revue bibliographique sur les méthodes d'analyse des ...¨res, notamment la cellulose, les...
TRANSCRIPT

BASE Biotechnol. Agron. Soc. Environ.201115(1),165-182 Le Point sur :
Revuebibliographiquesurlesméthodesd’analysedespolysaccharidesstructurauxdesbiomasseslignocellulosiquesBrunoGodin(1,3),RichardAgneessens(2),SébastienGofflot(2),StéphaneLamaudière(3),GeorgesSinnaeve(2),PatrickA.Gerin(3),JérômeDelcarte(1)(1)CentrewallondeRecherchesagronomiques(CRA-W).DépartementValorisationdesProductions.UnitéBiomasse,BioproduitsetÉnergies.ChausséedeNamur,146.B-5030Gembloux(Belgique).E-mail:[email protected](2)CentrewallondeRecherchesagronomiques(CRA-W).DépartementValorisationdesProductions.UnitéTechnologiesdelaTransformationdesProduits.RueduSerpont,100.B-6800Libramont(Belgique).(3)UniversitécatholiquedeLouvain(UCL).UnitédeGéniebiologique.CroixduSud,2/19.B-1348Louvain-la-Neuve(Belgique).
Reçule21mars2009,acceptéle7juin2010.
Lespolysaccharidesstructurauxetlaligninereprésententl’essentieldesbiomasseslignocellulosiques.Lacompositiondecespolymères,notammentlacellulose,leshémicellulosesetlespectines,peutêtreeffectuéepardiversesméthodesglobaleset/ousélectivesdecespolymères.Lesméthodesglobalesétudiéesdanscettesynthèsesontcellesde«VanSoest»,«Prosky»,«Uppsala»,«Selvendran»etdes«polysaccharidesnonamylacés».Afindedistinguerlesdifférences,lesavantagesetlesinconvénientsdecesméthodesglobales,celles-cisontdécritesetcomparées.LaméthodedeVanSoestestcelleprésentantlemeilleurcompromisentre lesdonnéesobtenuessur lanaturedespolysaccharidesstructurauxconstituant lesbiomasseslignocellulosiquesetletempsd’analysepourparveniràobtenircetteinformation.Enadaptantcetteméthode,ellepourraitapporterdesdonnéessurlaquantitédepectines(expriméesenacidegalacturonique),surlacompositionenmonosaccharidesdespolysaccharidesstructurauxetsurlaquantitéd’hydratesdecarbonenonstructurauxdesbiomasseslignocellulosiques.Quelquesméthodesdedosagesélectivesd’unoudeuxpolysaccharidesstructurauxcommelacellulose,lesxyloglucanes,lesxylanes,lesβ-glucanes,lesmannanesetlespectinessontégalementprésentéesdanscettesynthèse.Mots-clés.Cellulose,hémicelluloses,pectines,VanSoest,Prosky,polysaccharidesnonamylacés,Uppsala,Selvendran.
Review on analytical methods for lignocellulosic biomass structural polysaccharides. Structural polysaccharides andligninrepresentthemajorpartofthelignocellulosicbiomass.Thecharacterizationofthesestructuralpolymers,likecellulose,hemicelluloses and pectins, can be carried out by various global and/or polymer-selectivemethods.The globalmethodsstudiedinthisreviewarethoseof“VanSoest”,“Prosky”,“Uppsala”,“Selvendran”and“non-starchpolysaccharides”.Theseglobalmethodsaredescribedandcomparedinordertoenlightentheirdifferences,advantagesanddisadvantages.TheVanSoestmethodistheonepresentingthebestcompromisebetweenthedataobtainedonthenatureofstructuralpolysaccharidesconstitutingthelignocellulosicbiomassandthetimeofanalysisrequiredtoobtainthispieceofinformation.Byadaptingthismethod,itcouldalsogiveinformationonthequantityofpectins(expressedasgalacturonicacid),onthemonosaccharidiccompositionofthestructuralpolysaccharidesandonthequantityofnonstructuralcarbohydratesofthelignocellulosicbiomass.Somemethodsselectivetooneortwostructuralpolysaccharideslikecellulose,xyloglucans,xylans,β-glucans,mannanesandpectinsarealsopresentedinthisreview.Keywords.Cellulose,hemicelluloses,pectins,VanSoest,Prosky,non-starchpolysaccharides,Uppsala,Selvendran.
1. IntRoductIon
Les hydrates de carbone des biomasses lignocellu-losiques sont séparés en deux catégories: leshydrates de carbone non structuraux comme lesmonosaccharides,lesoligosaccharidesetlespolysac-
charides de réserves, et les hydrates de carbonestructuraux (dont la dénomination plus communede «polysaccharides structuraux» est utilisée parla suite dans cet article) (Southgate, 1995; Carpitaet al., 2000). La paroi cellulaire des biomasseslignocellulosiques est constituée d’une grande

166 Biotechnol. Agron. Soc. Environ. 201115(1),165-182 GodinB.,AgneessensR.,GofflotS.etal.
diversitédecespolysaccharidesstructurauxquisontdivisés en différentes catégories: la cellulose, leshémicelluloses (xyloglucanes, xylanes, β-glucaneset mannanes) et les pectines (homogalacturonanes,xylogalacturonanes, rhamnogalacturonanes type Iet type II) (Carpita et al., 2000). Ces composéssont liésàdesprotéinespariétales (richesenacidesaminés, soit de type hydroxyproline, soit de typethréonine-hydroxyproline, soit de typeglycine) et àdespolyphénols,plusparticulièrementlaligninequiest un polymère constitué d’unités phénylpropanestelles que les alcools de type p-hydroxyphényle(dont l’alcool p-coumarylique), syringyle (dontl’alcool synapilique) et guaïacyle (dont l’alcoolconiférylique) (Carpita et al., 2000; Grabber,2005).Lespolysaccharides structurauxet la ligninesont les constituants les plus abondants dans laparoi cellulaire (Ebringerová et al., 2005). Tousles constituants des parois végétales des biomasseslignocellulosiques(Figure 1)sontqualifiésdefibreset plus spécifiquement de fibres alimentaires dansle domaine de la nutrition (FAO, 2007;Ampuero,2008).LaFAOdéfinitlesfibresalimentairescomme«des polymères glucidiques avec un degré depolymérisation(DP)noninférieuràtrois,quinesontnidigérésniabsorbésdansl’intestingrêle.Undegrédepolymérisationnon inférieurà troisestdestinéàexclure lesmono- et disaccharides et non à refléterle DP moyen du mélange. Si elles sont d’originevégétale,lesfibresalimentairespeuventcomprendredes fractions de lignine et/ou d’autres composantss’ilssontassociésavecdespolysaccharidesdanslesparoiscellulairesvégétalesetsicescomposantssontquantifiésparlaméthoded’analysegravimétriquequi
a été adoptée pour l’analyse des fibres alimentaires(AOAC)»(FAO,2007).
Deux catégories de méthodes existent afin decaractériserlanaturedespolysaccharidesstructurauxdes biomasses lignocellulosiques: les méthodesglobalesetlesméthodessélectives.
Lesméthodesglobalespermettentdecaractériserdesensemblesdepolysaccharidesstructurauxsansendistinguerlanaturedanschaqueensembleisolé.Cespolysaccharidessontséparéssoitsousformedefibresalimentairestotales,soitdivisésenfibresalimentairessolublesetinsolublesdansl’eau.Ilfautnoterquelalignine,lesprotéinespariétales,lestanninsetd’autrescomposants liés aux polysaccharides structuraux setrouvent également dans les fibres alimentaires carils font partie intégrante de ces fractions de fibresalimentaires (Dorleans et al., 1996; Carpita et al.,2000;FAO,2007).LesprincipalesméthodesglobalessontlaméthodeauxdétergentsneutreetacidedeVanSoestetal.(1985),laméthodedesfibresalimentairesdeProskyetal.(1992),laméthodedespolysaccharidesnon amylacés d’Englyst et al. (1994), la méthodedes fibres alimentaires d’Uppsala (Theander et al.,1995) et la méthode alcaline de Selvendran et al.(1985).
Les méthodes sélectives isolent sélectivementun ou deux polysaccharides structuraux, comme lacellulose(Sunetal.,1995), lesxylanes(Izydorczyket al., 1998a; Izydorczyk et al., 1998b; Genestie,2006), les xyloglucanes (Cutillas-Iturralde et al.,1998), les β-glucanes (Izydorczyk et al., 1998a;Izydorczyketal.,1998b),lesmannanes(Selvendranetal.,1985)etlespectines(Selvendranetal.,1985;LeGoffetal.,2001).
L’objectif de cette synthèse est de déterminer laméthode présentant le meilleur compromis entreles données obtenues sur la nature (la structureet la composition en monosaccharides) despolysaccharides structuraux constituant la biomasselignocellulosiqueetletempsd’analysepourparveniràobtenircetteinformation.Leshydratesdecarbonenon structuraux représentent une des interférencesmajeures lors de l’analyse des polysaccharidesstructuraux des biomasses lignocellulosiques. Lapurificationdespolysaccharidesstructurauxparuneméthode suffisamment sélective est doncnécessairepourdéterminerlanaturedecespolysaccharidesdesbiomasseslignocellulosiques.
Lesméthodesglobales deVanSoest, deProsky,des polysaccharides non amylacés, d’Uppsala et deSelvendranserontprésentéesetcomparéesdanscetterevue.
Lesméthodessélectivesàlacellulose,auxxylanes,auxxyloglucanes,auxβ-glucanes,auxmannanesetauxpectinesdesbiomasseslignocellulosiquesserontégalementprésentéesdanscetterevue.
Figure 1. Organisation des polysaccharides structurauxet de la lignine dans les parois cellulaires des biomasseslignocellulosiques (adapté de Zhang, 2008)— Structural polysaccharides and lignin organization in lignocellulosic biomass cell walls (adapted from Zhang, 2008).
Paroi cellulaire
Cellule végétale
Macrofibrille
Microfibrille
Lignine
Cellulose
Hémicelluloseset pectines

