comment définir et classer les maladies inflammatoires...
TRANSCRIPT

http://france.elsevier.com/direct/REVRHU/
Revue du Rhumatisme 74 (2007) 714–725
Comment définir et classer les maladies inflammatoires ?
How to define and to classify inflammatory systemic diseases?
Jean Sibilia
Service de rhumatologie, centre national de référence des maladies auto-immunes systémiques rares, hôpital de Hautepierre, CHU de Strasbourg,1, avenue Molière, 67098 Strasbourg cedex, France
Reçu le 21 juin 2007 ; accepté le 3 juillet 2007Disponible sur internet le 25 juillet 2007
Mots clés : Maladies auto-inflammatoires ; Maladies auto-immunes ; Lupus ; Syndrome de Gougerot-Sjögren ; Critères ; Signature génique ; Cytokines
Keywords: IMID; Auto-immune disease; Systemic lupus; Sjögren’s syndrome; Criteria; Signature; Cytokines
1. Introduction
De nombreuses maladies inflammatoires sont liées à desmécanismes considérés comme dysimmunitaires. Ces affec-tions récemment regroupées sous le terme d’IMID (immunemediated inflammatory diseases) comprennent trois grandesentités nosologiques [1] :
● les maladies auto-immunes systémiques (non spécifiquesd’organe) et localisées (spécifiques d’organe) ;
● les maladies auto-inflammatoires ;● les affections inflammatoires de mécanisme indéterminécomprenant, notamment, des affections iatrogènes ou para-néoplasiques dont le mécanisme n’est pas auto-immun.
Ces maladies inflammatoires étaient définies jusqu’à présentpar des critères cliniques et biologiques validés par l’usage etl’avis d’experts. Cependant, leur polymorphisme suggèrequ’une même entité puisse être liée à des mécanismes molécu-laires différents. À titre d’exemple, la polyarthrite rhumatoïde(PR) est une maladie articulaire parfois extrêmement agressiveou inversement une forme bénigne non destructrice. S’agit-ilde la même affection ou d’affections différentes ? Jusqu’à pré-sent, il était difficile d’envisager « d’affiner » la classificationde ces maladies, mais de nouveaux outils immunitaires et/ou
Adresse e-mail : [email protected] (J. Sibilia).
1169-8330/$ - see front matter © 2007 Elsevier Masson SAS. Tous droits réservésdoi:10.1016/j.rhum.2007.07.003
moléculaires permettent maintenant une nouvelle approchenosologique. Au-delà du progrès conceptuel, cette nouvelleclassification permettra d’adapter les stratégies thérapeutiquesen utilisant des molécules ciblées pour chaque forme de mala-die, l’objectif le plus ambitieux étant de pouvoir disposer du« bon traitement pour le bon patient » [2,3].
2. Pourquoi envisager de nouveaux critèresdans les maladies immunitaires ?
Avant d’envisager de nouveaux critères des maladies immu-nitaires, il faut s’interroger sur leurs objectifs. Les critèresnosologiques vont-ils remplacer les critères pragmatiques ?
2.1. De nouveaux critères pragmatiques diagnostiques,pronostiques et évolutifs
Le polymorphisme de ces affections immunitaires chroni-ques rend l’utilisation des critères diagnostiques « classiques »très difficile. En pratique, ils sont souvent remplacés par desarbres décisionnels, qui sont également difficiles à constituercompte tenu de la diversité des maladies. Néanmoins, des cri-tères diagnostiques peuvent être utiles quand il existe un mar-queur biologique spécifique. L’un des exemples est le syn-drome des antiphospholipides (SAPL) dont la classification aété revue récemment avec l’intégration des anticorps anti-β2-glycoprotéine-1 comme critères diagnostiques biologiques [4](Tableau 1).
.

Tableau 1Les critères révisés du syndrome des antiphospholipides (Miyaris et al. 2006).Le diagnostic est évoqué s’il existe l’association d’un signe clinique et d’unsigne biologique à condition que les critères biologiques aient été vérifiés (à12 semaines d’intervalle) et qu’il n’y ait pas plus de cinq ans entre les signescliniques et biologiques
Critères cliniquesThromboses veineuses et/ou artériellesComplications obstétricales :
une ou plusieurs pertes fœtales (après la dixième semaine de gestation) sansqu'il existe d'anomalie morphologique du fœtusun ou plusieurs prématurés (avant la 34e semaine) liés àune éclampsie ou prééclampsieune insuffisance placentaire
trois ou plus de fausses couches précoces (avant la dixième semaine degestation) sans anomalie anatomique, hormonale ou chromosomique connue
Critères biologiquesAnticoagulant lupique détecté à deux reprises à au moins 12 semainesd'intervalle selon les recommandations de l'International Society of Thromboseand HaemostasisAnticorps anticardiolipine d'isotype IgG et/ou IgM détectés à deux reprises àau moins 12 semaines d'intervalle à titre élevé (> 40 UGPL ou UMPL ou > au99e percentile) mesurés par un Elisa standardisé.Anticorps anti-β2glycoprotéine 1 d'isotype IgG et/ou IgM détectés à deuxreprises à au moins 12 semaines d'intervalle à titre élevé (> 99e percentile) parun Elisa standardisé
J. Sibilia / Revue du Rhumatisme 74 (2007) 714–725 715
Dans un avenir proche, des critères moléculaires (protéomi-ques ou transcriptomiques) pourront être associés aux critèresclinicobiologiques classiques pour renforcer leur spécificité.Néanmoins, pour l’instant, même dans des affections héréditai-res comme la fièvre méditerranéenne familiale, le diagnosticreste clinique en raison de l’hétérogénéité des mutations dugène MEFV (pyrine).
Les critères pronostiques, très utilisés en hématologie et encancérologie, commencent à être évalués dans les maladiesimmunitaires. Deux exemples peuvent être cités :
● les critères de Leiden permettent de définir un rhumatismeinflammatoire d’évolution chronique et/ou érosive. Un scoreélevé suggère un risque évolutif qui peut justifier une priseen charge plus agressive [5] ;
● les critères FFS (five factors severity) permettent de préciserla stratégie thérapeutique, comme cela a été démontré dansles vascularites. Ainsi, un score FFS supérieur ou égal à 1dans les vascularites à anticorps anticytoplasmes des poly-nucléaires (ANCA), justifie un traitement immunosuppres-seur en complément de la corticothérapie [6,7].
Des critères d’activité ont été définis pour différentes mala-dies immunitaires comme la PR (DAS), les spondylarthropa-thies (BASDAI), le lupus (BILAG, SLEDAI…). Il serait justi-fié d’en déterminer dans d’autres maladies systémiques commele syndrome de Gougerot-Sjögren (SGS) et les sclérodermies,mais le polymorphisme de ces affections complique leur iden-tification et leur validation. Ces critères ont un réel intérêt pra-tique pour le suivi et le traitement de ces patients, mais n’ontpas d’intérêt pour le diagnostic ou la classification de ces mala-dies.
Si la recherche de critères pragmatiques est un objectif trèsutile, l’identification de critères de classification adaptés aux
progrès nosologiques est aussi d’une importance capitale.Mieux classer, c’est souvent mieux comprendre car un regrou-pement nosologique original fait souvent émerger un conceptnouveau, comme le suggère l’histoire des spondylarthropathies(SpA), du syndrome SAPHO ou, plus récemment, des maladiesauto-inflammatoires.
2.2. De nouveaux critères de classification nosologique
L’intérêt des critères de classification « classiques » n’estpas diagnostique, car ils sont souvent pris en défaut dans lesformes débutantes incomplètes, mais ils permettent de définirdes groupes homogènes de patients, ce qui est extrêmementutile en recherche clinique et dans l’évaluation des traitements.
