thÈse - vetagro sup iii) la défense cellulaire antioxydante...
TRANSCRIPT

1
VETAGRO SUP
CAMPUS VETERINAIRE DE LYON
Année 2015 - Thèse n°
IMPLICATION DU STRESS OXYDANT DANS PLUSIEURS AFFECTIONS DU CHEVAL ATHLETE : REVUE
BIBLIOGRAPHIQUE.
THÈSE
Présentée à l’UNIVERSITÉ CLAUDE-BERNARD - LYON I (Médecine - Pharmacie)
et soutenue publiquement le 12 juin 2015 pour obtenir le grade de Docteur Vétérinaire
par
Benjamin DUBOIS Née le 3 Juin 1990
à COMPIÈGNE

2
3
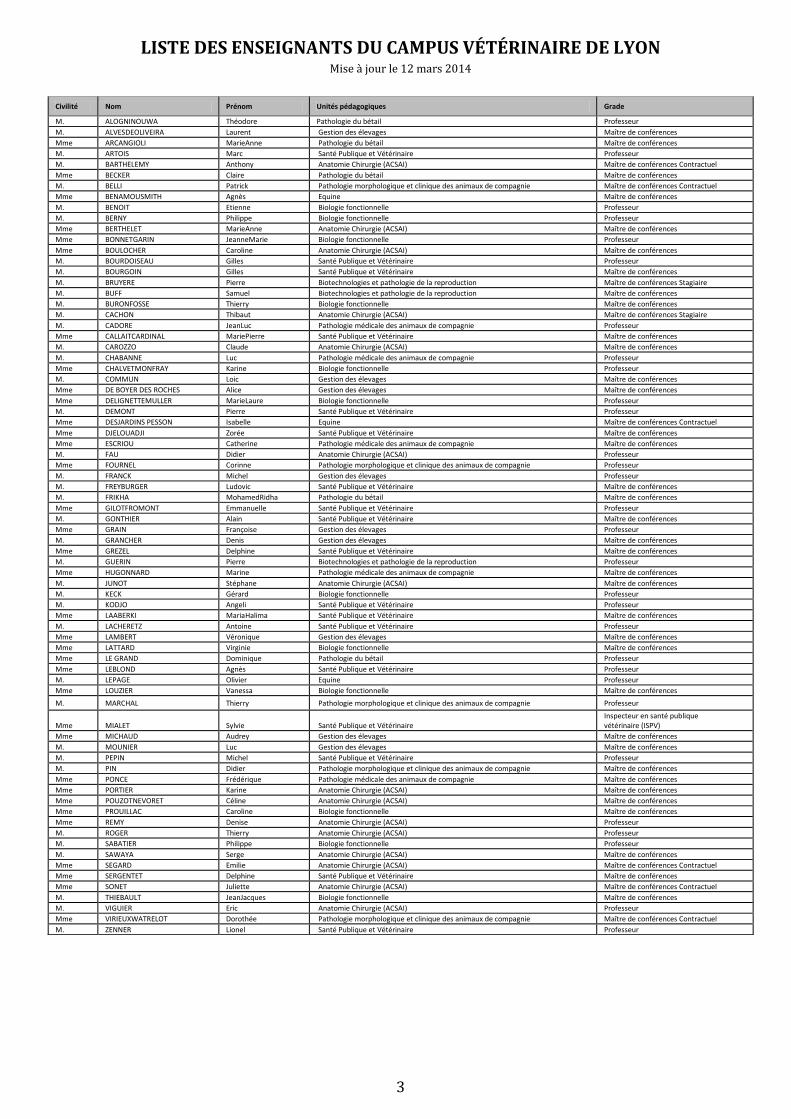
LISTE DES ENSEIGNANTS DU CAMPUS VÉTÉRINAIRE DE LYON Mise à jour le 12 mars 2014
Civilité Nom Prénom Unités pédagogiques Grade
M. ALOGNINOUWA Théodore Pathologie du bétail Professeur
M. ALVESDEOLIVEIRA Laurent Gestion des élevages Maître de conférences
Mme ARCANGIOLI MarieAnne Pathologie du bétail Maître de conférences
M. ARTOIS Marc Santé Publique et Vétérinaire Professeur
M. BARTHELEMY Anthony Anatomie Chirurgie (ACSAI) Maître de conférences Contractuel
Mme BECKER Claire Pathologie du bétail Maître de conférences
M. BELLI Patrick Pathologie morphologique et clinique des animaux de compagnie Maître de conférences Contractuel
Mme BENAMOUSMITH Agnès Equine Maître de conférences
M. BENOIT Etienne Biologie fonctionnelle Professeur
M. BERNY Philippe Biologie fonctionnelle Professeur
Mme BERTHELET MarieAnne Anatomie Chirurgie (ACSAI) Maître de conférences
Mme BONNETGARIN JeanneMarie Biologie fonctionnelle Professeur
Mme BOULOCHER Caroline Anatomie Chirurgie (ACSAI) Maître de conférences
M. BOURDOISEAU Gilles Santé Publique et Vétérinaire Professeur
M. BOURGOIN Gilles Santé Publique et Vétérinaire Maître de conférences
M. BRUYERE Pierre Biotechnologies et pathologie de la reproduction Maître de conférences Stagiaire
M. BUFF Samuel Biotechnologies et pathologie de la reproduction Maître de conférences
M. BURONFOSSE Thierry Biologie fonctionnelle Maître de conférences
M. CACHON Thibaut Anatomie Chirurgie (ACSAI) Maître de conférences Stagiaire
M. CADORE JeanLuc Pathologie médicale des animaux de compagnie Professeur
Mme CALLAITCARDINAL MariePierre Santé Publique et Vétérinaire Maître de conférences
M. CAROZZO Claude Anatomie Chirurgie (ACSAI) Maître de conférences
M. CHABANNE Luc Pathologie médicale des animaux de compagnie Professeur
Mme CHALVETMONFRAY Karine Biologie fonctionnelle Professeur
M. COMMUN Loic Gestion des élevages Maître de conférences
Mme DE BOYER DES ROCHES Alice Gestion des élevages Maître de conférences
Mme DELIGNETTEMULLER MarieLaure Biologie fonctionnelle Professeur
M. DEMONT Pierre Santé Publique et Vétérinaire Professeur
Mme DESJARDINS PESSON Isabelle Equine Maître de conférences Contractuel
Mme DJELOUADJI Zorée Santé Publique et Vétérinaire Maître de conférences
Mme ESCRIOU Catherine Pathologie médicale des animaux de compagnie Maître de conférences
M. FAU Didier Anatomie Chirurgie (ACSAI) Professeur
Mme FOURNEL Corinne Pathologie morphologique et clinique des animaux de compagnie Professeur
M. FRANCK Michel Gestion des élevages Professeur
M. FREYBURGER Ludovic Santé Publique et Vétérinaire Maître de conférences
M. FRIKHA MohamedRidha Pathologie du bétail Maître de conférences
Mme GILOTFROMONT Emmanuelle Santé Publique et Vétérinaire Professeur
M. GONTHIER Alain Santé Publique et Vétérinaire Maître de conférences
Mme GRAIN Françoise Gestion des élevages Professeur
M. GRANCHER Denis Gestion des élevages Maître de conférences
Mme GREZEL Delphine Santé Publique et Vétérinaire Maître de conférences
M. GUERIN Pierre Biotechnologies et pathologie de la reproduction Professeur
Mme HUGONNARD Marine Pathologie médicale des animaux de compagnie Maître de conférences
M. JUNOT Stéphane Anatomie Chirurgie (ACSAI) Maître de conférences
M. KECK Gérard Biologie fonctionnelle Professeur
M. KODJO Angeli Santé Publique et Vétérinaire Professeur
Mme LAABERKI MariaHalima Santé Publique et Vétérinaire Maître de conférences
M. LACHERETZ Antoine Santé Publique et Vétérinaire Professeur
Mme LAMBERT Véronique Gestion des élevages Maître de conférences
Mme LATTARD Virginie Biologie fonctionnelle Maître de conférences
Mme LE GRAND Dominique Pathologie du bétail Professeur
Mme LEBLOND Agnès Santé Publique et Vétérinaire Professeur
M. LEPAGE Olivier Equine Professeur
Mme LOUZIER Vanessa Biologie fonctionnelle Maître de conférences
M. MARCHAL Thierry Pathologie morphologique et clinique des animaux de compagnie Professeur
Mme MIALET Sylvie Santé Publique et Vétérinaire Inspecteur en santé publique vétérinaire (ISPV)
Mme MICHAUD Audrey Gestion des élevages Maître de conférences
M. MOUNIER Luc Gestion des élevages Maître de conférences
M. PEPIN Michel Santé Publique et Vétérinaire Professeur
M. PIN Didier Pathologie morphologique et clinique des animaux de compagnie Maître de conférences
Mme PONCE Frédérique Pathologie médicale des animaux de compagnie Maître de conférences
Mme PORTIER Karine Anatomie Chirurgie (ACSAI) Maître de conférences
Mme POUZOTNEVORET Céline Anatomie Chirurgie (ACSAI) Maître de conférences
Mme PROUILLAC Caroline Biologie fonctionnelle Maître de conférences
Mme REMY Denise Anatomie Chirurgie (ACSAI) Professeur
M. ROGER Thierry Anatomie Chirurgie (ACSAI) Professeur
M. SABATIER Philippe Biologie fonctionnelle Professeur
M. SAWAYA Serge Anatomie Chirurgie (ACSAI) Maître de conférences
Mme SEGARD Emilie Anatomie Chirurgie (ACSAI) Maître de conférences Contractuel
Mme SERGENTET Delphine Santé Publique et Vétérinaire Maître de conférences
Mme SONET Juliette Anatomie Chirurgie (ACSAI) Maître de conférences Contractuel
M. THIEBAULT JeanJacques Biologie fonctionnelle Maître de conférences
M. VIGUIER Eric Anatomie Chirurgie (ACSAI) Professeur
Mme VIRIEUXWATRELOT Dorothée Pathologie morphologique et clinique des animaux de compagnie Maître de conférences Contractuel
M. ZENNER Lionel Santé Publique et Vétérinaire Professeur

4
REMERCIEMENTS
À Monsieur le Professeur Gilles RODE,
De la Faculté de Médecine de Lyon,
Qui nous a fait l’honneur d’accepter la présidence du jury de cette thèse,
Qu’il reçoive ici l’expression de nos hommages très respectueux.
À Monsieur le Professeur Jean-Jacques THIÉBAULT,
De VetAgro Sup, Campus Vétérinaire de Lyon,
Qui nous a fait l’honneur d’encadrer ce travail de thèse,
Qu’il trouve ici l’expression de notre sincère reconnaissance,
Sincères remerciements.
À Monsieur le Professeur Philippe BERNY,
De VetAgro Sup, Campus Vétérinaire de Lyon,
Qui nous a fait l’honneur de bien vouloir juger ce travail,
Qu’il trouve ici l’expression de notre sincère reconnaissance,
Sincères remerciements.

5
TABLE DES MATIÈRES
Listes des Figures……….………………………………………………………………………………………….… 11
Liste des Tableaux ………………………………………………………………………………………………....... 12
Liste des Abréviations ………………………………………………………………………………………...…… 13
Introduction ………………………………………………………………………………………………………….… 15
1ère Partie : Qu’est-ce que le stress oxydant ?........................................................................16
I) Pré-requis de chimie radicalaire.…………………………………………………………………………………………………..…... 16
Bref historique scientifique..…………………………………………………………………………………………………………...….. 16
A) Qu’est ce qu’un radical libre ? ……………………………………………………………………………………………. 16
1) La réaction d’oxydo-réduction et potentiel redox .………………………………………...…….….. 16
2) Mécanismes chimiques de formation des radicaux libre………………………………………….. 17
2.i Oxydation……………………………………………………………………………………………...… 17
2.ii Réduction……………………….………………………………………………………………………. 17
2.iii Fission Homolytique …….……………………………………………………………………..… 17
3) Réactivité des radicaux libres ………………..……………………………………………………………… 18
3.i Réaction entre un radical libre et une molécule non radicalaire .………………... 18
3.ii Réaction entre deux radicaux libres .………………………………………………………... 19
3.iii Notion de chaîne de transmission radicalaire …………………………………………. 19
4) Le cas de l’oxygène ………………………………………………………………………………………….…… 19
4.i Capacité oxydante de l’atome d’oxygène…..……………………………………………….. 19
4.ii Particularités de la molécule de dioxygène .………………………………………..……. 19
4.iii Stabilité et activation de la molécule de dioxygène .………………………..……..…. 20
B) Espèces réactives de l’oxygène ou dérivés de son métabolisme .………………………………………….. 20
1) Terminologie et classification des espèces réactives .…………...………………………………… 20
2) Espèces radicalaires .……………………………………………………………………………………….……. 22
2.i Anion superoxide O2°- …………………………………………………………………………….... 22
2.ii Radical hydroxyle HO° .………………………………………………………….………….…….. 22
2.iii Les radicaux peroxyle RO2° et alkoxyle RO° .………………………….………………… 23
2.iv Les radicaux carbonate CO3°- et bicarbonate HCO3° …….…………..……….………. 24
2.v Le monoxyde d’azote NO° …………………………………………………………………….…. 25
2.vi Le dioxyde d’azote NO2° ……………………………………………………………………...… 27
3) Espèces non-radicalaires .…………………………………………………………………………………...…. 27
3.i Le dioxygène singulet 1O2 ……...………………………………………………………………… 27
3.ii Le peroxyde d’hydrogène H2O2 ………………………………………………………………. 28
3.iii Le peroxynitrite ONOO- ………...………………………………….……………………………. 29
3.iv L’acide hypochloreux HOCl ………………………………………………………………….… 30
3.v L’ozone O₃…………………………………………………………………………………………….… 31
II) Principales sources des espèces réactives …….………………………………………………………………………………… 32
A) La mitochondrie…..…………………………………………………………………………………………………………….. 32
1) La chaîne respiratoire mitochondriale .……………………………………...…………………………… 32
1.i Quelques rappels…..……………………………………………………….………………………… 32
1.ii Production d’espèces réactives de l’oxygène par la chaîne respiratoire
mitochondriale……………………………………………………………………………….………………33
1.iii Stress oxydant et dysfonctionnement mitochondrial..….…………………………….35
2) Autres sources mitochondriales d’espèces réactives.………………….………………….…………36
B) Importance des métaux de transition ………………………………………………………………………………….36
1) Définition………………………………………………………………………………………………………………36

6
2) Réaction de Fenton et réaction de Haber – Weiss …………………………………………...………36
2.i Réaction de Fenton …………………………………………………………………………..………37
2.ii Réaction de Haber-Weiss ……………………………………………..…………………………37
2.iii Disponibilité des métaux de transition in vivo ……………...…………………………38
3) Complexes « iron-oxygen » ou « Fe-O » : les ions ferryles et ions perferryles...…….…… 38
C) Des enzymes du métabolisme et de l’inflammation..……………………………………………………….……39
1) Les cofacteurs enzymatiques flaviniques des oxydases : FAD et FMN……….….….……….39
2) Xanthine déshydrogénase (XDH) et xanthine oxydase (XO) …………………………….………40
3) Les NADPH-oxydases (Nox)……………………………………………….……………………………......… 40
4) Les oxydases des peroxysomes..….………………………………………….……………………………… 41
5) Des enzymes du métabolisme de l’acide arachidonique : les cyclooxygénases (COX).. 41
6) La myéloperoxydase (MPO) ………………………………………………….………………………...……..42
7) Les monoxyde d’azote synthases (NOs).…………………………………………………………….……43
D) Autres sources endogènes d’intérêt : les « auto-oxydations » ..……………………………………………..43
III) La défense cellulaire antioxydante ……………………………………………..…………………………………………………...44
A) Définition d’un antioxydant et stratégies de défense.…………………………………………………………… 44
1) Qu’est ce qu’un antioxydant ? ….………………………………………………………………………………44
2) Stratégies de défense ……………….……………………………………………………………………..………44
B) Antioxydants enzymatiques .……………………………………………………………………………………………….45
1) Les superoxyde dismutases (SOD).…….……………………………………………………………………45
2) Les glutathion peroxydases (GPx) ………..…………………………………………………………………47
3) Les glutathion réductases (GRD) .……………………………………………………………………………48
4) Les catalases (CAT) …………………………………..……………………………………………………………48
5) Les thiorédoxine réductases (TXNRD) .……………...……………………………………………………49
6) Les peroxyrédoxines (PRX) .…………………………...………………………………………………………49
7) Les paraoxonases (PON) .………………………..………………………………………………………………50
C) Antioxydants non-enzymatiques .……...…………………………………………………………………………………51
1) Antioxydants non-enzymatiques d’origine endogène.……………………………………………........... 51
1.i Glutathion (GSH/GSSG) .….…………………………………………………………………………51
1.ii La thiorédoxine (Trx) .………………………………………………………………………………52
1.iii Acide urique ….…………………………………………..……………………………………………52
1.iv Bilirubine .………….……………………………………………………………………………………53
1.v Protéines régulatrices, de transport et de séquestration des
métaux de transition ……...………………………………………………………………………...……54
1.vi Les protéines découplantes. ………..……………………………………………………………55
1.vii Le monoxyde d’azote NO° .………………………………………………………………………56
1.viii Autres antioxydants non-enzymatiques endogènes .....……………………..………56
2) Antioxydants non-enzymatiques d’origine nutritionnelle…….………………………….............. 57
2.i La vitamine E.……………………………………….……………………………………………………57
2.ii La vitamine C ...…………………………………….……………………………………………………58
2.iii Les caroténoïdes ..……………………………….……………………………………………………60
2.iv Les polyphénols .…...…………………………….……………………………………………………60
3) Les cofacteurs minéraux …………………………………….……………………………………………………61
3.i Le zinc ………………………………………………….……………………………………………………61
3.ii le sélénium ………………………………………….……………………………………………………61
IV) Dégâts liés à la présence des ERO..……………………………………………………..……………………………………………61
A) Stress oxydant et altérations biochimiques à l’échelle moléculaire .……………….………………………61
1) Stress oxydant et altérations des lipides .………………………………………….………………………61
1.i Peroxydation des acides gras polyinsaturés membranaires....………………………61
1.ii Oxydation du cholestérol .………………………………………………….………………………64
2) Stress oxydant et altérations des protéines et des acides aminés……..…..……………………65
2.i Oxydation des chaînes latérales………………………………………………..…………………66

7
2.ii Nitration.…………………………………………………………………………………………………67
2.iii Nitrosylation .…………………………………………………………………………………………67
2.iv Glucoxydation…………………………………………………………………………………………67
2.v Chlorination ……………………………………………………………………………………………68
2.vi Déshydrogénation, rupture et remaniement de la chaîne protéique…..………68
3) Stress oxydant et altérations de l’ADN.……………………………………………………………………68
B) Conséquences directes des altérations biochimique à l’échelle cellulaire………………………………70
1) Conséquences fonctionnelles des altérations des lipides membranaires...…………………70
2) Conséquences fonctionnelles des altérations des protéines...……………………………………71
2.i Dysfonctionnements enzymatiques..…………………………………………………………..71 2.ii Dysfonctionnement des anticorps et perte des propriétés antigéniques des protéines..………………………………………………………………………………………………..71 2.iii Dégradation des propriétés physicochimiques et biomécaniques des protéines.………………………………………………………………………………………………………72 2.iv Dysfonctionnement/modulation des récepteurs cellulaires, des signaux de transduction et perturbation des fonctions de régulation ……………………………...72 2.v Dysfonctionnement du transport protéique et du métabolisme énergétique........................................................................................................................................72
3) Conséquences fonctionnelles des altérations de l’ADN ….…………………………………………73
3.i ADN nucléaire...…………………………………………………………………………………………73
3.ii ADN mitochondrial..…………………………………………………………………………………74
C) Impact cellulaire et tissulaire des produits de dégradations du stress oxydant…...…………………74
V) Évaluation expérimentale du stress oxydant….…………………………………………………………………………………76
A) Principe de l’exploration du stress oxydant …………………………………………………………………………76
B) Moyens d’explorations ….…..……………………………………………………………………..…………………………76
1) Méthodes directes …….……………………………………………………………………………………………76
1.i RPE : Résonance Paramagnétique Électronique….………………………………………76
1.ii Chimioluminescence...………………………………………………………………………………78
2) Méthodes indirectes : témoins de la peroxydation lipidique ….…………………………………78
2.i Mesure de la fluidité membranaire érythrocytaire..…………….………………………78
2.ii Méthode des TBARS …………………………………………………………………………………80
2.iii Dosage des peroxydes lipidiques..………….………………………...………………………81
2.iv Dosage des aldéhydes produits de la peroxydation lipidique ….…………………81
2.v Dosage des hydrocarbures volatils expirés ..…………………………….……………..…83
2.vi Dosage des isoprostanes.………………………………………………………….………………83
2.vii Dosage des diènes conjugués ………………………………………………….………………84
3) Méthodes indirectes : témoins de l’altération des protéines …………………….………………84
3.i Détection des carbonyles protéiques.………………………………………….………………84
3.ii Détection des protéines et acides aminés nitratés….….………………….………………84
4) Méthodes indirectes : témoins de l’altération de l’ADN…………………………….………………84
5) Méthodes indirectes : Mesure des défenses antioxydantes ....……………………………………85
5.i Mesure de l’activité des antioxydants enzymatiques...…………………………………85
5.ii Dosage des antioxydants non enzymatiques …...…………………………………………87
5.iii Mesure de la capacité antioxydante globale……………………………….……...………89
6) Autres moyens de mesure du stress oxydant..…………………………………….…….………………91
6.i Dosage du NO° expiré et dans les tissus biologiques…………………….………………91
6.ii Dosage du CO° expiré ………………………………………………………………..………………92
6.iii Autres facteurs exhalés pouvant être mesurés….…………………………..……………92
6.iv Détection des anticorps dirigés contre les produits de dégradation…..…………92
C) Défis de l’exploration expérimentale du stress oxydant……..…………………………………….….…………92
IV) Rôles physiologiques et physiopathologiques généraux des espèces réactives de l’oxygène ...…………… 93
A) ERO et régulation des fonctions cellulaires physiologiques ……………………………………………………93
1) Production et fonctions régulatrices de NO° ………………………..……………………………………93

8
2) Production d’ERO par les cellules phagocytaires..……………………………………………………94
3) Production d’ERO par les cellules dans le signalement cellulaires……………………………94
4) Production d’ERO dans les mécanismes d’homéostasie...…………………………………………94
5) La production d’ERO pour la régulation de l’adhésion cellulaire …………………………..…96
6) La régulation redox de la réponse immunitaire..………………………………………………………96
7) Implication des ERO dans la vie et la mort de la cellule ………………………………………..…96
7.i ERO et cycle cellulaire …………………………………………………………………………...…96
7.ii ERO et apoptose ………………………………………………………………………………………96
8) Statut et maintien de l’équilibre redox au sein de la cellule et dans les
systèmes biologiques..…………………………………………………………………………………………………97
B) Stress oxydant et processus inflammatoires..…………………………………………………………………..……99
1) Quelques rappels sur l’inflammation..………………………………………………………………………99
2) Production d’ERO lors de l’inflammation….…………………………………………………..…...……100
3) ERO et phagocytose...……………………………………………………………………………………...………100
4) ERO et établissement du processus inflammatoire...…………………………………………………102
5) Action pro-inflammatoire des produits de dégradation du stress oxydant……….............103
6) Stress oxydant et ER-stress..……………………………………………………………………………………104
6.i ER-stress et perturbation du métabolisme calcique ……………………………………104
6.ii ER-stress et réponse UPR …………………………………………………………………………105
6.iii Bilan de l’ER-stress …………………………………………………………………………………105
C) Stress oxydant et ischémie-reperfusion …..……………………………………………………………………………105
1) Principales modalités de production d’ERO lors de l’ischémie-reperfusion .…………...….105
2) Phase d’ischémie : phase préparatrice .…………………………………………………………………….106
2.i Chute de la production d’ATP et accumulation de produits de dégradation …..106
2.ii État mitochondrial MTP ……………………………………………………………………………106
2.iii Conversion de la xanthine déshydrogénase (XDH) en xanthine
oxydase(XO)……………………………………………………………………………………………………107
3) Reperfusion et production d’ERO ……………………………………………………………….……………107
2ième Partie : Stress oxydant et physiopathologie équine chez le sujet athlète…..…109
I) Pré-requis de physiologie de l’effort …………………………………………………………………………….………………….…109
A) Consommation de dioxygène et effort physique .………………………………………………..……………….…109
1) Consommation en dioxygène et VO2max ……………………………………………………………………..109
1.i Définition de la consommation en dioxygène..………………………………………………109
1.ii Détermination expérimentale VO2max…………….……………………………………………109
1.iii PMA = puissance maximale aérobie..……………………………………………………….…109
2) Différents types d’effort selon la consommation en dioxygène ……………………………….…110
B) Métabolisme énergétique lors de l’effort…………………………………………………………………..……………110
1) Les voies métaboliques de production d’ATP durant l’effort..……………………………..………110
1.i Voie anaérobie alactique….……………………………………………………………………..……110
1.ii Voie anaérobie lactique…..………………………………………………………………………..…110
1.iii Voie aérobie ….………………………………………………………………………………………..…111
1.iv La dette en oxygène….…………………………………………………………………………………111
2) Types de fibres musculaires….……………………………………………………………………………………111
III) L’exercice physique génère du stress oxydant chez le cheval…………………………………………………………….…113
A) Mise en évidence d’un stress oxydant lors d’un effort physique….………………………………………….…113
1) Evidences expérimentales….…………………………………………………………………………………….…113
1.i Mise en évidence chez le cheval …………………………………………………………………….113
1.ii Qu’en est-il chez l’homme ?.……………………………………………………………………….…119
1.iii Bilan .………………………………………………………………………………………………………….120
2) Facteurs de variations…..…………………………………………………………………………………………….120
2.i Facteurs individuels de base : l’âge, le sexe et la race...……………………………………120

9
2.ii L’effort physique lui-même .……………………………………….………………………………….121
2.iii Régime alimentaire et la supplémentation....…………………………………………………..121
2.iv Paramètres d’ambiance : rythme journalier, altitude, température et
hygrométrie..……….…………………………………………….………………………………………..………121
3) Influence de l’entraînement ?..……………………………………………………………………………………..122
B) Sources principales d’ERO lors de l’effort physique….………………………………………………………………..123
1) La fuite électronique depuis la chaîne de phosphorylation oxydative mitochondriale :
« l’hypothèse mitochondriale ».…………………………..………………………………………………………….…123
2) Activation de mécanismes inflammatoires…...….………………………………………………………….…124
2.i L’effort induit une réaction inflammatoire à l’origine de la genèse d’ERO…….….…124
2.ii Production des ERO par les cellules inflammatoires ……………………………..…….…...124
3) L’hypoxémie artérielle induite par l’exercice..…………………………………………………………………125
3.i L’effort intense induit un déséquilibre entre la ventilation et la perfusion …………..125
3.ii Une production d’ERO selon les modalités de l’ischémie–reperfusion...………………126
4) L’acidose métabolique…….………………………………………………………………………………………………126
5) L’hyperthermie …………………………………………………………………………………………………………...…127
6) La calcémie…….…………………………………………………………………………………………………………..….127
7) Autres facteurs de variations d’intérêt…..……………………………………………………………………...…128
IV) Relations entre les affections organiques de l’athlète et le stress oxydant……………………………………………….…129
A) Pathologie musculaire……………………………………………………………………………………………………………….…129
1) Stress oxydant et fatigue musculaire lors d’un effort physique…….……………………………….……129
1.i Définition de la fatigue musculaire.……………………………….………………………………….…129
1.ii Fatigue musculaire et stress oxydant……………………………………………………….…………129
1.iii Bilan…………………………………………………….……………………………………………………….…131
2) Stress oxydant et rhabdomyolyse d’effort ………………….……………………………………………….……132
1.i Présentation ……………………………………………………..…………………………………………….…132
1.ii Tableau clinique et lésionnel ……………………………….………………………………………….…132
1.iii Physiopathologie et lien avec le stress oxydant.………...…………………………………….…133
1.iv Bilan ……………………………………………………………….…………………………………………….…134
B) Pathologie respiratoire ………………………………………………………………….……………………………………………134
1) La maladie obstructive chronique des petites voies respiratoires (RAO)………...…………………134
1.i Présentation générale ……………………………………………………………………….………………134
1.ii Tableau clinique et lésionnel ……………………………………………………………….……………135
1.iii Physiopathologie générale ………………………………………………………………………………135
1.iv Déséquilibre redox des chevaux RAO……………………………………………………..…………136
1.v Conséquences physiopathologiques ……………………………………………………….…………138
1.vi Bilan …………………………………………………………………………………………………….…………139
2) La maladie inflammatoire des voies respiratoires profondes chez les jeunes chevaux de courses
(IAD)…………………………………………………………………………………………………………………………………..140
2.i Présentation générale….....………………………………………………………………………………..…140
2.ii Tableau clinique et lésionnel..…..……………………………………………………………………….140
2.iii Physiopathologie et lien avec le stress oxydant………………………………………………….141
2.iv Bilan ……………………………………………………………………………………………………………….143
3) L’hémorragie pulmonaire induite par l’effort (HPIE)..……………………...……………………………….144
3.i Présentation……………………...……………………………………………………………………………….144
3.ii Physiopathologie théorique et lien avec le stress oxydant……..……………………………144
3.iii Bilan……………………………………………………………………………………….……………………….145
C) Pathologie articulaire : la maladie articulaire dégénérative équine……….….…………………………………….145
1) Pré-requis d’arthrologie..………………………………………………………………...……………………………….145
1.i Organisation d’une diarthrose……….…………………………………………………………………….145
1.ii Le cartilage articulaire..………………………..…………………………………………………………….146
1.iii Le liquide synovial……..…………………………………………………………………………………….147
1.iv La capsule articulaire………..………………………………………………………………………….......147

10
1.v L’os sous-chondral……….……………………………………………………………………………….......148
1.vi Sensibilité de l’articulation au stress oxydant et défenses
antioxydantes…………………………...…………………………………………….……………………………...148
2) La maladie dégénérative articulaire équine……………………………………………………………………...148
2.i Présentation générale………………………………………………………………………………………...148
2.ii Tableau lésionnel……………………………………………………………………….……………………..149
3) Physiopathologie de l’OA et lien avec le stress oxydant………..……………………….…………………..150
3.i Mise en évidence d’un stress oxydant lors d’OA……..………………………….………………..150
3.ii Sources prépondérantes d’ERO lors d’OA……..…………………………………….……………...151
3.iii Conséquences dans le développement de l’OA …….………………………………..…………..153
4) Bilan ………………………………………………………………………………………………………………………..……..156
D) Pathologie du développement osseux : l’ostéochondrose disséquante (OCD) ……...……………………..…..157
1) Présentation et importance dans l’espèce équine ……………………………..……………………………….157
2) Physiopathologie de l’ostéochondrose et lien avec le stress oxydant ……….…..…………………….157
2.i Éléments de physiopathologie générale ………………………………………………...…………....157
2.ii Données disponibles sur la place du stress oxydant dans la
Physiopathologie de l’OCD.….………….…………………………………………………………..…………...157
3) Bilan ………….……………………………………………………………………………………………………………………158
E) Pathologie tendineuse : la maladie tendineuse du tendon fléchisseur superficiel du doigt (SFDT)…....158
1) Pré-requis ……………………………………………………………………………………………………………………….158
1.i Organisation générale du tendon………………………………………………………………………….158
1.ii Terminologie des désordres tendineux ……………………………………………………….………159
2) Présentation de la maladie tendineuse du SDFT ………………………………………………………………..160
2.i Impact sur les performances équines …………………………………………………………….........160
2.ii Histologie des lésions …………………………………………………………………………………………161
3) Physiopathologie de la maladie tendineuse du SDFT et lien avec le stress oxydant ……………..162
3.i Physiopathologie comparée entre l’homme et le cheval ………………………………………..162
3.ii Sources prépondérante d’ERO au sein du SDFT lors de l’effort.……………………….........162
3.iii Conséquences du stress oxydant sur le SDFT.……………………………………………………..164
4) Bilan ………………………………………………………………………………………………………………………………..165
F) Pathologie hématologique : l’hémolyse intravasculaire induite par l’effort (HIE)…………….……………….166
1) Quelques rappels sur la sensibilité des hématies aux ERO …………………………………………..……..166
2) L’hémolyse intravasculaire induite par l’effort (HIE) ………………………………………………..………..166
2.i Présentation générale…………………………………………………………………………………………..166
2.ii Mise en évidence de l’HIE chez le cheval ……………………………………………………………..167
3) Physiopathologie de l’HIE et lien avec le stress oxydant …………………………………………………….167
4) Bilan..……………………………………………………………………………………………………………………………….168
Conclusion ……………………………...…………………………………………………………………………………………………………………….170
Bibliographie …………………………………….…………………………………………………………………………………………………………..171

11
TABLE DES ILLUSTRATIONS
Liste des Figures
Figure 1: Illustration de la fission ....................................................................................................................18
Figure 2: La chaîne respiratoire mitochondriale dite de « phosphorylation
oxydative ». ………………………………………………………………………...………………………………………….…33
Figure 3: Le cycle de l’ubiquinone ou coenzyme Q et ses différents états d’oxydation…….……34
Figure 4: Production d’ERO par la xanthine oxydase (XO)…………………………………………….….…40
Figure 5: Mécanisme d’action de la glutathion réductase (GRD)……………………………………….…48
Figure 6: États d’oxydation du glutathion ……………………………………………………………………….…51
Figure 7: Formule de l’tocophérol: le pôle hydrophile est vers la gauche, le pôle lipophile vers la droite……………………………………………………………………………………………………………….….…58
Figure 8: Formes de la vitamine C à différent pH et sa réaction avec les radicaux
libres…………………………………………………………………………………………………………………………..….…59
Figure 9: Exemple classique de propagation d’une chaîne de peroxydation
lipidique…………………………………………………………………………………………………………………......…..…62
Figure 10: Complexité réactionnelle de la peroxydation lipidique : peroxydation de l’acide
linoléique (LH)…………………………………………………………………………………………………………...…..….63
Figure 11: Vulnérabilité des atomes d’hydrogène dans la chaîne polyinsaturée de l’acide
gras………………………………………………………………………………………………………………………….….……64
Figure 12: Les acides aminés soufrés ………………………………………………………………………….……65
Figure 13: Les acides aminés basiques ………………………………………………………………………..……66
Figure 14: Les acides aminés aromatiques ………………………………………………………………….……66
Figure 15: Altérations de l’ADN entraînées par un stress oxydant………………………….…….….…69
Figure 16: Deux « spin traps » : DMPO et POBN……………………………………………………..…...….……77
Figure 17: 4-hydroxynonenal (4-HNE), acroléine et malondialdéhyde (MDA)………………….…82
Figure 18: Détermination de la vitesse initiale maximale. KM désigne la constante de Michaelis,
propre à l’enzyme…………………………………………………………………………………………………………......85
Figure 19 : Activation de la voie ARE-Nrf2 suite à un stress oxydant modéré……………………...95
Figure 20: Exemple de chaîne de détoxification ……………………………………………...……………....…98
Figure 21: Représentation schématique du « respiratory burst » avec l’activation de la NADPH
oxydase…………………………………………………………………………………………………………………………..101

12
Figure 22: Exemple de mécanisme d’activation des facteurs de transcription pro-
inflammatoires par le stress oxydant et les voies d’AP-1 (« activation protein-1 ») et NF-
…………………………………………………………………………………………………………………………….….103
Figure 23: Échanges croisés entre stress oxydant, ER-stress et la mitochondrie…………….…105
Figure 24: Modèle in vitro de Reid et al. (1993) : effet biphasique des ERO sur la force
musculaire produite………………………………………………………………………………………………………130
Figure 25: Bilan des mécanismes suspectés de régulation redox de la force produite lors de la
contraction musculaire……………………………………………………………………………………………….….131
Figure 26: Apoptose des lymphocytes induite par l’effort………………………………………………143
Figure 27: Coupe frontale schématique d’une diarthrose…………………………………………….…146
Figure 28: Schéma principal de l’action délétère des principaux ERO sur la matrice
cartilagineuse…………………………………………………………………………………………………………..……154
Figure 29: Structure schématique hiérarchique du SDFT……………………………………...…..……158
Liste des Tableaux
Tableau 1: Nomenclature des principales espèces réactives……………………………………………21
Tableau 2: Principales caractéristiques des trois types de fibres musculaire I, IIA, IIX chez le
cheval…………………………………………………………………………………………………………………………...112
Tableau 3: Tableau récapitulatif chronologique des principales études mettant en évidence un
stress oxydant lors d’un effort physique chez le cheval………………………………………………...…113

13
Liste Des Abbréviations 1O2 : dioxygène singulet 3O2 : dioxygène triplet
4-HNE : 4-hydroxynonénal -Toc° : radical alpha-tocophéryl
-TocH : alpha-tocophérol
GCS = -glutamylcystéine synthétase
μg : microgramme μmol: micromole 8-OH-dG : 8-hydroxydéoxyguanosine ADN : acide désoxyribonucléique ADP : adénosine diphosphate AGE: Advanced glycation end products/ Produits finaux de la glucoxydation AMP : adénosine monphosphate AP-1: Activated Protein-1
ARE: Antioxidant response element
ARN: acide ribonucléique ArO°: radical tocophéryl ArOH: alpha tocophérol Asc°- : radical ascorbyle
AscH2 : acide ascorbique/ascorbate
AST ou ASAT : aspartate aminotransférase ATP : adénosine triphosphate BAP : Biological Antioxidant Potential C : carbone Ca : calcium CaO2 : concentration artérielle en oxygène Car : caroténoïde CAT : catalase CK : créatine kinase CIC : Concours International Combiné
Cl : chlore Cl- : ion chlorure CLHP : Chromatographie Liquide Haute Performance ClO- : ion hypochlorite CO : monoxyde de carbone CO2 : dioxyde de carbone CO3°- : radical carbonate COX : cyclooxygénase Cu : cuivre CV : cheval/chevaux
CvO2 : concentration veineuse en oxygène
DC : débit cardiaque DDVP : déplacement dorsal du voile du
palais
ER: endoplasmic reticulum
ERN : espèces réactives de l’azote ERO : espèces réactives de l’oxygène FAD+ : flavine adénine dinucléotide, forme
Oxydée FADH2 : flavin adénine dinucléotide, forme réduite Fe2+ : ion ferreux Fe3+ : ion ferrique FEI : Fédération Équestre Internationale
FMN : flavine monnucléotide, forme oxydée FMNH2 : flavine monnucléotide, forme Réduite GAG : glycosaminoglycane GSH : glutathion réduit GLUC ou Glyc : glycémie GMPc : guanosine monophosphate
cyclique
GPx : glutathion peroxydase GRD : glutathion réductase GSSG : glutathion disulfide ou glutathion oxydé GSTr : glutathion-S-transférase
H+
H2O : eau H2O2 : peroxyde d’hydrogène H2S: sulfure d’hydrogène
Hb : hémoglobine HCl : acide chlorhydrique HCO3° : radical bicarbonate HDL: High Density Lipoprotein HIE: hémolyse intravasculaire induite par l’effort HIF-1: Hypoxia Inducible Factor-1 HO-1: Hème Oxygénase-1
HO2°: radical hydroperoxyle HOCl: acide hypochloreux HPIE : hémorragie pulmonaire
induite par l’effort
HSP : Heat Shock Proteins
Hz: Hertz
IAD : Inflammatory Airway Disease
IFN : interféron gamma
IGF-1: insulin-like growth factor-1 IL : interleukine IRE: iron-responsive element IRP: iron-regulatory protein kDa : kilodaltons Keap1 : kelch-like ECH-associated protein 1 kg : kilogramme km : kilomètre
LBA : lavage broncho-alvéolaire

14
LDL : Light Density Lipoprotein LDH : lactate deshydrogénase LOOH ou ROOH: hydroperoxyde lipidique Maf: facteur musculoaponeurotique MAPK: mitogen-activated-protein-kinase MDA : malondialdéhyde/aldéhyde dimalonique MIF: Macrophage Inhibitory Factor
mg: milligramme Mn : manganèse MTP: Mitochondrial Permeability
Transition
N : azote N2 : diazote NAD+ : nicotinamide adénine dinucléotide, forme oxydée NADH2 : nicotinamide adénine dinucléotide, forme réduite
NADPH : nicotinamide adénine dinucléotide phosphate, forme réduite NFAT: Nuclear Factor of Activated T cells
NF-κB : nuclear factor-kappa B NH3 : ammoniaque
nmol : nanomole NO° : oxyde nitrique/monoxyde d’azote NO2° : dioxyde d’azote NOs: monoxyde d’azote synthétase Nox : NADPH-oxydase
NQO-1: NADPH-quinone-oxydoréductase-1
Nrf2: nuclear erythroid-2 related factor
O : atome d’oxygène O2 : dioxygène O2°- : radical superoxyde O3 : ozone OA: ostéoarthrose
OCD : ostéochondrose disséquante
-OH : groupement alcool
OH° : radical hydroxyle ONOO- : ion peroxynitrite PAF : Platelet Activating Factor
PAL : phosphatases alcalines PaO2 : pression artérielle en dioxygène
PCARE : Pompe calcique ATP-dépendante
du réticulum endoplasmique.
PCR : polymerase chain reaction
PCr : phosphocréatine musculaire
PDGF: Platelet Derived Growth Factor
PEI: prélèvement post-effort immédiat
PGC-1: Peroxisome proliferator-activated receptor gamma coactivator-1 PGE2 : prostaglandine E2 PGF2α : prostaglandines F2α
PGH2 prostaglandine H2 PGG2 : prostaglandine G2 Pi : phosphate inorganique PI3-K: phosphatidylinositol 3-kinase PKC : protéine kinase C PMA : Puissance Maximale Aérobie PNN : polynucléaire neutrophile PON : paraoxonase PRDX : peroxyrédoxine
PSA : pur-sang anglais
Q ou QH : coenzyme Q/ubiquinone
RAGE: récepteur aux AGE
RAO : Recurrent Airway Obstruction
RMN : Résonance Magnétique Nucléaire RO°: radical alkoxyle ROO°: radical peroxyle ROMs : Reactive Oxygen Metabolites RPE : Résonance Paramagnétique Electronique SDFT: Superficial Digital Flexor Tendon
tendon fléchisseur superficiel du doigt
SH : groupement thiol SDH: succinate déshydrogénase SOD: Superoxyde Dismutase TBARS : ThioBarbituric Acid Reactive Substances TNF : Tumor Necrosis FactorAlpha TRE : tetradecanoylphorbol-13-acetate
-response-element
TRP : tocophérol transfert protein
Trx : thiorédoxine
TXNRD : thiorédoxine réductase
UCP : uncoupling protein
UI : Unité Internationale UPR: Unfolded Protein Response
VDAC : voltage-dependent anion channel VO2 : consommation en dioxygène par unité de temps VO2max : consommation maximale en dioxygène par unité de temps XDH : xanthine déshydrogénase XO : xanthine oxydase
Zn : zinc

15
Introduction
Bien que nécessaire pour tous les organismes aérobies, l’oxygène peut également se
réveler toxique. Cette toxicité porte un nom : le stress oxydant.
Le stress oxydant découle de ce l’on appelle le « paradoxe de l’oxygène ». En effet, une
fraction plus ou moins grande de cet oxygène n’entre pas dans le métabolisme énergétique
bénéfique aux individus mais génère une large famille de molécules pro-oxydantes
hyperréactives appelées espèces réactives de l’oxygène (ERO).
Les organismes possèdent des moyens de lutte intrinsèques contre ces ERO, les
antioxydants, et peuvent même utiliser leurs propriétés pour leurs propres fonctions
biologiques. Cependant, si ces espèces oxydantes sont produites en trop grande quantité,
l’équilibre cellulaire redox peut être rompu. L’organisme ne peut plus y faire face correctement
et voit des dégâts oxydatifs s’accumuler dans ses cellules.
Le cheval athlète semble être particulièrement confronté à ce phénomène. C’est
pourquoi on s’intéressera ici sur l’implication réelle du stress oxydant dans la physiopathologie
sportive en posant la problématique suivante : existe-t-il une causalité directe ou indirecte
entre la genèse d’ERO, notamment lors de l’effort, et des affections limitant les performances du
sportif équin ?
Dans le travail qui va suivre, on identifiera dans un premier temps les acteurs à l’origine
du stress oxydant ainsi que leur impact sur la cellule mammalienne et ses composants. On
détaillera également comment celle-ci les contrôle et les implique dans son métabolisme en
condition physiologique. Ce pré-requis est obligatoire pour comprendre les problématiques de
l’évaluation expérimentale du stress oxydant et ses mécanismes pathogéniques principaux.
La deuxième partie de l’exposé constitue le cœur de la problématique. Il a été choisi ici
de présenter plusieurs affections communes de l’athlète équin, sélectionnées parmi celles qui
affectent les performances sportives et qui se développent voire s’aggravent avec l’effort
physique. A la lueur des résultats expérimentaux présents dans la littérature, nous tenterons de
dégager pour chacune d’entre elles l’implication du stress oxydant dans leur pathogénie.

16
1ere partie : Qu’est-ce que le stress oxydant ?
Bref historique scientifique
La connaissance des radicaux libres et la notion de stress oxydant restaient largement
méconnues pour la communauté scientifique jusqu’aux travaux de Daniel Gilbert, de Rebecca
Gershman et de leur collègues en 1956. Comparant les effets toxiques d’un rayon ionisant à
ceux de niveau élevés de dioxygène sur des organismes aérobies, ils furent les premiers à
proposer que la toxicité de l’oxygène est due à la formation de radicaux libres (Gershman and
al., 1956).
Par la suite, le développement des techniques d’analyses biochimiques de plus en plus
précises (tels que l’électrophorèse et de la chromatographie), notamment dans l’étude des
protéines, ont permis de préciser les données sur le sujet. Les années 1980 ont vu l’avènement
des techniques de séquençage de l’ADN, d’ADN recombinant, ainsi que le développement de la
PCR (« polymerase chain reaction »), permettant de préciser à l’échelle nucléaire l’impact des
radicaux libres. Ce qui suit n’est qu’une modeste synthèse de plus de 60 années d’avancées et
de découvertes scientifiques.
Dans la présentation suivante on inclura souvent par extension dans les espèces
réactives de l’oxygène (ERO) les espèces réactives de l’azote (ERN), qui leur sont souvent
intimement liées dans les mécanismes réactionnels et physiopathologiques.
I) Pré-requis de chimie radicalaire
A) Qu’est ce qu’un radical libre ?
Les radicaux libres comprennent toute espèce moléculaire pouvant exister seule et
contenant un ou plusieurs électrons non apparié sur sa couche externe, c’est à dire un électron
célibataire (Halliwell and Guterridge, 2008). C’est la présence d’un électron célibataire confère
à ces molécules une grande instabilité (elles ne respectent pas la règle de l’octet).
1) La réaction d’oxydoréduction et potentiel redox E°
Les propriétés thermodynamique des radicaux et des ERO en général varient de manière
significative entre ceux qui ont des propriétés oxydantes très fortes (comme le radical
hydroxyle HO°) et des molécules ayant des propriétés plus réductrices (comme l’acide
ascorbique ou vitamine C) (Valko, 2011). L’étude et la caractérisation des propriétés
thermodynamiques de ces molécules permettent aux scientifiques de prédire une hiérarchie
des réactions impliquant les radicaux libres.
La valeur thermodynamique pour caractériser l’évolution d’une réaction
d’oxydoréduction est le potentiel d’oxydoréduction ou potentiel redox E°, exprimé en volt (V)
(Valko, 2011). Le potentiel redox E° d’un couple oxydant/réducteur dépend de sa tendance à
accepter/donner un électron par rapport à une électrode standard normale à hydrogène, ainsi
que des concentrations en espèces réduites et oxydées du couple redox dans le milieu (Raman
and Berry, 2011).

17
Par exemple, la réduction du composé « A » par un électron est liée à un potentiel redox
E° du couple A/A°- :
A(Oxydant) + 1 e- A°- (Reducteur) E°, couple Oxydant/Réducteur [A/A°-].
Ainsi une réaction d’oxydoréduction mettant en jeu deux couples oxydant/réducteur
[A(Ox)/A(Red)] et [B(ox)/B(red)] peut être écrite :
A(Ox) + B(Red) A(Red) + B(Ox)
Cependant, il faut préciser que ces données thermodynamiques E° ne sont pas
forcément suffisantes in vivo pour prévoir la production et la réaction des espèces réactives.
Par exemple, une réaction d’oxydoréduction peut être thermodynamiquement possible mais ne
pourra pas se produire dans le milieu simplement parce que sa vitesse de réaction (qui dépend
couramment de la concentration de chacune des espèces) est trop faible.
2) Mécanismes chimiques de formation des radicaux libres
2.i Oxydation
Un radical libre peut être formé par oxydation, qui désigne la perte d’un ou plusieurs
électrons (Halliwell and Gutteridge, 2008). Par exemple:
X X° + e- où X° est un radical cation.
Par extension, on associe aussi l’oxydation comme le gain en oxygène. Ainsi, un agent
oxydant oxyde une molécule chimique en retirant des électrons, de l’hydrogène ou encore en
donnant de l’oxygène.
2.ii Réduction
Également, un radical libre peut aussi être formé par gain d’un ou de plusieurs électrons,
réaction appelée réduction (Halliwell and Gutteridge, 2008). Par exemple:
Y + e- Y° où Y° est un radical anion.
Par extension, on associe aussi la réduction à la perte en oxygène ou le gain en
hydrogène. Ainsi, un agent réducteur réduit une molécule chimique en donnant des électrons,
de l’hydrogène ou encore en retirant de l’oxygène.
2.iii Fission Homolytique
La fission homolytique désigne la rupture d’une liaison chimique covalente où les deux
électrons de la liaison A—B sont répartis équitablement (Halliwell and Gutteridge, 2008). Il en
résulte deux radicaux libres contenant évidemment chacun un électron non apparié sur leur
couche externe. Par exemple :
A—B A° + B°.
L’énergie requise pour dissocier une liaison covalente peut être issue de la chaleur, de la
lumière ultraviolette ou d’une radiation ionisante par exemple. Par exemple, plusieurs liaisons
covalentes se rompent spontanément sous de très fortes températures, comme les liaisons C—
C, C—H ou C—O, par exemple à 450°C à 600°C (Halliwell and Gutteridge, 2008).

18
Ainsi, la fission homolytique d’une liaison covalente O—H de l’eau donnera un radical
hydrogène H° et un radical hydroxyle HO°. La fission homolytique s’oppose à la fission
hétérolytique où les deux électrons de la liaison sont captés par un des atomes. On obtient des
ions plutôt que des radicaux:
A—B A- + B+.
Ainsi pour l’eau on obtiendrait l’ion H+ et l’ion hydroxyde HO-.
Un cas particulier : la fission .
La fission concerne les hydrocarbures comme les lipides et consiste à couper une
liaison C—C d’un radical libre polymérique (comme un radical peroxyle) pour former un
nouveau radical avec une chaîne carbonée plus courte (figure 1).
Figure 1 : Illustration de la fission
La fission sera reprise dans le chapitre concernant la peroxydation des lipides.
3) Réactivité des radicaux libres
3.i Réaction entre un radical libre et une molécule non radicalaire
La réaction impliquant un radical libre et une molécule non radicalaire donne un
nouveau radical libre et éventuellement d’autres produits. Ces réactions sont regroupées en
deux grandes familles : les substitutions et les additions.
Substitutions
Une substitution est un transfert d’un atome ou d’un groupe d’atomes d’un centre réactif
vers l’autre. La déshydrogénation dans les organismes biologiques rentre dans cette famille.
Ainsi :
R1° + R2—H R1—H + R2°.
Additions
Une addition est l’ajout sur un atome d’un autre atome ou un autre radical. C’est ce qui
peut se passer sur une double liaison >C=C< :
R° + >C=C< >C(R)—C°<.
Nous remarquerons également que la cyclisation est également possible lorsqu’un
radical libre possède lui-même une double liaison >C=C<.

19
3.ii Réaction entre deux radicaux libres
Il en existe deux types : les dismutations et les combinaisons. Dans les deux cas, les
radicaux libres perdent leur nature radicalaire. Cependant, cela ne veut pas forcément dire
qu’ils perdent leurs potentialités oxydantes selon la nature des produits formés.
Dismutations
La dismutation est une réaction d’oxydoréduction dans laquelle un radical libre joue à la
fois le rôle de l’oxydant et celui du le réducteur, le meilleur exemple étant la dismutation du
radical superoxyde (revue plus tard):
2O2°- + 2H+ H2O2 + O2.
Combinaisons
La combinaison est la réunion de deux radicaux libres pour former une nouvelle
molécule en formant une liaison simple covalente :
R1° + R2° R1—R2.
3.iii Notion de chaîne de transmission radicalaire
On parle de chaîne de transmission radicalaire lors de la formation de nouveaux
radicaux libres à partir d’un radical « primaire » ayant réagit avec une molécule stable ; ces
nouveaux radicaux seront plus ou moins réactifs et pourront réagir à leur tour avec d’autres
molécules.
On distingue plusieurs étapes dans cette chaîne de réactions :
- l’initiation : processus de formation des radicaux libres « primaires » ;
- la propagation : réactions entre les radicaux libres et d’autres molécules
formant de nouveaux radicaux libres avec amplification de la chaîne réactionnelle.
- la terminaison : réactions entre radicaux libres (dismutation ou combinaison)
ou formation d’un radical libre particulièrement stable.
La peroxydation lipidique est un parfait exemple de cette notion de chaîne de
transmission radicalaire et sera revue plus tard.
4) Le cas de l’oxygène
4.i Capacité oxydante de l’atome d’oxygène
L’atome d’oxygène a des propriétés électroniques uniques. Six électrons sont présents
sur son orbite externe : deux doublets d’électrons et deux électrons non appariés. Afin de
respecter la règle de l’octet, il doit donc accepter deux électrons pour être stable : l’atome
d’oxygène est donc oxydant (Halliwell and Gutteridge, 2008) (Valko, 2011).
4.ii Particularités de la molécule de dioxygène
Selon le principe d’exclusion de Pauli, pour chaque doublet remplissant une orbitale les
électrons doivent être de spin antiparallèle, annulant leur champ magnétique respectif ; ainsi
dans une molécule stable la résultante magnétique est normalement nulle.
La molécule de dioxygène a deux atomes d’oxygène où chacun met en commun ses
électrons non appariés pour être stable. Or le dioxygène est une molécule dite

20
« paramagnétique » (Faraday, années 1840) car elle contient à l’état fondamental deux
électrons de spin parallèle. C’est donc un diradical libre, et pourtant une molécule chimique
inerte à l’état fondamental (Halliwell and Gutteridge, 2008).
4.iii Stabilité et activation de la molécule de dioxygène
Si on classe les atomes et les molécules en se basant sur le nombre d’électrons
célibataires et leur spin (notion de multiplicité des espèces chimiques), on obtient 3 classes
définies par la formule 2S+1 où S est la somme des spins des électrons célibataires (+1 ou –1):
- Les singulets : espèces ayant une multiplicité égale à 1; ainsi, les singulets ont
tous leurs électrons appariés ou deux électrons célibataires de spin antiparallèle ; c’est le cas de
la plupart des molécules organiques ;
- Les doublets : espèces ayant une multiplicité égale à 2 ; ainsi, les doublets ont
un électron célibataire ; c’est le cas pour nos radicaux libres ;
- Les triplets : espèces ayant une multiplicité égale à 3 ; ainsi les triplets ont deux
électrons célibataires à spin parallèle. C’est le cas de la molécule de dioxygène à l’état
fondamental (voir plus loin le paramagnétisme du dioxygène).
Or d’après les règles de restriction de spin (énoncées par Wigner en 1929):
- Les doublets réagissent généralement plus lentement entre eux ;
- Les singulets, sauf interdiction thermodynamique, réagissent
spontanément avec les doublets mais leur réaction avec les triplets impossible ;
- Les doublets réagissent lentement avec les triplets.
Ainsi, la molécule de dioxygène à l’état fondamental est un triplet et est donc inerte à
l’état fondamental envers les molécules organiques. Une activation est donc obligatoire et ce
essentiellement de trois manières (Halliwell and Gutteridge, 2008):
- l’absorption de suffisamment d’énergie pour inverser le spin de l’un des
électrons célibataires : le dioxygène triplet passe à l’état singulet.
- la réduction d’un dioxygène triplet en doublet ou singulet
- l’oxydation des molécules organiques en doublet et l’action ensuite du
dioxygène triplet sur ces substrats « activés ».
B) Espèces réactives de l’oxygène ou dérivés de son métabolisme
1) Terminologie et classification des espèces réactives
Le terme espèces réactives de l’oxygène (ERO) est un nom collectif qui inclut non
seulement les radicaux libres de l’oxygène mais aussi plusieurs espèces non radicalaires aux
capacités oxydantes et contenant un ou plusieurs atomes d’oxygène.
Ainsi, tous les radicaux de l’oxygène sont des espèces réactives de l’oxygène, mais toutes
les espèces réactives de l’oxygène ne sont pas des radicaux de l’oxygène (Halliwell and
Gutteridge, 2008). Le tableau 1 reprend la nomenclature des espèces réactives, incluant les
principales espèces réactives de l’oxygène (radicalaires et non radicalaires). Seront reprises par
la suite que celles qui sont les plus représentatives lors de l’étude du stress oxydant et
susceptibles d’être évoquées à de nombreuses reprises.
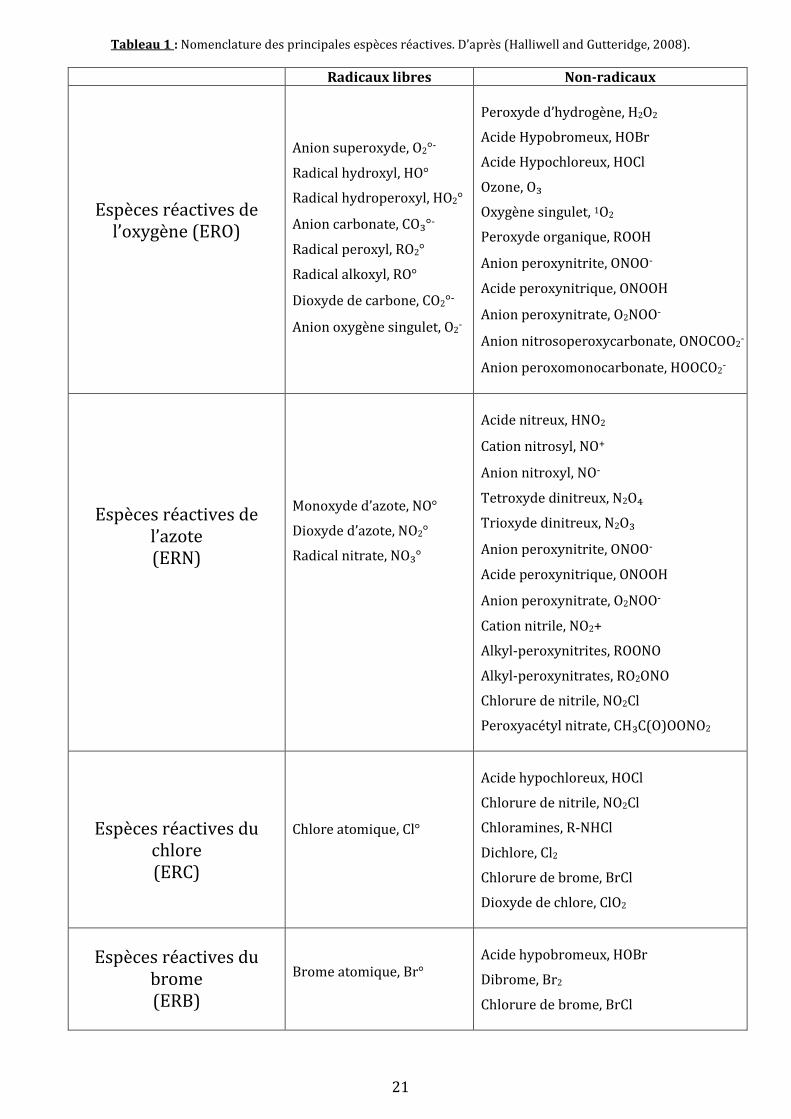
21
Tableau 1 : Nomenclature des principales espèces réactives. D’après (Halliwell and Gutteridge, 2008).
Radicaux libres Non-radicaux
Espèces réactives de l’oxygène (ERO)
Anion superoxyde, O2°-
Radical hydroxyl, HO°
Radical hydroperoxyl, HO2°
Anion carbonate, CO₃°-
Radical peroxyl, RO2°
Radical alkoxyl, RO°
Dioxyde de carbone, CO2°-
Anion oxygène singulet, O2-
Peroxyde d’hydrogène, H2O2
Acide Hypobromeux, HOBr
Acide Hypochloreux, HOCl
Ozone, O₃
Oxygène singulet, 1O2
Peroxyde organique, ROOH
Anion peroxynitrite, ONOO-
Acide peroxynitrique, ONOOH
Anion peroxynitrate, O2NOO-
Anion nitrosoperoxycarbonate, ONOCOO2-
Anion peroxomonocarbonate, HOOCO2-
Espèces réactives de l’azote (ERN)
Monoxyde d’azote, NO°
Dioxyde d’azote, NO2°
Radical nitrate, NO₃°
Acide nitreux, HNO2
Cation nitrosyl, NO+
Anion nitroxyl, NO-
Tetroxyde dinitreux, N2O₄
Trioxyde dinitreux, N2O₃
Anion peroxynitrite, ONOO-
Acide peroxynitrique, ONOOH
Anion peroxynitrate, O2NOO-
Cation nitrile, NO2+
Alkyl-peroxynitrites, ROONO
Alkyl-peroxynitrates, RO2ONO
Chlorure de nitrile, NO2Cl
Peroxyacétyl nitrate, CH₃C(O)OONO2
Espèces réactives du chlore (ERC)
Chlore atomique, Cl°
Acide hypochloreux, HOCl
Chlorure de nitrile, NO2Cl
Chloramines, R-NHCl
Dichlore, Cl2
Chlorure de brome, BrCl
Dioxyde de chlore, ClO2
Espèces réactives du brome (ERB)
Brome atomique, Br°
Acide hypobromeux, HOBr
Dibrome, Br2
Chlorure de brome, BrCl

22
2) Espèces radicalaires
2.i Anion superoxyde O2°-
L’anion superoxyde résulte de la réduction de la molécule de dioxygène triplet : cette
dernière accepte un électron qui vient alors se positionner sur une orbitale antiliante *
(Halliwell and Gutteridge, 2008).
Dans les tissus biologiques proches de la neutralité, la forme non protonée O2°- est
prédominante (le pKa du couple O2°-/HO2° étant approximativement égale à 4.8 en condition
physiologique). Or cette forme est ioniquement chargée et ne franchit pas les membranes
cellulaires, ce qui restreint a priori son pouvoir oxydant. Cependant il a été montré que des
pores membranaires protéiques laissant circuler les anions pourraient aussi faire entrer ou
sortir O2°- de la cellule (Halliwell and Gutteridge, 2008).
O2°- est bien moins réactif que le radical hydroxyle OH°; malgré des potentiels E° hauts
dans les conditions retrouvées in vivo, O2°- n’est pas un bon oxydant ni un bon réducteur. En
effet, les constantes de réaction sont généralement trop faibles dans les systèmes biologiques
(aux alentours de 102 mol/L/h). Ainsi la réactivité du radical superoxyde est quasiment
inexistante avec les acides nucléiques de l’ADN, les lipides et les acides aminés in vivo (Halliwell
and Gutteridge, 2008).
Cependant O2°- peut réagir rapidement avec d’autres radicaux comme NO°, certains
groupements enzymatiques contenant du soufre ou encore des radicaux phénoxyles générés
lors de la déshydrogénation du groupement alcool de la tyrosine. Il peut également réagir avec
le cytochrome c, la SOD (dont il est le substrat), la dopamine, la noradrénaline et l’acide
ascorbique (vitamine C) (Halliwell and Gutteridge, 2008).
Malgré cette réactivité limitée, O2°- a une durée de vie bien supérieure à des radicaux
plus réactif comme HO° ; son importance vient du fait qu’on considère le radical superoxyde
comme un radical « primaire » donnant naissance à des chaînes réactionnelles dans les tissus
biologiques (Valko, 2011).
Pour mémoire on retiendra en particulier que la réduction de l’anion superoxyde O2°-
donne l’anion peroxyde O22- qui n’est autre que la forme dibase du peroxyde d’hydrogène H2O2.
L’anion superoxyde peut également rejoindre un système de Fenton et favoriser alors la
formation du radical hydroxyle HO°, ERO également plus réactif in vivo. Enfin la réaction rapide
de l’ion superoxyde avec le monoxyde d’azote forme l’ion peroxynitrite ONOO-, très toxique
(Valko, 2011).
Dans les milieux biologiques, O2°- disparaît généralement rapidement par dismutation,
opérée par la superoxyde dismutase (SOD). La constante de cette réaction aux pH biologiques
en tant que telle est proche de zéro, et la dismutation ne se réalise qu’après protonation d’O2°-
en HO2°- qui réagit ensuite avec O2°-. Cette réaction est donc plus rapide en milieu fortement
acide souvent incompatible avec la vie (Halliwell en Gutteridge, 2008).
2.ii Radical libre hydroxyle HO°
Le radical hydroxyle HO° est un radical libre extrêmement oxydant. Quelques soient les
conditions expérimentales in vitro, son potentiel redox E° a des valeurs toujours très hautes et
fortement positives: c’est le radical de l’oxygène le plus réactif connu (Halliwell an Gutteridge,
2008).

23
Par ailleurs, sa durée de vie est très limitée (de l’ordre d’une nanoseconde en solution
aqueuse in vitro) (Valko, 2011). Certains auteurs considèrent d’ailleurs que cette grande
réactivité rend difficile l’idée que ce radical puisse migrer sur une distance suffisante pour
initier la peroxydation lipidique depuis les régions aqueuses où elles sont produites (Qian and
Buettner, 1999).
Il existe de nombreuses sources biochimiques produisant HO°. Dans les tissus
biologiques, la présence de métaux de transition libres (comme Fe2+ ou Cu2+) pourrait être une
des principales sources du radical hydroxyle in vivo (voir plus loin). HO° peut aussi être généré
par fission homolytique de l’H2O2 ou encore d’eau avec des rayons mais ces réactions ont
évidemment peu de chance d’avoir lieu in vivo, d’autant plus pour les affections qui nous
intéressent (Halliwell and Gutteridge, 2008).
s réactions impliquant HO° sont de trois types principaux (Halliwell and Gutteridge, 2008):
- Abstraction de l’hydrogène (ou déshydrogénation) :
Une illustration classique est la déshydrogénation de la fonction hydroxyle –OH des
alcools :
R—OH (alcool) + HO° R—O° (radical alkoxyle) + H2O.
Cette déshydrogénation concerne également les chaînes hydrogénées des
hydrocarbures (créant notamment des radicaux libres centrés sur le carbone):
R1—CH₃—R2 + °OH R1—C°H2—R2 (radical alkyle) + H2O.
- Addition :
La réaction de °OH avec des composés aromatiques (comme la guanine de l’ADN pour
former le radical 8-hydroxydesoxyguanine) est une réaction d’addition ; plus
simplement, l’ion hydroxyle peut s’ajouter aux doubles liaisons >C=C< (particulièrement
présentes sur les acides gras insaturés des lipides ou encore la thymine):
>C=C< + °OH >C(OH)—C°< (radical hydroxyalkyle)
- Transfert d’électrons :
Cette dernière modalité réactionnelle est notamment illustrée par la réaction de HO°
avec les ions carbonates (A), bicarbonates (B), l’ion nitrite (C) ou encore l’ion chlore (D) :
(A) CO₃2- + HO° CO₃°- (radical carbonate)+ HO-
(B) HCO₃- + HO° HCO₃° (radical bicarbonate) + HO-
(C) NO2- + HO° NO2° (dioxyde d’azote) + HO-
(D) Cl- + HO° Cl° (chlore atomique) + HO-
2.iii Les radicaux peroxyle RO2° et alkoxyle RO°
Les radicaux peroxyles et alkoxyles sont généralement de bons agents oxydants, ayant
des potentiels redox E° fortement positifs (Halliwell and Gutteridge, 2008). Dans une certaine
mesure, le radical hydroperoxyle HO2° (la forme protonée d’O2°-), peut également être
considéré comme un radical peroxyle.

