troubles minéraux et osseux de la maladie rénale … · dont la forme active de la vitamine d et...
TRANSCRIPT

REIN ET PATHOLOGIES
REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455 // 29
article reçu le 13 juin, accepté le 19 juin 2013.
© 2013 – Elsevier Masson SAS – Tous droits réservés.
RÉSUMÉ
Le maintien de la calcémie et de la phosphatémie dans des valeurs étroites est le résultat d’une régulation complexe faisant intervenir des échanges entre le rein, l’intestin et le squelette. Le rein joue un rôle essentiel dans cette régula-tion en permettant à court terme un maintien de l’équilibre phosphocalcique grâce à une adaptation de l’élimination rénale du calcium et des phosphates aux apports nutrition-nels. Cette adaptation est sous la dépendance d’hormones dont la forme active de la vitamine D et l’hormone parathy-roïdienne. Notre compréhension de la régulation du méta-bolisme phosphocalcique s’est considérablement enrichie et complexifiée ces dernières années grâce à la découverte de nouveaux acteurs dont le FGF23 et Klotho. La diminution progressive de la fonction rénale telle qu’elle est observée dans la maladie rénale chronique (MRC) s’accompagne de troubles minéraux et métaboliques importants regroupés sous le terme de « troubles minéraux et osseux de la mala-die rénale chronique ou CKD-MBD pour « chronic kidney disease - mineral and bone disorders ». Ces troubles sont caractérisés par des anomalies biochimiques du métabo-lisme phosphocalcique, du remodelage et de la minérali-sation osseuse et par le développement de calcifications vasculaires. L’hyperparathyroïdie secondaire à l’insuffisance rénale chronique, dont les mécanismes sont aujourd’hui en passe d’être mieux compris et dans lequel la rétention de phosphate joue un rôle important, est un déterminant phy-siopathologique essentiel dans la survenue des différentes anomalies des tissus osseux et des tissus mous. Des tra-vaux récents ont aussi démontré que les dérégulations du métabolisme des phosphates et du calcium sont fortement
Said Kamela,b,*, Tilman Druekea, Ziad Massya,c
Troubles minéraux et osseux de la maladie rénale chronique (TMO-MRC)
a INSERM U1088 « Mécanismes physiopathologiques et conséquences des calcifications vasculaires »Université de Picardie Jules-Verne – UFR de pharmacie1, rue des Louvels80037 Amiens cedex b Laboratoire de biochimieCentre hospitalier et universitaire d’AmiensPlace Victor-Pauchet80054 Amiens cedex 1 c Service de néphrologieHôpital Ambroise-Paré (AP-HP) – Hôpitaux universitaires Paris-OuestUniversité Versailles-Saint-Quentin (UVSQ)
* [email protected] ou [email protected]
associées à la mortalité et la morbidité cardiovasculaire du patient ayant une insuffisance rénale chronique. Dans cette revue, après un rappel sur la régulation de la calcémie et de la phosphatémie, nous décrirons dans un premier temps les mécanismes physiopathologiques à l’origine des troubles minéraux de la MRC et dans un second temps les conséquences de ces troubles minéraux sur le squelette et la paroi vasculaire. La biologie tenant une place importante dans le diagnostic et le suivi thérapeu-tique de ces atteintes, nous rappellerons les principales recommandations émises par la fondation internationale « Kidney disease improving global outcome (KDIGO) » sur les paramètres biologiques à mesurer.
Calcémie – phosphatémie – maladie rénale chronique – hyperparathyroïdie secondaire – ostéodystrophie rénale –
calcifications vasculaires.
SUMMARY
Chronic kidney disease - mineral and bone disorders (CKD-MBD)
Maintenance of calcemia and phosphatemia within a narrow physiolo-gical range is the result of a complex regulation based on exchanges of both calcium and phosphate between the kidney, the intestine and the skeleton. The kidney plays a crucial role in the balance of both ions by allowing a tightly adaptation between the renal elimination and the dietary intake of calcium and phosphate. Phosphocalcium homeostasis is largely regulated through an integrated hormonal system including parathyroid hormone and 1-25-dihydroxy-vitamin D. Our understanding of phosphocalcium metabolism has changed substantially in recent years thanks to the discovery of new actors such Fibroblast Growth Factor 23 and Klotho. The progressive reduction in the renal function as it is observed in the chronic kidney disease leads to serious disrup-tion of mineral metabolism responsible for several metabolic disorders gathered under the term of « Chronic Kidney Disease - Mineral and Bone Disorders (CKD-MBD) ». This term encompasses several abnormali-ties including altered levels of calcium, phosphate, PTH and vitamin D metabolites, disturbances in bone turnover and bone mineralization, and development of vascular calcifications. Secondary hyperparathy-roidism, whose mechanism is now better understood and whereby the phosphate retention plays a significant role, is one of the most impor-tant pathophysiological determinant involved in CKD-MBD. Numerous recent studies have shown that disturbances in mineral metabolism strongly contribute to the cardiovascular morbidity and mortality in CKD. In this review, we will first describe the main molecular mechanisms that contribute to regulate calcium and phosphate homeostasis and then we will focus on the pathogenesis of secondary hyperparathyroidism and its consequences on bone and vascular injuries. Finally, because biological measurements represent important tools in the diagnosis and the monitoring of CKD-MBD, we will point out the « Kidney disease improving global outcome (KDIGO) » recommendations on the biolo-gical parameters that can be measured.
Calcemia – phosphatemia – chronic kidney disease – secondary hyperparathyroidism – renal osteodystrophy – vascular calcification.

30 // REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455
1. Introduction
Par sa prévalence en augmentation constante et par ses complications cliniques dévastatrices, la maladie rénale chronique (MRC) est aujourd’hui un problème mondial de santé publique. La perte progressive de la fonction rénale est responsable d’une altération importante de la qualité de vie associée à une augmentation de la morbidité et de la mortalité cardiovasculaire. On estime en effet que 40 à 50 % des patients en insuffisance rénale terminale décèdent suites à des maladies cardiovasculaires. Cepen-dant, les mécanismes physiopathologiques à l’origine de ces atteintes cardiovasculaires ne sont pas encore tota-lement élucidés. La MRC est connue depuis longtemps pour aggraver certains facteurs de risques traditionnels de maladies cardiovasculaires comme le stress oxydant contribuant à l’augmentation de la rigidité vasculaire et de la pression artérielle, l’hyperlipidémie contribuant à la pathogénie de l’athérosclérose. Plus récemment, plusieurs études ont permis d’impliquer les troubles du métabolisme minéral et osseux dans ces complications cardiovasculaires. Le rein est en effet, avec le tissu osseux et l’intestin, l’un des principaux organes impliqué dans l’homéostasie du phosphate et du calcium. Ces trois organes agissent de concert pour réguler la calcémie et la phosphatémie et ainsi maintenir constant le pool de calcium et de phosphates dans les liquides extracellulaires et la teneur phosphocal-cique du squelette. Cet équilibre fait intervenir plusieurs facteurs hormonaux dont la forme active de la vitamine D (1,25 diOH vitamine D ou calcitriol) et l’hormone parathy-roïdienne (PTH) mais également des facteurs de décou-verte plus récente comme le FGF23 (fibroblast growth factor 23) et Klotho. L’altération progressive de la fonction rénale s’accompagne d’anomalies dans l’élimination du phosphate et du calcium à l’origine d’un déséquilibre du métabolisme phosphocalcique. Ces troubles métaboliques sont responsables de différents types d’atteintes osseuse et vasculaire qui peuvent survenir précocement dans l’évolution de la maladie rénale chronique. L’ensemble de ces désordres a été regroupé sous le terme de « troubles minéraux et osseux de la maladie rénale chronique » (TMO-MRC), le terme anglo-saxon est CKD-MBD pour « chronic kidney disease - mineral and bone disorders » par un panel d’experts qui ont émis des recommandations de la fondation « Kidney disease improving global outcome » (KDIGO). Ces troubles sont caractérisés par :1. des modifications de l’homéostasie des phosphates et du calcium ainsi que de ses éléments régulateurs, princi-palement la PTH et la 1,25 diOH vitamine D ;2. de profondes modifications dans la structure osseuse et/ou le remodelage osseux ;3. la survenue de calcifications vasculaires.L’objectif de cette revue est, après une description du métabolisme phosphocalcique et de ses principaux méca-nismes de régulation, de faire le point sur l’ensemble des manifestations biologiques et cliniques liées aux anoma-lies du métabolisme minéral au cours de la maladie rénale chronique et de comprendre comment ces troubles, en favorisant le développement des calcifications vasculaires, contribuent à la mortalité cardiovasculaire de l’IRC. Nous préciserons également quelle est la place de la biologie
dans le diagnostic et le suivi de ces troubles (voir aussi l’article « Physiologie du rein et bases physiopathologiques des maladies rénales » dans RFL « Reins et pathologies (1) », N° 451).
2. Rappel sur la régulation de la
calcémie et de la phosphatémie
Le calcium et les phosphates sont des ions indispen-sables à l’organisme. Ils jouent un rôle fondamental dans la minéralisation du squelette puisqu’ils constituent la base biochimique de l’hydroxyapatite [Ca10(PO4)6(OH)2], partie minérale qui confère à l’os sa rigidité. Au-delà de la minéralisation osseuse, le calcium et les phosphates interviennent également dans de nombreux processus biologiques tels que la coagulation sanguine, la contrac-tion musculaire, la conduction nerveuse pour le calcium, le métabolisme énergétique cellulaire, la synthèse de l’ADN, l’équilibre acido-basique pour les phosphates. En raison de leur importance pour l’organisme, des systèmes sont mis en jeu pour favoriser les échanges tissulaires de ces deux ions. L’homéostasie phosphocalcique fait référence à l’ensemble des mécanismes biologiques per-mettant de réguler et de maintenir constante la calcémie (2,2-2,6 mmol/L) et la phosphatémie chez l’adulte (0,8-1,4 mmol/L). Cette régulation vise avant tout à mainte-nir un équilibre dans la balance phosphocalcique, c’est-à-dire un équilibre entre, d’une part, les apports en calcium et phosphates provenant de l’alimentation et, d’autre part, les mécanismes mis en jeu dans l’organisme pour faciliter leur absorption, leur répartition dans les différents compartiments intra- et extracellulaires et leur élimination. L’équilibre de la balance phosphocalcique nécessite donc l’intervention de l’intestin par sa capacité à absorber les deux ions et du rein par sa capacité à les excréter. Le tissu osseux joue également un rôle dans cet équilibre car il permet de stocker le calcium et le phosphore et de les mettre à disposition en fonction des besoins par l’inter-médiaire du remodelage osseux. Les intestins et surtout les reins jouent un rôle majeur de régulation à court terme dans le maintien de la calcémie ionisée et de la phos-phatémie, tandis que le squelette assure l’homéostasie à moyen et long terme, en particulier lorsque des troubles de l’élimination du calcium et du phosphate surviennent comme dans la maladie rénale chronique.
