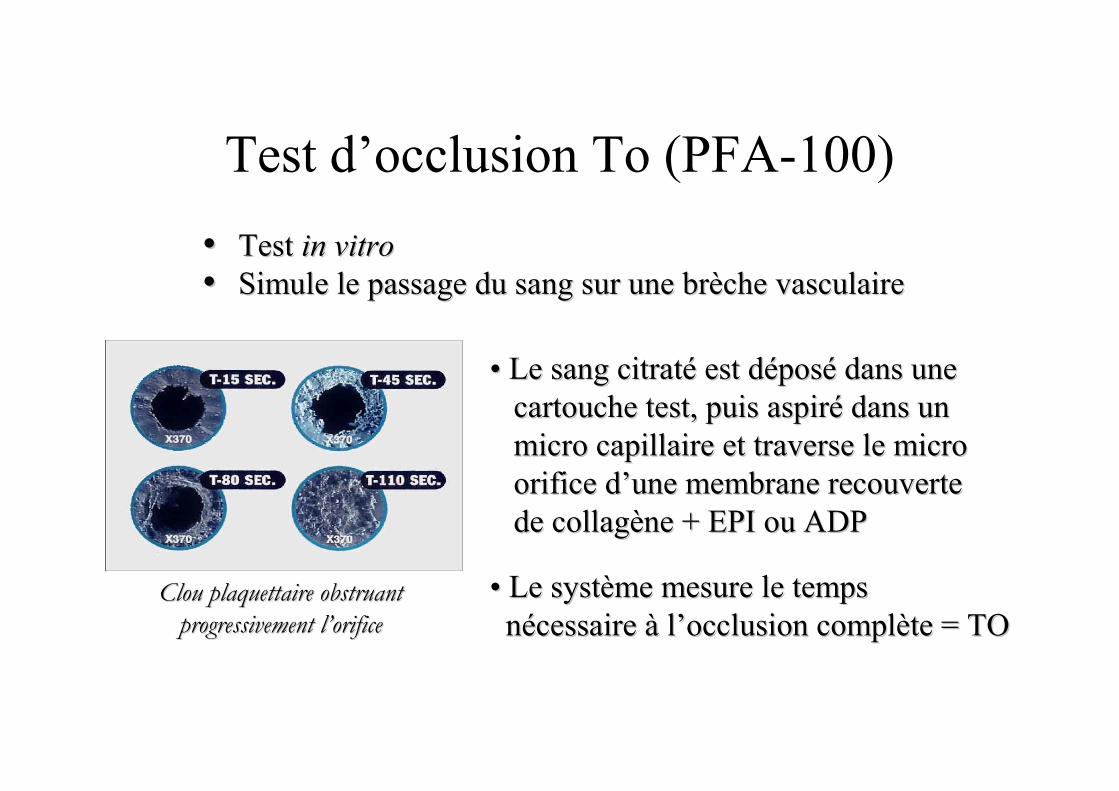
Test d’occlusion To (PFA-100)
•• Test Test in vitroin vitro
•• Simule le passage du sang sur une brSimule le passage du sang sur une brèèche vasculaireche vasculaire
•• Le sangLe sang citratcitratéé est dest dééposposéé dans une dans une cartouche test, puis aspircartouche test, puis aspiréé dans un dans un micro capillaire et traverse le micro micro capillaire et traverse le micro orifice dorifice d’’une membrane recouverte une membrane recouverte de collagde collagèène + EPI ou ADPne + EPI ou ADP
•• Le systLe systèème mesure le temps me mesure le temps nnéécessaire cessaire àà ll’’occlusion complocclusion complèète = TOte = TO
Clou plaquettaire obstruant Clou plaquettaire obstruant
progressivement lprogressivement l’’orificeorifice

• Conditions pré analytiques de la réalisation du test– Prélèvement veineux, pli du coude, aiguille 19G
– Aiguilles 21G ou 23G possible mais l’écoulement doit être facile et sans interruption
– TO ininterprétable si:• Ht<30% ou Hb<6g/dL
• Plaq< 70 G/L
– Réalisation du test dans un délai de 3h après prélèvement
Test d’occlusion To (PFA-100)

