td - anomalies chromosomiques - cdbn.fr · adulte risque augmenté d’hta et en cas de grossesse,...
TRANSCRIPT

UE 7 - Génétique Dr DORAY
Date : 12/03/2018 Plage horaire : Matin Promo : DFGSM3 2017/2018 Enseignant : Dr. DORAY Ronéistes : DAMOUR Ludivine / FONTAINE Cécile
TD - Anomalies chromosomiques
Observation 1
Mère de 22 ans et père de 25 ans : - 1ère grossesse : fausse couche (FC) à 3 mois - 2ème grossesse : l’échographie révèle à 31 SA un canal atrio-ventriculaire (CAV), une suspicion d’atrésie
duodénale, une langue protruse (qui sort) et un fémur court.
Quel diagnostic envisagez-vous ? Trisomie 21 : - Plusieurs malformations cardiaques mais la plus fréquente est le CAV dans la trisomie 21 - Atrésie duodénale se manifeste à l’échographie avec une image en double bulle : une bulle qui est
l’estomac et une autre qui est le duodénum, dilaté en amont de l’atrésie - Langue protruse car habituellement hypotonie bucco-faciale comme plus tard chez l’enfant trisomique 21 - Et même si pas de retard de croissance habituel, souvent les os longs (dont fémur) sont plus courts.
Comment poursuivez-vous les investigations ? On fait une amniocentèse, prélèvement de liquide amniotique, sur lequel on peut réaliser 2 examens : - Une FISH sur noyaux interphasiques - Un caryotype sur cellules amniotiques (cellules de la peau, de la vessie) cultivées
Il faut bien comprendre qu’on est à 31 SA, ce qui est un stade assez avancé de la grossesse. Il s’agit d’avoir un résultat rapidement. Un caryotype met une quinzaine de jours à être réalisé, c’est relativement long. La FISH interphasique permet d’avoir un résultat en 24h seulement. Attention, la FISH ne permet pas de poser le diagnostic de trisomie, il s’agit simplement d’un comptage de certains chromosomes.
FISH sur noyaux inter phasiques (résultat en 24h) :
En rouge les sondes spécifiques du 21 En vert les sondes spécifiques du 13
On observe donc 3 spots correspondant aux chromosomes 21, et donc une très probable trisomie 21.
Page ! sur !1 14

Caryotype (résultat en 15 jours) :
On voit donc qu’il n’y a que 2 chromosomes 21 libres et un troisième transloqué au chromosome 14.
Le caryotype confirme donc la tr isomie 21 par translocat ion déséquilibrée, qui se note donc : 46,XY,der(14 ;21)(q10;q10)+21
Ce genre de translocation (qui se trouve être robertsonienne) peut être de novo, mais il y’a de fortes chances qu’un des parents en soit également porteur. On ne se poserait pas cette question devant une T21 libre et homogène, qui serait sûrement de novo.
Une interruption de grossesse a été décidée à 33 SA. On réalise donc comme expliqué plus haut un caryotype des 2 parents. Celui du père est strictement normal, par contre, celui de la mère est effectivement porteur d’une translocation équilibrée 14/21.
Quels examens devez-vous demander en vue du conseil génétique ? On réalise les caryotypes des parents. Dès qu’on trouve une anomalie génétique chez un foetus, il faut toujours faire les caryotypes des parents. Il y a des situations où c’est moins important, trisomie 21 libre et homogène par exemple mais dans le cas de la translocation déséquilibrée c’est impératif parce qu’elle peut être apparue de novo ou être héritée d’une translocation équilibrée parentale.
Caryotype du père sans particularité.
Caryotype de la mère :
Le caryotype retrouve une translocation 14/21 équilibrée : 45,XX,der(14;21)(q10;q10).
