modélisation chimique de l’ozone et des oxydants...
TRANSCRIPT

Modélisation chimique de l’ozone
et des oxydants gazeux :problématique et état de l’art
Bernard Aumont, [email protected]
Laboratoire Interuniversitaire des Systèmes AtmosphériquesUMR CNRS 7583, Universités Paris 7 et Paris 12, 94010 Créteil
Décembre 2005
Le bilan de l’ozone et des photooxydants aux différentes échelles spatiales est principale-ment gouverné par des processus chimiques gazeux. Cette production de photooxydants estétroitement liée aux processus d’oxydation des composés organiques volatils et des oxydesd’azote. Ce chapitre présente l’état de l’art et les problèmes associés à la modélisation de cesystème. Il se focalise principalement sur les obstacles rencontrés pour décrire et modéliserl’oxydation gazeuse des composés organiques volatils (COV). L’influence de cette oxyda-tion constitue en effet l’une des principales incertitudes du bilan des photooxydants auxdifférentes échelles spatiales. La représentation aussi fidèle que possible de ce processus re-présente la principale difficulté et l’enjeu majeur du développement de schémas chimiques.

Table des matières
1 Développement des schémas chimiques . . . . . . . . . . . . . . . . . . . . . . 31.1 Des modèles numériques : pour quoi faire ? . . . . . . . . . . . . . . . . 31.2 Définition du schéma chimique . . . . . . . . . . . . . . . . . . . . . . . 51.3 Modélisation de l’oxydation gazeuse des composés organiques : en-
jeux et difficultés . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.4 Les étapes de l’oxydation des composés atmosphériques . . . . . . . . 101.5 Reconstitution des données cinétiques et mécanistiques . . . . . . . . . 101.6 Réduction des schémas : influence du milieu . . . . . . . . . . . . . . . 14
2 Schémas chimiques dédiés aux atmosphères continentales . . . . . . . . . . . 152.1 Regroupement en famille structurale . . . . . . . . . . . . . . . . . . . . 162.2 Les espèces de regroupement . . . . . . . . . . . . . . . . . . . . . . . . 172.3 Les espèces de remplacement . . . . . . . . . . . . . . . . . . . . . . . . 202.4 Equations bilans - opérateurs chimiques . . . . . . . . . . . . . . . . . . 22
3 Validation des schémas chimiques . . . . . . . . . . . . . . . . . . . . . . . . . 273.1 Comparaison modèles / mesures en laboratoire . . . . . . . . . . . . . 283.2 Comparaison modèles / mesures in situ . . . . . . . . . . . . . . . . . . 313.3 Inter comparaison des schémas chimiques et tests de sensibilité . . . . 33
2

MODÉLISATION CHIMIQUE DE L’OZONE 3
1. Développement des schémas chimiques
1.1. Des modèles numériques : pour quoi faire ?
L’une des finalités des études de chimie atmosphérique est d’évaluer la contribution desdifférents processus (émissions, transport, transformations physico-chimiques, dépôt) surla distribution des espèces. La diversité des paramètres à prendre en considération génèretoutefois un ensemble d’une grande complexité ; l’influence respective des différents para-mètres est, le plus souvent, difficilement caractérisable.
La démarche expérimentale "traditionnelle" quantifie l’influence des paramètres sur l’évo-lution d’un système particulier en renouvelant une expérience donnée et en modifiant, tourà tour, chaque paramètre. Cette approche expérimentale est impraticable pour l’étude dessystèmes atmosphériques : les paramètres de l’expérience ne sont pas contrôlés par l’expéri-mentateur et sont par essence non renouvelables. Les relations de causes à effets ne peuventdonc pas être directement quantifiées sur la seule base de l’expérimentation. L’intérêt desmodèles numériques est précisément de quantifier les relations causales entre paramètres,celles-ci étant le plus souvent inaccessibles par l’expérimentation in situ.
De façon très schématique, il est possible de distinguer deux grandes catégories de modèlesnumériques : les modèles de boîte et les modèles tridimensionnels de chimie/transport.
Les modèles de chimie/transportIl n’est guère possible de mesurer l’ensemble des espèces pertinentes en tout point de l’es-pace et à chaque instant. L’objectif principal des modèles de chimie/transport est de simulerl’évolution spatio-temporelle des concentrations. Ces modèles tentent ainsi de reproduire lesdifférents phénomènes agissant d’une part sur les quantités de matière en présence (émis-sion, dépôt, transformations chimiques) et d’autre part sur leurs distributions spatiales (ad-vection, diffusion turbulente, convection). Ces modèles sont développés et exploités aussibien en tant qu’outil diagnostique (exploitation et interprétation de données de terrain) quepronostique (prévision d’épisodes de pollution, étude d’impact, développement de stratégiede réduction des émissions)
La plupart des modèles tridimensionnels de chimie/transport (CTM) sont construits en sup-posant que les variables "météorologiques" (champs de vents, pression et température) in-fluencent les transformations chimiques et non réciproquement 1. Les paramètres météoro-logiques sont alors utilisés comme données d’entrée du modèle (on parle de forçage dusystème). En matière de chimie atmosphérique, le domaine modélisé varie typiquement del’échelle locale (par exemple l’échelle urbaine) à l’échelle continentale ou globale. Ce do-maine est habituellement discrétisé en cellules (maille) qui représentent l’unité de volume
1Le couplage entre chimie et dynamique de l’atmosphère doit toutefois être considéré pour certaines applications.C’est notamment le cas des modèles climatiques, pour lesquels la rétroaction de la chimie sur la circulation généraledes masses d’air doit être représentée via le transfert radiatif. C’est également le cas des modèles dédiés à l’étudedes systèmes nuageux, où les processus physico-chimiques associés aux interactions gaz/aérosols/gouttelettes etles mouvements verticaux des masses d’air sont étroitement interconnectés.

4 1. DÉVELOPPEMENT DES SCHÉMAS CHIMIQUES
sur laquelle les concentrations simulées sont moyennées. Le modèle repose sur la résolu-tion de l’équation de continuité dans laquelle les flux turbulents sont paramétrés comme unprocessus diffusif :
∂C∂t
= −∇uC +∇(
KCair∇ CCair
)+ P − L (1)
avec C la concentration (molécule.cm−3) de l’espèce considérée, u la vitesse du vent (cm.s−1),K (cm2.s−1) la matrice des coefficients de diffusion turbulente dans toutes les directions dutransport (les termes non diagonaux sont nuls), Cair la concentration moléculaire de l’air(molécule.cm−3), P et L (molécule.cm−3.s−1) respectivement les termes de production (chi-mie, émission) et consommation (chimie, dépôt).
Le système d’équations aux dérivées partielles est habituellement résolu par séparationd’opérateur. Cette méthode est en fait la seule méthode opérationnelle lorsque le systèmemodélisé implique de nombreuses espèces évoluant sur un maillage dense. Les opérateursassociés au transport (advection, diffusion) relient les concentrations d’une même espèceentre les différentes mailles ; ceux associés aux transformations chimiques couplent les concen-trations des différentes espèces en une maille. En pratique, ce système est intégré en sépa-rant la résolution du transport (advection, diffusion) de celle de la chimie (transformationphysico-chimique, émissions, dépôt). La contribution des différents processus, alors consi-dérés comme indépendants, est ainsi calculée d’une manière dissociée ; la variation totaleest estimée comme la somme de chaque contribution. La résolution des équations liées àchaque opérateur possède ses propres difficultés intrinsèques. L’approche par séparationd’opérateur permet ainsi (i) d’utiliser la méthode de résolution numérique (le solveur) laplus appropriée et (ii) le développement de modèles de chimie/transport présentant unestructure modulaire marquée.
De nombreux processus sont trop complexes pour pouvoir être traités directement dansles CTM à partir des lois physiques et/ou chimiques fondamentales. C’est notamment lecas de processus se déroulant sur de petites échelles spatiales et/ou temporelles comme laturbulence ou le développement de systèmes nuageux. Un traitement explicite de ces phé-nomènes de petite échelle implique une dimension des mailles du modèle comparable auxéchelles spatiales des processus représentés (de l’ordre de quelques mètres, voire inférieure).Ce raffinement du maillage pose deux types de problème : (i) des coûts de calcul totalementprohibitifs (ii) l’incapacité pour le modélisateur de renseigner l’ensemble des paramètrespertinents à l’échelle des mailles considérées. De même, les processus physico-chimiquesimpliquent une multitude d’espèces qu’il est exclu de représenter explicitement dans lesCTM, pour des raisons analogues à celles évoquées ci-dessus. Ces processus doivent ainsiêtre représentés de façon simplifiée dans le modèle, à l’aide de paramétrisations visant à re-présenter l’effet moyen du processus à l’échelle de la maille du modèle. Par exemple, dansl’équation 1, l’effet de la turbulence sur le mélange des masses d’air est représenté comme unprocessus diffusif. De même, il importe de développer des paramétrisations pour représen-ter, de façon simplifiée, l’impact des transformations chimiques sur les concentrations desespèces cibles.

MODÉLISATION CHIMIQUE DE L’OZONE 5
Le modèle de boîteCe type de modèle vise principalement à «élucider» le fonctionnement d’un système envi-ronnemental donné en étudiant, dans le détail, un processus ou un ensemble de processusdonnés. Il s’agit le plus souvent de modèles «conceptuels», présentant un caractère «explo-ratoire» marqué. L’approche repose sur une représentation «idéalisée» de situations envi-ronnementales pour lesquelles la contribution des processus étudiés est quantifiée.
La «boîte» représente une région de l’atmosphère (éventuellement un réacteur de labora-toire). Cette boîte est soumise à différents forçages extérieurs (émissions, dépôts, advection,rayonnement ...), fonction de la situation environnementale considérée. Grâce à leur struc-ture simple, ces modèles permettent une description exhaustive et explicite des processus,à travers un ensemble de relations physiques et chimiques reflétant l’état des connaissancessur ces processus. Par exemple, il est possible d’intégrer, dans ce type de modèle, des mé-canismes chimiques très détaillés, incluant plusieurs dizaines de milliers d’espèces ou deréactions. Les processus chimiques peuvent ainsi être analysés en quantifiant la réponse dusystème aux variations des forçages extérieurs et/ou en hiérarchisant leurs contributions re-latives dans l’évolution du système.
Ces modèles présentent des intérêts multiples. Ils sont souvent un préalable indispensablepour l’élaboration de paramétrisations à destination des CTM (voir paragraphe ci-dessus)et permettent d’identifier les processus pouvant raisonnablement être ignorés. Le modèlereflétant l’état des connaissances sur un processus (ou d’un ensemble de processus) donné,il permet également de tester le degré de compréhension de celui-ci lors de comparaisonmesure/modèle (en comparant par exemple les concentrations simulées et observées dansun réacteur). Enfin, de par leur caractère exploratoire, ce type de modèle conduit parfois àproposer de nouvelles hypothèses et de nouveaux paramètres à mesurer au laboratoire ousur le terrain afin de les vérifier (par exemple une liste d’espèces, la composition d’une phasecondensée, la valeur d’une constante physico-chimique ...).
Entre les modèles de boîte et les modèles de chimie/transport tridimensionnels, il existetoute une hiérarchie de modèles (modèles de colonne 1D vertical, ou modèles à 2 dimen-sions). Les sections suivantes se focalisent sur le développement de modules chimiques opé-rationnels (i.e. paramétrés et simplifiés) à destination des modèles de chimie/transport. L’ac-cent est principalement porté sur les difficultés rencontrées pour le développement de cesmodules, les différentes approches proposées dans la littérature pour pallier ces difficultéset les méthodes traditionnellement mises en œuvre pour évaluer leur fiabilité.
1.2. Définition du schéma chimique
Un système couplé d’équations différentielles ordinairesL’évolution des concentrations induite par les transformations chimiques est décrite par unsystème d’équations différentielles ordinaires (ODE) de la forme générale :
dCi
dt= Pc,i − Lc,i

6 1. DÉVELOPPEMENT DES SCHÉMAS CHIMIQUES
où Pc,i et Lc,i sont respectivement les termes de production et de consommation chimiqueassociés à chaque espèce i. Les espèces chimiques impliquées dans les mécanismes d’oxyda-tion atmosphérique interagissent les unes avec les autres. La chimie atmosphérique est donccaractérisée par des mécanismes indissociables, ce qui empêche de traiter l’évolution dechaque espèce de façon indépendante. Les termes Pc,i et Lc,i sont ainsi fonction des concen-trations Cj des différentes espèces ce qui entraine un "couplage" des différentes équations.Les termes de production/consommation associés aux différentes espèces chimiques sontdirectement déduits du "schéma chimique".
Schéma chimique : définitionUn schéma chimique est l’ensemble des réactions considérées, accompagnées des constantescinétiques et de leur dépendance en température, en pression et au rayonnement. Les réac-tions du schéma chimique sont établies de telle sorte que chacune d’entre elles se comportecomme une réaction élémentaire. Ceci permet en effet de déterminer directement, du jeu desréactions, les termes de production Pc,i et de consommation Lc,i associés à chaque espèce.
Schémas explicites et schémas réduitsLa démarche la plus rigoureuse et la plus naturelle consiste à écrire toutes les réactions élé-mentaires impliquées dans la transformation des différentes espèces. C’est ce que l’on ap-pelle des mécanismes explicites ou détaillés. Cette démarche conduit toutefois à des méca-nismes de grande dimension (plusieurs dizaines de milliers d’espèces) [Madronich and Cal-vert, 1989; Jenkin et al., 1997, 2003; Saunders et al., 2003]. Ces schémas peuvent aujourd’huifacilement être intégrés dans un modèle de boîte. Néanmoins, couplé à un modèle de chi-mie/transport tridimensionnel, le système résultant demeure trop lourd pour pouvoir êtretraité efficacement par les moyens de calcul disponibles actuellement. On est donc amené àconcevoir des schémas chimiques simplifiés et condensés, au prix d’une perte d’information.Ce point est développé ci-dessous.
Schéma inorganique et schéma organiqueEn matière de chimie gazeuse troposphérique, on distingue habituellement les transforma-tions n’impliquant que des espèces inorganiques (HOx, NOx, O3, SO2 ...) de celles impli-quant les composés organiques.
Pour les aspects purement inorganiques, le développement du schéma chimique ne poseguère de difficulté. En effet, la chimie inorganique est relativement bien établie. De plus, unedescription totalement explicite des transformations s’avère peu coûteuse : elle nécessite detenir compte d’une vingtaine d’espèces réagissant selon une trentaine de réactions. Cettedescription explicite est celle généralement retenue. Les schémas chimiques inorganiquesdes différents modèles disponibles dans la littérature sont ainsi très similaires. Par exemple,les mécanismes CB4 [Gery et al., 1989], SAPRC [Carter, 1990, 2000] et RADM et RACM [Sto-ckwell et al., 1990, 1997] traite la chimie des mêmes espèces inorganiques (14 composés : O3,O(1D), O(3P), NO, NO2, NO3, N2O5, HNO3, HONO, HNO4, OH, HO2, H2O2, CO).
Les principaux obstacles rencontrés pour développer le schéma chimique sont liés à la des-cription du processus d’oxydation des composés organiques. L’origine de ces difficultés et