Des critères de classification nosologiques permettraient deregrouper des pathologies considérées jusqu’alors comme trèsdifférentes ou, au contraire, de démembrer une entité trop poly-morphe. L’exemple le plus spectaculaire est celui des maladiesauto-inflammatoires. Une « dissection » physiopathologique(moléculaire) de ces maladies a permis d’identifier un grouped’affections assez homogène, connues depuis longtemps sousdes appellations très différentes (Tableau 2). La recherche decritères de classification nosologiques peut s’appliquer à la plu-part des IMID (immune mediated inflammatory diseases).
3. Critères de classification des maladies auto-immunes
Jusqu’à présent, les critères de classification des maladiesauto-immunes étaient fondés sur des arguments cliniques, bio-logiques et, éventuellement, d’autres anomalies morphologi-ques ou fonctionnelles. Une description de plus en plus précisede ces affections et une meilleure connaissance de leurs méca-nismes permettent d’envisager des critères différents.
3.1. Critères morphologiques
Pour certaines affections, une classification nosologiquereposant sur des critères « morphologiques » semble perfor-mante. Trois exemples peuvent être cités :
● la classification nosologique des rhumatismes inflammatoi-res débutants peut s’appuyer sur une analyse immunohisto-chimique de la membrane synoviale, ce qui est assezlogique, car il s’agit de l’organe « lésé ». Plusieurs travauxrécents ont démontré l’intérêt d’une analyse de la synovialepermettant de distinguer précocement une PR d’une SpA :○ dans la PR, des macrophages synoviaux (CD68+) sontprésents précocement (dès la phase préclinique) et corré-lés à l’activité et à la sévérité de l’atteinte articulaire [8].Il s’agit de macrophages « infiltrants » issus de monocy-tes marqués par les myeloïd related protéines (MRP) 8 et4 et non pas de macrophages « résidents » de la syno-viale (CD163+), comme dans les SpA. Dans la synoviterhumatoïde, les autres éléments différents des SpA sontla présence de cellules dendritiques (CD83+), la pré-sence de peptides citrullinés intracellulaires et de com-

Tableau 2Les différentes formes de syndromes auto-inflammatoires héréditaires
J. Sibilia / Revue du Rhumatisme 74 (2007) 714–725716
plexes de glycoprotéines cartilagineuses humaines(gp39) associés à des molécules HLA [9,10]. Cependant,dans un travail récent, il a été montré que la présence defibrine déiminée intracellulaire dans la synoviale n’estpas spécifique, observée aussi dans d’autres formes desynovites (SpA, arthrose), mais seuls les patients atteintsde PR produisent des autoanticorps antipeptides citrulli-nés [11] ;
○ dans les SpA, l’élément le plus caractéristique est la pré-sence de polynucléaires neutrophiles et surtout demacrophages synoviaux « résidents » (CD163+) corrélésà l’activité de la maladie, quelle que soit la forme cli-nique [12]. Il est intéressant d’observer que ces anoma-lies sont signalées dans toutes les formes cliniques derhumatisme psoriasique, même polyarticulaires, maispas de façon significative dans les SpA juvéniles [13].D’autres paramètres, comme l’hypervascularisation,caractérisent aussi les SpA, mais sont moins discrimi-nants [14].
Ainsi, si l’atteinte articulaire le permet, il serait possibled’envisager une analyse synoviale d’un rhumatisme inflam-matoire débutant. Reste à savoir si cette identification peut
avoir, dans l’état actuel des connaissances, une utilité autreque nosologique ;
● les vascularites primitives sont des affections polymorphes,mais la nouvelle classification de Chapel-Hill est unedémarche particulièrement rationnelle qui permet de définiravec beaucoup de clarté les principales vascularites primiti-ves. Cette classification est fondée sur la taille des vaisseauxlésés et sur les caractéristiques histologiques et/ou immuno-logiques des lésions. Cette classification intègre les ANCAqui ont une valeur diagnostique et pronostique importante.Ces anticorps sont associés à une expression phénotypiqueparticulière des vascularites. Ainsi, le syndrome de Churg etStrauss et la granulomatose de Wegener se caractérisent parune atteinte vasculaire inflammatoire plus sévère (souventrénale) pour les formes avec ANCA comparées à cellessans autoanticorps [15] (Fig. 1) ;
● le lupus cutané est une affection particulièrement poly-morphe classée habituellement en affections aiguës, subai-guës et chroniques. En fait, l’observation de formes histolo-giques sans atteinte de l’interface dermoépidermiquesuggère l’utilisation d’une classification plus morpholo-gique séparant les lupus cutanés en formes « classiques »
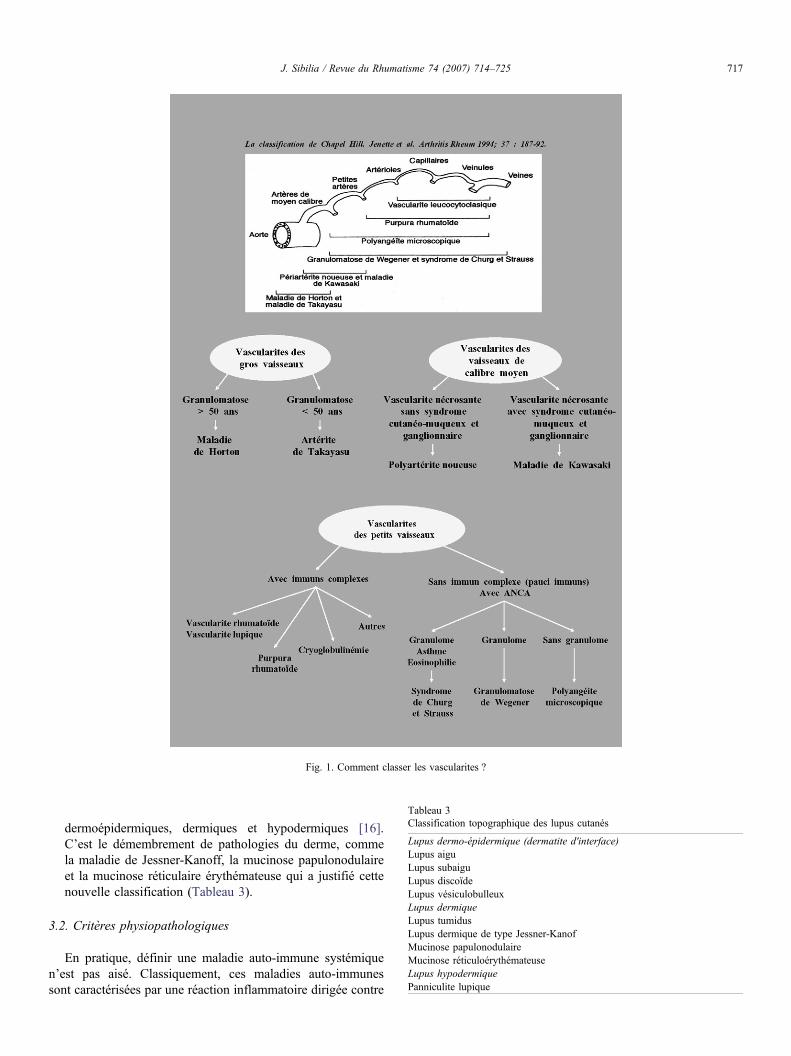
Fig. 1. Comment classer les vascularites ?
Tableau 3Classification topographique des lupus cutanés
Lupus dermo-épidermique (dermatite d'interface)Lupus aiguLupus subaiguLupus discoïdeLupus vésiculobulleuxLupus dermiqueLupus tumidusLupus dermique de type Jessner-KanofMucinose papulonodulaireMucinose réticuloérythémateuseLupus hypodermiquePanniculite lupique
J. Sibilia / Revue du Rhumatisme 74 (2007) 714–725 717
dermoépidermiques, dermiques et hypodermiques [16].C’est le démembrement de pathologies du derme, commela maladie de Jessner-Kanoff, la mucinose papulonodulaireet la mucinose réticulaire érythémateuse qui a justifié cettenouvelle classification (Tableau 3).
