thĒse de doctorat de l'universitĒ - …alaforgue.free.fr/these.pdf · vi-2) montage de...
TRANSCRIPT

THĒSE DE DOCTORAT DE L'UNIVERSITĒ
PARIS XII – VAL DE MARNE
Présentée par
Alexis LAFORGUE
Pour obtenir le titre de DOCTEUR DE L'UNIVERSITĒ PARIS XII – VAL DE MARNE
Spécialité : Polymères Fonctionnels
Sujet de thèse
SYNTHĒSE ET CARACTĒRISATION DE POLYMĒRES CONDUCTEURS
APPLICATION AU STOCKAGE DE L'ĒNERGIE
Soutenue le 19 Mars 2001 devant le jury composé de :
Mme Marina MASTRAGOSTINO Rapporteur
M. Daniel BELANGER Rapporteur
M. Bernard SEBILLE Examinateur
M. Patrick LAILLER Examinateur
M. Patrice SIMON Examinateur
M. Claude CHEVROT Examinateur
M. Jean-François FAUVARQUE Directeur de thèse
Thèse préparée au Laboratoire d'Electrochimie Industrielle du
Conservatoire National des Arts et Métiers (Paris)

Remerciements Je tiens tout d'abord à remercier Monsieur Jean-François Fauvarque, Professeur
titulaire de la Chaire d'Electrochimie Industrielle du C.N.A.M., pour m'avoir accueilli dans son laboratoire et m'avoir proposé ce travail de recherche passionnant.
Je remercie Monsieur Daniel Bélanger, Professeur de l'Université du Québec à
Montréal, et Madame Marina Mastragostino, Professeur de l'Université de Bologne, pour avoir bien voulu témoigner de l'intérêt qu'ils portent à ce travail en acceptant d'en être les rapporteurs.
Je remercie Patrick Lailler, Directeur du service de R&D de la société CEAC-Exide,
pour avoir mis à ma disposition son laboratoire, et pour avoir accepté de juger ce travail. Je tiens également à remercier Jean-François Sarrau, ingénieur de recherche, pour cette fructueuse collaboration tout au long du projet.
Je tiens à exprimer ma profonde gratitude à Monsieur Patrice Simon, Maître de
conférence au Laboratoire d'Electrochimie Industrielle du CNAM, pour avoir suivi quotidiennement ce travail avec attention et rigueur, tout en me laissant une grande liberté d'action et de choix.
Je souhaite remercier Monsieur Bernard Sébille, Professeur Émérite de l'Université
Paris XII - Val de Marne, de me faire l'honneur d'être examinateur de cette thèse et pour accepter de juger ce travail.
Je remercie tout spécialement Monsieur Jacques Bouet, ancien conseiller scientifique
chez SAFT, pour ses nombreux conseils, aussi avisés que ses calembours sont drôles. Je remercie chaleureusement tous les membres permanents du laboratoire et tous ceux
que j'y ai croisés et qui m'ont aidé : Christian, Jean-Luc, Muriel, Gérard, Hakim, M. Catonné, Emmanuel, Laurence, Aurélien, Isabelle, Nathaly, Marie-Thérèse, Marie-José, Brigitte, Florian, Jacques, Jojo, Jean-Gary, Jamila, et tous ceux que j'oublie … Leurs conseils, leur amitié et leur soutien m'ont été indispensables.
Je souhaite également remercier toutes les personnes du CNAM qui m'ont aidé dans
mon travail. Spéciale dédicace à Patron, Pierre-Louis, Laetitia, Gérard et Eric, parce qu'ils le
valent bien ! Et puis j'adresse un petit clin d'œil à ma famille et à ma future femme, Anne-Sophie.

Table des Matières
Introduction Générale 1
I) Etude bibliographique 3
I-1) Les polythiophènes 3
I-1-1) Théorie de la conduction dans les polymères conducteurs 3
I-1-2) Synthèse des polythiophènes 6
I-1-2-1) Polymérisations chimiques 6
I-1-2-2) Polymérisations électrochimiques 9
I-1-3) Propriétés des polythiophènes 12
I-1-3-1) Propriétés chimiques et optiques 12
I-1-3-2) Propriétés électrochimiques 18
I-1-4) Applications des polythiophènes 21
I-2) Les supercondensateurs 22
I-2-1) Les supercondensateurs à base de charbons actifs 23
I-2-2) Les supercondensateurs à base d'oxydes métalliques 25
I-2-3) Les supercondensateurs à base de polymères conducteurs 26
I-2-4) Modèles de capacité utilisés pour les polymères conducteurs 29
II) Partie expérimentale 31
II-1) Caractérisations chimiques 31
II-2) Caractérisations électrochimiques 31
II-2-1) Cellules électrochimiques 31
II-2-2) Techniques d'analyse électrochimiques 33
II-2-2-1) La voltampérométrie cyclique 33
II-2-2-2) Le cyclage galvanostatique 35
II-2-2-3) La spectroscopie d'impédance complexe 38

III) Synthèses 41
III-1) Synthèse des monomères 41
III-1-1) Principe 41
III-1-2) Procédure 42
III-2) Synthèse chimique du polythiophène 43
III-2-1) Principe 44
III-2-2) Procédure 44
III-3) Synthèse chimique du poly-4-fluorophénylthiophène (P-4-FPT) 45
III-3-1) Principe 45
III-3-2) Etude de la synthèse du PFPT 46
III-3-2-1) Etude de la température de polymérisation 46
III-3-2-2) Etude du rapport oxydant/monomère 48
III-3-2-3) Etude de la concentration des réactifs 50
III-3-2-4) Etude du temps de polymérisation 51
III-3-2-5) Conclusion de l'étude 53
III-3-3) Protocole de synthèse du P-4-FPT 53
III-4) Synthèse des autres polymères 54
IV) Mise en œuvre des polymères 55
IV-1) Influence de l'additif conducteur sur le dopage négatif 55
IV-2) Influence de la nature du liant sur le dopage négatif 57
IV-3) Etude du dopage positif 62
V) Caractérisation des polymères 66
V-1) Caractérisation de l'électrolyte 67
V-2) Caractérisation du Polythiophène (PTh) 70
V-2-1) Caractérisation chimique du PTh 70
V-2-2) Caractéristation électrochimique du PTh 71
V-2-2-1) Voltamétries cycliques des processus de dopage 71
V-2-2-2) Dopage négatif et réduction du solvant 73
V-2-2-3) Influence de l'anion sur le dopage positif du PTh 74
V-2-2-4) Cyclabilité du PTh en dopage positif 75

V-3) Caractérisation du PFPT 78
V-3-1) Caractérisation chimique du PFPT 78
V-3-2) Caractérisation des processus de dopage 81
V-3-2-1) Voltamétrie cyclique du dopage négatif 81
V-3-2-2) Cyclage galvanostatique du dopage négatif 86
V-3-2-3) Voltamétrie cyclique du dopage positif 90
V-3-2-4) Cyclage galvanostatique du dopage positif 92
V-3-3) Cyclabilité du dopage négatif 96
V-3-3-1) Cyclabilité en voltamétrie cyclique 96
V-3-3-2) Cyclage galvanostatique du dopage négatif 100
V-3-4) Cyclabilité du dopage positif 104
V-3-4-1) Cyclabilité en voltamétrie cyclique 104
V-3-4-2) Cyclage galvanostatique du dopage positif 108
V-3-5) Etude de la perte d'électroactivité du PFPT 113
V-3-5-1) Etude de l'électrolyte 113
V-3-5-2) Etude de l'électrode 115
V-4) Caractérisation des polyphénylthiophènes fluorés (PPTF) 118
V-4-1) Caractérisation chimique des PPTF 118
V-4-2) Caractérisation des dopages en voltamétrie cyclique 121
V-4-2-1) Dopage positif 121
V-4-2-2) Dopage négatif 124
V-4-3) Cyclabilité des processus de dopage 127
V-4-3-1) Dopage positif 127
V-4-3-2) Dopage négatif 129
VI) Application aux supercondensateurs 132
VI-1) Constitution des supercondensateurs 132
VI-1-1) Les matières actives 132
VI-1-1-1) L'électrode négative 132
VI-1-1-2) L'électrode positive 132
VI-1-2) Les collecteurs de courant 133
VI-1-3) Le séparateur 134
VI-1-4) L'électrolyte 135

VI-2) Montage de cellules-test de 4 cm2 135
VI-2-1) Les supercondensateurs polymères de type III 135
VI-2-1-1) Principe de fonctionnement 135
VI-2-1-2) Equilibrage des capacités 136
VI-2-1-3) Réversibilité coulombique 137
VI-2-1-4) Performances du système 138
VI-2-1-5) Spectre d'impédance électrochimique du système 141
VI-2-2) Les supercondensateurs hybrides polymère - carbone 142
VI-2-2-1) Principe de fonctionnement 142
VI-2-2-2) Les supercondensateurs Norit/PFPT 143
VI-2-2-3) Les supercondensateurs BP25/PFPT 146
VI-2-2-4) Spectres d'impédance des systèmes hybrides 150
VI-3) Assemblage de modules industriels 152
VI-3-1) Fabrication des électrodes 152
VI-3-2) Assemblage des modules 155
VI-3-3) Performances initiales des modules 155
VI-3-4) Cyclabilité des modules 156
Conclusion générale 158
Perspectives 161
Annexes
Annexe 1 : Protocoles de mise en oeuvre des liants 163
Annexe 2 : Spectres infrarouges des polymères 166
Annexe 3 : Notations de Wilson des vibrations du benzène 167
Annexe 4 : Caractéristiques des modules industriels 168
Références bibliographiques 171
Liste des figures 179
Liste des tableaux 187

Introduction générale
Depuis une vingtaine d'années, les polymères conducteurs électroniques sont l'objet de
nombreuses études, étant donné leur aptitude à passer réversiblement d'un état isolant à un
état conducteur, associé aussi dans la plupart des cas à une transition optique. Ainsi les
applications possibles sont nombreuses : ils peuvent être utilisés comme semi-conducteurs
organiques, matériaux électroluminescents, revêtements pour le blindage magnétique,
matières actives de stockage d'énergie, dispositifs électrochromes, senseurs, …
En particulier, le polythiophène (PTh) est intéressant car ses propriétés physico-
chimiques peuvent être modulées en greffant des substituants aux propriétés spécifiques sur
les chaînes macromoléculaires. En outre, cette famille de polymères conducteurs possède
deux domaines de conductivité différents selon la valeur du potentiel (domaines de dopage).
Un domaine positif dans lequel la conduction est réalisée par la délocalisation de charges
positives sur les chaînes, et un domaine négatif dans lequel la conduction est réalisée par la
délocalisation de charges négatives. Entre ces deux domaines se trouve une zone de potentiel
dans lequel les polythiophènes sont isolants électroniques.
Pour la plupart des applications utilisant les propriétés de conduction et/ou de passage
réversible de l'état conducteur à l'état isolant du polymère, c'est le domaine de dopage positif
qui est utilisé. En effet ce processus de dopage est plus stable et plus facile à réaliser.
Le dopage négatif est beaucoup moins stable, moins capacitif, et est plus difficile à
réaliser efficacement. C'est pourquoi ce type de dopage a été beaucoup moins étudié et les
applications qui nécessitent son utilisation spécifique par rapport au dopage positif sont rares.
L'une d'elles consiste à utiliser ce processus de dopage pour stocker de l'énergie dans des
électrodes négatives de supercondensateurs. Les propriétés demandées pour cette application
sont alors une capacité importante, une excellente réversibilité coulombique entre dopage et
dédopage ainsi qu'une grande cyclabilité. Le PTh lui-même ne permet pas d'obtenir de telles
propriétés car ce processus de dopage a lieu à des potentiels très négatifs où les milieux
électrolytiques ne sont pas stables. Une solution est de substituer le thiophène avec des
groupements électroattracteurs qui décalent les processus de dopage vers des potentiels plus
positifs. Dans cette optique, une équipe américaine a proposé il y a 6 ans une série de
polythiophènes synthétisés électrochimiquement et substitués par de tels groupements et dont
les performances étaient très intéressantes [1]. Cependant, ces synthèses électrochimiques ne
sont pas applicables à une échelle industrielle car les polymères sont déposés sur des substrats
conducteurs, ce qui ne peut être réalisé que sur des substrats de petite taille.
1

En revanche, les synthèses chimiques conduisent à des polymères sous forme de
poudres, ce qui permet une mise en œuvre dans des électrodes de grandes tailles, et rend un
éventuel passage à production industrielle plus simple. Le but de ce travail est donc de synthétiser chimiquement différents polythiophènes
substitués par des groupements électroattracteurs afin d'obtenir des processus de dopage
négatif ainsi que positifs facilement réalisables et stables.
Ce travail s'inscrit dans le cadre d'un projet européen (JOULE N° JOE3-CT97-0047) en
partenariat avec le Laboratoire de Chimie Physique de l'Université de Bologne (Italie), le
service de R&D de la société CEAC-Exide (France), la division Machine de la société
Arcotronics (Italie) et l'agence gouvernementale ENEA (Italie). Le but de ce projet est de
développer un prototype performant de supercondensateur à base de polymères conducteurs.
Dans ce cadre, le travail de synthèse et de sélection des polymères est préliminaire à un
travail de développement : mise en œuvre d'électrodes contenant ces polymères, mise au point
de systèmes de supercondensateurs en cellules-test de petites tailles et réalisation de modules
semi-industriels. - Le premier chapitre de ce travail est une revue bibliographique dont la première partie
est consacrée aux polythiophènes, décrivant leurs différents modes de synthèse, leurs
propriétés chimiques, électrochimiques et optiques ainsi que leurs principales applications. La
deuxième partie est consacrée à la présentation différents types de supercondensateurs.
- Le deuxième chapitre détaille les techniques expérimentales employées.
- Le troisième chapitre décrit les principes des synthèses chimiques des monomères et
des polymères, ainsi qu'une étude des paramètres macroscopiques de la polymérisation par le
chlorure ferrique.
- Le quatrième chapitre est consacré à la mise au point d'électrodes composites de
polymères conducteurs.
- Le cinquième chapitre décrit les propriétés chimiques et électrochimiques des
polymères, caractérisées à l'aide de différentes techniques spectrales, chimiques et
électrochimiques. Une étude de stabilité en cyclage est également détaillée à la fin de ce
chapitre.
- Le sixième chapitre est consacré à l'assemblage de supercondensateurs dans des
cellules test de 4 cm2 puis au passage à des modules industriels de n électrodes de 60 cm2.
- Les conclusions seront ensuite présentées avant d'évoquer les perspectives futures de
ce travail.
2

I) Etude bibliographique
I-1) Les polythiophènes
La découverte à la fin des années 1970 d'une conductivité presque équivalente à celle
des métaux pour du polyacétylène oxydé [2] a suscité un fort intérêt dans le monde entier et a
entraîné un nombre important d'études dans le domaine des systèmes π conjugués. Le
polyacétylène (PAc) étant relativement peu stable, la recherche s'est plutôt orientée vers les
polycycles aromatiques tels que le polyparaphénylène (PPP), la polyaniline (PANI), le
polypyrrole (PPy) ou encore le polythiophène (PTh) (cf. Figure I-1).
) )(()( NH
SS
n nn
)( Nn
PAc PPP PANI PPy PTh
NH
N
H
Figure I-1 : Représentation des principaux polymères conducteurs électroniques possédant un système π conjugué.
Le PTh a été particulièrement étudié pour sa stabilité à l'air et surtout la possibilité de
moduler ses propriétés chimiques, électrochimiques et spectrales en le substituant par des
groupements divers. Ce polymère possède en outre deux états conducteurs : oxydé (dopé
positivement) et réduit (dopé négativement). Le premier chapitre de cette étude
bibliographique est consacré au PTh et à ses dérivés. La première partie présente la théorie de
la conduction dans les polymères conducteurs. La deuxième partie détaille les différentes
méthodes de synthèse, leurs avantages et inconvénients. La troisième partie s'intéresse aux
propriétés chimiques, optiques et électrochimiques des polythiophènes. La quatrième partie
est consacrée aux applications potentielles de ces polymères.
I-1-1) Théorie de la conduction dans les polymères conducteurs
Les polymères conducteurs se caractérisent par une alternance de liaisons saturées et
insaturées le long de leur chaîne. Les atomes de carbone qui les composent sont donc hybridés
sp2, ce qui confère aux chaînes une structure plane [3,4]. Cette planéité va permettre le
3

recouvrement des orbitales Pz le long de la chaîne, ce qui va conduire à une hybridation de
type π.
Selon les principes d'exclusion de Pauli et de distorsion de Peierls, les états d'énergies
des différentes orbitales Pz vont se regrouper en bandes (cf. Figure I-2) : la bande de valence
qui regroupe les niveaux d'énergie occupés, et la bande conduction qui regroupe les niveaux
d'énergie inoccupés. Entre les deux bandes, il y a un gap de niveaux d'énergie interdits [5].
Figure I-2 : Niveaux d'énergie des orbitales π dans un polymère conducteur.
Ce schéma de bandes correspond à celui de matériaux isolants ou semi-conducteurs
(cf. Figure I-3).
Figure I-3 : Schéma de bande des matériaux isolants, semi-conducteurs et conducteurs.
4

Un matériau est dit isolant lorsque le gap entre sa bande de valence et sa bande de
conduction est supérieur à 5 eV. Les polythiophènes possèdent des gaps entre 1,7 et 2,3 eV et
font donc partie des matériaux semi-conducteurs. Pour obtenir une conduction dans un tel
matériau, il faut que les électrons passent de la bande de valence à la bande de conduction ; il
faut donc fournir une énergie d'excitation supérieure au gap.
Doper un polymère conducteur (par injection ou extraction d'électrons) consiste à créer
des défauts structuraux (électrons non appariés) appelés solitons. Ces solitons ne sont pas
stables et s'apparient pour donner des radicaux cations appelés polarons (cf. Figure I-4). Ces
polarons créent des états d'énergie, localisés dans la bande interdite (cf. Figure I-5). Le
déplacement de ces polarons sur la chaîne macromoléculaire crée la conduction.
][ SS
SS
S
SS
SS
S
⊕ ⊕
⊕
• Polaron (cation radical)
Bipolaron (dication)
configuration quinoïdale
Figure I-4 : Représentation des différents porteurs de charge dans les polythiophènes.
A partir d'une concentration limite de polarons sur la chaîne, ceux ci s'apparient de
nouveau pour donner des dications appelés bipolarons (cf. Figures I-4 et I-5) [6-10]. Ces
porteurs de charges conduisent la chaîne polymère à prendre une conformation quinoïdale
[11]. Pour des dopages importants, la conduction est donc principalement due à la
délocalisation des charges sous la forme de bipolarons. Les états d'énergie bipolaroniques se
regroupent en bandes situées dans le gap (cf. Figure I-5). La conductivité du polymère est
alors proche d'une conductivité métallique.
5

Figure I-5 : Schéma de bandes du dopage progressif d'un polymère conducteur.
I-1-2) Synthèse des polythiophènes
Il existe différentes voies de polymérisation chimiques et électrochimiques pour les
polythiophènes, étudiées et optimisées depuis une vingtaine d'années.
I-1-2-1) Polymérisations chimiques
Les premières publications de synthèses chimiques de polythiophènes datent du début
des années 80 [12-14]. Le principe de la synthèse est une polycondensation par couplage
organométallique de 2,5-dihalogénothiophène (principalement diiodé ou dibromé) en
présence de magnésium et catalysée par un métal de transition.
+ MgnTHF
NiCl2(dppe)S XX ( S )n
+ n MgX2n
La réaction se passe sous atmosphère inerte dans un solvant anhydre. Les rendements
peuvent atteindre 90% lorsque les conditions sont bien contrôlées. Les polymères obtenus
6

atteignent des conductivités de l'ordre de 1 à 10 S.cm-1 lorsqu'ils sont dopés. De plus, cette
méthode permet d'obtenir des polymères uniquement couplés en 2,5.
Cependant, l'étape d'insertion du magnésium dans la liaison Carbone-Halogène est
incontrôlable. Elle conduit ainsi à des Grignards possédant aléatoirement un ou deux
groupements organomagnésiens [13]. Dans le cas du PTh, il n'y a pas de problème de
régiorégularité du fait de la symétrie de la molécule. En revanche, pour les thiophènes
monosubstitués, le monomère est asymétrique et différents couplages structuraux peuvent se
produire (cf. Figure I-6).
)( SS
nS
R R
R
)( SS
nS
R
R
)( SS
nS
R
R
R
R
)( SS
nS
R
R R
Couplage Tête-Queue-Tête-Queue (TQ-TQ) Couplage Tête-Queue-Tête-Tête (TQ-TT)
Couplage Queue-Queue-Tête-Queue (QQ-TQ) Couplage Queue-Queue-Tête-Tête (QQ-TT)
Figure I-6 : Représentation des couplages possibles durant la polymérisation de polythiophènes substitués [4].
Les couplages TQ-TQ conduisent à des polymères réguliers et plans, qui présentent des
longueurs de conjugaison importantes et donc de bonnes conductivités. Les autres couplages
provoquent des répulsions stériques entre substituants, ce qui entraîne des distorsions de la
chaîne. Il s'en suit une perte de conjugaison électronique et ainsi une perte de conductivité
[4].
Pour éviter les couplages défectueux, Mc Cullough et al. ont mis au point un protocole
de synthèse permettant d'obtenir sélectivement un unique organomagnésien (cf. Figure I-7)
[15].
7

NiCl 2(dppp)S
BrRMgBr
EtOH / CHCl 3S
RNBS
- 40°CS
RLDA / THF
Br- 60°CS
RMgBr2.OEt2
BrLi S
R
BrBrMg
Figure I-7 : Méthode Mc Cullough pour la synthèse d'un organomagnésien sélectif, permettant l'obtention d'un polymère
régulier [15].
Ce protocole conduit à des polythiophènes possédant 100 % de couplages TQ-TQ, ce
qui permet d'obtenir des conductivités de l'ordre de 100 à 1000 S.cm-1 [16]. Le grand
inconvénient de cette méthode de préparation est le nombre important d'étapes de synthèses
qui limite les rendements (de l'ordre de 10% en masse) et augmente le coût des produits.
Une autre méthode très utilisée dans la synthèse des polythiophènes pour sa simplicité et
son efficacité est l'oxydation directe du monomère par le chlorure ferrique (FeCl3) [17-19].
Les rendements massiques de polymérisation atteignent souvent plus de 80% [20-21].
+ 2n FeCl 3CHCl 3 ( S )n
+ 2n FeCl 2n + 2n HClS
R
R
La réaction doit être effectuée sous atmosphère inerte (argon ou azote) et dans un
solvant rigoureusement anhydre. En effet, le chlorure ferrique est un oxydant très puissant et
donc très sensible [22-23]. Les principaux solvants utilisés sont le tétrachlorométhane, le
chloroforme, ou le dichlorométhane [20,24-26].
La réaction a besoin de FeCl3 solide. Les solvants dans lequel il est totalement dissous
ne permettent pas la polymérisation. Une étude de Niemi et al. montre que la partie soluble du
FeCl3 (0,1M dans le CHCl3) se trouve sous forme de dimères. Cette conformation inhibe le
caractère acide de Lewis du FeCl3 et le rend inerte chimiquement [27].
Le mécanisme de réaction de cette synthèse est assez controversé [26-27]. Cependant, le
mécanisme le plus généralement admis est l'oxydation des monomères par le FeCl3
conduisant à la formation de radicaux cations qui se couplent et donnent des dimères après
déprotonation. Le potentiel d'oxydation des dimères étant inférieur à celui des monomères
[20], ils sont favorablement réoxydés en radicaux cations et peuvent se coupler de nouveau
8

avec d'autres radicaux cations. Plus la chaîne formée est longue et plus son potentiel
d'oxydation est faible, ce qui favorise la réoxydation de celle-ci [20].
Cette réaction conduit donc à des polymères de haute masse moléculaire : les masses
moyennes en nombre (Mn) se situent typiquement entre 10000 et 100000 et les masses
moyennes en poids (Mp) entre 50000 et 300000 [24, 28-31]. Les indices de polydispersité (Ip
= Mp/Mn) sont relativement élevés, indiquant une dispersité de longueurs de chaîne.
Thermodynamiquement, les couplages en positions 2 et 5 par rapport au soufre sont
largement favorisés par rapport aux couplages en position 4 [20,23,31]. Cependant, la
virulence de cette réaction donne lieu à 15 à 30 % de couplages défectueux de différentes
sortes : des couplages 2,4 qui rompent la délocalisation électronique le long de la chaîne
polymère [30-31] (~ 5%), ainsi que des couplages 2,5 irréguliers (cf. Figure I-6) qui
engendrent des distorsions de la chaîne (10-25%) [32-33]. Pour éviter ces défauts de
régiorégularité, Andersson et al. ont mis au point un protocole dans lequel l'oxydant est ajouté
par petites fractions pour limiter la violence de la réaction et ainsi favoriser la régiosélectivité
thermodynamique [34-35]. Ils obtiennent ainsi des couplages réguliers TQ-TQ de plus de
90% [35].
Les polymères synthétisés par cette voie sont obtenus à l'état dopé, le potentiel
d'oxydation du monomère étant supérieur au potentiel de dopage du polymère [23,35]. Les
anions dopants sont principalement sous forme FeCl4- ou Cl- [36-37]. Il faut donc procéder à
un lavage avec différents solvants comme le méthanol ou l'acétonitrile pour extraire les ions
dopants et obtenir le polymère à l'état neutre. Généralement, après lavage il reste des quantités
en fer et en chlore inférieures à 1 % en poids [23-24].
D'autres méthodes chimiques de polymérisation ont été rapportées, telles que
l'oxydation par le CuClO4, en milieu organique [38] ou encore par le AsF5 en phase gazeuse
[39].
I-1-2-2) Polymérisations électrochimiques
Les méthodes électrochimiques de synthèse des polythiophènes sont très utilisées
[40-45]. Elles permettent une grande précision de contrôle de la réaction et donc des
propriétés des polymères obtenus.
9
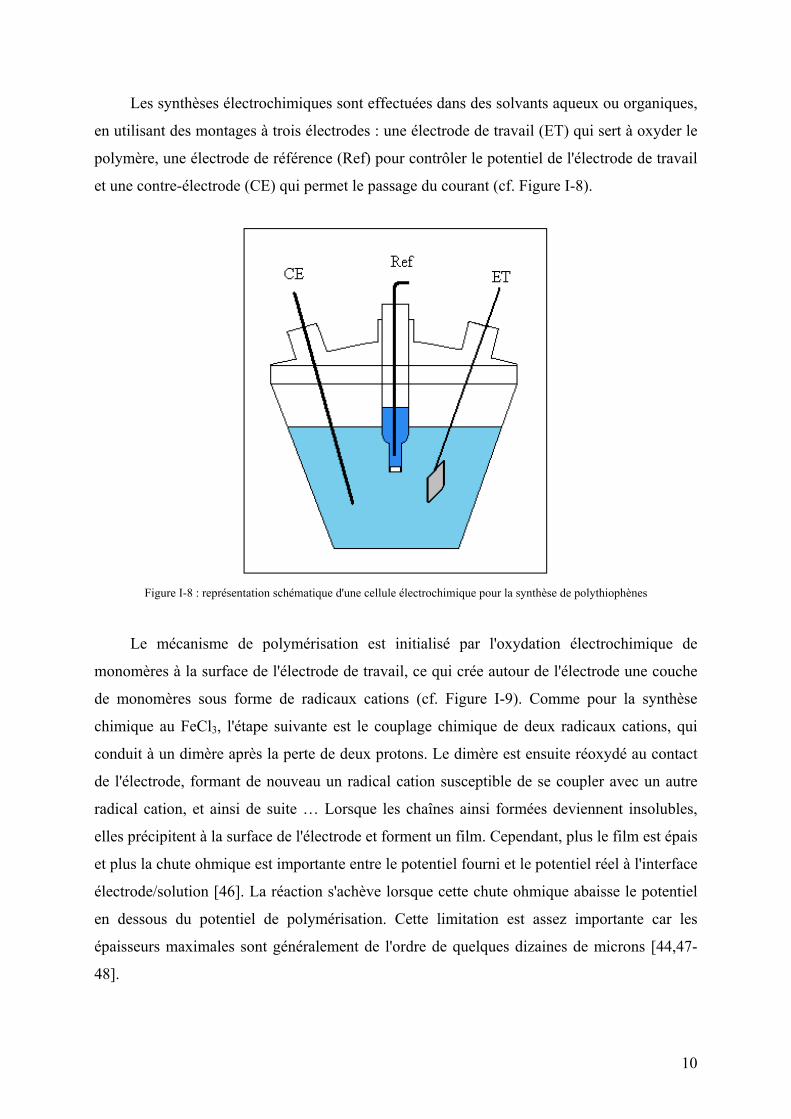
Les synthèses électrochimiques sont effectuées dans des solvants aqueux ou organiques,
en utilisant des montages à trois électrodes : une électrode de travail (ET) qui sert à oxyder le
polymère, une électrode de référence (Ref) pour contrôler le potentiel de l'électrode de travail
et une contre-électrode (CE) qui permet le passage du courant (cf. Figure I-8).
Figure I-8 : représentation schématique d'une cellule électrochimique pour la synthèse de polythiophènes
Le mécanisme de polymérisation est initialisé par l'oxydation électrochimique de
monomères à la surface de l'électrode de travail, ce qui crée autour de l'électrode une couche
de monomères sous forme de radicaux cations (cf. Figure I-9). Comme pour la synthèse
chimique au FeCl3, l'étape suivante est le couplage chimique de deux radicaux cations, qui
conduit à un dimère après la perte de deux protons. Le dimère est ensuite réoxydé au contact
de l'électrode, formant de nouveau un radical cation susceptible de se coupler avec un autre
radical cation, et ainsi de suite … Lorsque les chaînes ainsi formées deviennent insolubles,
elles précipitent à la surface de l'électrode et forment un film. Cependant, plus le film est épais
et plus la chute ohmique est importante entre le potentiel fourni et le potentiel réel à l'interface
électrode/solution [46]. La réaction s'achève lorsque cette chute ohmique abaisse le potentiel
en dessous du potentiel de polymérisation. Cette limitation est assez importante car les
épaisseurs maximales sont généralement de l'ordre de quelques dizaines de microns [44,47-
48].
10

.+
+
.- e -
2
+
.++
S
SH
H
S
S
S
S
S+ + 2 H
S
S - e -
S
S H
.+
S
S H .+S
+ ...
Figure I-9 : Mécanisme de polymérisation E (CE)n [40].
La structure des films de polymère dépend fortement des conditions de synthèse :
électrolyte, méthode électrochimique, substrat, … [40,49]
Différents substrats sont utilisés comme électrode de travail. Les substrats ITO (oxydes
d'étain et d'indium) déposés sur verres ainsi que des métaux nobles (platine et or) sont
préférablement utilisés pour l'étude des polymères ainsi que pour les caractérisations [41-43].
En revanche, pour obtenir des électrodes aux structures plus ouvertes, certains utilisent des
substrats carbonés qui présentent des structures fibrillaires [44-45].
Les polythiophènes présentant une forte hydrophobicité, ils sont rarement synthétisés en
milieu aqueux. On trouve dans la littérature des exemples de synthèse en milieu acide, mais
les polymères formés possèdent des longueurs de chaîne moins importantes que ceux
synthétisés en milieu organique [50]. Les milieux les plus souvent utilisés sont des solvants
aprotiques comme l'acétonitrile, le carbonate de propylène ou le nitrobenzène. Pour obtenir
des polymères de bonne qualité, il faut que l'électrolyte soit absolument anhydre [40].
De la même façon que pour les synthèses chimiques par oxydation, les polymères sont
synthétisés à l'état dopé. Les anions dopants ont donc une forte influence sur la morphologie
des polymères [40]. Dans la plupart des cas, l'ion dopant qui a servi à la polymérisation est
l'ion le plus adéquat pour doper ensuite le polymère, ce dernier ayant adapté sa structure à ce
dopant pendant sa formation [51].
11

Les méthodes électrochimiques les plus couramment employées pour la formation de
films de polymères à partir d'une solution de monomère sont la voltamétrie cyclique, la
chronopotentiométrie ou le maintien potentiostatique. Ces méthodes permettent de contrôler
très précisément la morphologie des polymères, ainsi que la masse et l'épaisseur déposée. La
voltamétrie cyclique est intéressante pour observer la progression de la réaction [47]. Le choix
du courant appliqué en chronopotentiométrie permet d'obtenir soit des films fins et
homogènes (faibles densités de courant), soit des structures nodulaires (fortes densités de
courant) [5,52-53]. La synthèse par maintien potentiostatique peut être effectuée à un seul
potentiel ou par étapes successives à différents potentiels et permet d'obtenir des films fins et
homogènes [8,48].
Le mécanisme des synthèses électrochimiques n'est pas sélectif au niveau des couplages
structuraux et les polythiophènes synthétisés par électrooxydation présentent 20% à 30% de
couplages défectueux avec une proportion importante de couplages 2,4 [4,33,54]. Ils
possèdent des degrés de cristallinité inférieurs aux polythiophènes synthétisés chimiquement
[54].
I-1-3) Propriétés des polythiophènes
I-1-3-1) Propriétés chimiques et optiques
Macroscopiquement, les polythiophènes sont des polymères principalement amorphes,
bien que des zones cristallines soient observées lorsque les polymères sont particulièrement
réguliers [52,55-57]. Comme il a été vu auparavant, ils se présentent sous différentes formes
selon la méthode de synthèse utilisée.
Les synthèses chimiques conduisent à des polymères sous forme de poudres très fines
dont les couleurs varient du rouge au noir. Ils sont infusibles et insolubles dans tous les
solvants aqueux ou organiques connus. En revanche, en greffant sur le noyau thiophène de
longues chaînes (alkyles ≥ butyl, alkoxy …), les polythiophènes peuvent être dissous dans
certains solvants, ce qui permet d'en faire des films plus ou moins épais, ou encore des gels
[22,58-62]. Les principales études structurales ont été effectuées sur ce type de polymères
solubles.
12

Les synthèses électrochimiques donnent lieu à des dépôts de polymères sur les
électrodes. Selon les conditions de synthèse, les films peuvent être plus ou moins épais, plus
ou moins homogènes macroscopiquement, plus ou moins ordonnés microscopiquement.
De nombreuses techniques sont utilisées pour caractériser les propriétés chimiques et
optiques des polythiophènes, ainsi que pour comprendre les mécanismes de conduction et les
paramètres qui influencent ces propriétés.
La chromatographie de perméation de gel (GPC) et la chromatographie d'exclusion
stérique (SEC) permettent de mesurer les masses moyennes en nombre (Mn) et en poids (Mp)
de ces polymères [24-25,28-29,46]. Cependant, les valeurs sont déterminées à l'aide de
courbes de calibration du polystyrène et sont donc approximatives. Selon les polymères et les
conditions de synthèse, Mn varie entre 1000 et 100000 et Mp entre 3000 et 500000. L'indice
de polydispersité (Ip = Mp/Mn) est souvent important, ce qui montre une dispersité de
longueurs de chaîne [46].
Les spectroscopies de vibration (infrarouge et Raman) permettent d'effectuer des
analyses structurales des polythiophènes [8,13-14,22,28,46-47,63-64]. Elles permettent
d'apprécier la régiorégularité du polymère ainsi que sa masse moyenne en mesurant la
différence d'aire sous les pics de vibration de déformation hors du plan des liaisons C-H
situées en position α et β du soufre [23,65]. En effet, un polymère régulier ayant un fort degré
de polymérisation possédera de rares liaisons C-H en α du soufre tandis qu'un polymère
pourvu de défauts structuraux (couplages 2,4) possédera des liaisons C-H en α du soufre en
quantités non négligeables.
Par ailleurs, lorsqu'elles sont utilisées in situ, ces techniques permettent de suivre les
changements structuraux qui interviennent lors du dopage des polymères : la délocalisation
des porteurs de charges ainsi que leur type (solitons, polarons, bipolarons) peuvent ainsi être
observés par le décalage des fréquences de vibration des liaisons C=C et C-C [8,63,66]. Ces
études ont mis en évidence la prépondérance de la conformation quinoïdale de la chaîne
lorsque le polymère est dopé [67]. La désactivation de certains polythiophènes lors de
dopages successifs a aussi été étudiée par spectroscopie in-situ [67-71]. Ces auteurs ont mis
en évidence l'apparition de bandes de vibration C-O et C=O, et ont proposé des mécanismes
de rupture de conjugaison par l'attaque d'eau ou des solvants organiques utilisés comme
électrolytes (acétonitrile, carbonate de propylène).
La spectroscopie d'absorption UV-visible est très efficace pour caractériser les
propriétés optiques des polythiophènes [18,24-25,29,34-35,54,72-74] et mesurer le gap
13

d'énergie correspondant à la transition π-π* dans ces polymères [22,32,62]. Certains
polythiophènes, comme les poly(3-alkoxy-4-méthylthiophène) possèdent d'intéressantes
propriétés de thermochromisme (cf. Figure I-10).
Figure I-10 : Phénomène de thermochromisme des poly(3-alkoxy-4-méthylthiophènes) [73].
Leur couleur naturelle à l'état solide est rouge-violet, ce qui correspond à une absorption
vers 550 nm. Cependant, lorsque ces polymères sont chauffés, une deuxième absorption
apparaît à 400 nm tandis que la première bande d'absorption disparaît. Ces polythiophènes
prennent alors une couleur jaune. Malgré un certain hystérésis, cette transition est réversible,
ce qui montre que ce phénomène n'est pas dû à une dégradation du polymère. Elle est plutôt
attribuée à un changement de conformation de la chaîne polymère, passant d'une structure
plane à température ambiante à une structure non planaire à haute température. Cette structure
non planaire induit aussi une baisse importante de la conductivité (augmentation du gap
14
d'énergie). Ces phénomènes de thermochromisme n'apparaissent que plus faiblement dans les
polyalkylthiophènes [28,62,73,75].
En revanche, des changements structuraux entraînant des modifications optiques
apparaissent pour la plupart des polythiophènes lors de leur dopage. La Figure I-11 montre le
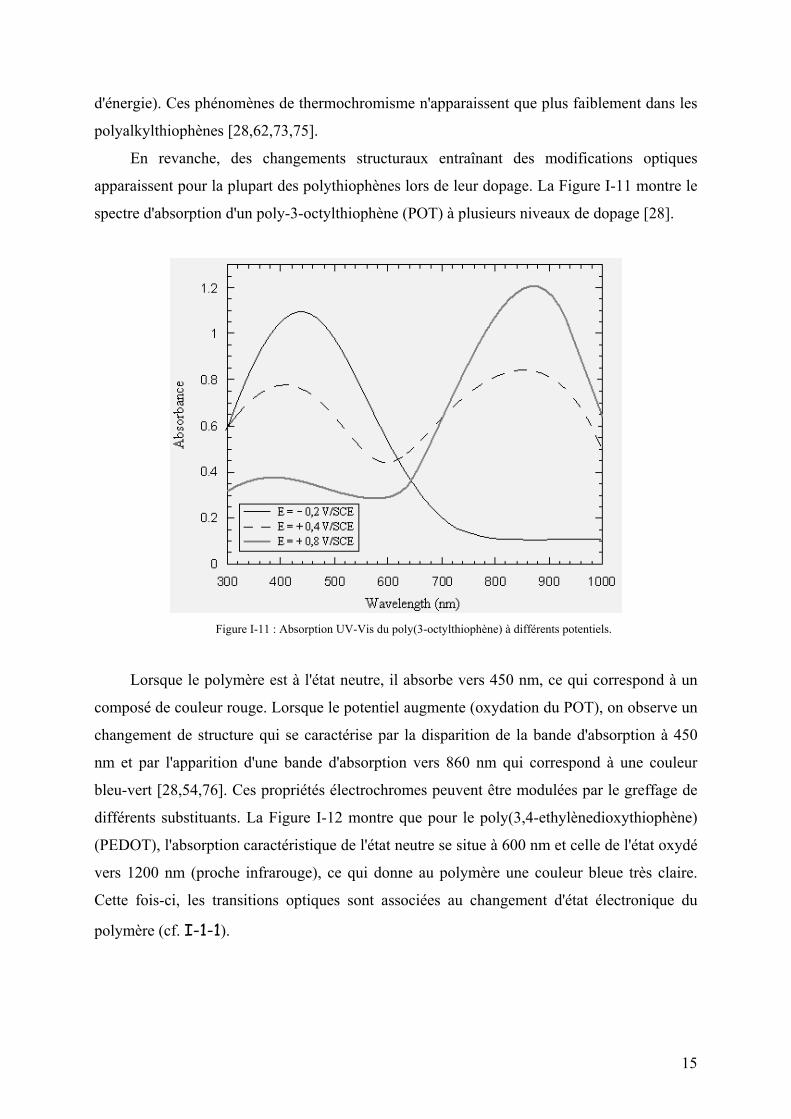
spectre d'absorption d'un poly-3-octylthiophène (POT) à plusieurs niveaux de dopage [28].
Figure I-11 : Absorption UV-Vis du poly(3-octylthiophène) à différents potentiels.
Lorsque le polymère est à l'état neutre, il absorbe vers 450 nm, ce qui correspond à un
composé de couleur rouge. Lorsque le potentiel augmente (oxydation du POT), on observe un
changement de structure qui se caractérise par la disparition de la bande d'absorption à 450
nm et par l'apparition d'une bande d'absorption vers 860 nm qui correspond à une couleur
bleu-vert [28,54,76]. Ces propriétés électrochromes peuvent être modulées par le greffage de
différents substituants. La Figure I-12 montre que pour le poly(3,4-ethylènedioxythiophène)
(PEDOT), l'absorption caractéristique de l'état neutre se situe à 600 nm et celle de l'état oxydé
vers 1200 nm (proche infrarouge), ce qui donne au polymère une couleur bleue très claire.
Cette fois-ci, les transitions optiques sont associées au changement d'état électronique du
polymère (cf. I-1-1).
15
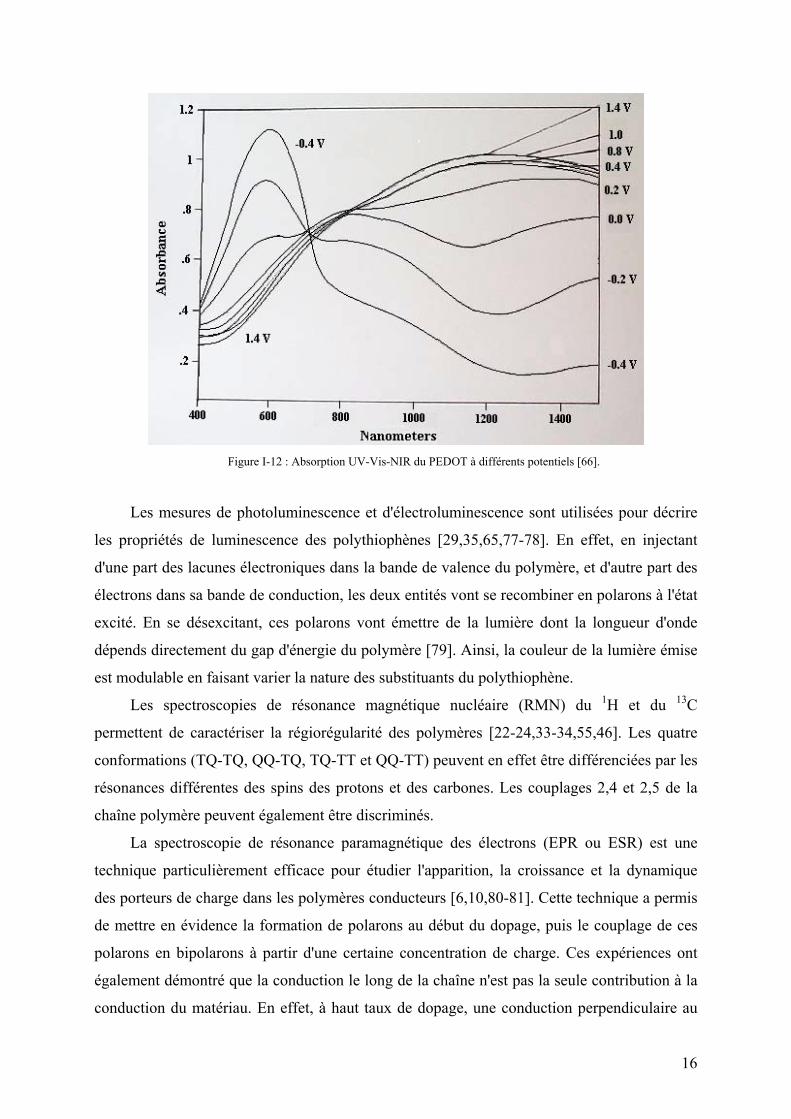
Figure I-12 : Absorption UV-Vis-NIR du PEDOT à différents potentiels [66].
Les mesures de photoluminescence et d'électroluminescence sont utilisées pour décrire
les propriétés de luminescence des polythiophènes [29,35,65,77-78]. En effet, en injectant
d'une part des lacunes électroniques dans la bande de valence du polymère, et d'autre part des
électrons dans sa bande de conduction, les deux entités vont se recombiner en polarons à l'état
excité. En se désexcitant, ces polarons vont émettre de la lumière dont la longueur d'onde
dépends directement du gap d'énergie du polymère [79]. Ainsi, la couleur de la lumière émise
est modulable en faisant varier la nature des substituants du polythiophène.
Les spectroscopies de résonance magnétique nucléaire (RMN) du 1H et du 13C
permettent de caractériser la régiorégularité des polymères [22-24,33-34,55,46]. Les quatre
conformations (TQ-TQ, QQ-TQ, TQ-TT et QQ-TT) peuvent en effet être différenciées par les
résonances différentes des spins des protons et des carbones. Les couplages 2,4 et 2,5 de la
chaîne polymère peuvent également être discriminés.
La spectroscopie de résonance paramagnétique des électrons (EPR ou ESR) est une
technique particulièrement efficace pour étudier l'apparition, la croissance et la dynamique
des porteurs de charge dans les polymères conducteurs [6,10,80-81]. Cette technique a permis
de mettre en évidence la formation de polarons au début du dopage, puis le couplage de ces
polarons en bipolarons à partir d'une certaine concentration de charge. Ces expériences ont
également démontré que la conduction le long de la chaîne n'est pas la seule contribution à la
conduction du matériau. En effet, à haut taux de dopage, une conduction perpendiculaire au
16

matériau (interchaîne) existe, bien que beaucoup plus faible que la conduction intrachaîne. Par
exemple, dans le PTh, pour une conductivité globale de 0,6 S.cm-1, la conductivité intrachaîne
a été calculée à 130 S.cm-1 pour une conductivité perpendiculaire de 0,006 S.cm-1 [80]. Cette
conduction interchaîne s'opère par transfert de charges entre les chaînes [82].
La diffraction de rayons X permet de déterminer les géométries intra et interchaînes des
polymères ainsi que de distinguer les zones amorphes et les zones cristallines [23-24,83-85].
Des modèles ont ainsi pu être proposés pour expliquer les structures des polythiophènes. Il en
ressort que plus les polymères sont réguliers et plus ils ont une structure plane. Cela permet
aux chaînes de s'empiler pour donner des arrangements cristallins de dimensions variables
selon la taille du substituant [23,54-55,86]. En revanche, les défauts de couplage α-β ainsi que
les défauts structuraux (TT-QQ, …) créent des distorsions des chaînes, ce qui limite
l'arrangement cristallin [4,86].
Les analyses thermiques (DSC, ATG) apportent des informations sur la tenue en
température des polymères [12,24,28]. Mis à part les décompositions de certains substituants
sensibles à la chaleur (alkoxy, acides carboxyliques, éthers) [23,87], les polythiophènes se
décomposent entre 200°C et 400°C selon la pureté du polymère, lorsqu'ils sont chauffés à
l'air. Sous atmosphère d'azote, leur décomposition apparaît à plus de 400°C [14]. Des analyses
plus fines (DSC modulée) permettent de déterminer des températures de transition vitreuse
pour les polythiophènes possédant de longues chaînes greffées [23]. Celles-ci varient entre -
20°C et 70°C selon la longueur des chaînes greffées. Les polythiophènes semicristallins
montrent des températures de fusion vers 150-200°C [56].
De nombreuses autres techniques d'analyse sont utilisées pour décrire la structure des
polythiophènes ainsi que pour comprendre les mécanismes de dopage et de conduction dans
ces polymères et toutes les propriétés induites.
La microbalance à quartz (EQCM) permet d'étudier les mécanismes d'électrodéposition
ainsi que le dopage des polythiophènes en analysant les variations de masse du polymère
[1,88-91].
Le microscope électronique à balayage permet d'obtenir des images de la surface des
polythiophènes à quelques micromètres près. On peut ainsi étudier les effets macroscopiques
des processus de dopage sur la géométrie des polymères (gonflement, dégradations) [44,52].
La microscopie à force atomique (AFM) permet d'obtenir des images de la surface des
polymères au nanomètre près. Les études d'électropolymérisation utilisant cette technique
montrent que le polymère se forme en nodules qui se rejoignent peu à peu pour former le film.
Cependant, la surface du film garde toujours une structure nodulaire avec des tailles variant
17

selon le polymère de 50 à 100 nm de diamètre [8,53,92-93]. Une étude AFM in-situ du
dopage du PMeT montrent que l'insertion d'ions dopants est accompagnée de modifications
de la structure du polymère avec un retard qui peut atteindre plusieurs minutes et qui
correspond à des phénomènes de relaxation des chaînes [94].
La spectroscopie photoélectronique UV (UPS) permet d'obtenir des informations sur les
énergies de liaison des polythiophènes (bande de valence), et donc indirectement sur la
structure chimique du polymère ainsi que sur l'état électronique des orbitales. Cependant,
cette technique nécessite la comparaison des spectres expérimentaux avec des spectres
théoriques obtenus par modélisation [62,95].
Les spectroscopies Mössbauer (129I, 57Fe, …) permettent d'identifier le degré
d'oxydation des ions dopants. Dans le cas de PTh et de PMeT dopés à l'iode, cette technique a
permis d'identifier deux sortes d'ions dopants : I-3 et I-5. Dans le cas d'un dopage au FeCl3, le
seul ion dopant est FeCl4- [37].
Il existe donc de nombreuses techniques permettant d'étudier les différentes propriétés
chimiques et optiques des polythiophènes. Cependant, leurs caractéristiques les plus
intéressantes et les plus étudiées sont les propriétés électrochimiques.
I-1-3-2) Propriétés électrochimiques
La principale caractéristique des polythiophènes est de posséder différents états
électroniques selon le potentiel auquel ils sont soumis. A l'état neutre, ils possèdent
d'intéressantes propriétés isolantes (conductivité σ ~10-11 S.cm-1) [12]. En revanche, une fois
dopés, ils changent de structure et deviennent d'excellents conducteurs électroniques (jusqu'à
103 S.cm-1) [16].
Le dopage des polythiophènes s'effectue soit en injectant, soit en arrachant des électrons
de la chaîne polymère. Il s'en suit une délocalisation de charges négatives (électrons) ou de
charges positives ("trous"). Plus la longueur de conjugaison est grande, plus la délocalisation
est importante et meilleure est la conductivité. En revanche, les couplages défectueux 2,4
formés au moment de la polymérisation sont aussi stables thermodynamiquement que les
couplages 2,5 et rompent la conjugaison [11]. La planéité de la chaîne est également très
importante pour obtenir une parfaite hybridation des orbitales π et donc une délocalisation
optimale des charges sur toute la longueur de la chaîne. Lorsque le polymère est dopé, des
18

ions viennent s'insérer le long des chaînes pour préserver localement l'électroneutralité. La
Figure I-13 représente les voltamogrammes classiques des dopages négatif et positif d'un
PTh.
-30
-20
-10
0
10
20
-2.5 -2 -1.5 -1 -0.5 0 0.5 1
i (m
A)
E (V/Ag+/Ag)
< >
Dopage positifDédopage négatif
Dopage négatif Dédopage positif
Figure I-13 : Voltamogramme représentant les dopages négatif et positif d'un polythiophène.
La vague de réduction à bas potentiel (cf. Equation I-1) correspond au dopage négatif
du polymère, c'est à dire à l'injection d'électrons dans les chaînes polymères. Pour maintenir
l'électroneutralité, des cations de l'électrolyte s'insèrent dans l'électrode. Le polymère devient
alors conducteur. On dit qu'il est dopé n.
Polym(0) + e- + C+ Polym-,C+ (Eq. I-1)
Le pic d'oxydation associé correspond au dédopage négatif du polymère
(cf. Equation I-1), c'est à dire à l'extraction des électrons injectés durant le dopage. Les
cations insérés dans l'électrode se désinsèrent alors. Le polymère revient à son état neutre et
isolant.
Le pic d'oxydation à haut potentiel (cf. Equation I-2) correspond à l'extraction
d'électrons des chaînes polymères et à l'insertion d'anions le long de celles-ci pour préserver
19

l'électroneutralité. Le polymère change de structure et une conduction se crée par la
délocalisation de charges positives le long des chaînes.
Polym(0) + A- Polym+,A- + e- (Eq. I-2)
Le pic de réduction correspond à la réinjection des électrons dans les chaînes et au rejet
dans l'électrolyte des anions insérés lors du dopage (cf. Equation I-2) . Le polymère revient
alors à son état neutre et isolant électronique.
Des études ont montré que la limitation cinétique des dopages était la diffusion des ions
le long des chaînes plutôt que la délocalisation électronique des charges sur celles-ci [96-97].
Les polythiophènes synthétisés par oxydation (FeCl3 ou électrooxydation) sont obtenus
dopés p, ce qui prépare leur structure à cet état oxydé [51]. Ce dopage est donc facilement
réalisable et possède une bonne réversibilité coulombique [40].
Le taux dopage maximum théorique est d'une charge pour une unité monomère.
Cependant, ce taux n'est jamais atteint, et on observe généralement une limite de dopage d'une
charge pour environ trois unités monomères (taux de 0,3) [55,71,92,98-100]. Cela correspond
à des quantités d'électricité de l'ordre de 40 à 50 mAh/g selon la masse molaire des
polythiophènes.
En revanche, le dopage négatif est beaucoup plus difficile à obtenir [40]. Il est
également moins réversible, moins stable que le dopage positif et les quantités d'électricités
mises en jeu sont largement inférieures [101].
Des études ont donc été menées pour obtenir des dérivés du PTh possédant une bonne
électroactivité négative [1,44,102-106]. Dans cette optique, les recherches se sont orientées
vers les polythiophènes substitués par des dérivés phénylés. Ces substituants possèdent en
effet un caractère attracteur d'électrons, ce qui déplace les processus de dopage vers des
potentiels plus positifs. Le dopage négatif devient ainsi plus facilement réalisable.
Les techniques utilisées pour caractériser les propriétés électrochimiques des
polythiophènes sont diverses.
La voltampérométrie cyclique ou voltamétrie cyclique (CV) permet de situer les
potentiels des processus de dopage, de quantifier les quantités d'électricité mises en jeu,
20

d'obtenir des informations sur les processus limitatifs du dopage (diffusion des ions, transfert
électronique) ainsi que sur la cinétique des réactions [42-43,45-46,89-90,96,103,105,107-
109].
Le cyclage galvanostatique permet, en parallèle avec la CV, de tester les variations des
propriétés de dopage des polythiophènes de lors de multiples cycles de dopage/dédopage et
d'utiliser le modèle électrostatique pour décrire le dopage des polymères [44,110] (cf. I-2-4).
La spectroscopie d'impédance complexe (EIS) permet d'obtenir des informations sur les
propriétés des phénomènes de conduction et de diffusion dans les polythiophènes
[96,111-117]. Les résultats de EIS sont cependant assez délicats à interpréter, car il faut les
comparer à des modèles de circuits électriques théoriques qui peuvent être très complexes
(cf. II-2-2-3).
Les analyses de conductivité in-situ sont très intéressantes pour étudier les mécanismes
de conduction au cours du dopage des polymères [5,66,90,99].
I-1-3) Applications des polythiophènes
Les propriétés isolantes des polythiophènes à l'état neutre ainsi que leurs excellentes
propriétés de tenue à la température en font de bons matériaux pour les revêtements
antistatiques ainsi que pour le blindage électromagnétique [6].
Leurs propriétés de semi-conducteurs sont étudiées dans des systèmes de jonctions p-n
et de jonctions métal-semiconducteur [40-41]
L'application des propriétés d'électroluminescence des polythiophènes est beaucoup
étudiée dans des systèmes de diodes électroluminescentes et de systèmes photovoltaïques, en
parallèle avec les polyparaphénylènes vinylènes (PPV) [41,78,118-120].
Les propriétés optiques sont intéressantes pour les applications dans des systèmes
électrochromes (écrans plats, verres photosensibles, …) [40-41,121].
Les quantités d'électricité que les polythiophènes sont capables d'incorporer lors de leur
dopage (50 mAh/g pour le dopage positif et 30 mAh/g pour le dopage négatif) sont très
intéressantes pour les systèmes de stockage de l'énergie. Ils sont ainsi étudiés comme cathodes
dans des systèmes lithium [40,98,122-123]. Une des dernières applications envisagée pour les
polythiophènes est leur utilisation comme matières actives dans des électrodes de
supercondensateurs [21,89,102-105,124-127]. C'est sur quoi nous avons plus particulièrement
travaillé et que nous vous proposons de présenter maintenant.
21