Page ! sur !2 14
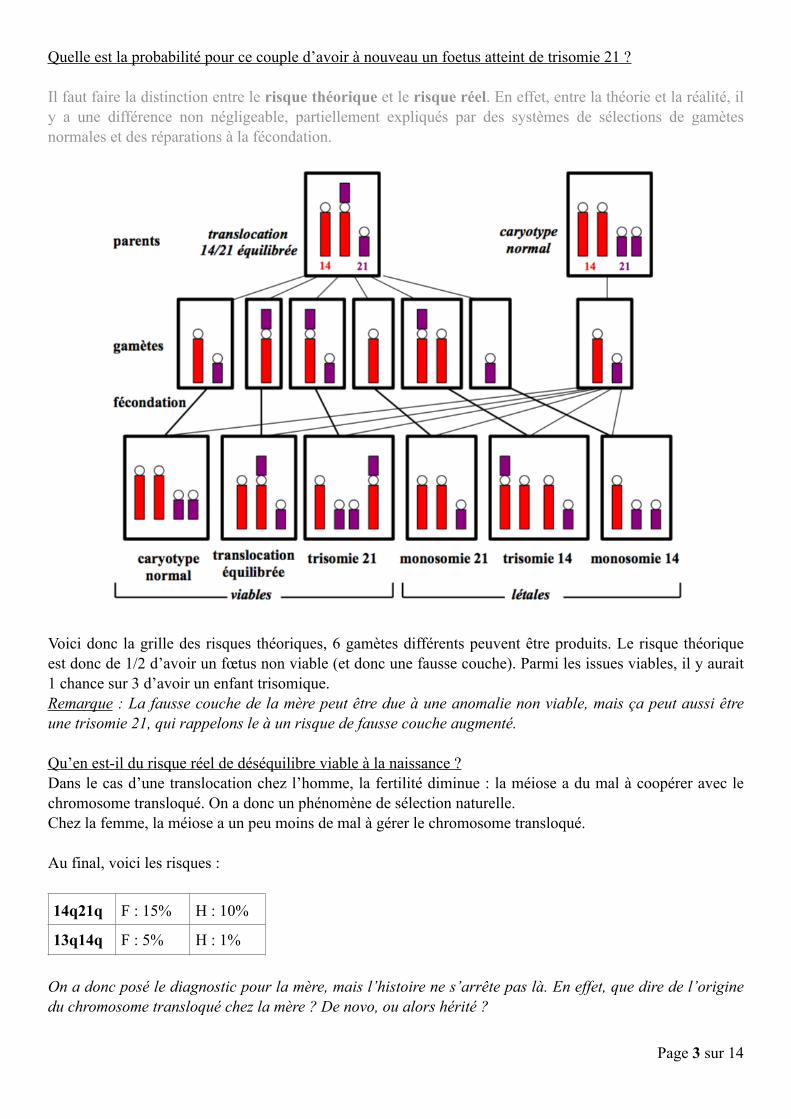
Quelle est la probabilité pour ce couple d’avoir à nouveau un foetus atteint de trisomie 21 ?
Il faut faire la distinction entre le risque théorique et le risque réel. En effet, entre la théorie et la réalité, il y a une différence non négligeable, partiellement expliqués par des systèmes de sélections de gamètes normales et des réparations à la fécondation.
Voici donc la grille des risques théoriques, 6 gamètes différents peuvent être produits. Le risque théorique est donc de 1/2 d’avoir un fœtus non viable (et donc une fausse couche). Parmi les issues viables, il y aurait 1 chance sur 3 d’avoir un enfant trisomique. Remarque : La fausse couche de la mère peut être due à une anomalie non viable, mais ça peut aussi être une trisomie 21, qui rappelons le à un risque de fausse couche augmenté.
Qu’en est-il du risque réel de déséquilibre viable à la naissance ? Dans le cas d’une translocation chez l’homme, la fertilité diminue : la méiose a du mal à coopérer avec le chromosome transloqué. On a donc un phénomène de sélection naturelle. Chez la femme, la méiose a un peu moins de mal à gérer le chromosome transloqué.
Au final, voici les risques :
On a donc posé le diagnostic pour la mère, mais l’histoire ne s’arrête pas là. En effet, que dire de l’origine du chromosome transloqué chez la mère ? De novo, ou alors hérité ?
14q21q F : 15% H : 10%
13q14q F : 5% H : 1%
Page ! sur !3 14

L’enquête familiale : Dans le cas présent, un oncle de la mère avait aussi consulté pour oligoasthénospermie, et 2 FC précoces chez sa conjointe. Le caryotype sanguin a révélé, oh surprise, une translocation 14/21 équilibrée. Cependant, reprochant à sa famille d’être « tous des cons », il n’a pas été question d’informer qui que ce soit d’autre pouvant aussi être porteur de cette anomalie. Ce qui mène à notre prochain sujet : gènes, famille et secret, la genèse de la discorde.
Secret médical et famille : Dans un passé récent, le secret médical était absolu. Seule la personne testée pouvait causer. Mais si elle refuse ? Se trouve t-on dans la terrible situation de non assistance à personne en danger ? On se le demande ! Depuis le 7 juillet 2011, avant tout examen génétique, le médecin explique au patient les risques pour sa famille s’il ne l’informe pas, et prévoit avec lui et par écrit les modalités de l’information familiale.