2.1. Régulation de la calcémieLe corps adulte renferme environ 1 000 g de calcium dont 99 % se trouvent dans le squelette. La calcémie totale se répartit en une fraction liée aux protéines plasmatiques (de l’ordre de 40 %) et une fraction non liée aux protéines ou diffusible (de l’ordre de 60 %), principalement com-posée de calcium ionisé représentant approximativement 50 % du calcium total. La concentration physiologique de calcium ionisé dans le sang circulant est comprise entre 1,1 et 1,35 mmol/L. Seule cette fraction représente le calcium « biologiquement actif ». Chaque jour, des mou-vements calciques ont lieu entre les compartiments extra-cellulaires et les tissus permettant d’absorber (l’intestin), d’éliminer (le rein) ou de stocker (le tissu osseux)
REIN ET PATHOLOGIES
REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455 // 31
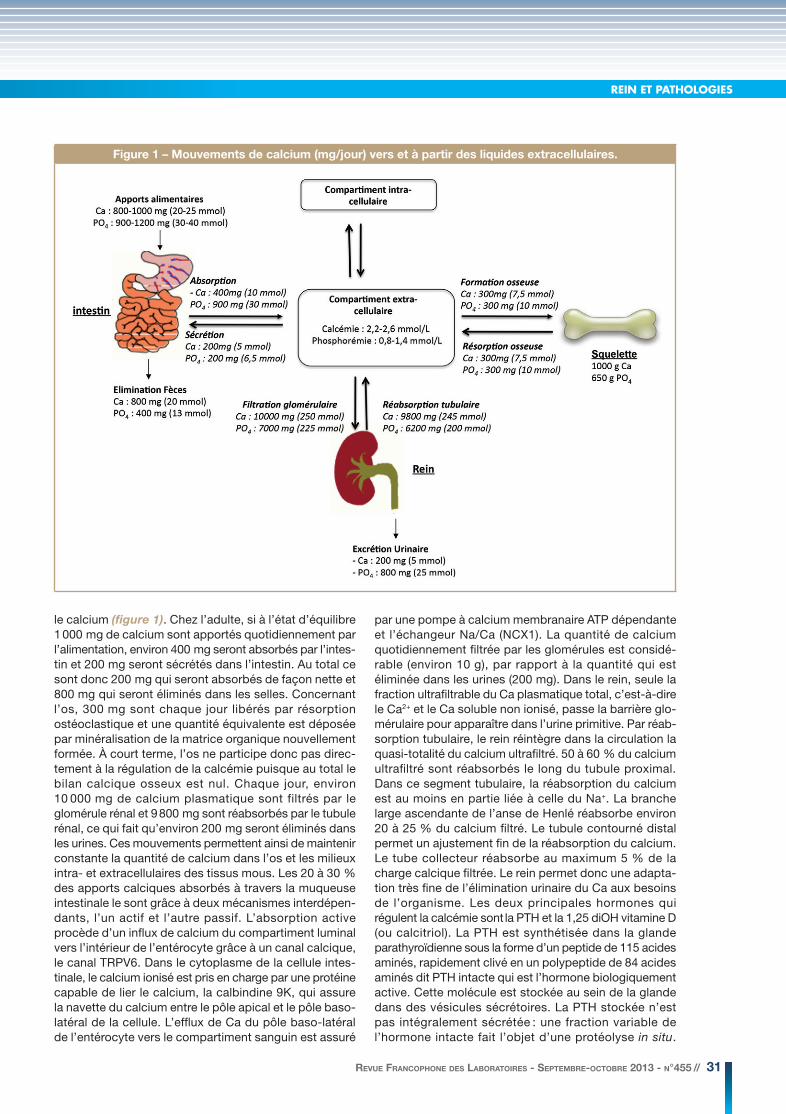
le calcium (figure 1). Chez l’adulte, si à l’état d’équilibre 1 000 mg de calcium sont apportés quotidiennement par l’alimentation, environ 400 mg seront absorbés par l’intes-tin et 200 mg seront sécrétés dans l’intestin. Au total ce sont donc 200 mg qui seront absorbés de façon nette et 800 mg qui seront éliminés dans les selles. Concernant l’os, 300 mg sont chaque jour libérés par résorption ostéoclastique et une quantité équivalente est déposée par minéralisation de la matrice organique nouvellement formée. À court terme, l’os ne participe donc pas direc-tement à la régulation de la calcémie puisque au total le bilan calcique osseux est nul. Chaque jour, environ 10 000 mg de calcium plasmatique sont filtrés par le glomérule rénal et 9 800 mg sont réabsorbés par le tubule rénal, ce qui fait qu’environ 200 mg seront éliminés dans les urines. Ces mouvements permettent ainsi de maintenir constante la quantité de calcium dans l’os et les milieux intra- et extracellulaires des tissus mous. Les 20 à 30 % des apports calciques absorbés à travers la muqueuse intestinale le sont grâce à deux mécanismes interdépen-dants, l’un actif et l’autre passif. L’absorption active procède d’un influx de calcium du compartiment luminal vers l’intérieur de l’entérocyte grâce à un canal calcique, le canal TRPV6. Dans le cytoplasme de la cellule intes-tinale, le calcium ionisé est pris en charge par une protéine capable de lier le calcium, la calbindine 9K, qui assure la navette du calcium entre le pôle apical et le pôle baso-latéral de la cellule. L’efflux de Ca du pôle baso-latéral de l’entérocyte vers le compartiment sanguin est assuré
par une pompe à calcium membranaire ATP dépendante et l’échangeur Na/Ca (NCX1). La quantité de calcium quotidiennement filtrée par les glomérules est considé-rable (environ 10 g), par rapport à la quantité qui est éliminée dans les urines (200 mg). Dans le rein, seule la fraction ultrafiltrable du Ca plasmatique total, c’est-à-dire le Ca2+ et le Ca soluble non ionisé, passe la barrière glo-mérulaire pour apparaître dans l’urine primitive. Par réab-sorption tubulaire, le rein réintègre dans la circulation la quasi-totalité du calcium ultrafiltré. 50 à 60 % du calcium ultrafiltré sont réabsorbés le long du tubule proximal. Dans ce segment tubulaire, la réabsorption du calcium est au moins en partie liée à celle du Na+. La branche large ascendante de l’anse de Henlé réabsorbe environ 20 à 25 % du calcium filtré. Le tubule contourné distal permet un ajustement fin de la réabsorption du calcium. Le tube collecteur réabsorbe au maximum 5 % de la charge calcique filtrée. Le rein permet donc une adapta-tion très fine de l’élimination urinaire du Ca aux besoins de l’organisme. Les deux principales hormones qui régulent la calcémie sont la PTH et la 1,25 diOH vitamine D (ou calcitriol). La PTH est synthétisée dans la glande parathyroïdienne sous la forme d’un peptide de 115 acides aminés, rapidement clivé en un polypeptide de 84 acides aminés dit PTH intacte qui est l’hormone biologiquement active. Cette molécule est stockée au sein de la glande dans des vésicules sécrétoires. La PTH stockée n’est pas intégralement sécrétée : une fraction variable de l’hormone intacte fait l’objet d’une protéolyse in situ.
Figure 1 – Mouvements de calcium (mg/jour) vers et à partir des liquides extracellulaires.

32 // REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455
La PTH exerce un puissant effet hypercalcé-miant en stimulant la libération de calcium à partir de l’os, la réabsorption rénale du calcium, et l’absorption intestinale du calcium par un effet indirect via la synthèse de la 1,25 diOH vitamine D. L’autre hormone importante dans la régulation de la calcémie est justement celle-ci. La vitamine D native a deux origines : l’une exogène, alimentaire et l’autre endogène, par photosynthèse cutanée à partir d’un pré-curseur, le 7-déhydrocholestérol. La photo-synthèse cutanée est liée à une action des rayons UVB (290-315 nm) solaires qui per-mettent la transformation du 7-déhydrocho-lestérol présent dans les téguments de la peau en vitamine D3. Celle-ci passe dans la circu-lation où elle est transportée jusqu’au foie grâce à une protéine porteuse, la « vitamin D- binding protein » (VDBP). Dans cet organe, elle est hydroxylée en position 25 par la 25-hydroxylase-cytochrome P 450 et ainsi transformée en 25-OH vitamine D (ou calcidiol) qui représente une forme de stockage de la vitamine D. La vitamine D d’origine cutanée représente la majeure partie de la 25-OH vita-mine D circulante. Ceci explique le fait que ses concentrations plasmatiques varient avec
les saisons et la latitude et donc le degré d’ensoleillement. La 25-OH vitamine D est transportée du foie vers le rein où elle est hydroxylée en position 1 par la 25-hydroxy-vitamine D-1-α-hydroxylase en 1,25 diOH vitamine D qui représente la forme biologiquement active de la vitamine D. Le rein est à l’origine de l’essentiel de la 1,25 diOH vita-mine D circulante, néanmoins certaines cellules de l’orga-nisme telles que les monocytes, les kératinocytes ou les cellules placentaires la produisent également. La 25-OH vitamine D peut être hydroxylée en position 24 dans le rein par une 24-hydroxylase. La 24-25 [OH]2 vitamine D ne joue pas de rôle dans la régulation de la calcémie. L’hydroxylation de la 25 OH vitamine D par la 1-α-hydroxylase rénale est, comme nous l’avons vu précédemment, régulée positivement par la PTH dont la sécrétion est induite par un abaissement du Ca2+ plasmatique. Par ailleurs, comme nous le verrons plus loin, le FGF23 est un puissant inhibi-teur de l’activité de la 1-α-hydroxylase rénale, ce qui entraîne une diminution de la concentration sérique du calcitriol. La vitamine D est hypercalcémiante. Sa principale action biologique s’exerce au niveau intestinal en stimulant l’ab-sorption du calcium. La vitamine D stimule l’expression et la synthèse des protéines assurant le transport du calcium dans l’entérocyte et son extrusion dans le compartiment sanguin dont les canaux TRPV6, la calbindine 9K et la pompe à calcium ATP dépendante. La 1,25 diOH vita-mine D,par un effet direct sur l’ostéoclaste et indirect via les ostéoblastes, augmente la différenciation ostéoclas-tique et donc la résorption osseuse. Par un effet indirect, elle stimule également la formation et la minéralisation osseuses. Enfin, le calcitriol agit directement sur les glandes parathyroïdiennes en inhibant la synthèse de l’ARN messager de la PTH, et de ce fait exerce un rétro-contrôle négatif.
