chapitre 3 introduction aux mÉthodes de ... -...
TRANSCRIPT

Chapitre 3
INTRODUCTIONAUX MÉTHODES DE
MINIMISATION D’ÉNERGIE
3.1 REPRÉSENTATION EMPIRIQUE DE L'ÉNERGIE MOLÉCULAIRE
La stabilité de la structure tridimensionnelle d'une molécule est déterminée par lesinteractions intramoléculaires et les interactions avec le milieu extérieur (solvant). Larecherche des conformations stables d'une molécule consiste à déterminer les minima del'énergie globale d'interaction. Cette énergie peut être calculée par des méthodesquantiques ou semi-empiriques généralement longues et onéreuses. Pour faciliterles calculs, on considère généralement que le terme variable de cette énergie dépend de laconstruction de la molécule et de l'arrangement de ses atomes: c'est le principe desméthodes empiriques (mécanique moléculaire, dynamique moléculaire). Dans laplupart de ces méthodes, il n'est pas tenu compte des interactions avec le solvant, maisuniquement des interactions entre les atomes constitutifs de la molécule. La recherched'une conformation consiste alors à faire une minimisation de l'énergie intramoléculaire.Cette énergie potentielle est fractionnée en un certain nombre de termes additifsindépendants. Chacun de ces termes est représenté par une fonction analytique simplejustifiée par des calculs quantiques et incluant des paramètres empiriques.

48
A) Méthodes quantiques et semi-empiriquesEn mécanique quantique on se préoccupe de la distribution des électrons
(orbitales) dans l'espace. Les meilleurs programmes comportent des processusd'optimisation de la géométrie. L'objectif de la mécanique quantique est principalementde déterminer l'énergie et la distribution électronique. Ainsi les énergies moléculairessont calculées en utilisant l'équation de Schrödinger avec le formalisme des orbitalesmoléculaire (MO). L'équation de Schrödinger d'un système moléculaire peut êtrerésolue sans approximation (ab initio), ou en introduisant des approximations (semi-empirique). Plusieurs logiciels sont disponibles via QCPE (Quantum Chemical ProgramExchange) de University of Indiana.
Programmes ab initio: GAUSSIAN (Pople, 1986) (avec Hartree-Fock; équations de Roothaan HONDO sans paramétrisation pour tous les électrons)Utile aussi pour un traitement local d’une molécule.
GTO: “Gaussian Basic Set” (plus facile à manipuler par ordinateur que STO).STO-3G: “Slater Type Orbitals” (combinaison linéaire mais faut 3 gaussiennes pour unebonne approximation).
Programmes semi-empiriques: AM1 (Dewar, 1985)(on considère que les électrons MNDO (“Modified Neglect Dif. Overlap”)de valence) CNDO (1er, Pople)- approx. valentielle INDO (2ème, “intermediate”)- négligence partielle MINDO (Deward, “modified”)- remplacement PCILO- bases d’orbitales de Slater ZINDO (Zerner + métaux transitions)
Il y a aussi des "packages" comme AMPAC et MOPAC. Ce dernier inclut AM1,MNDO et MINDO. Par les méthodes MO, les énergies peuvent être extraites via leformalisme SCF ("Self Consistent Field") ou par une méthode de perturbation commePCILO. Notons qu'en mécanique moléculaire, les calculs sont plus rapide (~10X) carl'énergie obtenue provient d'une sommation simple de chacune des coordonnéesatomiques de la molécule sans passer par une diagonalisation matricielle.
Consultez le Chapitre 9, section 9.3 pour plus de détail sur les calculs quantiqueset semi-empiriques.

49
Le champ auto-cohérent (“SCF”)
En chimie quantique il faut résoudre l’équation de Schrödinger. En mécaniquemoléculaire cependant, il faut faire des approximations. Une d’elles est l’approximationde Born-Oppenheimer (voir pp. 50, 80 et 84).
Pour les systèmes polyélectroniques, l’équation de Scrödinger n’est pas résoluecar on traite ici d’un problème à N-corps. Il faut donc aussi faire des approximationsorbitalaires en tenant compte de chaque électron de façon indépendante. On parle alorsd’un développement linéaire de combinaisons d’orbitales atomiques (“LCAO”) pourchaque électrons.
Un potentiel Hartree-Fock conciste à manipuler en jeu d’orbitales sansparamétrisation. Le calcul s’arrête lorsque la convergence est jugée satisfaisante. On ditque le processus en cours est auto-cohérent (“Self-Consistent Field”).
Hartree-Fock
aproxim. LCAO
non-SCF SCFnéglige les néglige pasrépulsions les répulsionscoulombiennes coulombiennes
OM de Hückel étendue ab initio
aproxim. σ, π INDOtoutes les valences
OM de Hückel CNDOintégrale de recouvrementpas négligé pour tous lesélectrons de valence aproxim. σ, π
PPPsemi-empirique

50
Par l’approximation de Bohn-Oppenheimer on suppose que les noyauxbougent beaucoup plus lentement que les électrons et on les considère alors comme desmasses fixes. Ce qui sépare l’équation de Shrödinger en deux termes (voir pp. 80 et84).Faut alors une solution approximative selon:
1) la méthode de Hückel qui traite que des électrons π ou 2) le “Self-ConsistentField” (voir p. 64). Dans la méthode de Hückel on parle alors de LCAO (“LinearComb. Atom. Orb.”), VB et MO.
D'autre part, la mécanique moléculaire, appelée parfois "calcul de champ de forceempirique", permet le calcul de la structure et de l'énergie d'entités moléculaires, enprenant le contre-pied de toutes les méthodes quantiques.
B) Méthodes empiriquesEn mécanique moléculaire on considère donc une structure moléculaire comme
étant composée de billes et de ressorts (forces harmoniques) associés à une série defonctions de potentiel. La somme de ces fonctions est exprimée sous la forme d'unchamp de force ("force field") moléculaire.
Etotal = El iaison + Eangle + Etorsion + Evan der Waals + Eélectro + Epont-H
Chacun de ces termes possédant une position d'équilibre préférentielle (longueurde liaison, angle de liaison...). La recherche de l'énergie minimale par optimisation de lagéométrie joue un rôle primordial. L'énergie de la molécule est exprimée sous la formed'une somme de contributions associées aux écarts de la structure par rapport à desparamètres structuraux de référence. A cet égard, la mécanique moléculaireressemble aux modèles de type "tiges et boules", mais elle est beaucoup plusquantitative.
L'idée directrice de cette méthode est d’établir, par le choix des fonctionsénergétiques et des paramètres qu'elles contiennent, un modèle mathématique, le "champde force", qui représente aussi bien que possible les variations de l'énergie potentielleavec la géométrie moléculaire. Cependant, il n'existe pas encore de modèle uniquepermettant de simuler tous les aspects du comportement moléculaire, mais un ensemblede modèles.
C) Types de champ de forceLe "champ de force" est une expression que la mécanique moléculaire a
empruntée à la spectroscopie, en modifiant légèrement sa signification. En effet, il estimportant de remarquer qu'un champ de force décrivant correctement les spectres de

51
vibration de molécules est souvent impropre à décrire les structures moléculaires et viceversa. Ce n’est donc pas une fonction arbitraire de coordonnées cartésiennes, mais bienune somme de contributions d’atomes liés et d’interactions non-liantes.
Trois aspects dans un champ de force: 1) électrostat. 2) van der Waals 3) valence
Champ de force: - Un champ de force consiste dans un ensemble defonctions d'énergie potentielle ("analytical energy func-tion") associée à une série de paramètres numériques,obtenus expérimentalement (IR, micro-onde...) ou évaluésthéoriquement. C’est donc une énergie potentielle multi-dimentionnelle; c’est-à-dire une somme de contributionsliantes et non-liantes à N-corps.- Pour une seule molécule, on peut compter plusieursmilliers de ces contributions individuelles.- Le développement d'un nouveau champ de force ou deparamètres (paramétrisation), demeure un défi majeurpour les fonctions chimiques complexes, i.e., cétoneconjugué, phosphonate, sulfate...- De plus, la compatibilité d'un système de champ de forceà un autre n'est pas souvent possible.- La performance d’un champ de force peut être reliée àcertains paramètres.
TYPES DE "CHAMP DE FORCE EMPIRIQUE" CLASSE I CLASSE IIMM2 (Allinger) MM3 (Allinger)AMBER (Kollman) CFF91 (Hagler)CVFF de Discover® (Hagler) MMFF94 (Merck)
Dans un champ de classe II, il y a des nouveaux termes croisés pour tenir compted’interactions mixtes.

52
Chimie quantique: ➠• on traite que des électrons mais pas des noyauxdonc on regarde la structure électronique et les noyaux restent fixes• les électrons sont localisés sur des liaisons
Méthodes semi-empiriques: ➠• on regarde seulement les électrons de valence
• avec un Hamiltonien plus simpleLa chimie quantique donc couvre:
• réarrangements
• intermédiaires de réaction
• états de transition
• réactions de transfert
☞ Information excellente sur les prop. électroniques et géométriques des molécules.Mais pour systèmes biol. plus complexes, la MM représente une alternative.Cependant, il faut alors des paramètres structuraux.
Mécanique moléculaire: ➠• on positionne les atomes avec une distribution implicite des électrons.• mais on ne peut pas modéliser, par exemple, un transfert de protonsni une tautomérisation car les longueurs de liaison changent.
En 1ère approximation: MM ➟ info. conformation
MQ ➟ info. structure électronique
En MM il faut: types de liaison/propriétés ➟ transférable
fragments moléculaires ➟ transférable
On compare ensuite les ∆E relatives avec la structure électronique venant de MQ.
En MM on suppose des forces pour décrire une surface d’énergie potentielle (“PES”).

53
SCF: "Self-consistent Field" (voir aussi p. 64 et Chapitre 9 sur HyperChem)
• méthode semi-empirique (Pariser, Parr, Pople, 1953)
• en essence il faut converger à un résultat (solution de l’équation de Schrödinger) même s’il n’est pas exact, en autant qu’il reste auto-cohérent.
• aucune méthode n’est meilleure qu’une autre: seule compte la validité de l’approximation dans le cadre du système moléculaire étudié.• faut une solution identique d’une itération à l’autre.
• solution meilleure que via une matrice simple diagonalisée par la méthode de Hückel, même si on sous-estime les répulsions des électrons.• paramétrisé pour reproduire les fréquences de vibration et les résultats géométriques, plus thermodynamiques.• distribution électronique optimisée par rapport au potentiel énergétique du noyau à cause de l'approximation Born-Oppenheimer (voir pp. 50 et 80)
DFT: "Density Fonctional Theory"• développé aussi chez Salahub (U de M)
• méthode quantique mais moins exigeante en temps de calcul.
• base utile pour les métaux de transitions
AM1: "Austin Method"• Hamiltonien de Dewar
• calcul "close shell" ➟ restricted RHF (Hartree-Fock)- mol. organiques
• calcul "open shell" ➟ unrestricted UHF (ab initio)- radicaux...
(Preuve faite avec des molécules organiques)
MOPAC: même Hamiltonien que AM1

54
moins complet
AMPAC: • traite aussi les électrons ➟ pas via champ de force
• s'occupe des électrons avec un programme d'orbitales moléculaires
• peut regarder les cyclisations radicalaires si pas trop d'effets stériques.
Temps CPU:Quantique: N4 N= nombre d'orbitalesSemi-empirique: N3
Empirique (MM): n2 n= nombre d'atomes
Champs de force: Westheimer 1946...(les développeurs) von R. Schleyer, Lifson 1968...
Allinger 1971...Kollman 1975...
Parmi les champs de force les plus utilisés et les mieux éprouvés citons:
MM2 (Allinger, 1976): hydrocarbones, fonctions simples avec hétéroatomes (MODEL)MM3 (Allinger, 1990) MM2 avec polarisation du lien C-H (nouvelle version)AMBER (Kollman, 1981): peptides et acides nucléiques (MACROMODEL)CHARMM (Karplus, 1983): molécules biologiques, macromolécules, interactions,
petites molécules et macromolécules (QUANTA)ECEPP (Scheraga, 1983): peptides et protéines.CVFF (Hagler, 1985) dynamique moléculaire (INSIGHT/DISCOVER)MAXIMIN2 (Clark, 1988): produits organiques et biologiques (SYBYL)DREIDING-II (Goddard III, 1990): produits org., peptides et protéines (BIOGRAF)
Donc, pour un CF en MM, on ignore le mouvement des électrons, on calcule l’énergiedu système en fonction des noyaux fixes (approx. BO).

55
Le champ de force MM2Ce champ de force pour la mécanique moléculaire a été développé par Allinger
dès 1976 et c'est le plus utilisé par la communauté des chimistes organiciens. Pourtraiter des molécules aromatiques avec des électrons π, la variante MMP2 a étédéveloppée. Le programme présente la particularité de traiter séparément les systèmes àélectrons π et les systèmes à électrons σ. Deux calculs séparés sont effectués: l'un sur le
système π par la méthode SCF ("Self-Consistent Field") faisant appel à la théorie
quantique, l'autre sur le système σ par la mécanique moléculaire. Avec le calcul SCF, onconsidère que l'aspect stérique des systèmes aromatiques en leur attribuant desparamètres qui leur imposent de garder leur planérité due à la délocalisation des électronsπ. L'énergie minimisée est donc la somme de ces deux énergies.
MM2: “aspect dipôles” AMBER: “aspect charges”
Le champ de force AMBER("Assisted Method Building and Energy Refinement")AMBER est un logiciel de mécanique moléculaire mis
au point par Kollman (UCSF). Il utilise les coordonnéescartésiennes des atomes de la molécule et permet d'effectuer descalculs aussi bien sur des protéines que sur des acides nucléiques.Les paramètres (résultats de calculs ab initio sur des moléculesmodèles) sont regroupés dans une bibliothèque utilisable par leprogramme. Ce logiciel fonctionne par modules.
EXEMPLE DU CHAMP DE FORCE DE AMBER(la première page seulement, consultez aussi l’Annexe B)
N.B. AMBER possède deux modes:1) United Atoms: Considère pas les hydrogènes non-polaires (CH, CH2, CH3)
mais seulement les ponts hydrogèneavec les hétéroatomes.
2) All Atoms: Paramétrisation détaillée pour les acides aminés et les acides nucléiques incluant les charges. Tous les atomes sont alors pris en compte.

56

57
MM2: Allinger, Schleyer ➟Champ de force qui néglige les fréquences devibrations mais tient compte des interactionsnon-liantes et donne de bons résultats géom.
MM1 (1971), MM2 (1985) 8 ans pour C-H alcanes...+3ans pour C-O +
C=O pas encore complet (voir MM3)(MM2 modifié dans MacroModel)
- souci du détail et corrélation avec les données expérimentales- reste encore le meilleur champ de force mais ne possède pas les
valeurs électrostatiques des peptides ➟ AMBER (Kollman)- inclut calculs: angle-liaison 1-3 et angle-torsion 1-4- basé sur BDNR (à venir, voir p. 91)- pas de traitement explicite des ponts-hydrogène (faut utiliser MM3)- utilise "bond-dipole model" tandis que d'autre champ de force utilise "point-charge model" et le "atom partial charge model" (comme MacroModel, voir l’Annexe B)- MM2 sous-estime la barrière rotationnelle d'environ 0.6 Kcal; faut donc un facteur de correction
MMX: Gajewski, Gilbert 6 ans de développementSteliou (PC-Model)
- inclut les métaux- correction pour les interactions 1-3 électrostatiques- "open shell" VESCF ("Variable Electronegativity Self-consistent Field")- voir Liotta, Ed., Adv. Mol. Modeling, Vol. 2 1990 (Jay)
MMP2: - effort pour incorporer les variations de struct. électroniques dans MM2
- calcul des électrons π ➟ produits aromatiques et conjugués
- liaison sp2 -sp2 = 1.34 Å ➟ mais différent si aromatique

58
- faut le "bond order" pour pouvoir évaluer la barrière de rotation
Le “bond order”, c’est une mesure de la probabilité de trouver des électrons π.Bond Order: 0 = pas de liaison
1 = liaison simple2 = liaison double3 = liaison triple
C'est aussi une façon de partitionner les électrons et de les corréler avec les longueurs deliaisons.
P = Σ Ci•Cj ➟ (matrice de densité) x (matrice de recouvrement)
(voir p. 64)(population de Mulliken)
Énergie de rotation?
OMeMeO
Dans MacroModel on a un MMP2 différent (voir l’Annexe B).
Pour l’atome Csp2, C. Still a ajouté 7-10 autres termes pour tenir compte des systèmesallyliques.
Csp3 - Csp2 - Csp2 - Csp3
1 2 3 4
si C à C2 + ... autres termes
Si O à C2 + ... d’autres termes différents

59
Donc on calcule pas spécifiquement les électrons π mais l’ensemble reste quand mêmeplus flexible que MM2.
En plus, on traite les facteurs électrostatiques par "atom partial charge model".
Base de MM2 en mécanique moléculaire:
- champ de force (de valence)
- pas d'intermédiaires réactionnels
- atomes non-liants? pot. Lennard-Joneset pot. Buckingham(voir p. 73)
Un autre abus:non-additivité des groupements fonctionnels.Ex 1.: C--C et C--OH ont des paramètres différents de C--C--OH
Ex. 2: Comment minimiser une β-lactame (antibiotique)?
C C C C N
Donc tous des "Atom Type"différents
+ 29
- 30
. 4832Csp
2
Csp
Pour tenir compte des différents types d’hybridations.

60
Atom types:

61
MM3: Allinger JACS 111 , 1106 (1989)JACS 112 , 8293 et 8307 (1990)JACS 113 , 4505 (1991)
1. polarisation du lien C-H2. terme spécial (type V der W) ajouté aux ponts hydrogène; traitement encore faible mais l'énergie d'interaction est mieux estimée que MM2 par environ 20%3. cycle thermodynamique (FEPT, "Free Energy Perturbation Technique")4. MM2 inclus des paires d'électrons libres pour C-O, C=O
- artefact- complique le traitement des spectres de vibration- on ne savait pas comment tenir compte des termes V der W et torsion du lien C-O
Ex.: lien C-O-R éther > C-OH alcool (en MM2)ce qui est contraire à l'expérience car l'effet électropositif de Hfait que C-OH (1.413Å) > C-OR- alors en MM3 on traite le C-O avec le même V der W que C=O (sphère)
5. dans R-NH2 : on ne tient plus compte du doublet, donc plus de problème d'inversion de N
6. pour C=O : on optimise mieux les cétones cycliques7. traite mieux les acétals8. on a modifié le potentiel "stretch-torsion" (élongation-torsion)9. bon résultats avec les molécules tendues et les têtes de pont (bicycles...)
En résumé, les codes semi-empiriques (MNDO, MINDO, MOPAC, AMPAC)peuvent aussi résoudre l’équation de mouvement des atomes mais il faut faire plusieursapproximations des intégrales nécessaires à l’aide de fonctions empiriques. Lamécanique moléculaire consiste en une solution de cette équation en utilisant unajustement empirique de la surface totale de l’énergie potentielle. Ainsi un lien covalentpeut être considéré, dans une première approximation, comme un oscillateurharmonique à la fois dans la théorie quantique et dans la théorie classique. Lamécanique moléculaire ignore la composante temps dans l’évolution du système et sefocalise surtout dans une recherche des géométries particulières des molécules et leursénergies associées. Les coordonnées actuelles d’une molécule, combinées aux donnéesd’un champ de force, constituent une expression de l’énergie ou fonction cible(“target function”) de la molécule.