Le médecin remet donc au patient un document écrit et signé : - Reprenant l’information médicale - Rédigé de manière loyale, claire, et approprié - Sauf si le patient demande par écrit à être tenu dans l’ignorance
Le patient atteste de cette remise, et est tenu d’informer les membres de sa famille connus et concernés lorsque des mesures de soins ou de préventions sont possibles.
Mais puisque la vie n’est qu’une succession d’imprévus, que faire devant un patient qui souhaite être tenu dans l’ignorance du diagnostic, ou ne veut pas informer lui-même sa famille ?
- Il demande par écrit au médecin de procéder à cette information - Il lui communique les coordonnées des personnes concernées identifiées - Le médecin informe ces personnes de l’utilité d’une consultation génétique - Il ne communique ni le nom du proposant, ni le diagnostic, ni les risques - Il informe les médecins de ses apparentés de l’affection génétique en cause.
On se retrouve parfois dans la situation où on reçoit une lettre du CHU de Marseille, ville où on sait pertinemment qu’un oncle à nous y réside. On nage dans l’hypocrisie, mais dans le respect du secret médical. Doit-on créer un CHU Anonyme ? Peut-on éthiquement faire des canulars à ce sujet ?
Observation 2
On est confronté à la naissance sans particularité d’un nouveau-né de sexe féminin. - Poids : 2800g, taille : 49cm, PC : 35cm - Examen neurologique normal - Morphologie faciale : micrognathisme, cou un peu large - Œdème blanc, mou, indolore, du dos des pieds et du dos des mains
Ce tableau nous fait penser à un syndrome de Turner.
L’examen psychomoteur est normal, elle marche à 12 mois. Il y a par contre un retard statural postnatal, un taille à -2,5 DS à 2 ans.
Quel diagnostic devez vous suspecter ? Tous les éléments semblent évoquer un syndrome de Turner. Quel examen réalisez-vous ? Un caryotype standard sur prélèvement sanguin.
Page ! sur !4 14

Le caryotype confirme une monosomie X (45,X), c’est-à-dire un syndrome de Turner.
Quel suivi proposez-vous ? Il faut réaliser :
- Un bilan cardiaque : échographie du coeur à la recherche d’une malformation congénitale touchant plutôt le cœur gauche (ex : une sténose de l’isthme aortique)
- Un bilan rénal : rein en fer à cheval, agénésie rénale unilatérale - Un suivi ORL : otites séro-muqueuses à répétition pouvant endommager l’audition - Un suivi ophtalmologique : souvent de la myopie, à vérifier pour la scolarisation
On propose un traitement par hormone de croissance à partir de 3 ans jusqu’à la puberté, plus on le propose tôt plus le traitement sera efficace. On démarre à la puberté (11 ans environ) un traitement oestroprogestatif pour traiter les anomalies de maintien des ovaires (impubérisme + infertilité).
A l’adolescence : - Supplémentation vitamino-calcique - Suivi cardio-vasculaire : même si tout est normal à la naissance, il faut surveiller ce point car à l’âge
adulte risque augmenté d’HTA et en cas de grossesse, risque de dissection aortique.
Il n’y a pas d’ambiguïté sexuelle dans le syndrome de Turner.
NDR : Doray a clairement dit que le Syndrome de Turner risquait fortement de tomber à l’examen.
Images typiques du syndrome de Tuner (cf. pdf pour plus d’images)
Page ! sur !5 14

Observation 3
Consultation pour une deuxième grossesse d’un couple non apparenté. La mère, 31 ans, et le père, 32 ans, ont un premier enfant en bonne santé. On découvre à 12 semaines une hyperclarté nucale.
A quelle anomalie chromosomique pensez-vous ? Trisomie 21 ou Syndrome de Turner. Que proposez vous sur le plan cytogénétique ? Un prélèvement des villosités choriales = PBT (Ponction Biopsie de Trophoblaste. On est à 12 SA. On ne peut pas réaliser d’amniocentèse, pour la simple et bonne raison que le liquide amniotique n’existe pas. On réalise donc une biopsie des villosités choriales. A la différence de l’amniocentèse, il ne s’agit pas de cellules du foetus mais de cellules du placenta !
Le caryotype est normal, 46,XX !
Quel suivi proposez-vous ? Suivi échographique renforcé On découvre ensuite à 19 SA une malformation cardiaque conotrocale de type tétralogie de Fallot.
