b. etude de la surtension de diffusion 1. … · electrochimie et applications 143 b. etude de la...
TRANSCRIPT

Electrochimie et applications 143
B. ETUDE DE LA SURTENSION DE DIFFUSION
1. Installation d'un régime de diffusion pure
Pour obtenir cette situation, il est nécessaire que les substances (ions) qui
réagissent à l'électrode soient transportées uniquement par diffusion.
D'après l'expression trouvée précédemment pour le flux total relatif à l'espèce , il faut
que J migration et J convection soient nuls.
Il en résulte que :
- la vitesse de déplacement de l'électrolyte doit être nulle. En réalité, l'électrolyte peut
être en mouvement stationnaire et produire un régime de diffusion convective; mais
nous n'envisagerons pas ce cas dans le présent développement.
- le nombre de transport t doit être nul. Pour cela, le courant de migration est assuré
par un électrolyte appelé électrolyte support
tel que Csupport C et usupport u t 0
De plus, l'électrolyte support (ou indifférent) ne doit pas réagir dans le domaine de
potentiel étudié.
Seule la substance (ion) transportée par diffusion peut s'oxyder ou se réduire à
l'électrode.
2. Calcul de la surtension de diffusion en régime stationnaire
Cette surtension intervient lorsque l'étape limitative est le transport de matière par
diffusion.
Les substances électroactives consommées ou produites à l'électrode sont freinées dans
leur mouvement d'approche ou de départ. C’est ce transfert de matière qui détermine la
vitesse de la réaction globale. Les étapes de décharge, de cristallisation, de réaction
chimique (plus rapides) sont à l'équilibre (ou n'interviennent pas dans le mécanisme
réactionnel).
Dans ces conditions, on peut appliquer la relation de Nernst pour le calcul des tensions
d'électrodes, même si celles-ci sont parcourues par un courant.
Il faudra cependant tenir compte dans ce calcul, des concentrations en
substances actives régnant au voisinage de l'électrode, et non de celles qui
existent au sein de la solution.
La surtension de diffusion est alors définie comme étant la différence :
d = Ei - Eéq
(différence entre la tension d'équilibre à courant nul et la tension Ei calculée par la
formule de Nernst pour la concentration en ions indicateurs au voisinage immédiat de la
surface de l'électrode).

Electrochimie et applications 144
Pour une substance électroactive quelconque, on a :
E éq = E° + R T
n F . ln a a = activité de au sein de la solution
Rappelons que est positif pour les réactifs situés du côté de l'oxydant et que, d'une
façon générale, E éq est donné par :
E éq = E° + R T
n F . . ln a
Si un courant i circule dans le système :
C = C (i,t) C
a = a (i,t) a
La tension devient :
E(i,t) = E° + R T
n F.ln a (i,t)
et = Ei - Eéq = R T
n F.ln
a i t
a
( , )
en général : d = RT
nF. .ln
a i t
a
( , )
Exemple : Fe2+ Fe 3+ + e
E éq = E° + RT
F
a
a. ln
3
2
d = R T
F.ln
.
.
a a
a a
3 2
2 3
Dans la suite du développement, nous conviendrons de remplacer les activités a par les
concentrations C .

Electrochimie et applications 145
Diffusion en présence d'un excès d'électrolyte support
Il existe plusieurs manières de faire apparaître un gradient de concentration au voisinage
d'une électrode:
* Diffusion sans migration (électrolyte support) ;
* Les ions sont produits par une réaction lente antécédente ou postérieure à la
décharge.
Nous envisagerons le premier cas, en régime stationnaire.
Exemples : une solution d'ions cuivriques Cu2+ dans un excès d'acide sulfurique H2SO4
Solution CuSO4 0,01 M
H2SO4 1 M
ou une solution de Cu2+ dans un excès de KCl.
Dans ces exemples les ions H+ et K + ne se déchargent pas à la cathode, mais ils sont
plus mobiles et plus concentrés que les ions Cu2+.
Si le courant augmente, on a enrichissement en Cu2+ au voisinage de l'anode et
épuisement en Cu2+ au voisinage de la cathode.
A C
CCu++
CCu++
La réaction de dépôt cathodique comporte 3 étapes :
Cu Cu
Cu2+
A
ANODE OXYDATION
CATHODE REDUCTION
i+ i
Cu Cu2+ + 2e Cu2+ + 2e Cu
CuSO4 0,01 M H2SO4 1 M

