resume des caracteristiques du...
TRANSCRIPT

ANSM-TAKEDA RCP Brigatinib 180 mg 1
RESUME DES CARACTERISTIQUES DU PRODUIT
1. DENOMINATION DU MEDICAMENT
BRIGATINIB 180 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Brigatinib ........................................................................................................................................ 180 mg
Pour un comprimé pelliculé
Excipient(s) à effet notoire : chaque comprimé pelliculé contient 336 mg de lactose monohydraté.
Pour la liste complète des excipients, voir rubrique 6.1.
3. FORME PHARMACEUTIQUE
Comprimé pelliculé (comprimé). Comprimé pelliculé ovale, blanc à blanc cassé, d'environ 19 mm de longueur, comportant la mention « U13 » gravée sur une face et aucune mention sur l'autre.
4. DONNEES CLINIQUES
4.1. Indications thérapeutiques
Brigatinib est indiqué dans le traitement des patients adultes non éligibles à un essai clinique en cours, atteints d’un cancer broncho-pulmonaire non à petites cellules (CBNPC) avancé présentant un réarrangement du gène ALK (ALK positif), prétraités par crizotinib et pour lesquels il n’existe pas d’alternative thérapeutique appropriée disponible.
4.2. Posologie et mode d'administration
Le traitement par brigatinib devra être instauré et supervisé par un médecin ayant l'expérience de l'utilisation des médicaments anticancéreux. Le statut de CBNPC ALK-positif devra être connu avant l'instauration du traitement par brigatinib. La recherche de la translocation ALK doit être effectuée par une plateforme hospitalière de génétique moléculaire du cancer validée par l’Inca pour sélectionner les patients pouvant être traités par brigatinib.
Posologie
La dose initiale recommandée de brigatinib est de 90 mg par voie orale, une fois par jour pendant les 7 premiers jours, puis de 180 mg une fois par jour tous les jours. Le traitement doit être poursuivi aussi longtemps que le rapport bénéfice risque reste favorable pour le patient. En cas d’interruption du traitement par brigatinib pendant une période de 14 jours ou plus, pour des raisons autres que la survenue d’effets indésirables, le traitement devra être réduit à 90 mg par jour pendant 7 jours avant d’augmenter la dose jusqu’à celle préalablement tolérée. En cas d'oubli d’une prise ou de vomissements survenant immédiatement après la prise du traitement, le patient ne doit pas prendre la dose oubliée, mais prendre la dose prescrite suivante à l’heure programmée. Adaptations posologiques Une interruption de traitement et/ou une réduction de la posologie peuvent être nécessaires en fonction l’évaluation individuelle de la tolérance. Lorsqu'une réduction de la posologie est nécessaire, la dose de brigatinib doit être réduite par paliers de 60 mg ou 30 mg par jour (Cf. tableau 1). Toute réduction de la posologie est définitive, un retour à la dose recommandée de 180mg/j n’est pas possible.

ANSM-TAKEDA RCP Brigatinib 180 mg 2
La dose journalière ne doit pas excéder 180 mg ni être inférieure à 60 mg. Le traitement sera définitivement arrêté chez les patients qui ne tolèrent pas la dose de 60 mg par jour. Les niveaux des modifications de doses de brigatinib sont résumés dans le Tableau 1. Tableau 1 : Paliers de dose de brigatinib
Posologie Paliers de dose de brigatinib
Palier 1 Palier 2 Palier 3
90 mg par jour
(7 premiers jours)
Diminuer à 60 mg par jour Arrêt définitif Non-applicable
180 mg par jour
Diminuer à 120 mg par jour
diminuer 90 mg par jour
60 mg par jour
Les recommandations de réduction de la posologie, d’interruption ou d’arrêt de brigatinib en cas de survenue d’effets indésirables sont résumées dans le tableau 2 ci-dessous. Tableau 2 : Recommandations d’adaptation de la poso logie en cas d’effets indésirables
Effet indésirable
Sévérité* Modification de la posologie
Pneumopathie
Grade 1 • Si survenue au cours des 7 premiers jours de traitement
- suspendre le traitement par brigatinib jusqu'à la récupération de l'état initial,
puis
- reprendre au même niveau de dose et ne pas augmenter à la dose de 180 mg une fois par jour.
• Si survenue au-delà des 7 premiers jours du traitement,
- suspendre le traitement par brigatinib jusqu'à la récupération de l'état initial,
puis
- reprendre au même niveau de dose.
• Arrêt immédiat et définitif du traitement par brigatinib en cas de récidive (quel que soit le grade de l'épisode précédent).
Grade 2 • Si survenue au cours des 7 premiers jours de traitement
- suspendre le traitement par brigatinib jusqu'à la récupération de l'état initial,
puis
- reprendre au palier de dose inferieur et ne pas augmenter à la dose de 180 mg une fois par jour.
• Si survenue au-delà des 7 premiers jours de traitement
- suspendre le traitement par brigatinib jusqu'à la récupération de l'état initial,
puis
- reprendre au palier de dose inferieur et ne pas augmenter à la dose de 180 mg une fois par jour (cf. tableau 1).
• Arrêt immédiat et définitif du traitement par brigatinib en cas de récidive (quel que soit le grade de l'épisode précédent).
Grade 3 ou 4 Arrêt immédiat et définitif du traitement par briga tinib
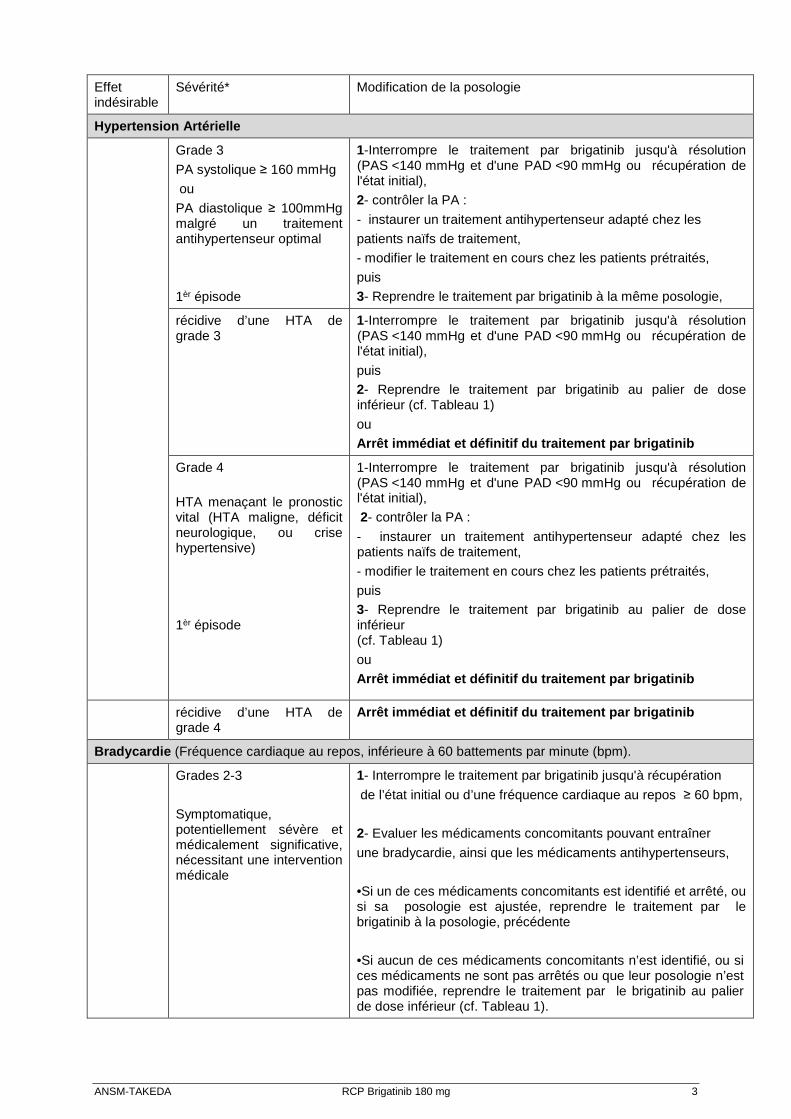
ANSM-TAKEDA RCP Brigatinib 180 mg 3
Effet indésirable
Sévérité* Modification de la posologie
Hypertension Artérielle
Grade 3
PA systolique ≥ 160 mmHg
ou
PA diastolique ≥ 100mmHg malgré un traitement antihypertenseur optimal 1èr épisode
1-Interrompre le traitement par brigatinib jusqu'à résolution (PAS <140 mmHg et d'une PAD <90 mmHg ou récupération de l'état initial),
2- contrôler la PA :
- instaurer un traitement antihypertenseur adapté chez les
patients naïfs de traitement,
- modifier le traitement en cours chez les patients prétraités,
puis
3- Reprendre le traitement par brigatinib à la même posologie,
récidive d’une HTA de grade 3
1-Interrompre le traitement par brigatinib jusqu'à résolution (PAS <140 mmHg et d'une PAD <90 mmHg ou récupération de l'état initial),
puis 2- Reprendre le traitement par brigatinib au palier de dose inférieur (cf. Tableau 1)
ou Arrêt immédiat et définitif du traitement par brigatinib
Grade 4
HTA menaçant le pronostic vital (HTA maligne, déficit neurologique, ou crise hypertensive)
1èr épisode
1-Interrompre le traitement par brigatinib jusqu'à résolution (PAS <140 mmHg et d'une PAD <90 mmHg ou récupération de l'état initial),
2- contrôler la PA :
- instaurer un traitement antihypertenseur adapté chez les patients naïfs de traitement,
- modifier le traitement en cours chez les patients prétraités,
puis 3- Reprendre le traitement par brigatinib au palier de dose inférieur (cf. Tableau 1)
ou Arrêt immédiat et définitif du traitement par brigatinib
récidive d’une HTA de grade 4
Arrêt immédiat et définitif du traitement par brigatinib
Bradycardie (Fréquence cardiaque au repos, inférieure à 60 battements par minute (bpm).
Grades 2-3
Symptomatique, potentiellement sévère et médicalement significative, nécessitant une intervention médicale
1- Interrompre le traitement par brigatinib jusqu'à récupération
de l’état initial ou d’une fréquence cardiaque au repos ≥ 60 bpm,
2- Evaluer les médicaments concomitants pouvant entraîner
une bradycardie, ainsi que les médicaments antihypertenseurs,
•Si un de ces médicaments concomitants est identifié et arrêté, ou si sa posologie est ajustée, reprendre le traitement par le brigatinib à la posologie, précédente
•Si aucun de ces médicaments concomitants n’est identifié, ou si ces médicaments ne sont pas arrêtés ou que leur posologie n’est pas modifiée, reprendre le traitement par le brigatinib au palier de dose inférieur (cf. Tableau 1).

