ch02_pg025_064_fr.qxd
TRANSCRIPT

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 1/40
C h a p i t r e 2
Composition et structure des protéines
L es protéines sont les macromolécules douées des propriétés les plus variées dessystèmes vivants où elles exercent des fonctions cruciales dans pratiquement
tous les processus biologiques. Elles jouent le rôle de catalyseurs, transportent etmettent en réserve d’autres molécules telles que l’oxygène, apportent un supportmécanique ou une protection immunitaire, créent le mouvement, transmettent lesinflux nerveux et contrôlent la croissance et la différenciation. En fait, la plusgrande partie de ce livre sera consacrée à la compréhension de ce que font les pro-téines et sur la façon dont elles y parviennent.
Plusieurs propriétés clé permettent aux protéines de participer à un champ aussiétendu de fonctions.
1. Les protéines sont des polymères linéaires construits à partir d’unités monomériquesappelées aminoacides qui sont liés l’un à la suite de l’autre. En particulier, les pro-téines se reploient spontanément en structures tridimensionnelles déterminées par
la séquence des aminoacides du polymère protéique. La fonction des protéines estdirectement dépendante de cette structure tridimensionnelle (Figure 2.1). Ainsi, les protéines sont l’incarnation de la transition d’un monde de séquences à une dimensionvers un monde de molécules à trois dimensions capables d’activités diverses.
2. Les protéines contiennent un large éventail de groupes fonctionnels . Parmi ces groupesfonctionnels, figurent des groupes alcool, thiol, thioéther, acide carboxylique,carboxamide et toute une série de groupes basiques. La plupart de ces groupessont réactifs chimiquement. Combiné dans des séquences diverses, ce réseau degroupes fonctionnels explique le large spectre de fonctions des protéines. Par
2.1 Les protéines sont construites à partir
d’un répertoire de 20 aminoacides
2.2 Structure primaire : les aminoacides sont
unis par des liaisons peptidiques pour
former des chaînes polypeptidiques
2.3 Structure secondaire: les chaînes
polypeptidiques peuvent se reployer
en structures régulières telles que
l’hélice alpha, le feuillet bêta, les coudes
et les boucles
2.4 Structure tertiaire : les protéines
hydrosolubles se reploienten des structures compactes présentant
des cores non polaires
2.5 Structure quaternaire: les chaînes
polypeptidiques peuvent s’assembler
en des structures multi-sous-unitaires
2.6 La séquence des aminoacides
d’une protéine détermine sa structure
tridimensionnelle
P l a n
2 5
Structuresecondaire
Leu
Tyr
Gln
Leu
Glu
Asn
Tyr
Glu
N
C
Structureprimaire
Structuretertiaire
Structurequaternaire
Leu
Cristaux d’insuline humaine. L’insuline est une hormone protéiqueessentielle pour le maintien du taux de glucose sanguin à des niveauxappropriés. (Ci-dessous) Une chaîne formée par une séquence spécifiqued’aminoacides (la structure primaire) définit une protéine
comme l’insuline. Cette chaîne se reploie en une structure bien définie(la structure tertiaire), dans ce cas une seule molécule d’insuline.De telles structures peuvent s’assembler avec d’autres chaînes pour formerdes réseaux tels que le complexe des six molécules d’insuline représentéà l’extrême droite (la structure quaternaire). Il est souvent possiblede conduire ces réseaux à former des cristaux bien définis (photographiede gauche), ce qui permet une description détaillée de ces structures.[Photographie de Alfred Pasieka/Peter Arnold.]

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 2/40
exemple, les propriétés réactives des enzymes (protéines qui catalysent de façonspécifique les réactions chimiques au sein des systèmes biologiques) sont essen-tielles à leur fonction, (voir Chapitres 8 à 10).
3. Les protéines peuvent interagir avec d’autres protéines, ainsi qu’avec d’autresmacromolécules biologiques pour former des ensembles complexes. Au sein de ces der-niers, elles peuvent agir de façon synergique pour générer des propriétés nouvellesdont les protéines individuelles peuvent être dépourvues (Figure 2.2). Desexemples de ces ensembles comprennent les machines macromoléculaires qui répli-quent le DNA, transmettent les signaux au sein des cellules et assurent un grandnombre d’autres processus essentiels.
4. Certaines protéines sont très rigides, tandis que d’autres présentent une flexibilité
considérable. Les unités rigides peuvent constituer les éléments structuraux ducytosquelette (échafaudage interne des cellules) ou du tissu conjonctif. Les pro-téines ayant une certaine flexibilité peuvent au contraire jouer le rôle de charnières,de ressorts ou de leviers, essentiels à la fonction des protéines, à l’assemblage desprotéines avec d’autres protéines ou d’autres molécules dans des unités complexes,ou encore à la transmission de l’information à l’intérieur des cellules ou entre cesdernières (Figure 2.3).
2 6
CHAPITRE 2 Composition et structuredes protéines
Figure 2.1 La structure dictela fonction. Un composant
protéique de la machinerie de réplicationdu DNA entoure un segmentde la double hélice de DNA représentécomme un cylindre. La protéine qui estformée de deux sous-unités identiques(en rouge et jaune) se comporte commeun clamp qui permet à de grandssegments de DNA d’être copiés sans quela machinerie de réplication se dissociedu DNA. [Dessiné d’après 2POL.pdb.]
DNA
Figure 2.2 Assemblage complexed’une protéine. Une micrographieélectronique d’une coupe du musclede vol d’un insecte montre un réseauhexagonal de deux types de filamentsprotéiques. [Autorisation du Dr MichaelReedy.]
Figure 2.3 Flexibilité et fonction. Après fixation de fer, la protéine lactoferrine subitun changement substantiel de conformation qui permet à d’autres molécules de faire
la distinction entre les formes dépourvues de fer et les formes liées au fer.[Dessiné d’après ILFM.pdp et ILFG.pdb.]
Fer
5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 3/40
2.1 Les protéines sont construites à partir d’un répertoirede 20 aminoacides
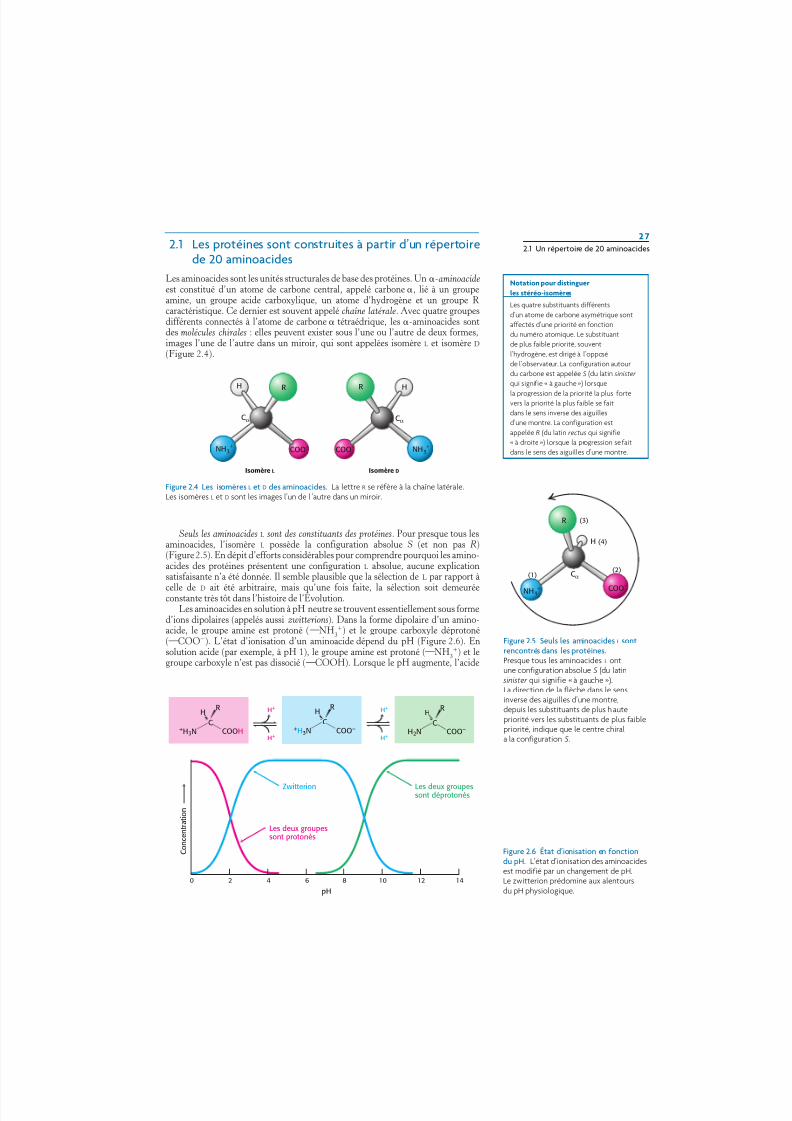
Les aminoacides sont les unités structurales de base des protéines. Un -aminoacideest constitué d’un atome de carbone central, appelé carbone , lié à un groupeamine, un groupe acide carboxylique, un atome d’hydrogène et un groupe R caractéristique. Ce dernier est souvent appelé chaîne latérale. Avec quatre groupesdifférents connectés à l’atome de carbone tétraédrique, les -aminoacides sontdes molécules chirales : elles peuvent exister sous l’une ou l’autre de deux formes,images l’une de l’autre dans un miroir, qui sont appelées isomère L et isomère D
(Figure 2.4).
Seuls les aminoacides L sont des constituants des protéines. Pour presque tous lesaminoacides, l’isomère L possède la configuration absolue S (et non pas R)(Figure 2.5). En dépit d’efforts considérables pour comprendre pourquoi les amino-acides des protéines présentent une configuration L absolue, aucune explicationsatisfaisante n’a été donnée. Il semble plausible que la sélection de L par rapport à
celle de D ait été arbitraire, mais qu’une fois faite, la sélection soit demeuréeconstante très tôt dans l’histoire de l’Évolution.Les aminoacides en solution à pH neutre se trouvent essentiellement sous forme
d’ions dipolaires (appelés aussi zwitterions). Dans la forme dipolaire d’un amino-acide, le groupe amine est protoné (¬NH3
) et le groupe carboxyle déprotoné(¬COO). L’état d’ionisation d’un aminoacide dépend du pH (Figure 2.6). Ensolution acide (par exemple, à pH 1), le groupe amine est protoné (¬NH3
) et legroupe carboxyle n’est pas dissocié (¬COOH). Lorsque le pH augmente, l’acide
2 7
2.1 Un répertoire de 20 aminoacides
Figure 2.4 Les isomères L et D des aminoacides. La lettre R se réfère à la chaîne latérale.Les isomères L et D sont les images l’un de l ’autre dans un miroir.
COO− COO−
RR H
H
Cα
Cα
NH3+NH3
+
Isomère L Isomère D
Figure 2.5 Seuls les aminoacides L sontrencontrés dans les protéines.Presque tous les aminoacides L ontune configuration absolue S (du latin
sinister qui signifie « à gauche »).La direction de la flèche dans le sensinverse des aiguilles d’une montre,depuis les substituants de plus hautepriorité vers les substituants de plus faiblepriorité, indique que le centre chirala la configuration S.
COO−
R
H (4)
(1)(2)
(3)
Cα
NH3+
Figure 2.6 État d’ionisation en fonctiondu pH. L’état d’ionisation des aminoacidesest modifié par un changement de pH.Le zwitterion prédomine aux alentoursdu pH physiologique.
0 2 4 6 8 10 12 14
C o n c e n t r a t i o n
pH
Zwitterion
Les deux groupessont protonés
Les deux groupessont déprotonés
H+
H+COOH +H3N+H3N COO– H2N COO–
H+
H+
RH
C
RH
C
RH
C
Notation pour distinguer
les stéréo-isomères
Les quatre substituants différents
d’un atome de carbone asymétrique sont
affectés d’une priorité en fonction
du numéro atomique. Le substituant
de plus faible priorité, souvent
l’hydrogène, est dirigé à l’opposé
de l’observateur. La configuration autour
du carbone est appelée S (du latin sinister
qui signifie « à gauche») lorsque
la progression de la priorité la plus fortevers la priorité la plus faible se fait
dans le sens inverse des aiguilles
d’une montre. La configuration est
appelée R (du latin rectus qui signifie
« à droite ») lorsque la progression se fait
dans le sens des aiguilles d’une montre.