I-2) Les supercondensateurs
Il existe différents types de systèmes électrochimiques de stockage de l'énergie. Ils se
divisent en deux grandes familles : les accumulateurs, qui possèdent une forte densité
d'énergie et une faible densité de puissance (NiMH, NiCd, Li-ion, Pb-PbO2, …), et les
condensateurs diélectriques, qui au contraire possèdent une forte densité de puissance et une
faible densité d'énergie. Les supercondensateurs (SC), apparus depuis une vingtaine d'années,
sont des systèmes dont les caractéristiques se situent entre celles des accumulateurs et celles
des condensateurs, comme le montre la Figure I-14.
Figure I-14 : Diagramme de Ragone des différents systèmes électrochimiques de stockage d'énergie [125]
Ils présentent ainsi un grand intérêt, possédant une puissance 10 à 100 fois supérieure à
celle des accumulateurs et une énergie 10 à 100 fois supérieure à celle des condensateurs
diélectriques. Les principales applications possibles pour ces systèmes sont le démarrage
automobile, et plus généralement l'apport de puissance nécessaire à l'alimentation des
fonctions électriques de plus en plus nombreuses dans les véhicules. Ils sont aussi étudiés
pour la traction automobile des véhicules hybrides. D'autres applications intéressantes
22

concernent la sauvegarde de mémoire informatique, le support et la compensation de
puissance ainsi que de nombreuses applications militaires et spatiales.
Il existe trois types de supercondensateurs : les supercondensateurs à base de charbons
actifs, les supercondensateurs à base d'oxydes métalliques et les supercondensateurs à base de
polymères conducteurs électroniques.
I-2-1) Les supercondensateurs à base de charbons actifs
Ce premier type de supercondensateurs utilise comme matière active des charbons
activés. Le stockage d'énergie est réalisé par l'organisation de charges d'espace à la surface de
l'électrode (cf. Figure I-15).
Figure I-15 : Principe de fonctionnement d'un supercondensateur à base de charbons actifs
Il s'agit donc uniquement d'interactions électrostatiques entre les ions de l'électrolyte et
les charges de surface des électrodes, comme pour les condensateurs diélectriques. Les
charbons actifs possèdent des surfaces BET de 1000 à 2500 m2/g, ce qui permet d'obtenir une
surface de contact très importante entre l'électrode et l'électrolyte. Ces charbons possèdent
ainsi des capacités de 100 à 200 F/g en milieu aqueux et de 50 à 150 F/g en milieu organique.
En théorie, leurs surfaces BET devraient permettre à ces carbones d'atteindre des capacités
supérieures mais la solvatation des ions dans l'électrolyte limite l'accès à la totalité de la
porosité.
23

Ces supercondensateurs fonctionnent sur le principe de la double-couche
électrochimique [128], l'interface électrode/électrolyte fonctionnant comme un condensateur
diélectrique. Lorsqu'on applique une différence de potentiel, des charges vont s'accumuler de
part et d'autre de cet interface : des charges électroniques du côté de l'électrode et des charges
ioniques du côté de l'électrolyte (cf. Figure I-16).
Figure I-16 : Double couche électrochimique selon Gouy-Chapman-Stern [129].
La première couche ionique, constituée des ions de charge opposée à l'électrode, est
appelée la couche de Helmholtz. Juste au-delà de cette couche, on trouve la couche diffuse de
Gouy-Chapman. Elle est composée des co-ions qui accompagnent les ions de la première
couche. L'ensemble de ces deux couches forme la double couche électrochimique, d'une
dizaine d'Å d'épaisseur.
Des études ont montré que les sels qui permettent d'obtenir les électrolytes organiques
les plus conducteurs sont les sels d'ammonium quaternaires [130-133], qui conviennent par
ailleurs parfaitement au dopage des polythiophènes.
Les charbons actifs se présentent sous différentes formes (poudres, fibres, tissus,
carbone vitreux) et leur grand avantage est leur faible coût lié à une production industrielle.
Ils sont en effet déjà connus et utilisés pour de nombreuses autres applications (dépollution,
filtrage, catalyse hétérogène) [134]. Les SC carbone/carbone sont les supercondensateurs les
plus développés industriellement et il existe une panoplie de brevets déposés depuis une
vingtaine d'années : citons par exemple Matsushita (Panasonic) [135-137], Asachi Glass [130]
24
et NEC [138] au Japon, Maxwell [139-140] et Polystor aux Etats-Unis, Econd, ESMA et
ELIT en Russie, Cap XX en Australie, SuperFarad en Suède, EPCOS (Matsushita & Siemens)
en Allemagne, SAFT en France [141-142], …
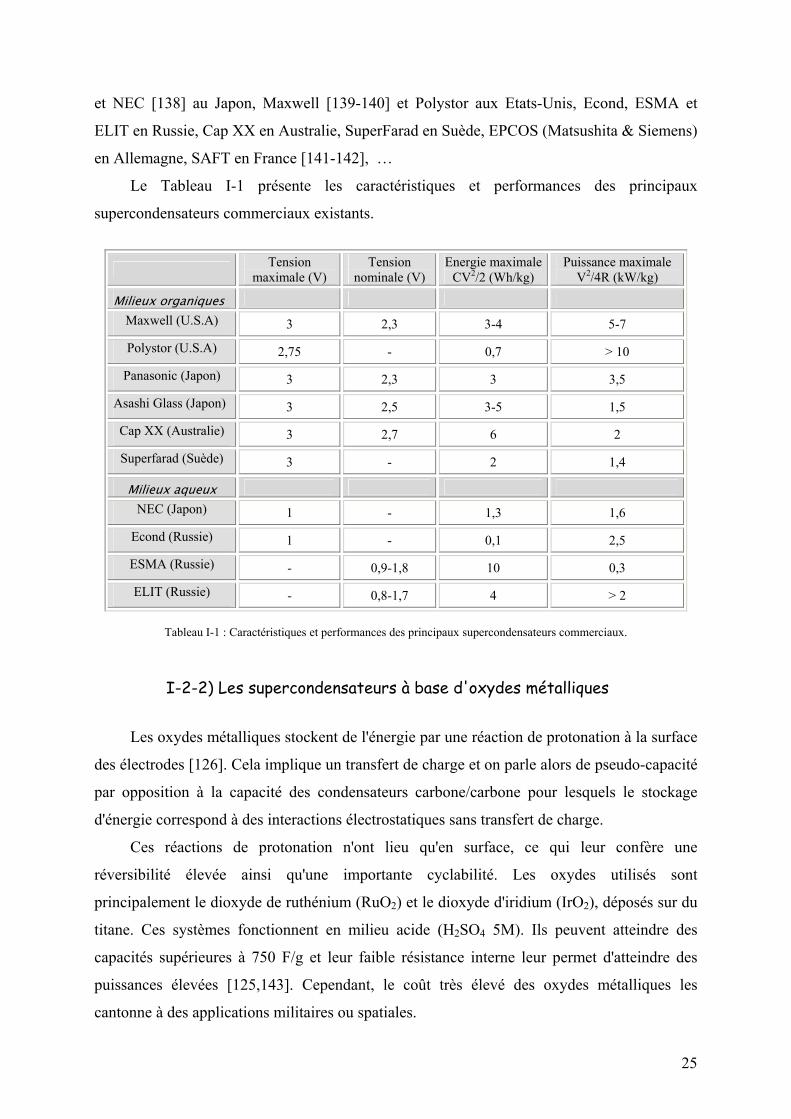
Le Tableau I-1 présente les caractéristiques et performances des principaux
supercondensateurs commerciaux existants.
Tension maximale (V)
Tension nominale (V)
Energie maximale CV2/2 (Wh/kg)
Puissance maximale V2/4R (kW/kg)
Milieux organiques Maxwell (U.S.A) 3 2,3 3-4 5-7
Polystor (U.S.A) 2,75 - 0,7 > 10
Panasonic (Japon) 3 2,3 3 3,5
Asashi Glass (Japon) 3 2,5 3-5 1,5
Cap XX (Australie) 3 2,7 6 2
Superfarad (Suède) 3 - 2 1,4
Milieux aqueux NEC (Japon) 1 - 1,3 1,6
Econd (Russie) 1 - 0,1 2,5
ESMA (Russie) - 0,9-1,8 10 0,3
ELIT (Russie) - 0,8-1,7 4 > 2
Tableau I-1 : Caractéristiques et performances des principaux supercondensateurs commerciaux.
I-2-2) Les supercondensateurs à base d'oxydes métalliques
Les oxydes métalliques stockent de l'énergie par une réaction de protonation à la surface
des électrodes [126]. Cela implique un transfert de charge et on parle alors de pseudo-capacité
par opposition à la capacité des condensateurs carbone/carbone pour lesquels le stockage
d'énergie correspond à des interactions électrostatiques sans transfert de charge.
Ces réactions de protonation n'ont lieu qu'en surface, ce qui leur confère une
réversibilité élevée ainsi qu'une importante cyclabilité. Les oxydes utilisés sont
principalement le dioxyde de ruthénium (RuO2) et le dioxyde d'iridium (IrO2), déposés sur du
titane. Ces systèmes fonctionnent en milieu acide (H2SO4 5M). Ils peuvent atteindre des
capacités supérieures à 750 F/g et leur faible résistance interne leur permet d'atteindre des
puissances élevées [125,143]. Cependant, le coût très élevé des oxydes métalliques les
cantonne à des applications militaires ou spatiales.
25
I-2-3) Les supercondensateurs à base de polymères conducteurs
Les supercondensateurs à base de polymères conducteurs stockent de l'énergie par des
processus de dopage des polymères (cf. I-1-3-2). Ceux-ci possèdent des capacités entre 200
et 300 F/g. Cependant, le dopage impliquant un transfert de charge, il s'agit encore une fois de
pseudo-capacité. Ces systèmes de supercondensateurs sont les plus récents et ne sont pas
encore au niveau du développement industriel car de nombreux problèmes subsistent, parmi
lesquels le coût de synthèse, la mise en œuvre, la cyclabilité …
Il existe trois types d'architectures possibles selon que l'un ou que les deux processus de
dopage (positif et négatif) sont utilisés pour stocker l'énergie.
Le système de type I ne met en jeu que le dopage positif des polymères. Les polymères
utilisés sont alors le polypyrrole [144-145], la polyaniline [146], ou le poly(3,4-
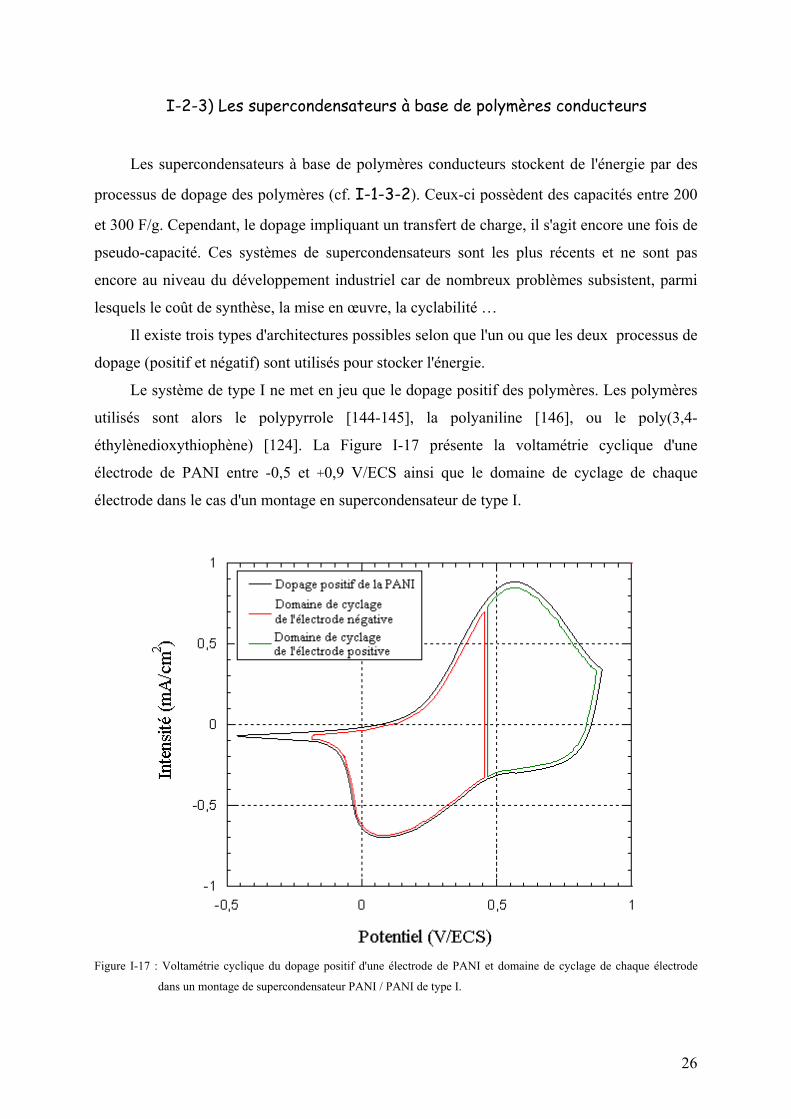
éthylènedioxythiophène) [124]. La Figure I-17 présente la voltamétrie cyclique d'une
électrode de PANI entre -0,5 et +0,9 V/ECS ainsi que le domaine de cyclage de chaque
électrode dans le cas d'un montage en supercondensateur de type I.
Figure I-17 : Voltamétrie cyclique du dopage positif d'une électrode de PANI et domaine de cyclage de chaque électrode
dans un montage de supercondensateur PANI / PANI de type I.
26
Lorsque le système est déchargé, chacune des électrodes est à moitié chargée. Lors de
la charge, l'électrode positive se dope totalement pendant que l'électrode négative se dédope.
Lorsque le système est chargé, la positive est totalement dopée tandis que la négative est
totalement dédopée. Cela représente un inconvénient car l'électrode négative est alors assez
résistive et la chute ohmique au début de la décharge du système est importante.
La tension aux bornes est d'environ 1 V lorsque le système est chargé. Ces systèmes
peuvent donc être assemblés soit en milieu organique, soit en milieu aqueux [146]. Leurs
performances sont toutefois limitées par la faible tension. D'autre part, ce concept ne permet
d'utiliser que la moitié de la capacité de charge des électrodes (cf. Figure I-17).
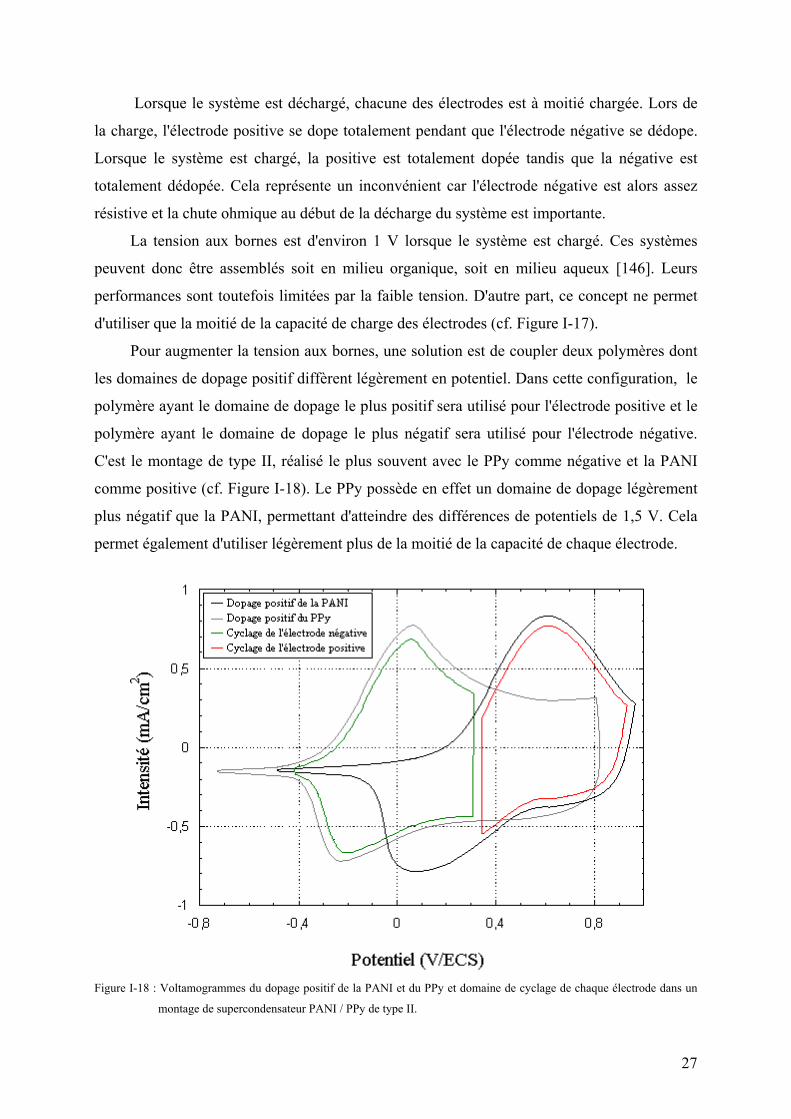
Pour augmenter la tension aux bornes, une solution est de coupler deux polymères dont
les domaines de dopage positif diffèrent légèrement en potentiel. Dans cette configuration, le
polymère ayant le domaine de dopage le plus positif sera utilisé pour l'électrode positive et le
polymère ayant le domaine de dopage le plus négatif sera utilisé pour l'électrode négative.
C'est le montage de type II, réalisé le plus souvent avec le PPy comme négative et la PANI
comme positive (cf. Figure I-18). Le PPy possède en effet un domaine de dopage légèrement
plus négatif que la PANI, permettant d'atteindre des différences de potentiels de 1,5 V. Cela
permet également d'utiliser légèrement plus de la moitié de la capacité de chaque électrode.
Figure I-18 : Voltamogrammes du dopage positif de la PANI et du PPy et domaine de cyclage de chaque électrode dans un
montage de supercondensateur PANI / PPy de type II.
27
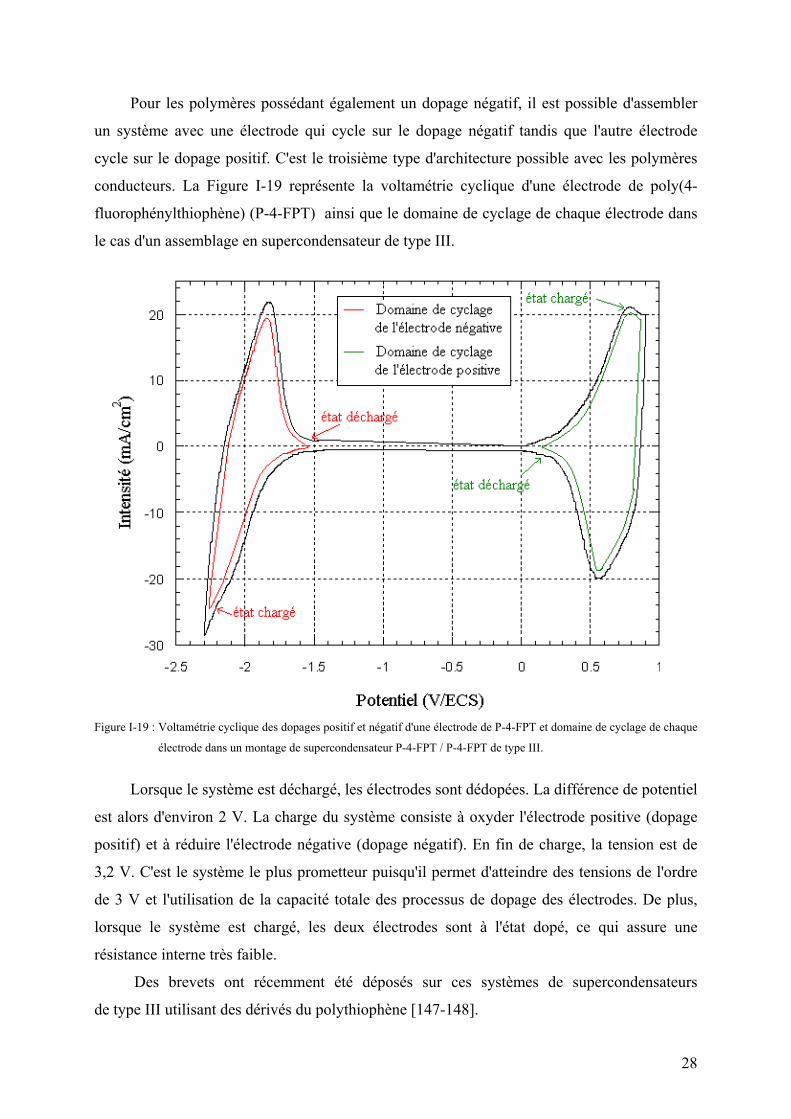
Pour les polymères possédant également un dopage négatif, il est possible d'assembler
un système avec une électrode qui cycle sur le dopage négatif tandis que l'autre électrode
cycle sur le dopage positif. C'est le troisième type d'architecture possible avec les polymères
conducteurs. La Figure I-19 représente la voltamétrie cyclique d'une électrode de poly(4-
fluorophénylthiophène) (P-4-FPT) ainsi que le domaine de cyclage de chaque électrode dans
le cas d'un assemblage en supercondensateur de type III.
Figure I-19 : Voltamétrie cyclique des dopages positif et négatif d'une électrode de P-4-FPT et domaine de cyclage de chaque
électrode dans un montage de supercondensateur P-4-FPT / P-4-FPT de type III.
Lorsque le système est déchargé, les électrodes sont dédopées. La différence de potentiel
est alors d'environ 2 V. La charge du système consiste à oxyder l'électrode positive (dopage
positif) et à réduire l'électrode négative (dopage négatif). En fin de charge, la tension est de
3,2 V. C'est le système le plus prometteur puisqu'il permet d'atteindre des tensions de l'ordre
de 3 V et l'utilisation de la capacité totale des processus de dopage des électrodes. De plus,
lorsque le système est chargé, les deux électrodes sont à l'état dopé, ce qui assure une
résistance interne très faible.
Des brevets ont récemment été déposés sur ces systèmes de supercondensateurs
de type III utilisant des dérivés du polythiophène [147-148].
28

I-2-4) Modèles de capacité utilisés pour les polymères conducteurs
Si le dopage des polymères conducteurs correspond à un transfert de charge avec une
modification de la structure du polymère, ses caractéristiques s'apparentent à celles de
l'accumulation électrostatique de charges de surface dans un condensateur diélectrique [128].
En effet, dans le processus de dopage, ce sont des interactions électrostatiques qui
interviennent entre les charges de la chaîne polymère et les ions dopants, tout comme lors de
la charge d'un condensateur diélectrique. En revanche le dopage n'intervient que dans un
domaine précis de potentiel. Il y a alors un changement de structure et un changement des
propriétés du matériau. C'est pourquoi, le dopage d'un polymère peut être considéré
globalement comme un transfert de charge et caractérisé par le couple redox Polym/Polymn+
(dopage positif) ou Polym/Polymn- (dopage négatif).
Il existe deux modèles développés pour exprimer la quantité d'électricité mise en jeu
dans un processus de stockage d'énergie, selon que le système est un accumulateur ou un
condensateur.
Dans le cas d'un accumulateur, on emploie la formule Q = i.t (en Coulomb = A.s). La
capacité spécifique d'un matériau d'électrode d'accumulateur est alors exprimée en Ah/kg
(ou mAh/g). Dans le cas d'un condensateur, la quantité de charge accumulée dépend de la
tension appliquée. Il faut donc prendre en compte la tension dans le calcul du stockage de
l'énergie. On utilise alors la formule C = Q/V (en Farad = A.s.V-1), et la capacité diélectrique
spécifique d'un matériau d'électrode est exprimée en F/g.
Dans la littérature, les caractéristiques et les performances des supercondensateurs à
base de polymères conducteurs sont rapportées aussi bien en se servant du modèle
"accumulateur" [39,48,52,89,105] que du modèle "condensateur" pour pouvoir les comparer
aux supercondensateurs à base de carbone [21,110,127]. Dans ce dernier cas, le terme
"capacité" étant utilisé pour indiquer la capacité électrostatique, on emploie le terme de
"pseudocapacité", exprimée également en F/g.
29

Les polythiophènes ont donc reçu une grande attention ces vingt dernières années.
Différentes voies de synthèse existent, chacune présentant ses avantages et ses inconvénients.
La possibilité de moduler les propriétés chimiques, optiques et électrochimiques de ces
polymères a été étudiée à l'aide de nombreuses techniques d'analyse.
Les applications potentielles de ces polymères sont assez importantes, dans des
domaines aussi variés que la protection antistatique, les diodes électroluminescentes, les
écrans plats ou le stockage de l'énergie.
Cependant, certaines applications nécessitent de bonnes propriétés de dopage négatif,
domaine encore peu étudié du fait de sa mauvaise stabilité par rapport au dopage positif.
C'est donc dans cette direction que se sont orientées nos recherches, afin de pouvoir
obtenir des électrodes négatives fiables et de les mettre en œuvre dans des
supercondensateurs de type III.
30

II) Partie expérimentale
II-1) Caractérisations chimiques
Les caractérisations par spectroscopie infrarouge ont été effectuées avec un spectromètre
Bruker Equinox IFS55 à transformée de Fourier couplé à un PC avec le logiciel OPUS 2.2.
Les spectres ont été enregistrés en utilisant les techniques de "Golden Gate" et de réflexion
diffuse.
Les spectres UV-Visible ont été enregistrés sur un spectromètre Kontron UVICON 930.
II-2) Caractérisations électrochimiques
II-2-1) Cellules électrochimiques
Pour la caractérisation de leurs propriétés électrochimiques, les polymères ont été mis en
œuvre dans des électrodes et testés dans des cellules électrochimiques à trois électrodes : une
électrode de travail, une contre-électrode et une électrode de référence. Le courant passe entre
l'électrode de travail et la contre-électrode, la référence servant à contrôler du potentiel.
L'électrode de travail est l'électrode contenant le polymère à caractériser.
L'électrode de référence est constituée d'un fil d'argent plongeant dans une solution
10-2 M de nitrate d'argent dans l'acétonitrile. Le couple électrochimique est Ag+/Ag. Le
potentiel de cette électrode, stable dans les milieux organiques, se situe à +0,29 V/ECS.
Plusieurs types de contre-électrodes ont été utilisés : une électrode de platine de surface
4 cm2 pour les différentes études en voltamétrie cyclique et une électrode surcapacitive de
charbon actif, pouvant stocker une grande quantité d'électricité pour les tests de cyclabilité.
L'utilisation de cette dernière électrode permet de limiter la variation en potentiel de la contre-
électrode et d'éviter ainsi la dégradation de l'électrolyte sur cette électrode.
Deux types de montages en cellules à trois électrodes ont été utilisés :
- le premier montage (cf. Figure II-1) est une cellule électrochimique classique, utilisée
pour les tests de voltamétrie cyclique. Les électrodes de 4 cm2 sont plongées dans
l'électrolyte, la référence étant placée entre les deux autres.
31

Figure II-1 : Cellule électrochimique de type I, utilisée pour les mesures classiques de voltampérométrie cyclique.
- le deuxième montage (cf. Figure II-2) est constitué de deux électrodes de 4 cm²
(électrode de travail et contre-électrode) pressées l'une contre l'autre entre deux cales de
téflon. Entre les deux électrodes est placé un séparateur (conducteur ionique et isolant
électronique) pour éviter les courts-circuits. Le montage est maintenu sous pression à l'aide de
deux pinces en acier inox (assurant une pression d'environ 2 bars/cm2). La référence est alors
placée au-dessus des électrodes, un emplacement aménagé dans les cales de téflon permettant
d'approcher la référence au plus près des électrodes. L'ensemble est plongé dans l'électrolyte.
Ce montage permet de tester les électrodes sous compression et avec un séparateur, c'est à
dire dans les conditions réelles de fonctionnement dans un système de type
supercondensateur. Il permet également de minimiser la chute ohmique existant entre
l'électrode de travail et la contre-électrode.
32

Figure II-2 : Cellule électrochimique de type II, avec les électrodes maintenues sous compression.
Le dopage des polymères étant très sensible à l'eau, les caractérisations ont été réalisées
sous atmosphère d'argon déshydraté. Cependant, l'objectif de ce travail étant une application
industrielle potentielle, les solvants n'ont pas été préalablement repurifiés et redéshydratés,
pour limiter les coûts de mise en œuvre des systèmes. Les teneurs en eau ont été mesurées à
l'aide d'un titrateur Karl Fischer, et sont de l'ordre de 0,08 % en volume dans l'acétonitrile et
de 0,04 % en volume dans le carbonate de propylène.
II-2-2) Techniques d'analyse électrochimique
II-2-2-1) La voltampérométrie cyclique
La voltampérométrie cyclique, plus communément appelée voltamétrie cyclique (CV),
est une des méthodes électrochimiques les plus efficaces pour caractériser les réactions de
transfert de charges. Elle consiste à imposer une rampe linéaire en potentiel avec une vitesse
de balayage positive ou négative, et à enregistrer l'intensité du courant. Le montage
généralement utilisé est un montage à trois électrodes. La Figure II-3 représente une courbe
33
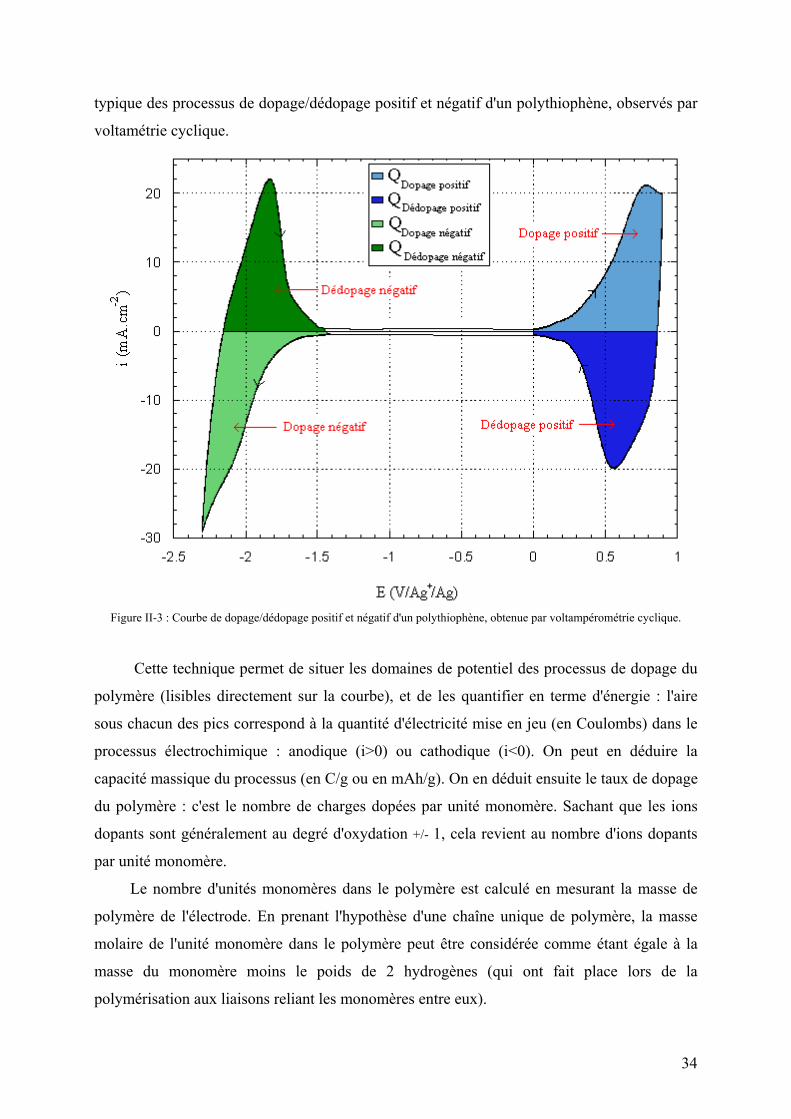
typique des processus de dopage/dédopage positif et négatif d'un polythiophène, observés par
voltamétrie cyclique.
Figure II-3 : Courbe de dopage/dédopage positif et négatif d'un polythiophène, obtenue par voltampérométrie cyclique.
Cette technique permet de situer les domaines de potentiel des processus de dopage du
polymère (lisibles directement sur la courbe), et de les quantifier en terme d'énergie : l'aire
sous chacun des pics correspond à la quantité d'électricité mise en jeu (en Coulombs) dans le
processus électrochimique : anodique (i>0) ou cathodique (i<0). On peut en déduire la
capacité massique du processus (en C/g ou en mAh/g). On en déduit ensuite le taux de dopage
du polymère : c'est le nombre de charges dopées par unité monomère. Sachant que les ions
dopants sont généralement au degré d'oxydation +/- 1, cela revient au nombre d'ions dopants
par unité monomère.
Le nombre d'unités monomères dans le polymère est calculé en mesurant la masse de
polymère de l'électrode. En prenant l'hypothèse d'une chaîne unique de polymère, la masse
molaire de l'unité monomère dans le polymère peut être considérée comme étant égale à la
masse du monomère moins le poids de 2 hydrogènes (qui ont fait place lors de la
polymérisation aux liaisons reliant les monomères entre eux).
34

Pour le polythiophène par exemple, Mmonomère = 84 g.moL-1 et donc Mu.m. = 82 g.moL-1.
Le nombre d'unités monomères présentes dans l'électrode est alors calculé d'après la
formule :
u.m.a
u.m. MN m n ×= , m étant la masse du polymère sur l'électrode et
Na le nombre d'Avogadro (6,02.1023 atomes.mol-1).
Un électron ayant une charge de 1,6.10-19 C, le taux de dopage (en électron par unité
monomère) est alors calculé selon la formule :
nQ τ 19-1,6.10.m.u
dopé
×= avec Qdopé en coulombs.
Des capacités de dopage et de dédopage, il est possible de déduire la réversibilité
coulombique du processus de dopage : dopé
dédopé
QQ =η
Pour la voltamétrie cyclique, nous avons utilisé un potentiostat EG&G PAR modèle
273A couplé à un PC avec le logiciel M270 pour l'aquisition des données.
II-2-2-2) Le cyclage galvanostatique
Une mesure galvanostatique consiste à imposer un courant constant à une électrode et à
suivre sa réponse en potentiel au cours du temps. Pour effectuer un cyclage galvanostatique, il
faut fixer deux valeurs limites de potentiel entre lesquelles l'électrode va cycler un nombre
défini de fois. On utilise soit un montage à trois électrodes (deux électrodes plus une
référence) et on obtient indépendamment la réponse en tension de chacune des électrodes par
rapport à l'électrode de référence, soit un montage à deux électrodes (on court-circuite contre-
électrode et référence) et on obtient la réponse du système entier (∆E = E+ - E-), ce qui permet
d'obtenir directement les caractéristiques générales de celui-ci.
La Figure II-4 représente la courbe typique de dopage et dédopage positif d'un
polythiophène entre +0,15 et +0,9 V/Ref (montage à trois électrodes).
35

Figure II-4 : Courbe de dopage/dédopage positif d'un polythiophène, obtenue par cyclage galvanostatique.
Cette courbe permet d'avoir accès à plusieurs caractéristiques importantes du système : - le domaine de cyclage en potentiel, lisible directement sur la courbe,
- la capacité dopée et dédopée (en Coulomb ou en mAh), qui est proportionnelle à la
durée du dopage ou du dédopage (Q = i.t). On peut en déduire la réversibilité coulombique :
dopé
dédopé
QQ =η
- la capacité électrostatique (en Farad) de l'électrode ou du système, déterminée à partir
de la pente de la partie linéaire de la courbe, selon la formule suivante :
- Q = i .t → dQ = i .dt Q en A.s
- Q = C .V → dQ = C .dV C en Farad
→ C .dV = i .dt → pi
dtdVi C == p : pente de la courbe
36

- la résistance interne du système, mesurable par la chute de potentiel, dite chute
ohmique (U = Ri), lors du basculement entre la fin de charge (+i) et le début de décharge (-i)
ou inversement (R = ∆U/∆i). Cette résistance est généralement traduite en résistance
surfacique Rs = R×S (Ω.cm2) qui permet de s'affranchir des dimensions spatiales et de
comparer ainsi des systèmes de différentes tailles.
Ces caractéristiques du système permettent d'obtenir des caractéristiques communes à
tous les systèmes de stockage d'énergie, l'énergie maximale et la puissance maximale :
- l'énergie maximale est obtenue d'après la formule :
)UC(21 2max max E ×= exprimée en W.s (J)
Umax est la tension du système à la fin de la charge (en Volts) et C est la capacité du système
(exprimée en Farad), mesurée au cours de la décharge.
- la puissance maximale est calculée à partir de la formule :
R4U
2max 0P
×= exprimée en W
U0 est le potentiel du système au début de la décharge (en V) et R est la résistance interne du
système (en Ω). Cette puissance est théorique et ne peut pas être atteinte : elle représente la
puissance que délivrerait le système s'il était totalement déchargé en un temps de 0 seconde.
Ces valeurs d'énergie et de puissance, bien que très utiles pour la comparaison des
systèmes, ne rendent pas compte des caractéristiques réelles du supercondensateur. Pour cela,
on utilise d'autres mesures d'énergie et de puissance :
- l'énergie réelle Eréelle = U.i.t (en W.s),
U est la tension moyenne de décharge, calculée selon la formule : 2 U U U minmax += , et t est
le temps de décharge du système,
- la puissance réelle Préelle = t
Eréelle (en W).
37

Le cyclage galvanostatique a été effectué sur un potentiostat VMP (Versatile
MultiPotentiostat) de Biologic Technologies, permettant un échantillonage avec une précision
de 20 ms. Ce potentiostat était couplé à un PC avec une interface graphique permettant de
suivre le cyclage ainsi que de modifier les paramètres en temps réel.
II-2-2-3) La spectroscopie d'impédance complexe
La spectroscopie d’impédance permet de différencier les divers phénomènes
élémentaires sur la base de leurs constantes de temps respectives. En pratique, il s’agit de se
placer à un potentiel stationnaire E et d’appliquer une surimposition sinusoïdale ( de
faible amplitude et de pulsation ω .
s )Eδ
Chaque processus perturbé revient à l’état stationnaire avec son propre temps de
réponse. Ainsi les réactions partielles se produisant à l’interface électrode/électrolyte peuvent
donc être différenciées les unes des autres : les phénomènes électrochimiques rapides sont
sollicités dans le domaine des hautes fréquences (transfert de charges), les phénomènes lents
apparaissent dans le domaine des basses fréquences (transports de matière : réactions de
diffusions et d’adsorption).
En appliquant une perturbation )E(δ au potentiel stationnaire E , il vient : s
EEsE δ+= avec )tjexp(EE ωδ=δ
On obtient alors une réponse en courant, après un temps de relaxation, de même
pulsation ω
Ιδ+Ι=Ι s avec )tjexp( ϕ+ωΙδ=Ιδ
Pour chaque fréquence, la fonction de transfert appelée impédance complexe )(ωΖ est
alors égale au rapport de la perturbation en potentiel sur la réponse en courant.
)()(RE)( mje ΖΙ+Ζ=Ιδ
δ=ωΖ
38

La représentation de la variation de l’impédance se fait généralement dans le plan de
Nyquist : en fonction de R)(m ΖΙ− )(e Ζ .
Le Tableau II-1 résume quelques exemples de simulations électriques de différentes
associations de composants classiques.
Elément Impédance Circuit équivalent Diagramme d’impédance
dans le plan de Nyquist
Résistance R
(1)
Z = Z’= R
0
R
-Z" (Ω)
Z' (Ω)
Condensateur C
(2)
Z = ω
=jC1"Z
ωC
0
-Z" (Ω)
Z' (Ω)
0
R, C en série
(3) ω
+=+=jC1RZZZ "'
0 R
-Z" (Ω)
Z' (Ω)
ω
0
R, C en parallèle (4)
(transfert de charge)
ω+= jCR1
Z1
0
0
-Z" (Ω)
Z' (Ω)
ω
Tableau II-1 : Spectres d’impédance complexe de quelques circuits électriques simples
La Figure II-5 (a) représente le spectre d'impédance dans le plan de Nyquist d'un
supercondensateur à base de polymère conducteur. Ce spectre peut être modélisé par une
combinaison de circuits électriques. La Figure II-5 (b) représente un des circuits équivalents
permettant de décrire le système.
39

Figure II-5 : Diagramme d'impédance d'un supercondensateur polymère conducteur et son circuit équivalent.
A haute fréquence, la réponse comprend uniquement les mouvements purement
électroniques. La résistance série englobe donc la résistance de l'électrolyte, la résistance de
contact à l'interface collecteur de courant / matière active et la résistance intrinsèque de la
matière active. Les collecteurs de courant étant métalliques, leur résistance est négligeable.
Lorsque la fréquence diminue, on distingue deux phénomènes : le transfert de charge lié
au passage des électrons lors de la réaction redox, qui se caractérise par un demi-cercle, ainsi
que la diffusion des ions dans l'électrolyte au sein de l'électrode, qui se traduit par une droite
d'angle 45° dite de Warburg.
A basse fréquence, on observe une droite presque verticale, représentative de la capacité
du système.
Pour la spectroscopie d'impédance complexe, nous avons utilisé un analyseur de
fréquence Schlumberger Solartron 1255 et un potenstiostat Schlumberger Solartron 1286
contrôlés par un PC avec le logiciel ZPlot.
40

III) Synthèse des polymères
Les polymères conducteurs qui sont présentés dans la littérature sont généralement
synthétisés par voie électrochimique [40-45]. Ils se présentent alors sous forme de films
déposés sur des substrats conducteurs. Cela permet de contrôler très précisément les
conditions de polymérisation ainsi que la masse de polymère déposée. Les synthèses
électrochimiques permettent également d'effectuer les polymérisations dans le même milieu
que celui de l'utilisation future du polymère (cf. I-1-2).
L'électropolymérisation ne permet cependant pas de réaliser des électrodes de grande
surface ni même des électrodes double-face (polymère déposé des deux côtés du substrat
conducteur) à cause de difficultés de mise en œuvre.
Pour ces raisons, nous avons décidé de synthétiser les polymères par voie chimique pour
obtenir des poudres plus facilement manipulables.
III-1) Synthèse des monomères
Le thiophène et le 2,5-dibromothiophène sont des produits commerciaux de bonne
pureté et qui ne nécessitent pas de purification supplémentaire avant de procéder à la
polymérisation.
Par contre, les dérivés phénylés du thiophène n'existent pas sur le marché. Ils sont
synthétisés à partir du 3-bromothiophène et du dérivé bromophénylé correspondant,
disponibles commercialement.
III-1-1) Principe
Le principe de cette synthèse est un couplage organomagnésien catalysé par un métal de
transition. C'est une synthèse très utilisée pour obtenir des dérivés du thiophène [149-151].
Elle s'effectue en deux étapes :
41

- insertion du magnésium dans la liaison Carbone - Halogène du dérivé bromophénylé :
+ MgTHF
BrF F
Mg Br
- couplage organomagnésien catalysé par le 1,3-bis(diphénylphosphinopropane)
dichloronickel (II).
+ THF
NiCl 2(dppp)+ Mg Br2S
Br
F
S
FMg Br
Pour la synthèse du 4-fluorophénylthiophène (4-FPT), pour simplifier la procédure de
synthèse, nous avons utilisé un organomagnésien commercial, proposé en solution molaire
dans le THF.
La synthèse doit être effectuée sous atmosphère inerte et en milieu anhydre : en présence
d'eau ou d'air, le magnésium réagit pour donner de la magnésie qui n'est plus réactive.
III-1-2) Procédure
Toute la verrerie utilisée dans l'expérience est préalablement rincée à l'acétone, séchée et
placée sous atmosphère d'argon.
On place un ballon bicol de 100 ml surmonté d'un réfrigérant sous flux d'argon et on y
introduit 25 mmol de magnésium sec en copeaux et 50 ml de THF anhydre. On agite et on
ajoute 25 mmol de bromobenzène fluoré à l'aide d'une ampoule de chargement. On chauffe
légèrement (30°C) pour amorcer la réaction. Celle-ci est exothermique et dure environ 1
heure.
Dans un tricol de 100 ml placé en boite à gant sous atmosphère d'argon, on introduit 25
mmol de 3-bromothiophène et 100 µmol de catalyseur NiCl2(dppp).
Le tricol est sorti de la boite à gants et placé sous flux d'argon sec et est refroidi à -10 °C
par un mélange de glace pillée et de NaCl.
42

Le grignard est transféré du bicol au tricol à l'aide d'une seringue de 100 ml pour éviter
le contact avec l'air. La réaction est poursuivie pendant une dizaine d'heures à -10°C et sous
agitation. On ajoute ensuite 30 ml d'HCl 1M pour terminer la réaction et neutraliser le réactif
restant. On ajoute de l'eau pour neutraliser le milieu puis on récupère la phase organique qu'on
lave encore plusieurs fois avec de l'eau. On évapore ensuite le THF sous vide au rotavapor et
on récupère une poudre de couleur marron beige.
Les différents monomères sont ensuite purifiés de différentes manières :
- le 4-FPT est purifié dans un mélange méthanol / eau (solvant / non-solvant) ; on le
dissout d'abord dans le méthanol puis on le précipite en ajoutant la même quantité d'eau.
Toutes les impuretés restent alors dans le méthanol. Le 4-FPT est ensuite filtré pour donner
une poudre légèrement jaune. Il peut ensuite éventuellement être sublimé et donne des
cristaux blancs translucides.
- les autres monomères multifluorés sont recristallisés dans de l'hexane et donnent des
poudres blanches.
La pureté des monomères est vérifiée par mesure du point de fusion :
- 85-86 °C pour le 4-FPT (85-87 °C dans la littérature [43]),
- 34-35 °C pour le 3,4-DFPT (33-34 °C dans la littérature [105]),
- 47-48 °C pour le 3,5-DFPT (48-49 °C dans la littérature [105]),
- 30,5-31 °C pour le 3,4,5-TFPT (31-32 °C dans la littérature [105]).
Les rendements de synthèse varient :
- 92 % pour le 4-FPT (79 % après sublimation),
- 71 % pour le 3,4-DFPT et le 3,4,5-TFPT,
- 58 % pour le 3,5-DFPT.
III-2) Synthèse chimique du polythiophène (PTh)
Le PTh peut être synthétisé chimiquement de différentes manières. Les deux voies de
synthèse les plus connues sont l'oxydation du thiophène ou du bithiophène par le chlorure
ferrique [17] et la polycondensation du 2,5-dibromothiophène [12-14].
Le PTh obtenu par oxydation directe est un produit commercial mais ne possède pas de
bonnes propriétés électrochimiques. Nous avons donc décidé de le synthétiser par
polycondensation. Cette réaction est du même type que la synthèse des monomères : c'est un
couplage organomagnésien catalysé par un métal de transition.
43

III-2-1) Principe
La première étape est l'insertion du magnésium dans la liaison Carbone – Halogène :
S BrBr + THF
S Mg BrBrMg
La deuxième étape est le couplage organomagnésien catalysé par le 1,3-bis
(diphénylphosphinoéthane) dicholoronickel(II) :
nTHF
NiCl 2(dppe)S Mg BrBr ( S )n+ n MgBr2
Cette réaction, comme celle de préparation des monomères, doit être effectuée sous
atmosphère inerte, en milieu totalement anhydre. En revanche, elle ne nécessite pas de basses
températures.
III-2-2) Procédure
La verrerie est préalablement rincée à l'acétone, séchée et placée sous atmosphère
d'argon sec.
Dans un ballon tricol surmonté d'un réfrigérant, le tout placé sous flux d'argon, on
introduit 6 g de magnésium sec en copeaux. On ajoute ensuite 200 ml de THF parfaitement
anhydre et 28 ml de 2,5-dibromothiophène, en se servant d'une seringue pour éviter les
contacts avec l'air. On agite vivement le mélange et on chauffe légèrement pour amorcer la
formation du Grignard. La réaction est exothermique et dure environ 30 minutes.
Lorsque la température est revenue à 22°C (température ambiante), on ajoute
précautionneusement le catalyseur. Celui-ci a été mis en suspension dans 40 ml de THF, dans
44

une ampoule de chargement sous flux d'argon. La réaction commence immédiatement et très
vivement (forte exothermie). La couleur rouge sombre du polymère apparaît immédiatement.
Au bout d'une heure et demi, la température commence à redescendre signifiant que la
polymérisation est achevée.
On filtre alors sur un fritté et on récupère une poudre rouge. Celle-ci est lavée plusieurs
fois avec un mélange 50/50 de méthanol et H2SO4 puis avec du méthanol.
Le rendement de synthèse varie entre 92 et 95% en masse.
III-3) Etude de la synthèse du poly-4-fluorophénylthiophène (P-4-FPT)
III-3-1) Principe
Le principe de la synthèse chimique du P-4-FPT est une oxydation directe du monomère
par le chlorure ferrique dans le chloroforme.
+ 2n FeCl 3CHCl 3 ( S )n
+ 2n FeCl 2n + 2n HClS
F F
La réaction doit être effectuée sous atmosphère inerte (argon ou azote) et anhydre car
l'oxydant utilisé est très sensible à l'eau.
Le polymère est synthétisé à l'état dopé positivement puisque le potentiel de
polymérisation est supérieur au potentiel de dopage positif du polymère. Il faut donc dédoper
le polymère après synthèse pour obtenir un matériau neutre, en le lavant au méthanol et à
l'eau.
45

III-3-2) Etude de la synthèse du P-4-FPT
Cette synthèse a été mise au point au Laboratoire d'Electrochimie Industrielle du
C.N.A.M. en 1996 mais n'a pas été optimisée, ce qui a conduit à des résultats très variables du
point de vue de la qualité électrochimique des polymères synthétisés [152]. Nous avons donc
étudié les différents paramètres macroscopiques de cette synthèse pour déterminer dans quelle
mesure ils affectent les propriétés électrochimiques du polymère. Les paramètres principaux
sont la température, la concentration des réactifs, le temps de synthèse et le rapport des
réactifs.
Les polymères synthétisés ont été caractérisés par voltamétrie cyclique. L'électrode de
travail est donc constituée du polymère (15 mg) mélangé à du graphite conducteur (15 mg), le
tout placé entre deux morceaux de tissus de carbone conducteur et encapsulé dans une grille
d'acier inoxydable. La contre-électrode est une électrode de platine de surface 1 cm2.
L'électrolyte utilisé est le tetrafluoroborate de tetraéthylammonium (NEt4+
,BF4-) 1M dans
l'acétonitrile. La vitesse de balayage est de 5 mV/s.
III-3-2-1) Etude de la température de polymérisation
Nous avons fait varier la température de polymérisation de 0°C à 60°C, qui semble être
la température maximale pour éviter l'évaporation du chloroforme.
La Figure III-1 présente les voltamogrammes des polymères synthétisés à différentes
températures. Nous avons rassemblé les principales données expérimentales obtenues dans le
Tableau III-1. Les quantités d'électricités ont été calculées d'après les aires qui se situent sous
les pics de dopage (cf. II-2-2-1).
46

-10
-5
0
5
-2.5 -2 -1.5 -1 -0.5 0 0.5 1
T = 0°C
T = 60°CT = 35°CT = 23°C
i (m
A/c
m2 )
E (V/Ag+/Ag) Figure III-1 : Voltamogrammes des P-4-FPT synthétisés à différentes températures. Electrolyte : acétonitrile 1M NEt4BF4
v = 5 mV/s ; Composition de l'électrode : 15 mg P-4-FPT, 15 mg NA encapsulés dans un déployé d'acier inox.
Température (°C) Dopage p (C) Dédopage p (C) ηp (%) Dopage n (C) Dédopage n (C) ηn (%) Rdt (%)
0 2,35 -1,74 74,0 -4,41 1,19 27,0 81,3
23 2,08 -1,68 80,8 -4,17 1,36 32,6 79,8
35 2,20 -1,61 73,2 -4,94 1,21 24,5 86,1
60 1,95 -1,31 67,2 -5,26 0,96 18,3 80,0
Tableau III-1 : Principales données tirées de la Figure III-1 ainsi que rendement massique de polymérisation.
On peut remarquer une déficience des processus de dopage du polymère synthétisé à
60°C : une réversibilité coulombique plus faible et des quantités d'électricité dédopées
inférieures. Cela tend à prouver que les températures de polymérisation élevées sont néfastes
pour l'électroactivité du polymère.
Par ailleurs la température n'influence pas le rendement de la synthèse qui est toujours
de l'ordre de 80% en masse, comme cela avait déjà été observé avec d'autres dérivés du PTh
[20].
Une température ambiante (23°C) paraît donc satisfaisante pour cette synthèse.
47

III-3-2-2) Etude du rapport oxydant/monomère
Le rapport théorique de la polymérisation est de 2,33 unités oxydantes pour 1 unité
monomère, si l'on considère que le polymère est synthétisé à l'état dopé à hauteur d'une
charge pour trois unités monomères.
( S )n
R2 n FeCl 3
S
0,33 n FeCl 3
R
Monomère Polymère neutre Polymère dopé tous les trois motifs
( S )nR
n [ ] 0,33 +
, 0,33 FeCl4-
Cependant, Sugimoto et al. ont mis au point un protocole de polymérisation chimique du
polythiophène par le chlorure ferrique avec une optimisation du rapport oxydant / monomère
à 4 [17]. Une explication de cet excès a été proposée par Niemi et al. [27] mettant en cause
d'une part l'inertie chimique de la phase soluble du chlorure ferrique (sous forme dimérisée
Fe2Cl6), et d'autre part la complexation d'une partie de la phase solide du FeCl3 par l'acide
chlorhydrique formé par la réaction (probablement sous forme FeCl4-, H+). Il nous a paru
intéressant de vérifier si cette théorie était vérifiée pour la polymérisation du 4-FPT. Nous
avons donc fait varier ce rapport de part et d'autre du rapport théorique.
La Figure III-2 présente la variation du rendement de synthèse en fonction du rapport
RFeCl3/4-FPT.
20
30
40
50
60
70
80
90
0 2 4 6 8
Rend
emen
t (%
)
Rapport FeCl3/FPT
10
Figure III-2 : Rendement massique de synthèse en fonction du rapport entre les réactifs
48
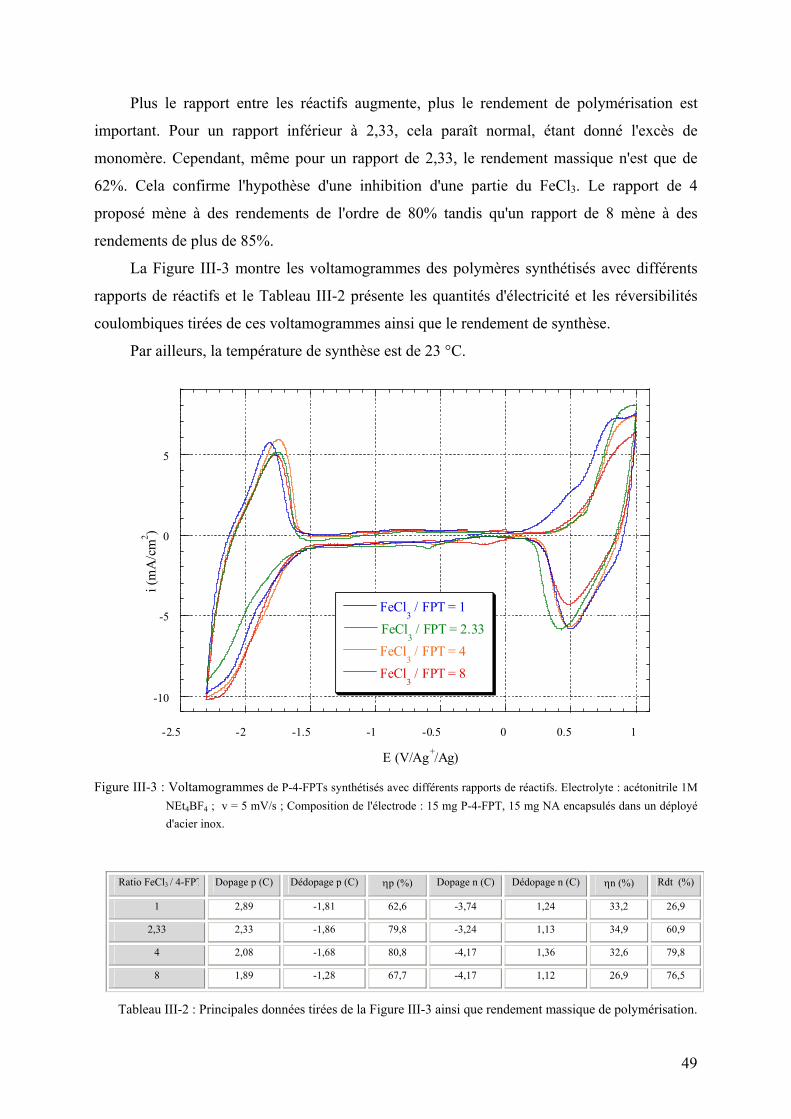
Plus le rapport entre les réactifs augmente, plus le rendement de polymérisation est
important. Pour un rapport inférieur à 2,33, cela paraît normal, étant donné l'excès de
monomère. Cependant, même pour un rapport de 2,33, le rendement massique n'est que de
62%. Cela confirme l'hypothèse d'une inhibition d'une partie du FeCl3. Le rapport de 4
proposé mène à des rendements de l'ordre de 80% tandis qu'un rapport de 8 mène à des
rendements de plus de 85%.
La Figure III-3 montre les voltamogrammes des polymères synthétisés avec différents
rapports de réactifs et le Tableau III-2 présente les quantités d'électricité et les réversibilités
coulombiques tirées de ces voltamogrammes ainsi que le rendement de synthèse.
Par ailleurs, la température de synthèse est de 23 °C.
-10
-5
0
5
-2.5 -2 -1.5 -1 -0.5 0 0.5 1
FeCl3 / FPT = 4
FeCl3 / FPT = 8
FeCl3 / FPT = 1
FeCl3 / FPT = 2.33
i (m
A/c
m2 )
E (V/Ag+/Ag) Figure III-3 : Voltamogrammes de P-4-FPTs synthétisés avec différents rapports de réactifs. Electrolyte : acétonitrile 1M
NEt4BF4 ; v = 5 mV/s ; Composition de l'électrode : 15 mg P-4-FPT, 15 mg NA encapsulés dans un déployé d'acier inox.
Ratio FeCl3 / 4-FPT Dopage p (C) Dédopage p (C) ηp (%) Dopage n (C) Dédopage n (C) ηn (%) Rdt (%)
1 2,89 -1,81 62,6 -3,74 1,24 33,2 26,9
2,33 2,33 -1,86 79,8 -3,24 1,13 34,9 60,9
4 2,08 -1,68 80,8 -4,17 1,36 32,6 79,8
8 1,89 -1,28 67,7 -4,17 1,12 26,9 76,5
Tableau III-2 : Principales données tirées de la Figure III-3 ainsi que rendement massique de polymérisation.
49
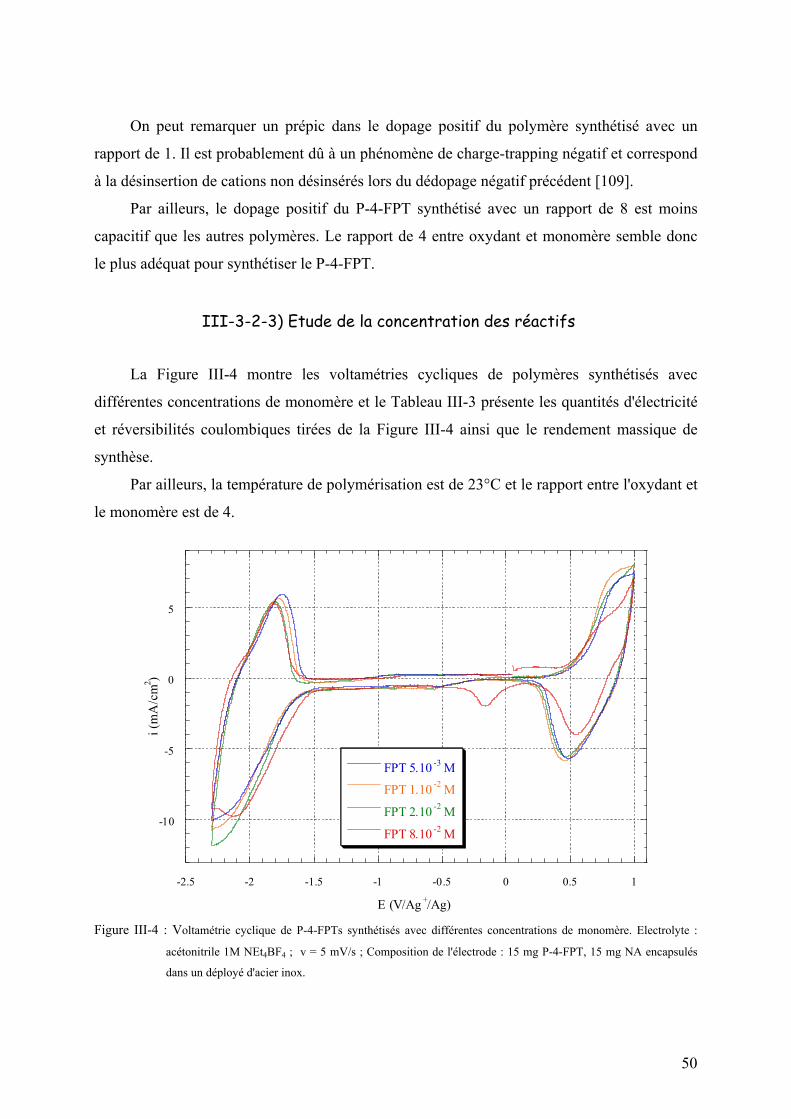
On peut remarquer un prépic dans le dopage positif du polymère synthétisé avec un
rapport de 1. Il est probablement dû à un phénomène de charge-trapping négatif et correspond
à la désinsertion de cations non désinsérés lors du dédopage négatif précédent [109].
Par ailleurs, le dopage positif du P-4-FPT synthétisé avec un rapport de 8 est moins
capacitif que les autres polymères. Le rapport de 4 entre oxydant et monomère semble donc
le plus adéquat pour synthétiser le P-4-FPT.
III-3-2-3) Etude de la concentration des réactifs
La Figure III-4 montre les voltamétries cycliques de polymères synthétisés avec
différentes concentrations de monomère et le Tableau III-3 présente les quantités d'électricité
et réversibilités coulombiques tirées de la Figure III-4 ainsi que le rendement massique de
synthèse.
Par ailleurs, la température de polymérisation est de 23°C et le rapport entre l'oxydant et
le monomère est de 4.
-10
-5
0
5
-2.5 -2 -1.5 -1 -0.5 0 0.5 1
FPT 2.10 -2 M
FPT 1.10 -2 M
FPT 5.10 -3 M
FPT 8.10 -2 M
i (m
A/c
m2 )
E (V/Ag +/Ag)
Figure III-4 : Voltamétrie cyclique de P-4-FPTs synthétisés avec différentes concentrations de monomère. Electrolyte :
acétonitrile 1M NEt4BF4 ; v = 5 mV/s ; Composition de l'électrode : 15 mg P-4-FPT, 15 mg NA encapsulés
dans un déployé d'acier inox.
50