ANSM-TAKEDA RCP Brigatinib 180 mg 4
Effet indésirable
Sévérité* Modification de la posologie
Grade 4
conséquences menaçant le pronostic vital, indication d'intervention urgente
1- Interrompre le traitement par brigatinib jusqu'à récupération
d’une fréquence cardiaque au repos ≥ 60 bpm,
2- Evaluer les médicaments concomitants pouvant entraîner
une bradycardie, ainsi que les médicaments antihypertenseurs
Si un de ces médicaments concomitants est identifié et arrêté, ou si sa posologie est ajustée, reprendre le traitement par le brigatinib au pallier de dose inférieur (cf. Tableau 1),
3- Arrêt immédiat et définitif du traitement par br igatinib , si aucun médicament concomitant n’est identifié.
4- Arrêt immédiat et définitif du traitement par br igatinib en cas de récidive (avec épisode précédent de grade ≥ 3).
Elévation du taux de Créatine Phosphokinase (CPK)
Grade 3 (> 5,0 × LSN)
1èr épisode
1- Interrompre le traitement par brigatinib jusqu’à résolution à un Grade ≤ 1 (≤2.5 × LSN) ou à la valeur initiale,
puis 2- Reprendre le traitement par brigatinib à la même posologie
récidive d’un grade 3
ou
Grade 4 (> 10,0 × LSN)
1- Interrompre le traitement par brigatinib jusqu’à résolution à un Grade ≤ 1 (≤2.5 × LSN) ou à la valeur initiale,
puis
2- Reprendre le traitement par brigatinib au palier de dose inférieur (cf. Tableau 1)
Elévation des taux de lipase ou d'amylase
Grade 3 (> 2,0 × LSN)
1èr épisode
1- Interrompre le traitement par brigatinib jusqu’à résolution à un Grade ≤ 1 (≤ 1.5 × LSN) ou à la valeur initiale,
puis
2- Reprendre le traitement par brigatinib à la même posologie
récidive d’un grade 3
ou
Grade 4 (> 5,0 × LSN)
1- Interrompre le traitement par brigatinib jusqu’à résolution à un Grade ≤ 1 (≤ 1.5 × LSN) ou à la valeur initiale,
puis 2- Reprendre le traitement par brigatinib au palier de dose
inférieur (cf. Tableau 1).
Elévation des enzymes hépatiques
Elévation des ALAT ou des ASAT de grade ≥ 3 (> 5,0 x LSN) et de la bilirubine totale ≤ 2 x LSN
1- Interrompre le traitement par brigatinib jusqu'à un retour aux valeurs de référence ou à des valeurs inférieures ou égales à 3 fois la LSN,
puis
2- Reprendre le traitement par brigatinib au palier de dose inférieur (cf. Tableau 1)
Elévation des ASAT ou des ALAT de Grade ≥ 2, (> 3 x LSN) et un taux de bilirubine totale de Grade ≥ 2
Arrêt immédiat et définitif du traitement par briga tinib .

ANSM-TAKEDA RCP Brigatinib 180 mg 5
Effet indésirable
Sévérité* Modification de la posologie
(> 1,5 x LSN) (en l’absence de cholestase ou d’hémolyse)
Hyperglycémie
Grade 3 (> 250 mg/dL ou 13,9 mmol/L) et plus
1-Interrompre le traitement par brigatinib jusqu'à résolution ou récupération de l'état initial,
2- contrôler l’équilibre glycémique :
- instaurer un traitement adapté chez les patients naïfs de traitement,
- modifier le traitement en cours chez les patients prétraités,
puis
3- Reprendre le traitement par brigatinib au palier de dose inférieur (cf. Tableau 1)
ou Arrêt immédiat et définitif du traitement par brigatinib
Troubles visuels
Grade 2 ou 3 1- Interrompre le traitement par brigatinib jusqu’à résolution à un Grade ≤ 1 ou récupération de l'état initial),
puis
2- reprendre le traitement par brigatinib au palier de dose inférieur (cf. Tableau 1).
Grade 4 Arrêt immédiat et définitif du traitement par briga tinib .
Autres effets indésirables
Grade 3
1èr épisode
1-Interrompre le traitement par brigatinib jusqu'à la récupération de l'état initial
puis
2- reprendre le traitement par brigatinib à la même posologie
Grade 4
1èr épisode
1-Interrompre le traitement par brigatinib jusqu'à la récupération de l'état initial
puis
2- reprendre le traitement par brigatinib au palier de dose inférieur (cf. Tableau 1)
récidive d’un grade ≥ 3 1-Interrompre le traitement par brigatinib jusqu'à la récupération de l'état initial
puis
2- reprendre le traitement par brigatinib au palier de dose inférieur (cf. Tableau 1)
ou Arrêt immédiat et définitif du traitement par briga tinib
bpm = battements par minute; PAD = pression artérielle diastolique ; FC = fréquence cardiaque ; PAS = pression artérielle systolique ; LSN = limite supérieure de la normale * Grades établis selon les critères communs de terminologie des évènements indésirables, établis par l’Institut national du cancer. Version 4.0 (NCI CTCAE v4).

ANSM-TAKEDA RCP Brigatinib 180 mg 6
Populations particulières
Sujets âgés Les données limitées sur la tolérance et l'efficacité de brigatinib chez les patients âgés d'au moins 65 ans suggèrent qu'une adaptation posologique n'est pas nécessaire chez les sujets âgés (voir rubrique 5.2). On ne dispose d'aucune donnée chez les patients âgés d'au moins 85 ans. Insuffisants hépatiques Aucune adaptation posologique de brigatinib n'est nécessaire chez les patients présentant une insuffisance hépatique légère (score de Child-Pugh A) ou modérée (score de Child-Pugh B). Une dose initiale réduite de 60 mg une fois par jour pendant les 7 premiers jours, puis 120 mg une fois par jour est recommandée pour les patients présentant une insuffisance hépatique sévère (score de Child-Pugh C) (voir rubrique 5.2). Insuffisants rénaux Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère ou modérée (débit de filtration glomérulaire estimé (DFGe) ≥ 30 mL/min/1,73 m²). Une dose initiale réduite de 60 mg une fois par jour pendant les 7 premiers jours, puis 90 mg une fois par jour est recommandée pour les patients présentant une insuffisance rénale sévère (DFGe < 30 mL/min/1,73 m) (voir rubrique 5.2). Population pédiatrique La tolérance et l'efficacité de brigatinib n'ont pas été établies chez les patients âgés de moins de 18 ans. Aucune donnée n'est disponible.
Mode d’administration
Brigatinib est administré par voie orale. Les comprimés doivent être avalés en entier avec de l’eau. Les comprimés peuvent être pris avec ou sans aliments. Le pamplemousse ou le jus de pamplemousse, susceptible d’augmenter les concentrations plasmatiques du brigatinib, doit être évité (voir rubrique 4.5). Les patients doivent veiller à ne pas avaler la capsule de dessiccant présente dans le flacon.
4.3. Contre-indications
Hypersensibilité au(x) substance(s) active(s) ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'empl oi
Compte-tenu du profil de tolérance du brigatinib (cf. Effets indésirables), il n’est pas recommandé de débuter un traitement chez les patients qui présentent :
- Une hypersensibilité au brigatinib ou à l’un des excipients, - Une toxicité non résolue de grade 3 ou 4 d’un traitement antérieur, - Une hypertension artérielle non traitée et/ou non contrôlée (systolique ≥ 160 mmHg,
diastolique ≥ 100 mmHg), - Un antécédent de pneumopathie interstitielle.
Les examens suivants devront être réalisés avant le début du traitement et au cours du traitement par brigatinib : Avant le début du traitement, il est indispensable de réaliser :
• Une recherche de la translocation ALK effectuée par une plateforme hospitalière de génétique moléculaire des cancers validée par l’Inca,
• Un bilan biologique sanguin comprenant: numération formule sanguine et taux de plaquettes, clairance de la créatinine, ionogramme avec dosage de la glycémie à jeûn, transaminases et phosphatases alcalines, taux de bilirubine sérique, taux des CPK et lipasémie,
• Un examen ophtalmologique de référence, • Une imagerie pulmonaire, • Un électrocardiogramme, un contrôle de la pression artérielle, • Un test sérologique de grossesse le cas échéant.

