rapport annuel 2012 - edqm
TRANSCRIPT

Cop
yrig
ht ©
201
3 D
EQ
M, C
onse
il de
l’E
urop
e. T
ous
droi
ts ré
serv
és. D
RPD
-13-
07. P
hoto
grap
hies
: C
andi
ce Im
bert
- Sh
utte
rsto
ck -
Foto
lia
RAPPORT ANNUEL 2012
Prem
s 579
13

RAPPORT ANNUEL 2012
Cop
yrig
ht ©
201
3 D
EQ
M, C
onse
il de
l’E
urop
e. T
ous
droi
ts ré
serv
és. D
RPD
-13-
07. P
hoto
grap
hies
: C
andi
ce Im
bert
- Sh
utte
rsto
ck -
Foto
lia

Comme chaque année, la publication du rapport annuel est l’occasion de nous retourner vers les 12 mois écoulés pour tirer le bilan de nos activités et réfléchir à tout ce qui s’est passé et tout ce que nous avons accompli. En 2012, la poursuite de la consolidation des normes et références qui constituent la Pharmacopée Européenne (Ph. Eur.) – textes de pharmacopée et étalons de référence associés – est restée l’un des objectifs majeurs de la DEQM.
La procédure P4 – une procédure dédiée aux substances encore sous brevet, où les monographies sont élaborées en étroite collaboration avec les autorités réglementaires et les innovateurs concernés – a encore fait la preuve de son utilité. Depuis ses débuts, elle a permis d’adopter plus de 60 monographies, couvrant des substances définies chimiquement ou des substances biologiques, dont 5 finalisées au cours de l’année 2012. La Commission européenne de Pharmacopée a poursuivi les travaux qu’elle a engagés pour faciliter la mise en œuvre d’une nouvelle philosophie de la qualité dans le développement et la production de médicaments, en adoptant un chapitre général, d’application non obligatoire, sur la démonstration de l’uniformité des préparations unidoses sur des échantillons de grande taille (2.9.47). Elle a également identifié le besoin d’examiner et tester la faisabilité de monographies portant sur des produits finis spécifiques et a engagé, à cet effet, un projet pilote d’élaboration de « monographies de produits finis » par un groupe de travail spécifique, exclusivement composé de représentants des autorités compétentes. Ce groupe doit travailler sur un double concept : d’une part un produit multi-sources, d’autre part, en étroite collaboration avec l’innovateur, un produit encore couvert par la propriété industrielle. Les résultats de ces travaux feront l’objet d’une évaluation en 2014. Dans le domaine des vaccins vétérinaires, la Commission a adopté la révision de quelque 80 monographies de vaccins, mises en conformité avec les dispositions de deux guidelines VICH récemment adoptés. Enfin, les efforts accomplis en matière d’essais sur animaux en faveur des 3R (Replacement, Reduction, Refinement), tant par la Commission que dans le cadre du Programme de Standardisation Biologique, ont permis de réaliser de nouveaux progrès, largement appréciés et reconnus par l’ensemble des acteurs concernés.
Au niveau international, la coopération de la Ph. Eur. avec les pharmacopées du Japon et des États-Unis s’est poursuivie dans le cadre du Groupe de Discussion des Pharmacopées (GDP), initiative informelle qui vise à l’harmonisation internationale des exigences de pharmacopée. La Ph. Eur. a aussi apporté un important soutien à une initiative de l’Organisation Mondiale de la Santé (OMS) en faveur d’une harmonisation des pharmacopées à l’échelle mondiale. Dans ce contexte, la décision a été prise par une assemblée mondiale des pharmacopées d’adopter des politiques et procédures harmonisées en matière, par exemple, d’élaboration de monographies, d’interaction
2 ◗ Avant-propos
5 ◗ LA DEQM en bref: valeurs, objectifs, activités
7 ◗ 1. LES ACTIVITÉS FONDAMENTALES
7 ◗ 1.1 La Pharmacopée Européenne (Ph. Eur.)
13 ◗ 1.2 Etalons de référence
15 ◗ 1.3 Activités du laboratoire
16 ◗ 1.4 Certification de conformité aux monographies de la Pharmacopée Européenne
18 ◗ 1.5 Réseau des OMCL
25 ◗ 1.6 Transfusion sanguine et transplantation d'organes
27 ◗ 1.7 Activités liées au suivi pharmaceutique et à la lutte contre la contrefaçon
30 ◗ 1.8 Cosmétiques, emballages alimentaires et pharmaceutiques
32 ◗ 2. LES ACTIVITÉS SUPPORT
32 ◗ 2.1 Système de management de la qualité
32 ◗ 2.2 Activités liées aux technologies de l'information et aux publications
35 ◗ 2.3 Communication, événements marquants
40 ◗ 2.4 Collaboration internationale
42 ◗ Liste des comités de la EDQM
Avant-propos

avec les acteurs concernés et de collaboration entre pharmacopées. Le Secrétariat de la Commission européenne de Pharmacopée s’est porté volontaire pour participer à la compilation de ce document, actuellement connu sous le titre « bonnes pratiques de pharmacopée », afin que les différentes pharmacopées européennes parlent d’une seule voix.
La « procédure de certification de conformité aux monographies de la Pharmacopée Européenne » a célébré le 20ème anniversaire d’une activité qui fut au départ engagée, en 1992, comme une phase pilote de deux ans. L’industrie et les autorités compétentes de plusieurs continents ont, à cette occasion, exprimé l’intérêt qu’ils portent à la procédure, en soulignant la valeur ajoutée et l’aide qu’elle apporte aux autorités compétentes pour faire le meilleur usage des ressources dont elles disposent, dans le cadre de la procédure d’autorisation de mise sur le marché. Les résultats des inspections BPF réalisées en 2012 sur des sites de fabrication de substances actives couvertes par des certificats de conformité (CEP) témoignent de l’utilité des inspections sur site et, une fois encore, de la pertinence des critères définis au niveau européen pour la sélection des sites, selon une approche d’évaluation du risque. Ils ont également montré la nécessité de contrôles de suivi plus fréquents pour suivre sur la durée la conformité aux BPF.
Le Réseau européen des Laboratoires officiels de contrôle des médicaments, outre ses activités « de routine » telles que la maintenance du système qualité commun, le programme d’essais d’aptitude (PTS pour proficiency testing scheme), la surveillance du marché et la libération officielle des lots par les autorités de contrôle, apporte nombre de contributions importantes dans d’autres domaines tels que la lutte contre les médicaments et substances actives contrefaits/illégaux, le contrôle des préparations pharmaceutiques non soumises à autorisation ou les produits de thérapie génique.
Dans le domaine de la transfusion sanguine, la préparation de la 17ème édition du « Guide pour la préparation, l’utilisation et l’assurance de qualité des composants sanguins », qui sera publié mi-2013, a été fondamentale. Quant au guide relatif à la transplantation d’organes (Guide to Safety and Quality of Organs for Transplantation), qui compile des lignes directrices couvrant les différents aspects du don d’organes et du processus de transplantation, de l’évaluation du facteur risque lié au donneur à la transmission de maladies, sa 5èmeédition a également été finalisée.
En matière de soins pharmaceutiques, le rapport “Policies and practices for a safer, more responsible and cost-effective health system” a été publié, et il a été cité dans les conclusions d’un sommet ministériel organisé à Amsterdam sur le thème « The benefits of responsible use of medicines: setting policies for better and cost-effective healthcare ».
Dans le cadre de la lutte contre la contrefaçon des médicaments falsifiés/contrefaits, la DEQM a consacré une importante part de ses activités à poursuivre la promotion de la Convention MEDICRIME du Conseil de l’Europe. Celle-ci, à la fin de l’année, avait été signée par 22 états et ratifiée par un état. Elle deviendra opérationnelle lorsqu’elle aura été ratifiée par 5 états. La DEQM a aussi poursuivi le développement d’eTACT, un système de sérialisation de masse des médicaments, grâce aux retours nombreux recueillis auprès de l’ensemble des acteurs de la chaîne de distribution des médicaments. La DEQM attache une importance particulière à certaines des spécificités de ce projet :
l’idée d’une gouvernance publique, l’interopérabilité avec les systèmes nationaux existants, la flexibilité et la responsabilisation du patient, à travers la possibilité dont il dispose, de vérifier l’authenticité de ses propres médicaments.
Le Réseau européen des laboratoires officiels de contrôle des cosmétiques (OCCL), pour sa part, a poursuivi sa campagne de surveillance du marché sur les cosmétiques décoratifs et engagé une autre étude sur les cosmétiques destinés aux enfants ou utilisés chez les enfants. Le Comité des Ministres du Conseil de l’Europe a adopté en avril 2012 une résolution sur les critères de sécurité applicables aux produits cosmétiques destinés aux jeunes enfants. Dans le domaine des emballages alimentaires, une nouvelle résolution relative aux métaux et alliages entrant dans la composition des matériaux pour contact alimentaire a été rédigée. Cette résolution annule et remplace les lignes directrices publiées voici 10 ans. Son adoption est prévue pour 2013.
Enfin, la DEQM est fière d’avoir étendu le périmètre de la certification ISO 9001. L’audit réalisé par l’Afnor - l’organisme français de certification - couvrait jusqu’en 2012 un certain nombre d’activités déjà certifiées les années précédentes : certification de conformité, études de surveillance des marchés, conduites sous l’égide de la DEQM, procédure OCABR pour la libération officielle des lots de médicaments immunologiques humains (produits sanguins et vaccins) par les autorités de contrôle. En 2012, pour la première fois, la certification a été étendue au processus d’élaboration des monographies. L’élaboration, la révision, la correction et la suppression des textes de la Pharmacopée Européenne, la publication en format imprimé et électronique et la distribution sont désormais certifiées ISO 9001.
Par ailleurs, l’organisme d’accréditation belge, Belac, a conduit en décembre 2012 un audit très poussé des activités du Laboratoire de la DEQM selon l’ISO/CEI 17025:2005. Le certificat correspondant est attendu début 2013.
Dans l’ensemble, 2012 aura été pour la DEQM une nouvelle année de défis et de succès. En témoignent la décision prise par l’Ukraine de ratifier la Convention relative à l’élaboration d’une Pharmacopée Européenne, en décembre 2012, et les demandes déposées par la République de Guinée et par Singapour pour obtenir le statut d’observateur, qui leur a aussi été accordé en 2012. Elles constituent une reconnaissance de l’importance des activités de la DEQM pour la protection de la santé publique en Europe et au-delà. Néanmoins, rien de tout cela n’aurait pu être réalisé en 2012 sans le soutien et le dévouement des nombreux experts désignés par les 37 états signataires de la Convention. Leur compétence et leur enthousiasme sont essentiels aux travaux de la Commission européenne de Pharmacopée et de ses Groupes d’Experts et Groupes de Travail, ainsi que des Comités et Groupes d’Experts qui œuvrent dans les domaines de la transfusion sanguine, de la transplantation d’organes, des produits et pratiques pharmaceutiques, de la protection de la santé des consommateurs, du Réseau OMCL et de la Certification. Je voudrais saisir l’occasion qui m’est donnée ici, de leur exprimer à tous notre sincère gratitude.
Susanne Keitel Directrice
3

4

« LA DEQM EN BREF » Valeurs, objectifs, activités
5
■ La Direction Européenne de la Qualité du Médicament & Soins de Santé (DEQM) : une Direction du Conseil de l’Europe
Le but premier du Conseil de l’Europe est de créer sur l’ensemble du continent un espace démocratique et juridique commun, en veillant au respect de valeurs fondamentales : les droits de l’homme, la démocratie et la prééminence du droit.
Droits de l’homme ... Démocratie ... État de droit
Fondements d’une société tolérante et civilisée, ces valeurs sont indispensables à la stabilité, la croissance économique et la cohésion sociale de l’Europe. Elles nous guident dans la recherche de solutions communes à des problèmes majeurs : terrorisme, criminalité organisée et corruption, cybercriminalité, bioéthique et clonage, racisme et préjugés, violences à l’égard des femmes et des enfants, traite des êtres humains. La coopération de tous les États membres est l’unique moyen de répondre aux grandes questions de notre temps.
Objectifs du Conseil de l’Europe
• Défendrelesdroitsdel’homme,ladémocratiepluralisteetla prééminence du droit,
• favoriser la prise de conscience et lamise en valeur del’identité culturelle de l’Europe et de sa diversité,
• rechercherdessolutionscommunesauxproblèmesdenossociétés,
• développerlastabilitédémocratiqueenEuropeensoutenantles réformes politiques, législatives et constitutionnelles.
■ Mission de la DEQMLa mission de la DEQM est d’œuvrer pour le droit humain fondamental que constitue l’accès à des médicaments et soins de santé de qualité et de contribuer à la promotion et la protection de la santé humaine et animale par différents moyens :
• elleétablitetpubliedesnormesofficiellesenmatièredefabrication et de contrôle qualité des médicaments, normes applicables dans tous les pays signataires de la Convention relative à l’élaboration d’une Pharmacopée Européenne et au-delà,
• elle veille à l’application de ces normes officielles auxsubstances utilisées pour la fabrication des médicaments,
• elleassure lacoordinationd’unRéseaudes laboratoiresofficielsdecontrôledesmédicaments,afind’établirdes
coopérations et des partages de compétences entre les États membres et optimiser l’utilisation des ressources disponibles,
• elleétablitdesnormesdequalitéetassurelapromotiondepratiques éthiques concernant :
- la collecte, la préparation, la conservation et l’utilisation des composants sanguins, en relation avec la médecine transfusionnelle,
- la transplantation d’organes, de tissus et de cellules,
• elle collabore avec des organisations nationales etinternationales dans la perspective d’éliminer les médicaments et produits médicaux illégaux et contrefaits,
• elledéfinitdespolitiquesetdesapprochesmodèlespourunbon usage des médicaments en Europe, via notamment des lignes directrices relatives au suivi pharmaceutique,
• elleétablitdesnormesetcoordonnedescontrôlessurlesproduits cosmétiques et les emballages alimentaires.
La Direction européenne de la Qualité du Médica-ment & Soins de Santé (DEQM)
La DEQM, dont les origines remontent à 1964, est devenue, aufildutemps,uneDirectionduConseilde l’Europe.En2012, elle comptait 260 employés à temps plein et était structurée en 9 entités administratives.
Mise en place en vertu de l’article 9 de la Convention relative à l’élaboration d’une Pharmacopée Européenne, signée en 1964 par 8 États membres du Conseil de l’Europe en vue de créer une Pharmacopée Européenne commune, elle a longtemps été connue comme le « Secrétariat de la Pharmacopée Européenne ». Puis cette entité administrative du Conseil de l’Europe a vu son appellation évoluer pour traduire au plus prèslesmissionsquiluiontpeuàpeuétéconfiées.

6
1. LES ACTIVITÉS FONDAMENTALES
7
Objet
Le but de la Pharmacopée Européenne (Ph. Eur.) est de promouvoir la santé publique par la mise à disposition de normes qualité communes et reconnues. L’existence de ces normes assure la qualité des médicaments et de leurs composants et en facilite la libre circulation en Europe. Les monographies et autres textes de la Ph. Eur. sont conçus pour répondre aux besoins des autorités réglementaires, des fabricants de matières premières et de médicaments et des différents acteurs du contrôle de la qualité des médicaments et de leurs constituants.
La Ph. Eur. est régie par la Commission européenne de Pharmacopée, qui supervise les travaux de plus de 70 groupes de travail et groupes d’experts. La Commission est composée de délégations des 38 parties signataires de la Convention relative à l’élaboration d’une Pharmacopée Européenne, auxquels s’ajoutent 24 observateurs.
La Pharmacopée Européenne est largement utilisée au niveau international. La Commission travaille en étroite collaboration avectouslesutilisateursdelaPharmacopée,afindemieuxsatisfaire leurs besoins et de faciliter leur coopération.
Une référence officielle au service de la santé publique
Les normes qualité de la Ph. Eur. font non seulement partie des exigences du dossier d’autorisation de mise sur le marché des médicaments, mais elles sont aussi juridiquement contraignantes pendant tout le cycle de vie d’un médicament. Elles garantissent l’accès à des médicaments de même qualité à travers toute l’Europe.
Tous les producteurs de médicaments et/ou de substances pour usage pharmaceutique sont donc tenus d’appliquer ces normes qualité pour commercialiser leurs produits dans les États signataires de la Convention.
Un champ d’application qui couvre l’ensemble des questions de santé publique
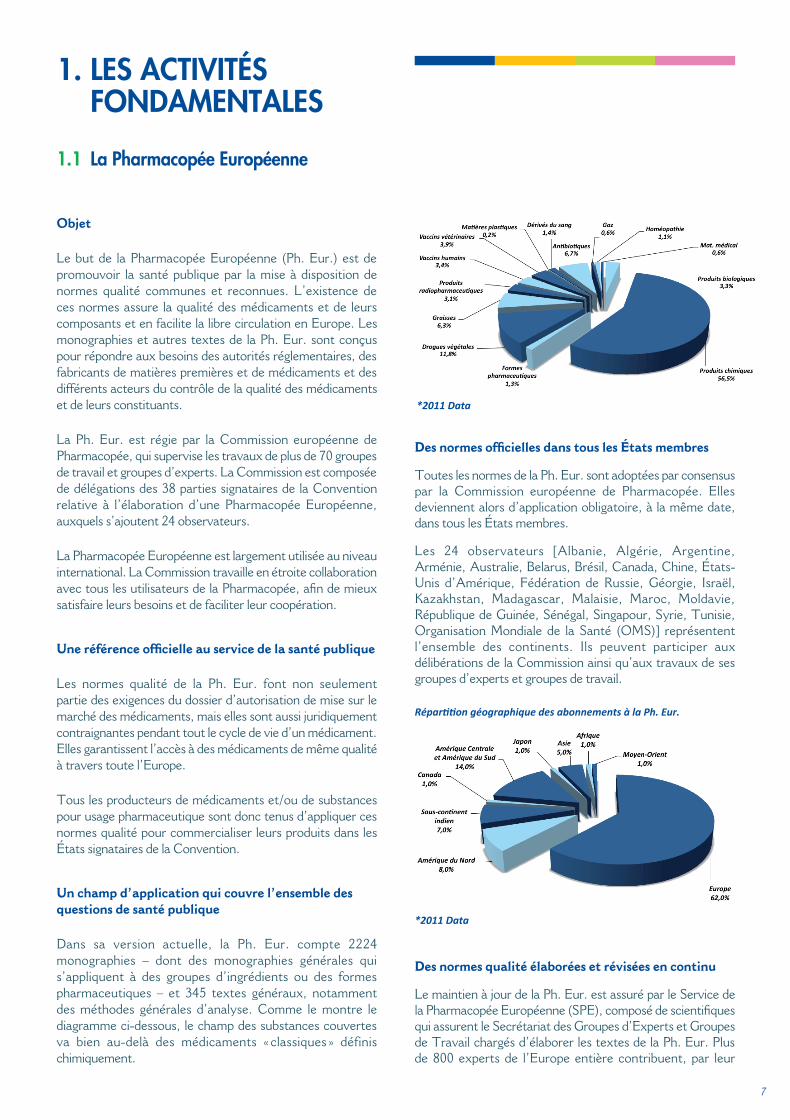
Dans sa version actuelle, la Ph. Eur. compte 2224 monographies – dont des monographies générales qui s’appliquent à des groupes d’ingrédients ou des formes pharmaceutiques – et 345 textes généraux, notamment des méthodes générales d’analyse. Comme le montre le diagramme ci-dessous, le champ des substances couvertes va bien au-delà des médicaments « classiques » définis chimiquement.
Des normes officielles dans tous les États membres
Toutes les normes de la Ph. Eur. sont adoptées par consensus par la Commission européenne de Pharmacopée. Elles deviennent alors d’application obligatoire, à la même date, dans tous les États membres.
Les 24 observateurs [Albanie, Algérie, Argentine, Arménie, Australie, Belarus, Brésil, Canada, Chine, États-Unis d’Amérique, Fédération de Russie, Géorgie, Israël, Kazakhstan, Madagascar, Malaisie, Maroc, Moldavie, République de Guinée, Sénégal, Singapour, Syrie, Tunisie, Organisation Mondiale de la Santé (OMS)] représentent l’ensemble des continents. Ils peuvent participer aux délibérations de la Commission ainsi qu’aux travaux de ses groupes d’experts et groupes de travail.
Des normes qualité élaborées et révisées en continu
Le maintien à jour de la Ph. Eur. est assuré par le Service de laPharmacopéeEuropéenne(SPE),composédescientifiquesqui assurent le Secrétariat des Groupes d’Experts et Groupes de Travail chargés d’élaborer les textes de la Ph. Eur. Plus de 800 experts de l’Europe entière contribuent, par leur
1.1 La Pharmacopée Européenne
Répartition géographique des abonnements à la Ph. Eur.
*2011 Data
*2011 Data