Test d’occlusion To (PFA-100)
• Valeurs normales– TO/CEI: 95 et 170 sec (moy 133 sec)
– TO/CADP: 70 et 120 sec (moy 92 sec)
• Variations physiologiques:– TO nouv né < TO enfant/adulte
– TO groupe O > TO autres groupes sg

Test d’occlusion To (PFA-100) et maladie de Willebrand
• 52 enfants (6 mois-18ans)
• Comparaison Se et Sp TS et PFA-100 (Col/EPI):– Anomalie qualitative plaquettaire:
• TS: Se=37% Sp=88%
• PFA-100: Se=100% Sp=97%
– Maladie de Willebrand:• TS: Se=17%
• PFA-100: Se=100%
Comparaison of PFA-100 and Bleeding Time Testing in Pediatric Patients With SuspectedHemorrhagic ProblemsCariappa et al. J Pediatr Hematol Oncol; 2003; 25: 474-9

• Avantages par rapport au TS: – Meilleure Se pour thrombopathies majeures constitutionelles
(Glanzmann, Bernard-Soulier, Sd aspirine-like)
– Meilleure Se pour formes sévères de Willebrand (type 3, type 1)
– Fait parti des tests de réponse au DDAVP– Non opérateur dépendant
• Limites : – N’explore pas les facteurs vasculaires– Moins sensible pour les thrombopathies sécrétoires modérées
Test d’occlusion To (PFA-100)

Thrombopénies

Thrombopénies
1. Vraie thrombopénie ?– Frottis � agrégats plaquettaires– Prélèvement sur citrate
2. Atteinte des autres lignées ? (orientation diagnostic)– GR : schizocytes, anisocytose, anémie,…– GB : blastes, granulations anormales, …– Plq : morphologie des plaquettes
3. Origine centrale ou périphérique ?– myélogramme

• Origines centrales– congénitale :
� May Hegglin : plq géantes, inclusions basophiles
dans les neutrophiles (corps de Döhle)
� Wiskott Aldrich : petites plq, immunodépression, eczéma
– acquise: aplasie, hémopathies malignes, métastases, intox. alcoolique aiguë, benzène, médicaments,carence en
folate
Thrombopénies

Thrombopénies
• Origines périphériques- Dilution
� Transfusions massives- Consommation
� CIVD� MAT (Purpura Thrombotique
Thrombocytopénique , Syndrome Hémolytique et Urémique)
- Par anomalie de répartition :� Hypersplénisme

Thrombopénies
• Origines périphériques- immunologique
� Purpura Thrombopénique Auto-Immun (PTAI)
- infections,MAI, hémopathies
- Purpura Thrombopénique Idiopathique
� immuno-allergique
� allo-immunisation : thrompopénies néonatales (allo-Acanti-PLT d’origine maternelle) et thrombopénies post-transfusionnelles

Thrombopathies

• Acquises : - associées aux SMP, LA, Dysglobulinémies, IHC, IRC,…
- médicamenteuses
• Constitutionnelles : rares (1/10 Millions)� Dystrophie de Bernard-Soulier : déficit GP Ib-IX (adhésion)
� Thrombasthénie de Glanzmann : déficit GP IIb-IIIa (agrégation)
� Maladie du pool vide : diminution contenu granules denses
� Syndrome des Plaquettes grises : diminution contenu granules α
� Déficit en Cyclo-oxygénase
Thrombopathies