4-FPT(Mol/L) Dopage p (C) Dédopage p (C) ηp (%) Dopage n (C) Dédopage n (C) ηn (%) Rdt (%)
5.10-3 2,41 -1,86 77,2 -4,27 1,24 29,0 88,7
10-2 2,08 -1,68 80,8 -4,17 1,36 32,6 79,8
2.10-2 2,32 -1,71 73,7 -4,82 1,06 22,0 80,0
8.10-2 1,98 -1,01 51,0 -4,51 1,12 24,8 82,0
Tableau III-3 : Principales données tirées de la Figure III-4 ainsi que rendement massique de polymérisation.
Pour le dopage négatif, la variation de ce paramètre ne semble pas altérer énormément
l'électroactivité des polymères. Cependant, on peut remarquer une électroactivité déficiente
pour le dopage positif du polymère synthétisé avec une concentration de 8.10-2 M. Il semble
donc qu'il y ait un seuil de concentration à ne pas dépasser.
Par ailleurs, la concentration des réactifs ne fait pas varier le rendement de synthèse des
polymères.
La concentration en monomère choisie est donc 2.10-2 M.
III-3-2-4) Etude du temps de polymérisation
Les temps de polymérisation généralement rencontrés dans la littérature sont d'environ
une demi-heure. Notamment, une étude de synthèse du polypyrrole montre que la
conductivité du polymère baisse avec le temps de polymérisation [153]. La Figure III-5
présente les voltamogrammes des P-4-FPT synthétisés avec différentes durées de
polymérisation et le Tableau III-4 montre les principales données tirées de ces courbes.
La température de synthèse est de 23°C, le rapport entre les réactifs de 4 et la
concentration initiale en monomère de 2.10-2 M.
51

-10
-5
0
5
-2.5 -2 -1.5 -1 -0.5 0 0.5 1
t = 20 h
t = 30 mnt = 1 ht = 2 h
i (m
A/c
m2 )
E (V/Ag+/Ag)
Figure III-5 : Voltamétrie cyclique de P-4-FPT synthétisé avec différents temps de polymérisation. Electrolyte : acétonitrile
1M NEt4BF4 ; v = 5 mV/s ; Composition de l'électrode : 15 mg P-4-FPT, 15 mg NA encapsulés dans un
déployé d'acier inox.
Temps de synthèse Dopage p (C) Dédopage p (C) ηp (%) Dopage n (C) Dédopage n (C) ηn (%) Rdt (%)
30 min 2,38 -1,58 65,8 -3,43 1,16 33,8 67,0
1 h 2,28 -1,54 66,4 -3,22 1,02 31,7 79,4
2 h 2,53 -1,87 73,9 -3,57 1,10 30,8 79,2
20 h 2,08 -1,68 80,8 -4,17 1,36 32,6 79,8
Tableau III-4 : Principales données tirées de la Figure III-5 ainsi que rendement massique de polymérisation.
Le rendement de synthèse est de 67% pour 30 minutes de polymérisation et de 79,4%
pour une heure de polymérisation. Cela montre que le processus de polymérisation arrive
pratiquement à terme en une heure. Cependant, des réarrangements moléculaires inter et intra-
chaînes sont toujours possibles ensuite, étant donné l'excès d'oxydant. D'après les quantités
d'électricités dédopées par les polymères, ces réarrangements semblent bénéfiques aux
propriétés électrochimiques du P-4-FPT. Par ailleurs, plus le temps de polymérisation
augmente et plus la réversibilité du dopage positif du polymère augmente. Cela signifie que
plus le temps de polymérisation est long et plus la structure du polymère est ouverte.
Une durée de synthèse de 20 heures semble donc plus intéressante.
52

III-3-2-5) Conclusion de l'étude
Sur la base de critères électrochimiques (quantité d'électricité fournie lors du dédopage
positif et négatif, réversibilité) et chimiques (rendement de polymérisation), nous pouvons
proposer un protocole amélioré pour la synthèse du P-4-FPT :
- une température ambiante si elle n'excède pas les 30°C,
- une concentration en monomère de 2.10-2 mol/L,
- une durée de synthèse de 20 heures,
- un rapport oxydant / monomère de 4.
La pureté du monomère ainsi que les conditions anhydres du milieu sont aussi très
importantes pour cette synthèse.
III-3-3) Protocole de synthèse du P-4-FPT
Cette procédure permet d'obtenir une quantité d'environ 2g de P-4-FPT. La verrerie est
préalablement rincée à l'acétone, séchée et placée sous atmosphère d'argon.
On pèse 9,10 g de FeCl3 dans un ballon bicol de 2 L surmonté d'un réfrigérant et d'une
ampoule de chargement de 100 mL faisant également office d'entrée d'argon. On y ajoute
1,3 L de chloroforme (CHCl3) préalablement séché sur tamis moléculaire 4 µm. La
suspension de FeCl3 est agitée afin d'obtenir un milieu oxydant homogène.
2,5 g de monomère sont dissous dans 100 mL de chloroforme, puis placé dans l'ampoule
de chargement. Le monomère est ensuite ajouté rapidement au milieu oxydant. Ainsi, durant
tout le temps de l'ajout, l'oxydant est en excès par rapport au monomère.
On laisse la réaction se poursuivre pendant 20 heures. Elle est stoppée par l'ajout de
200 mL de méthanol. Celui-ci neutralise l'oxydant en excès et dédope partiellement le
polymère.
La poudre est ensuite filtrée sur fritté et lavé plusieurs fois au méthanol et à l'eau pour la
dédoper totalement. Le lavage est estimé suffisant lorsque le filtrat est limpide.
Le polymère est séché 24 heures à 120°C. Il se présente sous la forme d'une fine poudre
de couleur marron.
53

III-4) Synthèse des autres polymères
Les différents 3-phénylthiophènes fluorés ont été polymérisés suivant la même
procédure que le 4-FPT, avec des rendements à peu près semblables :
- 81% pour le P-3,4-DFPT,
- 77% pour le P-3,5-DFPT,
- 79% pour le P-3,4,5-TFPT.
54

IV) Mise en œuvre des polymères
Les polymères synthétisés ont été mis en œuvre dans des électrodes pour permettre leur
caractérisation électrochimique. La mise en œuvre des électrodes consiste à fixer les matières
actives sur un collecteur de courant en métal déployé à l'aide d'un liant. Les polymères
présentant des caractéristiques isolantes électroniques lorsqu'ils sont à l'état neutre, il est
nécessaire de leur adjoindre dans l'électrode un conducteur pour assurer la percolation
électronique et assister le dopage des polymères.
Dans la technique de fabrication précédemment développée au laboratoire [152], le
polymère était lié à un déployé d'acier inox à l'aide d'un liant cellulosique, la
carboxyméthylcellulose (CMC).
Dans cette partie du travail, une étude approfondie de la mise en œuvre des polymères a
été entreprise. Différents conducteurs électroniques ainsi que différents liants ont été testés.
Le dopage négatif des polymères étant le plus délicat, nous avons choisi de mener cette étude
sur celui-ci, puis de vérifier et éventuellement d'adapter la composition obtenue pour le
dopage positif.
IV-1) Influence de l'additif conducteur sur le dopage négatif
Différents types de conducteurs électroniques ont été testés : des graphites sphériques de
différents diamètres (2 µm, 44 µm et 100 µm), un graphite de forme discoïdale (GMP), ainsi
que le noir d'acétylène et des fibres de graphite.
Les graphites sphériques tels que le UF2 (diamètre 2µm), le SFG44 (diamètre 44µm) et
le BNS100 (diamètre 100 µm), sont fabriqués industriellement par la société Lonza. Le GMP
est un graphite fabriqué à l'échelle laboratoire par le CEA. Les fibres de graphites proviennent
de la société Carbone-Lorraine et ont un diamètre moyen de 6µm pour une longueur de 2mm.
Elles sont finement broyées avant d'être utilisées. Le noir d'acétylène est un noir de carbone
fabriqué industriellement par la société Hœchst et qui possède une structure fibreuse.
Ces conducteurs ont été caractérisés par leur efficacité à assister le dopage des
polymères, c'est à dire par la capacité déchargée par le polymère lors du dédopage lorsqu'il est
associé au conducteur. Pour cela, nous avons utilisé la voltamétrie cyclique avec un montage
à trois électrodes, dans une cellule électrochimique de type I (cf. II-2-1).
55

Lors de cette étude, le liant utilisé pour la mise en œuvre est la CMC. Les électrodes
sont réalisées selon le protocole présenté en Annexe 1.
La Figure IV-1 présente les capacités dédopées par le P-4-FPT après un dopage négatif,
en fonction de la teneur en différents additifs conducteurs. Le dopage a été effectué par
voltamétrie cyclique à 20 mV/s entre -1,4 et -2,0 V/Ref dans une solution d'acétonitrile 1M
NEt4CF3SO3. La composition d'électrode utilisée est de x % en additif conducteur, (x-90)% en
polymère et de 10% en liant. Le collecteur de courant utilisé est un déployé d'inox AISI 316L
de surface 4 cm2.
0
1
2
3
4
5
6
7
8
0 20 40 60 80 100
Graphite 2 µmGraphite 44 µmGraphite 100 µmGraphite GMPNoir d'acétylèneFibres de graphiteP-4-FPT seul
Cap
acité
déd
opée
(mA
h/g PF
PT)
Teneur en conducteur (%) Figure IV-1 : Capacités partielles dédopées par le P-4-FPT après un dopage négatif effectué en voltamétrie cyclique entre -
1.4 et -2V/Ag+/Ag dans une solution d'acétonitrile 1M NEt4CF3SO3 ; v = 20mV/s ; Composition de l'électrode:
(x-90)% P-4-FPT, x % additif conducteur et 10% CMC ; Collecteur de courant en inox AISI 316L.
La Figure montre que le P-4-FPT, lorsqu'il n'est pas associé à un conducteur
électronique, est assez résistif et les chutes ohmiques au sein de l'électrode empêchent le
polymère de se doper efficacement. Il a alors une capacité de 1,6 mAh/g.
Les fibres de graphites ne se mélangent pas de façon homogène avec les poudres de
polymère. Leur utilisation est donc assez difficile. Cela entraîne une mauvaise percolation
électronique au sein des électrodes empâtées et explique donc les faibles valeurs des capacités
mesurées.
56

Les graphites sphériques ne permettent une bonne conduction électronique entre les
chaînes de polymère et le collecteur de courant que lorsqu'ils sont présents dans l'électrode en
importantes quantités. La taille des particules n'influe que très peu sur la conduction obtenue.
Les deux additifs les plus intéressants sont le noir d'acétylène et le graphite GMP. Leurs
formes géométriques sont adaptées à la morphologie des polymères, ce qui permet d'obtenir
une bonne percolation électronique même lorsqu'ils sont présents en faibles quantités. Le
drainage électronique au sein de l'électrode étant amélioré, la quantité de polymère qui
participe aux réactions faradiques augmente, ce qui a pour effet d'améliorer la capacité
délivrée par l'électrode. Avec le GMP, la percolation apparaît à partir d'un seuil de 20% et la
conduction ne cesse de croître ensuite pour atteindre un optimum pour une teneur de 80%.
Avec le noir d'acétylène, une percolation électronique se met en place dès l'ajout de 10% de
conducteur. La courbe présente ensuite un plateau entre 20 et 45% puis une augmentation
pour atteindre à 80% la même valeur obtenue avec le GMP à 80% dans l'électrode.
La Figure IV-1 montre donc clairement que l'ajout de graphite GMP ou de noir
d'acétylène au polymère conducteur améliore notablement la capacité massique délivrée par le
polymère. Pour la caractérisation des polymères, la composition d'électrode qui paraît la plus
satisfaisante est :
- P-4-FPT 10%,
- NA ou GMP 80%,
- CMC 10%.
IV-2) Influence de la nature du liant sur le dopage négatif
Différents liants ont été testés pour obtenir le meilleur accrochage des matières actives
sur le collecteur de courant : la carboxyméthylcellulose (CMC), le fluorure de polyvinylidène
(PVdF) et le polytetrafluoroéthylène (PTFE ou Téflon). La mise en œuvre de chacun de ces
liants est différente et est précisément détaillée en Annexe 1.
La CMC, dissoute dans de l'eau, conduit à l'obtention d'un gel auquel sont ajoutées les
poudres de polymère et de conducteur préalablement mélangées. L'eau est ensuite évaporée à
110°C et on obtient une pâte liquide. Celle-ci est enduite sur le collecteur de courant.
57

Pour le PVdF, la mise en œuvre est similaire mais il faut dissoudre le liant dans de la
N-méthylpyrrolidinone (NMP).
Le PTFE se trouve sous forme dispersée dans une solution aqueuse. Les poudres de
matières actives sont mélangées et mises en suspension dans de l'éthanol. Le téflon est ensuite
ajouté sous agitation. Sous l'effet de l'éthanol, le téflon fibrille et forme un réseau
tridimensionnel qui enserre les poudres. Après l'évaporation de l'éthanol obtenue en chauffant
le mélange à 80°C, on obtient une pâte solide. Un malaxage supplémentaire permet une
meilleure fibrillation des chaînes de téflon et on obtient ainsi de meilleures propriétés
mécaniques de la pâte. Il est ensuite possible d'étaler cette pâte très finement et de la laminer
sur le collecteur de courant.
Le PVdF et le PTFE confèrent une bonne tenue mécanique à l'électrode dès 5% en
masse [168], à la différence de la CMC qui requiert une teneur minimale de 10% pour
apporter une tenue mécanique suffisante.
La première partie de cette étude a été menée parallèlement à l'étude précédente sur
l'influence de l'additif conducteur. La composition initiale, mise au point au laboratoire par
J. Gonzalez [152] a donc été prise comme point de départ. Cette composition est la suivante :
30% de polymère, 60% de graphite SFG44 et 10% de CMC.
Pour cette étude, un montage à trois électrodes a été utilisé dans une cellule de type I
(cf. II-2-1). La Figure IV-2 présente les voltamogrammes du dopage négatif d'une électrode
de P-4-FPT fixée sur un déployé d'aluminium de 4 cm2 à l'aide de chacun des liants. La
vitesse de balayage est de 20 mV/s et l'électrolyte une solution d'acétonitrile 1M NEt4CF3SO3.
Les électrodes sont composées de 60% de graphite SFG44, de 35% de P-4-FPT (30%
dans le cas de la CMC), et de 5% de liant (10% dans le cas de la CMC).
58
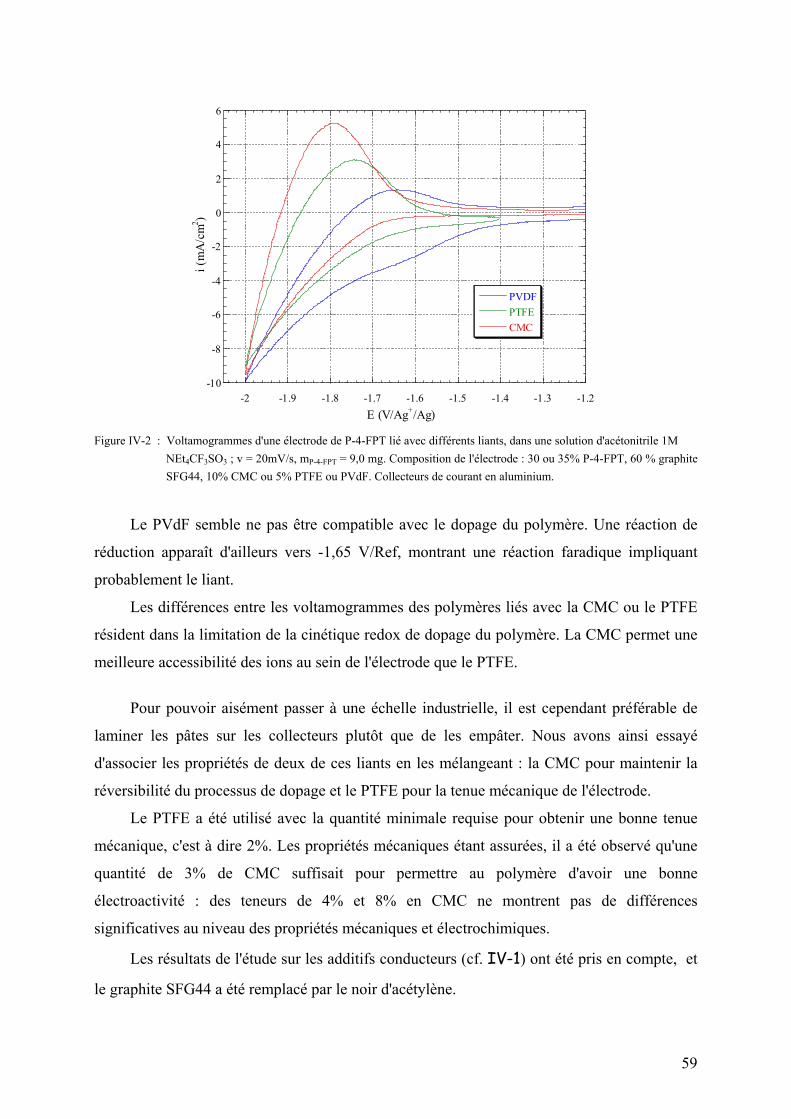
-10
-8
-6
-4
-2
0
2
4
6
-2 -1.9 -1.8 -1.7 -1.6 -1.5 -1.4 -1.3 -1.2
PVDFPTFECMC
i (m
A/c
m2 )
E (V/Ag+/Ag)
Figure IV-2 : Voltamogrammes d'une électrode de P-4-FPT lié avec différents liants, dans une solution d'acétonitrile 1M NEt4CF3SO3 ; v = 20mV/s, mP-4-FPT = 9,0 mg. Composition de l'électrode : 30 ou 35% P-4-FPT, 60 % graphite SFG44, 10% CMC ou 5% PTFE ou PVdF. Collecteurs de courant en aluminium.
Le PVdF semble ne pas être compatible avec le dopage du polymère. Une réaction de
réduction apparaît d'ailleurs vers -1,65 V/Ref, montrant une réaction faradique impliquant
probablement le liant.
Les différences entre les voltamogrammes des polymères liés avec la CMC ou le PTFE
résident dans la limitation de la cinétique redox de dopage du polymère. La CMC permet une
meilleure accessibilité des ions au sein de l'électrode que le PTFE.
Pour pouvoir aisément passer à une échelle industrielle, il est cependant préférable de
laminer les pâtes sur les collecteurs plutôt que de les empâter. Nous avons ainsi essayé
d'associer les propriétés de deux de ces liants en les mélangeant : la CMC pour maintenir la
réversibilité du processus de dopage et le PTFE pour la tenue mécanique de l'électrode.
Le PTFE a été utilisé avec la quantité minimale requise pour obtenir une bonne tenue
mécanique, c'est à dire 2%. Les propriétés mécaniques étant assurées, il a été observé qu'une
quantité de 3% de CMC suffisait pour permettre au polymère d'avoir une bonne
électroactivité : des teneurs de 4% et 8% en CMC ne montrent pas de différences
significatives au niveau des propriétés mécaniques et électrochimiques.
Les résultats de l'étude sur les additifs conducteurs (cf. IV-1) ont été pris en compte, et
le graphite SFG44 a été remplacé par le noir d'acétylène.
59
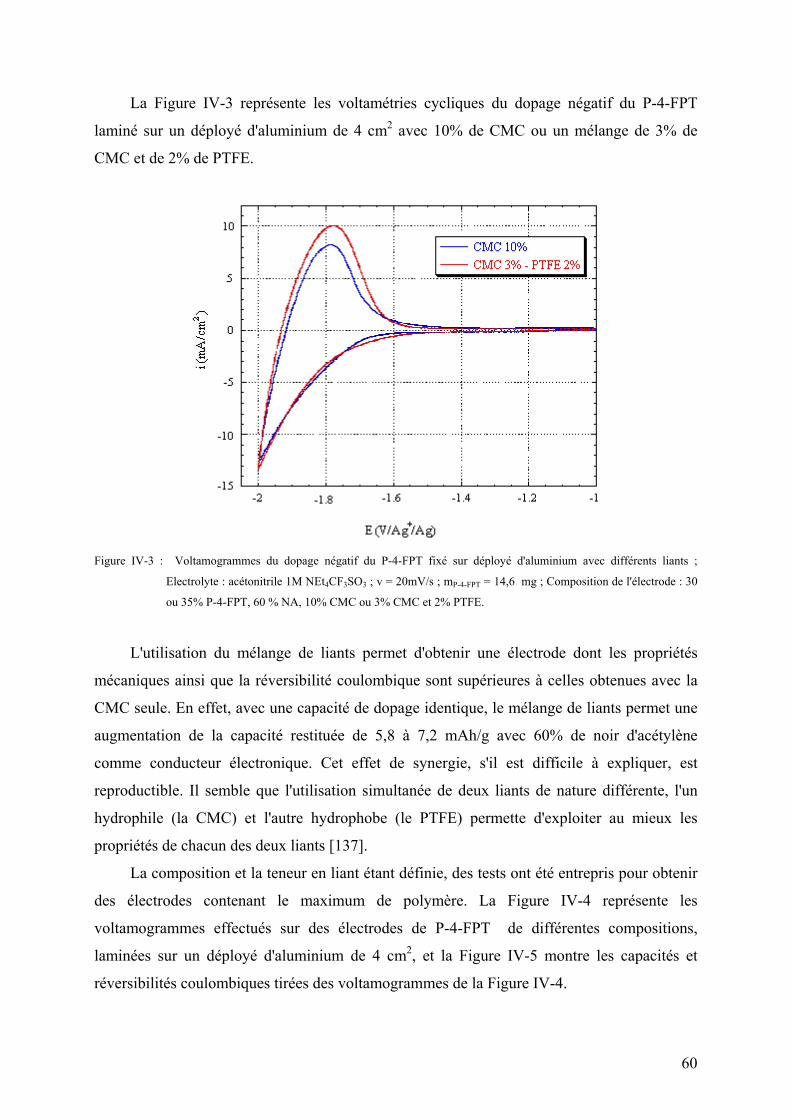
La Figure IV-3 représente les voltamétries cycliques du dopage négatif du P-4-FPT
laminé sur un déployé d'aluminium de 4 cm2 avec 10% de CMC ou un mélange de 3% de
CMC et de 2% de PTFE.
Figure IV-3 : Voltamogrammes du dopage négatif du P-4-FPT fixé sur déployé d'aluminium avec différents liants ;
Electrolyte : acétonitrile 1M NEt4CF3SO3 ; v = 20mV/s ; mP-4-FPT = 14,6 mg ; Composition de l'électrode : 30
ou 35% P-4-FPT, 60 % NA, 10% CMC ou 3% CMC et 2% PTFE.
L'utilisation du mélange de liants permet d'obtenir une électrode dont les propriétés
mécaniques ainsi que la réversibilité coulombique sont supérieures à celles obtenues avec la
CMC seule. En effet, avec une capacité de dopage identique, le mélange de liants permet une
augmentation de la capacité restituée de 5,8 à 7,2 mAh/g avec 60% de noir d'acétylène
comme conducteur électronique. Cet effet de synergie, s'il est difficile à expliquer, est
reproductible. Il semble que l'utilisation simultanée de deux liants de nature différente, l'un
hydrophile (la CMC) et l'autre hydrophobe (le PTFE) permette d'exploiter au mieux les
propriétés de chacun des deux liants [137].
La composition et la teneur en liant étant définie, des tests ont été entrepris pour obtenir
des électrodes contenant le maximum de polymère. La Figure IV-4 représente les
voltamogrammes effectués sur des électrodes de P-4-FPT de différentes compositions,
laminées sur un déployé d'aluminium de 4 cm2, et la Figure IV-5 montre les capacités et
réversibilités coulombiques tirées des voltamogrammes de la Figure IV-4.
60

-20
-15
-10
-5
0
5
10
15
-2 -1.8 -1.6 -1.4 -1.2 -1
80% P-4-FPT ; 15% NA75% P-4-FPT ; 20% NA70% P-4-FPT ; 25% NA65% P-4-FPT ; 30% NA30% P-4-FPT ; 65% NA
I (m
A/c
m2 )
E (V/Ag+/Ag)
Figure IV-4 : Voltamogrammes du dopage négatif du P-4-FPT avec différentes compositions d'électrode, dans une solution
d'acétonitrile 1M NEt4CF3SO3 ; v = 20mV/s, mP-4-FPT = 20,2 mg ; Composition de l'électrode : X% P-4-FPT,
(95-X%) NA, 3% CMC et 2% PTFE ; Collecteurs de courant en aluminium de 4 cm2.
-10
-5
0
5
10
65
70
75
80
85
90
10 20 30 40 50 60 70
Qch
arge
(mA
h/g)
Qdé
char
ge (m
Ah/
g)R
éversibilité coulombique (%
)
Teneur en noir d'acétylène (%)
Figure IV-5 : Capacités et réversibilités coulombiques associées à la Figure IV-4.
Sur la Figure IV-5, la percolation électronique apparaît clairement à partir d'une teneur
de 30% en NA. L'amélioration obtenue si l'on augmente encore cette teneur est très faible. En
dessous de cette valeur, plus la teneur en NA diminue, et plus l'électroactivité du polymère
diminue. Ceci est probablement dû à une perte de conduction et de drainage électronique au
61

sein de l'électrode. Il y a alors un étalement du pic de dédopage dû aux chutes ohmiques ainsi
qu'une perte de capacité dopée et dédopée dû à une mauvaise accessibilité des électrons aux
chaînes polymères.
Pour la caractérisation des polymères, la composition pour l'électrode négative a donc
été fixée à :
- 30 % de polymère,
- 65 % de noir d'acétylène,
- 5 % de liant dont 3 % de CMC et 2 % de PTFE.
Cette composition d'électrode garantit une très bonne percolation électronique et de ce
fait une bonne conductivité dans l'électrode, ce qui permet le dopage de la totalité du
polymère.
En revanche, pour la fabrication d'électrodes de supercondensateurs, la teneur en
polymère doit être maximale, car la quantité d'additif conducteur augmente la masse de
l'électrode. Il y a donc un compromis à trouver entre l'amélioration de la capacité massique du
polymère et la contribution de l'additif à la masse de matière active (polymère + additif
conducteur + liant). La composition retenue est alors :
- 65 % de polymère,
- 30 % de noir d'acétylène,
- 5 % de liant dont 3 % de CMC et 2 % de PTFE.
En effet, la teneur de 30% en NA est la quantité minimale requise pour obtenir la
percolation électronique au sein de l'électrode. Pour des teneurs inférieures, la baisse de
capacité et de conduction est significative.
IV-3) Etude du dopage positif
Une étude a ensuite été entreprise pour adapter ces résultats au dopage positif du
polymère. La Figure IV-6 représente le dopage positif du P-4-FPT lié sur un déployé d'inox
AISI 316L avec 3% de CMC et 2% de PTFE, pour différentes compositions d'électrodes. La
Figure IV-7 montre les capacités et réversibilités coulombiques tirées des voltamogrammes de
la Figure IV-6.
62
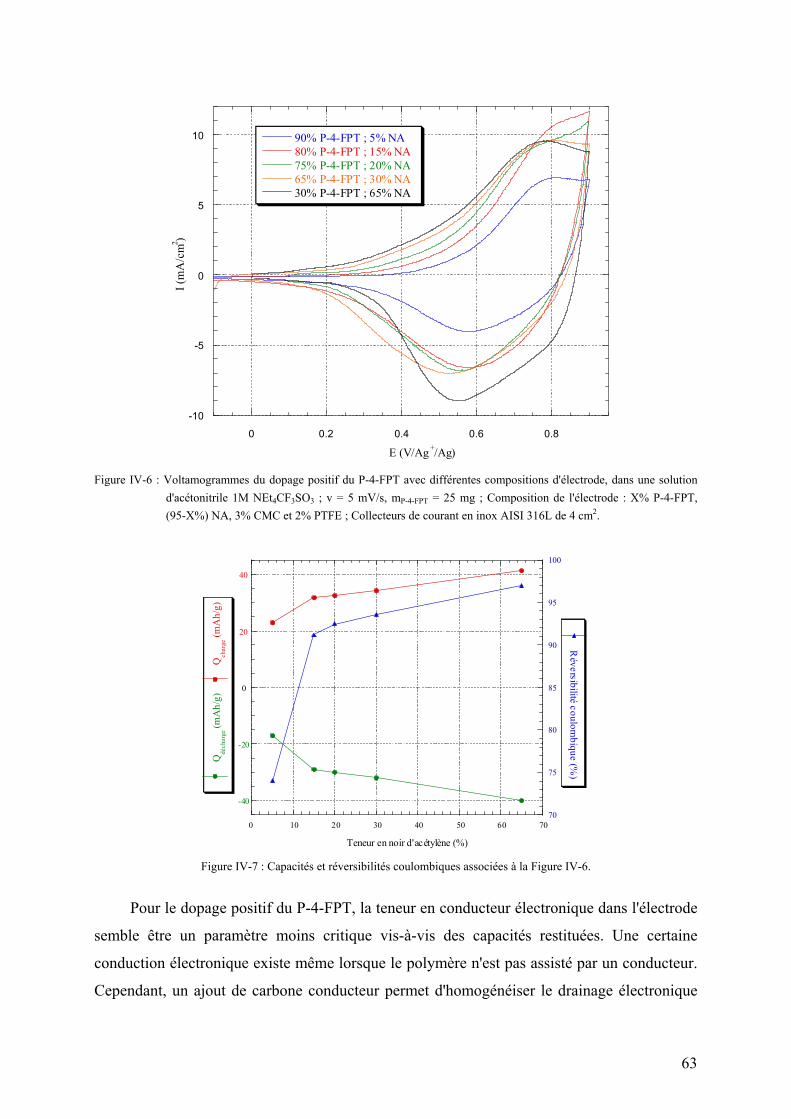
-10
-5
0
5
10
0 0.2 0.4 0.6 0.8
90% P-4-FPT ; 5% NA80% P-4-FPT ; 15% NA75% P-4-FPT ; 20% NA65% P-4-FPT ; 30% NA30% P-4-FPT ; 65% NA
I (m
A/c
m2 )
E (V/Ag +/Ag)
Figure IV-6 : Voltamogrammes du dopage positif du P-4-FPT avec différentes compositions d'électrode, dans une solution d'acétonitrile 1M NEt4CF3SO3 ; v = 5 mV/s, mP-4-FPT = 25 mg ; Composition de l'électrode : X% P-4-FPT, (95-X%) NA, 3% CMC et 2% PTFE ; Collecteurs de courant en inox AISI 316L de 4 cm2.
-40
-20
0
20
40
70
75
80
85
90
95
100
0 10 20 30 40 50 60 70
Qch
arge
(mA
h/g)
Qdé
char
ge (m
Ah/
g)R
éversibilité coulombique (%
)
Teneur en noir d'acétylène (%)
Figure IV-7 : Capacités et réversibilités coulombiques associées à la Figure IV-6.
Pour le dopage positif du P-4-FPT, la teneur en conducteur électronique dans l'électrode
semble être un paramètre moins critique vis-à-vis des capacités restituées. Une certaine
conduction électronique existe même lorsque le polymère n'est pas assisté par un conducteur.
Cependant, un ajout de carbone conducteur permet d'homogénéiser le drainage électronique
63
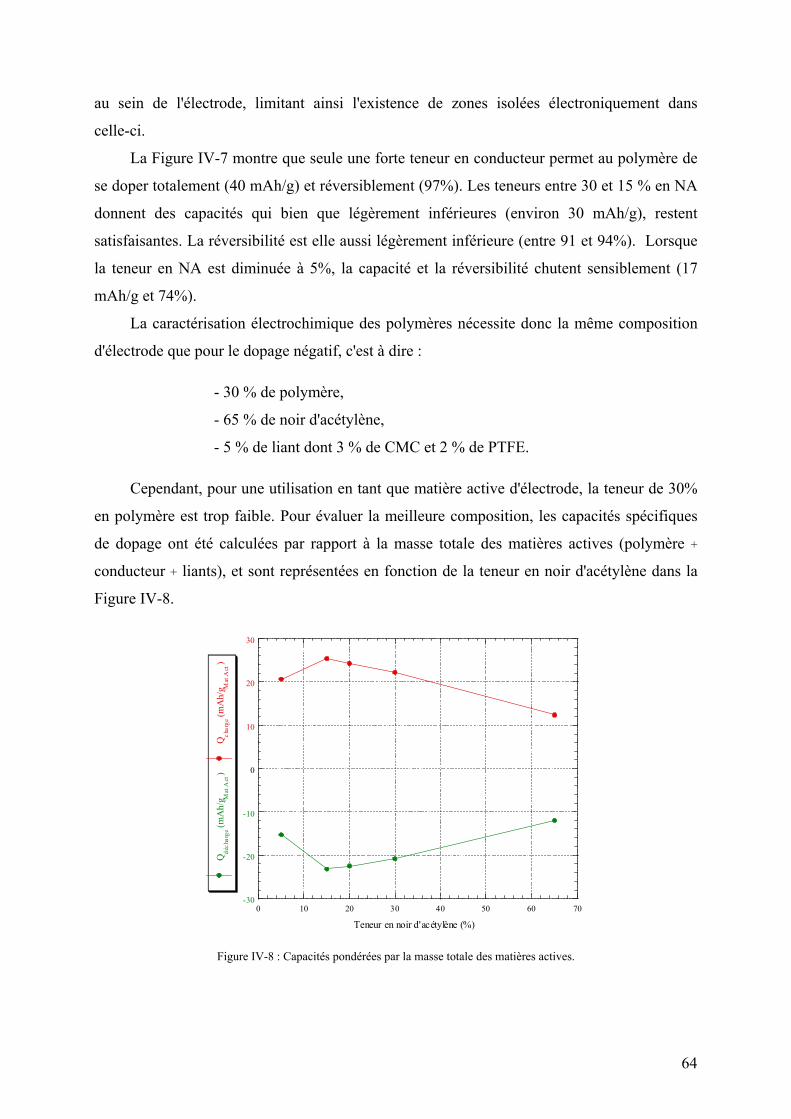
au sein de l'électrode, limitant ainsi l'existence de zones isolées électroniquement dans
celle-ci.
La Figure IV-7 montre que seule une forte teneur en conducteur permet au polymère de
se doper totalement (40 mAh/g) et réversiblement (97%). Les teneurs entre 30 et 15 % en NA
donnent des capacités qui bien que légèrement inférieures (environ 30 mAh/g), restent
satisfaisantes. La réversibilité est elle aussi légèrement inférieure (entre 91 et 94%). Lorsque
la teneur en NA est diminuée à 5%, la capacité et la réversibilité chutent sensiblement (17
mAh/g et 74%).
La caractérisation électrochimique des polymères nécessite donc la même composition
d'électrode que pour le dopage négatif, c'est à dire :
- 30 % de polymère,
- 65 % de noir d'acétylène,
- 5 % de liant dont 3 % de CMC et 2 % de PTFE.
Cependant, pour une utilisation en tant que matière active d'électrode, la teneur de 30%
en polymère est trop faible. Pour évaluer la meilleure composition, les capacités spécifiques
de dopage ont été calculées par rapport à la masse totale des matières actives (polymère +
conducteur + liants), et sont représentées en fonction de la teneur en noir d'acétylène dans la
Figure IV-8.
-30
-20
-10
0
10
20
30
0 10 20 30 40 50 60 70
Qch
arge
(mA
h/g M
at.A
ct )Q
déch
arge
(mA
h/g M
at.A
ct )
Teneur en noir d'acétylène (%)
Figure IV-8 : Capacités pondérées par la masse totale des matières actives.
64

Le meilleur compromis entre capacité et teneur en conducteur est donc :
- 80 % de polymère,
- 15 % de noir d'acétylène,
- 5 % de liant dont 3 % de CMC et 2 % de PTFE.
65

V) Caractérisation des polymères
Le polythiophène et les dérivés synthétisés dans ce travail se présentent sous forme de
poudres amorphes très fines avec des couleurs variant du rouge au marron. Ils sont tout à fait
stables à l'air.
Ces polymères sont totalement insolubles dans les solvants connus, organiques ou
aqueux. Leur caractérisation chimique est donc très restreinte : on ne peut pas déterminer de
masse en nombre ou en poids à partir des techniques chromatographiques classiques ou de la
spectrométrie de masse. Il est aussi très difficile et très long de déterminer précisément la
régiorégularité de leur structure (taux de greffages tête-à-tête et tête-à-queue) car il faut pour
cela employer la RMN en phase solide. Nous avons cependant effectué une analyse
élémentaire des poudres pour vérifier les teneurs respectives des atomes composant les
polymères et pour quantifier les principales impuretés présentes dans les poudres (le fer et le
chlore). Les spectres infrarouges des composés ont également été enregistrés.
La principale caractérisation des polymères a été faite en utilisant des méthodes
électrochimiques : la voltamétrie cyclique, la spectroscopie d'impédance et le cyclage
galvanostatique. Ces méthodes ont permis de caractériser l'électroactivité des polymères, c'est
à dire les processus positifs et négatifs de dopage. Cette étude étant destinée à la sélection de
polymères pour une application en tant que matières actives dans des électrodes pour
supercondensateurs, les grandeurs caractéristiques à mesurer pour ces polymères sont d'une
part la quantité d'électricité stockée/restituée lors du dopage ainsi que la réversibilité associée,
et d'autre part les potentiels de dopage, qui déterminent la tension aux bornes des
supercondensateurs. La stabilité de ces processus au cours d'un nombre important de cycles de
dopage/dédopage est aussi très importante car elle détermine la durée de vie du système.
Ces trois caractéristiques de l'électroactivité des polymères dépendent fortement du
milieu dans lequel le polymère est étudié. Des études sur ce sujet ont montré que des solvants
organiques aprotiques (comme l'acétonitrile, le carbonate de propylène, le sulfolane ou encore
la γ–butyrolactone) favorisaient le dopage de ces polymères [40,108,127]. Les solvants
protiques sont en effet susceptibles d'attaquer les polymères lorsque ceux-ci sont à l'état dopé
[154]. Il a également été montré que les sels d'ammonium dopaient les polythiophènes de
manière très efficace [87,92]. Nous avons donc décidé de caractériser les polymères dans
l'acétonitrile, solvant qui permet d'obtenir des électrolytes de bonne conductivité (30 mS.cm-1
à 25°C) et qui possède un domaine de stabilité en potentiel très étendu.
66
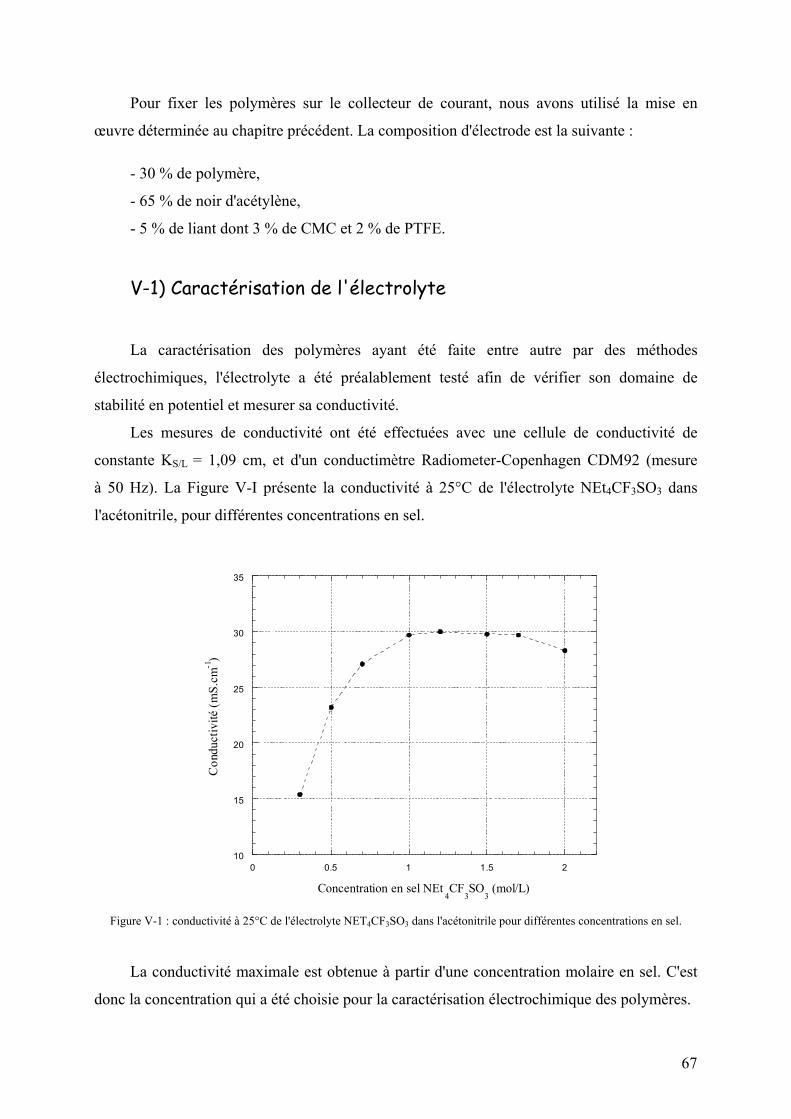
Pour fixer les polymères sur le collecteur de courant, nous avons utilisé la mise en
œuvre déterminée au chapitre précédent. La composition d'électrode est la suivante : - 30 % de polymère,
- 65 % de noir d'acétylène,
- 5 % de liant dont 3 % de CMC et 2 % de PTFE.
V-1) Caractérisation de l'électrolyte
La caractérisation des polymères ayant été faite entre autre par des méthodes
électrochimiques, l'électrolyte a été préalablement testé afin de vérifier son domaine de
stabilité en potentiel et mesurer sa conductivité.
Les mesures de conductivité ont été effectuées avec une cellule de conductivité de
constante KS/L = 1,09 cm, et d'un conductimètre Radiometer-Copenhagen CDM92 (mesure
à 50 Hz). La Figure V-I présente la conductivité à 25°C de l'électrolyte NEt4CF3SO3 dans
l'acétonitrile, pour différentes concentrations en sel.
10
15
20
25
30
35
0 0.5 1 1.5 2
Con
duct
ivité
(mS.
cm-1
)
Concentration en sel NEt4CF
3SO
3 (mol/L)
Figure V-1 : conductivité à 25°C de l'électrolyte NET4CF3SO3 dans l'acétonitrile pour différentes concentrations en sel.
La conductivité maximale est obtenue à partir d'une concentration molaire en sel. C'est
donc la concentration qui a été choisie pour la caractérisation électrochimique des polymères.
67

Le domaine de stabilité en potentiel de l'électrolyte a été testé pour une concentration
molaire en sel dans une cellule à trois électrodes de type I (cf. II-2-1). La Figure V-2 (a)
présente le voltamogramme effectué à 20 mV/s d'une électrode en platine.
-40
-30
-20
-10
0
10
20
30
40
-3 -2 -1 0 1 2 3
i (m
A/c
m2 )
E (V/Ag+/Ag) Figure V-2 (a) : Voltamogramme d'une électrode de platine dans l'électrolyte NET4CF3SO3 1M dans CH3CN.
Le domaine de stabilité en potentiel de l'électrolyte est important : l'oxydation de
l'électrolyte apparaît à +2,7 V/Ref tandis que sa réduction débute à -2,6 V/Ref.
Par ailleurs, il est nécessaire de vérifier le domaine de stabilité du collecteur de courant
dans cet électrolyte. La Figure V-2 (b) présente les voltamogrammes à 20 mV/s d'une
électrode en acier inoxydable AISI 316L, au premier et au 100ème cycle. Cet acier est connu
pour sa bonne résistance à la corrosion en milieu aqueux.
68

-60
-40
-20
0
20
40
-3 -2 -1 0 1
1er cycle100èmecycle
i (µA
/cm
2 )
E (V/Ag+/Ag)
Figure V-2 (b) : 100 cycles de voltamétrie cyclique d'une électrode d'inox AISI 316L dans l'électrolyte NET4CF3SO3 1M
dans l'acétonitrile.
Le voltamogramme du premier cycle montre une oxydation de l'acier qui débute à
+0,85 V/Ref avec un courant résiduel de 45 µA/cm2 à +1,3 V/Ref. Les produits d'oxydation
sont réduits vers -2,5 V/Ref. La réduction observée vers -2,0 V/Ref comprend aussi
certainement une part de réduction de l'électrolyte, les quantités d'électricité mises en jeu étant
supérieures à celles de l'oxydation du collecteur.
L'oxydation du collecteur est accompagnée d'une passivation et a tendance à diminuer
avec les cycles, pour n'atteindre, au 100ème cycle plus que 30 µA/cm2 à +1,3 V/Ref et
3 µA/cm2 à +1,0 V/Ref. La réduction suit la même décroissance et le courant résiduel au
100ème cycle est de 10 µA/cm2 à -2,0 V/Ref.
Cet électrolyte est donc stable entre -2,0 V/Ref et +1,0 V/Ref, si l'on admet un courant
résiduel 10 µA/cm2.
69

V-2) Caractérisation du polythiophène (PTh)
V-2-1) Caractérisation chimique du PTh
Le PTh a été polymérisé par polycondensation de 2,5-dibromothiophène (cf. III-2). Il
a ensuite été lavé pour éliminer le catalyseur ainsi que les autres produits de la synthèse. Sa
pureté a été évaluée par analyse élémentaire de ses constituants ainsi que par spectroscopie
infrarouge.
Le Tableau V-1 présente les résultats des analyses élémentaires. Sont indiquées la
quantité théorique calculée ainsi que la quantité mesurée dans l'échantillon.
C S H O Pureté
Calculé 58,54 39,02 2,44 0 100
Mesuré 57,26 38,98 2,03 1,57 98,27
Tableau V-1 : Analyse élémentaire du PTh synthétisé par polycondensation (valeurs en pourcentages massiques).
Le polymère est obtenu avec plus de 98% de pureté. La principale impureté est
l'oxygène, contenu à 1,57% dans le polymère.
La Figure V-3 représente le spectre infrarouge du PTh entre 600 et 3300 cm-1 et le
Tableau V-2 présente l'attribution des principaux pics du spectre [155-156].
0
0.05
0.1
0.15
0.2
0.25
0.3
10001500200025003000
Abs
orpt
ion
Longueur d'onde (cm-1)400
Figure V-3 : Spectre infrarouge du PTh entre 400 et 3200 cm-1.
70

Fréquence (cm-1) Attribution 3066 f νC-H 1508 m νC=C (asym) 1413 TF νC=C (sym) 1338 F ep νC-C 1245 F ep - 1211 F ep δC-H β 1149 TF δC-H β 1043 F δC-H β 836 tf γC-H α 786 TF γC-H β 725 f γC-H α 694 f déformation C-S-C 617 f -
Tableau V-2 : Attribution des principaux pics du spectre infrarouge du PTh.
Intensités : TF : très forte, F : forte, m : moyenne, f : faible, ep : épaulement. Notations des vibrations : ν : vibrations de valence, δ : vibrations de déformation dans le plan, γ : vibrations de déformation
hors du plan.
Le spectre est conforme à ceux de la littérature [13-14,69]. Le pic situé à 786 cm-1
correspond à la déformation hors du plan des liaisons C-H placées en β du soufre tandis que
les pics situés à 725 et 836 cm-1 correspondent aux mêmes modes pour les liaisons C-H
placées en α du soufre, et qui sont donc des bouts de chaînes [14,47,157-158]. La faible
intensité de ces pics montre que le polymère possède de fortes masses moléculaires.
Le spectre ne montre pas de pics attribuables à des liaisons C=O (entre 1700 et 1750
pour des carbonyles conjugués) qui auraient pu être présents dans le polymère [155]. La
teneur en oxygène est donc probablement due à des molécules d'oxygène ou d'eau adsorbées à
la surface de l'échantillon.
V-2-2) Caractérisation électrochimique du PTh
V-2-2-1) Voltamétries cycliques des processus de dopage
Les Figures V-4 et V-5 présentent les voltamogrammes des dopages négatif et positif du
PTh dans l'électrolyte NEt4+,CF3SO3
- 1M dans l'acétonitrile. Ces caractérisations ont été
effectuées dans une cellule de type I (à trois électrodes) avec une contre-électrode de platine
de surface 4 cm2 (cf. II-2-1).
71

-12
-8
-4
0
4
-2.6 -2.4 -2.2 -2 -1.8 -1.6 -1.4
i (m
A/c
m2 )
E (V/Ag+/Ag)
-10
-5
0
5
10
15
20
-0.2 0 0.2 0.4 0.6 0.8 1
i (m
A/c
m2 )
E (V/Ag+/Ag) Figure V-4 : Voltamogramme représentant le dopage
négatif du PTh dans une solution d'acétonitrile 1M
NEt4CF3SO3 ; v=20mV/s, mPTh = 15 mg. Composition de
l'électrode : 30% PTh, 65% NA, 3% CMC et 2% PTFE.
Collecteur de courant en inox AISI 316L.
Figure V-5 : Voltamogramme représentant le dopage
positif du PTh dans une solution d'acétonitrile 1M
NEt4CF3SO3 ; v=20mV/s, mPTh = 5,1 mg. Composition de
l'électrode : 30% PTh, 65% NA, 3% CMC et 2% PTFE.
Collecteur de courant en inox AISI 316L.
Le dopage négatif du polymère correspond à sa réduction, c'est à dire à l'injection
d'électrons dans ses chaînes. Pour maintenir l'électroneutralité, des cations de l'électrolyte
s'insèrent dans l'électrode (Equation. (V-1)). Il y a un changement de structure du polymère
qui devient alors conducteur.
PTh + e- + NEt4+ PTh-,NEt4
+ Eq. (V-1)
Le dopage négatif se situe à des potentiels très négatifs, typiquement vers -2,2 V/Ref.
Dans ce cas, il n'est pas possible d'obtenir le pic de dopage car celui-ci vient se confondre
avec la courbe de réduction du solvant vers -2,5V/Ref. Le dédopage correspond à l'extraction
des électrons injectés dans les chaînes polymères durant le dopage ainsi qu'à la désinsertion
des cations. Le polymère revient à son état neutre et isolant. Le dédopage s'étend de
-2,15V/Ref à -1,7V/Ref avec un pic à -1,78V/Ref.
Lors du dopage positif du polymère, c'est à dire son oxydation, les électrons sont
arrachés du polymère et ce sont des anions qui s'insèrent pour préserver l'électroneutralité,
selon l'équation (V-2).
PTh + CF3SO3- PTh+,CF3SO3
- + e- Eq. (V-2) Le pic d'oxydation débute à +0,4 V/Ref et atteint son maximum à +0,87 V/Ref. Le
dédopage s'étend sur 900 mV entre +0,9 et +0V/Ref. D'après ces courbes, la capacité chargée
72
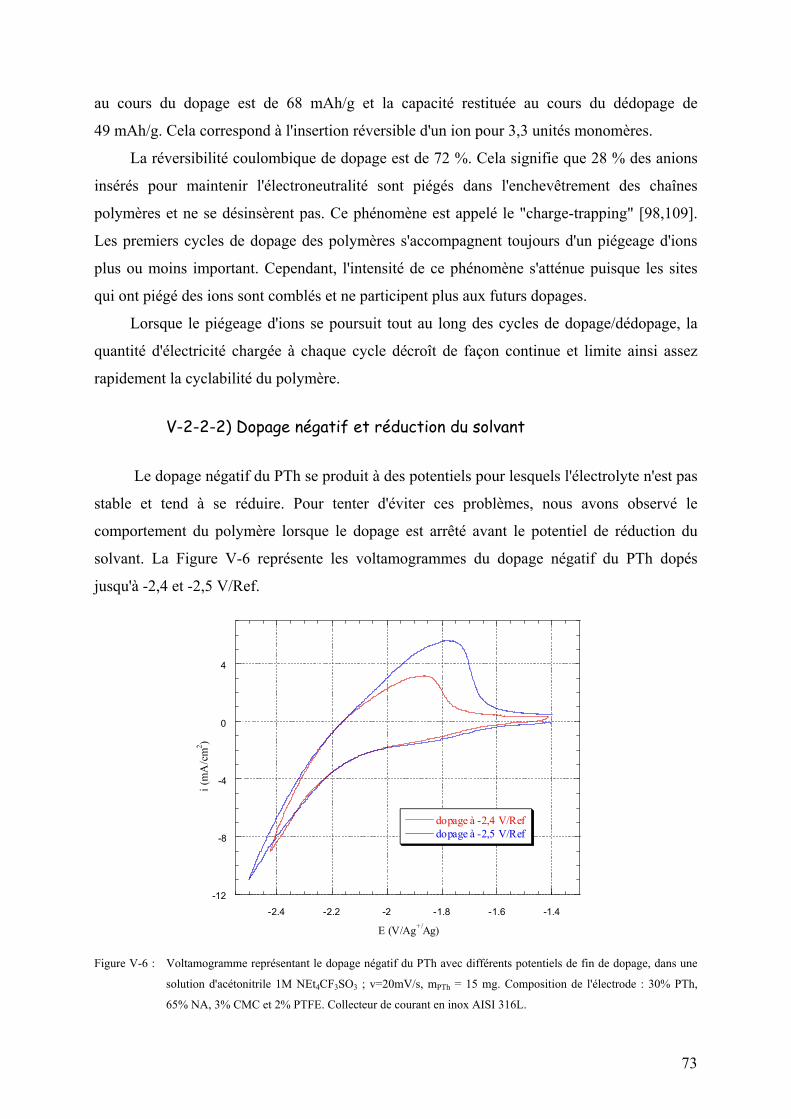
au cours du dopage est de 68 mAh/g et la capacité restituée au cours du dédopage de
49 mAh/g. Cela correspond à l'insertion réversible d'un ion pour 3,3 unités monomères.
La réversibilité coulombique de dopage est de 72 %. Cela signifie que 28 % des anions
insérés pour maintenir l'électroneutralité sont piégés dans l'enchevêtrement des chaînes
polymères et ne se désinsèrent pas. Ce phénomène est appelé le "charge-trapping" [98,109].
Les premiers cycles de dopage des polymères s'accompagnent toujours d'un piégeage d'ions
plus ou moins important. Cependant, l'intensité de ce phénomène s'atténue puisque les sites
qui ont piégé des ions sont comblés et ne participent plus aux futurs dopages.
Lorsque le piégeage d'ions se poursuit tout au long des cycles de dopage/dédopage, la
quantité d'électricité chargée à chaque cycle décroît de façon continue et limite ainsi assez
rapidement la cyclabilité du polymère.
V-2-2-2) Dopage négatif et réduction du solvant
Le dopage négatif du PTh se produit à des potentiels pour lesquels l'électrolyte n'est pas
stable et tend à se réduire. Pour tenter d'éviter ces problèmes, nous avons observé le
comportement du polymère lorsque le dopage est arrêté avant le potentiel de réduction du
solvant. La Figure V-6 représente les voltamogrammes du dopage négatif du PTh dopés
jusqu'à -2,4 et -2,5 V/Ref.
-12
-8
-4
0
4
-2.4 -2.2 -2 -1.8 -1.6 -1.4
dopage à -2,4 V/Refdopage à -2,5 V/Ref
i (m
A/c
m2 )
E (V/Ag+/Ag)
Figure V-6 : Voltamogramme représentant le dopage négatif du PTh avec différents potentiels de fin de dopage, dans une
solution d'acétonitrile 1M NEt4CF3SO3 ; v=20mV/s, mPTh = 15 mg. Composition de l'électrode : 30% PTh,
65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
73
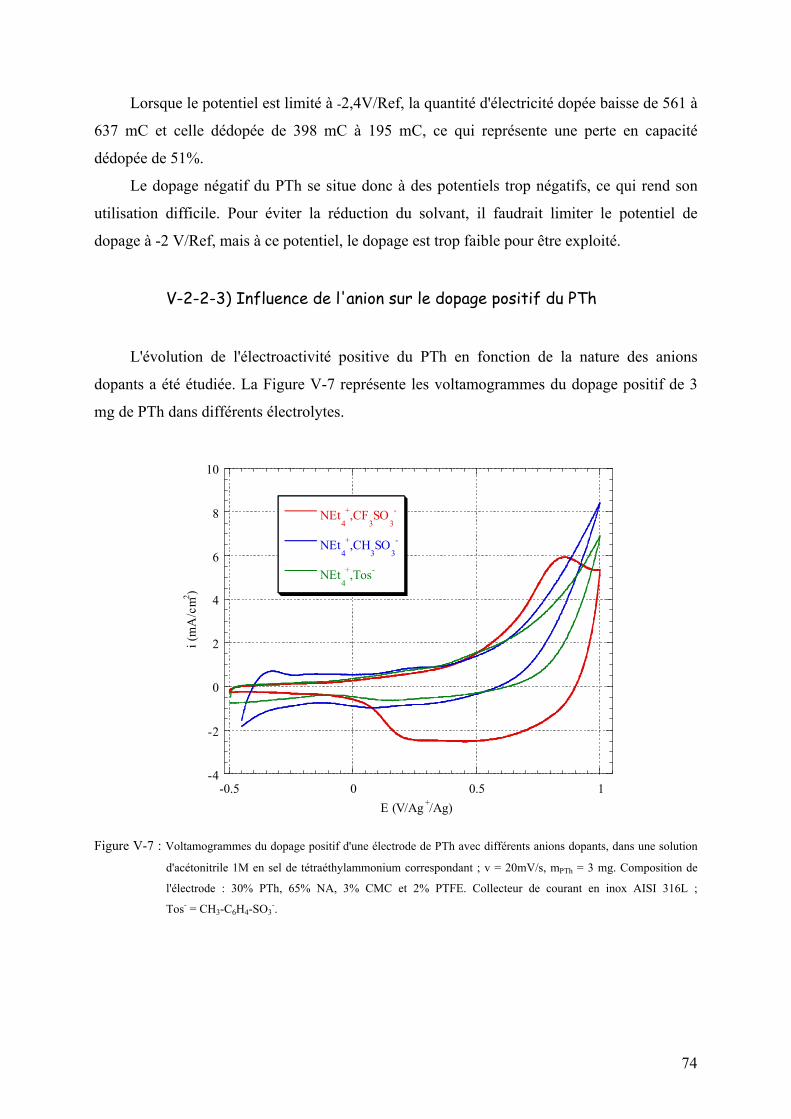
Lorsque le potentiel est limité à -2,4V/Ref, la quantité d'électricité dopée baisse de 561 à
637 mC et celle dédopée de 398 mC à 195 mC, ce qui représente une perte en capacité
dédopée de 51%.
Le dopage négatif du PTh se situe donc à des potentiels trop négatifs, ce qui rend son
utilisation difficile. Pour éviter la réduction du solvant, il faudrait limiter le potentiel de
dopage à -2 V/Ref, mais à ce potentiel, le dopage est trop faible pour être exploité.
V-2-2-3) Influence de l'anion sur le dopage positif du PTh
L'évolution de l'électroactivité positive du PTh en fonction de la nature des anions
dopants a été étudiée. La Figure V-7 représente les voltamogrammes du dopage positif de 3
mg de PTh dans différents électrolytes.
-4
-2
0
2
4
6
8
10
-0.5 0 0.5 1
NEt4+,CF
3SO
3-
NEt4+,CH
3SO
3-
NEt4+,Tos-
i (m
A/c
m2 )
E (V/Ag +/Ag)
Figure V-7 : Voltamogrammes du dopage positif d'une électrode de PTh avec différents anions dopants, dans une solution
d'acétonitrile 1M en sel de tétraéthylammonium correspondant ; v = 20mV/s, mPTh = 3 mg. Composition de
l'électrode : 30% PTh, 65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L ;
Tos- = CH3-C6H4-SO3-.
74