Revuedesméthodesd’analysedespolysaccharidesstructuraux 167
2. descRIPtIon des Méthodes d’analyse des PolysacchaRIdes stRuctuRaux des bIoMasses lIgnocellulosIques
2.1. Méthode aux détergents neutre et acide de Van soest
LaméthodeauxdétergentsneutreetacidedeVanSoestet al. (1985) (Figure 2,tableaux 1 et 2) fractionne
les fibres de la biomasse lignocellulosique par desextractionssuccessivesparvoiechimiqueetquantifiepargravimétrielesfibresinsolublessèchesrécupéréesaprèsfiltration.
LaméthodedeVanSoest(VanSoestetal.,1985;Van Soest et al., 1991; Mertens, 2002; Ampuero,2008) permet d’extraire les hydrates de carbone nonstructuraux,lespectines,lesgommes,lesmucilages1,lestanninssolublesàpHneutre,leslipides,lesprotéinessolubles au détergent neutre et certaines cendres à
Figure 2.SchémadufractionnementetdescomposésdosésdanslaméthodedeVanSoest—Diagram of the splitting up and the quantified compounds in the Van Soest method.
*composénonquantifié—not quantified component.
Fraction insolubleInsoluble fraction
Filtration Calcination
Fraction solubleSoluble fraction
Fraction volatileVolatile fraction
Méthode de dosage analytiqueAnalytical quantifying method
168 Biotechnol. Agron. Soc. Environ. 201115(1),165-182 GodinB.,AgneessensR.,GofflotS.etal.
l’aide d’une solution de détergent neutre, en excès,agissant sur l’échantillon pendant 1h à 100°C avecunpHde7±0,05.CettesolutiondedétergentneutreestcomposéedeNa2HPO4,de tétraboratedesodium,de 2-éthoxyéthanol (qui extrait l’amidon), d’EDTAdesodium(quiextraitlespectines),delaurylesulfatede sodium (qui extrait les protéines et les lipides) etde sulfite de sodium (qui hydrolyse les protéines).
La fraction extraite est séparée des fibres insolublesau détergent neutre (NDF) par filtration. La fractionNDF est constituée de cellulose, d’hémicelluloses,de lignine, de tannins insolubles à pH neutre, deprotéinesinsolublesaudétergentneutreetdecertainescendres.
Parallèlement (ou séquentiellement) à l’actiondu détergent neutre, une solution de détergent acide,en excès, permet d’extraire les hémicelluloses, lestanninssolublesenmilieuacide(présentsentrèsfaiblequantité), laligninesolubleenmilieuacide(présenteen très faible quantité), les protéines solubles audétergentacideetcertainescendres.Cettesolutionde
1D’aprèsSouthgate(1995),lesmotsgommeetmucilageontunesignificationspécifiqueetilscorrespondentàdesstructuresmoléculaires.
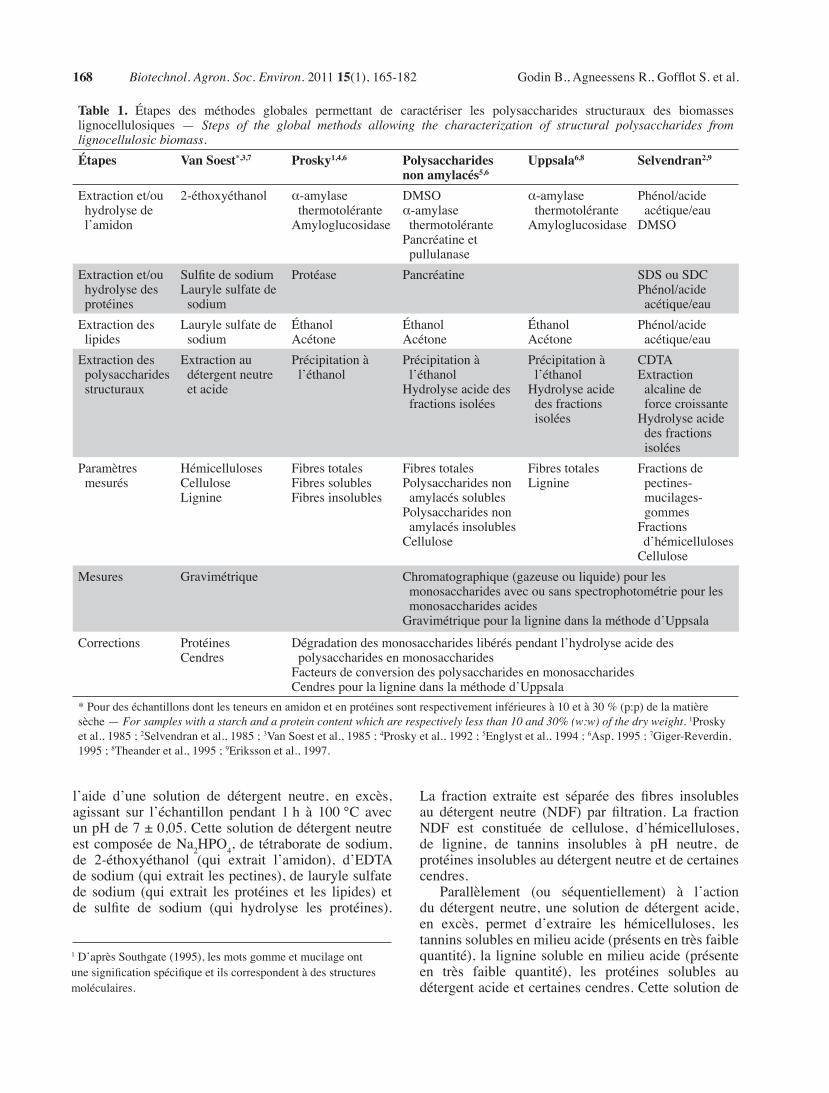
table 1. Étapes des méthodes globales permettant de caractériser les polysaccharides structuraux des biomasseslignocellulosiques — Steps of the global methods allowing the characterization of structural polysaccharides from lignocellulosic biomass.étapes Van soest*,3,7 Prosky1,4,6 Polysaccharides
non amylacés5,6uppsala6,8 selvendran2,9
Extractionet/ouhydrolysedel’amidon
2-éthoxyéthanol α-amylasethermotoléranteAmyloglucosidase
DMSOα-amylasethermotolérantePancréatineetpullulanase
α-amylasethermotoléranteAmyloglucosidase
Phénol/acideacétique/eauDMSO
Extractionet/ouhydrolysedesprotéines
SulfitedesodiumLaurylesulfatedesodium
Protéase Pancréatine SDSouSDCPhénol/acideacétique/eau
Extractiondeslipides
Laurylesulfatedesodium
ÉthanolAcétone
ÉthanolAcétone
ÉthanolAcétone
Phénol/acideacétique/eau
Extractiondespolysaccharidesstructuraux
Extractionaudétergentneutreetacide
Précipitationàl’éthanol
Précipitationàl’éthanolHydrolyseacidedesfractionsisolées
Précipitationàl’éthanolHydrolyseacidedesfractionsisolées
CDTAExtractionalcalinedeforcecroissanteHydrolyseacidedesfractionsisolées
Paramètresmesurés
HémicellulosesCelluloseLignine
FibrestotalesFibressolublesFibresinsolubles
FibrestotalesPolysaccharidesnonamylacéssolublesPolysaccharidesnonamylacésinsolublesCellulose
FibrestotalesLignine
Fractionsdepectines-mucilages-gommesFractionsd’hémicellulosesCellulose
Mesures Gravimétrique Chromatographique(gazeuseouliquide)pourlesmonosaccharidesavecousansspectrophotométriepourlesmonosaccharidesacidesGravimétriquepourlaligninedanslaméthoded’Uppsala
Corrections ProtéinesCendres
Dégradationdesmonosaccharideslibéréspendantl’hydrolyseacidedespolysaccharidesenmonosaccharidesFacteursdeconversiondespolysaccharidesenmonosaccharidesCendrespourlaligninedanslaméthoded’Uppsala
*Pourdeséchantillonsdontlesteneursenamidonetenprotéinessontrespectivementinférieuresà10età30%(p:p)delamatièresèche—For samples with a starch and a protein content which are respectively less than 10 and 30% (w:w) of the dry weight.1Proskyetal.,1985;2Selvendranetal.,1985;3VanSoestetal.,1985;4Proskyetal.,1992;5Englystetal.,1994;6Asp,1995;7Giger-Reverdin,1995;8Theanderetal.,1995;9Erikssonetal.,1997.