Electrochimie et applications 146
transfert de charges
Cu2+ + 2e Cu
transport incorporation dans le réseau cristallin
de la solution
vers la cathode
ELECTRODE
DC
H
COUCHE
DE DIFFUSION
épaisseur variable
selon agitation
de qqc 0.01 mm
à 1 mm
SOLUTION ELECTROLYTIQUE
Double
Couche Helmoltz
qqc Angströms
a) Cas de l'appauvrissement à la cathode
Pour la réaction de réduction : Cu2+ + 2 e Cu
Cu2 est positif et i est négatif par convention cath .
on a donc dans ce cas i. 0
Si : C = la concentration de la matière qui diffuse
x = la distance mesurée dans la direction de la diffusion (direction perpendiculaire
à l'électrode dans le cas d'une électrode plane).
et si on suppose que la couche de diffusion a une épaisseur diff constante, que le
gradient de concentration est linéaire et constant en état de régime,
C
Co
diff x
ELECTRODE
épaisseur de la couche
C

Electrochimie et applications 147
L'application de la première loi de Fick donne :
J =
i.
- D - DnF
grad C
dC
dx. .
dC
dx
C C
= - où C = C pour x = 0
i.
nF = D .
C C
ou (1)
Dans cette expression, i est bien négatif car C Co
i est donc une image de C (puisque tout le reste est constant).
Si C = 0, tous les ions arrivant à l'électrode sont instantanément déchargés et toute
augmentation du potentiel de l'électrode Ei (ou de ) ne pourra plus provoquer une
augmentation du courant.
Ce courant maximum est appelé courant limite.
(2)
id, est une mesure de C si D est constant
Comme d
R T
nF
C i t
C. ln
( , )
, il faut évaluer
C i t
C
( , )
pour obtenir la formule qui lie le
courant à la tension Ei appliquée.
Ce rapport peut être tiré des équations (1) et (2):
i
i
C C
C
C
Cd ,
1 ou encore :
C
C
i
id
1
,
i = nF
. ( )
D
C C
inF
D
Cd ,
. .

Electrochimie et applications 148
d'où d doit rester 0 pour une réduction et id 0 et i id
Dans la précédente formule, on remarque que si i tend vers id, la surtension d tend vers
l'infini. En réalité, cette surtension sera limitée par une autre réaction, par exemple la
décomposition de l'eau.
En général, si plusieurs substances diffusent simultanément, on peut écrire:
d
d
R T
nF
i
i. . ln
,
1
Dans l'exemple du système oxydo-réducteur:
Fe3+ + e Fe2+
on a: 2 = - 1 et 3 = 1
Ce qui donne: d
d
d
R T
F
i
i
i
i
. ln,
,
1
1
3
2
avec id,3 courant limite de diffusion lorsque la concentration en [Fe3+] à l'électrode
devient nulle, il s'agit alors d'un courant cathodique donc négatif.
id,2 courant limite de diffusion lorsque la concentration en [Fe2+] à l'électrode
devient nulle, c'est le cas de l'oxydation de Fe2+ en Fe3+, le courant de
diffusion correspondant est donc positif.
d
d
R T
nF
i
i. ln
,
1
id,3
id,2
E/ENH
i+
i

Electrochimie et applications 149
Pour l'exemple de départ, pour lequel une seule espèce diffuse: Cu2+ + 2e Cu
On peut aussi écrire la relation entre le courant et la surtension de la façon suivante:
Pour i tendant vers id, la surtension d tend vers l'infini.
Si plusieurs substances peuvent diffuser et se réduire successivement, on a:
id Cu
2 + U(V/ECS)
i
i i . (1 e ) d
.nF
R T
d
i
d
id2
id1 E(V/ECS)
i
id