3.2. Critères physiopathologiques
En pratique, définir une maladie auto-immune systémiquen’est pas aisé. Classiquement, ces maladies auto-immunessont caractérisées par une réaction inflammatoire dirigée contre

J. Sibilia / Revue du Rhumatisme 74 (2007) 714–725718
des constituants cellulaires ou tissulaires, liée à une réponseimmunitaire anormale définie comme « autoagressive ». Cetteréaction est la conséquence d’une rupture de tolérance qui setraduit par un dialogue « aberrant » entre les cellules présenta-trices de l’antigène (cellules dendritiques) et les lymphocytes Tet B. Elle s’associe, dans la plupart des cas, à la productiond’autoanticorps qui peuvent être des marqueurs diagnostiquesou, parfois aussi, des acteurs pathogènes détectés plusieursmois ou années avant l’apparition des signes cliniques [17].Cette définition n’est pas d’un grand apport pour la pratiquequotidienne, en dehors des autoanticorps qui sont souvent uti-lisés comme critères de classification. Les progrès physiopa-thologiques permettent maintenant d’envisager une classifica-tion plus nosologique qui pourrait s’appuyer sur quelquesavances conceptuelles.
3.2.1. Identification de maladies auto-immunes monogéniquesLa grande majorité des affections auto-immunes est polygé-
nique, mais il existe quelques exemples d’affections monogé-niques [18]. À ce jour, les trois principales sont des affectionstrès rares caractérisées par un défaut de régulation des lympho-cytes T.
● Le syndrome IPEX (immune dysregulation polyendocrino-pathy, entheropathy, X-linked) se caractérise dès l’enfancepar une atteinte digestive souvent sévère, un diabète detype I, une thyroïdite et des cytopénies auto-immunes (ané-mie hémolytique et thrombopénie). Ce syndrome est lié àdes mutations du gène FOX P3 qui code pour l’ADN-binding protein scurfin nécessaire à l’activité des lymphocy-tes T régulateurs CD4+, CD25+ [19,20] ;
● le syndrome ALPS (autoimmune lymphoproliferative syn-drome) se caractérise par des mutations du système FAS/FAS Ligand régulant l’apoptose lymphocytaire. Ce syn-drome, dont il existe plusieurs formes, se caractérise globa-lement par des manifestations auto-immunes cliniques etbiologiques (cytopénies) et l’apparition d’une lymphoproli-fération CD4–/CD8– [21] ;
● le syndrome APECED (autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy syndrome) appelé aussiAPS-1 (autoimmune polyendocrine syndrome-1) est caracté-risé par les mutations du gène AIRE (auto-immune regulatorproteine) qui intervient dans l’éducation thymique des lym-phocytes T régulateurs. Ce syndrome se caractérise par desmanifestations auto-immunes essentiellement endocrinien-nes parfois associées à une candidose chronique [22].
Ces différentes affections sont extrêmement rares, maisl’étude de leur mécanisme a permis de mieux comprendre lefonctionnement du système immunitaire dans les maladiesauto-immunes.
3.2.2. Identification d’une « signature » cytokiniquedes maladies auto-immunes
Les maladies auto-immunes sont caractérisées par une réac-tion immunitaire avec un certain « profil » cytokinique. La
mise en évidence d’une « signature cytokinique » a été un pro-grès conceptuel intéressant dans le lupus [23–25]. Dans cetteaffection, plusieurs travaux ont démontré le rôle de l’interféronde type I (α) qui est une cytokine produite par les cellules den-dritiques plasmacytoïdes capables de moduler la réponseimmunitaire en activant les lymphocytes T et B autoréactifs.En fait, cette signature interféron n’est probablement pas spé-cifique du lupus, mais traduit l’initiation de certaines maladiesauto-immunes, comme le SGS et les thyroïdites, par l’activa-tion des cellules dendritiques [26].
À l’inverse, d’autres affections auto-immunes, comme laPR, dépendraient du TNF-α dont l’un des effets pourrait êtred’inhiber la synthèse d’IFNα/β. Cette hypothèse permettraitd’expliquer l’apparition de maladies auto-immunes (lupus)dépendantes de l’IFN-α/β sous anti-TNF [27]. Ainsi, il y auraitune régulation inhibitrice réciproque entre la production deTNF-α et d’IFN-α/β, justifiant une classification en maladiesauto-immunes dépendantes de l’IFN et en maladies auto-immunes dépendantes du TNF-α (Fig. 2).
En réalité, la situation est plus complexe car le TNF-α et lesinterférons de type 1 ont des effets immunologiques qui peu-vent varier en fonction de leur origine cellulaire, de leurconcentration et du moment auquel ils sont produits au coursde la maladie [28]. Un des exemples les plus simples pourillustrer cette complexité est l’existence d’affections associantdes signes de PR ou de lupus (appelées rhupus [rheumatoidarthritis and lupus]).
3.2.3. Identification de maladies dysimmunitaires associantdes phénomènes d’allo- et d’auto-immunisation maternofœtale
Dans certaines affections considérées comme des allo-immunisations maternofœtales, il a été décrit des phénomènesd’auto-immunisation dont le rôle pathogène est discuté. Troisexemples peuvent être décrits :
● les néphropathies extramembraneuses néonatales sont liéesà des IgG anti-NEP (neutral endopeptidase). Ces autoanti-corps sont la conséquence d’une allo-immunisation mater-nelle contre la NEP syncitiotrophoblastique qui est absentechez la mère, mais présente dans les podocytes du rein del’enfant. Ces anticorps d’isotype IgG vont passer la barrièrefœtoplacentaire et se déposer en entraînant une néphropathieextramembraneuse fœtale ;
● dans l’arthrogrypose congénitale, la mère qui est saine a desautoanticorps antirécepteurs de l’acéthylcholine fœtale. Pen-dant la grossesse, ces autoanticorps passent chez le fœtusinduisant une myasthénie et des contractures à l’origine del’arthrogrypose [29,30] ;
● l’hémochromatose néonatale est une affection rare qui pour-rait être une affection liée à une allo-immunisation materno-fœtale contre un antigène inconnu. Néanmoins, dans certai-nes formes, il a été observé la présence d’autoanticorps, enparticulier anti-Ro/SS-A [31]. Dans une série récente de 15patientes ayant donné naissance à des enfants avec unehémochromatose néonatale, deux d’entre elles avaient unlupus et une troisième des signes biologiques d’auto-

Fig. 2. Le concept « séparateur » de la classification nosologique des IMID (immune mediated inflammatory diseases). Les affections auto-inflammatoires peuvent secaractériser par une « signature » cytokinique soit de type IL-1, soit de type TNF, ce qui correspond aux mécanismes moléculaires de ces maladies. Les affectionsauto-immunes pourraient aussi se caractériser par une « signature » cytokinique reposant essentiellement sur une balance TNF/IFN de type 1. Cette hypothèseséduisante est trop caricaturale, ne reprenant pas de façon suffisamment pertinente les mécanismes cytokiniques plus complexes des maladies auto-immunes.
J. Sibilia / Revue du Rhumatisme 74 (2007) 714–725 719
immunité [32]. Dans cette étude ouverte, il est intéressantd’observer l’efficacité des immunoglobulines intraveineusesqui est un argument complémentaire en faveur de l’originedysimmunitaire de cette complication rare.
Dans certaines affections auto-immunes, il a été décrit desphénomènes d’allo-immunistion potentiellement pathologi-ques. Dans la dermatomyosite et la sclérodermie systémiqueet même le lupus systémique et le SGS, des cellules d’originefœtale sont encore détectables, plus de 20 ans après l’accou-chement, dans la circulation et les tissus maternels [33–35].Ce microchimérisme a été considéré initialement comme unfacteur initiateur ou amplificateur de ces maladies auto-immunes par analogie avec la réaction du greffon contrel’hôte qui est un modèle d’allo-immunisation. Ce phénomène,appelé microchimérisme, qui peut être observé chez des fem-mes sans affection auto-immune, n’est peut-être pas pathogèneet pourrait être une réponse « réparatrice » [36].
L’hypothèse d’un regroupement des affections auto-immunes comportant une allo-immunisation est une piste inté-ressante qui pourrait justifier une classification nosologiquespécifique.