Revuedesméthodesd’analysedespolysaccharidesstructuraux 169
détergentacideestcomposéedebromuredetriméthyl-cétyl-ammoniumetdeH2SO41Netagitpendant1hà 100°C. La fraction extraite est séparée des fibresinsolublesaudétergentacide(ADF)parfiltration.LafractionADF est constituée de cellulose, de lignine
insoluble en milieu acide, de tannins insolubles enmilieuacide,desprotéinesinsolublesaudétergentacideetdecertainescendres.LadifférenceentrelesNDFetlesADF(encorrigeantparlateneurenprotéinesetencendres)correspondàlaquantitéd’hémicelluloses,de
table 2. Aptitudedesméthodesàl’analysededifférentsconstituantsdesbiomasseslignocellulosiquesetplusspécialementleshydratesdecarbone—Aptitude of the global methods to analyze various lignocellulosic biomass components and more specifically carbohydrates.
MéthodeVan soest *,3,7 Prosky 1,4,6 Polysaccharides non amylacés 5,6 uppsala 7,8 selvendran 2,9
hydrates de carbone Monosaccharides
GlucoseFructose Disaccharides
Saccharose Oligosaccharides
Fructooligosaccharidesα-Galactooligosaccharides Polysaccharides
PolyfructanesPectines d b b b bMucilagesetgommes d b b b bAmidonnonrésistantAmidonrésistant1(RS1)et2(RS2)Amidonrésistant3(RS3) b bHémicelluloses b b b b aCellulose a b a b aProtéinesProtéinesinsolublesaudétergentneutreounonenzymatiquementhydrolysables
c c
PolyphénolsTannins c cLignine c c aa:composéentièrementisolédefaçonsélectiveetdoséparcetteméthode—compound which is entirely isolated in a selective way and quantified by this method;b:composéentièrementisolédefaçonnonsélectiveetcomprisdansledosagedesfibresalimentaires(pourlesméthodedeVanSoest,deProskyetd’Uppsala),despolysaccharidesnonamylacés(pourlaméthodedespolysaccharidesnonamylacés)oudesfractionsdepectines-mucilages-gommes(pourlaméthodedeSelvendran)parcetteméthode—compound which is entirely isolated in a non selective way and quantified with the other dietary fibers (for the Van Soest, Prosky and Uppsala method), the other non starch polysaccharides (for the non-starch polysaccharides method) or the pectins-mucilages-gums fractions (for the Selvendran method) by this method;c:composépartiellementisolédefaçonnonsélectiveetcomprisdansledosagedesfibresalimentairesparcetteméthode—compound which is partially isolated in a non selective way and quantified with the other dietary fibers by this method;d:composénondoséparlaméthodedontlefractionnementsélectifestcependantréaliséetdontledosageestpossibleenadaptantlaméthode—compound which is not quantified by this method but it is isolated in a selective way and could be quantified by adapting this method;*pourdeséchantillonsdontlesteneursenamidonetenprotéinessontrespectivementinférieuresà10età30%(p:p)delamatièresèchedel’échantillon—for samples with a starch and a protein content which are respectively less than 10 and 30% (w:w) of the dry weight.1Proskyetal.,1985;2Selvendranetal.,1985;3VanSoestetal.,1985;4Proskyetal.,1992;5Englystetal.,1994;6Asp,1995;7Giger-Reverdin,1995;8Theanderetal.,1995;9Erikssonetal.,1997.

170 Biotechnol. Agron. Soc. Environ. 201115(1),165-182 GodinB.,AgneessensR.,GofflotS.etal.
ligninesolubleenmilieuacideetdetanninssolublesenmilieuacide.
LacelluloseetlesprotéinesinsolublesaudétergentacidesontextraitesentraitantlafractionADFauH2SO472%pendant3hà20-23°C.Lafractionextraiteestséparéedurésidu insolubleparfiltration.Laquantitédecellulosecorrespondàlapertedemasseinsolublede la fraction ADF (en corrigeant par la teneur enprotéines)durantcettehydrolyse.Lerésiduinsolubleest composéde lignine insoluble enmilieu acide,detanninsinsolublesenmilieuacideetdecendres.
Lalignineinsolubleenmilieuacideetlestanninsinsolubles en milieu acide (présents en très faiblequantité) sont volatilisés par calcination. Le résiduinsoluble obtenu après calcination est composé decendres.Lalignineinsolubleenmilieuacideaveclestanninsinsolublesenmilieuacidesontquantifiésparlapertedemassedurantlacalcination.
Afin de limiter l’incertitude sur la quantificationdes résidus insolubles, les valeurs gravimétriquesobtenues pour tous les résidus insolubles doiventêtre corrigées par la teneur en protéines et encendres. Les protéines sont quantifiées par unmicro-Kjeldahl ou une analyse élémentaire sur basede l’azote en appliquant un facteur de conversionN(gN)x6,25(gprotéines.gN
-1)(AOAC,1990).Lescendressontdoséesparcalcinationà525°C(VanSoestetal.,1985).Lesquantitésdecelluloseetd’hémicellulosespeuvent être respectivement surestimées et sous-estimées dans le cas d’une contamination des ADFpar des hémicelluloses. Cette contamination peutatteindre la valeur de 15,4% des polysaccharidesstructuraux desADF (Morrison, 1980; Jung, 1997).Les quantités de cellulose et d’hémicelluloses desbiomasses lignocellulosiques possédant une teneurimportante en protéines sont surestimées si la valeurrespectivementdesNDFetdesADFn’estpascorrigéeparleursteneursenprotéines(Jung,1997).Laquantitéd’hémicelluloses des biomasses lignocellulosiquespossédantune teneur importanteen lignine solubleàpH acide est surestimée car cette lignine soluble estcontenuedans lesNDF,maisellen’estpascontenuedanslesADF(Jung,1997).LesADFn’ayantpassubidetraitementaudétergentneutrecontiennentcertainespectinesquinesontpascontenuesdanslesADFayantsubiun traitementpréalable audétergentneutre.LeshémicellulosesdesADFn’ayantpassubidetraitementaudétergentneutresontsous-estiméessilabiomasselignocellulosique possède une teneur importante enpectines(Ampuero,2008).
Laméthode deVanSoest ne s’applique qu’à deséchantillonsdont les teneursenamidon,enprotéineset en lipides sont respectivement inférieures à 10,à 30 et à 10% (p:p) de la matière sèche. Les NDFsont surestimées lorsque l’échantillon contient tropd’amidonetdeprotéines.S’ilcontienttropdelipides,le
détergentneutreouacides’associepréférentiellementàlaphaselipidiqueetperddesonefficacité,donnantdesvaleursbiaiséespourlesNDFetlesADF(VanSoestetal.,1985;VanSoestetal.,1991;Giger-Reverdin,1995). Dans le cas d’un excès de lipides, il suffitd’extraire préalablement les lipides de l’échantillonpour pouvoir appliquer la méthode de Van Soest.Dans le cas d’un excès d’amidon et de protéines,des α-amylases et des protéases sont respectivementutilisées (Giger-Reverdin, 1995). Différentes autresvariantes de la méthode aux détergents neutre etacide de Van Soest ont été développées: ajoutd’α-amylases thermotolérantes au détergent neutrepour hydrolyser l’amidon (Mertens, 2002), ajout deprotéasespréalablementàl’attaqueaudétergentneutrepourhydrolyser lesprotéines (Dorleansetal.,1996),utilisationdequantitésplusfaiblesdedétergentneutrepour utiliser un moindre excès de détergent neutre(Chaietal.,1998).
2.2. Méthode des fibres alimentaires de Prosky
LaméthodedesfibresalimentairesdeProsky(Proskyetal.,1985;Proskyetal.,1992)(Figure 3,tableaux 1et 2) est basée sur des hydrolyses enzymatiquessuccessives de la biomasse lignocellulosique afind’extrairelesmoléculesquinefontpaspartiedesfibresalimentaires. Les fibres alimentaires sont quantifiéespargravimétrie.
LaméthodedeProsky(Proskyetal.,1985;Proskyetal.,1992;Asp,1995;Englystetal.,1996;McClearyetal.,2002;FAO,2007)commenceparuneextractiondeslipides,sil’échantillonencontientplusde10%(p:p).Cetteméthode permet d’extraire ou d’hydrolyser lesmono/di/oligo-saccharides,lesfructanes,lesgommes,lesmucilages,l’amidonnonrésistant(accessibleetnonrésistantàl’hydrolyseparl’α-amylasepancréatique),le RS1 (amidon inaccessible physiquement à l’α-amylasepancréatique),leRS2(amidonnongélatiniséet fortement résistant à l’hydrolyse par l’α-amylasepancréatique), les tannins solubles à pH neutre, lesprotéinesenzymatiquementhydrolysablesetcertainescendres à l’aide de, successivement, une α-amylasethermotoléranteà100°Cpendant30min,uneprotéaseà 60°C pendant 30min et une amyloglycosidase à60°Cpendant60min.Lesfibresalimentaires totalessont isolées par une précipitation à l’éthanol aqueux78% (v:v) suivie d’une filtration sur diatomite. Lesfibres alimentaires totales insolubles sont ensuiterincéesdeuxfoisàl’éthanolaqueux95%(v:v)etdeuxfoisàl’acétone.Lesétapesdeprécipitationàl’éthanol,derinçageàl’éthanoletàl’acétoneextraientleslipides.Les fibres alimentaires totales sont composées decellulose,d’hémicelluloses,duRS3(amidongélatiniséet fortement résistant à l’hydrolysepar l’α-amylase),despectines(saufcellesquisonttrèshydrosolubles),

Revuedesméthodesd’analysedespolysaccharidesstructuraux 171
desgommes(saufcellesquisonttrèshydrosolubles),desmucilages(saufceuxquisonttrèshydrosolubles),de la lignine,des tannins insolublesàpHneutre,desprotéines non enzymatiquement hydrolysables et decendres.
Afindelimiterl’incertitudesurlerésultatdesfibresalimentaires totales, chaque analyse est effectuéeendouble.Sur l’unedes répétitions, l’azote est dosépar un micro-Kjeldahl ou une analyse élémentairepourcorrigerlecontenuenprotéinesdanslafractioninsoluble, en appliquant un facteur de conversionN(gN)x6,25(gprotéines.gN
-1)(AOAC,1990).Surl’autrerépétition,lescendressontquantifiéesparcalcinationà 525°C dans la fraction insoluble. Le mêmeprotocole est appliqué en parallèle sur deux témoinsblancs (exempt d’échantillon) pour déterminer lescontributionsgravimétriquesdesdifférentessolutionsutilisées(Proskyetal.,1985;Proskyetal.,1992).
DansuneautrevariantedelaméthodedeProsky,les fibres alimentaires totales sont séparées en fibresalimentaires insolubles et solubles dans l’eau à pHneutre. Au lieu de réaliser l’étape de précipitationà l’éthanol aqueux 78% (v:v), la solution estdirectementfiltréesurdiatomiteafinderécupérer lesfibresalimentairessolublesdanslefiltratetlesfibresalimentairesinsolublesdanslerésiduinsoluble(Proskyetal.,1992).Dansuneautrevariantedelaméthodede
Prosky, le tempsd’analyseestréduitendiminuant levolumetotaldefiltrationpar l’utilisationdevolumesderéactifsplusfaiblesetenn’ajustantpaslepHlorsdel’utilisationdelaprotéase(Leeetal.,1992).
2.3. Méthode de dosage des polysaccharides non amylacés (nsP) d’englyst
La méthode de dosage des polysaccharides nonamylacés d’Englyst et al. (1994) (Figure 4,tableaux 1 et 2) extrait séquentiellement par voieenzymatique et chimique différentesmolécules de labiomasse lignocellulosiquequinefontpaspartiedespolysaccharides non amylacés. Les polysaccharidesnon amylacés sont quantifiés par chromatographie etavecousansspectrophotométrieaprèshydrolyseacide.
La méthode des polysaccharides non amylacés(Englystetal.,1994;Asp,1995;Englystetal.,1996)commenceparuneextractiondeslipides,sil’échantillonen contient plus de 10% (p:p). Cette méthodepermet d’extraire ou d’hydrolyser lesmono/di/oligo-saccharides, les fructanes, l’amidon non résistant, leRS1,leRS2,leRS3,lestanninssolublesàpHneutre,lesprotéinesenzymatiquementhydrolysablesetcertainescendresàl’aide,successivement,dudiméthylsulfoxide(DMSO) pendant 30min à 100°C, une α-amylasethermotolérante à 100°C pendant 10min et un
Figure 3.SchémadufractionnementetdescomposésdosésdanslaméthodedeProsky—Diagram of the splitting up and the quantified compounds in the Prosky method.
*composénonquantifié—not quantified component.
Filtration
Fraction insolubleInsoluble fraction
Fraction solubleSoluble fraction
Méthode de dosage analytiqueAnalytical quantifying method