8
expertise et leurs connaissances, au processus d’élaboration. Ces textes nécessitent d’être mis à jour en permanence, comme lemontre la figure ci-dessous, pour tenir comptedes évolutions et des nouvelles exigences scientifiques, réglementaires ou autres.
thérapeutiques. Sur les 36 États signataires de la Convention relative à l’élaboration d’une Pharmacopée Européenne, 21 étaient représentés.
Différents sujets ont été abordés :
• La stratégieàmoyenet long termede laPharmacopéeEuropéenne dans le domaine des produits biologiques et des produitsdéfinischimiquement;
• Le suivi des actions proposées et décidées suite à laconférence organisée par la DEQM en novembre 2011 sur le thème « Formulaire européen des formulations pédiatriques ». Les ANP ont très favorablement accueilli la propositiond’élaboreruntelformulaire;
• La poursuite éventuelle de l’action engagée sur la« deuxième identification » qui figure dans certaines monographies spécifiques, sous différents aspects : champ d’application, contenu des exigences, maintien de la rubrique. Ces discussions ont conduit à la décision, approuvée par la Commission en juin 2012, de créer un GroupedeTravail«Deuxièmeidentification».Lepremierobjectif de ce Groupe sera de préparer un document guide, définissantlescritèresd’inclusiondanslesmonographiesd’une deuxième série d’identifications, exclusivement destiné aux pharmacies. Le Groupe passera également en revuelesméthodesetinstrumentsdisponiblesàcettefindanslesofficines.
L’harmonisation internationale au sein du GDP
La mondialisation et l’expansion du commerce international dans le domaine du médicament renforcent la nécessité de développer des normes qualité ayant une portée mondiale. Ces normes constituent un instrument vital pour l’autorisation de mise sur le marché, la surveillance du marché, la libre circulation et le libre-échange des médicaments d’un pays ou d’une région à l’autre.
La Pharmacopée Européenne s’est engagée, aux côtés de la Pharmacopée Japonaise et de la Pharmacopée des États-Unis, dans un processus d’harmonisation des méthodes générales et des monographies d’excipients conduit au sein d’une structure informelle : Le Groupe de Discussion des Pharmacopées (GDP). Des informations sur les textes harmonisésfigurentdanslechapitregénéralHarmonisation des pharmacopées (5.8) de la Ph. Eur. ainsi que sur la page « Harmonisation internationale » du site internet de la DEQM.
Le GDP, après examen des propositions émanant des associations nationales de fabricants de produits pharmaceutiques et d’excipients, sélectionne les méthodes générales d’analyse et les monographies d’excipients à inscrire à son programme de travail.
À ce jour, 28 des 35 chapitres généraux et 43 des 62 monographies d’excipients inscrits au programme de travail ont été harmonisés.
Le Service de la Pharmacopée Européenne compte également des traducteurs, qui assurent la traduction de la Ph. Eur. en anglaisetenfrançais,lesdeuxlanguesofficiellesduConseilde l’Europe. La Ph. Eur. est également traduite en espagnol en collaboration avec les autorités espagnoles. Sa traduction vers d’autres langues nationales de pays signataires de la Convention (allemand, hongrois ou polonais par exemple) est effectuée sous la responsabilité de chaque pays concerné.
Processus de consultation publique
Afin d’optimiser l’interaction entre la Commission européenne de Pharmacopée et ses utilisateurs, en leur accordant davantage de temps pour commenter les projets de texte et en élargissant l’accès à toutes les parties intéressées, à l’échelle mondiale, Pharmeuropa – le forum de la Pharmacopée Européenne – est devenu une publication exclusivement électronique accessible gratuitement en ligne.
Les textes sont postés en continu, mais le principe des quatre numéros annuels, avec quatre dates butoirs correspondantes pour les commentaires, est resté inchangé, de même que les canaux et procédures de transmission des commentaires sur les projets de texte publiés (voir section 2.2 Activités liées aux technologies de l’information et aux publications, page 33).
Échanges et discussions avec les Autorités Nationales de Pharmacopée (ANP) membres de la Commission européenne de Pharmacopée
La réunion annuelle des Autorités Nationales de Pharmacopée des États membres de la Pharmacopée Européenne s’est tenue à Berne (Suisse) en mai 2012. Cette rencontre était accueillie par Swissmedic, l’agence suisse des produits
Des normes de qualité qui nécessitent d'être régulièrement revues et révisées

9
Monographies de produits finis
Jusqu’à sa 8ème édition, la Pharmacopée Européenne ne comportaitpasdemonographiesdeproduitsfinis,àquelquesexceptions près, telles que celles des immunosérums pour usage humain, des immunosérums pour usage vétérinaire, de certaines préparations biologiques comme les préparations d’insuline, des préparations radiopharmaceutiques, des vaccins pour usage humain et des vaccins pour usage vétérinaire.
Pourlesproduitsfinis,l’harmonisationetlastandardisationentre les États membres de la Ph. Eur. étaient jusqu’ici traitées de deux façons : d’une part l’élaboration de monographies générales de formes pharmaceutiques, établissant les éléments communs à toutes les préparations quirelèventdelaformepharmaceutiqueconsidérée;d’autrepart le développement de méthodes d’essai normalisées à utiliser pour le contrôle des produits finis. La présencedans la Ph. Eur. de ces monographies générales et de ces méthodes d’essai constitue pour les Autorités compétentes et les fabricants de médicaments une base commune pour la préparation et l’évaluation des dossiers d’autorisation de mise sur le marché (l’AMM). Une monographie générale Préparations pharmaceutiques (2619) a par ailleurs été adoptée en 2012. Cette monographie est conçue comme une référence en matière de normes applicables aux substances actives, excipients et formes pharmaceutiques, dans le cadre de la fabrication/préparation de produits pharmaceutiques, mais non comme un guide sur les modalités de leur fabrication,laquellefaitl’objetdetextesspécifiquescouvrantles méthodes de fabrication et les contrôles associés.
La Commission a toutefois, pris la décision, lors de sa 144ème session, de reconsidérer cette politique et de mettre en place un projet pilote sur l’élaboration de monographies de produitsfinis.L’objectifdeceprojet,confiéàungroupedetravail spécial, est de réaliser une étude de faisabilité, sur la basededeuxmonographiesdeproduitsfiniscontenantdessubstancesactivesdéfinieschimiquement(l’uneissued’unesource unique, l’autremulti-source).À la fin de la phasepilote, la Commission décidera s’il y a lieu ou non d’élaborer enroutinedesmonographiesdeproduitsfinis.
Procédure P4 : un succès confirmé
La procédure P4 concerne les substances pour lesquelles un seulfabricantaétéidentifié.Elleestgénéralementappliquéeà des substances encore sous brevet, avec un potentiel élevé de futurs génériques. Les projets de monographies reposent sur les substances qui entrent dans la composition de médicaments ayant été autorisés par des Autorités compétentes de pays signataires de la Convention de la Pharmacopée Européenne, normalement au sein de l’UE.
A ce jour, 61 monographies P4 et P4Bio ont été adoptées par la Commission Européenne de Pharmacopée, dont 5 en 2012 : Ciclésonide (2703), Pémétrexed disodique heptahydraté (2637), Chlorhydrate d’atomoxétine (2640), Dutastéride (2641) et Facteur VIIa de coagulation humain (ADNr) (2534).
La Ph. Eur. et l’approche PAT/QbD
Un groupe de travail a été créé pour assurer le suivi des évolutions techniques dans le domaine du contrôle analytique des procédés (PAT pour Process Analytical Technology) et de la qualité intégrée à la conception (QbD pour Quality by Design). Ce groupe PAT travaille en étroite collaboration avec celui de l’Agence européenne des Médicaments (EMA). L’un des objectifs majeurs de la création ou révision de textes généraux en rapport avec la PAT dans la Ph. Eur. est de faciliter et encourager l’application de stratégies de contrôle améliorées, faisant intervenir la PAT et la libération en temps réel.
Les travaux du groupe PAT ont conduit à l’élaboration d’un nouveau chapitre général Démonstration de l’uniformité des préparations unidoses à partir d’échantillons de grande taille (2.9.47), adopté par la Commission en 2012. Ce chapitre décrit une méthode alternative et optionnelle qui peut être utilisée par les demandeurs d’AMM lorsque, par exemple, ils disposent de données acquises en ligne par des technologies de contrôle des procédés de type non destructif, comme la spectroscopie dans le proche infrarouge et peut donc remplacer l’essai conventionnel d’uniformité des préparations unidoses. Le groupe de Travail a également révisé le chapitre Spectroscopie dans le proche-infrarouge (2.2.40) pour y introduire des concepts, tels que celui des mesures en ligne, en rapport avec le contrôle des procédés. Cette révision a été menée en étroite concertation avec le groupe de travail « Qualité » mixte CVMP/CHMP de l’EMA, et le texte sera utilisé en conjonction avec celui de l’EMA « Guideline on the use of near infrared spectroscopy by the pharmaceutical industry and the data requirements for new submissions and variations », quidoitêtrefinaliséen2013.Actuellement, leGroupedeTravail PAT travaille à la révision du chapitre spectrométrie Raman (2.2.48).
Une réflexion est également en cours sur la nécessité d’élaborer de nouveaux chapitres généraux, par exemple, sur l’imagerie proche infrarouge, la spectroscopie térahertz et l’acoustique.
Le Groupe de travail NIR travaille actuellement à l’élaboration d’un chapitre général sur les Méthodes chimiométriques appliquées aux données analytiques (5.21).
Métaux lourds
L’adoption d’une note explicative de l’EMA relative aux limites de teneur en résidus de catalyseurs ou réactifs métalliques a conduit la Commission européenne de Pharmacopée à prendre la décision d’élaborer un nouveau chapitre général Résidus de catalyseurs ou de réactifs métalliques (5.20), qui reprend la note explicative de l’EMA. Pour aider les utilisateurs à mettre ce chapitre en application, une méthode générale Dosage des résidus de catalyseurs ou de réactifs métalliques (2.4.20) a également été élaborée. Elle décrit l’approche générale à suivre pour le dosage des résidus de catalyseurs ou réactifs métalliques dans les substances pour usage pharmaceutique et les utilisateurs peuvent l’appliquer

10
lorsque cela est possible. Les deux textes ont été adoptés par la Commission européenne de Pharmacopée et publiés dans la Ph. Eur.
Lorsque le guideline sur les impuretés métalliques en cours d’élaboration dans le cadre ICH Q3D aura été adopté, il est envisagé qu’il remplace l’actuelle note explicative de l’EMA sur les limites de teneur en résidus de catalyseurs ou réactifs métalliques. En conséquence, le chapitre Résidus de catalyseurs ou de réactifs métalliques (5.20) de la Ph. Eur. sera alors révisé et mis à jour, de même que la méthode Dosage des résidus de catalyseurs ou de réactifs métalliques (2.4.20) s’il y a lieu.
Progrès en matière d’essais sur animaux
Lors de sa 142ème session, la Commission européenne de Pharmacopée a décidé d’harmoniser les textes de la Pharmacopée Européenne relatifs aux vaccins vétérinaires et ce faisant d’en améliorer la cohérence. Cette décision marquait l’aboutissement de plusieurs années d’un travail ambitieux et de grande ampleur qui avait été engagé dans le cadre de l’harmonisation avec les guidelines 41 (essai de retour à la virulence) et 44 (essais d’innocuité de développement) du VICH, entrés en vigueur en 2008 et 2009. Cette entreprise concernait environ 80 monographies de vaccins de la Ph. Eur., la méthode générale Vaccins pour usage vétérinaire (0062), et 2 chapitres généraux : Evaluation de l’innocuité des vaccins et immunosérums vétérinaires (5.2.6) et Evaluation de l’innocuité de chaque lot d’immunosérums pour usage vétérinaire (5.2.9), tous publiés pour enquête publique dans Pharmeuropa 23.1 et adoptés.
Allant au-delà de l’harmonisation avec le VICH, pour être en cohérence avec la réglementation européenne, la Pharmacopée Européenne a étendu l’harmonisation à toutes ses monographies de vaccins vétérinaires, y compris ceux destinés à des espèces hors champ d’application des guidelines VICH. Les essais d’innocuité et d’accroissement de la virulence effectués en cours de développement ont ainsi étéharmonisés,cequiréduirasignificativement lenombred’animaux utilisés.
Une révision de la monographie générale Vaccins pour usage vétérinaire (0062)aétéeffectuéeafind’ysupprimerlesessaisd’innocuité réalisés sur chaque lot chez l’animal cible (TABST,
Target Animal Batch Safety Tests), sauf « circonstances particulières » nécessitant la réalisation ponctuelle d’essais supplémentaires et notamment des essais d’innocuité.
Pour aller plus loin dans le sens des 3R (Replacement, Reduction, Refinement) en matière d’essais sur animaux, la Commission européenne de Pharmacopée a également adopté la suppression du TABST dans toutes les monographies de vaccins vétérinaires. Cette suppression est un nouveau pas en avant par rapport à l’option, proposée depuis 2004, qui permettait une dispense du TABST pour les vaccins bien établis.
Toujours dans le contexte des 3R, une révision de la monographie Vaccin rabique inactivé pour usage vétérinaire (0451) a été effectuée afin de mieux détailler le titrage sérologique qui doit être utilisé si possible comme alternative, moins stressante pour les animaux, au titrage d’activité sur animaux vivants.
Lors de la 143ème session, suite à l’adoption du chapitre Toxine coquelucheuse résiduelle et irréversibilité de l’anatoxine coquelucheuse (2.6.33), la Commission a adopté neuf monographies de vaccins révisées pour faire référence à ce nouveau chapitre général. Le protocole décrit dans le nouveau chapitre, qui résulte d’une étude menée dans le cadre du Programme de standardisation biologique (BSP), facilitera la standardisation de la méthode et réduira l’utilisation inutile d’animaux.
En bref, dans la Pharmacopée Européenne, aucun animal n’est plus utilisé actuellement aux fins de contrôle des médicaments dérivés de sang ou de plasma humain. Pour les vaccins humains et vétérinaires, de nombreux essais in vivo ont été remplacés par des méthodes in vitro. Quant aux titrages in vivo toujours en vigueur, différentes stratégies sont utilisées pour en encourager la réduction et l’allègement, par exemple les titrages sérologiques ou à une seule dilution pour les vaccins diphtérique, tétanique, coquelucheux acellulaire et rabique (vétérinaires ou humains).

11
Ces décisions sont pleinement conformes à la mission de la DEQM de promouvoir et protéger la santé humaine et animale et à l’engagement de la DEQM de respecter le principe des 3R.
La Directive 2010/63/UE du Parlement européen et du Conseil,relativeàlaprotectiondesanimauxutilisésàdesfinsscientifiquesayantpriseffetle1er janvier 2013, la Commission européenne de Pharmacopée a procédé lors de sa 144ème session, à une évaluation des textes de la Pharmacopée qui recommandent des alternatives à l’expérimentation animale,afindemettrecetteinformationàladispositiondesutilisateurs.
Quelques chiffres clés pour 2012
La Commission européenne de Pharmacopée, au cours de ses trois sessions de 2012, a adopté les textes suivants :
43 nouvelles monographies
• Substances chimiques et préparations biologiques: 5monographies élaborées dans le cadre des procédures P4 et P4Bio [Ciclésonide (2703), Pémétrexed disodique heptahydraté (2637), Chlorhydrate d’atomoxétine (2640), Dutastéride (2641), Facteur VIIa de coagulation humain (ADNr) (2534)], et 12 monographies élaborées par les procédures classiques [Atovaquone (2192), Oxcarbazépine (2577), Amidon hydroxypropylé prégélatinisé (2645), Follitropine (2285), Solution concentrée de follitropine (2286), Hémitartrate d’alimémazine (2650), Desloratadine (2570), Diacéréine (2409), Sulfate d’abacavir (2589), Anastrozole (2406), Risédronate sodique (2572), Hydrogénotartrate de rivastigmine (2630)].
• Préparations pharmaceutiques (2619) : une monographie générale destinée à fournir une référence en matière de normes applicables aux substances actives, excipients et formes pharmaceutiques dans le cadre de la fabrication/préparation de produits pharmaceutiques.
• Drogues végétales, préparations à base de droguesvégétales, et drogues végétales issues de la médecine traditionnelle chinoise : 14 monographies [Tige de Clematis armandii (2463), Fleur de Magnolia officinalis (2568), Racine et rhizome de Salvia miltiorrhiza (2663), Feuille de cassis (2528), Extrait de palmier de Floride (2579), Ecorce de Fraxinus rhynchophylla (2452), Fleur de sophora (2639), Partie aérienne d’Eclipta prostrata (2564), Ecorce d’Eucommia (2412), Epicarpe et mésocarpe de mandarine (2430), Racine de Polygonum multiflorum (2433), Extrait mou titré de piment de Cayenne (2529), Rhizome de curcuma (2543), Rhizome de Belamcanda chinensis (2561).
• Vaccins:5monographies[Vaccin vivant oral de la salmonellose à Salmonella Enteritidis pour le poulet (2520), Vaccin vivant oral de la salmonellose à Salmonella Typhimurium pour le poulet (2521), Vaccin vivant de la rhinotrachéite infectieuse pour la dinde (2461), Vaccin vivant de Bordetella bronchiseptica pour le chien (2525), Vaccin inactivé de la yersiniose pour salmonidés (1950)].
• Composésradioactifs:3monographies[Solution de chlorure de gallium (68Ga) pour radiomarquage (2464), Solution injectable d’alovudine (18F) (2460), Solution injectable de fluoromisonidazole (18F) (2459)], plus une monographie portant sur un précurseur de préparations radiopharmaceutiques [Succimer pour préparations radiopharmaceutiques (2545)].
• Unemonographieportantsurungazpropulseur [Norflurane (2257)] et la monographie générale Préparations vétérinaires semi-solides pour usage oral (2638).
Cinq nouveaux chapitres généraux
• Uneméthoded’analyse«Toxine coquelucheuse résiduelle et irréversibilité de l’anatoxine coquelucheuse (2.6.33) ».
• Uneméthode d’analyse pour les composés radioactifs:Détection et mesure de la radioactivité (2.2.66).

12
254 textes révisés
• Les sections Production et Substances apparentéesde la monographie générale Substances pour usage pharmaceutique (2034) ont été mises à jour suite à l’élaboration des méthodes générales Méthanesulfonate de méthyle, d’éthyle et d’isopropyle dans l’acide méthanesulfonique (2.5.37) et Méthanesulfonate de méthyle, d’éthyle et d’isopropyle dans les substances actives (2.5.38). Cette révision, ainsi que l’adoption des trois méthodes générales portant sur le dosage des méthanesulfonates de méthyle, d’éthyle et d’isopropyle, ont conduit à réviser la section PRODUCTION de 11 monographies des sels de mésilate de substances actives.
• Chapitre général Spectrophotométrie dans le proche infrarouge (2.2.40).
• ChapitreHarmonisation des Pharmacopées (5.8). Par souci de transparence du degré d’harmonisation dans les textes signés par le GDP et pour aider les utilisateurs, les éléments non harmonisés sont signalés par des losanges noirs (◊◊) et les exigences locales par des losanges blancs (◊◊).dans les textes correspondants de la Ph. Eur.
Le Programme de Standardisation Biologique (PSB)
Le Programme de Standardisation Biologique (PSB), initiative conjointe de la DEQM et de la Commission européenne, poursuit différents objectifs en matière de standardisation des produits biologiques : établissement de matériels biologiques de référence, développement et validation de nouvelles méthodes d’analyse, validation de méthodes alternatives fondées sur le concept des 3R.
Àcettefin,desétudescollaborativessontorganiséesaveclaparticipation de tous les partenaires intéressés, notamment les laboratoiresofficielsdecontrôledesmédicaments (lesOMCL) et les fabricants. Chaque fois que possible, elles sont conduitesconjointementavecl’OMSafind’économiserlesressources des laboratoires participants. Depuis le début du programme, en 1992, 117 projets BSP ont été engagés. En 2012, 16 projets ont été poursuivis dans différents domaines :
• vaccinspourusagehumain:5projets
• vaccinspourusagevétérinaire:1projet
• produitsdérivésduplasma:5projets
• produitsbiotechnologiques:5projets
Parmi ces projets, 5 étaient consacrés à l’établissement de méthodes alternatives pour remplacer des essais sur animaux, 2 au développement de méthodes nouvelles ou améliorées de dosage et 11 à l’établissement de matériels de référence destinés à l’analyse de produits biologiques (2 des projets portaient à la fois sur le développement de nouvelles méthodes de dosage et l’établissement de matériels de référence).
Ces travaux se sont traduits par l’établissement de 2 lots de remplacement d’étalons de référence existants (somatropine et endotoxine), de 2 nouveaux étalons de référence d’allergènes recombinants majeurs (Bet v 1 et Phl p 5a) et de 3 nouveaux Réactifs Biologiques de Référence (RBR) utilisés pour le dosage des vaccins de l’hépatite A (voir section 1.2, Matériels biologiques de référence). Le projet concernant l’établissement de l’étalon d’endotoxine a été conduit conjointement avec l’OMS et l’USP. Les étalons de la Ph. Eur., de l’OMS et de l’USP ainsi établis ont été préparés à partir du même matériel de départ et possèdent les mêmes teneurs assignées.
Les étalons de référence des allergènes recombinants majeurs Bet v 1 (pollen de bouleau) et Phl p 5a (pollen de phéole des prés) constituent les premiers étalons de cette nature jamais établis et sont uniques au monde. Ils seront d’une grande utilité pour améliorer la standardisation des extraits produits à partir decesdeuxallergènes;cetyped’extraitsesttrèslargementutilisé pour la désensibilisation des patients allergiques.
Des efforts considérables ont à nouveau été déployés en 2012 pour l’application du concept des 3R dans le domaine du contrôle de la qualité des produits biologiques. Un tiers environ de l’ensemble des projets BSP ont été consacrés à la validation de méthodes alternatives aux essais sur animaux. Un projet, portant sur la validation d’une méthode in vitro pour l’évaluation de l’activité des vaccins de l’hépatite A, a été mené à terme. Deux nouveaux projets ont par ailleurs été démarrés pour remplacer des essais sur animaux figurantencoredanslaPh.Eur.,l’épreuvedesensibilisationà l’histamine, utilisée pour détecter la présence de toxine coquelucheuse résiduelle dans les vaccins coquelucheux acellulaires et le test NIH, une épreuve virulente directe servant à déterminer l’activité des vaccins rabiques. Un précédent projet du BSP avait déjà permis de remplacer le test NIH par un titrage sérologique pour la libération des lots de vaccin rabique inactivé à usage vétérinaire. L’objectif de ce nouveau projet était de remplacer complètement le test NIH, qui est toujours utilisé pour l’étalonnage des matériels de référence et pour les essais de stabilité des vaccins antirabiques.
L’établissement de réactifs biologiques de référence (RBR) pour l’application de méthodes alternatives peut également être considéré comme un aspect des efforts consacrés aux 3R danslecadreduBSP.Sanscesréactifsspécifiques,quidansla plupart des cas ne sont pas disponibles dans le commerce, les méthodes conformes aux 3R seraient en effet impossibles à appliquer.
Les efforts déployés par la DEQM, en particulier dans le cadre du BSP, pour élaborer, valider et mettre en application des méthodes conformes aux 3R sont largement reconnus, notamment par le Partenariat européen pour la promotion des méthodes de substitution à l’expérimentation animale (EPAA) - une initiative commune au plus haut niveau de la Commission européenne et de l’industrie. La DEQM est donc représentée au sein du Comité directeur du projet « Vaccins » de l’EPAA ainsi que dans le Comité technique et des études futures de l’EPAA doivent être conduites par le BSP.