La PTH circulante représente donc un mélange hétéro-gène de plusieurs fragments peptidiques dont l’hormone intacte qui a une demi-vie plasmatique brève (concen-tration normale : 10-60 pg/L) et des fragments provenant de la protéolyse de la molécule entière, qui peuvent avoir par eux-mêmes une action physiologique. Le Ca2+ plas-matique module directement la synthèse d’ARNm et la sécrétion de PTH par les cellules parathyroïdiennes. Ces dernières possèdent en effet un récepteur sensible au calcium (CaSR) capable de détecter les variations locales de la calcémie. Une diminution de la calcémie s’accom-pagne d’une augmentation de la sécrétion de PTH tandis qu’en cas d’élévation de la calcémie, le calcium ionisé se lie alors au CaSR et inhibe la sécrétion de PTH. In vivo et in vitro, il existe une relation sigmoïdale inverse entre la concentration de calcium ionisé (Ca2+) et la sécrétion de PTH (figure 2). Le taux de sécrétion maximale repré-sente la réserve de sécrétion des glandes parathyroïdes. La concentration de calcium ionisé plasmatique pour laquelle la sécrétion de PTH représente la moitié de la suppression maximale définit le point moyen de sécrétion de PTH (« calcium set point »). La position du point moyen de sécrétion de PTH dans la zone de plus grande pente de la relation sigmoïdale inverse, montre que de faibles variations de la calcémie ionisée sont susceptibles d’en-traîner de fortes variations de la sécrétion de PTH, per-mettant ainsi de maintenir la calcémie ionisée à l’intérieur de valeurs étroites. Il existe d’autres déterminants de la sécrétion de PTH, ils sont d’une importance variable. Parmi ceux-ci, on peut citer le calcitriol qui exerce un effet inhibiteur direct sur la transcription de la PTH. À l’inverse, le phosphate stimule la sécrétion de PTH, possiblement par un effet direct sur les cellules parathy-roïdiennes par un mécanisme non encore élucidé.
Figure 2 – Relation sigmoïdale inverse entre la calcémie ionisée
et la sécrétion de PTH.
Le set point de calcium est défini comme la valeur de la calcémie requise pour induire 50 % de la PTH maximale. Le set point est augmenté dans la maladie rénale chronique et on observe un déplacement de la courbe calcium/PTH vers la droite, ce qui pourrait contribuer à l’augmentation de la concentration sérique de PTH malgré des valeurs élevées de calcémie.

REIN ET PATHOLOGIES
REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455 // 33
En résumé, la régulation hormonale de la calcémie néces-site l’action concertée de la PTH et de la vitamine D (figure 3). La diminution de la calcémie entraîne une inactivation du CaSR parathyroïdien et une sécrétion accrue de PTH. En retour la PTH agit sur ses récepteurs situés dans l’os et le rein, ce qui permet de restaurer la calcémie par stimulation de la résorption osseuse ostéo-clastique et augmentation de la réabsorption tubulaire rénale du calcium. Dans le rein, la PTH agit également en stimulant la synthèse du calcitriol qui en agissant sur ses récepteurs présents dans l’intestin et l’os contribue à restaurer la calcémie en favorisant l’absorption intes-tinale du calcium et en stimulant la résorption osseuse ostéoclastique.
2.2. Régulation de la phosphatémieLes phosphates osseux représentent 85 % du contenu en phosphate de l’organisme. Dans le plasma, les phos-phates existent sous deux formes, organique et minérale. Les phosphates organiques sont représentés par les phos-pholipides et les esters phosphoriques (ATP, ADP…). 85 % des phosphates présents dans le plasma sont cependant sous forme inorganique, notamment sous forme d’ions de l’acide orthophosphorique. La phosphatémie physiologique fluctue davantage que la calcémie, en fonction notam-ment des apports alimentaires. Pour un apport moyen
d’environ 1 200 mg, environ 70 % des phosphates ingérés sont absorbés et 30 % sont éliminés dans les selles (figure 1). Les phosphates sont absorbés dans l’intestin grêle par une voie passive majoritaire (85 %), et une voie active (15 %) impliquant en particulier le co-transporteur sodium- phosphate NPT2b. Des données récentes semblent cepen-dant suggérer une part plus importante (50 %) de la voie active. Le phosphate étant relativement abondant dans l’alimentation, l’organisme doit pouvoir l’éliminer. Ce rôle revient notamment au rein. Plus de 90 % des phosphates circulants sont filtrés au niveau du glomérule, puis le tubule rénal équilibre le bilan en réabsorbant plus ou moins les phosphates filtrés selon les besoins de l’organisme. L’excrétion urinaire de phosphates est comprise entre 750 et 1 000 mg/j. La réabsorption a essentiellement lieu dans le tubule proximal par un processus secondairement actif impliquant essentiellement deux co-transporteurs sodium-phosphate exprimés au pôle apical des cellules tubulaires, NPT2a et NPT2c. Le transporteur NPT1, également exprimé dans le tubule proximal, transporte les phosphates, mais aussi des anions organiques. Comme pour la calcémie, le tissu osseux ne participe pas directement au contrôle de la phosphatémie à court terme puisque chaque jour environ 300 mg de phosphore sont libérés par résorption ostéo-clastique et autant soit 300 mg sont déposés sur les fibres de collagène de la matrice osseuse.
Figure 3 – Schéma résumant la régulation hormonale de la calcémie.
Une diminution de la calcémie entraîne une inactivation du récepteur sensible au calcium (CaSR) dans les cellules de la glande parathyroïdienne et une libération de la PTH. La PTH agit sur ses récepteurs (PTHR) situés dans l’os et le rein, ce qui permet de restaurer la calcémie par stimulation de la résorption osseuse et augmentation de la réabsorption tubulaire rénale du calcium. Dans le rein, la PTH agit également en stimulant la sécrétion du calcitriol (1,25 diOH vit D) qui en agissant sur ses récepteurs (VDR) présents dans l’intestin et l’os contribue à restaurer la calcémie en favorisant l’absorption intestinale du calcium et en stimulant la résorption osseuse ostéoclastique.

34 // REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455
La régulation hormonale de la phosphatémie, moins étudiée que la celle de la calcémie, est restée longtemps inconnue. Parmi les différents mécanismes impliqués dans la régulation de l’homéostasie du phosphore, le contrôle de la réabsorption rénale des phosphates par action au niveau des co-transpor-teurs sodium-phosphate dans le tubule proximal joue un rôle particulièrement important. L’importance de ces co-transpor-teurs a été démontrée dans une étude réalisée chez la souris pour laquelle le gène codant le co-transporteur NPT2a a été invalidé. Ces souris présentent une diminution d’environ 70 % de la réabsorption rénale du phosphate ce qui conduit à une hypophosphatémie sévère. Le rôle capital des co-transpor-teurs sodium-phosphate a été confirmé par l’élucidation de l’origine moléculaire du diabète phosphaté héréditaire avec hypercalciurie, caractérisé par une mutation du gène codant le co-transporteur sodium-phosphate NPT2c, impliqué comme nous l’avons vu précédemment dans la réabsorption rénale du phosphate. Dans le même temps, plusieurs pathologies génétiques caractérisées par une hypophosphatémie sévère et une diminution des concentrations sériques en calcitriol ont été étudiées. Il s’agit du rachitisme hypophosphatémique autosomique dominant, du rachitisme hypophosphatémique autosomique récessif et du rachitisme hypophosphatémique lié à l’X. Pour ces trois pathologies, des mutations soit du gène codant le FGF23 lui-même, soit une surexpression du FGF23 liée à la mutation de gènes régulant le FGF23 ont été mises en évidence. Dans les tumeurs ostéomalaciantes, également caractérisées par une hypophosphatémie et une diminution de la calcitriolémie, le FGF23 a été identifié comme le principal agent causal de la pathologie. Ces études ont permis de démontrer que le FGF23 constitue un facteur clef dans la régulation de la phosphatémie et de la calcitriolémie. Ces données ont été par la suite confirmées par des études complémentaires, comme l’invalidation chez la souris du gène du FGF23 qui est responsable d’une calcinose tumorale avec hyperphosphatémie, d’une augmentation de la calcitriolémie et d’une augmentation de la capacité des reins à réabsorber le phosphate. Le FGF23 est une protéine de 251 acides aminés d’origine osseuse, sécrétée par les ostéocytes. Il possède un domaine homologue au FGF dans la région N-terminale et du fait de cette homologie se fixe sur les récepteurs du FGF (FGF-R), probablement de façon préférentielle sur FGF-R1. C’est pour ces raisons qu’il appartient à la famille des FGFs. Cependant, il se distingue des autres membres de la famille du FGF par son mode d’action. Le FGF23, à la différence des autres membres de cette famille agit principalement par voie endocrine, ce qui fait de cette molécule une hormone. L’autre particularité du FGF23 est de nécessiter pour son action bio-logique la protéine membranaire Klotho, qui interagit avec FGF-R. La protéine Klotho joue ici le rôle de co-récepteur et confère une spécificité à l’interaction entre FGF23 et son récepteur. Une forme soluble de Klotho provenant soit de la protéolyse du domaine extra-membranaire du co-récepteur, soit d’un variant d’épissage alternatif, existe et est retrouvée au niveau sanguin et urinaire. Le FGF23 diminue la capacité de réabsorption rénale du phosphate en inhibant l’expression des co-transporteurs sodium-phosphate NPT2a et NPT2c présents au niveau de la bordure en brosse des tubules proximaux. Par ailleurs, dans le tubule proximal, le FGF23 inhibe puissamment l’expression de l’enzyme 1-α-hydroxylase entraînant une baisse de la synthèse de calcitriol. En diminuant
