
UNIVERSITE OUAGA I Pr Joseph KI-ZERBO ---------------
École Doctorale Sciences et Technologies
--------------- Laboratoire de Biologie et de Génétique
Moléculaires (LABIOGENE)
Thèse de Doctorat Unique (PhD)
Présentée par
OUATTARA Abdoul Karim
Pour obtenir le grade de
Docteur de l’Université Ouaga I Professeur Joseph KI-ZERBO
Option : Sciences Appliquées
Spécialité : Biologie Moléculaire/Génétique
Polymorphisme du gène de la Glucose-6-phosphate
déshydrogénase et contrôle génétique des formes
asymptomatique et symptomatique de l’infection à
Plasmodium falciparum au Burkina Faso
Soutenue le, 07 Mars 2017 devant le jury composé de :
Président : David MODIANO, Professeur Titulaire, Université de Rome I, La Sapienza
Membres :
Simon A. AKPONA, Professeur Titulaire, Université de Parakou, Bénin (Rapporteur)
Jean Didier ZONGO, Professeur Titulaire, Université Ouaga I Pr Joseph KI-ZERBO (Rapporteur)
Yves TRAORE, Professeur Titulaire, Université Ouaga I Pr Joseph KI-ZERBO
Jacques SIMPORE, Professeur Titulaire, Université Ouaga I Pr Joseph KI-ZERBO (Directeur de thèse)
Simplice Damintoti KAROU, Maître de conférences, Université de Lomé, Togo

Thèse de Doctorat Unique OUATTARA Abdoul Karim i
DEDICACES
Je dédie ce travail :
A mon père Souleymane OUATTARA et à ma mère Biba DOUMBIA
A mes frères et sœur Ramata, Cheick Ahmed et Sehnia Kahdyatou
A mes cousins Diakalia OUATTARA et Tchèdjan TRAORE
Madeleine TRAORE, Léa pépin GARANE, Jocelyne
OUATTARA
tous ceux ou toutes celles qui ont accepté librement et
volontairement de participer à cette étude

Thèse de Doctorat Unique OUATTARA Abdoul Karim ii
REMERCIEMENTS
Ce travail a été réalisé au Laboratoire de Biologie Moléculaire et de Génétique (LABIOGENE de
l’UFR-SVT), au Centre de Recherche Biomoléculaire Pietro Annigoni (CERBA) et à l’Hôpital
Saint Camille de Ouagadougou (HOSCO).
Je tiens à exprimer ma profonde gratitude à mon Directeur de thèse, le Pr Jacques
SIMPORE, Professeur titulaire de Biologie moléculaire et de génétique (Université Ouaga
I Pr Joseph KI-ZERBO) Responsable du Laboratoire de Biologie Moléculaire et de Génétique
(LABIOGENE) labélisé Centre d’Excellence UEMOA, Directeur du Centre de Recherche
Biomoléculaire Pietro Annigoni (CERBA) érigé en Laboratoire National de Référence pour le
HPV (LNR-HPV), Recteur de l’Université Saint Thomas d’Aquin (USTA), Officier de l’ordre
National, Officier des Palmes Académiques, Chevalier international des Palmes Académiques du
CAMES, Membre de l’Académie Africaine des sciences, des arts et des lettres du Burkina Faso,
Membre de l’Académie Africaine des Sciences (AAS); Membre du Comité d’éthique et de
déontologie du CAMES. Merci pour ce thème de recherche pertinent que vous m’avez proposé,
pour votre encadrement exceptionnel, vos précieux conseils, vos encouragements et votre
disponibilité malgré vos multiples fonctions ;
Je remercie le Pr David MODIANO (Rapporteur), Professeur titulaire de
parasitologie (Université de Rome I, La Sapienza), pour avoir accepté de lire, critiquer, instruire
et présidé le jury de cette Thèse. Merci pour votre contribution à l’amélioration de ce document.
Trouvez ici l’expression de toute ma gratitude.
Je remercie le Pr SIMON A. AKPONA (Rapporteur), Professeur titulaire de
Biochimie (Université de Parakou, Bénin), pour avoir accepté d’instruire et de faire partie du
jury de cette thèse. Vous avez accepté de suivre et de juger ce travail malgré vos multiples
occupations. Recevez toute notre profonde gratitude.
Je remercie le Pr Jean-Didier ZONGO (Rapporteur), Professeur titulaire de
génétique et amélioration des plantes (Université Ouaga I Pr Joseph KI-ZERBO), pour avoir
accepté de lire, critiquer et instruire et de faire partie du jury pour juger cette Thèse. Merci pour
votre contribution à l’amélioration de ce document. Trouvez ici l’expression de toute ma
gratitude.
Je remercie le Professeur Yves TRAORE, Professeur Titulaire en Immunologie
(Université Ouaga I Pr Joseph KI-ZERBO), pour avoir accepté de faire partie du jury. Vous avez
accepté de suivre et de juger ce travail malgré vos multiples occupations. Recevez toute notre
profonde gratitude.

Thèse de Doctorat Unique OUATTARA Abdoul Karim iii
Je remercie le Dr Simplice Damintoti KAROU, Maître de Conférences en
Biochimie-Microbiologie (Université de Lomé, Togo), pour avoir accepté de faire partie du
jury. Vous avez accepté de suivre et de juger ce travail malgré vos multiples occupations. Recevez
toute notre profonde gratitude.
Je remercie les Dr Diane Djénéba OUERMI, Wenkuuni Florencia DJIGMA, Issoufou
TAO, Théodora Mahoukèdè ZOHONCON, Tegwindé Rébéca COMPAORE, Birama
DIARRA pour leurs contributions à l’amélioration de la qualité de ce document et
particulièrement le Dr Cyrille BISSEYE pour son soutien inconditionnel, sa compréhension et
ses encouragements pendant toutes ces années ;
Je remercie le Père Albert Théophane YONLI, Rémy MORET, Bapio Valérie Jean
Télesphore Elvira BAZIE, Pouiré YAMEOGO et toute l’équipe du CERBA/LABIOGENE et de
l’HOSCO, pour leur disponibilité lors de la collecte de nos échantillons.
Je remercie Ragnagnewendé Serge Théophile SOUBEIGA, Maléki ASSIH, Lassina
TRAORE, Damehan TCHELOUGOU et tous les camarades du Master de BioGeMA toute
promotion confondue pour leurs soutiens lors des manipulations et/ou pendant la rédaction
de cette thèse ainsi que pour les remarques et suggestions constructives à chaque fois que c’était
nécessaire.
La réalisation de l’ensemble des travaux de la présente thèse a été rendue possible grâce
à l’appui financier de la Bourse Nationale Burkinabè de 3ème cycle octroyée par le Centre
national de l’Information, de l’Orientation Scolaire et Professionnelle et des Bourses (CIOPB)
et des partenaires techniques et financiers du CERBA.LABIOGENE à savoir :
L’Union Economique et Monétaire Ouest Africaine (UEMOA) à travers son programme
d'appui et de développement des centres d'excellence régionaux et son soutien à la formation et
la recherche de l’excellence. Que ces donateurs soient assurés de mes sincères remerciements.
La Conférence Épiscopale Italienne (C.E.I.) pour le soutien financier dans l’achat des
multiples réactifs pour les travaux de cette thèse.
A tous mes camarades, frères et amis pour vos encouragements et votre indéfectible
soutien. Merci à tous !

Thèse de Doctorat Unique OUATTARA Abdoul Karim iv
TABLE DES MATIERES
DEDICACES .............................................................................................................................. i
REMERCIEMENTS .................................................................................................................. ii
TABLE DES MATIERES ........................................................................................................ iv
LISTE DES FIGURES .............................................................................................................. ix
LISTE DES TABLEAUX ......................................................................................................... xi
SIGLES ET ABREVIATIONS ................................................................................................ xii
RESUME ................................................................................................................................. xiv
ABSTRACT ............................................................................................................................ xiv
INTRODUCTION ...................................................................................................................... 1
OBJECTIFS DE LA THESE ..................................................................................................... 4
Objectif principal .................................................................................................................... 4
Objectifs spécifiques ............................................................................................................... 4
CHAPITRE I : GENERALITES SUR LE PALUDISME ......................................................... 5
I.1 Définition et Historique ........................................................................................................ 5
I.1.1 Définition ....................................................................................................................... 5
I.1.2 Historique ....................................................................................................................... 5
I.2 Epidémiologie du paludisme ................................................................................................. 6
I.2.1 Estimations mondiales .................................................................................................... 6
I.2.2 Répartition géographique ............................................................................................... 7
I.3 Cycle de vie de Plasmodium falciparum .............................................................................. 8
I.3.1 Le vecteur ....................................................................................................................... 8
I.3.2 L’agent pathogène .......................................................................................................... 9
I.3.3 Phase asexué chez l’homme ......................................................................................... 10
I.3.4 Phase sexuée chez le moustique ou sporogonie ........................................................... 13
I.4 Aspect génétique de Plasmodium falciparum ..................................................................... 14
I.4.1 Organisation du génome ............................................................................................... 14

Thèse de Doctorat Unique OUATTARA Abdoul Karim v
I.4.2 Architecture nucléaire des gènes var ............................................................................ 14
I.5 Manifestations cliniques du paludisme ............................................................................... 16
I.5.1 Accès palustre simple ................................................................................................... 17
I.5.2 Accès palustre grave ..................................................................................................... 17
I.5.3 Physiopathologie du paludisme .................................................................................... 17
I.6 Paludisme et immunité ........................................................................................................ 21
I.7 Prise en charge thérapeutique ............................................................................................. 22
I.7.1 Les traitements antipaludiques ..................................................................................... 22
I.7.2 Résistances aux médicaments antipaludiques .............................................................. 22
I.7.2.1 Facteurs liés à la résistance aux antipaludiques ..................................................... 23
I.7.2.2 Les marqueurs moléculaires de résistance ............................................................. 24
I.8 Paludisme et vaccin ............................................................................................................. 26
I.9 Diagnostic biologique du paludisme ................................................................................... 28
I.9.1 Diagnostic microscopique direct par frottis sanguin et goutte épaisse ........................ 28
I.9.2 Détection d’Antigènes palustres par tests Rapide d’Orientation diagnostic (TROD) . 29
I.9.3 Détection des acides nucléiques par les techniques d’amplification génique .............. 29
CHAPITRE II : HEMOGLOBINOPATHIES ET DEFICIT EN G-6-PD ............................... 30
II.1 Les hémoglobinopathies .................................................................................................... 30
II.1.1 Définitions ................................................................................................................... 30
II.1.2 L’hémoglobinose S ..................................................................................................... 31
II.1.2.1 Epidémiologie ...................................................................................................... 31
II.1.2.2 Physiopathologie .................................................................................................. 32
II.1.3.3 Rapport entre l’hémoglobine S et le Paludisme ................................................... 34
II.1.4 L’hémoglobine C ........................................................................................................ 35
II.1.4.1 Epidémiologie ...................................................................................................... 35
II.1.4.2 Physiopathologie .................................................................................................. 35
II.1.4.3 Rapport entre l’hémoglobine C et le Paludisme ................................................... 36

Thèse de Doctorat Unique OUATTARA Abdoul Karim vi
II.1.5 Les techniques de diagnostic des hémoglobines anormales ....................................... 37
II.1.5.1 Les techniques d’électrophorèse et de chromatographie ...................................... 37
II.1.5.2 Les techniques de biologie moléculaire ............................................................... 37
II.2 Le déficit en G-6-PD ......................................................................................................... 38
II.2.1 Définition .................................................................................................................... 38
II.2.2 Historique .................................................................................................................... 38
II.2.3 Epidémiologie ............................................................................................................. 39
II.2.4 Manifestations cliniques ............................................................................................. 41
II.2.5 Aspects génétiques ...................................................................................................... 42
II.2.6 Polymorphismes de la G-6-PD ................................................................................... 43
II.2.7 Rôle de la glucose-6-phosphate déshydrogénase ........................................................ 45
II.2.8 Déficit en G-6-PD et paludisme .................................................................................. 47
II.2.9 Techniques de diagnostic de la déficience en G-6-PD ............................................... 48
II.2.9.1 Génotypage par PCR/RFLP ................................................................................. 49
II.2.9.2 Génotypage par PCR en temps réel ...................................................................... 49
CHAPITRE III. MATERIEL ET METHODES ...................................................................... 50
III.1 Cadre d’étude ................................................................................................................... 50
III.2 Sites de collecte ................................................................................................................ 50
III.3 Méthodologie de la première étude .................................................................................. 51
III.3.1 Type et période d’étude ............................................................................................. 51
III.3.2 Population d’étude ..................................................................................................... 51
III.3.3 Echantillonnage ......................................................................................................... 52
III.3.3.1 Taille de l’échantillon ......................................................................................... 52
III.3.3.2 Prélèvements sanguins ........................................................................................ 52
III.3.4 Analyses biologiques ................................................................................................. 53
III.3.4.1 La numération formule sanguine ........................................................................ 53
III.3.4.2 L’électrophorèse de l’hémoglobine .................................................................... 53

Thèse de Doctorat Unique OUATTARA Abdoul Karim vii
III.3.5 Recherche des mutations responsables de la déficience en G-6-PD ......................... 54
III.3.5.1 Extraction de l’ADN ........................................................................................... 54
III.3.5.2 Génotypage par PCR en temps réel .................................................................... 55
III.3.5.3 Génotypage par PCR-RFLP ................................................................................ 56
III.4 Méthodologie de la deuxième étude ................................................................................. 58
III.4.1 Type et période d’étude ............................................................................................. 58
III.4.2 Population d’étude ..................................................................................................... 58
III.4.3 Echantillonnage ......................................................................................................... 58
III.4.3.1 Taille de l’échantillon ......................................................................................... 58
III.4.3.2 Prélèvements ....................................................................................................... 58
II.4.4 Détection de Plasmodium falciparum ......................................................................... 59
III.4.4.1 Le TROD SD Bioline Malaria Ag P.f/Pan .......................................................... 59
III.4.4.2 La goutte épaisse ................................................................................................. 59
III.4.5 Analyses biologiques ................................................................................................. 60
III.4.5.1 La numération formule sanguine ........................................................................ 60
III.4.5.2 L’électrophorèse de l’hémoglobine .................................................................... 60
III.4.6 Recherche des mutations responsables de la déficience en G-6-PD ......................... 61
III.4.6. 1 Extraction de l’ADN ....................................................................................... 61
III.4.4.2 Le génotypage par PCR classique ....................................................................... 61
III.6 L’analyse statistique ......................................................................................................... 64
III.7 Les considérations éthiques .............................................................................................. 64
CHAPITRE IV : RESULTATS ............................................................................................... 65
IV.1 Résultats des travaux de la première étude ...................................................................... 65
IV.1.1 Contexte et but de la première étude ......................................................................... 65
IV.1.2 Objectif de l’étude ..................................................................................................... 65
IV.1.3 Marqueurs génétiques et paludisme asymptomatique ............................................... 65
IV.1.3.1 Caractéristiques socio-démographiques ............................................................. 65

Thèse de Doctorat Unique OUATTARA Abdoul Karim viii
IV.1.3.2 Prévalence de la déficience en G-6-PD et des génotypes de l’hémoglobine ...... 66
IV.1.3.3 Corrélation entre les polymorphismes des gènes G-6-PD et HBB et le paludisme
asymptomatique ................................................................................................................ 68
IV.1.3.4 Conclusion partielle ............................................................................................ 71
IV.2 Résultats des travaux de la deuxième étude ..................................................................... 72
IV.2.1 Contexte et but de la deuxième étude ........................................................................ 72
IV.2.2 Objectif de la deuxième étude ................................................................................... 72
IV.2.3 Marqueurs génétiques et paludisme symptomatique ................................................. 72
IV.2.3.1 Caractéristiques sociodémographiques et cliniques ........................................... 72
IV.2.3.2 Prévalence du déficit en G-6-PD et des génotypes de l’hémoglobine ................ 73
IV.2.3.3 Corrélation entre les polymorphismes des gènes G-6-PD et HBB et le paludisme
symptomatique .................................................................................................................. 74
CHAPITRE V : DISCUSSION GENERALE .......................................................................... 76
V.1 Marqueurs génétiques et Paludisme asymptomatique ................................................... 76
V.2 Marqueurs génétiques et Paludisme symptomatique ..................................................... 80
Conclusion générale .............................................................................................................. 84
Perspectives .............................................................................................................................. 85
VI. REFERENCES BIBLIOGRAPHIQUES ........................................................................... 86
ANNEXES ............................................................................................................................... 88

Thèse de Doctorat Unique OUATTARA Abdoul Karim ix
LISTE DES FIGURES
Figure 1 : Fréquence des différents SNPs G-6-PDA- en Afrique ............................................. 2
Figure 2 : Différentes zones de transmission du paludisme dans le monde .............................. 6
Figure 3 : Anopheles gambiae ................................................................................................... 9
Figure 4 : Processus d’infection des hépatocytes par les sporozoïtes de Plasmodium ........... 11
Figure 5 : Processus d’invasion des érythrocytes par les mérozoïtes de Plasmodium falciparum
.................................................................................................................................................. 12
Figure 6 : Cycle de vie de Plasmodium falciparum ................................................................ 13
Figure 7 : Organisation des gènes var de Plasmodium falciparum ......................................... 15
Figure 8 : Caractéristiques des gènes var ................................................................................ 16
Figure 9 : Les bases moléculaires de la cytoadhérence ........................................................... 18
Figure 10 : Séquestration des hématies parasitées .................................................................. 19
Figure 11 : L’antigène vaccinal RTS,S. .................................................................................. 28
Figure 12 : Distribution mondiale de l’hémoglobine S ........................................................... 32
Figure 13 : Mécanisme physiopathologique de base de la drépanocytose. ............................. 33
Figure 14 : Répartition de l’hémoglobine C dans le monde .................................................... 35
Figure 15 : Prévalence du déficit en G-6-PD et distribution mondiale des différents variants
déficitaires ................................................................................................................................ 40
Figure 16 : Organisation du gène G-6-PD humain .................................................................. 42
Figure 17 : Expression de la déficience en G-6-PD chez les hommes (A) et inactivation
aléatoire du chromosome X chez les femmes (B). ................................................................... 43
Figure 18 : Répartition des mutations fréquentes le long de la séquence codante du gène G6PD
.................................................................................................................................................. 45
Figure 19 : Rôle de la G-6-PD dans la prévention du stress oxydatif ..................................... 46
Figure 20 : Relation triangulaire entre la primaquine, la G-6-PD et Plasmodium falciparum 47
Figure 21 : Situation géographique de Koubri et différents districts sanitaires de la ville de
Ouagadougou adapté de ........................................................................................................... 51
Figure 22 : Automate CELL-DYN Ruby ................................................................................ 53
Figure 23 : Appareil d’électrophorèse de type HELENA ....................................................... 54
Figure 24 : Appareil 7500 Fast Real Time PCR system (Applied Biosystems, USA) ........... 55

Thèse de Doctorat Unique OUATTARA Abdoul Karim x
Figure 25 : Thermocycleur GeneAmp PCR system 9700 (Applied Biosystems, USA) ......... 57
Figure 26 : Une goutte épaisse ................................................................................................ 59
Figure 27 : Automate ABX micro 60 ...................................................................................... 60
Figure 28 : Cuve et générateur pendant la migration .............................................................. 61
Figure 29 : Dispositif d’électrophorèse des amplicons G-6-PD (BioRad, USA).................... 62
Figure 30 : GENEFLASH ....................................................................................................... 63
Figure 31 : Table d’interprétation des bandes après l’électrophorèse sur gel d’agarose ........ 64
Figure 32 : Résultats du génotypage de la mutation G202A par PCR en temps reel .............. 66
Figure 33 : Fragments G680T et T968C après amplification PCR ......................................... 67
Figure 34 : Les différentes bandes après l’électrophorèse ...................................................... 73

Thèse de Doctorat Unique OUATTARA Abdoul Karim xi
LISTE DES TABLEAUX
Tableau I : Classification OMS des variants enzymatiques de la G-6-PD ............................. 41
Tableau II : Prévalence des 4 combinaisons de mutations responsables de la déficience en G-
6-PD ......................................................................................................................................... 67
Tableau III : Répartition des génotypes de l’hémoglobine selon l’âge, le sexe et le statut G-6-
PD ............................................................................................................................................. 68
Tableau IV : Prévalence de l’infection à Plasmodium falciparum et moyenne géométrique de
la parasitémie chez les personnes infectées en fonction de la déficience en G-6-PD .............. 70
Tableau V : L’effet des génotypes de l’hémoglobine sur l’infection à Plasmodium falciparum
.................................................................................................................................................. 71
Tableau VII : Caractéristiques de la population d’étude selon les génotypes HBB et fréquence
des allèles HbS et HbC ............................................................................................................. 74

Thèse de Doctorat Unique OUATTARA Abdoul Karim xii
SIGLES ET ABREVIATIONS
µL : Microlitre
ADN : Acide Désoxyribonucléotide
AM-L : Artéméther-Lumefantrine
AQ : Amodiaquine
AS : Artésunate
CCMH : Concentration Corpusculaire Moyenne en Hémoglobine
CEI : Conférence Episcopale Italienne
CERBA : Centre de Recherche Biomoléculaire Pietro Annigoni
CIDR : Cysteine-rich Interdomain Region
CIOSPB : Centre national de l’Information, de l’Orientation Scolaire et Professionnelle et des Bourses
CLHP : Chromatographie Liquide Haute Performance
CR : Récepteur de Complément
CS : Circumsporozoïte
CSA : Chondroïtine Sulfate
CSPS : Centre de Santé Primaire et de Promotion Sociale
DARC : Duffy Antigen Receptor for Chemokines
DBL : Duffy binding Like
DC : Domaine Cassette
dL : Décilitre
ED/ST : Ecole Doctorale/Sciences et Technologies
EPCR : Endothelial Protein C Receptor
G-6-PD : Glucose-6-phosphate déshydrogénase
GPI : Glycosylphosphatidylinositol
GR : Globule Rouge
Hb : Hémoglobine
HOSCO : Hôpital Saint Camille de Ouagadougou
HS : héparanes sulfates
HSPG : héparanes Sulfates Protéoglycanes
Ig : Immunoglobuline

Thèse de Doctorat Unique OUATTARA Abdoul Karim xiii
IL : Interleukine
KAH-RP : Knob-Associated Histidine-Rich Protein
Kb : Kilobase
LABIOGENE : Laboratoire de Biologie Moléculaire et de Génétique
MAPSS : MultiAngle Polarized Scatter Separation
Mb : Mégabases
MESA : Mature P. falciparum-infected Erythrocytes Surface Antigen
mL : Millilitre
MSP : Merozoite Surface Protein
NADP : Nicotinamide Adénosine Dinucléotide Phosphate
NK : Natural Killer
OMS : Organisation Mondiale de la Santé
pb : Paire de Bases
PCR : Polymerase Chain Reaction
pfEMP1 : Plasmodium falciparum Erythrocyte Membrane Protein-1
pH : potentiel d'Hydrogène
pLDH : Plasmodium Lactate Déshydrogénase
RESA : Ring-infected Erythrocyte Surface Antigen
RFLP : Restriction Fragment Length Polymorphism
SDS : Sodium-Dodecyl-Sulfate
SNPs : Single Nucleotide Polymorphism
TBE : Tris-Borate-EDTA
TNF : Tumor Necrosis Factor
TRAP : Thrombospondin-Related Anonymous Protein
TROD : Test Rapide d’Orientation Diagnostic
TSP : Thrombospondine
UEMOA : Union Economique et Monétaire Ouest Africaine
UTR : UnTranslated Regions
UV : Ultra-Violet

Thèse de Doctorat Unique OUATTARA Abdoul Karim xiv
RESUME
Le déficit en G-6-PD et les anomalies de l’hémoglobine ont une fréquence relativement élevée
dans les zones d’endémicité palustre. Ils conféreraient une certaine protection contre les formes
sévères du paludisme bien que les mécanismes ne soient pas complètement élucidés. Dans cette
étude nous avons réalisé le génotypage de cinq (05) SNPs du gène G-6-PD chez des personnes
présentant des formes asymptomatique ou symptomatique du paludisme au Burkina Faso.
Deux cent personnes vivant dans une communauté rurale du Burkina Faso, où le paludisme est
endémique et 182 patients souffrant de paludisme ont fait l’objet d’un génotypage par PCR en
temps réel, PCR/RFLP, ou par PCR classique à l’aide du kit GENESPARK G6PD African pour
les mutations G202A, A376G, A542T, G680T et T968C impliquées dans le déficit en G-6-PD.
Les génotypes de l’hémoglobine ont été déterminés par électrophorèse chez 325 individus.
La prévalence du déficit en G-6-PD était de 9,7% (37/382). On notait une fréquence
significativement plus élevée d’hémizygotes que d’homozygotes pour le déficit (14,8% vs 5,3%
; p = 0,002). L’électrophorèse de l’hémoglobine a révélé 75,4% (245/325) d’individus HbAA
et 21,8 % (71/325) d’individus HbAC contre 4,6% (15/325) de sujets HbAS avec 1,2% (4/325)
d’individus double hétérozygote HbSC. Bien que les variants G-6-PD A- Betica Selma
(376G/968T) et Santamaria (376G/542T) aient été détectés dans cette étude, le variant
202A/376G a été retrouvé dans 94,6% des cas de déficit. La parasitémie asymptomatique à
Plasmodium falciparum était significativement plus élevée chez les personnes non déficientes
en G-6-PD comparativement aux personnes hémizygotes/homozygotes (p < 0,001) et celles
hétérozygotes (p < 0,001). Cependant, aucune corrélation entre le déficit en G-6-PD, les
hémoglobinoses et l’infection symptomatique du paludisme n’a été observée dans cette étude.
Ces travaux montrent que le variant G-6-PDA- 202A/376G est associé à la protection contre le
paludisme asymptomatique au Burkina Faso. Ils confirment que le portage des hémoglobines S
ou C ne protège pas contre les infections palustres. Contrairement aux études antérieures de
génotypage, elle démontre pour la première fois l’existence du variant A- Betica Selma
(376G/968C) au Burkina Faso et suggère une investigation plus approfondie pour déterminer à
l’échelle nationale la fréquence de cette mutation.
Mots clés : G-6-PD, hémoglobinopathies, paludisme, SNPs, Burkina Faso

Thèse de Doctorat Unique OUATTARA Abdoul Karim xv
ABSTRACT
The G-6-PD deficiency and hemoglobin abnormalities occur with relatively high frequencies
in malaria-endemic areas. They confer protection against severe forms of malaria although the
mechanisms are not completely understood. In this study we genotyped five (05) Single
Nucleotide Polymorphisms (SNPs) of the G-6-PD gene in people with asymptomatic or
symptomatic forms of malaria in Burkina Faso.
Two hundred (200) individuals living in a rural community in Burkina Faso, where malaria is
endemic and 182 malaria patients were genotyped by real-time PCR, PCR/RFLP or
conventional PCR using the kit GENESPARK G6PD African for mutations G202A, A376G,
A542T, G680T and T968C involved in the G-6-PD deficiency. Genotypes of hemoglobin have
been determined by electrophoresis in 325 individuals.
The prevalence of the G-6-PD deficiency was 9.7% (37/382). Hemizygous males were
significantly higher than homozygous females (14.8% vs. 5.3%, p = 0.002). Hemoglobin
electrophoresis revealed 75.4% (245/325) of HbAA individuals and 21.8% (71/325) of HbAC
subjects against 4.6% (15/325) of HbAS subjects and 1.2% (4/325) of double heterozygous
HbSC. Although the G-6-PD variants A- Santamaria (376G/542T) and Betica Selma
(376G/968T) were detected in this study, the 202A/376G variant was found in 94.6% of the
deficiency. Plasmodium falciparum asymptomatic parasitaemia was significantly higher in G-
6-PD non-deficients individuals compared to hemizygous/homozygous subjects (p <0.001) or
heterozygous females (p <0.001). However, there was no correlation between the G-6-PD
deficiency or haemoglobinopathies and symptomatic malaria infection in this study.
This study shows that the G-6-PDA -202A/376G variant is associated with protection against
asymptomatic malaria in Burkina Faso. As opposed to previous genotyping studies carried out
in Burkina Faso, it demonstrates for the first time the existence of the G-6-PDA- Betica Selma
(376G/968C) variant in Burkina Faso and suggests further investigation at the national level
and in specific ethnic groups to determine more accurately the frequency of that mutation.
Key words: G-6-PD, haemoglobinopathies, malaria, SNPs, Burkina Faso

INTRODUCTION

Thèse de Doctorat Unique OUATTARA Abdoul Karim 1
INTRODUCTION
Le paludisme demeure l’une des maladies parasitaires les plus mortelles dans le monde en dépit
des multiples efforts de prévention et de contrôle. Il est beaucoup plus répandu dans les pays
tropicaux et subtropicaux en développement. Les femmes enceintes notamment les primipares,
et les enfants de moins de cinq ans constituent les groupes les plus touchés par la morbidité et
la mortalité palustre. Selon l’Organisation mondiale de la santé (OMS), 198 millions de cas de
paludisme et 584 000 décès associés à la maladie ont été enregistrés au niveau mondial en 2013
(OMS, 2014b). Au Burkina Faso, le paludisme est fortement endémique avec une augmentation
de la transmission pendant la saison des pluies. On estime que plus de la moitié de tous les cas
de fièvres seraient attribuables au paludisme au cours de la saison des pluies (BISOFFI et al.,
2010). En 2013, le paludisme demeurait la principale cause de consultations (46,5%),
d’hospitalisations (61,5%) et de décès (30,5%) dans les formations sanitaires (Ministère de la
Santé/Système National d’Information Sanitaires du Burkina-Faso, 2013).
Le paludisme est également considéré comme la plus forte pression de sélection naturelle dans
l’histoire récente du génome humain. Le maintien de certaines anomalies génétiques comme le
déficit en Glucose-6-phosphate déshydrogénase (G-6-PD) et les hémoglobinopathies à des
fréquences relativement élevées, serait due à l’avantage sélectif qu’elles confèrent
généralement à l’état hétérozygote aux individus vivant en zone d’endémie palustre (HOWES
et al., 2013a).
L’hémoglobine S (HbS) responsable de la drépanocytose est sûrement l’hémoglobinopathie
grave la plus fréquente dans le monde (PIEL et al., 2015). La maladie affecterait quelques 20
à 25 millions de personnes dans le monde avec les plus fortes prévalences du trait
drépanocytaire observées en Afrique du fait de l’endémicité du paludisme (BOOTH et al.,
2010). Les individus porteurs de l’allèle HbS développent une anémie falciforme, ou
drépanocytose, dont les symptômes les plus courants sont des accidents vasculaires cérébraux,
des douleurs articulaires, des insuffisances rénales et hépatiques. Sans traitement, ce qui est
souvent le cas dans les pays à faibles revenus, la grande majorité des enfants drépanocytaires
(50 à 80%) meurent avant l’âge de 5 ans (PIEL et al., 2015). L’hémoglobine C (HbC) s’observe
essentiellement chez les populations d’ascendance noire. Le gène βc semble avoir son origine
dans la zone ouest de l'Afrique (PIEL et al., 2013b).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 2
Contrairement à la drépanocytose, l’HbC ne provoque pas de polymérisation intracellulaire
linéaire au niveau des zones intravasculaires à faible tension d'oxygène. La distribution
géographique du déficit en G-6-PD et l’analyse haplotypique du locus G-6-PD indiquent une
sélection positive récente sous la pression du paludisme (HOWES et al., 2013b). Le gène de la
G-6-PD présente une importante hétérogénéité allélique, qui se manifeste par des expressions
biochimiques et cliniques variables. En effet plusieurs variants génétiques du déficit en G-6-
PD avec des fréquences polymorphiques sont connus dans la sous-région Ouest africaine
(Figure 1). Parmi les déficits enzymatiques touchant la lignée érythrocytaire, le déficit en G-6-
PD reste le plus significatif de par sa fréquence (plus de 400 millions de personnes dans le
monde) et ses implications cliniques. Il s’agit d’une pathologie génétique liée au chromosome
X avec des expressions cliniques variables à l’état hétérozygote et pouvant poser de graves
problèmes, notamment dans le traitement du paludisme (ALLAHVERDIYEV et al., 2012).
Figure 1 : Fréquence des différents SNPs G-6-PDA- en Afrique (HOWES et al., 2013b)

Thèse de Doctorat Unique OUATTARA Abdoul Karim 3
En effet, la prise de certains médicaments (dapsone, cotrimoxazole, primaquine...), la
consommation de certains aliments (fèves), et une variété d’infections (hépatites, fièvre
typhoïde, paludisme) entraînent chez les personnes déficientes en G-6-PD une anémie (baisse
du taux de globule rouge dans le sang) dite hémolytique (destruction des globules rouges)
d’intensité et de gravité variables, nécessitant parfois des transfusions sanguines en urgence
(HOWES et al., 2013a).
Il a été observé que la sévérité et la fréquence du déficit enzymatique variait d'un groupe
ethnique à l'autre et que les propriétés biochimiques de l'enzyme présentaient des différences
selon le groupe ethnique où elles étaient étudiées (Modiano et al., 2001a). La G-6-PDA-
(202A/37G) est le variant génétique responsable du déficit en G-6-PD le plus répandu et le plus
étudié en Afrique subsaharienne (HOWES et al., 2013b). Des études menées dans la sous-
région Ouest-africaine ont rapporté d’autres variants déficitaires tels que la G-6-PD Santamaria
(376G/542T) et la G-6-PD Betica-Selma (376G/968C) avec des fréquences relativement
élevées dans certaines populations (DE ARAUJO et al., 2006; CLARK et al., 2009b). La
plupart des quelques rares études de génotypage faites sur la déficience en G-6-PD au Burkina
Faso ont recherché uniquement la mutation G202A associée à la G-6-PDA (MODIANO et al.,
2001a). Cette approche peut entrainer une sous-estimation de la prévalence réelle du déficit en
G-6-PD au Burkina Faso. Les différentes études antérieures d'association génétiques entre le
déficit en G-6-PD et le paludisme (notamment le paludisme grave) présentent également des
divergences au regard de la force et de la spécificité de l’effet protecteur. Quelle est donc la
fréquence des autres variants impliqués dans le déficit en G-6-PD au Burkina Faso ? Quel est
l’effet du déficit en G-6-PD et des hémoglobines S et C sur la parasitémie à Plasmodium
falciparum ?
Pour répondre à ces questions, les principaux variants déficitaires de la G-6-PD présentant des
fréquences polymorphiques en Afrique de l’Ouest ont été recherchées afin d’évaluer leur
prévalence réelle au Burkina Faso. La détermination de la prévalence des hémoglobines S et C
dans la population d’étude ainsi que la corrélation entre le déficit en G-6-PD ou ces anomalies
de l’hémoglobine et le paludisme ont également fait l’objet de notre étude.

OBJECTIFS
DE LA THESE

Thèse de Doctorat Unique OUATTARA Abdoul Karim 4
OBJECTIFS DE LA THESE
Objectif principal
Etudier le polymorphisme génétique du déficit en G-6-PD et de l’hémoglobine chez des
personnes vivant en zone endémique du paludisme au Burkina Faso.
Objectifs spécifiques
Estimer la prévalence des hémoglobines S et C et des variants génétiques impliqués
dans le déficit en G-6-PD en zone endémique du paludisme au Burkina Faso.
Déterminer la corrélation entre ces anomalies génétiques et le paludisme
asymptomatique en zone rurale au Burkina Faso.
Evaluer l’effet de ces polymorphismes génétiques sur l’infection symptomatique à
Plasmodium falciparum dans la ville de Ouagadougou au Burkina Faso.

REVUE
BIBLIOGRAPHIQUE

Thèse de Doctorat Unique OUATTARA Abdoul Karim 5
CHAPITRE I : GENERALITES SUR LE PALUDISME
I.1 Définition et Historique
I.1.1 Définition
Le paludisme (du latin palus, marécage) aussi appelé malaria (de l'italien mal'aria, mauvais
air) est une parasitose due à un protozoaire du genre Plasmodium transmis à l’homme par la
piqure du moustique femelle du genre Anophèle, provoquant des fièvres intermittentes
(MISBAHI, 2013). Un cas de paludisme se définit comme tout individu chez qui la présence
de parasites du paludisme a été confirmée par un diagnostic en laboratoire de qualité contrôlée,
que le sujet manifeste ou non des symptômes cliniques (OMS, 2014a).
I.1.2 Historique
L’existence de fièvres particulières, spécialement fréquentes dans les zones marécageuses, est
connue depuis la plus haute antiquité. Le parasite responsable du paludisme autrefois appelé
Laverania malariae aujourd’hui connu sous le nom de Plasmodium falciparum, a été découvert
en 1880 par le chirurgien militaire français Alphonse Laveran dans le sang de patients
présentant des fièvres intermittentes (MISBAHI, 2013). En 1897, le scientifique William
MacCallum découvre les stades sanguins sexués chez des oiseaux infectés par Haemoproteus
columbae. Dans la même année, le médecin britannique Sir Ronald Ross a montré par la suite
que la transmission du paludisme des oiseaux se fait par des moustiques. Entre 1898 et 1900,
les travaux des scientifiques italiens Giovanni Battista Grassi, Amico Bignami, Giuseppe
Bastianelli, Angelo Celli, Camillo Golgi et Ettore Marchiafava ont démontré que la
transmission du paludisme humain se fait également par des moustiques. La phase de division
dans le foie précédant la phase érythrocytaire a été décrite en 1947 par Henry Shortt et Cyril
Garnham (COX, 2010). Trente ans après ces découvertes William Trager et J.B. Jensen ont
développé une technique de culture en continue des parasites responsables du paludisme
(TRAGER et JENSEN, 1976). Depuis 2002, la séquence complète du génome de Plasmodium
falciparum est connue (GARDNER et al., 2002).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 6
I.2 Epidémiologie du paludisme
I.2.1 Estimations mondiales
Le paludisme (malaria en anglais) est une infection parasitaire vectorielle essentiellement
présente dans les régions tropicales et subtropicales d’Amérique du Sud, d’Afrique
subsaharienne et d’Asie du Sud-Est (ARGY et HOUZE, 2014). Au niveau mondial, la
population susceptible d'être infectée par le parasite et de développer la maladie s'élève à 3,2
milliards, et le risque est élevé (plus d'une chance sur 1 000 de contracter le paludisme au cours
d'une année) pour 1,2 milliard de personnes (Figure 2). Selon les dernières estimations de
l’OMS, 198 millions [124 - 283 millions] de cas de paludisme et 584 000 [367 000 - 755 000]
décès associés à cette maladie ont été enregistrés au cours de l’année 2013. Environ 90% des
décès au niveau mondial ont été recensés dans la région africaine de l’OMS. La plupart (78%)
étant des enfants de moins de 5 ans (OMS, 2014b).
Figure 2 : Différentes zones de transmission du paludisme dans le monde (OMS, 2014b)
Les chiffres du rapport OMS 2014 montrent clairement que le paludisme demeure encore un
problème majeur de santé publique surtout dans les pays tropicaux et subtropicaux en
développement ; avec la majorité des cas en Afrique sub-saharienne. En 2015, la plupart des
cas ont été enregistrés dans la région Afrique (88 %), loin devant la région Asie du Sud-Est (10
%) et la région Méditerranée orientale (2 %).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 7
Cependant le renforcement des mesures de lutte et de prévention a permis de réduire de façon
spectaculaire la charge palustre dans certaines régions. Au niveau mondial, la baisse du nombre
de décès dus au paludisme est estimée à 48 %, de 839 000 décès en 2000 [653 000 - 1,1 million]
à 438 000 en 2015 [236 000 – 635 000]. Cette baisse est plus prononcée chez les enfants de
moins de 5 ans dans la région africaine où le paludisme qui était la première cause de mortalité
infantile, apparaît au quatrième rang en 2015 avec 10 % des décès à l’échelle du continent
(OMS, 2015b).
I.2.2 Répartition géographique
Le paludisme sévit actuellement dans la ceinture de pauvreté tropicale et subtropicale du
monde. La répartition géographique de l’épidémiologie du paludisme est très hétérogène et
extrêmement variable d’un continent à l’autre, d’un pays à l’autre, mais également au sein d’un
même pays. Cette répartition géographique de la maladie est fortement influencée par la
distribution des anophèles vecteurs, la capacité vectorielle, les caractéristiques biologiques des
différentes espèces de Plasmodium, les conditions climatiques etc. (GETHING et al., 2011).
En zone intertropicale, chaude et humide, abondent les anophèles capables d’assurer en
permanence la transmission des hématozoaires. Le paludisme essentiellement à Plasmodium
falciparum, y est donc endémique. Selon l’intensité de l’impaludation on distingue des zones
holo-endémiques, hyper-endémiques, méso-endémiques et hypo-endémiques, mais avec une
transmission qui a lieu tous les ans. Des poussées surviennent à la saison des pluies quand
pullulent les anophèles. Le terme holoendémique est appliqué au paludisme dans les zones où
la prévalence de l'infection chez les enfants de 2 à 9 ans est supérieure à 75%. Le paludisme est
considéré comme hyper-endémique dans les zones où la prévalence de l’infection se situe entre
50% et 75%. Lorsque la prévalence de l’infection est comprise entre 11% et 50% dans une
région donnée, le paludisme est dit méso endémique, et hypo endémique pour désigner une
zone où la transmission de l’infection est inférieure à 10% (HAY et al., 2008; MENDIS et al.,
2009). En Europe, le paludisme a été éradiqué, il a disparu des anciens foyers tandis qu’en Asie
il sévit par foyers limités avec une intensité variable. L’Amérique du Nord est indemne de
paludisme, par contre on le retrouve en Amérique Centrale et du Sud. En Océanie certaines îles
comme la Nouvelle-Guinée, l’île Salomon et le Vanuatu sont atteintes. Le développement des
5 espèces de Plasmodium inféodées à l’homme est possible partout où il y a des vecteurs
compétents et où les conditions climatiques permettent l’accomplissement de leur cycle
sporogonique.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 8
La répartition géographique des différentes espèces de Plasmodium peut se résumer ainsi (HAY
et al., 2009; BROOKER et al., 2009) :
P. falciparum est largement répandu dans toute la ceinture tropicale du globe avec une forte
prédominance en Afrique subsaharienne. En effet le développement du cycle chez le moustique
se fait à une température supérieure à 18°C, d’où l’absence de cet hématozoaire dans les
montagnes tropicales et dans les régions tempérées.
P. malariae peut se développer sous les tropiques comme en zone tempérée mais aujourd'hui
sa distribution est sporadique et surtout tropicale (Afrique).
P. ovale est essentiellement observé en Afrique tropicale mais il est aussi signalé en Asie du
sud-est (Vietnam).
P. vivax sévit en zone tropicale et subtropicale (surtout en Amérique centrale et du Sud, en Asie
du Sud-Est, en Afrique de l’Est, au Proche et Moyen – Orient). L’invasion de l’hématie par
cette espèce nécessite la présence du récepteur de l’antigène Duffy (RAHIMI et al., 2014). P.
vivax est pratiquement absent en Afrique de l’Ouest car la majorité des populations ne porte
pas le facteur Duffy. Quant à la cinquième espèce,
Plasmodium knowlesi (SINGH et al., 2004), il sévit en Asie du Sud-Est, principalement dans
les zones forestières où vivent les singes macaques et les anophèles qui piquent autant les singes
que les hommes. (SABBATANI et al., 2010).
I.3 Cycle de vie de Plasmodium falciparum
Le cycle évolutif des parasites responsables du paludisme est très complexe et se déroule chez
deux hôtes (COWMAN et al., 2012). Il peut être divisé en deux grandes phases : une phase
asexuée chez l’hôte vertébré ou hôte intermédiaire (l’homme par exemple) et une phase sexuée
chez le moustique vecteur (hôte définitif).
I.3.1 Agent vecteur
La transmission du paludisme fait intervenir des facteurs géographiques et climatiques
favorables au développement des moustiques vecteurs. Un vecteur n’est pas une simple
seringue récupérant un agent pathogène chez un vertébré pour l’injecter à un autre. C’est un
point de passage obligatoire pour la diffusion de l’agent pathogène qui va soit « simplement »
s’y multiplier (virus) ou y assurer une part de son cycle (parasites) (PAGES et al., 2007).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 9
L’intensité de la transmission dépend de facteurs liés au parasite, au vecteur, à l’hôte humain et
à l’environnement (LYIMO et al., 2012). Les femelles de certaines espèces d’anophèles chez
qui s’effectue le cycle sexué des plasmodies assurent seules la transmission du paludisme
d’homme à homme par leur piqûre, principalement entre le crépuscule et le petit matin. En
Afrique trois principales espèces de moustiques sont responsables de la transmission du
paludisme à l’homme. Il s’agit des espèces Anopheles gambiae (Figure 3), Anopheles
arabiensis et Anopheles funestus (SINKA et al., 2012).
Figure 3 : Anopheles gambiae (PAGES et al., 2007)
I.3.2 Agent pathogène
Les parasites du paludisme sont des protozoaires unicellulaires qui se développent à la fois chez
le moustique vecteur (multiplication sexuée ou sporogonique) et chez l’homme (multiplication
asexuée ou schizogonique). Au cours de leur cycle de vie, les plasmodies changent sans cesse
d’aspect et de taille, par suite d’alternance de phases de croissance et de phases de divisons
nucléaire et cytoplasmique (MILLER et al., 2013). Ils se développent de façon indépendante
dans le globule rouge et modifie cette cellule de sorte à en tirer leurs nutriments. On distingue
cinq principales espèces de Plasmodium responsables du paludisme chez l’homme :
Plasmodium falciparum, responsable de la fièvre tierce maligne,
Plasmodium vivax, responsable de la fièvre tierce bénigne avec une distribution plus
étendue que P. falciparum, sauf en Afrique subsaharienne,