ANSM-TAKEDA RCP Brigatinib 180 mg 7
Il est recommandé de ne débuter le traitement que s i :
• ASAT/ALAT : o 3 x LSN en l’absence de métastases hépatiques, o 5 x LSN en présence de métastases hépatiques,
• Bilirubine totale < 1,5 x LSN (sauf en cas d’obstruction biliaire documenté), ou • Un taux de polynucléaires neutrophiles > 1.5 G/L, ou, • Un taux de plaquettes > 75 G/L ou, • Un taux d’hémoglobine > 80 g/L, • Un ECG normal (en particulier : absence de troubles du rythme, ou de la conduction).
Pendant le traitement :
• Un bilan biologique sanguin devra être effectué tous les mois, comprenant : numération formule sanguine et taux de plaquettes, clairance de la créatinine, ionogramme, glycémie à jeûn, transaminases, bilirubine sérique,
• Une surveillance de l’équilibre tensionnel devra être réalisée mensuellement, • Un électrocardiogramme devra être effectué le cas échéant, si cela est cliniquement indiqué,
• En cas de survenue d’une dyspnée, d’une toux, ou de nouvelle anomalie radiologique, effectuer une tomodensitométrie haute résolution (TDM-HR) du thorax.
• Un examen ophtalmologique devra être réalisé, si cela est cliniquement indiqué, en particulier si le patient présente des troubles visuels.
Certains effets indésirables sont particulièrement à prendre en considération : Effets indésirables pulmonaires Des effets indésirables pulmonaires sévères, menaçant le pronostic vital et fatals, à type de pneumonie, pneumopathie, dyspnée, embolie pulmonaire, détresse respiratoire, y compris des effets ayant des manifestations compatibles avec une pneumopathie, peuvent survenir chez les patients traités par brigatinib (voir rubrique 4.8). La plupart des effets indésirables pulmonaires ont été observés au cours des 7 premiers jours de traitement. Les effets indésirables pulmonaires de grade 1-2 ont disparu à l'interruption du traitement ou après modification de la posologie. L'âge croissant et le délai plus court (moins de 7 jours) entre la dernière dose de crizotinib et la première dose de brigatinib ont été indépendamment associés à une augmentation du taux de ces effets indésirables pulmonaires. Ces facteurs doivent être pris en considération lors de l'instauration d'un traitement par brigatinib. Les patients ayant des antécédents de pneumopathie ou de pneumopathie induite par les médicaments ont été exclus de l’étude pivotale. Certains patients présentent une pneumopathie ultérieurement au cours du traitement par brigatinib. Les patients doivent faire l'objet d'une surveillance clinique mensuelle pour détecter l'apparition de nouveaux symptômes respiratoires ou une aggravation de ces symptômes (par exemple, dyspnée, toux, etc.), notamment pendant la première semaine de traitement. Chez un patient présentant une aggravation des symptômes respiratoires, la recherche de pneumopathie doit être effectuée rapidement. En cas de suspicion de pneumopathie, il convient de suspendre le traitement par brigatinib et d'éliminer les autres étiologies (par exemple, embolie pulmonaire, progression tumorale, pneumonie infectieuse, etc.). La posologie doit être modifiée en conséquence (voir rubrique 4.2). Hypertension artérielle Une hypertension artérielle est apparue chez des patients traités par brigatinib (voir rubrique 4.8). La pression artérielle doit être bien équilibrée avant l’instauration du traitement. Une détection précoce et une prise en charge efficace de l’hypertension sont importantes pour limiter la nécessité de réductions de la dose de brigatinib et d’interruptions du traitement. La pression artérielle devra être surveillée mensuellement pendant le traitement. L'hypertension artérielle doit être traitée conformément aux directives standard pour contrôler la pression artérielle. La fréquence cardiaque devra être surveillée plus fréquemment en cas d'association à des agents antihypertenseurs bradycardisants. En cas d'hypertension artérielle sévère (≥ grade 3), le traitement par brigatinib doit être suspendu jusqu'à résolution. La posologie doit être modifiée en conséquence (voir rubrique 4.2).

ANSM-TAKEDA RCP Brigatinib 180 mg 8
Bradycardie Une bradycardie est apparue chez des patients traités par brigatinib (voir rubrique 4.8). La prudence s'impose lors de l'administration de brigatinib en association à d'autres agents bradycardisants. La fréquence cardiaque et la pression artérielle devront être surveillées régulièrement. En cas de bradycardie symptomatique, le traitement par brigatinib doit être suspendu et les médicaments concomitants connus pour provoquer une bradycardie doivent être évalués. Dès la récupération, la posologie doit être modifiée en conséquence (voir rubrique 4.2). En cas de bradycardie menaçant le pronostic vital, si aucun médicament concomitant contributeur n'est identifié ou en cas de récidive, le traitement par brigatinib doit être arrêté (voir rubrique 4.2). Troubles visuels Des effets indésirables à type de vision floue, diplopie, baisse de l’acuité visuelle, blépharospasme sont apparus chez des patients traités par brigatinib (voir rubrique 4.8). Il doit être conseillé aux patients de déclarer tous leurs éventuels symptômes visuels. En cas d'apparition ou d'aggravation de symptômes visuels sévères, un bilan ophtalmologique et une diminution posologique doivent être envisagés (voir rubrique 4.2). Élévation des taux de créatine phosphokinase (CPK) Des élévations des taux de CPK sont apparues chez des patients traités par brigatinib (voir rubrique 4.8). Il doit être conseillé aux patients de déclarer toute douleur, sensibilité douloureuse ou faiblesse musculaire inexpliquée. Les taux de CPK devront être surveillés régulièrement pendant le traitement par brigatinib. En fonction de la sévérité de l'élévation des taux de CPK, le traitement par brigatinib doit être suspendu et la posologie modifiée en conséquence (voir rubrique 4.2). Élévations des enzymes pancréatiques Des élévations des taux d'amylase et de lipase sont apparues chez des patients traités par brigatinib (voir rubrique 4.8). Les taux d'amylase et de lipase devront être surveillés régulièrement pendant le traitement par brigatinib. En fonction de la sévérité des anomalies biologiques, le traitement par brigatinib doit être suspendu et la posologie modifiée en conséquence (voir rubrique 4.2). Élévations des enzymes hépatiques Des élévations des taux d’enzymes hépatiques (aspartate aminotransférase et alanine aminotransférase) sont apparues chez des patients traités par brigatinib (voir rubrique 4.8). Les taux d’enzymes hépatiques devront être surveillés régulièrement pendant le traitement. En fonction de la sévérité des anomalies biologiques, le traitement doit être suspendu et la posologie modifiée en conséquence (voir rubrique 4.2). Hyperglycémie La glycémie à jeun doit être évaluée avant l’initiation du traitement par brigatinib et surveillée de manière mensuelle par la suite notamment, chez le patient diabétique ou sous corticothérapie. Un traitement adéquat doit être initié ou optimisé si besoin. En l’absence d’un contrôle satisfaisant de la glycémie, une interruption du traitement, une adaptation posologique ou l’arrêt du traitement peut être nécessaire (voir rubrique 4.2, tableau 2). Interactions médicamenteuses L'utilisation concomitante de Brigatinib avec des inhibiteurs puissants du CYP3A doit être évitée. Si l’utilisation concomitante d’inhibiteurs puissants du CYP3A ne peut être évitée, la dose de brigatinib doit être réduite de 180 mg à 90 mg, ou de 90 mg à 60 mg. Après l’interruption d’un inhibiteur puissant du CYP3A, brigatinib doit être repris au niveau de dose toléré avant l’initiation de l’inhibiteur puissant du CYP3A. L'utilisation concomitante de brigatinib avec des inducteurs puissants ou modérés du CYP3A doit être évitée. Lactose Brigatinib contient du lactose monohydraté. Les patients présentant des maladies héréditaires rares d'intolérance au galactose, de déficit en lactase de Lapp ou un syndrome de malabsorption du glucose et du galactose ne doivent pas prendre ce médicament.