A quel type de pathologie pensez-vous ? Le tableau est évocateur d’un Syndrome de Di George Aussi appelé Syndrome vélo-cardio-facial :
- Vélo (pour voile du palais) : soit il y a une fente, soit une insuffisance, la voix sera nasonnée - Cardio : cardiopathies cono-trocales - Facial : dysmorphie = petite bouche, petites oreilles, nez…
Comment la confirmez-vous ? On réalise une FISH ! Le Syndrome de Di George est une microdélétion de type 22q11.2. On va donc utiliser des sondes complémentaires de cette région.
La FISH confirme le diagnostic de syndrome vélo-cardio-facial.
Page ! sur !6 14

Le couple prend la décision de faire une IMG à 21 SA.
Quels conseils génétiques donnez-vous ? Il faut réaliser un caryotype des parents + une FISH 22q11. - Dans 85% des cas, les caryotypes parentaux sont normaux. Le risque d’anomalie chromosomique pour
une autre grossesse n’est que de 1%. On propose un DPN cytogénétique. Le risque pour leur premier enfant est équivalent à celui de la population générale.
- Dans 15% des cas, la délétion est héritée d’un des parents. Dans ce cas, le risque à la prochaine grossesse est de 50% ➔ DPN cytogénétique. Pour leur premier enfant, un examen cytogénétique ne sera réalisé que s’il y a présence de signes cliniques.
Observation 4
Une échographie montre à 32 SA un hydramnios et une malformation cardiaque conotrocale. Le caryotype sur sang fœtal est 46,XX. Le caryotype sur sang est plus rapide à réaliser que celui sur liquide amniotique, mais il faut un stade de développement suffisamment avancé pour pouvoir le faire. La FISH montre une délétion 22q11. Le caryotype et la FISH des parents montrent :
- Père : 46,XY - Mère : 46,XX,del22q11
La mère a eu des difficultés scolaires, une CIV, une voix nasonnée.
On est donc face à un diagnostic prénatal tardif. Devant l’importante variabilité phénotypique de la délétion 22q11.2, on est incapable de dire aux parents comment l’enfant sera d’un point de vue cognitif. On se retrouve également face à la culpabilité parentale d’avoir refilé son anomalie.
Il y a la possibilité technique d’un DPN cytogénétique ultérieur. Il y a aussi le point de vue éthique à considérer : à la base, le couple vient pour leur progéniture, et finit avec un diagnostic pour la mère également.
Observation 5