Agrégamétrie photométrique
• Mise en évidence de l’agrégation par photométrie• Après ajout d’agonistes
� ADPagrégation dépendant de fib/GP IIb IIIa
� Ristocétineagrégation dépendant de GP Ib / vWf
� Collagèneagrégation dépendant de la dégranulation et de la production de thromboxane A2
� Ac. Arachidonique = suspicion de pathologie de ladégranulation
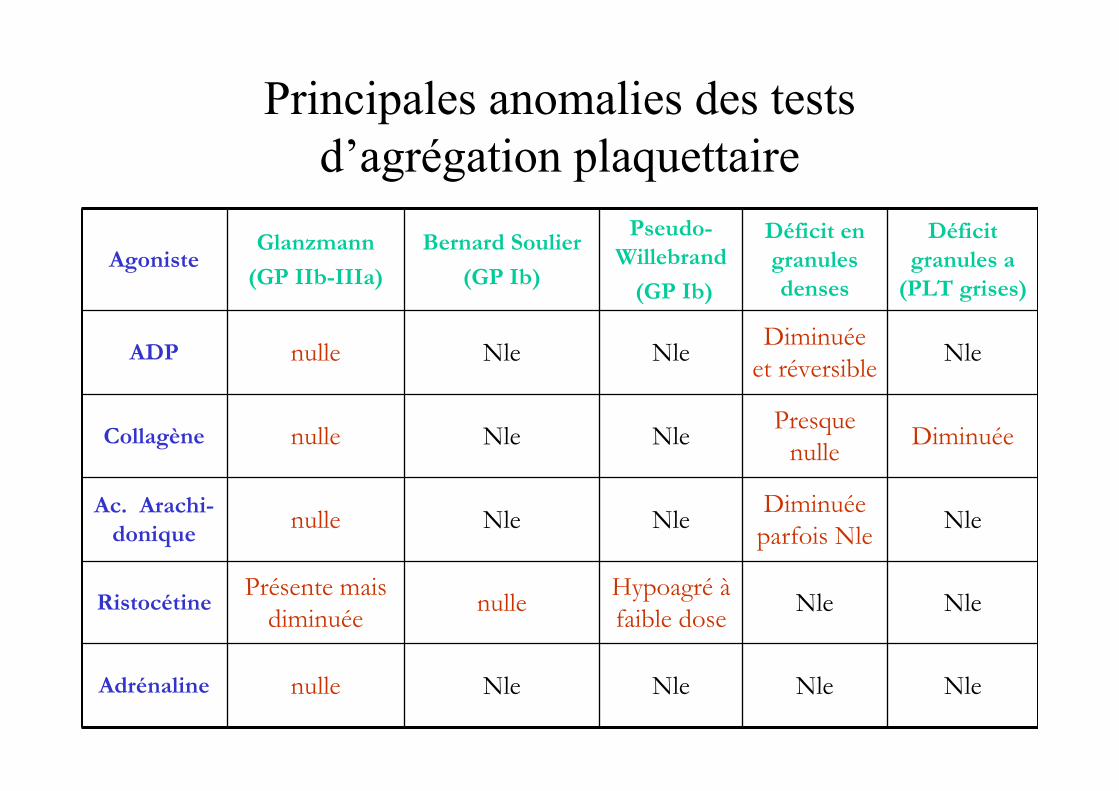
Principales anomalies des tests d’agrégation plaquettaire
AgonisteGlanzmann
(GP IIb-IIIa)
Bernard Soulier
(GP Ib)
Pseudo-
Willebrand
(GP Ib)
Déficit en
granules
denses
Déficit
granules a
(PLT grises)
ADP nulle Nle NleDiminuée
et réversibleNle
Collagène nulle Nle NlePresque
nulleDiminuée
Ac. Arachi-
doniquenulle Nle Nle
Diminuée
parfois NleNle
RistocétinePrésente mais
diminuéenulle
Hypoagré à
faible doseNle Nle
Adrénaline nulle Nle Nle Nle Nle
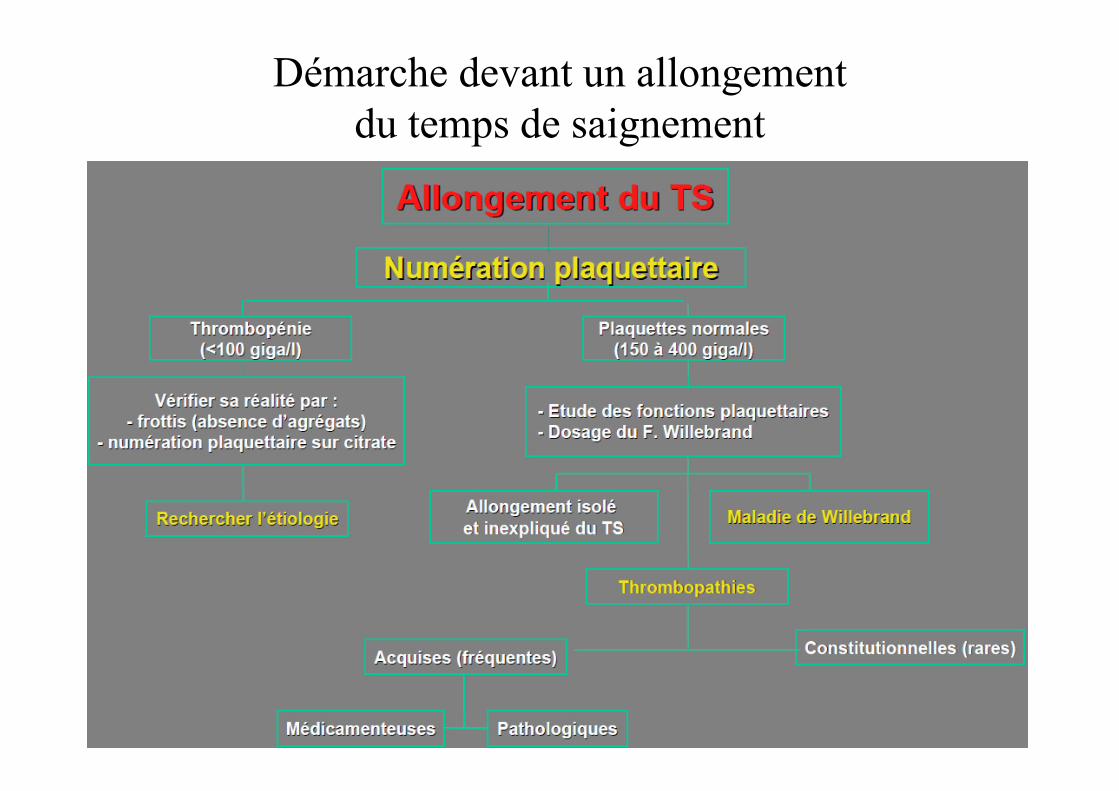
Démarche devant un allongement du temps de saignement