24
Remarquons que les radicaux alkoxyles et peroxyles aromatiques sont moins réactifs
que les formes aliphatiques. En effet les électrons se délocalisent sur l’anneau benzène ce qui
procure une certaine stabilité au radical.
La chaîne réactionnelle de peroxydation lipidique est un mécanisme de formation
courant de ces radicaux. Dans un premier temps un oxydant fort X° (tel que HO°) réalise
l’abstraction d’un atome d’hydrogène (déshydrogénation) fixé sur un atome de carbone (liaison
C—H) d’un alcane ou autre chaîne carbonée et favorise ainsi la formation d’un radical centré
sur un atome de carbone :
R° + R1—H R—H + R1°, où R1° est un radical centré sur le carbone
Puis ce radical R°, à présent activé en doublet, peut ensuite venir réagir avec le dioxygène
environnant (triplet) pour former un radical peroxyle:
R1° + O2 R1O2° (radical peroxyle)
Ces radicaux ont eux-mêmes la capacité de déshydrogéner facilement d’autres molécules
et le cycle recommence :
R1O2° + R2—H R1OOH + R2°, où R2° est un nouveau radical centré sur le carbone
La diversité et la réactivité des radicaux peroxyles et alkoxyles leur confèrent une place
importante dans la phase de propagation de la peroxydation lipidique.
RO2° et RO° peuvent également être générés par la décomposition des peroxydes
organiques (ROOH). Normalement stables dans l’organisme et à température ambiante,
l’addition de métaux de transition permet leur décomposition et constitue un des principaux
démarrages de la peroxydation lipidique (Halliwell and Gutteridge, 2008). Par exemple :
ROOH + Fe3+ RO2° + Fe2+ + H+
ROOH + Fe2+ RO° + OH- + Fe3+
Les radicaux peroxyles peuvent aussi réagir entre eux en générant l’oxygène singulet 1O2
selon le mécanisme de Russel (Halliwell and Gutteridge, 2008):
>CHOO° + >CHOO° >CHOH + >C=O + 1O2
Enfin, les peroxydes lipidiques peuvent aussi directement réagir avec HO2° pour former
RO2° :
HO2° + ROOH RO2° + H2O2
Cette réaction n’est cependant probablement pas prépondérante in vivo car comme nous
l’avons vu précédemment, la forme protonée du radical superoxyde n’est pas prépondérante
dans les organismes.
2.iv Le radical carbonate CO₃°- et bicarbonate HCO₃°
Les radicaux HCO₃° et carbonate CO₃°- sont essentiellement issus de la réaction
impliquant le radical hydroxyle OH° avec les ions bicarbonate HCO₃- et carbonate CO₃2-
respectivement (Halliwell and Guterridge, 2008):

25
CO₃2- + HO° CO₃°- + OH- et HCO₃- + HO° HCO₃° + H2O
(Tenant compte bien sûr que CO₃°- + H+ = HCO₃°).
Les constantes de réaction sont parmi les plus faibles avec HO° mais ces réactions sont
rendues possibles grâce aux concentrations élevées d’HCO₃- in vivo (25 mmol/L d’HCO₃- dans le
plasma en moyenne).
Ces radicaux peuvent également être des produits de la réaction entre l’ion peroxynitrite
ONOO- et le dioxyde de carbone CO2 in vivo.
CO₃°- peut également être issu du peroxomonocarbonate HCO₄-(HOOCO2-) formé par
l’interaction du peroxyde d’hydrogène avec HO₃- (ou CO2). HCO₄-(HOOCO2-) peut alors être
réduit, réduction entraînant la formation de CO₃°-. Cette réaction ne sera pas plus détaillée
(Halliwell and Guterridge, 2008).
Le radical carbonate est moins dangereux que le radical hydroxyle HO° pour les lipides,
les protéines et l’ADN mais peut tout de même oxyder plusieurs molécules biologiques comme
l’acide hyaluronique par exemple. Il est également capable de déshydrogéner plusieurs acides
aminés comme la cystéine, la tyrosine ou encore NAD(P)H, et peut également oxyder
l’ascorbate, la méthionine ou la guanine (Halliwell and Gutteridge, 2008). Avec NAD(P)H, le
radical carbonate est notamment une source de radical superoxyde O2° - après réaction avec
NADH :
NADH + CO₃°- NAD° + HCO₃- puis NAD° + O2 NAD+ + O2°-.
Bien que la formation de ces radicaux reste possible in vivo, les auteurs Halliwell et
Gutteridge (2008) indiquent que de plus amples recherches méritent d’être réalisées afin de
juger de leur importance dans le stress oxydant in vivo.
2.v Le monoxyde d’azote NO°
Le monoxyde d’azote peut à la fois être considéré comme une espèce radicalaire réactive
de l’oxygène (ERO radicalaire) et de l’azote (ERN radicalaire). NO° contient un électron non
apparié sur l’orbitale anti-liante 2π*y de l’azote qui en fait un radical libre (Valko, 2011)
(Halliwell et Gutteridge, 2008), relativement stable. NO° réagit seulement avec les molécules
ayant une orbitale avec un électron non apparié, c’est à dire les radicaux libres et certains
métaux de transition (Beckman and Koppenol, 1996).
In vitro le monoxyde d’azote est un gaz incolore modérément soluble dans l’eau mais
très soluble dans les solvants organiques. In vivo, NO° peut traverser les membranes cellulaires
et diffuse facilement d’une cellule à une autre (Halliwell et Gutteridge, 2008).
Dans les tissus biologiques, la synthèse du monoxyde d’azote est majoritairement
d’origine enzymatique (pour ne pas dire toute cette synthèse), produit par les NO-synthases ou
NOs (regroupant classiquement la nNOs/NOs neuronale, la iNOs/NOs inductible et la
eNOS/NOS endothéliale), à partir de la L-arginine. Bien que relativement stable, la demi-vie in
vivo du NO° est courte car ce dernier est rapidement capté par l’oxyhémoglobine contenue dans
les hématies (Beckman and Koppenol, 1996) selon la réaction:
NO° + Hb-O₂ (oxyhémoglobine) NO₃- + met-Hb (méthémoglobine).

26
Cette captation rapide vient de l’affinité du NO° pour Fe2+ (et les autres métaux de
transition, avec lequel il établit une liaison de coordination (liaison covalente où les deux
électrons échangés proviennent du même atome) pour former un complexe [Métal-nitrosyle] :
c’est la M-nitrosylation.
En d’autres termes, en condition physiologique, la demi-vie du monoxyde d’azote est
largement déterminée par le temps requis par la molécule pour atteindre un vaisseau sanguin.
Cette captation est à l’origine d’un gradient de concentration entre le site de production du NO°
et les vaisseaux sanguins (Beckman and Koppenol, 1996).
Le monoxyde d’azote fait parti des radicaux libres les moins puissants et réagit
lentement avec la plupart des molécules biologiques, incluant les groupements thiols (Halliwell
et Gutteridge, 2008). Ainsi, pour générer des thionitrites (S-nitrosylation) à partir des
groupements thiols (ou S-nitrosothiols RS—NO) NO° doit d’abord former l’ion peroxynitrite
ONOO- ou N₂O₃ (trioxyde d’azote, plus puissant).
Également, les thionitrites peuvent être formés par addition de NO° à des radicaux
thyiles: c’est la S-nitrosylation. Ainsi :
RS° + NO° RS—NO.
Cette réaction de S-nitrosylation a son importance car elle entre dans la régulation de certaines
protéines.
Bien que la réaction entre le monoxyde d’azote NO° et la plupart des biomolécules est
généralement lente, celle avec les radicaux libres et en particulier avec l’ion superoxyde est
incroyablement rapide (Gutteridge and Halliwell, 2000). La forte diffusion et la relative stabilité
du monoxyde d’azote NO° lui permettent d’atteindre le cœur de la mitochondrie des cellules et
de réagir avec O2°- pour former rapidement l’ion peroxynitrite ONOO-, au fort potentiel
oxydant (Alcaraz et al., 2013) :
NO° + O2°- ONOO-.
NO° est la seule molécule produite en concentration suffisante pour entrer en
compétition avec la superoxyde dismutase afin de réagir avec l’ion superoxyde (Beckman and
Koppenol, 1996).
NO° est aussi un puissant inhibiteur des chaînes réactionnelles comme la peroxydation
lipidique. En effet, NO° anéantit rapidement HO° (A) et les radicaux peroxyles (B) selon les
réactions (Halliwell et Gutteridge, 2008):
(A) NO° + OH° HNO2 (acide nitreux)
(B) RO2° + NO° ROONO.
L’ajout d’un électron (réduction) donne l’anion nitroxynile NO-, qui existe le plus
souvent sous forme protonée HNO in vivo ; HNO a une durée de vie courte et réagit facilement
avec le dioxygène pour former le peroxynitrite ONOO- (A) ou réagir avec NO° pour former le
radical hyponitrite ONNO°- (Halliwell et Gutteridge, 2008):
(A) NO- + O2 ONOO- (ion peroxynitrite)
(B) NO- +NO° ONNO°- (radical hypoxynitrite)

27
Cependant, si la formation du radical hypoxynitrite est possible dans les tissus
biologiques, son impact physiologique ou même pathologique n’est pas connu (Halliwell et
Gutteridge, 2008).
NO° est impliqué dans de très nombreux mécanismes physiologiques et
physiopathologiques dont les limites ne sont pas établies à ce jour.
2.vi Le dioxyde d’azote NO2°
In vitro le dioxyde d’azote est un gaz marron, mais aussi un radical libre aux
potentialités oxydantes bien plus fortes que NO° (Halliwell and Gutteridge, 2008). C’est un
polluant atmosphérique bien connu agissant comme de nombreux autres radicaux (par
addition, déshydrogénation et substitution). Il se forme notamment lorsque du monoxyde
d’azote NO° est exposé directement au dioxygène :
2NO° + O2 2NO2°.
In vivo les concentrations en dioxygène dissout ne sont cependant pas suffisantes pour
que la réaction soit quantitativement importante (Halliwell and Gutteridge, 2008) (Beckman
and Koppenol, 1996).
Cependant, un milieu organique hydrophobe (comme l’intérieur des membranes
cellulaires ou des lipoprotéines) pourrait y être plus propice puisqu’à la fois le NO° et le
dioxygène peuvent s’y concentrer. Le rôle biologique de NO2° et son impact réel est incertain,
mais sa toxicité suscite l’interrogation des chercheurs in vivo (Beckman and Koppenol, 1996).
3) Espèces non radicalaires
3.i Le dioxygène singulet 1O2
Le dioxygène singulet 1O2 provient de l’activation de la molécule de dioxygène triplet 3O2. C’est un état métastable se caractérisant par une configuration électronique particulière
dont il existe deux formes notées 1ΔgO2 (dans laquelle deux électrons de spin opposés se
retrouvent sur une même orbitale antiliante *) et 1Σg+O2 (dans laquelle les deux électrons de
spins opposés se retrouvent sur deux orbitales antiliantes * différentes) (Halliwell and
Gutteridge, 2008).
1Σg+O2 est la forme la plus réactive de l’oxygène singulet (énergie d’excitation égale à
157kJ au-dessus de l’état de base contre 93.6kJ pour 1ΔgO2). Elle se convertit rapidement en la
première, c’est donc le plus souvent 1ΔgO2 qui est considéré dans les systèmes biologiques.
Dans ces deux formes, on n’observe pas la même restriction de spin comme chez le dioxygène
triplet, ce qui leur confère une capacité d’oxydation bien supérieure.
L’origine de l’énergie d’activation du dioxygène triplet 3O2 en dioxygène singulet 1O2 est
diverse. Dans les organismes vivants, elle est le plus souvent générée par photosensibilisation :
l’énergie d’excitation d’une molécule photosensibilatrice P* (pouvant absorber une énergie
photonique E= h, où h est la constante de Planck et la fréquence du photon) peut être
ensuite transmise à du dioxygène triplet 3O2 dans le voisinage et former l’oxygène singulet 1O2
alors que P retourne à son état de base (Halliwell and Gutteridge, 2008):
P + h P*
P* + 3O2 P + 1O2

28
La riboflavine (vitamine B2) et ses dérivés flaviniques, la bilirubine, les porphyrines ou
encore le rétinal (une forme de la vitamine A) font partie de ces molécules
photosensibilisatrices (P) chez les mammifères. Les organes exposés sont aussi ceux
particulièrement exposés à la lumière comme la peau ou les yeux. On ne retrouvera donc pas
beaucoup 1O2 dans la suite de notre propos.
Il existe également d’autres sources de l’oxygène singulet in vivo: la réaction de Russel
durant la peroxydation lipidique évidemment, la réaction entre les acides hypochloreux (A) et
hypobromeux (B) avec le peroxyde d’hydrogène, mais aussi la réaction de l’ozone avec
certaines biomolécules par exemple (Halliwell and Gutteridge, 2008) :
(A) ClO- + H2O2 1O2 + H2O + Cl-
(B) BrO- + H2O2 1O2 + H2O + Br-
La réaction (A) est notamment retrouvée in vivo lorsque l’ion hypochlorite (ClO-) produit par la
myéloperoxydase est mis en contact avec le peroxyde d’hydrogène H2O2.
L’oxygène singulet interagit avec les biomolécules de deux manières (Halliwell and
Gutteridge, 2008):
- soit il libère son énergie d’excitation à la molécule au contact et retourne à son
niveau basal (le dioxygène 3O2) ;
- soit il réagit chimiquement avec elle. A titre d’exemple, l’oxygène singulet réagit
rapidement avec des composants contenant des doubles liaisons >C=C< (réaction d’addition),
formant alors des hydroperoxydes, par exemple :
RH—CH=CH2 + 1O2 R=CH-CH2OOH (peroxide organique)
Les molécules contenant deux doubles liaisons >C=C< séparées par une simple liaison
C—C (doubles liaisons conjuguées) réagissent notamment avec 1O2 pour donner des
endoperoxydes (molécule hétérocyclique contenant un résidu peroxyde –O—O– dans son
cycle). De nombreuses molécules peuvent également être formées par cyclisation ou encore
fragmentation et implique 1O2 (Halliwell and Gutteridge, 2008).
3.ii Le peroxyde d’hydrogène H2O2
Le peroxyde d’hydrogène est la forme protonée de l’ion peroxyde O22-, lui même issue de
la réduction du dioxygène basal par deux électrons. Comme O2°-, c’est un précurseur pour
d’autres espèces réactives.
Le peroxyde d’hydrogène H2O2 est issue de la double réduction de l’oxygène singulet ; on
peut décomposer cette réaction en deux étapes :
O2 + 1 e- O2°- puis O2°- + 1 e- O22- (+ 2H+ H2O2)
H2O2 est également le produit de la dismutation du radical superoxyde (par la SOD):
2O2°-+2H+ H2O2 + O2
Le métabolisme et la formation du peroxyde d’hydrogène H2O2 est donc extrêmement lié à celui
du radical superoxyde O2°-.

29
H2O2 est une molécule relativement stable in vivo et a la capacité de traverser les
membranes cellulaires et donc d’agir à distance du tissu producteur. De plus, H2O2 peut passer
par les aquaporines membranaires des cellules au même titre que l’eau (Halliwell and
Gutteridge, 2008).
La mitochondrie est un producteur majeur de peroxyde d’hydrogène grâce à la chaîne de
phosphorylation oxydative mais aussi à la monoamine oxydase (présente dans la membrane
mitochondriale externe). Les monoamines oxydases catalysent par exemple la réaction
suivante:
R1CH2NHR2+ H2O + O2 RCHO + R2NH2 + H2O2
De nombreuses autres oxydases produisent également H2O2, incluant la xanthine
oxydase, l’urate oxydase, la monoamine et les D-amino acides oxydases, la glucose oxydase…
pour ne citer qu’elles.
Tous les tissus en produisent à des taux heureusement non-toxiques et compris entre
10-7 à 10-8 mol/L en moyenne. Dans la matrice mitochondriale, la concentration en H2O2 a été
estimée à 5 nmol/L et ce de manière similaire entre les tissus (Halliwell and Gutteridge, 2008).
Le peroxyde d’hydrogène est donc particulièrement présent dans le milieu intracellulaire et
extracellulaire.
Pourtant, H2O2 est un oxydant faiblement réactif seul envers la majorité des molécules
biologiques. C’est peut être cette faible réactivité qui lui permet d’être retrouvé dans plusieurs
mécanismes de communication cellulaire. H2O2 est tout de même capable d’inactiver par
oxydation de groupements thiols les sites catalytiques de quelques enzymes directement
(incluant l’enzyme glycolytique G3PDH, des phosphatases et des caspases impliqués dans le
mécanisme de l’apoptose) (Halliwell and Gutteridge, 2008).
Comment expliquer alors l’oxydation des lipides, des protéines et de l’ADN par le
peroxyde d’hydrogène in vivo? Selon Halliwell et Guterridge (2008), la toxicité du peroxyde
d’hydrogène s’exprime lors de la présence de métaux de transition et surtout de l’ion fer Fe2+
(possiblement de l’ion cuivre Cu2+ également) dans l’environnement biologique lui permettant
de former des radicaux libres plus réactifs tels que HO°. H2O2 peut également permettre la
formation d’HOCl par la myéloperoxydase présente dans les granules azurophiles des
neutrophiles (Deaton and Marlin, 2003).
3.iii Le peroxynitrite ONOO-
Le peroxynitrite est une espèce réactive qui est très toxique pour de nombreuses
molécules biologiques, assurant une action potentiellement très néfaste lors de sa présence
dans les tissus vivants.
Les auteurs considèrent souvent que c’est la forme toxique de NO° (Beckman and
Koppenol, 1996). Déjà évoqué précédemment, le peroxynitrite naît principalement in vivo de la
forte réactivité entre NO° et O2°- (Halliwell and Gutteridge, 2008):
NO° + O2°- ONOO-
(k > 6 x 109 mol/L/s, avec k la constante de cette réaction)

30
Cette réaction a une constante de réaction bien plus élevée que la M-nitrosylation de
NO° avec Fe2+ de l’hème de l’hémoglobine ou même de la dismutation du radical superoxyde
par la SOD. ONOO- peut également être formé par la réaction entre NO- et le dioxygène O2 mais
cette situation est peu fréquente in vivo.
Aux pH biologiques (aux alentours de 7.4), le peroxynitrite est sous sa forme protonée
ONOOH (acide peroxynitreux), molécule très stable qui diffuse bien au travers des membranes
et dans les tissus biologiques (Beckman and Koppenol, 1996). Elle s’attaque à de nombreuses
molécules, assurant une toxicité large. Outre la réaction avec les molécules environnantes,
l’acide peroxynitreux peut également se transformer en nitrate par isomérisation :
ONOO- + H+ ONOOH = NO₃- + H+.
Les mécanismes biochimiques impliquant le peroxynitrite sont très complexes. Cet acide
peroxynitreux entraîne l’oxydation, la nitration ou encore la nitrosylation de nombreuses
protéines, lipides (oxydation ou nitration principalement), ADN (rupture dans la double hélice,
nitration ou désamination de ses bases azotées). On retiendra que dans le cas des protéines
contenant plusieurs acides aminés aromatiques (comme la tyrosine, la phénylalanine ou encore
le tryptophane), certains peuvent être nitratés (ajout d’un radical –NO2) et d’autres oxydés (par
exemple les résidus –SH de la cystéine, ou encore la méthionine) (Halliwell and Gutteridge,
2008).
Le peroxynitrite s’attaque également rapidement aux métaux de transition comme les
résidus contenant du fer ; ainsi, l’hémoglobine est également rapidement convertie en
méthémoglobine (M-nitrosylation), plus rapidement encore qu’avec NO° ; il s’attaque aussi
au « doigt de zinc » (le zinc étant un métal de transition), essentiels à la structure tertiaire et
quaternaire de nombreuses protéines (Beckman and Koppenol, 1996).
Certains auteurs pensent que le peroxynitrite pourrait également mener à
l’hydroxylation de composés aromatiques, faisant naître l’idée que ONOOH pourrait se
dissocier et former le radical °OH et NO2° (Kaur et al, 1997). Ces interprétations théoriques
semblent peu importantes en réalité et les études in vitro montrent qu’à pH 7.4, ONOO°- peut
effectivement libérer du °OH, mais en quantité très limitée (Kaur et al, 1997). Selon Halliwell et
Gutteridge (2008), on s’accorde actuellement pour dire qu’il est thermodynamiquement peu
probable que cette réaction ait lieu in vivo.
Aux concentrations physiologiques, l’ion peroxynitrite peut également réagir avec le
dioxyde de carbone CO2 (ou les ions bicarbonates et carbonates) et former l’anion
nitrosoperoxycarbonate ONOOCO2- (Valko, 2011) (Pavlovic, 2007). Cet ion peut, comme
ONOOH, réaliser des nitrations et des oxydations et serait particulièrement lié à la
physiopathologie de plusieurs affections impliquant le peroxynitrite. Si le
nitrosoperoxycarbonate peut se décomposer en NO₃- et en CO2, il peut également subir une
fission homolytique en générant NO2° et le radical carbonate CO₃°- ; on ne sait pas actuellement
si cette fission peut se passer in vivo.
3.iv L’acide hypochloreux HOCl
L’acide hypochloreux ou hypoclorite sous forme ionique (ClO-) est bien connu du grand
public par son utilisation comme désinfectant en forte concentration sous le nom « d’eau de
Javel ». Dans les organismes vivants, l’acide hypochloreux est produit par une enzyme contenue
dans les cellules phagocytaires, la myeloperoxydase (MPO) (Halliwell and Gutteridge, 2008).

31
Cette dernière a la faculté de catalyser la réaction suivante:
H2O2 +Cl HOCl+ OH-.
C’est un acide faible, ayant un pKa approximativement égal à 7.5 : dans les fluides
biologiques, la moitié de la molécule est sous forme ionique et l’autre moitié est sous forme
protonée. L’acide hypochloreux HOCl diffuse très bien au travers des membranes, entraînant
des dégâts sur les protéines et les lipides.
C’est un oxydant très puissant. L’hypochlorite réagit rapidement avec une grande variété
de protéines (notamment les groupes thiols –SH), les bases azotées de l’ADN et des lipides. Sa
toxicité inclut aussi d’autres radicaux libres dont les chaînes réactionnelles ne sont pas toutes
bien connues. Pour illustration, sa réaction avec des amines produit des chlorures d’amines ou
chloramines (R—NHCl), molécules également oxydantes :
R—NH2 + ClOH R—NHCl + H2O.
De plus, les chloramines peuvent également réagir avec O2°- pour générer des radicaux
de l’azote :
RNHCl + O2°- RNH° + Cl- + O2.
Ces radicaux RNH° peuvent aussi oxyder divers acides aminés et antioxydant comme
l’ascorbate par exemple mais de manière bien plus lente que HOCl.
Le radical hydroxyle HO° peut être produit lors de la réaction entre l’acide hypochloreux
et le radical superoxyde O2°- :
HOCl + O2°- O2 + Cl- + HO°.
Cette dernière réaction nous renvoie également au rôle central d’O2°- qui, malgré sa faible
réactivité, est le père de plusieurs espèces réactives encore plus toxiques.
L’acide hypochloreux peut également réagir avec l’ion fer Fe2+ et ses formes complexées
(chélates de fer), générant également le radical hydroxyle HO°:
Fe2+ +HOCl Fe3+ + HO° + Cl-.
HOCl a peut également réaliser l’addition sur une double liaison >C=C< pour former des
chlorhydrines :
>C=C< + HOCl >C(OH)—C(Cl)<
Ces réactions d’addition concernent particulièrement les acides gras insaturés présents sur les
phospholipides des membranes cellulaires alors transformés en chlorhydrines (Halliwell and
Guterridge, 2008).
3.v L’ozone O₃
L’ozone O₃ est un gaz triatomique irritant très peu soluble dans l’eau. S’il joue un rôle
« antioxydant » dans la haute atmosphère en filtrant les rayons ultraviolets, produit dans la
basse atmosphère, l’ozone est un polluant atmosphérique toxique et un puissant agent oxydant
(Halliwell and Gutteridge, 2008).
Brièvement, l’ozone inhalé est toxique pour le surfactant pulmonaire en altérant ses
lipides et protéines constitutives. Il réagit avec les acides gras polyinsaturés que l’on retrouve

32
dans les lipides du surfactant pulmonaire pour produire des lipides ozonide en absence d’eau
(peu probable dans les poumons in vivo), des aldéhydes cytotoxiques et du peroxyde
d’hydrogène en présence d’eau (Sagai and Bocci, 2011).
L’ozone oxyde également des protéines contenant des groupements thiols mais aussi
des acides aminés comme la tyrosine, le tryptophane, l’histidine et la méthionine.
En privant les poumons de leur surfactant, O₃ peut alors induire une inflammation
pulmonaire en activant les macrophages pulmonaires et attirant les neutrophiles vers les
poumons, à leur tour générateur de radicaux libres. En solution aqueuse, l’ozone se décompose
notamment en HO° mais ce processus aux pH physiologiques est très lent (favorisé au contraire
par des pH alcalins). Enfin, la réaction de l’ozone avec plusieurs molécules comme NAD(P)H, la
cystéine, l’albumine, la méthionine, l’acide urique ou le glutathion réduit produit de l’oxygène
singulet 1O2 qui peut alors à son tour oxyder d’autre molécules.
Bien qu’on parle le plus souvent de l’ozone exogène comme un polluant
environnemental, il semblerait également que de l’ozone soit produit in vivo, notamment lors
du burst oxydatif des neutrophiles activés (Sagai and Bocci, 2011). De plus amples recherches
méritent d’être envisagées à propos des voies de genèse de l’ozone chez les organismes vivants.
II) Principales sources des espèces réactives
Il existe de nombreuses sources de radicaux libres et d’espèces réactives de l’oxygène.
Elles sont classées en deux catégories, les sources endogènes, et les sources exogènes. Pour la
suite de notre propos et en accord avec notre problématique, nous ne décrirons pas de sources
exogènes (électromagnétisme, pesticides, médicaments…) mais nous focaliserons sur les
principales sources endogènes.
A) La mitochondrie
La mitochondrie est un organite au cœur du métabolisme énergétique de la cellule. Elle
est la « machine » de synthèse de l’ATP en condition aérobie. Si cette production constitue son
rôle le plus connu, la mitochondrie est aussi impliquée dans d’autres mécanismes comme
l’homéostasie calcique, l’apoptose et bien sûr la production d’espèces réactives de l’oxygène
(ERO) (Alcaraz et al., 2013).
1) La chaîne respiratoire mitochondriale
1.i Quelques rappels
Les mitochondries sont des organites intracellulaires possédant une double membrane,
définissant un espace intermembranaire et un compartiment matriciel interne.
La membrane externe est particulièrement « poreuse » et contient quasiment pour
moitié des protéines (canalaires principalement, nommées « porines ») et l’autre moitié des
lipides membranaires : cette porosité assure l’échange des métabolites et des déchets avec le
cytosol de la cellule.
On considère que la membrane interne contient 20% de protéines, dont les complexes
de la chaîne de phosphorylation oxydative, et n’est perméable quasiment qu’à l’O2, au CO2 et à
H2O. Cette perméabilité très sélective permet l’établissement du gradient protonique (H+)
nécessaire au fonctionnement des ATP synthases. La production au cours de la glycolyse et du

33
cycle de Krebs de coenzymes réduits comme NADH et FADH2 est la source d’électrons
nécessaires au fonctionnement de la chaîne.
Dans cette chaîne respiratoire mitochondriale, le transport électronique est opéré par
quatre complexes enzymatiques (I, II, III et IV) (Halliwell and Gutteridge, 2008) (Alcaraz et al.,
2013). Ces complexes contiennent tous des centres d’oxydoréduction : centres Fe-S, ions cuivre,
ions fers (hèmes), quinones, flavines. La synthèse d’ATP à partir d’ADP par l’ATP-synthase est
située au niveau du complexe V ou ATP synthases. C’est le passage des électrons de complexe
en complexe le long de la chaîne respiratoire qui crée le gradient de protons de part et d’autre
de la membrane mitochondriale interne.
Figure 2 : La chaîne respiratoire mitochondriale dite de « phosphorylation oxydative ». Q : coenzyme Q ou
ubiquinone ; CytC : cytochrome c. Modifié d’après (Alcaraz et al., 2013).
1.ii Production d’espèces réactives de l’oxygène par la chaîne respiratoire
mitochondriale
Le transport des électrons dans la chaîne mitochondriale est reconnu comme la source
principale d’ERO dans les cellules animales en aérobiose (Halliwell and Gutteridge, 2008).
Bien que la cytochrome oxydase ne produit pas d’ERO, certains composants de la chaîne
mitochondriale qui la précèdent font fuir les électrons de manière constante, réduisant
directement l’O2 ambiant en ion superoxyde O2°- au lieu d’être transmis aux composants
suivants (Halliwell and Gutteridge, 2008) (Wu et al., 2011).
O2°- peut à son tour être réduit en H2O2 par la fuite en électrons. L’impact peut donc être
grand, connaissant l’importance du radical superoxyde dans la genèse de nombreux radicaux
(Wu et al., 2011).
On considère généralement que sous atmosphère normale, 1 à 3% de l’O2 réduit dans la
mitochondrie peut former O2°-. Cependant, selon les auteurs, il semblerait que ces chiffres
pourraient être revus à la baisse. En effet, trois facteurs s’opposent naturellement à cette fuite
d’électrons (Halliwell and Gutteridge, 2008):

34
- La concentration en O2 intra-mitochondrial est relativement plus faible que dans
le cytosol,
- L’organisation structurale de la chaîne mitochondriale favorise le transfert des
électrons plutôt que leur fuite,
- La présence de protéines découplantes.
Ces fuites se situent principalement au niveau des complexes I et III, et parfois au niveau
du complexe II. Les modalités exactes de fuite des électrons sont complexes et ne seront pas
détaillées : cependant on sait qu’elles impliquent notamment l’ubiquinone au niveau du
complexe III.
L’ubiquinone ou coenzyme Q est un dérivé benzoquinonique avec une longue chaîne
latérale lipophile qui lui permet de s’insérer dans les membranes cellulaires. Le cycle benzène
porte deux fonctions cétones (>C=O, sur l’ubiquinone) qui peuvent être réduites en fonction
alcools (sur l’ubiquinol). Comme on peut le voir sur la figure suivante, les électrons sont
toujours transférés un par un et un radical intermédiaire ubisemiquinone Q°- est donc créé lors
de cette réduction.
Figure 3 : Le cycle de l’ubiquinone ou coenzyme Q et ses différents états d’oxydation.
C’est ce radical Q° qui peut réagir avec O2 et alors former O2°-, assurant la fuite des
électrons de la chaîne mitochondriale:
Q°- + O2 O2°- .
Plus la teneur en oxygène du milieu est importante et plus la production en O2°- est forte
(Halliwell and Gutteridge, 2008) (Wu et al., 2011), aspect à souligner dans le cadre sportif (base
de « l’hypothèse mitochondriale » qui sera revue en deuxième partie). De plus, selon les
espèces ou les tissus concernés, le taux de production d’ion superoxyde varie également. Pour
exemple une mitochondrie provenant du cœur d’un rat produit, dans des milieux identiques,

35
plus de radicaux que celle provenant de son foie, de même que les mitochondries aviaires
produisent généralement moins de radicaux que celles du rat (Halliwell and Gutteridge, 2008).
Les ions superoxydes O2°- sont relâchés primitivement dans la matrice mitochondriale
ainsi que dans l’espace intermembranaire (ceux produit par le complexe III). En conditions
physiologiques, ils sont en grande partie transformés par les superoxyde dismutases présentes
dans les deux compartiments (Wu et al., 2011).
1.iii Stress oxydant et dysfonctionnement mitochondrial
Si dans la physiologie normale la fuite en électrons est bien contrôlée par la
mitochondrie, en cas de stress oxydant, tout le métabolisme énergétique peut être atteint. De
puissants ERO comme le peroxynitrite ou le radical hydroxyle HO° peuvent être formés à
proximité des complexes respiratoires et endommager la mitochondrie. L’atteinte des
membranes lipidiques mitochondriales favorisent la désorganisation structurale et la
diminution de la fluidité de mouvements des complexes et des transporteurs d’électrons les
uns par rapport aux autres (voir partie IV)A)I)).
Les complexes de la chaîne respiratoire mitochondriale, de nature protéique, sont
également des cibles pour les ERO ; de plus, ils sont souvent complexés avec des métaux de
transition, cibles et des générateurs des radicaux libres. Les complexes protéiques I et III sont
particulièrement concernés (Halliwell et Gutteridge, 2008).
Par ailleurs, on sait que l’excès de NO° inhibe la cytochrome oxydase, favorisant
également la formation de O2°- depuis la chaîne respiratoire (Wu et al., 2011).
Le stress oxydant altère également les protéines VDAC (« voltage-dependent anion
channel ») et la prohibitine. VDAC est un composant majeur de la membrane mitochondriale
externe et du « complexe de perméabilité transitoire », qui régule le transport des ions et des
métabolites dedans et en dehors de la mitochondrie ; l’augmentation des dégâts oxydatifs sur
VDAC pourrait altérer ce flux bidirectionnel, entraînant une dérégulation et une diminution de
la phosphorylation oxydative i.e un dysfonctionnement mitochondrial (Wu et al., 2011).
Dans les conditions normales, la prohibitine fait office de protéine chaperonne et
empêche la désorganisation structurelle des protéines mitochondriales ; son altération par le
stress oxydant pourrait ainsi inhiber cette fonction et aggraver le dysfonctionnement
mitochondrial (Wu et al., 2011).
Ainsi, lors du dysfonctionnement mitochondrial, la fuite d’électrons s’accroît dans la
mitochondrie et au sein de la cellule, le métabolisme énergétique diminue créant un cercle
vicieux défavorable à la cellule.
Le dysfonctionnement mitochondrial est impliqué dans plusieurs mécanismes
inflammatoires. Pour mémoire, TNF- peut entraîner un dysfonctionnement mitochondrial à
l’origine d’une surproduction d’espèces réactives de l’oxygène et d’une baisse de la synthèse en
protéines mitochondriales (notamment en cytochrome c et cytochrome c-oxydase) (Taha,
2011).
Enfin, l’inflammasome est un complexe protéique oligomérique impliqué dans
l’immunité innée non spécifique et est notamment exprimé dans les cellules de la lignée
granulocytaire. Des travaux récents suggèrent que la mitochondrie est impliquée dans
l’intégration et le relai de différents signaux extracellulaires à l’inflammasome. En effet, une

36
mitochondrie a fortiori dysfonctionnelle produit des ERO en plus grande quantité et
stimuleraient l’activation de l’inflammasome (Alcaraz et al., 2013). L’inflammasome stimule
notamment l’activation de la caspase-1 dans l’apoptose et favorise la production et la sécrétion
de cytokines pro-inflammatoires.
2) Autres sources mitochondriales d’espèces réactives
Pour mémoire, on retiendra que la chaîne respiratoire mitochondriale n’est pas la seule
productrice d’ERO dans la mitochondrie. La monoamine oxydase est également une source
d’H2O2 et est située dans la membrane mitochondriale externe (Halliwell and Gutteridge, 2008).
Cette enzyme est une flavoprotéine qui catalyse la réaction :
R—NH2 (monoamine) + O2 +H2O -> R-CHO (aldéhyde) + H2O2 + NH₃.
Il existe deux types de monoamine oxydase: MAO-A (qui oxyde de préférence les amines
hydroxylées) et MAO-B (qui oxyde de préférence les amines non hydroxylées). Eles sont une
source importante d’H2O2, particulièrement dans le cerveau.
Plusieurs oxydases flaviniques mitochondriales pourraient également être responsables
de la synthèse d’ERO depuis la mitochondrie. Cette production reste anecdotique.
B) Importance des métaux de transitions
1) Définition
Le terme « métaux de transition » désigne une famille particulière d’éléments du tableau
périodique de Mendeleïev.
La définition que nous adopterons tout au long de ce travail correspond à celle choisie
par Halliwell et Gutteridge (2008) : « les métaux de transition correspondent à l’ensemble des
éléments présents dans la première ligne du bloc d du tableau périodique des éléments, contenant
des électrons non appariés dans leur atome et/ou ions ». Les métaux sont donc des radicaux
libres. La seule exception à cette définition est le zinc.
Les métaux de transition que l’on considère habituellement sont le chrome, le nickel, le
cobalt, le titane, le vanadium et bien sûr le fer et le cuivre.
Les fonctions biologiques que ces métaux assurent proviennent évidemment de leur
capacité à accepter et transférer des électrons libres. Cette capacité leur permet de vaincre la
restriction de spin (Principe de Pauli) qui empêche l’action directe du dioxygène triplet.
Nous nous attarderons particulièrement sur le fer chez les mammifères.
2) Réaction de Fenton et réaction de Haber – Weiss
Le fer est un composant bien connu des mécanismes biologiques d’oxydation par des
radicaux libres. Par ses propriétés de transfert d’électrons, le fer se voit donner des capacités
catalytiques dans de nombreuses réactions biochimiques.
Pendant longtemps, son implication dans genèse d’ERO était théorisée par deux
réactions : la réaction de Haber Weiss et la réaction de Fenton (Qian and Buettner, 1999).
Comme nous le verrons le long de cette présentation, ces réactions au sein de l’organisme sont
controversées au sein de la communauté scientifique.

37
2.i Réaction de Fenton
Le modèle de la réaction de Fenton illustre la toxicité du peroxyde d’hydrogène par la
production de radical hydroxyle HO° en présence d’un métal de transition (comme Fe2+ ou
Cu2+)(Gutteridge and Halliwell, 2000) et est classiquement écrit comme suit:
Fe2+ + H2O2 Fe3+ +OH- + HO°
(puis réaction possible Fe3+ + O2°- Fe2 + O2)
C’est également possible avec un peroxyde organique ROOH :
Fe2+ + ROOH ROO° (radical alkoxyle) + OH- + Fe3+
De nombreuses autres réactions sont possibles dans un système de Fenton et dépendent
largement des conditions initiales. De même, in vitro il a été montré que si un excès de fer libre
favorise le stress oxydant, une carence le peut également.
La prépondérance de la réaction de Fenton pourrait être remise en cause in vivo dans
l’organisme. Par exemple, contrairement à ce que l’on a longtemps pensé, il n’est pas forcément
nécessaire que du peroxyde d’hydrogène H2O2 soit présent dans le milieu pour qu’une réaction
de Fenton se passe ; en effet, une telle réaction pourrait également se mettre en place en
présence d’O2 et d’un métal de transition en solution in vitro. Cela n’a cependant pas été mis en
évidence in vivo pour l’instant et ne sera donc pas détaillé (Qian and Buettner, 1999). De plus,
le ratio [O2]/[H2O2] dans un milieu biologique est généralement supérieur à 1000. Enfin,
comme pour la réaction qui va suivre, la faible disponibilité en métaux de transition libre
diminue sa probabilité d’apparition in vivo.
La réaction de Fenton avec H2O2 préexistant reste possible et explique la toxicité du
peroxyde d’hydrogène ; cependant elle ne constituerait qu’un initiateur mineur des oxydations
radicalaires biologiques, comme la peroxydation lipidique (Qian and Buettner, 1999).
2.ii Réaction de Haber-Weiss
La réaction de Haber-Wess résume la réaction de réduction du peroxyde d’hydrogène
par le radical superoxyde pour former le radical hydroxyle HO° (Qian and Buettner, 1999) :
O2°- + H2O2 O2 + HO- + HO°
Cette réaction est thermodynamiquement possible mais est très lente en l’absence de
catalyseur, avec une constante de réaction dans les solutions aqueuses proche de 0 (Halliwell
and Gutteridge, 2008). Elle nécessite la présence de métaux de transition comme catalyseur, et
notamment de Fe3+ ; la réaction se décompose alors ainsi:
Fe3+ + O2°- Fe2+ + O2
puis
Fe2+ + H2O2 HO° + OH- + Fe3+
(réaction de Fenton)
Aujourd’hui, les scientifiques tendent à remettre en cause cette réaction in vivo, tant la
durée de vie et la concentration en O2°- est faible, mais aussi le manque de disponibilité de Fe2+
(paragraphe 2.iii qui suit) ; cependant l’électron pourrait provenir d’autres réducteurs comme

38
l’ascorbate, les cystéines, des flavoprotéines ou encore des quinones et semiquinones (Halliwell
and Gutteridge, 2008).
2.iii Disponibilité des métaux de transition in vivo
Importantes in vitro, il semble que ces réactions occupent une place moins importante in
vivo dans la genèse de nouveaux radicaux libres (Gutteridge and Halliwell, 2000).
D’un point de vue thermodynamique, les réactions de Fenton et d’Haber-Weiss
(catalysées par un métal de transition) peuvent facilement initier des oxydations dans les
systèmes biologiques. Cependant, les stocks d’ions fer libres dans la cellule saine sont très
faibles (Qian and Buettner, 1999) ; en effet, le fer est complexé dans la quasi totalité des
composés.
In vivo, la constante de réaction de la réaction de Fenton entre Fe2+ et H2O2 est ainsi
assez faible (à peu près 103–105 mol/L/s) alors que la réaction d’oxydation de Fe2+ par O2°- a
une constante de réaction de l’ordre de 106 à 107 mol/L/s dans les conditions physiologiques.
Ainsi les études actuelles supportent plutôt l’hypothèse des complexes « Fe-O » (voir
point suivant) pour expliquer l’implication du Fe2+ dans l’enclenchement des mécanismes
oxydatifs in vivo.
Pour autant, des études suggèrent que le stress oxydant génère des ions fers libre dans
les cellules (Srinivasan et al, 2000). Cette constatation serait également vraie pour les
mammifères (Keyer et al., 1994), où la dégradation de molécules comme la ferritine (Biemond
et al., 1984), l’hème de l’hémoglobine (Mohanty et al., 2013), et les cluster Fe-S des protéines
participerait à l’augmentation du pool de fer libre dans la cellule et donc à l’augmentation du
nombre de réactions de Fenton (par augmentation de la constante de réaction). Nous
remarquerons que de la même manière, les ERO peuvent dégrader des protéines comme la
céruloplasmine qui contiennent du cuivre (Halliwell and Gutteridge, 2008).
En accord avec notre sujet, il convient de souligner que les traumas et l’inflammation en
général peuvent également être responsables de la libération d’ions métalliques catalysant les
réactions de Fenton (Gutteridge and Halliwell, 2000).
Selon plusieurs auteurs dont Halliwell et Gutteridge (2000), il est clair que les molécules
liées aux métaux de transition, que le lien soit fort ou faible, dictent le déroulement de la
réaction de Fenton. Les études à ce sujet dépassent largement notre cadre de travail.
3) Complexes « iron-oxygen » ou « Fe-O » : les ions ferryles et ions perferryles.
Un autre modèle d’oxydation par des radicaux libres initiés par le fer autrement que la
réaction de Haber-Weiss a également été proposé dans le cadre de l’étude du système de
Fenton (Qian and Buettner, 1999): les complexes intermédiaires « Fe-O » i.e. les ions ferryles
et perferryles.
La nature exacte des espèces initiatrices est inconnue. Ce modèle peut également
s’appliquer à d’autres métaux de transition, comme le cuivre. Les ions ferryles peuvent être
formés de deux manières (Venkataraman et al., 2004).

39
Premièrement, sous certaines conditions de pH et de polarité, Fe2+ et H2O2 peuvent
entraîner la formation de l’ion ferryle plutôt qu’HO° comme dans la réaction « classique » de
Fenton (Qian and Buettner, 1999):
Fe2+ + H2O2 Fe2+–O (ion ferryle) + H2O.
La deuxième situation est la réaction d’un ion perferryle avec Fe2+ pour former un ion
ferryle:
Fe2+ + Fe2+–O2 (ion perferryle) Fe2+-O2-Fe2+ 2Fe2+–O (ion ferryle).
L’ion perferryle est un produit intermédiaire qui produit soit Fe2+/O2 soit Fe3+/O2°- :
Fe2+ + O2 = [Fe2+–O2 Fe3+–O2°-] (ion perferryle) = Fe3+ + O2°-
Ainsi, à cause de leur haute affinité électronique, ces complexes intermédiaires (ions
ferryles et perferryles) pourraient être des oxydants très puissants dans les systèmes
biologiques, aussi forts que HO°, et ainsi expliquer l’origine du stress oxydant à la libération de
métaux de transition dans les milieux biologiques.
Cette thèse est supportée notamment par le fait que le fer Fe2+ peut avoir une durée de
vie très limitée (de la seconde à quelques minutes selon les conditions) dans une solution
aqueuse correctement aérée (Cohen and Sinet, 1980). Selon Cohen et Sinet (1980), le taux
d’oxydation sur des biomolécules par ces complexes « Fe-O2 » pourrait être jusqu’à 108 fois plus
rapide que la réaction de Fenton si tous les Fe2+ du milieu étaient impliqués, ce qui n’est
sûrement pas le cas dans conditions in vivo.
Cependant, même en divisant par deux la concentration en fer réagissant pour former
ces complexes, ce taux d’oxydation resterait toujours 106 fois supérieur à la réaction de Fenton
(Qian and Buettner, 1999).
Enfin, selon cette hypothèse des complexes « Fe-O », O2°- ne serait plus nécessaire pour
réduire le Fe3+ comme souvent dans un système de Fenton et cette réduction pourrait être
réalisée par des réducteurs cellulaires, comme certains antioxydants tels l’acide ascorbique ou
le glutathion réduit GSH (Qian and Buettner, 1999).
C) Des enzymes du métabolisme et de l’inflammation
1) Les cofacteurs enzymatiques flaviniques des oxydases: FAD et FMN
La flavine adénine dinucléotide (forme oxydée FAD/ forme réduite FADH2) et la flavine
mononucléotide (FMN/FMNH2) sont des flavoprotéines intervenant comme co-facteurs dans le
mécanisme catalytique de nombreuses oxydoréductases. EIles sont issues du métabolisme de la
vitamine B2 d’où provient le noyau azoté isoalloxasine. Ce noyau permet à la molécule
d’accepter un à deux électrons qui peuvent être libérés rapidement.
Ces éléments sont toujours liés in vivo à des complexes protéiques. Lors de certaines de
ces catalyses, notamment avec les oxydases flaviniques qui utilisent O2 comme accepteur
d’électrons, ces cofacteurs réduisent le dioxygène (ajout d’un électron) en radical superoxyde
O2°- ou plus (ajout de deux électrons) en peroxyde d’hydrogène H2O2.

40
2) Xanthine déshydrogénase (XDH) et xanthine oxydase (XO)
La xanthine déshydrogénase (XDH) est une enzyme cytosolique qui appartient à la
famille des hydroxylases contenant du molybdène et est impliquée dans le métabolisme des
purines (Halliwell and Gutteridge, 2008).
Les purines comprennent l’ensemble des composés chimiques constitués d’un noyau
pyrimidine fusionné avec un noyau imidazole. C’est une protéine homodimérique qui catalyse
la transformation de l’hypoxanthine en xanthine puis en acide urique en réduisant
simultanément le NAD+ en NADH. Selon le métabolisme normal de la XDH, il n’y a pas de
production d’ERO. Cette enzyme est prédominante dans les tissus sains in vivo
comparativement à la xanthine oxydase.
Or la xanthine deshydrogénase (XDH) peut être transformée en xanthine oxydase (XO)
par oxydation de plusieurs groupements thiols (-SH) ou par clivage protéolytique, situation
pouvant être rencontrée en condition de stress oxydant au niveau d’un tissu (notamment lors
d’ischémie-reperfusion). Ces modifications biochimiques mènent à des modifications
conformationnelles de l’enzyme au niveau du site de fixation du FAD qui bloque l’accès du
NAD+ au site actif et ainsi mène au transfert des électrons au dioxygène O2 (Halliwell and
Gutteridge, 2008) :
RH + H2O + 2 O2 ROH + 2 O2°- + 2 H+
Figure 4 : Production d’ERO par la xanthine oxydase (XO).
Une autre réaction catalysée par la xanthine oxydase est la décomposition des S-
nitrosothiols en NO° (pouvant alors réagir avec le radical superoxyde pour former du
peroxynitrite). Dans une situation d’hypoxie, XO peut également réduire les nitrates en nitrites
et les nitrites en NO°.
3) La NADPH-oxydase (Nox)
La nicotinamide adénine dinucléotide phosphate oxydase (ou NADPH-oxydase) est un
complexe enzymatique présent dans les membranes plasmatiques des neutrophiles et des
macrophages mais est aussi retrouvée dans les cellules endothéliales et les cellules musculaires
lisses (Jairam et al., 2012). Son rôle principal est de générer le radical superoxyde O2°- en
transférant des électrons du NADPH au dioxygène O2:
2 O2 + NADPH 2 O2°- + NAD(P) + + H+

41
Le complexe NADPH-oxydase consiste essentiellement en deux sous-unités
(hétérodimère) peptidiques membranaires de 22kD (la sous-unité ) et 91 kDa (la sous-unité
, contenant la portion catalytique de l’enzyme), ainsi qu’un peptide régulateur nommé Rac ; la
partie cytosolique est constituée de plusieurs petites protéines (ou facteurs cytosoliques)
régulées par l’état redox intracellulaire (Henrotin, 2003).
Sept enzymes différentes sont connues dans le groupe des NADPH-oxydases. Le
fonctionnement enzymatique diffère d’une enzyme à une autre, mais cela reste encore peu
connu. Nox2 est la plus étudiée et est présente dans les cellules phagocytaires. Elle est activée
lors du « burst oxydatif » de la phagocytose au même titre que la myéloperoxydase (Halliwell
and Gutteridge, 2008).
Le complexe NADPH-oxydase peut également être inhibé par le monoxyde d’azote NO°
(Henrotin, 2003).
4) Les oxydases des peroxysomes
Les peroxysomes sont des organites retrouvés dans pratiquement toutes les cellules de
l’organisme à l’exception des hématies. Ils sont constitués d’une membrane cytosolique
phospholipidique simple et d’un contenu de composition très différente de celle du cytosol,
dont de nombreuses oxydases (glycolate oxydase, urate-oxydase, acyl-CoA, etc…) et des
antioxydants enzymatiques comme la catalase (Halliwell and Gutteridge, 2008).
Si leur fonction principale est la -oxydation des acides gras dans la majorité des
cellules de l’organisme, ils interviennent également dans la synthèse du cholestérol et des
acides biliaires au niveau des hépatocytes.
Les oxydases sont des enzymes utilisant le dioxygène O2 comme accepteur d’électrons
pour leur catalyse. Les enzymes oxydases enlèvent des atomes d'hydrogène libres à des
substrats organiques spécifiques potentiellement dangereux pour la cellule. La
déshydrogénation de ces molécules produit également du peroxyde d’hydrogène :
R—H2 + O2 → R + H2O2
La catalase détoxifie ensuite H2O2 et évite que sa concentration dépasse des seuils
dangereux pour la cellule en condition physiologique (le peroxyde d’hydrogène diffusant en
partie hors des peroxysomes) ; elle est par ailleurs plus abondante que les oxydases dans les
peroxysomes (Halliwell and Gutteridge, 2008).
5) Des enzymes du métabolisme de l’acide arachidonique : les cyclooxygénases
(COX).
Les cyclooxygénases (COX) sont les enzymes responsables de la production des
prostaglandines et particulièrement de la prostaglandine H2 (PGH2), à partir de l’acide
arachidonique. La prostaglandine PGH2 est un précurseur instable des prostanoïdes, qui
regroupent les prostaglandines, les prostacyclines et le thromboxanes.
Dans la cellule, les cyclooxygénases sont situées sur la membrane luminale du réticulum
endoplasmique (RE) ainsi que sur les membranes internes et externes de l’enveloppe nucléaire.
Il existe deux isoformes de COX : COX1 et COX2. COX1 est constitutive et ubiquiste alors que
COX2 est inductible et mobilisée notamment pendant une inflammation. Ces deux isoformes
obéissent au même mécanisme réactionnel.

42
Deux étapes enzymatiques mènent à la production de PGH2 :
- la cyclisation et l’oxygénation de l’acide arachidonique qui forme un
hydroperoxyde cyclique : la prostaglandine G2 (PGG2) ;
- la peroxydation de PGG2 en PGH2 par un site peroxydase.
Les effets potentiellement délétères des COX concernent le site peroxydase hémique
pour la production d’ERO. Après la réduction de PGG2 en PGH2, le retour à l’état initial de l’état
redox du site actif se fait par deux oxydations mono-électroniques successives. Ces oxydations
passent notamment par la formation du radical tyrosyle, radical qui peut réagir avec des
molécules environnantes telles que les phénols, les amines aromatiques, les polyènes et pour
former de nouveaux radicaux libres. (Halliwell and Gutteridge, 2008) (Jungbluth, 2008).
Ces nombreux radicaux formés lors de ces étapes sont souvent des radicaux libres
centrés sur le carbone, qui peuvent alors initier la peroxydation lipidique en formant des
radicaux peroxyles comme nous l’avons vu précédemment.
6) La myéloperoxydase (MPO)
La myéloperoxydase (MPO) est une enzyme retrouvée abondamment dans les
polynucléaires neutrophiles et les macrophages (à plus faible concentration chez ces derniers)
(Jairam et al., 2012). On la retrouve également dans les cellules de la lignée blanche. On
considère qu’elle représente jusqu’à 5% des protéines des neutrophiles (Halliwell and
Gutteridge, 2008).
C’est un dimère glycoprotéique (contenant chacun un noyau hémique) du lysosome
stocké dans les granules azurophiles des neutrophiles (Jairam et al., 2012). La MPO possède
une double activité peroxydase et chlorase.
La myéloperoxydase est capable d’oxyder un grand nombre de substrats (faible
spécificité), en utilisant le peroxyde d’hydrogène pour produire d’autres espèces réactives de
l’oxygène (Halliwell and Gutteridge, 2008). Cette faible spécifité vient en grande partie de la
formation de radicaux ferryles au niveau de son site hémique catalytique (voir partie II.B.3 sur
les métaux de transition). Elle peut également former le radical dioxyde d’azote NO2°, oxydant
très puissant.
Durant le « burst » oxydatif lors de la phagocytose, la MPO est activée et catalyse la
production d’acide hypochloreux (HOCl) à partir du peroxyde d’hydrogène et l’ion chlore Cl-
suivant la réaction (Jairam et al., 2012):
Cl- + H+ + H2O2 HOCl + H2O
L’hypochlorite ainsi produit peut également entraîner la formation d’autres espèces
réactives comme l’oxygène singulet (en réagissant avec le peroxyde d’hydrogène H2O2) ou le
chlorure de nitrile NO2Cl (en réagissant avec l’ion nitrite NO2-).
7) Les monoxyde d’azote synthases (NOs)
Présents dans la plupart des cellules chez les mammifères, les monoxydes d’azote
synthases (NOs) sont des dimères protéiques hémiques de la classe des mono-oxygénases qui
catalysent la formation de monoxyde d’azote NO°, d’eau et de L-citrulline à partir de l’acide
aminée L-arginine, en présence de NADPH (donneur d’électrons), de tétrahydrobiopterine
(cofacteurs essentiels) et de dioxygène (Halliwell and Gutteridge, 2008) :

43
R=NH (L-arginine) + 3 e- + 3 H+ + 2 O2 R=O (L-citrulline) + 2 HO2 + NO°
Quand les concentrations en cofacteurs (NADPH et tetrahydrobiopterine) et en L-
arginine sont faibles, les NOs peuvent produire des radicaux superoxydes O2°- et/ou du
peroxyde d’hydrogène ; du peroxynitrite se forme alors facilement dans l’environnement des
NOs (par réaction entre le radical superoxyde et le monoxyde d’azote).
On distingue trois isoformes des NOs (Henrotin, 2003). Deux sont exprimées de manière
constitutive, la NOs endothéliale (eNOS ou NOs3) et la NOs neuronale (nNOs ou NOs1), et une
est inductible, la NOs inductible (iNOs ou NOs2).
La nNOS est localisée dans le cytosol des cellules du système nerveux central. Elle
produit du monoxyde d’azote de manière constitutive mais peut up-réguler sa production en
réponse à l’action de cytokines pro-inflammatoires. Son activité est dépendante de la
concentration calcique intracellulaire (Jungbluth, 2008).
La eNOS est constitutive de nombreuses cellules dont les cellules endothéliales et les
plaquettes. Comme nNOS, son activité dépend également de la concentration calcique
intracellulaire (Jungbluth, 2008).
La iNOS est présente dans le cytosol des hépatocytes, des cellules des îlots
pancréatiques, des polynucléaires neutrophiles, des macrophages et bien sûr des cellules
endothéliales. Elle est régulée au niveau de son gène par de nombreux facteurs de croissance,
cytokines inflammatoires et endotoxines. En effet, la production de NO° est notamment
stimulée dans différentes situations inflammatoires par l’interleukine IL-1, le TNF,
l’interféron IFN ou encore le LPS des bactéries Gram - ; son expression est cependant inhibée
par TGF-, IL-4, IL-10 et IL-13 (Henrotin, 2003). Son activité n’est pas dépendante de la
concentration calcique intracellulaire contrairement aux deux autres isoformes (Jungbluth,
2008).
D) Autres sources endogènes d’intérêt : les « auto-oxydations »
De nombreuses molécules biologiquement essentielles sont oxydées par le dioxygène O2
avec production de radicaux superoxydes. Elles incluent notamment les catécholamines
(adrénaline et noradrénaline par exemple), le glycéraldéhyde, la dopamine et
dihydroxyphénylalanine (L-DOPA) et divers composés thiols dont la cystéine et des
tétrahydroptéridines (Halliwell and Gutteridge, 2008).
En général, ces oxydations commencent doucement mais, dès que suffisamment de
radicaux ont été produits, des réactions en chaîne se mettent en place, accroissant la genèse
d’O2°-. La molécule de dioxygène triplet O2 étant peu réactive, on a peu élucidé l’origine de ces
«auto-oxydations». Il semblerait que leur fréquence augmente en présence de métaux de
transition qui joueraient un rôle de catalyse. En effet, on constate qu’in vitro il est impossible de
les reproduire dans des solutions entièrement dénuées de ces métaux (Halliwell and
Gutteridge, 2008).

44
III) La défense cellulaire antioxydante
A) Définition d’un antioxydant et stratégies de défense
1) Qu’est ce qu’un antioxydant ?
La définition d’antioxydant n’est pas aussi claire qu’il y paraît. Selon Halliwell et
Gutteridge (2000 et 2008) :
« un antioxydant est toute substance qui, lorsqu’elle est présente en faible concentration par
rapport au substrat environnant pouvant être oxydé, retarde ou empêche de façon significative
l’oxydation de ce même substrat ».
On inclut dans ce « substrat oxydable » quasiment toutes les molécules présentes in vivo.
Comme toute définition, elle peut sembler imparfaite, et n’inclut pas par exemple les
inhibiteurs de la genèse des ROS. Cette définition peut donc être simplifiée comme :
« Toute substance qui retarde, empêche ou répare les dégâts oxydatifs d’une molécule cible ».
2) Stratégies de défense
Les cellules d’un animal ne peuvent pas, comme certaines bactéries, éviter directement
les espèces réactives. Les eucaryotes animaux ont alors adopté des stratégies de défense contre
le stress oxydant (Halliwell and Gutteridge, 2008) dont les principales sont :
- des entités qui catalysent le retrait, la dégradation des espèces réactives ou leur
conversion en ER moins réactifs comme les enzymes superoxyde dismutases (SOD), les
catalases (CAT), les glutathion peroxydases (GPx) ou les peroxyréductases.
- des entités qui ralentissent la formation des espèces réactives comme les protéines
découplantes mitochondriales et les molécules chélatrices des métaux de transition par
exemple.
- Le transfert de l’énergie d’excitation de l’oxygène singulet à une autre molécule
comme les caroténoïdes.
- Le remplacement des molécules sensibles au stress oxydant par des molécules plus
résistantes.
- des entités se dégradant au contact des espèces réactives pour en sauver d’autres plus
importantes ; citons par exemple le glutathion (GSH), -tocophérol (vitamine E), la bilirubine,
l’acide ascorbique (vitamine C), l’acide urique ou encore l’albumine.
De manière similaire à l’équilibre du pH, les systèmes biologiques régulent ainsi dans de
faibles intervalles l’état redox de la cellule. Cet équilibre s’illustre notamment par celui de
plusieurs antioxydants non enzymatiques qui sont autant de couples oxydant/réducteur (par
exemple Asc°-/AscH- pour la vitamine C, GSSG/GSH pour le gluthation ...).Ainsi, c’est cette
capacité d’échange des électrons (et des atomes d’hydrogène) entre les molécules
antioxydantes et les radicaux mais aussi la reconstitution des défenses qui déterminent la
capacité redox des systèmes biologiques et la réponse globale au stress oxydant.

45
B) Antioxydants enzymatiques
1) Les superoxyde dismutases (SOD)
Les superoxyde dismutases (SOD) sont des métalloprotéines enzymatiques très
importantes impliquées dans le catabolisme d’O2°- chez les organismes vivants. Grâce à elles, la
vitesse de la réaction de dismutation du radical superoxyde est mille fois plus rapide que
spontanément:
2 O2°- H2O2 + O2.
Bien que produisant du peroxyde d’hydrogène, les SOD sont des enzymes antioxydantes
car elles empêchent la genèse de radicaux plus fort et issus du radical superoxyde tel que HO°.
In vitro, la SOD réagit cependant moins rapidement que le monoxyde d’azote avec
l’anion superoxyde. In vivo, au sein de la mitochondrie, la concentration en monoxyde d’azote
est à peu près égale à celle en superoxyde dismutase, l’ion superoxyde produit réagira donc
plus volontiers avec le NO° pour former l’ion peroxynitrite (Beckman and Koppenol, 1996).
Cependant dans une cellule normale, les taux en SOD sont généralement suffisants pour éviter
un déséquilibre vers la réaction entre NO° et l’ion superoxyde (Gutteridge and Halliwell, 2000)
(Beckman and Koppenol, 1996). Combinée à la réaction rapide du NO° avec l’oxyhémoglobine,
l’action de la superoxyde dismutase réduit de façon significative la formation d’ion
peroxynitrite. Des auteurs considèrent même que les SOD permettent à NO° de rester actif et
d’assurer son rôle de messager sans réagir avec les ERO (Regan et al., 2005).
Elles sont présentes dans presque toutes les cellules eucaryotes (Halliwell and
Gutteridge, 2008) et trois types ont été identifiés chez les mammifères: SOD1, SOD2 et SOD3.
La CuZnSOD ou SOD1 a été découverte en 1968 par McCord et Fridovich (McCord and
al., 1968) comme un contaminant in vitro dans leur étude sur le cytochrome c. Si on a
longtemps considéré que cette enzyme était un transporteur du cuivre, il est avéré que la
CuZnSOD contribue largement au stock du cuivre intracellulaire (Gutteridge and Halliwell,
2000).
Cependant, Touati et al. (1989) ont été les premiers à montrer l’importance catalytique
in vivo de la SOD chez des E. Coli mutantes privées de l’expression de MnSOD et de FeSOD ; ces
dernières étaient viables mais malades.
Dans les cellules animales, SOD1 est surtout présente dans le cytosol, mais il apparaîtrait
qu’elle soit aussi présente dans les lysosomes, le noyau et l’espace intermembranaire de la
mitochondrie. Il est également possible que de la SOD1 en petite quantité soit présente dans les
peroxysomes. Elle est composée de deux sous-unités protéiques chacune contenant un ion
cuivre et un ion zinc et a une masse moléculaire d’à peu près 32 kDa. L’ion zinc Zn2+ ne fait pas
partie intégrante du site catalytique mais aide à la stabilisation de l’enzyme (Halliwell and
Guterridge, 2008). Par contre, l’ion cuivre catalyse directement la réaction de dismutation en
alternant oxydation et réduction du radical superoxyde:
Cu2+ + O2°- Cu+ + O2
puis
Cu+ + O2°- + 2 H+ Cu2++ H2O2.

46
L’enzyme tire également une partie de son efficacité de sa charge ionique : seul l’entrée
du site actif est chargée positivement contrairement au reste de la protéine. O2°- est ainsi
directement orienté vers lui (Regan et al., 2005).Le peroxyde d’hydrogène, produit de la
réaction de dismutation peut lui aussi réagir avec l’ion cuivre du site (qui est un métal de
transition) et générer des radicaux libres depuis le site actif ; cette situation est cependant peu
rencontrée dans les organismes vivants (Halliwell and Gutteridge, 2008).
La SOD2 ou MnSOD a pour la première fois été isolée dans la bactérie E.Coli. Son
importance pour les organismes a premièrement été établie chez des levures privées en SOD2
dont la croissance est bien plus faible que chez les levures sauvages en conditions aérobies
(Srinivasan et al., 2000). De même, les souris dépourvues de SOD2 sont très malades, avec un
phénotype plus grave chez les individus homozygotes : les jeunes souris souffrent de
défaillances cardiaques (cardiomyopathies dilatées essentiellement, se développant dans les 10
premiers jours de la vie de l’animal) (Li et al., 1995), mais aussi des neurodégénérescences,
d’anémies sévères et un fort taux de mortinatalité (Lebovitz et al., 1996). Enfin, des dégâts
sévères témoignant du stress oxydant sur les mitochondries des cardiomyocytes et d’autres
tissus avec une diminution de l’activité du complexe I de la chaîne mitochondriale, de la
succinate déshydrogénase et de l’aconitase, ainsi que l’élévation du dégât oxydatif sur l’ADN
mitochondrial sont constatés chez ces animaux.
Ces résultats témoignent du rôle essentiel de la MnSOD dans le maintien des fonctions
mitochondriales (Melov et al., 1999). De même, les souris défaillantes en SOD2 seulement dans
les cellules de la lignée hématopoïétique vivent plus longtemps que les précédentes mais
présentent une dégradation de l’hème plus rapide et une baisse significative de la déformabilité
érythrocytaire (Mohanty et al., 2013), indiquant une augmentation du stress oxydant du
globule rouge comparé au groupe contrôle.
Chez les cellules animales, la SOD2 est largement (pour ne pas dire entièrement) située
dans la mitochondrie au sein de la membrane interne (Regan et al., 2005) (Jairam et al., 2012).
Dans la lignée hématopoïétique, elle est retrouvée dans les cellules précurseurs des hématies,
mais pas dans les hématies matures (Mohanty et al., 2013).
SOD2 est un tétramère dont chaque sous-unité contient du manganèse Mn3+ sur son site
actif. La proportion de SOD2 par rapport à la SOD1 varie selon les tissus en lien avec le nombre
de mitochondries par cellule et de l’espèce. Pour illustration, 10% de l’activité de la SOD dans le
foie est réalisée par la SOD2 (Halliwell and Gutteridge, 2008).
La MnSOD catalyse exactement la même réaction que CuZnSOD, l’ion manganèse ayant
le rôle du cuivre :
Mn3+ + O2°- Mn2+ + O2
Puis
Mn2+ + O2°- + 2 H+ Mn3+ + H2O2.
À pH égal à 7, les constantes de la réaction de dismutation de l’oxygène singulet
catalysée par SOD1 et SOD2 sont semblables. Pourtant SOD2 voit ses constantes de réaction
diminuer quand le pH devient plus basique pour devenir très faible à un pH supérieur à 7.8.
La SOD3 ou SOD extracellulaire (EC-SOD) est le principal antioxydant des milieux
extracellulaires (le liquide synovial par exemple) (Halliwell and Gutteridge, 2008) (Jairam et al.,
2012) (Regan and al.,2005). C’est une enzyme glycoprotéique tétramérique avec un site de

47
fixation pour l’héparine chargé positivement sur sa partie C-terminale. Le site actif contient du
cuivre et du zinc et est analogue à SOD1.
On retrouve notamment SOD3 dans la matrice extracellulaire de nombreux tissus
interstitiels, accolée aux molécules de collagène et aux protéoglycanes chargés négativement. A
cette place, cette enzyme protège directement les protéines et les macromolécules vulnérables
de la matrice extracellulaire. C’est particulièrement vrai pour le tissu pulmonaire, le tissu
nerveux, les vaisseaux, le cartilage, les tendons et les ligaments (Jairam et al., 2012) (Regan and
al.,2005).
2) Les glutathion peroxydases (GPx)
La glutathion peroxydase (GPx) est une sélénoprotéine ayant fonction d'enzyme, formée
de quatre sous-unités identiques (homotétramère) contenant chacune un atome de sélénium
incorporé dans une molécule de sélénocystéine (Raman and Berry, 2011). Contrairement à un
grand nombre de peroxydases, elles ne sont pas héminiques.
Elle est bien distribuée dans tous les tissus chez les animaux (Halliwell and Gutteridge,
2008). La glutathion peroxydase est présente dans les liquides extracellulaires et dans les
cellules au niveau du cytosol et des mitochondries.
La GPx catalyse la réduction du peroxyde d’hydrogène (A) en oxydant deux molécules de
glutathion GSH réduites en GSSG. Elle assure plus largement la transformation des
hydroperoxydes organiques, lipidiques notamment de type ROOH, en alcools (ROH)
(B) (Halliwell and Gutteridge, 2008) (Raman and Berry, 2011):
(A) 2 GSH + H2O2 GSSG +2 H2O
(B) 2 GSH + ROOH GSSG + ROH + H2O
Concernant la réaction (B), il faut remarquer que la GPx (sauf GPx4) ne peut pas agir sur
les peroxydes organiques estérifiés aux lipides membranaires : les acides gras estérifiés
doivent donc d’abord être relâchés dans le cytosol par une lipase (Raman and Berry, 2011).
Au moins cinq types de GPx existent (Raman and Berry, 2011) (Halliwell and Gutteridge,
2008) : GPx1, GPx2, GPx3, GPx4 et GPx6.
GPx1 est la plus abondante et représente une fraction très importante du pool total de
sélénium en circulation (au sein des hématies) chez les mammifères. Cette enzyme est détectée
dans le cytoplasme mais aussi dans la mitochondrie de nombreux types cellulaires. GPx2, aussi
connue comme la « GPx gastro-intestinale », est une enzyme cytosolique spécifique des cellules
épithéliales et abonde en effet au niveau des intestins. GPx3 est uniquement extracellulaire et a
une spécificité de substrat plus large que ses homologues ; on la retrouve dans tous les fluides
biologiques (dont le lait, le liquide séminal, le liquide amniotique, l’humeur aqueuse de l’œil ou
encore le surfactant pulmonaire) (Halliwell and Gutteridge, 2008). Elle est aussi
particulièrement abondante dans les reins et en quantité moins importante dans le plasma.
Enfin, GPx6 est très proche dans sa structure avec GPx3 mais est seulement exprimée dans le
système olfactif.
Par ailleurs, GPx4 est une enzyme structurellement et fonctionnellement différente des
autres GPx car c’est une enzyme monomérique et peut directement réduire les hydroperoxydes

48
lipidiques membranaires encore présents dans la membrane. Il existe trois isoformes de la
GPx4 : une cytosolique, une mitochondriale et une nucléaire.
D’autres enzymes GPx (GPx5, GPx7 et GPx8) sont également connues mais ne sont pas
des sélénoprotéines.
3) Les glutathion réductases (GRD)
La glutathion réductase permet de recycler le GSH en réduisant GSSG, produisant deux
molécules de GSH (Raman and Berry, 2011).
C’est une flavoenzyme homodimérique ; chaque sous-unité contient du FAD au niveau
de son site actif. Le NADPH réduit d’abord le FAD en FADH2. FADH2 est ensuite une source
d’électrons sur un pont disulfide –S—S– de l’enzyme qui réduit à son tour le GSSG en GSH
(Halliwell and Gutteridge, 2008) (figure 5).
Figure 5 : Mécanisme d’action de la glutathion réductase (GRD)
La glutathion réductase permet de maintenir un ratio GSH/GSSG élevé dans les cellules ;
on estime aujourd’hui que plus de 90% du stock en glutathion est recyclé (Halliwell and
Gutteridge, 2008) (Raman and Berry, 2011).
4) Les catalases (CAT)
La catalase (CAT) est une enzyme dont la fonction principale est de catalyser la
décomposition par dismutation du peroxyde d’hydrogène en eau et en dioxygène (Powers and
Jackson, 2008) :
2 H2O2 H2O + O2
Elle n’utilise pas de cofacteurs enzymatiques et est composée de quatre sous-unités
contenant chacun un hème (Halliwell and Gutteridge, 2008).
Présente dans tous les organes, la catalase est particulièrement concentrée dans le foie.
Curieusement, le cerveau, le cœur et les muscles squelettiques ont les taux les moins élevés en
catalase. Dans la cellule, elle est majoritairement concentrée dans les peroxysomes où elle
régule la production de H2O2 issue des enzymes oxydases et empêche ainsi sa diffusion hors de
l’organite. La catalase est également présente à l’état libre dans le plasma.
La mitochondrie ne contient quasiment pas de catalase. Dans les hématies, la CAT
protège la membrane plasmique et les tissus traversés du peroxyde d’hydrogène produit par la
dismutation du radical superoxyde, lui-même issu des auto-oxydations de l’hémoglobine
(Halliwell and Gutteridge, 2008).
La catalase (CAT), la glutathion peroxydase (GPx), les peroxyréductases (Prx) et la
superoxyde dismutase (SOD) sont étroitement liées dans leurs actions en assurant la
destruction du peroxyde d’hydrogène produit par dismutation du radical superoxyde.