la calcitriolémie, le FGF23 diminue l’absorption intestinale du phosphate, ce qui majore son action hypophosphatémiante. À l’instar du calcium qui régule la sécrétion de la PTH par l’intermédiaire du récepteur sensible au calcium présent sur les glandes parathyroïdiennes, il a été suggéré que la sécré-tion du FGF23 était sous la dépendance du phosphate. À ce jour, aucun récepteur capable de détecter les variations de la phosphatémie n’a été découvert. Cependant, plusieurs facteurs systémiques capables de réguler la sécrétion du FGF23 ont été mis en évidence. Le calcitriol, par l’intermé-diaire d’éléments de réponse à la vitamine D présents dans le promoteur du gène codant le FGF23 stimule sa produc-tion. La synthèse/sécrétion du FGF23 ou son activité sont en revanche inhibées par des mécanismes encore inconnus impliquant des facteurs synthétisés par l’ostéocyte comme la protéine PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) et la protéine DMP1 (dentin matrix protein 1). La PTH sérique, qui joue comme nous l’avons vu un rôle central dans le contrôle de la calcé-mie, joue également un rôle important dans la régulation de la phosphatémie. L’augmentation de la concentration sérique de PTH en agissant sur les co-transporteurs sodium-phos-phate diminue la réabsorption rénale du phosphate et donc la phosphatémie. Inversement, une diminution de la PTH sérique s’accompagne d’une augmentation de la réabsorp-tion des phosphates et donc de la phosphatémie. La PTH a des effets opposés à ceux du FGF23 sur la synthèse rénale du calcitriol. L’augmentation de la PTH sérique stimule sa synthèse alors que l’augmentation du FGF23 la diminue. Il est à noter également que les glandes parathyroïdiennes expriment Klotho et constituent donc une cible du FGF23. FGF23 inhibe l’expression et la sécrétion de PTH et à l’inverse la PTH stimule l’expression du FGF23, suggérant l’existence d’un système de rétrocontrôle négatif entre FGF23 et la PTH. Il existe donc une interrelation sophistiquée entre l’homéos-tasie du calcium et celle du phosphate.En résumé (figure 4), l’action biologique du FGF23 sur le contrôle de la phosphatémie s’exerce principalement au niveau rénal. L’augmentation des apports en phosphate et l’hyperphosphatémie sont à l’origine d’une libération du FGF23 par les ostéocytes. Le FGF23 en se fixant sur le complexe formé par l’association du FGF-R avec Klotho inhibe la production des co-transporteurs sodium phosphate NPT2a et NPT2c présents dans le tubule proximal, ce qui a pour effet de diminuer la réabsorption rénale du phosphate et donc la phosphatémie. Dans le même temps, le FGF23 inhibe l’activité de la 1-α-hydroxylase rénale diminuant ainsi la synthèse de calcitriol et donc l’absorption du phosphate au niveau intestinal ce qui contribue à diminuer la phosphatémie.
3. Troubles minéraux associés
à la maladie rénale chronique :
l’hyperparathyroïdie secondaire
L’hyperparathyroïdie secondaire survient systématique-ment chez les patients qui souffrent d’insuffisance rénale chronique et constitue la manifestation biologique la plus marquante des TMO-MRC. Elle associe initialement une PTH élevée, une hyperphosphatémie, une hypocalcémie et un déficit en calcitriol. Dans les formes plus sévères

REIN ET PATHOLOGIES
REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455 // 35
Figure 4 – Action biologique du FGF23 au niveau rénal.
L’hyperphosphatémie ou l’augmentation des apports de phosphate est à l’origine d’une libération du FGF23 par les ostéocytes du tissu osseux. Le FGF23 en se fixant sur le complexe formé par l’association FGF-R/Klotho inhibe l’expression des co-transporteurs sodium/phosphate NPT2a et NPT2c présents dans les tubules proximaux, ce qui a pour effet de diminuer la réabsorption rénale du phosphate et donc la phosphatémie. Dans le même temps, le FGF23 inhibe l’activité de la 1-α-hydroxylase rénale diminuant ainsi le calcitriol et l’absorption du phosphate au niveau intestinal, ce qui contribue à diminuer la phosphatémie.
Figure 5 – Schéma simplifié des différents événements moléculaires possibles
conduisant à l’hyperparathyroïdie secondaire.

36 // REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455
L’hyperphosphatémie et la diminution de la synthèse de calcitriol ont plusieurs conséquences. La diminution de la calcitriolémie entraîne tout d’abord une diminution de l’absorption intestinale des phosphates, ce qui limiterait les effets délétères de l’hyperphosphatémie et serait donc bénéfique dans cette condition. Mais c’est surtout sur la sécrétion de PTH par les glandes parathyroïdiennes que les conséquences vont porter. Tout d’abord, l’hyperphos-phatémie pourrait être à l’origine d’une augmentation des concentrations intracellulaires de phosphate au niveau des cellules parathyroïdiennes, ce qui pourrait entraîner une augmentation de la durée de vie des ARNm de la PTH et une augmentation de la prolifération des cellules parathyroïdiennes. L’hyperphosphatémie est aussi à l’origine d’une réduction de la calcémie, dans le but d’éviter une augmentation du produit phosphocalcique (PxCa), en entraînant la formation de complexes avec les phosphates. Cette réduction de la calcémie est accentuée par le déficit en production de vitamine D active qui dimi-nue l’absorption intestinale et la réabsorption tubulaire du calcium. Afin de normaliser la calcémie, les glandes parathyroïdiennes via le CaSR augmentent la synthèse et la sécrétion d’hormone parathyroïdienne (PTH). La synthèse de PTH est aussi majorée par la levée de l’effet freinateur liée à l’action génomique du calcitriol sur les glandes parathyroïdiennes. Tous ces mécanismes biolo-giques conduisent donc du fait de la progression de l’IRC à une augmentation de la PTH. Ce mécanisme pathogé-nique de l’hyperparathyroïdie secondaire plaçant le FGF23 au centre d’un processus conduisant à la réaction para-thyroïdienne n’est pas encore consensuel. D’autres méca-nismes ont été rapportés conférant un rôle pivot à la diminution d’expression rénale de Klotho qui survient précocement dans la maladie et qui serait responsable d’une résistance à l’action du FGF23 et donc d’une aug-mentation de la production du FGF23 et de la PTH. Quel qu’en soit le mécanisme, l’augmentation de la synthèse de PTH est une conséquence adéquate du point de vue du métabolisme phosphocalcique. Elle tend à corriger d’une part l’hypocalcémie, par augmentation de la réab-sorption et diminution de l’excrétion rénale du calcium et par stimulation de la résorption osseuse, d’autre part l’hyperphosphatémie par inhibition de la réabsorption et augmentation de l’excrétion rénale des phosphates et enfin le déficit en vitamine D active par augmentation de son hydroxylation en position 1alpha, ce qui augmente l’absorption digestive du calcium, mais aussi des phos-phates. Son action la plus importante est donc le maintien d’un niveau de calcium ionisé plasmatique aussi normal que possible, malgré une diminution progressive de la fonction rénale. L’augmentation de la PTH observée au début de la MRC est due essentiellement à une augmen-tation des capacités de synthèse et de sécrétion des cellules parathyroïdiennes en réponse à l’hypocalcémie. Au stade plus avancé de la maladie, l’IRC est caractérisée par une diminution voir une perte de l’expression des récepteurs participant à la régulation du métabolisme phosphocalcique. C’est ainsi que le CaSR, le récepteur de la vitamine D (VDR) ou encore le FGF-R et son coré-cepteur Klotho sont tous diminués dans le tissu parathy-roïdien. Ces anomalies sont à l’origine d’un déplacement
d’hyperparathyroïdie, la calcémie s’élève progressivement. Les mécanismes physiopathologiques à l’origine de cette hyperparathyroïdie secondaire associent plusieurs fac-teurs dont la cinétique d’apparition n’est pas encore clairement identifiée. Une question essentielle est en effet de savoir quels sont les troubles du métabolisme phos-phocalcique qui peuvent survenir au tout début de la maladie ? En effet aucune modification dans les concen-trations sériques de calcium et de phosphate n’est mise en évidence tant que le débit de filtration glomérulaire reste supérieur à 30-40 mL/min. Dans ces conditions, il apparaît difficile de connaître exactement quelle est la cinétique des modifications biochimiques à l’origine de l’augmentation de la sécrétion de PTH et du développe-ment de l’hyperparathyroïdie secondaire. La découverte du couple FGF23/Klotho a grandement amélioré nos connaissances sur le métabolisme phosphocalcique et plusieurs scénarios expliquant les différents mécanismes physiopathologiques conduisant à l’augmentation de la PTH ont émergé ces dernières années. Il est possible qu’au début de l’apparition des troubles, le phosphate joue un rôle important (figure 5). Au stade initial d’IRC, l’altération de la fonction d’excrétion rénale entraîne une rétention de phosphate et donc une tendance à l’hyper-phosphatémie. L’organisme compense cette tendance hyperphosphatémique en diminuant le seuil de réabsorp-tion tubulaire rénale des phosphates grâce à une aug-mentation de la sécrétion de FGF23 par le tissu osseux. Cette augmentation du FGF23 est en effet détectée dès les stades précoces de l’IRC, suggérant ainsi une action biologique précoce du FGF23 sur la phosphaturie par diminution de la réabsorption tubulaire des phosphates. À ce stade, la phosphatémie reste normale, voire basse. L’augmentation du FGF23 dans les débuts de la MRC pourrait donc être considérée comme un système com-pensatoire luttant contre la rétention des phosphates. Les concentrations physiologiques de FGF23 s’élèvent dès les stades 2 à 4 de la MRC et ceci même chez des patients sans signes cliniques d’atteinte rénale. Elles atteignent généralement des valeurs beaucoup plus éle-vées au stade 5D. L’augmentation du FGF23 sérique résulte à la fois d’une augmentation de synthèse par les ostéocytes, mais également d’une rétention de FGF23 par un défaut d’élimination rénale, ce qui explique peut-être les concentrations extrêmement élevées observées au stade terminal de la MRC. Les principaux stimuli phy-siologiques connus de la sécrétion de FGF23 sont le calcitriol et des apports élevés en phosphate. L’hyper-phosphatémie persistante et non les variations rapides et intermittentes de la phosphatémie constituent égale-ment un stimulus pour la sécrétion de FGF23. Lorsque le débit de filtration glomérulaire devient inférieur à 30 mL/min, le dépassement des capacités excrétoires du rein provoque une hyperphosphatémie significative non compensée qui accentue l’élévation des concentra-tions de FGF23. Comme il a été vu précédemment, une des actions physiologiques du FGF23 est de réduire la calcitriolémie en inhibant la 1-α-hydroxylase rénale. Cette baisse de la calcitriolémie est accentuée par la réduction de la masse néphronique qui contribue probablement à la diminution de l’activité de la 1-α-hydroxylase rénale.