Anomalie de la coagulation
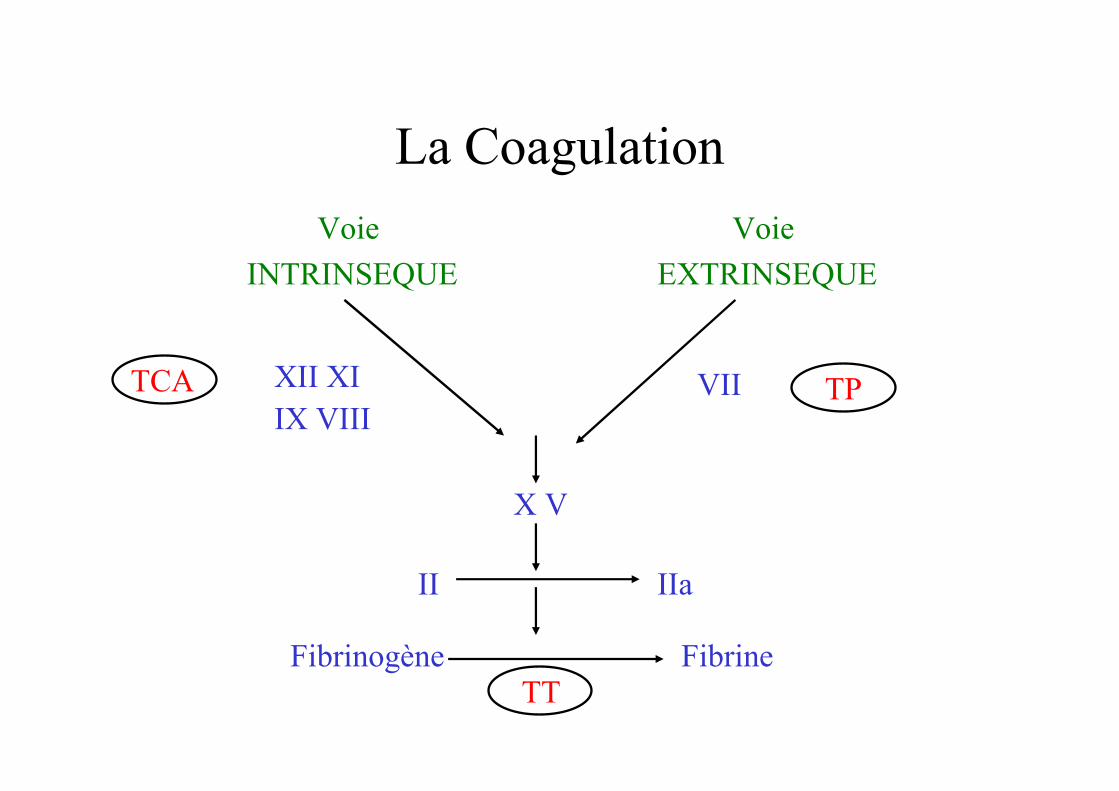
La Coagulation
Voie
INTRINSEQUE
Voie
EXTRINSEQUE
XII XIIX VIII
TCA VII TP
X V
II IIa
Fibrinogène FibrineTT