Thèse de Doctorat Unique OUATTARA Abdoul Karim 10
Plasmodium ovale, responsable de la fièvre tierce bénigne, Plasmodium malariae,
responsable de la fièvre quarte bénigne,
Et Plasmodium knowlesi, responsable du paludisme du singe, a été retrouvée comme
infection humaine à fièvre quarte dans quelques pays d'Asie du sud-Est.
I.3.3 Phase asexué chez l’homme
Le Plasmodium est transmis à l'homme par la piqûre de l’anophèle femelle lors de la prise de
son repas sanguin (CORNELIO et SERIANO, 2011; BOUSEMA et al., 2014). Un petit nombre
des stades infectieux sporozoïtes (10-100) contenus dans ses glandes salivaires sont alors
injectés avec la salive dans la circulation sanguine de l’homme. Les sporozoïtes migrent
activement à travers la peau, puis gagnent la circulation sanguine pour atteindre rapidement le
foie (Figure 4). Ils traversent ensuite la barrière sinusoïdale hépatique, migrent à travers
plusieurs cellules puis finalement pénètrent dans un hépatocyte en formant une vacuole
parasitophore. C’est au sein de ce compartiment membranaire particulier que les sporozoïtes se
différencient en formes exo-érythrocytaires (schizontes hépatiques).
Schizogonie hépatique ou exoérythrocytaire
Il s’agit du cycle de reproduction asexuée qui se déroule dans les hépatocytes parasités. Le
parasite se développera et se divisera dans les cellules du foie pendant 8-10 jours, pour former
une masse (le schizonte) contenant plusieurs milliers de cellules filles (les mérozoïtes). Les
mérozoïtes seront libérées à partir du foie dans la circulation sanguine, où ils envahiront
rapidement les érythrocytes (sous-phase érythrocytaire). Chaque schizonte libère environ 10
000 à 30 000 mérozoïtes chez P. falciparum (CROMPTON et al., 2010; ARUMUGAM et al.,
2014), et entre 2 000 et 15 000 pour les trois autres espèces de Plasmodium humains. La
schizogonie se déclenche immédiatement dans tous les hépatocytes parasités pour les espèces
P. malariae et P. falciparum. Par contre elle peut être retardée dans certains hépatocytes qui
restent en attente (formes dormantes ou hypnozoïtes) pendant des mois voire même des années
avant le développement de la schizogonie exoérythrocytaire pour les espèces P. vivax et P.
ovale (CORNELIO et SERIANO, 2011). A chaque phase du cycle, le parasite prend une forme
caractéristique et exprime à sa surface des produits spécifiques, adaptés à la prochaine
interaction avec l’hôte (WRIGHT et RAYNER, 2014). Dans les conditions naturelles de
transmission, le développement pré-érythrocytaire de Plasmodium est une phase initiale,
obligatoire et asymptomatique de l’infection. Il représente donc une cible idéale pour des
approches antipaludiques prophylactiques.
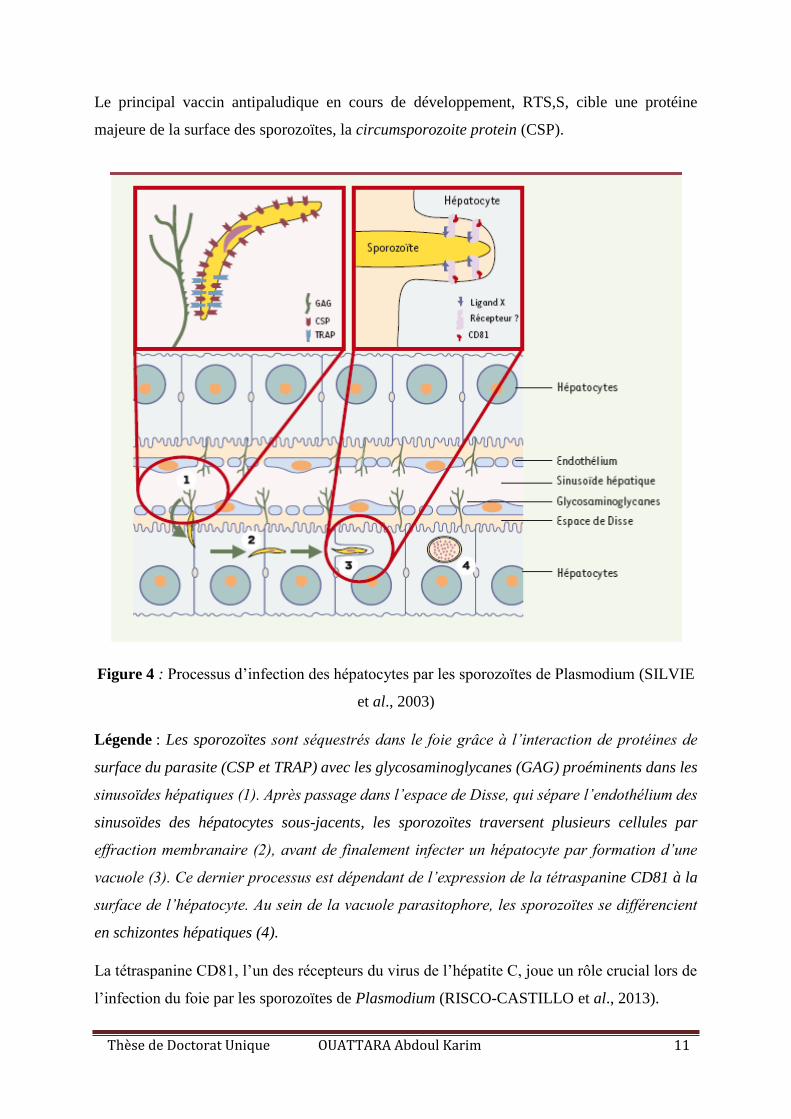
Thèse de Doctorat Unique OUATTARA Abdoul Karim 11
Le principal vaccin antipaludique en cours de développement, RTS,S, cible une protéine
majeure de la surface des sporozoïtes, la circumsporozoite protein (CSP).
Figure 4 : Processus d’infection des hépatocytes par les sporozoïtes de Plasmodium (SILVIE
et al., 2003)
Légende : Les sporozoïtes sont séquestrés dans le foie grâce à l’interaction de protéines de
surface du parasite (CSP et TRAP) avec les glycosaminoglycanes (GAG) proéminents dans les
sinusoïdes hépatiques (1). Après passage dans l’espace de Disse, qui sépare l’endothélium des
sinusoïdes des hépatocytes sous-jacents, les sporozoïtes traversent plusieurs cellules par
effraction membranaire (2), avant de finalement infecter un hépatocyte par formation d’une
vacuole (3). Ce dernier processus est dépendant de l’expression de la tétraspanine CD81 à la
surface de l’hépatocyte. Au sein de la vacuole parasitophore, les sporozoïtes se différencient
en schizontes hépatiques (4).
La tétraspanine CD81, l’un des récepteurs du virus de l’hépatite C, joue un rôle crucial lors de
l’infection du foie par les sporozoïtes de Plasmodium (RISCO-CASTILLO et al., 2013).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 12
Schizogonie érythrocytaire ou intra-érythrocytaire
Cette phase commence par l’invasion aléatoire des hématies saines par des formes ovoïdes
d’environ 1 µm de long appelées mérozoïtes, libérées suite à la rupture des schizontes
hépatiques. Il s’agit d’un processus complexe faisant intervenir des mécanismes d’interactions
fines hôte-parasite de type récepteurs-ligands présents à la surface des cellules hôte et parasite
(WRIGHT et RAYNER, 2014). L’invasion des hématies qui se fait grâce à un processus
parasitaire actif (endocytose) peut être décomposée en plusieurs phases (Figure 5) : (i) La phase
de contact ente le manteau glycoprotéique du mérozoïte et la surface de l’hématie, (ii) Une
phase de réorientation du mérozoïte pour que son pôle apical se retrouve perpendiculaire à la
surface de l’érythrocyte ; (iii) Une phase de formation de la jonction et début de la décharge
des rhoptries ; (iv) La phase de pénétration du mérozoïte survient alors par endocytose à travers
la jonction serrée avec formation de la vacuole parasitophore.
L'ensemble du processus d'invasion est complexe, mais il est également extrêmement rapide.
Des études ont montré que ce processus est bouclé en moins de deux minutes après la libération
des mérozoïtes (WRIGHT et RAYNER, 2014). En effet pour survivre le parasite doit réduire
son temps d’exposition aux éléments du système immunitaire de l’hôte. Au sein de chaque
hématie envahie le mérozoïte se développera ensuite sous forme d’anneau (trophozoïte jeune),
de trophozoïte mature, de schizonte jeune (nombre variable de noyaux) et de schizonte mature
à nombre de noyaux définis. Le cycle érythrocytaire asexué durera 48 heures, et sera complétée
par la rupture du schizonte et la libération de nouveaux mérozoïtes qui envahiront d’autres
érythrocytes non infectés (Figure 8). La densité parasitaire au cours de cette phase peut souvent
excéder 50 000 hématies infectés/µL de sang (CROMPTON et al., 2014).
Figure 5 : Processus d’invasion des érythrocytes par les mérozoïtes de Plasmodium falciparum
(WRIGHT et RAYNER, 2014)
Thèse de Doctorat Unique OUATTARA Abdoul Karim 13
C’est au cours du cycle érythrocytaire asexuée que se produiront les symptômes cliniques du
paludisme (fièvre, frissons, troubles de la conscience, etc.). Au cours de l'infection les
mérozoïtes remodèlent considérablement la membrane du globule rouge pour l’expression de
plusieurs de leurs protéines de surface à savoir principalement PfEMP1 (CROMPTON et al.,
2014). Il faut noter que l’infection de l’érythrocyte humain par Plasmodium vivax nécessite
obligatoirement la présence d’un antigène à la surface des globules rouges appelé, « Duffy
Antigen Receptor for Chemokines (DARC). P. vivax et P. knowlesi pénètrent dans les hématies
uniquement par l’interaction de leur « Duffy-Binding-Like domain » (DBL) avec le récepteur
DARC (COWMAN et al., 2012).
I.3.4 Phase sexuée chez le moustique ou sporogonie
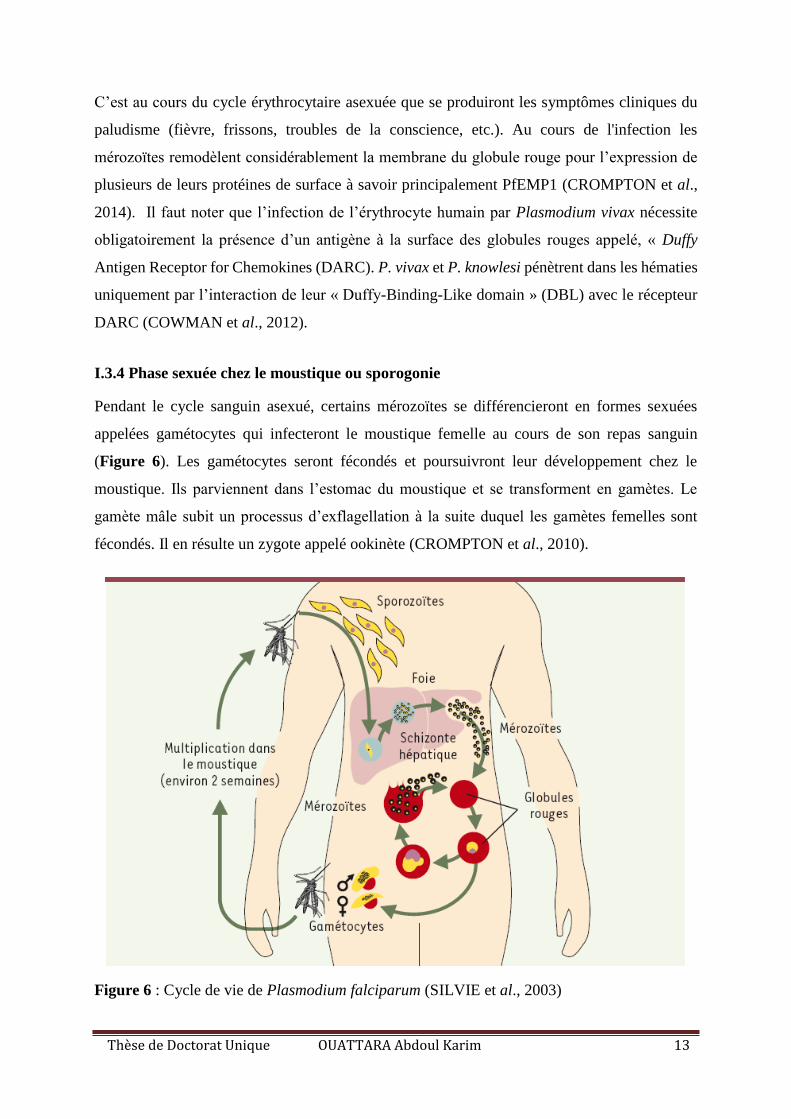
Pendant le cycle sanguin asexué, certains mérozoïtes se différencieront en formes sexuées
appelées gamétocytes qui infecteront le moustique femelle au cours de son repas sanguin
(Figure 6). Les gamétocytes seront fécondés et poursuivront leur développement chez le
moustique. Ils parviennent dans l’estomac du moustique et se transforment en gamètes. Le
gamète mâle subit un processus d’exflagellation à la suite duquel les gamètes femelles sont
fécondés. Il en résulte un zygote appelé ookinète (CROMPTON et al., 2010).
Figure 6 : Cycle de vie de Plasmodium falciparum (SILVIE et al., 2003)

Thèse de Doctorat Unique OUATTARA Abdoul Karim 14
Environ 24 heures après la formation du zygote, l’ookinète mature mobile traverse d’abord la
matrice péritrophique, et les cellules épithéliales de l'intestin moyen, avant de se différencier en
oocyste sous la membrane basale de l'épithélium de l'intestin. Cette phase diploïde est brève et
s’achève par une division méiotique laquelle est suivie de plusieurs mitoses aboutissant au
développement de sporoblastes, puis de sporozoïtes après une dizaine de jours. L’éclatement
de l’oocyste libère ces éléments haploïdes (5000 à 10 000 sporozoïtes) mobiles dans
l’hémolymphe. Les sporozoïtes migreront ensuite dans les glandes salivaires du moustique,
prêts à infecter un autre hôte humain.
I.4 Aspect génétique de Plasmodium falciparum
I.4.1 Organisation du génome
La séquence du génome de Plasmodium falciparum est connue depuis 2002. Les premiers
travaux ont décrit le génome nucléaire du clone 3D7 comme étant composé de 22,8 mégabases
(Mb) réparties en 14 chromosomes dont la taille varie de 0,643 à 3,29 Mb et codant pour environ
5 300 gènes d’où une densité moyenne d’environ un gène pour 4338 paires de bases (pb)
(GARDNER et al., 2002). En plus de ce génome nucléaire on distingue 6 Kb et 35 Kb d’ADN
circulaire localisé respectivement au niveau des mitochondries et des apicoplastes (WEBSTER
et MCFADDEN, 2014). Les gènes impliqués dans la variation antigénique sont concentrés dans
les régions subtélomériques des chromosomes. La composition en nucléotides présente une
forte richesse en adénines (A) et en thymines (T), en moyenne de 80,6% du génome voire plus
de 90% pour les régions intergéniques et les introns. La taille moyenne des gènes de
Plasmodium falciparum est de 2,3 kb sans les introns présents à 54% (GARDNER et al., 2002).
Après près de 9 ans d’efforts coordonnés, la séquence complète du génome a été définie comme
haploïde avec une taille de 23,26 Mb, contenant 6372 gènes et codant pour 5524 protéines
(génome version : 06-01-2010, http://plasmodb.org/plasmo/) (LE ROCH et al., 2012).
Plasmodium falciparum possède une machinerie typique des eucaryotes capable d’engendrer
les modifications et le remodelage de la chromatine pour contrôler la régulation
transcriptionnelle. Sa caractéristique majeure est d’être constamment capable de varier son
expression génique pour produire des formes phénotypiques différentes et ainsi contourner les
réponses immunitaires de l’hôte.
I.4.2 Architecture nucléaire des gènes var
La virulence de Plasmodium falciparum est principalement liée à l’adhérence de globules
rouges parasités à l’endothélium et entre eux, pour former des rosettes.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 15
Cette adhérence est le fait de la protéine de surface Plasmodium falciparum erythrocyte
membrane protein-1 (PfEMP1) (SCHERF et al., 2008). Dans le génome de P. falciparum, la
protéine PfEMP1 est codée par une soixantaine de gènes appartenant à la famille des gènes var.
Ce groupe de gènes, principalement situé dans les régions subtélomériques du chromosome, est
caractérisé par une fréquence élevée de recombinaison génique à l’origine d’une grande
diversification du répertoire des gènes var (Figure 7). Ils sont séparés du télomère par des
séquences répétitives (rep20 ou TARE) en nombre variable (1 à 6) et il y a à leur proximité
immédiate d’autres familles multigéniques (rif, stevor, Pf60). La conséquence directe de ce
polymorphisme génique est une grande variation antigénique de PfEMP1 qui permet au parasite
d’échapper au système immunitaire de l’hôte et d’avoir un grand nombre de phénotype
d’adhésion associé à la séquestration parasitaire (LABIE, 2005; ARGY et HOUZE, 2014).
Cette variation antigénique et phénotypique de PfEMP1 est associée à la composition variable
du domaine d’adhésion de la protéine en son extrémité N-terminal. En effet cette extrémité est
composée d’une succession de domaines protéiques tels que les domaines Duffy binding-like
(DBL) et les régions inter-domaine riche en cystéine (Cysteine-Rich Interdomain Region ou
CIDR).
Figure 7 : Organisation des gènes var de Plasmodium falciparum (SCHERF et al., 2008)
Légende : Génome haploïde organisé en 14 chromosomes linéaires. Les régions
subtélomériques des 14 chromosomes linéaires ont une organisation globale commune, avec
des répétitions non codantes de taille variable appelée TARE 1-6 situé à côté de répétitions
télomériques. Cette région est suivie par un membre de la famille des gènes var, souvent en
combinaison avec un deuxième gène var transcrit dans la direction opposée (vers le télomère).
Plusieurs groupes clusters de gènes var en tandem se trouvent dans des positions
chromosomiques centrales. Les gènes Rif sont souvent entrecoupés de gènes var.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 16
Dans différents génomes de souches de P. falciparum, des études d’alignement de 399
séquences protéiques de PfEMP1 des différents groupes cités précédemment ont mis en
évidence 23 domaines protéiques conservées appelés domaine cassette (DC) dont certains
comme DC8, DC13, DC4 et DC5 sont devenus des candidats potentiels pour l’élaboration d’un
vaccin (ARGY et HOUZE, 2014). Les caractéristiques des gènes var sont présentées dans la
figure 8 ci-dessous.
Figure 8 : Caractéristiques des gènes var (SCHERF et al., 2008)
Légende : Les caractéristiques communes entre les membres de la famille des gènes var sont
présentées. Tous les membres contiennent deux exons. Le domaine polymorphique
extracellulaire (exon 1) contient un nombre variable de Duffy-Binding-Like (DBL) domaines
adhésifs, qui peuvent être classés en cinq catégories principales (alpha à epsilon) ainsi que les
régions entre les domaines riches en cystéine. Le domaine intracellulaire (exon 2) code pour
un segment semi-terminal conservé d'acides aminés.
I.5 Manifestations cliniques du paludisme
L’accès palustre à P. falciparum se présente dans la grande majorité des cas par un tableau
infectieux non spécifique d’aspect pseudo-grippal caractérisé par de la fièvre, des frissons et
des douleurs abdominales. Néanmoins, dans certaines populations avec des facteurs de risque
associés (âge extrême, malnutrition, immunodépression) ou considérées comme non immunes
vis-à-vis du parasite (enfants < 5 ans, femme enceinte et voyageurs), l’infection par P.
falciparum peut conduire à un accès palustre grave (ARGY et HOUZE, 2014).
Les symptômes apparaissent au bout de 7 jours ou plus (généralement 10 à 15 jours) après la
piqûre infectante du moustique. De manière opérationnelle, le paludisme est aujourd’hui classifié
sous deux formes cliniques à savoir le paludisme simple et le paludisme grave.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 17
I.5.1 Accès palustre simple
Le paludisme simple se définit par une fièvre (température axillaire supérieure ou égale à
37,5°C ou antécédent de corps chaud dans les 72 dernières heures) et la mise en évidence du
plasmodium dans le sang par un examen microscopique (goutte épaisse/frottis sanguin) ou par
un Test Rapide d’Orientation diagnostic (TROD) ; et une absence de signe de gravité.
I.5.2 Accès palustre grave
La définition du paludisme grave donnée par l’OMS a évolué de 1986 à 2010. La définition la
plus utilisée date de 2000 et sépare l’enfant de l’adulte (LAURENT et al., 2014). Le paludisme
grave est ainsi défini par un frottis/goutte épaisse positifs à des formes asexuées de P.
falciparum, associée à au moins un critère clinique ou biologique de gravité tels que l’atteinte
neurologique, l’état de choc, une détresse respiratoire aiguë, l’hypoglycémie, l’acidose
métabolique, l’anémie sévère (Hb < 5 g/dL), l’hémoglobinurie, l’insuffisance rénale (créatinine
sérique > 265 µmol/L), un saignement spontané ou la présence d’une hyperparasitémie
(LAURENT et al., 2014).
I.5.3 Physiopathologie du paludisme
La physiopathologie du paludisme est encore incomplètement comprise. Les principaux
mécanismes impliquent l’hôte et le parasite dans des interactions complexes dont l’élément
central et singulier est le globule rouge parasité (LAURENT et al., 2014). Les manifestations
cliniques du paludisme sont directement ou indirectement liées à la schizogonie érythrocytaire,
alors que la schizogonie hépatique est asymptomatique (CHAN et al., 2014). Leur gravité
dépend, de l’espèce plasmodiale, de la densité parasitaire, du degré de prémunition de l’hôte.
La fièvre est due à l’éclatement des schizontes qui libèrent dans le torrent circulatoire du
pigment malarique appelé hémozoïne ; celui-ci se comporte comme une substance pyrétogène
et stimule la production des cytokines pro-inflammatoires comme le Tumor Necrosis Factor
(TNF) et l’interleukine 1. Les mécanismes immunitaires s’initient rapidement avec la
mobilisation de l’immunité innée ; et l’immunité adaptative est opérationnelle dans les 10 jours.
Les principaux acteurs sont les neutrophiles, les monocytes et les cellules Natural Killer (NK)
qui à leur tour produisent des cytokines, elles-mêmes capables de recruter des monocytes et
d’activer les neutrophiles (LAURENT et al., 2014). Si l’éclatement des schizontes est
asynchrone, la fièvre est irrégulière ou apparemment continue ; s’il est synchrone, la fièvre est
intermittente, quotidienne, tierce ou quarte selon la périodicité de la schizogonie (24, 48 ou 72
heures). L’anémie résulte avant tout de la lyse des hématies (éclatement des schizontes,
érythrophagocytose).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 18
Les hématies parasitées par P. falciparum ont une déformabilité modifiée dès le stade
trophozoïte jeune, et plus le parasite est mature, plus l’hématie devient rigide. La splénomégalie
et l’hépatomégalie, habituelles au bout d’un certain temps d’évolution, témoignent de
l’hyperactivité et de la congestion de ses organes (AUTINO et al., 2012). La rate dont le rôle
est capital, grâce à la microcirculation (avec l’existence d’une double circulation sanguine
filtrante/non filtrante) scrute pluri-quotidiennement la déformabilité érythrocytaire et retient les
hématies trop rigides, celles parasitées par P. falciparum. Le foie intervient par l’activité
phagocytaire des cellules de kupffer et par la transformation de l’hémoglobine libérée en
bilirubine libre, d’où le subictère. D’un point de vue physiopathologique, l’accès grave serait
associé à un phénomène de séquestration des hématies parasitées dans les capillaires des
organes, particulièrement le cerveau. La séquestration concerne surtout les formes matures des
hématies parasitées (trophozoïte et schizonte) et se décompose schématiquement en trois
mécanismes : la cytoadhérence, le phénomène de rosetting et l’autoagglutination.
La cytoadhérence correspond à l’adhérence des globules rouges parasités aux cellules
endothéliales de l’hôte via des structures appelées «knobs» situées à la surface du globule rouge
parasité, principalement dans les capillaires de la microcirculation viscérale (ARGY et
HOUZE, 2014). Elle implique de multiples interactions entre les ligands de l’hématie parasitée
et les récepteurs endothéliaux (Figure 9).
Figure 9 : Les bases moléculaires de la cytoadhérence (source : http://www.u-bordeaux2-
medtrop.org/doc/COURS/Cours_pdf_2014-2015/Paludisme%20grave_Pr%20Saissy.pdf)

Thèse de Doctorat Unique OUATTARA Abdoul Karim 19
Au cours du cycle intra-érythrocytaire, la prolifération et la maturation des formes parasitaires
sont accompagnées par la production de protéines parasitaires (Ring-infected Erythrocyte
Surface Antigen (RESA), Knob-Associated Histidine-Rich Protein (KAH-RP), Mature P.
falciparum-infected Erythrocytes Surface Antigen (MESA)) qui interagissent avec des
structures membranaires de l’érythrocyte (actine, spectrine) pour former les knobs. Ces
complexes protéiques parasitaires permettent aux stades matures de P. falciparum d’échapper
à la clairance splénique par séquestration dans les capillaires et veinules post-capillaires de
différents organes de l’hôte mais principalement au niveau cérébral (MILLER et al., 2013). Ce
phénomène de séquestration parasitaire, lié à la cytoadhérence, a pour conséquence l’activation
d’une cascade de phénomènes biologiques à l’origine de la pathogenèse de l’atteinte cérébrale
de l’accès palustre grave (ARGY et HOUZE, 2014).
La capacité des hématies parasitées à lier des hématies non parasitées conduit à la formation de
rosettes (rosetting), et l’autoagglutination correspond à l’adhérence entre plusieurs hématies
parasitées (LAURENT et al., 2014). La cytoadhérence à l’endothélium ainsi que le rosetting
réduisent la lumière des capillaires entraînant une occlusion du flux sanguin tissulaire et une
hypoperfusion d’organes (Figure 10).
Figure 10 : Séquestration des hématies parasitées (source : http://www.u-bordeaux2-
medtrop.org/doc/COURS/Cours_pdf_2014-2015/Paludisme%20grave_Pr%20Saissy.pdf)

