spectroscopie et réactivité des complexes. a...
TRANSCRIPT

Spectroscopie et réactivité des complexes.ANNÉE 2013-2014
Martin VÉROT

Le sujet est composé de plusieurs parties largement indépendantes.
TABLE DES MATIÈRESA. Une liaison quintuple entre deux atomes de molybdène 2
A.I Questions générales . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2A.II Analyse de la liaison molybdène-molybdène . . . . . . . . . . . . . . . . . . . . . . . 2A.III Analyse de l’indice de liaison pour certains complexes de molybdène . . . . . . . . . 3A.IV Analyse de l’indice de liaison pour le complexe 4 . . . . . . . . . . . . . . . . . . . . . 4
B. Des règles de Wade/Mingos aux ions de Zintl 6B.I B6H2−
6 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6B.II Les règles de Wade/Mingos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6B.III Harmoniques sphériques de vecteurs de surface . . . . . . . . . . . . . . . . . . . . . 7
B.III.1 Cas des orbitales s . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7B.III.2 Cas des orbitales p de surface . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
B.IV Analyse des liaisons dans le cas de cluster closo . . . . . . . . . . . . . . . . . . . . . . 11B.IV.1 Théorème d’Euler-Descartes . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11B.IV.2 Nombre d’orbitales liantes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
B.V Analyse des liaisons dans le cas de cluster nido . . . . . . . . . . . . . . . . . . . . . . 12B.VI L’ion [Pt@Pb12]
2− . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
C. Luminescence du vanadium dans une matrice de blende 15C.I Structure cristalline . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15C.II Spectre d’émission . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15C.III Paramètres de Racah . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
D. Physico-chimie des complexes carbéniques de métaux de transition 19D.I Catalyse de métathèse des oléfines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
D.I.1 Dissociation de la phosphine . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20D.I.2 Compétition entre dissociation de PCy3 et coordination de 1’o1éfine . . . . . 21
A Problème A. 28
B Problème B. 28
Toutes les réponses devront être justifiées et les résultats mis en valeur.
1 / 28

A. UNE LIAISON QUINTUPLE ENTRE DEUX ATOMESDE MOLYBDÈNE
(Inspiré de Angew. Chem. Int. Ed., 2013, 52, 3227–3231)
Cette année, l’équipe d’Ernesto Carmona a pu isoler un complexe binucléaire de molybdènepossédant formellement une liaison quintuple entre les atomes de molybdène. Ce type de liaisonest très intéressant car sa réactivité est encore mal connue.
Mo Mo
N N N N
DippDipp
Dipp
Dipp
x
y
z
4
Figure 1 – Complexe présentant une « liaison quintuple ». Dipp : 2,6-diisopropylphényl.
Dans un premier temps, nous allons étudier l’interaction entre les atomes de molybdène etceux du ligand azoté, puis nous allons prendre en compte le rôle de la partie aromatique.
A.I Questions générales
A.1. Quel est l’origine du nom molybdène ?
A.2. Donner une utilisation de ce métal.
A.3. En quelle année (à 50 ans près) a été découvert le molybdène ?
A.4. Représenter le 2,6-diisopropylphényl.
A.II Analyse de la liaison molybdène-molybdène
A.5. Donner la configuration électronique fondamentale d’un atome de molybdène. Quelle parti-cularité a cette configuration électronique ?
A.6. Quel est le groupe de symétrie du complexe 4 ?
A.7. Donner les étiquettes de symétrie associées aux 5 orbitales d d’un seul molybdène dans legroupe de symétrie du complexe 4.
On va étudier le fragment Mo-Mo.
A.8. À priori, pour la création du fragment on devrait avoir un recouvrement entre 4 orbitales demême symétrie, expliquer pourquoi on peut se limiter à 2 interactions entre 2 orbitales.
A.9. Représenter les orbitales de symétrie de Mo2. Donner les étiquettes de symétrie des 10 orbi-tales obtenues dans le groupe de symétrie du complexe 4.
2 / 28

A.10. Donner le type de recouvrement entre les différentes orbitales (σ, π, δ, γ).A.11. Compléter la figure 21 donnée en annexe en précisant les étiquettes de symétrie des diffé-
rentes orbitales.A.12. Prédire l’indice de liaison pour Mo(0)-Mo(0), Mo(I)-Mo(I), Mo(II)-Mo(II). Commenter.
A.III Analyse de l’indice de liaison pour certains complexes de molybdène
Le groupe de Carmona a synthétisé une suite de complexes faisant intervenir des ligands de typeformamidinate.
Mo Mo
NN
NNOO
O O
Dipp
DippDipp
Dipp
1) LiMe, THF, − 10 °C
2) Toluène, 100 °C
3) H2, THF, 20 °C
Mo Mo
NN
NN
Dipp
DippDipp
Dipp
H
HTHF
THF
1 2
Mo Mo
NN
NN
Dipp
DippDipp
Dipp
H
HTHF
THF
3
Mo Mo
NN
NN
Dipp
DippDipp
Dipp
2
hν (C6H12), 25 °C
THF, 25 °C+H2
Figure 2 – Série de complexes synthétisés par le groupe de Carmona.
Pour les ligands formamidinates, une charge est délocalisée sur l’enchaînement N-C-N.A.13. De quelle manière se coordonne le ligand formamidinate ? Comment le noterai-t-on dans la
formule du complexe ?A.14. Dans le formalisme de Green, quel est le type du ligand acétate ?A.15. De même, quel est le type du ligand formamidinate dans le formalisme de Green ?A.16. En déduire le degré d’oxydation de chaque atome de molybdène au sein du complexe 1.A.17. La représentation de Lewis pour la liaison Mo-Mo dans le complexe 1 correspond-t-elle au
résultat de la question A.12 ?A.18. Faire la même analyse pour le complexe 2 : donner le type des ligands hydrures et THF dans
le formalisme de Green. Commenter la représentation de Lewis donnée pour le complexe 2par rapport au résultat de la question A.12.
A.19. À quel grand type de réaction correspond le passage de 2 à 3 ?A.20. En déduire le degré d’oxydation de chaque atome de molybdène dans le complexe 3 et
commenter par rapport au résultat de la question A.12.
On peut obtenir le complexe 4 à partir du complexe 2 ou du complexe 3.
3 / 28
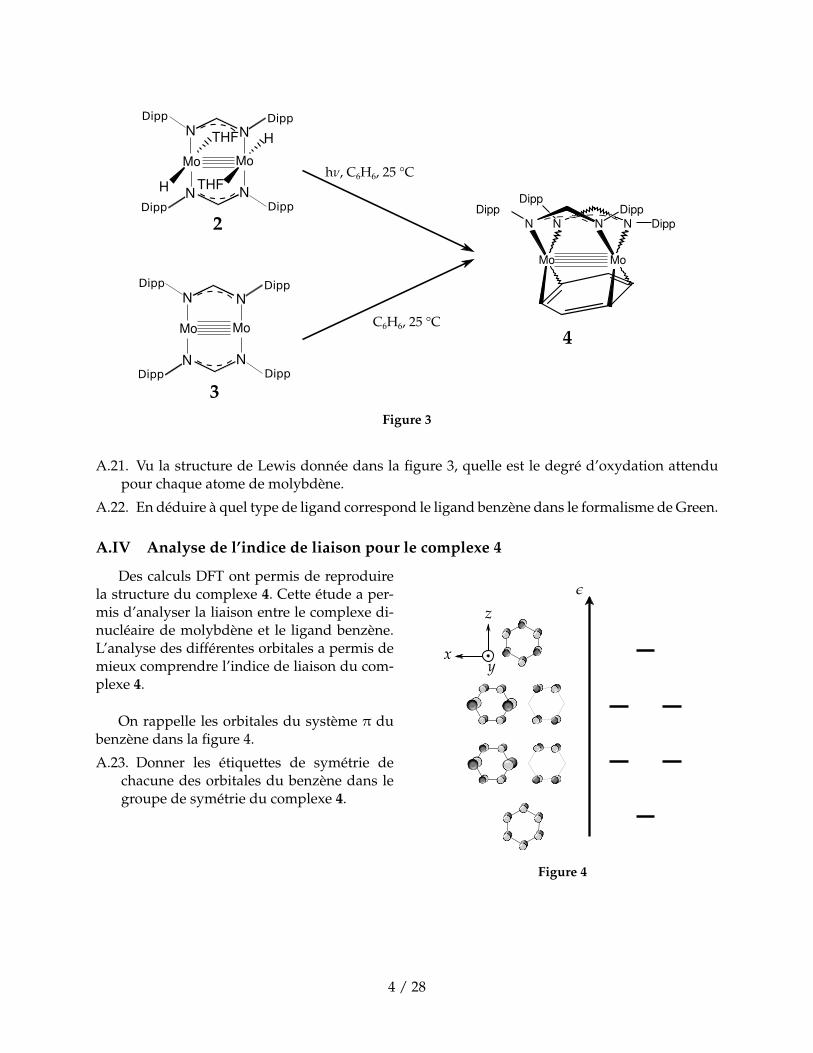
Mo Mo
N N N N
DippDipp
Dipp
Dipp
Mo Mo
NN
NN
Dipp
DippDipp
Dipp
H
HTHF
THF
2
3
Mo Mo
NN
NN
Dipp
DippDipp
Dipp
C6H6, 25 °C
hν, C6H6, 25 °C
4
Figure 3
A.21. Vu la structure de Lewis donnée dans la figure 3, quelle est le degré d’oxydation attendupour chaque atome de molybdène.
A.22. En déduire à quel type de ligand correspond le ligand benzène dans le formalisme de Green.
A.IV Analyse de l’indice de liaison pour le complexe 4
xy
z
Figure 4
Des calculs DFT ont permis de reproduirela structure du complexe 4. Cette étude a per-mis d’analyser la liaison entre le complexe di-nucléaire de molybdène et le ligand benzène.L’analyse des différentes orbitales a permis demieux comprendre l’indice de liaison du com-plexe 4.
On rappelle les orbitales du système π dubenzène dans la figure 4.
A.23. Donner les étiquettes de symétrie dechacune des orbitales du benzène dans legroupe de symétrie du complexe 4.
4 / 28