Il est question ici de la troisième grossesse d’un couple non apparenté. La mère, 41 ans, et le père, 44 ans ont 2 enfants en bonne santé. On découvre à 15 SA un RCIU sévère. Le contrôle à 18 SA montre une RCIU, une microcéphalie, un profil particulier, et une fente labiale bilatérale.
Que proposez-vous sur le plan cytogénétique ? Une amniocentèse pour réaliser un caryotype.
En observant bien le caryotype (et en s’aidant de la flèche rouge), on voit un chromosome 4 un peu court.
Tout ceci semble évoquer un Syndrome de Wolf-Hirschhorn.
Page ! sur !7 14
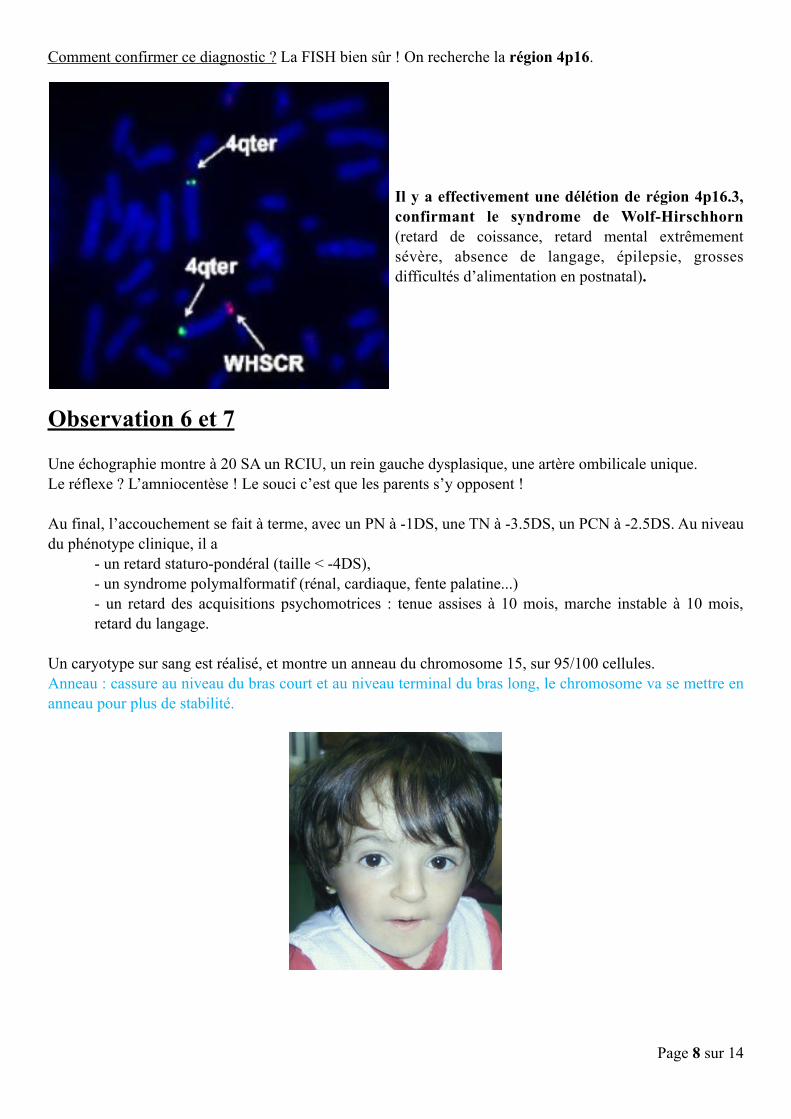
Comment confirmer ce diagnostic ? La FISH bien sûr ! On recherche la région 4p16.
Il y a effectivement une délétion de région 4p16.3, confirmant le syndrome de Wolf-Hirschhorn (retard de coissance, retard mental extrêmement sévère, absence de langage, épilepsie, grosses difficultés d’alimentation en postnatal).
Observation 6 et 7
Une échographie montre à 20 SA un RCIU, un rein gauche dysplasique, une artère ombilicale unique. Le réflexe ? L’amniocentèse ! Le souci c’est que les parents s’y opposent !
Au final, l’accouchement se fait à terme, avec un PN à -1DS, une TN à -3.5DS, un PCN à -2.5DS. Au niveau du phénotype clinique, il a
- un retard staturo-pondéral (taille < -4DS), - un syndrome polymalformatif (rénal, cardiaque, fente palatine...) - un retard des acquisitions psychomotrices : tenue assises à 10 mois, marche instable à 10 mois, retard du langage.
Un caryotype sur sang est réalisé, et montre un anneau du chromosome 15, sur 95/100 cellules. Anneau : cassure au niveau du bras court et au niveau terminal du bras long, le chromosome va se mettre en anneau pour plus de stabilité.
!
Page ! sur !8 14

Un patient d’origine turque vient en consultation quelques mois après, âgé de 34 ans consulte pour infertilité et petite taille (non familiale). Il n’y a pas de syndrome dysmorphie, pas de déficit mental. Il y’a une azoospermie. Le caryotype sur sang montre anneau du chromosome 15, sur 95/100 cellules.
!
On est face à 2 cas avec la même anomalie. Qu’est ce qui explique une si grande variabilité génétique ?
! Il se trouve que dans le premier cas, il y’a eu une délétion de la région subtélomérique (= régions très riche en gènes) du bras long du chromosome 15, et pas dans le deuxième cas.
Les anneaux :
On observe une variabilité extrême du phénotype (instabilité mitotique et méiotique). Il y’a souvent un petite taille. La stérilité masculine est courante, tandis que la fertilité féminine est à priori normale.