4. Maladies auto-inflammatoires
Ces affections se définissent par une agression tissulaire liéeà l’activation excessive de l’immunité innée, indépendammentde l’immunité adaptative (lymphocytaire) (Tableau 2). Ceconcept original repose sur le fait qu’une réaction immunitaireest mise en jeu par un signal « danger » qui doit être détecté etsi possible, éliminé le plus précocement possible par notreimmunité non spécifique (innée) [37]. Ce n’est que dans undeuxième temps, si cela est nécessaire, que l’immunité adapta-tive est mise en jeu, permettant ainsi une mémorisation del’agresseur.
4.1. Extension du concept de maladies auto-inflammatoiresmonogéniques à des affections polygéniques
Le concept « originel » des maladies auto-inflammatoires aété défini par l’existence d’anomalies génétiques à transmissionmendelienne interférant avec une voie cytokinique générique(en particulier celles de l’IL-1 et du TNF-α) ou avec des récep-teurs intervenant dans la détection de micro-organismes ou dedébris microbiens (pathogen associated molecular patterns ouPAMPS) [38–40]. Cette définition a permis d’identifier unedizaine de formes monogéniques qui étaient, pour la plupart,des fièvres périodiques héréditaires comme la fièvre méditerra-néenne familiale ou la fièvre familiale hibernienne appeléeaujourd’hui syndrome TRAPS (TNF necrosis factor receptor–associated periodic fever syndrome) (Tableau 2). Ce progrèsconceptuel a été une avancée majeure permettant l’utilisationde traitements immunomodulateurs adaptés [2,3,41,42].
Au-delà des formes monogéniques, il existe des formespolygéniques multifactorielles ayant de nombreuses similitu-des. Pour illustrer ce point, deux exemples originaux peuventêtre décrits.
4.2. Exemple de la goutte
La goutte est une affection inflammatoire caricaturale dontle mécanisme vient d’être décodé. Les cristaux d’urate desodium sont détectés par les cellules de l’immunité innéecomme un signal de « danger » [43]. Ces cristaux se fixentdirectement sur un récepteur de l’immunité innée de la familledes NLR. Ce récepteur NALP3 (cryopyrine) est celui dont lesmutations sont responsables des cryopyrinopathies monogéni-ques (CINCA, syndrome de Muckles-Wells, urticaire familialeau froid) [44]. Dans la goutte, il n’y a pas d’anomalie géné-tique de ce récepteur, mais une activation « inconsidérée »aboutissant à la production excessive d’interleukine-1. Même

J. Sibilia / Revue du Rhumatisme 74 (2007) 714–725720
s’il s’agit d’un progrès conceptuel majeur, il reste encore àcomprendre le rôle de phénomènes associés (comme les infec-tions microbiennes ou d’autres stimulations) capables d’induireles crises.
4.3. Exemple des syndromes neutrophiliques
Le concept de syndrome neutrophilique pourrait permettrede regrouper un certain nombre d’affections« auto-inflammatoires » dont le trait d’union pourrait être lepolynucléaire neutrophile (PNN). Cette « nébuleuse » des syn-dromes auto-inflammatoires neutrophiliques pourrait inclureles dermatoses neutrophiliques (en particulier le syndrome deSweet et le pyoderma gangrenosum), mais aussi certaines for-mes de maladie de Crohn, de SpA, de psoriasis, de polychon-drite et de maladie de Behçet (Fig. 3). Les arguments suggérantla pertinence de ce regroupement nosologique sont épidémio-logiques, cliniques et génétiques :
● ces affections peuvent être associées chez un même individuou dans une même famille avec une fréquence qui n’est pasliée au hasard de la rencontre d’affections assez fréquentes.De nombreuses associations pourraient être citées, en parti-culier celles des dermatoses neutrophiliques avec la maladiede Crohn et de la maladie de Crohn avec le psoriasis et lesspondylarthropathies ;
● il existe des manifestations communes systémiques (fièvre),cutanées (dermatose neutrophilique) et articulaires caractéri-sées par la présence de polynucléaires neutrophiles. Ces élé-ments suggèrent une réaction inflammatoire à une agressionmicrobienne originale ;
Fig. 3. La « nébuleuse » des affections auto-inflammatoires neutrophiliques. Cesépidémiologiques : ces affections peuvent s’associer avec une fréquence inhabitucommunes surtout articulaires et cutanées et parfois des signes systémiques (fièvre…des polynucléaires neutrophiles observées dans les lésions cutanées, osseuses ou vneutrophiliques monogéniques appelées PAPA (pyogenic arthritis, pyoderma ganrécurrentes, dysérythropoïèse et dermatoses neutrophiliques) liés à des anomalies génParmi les affections de cette nébuleuse, la maladie de Crohn se caractérise (dans 20 àinflammatoire intestinale aux agents bactériens. D’autres facteurs génétiques (HLA Bpourraient déterminer leur expression phénotypique.
● différents arguments génétiques plaident aussi pour ceregroupement nosologique avec, en particulier, deux syn-dromes auto-inflammatoires « neutrophiliques » monogéni-ques. Le PAPA (pyogenic sterile arthritis, pyoderma gan-grenosum and acne-syndrome) est une affection auto-inflammatoire liée à la mutation du gène CD2BPI/PSTPIT1(CD2-binding protein/proline-serine-treonine-phosphatase-interacting protein) qui intervient dans la régulation desneutrophiles et des monocytes [45–48]. Le syndrome deMAJEED est une affection auto-inflammatoire caractériséepar une ostéite récurrente multifocale, une dysérythropoïèseet une dermatose neutrophilique de type Sweet. Cette mala-die est liée à une mutation homozygote du gène LPIN2 [47],mais d’autres anomalies génomiques existent peut-être[49] ;
● un des meilleurs exemples pour illustrer le rôle des facteursgénétiques est la maladie de Crohn [50]. Près d’un tiers desformes est lié à des mutations du gène NOD2/CARD15, cequi se traduit par une réponse inflammatoire aberrante descellules dendritiques, des macrophages et des cellules dePaneth intestinales en réponse au muranyl–dipeptide bacté-rien [51–53]. Cette dérégulation de la réponse aux agentsbactériens implique très vraisemblablement une anomaliede la voie NF–KB [54–56]. Il a été démontré récemmentqu’une inhibition de cette voie (par un blocage de NEMO)induit l’apoptose des cellules épithéliales intestinales et undéfaut d’expression des peptides antibactériens. En consé-quence, il y a une rupture de la barrière épithéliale et uneprolifération bactérienne anormale qui peut avoir différentesconséquences pathologiques [55,56]. Certaines bactériescomme des colibacilles vont pouvoir adhérer aux cellules
affections multifactorielles ont en commun différents facteurs : a) facteurselle ; b) caractérisation phénotypique : ces affections ont des manifestations) ; c) facteurs immunologiques : le trait d’union est l’existence de lésions liées àiscérales ; d) facteurs génétiques : il existe deux maladies auto-inflammatoiresgrenosum, severe cystic acne) et syndrome de MAJEED (ostéites aseptiquesomiques connues qui ont des similitudes avec les affections de cette nébuleuse.30 % des cas) par des mutations du gène NOD2/CARD15 qui régule la réponse27, TNF-R1) ne sont pas responsables du déclenchement de ces affections, mais

J. Sibilia / Revue du Rhumatisme 74 (2007) 714–725 721
épithéliales intestinales car elles expriment spécifiquementune molécule d’adhésion appelée (CEACAM6) [carcinoem-bryonic antigen-related cell adhesion molecule 6] dontl’expression est induite par le TNF et l’IFN-γ [57]. D’autresfacteurs génétiques de susceptibilité interviennent ; en parti-culier, ATG16L1 qui code pour une protéine impliquéedans l’autophagie de bactéries intracellulaires comme lessalmonelles [58–60]. L’importance de l’autophagie estconfirmée par la découverte d’un nouveau gène de suscep-tibilité intervenant dans la régulation de ce phénomène phy-siologique [60] ;
● ainsi, dans les maladies auto-inflammatoires, le poids de lagénétique est important, modulant plus particulièrement laréponse immunitaire innée aux agents microbiens, maisd’autres anomalies peuvent être à l’origine de particularitésphénotypiques caractérisant certaines entités de cette nébu-leuse. À titre d’exemple, HLA B27 n’est probablement pasun gène majeur de la régulation de la réponse immunitaire,mais un gène qui peut expliquer l’expression enthésiopa-thique de ces affections [61,62]. De même, des anomaliesdes gènes impliqués dans les maladies auto-inflammatoiresmonogéniques (gènes MEFV, TNF RS1) ont été observéesdans des maladies auto-immunes comme la PR et le lupus,ce qui pourrait conférer à ces affections un profil phénoty-pique particulier [63].