On supposera par la suite que seules les orbitales qui donnent naissance aux orbitales 1 à4 évoluent énergétiquement lors de l’interaction entre les deux fragments. (Elles sont entouréesfigure 5.)
1
2
3
4
Figure 5 – Interaction entre le fragment Mo2L2 et le ligand benzène.
A.24. Quelles sont les étiquettes de symétrie des orbitales entourées qui interagissent ?A.25. Représenter schématiquement les quatre orbitales 1 à 4 de la figure 5.A.26. Commenter la nouvelle distribution des électrons dans le complexe 4.A.27. Qualifier le ligand benzène en terme de ligand donneur et/ou accepteur.A.28. Quel autre ligand présente les mêmes caractéristiques ? Quel est le nom du modèle permet-
tant de décrire la liaison pour cet analogue ?A.29. Combien y a-t-il d’électrons dans des orbitales à caractère essentiellement métallique ?A.30. Parmi ces orbitales à caractère métallique, combien sont liantes, non liantes et antiliantes ?A.31. En déduire l’indice de liaison pour la liaison Mo-Mo. Donner une formule de Lewis en
accord avec votre analyse.A.32. Si celle-ci diffère de celle donnée à la figure 1, expliquer l’origine de la différence. Le degré
d’oxydation des atomes de molybdène a-t-il changé ?
Complexe 2 3 4dMo-Mo (Å) 2,089 2,033 2,106
Tableau 1 – Distance entre les atomes de molybdène au sein des différents complexes (figure 3).Ces mesures ont été faites grâce aux structures cristallographiques.
A.33. Expliquer l’évolution des distances Mo-Mo données au tableau 1.
5 / 28

B. DES RÈGLES DE WADE/MINGOS AUX IONS DEZINTL
(Inspiré de Inorg. Chem., 1981, 20, 563–571 ; Polyhedron, 1984, 3, 1299–1306 ; Inorg. Chem., 1982, 21,2297–2302 ; Angew. Chem. 2004, 116, 2184–2186)
Tous les clusters de bore de formule BnHn+mq− adoptent des structures basées sur des polyèdres
réguliers avec des faces triangulaires.Les règles de Wade/Mingos ont été développées dans les années 70, elles différencient les
skeleton electron pairs (SEP) des autres liaisons au sein des cluster. Elles ont permis de grandesavancées dans la compréhension de leur structure. Par la suite, ces règles ont été étendues auxclusters incluant du carbone et des métaux de transition.
B.I B6H2−6
B.1. Combien y’a-t-il d’électrons de valence pour B6H62− ? (n = 6, m = 0, q = 2)
B.2. Sachant qu’il y a 6 liaisons BH au sein de ce cluster, combien reste-il d’électrons pour formerla cage de bore ?
B.3. Tous ces électrons servent à former des SEP, en déduire leur nombre et faire un lien simpleentre le nombre de SEP et le nombre de sommets n dans le cas des clusters closo.
B.II Les règles de Wade/Mingos
Les règles de Wade font le lien entre le nombre de SEP et la structure.
Décompte Nom Structure anticipéeélectronique4n− 2 Closo bicoiffée polyèdre closo à n− 2 sommets avec 2 faces coiffées4n Closo coiffée polyèdre closo à n− 1 sommets avec 1 faces coiffées4n + 2 Closo polyèdre closo à n sommets4n + 4 Nido polyèdre closo à n + 1 sommets et un sommet manquant
(celui en rouge sur la figure du tableau 3 à droite)4n + 6 Arachno polyèdre closo à n + 2 sommets et deux sommets manquants
(celui en rouge et celui en vert sur la figure du tableau 3 à droite)4n + 8 Hypho polyèdre closo à n + 3 sommets et trois sommets manquants4n + 10 Klado polyèdre closo à n + 4 sommets et quatre sommets manquants
Tableau 2 – Règles de Wade pour la prédiction des structures des différents clusters.
Les polyèdres correspondants sont donnés dans le tableau 3.
B.4. À l’aide des règles de Wade, donner la structure de B6H62−.
6 / 28

Nombre Polyèdrede sommets4 Tetraèdre5 Bipyramide trigonale6 Octaèdre7 Diamant pentagonal8 Disphénoïde adouci9 Prisme triangulaire triaugmenté10 Diamant carré gyroallongé11 Octadécaèdre12 Icosaèdre
Tableau 3 – Structures des différents polyèdres et leur nom.
B.III Harmoniques sphériques de vecteurs de surface
La justification théorique des règles de Wade a pris une dizaine d’années. De nombreux calculsont été effectués sur différents composés, mais c’est A.J. Stone qui a réussi à donner la justificationthéorique la plus convaincante grâce aux harmoniques sphériques de vecteurs de surface. Commetoutes les structures dérivent de polyèdres réguliers, on peut prendre comme approximation queles n atomes de bore sont placés sur la surface d’une sphère de rayon R.
B.III.1 Cas des orbitales s
Le principe repose sur la possibilité de pouvoir décrire les orbitales moléculaires du cluster sous laforme de combinaison linéaire d’orbitales atomiques pour lesquelles les coefficients correspondentà la valeur des harmoniques sphériques au point considéré.
ϕ`,m = ∑j
ci,jχj = ∑j
Y`,m(θj, φj
)χj (1)
où Y`,m est une harmonique sphérique et θj et φj sont les positions angulaires de l’atome j sur lasphère, ces angles sont représentés à la fin du sujet. Les orbitales ϕ`,m ne sont pas normées.
B.5. Donner l’expression de l’énergie εσ`,m des orbitales ϕ`,m en fonction de Y`,m(θj, φj
), Si,j =
7 / 28

⟨σi|σj
⟩et hi,j =
⟨σiHσj
⟩où σi et σj sont des orbitales 1s des atomes de bore placées aux
positions i et j.
On définit ensuite l’énergie moyenne des orbitales de même ` :
εσ` =∑m
εσ`,m‖ϕ`,m‖
∑m‖ϕ`,m‖
(3)
Sachant que :
∑m
Y`,m (θi, φi)Y`,m(θj, φj
)=
2`+ 14π
P`(cos ωi,j
)(4)
où P` est le polynôme de Legendre de degré `, et ωi,j est la séparation angulaire entre deux atomes.
B.6. Exprimer la valeur de εσ` en fonction de P`, ωi,j, hi,j et Si,j.
On suppose que :
Si,j = δi,j hi,j =
ασ si i = jβσ si i est voisin de j0 sinon
(6)
B.7. À quoi correspondent ces hypothèses ?
B.8. Montrer que si on considère que ωi,j est indépendant de la paire d’atomes voisins considérés,alors :
εσ` = ασ +2en
βσP` (cos ω) (7)
avec e le nombre d’arêtes du polygone à n faces, on rappelle que P`(1) = 1.
L’énergie de ces différentes orbitales en fonction de n est donnée à la figure 6.
B.9. Donner la valeur de ω pour n = 2, n = 3, n = 4, n = 6.
B.10. Expliquer le choix de la notation Sσs , Pσ
s , Dσs , etc.
B.11. À partir d’un décompte électronique et de la dégénérescence des orbitales Lσs (L = S, P, D, F),
expliquer pourquoi il n’est pas nécessaire de tracer les orbitales Dσs pour n 6 4, les orbitales
Fσs pour n 6 9, etc.
8 / 28