Observation 8
Une mère a eu des grossesses sans particularités, avec 2 enfants en bonne santé. Cependant, la troisième grossesse montre le tableau suivant :
- Naissance à terme, PN = 2100g, TN = 46 cm, PC = 32 cm - Difficultés d’alimentation, troubles de sucions-déglutition
Page ! sur !9 14
- Sténose aortique supra-valvulaire à l’échographie - Hypercalcémie - Dysmorphie faciale, difficultés scolaires
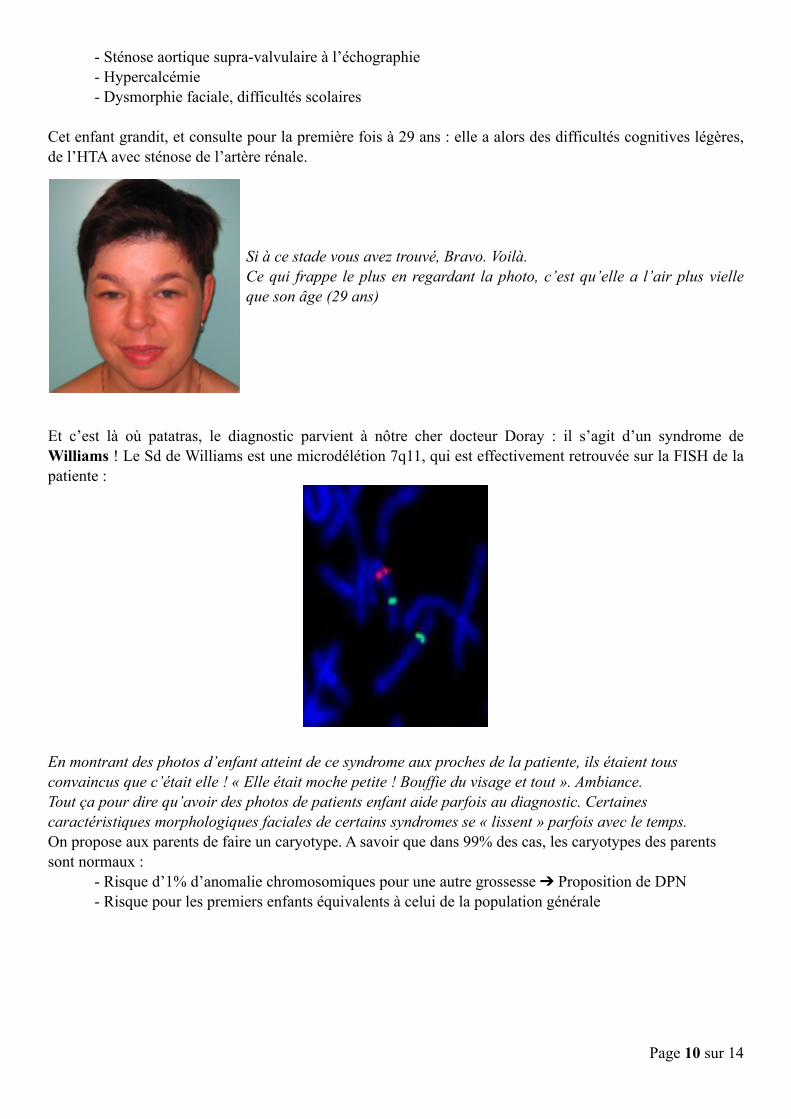
Cet enfant grandit, et consulte pour la première fois à 29 ans : elle a alors des difficultés cognitives légères, de l’HTA avec sténose de l’artère rénale.
Si à ce stade vous avez trouvé, Bravo. Voilà. Ce qui frappe le plus en regardant la photo, c’est qu’elle a l’air plus vielle que son âge (29 ans)
Et c’est là où patatras, le diagnostic parvient à nôtre cher docteur Doray : il s’agit d’un syndrome de Williams ! Le Sd de Williams est une microdélétion 7q11, qui est effectivement retrouvée sur la FISH de la patiente :
!
En montrant des photos d’enfant atteint de ce syndrome aux proches de la patiente, ils étaient tous convaincus que c’était elle ! « Elle était moche petite ! Bouffie du visage et tout ». Ambiance. Tout ça pour dire qu’avoir des photos de patients enfant aide parfois au diagnostic. Certaines caractéristiques morphologiques faciales de certains syndromes se « lissent » parfois avec le temps. On propose aux parents de faire un caryotype. A savoir que dans 99% des cas, les caryotypes des parents sont normaux :
- Risque d’1% d’anomalie chromosomiques pour une autre grossesse ➔ Proposition de DPN - Risque pour les premiers enfants équivalents à celui de la population générale
Page ! sur !10 14

!
D’un point de vue cognitif, un enfant atteint du syndrome de Williams aura beaucoup de problème de représentation visuelle dans l’espace. On voit qu’à QI égal, un trisomique réussit son dessin de vélo, contrairement au Williams. La prise en charge par un orthoptiste pour la rééducation visuelle apporte cependant une réelle amélioration.
L’intérêt du diagnostic réside dans l’explication concrète des difficultés à la famille, le suivi médical (HTA, sténose artérielle, AVC)
Observation 9
Une grossesse marquée par un hydramnios et une urétérohydronéphrose à 24 SA, avec un caryotype sur liquide amniotique 46,XX, aboutit à une naissance à 38 SA avec hypotrophie et dysmorphie faciale, ainsi qu’une atrésie de l’œsophage. Un bilan malformatif est réalisé, et montre une atrésie duodénale, communication interventriculaire.