Les trois anions testés sont le trifluorométhanesulfonate (CF3SO3-), le méthanesulfonate
(CH3SO3-) et le paratoluènesulfonate (ou tosylate = CH3-C6H4-SO3
-). La comparaison des
quantités d'électricité chargées et déchargées permet de conclure que seul l'anion CF3SO3-
permet un dopage efficace et réversible du polymère. Les ions méthanesulfonate (CH3SO3-) et
tosylate (CH3-C6H4-SO3-) ne s'insérent pas de manière efficace dans l'électrode et il n'y a
qu'une très faible désinsertion au cours du dédopage. Les interactions stériques causées par la
taille importante de l'anion tosylate expliquent la mauvaise insertion de ces ions le long des
chaînes polymères. Concernant l'ion méthanesulfonate, l'hypothèse la plus probable pour
expliquer la faible électroactivité du PTh est l'existence d'une forte interaction entre la paire
d'ions composant le sel, ce qui aurait pour conséquence le dopage plus difficile de ce
polymère.
V-2-2-4) Cyclabilité du PTh en dopage positif
Le dopage positif du PTh étant très intéressant au point de vue de la quantité d'électricité
dopée, sa stabilité a été testée en voltamétrie cyclique sur un nombre de cycles important. Les
Figures V-8 et V-9 représentent les voltamogrammes du cyclage du PTh à 20 mV/s, dans
l'électrolyte NEt4+,CF3SO3
- 1M dans l'acétonitrile, La contre-électrode utilisée dans ce test est
une électrode surcapacitive de charbon actif empâté sur une mousse de nickel, ce qui permet
de limiter la variation en potentiel de cette électrode, et ainsi d'éviter la dégradation de
l'électrolyte. La Figure V-10 représente la variation des capacités chargées et déchargées ainsi
que la réversibilité coulombique de dopage, en fonction du nombre de cycles effectués aux
Figures V-8 et V-9.
75
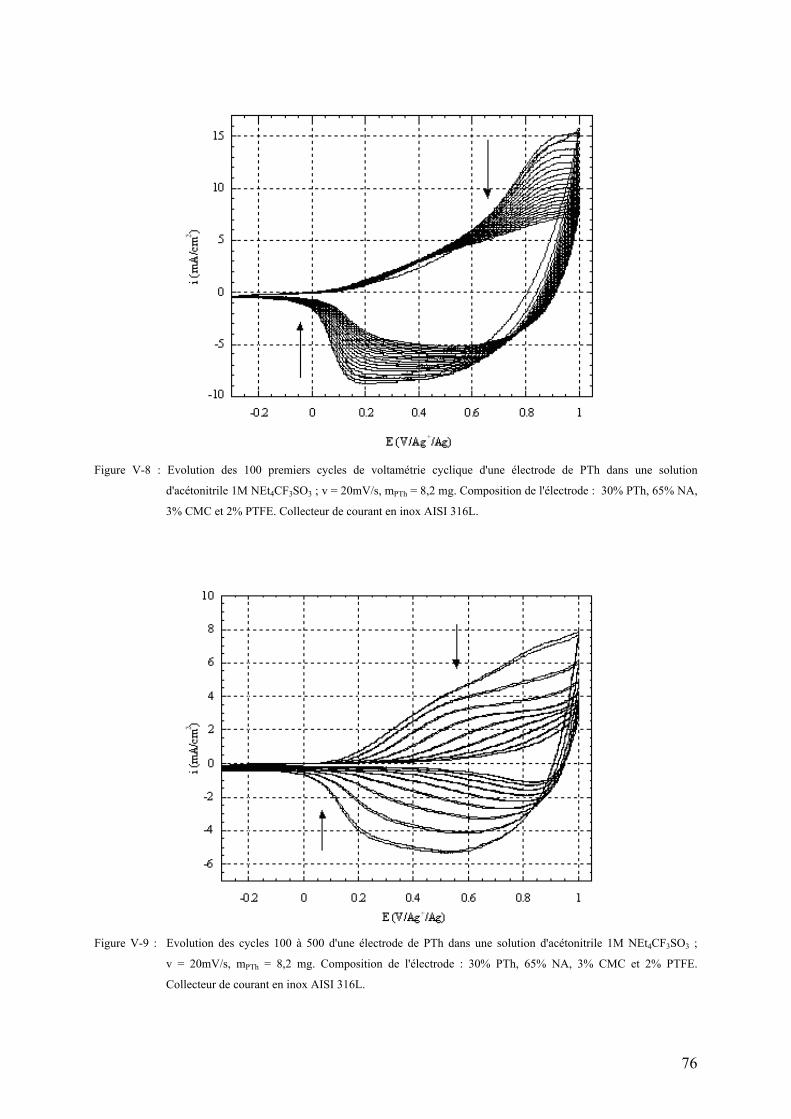
Figure V-8 : Evolution des 100 premiers cycles de voltamétrie cyclique d'une électrode de PTh dans une solution
d'acétonitrile 1M NEt4CF3SO3 ; v = 20mV/s, mPTh = 8,2 mg. Composition de l'électrode : 30% PTh, 65% NA,
3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
Figure V-9 : Evolution des cycles 100 à 500 d'une électrode de PTh dans une solution d'acétonitrile 1M NEt4CF3SO3 ;
v = 20mV/s, mPTh = 8,2 mg. Composition de l'électrode : 30% PTh, 65% NA, 3% CMC et 2% PTFE.
Collecteur de courant en inox AISI 316L.
76

-50
0
50
0
20
40
60
80
100
0 100 200 300 400 500
Qch
arge
(mA
h/g)
Qdé
char
ge (m
Ah/
g)R
éversibilité coulombique (%
)
Cycles
-10
-20
-30
-40
10
20
30
40
Figure V-10 : Evolutions des quantités d'électricité dopées et dédopées ainsi que de la réversibilité coulombique au cours
des cycles représentés sur les Figures V-8 et V-9.
On peut remarquer une rapide décroissance des quantités d'électricité associées à la
charge et à la décharge du polymère. Après 500 cycles, la capacité en charge du
polythiophène n'est plus que de 3,7 mAh/g pour une capacité de décharge de 2,9 mAh/g. Par
ailleurs, la réversibilité de dopage décroît au cours des cycles, ce qui montre une
augmentation du "charge-trapping".
Le PTh est donc capable de se doper négativement (réduction) et positivement
(oxydation). Cependant, sa réduction a lieu à des potentiels très négatifs, et la réduction de
l'électrolyte se produit simultanément, ce qui rend son exploitation difficile. En revanche, la
valeur du potentiel de dopage positif se situant dans le domaine de stabilité de l'électrolyte,
ce type de problème n'est pas rencontré. La capacité du dopage positif atteint près de 50
mAh/g. Cependant, ce dopage montre une mauvaise stabilité en cyclage, avec une perte de
92 % de capacité en 500 cycles.
Le montage de supercondensateurs de type III nécessitant un polymère qui possède un
dopage négatif stable, nous avons testé un dérivé du PTh possédant un substituant
fluorophényl, le P-4-FPT. Ce substituant a un effet électroattracteur qui décale les processus
de dopage vers des potentiels plus positifs, ce qui devrait placer le dopage négatif dans le
domaine de stabilité du solvant [1]. Par ailleurs, le greffage de substituants sur le
polythiophène permet de stabiliser les dopages positifs et négatifs [159].
77

V-3) Caractérisation du Poly-4-fluorophénylthiophène (P-4-FPT)
V-3-1) Caractérisation chimique du P-4-FPT
Le P-4-FPT se présente sous la forme d'une poudre marron très fine. C'est un polymère
amorphe insoluble et infusible (des mesures de DSC ont été menées jusqu'à 250°C à l'air sans
observer de transition thermique ni de dégradation). Son insolubilité limite les techniques de
caractérisation. Cependant, sa pureté a été appréciée par l'analyse élémentaire de ses
composants et une caractérisation par spectroscopie infrarouge a été faite.
Le Tableau V-3 présente les résultats des analyses élémentaires effectuées après lavage
et séchage du polymère. Le tableau indique la quantité théorique calculée ainsi que la quantité
mesurée dans l'échantillon. Les deux principales impuretés liées au mode de synthèse (le
chlore et le fer) ont été analysées.
C H S F Fe Cl Pureté
Calculé 68,18 2,84 18,18 10,79 0 0 100
Mesuré 67,13 2,83 18,05 10,25 0,25 1,49 98,26
Tableau V-3 : Analyse élémentaire du P-4-FPT (valeurs en pourcentages massiques).
Le P-4-FPT a été obtenu avec plus de 98% de pureté. Les deux seules impuretés
présentes en quantités non négligeables sont le fer et le chlore, le total des pourcentages
incluant ces éléments donnant 100%.
La Figure V-11 montre le spectre infrarouge du P-4-FPT et le Tableau V-4 présente
l'attribution des pics [155-156,160].
78
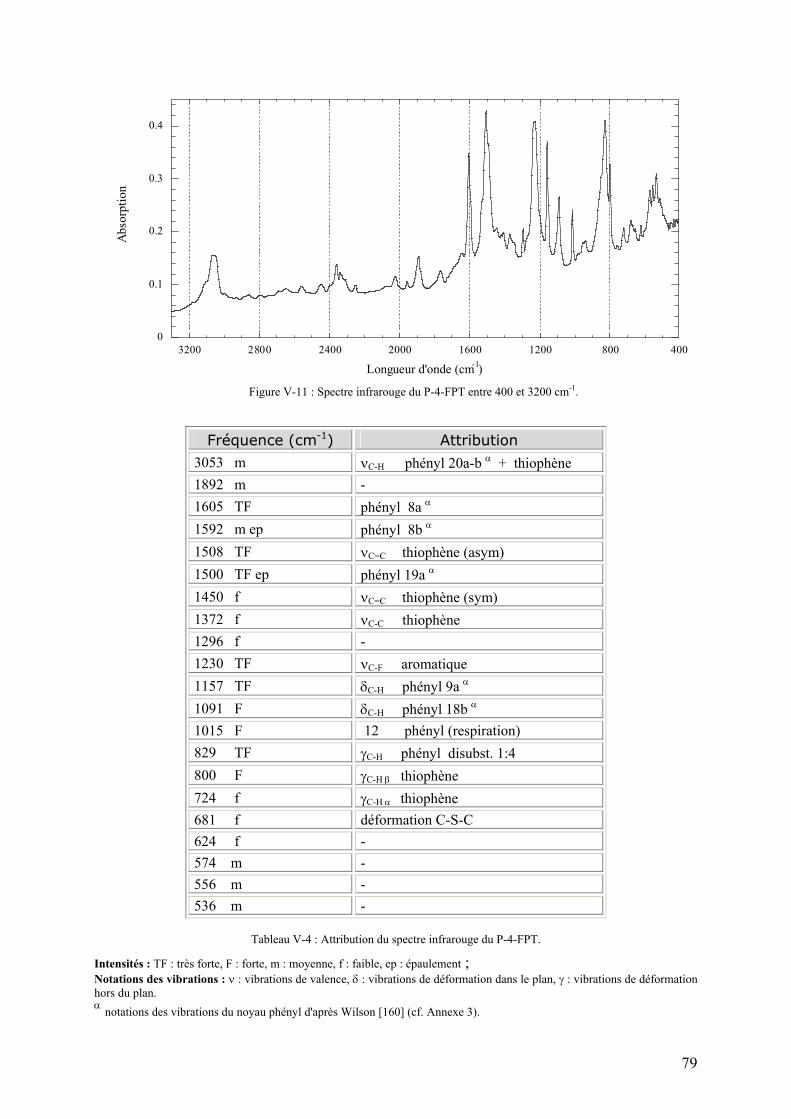
0
0.1
0.2
0.3
0.4
800120016002000240028003200
Abs
orpt
ion
Longueur d'onde (cm-1)400
Figure V-11 : Spectre infrarouge du P-4-FPT entre 400 et 3200 cm-1.
Fréquence (cm-1) Attribution 3053 m νC-H phényl 20a-b α + thiophène 1892 m - 1605 TF phényl 8a α 1592 m ep phényl 8b α 1508 TF νC=C thiophène (asym) 1500 TF ep phényl 19a α 1450 f νC=C thiophène (sym) 1372 f νC-C thiophène 1296 f - 1230 TF νC-F aromatique 1157 TF δC-H phényl 9a α 1091 F δC-H phényl 18b α 1015 F 12 phényl (respiration) 829 TF γC-H phényl disubst. 1:4 800 F γC-H β thiophène 724 f γC-H α thiophène 681 f déformation C-S-C 624 f - 574 m - 556 m - 536 m -
Tableau V-4 : Attribution du spectre infrarouge du P-4-FPT.
Intensités : TF : très forte, F : forte, m : moyenne, f : faible, ep : épaulement ; Notations des vibrations : ν : vibrations de valence, δ : vibrations de déformation dans le plan, γ : vibrations de déformation hors du plan. α notations des vibrations du noyau phényl d'après Wilson [160] (cf. Annexe 3).
79

L'attribution des principaux pics a été faite. Les principales vibrations de déformation
des noyaux phényl et thiophène sont visibles, ainsi que la bande de vibration de valence de la
liaison C-F. Le degré de polymérisation peut être apprécié en mesurant la différence d'aire
sous les pics de vibration de déformation hors du plan (γ) des liaisons C-H situées en position
α et β du soufre [23,65] (cf. I-1-3-1). Cependant, l'attribution des pics de vibration des ces
deux liaisons est délicate et les auteurs ne sont pas toujours en accord sur ce point [14,23-
24,28,47,64]. Pour pouvoir discriminer ces deux bandes de vibration, le spectre du monomère
4-FPT a été enregistré (cf. Figure V-12) et une attribution précise de la région 650-900 cm-1 a
été effectuée (cf. Tableau V-5).
0
0.5
1
1.5
2
2.5
3
3.5
650700750800850900
4-FPTP-4-FPT
Abs
.
Longueur d'onde (cm-1)
Figure V-12 : Spectre infrarouge du P-4-FPT et du 4-FPT entre 650 et 900 cm-1.
4-FPT P-4-FPT Attribution 864 f 865 tf (ep) 836 F 829 TF γC-H phényl disubstitué en 1:4 797 tf 800 F γC-H β thiophène 772 TF γC-H α thiophène 709 m 724 f γC-H α thiophène 688 f 681 f C-S-C déformation
Tableau V-5 : Attribution du spectre infrarouge du 4-FPT et du P-4-FPT.
Le pic de vibration situé à 836 cm-1 pour le 4-FPT et à 829 cm-1 pour le P-4-FPT est
caractéristique des vibrations de déformation hors du plan des protons d'un noyau phényl
80

disubstitué en 1:4 [155]. Ce pic apparaît plus étalé sur le spectre du polymère à cause des
interactions stériques entre les chaînes polymères.
Le pic apparaissant à 772 cm-1 pour le monomère, disparaît totalement sur le spectre du
polymère. Il a donc été attribué aux liaisons C-H en α du soufre, remplacées dans le polymère
par des liaisons C-C. La disparition totale de ce pic dans le spectre du P-4-FPT signifie que
celui-ci possède de hautes masses moléculaires.
Un pic de plus faible intensité, situé à 709 cm-1 dans le spectre du 4-FPT, est également
attribué aux vibrations de déformation hors du plan des liaisons C-H en α du soufre. Le
spectre du polymère possède un pic très faible et très étalé à 724 cm-1, qui correspond à la
même vibration, décalée par les interactions stériques de la structure polymère. Ce pic est
caractéristique de couplages défectueux 2,4 généralement observés à hauteur de 5% dans les
polymères synthétisés au chlorure ferrique (cf. I-1-2-1).
Le pic situé à 797 cm-1 pour le 4-FPT et à 800 cm-1 pour le P-4-FPT a été attribué aux
liaisons C-H en β du soufre.
V-3-2) Caractéristiques des processus de dopage
Pour ces expériences, de la même façon qu'avec le PTh, le P-4-FPT a été mis en oeuvre
dans des électrodes de composition 30% polymère, 65% noir d'acétylène, 3% CMC et 2%
PTFE. Les matières actives ont été laminées sur un micro-déployé d'acier inoxydable AISI
316L (Delker). Les tests ont été effectués dans une cellule de type I (trois électrodes) avec une
contre-électrode de charbon actif surcapacitive laminée sur un déployé métallique identique
(cf. II-2-1).
V-3-2-1 ) Voltamétrie cyclique du dopage négatif
La Figure V-13 représente les voltamogrammes des huit premiers cycles du dopage
négatif du P-4-FPT, effectués à 20mV/s dans un électrolyte NEt4+
,CF3SO3- 1M dans
l'acétonitrile ; la Figure V-14 montre l'évolution des quantités d'électricité mises en jeu durant
ces cycles ainsi que l'évolution de la réversibilité coulombique du processus.
81

-30
-20
-10
0
10
-2 -1.9 -1.8 -1.7 -1.6 -1.5 -1.4
i (m
A/c
m2 )
E (V/Ag +/Ag)
1er cycle
1er cycle
8e cycle
8e cycle
Figure V-13 : Voltamogramme représentant les huit premiers cycles d'une électrode de P-4-FPT dans une solution
d'acétonitrile 1M NEt4CF3SO3 ; v = 20mV/s, mP-4-FPT = 19,5 mg. Composition de l'électrode : 30% P-4-FPT,
65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
-25
-20
-15
-10
-5
0
5
10
0
20
40
60
80
100
1 2 3 4 5 6 7 8
Qch
arge
(mA
h/g)
Qdé
char
ge (m
Ah/
g)R
éversibilité coulombique (%
)
Cycles
Figure V-14 : Evolutions des quantités d'électricité dopées et dédopées et de la réversibilité coulombique du dopage au
cours des cycles représentés sur la Figure V-13.
82

La vague de réduction correspond au dopage négatif du P-4-FPT, c'est à dire à l'injection
d'électrons dans les chaînes polymères (cf. Eq. (V-3)). Pour maintenir l'électroneutralité, des
cations de l'électrolyte s'insèrent dans l'électrode. Le polymère devient alors conducteur.
P-4-FPT (0) + e- + NEt4+ P-4-FPT -, NEt4
+ Eq. (V-3)
Le pic de réoxydation correspond au dédopage du P-4-FPT, c'est à dire à l'extraction
des électrons injectés durant le dopage. Les cations insérés dans l'électrode se désinsérent
alors. Le polymère revient à son état neutre et isolant.
Il est nécessaire d'effectuer quelques cycles pour arriver à un équilibre stable entre la
quantité d'électricité chargée et celle déchargée. Pendant cette étape d'activation, les
aménagements stériques effectués par le polymère pour accueillir les cations dopants sont
importants. Pour adoucir les contraintes stériques subies par le polymère durant les premiers
cycles de dopage, la limite inférieure de potentiel a été fixée à -2 V/Ref, pour avoir un dopage
partiel.
D'après la Figure V-13 que la vague de réduction pour le premier cycle apparaît plus tôt
en potentiel que pour les cycles suivants. Cela signifie que lors du premier dopage, les sites
les plus électroactifs qui réduits. La quantité d'électricité chargée pendant ce premier cycle est
de 22,3 mAh/g. Cependant, la faible réoxydation (4 mAh/g) montre que les cations insérés
vont être pour la plupart piégés dans ces sites. Les sites qui ont piégé un cation ne sont alors
plus disponibles et lors du deuxième cycle, la quantité d'électricité chargée chute à 8,5 mAh/g.
Celle-ci diminue encore au cours des cycles suivants mais beaucoup plus faiblement.
La Figure V-14 montre que la capacité en décharge du polymère augmente en même
temps que celle en charge diminue. Elle passe de 4 mAh/g au premier cycle à 7 mAh/g au
8ème cycle. Au fur et à mesure que les sites piégeurs sont comblés, la proportion de sites
réversibles augmente, ce qui entraîne une augmentation de la réversibilité coulombique du
processus de 18 % à 91 % en 8 cycles. On considère que l'activation du polymère est achevée
lorsque les quantités d'électricité dopées et dédopées sont stables.
Cette étape d'activation a permis à la structure du polymère de s'ouvrir et lors du dopage
complet du polymère, les contraintes stériques seront moindres.
La Figure V-15 représente le dopage négatif complet du P-4-FPT, obtenu après l'étape
d'activation.
83
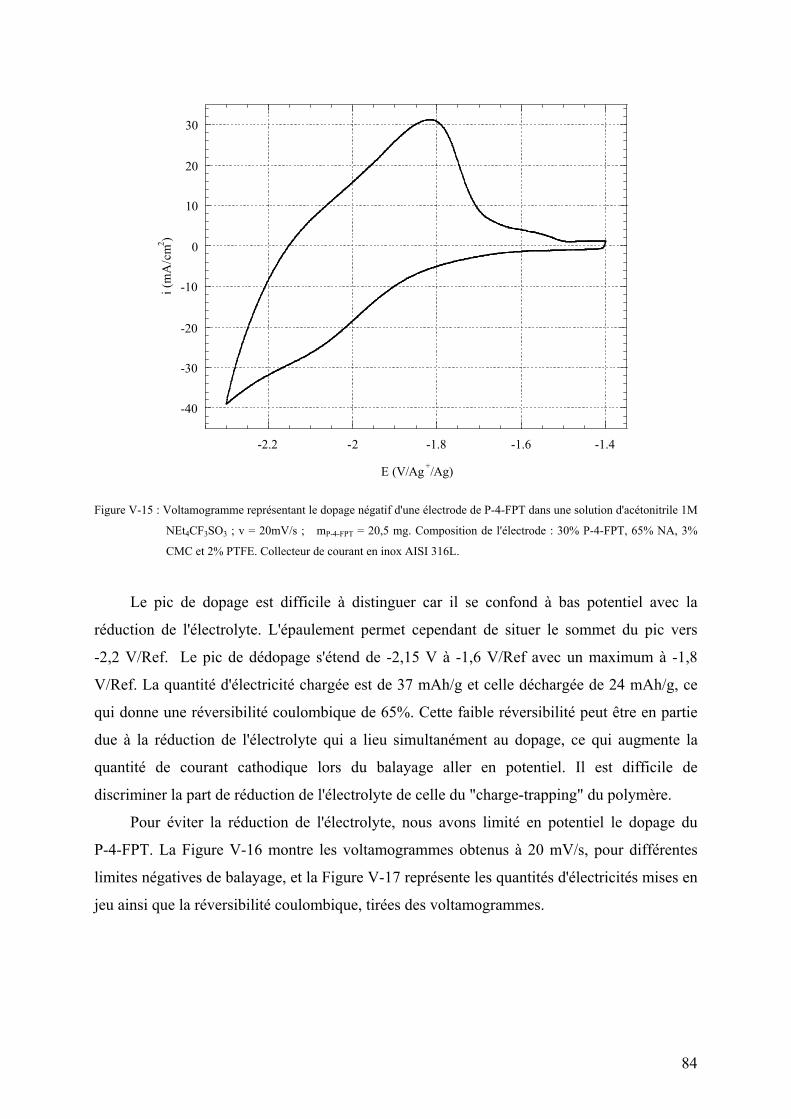
-40
-30
-20
-10
0
10
20
30
-2.2 -2 -1.8 -1.6 -1.4
i (m
A/c
m2 )
E (V/Ag +/Ag)
Figure V-15 : Voltamogramme représentant le dopage négatif d'une électrode de P-4-FPT dans une solution d'acétonitrile 1M
NEt4CF3SO3 ; v = 20mV/s ; mP-4-FPT = 20,5 mg. Composition de l'électrode : 30% P-4-FPT, 65% NA, 3%
CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
Le pic de dopage est difficile à distinguer car il se confond à bas potentiel avec la
réduction de l'électrolyte. L'épaulement permet cependant de situer le sommet du pic vers
-2,2 V/Ref. Le pic de dédopage s'étend de -2,15 V à -1,6 V/Ref avec un maximum à -1,8
V/Ref. La quantité d'électricité chargée est de 37 mAh/g et celle déchargée de 24 mAh/g, ce
qui donne une réversibilité coulombique de 65%. Cette faible réversibilité peut être en partie
due à la réduction de l'électrolyte qui a lieu simultanément au dopage, ce qui augmente la
quantité de courant cathodique lors du balayage aller en potentiel. Il est difficile de
discriminer la part de réduction de l'électrolyte de celle du "charge-trapping" du polymère.
Pour éviter la réduction de l'électrolyte, nous avons limité en potentiel le dopage du
P-4-FPT. La Figure V-16 montre les voltamogrammes obtenus à 20 mV/s, pour différentes
limites négatives de balayage, et la Figure V-17 représente les quantités d'électricités mises en
jeu ainsi que la réversibilité coulombique, tirées des voltamogrammes.
84

-40
-30
-20
-10
0
10
20
30
-2.2 -2 -1.8 -1.6 -1.4
dopage à -2,3 V/Refdopage à -2,2 V/Refdopage à -2,1 V/Refdopage à -2,0 V/Refdopage à -1,9 V/Ref
i (m
A/c
m2 )
E (V/Ag+/Ag)
Figure V-16 : Voltamogrammes d'une électrode de P-4-FPT pour différentes limites de potentiel de dopage dans une solution
d'acétonitrile 1M NEt4CF3SO3 ; v = 20mV/s, mP-4-FPT = 20,5 mg. Composition de l'électrode : 30% P-4-FPT,
65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
-40
-30
-20
-10
0
10
20
30
50
60
70
80
90
100
-2.3 -2.2 -2.1 -2 -1.9
Qch
arge
(mA
h/g)
Qdé
char
ge (m
Ah/
g)
Réversibilité coulom
bique (%)
Potentiel maximal de dopage (V/Ag+/Ag)
Figure V-17 : Evolutions des quantités d'électricité dopées et dédopées, ainsi que de la réversibilité coulombique du dopage
des voltamogrammes représentés sur la Figure V-16.
La Figure V-17 met en évidence que le dopage négatif du polymère se fait
progressivement selon le potentiel auquel il est soumis. Cela signifie qu'il y a une distribution
85

en potentiel de seuils de dopage différents, qui résulte de deux phénomènes différents,
développés ci-dessous :
- L'électrode est constituée d'un réseau tridimensionnel de chaînes polymères
enchevêtrées. L'insertion des cations s'accompagne de différentes contraintes diffusionnelles
selon l'emplacement plus ou moins profond des sites de dopage. Ces contraintes
diffusionnelles se concrétisent par un gradient de chutes ohmiques lors du dopage, qui donne
lieu à un dopage étalé en potentiel.
- Le polymère est synthétisé à l'aide d'un oxydant très puissant, entraînant une
polymérisation violente, rapide et incontrôlée. Cette réaction conduit à un polymère composé
d'une multitude de chaînes de longueurs différentes. La délocalisation électronique est donc
différente pour chaque longueur de chaîne, ce qui confère à chacune d'entre elles un potentiel
de dopage légèrement différent, qui se traduit par un pic de dopage général étendu sur une
large plage de potentiels.
La Figure V-17 montre également que plus la limite de dopage est négative, plus la
réversibilité coulombique baisse. Ce phénomène peut être expliqué soit par une augmentation
du "charge-trapping" lorsque le polymère est dopé plus profondément, soit par la réduction du
solvant, qui est de plus en plus importante au fur et à mesure que le potentiel de fin de dopage
devient négatif.
V-3-2-2) Cyclage galvanostatique du dopage négatif
Le dopage négatif du P-4-FPT a également été testé par cyclage galvanostatique. Cette
méthode permet d'obtenir des informations sur le dopage du polymère en considérant le
modèle électrostatique (cf. I-2-4).
Pour le cyclage galvanostatique, le P-4-FPT a été mis en œuvre dans une cellule de
type II (cf. II-2-1). La matière active est composée de 30% de polymère, 65% de noir
d'acétylène, 3% de CMC et 2% de PTFE. Le collecteur de courant est un micro-déployé
d'inox AISI 316L de 4 cm2. La contre-électrode surcapacitive utilisée est composée de
charbon actif laminé sur un collecteur de courant de même nature.
Avant de procéder au cyclage galvanostatique du polymère, il est nécessaire d'effectuer
une activation du dopage négatif. Pour cela, nous avons effectué 10 cycles de voltamétrie
cyclique à 20 mV/s entre -1,3 et -2,0 V/Ref.
86
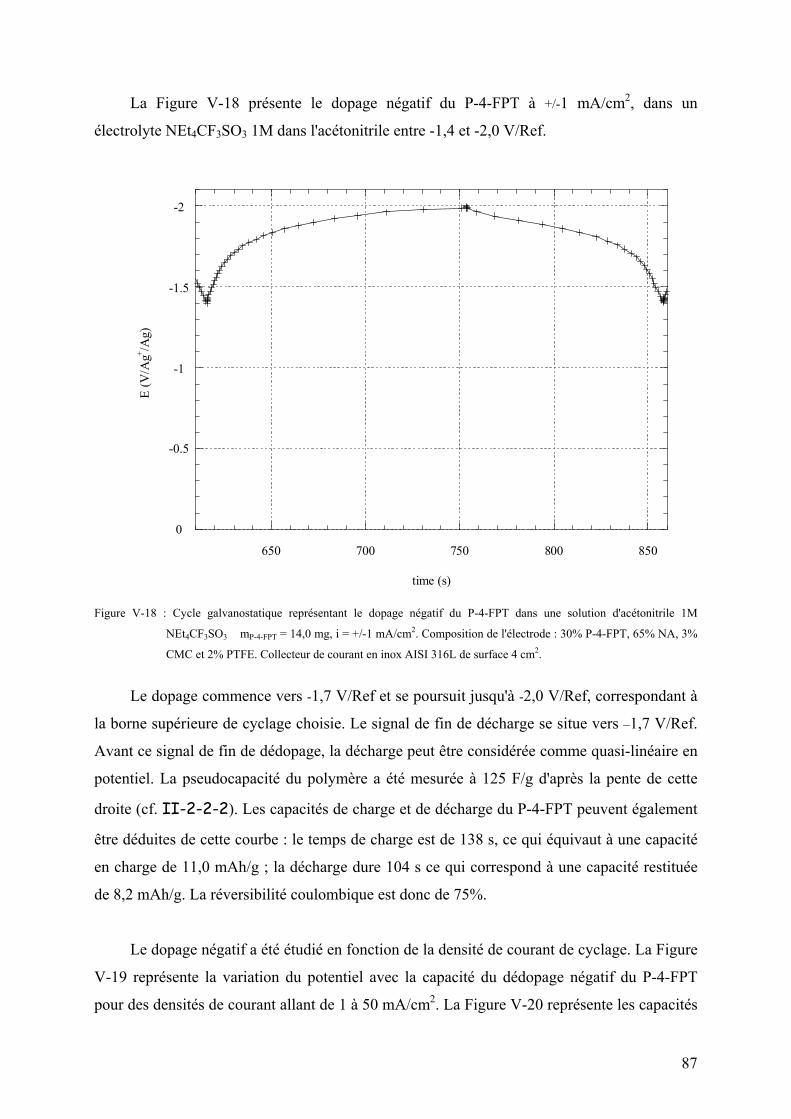
La Figure V-18 présente le dopage négatif du P-4-FPT à +/-1 mA/cm2, dans un
électrolyte NEt4CF3SO3 1M dans l'acétonitrile entre -1,4 et -2,0 V/Ref.
-2
-1.5
-1
-0.5
0
650 700 750 800 850
E (V
/Ag+ /A
g)
time (s)
Figure V-18 : Cycle galvanostatique représentant le dopage négatif du P-4-FPT dans une solution d'acétonitrile 1M
NEt4CF3SO3 mP-4-FPT = 14,0 mg, i = +/-1 mA/cm2. Composition de l'électrode : 30% P-4-FPT, 65% NA, 3%
CMC et 2% PTFE. Collecteur de courant en inox AISI 316L de surface 4 cm2.
Le dopage commence vers -1,7 V/Ref et se poursuit jusqu'à -2,0 V/Ref, correspondant à
la borne supérieure de cyclage choisie. Le signal de fin de décharge se situe vers –1,7 V/Ref.
Avant ce signal de fin de dédopage, la décharge peut être considérée comme quasi-linéaire en
potentiel. La pseudocapacité du polymère a été mesurée à 125 F/g d'après la pente de cette
droite (cf. II-2-2-2). Les capacités de charge et de décharge du P-4-FPT peuvent également
être déduites de cette courbe : le temps de charge est de 138 s, ce qui équivaut à une capacité
en charge de 11,0 mAh/g ; la décharge dure 104 s ce qui correspond à une capacité restituée
de 8,2 mAh/g. La réversibilité coulombique est donc de 75%.
Le dopage négatif a été étudié en fonction de la densité de courant de cyclage. La Figure
V-19 représente la variation du potentiel avec la capacité du dédopage négatif du P-4-FPT
pour des densités de courant allant de 1 à 50 mA/cm2. La Figure V-20 représente les capacités
87
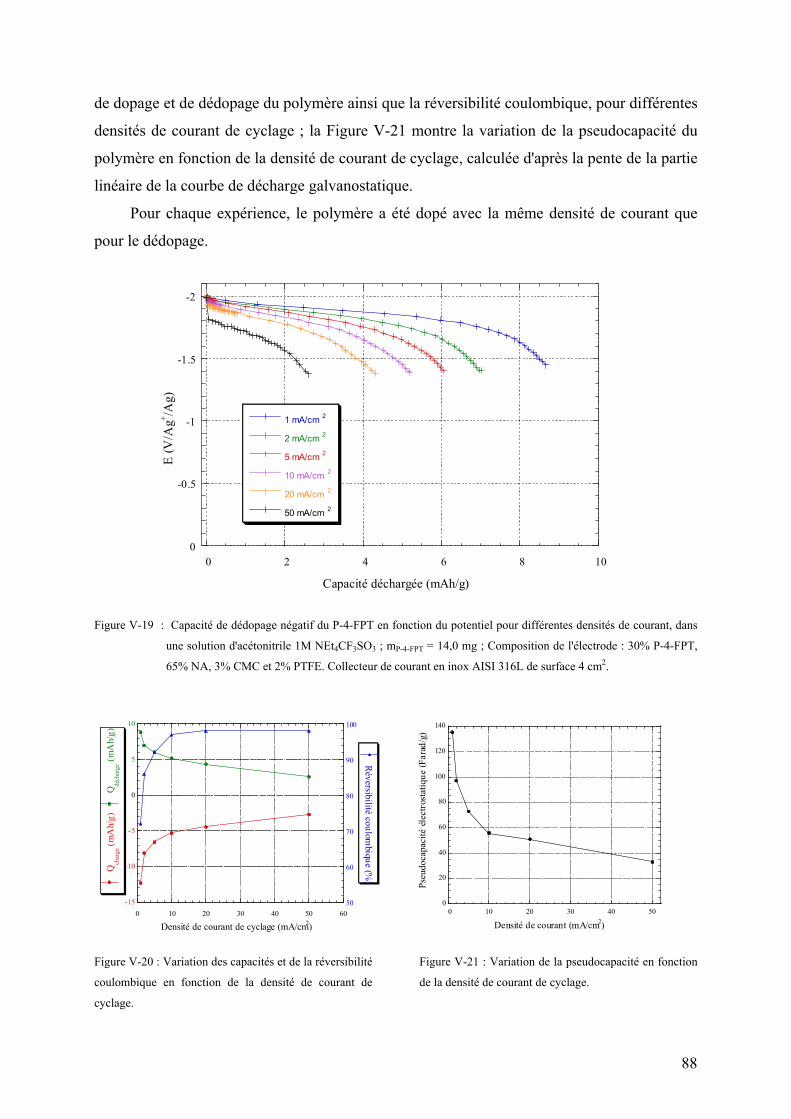
de dopage et de dédopage du polymère ainsi que la réversibilité coulombique, pour différentes
densités de courant de cyclage ; la Figure V-21 montre la variation de la pseudocapacité du
polymère en fonction de la densité de courant de cyclage, calculée d'après la pente de la partie
linéaire de la courbe de décharge galvanostatique.
Pour chaque expérience, le polymère a été dopé avec la même densité de courant que
pour le dédopage.
-2
-1.5
-1
-0.5
00 2 4 6 8 10
1 mA/cm 2
2 mA/cm 2
5 mA/cm 2
10 mA/cm 2
20 mA/cm 2
50 mA/cm 2
E (V
/Ag+ /A
g)
Capacité déchargée (mAh/g)
Figure V-19 : Capacité de dédopage négatif du P-4-FPT en fonction du potentiel pour différentes densités de courant, dans
une solution d'acétonitrile 1M NEt4CF3SO3 ; mP-4-FPT = 14,0 mg ; Composition de l'électrode : 30% P-4-FPT,
65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L de surface 4 cm2.
-15
-10
-5
0
5
10
50
60
70
80
90
100
0 10 20 30 40 50 60
Qch
arge
(m
Ah/
g)Q
déch
arge
(m
Ah/
g)
Réversibilité coulom
bique (%)
Densité de courant de cyclage (mA/cm2)
0
20
40
60
80
100
120
140
0 10 20 30 40 50
Pseu
doca
paci
té é
lect
rost
atiq
ue (F
arad
/g)
Densité de courant (mA/cm2) Figure V-20 : Variation des capacités et de la réversibilité
coulombique en fonction de la densité de courant de
cyclage.
Figure V-21 : Variation de la pseudocapacité en fonction
de la densité de courant de cyclage.
88

Le signal de fin de dédopage se situe toujours vers -1,6 V/Ref, conformément à ce qui
avait été observé en voltamétrie cyclique (cf. V-3-2-1).
On observe sur la FigureV-19 une baisse de capacité lorsque la densité de courant
augmente. En revanche, la Figure V-20 montre que cette baisse de capacité est associée à une
augmentation de la réversibilité. Cela tend à prouver que cette baisse n'est pas due à un
phénomène croissant de "charge-trapping" mais plutôt à un dopage de plus en plus superficiel
du polymère lorsque la densité de courant augmente.
Le phénomène limitatif entraînant la baisse de capacité lorsque la densité de courant
augmente est soit la chute ohmique dans l'électrode, soit la cinétique de diffusion des ions
dopants dans l'électrode. En effet, plus la densité de courant est importante et plus cette
cinétique est limitative par rapport à celle du dopage. Ce dernier est donc de plus en plus
superficiel. Le modèle électrostatique du dopage (cf. Figure V-21) arrive aux mêmes
conclusions : la pseudocapacité du polymère chute lorsque la densité de courant augmente, ce
qui signifie un dopage plus superficiel.
La Figure V-20 montre que le maximum de réversibilité coulombique est pratiquement
atteint à partir d'une densité de courant de 10 mA/cm2, ce qui correspond à une capacité de
charge de 5 mAh/g, c'est à dire à l'insertion d'un ion pour 38 unités monomère. Au-delà de
cette profondeur de dopage, des ions restent piégés dans le polymère et la réversibilité baisse.
En résumé, le dopage négatif du P-4-FPT nécessite une activation d'environ 10 cycles,
au cours de laquelle les sites susceptibles de piéger des charges se comblent tandis que la
structure du polymère s'aménage pour permettre l'insertion des cations de l'électrolyte.
Le P-4-FPT peut doper négativement une capacité de 40 mAh/g, mais avec une très
faible réversibilité coulombique, ne restituent alors que 25 mAh/g. En revanche, s'il est dopé
plus superficiellement, la réversibilité coulombique augmente. Pour obtenir 90 % de
réversibilité, il faut doper le polymère à 5 mAh/g, ce qui est assez faible.
Selon le modèle électrostatique du dopage négatif du P-4-FPT, celui-ci possède une
pseudocapacité de 125 Farad/g. Cette valeur est de l'ordre de celles obtenues avec les
charbons actifs les plus capacitifs. La perte en capacité survenant lorsque le polymère est
dopé avec de fortes densités de courant peut cependant être une limitation à la puissance des
dispositifs de stockage d'énergie utilisant le polymère.
89

V-3-2-3) Voltamétrie cyclique du dopage positif
La Figure V-22 représente le voltamogramme du dopage positif du P-4-FPT à 20 mV/s
dans un électrolyte NEt4CF3SO3 1M dans l'acétonitrile.
-20
-15
-10
-5
0
5
10
15
20
0 0.2 0.4 0.6 0.8
i (m
A/c
m2 )
E (V/Ag+/Ag)
Figure V-22 : Voltamogramme représentant le dopage positif du P-4-FPT dans une solution d'acétonitrile 1M NEt4CF3SO3 ;
v = 20mV/s, mP-4-FPT = 11,9 mg. Composition de l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE.
Collecteur de courant en inox AISI 316L.
Le pic d'oxydation correspond à l'extraction d'électrons des chaînes polymères et à
l'insertion d'anions le long de celles-ci pour préserver l'électroneutralité (cf. Eq. (V-4)). Le
polymère change de structure et une conduction se crée par la délocalisation de charges
positives le long des chaînes.
P-4-FPT + CF3SO3- P-4-FPT
+,CF3SO3- + e
- Eq. (V-4)
Le pic de réduction correspond à la réinjection des électrons dans les chaînes et au rejet
dans l'électrolyte des anions insérés lors du dopage. Le polymère revient alors à son état
neutre et isolant électronique.
90
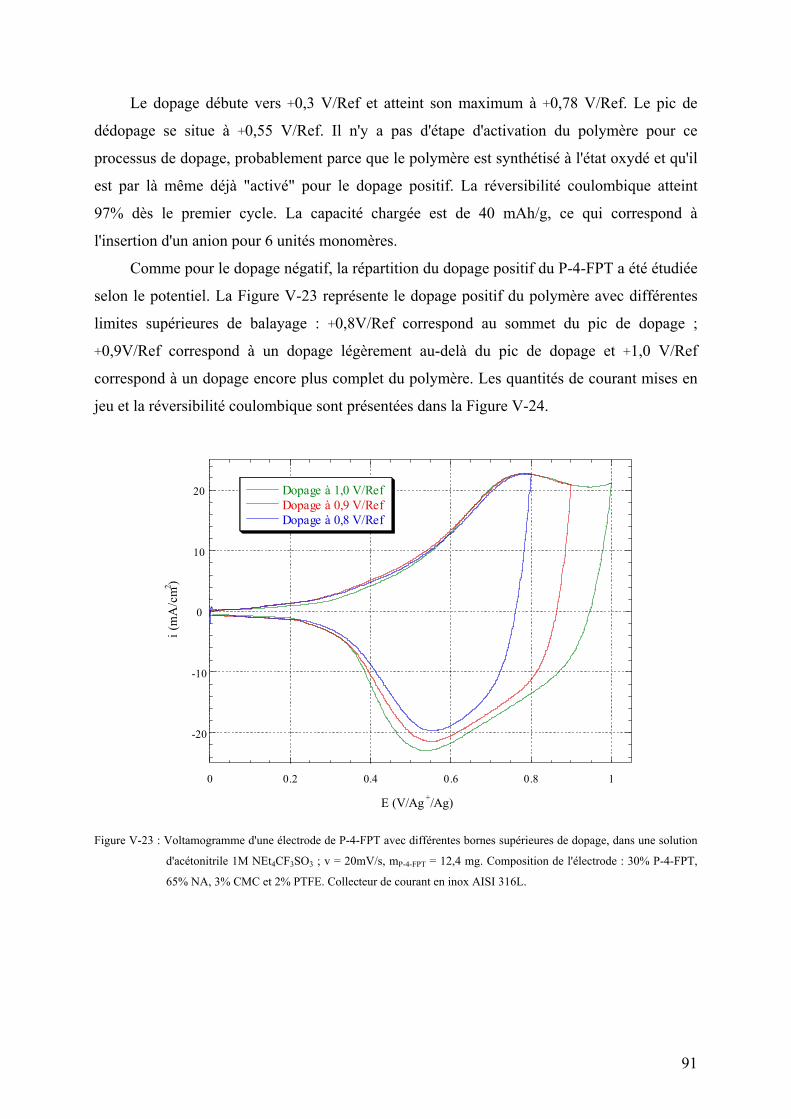
Le dopage débute vers +0,3 V/Ref et atteint son maximum à +0,78 V/Ref. Le pic de
dédopage se situe à +0,55 V/Ref. Il n'y a pas d'étape d'activation du polymère pour ce
processus de dopage, probablement parce que le polymère est synthétisé à l'état oxydé et qu'il
est par là même déjà "activé" pour le dopage positif. La réversibilité coulombique atteint
97% dès le premier cycle. La capacité chargée est de 40 mAh/g, ce qui correspond à
l'insertion d'un anion pour 6 unités monomères.
Comme pour le dopage négatif, la répartition du dopage positif du P-4-FPT a été étudiée
selon le potentiel. La Figure V-23 représente le dopage positif du polymère avec différentes
limites supérieures de balayage : +0,8V/Ref correspond au sommet du pic de dopage ;
+0,9V/Ref correspond à un dopage légèrement au-delà du pic de dopage et +1,0 V/Ref
correspond à un dopage encore plus complet du polymère. Les quantités de courant mises en
jeu et la réversibilité coulombique sont présentées dans la Figure V-24.
-20
-10
0
10
20
0 0.2 0.4 0.6 0.8 1
Dopage à 1,0 V/RefDopage à 0,9 V/RefDopage à 0,8 V/Ref
i (m
A/c
m2 )
E (V/Ag +/Ag)
Figure V-23 : Voltamogramme d'une électrode de P-4-FPT avec différentes bornes supérieures de dopage, dans une solution
d'acétonitrile 1M NEt4CF3SO3 ; v = 20mV/s, mP-4-FPT = 12,4 mg. Composition de l'électrode : 30% P-4-FPT,
65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
91
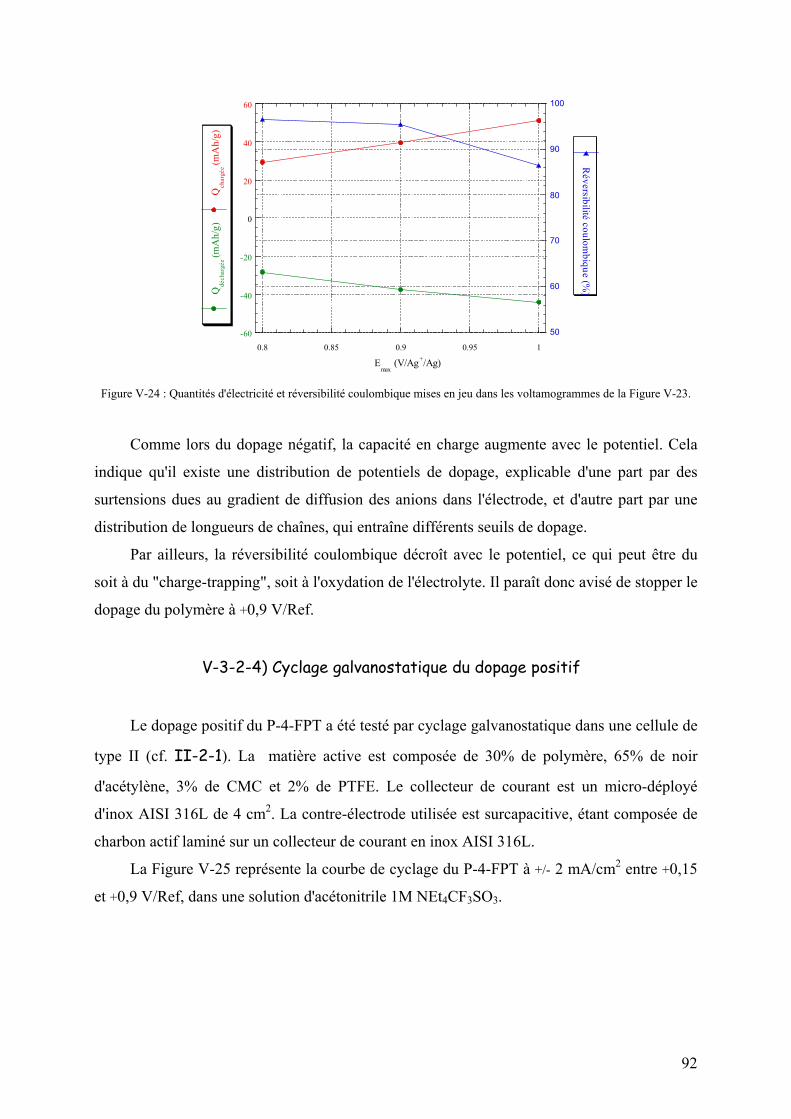
-60
-40
-20
0
20
40
60
50
60
70
80
90
100
0.8 0.85 0.9 0.95 1
Qch
argé
e (mA
h/g)
Qdé
char
gée (m
Ah/
g)R
éversibilité coulombique (%
)
Emax
(V/Ag+/Ag)
Figure V-24 : Quantités d'électricité et réversibilité coulombique mises en jeu dans les voltamogrammes de la Figure V-23.
Comme lors du dopage négatif, la capacité en charge augmente avec le potentiel. Cela
indique qu'il existe une distribution de potentiels de dopage, explicable d'une part par des
surtensions dues au gradient de diffusion des anions dans l'électrode, et d'autre part par une
distribution de longueurs de chaînes, qui entraîne différents seuils de dopage.
Par ailleurs, la réversibilité coulombique décroît avec le potentiel, ce qui peut être du
soit à du "charge-trapping", soit à l'oxydation de l'électrolyte. Il paraît donc avisé de stopper le
dopage du polymère à +0,9 V/Ref.
V-3-2-4) Cyclage galvanostatique du dopage positif
Le dopage positif du P-4-FPT a été testé par cyclage galvanostatique dans une cellule de
type II (cf. II-2-1). La matière active est composée de 30% de polymère, 65% de noir
d'acétylène, 3% de CMC et 2% de PTFE. Le collecteur de courant est un micro-déployé
d'inox AISI 316L de 4 cm2. La contre-électrode utilisée est surcapacitive, étant composée de
charbon actif laminé sur un collecteur de courant en inox AISI 316L.
La Figure V-25 représente la courbe de cyclage du P-4-FPT à +/- 2 mA/cm2 entre +0,15
et +0,9 V/Ref, dans une solution d'acétonitrile 1M NEt4CF3SO3.
92

-1
-0.5
0
0.5
1
800 900 1000 1100 1200 1300
E (V
/Ag+ /A
g)
Temps (s)
Figure V-25 : Cyclage galvanostatique du dopage positif du P-4-FPT dans une solution d'acétonitrile 1M NEt4CF3SO3 ;
mP-4-FPT = 15,8 mg., i = +/-2 mA/cm2. Composition de l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2%
PTFE. Collecteur de courant en inox AISI 316L.
Le dopage commence à environ +0,5 V/Ref et se poursuit quasi-linéairement jusqu'à
+0,9 V/Ref. En mesurant la pente de la partie linéaire de charge, on peut calculer la
pseudocapacité du polymère à 288 Farad/g. Le dédopage s'étend de +0,9 V/Ref à +0,5 V/Ref
potentiel auquel le signal de fin de dédopage est observé. La décharge est linéaire entre
+0,8 V/Ref et +0,5 V/Ref. La pseudocapacité délivrée, calculée à partir de la pente de la partie
linéaire de la décharge, est de 259 Farad/g. Entre +0,9 V/Ref et +0,8 V/Ref , le potentiel est
légèrement supérieur au domaine de dédopage du polymère et la pseudocapacité est plus
faible.
Les quantités d'électricité mises en jeu sont de 30,2 mAh/g pour le dopage et de
27,7 mAh/g pour le dédopage, ce qui correspond à une réversibilité coulombique de 91,7%.
Le dopage positif a également été étudié avec différentes densités de courant. La
Figure V-26 représente la variation du potentiel en fonction de la capacité dédopée pour
différentes densités de courant. Le polymère a été dopé avec la même densité de courant que
celle utilisée pour le dédopage. La Figure V-27 représente les capacités et la reversibilité
coulombique associées aux processus de dopage positif du P-4-FPT à différentes densités de
93
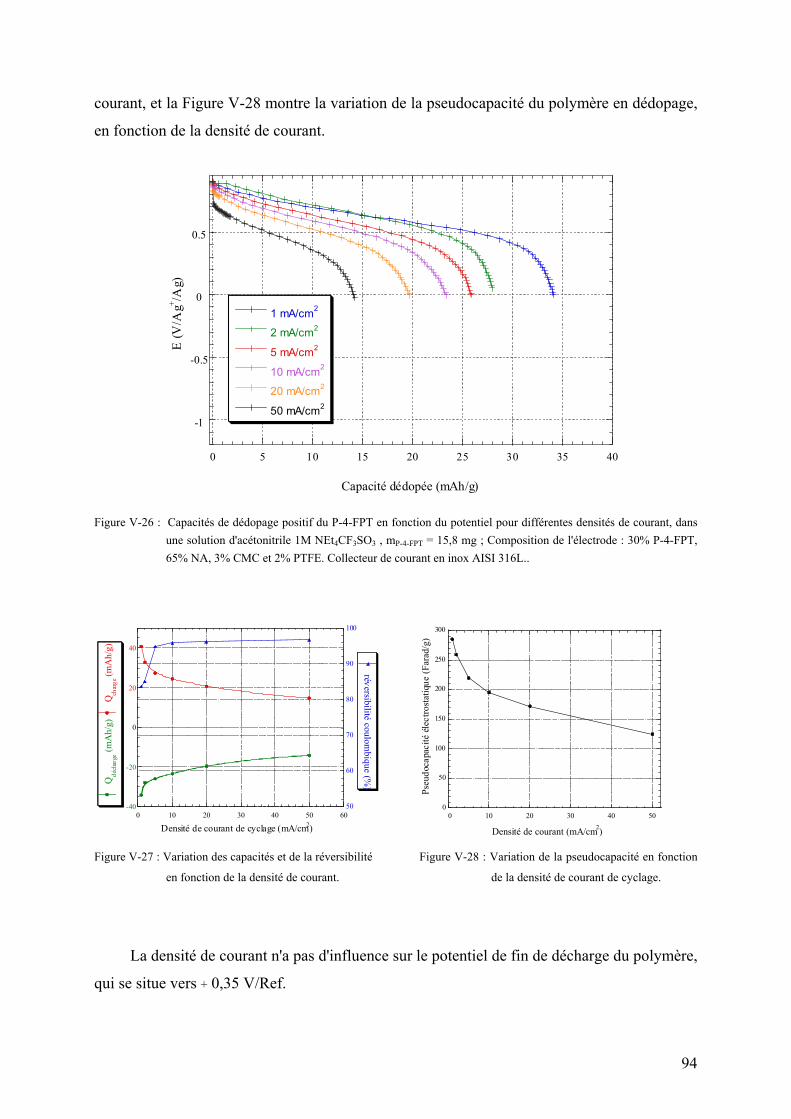
courant, et la Figure V-28 montre la variation de la pseudocapacité du polymère en dédopage,
en fonction de la densité de courant.
-1
-0.5
0
0.5
0 5 10 15 20 25 30 35 40
1 mA/cm2
2 mA/cm2
5 mA/cm2
10 mA/cm2
20 mA/cm2
50 mA/cm2
E (V
/Ag+ /A
g)
Capacité dédopée (mAh/g)
Figure V-26 : Capacités de dédopage positif du P-4-FPT en fonction du potentiel pour différentes densités de courant, dans
une solution d'acétonitrile 1M NEt4CF3SO3 , mP-4-FPT = 15,8 mg ; Composition de l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L..
-40
-20
0
20
40
50
60
70
80
90
100
0 10 20 30 40 50 60
Qch
arge
(mA
h/g)
Qdé
char
ge (m
Ah/
g)
réversibilité coulombique (%
)
Densité de courant de cyclage (mA/cm2)
0
50
100
150
200
250
300
0 10 20 30 40 50
Pseu
doca
paci
té é
lect
rost
atiq
ue (F
arad
/g)
Densité de courant (mA/cm2) Figure V-27 : Variation des capacités et de la réversibilité
en fonction de la densité de courant.
Figure V-28 : Variation de la pseudocapacité en fonction
de la densité de courant de cyclage.
La densité de courant n'a pas d'influence sur le potentiel de fin de décharge du polymère,
qui se situe vers + 0,35 V/Ref.
94