Figure 6 – Énergie des orbitales générées par les orbitales s des atomes de bore en fonction de nou de ω.
B.III.2 Cas des orbitales p de surface
Après avoir considéré les orbitales s, on doit maintenant considérer les orbitales p. L’analyse desorbitales p est beaucoup plus technique car elles ont elles même des plans nodaux ce qui rend plusdifficile l’analyse. De plus, on ne s’intéresse qu’aux orbitales p qui sont tangentes à la sphère. Lesautres orbitales ne créant pas de liaisons σ entre les atomes sur la surface de la cage. Au lieu deconsidérer des points sur une sphère, on va maintenant considérer des vecteurs sur la surface de lasphère. L’orientation de chaque vecteur indique la direction des orbitales p à considérer. La normedes vecteurs donne l’amplitude du coefficient au point considéré. Pour chaque Y`,m, on obtientdeux harmoniques sphériques de vecteurs de surface Lπ
p et Lπp . Ces harmoniques sphériques sont
obtenues à partir du gradient des harmoniques sphériques au point considéré.
Figure 7 – Représentation des harmoniques sphériques de vecteurs de surface pour ` = 1 et ` = 2.À gauche on a représenté Lπ
p et à droite Lπp
B.12. Sur la figure 22 donnée en annexe, représenter les orbitales p correspondant aux portionsd’harmoniques sphériques de vecteurs de surface pour les cas (a) à (f).
B.13. Indiquer si le recouvrement est liant ou antiliant dans la direction de la ligne de champ (entrait plein sur la figure 22) et la direction perpendiculaire si indiquée (équipotentielle en traits
9 / 28

pointillés sur la figure 22) pour les cas (a) à (f).
Des calculs similaires à ceux effectués dans la partie B.III.1 donnent l’expression des énergies pourles différentes orbitales moléculaires :
ε(
Lπp
)= απ +
2en
wπ` (ω) (12)
ε(
Lπp
)= απ +
2en
wπ` (ω) (13)
où wπ` et wπ
` sont des fonctions de βσp et βπ
p qui correspondent à des recouvrements de type σ ou π
entre orbitales p de surface.
Figure 8 – Énergie des harmoniques sphériques de vecteurs de surface en fonction de n.
B.14. Pourquoi n’existe-il pas d’orbitale Sπp et Sπ
p ?
B.15. Quelles groupes d’orbitales sont liantes ? De même quelles sont les orbitales antiliantes ?
10 / 28

B.IV Analyse des liaisons dans le cas de cluster closo
Dans les deux cas, les deux facteurs essentiels sont le pré-facteur en 2en et la séparation angulaire
entre les différents sommets du polyèdre. Il faut donc chercher à faire le lien entre le nombre desommets n, de faces f et d’arêtes e.
B.IV.1 Théorème d’Euler-Descartes
On cherche à faire un lien entre n, f et e. Pour cela, on admettra que la démonstration faite sur uncube reste valide quel que soit le polyèdre régulier.
1. On aplatit le polyèdre en écartant vers l’extérieur les côtés d’une face. On obtient alors ungraphe plan dont les nœuds sont les sommets et les arcs sont les arêtes déformées.
2. On trace ensuite des arêtes entre les faces qui ne sont pas déjà triangulaires jusqu’à ce qu’ilne reste plus que des faces triangulaires.
3. On supprime ensuite un à un tous les triangles qui comportent une seul arête aux frontièresextérieures du graphe.
4. On supprime ensuite un à un tous les triangles qui comportent deux arêtes aux frontièresextérieures de notre graphe. Cela revient à supprimer un sommet.
5. On répète les deux opérations précédentes jusqu’à ce qu’il ne reste plus qu’un triangle.
Figure 9 – Premières étapes de la démonstration pour un carré.
B.16. Pour les étapes 2) 3) et 4), la quantité n + f − e est-elle changée ? Justifier pour chaque étape.
B.17. Sachant que lorsqu’il ne reste plus qu’un triangle on compte la face extérieure et la faceintérieure, en déduire la relation
n + f − e = 2 (14)
B.18. En déduire la manière de maximiser le pré-facteur 2en pour maximiser le caractère liant des
orbitales.
11 / 28

B.IV.2 Nombre d’orbitales liantes
Il faut maintenant prendre en compte l’hybridation entre les orbitales s et p. Par symétrie, lesorbitales Lπ
p ont un recouvrement nul avec les orbitales Lπp et Lσ
s . Par contre, le recouvrement entreles orbitales Lσ
s et L′πp est non nul si L = L′.
B.19. À l’aide d’un raisonnement qualitatif, pour chaque interaction entre Lσs et Lπ
p , dessiner unschéma d’interaction sur un diagramme énergétique. Commenter l’apparition de groupesd’orbitales liantes, non liantes ou antiliantes.
B.20. En déduire qu’il y a n + 1 orbitales liantes, ce sont les skeletal eletron pairs. Comparer cerésultat à votre résultat de la question B.3.
B.21. En déduire que l’on vient de démontrer les règles de Wade dans le cas des boranes closo deformule BnHn
2−.
B.V Analyse des liaisons dans le cas de cluster nido
Pour les clusters de type nido, on part de la structure closo avec n pair et on enlève un atome situéà θ = 0, φ = 0 situé sur l’axe z. On passe alors de la symétrie sphérique Rh à la symétrie C∞,v.
orbital de cluster Rh C∞,v
Sσ Sg Σ+
Pσ Pu Σ+ ⊕ E1
Dσ Dg Σ+ ⊕ E1 ⊕ E2
Lσ Lg/u Σ+ ⊕ E1 ⊕ E2 ⊕ · · · ⊕ E`
Pπ Pu Σ+ ⊕ E1
Pπ Pg Σ− ⊕ E1
Dπ Dg Σ+ ⊕ E1 ⊕ E2
Dπ Du Σ− ⊕ E1 ⊕ E2
Lπ Lg/u Σ+ ⊕ E1 ⊕ E2 ⊕ · · · ⊕ E`
Lπ Lu/g Σ− ⊕ E1 ⊕ E2 ⊕ · · · ⊕ E`
Tableau 4 – Descente en symétrie des orbitales lors du passage de la structure closo à la structurenido
En enlevant un sommet, on enlève 3 orbitales atomiques, donc trois orbitales de cluster.
B.22. Donner la symétrie de ces orbitales.
L’orbitale s enlevée supprime une orbitale de cluster Lσ inoccupée.
B.23. Déduire de la question précédente que le nombre de représentations irréductibles de typeE1 générées pour un cluster nido est impair.
Bien que l’on ait perdu la symétrie sphérique, on doit avoir autant d’orbitales de cluster Lπ qued’orbitales Lπ.
12 / 28