172 Biotechnol. Agron. Soc. Environ. 201115(1),165-182 GodinB.,AgneessensR.,GofflotS.etal.
mélange de pancréatine et de pullulanase pendant30min à 50°C. Les polysaccharides non amylacéstotaux sont isolés par une précipitation à l’éthanolaqueux80%(v:v)àpH2suivied’unecentrifugation.Les polysaccharides non amylacés totaux insolublessont ensuite rincés une fois à l’éthanol aqueux85%(v:v)acidifié,unefoisàl’éthanolaqueux99,5%(v:v)et une fois à l’acétone.Les étapes de précipitation àl’éthanol,derinçageàl’éthanoletàl’acétoneextraientles lipides.Lespolysaccharidesnon amylacés totauxsont composés de cellulose, d’hémicelluloses, despectines(saufcellesquisonttrèshydrosolubles),desgommes(saufcellesquisonttrèshydrosolubles),desmucilages (sauf ceux qui sont très hydrosolubles),de la lignine,des tannins insolublesàpHneutre,des
protéines non enzymatiquement hydrolysables et decendres.
Les polysaccharides non amylacés obtenus sonthydrolysés en monosaccharides à l’aide d’H2SO412M pendant 30min à 35°C. De l’eau est ensuiteajoutée afin de diluer le H2SO4 jusqu’à 2M etde poursuivre l’hydrolyse des polysaccharidesnon amylacés en monosaccharides pendant 1h à100°C. Les monosaccharides sont quantifiés soitpar HPLC, soit par GC pour les monosaccharidesneutres en les dérivatisant en acétate d’alditol et parspectrophotométrie pour les monosaccharides acides(acides uroniques) en les transformant en acide5-formyl-2-furoïque.Cetacideestdoséaprèsréactionavec du 3,5-diméthylphénol en faisant la différence
Figure 4. Schémadufractionnementetdescomposésdosésdanslaméthodedespolysaccharidesnonamylacés—Diagram of the splitting up and the quantified compounds in the non-starch polysaccharides method.
*composénonquantifié—not quantified component.
Fraction insolubleInsoluble fraction
Centrifugation
Fraction solubleSoluble fraction
Méthode de dosage analytiqueAnalytical quantifying method

Revuedesméthodesd’analysedespolysaccharidesstructuraux 173
entrel’absorbanceà450nmetà400nm.Lasommedetouslesmonosaccharidesdéterminésaprèshydrolysecorrespondauxpolysaccharidesnonamylacés(Englystetal.,1994).
Lacellulosepeutaussiêtrequantifiéeeneffectuantle même protocole en double.Afin d’hydrolyser lespolysaccharidesnoncellulosiques,pourl’unedesdeuxanalyses, le culot correspondant aux polysaccharidesnon amylacés totaux est soumis à une étaped’hydrolyseauH2SO42Mpendant60minà100°C.Le glucose libéré lors de cette étape est analysé parchromatographie gazeuse ou liquide. La quantitéde cellulose correspond à la différence du glucoseentre l’échantillon «normal» (glucose venant despolysaccharidesnonamylacéstotaux)etl’échantillonayantsubiletraitementalternatif(glucosevenantdespolysaccharides non cellulosiques) (Englyst et al.,1994).
La dégradation des monosaccharides libéréspendant l’hydrolyse acide des polysaccharides enmonosaccharides analysés par GC, la dégradationdes monosaccharides libérés pendant l’hydrolyseacide des polysaccharides en monosaccharidesanalysésparHPLCet le rendementdedérivatisationdes monosaccharides neutres analysés par GC sontdéterminés enutilisant comme référenceunmélangede standards de monosaccharides dans du H2SO42M.Cemélange de standards est traité en parallèleavec les échantillons issus de l’étape de dilution duH2SO4concentré.Unstandardinterneestajoutéaprèsl’hydrolyse au H2SO4 dilué (Englyst et al., 1994).La quantification des polysaccharides non amylacésprend également en compte le facteur de conversiondes polysaccharides enmonosaccharides (gain d’unemolécule d’eau par l’hydrolyse), à savoir 89%pour les monosaccharides neutres et 91% pour lesmonosaccharidesacides(Englystetal.,1994).
Dans une variante de la méthode de dosage despolysaccharidesnonamylacés,lespolysaccharidesnonamylacés totauxsont séparésenpolysaccharidesnonamylacésinsolublesetsolublesdansl’eauàpHneutre.Aulieuderéaliserl’étapedeprécipitationàl’éthanolaqueux80%(v:v)àpH2,lasolutionestdirectementcentrifugéeafinderécupérer lespolysaccharidesnonamylacéssolublesdanslefiltratetlespolysaccharidesnon amylacés insolubles dans le résidu insoluble(Englystetal.,1994).
2.4. Méthode des fibres alimentaires d’uppsala
Laméthodedesfibresalimentairesd’Uppsala(Theanderet al., 1995) (Figure 5, tableaux 1 et 2) extraitséquentiellement par voie enzymatique et chimiquedifférentesmoléculesdelabiomasselignocellulosiquequinefontpaspartiedesfibresalimentaires.Lesfibresalimentaires sont quantifiées par chromatographie,
spectrophotométrie et gravimétrie après hydrolyseacide.
La méthode des fibres alimentaires d’Uppsala(Theander et al., 1995; Asp, 1995; Englyst et al.,1996;FAO,2007)commenceparuneextractiondeslipides,sil’échantillonencontientplusde5%(p:p).Cetteméthode permet d’extraire ou d’hydrolyser lesmono/di/oligo-saccharides, les fructanes, l’amidonnonrésistant,leRS1,leRS2,lestanninssolublesàpHneutreetcertainescendresàl’aidede,successivement,une α-amylase thermotolérante à 100°C pendant60minetuneamyloglycosidaseà60°Cpendant6h.Les fibres alimentaires totales sont isolées par uneprécipitation à l’éthanol aqueux 80% (v:v) suivied’une centrifugation. Les fibres alimentaires totalesinsolubles sont ensuite rincées une fois à l’éthanolaqueux99,5%(v:v)etdeuxfoisàl’acétone.Lesétapesdeprécipitationàl’éthanol,derinçageàl’éthanoletàl’acétoneextraient les lipides.Lesfibresalimentairestotalessontcomposéesdecellulose,d’hémicelluloses,du RS3, des pectines (sauf celles qui sont trèshydrosolubles),desgommes(saufcellesquisonttrèshydrosolubles),desmucilages(saufceuxquisonttrèshydrosolubles),delalignine,destanninsinsolublesàpHneutre,desprotéinesetdescendres.
Lesfibres alimentaires obtenues sont hydrolyséesen monosaccharides à l’aide d’H2SO4 12M pendant60minà30°C.Del’eauestensuiteajoutéeafindediluerleH2SO4jusqu’à0,47Metdepoursuivrel’hydrolysedes fibres alimentaires en monosaccharides pendant1hà125°C.Lasolutionestfiltréeafinderécupérerlesmonosaccharidesdanslefiltrat.Lesmonosaccharidesdu filtrat sont quantifiés par GC en les dérivatisanten acétate d’alditol, alors que les monosaccharidesuroniques (acides uroniques) sont quantifiés parspectrophotométrie selon le même protocole quedans la méthode des polysaccharides non amylacés.Le résidu insoluble de filtration permet de quantifierpar gravimétrie la lignine insoluble à pH acide avecles tannins insolubles à pH acide (présents en trèsfaiblequantité),quiformentensemblela«ligninedeKlason».LalignineinsolubleàpHacideetlestanninsinsolublesàpHacidesontvolatilisésparcalcination.Lerésiduinsolubleobtenuaprèscalcinationestcomposédecendres.Lalignineinsolubleenmilieuacideetlestannins insolublesenmilieuacidesontquantifiésparla perte demasse durant la calcination à 500°C.Lasommedetouslesmonosaccharidesdéterminésaprèshydrolyse et de la lignine deKlason correspond auxfibresalimentaires(Theanderetal.,1995).
La dégradation des monosaccharides libéréspendant l’hydrolyse acide des polysaccharides enmonosaccharidesanalysésparGCetlerendementdedérivatisation des monosaccharides neutres analysésparGCsontdéterminésenutilisantcommeréférenceunmélangedestandardsdesmonosaccharidesdansdu

