mobilité moléculaire d'élastomères réticulés étirés...
TRANSCRIPT

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
37
2
"Mobilité moléculaire d'élastomères réticulés étirés au-dessus de la
température de transition vitreuse"

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
38
1 Introduction .............................................................39 2 Contexte ...................................................................39
2.1 Les mouvements de relaxation .................................................................................39 2.2 Les différentes théories.............................................................................................40 2.3 L'impact de la déformation.......................................................................................40 2.4 Quelques résultats existants......................................................................................40 2.5 Conclusion................................................................................................................41
3 Présentation des conditions expérimentales ............41
3.1 Les matériaux ...........................................................................................................41 3.2 La spectrométrie mécanique.....................................................................................42 3.3 Le banc de diffusion des Rayons-X..........................................................................45 3.4 Les autres techniques................................................................................................46
4 Les Résultats ............................................................46
4.1 La caractérisation microstructurale des matériaux non-déformés............................46 4.1.1 Caractérisation du réseau par mesures de gonflement .....................................46 4.1.2 L'influence de la déformation sur l'entropie des matériaux..............................47
4.2 L'effet de la déformation sur la microstructure: mesure de WAXS .........................49 4.2.1 L'impact de la déformation sur l'arrangement amorphe ...................................50 4.2.2 L'impact de la déformation sur l'orientation des chaînes .................................52
5 L'influence de la déformation sur la mobilité moléculaire ..................................................................55 6 Discussion des résultats ...........................................60
6.1 La littérature et la déformation sous Tg ...................................................................60 6.2 La littérature et la déformation au-dessus de Tg ......................................................60 6.3 Les résultats de l'étude..............................................................................................61
6.3.1 D'un point de vue thermodynamique................................................................61 6.3.2 Discussion sur les paramètres d'activation .......................................................62 6.3.3 Une contradiction avec la théorie de Gibbs et DiMarzio? ...............................63
7 Conclusion ................................................................64

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
39
1 Introduction
Ce chapitre a pour but d'étudier l'effet de l'étirement des chaînes sur la mobilité moléculaire dans les polymères amorphes. L'utilisation d'un élastomère réticulé permet de modifier la conformation des chaînes par le biais d'une élongation uniaxiale. Cette modification de la conformation est figée par refroidissement du système depuis l'état d'équilibre thermodynamique au-dessus de Tg jusqu'à l'état vitreux. Après une présentation de quelques résultats de la littérature et du dispositif expérimental utilisé, l'effet de l'allongement est étudié de façon qualitative et quantitative par l'intermédiaire d'essais mécaniques et de diffusion des rayons-X. En second lieu, l'impact de l'allongement des chaînes sur la mobilité moléculaire est testé au moyen d'essais de spectrométrie mécanique.
2 Contexte
Un certain nombre d'idées ont déjà été évoquées au cours de la revue bibliographique de cette étude (chapitre 1). Il reste toutefois à préciser quelques points qui concernent le thème particulier de l'impact d'une déformation au-dessus de la zone de transition vitreuse, traité dans ce chapitre.
2.1 Les mouvements de relaxation
Un grand nombre de mouvements se produisent dans les polymères à différentes échelles de longueur et de temps en fonction de la température. Les processus rapides associés à l'échelle du nanomètre sont généralement observés par l'intermédiaire de mesures de diffusion quasi-élastique des neutrons, ou de spectroscopie Raman à basses fréquences [MER96]. Ces mouvements localisés sont à l'origine de la relaxation secondaire β, alors que les processus de la relaxation α sont liés à des mouvements coopératifs à grande échelle. Cette relaxation est associée à la température de transition vitreuse [GAU00]. Les modes de Rouse et de reptation déterminent la mobilité pour des échelles de temps et de distance plus grandes [ROL97]. L'ensemble de ces mouvements est affecté par la nature de la chaîne macromoléculaire, sa flexibilité, l'encombrement stérique des groupes pendants et l'état structural du polymère. L'effet de l'état microstructural sur la mobilité a déjà fait l'objet de plusieurs études. Il existe différentes méthodes pour modifier la microstructure1 d'un polymère: les procédures de recuit, le vieillissement physique ou bien la déformation plastique. Il a été montré par Oleynik que le mécanisme de déformation a un impact complètement différent sur le système lorsqu'il est réalisé au-dessous ou au-dessus de la température de transition vitreuse [OLE89].
1 Le terme microstructure peut bien souvent apparaître ambigu. Dans le cadre de notre étude, il fait référence en fonction du contexte, aussi bien à l'arrangement moléculaire qu'à l'architecture des chaînes.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
40
2.2 Les différentes théories
Parmi toutes les théories qui décrivent le comportement des matériaux amorphes au voisinage de leur transition vitreuse, la théorie du volume libre établit le lien entre les phénomènes visco-élastiques, le temps et la température. Elle donne une relation entre les coefficients d'expansion au-dessus de Tg, mais pêche dans la description des mouvements au niveau moléculaire. Cette théorie est applicable au-dessus de Tg, mais est très souvent utilisée de manière impropre pour des températures inférieures. Les théories cinétiques quant à elles décrivent quantitativement les décalages de Tg avec le temps. Elles permettent une détermination des capacités calorifiques mais ne prédisent pas les valeurs de Tg pour des échelles de temps infinis. Les théories thermodynamiques prédisent des variations de Tg avec le poids moléculaire, la densité de nœuds de réticulation, la température de la transition, sans la définir de façon précise [SPE92].
2.3 L'impact de la déformation
Pour pouvoir considérer l'impact d'une déformation sur la microstructure d'un système amorphe, il est important d'établir une distinction claire entre l'approche macroscopique des modèles thermodynamiques, qui évalue les contributions énergétiques et entropiques de la contrainte et l'approche locale des modèles qui s'intéressent aux variations de mobilité en fonction de l'énergie et de l'entropie d'activation. Les modèles globaux calculent la variation de l'énergie interne et de l'entropie au cours d'une transformation thermodynamique d'un état non-déformé vers un état déformé. Le comportement thermo-mécanique des élastomères a été le sujet de nombreuses études par l'intermédiaire de mesures thermo-élastiques complexes et difficiles à mettre en oeuvre [SHE69], [LYO87], [MOS87]. Ces mesures ont tenté d'établir une corrélation entre la contrainte ou la déformation avec des variations d'entropie et plus rarement d'enthalpie. Mais aucune information sur la mobilité moléculaire ne peut être réellement extraite à partir de telles considérations.
2.4 Quelques résultats existants
Au cours de mesures calorimétriques sur des élastomères soumis à une extension uniaxiale, un effet de compétition a été mis en évidence entre une augmentation de l'entropie de vibration (liée au changement de volume avec la déformation) et une diminution de l'entropie de configuration (liée au changement de forme avec la déformation). Dans tous les cas, il est important de considérer que les changements de volume restent toujours très faibles, du fait du caractère incompressible du matériau au-dessus de Tg. Dans son étude [FRO69], Frosini a effectué un calcul de la variation d'énergie interne au cours de la transformation thermodynamique, indépendamment de toute considération sur la mobilité moléculaire. Il a utilisé les équations générales d'état pour un réseau polymère gaussien en décomposant la force élastique totale en deux composantes énergétique et entropique. Il a ainsi pu montrer que la contribution énergétique était pratiquement indépendante du taux d'élongation. Outre ces considérations thermodynamiques, il faut étudier les variations de mobilité en fonction de l'entropie d'activation et de l'énergie d'activation, sur le modèle du

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
41
formalisme de Starkweather par exemple [STA91-1], [STA81]. Le point de départ de ce modèle est la théorie de la vitesse de réaction absolue, équivalente à une fonction d'Arrhénius:
exp exp2
A AS HkTfh k kTπ
∆ ∆⎛ ⎞ ⎛ ⎞= −⎜ ⎟ ⎜ ⎟⎝ ⎠ ⎝ ⎠
Dans cette expression, ∆HA est l'enthalpie d'activation; ∆SA est l'entropie d'activation; T est la température; k est la constante de Boltzmann et h est la constante de Planck. Sur la base de ce formalisme, il est possible de tester si la déformation a un impact sur les paramètres ∆HA et ∆SA. Peu d'études se sont intéressées à l'effet de la déformation sur la mobilité moléculaire. Diaz-Calleja a cherché à tester la validité de la théorie du volume libre pour des réseaux étirés. Ce faisant, il a observé un décalage du pic du facteur de perte tan δ pour des réseaux d'amorphes réticulés étirés [DIA90], [DIA87]. Il a montré la validité de l'utilisation d'une loi de compensation et de la théorie du volume libre et a émis l'hypothèse que ses observations étaient le résultat de la compétition entre une augmentation du volume libre et une diminution de l'entropie de conformation provoquées par la déformation. Il conclut finalement que l'augmentation de l'entropie d'activation avec une augmentation de la déformation est en accord avec la diminution de l'entropie de conformation de la chaîne.