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 4/40
carboxylique est le premier groupe à donner un proton, étant donné que son pK a estvoisin de 2. La forme dipolaire persiste jusqu’à ce que le pH s’approche de 9, zonedans laquelle le groupe amine protoné perd un proton.
Vingt types de chaînes latérales, qui se différencient par leur dimension, leur forme, leur charge, leur capacité à contracter des liaisons hydrogène, leur caractèrehydrophobe et leur réactivité chimique, sont communément rencontrées dans les pro-téines. En fait, toutes les protéines de toutes les espèces de bactéries, d’archae-bactéries et d’eucaryotes, sont construites à partir du même groupe de 20 aminoacidesavec seulement quelques exceptions. Cet alphabet fondamental pour la construc-tion des protéines date de plusieurs milliards d’années. L’étendue remarquable defonctions, dans lesquelles les protéines interviennent, résulte de la diversité et del’universalité de ces 20 types d’éléments fondamentaux. La compréhension de lafaçon dont cet alphabet est utilisé pour créer les structures tridimensionnelles com-plexes qui permettent aux protéines d’effectuer tant de processus biologiques est un
aspect excitant de la biochimie, et l’un de ceux sur lesquels nous reviendrons dansla Section 2.6.Examinons le répertoire d’aminoacides. Le plus simple est la glycine qui possède
un seul atome d’hydrogène comme chaîne latérale. Avec deux atomes d’hydrogèneliés à l’atome de carbone , la glycine est le seul aminoacide à ne pas être chiral.L’alanine, le plus simple des aminoacides après la glycine, possède un groupeméthyle (¬CH3) comme chaîne latérale (Figure 2.7).
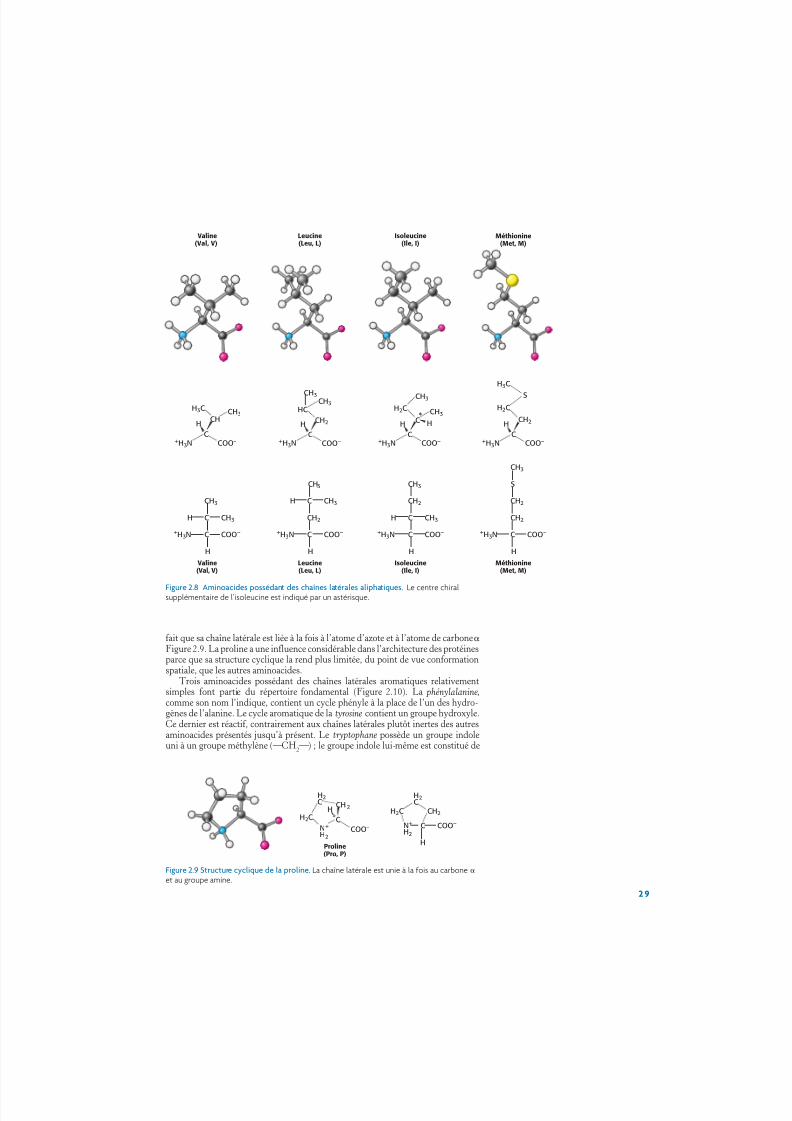
Des chaînes latérales hydrocarbonées plus longues sont rencontrées dans lavaline, la leucine et l’isoleucine (Figure 2.8). La méthionine présente une chaîne laté-rale essentiellement aliphatique, mais qui inclut un groupe thioéther (¬S¬). La
chaîne latérale de l’isoleucine contient un centre chiral supplémentaire. Seul l’iso-mère représenté dans la Figure 2.8 est rencontré dans les protéines. Les chaînes laté-rales aliphatiques les plus longues sont hydrophobes, c’est-à-dire qu’elles tendent às’agglomérer les unes avec les autres au contact de l’eau. Les structures tridimen-sionnelles des protéines hydrosolubles sont stabilisées par cette tendance desgroupes hydrophobes à venir au contact les uns des autres, appelée effet hydrophobe(p. 9). Les dimensions et les formes différentes de ces chaînes latérales hydro-carbonées leur permettent de se rassembler pour former des structures compactesprésentant peu d’espaces vides. La proline possède, elle aussi, une chaîne latérale ali-phatique, mais elle diffère des autres membres du groupe des 20 aminoacides par le
2 8
CHAPITRE 2 Composition et structuredes protéines
Figure 2.7 Structures de la glycineet de l’alanine. (En haut) Les modèleséclatés présentent la dispositiondes atomes et des liaisons dans l’espace.(Au milieu) Les formules stéréochimiquesréalistes montrent la dispositiongéométrique des liaisons autourdes atomes (voir p. 21).(En bas) Dans un souci de simplicité,les projections de Fischer présententtoutes les liaisons comme perpendiculaires(voir p. 22).
Glycine(Gly, G)
Alanine(Ala, A)
Glycine(Gly, G)
Alanine(Ala, A)
COO–
H
COO–
CH3
H
C
H
COO–+H3N
+H3N
+H3N
+H3N
CH3
C
H
COO–
HC
H
C
5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 5/40
Valine(Val, V)
Leucine(Leu, L)
Isoleucine(Ile, I)
Méthionine(Met, M)
Valine(Val, V)
Méthionine(Met, M)
Isoleucine(Ile, I)
Leucine(Leu, L)
COO–
CH2
H2C
S
H3C
+H3NCOO–
CH2
HCCH3
CH3
+H3NCOO–
CHH3C CH3
+H3N
C
C
H
COO–+H3N
CH3
CH3H
+H3N
CH2
C
H
COO–
C
CH3
H CH3
+H3N
CH2
C
H
COO–
CH2
S
CH3
+H3N
C
C
H
COO–
CH2
CH3H
CH3
COO–
C
H2C CH3
CH3
H
+H3N
*H
CH
CH
CH
C
fait que sa chaîne latérale est liée à la fois à l’atome d’azote et à l’atome de carbone Figure 2.9. La proline a une influence considérable dans l’architecture des protéinesparce que sa structure cyclique la rend plus limitée, du point de vue conformationspatiale, que les autres aminoacides.
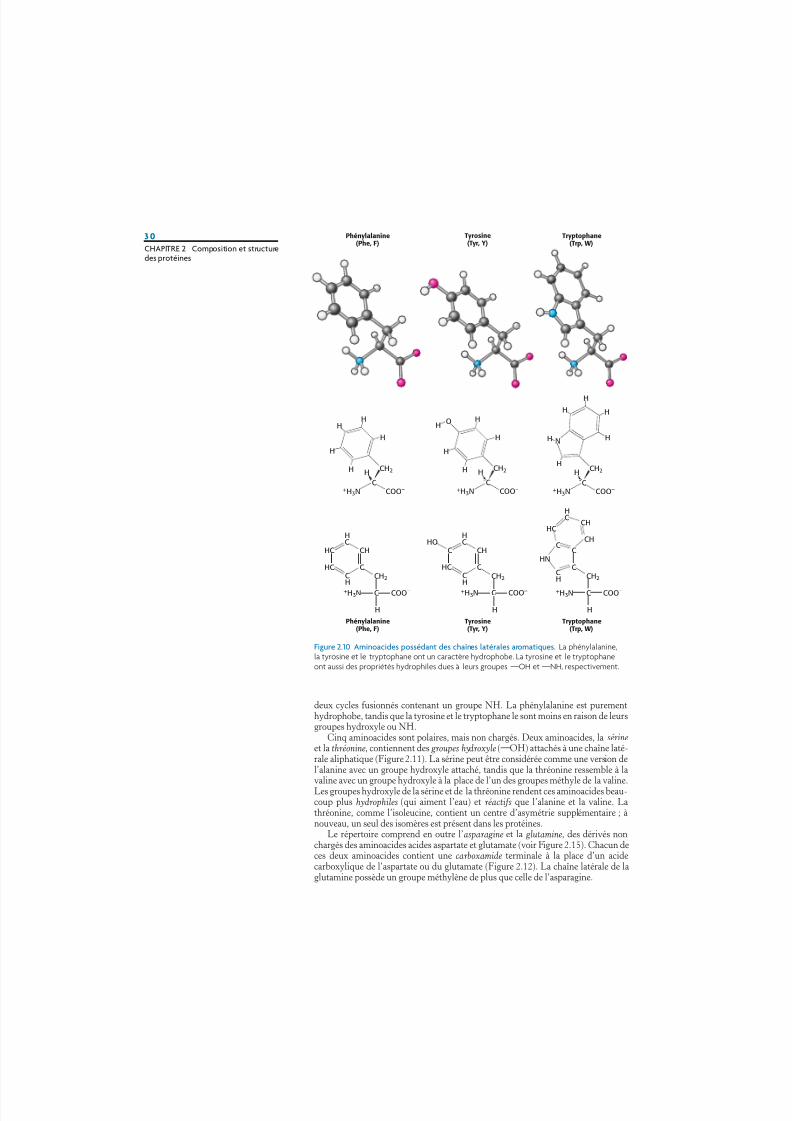
Trois aminoacides possédant des chaînes latérales aromatiques relativementsimples font partie du répertoire fondamental (Figure 2.10). La phénylalanine,comme son nom l’indique, contient un cycle phényle à la place de l’un des hydro-gènes de l’alanine. Le cycle aromatique de la tyrosine contient un groupe hydroxyle.Ce dernier est réactif, contrairement aux chaînes latérales plutôt inertes des autresaminoacides présentés jusqu’à présent. Le tryptophane possède un groupe indoleuni à un groupe méthylène (¬CH2¬) ; le groupe indole lui-même est constitué de
Figure 2.8 Aminoacides possédant des chaînes latérales aliphatiques. Le centre chiralsupplémentaire de l’isoleucine est indiqué par un astérisque.
Figure 2.9 Structure cyclique de la proline. La chaîne latérale est unie à la fois au carbone et au groupe amine.
Proline(Pro, P)
N+
H2COO–
HCH2
H2C
H2C
H2C
C
H
COO–N+
H2
CH2H2CC
2 9
5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 6/40
deux cycles fusionnés contenant un groupe NH. La phénylalanine est purementhydrophobe, tandis que la tyrosine et le tryptophane le sont moins en raison de leursgroupes hydroxyle ou NH.
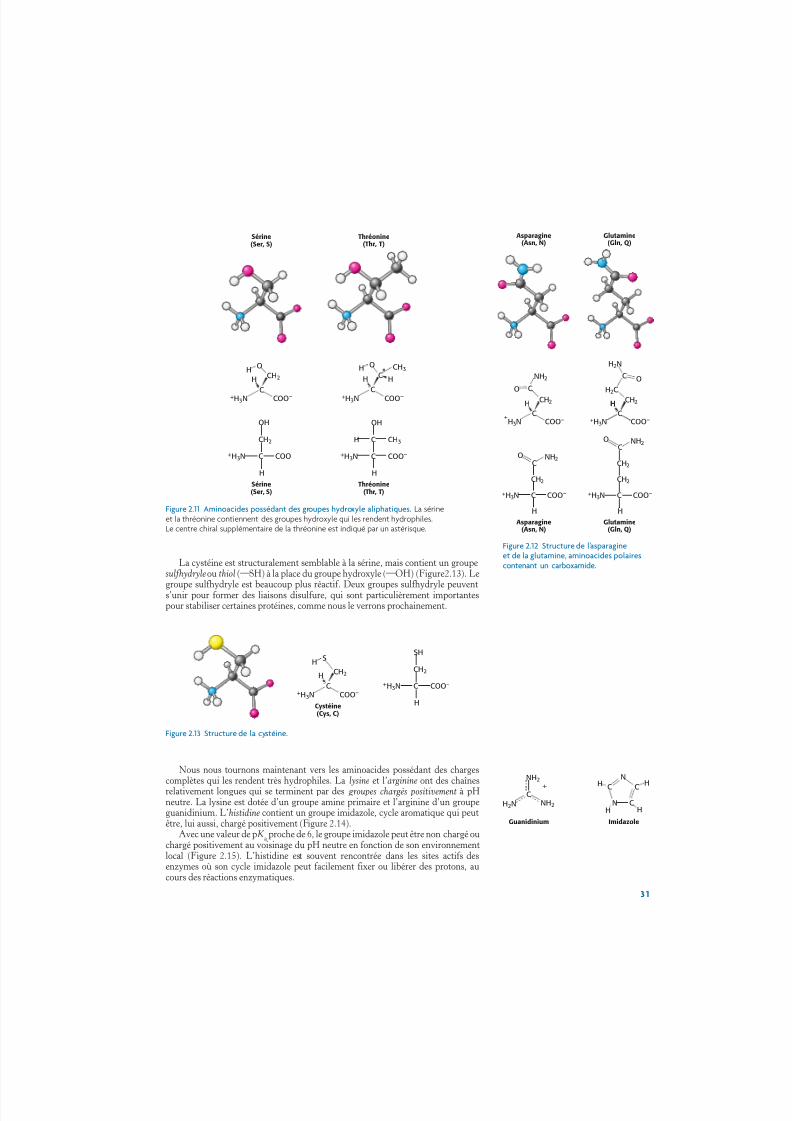
Cinq aminoacides sont polaires, mais non chargés. Deux aminoacides, la sérineet la thréonine, contiennent des groupes hydroxyle (¬OH) attachés à une chaîne laté-rale aliphatique (Figure 2.11). La sérine peut être considérée comme une version del’alanine avec un groupe hydroxyle attaché, tandis que la thréonine ressemble à la
valine avec un groupe hydroxyle à la place de l’un des groupes méthyle de la valine.Les groupes hydroxyle de la sérine et de la thréonine rendent ces aminoacides beau-coup plus hydrophiles (qui aiment l’eau) et réactifs que l’alanine et la valine. Lathréonine, comme l’isoleucine, contient un centre d’asymétrie supplémentaire ; ànouveau, un seul des isomères est présent dans les protéines.
Le répertoire comprend en outre l’asparagine et la glutamine, des dérivés nonchargés des aminoacides acides aspartate et glutamate (voir Figure 2.15). Chacun deces deux aminoacides contient une carboxamide terminale à la place d’un acidecarboxylique de l’aspartate ou du glutamate (Figure 2.12). La chaîne latérale de laglutamine possède un groupe méthylène de plus que celle de l’asparagine.
3 0
CHAPITRE 2 Composition et structuredes protéines
Figure 2.10 Aminoacides possédant des chaînes latérales aromatiques. La phénylalanine,la tyrosine et le tryptophane ont un caractère hydrophobe. La tyrosine et le tryptophaneont aussi des propriétés hydrophiles dues à leurs groupes¬OH et¬NH, respectivement.
Phénylalanine(Phe, F)
Tyrosine(Tyr, Y)
Tryptophane(Trp, W)
Phénylalanine(Phe, F)
Tyrosine(Tyr, Y)
Tryptophane(Trp, W)
+H3N COO–
CH2
OHH
H
H
H
+H3N COO–
CH2
H
H
H
H
H
+H3N COO–
CH2
NH
H
H
H
H
H
CH2
C
H
COO–+H3N
C
CH
HC
HC
HCCH
CH2
C
H
COO–+H3N
C
CH
HC
C
HCCH
HO
CH2
C
H
COO–+H3N
C
CC
HN
CH
CH
CH
HC
HC
HC
HC
H
C
5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 7/40
La cystéine est structuralement semblable à la sérine, mais contient un groupesulfhydryle ou thiol (¬SH) à la place du groupe hydroxyle (¬OH) (Figure2.13). Le
groupe sulfhydryle est beaucoup plus réactif. Deux groupes sulfhydryle peuvents’unir pour former des liaisons disulfure, qui sont particulièrement importantespour stabiliser certaines protéines, comme nous le verrons prochainement.
3 1
Figure 2.11 Aminoacides possédant des groupes hydroxyle aliphatiques. La sérineet la thréonine contiennent des groupes hydroxyle qui les rendent hydrophiles.Le centre chiral supplémentaire de la thréonine est indiqué par un astérisque.
Sérine(Ser, S)
Thréonine(Thr, T)
Sérine(Ser, S)
Thréonine(Thr, T)
+H3N COO–
CH2
OH
CH2
C
H
COO–+H3N
OH
C
C
H
COO–+H3N
OH
CH3H
+H3N COO–
COH CH3
H*
H
C
H
C
Figure 2.13 Structure de la cystéine.
Cystéine(Cys, C)
+H3N COO–
CH2
SHCH2
C
H
COO–+H3N
SH
HC
Guanidinium
NH2
CNH2H2N
+
N C
CN
C
H
H H
H
Imidazole
Figure 2.12 Structure de l’asparagineet de la glutamine, aminoacides polairescontenant un carboxamide.
Asparagine(Asn, N)
Glutamine(Gln, Q)
Asparagine(Asn, N)
Glutamine(Gln, Q)
+H3N COO–
CH2C
NH2
O
+H3N COO–
CH2H2C
C O
H2N
CH2
C
H
COO–+H3N
CO NH2
CH2
C
H
COO–+H3N
CH2
CO NH2
HC
HH
C
Nous nous tournons maintenant vers les aminoacides possédant des charges
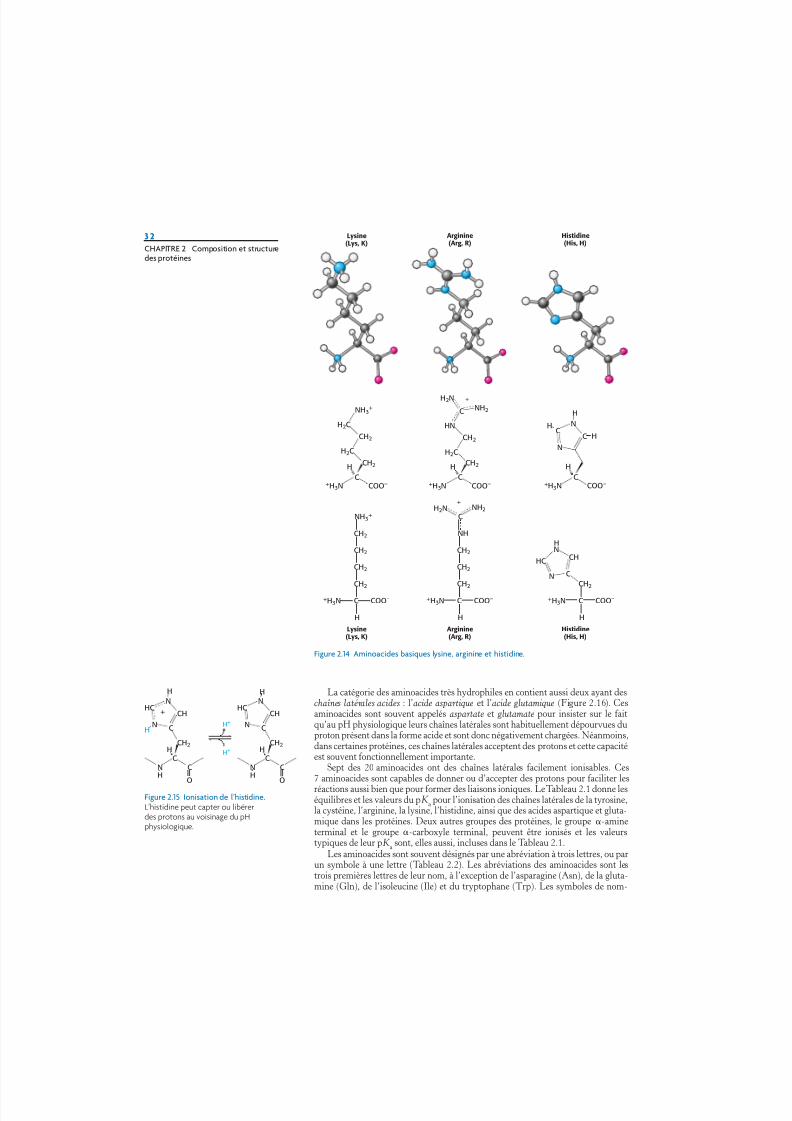
complètes qui les rendent très hydrophiles. La lysine et l’arginine ont des chaînesrelativement longues qui se terminent par des groupes chargés positivement à pHneutre. La lysine est dotée d’un groupe amine primaire et l’arginine d’un groupeguanidinium. L’histidine contient un groupe imidazole, cycle aromatique qui peutêtre, lui aussi, chargé positivement (Figure 2.14).
Avec une valeur de pK a proche de 6, le groupe imidazole peut être non chargé ouchargé positivement au voisinage du pH neutre en fonction de son environnementlocal (Figure 2.15). L’histidine est souvent rencontrée dans les sites actifs desenzymes où son cycle imidazole peut facilement fixer ou libérer des protons, aucours des réactions enzymatiques.
5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 8/40
La catégorie des aminoacides très hydrophiles en contient aussi deux ayant deschaînes latérales acides : l’acide aspartique et l’acide glutamique (Figure 2.16). Cesaminoacides sont souvent appelés aspartate et glutamate pour insister sur le faitqu’au pH physiologique leurs chaînes latérales sont habituellement dépourvues duproton présent dans la forme acide et sont donc négativement chargées. Néanmoins,dans certaines protéines, ces chaînes latérales acceptent des protons et cette capacitéest souvent fonctionnellement importante.
Sept des 20 aminoacides ont des chaînes latérales facilement ionisables. Ces
7 aminoacides sont capables de donner ou d’accepter des protons pour faciliter lesréactions aussi bien que pour former des liaisons ioniques. Le Tableau 2.1 donne leséquilibres et les valeurs du pK a pour l’ionisation des chaînes latérales de la tyrosine,la cystéine, l’arginine, la lysine, l’histidine, ainsi que des acides aspartique et gluta-mique dans les protéines. Deux autres groupes des protéines, le groupe -amineterminal et le groupe -carboxyle terminal, peuvent être ionisés et les valeurstypiques de leur pK a sont, elles aussi, incluses dans le Tableau 2.1.
Les aminoacides sont souvent désignés par une abréviation à trois lettres, ou parun symbole à une lettre (Tableau 2.2). Les abréviations des aminoacides sont lestrois premières lettres de leur nom, à l’exception de l’asparagine (Asn), de la gluta-mine (Gln), de l’isoleucine (Ile) et du tryptophane (Trp). Les symboles de nom-
3 2
CHAPITRE 2 Composition et structuredes protéines
Figure 2.14 Aminoacides basiques lysine, arginine et histidine.
Lysine(Lys, K)
Arginine(Arg, R)
Histidine(His, H)
Lysine(Lys, K)
Arginine(Arg, R)
Histidine(His, H)
+
+
H3N COO–
CH2
H2C
CH2
H2C
NH3
+H3N COO–
CH2
H2C
CH2
HN
C
H2NNH2
+
CH2
C
H
COO–+H3N
CH2
CH2
CH2
NH3+
CH2
C
H
COO–+H3N
CH2
CH2
NH
CH2N NH2
+
CH2
C
H
COO–+H3N
C
CHN
HC
N
H
+H3N COO–
C
NC
N
H
HH
HC
HC
HC
Figure 2.15 Ionisation de l’histidine.L’histidine peut capter ou libérerdes protons au voisinage du pHphysiologique.
+
CN C
H
O
H
CH2
C
CH
NHC
N
CN C
H
O
H
CH2
C
CH
NHC
NH
H H
H+
H+

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 9/40
breux aminoacides sont souvent la première lettre de leur nom (par exemple G pourla glycine et L pour la leucine) ; les autres symboles ont été admis par convention.Ces abréviations et symboles font partie du vocabulaire courant des biochimistes.
Comment ce répertoire particulier d’aminoacides s’est-il transformé enunités structurales de base des protéines ? Tout d’abord, bien que formant
un groupe, les aminoacides sont très divers ; leurs propriétés structurales et chi-miques sont très variées, conférant ainsi aux protéines l’universalité qui leur permetd’assumer un grand nombre de rôles fonctionnels. Deuxièmement, beaucoup de cesaminoacides ont eu vraisemblablement pour origine des réactions prébiotiques,c’est-à-dire des réactions qui se sont passées avant l’origine de la vie. Enfin, les autresaminoacides potentiels ont pu, tout simplement, être trop réactifs pour faire partie du
3 3
2.1 Un répertoire de 20 aminoacides
TABLEAU 2.1 Valeurs typiques du pK a des groupes ionisables dans les protéines
Groupe Acide Base pK a* typique
Groupe -carboxyle terminal 3,1
Acide aspartique 4,1Acide glutamique
Histidine 6,0
Groupe -amine terminal 8,0
Cystéine 8,3
Tyrosine 10,9
Lysine 10,8
Arginine 12,5
* Les valeurs du pK a
dépendent de la température, de la force ionique et du micro-environnement du groupe ionisable.
O
OHC
O
OHC
+N
N
H
H
SH
+
NH
HH
+N
H
HH
OH
+
N
N
N HH
H
HC
H
O
OC
–
O
OC
–
S–
N
NH
N HH
N HH
O–
N
N
N HH
H
CH
Figure 2.16 Aminoacides possédantdes chaînes latérales carboxylate.
Aspartate(Asp, D)
Glutamate(Glu, E)
Aspartate(Asp, D)
Glutamate(Glu, E)
+
H3N COO–
CH2
C
O
O–
CH2
C
H
COO–+H3N
CH2
CO O
–
CH2
C
H
COO–+H3N
CO O
–
+
H3N COO–
CH2
C
C O
O–
HC
HC
H2
TABLEAU 3.2 Abréviations des aminoacides
Abréviation Abréviation Abréviation AbréviationAminoacide à trois lettres à une lettre Aminoacide à trois lettres à une lettre
Alanine Ala A Méthionine Met M
Arginine Arg R Phénylalanine Phe F
Asparagine Asn N Proline Pro P
Acide aspartique Asp D Sérine Ser S
Cystéine Cys C Thréonine Thr T
Glutamine Gln Q Tryptophane Trp W
Acide glutamique Glu E Tyrosine Tyr Y
Glycine Gly G Valine Val V
Histidine His H Asparagine ou Asx B
Isoleucine Ile I acide aspartique
Leucine Leu L Glutamine ou Glx Z
Lysine Lys K acide glutamique