62
MM2 vs MMX1. Système π
a. MMX contient un type pour le carbone aromatique afin d’approximer la géométrie du groupement phényle, i.e. un lien C-C d’une distance de 1.40Åb. MMX utilise les calculs du format “open shell” VESCF (“Variable Electronegativity Self Consistent Field”). Généralement, ceci permet de calculerles chaleurs de formation à ±1 Kcal/mol des valeurs expérimentales.
2. Groupements polaires et paires d’électronsa. Dans MM2, seulement les oxygènes des alcools et des éthers ont des paires d’électrons. Dans MMX, tous les atomes possèdent des électrons non-pairés. Les atomes qui participent aux systèmes π on une paire de moins que nécessaire.Par exemple, l’azote des amides et l’oxygène carbonylé des esters ont respectivement zéro et une paire libre d’électrons. D’autre part, l’azote des imines et l’oxygène des carbonyles ont respectivement une et deux paires d’électrons libres.
3. Complexes de métaux de transitiona. Les longueurs de liaison sont fonction du rayon covalent du métal. La constante de force du lien (“stretching”) est la même pour tous les ligands, soit 2mdyne/Å), quel que soit le métal.b. Les potentiels d’angle (“bending”) sont enlevés lorsque l’atome central est unmétal.c. Les interactions de type 1-3 sont ignorées.d. Les potentiels de torsion impliquant le métal comme un des deux atomes centraux sont ignorés.e. Une coordination π est obtenue en invoquant un terme attractif en 1/r2 entre lemétal et le ligand.f. Avec les métaux saturés, le rayon covalent est un bon indice de distances métal-ligand.g. MMX traite les métaux comme saturés, insaturés ou plus grand que dix-huit électrons. Dans le cas de métaux insaturés coordonnés, les distances de liaison sont automatiquement corrigées en soustrayant 0.2Å. Dans le cas de métaux ayant plus de dix-huit électrons, 0.2Å est alors ajouté à la distance métal-ligand.

63
4. Ponts hydrogènea. Dans MM2, les ponts hydrogène résultent d’interactions dipôle-dipôle.b. Dans MMX, les ponts hydrogène résultent à la fois d’interactions charge-charge et d’interaction attractives (1/r2) entre paires d’électrons libres (rayon de van der Waals fixé à 0.2Å) et des atomes d’hydrogène sélectionnés.
5. États de transitiona. MMX reconnaît l’état de transition d’atomes pour onze types de liaison dans des états de transition:
C*-C* ➟ liens formants dans les réactions de Diels-Alder ou des réarrangements de Cope.
C#-C# ➟ liens formants dans les réactions de Diels-Alder ou liens brisants dans les réarrangements de Cope.
C-H* ➟ liens C-H allyliques des réactions de type ène.
C*-H* ➟ liens C-H formants dans les réactions ène ou d’hydroborations
C#-B# ➟ liens C-B formants dans l’hydroboration
C*-C$ ➟ liens formants dans les additions de nitrile oxyde spC sur sp2C
C#-C# ➟ liens formants dans les réarrangements de Claisen
C*-C% ➟ carbone nucléophile dans les réactions SN2
C%-I% ➟ groupe partant dans les réactions SN2
C%-O# ➟ oxygène nucléophile dans les réactions SN2
C#-N# ➟ liens brisants dans les réarrangements de type Aza-Cope.
b. Les atomes de carbone qui ne subissent pas de changement d’hybridation sont normalement traités comme des radicaux [i.e. type 29 avec une distance de liaison de 1.40Å pour C(29)-C(29)].c. Les “bond order” sont générés par une routine de départ (“input”).
N.B.: Il est parfois préférable d’utiliser AMPAC (calculs semi-empiriques) qui,pour ces états, tient compte des différents types d’électrons.

64
DESCRIPTION GÉNÉRALE DE LA MÉTHODE MMP2
1. Pour une géométrie initiale, calcul SCF (“Self-consistent Field”) est effectué sur lesystème π. Si le système est non-planaire, il est aplati dans une forme planaire.
2. Pour les coefficients des fonctions d’onde obtenues, les “bond orders” sont calculésen utilisant la relation suivante: Pij = Σ nrCri•Crjou la somme tient compte de tous les orbitales moléculaires (OM) r; nr étant le nombred’électrons dans les OM r, Cri étant le coefficient de l’atome i dans OM r et Crj étantle coefficient de l’atome j dans OM r.
3. On suppose une relation linéaire entre le “bond order” du lien et chacune des troisquantités suivantes:
a) la constante de force de liaison (“stretching”)b) la longueur “naturelle” de la liaisonc) la barrière rotationnelle (“torsional”).
En utilisant cette relation, les constantes de force requises sont calculées et utilisées dansle champ de force de la mécanique moléculaire.
4. L’utilisation d’une relation linéaire entre le “bond order” et la longueur naturelle de laliaison et entre le “bond order” et la constante de force de la liaison, donne de bonsrésultats pour les systèmes planaires. Le problème de la fonction de potentielle detorsion est traitée en utilisant une quantité appelée l’énergie de conjugaison, Ec,définie comme étant: Ec = kPij•Bijavec k comme paramètre ajustable et permettant à l’énergie de conjugaison d’atteindrezéro lorsque les atomes se séparent. Pi j est le “bond order” entre les atomes i et j, etB i j est l’intégral de résonance entre les atomes i et j. Quand la liaison est tordue, lesdeux termes Pi j et B i j tendent aussi vers zéro. Ce qui revient à dire que lerecouvrement est maximal si les orbitales sont coplanaires et minimal si elles sontperpendiculaires.
5. La méthode MMP2 utilise comme calcul le protocole VESCF (“VariableElectonegativity Self-consistent Field”). Au lieu d’utiliser la valeur atomique dupotentiel d’ionisation de l’orbitale p du carbone, elle utilise une valeur qui estdépendante des subtituents attachés au carbone, ainsi que de la densité électronique π aucarbone.

65
LE CHAMP DE FORCE POUR DES MOLÉCULES CONTENANTDES ÉLECTRONS π DÉLOCALISÉS
Le champ de force MMX tient compte des électrons π car en mécanique moléculaire, lesélectrons de liaison ne sont pas isolés mais subissent de part et d’autre des forcesélectrostatiques.
(caractère double plus prononcé ici)1.67Å
1.33Å
1.67Å 1.33Å
1.33Å
1.4Å 1.4Å (à cause de la délocalisation)
1.34Å ("bond order" 2)
1.58Å ("bond order " 1)Modèle de Kékulé

66
CALCUL DES ATOMES π DANS MMP2Un * désigne les atomes de type π .
X
O
R
O
N
XR
RX
R X
O O
O*
***
** *
*
*
*
* **
**
*
**
**
*
*
*
*
**
*
**
*
*
*
*
*
*
*
*
*
*
**
*
***
ESTER D'ACIDEAROMATIQUE
ESTER DEPHÉNOL
pas de π sur celui-ci
mais pas ici
un π ici
mais pas ici
un π ici
Les atomes et groupes spécifiques:Tous les N avec une paire d’électrons délocalisée ne doivent PAS avoir de paires libres et
doivent être des atomes de type 9 (sp2).Une imine N , qui donne un électron à un lien π, est un atome de type 37 et n’a PAS de paire
libre.L’azote d’un ammonium est de type 41, mais peut aussi être utilisé comme un atome π
donnant un électron au système π comme un ion immonium ou un N de nitro.Les groupes nitro sont de type 31 N+, avec un type 7 O, et un type 32 O-. Pour un calcul π
on inclue seulement le N+ et le type 7 O dans le calcul.Un oxygène avec une paire d’électron délocalisée doit avoir seulement une paire libre et doit
être un atome de type 6.Les groupes carboxylates: le lien carbonyle C-O- ne doit PAS être inclus dans le calcul π;
il faut inclure seulement le carbonyle C et le =O.

67
Objectif d'une bonne science: concevoir (design) des règles simples et des équations pour surmonter le grand nombre d'orbitales.
Donc dans un champ de force il faut utiliser des équations simples et qui sont fiables.
KB : lien simple, 5 mdyne/Å (1 mdyne/Å = 143.88 Kcal/mol/Å2)lien double, 10lien triple, 15
Kθ : dans MM2 il faut un "scaling factor" de 0.6 pour le "bending force" car sous-estime le terme torsionnel.
KTOR : barrière 3-fold (C2H6), utilise V3barrière 2-fold (C2H4), utilise V2
Harmonique: E = K/2 (r - ro)2
Morse: E = Do [e-α(r -ro) - 1]2
Cubique: E = K/2 (r - ro)2 . [1 + d(r -ro)]Do = énergie du lien; α = (K/2Do)1/2; d = -2 (relation cubique)
Paramètres: une trentaine de "atom type" différents- génériques: R1-Csp3-R2 avec même “set” de KB, Kθ, θ
R3-Csp3-Osp3-R4 avec même “set” Kϕ, n...- spéciaux: Csp3-Csp3-H avec un angle qui "override" le cas
générique R1-Csp3-R2
S'il faut apporter une pénalité on a alors une perte de précision et une géométrie approximative, ce qui est regrettable (faut faire des compromis).
pour les Csp3-Csp3 : ro = 1.53ÅKB/2 = 300 Kcal/mol/Å2
pour les Csp2=Csp2 ro = 1.33ÅKB/2 = 700 Kcal/mol/Å2
avec butadiène: Csp2=Csp2-Csp2=Csp2 lien central: ro = 1.51Å et KB/2 = 340

68
Composante électrostatique: approximation ε = rij (dépendant de la distance)- plus complexe et moins transférable que V der WEélectro = terme monopôle + terme dipolaire + terme quadrupolaire
Loi de Coulomb: applicable pour deux charges dans le vide en milieu homogène.
Mais pour une protéine: mol. pas homogène et milieu pas homogèneε? intérieur?; approximation: charge q propor. 1/r
molécules ioniques (peptides):• simulation dans le VIDE - interactions intra. mol. fortes + ponts salins• simulation dans EAU - dimin. des interactions électrostat. - spécialement avec groupes ioniques - structure plus relaxée - permet séparation et solvatation des groupes chargés (donc plus éloignés) voir: Pettitt, J. Amer. Chem. Soc. 113 , 67 (1991)
Ponts hydrogène:- analyse de Lennard-Jones (voir p. 72)
E = [C/d8 - P/d6] .fi . fj- distance optimale dépend de l'orientation relative des atomes- on utilise une fonction 10-12
RO
H
O
R
H
- linéarité importante- pas possible avec petits cycles
Problème de 3 corps (“3-body effect”):E = E12 + E13 + E23mais aussi E123 + Epolarisation

69
D) Formule empirique du champ de force*Une étape importante dans l'étude et la compréhension des géométries
moléculaires a été de décrire et de pouvoir simuler leurs propriétés structurales eténergétiques. L'évolution des puissants outils de calcul a permis un développementspectaculaire dans le domaine des calculs énergétiques. Le problème a donc consisté àchoisir une fonction potentielle (ou champ de force) analytiquement simple qui représenteles coordonnées des N atomes de la molécule (3N variables). Cette fonction doit êtresimple, pour pouvoir être calculée rapidement, et suffisamment précise pour simuler defaçon acceptable les propriétés structurales et thermodynamiques des macromolécules.De tels champs de force sont apparus au début des années 1970 et continuent à évolueraujourd'hui.
Un champ de force est constitué de plusieurs fonctions d'énergie potentielle quidécrivent les interactions intramoléculaires entre atomes liés et non-liés. Les interactionsentre atomes liés correspondent à des énergies de déformation des liaisons, des angles devalence, des angles dièdres impropres et aux énergies de torsion. Les interactions entreatomes non-liés sont représentées par les énergies de Van der Waals et électrostatiquesainsi que par l'énergie des liaisons hydrogène.
Etotal = Eliés + Enon-liés
a) Énergies d'interaction entre atomes liés
1) Énergie de déformation du squeletteLes potentiels utilisés sont du même type que ceux utilisés en analyse
vibrationnelle. La déformation du squelette est décrite à l'aide des termes suivants:- Élongation des liaisons et distorsion des angles de valence
Ces déformations sont en général faibles, de l'ordre de 0.05 Å pour les longueursde liaison et de quelques degrés pour les angles de valence. Les fonctions d'énergiepotentielle utilisées pour décrire ces déformations sont des fonctions harmoniques:
E = 12
KB
b − b o( )2
liaison∑
et
12 Kθ θ − θ o( )
2
anglevalence
∑
_________________________________* Cette section est extraire en grande partie de la thèse de Ph.D. de B. Stawarz (Orléans) 1990.

70
Une fonction harmonique est sinusoïdale (ou cosinus) en fonction du temps. Pour lesanglo-saxons, c'est une fonction au carré ou cubique. Cette équation est très utilisée danstous les champs de force et est valide si le lien n'est pas trop long. Au besoin on peutajouter à cette expression un terme en (r–ro)3.
L'indice o représente les valeurs standards des longueurs de liaison et des anglesde valence. Les constantes KB et Kθ sont les constantes de force dérivées de l'analysevibrationnelle de molécules modèles (spectroscopie IR, Raman). Elles sont respectivement de l'ordre de 400 kJ.mol -1.Å-2 et de 40 kJ.mol -1.deg-2
.- Déformation des angles dièdres impropres
Le potentiel d'interaction lié aux angles dièdres impropres exprime la déformationd'un groupe d'atomes par rapport à une conformation donnée. Un angle impropre estdéfini par trois atomes X, Y, Z liés à un même quatrième atome A. Cet atome est soit unatome de type sp2, soit un atome de type sp3. Dans le cas d'un atome A de type sp2, lesquatre atomes sont maintenus dans une configuration plane (ζο = 00). Pour un atome A
de type sp3, les quatre atomes sont maintenus dans une configuration tétraédrique (ζο =35.26ο). Cette déformation décrit donc les sorties de plans de certains atomes, nondescriptibles par des angles dièdres. Le potentiel est exprimé sous une formeharmonique:
E = 12
, Kζ
ζ − ζ o( )2
∑

71
2) Énergie de torsionL'énergie de torsion (angle dièdre) associée à la rotation autour d'une liaison BC
définie par quatre atomes consécutifs ABCD est exprimée sous la forme d'une fonctionpériodique développée en série de Fourier tronquée:
E = 12
Kϕ 1 + cos(nϕ − δ )[ ]angle de
torsion
∑
La constante de torsion Kϕ (barrière de rotation) est de l'ordre de 40 à 70
kJ.mol-1. La périodicité de la rotation est égale à n = 2 (sp2) ou à n = 3 (sp3) et δ estl'angle de phase.
Etorsion
=V
12
(1 + cos θ) +V
22
(1 − cos 2 θ) +V
33
(1 + cos 3 θ)
(polynôme de Lagrange)À compléter:
0° 180° 360° 0° 180° 360°V1, V3 ≠ 0, V2 = 0 V1, V3 = 0, V2≠ 0
symétrie d'ordre 2 du potentielde torsion pour Csp2
La combinaison des fonctions de liaison, d'angle et de torsion d'angle s'appellele champ de force de valence.

72
V = des paramètres empiriques pour interpréter les valeurs expérimentales.Ceci dit, un champ de force de valences demeure simple et rapide en temps de calcul.

73
b) Énergies d'interaction entre atomes non-liés
1) Interactions de Van der WaalsEntre deux atomes neutres i et j , les interactions entre dipôles fluctuants sont à
l'origine d'une énergie attractive qui varie en -1/rij6. Cette énergie est encore appeléeénergie de dispersion de London dans la mesure où son expression fait intervenir l'indicede réfraction du milieu. Sous une forme abrégée on écrit:
Edisp = -Cij / rij6Les coefficients Cij sont établis pour les différentes paires d'atomes présents dans lamolécule. Leur valeur peut varier suivant les auteurs et poser donc des problèmes deparamétrisation qu'il faut garder en mémoire dans le calcul d'énergie.
A ce terme attractif en -1/rij6 on associe un terme répulsif en Dij/rij12 traduisantle recouvrement des nuages électroniques à courtes distances. L'ensemble représentel'énergie de Van der Waals (fonction de Lennard-Jones):
E = 12
D
r12− C
r6
paires
∑
terme terme attractif,répulsif de dispersionLennard-Jones de London(Van der Waals)
La fonction de Lennard-Jones (L-J), ditecouramment potentiel "6-12", est laplus souvent utilisée dans les programmesde modélisation moléculaire. On peutcependant rencontrer quelques variantescomme la fonction de Buckingham oude Morse. Pour les molécules plus largeson utilise L-J , si non Buckingham est préférable.

74
Rappelons que la fonction d’énergiepotentielle d'une distance interatom-ique r, utilisée pour décrire l’élon-gation des liaisons est décrite par unefonction de type Morse. L'énergieminimale du lien est à ro (équilibre).Dans ce type de représentation, unefonction de potentiel simple obéit à laloi de Hooke par l'expression:
V = k2 (r − r o)
2
Les champs de forces CFF91 et AMBER utilisent une fonction harmonique similaire.
V = potentiel d'énergie (fonction harmonique)k = constante (kJ/mol/Å2)
La force électrostatique et de Van der Waals agissant sur chaque atome est doncune somme sur toutes les paires d'atomes non-liés. L'évaluation de tous les termesd'énergie va donc nécessiter un nombre d'opérations proportionnel à N2, ce qui est trèscoûteux en calcul pour les grands systèmes. Pour résoudre ce problème, on limite laportée du potentiel à une certaine distance (8 à 10 Å). La somme ne se fait plus sur toutesles paires ,mais sur les voisins et l'on passe alors de N2 opérations à N< n voisins>opérations (voisins compris dans une sphère de 8 à 10 Å). D'où l'introduction d'unefonction de coupure ou "switch".
Il faut donc imposer un Rcut ~ 9 Å. Lepotentiel de Van der Waals montre unminimum large. Cette fonction doit aussitenir compte des répulsions stériques entreles atomes liés. Ainsi il faut parfois inclureun terme "croisé". Ce terme tient compte àla fois de la longueur de liaison et de l'anglede liaison. Si θ augmente, la distance rdiminue.

75
Rappelons que:"stretching" A12
K(r - ro)2 B C
(terme d'allongement) "bending" 12
K'(θ - θo)2
(terme de variation d'angle de liaison)Il y a aussi le champ de force de Urey-Bradley pour les interactions 1-3 (deux
atomes liés à un atome commun). Le nombre d'interactions non-liantes augmente avec lecarré du nombre d'atomes: N(N-1)/2. Ainsi, pour 3000 atomes il y a 4.5 millionsd'interactions possible!
2) Interactions électrostatiquesDans l'approximation du monopôle, la molécule est décrite par des charges
ponctuelles qij centrées sur les atomes. Ces charges sont généralement déterminées pardes calculs ab initio ou semi-empiriques (“package” AMPAC/MOPAC). L'énergieélectrostatique est représentée par la relation suivante:
Loi de Coulomb: E propor. à 1/r (dist-dépendent)
E = 14πε
.q
iq
jr
ij
∑
Avec εr = permittivité relative et εο = permittivité du vide ( =1).Il faut déterminer une distribution de charges partielles compatible avec les
moments dipolaires des liaisons, le moment dipolaire total et la charge globale de la
molécule. L'évaluation des charges π peut se faire par la méthode de Hückel et le calcul
des charges σ est effectué par la méthode Del Ré (charges partielles). Le choix de la
valeur de la permittivité ε est délicat dans la mesure où il s'agit d'une grandeurmicroscopique qui peut être notablement différente de la grandeur macroscopique. Pourl'intérieur des molécules qui ne sont pas en contact avec l'eau, il est courant de prendre
des valeurs de εr de 2 à 4. Pour les paires d'atomes situées vers l'intérieur, on a
proposé des fonctions de variation de εr avec la distance r. Dans un environnement
aqueux (effet d’écran des charges par le solvant), εr peut atteindre la valeur macro-scopique de 80 pour r voisin de 7 à 10 Å.