ANSM-TAKEDA RCP Brigatinib 180 mg 9
4.5. Interactions avec d'autres médicaments et autr es formes d'interactions
Agents susceptibles d'augmenter les concentrations plasmatiques de brigatinib Inhibiteurs de CYP3A Des études in vitro ont démontré que le brigatinib est un substrat du CYP3A4/5. Chez les sujets sains, la co-administration de plusieurs doses biquotidiennes de 200 mg d'itraconazole, un inhibiteur puissant du CYP3A, avec une dose unique de 90 mg de brigatinib a augmenté de 21 % la Cmax du brigatinib, de 101 % l'ASC0--INF (2 fois) et de 82 % l'ASC0--120 (< 2 fois), par rapport à une dose de 90 mg de brigatinib administrée seule. L'utilisation concomitante d'inhibiteurs puissants du CYP3A et de brigatinib, y compris notamment certains antiviraux (par exemple, indinavir, nelfinavir, ritonavir, saquinavir), des antibiotiques macrolides par exemple, clarithromycine, télithromycine, troléandomycine), des antifongiques (par exemple, kétoconazole, voriconazole), le mibéfradil et le néfazodone doit être évitée. Si l’utilisation concomittante d’inhibiteurs puissants du CYP3A ne peut être évitée, la dose de brigatinib doit être réduite de 50 % (par exemple de 180 mg à 90 mg, ou de 90 mg à 60 mg, et les réductions de dose pourront aller jusqu'à 30 mg). Après l'arrêt d’un inhibiteur puissant du CYP3A, brigatinib doit être repris au niveau de dose toléré avant l’initiation de l’inhibiteur puissant du CYP3A après une période adaptée à la demi-vie de chaque molécule. Les inhibiteurs modérés du CYP3A4 (par exemple diltiazem et vérapamil) sont susceptibles d’augmenter l’ASC de brigatinib d’environ 40 % d’après des simulations réalisées à partir d’un modèle pharmacocinétique physiologique. Aucun ajustement de dose n’est requis pour brigatinib en association avec les inhibiteurs modérés du CYP3A. Les patients doivent être étroitement surveillés quand Brigatinib est co-administré les inhibiteurs modérés du CYP3A. La consommation de pamplemousse ou de jus de pamplemousse est également susceptible d'augmenter les concentrations plasmatiques du brigatinib et doit être évitée (voir rubrique 4.2). Inhibiteurs de CYP2C8 Des études in vitro ont démontré que le brigatinib est un substrat du CYP2C8. Chez les sujets sains, la co-administration de plusieurs doses biquotidiennes de 600 mg de gemfibrozil, un inhibiteur puissant du CYP2C8, avec une dose unique de 90 mg de brigatinib a diminué de 41 % la Cmax du brigatinib, de 12 % l'ASC0--INF et de 15 % l'ASC0--120, par rapport à une dose de 90 mg de brigatinib administrée seule. L'effet du gemfibrozil sur la pharmacocinétique du brigatinib n’est pas cliniquement significatif et le mécanisme sous-jacent de la diminution de l'exposition du brigatinib est inconnu. Aucun ajustement posologique n’est requis lors de la co-administration avec les inhibiteurs puissants du CYP2C8. Inhibiteurs de P-gp et BCRP Brigatinib est un substrat de la P-glycoprotéine (P-gp) et de la protéine de résistance du cancer du sein (BCRP, Breast Cancer Resistance Protein) in vitro. Etant donné la solubilité et la perméabilité élevées de brigatinib, l’inhibition de la P-gp et de la BCRP ne devrait pas avoir un effet cliniquement significatif sur l’exposition systémique au brigatinib. Aucun ajustement posologique n’est requis lors de la co-administration de brigatinib avec les inhibiteurs de la P-gp et de la BCRP. Agents susceptibles de diminuer les concentrations plasmatiques de brigatinib Inducteurs de CYP3A Chez les sujets sains, la co-administration de plusieurs doses de 600 mg par jour de rifampicine, un inducteur puissant du CYP3A, avec une dose unique de 180 mg de brigatinib a diminué de 60% la Cmax du brigatinib, de 80 % l'ASC0--INF (5 fois) et de 80 % l'ASC0--120 (5 fois), par rapport à une dose de 180 mg de brigatinib administrée seule. L'utilisation concomitante de brigatinib et d'inducteurs puissants de CYP3A, y compris notamment, la rifampicine, la carbamazépine, la phénytoïne, la rifabutine, le phénobarbital et le millepertuis doit être évitée. Les inducteurs modérés du CYP3A sont susceptibles de diminuer l’ASC de brigatinib d’environ 50 % d’après des simulations réalisées à partir d’un modèle pharmacocinétique physiologique. L’utilisation concomitante d’inducteurs modérés du CYP3A et de brigatinib, y compris notamment l’efavirenz, le modafinil, le bosentan, l’étravirine et la nafcilline, doit être évitée.

ANSM-TAKEDA RCP Brigatinib 180 mg 10
Agents dont les concentrations plasmatiques sont susceptibles d'être modifiées par le brigatinib Substrats de CYP3A Des études in vitro dans les hépatocytes ont montré que le brigatinib est un inducteur du CYP3A4. Aucune étude clinique d'interactions médicamenteuses avec des substrats sensibles au CYP3A n'a été effectuée. Brigatinib est susceptible de diminuer les concentrations plasmatiques de médicaments co-administrés qui sont surtout métabolisés par le CYP3A. L'utilisation concomitante de brigatinib et de substrats du CYP3A ayant des indices thérapeutiques étroits (par exemple l’alfentanil, le fentanyl, la quinidine, la cyclosporine, le sirolimus et le tacrolimus) doit être évitée en raison de la potentielle diminution de leur efficacité. Brigatinib est susceptible d’induire d’autres enzymes et d’autres transporteurs (par exemple CYP2C, P-gp) par le biais du même mécanisme responsable de l’induction du CYP3A (par exemple l’activation du récepteur pregnane X receptor). Substrats de transporteurs La co-administration de brigatinib avec des substrats de P-gp, (par exemple, digoxine, dabigatran, colchicine, pravastatine), BCRP (par exemple, méthotrexate, rosuvastatine, sulfasalazine), de transporteur 1 de cations organiques (OCT1), de protéines d'extrusion de multimédicaments et toxines 1(MATE1) et 2K (MATE2K), est susceptible d'augmenter leurs concentrations plasmatiques. Les patients doivent être étroitement surveillés quand brigatinib est co-administré par les substrats de ces transporteurs ayant des indices thérapeutiques étroits (par exemple digoxine, dabigatran, méthotrexate).
4.6. Fertilité, grossesse et allaitement
Contraception
Femmes en âge de procréer Les femmes susceptibles de procréer doivent avoir recours à une méthode efficace de contraception (non hormonale) pendant le traitement par brigatinib et la poursuivre pendant au moins 4 mois après la dernière prise. Hommes En raison du potentiel de génotoxicité, les hommes ayant des partenaires susceptibles de procréer doivent utiliser une méthode efficace de contraception pendant toute la durée du traitement par brigatinib et la poursuivre pendant au moins 4 mois après la dernière prise. Les hommes envisageant de concevoir un enfant doivent prévoir de faire congeler leur sperme avant le début du traitement. Aucun don de sperme ne doit être envisagé tout au long du traitement et jusqu’à 4 mois après l’arrêt du traitement.
Grossesse
Brigatinib est très probablement nocif pour le fœtus lorsqu'il est administré chez la femme enceinte. Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3). Il n’existe pas de données sur l'utilisation de brigatinib chez la femme enceinte. Brigatinib ne doit pas être utilisé pendant la grossesse à moins que la situation clinique de la femme ne justifie le traitement. Si la patiente est enceinte ou débute une grossesse pendant l'utilisation de brigatinib, elle devra être avertie des risques pour le fœtus.
Allaitement
On ne sait pas si brigatinib est excrété dans le lait maternel. Les données disponibles ne permettent pas d'exclure une excrétion potentielle dans le lait maternel. L’allaitement doit être interrompu au cours du traitement avec brigatinib.
Fertilité
Sur la base des résultats sur les organes reproducteurs mâles chez l’animal, le brigatinib peut provoquer une baisse de la fertilité chez les hommes (voir rubrique 5.3). La signification clinique exacte de ces données pour la fertilité humaine est inconnue.

ANSM-TAKEDA RCP Brigatinib 180 mg 11
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Brigatinib a une influence mineure sur l’aptitude à conduire des véhicules et à utiliser des machines. Cependant, la prudence s'impose lors de la conduite de véhicules ou l'utilisation de machines, puisque les patients sont susceptibles de présenter des troubles visuels, des sensations vertigineuses ou une fatigue lors de la prise de brigatinib.
4.8. Effets indésirables
Résumé du profil de tolérance
Les données décrites ci-dessous reflètent l’exposition au brigatinib de 456 patients traités dans la cadre d’une étude de phase II (ALTA) et d’une étude de phase I/II d’escalade de dose (étude 101). Dans la population traitée par brigatinib, 219 patients atteints d’un cancer du poumon non à petites cellules (CPNPC) ALK-positif à un stade localement avancé ou métastatique, ayant progressé sous crizotinib ont été traités dans le cadre de l’étude de phase II. La posologie reçue par les patients était de 90 mg à 180 mg, une fois par jour en continu. Les patients ont été randomisés selon un rapport de 1/1 pour recevoir un traitement par brigatinib à dose de 90 mg une fois par jour en continu (schéma posologique à 90 mg) ou à une dose de 180 mg une fois par jour avec 7 jours préliminaires à 90 mg une fois par jour (schéma posologique à 180 mg, recommandé en pratique). Cent-trente-sept (137) patients présentant des affections malignes avancées ont été traités dans l’étude 101, multicentrique, ouverte de phase I/II d'escalade de dose. Les effets indésirables les plus fréquents (≥ 25 %) rapportés chez les patients traités par brigatinib au schéma posologique recommandé étaient : hyperglycémie, élévation des taux d’ASAT, anémie, nausées, augmentation des taux de lipase, augmentation du nombre de lymphocytes, élévation des taux d’ALAT, diarrhées, augmentation des taux d’amylase, fatigue, toux, augmentation des taux sanguins de créatine phosphokinase, céphalées, augmentation des taux de phosphatases alcalines, hypophosphatémie, augmentation du temps de céphaline activée (TCA), éruption cutanée, vomissements, dyspnée et hypertension. Dans l’étude ALTA, les effets indésirables graves les plus fréquents, rapportés chez au moins 2 % des patients, étaient une pneumonie (5,0 %) et une pneumopathie (5,0 %).
Tableau répertoriant les effets indésirables
Les effets indésirables rapportés dans l'étude ALTA et l’étude 101 au schéma posologique recommandé sont présentés dans le Tableau 3 et sont répertoriés par classe de système d'organes, terme préférentiel et fréquence. Les catégories de fréquence sont les suivantes : très fréquent (≥ 1/10), fréquent (≥ 1/100 à <1/10) et peu fréquent (≥ 1/1 000 à < 1/100). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre de fréquence. Tableau 3 : Effets indésirables rapportés chez les patients traités par brigatinib dans l'étude ALTA et l’étude 101 [Common Terminology Criteria fo r Adverse Events (critères communs de terminologie pour les effets indésirables) – (CTCAE ) version 4.0] Classe de système d'organes
Fréquence Effets indésirables † de tous grades
Effets indésirables de grade 3-4
Infections et infestations
Très fréquent Infection des voies respiratoires supérieures Pneumoniea
Fréquent Pneumoniea Affections hématologiques et du système lymphatique
Très fréquent Anémie Diminution du nombre de lymphocytes Diminution du nombre de globules blancs Augmentation du TCA Diminution du nombre de plaquettes
Diminution du nombre de lymphocytes
Fréquent Anémie Diminution du nombre de lymphocytes Augmentation du TCA