13
Croissance du portfolio en 2012
Portfolio
La collection comporte actuellement 2537 produits. Elle fait l’objetd’unmonitoragerégulierpourvérificationdel’aptitudeàl’emploidesétalons;490lotsontainsiétéexaminésen2012.
■ Les étalons de référence de la Pharmacopée Européenne
Pourquoi des étalons de référence ?
La plupart des essais et dosages décrits dans la Ph. Eur. prescrivent l’utilisation des étalons de référence officiels de la Ph. Eur., c’est-à-dire des échantillons soigneusement caractérisés de substances destinées au contrôle de la qualité.
Les étalons de référence de la Ph. Eur. sont établis par la DEQM et adoptés par la Commission européenne de Pharmacopée.
Substances Chimiques de Référence (SCR)
En 2012, 297 SCR ont été établies, dont 75 étalons de dosage et 73 mélanges. Un aperçu des SCR établies sur la période 2007-2012 est présenté ci-dessous.
Etablissement de SCR
1.2 Etalons de référence

14
Évolution du nombre de commandes traitées /expéditions d’étalons de référence de la Ph. Eur. en 2012
■ Les activités conduites par la DEQM pour le compte de l’OMS
Substances Chimiques de Référence Internationales (SCRI)
Depuis 2010, la DEQM est responsable de l’établissement, du monitorage et de la distribution des SCRI, étalons de référence de l’OMS prescrits dans la Pharmacopée Internationale, qui est publiée par l’OMS et utilisée à l’échelle mondiale.
17 études d’établissement de SCRI ont été menées à terme en 2012 : pyriméthamine SCRI 1, éthylsuccinate d’érythromycine SCRI 1, ciprofloxacine SCRI 1, isétionate de pentamidine SCRI 1, spectre de référence du chlorhydrate de proguanil SCRI 1, niridazole SCRI 1, azobenzène SCRI 1, aténolol SCRI 1, clofazimine SCRI 1, dacarbazine SCRI 1, triéthiodure de gallamine SCRI 1, glibenclamide SCRI 1, phénobarbital SCRI 1, sulfate de salbutamol SCRI 1, spironolactone SCRI 1, tiabendazole SCRI 1, chlorhydrate de vérapamil SCRI 1.
Matériels biologiques de référence
En 2012, les études collaboratives internationales réalisées dans le cadre du Programme de Standardisation Biologique ont conduit à l’adoption par la Commission européenne de Pharmacopée de 4 étalons de référence – Préparations Biologiques de Référence (PBR) et Substances Chimiques de Référence (SCR) – destinés à l’analyse de produits biologiques :
• 2 lots de remplacement: somatropine SCR (lot 3) etendotoxine PBR (lot 5),
• 2nouvellesSCR,rBetv1etrPhlp5a,représentanttoutesdeux des allergènes recombinants majeurs, utilisées pour le dosage d’extraits allergènes à partir du pollen de bouleau (Betula verrucosa) et de phéole des prés (Pleum pratense).
Lors de sa session de novembre, la Commission européenne de Pharmacopée a approuvé la proposition de création d’une nouvelle classe de matériels de référence, les Réactifs biologiques de Référence (RBR) de la Ph. Eur. Les RBR sont des matériels de référence établis par la DEQM, ils possèdent un statut officiel (ils sont adoptés par la Commission européenne de Pharmacopée et cités en référence dans la Ph. Eur.) et ils sont nécessaires à la réalisation de certains essais de la Ph. Eur. (en général des dosages) sans être cependant considérés comme des étalons de référence. Les RBR sont particulièrement utiles pour l’application de méthodes alternatives remplaçant des essais sur animaux, par exemple des méthodes ELISA. Les RBR sont distribués par la DEQM.
Lors de la même session, la Commission européenne de Pharmacopée a adopté 3 RBR destinés au titrage des vaccins de l’hépatite A par ELISA (réactif de capture, anticorps de détection primaire et anticorps de détection secondaire). Les trois RBR seront utilisés pour remplacer le titrage in vivo des vaccins de l’hépatite A par un titrage in vitro ne nécessitant pas d’animaux.
Laplanification rigoureuseet lacoordinationdesactivitésinter-services ont facilité la réalisation de l’objectif visé : garantir à tout moment la disponibilité d’au moins 98 % des étalons de la collection.
Disponibilité du portfolio de SCR en 2012
Répartition géographique des commandes/ventes d’étalons de la Ph. Eur. en 2012
Les régions et pays hors zone géographique de l’Europe, représentent plus d’un tiers de l’ensemble des ventes d’étalons, en nombre d’ampoules distribuées.

15
Étalons internationaux d’antibiotiques (ISA)
Depuis mai 2006, la DEQM est responsable de l’établissement, du stockage et de la distribution des étalons ISA.
Les ISA jouent un rôle essentiel pour la standardisation et le contrôle qualité des substances et médicaments antibiotiques. Ils servent à la réalisation des titrages microbiologiques réalisés dans le cadre du contrôle qualité.
En 2012, le 3ème étalon international de néomycine et le 2ème étalon international de néomycine B ont été établis et des travaux ont été engagés pour le remplacement de l’actuel étalon de bléomycine.
Le Service Laboratoire de la DEQM (SLab) se compose d’une division « Chimie Analytique » et d’une section « Biologie ». En 2012, trois importants projets ont été poursuivis : déploiement total de LIMS, introduction d’une RMN et préparation de l’accréditation ISO 17025. Dans le même temps, le SLab a contribué au développement et au maintien à jour des normes et étalons de la Ph. Eur., ainsi qu’aux programmes d’essais d’aptitude (PTS) et de surveillance du marché (MSS) conduits dans le cadre du Réseau des OMCL, et à l‘établissement d’étalons de référence pour le compte de l’OMS.
Contribution aux travaux de la Pharmacopée Européenne
Normes documentaires :97rapportsd’étudesscientifiquesont été produits à la demande des différents groupes d’experts delaPh.Eur.;10projetsP4(dont2projetsP4Bio)ontétéconduits;16étudesontétéréaliséesdanslecadreduBSP.
Etalons matériels : 297 rapports d’établissement de SCR ont été adoptés par la Commission européenne de Pharmacopée (voir section 1.2 Etalons de référence, page 13).
Contribution aux études PTS/MSS du Réseau des OMCL
LeSLabaréalisédesétudesscientifiquesdanslecadredesprogrammes PTS et MSS du Réseau des OMCL (voir section 1.5 Réseau des OMCL, page 18).
Collaboration avec l’OMS
LeSLabaégalementréalisédesétudesscientifiquesdanslecadre du programme d’établissement de SCRI de l’OMS, en relation avec la Pharmacopée internationale, ainsi que du pro-gramme ISA (voir section 1.2 Etalons de référence, page 13).
Un objectif constant : améliorer l’efficience et la qualité
Différentes initiatives sont en cours dans les domaines suivants :
• Maintienetdéveloppementdesactivitésfondamentales,
• accroissementdurendementduprogrammedemonitorage des SCR,
• conformitéauxdispositionsdel’ISO17025.
Des mesures ont été prises pour inscrire la contribution du SLab dans une démarche durable à moyen terme :
• Introductiondenouvellestechniques:uninstrumentderésonance magnétique nucléaire (RMN) a été installé en 2012,
• renouvellementdeséquipements(détecteurUV,spectrophotomètre infrarouge, système CPG à espace de tête).
1.3 Activités du laboratoire
Répartition géographique des commandes/ventes de SCRI en 2012
24SCRIontfaitl’objetd’unmonitoragepourconfirmationdeleur aptitude à l’emploi.
Par ailleurs, les résultats des travaux analytiques visant à optimiser la méthode CL utilisée pour l’essai des substances apparentées de l’artémisinine ont été présentés au Comité d’Experts de l’OMS, qui a décidé d’adopter les propositions dulaboratoiredelaDEQMetdemodifierlamonographiedel’artémisinine en conséquence.

16
Objet de la procédure de certification
Laprocéduredecertificationdeconformitéauxmonographiesde la Pharmacopée Européenne (CEP), établie en 1994, répond à plusieurs objectifs :
• Garantirquelessubstancesutiliséesdanslaproductiondemédicaments sont de qualité conforme aux normes de la Ph. Eur. et, par voie de conséquence, aux exigences de la législation pharmaceutique de l’Union européenne et des États membres de la Ph. Eur.,
• contribuer à lamise à jour continue de la Ph. Eur. enévaluant si ses normes reflètent toujours la qualité des substances présentes sur le marché ; les informationsrecueilliessurlesvoiesdesynthèseetprofilsd’impuretésexistants permettent à la Ph. Eur. d’améliorer constamment laqualitédesesnormes;cettesynergieentrePh.Eur.etactivitésdecertificationestdelaplushauteimportance,car elle est le moyen de maintenir les normes à jour de l’évolution des produits présents sur le marché,
• Faciliter la gestion des demandes d’autorisation demise sur le marché au niveau européen, grâce à une évaluation centralisée de la qualité des substances à usage pharmaceutique et ainsi réduire la charge de travail des autorités et de l’industrie.
Quelles substances vise la procédure de certification ?
SelonlaprocédureofficielledécritedanslaRésolutionAP-CSP(07) 1, à laquelle font référence les directives communautaires 2001/83/CE et 2001/82/CE amendées ainsi que la directive 2003/63/CE, les demandes de certificats de conformité concernent les fabricants ou fournisseurs de principes actifs ou excipients pharmaceutiques, ou de produits à base de plantes utilisés dans la production ou la préparation de produits pharmaceutiques couverts par une monographie de la Ph. Eur. La procédure couvre également toute substance comportant un risque de transmission d’encéphalopathie spongiforme transmissible (EST).
Programme d’inspection par la DEQM de sites de fabrication de substances actives
L’évaluation documentaire des données soumises à la DEQM danslecadredesdemandesdecertificatsestcomplétéeparun programme d’inspection. L’objectif de ce programme est devérifierlaconformitédessitesdefabrication/distributioncouvertspar lescertificats(CEP)auxdossierssoumisà laDEQM ainsi qu’aux Bonnes Pratiques de Fabrication (BPF, telles qu’établies dans le Volume 4 de la Réglementation du médicamentauseindel’UE).LaDivisionCertificationdelaDEQM est responsable de l’organisation des inspections et des suites à donner, notamment quant aux CEP ou demandes de CEP concernés et à la communication avec les autorités
compétentes. Le programme annuel d’inspection, élaboré en fonction des priorités recommandées par l’EMA/UE, est adoptépar leComitéDirecteurde laCertification, aprèsconsultation des autorités des États membres et du Groupe de travail des inspecteurs BPF/BPD de l’EMA.
Evaluation des demandes de CEP : quelques chiffres
clés pour 2012
En 2012, 260 nouveaux CEP et 950 CEP révisés ont été délivrés. Par ailleurs, un nombre accru de demandes de révisionont été approuvées sansdélivrancede certificatsrévisés (520), conformément à la politique en vigueur pour lesnotificationsoulesrévisionsmineuressansincidencesurlaqualitédelasubstancefinaleousurlecontenuduCEP.Acesdemandes de révision adressées à la DEQM se sont ajoutées les mises à jour de CEP rendues nécessaires par la publication de monographies révisées dans des suppléments de la Ph. Eur.
Au cours de l’année 2012, plus de 82% des nouvelles demandes et des révisions ont été traitées dans les délais établis.
Le nombre de CEP en cours de validité – qu’ils couvrent la pureté chimique, le risque EST ou les préparations végétales – s’élève à ce jour à plus de 3500.
Le programme d’inspection de la DEQM : quelques chiffres clés pour 2012
En 2012, 32 inspections sur site ont été effectuées, principalement en Asie, avec la participation d’inspecteurs de différentes agences nationales. Trois sites ont refusé d’être inspectés suite à l’annonce d’une inspection, ce qui a entraîné la suspension immédiate des CEP concernés. Le partage d’informations avec les corps d’inspection des États membres et partenaires a permis de couvrir 25 autres sites. Sur l’ensemble des inspections effectuées, des non-conformités auxBPFontétéconstatéespour13firmesetontconduitàla
1.4 Certification de conformité aux monographies de la Pharmacopée Européenne
Evolution du nombre de nouvelles demandes de CEP

17
Répartition géographique des sites inspectés en 2012
Programme international d’inspection pour les substances actives
Depuis 2008, la DEQM participe à un programme international d’inspection des sites de production de substances actives, une initiative de l’EMA. Ce programme, qui implique les autorités d’inspection des États-Unis, d’Australie, de l’OMS (depuis 2012) et de certains pays européens, ainsi que la DEQM, vise à optimiser l’utilisation des ressources grâce à deséchangesd’informations (planificationdes inspectionset rapports d’inspections) et à la réalisation d’inspections
conjointes pour les sites de fabrication de substances actives d’intérêt commun. En 2012, en plus des échanges réguliers de rapports d’inspection et d’autres informations, la DEQM a effectué une inspection conjointe avec l’USFDA (United States Food and Drug Administration) sur un site de fabrication situé en Inde. Elle a également apporté une contribution active aux activités du Pharmaceutical Inspection Co-operation Scheme (PIC/S).
suspension ou au retrait de CEP ou à la clôture de demandes de CEP. Ces chiffres témoignent, une fois encore, de la pertinence des critères appliqués pour la sélection des sites à inspecter sur la base d’une approche d’analyse du risque. Par ailleurs, en 2012, les inspections sur site ont comporté une forte proportion de réinspections (>50%). Globalement, environ 60% des firmes soumises à une réinspection par la DEQM se sont avérées non conformes. Cette situation montrelepeud’empressementd’uncertainnombredefirmesà maintenir la conformité aux BPF.
Depuis les débuts du programme, la DEQM a réalisé 283 inspections et réinspections. Sur ces 283 inspections, 229 ont été effectuées en dehors de l’Espace Economique Européen (EEE). La vaste majorité des sites inspectés depuis 2003 se situent d’ailleurs hors EEE.

18
pour ledépistagedesmédicamentsfalsifiés, lemonitoragedes médicaments stockés, le contrôle des préparations non soumises à l’AMM et le contrôle de la qualité des substances actives sur le marché européen.
Depuis juillet 2012, des règles ont été mises en place pour le maintiendel’appartenanceauRéseau;ellesdéfinissentlesobligations des membres du Réseau et instituent un suivi de leur participation. Trois niveaux d’appartenance au GEON sont proposés : membre à part entière, membre associé, membre à participation limitée. La liste des membres est publiée sur le site internet de la DEQM.
Activités du Réseau général des OMCL
Systèmes de Management de la Qualité
Les activités suivantes ont été conduites en 2012 dans le cadre du programme de management de la qualité (MQ) du Réseau OMCL, sous la coordination de la DEQM.
Audits mutuels communs et visites mutuelles communes
En 2012, onze audits mutuels communs (MJA) et une visite mutuelle commune (MJV) ont été effectués sur des sites d’OMCL. Deux des MJA ont été effectués en collaboration avec les organes nationaux d’accréditation concernés.
Activités de formation au sein des OMCL
La DEQM a organisé en 2012 trois visites de formation, portant sur des méthodes d’analyse physico-chimiques et biologiques. Par ailleurs, un atelier a été organisé dans les locaux de la DEQM à l’intention des auditeurs techniques,
Introduction
Le Réseau européen général (GEON) des laboratoires officielsdecontrôledesmédicaments(OMCL), institué le26 mai 1994 par décision de la Commission Européenne (CE) et du Conseil de l’Europe (CoE), est ouvert à l’ensemble des pays signataires de la Convention relative à l’élaboration d’une Pharmacopée Européenne, ainsi qu’aux observateurs auprès de la Commission européenne de Pharmacopée, sous réserve qu’ils répondent à un certain nombre de critères (indépendance,financementpublic,applicationdelaPh.Eur.comme référence commune, conformité des laboratoires à la norme ISO/CEI 17025, etc.). Depuis 1995, la DEQM assume la coordination du Réseau et la responsabilité de son organisation et de son développement. Les activités de coordinationde laDEQMdanscedomainesontcertifiéesISO 9001 par l’AFNOR, l’organisme français d’accréditation, depuis 2010.
DanslecontextedesOMCL,le«Réseau»signifielamiseencommun de savoir-faire au sein d’une communauté d’experts, le partage du travail et la reconnaissance mutuelle des résultats expérimentaux obtenus sur la base de procédures approuvées collectivement et, par conséquent, une meilleure gestion des ressources et des coûts associés au contrôle des médicaments. Pour les autorités compétentes nationales, il est le moyen d’éviter la duplication des travaux et d’accéder à des technologies de pointe et des procédures d’analyse sélectives.
Outre les activités fondamentales du Réseau, de nouvelles initiatives ont été lancées au cours des années récentes et de nouveaux programmes ont été mis en place, en particulier
1.5 Réseau des OMCL

19
Les deux réunions tenues par le groupe de travail en 2012 ont conduitàlafinalisationd’undocumentdepolitiquegénéraledu Réseau “API Surveillance – Position Paper for OMCLs”, qui porte sur la contribution du Réseau OMCL à la surveillance du marché européen pour les substances pharmaceutiques actives. Une page consacrée aux travaux du groupe de travail, sur le site internet de la DEQM, est en préparation. Le document de politique générale sera publié sur cette page.
Un autre sujet de discussion, en 2012, a été la question de la réorientationduprojet«fingerprint».Ceprojet,dontl’objectifest de déterminer l’authenticité et la source des substances pharmaceutiques actives via différentes mesures permettant uneidentificationuniquedecessubstances(techniquesdefingerprinting, analyse chimiométrique, par exemple), a été conduit au cours des deux dernières années en collaboration avec des fabricants pharmaceutiques. La réorientation envisagée vise à accroître le rôle des OMCL dans le programme. Une étude de surveillance du marché (MSS) pilote, portant sur un groupe sélectionné de substances, est programmée pour 2013.
Groupe de travail sur les médicaments contrefaits/illégaux
Ce groupe de travail, créé à la suite du premier « Symposium des OMCL sur la contrefaçon » s’est réuni 2 fois en 2012, en vue de la préparation et la réalisation d’une première MSS sur des produits illégaux (MSS-IP), ciblant des compléments alimentaires à effet amincissant annoncé. Une seconde MSS de ce type est programmée pour 2013. A l’actif du groupe est également à mentionner l’organisation d’une première session de formation technique à l’intention des membres du Réseau OMCL, organisée conjointement à Varsovie, les 4-5 octobre 2012, par l’OMCL polonais et la DEQM. Les retours positifs des participants ont conduit à organiser une deuxième session en 2013.
Legroupeconstitueégalementl’assisescientifiqued’autresactivités du Réseau dans le domaine des médicaments falsifiés.L’uned’ellesestl’harmonisationdesrapportsd’essaisurlesmédicamentsfalsifiésoucontrefaitsqui,depuis2006,sont rassemblés dans une base commune, d’accès restreint aux membres du Réseau. Il est prévu, dans les années à venir, d’approfondir le partage des expertises au sein du Réseau et d’étendre la communication à d’autres partenaires également impliqués dans la lutte contre les médicaments contrefaits (douanes, police, autorités de santé).
Par ailleurs, une étude « SUP » (Suspicious Unknown Product) a été conduite en 2012. Des échantillons inconnus ont été envoyéspouridentification(substancesactives:JWH-081et butylone) et 20 participants ont fourni des résultats. Ce programme, organisé par la DEQM, vise à évaluer la capacité desOMCLduRéseauàidentifier(etsipossiblequantifier)lessubstances actives inconnues dans un échantillon sélectionné.
Les activités du Groupe sont décrites dans le détail sur le site internet de la DEQM, sur lequel se trouvent également des liens vers les sites des autorités nationales actives dans ce domaine.
afin de leur permettre d’échanger leurs expériences, de s’informer sur les nouveaux guidelines relatifs au management de la qualité et d’harmoniser les exigences appliquées lors des audits.
Coopération avec l’EA
A l’initiative de la DEQM, des contacts ont été ré-établis en 2012 avec l’EA (European Co-operation for Accreditation) en vue de la réalisation d’audits communs entre EDQM/MJA et organismes nationaux d’accréditation. L’objectif à long terme de cette initiative est la reconnaissance mutuelle des résultats d’audits.
Guidelines relatifs au management de la qualité
De nouveaux guidelines OMCL ont été adoptés par correspondanceet confirmés lorsde l’assembléeannuellede juin 2012 : un guideline sur la gestion des réactifs (PA/PH/OMCL (11) 157 5R) et un guideline sur la manipulation et l’utilisation des étalons de référence dans le réseau OMCL (PA/PH/OMCL (11) 204 3R). Le guideline sur la qualification des titrateurs automatiques a été révisé et est entré en vigueur dans sa nouvelle version (PA/PH/OMCL (07) 108 4) le 1er mai 2012.Le guideline en vigueur sur l’évaluation et le compte-rendu des résultats est en cours de réexamen. De nouveaux guidelines sont en cours d’élaboration sur la qualification des balances et sur la qualification des colonnes analytiques et il est prévu d’élaborer en 2013 un nouveau guideline sur l’étalonnage de pH-mètres, qui sera utilisé comme base de révision du chapitre général de la Ph. Eur. Détermination potentiométrique du pH (2.2.3).
Lors de la réunion annuelle du Réseau des OMCL, à Copenhague, il a été décidé de ne plus publier le « Quality Management book» ; lesversionsenvigueurdesdifférentsguidelines MQ sont en effet disponibles individuellement sur le site internet de la DEQM ainsi que les documents importants en matière d’assurance qualité (AQ).
Groupe de travail sur les substances actives
La priorité du groupe de travail sur les substances actives est de mettre en lumière la contribution des OMCL au contrôle de la qualité des substances pharmaceutiques sur le marché européen. Le renforcement du contrôle des substances actives est également un objectif majeur des autorités nationales compétentes européennes, ainsi que de l’EMA et le sommet international des Heads of Medicines Regulatory Agencies, en 2008, a également mis l’accent sur la nécessité de ce renforcement. La question du contrôle des substances actives se pose dans un contexte de mondialisation de la fabrication et de l’échange des substances pharmaceutiques. L’entrée en vigueur en 2013 de nouvelles dispositions réglementaires de l’UE (Directive 2011/62/EU, dite directive sur lesmédicaments falsifiés) est un autre élémentqui, àl’avenir, rendra nécessaire une implication accrue des OMCL dans la surveillance du marché européen pour les substances pharmaceutiques actives.