5. Lien entre les maladies auto-immunes et les maladiesauto-inflammatoires
A priori, il existe une séparation nosologique entre ces deuxentités, les unes liées à l’immunité innée et les autres à l’immu-nité adaptative (lymphocytaire). En réalité, ces affectionsinflammatoires se répartissent sur un spectre allant des mala-dies auto-inflammatoires monogéniques (sans signes d’auto-
Fig. 4. Le concept « unificateur » de la classification nosologique des IMID (monogéniques auto-inflammatoires et auto-immunes et des affections polygéniqphénomènes auto-inflammatoires (liés à l’immunité innée) et des phénomènes auto
immunité) aux affections auto-immunes monogéniques (sansintervention de l’immunité innée) [64] (Fig. 4). La plupart deces affections sont multifactorielles et multigéniques, déclen-chées par des facteurs d’environnement. Ainsi, une maladieauto-immune peut comporter une ou des « facettes » auto-inflammatoires expliquant son initiation, mais aussi certaineslésions tissulaires (Fig. 5).
5.1. Chronologie de l’histoire naturelle des maladies auto-immunes
● Le déclenchement d’une maladie auto-immune pourrait êtrelié à des facteurs d’environnement qui stimulent l’immunitéinnée locale [65]. Ce sont ces facteurs (probablement hété-rogènes pour chaque maladie) qui expliquent en partie letropisme d’organes de certaines maladies auto-immunes.Cette activation locale met en jeu les cellules de l’immunitéinnée (macrophages, cellules dendritiques, polynucléaires),mais aussi les cellules résidentes, comme les synoviocytesdans la PR et les cellules épithéliales glandulaires dans leSGS ;
● la pérennisation de ces maladies met en jeu des phénomènesauto-immuns qui peuvent être entretenus par des facteursd’environnement (en particulier microbiens) expliquantl’évolution parfois récurrente de ces maladies. Les lésionstissulaires sont liées à la conjonction de phénomènes auto-immuns spécifiques (autoanticorps, cytotoxicité lymphocy-taire) et de phénomènes auto-inflammatoires non spécifi-ques (agressions enzymatiques et cytokiniques) ;
● une des grandes difficultés est l’étude de la chronologie deces phénomènes, et cela, pour deux raisons :○ les modèles animaux sont « artificiels », car ils ne per-mettent qu’une approche très spécifique et donc partiellede la maladie humaine. En fait, la plupart des modèles
immune mediated inflammatory diseases). Ce spectre inclut des affectionsues de mécanisme intriqué associant, dans des proportions variables, des-immuns (liés à l’immunité lymphocytaire adaptative).

Fig. 5. Schéma synthétique de la pathogénie des maladies auto-immunes et auto-inflammatoires. Dans ces deux types de maladies, l’initiation est liée à une agression(signal « danger ») qui active l’immunité innée expliquant en partie le tropisme d’organe de la plupart de ces affections. Dans les maladies auto-inflammatoires, enraison d’anomalies monogéniques ou polygéniques, la réaction inflammatoire s’amplifie, créant des lésions caractéristiques de la maladie. Ces lésions sontsusceptibles d’induire l’apparition de néo-autoantigènes éventuellement responsables d’une réaction immunitaire lymphocytaire secondaire habituellement sansproduction d’autoanticorps. Dans les maladies auto-immunes, la réaction immunitaire innée initiale va stimuler préférentiellement les cellules dendritiques et lescellules résidentes tissulaires. Cette réaction va s’associer rapidement à une réaction auto-immune lymphocytaire qui va être amplifiée par le relargage de néo-antigènes tissulaires à l’origine de la production d’autoanticorps. Dans ces affections, différents facteurs génomiques peuvent conditionner l’apparition demanifestations clinicobiologiques ou l’apparition de complications. Ces facteurs génétiques ne sont pas inducteurs, mais ils peuvent modifier l’expressionphénotypique de ces maladies et l’apparition de certaines anomalies biologiques (autoanticorps).
J. Sibilia / Revue du Rhumatisme 74 (2007) 714–725722
murins, caractérisés par la délétion ou la surexpressiond’un gène, miment une maladie inflammatoire humainemonogénique, et cela, sans tenir compte de toutes lesspécificités transcriptionnelles et traductionnelles quiexistent dans les tissus humains ;
○ les affections auto-immunes humaines ne s’exprimentcliniquement qu’à une phase tardive de l’histoire de laréponse immunitaire. De nombreux exemples témoi-gnent de la présence d’autoanticorps spécifiques dansle lupus ou la PR, plusieurs années avant l’apparitiondes premiers signes cliniques [17]. En conséquence, lapathogénie de ces maladies est décrite en fonction dece qui est observable, c’est-à-dire en faisant abstraction,sauf cas particulier, de toute la phase infraclinique oupaucisymptômatique initiale. La prise en compte de cesdifficultés a permis une analyse plus pertinente desmaladies auto-immunes, avec la confirmation, un peusurprenante, du rôle important de l’immunité innée. Le« poids » de l’immunité innée peut être illustré par deuxmaladies auto-immunes emblématiques.
5.1.1. Exemple du lupus
5.1.1.1. Rôle de l’immunité innée : hypothèse TLR et… hypo-thèse NALP. Le lupus est considéré classiquement comme unemaladie auto-immune liée à une autoréactivité lymphocytaire Tet B. En fait, il existe de nombreux arguments en faveur d’unedérégulation, probablement initiatrice de l’immunité innée.
L’une des observations les plus curieuses est une prévalenceinattendue de lupus dans les granulomatoses septiques congé-nitales (mutation des gènes NADP-oxydases) qui sont des défi-cits primitifs de l’immunité innée [66,67]. Un des points lesplus difficiles est de comprendre l’enchaînement des phénomè-nes initiant la réaction auto-immune, mais de nombreux élé-ments récents permettent de proposer un schéma séduisant.
Des agressions (virus, ultraviolets…) induisent, surtout dansla peau, des lésions et l’activation des cellules « résidentes »(kératinocytes) [68]. Les cellules dendritiques plasmacytoïdesde « proximité » vont capter les débris cellulaires et des acidesnucléiques indépendamment de l’activation des récepteursTLR. Ces cellules dendritiques vont alors produire de l’IFNde type I.
Cette première étape est amplifiée par la coactivation descellules dendritiques par des ligands endogènes (surtout desacides nucléiques) et exogènes qui activent les TLR endoso-maux (TLR3, 7, 9), ainsi que par des immuns complexes quise fixent sur les récepteurs Fcγ-R [69–72]. Cette amplificationaboutit aussi à la synthèse d’IFN de type I.
Ces phénomènes d’activation sont amplifiés par l’accumu-lation de débris cellulaires observés au cours du lupus. Cedéfaut d’élimination des débris (endogènes et exogènes)s’explique par des anomalies de l’immunité innée (déficit encomplément, anomalie des Dnases, polymorphisme des Fcγ-Rmacrophagiques, anomalie de production des protéines de laphase aiguë de l’inflammation [CRP]) [73–77].

J. Sibilia / Revue du Rhumatisme 74 (2007) 714–725 723
Cette libération de débris microbiens peut activer directe-ment des LB autoréactifs [78] et même les LT [79].