B.24. En déduire qu’au moins une orbitale est strictement non liante (en fait l’égalité est stricte).
En pratique, l’ouverture du cluster de closo à nido s’accompagne de la stabilisation de l’orbitale quiétait strictement non-liante (grâce à la présence d’atomes d’hydrogènes par exemple).
B.25. En déduire le nombre de skeletal electron pairs pour les cluster de type nido à p = n − 1sommets issus du polyèdre closo à n sommets.
On vient alors de démontrer les règles de Wade pour les cluster nido. Un raisonnement analoguepermet d’en déduire que pour un cluster arachno à q = n− 2 sommets, on a q + 3 skeleton electronpairs.De même, il est possible d’étendre le concept d’harmoniques sphériques de vecteurs de surface àdes orbitales de symétrie d pour engendrer des orbitales de cluster Lσ
d , Lπd , Lδ
d afin de reproduire ledécompte électroniques des clusters métalliques.
B.VI L’ion [Pt@Pb12]2−
L’ion [Pt@Pb12]2− est préparé à partir d’un ion de Zintl [Pb9]
4− et de [Pt(PPh3)4] dans l’éthylènediamine en présence d’ions potassium complexés par le (2,2,2)-crypt. Il forme une solution decoloration intense marron-verte. L’atome de platine occupe le centre d’un polyèdre formé par lesatomes de plomb.Les règles de Wade/Mingos ont été initialement développées pour les complexes de bore BnHn+m
q−
et de carboranes.
B.26. Combien y’a-t-il d’électrons de valence par couples d’atomes BH ? Idem pour le plomb et lecarbone. Expliquer pourquoi les règles de Wade/Mingos sont transposables aux clusters deplomb, de carbone ou aux carboranes (cluster de carbone, de bore et d’hydrogène).
B.27. Calculer le nombre d’électrons de valence et prévoir la structure de l’ion de Zintl [Pb9]4−
(closo, nido, arachno du polyèdre correspondant).
B.28. Faire de même pour [Pb12]2−.
B.29. À l’aide de la formule des rotations, donner la représentation réductible des orbitales d duplatine dans le groupe de symétrie de la molécule [Pb12]
2−.
B.30. Décomposer Γd en somme de représentations irréductibles et commenter le résultat.
B.31. À l’aide d’un décompte électronique, expliquer pourquoi la présence de l’atome de platinene change pas la structure de [Pb12]
2−.
13 / 28
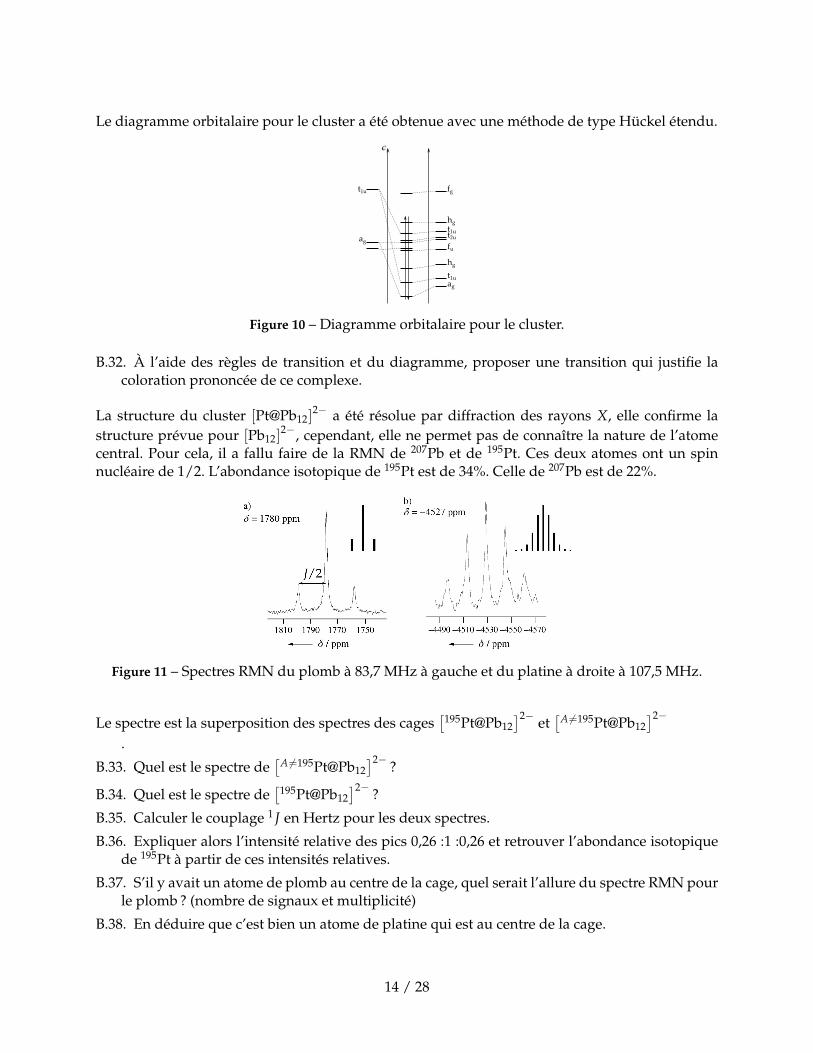
Le diagramme orbitalaire pour le cluster a été obtenue avec une méthode de type Hückel étendu.
agt1u
t2ufu
hg
fg
hg
t1uag
t1u
Figure 10 – Diagramme orbitalaire pour le cluster.
B.32. À l’aide des règles de transition et du diagramme, proposer une transition qui justifie lacoloration prononcée de ce complexe.
La structure du cluster [Pt@Pb12]2− a été résolue par diffraction des rayons X, elle confirme la
structure prévue pour [Pb12]2−, cependant, elle ne permet pas de connaître la nature de l’atome
central. Pour cela, il a fallu faire de la RMN de 207Pb et de 195Pt. Ces deux atomes ont un spinnucléaire de 1/2. L’abondance isotopique de 195Pt est de 34%. Celle de 207Pb est de 22%.
Figure 11 – Spectres RMN du plomb à 83,7 MHz à gauche et du platine à droite à 107,5 MHz.
Le spectre est la superposition des spectres des cages[195Pt@Pb12
]2− et[A 6=195Pt@Pb12
]2−
.
B.33. Quel est le spectre de[A 6=195Pt@Pb12
]2− ?
B.34. Quel est le spectre de[195Pt@Pb12
]2− ?
B.35. Calculer le couplage 1 J en Hertz pour les deux spectres.
B.36. Expliquer alors l’intensité relative des pics 0,26 :1 :0,26 et retrouver l’abondance isotopiquede 195Pt à partir de ces intensités relatives.
B.37. S’il y avait un atome de plomb au centre de la cage, quel serait l’allure du spectre RMN pourle plomb ? (nombre de signaux et multiplicité)
B.38. En déduire que c’est bien un atome de platine qui est au centre de la cage.
14 / 28
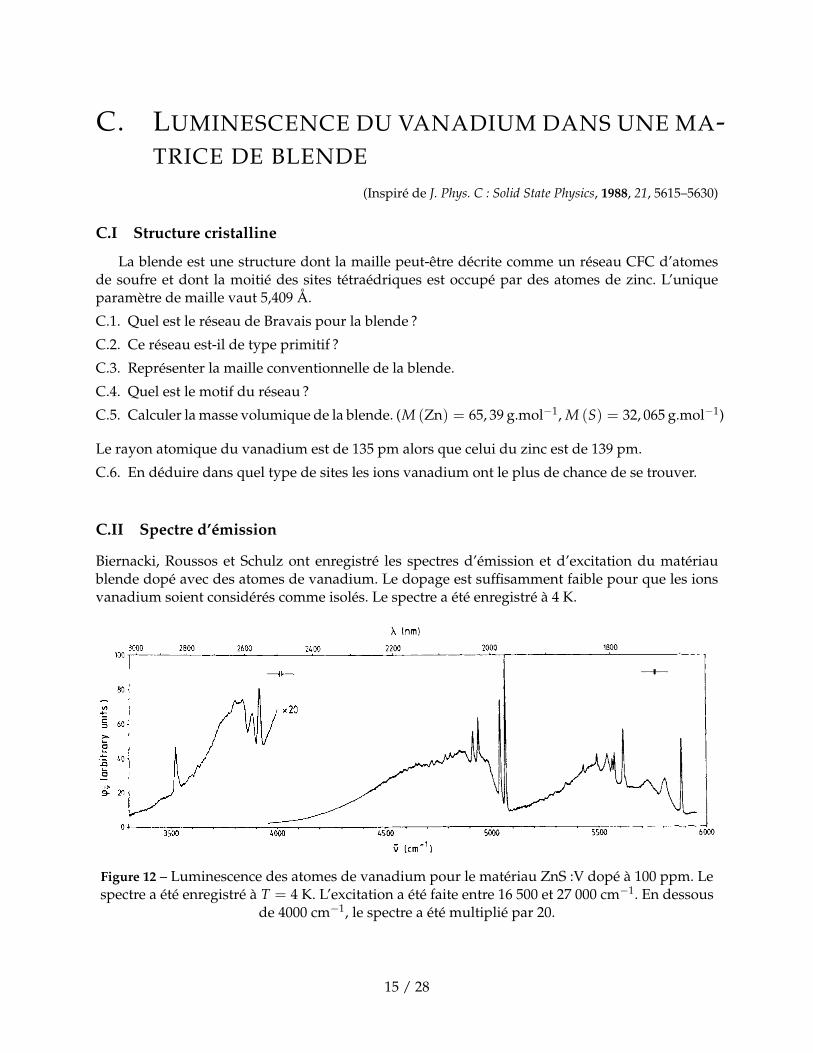
C. LUMINESCENCE DU VANADIUM DANS UNE MA-TRICE DE BLENDE
(Inspiré de J. Phys. C : Solid State Physics, 1988, 21, 5615–5630)
C.I Structure cristalline
La blende est une structure dont la maille peut-être décrite comme un réseau CFC d’atomesde soufre et dont la moitié des sites tétraédriques est occupé par des atomes de zinc. L’uniqueparamètre de maille vaut 5,409 Å.
C.1. Quel est le réseau de Bravais pour la blende ?
C.2. Ce réseau est-il de type primitif ?
C.3. Représenter la maille conventionnelle de la blende.
C.4. Quel est le motif du réseau ?
C.5. Calculer la masse volumique de la blende. (M (Zn) = 65, 39 g.mol−1, M (S) = 32, 065 g.mol−1)
Le rayon atomique du vanadium est de 135 pm alors que celui du zinc est de 139 pm.
C.6. En déduire dans quel type de sites les ions vanadium ont le plus de chance de se trouver.
C.II Spectre d’émission
Biernacki, Roussos et Schulz ont enregistré les spectres d’émission et d’excitation du matériaublende dopé avec des atomes de vanadium. Le dopage est suffisamment faible pour que les ionsvanadium soient considérés comme isolés. Le spectre a été enregistré à 4 K.
Figure 12 – Luminescence des atomes de vanadium pour le matériau ZnS :V dopé à 100 ppm. Lespectre a été enregistré à T = 4 K. L’excitation a été faite entre 16 500 et 27 000 cm−1. En dessous
de 4000 cm−1, le spectre a été multiplié par 20.
15 / 28
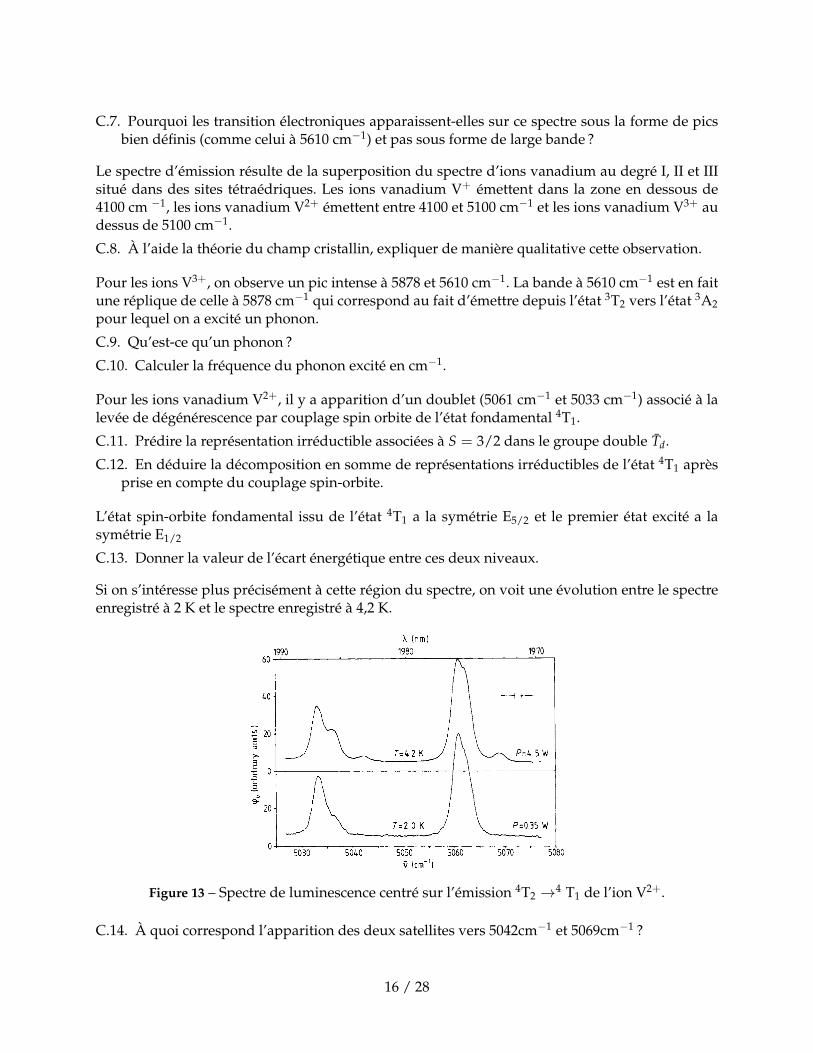
C.7. Pourquoi les transition électroniques apparaissent-elles sur ce spectre sous la forme de picsbien définis (comme celui à 5610 cm−1) et pas sous forme de large bande ?
Le spectre d’émission résulte de la superposition du spectre d’ions vanadium au degré I, II et IIIsitué dans des sites tétraédriques. Les ions vanadium V+ émettent dans la zone en dessous de4100 cm −1, les ions vanadium V2+ émettent entre 4100 et 5100 cm−1 et les ions vanadium V3+ audessus de 5100 cm−1.
C.8. À l’aide la théorie du champ cristallin, expliquer de manière qualitative cette observation.
Pour les ions V3+, on observe un pic intense à 5878 et 5610 cm−1. La bande à 5610 cm−1 est en faitune réplique de celle à 5878 cm−1 qui correspond au fait d’émettre depuis l’état 3T2 vers l’état 3A2pour lequel on a excité un phonon.
C.9. Qu’est-ce qu’un phonon ?
C.10. Calculer la fréquence du phonon excité en cm−1.
Pour les ions vanadium V2+, il y a apparition d’un doublet (5061 cm−1 et 5033 cm−1) associé à lalevée de dégénérescence par couplage spin orbite de l’état fondamental 4T1.
C.11. Prédire la représentation irréductible associées à S = 3/2 dans le groupe double Td.
C.12. En déduire la décomposition en somme de représentations irréductibles de l’état 4T1 aprèsprise en compte du couplage spin-orbite.
L’état spin-orbite fondamental issu de l’état 4T1 a la symétrie E5/2 et le premier état excité a lasymétrie E1/2
C.13. Donner la valeur de l’écart énergétique entre ces deux niveaux.
Si on s’intéresse plus précisément à cette région du spectre, on voit une évolution entre le spectreenregistré à 2 K et le spectre enregistré à 4,2 K.
Figure 13 – Spectre de luminescence centré sur l’émission 4T2 →4 T1 de l’ion V2+.
C.14. À quoi correspond l’apparition des deux satellites vers 5042cm−1 et 5069cm−1 ?
16 / 28