Thèse de Doctorat Unique OUATTARA Abdoul Karim 20
L’obstruction des capillaires et la production de toxines parasitaires
(glycosylphosphatidylinositol [GPI]) induisent également une réaction inflammatoire locale
liée au recrutement et à l’activation de polynucléaires neutrophiles, monocytes et plaquettes.
La production par ces cellules immunitaires de médiateurs pro-inflammatoires tels que le TNF-
α, l’interleukine-1 (IL-1) et l’IL-6 participe à la pathogenèse de l’accès grave via
l’augmentation de l’expression de récepteurs endothéliaux impliqués dans la séquestration
parasitaire et par l’altération du métabolisme du monoxyde d’azote qui joue un rôle dans
l’homéostasie de la barrière hémato-encéphalique. Enfin, l’activation des cellules endothéliales
après cytoadhérence parasitaire est un autre mécanisme physiopathologique de l’accès grave
(ARGY et HOUZE, 2014).
À l’heure actuelle, une douzaine de récepteurs de l’hôte ont été identifiés comme intervenant
dans la séquestration parasitaire : héparanes sulfates (HS), récepteur de complément 1 (CR1),
antigène du groupe sanguin (ABO), chondroïtine sulfate (CSA), PECAM/CD31, ICAM-1,
CD36, thrombospondine (TSP), VCAM-1, E-sélectine, immunoglobuline (Ig) non immune et
le récepteur à la protéine C (Endothelial Protein C Receptor ou EPCR) (GAY et al., 2012,
TURNER et al., 2013). Même si l’ensemble de ces récepteurs ont été décrits dans l’accès grave,
P. falciparum ne possède pas un phénotype d’adhésion pour tous ces récepteurs et, par
conséquent, ils n’ont pas tous la même importance en fonction de la présentation clinique et
biologique de l’accès palustre.
Alors que les récepteurs HS et CSA ont plus particulièrement été décrits dans le paludisme
gestationnel, ABO, CR1 et Ig semblent être plus impliqués dans le phénomène de rosetting.
CD36 est un récepteur constitutif de l’endothélium dont le rôle est controversé. ICAM-1 et
EPCR semble être des récepteurs potentiellement impliqués dans l’accès grave et plus
particulièrement dans l’atteinte cérébrale chez l’enfant de moins de 5 ans en Afrique
subsaharienne(ARGY et HOUZE, 2014). Les différents rôles de la rate sont actuellement
connus. Les globules rouges lors de leur passage dans la rate sont phagocytés par les
macrophages de la pulpe rouge si leur surface est altérée ou opsonisée. La rate a par ailleurs un
rôle mécanique de rétention d’intensité variable, notamment des formes jeunes, qui
contribueraient à réduire la biomasse parasitaire circulante. La rate joue aussi un rôle important
dans la clairance parasitaire sous traitement antipaludique, particulièrement lorsque le
traitement comporte un dérivé de l’artémisinine (artésunate, dihydroartémisinine) (LAURENT
et al., 2014).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 21
I.6 Paludisme et immunité
La réponse immunitaire dirigée contre Plasmodium falciparum, agent responsable des cas les
plus mortels de paludisme chez l’homme, est le résultat de plusieurs milliers d’années de
coévolution entre le parasite et son hôte (CROMPTON et al., 2014). Au cours de l’infection
par le plasmodium le système immunitaire de l’hôte se déploie contre le parasite lors des
différents stades de son cycle de vie. Par convention, les réponses immunitaires au paludisme
sont regroupées en réponses pré-érythrocytaires et en réponses érythrocytaires (CROMPTON
et al., 2014). L’immunité pré-érythrocytaire est généralement constituée de réponses cellulaires
contre les hépatocytes infectés, qui inhibent le développement du parasite intracellulaire par
l'induction d'intermédiaires réactifs de l'azote (STANISIC et al., 2013). Au cours de la phase
érythrocytaire, l’invasion des hématies par le parasite induit des modifications importantes de
leur surface membranaire, qui résultent en l’exposition de multiples molécules parasitaires. Au
cours de sa maturation le parasite produit un certain nombre de molécules impliquées dans la
modulation des processus pathogéniques, en grande partie à travers leurs effets sur le système
immunitaire (LANGHORNE et al., 2008). La détection de l’infection par le système
immunitaire permet de limiter la fréquence des cellules infectées circulantes, ainsi que les
manifestations cliniques. En effet, les infections plasmodiales multiples dès la petite enfance
dans les zones d’endémies stable, induisent une immunité non stérilisante qui confère une
protection contre les manifestations cliniques du paludisme sans empêcher les réinfections et
un portage intermittent du parasite. L’acquisition de cette immunité naturelle souvent appelée
« prémunition », nécessite plusieurs années d’exposition et elle disparait après quelques années
sans infection. Cette protection contre le « paludisme maladie » est le résultat d’une immunité
cellulaire et humorale complexe liée à la fois aux stimulations par les divers stades du parasite
et au polymorphisme antigénique des populations de Plasmodium circulant entre les anophèles
et l’homme dans une zone d’endémie (CROMPTON et al., 2014). Il a été rapporté que les
adultes gambiens naturellement exposés au Plasmodium présentaient de fortes réponses
cytokiniques Th1 et Th2 contre les formes sauvages et mutantes des protéines et peptides
MSP119 ainsi que des différences interethniques sur l’incidence du paludisme (Cyrille Bisseye,
2011). Des mécanismes d’inhibition cellulaire anticorps dépendant aboutissant à la production
de cytokines et de monoxyde d’azote, inhibant la multiplication érythrocytaire du parasite, sont
également en cause (PAWAR, 2014). Cependant, une activation excessive et inappropriée du
système immunitaire peut contribuer à l’apparition des formes sévères de la maladie.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 22
I.7 Prise en charge thérapeutique
I.7.1 Traitements antipaludiques
Les premiers traitements de synthèse, les amino-8-quinoléines, apparaissent peu après la
Première Guerre Mondiale (plasmoquine, 1926). En 1943, les Américains modifient la
composition chimique du Resochin (développé par une firme pharmaceutique allemande en
1934) et obtiennent la chloroquine (PULL et al., 2013). La production massive de chloroquine,
vers 1945, a permis de disposer d’un traitement efficace, peu toxique et très bon marché. Son
utilisation pour le traitement des accès ou en chimioprophylaxie a constitué un des deux piliers
de la campagne d’éradication du paludisme des années 1950-1970 avec la lutte antivectorielle
au moyen de dichloro-diphényltrichloroéthane (DDT) (MENARD et al., 2013). Les premiers
cas de résistance à la chloroquine ont été rapportés en plusieurs points du monde entre 1957 et
1960 (Colombie et Venezuela, Cambodge et Thaïlande, Papouasie Nouvelle Guinée,
Philippines). La résistance s’est peu à peu propagée à toutes les zones d’endémie. La marche
infernale de la sélection médicamenteuse s’est poursuivie après le déploiement des
médicaments qui ont remplacé la chloroquine : résistance à l’amodiaquine, aux antifolates, à la
méfloquine (MENARD et al., 2013; PULL et al., 2013). La montée des multirésistances a
amené l’OMS à préconiser, il y a une dizaine d’années, l’utilisation de combinaisons
comprenant un dérivé de l’artémisinine (ACT, Artemisinin Combination Therapy). Ces
combinaisons sont maintenant recommandées dans la plupart des pays d’endémie, mais leur
accès est encore loin d’être général. Aujourd’hui, une des préoccupations majeures est que l’on
assiste en Asie du Sud-Est aux premiers signes d’émergence de parasites résistants aux dérivés
des artémisinines. Aucune solution thérapeutique de remplacement n’est actuellement
disponible. La situation devient donc critique, l’urgence étant d’enrayer la dissémination des
parasites résistants (MENARD et al., 2013).
I.7.2 Résistances aux médicaments antipaludiques
L’OMS a défini en 1965 et 1973 la résistance comme la capacité du parasite à survivre et/ou à
se multiplier en dépit de l’administration et de l’absorption d’un médicament donné à doses
égales ou supérieures à celles habituellement recommandées mais dans les limites de la
tolérance du malade (PRADINES et al., 2010). Il a été ajouté en 1986 que la forme active du
médicament devait pouvoir atteindre le parasite ou accéder à l’intérieur de l’érythrocyte infecté
pendant la durée nécessaire à son action normale.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 23
Il s’agissait de tenir compte du fait que les individus pouvaient différer par leur capacité à
métaboliser les antipaludiques comme les sulfonamides et les sulfones, que les molécules
antipaludiques pouvaient se lier fortement aux protéines plasmatiques et que des médicaments
administrés de façon simultanée pouvaient avoir un effet antagoniste sur l’efficacité de
l’antipaludique (Par exemple l’acide folique peut altérer l’effet de la sulfadoxine–
pyriméthamine). Pour des raisons historiques et pratiques, la définition de la résistance est donc
essentiellement clinique et parasitologique (PRADINES et al., 2010).
I.7.2.1 Facteurs liés à la résistance aux antipaludiques
Malgré les efforts déployés pour la découverte de nouveaux médicaments antiplasmodiaux et
la mise en place effective par les systèmes de santé de combinaisons thérapeutiques pour le
traitement antipaludique, P. falciparum s’adapte en permanence et développe des résistances,
y compris contre les ACT. Ceci s’explique d’abord par la grande diversité génétique de P.
falciparum due à un taux élevé de mutations dans son génome et par les masses très importantes
de parasites portées par les individus infectés. Même si les mutations capables de conférer une
résistance à un nouveau médicament sont extrêmement rares et peu probables, le nombre élevé
de parasites infectant les humains fait que ces mutations finissent par apparaître et par être
sélectionnées par la pression médicamenteuse (PRADINES et al., 2010). Les erreurs de
réplication de l’ADN dans les cellules introduisent des mutations au hasard dans le génome et
permettent le processus d’évolution. Ces mutations sont à l’origine de la grande variabilité
génétique de P. falciparum et lorsque celles-ci ne sont ni létales pour le parasite, ni silencieuses,
elles peuvent dans certains cas avantager sa survie en lui permettant par exemple d’échapper au
système immunitaire de son hôte, de supporter la présence de molécules toxiques dans son
environnement ou de se multiplier plus rapidement que d’autres clones (LE BRAS, 2006,
PRADINES et al., 2010). Certaines mutations permettent au parasite de survivre en présence
d’un antipaludique pour lequel il devient résistant. La mutation est ensuite transmise à ses
descendants, générant ainsi une population capable de résister à une molécule.
La fréquence des mutations et la vitesse à laquelle les résistances se développent dépendent des
caractéristiques de la molécule utilisée, du contexte épidémiologique (intensité de la
transmission) et de la façon dont les médicaments sont utilisés. Toutefois le phénotype de
résistance acquis après mutation n’est pas toujours un avantage en l’absence de pression
médicamenteuse.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 24
Ainsi, les autres facteurs favorisant l’émergence de résistances sont i) une mauvaise utilisation
des antipaludiques par les individus infectés (automédications abusives, mauvaise observance)
conduisant à des traitements incomplets, ii) une indisponibilité des médicaments efficaces ou
le déploiement inadéquat des médicaments sous forme de monothérapies et iii) la
consommation de contrefaçons sous dosées, facteurs permettant à des parasites viables de
survivre à des concentrations sub-optimales d’antipaludiques et d’être sélectionnés pour leur
aptitude à résister (PRADINES et al., 2010).
I.7.2.2 Marqueurs moléculaires de résistance
Il s’agit d’un ensemble de marqueurs impliqués dans les mécanismes moléculaires de
résistance. Pertinents et spécifiques pour prédire le niveau de résistance d’une population
parasitaire aux antipaludiques, ils ont une place de choix dans la surveillance de l’activité de tel
ou tel antipaludique. Certains marqueurs sont associés à un défaut d’accumulation des
pharmacophores au niveau de la cible parasitaire, d’autres à une modification de la cible
parasitaire (MENARD et al., 2013).
Les principaux marqueurs moléculaires utilisés dans la surveillance de l’efficacité des
antipaludiques sont les suivants :
P. falciparum chloroquine transporter (Pfcrt) : Ce gène situé sur le chromosome 7
code pour un transporteur membranaire de la vacuole digestive. La mutation sur le
codon K76T, associée à sept autres points de mutation, permet au parasite de limiter
l’accumulation de chloroquine dans sa vacuole digestive, où elle exerce son action
inhibitrice. Pfcrt est également impliqué dans la baisse de sensibilité du parasite à
l’amodiaquine et à la quinine. Dans les zones où les allèles de résistance ne sont pas
fixés, on observe une augmentation de la fréquence de l’allèle sauvage après abandon
de la chloroquine. L’analyse de ce locus renseigne sur la pression médicamenteuse
exercée au sein des populations (MENARD et al., 2013).
P. falciparum multi-drug resistance 1 (Pfmdr-1) : Situé sur le chromosome 5, ce gène
code pour un transporteur de type ABC (ATP binding cassette). La protéine PfMDR-1
est impliquée dans la modulation de la sensibilité à de multiples antipaludiques et, plus
particulièrement, dans l’efflux des antipaludiques hydrophobes (MENARD et al.,
2013).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 25
Les mécanismes de résistance sont liés : (1) soit à des phénomènes de duplication,
entraînant une augmentation de l’expression de la protéine et la résistance aux aryl-
amino-alcool (comme la méfloquine ou la luméfantrine) et une baisse de sensibilité aux
dérivés de l’artémisinine (mais sans lien statistiquement établi avec l’efficacité clinique
des ACT) ; (2) soit à l’apparition de mutations au niveau des codons N86Y, Y184F,
S1034C, N1042D et D1246Y, entraînant une altération de sensibilité des parasites à
certains antipaludiques comme les amino-4-quinoléines. Il existe un effet antagoniste
entre la sensibilité à la chloroquine et à la méfloquine : la mutation 86Y diminue la
sensibilité des parasites à la chloroquine, mais augmente celle de la méfloquine. De
même, l’augmentation du nombre de copies du gène (86N) augmente la résistance à la
méfloquine et à l’inverse, elle accroît la sensibilité à la chloroquine.
P. falciparum dihydrofolate reductase (Pfdhfr) : Ce gène, situé sur chromosome 4,
code pour une enzyme intervenant dans la voie de synthèse des folates. Elle est la cible
des médicaments anti-folates (pyriméthamine, par exemple) qui, en inhibant son activité
enzymatique, entraînent le blocage de la synthèse des pyrimidines et la réplication de
l’ADN parasitaire. L’accumulation de plusieurs mutations spécifiques au sein de cette
protéine (codons N50R, C51I, S108N et I164L) entraînent la résistance clinique des
parasites à l’action des anti-folates (MENARD et al., 2013).
P. falciparum dihydropteroate synthase (Pfdhps) : La dihydroptéroate synthase
(DHPS) est une autre enzyme intervenant dans la synthèse des folates (le gène
correspondant est situé sur le chromosome 8). Elle est inhibée par les sulfamides. Les
mutations se situant au niveau des codons S436A/F, K437G, K540E, A581G, A613S/T
confèrent une résistance à la sulfadoxine. L’analyse groupée des mutations au niveau
des gènes Pfdhfr et Pfdhps permet de prévoir l’efficacité clinique de l’association
sulfadoxine-pyriméthamine, largement utilisée en Afrique chez la femme enceinte en
Traitement Préventif Intermittent (TPI) ou en association avec l’artésunate en traitement
curatif (MENARD et al., 2013).
P. falciparum cytochrome b (Pfcytb) : Porté par le génome mitochondrial, le gène cytb
code pour le complexe cytochrome bc1 intervenant dans le transport des électrons et la
synthèse de l’ATP. Il est la cible de l’atovaquone. Les mutations au niveau du codon
Y268N/S/C diminuent l’efficacité théorique de l’association atovaquone/proguanil
actuellement largement utilisée en chimioprophylaxie par les voyageurs.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 26
Aucune mutation n’a pu être mise en évidence sur des isolats sauvages. Les mutants
retrouvés ont tous été identifiés chez des patients au décours d’un traitement par
l’atovaquone (sélection intra-hôte), et entraînent une diminution importante de la
sensibilité du parasite à l’atovaquone (MENARD et al., 2013).
P. falciparum sodium/hydrogen exchanger gene (Pfnhe-1) : Le gène Pfnhe-1
(chromosome 13) code pour une protéine impliquée dans les mécanismes d’homéostasie
parasitaire comme la régulation du pH, le volume et la composition ionique du
cytoplasme. Il a été en particulier montré que le niveau d’expression de la protéine
PfNHE influençait la sensibilité des parasites à la quinine en liaison avec d’autres
facteurs, et que certains allèles du microsatellite intragénique ms4760 étaient associés à
des diminutions de sensibilité in vitro d’isolats ou de clones de P. falciparum adaptés
en culture (MENARD et al., 2013).
I.8 Paludisme et vaccin
La mise au point d’un vaccin efficace contre le paludisme a connu une avancée remarquable au
cours de ces dix dernières années. Cependant, de nos jours aucun vaccin présentant une
efficacité suffisante et durable n’est disponible. Cela est dû aux nombreuses difficultés
rencontrées, notamment la grande variabilité génétique avec des antigènes de surface variables
du parasite, la complexité de son cycle de vie avec plusieurs étapes et des antigènes multiples
et différents à chaque étape et un taux de réplication élevé avec une évolution rapide de la
stratégie d’échappement au système immunitaire par le parasite (MECHAIN, 2015). Les
antigènes exprimés sont souvent polymorphes d’un clone parasitaire à l’autre et certains
antigènes comme ceux codés par des gènes var sont polymorphes au sein d’un même clone
parasitaire. Par ailleurs, un essai vaccinal a montré que les réponses immunes induites pouvaient
sélectionner des parasites exprimant un variant antigénique différent de celui utilisé pour
immuniser (ROGIER et al., 2006). Les candidats vaccins se distinguent d’abord par les stades
parasitaires au niveau desquels les antigènes sont exprimés. De ces stades dépendent l’effet
attendu du vaccin et le type de réponse immune susceptible d’être protectrice. On distingue :
Les Vaccins contre les stades pré-érythrocytaires qui doivent induire des réponses
immunes visant les sporozoïtes ou les schizontes hépatiques dans le but d’empêcher
toute libération de mérozoïtes dans le sang. Pour induire une immunité chez des
individus non-immuns, l’efficacité de ce type de vaccin doit être de 100 % (ROGIER et
al., 2006).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 27
Les vaccins contre les stades érythrocytaires asexués visant soient à empêcher
l’invasion des hématies et donc à contrôler les densités plasmodiales circulantes, soit à
empêcher l’évolution des infections vers les formes cliniques et potentiellement graves
de la maladie. La mise au point d’un tel vaccin se heurte à différents obstacles : les
difficultés d’évaluation expérimentale chez l’homme, l’absence de modèle animal
pertinent et l’absence de corrélation immunologique de la protection.
Les Vaccins bloquant la transmission ou altruiste ne visant pas à protéger l’individu
vacciné mais à limiter la transmission des parasites de l’homme au vecteur, et
secondairement du vecteur à l’homme. Si la couverture ou l’efficacité vaccinale ne sont
pas totales, un petit nombre d’individus infectés suffirait à assurer la transmission du
paludisme (ROGIER et al., 2006).
Dans sa stratégie mondiale de lutte contre le paludisme 2016-2030, l’OMS vise désormais à
réduire les taux de mortalité et l’incidence du paludisme d’au moins 90 % d’ici à 2030. Dans
cette nouvelle stratégie, la place des vaccins antipaludiques devrait être importante, non pas de
façon exclusive mais bien de façon complémentaire aux autres moyens de lutte disponibles,
préventifs, diagnostiques et thérapeutiques(OMS, 2015a). Actuellement, plusieurs candidats
vaccins préventifs contre les infections à P. falciparum et à P. vivax sont en développement. En
2015, 18 candidats vaccins, avec des modes d’action différents, sont au stade d’essais cliniques
(MORENO et JOYNER, 2015). Deux d’entre eux ciblent P. vivax en tenant compte du stade
hypnozoïte. Les 16 autres visent P. falciparum seul. Le plus avancé d’entre eux, est le vaccin
MosquirixTM ou RTS, S/AS01 (Figure 11). Il s’agit d’un vaccin antigénique recombinant, sous
forme de particules de type virus non infectieux produites par des cellules de levure
(Saccharomyces cerevisiae) par génie génétique (technologie de l’ADN recombinant). Le nom
de ce vaccin inactivé renvoie à sa composition (COHEN et al., 2010; MECHAIN, 2015) :
R : région centrale répétée de la protéine de surface du sporozoïte de P. falciparum
(circumsporozoite protein) ;
T : et sa région carboxy-terminale complète qui contient plusieurs épitopes de cellule T ;
S, fusionnées avec l’antigène de surface de l’hépatite B ;
S/ : combinée (Co-expression dans les cellules de levure et auto-assemblage) avec l’antigène
de surface de l’hépatite B (libre) ;
AS01 : formulée avec un adjuvant contenant des liposomes, un lipide A mono-phosphorylé
(MPL) et une saponine (la QS21).
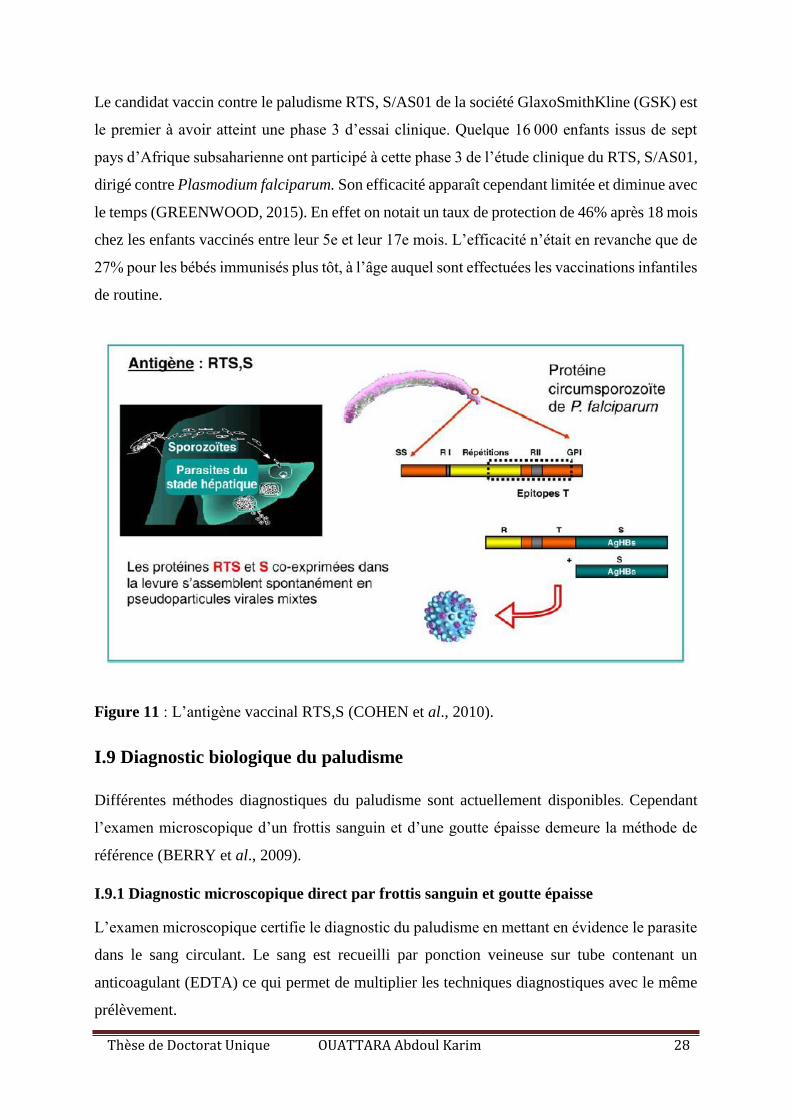
Thèse de Doctorat Unique OUATTARA Abdoul Karim 28
Le candidat vaccin contre le paludisme RTS, S/AS01 de la société GlaxoSmithKline (GSK) est
le premier à avoir atteint une phase 3 d’essai clinique. Quelque 16 000 enfants issus de sept
pays d’Afrique subsaharienne ont participé à cette phase 3 de l’étude clinique du RTS, S/AS01,
dirigé contre Plasmodium falciparum. Son efficacité apparaît cependant limitée et diminue avec
le temps (GREENWOOD, 2015). En effet on notait un taux de protection de 46% après 18 mois
chez les enfants vaccinés entre leur 5e et leur 17e mois. L’efficacité n’était en revanche que de
27% pour les bébés immunisés plus tôt, à l’âge auquel sont effectuées les vaccinations infantiles
de routine.
Figure 11 : L’antigène vaccinal RTS,S (COHEN et al., 2010).
I.9 Diagnostic biologique du paludisme
Différentes méthodes diagnostiques du paludisme sont actuellement disponibles. Cependant
l’examen microscopique d’un frottis sanguin et d’une goutte épaisse demeure la méthode de
référence (BERRY et al., 2009).
I.9.1 Diagnostic microscopique direct par frottis sanguin et goutte épaisse
L’examen microscopique certifie le diagnostic du paludisme en mettant en évidence le parasite
dans le sang circulant. Le sang est recueilli par ponction veineuse sur tube contenant un
anticoagulant (EDTA) ce qui permet de multiplier les techniques diagnostiques avec le même
prélèvement.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 29
Les étalements peuvent également être réalisés à partir d’un prélèvement capillaire par piqûre
au bout du doigt ou du talon pour les bébés. Quelques gouttes de sang sont déposées sur une
lame porte-objet pour confectionner une goutte épaisse, et un frottis mince. La lame est ensuite
séchée pendant 30 minutes, colorée au Giemsa à 10 % et lue au microscope optique à la lumière
électrique. C’est une technique de concentration, car la quantité de sang examinée est 20 à 30
fois plus élevée que lors du frottis sanguin. Sa sensibilité va de 10 à 20 parasites/µL (BERRY
et al., 2009). Le Frottis sanguin permet d’identifier l’espèce plasmodiale en cause à partir des
critères morphologiques des parasites et des hématies parasitées et de calculer la parasitémie,
exprimée en pourcentage d’hématies parasitées, très utile en cas d’infection par P. falciparum.
Sa sensibilité se situe entre 100-300 parasites/μL mais nécessite une lecture attentive pendant
au moins 20 minutes.
I.9.2 Détection d’Antigènes palustres par tests Rapide d’Orientation diagnostic (TROD)
Les TROD reposent sur le principe de l’immuno-chromatographie en utilisant des bandelettes
sensibilisées par des anticorps monoclonaux spécifiques détectant des antigènes plasmodiaux.
Ils sont réalisés avec une goutte de sang déposée sur une bandelette et ne nécessitent aucun
appareillage. La plupart de ces tests permettent la mise en évidence de l’Histidin-Rich Protein
2 (HRP2) spécifique de P. falciparum ; mais certains permettent la mise en évidence de la
Plasmodium Lactate Déshydrogénase (pLDH) commune aux quatre espèces plasmodiales. La
sensibilité et la spécificité revendiquées par les fabricants de ces tests sont comparables. Les
tests associent souvent sur une même bandelette plusieurs systèmes de détection, ajoutant alors
à la recherche de P. falciparum celle de P. vivax et/ou des trois autres espèces (BERRY et al.,
2009).
I.9.3 Détection des acides nucléiques par les techniques d’amplification génique
L’amplification génique par PCR est la technique la plus utilisée. C’est la technique la plus
sensible qui permet de détecter de très faibles parasitémies de l’ordre de 0.3 parasite/μL de sang
avec une possibilité de quantification de l’ADN plasmodial en utilisant la PCR quantitative. La
PCR a également une excellente valeur prédictive négative avec une spécificité absolue si elle
est réalisée dans de bonnes conditions. L’amplification du gène codant pour la petite sous unité
18S de l’ARN ribosomal permet aussi l’identification des espèces plasmodiales en cause en
utilisant une PCR nichée (BERRY et al., 2009).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 30
CHAPITRE II : HEMOGLOBINOPATHIES ET DEFICIT EN G-6-PD
Le paludisme est considéré comme la plus forte pression de sélection naturelle dans l’histoire
récente du génome humain. Il est supposé que le paludisme se soit réellement installé lors des
changements d’habitude de vie correspondant au passage de la chasse et de la cueillette vers les
pratiques agricoles et la sédentarisation il y a environ 10000 ans (KWIATKOWSKI, 2005;
HEDRICK, 2011). Des études micro et macroépidemiologiques ont montré l’existence d’un
chevauchement entre les distributions géographiques du paludisme à Plasmodium falciparum
et celles de certaines anomalies génétiques du globule rouge tels que les thalassémies, les
hémoglobinopathies (HbS, HbC et HbE), des perturbations métaboliques (en particulier le
déficit en G-6-PD) ou des anomalies de protéines membranaires.
Les mutations responsables de ces maladies n’entrainent généralement pas de manifestations
cliniques majeures à l’état homozygote, mais leur maintien au sein des populations témoigne
de l’avantage sélectif qu’elles confèrent à l’état hétérozygote aux individus vivant en zone
d’endémie palustre (BAUDUER, 2012). Dans ce chapitre nous insisterons sur les
hémoglobinoses S et C et la déficience en G-6-PD.
II.1 Hémoglobinopathies
Haldane a été le premier à proposer en 1949, d’une part, l’influence du paludisme sur le génome
humain, et, d’autre part, l’effet protecteur contre le paludisme de variants génétiques
provoquant la réduction ou l’absence d’une ou plusieurs sous-unité(s) de l’hémoglobine,
causant ainsi les thalassémies (HEDRICK, 2011).
De la même manière, des variants génétiques causant d’autres hémoglobinopathies ont été
ensuite associés à la résistance au paludisme (ALLISON, 1964) et des analyses récentes
indiquent une sélection balancée de ces variants, sous la pression positive du paludisme et celle
négative de l’hémoglobinopathie (HEDRICK, 2012).
II.1.1 Définition
Les hémoglobinopathies sont des maladies génétiques liées à un trouble qualitatif
(hémoglobinoses) ou quantitatif (thalassémies) de l’hémoglobine avec remplacement des sous-
unités protéiques composant l’hémoglobine par des protéines mutantes (LABIE et ELION,
2005).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 31
II.1.2 Hémoglobinose S (HbS)
Les hémoglobines anormales ont été les variants protéiques les mieux décrits depuis la mise en
évidence en 1949 du premier d’entre eux, l’HbS, responsable de la drépanocytose. On en
compte actuellement plusieurs centaines, variants d’une ou de l’autre des chaînes α ou β de
l’hémoglobine. Certains sont silencieux, d’autres s’accompagnent d’anomalies fonctionnelles
rendant compte de la pathologie observée (LABIE et ELION, 2005). La forme βs de
l’hémoglobine est provoquée par une mutation génétique unique de « transversion » du sixième
codon du premier exon du gène de la globine β6 (GAG GTG) donnant comme conséquence
la substitution de l’acide glutamique par la valine (β6Glu->Val) (HEDRICK, 2011).
II.1.2.1 Epidémiologie
La drépanocytose est sûrement l’hémoglobinopathie grave la plus fréquente (DELICAT-
LOEMBET et al., 2014, PIEL et al., 2015). Son épidémiologie est bien connue, chez les
populations d’origine africaine, mais aussi au Moyen-Orient, en Inde ainsi qu’au niveau des
pays du pourtour méditerranéen (Figure 12). La maladie affecterait quelques 12 500 personnes
au Royaume-Uni et 20 à 25 millions de personnes dans le monde (BOOTH et al., 2010). Elle
touche principalement l’Afrique où les plus fortes prévalences du trait drépanocytaire (10% à
40% de la population) surviennent entre les latitudes 15° Nord et 20° Sud (AGASA et al., 2010).
On estime qu’environ 275 000 enfants naissent chaque année en Afrique (approximativement
85% du taux mondial) avec une anomalie majeure de l’hémoglobine dont la plus fréquente est
celle de la drépanocytose (LIN et al., 2015). La prévalence de ses anomalies augmente chaque
année du fait de la croissance de la population et pourrait toucher plus de 400 000 enfants dans
le monde d’ici à 2050 (PIEL et al., 2015). Sans traitement, qui est rarement disponible dans les
pays à faibles revenus, la grande majorité des enfants drépanocytaires (50 à 80%) meurent avant
l’âge de 5 ans (PIEL et al., 2013a; MULUMBA et WILSON, 2015). Cette maladie génétique
du sang prédomine en Afrique centrale, où 30% de la population est drépanocytaire. Dans
certaines parties de l’Afrique subsaharienne, la drépanocytose touche jusqu’à 2 % des nouveau-
nés (SIMPORE et al., 2002b). La fréquence du trait drépanocytaire, c'est-à-dire le pourcentage
de porteurs sains qui n’ont hérité du gène mutant que d’un seul des parents, atteint 10 à 40 %
en Afrique équatoriale, 1 à 2 % sur la côte de l’Afrique du Nord et moins de 1 % en Afrique du
Sud. Dans les pays d’Afrique de l’Ouest (Ghana, Burkina Faso et Nigéria), la fréquence du trait
drépanocytaire atteint 15 à 30 %.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 32
Dans les pays où la prévalence du trait drépanocytaire est supérieure à 20%, la maladie affecte
environ 2% de la population (AIDOO et al., 2002). Au Burkina Faso, la drépanocytose sous ses
diverses formes graves (HbSS ou HbSC) touche 8,4% des enfants qui vont pour les
consultations médicales dans les structures sanitaires (SIMPORE et al., 2002b) et 1,2% des
enfants qui vont à l’école (SIMPORE et al., 2002a).
Figure 12 : Distribution mondiale de l’hémoglobine S (PIEL et al., 2010)
II.1.2.2 Physiopathologie
Le mécanisme physiopathologique de base de la drépanocytose a été très précisément décrit,
centré sur la polymérisation de l’HbS désoxygénée et les déformations cellulaires subséquentes
observées chez les homozygotes SS (Figure 13). Les molécules d’hémoglobine drépanocytaire
(HbS) ont la propriété, sous leur forme désoxygénée, de polymériser pour former des fibres qui
déforment l’érythrocyte en lui donnant une forme caractéristique en faucille, le drépanocyte.
La polymérisation des molécules d’HbS déforme la cellule, la fragilise et la rigidifie. Le
drépanocyte a comme particularité d’avoir perdu ses propriétés rhéologiques de déformabilité
et d’élasticité nécessaires pour passer à l’intérieur des petits vaisseaux de l’organisme, et d’être
plus rapidement détruit qu’un globule rouge normal, ce qui rend compte de l’anémie
hémolytique (KOSSOROTOFF et al., 2014). Les individus porteurs de l’allèle HbS
développent une anémie falciforme, ou drépanocytose, dont les symptômes les plus courants
sont des accidents vasculaires cérébraux, des douleurs articulaires, des insuffisances rénales et
hépatiques.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 33
Si les symptômes restent limités chez les individus hétérozygotes HbAS, qui portent l’allèle
mutant HbS et l’allèle sain HbA, l’homozygotie est fortement délétère. Les syndromes
drépanocytaires majeurs recouvrent différents génotypes selon que l’HbS est la seule
hémoglobine anormale présente (patients homozygotes SS et hétérozygotes composites S/β0),
ou associée à d’autres hémoglobines pathologiques (patients SC et S/β+). Les formes SS et S/β0
sont les plus sévères. Il existe en effet une production résiduelle d’hémoglobine normale chez
les patients S/β+ qui limite la polymérisation de l’HbS. Les complications observées chez les
patients atteints de syndromes drépanocytaires majeurs font intervenir plusieurs mécanismes :
d’une part, des phénomènes « viscosité-vaso-occlusion » (Crises Vaso-Occlusives [CVO]
osseuses ou d’organes avec occlusion des micro-vaisseaux) et, d’autre part, des phénomènes «
hémolyse-dysfonction endothéliale » (hyper-tension artérielle pulmonaire, priapisme).
L’atteinte vasculaire au niveau cérébral est aussi fréquente chez les enfants drépanocytaires
homozygotes. Elle peut par ses conséquences être source d’handicap sévère, d’où l’importance
de son dépistage et de sa prise en charge (KOSSOROTOFF et al., 2014).
Figure 13 : Mécanisme physiopathologique de base de la drépanocytose (LABIE et ELION,
2005).
Légende : La mutation au 6e codon du gène b-globine conduit à la substitution d’un acide
glutamique par une valine et à une hémoglobine anormale : l’HbS. À basse pression en
oxygène, la désoxy-HbS polymérise et entraîne une déformation, une rigidification et une
fragilisation cellulaires, responsables de l’anémie hémolytique et de la vaso-occlusion.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 34
II.1.3.3 Rapport entre l’hémoglobine S et le Paludisme
La superposition, en Afrique, des zones de paludisme sévère et de celles où la mutation
drépanocytaire est endémique est connue depuis longtemps (LABIE et ELION, 2010). Le rôle
sélectif du Plasmodium a été évoqué, procurant un avantage de survie aux hétérozygotes AS
alors que les malades SS mouraient le plus souvent de façon prématurée. Ce phénomène de
« polymorphisme équilibré » a été formalisé par Allison en 1964. En effet, les individus
porteurs de l’hémoglobine normale (AA) qui avaient le paludisme, étaient plus malades et
mouraient davantage que les porteurs de la mutation (AS). Donc, au fil des siècles et des
générations, cette mutation, qui était très rare au départ, est devenue plus fréquente.
C’est d’ailleurs parce que le paludisme est très présent en Afrique, que la drépanocytose touche
beaucoup les Africains. La drépanocytose est ainsi répandue parce qu'à l'état hétérozygote, la
présence du gène drépanocytaire contribue à protéger son porteur du paludisme, et lui procure
donc un avantage sélectif par rapport aux porteurs des gènes normaux AA, qui eux sont
vulnérables au Plasmodium.
L’hémoglobine S à l’état hétérozygote ne prévient pas l’infection à Plasmodium mais réduit
plutôt le risque de développer les formes graves de la maladie (MANGANO et al., 2015). En
effet les individus HbAS ont généralement une densité parasitaire significativement plus faible
par rapport aux individus de génotype HbAA (TRAVASSOS et al., 2015). Plusieurs études se
sont intéressées aux mécanismes de protection de l’hémoglobine S contre le paludisme.
L’implication de deux facteurs semble cependant dominer à savoir l’élimination précoce des
globules rouges falciformes infectés par le plasmodium et l’entrave à la croissance du parasite
dans l’hématie falciforme (LELLIOTT et al., 2015).
In vitro, il a été démontré qu’à pression d’oxygène réduite le développement du parasite se fait
mal dans le globule rouge (GR) AS, où s’observe une diminution de la densité parasitaire. Ces
GR AS seraient captés préférentiellement par la rate. De plus dans les GR AS il y aurait
interférence dans le remodelage des filaments d’actine de l’hôte par le parasite pour la formation
du sillon de Maurer nécessaire à ses trafics protéiques entre le milieu intra et extracellulaire
(CYRKLAFF et al., 2011 ; KILIAN et al., 2015).
Les sujets HbS exprimeraient également des titres élevés d’anticorps IgG contre les protéines
de surface de P. falciparum que les sujets HbAA d’où leur élimination précoce par le système
immunitaire (VERRA et al., 2007).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 35
II.1.4 Hémoglobine C
L'hémoglobine C (Hb C) est l'une des variantes les plus courantes d'hémoglobine structurelles
dans la population humaine. La forme βc de l’hémoglobine est induite par la mutation génétique
de « transition » du sixième codon du premier exon du gène de la globine β6 (GAG
AAG) donnant comme conséquence la substitution de l’acide glutamique par la lysine (β6Glu-
>Lys).
II.1.4.1 Epidémiologie
L’hémoglobine C s’observe essentiellement chez les sujets d’ascendance noire. Le gène βc
semble avoir son origine dans la zone ouest de l'Afrique. L’hémoglobine C se trouve dans
diverses populations d'Afrique, d'Europe du Sud et en Amérique centrale (Figure 14), bien que
sa distribution allélique exacte à travers ces diverses populations reste encore à préciser (PIEL
et al., 2013b). Aux États-Unis, 2-3% des américains d’origine africaine sont hétérozygotes pour
l'Hb C, et environ 1 sur 5000 sont homozygotes. Les plus hautes fréquences de l’hémoglobine
C ont été rapporté en Afrique de l’Ouest principalement au Burkina Fao, au Ghana et au Togo
avec des fréquences alléliques supérieures à 15% (PIEL et al., 2013b).
Figure 14 : Répartition de l’hémoglobine C dans le monde (PIEL et al., 2013b)
II.1.4.2 Physiopathologie
L'hémoglobine C est moins soluble que l'hémoglobine A dans les globules rouges. En effet
l’hémoglobine C est susceptible d’interactions électrostatiques entre les groupes de ß6-lysyl
chargés positivement et les groupes chargés négativement des molécules adjacentes.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 36
Il en résulte la formation de cristaux, pouvant conduire à une augmentation de la viscosité
sanguine et une rigidité cellulaire avec une diminution de la longévité des globules rouges.
Contrairement à la drépanocytose, l’HbC ne provoque pas de polymérisation intracellulaire
linéaire au niveau des zones intravasculaires de faible tension d'oxygène. Ainsi, bien qu'on
observe une légère déformabilité des globules rouges associée à la variante Hb C, cela
n’entraine pas de vaso-occlusion. Les états hétérozygotes aussi bien qu’homozygotes peuvent
induire une déshydratation des érythrocytes, et une Concentration Corpusculaire Moyenne en
Hémoglobine élevée (CCMH) peut être notée sur un hémogramme complet. Les individus
homozygotes porteurs de l’HbC développent une splénomégalie, des calculs biliaires et une
anémie modérée, tandis que les hétérozygotes sont asymptomatiques. Les patients atteints du
trait de l'hémoglobine C (HbAC) sont phénotypiquement normale sans symptômes
cliniquement évidents. Bien que les complications cliniques de la maladie de l'hémoglobine C
ne soient pas graves, l’association avec d'autres hémoglobinopathies telles que l'hémoglobine
S peut avoir des conséquences importantes.
II.1.4.3 Rapport entre l’hémoglobine C et le Paludisme
L’hémoglobine C aurait été sélectionnée sous la pression positive du paludisme à travers
l’Afrique de l’Ouest, principalement au Burkina Faso, au Ghana et au Togo où on observe des
fréquences élevées de cette mutation (PIEL et al., 2015). En effet un certain nombre d'études
suggèrent que l'hémoglobine C à l’instar de l’hémoglobine S, protège contre le paludisme en
fournissant un avantage évolutif sélectif à des gens qui expriment cette variante de
l'hémoglobine dans les régions où cette maladie est endémique (MODIANO et al., 2008).
Des études in vitro ont montré la baisse des taux de multiplication des parasites dans les HbCC
en comparaison aux globules rouges HbAA. Ainsi l'effet inhibiteur de l'Hb C sur la parasitémie
serait dû à une faible expression de PfEMP1 impliqué dans les phénomènes de cytoadhérence
et de rosetting et à une stimulation des réponses immunitaires dès les premiers stades de
l’infection (VERRA et al., 2007). Plusieurs études ont démontré la protection de l’HbC contre
le paludisme au Mali (TRAVASSOS et al., 2015), au Burkina Faso (MODIANO et al., 2001b;
MODIANO et al., 2008), et au Ghana (GHANSAH et al., 2012). Modiano et collaborateurs
dans leur étude cas-témoins au Burkina Faso ont trouvé une forte association entre la résistance
au paludisme clinique et de la présence de la variante Hb C à la fois à l’état hétérozygote (29%
de réduction du risque) qu’à l'état homozygote (93% de réduction du risque). Quelques études
cas-témoins et des études transversales n’ont rapporté aucune association entre Hb C et une
incidence réduite de la parasitémie (AMOAKO et al., 2014).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 37
Les fréquences élevées de l’allèle C observées en Afrique de l’Ouest ainsi que l’absence de
pathologies majeures chez les sujets HbAC et HbCC en comparaison aux sujets HbSS et HbSC,
suggère qu’à long terme sous la pression du paludisme on assistera à un remplacement de
l’allèle S par l’allèle C dans cette zone (MODIANO et al., 2001b).
II.1.5 Techniques de diagnostic des hémoglobines anormales
II.1.5.1 Techniques d’électrophorèse et de chromatographie
La plupart des hémoglobines anormales (mais non toutes) ont une charge électrique différente
de celle de l’hémoglobine adulte normale. En soumettant une hémoglobine suspecte à une
migration électrophorétique, on peut reconnaître sa migration anormale et découvrir ainsi la
cause d’une anémie. La technique utilisée pour le dépistage est l’électrophorèse sur acétate de
cellulose à pH alcalin dans laquelle les hémoglobines migrent vers l’anode. Elle tend à être
remplacée par l’électrophorèse capillaire, rapide et automatisable (CAQUET, 2010). L’HbS
migre sur acétate de cellulose en pH alcalin, entre l’hémoglobine A et l’hémoglobineA2 de la
même manière que de nombreux autres mutants (comme D ou G, voir figure ci-dessous). Il est
donc nécessaire, pour bien l’identifier, d’avoir recours à un second critère comme
l’électrophorèse sur gel d’agar à pH acide. L’HbC migre plus lentement que la S en gel d’agar
à pH acide. De l’anode vers la cathode on trouve donc les HbA, S et C. Dans ce système, HbS
se détache nettement. Ces deux méthodes sont complétées par un test de solubilité (ou test
d’Itano) mettant en évidence la polymérisation de l’hémoglobine S qui précipite en présence de
dithionite de sodium alors que les autres hémoglobines restent en solution. La Chromatographie
Liquide Haute Performance (CLHP) permet d’identifier avec précision l’homozygotie pour
l’HbS, l’hétérozygotie composite pour l’HbS et l’HbC (HbSC), l’hétérozygotie composite pour
l’HbS et la β-thalassémie.
II.1.5.2 Techniques de biologie moléculaire
Une variété de techniques basées sur l’amplification de l’ADN génomique par PCR ont été
développées pour le diagnostic des mutations du gène de la beta globine. Le matériel de base
utilisé pour l’extraction d’ADN est le plus souvent du sang total prélevé sur tube EDTA. La
recherche d’une mutation connue peut être réalisée, en alternative au séquençage, par une PCR-
RFLP, par PCR en temps réel avec révélation par sondes fluorescentes spécifiques ou encore
par la technique de reverse dot-blot (OLD, 2003).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 38
II.2 Déficit en G-6-PD
La Glucose-6-phosphate déshydrogénase est une enzyme ubiquitaire présente dans le
cytoplasme de toutes les cellules. Elle catalyse la première étape de la voie des pentoses
phosphates qui génère le Nicotinamide Adénosine Dinucléotide Phosphate réduit (NADPH),
coenzyme de la glutathion-réductase (HOWES et al., 2013a).
Le déficit en G-6PD touche principalement la lignée érythrocytaire. Tout au long de sa vie, le
globule rouge est soumis à des agents oxydants menaçant l’intégrité de sa membrane et de
l’hémoglobine. La lutte contre ces agressions oxydantes se fait grâce à l’action de la glutathion-
réductase en présence de NADPH (MEGARBANE, 2008). Cependant, contrairement aux
cellules des autres tissus qui possèdent de nombreuses voies de régénération de la NADPH,
pour les globules rouges qui manquent de noyau, de mitochondrie et d’autres organites, la G-
6-PD est particulièrement importante dans la lutte contre le stress oxydatif (CAPPELLINI et
FIORELLI, 2008). En effet dans les érythrocytes, la voie des pentoses phosphates avec pour
enzyme clé la G-6-PD, demeure la seule source de production de NADPH, coenzyme essentiel
à la lutte contre les agressions oxydantes.
II.2.1 Définition
Le déficit en G-6-PD est une pathologie génétique liée au chromosome X due à un déficit en
G-6-PD, une enzyme indispensable à la survie des globules rouges (FUZIER, 2015).
II.2.2 Historique
Les premiers éléments rapportés sur cette anomalie génétique datent de l’antiquité avec
Pythagore interdisant à ses élèves de manger les fèves (Vicia faba), probablement pour leur
effet potentiellement pathogène (HOWES et al., 2013a).
À partir de la fin du XIXe siècle, des cas de plus en plus nombreux d’accidents liés à leur
ingestion, voire à l’inhalation de leurs pollens, sont rapportés dans la littérature (WAJCMAN
et GALACTEROS, 2004).
En 1952, lors de campagnes militaires américaines en Asie, des études au chrome-51 avaient
permis de démontrer qu’une hémolyse corpusculaire se produisait lorsque des globules rouges
provenant de sujets sensibles étaient injectés à des sujets normaux recevant de la primaquine, à
la différence des globules rouges normaux injectés à des sujets sensibles à la primaquine
(HOWES et al., 2013a).
En 1956, Carson découvre que les globules rouges pathologiques présentaient un faible taux
d’activité de la G-6-PD (CARSON, 1956).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 39
En 1958, Childs détecte l’anomalie génétique responsable sur le chromosome X, expliquant
pourquoi la transmission de ce déficit touche principalement les hommes (CHILDS, 1958).
En 1959, Beutler décrit le mécanisme biochimique de l’anémie hémolytique après la prise de
médicaments oxydants (BEUTLER, 1959).
En 1986, le gène du déficit en G6PD est cloné et séquencé (MARTINI et al., 1986)
En 1989, l’OMS publie dans son bulletin la première étude synthétique de ce déficit, réalisée
avec le concours de biochimistes, d’hématologues et de pédiatres.
Depuis, les travaux sur cette enzymopathie se sont multipliés afin de mieux comprendre les
aspects génétiques, moléculaires et physiopathologiques en cause.
II.2.3 Epidémiologie
Le déficit en G-6-PD est l’enzymopathie héréditaire la plus fréquente dans le monde. On estime
à environ 470 millions de personnes affectées par la maladie soit près de 5% de la population
mondiale (HO et JOHN, 2015). Sa répartition mondiale n’est pas le fait du hasard. Elle reflète
le seul avantage conféré par la maladie aux patients atteints : une résistance accrue au
paludisme. Cette hypothèse, désormais communément admise, a longtemps été suggérée par
la remarquable concordance de répartition géographique entre les zones d’endémie du
paludisme et du déficit en G-6-PD (HOWES et al., 2013a). La fréquence des mutations varie
considérablement entre les différentes populations (Figure 15). Sa prévalence est surtout élevée
dans les régions tropicales, environ 25% de la population. Parmi les nombreux variants
moléculaires connus du déficit en G-6-PD, le plus répandu est la G-6-PDA‐ avec une prévalence
de 11% à 13% chez les afro-américains (HO et JOHN, 2015). Il concerne 90 % des patients
déficitaires en Afrique tropicale.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 40
Figure 15 : Prévalence du déficit en G-6-PD et distribution mondiale des différents variants
déficitaires (LUZZATTO et al., 2016)
Du fait des migrations de populations, la prévalence du déficit en G-6-PD est en nette
augmentation dans certains pays d’Europe du Nord, en Amérique du Nord et du Sud. Il est
également fréquent là où l’on trouve des personnes originaires d’Afrique tropicale : en
Amérique du Nord, du Sud et dans les Antilles. Mais on le rencontre aussi en Italie, aux Îles
Canaries, en Espagne, au Portugal, dans le Moyen‐Orient et le Proche‐Orient
(ALLAHVERDIYEV et al., 2012).
Le deuxième variant le plus répandu est la G-6-PD Méditerranéen (G-6-PDMed) avec une
prévalence de l’ordre de 2 à 20% dans différentes populations, exception faite des Juifs kurdes
où sa prévalence est estimée à 70% (PETERS et VAN NOORDEN, 2009). Il est présent dans
tous les pays du pourtour de la Méditerranée. Il est également répandu au Moyen‐Orient. C’est
quasiment le seul variant présent chez les Juifs kurdes ainsi qu’en Inde et en Indonésie. Dans
de nombreuses populations, on note la coexistence des variant G-6-PDA‐ et G-6-PDMed : c’est
le cas des pays du Golfe Persique (HOWES et al., 2013a).
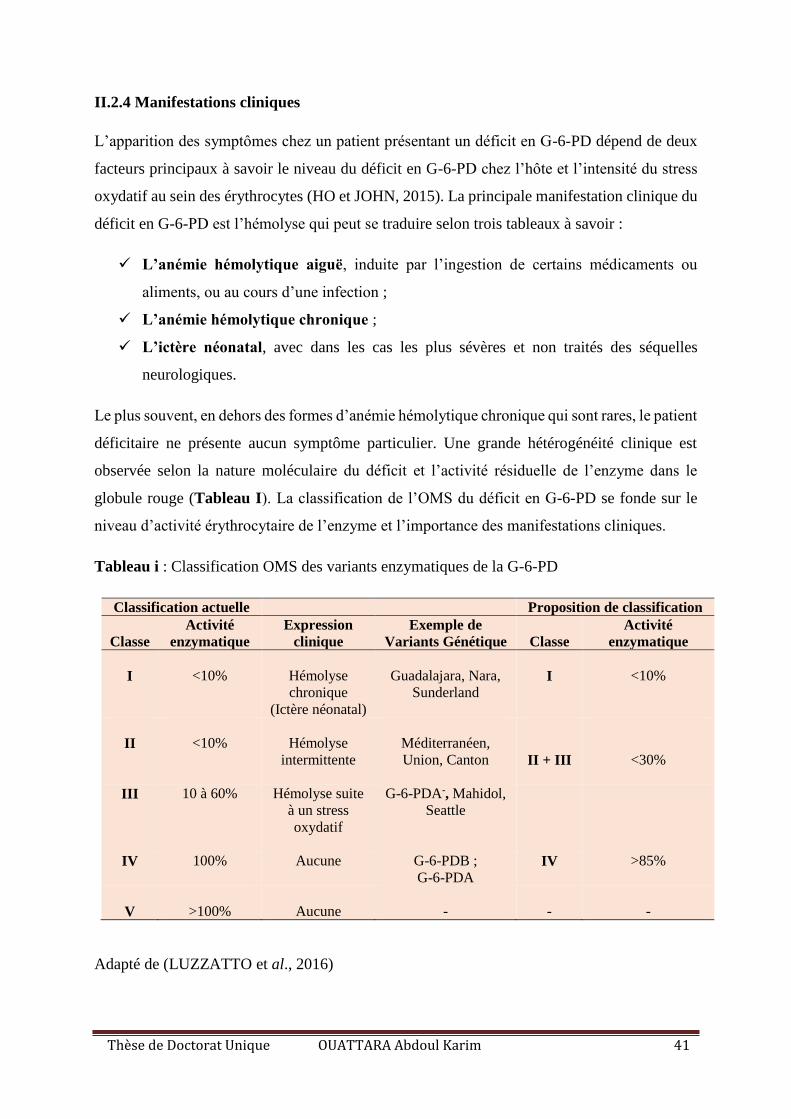
Thèse de Doctorat Unique OUATTARA Abdoul Karim 41
II.2.4 Manifestations cliniques
L’apparition des symptômes chez un patient présentant un déficit en G-6-PD dépend de deux
facteurs principaux à savoir le niveau du déficit en G-6-PD chez l’hôte et l’intensité du stress
oxydatif au sein des érythrocytes (HO et JOHN, 2015). La principale manifestation clinique du
déficit en G-6-PD est l’hémolyse qui peut se traduire selon trois tableaux à savoir :
L’anémie hémolytique aiguë, induite par l’ingestion de certains médicaments ou
aliments, ou au cours d’une infection ;
L’anémie hémolytique chronique ;
L’ictère néonatal, avec dans les cas les plus sévères et non traités des séquelles
neurologiques.
Le plus souvent, en dehors des formes d’anémie hémolytique chronique qui sont rares, le patient
déficitaire ne présente aucun symptôme particulier. Une grande hétérogénéité clinique est
observée selon la nature moléculaire du déficit et l’activité résiduelle de l’enzyme dans le
globule rouge (Tableau I). La classification de l’OMS du déficit en G-6-PD se fonde sur le
niveau d’activité érythrocytaire de l’enzyme et l’importance des manifestations cliniques.
Tableau i : Classification OMS des variants enzymatiques de la G-6-PD
Classification actuelle Proposition de classification
Classe
Activité
enzymatique
Expression
clinique
Exemple de
Variants Génétique
Classe
Activité
enzymatique
I
<10%
Hémolyse
chronique
(Ictère néonatal)
Guadalajara, Nara,
Sunderland
I
<10%
II
<10%
Hémolyse
intermittente
Méditerranéen,
Union, Canton
II + III
<30%
III 10 à 60% Hémolyse suite
à un stress
oxydatif
G-6-PDA-, Mahidol,
Seattle
IV 100% Aucune G-6-PDB ;
G-6-PDA IV >85%
V
>100%
Aucune
-
-
-
Adapté de (LUZZATTO et al., 2016)