REIN ET PATHOLOGIES
REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455 // 37
ment sur une analyse histomorphométrique osseuse qui nécessite une biopsie mais celle-ci n’est plus effectuée en routine clinique. À l’heure actuelle, une biopsie osseuse doit être pratiquée chez des patients insuffisants rénaux ayant des fractures pathologiques et/ou une hypophos-phatémie ou hypercalcémie inexpliquées. Afin de faciliter le diagnostic des différentes formes d’atteinte osseuse au cours de l’IRC, une nouvelle classification notée TMV (tableau I) a été récemment proposée par le groupe KDIGO. Ces atteintes sont caractérisées en fonction du turnover osseux (T), du niveau de minéralisation (M) et du volume osseux (V). L’échantillon biopsique est habi-tuellement prélevé au niveau de la crête iliaque antérieure après un double marquage à la tétracycline : on admi-nistre à deux reprises au patient de la tétracycline per os à 15 jours d’intervalle. La tétracycline administrée va se fixer sur le front de minéralisation sous la forme d’un dépôt jaune fluorescent. Le double marquage permet ainsi d’évaluer la vitesse de minéralisation en mesurant la distance séparant les deux fronts de minéralisation et donne donc une indication du taux de formation osseuse ou du turnover (remodelage). L’ostéite fibreuse se déve-loppe sous la dépendance de l’hyperparathyroïdie et est causée principalement par une exposition prolongée à des concentrations élevées de PTH. L’hyperparathyroïdie induit des modifications histologiques notamment une fibrose médullaire, et une augmentation du nombre et de l’activité des ostéoblastes et des ostéoclastes. C’est une pathologie au cours de laquelle le remodelage osseux est accéléré de façon inadéquate, mais qui ne provoque pas de troubles de la minéralisation. Cependant, les cellules osseuses sont dans l’incapacité de produire un os normal. L’os cortical, normalement composé de lamelles osseuses concentriques, se trouve remplacé par un tissu mal structuré. L’augmentation de la résorption osseuse entraîne l’apparition de cavités, et une fibrose de la moelle osseuse peut être observée, d’abord vers l’extérieur, et dans les cas les plus sévères sur toute la surface de la moelle. Histologiquement, l’ostéodystrophie mixte combine des lésions de type ostéite fibreuse avec augmentation du remodelage osseux, et des troubles de la minéralisation proches de ceux rencontrés au cours de l’ostéomalacie. Dans ce cas, la fibrose médullaire reste discrète voir absente et le remodelage osseux est moins augmenté que dans le cas de l’ostéite fibreuse. L’ostéo-malacie pure est caractérisée par une augmentation de la trame protéique osseuse non minéralisée. Ce défaut de minéralisation entraîne la formation d’une structure osseuse faible et instable et a été pendant longtemps la conséquence soit d’une carence en vitamine D soit d’une intoxication aluminique. La supplémentation en vitamine D, largement utilisée aujourd’hui dans la prise en
du set point du calcium pour la PTH (valeur de calcémie nécessaire pour induire une sécrétion de 50 % de la sécrétion de PTH maximale) ce qui expliquerait l’aug-mentation de la concentration sérique de PTH malgré des valeurs normales ou élevées de la calcémie. Certaines toxines urémiques qui s’accumulent du fait de l’insuffi-sance rénale chronique diminuent le nombre et l’activité du VDR parathyroïdien, le rendant alors moins sensible au calcitriol. La diminution d’expression du VDR s’accom-pagne également d’une hyperplasie parathyroïdienne par augmentation de la prolifération cellulaire et la formation d’abord de nodules hyperplasiques puis de véritables adénomes autonomisés. Ainsi, à ce stade de la maladie, le traitement par les dérivés actifs de la vitamine D ne permet plus de freiner la sécrétion excessive de PTH de façon efficace. La diminution de l’expression du VDR parathyroïdien étant plus marquée dans les zones d’hy-perplasie nodulaire que dans les zones d’hyperplasie diffuse, celle-ci pourrait jouer un rôle dans le développe-ment de ces formes de croissance tumorale bénigne qu’on appelle aussi l’hyperparathyroïdie « tertiaire ».
4. Désordres osseux liés
aux troubles minéraux au cours
de la maladie rénale chronique
Le tissu osseux est une des principales cibles des troubles minéraux et de l’hyperparathyroïdie secondaire observés au cours de la maladie rénale chronique. Si la réaction parathyroïdienne consécutive à la diminution du débit de filtration glomérulaire peut être considérée comme une situation d’adaptation de l’organisme au maintien de l’homéostasie phosphocalcique, il est clair que celle-ci s’effectue aux dépens du tissu osseux qui constitue la principale réserve en calcium et phosphate de l’orga-nisme. La dérégulation des différents éléments impliqués dans le métabolisme phosphocalcique dont la PTH et la vitamine D aboutit dans certains cas à un ensemble de manifestations cliniques complexes liées principalement à des modifications structurelles osseuses portant à la fois sur le niveau de remodelage osseux (turnover), la minéralisation et la masse osseuse. L’ensemble de ces modifications osseuses se développe au fur et à mesure de l’évolution de la MRC et a été regroupé sous le terme d’ostéodystrophie rénale. Parmi les atteintes osseuses, on distingue les atteintes liées à une accélération du remo-delage osseux (ostéite fibreuse, ostéodystrophie mixte, ostéoporose), les atteintes liées à un ralentissement du remodelage osseux (os adynamique) et les atteintes liées à un trouble pur de la minéralisation (ostéomalacie). Le diagnostic de ces différentes atteintes reposera idéale-
Tableau I – Critères de diagnostic différentiel des différentes formes d’atteinte osseuse
au cours de l’insuffisance rénale chronique.
Ostéite fibreuse Ostéodystrophie mixte Os adynamique Ostéomalacie
Turnover Normal à élevé Elevé Bas Bas
Minéralisation Normale Anormale Normale Anormale
Volume osseux Faible à élevé Faible à normal Faible à moyen Faible à moyen

38 // REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455
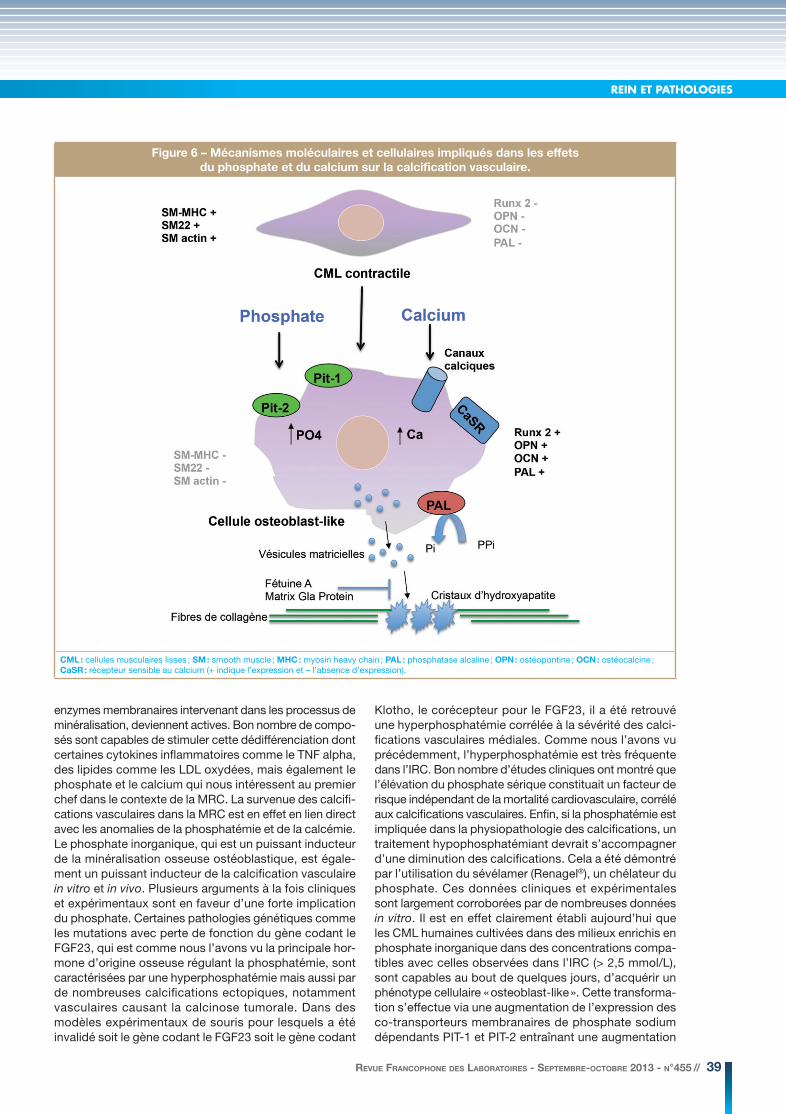
du remodelage vasculaire caractérisée par l’apparition de calcifications vasculaires. Ces calcifications ne sont pas spécifiques de la MRC, mais plusieurs études ont démontré que les patients insuffisants rénaux et plus particulièrement les patients hémodialysés présentaient un risque de calcification vasculaire jusqu’à 5 fois plus élevé comparativement à des sujets appariés pour l’âge, avec une fonction rénale normale. Les calcifications vas-culaires consistent en des dépôts de cristaux associant calcium et phosphate sous la forme d’hydroxyapatite sur la matrice extracellulaire des parois artérielles. D’un point de vue histologique et en fonction de leur localisation, on distingue trois grands types de calcifications vascu-laires. Les calcifications de l’intima qui accompagnent la formation de la plaque athéromateuse, les calcifications de la média (ou maladie de Mönckeberg) observées plus fréquemment au cours de la MRC mais également chez les patients diabétiques et enfin les calcifications valvulaires (valves mitrale et aortique). Les calcifications intimales des coronaires sont corrélées positivement avec la taille des plaques d’athérosclérose. Elles pourraient augmenter le risque d’accident ischémique coronarien. Les calcifica-tions médiales sont associées à une augmentation de la rigidité vasculaire qui à terme peut être à l’origine d’une hypertension artérielle, d’une hypertrophie ventriculaire gauche, d’anomalies de la perfusion coronarienne et d’une insuffisance cardiaque. Quelle que soit leur localisation, les calcifications vasculaires contribuent fortement à l’augmentation du risque de mortalité cardiovasculaire au cours de la maladie rénale chronique. Il existe aujourd’hui plusieurs outils diagnostiques permettant de mettre en évidence les calcifications vasculaires dont la radiogra-phie standard et d’autres techniques d’imagerie plus performantes comme l’échographie, le scanner spiralé et la scanographie à canon d’électrons. En revanche, il n’existe pas encore de marqueurs biologiques spécifiques des calcifications, bien que comme nous le verrons plus loin, plusieurs molécules candidates soient à l’étude. Les calcifications vasculaires ont longtemps été considérées comme résultant d’un dépôt passif de calcium et de phosphate liés à l’augmentation du produit phosphocal-cique sanguin. Les études réalisées au cours de ces dix dernières années ont permis de préciser les mécanismes impliqués et ont au contraire démontré que les calcifica-tions vasculaires sont le fait d’un mécanisme cellulaire actif étroitement régulé ressemblant à l’ostéogenèse. Sous l’effet de différents stimuli et dans des conditions appropriées, les cellules musculaires lisses vasculaires (CML) dérivées de la média se dédifférencient en cellules de type ostéoblastique capables d’initier la synthèse d’une matrice extracellulaire qui sera secondairement minéralisée au sein de la paroi artérielle (figure 6). Les CML qui se dédifférencient perdent progressivement leur phénotype contractile initial, avec notamment la perte d’expression de certains marqueurs caractéristiques comme l’alpha actine, la protéine SM22 ou encore la chaîne lourde de la myosine. Dans le même temps, l’expression de facteurs de transcription spécifiques des cellules ostéoformatrices comme CBFA1/RUNX2 est rapidement stimulé et sous l’action de ces facteurs, des protéines ostéoblastiques comme la phosphatase alcaline, une des principales