2.5 Conclusion Dans cette étude, nous allons tenter d'apporter des éléments de réponse à la question encore en suspend dans la littérature qui concerne l'impact d'une modification de l'entropie du système sur sa mobilité moléculaire. Pour cela, deux élastomères amorphes réticulés ont été étirés à une température supérieure à leur température de transition vitreuse, autorisés à relaxer puis refroidis sous Tg à un allongement maintenu constant. Un copolymère styrène-butadiène (SBR) et un copolymère nitrile butadiène (NBR) ont été étudiés pour tester les effets de la structure moléculaire. Le NBR a subi un traitement de recuit pour tester l'effet d'une variation du taux de réticulation.
3 Présentation des conditions expérimentales
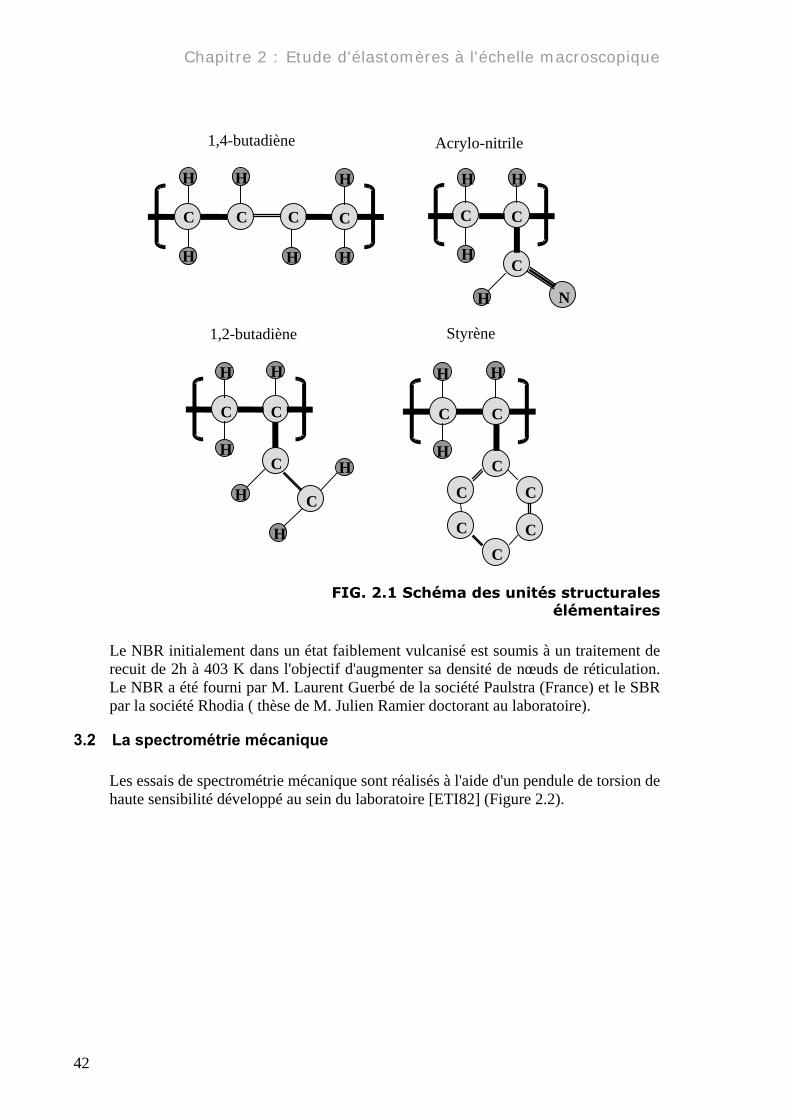
3.1 Les matériaux Le SBR et le NBR sont des élastomères thermoplastiques, qui ne cristallisent pas même sous l'effet de la déformation, comme peut le faire le caoutchouc naturel par exemple. Ils ont des températures de transition α autour de 263 K pour le SBR et 243 K pour le NBR, mesurées par spectroscopie mécanique à 1Hz et une rampe de température de 1K.min-1. Les élastomères synthétiques utilisés dans cette étude sont des copolymères statistiques composés de 1,2-butadiène (55%), 1,4-butadiène (20%), styrène (25%) pour le SBR et butadiène (72%), acrylo-nitrile (28%) pour le NBR (Figure 2.1). Les densités ont été mesurées à l'aide d'un pycnomètre à hélium: ρSBR=1,031g.cm-3 et ρ NBR=1,034 g.cm-3.
Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
42
N
C
C C
H
H
H
H
Acrylo - nitrile
H H
C C C C
H
H H H
1,4 - butadiène
C
C C
H
H
C
H
H
H
H
1,2 - butadiène
C
C C
C
C C
C
C
H
H
H
Styrène
FIG. 2.1 Schéma des unités structurales élémentaires
Le NBR initialement dans un état faiblement vulcanisé est soumis à un traitement de recuit de 2h à 403 K dans l'objectif d'augmenter sa densité de nœuds de réticulation. Le NBR a été fourni par M. Laurent Guerbé de la société Paulstra (France) et le SBR par la société Rhodia ( thèse de M. Julien Ramier doctorant au laboratoire).
3.2 La spectrométrie mécanique
Les essais de spectrométrie mécanique sont réalisés à l'aide d'un pendule de torsion de haute sensibilité développé au sein du laboratoire [ETI82] (Figure 2.2).

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
43
1
2
3
4
5 6
7
FIG. 2.2 Schéma du pendule de torsion: échantillon (1); four (2); Cryostat (3); miroir
(4); aimant (5); bobines de Helmholtz (6); masse morte (7).
L'appareillage initial a été modifié pour pouvoir maintenir une déformation constante tout au long du test (Figure 2.3).
8
FIG. 2.3 Modification du dispositif expérimental initial: élévateur de laboratoire fixé à la masse
mécanique du système (8).

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
44
Le poids mort qui compense le poids du mors supérieur et de l'échantillon pour assurer un essai de torsion pure est remplacé par un élévateur de laboratoire fixé à la masse mécanique du système. Le montage permet de faire varier la longueur de l'échantillon et de maintenir la valeur de l'allongement au cours de la relaxation des contraintes, et du refroidissement sous Tg. Tous les échantillons sont dans un état d'équilibre avant le refroidissement sous Tg qui s'effectue à une vitesse de 3K.min-1. La mise en place de l'échantillon est extrêmement délicate. Il faut à la fois s'assurer que l'éprouvette possède une géométrie après étirement qui soit compatible avec un essai de spectrométrie mécanique (parallélépipédique), et pouvoir étirer le matériau jusqu'à des taux importants (300%) à faible vitesse d'allongement de surcroît et maintenir la déformation sans rupture de l'échantillon un temps nécessaire à la relaxation du système. Le protocole est représenté sur la figure 2.4. Une éprouvette de traction avec réduction de section est prélevée par matriçage dans une plaque d'élastomère. Une plaque métallique est collée (colle cyanolite) sur les têtes des éprouvettes de traction pour faciliter leur maintien dans les mors du pendule. Le montage est laissé une nuit à l'ambiante (300K) pour assurer le séchage. L'ensemble est fixé aux mors. La zone de jonction entre la plaque et l'échantillon est un point faible du montage où se concentrent les contraintes, et où l'agressivité de la colle vis-à-vis de l'élastomère peut conduire à des déchirures du matériau. Les dimensions finales de l'éprouvette sont déterminées à partir de l'allongement uniaxial, et d'une hypothèse de déformation à volume constant.
1
2
3
mors
P laque de m é tal
échantillon
échantillon
colle
écrou / boulon
FIG. 2.4 Schéma du montage de l'éprouvette sur les mors du pendule de torsion.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
45
Les mesures isochrones sur le pendule sont effectuées à une fréquence de 1Hz et une rampe de montée en température de 1K.min-1 à partir de 90K jusqu'au-dessus de Tg (Figure 2.5). Pour la construction des courbes maîtresses, une rampe de température est imposée au système par palier de 2K, et le module complexe du matériau est mesuré pour chaque température à différentes fréquences comprises entre 0.03 et 3Hz.