MODÉLISATION CHIMIQUE DE L’OZONE 7
6 7carbon number
3 4 5102
103
104
105
106
nu
mb
er o
f sp
ecie
s o
r rea
ctio
ns 107
n-alkane seriesi-alkane series1-alkene seriesisoprene
Figure 1: Nombres d’espèces (symboles blancs) et de réactions (symboles gris) nécessaire pour décrire explicite-ment l’oxydation des COV. [D’après Aumont et al., 2005].
les contraintes qu’elles induisent sur le développement des schémas sont présentées dans lasection suivante.
1.3. Modélisation de l’oxydation gazeuse des composés organiques : enjeux etdifficultés
Des schémas chimiques de grande dimensionContrairement aux transformations inorganiques, les mécanismes impliqués dans l’oxyda-tion des composés organiques impliquent une myriade d’intermédiaires. La figure 1 pré-sente le nombre d’espèces nécessaire pour décrire de façon explicite l’oxydation complèted’un composé donné. Le nombre d’intermédiaires croît de façon exponentielle avec le nombred’atomes de carbone du composé parent (i.e. le composé émis). Le nombre d’intermédiairesest de l’ordre du million d’espèces pour un composé en C8 comme l’octane.
Plusieurs centaines de composés organiques volatiles (COV) sont émis dans l’atmosphère et,en toute rigueur, le schéma chimique devrait expliciter en détail les mécanismes d’oxydationde chaque composé. Cependant, même dans le cadre de simulation 0D (modèle de boîte),l’intégration d’un schéma chimique de cette nature demeure aujourd’hui encore ingérable.
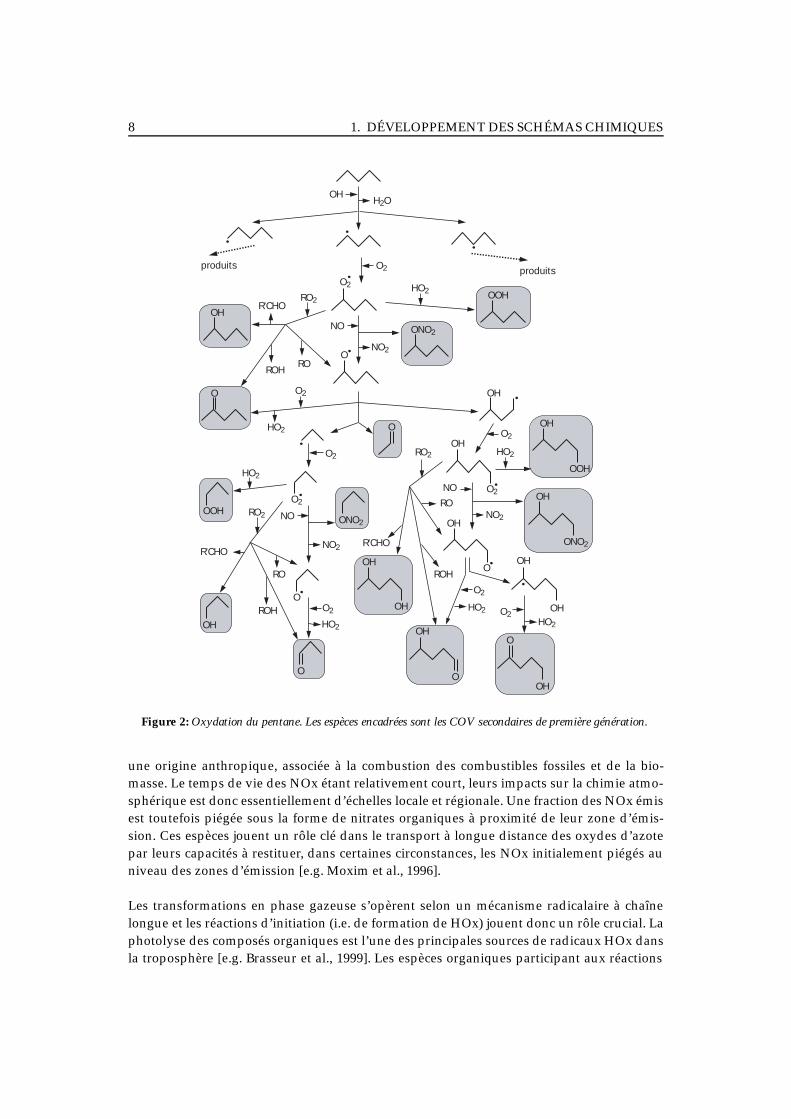
Oxydation des COV : quelles informations chimiques retenir ?La figure 2 présente les premières étapes associées à l’oxydation du pentane. Ce schémamet en évidence le grand nombre de branchements possibles au niveau de la plupart desintermédiaires de réaction. La nature et la distribution des espèces secondaires produitesdépendent directement de ces branchements. Certaines de ces espèces jouent un rôle centraldans la chimie de la troposphère aux différentes échelles.
Par exemple, les nitrates organiques (RONO2) sont un vecteur du transport à longue dis-tance des oxydes d’azote [Roberts, 1990; Moxim et al., 1996; Brasseur et al., 1999]. A l’échelleglobale, on estime que les oxydes d’azote (NOx) émis dans l’atmosphère ont principalement
8 1. DÉVELOPPEMENT DES SCHÉMAS CHIMIQUES
O2
OH
OH
OH
O
O2
O
OH
ONO2
OOH
OH
OOH
O
OH
OH
O
OH
O
OH
OOH
OH
ONO2
OH
O
O
OH
OOHONO2
OHH2O
NO
ROROH
R’CHO
ROH
R’CHO
NO
NO
R’CHO
ROH
RO
RO
produitsproduits
HO2
HO2
NO2
NO2
HO2
O2
O2
RO2
RO2
HO2
O2
O2
O2
HO2
NO2
O2
HO2
RO2
HO2
O2
O2
Figure 2: Oxydation du pentane. Les espèces encadrées sont les COV secondaires de première génération.
une origine anthropique, associée à la combustion des combustibles fossiles et de la bio-masse. Le temps de vie des NOx étant relativement court, leurs impacts sur la chimie atmo-sphérique est donc essentiellement d’échelles locale et régionale. Une fraction des NOx émisest toutefois piégée sous la forme de nitrates organiques à proximité de leur zone d’émis-sion. Ces espèces jouent un rôle clé dans le transport à longue distance des oxydes d’azotepar leurs capacités à restituer, dans certaines circonstances, les NOx initialement piégés auniveau des zones d’émission [e.g. Moxim et al., 1996].
Les transformations en phase gazeuse s’opèrent selon un mécanisme radicalaire à chaînelongue et les réactions d’initiation (i.e. de formation de HOx) jouent donc un rôle crucial. Laphotolyse des composés organiques est l’une des principales sources de radicaux HOx dansla troposphère [e.g. Brasseur et al., 1999]. Les espèces organiques participant aux réactions

MODÉLISATION CHIMIQUE DE L’OZONE 9
d’initiation sont essentiellement des composés carbonylés secondaires (i.e. issu de l’oxyda-tion des COV parents). La photodissociation de ces composés carbonylés représente notam-ment la principale source primaire de radicaux HOx en zones urbaines et péri urbaines [e.g.Seinfeld and Pandis, 1998; Aumont et al., 2003]. La photolyse des composés carbonylés estégalement suspectée d’être une source significative de radicaux dans la haute troposphère[Wennberg et al., 1998; Muller and Brasseur, 1999]. De même, les hydroperoxydes contri-buent au transport et à la redistribution des HOx, en piégeant les radicaux peroxyles aumoment de leur formation (RO2+HO2 → ROOH) et en restituant les HOx par photolyse(ROOH+hν → RO+OH) [Prather and Jacob, 1997; Muller and Brasseur, 1999].
Les COV secondaires présentent également des réactivités très différentes avec OH. Cetteréactivité dépend notamment des fonctionnalités portées par les composés organiques. Ladistribution des COV secondaires conditionne donc directement la réactivité de la massed’air lors de son vieillissement. La réactivité des COV secondaires est un paramètre parti-culièrement sensible à l’échelle régionale, lorsque (1) la réactivité de la masse d’air vis-à-visdes OH n’est plus totalement contrôlée par les composés primaires, (2) les concentrations deNOx sont suffisamment élevées pour maintenir le système dans un régime de type saturé enNOx.
La nature des séquences réactionnelles impliquées dépend également des branchements(c’est-à-dire la proportion d’une espèce qui évolue selon une voie déterminée). En ce quiconcerne la formation de l’ozone, les chaînons clés sont : la conversion de NO en NO2(contrôle la production nette d’ozone), les puits et sources de NOx (contrôlent le régimechimique) et les sources et puits de HOx (contrôlent l’efficacité des transformations). Cesdifférents chaînons caractéristiques sont reportés sur la figure 2 pour l’exemple du pentane.L’impact du pentane (en fait de chaque COV) sur la réactivité de la troposphère dépend doncdirectement des chemins réactionnels suivis lors de son oxydation.
Les rapports de branchement sont en fait des rapports de vitesse de réaction. Ils sont fonctiondes conditions physiques (température, pression) et chimiques (rapport de concentration desdifférents réactifs) ; leurs valeurs varient d’une situation à l’autre. L’évaluation précise desrapports de branchement (et donc des produits finaux) nécessite donc de conserver des sché-mas d’oxydation détaillés.
Enfin, la contribution d’un COV particulier sur le bilan total des NOx, des HOx ou de l’ozoneest généralement faible, quel que soit le composé considéré. C’est la somme de ces "petitescontributions" qu’il faut en pratique totaliser. Il est donc nécessaire, en toute rigueur, d’éva-luer précisément l’ensemble de ces branchements pour chaque COV.
Les contraintes liées au développement des schémas chimiquesLa représentation dans les schémas chimiques des mécanismes induits par l’oxydation descomposés organiques est donc soumise à deux contraintes fortes :– le nombre d’espèces et de réactions impliquées dépasse très largement celui pour les-
quelles des données cinétiques et mécanistiques expérimentales sont disponibles. Il fautdonc pallier le manque de données concernant l’oxydation des composés organiques ce

10 1. DÉVELOPPEMENT DES SCHÉMAS CHIMIQUES
qui impose le développement d’approches permettant d’estimer les paramètres mécon-nus.
– une description exhaustive conduit à des schémas de très grande dimension dont l’inté-gration au modèle de transport 3D est à exclure. Il faut donc développer des méthodespermettant de simplifier le schéma chimique organique afin d’en réduire sa dimension.
Différentes approches ont été proposées pour répondre à ces deux contraintes. Les choixet les priorités effectuées par les différents auteurs expliquent la diversité des mécanismesdisponibles dans la littérature. Une même base de données chimiques a ainsi conduit audéveloppement de schémas chimiques très différents, bien que tous dédiés à la simulationdes mêmes processus.
1.4. Les étapes de l’oxydation des composés atmosphériques
L’oxydation atmosphérique des COV est une oxydation progressive. Les étapes de cette oxy-dation peuvent être décrites par un nombre limité de réactions types, répétés de nombreusesfois jusqu’à l’oxydation complète du composé parent. A quelques exceptions près, ces étapespeuvent être résumées comme suit (cf. figure 3) :– l’initiation de la chaîne radicalaire organique par réaction avec OH, NO3, O3 ou par rup-
ture d’une liaison après absorption d’un photon. Cette première étape conduit générale-ment à la formation d’un radical peroxy RO2.
– les réactions des radicaux peroxy avec NO, NO2, NO3, HO2 ou avec d’autres RO2. Cesréactions conduisent soit à la formation d’un composé organique secondaire stable (i.e.non radicalaire), soit à la formation d’un radical alkoxy (RO).
– les réactions des radicaux alkoxy avec O2, de décomposition unimoléculaire (rupture d’uneliaison C-C) ou d’isomérisation. Ces transformations conduisent soit à la formation d’unCOV secondaire stable, soit à la formation d’un radical RO2.
De façon schématique, décrire les mécanismes d’oxydation des COV revient donc à docu-menter les réactions types, ci-dessus, pour les différentes structures moléculaires possibles.De nombreuses données expérimentales sont aujourd’hui disponibles concernant l’oxyda-tion des COV dans l’atmosphère. Des compilations de ces données, accompagnées d’éva-luations et de recommandations, sont régulièrement publiées par la NASA et l’IUPAC [e.g.DeMore et al., 1997; Atkinson et al., 1999], ainsi que dans des articles de synthèse [e.g. At-kinson, 1994; Atkinson et al., 2003, 2004, 2005; Orlando et al., 2003; Mellouki et al., 2003; Cal-vert et al., 2000]. Certaines étapes de l’oxydation des COV sont aujourd’hui bien documen-tées, comme par exemple les réactions COV+OH. En revanche, les données expérimentalesconcernant les réactions des alkoxy restent limitées ; en particulier pour les composés destructures moléculaires complexes (composés fonctionnalisés et/ou possédant de longueschaînes carbonées).
1.5. Reconstitution des données cinétiques et mécanistiques
Les relations de structure/réactivité (SAR)D’après la figure 1, il est clair que le nombre de paramètres cinétiques et mécanistiques à do-cumenter pour décrire complètement l’oxydation des COV excède très largement le nombre

MODÉLISATION CHIMIQUE DE L’OZONE 11
composé parent(primaire)
chimieCOV+oxydant
chimie desalkoxy (RO)
déc
om
po
siti
on
iso
mér
isat
ion
OH
NO3
O3
NONO3
RO2
HO
2 - N
O -
RO
2
O2
1ère générationde produits
CO2
chimie desperoxy (RO2)
chimieCOV+oxydant
chimie desalkoxy (RO)
déc
om
po
siti
on
iso
mér
isat
ion
OH
NO3
O3
NONO3
RO2
HO
2 - N
O -
RO
2O2
chimie desperoxy (RO2)
chimieCOV+oxydant
chimie desalkoxy (RO)
déc
om
po
siti
on
iso
mér
isat
ion
OH
NO3
O3
NONO3
RO2
chimie desperoxy (RO2)
2ième générationde produits
Figure 3: Répétition des trois principales étapes de l’oxydation des COV dans la troposphère jusqu’aux produitsfinaux d’oxydation. [D’après Aumont et al., 2005]
de données expérimentales disponibles. Pour la plupart des espèces, les constantes ciné-tiques et les mécanismes réactionnels doivent donc être estimés sur la base de leur structuremoléculaire. Les méthodes d’estimation proposées dans la littérature reposent pour la plu-part sur des approches de type "contribution de groupe". Une molécule donnée est scindéeen groupes élémentaires (par exemple les différents atomes de carbone et leurs substituants)dont la contribution à la réactivité totale est attribuée de façon empirique ou semi-empirique(par exemple sur la base de propriétés thermodynamiques).
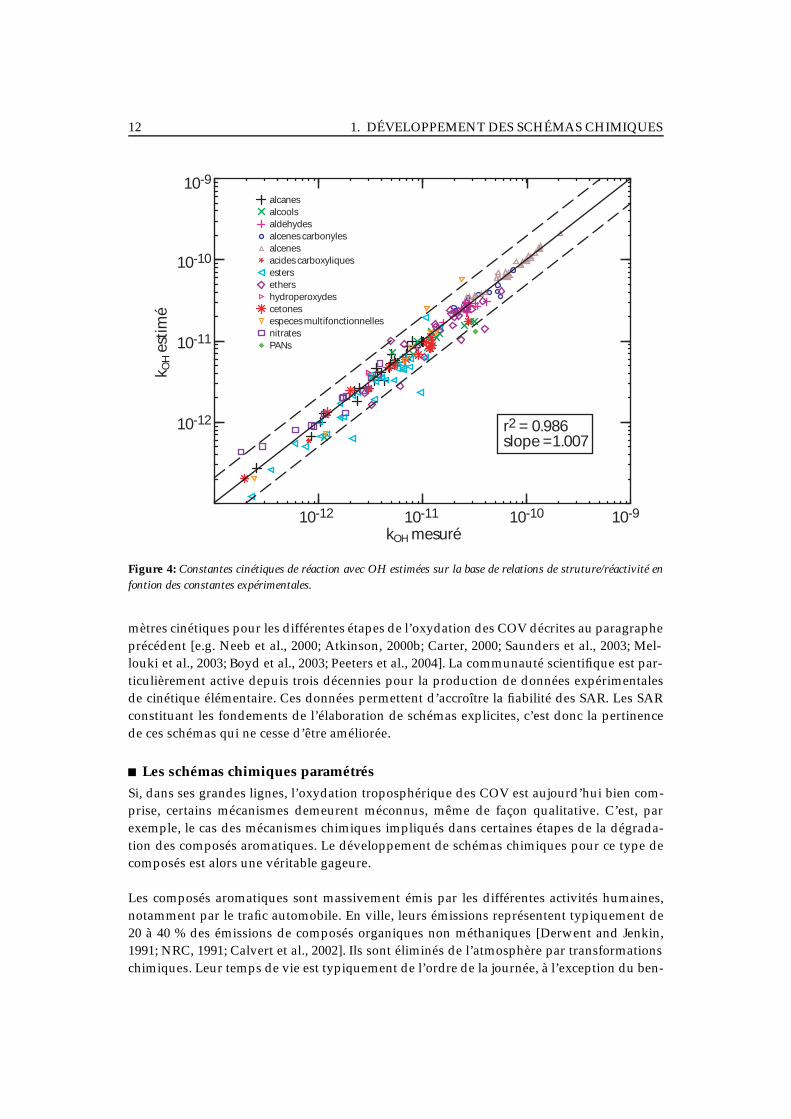
Les relations de structure/réactivité présupposent naturellement l’acquisition d’un nombreminimum d’informations expérimentales. Leur fiabilité dépend donc largement du nombrede données disponibles. Par exemple, on dispose aujourd’hui d’une large base de donnéessur les constantes cinétiques des réactions COV+OH (≈ 500 COV étudiés) [Atkinson, 2000b].Cette base a notamment permis l’établissement de relations simples de structure/réactivité,permettant de prédire 90 % des observations avec une incertitude inférieure à un facteur 2[Kwok and Atkinson, 1995; Atkinson, 2000b]. Ce point est illustré sur la figure 4, qui présenteles constantes cinétiques estimées sur la base de SAR en fonction des constantes expérimen-tales pour différentes familles de composés d’intérêt atmosphérique. A l’inverse, pour l’écri-ture de la réactivité des radicaux peroxy acyl (RCO3), les seules informations expérimentalesdisponibles sont relatives au radical CH3C(O)O2. Il faut donc attribuer à l’ensemble des radi-caux RCO3 la réactivité de CH3C(O)O2, ce qui constitue la plus simple (mais probablementla moins fiable) des relations de structure/réactivité.
Il existe aujourd’hui des relations de structure/réactivité permettant d’estimer les para-
12 1. DÉVELOPPEMENT DES SCHÉMAS CHIMIQUES
10-12 10-11 10-10 10-9
kOH mesuré
alcanesalcoolsaldehydesalcenes carbonylesalcenesacides carboxyliquesestersethershydroperoxydescetonesespeces multifonctionnellesnitratesPANs
r2 = 0.986slope =1.007
10-12
10-11
10-10
10-9
kO
H e
stim
é
Figure 4: Constantes cinétiques de réaction avec OH estimées sur la base de relations de struture/réactivité enfontion des constantes expérimentales.
mètres cinétiques pour les différentes étapes de l’oxydation des COV décrites au paragrapheprécédent [e.g. Neeb et al., 2000; Atkinson, 2000b; Carter, 2000; Saunders et al., 2003; Mel-louki et al., 2003; Boyd et al., 2003; Peeters et al., 2004]. La communauté scientifique est par-ticulièrement active depuis trois décennies pour la production de données expérimentalesde cinétique élémentaire. Ces données permettent d’accroître la fiabilité des SAR. Les SARconstituant les fondements de l’élaboration de schémas explicites, c’est donc la pertinencede ces schémas qui ne cesse d’être améliorée.
Les schémas chimiques paramétrésSi, dans ses grandes lignes, l’oxydation troposphérique des COV est aujourd’hui bien com-prise, certains mécanismes demeurent méconnus, même de façon qualitative. C’est, parexemple, le cas des mécanismes chimiques impliqués dans certaines étapes de la dégrada-tion des composés aromatiques. Le développement de schémas chimiques pour ce type decomposés est alors une véritable gageure.
Les composés aromatiques sont massivement émis par les différentes activités humaines,notamment par le trafic automobile. En ville, leurs émissions représentent typiquement de20 à 40 % des émissions de composés organiques non méthaniques [Derwent and Jenkin,1991; NRC, 1991; Calvert et al., 2002]. Ils sont éliminés de l’atmosphère par transformationschimiques. Leur temps de vie est typiquement de l’ordre de la journée, à l’exception du ben-