charge thérapeutique de l’IRC et la diminution de l’utili-sation des chélateurs de phosphates à base d’hydroxyde d’aluminium, a permis de diminuer considérablement l’incidence de ce défaut de minéralisation osseuse. Dans l’IRC, l’ostéomalacie pure est devenue extrêmement rare et est le plus souvent associée à l’hyperparathyroïdie secondaire (ostéodystrophie mixte). Plus récemment, il a été montré que la sclérostine, une protéine produite par l’ostéocyte et qui est capable de réguler un certain nombre de protéines impliquées dans la minéralisation osseuse, était particulièrement élevée chez les patients présentant une IRC. De futures études préciseront cer-tainement son rôle et son implication. L’os adynamique se caractérise par une diminution du remodelage osseux, aussi bien au niveau de la formation que de la résorption. Les ostéoblastes et les ostéoclastes voient leur nombre et leur activité diminués. Le diagnostic précis nécessite une biopsie avec un double marquage à la tétracycline. Plusieurs mécanismes physiopathologiques peuvent être impliqués. Le plus souvent, l’hypoparathyroïdie induite soit chirurgicalement, soit par l’administration de doses excessives de chélateurs de phosphate calciques ou de vitamine D active en est la cause. Cependant, de nom-breux autres facteurs circulants peuvent être à l’origine d’une diminution de la formation osseuse. Là encore, la sclérostine, un puissant inhibiteur de l’activité ostéoblas-tique, qui comme on l’a vu est élevée chez les patients en IRC, a été suggérée comme étant potentiellement impliquée. Il est à noter que la diminution de la formation osseuse chez les patients présentant un os adynamique peut s’accompagner d’une augmentation de la calcémie et de la phosphatémie, puisque ces ions sont proportion-nellement moins mobilisés par le tissu osseux, le risque de fracture étant par ailleurs également augmenté.Au total, la nature de l’atteinte osseuse initiale liée à la MRC n’est pas encore définie avec précision. Des don-nées récentes, chez l’animal et chez l’homme suggèrent une atteinte initiale de type os adynamique secondaire à l’accumulation des toxines urémiques qui jouent pro-bablement un rôle dans la genèse des modifications structurales et fonctionnelles et dans les modifications d’expression des récepteurs comme le récepteur à la PTH. L’augmentation de la PTH ne serait que secondaire pour vaincre ces effets et l’apparition de l’ostéite fibreuse ne se manifeste que tardivement vers les stades avancés de la MRC. Les places du FGF23, de la sclérostine, et de Klotho dans ce scénario sont en cours de détermination.
5. Désordres vasculaires
liés aux troubles minéraux
dans la maladie rénale chronique :
les calcifications vasculaires
La maladie rénale chronique est associée à une forte augmentation du risque de mortalité et de morbidité car-diovasculaires. Plusieurs altérations du système cardio-vasculaire sont retrouvées dès les stades 2 à 4 de la MRC dont une dysfonction endothéliale, une rigidité vasculaire, une hypertrophie ventriculaire gauche et une anomalie
REIN ET PATHOLOGIES
REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455 // 39
Klotho, le corécepteur pour le FGF23, il a été retrouvé une hyperphosphatémie corrélée à la sévérité des calci-fications vasculaires médiales. Comme nous l’avons vu précédemment, l’hyperphosphatémie est très fréquente dans l’IRC. Bon nombre d’études cliniques ont montré que l’élévation du phosphate sérique constituait un facteur de risque indépendant de la mortalité cardiovasculaire, corrélé aux calcifications vasculaires. Enfin, si la phosphatémie est impliquée dans la physiopathologie des calcifications, un traitement hypophosphatémiant devrait s’accompagner d’une diminution des calcifications. Cela a été démontré par l’utilisation du sévélamer (Renagel®), un chélateur du phosphate. Ces données cliniques et expérimentales sont largement corroborées par de nombreuses données in vitro. Il est en effet clairement établi aujourd’hui que les CML humaines cultivées dans des milieux enrichis en phosphate inorganique dans des concentrations compa-tibles avec celles observées dans l’IRC (> 2,5 mmol/L), sont capables au bout de quelques jours, d’acquérir un phénotype cellulaire « osteoblast-like ». Cette transforma-tion s’effectue via une augmentation de l’expression des co-transporteurs membranaires de phosphate sodium dépendants PIT-1 et PIT-2 entraînant une augmentation
enzymes membranaires intervenant dans les processus de minéralisation, deviennent actives. Bon nombre de compo-sés sont capables de stimuler cette dédifférenciation dont certaines cytokines inflammatoires comme le TNF alpha, des lipides comme les LDL oxydées, mais également le phosphate et le calcium qui nous intéressent au premier chef dans le contexte de la MRC. La survenue des calcifi-cations vasculaires dans la MRC est en effet en lien direct avec les anomalies de la phosphatémie et de la calcémie. Le phosphate inorganique, qui est un puissant inducteur de la minéralisation osseuse ostéoblastique, est égale-ment un puissant inducteur de la calcification vasculaire in vitro et in vivo. Plusieurs arguments à la fois cliniques et expérimentaux sont en faveur d’une forte implication du phosphate. Certaines pathologies génétiques comme les mutations avec perte de fonction du gène codant le FGF23, qui est comme nous l’avons vu la principale hor-mone d’origine osseuse régulant la phosphatémie, sont caractérisées par une hyperphosphatémie mais aussi par de nombreuses calcifications ectopiques, notamment vasculaires causant la calcinose tumorale. Dans des modèles expérimentaux de souris pour lesquels a été invalidé soit le gène codant le FGF23 soit le gène codant
Figure 6 – Mécanismes moléculaires et cellulaires impliqués dans les effets
du phosphate et du calcium sur la calcification vasculaire.
CML : cellules musculaires lisses ; SM : smooth muscle ; MHC : myosin heavy chain ; PAL : phosphatase alcaline ; OPN : ostéopontine ; OCN : ostéocalcine ; CaSR : récepteur sensible au calcium (+ indique l’expression et – l’absence d’expression).

40 // REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455
naturels agissant soit via la circulation sanguine soit localement. L’un des plus importants de ces inhibiteurs est la fétuine A, une protéine synthétisée par le foie et présente dans le sérum à des concentrations normales comprises entre 0,5 et 1 g/L. La fonction de la fétuine A est de s’opposer à la formation et au développement des cristaux d’hydroxyapatite ; dans le sérum, elle forme des complexes circulants avec le calcium, le phosphate et d’autres protéines, consistant en de petites sphères colloïdales solubles appelés CPP ou particules calcipro-téiques. Ces particules sont ensuite éliminées par des macrophages. Ce système permet d’éliminer les excès de calcium et de phosphate tel qu’ils sont observés au cours des anomalies de l’homéostasie phosphocalcique de la MRC et donc de prévenir la formation de calci-fications ectopiques. Le rôle évident dans la calcifica-tion de la fétuine A a été démontré dans un modèle de souris déficiente en cette protéine. Par ailleurs, chez les patients hémodialysés, la diminution des concentrations sériques de fétuine A est corrélée avec la calcification et la mortalité cardiovasculaire, suggérant ainsi qu’une diminution de la capacité de formation des CPP induite par la diminution de la fétuine A pourrait contribuer à la survenue des calcifications. D’autres inhibiteurs agissant localement sont également impliqués ; il s’agit d’une part de la MGP (matrix GLA protein) dont l’expression est modulée par le calcium, et d’autre part de l’ostéopon-tine et du pyrophosphate. Au travers de ces données, il apparaît clair qu’à la fois le phosphate et le calcium, agissant seuls ou ensemble, sont capables d’activer les mécanismes cellulaires et moléculaires responsables des calcifications vasculaires au cours de la MRC. Dans cette revue, nous nous sommes focalisés sur ces deux éléments en raison principalement de leurs larges ano-malies rencontrées au cours de la MRC, mais ce ne sont probablement pas les seuls impliqués. D’autres facteurs, comme l’inflammation et le stress oxydant, pour ne citer que ces deux-là, qui sont également excessivement actifs dans la MRC, contribuent à la survenue des cal-cifications vasculaires.