76
3) Énergie de liaison hydrogèneLa liaison hydrogène est une interaction d'importance intermédiaire (8 à 20
kJ/mol) entre un hydrogène déficient en électron et un atome de forte densité électroniqueportant un doublet d'électrons libres. Le modèle électrostatique, vrai lorsque la distanceA-B est grande, n'est pas suffisant pour décrire ces interactions particulières. A pluscourte distance, les phénomènes de répulsion et de délocalisation électroniqueinterviennent. Plusieurs types de fonctions d'énergie potentielle ont été développés pourtenir compte de la directivité de la liaison hydrogène. Actuellement, les fonctions les plusutilisées permettant d'exprimer ces interactions dans des systèmes moléculairesimportants sont souvent simplifiées:
La fonction "10-12" Eh = A/rij12 - B/rij10
La fonction "6-12" Eh = A'/rij12 - B'/rij6
Les coefficients A, B, A', B' sont spécifiques des liaisons hydrogène et l'interactionélectrostatique est calculée par la loi de Coulomb classique.
L'énergie totale d'interaction est la somme de toutes ces énergies potentielles(stériques) pour tous les atomes de la molécule.
Etotal = Eb + Eθ + Eζ + Etor + Evw + Eel +EhÉquation complète d'un champ de force se décompose ainsi:
Etotal = Evalence + Enon-liant + Eutilisateur+ Econst
Evderw + Ecoulomb + Epont-H
Ediagonal + Etermes croisésCF plus évolué comme CVFF
Eliens + Eangles + Etorsion + Einversion
Tout champ de force consiste donc en des fonctions multi-variables qui doiventêtre minimisées en ajoutant des paramètres pour reproduire les évidences expérimentales.

77
Problèmes résiduels:- mieux traiter la constante diélectrique- meilleurs paramètres électrostatiques- polarisibilité (déformation du nuage d'électrons en fonction de la conformation)- rôle du solvant
Représentation graphique des fonctions du champ de force:

78
DISCOVER®, Biosym Technologies, 1992
Représentation sous forme de pictogrammes:

79
c) Autres considérationsPour rendre compte des phénomènes de couplage des mouvements de
déformation de liaison et de déformation des angles de valence, Allinger a introduit unterme croisé ("cross term") dans le programme MM2. Il faut aussi tenir compte dedéformation "hors plan", dont l'énergie est associée à la déformation de la géométrie dela liaison des atomes sp2:
Ehp = 1/2 Khp d2 où d est la distance entre l'atome central et le plan de ses substituants, Khp la constante de déformation "hors plan".
d) Paramétrisation- Paramètres de références: les fonctions d'énergie décrites précédemment
contiennent de nombreux paramètres (Kb , Kθ , moments dipolaires, barrièresrotationnelles, etc...) qui sont ajustés généralement à partir de données expérimentalesdéduites de l'analyse de composés modèles (spectroscopie vibrationnelle,cristallographie, thermodynamique, etc...) ou dans quelques cas déduits de calculs demécanique quantique. Ces paramètres sont optimisés de façon à ce que les propriétéscalculées (géométries, énergies, chaleurs de formation...) se rapprochent le plus possibledes propriétés expérimentales des composés modèles de la banque de données.
- Paramètres de substitution: les banques de données des différents champs deforces sont généralement très limitées. L'absence de paramètres de référence enparticulier pour les systèmes hétérocycliques nécessite un choix de paramètres desubstitution ou paramètres ad hoc.
e) ConclusionLe principal défaut des champs de force vient de la description de charges
ponctuelles fixes. De plus le traitement de l'énergie électrostatique est discutable. Onnéglige les effets dus aux charges distribuées en volume et l'évolution de cettedistribution en fonction de la conformation. La polarisation est ignorée ainsi que leseffets à longue portée. Les autres termes sont une approximation satisfaisante.
On suppose que chaque type de fonction de potentiel est en principe transférabled'une molécule à l'autre. Puisque qu'il n'y a pas de règles absolues concernant lenombre et le type de fonctions d'énergie de potentiel utilisables, on retrouve un grandnombre différent de champ de force en mécanique moléculaire.
1ère loi de la thermodynamique: chimie physique:conservation de l’énergie, donc ∆G = ∆H - T∆Ssi Epot change, Ecin change aussi.

80
Minimum local vs minimum global:
MM est basée sur la MQ classique plus l'approximation de Born-Oppenheimer.Les électrons sont plusieurs milliers de fois plus légers que les noyaux et enconséquence se déplacent beaucoup plus vite. L’approximation de Born-Oppenheimerstipule que le mouvement des électrons peut être découplé de celui des noyaux,conduisant à deux équations séparées. On ignore donc les électrons et les atomes sonttraités comme des points de masse chargés avec une taille définie. Le mouvement desélectrons dépend donc paramétriquement des positions des noyaux.
• itération successive d'une mol. initiale ➟conduit à une géométrie optimisée
• faut des paramètres fiables (ceux des peptides fonctionnent pas avec ac. nuclé.)
• BUT ➟ trouver un minimum GLOBAL- chaque paramètre qui détermine la géométrie initiale est modifié par incrément jusqu'à l'obtention d'un min. LOCAL- faut ensuite une DM pour franchir certaines barrières (Ecinétique sous forme d'augm. de T° )
Méc. Mol. : statistiqueMéc. Vib. : énergétique(on ignore inter. non-liantes)
Minima locaux: - quasi inévitable- car la longueur de liaison et l’angle de liaison sont rapidement optimisés au stade initial de la minimisation;- mais quand eux ont atteint leur valeur d'équilibre, d'autres mouvements torsionnels sont inhibés puisque pour réduire d'avantage l'énergie totale du système il faut pouvoir bouger plusieurs atomes;- on est donc piégé!
Limitation du champ de force:
MM ☞ basée sur des méthodes empiriques, applicablesà des classes de composés avec des valeurs exp.adéquates pour permettre la paramétrisation.
- pas de valeurs de ∆H disponibles pour des systèmes d'on les angles varient de plus de 20° (déformation).- cycles à 3, 4...: résultats difficile à reproduire

81
Problème du temps de calcul et d'espace mémoire:
1. Dans un dièdre on suppose 3 minima- donc pour 3 liaisons: 33 = 27 conformations possibles
2. - pour 7 dièdres avec une résolution de 60° il faut 360°/60° = 66N = nombre de conform. par paire en rech. conformationnelle (“search”)67 = 279,936 géom. (conform.) de départ possible!
3. - ainsi pour l'acide arachidonique avec 15 liaisons simples/rotation315 = 14,348,907 minima possibles (3 minima par dièdre)
4. - pour l'enképhaline (5 ac. aminés)324 = 1011 possibilités (se réduit à moins de 106 si on regarde à ± 3 Kcal du min. local)- en effectuant une rech. conform. (multiconformère) via Monte Carlo
avec une variation de 30° ➟ - génère environ 600 géom. de base et conduit à une dizaine de struc. uniques
TYR-GLY-GLY-PHE-MET avec ± 20 Kcal/mol (min. locaux) en environ 5h CPU- Scheraga, J. Mol. Biol. 196 , 697 (1987)
5. - encore l'enképhaline (5 ac. aminés, 75 atomes)- storage nécessaire = 10N N = variables, fonctions objectives
N = n2/2 n = no. atomes (quadratique)- donc (75)2/2 = 2,812 variables 10N = 28,120 nombres réels qu'il faut stocker
ou encore: besoin storage ou espace mémoire nécessaire = 5n2
5n2 = 28,000
6. pour un peptide avec 15 rotations (liens)
- chacune de 360° à chaque 10° : 36 calculs/lien ➟3615 conform. possibles!
- si on calcule 1 conform./sec: 3615 sec = 700 trillions d'annéesnécessaire pour faire le calcul!

82
Méthode combinatoire ➟ permet d'apprécier la complexité conformationnelledes problèmes.
7. molécule avec 6 degrés libres de torsion et avec une variation de 5°360°/5° = 72
[360°/N]R ➟ [72]6 = 139,314,069,504conform. possibles à examiner en incrémentant de 5°
8. nombre de paire d’interactions à calculer par confor. = N(N - 1)/2 = N2/2- pour chlorpromazine (anti-dépressif): 42 atomes ➟ 42(42-1)/2 = 861
interactions énergétiquesà considérer pour chaqueconform. théorique.
- si on calcule une coordonnée/microseconde/5° d'incrément: 726•N•N(N-1)/2 = 1,399,409.7 heures (pour N = 42)
= 58,307.7 jours = 159.7 années!
Faut donc avoir à notre disposition, pour chaque champ de force, une série de bons algorithmes de minimisation: SD, BDCG, FMNR, PRCG,...
Autres problèmes du minimum global:
Avec le butane au hasard: minim. en incrémentant graduellement l'angle de torsion;-résultat: piégé dans une conform. gauche (min. local);- faudrait plutôt faire un "Rigid Rotor Search" (RRHO) dans un premier temps
pour calculer une énergie stérique initiale, et ensuite minim.➟trouverait plus rapidement la conform. la plus basse en énergie (logiciel NEMESIS);- on pourrait aussi offrir la possibilité de relaxer complètement la molécule en relâchant le dièdre fixe (“dihedral driver”) pour se diriger vers le vrai minimum.- On voit donc qu'il ne faut pas nécessairement que le modèle (molécule) soit complexe pour déjà causer des problèmes de recherche du minimum global.
Erreurs les plus communes:- abus du dièdre fixe, même avec relaxation- faut toujours tenir compte du principe de micro-réversibilité

83
3.2 LES MÉTHODES DE LA MÉCANIQUE MOLÉCULAIRE
La géométrie d'une molécule est stabilisée par un ensemble de forces exercées surchaque atome et entre eux. La mécanique moléculaire essaie de reproduire une surfaced'énergie de potentiel correspondant aux mouvements de tous les atomes d'unemolécule.
Une minimisation d'énergie en mécanique moléculaire implique une computationsuccessive et interactive d'une molécule à partir d'une conformation initiale, laquelle estsoumise à une optimisation géométrique complète. Tous les paramètres définissant lagéométrie du système sont systématiquement modifiés par petits "incréments" jusqu'à ceque l'énergie structurale moyenne atteigne un minimum local*. Le but de l'exercice estdonc d'atteindre un minimum local sur la surface de potentiel dans un temps minimum.Cependant, aucune méthode peut garantir de façon absolue l'obtention de la plus basseénergie, c'est-à-dire le minimum global. Presque toutes les méthodes de minimisationont un point commun: on commence à un endroit donné de l'hypersurface énergie-coordonnées et on accède au minimum local le plus proche.
Potentiel énergétique Dynamique moléculaire____________________* Un point x sera dit minimum local d'une fonction F(x) s'il existe d>0 tel que pour tout x' dont la
distance à x est inférieure à d, F(x)<F(x'). En ce point, la dérivée 1ère est nulle et la 2ème positive.

84
Un calcul de mécanique moléculaire, ou de champ de force, est basé sur unmodèle simple de mécanique classique de la structure moléculaire. Il y a peu ou pas designification physique comme tel des paramètres et énergies obtenus de ces calculs.Néanmoins, la mécanique moléculaire peut être d'une utilité pour certains types demolécules puisque c'est la somme des composantes énergétiques qui est importante. Leproblème des "minimums locaux" est l'inconvénient majeur de ce type de méthode. Pourse rapprocher le plus possible du minimum absolu, le choix de la structure de départ estessentiel. Ce choix doit être effectué judicieusement. En effet, presque toute lesméthodes de minimisation ont un point en commun: commençant à un endroit donné del’hypersurface énergie-coordonnées, elles ne peuvent que déterminer le minimum local leplus proche.
Plus souvent qu'autrement, on risque d'être piégé dans un minimum local. Pouren sortir on peut faire appel à la dynamique moléculaire à une température donnée etensuite on minimise individuellement chacune des structures ou conformères obtenus.Cependant, la méthode dite de "géométrie des distances", combinée à la mécaniquemoléculaire donne habituellement de meilleurs résultats (voir Chapitre 6).
Rappelons qu’en mécanique moléculaire on traite la molécule comme un ensembled'atomes gouverné par une série de fonctions de potentiel, tel qu'illustré par l'énergie deliaison chimique. Cependant il faut introduire une approximation. C'est l'approximationde Born-Oppenheimer en mécanique quantique (revoir p. 49). Elle stipule quel'équation de Schrödinger pour une molécule peut se diviser en une partie décrivant lespositions des électrons et un autre partie tenant compte des mouvements des noyaux, etque ces deux types de mouvements peuvent être étudier indépendamment.
ψΤ = ψΕ + ψΝ MQ traite les électrons; MM traite les noyaux (fixes)Par conséquent, en mécanique moléculaire, seuls sont pris en compte les
mouvements des noyaux alors les électrons ne sont pas explicitement examinés. Onsuppose qu'ils vont trouver une distribution optimale autour des noyaux atomiques.
La première étape de calcul en mécanique moléculaire est de déterminer lesdistances interatomiques, les angles et les angles de torsion de la géométrie de départ.Ces valeurs sont ensuite utilisées dans différentes fonctions de potentiel pour calculer uneénergie interne (stérique) initiale qui est spécifique au champ de force utilisé.

85
Parmi les avantages de la mécanique moléculaire citons le prix de calcul. Eneffet, le coût d'un calcul ab initio varie avec n4 où n est le nombre de fonctions de base(orbitales); cette méthode est donc réservée aux molécules de petites dimensions. Lesméthodes semi-empiriques nécessitent un temps de calcul proportionnel à n2 ou à n3,tandis que le temps de calcul en mécanique moléculaire est fonction de m2 où m est lenombre d'atomes. Dans une recherche conformationnelle on a la relation suivante:N = (360/étapes)n liaisons . Ou N est le nombre de conformations qui doit être examinerdans une recherche exhaustive.
Algorithmes de minimisation*La fonction énergie interne, définie précédemment, est une fonction de 3N
variables, où N est le nombre d'atomes du système étudié. C'est cette fonctionobjective F(x) qu'il faut minimiser où x représente l'ensemble des coordonnées de lamolécule. Une telle fonction présente, dans le cas général, un minimum absolu ou globalet un grand nombre de minima relatifs ou locaux. Il n'existe pas de méthodesanalytiques ou numériques permettant de déterminer la position de x du minimumabsolu: les algorithmes que nous allons citer nous conduisent dans le meilleur des casdans un minimum relatif de faible énergie et l'on ne peut qu'espérer que ce minimum nesoit pas trop éloigné en énergie du minimum absolu. Il faut donc choisir entre lesdifférentes méthodes (minimiseurs) qui différent par leur efficacité, leur rayon deconvergence, leur convergence vers le minimum et le nombre d'opérations qu'ellesdemandent.
Le concept de base de la plupart des algorithmes est la recherche itérative duminimum le long d'axes particuliers, comme le repère cartésien, ou plus généraux: un telalgorithme est dit "de recherche de ligne". La minimisation de l'énergie potentielle d'unemolécule consiste donc à résoudre un problème d'optimisation non-linéaire à plusieursvariables indépendantes. Il s'agit d'optimiser la fonction objective F(x) et de trouver unnouveau jeu de coordonnées x* tel que F(x*) soit inférieur à F(x). Il est cependantnécessaire d'introduire une estimation initiale xo qui, dans le cas de la minimisation del'énergie potentielle d'une molécule, sera le jeu de coordonnées brutes. On recherche le__________________* Inspiré en grande partie du résumé de D. Housset (Paris) 1989, et de l’article de J.S. Lomas dans
L’Actualité chimique, mars 1986.

86
minimum dans la direction s de la kième itération. Les diverses méthodes ne diffèrentque par le choix de la direction sk et la détermination du minimum dans une directiondonnée. Il existe de nombreux algorithmes de minimisation pour les fonctions à 3Nvariables où N est le nombre d’atomes du système à étudier.
Quatre d'entre eux sont plus couramment utilisés:1er ordre: • Méthode de la plus grande pente ("steepest descent")
(1ère dérivée) • Gradient conjugué
2ème ordre: • Newton Raphson
(2ème dérivée) • Recuit simulé (une méthode à part)
Principe de baseA partir d'une géométrie très approximative, on recherche le jeu de coordonnées
cartésiennes qui réduit à son minimum la somme de toutes les contributionsénergétiques dues aux déformations des coordonnées internes et aux interactions entreatomes non-liés. En principe, il suffit de prendre la dérivée première de l'énergiestérique par rapport à chacun des degrés de liberté de la molécule et de trouver l'endroitsur l'hypersurface énergétique où, pour chaque coordonnée ri, (dE/dri) = 0. Lesprocédures pour atteindre ce but sont de deux types: les unes utilisent uniquement lapente de la surface (dérivée première), les autres, à la fois cette pente et la courbure de lasurface (les dérivées première et seconde).
Presque toutes les méthodes de minimisation ont au moins un point en commun:on commence en un endroit donné de l'hypersurface et on descend vers le minimum leplus proche, sans savoir si ce minimum est local ou absolu. On doit donc présenter àl'ordinateur plusieurs conformations de départ, sous forme de coordonnées internes, ens'inspirant de modèles moléculaires. Les méthodes utilisent des algorithmes itéractifs.
La méthode de "steepest descent"Le premier programme de minimisation pouvant effectuer une optimisation de
géométrie est dû à Wiberg (1965) et utilise la méthode de la plus grande pente(steepest-descent). Après avoir calculé l'énergie correspondant à une géométrie

87
initiale, on déplace chaque atomeindividuellement selon ses trois coordonnéescartésiennes et l'on recalcule l'énergie aprèschaque déplacement. Ceci revient à calculer ladérivée première uniquement. Ensuite ondéplace tous les atomes sur une distance quidépend de (dE/dri), et ainsi de suite.Cet algorithme suivra donc la direction imposéepar les forces interatomiques dominantes.C'est pourquoi il se révèle très efficace pour supprimer les mauvais contacts ou lesprincipaux problèmes stéréochimiques qui existent dans les coordonnées brutes d'unestructure cristalline ou modélisée, tout en perturbant très peu cette dernière. Cependantcette méthode aléatoire est généralement longue vers la fin de chaque cycle deminimisation et la convergence devient très lente au-delà des premiers cycles(phénomènes oscillants, remontée d'énergie).
En fait, la méthode consiste à rechercher la direction de plus grande pente aucours de laquelle la fonction objective F(x) décroît le plus rapidement. La directionsuivie sera celle indiquée par l'opposé du gradient d'énergie, c'est-à-dire ladirection de la plus grande pente de la fonction d'énergie, qui est la direction danslaquelle l'énergie diminue le plus vite, du moins localement. Puisque cette méthodepermet de décroître rapidement la valeur de la fonction, elle est généralement utilisée pourajuster les contacts interatomiques forts d'une structure initiale.
Dans la méthode "steepest-descent" on peut définir un pas (déplacement) α, c'est-à-dire un changement de variable ou de coordonnées, ou un angle de rotation autourd'une liaison. On peut ainsi avoir N variables.Par exemple on fait varier de 10° un angle (pasde 10°) et on examine l'effet sur l'énergie. Onrecherche une diminution d'énergie.Si l'énergie diminue on augmente α car on aplus de chance d'aller vers un minimum.Si d'autre part l'énergie augmente, on diminueα en examinant des changements plus petits.