De la même façon que pour le dopage négatif, il y a une baisse de capacité restituée et
une augmentation de la réversibilité coulombique lorsque la densité de courant augmente, ce
qui signifie un dopage plus superficiel du polymère lorsqu'il est chargé avec une intensité
importante. Ce phénomène est dû soit à la chute ohmique dans l'électrode, soit à une
limitation du dopage par la cinétique de diffusion des ions au sein de l'électrode.
Cette baisse est confirmée par le modèle électrostatique de description du dopage du
polymère (cf. Figure V-28).
La Figure V-27 montre que le maximum de réversibilité coulombique est pratiquement
atteint à partir d'une densité de courant de 5 mA/cm2, ce qui correspond à une capacité de
charge de 27 mAh/g, c'est à dire à l'insertion d'un ion pour 8,2 unités monomère. Au-delà de
cette profondeur de dopage, c'est à dire pour des densités de charge inférieures, le phénomène
de charge-trapping devient de plus en plus prépondérant.
Le dopage positif du P-4-FPT est donc particulièrement capacitif. La quantité
d'électricité stockée peut aller jusqu'à près de 50 mAh/g, mais à cette profondeur de dopage,
on observe du "charge-trapping". Il est préférable de charger le polymère jusqu'à 40 mAh/g,
ce qui permet d'avoir plus de 95 % de réversibilité coulombique. Par ailleurs, en appliquant
le modèle électrostatique au dopage du polymère, on arrive à des valeurs tout à fait
remarquables de 260 Farad/g. Les charbons actifs qui développent les plus importantes
surfaces spécifiques (2500 m2/g), possèdent en milieu organique des capacités de
150 Farad/g au maximum.
Il y a donc une réelle potentialité pour l'application des polymères au stockage de
l'énergie dans des systèmes de supercondensateurs. Cependant, la perte en capacité
survenant lorsque le polymère est dopé avec de fortes densités de courant peut représenter
une limitation à la puissance des dispositifs de stockage d'énergie utilisant le polymère.
Il est néanmoins important de vérifier la stabilité des processus de dopage au cours de
nombreux cycles de dopage/dédopage, ce qui va conditionner la durée de vie des
supercondensateurs.
95

V-3-3) Cyclabilité du dopage négatif
L'évolution de l'électroactivité du P-4-FPT en cyclage a été étudiée à l'aide de la
voltamétrie cyclique et du cyclage galvanostatique.
Le polymère a été testé dans des cellules à trois électrodes de type II avec des contre-
électrodes surcapacitives de charbon actif pour éviter la dégradation de l'électrolyte sur cette
électrode (cf. II-2-1).
V-3-3-1) Cyclabilité en voltamétrie cyclique
La Figure V-29 représente l'évolution des vingt premiers cycles de voltamétrie cyclique
du dopage négatif complet du P-4-FPT (jusqu'à -2,3 V/Ref), dans l'électrolyte NEt4CF3SO3
1M dans l'acétonitrile.
Figure V-29 : Evolution des 20 premiers cycles de voltamétrie cyclique d'une électrode de P-4-FPT dans une solution
d'acétonitrile 1M NEt4CF3SO3 ; v = 20mV/s, mP-4-FPT = 16,5 mg. Composition de l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
L'électroactivité chute rapidement au cours des 20 cycles. Cependant, il est intéressant
de noter que l'allure générale du voltamogramme ne change pas : les potentiels de pic restent
les mêmes, seule l'intensité diminue. Cela signifie que la dégradation ne conduit pas à une
modification du couple redox, et donc n'implique pas les substituants. Dans le cas contraire, il
96
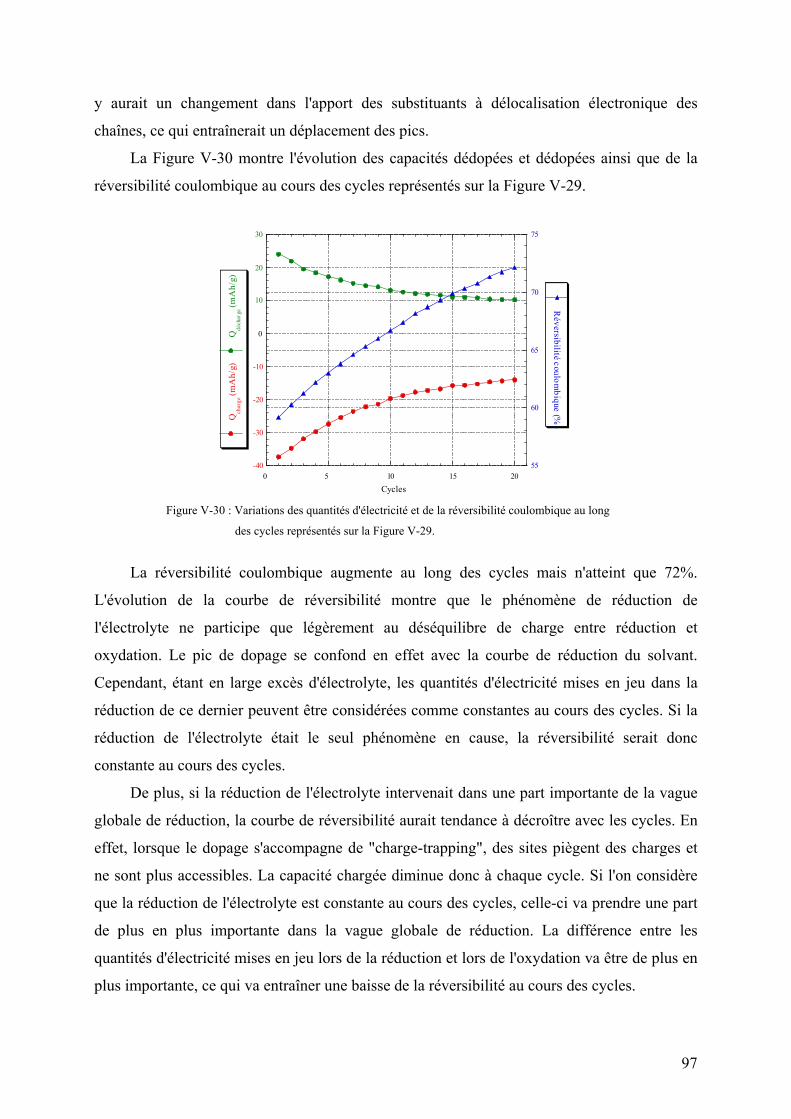
y aurait un changement dans l'apport des substituants à délocalisation électronique des
chaînes, ce qui entraînerait un déplacement des pics.
La Figure V-30 montre l'évolution des capacités dédopées et dédopées ainsi que de la
réversibilité coulombique au cours des cycles représentés sur la Figure V-29.
-40
-30
-20
-10
0
10
20
30
55
60
65
70
75
0 5 10 15 20
Qch
arge
(mA
h/g)
Qdé
char
ge (m
Ah/
g)R
éversibilité coulombique (%
)
Cycles
Figure V-30 : Variations des quantités d'électricité et de la réversibilité coulombique au long
des cycles représentés sur la Figure V-29.
La réversibilité coulombique augmente au long des cycles mais n'atteint que 72%.
L'évolution de la courbe de réversibilité montre que le phénomène de réduction de
l'électrolyte ne participe que légèrement au déséquilibre de charge entre réduction et
oxydation. Le pic de dopage se confond en effet avec la courbe de réduction du solvant.
Cependant, étant en large excès d'électrolyte, les quantités d'électricité mises en jeu dans la
réduction de ce dernier peuvent être considérées comme constantes au cours des cycles. Si la
réduction de l'électrolyte était le seul phénomène en cause, la réversibilité serait donc
constante au cours des cycles.
De plus, si la réduction de l'électrolyte intervenait dans une part importante de la vague
globale de réduction, la courbe de réversibilité aurait tendance à décroître avec les cycles. En
effet, lorsque le dopage s'accompagne de "charge-trapping", des sites piègent des charges et
ne sont plus accessibles. La capacité chargée diminue donc à chaque cycle. Si l'on considère
que la réduction de l'électrolyte est constante au cours des cycles, celle-ci va prendre une part
de plus en plus importante dans la vague globale de réduction. La différence entre les
quantités d'électricité mises en jeu lors de la réduction et lors de l'oxydation va être de plus en
plus importante, ce qui va entraîner une baisse de la réversibilité au cours des cycles.
97

Or ce n'est pas le cas, ce qui signifie que le principal phénomène qui limite la
réversibilité est le "charge-trapping".
Pour éviter ces problèmes de "charge-trapping" et de réduction du solvant, la borne
inférieure de potentiel à été limitée à -2,0 V/Ref. Les Figures V-31 et V-32 montrent
l'évolution des voltamogrammes du dopage négatif du P-4-FPT réalisés à 20 mV/s de -1,5 V à
-2,0 V/Ref durant les 100 premiers cycles (Figure V-31) puis entre les cycles 100 et 500
(Figure V-32).
Figure V-31 : Evolution des 100 premiers cycles de voltamétrie cyclique d'une électrode de P-4-FPT dans une solution
d'acétonitrile 1M NEt4CF3SO3 ; v = 20mV/s, mP-4-FPT = 14,8 mg. Composition de l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
98

Figure V-32 : Evolution des cycles 100 à 500 d'une électrode de P-4-FPT dans une solution d'acétonitrile 1M NEt4CF3SO3 ;
v = 20mV/s, mP-4-FPT = 14,8 mg. Composition de l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
Le dopage est beaucoup plus superficiel, ce qui permet une bien meilleure stabilité du
processus, qui décroît tout de même continuellement. Cela indique qu'une grande part de la
dégradation en capacité observée précédemment (cf. Figure V-29) est causée par le dopage
profond du polymère.
La Figure V-33 présente l'évolution de la réversibilité et des capacités de dopage et
dédopage au cours des cycles représentés sur les Figures V-31 et V-32.
-8
-6
-4
-2
0
2
4
6
8
90
92
94
96
98
100
0 100 200 300 400 500
Qch
arge
(mA
h/g)
Qdé
char
ge (m
Ah/
g)R
éversibilité coulmobique (%
)
Cycles
Figure V-33 : Evolution des quantités d'électricité et de la réversibilité des cycles effectués sur les Figures V-31 et V-32.
99

La baisse de l'électroactivité négative du P-4-FPT se situe essentiellement dans les 100
premiers cycles. Comme pour le cyclage profond du polymère, il n'y a pas de changement
dans les potentiels de dopage et de dédopage. Au 500ème cycle, on a une perte en capacitance
restituée de 25% par rapport au 10ème cycle. La réversibilité coulombique augmente
légèrement dans les 50 premiers cycles puis se stabilise à plus de 96%.
La limitation du potentiel de dopage à -2 V/Ref entraîne une amélioration importante de
la stabilité du processus au cours des cycles de même que de la réversibilité coulombique de
celui-ci. Cependant, la capacité du polymère s'en trouve très diminuée (6 mAh/g au lieu de
24 mAh/g).
V-3-3-2) Cyclage galvanostatique du dopage négatif
La cyclabilité du P-4-FPT a également été étudiée lors de cyclages galvanostatiques. Le
polymère a été testé dans le même type de montage que précédemment, avec toujours une
contre-électrode surcapacitive de charbon actif. Différentes densités de courant de cyclage et
différentes limites de dopage en potentiel ont été utilisées pour apprécier la cyclabilité du
polymère à différentes profondeurs de charge.
La Figure V-34 représente la variation au cours des cycles de la capacité du P-4-FPT
dopé avec deux densités de courant différentes de -1,4 V/Ref à -2,0 V/Ref. La densité de
courant de 1 mA/cm2 est considérée comme étant suffisamment faible pour permettre un
dopage lent et profond du polymère. En revanche, le cyclage à 20 mA/cm2 conduit à un
dopage plus rapide et donc plus superficiel. La valeur limite de potentiel a été choisie à
-2,0 V/Ref d'après les résultats de voltamétrie cyclique, pour conférer au polymère une
cyclabilité appréciable.
100

0
2
4
6
8
10
0 200 400 600 800 1000
i = +/- 1 mA/cm 2
i = +/- 20 mA/cm 2
Cap
acité
déd
opée
(mA
h/g)
Cycles Figure V-34 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une électrode de P-4-FPT avec
différentes densités de courant, dans une solution d'acétonitrile 1M NEt4CF3SO3 ; Composition de l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
Les quantités d'électricité déchargées au début du cyclage sont assez différentes selon la
densité de courant de cyclage, comme il avait été montré en Figure V-19 : 9,4 mAh/g à
1 mA/cm2 et 5,3 mAh/g pour 20 mA/cm2. Cela signifie bien que le dopage à 20 mA/cm2 est
plus superficiel que le dopage à 1 mA/cm2. Tout comme en voltamétrie cyclique, il y a une
baisse progressive des capacitances déchargées au cours des cycles, mais avec des profils de
dégradation très différents selon la densité de courant. Pour le dopage profond obtenu à faible
courant de cyclage, la chute est très rapide dans les 400 premiers cycles, passant de 9,4 à 2,4
mAh/g, puis se stabilise pour arriver à 1,5 mAh/g à 1000 cycles. Cela représente une baisse de
85% en 1000 cycles, dont 75% dans les 100 premiers cycles. Par ailleurs, quand le polymère
est dopé plus superficiellement, la dégradation est régulière et beaucoup moins forte. En 1000
cycles, le polymère passe d'une capacité de 5,3 à 4,5 mAh/g ce qui représente une dégradation
de 14%.
Pour les deux expériences, la réversibilité est restée à peu près constante : entre 72% et
76% pour le cyclage à 1 mA/cm2 et entre 95% et 98% pour le cyclage à 20 mA/cm2. Le
"charge-trapping" peut donc expliquer une part de la mauvaise cyclabilité du P-4-FPT, mais
pas en totalité : en effet, la Figure V-32 nous montre qu'il existe un dopage relativement
réversible d'au moins 4,5 mAh/g. Si le piégeage d'ions était la seule cause de la dégradation,
le dopage à 1 mA/cm2 se stabiliserait lui aussi vers 5 mAh/g pour arriver vers 4,5 mAh/g à
101

1000 cycles. Cependant, il continue de décroître jusqu'à 2 mAh/g. Cela met en évidence une
modification au niveau de l'électrode, amplifiée à faible densité de courant, provenant soit du
polymère lui-même (rupture de la délocalisation électronique), soit d'une modification de la
pâte dans laquelle celui-ci est mis en œuvre (dégradation du liant, fermeture de la porosité,
désolidarisation de la matière active du collecteur de courant, …).
Le cyclage galvanostatique nous permet aussi d'apprécier le dopage du polymère selon
le modèle électrostatique. La Figure V-35 représente l'évolution de la pseudocapacité restituée
par le P-4-FPT au cours de la même expérience que pour la figure précédente.
0
20
40
60
80
100
120
0 200 400 600 800 1000
i = +/- 1 mA/cm2
i = +/- 20 mA/cm2
Pseu
doca
paci
té d
édop
ée (F
arad
/g)
Cycles Figure V-35 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une électrode de P-4-FPT avec
différentes densités de courant, dans une solution d'acétonitrile 1M NEt4CF3SO3 ; Composition de
l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
Le modèle électrostatique du dopage mène aux mêmes conclusions que le modèle
classique de transfert de charge, les courbes étant pratiquement équivalentes.
Nous avons ensuite étudié l'influence de la limitation en potentiel sur le dopage du
polymère. La Figure V-36 représente l'évolution des capacités de dédopage au cours des
cycles, lorsque le polymère est dopé à -2,0 V/Ref ou -1,9 V/Ref. La dernière limite
correspond en voltamétrie cyclique à un dopage très léger (3,1 mAh/g) et très réversible
(97%), ce qui signifie un piégeage d'ions presque nul. La densité de courant utilisée est de
1 mA/cm2.
102
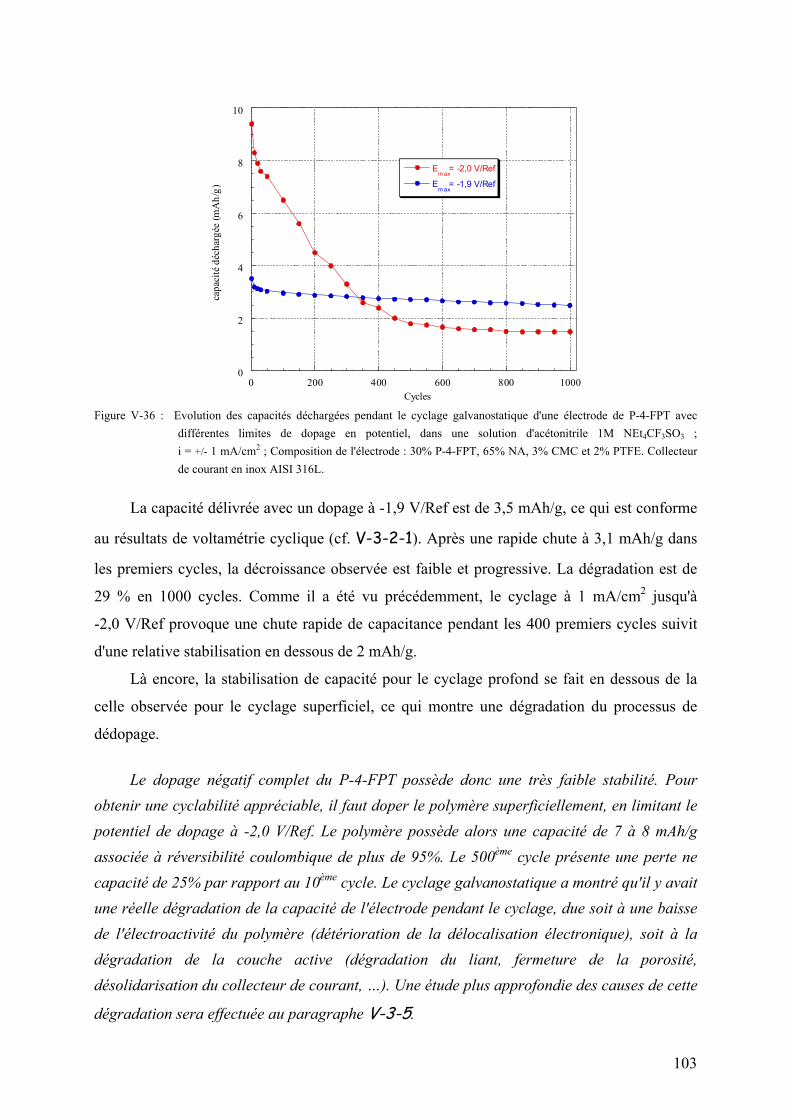
0
2
4
6
8
10
0 200 400 600 800 1000
Em ax
= -2,0 V/Ref
Em ax
= -1,9 V/Ref
capa
cité
déc
harg
ée (m
Ah/
g)
Cycles Figure V-36 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une électrode de P-4-FPT avec
différentes limites de dopage en potentiel, dans une solution d'acétonitrile 1M NEt4CF3SO3 ; i = +/- 1 mA/cm2 ; Composition de l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
La capacité délivrée avec un dopage à -1,9 V/Ref est de 3,5 mAh/g, ce qui est conforme
au résultats de voltamétrie cyclique (cf. V-3-2-1). Après une rapide chute à 3,1 mAh/g dans
les premiers cycles, la décroissance observée est faible et progressive. La dégradation est de
29 % en 1000 cycles. Comme il a été vu précédemment, le cyclage à 1 mA/cm2 jusqu'à
-2,0 V/Ref provoque une chute rapide de capacitance pendant les 400 premiers cycles suivit
d'une relative stabilisation en dessous de 2 mAh/g.
Là encore, la stabilisation de capacité pour le cyclage profond se fait en dessous de la
celle observée pour le cyclage superficiel, ce qui montre une dégradation du processus de
dédopage.
Le dopage négatif complet du P-4-FPT possède donc une très faible stabilité. Pour
obtenir une cyclabilité appréciable, il faut doper le polymère superficiellement, en limitant le potentiel de dopage à -2,0 V/Ref. Le polymère possède alors une capacité de 7 à 8 mAh/g associée à réversibilité coulombique de plus de 95%. Le 500ème cycle présente une perte ne capacité de 25% par rapport au 10ème cycle. Le cyclage galvanostatique a montré qu'il y avait une réelle dégradation de la capacité de l'électrode pendant le cyclage, due soit à une baisse de l'électroactivité du polymère (détérioration de la délocalisation électronique), soit à la dégradation de la couche active (dégradation du liant, fermeture de la porosité, désolidarisation du collecteur de courant, …). Une étude plus approfondie des causes de cette
dégradation sera effectuée au paragraphe V-3-5.
103

V-3-4) Cyclabilité du dopage positif
Le dopage positif a été étudié de la même façon que le dopage négatif pour évaluer la
cyclabilité à différentes profondeurs de dopage. Le P-4-FPT a été testé dans des cellules de
type II avec des contre-électrodes surcapacitives en charbon actif pour limiter la variation de
potentiel de cet électrode (cf. II-2-1).
V-3-4-1) Dopage positif en voltamétrie cyclique
Les Figures V-37 et V-38 représentent l'évolution des voltamogrammes du dopage
positif du P-4-FPT, effectuées à 20 mV/s entre 0 et +0,9 V/Ref, pendant les 100 premiers
cycles et entre les cycles 100 et 500 respectivement. La borne supérieure de potentiel a été
fixée à +0,9 V/Ref d'après les voltamogrammes obtenus précédemment (cf. V-3-2-3).
Figure V-37 : Evolution des 100 premiers cycles de voltamétrie cyclique d'une électrode de P-4-FPT dans une solution
d'acétonitrile 1M NEt4CF3SO3 ; v = 20mV/s, mP-4-FPT = 11,9 mg. Composition de l'électrode : 30% P-4-FPT,
65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
104
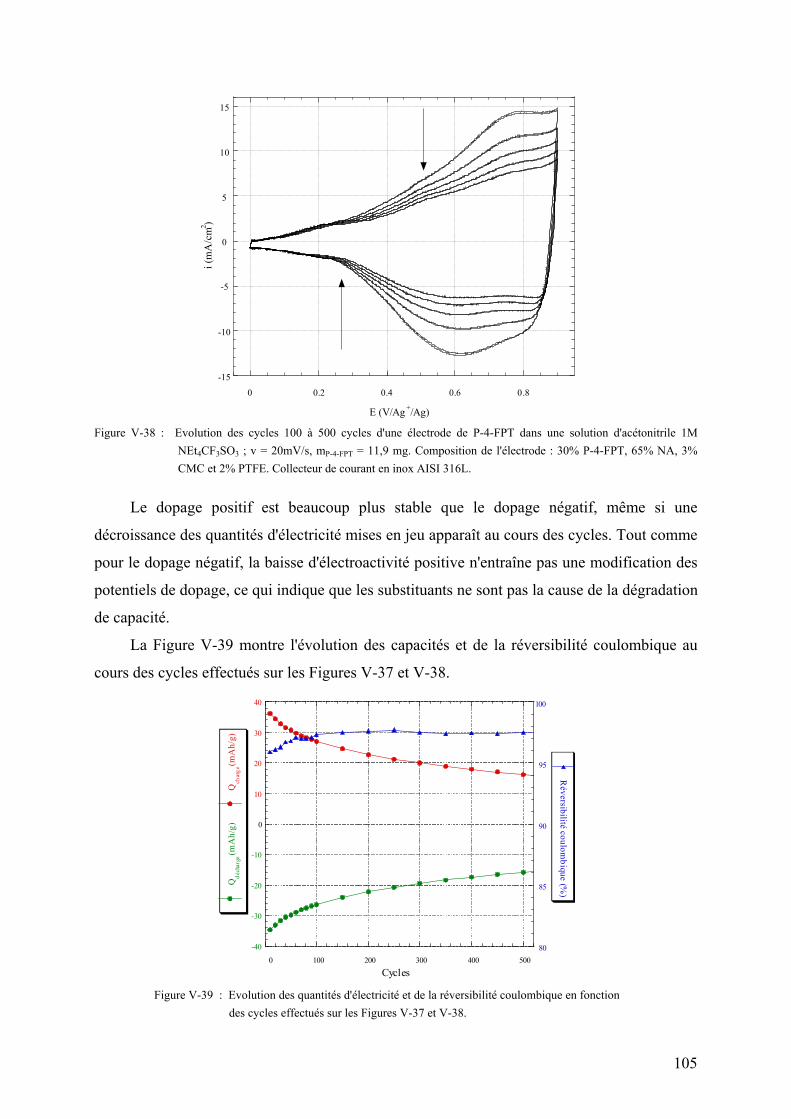
-15
-10
-5
0
5
10
15
0 0.2 0.4 0.6 0.8
i (m
A/c
m2 )
E (V/Ag +/Ag) Figure V-38 : Evolution des cycles 100 à 500 cycles d'une électrode de P-4-FPT dans une solution d'acétonitrile 1M
NEt4CF3SO3 ; v = 20mV/s, mP-4-FPT = 11,9 mg. Composition de l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
Le dopage positif est beaucoup plus stable que le dopage négatif, même si une
décroissance des quantités d'électricité mises en jeu apparaît au cours des cycles. Tout comme
pour le dopage négatif, la baisse d'électroactivité positive n'entraîne pas une modification des
potentiels de dopage, ce qui indique que les substituants ne sont pas la cause de la dégradation
de capacité.
La Figure V-39 montre l'évolution des capacités et de la réversibilité coulombique au
cours des cycles effectués sur les Figures V-37 et V-38.
-40
-30
-20
-10
0
10
20
30
40
80
85
90
95
100
0 100 200 300 400 500
Qch
arge
(mA
h/g)
Qdé
char
ge (m
Ah/
g)R
éversibilité coulombique (%
)
Cycles Figure V-39 : Evolution des quantités d'électricité et de la réversibilité coulombique en fonction
des cycles effectués sur les Figures V-37 et V-38.
105

Les quantités d'électricité chargées baissent progressivement au cours des cycles, avec
une atténuation de la dégradation au fur et à mesure du cyclage. La réversibilité augmente
légèrement de 96% à environ 98%, ce qui montre que le "charge-trapping" n'est pas la cause
de la décroissance d'électroactivité. La perte de capacité en dédopage est de 54 % en 500
cycles, passant de 35 mAh/g à 16 mAh/g.
La cyclabilité du dopage positif du P-4-FPT a également été testé avec la composition
d'électrode optimisée pour les systèmes de stockage d'énergie (cf. IV-3) : 80% P-4-FPT, 15%
NA, 3% CMC et 2% PTFE. Les Figures V-40 et V-41 représentent les voltamétries cycliques
d'une telle électrode, effectuées à 20 mV/s de -0,3 à +1,1 V/Ref. La borne supérieure de
potentiel a été fixée d'après des expériences préliminaires qui ont montré l'existence d'une
chute ohmique importante due à la faible teneur en NA.
Figure V-40 : Evolution des 1000 premiers cycles d'une électrode de P-4-FPT dans une solution d'acétonitrile 1M
NEt4CF3SO3 ; v = 20mV/s, mP-4-FPT = 36,2 mg. Composition de l'électrode : 80% P-4-FPT, 15% NA, 3% CMC
et 2% PTFE. Collecteur de courant en inox AISI 316L.
106

Figure V-41 : Evolution des cycles 1000 à 10000 d'une électrode de P-4-FPT dans une solution d'acétonitrile 1M
NEt4CF3SO3 ; v = 20mV/s, mP-4-FPT = 36,2 mg. Composition de l'électrode : 80% P-4-FPT, 15% NA, 3%
CMC et 2% PTFE. Collecteur de courant en inox AISI 316L.
Les potentiels de dopage et de dédopage sont très différents de ceux obtenus auparavant.
Ceci est dû d'une part au fait que l'électrode est moins conductrice que lorsque la teneur en
conducteur est de 65 %. Cela provoque des chutes ohmiques au sein de l'électrode qui font
que le potentiel réel au niveau de l'électrode est inférieur au potentiel que l'on observe. D'autre
part, l'électrode contient trois fois plus de polymère pour la même surface, ce qui donne une
électrode plus épaisse. Il y a donc un gradient de contraintes diffusionelles pour les ions
dopants beaucoup plus important que lorsqu'on a des couches fines de matière active, et cela
entraîne des chutes ohmiques plus importantes.
La Figure V-42 montre l'évolution des capacités dopées et dédopées ainsi que la
réversibilité coulombique au cours du cyclage représenté sur les Figures V-40 et V-41.
107
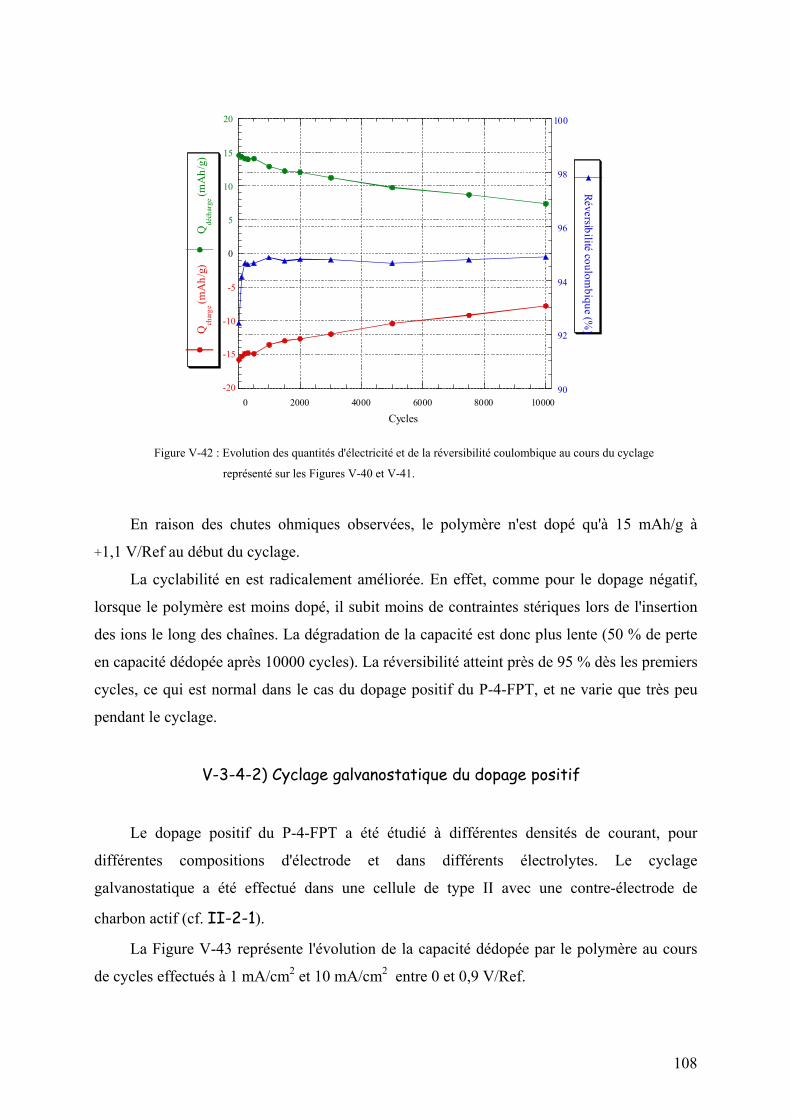
-20
-15
-10
-5
0
5
10
15
20
90
92
94
96
98
100
0 2000 4000 6000 8000 10000
Qch
arge
(mA
h/g)
Qdé
char
ge (m
Ah/
g)Réversibilité coulom
bique (%)
Cycles
Figure V-42 : Evolution des quantités d'électricité et de la réversibilité coulombique au cours du cyclage
représenté sur les Figures V-40 et V-41.
En raison des chutes ohmiques observées, le polymère n'est dopé qu'à 15 mAh/g à
+1,1 V/Ref au début du cyclage.
La cyclabilité en est radicalement améliorée. En effet, comme pour le dopage négatif,
lorsque le polymère est moins dopé, il subit moins de contraintes stériques lors de l'insertion
des ions le long des chaînes. La dégradation de la capacité est donc plus lente (50 % de perte
en capacité dédopée après 10000 cycles). La réversibilité atteint près de 95 % dès les premiers
cycles, ce qui est normal dans le cas du dopage positif du P-4-FPT, et ne varie que très peu
pendant le cyclage.
V-3-4-2) Cyclage galvanostatique du dopage positif
Le dopage positif du P-4-FPT a été étudié à différentes densités de courant, pour
différentes compositions d'électrode et dans différents électrolytes. Le cyclage
galvanostatique a été effectué dans une cellule de type II avec une contre-électrode de
charbon actif (cf. II-2-1).
La Figure V-43 représente l'évolution de la capacité dédopée par le polymère au cours
de cycles effectués à 1 mA/cm2 et 10 mA/cm2 entre 0 et 0,9 V/Ref.
108

0
5
10
15
20
25
30
35
40
0 200 400 600 800 1000
i = +/- 1 mA/cm 2
i = +/- 10 mA/cm 2
Cap
acité
déd
opée
(mA
h/g)
Cycles Figure V-43 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une électrode de P-4-FPT avec
différentes densités de courant, dans une solution d'acétonitrile 1M NEt4CF3SO3 ; Emax = 0,9 V/Ref ; Composition de l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE ; Collecteur de courant en inox AISI 316L.
Le dopage du P-4-FPT est de 37 mAh/g au premier cycle lorsque la densité de courant
est de 1 mA/cm2 et 24 mAh/g lorsque celle-ci est de 10 mA/cm2. Cela montre, comme pour le
dopage négatif, que plus la densité de courant est importante et plus le dopage est superficiel.
Cependant, pour le dopage à 1 mA/cm2, la figure montre une très forte décroissance de
la capacité dès le début du cyclage, avec une relative stabilisation à partir du 200ème cycle. En
1000 cycles, la perte en capacité est de 96% avec 78% dans les cent premiers cycles. Le
cyclage à 10 mA/cm2 montre quant à lui une cyclabilité nettement meilleure avec une perte de
37% en 1000 cycles, perte qui est régulière au cours du cyclage.
Des résultats similaires ont été publiés pour le dopage de polythiophènes limité ou non
en profondeur de charge [64,68].
La réversibilité coulombique du cyclage à 1 mA/cm2 est inhabituellement faible pour un
dopage positif : elle augmente de 70% à 85% dans les trente premiers cycles puis croît
progressivement jusqu'à 91%. Pour le dopage à 10 mA/cm2, celle-ci augmente
progressivement de 98 % à 98,7 %. Cette différence peut être due à du "charge-trapping",
mais aussi à une réaction d'oxydation du solvant ou de l'électrode qui dégraderait le polymère.
Le milieu de référence de ce travail est une solution 1M NEt4CF3SO3 dans l'acétonitrile,
cet électrolyte permettant un bon dopage positif comme négatif des polymères. Cependant, le
NEt4BF4 est un sel qui permet également un très bon dopage positif [44]. Il a donc été testé
109

pour apprécier l'influence du dopage par un anion plus petit. La Figure V-44 représente
l'évolution de la capacité dédopée par le polymère lorsqu'il est dopé avec CF3SO3- ou BF4
- 1M
dans l'acétonitrile.
0
5
10
15
20
25
30
35
40
0 200 400 600 800 1000
NEt4CF
3SO
3
NEt4BF
4
Cap
acité
déd
opée
(mA
h/g)
Cycles Figure V-44 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une électrode de P-4-FPT avec
différents sels 1M dans l'acétonitrile ; i = +/-1 mA/cm2 ; Emax = 0,9 V/Ref ; Composition de l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE ; Collecteur de courant en inox AISI 316L.
Les capacités de départ sont très proches : 37 mAh/g pour le CF3SO3
- et 34 mAh/g pour
le BF4-. Cela signifie que pour ces deux anions, la différence de taille n'influence pas
significativement la quantité d'électricité stockée.
Quelque soit le sel, il y a toujours une forte dégradation de la capacité du polymère au
cours des cycles. Cependant, celle-ci est moins importante pour le BF4-, ce qui est
probablement dû à sa taille, deux fois plus petite que le CF3SO3-. Ainsi pour une même
quantité d'électricité stockée, le stress stérique provoqué est moindre pour l'ion le plus petit.
Certains travaux ont attribué la désactivation anodique des polythiophènes à l'attaque
oxydative des chaînes polymères par les solvants (eau, acétonitrile et carbonate de propylène)
[69-71]. Le carbonate de propylène (PC) est un solvant connu pour son large domaine de
stabilité en potentiel [52,127]. Il est souvent utilisé pour les caractérisations de polymères
conducteurs, malgré sa moindre conductivité par rapport à l'acétonitrile, due à sa forte
viscosité. D'après Jiaxiong Wang, le PC permettrait une bien meilleure cyclabilité que
l'acétonitrile, l'attaque du polymère par ce solvant n'apparaissant qu'au début du cyclage
110

[70]. Le dopage positif du P-4-FPT a donc été testé dans ce solvant. La Figure V-45
représente l'évolution des capacités déchargées au cours d'un cyclage effectué à 1 mA/cm2
dans chacun des solvants. Le sel utilisé est le NEt4BF4 en concentration molaire.
0
5
10
15
20
25
30
35
40
0 200 400 600 800 1000
Carbonate de propylène
Acétonitrile
Cap
acité
déd
opée
(mA
h/g)
Cycles Figure V-45 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une électrode de P-4-FPT dans
différents solvants, avec le sel NEt4BF4 1M ; i = +/- 1 mA/cm2 ; Emax = 0,9 V/Ref ; Composition de
l'électrode : 30% P-4-FPT, 65% NA, 3% CMC et 2% PTFE ; Collecteur de courant en inox AISI 316L.
Le dopage du P-4-FPT présente une légère activation dans le PC, durant 5 cycles,
augmentant de 31 à 37 mAh/g. La dégradation de la capacité du polymère est ensuite
identique, quelque soit le solvant. Les résultats de Wang ne sont donc pas confirmés dans nos
conditions expérimentales.
Le P-4-FPT étant susceptible d'être utilisé dans des électrodes pour supercondensateurs,
son électroactivité a été testée pour des teneurs élevées dans l'électrode. La Figure V-46
représente l'évolution au cours des cycles de la capacité du P-4-FPT dans des électrodes de
composition 30% ou 80% en polymère.
111

0
5
10
15
20
25
30
35
0 200 400 600 800 1000
PFPT à 80%
PFPT à 30%
Cap
acité
déd
opée
(mA
h/g)
Cycles Figure V-46 : Evolution des capacités dédopées pendant le cyclage galvanostatique d'une électrode de P-4-FPT pour
différentes compositions d'électrode : 30 ou 80% P-4-FPT, respectivement 65 ou 15% NA, 3% CMC et 2% PTFE ; électrolyte : acétonitrile 1M NEt4BF4 ; i = +/-1 mA/cm2 ; collecteur de courant en inox AISI 316L.
Le dopage de l'électrode à 80 % de polymère montre une capacité de décharge de
23 mAh/g contre 34 mAh/g pour l'électrode contenant 30% de P-4-FPT. Cela semble signifier
que le dopage est plus superficiel car la dégradation durant le cyclage est assez importante.
Cependant, cela peut aussi indiquer que la capacité atteinte est limitée par la percolation
électronique au sein de l'électrode. La légère différence de cyclabilité résiderait alors dans la
différence de stress stérique due à l'insertion d'ions dans l'électrode.
Le dopage positif du P-4-FPT possède donc une stabilité en cyclage assez faible avec
une perte en capacité de 54% en 500 cycles, passant de 37 mAh/g à 16 mAh/g. La stabilité es
en revanche améliorée par rapport au PTh, pour lequel la perte en capacité est de 92% en
500 cycles dans les mêmes conditions (cf. V-2-2-4).
On observe une forte amélioration de la cyclabilité lorsque le dopage est superficiel.
Ainsi, 10000 cycles ont été obtenus en voltamétrie cyclique avec une perte de 50% en
capacité lorsque le dopage débute à 15 mAh/g. Le cyclage galvanostatique a été utilisé pour
étudier le dopage dans différents milieux. Le solvant ne semble pas influencer la désactivation
progressive du polymère. En revanche, une légère amélioration a été relevée lorsque le
polymère est dopé avec l'anion BF4 -, de taille plus petite que CF3SO3
-. Cela signifie qu'une
part de la désactivation peut être liée au stress généré par l'insertion des ions dans
112

l'électrode. Pour ces expériences, le modèle électrostatique de dopage du P-4-FPT n'apporte
aucune information supplémentaire sur la cyclabilité.
V–3-5) Etude de la perte d'électroactivité du PFPT
La perte d'électroactivité est un problème important pour les polymères conducteurs et
leurs éventuelles applications. Elle est le plus souvent due à une rupture de la délocalisation
électronique le long du polymère, soit par la coupure des chaînes, un réarrangement
moléculaire ou une attaque chimique de la chaîne. Plusieurs hypothèses ont été proposées
pour expliquer ce phénomène, mettant principalement en cause les attaques nucléophiles des
solvants ou de l'eau, contenue même à très faible teneur dans les électrolytes [5,64,68-
71,154,161]. Ces attaques conduisent à des greffages de molécules en position 4 du thiophène,
puis à des réarrangements qui interrompent délocalisation électronique et "désactivent" le
polymère.
Nous avons utilisé différentes techniques électrochimiques (voltamétrie cyclique,
cyclage galvanostatique) et spectrales (IR, UV-Visible) ainsi que des mesures d'analyse
élémentaire et de teneur en eau (titrage Karl Fischer), pour tenter d'expliquer le mécanisme de
dégradation du P-4-FPT observé dans nos conditions de cyclage. Ces travaux ont été orientés
dans deux directions : l'analyse des composés eux mêmes et l'analyse de l'électrolyte dans
lequel le polymère a été cyclé. Pour étudier le polymère, nous avons utilisé les techniques
électrochimiques, l'analyse élémentaire, ainsi que la spectroscopie infrarouge. Pour étudier
l'électrolyte, nous avons utilisé l'analyse élémentaire ainsi que la spectroscopie UV-Visible.
V-3-5-1) Etude de l'électrolyte
Cette étude a été effectuée avec l'électrolyte NEt4BF4 1M dans l'acétonitrile. Lors du
cyclage du polymère, l'électrolyte prend une teinte jaune plus ou moins prononcée, venant du
polymère lui-même et qui peut être due soit à des oligomères solubilisés ou d'autres produits
de dégradation, soit à des ions FeCl4- ou Cl- libérés lors du cyclage. L'électrolyte a donc été
analysé par spectroscopie UV-visible. La Figure V-47 représente le spectre UV-visible de
l'électrolyte à son état initial (avant cyclage) puis à différents stades du cyclage du polymère.
Pour cette expérience, le polymère a été cyclé sur son domaine de dopage positif.
113
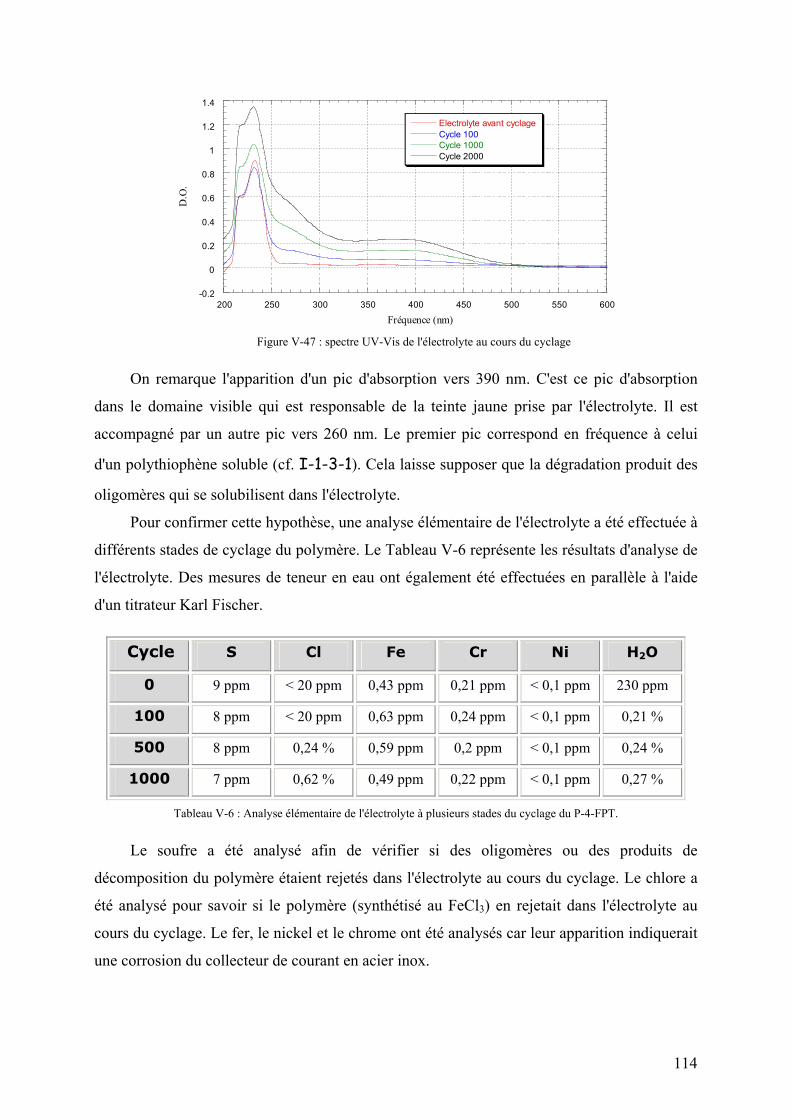
-0.2
0
0.2
0.4
0.6
0.8
1
1.2
1.4
200 250 300 350 400 450 500 550 600
Electrolyte avant cyclageCycle 100Cycle 1000Cycle 2000
D.O
.
Fréquence (nm)
Figure V-47 : spectre UV-Vis de l'électrolyte au cours du cyclage
On remarque l'apparition d'un pic d'absorption vers 390 nm. C'est ce pic d'absorption
dans le domaine visible qui est responsable de la teinte jaune prise par l'électrolyte. Il est
accompagné par un autre pic vers 260 nm. Le premier pic correspond en fréquence à celui
d'un polythiophène soluble (cf. I-1-3-1). Cela laisse supposer que la dégradation produit des
oligomères qui se solubilisent dans l'électrolyte.
Pour confirmer cette hypothèse, une analyse élémentaire de l'électrolyte a été effectuée à
différents stades de cyclage du polymère. Le Tableau V-6 représente les résultats d'analyse de
l'électrolyte. Des mesures de teneur en eau ont également été effectuées en parallèle à l'aide
d'un titrateur Karl Fischer.
Cycle S Cl Fe Cr Ni H2O
0 9 ppm < 20 ppm 0,43 ppm 0,21 ppm < 0,1 ppm 230 ppm
100 8 ppm < 20 ppm 0,63 ppm 0,24 ppm < 0,1 ppm 0,21 %
500 8 ppm 0,24 % 0,59 ppm 0,2 ppm < 0,1 ppm 0,24 %
1000 7 ppm 0,62 % 0,49 ppm 0,22 ppm < 0,1 ppm 0,27 %
Tableau V-6 : Analyse élémentaire de l'électrolyte à plusieurs stades du cyclage du P-4-FPT.
Le soufre a été analysé afin de vérifier si des oligomères ou des produits de
décomposition du polymère étaient rejetés dans l'électrolyte au cours du cyclage. Le chlore a
été analysé pour savoir si le polymère (synthétisé au FeCl3) en rejetait dans l'électrolyte au
cours du cyclage. Le fer, le nickel et le chrome ont été analysés car leur apparition indiquerait
une corrosion du collecteur de courant en acier inox.
114

Les résultats d'analyse élémentaire ne confirment pas l'hypothèse de relargage
d'oligomères dans l'électrolyte lors du cyclage du polymère, le soufre restant à des teneurs de
quelques ppm.
En revanche, le polymère rejète du chlore qui passe de quelques ppm à 0,62 % au bout
de 1000 cycles. Cependant, il ne semble pas corroder le collecteur de courant, les teneurs en
métaux restant très faibles tout au long du cyclage.
Par ailleurs, la teneur en fer n'augmentant pas dans l'électrolyte, cela montre que l'ion
dopant du polymère lors de la synthèse est Cl- et non FeCl4-, contrairement à ce qui a été
publié [36-37].
Les mesures de teneur en eau montrent que l'électrolyte possède une forte reprise en eau
dès le début du cyclage, passant de 230 ppm à 2100 ppm au cours des cent premiers cycles.
Ensuite, cette teneur en eau se stabilise, n'augmentant plus que de 600 ppm en 5000 cycles.
L'hypothèse la plus probable, en l'absence de rejet d'oligomères dans l'électrolyte, est
l'attaque des chaînes polymères par des solvants ou plus probablement l'eau, présente en
quantité non négligeable dans l'électrolyte. La teinte jaune prise par l'électrolyte serait alors
due aux produits de dégradation de ces réactions. Pour vérifier cette hypothèse, l'étude de
l'électrode est nécessaire.
V-3-5-2) Etude de l'électrode Pour étudier plus directement la dégradation du P-4-FPT, celui-ci a été mis en œuvre
dans des électrodes sans liant. La poudre de polymère, sans additif conducteur, est placée dans
un sachet de tissu de carbone conducteur, un déployé métallique servant de collecteur de
courant (cf. Figure V-48).
Figure V-48 : Schéma d'une électrode de poudre de polymère.
115
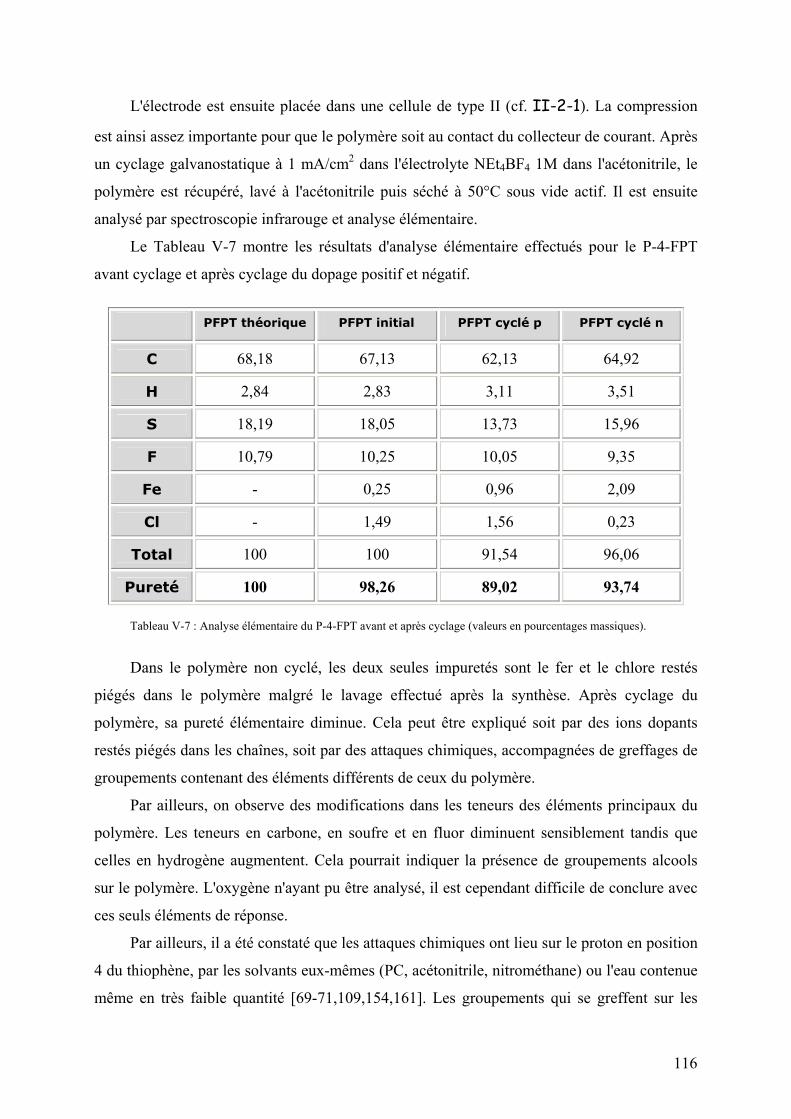
L'électrode est ensuite placée dans une cellule de type II (cf. II-2-1). La compression
est ainsi assez importante pour que le polymère soit au contact du collecteur de courant. Après
un cyclage galvanostatique à 1 mA/cm2 dans l'électrolyte NEt4BF4 1M dans l'acétonitrile, le
polymère est récupéré, lavé à l'acétonitrile puis séché à 50°C sous vide actif. Il est ensuite
analysé par spectroscopie infrarouge et analyse élémentaire.
Le Tableau V-7 montre les résultats d'analyse élémentaire effectués pour le P-4-FPT
avant cyclage et après cyclage du dopage positif et négatif.
PFPT théorique PFPT initial PFPT cyclé p PFPT cyclé n
C 68,18 67,13 62,13 64,92
H 2,84 2,83 3,11 3,51
S 18,19 18,05 13,73 15,96
F 10,79 10,25 10,05 9,35
Fe - 0,25 0,96 2,09
Cl - 1,49 1,56 0,23
Total 100 100 91,54 96,06
Pureté 100 98,26 89,02 93,74 Tableau V-7 : Analyse élémentaire du P-4-FPT avant et après cyclage (valeurs en pourcentages massiques).
Dans le polymère non cyclé, les deux seules impuretés sont le fer et le chlore restés
piégés dans le polymère malgré le lavage effectué après la synthèse. Après cyclage du
polymère, sa pureté élémentaire diminue. Cela peut être expliqué soit par des ions dopants
restés piégés dans les chaînes, soit par des attaques chimiques, accompagnées de greffages de
groupements contenant des éléments différents de ceux du polymère.
Par ailleurs, on observe des modifications dans les teneurs des éléments principaux du
polymère. Les teneurs en carbone, en soufre et en fluor diminuent sensiblement tandis que
celles en hydrogène augmentent. Cela pourrait indiquer la présence de groupements alcools
sur le polymère. L'oxygène n'ayant pu être analysé, il est cependant difficile de conclure avec
ces seuls éléments de réponse.
Par ailleurs, il a été constaté que les attaques chimiques ont lieu sur le proton en position
4 du thiophène, par les solvants eux-mêmes (PC, acétonitrile, nitrométhane) ou l'eau contenue
même en très faible quantité [69-71,109,154,161]. Les groupements qui se greffent sur les
116
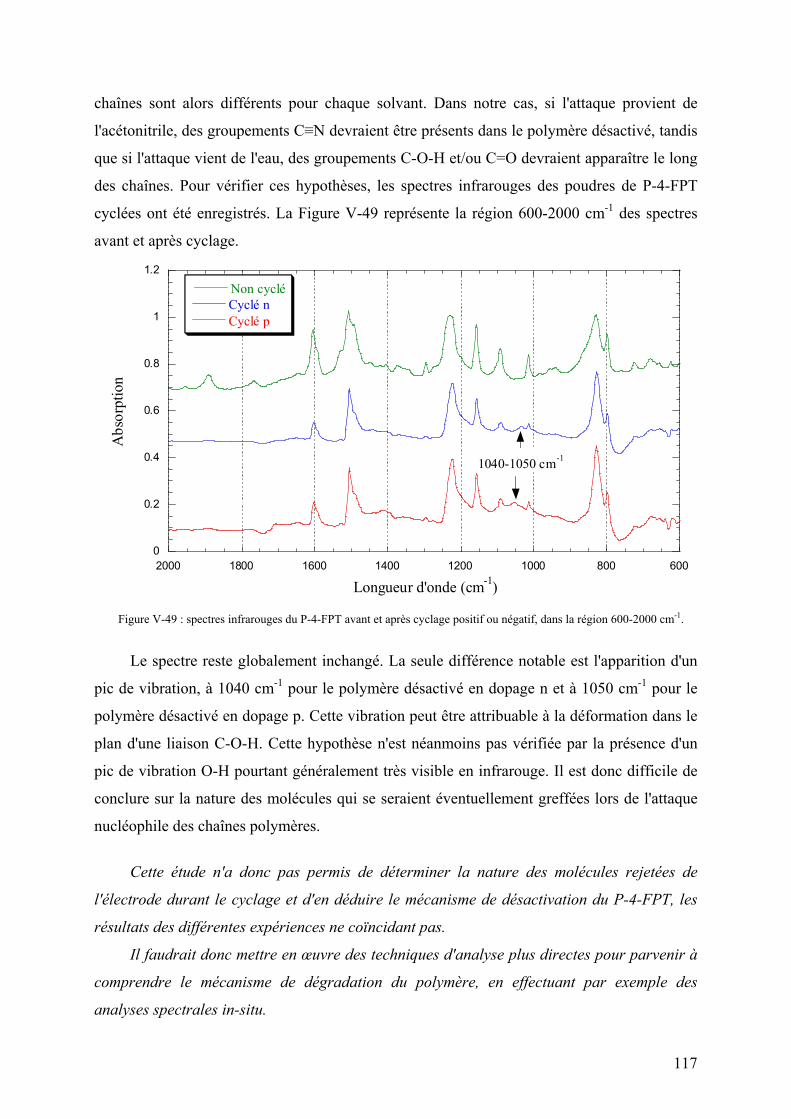
chaînes sont alors différents pour chaque solvant. Dans notre cas, si l'attaque provient de
l'acétonitrile, des groupements C≡N devraient être présents dans le polymère désactivé, tandis
que si l'attaque vient de l'eau, des groupements C-O-H et/ou C=O devraient apparaître le long
des chaînes. Pour vérifier ces hypothèses, les spectres infrarouges des poudres de P-4-FPT
cyclées ont été enregistrés. La Figure V-49 représente la région 600-2000 cm-1 des spectres
avant et après cyclage.
0
0.2
0.4
0.6
0.8
1
1.2
600800100012001400160018002000
Non cyclé
Cyclé pCyclé n
Abs
orpt
ion
Longueur d'onde (cm-1)
1040-1050 cm-1
Figure V-49 : spectres infrarouges du P-4-FPT avant et après cyclage positif ou négatif, dans la région 600-2000 cm-1.
Le spectre reste globalement inchangé. La seule différence notable est l'apparition d'un
pic de vibration, à 1040 cm-1 pour le polymère désactivé en dopage n et à 1050 cm-1 pour le
polymère désactivé en dopage p. Cette vibration peut être attribuable à la déformation dans le
plan d'une liaison C-O-H. Cette hypothèse n'est néanmoins pas vérifiée par la présence d'un
pic de vibration O-H pourtant généralement très visible en infrarouge. Il est donc difficile de
conclure sur la nature des molécules qui se seraient éventuellement greffées lors de l'attaque
nucléophile des chaînes polymères.
Cette étude n'a donc pas permis de déterminer la nature des molécules rejetées de
l'électrode durant le cyclage et d'en déduire le mécanisme de désactivation du P-4-FPT, les
résultats des différentes expériences ne coïncidant pas.
Il faudrait donc mettre en œuvre des techniques d'analyse plus directes pour parvenir à
comprendre le mécanisme de dégradation du polymère, en effectuant par exemple des
analyses spectrales in-situ.
117