6. Place de la biologie dans
le diagnostic et le suivi des troubles
minéraux et osseux associés
à la maladie rénale chronique :
les recommandations KDIGO
L’ensemble des données présentées dans cette revue démontre clairement que la dérégulation de l’homéostasie phosphocalcique dans la MRC est directement respon-sable, via des mécanismes complexes, d’un ensemble de troubles caractérisés par des atteintes osseuses et cardiovasculaires, souvent graves. Le contrôle des ano-malies du métabolisme phosphocalcique et l’utilisation de mesures correctives appropriées sont donc capitales dans la prévention de ces pathologies. Ceci a fait l’objet en 2009 d’un ensemble de recommandations, émises par un panel d’experts, appelées « Kidney disease impro-ving global outcomes (KDIGO) ». Ces recommandations
des concentrations intracellulaires de phosphates qui aboutit à son tour à une augmentation de l’expression de CBFA1/RUNX2, le facteur de transcription spéci-fique de l’ostéoblaste. L’augmentation de l’expression de CBFA1/RUNX2 induite par le phosphate est proba-blement médiée par d’autres protéines comme la BMP2 (bone morphogenetic protein 2) et la transglutaminase. Une fois transformée en cellule osteoblast-like, la CML accumule des vésicules matricielles contenant du phos-phate et du calcium qui seront relarguées dans le milieu extracellulaire au niveau des sites de nucléation et de minéralisation. Enfin, il est à noter que l’augmentation de la concentration extracellulaire en phosphate est capable d’induire directement l’apoptose des CML, un processus susceptible par lui-même d’accélérer le dépôt de cristaux d’hydroxyapatite sur la matrice extracellulaire. Au vu de ces données, il apparaît clair que l’hyperphosphatémie constitue l’un des principaux inducteurs de la calcification vasculaire, d’autant que c’est une situation fréquente au cours de la MRC aux stades avancés.Qu’en est-il du calcium ? Plusieurs études ont rapporté un lien entre l’élévation de la calcémie et les calcifi-cations vasculaires. Il est vrai cependant que dans la MRC l’hypercalcémie est une situation plus rare que l’hyperphosphatémie. Néanmoins plusieurs situations peuvent conduire à des épisodes d’hypercalcémie. La carence en vitamine D et l’hyperparathyroïdie secon-daire consécutif à la diminution du débit de filtration glomérulaire sont traitées par l’utilisation de différents agonistes du récepteur à la vitamine D capables de stimuler l’absorption intestinale du calcium. L’hyper-phosphatémie est souvent traitée par des chélateurs de phosphate contenant du calcium (acétate ou carbonate de calcium). Ces différentes substances thérapeutiques peuvent être à l’origine d’épisodes hypercalcémiques. Une hypercalcémie peut aussi survenir dans le cadre de l’ostéopathie adynamique, comme nous l’avons vu plus haut. L’hypercalcémie n’est pas le seul facteur impliqué dans l’exposition des cellules à de fortes concentrations extracellulaires en calcium. Dans les parois vasculaires, le calcium peut être libéré après nécrose ou apoptose des cellules, conduisant à une élévation de la concen-tration extracellulaire locale en calcium. Les mécanismes responsables d’une induction de la calcification par les CML exposées à de fortes concentrations en calcium extracellulaire sont moins bien établis que pour les phosphates. Il semblerait néanmoins que le calcium soit capable d’induire l’apoptose des CML avec formation de corps apoptotiques capables d’initier les proces-sus de calcification extracellulaire. Un mécanisme qui serait fortement potentialisé par le phosphate et qui impliquerait plusieurs systèmes capables de moduler les mouvements calciques au niveau des CML dont des canaux calciques voltage dépendants mais surtout le CaSR. Ce dernier est également exprimé dans les CML. Son rôle dans la calcification vasculaire serait plutôt protecteur et son activation s’opposerait à l’effet pro-calcifiant d’une augmentation extracellulaire de calcium par des mécanismes qui restent à préciser. Pour être complet sur les mécanismes des calcifications vascu-laires, il faut citer l’existence de plusieurs inhibiteurs

REIN ET PATHOLOGIES
REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455 // 41
procéder à la mesure de l’activité phosphatase alcaline totale tous les ans et ceci dès le stade 4, avec une fré-quence plus élevée en cas d’augmentation de la PTH. Dans certaines circonstances, la mesure de la phosphatase alcaline osseuse pourra être effectuée malgré son coût beaucoup plus élevé. En revanche, l’utilisation des autres marqueurs de résorption et formation osseuse actuelle-ment disponibles n’est pas recommandée en raison de leur métabolisme rénal. Les recommandations précisent également que l’atteinte osseuse peut être appréciée par la concentration de PTH sérique, bien que ce paramètre ne constitue pas à proprement parler un reflet direct du remodelage osseux.Ces recommandations semblent avoir fait l’objet d’un large consensus et définissent mieux les aspects dia-gnostiques des désordres du métabolisme phospho-calcique dans la MRC. Elles présentent néanmoins certaines limites. L’évaluation du remodelage osseux chez le patient en IRC est capitale compte tenu du reten-tissement sur le squelette de la MRC et des désordres du métabolisme phosphocalcique qui lui sont associés. La suggestion d’utiliser plus souvent que par le passé, la biopsie osseuse et l’analyse histomorphométrique pour apprécier le remodelage osseux est difficilement réalisable en routine en raison de son caractère inva-sif mais également du fait d’un nombre restreint de laboratoires capables d’analyser les échantillons de biopsie. L’utilisation de marqueurs biologiques s’avère donc indispensable. La recommandation d’utiliser la PTH pour évaluer le remodelage osseux est très limi-tative, en particulier dans l’évaluation de l’ostéopathie adynamique caractérisée par un niveau de remodelage osseux bas sans commune mesure avec les valeurs de PTH qui peuvent être normales, voire élevées. La mesure de la phosphatase alcaline totale, bien que facilement réalisée sur de nombreux automates et à des coûts modérés, manque de sensibilité et ne présente d’intérêt que dans des situations cliniques caractérisées par un très haut niveau de remodelage osseux. On lui préférera bien sûr la mesure de la phosphatase alcaline osseuse, plus spécifique et plus sensible pour évaluer la formation osseuse. II faut cependant noter qu’en raison de réac-tions croisées avec la phosphatase alcaline d’origine hépatique, ce marqueur ne peut pas être utilisé chez des sujets porteurs d’une maladie hépatique chronique. Par ailleurs, la phosphatase alcaline osseuse ne constitue qu’un reflet de l’activité ostéoblastique, or les anomalies du remodelage osseux de la MRC portent non seulement sur l’activité ostéoblastique mais également sur l’activité ostéoclastique. Il serait donc très utile de disposer d’un marqueur de résorption osseuse. Les marqueurs dispo-nibles actuellement, dont les produits de dégradation du collagène de type I comme la désoxypyridinoline (DPD) et les télopeptides associés (CTX, NTX et ICTP) ne peuvent être recommandés, principalement en raison d’un métabolisme rénal important. Des études récentes suggèrent cependant que pour le CTX, sa mesure chez les patients hémodialysés peut apporter certains rensei-gnements cliniques. Une des recommandations impor-tantes des KDIGO est la mesure des concentrations de 25 OH vitamine D dès le stade 3 et ceci bien sûr
portent sur le diagnostic des anomalies biochimiques de l’homéostasie phosphocalcique, sur le diagnostic des atteintes osseuses et des calcifications vasculaires, sur les traitements visant à diminuer l’hyperphosphaté-mie, à normaliser la calcémie, à corriger les anomalies de la PTH et à limiter l’atteinte osseuse, et enfin sur les mesures spécifiques concernant les troubles minéraux et osseux du sujet transplanté. Nous ne rappellerons ici que les principales recommandations qui concernent le diagnostic et le suivi biologique des troubles minéraux et osseux de la MRC. Pour le diagnostic des anomalies biochimiques de l’homéostasie phosphocalcique, il est recommandé d’effectuer un bilan standard comprenant les mesures de la calcémie, de la phosphatémie, de la PTH, des phosphatases alcalines totales et de la 25 OH vitamine D. La fréquence de ces mesures dépendra du stade de la MRC. Il est ainsi recommandé d’effectuer une mesure de la calcémie et de la phosphatémie tous les 6-12 mois au stade 3, tous les 3-6 mois au stade 4 et tous les 1-3 mois aux stades 5 et 5D. L’interprétation du bilan phosphocalcique ne se fera que sur les valeurs de calcémie et de phosphatémie et non sur leur produit (CaxP) qui n’apporte rien à l’évaluation des risques associés au métabolisme minéral. La calcémie pourra être rendue sous sa forme totale ou après correction par l’albumine. Les objectifs thérapeutiques en cas d’hyperphosphaté-mie sont de maintenir la phosphatémie dans les normes du laboratoire aux stades 3-5 de l’IRC et de tendre vers cette normale au stade 5D. Pour la calcémie, celle-ci doit être maintenue dans les normes du laboratoire aux stades 3-5D. La mesure des concentrations sériques de PTH s’effectuera tous les 6-12 mois au stade 4 et tous les 3-6 mois aux stades 5 et 5D. Les valeurs optimales de PTH aux stades 3-5 ne sont pas connues mais il est recommandé si les valeurs sont supérieures à la limite supérieure de la trousse de dosage, de rechercher et de corriger une hyperphosphatémie, une hypocalcémie et une carence en vitamine D. Au stade 5 D, il est recommandé de maintenir la PTH entre 2 et 9 fois la limite supérieure de la trousse utilisée. Dans tous les cas, le biologiste doit informer le clinicien de la méthode utilisée et des éventuels changements de trousses. L’expression des valeurs de PTH du patient en multiples de la limite supérieure de la normale permet de s’affranchir de l’importante variabilité inter-méthodes. En effet, des différences allant jusqu’à un facteur 4 peuvent être observées entre les concentra-tions mesurées avec les différents kits disponibles sur le marché rendant impossible l’utilisation de valeurs seuils identiques quelle que soit la technique de dosage utili-sée. Dès le stade 3, en raison de son rôle central dans le contrôle du métabolisme phosphocalcique, il est suggéré d’effectuer une mesure de la vitamine D (25 OH vitamine D ou 25OH-D). En cas de supplémentation visant à corriger une déficience ou une insuffisance en vitamine D, cette mesure sera répétée pour vérifier l’éventuelle réplétion. Il n’existe pas de consensus absolu sur les valeurs de référence de la 25 OH vitamine D et les KDIGO n’ont pas pris de position sur les cibles idéales à atteindre chez l’insuffisant rénal mais recommandent de se baser sur les cibles utilisées dans la population générale. Pour apprécier le niveau de remodelage osseux, il est recommandé de