300K
WAXS Gonflement
Déformation Relaxation WAXS DMA
1K.min-1 ≠freq.
temps
T
90K
FIG. 2.5 Schéma de la procédure expérimentale
3.3 Le banc de diffusion des Rayons-X
Les Rayons-X sont collimatés de manière ponctuelle et parallèle grâce à un système optique basé sur des miroirs de Göbels et développé par la société XENOX. Le système assure l'émission d'un faisceau monochromatique de la raie K-α du cuivre (λ= 1,54Å). L'échantillon est étiré à l'aide d'un dispositif manuel de traction. Une plaque en cuivre percée d'un trou (diamètre 3mm) est placée juste devant l'échantillon. Cette plaque ne laisse passer que le faisceau incident qui traverse ensuite l'échantillon, et écrante ainsi la diffusion du faisceau incident par l'air, qui provoque un bruit sur le spectre enregistré (figure 2.6). Le détecteur est placé à 7 cm de l'échantillon, ce qui correspond à une diffusion des Rayons-X aux grands angles (3 nm-1 < q < 24 nm-1). Les spectres 2-D sont enregistrés par une caméra ccd Princeton Instrument PI-SCX et les images sont traitées à l'aide du logiciel "winview" puis intégrées grâce au programme Fit2D développé à L'ESRF de Grenoble. Les données obtenues sont corrigées en prenant en compte le fond continu et le bruit du détecteur.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
46
8
7
6
5 4
3 2
1
FIG. 2.6 Schéma du montage de diffusion des Rayons-X: faisceau incident (1), écran en cuivre percé (2), dispositif de traction (3), échantillon (4), faisceau transmis (5), faisceau diffusé (6),
arrêt de faisceau (7), détecteur bi-dimensionnel (8).
3.4 Les autres techniques
Le comportement mécanique en tension du matériau est obtenu à l'aide d'une machine de traction MTS à la température ambiante (300K) et une vitesse de déformation de 0,4 s-1. Les mesures de gonflement sont effectuées dans du dichlorométhane, qui présente des paramètres d'interaction χ de 0,47 et 0,31 avec le SBR et le NBR respectivement [BRA99].
Les élastomères thermoplastiques réticulés, étirés à une température supérieure à la température de transition vitreuse, sont dans un étatd'équilibre thermodynamique. La présence des noeuds de réticulation entreles chaînes permet de maintenir la déformation du système et de l'autoriser à relaxer dans son état allongé avant toute caractérisation ultérieure.
4 Les Résultats
4.1 La caractérisation microstructurale des matériaux non-déformés
4.1.1 Caractérisation du réseau par mesures de gonflement Les résultats du gonflement sont présentés dans le tableau 2.1. La détermination de la densité de chaînes actives est faite sur la base de l'équation de Flory-Rehner:

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
47
2 1/3 22 2 2 2ln(1 )
2nV υυ υ χυ υ⎡ ⎤⎡ ⎤− − + + = −⎣ ⎦ ⎢ ⎥⎣ ⎦
Avec ν2 la fraction volumique de polymère dans la masse gonflée; V le volume molaire du solvant; χ le paramètre d'interaction de Flory-Huggins et n la densité de chaînes actives. Cette équation associe au mélange du polymère et du solvant un changement d'entropie positif et à la réduction du nombre de configurations possibles ainsi qu'à la chaleur du mélange entre polymère et solvant, un changement d'entropie négatif [SPE92], [BRA99].
Tableau 2.1: Résultats des mesures de gonflement du SBR et des NBR dans le
dichlorométhane.
Matériau
χ
état
Densité de chaînes actives
(mol.cm-3)
Nombre moyen de
monomères entre noeuds
SBR 0,47 Initial 9.18 x 10-5 235 NBR 0,31 Initial 1.80 x 10-4 103 NBR 0,31 Recuit 2.09 x 10-4 88
Dans les états initiaux, le SBR et le NBR présentent des masses molaires différentes entre nœuds d'enchevêtrement. Cette différence est encore accentuée par le traitement thermique qui conduit à une augmentation de la densité de chaînes actives.
4.1.2 L'influence de la déformation sur l'entropie des matériaux
Le premier objectif est d'estimer l'impact de la déformation sur l'entropie de conformation du matériau. Le comportement mécanique en traction du matériau est présenté sur la Figure 2.7, qui montre l'évolution de la contrainte nominale en fonction de la déformation nominale pour le SBR et le NBR dans son état initial et son état recuit.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
48
0
0.5
1
1.5
2
2.5
3
0 0.5 1 1.5 2 2.5 3 3.5Déformation nominale (l/lo)
Con
train
te n
omin
ale
(MPa
)
SBR
NBR
NBR recuit
FIG. 2.7 Contrainte nominale en fonction de la déformation nominale pour le SBR, le NBR initial
et le NBR recuit. Essais effectués à une vitesse de déformation de 0,4s-1 à la température
ambiante (300 K).
Ces résultats confirment la plus faible densité de nœuds de réticulation du SBR, qui présente une valeur de module à l'origine plus faible. Le traitement de recuit augmente, comme prévu, la valeur du module du NBR et diminue son allongement à rupture. Pour comprendre les effets d'une déformation macroscopique sur les arrangements locaux des macromolécules, il peut être intéressant d'utiliser la théorie de l'élasticité caoutchoutique. Dans son formalisme le plus classique et le plus simple, le comportement en déformation d'un réseau idéal de chaînes gaussiennes est donné par [VER95]:
( )2 1
C
RTMρσ λ λ−= −
Avec λ=l/l0 le taux de déformation uniaxiale; l la longueur de l'échantillon au cours de la déformation; l0 la longueur initiale; σ la contrainte nominale; ρ la densité de l'élastomère (g.cm-3); Mc la masse molaire entre nœuds d'enchevêtrement (g.mol-1); T la température (K) et R la constante des gaz (J.K-1.mol-1). Avec ce modèle il est possible d'établir un lien entre l'allongement et une modification de l'entropie de conformation. Il faut pour cela considérer la fonction d' énergie libre du matériau: F U TS= − En négligeant dans cette expression l'impact de la contribution de l'énergie interne, on obtient pour une traction uniaxiale:
2 2 32
nRS λλ
⎛ ⎞∆ ≈ + −⎜ ⎟⎝ ⎠
Avec n= ρ/MC (mol.cm-3).
Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
49
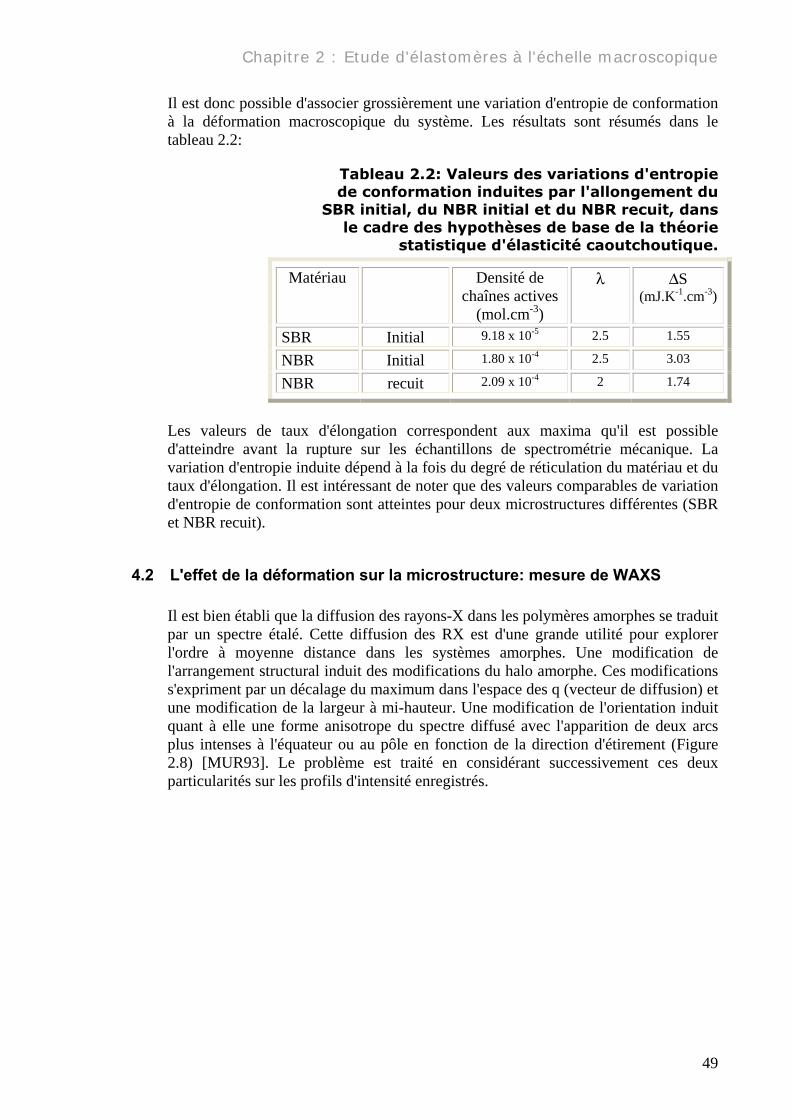
Il est donc possible d'associer grossièrement une variation d'entropie de conformation à la déformation macroscopique du système. Les résultats sont résumés dans le tableau 2.2:
Tableau 2.2: Valeurs des variations d'entropie de conformation induites par l'allongement du
SBR initial, du NBR initial et du NBR recuit, dans le cadre des hypothèses de base de la théorie
statistique d'élasticité caoutchoutique.