V-4) Caractérisation des polyphénylthiophènes fluorés (PPTF)
Une solution pour obtenir des polymères plus stables en dopage négatif que le P-4-FPT
est de greffer sur le noyau phényl d'autres fluors pour augmenter l'effet électroattracteur du
substituant. Différents polyphénylthiophènes fluorés (PPTF) ont ainsi été synthétisés selon le
même protocole que pour le P-4-FPT, avec des rendements plus ou moins importants selon la
difficulté de purification du monomère (cf. III-1 et III-4).
Les PPTF (cf. Figure V-50) se présentent sous forme de poudres très fines de couleur
rouge à rouge sombre.
Figure V-50 : Formules chimiques des différents PPTF
V-4-1) Caractérisation chimique des PPTF
La caractérisation chimique des polymères a été effectuée par analyse élémentaire et par
spectroscopie infrarouge. Le Tableau V-8 présente les résultats d'analyse élémentaire de
chacun des PPTF, indiquant la quantité théorique calculée ainsi que la quantité mesurée dans
l'échantillon. Les deux principales impuretés dues au mode de synthèse (le chlore et le fer) ont
été quantifiées. Les polymères sont obtenus avec des puretés de plus de 96 %. Le fer se trouve
toujours à des teneurs de moins de 0,5 %. Le chlore est la principale impureté des polymères
avec des teneurs variant entre 1 et 3 %.
118

Calculé Mesuré
P-4-FPT P-3,4-DFPT P-3,5-DFPT P-3,4,5-TFPT
C
68,18 67,13
61,86 61,47
61,86 61,13
56,6 55,15
H
2,84 2,83
2,06 1,92
2,06 1,89
1,42 1,63
S
18,18 18,05
16,49 16,25
16,49 16,53
15,09 14,05
F
10,79 10,25
19,59 18,28
19,59 18,69
26,89 25,46
Fe 0,25 0,32 0,41 0,46 Cl 1,49 2,22 1,26 2,46
Pureté 98,26 97,92 98,24 96,29
Tableau V-8 : Analyses élémentaires des PPTF (valeurs en pourcentages massiques)
Les spectres IR des polymères sont présentés en Annexe 2. Ces spectres sont assez
complexes car les noyaux thiophène et phényl des polymères ont des fréquences de vibration
assez nombreuses et assez proches. De plus, leur proximité ainsi que l'enchaînement de
thiophènes dans la chaîne polymère affecte les fréquences des vibrations. Ces spectres sont en
accord avec ceux présentés dans la littérature pour les PPTF synthétisés électrochimiquement
[105].
Une proposition d'attribution des principaux pics est présentée dans le Tableau V-9. Les
vibrations des noyaux phényls ont été reportées en utilisant les notations de Wilson [160]. Les
principales vibrations des noyaux aromatiques ont été attribuées ainsi que celles des fluors
aromatiques [155-156].
Les différents PPTF se distinguent aisément les uns des autres par leurs vibration de
valence des liaisons C-F dans la zone 1200-1300 cm-1, ainsi que par les vibrations de
déformation hors du plan des liaisons C-H phényliques entre 840 et 900 cm-1.
Les vibrations de déformation hors du plan des liaisons C-H du thiophène (γC-H)
apparaissent entre 720 et 830 cm-1. Les pics de vibration des liaisons C-H en α du soufre,
représentatifs de couplages défectueux en 2,4 ou de bouts de chaînes, ne possèdent que de
faibles intensités. Cela indique que les polymères possèdent de hautes masses moléculaires
[23].
119

P-4-FPT P-3,4-DFPT P-3,5-DFPT P-3,4,5-TFPT Attribution 3053 m 3074 m 3091 m 3079 f νC-H phényl 20a-b α + thiophène 1892 m 1895 tf - - - 1605 TF 1603 F 1621 TF 1615 TF phényl 8a α 1592 m ep 1560 f ep 1591 TF 1586 m phényl 8b α 1508 TF 1517 TF 1521 f 1528 TF νC=C thiophène (asym) 1500 TF ep 1500 F ep - 1499 F ep phényl 19a α - - 1440 m ep - - 1450 f 1431 F 1433 TF 1434 F νC=C thiophène (sym) - - 1385 m 1386 m ep - 1372 f 1373 f 1366 m 1370 m νC-C thiophène 1296 f - - - - - 1275 TF 1274 F 1294 m νC-F aromatique 1230 TF - - 1242 F νC-F aromatique - 1209 F 1217 f 1207 m νC-F aromatique 1157 TF 1180 m 1190 F - δC-H phényl 9a α - 1117 F 1120 TF 1123 f δC-H thiophène 1091 F 1086 f - 1086 f δC-H phényl 18b α 1015 F - - 1046 TF phényl 12 (respiration) α - 947 f 988 TF - phényl 1 (respiration) α - 875 m - - γC-H phényl trisubst. 1:2:4 (1H) - - 858 TF ep - γC-H phényl trisubst. 1:3:5 - - - 852 F ep γC-H phényl tetrasubst. 1:3:4:5 - - 847 TF - γC-H phényl trisubst. 1:3:5 - - - 844 F γC-H phényl tetrasubst. 1:3:4:5 - 830 m - - γC-H α thiophène ? 829 TF - - - γC-H phényl disubst. 1:4 - 821 F - - γC-H phényl trisubst. 1:2:4 (2H) 800 F 771 TF 758 m 808 m ep γC-H β thiophène 724 f - - 731 m γC-H α thiophène - 709 f - 708 f déformation C-S-C 681 f 681 f 689 m - déformation C-S-C - - 670 m - γC-H phényl trisubst. 1:3:5 624 f 625 f - 643 f - - 602 m - 599 f - 574 m 573 m - - - 556 m - - - - 536 m - 532 f - - - - 510 m - -
Tableau V-9 : Attribution des spectre infrarouge des PPTF.
Intensités : TF : très forte, F : forte, m : moyenne, f : faible, ep : épaulement ; Notations des vibrations : ν : vibrations de valence, δ : vibrations de déformation dans le plan, γ : vibrations de déformation hors du plan. α notations des vibrations du noyau phényl d'après Wilson [160] (cf. Annexe 3).
120
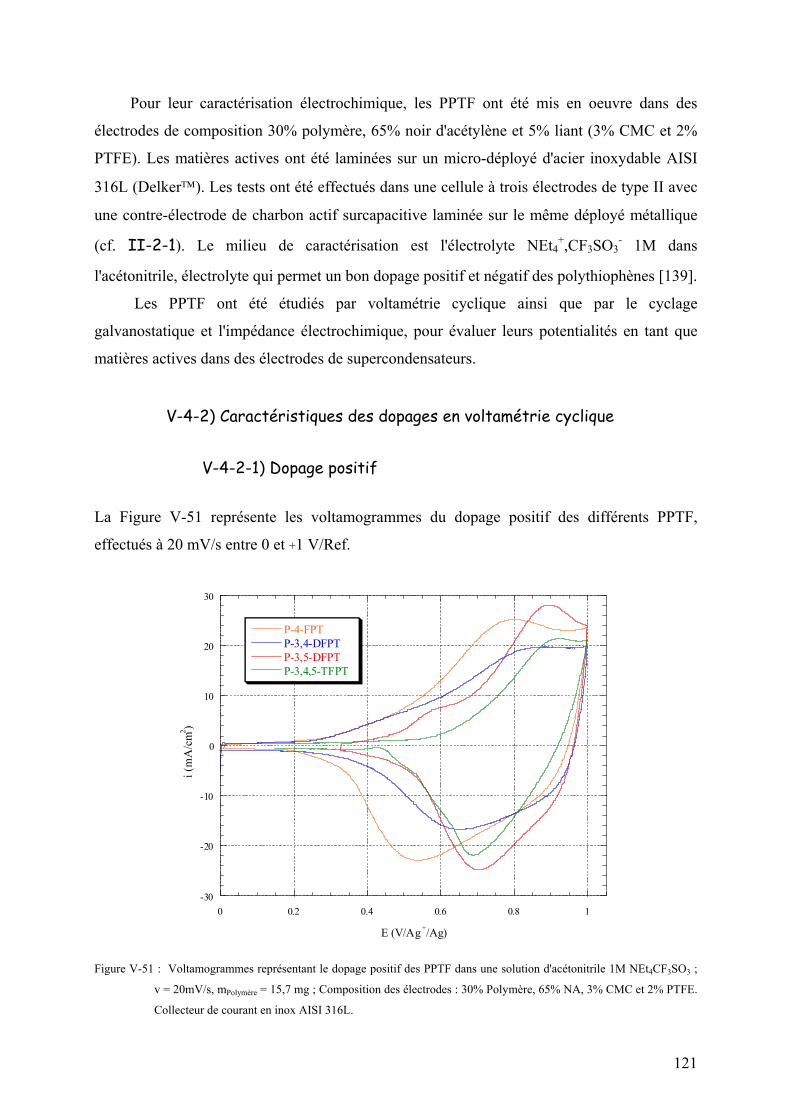
Pour leur caractérisation électrochimique, les PPTF ont été mis en oeuvre dans des
électrodes de composition 30% polymère, 65% noir d'acétylène et 5% liant (3% CMC et 2%
PTFE). Les matières actives ont été laminées sur un micro-déployé d'acier inoxydable AISI
316L (Delker). Les tests ont été effectués dans une cellule à trois électrodes de type II avec
une contre-électrode de charbon actif surcapacitive laminée sur le même déployé métallique
(cf. II-2-1). Le milieu de caractérisation est l'électrolyte NEt4+,CF3SO3
- 1M dans
l'acétonitrile, électrolyte qui permet un bon dopage positif et négatif des polythiophènes [139].
Les PPTF ont été étudiés par voltamétrie cyclique ainsi que par le cyclage
galvanostatique et l'impédance électrochimique, pour évaluer leurs potentialités en tant que
matières actives dans des électrodes de supercondensateurs.
V-4-2) Caractéristiques des dopages en voltamétrie cyclique
V-4-2-1) Dopage positif
La Figure V-51 représente les voltamogrammes du dopage positif des différents PPTF,
effectués à 20 mV/s entre 0 et +1 V/Ref.
-30
-20
-10
0
10
20
30
0 0.2 0.4 0.6 0.8 1
P-4-FPTP-3,4-DFPTP-3,5-DFPTP-3,4,5-TFPT
i (m
A/c
m2 )
E (V/Ag+/Ag)
Figure V-51 : Voltamogrammes représentant le dopage positif des PPTF dans une solution d'acétonitrile 1M NEt4CF3SO3 ;
v = 20mV/s, mPolymère = 15,7 mg ; Composition des électrodes : 30% Polymère, 65% NA, 3% CMC et 2% PTFE.
Collecteur de courant en inox AISI 316L.
121

Les différences se situent tant au niveau des potentiels de dopage que des quantités
d'électricité mises en jeu dans le processus. Les potentiels de pics de dopage rangés par ordre
croissant sont :
PFPT > P-3,4-DFPT > P-3,5-DFPT > P-3,4,5-TFPT.
Au niveau des pics de dédopage, l'ordre est quasiment le même :
PFPT > P-3,4-DFPT > P-3,4,5-TFPT > P-3,5-DFPT.
Ces différences sont liées à l'effet attracteur d'électrons plus ou moins marqué des
substituants du cycle phényl. Pour évaluer les effets dus aux substituants d'un noyau
phénylique, une méthode consiste à calculer la constante de substitution de Hammett (σ) qui
représente le caractère attracteur ou donneur d'électron d'un substituant, que l'effet soit
inductif ou de résonance [1,105,162-164]. Plus la valeur de cette constante est élevée et plus
le substituant possède un caractère attracteur.
D'après Hammett [162], la valeur des constantes pour les fluors d'un cycle phényl sont :
- σpara = 0,06
- σméta = 0,34
Pour les molécules qui possèdent plusieurs substituants, il est possible d'additionner les
constantes en considérant qu'il n'y a pas d'interactions intramoléculaires [165].
Les constantes de Hammett associées à nos polymères sont alors :
- σPFPT = 0,06
- σP-3,4-DFPT = 0,4
- σP-3,5-DFPT = 0,70
- σP-3,4,5-TFPT = 0,74
L'influence électronique des substituants sur les caractéristiques de dopage des
polymères peut alors être quantifiée. La Figure V-52 montre les potentiels de pic de dopage et
de dédopage des polymères (cf. Figure V-51) en fonction de la constante de Hammett
associée à l'effet attracteur de leur substituant.
122
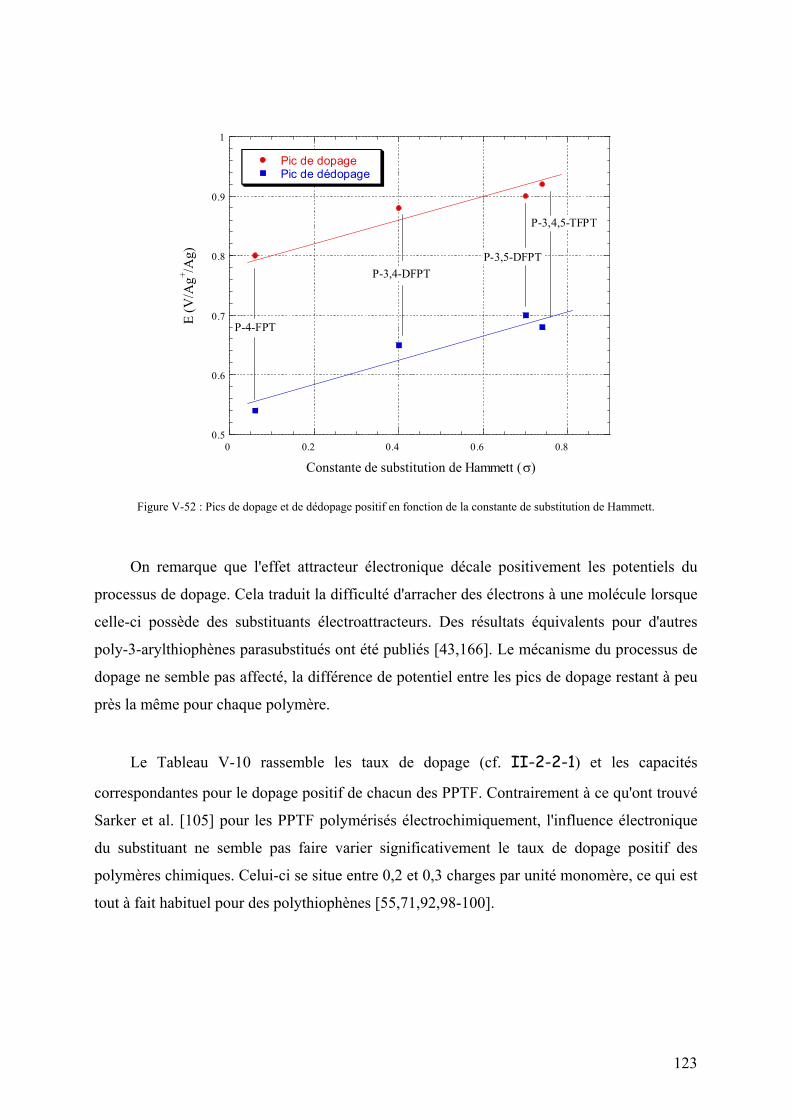
0.5
0.6
0.7
0.8
0.9
1
0 0.2 0.4 0.6 0.8
Pic de dopage Pic de dédopage
E (V
/Ag+ /A
g)
Constante de substitution de Hammett (σ)
P-4-FPT
P-3,4-DFPTP-3,5-DFPT
P-3,4,5-TFPT
Figure V-52 : Pics de dopage et de dédopage positif en fonction de la constante de substitution de Hammett.
On remarque que l'effet attracteur électronique décale positivement les potentiels du
processus de dopage. Cela traduit la difficulté d'arracher des électrons à une molécule lorsque
celle-ci possède des substituants électroattracteurs. Des résultats équivalents pour d'autres
poly-3-arylthiophènes parasubstitués ont été publiés [43,166]. Le mécanisme du processus de
dopage ne semble pas affecté, la différence de potentiel entre les pics de dopage restant à peu
près la même pour chaque polymère.
Le Tableau V-10 rassemble les taux de dopage (cf. II-2-2-1) et les capacités
correspondantes pour le dopage positif de chacun des PPTF. Contrairement à ce qu'ont trouvé
Sarker et al. [105] pour les PPTF polymérisés électrochimiquement, l'influence électronique
du substituant ne semble pas faire varier significativement le taux de dopage positif des
polymères chimiques. Celui-ci se situe entre 0,2 et 0,3 charges par unité monomère, ce qui est
tout à fait habituel pour des polythiophènes [55,71,92,98-100].
123

Polymère Taux de dopage
positif α Capacité positive
(mAh/g) α
P-4-FPT 0,26 40
P-3,4-DFPT 0,23 31,8
P-3,5-DFPT 0,25 34,6
P-3,4,5-TFPT 0,23 29,1
Tableau V-10 : Taux de dopage positif et capacité massique des PPTF. α valeurs mesurées d'après le pic de dédopage au premier cycle.
Les capacités massiques sont assez différentes pour des taux de dopage pourtant à peu
près équivalents. Les fluors sont en effet des substituants lourds et leur poids a une influence
non négligeable sur la capacité des polymères. Celle-ci passe ainsi de 40 mAh/g pour le
polymère possédant un seul fluor à 29,1 mAh/g pour celui qui en possède trois.
La constante de Hammett évalue efficacement l'effet plus ou moins électroattracteur des
substituants des PPTF. Elle permet ainsi de vérifier le déplacement attendu du dopage positif
vers des potentiels plus anodiques lorsque les substituants possèdent un effet électroattracteur
plus important. En revanche, aucune influence de l'effet électronique des substituants n'a été
clairement observée sur le taux de dopage des PPTF. Par ailleurs, la masse des fluors
influence la capacité massique des polymères, limitant ainsi l'intérêt de ceux qui en possèdent
plusieurs.
V-4-2-2) Dopage négatif
Comme il a été observé pour le P-4-FPT, le dopage négatif de chacun des PPTF
nécessite un certain nombre de cycles d'activation pendant lesquels sont observées une baisse
de capacité du dopage et une augmentation de celle du dédopage (cf. V-3-2-1). Chaque
polymère a été activé en voltamétrie cyclique par un dopage partiel effectué en limitant le
potentiel de dopage pour obtenir un taux de dopage entre 0,04 et 0,05 électrons par unité
monomère (e-/um). Le Tableau V-11 montre que le nombre de cycles nécessaires avant la
stabilisation de la réversibilité coulombique est différent pour chaque polymère.
124
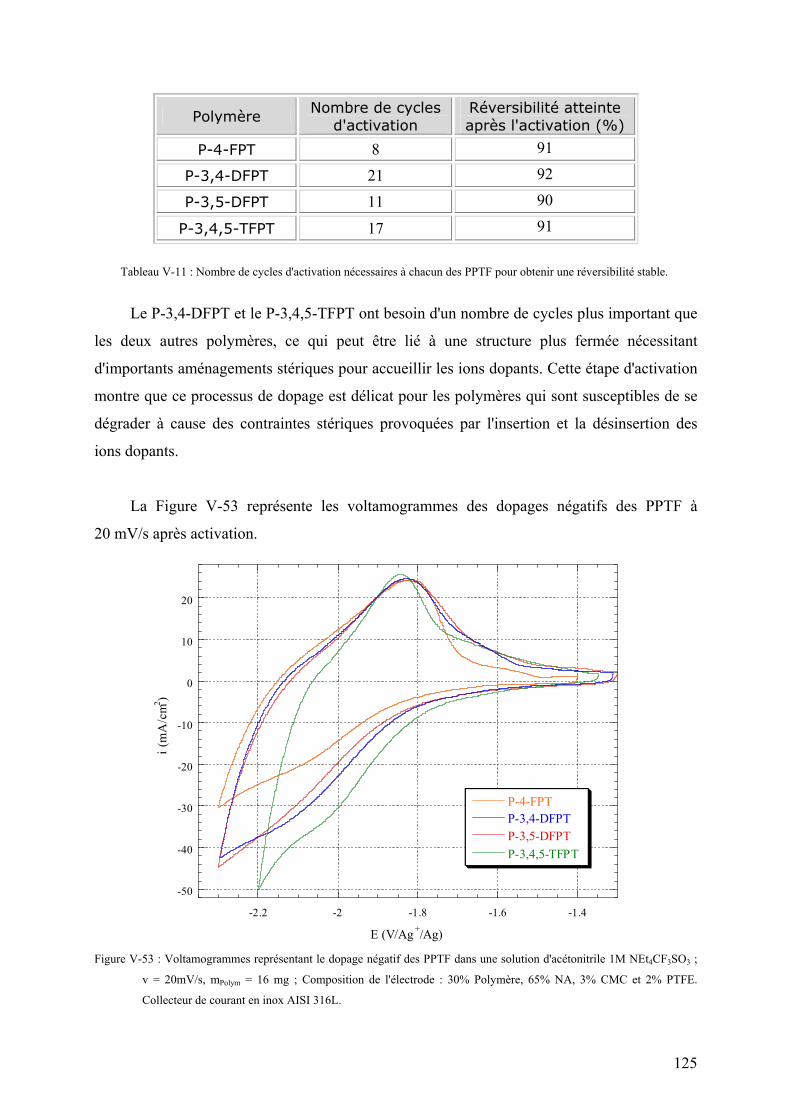
Polymère Nombre de cycles
d'activation Réversibilité atteinte après l'activation (%)
P-4-FPT 8 91
P-3,4-DFPT 21 92
P-3,5-DFPT 11 90
P-3,4,5-TFPT 17 91
Tableau V-11 : Nombre de cycles d'activation nécessaires à chacun des PPTF pour obtenir une réversibilité stable.
Le P-3,4-DFPT et le P-3,4,5-TFPT ont besoin d'un nombre de cycles plus important que
les deux autres polymères, ce qui peut être lié à une structure plus fermée nécessitant
d'importants aménagements stériques pour accueillir les ions dopants. Cette étape d'activation
montre que ce processus de dopage est délicat pour les polymères qui sont susceptibles de se
dégrader à cause des contraintes stériques provoquées par l'insertion et la désinsertion des
ions dopants.
La Figure V-53 représente les voltamogrammes des dopages négatifs des PPTF à
20 mV/s après activation.
-50
-40
-30
-20
-10
0
10
20
-2.2 -2 -1.8 -1.6 -1.4
P-4-FPTP-3,4-DFPTP-3,5-DFPTP-3,4,5-TFPT
i (m
A/c
m2 )
E (V/Ag +/Ag)
Figure V-53 : Voltamogrammes représentant le dopage négatif des PPTF dans une solution d'acétonitrile 1M NEt4CF3SO3 ;
v = 20mV/s, mPolym = 16 mg ; Composition de l'électrode : 30% Polymère, 65% NA, 3% CMC et 2% PTFE.
Collecteur de courant en inox AISI 316L.
125

Comme pour le dopage positif, on observe l'effet plus ou moins électroattracteur des
substituants, ce qui provoque le déplacement du processus de dopage vers des potentiels plus
positifs. Cependant, l'effet est moins important pour ce processus de dopage que pour le
processus positif. Le dédopage se situe à peu près au même potentiel pour tous les polymères
(cf. Figure V-54).
-2.3
-2.2
-2.1
-2
-1.9
-1.8
0 0.2 0.4 0.6 0.8
Pic de dopage
Pic de dédopage
E (V
/Ag+ /A
g)
Constante de Hammett (σ)
P-4-FPT
P-3,4-DFPT
P-3,5-DFPT
P-3,4,5-TFPT
II
II
Figure V-54 : Pics de dopage et de dédopage négatif en fonction de la constante de substitution de Hammett.
Par ailleurs, la Figure V-53 montre que la réversibilité coulombique du processus de
dopage négatif est faible pour tous les polymères : 65% pour le P-4-FPT, 44% pour le P-3,4-
DFPT, 46% pour le P-3,5-DFPT et 42% pour le P-3,4,5-TFPT.
Cette mauvaise réversibilité peut être expliquée par différents phénomènes comme cela
a été vu auparavant (cf. V-3-2-1) : réduction de l'électrolyte ou "charge-trapping".
Le Tableau V-12 présente les taux de dopage négatif ainsi que les capacités associées
pour chacun des polymères.
Polymère Taux de dopage
négatif α Capacité négative
(mAh/g) α
P-4-FPT 0,16 24,1
P-3,4-DFPT 0,2 27,3
P-3,5-DFPT 0,2 27,5
P-3,4,5-TFPT 0,17 21,7
Tableau V-12 : Taux de dopage négatif et capacité massique des PPTF. α valeurs mesurées d'après le pic de dédopage au premier cycle.
126

Le taux de dopage varie entre 0,16 et 0,2 e-/um selon les polymères, à priori
indépendamment de l'effet électroattracteur des substituants, comme cela a été vu pour le
dopage positif. Cela mène à des capacités massiques entre 21 et 28 mAh/g. De la même façon
que pour le dopage positif, la masse des substituants a une influence non négligeable sur la
capacité massique des polymères.
L'effet plus ou moins électroattracteur des différents substituants est aisément relié avec
les valeurs des potentiels de dopage positif et négatif des polymères, en utilisant les
constantes de substitution de Hammett. Les taux de dopage varient entre 0,23 et 0,26 pour le
dopage positif, et entre 0,16 et 0,2 pour le dopage négatif, indépendamment des effets
électroattracteur des substituants. Le dopage négatif possède une faible réversibilité
coulombique quelque soit le polymère, ce qui indique un piégeage important d'ions dans les
polymères.
V-4-3) Cyclabilité des processus de dopage
La raison principale de la synthèse de nouveaux polyphénylthiophènes fluorés est
d'obtenir des polymères plus stables en dopage et notamment en dopage négatif par la
stabilisation des charges négatives dans les noyaux phényls. Des tests de cyclabilité ont donc
été réalisés par voltamétrie cyclique ainsi que par cyclage galvanostatique, dans l'électrolyte
NEt4CF3SO3 1M dans l'acétonitrile.
V-4-3-1) Dopage positif
La Figure V-55 présente l'évolution du taux de dopage positif des polymères au cours
des cycles effectués en voltamétrie cyclique à 20 mV/s.
127

0
0.05
0.1
0.15
0.2
0.25
0 100 200 300 400 500
P-4-FPTP-3,4-DFPTP-3,5-DFPTP-3,4,5-TFPT
Taux
de
dopa
ge
Cycles Figure V-55 : Evolution du taux de dopage positif des PPTF au cours des cycles effectués en voltamétrie
cyclique à 20 mV/s. Electrolyte NEt4CF3SO3 1M dans l'acétonitrile.
Les polymères présentent tous un profil de dégradation durant le cyclage, plus accentué
dans les 50 premiers cycles, sans stabilisation sur au moins 500 cycles. Le greffage de
substituants plus électroattracteurs que le parafluorophényl n'améliore pas la stabilité des
polymères sur ce processus de dopage. Celle-ci est au contraire plus faible avec les polymères
polyfluorés. Cela peut être dû au décalage des domaines de dopage vers des potentiels plus
positifs.
La Figure V-56 présente l'évolution du taux de dopage positif des polymères au cours
des cycles effectués en cyclage galvanostatique à +/-1 mA/cm2.
0
0.05
0.1
0.15
0.2
0.25
0 200 400 600 800 1000
P-4-FPTP-3,4-DFPTP-3,5-DFPTP-3,4,5-TFPT
Taux
de
dopa
ge
Cycles
Figure V-56 : Evolution du taux de dopage positif des PPTF au cours des cycles effectués en cyclage
galvanostatique à +/- 1mA/cm2. Electrolyte NEt4CF3SO3 1M dans l'acétonitrile.
128

La Figure V-56 montre les mêmes profils de dégradation que le cyclage en voltamétrie
cyclique, mais avec une décroissance bien plus rapide. Pourtant, les taux de dopage des
polymères sont du même ordre de grandeur dans les deux expériences, ce qui montre que la
profondeur de dopage du polymère n'est pas la seule cause de dégradation de l'électroactivité
des PPTF.
Pour le dopage positif, la substitution du noyau thiophène par des groupements
électroattracteurs n'est pas favorable à la stabilité du processus. Cela peut s'expliquer par le
fait que le processus de dopage est décalé vers des potentiels plus positifs, ce qui peut
entraîner des problèmes d'oxydation du solvant et des réactions qui dégradent les polymères.
V-4-3-2) Dopage négatif
La stabilité du dopage complet des PPTF a été testé par voltamétrie cyclique. La Figure
V-57 présente l'évolution du taux de dopage des polymères en fonction du nombre de cycles
effectués à 20 mV/s entre -1,5 et -2,3 V/Ref dans l'électrolyte NEt4CF3SO3 1M dans
l'acétonitrile. Ce cyclage a été effectué après avoir activé les polymères.
0
0.05
0.1
0.15
0.2
0 5 10 15 20
P-4-FPTP-3,4-DFPTP-3,5-DFPTP-3,4,5-TFPT
Taux
de
dopa
ge
Cycles
Figure V-57 : Evolution du taux de dopage négatif complet des PPTF au cours des cycles effectués
en voltamétrie cyclique à 20 mV/s. Electrolyte NEt4CF3SO3 1M dans l'acétonitrile.
129
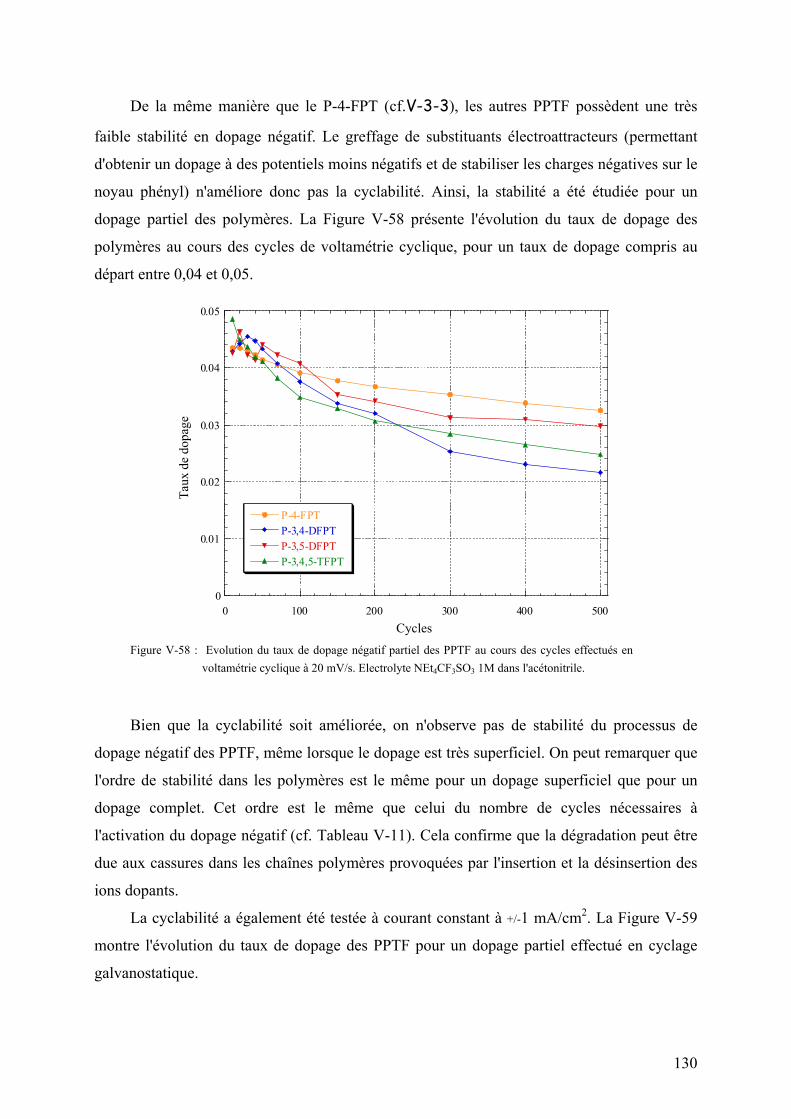
De la même manière que le P-4-FPT (cf.V-3-3), les autres PPTF possèdent une très
faible stabilité en dopage négatif. Le greffage de substituants électroattracteurs (permettant
d'obtenir un dopage à des potentiels moins négatifs et de stabiliser les charges négatives sur le
noyau phényl) n'améliore donc pas la cyclabilité. Ainsi, la stabilité a été étudiée pour un
dopage partiel des polymères. La Figure V-58 présente l'évolution du taux de dopage des
polymères au cours des cycles de voltamétrie cyclique, pour un taux de dopage compris au
départ entre 0,04 et 0,05.
0
0.01
0.02
0.03
0.04
0.05
0 100 200 300 400 500
P-4-FPTP-3,4-DFPTP-3,5-DFPTP-3,4,5-TFPT
Taux
de
dopa
ge
Cycles
Figure V-58 : Evolution du taux de dopage négatif partiel des PPTF au cours des cycles effectués en voltamétrie cyclique à 20 mV/s. Electrolyte NEt4CF3SO3 1M dans l'acétonitrile.
Bien que la cyclabilité soit améliorée, on n'observe pas de stabilité du processus de
dopage négatif des PPTF, même lorsque le dopage est très superficiel. On peut remarquer que
l'ordre de stabilité dans les polymères est le même pour un dopage superficiel que pour un
dopage complet. Cet ordre est le même que celui du nombre de cycles nécessaires à
l'activation du dopage négatif (cf. Tableau V-11). Cela confirme que la dégradation peut être
due aux cassures dans les chaînes polymères provoquées par l'insertion et la désinsertion des
ions dopants.
La cyclabilité a également été testée à courant constant à +/-1 mA/cm2. La Figure V-59
montre l'évolution du taux de dopage des PPTF pour un dopage partiel effectué en cyclage
galvanostatique.
130

0
0.01
0.02
0.03
0.04
0.05
0 200 400 600 800 1000
P-4-FPTP-3,4-DFPTP-3,5-DFPTP-3,4,5-TFPT
Taux
de
dopa
ge
Cycles
Figure V-59 : Evolution du taux de dopage négatif partiel des PPTF au cours des cycles effectués en cyclage galvanostatique à +/- 1mA/cm2. Electrolyte NEt4CF3SO3 1M dans l'acétonitrile.
De la même manière que pour le dopage positif, le cyclage à courant constant dégrade
les PPTF plus rapidement que la voltamétrie cyclique.
Tous les PPTF possèdent un profil de dégradation au cours des cycles, plus ou moins
accentué selon le polymère.
Pour le dopage positif, c'est le P-4-FPT qui se dégrade le moins rapidement. C'est donc
le polymère qui sera utilisé pour l'assemblage de supercondensateurs.
La stabilité du dopage négatif n'est pas améliorée par la substitution du thiophène par
des groupements plus électroattraceurs que le 4-fluorophényl. Par ailleurs, plus le polymère
nécessite un nombre de cycles d'activation important et plus il se dégrade rapidement. Cela
met en évidence que plus la structure est fermée et plus la dégradation est importante. Cela
indique que la dégradation des polymères est certainement due à des cassures de chaînes
provoquées par les contraintes stériques associées à l'insertion et à la désinsertion des ions
dopants.
131

VI) Application aux supercondensateurs
VI-1) Constitution des supercondensateurs
VI-1-1) Les matières actives
VI-1-1-1) L'électrode négative
Les polymères conducteurs synthétisés et caractérisés précédemment ne présentent pas
d'assez bonnes caractéristiques de dopage négatif pour pouvoir être utilisés dans des
électrodes négatives de supercondensateurs : leur dopage complet entraîne une dégradation
irréversible en quelques dizaines de cycles et ils ne possèdent une cyclabilité relative que
lorsqu'ils ne sont dopés qu'à 5-7 mAh/g. Ils ont été cependant testés pour évaluer leurs
performances dans des systèmes de supercondensateurs de type III.
D'autre part, nous avons développé un nouveau concept de supercondensateurs hybrides
en utilisant une électrode positive de polymère conducteur et une électrode négative de
charbon actif (cf. I-2-1).
Les charbons ont été choisis sous forme de poudre pour pouvoir adapter la même
technologie de mise en œuvre pour les deux électrodes. Plusieurs charbons ont été étudiés : le
Norit SX Ultra de la société Norit, ainsi que le BP25 de la société Spectracorp.
La mise en œuvre des charbons actifs est la même que pour les polymères conducteurs.
Les charbons actifs étant plus conducteurs que les polymères à l'état neutre, l'ajout de
graphites conducteurs n'est pas nécessaire. Cependant, un ajout en faible quantité permet de
diminuer la résistance surfacique.
VI-1-1-2) L'électrode positive
Les polymères synthétisés possèdent de bonnes caractéristiques électrochimiques de
dopage positif. Des capacités de 40 mAh/g - 260 F/g ont été obtenues en début de cyclage
pour le P-4-FPT. Ce polymère a donc été utilisé en tant que matière active dans les électrodes
positives.
Pour cela, il a fallu adapter sa synthèse à une échelle semi-industrielle pour en obtenir
quelques kilogrammes, ce qui a été fait en collaboration avec une société de chimie fine
(SERATEC). Il a fallu adapter la procédure de synthèse :
132

- Remplacement du chloroforme par le dichlorométhane (CHCl2) moins cher et
moins toxique.
→ les polymères synthétisés dans ce solvant ont les mêmes caractéristiques que
ceux préparés dans le chloroforme.
- Augmentation de la concentration du monomère à 4.10-2 mol/L pour limiter les
quantités de solvants utilisés.
→ cela n'a pas entraîné de conséquences néfastes sur l'électroactivité des
polymères comme on avait pu le voir à 8.10-2 mol/L (cf. III-3-2-2).
VI-1-2) Les collecteurs de courant
Les collecteurs de courant métalliques utilisés possèdent des conductivités électriques
élevées. Ils présentent néanmoins plusieurs inconvénients : ils sont lourds, ce qui pénalise la
puissance et l'énergie des systèmes, et rigides, ce qui peut entraîner des court-circuits.
L'aluminium donne parfois de très bons résultats mais ceux-ci ne sont pas reproductibles.
Nous lui avons préféré l'acier inoxydable, plus lourd et moins conducteur, mais conduisant à
des performances reproductibles comme il a été montré dans les chapitres précédents.
Pour limiter le poids des collecteurs, des feuilles de métaux déployés ont été utilisées.
Elles assurent un bon drainage électronique, et permettent un meilleur accrochage des
matières actives que des feuilles métalliques. Deux types d'aciers inoxydables ont été testés: le
AISI 304 et le AISI 316L. Ces deux aciers sont connus pour leur bonne résistance à la
corrosion. Le AISI 316L contient du molybdène, ce qui lui donne une résistance
supplémentaire contre la corrosion en milieux chlorés. La Figure VI-1 présente les
voltamogrammes effectués sur les deux déployés, dans une solution 1M NEt4BF4 dans le PC.
133
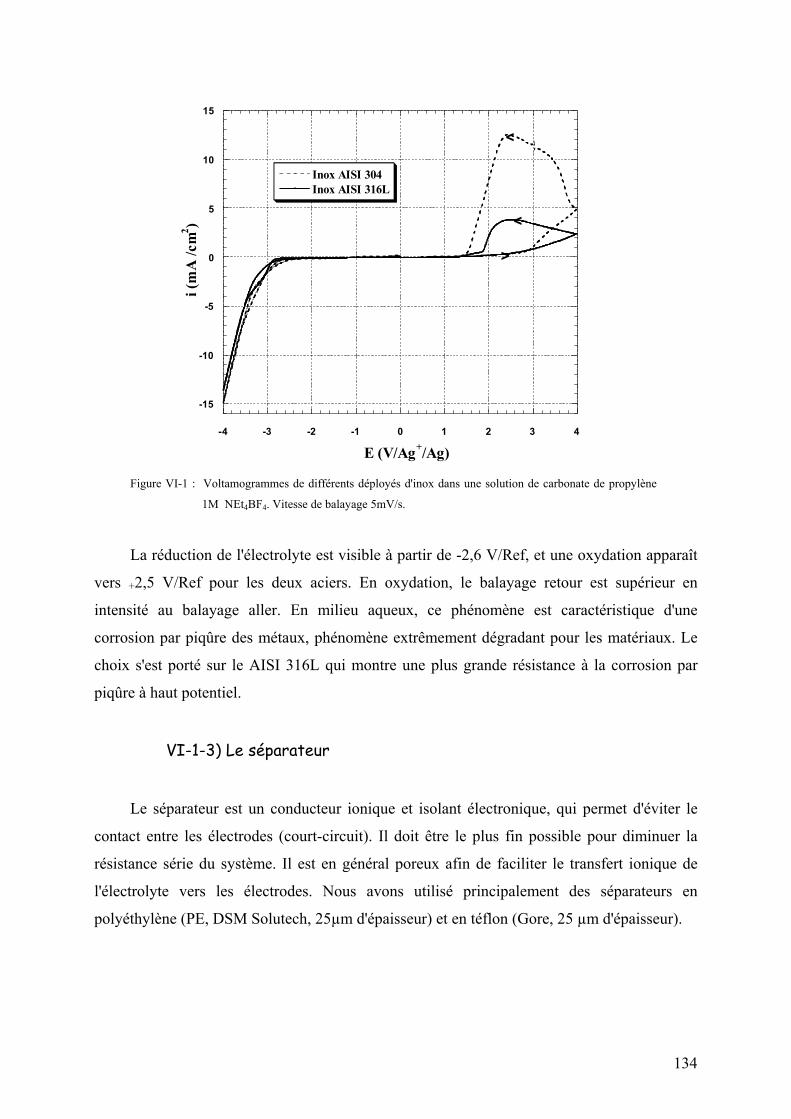
-15
-10
-5
0
5
10
15
-4 -3 -2 -1 0 1 2 3 4
Inox AISI 304 Inox AISI 316L
i (m
A /c
m2 )
E (V/Ag+/Ag)
<
>
<
Figure VI-1 : Voltamogrammes de différents déployés d'inox dans une solution de carbonate de propylène
1M NEt4BF4. Vitesse de balayage 5mV/s.
La réduction de l'électrolyte est visible à partir de -2,6 V/Ref, et une oxydation apparaît
vers +2,5 V/Ref pour les deux aciers. En oxydation, le balayage retour est supérieur en
intensité au balayage aller. En milieu aqueux, ce phénomène est caractéristique d'une
corrosion par piqûre des métaux, phénomène extrêmement dégradant pour les matériaux. Le
choix s'est porté sur le AISI 316L qui montre une plus grande résistance à la corrosion par
piqûre à haut potentiel.
VI-1-3) Le séparateur
Le séparateur est un conducteur ionique et isolant électronique, qui permet d'éviter le
contact entre les électrodes (court-circuit). Il doit être le plus fin possible pour diminuer la
résistance série du système. Il est en général poreux afin de faciliter le transfert ionique de
l'électrolyte vers les électrodes. Nous avons utilisé principalement des séparateurs en
polyéthylène (PE, DSM Solutech, 25µm d'épaisseur) et en téflon (Gore, 25 µm d'épaisseur).
134

VI-1-4) L'électrolyte
Le seul électrolyte qui permette un bon dopage du P-4-FPT en positif comme en négatif
est le NEt4CF3SO3 1M dans l'acétonitrile. Il sera donc utilisé dans les systèmes
P-4-FPT/P-4-FPT.
En revanche, pour les systèmes qui n'utilisent le polymère que pour l'électrode positive,
nous avons utilisé un électrolyte composé de NEt4BF4 1M dans le PC, d'après les résultats du
Laboratoire de Chimie Physique de Bologne, dans le cadre du projet européen [127]. En effet,
la dégradation du P-4-FPT dans cet électrolyte est moins importante que dans le NEt4CF3SO3
1M dans l'acétonitrile. Le PC a également été préféré à l'acétonitrile pour sa stabilité en
température et sa moindre nocivité.
VI-2) Montage de cellules-test de 4 cm2
Les polymères sont testés en cellules-test comportant deux électrodes de 4 cm2 séparées
par une feuille de PE ou de téflon. Les électrodes sont compressées par deux pinces en acier,
et sont plongées dans l'électrolyte. Une électrode de référence est placée au-dessus du
système. Ce montage est identique au montage de type II utilisé pour les caractérisations des
polymères (cf. II-2-1).
Les techniques utilisées pour caractériser les systèmes sont le cyclage galvanostatique
ainsi que la spectroscopie d'impédance, techniques permettant de mesurer les caractéristiques
initiales des systèmes et également de suivre leurs évolutions au cours de l'utilisation.
VI-2-1) Les supercondensateurs polymères de type III
VI-2-1-1) Principe de fonctionnement
Lorsque qu'un système composé de deux polymères conducteurs se charge, le polymère
de l'électrode négative se dope négativement (injection d'électrons et insertion de cations le
long des chaînes polymères) tandis que le polymère de l'électrode positive se dope
positivement (extraction d'électrons et insertion d'anions le long des chaînes polymères)
(cf. Figure VI-2 et Equations (VI-1) et (VI-2)).
135

P-4-FPT + e- + NEt4+ P-4-FPT-,NEt4
+ Eq.(VI-1)
P-4-FPT + CF3SO3- P-4-FPT+,CF3SO3
- + e- Eq.(VI-2)
Il en résulte que la tension de fin de charge du système va être égale à la différence entre
les deux potentiels de fin de dopage des polymères, tandis que la tension de fin de décharge
sera la différence entre les deux potentiels de fin de dédopage des polymères. Dans le cas d'un
système P-4-FPT/P-4-FPT, si le dopage de l'électrode positive est arrêté à +1,0 V/Ref et le
dopage de la négative à -2,0 V/Ref pour obtenir une certaine cyclabilité, le système cyclera
entre 2,0 V et 3,0 V.
Figure VI-2 : principe de fonctionnement d'un supercondensateur polymère/polymère
VI-2-1-2) Equilibrage des capacités
L'une des difficultés rencontrées pour assembler un système à base de polymères
conducteurs réside dans l'équilibrage des capacitances des deux électrodes. En effet, si les
électrodes ont des capacitances différentes, l'électrode la moins capacitive va subir une
136
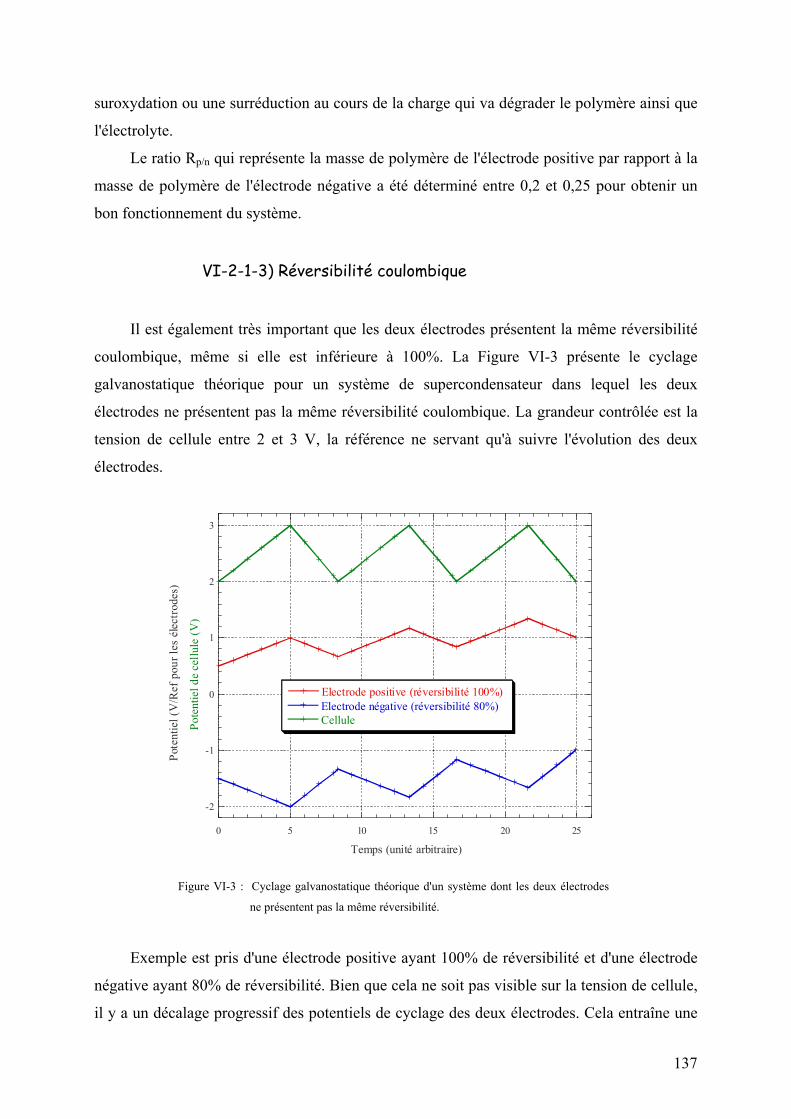
suroxydation ou une surréduction au cours de la charge qui va dégrader le polymère ainsi que
l'électrolyte.
Le ratio Rp/n qui représente la masse de polymère de l'électrode positive par rapport à la
masse de polymère de l'électrode négative a été déterminé entre 0,2 et 0,25 pour obtenir un
bon fonctionnement du système.
VI-2-1-3) Réversibilité coulombique
Il est également très important que les deux électrodes présentent la même réversibilité
coulombique, même si elle est inférieure à 100%. La Figure VI-3 présente le cyclage
galvanostatique théorique pour un système de supercondensateur dans lequel les deux
électrodes ne présentent pas la même réversibilité coulombique. La grandeur contrôlée est la
tension de cellule entre 2 et 3 V, la référence ne servant qu'à suivre l'évolution des deux
électrodes.
-2
-1
0
1
2
3
0 5 10 15 20 25
Electrode positive (réversibilité 100%)Electrode négative (réversibilité 80%)Cellule
Pote
ntie
l (V
/Ref
pou
r les
éle
ctro
des)
Temps (unité arbitraire)
Pote
ntie
l de
cellu
le (V
)
Figure VI-3 : Cyclage galvanostatique théorique d'un système dont les deux électrodes
ne présentent pas la même réversibilité.
Exemple est pris d'une électrode positive ayant 100% de réversibilité et d'une électrode
négative ayant 80% de réversibilité. Bien que cela ne soit pas visible sur la tension de cellule,
il y a un décalage progressif des potentiels de cyclage des deux électrodes. Cela entraîne une
137
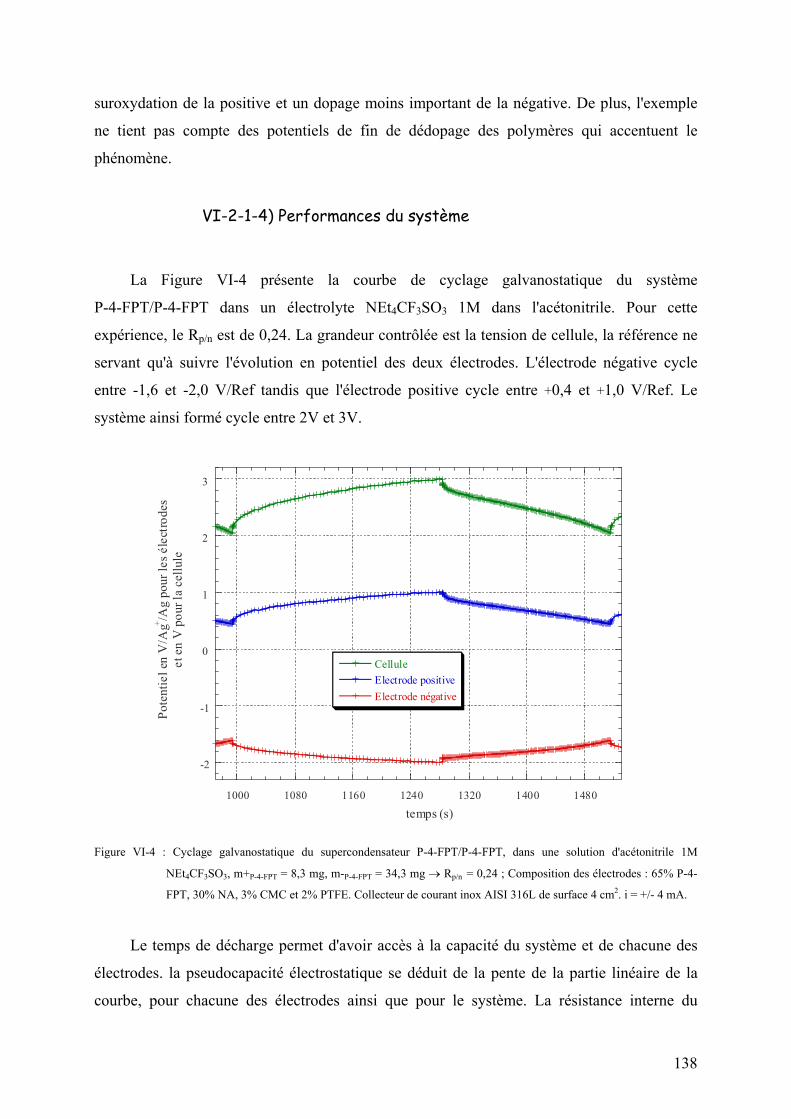
suroxydation de la positive et un dopage moins important de la négative. De plus, l'exemple
ne tient pas compte des potentiels de fin de dédopage des polymères qui accentuent le
phénomène.
VI-2-1-4) Performances du système
La Figure VI-4 présente la courbe de cyclage galvanostatique du système
P-4-FPT/P-4-FPT dans un électrolyte NEt4CF3SO3 1M dans l'acétonitrile. Pour cette
expérience, le Rp/n est de 0,24. La grandeur contrôlée est la tension de cellule, la référence ne
servant qu'à suivre l'évolution en potentiel des deux électrodes. L'électrode négative cycle
entre -1,6 et -2,0 V/Ref tandis que l'électrode positive cycle entre +0,4 et +1,0 V/Ref. Le
système ainsi formé cycle entre 2V et 3V.
-2
-1
0
1
2
3
1000 1080 1160 1240 1320 1400 1480
Electrode négativeElectrode positiveCellule
Pote
ntie
l en
V/A
g+ /Ag
pour
les é
lect
rode
set
en
V p
our l
a ce
llule
temps (s)
Figure VI-4 : Cyclage galvanostatique du supercondensateur P-4-FPT/P-4-FPT, dans une solution d'acétonitrile 1M
NEt4CF3SO3, m+P-4-FPT = 8,3 mg, m-P-4-FPT = 34,3 mg → Rp/n = 0,24 ; Composition des électrodes : 65% P-4-
FPT, 30% NA, 3% CMC et 2% PTFE. Collecteur de courant inox AISI 316L de surface 4 cm2. i = +/- 4 mA.
Le temps de décharge permet d'avoir accès à la capacité du système et de chacune des
électrodes. la pseudocapacité électrostatique se déduit de la pente de la partie linéaire de la
courbe, pour chacune des électrodes ainsi que pour le système. La résistance interne du
138

système est calculée à partir de la chute ohmique mesurée entre la fin de la charge et le début
de la décharge (cf. II-2-2-2).
Au début du cyclage, les capacités obtenues sont de 7,5 mAh/g pour la négative et de
31 mAh/g pour la positive, et les capacités "électrostatiques" sont respectivement de 104 F/g
et 265 F/g. La réversibilité coulombique du système est de 80,6%, ce qui laisse supposer
qu'elle n'est pas la même pour chacune des électrodes. La résistance interne calculée mesurée
d'après la chute ohmique de début de décharge pour le système est de 6 Ω.cm2 à 50 Hz.
On peut également déduire des courbes galvanostatiques des performances en termes
d'énergie et de puissance maximales. Pour ce système, l'énergie maximale obtenue est de
40 Wh/kg de P-4-FPT et la puissance maximale de 21 kW/kg de P-4-FPT.
Dans les systèmes de stockage d'énergie, il est généralement constaté qu'un facteur
d'environ 0,2 existe entre le poids des couches actives (matières actives + conducteur
électronique + liant) et celui du système complet. En considérant que le P-4-FPT est présent à
65% dans les électrodes, ces valeurs permettent d'estimer qu'un supercondensateur
P-4-FPT/P-4-FPT développerait une puissance d'environ 3 kW/kg pour une énergie de
5 Wh/kg.
Ces résultats sont tout à fait intéressants, comparés aux meilleurs systèmes commerciaux
de supercondensateurs carbone/carbone qui développent des puissances comprises entre
5 et 7 kW/kg associés à des énergies comprises entre 2 et 4 Wh/kg (cf. I-2-1).
Ce système a été testé sur quelques dizaines de cycles, pour évaluer la stabilité du
processus de dopage des deux électrodes lorsqu'elles sont mises en œuvre dans un système de
supercondensateur. La Figure VI-5 présente l'évolution des capacités déchargées par chacune
des électrodes durant le cyclage.
139
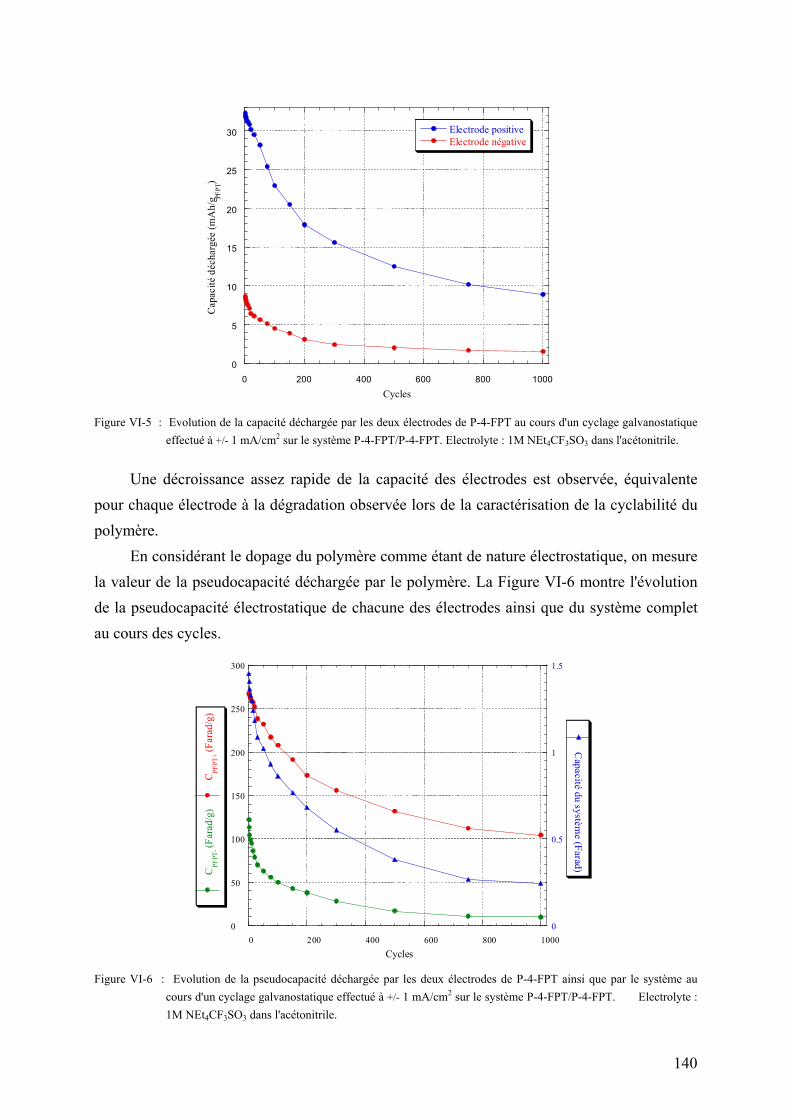
0
5
10
15
20
25
30
0 200 400 600 800 1000
Electrode positiveElectrode négative
Cap
acité
déc
harg
ée (m
Ah/
g PFPT
)
Cycles
Figure VI-5 : Evolution de la capacité déchargée par les deux électrodes de P-4-FPT au cours d'un cyclage galvanostatique
effectué à +/- 1 mA/cm2 sur le système P-4-FPT/P-4-FPT. Electrolyte : 1M NEt4CF3SO3 dans l'acétonitrile.
Une décroissance assez rapide de la capacité des électrodes est observée, équivalente
pour chaque électrode à la dégradation observée lors de la caractérisation de la cyclabilité du polymère.
En considérant le dopage du polymère comme étant de nature électrostatique, on mesure la valeur de la pseudocapacité déchargée par le polymère. La Figure VI-6 montre l'évolution de la pseudocapacité électrostatique de chacune des électrodes ainsi que du système complet au cours des cycles.
0
50
100
150
200
250
300
0
0.5
1
1.5
0 200 400 600 800 1000
CPF
PT+ (F
arad
/g)
CPF
PT- (F
arad
/g)
Capacité du systèm
e (Farad)
Cycles
Figure VI-6 : Evolution de la pseudocapacité déchargée par les deux électrodes de P-4-FPT ainsi que par le système au cours d'un cyclage galvanostatique effectué à +/- 1 mA/cm2 sur le système P-4-FPT/P-4-FPT. Electrolyte : 1M NEt4CF3SO3 dans l'acétonitrile.
140