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 10/40
répertoire. Par exemple, des aminoacides tels que l’homosérine et l’homocystéinetendent à donner des formes cycliques à cinq atomes qui limitent leur utilisationdans les protéines ; les aminoacides de structure alternative rencontrés dans les pro-téines, la sérine et la cystéine, ne se cyclisent pas facilement, car leurs formescycliques sont trop petites (Figure 2.17).
2.2 Structure primaire : les aminoacides sont unispar des liaisons peptidiques pour former des chaînespolypeptidiques
Les protéines sont des polymères linéaires formés par union du groupe -carboxyle
d’un aminoacide au groupe -amine d’un autre aminoacide. Ce type de liaison estappelé liaison peptidique (mais aussi liaison amide). La formation d’un dipeptide àpartir de deux aminoacides est accompagnée de la perte d’une molécule d’eau(Figure 2.18). L’équilibre de cette réaction est très en faveur de l’hydrolyse et nonpas de la synthèse, dans la plupart des conditions. Par conséquent, la biosynthèsedes liaisons peptidiques nécessite un apport d’énergie libre. Néanmoins, les liaisonspeptidiques sont très stables cinétiquement parce que leur vitesse d’hydrolyse estextrêmement lente ; leur durée de vie en solution aqueuse, en l’absence de cataly-seur, approche 1 000 ans.
Une chaîne polypeptidique est formée par de nombreux aminoacides unis par desliaisons peptidiques ; dans un polypeptide, chaque unité dérivée d’un aminoacideest appelée résidu. Une chaîne polypeptidique a une polarité, parce que ses extrémitéssont différentes, un groupe -amine est présent à une extrémité et un groupe -car-boxyle à l’autre. Par convention, l’extrémité aminée est prise comme origine de lachaîne polypeptidique et la séquence des aminoacides d’une chaîne polypeptidiqueest alors écrite à partir du résidu qui possède le groupe amine (résidu N-terminal).
3 4
CHAPITRE 2 Composition et structuredes protéines
Figure 2.17 Réactivitéindésirable des aminoacides.Certains aminoacides ne sontpas adaptés aux protéinesen raison d’une cyclisationindésirable. L’homosérinepeut se cycliser pour formerun cycle stable à cinq atomessusceptible de conduireà un clivage de la liaisonpeptidique. La cyclisationde la sérine conduiraità la formation d’un cycletendu à quatre atomeset donc non favorable. X peutêtre un groupe amine
d’un aminoacide voisinou un autre groupepotentiellement libre.
HX+
HX+
Homosérine
Sérine
CC
X
O
CC
O
H2C
H2C
H
O
H2C
H
H2C
OH
CC
X
O
H2C
HO
H
CC
H2C
H O
O
HN
HN
HN
HN
Figure 2.18 Formation de la liaison peptidique. La liaison de deux aminoacides est
accompagnée de la perte d’une molécule d’eau.
–+ H2O
Liaison peptidique
+H3NC
C
O
O
R1H
+H3NC
C
O
O
R2H
+H3NC
C
O
R1HN
CC
O
O
H
R2H
+–
–

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 11/40
Ainsi, dans le pentapeptide Tyr-Gly-Gly-Phe-Leu (YGGFL), la tyrosine estle résidu N-terminal et la leucine est le résidu qui présente le groupe carboxyletermin al (C-terminal) (Figure 2.19). Leu-Phe-Gly-Gly-Tyr (LFGGY) est un pen-tapeptide différent, ayant des propriétés chimiques différentes.
Une chaîne polypeptidique est constituée d’une partie répétitive régulière, appe-lée chaîne principale ou squelette, et d’une partie variable, comportant des chaînes
latérales spécifiques (Figure 2.20). Le squelette polypeptidique est riche en liaisonshydrogène potentielles. Chaque résidu contient un groupe carbonyle (C“O), qui estun bon accepteur de liaisons hydrogène et, à l’exception de la proline, un groupe NH,qui est un bon donneur de liaisons hydrogène. Ces groupes entrent en interactionl’un avec l’autre, ainsi qu’avec les groupes fonctionnels des chaînes latérales pour sta-biliser des structures particulières, comme il en sera discuté Section 2.3.
La plupart des chaînes polypeptidiques naturelles contiennent entre 50 et2 000 résidus aminoacide et sont habituellement appelées protéines. La plus grandeprotéine connue est la protéine musculaire titine, qui est constituée de plus de27 000 aminoacides. Les peptides constitués d’un plus petit nombre d’aminoacidessont appelés oligopeptides, ou simplement peptides. Le poids moléculaire moyend’un résidu aminoacide est de 110 g mol1 environ ; le poids moléculaire de la plu-part des protéines est donc compris entre 5 500 et 220 000 g mol1. Nous pouvonsaussi nous référer à la masse d’une protéine, qui est exprimée en daltons ; un daltonest égal à une unité de masse atomique. Une protéine d’un poids moléculaire de50 000 a une masse de 50 000 daltons, ou 50 kd (kilodaltons).
Dans certaines protéines, la chaîne polypeptidique linéaire présente des liaisonscroisées. Les plus habituelles sont les liaisons disulfure, formées par l’oxydation d’un
3 5
2.2 Structure primaire
Dalton
Unité de masse identique à la masse
d’un atome d’hydrogène. Ainsi dénommée
en référence à John Dalton (1766-1844)
qui a développé la théorie atomique
de la matière.
Kilodalton (kd)
Unité de masse égale à 1 000 daltons.
Figure 2.19 La séquence des aminoacides a une direction. Ce schéma du pentapeptideTyr-Gly-Gly-Phe-Leu (YGGFL) montre sa séquence de l’extrémité N-terminale vers l’extrémitéC-terminale. Ce pentapeptide, la Leu-enképhaline, est un peptide opioïde qui modulela perception de la douleur. Le pentapeptide inverse, Leu-Phe-Gly-Gly-Tyr (LFGGY) estune molécule différente qui n’a pas un tel effet.
LeuPheGly Gly Tyr
RésiduN-terminal
RésiduC-terminal
+H3N CN
C
O
HNH
CN
CC C
C
H H
O
HNH
HH
O
O
C
O
OCH
H2CH
H2CH2C
OH
HCCH3
CH3
CH
–
Figure 2.20 Constituants d’une chaîne polypeptidique. Une chaîne polypeptidique estconstituée d’un squelette constant (représenté en noir) et de chaînes latérales variables(représentées en vert).
CC CO
C
O
CC
O
HR1
H
HR3
R2
CC
O
CC
OH
HR5
R4
NH
HN
HN
HN
NH

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 12/40
couple de résidus cystéine (Figure 2.21). Le bloc formé par la liaison de deux cys-téines est appelé cystine. Les protéines extracellulaires ont souvent plusieurs liai-sons disulfure, tandis que les protéines intracellulaires n’en ont habituellement pas.Rarement, des liaisons croisées autres que des liaisons disulfure et dérivées d’autreschaînes latérales sont présentes dans les protéines. Par exemple, les fibres de colla-gène du tissu conjonctif sont renforcées par ces autres types de liaisons croisées, demême que la fibrine des caillots sanguins.
Les protéines ont des séquences spécifiques d’aminoacidesdéterminées par les gènesEn 1953, Frederick Sanger a déterminé la séquence des aminoacides de l’insuline,une hormone protéique (Figure 2.22). Ce travail a été un événement marquant de labiochimie, car il a montré pour la première fois qu’une protéine a une séquence d’amino-acides parfaitement définie constituée uniquement d’aminoacides L unis par des liaisons peptidiques. Cette découverte a incité d’autres scientifiques à effectuer l’étude de laséquence de toute une série de protéines. À l’heure actuelle les séquences complètesd’aminoacides de plus de 2 000 000 de protéines sont connues. Le fait marquant estque chaque protéine a une séquence unique d’aminoacides, parfaitement définie . Laséquence des aminoacides d’une protéine est appelée structure primaire.
Une série d’études décisives à la fin des années 1950 et au début des années 1960a révélé que la séquence des aminoacides des protéines était déterminée par laséquence nucléotidique des gènes. La séquence des nucléotides du DNA déterminela séquence complémentaire des nucléotides du RNA qui à son tour, détermine laséquence des aminoacides d’une protéine. En particulier, chacun des vingt amino-acides du répertoire est codé par une ou plusieurs séquences spécifiques de troisnucléotides (Section 5.5).
Il est important de connaître la séquence d’aminoacides d’une protéine pourplusieurs raisons. Tout d’abord, la connaissance de la séquence d’une protéine est
3 6
CHAPITRE 2 Composition et structuredes protéines
Figure 2.21 Liaisons croisées. La formation d’une liaison disulfure à partir de deux résiduscystéine est une réaction d’oxydation.
+ 2 e–+2 H+Oxydation
Réduction
Cystéine
Cystéine
Cystine
NC
C
CH2
S
H
OH
H
NC
C
H2C
S
H
OH
H
NC
C
CH2
S
H
OH
NC
C
H2C
S
H
OH
Figure 2.22 Séquence d’aminoacides de l’insuline bovine.
Gly-Ile-Val-Glu-Gln-Cys-Cys-Ala-Ser-Val-Cys-Ser-Leu-Tyr-Gln-Leu-Glu-Asn-Tyr-Cys-AsnChaîne A
Chaîne B
Phe-Val-Asn-Gln-His-Leu-Cys-Gly-Ser-His-Leu-Val-Glu-Ala-Leu-Tyr-Leu-Val-Cys-Gly-Glu-Arg-Gly-Phe-Phe-Tyr-Thr-Pro-Lys-Ala
5 10 15 21
5 10 15 20 25 30
S
S
S S
S
S

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 13/40
souvent essentielle pour élucider le mécanisme d’action de cette dernière (parexemple, le mécanisme catalytique d’un enzyme). En outre, des protéines possé-dant de nouvelles propriétés peuvent être créées en modifiant la séquence de pro-téines connues. Deuxièmement, la séquence d’aminoacides détermine la structuretridimensionnelle des protéines. La séquence d’aminoacides représente le lienentre le message génétique du DNA et la structure tridimensionnelle des pro-téines, qui leur confère une fonction biologique. Les analyses des relations entre laséquence d’aminoacides et la structure tridimensionnelle des protéines permettentde découvrir les règles qui gouvernent le reploiement des chaînes polypeptidiques.Troisièmement, la détermination de la séquence fait partie des investigations depathologie moléculaire, domaine de la médecine en croissance rapide. En effet, desmodifications de la séquence d’aminoacides peuvent conduire à une fonctionanormale de la protéine et à un état pathologique. Des maladies graves, et parfoismortelles, telles que l’anémie falciforme (ou drépanocytose p. 195) ou la muco-
viscidose, peuvent résulter du changement d’un seul aminoacide dans une pro-téine. Quatrièmement, la séquence d’une protéine révèle beaucoup d’informationspermettant de retracer l’histoire de son évolution (Chapitre 6). Les protéines ne seressemblent par la séquence de leurs aminoacides, que lorsqu’elles ont un ancêtrecommun. Par conséquent, les événements moléculaires de l’Évolution peuvent êtreretracés à partir des séquences des aminoacides. La paléontologie moléculaire estun domaine de recherche en expansion.
Les chaînes polypeptidiques sont flexibles bien que limitéesdans leur conformationL’examen de la géométrie du squelette des protéines révèle plusieurs caractèresimportants. Tout d’abord, la liaison peptidique est essentiellement plane (Figure 2.23).Ainsi, pour un couple d’aminoacides unis par une liaison peptidique, six atomes sesituent dans un même plan : l’atome de carbone et le groupe CO du premier ami-noacide, le groupe NH et le carbone du deuxième aminoacide. La nature de la liai-son chimique au sein d’un peptide rend compte du caractère plan de la liaison. Laliaison peptidique a un important caractère de double liaison qui empêche la rotationautour d’elle-même et contraint ainsi la conformation du squelette du peptide.
3 7
2.2 Structure primaire
Figure 2.23 Les liaisons peptidiques sont planes. Dans un couple d’aminoacides liés,six atomes (C
, C, O, N, H et C
) sont dans un plan. Les chaînes latérales sont représentées
sous forme de boules vertes.
N
H
O
CαC
Cα
CC
NC
H
O
CC
N+
C
H
O–
Structures résonantes de la liaison peptidique
Ce caractère de double liaison s’exprime aussi dans la longueur de la liaisonentre les groupes CO et NH. La distance C¬N dans une liaison peptidique esttypiquement 1,32 Å, ce qui se situe entre la valeur attendue pour une simple liaison

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 14/40
C¬N (1,49 Å) et une double liaison C“N (1,27 Å), comme le montre laFigure 2.24. Finalement, la liaison peptidique n’est pas chargée, ce qui rend pos-sible la formation de structures globulaires très compactes par les polymèresd’aminoacides liés par des liaisons peptidiques.
Deux configurations sont possibles pour une liaison peptidique plane. Dans laconfiguration trans, les deux atomes de carbone sont situés sur des côtés opposésde la liaison peptidique. Dans la configuration cis ces groupes sont du même côté dela liaison peptidique. Presque toutes les liaisons peptidiques sont trans. Cette préfé-rence de trans sur cis peut être expliquée par le fait que les chocs stériques entregroupes attachés aux atomes de carbone qui gênent la formation de la forme cis,ne se produisent pas dans la configuration trans (Figure 2.25). Les liaisons pepti-diques cis les plus communes sont de loin les liaisons X¬Pro. De telles liaisons pré-sentent une préférence réduite pour la configuration trans parce que l’azote de laproline est lié à deux atomes de carbone tétraédriques, ce qui limite les différencesstériques entre les formes trans et cis (Figure 2.26).
3 8
CHAPITRE 2 Composition et structuredes protéines
Figure 2.24 Longueurs typiques de liaison, au sein d’une unité peptidique. L’unitépeptidique est représentée dans la configuration trans.
1 , 3 2 Å
1,24 Å
1 ,4 5 Å 1 ,5 1 Å
H
Cα
CαC
N
1,0 Å
O
Figure 2.25 Liaisons peptidiques trans et cis. La forme trans est fortement favoriséeen raison de l’encombrement stérique qui apparaît dans la forme cis.
Trans Cis
Figure 2.26 Liaisons X¬Pro trans et cis. Les énergies de ces formes sont similairesparce qu’un encombrement stérique apparaît dans les deux formes.
Trans Cis

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 15/40
Contrastant avec la liaison peptidique les liaisons entre les groupes amine et l’atomede carbone et entre l’atome de carbone et le groupe carbonyle sont des liaisonssimples typiques. Les deux unités peptidiques rigides adjacentes peuvent tournerautour de ces liaisons et prendre diverses orientations. Cette liberté de rotation de deuxliaisons de chaque résidu aminoacide permet aux protéines de se reployer de nombreusesmanières différentes. Les rotations selon ces liaisons peuvent être définies par des anglesde torsion (Figure 2.27). L’angle de rotation autour de la liaison entre l’azote et l’atomede carbone est appelé phi (). L’angle de rotation autour de la liaison entre le carbone et l’atome de carbone du carbonyle est appelé psi (). Une rotation dans le senshoraire autour de l’une ou l’autre liaison, observée de l’atome d’azote vers l’atome decarbone ou du groupe carbonyle vers l’atome de carbone correspond à une valeurpositive. Les angles et déterminent la configuration de la chaîne polypeptidique.
Est-ce que toutes les combinaisons de et sont possibles ? GopalasamudramRamachandran a pu montrer que beaucoup de combinaisons sont interdites à cause
de collisions stériques entre atomes. Les valeurs permises peuvent être visualiséessur un graphe bidimensionnel appelé diagramme de Ramachandran (Figure 2.28).Les trois quarts des combinaisons possibles de et sont simplement exclues àcause de chocs stériques locaux. Les empêchements stériques, et le fait que deux atomesne peuvent pas être à la même place au même moment peuvent représenter un principe puissant d’organisation.
3 9
2.2 Structure primaire
Angle de torsion
Mesure de la rotation autour d’une liaison,
allant habituellement de 180° à 180°.
Les angles de torsion sont parfois appelés
angles dièdres.
Figure 2.27 Rotation autour des liaisons dans un polypeptide. La structure de chaqueaminoacide d’un polypeptide peut être ajustée par rotation autour de deux simples liaisons.(A) Phi () est l’angle de rotation autour de la liaison entre l’atome d’azote et l’atomede carbone, tandis que psi () est l’angle de rotation autour de la liaison entre l’atomede carbone et l’atome de carbone du carbonyle. (B) Vue de haut en bas de l a liaisonentre l’atome d’azote et l’atome de carbone , montrant comment est mesuré.(C) Vue de haut en bas de la liaison entre l ’atome de carbone et l’atome de carbonedu carbonyle montrant comment est déterminé.
= +85° = −80°
(B) (C)
NC
CO
RHN
CC
OH
RH
NH H
CC
O
RH
(A)
Figure 2.28 Diagramme de Ramachandran montrant les valeurs de et de . Toutesles valeurs de et de ne peuvent pas être prises sans qu’il y ait des collisionsentre les atomes. Les régions les plus favorables sont représentées en vert foncé, les régionslimite sont représentées en ver t clair. La structure de droite est défavorable en raisond’empêchements stériques.
+180
+180
120
120
60
600
0
−60
−60
−120
−120−180
−180
( = 90°, = −90° )Défavorisé