C.15. En déduire l’écart d’énergie entre l’état de plus basse énergie et le dernier état excité issusde l’état 4T2 après pris en compte du couplage spin-orbite.
On s’intéresse maintenant à la bande vers 3900 cm−1 de l’ion V+ pour lequel on a un champ faible.
C.16. À l’aide des diagrammes de Tanabe-Tsugano, donner la transition associée à cette bande(état initial et état final). Justifier le diagramme de Tanabe-Tsugano utilisé.
C.III Paramètres de Racah
Les paramètres de Racah permettent de prédire la position énergétique des différents niveaux del’ion libre vanadium. Pour l’ion V2+, si on prend pour référence l’énergie du terme 4F, l’énergiedes différents niveaux s’exprime de la manière suivante :
E(
4P)= 15B− 10α (16)
E(2G
)= 4B + 3C + 8α (17)
E(2P)= 9B + 3C− 10α (18)
E(2Da,b
)= 20B + 5C∓
√193B2 + 8BC + 4C2 − 6α (19)
E(2H
)= 9B + 3C + 18α (20)
E(2G
)= 24B + 3C (21)
Les énergies expérimentales ont été mesurées par Shadmi en 1969 (tableau 5).
État Énergie (cm−1)4P 11 330,52G 11 751,62P 15 222,82Da 16 019,82H 16 564,62F 27 441,52Db 41 971,7
Tableau 5 – Énergie expérimentale pour l’ion V2+.
C.17. Que décrit le terme B ?
Le terme C quantifie la même grandeur que B. Le terme α prend en compte une perturbation dudeuxième ordre entre les configurations électroniques qui diffèrent de deux électrons.
C.18. Calculer la valeur des paramètres des paramètres B et C en supposant la valeur de α nulle.Pour cela, utiliser les énergies des états 4P et 2G.
C.19. Avec ce jeu de paramètre, calculer l’énergie des états 2P et 2H. Commenter.
C.20. Recalculer de nouvelles valeurs de B′, C′ et α 6= 0 à partir des énergies des niveaux 4P, 2G etde la différence d’énergie E
(2H)− E
(2P).
17 / 28

C.21. Comparer l’erreur commise sur l’énergie de l’état 2H avec les deux jeux de paramètres.Commenter.
Le spectre d’excitation de l’ion V2+ présente deux bandes : une à 5030 cm−1et l’autre à 9300 cm−1.
C.22. À l’aide d’un diagramme de Tanabe-Tsugano et sachant que l’on est en champ faible, trouverla valeur de ∆O et de BZnS:V pour les ions vanadium V2+ dans le matériau.
C.23. Calculer le paramètre néphélauxétique. Commenter.
18 / 28