20
Groupe de contrôle des OMCL sur les préparations pharmaceutiques non soumises à AMM
Les principaux objectifs du groupe sont de valoriser l’importante contribution des OMCL au contrôle de la qualité des préparations pharmaceutiques non soumises à AMM et de fournir des lignes directrices sur les stratégies d’échantillonnage, la sélection de méthodes d’essai et, le cas échéant,lafixationdespécifications.LesrésultatsdelaMSSportant sur des préparations pédiatriques non soumises à AMM (capsules et suppositoires), décidée lors de la première réunion du Groupe en décembre 2011, seront disponibles dans le courant du 4ème trimestre 2013.
Groupe de travail sur les stocks constitués de produits biologiques
Après l’élaboration en 2009 d’un premier guideline technique décrivant la contribution des OMCL à la surveillance des stocks constitués de médicaments (document PA/PH/OMCL (09) 94 3R, disponible sur le site de la DEQM), un groupe de travail a été constitué à l’été 2011, suite à une décision prise lors de l’assemblée annuelle du Réseau OMCL, à Düsseldorf, dans l’objectif d’adapter le document actuel auxspécificitésdesproduitsbiologiquesdesstocksnationaux(vaccins et immunosérums). Le document révisé a été adopté lors de l’assemblée du Réseau OMCL de juin 2012 et publié sur le site internet de la DEQM. Le guideline révisé étant entré en application, il a été décidé de ne plus tenir de réunions régulières du Groupe, jusqu’à ce qu’en existe à nouveau le besoin.
Groupe de travail sur les produits de thérapie génique (GTP)
Le Groupe de travail GTP des OMCL a été mis en place en 2008afindepréparerlesOMCLaurôlequ’ilssontappelésàjouer dans la surveillance de la qualité des produits de thérapie génique. Il a pour objectif de favoriser la collaboration entre les OMCL travaillant dans le domaine des thérapies géniques, afindegagnerdutempsetd’économiserdesressourcesparun partage des connaissances et des technologies. En 2012, trois nouveaux membres se sont joints au Groupe : les OMCL autrichienne, belge et canadienne.
Lors de la 4ème réunion du groupe en décembre 2012, le programme de travail a été passé en revue.
En 2012, 3 études collaboratives ont été engagées sur des virus adéno-associés, dans l’objectif de valider la transférabilité des méthodes suivantes : détermination du titre en particules physiques par ELISA, détermination du titre en génome viral par PCR (polymerase chain reaction) quantitative et de la pureté par électrophorèse en gel de by polyacrylamide. L'étude ELISA a été menée à terme et sera publiée en 2013 dans Pharmeuropa Bio and Scientific Notes. Des travaux complémentaires ont également été conduits sur une méthode d’électrophorèse capillaire (EC) destinée à la détermination de la concentration et de la topologie de l’ADN. La prochaine réunion du Groupe de travail est prévue à l’automne 2013, dans les locaux de l’OMCL suisse Swissmedic, à Berne.
17ème assemblée annuelle du Réseau européen général des laboratoires officiels de contrôle des médicaments (GEON)
La 17ème assemblée annuelle du Réseau européen général des OMCL s’est tenue à Copenhague du 11 au 15 juin 2012. Elle était organisée avec l’aide et le soutien de l’Autorité danoise en matière de santé et de médicaments, pendant la présidence danoise de l’Union Européenne. Elle s’est déroulée en présence de 220 experts, venus de 34 pays et représentants 48 institutions, réunis pour partager leur expérience et discuterde thèmesd’intérêt communafindecoordonneret d’harmoniser leurs efforts en faveur de la protection de la santé humaine et animale en Europe.
Lors de huit sessions individuelles, des discussions entre experts se sont déroulées sur le contrôle en laboratoire des substances actives, des produits pharmaceutiques, des produitsbiotechnologiquesetsurlalibérationofficielle,parles autorités de contrôle, des lots de vaccins humains, de dérivés du sang et du plasma humains et des médicaments immunologiques à usage vétérinaire. Le Groupe de Travail sur les médicaments contrefaits/illégaux s’est par ailleurs réuni pour la première fois à l’occasion de cette assemblée.
Assemblée annuelle des OMCL - session générale/ textes de politique générale
Divers sujets ont été abordés lors de la session générale de l’assemblée annuelle, qui était ouverte à tous les membres du Réseau (membres à part entière, membres associés et membres à participation limitée) :
• Une nouvelle procédure relative au maintien del’appartenance au Réseau OMCL a été adoptée en séance plénière. Ce document, qui a été publié sur le site de la DEQM comme Annexe 5 au Mandat du GEON, introduit un certain nombre de mécanismes de suivi des membres du Réseau et permet de suspendre ou limiter leur appartenance au Réseau.
• Lamiseenapplicationdelaprocédured’appartenanceauRéseau a un impact sur plusieurs importants documents depolitiquegénéraleduRéseau;lemandatduGEONetcertaines de ses Annexes en particulier, ont été révisés en conséquence. Ces mises à jour sont disponibles sur le site internet de la DEQM.
• Une procédure interne de gestion de crise, qui devraitpermettreauRéseauOMCLderéagirefficacementdansdes situations de mise en danger de la santé des patients suite à des problèmes de qualité de médicaments, a été présentéeetdiscutée;cetteprocédureadepuisétéfinaliséeet adoptée par le Réseau.
• Il a également été question d’accentuer les efforts duRéseau en matière de surveillance du marché des substances actives et, dans la même perspective, de renforcer sa collaboration avec d’autres acteurs concernés, notamment les évaluateurs qualité et les inspecteurs BPF des Autorités nationales compétentes en Europe).

21
• Dessujetsplusspécifiquesontaussiétéabordés,commela contribution des OMCL au contrôle de la qualité des produits radiopharmaceutiques, des cosmétiques et des dispositifs médicaux. Dans ces différents domaines, la valeur ajoutée apportée par les contrôles de laboratoire a été démontrée de manière convaincante.
Programme d’essais d’aptitude (PTS)
Au fil des années, les PTS ont pris de l’ampleur jusqu’à devenir un programme régulier du réseau OMCL. En 2012, 5 études ont été organisées dans le domaine physico-chimique, avec la participation moyenne de 42 laboratoires de contrôle nationaux et 31 autres laboratoires de contrôle pharmaceutique relevant du secteur privé – industrie ou secteur hospitalier. Dans le domaine biologique, 3 études ont été organisées, avec la participation moyenne de 21 laboratoires (10 OMCL et 11 du secteur privé).
Deux nouvelles études ont également été organisées dans le cadre du 5ème programme PTS établi avec l’OMS, l’une sur l’essai de dissolution et l’autre sur les dosages réalisés par chromatographie. Ces études sont conduites, en moyenne, par une soixantaine de laboratoires de contrôle gouvernementaux exerçant dans les six régions de l’OMS (Afrique, Amériques, Méditerranée orientale, Europe, Asie duSud-Est,Pacifiqueoccidental).
Études générales de surveillance du marché
Les études de surveillance du marché (MSS) ont pour objectif de surveiller la qualité des médicaments commercialisés sur le marchéeuropéen.En2012,desMSSontétéfinaliséespourles formes orales d’acide acétylsalicylique, pour le clopidogrel (substance active et comprimés) et pour les substances actives et médicaments de la catégorie des sels de mésilates. Ces campagnes de contrôle apportent une vision d’ensemble de la qualité des produits présents sur le marché européen, pour une catégorie de produit donnée. Elles peuvent, le cas échéant, conduire à la révision des monographies et/ou chapitres généraux/méthodes concernés, ainsi qu’à des actions spécifiquesdesautoritésd’enregistrementoudesurveillance.
Une MSS « atypique » a également été lancée en 2012, voir pour plus de détail la section consacrée plus haut au « Groupe de travail sur les médicaments contrefaits/illégaux », ainsi que la section 1.7 Activités liées au suivi pharmaceutique et à la lutte contre la contrefaçon, page 19).
CombiStats
En 1999, la DEQM s’est engagée dans le développement d’un programme informatique destiné à l’évaluation statistique des titrages biologiques par dilution, tels que définis dans le chapitre 5.3 de la Ph. Eur. Jusqu’alors, la plupart des laboratoires du réseau OMCL utilisaient des logiciels qu’ils avaient eux-mêmes développés, d’où une forte demande pour un programme collectif qui permettrait d’harmoniser la présentation et l’analyse des résultats de titrages. L’absence
de logiciel approprié sur le marché a conduit au développement de CombiStats™, que le réseau utilise maintenant depuis l’année 2000 à la satisfaction générale. L’accès à ce logiciel était initialement réservé aux OMCL, mais, depuis novembre 2005, les autres laboratoires (non-OMCL) peuvent également obtenir une licence utilisateur. Le nombre des utilisateurs ne cesse d’augmenter depuis que le logiciel est d’accès public.
Deux sessions de formation, ouvertes à des participants de l’industrie et du secteur privé, ont été organisées en mars et novembre 2012. Une Version 5.0 de CombiStats est en développement depuis 2012 et devrait être disponible en 2013.
En décembre 2012, sur l’ensemble des licences délivrées, 14 % l’ont été à des laboratoires OMCL (de 24 pays) et 86 % à des utilisateurs non-OMCL (de 40 pays). Le diagramme ci- dessous montre que la moitié environ des licences non-OMCL ont été délivrées au sein de l’UE, l’autre moitié se répartissant sur le reste du monde, notamment des pays non-européens tels que l’Argentine, l’Australie, le Brésil, le Canada, la Chine, l’Égypte, l’Inde, l’Indonésie, Israël, le Japon, la Malaisie, le Mexique, l’Afrique du Sud, la Corée du Sud, la Syrie, Taïwan, la Tunisie, l’Uruguay, les USA. CombiStats™ est donc devenu une référence internationale dans son domaine, et contribue à la reconnaissance mutuelle des données et des résultats d’analyse.
Activités concernant spécifiquement les pays de l’UE/EEE
Répartition géographique des licences Combistats™ (en%)

22
Programme de contrôle des produits autorisés via les procédures de reconnaissance mutuelle (PRM) ou décentralisée (PDC)
Un dispositif commun de surveillance du marché existe également pour les produits PRM/PDC.Mis en place fin2010, sur la base du volontariat, à l’initiative de membres du réseau OMCL des pays de l’espace économique européen (EEE) et de la DEQM, il s’est largement développé depuis. En évitant la duplication des essais réalisés sur un même produit dans différents États membres, le dispositif assure une approche coordonnée et efficiente de la surveillancepost-commercialisation.
En 2012 a été conduit le 8ème programme régulier de surveillance du marché des médicaments autorisés par les procédures PRM/PDC au sein de l’EEE. Le nombre de projets inscrits au programme – plus de 800 – était plus élevé que pendant les trois années précédentes. Le nombre des participants par programme s’est stabilisé au cours des dernières années autour d’environ 25 OMCL.
Environ 10 % des échantillons contrôlés dans le cadre du programme provenaient d’un État membre ou d’un OMCL non impliqué dans le contrôle. Ceci témoigne de la valeur ajoutée du programme de surveillance en termes de partage des travaux. Dans 3 % des cas, des défauts de nature réglementaire (méthode d’essai insuffisamment détaillée, formuledecalculerronéedanslaSOP,...)ontétéidentifiéslors du contrôle des produits et, dans 2 % des cas, au moins un résultathors spécificationaétédétecté.Ces résultatsattestent de la bonne qualité des médicaments présents sur le marché européen. Environ 5 % des produits contrôlés étaient des produits à usage vétérinaire et 3 % des produits
Nombre de produits testés dans le cadre des programmes CAP 2007-2012
Le programme 2012 portait sur 33 médicaments à usage humain (11 produits biologiques, 22 produits chimiques) et 8 médicaments à usage vétérinaire (4 produits immunobiologiques, 4 produits chimiques). Le contrôle des substances actives, en plus de celui de la forme pharmaceutiquefinie,aétéréalisépour2produits.Lenombretotal de produits contrôlés (41) se situe dans l’ordre de grandeur optimal compte tenu des capacités opérationnelles du réseau OMCL.
Une procédure CAP normalisée pour les futurs programmes d’essai portant sur des génériques a été élaborée sur la base de l’expérience acquise lors du programme expérimental conduit en 2011 sur le clopidogrel. Cette procédure est une adaptation de l’actuelle procédure CAP d’échantillonnage et d’analyse.
Les activités de coordination assurées par la DEQM pour le programme d’échantillonnage et d’analyse CAP sont certifiéesISO9001depuis2010.Unauditdesuiviannuels’estdéroulé avec succès en décembre 2012.
Surveillance du marché des produits autorisés par la voie centralisée (CAP)
Le programme d’échantillonnage et de contrôle des produits autorisés par voie centralisée (CAP) s’est poursuivi avec succès en 2012 et entre dans sa 14ème année. Le programme CAP est conduit sur la base d’un contrat entre l’EMA, qui le parraine et en assume la responsabilité globale, et la DEQM, qui coordonne les opérations d’échantillonnage et de contrôle sur la base des informations fournies par les détenteurs d’AMM à la demande de l’EMA. Il concerne à la fois les médicaments à usage humain et les médicaments à usage vétérinaire.

23
biologiques, ce qui correspond à la répartition générale des types de produits enregistrés via les procédures PRM/PDC. En 2012, pour la première fois, les produits PDC inscrits au programme ont été plus nombreux que les produits PRM.
La procédure générale « Co-operation in post-marketing surveillance of Mutual Recognition/Decentralised Procedure Products » (PA/PH/OMCL (06) 116) a fait l’objet de modifications concernant la section où sont décrites les actions de suivi à mettre en place lorsque des non-conformités sont constatées. La version la plus récente de la procédure a été postée sur le site internet de la DEQM en septembre 2012.
Le développement d’une base de données interne au Réseau servant à organiser le contrôle des produits PRM/PDC (planification,échantillonnage,rapport)s’estpoursuivi.Autotal, 11 améliorations ont été apportées à la base en 2012, à l’initiative tant des OMCL utilisateurs du système que du Secrétariat de la DEQM. La base de données a été étendue de façon à prendre en compte les activités de contrôle réalisées sur des substances actives indépendamment du type d’AMM du médicament correspondant. Par ailleurs, une formation de rappel a été organisée en novembre 2012 à l’intention des utilisateursclésdelabasededonnées;unreprésentantdelaCroatie, pays en voie d’adhésion à l’UE, y participait pour la première fois.
Les activités de coordination assurées par la DEQM en matière de surveillance du marché des produits PRM/PDC sontcertifiéesISO9001.Unauditdesuiviannuels’estdérouléavec succès en décembre 2012.
Produits biologiques à usage humain : libération offi-cielle des lots par les autorités de contrôle (OCABR)
Le Réseau OCABR regroupe un ensemble d’OMCL intéressés à l’application harmonisée de l’article 114 de la Directive européenne 2001/83/CE modifiée. L’objectif du Réseau est de créer les conditions de la reconnaissance mutuelle des contrôles de libération des lots, prévue par la Directive. Cette coopération passe notamment par des échanges réguliers d’informations, par correspondance ou dans le cadre de réunions, et par l’élaboration et le maintien à jour de guidelines communs. L’existence d’une étroite interaction entre les membres du Réseau facilite grandement le partage du travail. Les activités du Réseau et la coopération dynamique de ses membres ont pour moteur leur aspiration commune à assurer une surveillance de haute qualité, reconnue, des vaccins et des médicaments dérivés du sang présents sur le marché de l’UE (et au-delà), et à trouver des alternatives aux essais sur animaux, selon l’approche des 3R.
Dans le cadre de l’assemblée annuelle de Copenhague, en juin 2012, des sessions parallèles se sont déroulées pour les produits du sang et les vaccins, et une session conjointe a permis d’examiner les points d’intérêt commun. Quelque 80 participants, issus de 24 États membres, y assistaient. Ils ont dressé le bilan des activités de l’année précédente et définides stratégiespour lapériodeàvenir.En2012,desreprésentants croates ont également assisté aux sessions, en tant qu’observateurs, en anticipation de l’accession de leur pays à l’UE en 2013.

24
Lors de cette session plénière, 3 des 6 postes que comporte le Comité consultatif ont été renouvelés. Le Comité comprend désormais des représentants élus de l’Allemagne, des Pays-Bas et de l’Italie pour la partie « Sang », et de la France, de la Belgique et du Royaume-Uni pour la partie « Vaccins ». Par ailleurs, les derniers éléments d’un protocole d’accord avec la Direction des produits biologiques et des thérapies génétiques de Santé Canada ont été mis en place, en vue de favoriser les activités de coopération dans le domaine de la libération des lots. Ce protocole d’accord a été ultérieurement signé par les deux parties, en juillet 2012, ouvrant la perspective d’une collaboration internationale encore renforcée avec les homologues canadiens.
Un certain nombre de guidelines et procédures internes concernant le fonctionnement du Réseau ont également été révisés. Le Comité consultatif poursuit ses efforts pour promouvoir sa contribution à la surveillance des médicaments biologiques et mettre en lumière l’importance qu’ont les retombées de ces activités sur d’autres branches du système réglementaire, comme l’évaluation des produits dans le cadre de l’AMM et les inspections.
D’autres réunions ont été consacrées, dans le courant de l’année, à divers sujets spécifiques, notamment une session de formation, très appréciée, sur l’OCABR pour les médicaments dérivés du sang, à laquelle ont assisté plus de 30 participants issus des OMCL, de l’industrie et des autorités réglementaires. Le séminaire annuel de formation sur le contrôle du vaccin poliomyélitique oral en vrac a une nouvelle fois réuni des participants de l’industrie et des OMCL et s’est avéré intéressant et fructueux. Un Groupe de Travail ayant pourmissiond’identifierdesstratégiesviablespourapporterà des auditeurs externes la démonstration de l’aptitude des laboratoires, lorsque des PTS ne sont pas disponibles, s’est également réuni.
Les évolutions décidées lors de l’assemblée de 2011 concernantlabasededonnéesOCABR,pourenaffinereten améliorer le fonctionnement, ont été menées à bien début 2012 et la base est désormais pleinement opérationnelle. Des mises à jour complémentaires sont prévues pour 2013.
Pour être plus facilement accessibles au public, tous les textes adoptés (guidelines-produits et procédures administratives) sont désormais exclusivement publiés en ligne et consultables à partir du site internet de la DEQM. Cette décision a été prise lors de l’assemblée annuelle du réseau OCABR. Les versions nouvelles ou révisées des guidelines seront publiées sur le site dans un délai d’un mois après leur date d’entrée en vigueur.
Médicaments immunologiques vétérinaires (MIV) : libération officielle des lots par les autorités de contrôle (OCABR), réseau de libération des lots vétérinaires (VBRN)
Lors de l’assemblée annuelle de Copenhague, 25 participants représentant 18 États membres ont pris part à la session consacrée au VBRN. Deux des quatre postes que compte le Comité consultatif ont été renouvelés et le Comité consultatif est désormais composé de représentants de l’Allemagne, de la Hongrie, de la Suisse et du Royaume-Uni. Comme à l’habitude, les différents États membres ont présenté des rapports annuels d’activité. Les progrès réalisés dans l’application harmonisée de l’Article 81 et de l’Article 82 de la Directive 2001/82/CE amendée, pour les médicaments vétérinaires,ontéténotés.Conformémentaumandatdéfinipar la Commission européenne dans ses recommandations sur la libération des lots de MIV, et à l’accord reçu du Comité vétérinaire pharmaceutique le 20/03/2007, le VBRN a poursuivi ses travaux de développement de guidelines communs pour l’application de l’OCABR. Une grille d’évaluation du risque a été adoptée, pour faciliter la prise de décision quant à la nécessité d’un contrôle OCABR, ainsi qu’une procédure sur les demandes temporaires d’OCABR applicables à des produits non inscrits à la liste restreinte, dans certainescirconstancesspécifiques.Uneprocéduregénéraleportant sur la révision de la liste restreinte des produits éligibles à l’OCABR a été approuvée lors de l’assemblée annuelle, puis ultérieurement adoptée après revue externe. Sadated’entréeenvigueuraétéfixéeau01janvier2013.Le Comité consultatif du VBRN a également poursuivi son action auprès du groupe européen des Chefs d’Agences du Médicament (HMA, pour Heads of Medicines Agency)afinde faire valoir la nécessité de consacrer des ressources au contrôle des MIV. Cette action a pris la forme, notamment, d’une présentation lors de la session consacrée aux produits vétérinaires de la réunion des HMA d’octobre 2012.
Le comité consultatif VBRN a rencontré des membres de l’association de fabricants IFAH-Europe en février 2012, pour discuter de sujets d’intérêt commun. La session plénière de l’assemblée annuelle a été l’occasion de diffuser auprès du Réseau général les informations qui se sont échangées.
L’ensemble des guidelines-produits et des procédures administratives adoptés, ainsi que les modèles de protocoles, sont téléchargeables sur le site internet de la DEQM. Les versions nouvelles ou révisées des guidelines seront publiées sur le site dans un délai d’un mois après leur date d’entrée en vigueur.