88
Le gradient est un vecteur qui indique le sens ou augmente l'énergie. C'est donc unevariation d'énergie en fonction des coordonnées (δE/δr). Si le gradient est positif, alorsl'énergie augmente avec r. Si le gradient est négatif, l'énergie diminue avec r. Doncune direction opposée au gradient correspond à une diminution d'énergie. On essaiealors d'avoir une descente rapide pour arriver au minimum.
Un gradient correspond donc à la dérivée première de l'énergie (δE/δr). Si ladérivée est positive, l'énergie augmente; si la dérivée est négative, l'énergie diminue. Onpeut aussi traiter la dérivée seconde (δ2E/δr2); elle nous renseigne sur la vitesse aveclaquelle le gradient augmente (vitesse vs accélération).
On peut aussi se limiter à un calcul direct d'énergie et regarder si elle augmenteou diminue. C'est la méthode SIMPLEX qui fait un seul calcul et fonctionne bien si ona 20 variables ou moins. La méthode consiste en essais successifs représentant unetriangulation de l’hyperespace, c'est-à-dire que l'on examine le sens et l'amplitude desvariations de la fonction dans différentes directions pour suivre celles qui mènent vers unminimum.
Néanmoins, il devient nécessaire de disposer d'une procédure qui part d'unestructure approchée et déplace les atomes vers leurs coordonnées optimales d'une façonmoins aléatoire. Afin d'accélérer la convergence, on peut utiliser l'information issue desitérations précédentes: cette idée conduit à l'algorithme du gradient conjugué.
La méthode du gradient conjuguéCette méthode, fondée sur le même principe que la précédente (direction opposée
au gradient d'énergie), prend également en compte les étapes précédentes, afin dedéterminer plus finement la direction et le pas. Pour une surface d'énergie quadratique,fonction de 3N variables, cette méthode converge en 3N pas. Elle conserve une bonneefficacité, mais est plus coûteuse en temps de calcul (un facteur 2 par rapport à "steepestdescent"). Le pas est ajusté à chaque cycle pour obtenir la meilleure diminutiond'énergie. L'intérêt de cet algorithme est d'éviter un comportement oscillatoire autour duminimum et d'accélérer la convergence. Il se révèle cependant moins efficace ou mêmeinutilisable (pas de convergence) pour des structures qui présentent beaucoup de mauvaiscontacts, telles que les structures moyennées sur la trajectoire d'une dynamiquemoléculaire.

89
La méthode Newton-RaphsonUne amélioration supplémentaire de la convergence peut encore être obtenue en
ayant recours à une approximation quadratique Q de la fonction F, obtenue pardéveloppement en série de Taylor. La méthode consiste à chercher à chaque pas leminimum du développement à l'ordre 2 de la fonction F. Cette méthode dite de"Newton-Raphson", a recours aux dérivées secondes. Maintenant on fait plutôtappel à cette technique d'optimisation. Elle évalue les dérivées secondes de l'énergiemoléculaire par rapport aux paramètres géométriques et converge donc plus rapidement.La programmation de cette procédure est nettement plus difficile que celle des méthodesdes dérivées premières seules, mais le gain en temps de calcul et en précision est siimportant que presque tous les programmes de mécanique moléculaire l'ont adoptée.Dans MacroModel version 5.5, l'optimiseur par défaut est PRCG. Le problèmecependant de cette méthode de minimisation en coordonnées cartésiennes est l'inversionde la matrice F, puisqu'elle est 6 fois singulière à cause des degrés de liberté de transitionet de rotation (seulement 3N-6 valeurs propres correspondent aux fréquencesvibrationnelles non nulles). Elle présente donc le grand inconvénient de nécessiterl'inversion d'une matrice d'ordre 3N*3N, ce qui en limite l'emploi à des systèmescontenant moins d'environ 300 atomes. Ce qui revient à résoudre un système de 3Néquations à 3N inconnues.
Ainsi la méthode calcule le gradient de lacourbe de potentiel à un point donné enévaluant l'énergie et ensuite en changeantlégèrement la géométrie d'une quantité δr.L'énergie est alors recalculée et la différenced'énergie entre les deux points est utiliséepour déterminer le gradient δE/δr. Onsuppose que la distance du minimum estproportionnelle à δE/δr et que la géométrieest altérée pour obtenir la prochainestructure, laquelle devrait être plus près duminimum (vide infra).

90
Le programme détermine donc non seulement la première dérivée, δE/δr, mais
aussi la seconde, δ2E/δr2. Ces secondes dérivées, ou constantes de force, témoignent dela courbure de la courbe de potentiel énergétique et peuvent ainsi être utilisées pourestimer la position du minimum. La courbe de la figure illustre le comportement de laméthode: on se dirige toujours vers l'extrémum le plus proche, qui peut être unminimum ou un maximum. On s’aperçoit donc du gros défaut de cette méthode: lerésultat global obtenu peut être aussi bien un minimum qu'un "col" ou "selle de cheval"de la surface de l'énergie. Pour remédier à ce défaut, certains programmes utilisentquelques étapes de "steepest descent" afin de revenir dans une région où la dérivéeseconde de l'énergie par rapport au vecteur propre est positive. On converge alorsrapidement vers un vrai minimum. Autrement, il ne faut l'utiliser que si l'on estsuffisamment proche du minimum.
En résumé, l'optimisation complète selon la méthode Newton-Raphson demandede calculer la matrice complète des dérivées secondes (matrice des constantes de force).Cette matrice contient 3N X 3N éléments pour une molécule de N atomes.Les dimensions 3N correspondent aux trois degrés de mouvement pour chaque atome.L'optimisation de la géométrie requière cependant seulement 3N-6 degrés de libertépuisque les trois translations et les trois rotations ne sont pas accompagnées par deschangements d'énergie. Les constantes de forces sont alors manipulées sous la formed'une matrice de dérivées secondes.
Revenons maintenant à l'inconvénient majeur de la méthode Newton-Raphson àmatrice complète, à savoir le temps nécessaire à l'inversion de la matrice et la quantitéd'informations qu'il faut mettre en mémoire. Son application est donc réservée aux petitssystèmes (maximum quelques centaines d'atomes), et dans la phase finale deminimisation. Cette méthode nécessite en effet beaucoup d'espace mémoire (table de2ème dérivés) et un temps de calcul proportionnel à N3. Pour les grands systèmes,l'algorithme ABNR ("Adapted Basis Set Newton-Raphson") permet de contourner ceproblème, tout en conservant les avantages des méthodes de second ordre.Cette méthode dite de “base adaptée” élimine la coopérativité entre les atomes et comporteseulement 9N éléments matriciels au lieu d’une matrice complète 3Nx3N=9N2 (FMNR).

91
Une simplification consiste à réduire lamatrice en sous-matrices, ou "blocs", le long de ladiagonale, chaque sous-matrice correspondant auxcoordonnées x, y, z d'un seul atome; on néglige lesblocs qui ne sont pas sur la diagonale. Cettetechnique est connue sous le nom de méthode enbloc diagonal ou "block diagonal Newton-Raphson" (BDNR) et requière seulement 9Néléments dans les calculs. Ainsi les atomes de mêmetypes se situent sur la diagonale. Dans ce cas,l'inversion est beaucoup plus rapide mais, enrevanche, l'amélioration de la géométrie à chaqueitération est plus faible. Selon la taille de lamolécule, la méthode à blocs peut être plus ou moinsefficace que la méthode à matrice complète; pour desmolécules importantes (de plus de 50 atomesenviron), on préfère la méthode à blocs.
Cependant, ce n'est qu'avec la matrice complète des dérivées secondes que l'on peutcalculer les valeurs propres, les fréquences vibrationnelles et, éventuellement, lesgrandeurs thermodynamiques associées.
La méthode du recuit simulé (“anneal”)Les méthodes que nous venons de décrire ont la particularité de faire décroître à
chaque pas la fonction F; ces méthodes ne peuvent donc pas échapper au minimum localproche de la structure de départ, et ont par conséquent, un rayon de convergence toujoursrestreint. La méthode de recuit simulé, développée par Kirkpatrick (1983), autorise lafonction F à augmenter momentanément afin de franchir des barrières d'énergie pourretomber dans un minimum plus profond. Le franchissement de ces barrières permetd'aller au delà des minima locaux au voisinage de la structure initiale pour explorer defaçon plus extensive l'espace conformationnel accessible, afin de découvrir des minimaplus profonds et plus éloignés de la structure initiale que les minima locaux.

92
Principe du recuit simulé
Le recuit simulé peut être entrepris par la méthode de Monte Carlo ou laméthode de dynamique moléculaire: dans les deux cas on introduit la notion detempérature, qui n'a pas ici un sens physique, mais qui donne l'ordre de grandeur desbarrières d'énergie que l'on s'autorise à franchir. Cette température peut varier dequelques centaines à quelques milliers de degrés Kelvin.
Estimation de la chaleur de formation (Model et PC-Model)Si une optimisation converge, cela veut dire que l'énergie d'une structure
demeure constante d'une itération à l'autre et que les dérivées premières sont presquenulles. Le programme va alors générer l'énergie interne (stérique) finale et lagéométrie optimisée de la molécule. Cette géométrie peut être utilisée pour calculer despropriétés comme le moment d'inertie et le moment dipolaire. La chaleur deformation, ∆H, peut aussi être calculée à partir de l'énergie stérique. Mais cetteapproche est valable seulement pour les alcanes et les cycloalcanes. Il ne fait pas avoird’hétéroatomes.
La chaleur de formation ne donne pas de renseignement sur les contraintes de lamolécule, notamment la formation de cycles. L'énergie intramoléculaire d'une moléculene donnant pas la chaleur de formation, il faut l'ajouter par incréments d'énergie degroupes ou de liaisons. Cette approche est cependant utile au chimiste pour les étudesthermodynamiques d'une réaction mais peu utilisée en modélisation par le modéliseur.

93
interactions liantes interactions non-liantesEtension (strain) Estérique (VderW)
Einterne = Eliaisons + Eangles + Etorsions + E VderW + Eélectrostat. + ...
= mesure de l'énergie intramoléculaire totale par rapport à une situation hypothétique.
= ∆ molécule réelle – molécule hypothétique (0° K) ayant des valeurs idéales ("naturelles") des liaisons et des angles.
La prochaine étape dans le calcul est de déterminer l'énergie de liaison("strain"), ce qui n'est pas particulier au programme de mécanique moléculaire mais quipeut être obtenue en utilisant les valeurs expérimentales de chaleur de formation.L'approche complète qui utilise les programmes de mécanique moléculaire pour estimerla chaleur de formation et les énergies de tension est résumée sur ce diagramme.
Rappel que: Estérique(VdW) non-liante = Etension(strain) liante = ∆H de formation (chaleur)donc Etot = Ein + Eex ; une partie déformation et une partie interaction
On pourrait supposer que Epot = ∆H, mais ne tient pas compte des vibrations!

94
L'énergie interne (stérique) calculée par le champ de force est convertie en chaleurde formation en ajoutant les incrémentations de groupe et de liaison de laparamétrisation. Ils n'ont pas de signification physique mais sont en accord avec lesdonnées expérimentales de chaleur de formation et sont alors utilisés en conjonction avecles énergies stériques par le champ de force.
On calcule l'énergie de tension de la même façon. Chaque incrément de groupepour une molécule est additionné ensemble pour donner une chaleur de formation sanstension, laquelle est ensuite soustraite de la chaleur de formation actuelle pour donnerl'énergie de tension. Le dernier point à considérer concerne les molécules ayant dessystèmes conjugués. À cette fin, un module π-MO est ajouté au programme (MMP2).
ConclusionEn somme, un minimiseur est une boîte noire mathématique qui demeure un outil
important pour la recherche d'un minimum d'énergie. En général, on utilise plusieursminimiseurs. On passe à un deuxième ou un troisième si ça ne converge pas assez vite.Tout dépend aussi du nombre de variables ou du nombre de variations d'angle de liaisonintroduit. En principe on calcul l'énergie du système avec des fonctions énergétiquesempiriques. Chaque logiciel à ces valeurs à lui, qui sont plus ou moins meilleures qu'unautre logiciel. En fin de compte, on va chercher un minimum qui converge et on va faireune vérification par "recuit" pour voir s'il n'y aurait pas un minimum d'énergie plus bas.
A ce titre, la modélisation moléculaire reste un moyen efficace de prédiction desconformations stable d'une structure moléculaire correspondant aux minima de sonénergie intramoléculaire. L'énergie calculée mesure la différence entre l'énergie de lastructure moléculaire considérée et celle d'une structure hypothétique dont toutes lescoordonnées internes prendraient leurs valeurs de référence. Sa valeur absolue dépenddu champ de force considéré, c'est-à-dire des fonctions d'énergie potentielle et desparamètres utilisés. Les valeurs de cette énergie sont toutefois très utiles lorsqu'il s'agitde comparer la stabilité relative des conformations ou des stéréoisomères d'une mêmemolécule.

95
Comparaison des structures moléculaires*La comparaison de deux molécules est un problème classique en modélisation
moléculaire. Il faut, sur la base de deux modèles structuraux préalablement optimisés,rechercher la ou les superpositions moléculaires ("molecular fitting") mettant enévidence le plus grand nombre de caractéristiques géométriques et structuralescommunes. La technique généralement adaptée consiste, quand cela est possible, àcomparer deux molécules dont l'une au moins possède une structure rigide pouvantservir de base à la superposition. La conformation de la molécule flexible est alorsajustée de façon à obtenir la superposition la plus intéressante. Il existe plusieursméthodes de superposition:
a) Superposition rigide ("rigid fitting")Les longueurs de liaison, angles de valence et angles de torsion des deux
molécules ne varient pas. Deux algorithmes de superposition rigide sont généralementutilisés: (i) celui de la méthodes des "moindres carrés" ("Least Squares Fitting Method")qui consiste à déterminer la superposition géométrique optimale obtenue en minimisant lasomme des carrés des distances entre chaque couple d'atomes spécifiés au préalable(minimum 3 couples); et (ii) l'algorithme de Ferro et Hermans (1977) qui nécessite lechoix d'un paramètre de pondération pour chaque paire d'atomes spécifiée. Cesparamètres de pondération permettent de privilégier l'ajustement de certaines pairesd'atomes par rapport à d'autres.
b) Superposition flexible ("flexible fitting")L'algorithme de superposition flexible explore l'espace conformationnel
accessible des deux molécules ou fragments moléculaires afin de trouver lesconformations donnant la meilleure superposition. En général, seuls les angles detorsion varient. La qualité d'une superposition flexible dépend de l'orientation initialedes deux molécules et du jeu de paramètres de pondération utilisé. Le "fitting" flexibleest idéal lorsqu'il s'agit de superposer deux molécules présentant un squelette identiqueet ne différant que par l'arrangement spatial de certains de leurs substituants.
_____________________* Extrait de la thèse de Ph.D. de E. Buisine (Orléans) 1990.

96
3.3 MINIMISEURS: CONSIDÉRATIONS THÉORIQUES
Stratégie générale de minimisation
La minimisation d’une molécule se fait en deux étapes:1. il faut définir une fonction cible ou objective (“target function”)2. la configuration est ajustée pour diminuer la valeur de cette fonction cible.
L’efficacité d’une minimisation est basée sur le temps nécessaire pour évaluer lafonction cible (ou le nombre de fonctions nécessaires) et le nombre d’ajustementsstructuraux (itérations) nécessaires pour converger vers un minimum.
La plupart des algorithmes de minimisation supposent que la surface énergétiqueest approximativement harmonique. Même pour les surfaces non-harmoniques, l’allurede la surface devient harmonique à la limite s’il y a convergence vers un minimum.
Ayant en main une fonction cible, qui définit une surface énergétique, unminimiseur doit déterminer à la fois la direction du minimum et la distance à parcourirdans cette direction pour atteindre le puits de valeur minimal.
Les dérivées dE/dx,y sont proportionnelles aux coordonnées de sorte que pluson est loin du minimum, plus grandes sont les valeurs des dérivées correspondantes.Une minimisation a convergé quand les dérivées (gradient) sont égales à zéro.
Les minimiseurs possèdent une composante majeure; c’est la recherche deligne (“line search”). Cette recherche de ligne permet de changer les valeurs descoordonnées vers une structure de plus basse énergie. Elle correspond à uneminimisation unidimensionnelle dans une direction donnée. Un résultat général d’unerecherche de ligne est que la dérivée au minimum doit être perpendiculaire (orthogonal) àla direction précédente et ne dépend pas de l’algorithme qui génère le vecteurdirectionnel.
Un attrait de la recherche de ligne est que l’on extrait toutes les énergies dans unedirection avant de se déplacer à la suivante.
Les figures suivantes représentent le cas simple de la fonction:
E(x,y) = x2 + 5y2

97
Certaines figures de cette partie du texte sont extraites du guideDISCOVER® Biosym Technologies, 1992.

98
Steepest descent (SD)Comme prévue, la recherche de ligne conduit à une nouvelle direction qui est
perpendiculaire au gradient précédent mais les directions oscillent en s’approchant duminimum. Ce comportement inefficace est une caractéristique de la méthode SD,particulièrement si la surface énergétique possède des vallées étroites.
La convergence est faible ou lente près du minimum car le gradient s’approchede zéro mais la méthode est robuste, même pour les systèmes qui sont loin d’une allureharmonique. Elle est donc recommandée pour les conformations qui sont loin duminimum comme c’est le cas de structures montées directement à l’écran ou de donnéescristallographiques non affinées.
Gradient conjugué (GC)Une raison pourquoi le SD est inefficace est que la méthode essaie de retracer à
chaque segment du trajet, le progrès fait dans l’itération précédente. Des recherches deligne successives permettent cependant de corriger pour cette déviation, mais puisquechaque déplacement doit être orthogonal au précédent, elles ne peuvent pas corrigerconvenablement les surfaces énergétiques non-orthogonales. La trajectoire se met alorsà osciller.
On utilise alors un algorithme qui produit une base complète de directionsmutuellement conjuguées de telle sorte que chaque étape successive raffinecontinuellement la direction vers le minimum. A la suite d’une recherche de ligne, lenouveau gradient est orthogonal à tous les gradients précédents et la prochaine directionest conjuguée à toutes les directions précédentes.

99
Le terme gradient conjuguée est un peut trompeur. En fait l’algorithme produitun ensemble de gradients mutuellement orthogonaux et un ensemble de directionsmutuellement conjuguées. L’algorithme de GC doit converger à chaque recherche deligne avant de pouvoir poursuivre une autre direction.
L’espace mémoire additionnelle nécessaire doit pouvoir stocker un vecteur de Néléments pour contenir les N composantes de l’ancien gradient. Dans un espacecartésien ceci correspond à 3N dérivées de l’énergie pour les coordonnées x, y, z dechaque atomes. Ceci rend la méthode CG une méthode de choix pour les structures quisont trop larges pour pouvoir stocker et manipuler une matrice de dérivées secondes,telle que la méthode Newton-Raphson.
Newton-Raphson (NR)Le gradient conjuguée améliore l’algorithme de base en minimisant seulement
dans les directions qui sont mutuellement conjuguées et le mouvement le long d’unedirection ne nuit pas au progrès accomplit dans les itérations précédentes. Pour unefonctions harmoniques, un nombre égal à N2 points indépendant est requis pourrésoudre numériquement une équation de N variables. Puisque qu’un gradientcorrespond a un vecteur de longueur N, le mieux que l’on puisse espérer est deconverger en N étapes avec un minimiseur basé sur un gradient.
Si par contre on veut exploiter l’information des dérivées secondes, un minimumpourrait converger en une seule étape puisque chaque dérivée seconde est une matrice detype NxN. Ceci est à la base d’un algorithme de minimisation à variables multiplescomme NR. En d’autres termes, la méthode NR, en plus d’utiliser un gradient qui aideà identifier une direction, utilise la “courbure” de la fonction (dérivée seconde) pourprédire où, le long du gradient, la fonction va changer de direction et passer par unminimum.
Puisque la matrice complète des dérivées secondes définit la courbure dans ladirection de chaque gradient, l’inverse de la matrice de dérivées secondes (matriceHessienne) peut être multipliée par le gradient pour obtenir le vecteur qui conduitdirectement au minimum le plus proche. Un minimum peut alors être obtenu sansrecherche de ligne et avec une simple évaluation du gradient et de la matrice de dérivéessecondes. De plus, la convergence est très efficace près du minimum.
Les désavantages de la méthode NR résident dans le fait qu’une matriceHessienne est difficile à dériver et demande beaucoup de temps de calcul. Si lastructures est loin du minimum, la minimisation peut devenir instable et peut conduite àun point encore plus loin du minimum. C’est le cas quand les forces sont grandes et lacourbure de la surface petite (“flat bottom well”). De plus, calculer, inverser et stockerune matrice NxN demandent une taille mémoire de l’ordre de 3N2 pour N atomes.