ANSM-TAKEDA RCP Brigatinib 180 mg 12
Classe de système d'organes
Fréquence Effets indésirables † de tous grades
Effets indésirables de grade 3-4
Troubles du métabolisme et de la nutrition
Très fréquent Hyperglycémie Diminution de l’appétit Hyponatrémie Hypokaliémie Hypomagnésémie Hypophosphatémie
Fréquent Hyperinsulinémieb Hypophosphatémie Hyperglycémie Hyponatrémie Hypokaliémie
Peu fréquent Diminution de l’appétit Troubles psychiatriques
Très fréquent Insomnie
Affections du système nerveux
Très fréquent Céphaléesc
Neuropathie périphériqued Sensations vertigineuses
Fréquent Disgueusie Neuropathie périphériqued Céphaléesc
Affections oculaires
Très fréquent Troubles visuelse
Fréquent Troubles de la visione
Affections cardiaques
Fréquent Bradycardief Prologation de l’intervalle QT sur l’électrocardiogramme Palpitations
Peu fréquent Prolongation de l’intervalle QT sur l’électrocardiogramme
Affections vasculaires
Très fréquent Hypertension
Fréquent Hypertension
Affections respiratoires, thoraciques et médiastinales
Très fréquent Toux Dyspnéeg
Fréquent Pneumopathieh Pneumopathieh Dyspnéeg
Affections gastro-intestinales
Très fréquent Nausée Diarrhée Vomissements Douleurs abdominalesi Constipation Augmentation du taux de lipase Augmentation du taux d’amylase Sécheresse buccale
Augmentation du taux de lipase
Fréquent Dyspepsie Stomatitej Flatulence
Douleurs abdominalesi Augmentation du taux d’amylase
Peu fréquent Pancréatite Nausée Dyspepsie Pancréatite Stomatite
Affections hépatobiliaires
Très fréquent Augmentation du taux d’ASAT Augmentation du taux d’ALAT Augmentation du taux de phosphatase alcaline
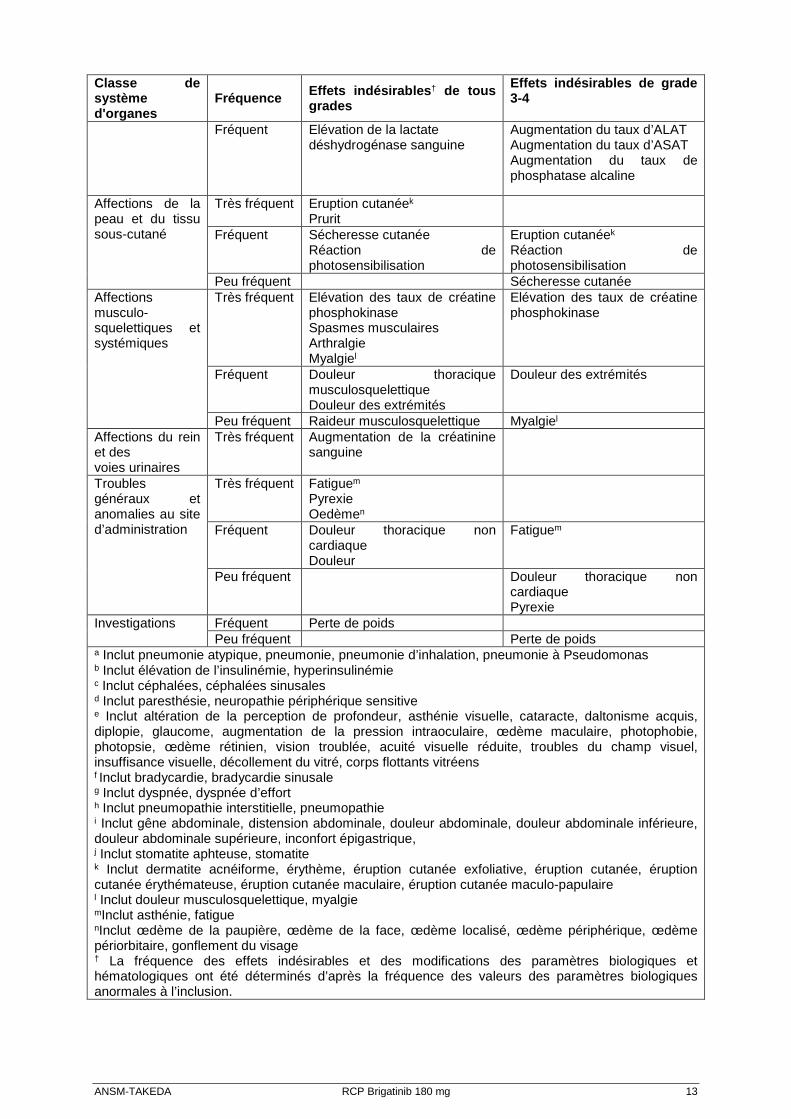
ANSM-TAKEDA RCP Brigatinib 180 mg 13
Classe de système d'organes
Fréquence Effets indésirables † de tous grades
Effets indésirables de grade 3-4
Fréquent Elévation de la lactate déshydrogénase sanguine
Augmentation du taux d’ALAT Augmentation du taux d’ASAT Augmentation du taux de phosphatase alcaline
Affections de la peau et du tissu sous-cutané
Très fréquent Eruption cutanéek Prurit
Fréquent Sécheresse cutanée Réaction de photosensibilisation
Eruption cutanéek Réaction de photosensibilisation
Peu fréquent Sécheresse cutanée Affections musculo-squelettiques et systémiques
Très fréquent Elévation des taux de créatine phosphokinase Spasmes musculaires Arthralgie Myalgiel
Elévation des taux de créatine phosphokinase
Fréquent Douleur thoracique musculosquelettique Douleur des extrémités
Douleur des extrémités
Peu fréquent Raideur musculosquelettique Myalgiel Affections du rein et des voies urinaires
Très fréquent Augmentation de la créatinine sanguine
Troubles généraux et anomalies au site d’administration
Très fréquent Fatiguem Pyrexie Oedèmen
Fréquent Douleur thoracique non cardiaque Douleur
Fatiguem
Peu fréquent Douleur thoracique non cardiaque Pyrexie
Investigations Fréquent Perte de poids Peu fréquent Perte de poids
a Inclut pneumonie atypique, pneumonie, pneumonie d’inhalation, pneumonie à Pseudomonas
b Inclut élévation de l’insulinémie, hyperinsulinémie c Inclut céphalées, céphalées sinusales d Inclut paresthésie, neuropathie périphérique sensitive e Inclut altération de la perception de profondeur, asthénie visuelle, cataracte, daltonisme acquis, diplopie, glaucome, augmentation de la pression intraoculaire, œdème maculaire, photophobie, photopsie, œdème rétinien, vision troublée, acuité visuelle réduite, troubles du champ visuel, insuffisance visuelle, décollement du vitré, corps flottants vitréens f Inclut bradycardie, bradycardie sinusale g Inclut dyspnée, dyspnée d’effort h Inclut pneumopathie interstitielle, pneumopathie i Inclut gêne abdominale, distension abdominale, douleur abdominale, douleur abdominale inférieure, douleur abdominale supérieure, inconfort épigastrique, j Inclut stomatite aphteuse, stomatite k Inclut dermatite acnéiforme, érythème, éruption cutanée exfoliative, éruption cutanée, éruption cutanée érythémateuse, éruption cutanée maculaire, éruption cutanée maculo-papulaire l Inclut douleur musculosquelettique, myalgie mInclut asthénie, fatigue nInclut œdème de la paupière, œdème de la face, œdème localisé, œdème périphérique, œdème périorbitaire, gonflement du visage † La fréquence des effets indésirables et des modifications des paramètres biologiques et hématologiques ont été déterminés d’après la fréquence des valeurs des paramètres biologiques anormales à l’inclusion.