25
Les résultats de l’enquête et les bonnes pratiques en matière de Gestion de l’Approvisionnement en Sang ont été présentés lorsd’unsymposiumscientifiqueàStrasbourgenoctobre2012.
Le projet « Comportements à risque ayant un impact sur la gestion des donneurs de sang et la sécurité transfusionnelle » est parvenu à son terme. Il avait débuté en février 2010, avec notamment la participation de l’UE, de l’EMA, du CEPCM (Centre européen de prévention et de contrôle des maladies), de l’EBA (European Blood Alliance), de la FDA, de Santé Canada, de la TGA et de l’OMS. Le groupe a analysé les données existantes provenant d’études épidémiologiques, d’études de modélisation des risques et de programmes de surveillance et a examiné les implications possibles d’un renoncement aux critères d’exclusion permanente du donneur qui sont actuellement en vigueur en cas de comportements à risque. Ces travaux ont été réalisés en coopération étroite avec d’autres comités directeurs et groupes d’experts du Conseil de l’Europe (bioéthique, transplantation et médicaments dérivés du sang). Un projet de résolution et un mémorandum technique ont été adoptés par le CD-P-TS et soumis au Comité des Ministres du Conseil de l’Europe.
La coopération interinstitutionnelle avec l’Union européenne a facilité la continuité d’un programme européen d’évaluation externe de la qualité, avec la participation volontaire des établissements de transfusion sanguine à des essais d’aptitude (B-PTS).
L’activité liée aux B-PTS vise à évaluer la performance des laboratoires en rapport avec les essais de dépistage servant à qualifierlesdonsdesangindividuels.Depuis2010,neufétudesB-PTS ont été organisées pour le contrôle des techniques d’amplificationdesacidesnucléiques(2étudessurl’hépatiteC(VHC)et2étudessurlesvirusdel’immunodéficiencehumaine(VIH)), la sérologie (2 études sur un antigène de surface de l’hépatite B (HBsAg) et l'étude sur les anticorps anti-VIH) et l’immunohématologie (2 études sur le groupage ABO et le phénotypage rhésus). Le bon accueil reçu par l’activité B-PTS auprès des établissements de transfusion sanguine entraîne un regain d’intérêt envers la participation au programme. Des mesures ont été prises pour poursuivre les programmes d’évaluation externe de la qualité.
Depuis 2012, le programme B-PTS a été complété par un programme pilote de visites/audits par les pairs dans les établissements de transfusion européens, appelé B-MJV (Blood Mutual Joint Visits, Visites mutuelles communes pour le Sang) et B-MJA (Blood Mutual Joint Audits, Audits mutuels
Transfusion sanguine
Le Comité européen sur la transfusion sanguine (CD-P-TS) s’est réuni à deux reprises, sous l’égide de la DEQM, en mars et en novembre 2012. Il a poursuivi ses travaux de préparation de la 17ème édition du « Guide pour la préparation, l’utilisation et l’assurance de qualité des composants sanguins », qui constitue uneréférenceessentielleenmatièrededéfinitiondesnormesàl’usage des services de transfusion sanguine. Ce guide servant de base à de nombreuses réglementations nationales en Europe et au-delà, le Conseil de l’Europe et l’Union européenne ont créé en 2012 un groupe de travail commun (composé d’experts issus des services de transfusion sanguine, des autorités de contrôle et des services d’inspection) dont l’objectif est de réviser le chapitre consacré aux Systèmes de qualité dans les établissements du sang et d’apporter un soutien aux experts du groupe de travail chargé de la révision du guide. Ce groupe de travail poursuivra ses travaux en 2013 pour traiter les aspects du guide liés aux BPF.
La coopération avec l’UE a également permis de débuter l’élaboration de lignes directrices communes sur les Systèmes de Management de la Qualité (SMQ) dans les établissements du sang (lignes directrices sur les « meilleures pratiques »), qui feront partie intégrante des prochaines éditions du « Guide pour la préparation, l’utilisation et l’assurance de qualité des composants sanguins » du Conseil de l’Europe.
La compilation des données relatives à la collecte, la qualificationetl’utilisationdescomposantssanguinsdanslespays européens et d’autres régions du monde jusqu’à l’année 2010 a été menée à son terme. Les rapports annuels pour 2009 et 2010, ainsi qu’une analyse de tendance portant sur les données collectées pour la période 2001-2008, sont sous presse.
La phase pilote d’une enquête sur des indicateurs de qualité destinés à optimiser l’utilisation clinique du sang s’est achevée fin2012.
Le groupe de travail ad hoc travaillant sur les questions de gestion de l’approvisionnement en sang (en rapport avec la pénurie récurrente de sang et de composants sanguins et les limitations qui en résultent en matière de thérapie transfusionnelle, et aussi l’hétérogénéité du niveau des dons dans les États membres) a validé, dans une étude pilote impliquant les banques de sang de quatre pays, un questionnaire d’auto-évaluation et a mis en place un questionnaire en ligne auquel 39 pays ont répondu.
1.6 Transfusion sanguine et transplantation d’organes

26
communs pour le Sang). Ce programme pilote se développera dans les années à venir, en appui de la mise en œuvre des systèmes de management de la qualité dans les établissements de transfusion sanguine.
Transplantation d’organes, de tissus et de cellules
Le Comité européen sur la transplantation d’organes (CD-P-TO) s’est réuni en mai 2012 à Strasbourg et en octobre 2012 à Budapest, à la veille de la 14ème Journée européenne du Don d’organes et de la Greffe organisée par le Conseil de l’Europe et les autorités hongroises.
Le CD-P-TO a poursuivi le développement de deux guides :
• Le Guide sur la sécurité et l’assurance de qualité de la transplantation d’organes (5ème édition), traite des différents aspects du don d’organes et du processus de transplantation, en couvrant des questions allant de l’évaluation des risques à la transmission de maladies. Il propose une compilation des informations à fournir aux professionnels de la transplantation avec un aperçu utile des progrès les plus récents dans le domaine;
• le nouveauGuide sur la qualité et la sécurité des tissus et cellules, fournit des informations et des conseils de qualité à tous les professionnels concernés par le don, les banques, la libération, la distribution et la transplantation de tissus et de cellules et ceux impliqués dans l’inspection des établissements concernés. La combinaison de toutes ces normes en un seul volume permettra d’optimiser la qualité et de réduire les risques de ces procédures complexes, ce qui améliorera le taux de réussite des applications cliniques des tissus et cellules.
Les deux guides seront soumis à une consultation publique au début de 2013 et disponibles à l’été 2013.
La 2ème édition de la compilation des conventions, des résolutions, des recommandations et de plusieurs rapports pertinents du Conseil de l’Europe sur la sécurité, la qualité et les questions éthiques relatives au prélèvement, à la conservation et à la transplantation d’organes, de tissus et de cellules a été publiée (voir section 2.2 Activités liées aux technologies de l’information et aux publications, page 33).
Newsletter Transplant, la publication des données internationales sur le don et la transplantation d’organes pour 2011, a été publiée en septembre 2012.
Dans le cadre de ses activités de promotion de la non commercialisation du don d’organes et de lutte contre le trafic d’organes, de tissus et de cellules, le CD-P-TO, en collaboration avec le Comité européen pour les problèmes criminels (CDPC) et le Comité de bioéthique (DH-BIO) du Conseil de l’Europe, a participé à l’élaboration d’une nouvelle conventiondedroitpénalpourluttercontreletraficd’organes.Le processus de rédaction s'est achevé en décembre 2012 et le textefinalserasoumisauComitédesMinistresduConseildel’Europe pour adoption au début de l’année 2013.
En juillet 2011, le Conseil de l’Europe a lancé un projet de collaboration de trois ans pour lutter contre les pénuries d’organes et améliorer l’accès aux services de transplantation
dans les états de la région de la Mer Noire membres du Conseil de l’Europe (Arménie, Azerbaïdjan, Bulgarie, Fédération de Russie, Géorgie, Moldavie, Roumanie, Turquie et Ukraine) en développant des programmes de dons et de transplantation répondant à des normes d’éthique et de sécurité. Les efforts se sont principalement orientés vers le développementdecadreslégislatifsefficacesetl’établissementd’autorités, de structures et de programmes nationaux de transplantation et vers l’analyse des pratiques cliniques en matière de don/transplantation au sein des hôpitaux des pays disposant de programmes de transplantation. Un conseil consultatif d’experts issus de pays disposant de systèmes de transplantation bien établis a assuré un suivi des avancées de ce nouveau projet et a proposé son aide. En mai 2012, tous les participants à ce projet se sont réunis à Strasbourg, réunion à laquelle ont participé les représentants permanents au Conseil de l’Europe de l’Arménie, l’Azerbaïdjan, la Géorgie, la Roumanie, la Fédération de Russie, la Turquie et l’Ukraine.
La mondialisation et le droit à la libre circulation des patients ont augmenté la probabilité d’inscriptions simultanées d’un même patient, pour le même type d’organe, sur les listes d’attente de différentes organisations d’échanges d’organes, quelle que soit la nation d’origine ou de résidence du patient. Le CD-P-TO, en collaboration avec le Comité de Bioéthique (DH-BIO) du Conseil de l’Europe, a étudié la question des inscriptions multiples pour déterminer si elles créent une situation d’inégalité de l’accès à la transplantation (selon les moyens et les ressources des individus) et empêchent un suivi clinique approprié des patients pendant la période d’attente.
La mobilité des citoyens par rapport aux frontières juridictionnelles est un phénomène probablement appelé à se banaliser et la possibilité de voir des personnes, en dehors de leur pays de résidence, qui décèdent dans des conditions permettant d’envisager un don ou qui nécessitent une transplantation, devrait se développer. Par conséquent, le CD-P-TO a également analysé, d’un point de vue juridique et/ou pratique, les approches actuellement utilisées par les États membres pour obtenir un consentement au don d’organe d’un donneur décédé, plus particulièrement quand le défunt est non résident. Le CD-P-TO a aussi étudié les pratiques de transplantation pour les non résidents dans les États membres, en particulier en ce qui concerne l’accès aux listes d’attente, la transplantation (y compris à partir d’un donneur vivant), l’allocation et l’aide sanitaire et sociale. Certains des résultats obtenus sont disponibles dans Newsletter Transplant 2012.
Les pays poursuivent leurs travaux en vue d’optimiser les programmes de dons de donneurs décédés et faire en sorte que le taux de dons de ce type, qui a déjà considérablement augmenté au cours des dernières années, continue sa progression. Dans aucun pays cependant, les organes provenant des seuls donneurs décédés ne parviennent à couvrir les besoins. Dans la plupart des centres de transplantation en Europe, le nombre d’inscrits sur liste d’attente dépasse largement le nombre d’organes disponibles. Les temps d’attente peuvent atteindre huit ans et de nombreux patients voient leur état se détériorer ou meurent dans l’attente d’un organe. Étant donné que la transplantation de reins de donneurs vivants est devenue une procédureefficacequisauvedesvies,leCD-P-TOatravailléà

27
1.7 Activités liées au suivi pharmaceutique et à la lutte contre la contrefaçon
Les activités relatives aux pratiques pharmaceutiques – à l'exception du système « eTACT » et de la base de données « Fingerprints » – sont conduites par des comités d’experts travaillant sous l’égide du Comité européen sur les produits et les soins pharmaceutiques (CD-P-PH). Elles ont pour objectif commun le développement et la promotion de bonnes pratiques de prise en charge pharmaceutique et la protection de la santé publique vis-à-vis des contrefaçons et autres médicaments illégaux (« infractions similaires »).
Bon usage des médicaments
Les autorités publiques, ainsi que les fabricants et les distributeurs, consacrent des ressources considérables pour assurerlaqualité,lasûretéetl’efficacitédesmédicaments.Cependant, le bon usage d’un médicament est tout aussi important que la qualité du produit pour qu’une médication produise un résultat optimal sur un patient donné. Le concept des soins pharmaceutiques s’entend comme un concept de qualité et une méthode de travail visant la délivrance responsable d’un traitement pharmaceutique dans le but d’obtenir des résultats précis qui améliorent la qualité de vie dupatient(voirladéfinitiondeHepleretStrand).LeComitéd’experts sur les normes de qualité et de sécurité relatives à la pratique et au suivi pharmaceutiques (CD-P-PH/PC) a élaboré des indicateurs scientifiques supplémentaires pourmesurer la qualité des soins pharmaceutiques en Europe. Ces
indicateurs couvrent la prestation d’actes ou de services par les professionnels de santé comme les médecins, les pharmaciens etlesinfirmièresetsontaxéssurlesrésultatsetsurlepatient.Les informations provenant des indicateurs seront d'une utilité pratique en matière de normalisation pour les décideurs et les associations professionnelles.
Des "études pilote" sur une séried’indicateurs spécifiquesd’application générale sont en cours dans plusieurs pays. Ces indicateurs couvrent la qualité des connaissances, la mise en œuvre des so ins pharmaceut iques et la cogestion des plans thérapeutiques entre patients et professionnels de santé. Ils sont présentés dans un rapport intitulé « Soins pharmaceutiques. Politiques et
pratiques pour un système de santé plus sûr, plus responsable etprésentantunbon rapport coût/efficacité (2012)».Ces"études pilotes" sont menées par huit institutions universitaires en Europe. Ce rapport recommande aux gouvernements de mettre en œuvre les soins pharmaceutiques dans leurs systèmes de santé nationaux en utilisant des indicateurs de base et de renforcer la participation des patients. Le rapport a été présenté aux Pays-Bas lors d'un Sommet ministériel intitulé « The responsible use of medicines, setting policies for better and cost-effective healthcare », le 3 octobre 2012. Les participants ont jugé ce rapport stimulant et conforme aux politiques mises en œuvre.
Les médicaments issus de processus industriels n’étant pas toujours adaptés aux besoins des patients, la préparation de médicaments en pharmacie reste une pratique importante. Le CD-P-PH/PC a rédigé la Résolution CM/ResAP(2011)1 du Comité des Ministres sur les exigences relatives à l’assurance de qualité et d’innocuité des médicaments préparés en pharmacie pour les besoins particuliers du patient. Dans le cadre de la stratégie de promotion de la mise en œuvre de cette résolution, deux webinaires ont été organisés, à l’intention des autorités compétentes (juin 2012) et des associations de pharmaciens (novembre 2012).
Face au succès croissant que connaissent en Europe les médecines traditionnelles étrangères, notamment la médecine traditionnelle chinoise (MTC), le CD-P-PH/PC a poursuivi son approche stratégique en vue de garantir en Europe une pratique sûre de la médecine traditionnelle chinoise. Une étude pilote a été menée auprès de patients et de consommateurs afindevaliderlesinformationsrelativesauxpratiquesdeMTCen Europe, de les aider dans leurs choix en matière de soins et de faciliter la communication avec leurs prestataires de soins. En outre, un concept de programme d’enseignement/formation des thérapeutes et des pharmaciens en Europe a été étudié.
1. Hepler, D.D. & Strand, L.M, Opportunities and Responsibilities in Pharmaceutical Care, Am.J.Pharm.Educ.,53, 7S-15S(1989).
l’élaboration d’une stratégie commune pour favoriser, en toute sécurité, les programmes de dons de donneurs vivants à travers l’Europe, tout en veillant à la bonne santé et à la sécurité des donneurs et en tenant compte de tous les aspects juridiques, sociologiques et psychologiques de ce type de don. Ainsi ont été élaborés deux projets de résolution sur « l’utilisation de reins de donneurs vivants pour la transplantation » et « l’établissement de registres/bases de données des donneurs vivants au niveau national/supranational ». Ces projets seront soumis au Comité des Ministres pour examen en 2013.
Le Conseil de l’Europe, qui étudie la question du don de sang de cordon depuis des années, a toujours été préoccupé par la prolifération des banques de sang de cordon privées qui recueillent et conservent du sang de cordon en vue d’un usage autologue. La recommandation Rec(2004)8 du Comité des Ministres aux États membres sur les banques de sang de cordon autologue et son exposé des motifs, recommandent que les États membres n’autorisent l’établissement et l’exploitation de banques de sang de cordon que sur la base de dons volontaires et altruistes. Plus généralement, ils recommandent que la création de banques de sang de cordon autologue et la promotion du don pour une utilisation autologue ne soient pas encouragées par les États membres ou leurs services de santé. Suite à cette recommandation, le CD-P-TO a élaboré et diffusé un questionnaire pour examiner la situation des banques de sang de cordon autologue en Europe.