Thèse de Doctorat Unique OUATTARA Abdoul Karim 42
II.2.5 Aspects génétiques
Le gène codant pour la G-6-PD est situé sur la partie télomérique du bras long du chromosome
X, en position q28. Il est localisé à proximité du gène responsable de l’hémophilie A. Il s’étend
sur environ 18 kb et comporte 13 exons et 12 introns (Figure 16). L’exon 13 a une taille
d'environ 800 nucléotides de long et contient le codon d'arrêt de traduction. La région codante
est divisée en 12 segments dont la taille varie entre 12 pb et 236 pb. Le premier exon ne contient
aucune séquence codante connue et l'intron 2 situé entre les exons 2 et 3 est extraordinairement
long, avec une taille d’environ 9857 pb (ALLAHVERDIYEV et al., 2012). Le rôle de cet intron
demeure encore inconnu bien qu’on le retrouve chez d’autres espèces animales autres que
l’homme.
Figure 16 : Organisation du gène G-6-PD humain (CAPPELLINI et FIORELLI, 2008)
Les bases génétiques et la grande variabilité de l’enzyme ont été établies. La séquence complète
du gène est connue. En effet il s’agit de l’un des gènes les plus polymorphiques du génome
humain avec au moins 186 mutations décrites (MINUCCI et al., 2012). L’analogie de séquence
du gène entre l’homme et la souris est de 87%. La plupart des dissemblances de séquence se
retrouve dans la région 3' UTR (UnTranslated Régions), constitué de 600 nucléotides en
moyenne et contient un seul site polyA (ALLAHVERDIYEV et al., 2012).
La maladie est liée au chromosome X avec une transmission récessive. Les garçons
hémizygotes et les filles homozygotes expriment totalement le déficit. La maladie touche donc
essentiellement les individus de sexe masculin (90 %) (Figure 17a) et sa transmission se fait
par les personnes de sexe féminin en général « porteuses saines ». Les filles hétérozygotes
présentent une forme variable, en général très modeste, du déficit du fait de l’inactivation
(Figure 17b) par lyonisation du chromosome X déficitaire (HO et JOHN, 2015).
La plupart des mutations du gène de la G-6-PD sont des mutations ponctuelles et de petites
délétions altérant la structure enzymatique. L’absence de mutations sévères indique qu’un
déficit absolu en G-6-PD pourrait être létal (LONGO et al., 2002).
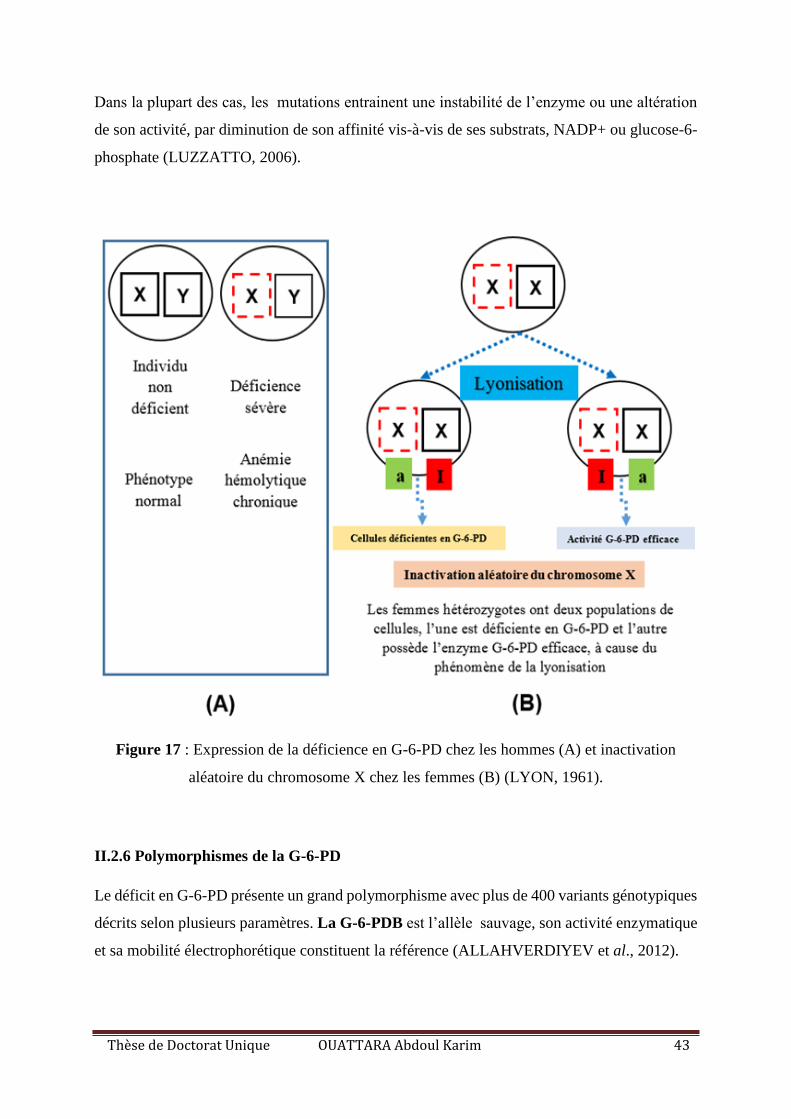
Thèse de Doctorat Unique OUATTARA Abdoul Karim 43
Dans la plupart des cas, les mutations entrainent une instabilité de l’enzyme ou une altération
de son activité, par diminution de son affinité vis-à-vis de ses substrats, NADP+ ou glucose-6-
phosphate (LUZZATTO, 2006).
Figure 17 : Expression de la déficience en G-6-PD chez les hommes (A) et inactivation
aléatoire du chromosome X chez les femmes (B) (LYON, 1961).
II.2.6 Polymorphismes de la G-6-PD
Le déficit en G-6-PD présente un grand polymorphisme avec plus de 400 variants génotypiques
décrits selon plusieurs paramètres. La G-6-PDB est l’allèle sauvage, son activité enzymatique
et sa mobilité électrophorétique constituent la référence (ALLAHVERDIYEV et al., 2012).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 44
Le variant G-6-PDA est observé avec une grande fréquence dans les populations d’ascendance
noire avec une prévalence de l’ordre de 20% à 40% chez les hommes africains et environ 20%
des hommes africains-américains sont touchés par cette anomalie génétique
(ALLAHVERDIYEV et al., 2012). Elle diffère du variant G-6-PDB par sa mobilité
électrophorétique plus anodique, par une substitution de nucléotide de l’adénine par la guanine
en position 376 ; ce qui résulte en une substitution de l’asparagine par l’acide aspartique au
niveau de la chaîne peptidique en position 126. Le variant G-6-PDA présente 90% de l’activité
enzymatique de la forme normale G-6-PDB.
Le variant G-6-PDA- retrouvé chez environ 11% des africains-américains, résulte d’une
seconde mutation sur un gène G-6-PDA. Il présente entre 10-20% de l’activité de la G-6-PDB
avec une mobilité électrophorétique similaire à la G-6-PDA (CARTER et al., 2011). La seconde
mutation couramment détectée sur le gène G-6-PDA est une substitution au niveau de l’exon 4
ou l’adénine remplace la guanine en position 202 sur la chaîne de nucléotides. Cela conduit au
remplacement de la valine par la méthionine en position 68 sur la séquence des acides aminés.
Cependant d’autres mutations autres que la mutation G202A sur l’exon 4 du gène conférant la
déficience en G-6-PD ont été identifiées en Afrique de l’Ouest. Ce sont les mutations A542T
sur l’exon 6, G680T sur l’exon 7 et T968C sur l’exon 9 du gène de la G-6-PD
(ALLAHVERDIYEV et al., 2012).
Le variant G-6-PD Méditerranéen : Ce variant répandu dans la région méditerranéenne et au
Moyen-Orient, a une même mobilité électrophorétique que la G-6-PDB. Sa demi-vie est de 8
jours et on assiste également à un déficit de synthèse. Il possède moins de 10% de l’activité de
la G-6-PDB. Il résulte de deux mutations : la cytosine (C) remplace la thymine (T) en position
563 sur la chaîne de nucléotides ce qui conduit au remplacement de la sérine par la
phénylalanine en position 188 sur la séquence des acides aminés. La seconde mutation est
silencieuse et se traduit par un remplacement de la cytosine par la thymine en position 1311.
D’autres variants A- tels les variants Canton, Mahidol, Seattle et Union existent dans le Sud-
Est asiatique et dans d’autres parties du monde (Figure 18).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 45
Figure 18 : Répartition des mutations fréquentes le long de la séquence codante du gène G6PD
(HOWES et al., 2013a).
Légende : Les exons sont présentés sous forme de cases numérotées de 2 à 13 de la gauche
vers la droite. Les cercles vides représentent des mutations qui causent des variants de classe
II ou III tandis que les cercles pleins indiquent des variants de classe I. Les carrés pleins sont
de petites délétions et la croix représente une mutation non-sens tandis que la lettre "F" désigne
une mutation du site d'épissage.
II.2.7 Rôle de la glucose-6-phosphate déshydrogénase
L’activité de la G-6-PD est nécessaire à la survie des hématies étant donné qu’elle catalyse la
première étape de la voie des pentoses phosphate, seule voie métabolique capable de générer
du pouvoir réducteur pour ces cellules dépourvues de mitochondries (HOWES et al., 2013a).
Elle transforme le glucose-6-phosphate en 6-phosphoglucono-δ-lactone, qui s’hydrolyse
classiquement en 6-phosphogluconate. Lors de cette réaction, une molécule de NADP+ est
réduite en NADPH, H+ (Figure 19). La seconde réaction de cette voie, transformation du 6-
phosphoglucono-δ-lactone en ribulose-5-phosphate, produit également du NADPH, mais, chez
les sujets déficitaires en G-6-PD, elle est totalement perturbée par le ralentissement de la
première étape. Le NADPH joue un rôle essentiel dans la réduction des agents oxydants, en
permettant dans le globule rouge en particulier, de maintenir le pool de glutathion réduit à un
niveau normal (HOWES et al., 2013a). Lorsque l’hématie possède une enzyme G-6-PD
efficace, elle résiste donc contre le stress oxydatif tandis qu’un déficit de l’enzyme G-6-PD
rend l’hématie vulnérable aux agressions oxydatives (molécules oxydantes, infections) avec
pour conséquences des anémies hémolytiques nécessitant parfois des transfusions sanguines.
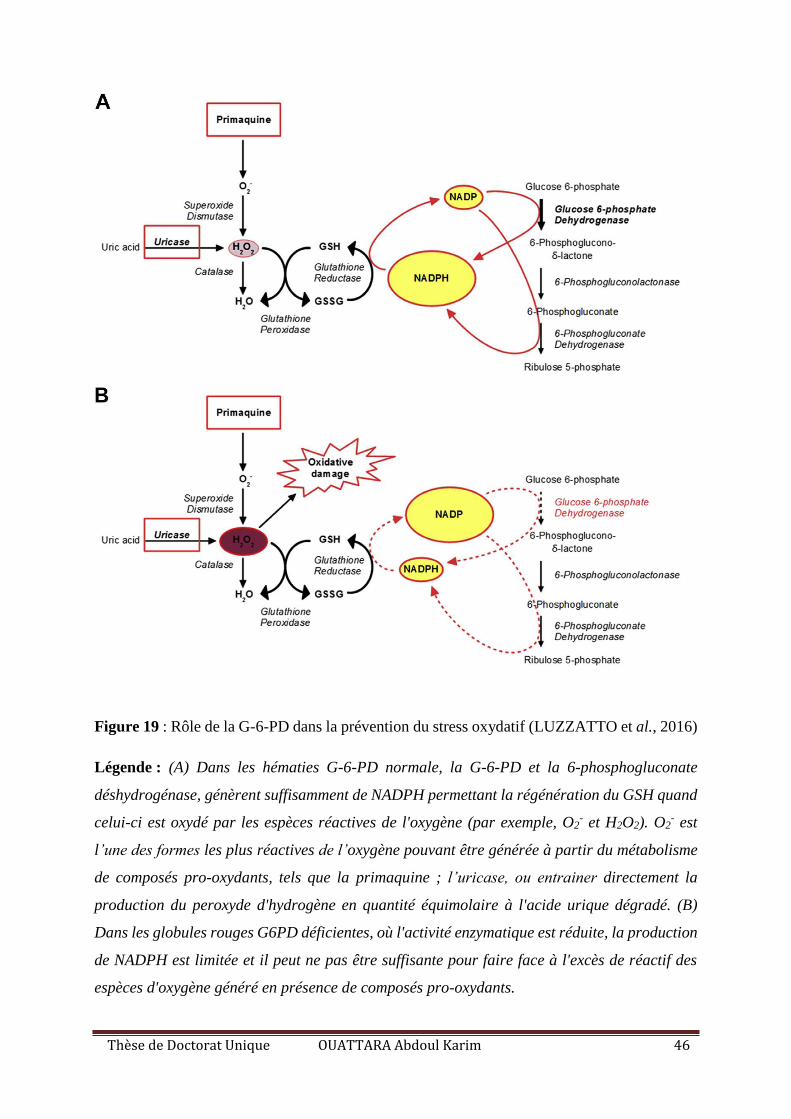
Thèse de Doctorat Unique OUATTARA Abdoul Karim 46
Figure 19 : Rôle de la G-6-PD dans la prévention du stress oxydatif (LUZZATTO et al., 2016)
Légende : (A) Dans les hématies G-6-PD normale, la G-6-PD et la 6-phosphogluconate
déshydrogénase, génèrent suffisamment de NADPH permettant la régénération du GSH quand
celui-ci est oxydé par les espèces réactives de l'oxygène (par exemple, O2- et H2O2). O2
- est
l’une des formes les plus réactives de l’oxygène pouvant être générée à partir du métabolisme
de composés pro-oxydants, tels que la primaquine ; l’uricase, ou entrainer directement la
production du peroxyde d'hydrogène en quantité équimolaire à l'acide urique dégradé. (B)
Dans les globules rouges G6PD déficientes, où l'activité enzymatique est réduite, la production
de NADPH est limitée et il peut ne pas être suffisante pour faire face à l'excès de réactif des
espèces d'oxygène généré en présence de composés pro-oxydants.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 47
II.2.8 Déficit en G-6-PD et paludisme
Il existe une forte relation entre le paludisme et le déficit en G6PD. L’hypothèse a été admise
que le déficit en G-6-PD confère une protection contre le paludisme sévère. Elle a longtemps
été suggérée par la remarquable concordance de la répartition géographique entre les zones
d’endémie du paludisme et celles du déficit en G-6-PD (HOWES et al., 2013a).Ces
observations révèlent deux faits importants. D’une part, les déficits en G-6-PD de classe II ou
III de l’OMS assurent une relative protection contre le paludisme, principalement contre les
infections à Plasmodium falciparum. En effet, les zones de déficit en G-6-PD de classe II et III
se superposent avec la zone d’infestation par Plasmodium falciparum (WAJCMAN et
GALACTEROS, 2004). D’autre part, l’utilisation des antipaludiques peut entrainer une anémie
hémolytique chez les patients présentant un déficit en G-6-PD (Figure 20).
Figure 20 : Relation triangulaire entre la primaquine, la G-6-PD et Plasmodium falciparum
(LUZZATTO et SENECA, 2014)
Dans diverses études épidémiologiques, il a été démontré que plus de 85% des variantes
génétiques ayant atteints des fréquences polymorphiques, sont enregistrées dans les populations
tropicales et subtropicales où le paludisme est endémique (ALLAHVERDIYEV et al., 2012).
En Afrique et Asie du sud-est, où l’infection à Plasmodium falciparum est endémique,
l’incidence du déficit en G-6-PD est estimée à plus de 10 % de la population.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 48
Au contraire, au Japon, au Nord de la Chine et en Europe du Nord, zones géographiques
historiquement indemnes de paludisme, l’incidence du déficit en G-6-PD est inférieure à 0,1
(MURA et al., 2009). Il existe également des incidences très variables entre les ethnies d’une
même région (Vietnam du Nord) selon qu’elles vivaient traditionnellement en dehors ou au sein
des zones d’endémie palustre (MATSUOKA et al., 2005). Les hommes hémizygotes déficients
développent moins la maladie que les individus porteurs de l’allèle sauvages du gène de la G-
6-PD. Généralement l’infection à Plasmodium falciparum n’est pas létale pour les personnes
exprimant le déficit en G-6-PD, probablement parce que le parasite prolifère moins dans leurs
érythrocytes (HOWES et al., 2013a).
Des expériences in vitro ont également montré que les hématies présentant une activité normale
en G-6-PD sont 2 à 80 fois plus susceptibles d’être infectées par Plasmodium falciparum.
Cependant les femmes hétérozygotes représentent un défi particulier en raison du phénomène
de la lyonisation. En conséquence, l'activité enzymatique de la G-6-PD peut varier en fonction
des proportions de cellules normales et déficientes qui sont inactivées chez chaque individu
(ALLAHVERDIYEV et al., 2012).
Les mécanismes de protection dans les études d’association du déficit en G-6-PD et le
paludisme demeurent controversés (JOHNSON et al., 2009). En effet les études sur
l’association, déficit en G-6-PD et paludisme grave, ont montré, soit une diminution du risque
de paludisme grave chez les hommes hémizygotes (GUINDO et al., 2007), soit une réduction
de ce risque (SIRUGO et al., 2014; UYOGA et al., 2015) ou sans association avec ce risque
chez les femmes hétérozygotes (GUINDO et al., 2007). D’autres études d’association ont
également rapporté des effets de protection du déficit en G-6-PD contre le paludisme cérébral
et une augmentation du risque du paludisme avec anémie sévère (SHAH et al., 2016). Les
études d’associations avec le paludisme simple ont été plus contradictoires, certaines montrant
un déficit en G-6-PD ayant un effet protecteur chez les femmes hétérozygotes (BIENZLE et
al., 1979; RUWENDE et al., 1995; CLARK et al., 2008), ou sans effet sur l'incidence du
paludisme simple chez les hommes hémizygotes ou les femmes hétérozygotes (ENEVOLD et
al., 2005). En revanche, d'autres études ont montré une augmentation de l'incidence du
paludisme simple chez les femmes hétérozygotes pour le déficit en G-6-PD (PARIKH et al.,
2004).
II.2.9 Techniques de diagnostic de la déficience en G-6-PD
Il existe plusieurs méthodes de diagnostic du déficit en G-6-PD telles que le Fluorescent spot
test aussi appelé « spot test de Beutler », le dosage spectrophotométrique de l’activité
enzymatique qui est le test de référence permettant la mise en évidence du déficit en G-6-PD.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 49
On distingue également la détection cytochimique des femmes hétérozygotes et les techniques
de biologie moléculaire que nous avons utilisées dans notre étude, qui décèlent la ou les
mutation(s) génétique(s) sur l'ADN et permettent ainsi de déterminer le variant en cause.
L’intérêt de cette technique est surtout de diagnostiquer le déficit en G-6-PD chez une femme
susceptible d’être porteuse hétérozygote mais chez qui le dosage spectrophotométrique de
l’activité enzymatique de la G-6-PD est négatif.
II.2.9.1 Génotypage par PCR/RFLP
Principe
La PCR-RFLP est basée sur la digestion enzymatique de l’ADN amplifié par PCR. Des
endonucléases de restriction spécifiques, reconnaissent et coupent l’ADN dans la région de la
mutation ponctuelle des produits PCR. Le type de SNP est facilement identifié en fonction de
la taille des fragments générés par la digestion enzymatique après migration sur gel
d’électrophorèse (OTA et al., 2007). En pratique, les régions contenant les sites de restriction
des mutations recherchées sont amplifiées par PCR à partir de l‘ADN génomique du patient à
tester grâce à des amorces spécifiques. Les produits PCR sont ensuite mis en présence des
enzymes de restriction appropriées.
II.2.9.2 Génotypage par PCR en temps réel
Principe
La particularité du système TaqManTM est d’exploiter l’activité 5’-3’ nucléase de l’ADN
polymérase qui permet d’hydrolyser la sonde hybridée à sa cible spécifique lors de l’étape
d’élongation des amorces (TSE et CAPEAU, 2003). Le principe est de construire deux sondes
TaqMan marquées avec deux fluorochromes différents, complémentaires respectivement de
l'allèle normal et de l'allèle muté, et de réaliser une PCR en présence de ces deux sondes. On va
donc détecter deux signaux différents correspondant aux produits de PCR de l'allèle normal et
de l'allèle muté, visibles par deux courbes d'amplification distinctes.
La sonde TaqMan est marquée de chaque côté par un fluorochrome (rapporteur et quencher),
de sorte que le rapporteur n'est fluorescent que lorsqu'il est séparé physiquement du quencher.
Lors de sa progression le long de la matrice pendant la phase d'élongation de la PCR, la Taq
polymérase dégrade l'extrémité 5' de la sonde lorsqu'elle la rencontre. Cela libère le rapporteur,
qui devient alors fluorescent.

MATERIEL
ET
METHODES

Thèse de Doctorat Unique OUATTARA Abdoul Karim 50
CHAPITRE III. MATERIEL ET METHODES
III.1 Cadre d’étude
Nos travaux de recherches ont été menés au Burkina Faso au Centre de Recherche
Biomoléculaire Pietro Annigoni (CERBA) et au Laboratoire de Biologie Moléculaire et de
Génétique (LABIOGENE) qui ont servi de cadre d’étude. Le Burkina Faso est un pays sahélien
enclavé situé au cœur de l’Afrique Occidentale. Il fait frontière avec le Mali, le Niger, le Bénin,
le Togo, le Ghana, et la Côte d’Ivoire. Il s’étend sur une superficie de 274 222 Km2 pour une
population de 17 880 386 habitants en 2014 (INSD, 2015). Ouagadougou sa capitale
administrative concentrait près de 1 915 102 habitants en 2012 (INSD, 2015). Au Burkina Faso
le paludisme est holoendémique avec la plupart des transmissions survenant pendant ou peu de
temps après la saison des pluies, de Juillet à Décembre (DIBOULO et al., 2016). En 2014 la
proportion du paludisme grave dans les principales causes de décès s’élevait à 26,2% (INSD,
2015).
Le LABIOGENE est un laboratoire de recherche de l’Ecole Doctorale Science et
Technologies (ED/ST) de l’Université Ouaga I Pr Joseph KI-ZERBO ayant reçu le label
de Centre d’Excellence de l’UEMOA.
Le CERBA est situé au secteur 51 de la ville de Ouagadougou. Il a été érigé en
Laboratoire National de Référence du HPV (LNR-HPV) depuis février 2015. Tout comme
l’Hôpital Saint Camille de Ouagadougou (HOSCO), il est dans le district sanitaire de
l’arrondissement 11. La capitale burkinabè compte 4 districts sanitaires (districts sanitaires de
Pissy, de l’ex secteur 30, de Kossodo et de Paul VI) (Figure 21). Dans le cadre d’un partenariat
solide, ces structures œuvrent à la promotion du développement de la santé au Burkina Faso et
en Afrique par la formation de jeunes médecins, pharmaciens et biologistes.
III.2 Sites de collecte
Les échantillons de la première étude ont été collectés de manière aléatoire dans la commune
rurale de Koubri au Burkina Faso. Koubri est un village du Burkina Faso située à 25 km au Sud
de la Capitale, dans la province du Kadiogo et dans la région du Centre. En ce qui concerne les
échantillons de la deuxième étude, la collecte a été réalisée dans trois centres de la ville de
Ouagadougou. Il s’est agi du centre médical de Samandin situé dans l’arrondissement N°1, de
l’Hôpital Saint Camille de Ouagadougou situé dans l’arrondissement N°5 et du CERBA situé
dans l’arrondissement N°11.
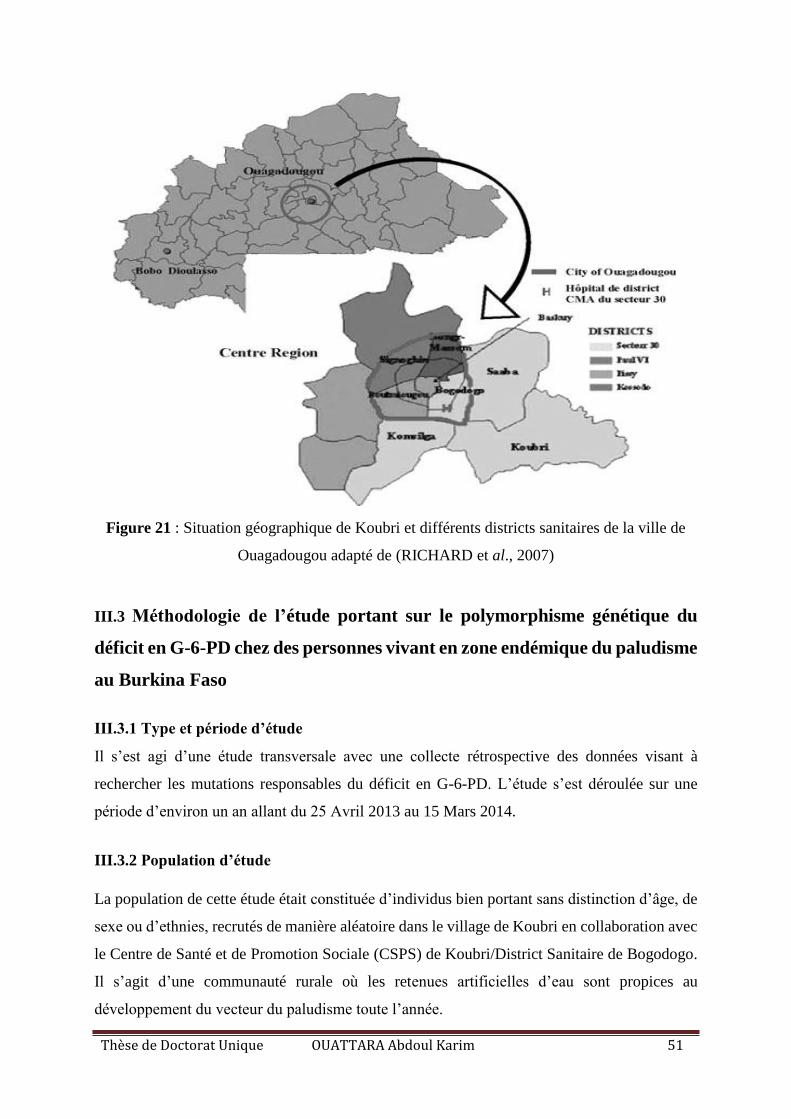
Thèse de Doctorat Unique OUATTARA Abdoul Karim 51
Figure 21 : Situation géographique de Koubri et différents districts sanitaires de la ville de
Ouagadougou adapté de (RICHARD et al., 2007)
III.3 Méthodologie de l’étude portant sur le polymorphisme génétique du
déficit en G-6-PD chez des personnes vivant en zone endémique du paludisme
au Burkina Faso
III.3.1 Type et période d’étude
Il s’est agi d’une étude transversale avec une collecte rétrospective des données visant à
rechercher les mutations responsables du déficit en G-6-PD. L’étude s’est déroulée sur une
période d’environ un an allant du 25 Avril 2013 au 15 Mars 2014.
III.3.2 Population d’étude
La population de cette étude était constituée d’individus bien portant sans distinction d’âge, de
sexe ou d’ethnies, recrutés de manière aléatoire dans le village de Koubri en collaboration avec
le Centre de Santé et de Promotion Sociale (CSPS) de Koubri/District Sanitaire de Bogodogo.
Il s’agit d’une communauté rurale où les retenues artificielles d’eau sont propices au
développement du vecteur du paludisme toute l’année.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 52
Pour être inclus dans l’étude les sujets majeurs devaient avoir pris connaissance de la fiche
d’information, l’avoir lu et compris puis approuver librement et volontairement la fiche
individuelle de consentement éclairé. Pour les sujets mineurs, le consentement éclairé des
parents ou tuteur légal étaient requis. Toute personne porteuse de toute forme de paludisme
grave selon les critères de l’OMS ou de toutes autres pathologies ou encore présentant une
allergie aux antipaludiques utilisés (Artésunate-Amodiaquine (AS+AQ) et Artéméther-
Lumefantrine (AM-L)) n’était pas inclus dans l’étude.
III.3.3 Echantillonnage
III.3.3.1 Taille de l’échantillon
Au total 200 échantillons ont été analysés par PCR en temps réel et PCR/RFLP pour la
recherche des mutations responsables du déficit en G-6-PD. La taille d'échantillon a été
déterminée par la formule de SWARTZ :
n = z² * p * (1-p) / m² = (1,96)² x 0,092 x (1- 0,092)/(0,05)² = 128 + 72 = 200
n : Taille d'échantillon minimale pour l'obtention de résultats significatifs pour un
événement et un niveau de risque fixé
z : Niveau de confiance (la valeur type du niveau de confiance de 95 % sera 1,96)
p : Fréquence du déficit en G-6-PD au Burkina Faso (0,09) (CARTER et al., 2011)
m : Marge d'erreur (généralement fixée à 5 %)
III.3.3.2 Prélèvements sanguins
Les échantillons sanguins veineux (environ 5 mL de sang par sujet adulte et 3 mL de sang par
enfant de moins de 5 ans) ont été prélevés sur des tubes imprégnés d’EDTA. Une partie du
prélèvement a été utilisée pour la recherche et la quantification des parasites du paludisme au
microscope optique et la réalisation des analyses hématologiques. L’autre partie a subi une
centrifugation à 15 000 rpm pendant 5mn pour séparer le plasma et le culot. Des aliquotes de
ces derniers ont été réalisés et conservés à -20°C. Le culot a été utilisé pour extraire l’ADN afin
de faire le génotypage de la déficience en G-6-PD.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 53
III.3.4 Analyses biologiques
III.3.4.1 Numération formule sanguine
Les paramètres hématologiques ont été déterminés à partir des prélèvements sanguins dans des
tubes EDTA, à l’aide d’un automate d’hématologie CELL-DYN Ruby (Abbott Laboratories,
Abbott Park, IL, USA) (Figure 22). L’hémogramme était réalisé dans le plus bref délai soit au
maximum 1 heure après le prélèvement. Pour le dosage de l'hémoglobine, l’automate CELL-
DYN Ruby utilise le principe de la spectrophotométrie. Après lyse des globules rouges,
l'hémoglobine est convertie en un chromogène stable. L'absorbance lue est directement
proportionnelle à la concentration en hémoglobine. La numération plaquettaire repose sur le
principe de l'analyse par diffraction-polarisation multi-angulaire (Multi-Angle Polarized Scatter
Separation ou MAPSS) en cytométrie de flux.
Figure 22 : Automate CELL-DYN Ruby
III.3.4.2 Electrophorèse de l’hémoglobine
La détermination du type d’hémoglobine a été faite sur la chaîne d’électrophorèse HELENA
(Helena Biosciences Europe, Queensway South, Gateshead Tyne and Wear NE II OSD)
(Figure 23) selon le protocole décrit par le fabriquant. L’hémolysât a été préparé en mélangeant
1 volume de sang total à 3 volumes du réactif d’hémolyse Helena (0,005 M d’EDTA et 0,01%
de cyanure de potassium). L'électrophorèse a été réalisée à 350 V pendant 25 minutes dans un
tampon Acide borique/Tris-EDTA (pH 8,4, force ionique = 0,035).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 54
Figure 23 : Appareil d’électrophorèse de type HELENA
III.3.5 Recherche des mutations responsables de la déficience en G-6-PD
III.3.5.1 Extraction de l’ADN
L’ADN génomique a été extrait à partir du culot de sang par la méthode du salting-out qui
comporte plusieurs étapes décrit dans le protocole à l’annexe I.
Cinq cent microlitres de culot de sang ont été mélangé à un tampon de lyse (annexe II) pour
faire éclater les hématies. Les débris cellulaires sont ensuite éliminés par centrifugation. A la
fin de la série de lavage à l’eau distillée, le culot cellulaire contenant les leucocytes est traité
par un mix contenant le Sodium Dodecyl Sulfate (SDS) et la protéinase K.
Le SDS permet l’élimination ou la séparation des lipides membranaires tandis que la protéinase
K entraine l’élimination des protéines de l’extrait cellulaire. Ces dernières seront par la suite
relarguées par l'intermédiaire de la force ionique du NaCl (5M).
La précipitation de l'ADN génomique est effectuée en utilisant une solution d'éthanol absolu
suivie deux lavages à l’éthanol 70% à froid. Les tubes sont ensuite séchés à bouchon ouvert
pour faire évaporé toute l’éthanol et l’ADN est enfin dissout dans un tampon d’élution et
conservé à -20°C.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 55
III.3.5.2 Génotypage par PCR en temps réel
Tous les échantillons ont d’abord fait l’objet d’un génotypage pour la mutation A376G et ceux
qui étaient hémizygotes, homozygotes ou hétérozygotes pour cette mutation ont ensuite subi un
génotypage pour les autres mutations G202A, A542T, G680T et T968C. En effet, les
échantillons ayant l’allèle sauvage (A) pour la mutation A376G sont du génotype B, et seuls
ceux ayant l’allèle mutant (G), de génotype A, peuvent être A- par accumulation d’une seconde
mutation.
Protocole de la PCR en temps réel
Préparation des échantillons pour l’amplification
La PCR en temps réel a été réalisée dans un volume réactionnel total de 25 µL comprenant 5
µL d’ADN (4ng/µL), 12,5 µL de TaqMan Universal PCR Master Mix (2X), 6,25 µL d’eau
stérile et 1,25 µL de SNP mix (40X). Les mutations A376G, G202A et A542T ont fait l’objet
d’un génotypage selon la méthode décrite par CLARK et collaborateurs en 2009 en utilisant les
sondes TaqMan (Applied Biosystem, Foster City, California, USA) suivantes : rs1050828 (G-
6-PD G202A), rs1050829 (G-6-PD A376G) et rs5030872 (G-6-PD A542T).
Programme d’amplification sur le 7500 Fast Real-Time PCR System
95° C pendant 10 minutes (Activation de la Taq polymérase)
92° C pendant 15 secondes (Dénaturation)
60° C pendant 1 minute (Hybridation)
Figure 24 : Appareil 7500 Fast Real Time PCR system (Applied Biosystems, USA)
40 cycles

Thèse de Doctorat Unique OUATTARA Abdoul Karim 56
III.3.5.3 Génotypage par PCR-RFLP
Protocole de la PCR-RFLP
Préparation des échantillons pour l’amplification
La PCR-RFLP a concerné les mutations G680T et T968C sur les échantillons
homo/hémizygotes ou hétérozygotes pour le SNP A376G. Elle a été réalisée selon la méthode
décrite par BEUTLER et al. (1989) dans un volume réactionnel de 25 µL composé de 12,5 µL
de AmpliTaq Gold® 360 Master Mix (2x), 1 µL d’enhancer, 3,125 µL d’amorces sens et anti-
sens (10µM) et 3,25 µL d’eau stérile et 2 µL d’ADN (50 - 100 ng/µL) de chaque échantillon.
Les couples d’amorces utilisées étaient les suivantes : 5’-ACATGTGGCCCCTGCACCAC-3’
(sens) ; 5’GTGACTGGCTCTGCCACCCTG-3’ (anti-sens) pour la G-6-PD (G680T) et 5’-
TCCCTGCACCCCAACTCAAC-3’ (sens) ; 5’-CCAGTTCTGCCTTGCTGGGC-3’ (anti-
sens) pour la G-6-PD (T968C).
Programme d’amplification sur le GeneAmp PCR system 9700
Pour la mutation G680T
95° C pendant 10 minutes (Activation de la Taq polymérase)
94° C pendant 45 secondes (Dénaturation)
69° C pendant 45 secondes (Hybridation) 30 Cycles Mutation G680T
72° C pendant 45 secondes (Elongation)
72° C pendant 7 minutes (Extension finale)
Pour la mutation T968C
95° C pendant 10 minutes (Activation de la Taq polymérase)
94° C pendant 45 secondes (Dénaturation)
65° C pendant 45 secondes (Hybridation) 30 Cycles Mutation T968C
72° C pendant 45 secondes (Elongation)
72° C pendant 7 minutes (Extension finale)

Thèse de Doctorat Unique OUATTARA Abdoul Karim 57
Figure 25 : Thermocycleur GeneAmp PCR system 9700 (Applied Biosystems, USA)
Digestion enzymatique et migration su gel d’agarose
Les produits PCR des mutations G680T et T968C ont été digérés en utilisant les enzymes de
restriction BstOI (source : Bacillus stearothermophilus O22, séquence reconnue : CCWGG) et
NciI (source : Neisseriacinerea ; séquence reconnue : CCSGG) respectivement (Promega no.
R6931, 10 U/µL (BstOI) ; no. R7061, 12 U/µL (NciI) ; Promega Corp., Madison, USA). La
digestion enzymatique a été effectuée pendant 16 heures à la température optimale (60° C pour
BstOI et 37° C pour NciI) dans un volume réactionnel total de 20 µL constitué dans l’ordre de :
16,3 µL d’eau stérile, 2 µL de RE 10X Buffer, 0,2 µL de BSA acétylé (10 µg/µL), 0,5 µL
d’enzyme et 1 µL de produit PCR. Les génotypes ont été déterminés après migration sur gel
d’agarose 2% à 120 Volt pendant 45 minutes et révélation sous UV en faisant un spectre
d’absorption d’environ 320 nm.
Pour la préparation du gel d’agarose 2%, un mélange de 4g d'agarose et 200 mL de Tris-Borate-
EDTA (TBE) 1X est mis à cuire à la micro-onde (2mn, 460W). On y ajoute ensuite 16 μL de
Bromure d’Ethidium (BET) après un léger refroidissement (10 mg/mL). Le gel d’agarose est
coulé délicatement dans le moule. Après la solidification du gel d’agarose à température
ambiante, le moule contenant le gel d’agarose solide est enfin placé dans la cuve à
électrophorèse contenant le tampon de migration. Pour la migration, 3 µL de bleu de charge est
ajouté aux amplicons d’ADN de chaque échantillon. Le dépôt des échantillons (5 µL du
mélange bleu de charge + amplicons d’ADN) sur le gel d’agarose est effectué en s’assurant que
le tampon de migration couvre entièrement le gel d’agarose de quelques millimètres.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 58
III.4 Méthodologie de l’étude portant sur l’hétérogénéité allélique du déficit
en G-6-PD chez des personnes malades du paludisme dans la ville de
Ouagadougou au Burkina Faso
III.4.1 Type et période d’étude
Il s’est agi d’une étude transversale visant à déterminer les variants génotypiques de
l’hémoglobine et du déficit en G-6-PD chez des personnes malades du paludisme. Elle s’est
déroulée sur une période allant du 27 Septembre 2014 au 05 Mars 2016.
III.4.2 Population d’étude
La population de la seconde étude était composée d’individus de plus d’un an, venant en
consultation de médecine générale dans les trois centres de santé retenus pour la collecte des
échantillons. Après suspicion du paludisme chez ces patients et leur orientation au laboratoire
pour les examens, ceux dont les Tests Rapides d’Orientation Diagnostic (TROD) du paludisme
étaient positifs ont été retenus pour l’étude avec leur consentement libre et éclairé sous réserve
d’une goutte épaisse positive. Les patients dont la goutte épaisse était négative ainsi que ceux
de moins d’un an n’ont pas été inclus dans l’étude. En effet la méthode d’électrophorèse à pH
alcalin utilisée au cours de l’étude est peu performante avant la première année de vie de
l’enfant car les Hb S, F, et A sont trop rapprochées.
III.4.3 Echantillonnage
III.4.3.1 Taille de l’échantillon
Pour cette seconde étude, un ensemble de 182 échantillons ont été analysés par PCR classique
et PCR en temps réel pour la détermination des mutations impliquées dans le déficit en G-6-
PD. La taille d'échantillon a été déterminée par la formule de SWARTZ : n = z² * p * (1-p) / m²
= (1,96) ² x 0,095 x (1- 0,095) / (0,05) ² = 132 + 50 avec p = fréquence du déficit en G-6-PD au
Burkina Faso (0,095) (OUATTARA et al., 2014).
III.4.3.2 Prélèvements
Nos échantillons étaient constitués de prélèvements sanguins veineux (5 mL de sang par sujet
adulte et 3 mL de sang par enfant) dans des tubes EDTA. Le prélèvement a été utilisé pour la
réalisation d’une Numération Formule Sanguine (NFS), d’une électrophorèse de l’hémoglobine
et d’une goutte épaisse. Par la suite, l’échantillon a été centrifugé à 15000 rpm pendant 5
minutes. Le plasma et le culot ont été séparés et conservés à -80°C. C’est le culot qui a été
utilisé pour extraire l’ADN afin de déterminer les mutations impliquées dans le déficit en G-6-
PD.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 59
II.4.4 Détection de Plasmodium falciparum
III.4.4.1 TROD SD Bioline Malaria Ag P.f/Pan
Les TROD ont été réalisés par piqûre du bout de doigt des patients orientés au laboratoire pour
suspicion du paludisme. Le Test Rapide d’Orientation Diagnostic SD BIOLINE Malaria Ag
P.f/Pan est un test immunologique, qualitatif et différentiel permettant la détection de l’antigène
HRP2 (Histidine-Rich Protein 2) spécifique à Plasmodium falciparum et de l’antigène pLDH
(Plasmodium Lactate Déshydrogénase) commune aux espèces de Plasmodium dans le sang
total humain. Le test détecte ces différents antigènes plasmodiaux par chromatographie de sang
total sur une membrane de nitrocellulose sur laquelle ont été fixés des anticorps monoclonaux
spécifiques des antigènes recherchés. La capture éventuelle est révélée simultanément par
d’autres anticorps monoclonaux couplés à de l’or colloïdal et présents dans le tampon de
migration. En cas de positivité, un sandwich « Ac monoclonal - Ag plasmodial- Ac monoclonal
marqué » est donc réalisé.
III.4.4.2 Goutte épaisse
Les lames de goutte épaisse (Figure 26), obtenu en déposant une goutte de sang au milieu d'une
lame de microscope et en l’étalant à l'aide du coin d'une autre lame jusqu'à l’obtention d’un
diamètre d’environ 10 à 15 mm, ont été colorées pendant 10 minutes dans une solution de
Giemsa à 10%. La lecture des lames séchées a été réalisée à l’aide d’un microscope optique à
l’objectif 100 avec l’huile à immersion. Les trophozoïtes ont été décomptés concomitamment
à 200 leucocytes et la densité parasitaire a été calculée suivant la formule :
Nombre de parasites relevés x 8000
Nombre de parasites/µL de sang =
Nombre de leucocytes comptés
Figure 26 : Une goutte épaisse