Les agressions tissulaires entraînent également la libérationd’autres cytokines de l’immunité innée, notamment du TNF-αet de l’IL-6, et cela, malgré l’importance de la productiond’IFN de type I [80–82]. Ces cytokines et d’autres phénomènesinflammatoires non spécifiques (protéases, radicaux libres)amplifient les lésions liées aux « agressions » lymphocytaires.
La phase initiale du lupus repose donc sur une activation del’immunité innée initialement TLR-indépendante, puis TLR-dépendante, aboutissant globalement à la production d’IFN detype I capable d’activer les lymphocytes T et B autoréactifs[73,83,84]. Le lupus est donc un exemple emblématique illus-trant la pertinence de la « TLR hypothèse » des maladies auto-immunes, mais une « NALP hypothèse » mettant en jeu d’autreséléments de l’immunité innée est aussi possible. Dans un travailrécent effectué chez des sujets souffrant d’affections auto-immunes multiples (dont souvent un vitiligo), il a été suggéréle rôle du gène NALP1 qui est une protéine fondamentale del’inflammasome, permettant la production d’IL-1 et d’IL-18[85]. Reste à déterminer précisément quelle est l’anomalie géné-tique en cause et quelle est sa conséquence fonctionnelle.
5.1.1.2. Conséquences nosologiques de cette nouvelle analysephysiopathologique du lupus. Le polymorphisme phénotypiquedu lupus est certainement lié à l’intrication de ces anomaliesimmunitaires pour lesquelles il existe différents scénariosmoléculaires. En effet, il existe des manifestations directementliées à des autoanticorps (cytopénies, neurolupus…) et d’autresmanifestations, souvent cutanées (lupus bulleux, vascularitesurticariennes), qui sont des lésions neutrophiliques liées à uneanomalie de l’immunité innée. Dans d’autres formes, la situa-tion est plus complexe. Le lupus cutané subaigu avec anti-Ro/SS-A est certainement l’un des meilleurs exemples pour illus-trer l’association d’anomalies de l’immunité innée et adaptative[86]. Dans cette forme de lupus, il y a une agression des cel-lules résidentes (kératinocytes) et, probablement, des cellulesdendritiques plasmacytoïdes cutanés par les ultraviolets et,éventuellement, d’autres facteurs (virus). L’activation de cescellules induit des lésions et la production de cytokines pro-inflammatoires (TNF-α) favorisant l’hyperexpression kératino-cytaire d’autoantigènes cutanés (Ro/SS-A) qui vont être captéspar des cellules dendritiques [81,87]. Cette situation va induireune réaction lymphocytaire auto-immune avec la productionlocale d’autoanticorps (anti-Ro/SS-A) [84]. C’est vraisembla-blement ce mécanisme assez stéréotypé qui est à l’origine del’aspect clinicobiologique très spécifique du lupus cutané sub-aigu avec anti-Ro/SS-A. Ainsi, une classification nosologiquemoderne du lupus pourrait permettre d’identifier des formesphénotypiques caractéristiques déterminées par un mécanismemoléculaire original. Dans l’état actuel des connaissances, lescytopénies lupiques, les néphropathies glomérulaires et certai-nes formes cutanées (lupus bulleux, lupus cutané subaigu…)pourraient entrer dans ce cadre. Cette nouvelle classificationpermettra d’identifier des groupes plus homogènes de patients,ce qui facilitera l’étude des facteurs immunogénétiques et,
peut-être aussi, de définir des stratégies thérapeutiques adap-tées à chacune de ces formes.
5.1.2. Exemple de la polyarthrite rhumatoïde
5.1.2.1. Rôle de l’immunité innée : exemple de la souris trans-génique TNFα +/+. La PR est la conséquence d’une activationdes lymphocytes T et B autoréactifs dirigés contre différentsautoantigènes, en particulier les peptides citrullinés, mais ellepourrait être initiée par une activation inappropriée de l’immu-nité innée synoviale. La principale difficulté est de comprendrecomment s’enchaînent ces différents phénomènes. Récemment,dans un modèle d’arthrite murine liée à la transfection duTNFα humain (h TNFα +/+), il a été démontré que pendantla phase préclinique (avant les arthrites) s’installe une réactioninflammatoire ténosynoviale faite essentiellement de PNN, demacrophages et de quelques lymphocytes T. Ce n’est que dansun deuxième temps qu’apparaissent une véritable proliférationsynoviale, puis des lésions ostéoarticulaires avec un infiltratlymphoïde de l’os sous-chondral fait de lymphocytes T et delymphocytes B. L’initiation de la maladie est donc induite parune activation de l’immunité innée, ce qui se traduit par la pro-duction de TNFα et d’interleukine-6 par les polynucléaires, lesmacrophages et les cellules ténosynoviales. Ce n’est qu’ulté-rieurement que se développe une réaction lymphocytaire diri-gée contre des autoantigènes produits par la réaction inflamma-toire intrasynoviale et/ou les lésions ostéoarticulaires [88].Reste à savoir quelles sont les similitudes entre ce modèle(qui est un modèle caricatural dépendant du TNF) et la PRhumaine, surtout, quels sont les facteurs initiateurs de l’activa-tion de l’immunité innée ?
5.1.2.2. Conséquences nosologiques de cette nouvelle analysephysiopathologique de la PR. Comme pour le lupus, il est doncpossible d’envisager une nouvelle classification de la PR dis-tinguant des formes de sévérité et d’évolution (articulaire etextra-articulaire). L’objectif est maintenant à une véritable dis-section moléculaire des différentes formes de la maladie et celapour adapter nos stratégies thérapeutiques.
6. Conclusion
Le concept d’IMID est une révolution dans l’approcheconceptuelle des maladies inflammatoires. Au-delà du clivagetraditionnel entre maladies auto-immunes et maladies auto-inflammatoires, il est nécessaire d’analyser, par une approcheplus physiopathologique, le rôle de l’immunité innée (liée à lapression de l’environnement) et le rôle de l’immunité lympho-cytaire adaptative (liée à des facteurs immunogénétiques).Cette nouvelle approche doit permettre de faire évoluer la clas-sification, et donc, la compréhension et le traitement de cesaffections. L’idéal serait donc d’avoir des critères de classifica-tion nosologique, utiles aux travaux de recherche fondamentaleet thérapeutique, et des critères pragmatiques, utiles pour lesuivi quotidien des patients.

[21
[22
[23
[24
[25
[26
[27
[28
[29
[30
[31
[32
[33
[34
[35
[36
[37
[38
[39
[40
[41
[42
[43
[44
J. Sibilia / Revue du Rhumatisme 74 (2007) 714–725724
Références
[1] Kuek A, Hazleman BL, Ostor AJ. Immune-mediated inflammatory disea-ses (IMIDs) and biologic therapy: a medical revolution. Postgrad Med J2007;83:251–60.
[2] Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J,Rubin BI, et al. Neonatal-onset multisystem inflammatory disease res-ponsive to interleukin-1beta inhibition. N Engl J Med 2006;355:581–92.
[3] Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting ofinnate immunity with Toll-like receptor agonists and antagonists. NatMed 2007;13:552–9.
[4] Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R,et al. International consensus statement on an update of the classificationcriteria for definite antiphospholipid syndrome (APS). J Thromb Hae-most 2006;4:295–306.
[5] Visser H, le Cessie S, Vos K, Breedveld FC, Hazes JM. How to diagnoserheumatoid arthritis early: a prediction model for persistent (erosive)arthritis. Arthritis Rheum 2002;46:357–65.
[6] Gayraud M, Guillevin L, le Toumelin P, Cohen P, Lhote F, Casassus P,et al. Long-term followup of polyarteritis nodosa, microscopic polyangii-tis, and Churg-Strauss syndrome: analysis of four prospective trials inclu-ding 278 patients. Arthritis Rheum 2001;44:666–75.
[7] Guillevin L, Lhote F, Gayraud M, Cohen P, Jarrousse B, Lortholary O,et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syn-drome. A prospective study in 342 patients. Medicine 1996;75:17–28.