49
Cependant, la CAT possède une affinité plus faible pour H2O2 que la GPx (Powers and Jackson,
2008).
5) Les thiorédoxine réductases (TXNRD)
Les thiorédoxines oxydées sont réduites in vivo chez les animaux par les thiorédoxine
réductases (TXNRD), une sélénoprotéine utilisant également FAD et NADPH pour sa catalyse
(similarités avec la glutathion réductase) (Halliwell and Gutteridge, 2008) (Raman and Berry,
2011):
Trx—S2 + NADPH + H+ NADP+ + Trx—(SH)2
Ces enzymes sont capables de catalyser la réduction de nombreux substrats, mais
dépendent du NADPH comme donneur d’électrons, qui sont d’abord transférés au groupe FAD
fixé à la protéine, donnés ensuite aux groupe dithiol N-terminal d’une sous-unité puis alors au
groupe sélényl-sulfide de l’autre sous-unité qui réduit enfin à son tour la thiorédoxine (Raman
and Berry, 2011).
Trois thiorédoxine réductases ont été mises en évidence chez les mammifères: TXNRD1
(cytosolique), TXNRD2 (mitochondriale) et TXNRD3 (testiculaire). Ces enzymes contiennent
deux sites sensibles à l’état rédox en terminaisons N- et C- qui interviennent dans la
conformation de l’enzyme active.
Le système Trx/TXNRD est complètement dépendant du sélénium chez les
mammifères ; pour mémoire ce n’est pas le cas chez les plantes, les levures ou encore les
bactéries. L’importance de ce système Trx/TXNRD pour les organismes est soulignée par les
études in vivo ayant montré que l’inhibition de l’expression des gènes codant pour TXNRD1 et
TXNRD2 est létale chez l’embryon de souris (Raman and Berry, 2011).
6) Les peroxyrédoxines (Prx)
Depuis le milieu des années 1990, la famille des peroxyrédoxines est une nouvelle
famille d’enzymes antioxydantes qui attire l’attention de la recherche. Jusqu’ici, 6 isoformes ont
été identifiées (Prx1 à Prx6), toutes participant à l’élimination du peroxyde d’hydrogène ainsi
que d’autres peroxydes organiques (Yuan et al., 2004) (Halliwell and Gutteridge, 2008).
Les peroxyrédoxines sont des peroxydases homodimériques utilisant comme donneur
d’électrons la thiorédoxine. Leur activité catalytique leur vient essentiellement de la présence
de résidus cystéines réactifs (thiols –SH) situés en région N-terminale et retrouvés au niveau du
site actif dans la structure tertiaire et quaternaire (Halliwell and Gutteridge, 2008). Durant leur
catalyse, elles oxydent un groupement –SH en acide sulphénique –SOH. Pour les
peroxyrédoxines 2-cys (contenant deux cystéines actives), cet acide sulfénique réagit avec un
autre –SH de la protéine pour donner un pont disulfide –S—S– qui sera ensuite réduit par la
thiorédoxine (Yuan et al., 2004) (Halliwell and Gutteridge, 2008).
Prx1 à Prx5 sont des 2-cys peroxyrédoxines et Prx6 est une 1-cys peroxyrédoxine
(mécanisme catabolique non détaillé ici). Prx1, Prx2 et Prx6 sont cytosoliques, Prx4 est
présente dans le réticulum endoplasmique et dans le milieu extracellulaire, et Prx5 est
retrouvée dans la mitochondrie et dans les peroxysomes. Prx5 est d’identification assez récente
chez les mammifères, et attire l’intérêt des chercheurs sur son rôle dans l’équilibre redox
mitochondrial et la protection de la chaîne respiratoire mitochondriale contre le stress
oxydant ; en effet l’augmentation de l’expression de la Prx5 est corrélée à une augmentation de

50
l’activité catalytique de la catalase (CAT) et de la glutathion peroxydase (GPx1). Cette
constatation suggère que Prx5 pourrait également protéger la CAT et la GPx de leur inactivation
par un stress oxydant (Yuan et al., 2004).
Les données actuelles suggèrent que les peroxyrédoxines sont probablement le système
de destruction et de recyclage du peroxyde d’hydrogène le plus important chez les animaux,
plus encore que la catalase ou la glutathion peroxydase (Halliwell and Gutteridge, 2008). Elles
seraient également impliquées dans la régulation du phénomène d’apoptose, la prolifération
cellulaire, la différenciation et dans l’expression génétique.
7) Les paraoxonases (PON)
Les paraoxonases (PON) sont des enzymes estérases Ca2+-dépendantes, découvertes
pour leur capacité à dégrader des composés organophosphorés (dont le paraoxon, forme
activée du parathion par le système du cytochrome P450) (Halliwell and Gutteridge, 2008).
La famille de ces enzymes contient trois membres différents, PON-1, PON-2 et PON-3.
Bien que toutes ayant une activité lactonase, ce sont des enzymes dont les fonctions ainsi que
leur implication au sein des tissus biologiques doivent être encore largement étudiées
(Précourt, 2011).
PON-1 est aussi fortement exprimée dans le foie et le sang, et son expression génique a
également été identifiée dans d’autres tissus comme le rein, le poumon et l’intestin. Le foie est
probablement le plus important producteur de PON-1, et celle-ci se retrouve associée aux
particules de HDL (High Density Lipoproteins) dans le sang (Précourt, 2011).
PON-1 trouve son potentiel antioxydant dans l’hydrolyse des lipides oxydés
biologiquement actifs générés dans les membranes des cellules endothéliales et dans les
lipoprotéines de faible densité (LDL, Low Density Lipoproteins) (Motta et al., 2009).
PON-2 est d’identification plus récente que PON-1 et moins connue (Précourt, 2011).
C’est une enzyme à l’activité lactonase probablement ubiquitaire, et retrouvée dans tous les
tissus sondés par la science dont les poumons, le foie, le cœur et les intestins. Néanmoins,
contrairement à PON-1, elle n’a pas d’activité paraoxonase, très peu d’activité estérase et n’est
pas présente dans les lipoprotéines. De plus, sa localisation est la membrane plasmique des
cellules, ce qui suggère une implication différente à PON-1. On suppose que la PON-2
interviendrait dans l’équilibre redox et la protection du réticulum endoplasmique (essentiel à
l’acquisition de la structure tertiaire et quaternaire des protéines) (Précourt, 2011).
Enfin, PON-3 est semblable à PON-1. Elle est également synthétisée par le foie et est
aussi fortement liée aux HDL dans la circulation (Précourt, 2011). C’est une protéine de 39kD
qui a une qu’une très faible activité paraoxonase et arylestérase et contribuerait de manière
moins efficace que PON-1 à la protection contre l’oxydation.
Si ces enzymes PON sont assez bien étudiées chez l’homme et notamment dans la
physiopathologie de l’athérosclérose (Précourt, 2011), il semble qu’elle n’occupe pas une place
prépondérante dans les études qui nous concernent ici.

51
C) Antioxydants non-enzymatiques
1) Antioxydants non-enzymatiques d’origine endogène
1.i Glutathion (GSH/GSSG)
Le glutathion ou gamma-L-glutamyl-L-cystéine est souvent considéré comme le
principal antioxydant non enzymatique intracellulaire (Valko, 2011). Outre la défense
antioxydante, il est impliqué dans de nombreuses fonctions comme la régulation de l’apoptose
et de l’immunité ainsi que des mécanismes de détoxification, mais aussi la modulation de la
prolifération cellulaire (Alcaraz et al., 2013) (Taha, 2011). Ensemble, le GSH et le TXN sont des
antioxydants intracellulaires centraux dans l’équilibre de l’état d’oxydation des cystéines, des
méthionines et des sélénocystéines et donc de la fonctionnalité des nombreuses protéines
redox-sensibles (Raman and Berry, 2011).
C’est un tripeptide formé de glycine, cystéine et de glutamate ; il appartient au groupe
des thiols parmi les plus abondantes dans la cellule, présent en concentrations millimolaires
(Raman and Berry, 2011).
Ce tripeptique possède une forme oxydée (GSSG) et une forme réduite (GSH). Ces deux
états forment le couple redox GSSG/GSH. La capacité antioxydante du glutathion réside dans la
présence d’un groupement thiol (–SH) présent sur la cystéine réduite (figure 6). La double
oxydation des cystéines thiols –SH lie deux molécules de GSH pour former le glutathion
disulfide GSSG (Halliwell and Gutteridge, 2008).
Figure 6 : États d’oxydation du glutathion
Cette réaction peut être spontanée en présence de composés électrophiles ou peut être
catalysée par des enzymes qui utilise le GSH comme donneur d’électrons, comme la glutathion
peroxydase (GPx), la glutarédoxine et la glutathion-S-transférase (Raman and Berry, 2011).
Dans les cellules, le recyclage du GSSG en GSH est assuré par la glutathion réductase (GRD).

52
Seul, le GSH réagit très bien avec de nombreux ERO comme les radicaux HO°, RO°, RO2°,
les radicaux centrés sur le carbone R°, ClO-, ONOO-, et l’oxygène singulet. La réaction avec les
radicaux libres forme le radical thyle GS° pouvant à son tour former de nouveaux radicaux
suivant de nombreuses réactions (Raman and Berry, 2011).
1.ii La thiorédoxine (Trx)
Les thiorédoxines sont des polypeptides d’une masse moléculaire d’approximativement
12 kDa largement distribués dans toutes les cellules des mammifères. Trx1, présente dans le
cytosol, et Trx2, dans la mitochondrie, ont été décrites (Halliwell and Gutteridge, 2008). Dans
les cellules, elles sont présentes en quantités similaires à légèrement inférieures à celles du
glutathion GSH (Raman and Berry, 2011).
Libre, c’est est un cofacteur enzymatique donneur d’électrons (réducteur) pour de
nombreuses enzymes comme la ribonucléotide réductase, les méthionine sulfoxyde
réductases (ces dernières sont essentielles à la réparation des dégâts oxydatifs sur les résidus
méthionine des protéines) et les peroxyrédoxines déjà présentées (Halliwell and Gutteridge,
2008).
Cependant, le motif thiorédoxine peut aussi être intégré dans la structure des enzymes,
comme pour la glutathion peroxydase (GPx). En effet, de nombreuses sélénoprotéines ont
également un fragment thiorédoxine dans leur structure secondaire et tertiaire ; il y a d’autres
« thioredoxine-like proteins » qui pourraient également agir en parallèle pour apporter du
substrat aux sélénoprotéines utilisant Trx (Raman and Berry, 2011).
Comme le glutathion, les thiorédoxines trouvent leurs qualités redox dans la présence de
groupements thiols (Halliwell and Gutteridge, 2008) (Raman and Berry, 2011). Leur potentiel
rédox est suffisemment négatif pour donner un fort potentiel réducteur à la molécule. Elles
contiennent deux groupes réduits –SH qui forment un pont disulfide à l’état oxydé (-S—S-):
Trx—(SH)2 + protéine—S2 Trx—S2 + protéine–(SH)2
À travers l’oxydation du site actif dithiol en un pont disulfide, les Trx médient ainsi
l’échange thiol-disulfide entre les protéines (comme le glutathion) ; elles contrôlent ainsi l’état
redox des cystéines dans les protéines cibles (Raman and Berry, 2011), et potentiellement
également leur activité.
Autant le GSH et la Trx sont des antioxydants intracellulaires centraux dans l’équilibre
de l’état d’oxydation et donc de la fonctionnalité des protéines contenant de la cystéine, de la
méthionine ou des séléno-cystéine, de manière directe ou indirecte comme cofacteur
enzymatique (Raman and Berry, 2011). La Trx pourrait également avoir d’autres fonctions
biologiques : il a notamment été montré qu’elle stimule également la croissance de plusieurs
types cellulaires en culture in vitro (Halliwell and Gutteridge, 2008).
1.iii Acide urique
L’acide urique est le produit de dégradation des composés puriques comme la xanthine
et l’hypoxanthine (voir partie I) C) 2)) (Villasante et al., 2010).
Aux pH sanguins proches de la neutralité, l’acide urique est sous forme anionique ion
urate (pKa aux alentours de 5.4) (Halliwell and Gutteridge, 2008). Il réagit avec plusieurs
espèces réactives aux potentialités oxydantes assez fortes tels que ROO°, HO°, ONOO-, NO2° et

53
l’oxygène singulet. Cette réactivité donne naissance au radical urate. Ce radical est relativement
stable grâce à la délocalisation de l’électron célibataire sur son noyau purine.
Ce radical urate est très bien réduit par l’acide ascorbique (vitamine C), intégrant bien
l’urate au sein de la défense antioxydante (Halliwell and Gutteridge, 2008). L’urate protège
également très bien les protéines de la nitration par le peroxynitrite ONOO- ; la réaction entre
l’urate et le peroxynitrite est complexe et peut être à l’origine de la naissance de plusieurs
espèces nitrosées pouvant se décomposer en NO° (Halliwell and Gutteridge, 2008) (Villasante
et al., 2010).
L’urate se fixe également très bien aux métaux de transition. Cette liaison diminue les
potentialités catalytiques de ces derniers. Cette propriété est très importante dans la protection
des membranes cellulaires contre la chaîne réactionnelle de peroxydation lipidiques (Halliwell
and Gutteridge, 2008).
L’augmentation rapide de l’acide urique durant l’effort est expliquée par la perte
d’adénine au travers de la dégradation de l’AMP en IMP et finalement en acide urique (Marlin et
al., 2002).
Pour mémoire, l’urate est normalement dégradée chez les mammifères par l’urate
oxydase, contenue dans les peroxysomes sauf chez l’homme et plusieurs primates, où l’urate
s’accumule à de plus fortes concentrations dans le sang et les fluides extracellulaires, voire à
cristalliser chez ces espèces (« goutte ») (Halliwell and Gutteridge, 2008).
C’est pour cette raison que sa potentialité antioxydante ne sera pas la même entre les
primates et les autres mammifères, notamment chez nos sujets sportifs équins.
1.iv Bilirubine
La bilirubine est le produit final de la dégradation de l’hème de plusieurs protéines
hémiques d’intérêt biologique comme l’hémoglobine des hématies et la myoglobine des
muscles. Transportée dans le sang, la bilirubine est conjuguée dans le foie et excrétée dans la
bile où elle quitte l’organisme. L’hème oxydase (HO) est une enzyme retrouvée dans le
réticulum endoplasmique, catalysant la dégradation de l’hème en biliverdine et libérant du fer à
l’état ionique Fe2+ et du monoxyde de carbone. La biliverdine est ensuite transformée en
bilirubine grâce à la biliverdine réductase présente dans le cytosol (Halliwell and Gutteridge,
2008) (Paredi et al., 2002).
Faiblement hydrosoble, la bilirubine est fortement liée aux protéines et lipoprotéines
plasmatiques et potentialise la défense antioxydante sanguine (notamment avec l’albumine)
(Halliwell and Gutteridge, 2008).
In vitro, on a montré que la bilirubine avait de puissantes propriétés antioxydantes
envers plusieurs espèces réactives comme le peroxynytrite, les radicaux peroxyles et alkoxyles,
ainsi que l’oxygène singulet (Halliwell and Gutteridge, 2008) (Paredi et al., 2002). Elle trouve
donc toute son expression dans la protection des membranes cellulaires contre la peroxydation
lipidique (notamment des cellules sanguines) et des protéines plasmatiques ; certains auteurs
pensent que la bilirubine serait un antioxydant efficace de l’oxydation protéique médiée par le
peroxynitrite et pourrait être plus efficace encore que la vitamine E dans la prévention de la
peroxydation lipidique (Paredi et al., 2002). La bilirubine est reconvertie en biliverdine par

54
certains ERO, qui est à nouveau réduite par la biliverdine réductase (Halliwell and Gutteridge,
2008).
Pour mémoire, la bilirubine est une molécule photosensibilisante qui peut entrer dans la
transformation du dioxygène triplet en dioxygène singulet, et devenir alors toxique (par
exemple chez un individu ictérique, dépourvu de poils et bien exposé au soleil, comme un
nourrisson chez l’homme) (Halliwell and Gutteridge, 2008). Cette particularité cependant est
peu encline à poser problème pour les animaux qui nous intéressent, en l’occurrence ici
l’espèce équine.
De plus amples recherches sont encore nécessaires pour définir l’implication de la
bilirubine dans les défenses antioxydantes in vivo aux concentrations physiologiques (Halliwell
and Gutteridge, 2008).
1.v Protéines régulatrices, de transport et de séquestration des métaux de
transition
Le cas du fer
La transferrine est une glycoprotéine de 79 kD synthétisée par le foie pouvant fixer
deux Fe3+ à pH 7.4 avec une affinité très forte à ce pH. Chez les mammifères en bonne santé, on
considère que 20 à 30% de la transferrine circulant est complexée avec des ions fer. De plus,
son turnover est très important, assurant le transport du fer efficacement aux cellules cibles. Le
plasma des mammifères a donc une forte capacité de fixation du fer libre, dont la concentration
est proche de zéro dans les conditions physiologiques (Halliwell and Gutteridge, 2008).
La ferritine est la protéine majoritaire de stockage intracellulaire du fer. C’est une
protéine en forme de coquille composée de 24 sous-unités ayant chacune une masse de 20 kDa.
Elle peut contenir jusqu’à 4 500 ions fer ; d’autres ions métalliques peuvent être également
présents, comme des ions cuivre. Si la majorité de la ferritine se trouve dans le cytosol de la
cellule, une partie est également présente dans les mitochondries (Halliwell and Gutteridge,
2008). En effet, il est aussi important de maintenir l’homéostasie du fer dans ces organites que
dans le cytosol car toute perturbation et libération de métaux de transition libres pourrait
entraîner une surproduction d’espèces radicalaires et alors provoquer un dysfonctionnement
mitochondrial (Wu et al., 2011) (Halliwell and Gutteridge, 2008).
Le fer entre dans la ferritine sous la forme Fe2+ puis est oxydé en Fe3+ ; le Fe3+ est ensuite
transformé en oxydes de fer, insolubles. La conformation de la cellule en coquille autour du fer
et l’insolubilité de cette forme de stockage empêche la réaction du fer avec les ERO (Halliwell
and Gutteridge, 2008) (Paredi et al., 2002). Il a également été démontré que, dans le cas de la
dégradation de l’hémoglobine, la libération de l’ion fer par l’hème oxydase-1 (HO-1) pourrait
induire la synthèse de ferritine, et que les facteurs d’activation de HO-1 pouvaient également
activer la synthèse de l’ARNm de la ferritine (Paredi et al., 2002).
Au sein de la cellule, la ferritine est clivée dans les lysosomes sous une autre forme
insoluble, l’hémosidérine. Ce pigment insoluble contient 25-30% du fer contenu dans
l’organisme. Il est localisé essentiellement dans les macrophages et trouve également sa
capacité antioxydante dans son insolubilité. L’hémosidérine est une forme de stockage au
même titre que la transferrine (Halliwell and Gutteridge, 2008).

55
Si le sang paraît être un tissu assez bien équipé en ce qui concerne les chélateurs des
métaux de transition, il n’en est pas de même dans tous les fluides extracellulaires et
notamment le liquide broncho-alvéolaire, plus pauvre en protéines. Toutefois, la lactoferrine
est également une protéine chélatrice du fer et est retrouvée dans plusieurs sécrétions comme
la salive, le mucus vaginal, le liquide séminal, les larmes, la bile, les sécrétions nasales, le lait …
mais aussi sécrétée par les neutrophiles durant l’inflammation. Elle y joue plusieurs rôles et on
pense qu’elle pourrait également limiter l’amplification du stress oxydant lié au « burst »
oxydatif en séquestrant le fer libre lors des processus inflammatoires (Halliwell and Gutteridge,
2008).
Brièvement, pour maintenir l’homéostasie du fer (sous forme Fe2+ surtout ), la cellule
utilise des protéines de régulation du fer (comme IRP1 et IRP2, pour « iron-regulatory protéines
1 et 2 ») qui contrôlent l’expression des gènes codant pour les protéines contenant du fer pour
leur fonctionnement (par exemple dans la mitochondrie l’aconitase ou la succinate
déshydrogénase) ou son transport (tranferrine et son récepteur membranaire) en se liant à un
élément nommé IRE (pour « iron-responsive element ») qui cible ensuite les ARNms (Wu et al.,
2011).
Les métaux de transition sont l’objet d’une homéostasie contrôlée et leur concentration
est limitée dans la cellule et dans le milieu extracellulaire. Des souris transgéniques déficitaires
en transferrine ont ainsi des stocks extracellulaires et intracellulaires plus élevés que des
souris normales et meurent très rapidement (Simpson et al., 1992).
Ainsi, la séquestration sous forme non catalytique du fer est aussi importante que des
molécules comme la SOD pour la survie des organismes.
Le cas du cuivre
Une fois capté par les intestins, le cuivre voyage des intestins vers les cellules cibles
principalement lié aux albumines et à une protéine de transport plus spécifique, la
transcuprine. Le foie intègre ensuite ce métal en le fixant à la céruloplasmine, protéine de
132 kDa, qui est ensuite sécrétée dans le plasma. Capable de fixer six à sept ions Cu2+, la
céruloplasmine a un rôle analogue à la transferrine dans le transport du cuivre, insoluble sous
cette forme. Dans les cellules, les enzymes utilisant le cuivre comme cofacteur de catalyse et les
métallothionéines (MT) assurent la majeure partie de la séquestration du cuivre intracellulaire
(Halliwell and Gutteridge, 2008).
Les méthallothionéines (MT) sont des protéines de faible poids moléculaire (environ
6.5 kDa), présentes dans tous les tissus sous différents isoformes qui ne seront pas détaillés ici.
Elles contiennent 20 à 30% de cystéines dans leur chaîne primaire et sont de fait d’importants
représentants du pool cellulaire en thiols. Les MT peuvent chacune fixer cinq à sept ions
métalliques comme les ions cuivre mais aussi des ions zinc, mercure, cadmium ou encore
argent (Halliwell and Gutteridge, 2008).
1.vi Les protéines découplantes
Les protéines découplantes ont été d’abord découvertes dans le tissu adipeux brun avec
la mise en évidence des protéines UCP-1 (pour « uncoupling protéine-1 »). Dans ce tissu, leur
présence dans la membrane mitochondriale interne permet le passage des protons vers
l’espace intermembranaire et ainsi annule le gradient protonique nécessaire au
fonctionnement des ATP-synthase ; toute l’énergie du transfert des électrons sur la chaîne

56
mitochondriale ne pouvant plus servir à la formation d’ATP, celle-ci est alors libérée sous forme
de chaleur.
Depuis, des protéines homologues à UCP1 ont été mises en évidence dans d’autres
tissus, comme UCP3 dans les muscles striés, en concentration moins importantes cependant. Il
a alors été proposé que ces dernières puissent agir comme des antioxydants (Valko, 2011)
(Halliwell and Gutteridge, 2008). En effet, en permettant la fuite de protons hors de la matrice
mitochondriale, elles ralentiraient le transfert électronique et par la même limiteraient en
conséquence leur fuite et la formation du radical superoxyde depuis la chaîne de
phosphorylation oxydative.
De plus, il semblerait que certains produits de la peroxydation lipidique (comme
l’alkénal 4-HNE dont nous reparlerons plus loin) puissent activer les protéines découplantes
UCP (Valko, 2011) (Hellsten et al., 2007).
1.vii Le monoxyde d’azote NO°
La capacité antioxydante du monoxyde d’azote NO° vient de sa forte affinité
réactionnelle avec les radicaux libres. Ainsi, cette propriété est bénéfique in vivo car elle permet
l’élimination des radicaux peroxyles : ainsi, le NO° est un puissant inhibiteur de la peroxydation
lipidique (Gutteridge and Halliwell, 2000).
On ajoutera également que le monoxyde d’azote NO° diminue également les propriétés
pro-oxydantes des protéines de l’hème. En effet, son affinité et sa capacité à se lier au fer
présent dans l’hème inhibe la formation de puissants radicaux tel qu’HO° (réaction de Fenton)
(Halliwell and Gutteridge, 2008).
Cet effet antioxydant reste relatif car la genèse d’ERO encore plus réactifs comme le
peroxynitrite modère cette dénomination.
1.viii Autres antioxydants non-enzymatiques endogènes
Il existe de nombreuses autres molécules endogènes aux propriétés antioxydantes dans
le sens qu’elles réagissent avec les ERO en produisant des composés moins toxiques. On
recense notamment in vivo l’acide lipoïque, toutes les protéines contenant des groupements
thiols, l’albumine, le glucose, le mannitol, -kéto-acides, l’acide hyaluronique, la glucosamine
(Halliwell and Gutteridge, 2008) (Villasante et al., 2010) …
Ainsi, le glucose et mannitol inactivent le radical hydroxyle HO° et pourraient ainsi
jouer un rôle protecteur de la glycation des protéines. Cette réactivité mérite d’être soulignée
dans le cadre de l’exercice physique et de l’évolution des potentialités antioxydantes du sang
(Halliwell and Gutteridge, 2008).
L’albumine est une protéine plasmatique chargée négativement pouvant fixer les ions
cuivre et fer présents dans le sang. Son importance est également liée à sa forte concentration
dans le sang. C’est aussi une « protéine sacrifice » qui est rapidement dégradée par des ERO
comme l’ion hypochlorite ClO- ou l’ion peroxynitrite ONOO-. Elle constitue une grosse partie du
pool thiols du sang et est également très liée à la bilirubine, potentialisant ses effets
antioxydants dans le sang (Villasante et al., 2010).
L’important turnover de l’albumine du sang pourrait être un élément en faveur de son
action antioxydante dans le sang (Halliwell and Gutteridge, 2008).

57
L’acide lipoïque, ou acide 1,2-dithiolane-3-pentanoïque, est un des cofacteurs essentiels
de la pyruvate deshydrogénase et de l’-cétoglutarate déshydrogénase qui interviennent dans
le cycle de Krebs. L’acide lipoïque contient des groupements soufrés définissant un état oxydé
(disulfide –S—S–) et un état réduit (dithiol, i.e 2 groupements –SH se faisant face) à la molécule.
Ces groupements lui confèrent ainsi un grand pouvoir anti-oxydant envers RO2°, HOCl, CO₃°,
NO2°, HO° et ONOO- (Halliwell and Gutteridge, 2008). Des études suggèrent également que
l’acide lipoïque pourrait favoriser la diminution de l’acide lactique après un effort chez le cheval
en relançant son métabolisme et stimuler la production de « heat-shock protéins » dans le
muscle (Zuo, 2002) (Kinnunen et al., 2009).
Les-kéto-acides, comme le pyruvate ou le -kétoglutarate (intermédiaire du cycle de
Krebs), ont une bonne réactivité avec H2O2, ONOO- et HOCl, confirmant ainsi leur rôle
antioxydant dans le milieu intracellulaire et notamment comme deuxième barrière à la toxicité
des peroxysomes (déjà contrôlée par la CAT) (Halliwell and Gutteridge, 2008).
La glucosamine, et particulièrement ses composés sulfatés possèdent des propriétés
antioxydantes en réagissant avec des ERO. Ils assurent notamment la protection des
protéoglycanes et des chondrocytes de l’articulation. Il semblerait également que la
glucosamine maintiendrait les niveaux de glutathion à l’état réduit (GSH) au sein de
l’articulation (Mendis et al., 2008).
L’acide hyaluronique assure un pouvoir antioxydant dans de nombreux systèmes, en
particulier au sein de l’articulation contre HO° (Kalaci and al., 2007) (Villasante et al., 2010)
(Halliwell and Gutteridge, 2008). Kalaci and al. (2007) rappelle que l’acide hyaluronique et un
de ses composants, l’acide D-glucuronique, ont le pouvoir de réduire la concentration en ERO
d’un milieu dans un modèle in vitro. On a également montré que l’acide hyaluronique avait
aussi la capacité de diminuer la synthèse d’IL-1 au sein d’une culture de chondrocytes bovins
(pouvoir anti-inflammatoire). Enfin, l’acide hyaluronique a le pouvoir de réduire les taux en
NO° dans une articulation atteinte d’ostéoarthrose (Kalaci and al., 2007).
Le haut poids moléculaire de l’acide hyaluronique joue beaucoup dans ses capacités
antioxydantes. Comme pour l’albumine, ces molécules doivent posséder un turnover important
pour rester des antioxydants, ce qui n’est pas forcément le cas en situation pathologique
d’ostéoarthrose par exemple. L’acide hyaluronique est dépolymérisée lors de son oxydation par
les ERO, diminuant la viscosité de la synovie (Halliwell and Gutteridge, 2008).
Enfin l’haptoglobine est une protéine de la phase aiguë de l’inflammation. Elle a la
capacité de fixer l’hémoglobine et l’hème libre, diminuant considérablement les activités pro-
oxydantes liées au fer contenu dans l’hème. On suggère actuellement que l’haptoglobine
possède une activité chaperonne en protégeant également d’autres protéines extracellulaires
contre les dégâts du stress oxydant (Halliwell and Gutteridge, 2008).
2) Antioxydants non-enzymatiques d’origine nutritionnelle
2.i La vitamine E
Le terme de « vitamine E » renvoie surtout à huit composés (de la famille des
tocophérols et des tocotriénols). Chacun de ces composés sont constitués d’une tête
aromatique chromanol avec une queue latérale isoprénoïde (saturée chez les tocophérols et
insaturée chez les tocotriénols). Les formes etdiffèrent selon la place et selon le
nombre de résidus méthyles sur le noyau aromatique (Johnson, 2006) (figure 7).

58
Figure 7 : Formule de l’tocophérol. Le pôle hydrophile est vers la gauche, le pôle lipophile vers la droite.
Dans les tissus des mammifères, la plus grande partie de la vitamine E se trouve dans la
bicouche phospholipidique de la membrane plasmatique ; la tête chromanol hydrophile est
localisée du côté extramembranaire et la queue est contenue au sein des autres acides gras des
phospholipides membranaires (Halliwell and Gutteridge, 2008). Pour illustration, la
composition membranaire de la vitamine E est comprise chez les mammifères entre 1/1000
([Vitamine E]/[Lipides membranaire]) dans la membrane des hématies, et 1/2000 dans les
membranes mitochondriales ou encore moins de 1/3000 dans les autres tissus.
La vitamine E est un bon protecteur des lipides contre la peroxydation lipidique. En
effet, elle réagit très rapidement avec les radicaux peroxyles lipidiques (LOO°) et coupe ainsi les
chaînes de propagation de la réaction (Halliwell and Gutteridge, 2008):
LOO° + TocH LOOH + Toc°
Le radical Toc° peut également réagir avec un autre LOO° et former des composés
non radicalaires et non toxiques qui pourront ensuite être dégradés par la cellule (Halliwell and
Gutteridge, 2008):
LOO° + Toc° TocOOL.
Ainsi la combinaison de ces deux réactions nous laisse conclure qu’une molécule d’-
tocophérol peut permettre la destruction de deux radicaux peroxyles. De plus, le radical -
tocophéryl est relativement peu réactif pour les cellules (Clarkson and Thompson, 2000). On
considère d’ailleurs souvent la vitamine E comme l’antioxydant liposoluble le plus important
dans l’organisme (Piercy et al., 2001) (Clarkson and Thompson, 2000) (Halliwell and
Gutteridge, 2008).
Il existe une assez bonne corrélation entre les concentrations en tocophérol dans le
muscle, le cœur, le foie avec celle dans le plasma ; elles sont très finement contrôlées,
notamment par les protéines qui lui sont associées, comme TRP (« tocophérol transfert
protein »).
Enfin, le recyclage (réduction) de la vitamine E oxydée par l’oxydation de l’acide
ascorbique est le mécanisme prépondérant in vivo (voir plus loin) (Halliwell and Gutteridge,
2008).
2.ii La vitamine C
La vitamine C ou acide L-ascorbique est un antioxydant hydrosoluble souvent considéré
comme le principal antioxydant des fluides extracellulaires. Elle possède une très bonne
réactivité envers plusieurs ERO comme le radical hydroxyle HO°, superoxyde O2°-, le dioxyde
d’azote NO2°, l’hypochlorite HOCl, l’oxygène singulet, l’ozone ou encore le trioxyde et le

59
tétroxyde d’azote. Elle est bien présente dans le sang mais aussi dans le surfactant pulmonaire
(Carr and Frei, 1999).
C’est un diacide contenant deux groupes hydroxyles pouvant s’ioniser selon les
variations du pH (figure 8). Or seule la forme anionique AscH- possède une action antioxydante
; cette forme est heureusement la plus abondante aux pH des systèmes biologiques (Valko,
2011).
Figure 8: Formes de la vitamine C à différent pH et sa réaction avec les radicaux libres. D’après (Valko,
2011).
En plus de réagir directement avec les ERO, la vitamine C permet la régénération
(réduction) d’autres antioxydants, tel que l’-tocophérol (vitamine E), le glutathion (sous forme
radicalaire GS°) et les autres protéines thiols (sous forme radicalaire thyles), l’urate (sous
forme radicalaire), et les -carotènes (sous forme radicalaire), en produisant le radical
ascorbyle (Carr and Frei, 1999).
Une des fonctions antioxydantes de la vitamine C est la régénération de la vitamine E, et
plus particulièrement la réaction d’oxydoréduction entre l’anion ascorbate AscH- et le radical α-
tocophéryle α-T-O° (Valko, 2011):
α-T-O° + AscH- Asc°- + α-T-OH
Cette réaction met en évidence que la reconstitution de l’α-tocophérol (Vitamine E)
génère aussi le radical ascorbyle Asc°- . L’ascorbate autant que son radical ascorbyle ont un
potentiel redox très faible, faisant de la vitamine C un excellent antioxydant.
On remarquera également que l’interaction entre l’ascorbate hydrophile et le radical -
tocophéryle pour régénérer l’-tocophérol permet aussi de déplacer les radicaux de la phase
lipidique (membrane plasmique) au cytosol ou au milieu extracellulaire, limitant la
peroxydation lipidique (Carr and Frei, 1999). Cette caractéristique jumelée à sa réactivité avec
les radicaux alkoxyles et peroxyles caractérise également le fort pouvoir protecteur de la
vitamine C sur les membranes cellulaires lipidiques et les lipoprotéines.
Le radical ascorbyle disparaît par dismutation en acide déhydroascorbique (DHA) et en
ascorbate. Il peut également être réduit par l’ascorbate ou encore l’acide lipoïque, mais aussi
par voie enzymatique avec la thioredoxine réductase ou la glutarédoxine. De même le radical
ascorbyle peut être réduit par la semihydroascorbate réductase en présence de NADPH (Carr
and Frei, 1999).

60
Enfin une autre importante fonction biologique de la vitamine C concerne son
interaction avec les métaux de transition, comme l’ion cuivre et l’ion fer, utilisés comme co-
substrat des oxygénases et des hydroxylases impliquées dans la synthèse du procollagène, de la
carnitine ou encore de certains neurotransmetteurs. Le rôle de l’ascorbate est de maintenir ces
métaux à l’état réduit pour assurer l’activité optimale de ces enzymes (Carr and Frei, 1999).
2.iii Les caroténoïdes
Les caroténoïdes forment un groupe de pigments colorés retrouvés et synthétisés dans
de nombreuses cellules végétales. C’est une très grande famille. Les caroténoïdes ont de
longues chaînes aliphatiques alternant simples et doubles liaisons permettant une
délocalisation des électrons le long de cette chaîne. On distingue classiquement les
caroténoïdes à chaîne oxygénée (comme la lutéine, la zéaxanthine ou encore la cryptoxanthine)
et ceux à chaîne non oxygénée (comme le lycopène). Les groupes terminaux ()
déterminent le nom final donné au caroténoïde en question. Certains caroténoïdes (surtout les
-carotènes) sont des précurseurs de la vitamine A (aussi appelée rétinol), présente au niveau
des yeux et de la peau (Halliwell and Gutteridge, 2008).
L’oxygène singulet, les radicaux peroxyles (réactions A, A’ et A’’) et alkyles sont les
cibles préférentielles des caroténoïdes (Car) (Clarkson and Thompson, 2000) (Zuo, 2002):
(A) Car + H+ + ROO° Car°+ + ROOH (transfert électronique)
Ou
(A’) CarH + ROO° Car°+ + ROOH (déshydrogénation)
Ou
(A’’) Car + ROO° [ROO—Car]° (addition)
On remarquera que les radicaux formés peuvent alors réagir avec d’autres ROO° pour
former des produits non toxiques. D’autres radicaux comme les radicaux thyles RS° ou HO°
peuvent également réagir avec les caroténoïdes (Clarkson and Thompson, 2000).
La délocalisation électronique sur la chaîne aliphatique stabilise les radicaux formés et
rend la réaction avec O2 très lente (aboutissant à la formation de nouveaux radicaux peroxyles).
In vivo, les capacités antioxydantes des caroténoïdes sont donc bien supérieures à leur capacité
pro-oxydante, notamment en présence d’un foyer de peroxydation lipidique (Halliwell and
Gutteridge, 2008).
2.iv Les polyphénols
Les polyphénols sont un vaste groupe de molécules possédant plus de deux groupes
aromatiques –OH et au moins un noyau aromatique benzène. Les polyphénols peuvent inhiber
la peroxydation lipidique et détruisent certaines espèces réactives telles que HO°et NO2° par
abstraction d’un de leurs atomes d’hydrogène. Certains peuvent réagir directement avec l’ion
superoxyde O2°-. On a longtemps pensé qu’ils pouvaient être de bon chélateurs des métaux de
transition, cela n’est peut-être pas vrai in vivo (Halliwell and Gutteridge, 2008).

61
3) Les cofacteurs minéraux
Certaines enzymes antioxydantes de l’organisme ne sont fonctionnelles que si
l’oligoélément est présent : le zinc (dans la SOD1), le manganèse (dans la SOD2), le fer (dans la
catalase), le cuivre (dans la SOD1) ou encore le sélénium (dans la glutathion peroxydase).
On reviendra sur deux d’entre eux dans les lignes qui suivent, le zinc et le sélénium.
3.i Le zinc
En plus de son rôle de cofacteur dans la SOD1, on suggère souvent que le zinc (qui n’est
pas un élément de transition) pourrait agir comme antioxydant en déplaçant le fer
(particulièrement ionique) des sites de fixation, inhibant alors la synthèse d’espèces réactives
rendue possible ou facilitée par sa présence (Halliwell and Gutteridge, 2008) .
3.iii Le sélénium
Le sélénium est un important composant de la glutathion peroxydase (GPx) et des
peroxyrédoxines. En effet, ces deux enzymes possèdent dans leur chaîne primaire un acide
aminé particulier : la L-sélénocystéine (21ième acide aminée, molécule analogue à la cystéine
avec une chaîne contenant l’atome de soufre remplacé par un atome de sélénium) qui en font
des sélénoprotéines (Raman and Berry, 2011).
Toutes les sélénoprotéines caractérisées par les scientifiques qui fonctionnent comme
des enzymes sont des oxydoréductases qui catalysent les réactions d’oxydoréduction
impliquant des groupements thiols et qui contiennent du sélénium dans leur site actif (Raman
and Berry, 2011).
Dans le cas des chevaux on précisera simplement que c’est un minéral largement
apporté par l’alimentation qui peut être déficiente chez certains animaux suivant la partie du
monde où ils pâturent et l’absence de supplémentation éventuelle (Haggett et al., 2010).
IV) Dégâts liés à la présence des ERO
A) Stress oxydant et altérations biochimiques à l’échelle moléculaire
1) Stress oxydant et altérations des lipides
1.i Peroxydation des acides gras polyinsaturés membranaires
Les phospholipides des membranes cellulaires sont particulièrement sensibles au stress
oxydant étant donné leur forte teneur en acides gras polyinsaturés (riches en double liaison
>C=C<) et la fréquence du motif -CH=CH—CH2—CH=CH- qui les rendent particulièrement
sensibles à l’attaque des ERO (Halliwell and Gutteridge, 2008). Les acides gras monoinsaturés
sont également présents dans les membranes mais sont moins réactifs avec les ERO, donc
moins concernés par le stress oxydant.
La peroxydation lipidique entraînée par l’action des radicaux libres est une réaction
spontanée en chaîne qui comporte trois étapes : l’initiation, la propagation et la
terminaison (Venkataraman et al., 2004) (Halliwell and Gutteridge, 2008). Une illustration
simple figure ci-dessous.

62
L—H + oxydant° L° + Oxydant—H (initiation)
L° + O2 LOO° (propagation 1)
LOO° + L—H LOOH + L° (propagation 2)
L° + L° produits non radicalaires (terminaison 1)
L° + LOO° produits non radicalaires (terminaison 2)
L—H représente un lipide insaturé, généralement un acide gras polyinsaturé. L° représente un
radical lipidique centré sur le carbone. Oxydant ° représente évidemment un radical aux
propriétés oxydatives sur L—H.
Phase d’initiation
Dans la réaction d’initiation, les auteurs rapportent qu’un radical ne peut être oxydant
pour un acide gras polyinsaturé que si son potentiel E° est supérieur à +600 mV (valeur
moyenne en condition physiologique in vitro) (Venkataraman et al., 2004). On considère
généralement que le radical hydroxyle HO°, les radicaux alkoxyles RO° et peroxyles ROO° (les
acteurs principaux de la propagation de la peroxydation lipidique), HOO°, et NO2° font parties
des espèces initiatrices majeures chez les mammifères.
Rappelons également que les métaux de transition libres comme le fer et le cuivre sont
considérés comme des facteurs d’initiation majeurs de la peroxydation lipidique in vivo (Wilcox
et al., 1993).
Phase de propagation
C’est la phase de propagation qui fait de la peroxydation lipidique une « chaîne
réactionnelle ». En effet, après la réaction d’initiation, les radicaux peroxyles et alkoxyles issus
des acides gras contribuent eux-mêmes à la chaîne de propagation (Jairam et al., 2012) (figure
9).
Figure 9 : Exemple classique de propagation d’une chaîne de peroxydation lipidique
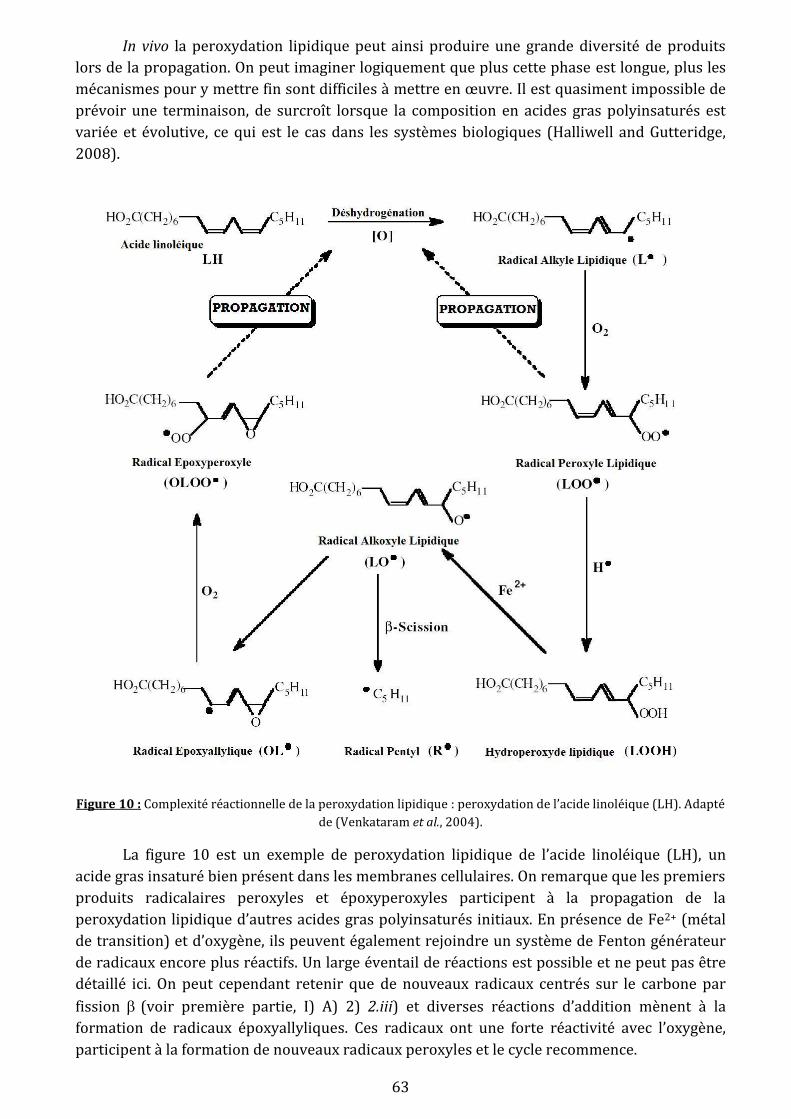
63
In vivo la peroxydation lipidique peut ainsi produire une grande diversité de produits
lors de la propagation. On peut imaginer logiquement que plus cette phase est longue, plus les
mécanismes pour y mettre fin sont difficiles à mettre en œuvre. Il est quasiment impossible de
prévoir une terminaison, de surcroît lorsque la composition en acides gras polyinsaturés est
variée et évolutive, ce qui est le cas dans les systèmes biologiques (Halliwell and Gutteridge,
2008).
Figure 10 : Complexité réactionnelle de la peroxydation lipidique : peroxydation de l’acide linoléique (LH). Adapté
de (Venkataram et al., 2004).
La figure 10 est un exemple de peroxydation lipidique de l’acide linoléique (LH), un
acide gras insaturé bien présent dans les membranes cellulaires. On remarque que les premiers
produits radicalaires peroxyles et époxyperoxyles participent à la propagation de la
peroxydation lipidique d’autres acides gras polyinsaturés initiaux. En présence de Fe2+ (métal
de transition) et d’oxygène, ils peuvent également rejoindre un système de Fenton générateur
de radicaux encore plus réactifs. Un large éventail de réactions est possible et ne peut pas être
détaillé ici. On peut cependant retenir que de nouveaux radicaux centrés sur le carbone par
fission (voir première partie, I) A) 2) 2.iii) et diverses réactions d’addition mènent à la
formation de radicaux époxyallyliques. Ces radicaux ont une forte réactivité avec l’oxygène,
participent à la formation de nouveaux radicaux peroxyles et le cycle recommence.

64
Venkataraman et al. (2004) ont montré in vivo que lors de la peroxydation lipidique des
acides gras polyinsaturés:
- La longueur de l’acide gras n’a pas d’impact sur la quantité de radicaux formés.
- La quantité de radicaux lipidiques générés par peroxydation lipidique augmente
si le nombre total d’atomes d’hydrogène en position bis-allylique (et donc le nombre de
doubles liaisons) augmente. Ces derniers sont en effet particulièrement vulnérables car
les liaisons C—H des carbones impliqués dans une double liaison C=C (hydrogènes bis-
allylique) sont plus faibles ce qui rend leur abstraction plus simple (figure 11).
Figure 11 : Vulnérabilité des atomes d’hydrogène dans la chaîne polyinsaturée de l’acide gras. D’après
(Venkataraman et al., 2004)
- Et plus important encore, le taux de peroxydation lipidique cellulaire croît de
manière exponentielle avec l’augmentation du nombre d’atome d’hydrogène en position
bis-allylique (Wagner et al., 1994).
Phase de terminaison
La phase de terminaison correspond à la phase de formation de molécules plus stables
non-radicalaires, obtenue le plus souvent par réaction entre deux radicaux libres ou grâce à
l’action d’un antioxydant.
Nous remarquerons par ailleurs que les hydroperoxydes lipidiques LOOH (qui
s’accumulent dans le cytoplasme des cellules lors de la peroxydation lipidique) sont
généralement assez instables en présence de métaux de transition et disposés à relancer une
peroxydation lipidique en régénérant des radicaux libres alkoxyles:
LOOH + Fe2+ LO° +HO- + Fe3+
puis
LO° + L1—H LOH + R1° et le cycle continue.
1.ii Oxydation du cholestérol
Le cholestérol présent dans les membranes plasmiques et dans les lipoprotéines peut
également être oxydé durant la peroxydation lipidique, en générant une grande variété de
produits regroupés sous le nom d’oxystérols.
Cette oxydation peut avoir lieu sur plusieurs sites ce qui explique entre autre la
complexité des produits générés. Les oxystérols (comme le 7-kéto-cholestérol) sont
notamment caractérisés par une solubilité plus grande dans l’eau, modifiant la cinétique et la
fluidité membranaire des cellules (Jairam et al., 2012).

65
La toxicité de ces produits est encore largement discutée au sein de la communauté
scientifique (Halliwell and Gutteridge, 2008).
2) Stress oxydant et altérations des protéines et des acides aminés
La biochimie de l’altération des protéines est beaucoup plus complexe que celle des
lipides, premièrement à cause du nombre d’acides aminés différents (plus de 20). Dans les
parties précédentes, nous avons déjà évoqué plusieurs exemples illustrant la réactivité des ERO
avec les acides aminés des protéines. Dans l’exposé qui suit, nous tenterons de retenir ce qui
illustre le mieux ces altérations oxydatives.
Comme vu précédemment, les ERO les plus réactifs avec les protéines sont le radical
hydroxyle HO° et le peroxynitrite ONOO-. L’ion superoxyde O2°- est le moins réactif des ERO
mais est suffisamment fort pour oxyder les groupements thiols (-SH) de la cystéine (Halliwell
and Guterridge, 2008).
Les ERO peuvent modifier les protéines suivant plusieurs modalités dont l’oxydation, la
nitrosylation, la nitration ou encore la chlorination d’acides aminés spécifiques, menant plus ou
moins directement à la modification de leur structure et donc à la perte de leur activité
biologique (Henrotin, 2003).
Les chaînes latérales de tous les acides aminés des protéines sont susceptibles de subir
l’oxydation des ERO (Stadtman, 2004). Ces résultats ont évidemment été obtenus in vitro, où la
quantité et la qualité des espèces réactives ainsi que les conditions expérimentales peuvent
être modifiées jusqu’à obtenir un résultat ; in vivo il est clair que tous les acides aminés ne sont
pas sensibles de la même manière au stress oxydant.
En effet, dans les conditions physiologiques, les acides aminés les plus sensibles aux ERO
rapportés par la littérature sont :
- les acides aminés soufrés : cystéine et méthionine
Figure 12 : Les acides aminés soufrés.
66
- les acides aminés basiques : arginine, histidine et lysine
Figure 13 : Les acides aminés basiques.
- les acides aminés aromatiques : tyrosine, phénylalanine et tryptophane
Figure 14 : Les acides aminés aromatiques.
Remarquons que la dégradation des protéines par le stress oxydant peut également être
indirecte, notamment en réagissant avec les produits de la peroxydation lipidique comme des
aldéhydes toxiques ou les radicaux peroxyles et ceux centrés sur le carbone.
2.i Oxydation des chaînes latérales
Les acides aminés soufrés sont particulièrement sensibles à l’oxydation : l’oxydation
du groupement thiols –SH de la cystéine peut mener à la formation de groupements sulféniques
(-SOH), sulfiniques (-SOOH), sulfoniques (SOOOH) ou à la formation de ponts disulfures (-S—S-
) entre deux cystéines. La méthionine peut également être oxydée en sulfoxyde de méthionine
(>S=O) ou en sulfone de méthionine (-S(=O)(=O)-)(Raman and Berry, 2011).

67
L’oxydation des acides aminés aromatiques (phénylalanine, tryptophane et tyrosine)
provoque l’ouverture du cycle aromatique ou son oxygénation (ajout de groupements
hydroxyde -OH). La tyrosine peut également former le radical tyrosyle sous l’action des ERO à
l’origine de ponts dityrosines qui modifient la structure tertiaire des protéines. Ce radical peut
alors réagir avec O2°- pour former l’hydroperoxyde de tyrosine (oxygénation du cycle)
(Halliwell and Gutteridge, 2008).
Les composés carbonylés se forment très tôt lors de l’altération oxydative des protéines.
Un composé carbonylé est un composé organique comportant une double liaison entre un
atome de carbone et un atome d’oxygène (fonction cétone >C=O ou aldéhydes -CH=O). Les
acides aminés les plus susceptibles de former des carbonyles sont les acides aminés basiques
(lysine, l’arginine et histidine). Ce type d’altération est souvent catalysé par des métaux de
transition. En effet, des ions comme Fe2+ ou Cu2+ peuvent se fixer sur les protéines et ainsi
faciliter l’attaque de H2O2 ou même du dioxygène directement (Reznick and Packer, 1994).
2.ii Nitration
La nitration correspond à l’attachement d’un groupe –NO2 (nitro-) à un composé, ici en
l’occurrence une chaîne latérale protéique. Cette liaison n’est généralement pas réversible
(Halliwell and Gutteridge, 2008).
Dans le contexte du stress oxydant, la nitration des protéines est en majeure partie
orchestrée par le peroxynitrite et le radical nitryle NO2°- au niveau des acides aminés
aromatiques comme la tyrosine (Alcaraz et al., 2013). Cette nitration a un impact sur les
qualités physico-chimiques de cet acide aminé, en échangeant l’hydrophobie de la tyrosine
pour l’hydrophilie de la nitrotyrosine, pouvant modifier la structure tertiaire de la protéine qui
la porte (l’extérieur de la protéine étant généralement hydrophile et l’intérieur hydrophobe
dans le cytosol, et inversement dans les membranes).
De plus, la nitration de la tyrosine diminue le pKa de son groupement alcool de 10 à 7.5
en moyenne, entraînant une importante ionisation aux pH biologiques de ces résidus (Halliwell
and Gutteridge, 2008).
2.iii Nitrosylation
La nitrosylation correspond à l’attachement d’un groupe –NO (nitroso-) à un résidu thiol
(S-nitrosylation) ou à un métal (M-nitrosylation). Cette liaison est généralement réversible.
Les groupements thiols de la cystéine peuvent subir une nitrosylation par le
peroxynitrite, le dioxyde ou le trioxyde d’azote (S-nitrosylation), et former un nitrosothiol (-S—
NO) ; cette réaction est plus rare avec le monoxyde d’azote car NO° réagit rarement
directement avec les groupements thiols –SH (Halliwell and Gutteridge, 2008).
Cette réaction, comme la nitration d’ailleurs, est particulièrement favorisée par la
colocation d’une source de radical superoxyde et de NO-synthases (favorable à la génèse de
peroxynitrite).
2.iv Glucoxydation
La glucoxydation d’une molécule regroupe une étape de glycation qui est suivie par une
étape d’oxydation des produits glyqués. Les produits issus de la glycation sont facilement
oxydés par de nombreuses espèces réactives, dont le radical hydroxyle HO° et le peroxynitrite
ONOO- pour donner les AGEs oxydés (AGEs pour « advanced glycation end products »).

68
La glycation est une réaction non enzymatique entre un sucre réducteur (par exemple le
glucose) et une protéine pour former une base de Schiff réversible (surtout avec de la lysine et
de l’arginine) (Andrades and Lorenzi, 2011) (Alcaraz et al., 2013). La glycation in vivo est un
phenomène très lent, et donc le phénomène global de glucoxydation touche essentiellement les
molécules très faiblement recyclées, comme le collagène (Halliwell and Gutteridge, 2008)
(Alcaraz et al., 2013).
La plupart des AGEs sont généralement très instables, très réactifs et difficiles à analyser
complètement. La pentosidine et la carboxyl-méthyl-lysine (CML) sont les plus connues car
elles sont assez stables pour être mesurées (Valko et al., 2007).
L’ensemble des mécanismes est complexe et ne sera pas détaillé.
2.v Chlorination
L’hypochloride HOCl/ClO- peut réagir rapidement avec le groupement amines (–NH2)
des acides aminés libres et des chaînes latérales aminés pour former les N- chloramines (R—
NHCl).
La chlorination des protéines résulte également de la modification des chaînes latérales
des acides aminés contenant des groupes fonctionnels tels que les groupements thiols –SH
(dans les bases soufrées), les ponts disulfides –SS–, le groupement hydroxyle –OH (dans la
tyrosine) ou encore le groupement indole du tryptophane (Olszowski et al., 2003).
2.vi Déshydrogénation, rupture et remaniement de la chaîne protéique
Pour mémoire, la liaison peptidique est une liaison covalente qui s’établit entre la
fonction carboxyle portée par le carbone d’un acide aminé et la fonction amine portée par le
carbone de l’acide aminé suivant.
Comme décrit précédemment, des ERO peuvent arracher les atomes d’hydrogène liés au
carbone de la chaîne polypeptidique (abstraction de –H) et créer des radicaux libres centrés
sur le carbone, qui peuvent à leur tour rentrer dans de nouvelles réactions radicalaires. Des
réarrangements peuvent également avoir lieu lors des réactions radicalaires en chaînes et
aboutir à la fracture de la chaîne polypeptidique ou encore à la formation de liaisons inter-
chaînes et/ou intra-chaînes, altérant de fait la structure de la protéine.
Enfin, certains aldéhydes issus de la peroxydation lipidique peuvent également être
ajoutés à la chaîne sur les acides aminés en situation de stress oxydant (Valko et al., 2007)
(Halliwell and Gutteridge, 2008).
3) Stress oxydant et altération de l’ADN
L’acide désoxyribonucléique (et l’acide ribonucléique par extension) est une molécule
très sensible au stress oxydant. La plupart des ERO sont en effet capables de réagir avec l’ADN,
aussi bien sur les bases azotées (puriques et pyrimidiques) que sur le ribose et le désoxyribose.
L’ensemble des types de dégâts oxydatifs sur l’ADN sont regroupés dans la figure 15.

69
Figure 15: Altérations de l’ADN entraînées par un stress oxydant. D’après (Favier, 2003) (Delattre et al., 2003)
(Webb and Twedt, 2008).
L'attaque radicalaire directe peut entraîner l'oxydation des bases, engendrant un très
grand nombre de bases modifiées : 8 oxo-guanine, 8 nitro-guanine, formamidopyrimidine, 8
oxo-adénine, formimido-uracile, 5 hydroxycytosine, 5 hydroxyméthyl-uracile, thymine-diol,
oxazolone (Favier, 2003) ... La guanine est une base azotée particulièrement sensible à ces
altérations (Alcaraz et al., 2013).
Aussi, les ERO peuvent s’attaquer à la liaison entre la base et le désoxyribose, créant un
site abasique, ou attaquer le sucre lui-même, créant une coupure de chaîne simple ou double
(rupture complète de la double hélice) (Favier, 2003).
L'attaque radicalaire des protéines qui entrant en contact avec l'ADN pour le protéger
(histones) ou pour le lire (enzymes et facteurs de la réplication ou de la transcription), peut
entraîner des pontages entre les protéines ou leurs adduits avec les bases azotées et former des
liaisons de type lysine-guanine ou tyrosine-thymine (Favier, 2003) (Delattre et al., 2003).
Par ailleurs, les chloramines et l’acide hypochloreux HOCl sont considérés comme des
perturbateurs des mécanismes de réparation de l’ADN dans le noyau (Halliwell and Gutteridge,
2008).
La sensibilité de l’ADN aux ERO s’explique également en partie par la présence de
nombreux métaux de transition (comme Fe, Cu, Zn, Mg, Ni, Cd…) fixés sur la molécule d’ADN en
raison de son caractère polyanionique. Toute production de radical superoxyde O2°- ou de
peroxyde d’hydrogène H2O2 est en effet susceptible de générer HO° par une réaction de Fenton
et d’amplifier ou d’orienter le profil de ces lésions le long de la double hélice (Favier, 2003)
(Valko et al., 2007).
L’ADN mitochondrial a une sensibilité particulière aux radicaux libres (Halliwell and
Guterridge, 2008). On considère généralement que l'ADN nucléaire présente une base modifiée
par les ERO sur 1,3.105 bases contre une base sur 8.103 bases dans l'ADN mitochondrial
(Favier, 2003).
En effet, l’ADN mitochondrial est au plus proche du principal site de production de
radicaux dans la cellule. Les radicaux les plus agressifs pour la mitochondrie sont le peroxyde
d’hydrogène et le peroxynitrite, formé localement au sein même de la machine mitochondriale
et attaquant directement son ADN (Valko, 2011). De plus, contrairement à l’ADN nucléaire,

70
l’ADN mitochondrial n’est pas enroulé autour d’histones ; il est donc particulièrement exposé. Il
a également été suggéré que les mécanismes de réparation de l’ADN mitochondrial, bien que
présents, étaient plus lents que dans le noyau (Halliwell and Guterridge, 2008).
Enfin, l’ADN mitochondrial, bien que codant pour un faible nombre de protéines (dont
sept sous-unités du complexe I, une du complexe III, trois du complexe IV et deux du complexe
V), ne comportent pas d’introns : les dommages entraînés par les radicaux libres sont alors plus
susceptibles d’affecter un gène que dans le cas de l’ADN nucléaire (Halliwell and Guterridge,
2008).
B) Conséquences directes des altérations biochimiques à l’échelle cellulaire
1) Conséquences fonctionnelles des altérations des lipides membranaires
La première conséquence de la peroxydation lipidique sur les cellules concerne la
perturbation de la structure et de la composition des membranes cellulaires. La peroxydation
des acides gras polyinsaturés des phospholipides membranaires fragilise leur chaîne carbonée
qui finit par casser. Cette altération dans la fonction de barrière de la bicouche
phospholipidique se manifeste généralement par une baisse de la fluidité membranaire et une
augmentation de sa perméabilité.
La diminution de la fluidité entraîne une rigidification de la membrane, dont la première
conséquence est la perte de la déformabilité globale de la cellule, pouvant entraîner la rupture
cellulaire suite à une contrainte mécanique normalement supportée par la structure
biphospholipidique. Ce phénomène est particulièrement palpable chez les hématies au passage
dans les plus petits capillaires (hémolyse). Les hématies, qui traversent les tissus producteurs
d’ERO sont particulièrement sensibles à cette peroxydation lipidique en situation de stress
oxydant (Portier, 2007).
Les protéines membranaires comme des enzymes, transporteurs et canaux ioniques, ou
encore des récepteurs sont sensibles aux modifications de la composition de la membrane ;
leur activité peut se retrouver modifiée par la baisse de la fluidté membranaire, pouvant avoir
un impact notable sur l’ensemble du métabolisme cellulaire. Par exemple, l’exposition de
cellules en culture ou de liposomes in vitro a pour conséquence l’altération des flux calciques
(Albano et al., 1991).
Dans le cadre de la membrane mitochondriale, la rigidification des membranes entraîne
une altération du mouvement latéral des protéines membranaires et notamment des
complexes de la chaîne de phosphorylation oxydative. Ce mécanisme entre également dans le
dysfonctionnement mitochondrial déjà évoqué plus tôt.
De même la rigidification membranaire trouble les mécanismes d’endocytose et
d’exocytose en fragilisant les vésicules qui transitent normalement dans le cytosol.
La déstabilisation des membranes intracellulaires des organites désorganise l’équilibre
intracellulaire et aboutit à une perte de la compartimentation cellulaire et de l’équilibre
physicochimique intracellulaire. Par exemple, la rupture du contenu des lysosomes libère des
hydrolases très toxiques dans le cytosol ; de même la libération de la matrice mitochondriale
active certains facteurs pro-apoptotiques (comme le cytochrome c et les caspases) (Halliwell
and Guterridge, 2008).

71
2) Conséquences fonctionnelles des altérations des protéines
Il n’existe pas de système de réparation des protéines connu, sauf quelques enzymes
dont l’action n’est que très ponctuelle ; on retiendra par exemple l’action conjointe de la
méthionine sulfoxyde réductase et des péroxyrédoxines qui permettent la réduction du
sulfoxyde de méthionine en méthionine.
Les protéines oxydées sont généralement détruites par les cellules comme mécanisme
de défense. Ce mécanisme est orchestré par les protéasomes et quelques protéases des
lysosymes. Les protéasomes sont des complexes enzymatiques multiprotéiques intracellulaires
ayant pourtant une action assez modeste. Dans certains cas, les protéines fortement dégradées
peuvent s’agréger entre elles sans pouvoir être éliminées par les protéasomes et s’accumulent
alors dans le cytoplasme. Cette accumulation peut éventuellement dérégler le métabolisme
cellulaire et mener à la mort de la cellule (Grune et al., 1997).
En effet, les protéines oxydées qui ne sont pas dégradées ont des propriétés biologiques
modifiées souvent dues à un changement conformationnel. Il est impossible de citer toutes les
conséquences fonctionnelles de l’attaque du stress pour la cellule oxydant sur les protéines ;
cependant certaines d’entre elles sont mentionnées ci-après.
2.i Dysfonctionnements enzymatiques
Les modalités d’altération des enzymes sont très variées. Bien sûr, l’oxydation d’acides
aminés comme la cystéine peut désorganiser la structure protéique, pouvant alors changer la
conformation des sites actifs et des sites modulateurs. De même, les nitrations mènent très
souvent à l’inactivation enzymatique : le meilleur exemple concerne la SOD elle-même (Alcaraz
et al., 2013).
De nombreuses enzymes contiennent des métaux de transition indispensables à leurs
fonctions biologiques. La dégradation de ces protéines peut notamment libérer dans le milieu
de ces métaux et exacerber le stress oxydant ambiant. L’interaction simple que peuvent
entretenir les métaux de transition avec les radicaux libres peut suffire à perturber le
fonctionnement enzymatique. Ainsi, si on reprend la SOD, on remarque que la dismutation
d’O2°- diminue également en présence de peroxynitrite et/ou de NO° en quantité suffisante par
un mécanisme de M-nitrozylation (Beckman and Koppenol, 1996).
Enfin, l’affinité du monoxyde d’azote pour les radicaux libres l’amène également à
inhiber de manière réversible les enzymes ayant des radicaux libres dans leur chaîne
réactionnelle comme la ribonucléotique réductase (utilisant le radical tyrosyle) (Beckman and
Koppenol, 1996). La ribonucléotide réductase intervient dans la synthèse de l’ADN ou de sa
réparation en produisant des désoxyribonucléotides di- ou triphosphates à partir de
ribonucléotides di- ou triphosphates.
2.ii Dysfonctionnement des anticorps et perte des propriétés antigéniques des protéines
Ce point a toute son importance dans l’entretien des mécanismes d’inflammation
chroniques sur des sites soumis à un stress oxydant où une modification des épitopes
cellulaires des récepteurs protéiques (reconnus alors étrangers) peut être constatée.
Sur le plan immunitaire, les AGEs peuvent également être reconnues par les monocytes,
les macrophages et d’autres cellules via des récepteurs spécifiques, les récepteurs RAGE,

72
pouvant activer une réponse inflammatoire inappropriée au sein du site de production des ERO
(Halliwell and Gutteridge, 2008).
2.iii Dégradation des propriétés physicochimiques et biomécaniques des protéines
Pour illustration, reprenons l’exemple de la nitration opérée par le peroxynitrite. Le
peroxynitrite est responsable de la nitration de la chaîne latérale de la tyrosine et de sa
conversion en nitrotyrosine. Il semble que les protéines structurelles soient les plus atteintes
par cette dégradation. Par exemple, chez les neurofilaments L de l’homme la nitration des
tyrosines en nitrotyrosines transforme des résidus normalement hydrophobes en résidus
hydrophiles chargés négativement, empêcheant alors la complexation des sous-unités
protéines en une longue structure polymère (Halliwell and Gutteridge, 2008).
De même, une étude a montré que le degré de nitration du collagène de type I était
corrélé à une réduction de sa déformabilité et de la capacité des fibroblastes à restaurer la
matrice collagénique ; la nitration entraînée par le peroxynitrite peut donc directement altérer
les propriétés biomécaniques des tissus conjonctifs (Alcaraz et al., 2013).
Enfin, la nitration du fibrinogène par le peroxynitrite peut également favoriser sa
transformation en fibrine, initiant alors la coagulation intravasculaire (Halliwell and
Gutteridge, 2008).
L’accumulation des AGEs dans les protéines est généralement irréversible sauf si la
protéine est dégradée. Pour illustration, au sein du collagène, les AGEs diminuent l’élasticité
globale du tissu et sa résistance à l’étirement (Halliwell and Gutteridge, 2008).
2.iv Dysfonctionnement/modulation des récepteurs cellulaires, des signaux de transduction et perturbation des fonctions de régulation
On retrouve des résidus cystéines « redox-sensibles » dans les récepteurs
membranaires, des protéines d’adhésion (notamment pour l’exocytose et endocytose du
réticulum endoplasmique et l’appareil de Golgi), des protéines du cytosquelette chez les
mammifères. Les domaines riches en cystéines au sein des récepteurs membranaires
permettent une régulation du métabolisme et de la croissance cellulaire par le potentiel redox
extracellulaire. Les récepteurs comme EGFR (« Epithelial Growth Factor Receptor ») et les
récepteurs NMDA (au N-méthyl-D-aspartate) en font partie. Dans le milieu intracellulaire aussi,
certaines kinases et des phosphatases impliquées dans la cascade transductionnelle ont des
sites sensibles à l’environnement redox qui module leur activité (Raman and Berry, 2011).
Par ailleurs, la formation de la nitrotyrosine est susceptible d’altérer la communication
cellulaire puisque de nombreuses protéines impliquées dans plusieurs signaux intracellulaires
utilisent la phosphorylation de la tyrosine pour leur activation (Loeser et al., 2002).
2.v Dysfonctionnement du transport protéique et du métabolisme énergétique
Chez les animaux, plusieurs études ont montré que le métabolisme énergétique pouvait
être une cible directe pour O2°- chez les mammifères.
Ainsi, l’aconitase (enzyme du cycle de Krebs transformant le citrate en isocitrate)
possède comme de nombreuses enzymes des groupements [Fe-S]. Ces propriétés la rendent
particulièrement sensible aux dégâts oxydatifs entraînés par la réaction de Fenton. Il a aussi été

73
reporté qu’O2°- pouvait inactiver la NADH déshydrogénase dans des mitochondries bovines in
vitro (Halliwell and Gutteridge, 2008).
Les protéines mitochondriales sont particulièrement exposées aux radicaux libres,
particulièrement lorsque un dysfonctionnement de la chaîne respiratoire est présent (voir
première partie, II) A) 1) 1.iii) et notamment le canal membranaire VDAC (pour « voltage-
dependent anion channel »). De plus, la glycoxydation des protéines dans la chaîne respiratoire
mitochondriale diminue l’efficacité du transport électronique, favorisant la fuite des électrons
(Halliwell and Gutteridge, 2008).
À des concentrations minimolaires, le monoxyde d’azote peut également inhiber la
cytochrome-c oxydase, ce qui peut amplifier la fuite des électrons de la chaîne respiratoire
mitochondriale (Beckman and Koppenol, 1996).
3) Conséquences fonctionnelles des altérations de l’ADN
3.i ADN nucléaire
L’attaque radicalaire de l'ADN est quotidienne puisque le nombre de lésions se formant
chaque jour dans une cellule au niveau de l’ADN est estimé à 104 en moyenne. Heureusement, la
fidélité de la séquence des 4.109 paires de bases de notre ADN (humain) cellulaire est
maintenue grâce à des systèmes de réparation perfectionnés, dont les principaux sont la
réparation par excision de base (BER) ou par excision de nucléotide (NER), couplées ou non à la
transcription, le système de réparation des mésappariements et la réparation par
recombinaison (Favier, 2003).
Cependant, les dégâts oxydatifs sur les bases azotées de l’ADN s’accumulent tout au long
de la vie cellulaire et peuvent influencer de manière significative la sénescence cellulaire. Par
exemple, les cellules sénescentes présentent jusqu’à 30% de guanine oxydée dans leur ADN et
quatre fois plus de 8-oxoguanine libre (Houben et al., 2008).
La plus belle illustration de ce phénomène concerne les télomères. Les télomères sont
des nucléoprotéines situés à la fin des brins des chromosomes. Les télomères perdent 20 à 200
paires de bases azotées à chaque mitose, entraînant une diminution de la taille des télomères
avec l’âge de la cellule. Cette réduction de la taille des télomères provient de l’incapacité des
ADN polymérases à répliquer entièrement l’ADN ; à la disparition des télomères, la cellule
arrête de se diviser et entre généralement en apoptose. Les télomères sont tous constitués de
plusieurs séquences d’ADN très riches en guanine et en cytosine. Cette richesse en guanine
rend les télomères particulièrement sensibles au stress oxydatif (Houben et al., 2008) (Alcaraz
et al., 2013). C’est une des modalités de sénescence cellulaire accélérée par le stress oxydant.
Chez les télomères encore, le raccourcissement est également accentué par la déficience
des mécanismes de réparation par rapport au reste de l’ADN (différence qui pourrait être
expliquée par la présence des protéines fixées sur les télomères) (Houben et al., 2008).
On imagine aisément que les altérations oxydatives de l’ADN induites par le stress
oxydant sont particulièrement impliquées dans le vieillissement des cellules de faible capacité
de renouvellement ou qui ne se divisent pas.
En cas de stress oxydant massif, les lésions non réparées comme les cassures et les
absences ponctuelles en bases azotées s’accumulent et vont perturber les mécanismes de
réplication de l'ADN et interférer avec la réplication et la transcription de l’ADN, entraînant

74
alors des erreurs de lecture ou de synthèse par les ADN polymérases qui aboutissent à une
mutation ponctuelle dans le génome (Favier, 2003).
Ainsi, les liaisons croisées (comme thymidine—tyrosine) interfèrent significativement
sur la réplication, la transcription, et la réparation de l'ADN (Delattre et al., 2003) ; une
impossibilité de copie de l'ADN en cas de dégradation oxydative sévère de l’ADN aboutira
inévitablement à l’apoptose (Favier, 2003). De même, des mutations par transversions GC
(guanine/cytosine) vers TA (thymine/adénine) sont souvent observées spontanément dans les
cellules cancéreuses (carcinogénèse).
3.ii ADN mitochondrial
Les conséquences des dégâts oxydatifs sur la double hélice et le mécanisme de formation
des mutations sur l’ADN mitochondrial sont similaires à ceux de l’ADN nucléaire. Cependant,
quelques précisions supplémentaires peuvent être apportées.
Chez l’homme il a été évalué que le taux de mutation de l’ADN mitochondrial est 20 fois
supérieur à celui de l’ADN nucléaire. Le défaut du système de réparation de l’ADN dans la
mitochondrie par rapport au noyau explique ce constat (Wu et al., 2011).
Les modifications dans l’ADN mitochondrial peuvent être responsables d’une libération
accrue d’ERO et entraîner un cercle vicieux entre la production d’ERO avec les dégâts oxydatifs.
Ce cercle vicieux mène rapidement à un déclin progressif des fonctions bioénergétiques des
cellules concernées, et alors à la mort cellulaire (Wu et al., 2011).
Le dysfonctionnement mitochondrial entraîné par les modifications de l’ADN
mitochondrial a été relié à la physiopathologie de nombreuses affections dégénératives
chroniques chez l’homme mais rarement chez l’animal. On manque également de preuves
solides pour corréler le taux de mutations de l’ADN mitochondrial avec le dysfonctionnement
de la chaîne respiratoire (Wu et al., 2011).
C) Impact cellulaire et tissulaire des produits de dégradation du stress
oxydant
La peroxydation lipidique initiée par les radicaux libres génère également un large
spectre d’aldéhydes réactifs. Ces aldéhydes, plus stables, amplifient les dégâts oxydatifs dans le
temps et à distance de leur point de genèse, même une fois la production d’ERO terminée, en
générant un mélange complexe de produits toxiques qui s’attaquent notamment aux protéines
environnantes. Ils peuvent notamment modifier les chaînes aminées en formant des radicaux
carbonylés (Jairam et al., 2012).
C’est particulièrement le cas pour le 4-hydroxynonenal (4-HNE), l’acroléine et le
malondialdéhyde (MDA). Très généralement retenus comme de simples marqueurs du stress
oxydant, on oublie souvent que dans les conditions physiologiques le MDA et le 4-HNE par
exemple modifient des protéines contenant de la lysine, les groupements imidazole de
l’histidine, l’arginine, la méthionine et les groupes sulfhydryles (C—S) des cystéines (Jairam et
al., 2012).
L’exemple du 4-HNE est particulièrement illustrateur du pouvoir délétère et de la
contribution toxique de ces aldéhydes au stress oxydant. Cette molécule est classée dans la
catégorie depuis 1993 comme « second messager toxique des radicaux libres » et pourrait être

75
impliquée dans la physiopathologie de nombreuses maladies, surtout chez l’homme (Zarcovic,
2003) (Dianzani, 2003), incluant l’athérosclérose, la maladie d’Alzheimer, la maladie de
Parkinson, la cirrhose du foie et plusieurs cancers. Sa haute réactivité provient
particulièrement de l’association d’une double liaison covalente C=C avec un groupement
carbonyle (>C=O, caractéristique des alkénals) (Schaur, 2003).
On pense que ces aldéhydes favorisaient le développement de plusieurs maladies où est
impliqué le stress oxydant en entretenant des réponses proinflammatoires et profibrogéniques
(Webb and Twedt, 2008) (Jairam et al., 2012). Dianzani (2003) rapporte ainsi qu’à de faibles
concentrations le 4-HNE a la capacité d’activer le chimiotactisme des neutrophiles ainsi que la
phospholipase C chez les hépatocytes et les neutrophiles mais aussi de bloquer le récepteur
PDGF-. Cette dernière constatation pourrait illustrer l’implication du 4-HNE dans la mise en
place et la régulation de la fibrose tissulaire.
D’autres auteurs comme Nakashima et al. (2003) ont trouvé des effets délétères du 4-
HNE dans la communication transductionnelle. Leurs résultats suggèrent qu’à des
concentrations pathologiques il réagit avec la surface de multiples types cellulaires et sites
intracellulaires, aboutissant à la suppression des fonctions cellulaires pour entraîner
l’apoptose. Ces observations sont également soutenues par Awasthi et al. (2003) qui ont
également montré in vitro que le 4-HNE induisait l’apoptose à des concentrations observées
lors d’un stress oxydant.
Enfin, le 4-HNE, pourrait également contribuer à la diminution du pool de glutathion
réduit en se conjuguant au GSH par double liaison covalente >C=C< (Forman et al., 2003).
Les isoprostanes générés in vivo par peroxydation des phospholipides, et plus
particulièrement de l’acide arachidonique, sont également biologiquement actifs. Suivant leur
nature chimique, ils peuvent être de puissants vasoconstricteurs ou vasodilatateurs. D’autres
encore peuvent entraîner la résorption osseuse ou altèrer l’aggrégation plaquettaire
normalement induite par l’ADP ou le contact avec le collagène. On attribue ces actions
biologiques diverses à leur similarité structurelle avec les éicosanoïdes (prostaglandines et
leukotriènes) qui leur permettraient de se fixer sur leurs récepteurs (Halliwell and Guterridge,
2008).
Pour mémoire, des dommages indirects sur l’ADN peuvent résulter de l'attaque des
aldéhydes issus de la peroxydation lipidique, formant des adduits sur les bases de l'ADN de
type MDA-guanine par exemple (Favier, 2003).