ANSM-TAKEDA RCP Brigatinib 180 mg 14
Description des effets indésirables sélectionnés Effets indésirables pulmonaires Dans l'étude ALTA, 14 (6,4 %) patients ont présenté des effets indésirables pulmonaires de tout grade, y compris pneumopathie, pneumonie et dyspnée, au début du traitement (dans les 9 jours, médiane : 2 jours) ; 2,7 % des patients ont présenté des effets indésirables pulmonaires de grade 3-4 et 1 patient (0,5 %) a présenté une pneumonie d’évolution fatale. Les patients ayant présenté des effets indésirables pulmonaires de grade ≥3 ont arrêté définitivement le traitement par brigatinib. 7 patients (3,2%) ont présenté des effets indésirables pulmonaires de grade 1-2 qui ont nécessité l’interruption temporaire du traitement par brigatinig chez six patients. Des effets indésirables pulmonaires précoces se sont également produits dans une étude d'escalade de doses chez des patients (N = 137) (étude 101), dont trois cas fatals (hypoxie, syndrome de détresse respiratoire aigüe et pneumonie). En outre, 2,3 % des patients de l’étude ALTA ont présenté une pneumopathie ultérieurement dans le traitement, dont 2 patients atteints de pneumopathie de grade 3 (voir rubriques 4.2 et 4.4) Sujets âgés Dans l’étude ALTA, 13,5 % des patients dont l’âge était ≥ 65 ans ont présenté un effet indésirable pulmonaire précoce alors que 4,2 % des patients dont l’âge était < 65 ans ont présenté un effet indésirable pulmonaire précoce. Hypertension artérielle Dans l'étude ALTA, une hypertension artérielle a été décrite chez 27 % des patients traités par brigatinib au schéma posologique à 180 mg, dont 8,2 % présentaient une hypertension artérielle de grade 3. Une diminution posologique pour hypertension artérielle a été effectuée dans 0,9 % des cas au schéma posologique à 180 mg. Deux patients (0,9%) ont arrêté temporairement le traitement. Aucune interruption définitive du traitement par brigatinib en relation avec la survenue d’une hypertension artérielle n’a été rapportée. Les pressions artérielles systolique et diastolique ont augmenté avec le temps chez tous les patients (voir rubriques 4.2 et 4.4). Bradycardie Dans l'étude ALTA, une bradycardie a été décrite chez 4,5 % des patients traités par brigatinib au schéma posologique à 180 mg. Des fréquences cardiaques inférieures à 50 battements par minute (bpm) ont été décrites chez 8,2 % des patients au schéma posologique à 180 mg (voir rubriques 4.2 et 4.4). Aucune bradycardie de grade ≥ 3 n’a été rapportée. Troubles visuels Dans l'étude ALTA, des troubles à type de vision floue, diplopie, baisse de l’acuité visuelle, blépharospasme ont été rapportés chez 16 % des patients traités par brigatinib au schéma posologique à 180 mg, dont deux effets indésirables de grade 3 (1,8 %), incluant un œdème maculaire et une cataracte. Une diminution posologique pour troubles visuels a été effectuée chez deux patients (1,8 %) au schéma posologique à 180 mg (voir rubriques 4.2 et 4.4). Un patient ayant présenté un effet indésirable de grade 3 (œdème maculaire) a arrêté temporairement le traitement par brigatinib puis a repris le traitement à une dose inférieure. Neuropathie périphérique Dans l’étude ALTA, les neuropathies périphériques, y compris les neuropathies périphériques sensitives (dont la paresthésie) ont été reportées chez 18 % des patients traités par brigatinib au schéma posologique à 180 mg. 75% ont conservé des séquelles jusqu'à la fin de leur suivi. Des neuropathies périphériques de grade ≥ 3 ont été rapportées pour 6 sur 219 patients (2,7%), un patient a arrêté temporairement et deux patients ont arrêté définitivement le traitement par brigatinib. Élévation des taux de créatine phosphokinase (CPK) Dans l'étude ALTA, des élévations des taux de créatine phosphokinase (CPK) ont été décrites chez 50 % des patients traités par brigatinib au schéma posologique à 180 mg. L'incidence des élévations de grade 3-4 des taux de CPK était de 14 %. Le délai médian avant l'apparition des élévations des taux de CPK était de 27 jours.

ANSM-TAKEDA RCP Brigatinib 180 mg 15
Une diminution posologique pour élévation des taux de CPK a été effectuée chez 6,4 % des patients au schéma posologique à 180 mg (voir rubriques 4.2 et 4.4). Parmi les patients ayant présenté une élévation des taux de CPK (tous schémas posologiques), 71,4% ont eu une résolution par diminution posologique. Aucun patient n’a arrêté définitivement le traitement par brigatinib. Élévations des enzymes pancréatiques Lors des dosages mensuels, dans l'étude ALTA, des augmentations des taux d'amylase et de lipase ont été décrites chez respectivement 41 % et 46 % des patients traités par brigatinib au schéma posologique à 180 mg. Pour les élévations de grade 3 ou 4, les incidences pour le taux d'amylase et le taux de lipase étaient respectivement de 7,3 % et 10 %. Les délais médians avant l'apparition des élévations des taux d'amylase et de lipase étaient respectivement de 16 jours et 29 jours. Une diminution posologique pour élévations des taux de lipase et d'amylase a été effectuée chez 1,8 % et 0,9 % des patients respectivement au schéma posologique à 180 mg (voir rubriques 4.2 et 4.4). Parmi les patients ayant présenté une élévation des taux d’amylase et de lipase (tous schémas posologiques), 2,7% ont arrêté temporairement le traitement par brigatinib. Aucun cas de pancréatite aigüe n’a été reporté et aucun patient n’a arrêté définitivement le traitement par brigatinib. Augmentation des taux d’enzymes hépatiques Lors des dosages mensuels, dans l’étude ALTA, des augmentations des taux d’ALAT et d’ASAT, suite à une analyse biologique, ont été reportées chez 46 % et 65 % des patients respectivement traités par brigatinib au schéma posologique à 180 mg. Pour les augmentations de grade 3 et 4, les incidences des taux d’ALAT et d’ASAT étaient respectivement de 4,5 % et de 2,7 %. Aucune diminution posologique n’a été effectuée suite à des augmentations des taux d’ALAT ou d’ASAT. Aucun patient n’a arrêté définitivement le traitement par brigatinib. Hyperglycémie Lors des dosages mensuels, dans l’étude ALTA, 67 % des patients ont présenté une hyperglycémie. Des hyperglycémies de grade 3 ont été rapportées chez 5,5 % des patients. Aucune diminution posologique n’a été effectuée suite à une hyperglycémie. Un patient a arrêté temporairement le traitement par brigatinib. Un cas de diabète a été reporté mais n’a pas nécessité d’adaptation posologique ni d’arrêt de traitement. Aucun patient n’a arrêté définitivement le traitement par brigatinib. Photosensibilité Des cas de photosensibilité à la lumière du jour ont été rapportés chez des patients traités par brigatinib. Il faut recommander aux patients d’éviter l’exposition au soleil, de porter des vêtements couvrants, d’utiliser une crème solaire et un baume pour les lèvres ayant un indice de protection maximal pendant toute la durée de votre traitement, et jusqu’à au moins 7 jours après l’arrêt du traitement.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration – voir PUT, section 3.1.
4.9. Surdosage
Il n'existe aucun antidote spécifique en cas de surdosage de brigatinib. Dans le cas d’un surdosage, surveiller le patient pour déceler tout signe ou symptôme de toxicité (voir rubrique 4.8) et la prise en charge doit être définie au cas par cas.

ANSM-TAKEDA RCP Brigatinib 180 mg 16
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : agent antinéoplasiqu e, inhibiteurs de la tyrosine kinase, code ATC : L01XE43 .
Mécanisme d’action
Le brigatinib est un inhibiteur de tyrosine kinase ciblant principalement ALK, ROS1 (c-ros oncogene 1) et IGF-1R (insulin--like growth factor 1 receptor, récepteur du facteur de croissance 1 de type insuline). Le brigatinib a inhibé l'autophosphorylation d'ALK et la phosphorylation médiée par ALK de la protéine de signalisation située en aval STAT3 dans les études in vitro et in vivo. Le brigatinib a inhibé la prolifération in vitro des lignées cellulaires exprimant les protéines de fusion EML4--ALK et NPM--ALK et a démontré une inhibition dose-dépendante de la croissance de xénogreffe de CBNPC EML4-ALK-positif chez la souris. Le brigatinib a inhibé la viabilité in vitro et in vivo des cellules exprimant les formes mutantes d’EML4-ALK associées à une résistance aux inhibiteurs d'ALK, y compris G1202R et L1196M.
Electrophysiologie cardiaque
Dans l’étude 101, le potentiel de prolongation de l’intervalle QT de brigatinib a été évalué chez 123 patients présentant des affections malignes avancées après une dose de 30 mg à 240 mg de brigatinib par jour. La modification moyenne maximum de l’intervalle QTcF (intervalle QT corrigé selon la méthode de Fridericia) depuis l’inclusion était inférieure à 10 msec. Une analyse de l’exposition et de l’intervalle QT a montré que la prolongation de l’intervalle QTc n’était pas dépendante de la concentration.
Efficacité et sécurité cliniques
Etude ALTA L'efficacité et la sécurité cliniques de brigatinib ont été évaluées dans une étude multicentrique randomisée (selon un rapport de 1/1), ouverte chez 222 patients adultes ayant un CBNPC ALK-positif localement avancé ou métastatique, dont la maladie a progressé sous crizotinib. Les critères d'éligibilité ont permis le recrutement de patients présentant un réarrangement d'ALK documenté sur la base d’un test validé, un score de performances ECOG de 0-2 et des traitements par chimiothérapie antérieurs. Par ailleurs, les patients présentant des métastases du système nerveux central (SNC) étaient inclus, à condition qu'elles soient stables sur le plan neurologique et ne nécessitent pas une augmentation de la dose de corticoïdes. Les patients ayant des antécédents de pneumopathie interstitielle ou de pneumopathie médicamenteuse ont été exclus. Les patients ont été randomisés selon un rapport de 1/1 pour recevoir un traitement par brigatinib à une dose de 90 mg une fois par jour (schéma posologique à 90 mg ; n = 112) ou à une dose de 180 mg une fois par jour avec 7- jours préliminaires à 90 mg une fois par jour (schéma posologique à 180 mg ; n = 110). La durée médiane du suivi était de 17,9 mois. La randomisation a été stratifiée en fonction des métastases cérébrales (présentes, absentes) et de la meilleure réponse précédente au traitement par crizotinib (réponse complète ou partielle, toute autre réponse/inconnue). Le principal critère d'évaluation a été le taux de réponse objective (TRO) selon les critères RECIST (Response Evaluation Criteria In Solid Tumors v1.1) évalué par l'investigateur. Les autres critères d'évaluation ont notamment inclus : TRO évalué par un comité d'examen indépendant (IRC, Independent Review Committee) ; délai avant la réponse ; survie sans progression (SSP) ; durée de la réponse (DdR) ; survie globale ; TRO intracrânienne et DdR intracrânienne, d'après l'évaluation d'un IRC.
Les données démographiques et les caractéristiques de la maladie à l'inclusion dans l'étude ALTA étaient : âge médian de 54 ans (intervalle : 18 à 82 ; 23 % de patients âgés d'au moins 65 ans), 67 % d’origine caucasienne et 31 % d’origine asiatique, 57 % de femmes, 36 % de patients avaient un score de performances ECOG de 0, 57 % un score de performances ECOG de 1 et 7 % un score de performance ECOG de 2, 60 % de patients n'avaient jamais fumé, 35 % des patients étaient d’anciens fumeurs et 5 % étaient fumeurs, 98 % de patients présentaient un stade IV, 97 % de patients avaient