D. PHYSICO-CHIMIE DES COMPLEXES CARBÉNIQUESDE MÉTAUX DE TRANSITION
(Inspiré de 2002 C)
L’objet de ce problème est d’étudier différentes propriétés qui résultent de l’interaction entreune molécule organique et un fragment MLn comportant un métal de transition M. qu’à l’influencede la coordination avec le fragment métallique.
D.I Catalyse de métathèse des oléfines
Ces dernières années, la réaction de métathèse des oléfines (alcènes), comme méthode de for-mation de liaison C=C, a connu un regain d’intérêt.
[Ru]
R1
R2
++
R1
R2
Figure 14
De nouveaux catalyseurs à base de ruthénium se sont montrés très efficaces. Ces derniers sontdes complexes carbéniques de formule générale :
L
Ru
Cl
Cl
PCy3
Ph
Cy = -C6H11
A : L = PCy3
B : L = N N
Figure 15
Dans le complexe B, un des ligands phosphine PCy3 a été substitué par un carbène stabiliséappelé carbène d’Arduengo. Ces derniers existent à l’état libre. Le catalyseur B a été synthétisétrès récemment et présente une activité catalytique bien plus importante que celle du catalyseurA plus ancien.
L’objet de cette partie est d’essayer de comprendre l’origine de la plus grande activité cataly-tique dont fait preuve B. Dans toute la suite nous ne considérerons pas la totalité du cycle cata-lytique, mais seulement les étapes de la phase d’initiation qui sont essentielles pour comprendrel’activité du catalyseur. Cette phase d’initiation conduit à la formation d’un intermédiaire métal-lacyclobutane I (figure 18) à partir duquel la réaction de métathèse se produit très rapidement.
19 / 28
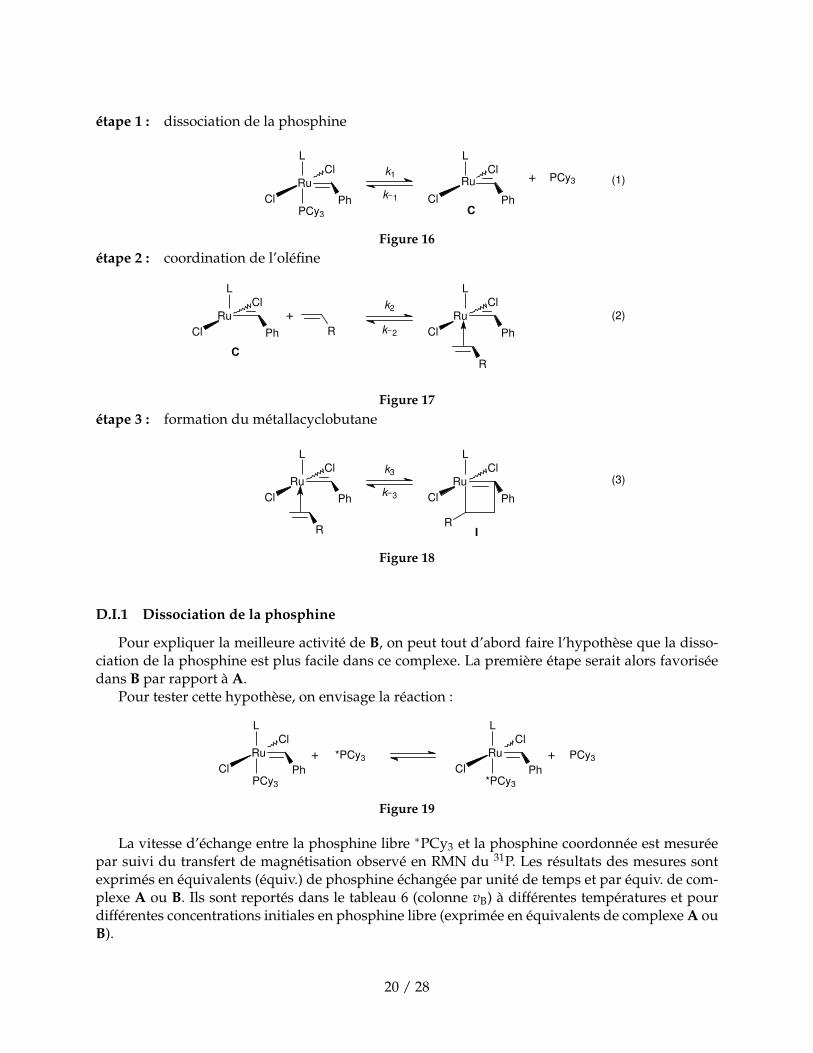
étape 1 : dissociation de la phosphine
L
PCy3
Ph
Ru
Cl
Cl k1
k−1
L
Ph
Ru
Cl
Cl+ PCy3 (1)
C
Figure 16étape 2 : coordination de l’oléfine
L
Ph
Ru
Cl
Cl
C
k2
k−2
+
R
L
Ph
Ru
Cl
Cl
R
(2)
Figure 17étape 3 : formation du métallacyclobutane
L
Ph
Ru
Cl
Cl
R
k3
k−3
L
Ph
Ru
Cl
Cl
R
(3)
I
Figure 18
D.I.1 Dissociation de la phosphine
Pour expliquer la meilleure activité de B, on peut tout d’abord faire l’hypothèse que la disso-ciation de la phosphine est plus facile dans ce complexe. La première étape serait alors favoriséedans B par rapport à A.
Pour tester cette hypothèse, on envisage la réaction :
L
PCy3
Ph
Ru
Cl
Cl
*PCy3+
L
Ph
Ru
Cl
Cl
*PCy3
PCy3+
Figure 19
La vitesse d’échange entre la phosphine libre ∗PCy3 et la phosphine coordonnée est mesuréepar suivi du transfert de magnétisation observé en RMN du 31P. Les résultats des mesures sontexprimés en équivalents (équiv.) de phosphine échangée par unité de temps et par équiv. de com-plexe A ou B. Ils sont reportés dans le tableau 6 (colonne vB) à différentes températures et pourdifférentes concentrations initiales en phosphine libre (exprimée en équivalents de complexe A ouB).
20 / 28

Complexe vB (s−1) équiv. PCy3 T (K)A 0,116 3 313A 0,381 3 323A 1,210 3 333A 3,560 3 343A 9,570 3 353B 0,040 1,5 343B 0,126 1,5 353B 0,355 1,5 363B 1,020 1,5 373B 0,121 5 353B 0,120 10 353
Tableau 6 – Vitesse pour la réaction d’échange entre la phosphine libre et la phosphinecoordonnée pour les deux complexes A et B à différentes températures et pour différents
rapports des concentrations initiales en réactifs.
D.1. De combien d’électrons est entouré le ruthénium dans l’intermédiaire C ? Quel est le degréd’oxydation de Ru ?
D.2. L’intermédiaire C est-il trés réactif ? En déduire que la vitesse vB est assimilable à k1 (éq. 1).
D.3. Calculer l’enthalpie d’activation ∆rH‡ de la réaction de dissociation de PCy3 pour les deuxcomplexes A et B.
D.4. Les valeurs des grandeurs des enthalpies d’activation permettent-elles d’expliquer la plusgrande activité catalytique de B par rapport à A ?
D.5. Montrer que les résultats expérimentaux pour B sont en accord avec un mécanisme dissocia-tif et non pas avec un mécanisme associatif selon lequel la phosphine libre se coordonneraitavant que l’autre ne parte.
D.I.2 Compétition entre dissociation de PCy3 et coordination de 1’o1éfine
Pour étudier la cinétique de la réactioni d’initiation, on fait réagir les complexes A et B avec desoléfines substituées par des groupes alkoxy : H2C=CH(OR). Le produit de réaction, formé demanière quantitative et rapide, est un carbène de Fischer L(PCy3)(Cl)2Ru=CHOR.En présence d’un excès d’oléfine, on peut suivre la disparition du complexe B par la diminutionde la hauteur de certains pics caractéristiques sur son spectre de RMN du proton. Dans le tableau7, on donne les résultats pour la réaction de B avec 15 équivalents de H2C=CH(OEt) à 308 K.
t (s) 0 250 500 750 1000 1250 1500 1750 2000I 163,9 146,3 130,6 116,7 104,2 93,0 83,0 74,10 66,2
Tableau 7 – Évolution temporelle de l’intensité (unité arbitraire) d’un pic caractéristique de B enRMN du proton au cours de la réaction avec 15 équivalents de H2C=CH(OEt).
21 / 28

D.6. Montrer que la disparition de B suit une cinétique du premier ordre.
D.7. Estimer la valeur de la constante de vitesse kobs.
D.8. Comparer la valeur de kobs à celle de k1 à la même température. Quelle conclusion peut-onen tirer ? ’
On suppose que toutes les étapes qui suivent la coordination de l’oléfine (étape 2) sont rapides.De plus, on se place en excès d’oléfine par rapport au complexe A ou B.
D.9. Expliquer pourquoi on peut négliger la réaction retour de constante de vitesse k2 dans l’équa-tion (2).
D.10. Expliquer la relation entre 1/kobs, [PCy3], [oléfine], k1, k2 et k−1. [PCy3] désigne la concen-tration en phosphine libre et [oléfine] la concentration en alcène non coordonné.
D.11. Expliquer en quoi k−1 [PCy3] et k2[oléfine] sont des grandeurs essentielles pour comprendrel’activité du catalyseur.
Pour différentes valeurs du rapport [PCy3]/[oléfine] on a pu mesurer la constante de vitesse kobspour les deux catalyseurs à une température donnée. Les résultats sont reportés dans le tableau 8.
Complexe [PCy3]/[oléfine] 1/kobs (s)A 0,0044 421A 0,0085 800A 0,017 1582A 0,025 2398B 0,112 353B 0,391 534B 0,733 629B 1,59 959B 2,29 1300
Tableau 8 – Variation de la constante de vitesse kobs avec le rapport [PCy3]/[oléfine] à T = 310 Kpour A et à T = 323 K pour B.
D.12. Montrer que l’expression déterminée précédemment est vérifiée.
D.13. En déduire pour chacun des catalyseurs la valeur du rapport k−1/k2.
D.14. Conclure quant à l’activité catalytique supérieure de B par rapport à A.
Questions bonus :– Qui sont les deux personnages représentés sur la couverture du sujet ? (Pour un goûter)– Qui a eu le prix Nobel de chimie cette année et pourquoi ? (Pour un bonbon)
22 / 28