100
Ainsi pour un système de 200 atomes, 120,000 “entrées” sont requises. Et l’Hessienpour un système de 10,000 atomes est déjà à la limite d’un super-ordinateur comme leCray-XMP.
Donc NR est réservé principalement pour les cas de convergence rapide avec unminimum bien défini et avec un RMS en dessous de 0.1 Kcal/mol/Å.
Méthode SD ("Steepest Descent") vs méthode GC (gradient conjugué):
en SD: - minimiseur toujours "down hill"- déplace chaque atome et calcule ∆E numériquement- variation rapide (dérivée élevée, pente élevée) au début mais ne sait pas bien la direction car cherche au hasard et recommence à nouveau -converge vite au début (facteurs stériques) mais évalue pas la pente précédente
- si pente plus faible, variation plus petite ➟ oscillation grande (petit pas) et converge mal au puits (dérivée faible)
- efficace seulement dans la plus grande pente ➟donc pas de convergence

101
en GC: - ici on tient compte du calcul précédent (dérivée première)
- mais pas au hasard➟descent rapidement en cherchant la ligne de la plus grande pente- sait mieux si se rapproche du minimum- trouve son chemin en regardant la position précédente- temps de calcul 2x plus long que SD- va rectifier sa trajectoire via dE/dr- converge mieux et plus vite même dans les pentes faibles
Une fois dans un minimum, on doit évaluer si on est sur un minimum local ou global.
En résumé:1. calcule ∆E initiale
2. calcule ∆E à deux déplacements le long d'une coordonnée
3. ajuste ("fit") la parabole pour ∆E
4. cherche le minimum de la parabole et calcule son ∆E minimum5. choisit une autre coordonnée et refait (2)6. arrête quand le changement est très faible
Pour une bonne convergence, vraie minimum: faut une 2ième dérivée (Hessien)positive et définie.
Gradient (pas harmonique, voir p. 73):E = k1(r - ro)2 + k2(r - ro)3 + ...dE/dr = 2k1(r -ro) + 3k2(r -ro)2 + ...d2E/dr2 = 2k1 + 6k2(r -ro) + ...
si r = ro; d2E/dr2 = 2k1 (cte)
quadratique: 2ième dérivée baisse la puissance à 0 = ctecubique: 2ième dérivée baisse la puissance à 1
3ième dérivée baisse la puissance à 0 = cte
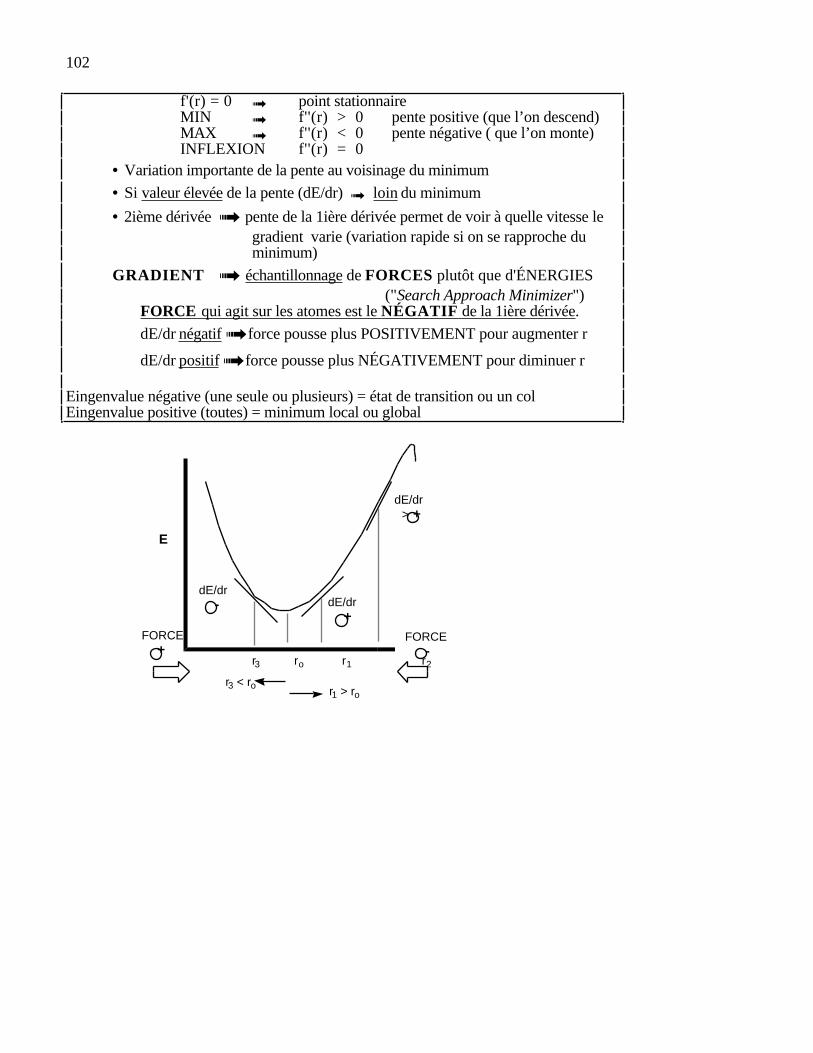
102
f'(r) = 0 ➟ point stationnaireMIN ➟ f''(r) > 0 pente positive (que l’on descend)MAX ➟ f''(r) < 0 pente négative ( que l’on monte)INFLEXION f''(r) = 0
• Variation importante de la pente au voisinage du minimum
• Si valeur élevée de la pente (dE/dr) ➟ loin du minimum
• 2ième dérivée ➟ pente de la 1ière dérivée permet de voir à quelle vitesse le gradient varie (variation rapide si on se rapproche du minimum)
GRADIENT ➟ échantillonnage de FORCES plutôt que d'ÉNERGIES("Search Approach Minimizer")
FORCE qui agit sur les atomes est le NÉGATIF de la 1ière dérivée.
dE/dr négatif ➟ force pousse plus POSITIVEMENT pour augmenter r
dE/dr positif ➟ force pousse plus NÉGATIVEMENT pour diminuer r
Eingenvalue négative (une seule ou plusieurs) = état de transition ou un colEingenvalue positive (toutes) = minimum local ou global
FORCE +
FORCE -
dE/dr - dE/dr
+
dE/dr > +
r3 ro r1 r2
E
r1 > ror3 < ro

103
∆X = -H -1 • gchangement de coord. = matrice Hessienne inverse • gradient (changement)
Notion d'inversion de matrice: ➟ NEWTON-RAPHSON (2ième dérivée)demande beaucoup plus d'espace mémoire cargarde la table de la 2ième dérivée
Faut prendre l'inverse de l'Hessien (2ième dérivée) pour satisfaire:H • H-1 = H-1 • H = I la matrice unitaire(en diagonale, tous des 1, le reste, tous des 0)
"Steepest descent": rapide mais ne converge pas car pas d'élément matriciel.
BDNR FMNR- Hessien plus simple - converge pas au début si loin du point sans sacrifier en convergence d'équilibre- diagonalisé par segment et - donc on peut faire SD/FMNR
on "colle" ➟ au complet - combinaison efficace mais faut pré-minimiser car FMNR requiert 9N2 éléments matriciels par interaction ("full Hessien metrix")- exigeant en calcul pour les protéines; passe plutôt par BDNR
- on néglige ceux (blocs) qui ne sont pas sur la diagonale A1: CH3; A2: CH2 par exemple...
1ière dérivée ➟ direction et vitesse "de tombée"
2ième dérivée ➟ distance de parcourt pour atteindre le minimumFaut donc avoir une 2ième dérivée (Hessien inverse) pour estimer si on est loin ou pasdu puits.

104
En gradient conjugué: ➟- le terme H-1 tombe (existe pas)
- suit l'équation en terme de ∆X, g (1ière dérivée) seulement; donc plus rapide
Reprenons:E(r) = k(r -ro)2
g = dE/dr = 2k(r - ro) = 2kr -2kro
H = d2E/dr2 = 2kindépendant de la position mais si on aun terme cubique, il faut une 3ième dérivéeH-1 = 1/2k∆X = -H-1 • g = -1/2k • 2k(r -ro)
= -(r -ro) = ro - r
si r > ro: ∆X négatif (force -)
si r < ro: ∆X positif (force +)(si r = ro; force = cte)
k > k'E
ro
k k'
ro
E
x x
"deep well" "shallow well"
- mais pas la même distance ∆X pour tomber sur le minimum- donc 2ième dérivée utile ici
1/2k < 1/2k' = H-1 ➟ matrice inverse (2ième dérivée)

105
1ière dérivée ➩ vitesse
2ième dérivée ➩ accélération
E = k1 (r - ro)2 + k2 (r - ro)3 k1 = 100 Kcal/mol/Åk2 = 10 Kcal/mol/Å
dE/dr = 2k1(r-ro) + 3k2(r-ro)2 ro = 2 Å
dE/dr = 200(r-2) + 30(r-2)2 (fait avec Mathematica)
d2E/dr2 = 2k1 + 6k1(r-ro)d2E/dr2 = 200 + 60(r-2) (donc linéaire)

106
E = k1 (r - ro)2 + k2 (r - ro)3 k1 = 100 Kcal/mol/Åk2 = -10 Kcal/mol/Å
dE/dr = 2k1(r-ro) + 3k2(r-ro)2 ro = 2 Å
dE/dr = 200(r-2) - 30(r-2)2 (fait avec Mathematica)
d2E/dr2 = 2k1 + 6k1(r-ro)d2E/dr2 = 200 - 60(r-2)

107
Quasi Newton-Raphson (QNR)
Une autre méthode à variables multiples a été développée par Fletcher et Powellen 1980. Elle consiste à calculer la matrice Hessienne numériquement en utilisantseulement l’information des dérivées premières plutôt que l’analyse par les dérivéessecondes. L’évaluation de la fonction d’énergie est plus rapide car les dérivéessecondes ne sont pas calculées analytiquement. Mais puisque que l’on garde unematrice Hessienne, la taille mémoire demeure une limitation.
Autres considérations
Une des étapes les plus importantes dans toute simulation sur ordinateur est depouvoir préparer adéquatement le système à l’étude. Ainsi, dans les stratégies deminimisation, les considérations suivantes sont à examinées:
1. L’importance de la structure de basse énergie obtenue et sa valeur énergique calculée.
2. Considérer les différences entropiques.3. Bien définir le système, et savoir s’il faut garder ou non les hydrogènes ou
un solvant.4. Savoir quand utiliser d’autres algorithmes de minimisation.5. Savoir quand ajouter des contraintes moléculaires et des restrictions.6. Utiliser les bons critères pour bien évaluer la convergence lors de la
minimisation.
De plus, le choix du bon algorithme dépend de deux facteurs: la taille dusystème à l’étude et le niveau d’optimisation désiré. Les algorithmes qui supposent unesurface énergétique quadratique (NR, CG) peuvent devenir instables si la molécule esttrop loin de ces limites. Il est souvent préférable de commencer par 10-100 étapes deSD et poursuivre ensuite par un GC ou un NR pour compléter la minimisation vers uneconvergence. Pour les molécules hautement distordues, la présence de termes croisés etde potentiels de liaison de type Morse dans le champ de force peut conduire à desproblèmes de convergence.
Des contraintes et des restrictions sont généralement utilisées pour contrôler etdiriger la minimisation. C’est le cas si on fait du “docking” enzyme-substrat. On peutaussi vouloir relaxer une structure cristalline. Il faut alors des artefacts comme latechnique d’enroulement sur une matrice fixe (“tethering”).

108
Un critère de convergence doit tenir compte qu’un minimum se définit commeétant un point ou les dérivées de la fonction sont nulles et la matrice des dérivéessecondes est positive et réelle. Pour cela, on utilise souvent le critère de RMS quidépend de l’objectif même de la minimisation. Ainsi on recherche des valeurs endessous de 0.5 Kcal/mol/Å.
Enfin, il faut noter que l’énergie (E) doit décroître monotoniquement durant uneminimisation mais pas nécessairement sa dérivée (dE/dr).
Calcul des fréquences de vibration
Pour calculer des fréquences de vibration, la structure doit déjà avoir étéminimisée et le gradient doit correspondre à zéro. Ceci ne signifie pas que laconformation de la molécule doit être dans un minimum, mais plutôt qu’elle doit êtredans une position stable sur la surface énergétique. Ce qui pourrait correspondre à uncol (“saddle point”) ou à un état de transition. Dans un col, au moins une valeur(eigenvalue) est négative. Si les fréquences sont réelles, on a affaire à un minimum; siune fréquence est imaginaire, on a plutôt un état de transition simple.
Le calcul des harmoniques des fréquences de vibration demande le stockage et ladiagonalisation d’une matrice de dérivées secondes avec les dimensions 3Nx3N, N étantle nombre d’atomes. Le travail atteint vite l’échelle N3 et devient prohibitif en temps decalcul. Les valeurs des fréquences obtenues sont valables uniquement pour desstructures bien minimisées et avec des RMS de l’ordre de 0.001 Kcal/mol/Å.
Le choix du champ force est primordial car généralement les champs de forcemettent l’emphase surtout sur les structures comme telles et leurs énergétiques plutôt quesur les fréquences de vibration. On peut ainsi facilement atteindre des erreurs de plus de100 cm-1 dans ce genre d’estimation. De plus, les torsions d’angle et les interactionsnon-liantes, qui dominent les modes de basses fréquences, sont fondamentalement non-harmoniques.
Il faut utiliser plus d’un champ de forceet ne pas avoir d’idées préconçues sur le système.Une petite différence d’énergie en MM n’est pas significative,c’est l’énergie totale qui compte et pas celle des composantes.

109
3.4 MINIMISEURS: CONSIDÉRATIONS PRATIQUES
Types de Champ de Force (MacroModel):
Lire le fichier.COM et regarder pour FFLD, MINI et/ou MDYN(voir Chapitre 8, Section 8.3)
Arg 1 = 1 MM2 2 MM3 3 AMBER (Amber94=4; MMFF de Merck=10; version 5.5) 5 OPLSA Jorgenson: paramètres non-liants + fonctions liantes
de AMBER
Types de minimiseurs (MacroModel):
Une grande partie du temps est "perdu" à calculer les dérivées de l'énergie par rapportaux coordonnées cartésiennes (surtout les distances non-liantes). Souvent il faut uneméthode de dérivation du type STREETT (voir Annexe B).
Arg 1 = 0 SD ➟ pré-minimisation
1 PRCG ➟ bon, mol. larges
2 FRCG ➟ moins bon
3 OSVM ➟ meilleur, "Variable Metric Method"
4 FMNR ➟ excellent si gradient faible, converge
9 TNCG ➟ excellent, mol. flexibles
SD: Steepest Descent
- utile pour struct. avec mauvaise géométrie au départ
- si la dérivée est élevée ➟ converge bien
- si la dérivée est faible ➟ converge mal- ne donne pas de col ("saddle point")- pas de convergence

110
PRCG: Polack-Ribière Conjugate Gradient
- par défaut sur MacroModel V. 4.5X (Unix)
- 100-250 atomes ➟ macromolécules, flexibles- réduit le gradient à 0.1 - 1.0 kJ/Å; difficile de faire mieux- 1ière dérivée boucle à chaque 3N itérations- devrait pas avoir de col ("saddle point")- conduit à un vrai minimum (bateau-croisé)
FRCG: Fletcher-Reeves Conjugate Gradient
- 1ière dérivée avec boucle qui reprend à 3N itérations- bonne méthode de minimisation- devrait pas conduire à un col ("saddle point")
BDNR: Block Diagonal Newton-Raphson
- pour les petites mol., semi-rigides- comme SD; converge bien loin du minim. mais mal si la pente est faible ("flat well")- mouvement par blocs (rassemble un gr. d'atomes à la fois)- pas utile pour les macromolécules- robuste et efficace pour les petites mol. et rapide (20 itérations pour un minim.)- utilise le SOR "Successive OverRelaxation Multiplier" (Annexe B)- matrice 3N x 3N; en diagonale faut 3N-6 valeurs propres positives + 6 valeurs propres nulles (si val. négatives: erreur ou incohérence sur le champ de force)
OSVM: Oren-Spedicato Variable Metric
- modifié par Fletcher-Powell- méthode quasi-Newton- converge en 3N-6 itérations mais lent: stockage du Hessien inverse - bon pour environ 250 atomes- possibilité d'un col ("saddle point")

111
FMNR: Full Metrix Newton-Raphson
- opposé à SD- converge bien si près du minimum (dérivée faible)- utile en combinaison avec SD (SD/FMNR)
donc pré-minim. avant ➟ RMS 0.1 kJ/mol- méthode de 2ième dérivée (Force = cte); permet de calculer fréq. de vibration
- faut utiliser "Line Search" ➟ cherche en double précision des interactions ("bouncing")- faut être près du minimum sur une surface de potentiel quadratique- excellent pour environ 200 atomes- résout 3N+6 équations (6 extras pour trans./rot.: voir Annexe B)- bonne méthode pour tomber sur un col ("saddle point") ou état de transition (bateau)
TNCG: Troncated Ponder-Richard Conjugate Gradient
- rapide- conduit au vrai minimum (chaise)- utilise une 2ième dérivée
RRHO: Rigid Rotor Harmonic Oscillator
- pour calculer les spectres vibrationnels et les prop. thermodynam.- pour une recherche de conformères et éviter les puits locaux- trouve plus rapidement des conformères de basse énergie
SCCG: Self-correcting Conjugate Gradient
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~En pratique:
< 200 atomes (flexible, acyclique): FMNR ou OSVM > plus efficace que PRCG > BDNR (SOR)> 200 atomes: TNCG en batchmin

112
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
MacroModel: coeur énergétique via des sous-routines ➟conduit à un "interaction ARRAY"
- identifie des “sets” (benzènoïde) et réajuste la longueur des liaisons.- "match" paramètres, champ de force et déplacements...(voir Annexe B)
Cas pratique de convergence: - trouver un point stationnaire (RMS = 0.003) - faire MTEST (calcul 2ième dérivée)
Exemple: construire le cis-4-méthylcyclohexanol
OH
CH3
ENERGY ➟ MM2 ➟ TNCG (rapide) ➟ STARTproduit mmodtmp.log (9.77 Kcal/mol avec RMS = 0.09)refaire avec MTEST et relire le fichier mmodtmp.log:
TRUE MIN avec RMS 0.01
ensuite construire le bateau en enlevant le méthyle et en déplaçant le carbone avec MXY. On remet à nouveau le méthyle en équatorial et on recalcule.
➟ FMNR + LSEARCHconverge sur un minimum "saddle point" (état de transition)avec MTEST ("saddle point" RMS = 0, avec 14 Kcal/mol)
refaire enfin avec ➟ PRCG + MTESTon rate le bateau pour tomber sur le bateau-croisé
TRUE MIN avec RMS 0.027

113
Recuit simulé:- pas une minimisation comme telle- "annealed" (analogie avec le procédé métallurgique de recuit)- explore un espace conformationnel- calcule la dérivée par rapport à un point- va donner la FORCE exercée sur les atomes
- faut ensuite sortir du point pour passer à un autre- on arrête le processus
- va fixer et changer vitesse des atomes
- temp. initiale 300° K ➟ passe 1000° K- on reprend le processus- voir si va tomber dans un minimum différent et meilleur
- à temp. ambiante: barrière de 0.6 Kcal (kT) est transparente aux
mouvements conformationnels ➟ peut être piégé!
"Anneal-Conformer":- développé par Wilson (1989)- utile pour trouver le minimum global d'un système complexe- marche bien si déjà près du minimum global- permet de trouver l'état énergétique de basse énergie- utile de minim. "HOST + GUEST" ensemble- dangereux avec des peptides car donne une hélice-α dans le vide pour maximiser les ponts hydrogène.
Calcul de ∆H:- une curiosité dans MM2 (PC-Model)- pas utile aux modélisateurs de macromolécules- minim. confinée au calcul d'énergie interne (∆H); peu de
contributions ∆S
- supposition dangereuse: Estérique reliée à ∆G (équilibre)
Etot. interne en MM ➟ état hypothétique immobile à 0° KEstrain = Etension (liant) , Esteric = EVan der W (non-liant)

114
- pour calculer ∆H de mol. (25° C), il faut ajouter des contributions: translation. rotation, (0.36 Kcal/mol par rot. à l’état gazeux). vibration. + E liaison
- valeurs de base en MM (paramètres) sont tirées d'expériences à T° non nulle; il y a déjà une contribution vibrationnelle- ∆H = E(MM) (valeur calculée) + Eliaison (incrément de groupes)
Attention: à temp. ambiante, la barrière de 0.6 Kcal (kT) est transparente auxmouvements moléculaires et on peut rester piégé si on fait pas une variation de T°.
Autres alternatives!
L’algorithme génétique (“genetic algorithm”) a été developpé ces dernièresannées pour permettre la superposition d’un ensemble de molécules flexibles et leurévaluation comme pharmacophore. Ce programme mimique le processus de l’évolutionen manipulant une collection de données structurales appelées chromosomes. Cetteapproche permet ainsi d’aligner une série de molécules actives afin d’identifier leséléments structuraux communs (“patterns”) et de l’appliquer aux pharmacophores quisont responsables de l’activité observée.Voir: Jones, Willett & Glen, J. Computer-Aided Mol. Design, 9 , 532-549 (1995).