MODÉLISATION CHIMIQUE DE L’OZONE 13
zène qui possède un temps de vie d’une dizaine de jours [Calvert et al., 2002]. L’impact di-rect des aromatiques se répercute ainsi principalement sur les échelles locale et régionale. Lacontribution des composés aromatiques au bilan de l’ozone dans le panache des grandes ag-glomérations est suspectée d’être de l’ordre de 30 à 40 % [Derwent and Jenkin, 1991; Derwentet al., 1996; Carter et al., 1994; Calvert et al., 2002]. Le schéma chimique décrivant l’évolutiondes composés aromatiques est donc un élément crucial des modèles opérationnels dédiésaux études de qualité de l’air.
Lorsque les mécanismes réactionnels demeurent méconnus, les modèles sont, pour la plu-part, développés en ayant recours à des schémas chimiques paramétrés. L’approche reposesur l’ajustement empirique du schéma chimique (c’est à dire des produits de réaction et deleurs coefficients stoechiométriques) de telle sorte que les résultats de simulation s’accordentsur les évolutions observées en Chambre de Simulation Atmosphérique (CSA). Par exemple,les espèces issues de l’addition de OH sur le cycle aromatique sont paramétrées, dans le mé-canisme SAPRC99 [Carter, 2000] selon le mécanisme :
OH + aromatique → adduit-OHadduit-OH + O2 → a1 (HO2 + crésol) + a2 (adduit-OH-O2)
(adduit-OH-O2) + NO → a3 (nitrates-aromatiques) + a4 (radicaux non caractérisés)(radicaux non caractérisés) → HO2 + α-dicarbonyls + (produits non caractérisés)
Dans ce mécanisme, les différents paramètres libres (les coefficients stoechiométriques ai etla réactivité des «produits non caractérisés») sont ajustés sur la base des observations enCSA. Trois composés dicarbonylés sont considérés (glyoxal, méthyl glyoxal et biacétyl). Lesproduits non caractérisés sont représentés par trois composés «modèles», dont la réactivitéest grossièrement calquée sur celles des espèces dicarbonylées insaturées et les rendementsajustés sur la base des observations. Avec autant de paramètres ajustables, les simulationsconvergent naturellement sur les observations en CSA, en particulier pour ce qui concernela production d’ozone. L’accord entre simulations et observations n’est cependant pas ungage d’exactitude du schéma chimique retenu. Le développement de schémas chimiquesparamétrés sur la base des mesures en CSA reste en effet sujette à caution [e.g. Dodge, 2000] :– les conditions imposées par l’expérience (notamment la limite de détection des instru-
ments) sont généralement sensiblement différentes des conditions atmosphériques. Cecipose un problème pour extrapoler les résultats de CSA à l’atmosphère.
– l’ajustement concerne préférentiellement l’ozone, et non d’autres composés (NOy, COVsecondaires notamment). La contribution des composés aromatiques sur le bilan des NOxet des HOx n’est donc que partiellement contrainte et peut également poser problème envue d’applications atmosphériques.
– les expériences sont habituellement conduites sur une période de temps limitée (typique-ment une journée). La contribution potentielle des composés organiques oxygénés issusdes étapes d’oxydation successives n’est ainsi que très partiellement prise en considéra-tion par le processus d’ajustement du mécanisme.
– les paramètres permettant de parfaitement qualifier l’expérience ne sont souvent pas oupeu caractérisés (rayonnement dans l’enceinte réactionnel, adsorption/désorption et trans-formation chimique aux parois). Ces incertitudes se répercutent directement sur le méca-nisme paramétré.
Les mécanismes opérationnels couramment utilisés dans les modèles tridimensionnels de

14 1. DÉVELOPPEMENT DES SCHÉMAS CHIMIQUES
qualité de l’air décrivent la chimie des aromatiques sont fondées sur une approche compa-rable, par ajustement sur les observations en CSA. C’est par exemple le cas du CB4 [Geryet al., 1989], RADM [Stockwell et al., 1990], RACM [Stockwell et al., 1997].
Si l’élucidation du mécanisme d’oxydation des composés aromatiques représente l’un desprincipaux défis de la communauté scientifique depuis une vingtaine d’années, d’autrespans de la chimie atmosphérique présentent des incertitudes analogues (c’est par exemplele cas de l’oxydation des terpènes, des composés oxygénés ou de certaines séquences réac-tionnelles comme l’ozonolyse des alcènes). L’utilisation de schémas chimiques paramétrésne se limite donc pas aux seuls composés aromatiques.
L’acquisition régulière de données de cinétique élémentaire permet peu à peu de contraindreles schémas chimiques paramétrés et d’accroître en conséquence leur fiabilité en diminuantle nombre de degrés de liberté. En particulier, de nombreux progrès ont été réalisés concer-nant le mécanisme d’oxydation des aromatiques. Des mécanismes de plus en plus détailléssont publiés dans la littérature [Wagner et al., 2003] et des protocoles sont aujourd’hui propo-sés pour développer le schéma d’oxydation des composés aromatiques sur une base systé-matique et cohérente à partir d’un ensemble de relations de type structure/réactivité [Jenkinet al., 2003; Bloss et al., 2005].
1.6. Réduction des schémas : influence du milieu
Nous avons vu précédemment qu’une description exhaustive des mécanismes d’oxydationdes COV conduit à des millions de composés. En raison des capacités des calculateurs ac-tuels, la prise en compte de l’ensemble des réactions est impossible dans les modèles tri-dimensionnels. Les schémas chimiques destinés à simuler les champs tridimensionnels desespèces sont donc systématiquement réduits. Actuellement, la dimension des schémas chi-miques destinés à être intégrés dans les modèles de chimie/transport doit être limitée àquelques centaines d’espèces. De plus, la recherche d’une représentation aussi simple quepossible des transformations chimiques constitue, en soit, une motivation à part en entièrepour le développement de modèles opérationnels. En effet, les sorties des modèles ne sontsouvent analysées qu’en regard de quelques espèces présentant un intérêt direct (ozone,NOx, OH, quelques précurseurs).
Influence du milieu sur la réduction des schémasDifférentes hypothèses et adaptations ad hoc sont souvent à l’origine des simplifications réa-lisées et l’ensemble est généralement formulé pour rendre compte de situations particulières.En pratique, les procédures de réduction doivent prendre en compte 2 principaux facteurs :(i) la grande diversité des COV émis et (ii) la variation rapide des concentrations de la plu-part des espèces clés de la chimie atmosphérique, en particulier des NOx (depuis des valeursoù la chimie de l’ozone est "limitée par les COV" jusqu’aux valeurs où elle est "limitée parles NOx"). Deux cas limites peuvent être considérés en premier lieu : les zones d’émissionsanthropiques intenses et la troposphère libre, éloignée de toute source intense d’émission.

MODÉLISATION CHIMIQUE DE L’OZONE 15
Les zones d’émissions anthropiques intensesCe cas correspond typiquement aux milieux urbains et péri-urbains et aux grandes zones in-dustrielles. Ces milieux sont caractérisés par des concentrations de NOx élevées (supérieuresà quelques ppb). La chimie des radicaux peroxy (RO2, HO2) est alors dominée par la réactionavec NO et leurs réactions de recombinaison sont négligeables. Il est, en conséquence, pos-sible de décrire "simplement" les schémas d’oxydation des COV. De plus, dans ces milieux,la chimie est principalement pilotée par la réactivité des COV primaires, ce qui autoriseune représentation simplifiée de la chimie induite par les COV secondaires. Néanmoins, lenombre de COV primaires à traiter reste élevé, ce qui impose d’effectuer des regroupementssous un nombre plus limité d’espèces représentatives. Les premiers schémas chimiques dé-diés à l’étude de la pollution urbaine ont couramment utilisé cette approche [e.g. Atkinsonet al., 1982]. Elle a toutefois été abandonnée, les échelles spatiales à traiter pour modéliserla formation des photooxydants s’étendant au-delà de l’échelle urbaine et impliquant undomaine où coexistent simultanément des milieux riches et pauvres en NOx.
La troposphère libre, éloignée de sources d’émissionLa chimie des atmosphères éloignées est principalement contrôlée par l’oxydation du mé-thane, du monoxyde de carbone et de quelques COV à temps de vie longs (éthane et acé-tone). Cette chimie de fond est aujourd’hui bien établie et documentée [DeMore et al., 1997;Atkinson et al., 1999, 2004]. Dans ces milieux, la concentration en NOx est faible (inférieure àquelques dizaines de ppt) et la chimie des radicaux peroxy doit être traitée en détail. Néan-moins, dans ce type d’environnement, le nombre de COV à gérer est faible et des schémasquasi-explicites peuvent être utilisés, la représentation des COV supérieurs étant traitée defaçon sommaire [e.g. Brasseur et al., 1998; Bey et al., 2001c].
La troposphère continentaleLa chimie de la troposphère continentale se trouve entre ces 2 cas limites. Pour ces milieux,aucune simplification simple ne peut être opérée ; la "compression" du schéma chimique estalors réalisée en effectuant des regroupements d’espèces. Ce point est abordé dans la sectionsuivante.
2. Schémas chimiques dédiés aux atmosphères continentales
La difficulté liée à la modélisation de ce type d’environnement tient au fait qu’aucune sim-plification chimique ne peut être aisément effectuée :– une multitude de COV, primaires comme secondaires, sont présents à des concentrations
significatives ;– la gamme des concentrations de NOx est extrêmement étendue, ce qui impose un traite-
ment détaillé de la chimie des radicaux peroxy.Dans ce cas, des regroupements intenses sont indispensables afin de donner au schéma chi-mique une taille raisonnable. Pour l’essentiel, deux types de regroupement sont utilisés dansla littérature pour représenter les différents COV : le regroupement en familles structurales

16 2. SCHÉMAS CHIMIQUES DÉDIÉS AUX ATMOSPHÈRES CONTINENTALES
PAR OLE ISOP TOL, XYL
DICARBONYLSALD2KETONES
PANETHMETHANE
FORM
CO
NOx, HOx O
8 PAR
1 OLE + 1 PAR
1 XYL + 1 PAR
1 ALD2 + 1 PAR
a b
Figure 5: a : Espèces retenues dans le mécanisme Carbon Bond (CB4) et leur relation "hiérarchique" [d’aprèsGery et al., 1989]. b : Représentation de quelques COV dans le mécanisme Carbon Bond.
et le regroupement en familles moléculaires. Le concept d’opérateur est également fréquem-ment utilisé pour regrouper la chimie des radicaux peroxy. Cette section vise à présenterles hypothèses et les approximations classiquement effectuées pour comprimer les schémaschimiques de quelques millions d’espèces à quelques centaines de COV. Cette compressiondu schéma induit une perte de précision et de représentativité qu’il importe de quantifier.Ce point est abordé à la section 3.3.
2.1. Regroupement en famille structurale
Le regroupement en familles structurales est basé sur une analyse de la structure des mo-lécules. La méthode consiste à scinder chaque molécule en autant de "composés" différentsque d’entités chimiques réactives qui la constituent. Les "composés" considérés ne sont pasdes espèces réelles mais des "variables chimiques" destinées à représenter la réactivité desdifférents groupes fonctionnels portés par la molécule considérée (par exemple carbone ali-phatique, oléfinique ...). Le schéma chimique est ensuite développé à partir de ces "variableschimiques". La sélection d’un nombre limité de variables représentatives permet de décrireun grand nombre de COV. L’approche conduit donc à des schémas chimiques très conden-sés. Ce type de regroupement a donné naissance à toute une famille de modèles, connus sousl’appellation de "Carbon Bond" [Whitten et al., 1980; Gery et al., 1989; Simonaitis et al., 1997].Dans la version 4 du "Carbon Bond" [Gery et al., 1988], l’ensemble de la chimie organiqueest représenté par 11 variables représentant les "structures moléculaires" stables (1 alcane, 2alcènes, 2 aromatiques, 1 biogénique, 4 carbonyles, 1 nitrate) et 6 "structures" radicalaires.Le schéma chimique complet renferme 32 espèces et 81 réactions. La figure 5 présente lesespèces majeures du mécanisme CB4 ainsi que quelques exemples de "découpage" de COVdans les différentes structures réactives du modèle.
L’approche par regroupement structural suppose que la réactivité d’un COV (et de l’en-semble de son mécanisme d’oxydation) dépend uniquement de son site réactionnel et nonde sa structure moléculaire. Toutefois, des paramètres comme la longueur de la chaîne car-bonée, la classe des radicaux, les groupes fonctionnels adjacents, la structure globale de la