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 16/40
La capacité des polymères biologiques comme les protéines de se reployer (ou sereplier) en structures bien définies est remarquable sur le plan thermodynamique.Un polymère non reployé prend la forme d’une série de boucles aléatoires : chaquecopie d’un même polymère non reployé aura une conformation différente.L’entropie favorable qui est associée à un mélange de nombreuses conformationss’oppose au reploiement, et doit être vaincue par des interactions favorisant la formereployée. Ainsi des polymères hautement flexibles ayant un grand nombre deconformations possibles ne peuvent pas se reployer en une structure unique. Enréalité, la rigidité de l’unité peptidique, et l’ensemble restreint des angles et permis,limitent le nombre des structures possibles pour la forme déployée, et ceci suffisamment pour permettre au reploiement des protéines de s’effectuer.
2.3 Structure secondaire : les chaînes polypeptidiquespeuvent se reployer en structures régulièrestelles que l’hélice alpha, le feuillet bêta, les coudeset les boucles
Une chaîne polypeptidique peut-elle se reployer en une structure régulièrementrépétitive ? En 1951, Linus Pauling et Robert Corey ont proposé deux structurespériodiques appelées hélice (hélice alpha) et feuillet plissé (feuillet plissé bêta).Par la suite, d’autres structures telles que le coude et la boucle oméga () ont étéidentifiées. Bien que non périodiques, ces structures courantes en coude ou enboucle sont bien définies et contribuent avec les hélices et les feuillets à la for-mation de la structure finale de la protéine.
Les hélices , les brins , et les coudes sont formés par une configuration régu-lière de liaisons hydrogène entre les groupes peptidiques N¬H et C“O des rési-dus d’aminoacides qui sont près les uns des autres dans la séquence linéaire. De telssegments repliés forment la structure secondaire.
L’hélice alpha est une structure enroulée stabilisée par des liaisonshydrogène intrachaîneDans leur évaluation des structures potentielles, Pauling et Corey se sontdemandé quelles étaient les conformations des peptides stériquement permises, etquelles étaient celles qui exploitaient le plus complètement la capacité des groupesNH et CO de la chaîne principale à former des liaisons hydrogène. La premièredes structures qu’ils ont proposées, l’hélice , est une structure en bâtonnet(Figure 2.29). La chaîne polypeptidique principale étroitement enroulée forme lapartie interne du bâtonnet et les chaînes latérales se disposent à l’extérieur en unarrangement hélicoïdal. L’hélice est stabilisée par des liaisons hydrogène entreles groupes NH et CO de la chaîne principale. En particulier, le groupe CO dechaque aminoacide contracte une liaison hydrogène avec le groupe NH de l’amino-acide situé quatre résidus plus en avant dans la séquence (Figure 2.30). Ainsi, àl’exception des aminoacides proches des extrémités d’une hélice , tous les groupesCO et NH de la chaîne principale sont unis par une liaison hydrogène . Chaque résiduest disposé par rapport au suivant selon une élévation (aussi appelée translation) de1,5 Å le long de l’axe de l’hélice et une rotation de 100 degrés, ce qui donne 3,6 rési-dus aminoacide par tour d’hélice. Ainsi, les aminoacides distants de trois ou quatre
résidus dans la séquence linéaire sont spatialement très proches l’un de l’autre dansune hélice . En revanche, les aminoacides distants de deux résidus dans laséquence linéaire sont situés sur des côtés opposés de l’hélice et, ainsi, ne peuventpas entrer en contact. Le pas de l’hélice , produit de la translation (1,5 Å) et dunombre de résidus par tour (3,6), est de 5,4 Å. Le sens d’enroulement d’une hélicepeut être droit (dans le sens des aiguilles d’une montre) ou gauche (dans le sensinverse des aiguilles d’une montre). Le diagramme de Ramachandran montre queles hélices droites et les hélices gauches figurent toutes deux parmi les conforma-tions permises (Figure 2.31). Cependant, les hélices droites sont énergétiquement
4 0
CHAPITRE 2 Composition et structuredes protéines
Sens d’enroulementDécrit la direction dans laquelle
une structure hélicoïdale tourne
par rapport à son axe. Si, vue vers le bas
suivant l’axe de l’hélice, la chaîne tourne
dans le sens des aiguilles d’une montre,
elle a un sens d’enroulement droit.
Si l’enroulement se fait dans le sens
inverse des aiguilles d’une montre, le sens
est gauche.

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 17/40
plus favorables en raison d’un encombrement stérique moindre entre les chaîneslatérales et la chaîne principale. Presque toutes les hélices rencontrées dans les pro-téines sont droites. Dans les représentations schématiques des protéines, les hélices sont représentées par des rubans enroulés ou des bâtonnets (Figure 2.32).
Pauling et Corey ont prédit la structure de l’hélice six ans avant qu’elle ne soitobservée grâce aux rayons X dans la structure de la myoglobine. L’élucidation de lastructure de l’hélice a été un événement marquant de la biologie moléculaire car ellea démontré que la conformation d’une chaîne polypeptidique pouvait être préditelorsque les propriétés de ses constituants étaient rigoureusement connues.
La proportion des hélices dans les protéines varie beaucoup, de zéro à presque100 %. Par exemple, environ 75 % des résidus de la ferritine, protéine qui contribue
Figure 2.29 Structure de l’hélice . (A) Représentation sous forme de ruban montrantles atomes de carbone et les chaînes latérales (en vert). (B) Une vue latérale d’un modèleéclaté décrit les liaisons hydrogène (lignes pointillées) entre les groupes NH et CO.(C) Une vue à partir d’une extrémité montre l’enroulement du squelette, ainsi que la partieintérieure de l’hélice et les chaînes latérales (en vert) qui se projettent vers l’extérieur.(D) Une vue d’un modèle compact d’un segment de C montre la partie interne étroitementcompactée de l’hélice.
(C)(B)
(D)
(A)
Figure 2.30 Schéma des liaisons hydrogène dans une hélice. Dans l’hélice , le groupeCO du résidu i forme une liaison hydrogène avec le groupe NH du résidu i 4.
NC
CN
CC
NC
CN
CC
NC
CN
H
HO
H O
H
OH
O
O
R i +2 R i +4H H H
R i +1
R i
H R i +3H
CC
HO
R i +5H
Figure 2.31 Diagramme de Ramachandranpour les hélices. Les hélices droiteset les hélices gauches se situent toutesdeux dans des régions de conformationspermises dans le diagrammede Ramachandran. Cependant,pratiquement toutes les hélices des protéines sont droites.
+180
+180
120
120
60
600
0
−60
−60
−120
−120−180
−180
Hélice droite(habituelle)
Hélice gauche(très rare)
4 1

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 18/40
à mettre le fer en réserve, sont engagés dans des hélices (Figure 2.33). À peu près 25 %de toutes les protéines solubles sont composées d’hélices connectées par des boucleset des coudes de la chaîne polypeptidique. Les hélices formées d’un seul tenant ontgénéralement moins de 45 Å de long. Un grand nombre de protéines qui traversentles membranes biologiques contiennent, elles aussi, des hélices .
Les feuillets bêta sont stabilisés par des liaisons hydrogèneentre les brins polypeptidiquesPauling et Corey ont proposé un autre motif structural périodique qu’ils ont appelé feuillet plissé ( parce qu’il était la seconde structure qu’ils élucidaient, l’hélice ayant été la première). Le feuillet plissé (ou plus simplement le feuillet ) diffèrenotablement de l’hélice qui a la forme d’un bâtonnet. Il est composé de deux ouplusieurs chaînes polypeptidiques appelées brins . Un brin , est presque totale-ment étiré, et non pas étroitement enroulé comme une hélice . Toute une série destructures étirées sont stériquement permises (Figure 2.34).
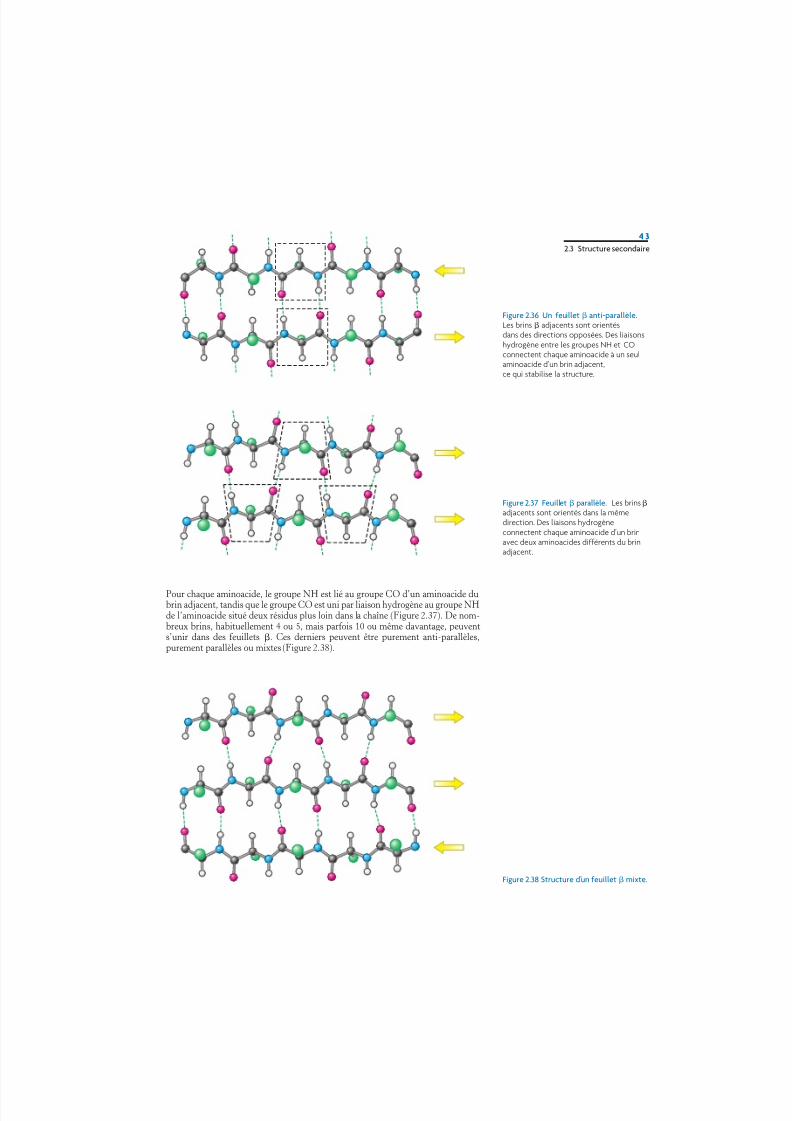
La distance entre les aminoacides adjacents le long d’un brin est approxima-tivement de 3,5 Å, contrastant avec la distance de 1,5 Å dans une hélice . Deplus, les chaînes latérales des aminoacides adjacents pointent dans des directionsopposées (Figure 2.35). Un feuillet plissé est constitué par l’union par des liai-sons hydrogène de deux ou de plusieurs brins situés les uns à côté des autres. Leschaînes adjacentes d’un feuillet peuvent être de sens opposé (feuillet anti-parallèle) ou de même sens (feuillet parallèle). Dans l’arrangement anti-parallèle,les groupes NH et CO de chaque aminoacide sont respectivement unis par des liai-sons hydrogène aux groupes CO et NH d’un partenaire de la chaîne adjacente(Figure 2.36). Dans l’arrangement parallèle, la disposition des liaisons hydrogèneest légèrement plus compliquée.
4 2
CHAPITRE 2 Composition et structuredes protéines
Figure 2.33 Une Protéineessentiellement-hélicoïdale.
La ferritine, protéine de mise en réservedu fer, est construite à partir d’un faisceaud’hélices . [Dessiné d’après 1AEW.pdb.]
Figure 2.32 Représentations schématiques des hélices. (A) Modèle éclaté.(B) Représentation sous forme d’un ruban. (C) Représentation sous forme d’un cylindre.
(A) (B) (C)
Figure 2.34 Diagramme de Ramachandranpour les brins . La zone rouge représenteles conformations stériquement permisesdes structures étirées semblables au brin .
+180
+180
120
120
60
600
0
−60
−60
−120
−120−180
−180
Brins bêta
Figure 2.35 Structure d’un brin . Les chaînes latérales (en vert) sont situées alternativementau-dessus et au-dessous du plan du brin.
5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 19/40
Pour chaque aminoacide, le groupe NH est lié au groupe CO d’un aminoacide dubrin adjacent, tandis que le groupe CO est uni par liaison hydrogène au groupe NHde l’aminoacide situé deux résidus plus loin dans la chaîne (Figure 2.37). De nom-breux brins, habituellement 4 ou 5, mais parfois 10 ou même davantage, peuvents’unir dans des feuillets . Ces derniers peuvent être purement anti-parallèles,purement parallèles ou mixtes (Figure 2.38).
4 3
2.3 Structure secondaire
Figure 2.36 Un feuillet anti-parallèle.Les brins adjacents sont orientésdans des directions opposées. Des liaisonshydrogène entre les groupes NH et COconnectent chaque aminoacide à un seulaminoacide d’un brin adjacent,ce qui stabilise la structure.
Figure 2.37 Feuillet parallèle. Les brinsadjacents sont orientés dans la mêmedirection. Des liaisons hydrogèneconnectent chaque aminoacide d’un brinavec deux aminoacides différents du brinadjacent.
Figure 2.38 Structure d’un feuillet mixte.

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 20/40
Dans les représentations schématiques, les brins sont habituellement repré-sentés par de larges flèches pointant dans la direction de l’extrémité C-terminalepour indiquer le type de feuillet formé, parallèle ou anti-parallèle. Plus structura-lement divers que les hélices , les feuillets peuvent être presque plats, mais laplupart d’entre eux adoptent une forme quelque peu tordue (Figure 2.39). Lefeuillet est un élément structural important de nombreuses protéines. Parexemple, les protéines qui fixent les acides gras, importantes pour le métabolismelipidique, sont construites presque entièrement de feuillets (Figure 2.40).
Les chaînes polypeptidiques peuvent changer de directionen formant des coudes ou des bouclesLa plupart des protéines ont des formes compactes, globulaires, dues à de fréquentschangements de direction de leurs chaînes polypeptidiques. Un certain nombre deces derniers sont effectués par un élément structural courant appelé coude (connuaussi sous le nom de coude ou de coude en épingle à cheveux) représenté dans laFigure 2.41. Dans de nombreux coudes, le groupe CO d’un résidu i d’un polypep-tide est uni par liaison hydrogène au groupe NH du résidu i 3. Cette interactionstabilise les changements abrupts de direction de la chaîne polypeptidique. Dansd’autres cas, des structures plus élaborées sont responsables des changements dedirection de la chaîne. Ces dernières sont appelées boucles ou parfois boucles (boucles oméga) pour suggérer leur forme d’ensemble. Contrairement aux hélices et
aux feuillets, les boucles n’ont pas de structures régulières périodiques. Néanmoins,les structures des boucles sont souvent rigides et bien définies (Figure 2.42). Lescoudes et les boucles se situent invariablement à la surface des protéines et participentdonc souvent aux interactions entre les protéines et d’autres molécules.
Figure 2.39 Feuillet tordu. (A) Modèleéclaté. (B) Modèle schématique.(C) La même vue schématique est montréeaprès une rotation de 90 degréspour illustrer cette torsion plus clairement.
(B) (C)(A)
Figure 2.40 Protéine richeen feuillets . Structured’une protéine qui fixe les acides gras.[Dessiné d’après 1FTP.pdb.]
Figure 2.41 Structure d’un coude inverse.Le groupe CO du résidu i de la chaînepolypeptidique est uni par liaisonhydrogène au groupe NH du résidu i 3afin de stabiliser le coude.
i
i + 1i + 2
i + 3
Figure 2.42 Boucles à la surfaced’une protéine. Cette partie
d’une molécule d’anticorps a des bouclesde surface (représentées en rouge)qui favorisent les interactions avec d’autresmolécules. [Dessiné d’après 7FTP.pdb.]
4 4

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 21/40
Les protéines fibreuses fournissent un support structuralpour les cellules et les tissusDes espèces particulières d’hélices sont présentes dans deux protéines l’-kératine etle collagène. Ces protéines forment de longues f ibres qui jouent un rôle structural.
L’-kératine, qui est le composant principal de la laine et des cheveux estconstituée de deux hélices droites enroulées l’une autour de l’autre pour formerune espèce de superhélice gauche appelée hélice surenroulée ou « coiled coil ».L’-kératine est membre d’une superfamille de protéines appelées protéines sur-enroulées (Figure 2.43). Dans ces protéines deux ou plusieurs hélices peuvents’enrouler l’une autour de l’autre pour former une structure très stable qui peutavoir une longueur de 1 000 Å (100 nm, ou 0,1 mm) ou plus. Il y a approximative-ment 60 membres de cette famille chez l’homme. Elle comprend les filamentsintermédiaires, protéines qui participent à la structure du cytosquelette (un écha-faudage interne de la cellule), et les protéines du muscle myosine et tropomyosine
(Section 34.2). Les membres de cette famille sont caractérisés par une région cen-trale de 300 aminoacides qui contient des répétitions imparfaites d’une séquencede sept aminoacides appelée répétition en heptade.
Les deux hélices de l’-kératine sont associées par des interactions faibles tellesles forces de van der Waals et les interactions ioniques. Ces interactions sont facili-tées par le fait que le surenroulement gauche modifie les deux hélices droites detelle sorte qu’il y a 3,5 résidus par tour au lieu de 3,6. Ainsi, la configuration desinteractions des chaînes latérales des aminoacides peut être répétée tous les septrésidus, formant ce qu’on appelle des répétitions en heptade. Deux hélices ayant detelles répétitions peuvent interagir l’une avec l’autre, si les répétitions sont complé-mentaires (Figure 2.44). Par exemple, les résidus répétés peuvent être hydrophobes,favorisant les interactions de van der Waals, ou bien posséder des charges opposées,permettant des interactions ioniques. De plus, les deux hélices peuvent être reliéespar des liaisons disulfure formées par des résidus cystéine voisins. La liaison deshélices est responsable des propriétés physiques de la laine, un exemple d’-kéra-tine. La laine est extensible et peut être étirée jusqu’à presque deux fois sa longueurparce que les hélices s’étirent en rompant les interactions f aibles entre hélices voi-sines. Cependant, les liaisons disulfure sont covalentes et résistent sans se casser ;elles permettent un retour de la fibre vers son état original une fois que l’étirement
est relâché. Le nombre de liaisons disulfure inter-chaînes définit plus complète-ment les propriétés des fibres. Les cheveux et la laine qui ont peu de liaisons inter-chaînes sont flexibles. Les cornes, griffes, et sabots ont plus de liaisonsinter-chaînes, et sont bien plus durs.
Un type différent d’hélice est présent dans le collagène, la protéine la plus abon-dante chez les mammifères. Le collagène est le composant fibreux majoritaire de lapeau, des os, des tendons, du cartilage, et des dents. Cette protéine extracellulaireest une molécule en forme de bâtonnet d’à peu près 3 000 Å de long et de seulement15 Å de diamètre. Elle contient trois chaînes polypeptidiques hélicoïdales, chacunelongue d’environ 1 000 résidus. Une glycine est présente tous les trois résidus de la
4 5
2.3 Structure secondaire
Figure 2.43 Une structure en hélice surenroulée. (A) Modèle compact. (B) Diagrammeen ruban. Les deux hélices s’enroulent l’une autour de l’autre pour former
une superhélice. De telles structures sont retrouvées dans beaucoup de protéines,par exemple les kératines des cheveux, des plumes, des griffes et des cornes.
[Dessiné d’après 1CIG.pdb.]
Figure 2.44 Répétitions en heptadedans une protéine surenroulée.
Un résidu sur sept (c’est-à-direchaque septième résidu) de chaque héliceest une leucine. Les deux hélices sontmaintenues ensemble par des interactionsde van der Waals principalemententre résidus leucine.[Dessiné d’après 2ZTA.pdb.]
Résidu
leucine (Leu)
Leu
Leu
Leu
Leu
Leu
Leu
Leu
C C
NN