42 // REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455
et diminution de la 25 OH vitamine D. Les mécanismes physiopathologiques de l’hyperactivité parathyroïdienne sont en passe d’être mieux compris, en particulier depuis la découverte d’acteurs clés que sont le FGF23 et Klotho. Elle pourrait être la conséquence de la rétention de phos-phate qui induit une augmentation précoce du FGF 23 ce qui favorise la diminution de synthèse du calcitriol par le rein et donc l’hypocalcémie. Ce schéma patho-génique n’est pas consensuel et d’autres hypothèses existent notamment celle donnant un rôle primordial à la diminution de l’expression rénale de Klotho. D’autres travaux de recherche seront nécessaires pour connaître la cinétique exacte des modifications moléculaires res-ponsables de l’augmentation de la PTH dans la MRC. L’hyperparathyroïdie secondaire peut être considérée, au moins à court terme, comme une réaction adéquate visant à compenser les troubles du métabolisme phos-phocalcique. À plus long terme, l’élévation de la PTH sérique et la résistance à son action dans les tissus cibles devient délétère. Elle est en particulier la cause d’alté-rations quantitatives et qualitatives du tissu osseux qui augmentent sa fragilité. Les troubles phosphocalciques qui accompagnent l’hyperparathyroïdie secondaire, contribuent également de façon majeure aux calcifica-tions vasculaires qui augmentent la mortalité cardiovas-culaire au cours de la MRC. Notre connaissance des mécanismes moléculaire et cellulaire impliqués dans les effets délétères osseux et vasculaires des troubles minéraux s’est considérablement améliorée ces der-nières années. Cette meilleure connaissance a eu des retentissements importants sur la prise en charge des troubles minéraux, ce qui a permis d’augmenter l’espé-rance de vie et d’améliorer la qualité de vie des patients. Les recommandations KDIGO, qui ont fait l’objet d’un large consensus, sont aujourd’hui admises. Elles portent entre autre sur le diagnostic et donnent au biologiste, dans son dialogue avec le clinicien, une place principale dans la prise en charge des troubles minéraux et osseux de la maladie rénale chronique.
Déclaration d’intérêts : les auteurs déclarent ne pas avoir de
conflits d’intérêts en relation avec cet article.
afin de diagnostiquer une éventuelle carence ou insuf-fisance. Après supplémentation chez les patients dia-lysés, il est également recommandé d’effectuer une mesure des concentrations de 25 OH vitamine D pour vérifier l’état de réplétion. Ces recommandations ne sont pas assorties de valeur seuil définissant l’insuf-fisance en vitamine D dans la MRC mais suggèrent de se référer aux valeurs proposées dans la popu-lation générale. De nombreux experts considèrent que les valeurs souhaitables de la 25 OH vitamine D sont comprises entre 75 et 150 nmol/L (30-60 ng/ml). Une concentration < 25 nmol/L (< 10 ng/mL) signe une carence et une concentration comprise entre 25 et 75 nmol/L (10-30 ng/mL) correspond à une insuffisance en vitamine D. Cependant, il n’y a pas encore de consensus général sur ces valeurs ce qui rend difficile l’exploration du statut en vitamine D au cours de la MRC, difficulté qui est accentuée par l’absence de standardisation au niveau des méthodes de dosage. Enfin, comme nous l’avons vu précédemment, les anomalies du métabolisme phospho-calcique sont à l’origine de calcifications vasculaires qui souvent aggravent le pronostic dans la MRC. Les KDIGO recommandent chez les patients aux stades 3-5D de rechercher ces calcifications par une radiographie simple de l’abdomen de profil et une échographie cardiaque. À ce jour, aucun marqueur biologique ne permet d’éva-luer la survenue de calcifications. Cependant, plusieurs molécules candidates peuvent d’ores et déjà être sug-gérées comme potentiellement intéressantes dont la fétuine A, la matrix GLA protein (MGP), l’ostéoproté-gérine ou encore le FGF23 et la forme soluble de Klo-tho. Les études à venir devront confirmer l’utilité de ces bio-marqueurs pour la détection des calcifications cardiovasculaires.
7. Conclusion
La maladie rénale chronique (MRC) est caractérisée par des troubles du métabolisme minéral qui conduisent à une hyperparathyroïdie secondaire associant hyper-phosphatémie, hypocalcémie, augmentation de la PTH
Pour en savoir plus Afin d’approfondir plus avant ses connaissances du sujet, le lecteur pourra consulter les revues générales récentes et articles référencés ci-dessous.
Mécanismes généraux de régulation du métabolisme phosphocalcique
Bacchetta J, Cochat P, Salusky IB. FGF23 et Klotho : les nouveaux incontournables du métabolisme phosphocalcique. Arch Pediat 2011; 18:686-95. Courbebaisse M, Souberbielle JC. Equilibre phosphocalcique : régulation
et exploration; Néphrol Thérap 2011;7:118-38. Peacock M. Calcium metabolism in health and disease. Clin J Am Soc
Nephrol 2010;5(Suppl1):S23-30. Quarles LD. Skeletal secretion of FGF23 regulates phosphate and vita-
min D metabolism. Nat Rev Endocrinol 2012;8:276-86. Vallet M, Tack I. Physiologie du calcium et des phosphates. Rev Rhum
Monographies 2012;79:203-9.
Physiopathologie de l’hyperparathyroïdie secondaire de la MRC Cunningham J, Locatelli F, Rodriguez M. Secondary hyperparathyroi-
dism: pathogenesis, disease progression, and therapeutic options. Clin J Am Soc Nephrol 2011;6:913-21. Hruska K, Mathew S, Lund R, et al. Hyperphosphatemia of chronic
kidney disease. Kidney Int 2008;74:148-57. Isakova T, Wolf MS. FGF23 or PTH: which comes in first in CKD. Kidney
Int 2010;78:947-9. Jonh GB, Cheng CY, Kuro-o M. Role of klotho in aging, phosphate metabolism
and CKD. Am J Kidney Dis, 2011, 58: 127-134. Jüppner H, Wolf M, Salusky IB. FGF23: more than a regulator of renal
phosphate handling. J Bone Mineral Res, 2010, 25: 2091-2097. Kuro-o M. Phosphate and klotho. Kidney Int, 2011, 79 (Suppl121): S20-S23. Mac Way F, Lessard M, Lafage-Proust MH. Pathophysiology of chronic
kidney disease-mineral and bone disorder. Joint Bone Spine 2012;79:544-9. Slatopolsky E. The intact nephron hypothesis: the concept and its
implication for phosphate management in CKD-related mineral and bone disorder. Kidney Int 2011;79(Suppl121):S3-S8.

REIN ET PATHOLOGIES
REVUE FRANCOPHONE DES LABORATOIRES - SEPTEMBRE-OCTOBRE 2013 - N°455 // 43
Troubles osseux de la MRC Andress DL. Adynamic bone in patients with chronic kidney disease.
Kidney Int 2008;73:1345-54. Cannata-Andia JB, Rodriguez Garcia M, Gomez Alonzo C. Osteoporosis
and adynamic bone in chronic kidney disease. J Nephrol 2013;26:73-80. Gordon PL, Frassetto LA. Management of osteoporosis in CKD stage
3 to 5. Am J Kidney Dis 2010;55:941-56. Moe S, Drüeke T, Cunningham J, et al. Definition, evaluation, and
classification of renal osteodystrophy: a position statement from kidney disease improving global outcome (KDIGO). Kidney Int 2006;69:1945-53.
Calcifications vasculaires Zhu D, Mackenzie NCW, Farquharson C, et al. Mechanism and clinical
consequences of vascular calcification. Font Endocrinol 2012;3:1-12. Shanahan C, Crouthamel MH, Kapustin A, et al.Arterial calcification in chronic
kidney disease: key roles for calcium and phosphate. Circ Res 2011;109:697-711. Giachelli CM. The emerging role of phosphate in vascular calcification.
Kidney Int 2009;75:890-7. Meiting W, Rementer C, Giachelli CM. Vascular calcification: an update
on mechanisms and challenges in treatment. Calc Tissue Int 2013;in press.
Exploration biologique des troubles minéraux et osseux de la maladie rénale chronique Kidney disease: improving global outcomes. KDIGO clinical practice
guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease–mineral and bone disorder (CKD–MBD), Kidney Int 2009;76(Suppl113):S1-S130.
Jean G, Chazot C. L’essentiel des nouvelles recommandations des Kidney disease: improving global outcomes (KDIGO) pour les désordres du métabolisme minéral et osseux à l’usage du clinicien francophone. Néphrol Thérap 2010;6:151-7. Souberbielle JC, Cavalier E, Jean G. Interpretation of serum parathy-
roid hormone concentrations in dialysis patients: what do the KDIGO guidelines change for the clinical laboratory? Clin Chem Lab Med 2010; 48:769-74. Delanaye P, Dubois BE, Jouret F, et al. Parathormone and bone specific
alkaline phosphatase for the follow-up of bone turnover in hemodialysis patients: Is it so simple. Clin Chim Acta 2013;417:35-8. Sardiwal S, Magnusson P, Goldsmitn DJA, et al. Bone alkaline
phosphatise in CKD-mineral bone disorder. Am J Kidney Dis 2013; in press.