Que devez-vous demander comme examen complémentaire et pourquoi ? Ici, on n’a pas vraiment d’intuition sur quoi que ce soit. Aujourd’hui, c’est le genre de situation où le CGH Array est notre sauveur. Mais c’est une technique récente. A l’époque du cas clinique, il a donc du se satisfaire d’un caryotype haute résolution, le premier caryotype étant normal. On réalise donc un caryotype haute résolution sur sang :
On observe une délétion du bras long du chromosome 12 ( à gauche)
Quels examens demandez-vous pour préciser ce diagnostic ? - Peinture du chromosome 12 par FISH - FISH du télomère 12q
Les parents s’interrogent à présent sur le risque de récidive sur une autre grossesse.
Page ! sur !11 14

Quoi fait-on ? A. On évite le regard des parents pour le reste de la consultation B. On leur suggère d’arrêter d’essayer de gratter des allocs C. On leur recommande Madame Irma D. On propose un caryotype aux parents
Si les caryotypes des parents sont normaux, le risque de récidive est dès lors très faible. On propose un DPN.
Observation 10
Un couple est adressé en consultation en raison d’une infertilité. Le bilan chez la femme est normal, il a déjà été réalisé pour une grossesse antérieure (d’une précédente union). Le spermogramme du conjoint, en parfaite santé, croyant que l'anomalie venait de sa femme parce que monsieur est "clean", montre une azoospermie complète, et à l’examen des OGE, des testicules de petite taille.
Quelle est votre hypothèse diagnostic ? Un syndrome de Klinefelter (47 XXY) !
! Il est à noter que le patient travaille à l’université en tant que chercheur, il n’y a pas de troubles cognitifs.