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 22/40
séquence d’aminoacides, et la séquence glycine-proline-hydroxyproline se répètefréquemment (Figure 2.45). L’hydroxyproline est un dérivé de la proline qui porteun groupe hydroxyle à la place de l’un des hydrogènes du cycle pyrrolidine.
L’hélice de collagène a des propriétés différentes des celle de l’hélice . Il n’y apas de liaisons hydrogène au sein d’une chaîne de collagène. Au contraire, l’hélice decollagène est stabilisée par répulsion stérique des cycles pyrrolidine des résidus proline ethydroxyproline (Figure 2.46). Les cycles pyrrolidine s’éloignent les uns des autresquand la chaîne polypeptidique prend sa forme hélicoïdale, qui a environ trois rési-dus par tour. Trois chaînes s’enroulent ensemble pour former un cable superhélicoï-dal qui est stabilisé par des liaisons hydrogène entre les chaînes. Les liaisonshydrogène se forment entre les groupes peptidiques des NH des résidus glycine etCO des résidus des autres chaînes. Les groupes hydroxyle des résidus hydroxypro-line participent aussi à la formation des liaisons hydrogène, et c’est l’absence desgroupes hydroxyle qui est responsable des symptômes du scorbut (Section 27.5).
L’intérieur du câble hélicoïdal triple-brin est très encombré, ce qui explique lanécessité de la présence d’une glycine à chaque troisième position sur chaque brin(Figure 2.47A) car le seul résidu qui peut « tenir » en position interne est la glycine. Lerésidu aminoacide de chaque côté de la glycine est situé à l’extérieur du câble, où il
y a de la place pour les volumineux cycles des résidus proline et hydroxyproline(Figure 2.47B).
2.4 Structure tertiaire : les protéines hydrosolublesse reploient en des structures compactesprésentant des cores non polaires
Examinons maintenant comment sont groupés les aminoacides dans une protéine.Des études cristallographiques par diffraction des rayons X ou par résonancemagnétique nucléaire (Section 3.6) ont révélé les structures tridimensionnelles
4 6
31-Gly-Pro-Met-Gly-Pro-Ser-Gly-Pro-Arg-22-Gly-Leu-Hyp-Gly-Pro-Hyp-Gly-Ala-Hyp-31-Gly-Pro-Gln-Gly-Phe-Gln-Gly-Pro-Hyp-40-Gly-Glu-Hyp-Gly-Glu-Hyp-Gly-Ala-Ser-49-Gly-Pro-Met-Gly-Pro-Arg-Gly-Pro-Hyp-58-Gly-Pro-Hyp-Gly-Lys-Asn-Gly-Asp-Asp-
Figure 2.45 Séquence d’aminoacidesd’une partie d’une chaîne de collagène.Un résidu sur trois est une glycine(c’est-à-dire chaque troisième résidu).La proline et l’hydroxyproline sont
également abondantes.
Figure 2.46 Conformation d’un seul brind’une triple hélice de collagène.
ProPro
ProProGly
Gly
Figure 2.47 Structure de la protéine collagène. (A) Modèle compactdu collagène. Chaque brin est représenté en une couleur différente.(B) Modèle en section du collagène. Chaque brin est lié aux deuxautres brins par des ponts hydrogène. L’atome de carbone d’un résidu glycine est repéré par un G. Un résidu sur trois doit êtreune glycine parce qu’il n’y a pas suf fisamment de placedans le centre de l’hélice.
(A)
GG
G
(B)

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 23/40
détaillées de milliers de protéines. Nous commencerons ici par une présentation dela myoglobine, première protéine à avoir été vue à un niveau de résolution atomique.
La myoglobine, le transporteur d’oxygène du muscle, est une chaîne polypepti-dique unique de 153 aminoacides (voir Chapitre 7). La capacité de la myoglobine àfixer l’oxygène dépend de la présence de l’hème, groupement prosthétique (c’est-à-dire auxiliaire) non polypeptidique, constitué d’une protoporphyrine IX et d’unatome de fer central. La myoglobine est une molécule extrêmement compacte. Ses
dimensions extérieures sont de 45 35 25 Å, soit un ordre de grandeur de moinsque la longueur qu’aurait la molécule si elle était totalement étirée (Figure 2.48).Environ 70 % de la chaîne principale est reployée en hélices, et la plus grande par-tie du reste de la chaîne forme des coudes et des boucles entre les hélices.
Le reploiement de la chaîne principale de la myoglobine, de même que celuid’autres protéines, est complexe et dépourvu de symétrie. La structure d’ensemblede la chaîne polypeptidique d’une protéine est considérée comme sa structure ter-tiaire. Un principe unificateur ressort de la distribution des chaînes latérales. Le faitmarquant est que la partie interne est constituée presque entièrement de résidus non polaires , tels que la leucine, la valine, la méthionine et la phénylalanine(Figure 2.49). Les résidus chargés, tels que l’aspartate, le glutamate, la lysine et
4 7
2.4 Structure tertiaire
Figure 2.49 Distributiondes aminoacides dans la myoglobine.
(A) Modèle compact de la myoglobine ;
les aminoacides hydrophobes sont en jaune,les aminoacides chargés, sont en bleuet les autres en blanc. Remarquez quela surface de la molécule contient beaucoupd’aminoacides chargés, ainsi que quelquesaminoacides hydrophobes (B). Remarquezsur cette vue en section que la plupartdes aminoacides hydrophobes sont situésà l’intérieur de la structure, tandis queles aminoacides chargés sont à la surfacede la protéine. [Dessiné d’après 1MBD.pdb.]
(B)(A)
Figure 2.48 Structure tridimensionnelle de la myoglobine. (A) Un diagrammmeen ruban montre que la protéine est constituée principalement d’hélices .
(B) Un modèle compact dans la même orientation montre à quel point la protéinereployée est étroitement empaquetée. Remarquez que le groupement hème est nichédans une crevasse de la protéine compacte avec seulement un côté exposé. Une héliceest dessinée en bleu pour permettre une comparaison des deux modèles structuraux.[Dessiné d’après 1A6N.pdb.]
(B)(B) Groupe hèmeGroupe hème(A)
Atome de ferGroupe hème
5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 24/40
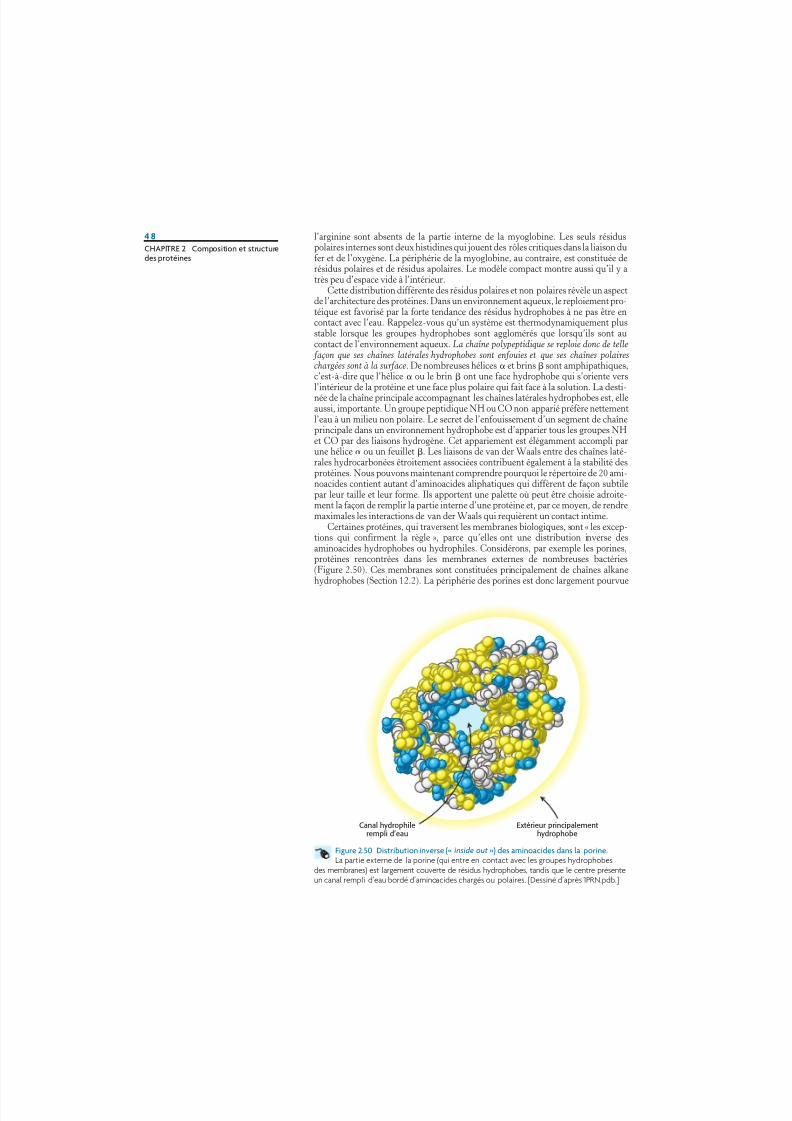
l’arginine sont absents de la partie interne de la myoglobine. Les seuls résiduspolaires internes sont deux histidines qui jouent des rôles critiques dans la liaison dufer et de l’oxygène. La périphérie de la myoglobine, au contraire, est constituée derésidus polaires et de résidus apolaires. Le modèle compact montre aussi qu’il y atrès peu d’espace vide à l’intérieur.
Cette distribution différente des résidus polaires et non polaires révèle un aspectde l’architecture des protéines. Dans un environnement aqueux, le reploiement pro-téique est favorisé par la forte tendance des résidus hydrophobes à ne pas être encontact avec l’eau. Rappelez-vous qu’un système est thermodynamiquement plusstable lorsque les groupes hydrophobes sont agglomérés que lorsqu’ils sont aucontact de l’environnement aqueux. La chaîne polypeptidique se reploie donc de telle façon que ses chaînes latérales hydrophobes sont enfouies et que ses chaînes polaireschargées sont à la surface. De nombreuses hélices et brins sont amphipathiques,c’est-à-dire que l’hélice ou le brin ont une face hydrophobe qui s’oriente vers
l’intérieur de la protéine et une face plus polaire qui fait face à la solution. La desti-née de la chaîne principale accompagnant les chaînes latérales hydrophobes est, elleaussi, importante. Un groupe peptidique NH ou CO non apparié préfère nettementl’eau à un milieu non polaire. Le secret de l’enfouissement d’un segment de chaîneprincipale dans un environnement hydrophobe est d’apparier tous les groupes NHet CO par des liaisons hydrogène. Cet appariement est élégamment accompli parune hélice ou un feuillet . Les liaisons de van der Waals entre des chaînes laté-rales hydrocarbonées étroitement associées contribuent également à la stabilité desprotéines. Nous pouvons maintenant comprendre pourquoi le répertoire de 20 ami-noacides contient autant d’aminoacides aliphatiques qui diffèrent de façon subtilepar leur taille et leur forme. Ils apportent une palette où peut être choisie adroite-ment la façon de remplir la partie interne d’une protéine et, par ce moyen, de rendremaximales les interactions de van der Waals qui requièrent un contact intime.
Certaines protéines, qui traversent les membranes biologiques, sont « les excep-tions qui confirment la règle », parce qu’elles ont une distribution inverse desaminoacides hydrophobes ou hydrophiles. Considérons, par exemple les porines,protéines rencontrées dans les membranes externes de nombreuses bactéries(Figure 2.50). Ces membranes sont constituées principalement de chaînes alkanehydrophobes (Section 12.2). La périphérie des porines est donc largement pourvue
Figure 2.50 Distribution inverse (« inside out ») des aminoacides dans la porine.La partie externe de la porine (qui entre en contact avec les groupes hydrophobes
des membranes) est largement couverte de résidus hydrophobes, tandis que le centre présenteun canal rempli d’eau bordé d’aminoacides chargés ou polaires. [Dessiné d’après 1PRN.pdb.]
Extérieur principalementhydrophobe
Canal hydrophilerempli d’eau
4 8
CHAPITRE 2 Composition et structuredes protéines

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 25/40
de résidus hydrophobes qui peuvent interagir avec les chaînes alkane environ-nantes. En revanche, le centre de la protéine contient de nombreux aminoacideschargés ou polaires délimitant un canal rempli d’eau qui passe par le milieu de laprotéine. Donc, étant donné que les porines fonctionnent dans des environnementshydrophobes, elles sont « inversées » par rapport aux protéines qui évoluent enmilieu aqueux.
Certaines combinaisons de structures secondaires sont présentes sur beaucoupde protéines et remplissent souvent des fonctions similaires. Ces combinaisons sontappelées motifs ou structures supersecondaires. Par exemple, une hélice séparéed’une autre hélice par un coude est appelée unité hélice-coude-hélice ; on latrouve sur beaucoup de protéines qui se fixent sur le DNA (Figure 2.51).
Certaines chaînes polypeptidiques se reploient en deux régions compactes, oudavantage, qui peuvent être unies par un segment flexible de la chaîne polypepti-dique, presque comme des perles sur un fil. Ces unités globulaires compactes,
appelées domaines, sont constituées d’environ 30 à 400 résidus aminoacide. Parexemple, la partie extracellulaire du CD4, protéine de la surface cellulaire de cer-taines cellules du système immunitaire auxquelles s’attache le virus de l’immuno-déficience humaine (VIH), est constituée de quatre domaines semblablesd’approximativement 100 aminoacides chacun (Figure 2.52). Les protéines peu-vent avoir des domaines communs même lorsque leurs structures d’ensemble sontdifférentes.
2.5 Structure quaternaire : les chaînes polypeptidiquespeuvent s’assembler en des structuresmulti-sous-unitaires
Quatre niveaux de structure sont fréquemment cités dans les discussions surl’architecture des protéines. Jusqu’à présent, nous n’en avons considéré que trois.La structure primaire est la séquence des aminoacides. La structure secondaire atrait aux relations dans l’espace des résidus aminoacide proches les uns des autresdans la séquence. Certains de ces arrangements sont réguliers et donnent nais-sance à une structure périodique. L’hélice et le feuillet sont des éléments destructure secondaire. La structure tertiaire se réfère aux relations dans l’espace desrésidus aminoacide éloignés dans la séquence, ainsi qu’à la distribution des liai-sons disulfure. Nous considérons maintenant les protéines quicontiennent plus d’une chaîne polypeptidique. Ces protéinesmontrent un quatrième niveau d’organisation structurale.Chaque chaîne polypeptidique d’une telle protéine est appeléesous-unité . La structure quaternaire se rapporte à la disposition
spatiale de chaque sous-unité et à la nature des contacts entreces dernières. Le type le plus simple de structure quaternaireest un dimère constitué de deux sous-unités identiques. Cetteorganisation est présente dans la protéine Cro qui se fixe auDNA, rencontrée dans un virus des bactéries appelé (Figure 2.53). Des structures quaternaires plus compliquéessont, elles aussi, habituelles. Par exemple, l’hémoglobinehumaine, protéine du sang qui transporte l’oxygène, estconstituée de deux sous-unités d’un certain type (désignées)et de deux sous-unités d’un autre type (désignées ), comme
Figure 2.51 Le motif hélice-coude-hélice, un élément de structure
supersecondaire. Les motifs hélice-coude-hélice sont présents dans beaucoupde protéines se fixant au DNA.[Dessiné d’après 1LMB.pdb.]
Hélice-coude-hélice
Figure 2.52 Domaines des protéines.La protéine de surface cellulaire
CD4 est constituée de quatre domainessemblables. [Dessiné d’après 1WIO.pdb.]
Figure 2.53. Une structure quaternaire. La protéine Crodu bactériophage est un dimère de sous-unités identiques.
[Dessiné d’après 5CRO.pdb.]
4 9

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 26/40
représenté dans la Figure 2.54. La molécule d’hémoglobine se présente donccomme un tétramère 22. De subtils changements dans la disposition relativedes sous-unités au sein d’une molécule d’hémoglobine permettent à cette dernièrede transporter l’oxygène des poumons vers les tissus avec une grande efficacité(Chapitre 7).
Les virus tirent le maximum d’une quantité limitée d’information génétique enformant des capsides qui utilisent le même type de sous-unités disposées de façonrépétitive en un réseau symétrique. La capside du rhinovirus, virus qui provoque lerhume banal, comprend 60 copies de chacune des quatre sous-unités différentes(Figure 2.55). Les sous-unités s’agglomèrent pour former une coque presque sphé-rique qui englobe le génome viral.
2.6 La séquence des aminoacides d’une protéinedétermine sa structure tridimensionnelle
Comment la structure tridimensionnelle élaborée des protéines est-elle produite ?
Le travail classique de Christian Anfinsen dans les années 1950 sur l’enzyme ribo-nucléase a révélé la relation existant entre la séquence des aminoacides d’une pro-téine et sa conformation. La ribonucléase est une chaîne polypeptidique uniqueconstituée de 124 résidus aminoacide où apparaissent quatre liaisons disulfure(Figure 2.56). L’idée d’Anfinsen était de détruire la structure tridimensionnelle del’enzyme, puis de déterminer quelles étaient les conditions nécessaires à la restaura-tion de cette structure.
Des agents tels que l’urée ou le chlorure de guanidine rompent efficacement lesliaisons non covalentes d’une protéine, bien que le mécanisme d’action de ces agentsne soit pas tota lement élucidé. Les liaisons disulfure peuvent être clivées réversible-ment par réduction par un réactif tel que le -mercaptoéthanol (Figure 2.57). Enprésence d’un large excès de -mercaptoéthanol, les disulfures (les cystines) sonttotalement transformés en sulfhydryles (cystéines).
En l’absence d’interactions croisées, la plupart des chaînes polypeptidiques pren-nent habituellement dans l’urée 8 M ou le chlorure de guanidine 6 M, une conforma-
Figure 2.54 Le tétramère22de l’hémoglobine humaine.
La structure des deux sous-unitésidentiques (rouge) est similaire mais pasidentique à celle des deux sous-unitésidentiques (jaune). La molécule contientquatre groupements hème (en gris avecl’atome de fer en violet). (A) Le diagrammeen ruban permet de souligner la similaritédes sous-unités et de montrer qu’elles sontcomposées principalement d’hélices .(B) Le model compact illustre la manièreselon laquelle les groupements hèmeoccupent des crevasses dans la protéine.[Dessiné d’après 1A3N.pdb.]
(B)(A)
Figure 2.55 Une structure quaternairecomplexe. La capside du rhinovirus humain,l’agent du rhume, contient 60 copiesde chacune des quatre sous-unités
(représentées en différentes couleurs).
Figure 2.56 Séquence des aminoacidesde la ribonucléase bovine. Les quatreliaisons disulfure sont représentéesen couleur. [D’après C.H.W. Hirs, S . Mooreet W.H. Stein. J. Biol. Chem. 235 (1960) :633-647.]
CC
M M Q N
Y N S S S A SA T
SS
DMHQREF KAAA
ET
K+H3N
KS
RNL T
KD
RC
K40P
V N T F V H E S L A D VQ
AV
CS Q
KNVA
CC
N T KQ NG
Y QS Y S TMSI TDR
E TGSS
K Y
PN
C A Y K T T Q A N K H I I V
ACE
GNP Y VPVHF DASV
C O
O100
124
90 120
30
1 20
80
10
70
60110
50
−
H2NC
O
NH2
Urée
HOCH2
H2C
SH
-Mercaptoéthanol
Chlorure de guanidine
NH2
CNH2H2N
Cl–+
5 0