Allongement du Temps de Quick
• Déficit en vitamine K– Nouveau-né (prématuré)– Carence d’apport– Trouble de l’absorption digestive– Antibiothérapies itératives
• Déficit en Facteur VII– Non exceptionnel (1/500 000)
• Anomalie du fibrinogène

• Valeur normale du nouveau né: 1,3 – 1,5 (témoin adulte)
• Etiologies:– Anticoagulants circulants (anti phospholipides)
• ↑ TCA non corrigé par plasma témoin
• Contexte d’infection virale
• Pas de risque hémorragique associé le plus souvent
– Déficit en facteur XII• Pas de risque hémorragique associé
– Maladie de Willebrand
Allongement du TCA

Allongement du TCA
• Etiologies:– Déficit congénital en facteur VIII (hémophilie A)
• Se du TCA varie en fonction de la sévérité du déficit:� 100% pour déficit sévère
� 99% pou déficit modéré
� 90% pour déficit mineur
– Déficit congénital en facteur IX (hémophilie B)
– Déficit congénital en facteur XI
– Anomalie du fibrinogène
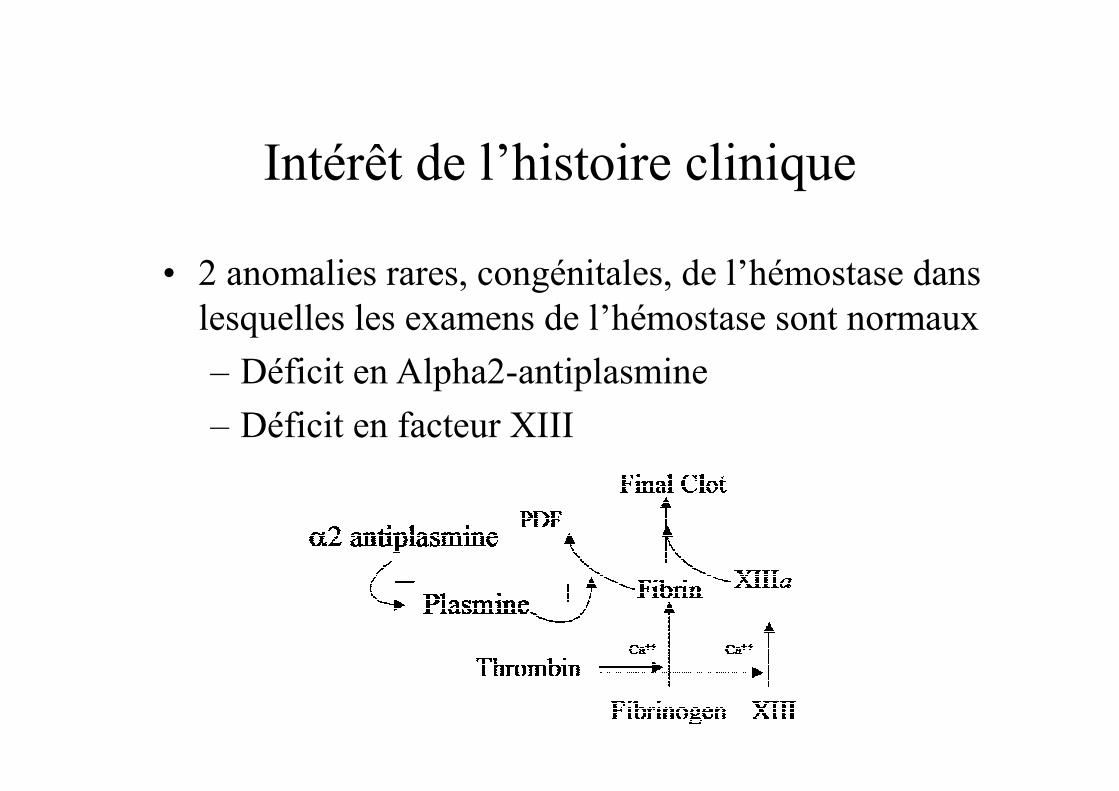
Intérêt de l’histoire clinique
• 2 anomalies rares, congénitales, de l’hémostase dans lesquelles les examens de l’hémostase sont normaux
– Déficit en Alpha2-antiplasmine
– Déficit en facteur XIII
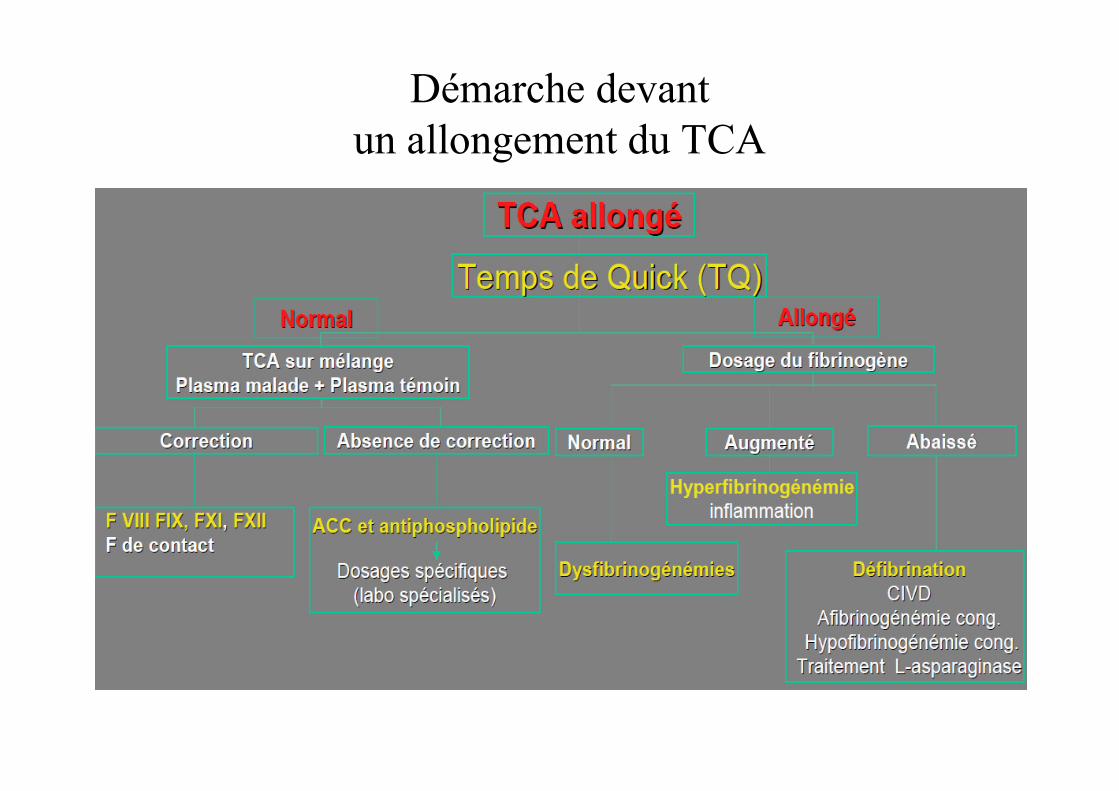
Démarche devant un allongement du TCA
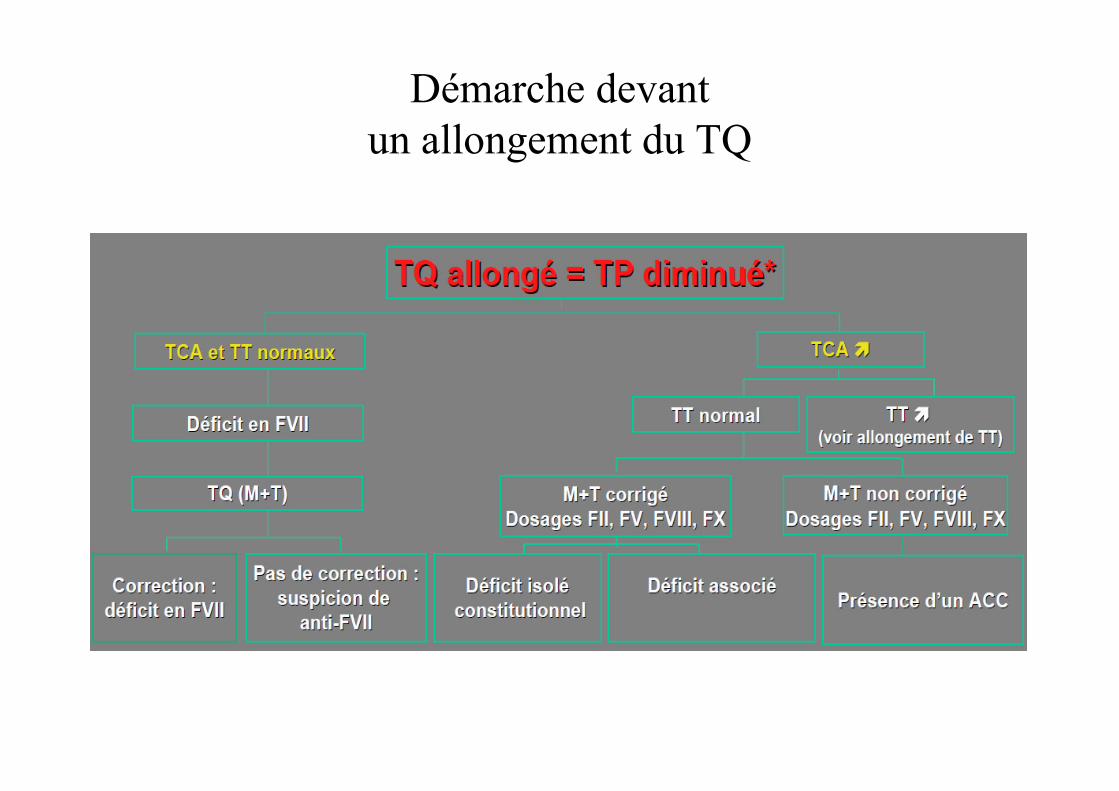
Démarche devant un allongement du TQ