Electrochimie et applications 150
b) Cas de l'enrichissement (exemple de la dissolution d'un métal à une anode)
Dans ce cas, le courant est positif (oxydation) et le coefficient stoechiométrique est
positif ( i. > 0).
Cu Cu 2+ + 2e
L'expression du courant reste la même que dans le cas de l'appauvrissement:
i = nF
. ( )
D
C C (1)
avec cependant C C
C ne peut augmenter indéfiniment, il existe une concentration limite C*qui peut, par
exemple, correspondre à la limite de solubilité du sel formé et i tend alors vers une limite
id ,*
.
3. Signification du courant limite ou courant de diffusion
Tous les ions arrivant à l'électrode sont déchargés instantanément, l'étape de transfert de
charge est donc beaucoup plus rapide que la diffusion des réactifs vers l'électrode.
Les courbes potentiocinétiques i = f ( d) sont caractéristiques et présentent un palier
appelé palier de diffusion.
Pour augmenter le courant limite, on peut:
- Augmenter la concentration des réactifs en solution C ;
- Diminuer l'épaisseur de la couche de diffusion , par agitation de
l’électrolyte ou rotation de l'électrode (on parle dans ce cas de diffusion
convective).
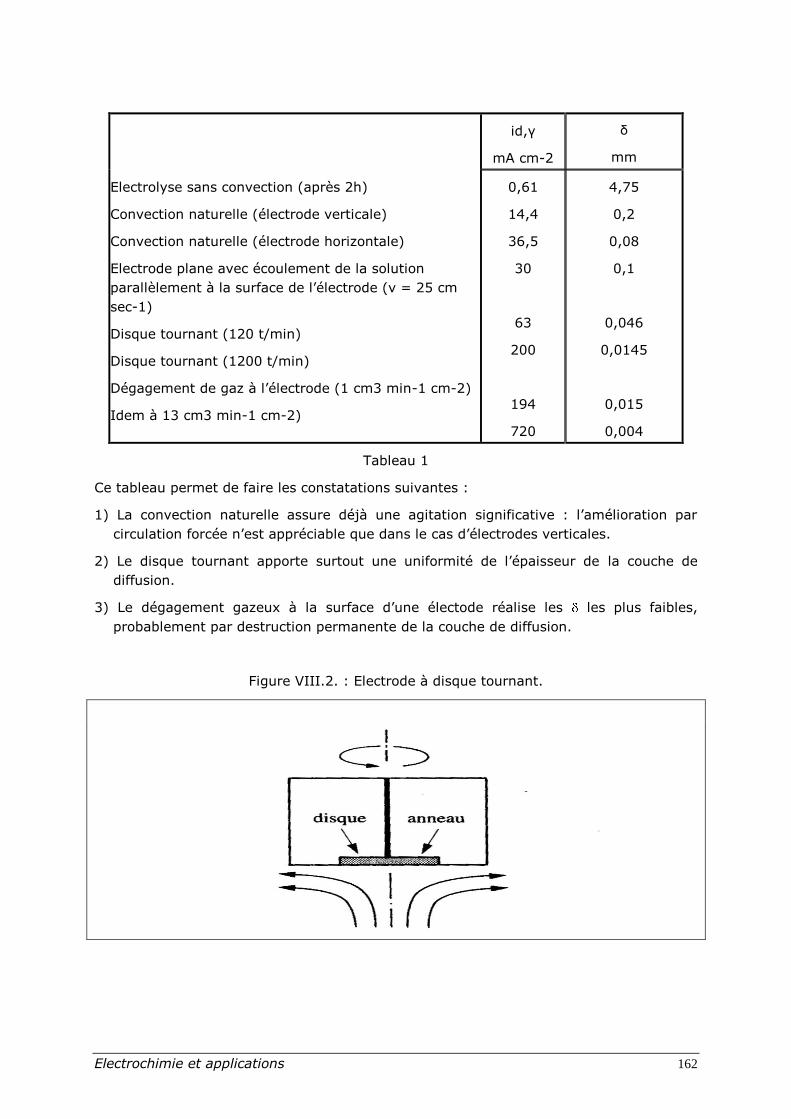
On donne au tableau I quelques valeurs de pour divers régimes hydrodynamiques.
id C
-E/ENH
i-
Electrochimie et applications 162
id,γ
mA cm-2
δ
mm
Electrolyse sans convection (après 2h)
Convection naturelle (électrode verticale)
Convection naturelle (électrode horizontale)
Electrode plane avec écoulement de la solution
parallèlement à la surface de l’électrode (v = 25 cm
sec-1)
Disque tournant (120 t/min)
Disque tournant (1200 t/min)
Dégagement de gaz à l’électrode (1 cm3 min-1 cm-2)
Idem à 13 cm3 min-1 cm-2)
0,61
14,4
36,5
30
63
200
194
720
4,75
0,2
0,08
0,1
0,046
0,0145
0,015
0,004
Tableau 1
Ce tableau permet de faire les constatations suivantes :
1) La convection naturelle assure déjà une agitation significative : l’amélioration par
circulation forcée n’est appréciable que dans le cas d’électrodes verticales.
2) Le disque tournant apporte surtout une uniformité de l’épaisseur de la couche de
diffusion.
3) Le dégagement gazeux à la surface d’une électode réalise les les plus faibles,
probablement par destruction permanente de la couche de diffusion.
Figure VIII.2. : Electrode à disque tournant.