MODÉLISATION CHIMIQUE DE L’OZONE 17
molécule interviennent parfois de façon déterminante sur la réactivité. De plus, avec unetelle méthode, les molécules organiques perdent intégralement leur individualité chimique.Ceci peut s’avérer suffisant lorsqu’il s’agit de simuler l’évolution chimique globale d’unemasse d’air ou l’évolution d’espèces telles que l’ozone. Néanmoins, l’approche s’avère to-talement inadaptée au suivi de composés organiques primaires et plus généralement à laquantification de chaînons chimiques.
2.2. Les espèces de regroupement
Ces regroupements sont appliqués dans la plupart des schémas chimiques destinés à êtreintégrés aux modèles de chimie/transport, comme par exemple SAPRC [Carter, 1990, 2000],RADM [Stockwell et al., 1990], RACM [Stockwell et al., 1997], MELCHIOR [Lattuati, 1997;Schmidt et al., 2001], EMEP [Simpson, 1992], MOCA [Aumont et al., 1996].
Principes de l’approcheLa méthode consiste à condenser le mécanisme en opérant un regroupement des compo-sés organiques de réactivité similaire. L’approche repose sur la détermination d’une espèce"moyenne" représentant le comportement d’une famille entière de composés. Par exemple,l’heptane et l’octane sont deux hydrocarbures primaires présentant une réactivité proche ;il est donc concevable de les regrouper sous une espèce unique représentant une réactivitémoyenne sans induire de biais significatif. L’espèce ainsi créée n’a pas de réalité physique ;elle est conçue de telle sorte que son évolution reflète le comportement moyen de la famillede composés. Cette entité virtuelle est qualifiée d’espèce de regroupement, d’espèce géné-ralisée ou encore d’espèce modèle. La réactivité des espèces de regroupement est calculée àpartir des constantes cinétiques et des mécanismes réactionnels des différentes espèces dugroupe.
Mise en oeuvreSoit par exemple les réactions avec OH d’un ensemble d’hydrocarbures HCi :
HC1 + oxydant → a1,1 prod1 + · · · + ai,1 prodi k1HCj + oxydant → a1,j prod1 + · · · + ai,j prodi kj
L’objectif des techniques de regroupement est de remplacer ce groupe de réactions par uneréaction unique de la forme :
REG + oxydant → ν1 prod1 + · · · + νi prodi kREG
où REG est l’espèce de regroupement utilisée comme substitut aux différents HCi. La dif-ficulté réside dans la détermination de la concentration CREG de l’espèce de regroupement,des coefficients stoechiométriques νj et de la constante cinétique kREG.
L’approche la plus directe consiste à regrouper une mole de HCi sous une mole de REG,soit en terme de concentration CREG = ΣCHCi. En égalant les vitesses de consommation

18 2. SCHÉMAS CHIMIQUES DÉDIÉS AUX ATMOSPHÈRES CONTINENTALES
(dCREG/dt = ΣdCHCi/dt), la constante cinétique associée à REG et les coefficients stoechio-métriques sont donnés par :
kREG = ∑ ki × CHCi
∑ CHCiνj =
∑ aij × CHCi
∑ CHCi
La difficulté est liée au fait que les concentrations relatives des différents HC varient dans letemps et dans l’espace (chaque HC ayant des constantes cinétiques différentes et la distribu-tion spatiale des sources étant hétérogène). Il en résulte que la constante cinétique kREG etles coefficients stœchiométriques sont, en toute rigueur, également des fonctions du tempset de l’espace. Ces paramètres devraient donc être constamment réactualisés au cours dutemps de la simulation dans chacune des mailles du modèle. Cette réactualisation est inen-visageable, la "mémoire" sur la distribution des HC étant perdue en raison du regroupement.En pratique, le schéma chimique est figé dans le temps et l’espace ; il n’est donc pas possiblede calculer rigoureusement la constante cinétique et les coefficients stœchiométriques de laréaction globale.
Afin de limiter les erreurs, le regroupement ne doit être exécuté que pour des espèces ayantdes constantes cinétiques voisines [Carter, 1988; Middleton et al., 1990]. L’application decette approche repose donc sur la recherche d’un optimum entre la précision du schémachimique (impliquant une multiplication des familles, donc d’espèces de regroupement) etsa dimension [e.g. Jaubertie, 1999; Junier et al., 2005]. Par exemple, dans le schéma RACM[Stockwell et al., 1997], les groupes sont définis de telle sorte que le rapport entre la constantede l’espèce la plus réactive et celle de la moins réactive d’une même famille n’excède pasun facteur 2. La totalité des alcanes est ainsi représentée par 3 espèces de regroupement,les constantes cinétiques (à 298k) avec OH fixant les frontières : (i) kOH < 3, 4.10−12, (ii)3, 4.10−12 < kOH < 6, 8.10−12, (iii) kOH > 6, 8.10−12 molécules−1.cm3.s−1.
Dépendance du schéma chimique aux inventaires d’émissionLa mise en œuvre d’espèces de regroupement nécessite un ensemble volumineux de don-nées (constantes cinétiques, mécanismes réactionnels et quantités de matière de chaque es-pèce). Cette approche est principalement utilisée pour le regroupement des hydrocarburesprimaires, les données fournies par les inventaires d’émission 2 étant utilisées pour pondérerla contribution de chaque composé dans l’espèce de regroupement correspondante [Carter,1988; Middleton et al., 1990; Stockwell et al., 1990; Kirchner, 2005] :
kREG = ∑ ki × ECOVi
∑ ECOVi
où ECOVi est l’émission du COVi sur le domaine considéré.
Les schémas chimiques disponibles dans la littérature ont été établis sur la base d’une spécia-tion fixée de VOC. Par exemple, la réactivité des espèces de regroupement du modèle RACMrepose directement sur l’inventaire des émissions annuelles des USA [Stockwell et al., 1990;
2L’application des modèles de chimie/transport nécessite l’utilisation d’inventaires d’émission renseignant, pourchaque espèce primaire, les flux émis dans chaque maille et à chaque instant par les différentes sources naturelles ouanthropogéniques. Ces inventaires sont directement fournis en tant que données d’entrée aux modèles, ou calculésà chaque instant par différents modules couplés au modèle de chimie/transport.

MODÉLISATION CHIMIQUE DE L’OZONE 19
Middleton et al., 1990]. En toute rigueur, les constantes cinétiques kREG et les coefficientsstoechiométriques νi devraient être systématiquement recalculés afin de refléter les condi-tions spécifiques locales de la zone modélisée [e.g. Jaubertie, 1999; Kirchner, 2005]. Toutefois,la complexité des procédures de regroupement rend cette réévaluation des différents para-mètres du schéma chimique particulièrement fastidieuse. Il en est de même pour la modifi-cation ou la réactualisation des constantes cinétiques (mise à jour du schéma). Les schémaschimiques sont ainsi fréquemment utilisés «en l’état», tels que publiés dans la littérature.
Facteurs d’agrégationAfin de minimiser les biais introduits par le regroupement, les données d’émission sont par-fois «corrigées» d’un facteur d’agrégation [Middleton et al., 1990]. Le principe est qu’unCOV donné peut être représenté par une espèce de remplacement (REG) de constante ciné-tique différente pourvu qu’un facteur d’agrégation α soit appliqué à l’émission de ce COV.L’émission de l’espèce généralisée (EREG) s’exprime alors à partir des émissions de chaqueCOV (ECOV) selon EREG = ∑ αi × ECOVi. Le facteur α permet donc de corriger l’écart entrela constante cinétique de l’espèce i et celle du composé modèle en jouant sur les quantitésémises :– si ki < kREG, l’émission de l’espèce de regroupement est plus faible que celle de l’espèce
correspondante (αi < 1),– si ki > kREG, l’émission de l’espèce de regroupement est plus forte que celle de l’espèce
correspondante (αi > 1).
La difficulté réside dans la définition d’un critère permettant d’évaluer le facteur α de fa-çon quantitative. L’hypothèse communément adoptée consiste à supposer que l’impact d’unCOV est proportionnel à la quantité de ce COV ayant réagi [Carter, 1988; Middleton et al.,1990], donc à définir α comme :
α =fraction du COVi vrai ayant reagi
fraction de l′espece de regroupement ayant reagie
La fraction de COV consommée chimiquement dépend de nombreux paramètres, notam-ment de la durée de la simulation, des dimensions du domaine considéré, des conditionsphysiques et chimiques. La valeur de α ne peut donc pas être déterminée de façon rigoureuseet sur la seule base des propriétés de chaque COV émis. Afin d’évaluer approximativementα, on se place habituellement dans le contexte d’un modèle de boîte (0D) évoluant entre uninstant initial et final, dans lequel on suppose que : (i) les COV sont initialement présents (ii)les COV ne sont éliminés que par réaction avec OH, (iii) il n’existe aucune source de COV.On montre que α peut alors s’exprimer sous la forme :
αi =1− exp(−ki × INTOH)
1 − exp(−kREG × INTOH)
où INTOH représente la concentration de OH intégrée sur la durée de la simulation : INTOH =∫COHdt. Le problème est alors déplacé vers l’évaluation du paramètre INTOH, ce para-
mètre dépendant également de la durée de la simulation et des conditions physiques et chi-miques. Pour des simulations de 1 à 2 jours, Middleton et al. [1990] recommande une valeurmoyenne de 110 ppt.min (soit une concentration 106 molécules.cm−3 intégrée sur 2 jours).

20 2. SCHÉMAS CHIMIQUES DÉDIÉS AUX ATMOSPHÈRES CONTINENTALES
Remarque : Les hypothèses émises pour définir le facteur d’agrégation α ont également étéproposées pour évaluer le facteur de pondération à attribuer à chaque COVi pour définirkREG (et les coefficients stoechiométriques) non pas à partir des quantités en présence maissur la base des quantités CR ayant réagi [Carter, 1988; Kirchner, 2005] soit :
kREG =∑ ki × CR
HCi
CRREG
= ∑ kiCHCi(1− exp(−ki × INTOH))∑ CHCi(1− exp(−ki × INTOH))
= ∑ αi × ki × CHCi
Σαi × CHCi
νj =∑ ai,j × CR
HCi
CRREG
=∑ αi × ai,j × CHCi
∑ αi × CHCi
Des facteurs d’agrégation ne reposant pas directement sur la réactivité du COVi mais surla conservation de la masse ont également été proposés. Par exemple, pour le modèle CHI-MERE (http ://euler.lmd.polytechnique.fr/chimere/), αi est défini comme :
αi =masse molaire du COVi vrai
masse molaire de l′espece de regroupement
Au sein d’une même famille, l’augmentation de la masse molaire est directement liée à l’aug-mentation de la chaîne carbonée donc de la réactivité des molécules. Comme la précédente,elle conduit donc de fait à (i) α > 1 pour les espèces les plus réactives, (ii) α < 1 pour lesespèces les moins réactives de la famille.
2.3. Les espèces de remplacement
Principe de l’approcheLa totalité des schémas chimiques implantés dans les modèles opérationnels de qualité del’air utilise cette approche. Avec cette approche, toutes les espèces d’un groupe donné deCOV sont substituées par une espèce réelle sélectionnée comme espèce de remplacement.Les espèces de remplacements sont surtout utilisées pour diminuer le nombre de COV se-condaires. Quelques schémas chimiques appliquent également cette approche aux COV pri-maires. Par exemple, EMEP [Simpson, 1992] et MELCHIOR [Lattuati, 1997; Schmidt et al.,2001] utilisent le butane comme espèce de remplacement à la famille des alcanes. Tout al-cane de chaîne carbonée supérieure à 3 est donc substitué dans le schéma par du butane. Ladéfinition des groupes de COV primaires utilise des principes analogues à ceux présentésci-dessus dans le contexte des espèces de regroupement (en terme de frontière des groupesou de facteur d’agrégation). Seule la mise en œuvre d’espèces de remplacement pour la re-présentation des COV secondaires est donc présentée ci-dessous.
Mise en œuvre pour les COV secondairesLe choix des composés utilisés comme espèces de remplacement résulte d’un compromisentre le nombre d’espèces à prendre en considération et la nécessité de couvrir au mieuxle comportement des différents COV. Dans les grandes lignes, les espèces de remplacementretenues varient peu d’un schéma à l’autre. Les traits principaux gouvernant la sélection deces espèces sont esquissés ci-dessous.

MODÉLISATION CHIMIQUE DE L’OZONE 21
Espèces carbonyléesL’oxydation des hydrocarbures conduit majoritairement à la production d’espèces carbony-lées (aldéhydes, cétones). Les premiers composés de la série des aldéhydes se distinguentsignificativement entre eux d’un point de vue cinétique et par la nature des produits is-sus de leur décomposition. Aussi, tous les schémas chimiques distinguent le formaldéhyde(HCHO) des aldéhydes supérieurs. Selon les schémas, l’acétaldéhyde (CH3CHO) ou le pro-panal (C2H5CHO) sont utilisés comme espèce de remplacement pour les aldéhydes supé-rieurs (e.g. CB4, RADM, SAPRC). De même, l’acétone étant significativement moins réactiveque les cétones supérieures, elle est souvent traitée de façon isolée ; la butanone est alors uti-lisée pour représenter les cétones supérieures (e.g. SAPRC).
La réactivité des espèces carbonylées est profondément modifiée lorsque la structure molé-culaire permet une délocalisation des électrons π de la liaison >C=O. L’oxydation des com-posés aromatiques et d’origine biogénique (isoprène en particulier) conduit à la formationd’espèces α-dicarbonylées (-CO-CO-) et/ou carbonylées insaturées (par exemple -C=C-CO-,-CO-C=C-CO). Le méthyl glyoxal CH3COCHO et le biacétyl (CH3COCOCH3) sont habi-tuellement utilisés pour représenter respectivement les structures RCOCHO et RCOCOR(e.g. RACM, Stockwell et al. [1997]). De même, les aldéhydes et les cétones insaturés sontrespectivement substitués par la méthacroléine (CH2=C(CH3)CHO) et la méthyl vinyl cé-tone (CH2=CHCOCH3), ces deux espèces étant massivement produites lors de l’oxydationde l’isoprène. Enfin, deux à trois COV sont également typiquement retenus pour représen-ter les espèces dicarbonylées insaturées (-COC=CCO-) issues de l’oxydation des composésaromatiques (e.g. RACM, SAPRC).
Autres COV secondaires oxygénésOutre les composés carbonylés, il importe également de représenter la formation des nitratesorganiques et des hydroperoxydes, du fait de leur rôle central sur le bilan des NOx et desHOx (voir section 1.3). Un nitrate organique est donc systématiquement retenu comme es-pèce de substitution, auquel s’ajoute un ou deux composés en remplacement des composésde type PAN (R-CO(OONO2)), issus de l’oxydation des aldéhydes retenus dans le schéma(cf. ci-dessus). De même, la formation des hydroperoxydes est typiquement représentée àl’aide d’une ou deux espèces de remplacement. Enfin, quelques espèces sont également uti-lisées comme espèces de substitution pour les composés aromatiques secondaires. Ainsi, lecrésol, le benzaldéhyde et le nitrophénol sont respectivement utilisés comme espèces de rem-placement des composés phénoliques, des aldéhydes aromatiques et des nitro-aromatiques.
Enfin, la nécessité de limiter autant que possible le jeu des espèces de remplacement conduitparfois à substituer une espèce par une autre d’une famille différente. Par exemple, les al-cools sont fréquemment substitués par le carbonyl correspondant (SAPRC90). Au final, cesont donc une quinzaine d’espèces qui sont utilisées en remplacement des millions de COVsecondaires formés lors de l’oxydation des COV.
Le problème du bilan de masseDans la majorité des cas, la substitution d’une espèce par son espèce de remplacement nepermet pas de conserver le bilan de masse. Par exemple, la substitution du butanal par le