Matériau Densité de chaînes actives
(mol.cm-3)
λ ∆S (mJ.K-1.cm-3)
SBR Initial 9.18 x 10-5 2.5 1.55
NBR Initial 1.80 x 10-4 2.5 3.03
NBR recuit 2.09 x 10-4 2 1.74
Les valeurs de taux d'élongation correspondent aux maxima qu'il est possible d'atteindre avant la rupture sur les échantillons de spectrométrie mécanique. La variation d'entropie induite dépend à la fois du degré de réticulation du matériau et du taux d'élongation. Il est intéressant de noter que des valeurs comparables de variation d'entropie de conformation sont atteintes pour deux microstructures différentes (SBR et NBR recuit).
4.2 L'effet de la déformation sur la microstructure: mesure de WAXS
Il est bien établi que la diffusion des rayons-X dans les polymères amorphes se traduit par un spectre étalé. Cette diffusion des RX est d'une grande utilité pour explorer l'ordre à moyenne distance dans les systèmes amorphes. Une modification de l'arrangement structural induit des modifications du halo amorphe. Ces modifications s'expriment par un décalage du maximum dans l'espace des q (vecteur de diffusion) et une modification de la largeur à mi-hauteur. Une modification de l'orientation induit quant à elle une forme anisotrope du spectre diffusé avec l'apparition de deux arcs plus intenses à l'équateur ou au pôle en fonction de la direction d'étirement (Figure 2.8) [MUR93]. Le problème est traité en considérant successivement ces deux particularités sur les profils d'intensité enregistrés.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
50
Direction d'étirement
FIG. 2.8 Image d'un spectre de diffusion anisotrope, obtenu par mesures de WAXS sur un
échantillon de NBR étiré de 250%.
4.2.1 L'impact de la déformation sur l'arrangement amorphe
Les spectres 2-D de diffusion des RX sont intégrés de façon radiale, comme le montre le schéma de la figure 2.9. Cette figure fait apparaître clairement la bosse amorphe, ainsi que deux anneaux de diffraction de faible intensité qui sont provoqués par le ZnO, charge couramment utilisée comme activateur de la réticulation des élastomères. Ces pics de ZnO peuvent être utilisés pour la calibration de la distance qui sépare l'échantillon du détecteur.
Equateur
Pôle ZnO
FIG. 2.9 Schéma de l'intégration radiale des spectres 2-D de WAXS. 1.intégration à
l'équateur, 2.intégration au pôle. La Figure 2.10 montre le résultat d'une intégration radiale sur l'exemple du spectre de diffusion du NBR dans son état initial pour différents taux d'élongation.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
51
0
4000
8000
12000
16000
5 7 9 11 13 15 17q (nm-1)
Inte
nsité
(u.a
.)
250%200%100%0%
équateur
pôle
FIG. 2.10 Intégration radiale du spectre de diffusion du NBR dans son état initial mesuré
pour différents taux d'élongation; intégration au méridien et à l'équateur.
Sur cette figure, il est possible de distinguer les intégrations à l'équateur et au pôle pour différents taux d'allongement (0%, 100%, 200%, 250%). La déformation a un effet faible sur le halo amorphe au pôle, alors qu'elle provoque une forte augmentation de l'intensité diffusée à l'équateur. Il est possible de faire ce genre d'observations sur l'ensemble des matériaux étudiés. Le Tableau 2.3 résume les résultats qui concernent le maximum du pic et sa largeur à mi-hauteur (FWHM) pour tous les échantillons étudiés.
Tableau 2.3: Synthèse des résultats de la position du pic et de la largeur à mi-hauteur
(FWHM) pour l'ensemble des matériaux étudiés.
SBR NBR NBR recuit FWHM
(q en nm-1)
Position du pic
(q en nm-1)
FWHM
(q en nm-1)
Position du pic
(q en nm-1)
FWHM
(q en nm-1)
Position du pic
(q en nm-1)
Equateur 0% 7,1 ± 0,2 12 ± 0,2 5 ± 0,2 13,2 ± 0,2 5,2 ± 0,2 12,9 ± 0,2 100% 7,2 ± 0,2 11,8 ± 0,2 5,3 ± 0,2 13,2 ± 0,2 5,2 ± 0,2 12,8 ± 0,2 200% 7,4 ± 0,2 11,9 ± 0,2 5 ± 0,2 13 ± 0,2 5,1 ± 0,2 12,9 ± 0,2 250% 7,3 ± 0,2 11,8 ± 0,2 5,1 ± 0,2 13 ± 0,2
Pôle 0% 7 ± 0,2 12,4 ± 0,2 5,4 ± 0,2 13,2 ± 0,2 4,9 ± 0,2 13,3 ± 0,2 100% 8 ± 0,2 12,4 ± 0,2 5,6 ± 0,2 13,2 ± 0,2 4,5 ± 0,2 13,2 ± 0,2 200% 7,5 ± 0,2 12,2 ± 0,2 5,4 ± 0,2 13,3 ± 0,2 5,4 ± 0,2 13,3 ± 0,2 250% 7,5 ± 0,2 12,1 ± 0,2 5,2 ± 0,2 13,3 ± 0,2

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
52
Il n'apparaît pas de décalage en q ni de variation de la largeur à mi-hauteur avec la déformation. Cette observation implique que ni la déformation ni le recuit (qui modifie la densité de réticulation) n'ont d'impact significatif sur la densité amorphe du matériau. Le résultat n'est pas surprenant dans la mesure où le matériau est incompressible au-dessus de sa température de transition vitreuse.
4.2.2 L'impact de la déformation sur l'orientation des chaînes
L'orientation amorphe peut être obtenue par le biais de la variation de l'intensité du halo amorphe sur une intégration azimutale. L'intégration azimutale, représentée schématiquement sur la Figure 2.11, se présente sous la forme d'un pic à l'équateur (angles de 0 et 180°) superposé à une ligne de base non nulle.
0°
FIG. 2.11 Schéma de l'intégration azimutale des spectres 2-D de WAXS
L'excès de diffusion de l'amorphe à l'équateur est du aux segments parallèles à l'axe des chaînes et l'intensité diffusée sous la ligne de base est liée à la partie amorphe non orientée et donc isotrope du matériau. La Figure 2.12 présente l'intensité azimutale diffusée, c'est à dire l'intégration de l'intensité autour du maximum de la bosse amorphe du matériau.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
53
0
2000
4000
6000
8000
10000
12000
14000
16000
0 50 100 150 200 250 300 350Angle azimuthal (Deg)
Inte
nsité
(a.
u.)
250 %200 %100 %0 %
FIG. 2.12 Intégration azimutale du spectre 2-D du SBR pour des taux d'élongation de 0 %, 100
%, 200 %, 250 %.