115
3.5 LES PRINCIPES DE LA DYNAMIQUE MOLÉCULAIRE
La dynamique moléculaire est une technique de modélisation sur ordinateur parlaquelle l'évolution en fonction du temps, ou trajectoire, d'une molécule est décrite parles principes de la mécanique classique Newtonienne. En dynamique moléculaire onessaie de simuler les mouvements intramoléculaires que l'on peut visualiser ensuite entemps réel.
La dynamique moléculaire consiste donc à simuler les mouvementsintramoléculaires au cours du temps. Ces mouvements correspondent à des vibrationsautour d'un minimum, ou au passage d'un minimum à un autre minimum d'énergie. Sil'énergie fournie virtuellement au système est suffisamment élevée, des barrièresénergétiques importantes peuvent être franchies. Les champs de force utilisés sontidentiques à ceux utilisés dans les méthodes de minimisation. Les forces calculées àpartir de l'énergie potentielle servent à résoudre les équations de Newton régissant lesmouvements du système. L'avantage majeur de la dynamique moléculaire est deconduire à des conformations qui dépendent moins de la structure initiale que cellesobtenues par mécanique moléculaire.
Depuis 1980, l'accès à des moyens informatiques de plus en plus puissants apermis de développer des méthodes de calcul de dynamique moléculaire qui ont undouble but:
1) Simuler les mouvements intramoléculaires et si possible aboutir à unedescription thermodynamique du système étudie (entropie, énergie libre, chaleurspécifique...).
2) Optimiser les structures d'une façon plus efficace en évitant plus facilement lesminima multiples en particulier lorsqu'on introduit dans le calcul un recuit simulé.L'échelle des temps actuelle est environ de 10-14 à 10-10 s par simulation.
Une structure aux rayons X représente une moyenne temporelle de structuresd'un système en mouvement continuel. D'autre part, la dynamique moléculaire simuleles mouvements instantanés et présents d'un système moléculaire. Pour cela, chaqueatome est traité comme une particule obéissant à la loi d'action de masse de Newton;F = ma. Des intégrations successives de cette équation en fonction du temps conduit àune trajectoire de l'atome en fonction du temps sous la forme d'une série de positionset de vitesses dans l'espace . L'analyse est effectuée par périodes de temps de l'ordre de

116
1-100 ps. Plusieurs mouvements sont alors effectués durant ces périodes. Si la périoded'évolution est plus courte (fraction de ps), la dynamique moléculaire démontre peu decohérence dans le déplacement des atomes. De plus, les mouvements moléculaires sontfréquemment interrompus par des collisions avec les groupements voisins, nuisant ainsià la trajectoire. Enfin, les calculs nécessitent une bonne puissance computationnelle etdes facilités graphiques adéquates. Ce qui permet une présentation rappelant uneanimation vidéo.
La dynamique moléculaire est aussi fort utilisée pour caractériser et identifier desmouvements préférentiels, aussi bien avec des petites molécules qu'avec des protéines.Le plus souvent avec les protéines il faut utiliser une simulation "restreinte", c'est-à-direcouplée à la méthode de "géométrie des distances" (en accord avec les données de RMN;voir Chapitre 6).
A) Principe de base*Lorsque l'on ignore les effets quantiques, chaque atome d'une molécule est
considéré comme une masse ponctuelle dont le mouvement dépend et est déterminé parl'ensemble des forces exercées par tous les autres atomes en fonction du temps. Lesintervalles de temps de chaque cycle de dynamique sont suffisamment courts, environ 1fs (10-15 s), pour que l'on puisse considérer les forces interatomiques constantes. Ildevient alors possible d'intégrer numériquement les équations de mouvement et dedéterminer ainsi la trajectoire des atomes de la molécule étudiée.
Les simulations de dynamique moléculaire introduisent donc la dimension dutemps dans l'étude des molécules. Cette évolution temporelle (trajectoire) d'unemolécule est décrite par la résolution des équations de mouvement de Newton. Lamolécule est donc perçue comme une entité dynamique dont les atomes modifient leurspositions spatiales au cours du temps. Ces changements sont influencés par leur énergiecinétique (1/2kBTo par degré de liberté, si la simulation a été réalisée à la température To)et la résultante des forces qui s'exercent sur eux par les atomes environnants.
Au temps t, chaque atome i, de vecteur position xi et de masse mi subira uneaccélération ai telle que selon la loi de Newton:__________________* Extrait en partie de la thèse de Ph.D. de L.A. Bach (Strasbourg) 1989.

117
F = ma = − dVdr = m
d2r
dt2
m = masse des atomesa = accélération (2ième dérivée)t = tempsr = coordonnées cartésiennes de l'atomeAinsi une molécule se comporte comme une structure dynamique ayant des
coordonnées atomiques qui vont changer en fonction du temps à cause de leur énergiecinétique et des forces exercées par les atomes environnants:
d2
Xi
dt 2= a
i=
F im i
; Fi
= ∂V∂X i
V = fonction de l'énergie potentielletotale d'interaction auquel est soumis la molécule
On peut alors intégrer les équations de mouvement et obtenir une trajectoirepour chaque atome en fonction du temps. Pour cela on utilise l'algorithme de précisionde 2e ordre de Verlet (en troncature de la série de Taylor). Sa validité est aisémenttestée à l'aide de la 3ième loi fondamentale de la thermodynamique qui implique quel'énergie totale d'un système doit être conservée au cours d'une simulation. En généralle potentiel d'interaction Vtotal utilisé est un potentiel empirique "complet". Ce qui veutdire qu'il contient les contributions des énergies de liaisons, d'angles de valence,d'angles dièdres et des interactions de Van der Waals et électrostatiques.
Afin de pouvoir tenir compte de toutes les interactions entre atomes le pas (steps)
d'intégration ∆t (algorithme de Verlet) doit être au moins 10 à 20 fois inférieur auxfréquences de vibration les plus élevées*. Ce qui est généralement de l'ordre de 1femtoseconde (1 fs = 10-15 s). Ceci restreint les longueurs des trajectoires de dynamiqueà quelques centaines de picosecondes (100 ps), à cause du temps prohibitif du calcul.._____________________* Pour les molécules organiques, le mouvement dont la fréquence est la plus élevée est l'élongation des
liaisons C-H (3600 cm-1 ). Ce qui correspond à une période de 10 fs.

118
Pour des temps plus courts, la simulation est beaucoup plus longue. De routine onutilise des temps de simulation de 20-30 ps (10-12s), donc 20,000-30,000 pas. Lessimulations peuvent être effectuées dans le vide ou en solution. Généralement endynamique moléculaire, on effectue une première minimisation partielle pour réduire unepartie de la tension du système. Ensuite d'autres forces peuvent être calculées.
Il faut se rappeler que la température d'une molécule ou agitation thermiquecorrespond à l'énergie cinétique des atomes de la molécule. On va supposer que la forceF (une fonction du temps) exercée sur les atomes est constante et on va intégrerl'équation de mouvement en fonction du temps pour avoir la position des atomes. Lavaleur de Vo est fixée au départ car on fixe la température et on va regarder l'évolution dusystème après un temps ∆t. Si le temps ∆t (un pas de 1x10-15 sec) est très court, laforce exercée demeure constante.
t = 0: A' Vo
ABC peuvent être reliés: C' Vo 1 fs à la fois, 106 fois B'
Vo
∆t (pas): x.....A' VV
x.....C'x....B'
V équil. durant 100 ps
à la fin: AC trajectoires des atomes A, B, C
B
On applique une force constante (entre les collisions) sur chaque atome, ce quichange leur position (momentum) durant un intervalle de temps discret, plus petit que lafréquence de vibration la plus élevée. L’énergie potentielle reste constante à l’intérieur decet intervalle. Ceci génère une trajectoire durant une période de 100 ps et nous renseignesur les propriétés de la molécule. Le tout est basé sur la mécanique statistique(déterministique).

119
On ajuste graduellement la température pour bien relaxer les molécules et on essaidonc de décrire le mouvement (V) des atomes. On peut introduire des contraintes dedistance entre A et B et la molécule va s'ajuster à ces contraintes. A chaque pas ( ∆t ) onva recalculer l'énergie et examiner dans quel sens va changer la valeur de l'énergie. Lescalculs ne tiennent pas compte du rôle du solvant et des contre-ions qui peuvent modifierla nature polaire de la molécule à l'étude.
Rappelons que le temps de calcul (∆t) doit être plus court que les fréquences demouvements moléculaires des liaisons. Donc on fait des pas ("steps") de l'ordre de 1 fs(10-15s) et on limite le temps ou durée (période) de parcourt de la trajectoire à environ10-100 ps (10-12s) pour les protéines à cause du temps de computation élevé. L'énergiecinétique du système est fournie par une simulation à 300 K ou à 600 K. On obtientalors une série de structures de départ (environ 100) que l'on peut ensuite minimiserindividuellement. En d'autre terme, en dynamique moléculaire on explore une surfaceconformationnelle, c'est-à-dire des points dans tout l’hyperespace. Cependant, lerésultat n'est pas nécessairement réaliste car les structures obtenues ne sont pasminimisées. On a donc une image de l'ensemble en fonction du temps ("time dependentpicture").
Ne pas confondre le pas = temps d'intégration en fs et
la durée ou période = temps de simulation et/ou d'équilibration en ps.
Par exemple, à 300 K on évalue les coordonnées x,y,z de chaque atome (leurforce) en calculant leurs interactions ou répulsions (pour chacun) à chaque fs pendant unepériode de temps de 10 ps. Durant ce temps de trajectoire, on a évalué 10,000 ∆Edifférents. Cependant, pour les peptides, ces temps ne sont pas assez longs car lestemps de calcul seraient trop longs autrement.
On pourrait comparer la mécanique moléculaire et la dynamique moléculaire de lafaçon suivante:

120
mécanique dynamiquemoléculaire moléculaire
B B
A C A C
On suit les potentiels Ici le système à beaucoupdes termes d'allongement plus de flexibilité. Si lesde liaisons et les variations déplacements font augmenterd'angle de liaison. l'énergie, on ajoute des cont-
raintes dans une autre direction.Si l'énergie diminue, on vaconserver cette valeur.
En dynamique moléculaire on va ainsi générer un grand nombre de conformations(environ 104) au voisinage d'un minimum. Il faut alors minimiser (BDNR par exemple)les valeurs les plus basses pour optimiser et faire redescende à nouveau l'énergie totaledu système.
Voici deux exemples de patron d'évolution d'une dynamique moléculaire:
Artefacts en DM:- “cut off” (non liant) trop petit- période d’équilibration trop courte- pas assez de molécules de solvant (dans la boite).

121
B) Applications possiblesUne application importante de la dynamique moléculaire est l'analyse des modes
normaux de vibration le long de la trajectoire. Une autre application est la recherche etl'optimisation des structures 3D d'après les données de la cristallographie et/ou de laRMN.
La mise en oeuvre de cette méthode requiert néanmoins des moyens de calculparticulièrement puissant (CRAY par exemple) et elle est coûteuse en temps et en argent.Elle se généralise cependant pour les études d'oligonucléotides, de peptides et de petitesprotéines.
C) Autres considérationsEn effet, un des problèmes de fond de la technique de dynamique moléculaire est
l'inadéquation entre l'échelle de temps des phénomènes physiques observés et celle dessimulations accessibles par la technologie actuelle. L'une des plus longues simulations(en absence de solvant et à 298 K) a durée 300 ps et a été réalisée sur la myoglobine(1423 atomes). Tandis que les échelles de temps des processus dynamiques observésvarient de la femtoseconde (e.g. l'élongation des liaisons covalentes) à la seconde, voireplus (e.g. la vibration des cycles aromatiques des résidus d'acides aminés).
Plusieurs efforts ont donc été faits pour prolonger la durée de simulations dedynamique moléculaire:
1) L'utilisation d'un potentiel d'interaction simplifié. Les interactions entreatomes non directement connectés sont représentées dans ce potentiel par uneénergie de répulsion de Van der Waals. Alors que dans les autres potentiels(AMBER, CHARMM,...) elles résultent de la somme des énergies de Van derWaals, électrostatique et de liaison hydrogène. Cette simplification raccourcit letemps de calcul consacré à l'évaluation de l'énergie totale du système au coursd'une simulation.2) La procédure SHAKE (van Gunsteren, Berendsen), qui néglige lesélongations des liaisons, ce qui permet d'allonger le pas d'intégration, jusqu'à 2,voire 4 fs. En effet, cet algorithme permet de fixer la géométrie de certainesliaisons (longueur du lien covalent) durant une dynamique moléculaire.3) Les simulations de dynamique moléculaire à haute température. Ellespermettent de franchir les barrières d'énergie élevées, et accélèrent leschangements de conformation.
Un autre problème de fond ce cette technique concerne la nature desconfigurations produites. En effet, en principe, on voudrait connaître la fonction dedistribution des coordonnées des atomes ainsi que l'évolution dans le temps de cettefonction. En pratique, les simulations de dynamique moléculaire engendrent un

122
ensemble de configurations qui sont supposées être une représentation adéquate de cettefonction de distribution. Autrement dit, même avec les super-ordinateurs les pluspuisants actuellement, on ne peut pas savoir si cette technique explore l'espaceconformationnel de façon représentative. Cet inconvénient peut néanmoins être en partiesurmonté de deux façons différentes.
Premièrement, on réduit l'espace à explorer par l'utilisation des informationsexpérimentales, notamment celles fournies par la RMN. Ce sont les simulations dedynamique moléculaire avec contraintes de distances ( 1H, 1H) et d'angles dièdres. Cesvaleurs sont estimées par l'effet Overhauser (NOE).
Deuxièmement, on élargit l'espace conformationnel exploré en simulant destrajectoires de dynamique moléculaire à haute température (e.g 1000 K). En effet, lesdurées de temps simulé (de l'ordre de 100 ps) à 300 K ne suffisent pas à conduire à deschangements conformationnels importants. Elles constituent avant tout un processusd'évolution et de relaxation structurales. Par contre, à température élevée, la molécule est"secouée" et possède une énergie cinétique suffisante pour franchir les barrièresd'énergie. Ceci lui permet aussi de s'échanger rapidement entre différents conformères.Le refroidissement et l'optimisation de ces structures (méthode du "recuit simulé")échantillonnent plus largement l'espace conformationnel. Ils conduisent aussi à unecollection de conformères différents. En pratique, le refroidissement de la structures'obtient par des simulations de dynamique moléculaire à 300 K. Elles sont réalisées àpartir des conformères provenant de la trajectoire de dynamique à haute température puisoptimisés.
D) Méthode de la matrice forcéeCette méthode dite de la matrice forcée est mieux connue sous son nom anglais de
“template forcing” (Biosym Technologies). Cette technique est utilisée pour analyserl’orientation relative des groupements fonctionnels communs à un ensemble de moléculesactives, comme les conformations des ligands d’un récepteur. En effet, cette méthodepermet d’évaluer si une molécule donnée peut adopter la conformation d’une moléculecible (dans la conformation “bio-active”) et ce, en quantifiant leur degré de recouvrementainsi que l’énergie requise pour effectuer cette superposition. Pour ce faire, une fonctionde pénalité, proportionnelle à la distance des moindres carrés entre les atomes choisispour la superposition, est ajoutée à la fonction d’énergie potentielle initiale. Il s’agitensuite de minimiser cette fonction de façon à obtenir un bon compromis entre la qualitéde la superposition (reflétée par la valeur du RMSD) et l’énergie totale de la moléculeétudiée.

123
Considérations de base et stratégie en dynamique moléculaire
L’étude des mouvements dynamiques des molécules est un phénomèneintuitivement attrayant pour le chimiste. Par exemple, la méthode de Monte Carlo (MC)déplace au hasard les atomes de telle façon à générer des conformations alternatives quine sont pas représentatives d’une trajectoire dépendante du temps, mais néanmoinséchantillonne un espace conformationnel d’une façon cohérente avec une énergiethermique donnée.
La dynamique moléculaire (DM) d’autre part, résout les équations de mouvementd’une molécule et représente une évolution temporelle des mouvements moléculaires(trajectoire) et permet, contrairement à MC, d’étudier les propriétés dépendantes dutemps comme la diffusion et le repliement moléculaire.
Un avantage de la DM est qu’elle ne se limite pas aux mouvements harmoniquesautour de simples minima. Elle permet en plus aux molécules de franchir des maximad’énergie et d’explorer d’autres conformations stables. Cependant, cette dépendance surle temps constitue une faiblesse de la méthode car les périodes de temps de simulationpraticables sont uniquement de l’ordre 10-12 à 10-9 s (picosec à nanosec).
La solution numérique des équations de mouvements (loi de Newton) dépend dedeux choses: la connaissance de la position r(t), la vitesse dr/dt, et son accélérationd2r/dt2. Les vitesses initiales sont assignées au hasard par une distribution de typeMaxwell-Boltzmann à une température donnée. Après que r (t + ∆t) est résolu, lescoordonnées originales sont remplacées par ces nouvelles coordonnées, les vitesses sontmises à jour et l’accélération est corrigée en calculant les gradients avec ces nouvellescoordonnées. Ce cycle de calcul est répété des milliers et même des millions de fois.L’algorithme de travail est celui de Verlet (1964) et sa version modifiée, le “leapfrogmethod”. Ainsi appelée car ici la vitesse est déphasée d’un demi pas (“time step”) parrapport aux positions.
Un paramètre clé dans l’algorithme de Verlet est l’intégration du pas (∆t). Eneffet, si la vibration la plus rapide d’un lien C-H à une périodicité de l’ordre de 10-14 s,alors le pas de la dynamique devrait être de l’ordre de 10-15 s (1 fs), ou moins. A unetempérature de 300 K ceci correspond à une vitesse initiale de 1500 m/s (0.015 Å/fs)pour des hydrogènes le long d’un vecteur les réunissants. Il faut éviter des pas tropgrands, ce qui causeraient un éclatement (“blow up”) du système. Ainsi en augmentantle pas de 2 à 4 fs, les atomes s’interpénètrent fortement et se repoussent ensuite à unevitesse pouvant atteindre 1Å/fs (105 m/s ou 360,000 km/hr !).