Comme pour le modèle de transfert de charge, la dégradation des performances du
système suit la dégradation de chacune des électrodes polymères.
VI-2-1-5) Spectre d'impédance électrochimique du système
La Figure VI-7 présente le spectre d'impédance électrochimique dans le plan de Nyquist
du système P-4-FPT / P-4-FPT entre 65 kHz et 10 mHz.
-30
-25
-20
-15
-10
-5
00 5 10 15 20 25 30
-Z" (
Ω.c
m2 )
Z' (Ω.cm2)
200 mHz
13 Hz
-10
-8
-6
-4
-2
00 2 4 6 8 10
-Z" (
Ω.c
m2 )
Z' (Ω.cm2)
13 Hz
1260 Hz
Agrandissement sur les hautes fréquences
Figure VI-7 : Spectre d'impédance dans le plan de Nyquist du système P-4-FPT / P-4-FPT entre 65 kHz et 200 mHz.
La résistance série du système est de 3,8 Ω.cm2. La boucle à haute fréquence correspond
au transfert de charge de la réaction de dopage du polymère. La droite de diffusion est assez
difficile à discerner. On observe ensuite la droite qui représente la capacité du système. En
extrapolant cette droite sur l'axe des réels, on détermine que la résistance interne du système
est de 6,2 Ω.cm2.
La stabilité ainsi que la capacité de l'électrode positive est plus importante que celle de
l'électrode négative, ce qui limite les performances du supercondensateur. C'est pourquoi, le
polymère a été remplacé dans les électrodes négatives par un charbon actif, plus stable.
141

VI-2-2) Les supercondensateurs hybrides polymère – carbone
Les polymères conducteurs ne possédant pas d'assez bonnes propriétés de dopage
négatif (faible capacitance, mauvaise cyclabilité), ils ont été remplacés dans les électrodes
négatives par des charbons actifs, qui possèdent une cyclabilité de plusieurs dizaines de
milliers de cycles.
VI-2-2-1) Principe de fonctionnement
Les supercondensateurs hybrides sont donc composés d'une électrode positive de
polymère conducteur et d'une électrode négative de charbon actif. Lorsque le système se
charge, le polymère conducteur se dope positivement pendant que le charbon actif se charge
négativement (cf. Figure VI-8).
Figure VI-8 : principe de fonctionnement d'un supercondensateur hybride carbone/polymère
En s'assurant du fonctionnement de l'électrode positive sur le domaine de potentiels du
dopage positif du polymère, il est alors possible de mettre en œuvre un système de générateur
électrochimique. Par ailleurs, les charbons actifs possédant une réversibilité de près de 100%,
il est impératif d'obtenir une réversibilité proche de l'unité pour le dopage positif du polymère
afin d'éviter la dérive des électrodes (cf. VI-2-1-2).
142
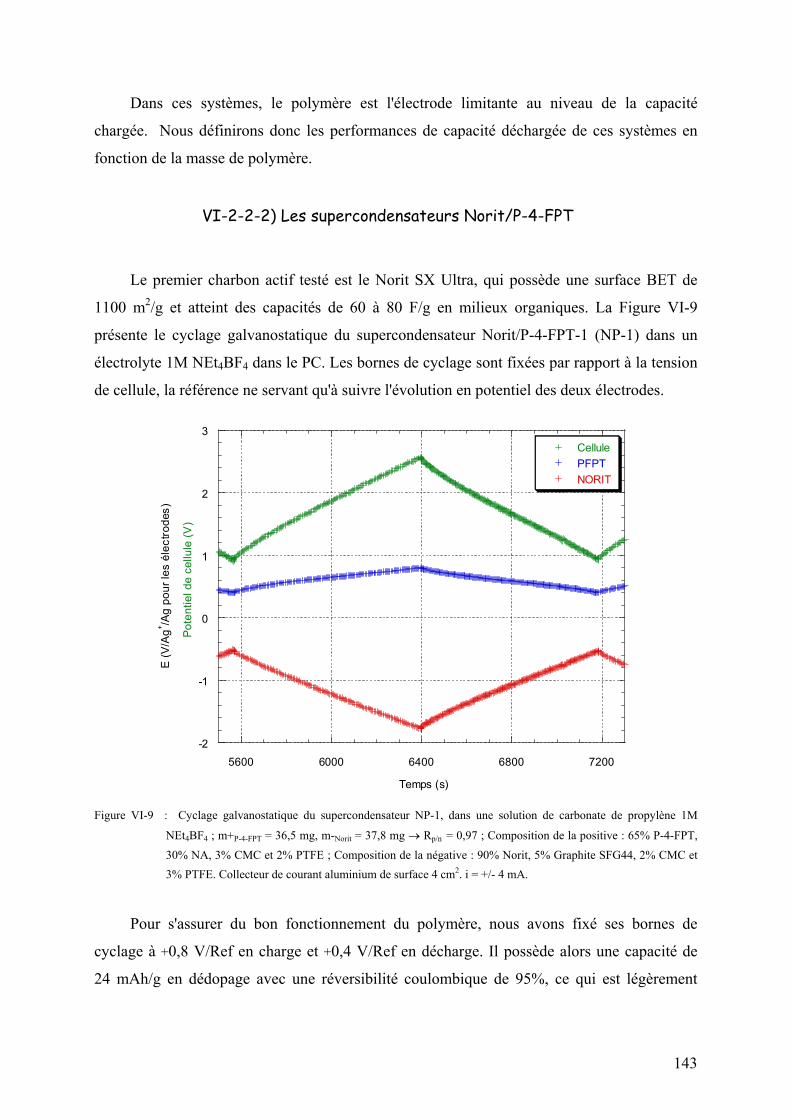
Dans ces systèmes, le polymère est l'électrode limitante au niveau de la capacité
chargée. Nous définirons donc les performances de capacité déchargée de ces systèmes en
fonction de la masse de polymère.
VI-2-2-2) Les supercondensateurs Norit/P-4-FPT
Le premier charbon actif testé est le Norit SX Ultra, qui possède une surface BET de
1100 m2/g et atteint des capacités de 60 à 80 F/g en milieux organiques. La Figure VI-9
présente le cyclage galvanostatique du supercondensateur Norit/P-4-FPT-1 (NP-1) dans un
électrolyte 1M NEt4BF4 dans le PC. Les bornes de cyclage sont fixées par rapport à la tension
de cellule, la référence ne servant qu'à suivre l'évolution en potentiel des deux électrodes.
-2
-1
0
1
2
3
5600 6000 6400 6800 7200
CellulePFPTNORIT
E (V
/Ag+ /A
g po
ur le
s él
ectro
des)
Temps (s)
Pote
ntie
l de
cellu
le (V
)
Figure VI-9 : Cyclage galvanostatique du supercondensateur NP-1, dans une solution de carbonate de propylène 1M
NEt4BF4 ; m+P-4-FPT = 36,5 mg, m-Norit = 37,8 mg → Rp/n = 0,97 ; Composition de la positive : 65% P-4-FPT,
30% NA, 3% CMC et 2% PTFE ; Composition de la négative : 90% Norit, 5% Graphite SFG44, 2% CMC et 3% PTFE. Collecteur de courant aluminium de surface 4 cm2. i = +/- 4 mA.
Pour s'assurer du bon fonctionnement du polymère, nous avons fixé ses bornes de
cyclage à +0,8 V/Ref en charge et +0,4 V/Ref en décharge. Il possède alors une capacité de
24 mAh/g en dédopage avec une réversibilité coulombique de 95%, ce qui est légèrement
143
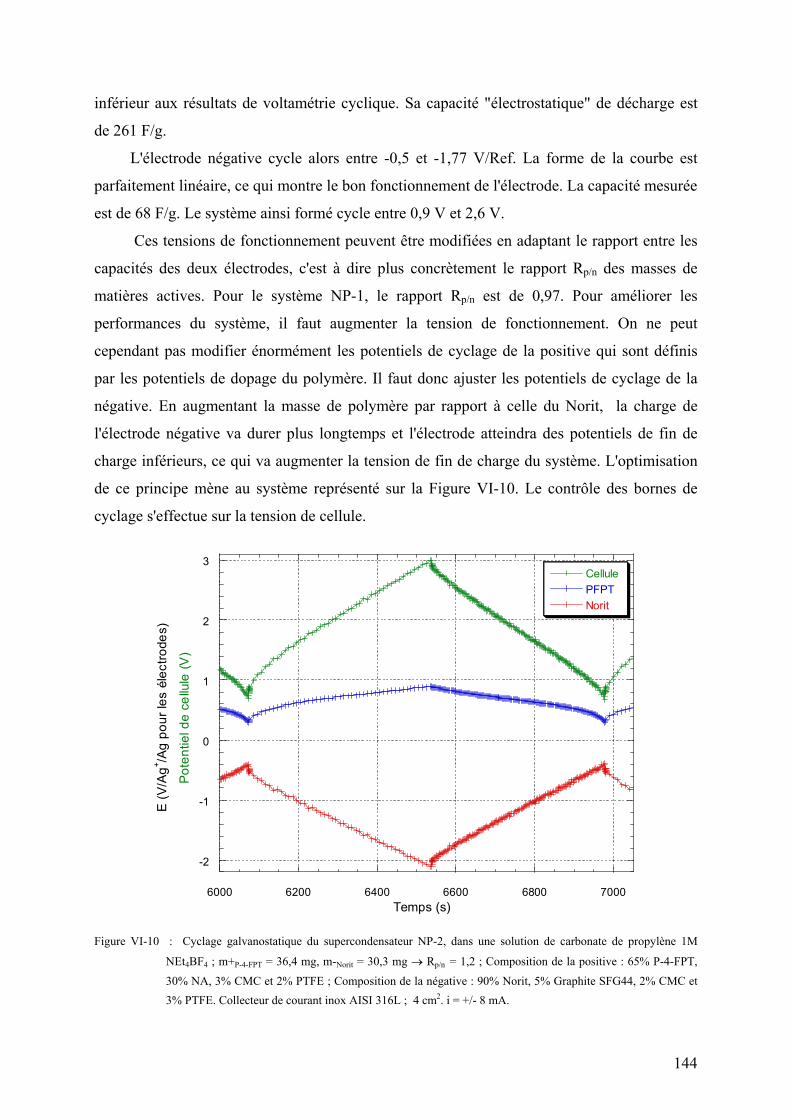
inférieur aux résultats de voltamétrie cyclique. Sa capacité "électrostatique" de décharge est
de 261 F/g.
L'électrode négative cycle alors entre -0,5 et -1,77 V/Ref. La forme de la courbe est
parfaitement linéaire, ce qui montre le bon fonctionnement de l'électrode. La capacité mesurée
est de 68 F/g. Le système ainsi formé cycle entre 0,9 V et 2,6 V.
Ces tensions de fonctionnement peuvent être modifiées en adaptant le rapport entre les
capacités des deux électrodes, c'est à dire plus concrètement le rapport Rp/n des masses de
matières actives. Pour le système NP-1, le rapport Rp/n est de 0,97. Pour améliorer les
performances du système, il faut augmenter la tension de fonctionnement. On ne peut
cependant pas modifier énormément les potentiels de cyclage de la positive qui sont définis
par les potentiels de dopage du polymère. Il faut donc ajuster les potentiels de cyclage de la
négative. En augmentant la masse de polymère par rapport à celle du Norit, la charge de
l'électrode négative va durer plus longtemps et l'électrode atteindra des potentiels de fin de
charge inférieurs, ce qui va augmenter la tension de fin de charge du système. L'optimisation
de ce principe mène au système représenté sur la Figure VI-10. Le contrôle des bornes de
cyclage s'effectue sur la tension de cellule.
-2
-1
0
1
2
3
6000 6200 6400 6600 6800 7000
CellulePFPTNorit
E (V
/Ag+ /A
g po
ur le
s él
ectro
des)
Temps (s)
Pote
ntie
l de
cellu
le (V
)
Figure VI-10 : Cyclage galvanostatique du supercondensateur NP-2, dans une solution de carbonate de propylène 1M
NEt4BF4 ; m+P-4-FPT = 36,4 mg, m-Norit = 30,3 mg → Rp/n = 1,2 ; Composition de la positive : 65% P-4-FPT,
30% NA, 3% CMC et 2% PTFE ; Composition de la négative : 90% Norit, 5% Graphite SFG44, 2% CMC et 3% PTFE. Collecteur de courant inox AISI 316L ; 4 cm2. i = +/- 8 mA.
144

L'électrode positive cycle entre 0,3 et 0,9 V/Ref, alors que l'électrode négative cycle
entre –0,4 V et –2,1 V/Ref. Des expériences ont montré qu'au-delà de –2,1 V/Ref, des
réactions faradiques apparaissent au niveau de l'électrode négative. Cette tension est donc la
limite maximale de charge pour la négative. Le système ainsi défini cycle entre 0,7 V et 3V.
Le polymère possède une capacité de 28,9 mAh/g avec une réversibilité de 94% en
début de cyclage. Sa capacité "électrostatique" est alors de 247 F/g et celle du Norit de 75 F/g.
La résistance interne du système est de 8,2 Ω.cm2 à 50 Hz.
L'énergie maximale du système est de 33,7 Wh/kg pour une puissance maximale de
11,2 kW/kg de matières actives (polymère et charbon actif). En tenant compte du facteur 0,2
et des différentes teneurs en matières actives des électrodes, on peut estimer qu'un système
complet de Norit/P-4-FPT présenterait une énergie maximale d'environ 5 Wh/kg et
développerait une puissance de 2 kW/kg.
Les performances de tels systèmes ont été testées sur un nombre de cycles important.
Les Figures VI-11 et VI-12 présentent l'évolution de la capacité chargée par le polymère ainsi
que des capacités électrostatiques de chacune des électrodes et du système au cours des
cycles.
0
5
10
15
20
25
30
0 1000 2000 3000 4000 5000
Cap
acité
déc
harg
ée (m
Ah/
g PFPT
)
Cycles
Figure VI-11 : Evolution de la capacité déchargée par le P-4-FPT lors du cyclage galvanostatique du supercondensateur
NP-2 ; i = +/- 20 mA.
145
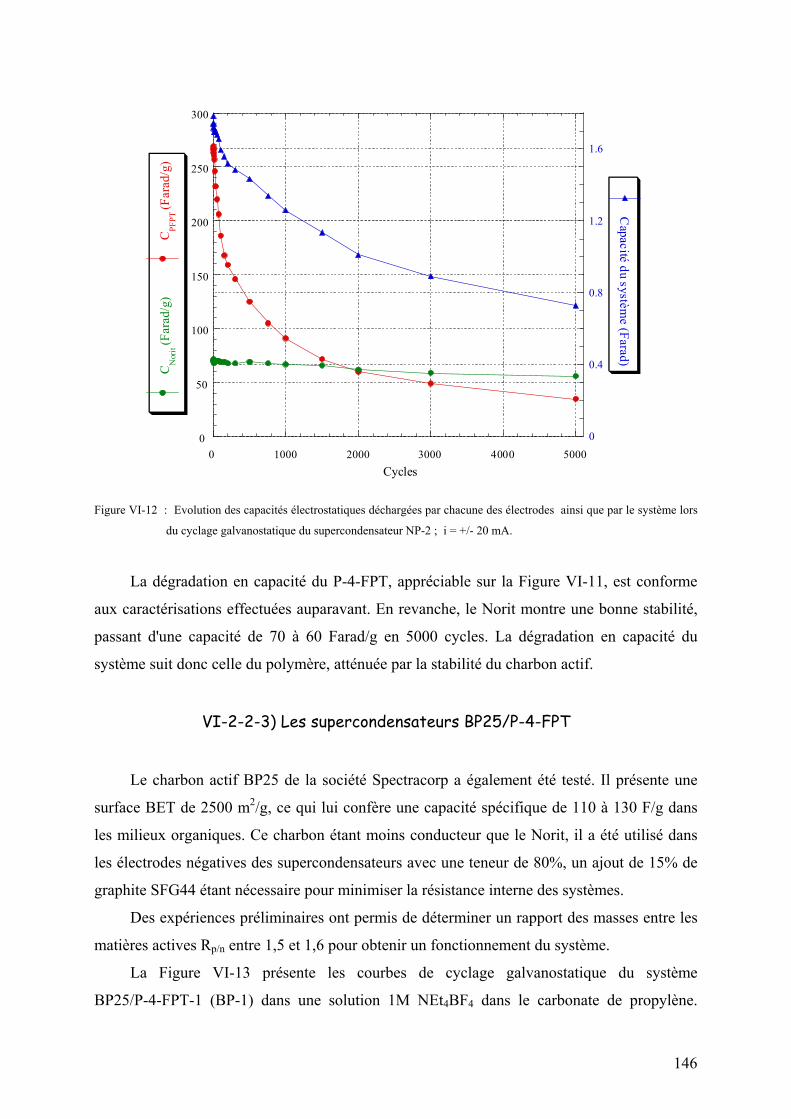
0
50
100
150
200
250
300
0
0.4
0.8
1.2
1.6
0 1000 2000 3000 4000 5000
CPF
PT (F
arad
/g)
CN
orit (F
arad
/g)
Capacité du systèm
e (Farad)
Cycles
Figure VI-12 : Evolution des capacités électrostatiques déchargées par chacune des électrodes ainsi que par le système lors
du cyclage galvanostatique du supercondensateur NP-2 ; i = +/- 20 mA.
La dégradation en capacité du P-4-FPT, appréciable sur la Figure VI-11, est conforme
aux caractérisations effectuées auparavant. En revanche, le Norit montre une bonne stabilité,
passant d'une capacité de 70 à 60 Farad/g en 5000 cycles. La dégradation en capacité du
système suit donc celle du polymère, atténuée par la stabilité du charbon actif.
VI-2-2-3) Les supercondensateurs BP25/P-4-FPT
Le charbon actif BP25 de la société Spectracorp a également été testé. Il présente une
surface BET de 2500 m2/g, ce qui lui confère une capacité spécifique de 110 à 130 F/g dans
les milieux organiques. Ce charbon étant moins conducteur que le Norit, il a été utilisé dans
les électrodes négatives des supercondensateurs avec une teneur de 80%, un ajout de 15% de
graphite SFG44 étant nécessaire pour minimiser la résistance interne des systèmes.
Des expériences préliminaires ont permis de déterminer un rapport des masses entre les
matières actives Rp/n entre 1,5 et 1,6 pour obtenir un fonctionnement du système.
La Figure VI-13 présente les courbes de cyclage galvanostatique du système
BP25/P-4-FPT-1 (BP-1) dans une solution 1M NEt4BF4 dans le carbonate de propylène.
146
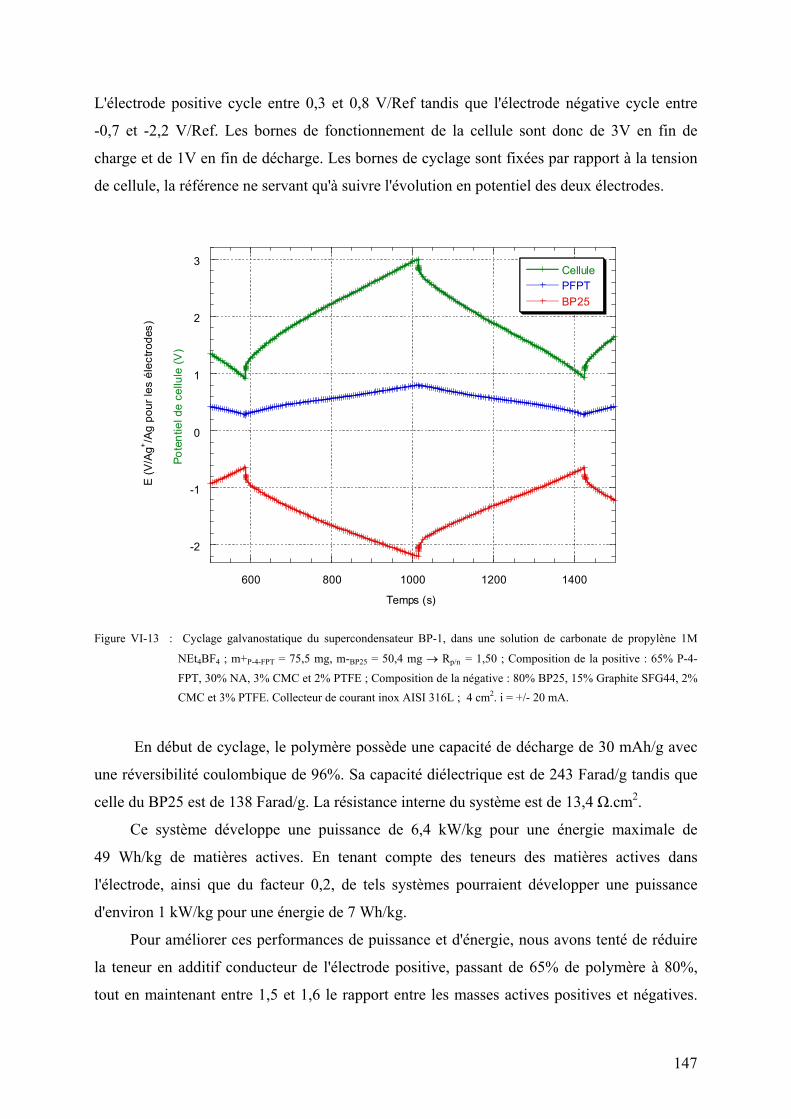
L'électrode positive cycle entre 0,3 et 0,8 V/Ref tandis que l'électrode négative cycle entre
-0,7 et -2,2 V/Ref. Les bornes de fonctionnement de la cellule sont donc de 3V en fin de
charge et de 1V en fin de décharge. Les bornes de cyclage sont fixées par rapport à la tension
de cellule, la référence ne servant qu'à suivre l'évolution en potentiel des deux électrodes.
-2
-1
0
1
2
3
600 800 1000 1200 1400
CellulePFPTBP25
Temps (s)
E (V
/Ag+ /A
g po
ur le
s él
ectro
des)
Pote
ntie
l de
cellu
le (V
)
Figure VI-13 : Cyclage galvanostatique du supercondensateur BP-1, dans une solution de carbonate de propylène 1M
NEt4BF4 ; m+P-4-FPT = 75,5 mg, m-BP25 = 50,4 mg → Rp/n = 1,50 ; Composition de la positive : 65% P-4-FPT, 30% NA, 3% CMC et 2% PTFE ; Composition de la négative : 80% BP25, 15% Graphite SFG44, 2% CMC et 3% PTFE. Collecteur de courant inox AISI 316L ; 4 cm2. i = +/- 20 mA.
En début de cyclage, le polymère possède une capacité de décharge de 30 mAh/g avec
une réversibilité coulombique de 96%. Sa capacité diélectrique est de 243 Farad/g tandis que
celle du BP25 est de 138 Farad/g. La résistance interne du système est de 13,4 Ω.cm2.
Ce système développe une puissance de 6,4 kW/kg pour une énergie maximale de
49 Wh/kg de matières actives. En tenant compte des teneurs des matières actives dans
l'électrode, ainsi que du facteur 0,2, de tels systèmes pourraient développer une puissance
d'environ 1 kW/kg pour une énergie de 7 Wh/kg.
Pour améliorer ces performances de puissance et d'énergie, nous avons tenté de réduire
la teneur en additif conducteur de l'électrode positive, passant de 65% de polymère à 80%,
tout en maintenant entre 1,5 et 1,6 le rapport entre les masses actives positives et négatives.
147
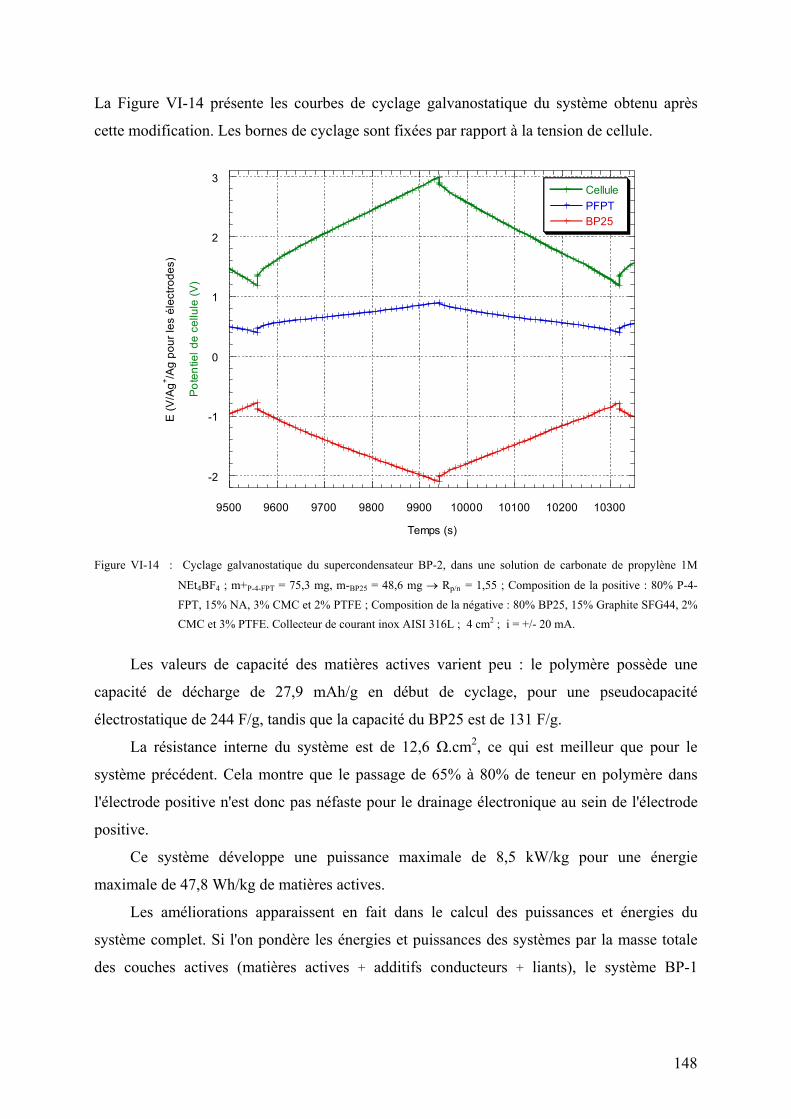
La Figure VI-14 présente les courbes de cyclage galvanostatique du système obtenu après
cette modification. Les bornes de cyclage sont fixées par rapport à la tension de cellule.
-2
-1
0
1
2
3
9500 9600 9700 9800 9900 10000 10100 10200 10300
CellulePFPTBP25
E (V
/Ag+ /A
g po
ur le
s él
ectro
des)
Temps (s)
Pote
ntie
l de
cellu
le (V
)
Figure VI-14 : Cyclage galvanostatique du supercondensateur BP-2, dans une solution de carbonate de propylène 1M
NEt4BF4 ; m+P-4-FPT = 75,3 mg, m-BP25 = 48,6 mg → Rp/n = 1,55 ; Composition de la positive : 80% P-4-
FPT, 15% NA, 3% CMC et 2% PTFE ; Composition de la négative : 80% BP25, 15% Graphite SFG44, 2% CMC et 3% PTFE. Collecteur de courant inox AISI 316L ; 4 cm2 ; i = +/- 20 mA.
Les valeurs de capacité des matières actives varient peu : le polymère possède une
capacité de décharge de 27,9 mAh/g en début de cyclage, pour une pseudocapacité
électrostatique de 244 F/g, tandis que la capacité du BP25 est de 131 F/g.
La résistance interne du système est de 12,6 Ω.cm2, ce qui est meilleur que pour le
système précédent. Cela montre que le passage de 65% à 80% de teneur en polymère dans
l'électrode positive n'est donc pas néfaste pour le drainage électronique au sein de l'électrode
positive.
Ce système développe une puissance maximale de 8,5 kW/kg pour une énergie
maximale de 47,8 Wh/kg de matières actives.
Les améliorations apparaissent en fait dans le calcul des puissances et énergies du
système complet. Si l'on pondère les énergies et puissances des systèmes par la masse totale
des couches actives (matières actives + additifs conducteurs + liants), le système BP-1
148
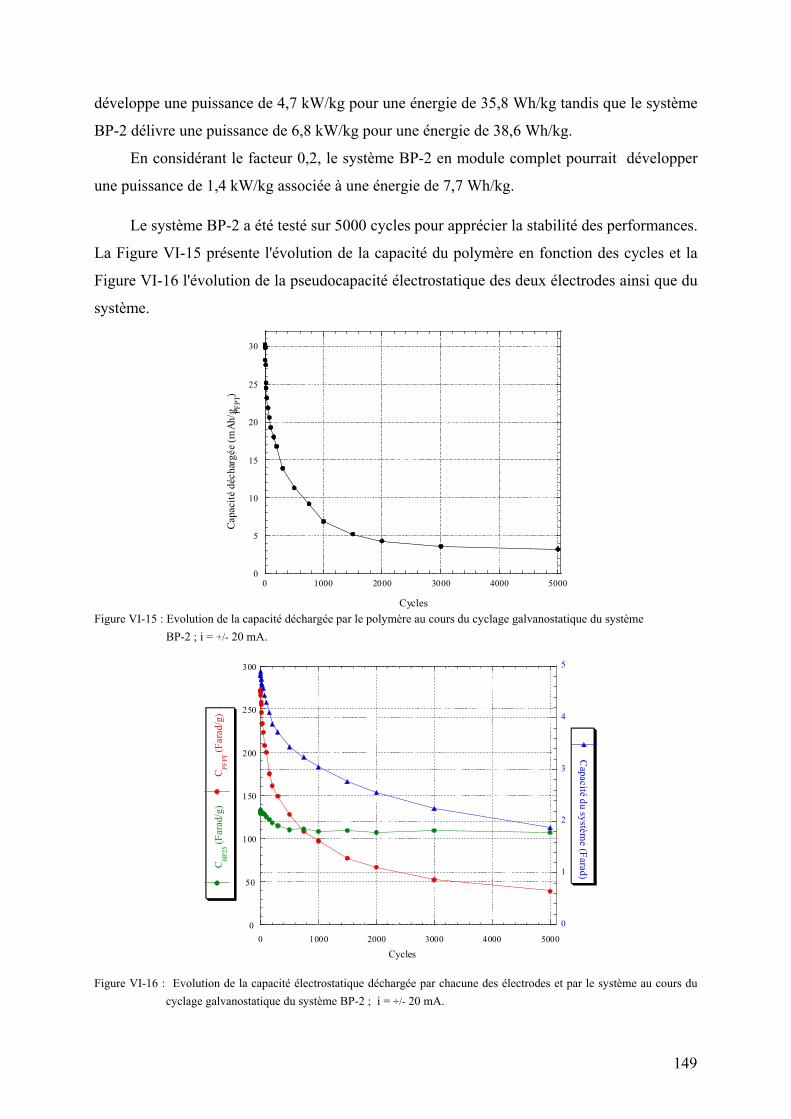
développe une puissance de 4,7 kW/kg pour une énergie de 35,8 Wh/kg tandis que le système
BP-2 délivre une puissance de 6,8 kW/kg pour une énergie de 38,6 Wh/kg.
En considérant le facteur 0,2, le système BP-2 en module complet pourrait développer
une puissance de 1,4 kW/kg associée à une énergie de 7,7 Wh/kg. Le système BP-2 a été testé sur 5000 cycles pour apprécier la stabilité des performances.
La Figure VI-15 présente l'évolution de la capacité du polymère en fonction des cycles et la
Figure VI-16 l'évolution de la pseudocapacité électrostatique des deux électrodes ainsi que du
système.
0
5
10
15
20
25
30
0 1000 2000 3000 4000 5000
Capa
cité
déc
harg
ée (m
Ah/g PF
PT)
Cycles Figure VI-15 : Evolution de la capacité déchargée par le polymère au cours du cyclage galvanostatique du système
BP-2 ; i = +/- 20 mA.
0
50
100
150
200
250
300
0
1
2
3
4
5
0 1000 2000 3000 4000 5000
CPF
PT (F
arad
/g)
CBP
25 (F
arad
/g)
Capacité du systèm
e (Farad)
Cycles
Figure VI-16 : Evolution de la capacité électrostatique déchargée par chacune des électrodes et par le système au cours du cyclage galvanostatique du système BP-2 ; i = +/- 20 mA.
149
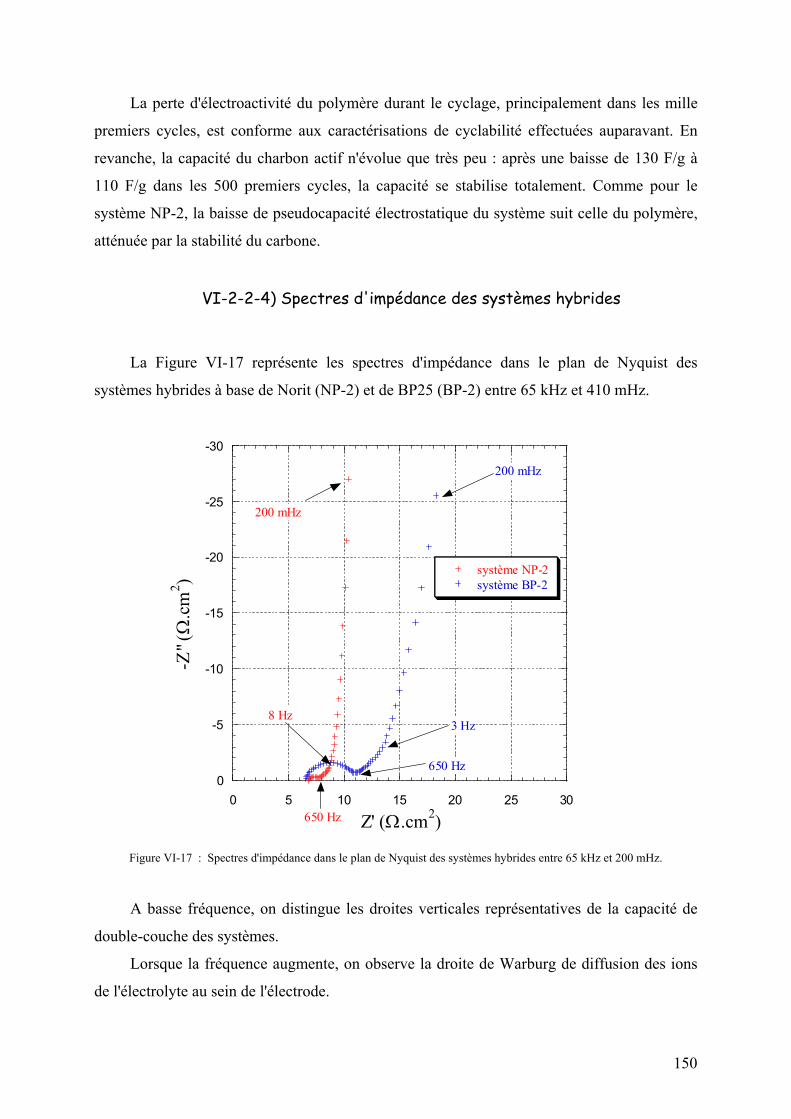
La perte d'électroactivité du polymère durant le cyclage, principalement dans les mille
premiers cycles, est conforme aux caractérisations de cyclabilité effectuées auparavant. En
revanche, la capacité du charbon actif n'évolue que très peu : après une baisse de 130 F/g à
110 F/g dans les 500 premiers cycles, la capacité se stabilise totalement. Comme pour le
système NP-2, la baisse de pseudocapacité électrostatique du système suit celle du polymère,
atténuée par la stabilité du carbone.
VI-2-2-4) Spectres d'impédance des systèmes hybrides
La Figure VI-17 représente les spectres d'impédance dans le plan de Nyquist des
systèmes hybrides à base de Norit (NP-2) et de BP25 (BP-2) entre 65 kHz et 410 mHz.
-30
-25
-20
-15
-10
-5
00 5 10 15 20 25 30
système NP-2système BP-2
-Z" (
Ω.c
m2 )
Z' (Ω.cm2)
8 Hz
200 mHz
200 mHz
3 Hz
650 Hz
650 Hz
Figure VI-17 : Spectres d'impédance dans le plan de Nyquist des systèmes hybrides entre 65 kHz et 200 mHz.
A basse fréquence, on distingue les droites verticales représentatives de la capacité de
double-couche des systèmes.
Lorsque la fréquence augmente, on observe la droite de Warburg de diffusion des ions
de l'électrolyte au sein de l'électrode.
150

A haute fréquence, les deux spectres sont différents. Sur celui du système NP-2, on
observe la boucle de transfert de charge de l'électrode de polymère (cf. VI-2-1-5). En
revanche, sur le spectre du système BP-2, cette boucle est beaucoup plus importante et est
attribuée à l'électrode de charbon actif. Dans les systèmes à double-couche électrochimiques,
ce genre de boucles est généralement attribué à un pseudo-transfert de charge [167].
Bien que les caractéristiques de dopage négatif des PPTF soient médiocres, les
performances d'un supercondensateur de type III P-4-FPT / P-4-FPT ont été évaluées en
cellules de 4 cm2. Les performances initiales sont assez intéressantes malgré la limitation à 7
mAh/g pour la négative. L'énergie maximale atteinte est de 40 Wh/kg pour une puissance de
21 kW/kg de P-4-FPT.
Nous avons ensuite étudié un nouveau concept de supercondensateurs hybrides
polymère/carbone. Les potentiels de cyclage du système sont déterminés par le ratio entre les
masses actives. Sous une tension de 3V, les énergies obtenues sont de 34 Wh/kg de matière
active pour le système NP, et de 48 Wh/kg pour le système BP. Les puissances respectives
atteintes sont de 11 kW/kg et 8,5 kW/kg de matières actives.
Les performances des divers systèmes réalisés suivent la dégradation du polymère au
cours des cycles, atténuée par la stabilité des charbons actifs dans le cas des systèmes
hybrides : pour le supercondensateur BP-2, les performances en capacité atteignent les deux
tiers des performances initiales après 5000 cycles.
151

VI-3) Assemblage de modules industriels
Dans le cadre du projet industriel européen SCOPE (contrat Joule JOE 3-CT-97-0047),
les systèmes hybrides BP25-PFPT ont été étudiés à une échelle semi-industrielle, en
collaboration avec la société CEAC-Exide.
Des modules multi-électrodes ont été assemblés en montage prismatique (cf. Figures
VI-18 et VI-19).
Figure VI-18 : montage prismatique multi-électrodes. Figure VI-19 : Module industriel.
VI-3-1) Fabrication des électrodes
Les principales caractéristiques des systèmes tests de 4 cm2 ont été conservées pour la
réalisation des modules industriels : mise en œuvre des matières actives, collecteurs de
courant en acier inox AISI 316L, séparateurs Gore (téflon, 25 µm d'épaisseur). Les seules
difficultés supplémentaires liées au changement d'échelle sont l'obtention de matières actives
homogènes sur 62 cm2, la mise en œuvre d'électrodes bifaces, ainsi que la connectique et
l'étanchéité du module.
152

D'importantes quantités de matières actives étant utilisées, le protocole de fabrication
des pâtes a été adapté au laboratoire de recherche de CEAC-Exide : le mélange des poudres
est effectué directement dans un malaxeur à 80°C et une quantité minimale d'alcool est
utilisée (150 ml). La CMC dissoute dans de l'eau est ensuite ajoutée, suivie du PTFE. Le
malaxeur est alors maintenu à 80 °C pendant environ 30 minutes. La pâte de carbone sortant
du malaxeur possède déjà de bonnes propriétés mécaniques, tandis que celle du polymère
nécessite un malaxage supplémentaire type "pâte feuilletée".
Le liant mis au point (cf. IV-2) permet de fabriquer des électrodes par co-laminage des
matières actives sur le collecteur de courant. La Figure VI-20 présente la procédure mise au
point pour laminer les pâtes à une épaisseur donnée.
Figure VI-20 : Procédure de laminage des pâtes à une épaisseur donnée.
La pâte humidifiée par de l'éthanol est laminée entre deux plaques de polypropylène de
1 mm d'épaisseur. De fines plaques de 11,5×5,4 cm (62 cm2) sont ensuite découpées.
Pour obtenir des électrodes identiques les unes aux autres ainsi que pour le contrôle du
ratio entre masses actives, il est nécessaire d'établir des courbes d'étalonnage masse/épaisseur.
Les Figures VI-21 et VI-22 représentent les courbes d'étalonnage du P-4-FPT et du BP25.
153

0
5
10
15
20
25
0 100 200 300 400 500 600
Mas
se d
e m
atiè
re ac
tive (
mg/
cm2 )
Epaisseur (µm)
0
5
10
15
20
25
0 100 200 300 400 500 600
Mas
se d
e m
atiè
re a
ctiv
e (m
g/cm
2 )
Epaisseur (µm) Figure VI-21 : droite d'étalonnage du P-4-FPT (80%). Figure VI-22 : droite d'étalonnage du BP25 (80%).
Pour avantager la puissance, les épaisseur d'électrodes ont été choisies les plus fines
possibles, pour pouvoir entrer un maximum d'électrodes dans le boîtier. Les pâtes les plus
fines obtenues avec le P-4-FPT ont une épaisseur de 280 µm, ce qui correspond à une densité
massique surfacique de 12 mg/cm2. En tenant compte du ratio Rp/n de 1,5, il faut donc obtenir
8 mg/cm2 pour les électrodes de BP25, ce qui correspond à une épaisseur de 220 µm.
Les pâtes sont ensuite co-laminées sur le collecteur de courant (déployé d'acier inox
AISI 316L) (cf. Figure VI-23).
Figure VI-23 : Procédure de co-laminage des pâtes à sur le collecteur de courant.
Pour cela, les pâtes de matière actives (encore légèrement humides) sont disposées sur le
collecteur, puis placées entre deux plaques de polypropylène de 5 mm d'épaisseur. Pour éviter
l'adhésion des matières actives sur les plaques, celles-ci sont recouvertes de papier
d'aluminium.
Les électrodes ainsi obtenues sont séchées sous vide actif dans une étuve à 60°C et sous
presse, pendant plusieurs heures.
154

VI-3-2) Assemblage des modules
Les électrodes sont placées dans des "sachets" de séparateur Gore (cf. Figure VI-24). Lorsque celles-ci sont assemblées, il y a donc deux épaisseurs de séparateur entre chaque électrode.
Après assemblage des électrodes, celles-ci sont entourées de quelques épaisseurs supplémentaires de séparateur pour éviter l'éventuel contact d'une électrode avec le boîtier métallique. Le stack est alors placé entre deux fines plaques de téflon ou de polyéthylène et introduites dans le boîtier en métal.
Une société de soudure réalise ensuite la fermeture du boîtier par soudure microplasma.
Le module est rempli d'électrolyte NEt4BF4 1M dans le PC. Pour un meilleur mouillage des électrodes, le boîtier est au préalable mis sous vide et le remplissage s'effectue en boite à gants sous atmosphère d'argon.
Figure VI-24 : électrode placée dans un
sachet de séparateur Gore.
VI-3-3) Performances initiales des modules
Les caractéristiques de trois prototypes semi-industriels réalisés au laboratoire de R&D
de CEAC-Exide sont présentées en Annexe 4. Les performances initiales des prototypes sont
résumées dans le Tableau VI-1.
PERFORMANCES INITIALES
Prototype Module 02 Module 03 Module 04
Energie maximale 3,13 Wh 3,25 Wh 3,25 Wh
Puissance maximale à 3V 90 W 97,8 W 112,5 W
Capacité 2500 F 2600 F 2600 F
Résistance (1000 Hz) 25 mΩ 23 mΩ 20 mΩ
Résistance surfacique 71 Ω.cm2 68 Ω.cm2 56 Ω.cm2
Energie spécifique 6,97 Wh/kg 7,49 Wh/kg 7,55 Wh/kg
Puissance spécifique 200,7 W/kg 225,3 W/kg 261,2 W/kg
Tableau VI-1 : performances des modules industriels
155

Les performances de chaque électrode ne sont pas accessibles étant donné l'impossibilité
de placer une électrode de référence.
Ces modules ont une capacité de l'ordre de 2500 F. L'énergie spécifique obtenue se situe
entre 7 et 7,6 Wh/kg, ce qui confirme bien les prévisions faites pour ce type de systèmes à
partir des résultats des cellules-test de 4 cm2 (cf. VI-2-2-3). Ces performances en énergie
sont bien supérieures à celles des supercondensateurs carbone/carbone actuels (cf. I-2-1).
Cependant, selon les résultats des cellules-test, ces performances se dégradent très rapidement
durant le cyclage.
La puissance spécifique obtenue sur ces modules est de l'ordre de 200-260 W/kg au lieu
des 1,4 kW/kg attendus. Cette différence s'explique par l'augmentation de la résistance interne
qui est 5 à 7 fois supérieure dans les modules par rapport aux cellules-test. Cette élévation de
la résistance est due au manque de contrôle de la compression dans ce type de modules.
Celle-ci garanti en effet le contact entre les matières actives et les collecteurs de courant. Les
performances en énergie montrent que la totalité des matières actives travaille, mais la
compression n'est pas assez importante pour obtenir une puissance importante.
VI-3-4) Cyclabilité des modules
La cyclabilité des modules a été testée par cyclage galvanostatique au service de R&D
de CEAC-Exide et à l'aide de différentes techniques de cyclages à l'ENEA (Italie). Les
performances en capacité diminuent au cours des cycles de la même manière qu'avec les
cellules test de 4 cm2, suivant probablement la dégradation du polymère.
En revanche, un module préparé au service de R&D de CEAC-Exide avec un autre
charbon actif de la société Spectracorp, le YP17 (110 Farad/g) a donné un résultat de
cyclabilité en cyclage galvanostatique entre intéressant comme on peut le voir sur la
Figure VI-25.
156

0
500
1000
1500
2000
0 1000 2000 3000 4000 5000 6000 7000 8000
Cap
acité
(Far
ad)
Cycles Figure VI-25 : Evolution de la capacité d'un module hybride P-4-FPT // NEt4BF4 1M /PC // YP17 au cours des cycles
effectués en cyclage galvanostatique à 5A entre 1,5 et 3V.
La capacité était de 2000 F au début du cyclage, mais a chuté à environ 1100 F vers le
200ème cycle (dégradation normale dans nos conditions). Des changements de régimes de
cyclage ont ensuite été effectués pendant 2000 cycles, alternant des maintiens
potentiostatiques plus ou moins longs et différentes densités de courant de cyclage. Pendant
ces changements de régimes, la capacité a oscillé plus ou moins entre 1000 F et 1200 F. Le
cyclage a ensuite été effectué à 5A (2 mA/cm2) pendant plus de 5000 cycles pratiquement
sans perte de capacité.
Ce résultat, difficilement explicable sans autres expériences, montre que le polymère a
fait preuve d'une remarquable stabilité provoquée par les changements du profil de cyclage.
C'est la première fois à notre connaissance qu'un prototype industriel utilisant des
polymères conducteurs est assemblé et démontre une excellente stabilité sur près de 8000
cycles.
157

Conclusion générale
L'étude de l'application des polymères conducteurs dans des systèmes de stockage
d'énergie connaît un regain d'intérêt depuis quelques années, principalement pour tenter de
mettre au point un supercondensateur fonctionnant sous 3V utilisant des dérivés du
polythiophène comme matières actives. La plupart des différents travaux rencontrés dans la
littérature font état de polymères synthétisés électrochimiquement sous forme de films,
déposés sur divers collecteurs de courant [40-45]. Ces synthèses sont cependant inapplicables
à un niveau de développement industriel. C'est pourquoi l'objectif de ce travail, au sein d'un
projet européen, était d'étudier les caractéristiques électrochimiques de polythiophènes
synthétisés chimiquement sous forme de poudres, et de les mettre en œuvre dans des
électrodes composites pour les utiliser en tant que matières actives dans des
supercondensateurs éventuellement transposables à une production industrielle.
D'autre part, si le dopage positif des polythiophènes possède de bonnes caractéristiques
au niveau de la capacité et de la stabilité, le dopage négatif est beaucoup plus difficile à
réaliser avec de bonnes propriétés. L'obtention d'un dopage négatif stable et capacitif est
pourtant un objectif incontournable pour la mise en œuvre de supercondensateurs de type III.
Le principal axe de recherche de ce travail était donc orienté vers l'électrode négative.
Les travaux de Rudge et al. [1,43,102] ont montré que des polythiophènes
(électrochimiques) substitués par des groupements électroattracteurs possédaient un dopage
négatif plus capacitif et plus stable que le polythiophène lui-même. Suivant ces travaux ainsi
que ceux de José Gonzalez obtenus au Laboratoire d'Electrochimie Industrielle du CNAM,
nous avons mis au point une polymérisation chimique de dérivés fluorophénylés du
polythiophène. Nous avons ensuite étudié la mise en œuvre de ces polymères dans des
électrodes composites. Pour ce faire, nous avons testé et mis au point un nouveau type de liant
à base de CMC et de PTFE, permettant un bon dopage des polythiophènes et une mise en
œuvre d'électrode par laminage des matières actives sur le collecteur de courant, procédé
aisément transposable à l'échelle industrielle.
Les polymères obtenus se présentent sous forme de poudres amorphes hydrophobes et
insolubles, ce qui traduit des masses molaires moyennes importantes. La caractérisation
chimique des polymères a été effectuée par analyse élémentaire et par spectroscopie
infrarouge. La majorité des pics de vibration a été identifiée.
158

La caractérisation électrochimique des processus de dopage des polymères a été
effectuée par voltamétrie cyclique et cyclage galvanostatique. L'effet plus ou moins
électroattracteur des substituants se reflète dans les potentiels de dopage des différents
polymères et a pu être mis en adéquation avec la théorie de Hammett.
Pour ces polymères, le dopage positif est relativement capacitif (40 mAh/g pour le
P-4-FPT) et très réversible. Cependant, la capacité décroît assez rapidement lors du cyclage
(54% de perte en 500 cycles).
Le dopage négatif est moins capacitif que le dopage positif (23,5 mAh/g pour le
P-3,5-DFPT) et nécessite une période d'activation qui varie entre 10 et 20 cycles selon les
polymères. D'autre part, malgré l'effet électroattracteur des substituants, sensé stabiliser la
chaîne polymère durant sa réduction, ce processus de dopage se dégrade très rapidement au
cours du cyclage s'il est effectué en totalité. En revanche, une stabilité plus importante peut
être atteinte en réalisant un dopage partiel du polymère (25% de perte en 500 cycles avec une
capacité initiale de 7 mAh/g).
Une études causes de la dégradation du polymère a été réalisée mais n'a pas donné les
résultats escomptés.
Compte tenu des faibles performances du dopage négatif de ces polymères,
l'assemblage de supercondensateurs de type III paraît impossible. Néanmoins, quelques
systèmes ont été réalisés en cellules de 4 cm2 pour évaluer les performances potentielles de
ces supercondensateurs. Le P-4-FPT a été choisi pour sa stabilité légèrement supérieure, pour
être utilisé comme matière active dans les deux électrodes. Les performances sont assez
intéressantes : l'énergie maximale obtenue est de 40 Wh/kg de P-4-FPT et la puissance
maximale de 21 kW/kg de P-4-FPT, avec une électrode négative limitée à 7 mAh/g.
Afin de tirer parti du dopage positif du P-4-FPT, assez performant, un nouveau concept
de supercondensateurs hybrides a été développé, comprenant un polymère conducteur en tant
qu'électrode positive et un charbon actif en tant qu'électrode négative. Forts de l'expérience du
laboratoire dans le domaine des supercondensateurs Carbone/Carbone [168,169-171], nous
avons choisi d'étudier ce concept avec deux charbons actifs : le Norit SX Ultra de la société
Norit (1100 m2/g - 60 F/g) et le BP25 de la société Spectracorp (2500 m2/g - 130 F/g). Nous
avons mis en évidence la nécessité de déterminer un ratio précis entre les matières actives
pour d'une part adapter strictement le domaine de cyclage de l'électrode positive au domaine
de dopage du P-4-FPT, et pour d'autre part abaisser au maximum le domaine de cyclage de
l'électrode négative afin d'atteindre le potentiel de cellule le plus important possible. Ce
concept fonctionne parfaitement et permet d'atteindre des tensions de plus de 3V. L'énergie
159

maximale du système obtenu avec le Norit est de 34 Wh/kg pour une puissance maximale de
11 kW/kg de matières actives. Avec le BP25, le système délivre une puissance de 8,5 kW/kg
pour une énergie de 48 Wh/kg de matières actives. Cependant, les performances des systèmes
suivent la dégradation du polymère, ce qui limite la cyclabilité à quelques milliers de cycles.
Nous avons décidé de développer les systèmes hybrides à l'échelle industrielle. Une
synthèse chimique du P-4-FPT en grande quantité a donc été mise au point, et n'a nécessité
que de légères adaptations de la synthèse de laboratoire.
Un protocole de fabrication d'électrodes a été mis au point en collaboration avec le
service de R&D de CEAC-Exide. Des modules de 2600 Farads ont été réalisés, avec des
performances initiales intéressantes : ils présentent des énergies spécifiques de l'ordre de
7,5 Wh/kg. Néanmoins, les puissances spécifiques n'atteignent que 250 W/kg, limitées par
une résistance interne importante due au manque de compression des électrodes.
C'est la première fois que des supercondensateurs en polymères conducteurs sont
réalisés à une échelle industrielle.
160

Perspectives
Les perspectives de ce travail suivent plusieurs axes de recherche.
D'un point de vue fondamental, il serait intéressant d'étudier la synthèse chimique de
ces polymères par les méthodes combinées de Mc Cullough et de Yamamoto (cf. I-1-2-1)
pour obtenir des polymères tout à fait réguliers, qui devraient posséder des propriétés de
dopage négatif améliorées par rapport à ceux synthétisés au chlorure ferrique.
L'étude de la désactivation du P-4-FPT au cours des cycles n'a pas permis de révéler les
mécanismes de dégradation, limitée par des techniques expérimentales trop indirectes. Il serait
intéressant de réaliser des expériences in-situ (spectroscopie IR et Raman, mesures de
conductivité) afin de mieux comprendre la désactivation des polymères.
D'un point de vue plus appliqué, le concept de supercondensateur hybride polymère
conducteur / charbon actif présente de réelles améliorations potentielles par rapport aux
performances des systèmes carbone/carbone actuels. Cependant, une étude poussée de
l'adhésion des matières actives sur les collecteurs de courant et de la compression interne
serait nécessaire pour améliorer la puissance des modules prismatiques. Une adaptation à des
modules spiralés est aussi envisageable, cette technologie permettant d'obtenir une
compression beaucoup plus importante.
Ce concept de supercondensateurs hybrides pourrait être appliqué à d'autres matières
actives : certains polymères possèdent d'excellentes stabilités en dopage positif ainsi que des
capacités supérieures au P-4-FPT, comme le poly-3-méthylthiophène (PMeT) ou le poly-3,4-
éthylènedioxythiophène (PEDOT). Des travaux sur ce sujet sont en cours au Laboratoire de
Chimie Physique de Bologne, utilisant des électrodes composites à base de PMeT. D'autre
part, des charbons actifs possédant de meilleures propriétés de charge électrostatique existent
[172].
Les systèmes résultants pourraient facilement dépasser des énergies de 10 Wh/kg tout
en développant des puissances de plus de 2 kW/kg avec une remarquable stabilité en cyclage.
161