La ligne de base qui doit correspondre à l'échantillon non déformé n'est pas complètement plate. Ceci indique qu'il existe une orientation préférentielle initiale au sein du matériau, qui est la conséquence du mode d'élaboration (calandrage) des plaques d'élastomères dans lesquelles les éprouvettes ont été prélevées. Les échantillons ont tous été étirés dans la direction d'alignement préférentiel des chaînes. De façon qualitative tout d'abord, il apparaît que l'étirement induit une augmentation de l'amplitude du pic associée à la partie amorphe orientée. Dans une approche plus quantitative, il est possible d'évaluer l'orientation induite par l'étirement grâce au facteur d'Herman défini de la manière suivante [VAN94]:
902
090
0
( ).cos ( ).sin( ).1 . 3. 12
( ).sin( ).X Ray
I df
I d−
⎡ ⎤Φ Φ Φ Φ⎢ ⎥
⎢ ⎥= −⎢ ⎥
Φ Φ Φ⎢ ⎥⎣ ⎦
∫
∫
Avec I(Φ) l'intensité diffusée et Φ l'angle azimutal. Cette expression est valable dans le cas où il est possible d'associer le halo équatorial aux distances inter-moléculaires perpendiculaires à la direction de la chaîne principale. Cette hypothèse semble raisonnable dans notre cas, où le maximum d'intensité diffusée se trouve à un angle 2θ de 18°, ce qui d'après la loi de Bragg donne une période moyenne de 4,9Å. Il est important de rappeler en outre que le facteur d'orientation déterminé expérimentalement fX-Ray est toujours inférieur au facteur f réel. En effet, comme le rappelle Van Aerle [VAN94], les polymères amorphes donnent lieu à une diffusion des rayons-X toujours étalée, et ce, même dans un état parfaitement orienté. La largeur azimutale intrinsèque de ces matériaux est essentiellement due à la courbure des segments diffusants. Une correction de cette sous-estimation du facteur d'orientation est possible grâce aux résultats de Pick [PIC80] à partir de l'expression:
. X rayf K f −=
Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
54
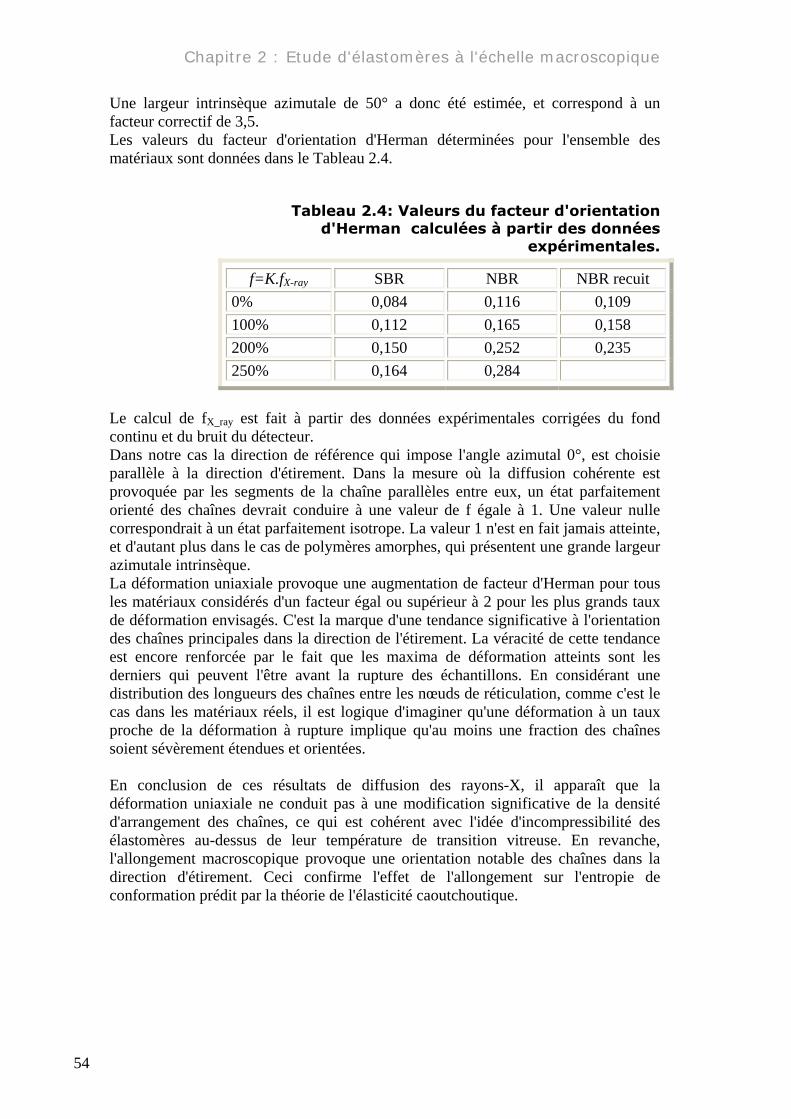
Une largeur intrinsèque azimutale de 50° a donc été estimée, et correspond à un facteur correctif de 3,5. Les valeurs du facteur d'orientation d'Herman déterminées pour l'ensemble des matériaux sont données dans le Tableau 2.4.
Tableau 2.4: Valeurs du facteur d'orientation d'Herman calculées à partir des données
expérimentales.
f=K.fX-ray SBR NBR NBR recuit 0% 0,084 0,116 0,109 100% 0,112 0,165 0,158 200% 0,150 0,252 0,235 250% 0,164 0,284
Le calcul de fX_ray est fait à partir des données expérimentales corrigées du fond continu et du bruit du détecteur. Dans notre cas la direction de référence qui impose l'angle azimutal 0°, est choisie parallèle à la direction d'étirement. Dans la mesure où la diffusion cohérente est provoquée par les segments de la chaîne parallèles entre eux, un état parfaitement orienté des chaînes devrait conduire à une valeur de f égale à 1. Une valeur nulle correspondrait à un état parfaitement isotrope. La valeur 1 n'est en fait jamais atteinte, et d'autant plus dans le cas de polymères amorphes, qui présentent une grande largeur azimutale intrinsèque. La déformation uniaxiale provoque une augmentation de facteur d'Herman pour tous les matériaux considérés d'un facteur égal ou supérieur à 2 pour les plus grands taux de déformation envisagés. C'est la marque d'une tendance significative à l'orientation des chaînes principales dans la direction de l'étirement. La véracité de cette tendance est encore renforcée par le fait que les maxima de déformation atteints sont les derniers qui peuvent l'être avant la rupture des échantillons. En considérant une distribution des longueurs des chaînes entre les nœuds de réticulation, comme c'est le cas dans les matériaux réels, il est logique d'imaginer qu'une déformation à un taux proche de la déformation à rupture implique qu'au moins une fraction des chaînes soient sévèrement étendues et orientées. En conclusion de ces résultats de diffusion des rayons-X, il apparaît que la déformation uniaxiale ne conduit pas à une modification significative de la densité d'arrangement des chaînes, ce qui est cohérent avec l'idée d'incompressibilité des élastomères au-dessus de leur température de transition vitreuse. En revanche, l'allongement macroscopique provoque une orientation notable des chaînes dans la direction d'étirement. Ceci confirme l'effet de l'allongement sur l'entropie de conformation prédit par la théorie de l'élasticité caoutchoutique.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
55
La sollicitation uniaxiale au-dessus de la température de transition vitreusedu matériau provoque un alignement significatif des chaînes dans ladirection de l'étirement, mais ne modifie pas la densité d'arrangement du système.
5 L'influence de la déformation sur la mobilité moléculaire
Le premier problème à résoudre consiste à déterminer le paramètre le plus représentatif et le plus fiable pour tester la mobilité moléculaire de nos systèmes. Les essais de spectrométrie mécanique donnent accès au module complexe en cisaillement de nos matériaux:
* ' . "G G i G= + Avec G' le module conservatif et G" le module dissipatif. Les valeurs de G' et G" sont déterminés avec une incertitude de 5% environ. Cette incertitude est liée à l'appareillage, au contrôle des conditions expérimentales et surtout à la difficulté d'évaluer les dimensions des échantillons. Nous faisons donc l'hypothèse que le module des matériaux à l'état vitreux est indépendant de la déformation à la température minimale envisagée au cours de chaque expérience. Dans ce cadre, les valeurs sont normées par rapport à la valeur obtenue pour G' à une température de 90K. Ceci permet d'augmenter la précision de la discussion comparative sur la mobilité moléculaire. Le rapport G"/G'=tan δ est habituellement utilisé comme indice de la mobilité moléculaire. En prenant comme premier exemple les résultats isochrones du NBR dans son état initial (Figure 2.13), il apparaît que pour des températures supérieures à la température de la relaxation principale α, le module conservatif G' mesuré au début du plateau caoutchoutique augmente avec le taux de déformation. Ceci est très bien expliqué par la théorie de l'élasticité caoutchoutique, comme étant une conséquence de l'alignement des chaînes entre les nœuds de réticulation sans modification de la masse moléculaire, donc une diminution de l'entropie de conformation. Cette observation nous permet également de vérifier la cohérence de notre procédure de normalisation.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
56
0.0001
0.001
0.01
0.1
1
80 130 180 230 280Température (K)
G' /
G' 9
0K
0.001
0.01
0.1
1
10
tang
ente
de
perte
200%
0%
200%
0%
G' / G' 90K
tan δ
FIG. 2.13 Module conservatif G' et frottement intérieur tan δ en fonction de la température
pour le NBR dans son état initial à deux taux de déformation 0% et 200%.