76
V) Évaluation expérimentale du stress oxydant
A) Principe de l’exploration du stress oxydant
Dans les systèmes biologiques, le stress oxydant est souvent caractérisé par les
paramètres suivants (Powers and Jackson, 2008):
- Une augmentation de la formation de radicaux libres ou d’autres oxydants
- Un déséquilibre du statut redox cellulaire
- Une diminution du pool d’antioxydants non-enzymatiques
- Une augmentation de l’activité des enzymes antioxydantes
- Une augmentation des molécules témoins des dégâts oxydatifs (par
exemple sur les lipides, les protéines ou encore l’ADN).
B) Moyens d’exploration
Les moyens d’exploration présentés dans les parties qui suivent regroupent les
principaux protocoles retrouvés dans la bibliographie ; cependant cette liste est évidemment
non-exhaustive.
1) Méthodes directes
1.i RPE : Résonance Paramagnétique Électronique
Le spectromètre à Résonance Paramagnétique Électronique (RPE) permet de mesurer
directement les radicaux libres. Cependant elle est très difficile à utiliser in vivo. Elle est
souvent associée aux techniques de « spin trapping » pour augmenter la sensibilité de la
technique (Alcaraz et al., 2013).
Schématiquement, la RPE désigne la capacité de certains électrons à absorber puis à
réémettre l’énergie d’un rayonnement électromagnétique lorsqu’ils sont placés dans un champ
magnétique précis (phénomène de résonance magnétique). Seuls les électrons non appariés,
présents dans les espèces chimiques radicalaires, les sels et métaux de transition, ont cette
propriété.
In vitro, la détection directe des radicaux lipidiques libres (dont les radicaux peroxyles et
alkoxyles formés durant la peroxydation lipidique de l’acide arachidonique et de l’acide
linoléique soumis à l’action de la lipooxygénase) utilise un système continu de RPE dit « fast-
flow » (Venkataraman et al., 2004).
La RPE « spin trapping » a pu être utilisée pour détecter une grande variété de radicaux
dans les cellules et les tissus (Venkataraman et al., 2004). Cette technologie utilise un composé
diamagnétique (le « spin trap ») qui réagit avec un radical libre pour former un radical plus
stable (durée de vie moins brève) et plus facilement détectable (le « spin adduct »). Dans les
systèmes biologiques, cette méthode permet notamment de détecter les radicaux hydroxyles et
superoxydes ainsi que de nombreux produits issus de la peroxydation lipidique (radicaux
peroxyles, alkoxyles et centrés sur le carbone).
Deux molécules utilisées comme « spin traps » sont le 5,5-Dimethyl-pyrroline-1-oxide
(DMPO) et l’alpha-(4-Pyridyl-1-oxide)-N-tert-butylnitrone (POBN) (figure 16).

77
Figure 16: Deux « spin traps » : DMPO et POBN. D’après (Venkataraman et al., 2004).
Le DMPO est très communément utilisé car il permet de capturer une grande variété de
radicaux libres (de l’azote, de l’oxygène, du soufre et des radicaux centrés sur le carbone
notamment), mais également parce que ses « spin adducts » ont une durée de vie plus grande et
se concentrent en quantité suffisante pour être correctement analysés (Buettner, 1987).
Le POBN « attrape » aussi des radicaux libres de l’oxygène mais la durée de vie des « spin
adducts » est bien plus courte qu’avec le DMPO ; par exemple, la durée de vie du « spin adduct »
issue de la réaction entre le POBN et HO° est d’environ 10 secondes. Cependant, le POBN trouve
son intérêt pour la détection des radicaux libres centrés sur le carbone, dont une partie des
produits de la peroxydation lipidique, dont les « spin adducts » ont une durée de vie plus longue
qu’avec DMPO (de quelques heures à quelques jours en solution aqueuse à pH neutre)
(Venkataraman et al., 2004). Pour cette dernière raison, POBN est souvent préféré à DMPO
pour l’étude de la peroxydation lipidique.
Pour mémoire, on peut combiner l’utilisation de la RPE avec la chromatographie liquide
et la spectrométrie de masse pour préciser la nature des « spin adducts ». En effet, avec la
technique de RPE standard on ne peut que connaître les types du radical (centré sur l’oxygène,
sur le carbone …) et la quantité de radicaux « attrapés » par POBN. Ainsi, la durée de vie des
radicaux de POBN permet d’utiliser d’autres procédés pour caractériser la nature des produits
radicalaires. Concernant la peroxydation lipidique, de nombreux radicaux lipidiques nés de la
fission peuvent ainsi être mis en évidence.
La présence de fer et métaux de transition dans les prélèvements biologiques est une
autre limite du « spin-trapping ». En effet, leur présence, particulièrement dans l’étude des
mécanismes de peroxydation lipidique, entraîne la formation de nouveaux radicaux libres (et
notamment des radicaux alkyles) qui peuvent alors anéantir ceux qui sont étudiés par RPE ; par
exemple :
POBN/L1° + L2° produits non radicalaires.
De plus, étant par définition une réaction en chaîne, arrêter la peroxydation lipidique à
un point précis du mécanisme étudié est très difficile (Venkataraman et al., 2004). Afin d’éviter
la destruction des produits de RPE issus du « spin-trapping » par d’autres radicaux libres, des
techniques d’extraction chimique ont également été utilisées pour notamment séparer les
lipides (phase organique) de la phase aqueuse où se trouve le fer. Ainsi, on ralentit le cycle de
peroxydation lipidique et limite également la destruction des POBN/L° ou des DMPO/L°. Les
techniques d’extraction diluent aussi les réactifs et les produits entre eux. Les « spin-adducts »

78
du DMPO post-extraction sont généralement stables pendant plus de 10 heures avec extraction
contre une vingtaine de minutes sans (Qian et al., 2000).
Enfin, associer la RPE « spin-trapping » à des techniques d’extraction permet de
différencier les radicaux selon les phases étudiées : par exemple on retrouvera les radicaux à
longue chaîne lipidique dans la phase organique alors que les plus petits fragments iront plutôt
dans la phase aqueuse. Evidemment, l’extraction permet de différencier les radicaux selon leur
nature plutôt hydrophile ou lipophile (Qian et al., 2000).
1.ii Chimioluminescence
La chimioluminescence utilise la propriété physique de certaines molécules excitées à
réémettre de la lumière en retournant à l’état basal. Certains ERO, tel que l’oxygène singulet, en
font partie. Elles peuvent être ainsi détectées directement dans les tissus et les organes sources
(Halliwell and Gutteridge, 2008). Pour l’oxygène singulet, deux types d’émission lumineuse
peuvent être produites : l’émission infrarouge monomol (1270 nm, la plus utilisée au
laboratoire) et l’émission dimol qui vient de la collision entre deux molécules d’oxygène
singulet (630 à 701nm).
D’autres réactions impliquant des radicaux peuvent également émettre de la lumière
comme la réaction de Fenton par exemple (en particulier entre le peroxyde d’hydrogène et les
protéines de l’hème), ou encore certaines impliquant le peroxynitrite (Halliwell and Gutteridge,
2008).
Cette technique permet une exploration et un suivi dynamique de la production de
certains ERO lors d’un phénomène biologique d’intérêt au sein des cellules, tissus et organes
isolés. Intéressante in vitro (notamment à l’échelle cellulaire avec le microscope), cette
technique d’exploration dynamique directe du stress oxydant est difficile à utiliser sur des
organismes vivants comme le cheval à l’effort.
2) Méthodes indirectes : témoins de la peroxydation lipidique
2.i Mesure de la fluidité membranaire érythrocytaire
Il existe plusieurs techniques de mesure de la fluidité membranaire comme la diffraction
des rayons X, la microscopie électronique, la microcalorimétrie différentielle, la diffusion de la
lumière, la résonance magnétique nucléaire (RMN) ainsi que les marqueurs paramagnétiques
et de fluorescence. Ne sont mentionnés ici que les détails techniques qui paraissent pertinents à
la compréhension de ce travail. Portier (2007) distingue dans son travail trois niveaux
d’analyse:
- Le niveau microscopique permet d’obtenir des informations sur chacuns des atomes
ou molécules constitutifs des lipides. La spectroscopie de résonance magnétique nucléaire
(spectroscopie RMN) est une technique utilisée.
La spectroscopie RMN fournit des informations qualitatives très fines sur la
coordination, la mobilité et l’environnement électronique immédiat des noyaux observés, ce
qui permet de remonter à la structure moléculaire du composé étudié et à la symétrie de la
phase dans laquelle il est inséré. Elle utilise la propriété de certains atomes (comme 1H, 13C, 17O
ou 31P) à absorber l’énergie électromagnétique puis à la relâcher lors d’une phase de relaxation.
Pour mémoire, l’IRM (pour imagerie par résonance magnétique) utilise également cette
propriété.

79
L’intégration des raies de RMN peut ainsi fournir des informations quantitatives sur la
molécule étudiée. Les phospholipides ne donnent en général qu’une raie unique dite 31P (seul
atome ayant un spin nucléaire) dont le déplacement chimique est caractéristique de la nature
de la tête polaire du lipide et des conditions du milieu (comme le pH). De fait, ces spectres 31P
n’apportent que des informations relatives à la tête polaire du lipide et non aux chaînes
latérales hydrophobes.
Dans la bicouche lipidique, les phospholipides possèdent des orientations privilégiées et
le passage d’une orientation à l’autre est souvent un phénomène lent qui peut être mesuré par
RMN. Les spectres 31P de ces systèmes sont donc plus ou moins larges suivant la mobilité des
phospholipides. La forme du spectre est ainsi caractéristique de la phase phospholipidique et
sa largeur à la base, appelée anisotropie de déplacement chimique (CSA), est reliée au degré de
mobilité des groupes phosphates (Portier, 2007).
- Le niveau sub-macroscopique où l’on étudie de manière globale la fluidité
membranaire à l’aide d’une sonde marquée « stable » et sa diffusion au sein de la membrane
plasmique pendant un temps d’étude donné. Cela permet éventuellement de mettre en
évidence une zone de rigidité membranaire, non spécifique. Pour ce faire, la polarisation de
fluorescence ou la résonance paramagnétique électronique (RPE) peuvent être utlisées.
RPE
Schématiquement, la RPE utilise une sonde paramagnétique (classiquement l’acide 16-
nitroxyde stéarique (16 NS)), constituée d’un acide gras saturé sur lequel est greffé au niveau
du carbone 16 un radical nitroxyle (-NO°), « piégeant » en son sein l’électron célibataire. Cette
sonde est un radical relativement stable et inoffensif pour la cellule. La sonde va ensuite se
localiser près du centre de la bicouche biphospholipidique pour explorer la région hydrophobe
de la membrane. On irradie alors l’échantillon avec une onde radio et on étudie le spectre émis
par la sonde.
Libre, la sonde émet un spectre propre ; incorporé dans la membrane, le spectre prend
une allure différente (en amplitude et en largeur de raie de résonance). L’analyse des spectres
obtenus met notamment en évidence un temps caractéristique de la technique appelé temps de
corrélation-relaxation. Ce temps de corrélation-relaxation Tc est inversement proportionnel à
la largeur de la raie de résonance mais surtout inversement proportionnel à la fluidité
membranaire.
La rotation de la sonde sur elle-même défini un cône ; Tc correspond au temps que met
la sonde pour parcourir un radian (57°) de rotation. Si le cône est très large, cela signifie que la
sonde a peu de contraintes: la sonde tourne très vite donc Tc est faible. Plus Tc est élevé, plus la
fluidité membranaire est faible et inversement (Portier, 2007).
Polarisation de fluorescence
Une molécule fluorescente (ou fluorophore) a la capacité d’absorber un photon d’une
énergie suffisante pour lui permettre d’atteindre un état d’excitation (plus particulièrement un
de ses électrons). Après un moment qui peut être plus ou moins long (noté ), cette molécule
rejoint son état basal en perdant de l’énergie sous forme de chaleur et de photons.
Sans rentrer dans des détails physiques qui sortiraient du cadre de notre travail, si on
excite un échantillon avec de la lumière polarisée linéairement, on remarque que la

80
fluorescence est, dans certains cas, également polarisée. Ainsi, lorsque de la lumière polarisée
est appliquée à un groupe de fluorophores orientés de manière aléatoire par rapport à elle, les
molécules qui pourront entrer en fluorescence seront celles qui seront disposées avec une
angulation comprise dans un intervalle défini par rapport à cette lumière polarisée. Si elles ne
sont pas en mouvements, la lumière qu’elles émettent sera elle aussi polarisée : le degré de
polarisation de la fluorescence est quantifié par une grandeur appelée anisotropie de
fluorescence, notée r.
Dans les protocoles expérimentaux, l'anisotropie de fluorescence est une mesure qui
indique le degré de liberté d'une molécule fluorescente, le degré de liberté étant vu comme la
facilité avec laquelle une molécule ou la partie fluorescente d'une molécule tourne sur elle-
même.
Plus précisément, l’anisotropie est une mesure de la dépolarisation de la fluorescence, et
ce dans la mesure où tout changement de direction pendant la durée de vie de l’état excité ()
provoquera une diminution de l’anisotropie. La dépolarisation de fluorescence peut être causée
par plusieurs phénomènes dont l'importance relative dépend de l'échantillon
étudié (mouvement brownien, vibration de torsion, transfert d'énergie ...), mais la principale
cause est la rotation par diffusion des fluorophores. La direction de l'émission de fluorescence
dépend de la liberté de rotation du fluorophore dans le milieu.
A partir des intensités de fluorescence émises parallèlement (Ipa) et
perpendiculairement au rayon incident (Ipe) on calcule alors le degré de polarisation (p) : p =
(Ipa - Ipe)/(Ipa + Ipe).
Ainsi, grâce à la polarisation de fluorescence on peut évaluer son degré de mouvement
de rotation dans une membrane plasmique et étudier la fluidité membranaire (Pigault, 2010).
- Le niveau macroscopique correspondant à la formation de domaines lipidiques plus
rigides (« ilôts ») et pouvant être atteint par des mesures calorimétriques ou mécaniques qui ne
seront pas détaillées.
Selon Portier (2007), la fluidité membranaire des érythrocytes pourrait être considérée
comme un marqueur direct du stress oxydant dans certaines conditions mais est moins
sensible que d’autres marqueurs du stress cellulaire tels que la mesure de la concentration
plasmatique en peroxydes lipidiques spécifiques.
2.ii Méthode des TBARS
La mesure des biomarqueurs la méthode des TBARS est l’un des premiers tests d’étude
du stress oxydant (Gutteridge and Halliwell, 2000) ; en effet, au début des années 1980, peu
d’autres méthodes simples étaient disponibles et surtout applicables à des prélèvements
biologiques. Son développement a notamment suivi l’analyse plus approfondie du rancissement
des graisses contenant des acides gras polyinsaturés dans l’industrie agro-alimentaire.
L’appellation TBARS vient de Thiobarbituric Acid Reactivity Reactive Substances. C’est
une méthode très simple consistant à chauffer le prélèvement en condition acide et le faire
réagir avec de l’acide 2-thiobarbiturique. L’obtention d’une solution rose signe un résultat
positif.

81
Cette technique fut appliquée à de nombreuses reprises en médecine, impliquant alors
les radicaux libres dans de très nombreux mécanismes physiopathologiques. Pourtant il n’a
jamais été clair sur ce que cette technique mesure réellement, et cela a pris de nombreuses
années avant que la communauté scientifique ne se rende compte du manque de spécificité et
de fiabilité de la technique. Ainsi, selon Halliwell et Gutteridge (2008), le TBARS test a
effectivement de la valeur pour évaluer l’oxydation globale de systèmes lipidiques définis
(aliments, microsomes …) mais ne peut pas être utilisé pour comparer les peroxydations dans
différents systèmes de compositions différentes en acides gras polyinsaturés, notamment des
prélèvements in vivo (différents acides gras polyinsaturés génèrent des niveaux en TBARS
différents).
Ce manque de spécificité est notamment illustré par le fait que les TBARS sont retrouvés
dans les fluides corporels et tissus en quantité augmentée chez n’importe quel individu
malade, indépendemment de la physiopathologie de la maladie (Gutteridge and Halliwell,
2000).
Ainsi, utilisée seule, la méthode de mesure des TBARS n’est pas fiable pour mesurer le
niveau de peroxydation lipidique dans les cellules, les tissus ou d’autres fluides corporels. De
plus, il faut bien rappeler que contrairement à ce que clament certaines études, la méthode des
TBARS ne mesure pas spécifiquement le malondialdéhyde (MDA) dans un échantillon (Halliwell
and Gutteridge, 2008). D’autres aldéhydes que le MDA peuvent également réagir avec l’acide
thiobarbiturique et produire des composés qui absorbent la lumière dans les mêmes longueurs
d’onde que le MDA.
De plus, la décomposition des peroxydes lipidiques pendant le test lui-même peut
masquer le contenu en MDA présent dans le prélèvement avant les mesures. Cette
décomposition est variable suivant la présence ou l’absence d’ions métalliques, dont les
concentrations sont difficilement contrôlées dans les protocoles utilisant ce test (Clarkson and
Thompson, 2000).
2.iii Dosage des peroxydes lipidiques
La peroxydation lipidique est un système complexe produisant de nombreux peroxydes
lipidiques. Il est possible de mettre en évidence et de quantifier un grand nombre de classes de
peroxydes par chromatographie liquide haute performance. Certains protocoles utilisent
également des enzymes de dégradation de ces peroxydes dont l’activité est ensuite mesurée
par colorimétrie par exemple.
Un autre test mesurant les hydroperoxydes dans leur globalité et évaluant le statut
global oxydant (d-ROMS test) a également été développé et sera revu plus tard.
2.iv Dosage des aldéhydes produits de la peroxydation lipidique
Comme nous l’avons vu précédemment, la peroxydation lipidique des acides gras
polyinsaturés génère de nombreux aldéhydes. Ces derniers, bien que très variés et en
concentration variable, peuvent être utilisés comme marqueurs du stress oxydant (Jairam et al.,
2012).
De nombreux α et β-aldéhydes insaturés ont été utilisés comme témoins du stress
oxydant dans les systèmes biologiques : on retiendra notamment l’acroléine, le 4-
hydroxynonenal (4-HNE) et le malondialdéhyde (MDA) (figure 17).

82
Figure 17 : 4-hydroxynonenal (4-HNE), acroléine et malondialdéhyde (MDA). Adapté de (Jairam et al., 2012).
Acroléine
L’acroléine est un polluant issu de la combustion de nombreuses matières organiques et
de plastiques (notamment la fumée de cigarette). Dans le cas du stress oxydant, c’est un
métabolite naturel de la peroxydation lipidique. L’oxydation de la thréonine (acide aminé,
homologue hydroxylé de la valine) par la myéloperoxydase (MPO) entraîne également une
augmentation de l’acroléine (Jairam et al., 2012).
C’est le plus réactif des trois. Comme pour le 4-HNE, il est aussi responsable de
l’oxydation des protéines en réagissant avec les groupements sulfhydryls (S-H) des cystéines,
des groupes imidazoles des histidines et mais aussi la lysine ; cette réactivité ne facilite pas son
utilisation dans les modèles biologiques pour évaluer le stress oxydant (Jairam et al., 2012).
4-hydroxynonenal (4-HNE)
Le 4-HNE appartient à la famille des alkénals. C’est un produit de la peroxydation
lipidique, en particulier des acides gras polyinsaturés du groupe des Ω6 (1ère double liaison
>C=C< sur le 6ième atome de carbone) comme l’acide linoléique et l’acide arachidonique,
particulièrement abondants dans les membranes cellulaires. C’est donc un marqueur d’intérêt
pour l’étude du stress oxydant dans les systèmes biologiques et les organismes vivants (Jairam
et al., 2012).
La pharmacocinétique de 4-HNE a été caractérisée. Lorsqu’il est administré par voie
intraveineuse ou intrapéritonéale, les métabolites du 4-HNE sont principalement excrétés dans
la bile et l’urine, avec un fort cycle entéro-hépatique. Une très faible quantité de métabolites est
liée aux protéines (Alary et al., 2003).
Deux groupes de métabolites existent : ceux liés à la conjugaison du 4-HNE avec le
GSH par la glutathion-S-transférase puis sa réduction en alcool par une réductase, ou son
oxydation par une déshydrogénase ; ceux lié à l’hydroxylation par le cytochrome P450 et son
éventuelle conjugaison à la GSH et la formation de conjugués d’acide mercapturique éliminés
par la voie biliaire (Alary et al., 2003).
Malondialdéhyde (MDA)
Dans les prélèvements frais, le malondialdéhyde (MDA) est formé principalement par la
peroxydation lipidique d’acides gras comme l’acide arachidonique (Jairam et al., 2012).

83
Le malondialdéhyde trouve son intérêt dans le fait qu’il indique que des radicaux lipidiques ont
été formés au sein de la membrane cellulaire (Alcaraz et al., 2013).
Si initialement on pensait que le MDA était mis en évidence par la technique des TBARS,
on sait aujourd’hui le doser spécifiquement avec ses produits de dégradation, notamment en
utilisant la chromatographie liquide haute performance (Jairam et al., 2012).
Par ailleurs la principale limite du dosage du malondialdéhyde est que les réactions
chimiques qui permettent sa détection sont influencées par la présence de fer dans l’échantillon
(Alcaraz et al., 2013).
2.v Dosage des hydrocarbures volatils expirés
L’éthane et le pentane sont deux hydrocarbures produits dans les tissus lors de la
peroxydation lipidique et pouvant être retrouvés dans l’air exhalé. L’éthane est notamment
issus de l’oxydation des acides gras Ω3 tel que l’acide linolénique, alors que le pentane dérive
plutôt de l’oxydation des Ω6 comme l’acide linoléique et l’acide arachidonique. On s’est parfois
un peu plus intéressé à l’éthane car sa mesure chromatographique sur gaz est plus facile et plus
rapide que les autres hydrocarbures (Paredi et al., 2002).
Le dosage du pentane et de l’éthane est particulièrement précis et répétable car les types
d’acides gras dont ils dérivent sont trouvés de manière prépondérante dans les membranes
cellulaires (Clarkson and Thompson, 2000).
Une des difficultés inhérentes à ces mesures dans l’air expiré consiste à éliminer les
hydrocarbures déjà présents dans l’espace mort des voies pulmonaires comme la trachée,
illustrant les difficultés techniques de recueil chez l’animal (Paredi et al., 2002). De plus, les
valeurs peuvent être affectées par la présence de dioxygène et de métaux de transition dans les
milieux utilisées pour les protocoles de dosage (Clarkson and Thompson, 2000). De même, les
valeurs de référence n’étant pas clairement établies, ni chez l’homme ni chez le cheval, ces
mesures, bien que non invasives, doivent être interprétées avec grande précaution.
2.vi Dosage des isoprostanes
Les isoprostanes sont des produits finaux, spécifiques de la peroxydation lipidique et
sont principalement issus de l’oxydation de l’acide arachidonique. Leur mesure représente
certainement la technique la plus spécifique d’évaluation de cette réaction en chaîne in vivo et
in vitro (Halliwell and Gutteridge, 2008).
De nombreux protocoles de mesure (notamment par chromatographie gazeuse et
liquide haute performance) ont été développés pour mesurer les isoprostanes et leurs
métabolites dans les urines et le plasma essentiellement (même à de très faibles
concentrations).
Bien sûr, comme tous les marqueurs du stress oxydant, ils ne sont pas parfaits. Un des
artéfacts classiques est que les isosprostanes peuvent aussi être produits par les plaquettes et
ce indépendamment du stress oxydant (Alcaraz et al., 2013). De plus, une mauvaise
conservation des prélèvements (particulièrement de plasma, moins fréquent pour l’urine) peut
entraîner une peroxydation lipidique faussant les dosages en isoprostanes (Halliwell and
Gutteridge, 2008).

84
Les isoprostanes font parti des alcanes ciblés lors d’étude du stress oxydatif affectant
plus spécifiquement les poumons et les voies respiratoires et peuvent aussi être également
mesurés dans l’air expiré (Paredi et al., 2002).
2.vii Dosage des diènes conjugués
Les diènes conjugués sont des diènes dont les deux doubles liaisons sont séparées par
une liaison simple selon le motif -CH=CH–CH2—CH2–C=CH-. Ils ont l’avantage d’absorber la
lumière ultraviolette à 230-235 nm, permettant des techniques simples de dosage par
spectrométrie.
Les diènes conjugués sont parmi les premiers produits de la peroxydation des acides
gras insaturés. Si certains considèrent leur mesure fiable et répétable, d’autres remettent en
cause la technique, en parti à cause de la grande fragilité des prélèvements. Le régime
alimentaire peut également modifier significativement la concentration tissulaire basale en
diènes conjugués, diminuant la spécificité de la technique concernant le stress oxydant
(Clarkson and Thompson, 2000).
3) Méthodes indirectes : témoins de l’altération des protéines
3.i Détection des carbonyles protéiques
Parmi toutes les modifications oxydatives des acides aminés des protéines, la formation
de résidus carbonylés est certainement le marqueur le plus précoce et une bonne mesure de
l’oxydation des protéines par les ERO (Reznick and Packer, 1994) (Valko et al., 2007). De
nombreuses méthodes et protocoles de mesures ont été développés (spectophotométrie,
ELISA ...) et varient selon les laboratoires. Le dosage des carbonyles protéiques est adapté
autant aux fluides qu’aux tissus biologiques.
Une méthode expérimentale courante consiste à utiliser le 2,4-dinitrophenylhydrazine
(DNPH, ou réactif de Brady, utilisé pour mettre en évidence les aldéhydes et les cétones) et
ensuite mesurer la concentration en protéines carbonylées par spectrophotométrie à la
longueur d’onde 370nm (Halliwell and Gutteridge, 2008) (Villasante et al., 2010).
Ce dosage peut également être suivi de l’identification de ces protéines afin de cibler les
régions et structures cellulaires affectées, pour plus de spécificité.
3.ii Détection des protéines et acides aminés nitratés
Le but théorique de la mesure des protéines nitratées est de montrer que le stress
oxydant suspecté implique la génèse de monoxyde d’azote, de la myéloperoxydase (MPO) mais
surtout du peroxynitrite (Alcaraz et al., 2013).
La 3-nitrotyrosine est généralement utilisée comme marqueur des dégâts oxydatifs lié à
l’action du peroxynitrite (Loeser et al., 2002). Dans les études concernant l’ostéoarthrose, le
dosage de la 3-nitrotyrosine au sein de la matrice collagénique est fréquent.
4) Méthodes indirectes : témoins de l’altération de l’ADN
Le 8-hydroxydéoxyguanosine (8-OH-dG) est une base azotée oxydée reflétant
l’hydroxylation de la guanosine. C’est le marqueur des dégâts oxydatifs de l’ADN le plus utilisé
et il représente les altérations de l’ADN que l’organisme ne peut pas réparer. Ce marqueur est
particulièrement bien filtré par le rein et concentré dans les urines. Ainsi un prélèvement
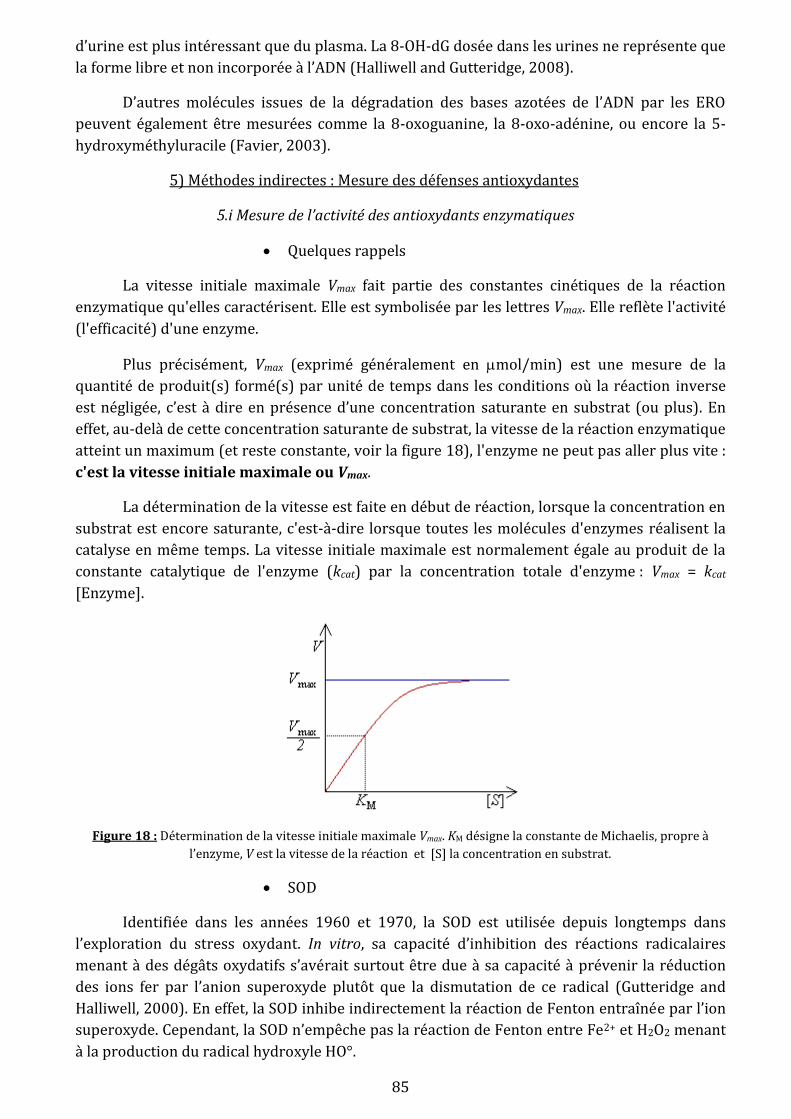
85
d’urine est plus intéressant que du plasma. La 8-OH-dG dosée dans les urines ne représente que
la forme libre et non incorporée à l’ADN (Halliwell and Gutteridge, 2008).
D’autres molécules issues de la dégradation des bases azotées de l’ADN par les ERO
peuvent également être mesurées comme la 8-oxoguanine, la 8-oxo-adénine, ou encore la 5-
hydroxyméthyluracile (Favier, 2003).
5) Méthodes indirectes : Mesure des défenses antioxydantes
5.i Mesure de l’activité des antioxydants enzymatiques
Quelques rappels
La vitesse initiale maximale Vmax fait partie des constantes cinétiques de la réaction
enzymatique qu'elles caractérisent. Elle est symbolisée par les lettres Vmax. Elle reflète l'activité
(l'efficacité) d'une enzyme.
Plus précisément, Vmax (exprimé généralement en mol/min) est une mesure de la
quantité de produit(s) formé(s) par unité de temps dans les conditions où la réaction inverse
est négligée, c’est à dire en présence d’une concentration saturante en substrat (ou plus). En
effet, au-delà de cette concentration saturante de substrat, la vitesse de la réaction enzymatique
atteint un maximum (et reste constante, voir la figure 18), l'enzyme ne peut pas aller plus vite :
c'est la vitesse initiale maximale ou Vmax.
La détermination de la vitesse est faite en début de réaction, lorsque la concentration en
substrat est encore saturante, c'est-à-dire lorsque toutes les molécules d'enzymes réalisent la
catalyse en même temps. La vitesse initiale maximale est normalement égale au produit de la
constante catalytique de l'enzyme (kcat) par la concentration totale d'enzyme : Vmax = kcat
[Enzyme].
Figure 18 : Détermination de la vitesse initiale maximale Vmax. KM désigne la constante de Michaelis, propre à
l’enzyme, V est la vitesse de la réaction et [S] la concentration en substrat.
SOD
Identifiée dans les années 1960 et 1970, la SOD est utilisée depuis longtemps dans
l’exploration du stress oxydant. In vitro, sa capacité d’inhibition des réactions radicalaires
menant à des dégâts oxydatifs s’avérait surtout être due à sa capacité à prévenir la réduction
des ions fer par l’anion superoxyde plutôt que la dismutation de ce radical (Gutteridge and
Halliwell, 2000). En effet, la SOD inhibe indirectement la réaction de Fenton entraînée par l’ion
superoxyde. Cependant, la SOD n’empêche pas la réaction de Fenton entre Fe2+ et H2O2 menant
à la production du radical hydroxyle HO°.

86
In vivo cependant, cela a peu d’importance selon les auteurs car le pool d’ions fer à l’état
libre dans la cellule reste très faible (Gutteridge and Halliwell, 2000).
Dans le cas du sang, on mesure généralement l’activité de la SOD intracellulaire et
extracellulaire (moins prépondérante) simultanément sans les différencier.
La SOD n’a pas d’inhibiteur (notamment allostérique) connu et catalyse un seul type de
substrat (en l’occurrence le radical superoxyde) sans avoir besoin de donneurs d’électron.
Ainsi, elle respecte parfaitement le modèle de Michaelis-Menten avec pour définition de
l’activité : Vmax = kcat [Enzyme].
En d’autres termes, toute variation de son activité est uniquement liée à la concentration
de l’enzyme, en l’occurrence dans le sang.
Pour autant, l’interprétation des variations de son activité varie selon les auteurs. En
effet, deux interprétations peuvent être données suite à une augmentation de la SOD après un
effort :
- Soit l’organisme synthétise de la SOD en réponse à l’exercice physique ; lors
d’effort de courte durée, cette hypothèse est improbable ;
- Soit les dommages tissulaires lors de l’effort entraînent la fuite de la SOD
(présente dans de nombreux tissus) vers le sang au même titre que la créatine kinase (CK) par
les muscles.
C’est la raison pour laquelle certains biochimistes considèrent d’ailleurs que la mesure
de l’activité de la SOD ne présente pas beaucoup d’intérêt dans l’exploration du stress oxydant.
GPx
La glutathion peroxydase (GPx) est une des rares enzymes séléno-dépendantes
antioxydantes pour laquelle une méthode de dosage a été mise au point et utilisée
fréquemment dans les études. On la dose généralement dans le plasma (faible activité) et les
hématies, mais aussi dans d’autres tissus comme dans les muscles striés, le myocarde ou encore
le foie.
On a démontré qu’il existe une relation linéaire entre l’activité de la GPx sanguine et la
teneur en sélénium dans le sang ou le plasma chez le cheval (Blackmore et al., 1982). Par
ailleurs, il convient de préciser que ce coefficient de corrélation ne permet pas toujours de
prédire une valeur exacte de la sélénémie sanguine pour chaque activité de la GPx, mais il est
possible de définir des groupes de chevaux (sélénémie insuffisante <50g/L, 50g/L <
marginale < 75 g/L et optimale >75 g/L) (Ludvikova et al., 2005).
Le dosage de l’activité de la GPx plasmatique est délicat car son activité ne représente
qu’1% de l’activité de la GPx érythrocytaire, majoritaire dans le sang. De plus son activité
diminue très rapidement ex vivo et les prélèvements nécessitent des conditions particulières de
réalisation et de transport (Blackmore et al., 1982) ; pour autant, la GPx plasmatique
représente la sélénémie instantanée.
A contrario la GPx érythrocytaire est stable 4 jours à température ambiante et permet de
quantifier les apports en sélénium sur l’ensemble de la durée de vie du globule rouge
(approximativement 120 jours chez le cheval).

87
L’étude des variations de l’activitée GPx érythrocytaire doit généralement être couplée à
celle de la concentration globale sanguine du glutathion réduit (GSH). En effet, l’activité de cette
enzyme dépend directement de l’apport en GSH (utilisé comme donneur d’électrons).
CAT
La mesure de l’activité de la CAT n’est pas facile notamment à cause du manque de
standardisation du protocole utilisé pour attester de l’activité de cette enzyme. De plus il
semblerait que les propriétés de cette enzyme n’obéissent pas à la loi de Michaelis Menten. Ces
difficultés se retrouvent dans les résultats controversés dans les études visant à montrer si
l’activité de la CAT augmente après un exercice (Powers and Jackson, 2008).
Par exemple, ces protocoles expérimentaux impliquent le suivi de la dégradation de son
substrat (en l’occurrence H2O2) au spectrophotomètre. La méthode de Aebi, développée en
1983, suit la diminution de l’absorbance dans le milieu expérimental (tampon phosphate et
extrait enzymatique) après addition de peroxyde d’hydrogène (Powers and Jackson, 2008).
Paraoxonase-1 (PON-1)
La capacité du sang à hydrolyser le paraoxon est souvent utilisée comme marqueur de
l’activité enzymatique de la paraoxonase-1. Des kits ELISA permettent la mesure sur le terrain
de la concentration plasmatique en paraoxonase-1. L’activité de cette enzyme peut également
être mesurée de manière plus sensible par fluorométrie et photométrie.
L’usage de ces kits est cependant peu retrouvé à ce jour dans la littérature équine.
5.ii Dosage des antioxydants non enzymatiques
Dosage du glutathion GSH, GSSG et la mesure du rapport GSH/GSSG
Le principe théorique de cette mesure repose sur le fait qu’en cas de stress oxydant, le
taux en glutathion disulfide GSSG augmente parallèlement à la production d’ERO. Lorsque la
concentration en GSSG dépasse les capacités de réduction de la glutathion réductase, du GSSG
est retrouvé dans le sang. GSSG est un marqueur assez précis du processus oxydatif (De
Moffarts et al., 2004b).
Le calcul du ratio GSH/GSSG (ou GSSG/GSH, le principe restant le même) permet de
s’affranchir de l’augmentation en GSH possible lors d’un effort physique (particulièrement
observé lors d’effort bref et intense) pour ne considérer que le déséquilibre entre la forme
oxydée et la forme réduite : c’est la définition même du stress oxydant appliqué au niveau du
glutathion.
Différentes techniques sont disponibles pour mesurer le taux en GSH et GSSG dans les
prélèvements biologiques comme la colométrie et la chromatographie liquide haute
performance.
Halliwell et Gutteridge (2008) rapportent que le rapport GSH/GSSG pourrait cependant
manquer de spécificité en raison d’une possibilité d’auto-oxydation du glutathion prélevé.

88
Dosage de la vitamine E
De nombreuses techniques ont été utilisées historiquement pour mesurer la vitamine E
dans les prélèvements biologiques. La chromatographie liquide haute performance (CLHP) est
actuellement la plus fiable et la plus utilisée pour isoler la vitamine E. Suivant les protocoles,
l’-tocophérol peut être mis en évidence seul, ou l’ensemble des tocophérols et de leurs
métabolites.
La technique de CLHP auquelle de nombreuses études font référence est celle de De
Leenheer (De Leenheer et al., 1979). Cette technique de chromatographie liquide haute
performance dite « en phase inversée » peut notamment être utilisée sur du plasma ou du
sérum.
Elle consiste à traiter l’échantillon avec un solvant organique comme l’éthanol qui a pour
but de « libérer » la vitamine E des structures biologiques dans lesquelles elle est intégrée
(membranes cellulaires notamment) et d’éliminer toutes les molécules insolubles dans le
solvant organique avec lesquelles la vitamine E peut potentiellement réagir (et qui restent dans
la phase aqueuse). Une phase de saponification (en utilisant de l’hydroxyde de potassium à
chaud) avant extraction peut également compléter le protocole : elle a pour but de récupérer
les éléments non saponifiables comme la vitamine E qui resteront dans le solvant organique
lors de l’extraction alors que les éléments saponifiés comme les sels d’acides gras ou le glycérol
resteront dans la phase aqueuse. On procède ensuite à l’extraction de la vitamine E avec de
l’hexane comme solvant d’extraction. Après centrifugation, le surnageant est récupéré pour
être évaporé à 40°C dans un courant d’azote.
L’analyse de l’extrait brut de vitamine E utilise alors la CLHP qui permet de séparer les
différente tocophérols et métabolites, qui sont ensuite dosées par électrochimie (méthode très
sensible), fluorescence, spectrométrie …
Les conditions de conservation des prélèvements influent grandement sur les résultats.
Le stockage à température ambiante peut augmenter les concentrations mesurées jusqu’à 5%.
L’hémolyse entraîne une diminution de plus de 30% de la quantité de vitamine E et le contact
prolongée avec le bouchon de caoutchouc jusqu’à 10%. Il semblerait que la lumière n’influence
pas les résultats (Craig et al., 1992).
Idéalement, il faut recueillir des échantillons non-hémolysés (sérum ou plasma),
conservés à la verticale pour éviter le contact avec le bouchon, dans un endroit réfrigéré, le
tube étant de taille la plus petite possible pour diminuer l’exposition à l’air (s’il n’y a pas
d’azote) ; l’analyse du sérum ou du plasma conservé au frais doit être réalisée dans les 72
heures. Si les prélèments ne sont pas analysés de suite, il faut les congeler à -80°C pour une
durée de conservation de presque 6 mois (Craig et al., 1992).
Dosage de la vitamine C
De la même manière que pour la vitamine E, des protocoles de mesure précis de la
vitamine C ont été mis au point, utilisant notamment la CLHP et la spectrophotométrie (Marlin
et al., 2002).
Dans une des études réalisées chez le cheval, Hargreaves et al. (2002) utilisent la
méthode de Schiiep (1987). Schématiquement, cette dernière consiste à faire réagir la vitamine
C présente dans le prélèvement avec de l’acide métaphosphorique (HPO3) pour former du 2,4-

89
dinitrophenylhydrazone de vitamine C analysée par CLHP et spectrophotométrie (à 520 nm de
longueur d’onde).
Une des limites du dosage repose sur le fait qu’une oxydation artéfactuelle de l’acide
ascorbique peut se produire au cours du prélèvement sanguin, sous l’effet de la lumière et
d’une température supérieure à 4°C. C’est pourquoi il est délicat de mesurer, de façon fiable, les
concentrations plasmatiques en acide ascorbique et hydroascorbate (Deaton et al., 2003).
5.iii Mesure de la capacité antioxydante globale
Voltamétrie cyclique
La voltampérométrie cyclique (ou voltamétrie cyclique) est une mesure électrochimique
basée sur une variation contrôlée du potentiel électrique appliqué à un échantillon. Le résultat
est ce qu’on appelle un diagramme de voltampérométrie cyclique où on mesure le cycle que
suit la variation du courant électrique par rapport à celle du potentiel appliqué (Halliwell and
Gutteridge, 2008).
Ce type de voltamétrie est utilisé pour étudier les propriétés redox de composés
chimiques dans une solution et identifier un grand nombre de composés aux potentiels
d’oxydation bien connus. Les diagrammes peuvent également être utilisés pour mesurer la
capacité réductrice totale des agents réducteurs présents (Halliwell and Gutteridge, 2008).
Très souvent, les antioxydants de faible poids moléculaire sont des agents réducteurs.
Encore peu utilisée chez nos animaux sportifs, la voltamétrie cyclique permet de mesurer la
concentration globale en agents réducteurs et donc avoir une approximation de la
concentration globale en antioxydants de faible poids moléculaire.
FRAP assay = ferric reducing antioxidant power assay
Le FRAP assay mesure la capacité des antioxydants plasmatiques (urate, vitamine E,
vitamine C, mais pas l’albumine) à réduire Fe3+ en Fe2+ à pH faible, par un test
spectrophotométrique.
Le plasma du patient est mélangé à une solution colorée (issue du mélange entre un
dérivé thiocyanate et du chlorure ferrique) (Celi et al., 2010). L’intensité de la décoloration
correspondant à la réduction de Fe3+ est mesurée par spectrophotométrie à 505 nm ; cette
intensité est proportionnelle à la capacité du plasma à réduire les ions ferriques. Les résultats
sont exprimés en mol/L d’ions ferreux.
Un prétraitement de l’échantillon avec de l’ascorbate oxydase peut être réalisé pour
déterminer la contribution jouée par la vitamine C dans la valeur obtenue. De par son principe,
ce test présume que la capacité réductrice du plasma équivaut à sa capacité antioxydante totale,
ce qui est évidemment contestable (le plasma contient des molécules comme l’albumine qui
jouent un rôle antioxydant en se dégradant au contact des ERO en déchets bien éliminés par
l’organisme, sans pour autant exercer un pouvoir réducteur direct) (voir première partie, III) C)
1)) (Halliwell and Gutteridge, 2008).
Pour mémoire le prélèvement de sang se fait sur un tube hépariné (Celi et al., 2010).

90
d-ROMS test
Il est également possible de doser la quantité globale des hydroperoxydes ROOH par le
d-ROMS test (ROMS pour « reactive oxygen metabolites »). In vivo, les hydroperoxydes de
l’organisme dosés sont issus de l’oxydation des lipides bien sûr, mais aussi des protéines et des
nucléotides.
C’est une méthode de mesure du statut global oxydant ; il ne permet pas de mesurer des
produits finaux mais permet d’évaluer le statut pro-oxydant en estimant la production
potentielle d’ERO. Par ailleurs, c’est une technique de colorimétrie qui requiert un grand apport
technique et matériel, difficilement adapté à des études de terrain (Clarkson and Thompson,
2000).
Cette technique colorimétrique fait réagir des radicaux hydroperoxyles et alkoxyles
produits par la réaction de Fenton (entre un métal de transition et les hydroperoxydes du
prélèvement) avec un indicateur coloré, le N-diethyl-paraphénylendiamine en milieu acide à
37°C. Le milieu se colore alors en rose. Par photométrie on dose la concentration en substrat
produit (proportionnelle à l’absorbance du milieu à la longueur d’onde 505 nm) et on exprime
les résultats en unités Caratelli (du nom du chimiste à l’origine de la technique) ; une unité
Caratelli correspond à 0.08 mg de peroxyde d’hydrogène par dL).
Ce test est approuvé chez le cheval, et il n’y a pas de différence significative des résultats
entre les prélèvements sanguins frais et réfrigérés à 4°C (jusqu’à 24h au moins) (Celi et al.,
2010). La technique peut donc être correctement appliquée par le vétérinaire de terrain et
gagne en popularité dans les études. Cependant, dans cette technique on dose un mélange sans
aucune indication sur les composés qui la composent. De même, si son manque de sensibilité
est acceptable pour des efforts très intenses, l’intérêt de cette étude est discutable pour des
efforts plus modestes.
TRAP = Total radical trapping antioxidant potential
TRAP est une technique de chimioluminescence qui de manière non spécifique
détermine la capacité antioxydante d’un prélèvement en mesurant le potentiel de capture des
radicaux peroxyles. Cette technique de mesure consiste à soumettre un échantillon à un flux
constant de radicaux peroxyles et à mesurer le temps d’épuisement des antoxydants présents
dans le milieu (Villasante et al., 2010) (Halliwell and Gutteridge, 2008).
En calibrant la mesure avec un antioxydant de concentration connue (généralement le
Trolox C, chacune pouvant capturer deux RO2°), une valeur de TRAP peut être obtenue et
exprimée en micromoles de radicaux peroxyles « capturés » dans un litre de solution. Dans le
plasma, les acteurs principaux de la valeur du TRAP sont généralement l’urate (35-65% de la
valeur chez l’homme), les protéines plasmatiques (10-50%), la vitamine C (jusqu’à 24%) et la
vitamine E (5-10%).
Elle est encore peu utilisée chez l’animal et les commentaires faits précédemment pour
le d-ROMS test s’appliquent également ici.
Barrière antioxydante thiols : SHp test
Dans les parties précédentes, on a souligné l’importance des thiols (comme la cystéine et
l’acide lipoïque), ces composés caractérisés par leurs groupements sulfhydryles (-SH) dans
l’équilibre redox des milieux biologiques de l’organisme.

91
Le SHp Test ou mesure de la « barrière antioxydante thiols » est basé sur l’habilité des
thiols à former un complexe coloré pouvant être mesuré par photométrie avec le réactif
d’Ellman (acide DNTB ou acide 5,5-dithio-bis-2-nitrobenzoique). Le titre en thiols dans le
prélèvement est directement proportionnel à l’intensité de la couleur détectée.
Chez les sujets normaux humains, les valeurs sont généralement comprises entre 450 et
650 mol/L et toute réduction signe un stress oxydant (Site internet Diacron International,
2015). Sauf mention contraire, aucunes normes n’ont encore été établies chez le cheval.
OXY-absorbent test
L’OXY-absorbent test s’intéresse à l’acide hypochloreux HClO (produit par la
myéloperoxydase). Ce test mesure la capacité d’un prélèvement de plasma à absorber les
attaques d’une solution d’acide hypochloreux. Les kits disponibles dans le commerce utilisent
un chromogène (indicateur coloré) et des mesures spectrophotométriques. La bilirubine,
l’acide urique, la vitamine C, la vitamine E, l’albumine et, en général, les complexes
macromoléculaires comme les glycoprotéines sont particulièrement mis en valeur dans ce test.
Chez les sujets sains, les valeurs de ce test sont supérieures à 350 mol/L, c’est à dire
que 1 mL de plasma est capable d’« absorber » 350 mol d’acide hypochloreux HOCl (Site
internet Diacron International, 2015).
6) Autres moyens de mesure du stress oxydant
L’air expiré par les animaux est composé d’une phase gazeuse qui contient du dioxygène,
du monoxyde d’azote NO°, du diazote, du dioxyde de carbone et un petit peu de monoxyde de
carbone, plus une phase contenant de la vapeur d’eau et des particules aérosols qui peuvent
être analysées après liquéfaction de la vapeur. Ainsi on a pu identifier des F2-isoprostanes, des
protéines modifiées par HNE, du malondialdéhyde, des protéines nitratées ainsi que du
peroxyde d’hydrogène dans cette dernière phase. Leurs taux peuvent ainsi être mesurés et
corrélés ou non avec un phénomène biologique d’intérêt et plus spécifiquement d’origine
pulmonaire pour refléter un stress oxydant ou un phénomène inflammatoire ; à ce jour ces
corrélations sont encore faites avec la plus grande prudence.
6.i Dosage du NO° expiré et dans les tissus biologiques
Le dosage du NO° dans l’air exhalé est souvent réalisé en même temps que les
marqueurs du stress oxydant dans les études ciblant les affections de la sphère pulmonaire.
Cependant, même s’il est lui même un radical libre, on ne le considère pas comme un marqueur
du stress oxydant mais simplement de l’inflammation dans la mesure où ce sont les cytokines
pro-inflammatoires qui activent la iNOS sont en majorité responsables de sa production. Sa
mesure reste intéressante car on sait que le NO° peut activer l’hème oxygénase-1 (HO-1) et
concourt donc à la production de CO (lors de la convertion de l’hème en biliverdine par HO-1) ;
des hauts niveaux de CO inactivent également iNOS (Paredi et al., 2002).
Le flux expiratoire, la fermeture du palais mou et l’espace mort sont autant de facteurs
qui peuvent également influencer les niveaux en NO. Des dispositifs sont mis en place pour
dissocier l’air des cavités nasales et de l’espace mort pulmonaire, souvent peu adapté au cheval.
Le dosage du monoxyde d’azote est ensuite généralement effectué par chimioluminescence
(Paredi et al., 2002).

92
La réaction très rapide du monoxyde d’azote avec des métalloprotéines (comme
l’hémoglobine ou la guanylate cyclase), le radical superoxyde mais aussi les groupements thiols
(comme sur la cystéine), sa faible solubilité et son peu de stabilité à de petites concentrations
rendent sa mesure dans les tissus et fluides biologiques difficile.
6.ii Dosage du monoxyde de carbone CO expiré
Le monoxyde de carbone CO est surtout produit par HO-1 lors du métabolisme de
l’hémoglobine et de la myoglobine en bilirubine. Dans les poumons, l’hème oxygénase-1 est
situé dans l’endothélium vasculaire pulmonaire et les macrophages alvéolaires, faisant du CO
un marqueur double du stress oxydant et de l’inflammation pulmonaire (d’autant plus que la
HO-1 est activée à la fois par les radicaux libres, comme NO°) mais aussi par les cytokines pro-
inflammatoires) (Paredi et al., 2002).
Ainsi, la mesure du CO pour caractériser le stress oxydant reste intéressante mais n’est
pas spécifique.
Dans la plupart des études, le CO est mesuré grâce à des méthodes électrochimiques. Les
capteurs utilisés aujourd’hui sont sélectifs et procurent des résultats reproductibles. Le CO peut
aussi être mesuré par spectrophotométrie laser et analyseurs infrarouges. La chromatographie
sur colonne de gaz est la méthode de référence.
Chez l’homme, les mesures de monoxyde de carbone peuvent être influencées par la
fumée de cigarette et si l’individu est un fumeur ou pas : ces problématiques se posent
évidemment peu chez nos animaux domestiques (Paredi et al., 2002).
6.iii Autres facteurs exhalés pouvant être mesurés
De nombreux autres composés expirés peuvent également être mesurés comme le
peroxyde d’hydrogène, d’autres produits de la peroxydation lipidique tels que le 8-isoprostane,
3-nitrotyrosine et les nitrosothiols.
Malheureusement, les protocoles et les valeurs normales sont encore trop faiblement
standardisés, les sites anatomo-histologiques des molécules étudiées ne peut pas être connue
et la contamination par l’environnement peut être grande. Enfin, l’utilité clinique de leur
mesure doit encore être confirmées (Paredi et al., 2002) (Cathcart et al., 2012).
6.iv Détection des anticorps dirigés contre les produits de dégradation
La présence de produits issus de la dégradation des protéines et des lipoprotéines par
les radicaux libres mène en partie à la formation d’anticorps. Ce phénomène est
particulièrement connu pour la peroxydation lipidique où des produits comme le 4-HNE se
lient et modifient les protéines ; les laboratoires possèdent un répertoire d’anticorps connus et
étudiés ensuite par ELISA, immunohistochimie et par immunotransfert (Jairam et al., 2012).
C) Défis de l’exploration du stress oxydant
La mise en évidence directe des radicaux libres étant difficile, de nombreuses méthodes
indirectes ont été proposées et basées sur la mesure de produits stables dérivant des effets des
ERO et ERN (« marqueurs du stress oxydant »), d’antioxydants, ou encore de l’activité des
enzymes protectrices.

93
Comme nous avons pu constater, tous les protocoles ont malheureusement leurs limites
et ne sont pas tous spécifiques du stress oxydant ni même d’un ERO ou ERN en particulier.
De plus, ces marqueurs ne sont pas forcément spécifiques du tissu dans lequel ils sont
issus, ce qui veut dire que si c’est l’articulation qui nous intéresse, on pourra réaliser des
prélèvements de liquide synovial, sans oublier que des marqueurs relatifs peuvent aussi se
retrouver dans l’urine ou le sang.
À cause du manque de spécificité et de sensibilité des marqueurs, on considère
actuellement qu’on ne peut pas se fier à l’utilisation d’un seul marqueur pour définir le stress
oxydant ; les études utilisent en généralement plusieurs au sein d’une même expérimentation.
Le statut antioxydant d’un individu comporte un grand nombre de facteurs de variation
qui seront repris plus loin dans le cas de l’exercice chez le cheval. Il n’y a souvent pas de
normes, et des études longitudinales (suivi d’une population dans le temps) sont souvent
requises pour s’affranchir de cette variabilité individuelle, mais souvent trop lourdes à mettre
en place chez nos athlètes équins.
Enfin, les modèles de genèse d’ERO in vitro, sont souvent trop loin de la complexité
biologique. Par exemple, la capacité d’un prélèvement biologiques isolé à inhiber des radicaux
libres tels que O2°- in vitro n’est pas forcément la même in vivo(variation de la capacité
antioxydante) (Alcaraz et al., 2013).
IV) Rôles physiologiques et physiopathologiques généraux des espèces
réactives de l’oxygène
A) ERO et régulation des fonctions cellulaires physiologiques
Un grand nombre de fonctions physiologiques repose sur des signaux redox faisant
intervenir des ERO (Valko et al., 2007).
Il est important de préciser que la synthèse d’espèces réactives codifiée, locale et limitée
n’entraîne pas nécessairement de déséquilibre délétère pour la cellule. Cependant, avant de
s’attaquer à l’implication éventuelle du stress oxydant dans la pathogénie d’une maladie, il
convient de souligner ces fonctions cellulaires physiologiques.
1) Production et fonctions régulatrices de NO°
L’exemple du NO° illustre parfaitement l’implication d’un radical libre dans les
mécanismes physiologiques normaux de l’organisme. La liste ci-dessous en regroupe un
échantillon et illustre la diversité de ses effets (Halliwell and Gutteridge, 2008):
- Système nerveux : élément de réponse aux acides aminés
excitateurs (glutamate en particulier) ; neurotransmission/neuromodulation ; plasticité
synaptique (le NO° permet notamment le maintien des connexions synaptiques d’intérêt,
jouant un rôle important dans la mémoire à long terme).
- Système vasculaire : contrôle de la pression artérielle (notamment
par la vasomotricité/propriétés vasodilatatrices) ; inhibition de l’agrégation plaquettaire ;
destruction de certains agents infectieux. La régulation du tonus vasculaire par NO° provient de
sa capacité à activer la guanylate cyclase et former du cGMP dans le milieu intracellulaire des
cellules musculaires lisses. Le NO° à se fixe en effet aux Fe2+ des centres hémiques de cette

94
enzyme qui entraîne une modification conformationnelle à l’origine de l’activation de l’enzyme.
Le cGMP module la fonction de nombreuses kinases, canaux ioniques et d’autres cibles pour
entraîner des phénomènes physiologiques dont les plus étudiés sont la régulation du tonus
musculaire des vaisseaux (vasomotricité) mais aussi l’inhibition de l’adhésion plaquettaire
(Valko et al., 2007) ;
- Système uro-génital : érection et le contrôle du tonus de la vessie
(muscle dextrusor) ;
- Système gastro-intestinal : relaxation musculaire des fibres lisses
du tube digestif lors du péristaltisme ;
- Système pulmonaire : régulation de la bronchomotricité ;
- Muscles squelettiques : régulation de la tonicité et de la contractilité
musculaire (voir plus loin);
- Cicatrisation tissulaire.
La production de monoxyde d’azote NO° varie selon le potentiel redox environnant :
alors que la nNOS et la eNOS sont exprimé de manière constitutive, la iNOS exprimée de
manière inductible suivant différentes stimulations (cytokines, LPS et autres agents) et fait
intervenir des mécanismes transductionnel (comme la voie NF-B dépendant de l’état redox de
la cellules (voir plus loin) (Valko et al., 2007).
2) Production d’ERO par les cellules phagocytaires
La production d’ERO par les Nox des cellules phagocytaires durant le « burst » oxydatif
de la phagocytose sera étudié en partie B)3) ; ce « burst » peut en effet être présent dans une
réponse physiologique comme pathologique dans les tissus concernés.
3) Production d’ERO par les cellules dans le signalement cellulaire
De nombreux types cellulaires non phagocytaires comme les fibroblastes, les cellules
musculaires lisses, les cardiomyocytes et les cellules endothéliales sont connus pour produire
des ERO avec la NADPH oxydases pour réguler les cascades de signalements
(transductionnelle) intracellulaires (Valko et al., 2007).
Par exemple, la thrombine, le PDGF (« platelet-derived growth factor »), l’angiotensine II
et le TNF- activent la production d’O2°- par les Nox dans les cellules musculaires lisses
(notamment vasculaires). Il en va de même pour IL-1, TNF-, l’angiotensine II et le PAF
(« platelet-activating factor ») chez les fibroblastes (Valko et al., 2007).
Comme pour la phagocytose, le schéma complexe d’implication des ERO dans les
cascades transductionnelles peut être commun à des mécanismes physiologiques comme
pathologiques.
4) Production d’ERO dans les mécanismes d’homéostasie
L’homéostasie de l’oxygène est préservée dans les organismes pluricellulaires par une
régulation fine de la quantité de globules rouges circulant et de respiration pulmonaire. Un des
modèles théoriques a proposé que les changements dans la concentration en dioxygène soient
détectés par la production de différents ERO et impliquent le cytochrome b (Valko et al., 2007).
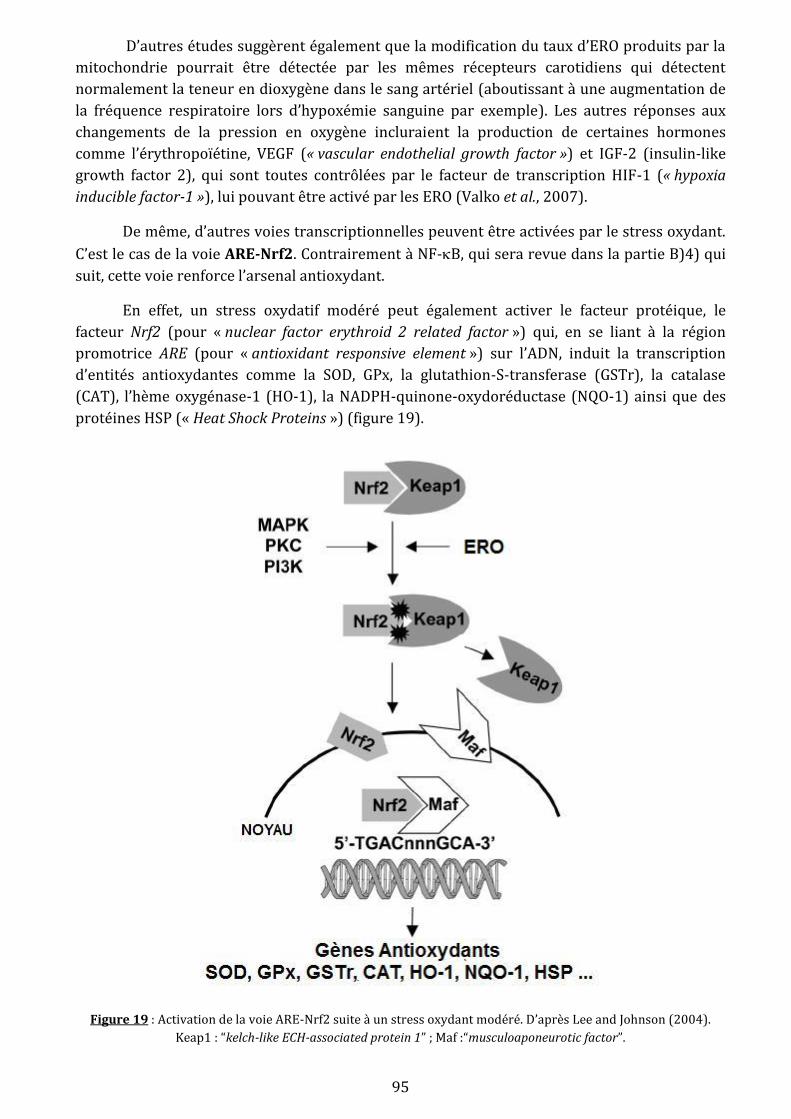
95
D’autres études suggèrent également que la modification du taux d’ERO produits par la
mitochondrie pourrait être détectée par les mêmes récepteurs carotidiens qui détectent
normalement la teneur en dioxygène dans le sang artériel (aboutissant à une augmentation de
la fréquence respiratoire lors d’hypoxémie sanguine par exemple). Les autres réponses aux
changements de la pression en oxygène incluraient la production de certaines hormones
comme l’érythropoïétine, VEGF (« vascular endothelial growth factor ») et IGF-2 (insulin-like
growth factor 2), qui sont toutes contrôlées par le facteur de transcription HIF-1 (« hypoxia
inducible factor-1 »), lui pouvant être activé par les ERO (Valko et al., 2007).
De même, d’autres voies transcriptionnelles peuvent être activées par le stress oxydant.
C’est le cas de la voie ARE-Nrf2. Contrairement à NF-B, qui sera revue dans la partie B)4) qui
suit, cette voie renforce l’arsenal antioxydant.
En effet, un stress oxydatif modéré peut également activer le facteur protéique, le
facteur Nrf2 (pour « nuclear factor erythroid 2 related factor ») qui, en se liant à la région
promotrice ARE (pour « antioxidant responsive element ») sur l’ADN, induit la transcription
d’entités antioxydantes comme la SOD, GPx, la glutathion-S-transferase (GSTr), la catalase
(CAT), l’hème oxygénase-1 (HO-1), la NADPH-quinone-oxydoréductase (NQO-1) ainsi que des
protéines HSP (« Heat Shock Proteins ») (figure 19).
Figure 19 : Activation de la voie ARE-Nrf2 suite à un stress oxydant modéré. D’après Lee and Johnson (2004).
Keap1 : “kelch-like ECH-associated protein 1” ; Maf :“musculoaponeurotic factor”.