174 Biotechnol. Agron. Soc. Environ. 201115(1),165-182 GodinB.,AgneessensR.,GofflotS.etal.
H2SO40,47M.Cemélangedestandardsest traitéenparallèleavecleséchantillonsissusdel’étapededilutionduH2SO4concentré.Unstandardinterneestajoutéàl’étaped’hydrolyseauH2SO4dilué.Laquantificationdespolysaccharidesnonamylacésprendégalementencompte le facteur de conversion des polysaccharidesenmonosaccharides (gain d’unemolécule d’eau parl’hydrolyse),àsavoir91%pourlesmonosaccharidesacides,88%pourlespentosesetlesdéoxyhexoseset90%pourlesautreshexoses(Theanderetal.,1995).Afin de limiter l’incertitude sur la quantificationde la lignine de Klason, la valeur gravimétrique durésidu insoluble obtenu après l’hydrolyse acide doitêtre corrigée par la teneur en protéines résiduellescontenuesdanscerésidu.Lesprotéinessontquantifiées
parunmicro-Kjeldahlouuneanalyseélémentairesurbasedel’azoteenappliquantunfacteurdeconversionN(gN)x6,25(gprotéines.gN)(Hatfieldetal.,1994).
2.5. Méthode alcaline de selvendran
La méthode alcaline de Selvendran et al. (1985)(Figure 6,tableaux 1et2)extrait séquentiellementparvoiechimiquedifférentesfractionsspécifiquesdela biomasse lignocellulosique. Les polysaccharidesstructuraux sont quantifiés par chromatographie etpar spectrophotométrie après hydrolyse acide. Cetteméthode se démarque de celles décrites ci-dessusdu fait qu’elle permet un fractionnement sélectifdes polysaccharides structuraux (plus de deux
Figure 5. Schémadufractionnementetdescomposésdosésdanslaméthoded’Uppsala—Diagram of the splitting up and the quantified compounds in the Uppsala method.
*composénonquantifié—not quantified component.
Fraction insolubleInsoluble fraction
Centrifugation
Fraction solubleSoluble fraction
Méthode de dosage analytiqueAnalytical quantifying method
Filtration Calcination
Fraction volatileVolatile fraction

Revuedesméthodesd’analysedespolysaccharidesstructuraux 175
polysaccharidesstructuraux)enfonctiondeleurnature.Lesprotéinesdel’échantillonsontd’abordéliminées
par un traitement au déoxycholate de sodium (SDC)aqueuxà1%(v:v)ouaudodécylsulphatedesodium
(SDS) aqueux à 1,5%, contenant 5mM deNa2S2O5(pourlimiterlaformationdeproduitsd’oxydationdespolyphénols)pendant5min.Lasolutionestfiltréeetcetteétapeestrépétéesurlerésiduinsoluble.Cerésidu
Figure 6. SchémadufractionnementetdescomposésdosésdanslaméthodedeSelvendran—Diagram of the splitting up and the quantified compounds in the Selvendran method.
*composénonquantifié—not quantified component; ** fractionnementinconnudelamolécule—unknown molecule splitting up.
Fraction insolubleInsoluble fraction
Centrifugation
Fraction solubleSoluble fraction
Méthode de dosage analytiqueAnalytical quantifying method
Filtration

176 Biotechnol. Agron. Soc. Environ. 201115(1),165-182 GodinB.,AgneessensR.,GofflotS.etal.
insolubleestbroyéàunetailleinférieureà3mmdansdu déoxycholate de sodium (SDC) aqueux à 0,5%(v:v)oududodécylsulphatedesodium(SDS)aqueuxà0,5%,contenant3mMdeNa2S2O5pendant5min.La solution est centrifugée et cette étape est répétéesur le résidu insoluble.Lesfiltrats et les surnageantsobtenusjusqu’àcetteétapecontiennentdesprotéines,despectinestrèshydrosolubles,certainescendres,descomposés intracellulaires comme les mono/di/oligo-saccharides.Lesprotéines résiduelles, les lipides, lespigments et une partie de l’amidon sont extraits durésiduinsolublepardeuxtraitementsavecunesolutionphénol/acide acétique/eau (2:1:1; p/v/v).La solutionestcentrifugéeetcetteétapeest répétéesur lerésiduinsoluble.L’amidonestextraitdurésiduinsolubleparduDMSOaqueuxà90%(v/v)à20°Cpendant16h.Le résidu insoluble est centrifugé et lavé six fois àl’eauafind’obtenir lescomposantspariétauxpurifiés(Selvendranetal.,1985).
Lespectinesetleshémicellulosesrichesenxylanessont extraites du résidu insoluble. Premièrement,ce dernier est traité au N,N,N’,N’-tétraacétatecyclohexanediamine (CDTA) de sodium 0,05M à21°C et à pH6,5 pendant 6h.LeCDTAde sodiumest un agent chélatant qui extrait principalement lessubstances pectiques de la lamellemitoyenne et qui,àunetempératurede20°C,estaussiefficacequedesagentschélatantsconventionnelsàhautetempérature.La solution est centrifugée et cette étape est répétéesurlerésiduinsoluble,maisladuréeàappliquerestde2h.Lesdeuxsurnageantscontenantdespectinesdelalamellemitoyennesontfiltrés,dialysésetconcentrés.Deuxièmement, le résidu insoluble est soumis auNa2CO3 0,05M et auNaBH4 20mM (pour protégerlespolysaccharidesdelaβ-élimination)pendant16hà 1°C. La solution est centrifugée et extraite unedeuxièmefoisauNa2CO30,05MetauNaBH420mMpendant3hà1°C.Lerésiduinsolublecorrespondauxcomposantspariétauxpurifiésetdépectinés.Lesdeuxsurnageantscontenantdespectinesdelaparoiprimaireet d’hémicelluloses riches en xylanes sont filtrés.LepHdecesfiltratsestajustéàpH5parde l’acideacétique afin de précipiter les hémicelluloses richesen xylanes.Après centrifugation, les surnageants depectinessontdialysésetconcentrés(Selvendranetal.,1985;Southgate,1995).
Leshémicelluloses(xylanesrésiduels,xyloglucanes,β-glucanesetmannanes)etlacellulosesontisoléesdurésidu insolubledes composantspariétauxpurifiés etdépectinés.Dansunpremiertemps,lerésiduinsolubleest soumisà laKOH1MdésoxygénéeetauNaBH410mMpendant2hà1°C.Lasolutionestfiltrée.LerésiduinsolubleestextraitunedeuxièmefoisàlaKOH1MdésoxygénéeetauNaBH410mMpendant2hà21°Csousazoteouargonetcettesolutionestfiltrée.Le pH des deux filtrats contenant les hémicelluloses
extraitesestajustéàpH5pardel’acideacétiqueafinde précipiter les hémicelluloses riches en xylanes.Ces solutions sont centrifugées afin d’obtenir leshémicelluloses riches en xylanes dans le culot etles hémicelluloses riches en xyloglucanes dans lesurnageant. Le surnageant avec les hémicellulosesrichesenxyloglucanesestdialyséetconcentré.Dansundeuxième temps, le résidu insolubleobtenu aprèsdeux traitementà laKOH1Mest soumisà laKOH4MdésoxygénéeetauNaBH410mMpendant2hà21°C sous argon ou azote. La solution est filtrée etlerésiduinsolubleestsoumisunedeuxièmefoisàlaKOH4MdésoxygénéeetauNaBH410mMpendant2hà21°Csousazoteouargonavecdel’acideborique3-4%.Lesdeuxfiltratscontenantdeshémicellulosessont filtrés. Le pH des filtrats est ajusté à pH5par de l’acide acétique. Ces nouveaux filtrats sontdialysésetconcentrés.Lepremierfiltratestcomposéd’hémicelluloses riches en xyloglucanes, alors quele deuxième filtrat est constitué d’hémicellulosesriches en mannanes. Le résidu insoluble obtenu estessentiellementcomposédecellulose,delignineetdecendres(Selvendranetal.,1985;Southgate,1995).
Aprèslesétapesdedépectinationdel’échantillon,ilpeutêtredélignifiépourrendreleréseaudecelluloseetd’hémicellulosesplusaccessibleàl’extractionalcaline(Genestie,2006).Ladélignificationalieuà70°CparduNaClO2etdel’acideminéralouorganiquepouruneduréeentre1et4hselonledegrédelignificationdel’échantillon.Cetteétapededélignificationaplusieurseffets sur l’échantillon analysé. Premièrement, ellen’estquepartielle.Elleestde60%sicetteétapedure2h. Deuxièmement, elle extrait de faibles quantités,aux environs 5 à 10%, des polysaccharides del’échantillon.Troisièmement,elleextraitpartiellementlesprotéinespariétales(Selvendranetal.,1985).
Lespolysaccharidesdetouteslesfractionsobtenuesetneutraliséessontcaractérisésetquantifiésensuivantle même protocole que dans la méthode d’Uppsala,c’est-à-dire à partir de l’hydrolyse acide au H2SO412M(Selvendranetal.,1985;Erikssonetal.,1997).
2.6. Méthodes sélectives d’un ou de deux polysaccharides structuraux des biomasses lignocellulosiques
Denombreusesméthodesd’analysesélectived’unoude deux polysaccharides structuraux des biomasseslignocellulosiquesexistentdanslalittérature.Certainesde ces méthodes d’analyse sont décrites ci-dessouspour lacellulose (Sunet al.,1995), lesxyloglucanes(Cutillas-Iturraldeetal.,1998),lesxylanes(Izydorczyketal.,1998a;Izydorczyketal.,1998b;Genestie,2006),lesβ-glucanes(Izydorczyketal.,1998a; Izydorczyketal.,1998b),lesmannanes(Selvendranetal.,1985)et