Mais cette augmentation affecte également la partie haute température du pic de frottement intérieur tan δ. Cet effet a été interprété à tort par le passé comme une modification de la mobilité moléculaire [DIA86], [DIA88], alors qu'il ne s'agit que d'une conséquence de la modification du module caoutchoutique. Cette observation est confirmée par le résultat du module dissipatif G" présenté sur la Figure 2.14, où aucun décalage n'est observé le long de l'axe des températures pour des échantillons étirés jusqu'à 200% (NBR) et 300% (SBR). Finalement, il apparaît que le module dissipatif G" est le seul paramètre valable pour caractériser l'évolution de la mobilité moléculaire dans notre contexte. En effet, tan δ intègre les effets de l'allongement sur le module caoutchoutique du matériau et ne rend pas uniquement compte de la mobilité moléculaire.
0.0001
0.001
0.01
0.1
1
100 120 140 160 180 200 220 240 260 280 300Température (K)
G"/
G' 11
0K
SBR (1, 2)NBR (1,2,3,4)
FIG. 2.14 Module dissipatif G" en fonction de la température pour le NBR initial [1(□) 0%; 2(■) 250%], le NBR recuit [3(○) 0%; 4(●) 200%] et
le SBR [1(∆) 0%; 2(▲)300%].
Aucun décalage le long de l'axe des températures n'est observé sous l'effet d'une déformation uniaxiale pour des matériaux avec des microstructures et des densités de
Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
57
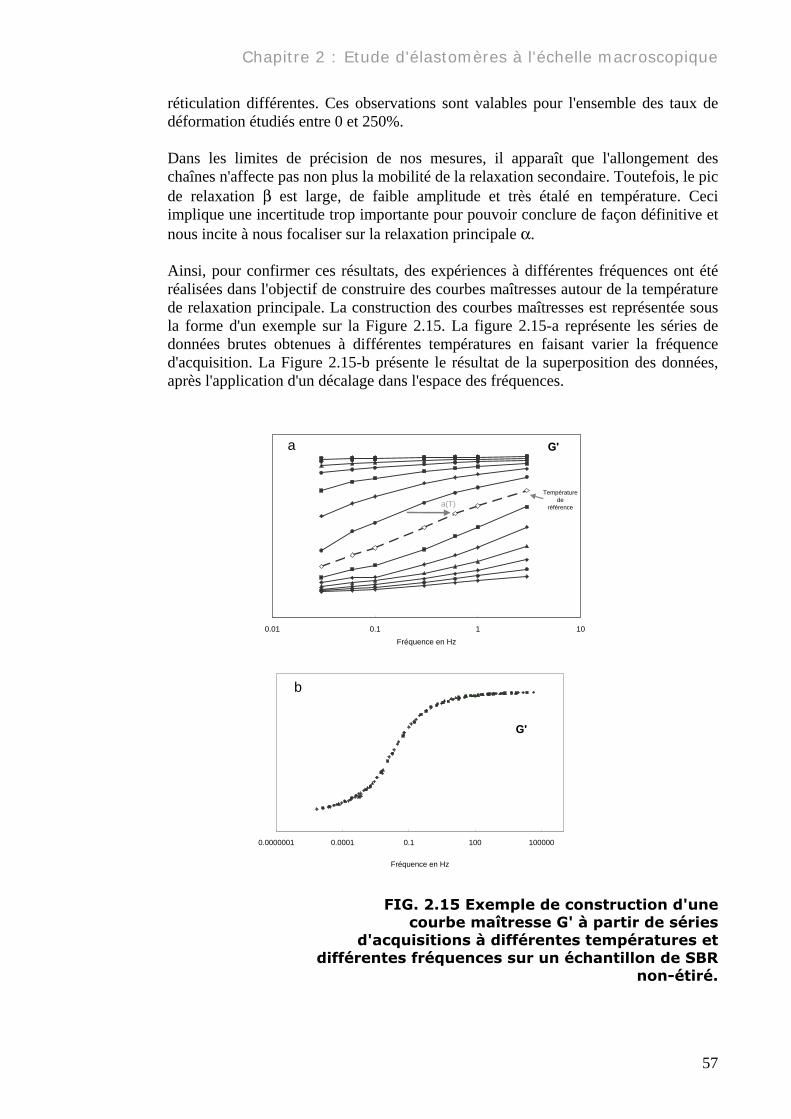
réticulation différentes. Ces observations sont valables pour l'ensemble des taux de déformation étudiés entre 0 et 250%. Dans les limites de précision de nos mesures, il apparaît que l'allongement des chaînes n'affecte pas non plus la mobilité de la relaxation secondaire. Toutefois, le pic de relaxation β est large, de faible amplitude et très étalé en température. Ceci implique une incertitude trop importante pour pouvoir conclure de façon définitive et nous incite à nous focaliser sur la relaxation principale α. Ainsi, pour confirmer ces résultats, des expériences à différentes fréquences ont été réalisées dans l'objectif de construire des courbes maîtresses autour de la température de relaxation principale. La construction des courbes maîtresses est représentée sous la forme d'un exemple sur la Figure 2.15. La figure 2.15-a représente les séries de données brutes obtenues à différentes températures en faisant varier la fréquence d'acquisition. La Figure 2.15-b présente le résultat de la superposition des données, après l'application d'un décalage dans l'espace des fréquences.
0.01 0.1 1 10Fréquence en Hz
a(T)
a G'
Température de
référence
0.0000001 0.0001 0.1 100 100000
Fréquence en Hz
b
G'
FIG. 2.15 Exemple de construction d'une courbe maîtresse G' à partir de séries
d'acquisitions à différentes températures et différentes fréquences sur un échantillon de SBR
non-étiré.
Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
58
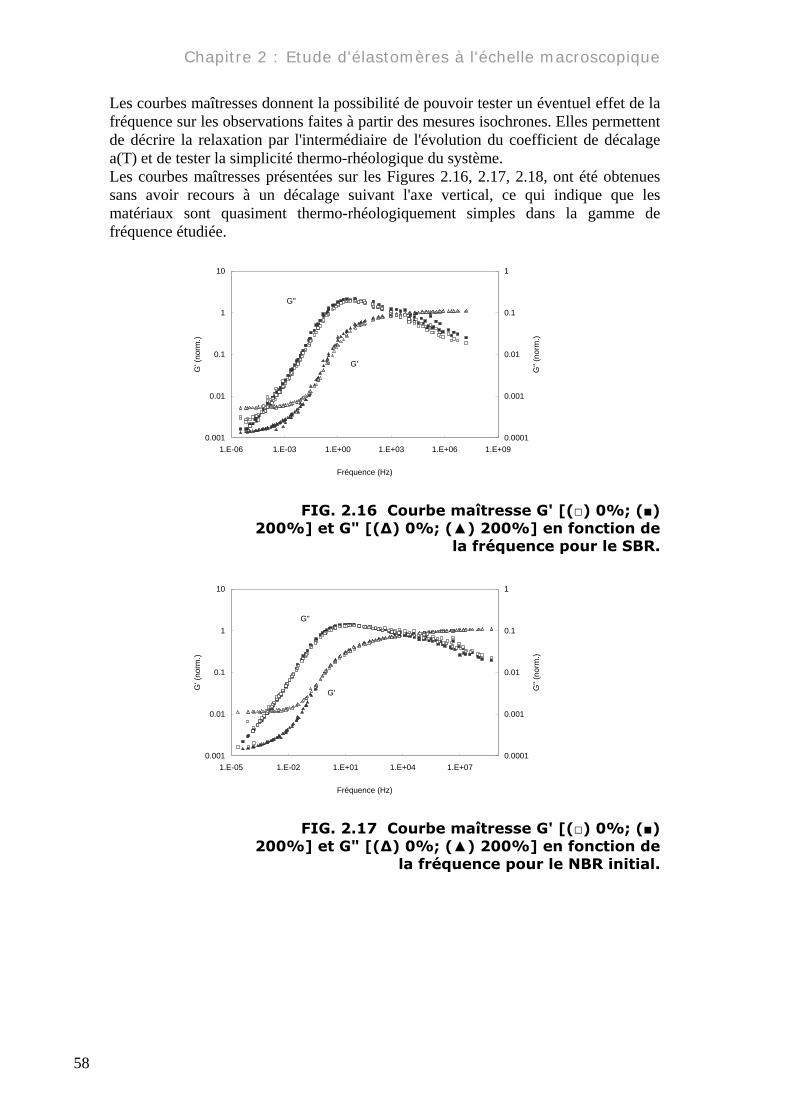
Les courbes maîtresses donnent la possibilité de pouvoir tester un éventuel effet de la fréquence sur les observations faites à partir des mesures isochrones. Elles permettent de décrire la relaxation par l'intermédiaire de l'évolution du coefficient de décalage a(T) et de tester la simplicité thermo-rhéologique du système. Les courbes maîtresses présentées sur les Figures 2.16, 2.17, 2.18, ont été obtenues sans avoir recours à un décalage suivant l'axe vertical, ce qui indique que les matériaux sont quasiment thermo-rhéologiquement simples dans la gamme de fréquence étudiée.