Thèse de Doctorat Unique OUATTARA Abdoul Karim 60
III.4.5 Analyses biologiques
III.4.5.1 Numération formule sanguine
Les paramètres hématologiques ont été déterminés à partir des prélèvements sanguins dans des
tubes EDTA, à l’aide d’un compteur hématologique ABX micros 60 (ABX Diagnostics,
Montpellier, France) (Figure 27). La numération formule sanguine était réalisée dans le plus
bref délai soit au maximum 1 heure après le prélèvement. Pour le dosage de l'hémoglobine,
l’automate ABX micros 60 utilise également la spectrophotométrie tandis que pour la
numération plaquettaire la mesure est réalisée par la méthode impédentielle (variation
d'impédence engendrée par le passage des plaquettes).
Figure 27 : Automate ABX micro 60
III.4.5.2 Electrophorèse de l’hémoglobine
La détermination du type d’hémoglobine a été faite à l’aide d’une électrophorèse à pH alcalin
sur une bande d’acétate de cellulose : CELLOGEL (5,7×14 cm) (Figure 28). Le tampon utilisé
était du tris-glycine pH 9,5. Pour le lavage, 4 mL de solution physiologique (NaCl à 0,9 %) ont
été ajoutés à 250 µL de culot globulaire de chaque échantillon. Le mélange a ensuite été
homogénéisé par retournements successifs puis centrifuger à environ 3000 rpm pendant 5
minutes. L'élimination du surnageant a été effectué à l’aide d’une pipette Pasteur en veillant à
aspirer la couche cellulaire supérieure pour éliminer les globules blancs. Les cellules ont enfin
été lysées avec 500 µL de saponine 1%. A l’aide de tubes capillaires, des spots d’échantillons
ont été déposés sur le CELLOGEL. La migration s’est effectuée en 60 minutes à 150 V en
moyenne.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 61
Figure 28 : Cuve et générateur pendant la migration
III.4.6 Recherche des mutations responsables de la déficience en G-6-PD
III.4.6. 1 Extraction de l’ADN
L’ADN génomique a été extrait à partir du culot de sang par la méthode du salting-out comme
précédemment décrit.
III.4.4.2 Génotypage par PCR classique
La PCR classique a été réalisée avec le kit GENESPARK G6PD African. La PCR a lieu dans
un volume réactionnel de 25 μL composé de 12,5 μL de PCR Multiplex Smart Mix (2x), 2 μL
de Primer Mixture (G-6-PD-African), 8,5 µL d’eau stérile et 2 μL d’ADN (50 - 100 ng/μL) de
chaque échantillon. Le mix, les amorces, l’eau stérile, et les contrôles sont fournis par le
fabricant. Les individus de sexe féminin présentant des mutations en position 202 et/ou 376 ont
été repris par PCR en temps réel pour les classer en homozygotes et hétérozygotes.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 62
Le programme d’amplification
L’amplification a été réalisée à l’aide du thermocycleur GeneAmp® PCR system 9700 suivant
le programme d’amplification présenté ci-dessous.
95° C pendant 15 minutes (Activation de l’ADN polymérase)
95° C pendant 20 secondes (Dénaturation)
60° C pendant 40 secondes (Hybridation) 30 Cycles
72° C pendant 1 minute (Elongation)
72° C pendant 7 minutes (Extension finale)
La migration sur gel d’agarose
Le produit PCR a subi une électrophorèse, sur gel d’agarose 2 % (4g d’agarose dans 200 µL de
Tris-Borate-EDTA (TBE 1X)) contenant 8 µL de BET, à 120 volts pendant 45 minutes.
L’électrophorèse a été réalisée dans un tampon TBE 1X ; le tampon étant légèrement basique
(pH = 8,2), l’ADN s’ionise sur ses groupements phosphates et migre vers l’anode (borne rouge
sur la figure 29 ci-dessous).
Figure 29 : Dispositif d’électrophorèse des amplicons G-6-PD (BioRad, USA)
G202A, A376G, A542T,
G680T, C563T et T968C

Thèse de Doctorat Unique OUATTARA Abdoul Karim 63
La révélation des bandes
Les bandes obtenues ont été révélées sous UV à 320 nm au moyen de l’appareil GENE FLASH
(Figure 30). Le BET fluorescent aux UV s’intercale entre les bases de l’ADN et permet la
visualisation des fragments dans le gel placé sous UV.
Figure 30 : GENEFLASH
Interprétation des résultats
Les bandes obtenues après migration sur gel d’agarose ont été interprétées au moyen d’une
table fournie par le fabriquant (Figure 31).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 64
Figure 31 : Table d’interprétation des bandes après l’électrophorèse sur gel d’agarose
III.6 Analyse statistique
Les données ont été ordonnées et enregistrées sur Microsoft Excel 2013 d’un micro-ordinateur.
Les analyses ont été effectuées à l’aide des logiciels Statistical Package for Social Sciences
(SPSS) 21.0 et Epi Info version 7. Le test de chi carré a été utilisé pour les comparaisons. La
différence a été considérée significative pour p < 0,05.
III.7 Considérations éthiques
La réalisation de la présente étude a été approuvée par le Comité d’éthique de Recherche en
Santé (CERS) du Burkina Faso (Délibération n° 2014-9-128). Les personnes adultes ont signé
les fiches de consentement de manière volontaire et anonyme (annexe III), et les consentements
éclairés des enfants ou mineurs ont été obtenus de leurs parents ou tuteurs légaux (annexe IV).
Tous les examens ont été pris en charge par l’étude. Les résultats de l’électrophorèse et de la
Numération Formule Sanguine ont été remis aux patients ou à leur médecin traitant pour leur
suivi médical. A la fin de la collecte après numérisation anonyme des données, les fiches de
consentement éclairé et de collectes ont été déposées dans les archives du
CERBA/LABIOGENE à l’abri de toute utilisation pouvant enfreindre les règles éthiques. Seuls
les investigateurs de l’étude ont accès à ces dossiers.

RESULTATS

Thèse de Doctorat Unique OUATTARA Abdoul Karim 65
CHAPITRE IV : RESULTATS
IV.1 Résultats des travaux portant sur le polymorphisme génétique du déficit
en G-6-PD chez des personnes vivant en zone endémique du paludisme au
Burkina Faso
IV.1.1 Bref rappel du contexte et but de l’étude
La revue de la littérature indique que le variant G-6-PDA- (202A/376G) est l’allèle déficient le
plus fréquent en Afrique subsaharienne. Des études récentes réalisées dans la sous-région ont
également montré la présence d’autres mutations ponctuelles telles que la G-6-PD Santamaria
(376G/542T), et la G-6-PD Betica-Selma (376G/968C) associées à la déficience avec des
fréquences relativement élevées. Au Burkina Faso, les rares études de génotypage se sont
intéressées uniquement au variant G-6-PDA- (202A/376G). Cette approche est restrictive et
pourrait donc entraîner une sous-estimation de la prévalence réelle du déficit en G-6-PD dans
nos populations. De plus plusieurs travaux ont démontré le rôle protecteur du déficit en G-6-
PD contre les formes sévères du paludisme. Quant à son rôle au niveau des formes
asymptomatiques très peu d’études s’y sont intéressées.
IV.1.2 Rappel de l’objectif de l’étude
L’objectif de l’étude était de déterminer par PCR en temps réel et par PCR/RFLP les génotypes
de quatre combinaisons de mutations impliquées dans le déficit en G-6-PD en corrélation avec
le paludisme asymptomatique en zone rurale au Burkina Faso.
IV.1.3 Marqueurs génétiques et paludisme asymptomatique
IV.1.3.1 Caractéristiques socio-démographiques de la population d’étude 1
Deux cents (200) personnes recrutées de façon aléatoire au CSPS de Koubri ont été inclus dans
cette étude. La population d’étude était constituée de 42,0% (84/200) d’hommes et de 58,0%
(116/200) de femmes. L’âge était compris entre 1 et 79 ans avec une moyenne de 19,7 ± 20,1
ans. Les enfants dont l’âge était inférieur à 5 ans représentaient 43,0% de la population d’étude
contre 57,0% d’individus qui avaient un âge supérieur ou égal à 5 ans.
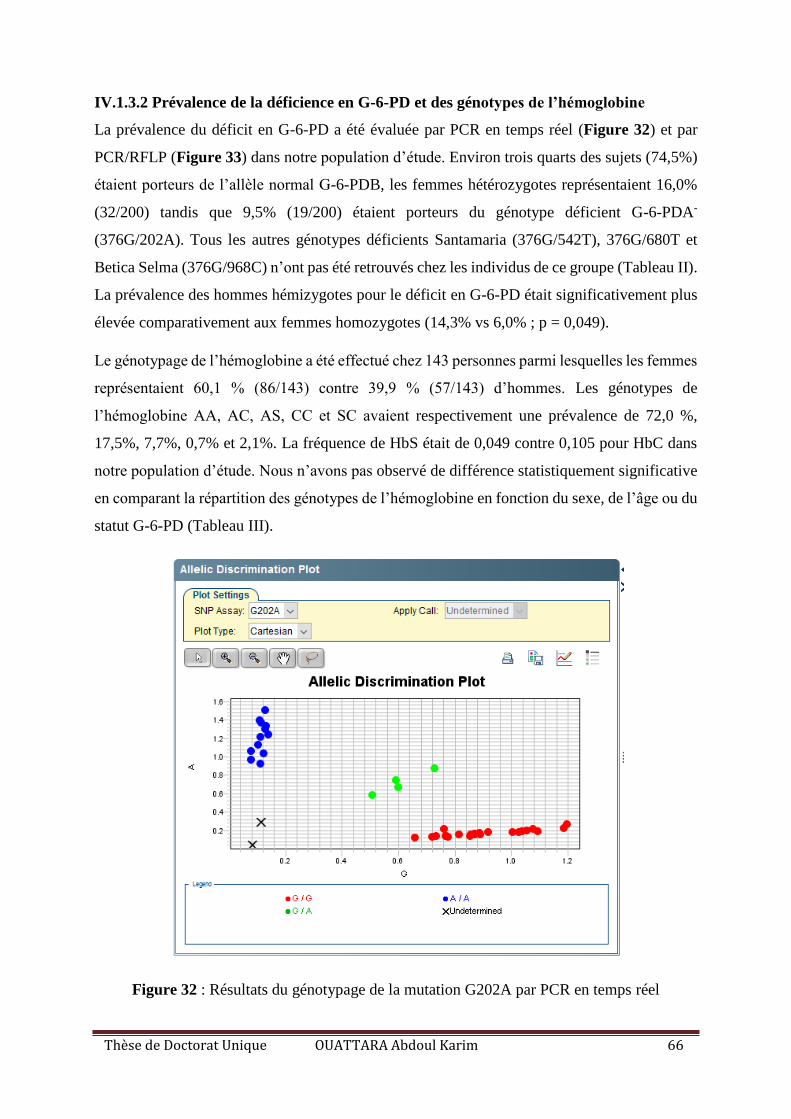
Thèse de Doctorat Unique OUATTARA Abdoul Karim 66
IV.1.3.2 Prévalence de la déficience en G-6-PD et des génotypes de l’hémoglobine
La prévalence du déficit en G-6-PD a été évaluée par PCR en temps réel (Figure 32) et par
PCR/RFLP (Figure 33) dans notre population d’étude. Environ trois quarts des sujets (74,5%)
étaient porteurs de l’allèle normal G-6-PDB, les femmes hétérozygotes représentaient 16,0%
(32/200) tandis que 9,5% (19/200) étaient porteurs du génotype déficient G-6-PDA-
(376G/202A). Tous les autres génotypes déficients Santamaria (376G/542T), 376G/680T et
Betica Selma (376G/968C) n’ont pas été retrouvés chez les individus de ce groupe (Tableau II).
La prévalence des hommes hémizygotes pour le déficit en G-6-PD était significativement plus
élevée comparativement aux femmes homozygotes (14,3% vs 6,0% ; p = 0,049).
Le génotypage de l’hémoglobine a été effectué chez 143 personnes parmi lesquelles les femmes
représentaient 60,1 % (86/143) contre 39,9 % (57/143) d’hommes. Les génotypes de
l’hémoglobine AA, AC, AS, CC et SC avaient respectivement une prévalence de 72,0 %,
17,5%, 7,7%, 0,7% et 2,1%. La fréquence de HbS était de 0,049 contre 0,105 pour HbC dans
notre population d’étude. Nous n’avons pas observé de différence statistiquement significative
en comparant la répartition des génotypes de l’hémoglobine en fonction du sexe, de l’âge ou du
statut G-6-PD (Tableau III).
Figure 32 : Résultats du génotypage de la mutation G202A par PCR en temps réel

Thèse de Doctorat Unique OUATTARA Abdoul Karim 67
Figure 33 : Fragments G680T et T968C après amplification PCR
On distingue les marqueurs de poids moléculaire (M) dans la première colonne à gauche. Les
colonnes 2 à 11 de la gauche vers la droite, montrent des fragments de 242 paires de bases
(pb) correspondant aux amplicons de l’exon 7 du gène G-6-PD (des échantillons E1 à E10) qui
comporte la mutation G680T, tandis que les colonnes 13 à 22 montrent des fragments de 282
paires de bases correspondant aux amplicons de l’exons 9 du gène G-6-PD qui comporte la
mutation T968C.
Tableau ii : Prévalence des 4 combinaisons de mutations responsables de la déficience en G-
6-PD
Genotypes G-6-PD N %
Hommes
84
-
202A/376G hémizygotes 12 14,3
376G/542T 0 0
376G/680T 0 0
376G/968C 0 0
G-6-PD normal 72 85,7
Femmes
116
-
202A/376G homozygotes 7 6,0
202A/376G hétérozygotes 32 27,6
376G/542T 0 0
376G/680T 0 0
376G/968C 0 0
G-6-PD normal 77 66,4
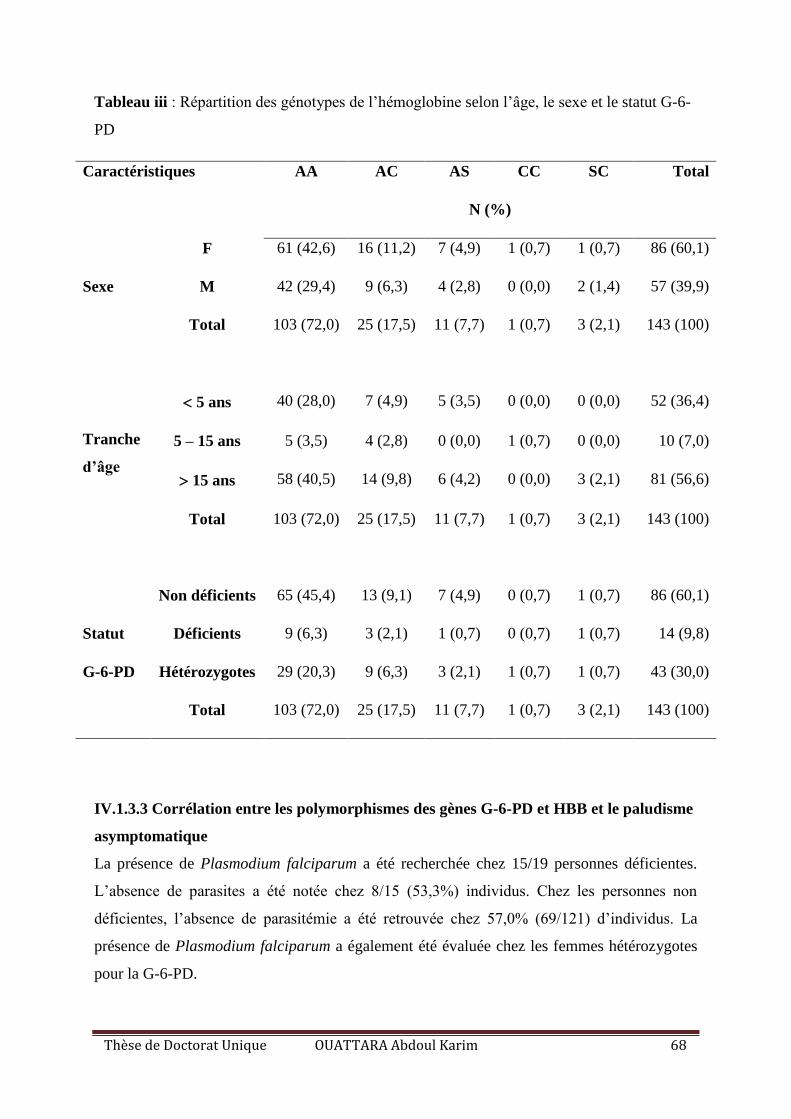
Thèse de Doctorat Unique OUATTARA Abdoul Karim 68
Tableau iii : Répartition des génotypes de l’hémoglobine selon l’âge, le sexe et le statut G-6-
PD
Caractéristiques AA AC AS CC SC Total
N (%)
Sexe
F 61 (42,6) 16 (11,2) 7 (4,9) 1 (0,7) 1 (0,7) 86 (60,1)
M 42 (29,4) 9 (6,3) 4 (2,8) 0 (0,0) 2 (1,4) 57 (39,9)
Total 103 (72,0) 25 (17,5) 11 (7,7) 1 (0,7) 3 (2,1) 143 (100)
Tranche
d’âge
5 ans 40 (28,0) 7 (4,9) 5 (3,5) 0 (0,0) 0 (0,0) 52 (36,4)
5 – 15 ans 5 (3,5) 4 (2,8) 0 (0,0) 1 (0,7) 0 (0,0) 10 (7,0)
15 ans 58 (40,5) 14 (9,8) 6 (4,2) 0 (0,0) 3 (2,1) 81 (56,6)
Total 103 (72,0) 25 (17,5) 11 (7,7) 1 (0,7) 3 (2,1) 143 (100)
Statut
G-6-PD
Non déficients 65 (45,4) 13 (9,1) 7 (4,9) 0 (0,7) 1 (0,7) 86 (60,1)
Déficients 9 (6,3) 3 (2,1) 1 (0,7) 0 (0,7) 1 (0,7) 14 (9,8)
Hétérozygotes 29 (20,3) 9 (6,3) 3 (2,1) 1 (0,7) 1 (0,7) 43 (30,0)
Total 103 (72,0) 25 (17,5) 11 (7,7) 1 (0,7) 3 (2,1) 143 (100)
IV.1.3.3 Corrélation entre les polymorphismes des gènes G-6-PD et HBB et le paludisme
asymptomatique
La présence de Plasmodium falciparum a été recherchée chez 15/19 personnes déficientes.
L’absence de parasites a été notée chez 8/15 (53,3%) individus. Chez les personnes non
déficientes, l’absence de parasitémie a été retrouvée chez 57,0% (69/121) d’individus. La
présence de Plasmodium falciparum a également été évaluée chez les femmes hétérozygotes
pour la G-6-PD.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 69
Aucune différence statistiquement significative n’a été trouvée en comparant la prévalence de
l’infection à Plasmodium falciparum entre les personnes déficientes et non déficientes d’une
part, et d’autre part entre les personnes non déficientes et les hétérozygotes. Cependant la
moyenne géométrique des parasites chez les personnes infectées des trois groupes était
significativement plus élevée chez les personnes non déficientes comparativement aux
personnes déficientes et chez les personnes non déficientes comparativement aux hétérozygotes
(Tableau IV).
La corrélation entre les génotypes de l’hémoglobine et la parasitémie est résumée dans le
tableau V. La prévalence de l’infection à Plasmodium falciparum était de 37,9% (39/103) chez
les personnes de génotype AA pour l’hémoglobine contre 55,0% (22/40) chez les personnes
porteuses d’allèles S et/ou C (AC, AS, CC, SC). La différence de l’infection n’était pas
statistiquement significative entre les individus de génotypes AA et les porteurs d’allèles S et/ou
C (p = 0,06).
L’analyse du taux de la parasitémie a concerné les 61 personnes de l’étude ayant subi le
génotypage pour l’hémoglobine et positives à l’infection à Plasmodium falciparum. Le degré
de parasitémie était similaire chez les personnes de génotypes AA comparativement aux
personnes porteuses de l’allèle S et/ou C (p 0,05).
Cependant bien que limité dans nos analyses statistiques par cette faible taille d’échantillon, on
notait une moyenne géométrique de la parasitémie relativement plus élevée chez les individus
HbAA par rapport aux personnes HbAC (965,301 vs 653,987 parasites/µL) et HbAS (965,301
vs 583,788 parasites/µL). Comparativement à ces trois groupes on notait également une
parasitémie plus basse chez les trois (03) individus doubles hétérozygotes SC de notre étude
(145,370 parasites par/µL) même si l’unique individu de génotype CC présentait une forte
parasitémie (16630,000).
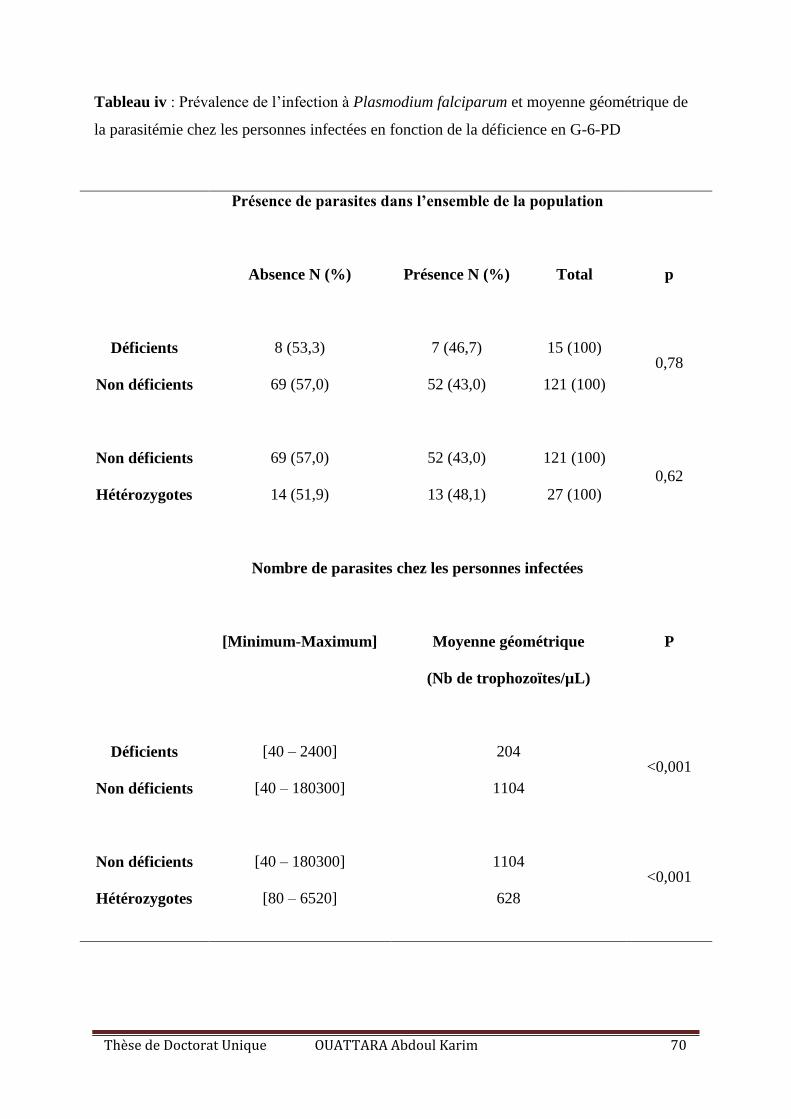
Thèse de Doctorat Unique OUATTARA Abdoul Karim 70
Tableau iv : Prévalence de l’infection à Plasmodium falciparum et moyenne géométrique de
la parasitémie chez les personnes infectées en fonction de la déficience en G-6-PD
Présence de parasites dans l’ensemble de la population
Absence N (%)
Présence N (%)
Total
p
Déficients 8 (53,3) 7 (46,7) 15 (100)
0,78
Non déficients 69 (57,0) 52 (43,0) 121 (100)
Non déficients 69 (57,0) 52 (43,0) 121 (100)
0,62
Hétérozygotes 14 (51,9) 13 (48,1) 27 (100)
Nombre de parasites chez les personnes infectées
[Minimum-Maximum] Moyenne géométrique
(Nb de trophozoïtes/µL)
P
Déficients [40 – 2400] 204
<0,001
Non déficients [40 – 180300] 1104
Non déficients [40 – 180300] 1104
<0,001
Hétérozygotes [80 – 6520] 628
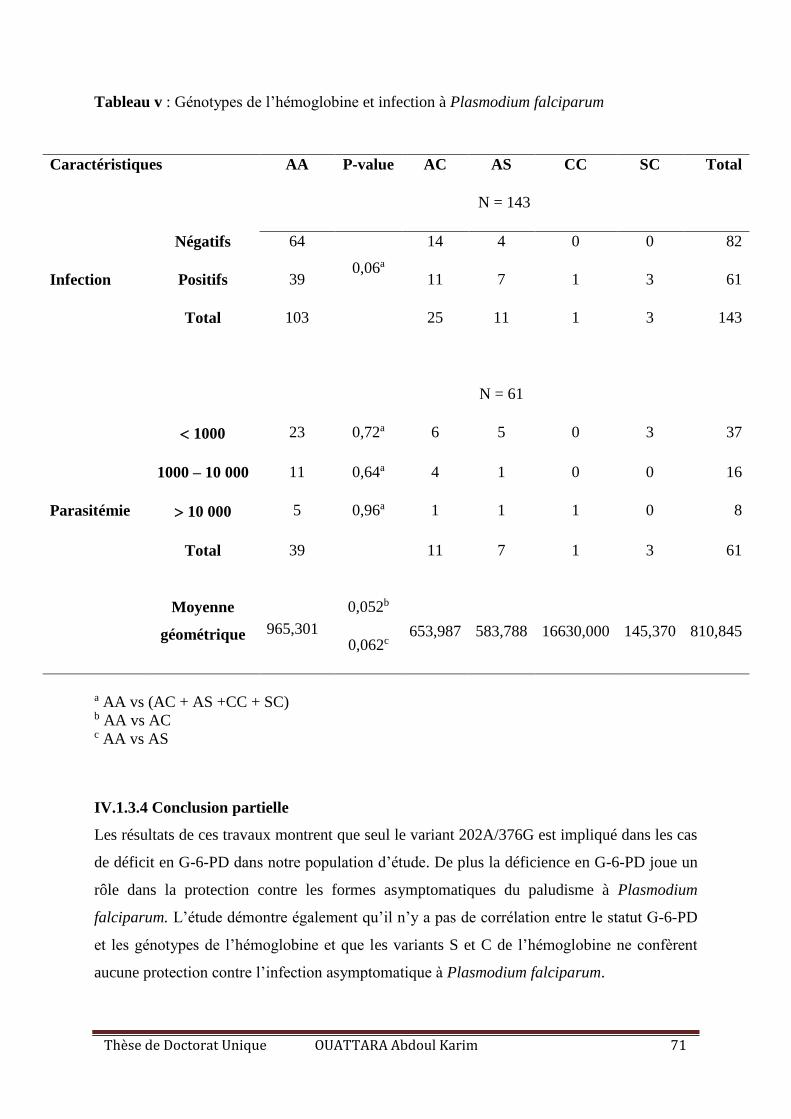
Thèse de Doctorat Unique OUATTARA Abdoul Karim 71
Tableau v : Génotypes de l’hémoglobine et infection à Plasmodium falciparum
Caractéristiques AA P-value AC AS CC SC Total
N = 143
Négatifs 64
0,06a
14 4 0 0 82
Infection Positifs 39 11 7 1 3 61
Total 103 25 11 1 3 143
N = 61
1000 23 0,72a 6 5 0 3 37
Parasitémie
1000 – 10 000 11 0,64a 4 1 0 0 16
10 000 5 0,96a 1 1 1 0 8
Total 39 11 7 1 3 61
Moyenne
géométrique
965,301
0,052b
0,062c
653,987
583,788
16630,000
145,370
810,845
a AA vs (AC + AS +CC + SC) b AA vs AC c AA vs AS
IV.1.3.4 Conclusion partielle
Les résultats de ces travaux montrent que seul le variant 202A/376G est impliqué dans les cas
de déficit en G-6-PD dans notre population d’étude. De plus la déficience en G-6-PD joue un
rôle dans la protection contre les formes asymptomatiques du paludisme à Plasmodium
falciparum. L’étude démontre également qu’il n’y a pas de corrélation entre le statut G-6-PD
et les génotypes de l’hémoglobine et que les variants S et C de l’hémoglobine ne confèrent
aucune protection contre l’infection asymptomatique à Plasmodium falciparum.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 72
IV.2 Résultats des travaux portant sur l’hétérogénéité allélique du déficit en
G-6-PD chez des personnes malades du paludisme dans la ville de
Ouagadougou au Burkina Faso
IV.2.1 Bref rappel du contexte et but de l’étude
Le gène responsable du déficit en G-6-PD est un gène très polymorphique avec les variants A-
202A/376G, Santamaria (376G/542T) et Betica Selma (376G/968T) connus dans les
populations d’Afrique de l’Ouest. Il conférerait une protection contre les formes sévères du
paludisme bien qu’il existe des divergences de point de vue quant aux mécanismes de la
protection entre les différentes études d’associations. Bien que les résultats de notre première
étude aient également démontré que seul le variant G-6-PD A- 202A/376 est impliqué dans les
cas de déficit en G-6-PD au Burkina Faso, une confirmation de l’absence des autres variants G-
6-PD A- dans nos populations demeure nécessaire.
IV.2.2 Rappel de l’objectif de l’étude
L’objectif de cette étude était de rechercher par PCR classique et PCR en temps réel six (06)
SNPs du gène G-6-PD en corrélation avec le paludisme symptomatique en zone urbaine au
Burkina Faso.
IV.2.3 Marqueurs génétiques et paludisme symptomatique
IV.2.3.1 Caractéristiques sociodémographiques et cliniques
Cette deuxième population d’étude était constituée de 50,5% (92/182) d’hommes et de 49,5%
(90/182) de femmes, âgés de 1 à 72 ans avec un âge moyen de 17,1 ± 13,9 ans. Les enfants de
moins de 5 ans représentaient 19,2% (35/182) de la population d’étude tandis qu’on notait
46,7% (85/182) d’individus de plus de 15 ans. L’analyse sociodémographique a montré que
les 182 patients inclus dans cette étude étaient de différents groupes ethniques. Les Mossis
représentaient l’ethnie majoritaire avec une proportion de 76,4% (139/182) d’individus dont les
deux parents étaient de ce groupe ethnique (Tableau VI). Il faut noter que la moitié du groupe
« Autres » (10/20) était constituée de groupes ethniques des pays de la sous-région avec une
prédominance des nigérians (6/10). Plus de 43% des patients de notre étude avaient commencé
un traitement avant la réalisation de la goutte épaisse. La population d’étude était en conformité
avec l’équilibre d’Hardy-Weinberg (p = 0,464).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 73
IV.2.3.2 Prévalence du déficit en G-6-PD et des génotypes de l’hémoglobine
L’analyse des résultats de la PCR classique a donné plus de 64% (58/90) de femmes porteuses
des mutations en position 202 et/ou 376. Vingt-cinq (25) d’entre elles ont été classées en
homozygotes et hétérozygotes par la PCR en temps réel. Le génotypage du déficit en G-6-PD
a révélé une prévalence de 78,6% d’individus G-6-PD normal (29,7% de génotype B, 13,2% de
génotype A, 20,9% BB, 12,1% BA et 2,7% AA) contre 9,9% de personnes
hémizygotes/homozygotes pour le déficit en G-6-PD (8,8% 202A/376G et 0,5% 376G/968C).
Il faut noter que les deux parents de l’individu G-6-PDA- (376G/968C) étaient Mossi tandis
que les parents de l’individu de sexe féminin porteur du variant G-6-PDA- Santamaria
(376G/542T) étaient du groupe ethnique Gouro de la Côte d’Ivoire. La figure 34 présente les
bandes des différentes variantes observées après électrophorèse. Les femmes hétérozygotes
représentaient 11,5% (6,6% BA- et 4,9% AA-) de la population d’étude. La prévalence des
hommes hémizygotes était significativement plus élevée que les femmes homozygotes pour le
déficit en G-6-PD (15,5% vs 4,4% avec p = 0,015). La prévalence des génotypes de
l’hémoglobine était estimée à 73,6%, 22,5% et 2,2% respectivement pour les génotypes AA,
AC et AS. La fréquence de l’allèle HbS était de 0,014 contre 0,126 pour l’allèle HbC. La
répartition de ces génotypes était similaire selon le sexe et le statut G-6-PD (Tableau VII).
Figure 34 : Les différentes bandes après l’électrophorèse (A) l’échantillon n°17 porte les
mutations 376G/968C (B) l’échantillon n°30 porte les mutations 202A/376G/542T. SM =
Standard Marker, MTC = Mutant Type Control, IC = Internal Control
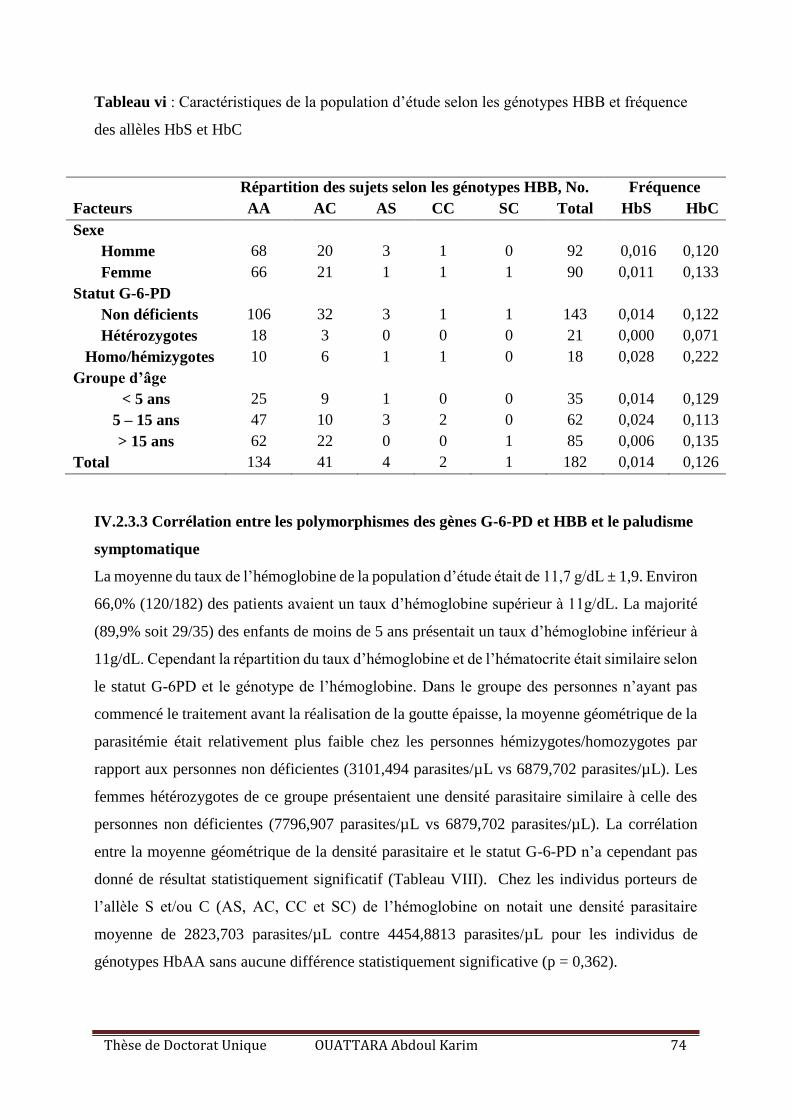
Thèse de Doctorat Unique OUATTARA Abdoul Karim 74
Tableau vi : Caractéristiques de la population d’étude selon les génotypes HBB et fréquence
des allèles HbS et HbC
Répartition des sujets selon les génotypes HBB, No. Fréquence
Facteurs AA AC AS CC SC Total HbS HbC
Sexe
Homme 68 20 3 1 0 92 0,016 0,120
Femme 66 21 1 1 1 90 0,011 0,133
Statut G-6-PD
Non déficients 106 32 3 1 1 143 0,014 0,122
Hétérozygotes 18 3 0 0 0 21 0,000 0,071
Homo/hémizygotes 10 6 1 1 0 18 0,028 0,222
Groupe d’âge
< 5 ans 25 9 1 0 0 35 0,014 0,129
5 – 15 ans 47 10 3 2 0 62 0,024 0,113
> 15 ans 62 22 0 0 1 85 0,006 0,135
Total 134 41 4 2 1 182 0,014 0,126
IV.2.3.3 Corrélation entre les polymorphismes des gènes G-6-PD et HBB et le paludisme
symptomatique
La moyenne du taux de l’hémoglobine de la population d’étude était de 11,7 g/dL ± 1,9. Environ
66,0% (120/182) des patients avaient un taux d’hémoglobine supérieur à 11g/dL. La majorité
(89,9% soit 29/35) des enfants de moins de 5 ans présentait un taux d’hémoglobine inférieur à
11g/dL. Cependant la répartition du taux d’hémoglobine et de l’hématocrite était similaire selon
le statut G-6PD et le génotype de l’hémoglobine. Dans le groupe des personnes n’ayant pas
commencé le traitement avant la réalisation de la goutte épaisse, la moyenne géométrique de la
parasitémie était relativement plus faible chez les personnes hémizygotes/homozygotes par
rapport aux personnes non déficientes (3101,494 parasites/µL vs 6879,702 parasites/µL). Les
femmes hétérozygotes de ce groupe présentaient une densité parasitaire similaire à celle des
personnes non déficientes (7796,907 parasites/µL vs 6879,702 parasites/µL). La corrélation
entre la moyenne géométrique de la densité parasitaire et le statut G-6-PD n’a cependant pas
donné de résultat statistiquement significatif (Tableau VIII). Chez les individus porteurs de
l’allèle S et/ou C (AS, AC, CC et SC) de l’hémoglobine on notait une densité parasitaire
moyenne de 2823,703 parasites/µL contre 4454,8813 parasites/µL pour les individus de
génotypes HbAA sans aucune différence statistiquement significative (p = 0,362).
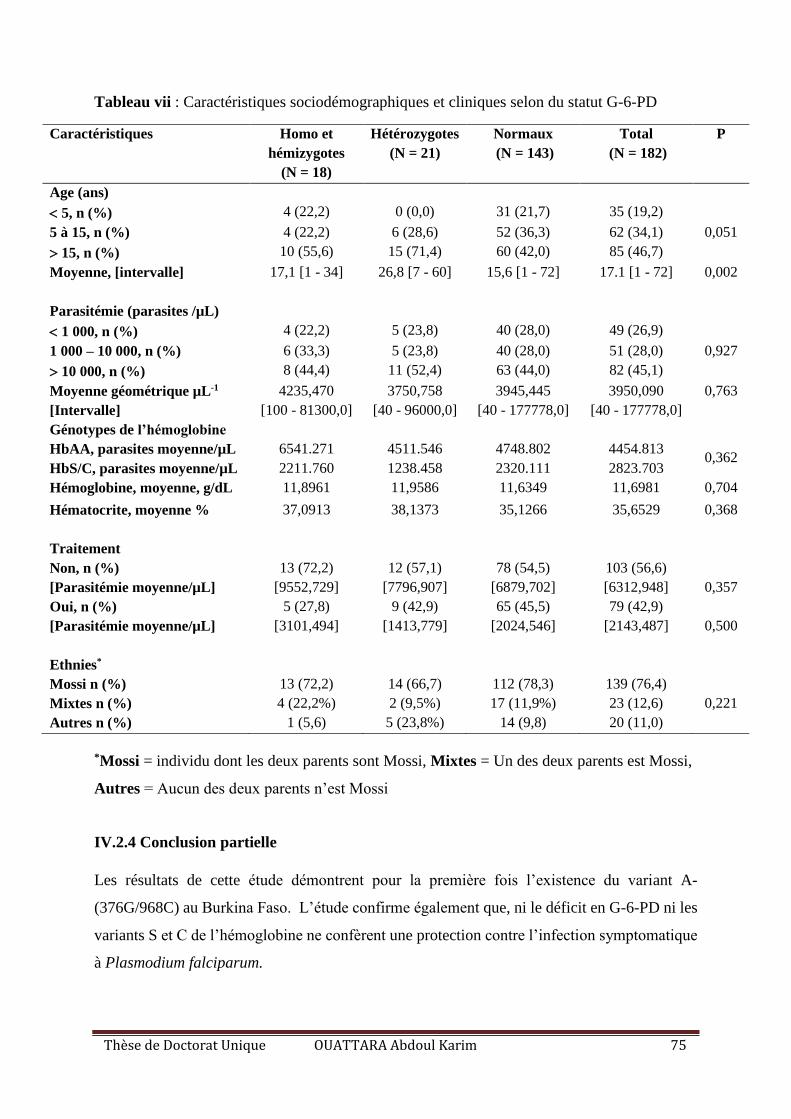
Thèse de Doctorat Unique OUATTARA Abdoul Karim 75
Tableau vii : Caractéristiques sociodémographiques et cliniques selon du statut G-6-PD
Caractéristiques Homo et
hémizygotes
(N = 18)
Hétérozygotes
(N = 21)
Normaux
(N = 143)
Total
(N = 182)
P
Age (ans)
5, n (%) 4 (22,2) 0 (0,0) 31 (21,7) 35 (19,2)
5 à 15, n (%) 4 (22,2) 6 (28,6) 52 (36,3) 62 (34,1) 0,051
15, n (%) 10 (55,6) 15 (71,4) 60 (42,0) 85 (46,7)
Moyenne, [intervalle] 17,1 [1 - 34] 26,8 [7 - 60] 15,6 [1 - 72] 17.1 [1 - 72] 0,002
Parasitémie (parasites /µL)
1 000, n (%) 4 (22,2) 5 (23,8) 40 (28,0) 49 (26,9)
1 000 – 10 000, n (%) 6 (33,3) 5 (23,8) 40 (28,0) 51 (28,0) 0,927
10 000, n (%) 8 (44,4) 11 (52,4) 63 (44,0) 82 (45,1)
Moyenne géométrique µL-1
[Intervalle]
4235,470
[100 - 81300,0]
3750,758
[40 - 96000,0]
3945,445
[40 - 177778,0]
3950,090
[40 - 177778,0]
0,763
Génotypes de l’hémoglobine
HbAA, parasites moyenne/µL 6541.271 4511.546 4748.802 4454.813
0,362 HbS/C, parasites moyenne/µL 2211.760 1238.458 2320.111 2823.703
Hémoglobine, moyenne, g/dL 11,8961 11,9586 11,6349 11,6981 0,704
Hématocrite, moyenne % 37,0913 38,1373 35,1266 35,6529 0,368
Traitement
Non, n (%)
[Parasitémie moyenne/µL]
13 (72,2)
[9552,729]
12 (57,1)
[7796,907]
78 (54,5)
[6879,702]
103 (56,6)
[6312,948]
0,357
Oui, n (%)
[Parasitémie moyenne/µL]
5 (27,8)
[3101,494]
9 (42,9)
[1413,779]
65 (45,5)
[2024,546]
79 (42,9)
[2143,487]
0,500
Ethnies*
Mossi n (%) 13 (72,2) 14 (66,7) 112 (78,3) 139 (76,4)
Mixtes n (%) 4 (22,2%) 2 (9,5%) 17 (11,9%) 23 (12,6) 0,221
Autres n (%) 1 (5,6) 5 (23,8%) 14 (9,8) 20 (11,0)
*Mossi = individu dont les deux parents sont Mossi, Mixtes = Un des deux parents est Mossi,
Autres = Aucun des deux parents n’est Mossi
IV.2.4 Conclusion partielle
Les résultats de cette étude démontrent pour la première fois l’existence du variant A-
(376G/968C) au Burkina Faso. L’étude confirme également que, ni le déficit en G-6-PD ni les
variants S et C de l’hémoglobine ne confèrent une protection contre l’infection symptomatique
à Plasmodium falciparum.