96
Pour mémoire, un stress oxydant modéré entraîne aussi l’activation de NFAT (« Nuclear
Factor of Activated T cells ») ou encore AP-1 (« Activated protein-1 ») (Sagai and Bocci, 2011).
5) La production d’ERO pour la régulation de l’adhésion cellulaire
L’adhésion cellulaire joue un rôle très important dans l’embryogénèse, la croissance
cellulaire, la différentiation, la cicatrisation et d’autres processus cellulaires.
L’expression des molécules adhésives est notamment stimulée par le LPS et différentes
cytokines comme le TNF-, IL-1 et IL-1, mais aussi les ERO, notamment chez les leucocytes
lors de la margination sur le site inflammatoire (Valko et al., 2007).
6) La régulation redox de la réponse immunitaire
La réponse immunitaire est un phénomène soumis au contrôle redox de
l’environnement des cellules. Ainsi, l’activation des lymphocytes T est augmentée de manière
significative en présence d’ERO ou par une modification brutale (diminution) du statut
réducteur du milieu (Valko et al., 2007).
Aussi, des concentrations physiologiques en peroxyde d’hydrogène ou en radicaux
superoxyde O2°- peuvent accroître la production en IL-2 de ces lymphocytes. On a également
des preuves que le statut redox intracellulaire modifie les fonctions phagocytaires des
macrophages (Valko et al., 2007).
7) Implication des ERO dans la vie et la mort de la cellule
7.i ERO et cycle cellulaire
Sans que cela ne soit pas clairement élucidé, des modifications de l’environnement redox
de la cellule sont étroitement liées au cycle cellulaire. Ainsi, un environnement réducteur est
typique de la prolifération cellulaire. La différenciation cellulaire voit un environnement plus
oxydant se mettre en place. En effet, des concentrations inférieures à 10 mol/L en peroxyde
d’hydrogène dans une culture cellulaire in vitro peut favoriser la multiplication des cellules
(Halliwell and Guterridge, 2008).
Enfin, un environnement encore plus oxydant est aussi typique d’une cellule en voie
d’apoptose (Valko, 2011) (partie suivante).
7.ii ERO et apoptose
Le programme de mort cellulaire (apoptose) est un mécanisme nécessaire lors de
l’embryogénèse, mais aussi pour la destruction des cellules dysfonctionnelles qui présentent un
danger pour l’intégrité de l’organisme (Valko et al., 2007) (Alcaraz et al., 2013). L’apoptose
représente un déséquilibre entre des signaux positifs de « vie » et les signaux négatifs (dont
l’augmentation des dégâts oxydatifs sur la cellule et son ADN).
Concernant les ERO, il est intéressant de constater que le peroxyde d’hydrogène est
toxique pour de nombreuses cellules entre 10 à 100 mol/L, entraînant l’apoptose cellulaire et
la nécrose pour des concentrations supérieures (Halliwell and Gutteridge, 2008).
L’accumulation des dégâts oxydatifs sur les lipides membranaires, l’ADN et les protéines
intracellulaires sont notamment « détectés » par des protéines signals comme Bcl-2 (une
protéine située dans la membrane mitochondriale externe) (Valko et al., 2007). Ainsi, la

97
modification des télomères des chromosomes par les ERO peut également enclencher le
processus d’apoptose (Alcaraz et al., 2013) (voir précédemment).
L’activation de Bcl-2 active à son tour la protéine Bax. Bax entraîne la formation de
pores dans la membrane mitochondriale interne entraînant une fuite intracellulaires du
cytochrome c. En utilisant l’énergie de l’ATP, le cytochrome c le lie à la protéine Apaf-1
(« apoptotic protease activating factor-1 »). Cette étape est suivie par l’aggrégation de ces
complexes pour former des « apoptosomes » qui activent enfin des protéases, les caspases
(Valko et al., 2007) (Taha, 2011). L’activation des caspases, et notamment de la caspase-9 (qui
active en chaîne les caspases-3 et -7), ainsi que d’autres protéases, entraîne la « digestion » des
protéines structurelles de la cellule.
L’apoptose peut également être entraînée directement par les radicaux libres, comme
NO°. Elle est associée à une diminution de la concentration en cardiolipine dans la membrane
mitochondriale interne (responsable normalement de sa forte imperméabilité aux protons) et
le relargage du cytochrome c par la mitochondrie. Fait intéressant, les cellules endothéliales
sont par ailleurs résistantes à l’induction de l’apoptose par NO° ; cette résistance est
notamment liée à de fort taux intracellulaires en GSH selon Valko (2011).
Il a également été montré in vitro que la mise en incubation de sarcolemmes
cytoplasmiques avec des radicaux superoxydes (afin de mimer une situation de stress oxydant)
peut entraîner une libération de Ca2+ dans le milieu. D’autres études ont également mis en
évidence in vitro que d’autres radicaux comme les radicaux HO° et le monoxyde d’azote NO°
interagissent avec les groupes sulfhydryls –SH des récepteurs à la ryanodine, qui à leur tour
favorisent la sortie du Ca2+ du sarcolemme. Ce relargage en Ca2+ dans le cytosol des cellules
peut à son tour activer des protéines comme la protéine kinase C (PKC) qui peut activer à son
tour les protéines MAPKs aboutissant à l’apoptose (Valko, 2011).
Par ailleurs, la voie extrinsèque (dit « signaux de mort ») de l’apoptose peut également
être ativée par la transcription de Fas (protéine transmembranaire appartenant à la famille des
récepteurs du TNF) et de son ligant Fas-L (Krüger et al.. 2014).
L’apoptose elle même peut être génératrice de radicaux libres (Gutteridge and Halliwell,
2000). En effet, la perte graduelle du cytochrome c depuis l’espace intermembranaire favorise
la formation de radical superoxyde, et ce de deux manières. La première est simple, le
cytochrome c est un antioxydant de O2°- ; la deuxième vient du fait que lorsque le cytochrome c
est libéré, le flux électronique entre les complexes III et IV n’est plus possible, et la fuite
électronique s’intensifie (Taha, 2011).
8) Statut et maintien de l’équilibre redox au sein de la cellule et dans les systèmes
biologiques
Dans l’organisme, chaque compartiment, qu’il soit intracellulaire ou extracellulaire, peut
schématiquement être caractérisé par un potentiel redox propre, régulé de manière
homéostatique. C’est ce qu’on appelle l’équilibre redox. L’efficacité du système de défense
antioxydante endogène repose aussi sur sa coordination, de façon à maintenir cet équilibre.
En effet, chaque membre de la défense antioxydante a un rôle précis à jouer dans une
chaîne de détoxification des ERO (figure 20).

98
Figure 20: Exemple de chaîne de détoxification. D’après (Zicker et al., 2006)
La perte ou la surexpression d’un des acteurs et une perte de coordination dans la
réponse peuvent avoir des conséquences pathologiques.
Ainsi les SOD prennent en charge les anions superoxydes, mais produisent du H2O2, qui
doit à son tour être pris en charge par la CAT ou la GPx. La disponibilité du GSH est par ailleurs
importante pour l’activité de la GPx et le maintien de la cellule dans un état redox approprié,
qui repose à son tour sur l’activité de différentes enzymes, dont la glutathion réductase.
Une illustration de l’importance du maintien du milieu redox concerne le réticulum
endoplasmique, lieu de synthèse de nombreuses protéines, où le milieu oxydant favorise
notamment la formation de ponts disulfides cystéine-cystéine et l’acquisition de la structure
secondaire et tertiaire de la protéine. On sait bien aujourd’hui que le GSH et la TXN sont
également présents dans ce réticulum. On rappellera également que les radicaux libres et le
maintien de l’équilibre du potentiel redox du réticulum endoplasmique sont étroitement liés à
l’homéostasie du calcium Ca2+ (voir également plus loin avec le stress du réticulum
endoplasmique) (Raman and Berry, 2011).
Évaluer l’équilibre oxydo-réducteur d’un système biologique défini n’est pas chose
aisée : comme vu précédemment, le potentiel redox E° dépend notamment de la concentration
de chacun des éléments oxydés et réduits d’un couple en question. Or la cellule est un milieu
ouvert, où des échanges se produisent, et où les molécules assurant cet équilibre sont variées.
En effet, dans les systèmes biologiques, le potentiel redox des couples majeurs
thiols/disulfide (GSH et Trx) est maintenu dans un état stable redox de non équilibre (Raman
and Berry, 2011). En d’autres mots, la moyenne E° des couples redox du GSH et du Trx (les
principaux antioxydants non-enzymatiques) en concentration dans les compartiments
intracellulaires n’est pas égale à zéro. Ces couples redox, avec ceux des couples cystéine-

99
thiols/cystéine-disulfides des protéines (impliqués ou non dans le signalement redox) ne sont
pas en équilibre. Cest constatations sont également vraies pour le compartiment
extracellulaire.
Les compartiments nucléaire et mitochondrial tendent par exemple à être plutôt réduits,
(et cela s’entend : la mitochondrie étant un milieu générateur de radicaux et le noyau contenant
l’ADN) alors que le compartiment extracellulaire est plutôt oxydé, ainsi que le cytosol, le
réticulum endoplasmique et les lysosomes (Raman and Berry, 2011). Comme décrit
précédemment, la thiorédoxine et le glutathion sont des éléments centraux des réseaux de
contrôle de l’équilibre redox. La GSH-réductase et la thiorédoxine-réductase réduisent leur pool
respectif, maintenant ainsi cet équilibre redox.
Enfin, même en condition physiologique, l’équilibre instable redox varie selon le cycle ou
l’état de multiplication et de différenciation de la cellule (voir ci-dessus), selon des mécanismes
encore à explorer où les radicaux libres auraient leur place ; des variations faibles de ce
potentiel redox sont suffisantes pour distinguer des cellules en prolifération de cellules en
apoptose (Raman and Berry, 2011).
Le facteur de transcription Nrf2 déjà évoqué précédemment (figure 20) est un acteur
majeur dans la régulation de la défense antioxydante de l’organisme, en grande partie via le
contrôle des niveaux de GSH. Il a été décrit dans de nombreux tissus. Nrf2 intervient dans la
coordination de la réponse antioxydante, via sa liaison à des sites ARE (« antioxidant response
element ») sur le promoteur des gènes cibles pour activer leur transcription.
Les facteurs de transcription PGC1- et PGC1- sont aussi fortement surexprimés suite à
un stress oxydant. Ces facteurs sont impliqués dans l’activation de la transcription de plusieurs
enzymes antioxydantes endogènes comme la CAT, la GPx et les SOD (Précourt, 2011) (Taha,
2011). On pense également que le stress oxydant entraîné par l’effort physique serait
également responsable de l’activation des PGC1 et de la réponse antioxydante (Précourt, 2011).
Les études actuelles suggèrent que le statut redox régulerait l’activation du facteur NF-
B et l’expression des gènes (notamment pro-inflammatoires) liés (Taha, 2011). Cette voie
d’activation par le stress oxydant dans le cas de l’inflammation sera reprise dans la partie qui
suit.
B) Stress oxydant et processus inflammatoires
1) Quelques rappels sur l’inflammation
L’inflammation est un processus complexe de réaction à une aggression tissulaire,
endogène (ischémie-reperfusion, processus dégénératif …) ou exogène (agent infectieux,
traumatisme ...) Chaque tissu et chaque espèce répondent à leur manière et plus ou moins
spécifiquement aux aggressions.
Contrôlée, l’inflammation est nécessaire ; incontrôlée elle peut rapidement être néfaste
pour l’organisme et être impliquée dans la pathogénie de nombreuses affections, où les ERO
jouent un rôle plus ou moins prépondérant.
Trois étapes sont généralement mises en évidence :
- La phase d’initiation: cette phase est notamment caractérisée par des modifications
vasculaires (vasodilatation locale, augmentation de la perméabilité vasculaire) et l’activation

100
des cellules endothéliales. Cette dernière activation permet la synthèse de molécules
d’adhérence et chimiotactiques permettant le recrutement des leucocytes sur le site de
l’inflammation.
- La phase de propagation: elle est illustrée par la pénétration (après margination,
adhésion et diapédèse) et l’activation des leucocytes dans les tissus lésés. La phagocytose des
agents infectieux et des éléments étrangers a lieu à ce moment.
- La phase de réparation: elle correspond à la restauration ad integrum du tissu ou à la
formation d’un tissu de cicatrisation. Les fibroblastes prolifèrent particulièrement durant cette
phase, produisant collagène et protéoglycanes notamment.
2) Production d’ERO lors de l’inflammation
La réaction inflammatoire s’accompagne d’une surproduction d’ERO dans le milieu
inflammatoire et au sein des cellules du tissu inflammé. On doit cette production
principalement à l’activation de plusieurs enzymes déjà évoquées (Jungbluth, 2008):
- la stimulation d’enzymes constitutives : les NOS endothéliales (et
éventuellement neuronales), la xanthine oxydoréductase (XOR), les cyclooxygénases-1 (COX 1),
les Nox1.
- l’activation d’enzymes inductibles : iNOS, COX2 et Nox2 principalement.
Les mécanismes d’activation incluent de nombreux facteurs tels que plusieurs cytokines,
les éicosanoïdes (leucotriènes et prostaglandines), les protéines du complément, amines
vasoactives (histamine, sérotonine), molécules d’adhérence … Cependant, ils demeurent encore
largement méconnus et complexes.
Si les principales cellules productrices d’ERO sont les cellules phagocytaires, il ne faut
pas oublier que d’autres cellules plus spécifiques du tissu génèrent des ERO lors de
l’inflammation : synoviocytes et chondrocytes dans le cartilage, fibroblastes, cellules
endothéliales … Elles seront mises en évidence lors de l’étude des affections qui nous
intéressent.
3) ERO et phagocytose
La phagocytose est un processus biologique réalisé par les cellules phagocytaires
(essentiellement les polynucléaires neutrophiles, les macrophages et dans une moindre mesure
les polynucléaires éosinophiles) ayant un rôle essentiel dans la défense anti-infectieuse mais
aussi dans la destruction de cellules défectueuses ou mortes.
Les neutrophiles au repos consomment très peu de dioxygène et ont un métabolisme
reposant surtout sur la glycolyse anaérobie avec leur stock de glycogène pour leur synthèse
d’ATP. Les macrophages ont eux plus de mitochondries et reposent plus sur le fonctionnement
de la chaîne mitochondriale. La phagocytose est notamment caractérisée par une explosion
respiratoire avec une surconsommation d’oxygène ou « respiratory burst », pouvant atteindre
10 à 20 fois la consommation basale chez les neutrophiles activés (Halliwell and Guterridge,
2008). Il est également soumis à de nombreuses influences intraspécifiques et intraspécifiques,
tissulaires et aux cytokines principalement.
Cette surconsommation en dioxygène est corrélée à une augmentation de la
consommation en glucose, essentiellement liée à la l’activation de la voie des pentoses

101
phosphate produisant du NADPH. L’oxydation du NADPH est indispensable à la production de
radical superoxyde par la NADPH oxydase (Nox) (Halliwell and Gutteridge, 2008) (figure
suivante).
Figure 21 : Représentation schématique du « respiratory burst » avec l’activation de la NADPH oxydase. D’après
(Halliwell and Gutteridge, 2008).
Les récepteurs membranaires TLR (“Toll Like Receptor ”), comme TLR2 et TLR4, peuvent
activer la NADPH-oxidase via d’autres molécules (comme MyD88, pour illustration) et ainsi
produire des radicaux libres lors la réponse inflammatoire et immunitaire (Sagai and Bocci,
2011). Chez les neutrophiles l’activation de la Nox2 est activée par l’isoforme rac2 alors que
c’est l’isoforme rac1qui active la Nox2 chez les macrophages (Valko et al., 2007).
La dismutation du radical superoxyde produit par Nox2 en peroxyde d’hydrogène est
favorisée par l’acidité du phagolysosome. La concentration en peroxyde d’hydrogène dans les
vésicules de phagocytose peut atteindre 100 mol/L (Valko et al., 2007).
D’autres enzymes complètent également le rôle de Nox dans la production d’ERO :
- La myéloperoxydase, présente dans les phagolysosomes et produisant
l’hypochloride HOCl.
- La iNOs, présente essentiellement dans les macrophages, essentielle à la
synthèse du peroxynitrite. En effet, on a pu montrer que les macrophages activés produisent du
NO° conjointement à l’ion superoxyde O2°-, aboutissant à la production locale d’ion
peroxynitrite (Beckman and Koppenol, 1996).
- La xanthine oxydase (XO), présente physiologiquement en quantité plus
importante dans les cellules endothéliales et les cellules phagocytaires et pouvant être activée
par les médiateurs classiques de l’inflammation.
Pour autant, cette surproduction d’ERO lors de la phagocytose peut être délétère pour
les cellules qui la produisent mais aussi les tissus infiltrés puisqu’une partie des radicaux et des
espèces oxydantes diffusent en dehors des membranes ou sont libérés de manière non
spécifique avec les déchets de la phagocytose ; également, la mort des phagocytes relâche dans
le milieu les contenus des granules avec leurs ERO ainsi que les enzymes qui les produisent.

102
4) ERO et établissement du processus inflammatoire
S’il est vrai que l’inflammation entraîne du stress oxydant, un stress oxydant peut
également entraîner une inflammation. En effet, on sait que les ERO agissent comme un second
messager dans plusieurs mécanismes transductionnels inflammatoires (Sagai and Bocci, 2011).
Les ERO modulent notamment l’activation de facteurs de transcription tels que NF-B, le
facteur inductible par l’hypoxie HIF ou encore AP-1 chez de nombreux types cellulaires
(cellules épithéliales bronchiques, cellules endothéliales, macrophages alvéolaires,
neutrophiles et les mastocytes) (Moran et al., 2011) (Bureau et al., 2000b) (Rahman, 2003).
Le facteur de transcription pro-inflammatoire NF-B est classiquement activé par la
liaison de facteurs comme les lipopolysaccharides (LPS) ou le TNF- à leurs récepteurs. Les
radicaux libres (ROS) peuvent eux aussi mener à la dégradation de IB et entraîner l’activation
du NF-B (figure 22) (Halliwell and Guterridge, 2008) (Précourt, 2011). Ceci déclenche une
cascade de signalisation menant à la dégradation d’IkB, la protéine inhibitrice du NF-kB. Une
fois libéré d’IB, le NF-B est transloqué au noyau où il se lie à l’ADN pour induire l’expression
de différents gènes pro-inflammatoires.
L’activation de NF-kB induit en effet l’expression des gènes codant pour l’IL-6, le TNF-,
MIP-1L-8, IL-1, COX-2 (à l’origine de la synthèse de PGE2 notamment) ou encore plusieurs
autres facteurs responsable de réponse immunitaire de type Th-2 (Précourt, 2011) (Lekeux et
al., 2000) (Sagai and Bocci, 2011). NF- mène également à l’augmentation de l’expression des
molécules d’adhésion comme l’E-sélectine, ICAM (pour « IntraCellular Adhesion Molecule ») et
VCAM-1 (pour « Vascular Cell Adhesion Molecule-1 ») et ainsi facilite l’adhérence des cellules
phagocytaires (Précourt, 2011).
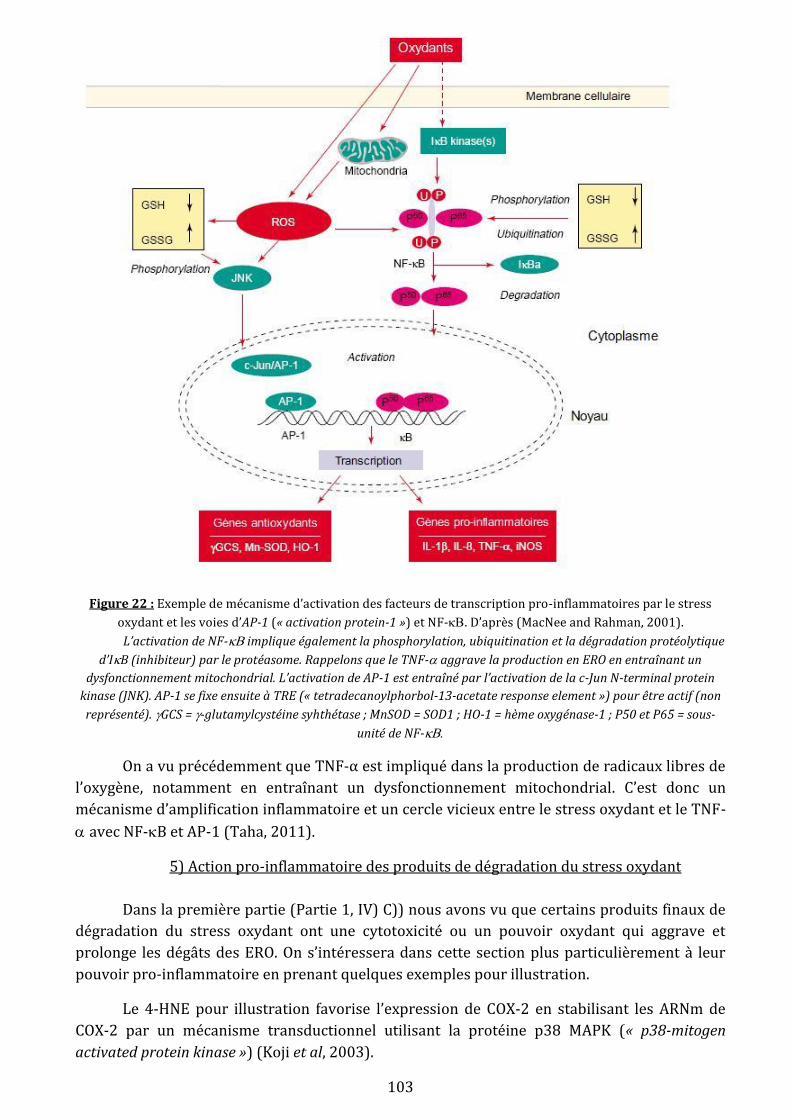
103
Figure 22 : Exemple de mécanisme d’activation des facteurs de transcription pro-inflammatoires par le stress
oxydant et les voies d’AP-1 (« activation protein-1 ») et NF-. D’après (MacNee and Rahman, 2001).
L’activation de NF- implique également la phosphorylation, ubiquitination et la dégradation protéolytique
d’IB (inhibiteur) par le protéasome. Rappelons que le TNF-aggrave la production en ERO en entraînant un
dysfonctionnement mitochondrial. L’activation de AP-1 est entraîné par l’activation de la c-Jun N-terminal protein
kinase (JNK). AP-1 se fixe ensuite à TRE (« tetradecanoylphorbol-13-acetate response element ») pour être actif (non
représenté).GCS = -glutamylcystéine syhthétase ; MnSOD = SOD1 ; HO-1 = hème oxygénase-1 ; P50 et P65 = sous-
unité de NF-
On a vu précédemment que TNF-α est impliqué dans la production de radicaux libres de
l’oxygène, notamment en entraînant un dysfonctionnement mitochondrial. C’est donc un
mécanisme d’amplification inflammatoire et un cercle vicieux entre le stress oxydant et le TNF-
avec NF-B et AP-1 (Taha, 2011).
5) Action pro-inflammatoire des produits de dégradation du stress oxydant
Dans la première partie (Partie 1, IV) C)) nous avons vu que certains produits finaux de
dégradation du stress oxydant ont une cytotoxicité ou un pouvoir oxydant qui aggrave et
prolonge les dégâts des ERO. On s’intéressera dans cette section plus particulièrement à leur
pouvoir pro-inflammatoire en prenant quelques exemples pour illustration.
Le 4-HNE pour illustration favorise l’expression de COX-2 en stabilisant les ARNm de
COX-2 par un mécanisme transductionnel utilisant la protéine p38 MAPK (« p38-mitogen
activated protein kinase ») (Koji et al, 2003).

104
Les produits issus de la glucoxydation favorisent également le maintien de
l’inflammation. Ainsi, la glycoxydation du collagène, notamment du cartilage articulaire,
augmente les propriétés chimiotactiques des polynucléaires neutrophiles (Alcaraz et al., 2013).
Les effets délétères pro-inflammatoires des produits de la glycoxydation sont aussi médiés par
des récepteurs. Au moins quatre récepteurs on été mis en évidence. On a montré que lorsque
l’un d’entre eux, le récepteur RAGE (pour « receptor of AGEs »), largement répandu dans les
tissus, est incubé avec des AGEs, on observe dans plusieurs types cellulaires (dont les
neutrophiles) une synthèse de radicaux libres accrue et l’activation du facteur NF-B (Sagai and
Bocci, 2011). Également, un modèle expérimental d’étude de l’insuffisance rénale chronique
chez l’homme a montré que les protéines fortement oxydées, dont les AGEs, agissent comme
des médiateurs du stress oxydant dans l’activation et l’amplification du « respiratory burst »
chez les monocytes et les macrophages (Witko-Sarsat et al., 1998).
De même, les produits issus de l’oxydation de l’ADN mitochondrial (comme les
guanosines déméthylées) peuvent activer le facteur NF-B (Collins, 2004).
Enfin, certains isoprostanes sont instables même à la température corporelle et peuvent
subir des réarrangements spontanés et former des prostaglandines biologiquement actives et
pro-inflammatoires (Halliwell and Gutteridge, 2008).
6) Stress oxydant et ER-stress
Le « stress du reticulum endoplasmique » ou ER-stress permet également de faire le lien
entre le stress oxydant, l’apoptose et de nombreux phénomènes inflammatoires. Certains
auteurs en parlent comme une entité clé dans la description de la physiopathologie de certaines
maladies, dont il convient de donner une brève définition dans cet exposé.
6.i ER-stress et perturbation du métabolisme calcique
En conditions physiologiques, le Ca2+ relargué par le réticulum endoplasmique (par
exemple lors d’une contraction musculaire dans le myocyte) est en partie capté par la
mitochondrie et favorise la chaîne respiratoire de phosphorylation oxydative.
Or on sait que l’état d’ouverture des canaux calciques du réticulum endoplasmique est
intimement lié à la présence de radicaux libres ainsi que l’état rédox du milieu (un
environnement trop oxydant favorisant leur ouverture) (Raman and Berry, 2011). En
l’occurrence, les ERO peuvent également s’attaquer à des canaux calciques comme des
récepteurs à la ryanodine (voir la partie précédente sur l’apoptose) ce qui exacerbe le relargage
de calcium intracellulaire (Zhang ,2009).
De même, lors de la présence d’un stress oxydant, le relargage massif de Ca2+ par le
réticulum endoplasmique dans le cytosol est également accompagné par l’augmentation de la
concentration en Ca2+ dans la mitochondrie (Taha, 2011). Des études récentes suggèrent que la
mitochondrie peut former des jonctions avec le réticulum endoplasmique, ce qui pourrait
fournir une plateforme d’interaction privilégiée entre les radicaux libres et le calcium (Zhang,
2009).
Il semblerait que l’hypercalcémie mitochondriale entraîne une modification dans la
conformation tri-dimentionnelle des complexes respiratoires, aggravant la fuite des électrons
de la chaîne mitochondriale (Zhang ,2009).

105
Un cercle auto-aggravant se met en place, pouvant éventuellement se terminer par
l’apoptose de la cellule : c’est l’ER-stress.
6.ii ER-stress et réponse UPR
L’ ER-stress est un phénomène caractérisé par une augmentation des radicaux libres au
sein du réticulum endoplasmique, accompagné d’une « unfolded protein response » ou réponse
UPR de la cellule (Précourt, 2011). Cet état a notamment pour conséquence l’accumulation de
protéines non ou mal-repliées dans le réticulum endoplasmique (d’où son nom), qui stimule
certaines réponses inflammatoires, pro-apoptotiques, mais aussi antioxydantes dans les étapes
précoces de l’ER-stress (Zhang, 2009).
6.iii Bilan de l’ER-stress
Figure 23 : Échanges croisés entre stress oxydant, ER-stress et la mitochondrie. D’après (Zhang, 2009).
Ainsi, le réticulum endoplasmique jouerait le rôle d’une plateforme d’interaction entre
les ERO, le calcium intracellulaire et la mitochondrie en modulant les réponses inflammatoires
et l’apoptose.
C) Stress oxydant et ischémie-reperfusion
1) Principales modalités de production d’ERO lors de l’ischémie- reperfusion
L’ischémie-reperfusion est un mécanisme physio-pathologique présent dans plusieurs
affections, notamment traumatiques, susceptibles d’être retrouvées pendant l’effort.

106
Si la mise en évidence de son implication directe n’est parfois pas si aisée, l’ischémie-
reperfusion est génératrice d’ERO suivant les modalités principales suivantes :
- l’activation de la xanthine oxydase lors de la phase d’ischémie,
- le dérèglement de la chaîne respiratoire mitochondriale lors de la phase précoce
de la reperfusion (dysfontionnement mitochondrial),
- le burst oxydatif des cellules phagocytaires lors de la phase tardive de la
reperfusion,
- le métabolisme de l’acide arachidonique.
L’ischémie-reperfusion est si efficace que les antioxydants comme la SOD ou GPX
(notamment mitochondriaux) sont souvent incapable de contrer la production massive d’ERO
(Andrades and Lorenzi, 2011).
2) Phase d’ischémie : phase préparatrice
2.i Chute de la production d’ATP et accumulation de produits de dégradation
La phase d’ischémie est particulièrement importante. Il a été montré qu’il existe une
corrélation positive entre sa durée et la sévérité des lésions, allant du dysfonctionnement
cellulaire (et mitochondrial) à la nécrose (Soffler, 2007).
Lors de la phase d’hypoxémie voire d’ischémie, la cellule ne peut plus utiliser le
dioxygène pour la chaîne respiratoire et la glycolyse est la seule manière de produire de l’ATP.
Cette situation mène rapidement à une surproduction de lactates à l’origine d’une acidose. De
plus, avec la baisse des substrats de la glycolyse (comme le glycogène), la production d’ATP
chute et une accumulation de ses produits de dégradation comme l’hypoxanthine se met en
place. L’ADP formé par la diminution de l’ATP est oxydé pour former de l’AMP, de l’adénosine,
de l’hypoxanthine et de la xanthine (Andrades and Lorenzi, 2011).
Des études ont également montré dans le myocarde et le cerveau en état d’ischémie une
accumulation d’acide arachidonique non estérifié, qui se poursuit au rétablissement de la
perfusion. Cocco et al. (1999) ont montré dans des mitochondries de cœur de bovin que l’acide
arachidonique, mais aussi d’autres acides gras à longues chaînes, représentent des médiateurs
de la synthèse d’espèces réactives. En effet, l’accumulation de calcium intracellulaire active des
phospholipases membranaires comme la phospholipase-A2 qui entraîne le relargage d’acide
arachidonique. Ce relargage active alors les cyclooxygénases et leur potentialité pro-
inflammatoires et pro-oxydantes (Al-Qudah and Al-Majali, 2008).
Enfin, la phase d’ischémie favorise le relargage de métaux de transition dans le milieu
suite à la désorganisation du métabolisme mitochondrial, métaux eux-même propices à la
génèse d’ERO (Halliwell and Guterridge, 2008).
2.ii État mitochondrial MTP
On a également découvert qu’après un épisode d’ischémie (et même après un trauma) la
concentration en Ca2+ extracellulaire chute drastiquement, et ce corrélé à une accumulation de
Ca2+ intracellulaire (Al-Qudah and Al-Majali, 2008), vraisemblablement selon les modalités de
l’ER-stress.
Ces modifications intracellulaires et la perturbation du métabolisme énergétique
causent l’ouverture des canaux MTP (pour « mitochondrial permeability transition » , canaux

107
voltage et Ca2+-dépendants). Celle-ci mène à l’augmentation de la perméabilité de la membrane
mitochondriale interne ayant pour conséquence la dissipation du gradient protonique (qui
permet le fonctionnement de la chaîne respiratoire) et le relargage dans le cytosol du
cytochrome c, de radicaux libres et de facteurs activant le processus d’apoptose (Halliwell and
Gutteridge, 2008) (Andrades and Lorenzi, 2011).
Cet état mitochondrial MTP accompagne le réarrangement de protéines membranaires
(comme la cyclophiline D ou la VDAC pour « Voltage Dependant Anion Transporter ») créant des
pores non spécifiques entre les membranes mitochondriales interne et externe (Halliwell and
Gutteridge, 2008). Ils permettent alors l’entrée de l’eau dans la mitochondrie, qui mène à la
rupture finale de l’organite et la mort cellulaire.
Remarquons que dans les cas graves d’ischémie-reperfusion, la libération massive des
radicaux libres et l’arrêt de la synthèse en ATP mène à la nécrose cellulaire sans laisser le
temps aux processus apoptotiques de se mettre en place (Andrades and Lorenzi, 2011).
Selon certains auteurs, cet état MTP pourrait, sous certaines circonstances comme au
début de l’ischémie, entraîner une production accrue d’O2°- en réarrangeant les transporteurs
des électrons présents au sein du complexe I de la chaîne (Halliwell and Gutteridge, 2008). En
effet, la production de radicaux libres peut résulter d’une augmentation de la consommation
mitochondriale en oxygène lors d’hypoxémie, en réponse à un déséquilibre entre le débit
d’oxygène et le transport des électrons au niveau de la chaîne respiratoire (Fluck, 2005).
2.iii Conversion de la xanthine déshydrogénase (XDH) en xanthine oxydase
(XO)
Dans un tissu sain, presque toutes les réactions d’oxydation de la xanthine et de
l’hypoxanthine en acide urique sont catalysées par la xanthine déshydrogénase (XDH) ; cette
catalyse ne mène pas à la genèse de radical superoxide.
Cependant, lors de l’ischémie, les XDH sont transformées en xanthine oxydases (XO) par
clivage protéolytique et oxydation de groupements thiols (Halliwell and Gutteridge, 2008). En
effet, le relargage intracellulaire de Ca2+ active des protéases à l’origine de ces clivages
(Andrades and Lorenzi, 2011). L’accumulation d’hypoxanthine lors de cette phase favorise
l’activité des XO et la formation en quantité importante de peroxyde d’hydrogène et de radical
superoxide.
3) Reperfusion et production d’ERO
Au cours de la phase précoce de la reperfusion, les cellules qui ne sont pas mortes mais
dont les mitochondries sont dysfonctionnelles sont à l’origine d’une forte production d’ERO
(Hermes et al., 2002). En plus du radical superoxyde, il a aussi été mis en évidence in vitro que
lors de la reperfusion le radical semiquinone (ou ubisemiquinone, radical issu de la
déshydrogénation de l’hydroquinone) et des radicaux libres de l’azote sont également produits
depuis la mitochondrie (Andrades and Lorenzi, 2011).
Plus tard, l’envahissement du site ischémié par les cellules phagocytaires du système
immunitaire (neutrophiles et macrophages principalement) sont également fortement
générateurs d’ERO et délabrantes pour le tissu concerné (Hermes et al., 2002) (Halliwell and
Gutteridge, 2008).

108
Enfin, le rétablissement de l’apport en glucose et en oxygène restaure les niveaux en ATP
nécessaires à la réalisation de l’apoptose « préparée » par la phase d’ischémie (Andrades and
Lorenzi, 2011).

109
2ième Partie : Stress oxydant et physiopathologie équine chez le sujet athlète.
I) Pré-requis de physiologie de l’effort
A) Consommation de dioxygène et effort physique
1) Consommation en dioxygène et VO2max
1.i Définition de la consommation en dioxygène
La consommation d’oxygène par l’organisme par unité de temps (notée VO2), se définie comme suit :
VO2 = DC x (CaO2 – CvO2), exprimé en mL/kg/min
Avec :
- DC = le debit cardiaque ;
- CaO2 = concentration artérielle en oxygène, déterminée par la ventilation, l’hémoglobinémie
et les échanges alvéolaires ;
- CvO2 = concentration veineuse en dioxygène, déterminée par la consommation tissulaire
(intensité de la voie aérobie, circulation veineuse).
1.ii Détermination expérimentale VO2max
Dans les études concernant le cheval, les animaux sont généralement équipés d’un
masque imperméable fixé sur le museau et équipé de valves pour permettre la sortie et l’entrée
d’air (De Moffarts et al., 2004a); le flux respiratoire de chaque narine est mesuré en utilisant un
pneumotachographe (unité L/s généralement). Un spectromètre de masse est utilisé pour
mesurer la concentration en O2 et en CO2 dans l’air inspiré et dans l’air expiré, et ce à chaque
cycle respiratoire. Ces mesures permettent d’approximer CaO2 et CvO2 et alors déterminer la
VO2.
En compilant chacune des valeurs de VO2 sur un graphe on peut déterminer la VO2max
pour chaque cheval.
La prise maximale en dioxygène, c’est à dire la VO2max, du cheval peut atteindre
jusqu’à 200 mL d’O2/kg/min, ce qui représente plus de deux à trois fois la VO2max de l’homme
(Minami et al., 2011). C’est une des raisons pour lesquelles le cheval est un excellent candidat
pour l’étude du stress oxydant en condition d’effort physique.
Lorsqu’on considère un athlète en particulier et ses performances, une valeur élevée de
la VO2max constitue une bonne adaptation à l’effort.
1.iii PMA = puissance maximale aérobie
Une valeur intéressante à souligner est la puissance maximale aérobie (PMA), c’est à
dire la plus petite puissance d’exercice qui entraîne la consommation maximale d’oxygène que
le sujet athlète est capable d’atteindre (VO2max). Ensemble la PMA et la VO2max représentent les
capacités maximales de distribution, de transport de l’oxygène par le sang et d’extraction de
l’oxygène par le muscle. Elles constituent ainsi la mesure simple de l’aptitude aérobie lors d’un
effort.

110
2) Différents types d’effort selon la consommation en dioxygène
Lors de l’effort sous-maximal, la VO2 augmente progressivement avant d’atteindre un
faux plateau dont la valeur limite est la VO2max. Un effort maximal est défini comme atteignant
VO2max. La puissance de travail peut être augmentée au-delà de ce niveau mais en sollicitant le
métabolisme anaérobie (effort supra-maximal). L’aptitude à réaliser un exercice supra-
maximal, c’est-à-dire supérieur à la puissance maximale aérobie, correspond à la notion de
résistance.
Par définition, un exercice sera dit infra-maximal si sa puissance est inférieure à la
puissance maximale aérobie (PMA). L’aptitude à prolonger ce type d’exercice correspond à
l’endurance (30-60% VO2max chez le cheval en général).
B) Métabolisme énergétique lors de l’effort
1) Les voies métaboliques de production d’ATP durant l’effort
1.i Voie anaérobie alactique
La filière anaérobie alactique (sans production de lactate), mise en jeu pour des
efforts intenses d’une durée inférieure à quelques dizaines de secondes, utilise la
phosphocréatine musculaire (PCr) dont les réserves sont très faibles mais rapidement
reconstituées (à partir des deux autres filières). Au repos, la mitochondrie utilise l’ATP formé
par la voie aérobie pour constituer des stocks de PCr suivant la réaction :
Cr + ATP PCr + ADP.
Au tout début de l’effort, où il faut fournir rapidement une grande quantité d’ATP pour la
contraction musculaire, l’ATP est reconstitué suivant la réaction inverse :
PCr + ADP Cr + ATP.
Le rendement énergétique est ici voisin de 100 % et cette filière, qui permet de
développer des puissances considérables, représente un système parfaitement adapté aux
variations importantes des besoins en ATP (Oppert et al., 2005).
1.ii Voie anaérobie lactique
La filière anaérobie lactique, mise en jeu pour des efforts intenses d’une durée
supérieure à 10-15 secondes, utilise le glycogène musculaire par la glycolyse anaérobie
aboutissant à la production de lactate. Cette filière est capable d’assurer des puissances
maximales plus faibles que la filière précédente.
La participation glycolytique est de plus en plus importante au fur et à mesure que la
puissance augmente, jusqu’à devenir exclusive pour des puissances proches de la puissance
aérobie maximale. Il faut noter que pour des puissances comprises entre 50 et 100 % de la
puissance maximale aérobie (infra-maximaux à maximaux), la cellule musculaire est
progressivement obligée de faire appel à la glycolyse anaérobie pour couvrir les besoins
énergétiques.
Entre 100 et 150 % de la puissance maximale aérobie (c’est-à-dire pour un -exercice en
résistance, supra-maximal), la glycolyse anaérobie devient prépondérante, la concentration de
lactates sanguins et musculaires augmente, limitant la durée de l’exercice. Au dessus de 150%

111
de la puissance maximale aérobie, les besoins en ATP sont tellement élevés que seule la filière
anaérobie alactique peut y répondre pendant un temps très court correspondant à
l’épuisement des stocks de phosphocréatine (Oppert et al., 2005).
1.iii Voie aérobie
La filière aérobie, mise en jeu pour des efforts prolongés au-delà de quelques minutes,
représente le système le plus important de fourniture de l’ATP, principalement à partir de
l’oxydation des substrats glucidiques (glycogène, glucose plasmatique) et lipidiques (acides
gras libres plasmatiques libérés par le tissu adipeux, triglycérides intramusculaires) au niveau
de la chaîne respiratoire mitochondriale. Ce dernier mécanisme présente l’avantage d’une
capacité énergétique importante grâce aux graisses de l’organisme.
La puissance maximale développée par cette filière, plus faible qu’avec les deux
premières, dépend directement de la capacité de l’organisme à fournir de l’oxygène aux
muscles d’une part et du rendement musculaire d’autre part.
Lors d’un exercice en endurance, la proportion de la dépense énergétique dérivée de
l’oxydation des lipides augmente au fur et à mesure que l’intensité de l’exercice augmente.
L’inverse se produit pour les glucides. En théorie, on considère généralement que le niveau le
plus élevé d’oxydation des lipides, en valeur relative, est observé pour des activités d’intensité
moyenne correspondant à 50-60 % de la VO2max (valeurs chez l’homme appliquées également
chez le cheval). La participation relative de l’un ou l’autre substrat dépend en grande partie de
la puissance développée : la -oxydation et l’utilisation des lipides sont prépondérantes pour
une puissance faible (Oppert et al., 2005).
1.iv La dette en oxygène
La « dette en oxygène » concerne essentiellement ces efforts brefs et intenses supra-
maximaux comme le sprint lorsque le muscle utilise plus d’énergie que le système aérobie ne
pourrait en fournir, notamment au début de l’effort.
Le début de l'exercice est anaérobique et l'adaptation ventilatoire se produit en 3 à 4
minutes. Ce retard dans la consommation d'oxygène par rapport aux besoins théoriques qui se
constituent au début de l'exercice est restitué après arrêt de l'effort grâce à une consommation
supérieure aux besoins. Ce remboursement d'oxygène est toujours supérieur à la dette
d'oxygène, car il comprend l'oxygène nécessaire à certains mécanismes (comme la
retransformation de l'acide lactique).
Ce délai de 3 minutes est nécessaire pour atteindre la consommation maximale
d’oxygène car le délai d’apparition de la consommation maximale d’oxygène par l’organisme
provient de l’inertie des adaptations cardio-vasculaires et tissulaires à l’effort: augmentation du
débit cardiaque, modification de la répartition de la masse sanguine corporelle, augmentation
du débit sanguin musculaire, activation des réactions d’oxydation de la fibre musculaire. Ce
delai peut être réduit avec l’entraînement.
2) Types de fibres musculaires
Les fibres musculaires sont principalement classées en trois types principaux dont le
métabolisme est différent :
- Les fibres lentes (« fibres rouges ») ou fibres de type I ;
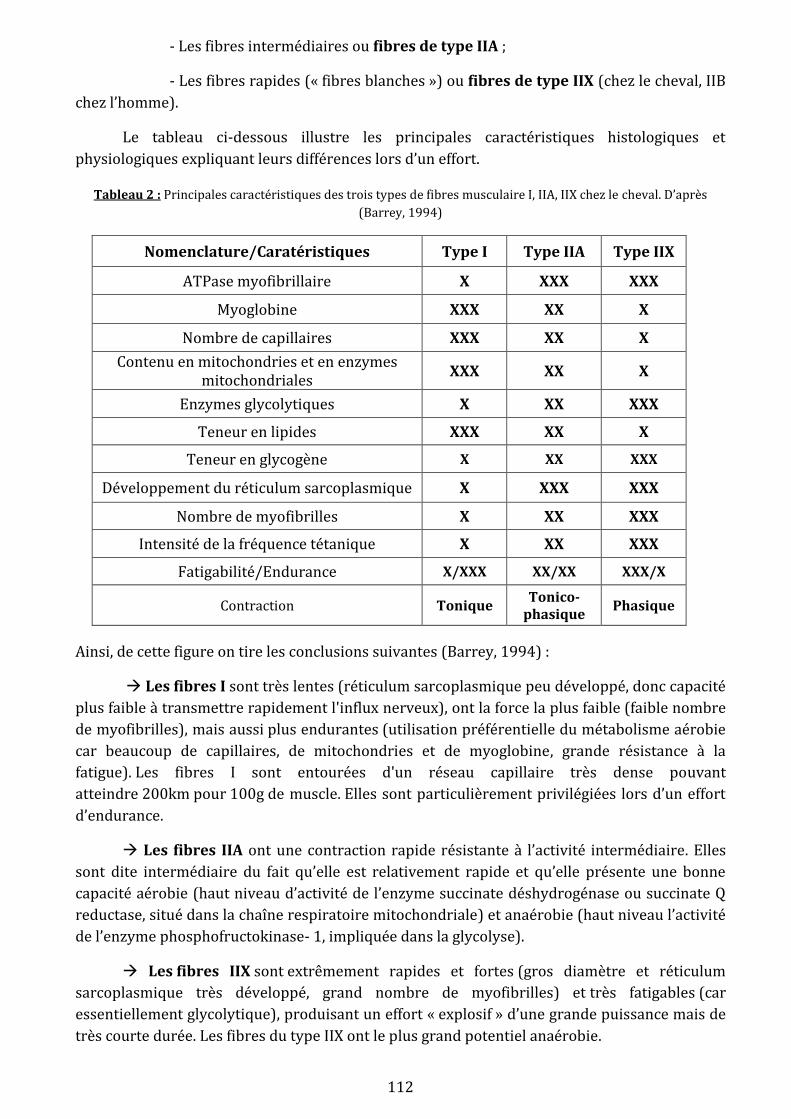
112
- Les fibres intermédiaires ou fibres de type IIA ;
- Les fibres rapides (« fibres blanches ») ou fibres de type IIX (chez le cheval, IIB
chez l’homme).
Le tableau ci-dessous illustre les principales caractéristiques histologiques et
physiologiques expliquant leurs différences lors d’un effort.
Tableau 2 : Principales caractéristiques des trois types de fibres musculaire I, IIA, IIX chez le cheval. D’après
(Barrey, 1994)
Nomenclature/Caratéristiques Type I Type IIA Type IIX
ATPase myofibrillaire X XXX XXX
Myoglobine XXX XX X
Nombre de capillaires XXX XX X
Contenu en mitochondries et en enzymes mitochondriales
XXX XX X
Enzymes glycolytiques X XX XXX
Teneur en lipides XXX XX X
Teneur en glycogène X XX XXX
Développement du réticulum sarcoplasmique X XXX XXX
Nombre de myofibrilles X XX XXX
Intensité de la fréquence tétanique X XX XXX
Fatigabilité/Endurance X/XXX XX/XX XXX/X
Contraction Tonique Tonico-
phasique Phasique
Ainsi, de cette figure on tire les conclusions suivantes (Barrey, 1994) :
Les fibres I sont très lentes (réticulum sarcoplasmique peu développé, donc capacité
plus faible à transmettre rapidement l'influx nerveux), ont la force la plus faible (faible nombre
de myofibrilles), mais aussi plus endurantes (utilisation préférentielle du métabolisme aérobie
car beaucoup de capillaires, de mitochondries et de myoglobine, grande résistance à la
fatigue). Les fibres I sont entourées d'un réseau capillaire très dense pouvant
atteindre 200km pour 100g de muscle. Elles sont particulièrement privilégiées lors d’un effort
d’endurance.
Les fibres IIA ont une contraction rapide résistante à l’activité intermédiaire. Elles
sont dite intermédiaire du fait qu’elle est relativement rapide et qu’elle présente une bonne
capacité aérobie (haut niveau d’activité de l’enzyme succinate déshydrogénase ou succinate Q
reductase, situé dans la chaîne respiratoire mitochondriale) et anaérobie (haut niveau l’activité
de l’enzyme phosphofructokinase- 1, impliquée dans la glycolyse).
Les fibres IIX sont extrêmement rapides et fortes (gros diamètre et réticulum
sarcoplasmique très développé, grand nombre de myofibrilles) et très fatigables (car
essentiellement glycolytique), produisant un effort « explosif » d’une grande puissance mais de
très courte durée. Les fibres du type IIX ont le plus grand potentiel anaérobie.
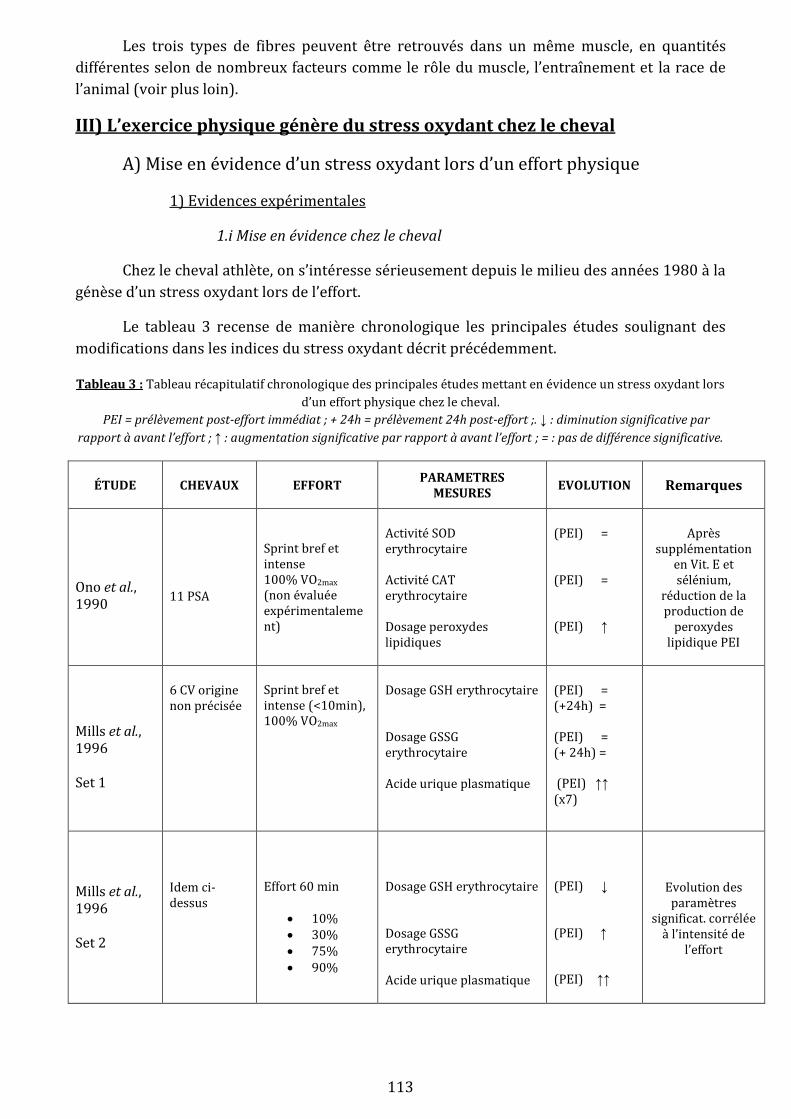
113
Les trois types de fibres peuvent être retrouvés dans un même muscle, en quantités
différentes selon de nombreux facteurs comme le rôle du muscle, l’entraînement et la race de
l’animal (voir plus loin).
III) L’exercice physique génère du stress oxydant chez le cheval
A) Mise en évidence d’un stress oxydant lors d’un effort physique
1) Evidences expérimentales
1.i Mise en évidence chez le cheval
Chez le cheval athlète, on s’intéresse sérieusement depuis le milieu des années 1980 à la
génèse d’un stress oxydant lors de l’effort.
Le tableau 3 recense de manière chronologique les principales études soulignant des
modifications dans les indices du stress oxydant décrit précédemment.
Tableau 3 : Tableau récapitulatif chronologique des principales études mettant en évidence un stress oxydant lors
d’un effort physique chez le cheval.
PEI = prélèvement post-effort immédiat ; + 24h = prélèvement 24h post-effort ;. ↓ : diminution significative par
rapport à avant l’effort ; ↑ : augmentation significative par rapport à avant l’effort ; = : pas de différence significative.
ÉTUDE
CHEVAUX
EFFORT PARAMETRES
MESURES EVOLUTION Remarques
Ono et al., 1990
11 PSA
Sprint bref et intense 100% VO2max (non évaluée expérimentalement)
Activité SOD erythrocytaire Activité CAT erythrocytaire Dosage peroxydes lipidiques
(PEI) = (PEI) = (PEI) ↑
Après supplémentation
en Vit. E et sélénium,
réduction de la production de
peroxydes lipidique PEI
Mills et al., 1996 Set 1
6 CV origine non précisée
Sprint bref et intense (<10min), 100% VO2max
Dosage GSH erythrocytaire Dosage GSSG erythrocytaire Acide urique plasmatique
(PEI) = (+24h) = (PEI) = (+ 24h) = (PEI) ↑↑ (x7)
Mills et al., 1996 Set 2
Idem ci-dessus
Effort 60 min
10% 30% 75% 90%
Dosage GSH erythrocytaire Dosage GSSG erythrocytaire Acide urique plasmatique
(PEI) ↓ (PEI) ↑
(PEI) ↑↑
Evolution des paramètres
significat. corrélée à l’intensité de
l’effort
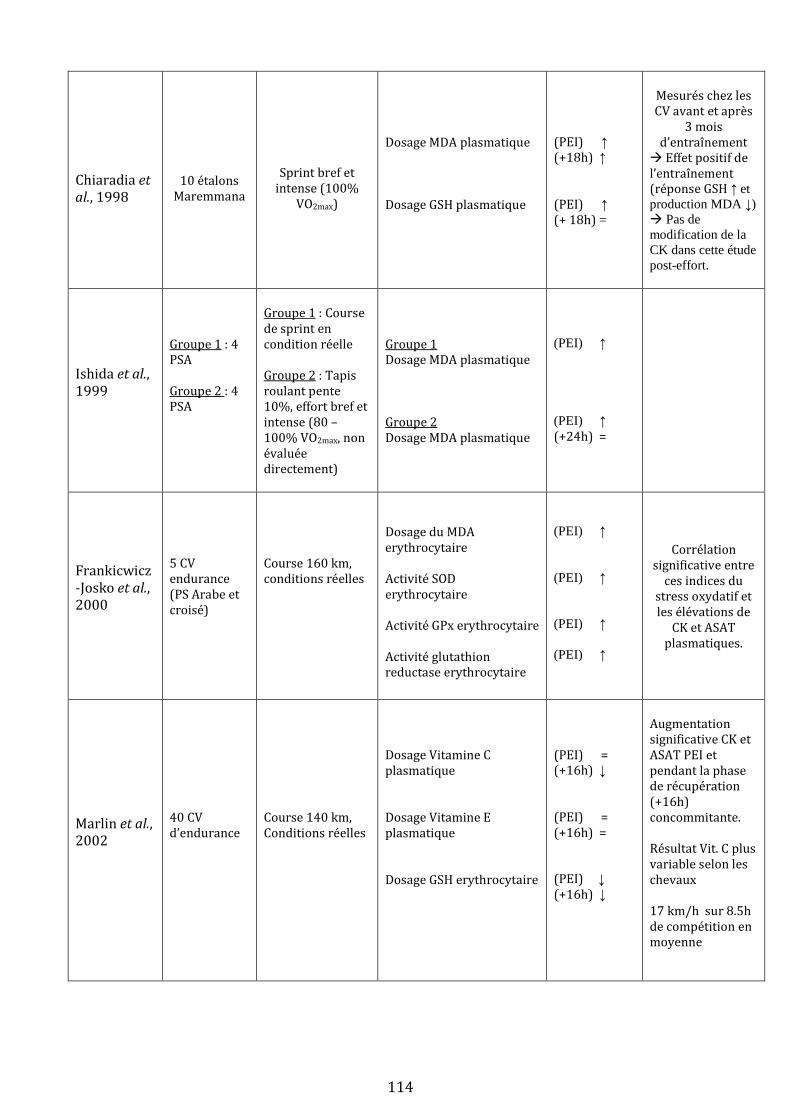
114
Chiaradia et al., 1998
10 étalons Maremmana
Sprint bref et
intense (100% VO2max)
Dosage MDA plasmatique Dosage GSH plasmatique
(PEI) ↑ (+18h) ↑ (PEI) ↑ (+ 18h) =
Mesurés chez les CV avant et après
3 mois d’entraînement Effet positif de l’entraînement (réponse GSH ↑ et
production MDA ↓)
Pas de
modification de la
CK dans cette étude
post-effort.
Ishida et al., 1999
Groupe 1 : 4 PSA Groupe 2 : 4 PSA
Groupe 1 : Course de sprint en condition réelle Groupe 2 : Tapis roulant pente 10%, effort bref et intense (80 – 100% VO2max, non évaluée directement)
Groupe 1 Dosage MDA plasmatique Groupe 2 Dosage MDA plasmatique
(PEI) ↑
(PEI) ↑
(+24h) =
Frankicwicz-Josko et al., 2000
5 CV endurance (PS Arabe et croisé)
Course 160 km, conditions réelles
Dosage du MDA erythrocytaire Activité SOD erythrocytaire Activité GPx erythrocytaire Activité glutathion reductase erythrocytaire
(PEI) ↑
(PEI) ↑
(PEI) ↑
(PEI) ↑
Corrélation significative entre
ces indices du stress oxydatif et les élévations de
CK et ASAT plasmatiques.
Marlin et al., 2002
40 CV d’endurance
Course 140 km, Conditions réelles
Dosage Vitamine C plasmatique Dosage Vitamine E plasmatique Dosage GSH erythrocytaire
(PEI) = (+16h) ↓
(PEI) = (+16h) =
(PEI) ↓ (+16h) ↓
Augmentation significative CK et ASAT PEI et pendant la phase de récupération (+16h) concommitante. Résultat Vit. C plus variable selon les chevaux 17 km/h sur 8.5h de compétition en moyenne
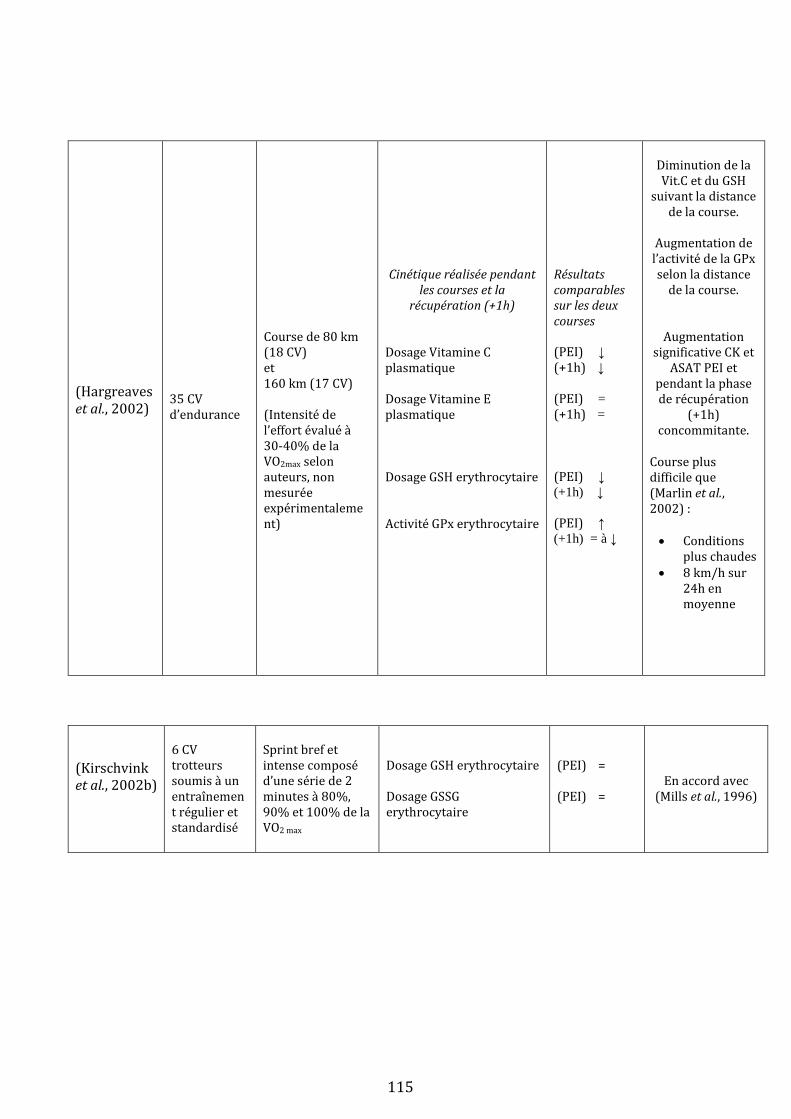
115
(Hargreaves et al., 2002)
35 CV d’endurance
Course de 80 km (18 CV) et 160 km (17 CV) (Intensité de l’effort évalué à 30-40% de la VO2max selon auteurs, non mesurée expérimentalement)
Cinétique réalisée pendant les courses et la
récupération (+1h) Dosage Vitamine C plasmatique Dosage Vitamine E plasmatique Dosage GSH erythrocytaire Activité GPx erythrocytaire
Résultats comparables sur les deux courses (PEI) ↓ (+1h) ↓
(PEI) = (+1h) =
(PEI) ↓
(+1h) ↓ (PEI) ↑
(+1h) = à ↓
Diminution de la Vit.C et du GSH
suivant la distance de la course.
Augmentation de l’activité de la GPx selon la distance
de la course.
Augmentation significative CK et
ASAT PEI et pendant la phase de récupération
(+1h) concommitante.
Course plus difficile que (Marlin et al., 2002) :
Conditions plus chaudes
8 km/h sur 24h en moyenne
(Kirschvink et al., 2002b)
6 CV trotteurs soumis à un entraînement régulier et standardisé
Sprint bref et intense composé d’une série de 2 minutes à 80%, 90% et 100% de la VO2 max
Dosage GSH erythrocytaire Dosage GSSG erythrocytaire
(PEI) = (PEI) =
En accord avec (Mills et al., 1996)

116
(De Moffarts et al., 2004a)
6 CV trotteurs français
Sprint (trot) bref et intense (pente 10%, vitesse maxi 36 km/h pendant 4 min atteint progressivement), après échauffement au pas.
Dosage Vitamine C plasmatique Dosage Vitamine E plasmatique Dosage Vitamine A plasmatique Dosage GSH érythrocytaire Dosage GSSG érythrocytaire Dosage de l’acide urique plasmatique Dosage cuivre plasmatique Dosage zinc plasmatique Dosage sélénium plasmatique Activité GPx érythrocytaire Activité de la SOD érythrocytaire
(PEI) ↑ (PEI) = (PEI) ↓ (PEI) ↓ (PEI) = (PEI) ↑
(PEI) = (PEI) = (PEI) = (PEI) = (PEI) =
Mesurés chez les CV avant, 1 mois et 2 mois après entraînement
régulier et standardisé.
Effet positif de
l’exercice et atténuation des
variations.
(Fazio et al., 2009)
10 PSA galopeurs femelles de 5 ans en bonne santé.
Effort bref et intense : Course officielle de galop sur 2100 mètres (sprint).
Mesure des d-ROMS Oxy-adsorbent Barrière antioxydante thiols (SHp) Dosage homocystéine plasmatique
Mise en évidence d’une régression linéaire significative entre les valeurs d’homocystéine sériques et la valeur des d-ROMS après la course
Les variations de l ‘homocystéine,
d-ROMS, Oxy-adsorbent et SHp
suggère le rôle important du
statut oxydant du cheval athlète selon les auteurs.
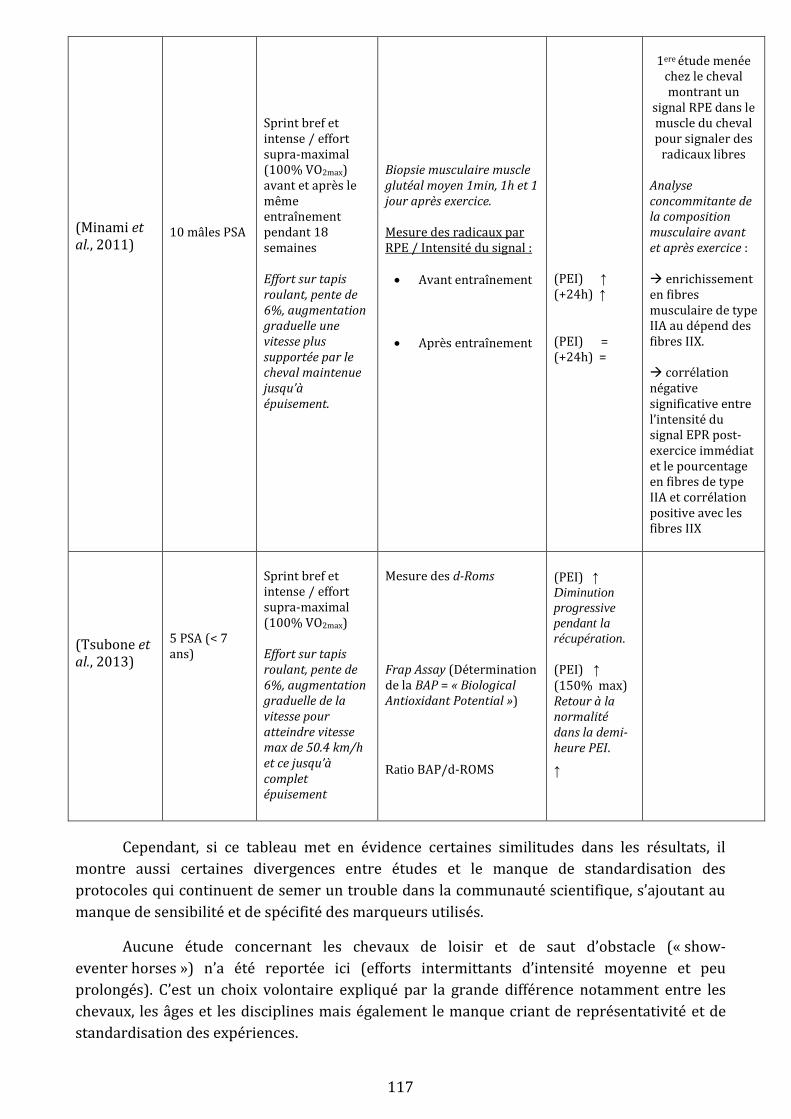
117
(Minami et al., 2011)
10 mâles PSA
Sprint bref et intense / effort supra-maximal (100% VO2max) avant et après le même entraînement pendant 18 semaines Effort sur tapis roulant, pente de 6%, augmentation graduelle une vitesse plus supportée par le cheval maintenue jusqu’à épuisement.
Biopsie musculaire muscle glutéal moyen 1min, 1h et 1 jour après exercice. Mesure des radicaux par RPE / Intensité du signal : Avant entraînement
Après entraînement
(PEI) ↑ (+24h) ↑ (PEI) = (+24h) =
1ere étude menée
chez le cheval montrant un
signal RPE dans le muscle du cheval pour signaler des
radicaux libres Analyse concommitante de la composition musculaire avant et après exercice : enrichissement en fibres musculaire de type IIA au dépend des fibres IIX. corrélation négative significative entre l’intensité du signal EPR post-exercice immédiat et le pourcentage en fibres de type IIA et corrélation positive avec les fibres IIX
(Tsubone et al., 2013)
5 PSA (< 7 ans)
Sprint bref et intense / effort supra-maximal (100% VO2max) Effort sur tapis roulant, pente de 6%, augmentation graduelle de la vitesse pour atteindre vitesse max de 50.4 km/h et ce jusqu’à complet épuisement
Mesure des d-Roms Frap Assay (Détermination de la BAP = « Biological Antioxidant Potential »)
Ratio BAP/d-ROMS
(PEI) ↑ Diminution
progressive
pendant la
récupération. (PEI) ↑
(150% max) Retour à la normalité dans la demi-heure PEI.
↑
Cependant, si ce tableau met en évidence certaines similitudes dans les résultats, il
montre aussi certaines divergences entre études et le manque de standardisation des
protocoles qui continuent de semer un trouble dans la communauté scientifique, s’ajoutant au
manque de sensibilité et de spécifité des marqueurs utilisés.
Aucune étude concernant les chevaux de loisir et de saut d’obstacle (« show-
eventer horses ») n’a été reportée ici (efforts intermittants d’intensité moyenne et peu
prolongés). C’est un choix volontaire expliqué par la grande différence notamment entre les
chevaux, les âges et les disciplines mais également le manque criant de représentativité et de
standardisation des expériences.

118
On s’appuie donc ici sur deux types d’exercice principaux : l’effort maximal à supra-
maximal observé sur un temps court et à une intensité supérieure à 80% de la VO2max du
cheval, et l’effort d’endurance à des intensités atours de 30% de la VO2max mais soutenue sur
des distances dépassant 100 kilomètres.
Certaines observations quant à l’analyse de ce tableau et des études contenues peuvent
ainsi être tirées.
Concernant l’endurance, Marlin et al. (2002) attribuent les différences des résultats
obtenus avec ceux de Hargreaves et al. (2002) à la difficulté environnementale de la course
(notamment température ambiante et le parcours) se répercutant directement sur les
performances moyennes des chevaux (de niveau sportif comparable) en précisant une vitesse
inférieure globalement (8 km/h sur 24 heures en moyenne contre 17 km/h sur 8.5 heures de
compétition en moyenne).
De même dans l’étude de Hargreaves et al. (2002), on constate curieusement une
augmentation de l’activité de la GPx avec l’augmentation de la distance et la prolongation de
l’effort malgré la diminution des concentrations en GSH érythrocytaire. Aussi la diminution de
l’activité de la GPx lors de la récupération (+1h) signe soit une réduction de l’apport en GSH
(observée dans cette étude mais aussi dans celle de Marlin et al. (2002)), soit une diminution de
l’activité de la glutathion réductase (non établie dans l’étude et également non observée dans
l’étude de Frankicwicz-Josko et al. (2000) où c’est au contraire une augmentation de l’activité
qui est constatée).
La stabilité des teneurs en vitamine E lors de l’effort d’endurance constatée chez
Hargreaves et al. (2002) et chez Marlin et al. (2002) peut être expliquée par la mobilisation
concommitante de cette dernière avec les acides gras des tissus adipeux (Rokitski et al., 1994).
La graisse est en effet une source majeure d’énergie pendant un effort d’endurance. De plus les
stocks en vitamine E sont régénérés grâce à la vitamine C, elle-même régénérée grâce au GSH.
Selon Hargreaves et al. (2002), la diminution des concentrations en vitamine C et du GSH
erythrocytaire constatée avec l’augmentation de la distance chez les chevaux d’endurance
indiquent une baisse de la capacité antioxydante relativement avancée et donc un stress
oxydant véritablement présent. Suivant ces constatations, la vitamine E fait partie des derniers
remparts contre le stress oxydant (sauf dans la phase aqueuse) après une production massive
d’ERO.
Les corrélations positives entre les marqueurs du stress oxydant et ceux de la lyse
musculaire (CK et ASAT essentiellement) chez les chevaux d’endurance (Ono et al., 1990)
(Hargreaves et al., 2002) (Frankicwicz-Josko et al., 2000) (Marlin et al., 2002), sont les plus
illustratrices d’un stress oxydant délétère durant l’effort chez le cheval, même si on ne peut pas
affirmer une relation de cause à effet directe sur le muscle.
Concernant les effort maximaux à supramaximaux, un premier résultat concernant le
GSH est intéressant. Lors d’un effort bref et intense, on peut observer une augmentation ou une
baisse de sa concentration plasmatique pendant l’effort et lors de la phase de récupération
suivant les études. Il n’y a pas de justification claire à ces variations. Il est possible que le GSH,
étant parmi les premières défenses antioxydantes, soit également la plus variable selon les
chevaux et leur niveau d’entraînement, qui peut différer selon les études.