0.001
0.01
0.1
1
10
1.E-06 1.E-03 1.E+00 1.E+03 1.E+06 1.E+09
Fréquence (Hz)
G' (
norm
.)
0.0001
0.001
0.01
0.1
1
G" (
norm
.)
G"
G'
FIG. 2.16 Courbe maîtresse G' [(□) 0%; (■) 200%] et G" [(∆) 0%; (▲) 200%] en fonction de
la fréquence pour le SBR.
0.001
0.01
0.1
1
10
1.E-05 1.E-02 1.E+01 1.E+04 1.E+07
Fréquence (Hz)
G' (
norm
.)
0.0001
0.001
0.01
0.1
1
G" (
norm
.)
G"
G'
FIG. 2.17 Courbe maîtresse G' [(□) 0%; (■) 200%] et G" [(∆) 0%; (▲) 200%] en fonction de
la fréquence pour le NBR initial.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
59
0.001
0.01
0.1
1
10
1.E-05 1.E-02 1.E+01 1.E+04 1.E+07
Fréquence (Hz)
G' (
norm
.)
0.0001
0.001
0.01
0.1
1
G" (
norm
.)
G"
G'
FIG. 2.18 Courbe maîtresse G' [(□) 0%; (■) 200%] et G" [(∆) 0%; (▲) 200%] en fonction de
la fréquence pour le NBR recuit.
Les courbes confirment l'influence de la déformation sur le module conservatif dans la gamme des basses fréquences et le fait qu'aucun décalage n'est observable sur le module de perte dans tous les cas envisagés. La déformation n'a pas d'influence sur la mobilité moléculaire sur une large gamme de fréquence, indépendamment de la microstructure ou de la densité de nœuds de réticulation. A partir des courbes d'Arrhénius présentées sur la Figure 2.19, qui présentent les valeurs du logarithme du temps de relaxation en fonction de 1000/T, il est possible de déduire les valeurs de l'énergie d'activation pour le NBR et le SBR. Ces courbes d'Arrhénius sont obtenues en considérant que ωτ=2πfτ=1 au maximum du pic de relaxation α.
-10
-5
0
5
10
15
20
3.4 3.6 3.8 4 4.2 4.4
1000/T (K-1)
log
τ
NBR initial
NBR recuit
SBR
Tg
Tg
FIG. 2.19 Diagramme d'Arrhénius représentant log τ en fonction de 1000/T. les flèches
représentent les valeurs de température de la transition α déterminée à une fréquence de 1Hz.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
60
L'alignement préférentiel des chaînes polymères dans la direction de lasollicitation n'a aucun effet sur les mouvements responsables de la relaxation α du matériau.
6 Discussion des résultats Il a été montré expérimentalement qu'une élongation uniaxiale d'élastomères réticulés induisait une orientation des chaînes macromoléculaires, mais n'avait pas d'effet sur la densité d'arrangement du système. Ces changements d'entropie de conformation n'ont pas d'effet sur la mobilité moléculaire, et ceci, indépendamment de la nature chimique des chaînes et de la densité de réticulation du matériau.
6.1 La littérature et la déformation sous Tg Il existe dans la littérature un certain nombre d'études sur l'effet d'un étirement au-dessous de la température de transition vitreuse qui ont conduit à des résultats contradictoires. Spiess, par le biais de mesure de RMN, a étudié l'orientation d'échantillons de polycarbonate Bisphenol-A amorphe étirés sous Tg. Il a ainsi pu montrer que l'étirement à froid menait à une immobilisation partielle de la mobilité des groupes phényls, du fait d'une densité d'arrangement des chaînes plus grande [WEI95]. L'évolution des processus de relaxation lents en fonction des états iso-configurationnels obtenus par déformation sous Tg a également fait l'objet d'étude par spectroscopie mécanique à basse fréquence [GAU00]. Les caractéristiques des relaxations vitreuses sont peu modifiées par la déformation anélastique, mais celle-ci induit une forte augmentation de mobilité sur la partie basse température du pic de relaxation principale α. Cette question sera évoquée en détail au cours du chapitre 4 de ce travail.
6.2 La littérature et la déformation au-dessus de Tg La littérature est moins riche en ce qui concerne les effets de l'orientation des chaînes sur la mobilité, lorsque le matériau est étiré au-dessus de la transition vitreuse. Shelby a étudié ces effets par l'intermédiaire de l'évolution des paramètres de vieillissement dans un polycarbonate Bisphenol-A étiré au-dessus de Tg puis refroidi dans l'état vitreux [SHE01], [SHE98-1]. Les mesures de vitesse de relaxation volumique et de fluage ont suggéré que l'orientation induite par un étirement dans le plateau caoutchoutique conduisait à une diminution significative de la mobilité α en parallèle avec une augmentation de la mobilité des mouvements associés à la relaxation β. Pour interpréter ces résultats, il est suggéré que l'alignement des chaînes provoque une augmentation de la taille des domaines de coopérativité des processus α, et une augmentation de la taille des zones de volume libre dans lesquelles les segments reliés au processus β relaxent.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
61
En ce qui concerne les résultats de spectrométrie mécanique, deux exemples ressortent de la littérature. Le premier concerne les travaux de Frosini, qui consiste en des mesures de module en oscillations libres [FRO69]. Dans son dispositif expérimental, il utilise un poids mort pour déformer les échantillons. Ceci implique qu'il ne peut s'assurer d'une déformation constante au cours du refroidissement et de l'augmentation du module qui lui est associée. Il a observé que l'alignement des chaînes conduisait à une augmentation des barrières énergétiques opposées aux mouvements, et un élargissement de la distribution des temps de relaxation. Dans des conditions expérimentales beaucoup plus proches de la présente étude, Diaz-Calleja a observé un décalage du pic de relaxation α vers les basses températures avec l'étirement [DIA86], [DIA88]. L'opposition entre nos observations et ces résultats de la littérature nous amène à la discussion qui suit.
6.3 Les résultats de l'étude
Il est important tout d'abord, de mentionner que l'utilisation d'un matériau réticulé est le seul moyen de maintenir les changements d'entropie induits par la déformation et d'atteindre un état d'équilibre avant le refroidissement du système sous Tg.