Revuedesméthodesd’analysedespolysaccharidesstructuraux 177
lespectines (Selvendranet al.,1985;LeGoffet al.,2001).
cellulose.LaméthodedeSunetal.(1995)isoleàpartirde paille de froment la cellulose en plusieurs étapesd’extractionchimique.Deuxdesétapesintermédiairespermettent d’isoler une fraction riche en pectines etunefractionricheenhémicelluloses.
Les composés intracellulaires, les hydrates decarbone non structuraux et une partie de lignine,d’hémicelluloses, des pectines et des protéines sontextraitsparuneétapedeprétraitementdel’échantillonàlaNaOH1,5%pendant144hà20°C.Lespectinesrésiduelles dans l’échantillon sont extraites par unagent chélatant, l’oxalate d’ammonium à 0,25%pendant4hà85°C.Laligninerésiduelleestéliminéepar une extraction au NaClO2 et à l’acide acétique(pHentre4,2et4,7)pendant24hà75°C(Sunetal.,1995). D’autres agents de délignification existent,commel’acideperacétiquequiestsélectifdelalignine(Teixeiraetal.,1999),leH2O2enmilieualcalin(Fangetal.,1999)et leKMnO4utiliséparVanSoestetal.(1985).LeKMnO4estunagentd’oxydationplusfortque leNaClO2 (Collingsetal.,1978).Afind’obtenirun résidu insoluble de cellulose purifiée contenantencore des cendres, les hémicelluloses résiduellessontextraitesàlaKOH24%etàl’acideborique2%pendant2hetà20°C(Sunetal.,1995).
xylanes et xyloglucanes. Lesméthodes deCutillas-Iturraldeetal. (1998)etdeGenestie (2006)purifientàpartir,respectivement,desparoisdekakietdessonsde céréale, les xylanes et les xyloglucanes par voiechimiqueetchromatographique.
L’échantillon est soumis à la délignification auNaClO240%etauH2SO40,3%pendant2hà70°C.LesxylanesetlesxyloglucanesduculotobtenusaprèscentrifugationsontextraitsàlaKOH1M,auNaBH420mMetsousatmosphèred’azotependant2hà60°C.Cettesuspensionestcentrifugéeafinderécupérer lesxylanes dans le surnageant et les xyloglucanes dansle culot. Les xyloglucanes du culot sont extraits parquatretraitementsàlaKOH4MetauNaBH420mMsous atmosphère d’azote pendant 24h à 20°C. Lasuspension résultante est centrifugée. Les quatresurnageants sont combinés pour obtenir la fractiondes xyloglucanes. Les fractions de xylanes et dexyloglucanessontpurifiéesparl’hydrolysedel’amidonetdesprotéinesparrespectivementdesglycohydrolases(α-amylase et pullulanase) et des protéases. Lesenzymessontdésactivéesetlasolutionestdialyséeetcentrifugéepourrécupérerlesurnageant.Lesxylanesoulesxyloglucanessontséparésdespectinesparunechromatographie préparative d’échange d’anions surune colonne de DEAE-Trisacryl (Cutillas-Iturraldeetal.,1998;Genestie,2006).
β-glucanes et xylanes. La méthode d’Izydorczyketal. (1998a et 1998b) extrait par voie enzymatiqueà partir de grains d’orge une fraction de β-glucaneset de xylanes. Les β-glucanes sont ensuite séparésdes xylanes par une précipitation sélective au(NH4)2SO4.
L’échantillon est traité dans de l’eau à 40°Cpendant30min.Ilestsoumisàdeuxcentrifugations.
Premièrement, lesprotéinesdumélangedesdeuxsurnageants contenant les β-glucanes et les xylaneshydrosolublessontdénaturéesenchauffantlasolutionà 95°C.L’élimination des protéines dumélange desdeuxsurnageantsesteffectuéeparfiltrationsurde ladiatomitesuivieparadsorptiondesprotéinesdufiltratsur de l’argile. Un nouveau surnageant est obtenupar centrifugation de la suspension. Ce surnageantest soumis à l’hydrolyse de l’amidon par de l’α-amylase pancréatique.Afin d’obtenir les β-glucanesetlesxylaneshydrosolublesàpurifier,lasolutionestdialyséepourenéliminerleshydratesdecarbonenonstructuraux, chauffée à 95°C pour en désactiver lesenzymesajoutéesetcentrifugées.
Deuxièmement, lesβ-glucaneset lesxylanesnonhydrosolublescontenusdansleculotinitialsontextraitsparvoiealcalineparunesolutionsaturéeenBa(OH)2contenant 1% (p/v) de NaBH4. Les β-glucanes etles xylanes sont séparés du résidu insoluble pardeux centrifugations. Les deux surnageants sontcombinésetneutraliséspardel’acideacétique.Cettesolution contenant les β-glucanes et les xylanes nonhydrosolubles est soumise à l’hydrolyse de l’amidonet des protéines par respectivement de l’α-amylasepancréatique et de la pronase. Afin d’obtenir lesβ-glucaneset lesxylanesnonhydrosolublespurifiés,cettesolutionestdialyséepourenéliminerleshydratesdecarbonenonstructurauxetlesprotéineshydrolysées,chauffée à 95°C pour en désactiver les enzymes etcentrifugée.
Les extraits de β-glucanes et de xylanes hydro-solubles et non hydrosolubles isolés et à purifiersubissent ensuite le même processus. Les extraitssecs sont dissous dans un tampon phosphate à pH7et du (NH4)2SO4 est lentement ajouté jusqu’à 28%de saturation. Après avoir laissé cette solution agirpendantunenuità5°C,elleestcentrifugéeafind’isolerleprécipité.Le(NH4)2SO4résidueldusurnageantestdialysé. Une fois le surnageant lyophilisé, le mêmeprocédé est répété mais en atteignant à chaque foisunpourcentage supérieurauprécédenten (NH4)2SO4et cela, jusqu’à saturation. Si le pourcentage de(NH4)2SO4 par rapport à la saturation est inférieurà 45%, alors seuls les β-glucanes sont précipités.Au-delà de 45%, les xylanes précipitent égalementet cela augmente avec le pourcentage de (NH4)2SO4par rapportà lasaturation(Izydorczyketal.,1998a;Izydorczyketal.,1998b).

178 Biotechnol. Agron. Soc. Environ. 201115(1),165-182 GodinB.,AgneessensR.,GofflotS.etal.
Mannanes. Pour des extraits d’hémicelluloseshydrosolublesoudeshémicellulosesextraitesparvoiealcalinevenantdeboisdegymnospermesetpurifiésdespectinesparunechromatographiepréparatived’échanged’anions, les mannanes peuvent être précipitéssélectivement par rapport aux autres polysaccharidesstructurauxparvoiechimique.
Lesmannanessontextraitsdansl’eauouàlaNaOHdiluée.Laprécipitationdesmannanesestensuiteréaliséepar du Ba(OH)2 0,03M. Le précipité est récupéréparcentrifugationet estpurifiépar lamêmeétapedeprécipitation que celle décrite ci-dessus. Le nouveauprécipité de mannanes obtenus par centrifugation estextraitàl’acideacétique2Metreprécipitéàl’éthanol(Selvendranetal.,1985).
Pectines.LaméthodedeLeGoffetal.(2001)fractionneles pectines de gousse de pois en trois catégories:solublesenmilieualcalin,solublesenmilieuacideetsolublesenmilieuneutre.
Les pectines sont déestérifiées en milieu alcalinparuntraitementàlaNaOH1Mpendant2hà4°C.Ce traitement permet également d’extraire la fractiondes substances, dont les pectines, solubles en milieualcalin.Afin de faire descendre le pH à 5, de l’acideacétiqueglacialestajoutéàlasolution.Lasuspensionrésultanteestensuitefiltréeafind’obtenirlafractiondessubstances,dontlespectines,extraitesenmilieualcalindans le filtrat. La fraction des substances, dont lespectines,solublesenmilieuacide,estextraitedurésiduinsolubleauHCl1Mpendant24hà80°C.Lasuspensionrésultanteestensuitecentrifugéeafind’obtenirdanslesurnageantlafractiondessubstances,dontlespectines,solubles enmilieu acide. La fraction des substances,dontlespectines,solublesenmilieuneutre,estextraitedurésiduinsolubleenramenantlepHà7.Cettefractionest centrifugée afin d’isoler les substances, dont lespectines,solublesenmilieuneutredanslesurnageant.Lestroisfractionsdepectinesextraitessontpurifiéesdesautressubstancesextraitescommelespolysaccharidesneutresparunechromatographiepréparatived’échanged’anionssurunecolonnedeDEAE-SepharoseCL-6B(LeGoffetal.,2001).
Lespolysaccharidesacides(lespectinesacidesetlesglucuronoxylanes) peuvent être précipités par rapportauxpolysaccharides neutres par une concentration enBa(OH)2de0,15M(Selvendranetal.,1985).
3. coMPaRaIson des Méthodes globales d’analyse des PolysacchaRIdes stRuctuRaux des bIoMasses lIgnocellulosIques
Les méthodes d’analyse des polysaccharidesstructuraux des biomasses lignocellulosiques décrites
ci-dessus sont basées sur un ou des objectifs précis àatteindre,qui sontpropresàchaqueméthode.Chaqueméthodeestfondéesuruneouplusieurshypothèsesetnégligecertainsfacteursquin’ontpasd’intérêtdansledomained’applicationpourlequelelleaétéinitialementoptimisée.
Les méthodes globales comme celles de VanSoest, de Prosky, des polysaccharides non amylacéset d’Uppsala permettent la quantification de fractionsinsolubles.Cesfractionsinsolublespeuventcontenirunou plusieurs types de polysaccharides structuraux desbiomasseslignocellulosiquesdontlanatureestconnue.Cesméthodesglobalesnerenseignentpassurlaquantitérelatived’untypedepolysaccharidestructuralparrapportauxautrestypesdepolysaccharidesstructurauxcontenusdanslamêmefractiondesbiomasseslignocellulosiques.Lesméthodesquantifiantlespolysaccharidesstructurauxdes biomasses lignocellulosiques par gravimétrie(méthodes de Van Soest et de Prosky) (tableau 1)ne donnent aucune information sur la composition enmonosaccharidesdespolysaccharidesstructuraux.CesdeuxméthodesselimitentsimplementàdéterminerlaquantitédefibresalimentairesenyintégrantlalignineetlestanninsinsolublesàpHneutre.Lesfractionsisoléescontiennent également des protéines insolubles audétergentneutre,desprotéines insolublesaudétergentacide ou non enzymatiquement hydrolysables, despertes de diatomite (dans la méthode de Prosky) etdes cendres (Prosky et al., 1985; Van Soest et al.,1985;VanSoestetal.,1991;Proskyetal.,1992),quel’analyseprendencomptepardesdosagesséparés.Pourles méthodes de quantification chromatographique etspectrophotométrique(méthodedespolysaccharidesnonamylacés,d’UppsalaetdeSelvendran)(tableau 1),lesvaleurs obtenues pour les polysaccharides structurauxdes biomasses lignocellulosiques sont corrigées parles facteurs de dégradation des monosaccharideslibérés pendant l’hydrolyse acide des polysaccharidesenmonosaccharides et les facteurs de conversion despolysaccharides en monosaccharides (Englyst et al.,1994;Theanderetal.,1995).
En plus de donner la nature desmonosaccharidesconstituantlespolysaccharidesstructurauxparchroma-tographie et spectrophotométrie, la méthode deSelvendranfractionnesélectivementlespolysaccharidesstructuraux des biomasses lignocellulosiques enfonction de leur nature, c’est-à-dire les pectines de lalamellemitoyenne,lespectinesdelaparoiprimaire,lesxylanes,lesxyloglucanes,lesmannanesetlacellulose(Selvendranetal.,1985).LesméthodesdeVanSoestetdespolysaccharidesnonamylacéspermettentégalementdequantifierlacellulose(VanSoestetal.,1991;Englystetal.,1994).
Selon la méthode globale choisie, les poly-saccharidesstructuraux isoléssontdifférentspourunebiomasselignocellulosiquedonnée(tableau 2).