22 2. SCHÉMAS CHIMIQUES DÉDIÉS AUX ATMOSPHÈRES CONTINENTALES
propanal induit la perte d’un atome de carbone. Les conséquences du non respect de laconservation de la masse de carbone doivent être relativisées en ce qui concerne la formationde l’ozone aux échelles locales et régionales, car :– les COV ne sont jamais en quantité "strictement limitante",– le régime limité en NOx est peu sensible aux COV,– le régime saturé en NOx est sensible à la réactivité de la matière organique (i.e. aux vitesses
de réaction COV+OH). Pour les COV secondaires, cette réactivité dépend principalementde la nature des fonctions.
Pour les COV secondaires, la priorité est donc donnée à la représentation des fonctions or-ganiques plutôt qu’au bilan du carbone. Cette approche entrave fortement la capacité de cesmodèles à être utilisés pour résoudre des questions scientifiques ne relevant pas directementdes problèmes des panaches d’ozone aux échelles locale et régionale.
Règles de décomposition et espèces multifonctionnellesL’oxydation des composés organiques conduit à la formation d’espèces multifonctionnelles.Leurs substitutions par des espèces de remplacement posent donc une difficulté supplé-mentaire : des règles de priorité doivent être établies entre les fonctions afin d’attribuer uneespèce de remplacement à chaque espèce multifonctionnelle considérée.
Les sources primaires de HOx jouent un rôle direct majeur dans la production d’ozone, aussibien en régime limité que saturé en NOx. La photolyse des aldéhydes est l’une des sourcesmajeures de radicaux. C’est de plus une fonction particulièrement réactive vis-à-vis du ra-dical OH. Le premier critère pris en compte pour opérer la substitution est donc la présenceou non du groupement aldéhydique [Carter, 1990; Stockwell et al., 1997; Kirchner, 2005]. Ilfaut de même définir une succession de règles de priorité pour les différentes fonctions as-sociées au squelette carboné. Dans le détail, il est parfois difficile de justifier la hiérarchiedes différentes priorités. Il est clair qu’elle possède au moins en partie un caractère arbitraireprononcé. La figure 6 présente l’exemple de la substitution de l’espèce réelle "CHO-CH2-CO-CHOH-COOH" selon les priorités et le jeu d’espèces de remplacement défini pour le schémaRACM [Kirchner, 2005]. L’application de ces règles de priorité conduit à simplement sub-stituer l’espèce réelle CHO-CH2-CO-CHOH-COOH par l’acétaldéhyde (CH3CHO), donc àignorer la présence des fonctions alcool, acide et cétone et à la perte de 3 atomes de carbone.Cette même espèce serait remplacée dans le schéma SAPRC [Carter, 1990] par "C2H5CHO+ 0.5 CH3COC2H5". Ceci résulte de l’utilisation, dans SAPRC, du propanal comme espècede remplacement des aldéhydes supérieurs et à la redistribution du carbone "résiduel" sousl’espèce de remplacement "butanone".
2.4. Equations bilans - opérateurs chimiques
Les COV sont oxydés au cours d’une suite de réactions élémentaires qui met en jeu unemultitude de radicaux peroxy (RO2) et alcoxy (RO) (cf. figure 3). Les constantes cinétiquesassociées aux transformations de ces intermédiaires étant le plus souvent méconnues, cer-taines d’entre-elles sont utilisées de façon générique pour un ensemble de réactions. Il s’ensuit un très grand nombre de réactions, formellement identiques, ne se distinguant que parla nature du groupement R. Ceci conduit à une grande redondance des chaînes d’oxydation

MODÉLISATION CHIMIQUE DE L’OZONE 23
O O O
OHOH
Aldehyde?
dicarbonyl?
-C(O)OOH?
peroxynitrate?
org.acid ?
Real species
1st priority
2nd priority
3th priority
4th priority
5th priority
mechanismspecies
>C=C< bond?
alcohol ?
ORA2KET HKET PAA PAN TPAN ALD MGLY MACR DCB UDD
>C=C< bond?
alcohol ?
methylglyoxal and other
a-carbonyl aldehydes
acetaldehyde and
higher analogs
unsatured PANs
peroxy acetyl nitrate &
higher satured PANs
peroxy acetic acid
and higher analogs
acetic acid and
higher acid
hydroxy ketone
ketones
unsaturated
monoadehydes
unsaturated
dicarbonyls
unsaturated
di-hydroxy dicarbonyls
yes
yes
yes
yes
yesyes yes
yes
yes
yes
no
no
no
no
no
no no
no
no
no
Figure 6: Regroupement des COV secondaires dans le modèle RACM. [D’après Kirchner, 2005].
en terme de bilan inorganique. La méthode des opérateurs chimiques, décrite dans cette sec-tion, tire profit de cette redondance pour condenser les schémas chimiques. Les opérateurschimiques sont utilisés, à des degrés divers, dans la plupart des schémas chimiques dédiés àla modélisation tridimensionnelle des photooxydants aux échelles locale et régionale [Geryet al., 1989; Stockwell et al., 1990, 1997; Aumont et al., 1996; Carter, 1990, 2000; Szopa et al.,2005].
Recherche d’une équation bilanL’étape cinétique limitante du processus d’oxydation des COV est l’initiation des chaînesradicalaires organiques. L’hypothèse de quasi-stationnarité peut ainsi être appliquée auxespèces intermédiaires radicalaires (radicaux RO2 et RO). Pour chaque COV, il est donc pos-sible de représenter l’ensemble des étapes élémentaires de l’oxydation sous la forme d’uneéquation bilan du type :
COV + Ox → α1COVsec1 + α2COVsec2 + · · ·+ β1NO + β2NO2 + β3HO2 + · · ·
où Ox est l’espèce initiant la chaîne radicalaire (OH, O3, NO3, éventuellement un photon),COVseci sont les composés organiques secondaires stables (i.e. non radicalaires) issus de lachaîne d’oxydation. Par exemple, l’ensemble des transformations associées à l’oxydation du

24 2. SCHÉMAS CHIMIQUES DÉDIÉS AUX ATMOSPHÈRES CONTINENTALES
pentane reporté figure 2 peut être représenté par une équation bilan du type :
C5H12 + OH → α1CH3COC3H7 + α2CH3COCH2CH2CH2(OH) + · · · + β1NO + β2NO2 + β3HO2 · · ·
Les coefficients stoechiométriques αi et β j indiqués dans l’équation bilan proviennent di-rectement des rapports de branchement des voies d’évolution des radicaux RO2 et RO. Cesrapports de branchement varient notablement en fonction du milieu chimique (cf. section1.3) ; il en est donc de même des coefficients stoechiométriques. Ceux-ci doivent donc êtreréévalués dans chaque maille du modèle et à chaque instant. Cette solution est évidem-ment coûteuse en temps calcul et n’apporte qu’un bénéfice limité par rapport à un schémachimique explicite (hormis la diminution de la raideur du système différentiel associé à l’in-tégration des radicaux RO).
Remarque : les coefficients stoechiométriques de l’équation bilan sont négatifs pour les es-pèces consommées. C’est par exemple le cas de NO, principalement converti en NO2 par lachaîne d’oxydation.
Principe des opérateurs chimiquesLa méthode développée par Carter [1990], dite méthode des "opérateurs", permet de s’af-franchir de la réévaluation périodique des coefficients stoechiométriques des équations bi-lans. Cette méthode repose sur le fait que les bilans inorganiques des chaînes d’oxydationsont peu variables d’un COV à l’autre : il s’agit pour l’essentiel de conversion NO → NO2et de production et/ou consommation de HO2. L’objectif de l’approche par opérateur est detraiter séparément le bilan organique, qui diffère d’un COV à l’autre, du bilan inorganique,commun à une multitude de COV. Ce bilan inorganique est piloté par quelques espècesmodèles appelées opérateurs et notées ci-dessous <opérateurs>. Avec cette approche, l’oxy-dation de chaque COV est représentée par une équation bilan de type :
COV+Ox→ a1 COVsec1 + a2 COVsec2 + · · · + an COVsecn︸ ︷︷ ︸produits organiques secondaires stables
+ b1 < operateur1 > +... + bn < operateurn >︸ ︷︷ ︸operateurs inorganiques
Exemple d’application des opérateurs : un cas académiqueLa mise en œuvre d’opérateurs chimiques est illustrée ci-dessous sur la base d’un exemplesimplifié. On considère deux hydrocarbures HC1 et HC2 dont l’évolution est schématique-ment représentée sur la figure 7. La réaction de chaque hydrocarbure avec OH conduit à laproduction d’un radical peroxy, respectivement R1O2 et R2O2. On suppose que les radicauxRiO2 évoluent uniquement selon deux réactions :– avec HO2 : la transformation conduit à la formation d’un hydropéroxyde (respectivement
R1OOH et R2OOH). La constante kHO2 est supposée indépendante du RO2 considéré.– avec NO : la transformation conduit à la formation d’un radical alkoxy (respectivement
R1O et R2O). La constante kNO est également supposée indépendante du RO2 considéré.On suppose enfin que chaque radical alkoxy RO libère, par réaction avec O2, un COV et HO2(cf. figure 7). Le bilan global de ces chaînes d’oxydation est alors :

MODÉLISATION CHIMIQUE DE L’OZONE 25
HC1 + OH R1O2 R1O COV1
R1OOH
O2
HO2
NO
NO2
HO2
kNO
kHO2
k1,OH
HC2 + OH R2O2 R2O COV2
R2OOH
O2
HO2
NO
NO2
HO2
kNO
kHO2
k2,OH
Figure 7: Représentation des chaînes d’oxydation avec des opérateurs. Le "cas académique" de l’oxydation dedeux hydrocarbures (HC).
HC1 + OH → αCOV1 + (1 − α)R1OOH + (1− α)NO2 − αNO + (2α − 1)HO2HC2 + OH → αCOV2 + (1 − α)R2OOH︸ ︷︷ ︸
Bilan organique
+ (1− α)NO2 − αNO + (2α − 1)HO2︸ ︷︷ ︸Bilan inorganique
avec
α =kNOCNO
(kNOCNO + kHO2CHO2)
Les deux chaînes d’oxydation sont donc identiques du point de vue du bilan inorganique.Elles ne se distinguent que par les vitesses d’initiation et la nature des COV produits. Ladifficulté est de représenter le coefficient α, qui découle directement des rapports de bran-chement au niveau des radicaux RO2. La méthode des opérateurs est un artifice destiné àreprésenter ce rapport de branchement. Elle consiste à introduire des espèces modèles (opé-rateurs chimiques) qui permettent une représentation condensée des différentes séquencesréactionnelles induites par les radicaux peroxy en terme d’espèces inorganiques.
Soit l’opérateur <RO2> destiné à représenter le bilan inorganique de HC1 et HC2. En négli-geant dans un premier temps le bilan de matière organique, on peut retranscrire la chaîned’oxydation de chaque HCi selon le schéma chimique :
(1) HCi + OH → <RO2> ki,OH(2) <RO2> + NO → NO2 + HO2 kNO(3) <RO2> + HO2 → kHO2
Dans ce schéma, le rapport de branchement de la voie (2) est exactement le coefficient α in-troduit ci-dessus. Pour chaque <RO2> consommé, le bilan des espèces inorganiques est doncα NO2, −α NO et (2α − 1) HO2, ce qui est strictement équivalent au bilan calculé ci-dessusavec la formulation complète. L’objectif de représentation fidèle des bilans inorganiques est

26 2. SCHÉMAS CHIMIQUES DÉDIÉS AUX ATMOSPHÈRES CONTINENTALES
donc atteint.
L’espèce <RO2> est une espèce modèle introduite uniquement pour traiter les transforma-tions des espèces inorganiques ; elle ne contient pas d’information relative à la structure dela chaîne carbonée du radical peroxy. Il est donc impossible de représenter l’évolution dela matière organique à travers l’évolution des opérateurs. Le bilan organique doit donc êtreintroduit au niveau de la réaction (1). Il est porté par l’espèce COVi et suppose donc uneconversion totale de RO2 en RO. La conservation du carbone entre HCi et COVi est doncreprésentée par :
(1) HCi + OH → <RO2> + COVi ki,OH
L’espèce COVi représente donc l’ensemble des espèces organiques formées par la chaîned’oxydation de HCi, c’est-à-dire non seulement les α COVi mais également les (1− α) RiOOH.Les espèces organiques formées ne résultant pas d’une conversion RO2 → RO (par exempleici RiOOH) n’apparaissent donc pas explicitement dans la réaction (1). Il importe toutefoisde représenter la réactivité induite par les hydroperoxydes (en particulier la photolyse). Uneespèce modèle supplémentaire est ainsi introduite dans le schéma, directement au niveau dela réaction (3) :
(3) <RO2> + HO2 → X-OOH kHO2
Le carbone étant déjà comptabilisé au niveau de la réaction (1), X-OOH est par définitionune espèce sans carbone dont la seule fonction est de représenter la réactivité de la fonctionhydroperoxyde -OOH.
L’approximation selon laquelle l’ensemble des espèces organiques formées résulte de laconversion RO2→RO recoupe plutôt bien la réalité. Dans de nombreux cas (milieux richesou moyennement riches en NO), la réaction avec NO est en effet largement majoritaire etl’essentiel du carbone se retrouve bien dans COVi. En outre, même lorsque la concentrationen NO devient faible, les réactions RO2+RO2 conduisent, selon les RO2 considérés, pour 30à 100% à des radicaux RO, donc à une conversion RO2→RO [Lesclaux, 1997]. Dans les situa-tions pauvres en NOx, la production d’hydroperoxyde par les réactions RO2+HO2 est éga-lement significative. Les hydroperoxydes réagissent par photolyse pour donner des alcoxy.Dans ce cas, la chaîne organique réelle RO2→ROOH→RO est, par construction, directementreprésentée dans le schéma chimique par RO2→RO. Le bilan organique est donc équivalenten terme de produits mais ils sont formés «plus tôt», après une seule étape d’initiation. Leshydroperoxydes réagissent également avec OH pour restituer majoritairement les radicauxRO2 ou directement des carbonyles (i.e. les COVi). Comme précédemment, le bilan orga-nique du schéma simplifié avec opérateur est équivalent au schéma explicite en terme deproduits, avec toutefois un décalage sur le temps requis pour leur production.
L’évolution des radicaux RO2 présentée figure 7 est simplifiée : seuls deux chemins réaction-nels sont considérés. En pratique, il faut écrire pour l’opérateur <RO2> autant de réactionsqu’il y a de chemins possibles (réactions avec NO, HO2, NO3, et les autres radicaux RO2).L’ajout de ces transformations repose sur les principes décrits ci-dessus : (i) la transforma-tion RO2→RO est supposée totale et le bilan organique de l’équation (1) n’est pas modifié,

MODÉLISATION CHIMIQUE DE L’OZONE 27
(ii) seul le bilan inorganique est considéré pour les transformations des opérateurs.
Le cas traité dans ce paragraphe est académique : la réaction initiale HC i+OH ne conduit à laformation que d’un seul radical RiO2. Le plus souvent, les chaînes d’oxydation conduisentà la formation de divers radicaux qui peuvent présenter des séquences réactionnelles dif-férentes. Il faut alors généraliser la méthode en créant autant d’opérateurs qu’il y a de sé-quences réactionnelles inorganiques distinctes puis en écrivant, pour chacun de ces opéra-teurs, l’équivalent des réactions (2) et (3) présentées dans cet exemple.
Bilan des approximations introduites pas la méthode des opérateursLa méthode des opérateurs permet donc une représentation très condensée des différentschemins impliqués dans l’évolution des radicaux RO2. L’application de cette méthode im-plique toutefois plusieurs hypothèses et/ou approximations :– les constantes cinétiques des réactions des opérateurs (réactions (2) et (3) dans l’exemple
précédent) ont une valeur unique quelle que soit la nature du radical R. Ce n’est évidem-ment pas le cas, en particulier pour les réactions RO2+RO2 pour lesquelles les constantescinétiques s’étalent sur 6 ordres de grandeur [Lightfoot et al., 1992]. Ceci conduit à regrou-per les radicaux en classes de réactivité et à créer pour chacune d’entre elles un opérateurparticulier. Le nombre de classes à introduire dépend naturellement d’un compromis entrela précision des résultats et la dimension du schéma chimique. Par exemple, trois opéra-teurs distincts sont utilisés dans le modèle SAPRC90 [Carter, 1990] pour représenter lestransformations inorganiques induites par les RO2 ; le modèle développé par Bey et al.[2001a] pour représenter finement la chimie en situation nocturne en utilise 29.
– les hydroperoxydes formés par les réactions RO2+HO2 sont représentés par (i) les COVissus d’une conversion totale RO2→RO et (ii) l’espèce chimique XOOH, sans masse car-bonée, représentative de la réactivité spécifiquement induite par la fonction -OOH.
– les COV secondaires formés par les réactions RO2+RO2 (cétones ou alcools) sont égale-ment représentés par les COV issus de la conversion totale RO2→RO (cétones ou aldé-hydes).
3. Validation des schémas chimiques
Nous avons vu dans les sections précédentes que le développement des schémas chimiquesimpose d’effectuer de nombreuses approximations et hypothèses, aussi bien pour pallier lemanque de données expérimentales que pour diminuer le nombre d’espèces et de réactionsimpliquées. Comme pour toute démarche de modélisation, il importe d’évaluer la fiabilitédes schémas chimiques, c’est-à-dire tester leur capacité à rendre compte des observations ef-fectuées pour différentes espèces cibles (O3, PAN, aldéhydes, radicaux...) et sous différentesconditions environnementales.
Pour valider spécifiquement les schémas chimiques, il est nécessaire de s’affranchir desprocessus liés aux émissions, au transport et au dépôt. L’expérience idéale permettant devalider les schémas serait ainsi une expérience de laboratoire dans laquelle (i) l’évolutiondes concentrations ne serait pilotée que par les processus chimiques gazeux (ii) les niveaux