Hémostase et ALR
• Coagulopathie = CI aux blocs périnerveux profonds, périmédullaires et aux blocs intéressant un territoire àvascularisation terminale.
• Quel que soit l’âge de l’enfant,examen clinique et anamnèse minutieuse sur les ATCD personnels et familiaux.
• Pas de bilan biologique systématique lorsque la marche est acquise et l’étape clinique totalement négative.
• Marche non acquise ou bilan biologique nécessaire, limiter le bilan à un TCA et une numération plaquettaire.
• Toute anomalie du bilan initial persistant après contrôle doit être explorée après avis éventuel de l'hémobiologiste.
Anesthésie loco-régionale en pédiatrie, RFE SFAR/ADARPEF2010

Thrombose
• Incidence très faible des thromboses chez l’enfant : 0.07 /100 000
• Peu de TV idiopathiques
• Facteurs acquis• Stase vasculaire (cathéter veineux central, immobilisation…)• Maladie inflammatoire sévère• Anticorps antiphospholipides
• Déficits héréditaires des inhibiteurs de la coagulation• ATIII (<85%)• Proteine C (<65%)• Proteine S (<65%)• Dysfibrinogenemie• facteur V leiden• Mutation du gène de la prothrombine

Conclusion
• Difficulté de la prescription du bilan d’hémostase en pédiatrie
• Importance de l’enquête anamnestique (intérêt de l’utilisation de questionnaires standardisés)
• La qualité du dialogue biologiste/clinicien est capitale pour orienter les investigations
• Fréquence élevée de la maladie de Willebrand