C2v E C2 σx σz
A1 1 1 1 1A2 1 1 −1 −1B1 1 −1 −1 1B2 1 −1 1 −1
Tableau 9 –h Table du groupe C2v pour le problème A.h
C4v E 2C4 C2 2σv 2σd
A1 1 1 1 1 1A2 1 1 1 −1 −1B1 1 −1 1 1 −1B2 1 −1 1 −1 1E 2 0 −2 0 0
I E 12C5 12C25 20C3 15C2
A 1 1 1 1 1
T1 3 2c5 2c35 0 − 1
T2 3 2c35 2c5 0 − 1
F 4 − 1 − 1 1 0
H 5 0 0 − 1 1
cmn = cosm
nπ
Ih E 12C5 12C25 20C3 15C2 i 12S 3
10 12S10 20S6 15σ
Ag 1 1 1 1 1 1 1 1 1 1T1g 3 2c5 2c35 0 − 1 3 2c5 2c35 0 − 1T2g 3 2c35 2c5 0 − 1 3 2c35 2c5 0 − 1Fg 4 − 1 − 1 1 0 4 − 1 − 1 1 0Hg 5 0 0 − 1 1 5 0 0 − 1 1Au 1 1 1 1 1 − 1 − 1 − 1 − 1 − 1T1u 3 2c5 2c35 0 − 1 − 3 − 2c5 − 2c35 0 1T2u 3 2c35 2c5 0 − 1 − 3 − 2c35 − 2c5 0 1Fu 4 − 1 − 1 1 0 − 4 1 1 − 1 0Hu 5 0 0 − 1 1 − 5 0 0 1 − 1
cmn = cos mn π
Ih Ag T1g T2g Fg Hg
Ag Ag T1g T2g Fg Hg
T1g Ag ⊕ { T1g} ⊕Hg Fg ⊕Hg T2g ⊕Fg ⊕Hg T1g ⊕T2g ⊕Fg ⊕Hg
T2g Ag ⊕ { T2g} ⊕Hg T1g ⊕Fg ⊕Hg T1g ⊕T2g ⊕Fg ⊕Hg
Fg Ag ⊕ { T1g} ⊕ { T2g} T1g ⊕T2g ⊕Fg ⊕ 2Hg
⊕Fg ⊕Hg
Hg Ag ⊕ { T1g} ⊕ { T2g} ⊕Fg
⊕ { Fg} ⊕ 2Hg
C2v E C2 σv(xz) σv(yz) problème D.A1 1 1 1 1 z x2, y2, z2
A2 1 1 −1 −1 xyB1 1 −1 1 −1 x xzB2 1 −1 −1 1 y yz
Tableau 10 –h Table du groupe C2v pour le problème D.h
23 / 28
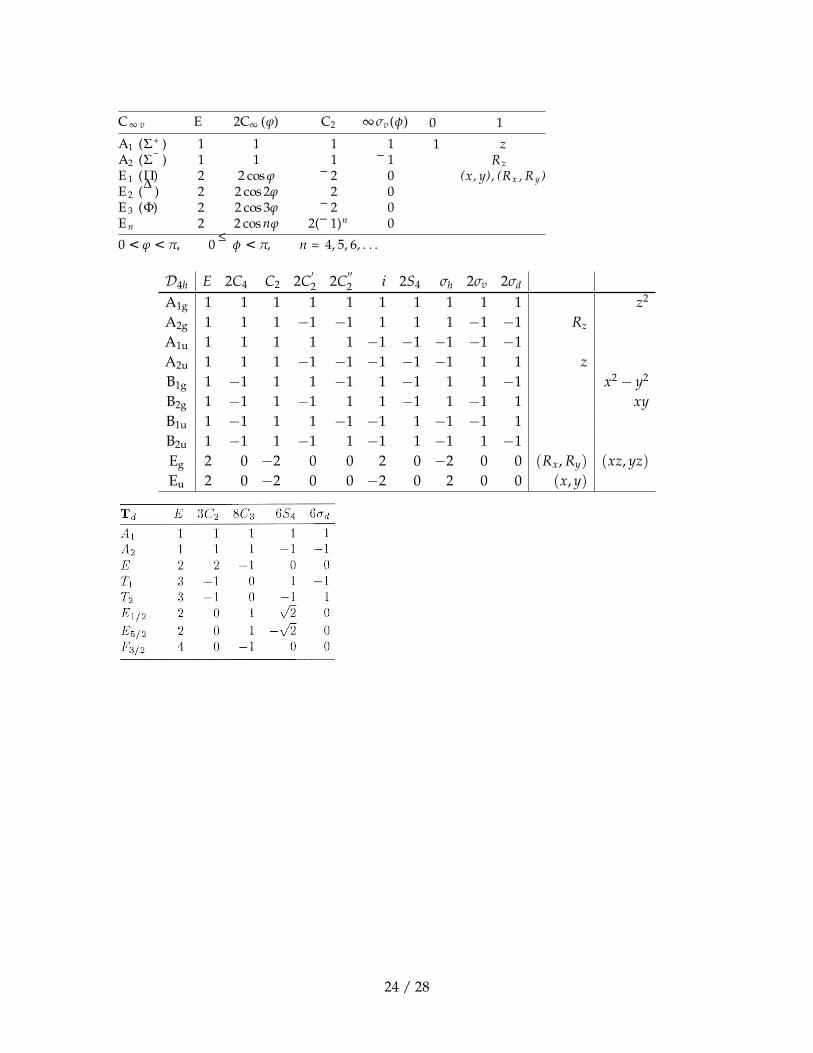
C∞ v E 2C∞ (ϕ) C2 ∞σv (φ)
A1 (Σ+ ) 1 1 1 1
A2 (Σ− ) 1 1 1 − 1
E 1 (Π) 2 2 cosϕ − 2 0
E 2 (Δ ) 2 2 cos 2ϕ 2 0
E 3 (Φ) 2 2 cos 3ϕ − 2 0
E n 2 2 cosnϕ 2(− 1)n 0
0 < ϕ < π, 0 ≤ φ < π, n = 4, 5, 6, . . .
0 1
1 zRz
(x, y), (Rx , Ry )
D4h E 2C4 C2 2C′2 2C
′′2 i 2S4 σh 2σv 2σd
A1g 1 1 1 1 1 1 1 1 1 1 z2
A2g 1 1 1 −1 −1 1 1 1 −1 −1 Rz
A1u 1 1 1 1 1 −1 −1 −1 −1 −1A2u 1 1 1 −1 −1 −1 −1 −1 1 1 zB1g 1 −1 1 1 −1 1 −1 1 1 −1 x2 − y2
B2g 1 −1 1 −1 1 1 −1 1 −1 1 xyB1u 1 −1 1 1 −1 −1 1 −1 −1 1B2u 1 −1 1 −1 1 −1 1 −1 1 −1Eg 2 0 −2 0 0 2 0 −2 0 0 (Rx, Ry) (xz, yz)Eu 2 0 −2 0 0 −2 0 2 0 0 (x, y)
24 / 28
25 / 28
26 / 28
1 1.0079
HHydrogen
3 6.941
LiLithium
11 22.990
NaSodium
19 39.098
KPotassium
37 85.468
RbRubidium
55 132.91
CsCesium
87 223
FrFrancium
4 9.0122
BeBeryllium
12 24.305
MgMagnesium
20 40.078
CaCalcium
38 87.62
SrStrontium
56 137.33
BaBarium
88 226
RaRadium
21 44.956
ScScandium
39 88.906
YYttrium
57-71
La..Lanthanide
89-103
Ac..Actinide
22 47.867
TiTitanium
40 91.224
ZrZirconium
72 178.49
HfHafnium
104 261
Rf
Rutherfordium
23 50.942
VVanadium
41 92.906
NbNiobium
73 180.95
TaTantalum
105 262
Db
Dubnium
24 51.996
CrChromium
42 95.94
MoMolybdenum
74 183.84
WTungsten
106 266
Sg
Seaborgium