28
Le Comité d’Experts chargé de la classification des médicaments relativement à leurs conditions de délivrance (CD-P-PH/PHO) émet chaque année des recommandations à l’intentiondesautoritésdesantésur laclassificationdesmédicaments en fonction de leur mode de délivrance (avec ou sans prescription) qui n’est pas harmonisée en Europe. Ses travaux visent à promouvoir la sécurité des patients et l’accessibilité des médicaments en Europe. L’examen de la révisionannuelledesrecommandationsdeclassificationpour2012 a été mené à bien. La révision sera publiée en 2013 sur la page dédiée du site web de la DEQM (http://www.edqm.eu/melclass/).
Protection de la santé publique contre les contrefaçons et autres médicaments illégaux
Le Comité directeur et le Comité d’experts sur la réduction des risques de santé publique liés à la contrefaçon des médicaments et à la criminalité connexe (CD-P-PH/CMED) mènent à bien leur programme de travail à travers la prévention multisectorielle et des stratégies de gestion des risques, en soutenant, par exemple, la mise en œuvre de législations pertinentes, le transfert des savoir-faire, des propositionsdepolitiquesspécifiquesetdesoutilspratiques.Impliqués dès le départ dans le processus de développement et l’adoption de la Convention MEDICRIME du Conseil de l’Europe, ces comités ont été chargés de contribuer au suivi de la Convention dès son entrée en vigueur.
Le Comité directeur et le Comité d’experts sur la réduction des risques de santé publique liés à la contrefaçon des médicaments et à la criminalité connexe (CD-P-PH/CMED) mènent à bien leur programme de travail à travers la prévention multisectorielle et des stratégies de gestion des risques, en soutenant, par exemple, la mise en œuvre de législations pertinentes, le transfert des savoir-faire, des propositionsdepolitiquesspécifiquesetdesoutilspratiques.Impliqués dès le départ dans le processus de développement et l’adoption de la Convention MEDICRIME du Conseil de l’Europe, ces comités ont été chargés de contribuer au suivi de la Convention dès son entrée en vigueur.
En janvier 2013, la convention MEDICRIME compte vingt-deux états signataires et elle est déjà ratifée par l'un de ces états.
Le 16 mai 2012, sous la présidence danoise du Conseil de l’Union européenne, une conférence co-organisée par le Conseil de l’Europe sur le thème « Combattre la contrefaçon des produits médicaux et les infractions similaires par l’intermédiaire d’instruments juridiques et de mesures pratiques » s’est déroulée à Copenhague. Elle a été suivie par des fonctionnaires et des experts d’États membres du Conseil de l’Europe et de la Convention relative à l’élaboration de la Pharmacopée Européenne et d’autres États. La conférence a permis de développer une approche stratégique en vue de soutenir la mise en œuvre des deux instruments juridiques complémentaires que sont la Convention MEDICRIME du Conseil de l’Europe et la directive européenne 2011/62/UE surles«médicamentsfalsifiés».LeCD-P-PHetleCD-P-PH/
CMEDontcontribuédemanièresignificativeauprogrammede cette conférence et leurs suggestions pratiques à l’intention desétatsfigurentenannexeauxconclusionsdelaconférence.
Dans le cadre de la plate-forme de formation de lutte contre le médicrime, soutenue par la DEQM, le CD-P-PH/CMED a co-organisé :
• Avec la Medicines and Healthcare Products Regulatory Authority (MHRA) du Royaume Uni, une session de formation régionale destinée aux fonctionnaires de Bulgarie, de Hongrie, de Pologne, de Roumanie, de la République slovaqueetdeTurquie,les27-29mars2012;
• Aveclesautoritésarméniennesdesanté,unesessiondeformation régionale destinée aux fonctionnaires d’Arménie, de Biélorussie, de Géorgie, de Moldavie, de la Fédération de Russieetd’Ukraine,les7-8novembre2012;
• Avec les autorités croates de réglementation desmédicaments, une session de formation régionale à l’intention des fonctionnaires d’Albanie, de Bosnie-Herzégovine, de Croatie, de Grèce, du Monténégro, de Serbie, de Slovénie et de l’ex-République yougoslave de Macédoine, les 4-5 décembre 2012, avec, pour la première fois,uneformationspécifiquesurlesréseauxmultisectorielsentre points de contact unique (PCU).
Grâce à deux réunions d’experts, tenues le 29 octobre et le 2 novembre 2012, le CD-P-PH/CMED a également pu développer et tester un modèle d’approche basique d’application générale pour les autorités d’Afrique de l’ouest souhaitant analyser la législation, les structures et les processus permettant de mieux combattre la contrefaçon des produits médicaux et des infractions similaires et protéger la santé publique.
Les Actes de l’atelier d’experts « Communication des risques pour la santé publique posés par la contrefaçon des produits médicaux et les infractions similaires » ont été publiés, grâce au soutien apporté par l’Agence italienne du médicament

29
(AIFA) dans les processus de publication, de rédaction et de production.
Le Comité d’experts a conclu un protocole d’étude qui sera mis à l’essai en 2013 et proposera une approche visant à mieuxidentifieretsuivrelessignauxindiquantdesdommagesprovoqués sur la santé des patients et des consommateurs par les produits médicaux contrefaits et les infractions similaires.
Le service eTACT d’un coup d’œil : intérêt et valeur ajoutée
de la sécurisation des médicaments dans le contexte des nouveaux règlements européens
Dans le cadre de sa stratégie globale pour contrer la contrefaçon, le Conseil de l’Europe et la DEQM ont en outre développé un projet de service anti-contrefaçon de traçabilité des médicaments. Le service eTACT vise à fournir un système de traçabilité et de sérialisation de masse qui puisse être utilisé par les autorités et par tous les intervenants (fabricants, distributeurs, professionnels de santé et patients) sur l’ensemble de la chaîne d’approvisionnement du médicament, au sein des 37 États membres de la Pharmacopée Européenne et au-delà.
Le service eTACT aidera à lutter contre la contrefaçon en adoptant une approche harmonisée dans l’ensemble des pays participants. Il s’agira d’un système flexible qui permettra d’améliorer le contrôle de la chaîne d’approvisionnement.
Lapossibilitéofferteauxpatientsdevérifier l’authenticitéde leurs médicaments est une caractéristique unique du projetdelaDEQMquicontribuerademanièresignificativeà renforcer la confiance du public envers la chaîne légaled’approvisionnement. La gouvernance publique d’un tel système est essentielle pour assurer un développement approprié et efficace du projet, en coordination avec les autorités de réglementation, et prévenir toute utilisation abusive des données (données commerciales, par exemple).
En 2012, le système informatique eTACT a été présenté à denombreusesautoritésetautrespartiesprenantesafinderecueillir des observations de toute l’Europe. Ceci permettra à la DEQM d’élaborer un document robuste établissant
les « contraintes métier et exigences client » d’un service à échelle réelle, dont la mise en œuvre devra être conforme aux exigences nationales et régionales à venir en matière de traçabilité des produits pharmaceutiques en Europe.
Des progrès ont également été réalisés en ce qui concerne la fonctionnalité du système, la gouvernance harmonisée des parties prenantes et la traçabilité, offrant ainsi aux autorités et aux patients un outil performant.
Référentiel « Fingerprints » des substances actives pharmaceutiques
La DEQM a également élaboré un projet de référentiel des « empreintes digitales » ou signatures, des substances actives pharmaceutiques servant à la fabrication des médicaments. A cet effet, la DEQM a collaboré avec un groupe de travail dédié du réseau OMCL. (Voir section 1.5 Réseau des OMCL, page 18).
En 2012, dans le cadre d’un "projet pilote", les OMCL impliqués ont réalisé un certain nombre d’études, en analysant différentes méthodes et différents échantillons fournis par les sociétés ayant adhéré au projet. En 2013, la DEQM et le réseau OMCL attaqueront une deuxième phase duprojet,enprofitantdel’examendelapremièrephasepourélargir le champ d’application à des méthodes d’analyse plus transversales, sur un plus grand nombre d’échantillons de substances actives cibles.

30
conformément aux normes internationales. Sous l’égide de la DEQM, des études analytiques collaboratives et des réunions d’experts sont organisées, auxquelles participe tout ou partie des laboratoires participants. La longue expérience acquiseavecleréseaudeslaboratoiresofficielsdecontrôledes médicaments (OMCL) a constitué un atout dans la phase de démarrage. Le réseau OCCL a établi des contacts étroits avec la Commission européenne (la DG SANCO et le Centre commun de recherche).
La tâche principale d’un OCCL est la vérification de la qualité des produits sur le marché. Dans le cadre d’une étude de surveillance du marché (MSS) lancée en 2011 (qui se poursuivrajusqu’àlafin2013),plusieurspaysontprélevédeséchantillons de produits cosmétiques décoratifs (maquillage, ombres à paupières, eye-liner, brillant à lèvres, etc.) pour en mesurer la teneur en certains métaux pouvant donner lieu à des problèmes de santé : l’antimoine, le cadmium, le chrome, le plomb, le mercure et le nickel. Même si la présence à l’état de traces de certains de ces métaux peut être inévitable pour desraisonstechniques,iln'estpasfixédelimitesmaximalesacceptables dans la plupart des pays. Les résultats de cette étude peuvent permettre d’établir des valeurs de référence communes pouvant servir aux autorités de surveillance.
La qualité des produits cosmétiques destinés à être utilisés sur ou par des enfants, et leur conformité aux réglementations de l’UE, ont été testées dans une MSS commencée en 2012 (poursuivie en 2013). La conformité aux réglementations européennes ou nationales pertinentes de shampooings, pommades, lotions pour le bain et de plusieurs autres types de produits sera contrôlée.
Pourvérifierlaperformancedeslaboratoiresdanslecontrôled’ingrédients cosmétiques spécifiques et veiller à ce que les résultats des essais soient comparables en Europe, des programmes d’essais d’aptitude (PTS) sont réalisés. En 2012, la gamme des produits comprenait les dentifrices, les défrisants et les crèmes dépilatoires. La quantité de diéthylène glycol a été déterminée dans les dentifrices, tandis que les défrisants et crèmes dépilatoires étaient contrôlés pour déterminer la teneur de l’acide thioglycolique, un ingrédient cosmétique.
Les produits solaires contiennent des substances filtrant ou bloquant la lumière UV et de nombreux produits indiquent un « facteur de protection solaire » (FPS). Une surveillanceefficacedumarchépourcesproduitscomprendlavérificationdecefacteur,pardesméthodesinvivoetinvitro appropriées. Pour examiner l’évolution des technologies en matière d’analyse et de surveillance dans ce domaine, un séminaire a été organisée en avril 2012 dans les locaux du CVUA (Chemischen und Veterinäruntersuchungsämter) de Karlsruhe (Allemagne). Le séminaire a permis d’assurer un suivi des sujets présentés à Montpellier (France) en 2011. Des experts de 12 pays européens y ont participé et ont convenu d’harmoniser les approches et de développer davantage les méthodologies.
1.8 Cosmétiques, emballages alimentaires et pharmaceutiques
Protection sanitaire des consommateurs
Depuis le 1er janvier 2009, la DEQM a intégré à ses activités, le renforcement de la protection sanitaire des consommateurs en Europe. Ses travaux dans ce domaine portent notamment sur la bonne utilisation des cosmétiques et sur les matériaux (d’emballage ou autre) en contact avec les aliments ou les médicaments.
Le programme de travail a été établi par le Comité de protection sanitaire du consommateur (CD-P-SC, Comité directeur), qui se compose de représentants de ministères nationaux de la santé. Plus de 200 experts provenant de 35 États membres ou observateurs auprès de la Convention relative à l’élaboration d’une Pharmacopée Européenne suivent les travaux ou y contribuent activement.
Leurs travaux sont axés sur le nouveau réseau européen des laboratoiresofficielsdecontrôledescosmétiques(OCCL)et sur le développement de normes harmonisées de qualité applicables aux matériaux d’emballage alimentaire ou pharmaceutique.
Deux comités d’experts subordonnés sont chargés de mettre enœuvre les travaux définis par leCD-P-SC: leComitéd’experts sur les produits cosmétiques (P-SC-COS) et le Comité d’experts sur les matériaux d’emballage alimentaires et pharmaceutiques (P-SC-EMB).
Contrôle des cosmétiques
Le réseau européen des OCCL nationaux, composé de membres volontaires, a été mis en place en 2010 pour mettre en commun compétences et ressources et pour améliorer le management de la qualité dans chaque laboratoire,

31
Cosmétiques pour les enfants de moins de trois ans
Les restrictions applicables aux produits cosmétiques destinés à être utilisés sur des enfants jusqu’à l’âge de trois ans font l’objet d’une nouvelle Résolution du Conseil de l’Europe adoptée le 14 mars 2012 [CM/ResAP (2012) 1]. Cette résolution et son document explicatif
supplémentaire destiné aux fabricants et aux évaluateurs sécurité ont été publiés en 2012 (« Safe Cosmetics for Young Children » - 1ère Édition). (Voir section 2.2 Activités liées aux technologies de l’information et aux publications, page 33).
Tatouages et maquillages permanents
Pour mettre en œuvre les recommandations de la Résolution AP (2008) 1 du Conseil de l’Europe sur les tatouages et les maquillages permanents, la compilation des exigences de sécurité et de documentation relatives aux tatouages et aux maquillages permanents est en préparation. Ce document devraitégalementêtrefinaliséetpubliéen2013.
Emballages alimentaires et pharmaceutiques
Une nouvelle résolution sur les métaux et alliages en contact avec les aliments a été rédigée. Elle fait référence aux exigences de qualité de matériaux comme le papier d’aluminium, les ustensiles de cuisine, les machines à café, etc... pour lesquels il n’existe aucune réglementation spécifiquedel’UE.IlestrecommandéauxÉtatsmembresduConseil de l’Europe de mettre en application des limites de migrationspécifiques(LMS)pourlesionsmétalliqueslibéréspar les matériaux en contact avec les denrées alimentaires et transférés aux aliments par les emballages ou récipients. Après l’adoption de cette résolution, prévue en 2013, ces exigences se substitueront aux lignes directrices correspondantes, publiées en 2002, relatives aux métaux et alliages utilisés comme matériaux destinés à entrer en contact avec des denrées alimentaires.
Les limites supérieures pour la libération d’ions métalliques ont été approuvées par les experts nationaux du P-SC-EMB. Par exemple, la limite maximale recommandée pour le nickel est de 0,14 mg/kg et de 0,010 mg/kg pour le plomb (concentrations mesurées dans les aliments). Des instructions détaillées quant au mode de réalisation des essais en laboratoire sont décrits dans un nouveau guide technique dont la publication est prévue en 2013.
En outre, le Comité d’experts P-SC-EMB a décidé de passer en revue les résolutions existantes et les documents techniques élaborés sous l’Accord partiel du Conseil de l’Europe dans le domaine social et de la santé publique (dissous le 31 décembre 2008). Ce travail, réparti entre plusieurs rapporteurs chargés de préparer des projets de dispositions sur des matériaux comme le liège, les résines échangeuses d’ions ou le papier et le carton, se poursuivra en 2013.
Enfin,unprojettransversalviseàlimiterl’utilisationd’encreset de colorants sur les récipients imprimés renfermant des médicaments ou de la nourriture. Une liste positive de substances sûres sera constituée. Elle pourra être consultée par les fabricants et les autorités de surveillance. Des critères généraux de qualité et des méthodes d’essai analytiques seront développés.

32
La DEQM maintient et étend sa certification ISO 9001.
À l’issue d’un audit complet de quatre jours, Afnor Certification (AFAQ) a prolongé le certificat ISO 9001 de la DEQM et élargi son
champ d’application aux activités de pharmacopée.
AFNOR Certification a certifié la DEQM selon les exigences de la norme ISO 9001:2008 pour les activités suivantes :
• « Évaluation des dossiers de demande (initiale, révision et renouvellement) de certificats de conformité aux monographies de la Pharmacopée Européenne, délivrance des certificats et gestion du programme d’inspection des sites de fabrication et des distributeurs associés,
• Programmation, mise en œuvre et coordination des études de surveillance du marché des médicaments autorisés par les procédures centralisée (CAP) et nationale (études MSS) ;
• Gestion de la base de données associée aux études de surveillance du marché des médicaments autorisés selon les procédures de reconnaissance mutuelle (MRP) et décentralisée (DCP) et gestion des interactions avec ses utilisateurs ;
2.1 Système de management de la qualité
2. LES ACTIVITÉS SUPPORT
• Coordination de l’élaboration et de la diffusion des lignes directrices relatives à la procédure OCABR pour la libération de lots de médicaments immunologiques (sang et vaccins) à usage humain, en vertu de la réglementation pharmaceutique, notamment les directives 2001/82/CE et 2001/83/CE telles qu’amendées et le Règlement 726/2004 (CE) pour les pays de l’UE ;
• Gestion de l’élaboration des révisions, des corrections et des suppressions des textes de la Pharmacopée Européenne, leur publication en format imprimé et électronique ainsi que leur distribution .
Le laboratoire de la DEQM (SLab) est audité par l’organisme belge d’accréditation (BELAC).
Afindegénérerdesdonnéesdehautequalité,SLabutiliseunsystème de Management de la Qualité qui répond à la norme ISO/IEC 17025:2005. Ce système a fait l’objet d’un audit approfondi réalisé par une équipe indépendante de quatre experts de l’organisme belge d’accréditation (BELAC) en décembre 2012. Le certificat d’accréditation ISO/IEC 17025:2005 devrait être attribué début 2013.

33
Un site internet (www.edqm.eu) toujours plus fréquenté
Un certain nombre de changements de conception, de mise en page et de contenu du site web de la DEQM ont été mis en œuvre en 2012. En juillet, l’ensemble du site a été remanié afindemieuxservirlesutilisateursetderenforcerl’identitévisuelle de la DEQM et du Conseil de l’Europe. En 2012, le site a attiré 32 774 visiteurs par mois, pour 80 157 visites (statistiques Google Analytics), soit une légère augmentation par rapport à 2011.
Plusieurs nouvelles pages web ont été créées (Protection sanitaire des consommateurs, eTACT, Medicrime, Soins pharmaceutiques, Alternatives à l’expérimentation animale, par exemple) pour promouvoir les travaux de la DEQM dans ces domaines et augmenter leur visibilité. Plusieurs autres sections et pagesont été révisées etmodernisées afindefaciliter l’accès des utilisateurs aux ressources de la DEQM. Les flux RSS ont également été améliorés pour proposer aux utilisateurs un meilleur suivi par thèmes.
Un service en ligne d’assistance aux utilisateurs (HELPDESK) incontournable
Le service HELPDESK est le premier point de contact pour obtenir des informations et de l’aide auprès de la DEQM. L’assistance ainsi fournie couvre les questions techniques ou scientifiquessurnosdifférentesactivités,ainsiquelesproduitsou services de la DEQM. Le service Helpdesk a traité 10 766 questions en 2012, ce qui représente une légère hausse en volume de 1%. La section des questions fréquemment posées (FAQ) via le HELPDESK est régulièrement mise à jour pour toujours couvrir l’ensemble des activités et répondre aux besoins changeants des utilisateurs.
Progiciel de gestion intégré (ERP, Enterprise Resource Planning)
La mise en œuvre d’un ERP standard s’est poursuivie en 2012 avec la mise en place du processus de vente et de distribution pour les publications de la DEQM et le système de gestion de la relation client (CRM). Elle se poursuivra en 2013 avec une
nouvelle boutique en ligne pour les publications et les étalons de référence. Le processus opérationnel de production, de vente et de distribution des étalons de référence sera déployé en 2013. Cette intégration permettra d’améliorer et d’uniformiser plus encore le service à la clientèle de la DEQM.
Des activités de publication dynamiques
L’unité Publications est principalement chargée de préparer la publication technique et administrative de la Pharmacopée Européenne,dePharmeuropa,PharmeuropaBio&ScientificNotes et des autres publications et guides techniques de la DEQM.
Trois suppléments de la 7ème édition de la Pharmacopée Européenne (7.6 à 7.8) ont été publiés en 2012, pour un total de 1 066 pages dans la version anglaise et 1 140 pages dans la version française. La 7ème édition (y compris le supplément 7.8) se compose de 2 205 monographies (dont les monographies relatives à des formes pharmaceutiques) et 344 textes généraux (dont les monographies générales et méthodes d’analyse).
La 1ère édition de « Safe Cosmetics for Young Children » a été publiée en 2012 et une version électronique est disponible sur le site de la DEQM. Ce guide, élaboré par le Comité d’experts sur les produits cosmétiques (P-SC-COS), stipule que les produits doivent être sans danger pour la santé des jeunes enfants et ne contenir que des ingrédients non toxiques et ainsi exclure les substances fortement allergènes ou les substances perturbant le système endocrinien. Les conservateurs ne devraient quant à eux n’être utilisés qu’aux concentrations minimalesefficaces.Desexpertsdans ledomaineont,enoutre, convenu de recommandations détaillées pour les crèmes et lotions pour bébés.
L’unité a également fourni son soutien pour la publication d’autres documents de la DEQM. En 2012, des documents traitant des domaines suivants :
• Sang et composants sanguins - «Safety, Quality, Training and Ethical Matters Concerning Preparation, Use and Quality Assurance : Council of Europe Resolutions, Recommendations and Conventions » (2012) - 1ère édition ;
• Organes,TissusetCellules-« Safety, Quality and Ethical Matters Concerning Procurement, Storage and Transplantation. Council of Europe Conventions, Recommendations, Resolutions and Reports » (2012) – 2ème édition ;
• Soinspharmaceutiques–« Policies and Practices for a Safer, More Responsible and Cost-effective Health System » (2012)
2.2 Activités liées aux technologies de l’information et aux publications
Blood and Blood Components
Safety, Quality, Training and
Ethical Matters Concerning Preparation,
Use and Quality Assurance
Council of Europe Resolutions,Recommendations and Convention
www.edqm.eu
Bloo
d an
d Bl
ood
Com
pone
nts
– P
repa
ratio
n, U
se a
nd Q
ualit
y A
ssur
ance
EDQ
M
1st Edition
1st E
ditio
nBlood and Blood Components – Safety, Quality, Training and Ethical Matters Concerning Preparation, Use and Quality Assurance
Council of Europe Resolutions,
Recommendations and Convention
1st Edition
The EDQM is a Directorate of the Council of Europe, an international organisation founded in 1949 that covers almost the entire continent of Europe. The Council of Europe aims to develop common democratic and legal principles based on the European Convention on Human Rights and other reference texts on the protection of individuals.
Organs, Tissues and Cells
Safety, Quality and Ethical Matters
Concerning Procurement, Storage
and Transplantation
Council of Europe Convention, Resolutions, Recommendations and Reports
2nd Edition