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 27/40
tion enroulée au hasard. Lorsque la ribonucléase est traitée par le -mercaptoéthanoldans l’urée 8 M, le produit est une chaîne polypeptidique totalement réduite, enrou-lée au hasard, dépourvue d’activité enzymatique. Quand une protéine est convertie enun peptide enroulé au hasard dénué d’activité normale, on dit qu’elle est dénaturée(Figure 2.58).
Anfinsen a ensuite fait l’observation fondamentale que la ribonucléase dénatu-rée, une fois libérée de l’urée et du -mercaptoéthanol par dialyse, récupère lente-ment une activité enzymatique. Il a immédiatement perçu la signification de cetteconstatation faite par hasard. Les groupes sulfhydryle de l’enzyme dénaturé sontréoxydés par l’air et l’enzyme se reploie spontanément en une forme catalytique-ment active. Des études détaillées ont montré que la presque totalité de l’activitéenzymatique de départ est récupérée lorsque les groupes sulfhydryle sont oxydésdans des conditions convenables. Toutes les propriétés physiques et chimiquesmesurées de l’enzyme reployé in vitro sont pratiquement identiques à celles del’enzyme natif. Ces expériences ont montré que l’information nécessaire à la déter-mination de la structure tridimensionnelle complète de la ribonucléase est contenue dansla séquence de ses aminoacides. Les études ultérieures d’autres protéines ont établi la
généralité de ce principe fondamental de la biochimie : la séquence détermine laconformation. La relation de dépendance de la conformation à la séquence est parti-culièrement significative en raison de l’intime rapport entre conformation et fonction.
Un résultat très différent a été obtenu lorsque la ribonucléase réduite était réoxy-dée alors qu’elle était encore en présence d’urée 8 M et que la préparation étaitensuite dialysée pour éliminer l’urée. La ribonucléase réoxydée de cette façonn’avait que 1 % de l’activité enzymatique de la protéine native. Pourquoi le résul-tat de cette expérience était-il si différent lorsque la ribonucléase réduite étaitréoxydée en présence ou en absence d’urée ? La raison en est que de mauvais appa-riements par liaisons disulfure se constituent en présence d’urée. Il y a 105 façonsdifférentes d’apparier huit cystéines pour former quatre liaisons disulfure ; uneseule de ces combinaisons est enzymatiquement active. Les 104 mauvais apparie-ments ont été appelés, d’un terme pittoresque, ribonucléase « brouillée ». Anfinsena ensuite montré que la ribonucléase brouillée se convertissait spontanément enribonucléase native, totalement active, lorsque des traces de -mercaptoéthanol
5 1
2.6 Séquence et structure
Figure 2.57 Rôle du -mercaptoéthanol dans la réduction des liaisons disulfure.Remarquez que, lorsque les disulfures sont réduits, le -mercaptoéthanol est oxydéet forme des dimères.
H
HS
S
S
SProtéine Protéine
Excès
CH2
H2C
SHO
H
CH2
H2C
SO
H CH2
H2C
SO
H
Figure 2.58 Réduction et dénaturation de la ribonucléase.
110
26
58
6584
124
Ribonucléase réduite dénaturéeRibonucléase native
95
40
95
26 72
65
110
58
1
8472
40
HS
HSHS
HS
SH
SH
HSHS
Urée 8 M et-mercaptoéthanol

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 28/40
étaient ajoutées à la solution aqueuse de la protéine (Figure 2.59). Le -mercaptoé-thanol ajouté catalysait le réarrangement des appariements par liaisons disulfure jusqu’à ce que la structure native ait été reconstituée, ce qui prend environ10 heures. Ce processus est uniquement favorisé par la diminution d’énergie libre quirésulte de la conversion des conformations brouillées en conformation native, stable, del’enzyme. Ainsi, les appariements par liaison disulfure natifs de la ribonucléasecontribuent à la stabilisation de la structure thermodynamiquement préférentielle.
Des expériences de reploiement similaires ont été réalisées avec beaucoupd’autres protéines. Dans de nombreux cas, la structure native peut être régénéréedans des conditions favorables. Cependant, pour d’autres protéines, le reploiementne s’effectue pas de façon efficace. Dans ces cas, les molécules de protéinesdéployées s’emmêlent les unes avec les autres pour former des agrégats. Au sein descellules, des protéines appelées chaperons bloquent ces interactions « illicites ».
Les aminoacides ont des propensions différentes à formerdes hélices alpha, des feuillets bêta ou des coudes bêta
Comment la séquence des aminoacides d’une protéine détermine-t-elle la structuretridimensionnelle de cette dernière ? Comment une chaîne polypeptidique déployéeacquiert-elle la forme de la protéine native ? Ces questions fondamentales en bio-chimie peuvent être approchées en posant tout d’abord une question plus simple :qu’est-ce qui détermine une séquence particulière d’une protéine à former unehélice , un brin ou un coude ? Une source d’indications peut être l’examen de lafréquence d’apparition de résidus aminoacide particuliers dans ces structuressecondaires (Tableau 2.3). Des résidus tels que l’alanine, le glutamate et la leucineon tendance à être présents dans les hélices , tandis que la valine et l’isoleucine ten-dent à être présentes dans les brins . La glycine, l’arginine et la proline ont unepropension à se situer dans les coudes.
Les études portant sur des protéines ou des peptides synthétiques ont révélécertaines raisons de ces préférences. L’hélice peut être considérée comme laconformation par défaut. Un branchement au niveau de l’atome de carbone ,comme dans la valine, la thréonine et l’isoleucine, tend à déstabiliser les hélices
en raison de conflits stériques. Ces résidus sont facilement acceptés dans lesbrins où leurs chaînes latérales se projettent hors du plan contenant la chaîne
5 2
Figure 2.59 Rétablissementde l’appariement correct par liaisonsdisulfure. La ribonucléase native peutse reformer à partir de la ribonucléase« brouillée » en présence d’une tracede -mercaptoéthanol.
1241
7284
95
58
4026
110
65
Ribonucléase native
40
95
26 72
65
110
58
1
84
Ribonucléase « brouillée »
Traces de-mercaptoéthanol
TABLEAU 2.3 Fréquences relatives des résidus aminoacide dans les structures secondaires
Aminoacide Hélice Feuillet Coude
Glu 1,59 0,52 1,01
Ala 1,41 0,72 0,82
Leu 1,34 1,22 0,57
Met 1,30 1,14 0,52
Gln 1,27 0,98 0,84
Lys 1,23 0,69 1,07
Arg 1,21 0,84 0,90
His 1,05 0,80 0,81
Val 0,90 1,87 0,41
Ile 1,09 1,67 0,47
Tyr 0,74 1,45 0,76
Cys 0,66 1,40 0,54
Trp 1,02 1,35 0,65
Phe 1,16 1,33 0,59
Thr 0,76 1,17 0,96Gly 0,43 0,58 1,77Asn 0,76 0,48 1,34Pro 0,34 0,31 1,32Ser 0,57 0,96 1,22Asp 0,99 0,39 1,24
R EMARQUE : Les aminoacides sont groupés selon leur préférence pour les hélices (groupe du haut), les feuillets (second
groupe) ou les coudes (troisième groupe).
D’après T.E. Creighton, Proteins : Structures and Molecular Properties, 2d ed. (W.H. Freeman and Company, 1992), p. 256.

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 29/40
principale. La sérine, l’aspartate et l’asparagine tendent à rompre les hélices , carleurs chaînes latérales contiennent des donneurs ou des accepteurs de liaisonshydrogène proches de la chaîne principale et ils entrent en compétition avec lesgroupes NH et CO de la chaîne principale. La proline tend à rompre les hélices autant que les brins car elle n’a pas de groupe NH et sa structure cyclique limitela valeur de à environ 60 degrés. La glycine s’insère facilement dans toutes lesstructures et, pour cette raison, elle ne favorise pas la formation d’une hélice enparticulier.
Peut-on prévoir la structure secondaire des protéines en utilisant cette connais-sance des préférences de conformation spatiale des résidus aminoacide ? Les prévi-sions de la structure secondaire adoptée par un segment de 6 résidus, ou moins, sesont montrées exactes dans 60 à 70 % environ des cas. Qu’est-cequi s’oppose à une prédiction plus précise ? Remarquez que lespréférences de conformation spatiale des résidus aminoacide ne
permettent pas la prédiction d’une structure unique (voirTableau 2.3). Par exemple, le glutamate, l’un des plus puissantsformateurs d’hélice, ne préfère l’hélice au brin que d’un fac-teur 2 seulement. Les rapports de préférence de la plupart desautres résidus sont plus faibles. En fait, on a montré que cer-taines séquences de pentapeptides ou d’hexapeptides adoptentune certaine structure dans une protéine et une structure entiè-rement différente dans une autre (Figure 2.60). Ainsi, desséquences données d’aminoacides ne déterminent pas une struc-ture secondaire de façon univoque. Des interactions tertiaires,c’est-à-dire des interactions entre résidus éloignés dans laséquence, peuvent être décisives pour déterminer la structuresecondaire de certains segments. Le contexte est souvent essen-tiel pour déterminer le devenir conformationnel. La confor-mation des protéines a évolué pour qu’elles puissent fonctionnerdans un environnement ou un contexte particulier. Des amé-liorations substantielles dans la prédiction des structuressecondaires peuvent être obtenues en étudiant des familles deséquences apparentées qui adoptent toutes des structuressemblables.
Le mauvais reploiement des protéines et leur agrégationsont associés a certaines maladies neurologiques
Jusqu’à une période récente, on croyait que toutes les maladies infectieusesétaient transmises par des microbes ou des virus. Une des grandes surprises
de la médecine moderne fut la reconnaissance de la transmission de certaines mala-dies infectieuses neurologiques par des agents dont la taille est de l’ordre de celle desvirus, mais qui sont constitués uniquement de protéines. Ces maladies compren-nent l’encéphalopathie spongiforme bovine (appelée communément maladie de lavache folle) et des maladies analogues des autres espèces animales, y compris lamaladie de Creutzfeldt-Jakob (CJD) chez l’homme, et la scrapie chez le mouton. Lesagents causant ces maladies sont appelés prions. Stanley Prusiner, personnalitéscientifique ayant joué un rôle de leader en proposant l’hypothèse que des maladiespouvaient être transmises par des protéines seules, a reçu le Prix Nobel de physio-logie et de médecine en 1997.
L’étude de ces agents infectieux révéla les caractéristiques suivantes :
1. L’agent transmissible consiste en des formes agrégées d’une protéine spécifique.Ces agrégats ont tout un éventail de poids moléculaires.
2. Ces agrégats de protéines sont résistants aux traitements par des agents quidégradent la plupart des protéines.
3. La protéine est en majeure partie ou totalement dérivée d’une protéine cellulairenormale, appelée PrP, qui est présente dans le cerveau.
5 3
2.6 Séquence et structure
Figure 2.60. Conformations alternatives d’une séquencepeptidique. De nombreuses séquences peuvent adopter
des conformations différentes dans des protéines différentes.Ici la séquence VDLLKN représentée en rouge prend la formed’une hélice dans un certain contexte protéique (à gauche)et celle d’un brin dans un autre (à droite).
[Dessiné d’après 3WRP.pdb (à gauche) et 2HLA.pdb (à droite).]

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 30/40
En quoi la structure de la forme agrégée de la protéine diffère-t-elle de celle dela protéine dans son état normal dans le cerveau ? La structure de la protéine cellu-laire normale PrP contient de grandes régions d’hélices et relativement peu destructures en feuillets . La structure de la forme de la protéine présente dans lescerveaux infectés, appelées PrPsc, n’a pas encore été déterminée à cause d’obstaclesdus à son insolubilité et à sa nature hétérogène. Cependant, un ensemble de raisonsindiquent que certaines régions de la protéine qui formaient des hélices ou descoudes ont été converties en conformations de type feuillet . Les feuillets d’uneprotéine s’associent souvent à ceux d’une autre protéine pour former des empile-ments de feuillets des deux protéines, pouvant aboutir à la formation d’agrégats.Ces agrégats protéiques fibreux sont souvent désignés comme formes amyloïdes.
La découverte, que l’agent infectieux des maladies à prion, est une forme agré-gée d’une protéine déjà présente dans le cerveau, a suggéré un modèle pour expli-quer la transmission de la maladie (Figure 2.61). Les agrégats de protéines
construits à partir de formes anormales de PrP agissent comme des centres denucléation sur lesquels d’autres molécules PrP s’attachent. Les maladies à prionpeuvent ainsi se transmettre d’un individu à un autre par le transfert d’un de cescentres de nucléation. C’est probablement ce qui s’est passé lors de l’épidémie demaladie de vache folle dans le Royaume Uni dans les années 1990. Le bétail nourriavec de la nourriture animale contenant des produits dérivés de bovins malades,devint malade à son tour.
Des fibres amyloïdes sont aussi présentes dans le cerveau de patients atteints decertaines maladies neurodégénératives non infectieuses telles que les maladiesd’Alzheimer ou de Parkinson. Par exemple les cerveaux de patients atteints demaladie d’Alzheimer contiennent des agrégats de protéines appelés plaques amy-loïdes constitués presque uniquement d’un polypeptide appelé A. Ce polypeptideest dérivé d’une protéine cellulaire, la protéine précurseur de l’amyloïde ou APP,après action de protéases spécifiques. Le polypeptide A a tendance à former desagrégats insolubles. En dépit des difficultés posées par l’insolubilité de la protéine,un modèle structural détaillé de A a été construit grâce à l’utilisation de techniquesde RMN (résonance magnétique nucléaire) qui peuvent être appliquées sur dessolides plutôt que sur des produits en solution. Comme on s’y attendait la structureest riche en feuillets qui s’assemblent pour former une vaste structure de feuilletsparallèles empilés (Figure 2.62).
5 4
CHAPITRE 2 Composition et structuredes protéines
Figure 2.61 Le modèle « protéine-seule »de transmission des maladies à prion.Un noyau constitué de protéinesde configuration anormale est agrandi
par addition de protéines provenantdu pool de protéines normales. Pool de PrP normales
Noyau de PrPSC
Figure 2.62 Une structure de fibresamyloïdes. Un modèle détaillédes fibrilles A‚ déduit d’études de RMNà l’état solide, montre que l’agrégationdes protéines est dû à la formationde grandes zones de feuillets parallèles.[D’après A. T. Petkova, Y. Ishii, J. J. Balbach,O. N. Antzukin, R. D. Leapman, F. Delagio,and R. Tycko, Proc, Natl. Acad. Sci. U.S.A.99 (2002) : 16742–16747.]
A x e
d e s f i b
r i l l e s