Tout comme le Sd de Turner, le Sd de Klinefelter ne comprend pas d’ambiguïtés sexuelles. Dans le syndrome de K. il y a un défaut de production de testostérone à la puberté et donc il faut supplémenter en testostérone pour avoir une virilisation normale à l'adolescence.
Observation 11
On a le cas clinique suivant : - Grossesse sans particularités - Naissance à terme, avec :
* Hypotrophie modérée * Hypotonie majeure, difficultés sévères d’alimentation * Cryptorchidie
- Consultation génétique à 4 ans en raison d’un retard des acquisitions : marche à 24 mois, retard du langage, et :
* Obésité avec poids à + 4DS, taille à -1.5DS (hyperphagie)
Page ! sur !12 14
* Dysmorphie : yeux en amande, acromicrie, cryptorchidie bilatérale modérée, hypo-pigmentation
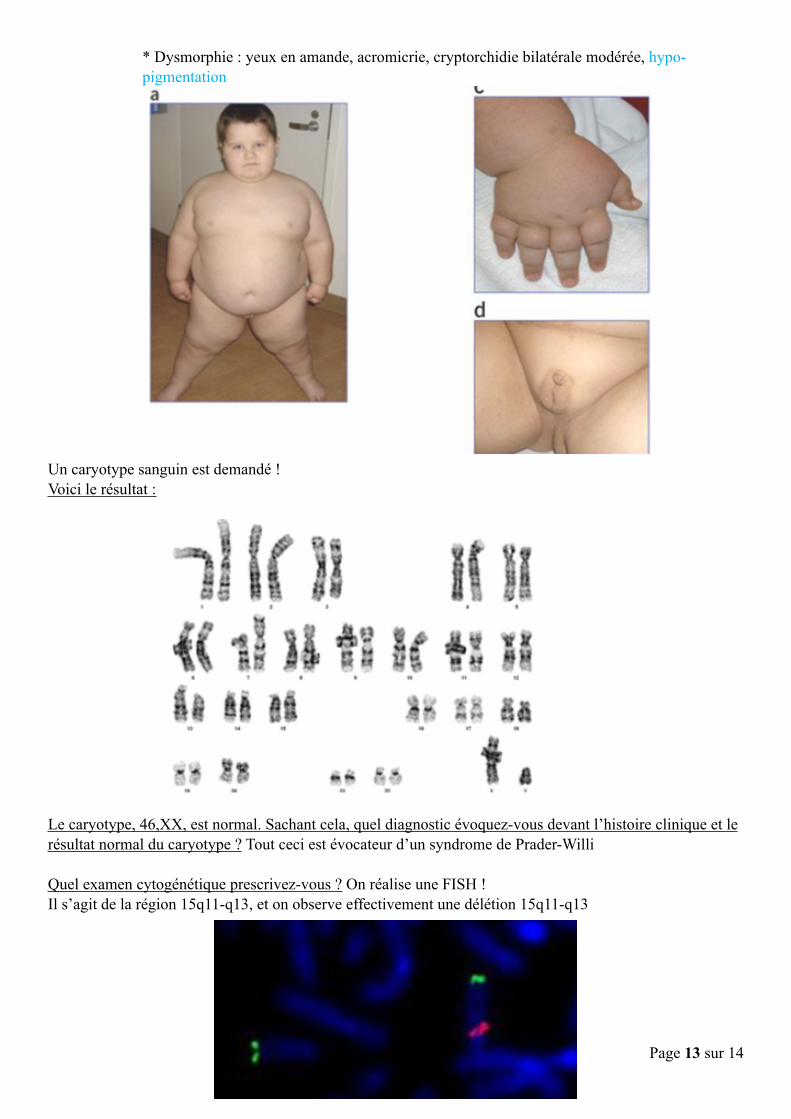
! Un caryotype sanguin est demandé ! Voici le résultat :
! Le caryotype, 46,XX, est normal. Sachant cela, quel diagnostic évoquez-vous devant l’histoire clinique et le résultat normal du caryotype ? Tout ceci est évocateur d’un syndrome de Prader-Willi
Quel examen cytogénétique prescrivez-vous ? On réalise une FISH ! Il s’agit de la région 15q11-q13, et on observe effectivement une délétion 15q11-q13
Page ! sur !13 14
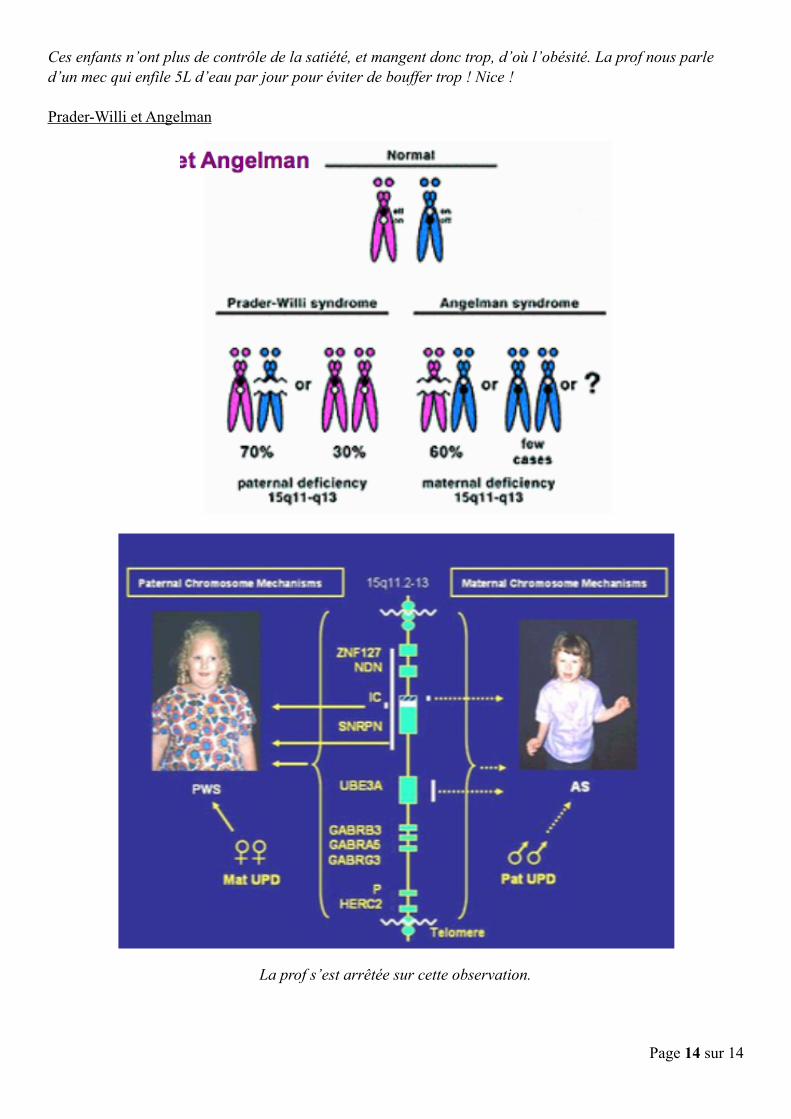
Ces enfants n’ont plus de contrôle de la satiété, et mangent donc trop, d’où l’obésité. La prof nous parle d’un mec qui enfile 5L d’eau par jour pour éviter de bouffer trop ! Nice !
Prader-Willi et Angelman
La prof s’est arrêtée sur cette observation.
Page ! sur !14 14