[8] Haringman JJ, Gerlag DM, Zwinderman AH, Smeets TJ, Kraan MC,Baeten D, et al. Synovial tissue macrophages: a sensitive biomarker forresponse to treatment in patients with rheumatoid arthritis. Ann RheumDis 2005;64:834–8.
[9] Kruithof E, Baeten D, De Rycke L, Vandooren B, Foell D, Roth J, et al.Synovial histopathology of psoriatic arthritis, both oligo- and polyarticu-lar, resembles spondyloarthropathy more than it does rheumatoid arthri-tis. Arthritis Res Ther 2005;7:R569–R580.
[10] Baeten D, Steenbakkers PG, Rijnders AM, Boots AM, Veys EM, DeKeyser F. Detection of major histocompatibility complex/human cartilagegp-39 complexes in rheumatoid arthritis synovitis as a specific and inde-pendent histologic marker. Arthritis Rheum 2004;50:444–51.
[11] Chapuy-Regaud S, Sebbag M, Baeten D, Clavel C, Foulquier C, De Key-ser F, et al. Fibrin deimination in synovial tissue is not specific for rheu-matoid arthritis but commonly occurs during synovitides. J Immunol2005;174:5057–64.
[12] Baeten D, Kruithof E, De Rycke L, Boots AM, Mielants H, Veys EM,et al. Infiltration of the synovial membrane with macrophage subsets andpolymorphonuclear cells reflects global disease activity in spondyloar-thropathy. Arthritis Res Ther 2005;7:R359–R369.
[13] Kruithof E, Van den Bossche V, De Rycke L, Vandooren B, Joos R,Canete JD, et al. Distinct synovial immunopathologic characteristics ofjuvenile-onset spondylarthritis and other forms of juvenile idiopathicarthritis. Arthritis Rheum 2006;54:2594–604.
[14] Baeten D, Kruithof E, De Rycke L, Vandooren B, Wyns B, Boullart L,et al. Diagnostic classification of spondylarthropathy and rheumatoidarthritis by synovial histopathology: a prospective study in 154 consecu-tive patients. Arthritis Rheum 2004;50:2931–41.
[15] Jennette JC, Falk RJ. Nosology of primary vasculitis. Curr Opin Rheu-matol 2007;19:10–6.
[16] Lipsker D, Mitschler A, Grosshans E, Cribier B. Could Jessner’s lym-phocytic infiltrate of the skin be a dermal variant of lupus erythemato-sus? An analysis of 210 cases. Dermatology 2006;213:15–22.
[17] Bizzaro N, Tozzoli R, Shoenfeld Y. Are we at a stage to predict auto-immune rheumatic diseases? Arthritis Rheum 2007;56:1736–44.
[18] Gregersen PK, Behrens TW. Genetics of autoimmune diseases-disordersof immune homeostasis. Nat Rev Genet 2006;7:917–28.
[19] Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, White-sell L, et al. The immune dysregulation, polyendocrinopathy, enteropa-thy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. NatGenet 2001;27:20–1.
[20] Nomura T, Sakaguchi S. Foxp3 and Aire in thymus-generated Treg cells:a link in self-tolerance. Nat Immunol 2007;8:333–4.
] Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F,Prieur AM, et al. Autoimmune lymphoproliferative syndrome with soma-tic Fas mutations. N Engl J Med 2004;351:1409–18.
] Gavanescu I, Kessler B, Ploegh H, Benoist C, Mathis D. Loss of Aire-dependent thymic expression of a peripheral tissue antigen renders it atarget of autoimmunity. Proc Natl Acad Sci USA 2007;104:4583–7.
] Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA,Espe KJ, et al. Interferon-inducible gene expression signature in periphe-ral blood cells of patients with severe lupus. Proc Natl Acad Sci USA2003;100:2610–5.
] Banchereau J, Pascual V, Palucka AK. Autoimmunity through cytokine-induced dendritic cell activation. Immunity 2004;20:539–50.
] Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction ofdendritic cell differentiation by IFN-alpha in systemic lupus erythemato-sus. Science 2001;294:1540–3.
] Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, LazureT, et al. Activation of IFN pathways and plasmacytoid dendritic cellrecruitment in target organs of primary Sjogren’s syndrome. Proc NatlAcad Sci USA 2006;103:2770–5.
] Aringer M, Steiner G, Graninger WB, Hofler E, Steiner CW, Smolen JS.Effects of short-term infliximab therapy on autoantibodies in systemiclupus erythematosus. Arthritis Rheum 2007;56:274–9.
] Alsaleh G, Messer L, Semaan N, Boulanger N, Gottenberg JE, Wachs-mann D, et al. BAFF synthesis by rheumatoid synoviocytes is positivelycontroled by integrin α5β1 stimulation and negatively regulated by TNFαand TLR ligands. Arthritis Rheum 2007 (in press).
] Riemersma S, Vincent A, Beeson D, Newland C, Hawke S, Vernet-derGarabedian B, et al. Association of arthrogryposis multiplex congenitawith maternal antibodies inhibiting fetal acetylcholine receptor function.J Clin Invest 1996;98:2358–63.
] Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, BensmanA, et al. Antenatal membranous glomerulonephritis due to anti-neutralendopeptidase antibodies. N Engl J Med 2002;346:2053–60.
] Schoenlebe J, Buyon JP, Zitelli BJ, Friedman D, Greco MA, Knisely AS.Neonatal hemochromatosis associated with maternal autoantibodiesagainst Ro/SS-A and La/SS-B ribonucleoproteins. Am J Dis Child1993;147:1072–5.
] Whitington PF, Hibbard JU. High-dose immunoglobulin during pre-gnancy for recurrent neonatal haemochromatosis. Lancet 2004;364:1690–8.
] Kremer Hovinga IC, Koopmans M, de Heer E, Bruijn JA, Bajema IM.Chimerism in systemic lupus erythematosus-three hypotheses. Rheumatol2007;46:200–8.
] Aractingi S, Sibilia J, Meignin V, Launay D, Hachulla E, Le Danff C,et al. Presence of microchimerism in labial salivary glands in systemicsclerosis but not in Sjogren’s syndrome. Arthritis Rheum 2002;46:1039–43.
] Sarkar K, Miller FW. Possible roles and determinants of microchimerismin autoimmune and other disorders. Autoimmun Rev 2004;3:454–63.
] Nguyen Huu S, Dubernard G, Aractingi S, Khosrotehrani K. Feto-maternal cell trafficking: a transfer of pregnancy associated progenitorcells. Stem Cell Rev 2006;2:111–6.
] Matzinger P. The danger model: a renewed sense of self. Science 2002;296:301–5.
] Grateau G. Clinical and genetic aspects of the hereditary periodic feversyndromes. Rheumatol 2004;43:410–5.
] McDermott MF. A common pathway in periodic fever syndromes.Trends Immunol 2004;25:457–60.
] Petrilli V, Papin S, Tschopp J. The inflammasome. Curr Biol 2005;15:R581.
] Hoffman HM, Patel DD. Genomic-based therapy: targeting interleukin-1for autoinflammatory diseases. Arthritis Rheum 2004;50:345–9.
] Lovell DJ, Bowyer SL, Solinger AM. Interleukin-1 blockade by anakinraimproves clinical symptoms in patients with neonatal-onset multisysteminflammatory disease. Arthritis Rheum 2005;52:1283–6.
] Shi Y, Evans JE, Rock KL. Molecular identification of a danger signalthat alerts the immune system to dying cells. Nature 2003;425:516–21.
] Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associateduric acid crystals activate the NALP3 inflammasome. Nature 2006;440:237–41.

[45
[46
[47
[48
[49
[50
[51
[52
[53
[54
[55
[56
[57
[58
[59
[60
[61
[62
[63
[64
[65
[66
[67
[68
[69
[70
[71
[72
[73
[74
[75
[76
[77
[78
[79
[80
[81
[82
[83
[84
[85
[86
[87
[88
J. Sibilia / Revue du Rhumatisme 74 (2007) 714–725 725
] Ferguson PJ, Bing X, Vasef MA, Ochoa LA, Mahgoub A, Waldsch-midt TJ, et al. A missense mutation in pstpip2 is associated with themurine autoinflammatory disorder chronic multifocal osteomyelitis.Bone 2006;38:41–7.