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 31/40
Comment est-ce que de tels agrégats conduisent à la mort des cellules qui leshébergent ? La réponse est toujours controversée. Une hypothèse est que les grandsagrégats ne sont pas toxiques, mais qu’au contraire les petits agrégats des mêmesprotéines seraient les coupables, peut-être en endommageant les membranes.
Le reploiement des protéines est un processus hautementcoopératif
Comme il a été mentionné précédemment, les protéines peuvent être dénaturées parla chaleur ou par des dénaturants chimiques, tels que l’urée ou le chlorure de guani-dine. Dans de nombreuses protéines, une comparaison du degré de déploiement enfonction de l’augmentation de la concentration du dénaturant révèle une transitionrelativement rapide de la forme reployée, ou native, vers la forme déployée ou déna-turée, ce qui suggère que seuls ces deux états conformationnels sont présents à untaux significatif (Figure 2.63). De même, une transition rapide est observée lors-qu’on élimine les dénaturants des protéines déployées ce qui permet aux protéinesde se reployer.
La transition brutale observable dans la Figure 2.63 suggère que le reploiementet le déploiement de la protéine est un processus « de tout ou rien » qui résulte d’unetransition coopérative. Par exemple, supposez qu’une protéine soit placée dans desconditions où une région de la structure de la protéine est thermodynamiquementinstable. Une partie de la structure reployée étant rompue, les interactions entrecette dernière et le reste de la protéine seront perdues. La perte de ces dernièresdéstabilisera alors le reste de la structure. Ainsi, des conditions qui conduisent à ladisparition d’une région de la structure de la protéine dénat urantvraisemblablement la protéine dans son ensemble. Les proprié-tés structurales des protéines apportent une compréhensionclaire de cette transition coopérative.
Les conséquences du reploiement coopératif peuvent êtreillustrées en considérant le contenu d’une solution d’une pro-téine dans des conditions correspondant à un état intermédiairede transition entre les formes reployées et déployées. Dans detelles conditions, la protéine est « demi-déployée ». Remarquez
que la solution ne contient pas de molécules demi-déployéesmais, au contraire, un mélange à parties égales de molécules tota-lement reployées et de molécules totalement déployées(Figure 2.64).
Bien que les protéines puissent sembler se comporter commeexistant seulement sous deux états, cette interprétation simpled’une existence à deux états est impossible au niveau atomique.Des structures intermédiaires instables, transitoires doiventexister entre les états natif et dénaturé (p. 56). La déterminationde la nature de ces structures intermédiaires est un domaineintensif de la recherche en biochimie.
Les protéines se reploient par stabilisation progressived’intermédiaires et non par une recherche au hasard
Comment une protéine effectue-t-elle la transition d’une structure déployée à laconformation unique de la forme native ? Une possibilité a priori, serait que toutesles conformations possibles soient explorées afin que soit trouvée celle qui est éner-
gétiquement la plus favorable. Combien de temps une telle recherche au hasardprendrait-elle ? Considérez une petite molécule de 100 résidus. Cyrus Levinthal acalculé que si chaque résidu peut assumer trois positions différentes, le nombre totalde structures est de 3100, soit 5 1047. S’il faut 1013 s pour convertir une structureen une autre, le temps total de recherche serait de 5 1047 1013 s, soit 5 1034 sou 1,6 1027 années. Manifestement, cela prendrait beaucoup trop de temps,même à une petite protéine, pour se reployer correctement en cherchant au hasardtoutes les conformations possibles ! L’énorme différence entre le temps calculé et letemps réel de reploiement est appelée paradoxe de Levinthal. Ce paradoxe révèleclairement que les protéines ne se replient pas en essayant chaque conformation
5 5
Figure 2.63. Transition d’un état reployéà un état déployé. La plupartdes protéines montrent une transition
rapide de la forme reployée vers la formedéployée à la suite d’un traitementpar des concentrations croissantesd’agents dénaturants.
0
100
[ P r o t é i n e d é p l o y é e ] , %
[Dénaturant]
Figure 2.64. Constituants d’une solution de protéinepartiellement dénaturée. Dans une solution de protéineà demi déployée, une moitié des molécules est totalementreployée et l’autre moitié totalement déployée.
Déployée
Reployée
50
0
100
[ P r o t é i n e d é p l o y é e ] , %
[Dénaturant]
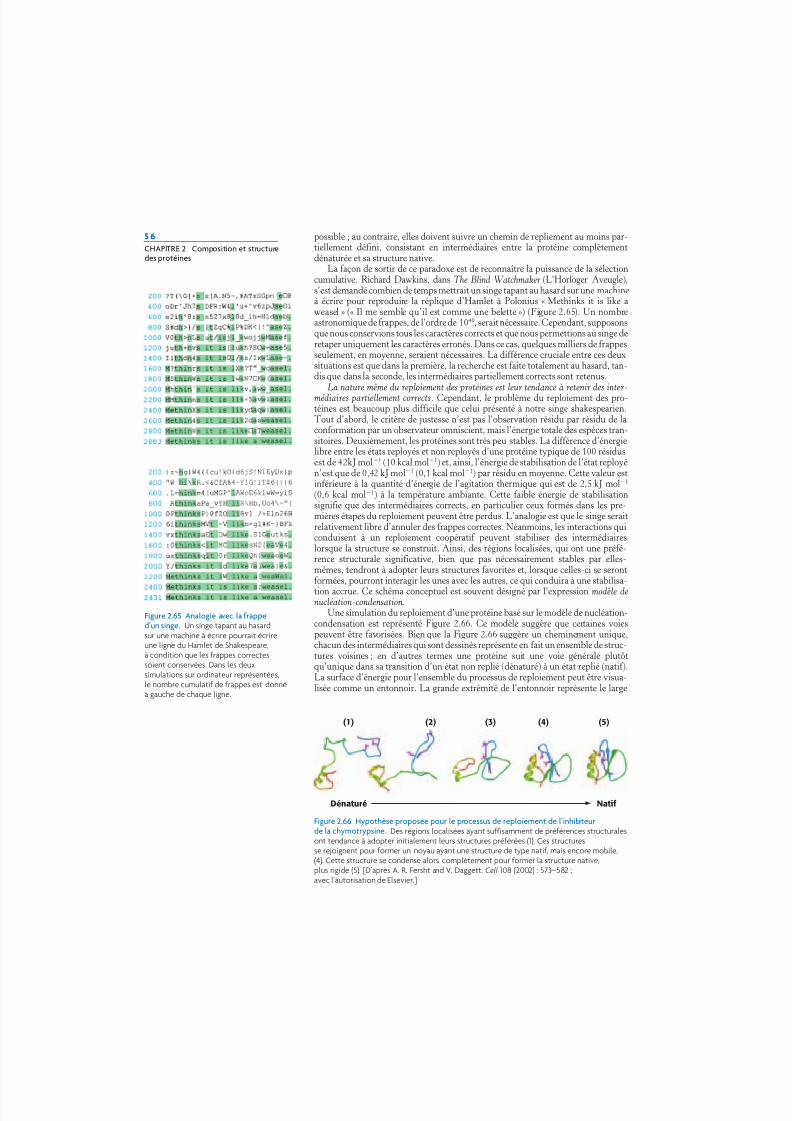
5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 32/40
possible ; au contraire, elles doivent suivre un chemin de repliement au moins par-tiellement défini, consistant en intermédiaires entre la protéine complètementdénaturée et sa structure native.
La façon de sortir de ce paradoxe est de reconnaître la puissance de la sélectioncumulative. Richard Dawkins, dans The Blind Watchmaker (L’Horloger Aveugle),s’est demandé combien de temps mettrait un singe tapant au hasard sur une machineà écrire pour reproduire la réplique d’Hamlet à Polonius « Methinks it is like aweasel » (« Il me semble qu’il est comme une belette ») (Figure 2.65). Un nombreastronomique de frappes, de l’ordre de 1040, serait nécessaire. Cependant, supposonsque nous conservions tous les caractères corrects et que nous permettions au singe deretaper uniquement les caractères erronés. Dans ce cas, quelques milliers de frappesseulement, en moyenne, seraient nécessaires. La différence cruciale entre ces deuxsituations est que dans la première, la recherche est faite totalement au hasard, tan-dis que dans la seconde, les intermédiaires partiellement corrects sont retenus.
La nature même du reploiement des protéines est leur tendance à retenir des inter-médiaires partiellement corrects. Cependant, le problème du reploiement des pro-téines est beaucoup plus difficile que celui présenté à notre singe shakespearien.Tout d’abord, le critère de justesse n’est pas l’observation résidu par résidu de laconformation par un observateur omniscient, mais l’énergie totale des espèces tran-sitoires. Deuxièmement, les protéines sont très peu stables. La différence d’énergielibre entre les états reployés et non reployés d’une protéine typique de 100 résidusest de 42kJ mol1 (10 kcal mol1) et, ainsi, l’énergie de stabilisation de l’état reployén’est que de 0,42 kJ mol1 (0,1 kcal mol1) par résidu en moyenne. Cette valeur estinférieure à la quantité d’énergie de l’agitation thermique qui est de 2,5 kJ mol1
(0,6 kcal mol1) à la température ambiante. Cette faible énergie de stabilisationsignifie que des intermédiaires corrects, en particulier ceux formés dans les pre-mières étapes du reploiement peuvent être perdus. L’analogie est que le singe seraitrelativement libre d’annuler des frappes correctes. Néanmoins, les interactions quiconduisent à un reploiement coopératif peuvent stabiliser des intermédiaireslorsque la structure se construit. Ainsi, des régions localisées, qui ont une préfé-rence structurale significative, bien que pas nécessairement stables par elles-mêmes, tendront à adopter leurs structures favorites et, lorsque celles-ci se serontformées, pourront interagir les unes avec les autres, ce qui conduira à une stabilisa-tion accrue. Ce schéma conceptuel est souvent désigné par l’expression modèle denucléation-condensation.
Une simulation du reploiement d’une protéine basé sur le modèle de nucléation-condensation est représenté Figure 2.66. Ce modèle suggère que certaines voiespeuvent être favorisées. Bien que la Figure 2.66 suggère un cheminement unique,chacun des intermédiaires qui sont dessinés représente en fait un ensemble de struc-tures voisines ; en d’autres termes une protéine suit une voie générale plutôtqu’unique dans sa transition d’un état non replié (dénaturé) à un état replié (natif).La surface d’énergie pour l’ensemble du processus de reploiement peut être visua-lisée comme un entonnoir. La grande extrémité de l’entonnoir représente le large
5 6
CHAPITRE 2 Composition et structuredes protéines
Figure 2.65 Analogie avec la frapped’un singe. Un singe tapant au hasardsur une machine à écrire pourrait écrireune ligne du Hamlet de Shakespeare,à condition que les frappes correctessoient conservées. Dans les deuxsimulations sur ordinateur représentées,le nombre cumulatif de frappes est donnéà gauche de chaque ligne.
Figure 2.66 Hypothèse proposée pour le processus de reploiement de l’inhibiteurde la chymotrypsine. Des régions localisées ayant suffisamment de préférences structuralesont tendance à adopter initialement leurs structures préférées (1). Ces structuresse rejoignent pour former un noyau ayant une structure de type natif, mais encore mobile,(4). Cette structure se condense alors complètement pour former la structure native,plus rigide (5). [D’après A. R. Fersht and V. Daggett. Cell 108 (2002) : 573–582 ;avec l’autorisation de Elsevier.]
Dénaturé
(1) (2) (3) (4) (5)
Natif

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 33/40
éventail des structures compatibles avec l’ensemble des molécules de protéinesdénaturées. Au fur et à mesure que l’énergie libre de la population de moléculesdécroît, les protéines se déplacent vers les régions plus étroites de l’entonnoir, etmoins de conformations y ont accès. A la sortie de l’entonnoir on ne trouve plus quela protéine reployée dans sa conformation parfaitement définie. De nombreux che-mins différents peuvent mener au même minimum d’énergie.
La prédiction de la structure tridimensionnelleà partir de la séquence demeure un grand problèmeLa prédiction de la structure tridimensionnelle à partir de la séquence s’est révéléeextrêmement difficile. Comme nous l’avons vu, la séquence locale semble nedéterminer que 60 à 70 % de la structure secondaire et des interactions à grande dis-tance sont nécessaires pour fixer la structure secondaire complète et la structuretertiaire.
Les chercheurs ont exploré deux approches f ondamentales différentes pour pré-dire la structure tridimensionnelle à partir de la séquence des aminoacides. La pre-mière est la prédiction ab initio « sans autre information », qui tend à prédire lereploiement d’une séquence d’aminoacides sans connaissance antérieure deséquences similaires de protéines dont les structures sont connues. On effectue surordinateur des calculs qui tendent à rendre minimale l’énergie libre de la structureà partir d’une séquence donnée d’aminoacides, ou encore à simuler le processus dereploiement. L’utilité de ces méthodes est limitée par le très grand nombre deconformations possibles, la faible stabilité des protéines et la subtile énergétique desinteractions faibles en solution aqueuse. La deuxième approche tire avantage denotre connaissance grandissante de la structure tridimensionnelle de nombreusesprotéines. Dans ces méthodes basées sur la connaissance, la séquence des aminoacidesd’une structure inconnue est examinée pour sa compatibilité avec les structures desprotéines connues ou des fragments de celles-ci. Lorsqu’une concordance significa-tive est détectée, la structure connue peut être utilisée comme modèle de départ. Lesméthodes basées sur la connaissance ont été à l’origine de nombreuses informationssur la conformation tridimensionnelle des protéines de séquence connue, mais destructure alors inconnue.
La modification et le clivage des protéines génèrentde nouvelles possibilités structurales
Les protéines sont susceptibles d’assumer des fonctions diverses unique-ment en s’appuyant sur les propriétés variées de leurs 20 aminoacides. Mais
en outre, de nombreuses protéines peuvent être modifiées de façon covalente par lafixation de groupes autres que les aminoacides, ce qui étend leurs fonctions(Figure 2.67). Par exemple, des groupes acétyle sont fixés à l’extrémité N-terminalede beaucoup de protéines, ce qui rend ces dernières plus résistantes à la dégradation.Comme nous l’avons discuté auparavant (p. 46) l’addition de groupes hydroxyle àde nombreux résidus proline stabilise les fibres de collagène nouvellement synthé-tisées. La signification biologique de cette modification est évidente dans le scor-
5 7
2.6 Séquence et structure
Figure 2.67 Touches de finition. Certaines modifications courantes et importantesdes chaînes latérales des aminoacides sont représentées.
NH
H2C
NH
H2CCH
–OOCCOO–
NH
H2CO
POO O
H2CCH
H2CN
HO C O
HN
OHOH2C
OH
NHHO
CO CH3
Hydroxyproline γ
-Carboxyglutamate Complexe ose-asparagine Phosphosérine
2–
C
O
H
C CC
O
H
CC
O
H
CC
O
H

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 34/40
but : un déficit de vitamine C se traduit par une hydroxylation insuffisante du col-lagène et les fibres de collagène anormal qui en résultent sont incapables de mainte-nir une résistance normale des tissus (Section 27.5). Un autre aminoacide spécialiséproduit par une touche de finition est le -carboxyglutamate. En cas de déficit envitamine K, une carboxylation insuffisante du glutamate de la prothrombine, pro-téine de la coagulation, peut conduire à des hémorragies (p. 295). De nombreusesprotéines, en particulier celles présentes à la surface des cellules ou qui sont sécré-tées, acquièrent des unités glucidiques fixées à des résidus asparagine spécifiques(voir Chapitre 11). L’addition de glucides rend les protéines plus hydrophiles etcapables de participer à des interactions avec d’autres protéines. À l’inverse, l’addi-tion d’un acide gras au groupe amine ou au groupe sulfhydryle d’une cystéine pro-duit une protéine plus hydrophobe.
De nombreuses hormones, telles que l’adrénaline, modifient l’activité desenzymes en stimulant la phosphorylation des aminoacides hydroxylés sérine et thréo-
nine ; la phosphosérine et la phosphothréonine sont les aminoacides modifiés les plusubiquitaires des protéines. Des facteurs de croissance, tels que l’insuline, agissent endéclenchant la phosphorylation du groupe hydroxyle des résidus tyrosine pour formerla phosphotyrosine. Les groupes phosphoryle de ces trois aminoacides modifiés sontaisément éliminés ; ils sont alors capables de se comporter en commutateurs réver-sibles dans la régulation des processus cellulaires. Le rôle de la phosphorylation dansla transduction des signaux sera discuté en détail dans le Chapitre 14.
Les modifications précédentes résultent de l’addition de groupes spécifiques à desaminoacides. D’autres groupes spéciaux sont créés par réaménagements chimiques deschaînes latérales et, parfois, du squelette peptidique. Par exemple, certaines médusesproduisent une protéine présentant une fluorescence verte (la GFP, Figure 2.68).
5 8
CHAPITRE 2 Composition et structuredes protéines
Figure 2.68 Réarrangement chimique dans la GFP. (A) Structure de la protéine GFP(de « green fluorescent protein », protéine à fluorescence verte). Le réarrangement
et l’oxydation de la séquence Ser-Tyr-Gly est à l’origine de la fluorescence. (B) Micrographiede fluorescence d’un embryon à quatre cellules (les contours des cellules sont dessinés)du ver rond Caenorhabditis elegans contenant la protéine PIE-1 marquée par la GFP.La protéine n’est exprimée que dans la cellule (à la partie supérieure) qui donnera naissanceà la lignée germinale. [(A) Dessiné d’après 1GFL.pdb ; (B) Autorisation du Dr Geraldine Seydoux.]
(A)
O2
Ser
Tyr
Gly
N
N
N
H
HO
C
O
O
HO
H
N
O
N
O
N
O
HH
H
CH2H
H
HO
HO
H
(B)

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 35/40
L’origine de la fluorescence est due à un groupe formé par le réarrangement etl’oxydation spontanée de la séquence Ser-Tyr-Gly au centre de la protéine. Cetteprotéine est d’une grande utilité pour les chercheurs comme marqueur au sein descellules (p. 89).
Enfin, de nombreuses protéines sont clivées et découpées après synthèse. Parexemple, les enzymes digestifs sont synthétisés sous forme de précurseurs inactifsqui peuvent être mis en réserve sans danger dans le pancréas. Après libération dansl’intestin, ces précurseurs sont activés par clivage d’une liaison peptidique (p. 289).Dans la coagulation du sang, le clivage d’une liaison peptidique convertit le fibrino-gène soluble en fibrine insoluble (p. 293). De nombreuses hormones polypepti-diques, telles que l’hormone adrénocorticotrope (ACTH), proviennent du clivaged’un grand précurseur protéique unique. De même, de nombreuses protéinesvirales sont produites par le clivage de grandes polyprotéines précurseurs. Nousrencontrerons de nombreux autres exemples de modification et de clivage comme
caractères essentiels de la formation et de la fonction des protéines. En fait, cestouches de finition expliquent une grande partie de la diversité, de la précision et del’élégance de l’action et de la régulation des protéines.
RésuméLa structure des protéines peut être décrite à quatre niveaux. La structure pri-maire se réfère à la séquence des aminoacides. La structure secondaire décrit laconformation adoptée par des régions localisées de la chaîne polypeptidique. Lastructure tertiaire décrit le reploiement d’ensemble de la chaîne polypeptidique.Enfin, la structure quaternaire a trait à l’association spécifique de plusieurschaînes polypeptidiques pour former des complexes multi-sous-unitaires.
2.1 Les protéines sont construites à partir d’un répertoire de 20 aminoacidesLes protéines sont des polymères linéaires d’aminoacides. Chaque aminoacideest constitué d’un atome de carbone tétraédrique central uni à un groupeamine, un groupe acide carboxylique, une chaîne latérale caractéristique et unatome d’hydrogène. Ces centres tétraédriques, à l’exception de celui de la gly-cine, présentent une chiralité ; seul l’isomère L existe dans les protéines natu-relles. Toutes les protéines naturelles sont construites à partir du même groupede 20 aminoacides. Les chaînes latérales de ces 20 éléments constitutifs diffè-rent considérablement par leur taille, leur forme et la présence de groupes fonc-tionnels. Ces derniers peuvent être regroupés de la façon suivante : (1) chaîneslatérales aliphatiques : glycine, alanine, valine, leucine, isoleucine, méthionineet proline ; (2) chaînes latérales aromatiques : phénylalanine, tyrosine et tryp-tophane ; (3) chaînes latérales aliphatiques contenant un hydroxyle : sérine etthréonine ; (4) cystéine contenant un sulfhydryle ; (5) chaînes latérales conte-nant un carboxamide : asparagine et glutamine (6) chaînes latérales basiques :lysine, arginine et histidine ; et (7) chaînes latérales acides : acide aspartique etacide glutamique. Ces regroupements sont quelque peu arbitraires et d’autressont possibles.
2.2 Structure primaire : les aminoacides sont unis par des liaisons peptidiquespour former des chaînes polypeptidiquesLes aminoacides d’un polypeptide sont unis par des liaisons amide formées
entre le groupe carboxyle d’un aminoacide, et le groupe amine du suivant.Cette liaison, appelée liaison peptidique a plusieurs propriétés importantes.Tout d’abord, elle est résistante à l’hydrolyse et donc les protéines sont remar-quablement stables du point de vue cinétique. Deuxièmement, la liaison pep-tidique est plane, étant donné que la liaison C¬N a un caractère important dedouble liaison. Troisièmement, chaque liaison peptidique possède à la fois undonneur de liaison hydrogène (le groupe NH) et un accepteur de liaison hydro-gène (le groupe CO). Les liaisons hydrogène entre ces groupes du squelettepolypeptidique représentent un des caractères spécifiques de la structure desprotéines. Enfin, la liaison peptidique n’est pas chargée, ce qui permet aux pro-
5 9
Résumé

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 36/40
téines de former des structures globulaires fortement compactes dont une partimportante du squelette polypeptidique est enfouie au sein de la protéine.Étant donné que les protéines sont des polymères linéaires, elles peuvent êtredécrites comme une séquence d’aminoacides. Cette dernière est écrite del’extrémité N-terminale vers l’extrémité C-terminale.
2.3 Structure secondaire : les chaînes polypeptidiques peuvent se reployeren structures régulières telles que l’hélice alpha, le feuillet bêta,les coudes et les bouclesDeux éléments majeurs de la structure secondaire sont l’hélice et le brin .Dans l’hélice , la chaîne polypeptidique s’enroule en un bâtonnet étroitementcompacté. Au sein de l’hélice, le groupe CO de chaque aminoacide est uni parune liaison hydrogène au groupe NH de l’aminoacide situé quatre résidus plusloin dans la chaîne polypeptidique. Dans le brin , la chaîne polypeptidique estpresque totalement étirée. Deux brins , ou davantage, connectés par des liai-sons hydrogène entre des NH et des CO s’assemblent pour former des feuillets. Les brins des feuillets peuvent être antiparallèles, parallèles, ou mixtes.
2.4 Structure tertiaire : les protéines hydrosolubles se reploient en structurescompactes présentant des cores non polairesLa structure compacte, asymétrique, qu’adopte chaque polypeptide, est appeléestructure tertiaire. Les structures tertiaires des protéines hydrosolubles ont encommun divers caractères : (1) une partie interne formée d’aminoacides présen-tant des chaînes latérales hydrophobes et (2) une surface constituée principale-ment d’aminoacides hydrophiles qui interagissent avec l’environnement aqueux.La force qui détermine la formation de la structure tertiaire des protéines hydro-solubles est due aux interactions hydrophobes entre les résidus de la régioninterne. Les protéines qui se situent dans l’environnement hydrophobe des mem-branes présentent une distribution inverse des aminoacides hydrophobes et desaminoacides hydrophiles. Dans ces protéines, les aminoacides hydrophobes sontà la surface pour entrer en interaction avec l’environnement lipidique, tandis queles groupes hydrophiles sont protégés de cet environnement en étant situés ausein de la protéine.
2.5 Structure quaternaire : les chaînes polypeptidiques peuvent s’assembleren des structures multi-sous-unitairesLes protéines constituées de plus d’une chaîne polypeptidique présentent unestructure quaternaire et chaque chaîne polypeptidique prise individuellementest appelée sous-unité. La structure quaternaire peut être aussi simple quedeux sous-unités identiques, ou aussi complexe que des douzaines de sous-unités différentes. Dans la plupart des cas, les sous-unités sont maintenuesensemble par des liaisons non covalentes.
2.6 La séquence des aminoacides d’une protéine détermine sa structuretridimensionnelleLa séquence des aminoacides détermine totalement la structure tridimension-nelle et, par conséquent, toutes les autres propriétés d’une protéine. Certainesprotéines peuvent être complètement déployées, puis reployées efficacementlorsqu’elles sont placées dans des conditions où la forme reployée de la protéineest stable. La séquence des aminoacides d’une protéine est déterminée par laséquence des bases d’une molécule de DNA. Cette information à une dimen-sion de la séquence est étendue au monde tridimensionnel par la capacité des
protéines à se reployer spontanément. Le reploiement des protéines est un pro-cessus hautement coopératif ; les intermédiaires structuraux entre les formesdéployées et les formes reployées ne s’accumulent pas.
La diversité des protéines est considérablement accrue par des modifica-tions covalentes. Ces dernières peuvent inclure des groupes fonctionnels nonprésents dans les 20 aminoacides. D’autres modifications sont importantespour la régulation de l’activité des protéines. Grâce à leur stabilité structurale,leur diversité et leur réactivité chimique, les protéines rendent possibles la plu-part des processus clé associés à la vie.
6 0
CHAPITRE 2 Composition et structuredes protéines