Electrochimie et applications 163
L'électrode à disque tournant est un dispositif souvent utilisé en électrochimie pour
déterminer si une réaction est limitée par la diffusion des réactifs. Ce système est en
effet fort intéressant et on montre que l'épaisseur de la couche de diffusion varie dans ce
cas inversément proportionnellement à la vitesse de rotation angulaire de l'électrode à
disque:
/1 avec vitesse de rotation du disque (trs/min)
Si la réaction étudiée est limitée par la diffusion, on a:
di
REMARQUE IMPORTANTE
Si on ne connait pas avec précision les coefficients de diffusion D des ions , il existe des
formules permettant de les calculer lorsque l'on connait la mobilité u de l'ion
correspondant ou la conductivité ionique . Ces formules, que nous ne démontrerons
pas, indiquent qu' il existe une relation directe entre mobilité des ions et coefficient de
diffusion:
Relation Einstein : relie le coefficient de diffusion et la mobilité des ions
Du RT
nF
.
Relation Nernst-Einstein : relie la conductivité ionique au coefficient de diffusion D
(.
).n F
RTD
2 2
4. Diffusion en régime non stationnaire
Comme nous l'avons signalé dans l'introduction relative à la cinétique, on exploite en
régime transitoire, des relations dans lesquelles on suit l'évolution au cours du temps de
l'une des deux variables électrochimiques (Eélect/réf, I ) lorsque l'on impose une valeur
constante à l'autre.
On distingue: * la chronoampérométie : courbes I = f (t) à E = constante
* la chronopotentiométrie : courbes E = f (t) à I = constante
Les lois relatives à ces phénomènes transitoires s'obtiennent à partir de la résolution de
la seconde loi de Fick :

Electrochimie et applications 164
C
tD C.
2
Dans le cas particulier de l' électrode plane, elle s'écrit:
C
tD
C
x
.
2
2
On peut résoudre l'équation en maintenant la tension d'électrode constante (électrolyse
potentiostatique) ou le courant d'électrolyse constant (électrolyse galvanostatique).
a) Electrolyse potentiostatique (chronoampérométrie)
On fixe E électrode/Réf = cste , or comme nous l'avons vu précédemment, la surtension
de diffusion vaut : d
RT
nF
C
C. ln . De ce fait, il faut nécessairement que C i t( , ) soit
constant.
Le problème revient donc à trouver, par intégration, la distribution des concentrations
C i t( , ) et à calculer le courant traversant l'électrode:
i nF
JnF
D
Cx t
xx
x
= . .( , )
( ).
0
0
L'intégration de l'équation de Fick avec la condition limite correspondant au potentiel
constant donne:
i
nF D C C t
tt
.
( , )
.
0
En particulier, pour des tensions appliquées suffisamment négatives, on tire:
i
nF DC
tt
..
La densité de courant est inversément proportionnelle à la racine carrée du temps
d'électrolyse et en particulier le courant limite (C 0) est proportionnel à la
concentration de l'ion au sein de la solution. On peut faire usage de cette propriété pour
déterminer la concentration de la solution en élément électroactif (chronoampérométrie).

Electrochimie et applications 165
Figure VIII.3. : Chronoampérométrie.
b) Electrolyse galvanostatique (chronopotentiométrie)
On maintient la densité de courant constante:
i cstenF
D grad Cx
. . ( )0
c'est-à-dire que cela revient à maintenir (grad C )x=0 constant.
En intégrant l'équation de Fick, on obtient à l'interface, pour x=0:
C t Ci t
nF D( , )
. .
.0
2
Pour un courant suffisant, la concentration C i t( , ) décroît avec le temps et devient nulle
après un temps appelé temps de transition (figure VIII.4.).
est égal à :
( ) . . .
.
nF D C
i
2
2
2
4
C
Ces équations constituent la base théorique de diverses méthodes d'étude de la cinétique
des réactions d'électrodes. En outre, l'expression de permet des applications analytiques
puisque est proportionnel au carré de la concentration de l'ion en solution
(chronopotentiométrie).
Pour une réaction électrochimique ayant lieu sans complication d'ordre cinétique, on montre
que :
i c k Cste
. .
C it
t X
C
t1 t2 t3 t4 t5
t1 t2 t3 t4 t5

Electrochimie et applications 166
C'est la relation de SAND
Figure VIII.5. : Chonopotentiométrie.
5. Application de la surtension de diffusion : la polarographie.
La polarographie est une méthode d'analyse qualitative et quantitative des ions en
solution, elle est basée sur la surtension de diffusion.
En réalité, on trace la courbe de polarisation potentiodynamique d'une électrode à goutte
de mercure dans une solution composée d'un électrolyte support et du ou des ions à
analyser en faible concentration (de 10-3 à 10-6 M).
La variable indépendante est le potentiel appliqué à l'électrode à goutte de mercure, on
fait varier linéairement ce potentiel en fonction du temps (balayage en tension). Le plus
souvent, on travaille en réduction et on applique des potentiels de plus en plus négatifs.
Le dispositif expérimental (Figure VIII.6.) simplifié est représenté ci-dessous:
Figure VIII.6. : Schéma de principe d’un polarographe.