28 3. VALIDATION DES SCHÉMAS CHIMIQUES
de concentration dans l’enceinte réactionnelle seraient comparables aux niveaux atmosphé-riques (iii) l’ensemble des paramètres (e.g. température, rayonnement ...) serait parfaitementconnu et maîtrisé (iv) les concentrations de toutes les espèces seraient mesurées à chaqueinstant et sur une période de temps comparable à celle requise pour atteindre l’oxydationcomplète des précurseurs. Cette expérience idéale est inconcevable : (i) un réacteur présentenécessairement des parois à l’origine de transformations hétérogènes indésirables (ii) des li-mitations d’ordre technologique ne permettent pas de mesurer l’ensemble des espèces cibleset/ou de simuler des niveaux représentatifs des conditions atmosphériques (iii) les échellesde temps requises pour l’oxydation complète des composés (de quelques jours à plusieursmois) ne sont guère compatibles avec l’expérimentation de laboratoire.
Il est donc en pratique difficile (voire impossible) d’évaluer rigoureusement l’exactitude desschémas chimiques. Différentes approches, présentant des avantages et des limitations parti-culières, sont menées pour valider les schémas chimiques. Bien que ces démarches ne soientpas pleinement satisfaisantes, la confrontation d’un schéma chimique aux «méthodes de va-lidation» décrites ci-dessous permet néanmoins de définir un certain niveau de confiancedans le dit schéma chimique.
3.1. Comparaison modèles / mesures en laboratoire
Les chambres de simulation atmosphériqueLes chambres de simulation atmosphérique (CSA) sont fréquemment utilisées pour testerla validité des schémas chimiques. Dans ce type d’expérience, un mélange réactionnel (parexemple COV/NOx/air) est réalisé dans une enceinte, puis soumis au rayonnement solaireou artificiel. L’évolution temporelle des concentrations de quelques espèces clés est enregis-trée (COV, NOx, O3 ...). Les concentrations simulées par le modèle dans les mêmes condi-tions sont ensuite comparées aux mesures. Les CSA s’approchent donc de l’expérience idéalepour évaluer la fiabilité des schémas chimiques :– les conditions de l’expérience sont maîtrisées et reproductibles,– le système n’évolue que sous l’influence des transformations chimiques.Le schéma chimique est fréquemment qualifié de «validé» lorsque les concentrations simu-lées par le modèle et observées dans l’enceinte réactionnelle convergent. Il existe actuelle-ment une vingtaine de CSA fournissant régulièrement des données pour tester la cohérencedes schémas chimiques gazeux [Dodge, 2000; Finlayson-Pitts and Pitts, 2000; Eurochamp,2004]. Les données collectées par quelques organismes sont en accès libre (http ://pah.cert.ucr.edu/carter/).
Les limitations des CSACependant, les données collectées dans les CSA sont entachées d’un certain nombre de «dé-fauts». Quelques exemples de limitations associées aux CSA sont brièvement exposés ci-dessous.
Les parois du réacteur entrainent l’existence de processus d’adsorption/désorption aux pa-rois du réacteur. Des espèces comme O3, HNO3, H2O2, HCHO ... se «déposent» aisément surles parois et peuvent par la suite être «réémises» au cours de l’expérience ou dans une expé-rience ultérieure. La désorption de NOx des parois est la principale cause de contamination

MODÉLISATION CHIMIQUE DE L’OZONE 29
Irradiation time (hr)0 2 4 6
0
0.1
0.2
O3
(pp
m)
0
0.1
0.2
0 2 4 6Irradiation time (hr)
O3
(pp
m)
Figure 8: Evolution des concentrations de O3 pour deux expériences en chambre de simulation atmosphériquerenfermant initialement un mélange CO+NOx. Courbe en pointillée : concentrations modélisées lorsque aucuneréaction aux parois du réacteur n’est considérée dans le modèle. Courbe continue : concentrations modéliséesaprès paramétrisation des réactions aux parois. [D’après Finlayson-Pitts and Pitts, 2000].
observée dans les CSA [Finlayson-Pitts and Pitts, 2000]. De même, les parois sont à l’originede transformations chimiques hétérogènes «parasites». En particulier, les concentrations deradicaux OH observées dans la phase gazeuse ne peuvent, le plus souvent, pas être expli-quées à partir des seules transformations gazeuses. Une source additionnelle de radicauxdoit être considérée ; elle est attribuée à l’existence de transformations hétérogènes aux pa-rois. La dismutation hétérogène de NO2 est l’une des sources suspectées, via la formationpuis la photolyse de l’acide nitreux [e.g. Finlayson-Pitts and Pitts, 2000] :
2 NO2 (+surface) → HNO3 + HONOHONO + hν → OH + NO
L’influence des «sources hétérogènes» de radicaux sur les expériences est illustrée sur la fi-gure 8. Leur contribution peut atteindre 10 à 50 % de la source totale de radicaux [Killus andWhitten, 1990]. L’expérience montre que l’intensité de ces effets de parois varie fortementd’une chambre à l’autre et, pour une chambre donnée, d’une expérience à l’autre. Il est doncparticulièrement délicat d’évaluer quantitativement la contribution de ces processus hété-rogènes au cours d’une expérience particulière. Cette contribution est typiquement évaluéeavec 50 % d’incertitude [Dodge, 2000]. L’influence des effets de parois ne peut ainsi être para-métrée de façon pleinement appropriée. En terme de validation, l’exploitation des donnéesde CSA pose donc un problème méthodologique, les divergences modèles/observations nepouvant être directement imputés au schéma chimique testé.
Pour évaluer l’exactitude d’un schéma chimique, les conditions expérimentales doivent êtreparfaitement définies. Toutefois, l’intensité et la distribution spectrale du rayonnement dansl’enceinte réactionnelle s’avèrent particulièrement difficile à caractériser, aussi bien pour lesréacteurs à irradiation naturelle qu’artificielle. Il en résulte donc une incertitude sur l’éva-luation des fréquences de photolyse pour l’expérience simulée. Par exemple, Dodge [2000]estime que les fréquences de photolyse de certaines espèces comme NO2 ou O3 sont en-tachées d’une incertitude de 20-30%, incertitude pouvant être supérieure pour d’autres es-pèces. S’ajoutent également les incertitudes liées à l’instrumentation utilisée pour réaliser la

30 3. VALIDATION DES SCHÉMAS CHIMIQUES
mesure des concentrations dans la chambre. Si celle-ci reste faible pour des espèces commel’ozone (±10 %), elle devient significative pour la mesure d’espèces comme le PAN (±20 %)ou les composés oxygénés (±30 %) [Dodge, 2000].
Un grand nombre d’espèces n’est pas mesuré en routine au cours des expériences en CSA.L’exactitude du schéma chimique ne peut donc être évaluée que pour quelques espèces,principalement les espèces «terminales» comme l’ozone. L’absence de mesures pour «vali-der» les concentrations simulées d’espèces clés comme les radicaux, les nitrates, les hydro-peroxydes... restreint évidemment la portée des tests réalisés.
La représentativité et l’extrapolation des mesures en CSA aux conditions qui prévalent pourles atmosphères réelles sont également sujettes à caution. Afin de pallier les problèmes asso-ciés aux limites de détection des instruments et de simplifier les stratégies expérimentales,les expériences sont souvent conduites sous des niveaux de concentration notablement plusélevés que dans l’atmosphère réelle. Les expériences sont également fréquemment conduitesen utilisant des rapports COV/NOx élevés afin de minimiser les effets de parois [e.g. Dodge,2000]. De plus, les expériences ne sont habituellement conduites que sur quelques heures,jusqu’à consommation des NOx (au-delà de cette période, la chimie est pilotée par la dé-sorption d’espèces azotées des parois). La fidélité des schémas chimiques n’est donc évaluéequ’en regard de leur capacité à reproduire la formation des photo-oxydants sur une échelletemporelle de l’ordre de la journée. Les processus impliqués sur des échelles de temps pluslongues soulèvent de nombreuses interrogations, notamment en regard du rôle des COVsecondaires dans la production de photooxydants et d’aérosols. La capacité des schémas ac-tuels à traiter ce type de question reste difficilement appréciable.
Enfin, nous avons vu à la section 1.5 que les CSA sont également utilisées pour paramétrerles schémas chimiques lorsque les mécanismes réactionnels sont méconnus. Il en résulte unecertaine ambiguïté concernant le statut de ces expériences, à la fois support de développe-ment et de validation. Afin de lever cette circularité dans la démarche méthodologique, ilimporte de s’affranchir autant que possible des expériences en CSA lors du développementdes schémas chimiques. Les progrès à venir en matière de développement de schémas chi-miques se situent vraisemblablement dans notre capacité à (i) développer des schémas chi-miques sur la seule base de données cinétiques ou mécanistiques expérimentales ou d’éva-luation théorique et (ii) à évaluer l’exactitude de ces schémas sur la base d’expérimentationsen atmosphère contrôlée.
Validation des schémas chimiques en CSABien qu’imparfaites, les CSA demeurent l’instrument le plus approprié pour évaluer l’exac-titude des schémas chimiques. Les imperfections évoquées ci-dessus sont de plus atténuéesdans les nouvelles générations de chambres (e.g. la chambre EUPHORE, http ://www.ceam.es),celle-ci présentant un grand rapport volume/surface et une forte instrumentation en routine.
Afin d’éviter la possibilité de biais systématiques induits par les limitations exposées ci-dessus, l’exercice de validation doit être réalisé sur la base d’une multitude d’expériences,réalisées dans différentes CSA présentant différents matériaux pour le revêtement du réac-

MODÉLISATION CHIMIQUE DE L’OZONE 31
0
100
200
300
400
500
600
> -50 %- 40 %
- 30 %- 20 % 10 % 50 %40 %30 %20 %
- 10 % 0 %> 60 %
No
mb
re d
'exp
érie
nce
s
t = 1 heure
t = 6 heures
Figure 9: Ecarts relatifs des valeurs simulées avec le schéma SAPRC99 aux observations en CSA après unepériode de 1 heure et de 6 heures. L’étude porte sur ≈ 1700 expériences et chaque point indique le nombrede simulations en accord avec l’expérience pour un pourcentage d’erreur donné. La grandeur ∆(O3 − NO)représente la quantité «O3 produit + NO oxydé». [D’après Carter, 2000].
teur et différents protocoles d’expérience. Le mécanisme SAPRC est probablement le schémaqui a fait l’objet de la validation la plus approfondie. La version SAPRC99 a par exemple ététestée sur plus de 1700 expériences, réalisées sur 7 chambres distinctes [Carter, 2000]. Laperformance globale de SAPRC99 à reproduire les observations est donnée figure 9. Cettefigure présente l’écart relatif du modèle à l’expérience à différents temps et pour une gran-deur ∆(O3 − NO) caractérisant la formation des photooxydants [Carter and Lurmann, 1991]selon :
∆(O3 − NO) = O3(produit) + NO(oxyde) =(
CtO3 − C0
O3
)+
(C0
NO − CtNO
)
où Ct et C0 représente respectivement les concentrations au temps t et au début de l’expé-rience 3. Après une période expérimentale de 6 heures, l’accord de SAPRC99 aux expériencesest de ±5% pour ≈ 1/3 des expériences et de ±15% pour ≈ 3/4 des expériences. 90 % desexpériences sont simulées dans une fourchette de ±25%. En moyenne, le schéma conduit àune très légère surestimation de ∆(O3 − NO) (4 %) après 6 heures de simulation.
3.2. Comparaison modèles / mesures in situ
La pertinence d’un modèle est naturellement évaluée par sa capacité à rendre compte duréel. La divergence des simulations et des observations témoigne en effet nécessairementd’une imperfection du modèle. Toutefois, les confrontations "expérience/théorie" présententde multiples difficultés méthodologiques et les comparaisons observation/simulation s’avèrent
3Lorsque les concentrations initiales de NO sont élevées, la production d’ozone dans la CSA demeure faible duranttoute la période de l’expérience. La concentration de O3 ne reflète alors guère l’activité photooxydante. Pour cessituations, la variable «O3 produit + NO oxydé» est plus pertinente

32 3. VALIDATION DES SCHÉMAS CHIMIQUES
souvent délicates à mener.
Les champs de concentration simulés sont le résultat de l’intégration simultanée de diversprocessus : dynamique, chimie, émissions, transfert radiatif ... Les processus sont représen-tés par des modules distincts, assemblés au sein du modèle complet de chimie/transport.Chaque module possède ses propres carences. Le modèle cumule ainsi les imperfections desdifférents modules. Si les comparaisons mesures/modèles permettent d’apprécier la cohé-rence globale du modèle, l’attribution des responsabilités de chaque module dans les écartsmesures/modèles finalement obtenus demeure souvent difficile à quantifier.
La variabilité spatio/temporelle des espèces est étroitement liée à leur temps de vie ou ré-sidence dans la troposphère. De façon très schématique, il est possible de classer les com-paraisons mesures/modèles selon trois grands cas de figure, fonction du temps de vie desespèces considérées.
les espèces à «longue» durée de vieCes espèces présentent une variabilité spatiale faible. La confrontation des résultats de simu-lations aux observations ne pose pas de problème méthodologique particulier et peut êtreréalisée de façon directe. C’est par exemple le cas du méthane, du CO ou même de l’ozone àl’échelle globale dans la troposphère libre.
les espèces à très «courte» durée de vieCes espèces sont localement en état d’équilibre dynamique. Pour ces espèces, la contribu-tion des processus de transport est négligeable et les niveaux de concentrations dépendentquasi-exclusivement des processus physico-chimiques. Pour ce type d’espèces, il est possiblede concevoir des confrontations modèle/observation à partir d’un ensemble de mesures réa-lisées localement. Par exemple, les HOx (OH + HO2 + RO2) possèdent un temps de vie del’ordre de quelques secondes. Les HOx sont donc dans un état d’équilibre dynamique, lesconcentrations étant régies par l’efficacité des sources (réaction d’initiation) sur celles despuits (réactions de terminaison). Une expérience de «fermeture chimique» peut ainsi êtreréalisée par la mesure des différents paramètres associés aux sources et puits de HOx : dis-tribution spectrale du rayonnement, concentrations des espèces photolysables et/ou par-ticipant à l’interconversion de HOx et/ou à leurs consommations (O3, NOx, COVs, H2O2...). Il est alors possible d’initialiser le modèle à partir de ces observations afin de simulerles concentrations d’équilibre des HOx. La représentativité du schéma chimique peut alorsêtre évaluée en comparant les concentrations de HOx ainsi simulées avec les concentrationsobservées [e.g. Volz et al., 2003; Konrad et al., 2003].
les espèces à durée de vie «intermédaire»Les comparaisons mesures/modèles posent des difficultés méthodologiques lorsque le tempsde vie de l’espèce cible considéré est «trop long» pour que le transport soit ignoré mais «tropcourt» pour que l’espèce ne présente pas une forte variabilité spatiale et/ou temporelle. Larésolution spatiale du modèle est en effet fixée par la dimension des mailles utilisées pour