DISCUSSION

Thèse de Doctorat Unique OUATTARA Abdoul Karim 76
CHAPITRE V : DISCUSSION GENERALE
La population générale de notre étude était constituée de 382 échantillons répartis en deux
groupes. Le premier groupe comptait 200 individus vivant dans une zone rurale où leur
exposition répétée au paludisme, ne prévient pas nécessairement l'infection, mais peut limiter
la densité et les symptômes du paludisme. Dans ce groupe, un génotypage par PCR en temps
réel pour les SNPs G202A, A376G, A542T et par PCR/RFLP pour les mutations G680T et
T968C impliquées dans le déficit en G-6-PD a été réalisé. Le second groupe était constitué de
182 patients malades du paludisme qui ont subi un génotypage par PCR classique et par PCR
en temps réel pour six mutations du gène G-6-PD.
V.1 Marqueurs génétiques et Paludisme asymptomatique
Les objectifs de cette étude étaient d’une part d’estimer la prévalence des génotypes de
l’hémoglobine et le polymorphisme génétique du déficit en G-6-PD au sein de notre population
d’étude et d’autre part de déterminer la corrélation entre ces anomalies génétiques et le
paludisme asymptomatique. En effet le paludisme asymptomatique reste un défi pour les
programmes de lutte contre le paludisme, car il influence de manière significative la dynamique
de la transmission de la maladie. Une compréhension approfondie de l'interaction entre hôte et
parasites dans le développement de différents tableaux cliniques est donc nécessaire. La
prévalence de la déficience en G-6-PD a été estimée chez 200 personnes recrutées dans une
communauté rurale (KOUBRI), en zone endémique du paludisme au Burkina Faso en
recherchant tous les génotypes déficients avec des fréquences polymorphiques connus dans la
sous-région. Il s’agit du variant déficient 202A/376G considéré comme le plus fréquent en
Afrique subsaharienne (BEUTLER et DUPARC, 2007), le variant Santamaria (376G/542T)
observé avec une fréquence élevée dans la population Sérère au Sénégal (DE ARAUJO et al.,
2006), le variant Betica Selma (376G/968C) considéré comme le variant déficient le plus
fréquent en Gambie (CLARK et al., 2009b) et le variant 376G/680T (BEUTLER et al., 1989).
Toutes les personnes déficientes de ce groupe étaient porteuses du variant G-6-PDA-
(202A/376G). Les résultats de notre étude sont similaires à ceux obtenus par Carter et al.,
(2011) dans six pays africains incluant le Burkina Faso, et confirme les résultats d’études
antérieures ayant montré que la G-6-PDA- (202A/376G) est le variant déficient le plus fréquent
en Afrique subsaharienne (HIRONO et BEUTLER, 1988, BEUTLER et DUPARC, 2007).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 77
Le test de l’équilibre de Hardy-Weinberg déterminé chez les femmes selon la méthode de
CARTER et al., (2011); a donné une valeur de p = 0,698, qui montre qu’il n’y a pas de
différence significative entre la distribution observée et la distribution théorique. Par
conséquent notre population d’étude était en équilibre de Hardy-Weinberg.
La prévalence de la déficience en G-6-PD de 9,5% observée dans notre étude est comparable à
celle de 9,0% rapportée par CARTER et al., (2011) dans six pays africains dont le Burkina
Faso. Cependant, elle est inférieure aux prévalences de 19,6% et 16,3% rapportées
respectivement par MODIANO et al., (2001a) chez les mossi et les Rimaibé et par SIMPORE
et al., (2007) dans la population générale au Burkina Faso. Ces variations pourraient s’expliquer
par la taille de la population d’étude et la méthode de diagnostic utilisée (PCR en temps réel,
PCR/RFLP ou mesure de l’activité enzymatique), mais aussi par la difficulté de distinguer les
femmes hétérozygotes déficientes et non déficientes (PETERS et VAN NOORDEN, 2009). Le
génotypage seul ne permettant pas de déterminer le statut déficient ou non déficient des femmes
hétérozygotes. Par conséquent la prévalence du déficit de 9,5% observée dans cette étude
pourrait être plus élevée, si l’on déterminait par le test enzymatique le statut des femmes
hétérozygotes, car elles présentent une variété d’expression phénotypique à cause de la
lyonisation. En effet, au Nigéria une étude a montré que sur 81 femmes hétérozygotes 53%
avaient une activité G-6-PD normal, 33% étaient intermédiaire et 14% étaient biochimiquement
déficient (MAY et al., 2000).
Dans notre étude la prévalence des hommes hémizygotes pour le déficit en G-6-PD était
significativement plus élevée comparée aux femmes homozygotes (14,3%). Ces résultats sont
comparables à ceux rapportés par SIMPORE et al., (2007) qui avaient observé 20,5% de
déficience chez les hommes et 12,3% chez les femmes. En effet, la déficience en G-6-PD étant
une enzymopathie liée au chromosome X, elle se manifeste chez les hommes hémizygotes et
les femmes homozygotes. La probabilité qu’une femme hérite de deux chromosomes X malades
est faible. Par conséquent les hommes sont beaucoup plus touchés par la maladie que les
femmes qui sont généralement des porteuses saines. Dans ce groupe la prévalence des
génotypes HbAA, HbAC et HbAS était respectivement de 72,0%, 17,5% et 7,7%. Ces
prévalences sont similaires aux 73.2%; 15.0%; et 8.2% rapportées respectivement pour les
sujets HbAA, HbAC et HbAS par BOUGOUMA et al., (2012) au Burkina Faso ainsi qu’aux
prévalences rapportées par de nombreuses études menées en Afrique de l’Ouest et notamment
au Burkina Faso où l’hémoglobine C a une prévalence relativement élevée (MODIANO et al.,
2001b; DIALLO et al., 2004).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 78
Cela pourrait s’expliquer par le fait que le gène βc trouve son origine dans la zone Ouest de
l’Afrique. La prévalence de l’infection était similaire chez les individus HbAA
comparativement aux individus porteurs d’allèles S et/ou C. Dans leur étude menée en 2008 au
Sénégal, VAFA et ses collaborateurs n’ont pas observé d’association entre le trait
drépanocytaire et l’infection asymptomatique à Plasmodium falciparum (VAFA et al., 2008).
En effet de nombreuses études d’associations ont montré que les allèles S et C de l’hémoglobine
ne conféraient pas une protection contre l’infection à Plasmodium mais plutôt contre la
progression de la maladie vers des formes sévères (AMOAKO et al., 2014; MANGANO et al.,
2015). En dépit de la faible taille de notre population qui a limitée certaines de nos analyses
statistiques nous avons pu observer une réduction de la charge parasitaire chez les sujets
porteurs d’allèles S et/ou C par rapport aux personnes HbAA. Ces résultats sont similaires aux
travaux de AKANBI, (2015) au Nigeria, TRAVASSOS et al., (2015) au Mali et MANGANO
et al., (2015) au Burkina Faso. MANGANO et collaborateurs ont observé que HbS à l’état
hétérozygote était associé à une réduction de 70% de la parasitémie à Plasmodium falciparum.
Les auteurs ont également noté une forte protection conférée par HbC à l’état homozygote.
Nous avons également étudié la corrélation entre le déficit en G-6-PD et le paludisme
asymptomatique. Dans ce groupe nous n’avons pas observé d’effet significatif du génotype de
la G-6-PD sur la prévalence de l’infection à Plasmodium falciparum. Cependant parmi les
personnes infectées, la parasitémie était significativement plus élevée chez les personnes non
déficientes par rapport aux personnes hémizygotes/homozygotes (p < 0,001) et les femmes
hétérozygotes (p < 0,001). Nos résultats sont comparables à ceux d’une étude faite au Gabon
ayant montré l’association entre déficience en G-6-PD et protection contre le paludisme
asymptomatique (MOMBO et al., 2003). Dans la même année au Gabon, MISSINOU et ses
collaborateurs ont démontré que les individus porteurs des mutations G-6-PD sont plus
susceptibles de faire une infection asymptomatique que les individus dépourvus de ces
polymorphismes (MISSINOU et al., 2003). En 2008 au Sénégal, VAFA et collaborateurs ont
également observé une densité parasitaire significativement plus faible chez les filles porteuses
de mutations G-6-PD comparativement à celles qui étaient G-6-PD normal (VAFA et al., 2008).
Ces résultats s’expliqueraient par le fait que la déficience en G-6-PD inhibe le développement
de Plasmodium falciparum. La quantité de G-6-PD décroit durant la vie de l’hématie rendant
les vielles hématies chez qui l’activité de la G-6-PD est en baisse, vulnérables au stress oxydatif
(RUWENDE et HILL, 1998).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 79
En effet, Plasmodium falciparum très sensible aux agressions oxydatives, préfère envahir les
jeunes hématies avec une haute activité en G-6-PD plutôt que les vielles hématies où l’activité
de l’enzyme est faible, d’où la protection de la déficience en G-6-PD contre le Plasmodium
(ALLISON et CLYDE, 1961).
A l’échelle cellulaire, cela a été confirmé par les données relatives aux femmes hétérozygotes
parasitées par le Plasmodium. LUZZATTO et ses collaborateurs ont montré en 1969, que chez
ces femmes, la fréquence des hématies G-6-PD déficientes parasitées était, suivant les
individus, 2 à 80 fois plus faible que celle des hématies parasitées possédant l’enzyme G-6-
PD efficace (LUZZATTO et al., 1969).
En effet les allèles des gènes portés par un des chromosomes X s’expriment dans certaines
cellules tandis que les allèles de l’autre chromosome X s’expriment dans d’autres cellules, d’où
la présence de deux populations d’hématies chez les femmes hétérozygotes pour le gène de la
G-6-PD. Le variant G-6-PDA- (202A/376G) entraine une diminution de l’activité de la G-6-PD
de 10 à 20% par rapport à l’enzyme normale G-6-PDB (BEUTLER, 1991). D’une part cette
déficience créée un environnement défavorable à la survie de Plasmodium falciparum qui est
très sensible au stress oxydatif (CLARK et al., 1989). D’autre part, lorsqu’elles sont parasitées,
elles sont plus phagocytées à un stade où le parasite n’a pas encore réalisé sa multiplication. Il
en résulte une inhibition de la prolifération de Plasmodium et donc un paludisme moins sévère.
Avec l’agglutination précoce des hématies parasitées, le déficit en G-6-PD protège contre le
paludisme (CAPPADORO et al., 1998).
Des études ont montré que la déficience en G-6-PD protège contre le paludisme (ALLISON et
CLYDE, 1961; LUZZATTO et al., 1969), mais aussi qu’elle peut poser de graves problèmes,
notamment lors du traitement de cette maladie. En effet certains médicaments contre le
paludisme comme la primaquine, peuvent induire des crises hémolytiques chez les personnes
déficientes en G-6-PD (HOWES et al., 2013a).Cependant nous n’avons pas observés de
différence significative en comparant la moyenne du taux d’hémoglobine et celle l’hématocrite
d’une part, chez les personnes hémizygotes/homozygotes pour le déficit en G-6-PD et non
déficientes en G-6-PD et d’autre part, par rapport aux personnes non déficientes et les
hétérozygotes. Ces résultats sont comparables aux travaux antérieurs qui n’ont pas observé
d’effet significatif du génotype de la G-6-PD sur le taux d’hémoglobine (ENEVOLD et al.,
2008; CARTER et al., 2011).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 80
Cela s’expliquerait par le fait que la déficience en G-6-PD est en général asymptomatique et
l’anémie hémolytique ne survient qu’en cas d’accident suite à l’ingestion de certains aliments
ou médicaments contre-indiqués pour les personnes déficientes en G-6-PD. Aussi la taille de
l’échantillon a également constitué une limite dans cette étude. Elle ne nous a pas permis de
faire la relation entre l’anémie et la déficience en G-6-PD chez les enfants et les adultes vivants
dans une zone d’endémie palustre.
Dans cette étude la relation entre la déficience en G-6-PD et le type d’hémoglobine n’a donné
aucune différence significative. Cependant dans une étude prospective transversale menée au
Sénégal en 2005, sur 319 personnes porteuses de l’hémoglobine S et 318 cas témoins appariés
selon l’âge et le sexe, DIOP et al. ont observé un déficit en G-6-PD significativement plus
élevé chez les drépanocytaires (21,6 %) que chez les témoins (12,3 %) (DIOP et al., 2005).
V.2 Marqueurs génétiques et Paludisme symptomatique
Les objectifs de notre deuxième étude étaient d’une part d’estimer la prévalence des génotypes
de l’hémoglobine afin de confirmer ou d’infirmer l’absence d’autres variants impliqués dans le
déficit en G-6-PD au Burkina Faso et d’autre part de déterminer la corrélation entre ces
anomalies génétiques et le paludisme symptomatique.
Il s’est agi de rechercher six (06) variants G-6-PDA- ainsi que la corrélation entre le déficit en
G-6-PD ou les génotypes de l’hémoglobine et le paludisme symptomatique chez 182 patients
souffrant de paludisme, recrutés dans la ville de Ouagadougou. Le déficit en G-6-PD présente
un grand polymorphisme avec de nombreux variants génotypiques connus en Afrique de
l’Ouest (MINUCCI et al., 2012). Ainsi le génotypage de six (06) SNPs impliqués dans cette
maladie génétique nous a permis d’évaluer la fréquence des différents variants déficients chez
des patients atteints de paludisme symptomatique. Il faut noter que le Kit GENESPARK G6PD
African (Immunospark, Rome, Italy) à travers une PCR classique suivie d’une électrophorèse
sans digestion enzymatique est commode et présente l’avantage de permettre le génotypage
simultané de six SNPs pour chaque échantillon. Cependant il présente une limite au niveau des
femmes car il indique la présence de la mutation sans permettre la distinction entre
homozygotes et hétérozygotes. La prévalence des homozygotes et hémizygotes pour le déficit
en G-6-PD était estimée à 9,9% (dans ce groupe. Cette prévalence est similaire à celles
rapportées par les études de génotypage au Burkina Faso et dans la sous-région (CARTER et
al., 2011, OUATTARA et al., 2014).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 81
Selon le genre, on observait une prévalence des hommes hémizygotes pour le déficit en G-6-
PD (15,2%) significativement plus élevée que les femmes homozygotes (4,4%) du fait que la
maladie soit liée au chromosome X (OUATTARA et al., 2014). Parmi les personnes atteintes
du déficit en G-6-PD dans cette étude, le variant G-6-PDA- (202A/376G) était le variant le plus
fréquent. En effet, il a été observé dans 88,9% des cas de déficit. Ces observations sont
conformes aux études antérieures de génotypage menées dans la sous-région montrant que ce
variant est le plus fréquent dans la plupart des populations africaines (LUZZATTO, 2012,
OUATTARA et al., 2014). Dans cette étude, le variant G-6-PD Betica Selma (376G/968C) a
été retrouvé chez un individu du groupe ethnique Mossi. Ce variant observé avec des fréquences
relativement élevées dans la population gambienne (CLARK et al., 2009b), est identifié pour
la première fois au Burkina Faso. Une étude au Mali a rapporté en 2014, une fréquence élevée
de ce variant chez les Fulani (6,1%) comparativement aux Dogons (0,0%) (MAIGA et al.,
2014). Bien que ce variant présente donc une faible prévalence au sein du groupe ethnique
Mossi, sa fréquence pourrait être bien plus élevée au Nord du pays où prédomine le groupe
ethnique Fulani qui est moins susceptible au paludisme avec une faible prévalence du variant
G-6-PDA- (202A/376G) comparativement aux autres groupes ethniques (MODIANO et al.,
2001a) . Une des femmes déficientes de l’ethnie Gouro (Côte d’Ivoire) de notre étude portait le
variant A- (202A/376G) sur l’un de ses chromosomes X et le variant Santamaria (376G/542T)
sur le second chromosome X avec une très faible parasitémie. Toutes ces observations
suggèrent que ces variants existent dans nos populations même s’ils surviennent avec des
fréquences relativement faibles dans certaines zones. Les plus hautes fréquences de la G-6-PD
Santamaria ont été rapportées dans la population Sérère au Sénégal (DE ARAUJO et al., 2006).
Les observations de la présente étude indiquent une sous-estimation de la prévalence réelle du
déficit en G-6-PD dans la plupart des études de génotypage et pourraient expliquer en partie les
différences de fréquences du déficit entre ces études de génotypage et les études de mesure de
l’activité enzymatique (MODIANO et al., 2001a, SIMPORE et al., 2007). Nos résultats
suggèrent l’existence du variant G-6-PD Santamaria (376G/542T) en Côte d’Ivoire avec une
fréquence relativement élevée au sein du groupe ethnique Gouro. Les Gouro sont une
population Mandingue d’Afrique de l’Ouest principalement établie au Centre-Ouest de la Côte
d’Ivoire. Ce groupe ethnique partage probablement une histoire commune avec certaines
populations mandingues du Burkina Faso. Nous émettons donc l’hypothèse d’une fréquence
relativement élevée du variant G-6-PD Santamaria au sein de certains groupes ethniques du
Sud-Ouest du Burkina Faso.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 82
Dans le cadre de cette étude, les patients ont subi une électrophorèse de l’hémoglobine. Un
syndrome drépanocytaire majeur (SC) a pu être mis en évidence chez 0,5% (1/182) des patients
et 2,2% (4/182) avaient le trait drépanocytaire AS. Les individus HbAC représentaient 22,5%
(41/182) de la population d’étude avec 1,1% (2/182) de sujets HbCC. La prévalence de
l’hémoglobine AC trouvée dans notre étude est comparable à celle (19,1%) trouvée par
SIMPORE et al. en 2007 et est supérieure à celle rapportée par KAFANDO et al. en 2005 chez
des nouveaux nés (15,4%) au Burkina Faso (KAFANDO et al., 2005). Des prévalences de
14,7% et de 13,0% d’hémoglobine AC ont été rapportées respectivement par AMOAKO et
collaborateurs au Ghana et TRAVASSOS et collaborateurs au Mali (AMOAKO et al., 2014;
TRAVASSOS et al., 2015). Toutes ces études montrent une fréquence élevée de l’hémoglobine
C dans la sous-région. En effet, l’Afrique de l’Ouest est située dans l’épicentre de
l’hémoglobine C (MODIANO et al., 2008).
Bien que des parasitémies réduites aient été observée dans certains cas (déficients vs Non
déficients en G-6-PD sans traitement ou sujets HbAA vs sujets HbS/HbC), l’ensemble des
analyses d’association entre le statut G-6-PD, les génotypes de l’hémoglobine et la densité
parasitaire, le taux d’hémoglobine ou l’hématocrite n’a pas donné de différences statistiquement
significatives. Nos résultats sont similaires à ceux rapportés par Carter et collaborateurs en 2011
dans six pays africains incluant le Burkina Faso (CARTER et al., 2011). Les auteurs n’avaient
pas observé d’effet significatif du génotype de la G-6-PD sur le taux d’hémoglobine et la
parasitémie. L’assortiment indépendant entre les génotypes de l’hémoglobine et le statut G-6-
PD est prévisible, du fait que les gènes respectifs soient sur différents chromosomes, et cela a
été rapporté dans une étude antérieure (LUZZATTO et ALLAN, 1968). Nos résultats
confirment également qu’il n’y a pas de protection contre l’infection palustre. En effet le déficit
en G-6-PD et les hémoglobinopathies S et C ne confèrent pas de protection contre l’infection à
Plasmodium falciparum mais pourraient plutôt permettre une évolution du paludisme vers la
guérison du malade. La protection par le déficit en G-6-PD contre les formes sévères ou contre
la mortalité palustre est connue, cependant le mécanisme de protection n’est pas complètement
élucidé (LUZZATTO, 2015). En 2009 dans une étude menée en Gambie, CLARK et al. n’ont
pas trouvé d’association entre le paludisme sévère et le seul variant 202A/376G (CLARK et al.,
2009a). Mais ce variant en pool avec d’autres allèles déficitaires révélait des signaux de
protection. En 2014 au Mali, MAIGA et ses collaborateurs n’ont pas trouvé de résultats
concluant sur l’effet protecteur des différents génotypes G-6-PD en corrélation avec le
paludisme simple (MAIGA et al., 2014).

Thèse de Doctorat Unique OUATTARA Abdoul Karim 83
Les auteurs ont même suggéré des signaux de risque accru de paludisme modéré chez les dogon
porteurs de la mutation 202A, notamment au niveau des femmes. Des effets de protection par
le déficit en G-6-PD contre le paludisme cérébral et une augmentation du risque du paludisme
avec anémie sévère ont été rapporté dans certains travaux sur la corrélation entre cette maladie
génétique et le paludisme sévère (MALARIA GENOMIC EPIDEMIOLOGY, 2014; SHAH et
al., 2016). L’hétérogénéité allélique de la G-6-PD, la complexité phénotypique et les difficultés
de classement des formes cliniques du paludisme sont autant de facteurs pouvant expliquer les
divergences entre les différentes études.
Cependant, la corrélation entre le déficit en G-6-PD et la protection contre le paludisme
asymptomatique a été rapportée par certains auteurs (MOMBO et al., 2003; OUATTARA et
al., 2014). L’avantage sélectif contre le paludisme des femmes hétérozygotes pour la G-6-PD a
été rapporté très tôt par BIENZYLE et al. (BIENZLE et al., 1972). Dans une étude cas témoins
menée en Tanzanie, il a été établi à travers un certain nombre de SNPs G-6-PD, que seules les
femmes hétérozygotes étaient protégées contre les formes sévères du paludisme
(MANJURANO et al., 2015). Une autre étude en Gambie avait abouti à la même conclusion
avec le variant G-6-PDA- (376G/968C) (SIRUGO et al., 2014).
Dans une autre récente étude de cohorte cas-témoins au Kenya, UYOGA et ses collaborateurs
ont montré que la clé de la protection contre les formes sévères du paludisme étaient les femmes
hétérozygotes pour le déficit en G-6-PD (UYOGA et al., 2015). Des paramètres comme le
traitement avant la réalisation de la goutte épaisse sont des facteurs qui peuvent également
influencer la densité parasitaire et biaiser les analyses d’associations.
En 2015 au Nigeria, IGBENEGHU et al. ont rapporté une forte protection des génotypes HbAS
et HbAC de l’hémoglobine contre le paludisme asymptomatique à Plasmodium falciparum
(IGBENEGHU et al., 2015). La protection des sujets HbAC contre les formes cliniques du
paludisme à Plasmodium falciparum a également été rapportée au Mali (TRAVASSOS et al.,
2015). Au cours de la même année, MANGANO et collaborateurs ont rapporté au Burkina Faso
que le génotype HbAS était associé à une réduction de 70% de la parasitémie à Plasmodium
falciparum contrairement aux sujets HbAC, bien qu’une forte protection fût également
observée chez les sujets HbCC et HbSC (MANGANO et al., 2015). Nos résultats suggèrent
que même si les hémoglobinopathies S et C protègent contre le paludisme grave comme le
déficit en G-6-PD, ils ne confèrent pas de protection contre les infections palustres.

CONCLUSION

Thèse de Doctorat Unique OUATTARA Abdoul Karim 84
Conclusion générale
Le déficit en G-6-PD est une pathologie génétique liée au chromosome X dont la sévérité
dépend du variant en cause. Les différents variants génétiques de cette anomalie sont
principalement le résultat d’une combinaison de mutations sur le gène G-6-PD. Cette étude
apporte la confirmation que la prévalence de la déficience en G-6-PD et des hémoglobines S et
C est relativement élevée au Burkina Faso du fait de l’endémicité du paludisme. Au cours de
nos travaux, aucune corrélation n’a été observée entre ces deux anomalies génétiques. Cet
assortiment indépendant entre les génotypes de l’hémoglobine et le statut G-6-PD serait
probablement dû au fait que leurs gènes respectifs sont sur des chromosomes différents. Le
déficit en G-6-PD étant lié au chromosome X, il touche beaucoup plus les hommes que les
femmes. Il faut noter cependant que les femmes hétérozygotes présentent des expressions
cliniques variables du fait de l’inactivation aléatoire du chromosome X. Les résultats de ces
travaux indiquent également que le portage d’hémoglobine S et/ou C ne confère aucun effet
protecteur contre l’infection palustre aussi bien symptomatique qu’asymptomatique. Ces
anomalies de l’hémoglobine protégeraient plutôt contre les formes sévères du paludisme.
L’étude a cependant démontré que le déficit en G-6-PD conférait une protection contre le
paludisme asymptomatique, probablement à travers une réduction du taux de parasites chez les
personnes hémizygotes/homozygotes et les femmes hétérozygotes. Tandis que la protection
contre l’infection symptomatique à Plasmodium falciparum (avec une complexité de
classification phénotypique) n’a pas été mise en évidence au cours de ses travaux.
Nos résultats confirment que le variant G-6-PDA- (202A/376G) est bien le variant déficitaire
le plus fréquent au Burkina Faso. Cependant contrairement aux études antérieures de
génotypage menées au Burkina Faso, cette étude a démontré pour la première fois l’existence
du variant G-6-PDA- (376G/968C). La G-6-PD Santamaria a également été mis en évidence
chez une femme de l’ethnie Gouro (Côte d’Ivoire). Le variant G-6-PD Santamaria (rs5030872 ;
G6PD c.542A>T) est un variant de classe II de l’OMS responsable d’une hémolyse
intermittente avec une activité enzymatique inférieure à 10%. Nos travaux suggèrent donc que
des variants comme la G-6-PD Betica Selma et peut être la G-6-PD Santamaria existent dans
nos populations. La faible taille de notre échantillon d’étude a limité certaines de nos analyses.
Une investigation plus poussée à grande échelle et dans les groupes ethniques spécifiques est
donc nécessaire pour une estimation réelle des variants impliqués dans le déficit en G-6-PD au
Burkina Faso et leur prise en compte dans les études d’associations.

PERSPECTIVES

Thèse de Doctorat Unique OUATTARA Abdoul Karim 85
Perspectives
Au regard de ce travail nous pouvons envisager les perspectives suivantes :
Etude de génotypage du même ordre sur une grande population afin de déterminer avec
plus de précision la prévalence réelle des variants G-6-PD Betica Selma et Santamaria
dans la population générale et les groupes ethniques spécifiques. Cette étude permettra
la mise en place d’une politique nationale du dépistage systématique du déficit en G-6-
PD au moins au sein des groupes les plus vulnérables en l’occurrence les femmes
enceintes et les enfants de moins de 5 ans.
Associer le phénotypage au génotypage pour déterminer le statut G-6-PD des femmes
hétérozygotes.
Faire un génotypage prenant en compte plusieurs SNPs G-6-PD avec des fréquences
polymorphiques tout en distinguant les formes cliniques du paludisme symptomatique
dans une étude d’association.
Elargir la taille de la population d’étude dans une étude d’association entre les
hémoglobinopathies et le paludisme pour une analyse plus approfondie.