6.3.1 D'un point de vue thermodynamique Dans un premier temps, les résultats de diffusion des RX n'ont pas montré de changement significatif de la densité d'arrangement des chaînes. Le coefficient de Poisson de nos matériaux vaut 0,5 au-dessus de Tg. Dans le cadre de la théorie du volume libre qui semble bien s'appliquer au-dessus de la zone de transition vitreuse, si aucune variation de volume n'intervient au cours de la déformation, aucune variation de la mobilité n'est à prévoir. L'absence de variation de la densité d'arrangement est à relier à la théorie de l'élasticité caoutchoutique, déjà très largement discutée dans la littérature et qui traite des modifications d'enthalpie et d'entropie de conformation d'un système sous élongation [MOS87], [GOD81]. Dans un second temps, il a été montré que la déformation conduisait à une modification significative de l'entropie de conformation à travers l'orientation des chaînes macromoléculaires. En considérant le caractère grande échelle des mouvements impliqués dans la relaxation α, il serait légitime d'envisager qu'une modification de l'orientation des chaînes eût un impact sur ses caractéristiques. Les expériences de diffusion de RX ont mis en évidence une augmentation du facteur d'Herman avec la déformation. De plus en considérant une large distribution des longueurs de chaînes entre nœuds de réticulation, il apparaît très probable que ce facteur représente une valeur moyenne entre des zones faiblement orientées et des zones qui le sont très fortement. Malgré l'existence de zones très fortement orientées, aucune évolution des caractéristiques du pic α n'est constatée. Il apparaît donc que la modification de l'entropie de conformation résultant de l'alignement des chaînes induit par la déformation uniaxiale n'a aucun impact sur la mobilité moléculaire.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
62
6.3.2 Discussion sur les paramètres d'activation L'objectif de cette discussion est d'établir s'il existe un lien possible entre l'entropie de conformation et l'entropie d'activation et si la déformation a un impact sur les paramètres d'activation. Dans le formalisme de Starkweather, une distinction est faite entre le facteur pré-exponentiel (lié à l'entropie d'activation) indicateur de la complexité de la relaxation et la part d'énergie interne reliée aux barrières énergétiques opposées aux mouvements moléculaires. Il est possible d'établir un lien entre l'enthalpie d'activation et l'entropie d'activation [STA91-1], [STA81]:
1 ln ln2a a
k TE RT T Sh fπ
⎡ ⎤⎛ ⎞⎛ ⎞= + + + ∆⎢ ⎥⎜ ⎟⎜ ⎟⎝ ⎠ ⎝ ⎠⎣ ⎦
Avec a aE H RT= ∆ + Le calcul de l'énergie d'activation de la relaxation donne accès à l'entropie d'activation et permet de déterminer son degré de coopérativité. En suivant les arguments développés par Starkweather, il est possible de déterminer l'entropie d'activation:
.exp .a a theoa
E ES
T−
∆ =
Dans cette expression Ea.exp est l'énergie d'activation déterminée à partir du diagramme d'Arhénius et Ea.theo est l'énergie d'activation théorique décrite par Starkweather, en considérant une entropie d'activation nulle pour la relaxation considérée:
[ ]. 22,922 ln( )a theoE RT T= + Avec T la température au maximum du module de perte obtenu à 1Hz. Les différents résultats concernant les variations d'entropie sont rassemblés dans le tableau 2.5:
Tableau 2.5: Synthèse des résultats concernant les entropies de conformation et d'activation des
différents matériaux.
∆Sactivation (kJ.K-1.mol-1)
∆Sconformation (J.K-1.mol-1)
Tα à 1Hz (K)
SBR initial 0,7 3,3.10-15 263 NBR initial 0,5 2,8.10-15 249 NBR recuit 0,5 5,8.10-15 249
Aucune comparaison n'est possible entre les valeurs d'entropie d'activation obtenues par l'analyse de Starkweather et les valeurs d'entropie de conformation obtenues à l'aide d'une forme simple de la théorie de l'élasticité caoutchoutique. Il apparaît qu'il n'y a pas de connexion directe possible entre l'entropie d'activation et l'entropie de conformation. Les valeurs déraisonnablement grandes de l'entropie d'activation sont le symbole du caractère coopératif de la relaxation α. Les valeurs sont du même ordre de grandeur que celles obtenues par ailleurs par Starkweather de 2kJ.K-1.mol-1 [STA91-2], mais ne peuvent pas être interprétées directement de façon physique, si ce n'est au moyen de

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
63
mouvements corrélés. L'analyse de Starkweather ne donne que des valeurs apparentes sur la base d'une hypothèse de mouvements microscopiques indépendants. Cette supposition n'est pas réaliste en ce qui concerne les polymères amorphes près de leur température de transition vitreuse. Enfin, il est possible de conclure que la déformation uniaxiale n'a pas d'effet ni sur la coopérativité de la relaxation principale α, ni sur les mouvements impliqués dans cette relaxation.
6.3.3 Une contradiction avec la théorie de Gibbs et DiMarzio? La théorie de Gibbs et DiMarzio prédit une transition vitreuse thermodynamique lorsque l'entropie de conformation est nulle [DIM64]. Du point de vue de l'entropie de conformation, une chaîne orientée doit avoir une entropie plus faible et la température de transition vitreuse effective d'échantillons orientés doit être augmentée. Les mouvements associés à la transition α sont aussi associés à la transition vitreuse aux basses fréquences. La théorie de Gibbs et DiMarzio prédit donc que sous l'effet d'un étirement, Tg, tout comme Tα, augmente [SHE98-2]:
( )( ) 2
( 1) 0
exp 2 32
g
g p
T GT C T
λ
λ
λ λ=
⎡ ⎤= + −⎢ ⎥∆⎢ ⎥⎣ ⎦
Avec λ le taux d'étirement uniaxial; G le module de cisaillement à la température T0 considérée et ∆Cp la variation de capacité calorifique entre l'état liquide et l'état vitreux. Les résultats de cette étude sont en désaccord avec ce modèle, mais les simulations par dynamique moléculaire effectuées sur des chaînes de SBR orientées peuvent aider à comprendre ce désaccord (Partie A-II). Les simulations donnent une vision plus précise des changements qui se produisent à l'échelle moléculaire au cours de l'étirement de l'élastomère et comment ces changements peuvent affecter les processus de relaxation. Il est généralement accepté que les processus de réorientation de chaînes peuvent être séparés en deux composantes. La première composante est due aux interactions inter-moléculaires et la seconde concerne les interactions intra-moléculaires. Les résultats de simulation suggèrent que les interactions inter-moléculaires ne sont pas modifiées de façon significative par l'étirement des chaînes. Ce résultat pourrait expliquer que les dépendances en temps et en température des processus de relaxation à grande échelle ne sont pas modifiés par l'orientation. Au contraire l'orientation et l'augmentation de la rigidité des chaînes influencent de façon significative les interactions intra-moléculaires. Les mouvements de vibration à petite échelle sont favorisés par l'alignement des chaînes. Par ailleurs, certains auteurs ont essayé d'établir un lien entre l'entropie d'activation et l'entropie de conformation à partir de résultats expérimentaux. Ainsi Diaz-Calleja a interprété ses observations sur la base du concept de température de compensation et a argumenté en faveur d'une diminution du nombre de mécanismes visco-élastiques avec l'allongement [DIA86], [DIA88]. L'effet de compensation s'exprime d'après lui par l'opposition entre une augmentation des barrières opposées aux mouvements browniens, une augmentation de l'entropie d'activation et une diminution de l'entropie de conformation. Nos observations quant à elles, s'opposent à toute mise en relation directe des deux types d'entropie.

Chapitre 2 : Etude d'élastomères à l'échelle macroscopique
64
7 Conclusion
La déformation uniaxiale au-dessus de Tg effectuée sur un SBR et deux NBR avec des densités de réticulation différentes, conduit à une modification de l'arrangement des chaînes. Ceci a été mis en évidence par l'intermédiaire de mesures de diffusion des rayons-X, ainsi qu'à l'aide d'une version simple de la théorie de l'élasticité caoutchoutique. Les modifications d'entropie de conformation du système dans son état d'équilibre peuvent être figées lors du passage dans l'état vitreux jusqu'à des températures inférieures à la transition β, grâce aux noeuds de réticulation au sein des matériaux. En considérant le bon indicateur de la mobilité moléculaire (G"), il a été démontré qu'une modification de l'entropie de conformation n'a pas d'effet sur la mobilité moléculaire. De plus, il apparaît qu'une distinction claire s'impose entre le paramètre thermodynamique "entropie de conformation" et le paramètre de relaxation "entropie d'activation". Finalement, ces observations amènent à une contradiction apparente avec le modèle de Gibbs et DiMarzio. Les modèles théoriques tels que la théorie du volume libre ne prévoient aucune modification de la mobilité moléculaire sans modification de volume. Ceci est conforme à nos observations expérimentales. Dans la mesure où une modification de l'entropie de conformation n'a pas d'effet sur la mobilité moléculaire associée à la relaxation principale, il semble que le paramètre énergétique prédomine pour expliquer les modifications de mobilité après déformation sous Tg (cf. chapitre 4). D'autre part, l'impossibilité de statuer à partir d'observations expérimentales, sur l'impact de l'alignement des chaînes sur la relaxation β et l'ensemble des mouvements locaux, nous pousse à mettre en place une étude de simulation par dynamique moléculaire et à considérer l'impact des interactions inter et intra-moléculaires sur la conformation de la chaîne.
La contradiction apparente entre les résultats expérimentaux et la théorie de Gibbs et Dimarzio nécessite d'étendre l'étude à une échelle plus locale.