MODÉLISATION CHIMIQUE DE L’OZONE 33
discrétiser l’espace. La quantité de matière de chaque espèce est donc moyennée sur le vo-lume d’une maille. A l’inverse, les mesures de terrain sont de nature ponctuelle. Les compa-raisons mesure/modèle ne sont ainsi pertinentes que si le milieu autour du point de mesureest suffisamment homogène pour autoriser ce transfert d’échelle. Par exemple, dans les mo-dèles tridimensionnels dédiés à l’étude de la pollution photooxydante aux échelles localeet régionale, la résolution horizontale du maillage est typiquement de l’ordre de quelqueskm2 avec, au voisinage du sol, une résolution verticale de plusieurs dizaines de mètres. Enzone urbaine, la forte hétérogénéité spatiale des sources d’émission induits des gradientsd’échelle nettement inférieure. Dans ces milieux (et avec ce type de modèle), la comparaisonmesure/modèle n’a de sens que si le point d’échantillonnage est représentatif du «compor-tement moyen» de la zone simulée. Les critères permettant d’évaluer de façon quantitativeet objective la représentativité de différents points de mesure dans un milieu présentantune forte hétérogénéité spatiale sont en pratique délicats à établir. L’évaluation des modèlessur la base d’observations in situ requiert une stratégie expérimentale spécifiquement dé-diée à ce type d’exercice, car elle nécessite de renseigner simultanément une multitude deparamètres sur un domaine vaste. Par exemple, le programme expérimental ESCOMPTE[Cros et al., 2004, 2005] a été réalisé dans le but de contraindre le maximum de paramètresafin de conduire une validation aussi rigoureuse que possible des modèles. La campagnes’est focalisée sur un domaine de 120 × 120 km, centré sur l’agglomération Marseillaise etl’étang de Berre. Elle a mobilisée plus d’une centaine de chercheurs et des moyens lourds :6 avions, 2 bateaux, 80 stations sol, des lâchers de ballons plafonnants, 20 intruments detélédétection, 3 radiosondages mobiles... Il est également possible d’apprécier la représen-tativité d’un modèle en utilisant de longue série temporelle sur un ensemble de sites demesure. Des tests statistiques sont ensuite mis en œuvre afin de comparer les concentra-tions de polluants prévues aux mesures disponibles, et évaluer ainsi les performances desmodèles. Cette approche est mise en œuvre pour tester le modèle CHIMERE (http ://eu-ler.lmd.polytechnique.fr/chimere/) sur la base d’un ensemble de stations de réseaux euro-péens [Schmidt et al., 2001] (http ://www.prevair.org/).
3.3. Inter comparaison des schémas chimiques et tests de sensibilité
Inter comparaison des schémas chimiquesUne approche permettant d’apprécier la cohérence des schémas chimiques consiste à lesconfronter les uns aux autres [Hough, 1988; Dodge, 1989; Olson et al., 1997; Kuhn et al.,1998; Jimenez et al., 2003]. Une même situation est modélisée par différents schémas chi-miques et les résultats sont comparés. Cette approche ne permet évidemment pas d’évaluerde façon absolue l’exactitude d’un schéma donné, aucun schéma ne pouvant être utilisécomme schéma de référence. Elle permet toutefois d’évaluer leur crédibilité relative en vued’applications.
Kuhn et al. [1998] ont comparé les résultats obtenus par 12 équipes scientifiques lors de lasimulation d’un jeu de scénarios imposés. Pour cette étude, différents scénarios représenta-tifs de situations environnementales allant du panache urbain à la troposphère libre ont étédéfinis puis soumis aux différentes équipes pour simulation. Chaque équipe réalise les si-mulations avec son propre schéma chimique (regroupement d’espèces, calcul des fréquences

34 3. VALIDATION DES SCHÉMAS CHIMIQUES
60
80
100
120
140
160
180
0 50 100
time (hours)
mix
ing
rati
o (p
pb
) O3
0 50 100
NO2
0
0.5
1.0
1.5
2.0
2.5
mix
ing
rati
o (p
pb
)
0 50 100 0 50 100
OH H2O2
0
0.1
0.2
0.3
0.4
0.5
mix
ing
rati
o (p
pt)
5
10
15
20
0
mix
ing
rati
o (p
pb
)
HCHO PAN
0 50 1000 50 100time (hours)
2
1
3
4
mix
ing
rati
o (p
pb
)
mix
ing
rati
o (p
pb
)
0
2
4
6
8
Figure 10: Profils de concentrations simulées par 12 schémas modèles pour un scénario de type panache urbain.[D’après Kuhn et al., 1998].
de photolyse...) et sous leur condition usuelle d’utilisation (solveur ...). Les résultats obtenuspour le scénario "panache urbain" sont reportés pour quelques espèces figure 10. L’étude deKuhn et al. [1998] montre que les concentrations d’ozone simulées par les différents sché-mas chimiques tendent à s’accorder à ±10%. Les divergences sont toutefois nettement plusélevées (typiquement plusieurs dizaines de pourcents) pour d’autres composés clés de lachimie troposphérique comme H2O2, HCHO, OH, PAN ... Des résultats comparables ont étéobtenus par Dodge [1989] et Jimenez et al. [2003].
Différents arguments expliquent la convergence observée sur les profils d’ozone simulés. Ledéveloppement des différents schémas s’appuie d’une part sur les mêmes bases de donnéeset il existe, d’autre part, un certain consensus entre les différentes équipes sur la façon dereprésenter les mécanismes chimiques méconnus ou incertains. Par exemple, de nombreuxschémas chimiques représentent de façon similaire l’oxydation des composés aromatiques,

MODÉLISATION CHIMIQUE DE L’OZONE 35
malgré les incertitudes actuelles sur ces transformations. De plus, les schémas chimiquesont été testés sur les mêmes données de CSA et, le plus souvent, ont été modifiés de façonà s’accorder sur celles-ci. Pour l’ozone, les différents schémas sont donc en quelque sortecontraints à converger vers les mêmes solutions.
La technique d’intégration (le solveur) utilisée pour résoudre l’évolution temporelle desconcentrations est également une source possible de divergence entre les différents modèles.L’étude de [Poppe et al., 2001] a toutefois montré que lorsque le scénario et le schéma chi-mique sont imposés lors d’un exercice d’inter comparaison, un excellent accord est observépour toutes les espèces du schéma chimique.
Comparaison schéma explicite – schéma réduitUne méthode couramment utilisée pour évaluer les erreurs induites par la réduction desschémas chimiques consiste à développer un schéma explicite puis à lui appliquer diffé-rentes procédures de réduction afin d’aboutir au schéma réduit que l’on souhaite tester.L’analyse des écarts entre les profils simulés permet de quantifier les erreurs générées parla procédure de réduction [Jenkin et al., 2002; Kirchner, 2005; Whitehouse et al., 2004]. Cetteapproche a été abondamment utilisée au LISA pour le développement et l’évaluation deschémas réduits [Aumont et al., 1996; Jaubertie, 1999; Szopa et al., 2005].
Tests de sensibilitésLes schémas explicites seraient une représentation fidèle des transformations atmosphé-riques si l’ensemble des réactions était connu avec une grande précision. Les différents pa-ramètres cinétiques (constantes de vitesse, coefficients stœchiométriques, énergies d’activa-tion, coefficients d’absorption, rendements quantiques ...) sont déterminés par l’expérienceavec une certaine gamme d’incertitude. Une approche permettant d’apprécier l’exactituded’un schéma donné consiste à évaluer comment se répercute l’incertitude associée à chaqueparamètre du schéma chimique sur la fiabilité des concentrations simulées.
Le coefficient de sensibilité sij des concentrations Ci simulées pour l’espèce i aux différentsparamètres pj du modèle est défini comme : sij = dCi/dpj. L’incertitude sur les profils si-mulés peut être évaluée en combinant le coefficient de sensibilité sij et l’incertitude sur leparamètre pj. Sur la base d’un développement de premier ordre en série de Taylor et en sup-posant que les paramètres sont indépendants, la variance sur les concentrations simulées σ2
Cipeut être estimée à partir de la variance σ2
pjassociée à chaque paramètre pj selon [Gao et al.,
1995] :σ2
Ci = σ2pj× s2
ij (2)
Compte tenu de la multitude des paramètres du modèle, une compensation significative deserreurs est toutefois fortement probable. L’incertitude globale de premier ordre calculée parla relation 2 donne donc une indication majorée sur la précision des concentrations simulées(voir l’approche de Monte Carlo ci-dessous). Ces tests de sensibilité sont néanmoins particu-lièrement efficaces pour identifier et hiérarchiser les paramètres du modèle dont l’incertitudese répercute significativement sur la précision des simulations. Dans ce but, la contributiondu paramètre pj à l’incertitude globale est estimée selon [Gao et al., 1995; Yang et al., 1995] :

36 3. VALIDATION DES SCHÉMAS CHIMIQUES
0 2 4 6 8 10 12Time (Hour)
0
0.05
0.10
0.15
0.20
0.25
Co
nce
ntr
atio
n (p
pm
)
Urban, ROG/NOx=6:1
0 2 4 6 8 10 12Time (Hour)
0
0.02
0.04
0.06
0.08
0.10
Co
nce
ntr
atio
n (p
pm
)
Rural, ROG/NOx=24:10.12
Figure 11: Concentrations moyennes d’ozone pour un scénario urbain (de rapport COV/NOx initial de 6 :1) etun scénario rural (de rapport COV/NOx initial de 24 :1) simulées avec une approche de Monte Carlo. Cercle :concentration moyenne de l’ensemble de résultats, barre : incertitude (1σ), courbe pointillée : profil obtenu avectous les paramètres fixés à leur valeur nominale. [D’après Gao et al., 1995].
Vij = σ2pj× s2
ij. Les tests de sensibilité réalisés par Gao et al. [1995] sur un cycle diurne in-diquent que la variance globale des concentrations d’ozone simulées est principalement at-tribuable aux paramètres (i) des réactions d’initiation/terminaison et/ou (ii) des réactionscontribuant aux bilans de NOx. Les incertitudes associées aux constantes cinétiques des ré-actions de photolyse de HCHO, O3, NO2, de la réaction OH+NO2 → HNO3, à la chimie duPAN et des aromatiques sont notamment observées comme des paramètres singulièrementdécisifs [Gao et al., 1995; Jiang et al., 1997] 4.
L’incertitude globale des concentrations simulées induites par les incertitudes associées auxdifférents paramètres du schéma chimique est généralement quantifiée sur la base d’uneanalyse de Monte Carlo [Gao et al., 1996; Yang et al., 1995, 1996]. L’incertitude sur chaqueparamètre du schéma chimique est caractérisée par une loi de distribution. La valeur desdifférents paramètres est attribuée par tirage aléatoire ; une simulation est réalisée avec cettedistribution singulière. Les simulations sont répétées de nombreuses fois en modifiant, àchaque fois, les valeurs attribuées aux jeux des paramètres. Les lois de distribution desconcentrations modélisées sont estimées à partir de ces différentes simulations. L’exactitudedes estimations augmente de façon asymptotique avec le nombre de simulation réalisée.Pour des schémas chimiques complexes, un grand nombre de simulation est nécessaire pours’assurer qu’un échantillonnage représentatif des différents paramètres a été réalisé. Des mé-thodes d’échantillonnage permettent de diminuer le nombre de simulations nécessaires à laconvergence de la moyenne et de l’écart type des concentrations [e.g. Gao et al., 1996; Berginet al., 1999]. La figure 11 présente quelques exemples de profils d’ozone obtenus par uneanalyse de Monte Carlo. Pour une période de 12 heures, l’écart type calculé sur les concen-trations maximales d’ozone est de l’ordre de 20 à 50 % [Gao et al., 1996; Yang et al., 1995,
4Outre l’incertitude induite par les paramètres chimiques, il importe également d’évaluer la sensibilité des concen-trations simulées aux incertitudes sur les paramètres physiques (hauteur de la couche de mélange, température,vitesse et direction du vent, vitesse de dépôt ...) et aux données d’entrées du modèle (intensité des émissions...).La modélisation adjointe est aujourd’hui utilisée pour la réalisation de ces tests de sensibilité dans les modèlestridimensionnels de chimie/transport [e.g. Menut, 2003]

MODÉLISATION CHIMIQUE DE L’OZONE 37
1996]. L’incertitude calculée sur les profils de H2O2 est significativement plus élevée (30 à200 %), de même pour le PAN (40 à 70 %). A l’inverse, l’incertitude associée aux concentra-tions de HNO3 et HCHO montrent une variabilité plus faible (15 à 30 %) [Gao et al., 1996].
Les tests de sensibilité fournissent une indication précieuse pour définir les réactions à étu-dier prioritairement au laboratoire afin d’augmenter la fiabilité des schémas chimiques. Tou-tefois, ces tests ne fournissent qu’une information partielle sur l’incertitude des schémas chi-miques. Les erreurs induites par le regroupement des espèces et des réactions ne sont pasincluses dans le calcul des incertitudes. De même, l’analyse ne porte que sur les réactionsou les produits de réactions identifiées. L’erreur causée par l’absence d’une réaction ou d’unproduit de réaction ne peut évidemment pas être quantifiée.

Bibliographie
Atkinson, R., Lloyd, A.C., and Winges, L. : An updated chemical mechanism for hydrocarbons/NOx/SO2 pho-tooxidations suitable for inclusion in atmospheric simulations models, Atmospheric Environment, 16, 1341-1355,1982.
Atkinson, R. : Gas-Phase Tropospheric Chemistry of Organic Compounds, Journal of Physical and Chemical ReferenceData, Monograph 2, 1994.
Atkinson, R., Baulch, D. L., Cox, R. A., Hampson, R. F., Kerr, J. A., and Rossi, M. J. : Evaluated kinetics and photoche-mical data for atmospheric chemistry, Organic species, Supplement VII, Journal of Physical and Chemical ReferenceData, 28, 191–392, 1999.
Atkinson, R. : Atmospheric chemistry of VOCs and NOx, Atmospheric Environment, 34, 2063-2101, 2000.
Atkinson, R. : Atmospheric oxidation, in Handbook of property estimation methods for chemicals, edited by R.S.Boethling, and D. Mackay, pp. 335-354, Lewis Publisher, London, 2000b.
Atkinson, R. and Arey, J. : Atmospheric degradation of volatile organic compounds, Chemical Review, 103, 4605–4638, 2003.
Atkinson, R., Baulch, D.L., Cox, R.A., Crowley, J.N., Hampson, R.F., Hynes, R.G., Jenkin, M.E., Rossi, M.J., and Troe,J. : Evaluated kinetic and photochemical data for atmospheric chemistry : Volume I - gas phase reactions of Ox,HOx, NOx and SOx species, Atmospheric Chemistry and Physics, 4, 1461-1738, 2004.
Atkinson, R., Baulch, D.L., Cox, R.A., Crowley, J.N., Hampson, R.F., Hynes, R.G., Jenkin, M.E., Rossi, M.J., and Troe,J. : Evaluated kinetic and photochemical data for atmospheric chemistry : Volume II reactions of organic species,Atmospheric Chemistry and Physics Discussions, 5, 6295-7168, 2005.
Aumont, B., Jaecker-Voirol, A., Martin, B., and Toupance, G. : Tests of some reduction hypotheses made in photo-chemical mechanisms, Atmospheric Environment, 30, 2061-2077, 1996.
Aumont, B., Chervier, F., and Laval, S. : Contribution of HONO sources to the NOx/HOx/O3 chemistry in thepolluted boundary layer, Atmospheric Environment, 37, 487-498, 2003.
Aumont, B., Szopa, S., and Madronich, S. : Modelling the evolution of organic carbon during its gas-phase tropos-pheric oxidation : development of an explicit model based on a self generating approach, Atmospheric Chemistryand Physics, 5, 2497-2517, 2005.
Bergin, M.S., Noblet, G.S., Petrini, K., Dhieux, J.R., Milford, J.B., and Harley, A.R. : Formal uncertainty analysis of alagrangian photochemical air pollution model, Environmental Science & Technology, 33, 1116-1126, 1999.
Bey, I., Aumont, B., and Toupance, G. : A modeling study of the nighttime radical chemistry in the lower conti-nental troposphere 1. Development of a detailed chemical mechanism including nighttime chemistry, Journal ofGeophysical Research, 106, 9959-9990, 2001a.
Bey, I., Jacob, D., Yantosca, R.M., Logan, J.A., Field, B.D., Fiore, A.M., Li, Q., Liu, H., and Mickeley, L.J. : Globalmodeling of tropospheric chemistry with assimilated meteorology : model description and evaluation, Journal ofGeophysical Research, 106, 23073-23096, 2001c.
Bloss, C., Wagner, V., Jenkin, M.E., Volkamer, R., Bloss, W.J., Lee, J.D., Heard, D.E., Wirtz, K., Martin-Reviejo, M.,Rea, G., Wenger, J.C., and Pilling, M.J. : Development of a detailed chemical mechanism (MCMv3.1) for theatmospheric oxidation of aromatic hydrocarbons, Atmospheric Chemistry and Physics, 5, 641-664, 2005.
Boyd, A.A., Flaud, P.M., Daugey, N., and Lesclaux, R. : Rate constant for RO2+HO2 reactions measured under alarge excess of HO2, Journal of Physical Chemistry A, 107, 818-821, 2003.
Brasseur, G.P., Hauglustaine, D.A., Walters, S., Rasch, P.J., Muller, J.-F., Granier, C., and Tie, X.X. : MOZART : a globalchemical transport model for ozone and related chemical tracers, Part 1. Model description, Journal of GeophysicalResearch, 103, 28265-28289, 1998.
Brasseur, G., Orlando, J.J., and Tyndall, G.S. : Atmospheric chemistry and global change, Oxford University Press, Ox-ford, 1999.
38