ANNEXES
162

Annexe 1 : Protocoles de fabrication de matières actives
Matière active à base de Carboxyméthylcellulose (CMC) :
La CMC est disponible sous forme d'un sel de sodium. Elle se trouve alors sous la forme
d'une poudre fine de haute densité. Elle est soluble dans les solvants aqueux.
Pour la mettre en œuvre comme liant d'électrodes, elle est pesée dans un bécher puis
dissoute dans de l'eau en proportion de 10 mg/ml, sous agitation et en chauffant légèrement
(30-40°C). Les poudres de matières actives et éventuellement d'additif conducteur sont
mélangées ensemble puis mises en suspension dans de l'eau dans des proportions adéquates.
La suspension est alors ajoutée à la solution de CMC et homogénéisée en agitant vivement à
l'aide d'un agitateur magnétique. L'eau est ensuite évaporée en menant la température du
mélange à 110°C, tout en maintenant l'agitation.
Après évaporation de la quasi-totalité de l'eau, on récupère une pâte de matières actives
qu'il est ensuite possible d'empâter sur un collecteur de courant.
Les électrodes ainsi fabriquées sont alors mises à sécher pendant 24 heures sous vide
actif et à une température de 80°C.
La teneur minimale de CMC nécessaire pour que l'électrode possède des propriétés
mécaniques satisfaisantes est de 10% en masse par rapport au poids des matières actives
(poudres + liant).
Matière active à base de fluorure de polyvinylidène (PVdF) :
Le PVdF est un liant polymère bien connu pour la mise en œuvre de cathodes dans les
systèmes lithium. C'est une poudre très fine qui est soluble dans certains solvants organiques
comme la N-méthylpyrrolidinone (NMP).
Sa mise en œuvre est du même type que pour la CMC : il faut le dissoudre dans de la
NMP, puis ajouter les poudres de matières actives, mélanger et évaporer le solvant à 130°C.
La pâte obtenue est liquide mais s'épaissit par une action mécanique (malaxage). Ainsi,
les chaînes de polymère s'entremêlent conférant ainsi de bonnes propriétés mécaniques à la
pâte. Il est ensuite aisé d'empâter celle-ci sur un collecteur de courant.
163

Afin de les sécher, les électrodes sont placées sous vide actif pendant 24 heures à une
température de 80°C.
La teneur en PVdF que nous avons utilisée est de 5% en masse, ce qui est suffisant pour
assurer une bonne tenue mécanique à l'électrode.
Matière active à base de polytetrafluoroéthylène (PTFE) :
Le PTFE est disponible sous forme d'une dispersion en solution aqueuse à 60% en
masse (Zonyl ou PTFE de Dupont de Nemours). Sa mise en œuvre est différente des deux
autres liants.
Les poudres de matières actives sont mélangées ensemble puis mises en suspension dans
de l'éthanol en quantité suffisante pour obtenir en agitant une bonne homogénéisation du
mélange. Ce solvant possède la propriété de faire fibriller le PTFE en déstabilisant les
tensioactifs utilisés pour la dispersion.
Le PTFE est pesé dans un bécher et ajouté ensuite à la suspension de poudres et le
bécher est soigneusement rincé à l'éthanol afin de s'assurer que la totalité du PTFE est bien
ajoutée.
Le mélange est mis sous vive agitation à une température de 90°C. Après évaporation
de l'éthanol, on récupère une pâte solide. Celle-ci doit être malaxée (façon "pâte feuilletée")
pour que le PTFE fibrille de manière encore plus efficace. Des alternances de séchage et
d'humidification à l'éthanol (pendant le malaxage) permettent aux chaînes polymères de
former un filet homogène autour des matières actives.
Il est ensuite possible de laminer la pâte sur le collecteur de courant, ce qui donne aux
électrodes une excellente tenue mécanique et permet de contrôler l'épaisseur des matières
actives.
Les électrodes sont séchées à 80°C sous vide actif pendant 24 heures.
Le PTFE permet d'obtenir de remarquables propriétés mécaniques avec une teneur de
2% en masse.
164

Matière active à base du mélange de liants CMC-PTFE :
Pour réaliser des électrodes en utilisant la CMC et le PTFE, les deux mises en œuvre
ont été combinées : la CMC est dissoute dans de l'eau, en proportion de 10 mg/ml, sous légère
agitation et une température de 30-40°C. Les poudres préalablement mélangées sont mises en
suspension dans de l'éthanol et ajoutées à la solution de CMC. Le PTFE préalablement pesé
est ensuite ajouté au mélange et ce dernier est placé sous vive agitation à 110°C pour évaporer
les solvants.
La pâte obtenue possède les propriétés mécaniques de la pâte de PTFE, et est travaillée
de la même manière, alternativement de séchée et humidifiée pour lui donner de bonnes
propriétés mécaniques. Les électrodes sont ensuite placées sous vide actif à 80°C pendant 24
heures.
Les teneurs que nous avons utilisées pour les liants sont de 3% pour la CMC et de 2% pour le PTFE.
Cette utilisation conjointe de la CMC et du PTFE pour la fabrication d'électrodes de
supercondensateurs à base de charbons actifs ou de polymères conducteurs, a fait l'objet d'une
demande de brevet européen [173].
165

Annexe 2 : Spectres infrarouges des polymères
0
0.5
1
1.5
2
2.5
3
3.5
4
800120016002000240028003200
Abs
orpt
ion
Longueur d'onde (cm-1)400
P-4-FPT
P-3,4-DFPT
P-3,5-DFPT
P-3,4,5-TFPT
166

Annexe 3 : Notations des vibrations du noyau benzène d’après Wilson [160].
167

Annexe 4 :
Caractéristiques des modules industriels
Module 2 :
Composition du module : - 2×24 électrodes de 60 cm2 (surface d'échange: 2820 cm2) - Electrode positive : P-4-FPT 80%, noir d'acétylène 15%, CMC 3%, PTFE 2%. - Electrode négative : BP25 80%, graphite SFG44 15%, CMC 3%, PTFE 2%. - Rapport Rp/n = 1,7 - Séparateur : 2×25 µm PTFE (Gore) - Collecteurs : micro-déployé d'acier inox AISI 316L (Delker 4SS-7-100AN). - Boîtier et connexions : acier inox.
COMPOSITION EN MASSE Masse (g) P-4-FPT 40,1 BP25 23,6 Noir d'acétylène 7,5 Graphite SFG44 4,4 Liant CMC 2,1 Liant PTFE 1,9 → Matières actives 79,6 Collecteurs 71,5 Séparateur PTFE 15,4 → Stack 166,5 Protection PE & PTFE 3,8 Connexions 13 Boîtier 118 → Module sans électrolyte 301,3 Electrolyte 147,2 → Module complet 448,5
168

Module 3 :
Composition du module : - 2×25 électrodes de 60 cm2 (surface d'échange: 2940 cm2) - Electrode positive : P-4-FPT 80%, noir d'acétylène 15%, CMC 3%, PTFE 2%. - Electrode négative : BP25 80%, graphite SFG44 15%, CMC 3%, PTFE 2%. - Rapport Rp/n = 1,58 - Séparateur : 2×25 µm PTFE (Gore) - Collecteurs : micro-déployé d'acier inox AISI 316L (Delker 4SS-7-100AN). - Boîtier et connexions : acier inox.
COMPOSITION EN MASSE Masse (g) P-4-FPT 37,6 BP25 23,8 Noir d'acétylène 7,1 Graphite SFG44 4,5 Liant CMC 2 Liant PTFE 1,8 → Matières actives 76,8 Collecteurs 74,5 Séparateur PTFE 16,1 → Stack 167,4 Protection PE & PTFE 4,3 Connexions 13 Boîtier 118 → Module sans électrolyte 302,7 Electrolyte 131,5 → Module complet 434,2
169
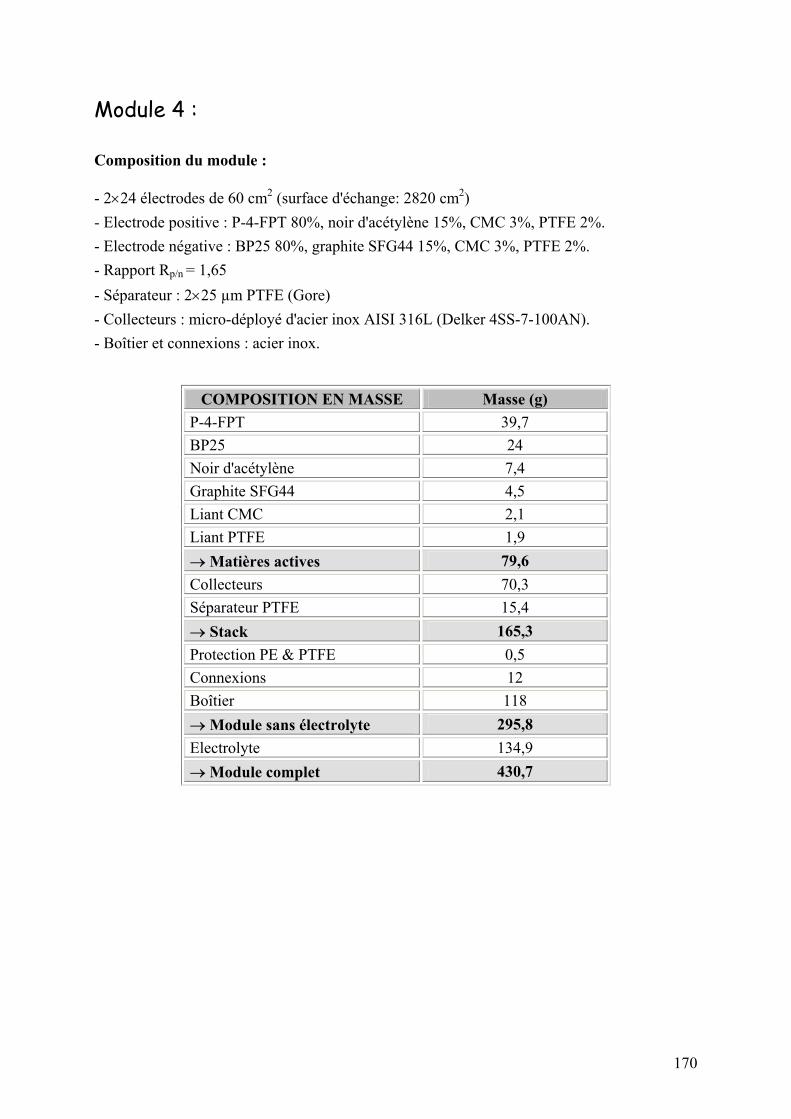
Module 4 :
Composition du module : - 2×24 électrodes de 60 cm2 (surface d'échange: 2820 cm2) - Electrode positive : P-4-FPT 80%, noir d'acétylène 15%, CMC 3%, PTFE 2%. - Electrode négative : BP25 80%, graphite SFG44 15%, CMC 3%, PTFE 2%. - Rapport Rp/n = 1,65 - Séparateur : 2×25 µm PTFE (Gore) - Collecteurs : micro-déployé d'acier inox AISI 316L (Delker 4SS-7-100AN). - Boîtier et connexions : acier inox.
COMPOSITION EN MASSE Masse (g) P-4-FPT 39,7 BP25 24 Noir d'acétylène 7,4 Graphite SFG44 4,5 Liant CMC 2,1 Liant PTFE 1,9 → Matières actives 79,6 Collecteurs 70,3 Séparateur PTFE 15,4 → Stack 165,3 Protection PE & PTFE 0,5 Connexions 12 Boîtier 118 → Module sans électrolyte 295,8 Electrolyte 134,9 → Module complet 430,7
170

Références Bibliographiques
[1] A.Rudge, I.Raistrick, S.Gottesfeld, J.P.Ferraris Electrochim. Acta 39 (1994) 273-287.
[2] C.K.Chiang, Y.W.Park, A.J.Heeger, H.Shirakawa, E.J.Louis, A.G.MacDiarmid, Phys. Rev.
Lett. 39 (1977) 1098.
[3] W.R.Salaneck, R.Lazzaroni, N.Sato, M.P.Keane, N.Correia, S.Lunell, Conjugated Polymeric
Materials : Opportunities in Electronics, Optoelectronics and Molecular Electronics. édité
par J.L.Brédas and R.R.Chance, NATO SERIES 182 (1990) 101-113.
[4] R.D. Mc Cullough, P.C. Ewbank, Handbook of Conducting Polymers 2nd Ed. édité par
T.A.Skotheim, R.L.Elsenbauer and J.R.Reynolds (1998) 225-258.
[5] P.G.Pickup, dans Modern Aspects of Electrochemistry, édité par R.E.White, J.Bockris et
B.E.Conway, Kluwer Academic / Plenum Publishers 33 (1999) 549-597.
[6] G.Inzelt, M.Pineri, J.W.Schultze, M.A. Vorotyntsev, Electrochim. Acta 45 (2000) 2403-2421.
[7] M.Trznadel, O.Chauvet, M.Lapkowski, A.Pron, Synthetic Metals (1999) 358.
[8] E.Lankinen, G.Sundholm, P.Talonen, H.Granö, F.Sundholm, J. Electroanal. Chem 460 (1999)
176-187.
[9] D.Fichou, G.Horowitz, F.Garnier, Synthetic Metals 39 (1990) 125-131.
[10] V.I.Krinichnyi, Synthetic Metals 108 (2000) 173-222.
[11] E.Yurtsever, Synthetic Metals 105 (1999) 179-183.
[12] T.Yamamoto, K.Saneshika, A.Yamamoto, J. Polym. Sci., Letters Ed. 18 (1980) 9-12.
[13] J.W.Lin, L.P.Dudek, J.P Polym. Sci., Polym. Chem. Ed., 18 (1980) 2869-2873.
[14] T.Yamamoto, K.Saneshika, A.Yamamoto, Bull. Chem. Soc. Jpn., 56 (1983) 1497-1502.
[15] R.D.McCullough, M.Jayaraman, J. Chem. Soc., Chem. Commun. (1995) 135-136.
[16] R.D.McCullough, S.P.Williams, S.Tristam-Nagle, M.Jayaraman, P.C.Ewbank, L.Miller,
Synthetic Metals 69 (1995) 279-282.
[17] R.Sugimoto, S.Takeda, H.B.Gu, K.Yoshino, Chemistry Express 1 (1986) 635.
[18] P.Buvat, P.Hourquebie, Synthetic Metals 101 (1999) 17-18.
[19] N.Sato, M.Rikukawa, K.Sanui, N.Ogata, Synthetic Metals 101 (1999) 132-133.
[20] S-H.Jin, H-J.Lee, Y-K.Sun, H-D.Kim, K-N.Koh, Y-S.Gal, D-K. Park, Eur. Polym. Journal
35 (1999) 89-94.
[21] A.Laforgue, P.Simon, C.Sarrazin, J.F.Fauvarque, Journal of Power Sources 80 (1999)
142-148.
[22] L.Kreja, W.Czervinski, J.Kurzawa, M.Kurzawa, Synthetic Metals 72 (1995) 153-158.
171

[23] X.Hu, L.Xu, Polymer 41 (2000) 9147-9154.
[24] F.Andreani, E.Salatelli, M.Lanzi, F.Bertinelli, A.M.Fichera, M.Gazzano, Polymer 41 (2000)
3147-3157.
[25] C.Lee, K.J. Kim, S.B.Rhee, Synthetic Metals 69 (1995) 295-296.
[26] T.Olinga, B.François, Synthetic Metals 69 (1995) 297.
[27] V.M.Niemi, P.Knuuttila, J.E.Österholm, J.Korvola, Polymer 33 (1992) 1559-1562.
[28] J.E.Österholm, J.Laakso, P.Nyholm, H.Isotalo, H.Stubb, O.Inganäs, W.R.Salaneck,
Synthetic Metals 28 (1989) 435-444.
[29] M.Kobashi, H.Takeuchi, Synthetic Metals 101 (1999) 585-586.
[30] (a) C.Sentein, B.Mouanda, A.Rosilio, C.Rosilio, J. Chim. Phys. 92 (1995) 983-986.
(b) J.M.Raimundo, E.Levillain, N.Gallego-Planas, J.Roncali, Electrochem. Commun. 2
(2000) 211-215.
[31] R.M.Souto Maior, K.Hinkelmann, H.Eckert, F.Wudl, Macromolecules 23 (1990) 1268-1279.
[32] J.Roncali, Chemical Reviews 97 (1997) 173-205.
[33] P.C.Stein, C.Botta, A.Bolognesi, M.Catellani, Synthetic Metals 69 (1995) 305-306.
[34] M.R.Andersson, D.Selse, M.Berggren, H.Järvinen, T.Hjertberg, O.Inganäs, O.Wennerström,
J.E.Österholm, Macromol. 27 (1994) 6503-6506.
[35] M.R.Andersson, W.Mammo, T.Olinga, M.Svensson, M.Theander, O.Inganäs, Synthetic
Metals 101 (1999) 11-12.
[36] S.Masubuchi, R.Imai, K.Yamazaki, S.Kazama, J.Takada, T.Matsuyama, Synthetic Metals
101 (1999) 594-595.
[37] S.Kitao, T.Matsuyama, M.Seto, Y.Maeda, S.Masubushi and S.Kazama, Synthetic Metals 69
(1995) 371-372.
[38] M.B.Inoue, E.F.Velasquez, M.Inoue, Synthetic Metals 24 (1988) 223-229.
[39] G.Koßmehl, G.Chatzitheodorou, Makromol. Chem., Rapid Commun. 2 (1981) 551-555.
[40] J.Roncali, Chemical Reviews 92 (1992) 711-781.
[41] K.Gurunathan, A.Vadivel Murugan, R.Marimuthu, U.P.Mulik, D.P. Amalnerkar, Materials
Chemistry and Physics 61 (1999) 173-191.
[42] G.Tourillon, F.Garnier, J. Electroanal. Chem., 161 (1984) 51-58.
[43] D.J.Guerrero, X.Ren, J.P.Ferraris, Chem. Mater. 6 (1994) 1437-1443.
[44] J.P.Ferraris, M.M.Eissa, I.D.Brotherston, D.C.Loveday, Chem. Mater. 10 (1998) 3528-3535.
[45] C.Arbizzani, M.Catellani, M.Mastragostino, M.G.Cerroni, J. Electroanal. Chem. 423 (1997)
23-28.
172

[46] B.Ballarin, R.Seeber, D.Tonelli, D.Andreani, P.Costa Bizzarri, C. Della Casa, E. Salatelli,
Synthetic Metals 88 (1997) 7-13.
[47] S.Aeiyach, A.Kone, M. Dieng, J.J.Aaron, P.C.Lacaze, J. Chem. Soc., Chem. Commun.
(1991) 822-824.
[48] H.Sarker, Y.Gofer, J.G.Killian, T.O.Poehler, P.C.Searson, Synthetic Metals 97 (1998) 1-6.
[49] H.Nguyen Cong, C.Sene, P.Chartier, J. Chim. Phys. 92 (1995) 979-982.
[50] S.Mu, S-M.Park, Synthetic Metals 69 (1995) 311-312.
[51] S.A.Chen, C.C.Tsai, Macromol. 26 (1993) 2234-2239
[52] Y.Gofer, J.G.Killian, H.Sarker, T.O.Poehler, P.C.Searson, J. Electroanal. Chem. 443 (1998)
103-115.
[53] O.A.Semenikhin, L.Jiang, T.Iyoda, K.Hashimoto, A.Fujishima, Synthetic Metals 110 (2000)
195-201.
[54] M.Leclerc, F.M. Diaz, G.Wegner, Makromol. Chem. 190 (1989) 3105-3116.
[55] F.Garnier, G.Tourillon, J.Y.Barraud, H.Dexpert, J. Mat. Science 20 (1985) 2687-2694.
[56] M.Shimomura, M.Kaga, N.Nakayama, S.Miyauchi, Synthetic Metals 69 (1995) 313.
[57] B.Pépin-Donat, B.Sixou, A.de Geyer, A.Viallat, L.Won Fah Hin, J. Chim. Phys. 95 (1998)
1225-1228.
[58] J.Li, K.Aoki, J. Electroanal. Chem. 453 (1998) 107-112.
[59] E.Rebourt, B.Pépin-Donat, E.Dinh, M.Nechtschein, J. Chim. Phys. 92 (1995) 775-778.
[60] B.Pépin-Donat, E.Rebourt, A. Viallat, J. Chim. Phys. 92 (1995) 779-782.
[61] J.Bras, S.Guillerez, B.Pépin-Donat, J. Chim. Phys. 95 (1998) 1161-1164.
[62] O.Inganäs, Indian Journal of Chemistry 33A (1994) 499-505.
[63] S.Garreau, G.Louarn, S.Lefrant, J.P.Buisson, G.Froyer, Synthetic Metals 101 (1999)
312-313.
[64] B.Rasch, W.Viekstich, J. Electroanal. Chem. 370 (1994) 109-117.
[65] J.L.Sauvajol, D.Chenouni, J.P.Lère-Porte, C.Chorro, B.Moukala, J.Petrissans, Synthetic
Metals 38 (1990) 1-12.
[66] M.Lapkowski, A.Pron, Synthetic Metals 110 (2000) 79-83.
[67] J.L.Brédas, Handbook of Conducting Polymers 1st Ed. édité par T.A.Skotheim (1986) 860-
907.
[68] B.Rasch, P.Novak, W.Vielstich, Synthetic Metals 41-43 (1991) 2963-2966.
[69] J.Wang, Electrochimica Acta, 39 (1994) 417-429.
[70] J.Wang, Electrochimica Acta, 42 (1997) 2545-2554.
173

[71] E.W.Tsai, S.Basak, J.P.Ruiz, J.R.Reynolds and K. Rajeshwar, J. Electrochem. Soc. 136-12
(1989).
[72] M.Lemaire, R.Garreau, D.Delabouglise, J.Roncali, H.K.Youssoufi, F.Garnier, New J. Chem.
14 (1990) 359-364.
[73] M.Leclerc, K.Faïd, Handbook of Conducting Polymers 2nd Ed. édité par T.A.Skotheim,
R.L.Elsenbauer and J.R.Reynolds (1998) 225-258.
[74] T.Yohannes, J.C.Carlberg, O.Inganäs, T.Solomon, Synthetic Metals 88 (1997) 15-21.
[75] O.Inganäs, W.R.Salaneck, J.E.Österholm, J.Laakso, Synthetic Metals 22 (1988) 395-401.
[76] M.Aizawa, S.Watanabe, H.Shinohara, H.Shirakawa, J. Chem. Soc., Chem. Commun. (1985)
264-265
[77] P.Barta, F.Cacialli, R.H.Friend, W.R.Salaneck, M.Zagorska, A.Pron, Synthetic Metals 101
(1999) 296-297.
[78] A.Bolognesi, G.Bajo, J.Paloheimo, T.Östergard, H.Stubb, Adv. Mater. 9-2 (1997) 121-123.
[79] J.L.Brédas, K.Cornil, F.Meyers,D.Beljonne, Handbook of Conducting Polymers 2nd Ed. édité
par T.A.Skotheim, R.L.Elsenbauer and J.R.Reynolds (1998) 225-258.
[80] M.Nechtschein, Handbook of Conducting Polymers 2nd Ed. édité par T.A.Skotheim,
R.L.Elsenbauer and J.R.Reynolds (1998) 141-164.
[81] M.Trznadel, O.Chauvet, M.Lapkowski, A.Pron, Synthetic Metals 101 (1999) 358.
[82] J.Fagerström, S.Stafström, Synthetic Metals 85 (1997) 1065-1068.
[83] K.E.Aasmundtveit, E.J.Samuelsen, C.Steinland, C.Meneghini, A.Filipponi, Synthetic Metals
101 (1999) 363-364.
[84] K.E.Aasmundtveit, E.J.Samuelsen, L.A.Pettersson, O.Inganäs, T.Johansson, R.Feidenhans,
Synthetic Metals 101 (1999) 561-564.
[85] K.E.Aasmundtveit, E.J.Samuelsen, O.Inganäs, L.A.A.Pettersson, T.Johansson, S.Ferrer,
Synthetic Metals 113 (2000) 93-97.
[86] K.E.Aasmundtveit, E.J.Samuelsen, K.Hoffmann, E.Bakken, P.H.J.Carlsen, Synthetic Metals
113 (2000) 7-18.
[87] P.Marque, J.Roncali, F.Garnier, J. Electroanal. Chem. 218 (1987) 107-118.
[88] M. Skompska, Electrochimica Acta 45 (2000) 3841-3850.
[89] J.P.Ferraris, M.M.Eissa, I.D.Brotherston, D.C.Loveday, A.A.Moxey, J. Electroanal. Chem.
459 (1998) 57-69.
[90] C.Visy, J.Kankare, J. Electroanal. Chem. 442 (1998) 175-188.
[91] A.R.Hillman, D.C.Loveday, M.J.Swann, S.Bruckenstein, C.P.Wilde, J. Chem. Soc., Faraday
Trans. 87 (1991) 2047-2053.
174

[92] H.Tang, L.Zhu, Y.Harima and K.Yamashita, Synthetic Metals 110 (2000) 105-113.
[93] J.Ding, W.E.Price, S.F.Ralph, G.G.Wallace, Synthetic Metals 110 (2000) 123-132.
[94] M.Costa, F.Chao, E.Museux, C.Tian, J. Chim. Phys. 92 (1995) 939-942.
[95] M.Lögdlund, P.Dannetun, W.R.Salaneck, Handbook of Conducting Polymers 2nd Ed. édité
par T.A.Skotheim, R.L.Elsenbauer and J.R.Reynolds (1998) 667-694.
[96] K.Doblhofer, K.Rajeshwar, Handbook of Conducting Polymers 2nd Ed., édité par
T.A.Skotheim, R.L.Elsenbauer and J.R.Reynolds (1998) 531-588.
[97] F.Mohammad, Synthetic Metals 99 (1999) 149-154.
[98] P.Novák, K.Müller, K.S.V.Santhanam, O.Haas, Chemical Reviews 97 (1997) 207-281.
[99] J.A.Reedijk, H.C.F.Martens, S.M.C.Van Bohemen, O.Hilt, H.B.Brom, M.A.J.Michels,
Synthetic Metals 101 (1999) 475-476.
[100] R.H.J.Schmitz, A.S.Hudson, K.Jüttner, Synthetic Metals 101 (1999) 102-103.
[101] L.M.Abrantes, J.P.Correia, J.Gonzalez, J. Chim. Phys. 95 (1998) 1172-1175.
[102] A.Rudge, J.Davey, I.Raistrick, S.Gottesfeld, J.P.Ferraris, J. Power Sources 47 (1994) 89.
[103] C.Arbizzani, M.Catellani, M.Mastragostino, C.Mingazzini, Electrochimica Acta 40 (1995)
1871-1876.
[104] C.Arbizzani, M.Mastragostino, F.Soavi, Electrochimica Acta 45 (2000) 2273-2278.
[105] H.Sarker, Y.Gofer, J.G.Killian, T.O.Poehler, P.C.Searson, Synthetic Metals 88 (1997) 179-
185.
[106] M.Sato, S.Tanaka, K.Kaeriyama, J.Chem. Soc., Chem. Commun. (1987) 1725-1726.
[107] K.M.Mangold, K.Jüttner, Synthetic Metals 101 (1999) 71-72.
[108] V.D.Pokhodenko, V.A.Krylov, N.V.Konoshchuk, Synthetic Metals 99 (1999) 91-95.
[109] G.Zotti, G.Schiavon, S.Zecchin, Synthetic Metals 72 (1995) 275-281.
[110] M.Mastragostino, R.Paraventi, A.Zanelli, J. Electrochem. Soc. 147 (2000) 3167-3170.
[111] C.Arbizzani, M.Mastragostino, L.Meneghello, Electrochimica Acta 41 (1996) 21-26.
[112] K.Jüttner, R.H.Schmitz, A.Hudson, Electrochimica Acta 44 (1999) 4177-4187.
[113] I.Betova, M.Bojinov, E.Lankinen, G. Sundholm, J. Electroanal. Chem. 472 (1999) 20-32.
[114] B.Ferloni, M.Mastragostino, L.Meneghello, Electrochimica Acta 41 (1996) 27-33.
[115] O.A.Semenikhin, E.V.Ovsyannikova, N.M.Alpatova, Z.A.Rotenberg, J. Electroanal. Chem.
463 (1999) 190-199.
[116] M.A.Vorotyntsev, J.P.Badiali, G. Inzelt, J. Electroanal. Chem. 472 (1999) 7-19.
[117] S.A.M.Refaey, G.Schwitzgebel, O.Schneider, Synthetic Metals 98 (1999) 183-192.
[118] G.Leising, S.Tasch, W.Graupner, Handbook of Conducting Polymers 2nd Ed. édité par
T.A.Skotheim, R.L.Elsenbauer and J.R.Reynolds (1998) 847-880.
175

[119] R.H.Friend, N.C.Greenham, Handbook of Conducting Polymers 2nd Ed. édité par
T.A.Skotheim, R.L.Elsenbauer and J.R.Reynolds (1998) 823-846.
[120] T.M.Brown, J.S.Kim, R.H.Friend, F.Cacialli, R.Daik, W.J.Feast, Synthetic Metals 111-112
(2000) 285-287.
[121] D.Kumar, R.C.Sharma, Eur. Polym. Journal 34 (1998) 1053-1060.
[122] S.Panero, E.Spila, B.Scrosati, J. Electroanal. Chem. 396 (1995) 385-389.
[123] S.Panero, P.Prosperi, B.Scrosati, Electrochimica Acta 31 (1986) 1597-1600.
[124] J.C.Carlberg, O.Inganäs, J. Electrochem. Soc. 144 (1997) 61-64.
[125] R.Kötz, M.Carlen, Electrochimica Acta 45 (2000) 2483-2498.
[126] A.Burke, J. Power Sources 91 (2000) 37-50.
[127] M.Mastragostino, C.Arbizzani, R.Paraventi and A. Zanelli, J. Electrochem. Soc. 147 (2000)
407-412.
[128] B.E.Conway, Electrochemical Supercapacitors, Kluwer Academic / Plenum Publishers
(1999).
[129] A.J.Bard, L.R.Faulkner, Electrochimie, principes, méthodes et applications, Ed. Masson,
1983.
[130] T.Morimoto, K.Hiratsuka, Y.Sanada, K.Kurihara, J. Power Sources 60 (1996) 239-247.
[131] J.P.Zheng, T.R.Jow, J. Electrochem. Soc. 144-7 (1997) 2417-2420.
[132] M.Ue, M.Takeda, M.Takehara, S.Mori, J. Electrochem. Soc. 144-8 (1997) 2684-2688.
[133] Makoto Ue, Proceedings of the 8th Int. Seminar on EDLCs and Similar Energy Storage
Devices, 7-9 December 1998, Deerfield Beach, Florida.
[134] (a) M.Chesneau, dans Le carbone dans tous ses états, Gordon Ed., Breach Science
Publishers (1997) 535.
(b) Site internet de la société Pica : www.pica.fr
[135] A.Nishino, A.Yoshida, I.Tanahashi (MATSUSHITA), US Patent 4,562,511 (1985).
[136] A.Yoshida, K.Imoto (MATSUSHITA), US Patent 5,150,283 (1992).
[137] K.Nakao, K.Shimizu, T.Yamaguchi (MATSUSHITA), European Patent WO 98/58397
(1998).
[138] T.Saito, J.Tabuchi, Y.Kibi, A.Oshi, (NEC Corp.), US Patent 5,303,118 (1994).
[139] C.J.Farahmandi, J.Dispennette, E.Blank, The Electrochemical Society Proceedings, 95-29
(1995) 187-197.
[140] C.J.Farahmandi, J.M.Dispennette (MAXWELL), US Patent 5,621,607 (1997).
[141] X.Andrieu, L.Josset, The Electrochemical Society Proceedings, 95-29 (1995) 181-186
176

[142] L.Moreau, D.Cesbron,A.Chaillet, C.Jehoulet et al., Proceedings of the 10th Int. Seminar on
EDLCs and Similar Energy Storage Devices, 4-6 décembre 2000, Deerfield Beach, Florida.
[143] J.P.Zengh, P.J.Cygan, T.R.Jow, J. Electrochem. Soc. 142 (1995) 2699.
[144] X.Andrieu, L.Josset, J.F.Fauvarque, J. Chim. Phys. 92 (1995) 879-882.
[145] T.F.Otero, I.Cantero, H.Grande, Electrochimica Acta 44 (1999) 2053-2059.
[146] D.Bélanger, X.Ren, J.Davey, F.Uribe, S.Gottesfeld, J. Electrochem. Soc. 147 (2000) 2923-
2929.
[147] J.P.Ferraris, S.Gottesfeld, A.J.Rudge, US Patent 5,527,640 (1996).
[148] J.Giaccai, Y.Gofer, J.G.Killian, T.O.Poehler, H.Sarker, P.Searson, US Patent 5,733,683
(1998).
[149] K.Tamao, S.Kodama, I.Nakajima, M.Kumada, A.Minato, K.Suzuki, Tetrahedron 38 (1982)
3347-3354.
[150] T.Sone, M.Inoue, K.Sato, Bull. Chem. Soc. Jpn. 61 (1988) 3779-3781.
[151] J.P. Monthéard, J.F.Delzant, M.Gazard, Synth. Commun. 14 (1984) 289-292.
[152] J.Gonzalez, Mémoire d'Ingénieur CNAM, soutenu le 22 juillet 1997, Paris.
[153] S.Michida, S.Miyita, A.Techagumpuch, Synthetic Metals 31 (1989) 311-318.
[154] F.Beck, U.Barsch, R.Michaelis, J. Electroanal. Chem. 351 (1993) 169-184.
[155] Pretsch, Clerc, Seibl and Simon, Tables of spectra data for structure determination of
organic coupounds Springer-Verlag 2nd Ed. (1989).
[156] (a) G.Socrates, Infrared Characteristic Group Frequencies 2nd Ed. Wiley publish. (1994).
(b) Se Jonny, Biophenil and Bismuth Intern. J. , Marcabral. Commun. (2000) se7-se7-7.
[157] X.Wang, G.Shi, Y.Liang, Electrochem. Commun. 1 (1999) 536-539.
[158] K. Meerholtz, J.Heinze, Electrochimica Acta 41 (1996) 1839-1854.
[159] A.Laforgue, P.Simon, J.F.Fauvarque, Proceedings of the 198th Meeting of the
Electrochemical Society, 22-27 Octobre 2000, Phoenix , Arizona.
[160] E.B.Wilson, J. Phys. Rev. 45 (1934) 706.
[161] U.Barsch, F.Beck, Electrochimica Acta 41 (1996) 1761-1771.
[162] L.P.Hammett, J. Am. Chem. Soc. 59 (1937) 96.
[163] M.J.S. Dewar, dans The electronic theory of organic chemistry, Oxford University Press
(1949).
[164] T.Lowry, K.S. Richarson, dans Mecanism and theory in organic chemistry, Harper & Row
(1976).
[165] P.Garcia, J.M.Pernaut, P.Hapiot, V.Wintgens, P.Valat, F.Garnier and D.Delabouglise, J.
Phys. Chem. 97 (1993) 513-516.
177

[166] E.Kinbara, Y.Kunugi, Y.Harima, K.Yamashita, Synthetic Metals 114 (2000) 295-303.
[167] X.Andrieu, G.Crépy, L.Josset, S.Y.Andersen, M.Hardgrave, V.Danel, Proceedings of the
3th Int. Seminar on EDLCs and Similar Energy Storage Devices, 6-8 December 1993
Deerfield Beach, Florida.
[168] L.Bonnefoi, thèse de l'Université Pierre et Marie Curie (Paris VI), soutenue le 14 décembre
1999, Paris.
[169] I.Bispo-Fonseca, thèse de l'Université Pierre et Marie Curie (Paris VI), soutenue le 15 mai
1998, Paris.
[170] J.Aggar, Mémoire d'ingénieur CNAM, 1996, Paris.
[171] C.Hiron, Mémoire d’ingénieur CNAM,1994, Paris.
[172] J.Gamby, P.L.Taberna, P.Simon, prochainement publié dans J. Electrochem. Soc.
[173] L.Bonnefoi, A.Laforgue, P.Simon, C.Sarrazin, J.F.Fauvarque (LEI CNAM), J.F.Sarrau,
P.Lailler (CEAC-Exide), Demande de Brevet Européen (1999).
178

Liste des figures
I) Etude bibliographique
Figure I-1 : Représentation des principaux polymères conducteurs électroniques possédant un
système π conjugué…………………………………………………………………………… 3
Figure I-2 : Niveaux d'énergie des orbitales π dans un polymère conducteur……………….. 4
Figure I-3 : Schéma de bande des matériaux isolants, semi-conducteurs et conducteurs…… 4
Figure I-4 : Représentation des différents porteurs de charge dans les polythiophènes…….. 5
Figure I-5 : Schéma de bandes du dopage progressif d'un polymère conducteur……………. 6
Figure I-6 : Représentation des couplages possibles durant la polymérisation de
polythiophènes substitués…………………………………………………………………….. 7
Figure I-7 : Méthode Mc Cullough pour la synthèse d'un organomagnésien sélectif
permettant l'obtention d'un polymère régulier………………………………………………. .. 8
Figure I-8 : représentation schématique d'une cellule électrochimique pour la synthèse de
polythiophènes………………………………………………………………………………. 10
Figure I-9 : Mécanisme de polymérisation E (CE)n ……………………………………….. 11
Figure I-10 : Phénomène de thermochromisme des poly(3-alkoxy-4-méthylthiophènes) ….14
Figure I-11 : Absorption UV-Vis du poly(3-octylthiophène) à différents potentiels………. 15
Figure I-12 : Absorption UV-Vis-NIR du PEDOT à différents potentiels ………………… 16
Figure I-13 : Voltamogramme représentant les dopages négatif et positif d'un
polythiophène……………………………………………………………………………….. 19
Figure I-14 : Diagramme de Ragone des différents systèmes électrochimiques de stockage
d'énergie ……………………………………………………………………………………. 22
Figure I-15 : Principe de fonctionnement d'un supercondensateur à base de charbons
actifs………………………………………………………………………………………… 23
Figure I-16 : Double couche électrochimique selon Gouy-Chapman-Stern ………………. 24
Figure I-17 : Voltamétrie cyclique du dopage positif d'une électrode de PANI et domaine de
cyclage de chaque électrode dans un montage de supercondensateur PANI / PANI de
type I………………………………………………………………………………………… 26
Figure I-18 : Voltamogrammes du dopage positif de la PANI et du PPy et domaine de
cyclage de chaque électrode dans un montage de supercondensateur PANI / PPy de
type II……………………………………………………………………………………….. 27
179

Figure I-19 : Voltamétrie cyclique des dopages positif et négatif d'une électrode de P-4-FPT
et domaine de cyclage de chaque électrode dans un montage de supercondensateur
P-4-FPT / P-4-FPT de type III………………………………………………………………. 28
II) Partie expérimentale
Figure II-1 : Cellule électrochimique de type I, utilisée pour les mesures classiques de
voltampérométrie cyclique…………………………………………………………………. .32
Figure II-2 : Cellule électrochimique de type II, avec les électrodes maintenues sous
compression…………………………………………………………………………………. 33
Figure II-3 : Courbe de dopage/dédopage positif et négatif d'un polythiophène, obtenue par
voltampérométrie cyclique………………………………………………………………….. 34
Figure II-4 : Courbe de dopage/dédopage positif d'un polythiophène, obtenue par cyclage
galvanostatique………………………………………………………………………………. 36
Figure II-5 : Diagramme d'impédance d'un supercondensateur polymère conducteur et son
circuit équivalent…………………………………………………………………………….. 40
III) Synthèses
Figure III-1 : Voltamogrammes des P-4-FPT synthétisés à différentes températures.…….. 47
Figure III-2 : Rendement massique de synthèse en fonction du rapport entre les
réactifs……………………………………………………………………………………….. 48
Figure III-3 : Voltamogrammes de P-4-FPTs synthétisés avec différents rapports de réactifs.
……………………………………………………………………………………………… 49
Figure III-4 : Voltamétrie cyclique de P-4-FPTs synthétisés avec différentes concentrations
de monomère. ……………………………………………………………………………….. 50
Figure III-5 : Voltamétrie cyclique de P-4-FPT synthétisé avec différents temps de
polymérisation. ……………………………………………………………………………… 52
IV) Mise en œuvre des polymères
Figure IV-1 : Capacités partielles dédopées par le P-4-FPT après un dopage négatif effectué
en voltamétrie cyclique entre -1.4 et -2V/Ag+/Ag dans une solution d'acétonitrile
1M NEt4CF3SO3 …………………………………….……………………………………. . 56
180

Figure IV-2 : Voltamogrammes d'une électrode de P-4-FPT lié avec différents liants, dans
une solution d'acétonitrile 1M NEt4CF3SO3 ……………………………………………….. 59
Figure IV-3 : Voltamogrammes du dopage négatif du P-4-FPT fixé sur déployé d'aluminium
avec différents liants ……………………………………………………………………… .. 60
Figure IV-4 : Voltamogrammes du dopage négatif du P-4-FPT avec différentes compositions
d'électrode, dans une solution d'acétonitrile 1M NEt4CF3SO3 ………………………………61
Figure IV-5 : Capacités et réversibilités coulombiques associées à la Figure IV-4. ……..…61
Figure IV-6 : Voltamogrammes du dopage positif du P-4-FPT avec différentes compositions
d'électrode, dans une solution d'acétonitrile 1M NEt4CF3SO3 ………………………………63
Figure IV-7 : Capacités et réversibilités coulombiques associées à la Figure IV-6. ………..63
Figure IV-8 : Capacités pondérées par la masse totale des matières actives………………...64
V) Caractérisation des polymères
Figure V-1 : conductivité à 25°C de l'électrolyte NET4CF3SO3 dans l'acétonitrile pour
différentes concentrations en sel……………………………………………………………...67
Figure V-2 (a) : Voltamogramme d'une électrode de platine dans l'électrolyte NET4CF3SO3
1M dans CH3CN……………………………………………………………………………...68
Figure V-2 (b) : 100 cycles de voltamétrie cyclique d'une électrode d'inox AISI 316L dans
l'électrolyte NET4CF3SO3 1M dans l'acétonitrile……………………………………………. 69
Figure V-3 : Spectre infrarouge du PTh entre 400 et 3200 cm-1………………………..….. 70
Figure V-4 : Voltamogramme représentant le dopage négatif du PTh dans une solution
d'acétonitrile 1M NEt4CF3SO3 ………………………………………………………… ….. 72
Figure V-5 : Voltamogramme représentant le dopage positif du PTh dans une solution
d'acétonitrile 1M NEt4CF3SO3 …………………………………………………………...….72
Figure V-6 : Voltamogramme représentant le dopage négatif du PTh avec différents
potentiels de fin de dopage, dans une solution d'acétonitrile 1M NEt4CF3SO3 ……………..73
Figure V-7 : Voltamogrammes du dopage positif d'une électrode de PTh avec différents
anions dopants, dans une solution d'acétonitrile 1M en sel de tétraéthylammonium
correspondant ……………………………………………………………………………..…74
Figure V-8 : Evolution des 100 premiers cycles de voltamétrie cyclique d'une électrode de
PTh dans une solution d'acétonitrile 1M NEt4CF3SO3 ……………………………………...76
Figure V-9 : Evolution des cycles 100 à 500 d'une électrode de PTh dans une solution
d'acétonitrile 1M NEt4CF3SO3 ………………………………………………………….….. 76
181

Figure V-10 : Evolutions des quantités d'électricité dopées et dédopées ainsi que de la
réversibilité coulombique au cours des cycles représentés sur les Figures V-8 et V-9…….. 77
Figure V-11 : Spectre infrarouge du P-4-FPT entre 400 et 3200 cm-1……………………... 79
Figure V-12 : Spectre infrarouge du P-4-FPT et du 4-FPT entre 650 et 900 cm-1………… 80
Figure V-13 : Voltamogramme représentant les huit premiers cycles d'une électrode de
P-4-FPT dans une solution d'acétonitrile 1M NEt4CF3SO3 ……………………………....… 82
Figure V-14 : Evolutions des quantités d'électricité dopées et dédopées et de la réversibilité
coulombique du dopage au cours des cycles représentés sur la Figure V-13……………… 82
Figure V-15 : Voltamogramme représentant le dopage négatif d'une électrode de P-4-FPT
dans une solution d'acétonitrile 1M NEt4CF3SO3 ………………………………….……… 84
Figure V-16 : Voltamogrammes d'une électrode de P-4-FPT pour différentes limites de
potentiel de dopage dans une solution d'acétonitrile 1M NEt4CF3SO3 ……………………. 85
Figure V-17 : Evolutions des quantités d'électricité dopées et dédopées, ainsi que de la
réversibilité coulombique du dopage des voltamogrammes représentés sur
la Figure V-16……………………………………………………………………………….. 85
Figure V-18 : Cycle galvanostatique représentant le dopage négatif du P-4-FPT dans une
solution d'acétonitrile 1M NEt4CF3SO3 ……………………………………………………. 87
Figure V-19 : Capacité de dédopage négatif du P-4-FPT en fonction du potentiel pour
différentes densités de courant, dans une solution d'acétonitrile 1M NEt4CF3SO3 ……… … 88
Figure V-20 : Variation des capacités et de la réversibilité coulombique en fonction de la
densité de courant de cyclage……………………………………………………………….. 88
Figure V-21 : Variation de la pseudocapacité en fonction de la densité de courant
de cyclage. ………………………………………………………………………………….. 88
Figure V-22 : Voltamogramme représentant le dopage positif du P-4-FPT dans une solution
d'acétonitrile 1M NEt4CF3SO3 ………………………………………………………………90
Figure V-23 : Voltamogramme d'une électrode de P-4-FPT avec différentes bornes
supérieures de dopage, dans une solution d'acétonitrile 1M NEt4CF3SO3 ………………..… 91
Figure V-24 : Quantités d'électricité et réversibilité coulombique mises en jeu dans les
voltamogrammes de la Figure V-23. ………………………………………………………. 92
Figure V-25 : Cyclage galvanostatique du dopage positif du P-4-FPT dans une solution
d'acétonitrile 1M NEt4CF3SO3 ……………………………………………………………… 93
Figure V-26 : Capacités de dédopage positif du P-4-FPT en fonction du potentiel pour
différentes densités de courant, dans une solution d'acétonitrile 1M NEt4CF3SO3 ………… 94
182

Figure V-27 : Variation des capacités et de la réversibilité en fonction de la densité de
courant……………………………………………………………………………………….. 94
Figure V-28 : Variation de la pseudocapacité en fonction de la densité de courant
de cyclage……………………………………………………………………………………. 94
Figure V-29 : Evolution des 20 premiers cycles de voltamétrie cyclique d'une électrode de P-
4-FPT dans une solution d'acétonitrile 1M NEt4CF3SO3 …………………………………… 96
Figure V-30 : Variations des quantités d'électricité et de la réversibilité coulombique au long
des cycles représentés sur la Figure V-29…………………………………………………… 97
Figure V-31 : Evolution des 100 premiers cycles de voltamétrie cyclique d'une électrode de
P-4-FPT dans une solution d'acétonitrile 1M NEt4CF3SO3 ………………………………… 98
Figure V-32 : Evolution des cycles 100 à 500 d'une électrode de P-4-FPT dans une solution
d'acétonitrile 1M NEt4CF3SO3 …………………………………………………………….... 99
Figure V-33 : Evolution des quantités d'électricité et de la réversibilité des cycles effectués
sur les Figures V-31 et V-32………………………………………………………………… 99
Figure V-34 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une
électrode de P-4-FPT avec différentes densités de courant, dans une solution d'acétonitrile 1M
NEt4CF3SO3 ……………………………………………………………………………….. 101
Figure V-35 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une
électrode de P-4-FPT avec différentes densités de courant, dans une solution d'acétonitrile 1M
NEt4CF3SO3 ………………………………………………………………………………...102
Figure V-36 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une
électrode de P-4-FPT avec différentes limites de dopage en potentiel, dans une solution
d'acétonitrile 1M NEt4CF3SO3 …………………………………………………………….. 103
Figure V-37 : Evolution des 100 premiers cycles de voltamétrie cyclique d'une électrode de
P-4-FPT dans une solution d'acétonitrile 1M NEt4CF3SO3 ……………………………….. 104
Figure V-38 : Evolution des cycles 100 à 500 cycles d'une électrode de P-4-FPT dans une
solution d'acétonitrile 1M NEt4CF3SO3 ………………………………………………….. 105
Figure V-39 : Evolution des quantités d'électricité et de la réversibilité coulombique en
fonction des cycles effectués sur les Figures V-37 et V-38……………………………….. 105
Figure V-40 : Evolution des 1000 premiers cycles d'une électrode de P-4-FPT dans une
solution d'acétonitrile 1M NEt4CF3SO3 ……………………………………………………. 106
Figure V-41 : Evolution des cycles 1000 à 10000 d'une électrode de P-4-FPT dans une
solution d'acétonitrile 1M NEt4CF3SO3 …………………………………………………… 107
183

Figure V-42 : Evolution des quantités d'électricité et de la réversibilité coulombique au cours
du cyclage représenté sur les Figures V-40 et V-41………………………………………. 108
Figure V-43 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une
électrode de P-4-FPT avec différentes densités de courant, dans une solution d'acétonitrile 1M
NEt4CF3SO3 ……………………………………………………………………………….. 109
Figure V-44 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une
électrode de P-4-FPT avec différents sels 1M dans l'acétonitrile …………………………. 110
Figure V-45 : Evolution des capacités déchargées pendant le cyclage galvanostatique d'une
électrode de P-4-FPT dans différents solvants, avec le sel NEt4BF4 1M ………………….. 111
Figure V-46 : Evolution des capacités dédopées pendant le cyclage galvanostatique d'une
électrode de P-4-FPT pour différentes compositions d'électrode ………………………….. 112
Figure V-47 : spectre UV-Vis de l'électrolyte au cours du cyclage……………………….. 114
Figure V-48 : Schéma d'une électrode de poudre de polymère……………………………. 115
Figure V-49 : spectres infrarouge du P-4-FPT avant et après cyclage positif ou négatif, dans
la région 600-2000 cm-1……………………………………………………………………. 117
Figure V-50 : Formules chimiques des différents PPTF………………………………….. 118
Figure V-51 : Voltamogrammes représentant le dopage positif des PPTF dans une solution
d'acétonitrile 1M NEt4CF3SO3 …………………………………………………………….. 121
Figure V-52 : Pics de dopage et de dédopage positif en fonction de la constante de
substitution de Hammett……………………………………………………………………. 123
Figure V-53 : Voltamogrammes représentant le dopage négatif des PPTF dans une solution
d'acétonitrile 1M NEt4CF3SO3 …………………………………………………………….. 125
Figure V-54 : Pics de dopage et de dédopage négatif en fonction de la constante de
substitution de Hammett……………………………………………………………………. 126
Figure V-55 : Evolution du taux de dopage positif des PPTF au cours des cycles effectués en
voltamétrie cyclique à 20 mV/s. …………………………………………………………… 128
Figure V-56 : Evolution du taux de dopage positif des PPTF au cours des cycles effectués en
cyclage galvanostatique à +/- 1mA/cm2…………………………………………………….. 128
Figure V-57 : Evolution du taux de dopage négatif complet des PPTF au cours des cycles
effectués en voltamétrie cyclique à 20 mV/s. …………………………………………..…. 129
Figure V-58 : Evolution du taux de dopage négatif partiel des PPTF au cours des cycles
effectués en voltamétrie cyclique à 20 mV/s………………………………………………. 130
Figure V-59 : Evolution du taux de dopage négatif partiel des PPTF au cours des cycles
effectués en cyclage galvanostatique à +/- 1mA/cm2. ……………………………………... 131
184

VI) Application aux supercondensateurs
Figure VI-1 : Voltamogrammes de différents déployés d'inox dans une solution de carbonate
de propylène 1M NEt4BF4…………………………………………………………………. 134
Figure VI-2 : principe de fonctionnement d'un supercondensateur polymère/polymère….. 136
Figure VI-3 : Cyclage galvanostatique théorique d'un système dont les deux électrodes ne
présentent pas la même réversibilité……………………………………………………….. 137
Figure VI-4 : Cyclage galvanostatique du supercondensateur P-4-FPT/P-4-FPT, dans une
solution d'acétonitrile 1M NEt4CF3SO3 ………………………………………………..….. 138
Figure VI-5 : Evolution de la capacité déchargée par les deux électrodes de P-4-FPT au cours
d'un cyclage galvanostatique effectué à +/- 1 mA/cm2 sur le système P-4-FPT/P-4-FPT…. 140
Figure VI-6 : Evolution de la pseudocapacité déchargée par les deux électrodes de P-4-FPT
ainsi que par le système au cours d'un cyclage galvanostatique effectué à +/- 1 mA/cm2 sur le
système P-4-FPT/P-4-FPT…………………………………………………………………. 140
Figure VI-7 : Spectre d'impédance dans le plan de Nyquist du système P-4-FPT / P-4-FPT
entre 65 kHz et 200 mHz…………………………………………………………………… 141
Figure VI-8 : principe de fonctionnement d'un supercondensateur hybride
carbone/polymère………………………………………………………………………….. 142
Figure VI-9 : Cyclage galvanostatique du supercondensateur NP-1, dans une solution de
carbonate de propylène 1M NEt4BF4 ……………………………………………………… 143
Figure VI-10 : Cyclage galvanostatique du supercondensateur NP-2, dans une solution de
carbonate de propylène 1M NEt4BF4 ……………………………………………………… 144
Figure VI-11 : Evolution de la capacité déchargée par le P-4-FPT lors du cyclage
galvanostatique du supercondensateur NP-2 ……………….…………………………… .. 145
Figure VI-12 : Evolution des capacités électrostatiques déchargées par chacune des
électrodes ainsi que par le système lors du cyclage galvanostatique du supercondensateur
NP-2 ……………………………………………………………………………………….. 146
Figure VI-13 : Cyclage galvanostatique du supercondensateur BP-1, dans une solution de
carbonate de propylène 1M NEt4BF4 ……………………………………………………… 147
Figure VI-14 : Cyclage galvanostatique du supercondensateur BP-2, dans une solution de
carbonate de propylène 1M NEt4BF4 ……………………………………………………… 148
Figure VI-15 : Evolution de la capacité déchargée par le polymère au cours du cyclage
galvanostatique du système BP-2 ………………………………………………………….. 149
185

Figure VI-16 : Evolution de la capacité électrostatique déchargée par chacune des électrodes
et par le système au cours du cyclage galvanostatique du système BP-2 ………………… 149
Figure VI-17 : Spectres d'impédance dans le plan de Nyquist des systèmes hybrides entre
65 kHz et 200 mHz…………………………………………………………………………. 150
Figure VI-18 : montage prismatique multi-électrodes.……………………………………. 152
Figure VI-19 : Module industriel………………………………………………………….. 152
Figure VI-20 : Procédure de laminage des pâtes à une épaisseur donnée………………… 153
Figure VI-21 : droite d'étalonnage du P-4-FPT (80%).…………………………………… 154
Figure VI-22 : droite d'étalonnage du BP25 (80%)……………………………………….. 154
Figure VI-23 : Procédure de co-laminage des pâtes à sur le collecteur de courant……….. 154
Figure VI-24 : électrode placée dans un sachet de séparateur Gore………………………. 155
Figure VI-25 : Evolution de la capacité d'un module hybride
P-4-FPT // NEt4BF4 1M /PC // YP17 au cours de cycles effectués en cyclage galvanostatique
à 5A entre 1,5 et 3V……………………………………………………………………….. . 157
186

Liste des tableaux
I) Etude bibliographique
Tableau I-1 : Caractéristiques et performances des principaux supercondensateurs
commerciaux………………………………………………………………………………… 25
II) Partie expérimentale
Tableau II-1 : Spectres d’impédance complexe de quelques circuits électriques simples…. 39
III) Synthèses
Tableau III-1 : Principales données tirées de la Figure III-1 ainsi que rendement massique de
polymérisation………………………………………………………………………………. 47
Tableau III-2 : Principales données tirées de la Figure III-3 ainsi que rendement massique de
polymérisation………………………………………………………………………………. 49
Tableau III-3 : Principales données tirées de la Figure III-4 ainsi que rendement massique de
polymérisation………………………………………………………………………………. 51
Tableau III-4 : Principales données tirées de la Figure III-5 ainsi que rendement massique de
polymérisation.……………………………………………………………………………… 52
V) Caractérisation des polymères
Tableau V-1 : Analyse élémentaire du PTh synthétisé par polycondensation .……………. 70
Tableau V-2 : Attribution des principaux pics du spectre infrarouge du PTh……………… 71
Tableau V-3 : Analyse élémentaire du P-4-FPT …………………………………………… 78
Tableau V-4 : Attribution du spectre infrarouge du P-4-FPT………………………………. 79
Tableau V-5 : Attribution du spectre infrarouge du 4-FPT et du P-4-FPT…………………. 80
Tableau V-6 : Analyse élémentaire de l'électrolyte à plusieurs stades du cyclage
du P-4-FPT…………………………………………………………………………………. 114
Tableau V-7 : Analyse élémentaire du P-4-FPT avant et après cyclage …………………. 116
Tableau V-8 : Analyses élémentaires des PPTF………………………………………….. 119
187

Tableau V-9 : Attribution des spectre infrarouge des PPTF………………………………. 120
Tableau V-10 : Taux de dopage positif et capacité massique des PPTF………………….. 124
Tableau V-11 : Nombre de cycles d'activation nécessaires à chacun des PPTF pour obtenir
une réversibilité stable…………………………………………………………………….. . 125
Tableau V-12 : Taux de dopage négatif et capacité massique des PPTF………………….. 126
VI) Application aux supercondensateurs
Tableau VI-1 : performances des modules industriels…………………………………….. 155
188