Revuedesméthodesd’analysedespolysaccharidesstructuraux 179
LaméthodedeVanSoestneprendpasencompteles pectines, les gommes et les mucilages dans lesfibresalimentaires,saufsilesADFn’ontpassubidetraitementaudétergentneutre.Danscecas,certainespectines sont comprises dans le dosage des ADF(Van Soest et al., 1991;Ampuero, 2008). Dans lecasdesméthodesdeProsky,despolysaccharidesnonamylacés et d’Uppsala, les pectines, les gommes etlesmucilagesfontpartiedesfibresalimentaires.Lesfibres alimentaires peuvent être séparées en fibressolublesetfibresinsolublesdansl’eau,maissansquelespolysaccharidesconstituantchaquefractionsoientisoléslesunsdesautres.Lespectines,lesmucilageset les gommes très hydrosolubles ne sont pasprécipitésparl’éthanoletnefontdoncpaspartiedesfibresalimentaires(Asp,1995).DanslaméthodedeSelvendran,lespectinessontextraitessélectivementen pectines de la lamellemitoyenne et en pectinesde la paroi primaire. Lesmucilages et les gommeshydrosolublesontunesolubilitédansl’eausimilaireaux pectines. Si cesmucilages et ces gommes sontprésentsdansunéchantillon,alorsilsseretrouverontfort probablement dans les fractions pectiques.Les mucilages et les gommes neutres peuvent êtreséparésdespectinesparchromatographied’échanged’anions. S’ils sont acides, leur séparation parrapportauxpectinesdépenddeleurteneurenacidesuroniques.
Dans la méthode des polysaccharides nonamylacés, la précipitation des fibres alimentaires àl’éthanol acidifié à pH2 conserve dans la fractionextraite les mono/di/oligo-saccharides et l’acidegalacturonique libre. Cela n’est pas le cas pour laprécipitationàl’éthanoldeProskyetd’UppsalaoùlepHestsupérieurà2(Englystetal.,1994).
L’amidon, polysaccharide non structural, estconsidérédedifférentesmanièresselonlesméthodes.LesméthodesdeVanSoest,despolysaccharidesnonamylacésetdeSelvendranéliminent tout l’amidon,alorsquecellesdeProskyetd’Uppsalahydrolysentl’amidonnonrésistant,RS1etRS2maispasleRS3quiestdonccomprisdanslesfibresalimentaires(VanSoestetal.,1985;VanSoestetal.,1991;Asp,1995;Englystetal.,1996;FAO,2007).
Lesméthodesglobalespeuventêtrerésuméesdelamanièresuivante:– LaméthodedeVanSoest(Figure 2)neprendpasen comptelespectines,lesgommesetlesmucilages,ne donne pas d’information sur la nature d’hémi- celluloses contenuesdans lesNDFet fournit des donnéescomme laquantitédecellulose,d’hémi- cellulosesetdelignineavecdestanninsinsolublesà pH acide. Les fractions de NDF et d’ADF ne contiennent pas de polysaccharides non struc- turaux. Cette méthode est utilisée dans le cadre
delaquantificationdesfibresalimentairespourles ruminants, ainsi que pour les non-ruminants si l’alimentnecontientpastropd’amidon.Danslecas contraire, la variante de Mertens (2002) de la méthodedeVanSoestestemployée(voir lepoint 2.1.pourplusdedétails).– LaméthodedeProsky(Figure 3)extraitdesfractions quicontiennentdespolysaccharidesstructuraux,des polysaccharides non structuraux (mucilages, gommes et amidon RS3) et d’autres composés commedelalignine,destannins,desprotéineset descendres.Ellenedonneaucuneinformationsur lastructure(cellulose,hémicellulosesetpectines)et la composition en monosaccharides des poly- saccharidesstructurauxisolésdesfibresalimentaires. Laseuleinformationdisponibleparcetteméthodeest le caractère soluble ou insoluble des fibres alimentairesdansl’eauàpHneutre.Cetteméthode estutiliséedans ledomainedudosagedesfibres alimentairesenalimentationhumaine.– La méthode des polysaccharides non amylacés (Figure 4)extraitdesfractionsquicontiennentdes polysaccharides structuraux, dose la cellulose, quantifielesmonosaccharidesconstituantlespoly- saccharides structuraux isolés et sépare les poly- saccharidesnonamylacéssolublesetinsolublesdans l’eauàpHneutre.Ellenepermetpasdedifférencier leshémicelluloses,lespectines,lesgommesetles mucilagesdanslesfractionsextraites.Cetteméthode estemployéeafindedoserlespolysaccharidesnon amylacéssansambigüité.– Laméthoded’Uppsala(Figure 5)isoledesfractions forméesdesmêmesconstituantsquecelledeProsky. Laméthoded’Uppsalaquantifielesmonosaccharides constituant les polysaccharides isolés. Dans la fractioninsoluble,cetteméthodedoselaligninede Klason. Elle ne permet pas de différencier la cellulose, les hémicelluloses, les pectines, les gommesetlesmucilagesdanslesfractionsextraites. Cetteméthodeestutiliséedanslecontextedesfibres alimentairesenalimentationhumaine.– La méthode de Selvendran (Figure 6) extrait sélectivement les pectines avec les mucilages et les gommes, les xylanes, les xyloglucanes, lesmannaneset lacellulose.Lespolysaccharides neutres peuvent être séparés des polysaccharides acidesparunechromatographied’échanged’anions. Elle permet de quantifier les monosaccharides constituantlespolysaccharidesstructurauxisolés. Cetteméthodeextraitsélectivementlesprincipaux typesdepolysaccharidesstructurauxetencaractérise la composition. Du fait du nombre important d’étapesd’extraction,l’analysed’unéchantillonest de longue durée, mais apporte une quantité d’information importante concernant les poly- saccharidesstructuraux.