119
De même, l’état d’hydratation des animaux n’a pas été mesuré précisément dans
chacune des études ce qui peut également expliqué ces résultats variables.
Pour autant, la mise en évidence de produits de dégradation du stress oxydant et
notamment de la peroxydation lipidique par des protocoles excluant le TBARS test semble
soutenir la thèse selon laquelle un stress oxydant est également généré durant un efforts
maximal chez le cheval (Chiaradia et al., 1998) (Ishida et al., 1999) (Minami et al., 2011)
(Tsubone et al., 2013).
Les variations de l ‘homocystéine, d-ROMS, Oxy-adsorbent et des résultats du SHp test
suggère le rôle important du statut oxydant du cheval athlète (Fazio et al., 2009). L’analyse
systématique du stress oxydant et de son impact sur les niveaux en homocystéine pourrait
contribuer à l’évaluation clinique des performances athlétiques du cheval.
Dans leur étude, Minami et al. (2011) attribuent leurs signaux RPE dérivant
essentiellement du métabolisme de l’ubiquinone (radicaux ubiquinone), sans pouvoir en
apporter la preuve, en se basant sur les résultats obtenus chez l’homme et le rat. Fait
intéressant, si aucune différence significative dans l’intensité des signaux RPE n’est constatée
post-effort chez les chevaux après 18 semaines d’entraînement, une augmentation significative
des taux en radicaux libres détectés par RPE au repos est constatée par rapport aux individus
témoins.
Ces dernières observations suggèrent qu’un entraînement régulier au long terme
entraîne une « accumulation » d’ERO dans l’organisme et un stress oxydatif chronique (Minami
et al., 2011).
1.ii Qu’en est-il chez l’homme ?
Alors qu’il existe un nombre limité de publications décrivant les modifications du statut
antioxidant et des marqueurs du stress oxydant pendant l’exercice chez le cheval, il existe un
nombre considérable de publications en médecine humaine pour de nombreuses disciplines
sportives.
Pour mémoire, les concentrations plasmatiques en acide ascorbique, -tocophérol et
acide urique de base sont considérablement plus hautes chez l’homme que chez le cheval
(Marlin et al., 2002). Par exemple des taux d’acide ascorbique plasmatique compris entre 43
mol/L à 176 mol/L ont été rapportés dans la littérature humaine, ce qui est plus élevé par
rapport au cheval (5 à 40 mol/L en moyenne). De même, les concentrations en -tocophérol
chez les athlètes masculins sont approximativement 3 fois plus hautes que chez leurs
homologues équins, et les taux plasmatiques en acide urique sont jusqu’à 10-20 fois plus
grands que ceux rapportés chez les chevaux (Marlin et al., 2002) (Raltson et al., 2012).
Cependant comme chez le cheval, les variations en GSH érythrocytaire de l’organisme
humain dépend des études et de l’effort considéré. Ainsi Laaksonen et al. (1999) ont rapporté
une diminution significative de la GSH plasmatique après 40 min d’exercice à 60% de la VO2max,
proche des résultats de Marlin et al. (2002) chez le cheval d’endurance, alors que Sastre et al.
(1992) n’ont trouvé aucune différence significative lors d’un effort progressif jusqu’à
épuisement.

120
De même, après 35 minutes de course à 60% de la VO2 max, Camus et al. (1994) n’ont
rapporté aucune différence significative dans les teneurs plasmatiques en GSH, GSSG et le ratio
GSH/GSSG.
Cette simple parenthèse rappelle simplement l’immense potentiel d’investigation et de
standardisation de la mesure du stress oxydant chez le cheval dans chacune de ses facettes
athlétiques.
1.iii Bilan
Le stress oxydant, selon la définition expérimentale, a été identifié dans plusieurs modes
d’exercices chez le cheval. Cependant la sévérité et les niveaux de dégâts tissulaires ne sont pas
toujours similaires entre les études, même celles qui utilisent des protocoles très proches.
2) Facteurs de variations
2.i Facteurs individuels de base : l’âge, le sexe et la race
Les facteurs individuels dit « de base » concernent essentiellement l’âge, le sexe et la
race du cheval.
À propos de l’âge, Smarsh and Williams (2013) ont montré que des pouliches yearlings
Standardbred (nouvelles à l’entraînement) avaient des marqueurs du stress oxydant moins
augmentés après un effort sub-maximal par rapport à des juments Standardbred de plus de dix
ans (non entraînées régulièrement) et une récupération plus rapide des taux basals.
Cette étude a l’avantage de souligner l’intérêt de la prise en compte de l’âge dans les
protocoles d’étude du stress oxydant chez l’athlète équin.
Les races de chevaux reflètent généralement l’effort physique vers lesquelles on les
destine. Il ne fait donc quasiment aucun doute que des différences existent entre chacun des
individus concernant le stress oxydant généré par l’effort. L’étude de Snow et Guy (1980), bien
que relativement ancienne, montre notamment une différence significative dans la composition
des fibres musculaires dans le muscle moyen glutéal entre de nombreuses races de chevaux
étudiées. Cette différence concerne notamment les races destinées au sprint et celles destinées
à de la course d’endurance, avec une proportion de fibre I plus importante chez les chevaux
d’endurance. Le métabolisme musculaire basal entre les races est donc différent, ce qui par
extension peut influencer la production en ERO.
Bien que non approfondie ici et au delà de la composition en fibres musculaires, il est
également possible que les défenses antioxydantes diffèrent selon les races, mais les
mécanismes et prédispositions génétiques restent encore trop peu étudiées pour en parler
dans cet exposé. On retiendra simplement que certains auteurs signalent notamment des
différences avant effort entre les taux sanguins en vitamine E entre les galopeurs et les chevaux
d’endurance (jusqu’à deux fois plus important chez les chevaux d’endurance) (Hargreaves et
al., 2002) (Chiaradia et al., 1998).
Enfin, concernant le sexe, les études manquent et aucune conclusion hative ne peut être
tirée concernant le cheval. Cependant, il est très probable que les états physiologiques propres
à chacun des sexes aient un effet sur le métabolisme basal et donc la production d’ERO.

121
2.ii L’effort physique lui-même
Théoriquement, il paraît vraisemblable que l’importance des dégâts causés par un stress
oxydant dépend du type d’effort considéré. C’est la raison pour laquelle pour chaque étude il
faudrait idéalement le définir précisément avant de conclure et comparer les résultats obtenus,
en prenant en compte la durée, l’intensité et la répétition de l’effort (entraînement ou non,
exercice continu ou répété en série…). L’intensité semble être encore le facteur le plus
important selon de nombreux auteurs (Halliwell and Gutteridge, 2008).
Les études précédentes tendent à montrer une plus grande importance du stress
oxydant induit par un effort d’endurance inframaximal intense et de longue durée par rapport à
un effort maximal ou supra-maximal. Chez l’homme, des dégâts oxydatifs affectant l’ADN sont
également mis en évidence après un effort d’endurance intense et de longue durée mais moins
fréquemment après un effort aigu et bref (type sprint) (Deaton and Marlin, 2003).
Halliwell et Guterridge (2008) rappellent également qu’il faut se méfier des résultats
parfois un peu trop « significatifs » dans les études impliquant un effort d’endurance où un
paramètre aussi simple que la déshydratation et la diminution du volume plasmatique peuvent
faire tendre certains marqueurs vers le haut. En effet, durant un effort d’endurance intense, en
plus de la splénocontraction le cheval peut perdre jusqu’à plus de 25 litres de fluides (urine et
sueur principalement) sur une course, entraînant une forte hémoconcentration.
2.iii Régime alimentaire et la supplémentation
Les questions de la supplémentation en antioxydants et du régime alimentaire sont en
dehors du cadre de cette thèse.
Cependant, il est nécessaire de l’évoquer comme facteur de variation possible des
résultats. Chez l’homme, on a notamment montré que la supplémentation en antioxydants
pouvait réduire les niveaux plasmatiques en CK après un marathon (Kaikkonen et al., 1998)
mais ces résultats restent inconstants entre les études.
Chez le cheval, nous remarquerons simplement que White et al. (2001) et Siciliano et al.
(1996) n’ont démontré aucune amélioration des niveaux en CK plasmatiques chez des
galopeurs supplémentés en vitamine C et vitamine E respectivement. Également, dans l’étude
de Hargreaves et al. (2002), 5 des 10 chevaux qui ont fini la course de 160 km ont reçu 20g/j
d’acide ascorbique per os les trois jours précédents la course sans qu’aucune différence
significative ne soit retrouvée entre les concentrations plasmatiques de cette vitamine, les
autres antioxydants érythrocytaires et les enzymes musculaires (CK et ASAT) des ces animaux
avec les autres chevaux non supplémentés.
Pour autant, Deaton et al. (2002) ont observé des modifications significatives de la
concentration plasmatique en acide ascorbique chez des chevaux supplémentés 24 heures
après exercice, sans qu’aucun lien clair ne soit établi et aucune précision quant à cette
supplémentation ne soit faite.
2.iv Paramètres d’ambiance : rythme journalier, altitude, température et
hygrométrie
Très tôt, on a suggéré que l’ambiance et les paramètres environnementaux dans
lesquelles le cheval réalise son effort physique ont une influence sur la production d’ERO

122
durant l’effort. Les efforts d’endurance, qui s’exercent dans la durée, sont particulièrement
concernés par leurs variations.
Piccione et al. (2012) suggèrent notamment que la capacité antioxydante de l’organisme
est affectée par le rythme circadien chez le cheval. Leurs résultats ont une importance toute
particulière étant donné que les athlètes équins internationaux voyagent, parfois sur de
grandes distances, et pourraient voir leurs défenses antioxydantes altérées par le décalage
horaire.
Les conditions d’hypoxie rencontrées en altitude peuvent évidemment majorer le stress
oxydant, retrouvant des similitudes avec le phénomène d’ischémie-reperfusion. Cependant en
ce qui concerne le cheval et les disciplines équestres qui l’intéressent, son importance ne sera
pas approfondie ici.
En comparant ses résultats avec d’autres études, Mills et al. (1996) suggèrent que les
paramètres biochimiques indicateurs du stress oxydant qui sont modifiés lors d’un effort
physique peuvent être exacerbés chez le cheval par de fortes températures et une importante
humidité ambiante. Le muscle peut s’adapter jusqu’à une certaine mesure et limiter les effets
de la température (haute essentiellement) en provoquant une réponse « heat-shock » qui ne
sera pas détaillée ici (Halliwell and Gutteridge, 2008).
3) Influence de l’entraînement ?
Chez le cheval, l’entraînement apporte différentes adaptations physiologiques dont les
principales rapportées sont (Minami et al., 2011) : l’hypertrophie musculaire, l’amélioration
des fonctions oxydative et glycolytique (notamment par les enzymes) du muscle, la
modification du contenu en fibre musculaire (selon la discipline) vers des fibres musculaires
oxydatives (I et IIA) mais aussi l’amélioration de la capacité antioxydante.
Siciliano et al. (1995) montrent que des chevaux ayant reçu un entraînement pendant
huit semaines avant un effort sub-maximal présentent des augmentations en CK et en ASAT
significativement diminuées par rapport aux individus non pré-conditionnés. De même, dans
leur étude, Hargreaves et al. (2002) ont montré une grande variabilité dans les valeurs de la CK
plasmatique chez les chevaux courant le 160 km, suggérant des différences dans la condition
physique des animaux selon les auteurs.
Si ces résultats ne peuvent pas à eux-mêmes montrer une diminution du stress oxydant
induit par l’effort, ils ouvrent la voie à son étude. Chez le cheval, il a été suggéré que la capacité
antioxydante de l’organisme pouvait augmenter avec l’entraînement comme chez l’homme
(Piccione et al., 2012).
De Moffarts et al. (2004a) ont mis en évidence une augmentation significative de la
concentration plasmatique en acide urique, en vitamine C, en sélénium, en cuivre ainsi que de
l’activité de la GPx mais surtout de la SOD grâce à l’entraînement chez des trotteurs soumis à un
effort standardisé. En effet, il est intéressant de constater que l’augmentation de l’activité de la
SOD libre et érythrocytaire au repos et de la VO2max grâce à l’exercice suggère clairement une
corrélation positive entre un marqueur antioxydant et un marqueur de la performance aérobie
dans l’espèce équine.

123
Autres données, Minami et al. (2011) ne mettent pas en évidence de différence
significative dans le signal EPR avant et après exercice chez les chevaux entraînés alors que
c’est le cas pour les chevaux non entraînés.
Tsubone et al., (2013) montrent une augmentation temporaire de la BAP limitée à la
période de l’effort maximal, avec un retour rapide à la valeur avant effort à la fin de l’exercice.
Ces résultats suggèrent qu’un effort répété (et donc par voie de conséquence l’entraînement)
pourrait améliorer la mobilisation des défenses antioxydantes lors d’un exercice physique.
En particulier, la modification de la composition en fibres musculaires, notamment vers
des fibres mettant plus en avant le métabolisme aérobie, peut entraîner une augmentation du
contenu mitochondrial et ainsi favoriser la production de radicaux libres (production a priori
compensée au moins en partie par l’augmentation de la défense antioxydante concomittante).
Dans l’étude de Minami et al. (2011), les résultats indiquent que l’entraînement favorise
la transformation des fibres de types IIX en fibres de type IIA, tranformation qui jouerait un
rôle important dans la diminution du stress oxydant après l’exercice selon les auteurs. Il n’y a
pas d’explication claire pour l’instant, notamment parce que la consommation mitochondriale
en dioxygène est plus importante chez les fibres de type IIA que chez les fibres musculaires de
type IIX. Les auteurs avancent l’idée que les défenses antioxydantes des mitochondries sont
différentes entre les types de fibres musculaires, meilleures chez les types IIA. Ces suppositions
sont par ailleurs en accord avec Hollander et al. (1999) qui montrent que le renforcement des
fibres musculaires en antioxydants par l’entraînement chez l’homme concerne surtout certains
types, en particulier les fibres IIA.
Par ailleurs, Minami et al. (2011) montrent une augmentation significative des taux en
radicaux libres détectés par RPE au repos. Cette étude suggère donc également qu’un
entraînement régulier au long terme entraîne une « accumulation » d’ERO et un stress oxydatif
chronique. Cet effet pervers de l’entraînement a également été rapporté chez l’homme, et
pourrait jouer dans la physiopathologie de plusieurs affections chez le sujet athlète.
B) Sources principales d’ERO lors de l’effort physique
Les ERO peuvent être produits selon plusieurs modalités et différentes sources
cellulaires. Certaines d’entre elles ont plus d’importance dans certains tissus que dans d’autres,
et varient selon les conditions environnementales d’exercice et le type d’effort.
1) La fuite électronique depuis la chaîne de phosphorylation oxydative
mitochondriale : « l’hypothèse mitochondriale »
Dans la première partie nous avons souligné que la chaîne de phosphorylation oxydative
possède un faible pourcentage de perte en électrons qui ne participe pas à la réduction en eau
(en général 1 à 3%).
Lors de l’effort, l’augmentation de la VO2 est également corrélée à une augmentation de
la production basale en ATP et donc a priori de la production en ERO (Halliwell and Gutteridge,
2008). En effet, la consommation basale en dioxygène de tout l’organisme est multipliée de 10 à
20 chez l’homme, contre plus de 30 fois chez le cheval (Hargreaves et al., 2002). Dans le muscle
cette consommation peut atteindre jusqu’à 100 fois la consommation basale (Li, 1999).

124
De plus, on considère généralement que le pourcentage de O2 converti en radical
superoxyde O2°- reste le même : de fait la production en O2°- est proportionnelle à la
consommation en dioxygène et donc à la VO2 : c’est le fondement de « l’hypothèse
mitochondriale » (Wu et al., 2011).
Cependant, certaines données in vitro mettent en doute ce rapport de proportionalité,
sans pour autant pouvoir complètement la rejeter. En effet, l’incapacité expérimentale d’isoler
une mitochondrie pendant et juste après l’effort pour étudier sa production empêche
notamment de rejeter en bloc cette hypothèse.
L’ « hypothèse mitochondriale » est principalement soutenue par des données indirectes
montrant l’altération de la mitochondrie après un effort (Li, 1999). Ces observations mettent en
évidence une augmentation de la perméabilité de la membrane mitochondriale interne.
Il est possible que l’exercice puisse entraîner l’apparition d’un phénomène de
découplement expliqué par l’altération de la membrane mitochondriale interne et de
l’hyperthermie (Li, 1999). De plus, la peroxydation lipidique et l’altération oxydative des
protéines de la chaîne respiratoire mitochondriale contribuent au relargage des ERO comme
expliqué dans la première partie (dysfonctionnement mitochondrial).
2) Activation de mécanismes inflammatoires
2.i L’effort induit une réaction inflammatoire à l’origine de la genèse d’ERO
Chez l’homme, courir un marathon augmente significativement le nombre de
neutrophiles circulant et peut entraîner un schéma physiologique et biochimique rappelant la
phase inflammatoire aiguë, comme de la fièvre, une chute du contenu plasmatique en zinc et en
fer, la transcription du TNF-, de l’IL-1, de l’IFN- ou encore de l’IL-6, IL-10 et de la protéine C-
réactive (Halliwell and Gutteridge, 2008) (Powers and Jackson, 2008). De pareilles
constatations ont également été établies chez le cheval, en particulier d’endurance (Cywinska et
al., 2010a).
Pour exemple, on a démontré que chez le cheval (comme chez l’homme), l’exercice peut
entraîner l’activation, la dégranulation des neutrophiles ainsi que le burst oxydatif,
accompagnées de l’augmentation significative de la concentration sanguine en
myéloperoxydase (MPO). C’est particulièrement vrai dans un exercice d’endurance intense
(160 km dans l’étude) (Art et al., 2006).
Nethery et al. (1999) rapportent également l’activation possible de la phospholipase A2
lors de la contraction musculaire, entraînant un relargage d’acide arachidonique et la genèse
d’ERO dans le muscle. De plus, cet acide arachidonique peut interférer directement avec la
chaîne de phosphorylation oxydative mitochondriale, aggravant la fuite électronique en plus de
l’activation des cycooxygénases (COX).
2.ii Production des ERO par les cellules inflammatoires
Cette production concerne en particulier la phase de récupération (Shute, 2004). Après
un effort physique, les neutrophiles et les macrophages, qui sont évidemment des sources
potentielles d’ERO, envahissent les fibres musculaires plus ou moins massivement selon
l’effort. Ces phagocytes, bien que nettoyant le muscle des cellules lésées, peuvent jouer un rôle
dans la destruction des fibres musculaires saines et un retard dans la cicatrisation tissulaire

125
lorque les ERO sont produits en quantité trop importante (Duarte et al., 1994) (Shute, 2004)
(Bestwick and Maffulli, 2004).
Dans l’étude de Duarte et al. (1994) menée chez des rats, on observe une nette
augmentation du contenu musculaire en GSH lors de la phase de récupération 96 heures post-
effort après blocage de la production de radicaux libres par les leucocytes (avec de la
colchicine) par rapport aux myocytes d’individus témoins soumis au même exercice. Ces
résultats vont en faveur d’une surproduction d’espèces réactives de l’oxygène par les cellules
phagocytaires lors de la phase de récupération.
Dans l’étude de Minami et al. (2011) chez le cheval, les auteurs retrouvent une
production d’ERO significativement augmentée 24 heures après l’effort par RPE, plus
importante chez les chevaux sans entraînement. Ils attribuent cette production d’ERO à
l’inflammation musculaire et l’intrusion de cellules phagocytaires dans le muscle post-exercice
sans cependant pouvoir le vérifier.
3) L’hypoxémie artérielle induite par l’exercice
3.i L’effort intense induit un déséquilibre entre la ventilation et la perfusion
L’« exercise-induced arterial hypoxemia » ou EIAH est bien décrite chez l’homme mais
aussi chez le cheval (Hopkins, 2006).
Pour autant, on ne connaît pas précisément les mécanismes sous-jacents de l’EIAH. On
constate que chez le sujet sportif, il existe une corrélation négative entre la PaO2 (pression
artérielle en dioxygène) et la VO2max de sorte que les sujets ayant la plus grande VO2max sont
aussi ceux qui connaissent la plus grande diminution de PaO2 lors d’un effort maximal. Cette
diminution de l’efficacité des échanges gazeux lors d’exercice à la VO2max est responsable de
l’augmentation du gradient alvéolo-artériel en dioxygène (Hopkins, 2006).
L’entraînement ne semble pas influencer le déséquilibre entre la perfusion et la
ventilation observé durant un exercice maximal chez l’homme (Hopkins, 2006).
Si la cause exacte n’est pas connue et probablement multifactorielle, l’œdème
pulmonaire interstitiel induit par l’effort pourrait en être une. Il se met en place lors d’un effort
conjointement à l’augmentation de la pression vasculaire pulmonaire. Des études réalisées chez
des porcs l’identifient clairement sur les coupes histologiques réalisées sur les individus ayant
réalisé un exercice par rapport à ceux étant restés au repos. Cependant le lien avec
l’augmentation du gradient alvéolo-artériel reste à confirmer (Hopkins, 2006).
De même l’hypothèse de shunts intra- ou extra-pulmonaires s’établissant durant
l’exercice ne peut être exclue. Un shunt est défini lorsque le sang entre dans le système artériel
pulmonaire sans rentrer en contact avec les portions ventilées du poumon. Si certaines études
mettent en évidence la mise en place de shunts intra-pulmonaires chez l’homme, cela n’a jamais
été montré chez le cheval (même si fortement suspecté) (Hopkins, 2006).
Si l’hypoxie artérielle induite par l’exercice est plus à même d’apparaître lors d’exercices
maximaux, la PaO2 diminue aussi lors d’exercices infra-maximaux/d’endurance lors
d’hyperventilation inadéquate. Le mécanisme n’est pas clair, et les auteurs avancent
l’éventualité d’une contribution du métabolisme des muscles respiratoires (qui consomment
jusqu’à 15% de la consommation en oxygène) comme aggravant la PaO2 lors hyperventilation
inadéquate (Hopkins, 2006).

126
3.ii Une production d’ERO selon les modalités de l’ischémie -reperfusion
Pour autant, même chez un individu a priori sain, l’exercice intense peut représenter une
situation répondant au schéma de l’ischémie-reperfusion pour de nombreux tissus (Cocco et al.,
1999) (Romer et al., 2006). L’effort provoque en effet une redistribution du flux sanguin de
certains territoires vers d’autres (comme les muscles) ; de plus la déshydratation peut
également entraîner une diminution du volume circulant et accentuer l’ischémie.
L’augmentation de l’acide urique dans le plasma après un effort reflète la perte d’ATP, la
dégradation des purines (adénosine, inosine) et la formation l’hypoxanthine (Kirschvink et al.,
2002a). Les exercices intenses entraînent une ischémie sévère au niveau du muscle où la
xanthine déshydrogénase est convertie en xanthine oxydase qui relargue des anions
superoxydes lors de la transformation de la xanthine en hypoxanthine et en acide urique.
Lors de l’exercice anaérobie, l’activation de la xanthine oxydase (XO) est probablement
le mécanisme prépondérant de production d’ERO, et ce en particulier dans les efforts supra-
maximaux où il y a un déséquilibre entre la synthèse et l’utilisation de l’ATP (Groussac, 2006).
Mills et al. (1997b) montrent dans leur étude une diminution significative de la production de
peroxydes lipidiques induite par l’effort par inhibition avec l’allopurinol de la xanthine oxydase
chez des chevaux soumis à un effort maximal.
4) L’acidose métabolique
L’acidose pourrait être un facteur important quant à la survenue du stress oxydant au
cours de l’effort anaérobie. En effet, les exercices anaérobies produisent de l’acide lactique qui
provoque un abaissement du pH sanguin. Cependant cette modalité de production est
controversée.
Chez l’homme, on considère que tant que l’exercice ne dépasse pas 50% de la VO2max, le
pH varie peu. Au delà, le pH sanguin diminue de manière significative pendant l’exercice
(Groussard, 2006). Chez le cheval, comme chez le chien, ces constatations pourraient être à
moduler pour les efforts infra-maxiamaux.
Théoriquement, l’acidose permettrait la production des radicaux libres. Les protons
favorisent notamment la réaction de dismutation du radical superoxyde O2°- en peroxyde
d’hydrogène, mais l’acidité rencontrée lors d’un effort n’est généralement pas suffisamment
importante pour permettre à cette réaction d’avoir lieu sans l’action de la SOD (Halliwell and
Guterridge, 2008).
Par ailleurs, Groussard et al. (2000) ont montré in vitro que l’ion lactate est un
antioxydant en réagissant avec l’anion superoxyde et le radical HO°. Au delà de cette
constatation de laboratoire, les auteurs précisent que la formation de lactates in vivo entraîne
aussi une acidose métabolique qui peut, elle, avoir un effet pro-oxydant. En effet, les protons
peuvent réagir avec l’anion superoxyde O2°- et former le radical perhydroxyle HO2° (plus
oxydant que la forme non protonée), mais aussi favoriser sa dismutation spontanée en
peroxyde d’hydrogène H2O2.
En effet, plusieurs modèles expérimentaux de peroxydation lipidique in vitro supportent
l’hypothèse que la cytotoxicité de l’acidose pour l’organisme passe par la génèse d’ERO (Siesjo
et al., 1985) (Bralet et al., 1991) (Fauconneau et al., 1993). Cependant des précautions doivent
être prises quant à l’interprétation de ces résultats concernant l’effort physique in vivo où

127
l’acidose est minimale comparée aux situations extrêmes auxquelles les tissus étudiés sont
soumis dans ces études (Bloomer and Cole, 2009).
Si de nombreuses études menées chez plusieurs espèces ont montré une augmentation
concomitante des lactates sanguins et des marqueurs du stress oxydant après un effort, il n’est
pas clair, même chez l’homme, que ce fort taux de lactates circulant dans le sang contribue au
stress oxydant généré par l’effort in vivo (Bloomer and Cole, 2009).
L’étude menée chez l’homme par Lovlin et al. (1987) semble mettre en évidence un lien
clair entre l’acidose lactique comme facteur favorisant de la génèse du stress oxydant in vivo.
Cependant la mesure du stress oxydant repose sur la technique des TBARS. Bloomer and Cole
(2009) n’ont eux pas trouvé une telle corrélation en dosant directement le MDA et les protéines
carbonylées chez des sportifs humains.
En bref, il existe des contradictions entre résultats in vivo et in vitro chez l’homme.
Malheureusement il n’existe pas de preuves chez le cheval non plus.
5) L’hyperthermie
L’organisme et le muscle en particulier sont exposés à des températures importantes
durant un effort (dépassant parfois 42°C au sein du muscle), volontier aggravées par les
conditions de l’environnement.
Lorsque la température corporelle augmente, la cellule musculaire striée squelettique
montre une baisse de l’efficacité de la chaîne de phosphorylation oxydative et une
augmentation de la demande en dioxygène O2 (Brook et al., 1971).
Zuo et al. (2002) ont démontré que l’hyperthermie constitue un stress (« heat stress ») et
accroît la production intra- et extra-cellulaire d’ERO dans la cellule musculaire striée
squelettique (en utilisant des myocytes de diaphragme de rat dans un modèle expérimental in
vitro). Leurs résultats suggèrent en effet que le « heat stress » stimule la production de radicaux
superoxydes O2°-, qui pourraient alors contribuer aux réponses physiologiques de défenses lors
d’exercice d’intensité sévère, mais également entraîner des dégâts oxydatifs pathologiques.
Les auteurs avancent plusieurs mécanismes de production possibles qui pourraient être
up-régulés lors d’un « heat stress ». Si le dysfonctionnement mitochondrial ne fait quasiment
aucun doute même s’il est n’est pas clairement compris, l’endothélium capillaire pourrait
répondre à ce stress en activant ses Nox membranaires selon Zuo et al. (2002).
Les iNOS pourraient être également up-régulées lors de l’hyperthermie induite par
l’effort, favorisant alors la formation du peroxynitrite, ce qui serait compatible avec les travaux
de Venturini et al. (1999).
Enfin les myocytes eux-mêmes pourraient contenir des Nox membranaires ou
cytosoliques influencées par l’hyperthermie, mais cela doit encore être étudié (Zuo, 2002).
6) La calcémie
Au sujet de la calcémie, un article de la littérature étudiant l’impact du flutter
diaphragmatique synchrone de chevaux d’endurance sur les paramètres de la peroxydation
lipidique apporte quelques informations intéressantes (Al-Qudah and Al-Majali, 2008).

128
Le flutter diaphragmatique synchrone est une affection caractérisée par une
augmentation de l’irritabilité neuromusculaire du diaphragme, causée par une hypersensitivité
du nerf phrénique. L’activité électrique du cœur lors de la contraction cardiaque stimule le nerf
phrénique présent au-dessus, entraînant une contraction du diaphragme à chaque battement.
Cela se manifeste essentiellement par des mouvements rythmiques unilatéraux ou bilatéraux
des flancs du cheval, et ce à la même fréquence que les battements du coeur.
La principale cause cette affection est la perte d’ions calciques Ca2+ dans la sueur et
l’urine durant l’effort à l’origine d’une hypocalcémie.
Les auteurs de cette étude ont constaté des marqueurs de la peroxydation lipidique
plasmatique (dosage du MDA et des peroxydes lipidiques dans le plasma) en quantité
supérieure chez les chevaux d’endurance éliminés pour flutter diaphragmatique synchrone que
les individus témoins. En particulier, les faibles teneurs en Ca2+ plasmatique étaient corrélées
avec les fortes teneurs en marqueurs de la peroxydation lipidique.
Les auteurs avancent même l’hypothèse selon laquelle l’effort intense perturbe
l’homéostasie calcique à l’origine même du stress oxydant. Peu de données supplémentaires
existent à ce sujet.
7) Autres facteurs de variations d’intérêt
L’auto-oxydation des catécholamines ou encore l’auto-oxydation de l’oxyhémoglobine en
méthémoglobine (mécanisme similaire pour la myoglobine dans le muscle) favoriseraient la
formation d’ERO lors de l’effort, mais dont l’impact reste encore largement à déterminer et
vraisemblablement mineurs par rapport à ceux présentés précédemment.
L’auto-oxydation des catécholamines renvoie à l’augmentation de leur concentration
plasmatique lors de l’effort. Lors d’un effort de type sprint, les taux peuvent être multipliés par
12 (Groussard, 2006). Leur auto-oxydation pourrait produire des ERO, mais c’est une
constatation théorique basée sur des données in vitro. Aucune étude chez le cheval ne permet
aujourd’hui d’affirmer l’existence de ce phénomène in vivo (Deaton et al., 2003).
Enfin, chez l’homme, il a été suggéré que le radical superoxyde pourrait également être
produit lors de l’auto-oxydation de l’oxyhémoglobine en méthémoglobine (Deaton et al., 2003).
En effet, la libération d’hémoglobine, dans le plasma, facilite son oxydation en methémoglobine
qui fonctionne comme une puissante peroxydase capable de favoriser l’apparition d’espèces
réactives de l’oxygène ; secondairement, l’intéraction des ERO formés avec ces hémoprotéines
fait apparaître des composés très oxydants comme la ferrylhémoglobine ou le radical ferryl.

129
IV) Relations entre les affections organiques de l’athlète et le stress oxydant
A) Pathologie musculaire
1) Stress oxydant et fatigue musculaire lors d’un effort physique
1.i Définition de la fatigue musculaire
La fatigue musculaire se définit comme une réduction de la force contractile musculaire
induite par l’exercice et réversible avec le repos (Zuo, 2002) (Powers and Jackson, 2008).
C’est un phénomène plurifactoriel : baisse de la concentration potassique, acidose
intracellulaire (notamment avec l’accumulation des lactates), modification de la sensibilité des
myofibrilles pour le calcium, augmentation de la concentration intracellulaire en phosphate
inorganique, hypoxémie, déplétion des substrats énergétiques comme l’ATP, la
phosphocréatinine, le glycogène … (Richardson et al., 1995) (Zuo, 2002) (Al-Qudah and Al-
Majali, 2008) (Jungbluth, 2008). Tous ces mécanismes ne sont pas clairement élucidés.
Selon plusieurs auteurs, le stress oxydant et la présence d’ERO pourrait en partie
contribuer à l’établissement de la fatigue musculaire et ralentir la récupération musculaire
(Zuo, 2002) (Shute, 2004).
1.ii Fatigue musculaire et stress oxydant
Effet biphasique des ERO sur la contractilité musculaire
En 1991, Barclay et al., ont montré in vitro que les ERO contribuent de manière
significative à la fatigue des muscles striés squelettiques à partir de prélèvements de muscle
solaire de rat et de muscle gastrocnémien de chien. Les auteurs ont en effet soumis leurs
prélèvements à des stimulations électriques de 70 Hz une fois toutes les minutes pendant une
heure. Dans les milieux expérimentaux additionnés de xanthine oxydase (XO), la force
développée par les prélèvements était au moins divisée par deux par rapport aux prélèvements
non-traités.
En se basant sur ces résultats et les données issues de leurs propres études, Reid et al.
(1993) ont proposé un modèle théorique pour expliquer la relation qui existe entre l’équilibre
redox et la force isométrique produite par le muscle en question. Le modèle théorique de Reid
et al. est ainsi basé sur le principe que l’homéostasie de l’état redox du muscle s’adapte pour
assurer la force musculaire optimale à l’effort (figure 24). Tout stress oxydant a
potentiellement un impact sur la contraction musculaire elle-même.
Ainsi, les ERO ont un effet biphasique sur la contraction musculaire. Un niveau faible en
ERO, autour de la production basale, est essentiel à la bonne contraction musculaire. Au delà du
niveau basal, le pouvoir antioxydant global limite les dégâts oxydatifs tout en permettant une
contraction musculaire plus forte jusqu’à atteindre un pic optimal (selon des mécanismes qui
ne sont pas clairement élucidés). Ainsi, la diminution expérimentale des niveaux basaux en ERO
dans le muscle in vitro au moyen d’antioxydants a pour résultat de diminuer significativement
la force musculaire produite par la fibre musculaire (Reid et al., 1993) (Reid, 2001). Au delà de
cet optimum, les ERO et le NO° ont des effets néfastes et limitent la force musculaire de manière
temps- et concentration- dépendante : c’est la fatigue musculaire.

130
Figure 24 : Modèle in vitro de Reid et al. (1993) : effet biphasique des ERO sur la force musculaire produite.
Adapté d’après Reid et al. (1993).
Le point 1 représente simplement le début de la courbe du muscle non-fatigué soumis à des agents oxydants ou
réducteurs. Le point 2 représente la force développée par un muscle après une phase de repos, sans ajout d’agents
oxydants ou de réducteurs dans le milieu. Le point 3 illustre la force produite par le muscle exposé à un taux faible
d’ERO. A contrario le point 4 illustre les effets délétères des ERO sur la force musculaire squelettique produite
(situation de stress oxydant).
Reid (2001) rappelle cependant qu’une telle courbe biphasique pourrait également être
proposée pour d’autres variables pouvant influencer la contraction musculaire et également
régulées de manière homéostatique comme l’osmolarité intracellulaire, le pH ou la température
de l’organisme.
Mécanismes sous-jascent au niveau du myocyte
McKenna et al. (2006) ont démontré, chez l’homme, l’impact du stress oxydant sur la
diminution de la concentration potassique intracellulaire due à la baisse d’activité des pompes
Na+/K+-ATPases. En effet, la supplémentation en N-acétyl-cystéine chez des sujets soumis à un
effort sub-maximal a permis de ralentir le dysfonctionnement des pompes Na+/K+-ATPAases et
ainsi de retarder l’apparition de la fatigue musculaire. Selon les auteurs, les ERO inhibent ces
pompes par l’oxydation de leurs fonctions thiols. Également, il semblerait que la CK et la pompe
calcique ATP-dépendante du réticulum endoplasmique (PCARE) aient une sensibilité
particulière aux ERO (Powers and Jackson, 2008) (Zuo, 2002).
Le monoxyde d’azote pourrait également jouer un rôle dans les mécanismes de
réduction de la force de contraction musculaire (Reid, 2001) (Powers and Jackson, 2008). Il est
clairement établi qu’in vitro, des fibres musculaires squelettiques isolées produisent de faibles
niveaux de NO° au repos et que la production augmente pendant les phases de contraction. En
plus de favoriser la perfusion du muscle, cette production endogène de NO° pourrait, comme
les autres ERO, moduler la contractilité musculaire.
Ainsi, en bloquant spécifiquement in vitro les NOs, la force de contraction submaximale
augmente ; au contraire des donneurs en NO° ont l’effet opposé. On considère que la moitié de
l’inhibition de la contractilité musculaire par NO° est médié par le GMPc dans la cellule ; l’autre
moitié proviendrait de l’action directe du NO° et plus particulièrement du peroxynitrite sur les
protéines redox-sensibles (Reid, 2001). Par ailleurs, on a retrouvé des (eNOS et nNOS) dans la

131
membrane du réticulum sarcoplasmique des myocytes (Zuo, 2002), suggérant l’association
entre la libération du calcium et la production de NO° lors de la contraction musculaire.
Selon plusieurs études, le monoxyde d’azote abaisse la sensibilité des myofibrilles au
Ca2+ en inhibant l’activation des filaments d’actine in vitro (PaO2 > 150 mmHg). In vivo (PaO2
autour de 10 mmHg dans le myocyte) d’autres auteurs ont montré qu’à des concentrations
inférieures à 1 mol/L, le NO° pouvait activer les récepteurs à la ryanodine du réticulum
endoplasmique (par S-nitrosylation d’une cystéine) qui libère alors du Ca2+. Ainsi, suivant la
concentration en monoxyde d’azote, la contractilité du muscle peut être activée ou au contraire
inhibée. Pourtant, de manière intéressante, la production en NO° n’a aucune influence sur la
force maximale à la tétanie de la fibre musculaire in vitro. NOS1 (nNOS) semblerait être
incriminée plus particulièrement (Powers and Jackson, 2008).
Enfin, Zuo (2002) rapporte d’autres effets biologiques du NO° sur le muscle pouvant
influencer ses performances comme la diminution de la consommation en dioxygène, ou
l’inhibition de la cytochrome c oxydase. Les mécanismes exacts et les cibles du NO° lors de la
contraction musculaire restent malheureusement très peu connus.
Attention cependant, si localement au niveau du myocyte le NO° peut effectivement
avoir des effets néfastes, il ne faut pas oublier qu’il NO° régule aussi l’apport sanguin au muscle
durant l’effort. Ainsi des tentatives expérimentales in vitro d’inhibition de cette synthèse ont
mené à une fatigue accrue par rapport aux muscles non traités (Powers and Jackson, 2008).
Figure 25 : Bilan des mécanismes suspectés de régulation redox de la force produite lors de la contraction
musculaire. D’après Powers and Jackson (2008). PCARE : Pompe calcique ATP-dépendante du réticulum endoplasmique.
1.iii Bilan
Même si les études qui s’intéressent au lien entre le stress oxydant et la fatigue
musculaire ont des résultats très variables, plusieurs d’entre elles sont en faveur d’une

132
implication active des ERO dans la régulation de la contractilité musculaire ; cependant, les
modes d’action des ERO (notamment lors de la libération du calcium par le réticulum
endoplasmique) restent encore largement incompris et des travaux complémentaires s’avèrent
nécessaires.
Pour l’instant, chez le cheval, il n’existe pas de preuve d’un effet positif de la
supplémentation en antioxydants sur l’installation de la fatigue musculaire. Cependant, au vu
des données actuelles, on peut imaginer que l’entraînement favorise la réponse antioxydante
du muscle, permettant ainsi d’atténuer la fatigue musculaire induite par l’effort (Reid et al.,
1992).
La fatigue musculaire s’accompagne souvent de microlésions à l’origine d’une réponse
inflammatoire et dont l’expression clinique la plus courante est la courbature. Dans les cas
graves, ces lésions entraînent la lyse des myocytes et du muscle dans sa globalité : c’est la
rhabdomyolyse d’effort.
2) Stress oxydant et rhabdomyolyse d’effort
1.i Présentation
La rhabdomyolyse d’effort aiguë est un syndrome qui s’explique par la lyse des cellules
musculaires striées squelettiques suite à un effort physique. Les cas sont généralement
sporadiques et causés par un effort intense inadapté. Le développement de cette affection est
influencé par divers facteurs comme le niveau d’entraînement, le sexe, l’âge, le tempérament
des animaux, la présence d’une boîterie et le régime alimentaire (Valberg., 2006).
Nous n’aborderons pas les cas chroniques, liés surtout à des prédispositions génétiques.
On considère en moyenne que 3% des chevaux athlètes soumis à un effort et à des
compétitions régulières ont eu au moins un épisode de rhabdomyolyse aiguë dans les 12
derniers mois. La prévalence est plus grande chez les PSA de course (galopeurs) et les
steeplechasers (6% en moyenne) ; cependant, chez les chevaux de polo, cette prévalence peut
atteindre 13% (Valberg., 2006). Lindholm (1987) rapporte que des individus jeunes peu
entraînés sont particulièrement sujets à la rhabdomyolyse aïgue, notamment en lien avec leur
faible niveau d’entraînement.
1.ii Tableau clinique et lésionnel
Les signes cliniques apparaissent en général rapidement 5 à 15 minutes après l’effort
(Lindholm, 1987). Les chevaux atteints de rhabdomyolyse aiguë présentent une myalgie, des
raideurs et des spasmes musculaires, en particulier au niveau des muscles glutéaux et des
membres postérieurs, une réticence à la marche ainsi qu’une myoglobinurie caractéristique. Un
état de choc peut également être observé dans les cas graves. Des épisodes subcliniques
peuvent aussi avoir lieu et ils sont également caractérisés par une douleur musculaire plus
modérée et une diminution des performances sportives (Valberg., 2006).
Les concentrations en CK, LDH et AST plasmatiques sont considérées comme
proportionnelles aux lésions musculaires. Chacune de ces trois enzymes atteint son pic
plasmatique à des moments différents (4-6, 12 et 24h respectivement). Lors de rhabdomyolyse
sévère, des déséquilibres électrolytiques plus ou moins importants sont constatés :
hyponatrémie, hypochlorémie, hypocalcémie, hyperkaliémie et hyperphosphatémie. C’est

133
généralement une alcalose métabolique qui est observée plutôt qu’une acidose, liée à
l’hypochlorémie (Valberg., 2006).
La rhabdomyolyse d’effort est une véritable nécrose musculaire (Valberg., 2006). Les
lésions microscopiques montrent des ruptures dans les myofibrilles, visibles quelques heures
après un effort. Après deux à trois jours, les structures musculaires lésées sont envahies par
des cellules phagocytaires. Quatre à six jours après l’effort, la régénération du muscle se met en
place avec l’activation des cellules souches musculaires et des cellules satellites ; cependant la
réparation du muscle est rarement ad integrum avec l’envahissement du site par les
fibroblastes aboutissant à de la fibrose (d’intensité variable selon l’importance des dégâts
musculaires) (McArdle and Jackson, 2000).
1.iii Physiopathologie et lien avec le stress oxydant
On a longtemps pensé chez l’homme comme chez le cheval que la rhabdomyolyse
d’effort est causée par l’accumulation d’acide lactique dans les muscles ainsi que l’acidose
qu’elle entraîne. On sait aujourd’hui que ce n’est pas le cas. D’autres études suggèrent que les
chevaux souffrant de rhabdomyolyse chronique pourraient montrer des altérations de la
régulation calcique par le réticulum endoplasmique ; rien de tel n’a été démontré pour les
épisodes aigus. Cependant, les microlésions musculaires peuvent se développer conjointement
à la fatigue musculaire où la régulation du calcique est également compromise (voir
précédemment) (Valberg., 2006) (Jungbluth, 2008).
Chez le cheval d’endurance, les études de Frankicwicz-Josko et al. (2000) et de
Hargreaves et al. (2002) ont mis en évidence des corrélations positives entre les marqueurs de
lyse des myocytes striés squelettiques (CK et ASAT essentiellement) et ceux du stress oxydant.
Ces résultats, également constatés chez l’homme, ont conduit les auteurs à fortement suspecter
l’implication du stress oxydant dans la lyse musculaire.
De manière surprenante, dans l’étude menée chez des chevaux de course de galop après
une épreuve (effort maximal), Chiaradia et al. (1998) ont certes mis en évidence un stress
oxydant induit par l’effort (voir 2ième Partie, A) 1)1.i) mais n’ont pas noté de modification
significative des taux plasmatiques de CK post-effort. Par ailleurs, les taux de LDH étaient eux
augmenté après et 18h après la course. Cependant, ce résultat concernant la CK est
relativement isolé chez les chevaux de course qui sont parmi les plus touchés par l’affection.
On a également supposé que l’apport insuffisant de vitamine E et de sélénium pourrait
être un facteur favorisant cette affection, et supporterait l’implication du stress oxydant dans la
pathogénie de la maladie. Chez un autre athlète, le chien de traîneau (effort d’endurance),
Piercy et al. (2001) ont montré qu’il n’y a pas de corrélation entre la concentration plasmatique
en vitamine E des individus et le risque de développer une rhabdomyolyse d’effort. De même
chez le cheval, la littérature indique que les chevaux affectés par la rhabdomyolyse aiguë ne
sont que très rarement carencés en vitamine E et en sélénium ; rien aujourd’hui ne permet
d’affirmer que la supplémentation en ces antioxydants pourraient leur être bénéfique
(Lindholm, 1987) (Valberg., 2006).
Loin d’exclure complètement le rôle des espèces réactives de l’oxygène dans la
physiopathologie de la rhabdomyolyse d’effort, il est possible que les mécanismes
pathogéniques des lésions musculaires induites au cours de l’effort (comme le stress oxydant,
mais aussi l’étirement ou les traumatismes par exemple) dépendent du type d’effort considéré.

134
1.iv Bilan
Si les études n’isolent pas précisément le stress oxydant dans les mécanismes primaires
de formation des lésions dans la rhabdomyolyse d’effort, il est assez clair que l’invasion des
fibres musculaires lésées par les cellules phagocytaires fait place à une libération massive
d’espèces réactives de l’oxygène, qui accélèrent la dégénérescence du tissu nécrotique et la
mise en place de la cicatrisation (McArdle and Jackson, 2000).
Par ailleurs, le stress oxydant supplémentaire entraîné par l’auto-oxydation de la
myoglobine est probablement responsable de manière primitive de la principale complication
de la myoglobinurie : l’insuffisance rénale aiguë (Valberg., 2006) (Boutaud and Roberts II,
2011). Pour mémoire, en corrélant les marqueurs du stress oxydant et ceux de l’insuffisance
rénale, el-Ashker et al. (2011) ont trouvé chez 31 chevaux arabes en rhabdomyolyse d’effort
une exacerbation du stress oxydant dans le rein, retrouvant des résultats proches avec ceux
chez l’homme.
B) Pathologie respiratoire
Les affections respiratoires du cheval sont d’importants facteurs d’intolérance à l’effort
et de baisse de performance chez le cheval (Soffler, 2007).
Les affections pulmonaires ont très souvent une composante inflammatoire forte,
chronique et compliquée, générant secondairement des ERO selon des modalités déjà décrites.
Dans cet exposé, nous tenterons surtout de cibler par la suite les mécanismes
physiopathologiques primitifs afin de discerner une genèse éventuelle et délétère d’espèces
réactives de l’oxygène, notamment en lien avec l’effort.
Trois affections retiennent plus particulièrement notre attention :
- La maladie obstructive chronique des petites voies respiratoires (RAO,
pour « recurrent airway obstruction ») ;
- La maladie inflammatoire des voies respiratoires profondes chez les
jeunes chevaux de courses (IAD, pour« pour inflammatory airway disease ») ;
- L’hémorragie pulmonaire induite par l’effort (HPIE).
1) La maladie obstructive chronique des petites voies respiratoires (RAO)
1.i Présentation générale
L’obstruction chronique des petites voies respiratoires ou « Recurrent Airway
Obstruction » (RAO) ou « heaves », plus communément appelée « pousse » (ou encore
« emphysème » par les cavaliers), est une maladie commune chez le cheval qui limite ses
performances sportives. Dans certaines parties du monde, on recense jusqu’à 55% des chevaux
atteints par cette affection. Cette affection allergique est moins susceptible de toucher de jeunes
chevaux de course mais plutôt des chevaux de sport et notamment de dressage, de saut ou de
concours complet. En effet, l’âge moyen de développement des signes cliniques est de 9 ans.
(Art et al., 1999) (Rush and Mair, 2004) (Moran et al., 2011).
Aucune prédilection de race ou de sexe n’a été mis en évidence ; cependant les auteurs
rapportent qu’il semblerait qu’une prédisposition génétique ait été observée sur le terrain
(Rush and Mair, 2004) (Moran et al., 2011).

135
L’obstruction des petites voies respiratoires (RAO) du cheval a des similarités avec
l’asthme de l’homme qui a été utilisée pendant longtemps comme modèle animal de référence
(Rush and Mair, 2004) (Deaton, 2006) (Moran et al., 2011). Deux formes cliniques ont été mises
en évidence chez le cheval, l’une plus susceptible d’apparaître au box et l’autre, plus
saisonnière, au pré ; elles ne seront pas dissociées dans le cadre de cet exposé.
Ce n’est donc pas l’effort physique qui est à l’origine de la maladie mais bien une
sensibilité allergique individuelle à des contaminants de l’environnement. Cependant compte
tenu de l’atteinte de la ventilation pulmonaire du cheval, il est intéressant de constater son
impact sur l’équilibre redox pendant l’effort.
1.ii Tableau clinique et lésionnel
Les signes cliniques apparaissent chez les chevaux sensibles suite à l’exposition à des
poussières de matières organiques diverses et allergènes souvent présents dans le foin et les
litières. Comme chez les asthmatiques humains, la sensibilité et la gravité des signes cliniques
varient selon les individus (Rush and Mair, 2004).
Le champignon Aspergillus fumigatus est considéré comme un des allergènes principaux
de cette affection. D’autres pathogènes sont également de potentiels allergènes, comme Faenia
rectivirgula (Moran et al., 2011).
La maladie est composée d’épisodes de crises d’obstructions pulmonaires aiguës suivies
par des périodes de rémission clinique. Si les chevaux en rémission clinique peuvent a priori
retourner à la compétition, les chevaux en crise éprouvent une souffrance musculaire et des
signes hypoxiques témoins d’une altération de la physiologie respiratoire normale lors de
l’effort (Kirschvink et al., 2002a) (Moran et al., 2011).
Les crises sont caractérisées par de la toux, du jetage nasal et une dyspnée expiratoire à
mixte (phase expiratoire allongée, douloureuse et laborieuse) ; l’auscultation pulmonaire met
souvent en évidence des sifflements (« wheezes »), des crépitements dans la trachée et dans les
poumons (signant la présence de mucus).
Les muscles abdominaux se contractent de manière active lors de l’expiration et
l’hypertrophie de ces muscles produit une ligne de pousse caractéristique. Pour les chevaux en
rémission, les insuffisances respiratoires peuvent être constatées chez les chevaux au repos,
mais la plupart des chevaux présentent de la toux à l’effort et une intolérance à l’exercice. Dans
les cas sévères, une sur-inflation des poumons due à la séquestration de l’air dans les poumons
est observée post-mortem (Rush and Mair, 2004).
1.iii Physiopathologie générale
Le schéma pathophysiologique global est (Rush and Mair, 2004) (Deaton, 2006) (Moran
et al., 2011) :
- une bronchiolite neutrophilique : souvent considérée comme une preuve
d’une hypersensibilité de type III (accumulation de complexes immuns et activation du
complément avec la libération d’anaphylatoxine C3a et C5a), elle est également associée à une
accumulation lymphocytaire péribronchique. L’exocytose des contenus des granules des
neutrophiles libère des enzymes et espèces réactives de l’oxygène dans le milieu extracellulaire
mais aussi des facteurs pro-inflammatoires. L’implication de certains lymphocytes T mémoires
entretiennent dans les cas chroniques l’afflux des neutrophiles dans les poumons lors des

136
phases de rémission clinique (Moran et al., 2011). Cependant, les facteurs d’initiation de cette
neutrophilie ne sont pas entièrement élucidés.
- une bronchoconstrition, particulièrement présente lors de crise aiguë. La
présence de neutrophiles dans la lumière des petites voies respiratoires favorise la
bronchoconstriction par contraction des muscles lisses pariétaux.
- la surproduction de mucus aggravant l’obstruction des petites voies
respiratoires. Elle est liée à l’hyperplasie des cellules caliciformes productrices du mucus et
l’hypertrophie des glandes mucosales. L’inflammation entraîne également une altération de la
mobilité muco-ciliaire et une modification de la composition et des qualités rhéologiques du
mucus, souvent plus visqueux chez le cheval RAO.
- une hyperréactivité bronchique à l’exposition d’un allergène, s’aggravant
crise après crise.
Au long terme, l’inflammation pulmonaire et l’obstruction chronique des bronches du
cheval entraînent des remaniements histologiques dont les principaux sont la fibrose sub-
épithéliale, la néovascularisation et l’accroissement de la perfusion, l’hyperplasie glandulaires
et l’hypertrophie de la musculature lisse bronchique.
Ces modifications histologiques des poumons ont des répercussions directes sur la
physiologie respiratoire : en effet les chevaux RAO ont une pression pleurale et une résistance
pulmonaire plus forte ainsi qu’une compliance pulmonaire et une PaO2 plus faible que les
chevaux sains (Soffler, 2007) (Moran et al., 2011).
1.iv Déséquilibre redox des chevaux RAO
Stress oxydant chez les chevaux RAO au repos
Le glutathion est une des premières défenses des poumons et est présent dans le
surfactant pulmonaire en concentration en moyenne 100 fois plus importante que dans les
autres fluides extracellulaires. Il semble que c’est aussi le meilleur marqueur systémique pour
évaluer l’impact de RAO sur l’importance des variations du stress oxydant (Soffler, 2007).
En 1999, Art et al. ont obtenu les premiers résultats suggérant qu’un stress oxydant est
associé aux désordres des voies aériennes profondes chez le cheval souffrant de RAO, en
montrant une diminution significative du ratio GSH/GSSG dans le surfactant pulmonaire des
chevaux RAO en crise par rapport aux sujets sains et aux chevaux RAO en rémission. Cette
étude met également en évidence une stabilité voire augmentation des teneurs en GSH dans le
surfactant pulmonaire lors de la crise, compatible avec une réponse antioxydante de
l’organisme et corroborant cette hypothèse.
Moran et al., 2011 rapportent des résultats similaires lors de crises (GSSG augmente et le
ratio GSH/GSSG diminue dans le surfactant pulmonaire) en précisant que ces variations sont
également corrélées de manière significative avec le comptage neutrophilique dans le liquide
de lavage broncho-alvéolaire (LBA). Deaton et al. (2005) ont également corrélé de la même
manière la neutrophilie et les taux en élastases du LBA avec une diminution de la concentration
en vitamine C (acide ascorbique) dans ce même prélèvement des chevaux RAO par rapport aux
chevaux sains inclus dans l’étude.

137
Chez l’homme asthmatique les taux d’H2O2 exhalé est proportionnel à la sévérité de
l’affection selon Cathcart et al., 2012. Les auteurs remarquent cependant qu’il semble exister
une différence interspécifique voire intraspécifique (étude chez le chien) concernant les
niveaux de peroxyde d’hydrogène présent dans l’air expiré. Ces taux après exposition à un
allergène sont variables dans l’espèce équine, tantôt augmentés chez les chevaux RAO (Deaton
et al., 2004), tantôt sans différence significative par rapport aux individus sains (Deaton et al.,
2005), laissant apparaître le manque de fiabilité de ce paramètre d’évaluation chez le cheval.
Si la sphère pulmonaire est impactée par un stress oxydant chez le cheval RAO (en
particulier lors de crise), il semble que l’organisme entier pâtit de cette situation. Deaton
(2006) rapporte une diminution du ratio GSH/GSSG plasmatique et de la concentration en
vitamine C plasmatique chez des chevaux RAO au repos en rémission.
Remarquons que ces résultats sont en désaccord avec ceux de Kirschvink et al. (2002b).
Ces derniers ont constaté une augmentation significative de la GSH plasmatique au repos chez
les chevaux en crise et les chevaux en rémission comparée aux individus sains. Les résultats
suggèrent également que le stress oxydant chez les chevaux RAO est corrélé avec
l’inflammation pulmonaire et éventuellement les remaniements histologiques (Kirschvink et
al., 2002b).
Plus récemment une augmentation significative de l’activité de la GPx et de la SOD
érythrocytaire a été montrée chez des chevaux RAO par rapport à des chevaux sains (sans
variation significative de la CAT et de la glutathion réductase) (Niedzwiedz and Jaworski,
2014).
Les neutrophiles présents dans la vascularisation pulmonaire chez les individus équins
RAO pourraient déstabiliser l’équilibre oxydant/antioxydant systémique en libérant des ERO
dans la circulation générale, comme pour les cas graves de COPD (« Chronic Obstructive
Pulmonary Disease ») chez l’homme (MacNee and Rahman, 2001). En effet, chez les chevaux
RAO, les neutrophiles sanguins circulant s’activent et produisent des radicaux superoxydes
après exposition à des poussières allergènes (Marr et al., 2002).
Concernant le monoxyde d’azote NO°, Costa et al. (2001) ont montré que l’expression
d’iNOs est supérieure dans les cellules épithéliales bronchiques des chevaux RAO comparée aux
chevaux sains, suggérant que le monoxyde d’azote NO° pourrait jouer un rôle dans
l’amplification des processus inflammatoires dans les voies respiratoires, même si les auteurs
indiquent que le NO° n’est probablement pas le médiateur majeur.
Stress oxydant chez les chevaux RAO lors d’un effort
Le stress oxydant entraîné par l’effort physique est responsable d’une diminution de la
quantité de GSH dans le surfactant pulmonaire du cheval sain, résultat retrouvé également chez
l’homme et chez le cheval. Au vu des résultats présentés précédemment chez des chevaux
malades au repos, cette constatation suggère que le sujet RAO pourraît être plus
particulièrement sensible au stress oxydant entraîné par l’effort (Kirschvink et al., 2002a).
Dans l’étude d’Art et al. (1999), les chevaux en rémission montrent une amélioration des
performances mais pas de différences significatives dans les marqueurs du stress oxydant par
rapport aux chevaux en crise. Également, Kirschvink et al. (2002b) n’ont pas obtenu de
différences significatives de la GSH plasmatique chez les chevaux RAO après un effort modéré.

138
Par ailleurs, dans une précédente étude menée par la même équipe, une élévation
significative des isoprostanes (le 8-épi-PGF2en l’occurrence) dans le plasma et le LBA des
chevaux RAO après un effort progressif sur tapis avait mis en lumière un stress oxydant induit
par l’exercice physique, plus fort chez les chevaux malades (Kirschvink et al., 2002a).
Chez les chevaux de Kirschvink et al. (2002b), l’acide urique est également produit en
quantité augmentée par l’exercice chez les animaux en crise et en rémission par rapport aux
individus sains ; or celui-ci est formée par la transformation de l’hypoxanthine par la XDH ou
par la XO. L’implication éventuelle de XO (activée par l’inflammation et l’hypoxie) dans la
genèse du stress oxydant n’a pas été élucidée par l’étude.
1.v Conséquences physiopathologiques
Ainsi, malgré plusieurs preuves de déséquilibre redox et d’une sensibilité plus grande au
stress oxydant chez le cheval RAO, les données restent limitées concernant le lien causal
éventuel entre le stress oxydant et l’aggravation des éléments lésionnels précédents chez cette
espèce (Deaton, 2006).
Cependant, quelques résultats issus de la littérature méritent d’être soulignés. En effet,
chez l’homme et la souris asthmatique, il a été montré que le stress oxydant favorisait la
contraction musculaire lisse entraînant la bronchoconstriction, l’hyperproduction de mucus
dans les bronches, l’hyperréactivité des voies respiratoires, la nécrose et l’exfoliation des
cellules épithéliales et l’augmentation de la perméabilité vasculaire (Moran et al., 2011).
Bronchoconstriction
Dans le cadre de leur étude chez le cheval RAO, Bureau et al. (2000b) avancent que les
cytokines pro-inflammatoires et les ERO pourraient indirectement altérer la réponse
contractile nerveuse de la musculature lisse des bronches du cheval ; une augmentation du
nombre et de l’intensité des contractions dues à l’activation des récepteurs cholinergiques
induite par des molécules oxydantes a déjà été documenté pour le muscle lisse trachéal du rat
(Ohrui et al., 1991). De même, un stress oxydant augmente la contractilité musculaire de la
cellule musculaire lisse bronchique humaine isolée in vitro (Cortijo et al., 1999).
Le 8-épi-PGF2 est un isoprostane marqueur de stress oxydatif dont la synthèse
pulmonaire est accrue chez les humains asthmatiques et les chevaux dit « poussifs » en crise
aiguë. Chez l’homme, il a été montré que ce métabolite de l’acide arachidonique entraînait une
bronchoconstriction in vitro et in vivo. Chez le cheval, l’étude de Kirschvink et al. (2001) a
montré son pouvoir bronchoconstricteur chez les chevaux poussifs mais pas chez les chevaux
sains (même si moins efficace que PGF2).
Les résultats de Kirschvink et al. (2001) semblent également indiquer que si les
prostaglandines produites par la voie des COX sont impliquées dans la bronchoconstriction lors
de la crise de pousse (PGE2 et PGF2par exemple), d’autres voies, différente de celle des COX,
générent des prostaglandines et des molécules bronchoconstrictrices. En effet, le stress
oxydant pourrait également produire des isoprostanes impliqués dans la bronchoconstriction
dont la signification reste encore à éclaircir. Cette hypothèse concorde d’ailleurs avec le fait que
la brochoconstriction pulmonaire répond très mal aux anti-inflammatoires non-stéroïdiens.
Cependant, si le 8-épi-PGF2est certes brochoconstricteur, il doit encore être considéré
comme un marqueur du stress oxydant plutôt qu’un acteur majeur lors de crises de pousse au

139
vu des connaissances actuelles chez le cheval. Des études antérieures in vitro ont montré que la
plus faible action du 8-épi-PGF2 par rapport à PGF2provient du fait que le 8-épi-PGF2 agit
de manière agoniste sur les récepteurs au tromboxane plutôt que sur ceux de
PGF2(Kirschvink et al., 2001)
Hyperréactivité aux allergènes
Chez les individus humains asthmatiques, il a été montré que les leucocytes pulmonaires
(en particuliers les neutrophiles et macrophages, dit « sensibilisés ») ont les capacités de
produire plus d’ERO que les leucocytes d’individus sains. De nombreuses études menées chez
l’homme montrent que les IgE peuvent entraîner un stress oxydant au sein de la population
leucocytaire à l’origine de l’activation de nombreux gènes codant des cytokines pro-
inflammatoires, comme IL-1, à l’origine de cette hyperréactivité des bronches
Il en est, très vraisemblablement, de même chez le cheval RAO selon Bureau et al.
(2000b), mais peu de données existent à ce sujet pour l’espèce équine. On ne sait pas non plus
si le stress oxydant induit par l’effort (plus fort chez les chevaux RAO) peut favoriser ces
mécanismes de sensibilisation aux allergènes par les IgE.
Inflammation pulmonaire neutrophilique
L’induction expérimentale d’une inflammation neutrophilique des bronches chez les
chevaux RAO par l’exposition d’allergènes n’entraîne pas forcément un stress oxydant
significatif selon les études (Deaton, 2006).
Par ailleurs, même si cela n’est pas suffisant pour établir un lien causal direct entre
l’inflammation neutrophile et les ERO, on sait que ces facteurs NF-B et AP-1 sont fortement
exprimés dans les cellules épithéliales bronchiques, les macrophages alvéolaires, les
neutrophiles et les mastocytes chez les chevaux RAO comme chez les humains asthmatiques
(Moran et al., 2011) (Bureau et al., 2000b).
Ces forts niveaux d’activation du facteur NF-B expliquent notamment l’importante
expression des cytokines pro-inflammatoires TNF-et d’IL-1 par les cellules récoltées lors du
lavage bronchoalvéolaire chez les chevaux RAO (Bureau et al., 2000b). Il a également été
montré que les cellules bronchiques isolées post mortem de chevaux RAO en crise ont des taux
d’expression en IL-1plus importants que ceux des chevaux sains ; cette expression est
évidemment plus forte en crise aiguë qu’en rémission clinique de la maladie (Bureau et al.,
2000a) (Bureau et al., 2000b).
1.vi Bilan
Chez l’homme asthmatique, le stress oxydant a été identifié dans les mécanismes
primaires qui précèdent le développement de l’inflammation allergique et les signes cliniques.
Cela n’a pas été montré chez le cheval RAO même si le rapprochement fréquent entre les deux
espèces nous laisse imaginer que des déséquilibres redox pourraient également contribuer au
développement de la maladie (Deaton, 2006) (Soffler, 2007) (Ahmad et al.. 2012).
Les résultats expérimentaux de la littérature montrent que les chevaux atteints
semblent avoir une production basale en ERO plus forte au repos que les individus sains et ce
en rémission comme en crise. L’organisme semble y répondre notamment en synthétisant plus
de GSH (résultats de Kirschvink et al. (2002b)).

140
Il ne fait presque aucun doute que les mécanismes physipathologiques de la RAO sont
multiples et prédominés par une inflammation neutrophilique. Cependant, si l’athlète équin
RAO peut être plus sensible aux ERO induit par un effort, on manque de preuves claires que
l’effort se surajoute à l’inflammation dans la production d’espèces réactive.
De même, on ne sait actuellement pas si les polynucléaires neutrophiles présents dans la
vascularisation pulmonaire et les voies respiratoires des chevaux RAO s’activent plus
facilement lors d’un effort que ceux des chevaux sains.
2) La maladie inflammatoire des voies respiratoires profondes chez les jeunes
chevaux de courses (IAD).
2.i Présentation générale
L’IAD ou « Inflammatory Airway Disease » touche 22 à 50% des purs-sangs anglais (PSA,
galopeurs) ainsi que des trotteurs et ambleurs. Dans leur étude, Wood et al. (2005), ont montré
une prévalence cumulée de 80% sur des PSA de 2 ans sur une saison. C’est une cause commune
d’altération des performances et d’interruption de l’entraînement (selon les études, l’arrêt
moyen pour IAD est compris entre 2 et 6 mois) (Rush and Mair, 2004) (Wood et al.. 2005).
C’est un véritable syndrome encore trop méconnu qui est sûrement la résultante de
nombreuses étiologies. Les étiologies proposées pour expliquer l’IAD incluent une composante
allergique avec l’inhalation de poussières, un stress pulmonaire récurrent (des saignements par
exemple), l’inhalation de polluants athmosphériques irritants pour l’épithélium pulmonaire
(sulfure d’hydrogène H2S, ammoniaque NH3 …) mais aussi des infections à répétition,
notamment virales (comme EHV-2) (Rush and Mair, 2004).
Bien que des facteurs génétiques soient suspectés, il semble que l’exercice favorise un
certain degré d’immunodépression pouvant aboutir au syndrome IAD, plus grave chez des
jeunes chevaux (Wood et al.. 2005) (Cywinska et al., 2013).
2.ii Tableau clinique et lésionnel
Au repos, l’examen physique est très souvent normal et la gravité des signes cliniques
faible. De la toux chronique et du jetage nasal mucoïde à muco-purulent sont souvent les seuls
signes cliniques visibles. À l’endoscopie, un exsudat muco-purulent est retrouvé dans le
pharynx, la trachée et les bronches (Wood et al.. 2005). Cette présence de mucus est
évidemment associée à une mauvaise ventilation entraînant une hypoxémie artérielle durant
l’effort maximal (Rush and Mair, 2004).
Le lavage bronchoalvéolaire met en évidence une inflammation mixte avec une forte
cellularité (Rush and Mair, 2004):
- une légère neutrophilie (15% des cellules en moyenne), lymphocytose et
monocytose (macrophages) ;
- une inflammation éosinophilique (5-60% des cellules) ;
- une quantité augmentée de mastocytes (>2% des cellules totales).
Cette inflammation pulmonaire est très compatible avec une réponse d’hypersensibilité
de Type I. Post-mortem, des granulomes éosinophiliques peuvent également être visualisés
(Rush and Mair, 2004).

141
2.iii Physiopathologie et lien avec le stress oxydant
Effort intense et l’immuno-dépression
Chez l’homme, des études épidémiologiques ont établi un lien direct entre l’effort
physique (endurance plus particulièrement) et les infections respiratoires supérieures se
développant après une épreuve (Fisher et al., 2011).
En effet, l’effort intense constitue un stress caractérisé par une neutrophilie et une
lymphopénie qui durent quelques heures à plusieurs jours après l’effort. Différentes sous-
populations lymphocytaires peuvent être concernées chez l’homme comme les lymphocytes T
CD3+, CD4+ (« T-helper cells », cruciaux dans l’activation des lymphocytes B) et CD8 + (Shute et
al., 2004). On observe également également une diminution de l’activité de nombreuses
enzymes impliquées dans le métabolisme énergétique des lymphocytes concernés (Hosoya et
al., 2004). Ainsi, cette lymphopénie contribuerait à expliquer la sensibilité virale et bactérienne
de l’athlète après un effort.
Cette théorie de la « fenêtre ouverte » (« open window ») après un effort semble
également s’appliquer chez le cheval. L’exercice intense et l’épuisement pendant une
compétition équestre peuvent également altérer l’immunité du cheval et accroître la
susceptibilité aux infections. Cette sensibilité varie suivant l’intensité de l’effort, sa fréquence et
les individus eux-mêmes (Cywinska et al., 2013).
Ainsi, l’IAD peut se développer après une infection virale lors des sessions
d’entraînement avec l’incapacité du système immunitaire de cibler et d’éliminer les virus et les
bactéries dans les petites voies respiratoires. Des herpesvirus équins (comme EHV-1, 2 et 4),
Streptococcus pneumoniae et Streptococcus equi zooepidemicus ont notamment été isolés dans
les petites bronches des chevaux atteints et qui pourraient jouer un rôle important dans la
physiopathologie de cette affection (Wood et al.. 2005).
Cywinska et al., (2013) ont montré que les cessions d’entraînement chez des PSA de
course (effort maximal) et des chevaux arabes (effort d’endurance) n’avaient aucune
répercussion significative sur la défense immunitaire innée (neutrophiles et monocytes). Une
étude précédente (Cywinska et al., 2010b) avait montré qu’après une course de 162 km (CEI
3*) d’endurance, une lymphopénie et une neutrophilie significative avec potentialisation du «
burst oxydatif » sont observées chez les chevaux. De plus, les lymphocytes étaient moins
réceptifs in vitro aux facteurs de multiplication cellulaires après l’effort. Nesse et al., (2002) ont
également montré que lors d’un effort maximal intense une diminution significative du taux de
prolifération et du nombre de lymphocytes circulant dans le sang étaient constatés entre 12 et
16 heures après une course de galop chez des chevaux PSA.
Ainsi, le type, l’intensité et la durée de l’effort ont une importance particulière chez le
cheval comme chez l’homme, établissant un seuil au delà duquel l’exercice physique devient
néfaste pour les défenses immunitaires, acquises en particulier.
Cependant, il semblerait que les défenses innées puissent également être altérées lors
d’efforts maximaux répétées, ou une réduction du burst oxydatif a été observée plusieurs
heures après une course chez des PSA (Raidal et al., 2000). D’autres résultats doivent encore
être obtenus à ce sujet.