Electrochimie et applications 167
La polarographie utilise une électrode à goutte de mercure (ou une microélectrode solide)
qui joue le plus souvent le rôle de cathode, et on s’arrange pour que les ions réductibles
(ou oxydables) parviennent à l’électrode à goutte sous le seul effet de la diffusion; dans
ce but, on incorpore à la solution un électrolyte support.
Dans ces conditions, nous verrons qu’il est possible d’obtenir des
renseignements d’ordre analytique (qualitatif et quantitatif) et d’ordre cinétique
(cinétique de diffusion de l’oxydant ou de réducteur et cinétique de la réaction
d’électrode).
Il se crée entre l’électrode à goutte de mercure et le sein de la solution un gradient de
concentration qui peut être défini en chaque point et à chaque instant. Tout le
développement théorique qui va suivre a pour but d’exprimer ce gradient sous forme
mathématique en fonction des divers paramètres opératoires.
L’électrode à goutte est constituée d’un capillaire d’où s’écoule le mercure sous l’effet de
la pesanteur ou d’une pression appliquée. En jouant sur les dimensions du capillaire, on
peut réaliser un régime de fines gouttelettes ( 0,01 cm2) qui se détachent
périodiquement (2 à 10 sec), on peut également régler la durée de vie d’une goutte à
l’aide d’un marteau ou d’un dispositif pneumatique à des valeurs comprises entre 0,1 et
quelques secondes.
L’intérêt de l’électrode à goutte est son renouvellement continu (électrode toujours
identique).
La caractéristique d’une électrode impolarisable est de conserver un potentiel constant
quels que soient le sens et l’intensité du courant qui la traverse; il s’agit généralement
soit d’une nappe de mercure, soit d’une électrode de référence qui offre l’avantage de
présenter un potentiel bien déterminé indépendant de la composition du milieu.
Sur la plupart des polarographes, le potentiel varie lentement et la courbe est enregistrée
automatiquement; la variation de potentiel est suffisamment lente pour que l’on puisse
considérer que la mesure est effectuée sur chaque goutte à potentiel appliqué constant.
Les polarogrammes obtenus de cette manière présentent des oscillations de courant.
Le tracé complet de la courbe i = f (E) s'appelle un polarogramme et il a l'allure reprise à
la figure ci-dessous. Cette figure correspond au polarogramme (Figure VIII.7.) tracé à
partir d'une solution contenant des ions Cu2+ et Tl+ .
Le courant correspondant au palier est le courant limite proportionnel à la concentration
de l'ion en solution. Le potentiel correspondant au point d'inflexion est appelé potentiel
de demi-onde et on montre qu'il est caractéristique d'un ion donné dans un milieu
support fixé.