124

125
En DM, le concept de température est fondamental puisque la vitesse estproportionnelle à la racine carrée de la température. En pratique ceci signifie quel’énergie cinétique du système correspond à 1/2kT. A l’équilibre, l’énergie potentielle etl’énergie cinétique doivent être égales. Donc en cours de DM, l’énergie cinétique estredistribuée en énergie potentielle. On a donc intérêt à effectuer des DM, soit àtempérature constante, soit à volume constant ou soit à pression constante.
Énergie cte, Volume cte: ➠ NVE (Discover), EVN (Biograf)processus adiabatique
Température cte, Volume cte: ➠ NVT (Discover), TVN (Biograf)processus isothermeensemble canonique (suit des règles)
Biograf utilise la théorie de Nosé-Hoover (1984) et la méthode de Berendsen (1984); un couplage faible à un bain thermique.
En MM comme en DM, les domaines d’application qui sont au-delà de lacapacités des méthodes classiques sont:
• transitions électroniques (abs. de photons)
• phénomènes de transport d’électrons
• formation et/ou clivage de liaisons
• transfert de protons (réaction acide-base)
Considérations plus pratiques:
Obéit à la loi de Newton: F = m•a = m•dV/dxforce dictée par Ecinétique, qui est reliée à (T°)3/2 NkBT = 1/2 Σ mivi2
➟ connaissant F et V, on peut déduire x (trajectoire)x (comment Ecinétique est redistribuée sur une molécule)
(Å)
_____________________________ trajectoire de x en f(t)20 80 t (psec)
position d'équilibre

126
- on traite un atome comme une particule répondant à l’éq. de Newton avec des FORCES calculées par les potentiels de MM
- on intègre l'équation de mouvement environ 105 fois en calculant la vitesse de chaque atomes dans un pas de 1 fs; ce qui conduit après 100 ps, à un trajectoire (10-15 x 105 = 10-10 sec = 100 ps)
- on suppose que durant 1 fs, Epot ne change pas (force interatomique reste cte) (dans 1 fs il y a environ 30 vibrations des liens C-C)
- pour chaque interaction: calcule la force F = -kx (oscillateur harmonique), x étant le déplacement de l'équilibre
- énergie est redistribuée sur la molécule après un certain temps (100 ps)- à la fin de chaque fs, on recalcule la vitesse (force et changement de vélocité)
par l’équation de Newton, F = ma- pour identifier des mouvements préférentiels, on peut ajouter des contraintes:
"restrained MD" plus “DG” ("distance geometry") ➟ pénalité artificielle
- on peut aussi garder la longueur des liens fixes par SHAKE- on obtient le mouvement de la molécule en fonction du temps: simulation de
"real time motion": si vrai minim. ➟ F sur atome = 0- pas basé sur une statistique au hasard car pas l'aspect aléatoire dans le calcul
➟ déterministique: mouvement (amplitude) obéit à la loi de Newton.
La DM a l'avantage qu'elle peut temporairement déplacer vers le haut unebarrière de potentiel et trouver une meilleure région de l'espace en terme d'énergie.
Autrement dit:- une nouvelle position et une vitesse (x1, v1) au temps t + ∆t est calculable
- à t + 2∆t: la nouvelle valeur de v, devient ve après le premier pas (timestep), pour le début du second pas
- et la position x1 calculée après le premier pas est utilisée pour calculer la nouvelle valeur de F et donc de a
- le dernier vn et a servent à calculer un nouveau vn+1 et xn+1, et ainsi de suite...- une expansion de Taylor est régit par l'algorithme de VERLET qui intègre
les équations de mouvement

127
- la MM cherche un ∆Emin (vide, solvant) tandis que la DM donne des changements de conformation sur une intervalle de t (vs T°)
V1
F1F2
V2
F3
V3
∆t∆t
321 ttt
Amplitude: 0.1-1000nm (1Å-100Å) ∆E: 0.1-100 Kcal/mol temps: 10-15-10-3 sec- dans une série de petites étapes (fs) et recalcule les positions des atomes et des mouvements internes durant ~ 10,000 pas (10 ps)
Recuit ("annealed"):
- chauffe de 60 K ➟ 600 Kdurant une période de 10 psensuitefait 35 ps de DM à 600 K
- finalement: recuit à 300 K durant 5 pspoursuit la DM durant 40 ps à 300 K
- donc Ti ➟ + ∆T (intervalle de 0.1 ps) ➟ Tf
- La majorité des mouvements ➟ C-H (car H léger) mais cette information n'est pas très utile.
- Alors on utilise SHAKE ➟ calcule ces mouvements et les gardent fixes.
DM à T° élevée: permet de balayer une grande portion de l’hypersurface conformat.
DM à T° basse: obtient plus de détail pour explorer la région autour de chaque minima.

128
3.6 LES SIMULATIONS DE MONTE CARLO
Les méthodes de Monte Carlo consistent en des simulations expérimentales deproblèmes mathématiques dans lesquelles des nombres aléatoires sont utilisés pourtrouver une solution qui peut ne pas être aléatoire. Historiquement, le développement decette méthode coïncide avec l'élaboration de la bombe atomique. En effet, la diffusionaléatoire de neutrons dans un matériau fissile était un problème majeur qui allait êtresoumis avec succès à l'expérimentation numérique. Vers 1948, Fermi, Metropolis etUlm appliquaient cette méthode à la chimie théorique et obtenaient des estimations desvaleurs propres de l'équation de Schrödinger par une méthode de Monte Carlo.
Cette procédure de Monte Carlo est une méthode stochastique (au hasard) quiconsiste à générer un enchaînement (série) de conformations où les propriétésthermodynamiques et structurales sont obtenues par moyennage. Il faut générer un trèsgrand nombre de conformations ou de configurations. Une distribution de Boltzmanndu type exp (–∆Ei/kT) permet ce moyennage. Contrairement à une minimisationd'énergie, l'approche Monte Carlo ne trouve pas un minimum d'énergie maiséchantillonne un ensemble d'états moléculaires avec des états énergétiques croissantsavec la température. Mais les états trop hauts en énergie auront peu de contributions surla stabilité du système car si l'énergie du système augmente, le terme de Boltzmanndevient petit. L'algorithme qui effectue cet échantillonnage est celui de Metropolis.
Par opposition à la dynamique moléculaire, en Monte Carlo on peut prendre dessauts plus grands pour optimiser. De plus, on peut effectuer des pas différents pour lesrotations, les translations, les coordonnées internes, et optimiser chacunindépendamment. La dynamique moléculaire est plutôt un processus déterministique.Il n'y a pas d'aspect aléatoire dans les calculs. Tout est déterminé au point initial dedépart et on laisse évoluer le système.
Dans les calculs du type Monte Carlo, le comportement dynamique d'unemolécule est simulé par des variations au hasard imposées aux système comme unemodification de rotation des angles dièdres. L'énergie de cette nouvelle entité est calculéeet conservée dans une nouvelle configuration si son énergie est inférieure à laconfiguration précédente. Si l'énergie est supérieure, l'algorithme Metropolis est utilisépour décider si cette nouvelle configuration est acceptée.

129
Ainsi, dans les procédures de Monte Carlo, le comportement de la molécule estsimulé par des modifications locales du système. Une perturbation locale peut parexemple être un petit déplacement de direction aléatoire d'un atome. La succession desmodifications aléatoires imposées à une structure tridimensionnelle initiale forme unensemble de configurations. La procédure d’échantillonnage peut être décrite de la façonsuivante: utilisant un générateur de nombres aléatoires, un atome choisi au hasard estdéplacé dans une direction aléatoire, aléatoirement suivant un des axes cartésiens choisiau hasard ou aléatoire suivant le coordonnées polaires. L'énergie d'une configurationobtenue à un moment donné est ensuite calculée. Si elle est inférieure à celle de laconfiguration précédente, la nouvelle configuration sera acceptée. Si par contre,l'énergie de la configuration est supérieure après les modifications aléatoires,l'algorithme Metropolis sert à déterminer si la nouvelle configuration était retenue ounon. La probabilité que la nouvelle structure soit gardée est son facteur de Boltzmann,qui vaut
B = exp (–∆Ei/kT).Autrement dit, un nombre I de l'intervalle [0,1], tiré au hasard, est comparé au
facteur de Boltzmann B. Si I est inférieur à B, la nouvelle configuration sera acceptée.Sinon, elle sera rejetée et l'ancienne configuration sera de nouveau soumise à une autreperturbation élémentaire. L'ensemble des configurations retenues est ainsiprogressivement construit et forme une chaîne de Markov. C'est une façon naturellede faire une recherche de l'espace conformationnel. Les structures obtenues peuvent êtrele point de départ pour une minimisation future.
Si sur un essai, le déplacement d'un atome est dans la même direction que le liencovalent, la surface énergétique sera "accentuée" et il y aura une probabilité élevée que cedéplacement ne sera pas accepté. Donc la surface sera plus "douce" dans la directionperpendiculaire au lien car alors des forces plus petites agissent sur l'atome. Un teldéplacement recevra une probabilité plus haute d'être acceptée.
A la fin, le conformère le plus bas en énergie est présumé être au minimumglobal. On peut aussi utiliser DM, mais c’est pas aussi efficace car une grande partie dutemps total de calcul explore des conformations non à l’équilibre et faudrait donc faire lasimulation beaucoup plus longtemps.

130
Voici l’organigramme de base:
Molécule rejetée si l’énergieest plus grande que ce queprédit la distribution.
Permet ainsi au système de“sauter” sur un point plushaut en énergie pour échapperaux minima locaux.
MC: stochastique (au hasard) sur un lien, un atome, pas tous à la fois. Si le temps decalcul est assez long, on aura peut-être le temps de voir (évaluer) toutes les conformationspossibles du système...si non!

131
Dynamique stochastique:- augmente la force du champ de force pour tenir compte de la friction- simule les propriétés du milieu (solvant)
MDBT "Berendsen Thermal Bath"MDCT "Stochastic Anderson Method"
Ftotal = mx (dynamique déterministique normale: au temps t=0, vitesse x=0gaussienne aléatoire)
ici: Ftotal = Fpotentiel -ζfrictionx + Faléatoire(t) (donc une partie stochastique)
1) dynamique de Langevin:- approximation du dipôle de Langevin (remplace eq. de Newton)- calcule les interactions électrostatiques- demande beaucoup de temps CPU- dynamique complexe- bain thermique simulé- avec un “water shell” autour d’un peptide
2) dynamique Brownienne:- comme Langevin mais on néglige les effets d'inertie- mx << Faléatoire- donc: x(t) = 1/ζ Fpotentiel + 1/ζ Faléatoire(t)
BOSS ("Biochemical & Organic Simulation Systems"):- développé par Jorgensen; pour les effets de solvant- calcule la charge partielle via TIP4P- pas de sens physique, purement opérationnel- choisir sa cte diélectrique- moment quadripolaire ?- propriétés des fluides ?
Dynamique par sauts ("Jump dynamics"):- comme un Monte Carlo- chaîne de Markov (cinétique chimique)- comme sur une grille, on fait des sauts...
Monte Carlo:- pas une dynamique comme telle- recherche de conformères {1,.......N}
i j; évolution n'est pas physique

132
- "random walk process"- au hasard (stochastique)- échantillonnage de l'espace conformationnel vers un minimum- développé par Scheraga (pour les peptides)
2 domaines:Conformation moléculaire Environnement moléculaire
-sélectionne au hasard un résidu i - avec solvant- bouge i au hasard selon les axes ∆Etranslation/∆Erotation- environ 106 fois- calcule Ei - avec soluté (8-9Å "cut off")
- si Ei < Einitial ➟ garde
- si Ei > Einitial ➟garde si e-∆E/kT (probabilité par dist. Boltzmann)
Le nombre de conformation possible = eno. degré de liberté
Algorithme Metropolis en résumé:
1. sélection au hasard d'un atome et déplacement au hasard de ∆x, ∆y, ∆z
2. calcule du changement ∆Epot. après le déplacement de l'atome
3. si ∆E < 0 ➟ accepte nouvelle conformation
4. si ∆E > 0 ➟ choisie un nombre i au hasard entre [0,1]
a) si exp(∆E/kT) < 1➟ accepte la nouvelle conformation
b) si exp(∆E/kT) > 1➟ garde la conformation originale etretourne à (1.)

133
• On obtient, après plus de 1000 boucles (steps), une collection de conformères énergétiquement favorables = ENSEMBLE (chaîne de MARKOV).
Il faut ensuite minimiser chacun des conformères par MM.• Tandis quand DM on obtient une MOYENNE en f(t).
La méthode Monte Carlo n'est pas vraiment une dynamique car le temps n'entre pasdans la formulation.
Formalisme MARKOV:- processus stochastique de non-équilibre
avec un temps long ➟ conduit à un comportement à l'équilibre
- mais n'obéit pas à une dist. de Boltzmann au début- converge ensuite vers une structure thermodyn. stable [voir Scheraga, PNAS 84, 6611 (1987)]
Comparaison entre SA vs MCM:SA: "Simulated-annealing" (Wilson) MCM: Monte Carlo + minimisation
- problème des minima multiples- MC traditionnel est inefficace à trouver le minimum global- faut combiner des étapes de minimisation après chaque recherche de MC pour pouvoir converger vers l'énergie la plus basse (T° cte à 300 K)
SA: - combine la recherche MC d'espace conformationnel à une T° initiale élevée (1400 K), suivi d’un processus de refroidissement- faut refroidir lentement pour ne pas être piégé dans une mauvaise valeur d'énergie (très critique)- n'utilise pas les critères de sélection de l’algorithme Metropolis- aucune minimisation durant les étapes- plus rapide que MCM (10X plus); mais dans l'eau c'est MCM!
- choix de T° ➟ fonction de la topologie de l'hypersurface- peut converger vers un puits d’énergie mais en manquant d’autres conformations potentiellement intéressantes (désavantage).
- SA toujours 5 Kcal > que MCM- MCM trouve une solution unique (SA souvent 2...)
- MCM fait à 300 K + minim. ➟ T° moins élevée

134
MCM: - plus efficace à converger (solution unique) vers un minimum global- mais ne peut pas franchir une barrière > 1 Kcal car fait à temp. ambiante- échantillonne un espace discret de variables et conduit à une hypersurface- moins critique que SA car trouve un minimum.- supérieure à DM pour des molécules larges [voir Scheraga, J. Compt. Chem. 12 , 594 (1991)]
MC-JBW/SD: - C. Still, JACS 117 , 8211-8219 (1995)- “Smart MC” avec “Jumping between wells”- une technique hybride (dans MacroModel v. 5.5)- 106 fois plus rapide qu’une dynam. stochastique simple
Avantage de MC et DM sur MM:- peut simuler des changements relatifs à ∆G via FEPT (voir Chapitre 4)- mais peut pas simuler les valeurs absolues de ∆S et ∆G
En minimisation:- important de faire des “run” multiples de configurations différentes de départ du système- “sampling” important- regarder les populations des conform. et prendre une moyenne des confor. et non pas un seule conformation
N/B: Le minimum global trouvé dépend souvant de la molécule de départ!
Parallèle entre dynamique et Monte Carlo:
DM: - info. propriétés de la molécule en fonction du temps- possible de prédire config. du système en f(t) passé ou futur- contribution cinétique avec un nombre constant de particules- peut extraire du calcul des propriétés thermodynamiques
MC: - pas de relation temporelle entre les config. successives (pas momentum)- résultat de la simulation est fonction du précurseur immédiat- énergie totale est déterminée directement en fonction de l’énergie potentielle- simule un ensemble canonique (N, V, T cte) voir p.125

135
Recherche conformationnelle à l’aide de la dynamique
Une limitation inhérente aux algorithmes classiques de minimisation est quenormalement ils localisent un minimum local proche de la conformation initiale, ce quin’est pas nécessairement le minimum global. Ils ne poussent donc pas le système au-delà des barrières d’énergie, mais descendent plutôt vers le puits le plus près.
Les vraies molécules trouvent naturellement leur conformation au minimumglobal en fluctuant autour d’un ensemble de conformations ayant des énergiesrapprochées. Le minimum global est ainsi échantillonné par simulation en MM et lenégatif du gradient du potentiel d’énergie correspond à la force présente. Ainsi la forceet la masse de chaque atome (loi de Newton) peuvent être intégrées numériquement pourprédire ou les atomes vont se déplacer dans un court intervalle de temps (pas). Enutilisant une succession de cet intervalle de temps, on peut construire une trajectoire dumouvement en fonction du temps.
Un ennui cependant de la dynamique est que plusieurs centaines et même milliersd’états conformationnels doivent être échantillonnés durant cette trajectoire. De plus, lephénomène d’intérêt peut très bien se produire dans une échelle de temps qui va de lamilliseconde à des minutes et même plus longtemps à 300 K. Actuellement, la DM estlimitée à une dizaine de nanosec.

136
Ainsi en partant du point a, le gradient initial amène la particule dans un niveauplus bas, mais l’énergie étant conservée, sa vitesse augmente et elle passe plus près duminimum. Le momentum fait fluctuer la particule, la faisant alors visiter plusieurspoints de la surface énergétique durant sa trajectoire. Cependant, si on arrête la particuleà un point précis durant la dynamique et on minimise, on trouve un seul point. Dans unsystème de plus haute dimension comme les molécules, plusieurs minima locauxdifférents sont ainsi identifiés. Mais ce nombre de minima est nettement inférieur aunombre de points échantillonnés durant la dynamique.
Une application: la vasopressineAvant de commencer une DM sur un système, on a intérêt à relaxer le système
pour éliminer les points chauds (“hot spots”) de haute énergie. La simulation estsouvent plus efficace que la dynamique pour relaxer les tensions initiales d’unemolécule, même si son RMS n’est que de 1 Kcal/mol/Å.
Les figures de cette partie du texte ont été extraites du guideDISCOVER® Biosym Technologies, 1992.