34
ont été publiés en anglais. En outre, l’Unité Publications a contribué à la publication de l’Organización Nacional de Trasplantes (ONT), « Newsletter Transplant ».
L’un des rôles importants de l’Unité Publications est d’assurer un style rédactionnel harmonisé dans toutes les publications via la relecture des textes à publier par des relecteurs scientifiques.Enoutre,l’unitéassurelemaintiendelistesdedonnées nécessaires pour les travaux de publication.
Les publications sur la transfusion sanguine et la transplantation d’organes disponibles sur un nouveau site (TOTS)
En mars, les publications concernant la transfusion sanguine et la transplantation d’organes ont été publiées en ligne, fournissant ainsi un accès électronique à quiconque ayant commandé les ouvrages papier. L’édition en ligne est proposée en plusieurs langues, certaines traductions étant d’ailleurs uniquement disponibles en ligne. Le site, réservé aux abonnés, est accessible à l’adresse : http://tots.edqm.eu/.
Pharmeuropa désormais accessible gratuitement en ligne
Les quatre numéros de Pharmeuropa publiés en 2012 contenaient 159 textes pour enquête publique. Un numéro dePharmeuropaBio&ScientificNotes,contenant9articlesscientifiquesenanglais,aétépublié.
Laversion en ligne est devenueofficielle en2012 avec lapublication de Pharmeuropa 24.1 dans la base de données « Textes pour commentaires ». La page d’accueil du site propose des liens vers des enquêtes publiques relatives à des projets de texte de la Pharmacopée Européenne ou à des questions de politique générale, vers les dernières annonces officiellesdemonographiesnouvellementadoptées,verslesactualités en matière d’harmonisation des pharmacopées, vers la tribune de lecteurs et un accès à trois bases de données :
• Textes pour commentaires, qui contiennent des propositions de monographies et de textes généraux nouveauxourévisésdestinésàfigurerdanslaPharmacopéeEuropéenneetsoumisauxcommentairesdupublic;
• Pharmeuropa Bio & Scientific Notes, qui contient toute l’actualité dans le domaine de la standardisation biologiqueetdesarticlesscientifiquesliésauxtravauxdelaPharmacopéeEuropéenne;
• Archives de Pharmeuropa, qui contiennent les numéros précédents de Pharmeuropa.
Il est possible de s’inscrire pour être averti par courrier électronique des derniers commentaires sur des projets de textes ajoutés au site. L’accès à Pharmeuropa en ligne est gratuit.Pourbénéficierd’unaccèscompletaucontenudusite, il est nécessaire d’être inscrit. Les autorités nationales ont un accès privilégié à un outil de commentaires qui leur permet de soumettre leurs commentaires directement dans la base de données « Textes pour commentaires ». Le site est disponible à l’adresse : http://pharmeuropa.edqm.eu/.
Termes normalisés
Depuis 1996, la DEQM publie des listes de termes normalisés qui proposent des vocabulaires harmonisés sur les formes pharmaceutiques, les voies d’administration et les emballages (notamment les récipients, les fermetures et les dispositifs d’administration). Ces listes ont initialement été rédigées en réponse à une demande de la Commission européenne. Elles couvrent des médicaments tant à usage humain que vétérinaire. Les termes normalisés sont utilisés dans le formulaire européen de demande d’autorisation de mise sur le marché, dans les Résumés des Caractéristiques du Produit (RCP), dans l’étiquetage des produits et dans des communications électroniques.
Des demandes de nouveaux Termes normalisés sont soumises à la DEQM par les autorités nationales, l’EMA ou l’UE, puis évaluées par le Groupe de Travail sur les Termes normalisés (STWP,StandardTermsWorkingParty),composéd’expertsélus par la Commission européenne de Pharmacopée. Le STWPseréunit3foisparanpourexaminerlesdemandesetproposerdenouveauxtermesetdéfinitionsouréviserdestermesetdéfinitionsexistants,sinécessaire.Lecaséchéant,il traite aussi les demandes par voie électronique afin de permettre des décisions entre les réunions et ainsi parvenir à des résolutions plus rapides. Tout terme normalisé accepté désignant un concept entièrement nouveau est ensuite envoyé à la Commission pour adoption par correspondance avant publication. Les termes publiés sont mis à la disposition des autorités nationales pour permettre à leurs experts de fournir des traductions qui seront soumises au Secrétariat de la DEQM pour publication.
En 2012, les premières traductions ukrainiennes ont été introduites dans la base de données, ce qui porte le nombre total de langues à 32 (albanais, allemand, anglais, bulgare, chinois,croate,danois,espagnol,estonien,finnois,français,grec, hongrois, islandais, italien, kazakh, letton, lituanien, macédonien, maltais, néerlandais, norvégien, polonais, portugais, roumain, serbe, slovaque, slovène, suédois, tchèque, turc et ukrainien).
Àlafinde2012,labasededonnéesdestermesnormaliséscomptait 771 termes uniques, dont 28 nouveaux termes ajoutés au cours de l’année, ainsi que plusieurs centaines de traductions soumises par les différentes autorités nationales.

35
2.3 Communication, événements marquants
l’outil a en outre permis de présenter sa flexibilité et son interopérabilité avec les systèmes nationaux existants dans la chaîne d’approvisionnement des médicaments.
Un troisième atelier, organisé à Strasbourg en janvier 2012 sur le thème « Alternatives au titrage d’activité de la leptospirose », a permis un partage d'informations et d'expériences sur les progrès récents dans ce domaine. Les idées avancées et les résultats obtenus ont été communiqués à la Commission européenne de Pharmacopée.
Conférences et symposiums internationaux
Pour célébrer le 20ème anniversaire de sa Procédure de Certification de Conformité aux Monographies de la Pharmacopée Européenne, la DEQM a organisé une conférence internationale en mars 2012 à Larnaca (Chypre). 150 délégués y ont participé, provenant de 35 pays et représentant toutes les régions du monde. Divers sujets ont été abordés, notamment l’évolution de la procédure de certification,lesprincipauxchangementsappliquésaucoursdesdeuxdernièresdécennies, l’utilisationdesCertificats(CEP) et le programme d’inspections. Des experts et des représentants des autorités de réglementation, dont Santé Canada, la TGA (Therapeutic Goods administration) australienne et l’EMA, ont fait part de leurs expériences et donné un aperçu de l’utilisation des CEP. Trois ateliers se sont également déroulés en parallèle et les commentaires recueillis faciliteront l’adaptation de la procédure de certificationetdesespolitiquespourmieuxrépondreauxbesoins actuels.
En mai 2012, une conférence internationale de haut niveau sur la Convention MEDICRIME a été organisée à Copenhague, au Danemark, en vue de sensibiliser à la Convention, d’en promouvoir la signature et de mettre en évidence l’importance de la coopération et du travail en réseau entre les autorités sanitaires, judiciaires et de police concernées par la lutte contre les produits médicaux falsifiés et les infractions similaires. La conférence étaitconjointement organisée par la DEQM et l’autorité danoise de santé et des médicaments, sous la présidence danoise du Conseil de l’Union européenne. (Voir section 1.7 Activités liées au suivi pharmaceutique et à la lutte contre la contrefaçon, page 27).
La DEQM a organisé une table ronde sur le thème «Identificationdesmédicamentsfalsifiés:commentmieuxprotéger les citoyens européens ? », à Bruxelles (Belgique) en juin 2012. À cette occasion, la DEQM a pu présenter le service eTACT à des journalistes, des ambassadeurs d’états membres de l’UE et des associations de patients, et attirer leur attention sur les discussions en cours au niveau européen sur la future architecture des systèmes de sérialisation de masse des médicaments visant à protéger la chaîne d’approvisionnement légale contre les médicaments contrefaits/falsifiés.
Communication avec les intervenants, nos partenaires et le grand public
La communication occupe une place stratégique au sein de l’environnement réglementaire. La DEQM diffuse des annonces importantes et communique sur les changements de politique, les lancements de produits et les nouvelles collaborations à travers des communiqués de presse réguliers. En 2012, 21 communiqués de presse ont été publiés et diffusés par courrier électronique dans le monde entier à travers divers médias, ainsi qu’aux autorités et aux associations partenaires. L’impact de ces communiqués de presse est régulièrement évalué grâce à un logiciel en ligne qui mesure le nombre defoisqu’unfichieresttéléchargéouaffiché.Ils’agitd’unprocessus en cours qui aide la DEQM à suivre régulièrement la façon dont l’information diffusée est reçue et utilisée par les journalistes.
Le bulletin d’information en ligne de la DEQM, est un bulletin mensuel gratuit qui résume les derniers événements, informations et nouvelles, avec des liens vers le site internet de la DEQM, permettant de récupérer les informations en question. Le nombre d’abonnés à ce service se porte désormais à plus de 12 600 et, en conjonction avec les flux RSS du site internet, les utilisateurs peuvent aisément être informés des nouveaux contenus en ligne via un résumé des grands titres.
Une année consacrée à des consultations sur des sujets d’actualité pour permettre un retour d’informations et mieux préparer les utilisateurs à relever les défis liés à la qualité
Dans le domaine de la communication technique et scientifique relative aux activités de la DEQM, divers événements se sont déroulés.
La DEQM a organisé plusieurs ateliers techniques en 2012
Deux ateliers techniques ont porté sur le projet eTACT de lutte contre la contrefaçon de la DEQM. Ils ont été organisésàStrasbourg(France)enjanvier2012etàSofia(Bulgarie) en novembre 2012. Au cours de discussions ouvertes, la DEQM a recueilli des commentaires importants et précieux, émanant des principales parties prenantes impliquées dans la chaîne d’approvisionnement, des autorités de réglementation des médicaments et des organisations de patients. Leurs observations ont permis à la DEQM deredéfinirleserviceenvuedesonadaptationàuneplusgrande échelle et de son futur déploiement. Les questions liées à la transparence, à la participation des patients et à la gouvernance ont été examinées. Une démonstration de

36
Le 11ème « Symposium international sur les étalons de référence pharmaceutiques », organisé par la DEQM, a eu lieu à Strasbourg en septembre 2012. Cet événement bisannuel a réuni 146 experts de 27 pays. Les séances plénières, ont couvert des sujets liés aux aspects réglementaires et logistiques, ou à l’utilisation et l’établissement des étalons de référence. Des séances ciblées, en petits groupes, ont égalementétéorganiséespourdiscuterplusspécifiquementdes étalons de référence de produits biologiques et des défis posés par la caractérisation des petites molécules. Uneprésentationdepostersetunetable-rondefiguraientégalement au programme, avec la participation de représentants des autorités, des pharmacopées et des associations de l’industrie.
Webinaires (séminaires via le web)
En 2012, la DEQM a organisé sept webinaires au total, sur différents sujets. L’intérêt de ces séminaires sur le web n’a cessé de croître et les échos recueillis ont été très positifs. Cette technologie a permis à la DEQM d’offrir des sessions de formation sur des sujets d’actualité à un nombre potentiellement illimité de personnes, partout dans le monde et d’atteindre ainsi un large public.
Trois webinaires sur le projet eTACT et deux autres dans le domaine des soins pharmaceutiques étaient adaptés à un public particulier (autorités nationales, associations de patients, pharmaciens hospitaliers) et portaient sur un sujet spécifique (Résolution CM/ResAP(2011)1 sur les exigences relatives à l’assurance de qualité et d’innocuité des médicaments préparés en pharmacie pour les besoins particuliers du patient). Les présentations et les informations données étaient ainsi pertinentes et ciblées.
Compte tenu de l’ampleur du sujet, deux webinaires distincts ont été organisés en mai sur les étalons de référence. La
première session portait sur les concepts généraux et les étalons de référence de la DEQM, tandis que la seconde, organisée en collaboration avec l’Organisation mondiale de la Santé (OMS), était axée sur les étalons de référence de la Pharmacopée internationale et d’autres étalons de travail (étalons de travail établis en interne).
Tous les webinaires ont été suivis d’une session questions-réponses entre tous les participants. Des copies des diapositives et les enregistrements des webinaires ont ensuite été mis à disposition.
Sessions de formation - approfondir les connaissances et partager les savoir-faire
En 2012, quatre sessions de formation sur la Pharmacopée Européenne ont été organisées (deux à Strasbourg (France) en mai et juillet 2012, une à Tel-Aviv (Israël) en juillet 2012 et une à Lisbonne (Portugal) en décembre 2012).
La première session portait sur les substances biologiques (peptides, vaccins humains, sang et produits issus des biotechnologies), le programme ayant été adapté aux besoins spécifiquesdesparticipantsactifsdansledomaine.
Les autres sessions étaient plus particulièrement axées sur les produits chimiques et, même si le format général de la formationn’apasétémodifié,leprogrammeaétérévisésurla base des retours d’information émanant des participants, des besoins en matière de formation et de l’évolution des tendances et règlements pertinents. De nombreux ateliers, discussions de groupe et études de cas ont en outre offert des possibilités d’interaction avec les participants.
La session de formation de Tel-Aviv était organisée à l’initiative et avec le soutien de l’Institut israélien de normalisation et de contrôle des produits pharmaceutiques

37
du ministère israélien de la santé. Pour la session de formation organisée à Lisbonne, la DEQM a reçu l'inestimable soutien de l’autorité nationale portugaise des médicaments et des produits de santé (INFARMED). La DEQM tient à remercier ces deux autorités qui, outre leur aide et leurs conseils logistiques, ont mis à disposition des salles de réunion et l’assistance du personnel technique.
Expositions et salons internationaux
La participation à des expositions et salons internationaux est un excellent moyen pour la DEQM de promouvoir ses activités, ses produits et ses services auprès d'un publicoud'unsecteurd’activité spécifiques.Cetteannéeencore, la DEQM a participé au Congrès des ingrédients pharmaceutiques (CPhI) à Shanghai (Chine) en juin 2012 et à Sao Paulo (Brésil) en août 2012. Le stand de la DEQM a suscité beaucoup de curiosité et a fourni à la DEQM une occasion unique de rencontrer les visiteurs, de renforcer ses réseaux avec des clients nouveaux ou existants et de développer ses relations avec les professionnels de l’industrie. Des brochures et des dépliants d’information actualisés et complètessurlesactivitésdelaDEQMontétédistribués;un grand nombre de ces documents étaient disponibles en chinois. Des consultations techniques en tête-à-tête sur la procéduredecertificationde laDEQMontégalementétéorganisées et des distributeurs locaux des publications de la DEQM ont apporté leur assistance sur les stands.
Partenariats au niveau international
Pour aider les autorités nationales et les professionnels à travers le monde, la DEQM a co-organisé un certain nombre d’événements présentés ci-après par ordre chronologique.
Collaboration internationale entre les pharmacopées du monde
La première « Rencontre internationale des pharmacopées du monde », qui réunit toutes les pharmacopées et secrétariats de pharmacopées connus de l’OMS, s’est tenue en février 2012, sous l’égide de l’OMS, dans le but de rapprocher les différentes pharmacopées et de discuter des façons envisageables de renforcer la collaboration et l’harmonisation.
Cette rencontre a été suivie, en octobre, d’une réunion publique entre les parties concernées, sous l’égide de l’OMS et de la FIP (Fédération internationale pharmaceutique). Cette réunion de deux jours, à Amsterdam, avait pour objectif principal de recueillir les commentaires des parties concernées sur leurs besoins en matière d’harmonisation des pharmacopées mondiales. Un premier projet de structure applicable à d’éventuelles futures Bonnes Pratiques de Pharmacopée (BPP, une compilation de politiques et d’approches harmonisées de l’élaboration de monographies) a été établi par le Comité d’experts de l’OMS sur les spécificationsrelativesauxpréparationspharmaceutiques,afindemontrerqu’ilprenaitdesmesuresconcrètesenvued’une harmonisation entre les pharmacopées mondiales.
Activités inter-gouvernementales
L’APEC(AsiaPacificEconomicCooperation)aorganiséenmars 2012, à Singapour, le « Life Science Innovation Forum - Regulatory Harmonisation Steering Committee Meeting ». La DEQM a été invitée à faire part de son approche globale en matière de lutte contre les médicaments contrefaits. L’objectif de cette réunion de l’APEC était de promouvoir une prise de conscience du besoin de médicaments de haute qualité et d’une chaîne d’approvisionnement intègre sur la scène mondiale. Plusieurs « feuilles de route » ont été proposées par les participants, dans le but de parvenir prochainement à une harmonisation dans le domaine de la sécurité des médicaments. Les participants à la réunion provenaient principalement des économies membres de l’APEC.
Autres collaborations intéressantes à travers le monde
Un atelier sur les impuretés des produits pharmaceutiques a eu lieu à Casablanca (Maroc) le 27 mars 2012. La DEQM y a présenté les étalons de référence de la Ph. Eur.
En mars 2012, l’Institute of Medicines (IOM) a invité la DEQM àassisteràuneréunionàWashington(USA)sur lafaçond’atténuer les répercussions sur la santé publique au niveau mondialdesmédicamentscontrefaits, falsifiésetdesous-qualité. La DEQM a présenté à cette occasion la Convention MEDICRIME du Conseil de l’Europe, qui a été bien accueillie par les participants. Tout au long de la réunion, la nécessité d’une collaboration et d’une harmonisation internationales dans le domaine de la lutte contre les médicaments contrefaits etfalsifiés,aétésoulignée.
Une conférence sur le contrôle de qualité des médicaments et dispositifs médicaux a eu lieu à Moscou (Fédération de Russie) en avril 2012. La DEQM y a présenté ses principales activités liées à la Ph. Eur., à savoir l’établissement de normes documentaires et de normes matérielles, ainsi que ses activités anti-contrefaçon. Dans l’ensemble, les participants ont souligné la nécessité d’un renforcement de la collaboration et de l’échange d’informations.
En mai 2012, la DEQM a participé à un atelier sur les substances actives à Shanghai (Chine) dans le cadre de la réunion annuelle DIA China. Lors de cet atelier co-organisé par le Département du Commerce et le SFDA américains, les présentations étaient principalement axées sur la réglementation des substances actives, à l’échelle du monde et, plus particulièrement, en Chine. Avec le soutien administratif de la DIA, 100 participants étaient présents, dont des représentants de l’industrie, la SFDA et certaines autorités locales chinoises. L’implication de la DEQM dans cet atelier a également permis, de souligner et de mettre en valeur, le travail effectué par la DEQM pour assurer la qualité desmédicamentsàtraverssesactivitésdecertification(tanten termes d’évaluation que d’inspection).
En juin, la DEQM a participé à un certain nombre de réunions à Shanghai (Chine) à l’occasion du Congrès CPhI China. Une réunion d’information parallèle était organisée sur une

38
demi-journée à l’intention des fabricants de substances actives. La réunion était organisée conjointement avec l’Organisation mondiale de la Santé (OMS), la SFDA (State Food and Drug Administration) chinoise et le CCPIE (China Centre for Pharmaceutical International Exchange). Le programme sur mesure avait pour but d’accroître la compréhension qu’ont les fabricantsdelaprocéduredecertificationdelaDEQMetdeson programme d’inspection, ainsi que du Programme OMS dePréqualificationdessubstancesactives.Enfin,uneréuniond’une journée a été organisée à la Faculté de Pharmacie de l’Université des Sciences et Technologies de Chine orientale, par la DEQM, l’OMS, la SFDA et CCPIE pour sensibiliser les participants à un large éventail de questions liées à la qualité des médicaments, comme par exemple l’assurance qualité, le ProgrammeOMSdePréqualificationdessubstancesactives,laprocéduredecertificationetlesinspectionsdelaDEQMet de l’USFDA. La rencontre a réuni environ 75 participants, provenant principalement de l’industrie et des universités.
La DEQM a fait une présentation plénière sur les étalons de référence de la Ph. Eur. lors de la 13ème conférence BERM (Biological and Environmental Reference Material), organisée par l’Agence internationale de l’énergie atomique (AIEA) à Vienne (Autriche), en juin 2012.
Un atelier consacré à la qualité des substances actives, aux BPF et aux pharmacopées a été organisé conjointement par le Projet commercial UE-Chine et la Chambre de commerce chinoise pour l’importation et l’exportation de médicaments et de produits de santé (CCCMHPIE) à Pékin, en juillet 2012. Cet atelier a constitué une source importante d’informations sur les nouveaux règlements de la Directive de l’UE sur les médicaments falsifiés et leur impact sur l’exportation de substances actives de la Chine vers l’Europe. Au cours dediscussionsanimées, laprocéduredecertificationde laDEQM et les inspections associées ont suscité un grand nombre de questions.
Uneconférencetechniquesur lesdéfisactuelsenmatièrede conformité réglementaire mondiale et la qualité des ingrédients pharmaceutiques a été organisée conjointement par la DEQM, l’Organisation mondiale de la Santé (OMS) et l’Indian Pharmaceutical Association (IPA). La procédure decertificationde laDEQM,notammentsonprogrammed’inspection des fabricants de substances actives, ainsi que la Pharmacopée Européenne et l’harmonisation internationale, figuraientenbonneplaceàl’ordredujour.Troistutorielspré-conférence ont été organisés. Couvrant la préparation d’un nouveau CEP, la demande de révision et l’inspection de la DEQM, ils étaient conçus pour fournir une compréhension plus approfondie des politiques et des procédures concernées. Organisée à Bombay (Inde) en septembre 2012, la conférence a réuni environ 90 participants représentant un vaste échantillon de l’industrie pharmaceutique indienne.
En octobre 2012, un symposium sur les pharmacopées et les étalons de référence a été organisé par la Société pharmaceutique de Corée, à Séoul, ainsi qu’un atelier conjoint KFDA/DEQM/USP sur les pharmacopées et les étalons
de référence à Osong (Corée). La DEQM a activement participé à ces deux événements en présentant ses principales activités liées à la Ph. Eur., à savoir l’établissement de normes documentaires et de normes matérielles.
Collaboration spécifique développée sous l’égide des autorités nationales et de l’OMS
En septembre 2012, en collaboration avec le Partnership for Safe Medicines (PSM) et l’Organisation mondiale de la Santé (OMS), le gouvernement indien a invité la DEQM à participer, à New Delhi, à un atelier consacré à la sécurité du patient et aux technologies d’authentification et de détection des médicaments. Cet atelier faisait suite à un séminaire de l’APEC organisé à Pékin (Chine) en septembre 2011 sur la sécurité des médicaments et les technologies de détection.Ilsedivisaitenquatresessionstechniques:«Défisde l’accessibilité à des médicaments de qualité et utilisation des technologies de détection », « Perspectives réglementaires sur la sécurité des patients », «Technologies de détection dans le cadred’une stratégie plus larged’authentificationde qualité des médicaments » et «Technologies de détection précoce pour des médicaments de meilleure qualité et rôle de la sérialisation ». Les participants ont pu obtenir de nombreuses informations sur les difficultés rencontrées par le gouvernement et les industries et sur la façon dont ils entendent renverser la tendance haussière des cas de contrefaçon.
La 15ème édition de l’ « International Conference of Drug Regulatory Authorities (ICDRA) », qui vise à promouvoir l’échange d’informations entre les autorités de réglementation des médicaments et le développement d’approches collaboratives sur des questions d’intérêt commun, s’est tenue à Tallinn (Estonie) en octobre 2012. La conférence, qui se déroule tous les deux ans, était organisée conjointement par l’agence estonienne des médicaments et l’Organisation mondiale de la Santé (OMS). Au cours du programme pré-ICDRA qu’elle co-organisait, la DEQM a fait plusieurs présentations sur des questions relatives aux principes actifs pharmaceutiques, comme par exemple le contrôle de la qualité des principes actifs, l’évaluation de la documentation dessubstancesactivesetlesproblèmesspécifiquesposésparles médicaments à base de plantes.
Collaboration internationale sur les demandes d’AMM de médicaments génériques
En décembre 2012, la DEQM a participé aux discussions de l’International Generic Drug Regulators Pilot Project (IGDRP) à Nanchang (Chine). L’IGDRP vise à étudier les possibilités de partage du travail et de reconnaissance mutuelle dans le domaine des demandes d’AMM pour des médicaments génériques. L’acceptation des CEP peut aider à réduire la charge de travail des organismes de réglementation dans le domaine des substances actives. La DEQM a également participé à la troisième conférence internationale sur les médicaments génériques qui se tenait au même endroit. Cette conférence, divisée en plusieurs sessions, a donné un aperçu