Electrochimie et applications 168
Figure VIII.7. Polarogramme de solutions contenant des ions Cu2+ et Tl+.
5.1. Recherche de l’expression générale du courant.
Les facteurs susceptibles de limiter le courant sont la vitesse de diffusion et la vitesse de
la réaction électrochimique. L’adsorption de substances à l’électrode peut également
jouer un rôle important que nous n’envisagerons pas dans le cadre de cette manipulation.
La résolution des équations de la diffusion avec les conditions initiales convenables et les
conditions aux limites tenant compte en particulier de la vitesse de la réaction
électrochimique, permet d’obtenir l’expression du courant en fonction du temps.
C'est une application de la chronoampérométrie, mais le problème est compliqué car la
surface de la goutte de mercure varie. La surface de l'électrode ainsi que la couche de
diffusion se renouvellent régulièrement. La durée de vie T d'une goutte est fonction du
diamètre du capillaire et de la pression exerçée sur le mercure.
Pour trouver l'expression du courant, il faut tenir compte de l'effet de diminution du
courant (proportionnel au gradient de concentration à la surface de l'électrode) et de
l'effet d'augmentation de la surface de la goutte: l'augmentation de la surface de la
goutte l'emporte sur la diminution du gradient de concentration et le courant global
augmente avec le temps.
a) Electrode plane de surface constante.
En chonoampérométrie, pour une tension E appliquée, le courant i est donc une fonction
décroissante du temps.
Electrochimie et applications 169
Pour un courant E suffisamment négatif, on obtient l’expression suivante du courant
limite
ia F C solution D
td
M en. . ( ).
.
/
/ /
1 2
1 2 1 2 Equation de COTRELL
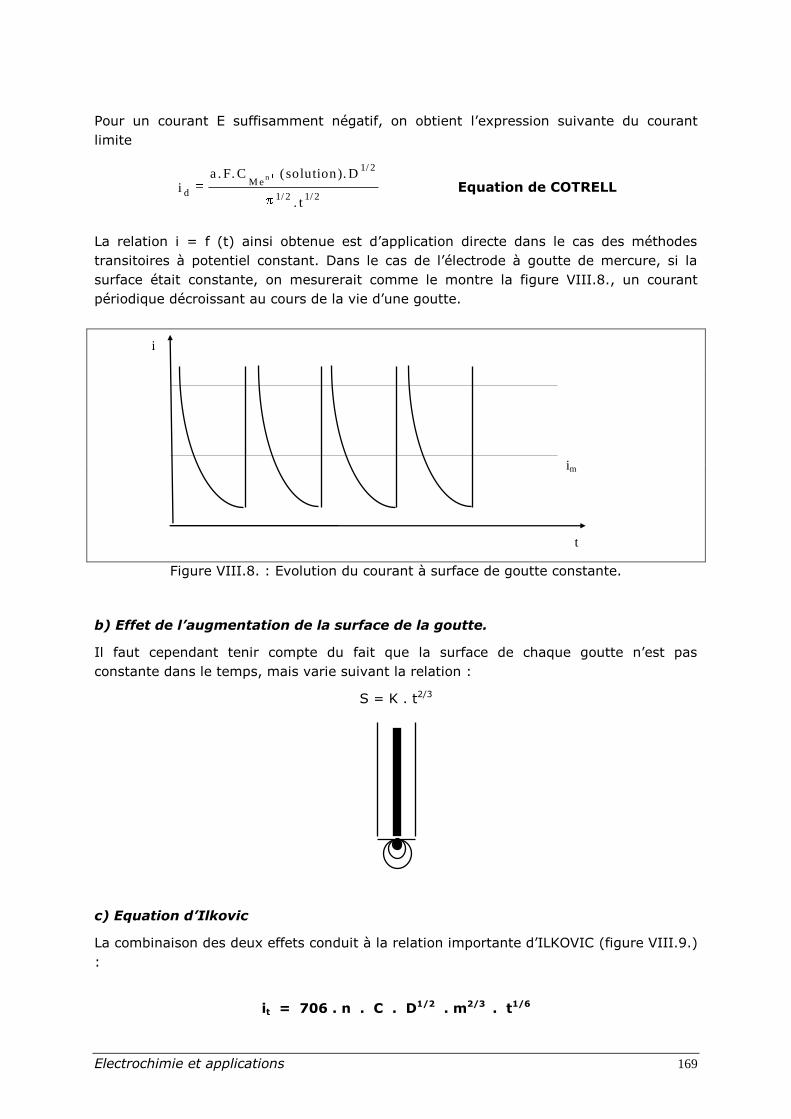
La relation i = f (t) ainsi obtenue est d’application directe dans le cas des méthodes
transitoires à potentiel constant. Dans le cas de l’électrode à goutte de mercure, si la
surface était constante, on mesurerait comme le montre la figure VIII.8., un courant
périodique décroissant au cours de la vie d’une goutte.
Figure VIII.8. : Evolution du courant à surface de goutte constante.
b) Effet de l’augmentation de la surface de la goutte.
Il faut cependant tenir compte du fait que la surface de chaque goutte n’est pas
constante dans le temps, mais varie suivant la relation :
S = K . t2/3
c) Equation d’Ilkovic
La combinaison des deux effets conduit à la relation importante d’ILKOVIC (figure VIII.9.)
:
it = 706 . n . C . D1/2 . m2/3 . t1/6
im
t
i

Electrochimie et applications 170
où it courant instantané ( A)
n nombre d’électrons échangés
C concentration (millimoles/litre)
m débit de mercure (mg/s)
t durée de vie d’une goutte (s)
(t = 0 au début de sa formation)
D coefficient de diffusion de l’ion en solution (cm2/s)
Figure VIII.9. : Evolution du courant lors du relevé d’un polarogramme.
Par intégration sur la durée de vie d’une goutte, le courant moyen est donné par :
iT
i dtm oy t
T1
0
. .
ou encre i moy = 607 . n . C . D1/2 . m2/3 . T1/6
T cadence de chute (sec).
d) Relation courant – Tension appliquée : i = f(E/Réf)
La courbe courant-tension enregistrée dans une solution constituée de l’électrolyte
support, mais exempte de substance active est représentée sur la figure VIII.10.
On peut constater que le domaine de tension utile est limité du côté anodique par la
dissolution anodique du mercure (0,3 V/Eréf) et du côté cathodique par la réduction du
cation de l’électrolyte support ou la décomposition du solvant avec dégagement d’H2.
i