142
Lymphopénie et stress oxydant induit par l’effort
Si les mécanismes impliqués dans l’immunodépression liés à l’effort impliquent la
réponse neuroendocrine avec des facteurs comme les catécholamines, le cortisol ou encore les
hormones de croissance, il existe également des preuves scientifiques pour relier le stress
oxydant induit par l’effort avec l’« open window » immunitaire après un effort (Fisher et al.,
2011).
Les lymphocytes peuvent générer de manière importante des ERO dans le milieu
intracellulaire suite à différents stimulus, jouant le plus souvent un rôle de second messager. In
vitro, des lymphocytes équins soumis à un stress oxydant fort (>200 mol/L d’H2O2) voient
leur réponse proliférative diminuer très fortement, alors que, comme dans d’autres types
cellulaires, une réponse proliférative est observée lors d’un stress oxydant faible (10 mol/L
d’H2O2) (Chiarada et al., 2002). Ces résultats sont en accord avec des études menées chez
l’homme. Si, de manière générale, l’exercice modéré a un effet globalement immunostimulateur,
un exercice physique intense peut-être suivi par différents degrés d’immunodépression voir
d’immunosuppression des lymphocytes (Shute et al., 2004).
De plus, des analyses in vitro suggèrent que les lymphocytes sont moins actifs et plus
sensibles aux ERO comme H2O2 pendant la phase de récupération (3h après effort maximal
chez l’homme) (Fisher et al., 2011). Or c’est également pendant cette période de récupération à
cause de la réoxygénation (et plus ou moins l’inflammation suivant l’effort considéré) qu’un du
stress oxydant peut se mettre se place. Leurs résultats indiquent par ailleurs que
l’entraînement peut aider à renforcer les défenses antioxydantes des lymphocytes et contribuer
à l’atténuation des lymphopénies observées.
Lors de l’effort, on pense que la lymphopénie induite provient de deux mécanismes
principaux :
- La distribution des lymphocytes dans les tissus et notamment le muscle.
- L’apoptose des lymphocytes, dépendant essentiellement de l’intensité de
l’effort.
L’importance respective de ces deux mécanismes dépendrait largement de l’effort
considéré, pouvant expliquer les variations et le temps de récupération du niveau normal en
lymphocytes circulants (Krüger et al., 2014).
L’apoptose fait partie de l’homéostasie normale des lymphocytes, notamment lors de la
maturation. Dans le cadre de l’exercice, cette apoptose semble impliquer le stress oxydant et la
protéines Bcl-2 (Krüger et al., 2014), (décrite en première partie, IV) A) 7). Quadrilatero et
Hoffman-Goetz ont également montré en 2004 que l’application de N-acétyl-cystéine empêche
l’apoptose de lymphocytes intestinaux suite à un effort intense chez le rat en maintenant des
niveaux importants en GSH intracellulaire.

143
Figure 26 : Apoptose des lymphocytes induite par l’effort. Adapté d’après (Krüger et al., 2014)
m : potentiel protonique mitochondrial ; FasL : Fas Ligant ; FasR : Fas Receptor ; GC : glucocorticoïdes ; GR :
récepteurs aux Glucocorticoïdes ; Cas : caspase.
2.iv Bilan
Le stress oxydant induit par l’effort pourrait contribuer à l’immunodéficience passagère
après l’effort (apoptose des lymphocytes et perte des compétences immunitaires) pour les
agressions (environnementales et infectieuses) que subissent les jeunes chevaux mis à
l’entraînement.
La mise en évidence de pathogènes viraux et bactériens dans les phases précoces de
l’IAD supporte l’idée que la « fenêtre ouverte » entraînée par l’effort contribue véritablement à
la mise en place primaire de l’affection. Cette dernière se complique ensuite par la mise en
place d’une inflammation pulmonaire également délétère pour les performances. N’oublions
pas que l’IAD peut également être considérée comme une forme précurseur du syndrome
allergique RAO.
Sur la base de l’exposé qui précède, le stress oxydant induit par l’effort pourrait
contribuer à la perte d’immunocompétence que subissent les jeunes chevaux mis à
l’entraînement, et ce même pour des efforts maximaux (contrairement aux données chez
l’homme). Cependant, la communauté scientifique s’accorde pour dire que d’autres facteurs,
parfois plus prépondérants (comme le cortisol et les autres agents neuro-endocriniens),
agissent de la même façon. De plus, chez l’homme au moins, l’importance de l’apoptose dans la
lymphopénie reste discutée (Krüger et al., 2014).

144
3) L’hémorragie pulmonaire induite par l’effort (HPIE)
3.i Présentation
L’hémorragie pulmonaire induite par l’effort (HPIE) est une affection courante du cheval
de course soumis à un effort submaximal à maximal. La prévalence réelle est difficile à établir et
les résultats dépendent largement de la méthode et des critères choisis pour conclure au
diagnostic d’HPIE.
Elle est le plus généralement à l’origine de contre-performances, associées ou non à un
l’épistaxis bilatéral pendant et juste après l’effort, qui se résoud quelques heures après la
course. L’épistaxis n’est présent que dans 0.1% à 9% des chevaux affectés, cette fréquence
variant selon les études. Pour mémoire, on observe une localisation préférentielle de
l’hémorragie pulmonaire à la zone dorso-caudale post-mortem (O’Callaghan et al., 1987).
Cependant, l’HPIE a récemment été identifiée comme une cause prépondérante de morts
subites pendant une course. Dans les cas extrêmes, la perte sanguine peut être si rapide et si
sévère qu’elle entraîne une défaillance cardio-pulmonaire (Lyle et al., 2012).
3.ii Physiopathologie théorique et lien avec le stress oxydant
Jamais clairement élucidée, la physiopathologie de l’HPIE est vraisemblablement
plurifactorielle.
Partant des observations post-mortem et la localisation des lésions pulmonaires, des
auteurs ont proposé très tôt une origine traumatique avec un stress mécanique locomoteur : les
poumons seraient soumis à des traumas répétés contre la colonne vertébrale par transmission
de l’onde choc de la foulée lors de l’effort (Clarke et al., 1985) (Schroter et al, 1998).
L’âge et l’historique de crises d’HPIE du cheval, ainsi que le degré d’inflammation
pulmonaire (incluant les chevaux souffrant d’IAD ou de RAO) sont également des facteurs
d’aggravation et de récidive fréquents (Derksen et al., 1992) (Couetil et al., 1999) (Newton and
Wood, 2002). Un PSA de 5 ans a ainsi plus de six fois plus de chance de développer l’affection
qu’un individu de 2 ans soumis au même effort en course (Couetil et al., 1999).
Les résultats de Cook et al. (1988) désignent l’obstruction des voies aériennes
supérieures (DDVP, affection laryngée …) comme une des causes principales d’HPIE entraînant
des variations de la pression intra-thoracique et de la pression transmurale entre les capillaires
et les alvéoles pulmonaires pendant l’effort menant à la rupture vasculaire et aux saignements.
Après chaque épisode d’HPIE un cycle inflammatoire auto-aggravant se met en place. La
présence de sang dans les voies respiratoires entraîne une inflammation pulmonaire
interstitielle, de la fibrose et de l’angiogénèse qui favorisent de futurs épisodes hémorragiques.
En plus d’être générateur d’ERO, l’inflammation des petites voies respiratoires augmente le
débit sanguin dans les petites bronches, prédisposant également les vaisseaux à la rupture et
aggravant l’hémorragie (Newton and Wood, 2002).
L’hypoxie induite par l’effort (voir seconde partie, III) B) 3)) pourrait être en partie
responsable de la fragilisation des vaisseaux et capillaires pulmonaires et semble être un
maillon clé dans la physiopathologie de l’affection selon West et al. (1993). Pour O’Callaghan et
al. (1987), les lésions primitives focales et subcliniques confinées aux lobules secondaires
évoluent défavorablement avec l’hypoxie induite par l’exercice maximal et l’obstruction

145
partielle des voies respiratoires. Mills et Higgins (1997a) ainsi que Derksen (1997) sont les
premiers à suggérer et à théoriser l’implication oxydative des ERO dans la physiopathologie de
l’HPIE. Les lésions vasculaires liées à l’augmentation de la pression pulmonaire et à l’hypoxie
lors d’un exercice intense maximal provoquent une flambée du stress oxydant aggravant le
mécanisme physipathologique par trois composantes principales :
- la fragilisation directe des vaisseaux bronchiques sanguins et des capillaires
alvéolaires ;
- la diminution de l’action vasodilatatrice du NO° au profit de la vasoconstriction
des artérioles bronchiques par des mécanismes non élucidés clairement par les auteurs
(transformation en peroxynitrite restant théoriquement possible) ;
- la diminution de la fluidité membranaire des hématies, entraînant un
ralentissement de l’écoulement sanguin dans les capillaires pulmonaires et donc une
augmentation de la pression vasculaire pulmonaire.
Certaines études réalisées chez d’autres espèces pourraient également apporter du
crédit au modèle de Mills, Higgins (1997a) et Dersen (1997). Chez le rat, notamment, il a été
montré que les ERO produits par la xanthine oxydase activée pendant la phase d’hypoxie
contribuent très significativement au développement de l’hypertension pulmonaire chronique
(Hoshikawa et al., 2001).
3.iii Bilan
L’hypothèse de l’implication du stress oxydant dans le mécanisme primaire de
l’hémorragie pulmonaire induite par l’effort a été émise dans la fin des années 90 ; si le modèle
proposé est alléchant, de nombreuses zones d’ombre doivent encore être éclaircies par la
science (Soffler, 2007).
Constatation plus probable, l’HPIE est un événement pro-inflammatoire et générateur de
stress oxydant secondaire. La forte concentration en hémosidérophages dans le liquide de
lavage broncho-alvéolaire (LBA) après un épisode de HPIE est là pour nous le rappeler.
Par ailleurs, Couetil et al. (1999) ont également montré que des chevaux souffrant
d’HPIE présentent une hypoxémie artérielle, une hypercapnie et un taux en lactates sanguins
supérieurs à ceux des individus témoins lors d’effort maximal. Plus que de redémontrer
l’intolérance à l’effort constatée cliniquement chez ces chevaux, cette étude nous laisse
imaginer théoriquement la genèse d’un stress oxydant général supérieur en aggravant
l’hypoxie déjà entraînée par un effort maximal à supra-maximal, phénomène par ailleurs déjà
remarqué pour les autres affections pulmonaires étudiées précédemment.
C) Pathologie articulaire : la maladie articulaire dégénérative équine
1) Pré-requis d’arthrologie
1.i Organisation générale d’une diarthrose
Une diarthrose est une articulation mobile caractérisée par la présence d’un cartilage
articulaire, de synovie et une capsule articulaire fibreuse entourant le tout (tapissée à
l’intérieur de la membrane synoviale) (figure 26).

146
Figure 27 : Coupe frontale schématique d’une diarthrose.
1.ii Le cartilage articulaire
Le cartilage articulaire assure le glissement entre les deux surfaces articulaires. C’est un
tissu avasculaire et non innervé, composé d’un contingent cellulaire (représenté par les
chondrocytes) et d’un contingent non-cellulaire, la matrice cartilagineuse.
La matrice, masse « sèche » du cartilage, est principalement représentée par le collagène
et en moindre importance par les protéoglycanes et les glycoprotéines. Le collagène de type II
est le collagène fibrillaire majeur du cartilage hyalin (90-95% du collagène cartilagineux).
Le collagène II constitue le maillage de la matrice extracellulaire, son architecture. Son
turnover est cependant très faible et la demi-vie du collagène de type II est estimée à plusieurs
centaines d’années ; on peut donc considérer qu’il n’est pas renouvelé au cours de la vie d’un
individu (Alcaraz et al., 2013).
Dans cette matrice vivent les chondrocytes qui représentent approximativement 5% du
volume du cartilage. Dans les conditions normales, les chondrocytes vivent dans une matrice
avasculaire, avec pour conséquence directe un faible apport en dioxygène. Le liquide synovial
est l’unique source d’approvisionnement pour ces cellules qui ont un métabolisme adapté à cet
environnement hypoxémique (Henrotin, 2003). Les chondrocytes équins restent
particulièrement résistant à l’hypoxie et peuvent survivre plus de 10 jours dans une
atmosphère avec moins de 1% de dioxygène. De même, leur consommation basale
physiologique est particulièrement faible (moins de 20.5 pmol O2/min/106 cellules in vitro)
(Schneider et al., 2005).
Les chondrocytes sont directement responsables de la synthèse des éléments de la
matrice qui les hébergent ; leur métabolisme est garant du bon équilibre fonctionnel de
l’ensemble du cartilage. On considère que les cellules sur la couche la plus superficielle sont

147
exposées à 5-7% d’oxygène alors que les chondrocytes des couches les plus profondes à moins
de 1% d’oxygène. Ces derniers ont donc un métabolisme anaérobie prédominant par rapport
aux couches les plus superficielles où la respiration aérobie est possible (Alcaraz et al., 2013).
1.iii Le liquide synovial
Le liquide synovial (ou synovie) est un liquide visqueux, légèrement ambré, au pH
faiblement alcalin. Ses fonctions les plus importantes sont la lubrification et la nutrition du
tissu articulaire. Il dérive essentiellement de l’ultrafiltration des vaisseaux sanguins de la
membrane synoviale, cependant les protéines de la synovie sont différentes de celles du plasma
sanguin.
L’albumine et les protéines de bas poids moléculaire jouent un rôle important dans sa
pression osmotique et traversent facilement l’endothélium vasculaire, tandis que le
fibrinogène, avec son poids moléculaire élevé, est retenu dans le sang (mis à part lors d’une
inflammation où la perméabilité vasculaire est augmentée).
Le liquide synovial équin contient également des cellules (moins de 500 cellules
nuclées/μL): 90% d’entre elles sont des cellules mononucléaires, surtout des synoviocytes, des
monocytes et des lymphocytes, et 10% sont des PNN. Le nombre de leucocytes diffère selon les
articulations chez le cheval (par exemple, pour un boulet on considère en moyenne: 3,9 x 108/l,
et le grasset : 1,2x108/l).
Il existe également une variation de la teneur en protéines et en fractions protéiques en
fonction de l’âge, du sexe et de l’intensité de l’entraînement du cheval (Schneider et al., 2007).
1.iv La capsule articulaire
La capsule articulaire se compose de deux couches : une paroi fibreuse externe et la
membrane synoviale.
La paroi fibreuse externe
La paroi fibreuse est constituée d’un tissu conjonctif fibreux, partiellement renforcé par
des ligaments, peu vascularisée et jointe au périoste. Elle contribue à stabiliser l’articulation. Le
tissu conjonctif est différent selon les régions: fibreux aux zones de traction, adipeux aux
endroits exposés à l’extension et à la flexion et aréolaire là où il y a une activité métabolique
accrue (couche cellulaire épaisse, tissu conjonctif lâche, riche en vaisseaux sanguins). La
circulation sanguine y est assurée par des capillaires fenêtrés (Schneider et al., 2007).
La membrane synoviale
La membrane synoviale ou synovium est la couche intérieure de la capsule articulaire
avec une intima synoviale (couche lâche de cellules plates sans membrane basale) et une sub-
intima (tissus conjonctif avec de nombreux capillaires fenêtrés). Dans l’intima, on distingue
deux phénotypes différents de synoviocytes :
- les synoviocytes de type A : Les synoviocytes A sont des macrophages ; ils
contribuent à maintenir l’articulation « propre ». En condition pathologique, ils participent
également aux réponses immunologiques. Les cellules sont sphériques et possèdent une
membrane riche en lamellipodes. Ces cellules se retrouvent préférentiellement dans la région
supérieure des villosités synoviales et sont beaucoup plus rares au niveau de leur pôle basal.

148
Elles sont riches en vacuoles et en lysosomes. Le nombre de synoviocytes A est
particulièrement élevé lors d’ostéoarthrose (OA).
- les synoviocytes de type B : Les synoviocytes B sont très proches des
fibroblastes. Ils sont responsables de la production des constituants du liquide synovial et de la
matrice extracellulaire de l’intima synoviale : collagène, fibronectine, acide hyaluronique... Ils
sont notamment caractérisés par un réticulum endoplasmique et un appareil de Golgi bien
développé par rapport aux synoviocytes A. Chez le cheval, les synoviocytes B ne possèdent pas
ces granules cytoplasmiques et sont comparables aux synoviocytes agranulaires trouvés chez
d’autres mammifères (cobaye, lapin et homme) (Schneider et al., 2007).
1.v L’os sous-chondral
L’os sous-chondral est un os compact situé au dessous du cartilage articulaire. Il joue
notamment une fonction d’amortissement en dissipant les forces de compression qui s’exercent
sur l’articulation (durant la foulée par exemple), protégeant par la même le cartilage des
contraintes mécaniques.
1.vi Sensibilité de l’articulation au stress oxydant et défenses antioxydantes
Des études immuno-histochimiques chez le rat ont montré que les principaux
antioxydants des chondrocytes sont la catalase et le système glutathion peroxydase/réductase.
Le GSH, la glutathion-S-transférase et la superoxyde dismutase (SOD) existent également mais
leur importance est plus faible dans ces défenses. Des études menées chez le rat n’ont pas
montré de différence significative dans la distribution des défenses enzymatiques selon l’âge
des individus (Alcaraz et al. (eds.), 2013).
Dans la synovie, l’acide hyaluronique joue un rôle essentiel qui sera revu plus loin. Pour
illustration, une injection exogène d’acide hyaluronique dans des genoux atteints d’OA chez
l’homme augmente significativement l’activité de la SOD et pourrait également inhiber la
production de monoxyde d’azote NO° (Kalaci and al., 2007).
Mis à part l’acide hyaluronique, il a également été montré chez le cheval que la
transferrine et la céruloplasmasmine constituaient une grosse partie de la défense
antioxydante de la synovie (Dimock et al., 2000). Leurs taux sinoviaux augmentent d’ailleurs
lors d’inflammation articulaire.
2) La maladie articulaire dégénérative équine
2.i Présentation générale
La maladie articulaire dégénérative équine (« equine joint disease ») ou ostéoarthrose
dégénérative (OA) est caractérisée par une dégradation de la matrice cartilagineuse et une
mort cellulaire qui mène à la perte progressive de l’intégrité du cartilage articulaire. C’est une
affection d’évolution lente.
L’étude de la pathologie de l’OA chez le cheval est un domaine de la recherche très actif.
Pouvant toucher de nombreuses articulations, c’est en effet une cause majeure de contre-
performance (boîterie) et de retraite anticipée du cheval athlète (Soffler, 2007) (Lamprecht and
Williams, 2012).
On ne comprend actuellement pas bien pourquoi certains chevaux déclarent des lésions
d’OA lors de régimes d’entraînement moins intenses que d’autres chevaux, et la sensibilité au

149
développement de l’OA n’est pas la même suivant les individus. De plus, on ne peut pas
actuellement prédire à partir de quelle intensité d’exercice et de quel niveau d’entraînement
des lésions apparaîtront pour une articulation donnée (en d’autres termes sa tolérance à
l’effort) (Firth, 2006).
2.ii Tableau lésionnel
Lors de l’exercice, l’OA est généralement initiée par une agression mécanique sur
l’articulation. Des débris de la matrice extracellulaire (constitué de collagène et de
protéoglycanes), sont libérés dans le liquide synovial, ce qui provoque une synovite. Dans les
stades précoces de la maladie, c’est elle qui est responsable des signes cliniques (douleur,
boîterie et réticence à la mobilisation de l’articulation atteinte).
Les débris de matrice cartilagineuse activent les cellules de l’inflammation mais aussi les
synoviocytes (type A et B). Les synoviocytes B sécrètent de nombreux facteurs pro- et anti-
inflammatoires (comme IL-1et TNF-), de chémoattraction, d’angiogénèse, de dégradation
matricielle, d’inhibition de la dégradation matricielle, d’ostéoclastogénèse et d’ostéogénèse
(Schneider et al., 2007). Les chondrocytes adoptent alors un phénotype pathologique : ces
derniers synthétisent à leur tour des médiateurs de l’inflammation ainsi que des enzymes
(principalement des métalloprotéinases), ainsi que d’autres métabolites toxiques pour les
constituants de la matrice cellulaire (dont des ERO). Cette réactivité aggrave les lésions initiales
et déclenche un phénomène d’amplification, empirant encore l’inflammation synoviale, et ce
dans un cercle vicieux complexe et délétère pour le cartilage (Alcaraz et al., 2013).
Progressivement le cartilage devient hypocellulaire (disparition des chondrocytes), et se
fragmente sous l’effet des contraintes pour disparaître (Jungbluth et al., 2008).
L’OA est une maladie qui implique non seulement le cartilage articulaire, mais aussi la
membrane synoviale, l’os sous-chondral et les tissus moux péri-articulaires.
Macroscopiquement, des remodelages caractéristiques apparaissent, variables selon la gravité
et la chronicité de l’affection :
- Dégradation cartilagineuse : fibrillation (apparition de fines crevasses sur la
surface du cartilage), excoriation et amincissement progressif de la couche jusqu’à laisser
apparaître l’os sous-chondral. Le cartilage perd également en fermeté et cohérence. Il est
égalemement moins élastique.
- Synovite : c’est généralement le premier signe observé et accompagné d’une
douleur articulaire. Elle se caractérise par une augmentation du volume de la synovie, qui
devient moins visqueuse et plus liquide. Cette dernière se charge en cellules de l’inflammation
et de protéines. Cette perte de viscosité du liquide synovial augmente les frictions entre les
deux faces cartilagineuses, elles-même déjà fragilisées.
- Croissance d’ostéophytes péri-articulaires, réduisant dans les cas graves
l’amplitude articulaire et pouvant être douloureux.
- Sclérose sous-chondrale : lors de l’OA, l’os sous-chondral devient sclérotique et
hypominéralisé. Des kystes sous-chondraux peuvent également se développer. La sclérose
sous-chondrale réduit la capacité de l’articulation à amortir les chocs, reportant ces contraintes
sur le cartilage. Il n’est cependant pas clair à ce jour si les mécanismes entraînant ce tableau

150
lésionnel dans l’os sous-chondral précèdent ou sont une conséquence de la dégradation
cartilagineuse.
- Capsulite : une synovite ou une capsulite aiguë consécutive à un traumatisme
peut à elle seule induire une dégénérescence articulaire. L’inflammation aiguë de la membrane
synoviale se caractérise par une congestion et une importante néovascularisation (due à la
sécrétion de VGEF) ; des hémorragies peuvent également être mises en évidence. Lorsque
l’inflammation devient chronique, la paroi fibreuse s’épaissit et perd en élasticité sous l’effet de
l’accroissement du nombre de fibroblastes sous l’intima, réduisant également la mobilité
articulaire (Schneider et al., 2007).
Bien qu’il soit généralement accepté que l’OA commence au niveau du cartilage
articulaire puis se propage à l’os sous-chondral et aux autres tissus synoviaux, il existe une
controverse quant au point d’origine de l’OA.
3) Physiopathologie de l’OA et lien avec le stress oxydant
3.i Mise en évidence d’un stress oxydant lors d’OA
Plusieurs marqueurs du stress oxydant peuvent être utilisés pour témoigner d’un stress
oxydant in vivo. Les plus communément utilisés sont les produits issus de la peroxydation
lipidique, la nitrotyrosine, les protéines carbonylées ainsi que le collagène nitraté et glucoxydé
(Henrotin, 2003). En particulier, le collagène glucoxydé est un marqueur d’accumulation des
dégâts oxydatifs dans le temps et donc aussi du vieillissement de l’articulation (voir également
première partie, IV) A) 2) 2.iv). Des techniques d’immunohistochimie peuvent également
permettre de localiser précisément les dépôts de ces marqueurs dans l’articulation.
L’exercice physique a un effet sur l’équilibre redox de l’articulation. Une étude récente
démontre notamment que la course chez des souris déficientes en SOD2 entraîne une
augmentation significative du stress oxydant dans l’articulation (Baur et al., 2011).
Chez l’homme, il a été montré à de nombreuses reprises qu’une surproduction d’ERO et
de monoxyde d’azote est corrélée à l’OA. En effet, de la nitrotyrosine a été détectée en quantité
significativement augmentée dans la synovie et le cartilage des articulations atteintes (Loeser
et al., 2002) (Kalaci and al., 2007). De même, une étude a montré que l’activité de la SOD est
diminuée de manière significative chez des individus atteints d’OA. L’activité de la SOD pourrait
donc être altérée lors d’OA, au moins chez l’homme (Kalaci and al., 2007).
Chez le cheval, des résultats similaires ont également été obtenus. On sait notamment
que comme chez l’homme, les neutrophiles activent le « respiratory burst » dans une
articulation inflammée expérimentalement (Soffler, 2007). Les chevaux ayant eu un trauma
articulaire (principalement à cause ou aggravé par la présence d’un fragment articulaire) ou
souffrant d’ostéochondrose disséquante (OCD) ont également des taux en protéines
carbonylées bien significativement supérieurs dans leur synovie à ceux des chevaux sains
(Dimock et al., 2000). Enfin, on note la présence de nitrotyrosine en quantité augmentée dans le
cartilage (Kim et al., 2003) et l’os sous-chondral (Van der Harst et al., 2006) chez les chevaux
atteints d’OA.
Contrairement à l’étude de Kim et al. (2003), Van der Harst et al. (2006) ont trouvé que
la nitrotyrosine était très présente dans l’os sous-chondral des chevaux atteints d’OA

151
débutantes et modérées. Ces résultats semblent indiquer l’importance du stress oxydant dans
cette couche au début du développement de la maladie.
3.ii Sources prépondérantes d’ERO lors d’OA
Différents mécanismes de production d’ERO ont été identifiés dans l’articulation lors du
développement de l’OA, pas toujours in vivo malheureusement. Cependant, les résultats de
plusieurs études indiquent que le stress oxydant pourrait être retrouvé au commencement de
la maladie dégénérative articulaire, avant que l’inflammation ne s’installe.
Forces et contraintes mécaniques
Des études réalisées in vitro montrent qu’une charge mécanique compressive excessive
augmente le stress oxydant dans le chondrocyte (Beecher et al., 2007) (Tomiyama et al., 2007).
Également, des chondrocytes humains exposés à des forces de cisaillement up-régulent la
synthèse d’ERO et l’activation d’iNOS, menant à la production conjointe de NO° et d’ions
superoxydes, pouvant évidemment entraîner la formation de peroxynitrite (Martin et al., 2004)
(Kalaci and al., 2007).
Fait intéressant, dans l’étude de Martin et al., cette synthèse d’ERO entraînée par des
forces de cisaillement est postivement corrélée à la disparition des chondrocytes par apoptose.
Martin et al. ont repris leur protocole de 2004 en 2006 et obtiennent les mêmes résultats ; ils
ont également montré que l’ajout d’antioxydants diminuait l’apoptose des chondrocytes induite
par les forces de cisaillement.
Ainsi, in vitro au moins, les forces compressives et de cisaillement entraînent la
production d’ERO aboutissant à un stress oxydant suffisant et responsable de l’apoptose des
chondrocytes, de manière primaire. Cela n’a jamais été montré in vivo. Healy et al. (2005)
ont essayé d’établir le mécanisme à l’origine de cette production. Loin d’y répondre
complètement, ils ont découvert que les forces de cisaillement peuvent supprimer l’activité de
la phosphatidylinositol 3-kinase (PI3-K). Or cette kinase réprime normalement le facteur Nrf2,
et donc la voie d’activation du facteur transcriptionnel ARE (figure 20).
Les chondrocytes ne sembleraient pas être les seuls à être liés au stress oxydant par ces
contraintes mécaniques, l’os sous-chondral pourrait l’être également: Yamamoto et al. (2005)
ont montré in vitro que l’augmentation de la charge mécanique sur des cellules ostéoblastique-
like mène à l’augmentation de la synthèse d’ERO et de l’activité de la SOD, dépendamment de la
force et du temps d’application de la contrainte. Les résultats indiquent également que le
dysfonctionnement mitochondrial et les filaments d’actine pourraient être impliqués dans le
mécanisme.
Phénomène d’ischémie-reperfusion articulaire lors de l’effort
Allen et al. (1989) ont été parmi les premiers à évoquer qu’une articulation enflammée
subit, lors du mouvement, des cycles d’ischémie-reperfusion courts et répétés. Il faut préciser
que leur étude concernait des cas d’arthrite rhumatoïde humaine où la synovite est souvent
très importante. Ce phénomène contribuerait à la persistance du processus inflammatoire et la
production d’ERO. Selon eux, cette production aurait pour principale origine la xanthine
oxydase présente dans les cellules endothéliales des vaisseaux bordant la membrane synoviale
de l’articulation, et qui prolifèrent lors d’OA (résultat précisé dans l’étude avec l’usage de
l’allopurinol).

152
Chez le cheval, un trauma aigu ou même chronique notamment lors d’un effort physique,
peut également altérer l’apport sanguin de la membrane synoviale, entraînant des cycles
d’ischémie-reperfusion au travers de l’œdème et de l’hypoxie passagère lors du mouvement
articulaire (Mcllwraith, 1996).
A priori, si un tel phénomène a lieu, les chondrocytes supporteraient mieux le
phénomène (vivant déjà proches de l’anaérobiose, avec moins de 7% d’O2 dans le liquide
synovial) que les synoviocytes (physiologiquement mieux irrigués avec plus de 12% d’O2).
In vitro, des cycles d’anoxie/ré-oxygénation (20 minutes d’anoxie puis réoxygénation)
sur des synoviocytes et des chondrocytes équins ont montré une production accrue de radicaux
libres de l’oxygène (détectés par RPE) par les synoviocytes mais pas par les chondrocytes
(Schneider et al., 2005). Les auteurs suggèrent que l’activité oxydante des synoviocytes
pourrait jouer un rôle important dans l’initiation de l’OA chez le cheval : ainsi au tout début de
l’OA les chondrocytes pourraient être dans un premier temps les victimes des ERO générés par
les synoviocytes, avant de construire leur propre réponse inflammatoire.
Malheureusement aujourd’hui, il n’y a pas d’étude menée chez le cheval qui permette de
cibler les mouvements qui génèrent le plus d’ERO suivant ce mécanisme physiopathologique.
Par ailleurs, dans une articulation déjà atteinte d’OA, la tension en oxygène dans le
liquide synovial subit des fluctuations à cause de l’accélération du métabolisme et les
altérations de la qualité du liquide synovial. En réponse aux variations de la pO2, les
chondrocytes « pathologiques » peuvent effectivement produire des ERO à des taux qui sont
normalement ceux générés par les cellules immunitaires comme les macrophages (Henrotin,
2003).
Production secondaire au cours de l’inflammation chronique
Une fois initié, l’inflammation articulaire voit ces cellules de la phagocytose (les
neutrophiles, les macrophages dont les synoviocytes A) s’activer et générer du stress oxydant.
Les mécanismes sont schématiquement les mêmes que pour les autres tissus et ont déjà été
évoqués précédemment (partie 2, I) et II)). Cependant bien que pouvant être retrouvés dans le
liquide synovial lors d’OA, il ne faut pas donner aux neutrophiles la même importance dans la
physiopathologie de la maladie dégénérative équine que celle de l’arthrite septique ou de
d’arthrite rhumatoïde chez l’homme.
L’activation des NADPH oxydase et les NOs (eNOS et iNOS en particulier) dans les
chondrocytes et les cellules endothéliales jouent une place prépondérante dans l’adoption du
profil pathologique de l’ostéoarthrose (Jungbluth et al., 2008) (Henrotin, 2003). Henrotin et al.
(2003) rapportent qu’une injection intra-articulaire de N-iminoéthyl-L-lysine, un inhibiteur
sélectif de la iNOS, réduisait la progression de l’érosion cartilagineuse dans un modèle canin
d’ostéoarthrose.
Le chondrocyte normal est capable de synthétiser la myéloperoxydase mais cette
production est faible dans une articulation saine. Lors d’OA, le taux en ARNm pour cette
enzyme augmente, ce qui suggère clairement que la production d’ClO- est également accrue
(Henrotin, 2003). Selon Olszowski et al., (2003) l’ ion hypochlorite fait partie des ERO les plus
toxiques pour une articulation.
Ainsi, des chondrocytes activés peuvent produire un large éventail d’espèces réactives,
tel que le monoxyde d’azote NO° (et du peroxynitrite), l’ion superoxyde O2°-, le radical

153
hydroxyle HO° et le peroxyde d’hydrogène H2O2 et ClO- (Henrotin, 2003) (Dai et al., 2006)
(Alcaraz et al., 2013).
Malgré des preuves multiples supportant l’hypothèse selon laquelle le NO° est
directement lié au développement initial de l’OA, les mécanismes sont complexes et pas
clairement connus, encore moins chez le cheval dont le profil inflammatoire pourrait présenter
des particularités propres à l’espèce. De plus, on ne sait pas si le NO° seul est suffisant pour
entrainer des dégâts dans l’articulation ou si les effets du NO° sont liés à sa réaction avec les
ERO pour former le peroxynitrite (Loeser et al., 2002) (Henrotin, 2003).
3.iii Conséquences dans le développement de l’OA
Le stress oxydant peut directement altérer de nombreuses biomolécules dans
l’articulation comme dans les autres tissus (voir partie 1, IV)) mais aussi de manière indirecte
en modifiant le métabolisme cellulaire et l’homéostasie articulaire. Ci-dessous sont regroupés
les effets délétères des ERO pouvant expliquer, en partie au moins, les remodelages lésionnels
ostéo-articulaires cités précédemment :
Destruction de l’acide hyaluronique
Comme souligné plus tôt (partie 1, III)C)1)1.viii), l’acide hyaluronique assure une partie
de la défense antioxydante de l’articulation ; son turnover et la phagocytose de ces métabolites
par les synoviocytes A assurent le maintien fonctionnel de la synovie.
En cas de stress oxydant, les ERO sont responsables de la baisse en viscosité de la
synovie en coupant l’acide hyaluronique en petits fragments. Cette baisse de la viscosité
augmente la friction entre les deux surfaces du cartilage et également élimine la capacité de
l’acide hyaluronique à inhiber la phagocytose ainsi que son « burst » oxydatif (Soffler, 2007).
Destruction de la matrice collagénique
Le collagène est une molécule sensible aux stress oxydant. L’exposition du collagène aux
ERO entraîne des coupures dans sa structure primaire qui libèrent de petits peptides contenant
la 4-hydroxyproline, souvent utilisée comme marqueur de cette dégradation (Alcaraz et al.,
2013).
L’ion hypochlorite (ClO-) généré par la myéloperoxydase cause la fragmentation du
collagène II articulaire pour de fortes concentrations. Par ailleurs, à des taux inférieurs, il peut
entraîner des désaminations et des chlorations des chaînes latérales (notamment de la tyrosine
en dichlorotyrosine). Les chloramines formées par la réaction entre une amine primaire et ClO-
n’entraînent pas de fragmentation directe du collagène mais augmentent de manière
significative sa sensibilité aux dégradations des collagénases pouvant être produites lors de
l’inflammation (Olszowski et al., 2003).
Comme nous l’avons vu précédemment, le collagène est une protéine particulièrement
sensible à la glycoxydation in vivo, principalement à cause de son faible turnover. Enfin, les
molécules glycoxydées ont un effet pro-inflammatoire en attirant les neutrophiles vers
l’articulation touchée (Alcaraz et al., 2013).

154
Figure 28: Schéma principal de l’action délétère des ERO sur la matrice cartilagineuse. Adapaté d’après (Alcaraz et
al., 2013).
Inhibition de la cicatrisation cartilagineuse
Il a été démontré que de fortes concentrations locales de monoxyde d’azote avaient des
effets néfastes sur l’articulation, tels que (Kalaci and al., 2007) (Alcaraz et al., 2013):
- inhibition la synthèse de protéoglycanes et de collagène ;
- inhibition de synthèse de l’antagoniste du récepteur de l’IL-1 ;
- inhibition de la prolifération chondrocytaire ;
- activation des métalloprotéinases.
IL-1 peut aussi inhiber la synthèse du collagène de type II dans une culture in vitro de
chondrocytes de lapin : une des hypothèses est l’inhibition de la prolyl-hydroxylase par NO°
(Henrotin, 2003).
In vitro, des études ont montré que le NO° est impliqué dans l’inhibition de la synthèse
en protéoglycanes et la sulfation de ces branches par IL-1. Il semblerait que la génèse en
radical superoxyde conjointement à NO° soit également nécessaire à l’inactivation de cette
synthèse. En effet, le traitement d’une culture in vitro de chondrocytes avec ONOO- diminue de
manière significative l’expression des gènes responsables de la production d’agrécanes
(protéoglycanes du tissu cartilagineux de masse moléculaire atteignant plusieurs milliers de
kDa) (Henrotin, 2003).
De plus, les ERO inhivent également la synthèse de la matrice cartilagineuse en inhibant
la sensibilité des chondrocytes aux facteurs de croissance : une étude menée chez des souris
incapables de synthétiser iNOS suggère que NO° est responsable au moins en partie de

155
l’insensibilité des chondrocytes à IGF-1 (« insuline growth factor-1 ») en inhibant la
phosphorylation du récepteur à IGF-1. Henrotin (2003) indique également que le monoxyde
d’azote inhibe également la migration des chondrocytes en modifiant le cytosquelette d’actine
et leurs attaches à la fibronectine.
Dysfonctionnement énergétique et mort des chondrocytes
En situation d’OA, les chondrocytes sont sensibles au dysfonctionnement mitochondrial
entraîné par des cytokines pro-inflammatoires comme le TNF-et IL-1. Ainsi, le
dysfonctionnement mitochondrial pourrait accélérer le mécanisme de dégradation du cartilage
et la mise en place de l’inflammation en accroissant la production d’ERO et entraîner l’apoptose
des chondrocytes endommagés (Henrotin, 2003). On a pu montrer que dans un modèle d’OA,
l’ADN mitochondrial des synoviocytes et des chondrocytes comportait un taux de mutation
significativement plus élevé que pour une articulation saine (Alcaraz et al., 2013).
La disparition des chondrocytes est une étape clé de la physiopathologie de l’OA. Autant
l’apoptose que la nécrose sont responsables de la perte du contingent cellulaire du cartilage, ne
favorisant pas la réhabilitation fonctionnelle d’un tissu qui cicatrise déjà très mal (Henrotin,
2003).
Kim et al. (2003) ont trouvé un pourcentage de chondrocytes apoptotiques avec un
marquage positif à la nitrotyrosine dans les articulations des chevaux atteints d’OA
significativement plus élevé que chez les individus sains. Ces résultats suggèrent l’interrelation
proche entre le stress oxydant lors d’OA dans l’apoptose des chondrocytes, et notamment
l’implication du peroxynitrite. Ces résultats sont en accord avec d’autres auteurs qui indiquent
que l’apoptose des chondrocytes est principalement déclenchée par le monoxyde d’azote
(Kalaci and al., 2007).
Dai et al. (2006) ont mené une étude in vitro avec des chondrocytes issus d’articulations
atteintes d’OA de rat et d’homme dont les résultats suggèrent qu’ IL-1 et le stress oxydant
entraînent l’apoptose prématurée des chondrocytes médiée en partie par l’expression de
cavéoline-1 sans que les mécanismes ne soient bien compris.
Une autre hypothèse suggère que les ERO ralentissent également la mise en place d’un
tissu cicatriciel en réduisant la capacité de migration et de prolifération des cellules
précurseurs des chondrocytes.
Il a en effet été démontré que le NO° inhibe cette migration en altérant la fibronectine et
l’actine du cytosquelette (Henrotin, 2003).
De plus, les dommages oxydatifs des chondrocytes entraînant cette sénescence
prématurée s’accompagnent d’une résistance à l’action de l’« insuline-like growth factor-1 »
(IGF-1) qui potentialise l’anabolisme cellulaire sur des chondrocytes normaux; les auteurs
soulignent néanmoins que le stress oxydant ne semble pas être le seul acteur de cette chondro-
résistance (Loeser et al., 2002).
In vitro, l’absence de réponse à IGF-1 des chondrocytes dans l’OA a été reliée à la
transformation de la tyrosine en nitrotyrosine de son récepteur par le peroxynitrite. Cette
modification empêche la phosphorylation du récepteur à IGF-1 qui l’active normalement après
fixation du facteur de croissance (Studer et al., 2000).

156
Mort des synoviocytes
Comme pour les chondrocytes le dérèglement mitochondrial des synoviocytes est
constaté lors d’OA et pourrait également stimuler une réponse pathologique aux cytokines pro-
inflammatoires, accélérant le mécanisme de dégradation du cartilage et l’entretien de
l’inflammation articulaire.
L’induction de l’apoptose des synoviocytes ostéoarthritiques semble associée à
l’expression de la protéine p53 dans la membrane synoviale ostéoarthritique in vivo et son
expression semble positivement corrélée à celle de la concentration en NO° (Schneider et al.,
2007).
Sclérose sous-chondrale
Le NO° semble aussi jouer un rôle dans la sclérose de l’os sous-chondral lors d’OA. Un
modèle expérimental ovin d’OA a montré qu’un donneur de NO° (le glycéryl trinitrate)
augmentait la sclérose de l’os (Cake et al., 2004). Ces résultats sont intéressants en complément
de ceux de Van der Harst et al. (2006) chez le cheval.
Cependant, à ce jour, on ne sait pas si le stress oxydant au niveau du cartilage articulaire
peut aussi affecter le métabolisme des cellules de l’os sous-chondral et ce de manière directe et
indirecte. De plus amples études doivent être réalisées pour confirmer que le stress oxydant
dans le cadre de l’OA contribue à la sclérose sous-chondrale.
4) Bilan
Des marqueurs du stress oxydant ont bien été mis en évidence chez le cheval atteint
d’OA, et ce très tôt dans la physiopathologie. Fait intéressant, on dispose de données qui nous
permettraient de situer le stress oxydant dans les mécanismes primaires de la maladie et ce en
confrontant les données in vivo avec les données in vitro obtenues dans plusieurs modèles d’OA,
dont le cheval.
Ils concernent notamment la réponse oxydative des synoviocytes et des chondrocytes
aux traumatismes articulaires, mais aussi l’importance de l’os sous-chondral dans la génèse
d’ERO au début de la maladie articulaire dégénérative équine.
On soulignera la corrélation positive entre la disparition des chondrocytes par apoptose
et la génèse des ERO. L’inflammation aggrave par la suite cette explosion oxydative par des
mécanismes que nous avons déjà passés en revue ; elle s’accompagne également du
dysfonctionnement pathologique objectif des chondrocytes et des synoviocytes ainsi que de
l’accumulation des produits de dégradation toxiques dans ce vase clos qu’est une articulation
inflammée.
En conclusion, il ne faut pas oublier non plus que les bienfaits de l’exercice sur les
articulations peuvent également passer par le stress oxydant en favorisant chez le cheval la
synthèse transitoire d’aggrécanes (mais aussi de chondroïtine sulfate) deux heures après un
trauma modéré (Lamprecht and Williams, 2012).

157
D) Ostéologie et croissance : l’ostéochondrose disséquante (OCD)
1) Présentation et importance dans l’espèce équine
La physiopathologie de l’ostéochondrose dissséquante (OCD) chez le cheval et d’autres
espèces de mammifères est très étudiée mais de nombreuses zones d’ombre demeurent.
Classiquement, l’ostéochondrose est primitivement une maladie développementale de
l’os et non du cartilage articulaire. Chez le cheval, l’OCD est décrite comme un arrêt focal de
l’ossification endochondrale pouvant entraîner la libération d’un fragment cartilagineux dans
l’articulation. L’ostéchondrose peut également intéresser l’os sous-chondral et notamment
former des kystes sous-chondraux.
Son importance chez le cheval vient essentiellement du fait que la présence de
fragments ostéochondraux dans l’articulation peut être à l’origine d’une gêne mécanique et
d’une inflammation articulaire douloureuse pour l’animal aboutissant à la maladie articulaire
dégénérative (Soffler, 2007).
2) Physiopathologie de l’ostéochondrose et lien avec le stress oxydant
2.i Éléments de physiopathologie générale
L’ostéochondrose est souvent considérée comme plurifactorielle et les gènes
potentiellement impliqués dans une prédisposition familiale sont toujours inconnus. Deux
hypothèses quant à la cause primaire de l’arrêt de l’ossification endochondrale ont été
proposées (Desjardin et al., 2014):
- L’hypothèse vasculaire : une interruption focale au niveau des canaux
vasculaires osseux et cartilagineux entraîne la formation de zones de nécrose et d’ossification
anormale.
- L’hypothèse dyschondroplasique : un déséquilibre de l’ossification
endonchondrale pur, d’origine chondrocytaire.
Histologiquement, on observe au sein des lésions chondroplasiques une forme anormale
des chondrocytes, la présence de chondrocytes arrêtés en phase pré-hypertrophique, une
matrice anormalement minéralisée, de la nécrose, une augmentation du collagène de type VI et
des zones focales de cartilage encore visibles dans l’os sous-chondral (Desjardin et al., 2014).
Ces constatations font ressortir l’idée que la zone problématique se situe dans la
jonction chondro-osseuse.
2.ii Données disponibles sur la place du stress oxydant dans la
physiopathologie de l’OCD
Elles sont très limitées. Cependant, Desjardin et al. (2014) ont comparé le protéome de
l’os sous-chondral et du cartilage de deux poulains trotteurs sains de 18 mois avec ceux de deux
poulains du même âge et de la même race atteints d’OCD sur la crête intermédiaire du tibia
distal. La microscopie électronique à transmission a ensuite été utilisée pour comparer la
structure du cartilage et de l’os sous-chondral.
Ensembles, les données concluent que la maturation chondrocytaire lors de l’ossification
endochondrale pourrait être altérée lors de l’ostéochondrose. En effet, un état d’« ER-

158
stress » (voir partie 2, I)B)6)) ainsi qu’un dysfonctionnement mitochondrial des chondrocytes
pourraient être impliqués dans la physiopathologie de l’OCD.
Or ces états sont très fortement liés au stress oxydant comme nous l’avons vu plus tôt.
3) Bilan
En conclusion, les auteurs considèrent qu’un dysfonctionnement mitochondrial et un
stress du reticulum endoplasmique dans les chondrocytes hypertrophiques de la couche
profonde font partie de la physiopathologie de l’OCD en empêcheant leur différenciation par
déficit énergétique. Cependant, rien ne permet d’en faire des facteurs causals primaires pour
l’instant.
Plus d’études sont encore nécessaires à ce sujet.
E) Tendons et ligaments : la maladie tendineuse du tendon fléchisseur
superficiel du doigt (SDFT)
1) Pré-requis
1.i Organisation générale du tendon
Revenons un instant sur la structure du tendon (figure 28).
Figure 29 : Structure schématique hiérarchique du SDFT. Daprès (Patterson-Kane and Rich, 2014).
(A) Longitudinalement les fibrilles de collagène au sein du fascicule (en forme de vague). (B) Transversalement, un
fascicule est avec une structure évoquant un hexagone (d’approximativement 1 mm de diamètre, comprenant
essentiellement des fibrilles de collagène, une matrice non-collagénique et des cellules fibroblastiques). Cette structure
nous montre combien les mouvements entre les fascicules est essentielle pour maintenir l’élasticité du SDFT et sa
fonction de stockage de l’énergie élastique. (C) La fibrille de collagène en vue longitudinale et son aspect en onde (D)
expliquée par l’agencement décalé d’un quart des molécules de collagène qui le composent (E). Chaque molécule de
collagène est une triple héliceF. Le collagène I, majoritaire dans le tendon adulte, a deux chaines et une chaîne
Les tendons sont classiquement composés :
- De collagène, principalement du collagène de type I (65 à 80% du poids sec), qui
donne au tendon sa résistance à l’étirement. Ces fibrilles de collagène sont de section ronde,
d’un diamètre compris entre 20 et 300 nm et mesurent de quelques millimètres à quelques
centimètres de long chez le cheval. Dans le SDFT du cheval, 4 à 5% du collagène est du

159
collagène III dans la région métacarpienne ; de petites quantité de collagène de types IV, V, IX,
X, et XII ont également été identifiées dans plusieurs régions incluant les sites d’insertion du
tendon sur l’os (enthèse) (Patterson-Kane and Firth, 2009). Le collagène du SDFT n’est
pratiquement pas renouvelé et sa demi-vie a été estimée à 197,5 années (contre 34 chez le
tendon du muscle extenseur commun du doigt). Ces valeurs illustrent le faible métabolisme des
ténocytes pour le collagène (Patterson-Kane and Rich, 2014).
- De l’élastine (1 à 2% du poids sec), ajoute de la flexibilité et donne au tendon ses
propriétés élastiques.
- D’une substance fondamentale, principalement composé d’eau (approximativement
60 à 80%) et protéoglycanes. Les protéoglycanes jouent un rôle dans la régulation du diamètre
des fibrilles de collagène et de l’espace interfibrillaire. Leur hydrophilie influence également
l’élasticité de la matrice et permet la diffusion des molécules et des cellules au sein du tendon
(Patterson-Kane and Firth, 2009).
- De cellules fibroblastiques, les ténoblastes et les ténocytes, disposés en rangées
parrallèles entre les faisceaux de collagène. Les ténoblastes sont des cellules tendineuses
immatures fusiformes ayant une forte activité métabolique. En vieillissant, les ténoblastes
deviennent des ténocytes ; ils s’allongent alors. Ténocytes et ténoblastes comptent pour 90 à
95% des cellules tendineuses ; le reste est constitué par des chondrocytes (bien présents au
niveau des enthèses), des synoviocytes et des cellules endothéliales. Les ténocytes étendent de
longs processus cytoplasmiques entre les rangées et sont liés les uns aux autres par des
jonctions « gap » et des jonctions adhérentes. Ce maillage facilite la coordination de la réponse
à l’étirement ainsi que l’échange de molécules, dont les radicaux libres en théorie (Patterson-
Kane and Firth, 2009).
- D’endotendon, de péritendon et d’épitendon qui sont des tissus interstitiels plus ou
moins lâches. Ils contiennent le contigent vasculaire, nerveux et lymphatique du tendon. Au
dessus de l’épitendon se trouve également le paratendon, plus dense. Entre le paratendon et
l’épitendon se trouve un espace contenant un fluide riche en mucopolysaccharides qui permet
la lubrification du tendon lors du mouvement (Abate et al., 2009).
- le SDFT possèdent également une gaine tendineuse synoviale au même titre que le
tendon extenseur commun du doigt au niveau du carpe ou encore la bourse bicipitale chez le
cheval (Abate et al., 2009).
- La jonction ostéo-tendineuse souvent appelée « enthèse », est une transition
progressive entre le tissu tendineux et tissu cartilagineux jusqu’à l’os lamellaire. C’est une zone
complexe et riche en récepteurs nerveux qui est soumise à un grand stress mécanique durant la
transmission de la force contractile musculaire au tendon (Abate et al., 2009).
1.ii Terminologie des désordres tendineux
Aujourd’hui étant donné la méconnaissance des phénomènes physiopathologiques des
affections du tendon, il est difficile de définir une terminologie précise. Même si certains termes
se croisent, on s’appuiera sur celle proposée par Dakin et al. (2014) :
- Le terme tendinopathie (« tendinopathy ») est utilisé pour désigner généralement tous
les troubles affectant les tendons, de la simple douleur chronique à la rupture. C’est un terme

160
général qui ne sous-entend aucun mécanisme pathogénique à l’origine des lésions et/ou des
signes cliniques constatés.
- Le terme tendinite (« tendonitis ») est utilisé pour désigner un tendon douloureux et
implique que la lésion tendineuse est accompagnée d’une réponse inflammatoire.
- Le terme tendinose (« tendinosis ») est utilisé pour désigner un tendon douloureux et
implique que la blessure tendineuse s’est développée comme conséquence d’un processus
dégénératif sans présence d’inflammation.
- Le terme maladie tendineuse (« tendon disease ») indique que la blessure s’est
développée comme conséquence de contraintes mécaniques cycliques et des effets de l’âge
résultant en de micro-blessures. Souvent exacerbée par une blessure de fatigue/surmenage du
tendon, la manifestation la plus commune chez le cheval est la lésion corticale centrale du
SDFT.
- Le terme blessure tendineuse (« tendon injury ») englobe la maladie tendineuse mais
aussi les conséquences d’une lésion traumatique (œdème ou tuméfaction cutanée par
exemple).
2) Présentation de la maladie tendineuse du SDFT
2.i Impact sur les performances équines
Les tendinopathies du tendon fléchisseur superficiel du doigt (SDFT) est une des causes
de boiterie les plus communes chez le cheval de course (particulièrement sur les membres
antérieurs) et est particulièrement étudiée. Ces affections prolongent sensiblement le temps de
retrait des courses du cheval (jusqu’à 18 mois) et augmentent significativement le taux de
retraite prématurée des athlètes (Patterson-Kane and Firth, 2009).
On a pu estimer que chez des galopeurs (toutes disciplines confondues), les lésions
tendineuses ont été rapportées dans 0.6 à 9 sur 1000 arrivées de course, avec des taux
d’incidence compris entre 11 et 30% sur une période de 1 à 10 ans selon les études (Patterson-
Kane and Firth, 2009).
Les données sont moins disponibles chez les chevaux de sport où les lésions du SDFT
sont également rapportées. Dans une étude de Singer et al. (2008) menée chez des chevaux de
sport au Royaume-Uni (majoritairement de saut d’obstacle), 3% des blessures dans les
compétitions d’une journée et 17% sur le Concours International Combiné (CIC ou concours
complet, niveau FEI) impliquaient des blessures tendineuses digitales (essentiellement des
fléchisseurs). Chez les chevaux à l’entraînement pour des épreuves de CIC, 43% était atteints
d’une lésion tendineuse ou ligamentaire (aiguë ou chronique) et 36% de ces lésions
impliquaient spécifiquement le SDFT.
Enfin, dans une étude impliquant 101 Dutch Warmbloods et 71 Standardbreds avec des
blessures tendineuses digitales, 29% et 32% respectivement de ces lésions impliquaient le
SDFT d’un des deux antérieurs (Van den Belt et al., 1994).
Les lésions tendineuses peuvent aller jusqu’à la rupture après un effort. Elles sont
généralement situées au niveau de la région moyenne du métacarpe/métatarse, mais peuvent
toucher l’ensemble du tendon.

161
2.ii Histologie des lésions
Sauf cas exceptionel, on considère aujourd’hui chez le cheval athlète que les
tendinopathies du SDFT proviennent généralement de l’inédaquation entre la charge appliquée
et sa capacité biomécanique à y résister, résultant souvent de l’accumulation des micro-lésions
tendineuses liées à un exercice incontrôlé et à l’âge ; c’est également observé chez l’homme
pour le tendon d’Achille (Abate et al., 2009) (Patterson-Kane and Firth, 2009) (Papalia et al.,
2013).
Selon ces considérations et notre terminologie, on est donc véritablement dans le cas
d’une maladie tendineuse du SDFT, dont l’epression aiguë résulte d’un processus de
surmenage du tendon.
Dans l’espèce équine comme dans l’espèce humaine, l’histologie rapporte bien souvent
une désorganisation, des zones de rupture ainsi que de discontinuité dans l’architecture des
fibrilles de collagène ; la composition change également (Abate et al., 2009) (Yuan et al., 2003).
On rapporte également des altérations de l’aspect microscopique ondulé du collagène, une
réduction du diamètre des fibrilles (Patterson-Kane and Rich, 2014). Le contenu en
glycosaminoglycanes (GAG) diminue également.
Des lésions de micro-traumas du cœur du SDFT sont aussi rapportées chez des chevaux
asymptomatiques. Ils sont notamment liés à l’augmentation de la quantité de collagène III avec
l’âge (moins résistant, en remplacement du collagène I), la dénaturation et la dépolymérisation
des fibrilles de collagène et l’augmentation de cathepsine B.
Chez l’homme, on rapporte aussi très souvent la présence de lésions dégénératives
s’illustrant de trois manières (Yan et al., 2003b) (Lian et al., 2007) (Abate et al., 2009):
- Une baisse de la cellularité avec la disparition des ténocytes par apoptose ;
- l’accumulation de graisse (dégénérescence graisseuse) et/ou de substance mucoïde
(dégénerescence mucoïde) dans la matrice tendineuse ; des calcifications et des métaplasies
fibro-cartilagineuses sont également fréquemment retrouvées dans les cas plus chroniques ;
- l’absence de cellules inflammatoires : cette dernière remarque appuie le terme de
tendinose plutôt que de tendinite pour désigner ces tendinopathies.
Sans pouvoir les exclure, les lésions dégénératives (graisseuses et mucoïdes) souvent
constatées chez l’homme, athlète comme non-athlète, n’ont pas clairement été rapportées pour
le SDFT du cheval. La disparition des ténocytes est suspectée et incluse dans la
physiopathologie de la maladie tendineuse du SDFT, sans avoir été étudiée spécifiquement chez
le cheval (Patterson-Kane and Firth, 2009), les auteurs s’appuyant volontairement sur les
résultats obtenus dans les modèles de laboratoires et chez l’homme.
Pourtant, malgré ces observations et d’autant plus dans le cas du SDFT, l’importance de
l’inflammation dans cette tendinopathie reste controversée au sein de la communauté
scientifique (Dakin et al., 2014). Si la manifestation de signes cliniques de l’inflammation est
assez claire pour tous les praticiens équins immédiatement après une lésion tendineuse, ces
signes ne sont pas aussi évidents lors de la phase chronique.
Par ailleurs, malgré l’absence de cellules de l’inflammation sur les lames histologiques,
des facteurs pro-inflammatoires ont été mis en évidence comme nous le verrons plus loin, chez

162
l’homme comme chez le cheval. Dans ce contexte et malgré ses détracteurs, le terme de
tendinite pourrait donc être conservé pour désigner cette affection.
3) Physiopathologie de la maladie tendineuse du SDFT et lien avec le stress
oxydant
3.i Physiopathologie comparée entre l’homme et le cheval
Comme nous l’avons précédemment souligné, les modifications pathologiques des
tendons suite à un usage répété est une cause significative de morbidité chez l’homme et chez
le cheval athlète (Dakin et al., 2014). Les blessures du tendon SDFT chez les athlètes équins est
le modèle animal classique des tendinopathies induites par l’exercice chez l’homme. Ces
dernières regroupent notamment celles du tendon d’Achille et de la coiffe du muscle supra-
épineux. C’est la raison pour laquelle de nombreux rapprochements seront réalisés entre les
résultats obtenus chez l’homme et ceux du cheval dans l’exposé qui va suivre.
Pour mémoire, le SDFT et le tendon d’Achille ont en commun la capacité d’accumuler
l’énergie élastique durant l’effort (Patterson-Kane and Firth, 2009) (Papalia et al., 2013)
(Dakin et al., 2014) (Patterson-Kane and Rich, 2014). Dans le cas du SDFT, l’accumulation
d’énergie élastique et motrice lors de la phase d’appui permet la flexion digitale et carpale
passive plus rapide durant le pivot et la phase de propulsion.
La disponibilité des tissus post-mortem chez les chevaux a facilité l’avancée des
connaissances dans la description des effets de l’exercice sur la structure ; a contrario l’accès
aux biopsies humaines a apporté un complément d’information quant à des situations plus
avancées et plus chroniques de la maladie (Patterson-Kane and Rich, 2014).
3.ii Sources prépondérante d’ERO au sein du SDFT lors de l’effort
Chez l’homme, l’étude de Wang et al. (2001) a montré une augmentation de la
production de peroxyrédoxine 5 par les cellules endothéliales et les ténocytes pathologiques
dans le tendon humain dégénératif in vivo, observations qui suggèrent un stress oxydant dans
les mécanismes de cette dégénérescence.
Malheureusement, la littérature disponible ne rapporte pas d’étude concluante
investigant des marqueurs du stress oxydant in vivo chez le SDFT du cheval lors de l’effort
(comme la nitrotyrosine dans le cas du collagène articulaire).
Selon divers auteurs, le tendon est soumis à un environnement oxydant durant l’effort
qui pourrait le prédisposer aux dégâts oxydatifs (Bestwick et al., 2004) (Longo et al., 2008). Les
espèces réactives proviendraient des tissus à proximité des tendons (le muscle par exemple),
mais également des tendons eux-mêmes par plusieurs mécanismes dont les principaux sont
présentés ci-dessous.
Ischémie-reperfusion
Un mécanisme d’ischémie reperfusion a très tôt été suspecté chez le cheval lors de
l’exercice physique. On a en effet évoqué la possibilité que la charge cyclique de chaque foulée
est associée à la compression des vaisseaux au point maximum de tension du tendon, suivie par
une réoxygénation rapide à la détente du tendon. (Goodship et al., 1994) (Longo et al., 2008).
L’anatomie vasculaire et microvasculaire du SDFT a été étudiée par Kraus-Hansen et al.
(1992). La présence d’un treillis intra-tendineux a été confirmée, avec une « artère

163
nourricière » décrite, associée à la bride radiale sur les membres antérieurs. Chez l’homme,
certains tendons comme le tendon d’Achille et celui de la coiffe du muscle supra-tendineux
présentent des régions particulièrement hypovascularisées (Schmidt-Rohlfing et al., 1992)
(Seitz et al., 2011). Elles n’ont pas été rapportées par Kraus-Hansen et al. (1992) sur le SFDT.
Les avis sur ces zones diffèrent selon les études : si Schmidt-Rohlfing et al., (1992) jugent
qu’il n’y a pas forcément de lien entre les régions hypovascularisées et les zones lésionnelles
pour le tendon d’Achille chez l’homme (où les lésions interviennent plus souvent dans la partie
moyenne, mieux vascularisée), Seitz et al. (2011) constatent que très souvent dans le tendon de
la coiffe du muscle supra-épineux les sites où les lésions tendineuses sont observées
concordent avec les zones hypovascularisées.
On sait aujourd’hui que les ténocytes équins dépendent du bon fonctionnement du
métabolisme énergétique aérobie (contrairement aux chondrocytes) (Birch et al., 1997)
(Patterson-Kane and Firth, 2009). Cependant, si des lésions dégénératives liées à l’hypoxie du
tendon ont été rapportées et caractérisées chez l’homme, il n’y a pas à ce jour d’étude similiaire
chez le cheval (Patterson-Kane and Firth, 2009). De même, aucune mesure directe de l’apport
sanguin ni des taux en dioxygène au sein du SDFT n’ont été réalisées chez le cheval à l’exercice
pour illustrer une hypoxémie lors de l’exercice au sein du tendon.
La théorie du dysfonctionnement endothélial conforte l’idée qu’un stress oxydant
comme celui induit par l’effort peut entraîner une vasoconstriction locale au niveau du tendon.
Cette dernière surpasserait même les pouvoirs vasodilatateurs du NO° et impliquerait les
cellules endothéliales dans la génèse d’ERO. Papalia et al. (2013) ont réalisé une synthèse de la
littérature (chez l’homme et chez l’animal, mais pas le cheval) pour mettre en lien un
dysfonctionnement des cellules endothéliales dans les facteurs primaires de la
pathophysiologie des tendinopathies. Pour l’instant il n’y a pas d’évidence claire que ce
dysfonctionnement endothélial a vraiment lieu pendant et après un effort.
Cette théorie est même contredite par l’étude menée in vivo chez le rat de Szomor et al.
(2006) qui souligne l’activation des iNOs et la surexpression des gènes des trois isoformes de
NOs dans le tendon de la coiffe du muscle supra-épineux suite à un usage chronique durant
l’effort, suggérant la production de NO° en réponse à un stress mécanique.
Hyperthermie
L’hyperthermie du tendon lors de l’exercice, qui peut atteindre 45°C, peut être
générateur d’ERO en stimulant leur production par les mitochondries des cellules endothéliales
et des ténocytes (voir partie 2, III)B) 5)) (Goodship et al., 1994).
Cette hyperthermie pourrait dépasser les températures atteintes dans le muscle pour
certains auteurs, notamment parce que l’énergie élastique stockée pendant la phase de
suspension n’est toujours entièrement libérée lors de la propulsion. De plus, cette
hyperthermie est aggravée au niveau du tendon par rapport au muscle étant donné le faible
approvisionnement en sang du tendon par rapport aux autres tissus ; ce dernier ne joue alors
que partiellement son rôle de fluide caloporteur dans la dissipation de la chaleur lors de l’effort
(Patterson-Kane and Firth, 2009).

164
Tendinopathie du SDFT et cytokines pro-inflammatoires
Si l’histologie met rarement en évidence des cellules de l’inflammation au sein des
tendons lésés, plusieurs études ont déjà rapporté l’importance des cytokines, notamment pro-
inflammatoires, dans l’homéostasie des tendons et des ligaments (Bestwick et al., 2004).
Les travaux de Millar et al. (2009) suggèrent que l’expression de ces cytokines
précocement après l’apparition des lésions tendineuses mais aussi lors de la cicatrisation
entraîne la production d’espèces réactives de l’oxygène favorisant les mécanismes
apoptotiques voire nécrotique. En effet, l’expression des cytokines pro-inflammatoires a été
montrée de manière significative chez des cellules tendineuses soumises à une contrainte
mécanique in vitro, mais également in vivo où IL-18, IL-15, IL-6 et MIF (pour « macrophage
migration inhibitory factor ») ont été mis identifiées sur les tendons du muscle supra-épineux
de rats soumis à un effort soutenu et répété. MIF est connu comme pouvant activer
l’expression génique d’autres facteurs pro-inflammatoires (comme IL-6 et TNF-. IL-18, IL-15
et IL-6 peuvent aussi promouvoir intensément la synthèse de radicaux libres par la
mitochondrie au même titre que le TNF-.
Chez le cheval, les échantillons de SDFT lésés sont positifs aux cytokines pro-
inflammatoires IL-1, IL-1, TNF- et IFN- alors qu’elles ne sont pas identifiées dans des
tendons sains ; ces constations évoquent la contributions des cytokines pro-inflammatoires
dans le développement de la maladie tendineuse du SDFT (Dakin et al., 2014).
In vitro, on a montré que les fibroblastes génèrent également des ERO en utilisant le
complexe NADPH oxydase après une blessure tendineuse, en réponse à ces cytokines pro-
inflammatoires tel que IL-1 et TNF- (Meier et al., 1989) (Goodship et al., 1994).
Ainsi, comme pour les chondrocytes dans une articulation atteinte d’OA, les ténocytes
pourraient adopter un profil pathologique lors de la maladie tendineuse du SDFT et produire
des espèces réactives de l’oxygène.
Les tissus fibreux supports du tendon comme le péritendon joueraient également un
rôle dans cette production, notamment durant un exercice physique. Très tôt, Langberg et al.
(1999) ont montré que le péritendon du tendon d’Achille de l’homme voit sa concentration en
PGE2 significativement augmenter durant l’effort. Des résultats identiques ont également été
obtenus pour le ligament fémoro-patellaire et le tendon d’Achille dans un modèle murin soumis
à de la course sur un tapis roulant (Zhang et al., 2010). Cependant le lien entre PGE2 et le stress
oxydant doit être approfondi. Par exemple, Dakin et al. (2012a et 2012b) soulignent que chez le
cheval PGE2 semblerait aussi jouer un rôle lors de la cicatrisation du tendon en contrant les
effets cataboliques d’IL-1, à fortes doses.
3.iii Conséquences du stress oxydant sur le SDFT
Dégradation directe de la matrice tendineuse
La tendinose (voire la rupture tendineuse) est un des effets secondaires classiques des
fluoroquinolones, chez le cheval comme chez l’homme. Cette toxicité est connue depuis
longtemps. Pouzaud et al. (2003) ont montré que les fluoroquinolones stimulent la production
de radicaux libres au sein du tendon, supportant l’implication du stress oxydatif dans les
mécanismes de leur toxicité.

165
Cette observation trouve son intérêt dans la démonstration de la sensibilité particulière
du tendon au stress oxydant et de sa potentielle implication dans la physiopathologie des
tendinopathies.
Les mécanismes de dégradation du collagène et des protéoglycanes aux ERO ont été
décrits tout au long de cet exposé et notamment en partie D) à propos de l’OA : ils ne seront
donc pas repris ici.
Apoptose du contigent cellulaire de la matrice tendineuse
Selon Yuan et al. (2002 et 2003), le stress oxydant (« oxidative stress induced
apoptosis ») est impliqué dans la dégénérescence et la disparition des cellules tenineuses. Chez
le cheval, Goodship et al. (1994) rapportent des études préliminaires montrant qu’en condition
de stress oxydant dans un milieu contenant entre 10 et 100 mol/L d’H2O2, la multiplication et
la croissance in vitro des ténocytes équins diminuent, réitérant des résultats également
retrouvés pour d’autres tissus.
Chez l’homme, Kim et al. (2014) montrent qu’H2O2 à de plus fortes concentrations
entraîne l’apoptose des ténocytes de la coiffe du tendon supra-épineux in vitro ; l’addition d’un
antioxydant, la cyanidine, inhibe cette mort cellulaire, retrouvant les résultats de Yan et al.
(2002).
Sauf mention contraire, aucune littérature n’existe chez le cheval concernant l’apoptose
des ténocytes équins et le stress oxydant entraîné par l’effort.
Par ailleurs d’autres scientifiques ont élaboré d’autres théories pour expliquer
l’apoptose des cellules tendineuse : Arnoczky et al. ont montré in vitro que des cycles répétés de
contraintes mécaniques sont associés à l’activation de « stress-activated protein kinases »
(SAPKs), qui existent dans de nombreuses cellules, dont les fibroblastes des tendons.
4) Bilan
De nombreuses théories ont essayé d’expliquer la physiopathologie des tendinopathies,
qui est très probablement multimodale.
Théoriquement, le stress oxydant lors de l’exercice pourrait contribuer à la fragilisation
du tendon et le prédisposer à la rupture de ses fibres de collagène. De plus, l’ensemble des
conditions (hyperthermie, ischémie-reperfusion et cytokines pro-inflammatoires et pro-
oxydantes) pourraient être réunies lors de l’effort pour générer des radicaux libres.
Cependant il n’a jamais été montré que l’effort entraîne un stress oxydant d’un exercice
responsable de la dégénérescence de la matrice tendineuse et de la mort des ténocytes chez le
SDFT du cheval.
Par ailleurs, connaissant l’impact des cytokines pro-inflammatoires sur l’anabolisme des
ténocytes et des cellules endothéliales, il est possible que dans les cas chroniques, la
production d’ERO par ces cellules puisse être suffisante pour perturber la cicatrisation au sein
du tendon lésé.
Avec le maigre niveau d’informations dont nous disposons aujourd’hui, on ne peut pas
inclure spécifiquement le stress oxydant comme cause primaire dans la physiopathologie des
tendinopathies du SDFT chez le cheval athlète.