ANSM-TAKEDA RCP Brigatinib 180 mg 17
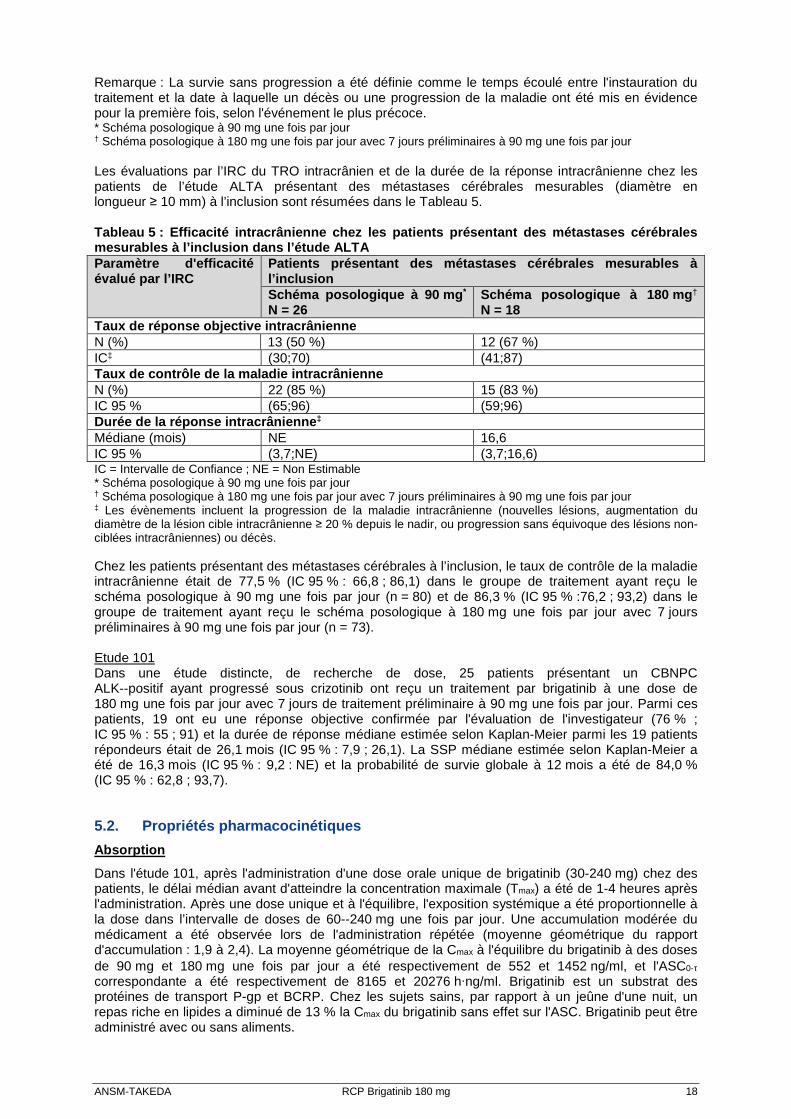
un adénocarcinome et 74 % avaient reçu une chimiothérapie antérieure. Les sites les plus fréquents de métastases extra thoraciques étaient le cerveau (69 %, dont 62 % ayant reçu une radiothérapie cérébrale antérieure), les os (39 %) et le foie (26 %). Les résultats d'efficacité de l'analyse de l'étude ALTA sont récapitulés dans le Tableau 4 et les courbes de Kaplan--Meier (KM) relatives aux évaluations de SSP effectuées par l'investigateur sont présentées en Figure 1. Tableau 4 : Résultats d'efficacité dans l'étude ALT A (population ITT) Paramètre d’Efficacité Évaluation de l'investigateur Évaluation de l’IRC
Schéma posologique à 90 mg * N = 112
Schéma posologique à 180 mg † N = 110
Schéma posologique à 90 mg * N = 112
Schéma posologique à 180 mg † N = 110
Taux de réponse objective N (%) 50 (45 %) 61 (55 %) 56 (50 %) 59 (54 %) IC‡ (34, 56) (44, 66) (40, 60) (44, 64) Délai avant la réponse Médian (mois) 1,8 1,9 1,8 1,9 Durée de la réponse Médiane (mois) 12,0 13,8 13,8 14,8 IC 95 % (9,2, 17,7) (10,2, 17,5) (7,4, 12,8) (12,6, NE) Survie sans progression Médiane (mois) 9,2 15,6 9,2 16,7 IC 95 % (7,4, 11,1) (11,1, 19,4) (7,4, NE) (11,6, NE) Survie globale Médiane (mois) NE 27,6 NA NA IC 95 % (20,2, NE) (27,6, NE) NA NA Probabilité de survie à 12 mois (%)
70,3 % 80,1 % NA NA
IC = Intervalle de Confiance ; NE = Non Estimable ; NA = Non Applicable * Schéma posologique à 90 mg une fois par jour
† Schéma posologique à 180 mg une fois par jour avec 7 jours préliminaires à 90 mg une fois par jour
‡ L'intervalle de confiance pour le TRO évalué par l'investigateur est de 97,5 % ; il est de 95 % pour le TRO évalué par l’IRC Figure 1 : Survie sans progression systémique évaluée par l'in vestigateur : population ITT par groupe de traitement (étude ALTA)
Abréviations : ITT = intention de traiter
ANSM-TAKEDA RCP Brigatinib 180 mg 18
Remarque : La survie sans progression a été définie comme le temps écoulé entre l'instauration du traitement et la date à laquelle un décès ou une progression de la maladie ont été mis en évidence pour la première fois, selon l'événement le plus précoce. * Schéma posologique à 90 mg une fois par jour
† Schéma posologique à 180 mg une fois par jour avec 7 jours préliminaires à 90 mg une fois par jour Les évaluations par l’IRC du TRO intracrânien et de la durée de la réponse intracrânienne chez les patients de l’étude ALTA présentant des métastases cérébrales mesurables (diamètre en longueur ≥ 10 mm) à l’inclusion sont résumées dans le Tableau 5. Tableau 5 : Efficacité intracrânienne chez les pati ents présentant des métastases cérébrales mesurables à l’inclusion dans l’étude ALTA Paramètre d'efficacité évalué par l’IRC
Patients présentant des métastases cérébrales mesur ables à l’inclusion Schéma posologique à 90 mg * N = 26
Schéma posologique à 180 mg † N = 18
Taux de réponse objective intracrânienne N (%) 13 (50 %) 12 (67 %) IC‡ (30;70) (41;87) Taux de contrôle de la maladie intracrânienne N (%) 22 (85 %) 15 (83 %) IC 95 % (65;96) (59;96) Durée de la réponse intracrânienne ‡ Médiane (mois) NE 16,6 IC 95 % (3,7;NE) (3,7;16,6) IC = Intervalle de Confiance ; NE = Non Estimable * Schéma posologique à 90 mg une fois par jour
† Schéma posologique à 180 mg une fois par jour avec 7 jours préliminaires à 90 mg une fois par jour ‡ Les évènements incluent la progression de la maladie intracrânienne (nouvelles lésions, augmentation du diamètre de la lésion cible intracrânienne ≥ 20 % depuis le nadir, ou progression sans équivoque des lésions non-ciblées intracrâniennes) ou décès. Chez les patients présentant des métastases cérébrales à l’inclusion, le taux de contrôle de la maladie intracrânienne était de 77,5 % (IC 95 % : 66,8 ; 86,1) dans le groupe de traitement ayant reçu le schéma posologique à 90 mg une fois par jour (n = 80) et de 86,3 % (IC 95 % :76,2 ; 93,2) dans le groupe de traitement ayant reçu le schéma posologique à 180 mg une fois par jour avec 7 jours préliminaires à 90 mg une fois par jour (n = 73). Etude 101 Dans une étude distincte, de recherche de dose, 25 patients présentant un CBNPC ALK--positif ayant progressé sous crizotinib ont reçu un traitement par brigatinib à une dose de 180 mg une fois par jour avec 7 jours de traitement préliminaire à 90 mg une fois par jour. Parmi ces patients, 19 ont eu une réponse objective confirmée par l'évaluation de l'investigateur (76 % ; IC 95 % : 55 ; 91) et la durée de réponse médiane estimée selon Kaplan-Meier parmi les 19 patients répondeurs était de 26,1 mois (IC 95 % : 7,9 ; 26,1). La SSP médiane estimée selon Kaplan-Meier a été de 16,3 mois (IC 95 % : 9,2 : NE) et la probabilité de survie globale à 12 mois a été de 84,0 % (IC 95 % : 62,8 ; 93,7).
5.2. Propriétés pharmacocinétiques
Absorption
Dans l'étude 101, après l'administration d'une dose orale unique de brigatinib (30-240 mg) chez des patients, le délai médian avant d'atteindre la concentration maximale (Tmax) a été de 1-4 heures après l'administration. Après une dose unique et à l'équilibre, l'exposition systémique a été proportionnelle à la dose dans l’intervalle de doses de 60--240 mg une fois par jour. Une accumulation modérée du médicament a été observée lors de l'administration répétée (moyenne géométrique du rapport d'accumulation : 1,9 à 2,4). La moyenne géométrique de la Cmax à l'équilibre du brigatinib à des doses de 90 mg et 180 mg une fois par jour a été respectivement de 552 et 1452 ng/ml, et l'ASC0-τ correspondante a été respectivement de 8165 et 20276 h·ng/ml. Brigatinib est un substrat des protéines de transport P-gp et BCRP. Chez les sujets sains, par rapport à un jeûne d'une nuit, un repas riche en lipides a diminué de 13 % la Cmax du brigatinib sans effet sur l'ASC. Brigatinib peut être administré avec ou sans aliments.