Electrochimie et applications 171
Figure VIII.10. : Domaine de tension cinétiquement élargi sur électrode de mercure.
Entre ces limites, on observe la présence d’un courant résiduel ou « capacitif », ic.
Ce courant de l’ordre de 0,1 A est dû au fait que l’interface Hg-solution agit comme un
condensateur et réclame un certain nombre de coulombs pour se charger (voir cours sur
la double couche). A -0,48 V/Eréf, le courant s’annule (point de charge nulle).
Lorsque l’on introduit en solution un cation ou une autre substance dissoute réductible à
l’électrode, on enregistre l’apparition d’une onde que nous allons mettre en équation.
Si la vitesse de la réaction électrochimique (transfert d’électrons) est
suffisamment grande, l’équilibre entre Men+ et Me(Hg) à la surface de l’électrode est
réalisé à tout moment et on peut écrire la relation de NERNST :
),0(
),0(ln.
)(
0
tC
tC
nF
RTEE
HgMe
Me
éq
n
où CM e
n concentration de l’ion réductible à la surface de l’électrode au
temps t, cette concentration dépend du potentiel appliqué
et est indépendante de t (voir développements précédents).
C Me(Hg (0,t) concentration du métal dans l’amalgame c-à-d à la surface
de la goutte.
Si le courant est limité par la diffusion, on a à la surface de l’électrode (x = 0) :
tCtCki nnMeMe
,),0(.

Electrochimie et applications 172
en particulier au courant limite :
i k C td M en. ( , )
De même, la proportion de Me dans l’amalgame est proportionnelle au courant et :
),0('.)(
tCkiHgMe
k et k’ sont deux facteurs de proportionnalité et d’après la relation d’ILKOVIC :
k n D m tox6071 2 2 3 1 6
. . . ./ / /
k est donc fonction de D ox
k’ est identique à k à part que D est remplacé par D’ coefficient de diffusion de Me dans le
mercure et le rapport k/k’ devient :
k
k
D
D' '
de sorte que :
),(.),0(. tCktCki nnMeMe
dMeiiktC n ).,0(
k
k
i
ii
tC
tCd
HgMe
Men '
.),0(
),0(
)(
i
ii
nF
RT
k
k
nF
RTEE
do
HgMeMeHgMeMenn ln.
'ln.
)(/)(/
i
ii
nF
RT
D
D
nF
RTEE
do
HgMeMeHgMeMenn ln.
'ln.
2)(/)(/
i
ii
nF
RTEE
d
HgMeMen ln.
2/1)(/
où id valeur limite du courant de diffusion
E1/2 potentiel de demi-onde défini comme étant la valeur du potentiel de
l’électrode à gouttes de Hg au point de l’onde où i = id/2.

Electrochimie et applications 173
Le courant limite id est une mesure de la concentration lorsque les autres
paramètres de la relation d’Ilkovic sont maintenus constants.
Le potentiel de demi-onde E1/2 est une constante indépendante de la
concentration et spécifique de l’ion réduit (ou oxydé) à l’électrode dans un
milieu support fixé.
Remarque
Pour les réactions électrochimiques limitées par le transfert de charge (systèmes lents), il
intervient dans l’expression des constantes de vitesses k, des coefficients de transfert
et ( + =1).
La valeur expérimentale de E1/2 est l’abscisse à l’origine de la droite obtenue en
portant le log i/(id-i) en fonction du potentiel E (Figure VIII.11.).
Figure VIII.11. : Potentiel de demi-onde.
En présence de plusieurs ions réductibles (Figure VIII.12.), les courants de diffusion
s’ajoutent et on observe une superposition des ondes.