25 54.938
MnManganese
43 96
TcTechnetium
75 186.21
ReRhenium
107 264
Bh
Bohrium
26 55.845
FeIron
44 101.07
RuRuthenium
76 190.23
OsOsmium
108 277
Hs
Hassium
27 58.933
CoCobalt
45 102.91
RhRhodium
77 192.22
IrIridium
109 268
Mt
Meitnerium
28 58.693
NiNickel
46 106.42
PdPalladium
78 195.08
PtPlatinum
110 281
Ds
Darmstadtium
29 63.546
CuCopper
47 107.87
AgSilver
79 196.97
AuGold
111 280
Rg
Roentgenium
30 65.39
ZnZinc
48 112.41
CdCadmium
80 200.59
HgMercury
112 285
Uub
Ununbium
31 69.723
GaGallium
13 26.982
AlAluminium
5 10.811
BBoron
49 114.82
InIndium
81 204.38
TlThallium
113 284
Uut
Ununtrium
6 12.011
CCarbon
14 28.086
SiSilicon
32 72.64
GeGermanium
50 118.71
SnTin
82 207.2
PbLead
114 289
Uuq
Ununquadium
7 14.007
NNitrogen
15 30.974
PPhosphorus
33 74.922
AsArsenic
51 121.76
SbAntimony
83 208.98
BiBismuth
115 288
Uup
Ununpentium
8 15.999
OOxygen
16 32.065
SSulphur
34 78.96
SeSelenium
52 127.6
TeTellurium
84 209
PoPolonium
116 293
Uuh
Ununhexium
9 18.998
FFluorine
17 35.453
ClChlorine
35 79.904
BrBromine
53 126.9
IIodine
85 210
AtAstatine
117 292
Uus
Ununseptium
10 20.180
NeNeon
2 4.0025
HeHelium
18 39.948
ArArgon
36 83.8
KrKrypton
54 131.29
XeXenon
86 222
RnRadon
118 294
Uuo
Ununoctium
1
2
3
4
5
6
7
57 138.91
LaLanthanum
58 140.12
CeCerium
59 140.91
PrPraseodymium
60 144.24
NdNeodymium
61 145
PmPromethium
62 150.36
SmSamarium
63 151.96
EuEuropium
64 157.25
GdGadolinium
65 158.93
TbTerbium
66 162.50
DyDysprosium
67 164.93
HoHolmium
68 167.26
ErErbium
69 168.93
TmThulium
70 173.04
YbYtterbium
71 174.97
LuLutetium
89 227
AcActinium
90 232.04
ThThorium
91 231.04
PaProtactinium
92 238.03
UUranium
93 237
Np
Neptunium
94 244
Pu
Plutonium
95 243
Am
Americium
96 247
Cm
Curium
97 247
Bk
Berkelium
98 251
Cf
Californium
99 252
Es
Einsteinium
100 257
Fm
Fermium
101 258
Md
Mendelevium
102 259
No
Nobelium
103 262
Lr
Lawrencium
Z mass
SymbolName
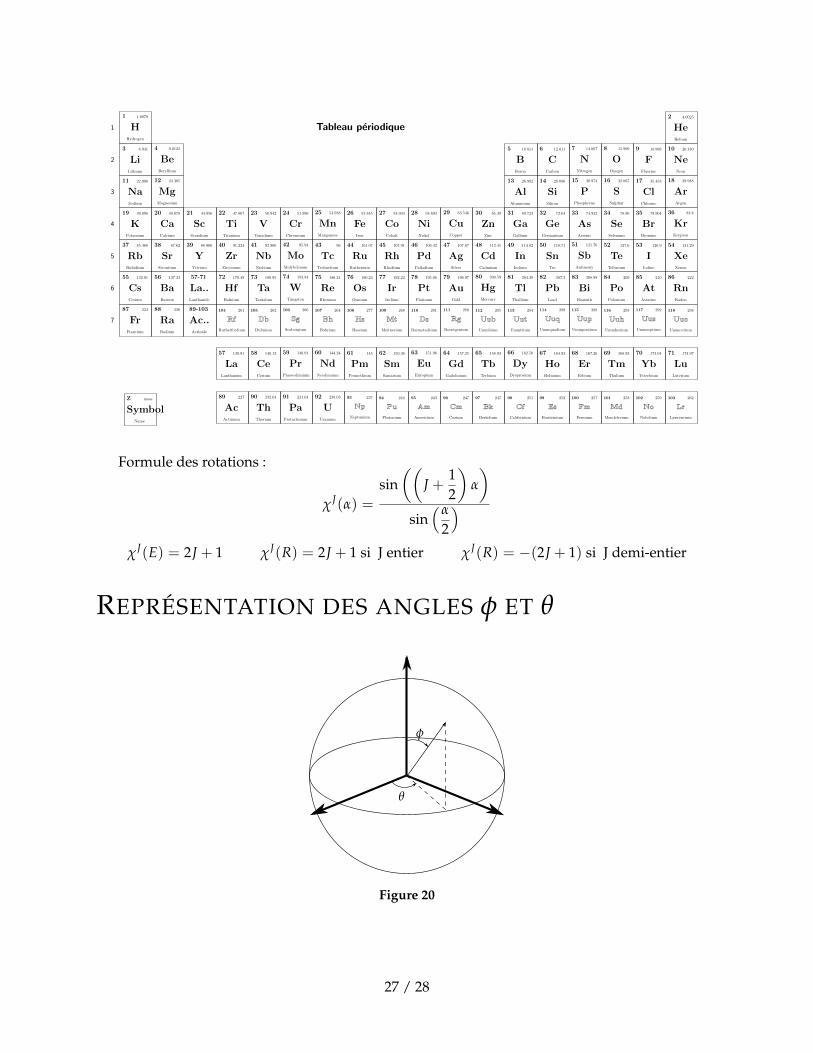
Tableau periodique
Formule des rotations :
χJ(α) =
sin((
J +12
)α
)sin(α
2
)χJ(E) = 2J + 1 χJ(R) = 2J + 1 si J entier χJ(R) = −(2J + 1) si J demi-entier
REPRÉSENTATION DES ANGLES φ ET θ
θ
φ
Figure 20
27 / 28
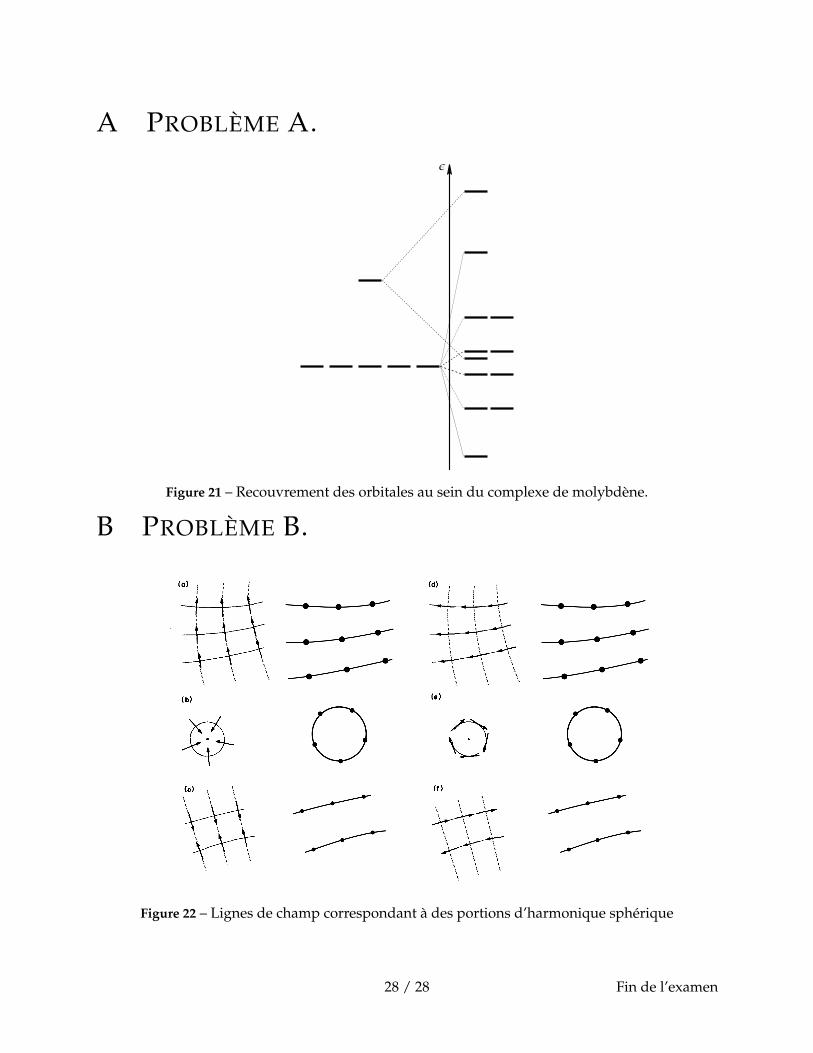
A PROBLÈME A.
Figure 21 – Recouvrement des orbitales au sein du complexe de molybdène.
B PROBLÈME B.
Figure 22 – Lignes de champ correspondant à des portions d’harmonique sphérique
28 / 28 Fin de l’examen