REFERENCES

Thèse de Doctorat Unique OUATTARA Abdoul Karim 86
VI. REFERENCES
AGASA, B., BOSUNGA, K., OPARA, A., TSHILUMBA, K., DUPONT, E., VERTONGEN,
F., COTTON, F. & GULBIS, B. 2010. Prevalence of sickle cell disease in a northeastern
region of the Democratic Republic of Congo: what impact on transfusion policy?
Transfus Med, 20, 62-5.
AIDOO, M., TERLOUW, D. J., KOLCZAK, M. S., MCELROY, P. D., TER KUILE, F. O.,
KARIUKI, S., NAHLEN, B. L., LAL, A. A. & UDHAYAKUMAR, V. 2002. Protective
effects of the sickle cell gene against malaria morbidity and mortality. Lancet, 359,
1311-2.
AKANBI, O. M. 2015. Influence of haemoglobin genotype on malaria parasite density and anti-
msp-119 IgG (antibody) response in pregnant women and birth weight of neonates.
FUTA Journal of Research in Sciences, 2015 (1): 46-54, 11, 46-54.
ALLAHVERDIYEV, A. M., BAGIROVA, M., KOC, R. C., ATES, S. C., BAYDAR, S. Y.,
YAMAN, S., ABAMOR, E. S., OZTEL, O. N. & ELCICEK, S. 2012. Glucose-6-
Phosphate Dehydrogenase Deficiency and Malaria: A Method to Detect Primaquine-
Induced Hemolysis in vitro.
ALLISON, A. C. 1964. Polymorphism and Natural Selection in Human Populations. Cold
Spring Harb Symp Quant Biol, 29, 137-49.
ALLISON, A. C. & CLYDE, D. F. 1961. Malaria in African children with deficient erythrocyte
glucose-6-phosphate dehydrogenase. Br Med J, 1, 1346-9.
AMOAKO, N., ASANTE, K. P., ADJEI, G., AWANDARE, G. A., BIMI, L. & OWUSU-
AGYEI, S. 2014. Associations between Red Cell Polymorphisms and
<italic>Plasmodium falciparum</italic> Infection in the Middle Belt of Ghana. PLoS
ONE, 9, e112868.
ARGY, N. & HOUZÉ, S. 2014. Paludisme grave : de la physiopathologie aux nouveautés
thérapeutiques. Journal des Anti-infectieux, 16, 13-17.
ARUMUGAM, T. U., ITO, D., TAKASHIMA, E., TACHIBANA, M., ISHINO, T., TORII, M.
& TSUBOI, T. 2014. Application of wheat germ cell-free protein expression system for
novel malaria vaccine candidate discovery. Expert Rev Vaccines, 13, 75-85.
AUTINO, B., CORBETT, Y., CASTELLI, F. & TARAMELLI, D. 2012. Pathogenesis of
malaria in tissues and blood. Mediterr J Hematol Infect Dis, 4, e2012061.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 87
BAUDUER, F. 2012. Red cell polymorphisms and malaria: an evolutionary approach. Bulletins
et mémoires de la Société d'anthropologie de Paris, 25, 55-64.
BERRY, A., IRIART, X. & MAGNAVAL, J.-F. 2009. Nouvelles méthodes de diagnostic du
paludisme. Revue Francophone des Laboratoires, 2009, 65-70.
BEUTLER, E. 1959. The hemolytic effect of primaquine and related compounds. Blood 14,
103-39.
BEUTLER, E. 1991. Glucose-6-phosphate dehydrogenase deficiency. N Engl J Med, 324, 169-
74.
BEUTLER, E. & DUPARC, S. 2007. Glucose-6-phosphate dehydrogenase deficiency and
antimalarial drug development. Am J Trop Med Hyg, 77, 779-89.
BEUTLER, E., KUHL, W., VIVES-CORRONS, J. L. & PRCHAL, J. T. 1989. Molecular
heterogeneity of glucose-6-phosphate dehydrogenase A. Blood, 74, 2550-5.
BIENZLE, U., GUGGENMOOS-HOLZMANN, I. & LUZZATTO, L. 1979. Malaria and
erythrocyte glucose-6-phosphate dehydrogenase variants in West Africa. Am J Trop
Med Hyg, 28, 619-21.
BIENZLE, U., LUCAS, A., AYENI, O. & LUZZATTO, L. 1972. GLUCOSE-6-PHOSPHATE
DEHYDROGENASE AND MALARIA: GREATER RESISTANCE OF FEMALES
HETEROZYGOUS FOR ENZYME DEFICIENCY AND OF MALES WITH NON-
DEFICIENT VARIANT. The Lancet, 299, 107-110.
BISOFFI, Z., SIRIMA, S. B., MENTEN, J., PATTARO, C., ANGHEBEN, A., GOBBI, F.,
TINTO, H., LODESANI, C., NEYA, B., GOBBO, M. & VAN DEN ENDE, J. 2010.
Accuracy of a rapid diagnostic test on the diagnosis of malaria infection and of malaria-
attributable fever during low and high transmission season in Burkina Faso. Malar J, 9,
192.
BOOTH, C., INUSA, B. & OBARO, S. K. 2010. Infection in sickle cell disease: a review. Int
J Infect Dis, 14, e2-e12.
BOUGOUMA, E. C., TIONO, A. B., OUÉDRAOGO, A., SOULAMA, I., DIARRA, A.,
YARO, J.-B., OUÉDRAOGO, E., SANON, S., KONATÉ, A. T., NÉBIÉ, I., WATSON,
N. L., SANZA, M., DUBE, T. J. & SIRIMA, S. B. 2012. Haemoglobin variants and
Plasmodium falciparum malaria in children under five years of age living in a high and
seasonal malaria transmission area of Burkina Faso. Malaria Journal, 11, 1-10.
BOUSEMA, T., OKELL, L., FELGER, I. & DRAKELEY, C. 2014. Asymptomatic malaria
infections: detectability, transmissibility and public health relevance. Nat Rev
Microbiol, 12, 833-40.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 88
BROOKER, S., KOLACZINSKI, J. H., GITONGA, C. W., NOOR, A. M. & SNOW, R. W.
2009. The use of schools for malaria surveillance and programme evaluation in Africa.
Malar J, 8, 231.
CAPPADORO, M., GIRIBALDI, G., O'BRIEN, E., TURRINI, F., MANNU, F., ULLIERS, D.,
SIMULA, G., LUZZATTO, L. & ARESE, P. 1998. Early phagocytosis of glucose-6-
phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium
falciparum may explain malaria protection in G6PD deficiency. Blood, 92, 2527-34.
CAPPELLINI, M. D. & FIORELLI, G. 2008. Glucose-6-phosphate dehydrogenase deficiency.
Lancet, 371, 64-74.
CAQUET, R. 2010. Hémoglobine (électrophorèse de l'). 250 examens de laboratoire (11e
édition). Paris: Elsevier Masson.
CARSON, P. E., FLANAGAN C. L, ICKES C. E & S., A. A. 1956. Enzymatic deficiency in
primaquine-sensitive erythrocytes. Science, 124, 484-5.
CARTER, N., PAMBA, A., DUPARC, S. & WAITUMBI, J. N. 2011. Frequency of glucose-
6-phosphate dehydrogenase deficiency in malaria patients from six African countries
enrolled in two randomized anti-malarial clinical trials. Malar J, 10, 241.
CHAN, J. A., FOWKES, F. J. & BEESON, J. G. 2014. Surface antigens of Plasmodium
falciparum-infected erythrocytes as immune targets and malaria vaccine candidates.
Cell Mol Life Sci, 71, 3633-57.
CHILDS, B., ZINKHAM, W., BROWNE, E. A., KIMBRO, E. L. & TORBERT, J. V. 1958. A
genetic study of a defect in glutathione metabolism of the erythrocyte. Bull Johns
Hopkins Hosp, 102, 21-37.
CLARK, I. A., CHAUDHRI, G. & COWDEN, W. B. 1989. Some roles of free radicals in
malaria. Free Radic Biol Med, 6, 315-21.
CLARK, T. D., GREENHOUSE, B., NJAMA-MEYA, D., NZARUBARA, B., MAITEKI-
SEBUGUZI, C., STAEDKE, S. G., SETO, E., KAMYA, M. R., ROSENTHAL, P. J. &
DORSEY, G. 2008. Factors determining the heterogeneity of malaria incidence in
children in Kampala, Uganda. J Infect Dis, 198, 393-400.
CLARK, T. G., FRY, A. E., AUBURN, S., CAMPINO, S., DIAKITE, M., GREEN, A.,
RICHARDSON, A., TEO, Y. Y., SMALL, K., WILSON, J., JALLOW, M., SISAY-
JOOF, F., PINDER, M., SABETI, P., KWIATKOWSKI, D. P. & ROCKETT, K. A.
2009a. Allelic heterogeneity of G6PD deficiency in West Africa and severe malaria
susceptibility. Eur J Hum Genet, 17, 1080-5.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 89
CLARK, T. G., FRY, A. E., AUBURN, S., CAMPINO, S., DIAKITE, M., GREEN, A.,
RICHARDSON, A., TEO, Y. Y., SMALL, K., WILSON, J., JALLOW, M., SISAY-
JOOF, F., PINDER, M., SABETI, P., KWIATKOWSKI, D. P. & ROCKETT, K. A.
2009b. Allelic heterogeneity of G6PD deficiency in West Africa and severe malaria
susceptibility. Eur J Hum Genet, 17, 1080-5.
COHEN, J., BENNS, S., VEKEMANS, J. & LEACH, A. 2010. Le candidat vaccin
antipaludique RTS,S/AS est entré en essais cliniques de phase III. Annales
Pharmaceutiques Françaises, 68, 370-379.
CORNELIO, C. O. & SERIANO, O. F. 2011. Malaria in South Sudan 1: introduction and
pathophysiology. Southern Sudan Medical Journal, 4.
COWMAN, A. F., BERRY, D. & BAUM, J. 2012. The cellular and molecular basis for malaria
parasite invasion of the human red blood cell. J Cell Biol, 198, 961-71.
COX, F. E. 2010. History of the discovery of the malaria parasites and their vectors. Parasit
Vectors, 3, 5.
CROMPTON, P. D., MOEBIUS, J., PORTUGAL, S., WAISBERG, M., HART, G., GARVER,
L. S., MILLER, L. H., BARILLAS-MURY, C. & PIERCE, S. K. 2014. Malaria
immunity in man and mosquito: insights into unsolved mysteries of a deadly infectious
disease. Annu Rev Immunol, 32, 157-87.
CROMPTON, P. D., PIERCE, S. K. & MILLER, L. H. 2010. Advances and challenges in
malaria vaccine development. J Clin Invest, 120, 4168-78.
CYRKLAFF, M., SANCHEZ, C. P., KILIAN, N., BISSEYE, C., SIMPORE, J.,
FRISCHKNECHT, F. & LANZER, M. 2011. Hemoglobins S and C interfere with actin
remodeling in Plasmodium falciparum-infected erythrocytes. Science, 334, 1283-6.
DE ARAUJO, C., MIGOT-NABIAS, F., GUITARD, J., PELLEAU, S., VULLIAMY, T. &
DUCROCQ, R. 2006. The role of the G6PD AEth376G/968C allele in glucose-6-
phosphate dehydrogenase deficiency in the seerer population of Senegal.
Haematologica, 91, 262-3.
DELICAT-LOEMBET, L. M., ELGUERO, E., ARNATHAU, C., DURAND, P., OLLOMO,
B., OSSARI, S., MEZUI-ME-NDONG, J., MBANG MBORO, T., BECQUART, P.,
NKOGHE, D., LEROY, E., SICA, L., GONZALEZ, J. P., PRUGNOLLE, F. &
RENAUD, F. 2014. Prevalence of the sickle cell trait in Gabon: a nationwide study.
Infect Genet Evol, 25, 52-6.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 90
DIALLO, D. A., DOUMBO, O. K., DICKO, A., GUINDO, A., COULIBALY, D.,
KAYENTAO, K., DJIMDE, A. A., THERA, M. A., FAIRHURST, R. M., PLOWE, C.
V. & WELLEMS, T. E. 2004. A comparison of anemia in hemoglobin C and normal
hemoglobin A children with Plasmodium falciparum malaria. Acta Trop, 90.
DIBOULO, E., SIÉ, A. & VOUNATSOU, P. 2016. Assessing the effects of malaria
interventions on the geographical distribution of parasitaemia risk in Burkina Faso.
Malar J, 15, 1-14.
DIOP, S., SENE, A., CISSE, M., TOURE, A. O., SOW, O., THIAM, D. & DIAKHATE, L.
2005. [Prevalence and morbidity of G6PD deficiency in sickle cell disease in the
homozygote]. Dakar Med, 50, 56-60.
ENEVOLD, A., LUSINGU, J. P., MMBANDO, B., ALIFRANGIS, M., LEMNGE, M. M.,
BYGBJERG, I. C., THEANDER, T. G. & VESTERGAARD, L. S. 2008. Reduced risk
of uncomplicated malaria episodes in children with alpha+-thalassemia in northeastern
Tanzania. Am J Trop Med Hyg, 78, 714-20.
ENEVOLD, A., VESTERGAARD, L. S., LUSINGU, J., DRAKELEY, C. J., LEMNGE, M.
M., THEANDER, T. G., BYGBJERG, I. C. & ALIFRANGIS, M. 2005. Rapid
screening for glucose-6-phosphate dehydrogenase deficiency and haemoglobin
polymorphisms in Africa by a simple high-throughput SSOP-ELISA method. Malar J,
4, 61.
FUZIER, V. 2015. Favisme: Favism disease, G6PD deficiency. Prise en charge des maladies
rares en anesthésie et analgésie obstétricales. Paris: Elsevier Masson.
GARDNER, M. J., HALL, N., FUNG, E., WHITE, O., BERRIMAN, M., HYMAN, R. W.,
CARLTON, J. M., PAIN, A., NELSON, K. E., BOWMAN, S., PAULSEN, I. T.,
JAMES, K., EISEN, J. A., RUTHERFORD, K., SALZBERG, S. L., CRAIG, A., KYES,
S., CHAN, M. S., NENE, V., SHALLOM, S. J., SUH, B., PETERSON, J., ANGIUOLI,
S., PERTEA, M., ALLEN, J., SELENGUT, J., HAFT, D., MATHER, M. W., VAIDYA,
A. B., MARTIN, D. M., FAIRLAMB, A. H., FRAUNHOLZ, M. J., ROOS, D. S.,
RALPH, S. A., MCFADDEN, G. I., CUMMINGS, L. M., SUBRAMANIAN, G. M.,
MUNGALL, C., VENTER, J. C., CARUCCI, D. J., HOFFMAN, S. L., NEWBOLD,
C., DAVIS, R. W., FRASER, C. M. & BARRELL, B. 2002. Genome sequence of the
human malaria parasite Plasmodium falciparum. Nature, 419, 498-511.
GAY, F., ZOUGBÉDÉ, S., N’DILIMABAKA, N., REBOLLO, A., MAZIER, D. & MORENO,
A. 2012. Cerebral malaria: What is known and what is on research. Revue Neurologique,
168, 239-256.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 91
GETHING, P. W., PATIL, A. P., SMITH, D. L., GUERRA, C. A., ELYAZAR, I. R.,
JOHNSTON, G. L., TATEM, A. J. & HAY, S. I. 2011. A new world malaria map:
Plasmodium falciparum endemicity in 2010. Malar J, 10, 378.
GHANSAH, A., ROCKETT, K. A., CLARK, T. G., WILSON, M. D., KORAM, K. A.,
ODURO, A. R., AMENGA-ETEGO, L., ANYORIGIYA, T., HODGSON, A.,
MILLIGAN, P., ROGERS, W. O. & KWIATKOWSKI, D. P. 2012. Haplotype analyses
of haemoglobin C and haemoglobin S and the dynamics of the evolutionary response to
malaria in Kassena-Nankana District of Ghana. PLoS One, 7, e34565.
GREENWOOD, B. M. 2015. Efficacy and safety of RTS,S/AS01 malaria vaccine with or
without a booster dose in infants and children in Africa: final results of a phase 3,
individually randomised, controlled trial. The Lancet, 386, 31-45.
GUINDO, A., FAIRHURST, R. M., DOUMBO, O. K., WELLEMS, T. E. & DIALLO, D. A.
2007. X-linked G6PD deficiency protects hemizygous males but not heterozygous
females against severe malaria. PLoS Med, 4, e66.
HAY, S. I., GUERRA, C. A., GETHING, P. W., PATIL, A. P., TATEM, A. J., NOOR, A. M.,
KABARIA, C. W., MANH, B. H., ELYAZAR, I. R., BROOKER, S., SMITH, D. L.,
MOYEED, R. A. & SNOW, R. W. 2009. A world malaria map: Plasmodium falciparum
endemicity in 2007. PLoS Med, 6, e1000048.
HAY, S. I., SMITH, D. L. & SNOW, R. W. 2008. Measuring malaria endemicity from intense
to interrupted transmission. Lancet Infect Dis, 8, 369-78.
HEDRICK, P. W. 2011. Population genetics of malaria resistance in humans. Heredity (Edinb),
107, 283-304.
HEDRICK, P. W. 2012. Resistance to malaria in humans: the impact of strong, recent selection.
Malar J, 11, 349.
HIRONO, A. & BEUTLER, E. 1988. Molecular cloning and nucleotide sequence of cDNA for
human glucose-6-phosphate dehydrogenase variant A(-). Proc Natl Acad Sci U S A, 85,
3951-4.
HO, L. & JOHN, R. M. 2015. Understanding and Managing Glucose-6-Phosphate
Dehydrogenase Deficiency. The Journal for Nurse Practitioners, 11, 443-450.
HOWES, R. E., BATTLE, K. E., SATYAGRAHA, A. W., BAIRD, J. K. & HAY, S. I. 2013a.
G6PD deficiency: global distribution, genetic variants and primaquine therapy. Adv
Parasitol, 81, 133-201.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 92
HOWES, R. E., DEWI, M., PIEL, F. B., MONTEIRO, W. M., BATTLE, K. E., MESSINA, J.
P., SAKUNTABHAI, A., SATYAGRAHA, A. W., WILLIAMS, T. N., BAIRD, J. K.
& HAY, S. I. 2013b. Spatial distribution of G6PD deficiency variants across malaria-
endemic regions. Malar J, 12, 418.
IGBENEGHU, C., OLISEKODIAKA, M. J., AKINOLA, F. F. S. & ODAIBO, A. B. 2015.
Impact of Haemoglobin Variants AS and AC on Asymptomatic Falciparum Malaria
among Adults in Iwo, Southwestern Nigeria. Scholars Journal of Applied Medical
Sciences, 3, 17-20.
INSD 2015. Annuaire Statitistique 2014.
http://www.insd.bf/n/contenu/pub_periodiques/annuaires_stat/Annuaires_stat_nationa
ux_BF/Annuaire_stat_2014.pdf. Consulté le 25/08/2016 à 10h 57 minutes.
JOHNSON, M. K., CLARK, T. D., NJAMA-MEYA, D., ROSENTHAL, P. J. & PARIKH, S.
2009. Impact of the method of G6PD deficiency assessment on genetic association
studies of malaria susceptibility. PLoS One, 4, e7246.
KAFANDO, E., SAWADOGO, M. & COTTON, F. 2005. Neonatal screening for sickle cell
disorders in Ouagadougou, Burkina Faso: a pilot study. J Med Screen, 12.
KILIAN, N., SRISMITH, S., DITTMER, M., OUERMI, D., BISSEYE, C., SIMPORE, J.,
CYRKLAFF, M., SANCHEZ, C. P. & LANZER, M. 2015. Hemoglobin S and C affect
protein export in Plasmodium falciparum-infected erythrocytes. Biol Open, 4, 400-10.
KOSSOROTOFF, M., GREVENT, D. & DE MONTALEMBERT, M. 2014. Drépanocytose et
atteinte vasculaire cérébrale chez l’enfant. Archives de Pédiatrie, 21, 404-414.
KWIATKOWSKI, D. P. 2005. How malaria has affected the human genome and what human
genetics can teach us about malaria. Am J Hum Genet, 77, 171-92.
LABIE, D. 2005. [Plasmodium's stratagems to be protected in the invaded organisms]. Med Sci
(Paris), 21, 700-2.
LABIE, D. & ELION, J. 2005. Bases moléculaires et physiopathologiques des maladies de
l'hémoglobine. EMC - Hématologie, 2, 220-239.
LABIE, D. & ELION, J. 2010. La drépanocytose : problème de l’Afrique. Médecine Tropicale,
70 449-453.
LANGHORNE, J., NDUNGU, F. M., SPONAAS, A. M. & MARSH, K. 2008. Immunity to
malaria: more questions than answers. Nat Immunol, 9, 725-32.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 93
LAURENT, V., HILLY, J., BEDEL, J., PLANQUETTE, B., LEGRIEL, S., TROCHÉ, G.,
GUEZENNEC, P., BÉDOS, J.-P. & BRUNEEL, F. 2014. Paludisme grave
d’importation de l’adulte. Journal Européen des Urgences et de Réanimation, 26, 97-
104.
LE ROCH, K. G., CHUNG, D. W. & PONTS, N. 2012. Genomics and integrated systems
biology in Plasmodium falciparum: a path to malaria control and eradication. Parasite
Immunol, 34, 50-60.
LELLIOTT, P. M., MCMORRAN, B. J., FOOTE, S. J. & BURGIO, G. 2015. The influence of
host genetics on erythrocytes and malaria infection: is there therapeutic potential? Malar
J, 14, 289.
LIN, M., YANG, L. Y., XIE, D. D., CHEN, J. T., NGUBA, S. M., EHAPO, C. S., ZHAN, X.
F., EYI, J. U., MATESA, R. A., OBONO, M. M., YANG, H., YANG, H. T. & CHENG,
J. D. 2015. G6PD Deficiency and Hemoglobinopathies: Molecular Epidemiological
Characteristics and Healthy Effects on Malaria Endemic Bioko Island, Equatorial
Guinea. PLoS One, 10, e0123991.
LONGO, L., VANEGAS, O. C., PATEL, M., ROSTI, V., LI, H., WAKA, J., MERGHOUB,
T., PANDOLFI, P. P., NOTARO, R., MANOVA, K. & LUZZATTO, L. 2002.
Maternally transmitted severe glucose 6-phosphate dehydrogenase deficiency is an
embryonic lethal. Embo J, 21, 4229-39.
LUZZATTO, L. 2006. Glucose 6-phosphate dehydrogenase deficiency: from genotype to
phenotype. Haematologica, 91, 1303-6.
LUZZATTO, L. 2012. Sickle cell anaemia and malaria. Mediterr J Hematol Infect Dis, 4,
e2012065.
LUZZATTO, L. 2015. G6PD deficiency: a polymorphism balanced by heterozygote advantage
against malaria. The Lancet Haematology, 2, e400-e401.
LUZZATTO, L. & ALLAN, N. C. 1968. Relationship between the Genes for Glucose-6-
phospahate dehydrogenase and for Haemoglobins in a Nigerian Population. Nature
Publishing Group, 219, 1041-1042.
LUZZATTO, L., NANNELLI, C. & NOTARO, R. 2016. Glucose-6-Phosphate Dehydrogenase
Deficiency. Hematol Oncol Clin North Am, 30, 373-93.
LUZZATTO, L. & SENECA, E. 2014. G6PD deficiency: a classic example of
pharmacogenetics with on-going clinical implications. Br J Haematol, 164, 469-80.
LUZZATTO, L., USANGA, F. A. & REDDY, S. 1969. Glucose-6-phosphate dehydrogenase
deficient red cells: resistance to infection by malarial parasites. Science, 164, 839-42.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 94
LYIMO, I. N., HAYDON, D. T., MBINA, K. F., DARAJA, A. A., MBEHELA, E. M., REEVE,
R. & FERGUSON, H. M. 2012. The fitness of African malaria vectors in the presence
and limitation of host behaviour. Malar J, 11, 425.
LYON, M. F. 1961. Gene action in the X-chromosome of the mouse (Mus musculus L.).
Nature, 190, 372-3.
MAIGA, B., DOLO, A., CAMPINO, S., SEPULVEDA, N., CORRAN, P., ROCKETT, K. A.,
TROYE-BLOMBERG, M., DOUMBO, O. K. & CLARK, T. G. 2014. Glucose-6-
phosphate dehydrogenase polymorphisms and susceptibility to mild malaria in Dogon
and Fulani, Mali. Malar J, 13, 270.
MALARIA GENOMIC EPIDEMIOLOGY, N. 2014. Reappraisal of known malaria resistance
loci in a large multicenter study. Nat Genet, 46, 1197-1204.
MANGANO, V. D., KABORE, Y., BOUGOUMA, E. C., VERRA, F., SEPULVEDA, N.,
BISSEYE, C., SANTOLAMAZZA, F., AVELLINO, P., TIONO, A. B., DIARRA, A.,
NEBIE, I., ROCKETT, K. A., SIRIMA, S. B., MODIANO, D. & MALARIA, G. E. N.
C. 2015. Novel Insights Into the Protective Role of Hemoglobin S and C Against
Plasmodium falciparum Parasitemia. J Infect Dis, 212, 626-34.
MANJURANO, A., SEPULVEDA, N., NADJM, B., MTOVE, G., WANGAI, H.,
MAXWELL, C., OLOMI, R., REYBURN, H., RILEY, E. M., DRAKELEY, C. J.,
CLARK, T. G. & MALARIA, G. E. N. C. 2015. African glucose-6-phosphate
dehydrogenase alleles associated with protection from severe malaria in heterozygous
females in Tanzania. PLoS Genet, 11, e1004960.
MARTINI, G., TONIOLO, D., VULLIAMY, T., LUZZATTO, L., DONO, R., VIGLIETTO,
G., PAONESSA, G., D'URSO, M. & PERSICO, M. G. 1986. Structural analysis of the
X-linked gene encoding human glucose 6-phosphate dehydrogenase. Embo J, 5, 1849-
55.
MATSUOKA, H., NGUON, C., KANBE, T., JALLOH, A., SATO, H., YOSHIDA, S., HIRAI,
M., ARAI, M., SOCHEAT, D. & KAWAMOTO, F. 2005. Glucose-6-phosphate
dehydrogenase (G6PD) mutations in Cambodia: G6PD Viangchan (871G>A) is the
most common variant in the Cambodian population. J Hum Genet, 50, 468-72.
MAY, J., MEYER, C. G., GROSSTERLINDEN, L., ADEMOWO, O. G., MOCKENHAUPT,
F. P., OLUMESE, P. E., FALUSI, A. G., LUZZATTO, L. & BIENZLE, U. 2000. Red
cell glucose-6-phosphate dehydrogenase status and pyruvate kinase activity in a
Nigerian population. Trop Med Int Health, 5, 119-23.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 95
MECHAIN, M. 2015. Un vaccin contre le paludisme à portée de main : quelle place dans la
stratégie mondiale ? Médecine thérapeutique / Pédiatrie, 18, 133-138.
MEGARBANE, B. 2008. Déficit en Glucose-6-phosphate déshydrogénase : Quant y penser et
quelles précautions prendre? . Elsevier Masson SAS, 399-406.
MENARD, D., ARIEY, F. & MERCEREAU-PUIJALON, O. 2013. Étude de la résistance de
Plasmodium falciparum aux antipaludiques au sein du réseau international des Instituts
Pasteur (RIIP-Palu). Med Sci (Paris), 29, 647-655, médecine/sciences, 17, 720-9.
MENDIS, K., RIETVELD, A., WARSAME, M., BOSMAN, A., GREENWOOD, B. &
WERNSDORFER, W. H. 2009. From malaria control to eradication: The WHO
perspective. Trop Med Int Health, 14, 802-9.
MILLER, L. H., ACKERMAN, H. C., SU, X. Z. & WELLEMS, T. E. 2013. Malaria biology
and disease pathogenesis: insights for new treatments. Nat Med, 19, 156-67.
MINUCCI, A., MORADKHANI, K., HWANG, M. J., ZUPPI, C., GIARDINA, B. &
CAPOLUONGO, E. 2012. Glucose-6-phosphate dehydrogenase (G6PD) mutations
database: review of the "old" and update of the new mutations. Blood Cells Mol Dis, 48,
154-65.
MISBAHI, H. 2013. Paludisme : mode d’action de la chloroquine et mécanisme de la
chloroquinorésistance. J Pharm Clin, 32, 143-53.
MISSINOU, M. A., LELL, B. & KREMSNER, P. G. 2003. Uncommon asymptomatic
Plasmodium falciparum infections in Gabonese children. Clin Infect Dis, 36, 1198-202.
MODIANO, D., BANCONE, G., CIMINELLI, B. M., POMPEI, F., BLOT, I., SIMPORE, J.
& MODIANO, G. 2008. Haemoglobin S and haemoglobin C: 'quick but costly' versus
'slow but gratis' genetic adaptations to Plasmodium falciparum malaria. Hum Mol
Genet, 17, 789-99.
MODIANO, D., LUONI, G., SIRIMA, B. S., LANFRANCOTTI, A., PETRARCA, V.,
CRUCIANI, F., SIMPORE, J., CIMINELLI, B. M., FOGLIETTA, E., GRISANTI, P.,
BIANCO, I., MODIANO, G. & COLUZZI, M. 2001a. The lower susceptibility to
Plasmodium falciparum malaria of Fulani of Burkina Faso (west Africa) is associated
with low frequencies of classic malaria-resistance genes. Trans R Soc Trop Med Hyg,
95, 149-52.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 96
MODIANO, D., LUONI, G., SIRIMA, B. S., SIMPORE, J., VERRA, F., KONATE, A.,
RASTRELLI, E., OLIVIERI, A., CALISSANO, C., PAGANOTTI, G. M.,
D'URBANO, L., SANOU, I., SAWADOGO, A., MODIANO, G. & COLUZZI, M.
2001b. Haemoglobin C protects against clinical Plasmodium falciparum malaria.
Nature, 414, 305-8.
MOMBO, L. E., NTOUMI, F., BISSEYE, C., OSSARI, S., LU, C. Y., NAGEL, R. L. &
KRISHNAMOORTHY, R. 2003. Human genetic polymorphisms and asymptomatic
Plasmodium falciparum malaria in Gabonese schoolchildren. Am J Trop Med Hyg, 68,
186-90.
MORENO, A. & JOYNER, C. 2015. Malaria vaccine clinical trials: what's on the horizon.
Current Opinion in Immunology, 35, 98-106.
MULUMBA, L. L. & WILSON, L. 2015. Sickle cell disease among children in Africa: An
integrative literature review and global recommendations. International Journal of
Africa Nursing Sciences, 3, 56-64.
MURA, M., SAIDI, R., WOLF, A., MOALIC, J. L. & OLIVER, M. 2009. [Congenital
hemolytic anemia due to glucose-6-phosphate dehydrogenase deficiency]. Med Trop
(Mars), 69, 551-5.
OLD, J. M. 2003. Screening and genetic diagnosis of haemoglobin disorders. Blood Reviews,
17, 43-53.
OMS 2013. World Malaria Report.
http://apps.who.int/iris/bitstream/10665/97008/1/9789241564694_eng.pdf
OMS 2014a. Surveillance épidémiologique en vue de l’élimination du paludisme : manuel
opérationnel.
OMS 2014b. World Malaria Report.
http://www.who.int/malaria/publications/world_malaria_report_2014/wmr-2014-no-
profiles.pdf
OMS 2015a. Global technical strategy for malaria 2016–2030. Geneva :World Health
Organization, 2015.
OMS 2015b. World Malaria Report.
http://apps.who.int/iris/bitstream/10665/200018/1/9789241565158_eng.pdf
OTA, M., FUKUSHIMA, H., KULSKI, J. K. & INOKO, H. 2007. Single nucleotide
polymorphism detection by polymerase chain reaction-restriction fragment length
polymorphism. Nat Protoc, 2, 2857-64.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 97
OUATTARA, A. K., BISSEYE, C., BAZIE, B. V., DIARRA, B., COMPAORE, T. R.,
DJIGMA, F., PIETRA, V., MORET, R. & SIMPORE, J. 2014. Glucose-6-phosphate
dehydrogenase (G6PD) deficiency is associated with asymptomatic malaria in a rural
community in Burkina Faso. Asian Pac J Trop Biomed, 4, 655-8.
OUÉDRAOGO, A., BOUGOUMA, E. C., DIARRA, A., KONATÉ, A. T., NÉBIÉ, I., TIONO,
A. B. & SIRIMA, S. B. 2008. Impact comparatif de trois schémas de prévention du
paludisme pendant la grossesse sur l’anémie maternelle, associée à l’infection palustre
au Burkina Faso. Médecine et Maladies Infectieuses, 38, 180-186.
PAGES, F., ORLANDI-PRADINES, E. & CORBEL, V. 2007. [Vectors of malaria: biology,
diversity, prevention, and individual protection]. Med Mal Infect, 37, 153-61.
PARIKH, S., DORSEY, G. & ROSENTHAL, P. J. 2004. Host polymorphisms and the
incidence of malaria in Ugandan children. Am J Trop Med Hyg, 71, 750-3.
PAWAR, A. 2014. Immune Mechanisms Involved in Malaria: A Review. IJCRR, 6, 10-14.
PETERS, A. L. & VAN NOORDEN, C. J. 2009. Glucose-6-phosphate dehydrogenase
deficiency and malaria: cytochemical detection of heterozygous G6PD deficiency in
women. J Histochem Cytochem, 57, 1003-11.
PIEL, F. B., ADAMKIEWICZ, T. V., AMENDAH, D., WILLIAMS, T. N., GUPTA, S. &
GROSSE, S. D. 2015. Observed and expected frequencies of structural hemoglobin
variants in newborn screening surveys in Africa and the Middle East: deviations from
Hardy-Weinberg equilibrium. Genet Med.
PIEL, F. B., HAY, S. I., GUPTA, S., WEATHERALL, D. J. & WILLIAMS, T. N. 2013a.
Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based
on demographics, excess mortality, and interventions. PLoS Med, 10, e1001484.
PIEL, F. B., HOWES, R. E., PATIL, A. P., NYANGIRI, O. A., GETHING, P. W., BHATT, S.,
WILLIAMS, T. N., WEATHERALL, D. J. & HAY, S. I. 2013b. The distribution of
haemoglobin C and its prevalence in newborns in Africa. Sci Rep, 3, 1671.
PIEL, F. B., PATIL, A. P., HOWES, R. E., NYANGIRI, O. A., GETHING, P. W., WILLIAMS,
T. N., WEATHERALL, D. J. & HAY, S. I. 2010. Global distribution of the sickle cell
gene and geographical confirmation of the malaria hypothesis. Nat Commun, 1, 104.
PRADINES, B., DORMOI, J., BRIOLANT, S., BOGREAU, H. & ROGIER, C. 2010. La
résistance aux antipaludiques. Revue Francophone des Laboratoires, 2010, 51-62.
PULL, L., BELLETTRE, X., MICHEL, J. F., BOUCHAUD, O. & SIRIEZ, J. Y. 2013.
[Treatment of severe and uncomplicated falciparum malaria in children in France]. Arch
Pediatr, 20, 1260-4.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 98
RAHIMI, B. A., THAKKINSTIAN, A., WHITE, N. J., SIRIVICHAYAKUL, C., DONDORP,
A. M. & CHOKEJINDACHAI, W. 2014. Severe vivax malaria: a systematic review
and meta-analysis of clinical studies since 1900. Malar J, 13, 481.
RICHARD, F., OUEDRAOGO, C., COMPAORE, J., DUBOURG, D. & DE BROUWERE, V.
2007. Reducing financial barriers to emergency obstetric care: experience of cost-
sharing mechanism in a district hospital in Burkina Faso. Trop Med Int Health, 12, 972-
81.
RISCO-CASTILLO, V., SON, O., FRANETICH, J. F., RUBINSTEIN, E., MAZIER, D. &
SILVIE, O. 2013. [Plasmodium sporozoite entry pathways during malaria liver
infection]. Biol Aujourdhui, 207, 219-29.
ROGIER, C., ORLANDI-PRADINES, E., FUSAÏ, T., PRADINES, B., BRIOLANT, S. &
ALMERAS, L. 2006. Vaccins contre le paludisme : perspectives et réalité. Médecine et
Maladies Infectieuses, 36, 414-422.
RUWENDE, C. & HILL, A. 1998. Glucose-6-phosphate dehydrogenase deficiency and
malaria. J Mol Med (Berl), 76, 581-8.
RUWENDE, C., KHOO, S. C., SNOW, R. W., YATES, S. N., KWIATKOWSKI, D., GUPTA,
S., WARN, P., ALLSOPP, C. E., GILBERT, S. C., PESCHU, N. & ET AL. 1995.
Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by
resistance to severe malaria. Nature, 376, 246-9.
SABBATANI, S., FIORINO, S. & MANFREDI, R. 2010. The emerging of the fifth malaria
parasite (Plasmodium knowlesi): a public health concern? Braz J Infect Dis, 14, 299-
309.
SCHERF, A., LOPEZ-RUBIO, J. J. & RIVIERE, L. 2008. Antigenic variation in Plasmodium
falciparum. Annu Rev Microbiol, 62, 445-70.
SHAH, S. S., ROCKETT, K. A., JALLOW, M., SISAY-JOOF, F., BOJANG, K. A., PINDER,
M., JEFFREYS, A., CRAIK, R., HUBBART, C., WELLEMS, T. E., KWIATKOWSKI,
D. P. & MALARIA, G. E. N. C. 2016. Heterogeneous alleles comprising G6PD
deficiency trait in West Africa exert contrasting effects on two major clinical
presentations of severe malaria. Malar J, 15, 13.
SILVIE, O., RUBINSTEIN, E., BOUCHEIX, C. & MAZIER, D. 2003. [CD81: a tetraspanin
implicated in Plasmodium infection]. Med Sci (Paris), 19, 169-71.
SIMPORE, J., ILBOUDO, D., DAMINTOTI, K., SAWADOGO, L., MARIA, E., BINET, S.,
NITIEMA, H., OUEDRAOGO, P., PIGNATELLI, S. & NIKIEMA, J. B. 2007.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 99
Glucose-6-phosphate dehydrogenase deficiency and sickle cell disease in Burkina Faso.
Pak J Biol Sci, 10, 409-14.
SIMPORE, J., PIGNATELLI, S., BARLATI, S. & MUSUMECI, S. 2002a. Biological and
clinical presentation of patients with hemoglobinopathies attending an urban hospital in
Ouagadougou: confirmation of the modification of the balance between Hb S and Hb C
in Burkina Faso. Hemoglobin, 26, 121-7.
SIMPORE, J., PIGNATELLI, S., BARLATI, S. & MUSUMECI, S. 2002b. Modification in the
frequency of Hb C and Hb S in Burkina Faso: an influence of migratory fluxes and
improvement of patient health care. Hemoglobin, 26, 113-20.
SINGH, B., KIM SUNG, L., MATUSOP, A., RADHAKRISHNAN, A., SHAMSUL, S. S.,
COX-SINGH, J., THOMAS, A. & CONWAY, D. J. 2004. A large focus of naturally
acquired Plasmodium knowlesi infections in human beings. Lancet, 363, 1017-24.
SINKA, M. E., BANGS, M. J., MANGUIN, S., RUBIO-PALIS, Y.,
CHAREONVIRIYAPHAP, T., COETZEE, M., MBOGO, C. M., HEMINGWAY, J.,
PATIL, A. P., TEMPERLEY, W. H., GETHING, P. W., KABARIA, C. W., BURKOT,
T. R., HARBACH, R. E. & HAY, S. I. 2012. A global map of dominant malaria vectors.
Parasit Vectors, 5, 69.
SIRUGO, G., PREDAZZI, I. M., BARTLETT, J., TACCONELLI, A., WALTHER, M. &
WILLIAMS, S. M. 2014. G6PD A- deficiency and severe malaria in The Gambia:
heterozygote advantage and possible homozygote disadvantage. Am J Trop Med Hyg,
90, 856-9.
STANISIC, D. I., BARRY, A. E. & GOOD, M. F. 2013. Escaping the immune system: How
the malaria parasite makes vaccine development a challenge. Trends Parasitol, 29, 612-
22.
TRAGER, W. & JENSEN, J. B. 1976. Human malaria parasites in continuous culture. Science,
193, 673-5.
TRAVASSOS, M. A., COULIBALY, D., LAURENS, M. B., DEMBELE, A., TOLO, Y.,
KONE, A. K., TRAORE, K., NIANGALY, A., GUINDO, A., WU, Y., BERRY, A. A.,
JACOB, C. G., TAKALA-HARRISON, S., ADAMS, M., SHRESTHA, B., MU, A. Z.,
KOURIBA, B., LYKE, K. E., DIALLO, D. A., DOUMBO, O. K., PLOWE, C. V. &
THERA, M. A. 2015. Hemoglobin C Trait Provides Protection From Clinical
Falciparum Malaria in Malian Children. J Infect Dis, 212, 1778-86.
TSE, C. & CAPEAU, J. 2003. [Real time PCR methodology for quantification of nucleic acids].
Ann Biol Clin (Paris), 61, 279-93.

Thèse de Doctorat Unique OUATTARA Abdoul Karim 100
TURNER, L., LAVSTSEN, T., BERGER, S. S., WANG, C. W., PETERSEN, J. E., AVRIL,
M., BRAZIER, A. J., FREETH, J., JESPERSEN, J. S., NIELSEN, M. A.,
MAGISTRADO, P., LUSINGU, J., SMITH, J. D., HIGGINS, M. K. & THEANDER,
T. G. 2013. Severe malaria is associated with parasite binding to endothelial protein C
receptor. Nature, 498, 502-5.
UYOGA, S., NDILA, C. M., MACHARIA, A. W., NYUTU, G., SHAH, S., PESHU, N.,
CLARKE, G. M., KWIATKOWSKI, D. P., ROCKETT, K. A. & WILLIAMS, T. N.
2015. Glucose-6-phosphate dehydrogenase deficiency and the risk of malaria and other
diseases in children in Kenya: a case-control and a cohort study. Lancet Haematol, 2,
e437-44.
VAFA, M., TROYE-BLOMBERG, M., ANCHANG, J., GARCIA, A. & MIGOT-NABIAS, F.
2008. Multiplicity of Plasmodium falciparum infection in asymptomatic children in
Senegal: relation to transmission, age and erythrocyte variants. Malar J, 7, 17.
VERRA, F., SIMPORE, J., WARIMWE, G. M., TETTEH, K. K., HOWARD, T., OSIER, F.
H., BANCONE, G., AVELLINO, P., BLOT, I., FEGAN, G., BULL, P. C., WILLIAMS,
T. N., CONWAY, D. J., MARSH, K. & MODIANO, D. 2007. Haemoglobin C and S
role in acquired immunity against Plasmodium falciparum malaria. PLoS One, 2, e978.
WAJCMAN, H. & GALACTEROS, F. 2004. [Glucose 6-phosphate dehydrogenase deficiency:
a protection against malaria and a risk for hemolytic accidents]. C R Biol, 327, 711-20.
WEBSTER, W. A. & MCFADDEN, G. I. 2014. From the genome to the phenome: tools to
understand the basic biology of Plasmodium falciparum. J Eukaryot Microbiol, 61, 655-
71.
WRIGHT, G. J. & RAYNER, J. C. 2014. Plasmodium falciparum erythrocyte invasion:
combining function with immune evasion. PLoS Pathog, 10, e1003943.

ANNEXES

ANNEXES
I. Protocole d’extraction par la méthode du Salting out
1. Ajouter 1 mL de tampon de lyse des hématies à 500 µL de culot de sang total (frais ou congelés).
Mélanger délicatement à l’aide de la micropipette pendant 15 secondes.
Passer au vortex, puis centrifugé à 13000 rpm pendant 1 min. Jeter le surnageant.
2. Répéter l’étape 1. Laver trois fois en ajoutant 1 mL H2O distillée (Passer au vortex et centrifuger à
13000 rpm pendant 1 minutes puis jeter le surnageant).
3. Reprendre le culot dans 370 μL du Mix (80μl de protéinase K-5X tampon, 30μl de protéinase K, 20
µL de SDS 20% et 240μl H2O et cela par échantillon), puis passer au vortex.
4. Incuber à 55 ° C pendant 15 min.
5. Ajouter 200 µL de NaCl 5M, passer au vortex pendant 15 secondes. Isoler les protéines précipitées
par centrifugation à 13000 rpm pendant 5 min.
6. Transférer 500 μL du surnageant dans de nouveaux tubes de 1,5 mL. Ajouter 1 mL d'éthanol absolu
(T° ambiante). Laissez précipiter l'ADN en agitant les tubes délicatement à la main. Isoler l'ADN
par centrifugation à 13000 rpm pendant 1 minute ; puis jeter le surnageant.
7. Laver deux fois avec 800 µL d'éthanol 70% frais (préalablement conservé à -20°C). Passer au
vortex, et centrifuger à 13000 rpm pendant 1 min puis jeter le surnageant.
8. Eliminer le reste d'éthanol en égouttant les tubes sur du papier absorbant, puis les sécher à bouchon
ouvert sur incubateur.
9. Dissoudre l'ADN dans 100 µL de Tampon d’élution (TE) et passer au vortex pendant 30
secondes puis incuber. Utilisez 2 µL d'ADN pour la quantification au Nanodrop. Conserver l’ADN
à -20°C. La pureté de l’ADN utilisé pour le génotypage était comprise entre 1,8 et 1,9.
Tampon de lyse pour l’extraction : Quantités et concentrations pour 1L
0.32 M Sucrose 109,5 g sucrose
5 mM MgCl2 10 mL 5M MgCl2
12 mM Tris-HCl 12 mL 1M Tris-HCl
1% Triton X-100 10 mL Triton X-100
Tampon 5X Protéinase K (0.375 M NaCl/ 0.12 M EDTA) [En liquide dans les bouteilles].

II. Certification De Consentement
J’ai été invité à participer à la recherche de Abdoul Karim OUATTARA du
…………………………………………………. J’ai appris qu’il impliquera une prise en charge des
examens biologiques à la prise de sang. J’ai été informé que les risques sont minimes et peuvent inclure
seulement, les malaises ou inconforts liés aux prélèvements sanguins. Je me rends compte qu’il ne puisse
y avoir aucun avantage pour moi personnellement et qu’aucune récompense ne me sera donnée. Il m’a
donné les noms, les adresses et les numéros de téléphone des chercheurs qui peuvent être facilement
contactés au besoin.
Je soussigné……………………………………………………………………………………...
Certifie avoir lu la notice d’information de l’étude « Paludisme au Burkina Faso : Prévalence, marqueurs
génétiques de résistance et polymorphismes des gènes MSP1 et MSP2 de Plasmodium falciparum »
J’ai eu l’opportunité de poser toutes les questions que je souhaitais et toutes les questions que j’ai posées
ont reçu des réponses satisfaisantes.
J’ai reçu une copie du document d’information aux donneurs de sang et de Certificat de consentement.
Je soussigne certifie d’accepter de participer à cette étude.
Nom et prénom(s) du patient :
Signature du patient :
Date (JJ/MM/AAAA) :
(Le patient doit signer, dater et écrire lui-même son propre nom)
Personne obtenant le consentement (Investigateur ou personne désigné par l’investigateur principal) :
Nom et prénom de l’Investigateur :
Signature de l’Investigateur
Date (JJ/MM/AAAA) :

Si le répondant ne sait pas lire
Témoin
Je suis témoin de la lecture de la notice d’Information de l’étude « Paludisme au Burkina Faso :
« Prévalence, marqueurs génétiques de résistance et polymorphismes des gènes MSP1 et MSP2 de
Plasmodium falciparum ». L’intéressé a eu l’opportunité de poser toutes les questions qu’il souhaitait et
toutes les questions qu’il a posées ont reçu des réponses satisfaisantes. J’atteste par ce document que je n’ai
aucune relation avec l’équipe de recherche, qu’elle soit contractuelle, professionnelle ou de toute autre
nature susceptible de réduire mon indépendance.
Je confirme que le répondant consent à participer volontairement à cette étude.
Nom et prénom(s) du témoin Nom et prénom(s) du patient
Signature du témoin : Application de l’index du patient :
Date (JJ/MM/AAAA) :
(Le témoin doit signer, dater et écrire lui-même son nom et le nom du donneur. Le donneur doit appliquer
lui-même son index à la place prévue à cet effet)

III. Liste des publications de cette Thèse
OUATTARA A. K, BISSEYE C, BAZIE B.V.J.T.E, DIARRA B, COMPAORE T. R, DJIGMA F. W,
PIETRA V, MORET R, SIMPORE J. (2014) Glucose-6-phosphate dehydrogenase (G6PD) deficiency is
associated with asymptomatic malaria in a rural community in Burkina Faso. Asian Pacific Journal
of Tropical Biomedicine. Volume 4, Issue 8, August 2014, Pages 655–658.
OUATTARA A. K, YAMEOGO P, DIARRA B, OBIRI-YEBOAH D, YONLI A. T, COMPAORE T. R,
SOUBEIGA R. S. T, DJIGMA F. W, SIMPORE J. (2016) Molecular heterogeneity of glucose-6-
phosphate dehydrogenase deficiency in Burkina Faso: G-6-PD Betica Selma and Santamaria in
people with symptomatic malaria in Ouagadougou. Mediterr J Hematol Infect Dis. Volume 8, Issue 1,
June 2016, e2016029.
Autres publications
ZOURE A. A, BAMBARA A. H, SAWADOGO A. Y, OUATTARA A. K, OUEDRAOGO M, TRAORE
S. S, BAKRI Y, SIMPORE J. (2017) Multiparity and Breast Cancer Risk Factor among Women in
Burkina Faso. Asian Pac J Cancer Prev. 17 (12), 5095-5099.
ZONGO A. W, DIAGBOUGA S, OUERMI D, T. COMPOARE T. R, OBIRI-YEBOAH D, OUATTARA
A. K, KIENTEGA T, SOUBEIGA S. T, YONLI A. T, DJIGMA F. W, ILBOUDO D. AND SIMPORE J.
(2016) Real Time PCR Assay in differentiating Entamoeba histolytica and Entamoeba dispar
infections in patients fecal samples attending St Camille hospital in Ouagadougou, Burkina Faso.
Curr Res Microbiol Biotechnol. 4(6): 938-943.
COMPAORE T. R, DIARRA B, ASSIH M, SOUBEIGA R. S. T, OUATTARA A. K, TCHELOUGOU
D, BISSEYE C, BAKOUAN D, COMPAORE IP. DEMBELE A, OBIRI-YEBOAH D, DJIGMA WF,
SIMPORE J. (2016) HBV/HIV co-infection and APOBEC3G polymorphisms in a population from
Burkina Faso. BMC Infectious Diseases. Volume 16, Issue 1, July 2016, Pages 1-7.
COMPAORE T. R, SOUBEIGA R. S. T, OUATTARA A. K, OBIRI-YEBOAH D, TCHELOUGOU D,
MAIGA M, ASSIH M, BISSEYE C, BAKOUAN D, COMPAORE IP, DEMBELE A, MARTINSON J,
SIMPORE J. (2016) APOBEC3G Variants and Protection against HIV-1 Infection in Burkina Faso.
PLoS ONE. Volume 11, January 2016.

Manuscrits soumis
Serge Théophile SOUBEIGA, Bapio Valéry Jean Télesphore Elvira BAZI, Tegwindé Rebeca
COMPAORE, Abdoul Karim OUATTARA, Albert Théophane YONLI, Arsène ZONGO, Lassina
TRAORE, Serge DIAGBOUGA, Jacques SIMPORE. Transmitted HIV-1 drug resistance in HAART-
treated pregnant women and their infants during PMCT in Ouagadougou, Burkina Faso (soumis
dans Mediterr J Hematol Infect Dis).
COMPAORE T. R, SOUBEIGA R. S. T, OUATTARA A. K, TCHELOUGOU D, BISSEYE C,
BAKOUAN D, COMPAORE IP, DEMBELE A, YONLI AT, OBIRI-YEBOAH D, DJIGMA WF,
SIMPORE J. (2016) Expression d’APOBEC 3G et infection au VIH-1 au Burkina Faso. (Soumis dans
Journal of Medical Virology).
IV. Communications Orales
Abdoul Karim OUATTARA, Cyrille BISSEYE, Bapio Valéry Jean Télesphore Elvira BAZIE, Birama
DIARRA, Tegwindé Rebeca COMPAORE, Florencia W. DJIGMA, Rémy MORET, Jacques SIMPORE.
Fréquence des mutations responsables de la déficience en glucose-6-phosphate-déshydrogénase et le
paludisme asymptomatique dans une communauté rurale au Burkina Faso. XVI ème Journées Portes
Ouvertes de l’Université Ouaga I Pr Joseph KI-ZERBO (JPO) du 25 au 30 Novembre 2013.
Abdoul Karim OUATTARA, Cyrille BISSEYE, Bapio, Valérie Jean Télesphore Elvira BAZIE, Birama
DIARRA, Rébéca COMPAORE, Florencia DJIGMA, Djeneba OUERMI ; Virginio PIETRA, Jacques
SIMPORE. Mutations responsables de la déficience en glucose-6-phosphate-déshydrogénase dans une
communauté rurale au Burkina Faso. XVII ème Journées des Sciences de la Santé de Bobo-Dioulasso
(JSSB) du 06 au 09 Mai 2014.
Abdoul Karim OUATTARA, Bapio Valéry Jean Télesphore Elvira BAZIE, Cyrille BISSEYE, Birama
DIARRA, Tegwindé Rebeca COMPAORE, Florencia W. DJIGMA, Rémy MORET, Jacques SIMPORE.
La déficience en Glucose-6-phosphate déshydrogénase (G-6-PD) est associée au paludisme
asymptomatique dans une communauté rurale au Burkina Faso. Colloque Scientifique International
de l’Université de Kara, Togo 12-16 Mai 2014.
Abdoul Karim OUATTARA, Pouiré YAMEOGO, Birama DIARRA, Dorcas OBIRI-YEBOAH, Albert
Théophane YONLI, Tegwindé Rebeca COMPAORE, Serge Théophile SOUBEIGA, Florencia Wenkuuni
DJIGMA, Jacques SIMPORE. Hétérogénéité allélique du déficit en Glucose-6-phosphate
déshydrogénase au Burkina Faso : La G-6-PD Betica Selma et Santamaria chez des personnes
atteintes de paludisme symptomatique à Ouagadougou. XVIIe édition des Journées Scientifiques
Internationales de Lomé (JSIL), Togo du 03 – 08 Octobre 2016.