Electrochimie et applications 174
Figure VIII.12. : Potentiels de solution pour une solution contenant plusieurs cations.
Les divers potentiels de demi-onde permettent de caractériser les ions en présence tandis
que les courants i’, i’’, i’’’ permettent de calculer les concentrations correspondantes.
La polarographie est une méthode d'analyse qualitative et quantitative puisque
le potentiel de demi-onde permet d'identifier le ou les ions présents dans la
solution et que le courant limite est proportionnel à la concentration de ces ions.
e) Maxima polarographiques.
Les polarogrammes présentent fréquemment un pic avant le palier de diffusion. La forme
et la taille des pics varient avec la nature de l’électrolyte support et les concentrations
employées. Ils peuvent être pointus ou arrondis, mais sont généralement reproductibles
pour un système donné.
Sous l’effet du courant d’électrolyse, il peut se créer une légère dissymétrie de la goutte,
due à un effet d’écran joué par le plan de section du capillaire. Cette dissymétrie est
d’autant plus importante que le milieu est peu conducteur et que le bas de la goutte est
plus négatif que le col.
Figure VIII.13. : Maxima polarographiques.
La tension interfaciale étant une fonction du potentiel, les valeurs de la tension
interfaciale sont différentes dans le haut et le bas de la goutte; ce phénomène peut
entraîner des mouvements d’agitation intenses et des valeurs anormalement élevées du
courant.
On dit alors qu’il y a apparition d’un maximum polarographique qu’il convient de faire
disparaître par addition à la solution d’un suppresseur de maximum. Il s’agit de
substances tensioactives telles que la gélatine, la tylose (méthylcellulose) qui s’adsorbent

Electrochimie et applications 175
sur l’électrode et suppriment les mouvements superficiels de la goutte de mercure (ils
rabotent la courbe électrocapillaire - voir cours d’électrochimie).
Figure VIII.14. : Effet d’un tensioactif sur la tension superficielle.
La ligne de base et le palier de diffusion ne sont pas toujours horizontaux. La figure
VIII.15. suivante montre la façon de déterminer le courant de diffusion avec précision
dans un tel cas.
Figure VIII.15. : Dérive de la ligne de base d’un polarogramme.
f) Limitations de la polarographie classique
Au contact électrode à goutte de mercure - solution électrolytique, il existe une double
couche électrochimique fonctionnant comme un condensateur et caractérisée par une
capacité; un courant capacitif de charge (ou de décharge) ic accompagne donc toujours

Electrochimie et applications 176
tout transfert de charge à l’interface; ce courant a été appelé précédemment courant
résiduel.
De plus, en polarographie classique, l’effet de ce courant parasite est encore accentué par
le fait que la surface de la goutte augmente dans le temps.
Le fait que la surface de la goutte de mercure change au cours du temps entraîne
l'apparition d'un courant capacitif de charge et de décharge de la double couche et on
mesure finalement iglob. = if + ic. Pour des faibles concentrations en ion à doser (+/-105
M), le courant capacitif devient du même ordre de grandeur que le courant faradique, il
est alors nécessaire de séparer d'une façon ou d'une autre ces deux composantes.
On travaille alors en polarographie impulsionnelle ou alternative.
Le courant faradique if est le seul à être lié à la réaction électrochimique et c’est le
courant utile pour l’analyse quantitative; or le courant mesuré en pratique est le courant
global iglob = if + ic.
La sensibilité de la méthode classique pour le dosage est dès lors limitée par le rapport :
parasiteCourant
utileCourant
i
i
c
f
Le tableau 2 donne les ordres de grandeurs de if et ic.
CONCENTRATION
(Moles/litre)
COURANT DE DIFFUSION
(ampère)
COURANT CAPACITIF (ou courant résiduel)
(ampère)
10-3 10-5 il varie
10-4 10-6 dans tous les cas
10-5 10-7 De
10-6 10-8 10-7 à 10-8
Tableau 2.
La polarographie classique est limitée du point de vue :
- sensibilité
- sélectivité - pouvoir de résolution
CDC
RP
ic
iF

Electrochimie et applications 177
- facteur d’interférence.
Les méthodes polarographiques évoluées sont basées sur la séparation des courants
faradiques et capacitifs pour permettre d’augmenter de manière importante les
possibilités de la polarographie; les deux méthodes les plus utilisées sont la
polarographie alternative et la polarographie impulsionnelle.
Polarographie à tension alternative surimposée
Cette méthode se fonde sur le fait que le courant capacitif est déphasé de 90° par rapport
à la tension, tandis que le courant faradique est déphasé d’un angle nettement plus faible
(45° au maximum pour un système parfaitement réversible). Les polarographes
correspondants permettent en général d’évaluer le courant faradique avec un courant
capacitif très atténué.
Polarographie à impulsions de potentiel surimposées
On superpose à une période fixée de la vie de la goutte et au balayage continu, des
impulsions de potentiel. Dans ce cas, le courant capacitif résultant de la charge de la
double couche décroît de façon exponentielle, tandis que le courant faradique décroît en
t1/2; après quelques millisecondes, seul le courant faradique subsiste.
La figure VIII.16. explicite ce que l’on vient d’exposer brièvement.

Electrochimie et applications 178
Figure VIII.16. : Principe de la polarographie impulsionnelle.
La polarographie impulsionnelle est actuellement la méthode polarographique la plus
utilisée.