166
F) Hématologie : hémolyse intravasculaire induite par l’effort (HIE)
1) Quelques rappels sur la sensibilité des hématies aux ERO
Bien que dépourvues de mitochondrie, les hématies ou érythrocytes sont
particulièrement sensibles aux radicaux libres de l’oxygène, sensibilité lié à leur interaction
constante avec l’oxygène, la traversée de milieux producteurs d’ERO comme le muscle, mais
aussi la production d’ERO par les cellules de l’inflammation activées par l’exercice. La présence
d’hémoglobine en grande quantité rend aussi les hématies particulièrement sensibles à son
auto-oxydation (Brun et al., 1998).
De plus, le stress oxydant peut également altérer l’homéostasie ionique des érythrocytes
en s’attaquant aux protéines membranaires, aggravant la rigidité membranaire de la cellule,
plus fragile au passage dans les capillaires (où elle se déforme normalement pour circuler)
(Bonilla et al., 2005).
Les globules rouges sont cependant équipés de plusieurs antioxydants intracellulaires
comme la catalase, la glutathion-peroxydase et la SOD1 afin de maintenir un équilibre redox
convenable dans cet environnement et limiter la rigidification de leur membrane (voir
également partie 1, IV) B) 1)).
Portier (2007) nous rappelle cependant que les hématies, dépourvues de noyaux et
véritables « sacs à hémoglobine », n’ont pas le moyen de réparer les dégâts oxydatifs causées
par les ERO, qui s’accumulent jusqu’à rupture de la membrane : c’est l’hémolyse.
2) L’hémolyse intravasculaire induite par l’effort (HIE)
2.i Présentation générale
L’hémolyse intravasculaire induite par l’effort (HIE) ou « anémie du sportif » est bien
décrite chez l’homme et une source potentielle de baisse des performances, particulièrement
dans les efforts d‘endurance où le métabolisme aérobie est prépondérant.
L’HIE est bien associée aà une destruction vraie des hématies (hémolyse) dans les
vaisseaux sanguins au cours de l’exercice physique. Elle doit être différenciée de :
- la « pseudo-anémie » ou « anémie de dilution » qui provient de l’augmentation
du volume plasmatique immédiatement après un effort physique, montrée chez
l’homme mais également chez le cheval (Bonilla et al., 2005).
- l ’« anémie ferropénique secondaire à l’exercice » qui est principalement
entraînée par des pertes en sang brut de manière plus chronique (hématurie et perte
intestinale notamment) ou lors de carence extrême en fer.Elle concerne beaucoup les
épreuves d’endurance chez l’homme mais est peu étudiée chez le cheval (Bonilla et al.,
2005) (Robinson et al., 2006).
Si l’HIE est également fortement suspectée dans l’espèce équine, la littérature
concernant l’HIE chez le cheval est très limitée (Pelligrini Massini et al., 2003) ; il semblerait
que son impact clinique sur le cheval soit moindre par rapport à l’homme, en tout cas sûrement
largement sous-étudié (Cywinska et al., 2011).

167
2.ii Mise en évidence de l’HIE chez le cheval
L’hémolyse intravasculaire après un effort a trois composantes paracliniques
principales : une augmentation de l’hémoglobinémie (hémoglobine libre dans le sang) et de
l’hémoglobinurie ainsi qu’une diminution de l’haptoglobinémie. Le taux sérique en fer libre
peut également entrer en compte dans la caractérisation de l’hémolyse.
L’haptoglobine est une globuline synthétisée par le foie ayant la capacité de fixer
l’hémoglobine libre dans le sang. En plus d’être un marqueur de la phase aiguë de
l’inflammation, l’haptoglobine a un intérêt dans l’étude de l’hémolyse intravasculaire aiguë. En
effet, son manque de sensibilité (chez l’homme comme chez le cheval) pour des épisodes
d’hémolyse modérée permet de conclure uniquement pour de fortes hémolyses en cas de
diminution de l’haptoglobinémie. En effet, ces variations ont été étudiées tout le long d’une
saison d’entraînement (effort modéré) chez des PSA (galopeurs) sans qu’aucune variation
significative ne soit notée (Willet and Blackmore, 1979).
Dans l’espèce équine, des preuves d’HIE ont été trouvées pour les chevaux d’endurance
(Murakami et al., 1974), mais aussi les chevaux de course (effort maximal) (Pelligrini Massini et
al., 2003) (Cywinska et al., 2011) (Kristensen et al., 2014), et des chevaux soumis à un effort
d’intensité progressif sur tapis roulant (infra-maximal à maximal) (Inoue et al., 2005). Lors
d’épreuve d’endurance on observe une augmentation significative dans le nombre d’hématies
en circulation et de l’hématocrite (mécanisme de splénocontraction notamment) en début de
course, suivi par une légère diminution suggérant une perte de globules rouges lors d’un
exercice prolongé.
Les études, malgré des résultats pouvant varier selon les auteurs, ont notamment mis en
évidence une hémoglobinurie augmentée (Murakami et al., 1974), l’élévation hémoglobinémie
(hémoglobine sanguine libre) (Pelligrini Massini et al., 2003) (Inoue et al., 2005) (Cywinska et
al., 2011) (Kristensen et al., 2014), et en fer libre dans le sang (Inoue et al., 2005), mais
également une baisse significative de l’haptoglobine sérique (Pelligrini Massini et al., 2003)
(Cywinska et al., 2011) (Kristensen et al., 2014) à la fin d’un effort.
Cependant chez le cheval, ces modifications sanguines n’ont pas clairement été corrélées
avec une baisse de la performance ou une altération de la santé de l’animal (Kristensen et al.,
2014). Enfin, les résultats indiquent que l’hémolyse intravasculaire apparaît plus volontiers
chez les juments (par ailleurs avec des taux en haptoglobine sérique plus élevés au repos)
(Cywinska et al., 2011).
3) Physiopathologie de l’HIE et lien avec le stress oxydant
L’exercice intense semble diminuer la fluidité membranaire du globule rouge chez le
cheval athlète. Cette diminution s’observe dès 15 minutes après l’arrêt de l’exercice et persiste
24 heures après (Portier, 2007).
Très tôt chez l’homme, on a découvert que des hématies issues d’un individu ayant
réalisé un effort intense sont plus sensibles au stress mécanique, osmotique et oxydant (Brun et
al., 1998) (Bonilla et al., 2005).
Plusieurs auteurs ont d’abord suggéré que le stress mécanique répété lors de la foulée
pouvait entraîner l’hémolyse des hématies (« foot-strike hemolysis »). Ce mécanisme, s’il existe,
ne peut pas expliquer seul la pathogénie de la maladie, car si chez l’homme les coureurs de

168
marathon et les cyclistes sont directement visés, l’HIE a également été mise en évidence chez
les nageurs (Bonilla et al., 2005) (Robinson et al., 2006).
Par ailleurs, la contraction musculaire elle même peut être à l’origine d’hémolyse intra-
vasculaire (Robinson et al., 2006) et pourrait concerner le cheval en particulier qui développe
de contractions particulièrement fortes lors d’un effort. Ce mécanisme est très peu étudié chez
l’espèce qui nous intéresse.
Loin de rentrer en opposition avec les hypothèses précédentes, la recherche s’est
penchée vers un autre mécanisme de fragilisation des érythrocytes et la dégradation de la
rhéologie du sang durant l’effort. Plusieurs facteurs peuvent entrer en cause, dont le stress
oxydant (Robinson et al., 2006).
Brun et al. (1998) rapportent que la baisse de la déformabilité est aussi accompagnée
d’une modification de la forme des hématies, alors appelées échinocytes (forme précédant
l’hémolyse).
Smith et al. (1995) ont étudié l’impact de l’effort sur les hématies chez des coureurs et
des cyclistes humains jusqu’à épuisement. Leurs résultats indiquent qu’un effort submaximal
(60-75% VO2max) entraînait des modifications significatives des globules rouges par le stress
oxydant.
Après une course à pied, la teneur en malondialdéhyde dans les membranes plasmiques
des hématies peut dépasser 45% du contenu normal avant un effort.
Chez des cyclistes soumis à un effort submaximal jusqu’à épuisement, les résultats ont
indiqué une phase de stress oxydant ayant pour conséquence des modifications structurales
oxydatives des protéines membranaires érythrocytaires et une rigidification de la membrane
cellulaire (peroxydation lipidique) (Brzeszczynska et al., 2008).
Cependant, d’autres facteurs contribuent à la rigidité de la membrane érythrocytaire
durant l’effort. Ainsi, la déshydratation entraîne une augmentation de la viscosité du sang qui
favorise les chocs et les traumas mécaniques sur les érythrocytes. Brun et al. (1998) ont montré
qu’à partir de 4 mmol/L, les lactates sanguins diminuent significativement la flexibilité des
érythrocytes ; des taux inférieurs peuvent également être responsables d’altération de cette
flexibilité. Enfin, le stress osmotique notamment entraîné par l’augmentation de l’osmolarité
sanguine durant l’effort contribue à cette fragilité membranaire (Robinson et al., 2006). En
effet, l’eau représente 62% du contenu des hématies (même si 25% est libre) et l’exercice est
susceptible d’accroître le pourcentage en eau libre ; or le pourcentage d’eau liée est associé à la
déformabilité des hématies (Brun et al., 1998).
4) Bilan
La théorie de l’implication du stress oxydant lors de l’hémolyse intravasculaire induite
par l’effort repose sur la rigidification et l’augmentation de la perméabilité de la membrane
plasmique des hématies.
Généralement les modifications membranaires des hématies attribuées au stress
oxydant induit par l’exercice sont moins graves que les modèles in vitro, et donc théoriquement
moins susceptible de générer des hémolyses intravasculaires. Comme nous venons de le voir,

169
s’il est clair que si le stress oxydant puisse être un acteur de fragilisation des hématies, il n’est
sûrement pas le mécanisme causal unique dans la physiopathologie de l’hémolyse
intravasculaire.
Malheureusement, on ne sait pas dire aujourd’hui quelle est la part du stress oxydant
dans la rigidification et la perméabilité des membranes plasmiques des érythrocytes par
rapport aux autres mécanismes lors de l’effort, chez l’homme comme chez le cheval.
Par ailleurs, comment expliquer la différence de gravité dans la clinique chez le cheval ?
Les hématies équines pourraient-elles être plus résistantes que les hématies humaines ? Les
hématies équines ont une tendance à l’aggrégation bien supérieure à l’homme ; il semble
également que le cheval possède des hématies naturellement plus déformables. D’ailleurs ces
deux propriétés pourraient être liées selon Baskurt et al. (1997) : en effet, l’hématie du cheval
doit pouvoir se déformer suffisamment pour former des surfaces parallèles durant
l’aggrégation.

170
CONCLUSION
La chimie radicalaire a conduit au fil des années à une vision manichéenne des espèces réactives de
l’oxygène : elles sont considérée comme une menace qui s’illustre par leur capacité à dégrader de
nombreuses molécules organiques piliers de l’intégrité des mammifères (protéines, lipides et acides
nucléiques). Ce « péril oxydant », l’organisme doit y faire face tous les jours par des mécanismes de défense
complexes et finement régulés. En cas de déséquilibre, c’est le stress oxydant, les tissus et l’organisme sont
en danger.
Cette réputation, associée à des techniques expérimentales limitées et peu spécifiques, a mené de
nombreux scientifiques à conclure de manière hasardeuse sur la réalité du stress oxydant dans les
phénomènes biologiques. En effet, dans une étude scientifique, la définition du stress oxydant est d’abords
expérimentale et dépend largement des marqueurs utilisés et de la confiance qu’on leur attribue.
La présente synthèse indique clairement que les espèces réactives de l’oxygène (ERO) sont aussi
impliquées physiologiquement dans la signalisation, dans l’homéostasie cellulaire (notamment redox) ainsi
que dans la formulation de nombreuses réponses ciblées et ponctuelles. L’excès seulement peut être
délétère, comme dans le cas de l’inflammation ou du phénomène d’ischémie-reperfusion ; la frontière entre
physiologie et pathologie est fine et variable suivant les conditions et l’individu considéré.
Chez le cheval athlète en particulier, l’analyse cumulée de la bibliographie utilisant plusieurs
marqueurs d’exploration sur différents types d’effort semble bien indiquer que l’exercice physique est
générateur d’espèces réactives de l’oxygène pouvant évoluer vers la genèse d’un état de stress oxydant pour
l’organisme.
La problématique de ce travail était d’établir dans quelle mesure cette production d’espèces
réactives de l’oxygène est reliée ou non à la mise en place d’affections limitant les performances de l’athlète
équin. Pour la plupart des affections étudiées, des corrélations positives in vitro et vivo ont été trouvées
entre la physiopathologie générale connue et les mécanismes d’action des ERO. À la lecture de la littérature,
on peut cependant assez confortablement établir que le stress oxydant n’est jamais un mécanisme
physiopathologique isolé dans la pathologie du sujet sportif mais plutôt un facteur de prédisposition ou
d’aggravation.
Notre méconnaissance des cibles exactes, indépendamment du métabolisme et de l’homéostasie
normale de la cellule, nous empêche toujours à ce jour d’établir un lien clair entre la production normale
d’espèces réactives de l’oxygène et le véritable stress oxydant. Enfin, le futur de l’exploration du stress
oxydant est évidemment lié au développement de protocoles expérimentaux mieux standardisés et plus
spécifiques mettant mieux en évidence les ERO au sein des tissus vivants pendant l’effort en temps réel.
Thèse de M. DUBOIS Benjamin

171
Bibliographie
Abate M., Gravare-Silbernagel K., Siljeholm C., Di Iorio A., De Amicis D., Salini V., Werner S., Paganelli R. (2009). Pathogenesis of tendinopathies: inflammation or degeneration? Arthritis Research and Therapy 11, (3), 235-250.
Ahmad A, Shameem M, Husain Q. (2012). Relation of oxidant/antioxidant imbalance with
disease progression in patients with asthma. Ann. Thorac. Med. 7, (4),226–232.
Alary J., F. Guéraud, J.-P. Cravedi. (2003). Fate of 4-hydroxynonenal in vivo: disposition and
metabolic pathways. Mol. Asp. Med. 24, (4-5), 213-218.
Albano E., Bellomo G., Parola M., Carini R., Dianzani M.U. (1991). Stimulation of lipid
peroxidation increases the intracellular calcium content of isolated hepatocytes. Biochim.
Biophys. Acta 1091, 310-316.
Alcaraz M. J., Gualillo O., Sánchez-Pernaute O. (2013). Studies on Arthritis and Joint Disorders.
Springer New York.
Allen R.E., Blake D.R., Nazhat N.B., Jones P. (1989). Superoxide radical generation by inflamed
human synovium after hypoxia. Lancet. 334, ( 8657), 282–283.
Al-Qudah K.M., Al-Majali A.M. (2008) Higher lipid peroxidation indices in horses eliminated from endurance race because of synchronous diaphragmatic flutter (thumps). Journal of Equine Veterinary Science 28, (10), 573–578.
Andrades M.E., Lorenzi R. (2011). Oxidative Stress in Cardiovascular Diseases. Oxidative Stress in Vertebrates and Invertebrates: Molecul Aspects of Cell Signaling, First Edition, Edited by Tahira Farooqui and Akhlaq A. Farooqui., pp 153 - 166.
Arnoczky SP, Tian T, Lavagnino M, et al (2002). Activation of stress-activated protein kinases
(SAPK) in tendon cells following cyclic strain: the effects of strain frequency, strain magnitude,
and cytosolic calcium. J Orthop Res 20, 947– 952.
Art T., Kirschvink N., Smith N., Pierre Lekeux P. (1999). Indices of oxidative stress in blood and
pulmonary epithelium lining fluid in horses suffering from recurrent airway obstruction.
Equine Veterinary Journal 31, (5), 397–401.
Art T., Franck T., Gangl M., Votion D., Kohnen S., Deby-Dupont G., Serteyn D. (2006). Plasma
concentrations of myeloperoxidase in endurance and 3-day event horses after a competition.
Equine Veterinary Journal 38, (36), 298–302.
Barclay J.K., Hansel M. (1991). Free radicals may contribute to oxidative skeletal muscle fatigue.
Can. J. Physiol. Pharmacol., 69, (2), 279-284.
E. Barrey. (1994). Propriétées contractiles des fibres musculaires et performance physique chez le cheval. INRA Productions animales, 7, (1), 41-53. Baskurt O.K., Farley R.A., Meiselman H.J. (1997). Erythrocyte aggregation tendency and cellular
properties in horse, human, and rat: a comparative study. American Journal of Physiology -
Heart and Circulatory Physiology 273,(6), 2604-2612.
Baur A., Henkel J., Bloch W., Treiber N., Scharffetter-Kochanek K., Brüggemann G.P., Niehoff A.
(2011). Effect of exercise on bone and articular cartilage in heterozygous manganese
superoxide dismutase (SOD2) deficient mice. Free Radic. Res. 45, (5), 550-558.

172
Beckman J.S., Koppenol W.H. (1996). Nitric oxide, superoxide, and peroxynitrite: the good, the
bad, and the ugly. American Journal of Physiology - Cell Physiology 40, (5), 1424-1437.
Beecher B.R., Martin J.A., Pedersen D.R. et al (2007) Antioxidants block cyclic loading induced
chondrocyte death. Iowa Orthop. J. 27, 1–8.
Bestwick C.S., N. Maffulli . (2004). Reactive oxygen species and tendinopathy: do they matter?
Br J Sports Med 38,(6), 672-674.
Biemond, P., H.G. Van Eijk, A.J. Swaak & J.F. Koster. (1984). Iron mobilization from ferritin by
superoxide derived from stimulated polymorphonuclear leukocytes. J. Clin. Invest. 73, 1576–
1579.
Birch, H.L., Rutter, G.A., Goodship, A.E. (1997). Oxidative energy metabolism in equine tendon
cells. Research in Veterinary Science 62, 93–97.
Blackmore D.J., Campbell C., Dant C., Holden J.E., Kent J.E. (1982). Selenium status of
Thoroughbreds in the United Kingdom, Equine Vet. J., 14, (2), 139-143.
Bloomer R. J., Bradford J. C. (2009). Relationship between blood lactate and oxidative stress
biomarkers following acute exercise. Open Sports Medicine Journal 3, (1) 44–48.
Bonilla J.F., Narváez R., Chuaire R.. (2005). Sports as a cause of oxidative stress and hemolysis.
Colombia Médica 36, (4).
Boutaud O., Roberts II L.J. (2011). Mechanism-based therapeutic approaches to
rhabdomyolysis-induced renal failure. Free Radic Biol Med. 51, (5), 1062-7.
Bralet J., Bouvier C., Schrieber L., Boquillon M. (1991). Effect of acidosis on lipid peroxidation in
brain slices. Brain Res. 539, (1), 175-177.
Brooks, G. A., K. J. Hittelman, J. A. Faulkner, R. E. Beyer (1971). Temperature, skeletal muscle
mitochondrial functions, and oxygen debt. Am J Physiol 220, (4), 1053-1059.
Brzeszczynska J., Pieniazek A., Gwozdzinski L., Gwozdzinski K., Jegier A. (2008). Structural
alterations of erythrocyte membrane components induced by exhaustive exercise. Appl. Physiol.
Nutr. Metab. 33, (6), 1223-1231.
Brun J.F, Khaled S., Raynaud E., Bouix D., Micallef J.P., Orsetti A (1998). The triphasic effects on
exercise on blood rheology: which relevance to physiology andpathophysiology. Clinical
Hemorheology and microcirculation 19, (2), 89-104.
Buettner G.R (1987). Spin trapping: ESR parameters of spin adducts. Free Radic Biol Med 3,
259–303.
Bureau F., Bonizzi G., Kirschvink N., Delhalle S., Desmecht D., Merville M.P., Bours V., Lekeux P.
(2000a). Correlation between nuclear factor-κB activity in bronchial brushing samples and lung
dysfunction in an animal model of asthma. Am J Respir Crit Care Med, 161, (4), 1314-1321.
Bureau F., Delhalle S., Bonizzi G., Fiévez L., Dogné S., Kirschvink N., Vanderplasschen A., Merville
M.P., Bours V., Lekeux P.(2000b). Mechanisms of persistent NF-kappa B activity in the bronchi
of an animal model of asthma. J. Immunol.165, (10), 5822-5830.

173
Cake M.A., Read R.A., Appleyard R.C., Hwa S.Y., Ghosh P.(2004). The nitric oxide donor glyceryl
trinitrate increases subchondral bone sclerosis and cartilage degeneration
following ovine meniscectomy. Osteoarthritis Cartilage. 12, (12), 974-981.
Camus, G., Felekidis, A., Pincemail, J., Deby-Dupont, G., Deby, C., Juchmes-Ferir, A., Lejeune, R.,
Lamy, M. (1994). Blood levels of reduced/oxidized glutathione and plasma concentration of
ascorbic acid during eccentric and concentric exercises of similar energy cost. Arch. Int. Physiol.
Biochim. Biophys 102, (1), 67-70.
Carr A., Frei B. (1999). Does vitamin C act as a pro-oxidant under physiological conditions? The
FASEB Journal 13, (9), 1007–1024.
Cathcart M.P., Love S., Hughes K.J. (2012). The application of exhaled breath gas and exhaled
breath condensate analysis in the investigation of the lower respiratory tract in veterinary
medicine: a review. The Veterinary Journal 191, (3), 282–291.
Celi P., M. Sullivan, D. Evans (2010). The stability of the reactive oxygen metabolites (d-ROMs)
and biological antioxidant potential (BAP) tests on stored horse blood. The Veterinary Journal
183, (2), 217–218.
Chiaradia, E., Avellini, L., Rueca, F., Spaterna, A., Porciello, F., Antonioni, M.T. and Gaiti, A.
(1998). Physical exercise, oxidative stress and muscle damage in racehorses. Comp. Biochem.
Physiol. Part B. 119, (4), 833-836.
Chiaradia E., Gaiti A., Scaringi L., Cornacchione P., Marconi P., Avellini L. (2002). Antioxidant systems and lymphocyte proliferation in the horse, sheep and dog. Vet. Res. 33, (6), 661–668. Clarke, A.F. (1985) Review of exercise induced pulmonary haemorrhage and its possible
relationship with mechanical stress. Equine vet. J. 17, (3), 166-172.
Clarkson P.M. and Thompson H.S (2000). Antioxidants: what role do they play in physical
activity and health? Am. J. Clin. Nutr. 72, (2 suppl), 637–646.
Cocco T., Marco Di P., Papa S., Lorusso M. (1999). Arachidonic acid interaction with the
mitochondrial electron transport chain promotes reactive oxygen species generation. Free
Radical Biology and Medicine 27, (1), 51–59.
Cohen, G., Sinet, P.M. Fenton's reagent - once more revisited. (1980). Chemical and Biochemical
Aspects of Superoxide and Superoxide Dismutase, Proc. Fed. European Biochem. Soc. Sym. 62, J.V.
Bannister and H.A.O. Hill (eds.), Elsevier, New York, NY, p.27-37
Collins L. V. (2004). Endogenously oxidized mitochondrial DNA induces in vivo and in vitro
inflammatory responses. Journal of Leukocyte Biology 75, (6), 995–1000.
Cook W.R., Williams R.M., Kirker-Head C.A., Verbridge D.J. (1988). Upper airway obstruction
(partial asphyxia) as the possible cause of exercise-induced pulmonary hemorrhage in the
horse: an hypothesis. J. equine vet. Sci. 8, (1), 11-26.
Cortijo J., Marti-Cabrera M., De la Asuncion J.G., Pallardo F.V., Esteras A., Bruseghini L., Vina J.,
Morcillo E.J. (1999). Contraction of human airways by oxidative stress protection by N-
acetylcysteine. Free Radical Biol. Med. 27, (3-4), 392-400.

174
Costa L.R., Seahorn T.L., Moore R.M., Oliver J.L., Hosgood G.L. (2001). Plasma and
bronchoalveolar fluid concentrations of nitric oxide and localization of nitric oxide synthesis in
the lungs of horses with summer pasture-associated obstructive pulmonary disease. Am. J. Vet.
Res. 62, (9), 1381–138.
Couetil L.L., Denicola D.B. (1999). Blood gas, plasma lactate and bronchoalveolar lavage
cytology analyses in racehorses with respiratory disease. Equine Vet. J. Suppl. 30, 77–82.
Craig A.M., Blythe L.L., Rowe K.E., Lassen E.D., Barrington R., Walker K.C. (1992). Variability of
-tocopherol values associated with procurement, storage, and freezing of equine serum and
plasma samples. American Journal of Veterinary Research, 53, (12), 2228-2234.
Cywinska A, Gorecka R, Szareska E, Witkowski L, Dziekan P, Schollenberger A. (2010a). Serum
amyloid A level as a potential indicator of the status of endurance horses. Eq. Vet. J. 38, 23–27.
Cywińska A., Wyszyńska Z., Górecka R., Szarska E., Witkowski L., Dziekan P., Winnicka A.,
Schollenberger A. (2010b). The effect of the 162 km endurance ride on equine peripheral blood
neutrophil and lymphocyte functions. Po.l J. Vet. Sci. 13, (2), 279-285.
Cywinska A., Szarska E., Kowalska A., Ostaszewski P., Schollenberger A. (2011). Gender
differences in exercise-induced intravascular haemolysis during race training in
thoroughbred horses. Res. Vet. Sci. 90, (1), 133-7
Cywinska A., Szarska E., Degorski A., Guzera M., Gorecka R., Strzelec K., Kowalik
S., Schollenberger A., Winnicka A. (2013). Blood phagocyte activity after race training sessions
in Thoroughbred and Arabian horses. Res Vet Sci. 95, (2), 459-64.
Dai S.M., Shan Z.Z., Nakamura H., Masuko-Hongo K., Kato T., Nishioka K., Yudoh K. (2006).
Catabolic stress induces features of chondrocyte senescence through overexpression of
caveolin 1: possible involvement of caveolin 1–induced down-regulation of articular
chondrocytes in the pathogenesis of osteoarthritis. Arthritis & Rheumatism 54, (3), 818–831.
Dakin, S.G., Dudhia, J., Werling, N.J., Werling, D., Abayasekara, D.R., Smith,R.K. (2012a). Inflamm-
aging and arachadonic acid metabolite differ-ences with stage of tendon disease. PLoS One 7,
(11), e48978.
Dakin S.G., Werling, D., Hibbert, A., Abayasekara, D.R., Young, N.J., Smith,R.K., Dudhia, J., (2012b).
Macrophage sub-populations and the lipoxinA4 receptor implicate active inflammation during
equine tendon repair. PLoS One 7, e32333.
Dakin S.G., Dudhia J, Smith R.K.W.(2014). Resolving an inflammatory concept: the importance of inflammation and resolution in tendinopathy. Veterinary Immunology and Immunopathology 158, (3-4), 121–127.
Deaton CM, Marlin DJ, Roberts CA, Smith N, Harris PA, Kelly FJ et al. (2002). Antioxidant
supplementation and pulmonary function at rest and exercise. Equine Veterinary Journal
Supplement 34: 58–65.
Deaton C.M., Marlin D.J. (2003) Exercise-associated oxidative stress. Clinical Techniques in Equine Practice 2, (3), 278 – 291. Deaton C.M., Marlin D.J., Smith N.C., Smith K.C., Newton R.J., Gower S.M., Cade S.M., Roberts C.A.,
Harris P.A., Schroter R.C., Kelly, F.J. (2004). Breath condensate hydrogen peroxide correlates

175
with both airway cytology and epithelial lining fluid ascorbic acid concentration in the horse.
Free Radical Research 38,(2), 201–208.
Deaton C.M., Marlin D.J., Smith N.C., Harris P.A., Dagleish M.P., Schroter R.C., Kelly F.J. (2005).
Effect of acute airway inflammation on the pulmonary antioxidant status. Experimental Lung
Research 31, (7), 653–670.
Deaton C.M. (2006). The role of oxidative stress in an equine model of human asthma. Maney
Online – Redox Report 11, (2), 46-52.
Delattre J., Durant G., Jardillier C., (2003). Biochimie pathologique, aspects moléculaires et
cellulaires. Édition Flammarion, pp 176.
De Leenheer A.P., De Bevere V.O., Claeys A.E (1979). Measurements of -, -, and - tocopherol in serum by liquid chromatography. Clinical Chemistry, 25, (3), 425-428.
De Moffarts B., Kirschvink N., Art T., Pincemail J., Michaux C., Cayeux K., Defraigne J.O., Lekeux P.
(2004a). Impact of training and exercise intensity on blood antioxidant markers in healthy
Standardbred horses. Equine and Comparative Exercise Physiology 1, (3), 211–220.
De Moffarts B., N. Kirschvink N., Lekeux P. (2004b). Evaluation et correction du stress oxydant. XXIème Journée d’Etude de la Belgian Equine Practitioners Society (BEPS), 6-14.
Derksen F.J., Slocombe R.F., Gray P.R., Robinson, N.E. (1992) Exercise induced pulmonary
haemorrhage in horses with experimentally induced allergic lung disease. Am. J. vet. Res. 53, (1),
15-21.
Derksen, F.J. (1997). Oxidant injury and nitric oxide: a role in exercise-induced pulmonary
haemorrhage. Equine Vet. J. 153, (2), 119-122.
Desjardin C., Chat S., Gilles M., Legendre R., Riviere J., Mata X., Balliau T., Esquerré D., Cribiu E.P.,
Betch J-M, Schibler L. (2014). Involvement of mitochondrial dysfunction and ER-stress in the
physiopathology of equine osteochondritis dissecans (OCD). Experimental and Molecular
Pathology 96, (3), 328–338.
Dianzani, M.U. (2003). 4-Hydroxynonenal from pathology to physiology. Mol. Asp. Med. 24, (4-
5), 263-272.
Dimock A.N., Siciliano P.D., McIlwraith C.W. (2000). Evidence supporting an increased presence
of reactive oxygen species in the diseased equine joint. Equine Vet. J. 32, (5), 439–43.
Duarte, J.A., Carvalho, F., Bastons, M.L., Soares, J.M.C., and Appell, H.J. (1994). Do invading
leucocytes contribute to the decrease in glutathione concentrations indicating oxidative stress
in exercised muscle, or are they important for the recovery ? Eur. J. Appl. Physiol. 68, (1), 48–53.
el-Ashker M.R. (2011). Acute kidney injury mediated by oxidative stress in Egyptian horses
with exertional rhabdomyolysis. Vet. Res. Commun 35, (5), 311-320.
Fauconneau B., Tallineau C., Huguet F., Guillard O., Piriou A. (1993). Iron and lactic acid-induced
lipid peroxidation in rat kidney homogenates and slices. Biochem. Mol. Biol. Int. 31, (3), 421-7.
Favier A. (2003). Le stress oxydant : intérêt conceptuel dans la compréhension des mécanismes
et potentiel thérapeutique. Act. Chim., novembre-décémbre 2003, 108-115.

176
Fazio, F., Casella, S., Giannetto, C. Caola, G., Piccione G. (2009). Serum homocysteine and
oxidative stress evaluation during exercise in horse. Pol. J. Vet. Sci. 12, (2), 169–174.
Firth E.C. (2006). The response of bone, articular cartilage and tendon to exercise in the horse.
Journal of Anatomy 208, (4), 513–526.
Fisher G., Schwartz D.D., Quindry J., Barberio M. D., Foster E. B., Jones K. W., Pascoe D.D. (2011).
Lymphocyte enzymatic antioxidant responses to oxidative stress following high-intensity
interval exercise. J Appl Physiol 110, (3), 730–737.
Forman H. J., D. A. Dickinson, K. E. Iles, (2003). 4 - Hydroxynonenal: A lipid degradation product
provided with cell regulatory functions. Mol. Asp. Med. 24, (4-5), 189-194.
Frankiewicz-Jozko, A. Szarska, E. (2000) Antioxidant level to exercise in the blood of endurance horses. Biol. Sport. 17, (3), 217-227.
Gerschman, R., D.L. Gilbert, S.W. Nye, P. Dwyer, W.O. Fenn. (1956). Oxygen poisoning and X-
irradiation: a mechanism in common. Science 119, (3097), 623–626.
Goodship A.E., Birch H.L., Wilson A.M. (1994). The pathobiology and repair of tendon and
ligament injury. Vet Clin North Am: Equine Practice 10, (2), 323–48.
Groussard C., Morel I., Chevanne M., Monnier M., Cillard J., Delamarche A. (2000). Free radical
scavenging and antioxidant effects of lactate ion: an in vitro study. J Appl Physiol. 89, (1), 169-
75.
Groussard C. (2006). Oxidative stress and anaerobic exercise. Science & Sports 21, 62 – 67
Grune T., Reinheckel T., Davies K.J.A. (1997). Degradation of oxidized proteins in mammalian
cells. FASEB J., 11, (7), 526-534.
Gutteridge J., Halliwell B. (2000). Free radicals and antioxidants in the year 2000: a historical
look to the future. Annals of the New York Academy of Sciences 899, (1), 136–147.
Haggett E., Magdesian K.G., Maas J., Pushner B., Higgins J., Fiack C. (2010). Whole blood
selenium concentrations in endurance horses. The Veterinary Journal 186, (2), 192–196.
Halliwell B., J. M. C. Gutteridge (2008). Free Radicals in Biology and Medicine. Fourth Edition.
Oxford University Press.
Hargreaves, B. J., Kronfeld D. S., Waldron J. N., Lopes M.A, Gay L.S., Saker K.E., Cooper W.L., Sklan
D.J., Harris P.A. (2002). Antioxidant status and muscle cell leakage during endurance exercise.
Equine Veterinary Journal 34, (34), 116–121.
Healy Z. R., Lee N.H, Gao X., Goldring M.B., Talalay P., Kensler T.W., Konstantopoulos K. (2005).
Divergent responses of chondrocytes and endothelial cells to shear stress: cross-talk among
COX-2, the phase 2 response, and apoptosis. Proceedings of the National Academy of Sciences of
the United States of America 102, (39), 14010–14015.
Hellsten .Ylva, Nielsen J.J., Lykkesfeldt J., Bruhn M., Silveira L., Pilegaard H., Bangsbo J. (2007).
Antioxidant supplementation enhances the exercise-induced increase in mitochondrial
uncoupling protein 3 and endothelial nitric oxide synthase mRNA content in human skeletal
muscle. Free Radical Biology and Medicine 43, (3), 353–361.

177
Henrotin Y. (2003). The role of reactive oxygen species in homeostasis and degradation of
cartilage. Osteoarthritis and Cartilage 11, (10), 747–755.
Hermes-Lima M., Zenteno-Savin T. (2002). Animal response to drastic changes in oxygen
availability and physiological oxidative stress. Comparative Biochemistry and Physiology Part C
133, 537 – 556.
Hollander J., Flebiq R., Gore M., Bejma, J., Ookawara T., Ohno H., Ji L.L. (1999). Superoxide
dismutase gene expression in skeletal muscle: fiber-specific adaptation to endurance training.
Am. J. Physiol. 277, (3 Pt 2), 856–862.
Hopkins S. R. (2006). Exercice Induced Arterial Hypoxemia: The role of ventilation-Perfusion
Inequality And Pulmonary Diffusion Limitation. Hypoxia and Exercise, pp. 17-30, edited by R.C.
Roach et al. Springer, New York.
Hosoya M., Inoue A., Kimura N., Arai T. (2004). Enzyme activities in some types of peripheral
leukocytes of thoroughbred race horses before and after races. Res. Vet. Sci. 77, (2), 101–104.
Hoshikawa Y., Ono S., Suzuki S., Tanita T., Chida M., Song C., Noda M., Tabata T., F. Voelkel
N.F., Fujimura S. (2001). Generation of oxidative stress contributes to the development of pulmonary
hypertension induced by hypoxia. J. Appl. Physiol. 90, (4), 1299–1306.
Houben J.M, Moonen H.J., Van Schooten F.J., Hageman G.J. (2008). Telomere length assessment:
biomarker of chronic oxidative stress? Free Radical Biology and Medicine 44, (3), 235–246.
Inoue, Y., Matsui, A., Asai, Y., Aoki, F., Matsui, T., Yano, H., 2005. Effect of exercise on iron
metabolism in horses. Biological Trace Element Research 107, (1), 33–41.
Ishida N., Hobo S., Takahashi T., Nanbo Y., Sato F., Hasegawa T., Mukoyama H. (1999).
Chronological changes in superoxide-scavenging ability and lipid peroxide concentration of
equine serum due to stress from exercise and transport. Equine vet. J., Suppl. 30, 430-433.
Jairam V., Uchida K., Narayanaswami V. (2012) Pathophysiology of lipoprotein oxidation. In
Lipoproteins - Role in Health and Diseases. Biochemistry, Genetics and Molecular Biology, édité
par Sasa Frank and Gerhard Kostner, chap. 16.
Johnson K. (2006). Effect of vitamin E supplementation on oxidative stress parameters
measured in exercising horses. Thèse de PhD. University of Florida.
Jungbluth G. (2008). Les espèces réactives de l’oxygène et leurs principales implications dans la
physiopathologie canine. Thèse de Médecine Vétérinaire, Lyon.
Kaikkonen J., Kosonen L., Nyyssonen K., Porkkala-Sarataho E., Salonen R., Korpela H., Salonen J.
T. (1998). Effect of combined coenzyme Q10 and D-α-tocopheryl acetate supplementation on
exercise-induced lipid peroxidation and muscular damage: a placebo-controlled double-blind
study in marathon runners. Free Radic. Res. 29, (1), 85-92.
Kalaci A., Yilmaz H.R., Aslan B., Söğüt S., Yanat A.N., Uz E. (2007). Effects of hyaluronan on nitric
oxide levels and superoxide dismutase activities in synovial fluid in knee osteoarthritis. Clin.
Rheumatol. 26, (8), 1306-1311.
Kaur H., Halliwell B (1997). Hydroxylation of salicylate and phenylalanine as assays for
hydroxyl radicals: a cautionary note visited for the third time. Free Radic Res. 27, (3), 239-244.

178
Kim D.Y., Taylor H.W., Moore R.M., et al (2003). Articular chondrocyte apoptosis in equine
osteoarthritis. Vet. J. 166, (1), 52–57.
Kim R.J., Hah Y.S., Sung C.M., Kang J.R., Park H.B. (2014). Do antioxidants inhibit oxidative-
stress-induced autophagy of tenofibroblasts ? J. Orthop. Res. 32, (7), 937-43.
Kinnunen S., Hyyppä S., Oksala N., Laaksonen D.E., Hannila M.L., Sen C.K., Atalay M. (2009). Α-
Lipoic acid supplementation enhances heat shock protein production and decreases post
exercise lactic acid concentrations in exercised standardbred trotters. Research in Veterinary
Science 87, (3), 462–467.
Kirschvink N., Bureau F., Art T., Lekeux P. (2001). Bronchoconstrictive properties of inhaled 8-
epi-PGF2alpha in healthy and heaves-susceptible horses. Veterinary Research 32, (5), 397–407.
Kirschvink N., Fievez L., Bougnet V., Art T., Degand G., Smith N., Marlin D., Roberts C., Harris
P., Lekeux P. (2002a). Effect of nutritional antioxidant supplementation on systemic and
pulmonary antioxidant status, airway inflammation and lung function in heaves-affected
horses. Equine Veterinary Journal, 34, (7), 705–712.
Kirschvink N., Smith N., Fievez L., Bougnet V., Art T., Degand G., Marlin D., Roberts C., Génicot
B., Lindsey P., Lekeux P.(2002b) Effect of chronic airway inflammation and exercise on
pulmonary and systemic antioxidant status of healthy and heaves-affected horses. Equine
Veterinary Journal 34, (6): 563–571.
Koji U., T. Kumagai. (2003). 4-Hydroxy-2-nonenalas a COX-2 Inducer. Mol. Asp. Med., Vol. 24, (4-
5), 213-218.
Kraus-Hansen, A.E., Fackelman, G.E., Becker, C., Williams, R.M., Pipers, F.S. (1992). Preliminary studies on the vascular anatomy of the equine superficial digital flexor tendon. Equine Veterinary Journal 24, (1), 46–51. Kristensen L., Buhl R., Nostell K., Bak L., Petersen E., Lindholm M., Jacobsen S. (2014). Acute exercise does not induce an acute phase response (APR) in Standardbred Trotters. Canadian Journal of Veterinary Research 78, (2), 97.
Krüger K., Mooren F.C. (2014). Exercise-induced leukocyte apoptosis. Exerc. Immunol. Rev. 20,
117-34.
Laaksonen D.E., Atalay M., Niskanen L., Uusitupa M., Kanninen O., Sen, C. K. (1999) Blood
glutathione homeostasis as a determinant of resting and exercise-induced oxidative stress in
young men. Redox Rep. 4, (1), 53-59.
Lamprecht E.D., Williams C.A. (2012). Biomarkers of antioxidant status, inflammation, and
cartilage metabolism are affected by acute intense exercise but not superoxide dismutase
supplementation in horses. Oxidative Medicine and Cellular Longevity 2012, 1–15.
Langberg H., Skovgaard D., Karamouzis M., Bulow J., Kjaer M. (1999). Metabolism and
inflammatory mediators in the peritendinous spacemeasured by microdialysis during
intermittent isometric exercise inhumans. J. Physiol. 515, (Pt 3), 919–927.
Lebovitz R. M., H. Zhang, H. Vogel, Cartwright J.Jr, Dionne L., Lu N., Huang S., Matzuk M.M.
(1996). Neurodegeneration, myocardial injury and perinatal death in mitochondrial superoxide
dismutase-deficient mice. Proc. Natl. Acad. Sci. US 93, (18), 9782–9787.

179
Lee J.M., Johnson J. A. (2004). An important role of Nrf2-ARE pathway in the cellular defense
mechanism. Journal of Biochemistry and Molecular Biology 37, (2), 139-143.
Li Y. T.T., Huang, E. J., Carlson, Melov S., Ursell P.C., Olson J.L., Noble L.J., Yoshimura M.P., Berger
C., Chan P.H., Wallace D.C., Epstein C.J. (1995). Dilated cardiomyopathy and neonatal lethality in
mutant mice lacking manganese SOD. Nature Genet. 11, (4), 376–381.
Li L.J. (1999). Antioxidants and Oxidative Stress in Exercise. Exp. Biol. Med. 222 (3), 283-292.
Lian O., Scott A., Engebretsen L., et al (2007). Excessive apoptosis in patellar tendinopathy in
athletes. Am J Sports Med 35, (4), 605-611.
Lindholm A. (1987). Pathophysiology of exercise induced diseases of the musculoskeletal system of the equine athlete. Equine Exercise Physiology II – Exercise Induced Musculoskeletal Diseases, pp 711-727.
Loeser R. F., Carlson C.S, Del Carlo M., Cole A. (2002). Detection of nitrotyrosine in aging and
osteoarthritic cartilage: correlation of oxidative damage with the presence of interleukin-1?
And with chondrocyte resistance to insulin-like growth factor 1. Arthritis & Rheumatism, 46,
(9), 2349–2357.
Longo U. G., F. Olivia, V. Denaro, N. Maffulli, (2008).Oxygen species and overuse tendinopathy in
athletes. Disability and Rehabilitation, 30, (20-22), 1563-1571.
Lovlin R., Cottle W., Pyke I., Kavanagh M., Belcastro A.N. (1987). Are indices of free radical
damage related to exercise intensity. Eur. J. Appl. Physiol. 56, (3), 313-316.
Ludvikova E., Pavlata L., Vyskocil M., Jahn P. (2005). Selenium status of horses in Czech Republic. Acta Vet. Brno, 2005, 74, (3), 369-375.
Lyle C.H., K.J.Blissitt, R.N. Kennedy, B.C. McGorum, J.R Newton, T.D.H. Parkin, A.Stirk, L.A. Boden
(2012). Risk factors for race-associated sudden death in Thoroughbred racehorses in the UK
(2000–2007). Equine Veterinary Journal 44, (4), 459-465.
Marr K.A., Lees P., Cunningham F.M. (2002). Antigen challenge increases adherence of circulating neutrophils in horses with chronic obstructive pulmonary disease. Equine. Vet. J. 34, (1), 65–70.
Martin J.A., Brown T., Heiner A. (2004) Post-traumatic osteoarthritis: the role of accelerated
chondrocyte senescence. Biorheology 41, (3-4), 479–491
Martin J.A., Buckwalter J.A. (2006). Post-traumatic osteoarthritis: the role of the stress induced
chondrocyte damage. Biorheology 43, (3-4), 517-21.
MacNee W., Rahman I. (2001) Is oxidative stress central to the pathogenesis of chronic obstructive pulmonary disease? Trends in Molecular Medicine, 7, (2), 55–62.
Marlin D.J., Fenn K., Smith N., Deaton C.D., Roberts C.A., Harris P.A., Dunster C., Kelly, F.J. (2002).
Changes in circulatory antioxidant status in horses during prolonged exercise. J. Nutr 132, (6
Suppl 2), 1622-1627.
McArdle A., J. Jackson M.J. (2000). Exercise, oxidative stress and ageing. J. Anat. 197, 539-541
McCord, J.M., I. Fridovich (1968). The reduction of cytochrome c by milk xanthine oxidase. J.
Biol. Chem. 243, (21), 5753–5760.

180
McKenna M.J., Medved I., Goodman C.A., Brown M.J., Bjorksten A.R., Murphy K.T., Petersen
A.C., Sostaric S., Gong X. (2006). N-acetylcysteine attenuates the decline in muscle Na+,K+-pump
activity and delays fatigue during prolonged exercise in humans. J Physiol. 1576, (Pt 1), 279-
288.
McIlwraith C.W. (1996). General pathophysiology of the joint and response to injury. C.W.
McIlwraith, G.W. Wayne (Eds.). Joint Disease in the Horse, W.B. Saunders, Philadelphia, Chapter
3, p. 43.
Meier B., Radeke H.H., Selle S., Younes M., Sies H., Resch K., Habermehl G.G. (1989). Human
Fibroblasts Release Reactive Oxygen Species in Response to Interleukin-1 or Tumour Necrosis
Factor-Alpha. Biochem. J. 263, (2), 539–545.
Melov S, Coskun P, Patel M, et al (1998). Mitochondrial disease in superoxide dismutase 2
mutant mice. Proc. Natl. Acad. Sci. U. S. A. 96, (3), 846–851.
Mendis E., Kim M.M., Rajapakse N., Kim S.K. (2008). Sulfated glucosamine inhibits oxidation of biomolecules in cells via a mechanism involving intracellular free radical scavenging. European Journal of Pharmacology 579, (1-3), 74–85.
Minami Y., Kawai M., Migita T.C., Hiraga A., Miyata H. (2011). Free radical formation after intensive exercise in thoroughbred skeletal muscles. Journal of Equine Science 22, (2), 21-28.
Millar N.L., Wei A. Q., Molloy T. J., Bonar F., Murrell G. A. C. (2009). Cytokines and apoptosis in
supraspinatus tendinopathy. J Bone Joint Surg 91-B, (3), 417-24.
Mills, P. C., Smith, N. C., Casas, I., Harris, P., Harris, R. C. & Marlin, D. J. (1996) Effects of exercise
intensity and environmental stress on indices of oxidative stress and iron homeostasis during
exercise in the horse. Eur. J. Appl. Physiol. 74, (1-2), 60-66
Mills P.C., Higgins A.J. (1997a). Oxidant injury, nitric oxide and pulmonary vascular function:
implications for the exercising horse. Vet. J. 153, (2), 125–148.
Mills, P.C., Smith, N. C., Harris, R. C., and Harris, P. (1997b). Effect of allopurinol on the
formation of reactive oxygen species during intense exercise in the horse. Res. Vet. Sci. 62, (1),
11–16.
Mohanty J.G., Nagababu E., Friedman J.S., Rifkind J.M. (2013). SOD2 deficiency in hematopoietic cells in mice results in reduced red blood cell deformability and increased heme degradation. Experimental Hematology 41,(3), 316–321.
Moran G., Folch H. (2011). Recurrent airway obstruction in horses–an allergic inflammation: a review. Veterinarni Medicina, 56, (1), 1–13.
Motta S., Letellier C., Ropert M., Motta C., Thiébault J.J. (2009). Protecting effect of vitamin E supplementation on submaximal exercise-induced oxidative stress in sedentary dogs as assessed by erythrocyte membrane fluidity and paraoxonase-1 activity. The Veterinary Journal 181, (3), 288–295.
Murakami, M. (1974). Haemolysis observed in continuous long distance running exercise in
horses. Experimental Reports of Equine Health Laboratory 11, 120– 127.

181
Nakashima I., Liu W., Akhand A.A., Takeda K., Kawamoto Y., Kato M., Suzuki H. et al. (2003). 4-
Hydroxynonenal triggers multistep signal transduction cascades for suppression of cellular
functions. Mol. Asp. Med. 24, (4-5), 231-238.
Nesse L.L., Johansen G.I., Blom A.K. (2002). Effects of racing on lymphocyte proliferation in horses. Am. J. Vet. Res. 63, (4), 528–530.
Nethery, D., D. Stofan, L. Callahan, A. DiMarco, and G. Supinski. Formation of reactive oxygen
species by the contracting diaphragm is PLA2 dependent. J. Appl. Physiol. 87, (1), 792-800.
Newton J. R., Wood J. L. N. (2002). Evidence of an association between inflammatory airway disease and EIPH in young thoroughbreds during training. Equine Veterinary Journal 34, (34), 417–424.
Niedzwiedz A., Jaworski Z. (2014). Oxidant-antioxidant status in the blood of horses with
symptomatic Recurrent Airway Obstruction (RAO). J. Vet. Intern. Med. 28, (6), 1845-1852.
O'Callaghan M.W., Pascoe J.R., Tyler W.S., Mason D.K. (1987) Exercise induced pulmonary
hemorrhage in the horse: results of a detailed clinical, post mortem and imaging study. II. Gross
lung pathology. Equine vet. J. 19, (5), 389-393.
Ohrui T., Sekizawa K., Yamauchi K., Ohkawara Y., Nakazawa H., Aikawa T., Sasaki H., Takishima
T. (1991). Chemical oxidant potentiates electrically and acetylcholine-induced contraction in
rat trachea: possible involvement of cholinesterase inhibition. J Pharmacol. Exp. Ther. , 259, (1),
371-376.
Olszowski S., Mak P., Olszowska E., Marcinkiewicz J. (2003). Collagen type II modification by
hypochlorite. Acta Biochim Pol. 50, (2), 471-479.
Ono K., Inui K., Hasegawa T., Matsuki N., Watanabe H., Takagi S., Hasegawa, A. (1990) The change of antioxidative enzyme activities in equine erythrocytes following exercise. Jap. J. vet. Sci. 52, (4), 759-765.
Oppert J.M., Simon C., Riviere D., Guezennec C.Y. (2005). Activité physique et santé, arguments
scientifique, piste pratique. Bulletin de la Société française de nutrition, Octobre 2005.
Papalia R., Moro L., Franceschi F., Albo E., D’Adamio S., Di Martino A., Vadala G., Faldini C.,
Denaro V. (2013). Endothelial dysfunction and tendinopathy: how far have we come ?
Musculoskelet. Surg. 97, (3),199–209.
Paredi P., Kharitonov S.A., Barnes P.J. (2002). Analysis of expired air for oxidation products. American Journal of Respiratory and Critical Care Medicine 166, (supplement 1), 31–37.
Patterson-Kane J.C., Firth E.C. (2009). The pathobiology of exercise-induced superficial digital flexor tendon injury in thoroughbred racehorses. The Veterinary Journal 181, (2), 79–89.
Patterson-Kane J. C., Rich T. (2014). Achilles tendon injuries in elite athletes: lessons in pathophysiology from their equine counterparts. ILAR Journal 55, (1), 86–99.
Pavlovic R., E. Santaniello (2007). Peroxynitrite and nitrosoperoxycarbonate, a tightly
connected oxidizing-nitrating couple in the reactive nitrogen-oxygen species family: new
perspectives for protection from radical-promoted injury by flavonoids. J. Pharm. Pharmacol.
59, (12), 1687-1695.

182
Pellegrini Masini A., Tedeschi D., Badagli P., Sighieri C., Lubas, G. (2003). Exercise induced
intravascular haemolysis in standardbred horses. Comparative and Clinical Pathology 12, (1),
45–48.
Piccione G, Giannetto C., Marafioti S., Faggio C., Alberghina D., Fazio F. (2012). Training-induced
modifications of circadian rhythmicity of peroxidative parameters in horses. Journal of Animal
Physiology and Animal Nutrition 96, (6), 978–984.
Piercy R.J., Hinchcliff K.W., Morley P.S., Di Silvestro R.A., Reinhart G.A., Nelson S.L. J., Schmidt K.E., Craig A.M. (2001). Vitamin E and exertional rhabdomyolysis during endurance sled dog racing. Neuromuscular Disorders 11, (3), 278–286.
Portier K. (2007). Effets de l‟oxygénation et de l‟exercice sur la fluidité membranaire de l’érythrocyte du cheval. Thèse de doctorat d’université. Université d’Auvergne.
Pouzaud F., Bernard-Beaubois K., Thevenin M., Warnet J.-M., Hayem G., Rat P. (2003). In vitro
discrimination of fluoroquinolones toxicity on tendon cells: involvement of oxidative stress. J.
Pharmacol. Exp. Ther. 308, (1), 394–402.
Powers S. K., Jackson M. J. (2008). Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiological Reviews 88(4): 1243–1276.
Précourt L.P. (2011). Rôles et régulation des enzymes antioxydantes paraoxonases au niveau
intestinal et implication dans les maladies inflammatoires de l’intestin. Thèse de Doctorat en
nutrition. Université de Montréal.
Qian S.Y., Buettner G.R. (1999). Iron and dioxygen chemistry is an important route to initiation
of biological free radical oxidations: an electron paramagnetic resonance spin trapping study.
Free Radic Biol Med 26, (11-12), 1447–1456.
Qian S.Y., Wang H.P., Schafer F.Q., Buettner G.R. (2000). EPR detection of lipid-derived radicals
from PUFA, LDL, and cell oxidations. Free Radic Biol Med 29, (6), 568–579.
Quadrilatero J., Hoffman-Goetz L. (2004). N-Acetyl-L-cysteine prevents exercise-induced
intestinal lymphocyte apoptosis by maintaining intracellular glutathione levels and reducing
mitochondrial membrane depolarization. Biochem Biophys Res Commun. 319, (3), 894-901.
Rahman I. (2003). Oxidative stress: chromatin remodeling and gene transcription in
inflammation and chronic lung diseases. Journal of Biochemistry and Molecular Biology 36, (1),
95-109.
Raidal S.L., Love D.M., Bailey G.D., Rose R.J. (2000). Effect of single bouts of moderate and high intensity exercise and training on equine peripheral blood neutrophil function. Res. Vet. Sci. 68, (2), 141–146.
Raltson S. and Stives M. (2012). Supplementation of ascorbic Acid in weanling horses following prolonged transportation. Animals 2, (2), 184-194.
Raman, Arjun V., and Marla J. Berry. (2011). Selenoproteins in cellular redox regulation and signaling. Oxidative Stress in Vertebrates and Invertebrates: Molecular Aspects of Cell Signaling, 195-208.
Regan E., Flannelly J., Bowler R., Tran K., Nicks M., Carbone B.D., Glueck D., Heijnen H., Mason R., Crapo J. (2005) Extracellular superoxide dismutase and oxidant damage in osteoarthritis. Arthritis & Rheumatism 52, (11), 3479–3491.

183
Reid M.B., Haack K.E., Franchek K.M., Valberg P.A., Kobzik L., West M.S. (1992). Reactive oxygen
in skeletal muscle I. intracellular oxidant kinetics and fatigue in vitro. J.Appl. Physiol. 73, (5),
1797-1804.
Reid MB, Khawli FA, Moody MR (1993). Reactive oxygen in skeletal muscle. III. Contractility of
unfatigued muscle. J Appl Physiol 75, 1081–1087.
Reid M.B. (2001). Plasticity in skeletal, cardiac, and smooth muscle. Invited Review: Redox modulation of skeletal muscle contraction: what we know and what we don’t. J. Appl. Physiol. 90, 724–731.
Reznick A. and Packer L. (1994). Oxidative damage to proteins: Spectrophotometric Method for
Carbonyl Assay. Methods in Enzymology 233, 357–363.
Richardson R. S., E. A. Noyszewski, K. F. Kendrick, J. S. Leigh, P. D. Wagner (1995). Myoglobin O2
desaturation during exercise. Evidence of limited O2 transport. J. Clin. Invest. 96, (4), 1916-1926.
Roach, Robert C., Peter D. Wagner, and Peter H. Hackett. (2006). Hypoxia and Exercise, volume 588. Springer.
Robinson Y., Cristancho E., Böning D. (2006). Intravascular hemolysis and mean red blood cell age in athletes. Med. Sci. Sports Exerc. 38, (3), 480-483.
Rokitski L., Logemann E., Sagredos N., Murphy W., Wetzel-Roth W., Keul J. (1994) Lipid peroxidation and antioxidative vitamins under extreme endurance stress. Acta Physiol. Scand. 151, (2), 149-158.
Romer L.M., Dempsey J.A., Eldridge A.L.M. (2006). Exercice induced arterial hypoxemia: consequence for locomotor muscle fatigue. Hypoxia and Exercise, pp 47-55, edited by R.C. Roach et al. Springer, New York.
Rush B., Mair T. (2004). Noninfectious Pulmonary diseases. Equine pulmonary diseases. 19, 189-
231, Blackwell Science Ltd.
Sagai M., Bocci V. (2011). Mechanisms of action involved in ozone therapy: is healing induced via a mild oxidative stress. Medical Gas Research 1:29.
Sastre J., Asensi M., Gasco E., Pallardo F. V., Ferrero J. A., Furukawa T., Vina, J. (1992). Exhaustive
physical exercise causes oxidation of glutathione status in blood: prevention by antioxidant
administration. Am. J. Physiol. 263, (5 Pt 2), 992-995.
Schmidt-Rohlfing B., Graf J., Schneider U., Niethard F.U. (1992). The blood supply of the Achilles
tendon. International Orthopaedics 16, 29–31.
Schneider N., Mouithys-Mickalad A.L., Lejeune J.P., Deby-Dupont G.P., Hoebeke M., Serteyn D.A. (2005) Synoviocytes, not chondrocytes, release free radicals after cycles of anoxia/re-oxygenation. Biochemical and Biophysical Research Communications 334, (2), 669–673.
Schneider N., Lejeune J.P., Deby-Dupont G., Serteyn D. (2007). Le rôle des synoviocytes dans
l’articulation diarthrodiale enflammée. Ann. Méd. Vét. 151, 24-43.
Schroter R.C., Marlin D.J., Denny E. (1998). Exercise-induced pulmonary haemorrhage (EIPH) in
horses results from locomotory impact induced trauma - a novel, unifying concept. Equine vet. J.
30, (3), 186-192.

184
Seitz A.L., McClure P.W., Finucane S., Boardman N.D., Michener L.A. (2011). Mechanisms of
rotator cuff tendinopathy: intrinsic, extrinsic, or both? Clin. Biomech. 26, (1), 1–12.
Shute M. (2004). Effect of Whey Protein Isolate on Oxidative Stress, Exercise Performance, and Immunity. Thèse de PhD. Virginia Polytechnic.
Siciliano P.D., Lawrence L.M., Danielsen K., Powell D.M., Thompson K.N. (1995). Effect of
conditioning and exercise type on serum creatine kinase and aspartate aminotransferase
activity. Equine vet. J., Suppl. 27, (18), 243-247.
Siciliano, P.D., Aparker A.L., Lawrence L. M. (1996) Effect of dietary vitamin E supplementation
on the integrity of skeletal muscle in exercised horses. J. Anim. Sci. 75, (6), 1553-1560.
Siesjo B.K., Bendek G., Koide T., Westerberg E., Wieloch T. (1985). Influence of acidosis on lipid
peroxidation in brain tissues in vitro. J. Cerebral. Blood. Flow. Metab. 5(2): 253-258.
Smarsh D.N., Williams C.A. (2013). The effects of acute exercise on oxidative stress in the
skeletal muscle and blood of yearling and mature horses. Journal of Equine Veterinary Science
33, (5), 324.
Simon M. C. (2006). Mitochondrial reactive oxygen species are required for hypoxic HIF alpha
stabilization. Hypoxia and Exercise, pp 165-170, edited by R.C. Roach et al. Springer, New York,
2006.
Singer E.R., Barnes, J., Saxby, F., Murray, J.K. (2008). Injuries in the event horse: training versus
competition. Veterinary Journal 175, (1), 76–81.
Smith J.A., Kolbuch-Braddon M., Gillam I., Telford R.D., Weidemann M.J. Changes in the
susceptibility of red blood cells to oxidative and osmotic stress following submaximal exercise.
Eur J Appl Physiol Occup Physiol. 70, (5), 427-436.
Snow D.H., Guy P.S. (1980). Muscle fibre composition of a number of limb muscles on different
types of horses. Res. Vet. Sci. 28, (2), 137-144.
Soffler C. (2007). Oxidative stress. Veterinary Clinics of North America: Equine Practice 23, (1), 135–157.
Srinivasan C, Liba A, Imlay J.A, Valentine J.S., Gralla EB. Yeast lacking superoxide dismutase(s)
show elevated levels of “free iron” as measured by whole cell electron paramagnetic resonance.
JBiol Chem. 275, (38), 29187–29192.
Stadtman, E. R. (2004). Role of oxidant species in aging. Curr. Med. Chem. 11, (9), 1105–1112.
Studer R.K., Levicoff E., Georgescu H., Miller L., Jaffurs D., Evans C.H.(2000). Nitric oxide inhibits
chondrocyte response to IGF-I: inhibition of IGF-IRbeta tyrosine phosphorylation. American
Journal of Physiology - Cell Physiology 279, (4), 961-969.
Taha R. (2011). Stress oxydatif, fonction mitochondriale et maladie inflammatoire de l’intestin.
Thèse de Doctorat en nutrition. Université de Montréal.
Tomiyama T., Fukuda K., Yamazaki K., Hashimoto K., Ueda H., Mori S., Hamanishi C. (2007).
Cyclic compression loaded on cartilage explants enhances the production of reactive oxygen
species. J. Rheumatol. 34, (3), 556–562.

185
Touati, D. 1989. The molecular genetics of superoxide dismutase in E. coli. Free Rad. Res.
Commun. 8: 1–9.
Tsubone H., Hanafusa M, Endo M., Manabe N., Hiraga A., Ohmura H., Aida H. (2013). Effect of
treadmill exercise and hydrogen-rich water intake on serum oxidative and anti-oxidative
metabolites in serum of thoroughbred horses. Journal of Equine Science 24, (1), 1-8.
Valberg S.J. (2006). Exertional Rhabdomyolisis. AAEP Proceedings – Muscle Disorders, 52, 365-372
Valko M., Leibfritz D., Moncol J., Cronin M.T., Mazur M., Telser J. (2007) Free radicals and antioxidants in normal physiological functions and human disease. The International Journal of Biochemistry & Cell Biology 39, (1), 44–84.
Valko M., Omova K.J. (2011). Free Radicals, Signal Transduction, and Human Disease. Oxidative Stress in Vertebrates and Invertebrates: Molecular Aspects of Cell Signaling, pp 17-32.
Van den Belt A.J., Dik K.J., Barneveld A. (1994). Ultrasonographic evaluation and longterm
follow-up of flexor tendonitis/desmitis in the metacarpal/metatarsal region in Dutch
Warmblood horses and Standardbred racehorses. Veterinary Quarterly Suppl. 2, 76–80.
Van der Harst M., Bull S., Brama P.A., Barneveld A.B., van Weeren P.R., van de Lest C. (2006).
Nitrite and nitrotyrosine concentrations in articular cartilage, subchondral bone, and
trabecular bone of normal juvenile, normal adult, and osteoarthritic adult equine
metacarpophalangeal joints. J. Rheumatol. 33, (8), 1662–1667.
Venkataraman S., Schafer F. Q., Buettner G. R. (2004) Detection of lipid radicals using EPR. Antioxidants and Redox Signaling 6, (3), 631–638.
Venturini, G., Colasanti M., Fioravanti E., Bianchini A., Ascenzi P. (1999). Direct effect of
temperature on the catalytic activity of nitric oxide synthases type I, II, and III. Nitric Oxide:
Biology and Chemitry 3, (5), 375-382.
Villasante A., Araneda O.F., Behn C., Galleguillos M., Adarmes H. (2010). Antioxidant capacity and oxidative damage determination in synovial fluid of chronically damaged equine metacarpophalangeal joint. Veterinary Research Communications 34, (2), 133–141.
Wang M.X., Wei A., Yuan J., Clippe A. Bernard A., Knoops B., Murrell G.A. (2001). Antioxidant
enzyme peroxiredoxin 5 is upregulated in degenerative human tendon. Biochem Biophys Res.
Commun. 284, (3), 667–73.
Wagner B.A., Buettner G.R., Burns C.P (1994). Free radical mediated peroxidation in cells:
Oxidizability is a function of cell lipid bis-allylic hydrogen content. Biochemistry 33, (15), 4449–
4453.
Webb C., Twedt D. (2008). Oxidative stress and liver disease. Veterinary Clinics of North America: Small Animal Practice 38, (1), 125–135.
West J.B., Mathieu-Costello O., Jones, J.H. Birks, E.K. Logemann R.B., Pascoe J.R., Tyler W.S.
(1993) Stress failure of pulmonary capillaries in racehorses with exercise-induced pulmonary
hemorrhage. J. appl. Physiol. 75, (3), 1097-1109
White A., Estrada M., Walker K., Wisnia P., Filgueira G., Valdes F., Araneda O., Behn C., Martinez
R. (2001) Role of exercise and ascorbate on plasma antioxidant capacity in Thoroughbred race
horses. Comp. Biochem. Physiol. A 128, (1), 99-104.

186
Wilcox A.L., Marnett L.J. Polyunsaturated fatty acid alkoxyl radicals exist as carbon-centered
epoxyallylic radicals: a key step in hydroperoxide-amplified lipid peroxidation. Chem Res
Toxicol 6, (4), 413–416.
Willett, K., Blackmore, D.J., 1979. Haptoglobin in the serum of thoroughbreds in training.
Research in Veterinary Science 26, (3), 308–314.
Witko-Sarsat V., Friedlander M., Nguyen Khoa T., Capeillère-Blandin C., Nguyen A.T., Canteloup
S., Dayer J.M., Jungers P., Drüeke T., Descamps-Latscha B. (1998). Advanced oxidation protein
products as novel mediators of inflammation and monocyte activation in chronic renal failure.
The Journal of Immunology 161, (5), 2524–2532.
Wood J.L.N., Newton J.R., Chanter N., Mumford J.A. (2005). Association between respiratory
disease and bacterial and viral infections in british racehorses. Journal of Clinical Microbiology
43, (1), 120–126.
Wu S.B., Wu Y.T, Ma Y.S, Yau-Huei Wei Y.H. (2011). Oxidative stress and its biochemical consequences in mitochondrial DNA mutation-associated diseases: implications of redox therapy for mitochondrial diseases. Oxidative Stress in Vertebrates and Invertebrates: Molecular Aspects of Cell Signaling, pp. 33-49.
Yamamoto N., Fukuda K., Matsushita T., Matsukawa M., Hara F., Hamanishi C. (2005) Cyclic
tensile stretch stimulates the release of reactive oxygen species from osteoblast-like cells.
Calcif. Tissue Int. 76, (6), 433-8.
Yuan J., Murrell G.A., Wei A.Q., Wang M.X. (2002). Apoptosis in rotator cuff tendonopathy. J
Orthop Res 20, (6),1372-1379.
Yuan J., Wang M.X., Murrell G.A.C. (2003). Cell death and tendinopathy. Clinics in Sports Medicine 22, (4), 693–701.
Yuan J., Murrell G.A., Trickett A., Landtmeters M., Knoops B., Wang M.X. (2004). Overexpression of antioxidant enzyme peroxiredoxin 5 protects human tendon cells against apoptosis and loss of cellular function during oxidative stress. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1693, (1), 37–45.
Zarkovic, N. (2003). 4-Hydroxynonenal as a bioactive marker of pathophysiological processes.
Mol. Asp. Med. 24, (4-5), 281-291.
Zhang, J., Wang, J.H. (2010). Production of PGE2 increases in tendons sub-jected to repetitive
mechanical loading and induces differentiation oftendon stem cells into non-tenocytes. J.
Orthop. Res. 28, (2), 198–203.
Zhang K. (2009). Integration of ER stress, oxidative stress and the inflammatory response in
health and disease. Int J Clin Exp Med 3, (1), 33-40.
Zicker S. C., Wedekind K.J, Jewell D.E. (2006). Antioxidants in veterinary nutrition. Veterinary Clinics of North America: Small Animal Practice 36, (6), 1183–1198.
Zuo, L. (2002) Molecular Mechanisms of Stress-Induced Reactive Oxygen Species Formation in Skeletal Muscle. Thèse de Phd en biophysique. Ohio State University.

187
Pages internet consultées
Diacron International
(Consultation le 23 mars 2015)
Diacron International - Home.
http://www.diacron.com
Claire PIGAULT, 2010
(Consultation le 9 Avril 2015)
ABSORPTION-FLUORESCENCE (module) http://unt-ori2.crihan.fr/unspf/2010_Strasbourg_Pigault_AbsorptionFluorescence/co/01_Abs_Fluo_web.html

188
NOM PRÉNOM : DUBOIS Benjamin
TITRE : IMPLICATION DU STRESS OXYDANT DANS PLUSIEURS AFFECTIONS DU CHEVAL ATHLÈTE
Thèse d’État de Doctorat Vétérinaire : Lyon, 12 Juin 2015
RÉSUMÉ : Les fondements biochimiques du stress oxydant reposent sur la production excessive d’espèces réactives de l’oxygène et de leur réactivité pour de nombreux composants cellulaires. Plusieurs mécanismes d’apparition ont été identifiés chez les mammifères. Certains d’entre eux sont notamment susceptibles d’intéresser le cheval athlète. Comme chez d’autres espèces, des études ont montré qu’un stress oxydant peut être induit par l’effort physique chez le cheval et pourrait entrer dans la physiopathologie de plusieurs affections limitant ses performances sportives. Ces dernières concernent essentiellement les muscles, les appareils cardiovasculaire et respiratoire, mais aussi les articulations, les os, les tendons et les ligaments. Par ailleurs, les limites et la difficulté de l’exploration expérimentale du stress oxydant doit également être pris en compte afin de faire le point sur son lien éventuel avec la physiopathologie de l’athlète équin.
MOTS CLÉS : - Cheval - Stress oxydatif
- Radicaux libres - Effort physique
- Performance - Physiopathologie
JURY :
Président : Monsieur le Professeur Gilles RODE
1er Assesseur : Monsieur le Professeur Jean-Jacques THIÉBAULT
2ème Assesseur : Monsieur le Professeur Philippe BERNY
DATE DE SOUTENANCE : 12 juin 2015
ADRESSE DE L’AUTEUR :
9 Chemin de Founsut 11110 Salles d’Aude FRANCE