137
Autres considérations
Quoique la méthode de Monte Carlo ait souvent été utilisée pour simuler des bainsde solvants (e.g. simulation de l'eau liquide), son algorithme a été adapté à l'étudeconformationnelle des molécules organiques et des peptides. Dans l'algorithme modifié,les longueurs de liaison et les angles de valence sont gardés fixes. Seuls les angles detorsion sont soumis au tirage aléatoire. Chaque configuration retenue est alorsreprésentée par un N-uplet de valeurs, où N est le nombre d'angles de torsion de lamolécule. On détermine ensuite les coordonnées cartésiennes de cette configuration àpartir des valeurs de ses dièdres. A titre d'exemple, pour un peptide ayant 20 angles derotation (variables), on va examiner au hasard ces valeurs, chacune va conduire à unnouveau conformère. Si le résultat est mauvais (structure inacceptable), on va refaire auhasard un deuxième tirage et obtenir un autre conformère. Si l'énergie augmente on larejette, sinon on la garde. On a donc un outil mathématique qui correspond à ungénérateur de nombres aléatoires . Pour 100 tirages on aura 100 valeurs aléatoires et onpeut faire ces calculs pour les 20 variables.
Par ailleurs, afin d'explorer l'espace conformationnel le plus large possible,d'autres innovations ont été introduites dans l'algorithme initial. Ce sont:
1) L'existence de plusieurs départs différents (chaînes de Markov distinctes)au cours d'une simulation.
2) La montée en température suivie de refroidissement progressif("recuit simulé") afin de sortir des minima locaux.
A) Un exemple d'application aux protéinesLa technique de Monte Carlo a été utilisée pour étudier l'inhibiteur pancréatique
bovin BPTI (57 acides aminés). Combinée aux simulations de dynamique moléculaire,elle a permis d'étudier la rotation du noyau aromatique du résidu Tyr-35. En effet, cemouvement se passe à l'échelle de temps de la microseconde (ms), trop longue pour êtresimulé par dynamique moléculaire. Pour cette même protéine globulaire, une autre étudea comparé l'efficacité des simulations de Monte Carlo et de dynamique moléculaire dansla génération de conformères de basses énergies. L'étude conclue que la procédure de

138
Monte Carlo serait 5 à 50 fois plus efficace que la dynamique moléculaire classique vis-à-vis ce critère.
B) Analyse conformationnelleDans une première approximation, on tient compte uniquement des forces
intramoléculaires pour calculer les propriétés conformationnelles d'une molécule.Une nouvelle approche maintenant consiste à développer des fonctions générant
des conformations multiples ("multiconformer search"). Plutôt que de se baser sur unetechnique de recherche systématique ("systematic search"), la méthode d'analyse utilisel'approche Monte Carlo (au hasard). Elle est basée sur une évaluation progressive desangles de torsion ("torsion search method"); développée par Still et Lipton [J. Comput.Chem. 11 , 440 (1990)]. Cette approche est plus rapide car elle n’optimise que lesangles dièdres plutôt que chaque angle de liaison. Ce sont les angles dièdres quifluctuent le plus lors d'une minimisation (voir MacroModel). Rappelons quecontrairement à une minimisation d'énergie, la méthode Monte Carlo ne cherche pas uneénergie minimum mais fait plutôt un échantillonnage d'un ensemble d'étatconformationnel, sondant des états énergétiques de plus haute énergie en augmentant latempérature.
Recherche conformationnelle:L’objectif est d’identifier des conformations préférentielles. Donc concernée par
la localisation des énergies minimales des structures. Tandis qu’une simulation MCgénère aussi beaucoup de structures qui ne sont pas dans des minima. Mais comme enDM, tous deux sont utilisées en statégie de recherche conformationnelle.
Le processus (via MC, DG ou recuit) est efficace si la temp. est élevée et si lasimulation (ps) longue. Pour des ∆G on utilise FEPT. Si le système est en équilibre,
MC (Métropolis) évalue ∆G de l’ensemble des conformères générés.
C) Application aux molécules cycliquesCette technique ne semble pas appropriée à l'étude des molécules cycliques. En
effet, pour une molécule linéaire, les combinaisons d'angles dièdres possibles peuventvarier à l'infini. A l'inverse, pour les systèmes cycliques, seules certaines combinaisonsfermeront le cycle. En d'autres termes, une combinaison arbitraire d'angles dièdresconduit le plus souvent à des structures non réalistes, par exemple, avec des longueursde liaison et d'angles de valence déraisonnables. Or, pendant une simulation de Monte

139
Carlo, les valeurs des dièdres des configurations retenues risquent très souvent d'être descombinaisons qui ne permettent pas d'obtenir une conformation fermée du cycle.
D) Évaluation de l'énergie de solvatationLa meilleure méthode est d'entourer le soluté de plusieurs centaines de molécules
de solvant et d'utiliser soit la dynamique moléculaire, soit la méthode Monte Carlo.Ainsi on offre une fraction des configurations du solvant disponible afin d'obtenir unemoyenne des énergies de solvatation. Pour estimer la polarisation, il faut une méthodede perturbation d'énergie libre. On utilise le programme OPLS de Jorgensen (efficaceen solvatation mais pas en analyse conformationnelle).
La modélisation et minimisation des hydrocarbures (non cycliques) ne présententpas trop de difficultés car il y a un minimum de charges et de polarisibilité associées à cesmolécules. On ne peut pas en dire autant des polypeptides et des acides nucléiques. Ilfaut donc en tenir compte et il faut aussi faire intervenir les interactions solvant-polypeptide. Hélas, ceci est souvent prohibitif et coûteux en terme de temps CPU. Il y acependant des travaux récents dans le domaine des calculs des couches d'hydratation("hydration shell").
E) Évaluation de ∆H et ∆STraditionnellement, les changements d'entropie sont calculés à partir des
moments d'inertie et des harmoniques de fréquences de vibration. Cependant, pour unsystème substrat-récepteur, les potentiels de surface sont plutôt anharmoniques.
potentiel harmonique le même potentiel irrégulierpour une molécule simple pour une molécule complexe
Pour obtenir de l'information sur l'énergie d'un système complexe, on effectueune dynamique moléculaire à température constante. Les atomes de la molécule vontsuivre une vitesse de mouvement au hasard, correspondant à l'énergie cinétique fournie

140
par la température (300 K ou 600 K) de simulation. En utilisant le champ de force de lamécanique moléculaire et la deuxième loi de Newton (F = ma), les positions des atomespourront être déterminées en fonction du temps. Au cours du temps de simulation, toutesles régions du potentiel seront explorées et des valeurs thermodynamiques moyennesseront enregistrées. S'il y a plusieurs puits de potentiel mais séparés par de largesbarrières énergétiques (donc infranchissable durant la simulation), on effectuera alorsd'autres simulations (à des temp. plus basses) pour chaque puits. Les résultats dessimulations donnent toujours une valeur du ∆H.
Pour évaluer le ∆S on utilise la méthode quasiharmonique de Karplus [JACS107 , 6103 (1985)]. Cette méthode suit les coordonnées internes (angles de liaison ettorsion) durant une simulation de dynamique moléculaire. Avec MacroModel, il faututiliser BATCHMIN.
Les différences de ∆S d'un lien varie selon différentes forces de liaison. Si laconstante de force est petite, alors la fréquence de vibration sera faible et l'entropie élevé.
Rappel:Interactions de Van der WaalsCe sont des forces d'attraction entre atomes ou groupes d'atomes non chargés et nonpolaires autres que celles causées par des ponts hydrogène. Ils sont causées par unphénomène de polarisation sur un atome en proximité d'un autre atome.Les contacts Van der Waals sont à des distances de moins de 4 Å .

141
Interactions hydrophobesElles sont le résultat d'une tendance qu'ont les composés ou groupements hydrophobes deformer des agrégats en milieu aqueux. Elles sont causées par une répulsion apparentequ'ont ces composés vis-à-vis l'eau. C'est un processus favorable entropiquement (∆S del'eau).La liaison hydrogèneElle est formée d'une association entre un atome électronégatif et un hydrogène lié sur unsecond atome relativement moins électronégatif. Les atomes qui participent généralementdans la formation de ponts hydrogène sont l'oxygène et l'azote. Il faut une dispositionlinéaire d’atomes. L'énergie d'une liaison hydrogène est relativement élevée, de l'ordre de5 Kcal/mol.
En mécanique moléculaire:On considère les atomes comme une collection de billes et la mécanique classiquedétermine leur position et leur vitesse à un moment donné dans le temps. On cherche unminimum sous un champ de force.
En dynamique moléculaire:Comme la position des atomes change par rapport aux autres, les forces qu’ellesexpérimentent changes aussi. Cependant, le pas (“time set”) doit être petit, de tel sorteque la position des atomes autour ne change pas durant cet incrément de temps. Onobtient une trajectoire.De plus, il faut suffisamment de molécules de solvant et il faut contrôler la température
de la simulation, avec la pression (∆G de Helmholtz) ou le volume (∆G de Gibbs)constant.
MMOD interfacé avec AMBERSYBYL “ MAXIMIN (Tripos)QUANTA “ CHARMBIOGRAF “ DREIDING
partie partiegraphique calcul
M

142
3.7 PRINCIPAUX LOGICIELS DE MANIPULATION GRAPHIQUE

143
3.8 DIX questions, DIX réponses pour les spécialistes en modélisation*
Question 1: Quelles sont les grandes catégories de méthodes de calcul par ordinateur disponibles en méthodologie de modélisation moléculaire?
Les catégories générales suivantes sont disponibles:a) interface graphique moléculaire sans optimisation de la géométrie moléculaireb) interface graphique moléculaire avec optimisation de la géométrie moléculairec) optimisation de la géométrie moléculaire via une multitude de tech. math.d) détermination des propriétés géométriques et électroniques des moléculese) simulation des effets de solvatation sur la conformation moléculairef) simulation des effets de température par dynamique moléculaire
Si l’utilisateur possède les coordonnées cartésiennes pour chaque atome de lamolécule, alors elles peuvent être compilées et le graphisme moléculaire peut êtreutilisé pour examiner la molécule. La molécule peut, à l’écran, être tournée etexaminée sous différents angles. Alternativement, l’utilisateur peut construire lamolécule à l’écran à l’aide d’un logiciel de modélisation et ensuite optimiser sagéométrie. Chacune de ces techniques possède des points forts et des faiblesses.Suivant la méthode mathématique employée, une multitude d’informationconcernant les propriétés géométriques et électroniques de la molécule peut êtreobtenue.
Question 2: Quels types de questions générales de nature biologique, pharmacologique ou médicinale peuvent être posés en modélisation moléculaire?
Les types suivants de questions peuvent avoir une réponse en modélisationmoléculaire:
a) le graphisme moléculaire permet la visualisation de macromolécules biologiques (protéines, lipides, carbohydrates, acides nucléiques)b) dans des circonstances bien définies, des calculs mathématiques permettent d’optimiser les géométries des macromolécules biologiquesc) les propriétés géométriques et électroniques précises peuvent être calculées rigoureusement que pour de petites molécules, comme des droguesd) les interactions intermoléculaires (drogue-récepteur ou enzyme-substrat) peuvent être simuléesf) la flexibilité moléculaire de molécules à des températures physiologiques peut être simulée par des calculs de dynamique moléculaire
____________________* Texte inspiré de Molecular Modeling Woorkshop, Queen’s University, Kingston, Ont. 1991.

144
g) les changements de conformation moléculaire en présence d’un solvant ou dans un environnement lipidique peuvent être simulésh) pour les protéines, des algorithmes statistiques peuvent être utilisés pour identifier les zones de structure secondaire (hélice-α, feuillet-β, tour-β)
Question 3: Quelles sont les différentes techniques mathématiques disponibles pour optimiser des géométries moléculaires?
Les méthodes mathématiques suivantes sont utilisées pour optimiser lesgéométries des molécules:
a) les calculs de mécanique moléculaire1) le mode “united atoms”: considère un CH3 ou des CH2 d’une chaîne latérale comme une entité atomique. Cette approche est bien acceptable etdonne d’assez bons résultats pour les macromolécules.2) le mode “all atoms”: considère tous les atomes.
b) les calculs de mécanique quantique1) par voie semi-empirique
méthode Hückel étendueméthode CNDOméthode INDOméthode MINDOméthode MNDOméthode AM1 (pour ponts hydrogène)méthode PM3 (pour ponts hydrogène)
2) par voie ab initiométhode STO-3Gméthode 3-21Gméthode 6-31Gméthode 6-31G** (nécessite un Cray)
Question 4: En quoi consiste les calculs de mécanique moléculaire?La mécanique moléculaire est une technique empirique de calcul basée sur leséquations classiques Newtoniennes de mouvement. On suppose que l’énergieglobale d’une molécule peut être décrite par une équation d’énergie potentiellemultidimensionnelle, appelée le champ de force, et qui témoigne du rétablissementdes forces agissant sur la molécule lorsque son état conformationnel énergétiqueminimal est perturbé.

145
Question 5: Quels sont les points forts et les faiblesses des calculs de mécanique moléculaire?
Les points forts sont les suivants:a) c’est mathématiquement simple et donc pas exigeant sur le plan calcul. En conséquence, la mécanique moléculaire ne demande pas des temps prohibitifs de calcul et peut quelques fois être effectuée sur des ordinateurs personnels (PC)b) on peut manipuler et traiter de larges molécules contenant même des milliers d’atomes avec une assez grande exactitudec) la technique fonctionne très bien pour les macromolécules biologiques comme les protéines et les acides nucléiques
Les points faibles sont les suivants:a) la mécanique moléculaire fournit aucune information sur les propriétés électroniques de la molécule à l’étudeb) la méthode n’est pas universellement transposable et les équations du champ de force doivent être “paramétrisées” pour chaque nouvelle classe de composésc) plusieurs programmes commerciaux de mécanique moléculaire ont des valeurs de paramétrisation qui sont discutables
Question 6: Quelles sont les caractéristiques particulières des différents champs de force commerciaux?
Le coeur d’un programme de mécanique moléculaire est son champ de force etplusieurs sont disponibles commercialement. Les champs de force les plusconnus et utilisés sont:
MM2 et MM3 (Allinger)AMBER (Kollman)CHARMM (Karplus, logiciel Quanta de Polygen)ECEPP (Scheraga)DREIDING (Goddard, logiciels Biograf/Polygraf de Molecular
Simulations))CVFF (Hagler, logiciels Insight/Discover de Biosym)
Cette liste n’est pas complète et d’autres champs de force sont aussi disponibles.Fondamentalement, tous les champs de force se ressemblent mais ils ont aussi desdifférences. MM2 (ou MM3) est le favori parmi les chimistes et fonctionneextrêmement bien pour les petites molécules. Les paramètres de MM2 pour lesprotéines et les acides nucléiques, quoique disponibles, ne sont pas aussi bienéprouvés que dans AMBER ou CHARMM. AMBER et CHARMM sont préféréspar les biochimistes et les biologistes et fonctionnent extrêmement bien pour les

146
protéines et les acides nucléiques. Comparé à MM2, ils sont moins fiables pourles petites molécules. AMBER est disponible en mode “united atoms” et en mode“all atoms”. Le champ de force ECEPP traite uniquement les protéines.
Question 7: En quoi consiste les calculs de mécanique quantique?La mécanique quantique est une technique mathématique rigoureuse basée surl’équation de Schrödinger. La solution de cette équation permet d’obtenir desinformations précises sur les propriétés géométriques et électroniques de lamolécule. Les calculs peuvent être de type ab initio ou semi-empirique. En abinitio on tient compte de tous les électrons de la molécule et on vise une solutionrigoureuse de l’Hamiltonien. Les calculs semi-empiriques traitent seulement lesélectrons de valence et utilisent un Hamiltonien plus simple ayant des facteurs decorrection basés sur des données expérimentales.
Question 8: Quels sont les points forts et les points faibles des calculs de mécanique quantique?
Les points forts sont:a) une information rigoureuse et de haute qualité sur les propriétés de la moléculeb) contrairement à la mécanique moléculaire, ici on est moins dépendant des paramètres empiriques et spécifiquesc) les programmes commerciaux de mécanique quantique sont excellents et donnent d’excellents résultats
Les faiblesses sont:a) la méthode est très exigeante en temps de calcul et demande de gros ordinateursb) actuellement (1992), la mécanique quantique est valable que pour des systèmes de moins de 40 atomes par ab initio et environ 100 atomes par des calculs semi-empiriquesc) l’interprétation des résultats est plus difficile et requière beaucoup plus d’expertise de la part de l’utilisateur que la mécanique moléculaire
Question 9: Quels sont les différences essentielles entre la mécanique moléculaire et lamécanique quantique?
La mécanique moléculaire (MM) traite des molécules de toutes tailles maisperforme mieux avec les molécules larges (100-20,000 atomes). La mécaniquequantique (MQ) est limitée aux molécules de moins de 100 atomes. La MM est“computationnellement” plus simple et peut être faite sur des PC. La MQ est trèsexigeante “computationnellement” et nécessite de gros ordinateurs. Les calculs de

147
MM peuvent être faits rapidement (quelques heures pour un problème de 1,000atomes) tandis que la MQ demande des temps prolongés de calcul (plusieurssemaines pour un problème de 75 atomes). La MM considère seulement lesnoyaux, donc pas de données sur les propriétés électroniques tandis que la MQfournie une information rigoureuse et utile sur l’état électronique de la molécule.La MM est basée sur les équations de mouvement de Newton tandis que la MQ estbasée sur l’équation de Schrödinger. La MM est tout indiquée pour lesmacromolécules. La MQ se prête mieux aux petites molécules comme lesdrogues.
Question 10: Si vous aviez à modéliser une molécule de taille intermédiaire (disons unoctapeptide de 110 atomes) dont vous ne connaissez rien sur sa structure,
pourriez-vous utiliser la mécanique moléculaire ou la mécanique quantique pourtirer de l’information sur sa conformation optimale?
NON, NON, NON! Il y a un problème de base en mathématique appelé lesminima multiples, ce qui veut dire qu’une telle molécule est tout simplement tropvolumineuse pour être résolue complètement en utilisant seulement des techniquesde calcul. De l’information additionnelle, soit par rayons X ou soit par desdonnées de RMN, est nécessaire.
Comparaison entre mécanique quantique et mécanique moléculaire
Avantage d’utiliser Mécanique Mécanique quantique moléculaire Simplicité conceptuelle •Vitesse de calcul sur ordinateur •Capacité de traiter des molécules larges •Inclusion des effets de solvatation •Analyse conformationnelle de molécules “normales” •Détermination des géométries et des énergies relatives •Généralité de paramétrisation •Applicable aux espèces de courte-vie •Traitement d’hybridation intermédiaire •Détermination des propriétés électroniques •Rigueur théorique •

148
Y a-t-il plusieursconformères rapprochés?
Faut t-il beaucoupde temps de calcul?
L'énergie libreest-elle requise?
La solvatationest-elle importante?
Géométrie de départ venant de rayons-X, etc.
MOLÉCULE
mécaniquemoléculaire
dynamiquemoléculaire ou Monte Carlo
mécanique quantique
La formation et/oula brisure de lienssont-ils important?
Manque-t-il desparamères au champ de force?
Est-ce que c'est pluspetit que 100 atomes?
Les charges sont-ellesimportantes?
Un diagramme pour choisir la bonne méthode de modélisation moléculaire.Une molécule de départ est essentielle et peut venir de plusieurs sources.Normalement, la mécanique moléculaire est une bonne approche à la modélisation.Cependant, si les réponses aux questions de gauche sont OUI, alors la mécaniquequantique est recommandée. Si les réponses aux question de droite sont OUI, alorsune simulation moléculaire utilisant un champ de force empirique serait plus appropriée.
Réf.: Lipkowitz et Boyd, Reviews in Computational ChemistryVCH Publishers, Inc. 1990.

149
Résumé de la MM sus forme de diagrammes:

150

151
2 sp2 → V2 augmente
2 sp3 → V3 “
aromatique → V1 “Pour reproduire les barrières de rotation mais pas facile des obtenir.

152
Abus en modélisation:consultez Lipkowitz, J. Chem. Educ. 72 , 1070 (1995).
Évaluation et limitations des champs de force:P. Kollman, Acc. Chem. Res. 29 , 462-469 (1996).
Limites:- exactitude requise (qual./quant.)- accessibilité des logiciels (+/- performant)- ressources d’ordinateurs (coût/model)- développement rapide de la méthodologie- impact du modèle dans le contexte industriel

153

154
3.9 MODÉLISATION ET PRÉDICTION EN CHIMIE ORGANIQUE
On reproduit ici quelques exemples inspirés des deux articles suivants, parus dansChemical Reviews (numéro spécial de novembre) 1993:
Eksterowicz et Houk“Transition-State Modeling with Empirical Force Fields”Chem. Rev. 93 , 2439-2461 (1993)
Lipkowitz et Peterson“Molecular Mechanics in Organic Synthesis”Chem. Rev. 93 , 2463-2486 (1993)
Ces articles contiennent aussi de nombreux tableaux de résultats à consulter.

155
Tetrahedron 34 ,3981 (1981)
Tetrahedron 40 ,2275 (1984)

156
Tetra. Lett. 31 ,3615 (1990)

157
J. Amer. Chem. Soc. 110 ,3328 (1988)

158
J. Amer. Chem. Soc. 114 ,1774 (1982)
Wender, Pure Appl. Chem. 61 , 469 (1989)
Olson, J.Amer. Chem. Soc.112 , 323 (1990)

159