MODÉLISATION CHIMIQUE DE L’OZONE 39
Calvert, J.G., Atkinson, R., Kerr, J.A., Madronich, S., Moortgat, G., Wallington, T.J., and Yarwood, G. : The mecha-nisms of atmospheric oxidation of the alkenes, Oxford University Press, New York, 2000.
Calvert, J.G., Atkinson, R., Becker, K.H., Kamens, R.M., Seinfeld, J.H., Wallington, T.J., and Yarwood, G. : The mecha-nisms of atmospheric oxidation of aromatic hydrocarbons, Oxford University Press, Oxford, 2002.
Carter, W.P.L. : Documentation for the SAPRC atmospheric photochemical mechanism. Preparation and emissionsprocessing programs for the implementation in airshed models. Report for the California Air Resources Board,Contract A5-122-32, Statewide Air Pollution Research Center, University of California, Riverside, CA, 1988.(http ://pah.cert.ucr.edu/ carter/bycarter.htm)
Carter, W.P.L. : A detailed mechanism for the gas-phase atmospheric reactions of organic compounds, AtmosphericEnvironment, 24A, 481-518, 1990.
Carter, W.P.L., and Lurmann, F.W. : Evaluation of a detailed gas-phase atmospheric reaction mechanism usingenvironmental chamber data, Atmospheric Environment, 25A, 2771-2806, 1991.
Carter, W.P.L. : Development of ozone reactivity scales for volatile organic compounds, Journal of Air & Waste Ma-nagement Association, 44, 881-899, 1994.
Carter, W.P.L. : Documentation of the SAPRC-99 chemical mechanism for the VOC reactivity assessment. Final Re-port to the California Air Resources Board under Contracts 92-329 and 95-308, Center of Environmental Researchand Technology, Riverside, 2000.
Cros, B., Durand, P., Cachier, H., Drobinski, P., Fréjafon, E., Kottmeier, C., Perros, P.E., Ponche, J.-L., Robin, D., Saïd,F., Toupance, G., and Wortham, H. : The ESCOMPTE program : an overview, Atmospheric Research, 69, 241-279,2004.
Cros, B., and Durand, P : Special Issue : Escompte, Atmospheric Research, 74, 1-595, 2005.
De More, W. B., Molina, M. J., Sander, S. P., Golden, D. M., Hampson, R. F., Kurylo, M. J., Howard, C. J., Kolb, C. E.,and Ravishankara, A. R. : Chemical Kinetics and photochemical data for use in stratospheric modeling, JPL Pub.97-4, Jet Propulsion Laboratory, California Institute of Technology, Pasadena, California, 1997.
Derwent, R.G., and Jenkin, M.E. : Hydrocarbons and the long range transport of ozone and PAN across Europe,Atmospheric Environment, 25A, 1661-1678, 1991.
Derwent, R.G., Jenkin, M.E., and Saunders, S.M. : Photochemical ozone creation potentials for a large number ofreactive hydrocarbons under European conditions, Atmospheric Environment, 30, 181-199, 1996.
Dodge, M.C. : A comparison of three photochemical oxidant mechanisms, Journal of Geophysical Research, 94, 5121-5136, 1989.
Dodge, M.C. : Chemical oxidant mechanisms for air quality modeling : critical review, Atmospheric Environment, 34,2103-2130, 2000.
Eurochamp : Integration of European Simulation Chambers for Investigating Atmospheric Processes,http ://www.eurochamp.org/, 2004.
Finlayson-Pitts, B.J., and Pitts, J.N., Chemistry of the upper and lower atmosphere, Academic Press, San Diego,2000.
Gao, D., Stockwell, W.R., and Milford, J.B. : First-order sensitivity and uncertaintiy analysis for regional-scale gas-phase chemical mechanism, Journal of Geophysical Research, 100, 23,153-23,166, 1995.
Gao, D., Stockwell, W.R., and Milford, J.B. : Global uncertainty analysis of a regional-scale gas-phase chemicalmechanism, Journal of Geophysical Research, 101, 9107-9119, 1996.
Gery, M., Whitten, G.Z.and Killus, J., and Dodge, M. : A photochemical kinetics mechanism for urban and regionalcomputer modeling., Journal of Geophysical Research, 94, 12 925–12 956, 1989.
Gery, M.W., Whitten, G.Z., and Killus, J.P. : Development and testing of the CBM-IV for urban and regional mode-ling, EPA-report, contract 68-02-41136, 1988.
Hough, A.M. : An intercomparison of mechanisms for the production of photochemical oxidants, Journal of Geophy-sical Research, 93, 3789-3812, 1988.
Jang, M., Czoschke, N.M., Lee, S., and Kamens, R.M. : Heterogeneous Atmospheric Organic Aerosol Production byInorganic Acid-Catalyzed Particle-Phase Reactions, Science, 298, 814-817, 2002.
Jaubertie, A. : Optimisation en taille et en raideur des schémas cinétiques utilisés en chimie atmosphérique, Thèsede l’Université Paris 12, 1999.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J. : The tropospheric degradation of volatile organic compounds : aprotocol for mechanism development, Atmospheric Environment, 31, 81-104, 1997.
Jenkin, M.E., Saunders, S.M., Derwent, R.G., and Pilling, M.J. : Development of a reduced speciated VOC degrada-tion mechanism for use in ozone models, Atmospheric Environment, 36, 4725-4734, 2002.
Jenkin, M.E., Saunders, S.M., Wagner, V., and Pilling, M.J. : Protocol for the development of the Master ChemicalMechanism, MCM v3 (Part B) : tropospheric degradation of aromatic volatile organic compounds, Atmospheric

40 BIBLIOGRAPHIE
Chemistry and Physics, 3, 181-193, 2003.
Jenkin, M.E. : Modelling the formation and composition of secondary organic aerosol from alpha- and beta-pineneozonolysis using MCM v3, Atmospheric Chemistry and Physics, 4, 1741-1757, 2004.
Jiang, W., Singleton, D.L., McLaren, R., and Hedley, M. : Sensitivity of ozone concentration to rate constants ina modified SAPRC90 chemical mechanism used for Canadian lower fraser valley ozone studies, AtmosphericEnvironment, 31, 1195-1208, 1997.
Jimenez, P., Baldasano, J.M., and Dabdub, D. : Comparison of photochemical mechanisms for air quality modeling,Atmospheric Environment, 37, 4179-4194, 2003.
Junier, M., Kirchner, F., Clappier, A., and Van den Bergh, H. : The chemical mechanism generation programme CHE-MATA - Part 2 : Comparison of four chemical mechanisms for mesoscale calculation of atmospheric pollution,Atmospheric Environment, 39, 1161-1171, 2005.
Killus, J.P., and Whitten, G.Z. : Background reactivity in smog chambers, International Journal of Chemical Kinetics,22, 547-575, 1990.
Kirchner, F. : The chemical mechanism generation programme CHEMATA-Part 1 : The programme and first appli-cations, Atmospheric Environment, 39, 1143-1159, 2005.
Konrad, S., Schmitz, T., Buers, H.-J., Houben, N., Mannschreck, K., Mihelcic, D., Musgen, P., Patz, H.-W., Holland, F.,Hofzumahaus, A., Schafer, H.-J., Schroder, S., Volz-Thomas, A., Moortgat, G., and Grossmann, D. : Hydrocarbonmeasurements at Pabstthum during the BERLIOZ campaign and modeling of free radicals, Journal of GeophysicalResearch, 108, 8251, doi :10.1029/2001JD000866, 2003.
Kuhn, M., Builtjes, P.J.H., Poppe, D., Simpson, D., Stockwell, W.R., Anderson-sköld, Y., Baart, A., Das, M., Fiedler,F., Hov, O., Kirchner, F., Makar, P.A., Milford, J.B., Roemer, M.G.M., Ruhnke, R., Strand, A., Vogel, B., and Vo-gel, H. : Intercomparaison of the gas-phase chemistry in several chemistry and transport models, AtmosphericEnvironment, 32, 693-702, 1998.
Kwok, E.S.C., and Atkinson, R. : Estimation of the hydroxyl radical reaction rate constants for gas-phase organiccompounds using a structure-reactivity relationship : an update, Atmospheric Environment, 29, 1685-1695, 1995.
Lattuati, M. : Impact des émissions européennes sur le bilan de l’ozone troposphérique à l’interface de l’Europeet de l’Atlantique nord : apport de la modélisation lagrangienne et des mesures en altitude, Université Paris 6,Paris, 1997.
Lesclaux, R. : Combination of peroxyl radicals in the gas phase, in Peroxyl radicals, edited by Z.B. Alfassi, pp. 81-112,John Wiley, New York, 1997.
Lightfoot, P.D., Cox, R.A., Crowley, J.N., Destriau, M., Jenkin, M.E., Moortgat, G., and Zabel, F. : Organic peroxyradicals : kinetics, spectroscopy, and tropospheric chemistry, Atmospheric Environment, 26, 1805-1961, 1992.
Madronich, S., and Calvert, J.G. : The NCAR master mechanism of the gas-phase chemistry-Version 2.0, pp. Rep.NCAR/TN-333+STR, National Center for atmospheric Research, Boulder, Colorado, 1989.
Madronich, S., and Calvert, J.G. : Permutation reactions of organic peroxy radicals in the troposphere, Journal ofGeophysical Research, 95, 5697-5715, 1990.
Mellouki, A., Le Bras, G., and Sidebottom, H. : Kinetics and mechanisms of the oxidation of oxygenated organiccompounds in the gas phase, Chemical Review, 103, 5077–5096, 2003.
Menut, L. : Adjoint modeling for atmospheric pollution process sensitivity at regional scale, Journal of GeophysicalResearch, 108, 8562, doi :10.1029/2002JD002549, 2003.
Middleton, P., Stockwell, W.R., and Carter, W.P.L. : Aggregation and analysis of volatile organic compound emis-sions for regional modeling, Atmospheric Environment, 24A, 1107-1133, 1990.
Moxim, W.J., Levy II, H., and Kasibhatla, P.S. : Simulated global tropospheric PAN : its transport and impact onNOx, Journal of Geophysical Research, 101, 12,621-12,638, 1996.
Müller, J.-F., and Brasseur, G. : Sources of upper tropospheric HOx : a three dimensional study, Journal of GeophysicalResearch, 104, 1705-1715, 1999.
National Research Council (NRC) : Rethinking the ozone problem in urban and regional air pollution, National AcademyPress, Washington, D.C., 1991.
Neeb, P. : Structure-reactivity based estimation of the rate constants for hydroxyl radical reactions with hydrocar-bons, Journal of Atmospheric Chemistry, 35, 295-315, 2000.
Olson, J., Prather, M.J., Berntsen, T., Carmichael, G.R., Chatfield, R.B., Connell, P., Derwent, D., Horowitz, L., Jin,S., Kanakidou, M.A., Kasibhatla, P.S., Kotamarthi, V.R., Kuhn, M., Law, K.S., Penner, J.E., Perliski, L., Sillman,S., Stordal, F., Thompson, A.M., and Wild, O. : Results from the Intergovernmental Panel on Climate Changephotochemical model intercomparison (photocomp), Journal of Geophysical Research, 102, 5979-5991, 1997.
Orlando, J. J., Tyndall, G. S., and Wallington, T. J. : The atmospheric chemistry of alkoxy radicals, Chemical Review,103, 4657–4689, 2003.
Peeters, J., Fantechi, G., and Vereecken, L. : A generalized structure-activity relationship for the decomposition of

MODÉLISATION CHIMIQUE DE L’OZONE 41
(substituted) alkoxy radicals, Journal of Atmospheric Chemistry, 48, 59-80, 2004.
Poppe, D., Aumont, B., Ervens, B., Geiger, H., Herrmann, H., Roth, E.P., Seidl, W., Stockwell, W.R., Vogel, B., Wagner,S., and Weise, D. : Scenario for modeling multiphase tropospheric chemistry, Journal of Atmospheric Chemistry, 40,77-86, 2001.
Prather, M.J., and Jacob, D.J. : A persistent imbalance in HOx and NOx photochemistry of the upper tropospheredriven by deep tropical convection, Geophysical Research Letters, 24, 3189-3192, 1997.
Roberts, J.M. : The atmospheric chemistry of organic nitrates, Atmospheric Environment, 24A, 243-287, 1990.
Saunders, S.M., Jenkin, M.E., Derwent, R.G., and Pilling, M.J. : Protocol for the development of the Master Che-mical Mechanism, MCM v3 (Part A) : tropospheric degradation of non-aromatic volatile organic compounds,Atmospheric Chemistry and Physics, 3, 161-180, 2003.
Schmidt, H., Derognat, C., Vautard, R., and Beekmann, M. : A comparison of simulated and observed ozone mixingratios for the summer of 1998 in western Europe, Atmospheric Environment, 35, 6277-6297, 2001.
Seinfeld, J.H., and Pandis, S.N. : Atmospheric chemistry and physics, Wiley, New York, 1998.
Simonaitis, R., Meagher, J.F., and Bailey, E.M. : Evaluation of the condensed carbon bond (CB-IV) mechanism againstsomg chamber data at low VOC and NOx concentrations, Atmospheric Environment, 31, 27-43, 1997.
Simpson, D. : Long period modelling of photochemical oxidants in Europe. Model calculations for July 1985, At-mospheric Environment, 26A, 1609-1634, 1992.
Stockwell, W.R., Middleton, P., Chang, J.S., and Tang, X. : The second generation regional acid deposition modelchemical mechanism for regional air quality modeling, Journal of Geophysical Research, 95, 16,343-16,367, 1990.
Stockwell, W.R., Kirchner, F., Kuhn, M., and Seefeld, S. : A new mechanism for regional atmospheric chemistrymodeling, Journal of Geophysical Research, 102, 25,847-25,879, 1997.
Szopa, S., Aumont, B., and Madronich, S. : Assessment of the reduction methods used to develop chemical schemes :building of a new chemical scheme for VOC oxidation suited to three-dimensional multiscale HOx-NOx-VOCchemistry simulations, Atmospheric Chemistry and Physics, 5, 2519-2538, 2005.
Volz-Thomas, A., Geiss, H., Hofzumahaus, A., and Becker, K.-H. : Introduction to Special Section : PhotochemistryExperiment in BERLIOZ, Journal of Geophysical Research, 108, 8252, doi :10.1029/2001JD002029, 2003.
Wagner, V., Jenkin, M.E., Saunders, S.M., Stanton, J., Wirtz, K., and Pilling, M.J. : Modelling of the photooxidation oftoluene : conceptual ideas for validating detailed mechanisms, Atmospheric Chemistry and Physics, 3, 89-106, 2003.
Wennberg, P.O., Hanisco, T.F., Jaeglé, L., Jacob, D.J., Hintsa, E.J., Lazendorf, E.J., Anderson, J.G., Gao, R.-S., Keim,E.R., Donnelly, S.G., Del Negro, L.A., Fahey, D.W., McKeen, S.A., Salawitch, R.J., Webster, C.R., May, R.D., Her-man, R.L., Proffitt, M.H., Margitan, J.J., Atlas, E.L., Schauffler, S.M., Flocke, F., McElroy, C.T., and Bui, T.P. :Hydrogen radicals, nitrogen radicals, and the production of O3 in the upper troposphere, Science, 279, 49-53,1998.
Whitehouse, L.E., Tomlin, A.S., and Pilling, M.J. : Systematic reduction of complex tropospheric chemical mecha-nisms, Part II : Lumping using a time-scale based approach, Atmospheric Chemistry and Physics, 4, 2057-2081,2004.
Whitten, G.Z., Hogo, H., and Killus, J.P. : The carbon bond mechanism : a condensed kinetic mechanism for photo-chemical smog, Environmental Science & Technology, 18, 280-287, 1980.
Yang, Y.-J., Stockwell, W.R., and Milford, J.B. : Uncertainties in incremental reactivities of volatile organic com-pounds, Environmental Science & Technology, 29, 1136-1345, 1995.
Yang, Y.-J., Stockwell, W.R., and Milford, J.B. : Effect of chemical product yield uncertainties on reactivities of VOCsand emissions from reformulated gazoline and methanol fuels, Environmental Science & Technology, 30, 1392-1397,1996.