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 37/40
Appendice 6 1
APPENDICE. Représentation des structures moléculaire II : protéines
Figure 2.69 Modèle compact du lysozyme. Remarquez commeles atomes sont étroitement empilés, avec très peu d’espace vide.Tous les atomes sont montrés à l’exception des atomesd’hydrogène. Les atomes d’hydrogène sont souvent omiscar leurs positions ne sont pas déterminées facilementpar cristallographie aux rayons X, et parce que cette omissionaméliore la clarté de la représentation de la structure.
Les scientifiques on développé des techniques puissantes pourla détermination des structures des protéines, comme nous leverrons dans le Chapitre 3. Dans la plupart des cas, ces tech-niques permettent de déterminer les positions des milliersd’atomes d’une protéine. Le résultat final d’une telle expé-rience comprend les coordonnées x, y, et z pour chaque atomede la structure. Ces dossiers de coordonnées sont compilésdans la Banque de Données des Protéines (http://www.rcsb.org/pdb/) à partir de laquelle elles peuvent être facile-ment téléchargées. Ces structures comprennent des milliers oumême des dizaines de milliers d’atomes. La complexité de pro-téines ayant des milliers d’atomes pose un problème sérieux àceux qui veulent décrire leur structure. Plusieurs types diffé-rents de représentations sont utilisés pour dépeindre les pro-téines, chacun avec ses forces et ses faiblesses. Les types quevous verrez le plus souvent dans ce livre sont les modèles com-pacts, les modèles éclatés, les modèles « fil de fer » (ou sque-lette), et les diagrammes en ruban. Quand cela sera approprié,nous noterons les caractères structuraux d’importance ou depertinence particulières dans la légende des illustrations.
Modèles compacts
Les modèles compacts sont les types de représentations lesplus réalistes. Chaque atome est montré comme une sphèredont la taille correspond au rayon de van der Waals de l’atome(p. 8). Les liaisons ne sont pas montrées explicitement maissont représentées par l’intersection des sphères apparentes
quand les atomes sont plus proches que la somme de leursrayons de van der Waals. Tous les atomes sont montrés, ycompris ceux qui forment le squelette et ceux des chaîneslatérales. Un modèle compact du lysozyme est représentéFigure 2.69.
Les modèles compacts rendent compte du peu d’espacelibre existant dans la structure d’une protéine, qui a toujoursbeaucoup d’atomes en contact de van der Waals les uns avecles autres. Ces modèles sont particulièrement utiles pourmontrer les changements de conformation spatiale d’uneprotéine dans des conditions différentes. Un désavantagedes modèles compacts est que les structures secondaire et ter-tiaire de la protéine sont difficiles à voir. Donc, ces modèlesne sont pas très utiles pour distinguer une protéine d’uneautre (beaucoup de modèles compacts de protéines se res-semblent énormément).
Modèles éclatés
Les modèles éclatés ne sont pas aussi réalistes que les modèlescompacts. Les atomes dépeints de manière réaliste occupentplus d’espace (car celui-ci est déterminé par leurs rayons devan der Waals), que les atomes représentés dans les modèleséclatés. Cependant, l’arrangement des liaisons est plus facileà voir parce que les liaisons sont explicitement représentéescomme des bâtonnets (Figure 2.70). Un modèle éclaté révèleune structure complexe plus clairement qu’un modèle com-pact. Cependant, la description est si compliquée que les
caractères structuraux tels que les hélices ou les sites defixation potentiels sont difficiles à voir.
Comme les modèles compacts et éclatés représentent lesstructures des protéines au niveau atomique, le grand nombre
d’atomes de ces structures complexes rend difficile la percep-tion des caractères structuraux pertinents. Aussi, des repré-sentations plus schématiques, comme les modèles « fil de fer »et les diagrammes en ruban, ont été développés pour la des-cription des structures macromoléculaires. Dans ces repré-sentations, la plupart ou la totalité des atomes ne sont pasmontrés explicitement.
Figure 2.70 Modèle éclaté du lysozyme. Les atomes d’hydrogènesont encore omis.

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 38/40
6 2 CHAPITRE 2 Composition et structure des protéines
Figure 2.71 Modèle fil de fer (ou en squelette) du lysozyme.
Figure 2.72 Diagramme en ruban du lysozyme. Les hélices sontreprésentées comme des rubans enroulés, et les brins commedes flèches. Les structures plus irrégulières sont représentéessous la forme de tubes fins.
Figure 2.73 Diagramme en ruban du lysozyme avec les particularitésimportantes. Quatre ponts disulfure et un résidu aspartatefonctionnellement important sont représentés sous la formede modèle éclaté.
Modèles « fil de fer » (ou en squelette)
Les modèles fil de fer montrent seulement le squelette desatomes d’une molécule de polypeptide ou même seulementl’atome de carbone de chaque aminoacide. Les atomes sontreliés par des lignes représentant les liaisons ; quand unique-ment les atomes de carbone sont représentés, ces lignesconnectent les atomes de carbone des aminoacides qui sontadjacents dans la séquence d’aminoacides (Figure 2.71).Dans ce livre, les modèles fil de fer montrent seulement leslignes qui connectent les atomes de carbone ; les autresatomes ne sont pas montrés.
Un modèle fil de fer montre l’aspect général d’une chaîne
polypeptidique bien mieux que les modèles compacts ouéclatés. Cependant, les éléments de structure secondaire sontencore difficiles à voir.
Diagrammes en ruban
Les diagrammes en ruban sont très schématiques et le plussouvent utilisés pour insister sur quelques aspects saillantsde la structure d’une protéine, telle que l’hélice (dessinéecomme un ruban enroulé ou un cylindre), le brin (uneflèche large), et les boucles (de simples lignes), pour donnerune vue claire et nette du reploiement général d’une pro-téine (Figure 2.72). Le diagramme en ruban permet deretracer la progression d’une chaîne polypeptidique etmontre facilement les éléments de structure secondaire.Ainsi, les diagrammes en ruban de protéines qui sont appa-rentées par divergence évolutive se ressemblent (voir
Figure 6.14), alors que les protéines non apparentées sontclairement distinctes.Dans ce livre, les rubans enroulés seront en général utilisés
pour dépeindre les hélices. Cependant, pour les protéines demembrane, qui sont souvent très complexes, des cylindresseront plutôt utilisés. Cette convention rendra aussi faciles àreconnaître les protéines membranaires avec leurs hélices transmembranaires (voir Figure 12.18).
Gardez cependant présent à l’esprit que l’apparence desdiagrammes en ruban est trompeuse. Comme nous l’avonsremarqué plus tôt, les structures des protéines sont très com-pactes et ont peu d’espace libre. La clarté des diagrammes enruban les rend particulièrement utiles comme cadres danslesquels ont peu mettre en lumière d’autres aspects de lastructure d’une protéine. Les sites actifs, les substrats, lesliaisons, et d’autres éléments de structure peuvent être inclussous une forme éclatée ou compacte au sein même d’un dia-gramme en ruban (Figure 2.73).
chaîne latérale (groupe R) (p.27) side chain (R group)
aminoacide L (p.27) L amino acid
ion dipolaire (zwitterion) (p.27) dipolar ion (zwitterion)
liaison peptidique (liaison amide) (p.34) peptide bond (amide bond)
Mots Clé
Brin β
Hélice α
Liaisons disulfure
Liaisons disulfure
Résiduaspartate
du site actif

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 39/40
Pour commencerRichardson, J. S. 1981. The anatomy and taxonomy of protein structure.
Adv. Protein Chem. 34:167–339.Doolittle, R. F. 1985. Proteins. Sci. Am. 253(4):88–99.Richards, F. M. 1991. The protein folding problem. Sci. Am. 264(1):
54–57.Weber, A. L., and Miller, S. L. 1981. Reasons for the occurrence of the
twenty coded protein amino acids. J. Mol. Evol. 17:273–284.
LivresPetsko, G.A., and Ringe, D. 2004. Protein Structure and Function. New
Science Press.Branden, C., and Tooze, J. 1999. Introduction to Protein Structure (2d
ed.). Garland.Perutz, M. F. 1992. Protein Structure: New Approaches to Disease and
Therapy.W. H. Freeman and Company.Creighton, T. E. 1992. Proteins: Structures and Molecular Principles (2d
ed.). W. H. Freeman and Company.Conformation des protéinesDobson, C.M. 2003. Protein folding and misfolding. Nature 426:
884–890.Richardson, J. S., Richardson, D. C., Tweedy, N. B., Gernert, K. M.,
Quinn, T. P., Hecht, M. H., Erickson, B. W., Yan, Y., McClain,R. D., Donlan, M. E., and Suries, M. C. 1992. Looking at proteins:Representations, folding, packing, and design. Biophys. J.
63:1186–1220.Chothia, C., and Finkelstein, A. V. 1990. The classification and origin
of protein folding patterns. Annu. Rev. Biochem. 59:1007–1039.
Hélices alpha, feuillets bêta, et coudesO’Neil, K. T., and DeGrado, W. F. 1990. A thermodynamic scale for
the helix-forming tendencies of the commonly occurring aminoacids. Science 250:646–651.
Zhang, C., and Kim, S. H. 2000. The anatomy of protein beta-sheettopology. J. Mol. Biol. 299:1075–1089.
Regan, L. 1994. Protein structure: Born to be beta. Curr. Biol.
4:656–658.Leszczynski, J. F., and Rose, G. D. 1986. Loops in globular proteins: A
novel category of secondary structure. Science 234:849–855.Srinivasan, R., and Rose, G. D. 1999. A physical basis for protein
secondary structure. Proc. Natl. Acad. Sci. U.S.A.
96:14258–14263.
DomainesBennett, M. J., Choe, S., and Eisenberg, D. 1994. Domain swapping:
Entangling alliances between proteins. Proc. Natl. Acad. Sci.
U.S.A. 91:3127–3131.Bergdoll, M., Eltis, L. D., Cameron, A. D., Dumas, P., and Bolin, J. T.
1998. All in the family: Structural and evolutionary relationships
among three modular proteins with diverse functions and variableassembly. Protein Sci. 7:1661–1670.
Hopfner, K. P., Kopetzki, E., Kresse, G. B., Bode, W., Huber, R., andEngh, R. A. 1998. New enzyme lineages by subdomain shuffling.Proc. Natl. Acad. Sci. U.S.A 95:9813–9818.
Ponting, C. P., Schultz, J., Copley, R. R., Andrade, M. A., and Bork, P.2000. Evolution of domain families. Adv. Protein Chem.
54:185–244.
Reploiement des protéinesCaughey, B., and Lansbury, P. T. 2003. Protofibrils, pores, fibrits, and
neurodegeneration: Separating the responsible protein aggregatesfrom innocent bystanders. Annu. Rev. Neurosci. 26: 267–298.
Daggett, V., and Fersht, A. R. 2003. Is there a unifying mechanism forprotein folding? Trends Biochem. Sci. 28:18–25.
Selkoe, D. J. 2003. Folding proteins in fatal ways . Nature 426:900–904.Anfinsen, C. B. 1973. Principles that govern the folding of protein
chains. Science 181:223–230.Baldwin, R. L., and Rose, G. D. 1999. Is protein folding hierarchic? I.
Local structure and peptide folding. Trends Biochem. Sci. 24:26–33.Baldwin, R. L., and Rose, G. D. 1999. Is protein folding hierarchic? II.
Folding intermediates and transition states. Trends Biochem. Sci.
24:77–83.Kuhlman, B., Dantas, G., Ireton, G. C., Varani, G., Stoddard, B. L.,
and Baker, D. 2003. Design of a novel globular protein with atomic-level accuracy. Science 302: 1364–1368.
Staley, J. P., and Kim, P. S. 1990. Role of a subdomain in the folding of bovine pancreatic trypsin inhibitor. Nature 344:685–688.
Modifications covalentes des protéinesKrishna, R. G., and Wold, F. 1993. Post-translational modification of
proteins. Adv. Enzymol. Relat. Areas. Mol. Biol. 67:265–298.Aletta, J. M., Cimato, T. R., and Ettinger, M. J. 1998. Protein methyl-
ation: A signal event in post-translational modification. Trends
Biochem. Sci. 23:89–91.Glazer, A. N., DeLange, R. J., and Sigman, D. S. 1975. Chemical
Modification of Proteins. North-Holland.Tsien, R. Y. 1998. The green fluorescent protein. Annu. Rev. Biochem.
67:509–544.
Représentations moléculaires graphiquesKraulis, P. 1991. MOLSCRIPT: A program to produce both detailed
and schematic plots of protein structures. J. Appl. Cryst.
24:946–950.Ferrin, T., Huang, C., Jarvis, L., and Langridge, R. 1988. The MIDAS
display system. J. Mol. Graphics 6:13–27.Richardson, D. C., and Richardson, J. S. 1994. Kinemages: Simple
macromolecular graphics for interactive teaching and publication.Trends Biochem. Sci. 19:135–138.
Choix de lectures
Choix de lectures 6 3
liaison disulfure (p.35) disulfide bond
structure primaire (p.36) primary structure
angle de torsion (p.39) torsion angle
angle phi () (p.39) phi ( ) angle
angle psi () (p.39) psi ( ) angle
diagramme de Ramachandran (p. 39) Ramachandran diagram
structure secondaire (p.40) secondary structure
hélice (p.40) helix
élévation (p.40) rise
translation (p.40) translation
feuillet plissé (p.42) pleated sheet
brin (p.42) strand
coude (coude ; coude en épingle à cheveux) (p.44) reverseturn ( turn ; hairpin turn)
structure surenroulée (p.45) coiled coil
répétitions en heptade (p. 45)heptad repeats
structure tertiaire (p.47) tertiary structure
motif (p.49) motif
structure supersecondaire (p. 49) supersecondary structure
domaine (p.49) domain
sous-unité (p.49) subunit
structure quaternaire (p.49) quaternary structure
prion (p.53) prion
transition coopérative (p.55) cooperative transition

5/17/2018 ch02_pg025_064_Fr.qxd - slidepdf.com
http://slidepdf.com/reader/full/ch02pg025064frqxd 40/40
6 4 CHAPITRE 2 Composition et structure des protéines
(A)
(a)
120°, 120°
(b)
180°, 0°
(c)
180°, 180°
(d)
0°, 180°
(e)
60°, 40°
(B) (C) (D) (E)
1. Forme et dimension. (a) La tropomyosine, protéine musculairede 70 kd, est constituée de deux brins en hélice enroulés l’unautour de l’autre. Quelle est la longueur de la molécule ? (b)Supposez qu’un segment de 40 résidus d’une protéine se reploieen une structure antiparallèle à deux brins avec un coude enépingle à cheveux de quatre résidus. Quelle est la plus longuedimension de ce motif ?
2. Isomères différents. La poly-L-leucine dans un solvant orga-nique tel que le dioxane est en hélice , tandis que la poly-L-isoleucine ne l’est pas. Pourquoi ces aminoacides, ayant le mêmenombre et le même type d’atomes, ont-ils des tendances diffé-rentes à former une hélice ?
3. De nouveau actif . Une mutation qui change un résidu alanineen une valine à l’intérieur d’une protéine conduit à la perte del’activité. Cependant, l’activité est récupérée lorsqu’une secondemutation à une position différente change un résidu isoleucineen glycine. Comment cette seconde mutation peut-elle conduireà la restauration de l’activité ?
4. Test d’échange. Un enzyme qui catalyse des réactionsd’échange disulfure-sulfhydryle, appelé protéine disulfure iso-mérase, a été isolé. La ribonucléase « brouillée » inactive est rapi-dement convertie en ribonucléase enzymatiquement active parcet enzyme. Par contre, l’insuline est rapidement inactivée par cemême enzyme. Qu’implique cette importante observation quantà la relation entre la séquence des aminoacides de l’insuline et sastructure tridimensionnelle ?
5. En étirant une cible. Une protéase est un enzyme qui catalysel’hydrolyse des liaisons peptidiques de protéines cible. Commentune protéase peut-elle se lier à une protéine cible de telle façonque la chaîne principale de cette dernière devienne complètementétirée dans la région de la liaison peptidique vulnérable ?
6. Souvent irremplaçable. La glycine est un résidu aminoacidetrès conservé dans l’évolution des protéines. Pourquoi ?
7. Partenaires potentiels. Identifiez les groupes d’une protéinequi peuvent former des liaisons hydrogène ou des liaisons élec-trostatiques avec une chaîne latérale arginine à pH 7.
8. Ondulations permanentes. La forme des cheveux est en partiedéterminée par le profil des liaisons disulfure de la kératine quiest leur protéine essentielle. Comment induire la formation deboucles de cheveux ?
9. La localisation est essentielle. Les protéines qui traversent lesmembranes biologiques contiennent souvent des hélices .
Étant donné que la partie interne des membranes est hautementhydrophobe (Section 12.2), prédire quel type d’aminoacide seraprésent dans une telle hélice. Pourquoi une hélice est-elle par-ticulièrement adaptée pour s’insérer dans l’environnementhydrophobe de la partie interne d’une membrane ?
10. Problèmes de stabilité . Les protéines sont très stables. Ladurée de vie d’une liaison peptidique en solution aqueuse estd’environ 1 000 ans. Cependant, l’énergie libre de l’hydrolysedes protéines est négative et très élevée. Comment pouvez-vousexpliquer la stabilité de la liaison peptidique, compte tenu du faitque l’hydrolyse libère autant d’énergie ?
11. Espèces mineures. Pour un aminoacide tel que l’alanine, l’es-pèce moléculaire majoritaire en solution à pH 7 est le zwitte-rion. En prenant une valeur de 8 pour le pK a du groupe amineet de 3 pour l’acide carboxylique, estimez le rapport de laconcentration de l’aminoacide neutre (avec l’acide carboxyliqueprotoné et le groupe amine neutre) à celle du zwitterion, à pH 7(voir p. 16).
12. Question de convention. Tous les aminoacides L ont la confi-guration absolue S à l’exception de la L-cystéine qui a la confi-guration R. Expliquez pourquoi la L-cystéine a la configurationabsolue R.
13. Message caché . Traduisez la séquence des aminoacides ci-après dans le code à une lettre : Glu-Leu-Val-Ile-Ser-Ile-Ser-Leu-Ile-Val-Ile-Asn-Gly-Ile-Asn-Leu-Ala-Ser-Val-Glu-Gly-Ala-Ser.
14. Qui arrive le premier ? Selon vous, les liaisons peptidiquesPro-X tendent-elles à prendre des conformations cis commecelles des liaisons X-Pro ? Pourquoi ou pourquoi pas ?
15. Appariement. Pour chacun des dérivés d’aminoacides repré-sentés ci-contre (A-E), trouver l’appariement des valeurs de etde (a-e).
Problèmes