180 Biotechnol. Agron. Soc. Environ. 201115(1),165-182 GodinB.,AgneessensR.,GofflotS.etal.
Le classement des méthodes en ordre croissantdes données qu’elles apportent sur la naturedes polysaccharides structuraux des biomasseslignocellulosiques est le suivant: la méthode deProsky,d’Uppsala,despolysaccharidesnonamylacés,deVanSoestetdeSelvendran.
Leclassementdesméthodesenordrecroissantdutemps d’analyse pour l’analyse des polysaccharidesstructuraux des biomasses lignocellulosiques est lesuivant: laméthode deProsky, des polysaccharidesnon amylacés, d’Uppsala, de Van Soest et deSelvendran. La méthode de Van Soest présente ànotreavislemeilleurcompromisentrel’analysedespolysaccharides structuraux constituant la biomasselignocellulosiqueetletempsd’analysepourparvenirà obtenir cette information. Elle permet d’isoler enun minimum de temps des fractions dont trois quisont majoritairement composées respectivementdes pectines-mucilages-gommes, d’hémicelluloseset de cellulose. La fraction contenant les pectines-mucilages-gommes n’est pas quantifiée par cetteméthode. Les pectines de cette fraction pourraientêtre séparées des mucilages et des gommes neutrespar chromatographie d’échange d’anions. Si lesgommesetlesmucilagessontacides,leurséparationpar rapport aux pectines dépend de leur teneur enacides uroniques. L’acide galacturonique, un acideuronique constituant majoritaire des pectines,pourrait être quantifié dans la fraction de pectinespurifiéesenappliquantleprotocoledequantificationdes acides uroniques par HPLC de la méthode despolysaccharidesnonamylacés.
Pourapporterdesdonnées sur lacompositionenmonosaccharides des polysaccharides structurauxdes biomasses lignocellulosiques, la méthode deVanSoestpourraitêtremiseenœuvrepourpréparerlesdifférentesfractionsquipourraientensuiteêtrehydro-lyséesenvued’unequantificationdesmonosaccharidespar chromatographie analytique (comme dans laméthode des polysaccharides non amylacés etd’Uppsala).LaméthodedeVanSoestpourrait aussiapporterdesdonnéessurleshydratesdecarbonenonstructuraux (mono/di/oligo-saccharides, amidon etfructanes)desbiomasseslignocellulosiques,siceux-cisontisoléspréalablementàl’extractionaudétergentneutre.
Afin de réaliser cette extraction des hydratesde carbone non structuraux des biomasses ligno-cellulosiques, le protocole de Hoebregs (1997) oucelui deMcCleary (2004) pourraient être adaptés etappliqués. Ces méthodes permettent d’extraire lesmono/di/oligo-saccharides, les fructanes et l’amidondes biomasses lignocellulosiques à l’aide d’eau àpHneutreà85°Cpendant10minselonleprotocolede Hoebregs ou à 80°C pendant 15min selon leprotocoledeMcCleary.
4. conclusIon
Vu la diversité et la complexité des polysaccharidesstructuraux contenus dans les biomasses ligno-cellulosiques, ladéterminationdeleurstructureetdeleur composition enmonosaccharides est à effectuerpar une méthode éliminant les hydrates de carbonenon structuraux ainsi que la lignine et caractérisantsélectivement les polysaccharides structuraux dontles fractions isolées sont hydrolysées, puis analyséespar chromatographie analytique. Les fractionsextraites peuvent éventuellement être purifiées parchromatographiepréparatived’échanged’anionsafindeséparerlespolysaccharidesneutresdespolysaccharidesacides. Pour les échantillons fortement lignifiés, afinde rendre le réseau de cellulose et d’hémicellulosesplus accessible à l’extraction des polysaccharidesstructuraux, il est préférable d’intégrer une étape dedélignificationdanslaméthodedefractionnement.
LaméthodedeVanSoestestlaméthodeprésentantànotreavis lemeilleurcompromisentre lesdonnéesobtenuessurlanaturedespolysaccharidesstructurauxconstituantlesbiomasseslignocellulosiquesetletempsd’analyse pour parvenir à obtenir cette information.En adaptant cette méthode, elle pourrait contribuerà apporter des données sur la quantité de pectines(expriméeenacidegalacturonique),surlacompositionenmonosaccharidesdespolysaccharidesstructurauxetsurleshydratesdecarbonenonstructuraux.
Actuellement, il n’existe pas assez de donnéespertinentes dans la littérature comparant l’utilisationdes méthodes globales présentées sur un mêmeéchantillon ou sur des échantillons semblables debiomasselignocellulosique.
liste des abréviations
ADF:fibresinsolublesaudétergentacideAOAC:AssociationofOfficialAnalyticalChemistsCDTA:N,N,N’,N’-tétraacétatecyclohexanediamineDMSO:diméthylsulfoxideGC:chromatographiegazeuseHPLC:chromatographieliquideàhauteperformanceNDF:fibresinsolublesaudétergentneutreSDC:déoxycholatedesodiumSDS:dodécylsulphatedesodiumRS1:amidonrésistantdetype1RS2:amidonrésistantdetype2RS3:amidonrésistantdetype3
bibliographie
AmpueroS., 2008. Détermination de la teneur en fibres dans les aliments pour animaux à ALP.Posieux,France:AgroscopeLiebefeld-Posieux(ALP).

Revuedesméthodesd’analysedespolysaccharidesstructuraux 181
AOAC, 1990. Official methods of analysis. 15th ed.Washington, DC, USA: Association of OfficialAnalyticalChemist.
AspN.-G.,1995.Dietaryfibreanalysis.Anoverview.Eur. J. Clin. Nutr.,49(Suppl.3),S42-S47.
CarpitaN. & McCannM., 2000. The cell wall. In:BuchananB.,GruissemW.&JonesR.,eds.Biochemistry and molecular biology of plants. Rockville, USA:AmericanSocietyofPlantPhysiologists,52-108.
ChaiW. & UdénP., 1998. An alternative oven methodcombined with different strengths in the analysis ofneutral detergent fibre. Anim. Feed Sci. Technol., 74,281-288.
CollingsG.,YokoyamaM.&BergenW., 1978. Lignin asdetermined by oxidation with sodium chlorite and acomparisonwithpermanganatelignin.J. Dairy Sci.,61,1156-1160.
Cutillas-IturraldeA., PeñaM., ZarraI. & LorencesE.,1998. Xyloglucan from persimmon fruit cell walls.Phytochemistry,48(4),607-610.
DorleansM.,MandranN.&SauvantD.,1996.Studyoftheuseofaproteasewith theVanSoestprocedure.Anim. Feed Sci. Technol.,61,129-136.
EbringerováA., HromádkováZ. & HeinzeT., 2005.Hemicellulose.Adv. Polym. Sci.,186,1-67.
EnglystH.,QuigleyM.&HudsonG.,1994.Determinationof dietary fibre as non-starch polysaccharides withgas-liquid chromatographic, high-performance liquidchromatographicorspectrophotometricmeasurementofconstituentsugars.Analyst,119,1497-1509.
EnglystH. & HudsonG., 1996. The classification andmeasurement of dietary carbohydrates. Food Chem.,57(1),15-21.
ErikssonI.,AnderssonR.&AmanP., 1997.Extraction ofpectic substances form dehulled rapeseed.Carbohydr. Res.,301,177-185.
FangJ. et al., 1999.Comparative study of hemicellulosesfrom wheat straw by alkali and hydrogen peroxideextractions.Polym. Degrad. Stab.,66,423-432.
FAO, 2007. Directives concernant l’utilisation des allégations relatives à la nutrition : projet de tableau des conditions applicables à la teneur en éléments nutritifs (Partie B : Fibres alimentaires) à l’étape 6.Rome:FAO.
GenestieB., 2006. Optimisation de la production d’arabinoxylooligosaccharides d’intérêt biologique à partir de sons de céréales : approches méthodologiques.Thèsededoctorat:UniversitédeLimoges(France).
Giger-ReverdinS., 1995.Review of themainmethods ofcellwall estimation: interest and limits for ruminants.Anim. Feed Sci. Technol.,55,295-334.
GrabberJ.,2005.Howdolignincomposition,structure,andcross-linking affect degradibility?Crop Sci., 45, 820-831.
HatfieldR. et al., 1994. A comparison of the insolubleresiduesproducedbytheKlasonligninandaciddetergentligninprocedures.J. Sci. Food Agric.,65,51-58.
HoebregsH., 1997. Fructans in foods and food products,ion-exchange chromatographic method: collaborativestudy.AOAC Int. J.,80(5),1029-1037.
IzydorczykM.,MacriL.&MacGregorA.,1998a.Structureand physiochemical properties of barley non-starchpolysaccharides - I. Water-extractable β-glucans andarabinoxylans.Carbohydr. Polym.,35,249-258.
IzydorczykM.,MacriL.&MacGregorA.,1998b.Structureand physiochemical properties of barley non-starchpolysaccharides - II. Alkali-extractable β-glucans andarabinoxylans.Carbohydr. Polym.,35,259-269.
JungH.-J.,1997.Analysisofforagefiberandcellwallsinruminantnutrition.J. Nutr.,127(5),810S-813S.
Le GoffA., RenardC., BonninE. & ThibaultJ.-F., 2001.Extraction, purification and chemical characterisationxylogalacturonans from pea hulls.Carbohydr. Polym.,45,325-334.
LeeS., ProskyL. & De VriesJ., 1992. Determination oftotal, soluble, and insolubles dietary fiber in foods-enzymatic-gravimetric method, MES-TRIS Buffer:collaborativestudy.AOAC Int. J.,75(3),395-416.
McClearyB. & MonaghanD., 2002. Measurement ofresistantstarch.AOAC Int. J.,85(2),665-675.
McClearyB. & RossiterP., 2004. Measurement of noveldietaryfibers.AOAC Int. J.,87(4),707-717.
MertensD., 2002. Gravimetric determination of amylase-treatedneutraldetergentfiberinfeedswithrefluxinginbeakersorcrucibles:collaborativestudy.AOAC Int. J.,85(6),1217-1240.
MorrisonI., 1980. Hemicellulosic contamination of aciddetergent residues and their replacement by celluloseresidues in cellwall analysis. J. Sci. Food Agric.,31,639-645.
ProskyL.etal.,1985.Determinationoftotaldietaryfiberinfoodsandfoodproducts:collaborativestudy.AOAC J.,68(4),677-679.
ProskyL. et al.., 1992. Determination of insoluble andsoluble dietary fiber in foods and food products:collaborativestudy.AOAC Int. J.,75(2),360-367.
SelvendranR.&O’NeillM.,1985.Isolationandanalysisofcellwallsfromplantmaterial.In:GlickD.,ed.Methods of biochemical analysis. Vol. 32. New York, USA:Wiley,25-153.
SouthgateD.,1995.Dietary fibre analysis.Cambridge,UK:TheRoyalSocietyofChemistry.
SunR.,LawtherM.&BanksW.,1995.Influenceofalkalinepre-treatments on the cell wall components of wheatstraw.Ind. Crops Prod.,4,127-145.
TeixeiraL.,LindenJ.&SchroederA., 1999.Alkaline andperacetic acid pretreatments of biomass for ethanolproduction. Appl. Biochem. Biotechnol., 77-79, 19-34.
TheanderO.etal.,1995.Totaldietaryfiberdeterminedasneutral sugar residuesuronicacid residues,andklasonlignin(theUppsalamethod):collaborativestudy.AOAC Int. J.,78(4),1030-1044.

182 Biotechnol. Agron. Soc. Environ. 201115(1),165-182 GodinB.,AgneessensR.,GofflotS.etal.
Van SoestP. & RobertsonJ., 1985. Analysis of forages and fibrous foods.Ithaca,USA:DepartmentofAnimalScience,CornellUniversity.
Van SoestP., RobertsonJ. & LewisB., 1991. Methodsfor dietary fiber, detergent fiber, and non-starchpolysaccharidesinrelationtoanimalnutrition.J. Dairy Sci.,74,3583-3597.
ZhangY.-H., 2008. Reviving the carbohydrate economyvia multi-product lignocellulose biorefineries. J. Ind. Microbiol. Biotechnol.,35,367-375.
(38réf.)