ANSM-TAKEDA RCP Brigatinib 180 mg 19
Distribution
Brigatinib se liait modérément (91 %) aux protéines plasmatiques humaines et la liaison n'était pas dépendante de la concentration. Le rapport de concentration sang/plasma est de 0,69. Chez les patients ayant reçu un traitement par brigatinib 180 mg une fois par jour, la moyenne géométrique du volume apparent de distribution (Vz/F) du brigatinib à l'équilibre a été de 153 l, indiquant une distribution modérée dans les tissus.
Biotransformation
Les études in vitro ont démontré que le brigatinib est principalement métabolisé par le CYP2C8 et le CYP3A4, et beaucoup moins par le CYP3A5. Après l'administration orale d'une dose unique de 180 mg de [14C] brigatinib chez des sujets sains, les deux voies majeures d'élimination métabolique ont été la N--déméthylation et la conjugaison à la cystéine. Dans les urines et fèces combinées, respectivement 48 %, 27 % et 9,1 % de la dose radioactive ont été excrétés sous forme de brigatinib inchangée, de N--desméthyl brigatinib (AP26123), et de conjugué de brigatinib à la cystéine. La forme inchangée de brigatinib a été le composant radioactif circulant majeur (92 %), avec l'AP26123 (3,5 %), le principal métabolite également observé in vitro. Chez les patients, à l'équilibre, l'ASC plasmatique de l'AP26123 a été < 10 % de l'exposition du brigatinib. Dans les essais cellulaires et de kinase in vitro, le métabolite AP26123 a inhibé ALK environ 3 fois moins que le brigatinib.
Élimination
Chez les patients ayant reçu un traitement par brigatinib 180 mg une fois par jour, la moyenne géométrique de la clairance orale apparente (CL/F) du brigatinib à l'équilibre a été de 13 l/h et la demi-vie d'élimination plasmatique moyenne a été de 25 h. Les fèces constituent la principale voie d'excrétion du brigatinib. Chez 6 sujets sains de sexe masculin ayant reçu une dose orale unique de 180 mg de [14C] brigatinib, 65 % de la dose administrée ont été éliminés dans les fèces et 25 % de la dose administrée ont été éliminés dans les urines. La forme inchangée du brigatinib a représenté respectivement 41 % et 86 % de la radioactivité totale dans les fèces et les urines, le reste étant des métabolites.
Populations spécifiques
Insuffisants hépatiques La pharmacocinétique du brigatinib a été caractérisée chez des patients présentant une fonction hépatique normale (n = 9), des patients atteints d’une insuffisance hépatique légère (score de Child-Pugh A, n = 6), des patients atteints d’une insuffisance hépatique modérée (score de Child-Pugh B, n = 6) et des patients atteints d’une insuffisance hépatique sévère (score de Child-Pugh C, n = 6). La pharmacocinétique de brigatinib était similaire chez les patients présentant une fonction hépatique normale et les patients atteints d’une insuffisance hépatique légère (score de Child-Pugh A) ou modérée (score de Child-Pugh B). L’ASC0-INF de brigatinib non lié était 37 % supérieure chez les patients atteints d’une insuffisance hépatique sévère (score de Child-Pugh C) en comparaison aux patients présentant une fonction hépatique normale (voir rubrique 4.2). Insuffisants rénaux La pharmacocinétique du brigatinib est similaire chez les patients présentant une fonction rénale normale et les patients atteints d’une insuffisance rénale légère ou modérée (eGFR ≥ 30 mL/min/1,73 m²) d’après les résultats d’analyses pharmacocinétiques de population. Dans une étude de pharmacocinétique, l’ASC0-INF de brigatinib non lié était 92 % supérieure chez les patients atteints d’une insuffisance rénale sévère (eGFR < 30 mL/min/1,73 m², n = 8) en comparaison aux patients présentant une fonction rénale normale (eGFR ≥ 90 mL/min/1,73 m², n = 8) (voir rubrique 4.2). Race et sexe Les analyses de pharmacocinétique de population ont montré que la race et le sexe n'avaient aucune interférence sur la pharmacocinétique du brigatinib.

ANSM-TAKEDA RCP Brigatinib 180 mg 20
Âge, poids corporel et concentrations d'albumine Les analyses de pharmacocinétique de population ont montré que le poids corporel, l'âge, et les concentrations d'albumine n'avaient aucune répercussion cliniquement significative sur la pharmacocinétique du brigatinib.
5.3. Données de sécurité préclinique
Les études de pharmacologie de sécurité évaluant le brigatinib n'ont pas indiqué de potentiel d'allongement de l'intervalle QT ou d'effets neurofonctionnels mais ont identifié un potentiel d'effets pulmonaires (modification de la fréquence respiratoire ; à 1-2 fois la Cmax humaine), d'effets cardiovasculaires (modification de la fréquence cardiaque et de la pression artérielle ; à 0,5 fois la Cmax humaine) et des effets rénaux (diminution de la fonction rénale ; à 1-2,5 fois la Cmax humaine) à la dose de 180 mg une fois par jour. Les effets indésirables observés chez l’animal à des niveaux d’exposition similaires aux niveaux d’exposition clinique avec une possible pertinence pour l’utilisation clinique étaient les suivants : système gastro-intestinal, moelle osseuse, yeux, testicules, foie, reins, os et cœur. Ces effets étaient généralement réversibles pendant la période de récupération sans traitement, cependant les effets oculaires et testiculaires ont été des exceptions en raison d’une absence de récupération. Aucune étude de carcinogenicité n'a été effectuée avec le brigatinib. Brigatinib n'a pas été mutagène in vitro dans le test d'Ames (mutation bactérienne inverse) ou les essais d'aberration chromosomique dans des cellules de mammifères mais a légèrement augmenté le nombre de micronoyaux dans un test de micronoyaux sur des cellules de moelle osseuse de rat. Le mécanisme d'induction de micronoyaux a été une ségrégation chromosomique anormale (aneugénicité) et non un effet clastogène sur les chromosomes. Cet effet a été observé à environ cinq fois l'exposition humaine à la dose de 180 mg une fois par jour. Brigatinib est susceptible d'avoir un effet négatif sur la fertilité masculine. Une toxicité testiculaire a été observée dans les études chez l'animal à doses itératives. Chez le rat, cette toxicité s'est manifestée par une diminution du poids des testicules, des vésicules séminales et de la prostate, ainsi que par une dégénérescence tubulaire testiculaire ; ces effets n'ont pas été réversibles pendant la période de récupération. Chez le singe, on a observé une diminution de la taille des testicules et des signes d'hypospermatogénèse à l'examen microscopique ; ces effets ont été réversibles pendant la période de récupération. Globalement, ces effets sur les organes reproducteurs chez le rat et le singe mâles sont apparus à des expositions ≥ 0,2- fois l'ASC observée chez les patients à la dose de 180 mg une fois par jour. Aucun effet indésirable apparent sur les organes reproducteurs de la femelle n'a été observé dans les études de toxicologie générale effectuées chez le rat et le singe. Dans une étude de développement embryofœtal dans laquelle des rates gestantes ont reçu des doses quotidiennes de brigatinib pendant l'organogenèse, des anomalies squelettiques liées à la dose ont été observées à des doses aussi faibles qu'environ 0,7- fois l'exposition humaine selon l'ASC à la dose de 180 mg une fois par jour. Ces anomalies se sont manifestées par une létalité embryonnaire, une diminution de la croissance fœtale et des variations squelettiques.
6. DONNEES PHARMACEUTIQUES
6.1. Liste des excipients
Noyau du comprimé Lactose monohydraté Cellulose microcristalline Carboxyméthylamidon sodique (type A) Silice colloïdale anhydre Stéarate de magnésium Pelliculage Talc Macrogol Alcool polyvinylique Dioxyde de titane

ANSM-TAKEDA RCP Brigatinib 180 mg 21
6.2. Incompatibilités
Sans objet.
6.3. Durée de conservation
2 ans
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Flacons en polyéthylène haute densité (PEHD) avec bouchon à visser en polypropylène en deux parties dotés d’un opercule scellé par induction, contenant 30 comprimés pelliculés avec une capsule en plastique PEHD contenant un dessiccant de type tamis moléculaire.
6.6. Précautions particulières d’élimination et de manipulation
Les patients doivent veiller à conserver la capsule de dessiccant dans le flacon et à ne pas l’avaler. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION TEMPORAIRE D’UTILISA TION
TAKEDA FRANCE 11-13 COURS VALMY IMMEUBLE PACIFIC 92800 PUTEAUX
8. NUMERO(S) D’AUTORISATION TEMPORAIRE D’UTILISATIO N
• 34009 589 011 8 8 : 30 comprimés en flacon PEHD
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
23/05/2018
10. DATE DE MISE A JOUR DU TEXTE
{JJ mois AAAA}
11. DOSIMETRIE
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARM ACEUTIQUES
Sans objet.
CONDITIONS DE PRESCRIPTION ET DE DELIVRANCE Médicament soumis à prescription hospitalière, réservé aux spécialistes en oncologie, ou hématologie ou aux médecins compétents en cancérologie.