Mediterranean Journal of Hematology and Infectious Diseases
Original Article
Molecular Heterogeneity of Glucose-6-Phosphate Dehydrogenase Deficiency in
Burkina Faso: G-6-PD Betica Selma and Santamaria in People with Symptomatic
Malaria in Ouagadougou
Abdoul Karim Ouattara1, Pouiré Yameogo1, Birama Diarra1, Dorcas Obiri-Yeboah2, Albert Yonli1, Tegwindé Rebeca
Compaore1, Serge Théophile Soubeiga1, Florencia Wenkuuni Djigma1 and Jacques Simpore1
1Biomolecular Research Center Pietro Annigoni (CERBA) LABIOGENE UFR/SVT, University of Ouagadougou BP 364
Ouagadougou, Burkina Faso. 2Department of Microbiology and Immunology, University of Cape Coast, Ghana.
Competing interests: The authors have declared that no competing interests exist.
Abstract. The G-6-PD deficiency has an important polymorphism with genotypic variants such as
202A/376G, 376G/542T and 376G/968T known in West African populations. It would confer
protection against severe forms of malaria although there are differences between the various
associations in different studies. In this study we genotyped six (06) variants of the G-6-PD gene
in people with symptomatic malaria in urban areas in Burkina Faso.
One hundred and eighty-two (182) patients who tested positive using rapid detection test and
microscopy were included in this study. A regular PCR with the GENESPARK G6PD African kit
was run followed by electrophoresis, allowing initially to genotype six SNPs (G202A, A376G,
A542T, G680T, C563T and T968C). Women carrying the mutations 202A and/or 376G were
further typed by real-time PCR using TaqMan probes rs1050828 and rs1050829.
In the study population the G-6-PD deficiency prevalence was 9.9%. In addition of G-6-PD A-
(202A/376G) variant, 376G/542T and 376G/968T variants were also detected. Hemoglobin
electrophoresis revealed that 22.5% (41/182) of the individuals had HbAC compared with2.2%
with HbAS and one individual had double heterozygous HbSC. There was no correlation between
the G-6-PD deficiency or haemoglobinopathies and symptomatic malaria infections in this study.
Our study confirms that the G-6-PD deficiency does not confer protection against Plasmodium
falciparum infections. As opposed to previous genotyping studies carried out in Burkina Faso, this
study shows for the first time the presence of the variant A- (376G/968C)and warrants further
investigation at the national level and in specific ethnic groups.
Citation: Ouattara A.K., Yameogo P., Diarra B., Obiri-Yeboah D., Yonli A., Compaore T.R., Soubeiga S.T., Djigma F.W., Simpore J.
Molecular heterogeneity of glucose-6-phosphate dehydrogenase deficiency in Burkina Faso: g-6-pd betica selma and santamaria in people with
symptomatic malaria in ouagadougou. Mediterr J Hematol Infect Dis 2016, 8(1): e2016029, DOI: http://dx.doi.org/10.4084/MJHID.2016.029
Published: June 15, 2016 Received: April 11, 2016 Accepted: May 25, 2016 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which
permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Correspondence to: Prof. Jacques SIMPORE. Biomolecular Research Center Pietro Annigoni (CERBA) LABIOGENE
UFR/SVT, University of Ouagadougou BP 364 Ouagadougou, Burkina Faso, West Africa. Tel: +226 50361232 / + 226
70230792. E-mail: [email protected]
Introduction. The G-6-PD deficiency and
haemoglobinopathies occur with high frequency in
sub-Saharan Africa due to the malaria endemicity.1
Indeed, the maintenance of these genetic
abnormalities usually asymptomatic in the
homozygous state, within populations,
demonstrates the selective advantage that they
confer in the heterozygous state to carriers against

severe malaria.2 S hemoglobin responsible for
sickle cell disease is probably the most common
serious of haemoglobinopathies in the world.
Hemoglobin C is found only in parts of West Africa
with the highest frequencies observed in Burkina
Faso.3 The beta S (βs) form of hemoglobin is caused
by a single gene mutation: a "transversion" of the β6
globin gene (GAG to GTG) sixth codon of the first
exon that result in the substitution of glutamic acid
with valine (β6Glu Val). As to the βc form, it is
induced by gene mutation "transition" of the β6
globin gene (GAG to AAG) sixth codon of the first
exon that resulted in the substitution of glutamic
acid for lysine (β6GluLys). Since the Haldane
hypothesis, several studies have demonstrated the
protective effect of hemoglobin S and C against
severe malaria although certain mechanisms remain
controversial.4-6
The G-6-PD deficiency is the most common
inherited enzymopathy, with over 400 million
carriers worldwide.7 It is a genetic X-linked
abnormality with various clinical expressions in
heterozygous female.8 In humans, the G-6-PD gene
is located in the telomeric region of the long arm of
the X chromosome in position q28. It spans about
18 kb and contains 13 exons and 12 introns, ranging
in size between 12 bp and 236 bp.9
The G-6-PD gene is highly polymorphic with
over 180 mutations described and at least 35 mutant
alleles with polymorphic frequencies. These
polymorphisms are relatively common in different
parts of the world.10,11 Over 85% of these mutations
are single nucleotide substitutions. The enzyme
deficiency is asymptomatic except in cases of
infections, ingestion of certain foods or oxidizing
molecules.11 The severity of the disease depends on
the genetic variant involved. G-6-PDB is the wild
allele. The G-6-PDA, a non-deficient variant, is the
result of a substitution of adenine for guanine in
position 376 of G-6-PD gene exon 5. It is faster than
G6PDB electrophoretically and it does not cause
hemolysis.
Most deficient variants G-6-PDA- are usually
due to a second mutation on the G-6-PDA gene.12
The most prevalent G-6-PDA- variant in sub-
Saharan Africa and the most studied is the G-6-
PDA-202A/376G. However, other variants like the
G-6-PDA-376G/542T, 376G/680T, 376G/968C
have been reported in some African populations,
including West African populations with relatively
high frequencies.13-15 Also in their study in Mali,
Maiga et al.15 observed an association of other G-6-
PD SNPs, including rs915942 and rs915941 with
asymptomatic malaria in Dogon women. The allelic
heterogeneity of the G-6-PD gene suggests the need
to consider a broad range of G-6-PD variants in
association studies. In this study we sought for six
(06) single nucleotide polymorphisms substitution
involved in the G-6-PD deficiency in patients with
symptomatic malaria consulting in three health
centers in the city of Ouagadougou in Burkina Faso.
Materials and Methods.
Setting and type of study: This is a prospective
study in which patients regardless of gender or
ethnic group were recruited in three health centers
in Ouagadougou, the capital of Burkina Faso, from
September 27 to November 10, 2014. Malaria
transmission is hyper-endemic and seasonal during
the rainy season from June to October.
Study Population: The study involved 182 patients
aged 1 to 72 years, attending Saint Camille Hospital
of Ouagadougou (HOSCO), the Medical Center of
Samandin and the Biomolecular Research Center
Pietro ANNIGONI (CERBA) of Ouagadougou.
Patients were sent to the laboratory when malaria
was suspected, and underwent a Rapid Diagnostic
Test using SD Bioline Malaria Ag Pf/Pan, after
which positive individuals were included with their
free and informed consent on condition that they are
also positive microscopy.
Sampling: The samples consisted of venous blood
samples (5 mL of blood per subject adult and child
by blood 3 mL) in EDTA tubes. Part of the sample
has been used for the realization of the Complete
Blood Count (CBC), hemoglobin electrophoresis
and thick blood. After that, the remainder of the
sample was centrifuged at 15000 rpm for 5 minutes
to separate the plasma from the pellet, aliquoted and
stored at -80 ° C for molecular analysis.
Hematologic and thick smear: Hematological
parameters were determined from blood samples
with EDTA, using a blood counter ABX Micros 60
(ABX Diagnostics, Montpellier, France).
Hemoglobin Genotyping was made by
electrophoresis in alkaline pH on a cellulose acetate
tape. Tris-glycine at pH 9.5 was used as a buffer.
The cells were washed and then lysed using 1%
saponin. The migration was carried out for 60
minutes at 200 V on average.
The blades of thick films were stained for 10
minutes in a solution of 10% Giemsa. The reading
was performed using an optical microscope

objective 100 under oil immersion and parasite
density positive slides was calculated and expressed
as the number of parasites/µL. In preparing the final
parasite density, trophozoites were counted
simultaneously with 200 leukocytes. Two
independent microscopists did quality control by a
repeated examination of the blades. In the case of
difference of more than 5% between the results of
three readings, the average of the two closest results
was then retained.
DNA extraction and genotyping of G-6-PD
deficient variants: Genomic DNA was extracted
from blood pellet by the standard salting-out
method.11 The purity and the final concentration of
DNA extracts were determined using the Biodrop
μLITE (Isogen Life Science N.V./S.A, Temse,
Belgium). All samples were initially genotyped by
standard PCR. The amplification was done using
the kit GENESPARK G6PD African
(Immunospark, Rome, Italy) followed by
electrophoresis on agarose gel 2% for six SNPs
(G202A, A376G, A542T, G680T, T968C and
C563T) involved in the G-6-PD deficiency.
PCR was performed in a reaction volume of 25
µL composed of 12.5 µL of Multiplex PCR smart
mix (2x), 2 µL Primer Mixture (G6PD African), 8.5
µL of sterile water and 2 µL of 'DNA fragment (50-
100 ng) of each sample. Electrophoresis was
performed at 100 V for 1 hour.
Female individuals with mutations in position
202 and/or 376 were further analyzed by real-time
PCR using TaqMan probes respectively (Applied
Biosystems, Foster City, California, USA) include:
rs1050829, rs1050828.12
Statistical Analyses: Data were analyzed using the
software Statistical Package for Social Sciences
(SPSS) 21.0 (IBM, Armonk, NY, USA) and
EpiInfoTM 7. Hardy-Weinberg equilibrium was
determined in women according to the method
described by Carter et al.16 Pearson's chi-square test
was used for categorical variables such as age
groups, parasite density groups. ANOVA was
employed in the comparison of hemoglobin and
hematocrit means between groups. Non-parametric
tests were used to compare the geometric mean of
the parasite density. The difference was significant
at p <0.05.
Ethical Considerations: The present study was
approved by the Ethics Committee on Health
Research of Burkina Faso (Deliberation No. 2014-
9-128). Written informed consent was obtained
from the adults and guardians of children.
Results. Demographics and Clinical Characteristics: Our
study population consisted of 50.5% (92/182) of
men and 49.5% (90/182) of women aged 1 to 72
years with a mean age of 17.1 ± 13.9 years.
Children under 5 years old accounted for 19.2%
(35/182) of the study population noted that while
46.7% (85/182) of individuals over 15 years. Socio-
demographic analysis showed that 182 patients
included in this study were derived from several
different ethnic groups. Mossi represented the
majority ethnic group with a proportion of 76.4%
(139/182) of individuals whose parents were of this
ethnic group (Table 1). Note that half of the group
"Others" (10/20) was made up of ethnic groups
from countries in the sub-region with a
predominance of Nigerians (6/10). Over 43% of
patients in our study had started treatment before
the completion of the thick smear test. The study
population was in following with Hardy-Weinberg
equilibrium (p = 0.464).
Prevalence of deficiency G-6-PD and genotypes of
hemoglobin: Conventional PCR analysis gave more
than 64% (58/90) of females carrying mutations at
position 202 and/or 376. Twenty-five of them were
classified as homozygous and heterozygous by real-
time PCR (Figure 1). The G-6-PD deficiency
genotyping showed a prevalence of 78.6% of
individuals with a normal G-6-PD (29.7% genotype
B, 13.2% of genotype A, 20.9% B/B, 12.1% B/A
and 2.7% A/A) against 9.9%
hemizygous/homozygous subjects (8.8%
202A/376G and 0.5% 376G/968C) (Table 2). Note
that both parents of the individual carrier of G-6-
PDA- (376G/968C) were Mossi while the parents
of the female carrying the G-6-PDA- (376G/542T)
variant were of the Gouro ethnic group of Ivory
Coast.
She had the particularity of bearing G-6-PDA-
(202A/376G) on one of the X chromosome and G-
6-PDA- (376G/542T) on the other X chromosome
with a parasite density of 40 parasites/µL. Figure
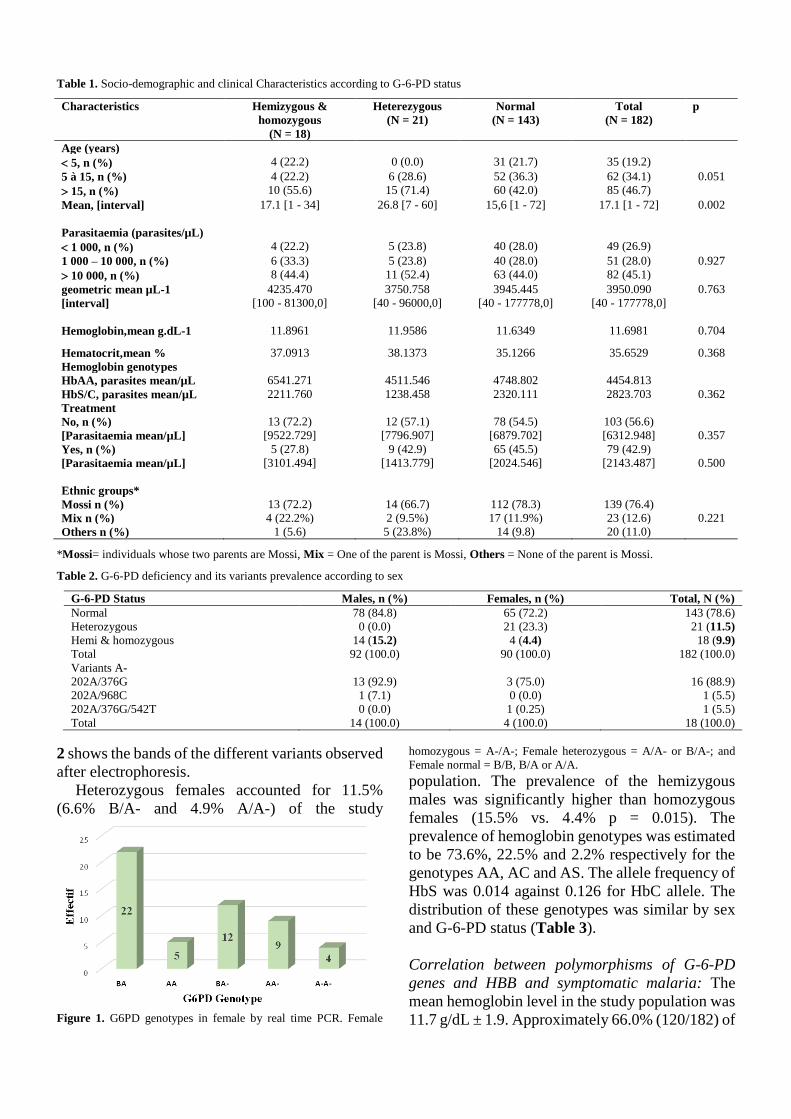
Table 1. Socio-demographic and clinical Characteristics according to G-6-PD status
Characteristics Hemizygous &
homozygous
(N = 18)
Heterezygous
(N = 21)
Normal
(N = 143)
Total
(N = 182)
p
Age (years)
5, n (%) 4 (22.2) 0 (0.0) 31 (21.7) 35 (19.2)
5 à 15, n (%) 4 (22.2) 6 (28.6) 52 (36.3) 62 (34.1) 0.051
15, n (%) 10 (55.6) 15 (71.4) 60 (42.0) 85 (46.7)
Mean, [interval] 17.1 [1 - 34] 26.8 [7 - 60] 15,6 [1 - 72] 17.1 [1 - 72] 0.002
Parasitaemia (parasites/µL)
1 000, n (%) 4 (22.2) 5 (23.8) 40 (28.0) 49 (26.9)
1 000 – 10 000, n (%) 6 (33.3) 5 (23.8) 40 (28.0) 51 (28.0) 0.927
10 000, n (%) 8 (44.4) 11 (52.4) 63 (44.0) 82 (45.1)
geometric mean µL-1
[interval]
4235.470
[100 - 81300,0]
3750.758
[40 - 96000,0]
3945.445
[40 - 177778,0]
3950.090
[40 - 177778,0]
0.763
Hemoglobin,mean g.dL-1 11.8961 11.9586 11.6349 11.6981 0.704
Hematocrit,mean % 37.0913 38.1373 35.1266 35.6529 0.368
Hemoglobin genotypes
HbAA, parasites mean/µL 6541.271 4511.546 4748.802 4454.813
0.362 HbS/C, parasites mean/µL 2211.760 1238.458 2320.111 2823.703
Treatment
No, n (%)
[Parasitaemia mean/µL]
13 (72.2)
[9522.729]
12 (57.1)
[7796.907]
78 (54.5)
[6879.702]
103 (56.6)
[6312.948]
0.357
Yes, n (%)
[Parasitaemia mean/µL]
5 (27.8)
[3101.494]
9 (42.9)
[1413.779]
65 (45.5)
[2024.546]
79 (42.9)
[2143.487]
0.500
Ethnic groups*
Mossi n (%) 13 (72.2) 14 (66.7) 112 (78.3) 139 (76.4)
Mix n (%) 4 (22.2%) 2 (9.5%) 17 (11.9%) 23 (12.6) 0.221
Others n (%) 1 (5.6) 5 (23.8%) 14 (9.8) 20 (11.0)
*Mossi= individuals whose two parents are Mossi, Mix = One of the parent is Mossi, Others = None of the parent is Mossi.
Table 2. G-6-PD deficiency and its variants prevalence according to sex
G-6-PD Status Males, n (%) Females, n (%) Total, N (%)
Normal 78 (84.8) 65 (72.2) 143 (78.6)
Heterozygous 0 (0.0) 21 (23.3) 21 (11.5)
Hemi & homozygous 14 (15.2) 4 (4.4) 18 (9.9)
Total 92 (100.0) 90 (100.0) 182 (100.0)
Variants A-
202A/376G 13 (92.9) 3 (75.0) 16 (88.9)
202A/968C 1 (7.1) 0 (0.0) 1 (5.5)
202A/376G/542T 0 (0.0) 1 (0.25) 1 (5.5)
Total 14 (100.0) 4 (100.0) 18 (100.0)
2 shows the bands of the different variants observed
after electrophoresis.
Heterozygous females accounted for 11.5%
(6.6% B/A- and 4.9% A/A-) of the study
Figure 1. G6PD genotypes in female by real time PCR. Female
homozygous = A-/A-; Female heterozygous = A/A- or B/A-; and
Female normal = B/B, B/A or A/A.
population. The prevalence of the hemizygous
males was significantly higher than homozygous
females (15.5% vs. 4.4% p = 0.015). The
prevalence of hemoglobin genotypes was estimated
to be 73.6%, 22.5% and 2.2% respectively for the
genotypes AA, AC and AS. The allele frequency of
HbS was 0.014 against 0.126 for HbC allele. The
distribution of these genotypes was similar by sex
and G-6-PD status (Table 3).
Correlation between polymorphisms of G-6-PD
genes and HBB and symptomatic malaria: The
mean hemoglobin level in the study population was
11.7 g/dL ± 1.9. Approximately 66.0% (120/182) of
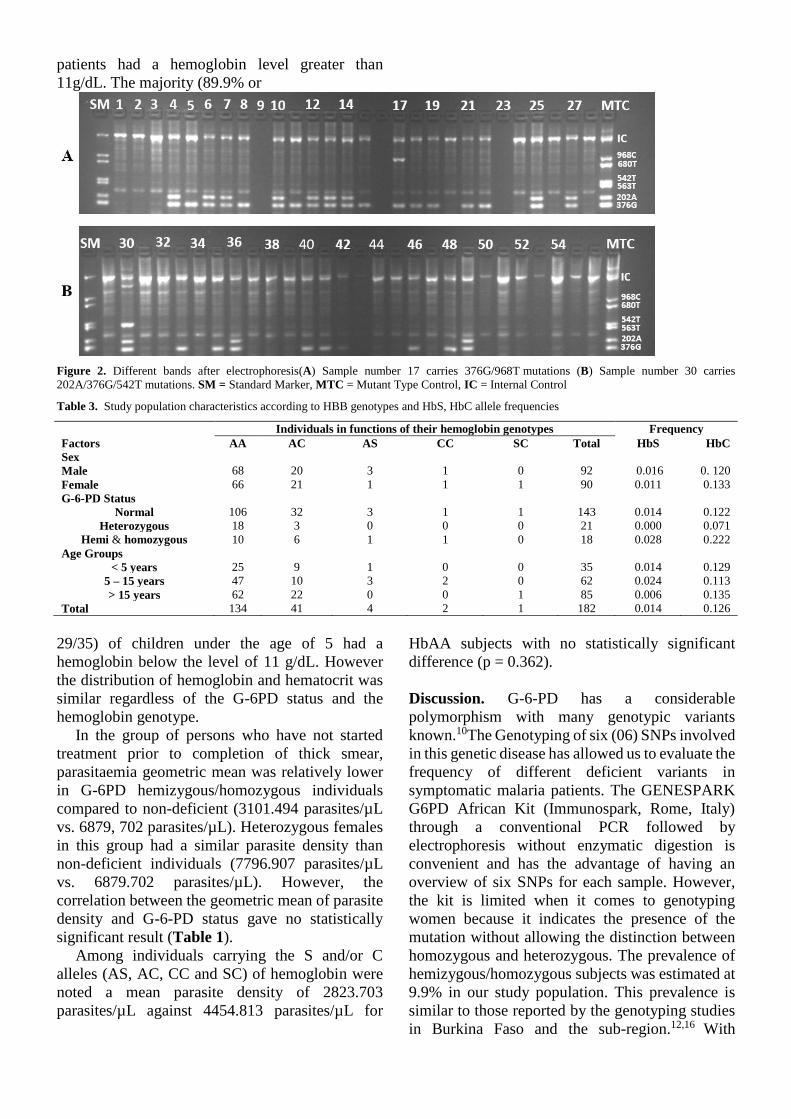
patients had a hemoglobin level greater than
11g/dL. The majority (89.9% or
Figure 2. Different bands after electrophoresis(A) Sample number 17 carries 376G/968T mutations (B) Sample number 30 carries
202A/376G/542T mutations. SM = Standard Marker, MTC = Mutant Type Control, IC = Internal Control
Table 3. Study population characteristics according to HBB genotypes and HbS, HbC allele frequencies
Individuals in functions of their hemoglobin genotypes Frequency
Factors AA AC AS CC SC Total HbS HbC
Sex
Male 68 20 3 1 0 92 0.016 0. 120
Female 66 21 1 1 1 90 0.011 0.133
G-6-PD Status
Normal 106 32 3 1 1 143 0.014 0.122
Heterozygous 18 3 0 0 0 21 0.000 0.071
Hemi & homozygous 10 6 1 1 0 18 0.028 0.222
Age Groups
< 5 years 25 9 1 0 0 35 0.014 0.129
5 – 15 years 47 10 3 2 0 62 0.024 0.113
> 15 years 62 22 0 0 1 85 0.006 0.135
Total 134 41 4 2 1 182 0.014 0.126
29/35) of children under the age of 5 had a
hemoglobin below the level of 11 g/dL. However
the distribution of hemoglobin and hematocrit was
similar regardless of the G-6PD status and the
hemoglobin genotype.
In the group of persons who have not started
treatment prior to completion of thick smear,
parasitaemia geometric mean was relatively lower
in G-6PD hemizygous/homozygous individuals
compared to non-deficient (3101.494 parasites/µL
vs. 6879, 702 parasites/µL). Heterozygous females
in this group had a similar parasite density than
non-deficient individuals (7796.907 parasites/µL
vs. 6879.702 parasites/µL). However, the
correlation between the geometric mean of parasite
density and G-6-PD status gave no statistically
significant result (Table 1).
Among individuals carrying the S and/or C
alleles (AS, AC, CC and SC) of hemoglobin were
noted a mean parasite density of 2823.703
parasites/µL against 4454.813 parasites/µL for
HbAA subjects with no statistically significant
difference (p = 0.362).
Discussion. G-6-PD has a considerable
polymorphism with many genotypic variants
known.10The Genotyping of six (06) SNPs involved
in this genetic disease has allowed us to evaluate the
frequency of different deficient variants in
symptomatic malaria patients. The GENESPARK
G6PD African Kit (Immunospark, Rome, Italy)
through a conventional PCR followed by
electrophoresis without enzymatic digestion is
convenient and has the advantage of having an
overview of six SNPs for each sample. However,
the kit is limited when it comes to genotyping
women because it indicates the presence of the
mutation without allowing the distinction between
homozygous and heterozygous. The prevalence of
hemizygous/homozygous subjects was estimated at
9.9% in our study population. This prevalence is
similar to those reported by the genotyping studies
in Burkina Faso and the sub-region.12,16 With

respect to gender, there was a significantly higher
prevalence of hemizygous male (15.5%) than
homozygous female (4.4%) since the disease is X-
linked.11Among people with G-6-PD deficiency in
this study, the G-6-PDA- (202A/376G) was the
most common variant observed in 88.9% of
deficiency cases. These observations are consistent
with previous genotyping studies.12,16 The G-6-PD
Betica Selma (376G/968C) was found in an
individual of the Mossi ethnic group. This variant
was observed in relatively high frequencies in the
Gambian population14, and in this study it is
identified for the first time in Burkina Faso. A study
in Mali reported a high frequency of this variant in
the Fulani (6.1%) compared to the Dogon (0.0%).15
A deficient woman of ethnic Gouro (Ivory
Coast) in our study shows the A- variants
(202A/376G) and Santamaria (376G/542T) on both
X chromosomes with very low parasitaemia. All
these observations suggest that these variants exist
in our populations even if they occur with relatively
low frequency in some areas. The highest
frequencies of the G-6-PD Santamaria were
reported in Sere population in Senegal.13
Our present results show that there is an
underestimation of genotypes real prevalence
causing the G-6-PD deficiency in Burkina Faso.
The latter could explain the different frequencies of
the deficit between genotyping studies and
enzymatic quantification studies in our
populations.1,17
As part of this study, patients underwent a
hemoglobin electrophoresis. A major sickle cell
syndrome (SC) was demonstrated in 0.5% (1/182)
of patients and 2.2% (4/182) had sickle cell trait
AS. The most detected hemoglobin genotype was
heterozygous AC present in 22.5% (41/182) of
patients with 1.1% (2/182) of CC homozygosity.
The prevalence of HbAC found in our study is
comparable to that (19.1%) found by Simporeetal.1
in 2007 and is higher than that found by Kafando et
al.18 in 2005 among newborns (15.4%). Prevalence
of 14.7% and 13.0% of the AC hemoglobin were
reported respectively by Amoako et al.19 in Ghana
and Travassos et al.20 in Mali. All these studies
show a higher rate of hemoglobin C in the sub-
region. Indeed, West Africa is the epicenter of
hemoglobin C.3
Although a reduced parasitaemia was observed
in some cases (G-6-PD deficient vs.G-6-PD non-
deficient individuals without treatment or HbAA
subjects vs. HbS/HbC subjects), all analyses of the
association between the G-6-PD status, hemoglobin
genotypes, parasite density, and rate of hemoglobin
or hematocrit gave no statistically significant
differences. Our results are similar to those reported
by Carter et al.16 in 2011 in six African countries
including Burkina Faso. The authors did not
observe significant effects of G-6-PD genotypes on
the hemoglobin and parasitaemia. The lack of
correlation between haemoglobins and G-6-PD
genotypes is expected, since respective genes are on
different chromosomes; and it has been reported by
a previous study.21 The latter results also confirm
that there is no protection against malaria
infections. Indeed, the G-6-PD deficiency or
haemoglobinopathies S and C did not offer
protection against Plasmodium falciparum
infections but may allow a favorable evolution of
malaria.
The protection of the G-6-PD deficiency against
severe form of malaria or malaria mortality is
known, but the mechanism of protection is not
entirely elucidated.22
In 2009 in a study in Gambia, Clark et al.14did
not find an association between severe malaria and
the 202A/376Gvariant only. However, pooling this
variant with other deficiency alleles revealed the
signal of protection. In 2014 in Mali, Maiga et
al.15found no conclusive results on the protective
effect of different G-6-PD genotypes correlated
with uncomplicated malaria. The authors also
suggest a higher risk of moderate malaria signs in
Dogon 202A mutation carriers, especially in
women. G-6-PD deficiency protective effects
against cerebral malaria and an increased risk of
severe malaria anemia have been reported in some
studies investigating the correlation between this
genetic disease and severe malaria.23,24 The allelic
heterogeneity of the G-6-PD, phenotypic
complexity and the difficulties of classification of
clinical forms of malaria are all factors that can
explain the differences between the different
studies. The correlation between G-6-PD
deficiency and protection against asymptomatic
malaria has been reported in the literature.12,25The
selective advantage against malaria of G-6-PD
heterozygous females has been early reported by
Bienzle et al.26 In a case-control study carried out in
Tanzania, it was established through the number of
G-6-PD SNPs, that only heterozygous women were
protected against severe forms of malaria.27
Another study in the Gambia had led to the same
conclusion with variant G-6-PDA- (376G/968C).28
In a case-control and cohort study in Kenya, Uyoga
et al. showed, comparing boys and girls a

significant protection from severe malaria among
G6PD c.202T heterozygous girls but no evidence
for protection among G6PD c.202T hemizygous
boys and homozygous girls (OR 1·18, 0·99-1·40;
p=0·056), thus the key to protection from severe
malaria could be the girls heterozygous for G6PD
deficiency.29
Our study population size was limited in
deepening the analysis. Parameters such as
treatment before completion of thick smears are
factors that may influence parasite density and bias
the association analyses.
In 2015 in Nigeria Igbeneghu et al.30 reported a
strong protection of HbAS and HbAC genotypes
against asymptomatic Plasmodium falciparum.
Protection of HbAC carriers against clinical forms
of Plasmodium falciparum malaria has also been
reported in Mali.20
During the same year, Mangano et al.5 reported
in Burkina Faso that HbAS genotype was
associated with a 70% reduction of parasite
Plasmodium falciparum unlike HbAC carriers,
although a strong protection was also observed in
HbCC and HbSC subjects. Our results suggest that
even if haemoglobinopathies S or C protect against
severe forms of malaria like G-6-PD deficiency,
they do not confer protection against Plasmodium
falciparum infections.
Conclusion. Our study confirms that the G-6-PDA-
variant (202A/376G), the most common in Burkina
Faso, does not confer protection against
Plasmodium falciparum malaria infections.
However, it shows that other variants such as
T968C and probably A542T exist in our population.
Further investigations are required in a larger
population with well certain ethnic groups for a real
estimate of the prevalence of Glucose-6-phosphate
dehydrogenase variants involved in a possible
association with the resistance to various kinds of
malaria.
Acknowledgment. Our sincere thanks to the entire
team of CERBA/LABIOGENE and the participants
of this study. Our gratitude also goes to the Italian
Episcopal Conference (CEI) and the West African
Economic and Monetary Union (WAEMU)
(through the PACER2 program) for their financial
support.
Project Foundation. This project was supported
by the West African Economic and Monetary
Union (WAEMU) through «le programme d'appui
et de développement des centres d'excellence
régionaux » and « soutien à la formation et la
recherche de l’excellence ».
Grant No. PACER II
References:
1. Simpore J, Ilboudo D, Damintoti K, Sawadogo L, Maria E, Binet S, Nitiema H, Ouedraogo P, Pignatelli S, Nikiema JB. Glucose-6-
phosphate dehydrogenase deficiency and sickle cell disease in
Burkina Faso. Pak J Biol Sci 2007, 10:409-414. http://dx.doi.org/10.3923/pjbs.2007.409.414 PMid:19069510
2. Hedrick PW. Population genetics of malaria resistance in humans. Heredity (Edinb) 2011, 107:283-304.
http://dx.doi.org/10.1038/hdy.2011.16 PMid:21427751
PMCid:PMC3182497
3. Modiano D, Bancone G, Ciminelli BM, Pompei F, Blot I, Simpore
J, Modiano G. Haemoglobin S and haemoglobin C: 'quick but
costly' versus 'slow but gratis' genetic adaptations to Plasmodium falciparum malaria. Hum Mol Genet 2008, 17:789-799.
http://dx.doi.org/10.1093/hmg/ddm350 PMid:18048408
4. Modiano D, Luoni G, Sirima BS, Simpore J, Verra F, Konate A, Rastrelli E, Olivieri A, Calissano C, Paganotti GM, D'Urbano L,
Sanou I, Sawadogo A, Modiano G, Coluzzi M. Haemoglobin C
protects against clinical Plasmodium falciparum malaria. Nature 2001, 414:305-308. http://dx.doi.org/10.1038/35104556
PMid:11713529
5. Mangano VD, Kabore Y, Bougouma EC, Verra F, Sepulveda N, Bisseye C, Santolamazza F, Avellino P, Tiono AB, Diarra A,
Nebie I, Rockett KA, Sirima SB, Modiano D, Malaria G. E. N.
Consortium. Novel Insights Into the Protective Role of Hemoglobin S and C Against Plasmodium falciparum
Parasitemia. J Infect Dis 2015, 212:626-634.
http://dx.doi.org/10.1093/infdis/jiv098 PMid:25712976
PMCid:PMC4512610
6. Luzzatto L. Sickle cell anaemia and malaria. Mediterr J Hematol
Infect Dis 2012, 4:e2012065. http://dx.doi.org/10.4084/mjhid.2012.065 PMid:23170194
PMCid:PMC3499995
7. Howes RE, Dewi M, Piel FB, Monteiro WM, Battle KE, Messina JP, Sakuntabhai A, Satyagraha AW, Williams TN, Baird JK, Hay
SI. Spatial distribution of G6PD deficiency variants across
malaria-endemic regions. Malar J 2013, 12:418. http://dx.doi.org/10.1186/1475-2875-12-418 PMid:24228846
PMCid:PMC3835423 8. Luzzatto L, Nannelli C, Notaro R. Glucose-6-Phosphate
Dehydrogenase Deficiency. Hematol Oncol Clin North Am 2016,
30:373-393. http://dx.doi.org/10.1016/j.hoc.2015.11.006
PMid:27040960
9. Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase
deficiency. The Lancet, 371:64-74. http://dx.doi.org/10.1016/S0140-6736(08)60073-2
PMid:18177777
10. Minucci A, Moradkhani K, Hwang MJ, Zuppi C, Giardina B,
Capoluongo E. Glucose-6-phosphate dehydrogenase (G6PD)
mutations database: review of the "old" and update of the new mutations. Blood Cells Mol Dis 2012, 48:154-165.
http://dx.doi.org/10.1016/j.bcmd.2012.01.001 PMid:22293322
11. Luzzatto L, Seneca E. G6PD deficiency: a classic example of pharmacogenetics with on-going clinical implications. Br J
Haematol 2014, 164:469-480.
http://dx.doi.org/10.1111/bjh.12665 PMid:24372186 PMCid:PMC4153881
12. Ouattara AK, Bisseye C, Bazie BV, Diarra B, Compaore TR,
Djigma F, Pietra V, Moret R, Simpore J. Glucose-6-phosphate
dehydrogenase (G6PD) deficiency is associated with
asymptomatic malaria in a rural community in Burkina Faso.
Asian Pac J Trop Biomed 2014, 4:655-658. http://dx.doi.org/10.12980/APJTB.4.2014APJTB-2014-0100
PMid:25183336 PMCid:PMC4037660

13. De Araujo C, Migot-Nabias F, Guitard J, Pelleau S, Vulliamy T, Ducrocq R. The role of the G6PD AEth376G/968C allele in
glucose-6-phosphate dehydrogenase deficiency in the seerer
population of Senegal. Haematologica 2006, 91:262-263.
PMid:16461316
14. Clark TG, Fry AE, Auburn S, Campino S, Diakite M, Green A,
Richardson A, Teo YY, Small K, Wilson J, Jallow M, Sisay-Joof F, Pinder M, Sabeti P, Kwiatkowski DP, Rockett KA. Allelic
heterogeneity of G6PD deficiency in West Africa and severe
malaria susceptibility. Eur J Hum Genet 2009, 17:1080-1085. http://dx.doi.org/10.1038/ejhg.2009.8 PMid:19223928
PMCid:PMC2986558
15. Maiga B, Dolo A, Campino S, Sepulveda N, Corran P, Rockett KA, Troye-Blomberg M, Doumbo OK, Clark TG. Glucose-6-
phosphate dehydrogenase polymorphisms and susceptibility to mild malaria in Dogon and Fulani, Mali. Malar J 2014, 13:270.
http://dx.doi.org/10.1186/1475-2875-13-270 PMid:25015414
PMCid:PMC4110528 16. Carter N, Pamba A, Duparc S, Waitumbi JN. Frequency of
glucose-6-phosphate dehydrogenase deficiency in malaria
patients from six African countries enrolled in two randomized anti-malarial clinical trials. Malar J 2011, 10:241.
http://dx.doi.org/10.1186/1475-2875-10-241 PMid:21849081
PMCid:PMC3188486 17. Modiano D, Luoni G, Sirima BS, Lanfrancotti A, Petrarca V,
Cruciani F, Simpore J, Ciminelli BM, Foglietta E, Grisanti P,
Bianco I, Modiano G, Coluzzi M. The lower susceptibility to Plasmodium falciparum malaria of Fulani of Burkina Faso (west
Africa) is associated with low frequencies of classic malaria-
resistance genes. Trans R Soc Trop Med Hyg 2001, 95:149-152. http://dx.doi.org/10.1016/S0035-9203(01)90141-5
18. Kafando E, Sawadogo M, Cotton F, Vertongen F, Gulbis B.
Neonatal screening for sickle cell disorders in Ouagadougou, Burkina Faso: a pilot study. J Med Screen 2005, 12:112-114.
http://dx.doi.org/10.1258/0969141054855300 PMid:16156939
19. Amoako N, Asante KP, Adjei G, Awandare GA, Bimi L, Owusu-Agyei S. Associations between Red Cell Polymorphisms and
Plasmodium falciparum infection in the Middle Belt of Ghana.
PLoS ONE 2014, 9:e112868.
http://dx.doi.org/10.1371/journal.pone.0112868
PMid:25470251 PMCid:PMC4254276
20. Travassos MA, Coulibaly D, Laurens MB, Dembele A, Tolo Y, Kone AK, Traore K, Niangaly A, Guindo A, Wu Y, Berry AA,
Jacob CG, Takala-Harrison S, Adams M, Shrestha B, Mu AZ,
Kouriba B, Lyke KE, Diallo DA, Doumbo OK, Plowe CV, Thera MA. Hemoglobin C Trait Provides Protection From Clinical
Falciparum Malaria in Malian Children. J Infect Dis 2015,
212:1778-1786. http://dx.doi.org/10.1093/infdis/jiv308 PMid:26019283 PMCid:PMC4633765
21. Luzzatto L, Allan NC. Relationship between the Genes for
Glucose-6-phospahate dehydrogenase and for Haemoglobins in a Nigerian Population. Nature Publishing Group1968, 219:1041-
1042.
22. Luzzatto L. G6PD deficiency: a polymorphism balanced by heterozygote advantage against malaria. The Lancet Haematology
2015, 2:e400-e401.
http://dx.doi.org/10.1016/S2352-3026(15)00191-X
23. Malaria Genomic Epidemiology Network. Reappraisal of known
malaria resistance loci in a large multicenter study. Nat Genet
2014, 46:1197-1204. http://dx.doi.org/10.1038/ng.3107 PMid:25261933 PMCid:PMC4617542
24. Shah SS, Rockett KA, Jallow M, Sisay-Joof F, Bojang KA, Pinder
M, Jeffreys A, Craik R, Hubbart C, Wellems TE, Kwiatkowski DP, Malaria G. E. N. Consortium. Heterogeneous alleles
comprising G6PD deficiency trait in West Africa exert contrasting effects on two major clinical presentations of severe malaria.
Malar J 2016, 15:13. http://dx.doi.org/10.1186/s12936-015-1045-
0 PMid:26738565 PMCid:PMC4704392 25. Mombo LE, Ntoumi F, Bisseye C, Ossari S, Lu CY, Nagel RL,
Krishnamoorthy R. Human genetic polymorphisms and
asymptomatic Plasmodium falciparum malaria in Gabonese schoolchildren. Am J Trop Med Hyg 2003, 68:186-190.
PMid:12641410
26. Bienzle U, Lucas A, Ayeni O, Luzzatto L. Glucose-6-phosphate dehydrogenase and malaria: greater resistance of females
heterozygous for enzyme deficiency and of males with non-
deficient variant. The Lancet 1972, 299:107-110. http://dx.doi.org/10.1016/S0140-6736(72)90676-9
27. Manjurano A, Sepulveda N, Nadjm B, Mtove G, Wangai H,
Maxwell C, Olomi R, Reyburn H, Riley EM, Drakeley CJ, Clark
TG, Malaria G. E. N. Consortium. African glucose-6-phosphate dehydrogenase alleles associated with protection from severe
malaria in heterozygous females in Tanzania. PLoS Genet 2015,
11:e1004960. http://dx.doi.org/10.1371/journal.pgen.1004960
PMid:25671784 PMCid:PMC4335500
28. Sirugo G, Predazzi IM, Bartlett J, Tacconelli A, Walther M,
Williams SM. G6PD A- deficiency and severe malaria in The Gambia: heterozygote advantage and possible homozygote
disadvantage. Am J Trop Med Hyg 2014, 90:856-859.
http://dx.doi.org/10.4269/ajtmh.13-0622 PMid:24615128 PMCid:PMC4015578
29. Uyoga S, Ndila CM, Macharia AW, Nyutu G, Shah S, Peshu N,
Clarke GM, Kwiatkowski DP, Rockett KA, Williams TN. Glucose-6-phosphate dehydrogenase deficiency and the risk of
malaria and other diseases in children in Kenya: a case-control and a cohort study. Lancet Haematol 2015, 2:e437-444.
http://dx.doi.org/10.1016/S2352-3026(15)00152-0
30. Igbeneghu C, Olisekodiaka MJ, Akinola FFS, Odaibo AB. Impact of Haemoglobin Variants AS and AC on Asymptomatic
Falciparum Malaria among Adults in Iwo, Southwestern Nigeria.
Scholars Journal of Applied Medical Sciences 2015, 3:1