39
de l’environnement réglementaire mondial des médicaments génériques, avecdesprésentations sur la certificationparle SFDA, Santé Canada, la DEQM et le bureau polonais de l’enregistrement des médicaments, des dispositifs médicaux et des produits biocides. Des présentations ont également été faites sur les exigences techniques applicables aux médicaments génériques en Chine, sur les dernières informations relatives aux inspections de la FDA en Chine et sur les nouveaux développements en matière d’exigences réglementaires pour les génériques et les substances actives aux États-Unis. Un certain nombre de présentations étaient en outre proposées par l’industrie sur leurs expériences en Chine et sur la manière de faciliter l’accès des entreprises chinoises au marché de l’UE et des États-Unis.
Communication et promotion des activités de transfusion sanguine
La Journée mondiale des donneurs de sang a été célébrée le 14 juin 2012, sous le thème « Chaque donneur de sang est un héros ». La campagne vise à encourager chacun à faire volontairement un don de sang pour maintenir un approvisionnement sûr et adéquat. Cet événement de portée mondiale, organisé par la Croix-Rouge coréenne et le Ministère de la Santé et des Affaires sociales de la République de Corée, se tenait à Séoul.
À cette occasion, la DEQM a publié sur son site web une carte interactive proposant des liens vers les sites des établissements du sang en Europe. Les visiteurs ont pu se connecter et découvrir quelles festivités étaient organisées dans leur pays et comment ils pouvaient s’impliquer. Un kit média était mis à la disposition de tous les États membres souhaitant l’utiliser pour leurs campagnes locales.
La DEQM a participé au 32ème congrès international de la Société internationale de transfusion sanguine (SITS) à Cancun (Mexique) en juillet 2012. Le congrès a attiré plus de 2400 délégués, dont la plupart étaient des professionnels impliqués dans les activités de médecine transfusionnelle. La DEQM a poursuivi la promotion de son « Guide pour la préparation, l’utilisation et l’assurance de qualité des composants sanguins » (16ème édition 2010) et les délégués ont pu s’informer sur les projets en cours, sur les recommandations et résolutions du Conseil de l’Europe ou encore sur les rapports de la DEQM sur la collecte, le contrôle et l’utilisation du sang et de ses composants en Europe. Des
supports en espagnol avaient spécialement été préparés pour l’événement.
En octobre 2012, la DEQM a organisé un symposium à Strasbourg (France) en partie réservé aux représentants des autorités et des services transfusionnels, sur le thème « Gestion de l’approvisionnement en sang ». Le programme couvrait des sujets comme le projet et l’enquête du Conseil de l’Europe sur la gestion de l’approvisionnement en sang, les différentes approches en la matière prises par les différents services de transfusion européens et un aperçu de quelques-uns des logiciels commerciaux disponibles pouvant aider à gérer la chaîne d’approvisionnement en sang. La réunion s’est terminée par une table ronde consacrée aux bonnes pratiques.
La DEQM a également soutenu l’Association de lutte contre la drépanocytose (DORYS) pour l’organisation de son 7ème
congrès sur la drépanocytose, en mai 2012 à Strasbourg (France).
Activités liées à la transplantation d’organes
Depuis 1998, le Conseil de l’Europe organise une Journée européenne du don d’organes et de la greffe en vue de promouvoir le don d’organes et la transplantation dans ses États membres. La journée, accueillie chaque année par un pays différent, a pour but de sensibiliser à l’importance du don et de la transplantation d’organes.
La DEQM a célébré la Journée européenne du don d’organes et de la greffe à Budapest (Hongrie) en octobre 2012. La journée était organisée par la société hongroise de transplantation et le stand d’information de la DEQM a permis au public de se procurer des brochures d’information en hongrois sur le don d’organes. Outre une conférence scientifique et une conférence de presse, de nombreusesactivités de plein air étaient organisées. Une cérémonie de remise de prix a permis de rendre hommage aux donneurs. La journée s’est terminée par un concert animé par des artistes hongrois de renom.
Visites
Cette année encore, la DEQM a ouvert ses portes aux différents groupes de visiteurs, permettant ainsi au grand public de mieux connaître et comprendre ses travaux.
Phot
o : D
avid
Bet
zing
er /
© C
onse
il de
l'Eur
ope

40
2.4 Collaboration internationale
À l’heure de la mondialisation, la collaboration internationale est d’une importance primordiale pour la DEQM dans l’optique de sa mission de protection de la santé publique. L’harmonisation des normes, l’échange d’informations et le partage du travail ne sont que quelques exemples des activités que la DEQM réalise en étroite collaboration avec ses partenaires et les intervenants au niveau international.
Réunions et visites bilatérales
Chine
La coopération fructueuse avec la Chine s’est poursuivie avec les visites de délégations de la State Food and Drug Administration (SFDA), des National Institutes for Food and Drug Control (NIFDC) et du China Pharmaceutical International Exchange Centre (CCPIE) à la DEQM en janvier et novembre. Les discussions ont porté sur l’expérience dans le domaine des exigences de qualité des médicaments sur le marché, la surveillance de la qualité et les mécanismes d’évaluation des médicaments commercialisés, le traitement des risques liés à la qualité et les systèmes de management de la qualité. La visite comprenait le laboratoire et les installations de production des étalons de référence de la DEQM.
Ukraine
En juillet de cette année, la DEQM a participé à un événement festif consacré au 20e anniversaire du centre scientifiqueukrainiendepharmacopéepour laqualitédesmédicaments. Au cours de cette manifestation organisée à Kharkov, le chef de l’administration d’État a réitéré la ferme volonté et l’intention du gouvernement ukrainien de signer la Convention relative à l’élaboration d’une Pharmacopée Européenne et ainsi devenir membre à part entière de la Ph. Eur., ce qui s’est effectivement produit le 17 décembre 2012.
Singapour
En juin 2012, l’administrateur général de la Health Sciences Authority (HSA) de Singapour a fait une visite de courtoisie avec une délégation du HSA pour discuter des travaux effectués à la DEQM. Singapour est le 24ème et plus récent observateur de la Pharmacopée Européenne. Étant responsable de la banque du sang de Singapour et de la réglementation des produits de santé, la HSA est très intéressée par un renforcement de la collaboration avec la DEQM dans les domaines des OMCL, des laboratoires cosmétiques et de la transfusion sanguine.
États-Unis d’Amérique
En 2012, la visite à la DEQM par le responsable de la Food and Drug Administration (FDA) américaine chargé de la
liaison avec l’EMA, a permis d’approfondir les relations entre les deux parties. Cette visite avait pour but de présenter les travaux de la FDA et de fournir des informations sur leur interaction avec la pharmacopée américaine (USP).
Organisation mondiale de la Santé (OMS)
Outre ses activités concernant les Substances Chimiques de Référence Internationales (SCRI) et les étalons ISA (International Standards for Antibiotics) décrites dans le chapitre 1.2, la DEQM a soutenu les activités normatives de l’OMS en contribuant activement aux travaux du Comité d’expertssurlesSpécificationsetauComitéd’expertssurlaStandardisation biologique.
Relations avec la Fédération internationale pharma-ceutique (FIP)
Les « tables rondes des intervenants de la FIP en conjonction avec le Sommet des Ministres d’Amsterdam », qui portaient sur l’établissement de meilleures politiques de santé présentantunrapportcoût-efficacitéplusrentable,sesonttenues à Amsterdam en octobre 2012. Sur les quatre tables rondes organisées en 3 jours, trois l’ont été avant le Sommet des Ministres dans le but de préparer des suggestions pour discussion. La quatrième table ronde, qui se déroulait le dernier jour, était axée sur l’innovation.
Dans l’ensemble, les tables rondes des intervenants de la FIP et le Sommet des ministres ont souligné l’importance et la nécessité de poursuivre l’action dans le domaine des soins pharmaceutiques. Les travaux de la DEQM/Conseil de l’Europe dans le domaine, ont été reconnus tout au long de la conférence et mentionnés dans les conclusions (voir section 1.7, page 27).
Élaboration de normes internationales
Organisation internationale de normalisation (ISO)
Depuis plusieurs années, la DEQM participe activement à l’élaboration d’un ensemble de cinq normes ISO destinées àharmoniser l’identificationdesmédicaments (IDMP)dupoint de vue réglementaire. Ces travaux ont été menés par le Groupe de travail 6 du Comité technique 215 de l’ISO. La DEQM a plus particulièrement apporté son expertise à la préparation de la norme ISO 11239 relative à la préparation de vocabulaires contrôlés pour les formes pharmaceutiques, les voies d’administration, les unités de présentation et les emballages, norme pour laquelle elle agit en qualité de rédacteur.
En octobre 2012, ces cinq normes IDMP ont été publiées en tant quenormes internationalesofficielles,marquantuneétape importante du projet IDMP.
Outre ses travaux au sein de l’ISO, la DEQM a participé à la rédaction d’un guide de mise en œuvre de l’IDMP avec

41
le groupe d’experts M5 de la Conférence internationale sur l’harmonisation (des exigences techniques pour l’enregistrement des produits pharmaceutiques à usage humain : ICH), d’où est né le projet IDMP. Là aussi, la DEQM a agi en qualité de rédacteur pour le module relatif à la norme ISO 11239.
Des téléconférences ont été régulièrement organisées tout au long de l’année, ainsi qu’un certain nombre de réunions en tête-à-tête aux États-Unis et au Japon, au cours desquelles a été favorisée la collaboration entre les régulateurs et les représentants de l’industrie du monde entier et notamment les membres (Europe, USA et Japon) et les observateurs (Suisse, Canada) de l’ICH. Lors d’une session de formation organiséedansleslocauxdelaFDAàWashingtonDCenmars 2012, des intervenants de la DEQM, de l’EMA et de la FDA américaine ont fourni, à l’intention des participants provenant d’organismes de réglementation et de l’industrie américaine, des rapports d’avancement du développement de normes et de l’élaboration du guide de mise en œuvre.
Conférence internationale sur l’harmonisation (ICH)
Le guideline Q3A (ICH) actuel classe les impuretés en solvants organiques, inorganiques et résiduels. Les guidelines
Q3A et Q3B traitent des exigences relatives aux impuretés organiques, tandis que le guideline Q3C couvre celles concernant les solvants résiduels. Un nouveau guideline, Q3D,est en coursdedéveloppementafindepréciser lesexigences relatives aux métaux, qui figurent parmi les impuretésinorganiquesdanslaclassificationICH.En2012,laDEQM a contribué à l’élaboration d’une version préliminaire de Q3D qui fera l’objet d’une enquête publique en 2013 en vued’unefinalisationduguidelineen2014.
La vaste expérience des inspecteurs de la DEQM en matière d’inspection des BPF chez les fabricants de substances actives a été reconnue par le comité directeur de l’ICH qui a invité la DEQM à participer en tant qu’observateur aux délibérations du groupe de travail sur la mise en œuvre de l’ICH Q7.
Conférences et ateliers
Des informations sur les conférences et ateliers internationaux organisés par la DEQM ou auxquels la DEQM a participé figurentdans la section2.3Communication, évènementsmarquants, page 35.

42
Commission européenne de Pharmacopée
La Commission a été créée en 1964 en application de la Convention relative à l’élaboration d’une Pharmacopée Européenne. Suite à la ratification de la Convention par l’Ukraine en décembre 2012, elle se compose désormais de 38 parties signataires de la Convention (37 États et l’Union européenne). Les 24 observateurs, originaires du monde entier, démontrent l’importance des travaux de la Commission européenne de Pharmacopée au niveau international. La Commission décide du programme de travail et adopte les normes de qualité pour tous les médicaments et leurs composants sur les territoires des États membres. Dix-neuf groupes d’experts permanents et 54 groupes de travail « ad hoc » mis en place par la Commission mènent à bienleprogrammedetravaildelaPh.Eur.Àlafindel’année2012,2569textescontenantdesspécificationsrelativesàlaqualité ont ainsi été élaborés, adoptés et mis en application. L’actualisation continue de ces textes permet de les maintenir àjourdesprogrèsscientifiquesettechniquesenmatièredeproduction et de contrôle qualité. La Ph. Eur., actuellement publiée dans sa 7e Édition, joue un rôle essentiel pour la protection de la santé publique. Elle est élaborée à l’usage des professionnels du monde du médicament, pour lesquels elle constitue une référence constante.
Comité directeur du programme de standardisation biologique (PSB)
Le PSB travaille à la normalisation des méthodes et outils relatifs au contrôle de la qualité des produits biologiques, en établissant des étalons de référence et en validant de nouvelles méthodes, en particulier celles inspirées par le concept dit des « 3R » (Replacement, Reduction, Refinement) en matière d’essais sur animaux. Ses activités sont encadrées par le Comité directeur du PSB.
Réseau des laboratoires officiels de contrôle des médicaments (OMCL), Comités consultatifs
Environ 35 pays participent depuis 1994 aux activités du réseau des OMCL, que coordonne la DEQM. Ce réseau a pour rôle de veiller à l’uniformité de la qualité des médicaments commercialisés dans les États membres et de contribuer à la reconnaissance mutuelle, entre ces États, des résultats des contrôles qualité effectués sur les médicaments. Les décisions importantes sont prises lors des assemblées plénières annuelles du réseau. Les comités consultatifs préparent le programme de travail annuel et veillent à sa mise en œuvre. Il existe deux niveaux de collaboration au sein du réseau :
• Les activitésd’intérêt général impliquant tous lesÉtatssignataires de la Convention et États observateurs. Ces activités générales couvrent les travaux relatifs aux systèmes de management de la qualité, comme les audits et les essais d’aptitude (PTS, Proficiency Testing Studies), ainsi que les études de surveillance du marché (MSS, Market Surveillance Studies). Elles sont préparées et suivies
par le Comité consultatif du réseau général des OMCL (AdGEON);
• Lesactivitésrestreintesauxpaysdel’UnionEuropéenne(UE)etde l’Espaceéconomiqueeuropéen(EEE) ;ellesconcernent les produits faisant l’objet d’une autorisation de mise sur le marché centralisée (CAP, Centrally Authorised Products), les produits autorisés selon la procédure de reconnaissance mutuelle ou la procédure décentralisée (PRM/PDC)et le systèmeofficiel de libérationdes lotspar les autorités de contrôle (OCABR) en vigueur pour les produits biologiques (humains et vétérinaires). La Suisse est également partie prenante de cette dernière activité. Pour les activités CAP et OCABR, des groupes consultatifs veillent entre deux réunions annuelles à la continuité du fonctionnementdechacundesréseauxspécifiques.
Les autorités européennes et nationales sont impliquées dans ces activités. Le réseau des OMCL participe également à des enquêtes sur les médicaments frauduleux.
Certification de conformité aux monographies de la Ph. Eur., comité directeur
Lesactivitésenrapportaveclacertificationdeconformitéaux monographies de la Ph. Eur. sont pilotées par un Comité directeur et deux Comités techniques consultatifs (CTC). Le Comité directeur est composé de représentants des autorités d’enregistrement et d’inspection. Il prend des décisions de politique générale, examine et commente les questions soulevées par les CTC, adopte les guidelines et le programme d’inspection et coordonne les questions entre les parties représentées. Il est en outre chargé de désigner les assesseurs ainsi que les membres des CTC et leurs présidents.
Un réseau de quelque 80 assesseurs et 30 inspecteurs nationaux participent aux travaux liés à l’évaluation des dossiers Qualité des substances actives et à l’inspection des sites de fabrication.
Comité européen sur la transfusion sanguine (CD-P-TS)
Ce Comité supervise les travaux d’un certain nombre de groupes de travail et de projets individuels, par exemple, la base de données européenne sur les réserves de sang congelé de groupes rares, le management des donneurs de sang, l’élaboration du Guide pour la préparation, l’utilisation et l’assurance de qualité des composants sanguins par un groupe de travail ad hoc et les travaux d’un autre groupe de travail ad hoc sur les comportements à risque ayant un impact sur le management des donneurs de sang.
Comité européen sur la transplantation d’organes (CD-P-TO)
Ce Comité met l’accent sur l’élaboration et la promotion du principe de non commercialisation du don d’organes, les mesuresderenforcementdelaluttecontreletraficd’organeset l’élaboration de normes éthiques exigeantes en matière de
Liste des comités de la DEQM

qualité et de sécurité dans le domaine de la transplantation d’organes. Les membres et observateurs de ce comité représentent 41 pays en Europe et au-delà. Il supervise les activités de plusieurs projets individuels couvrant des sujets comme le don de donneurs vivants, la transplantation pour les non résidents, les inscriptions multiples sur les listes d’attente de transplantation, les banques de sang de cordon autologue et la coopération entre États de la région de la Mer Noire en matière de transplantation.
Comité européen sur les produits et les soins pharmaceu-tiques (CD-P-PH)
Le Comité directeur supervise les programmes d’activités de plusieurs comités subordonnés :
• LeComitéd’Expertssurlaclassificationdesmédicamentsen matière de leur délivrance (CD-P-PH/PHO),
• le Comité d’experts sur les normes de qualité et desécurité relatives à la pratique et au suivi pharmaceutiques (CD-P-PH/PC),
• leComitéd’expertssurlaréductiondesrisquesdesantépublique liés à la contrefaçon des médicaments et à la criminalité connexe (CD-P-PH/CMED).
Comité de protection de la santé des consommateurs (CD-P-SC)
Le CD-P-SC est responsable de la gestion du programme de travail et de la prise de décisions dans les domaines des
cosmétiques et de l’emballage alimentaire et pharmaceutique. Les autorités de santé des 31 pays européens signataires de la Convention relative à l’élaboration d’une Pharmacopée Européenne peuvent contribuer aux travaux, ainsi que quatre observateurs (Arménie, Géorgie, Moldavie et Singapour).
Le Comité supervise deux groupes qui sont chargés de l’examen des aspects sanitaires et de l’évaluation des risques, et rédigent des rapports et des recommandations en matière d’approche réglementaire :
• LeComitéd’Experts sur les emballagesalimentairesetpharmaceutiques (P-SC-EMB), qui dispose de groupes de travail consacrés aux essais de migration des métaux et alliages, aux papiers et cartons et aux encres d’imprimerie,
• leComitéd’Expertssurlesproduitscosmétiques(P-SC-COS), qui coordonne les travaux du réseau des laboratoires officielsdecontrôledescosmétiques(OCCL).Lestravauxde ce réseau sont principalement axés sur le management de la qualité, les méthodes d’analyse et la reconnaissance mutuelle. Des essais d’aptitude et des études de surveillance du marché sont organisées dans le but d’améliorer la qualité desproduits cosmétiques sur lemarché.Àcettefin, leréseau OCCL interagit avec les autorités nationales de surveillance du marché, la Commission européenne (CE) et le Centre commun de recherche (CCR).
Outre les cosmétiques, le P-SC-COS dispose également d’un groupe de travail chargé des problèmes de santé liés aux tatouages et au maquillage permanent.

44
Notes

45
Notes

46
Notes

47
Direction Européenne de la Qualité du Médicament & Soins de santé (DEQM)
7, allée KastnerCS 30026F-67081 Strasbourg - FranceTél. : +33 (0)3 88 41 30 30Fax : +33 (0)3 88 41 27 71www.edqm.eu
Pour obtenir l’autorisation de reproduire une partie du présent rapport ou pour tout renseigne-ment, merci de passer par le service HelpDesk de la DEQM, Relations publiques : www.edqm.eu/hd
Copyright © 2013 DEQM, Conseil de l’Europe. Tous droits réservés. PRDD-13-07 Photographies : Shutterstock -Fotolia - Candice Imbert