] Shoham NG, Centola M, Mansfield E, Hull KM, Wood G, Wise CA,et al. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Medi-terranean fever and PAPA syndrome as disorders in the same pathway.Proc Natl Acad Sci USA 2003;100:13501–6.
] Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, et al.Homozygous mutations in LPIN2 are responsible for the syndrome ofchronic recurrent multifocal osteomyelitis and congenital dyserythropoie-tic anaemia (Majeed syndrome). J Med Genet 2005;42:551–7.
] Lindor NM, Arsenault TM, Solomon H, Seidman CE, McEvoy MT. Anew autosomal dominant disorder of pyogenic sterile arthritis, pyodermagangrenosum, and acne: PAPA syndrome. Mayo Clin Proc 1997;72:611–5.
] Golla A, Jansson A, Ramser J, Hellebrand H, Zahn R, Meitinger T, et al.Chronic recurrent multifocal osteomyelitis (CRMO): evidence for a sus-ceptibility gene located on chromosome 18q21.3-18q22. Eur J HumGenet 2002;10:217–21.
] Strober W, Fuss I, Mannon P. The fundamental basis of inflammatorybowel disease. J Clin Invest 2007;117:514–21.
] Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al.A frameshift mutation in NOD2 associated with susceptibility to Crohn’sdisease. Nature 2001;411:603–6.
] Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J,et al. Association of NOD2 leucine-rich repeat variants with susceptibi-lity to Crohn’s disease. Nature 2001;411:599–603.
] Korzenik JR. Is Crohn’s disease due to defective immunity? Gut 2007;56:2–5.
] Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, et al.Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity andIL-1beta processing. Science 2005;307:734–8.
] Nenci A, Becker C, Wullaert A, Gareus R, Van Loo G, Danese S, et al.Epithelial NEMO links innate immunity to chronic intestinal inflamma-tion. Nature 2007;446:557–61.
] Zaph C, Troy AE, Taylor BC, Berman-Booty LD, Guild KJ, Du Y, et al.Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immunehomeostasis. Nature 2007;446:552–6.
] Barnich N, Carvalho FA, Glasser AL, Darcha C, Jantscheff P, Allez M,et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, suppor-ting ileal mucosa colonization in Crohn disease. J Clin Invest 2007;117:1566–74.
] Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A,et al. Genome-wide association study identifies new susceptibility locifor Crohn disease and implicates autophagy in disease pathogenesis.Nat Genet 2007;39:596–604.
] Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. Agenome-wide association scan of nonsynonymous SNPs identifies a sus-ceptibility variant for Crohn disease in ATG16L1. Nat Genet 2007;39:207–11.
] Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fis-her SA, et al. Sequence variants in the autophagy gene IRGM and multi-ple other replicating loci contribute to Crohn’s disease susceptibility. NatGenet 2007;39:830–2.
] McGonagle D, Gibbon W, Emery P. Classification of inflammatoryarthritis by enthesitis. Lancet 1998;352:1137–40.
] Tan AL, Grainger AJ, Tanner SF, Emery P, McGonagle D. A high-resolution magnetic resonance imaging study of distal interphalangealjoint arthropathy in psoriatic arthritis and osteoarthritis : are they thesame? Arthritis Rheum 2006;54:1328–33.
] Ozen S, Bakkaloglu A, Yilmaz E, Duzova A, Balci B, Topaloglu R,et al. Mutations in the gene for familial Mediterranean fever: do they pre-dispose to inflammation? J Rheumatol 2003;30:2014–8.
] McGonagle D, McDermott MF. A proposed classification of the immu-nological diseases. Plos Med 2006;3:1242–8.
] Wucherpfennig KW. Mechanisms for the induction of autoimmunity byinfectious agents. J Clin Invest 2001;108:1097–104.
] Cobeta-Garcia JC, Domingo-Morera JA, Monteagudo-Saez I, Lopez-Longo FJ. Autosomal chronic granulomatous disease and systemic
lupus erythematosus with fatal outcome. Br J Rheumatol 1998;37:109–11.
] Winkelstein JA, Marino MC, Johnston Jr. RB, Boyle J, Curnutte J, Gal-lin JI, et al. Chronic granulomatous disease. Report on a national registryof 368 patients. Medicine 2000;79:155–69.
] van Zandbergen G, Solbach W, Laskay T. Apoptosis driven infection.Autoimmunity 2007;40:349–52.
] Blander JM, Medzhitov R. Toll-dependent selection of microbial anti-gens for presentation by dendritic cells. Nature 2006;440:808–12.
] Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M,Endres S, et al. Plasmacytoid dendritic cells control TLR7 sensitivity ofnaive B cells via type I IFN. J Immunol 2005;174:4043–50.
] Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2aand 2b autoantibodies in SLE. J Exp Med 2006;203:553–61.
] Ruprecht CR, Lanzavecchia A. Toll-like receptor stimulation as a thirdsignal required for activation of human naive B cells. Eur J Immunol2006;36:810–6.
] Erbacher AI, Ronnefarth VM, Decker P. Circulating nucleosomes due toa defective clearance of dying cells: consequences for systemic lupuserythematosus. Autoimmunity 2007;40:311–4.
] Vogt B, Fuhrnrohr B, Muller R, Sheriff A. CRP and the disposal ofdying cells : Consequences for systemic lupus erythematosus and rheu-matoid arthritis. Autoimmunity 2007;40:295–8.
] Sjowall C, Wettero J. Pathogenic implications for autoantibodies againstC-reactive protein and other acute phase proteins. Clin Chim Acta 2007;378:13–23.
] Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L,et al. FCGR3B copy number variation is associated with susceptibility tosystemic, but not organ-specific, autoimmunity. Nat Genet 2007;39:721–3.
] Patel VA, Longacre-Antoni A, Cvetanovic M, Lee DJ, Feng L, Fan H,et al. The affirmative response of the innate immune system to apoptoticcells. Autoimmunity 2007;40:274–80.
] Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ,Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dualengagement of IgM and Toll-like receptors. Nature 2002;416:603–7.
] Liew FY, Komai-Koma M, Xu D. A toll for T cell costimulation. AnnRheum Dis 2004;63(Suppl 2):ii76–ii78.
] Lee YH, Harley JB, Nath SK. Meta-analysis of TNF-alpha promoter -308 A/G polymorphism and SLE susceptibility. Eur J Hum Genet 2006;14:364–71.
] Zampieri S, Alaibac M, Iaccarino L, Rondinone R, Ghirardello A, Sarzi-Puttini P, et al. Tumour necrosis factor alpha is expressed in refractoryskin lesions from patients with subacute cutaneous lupus erythematosus.Ann Rheum Dis 2006;65:545–8.
] Vielhauer V, Mayadas TN. Functions of TNF and its Receptors in RenalDisease : Distinct Roles in Inflammatory Tissue Injury and ImmuneRegulation. Semin Nephrol 2007;27:286–308.
] Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon inductionin systemic autoimmunity. Nat Med 2007;13:543–51.
] Christensen SR, Shlomchik MJ. Regulation of lupus-related autoantibodyproduction and clinical disease by Toll-like receptors. Semin Immunol2007;19:11–23.
] Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC,et al. NALP1 in vitiligo-associated multiple autoimmune disease. N EnglJ Med 2007;356:1216–25.
] Sontheimer RD. Subacute cutaneous lupus erythematosus: 25-year evolu-tion of a prototypic subset (subphenotype) of lupus erythematosus defi-ned by characteristic cutaneous, pathological, immunological, and gene-tic findings. Autoimmun Rev 2005;4:253–63.
] Gerl V, Hostmann B, Johnen C, Waka A, Gerl M, Schumann F, et al.The intracellular 52-kd Ro/SSA autoantigen in keratinocytes is up-regulated by tumor necrosis factor alpha via tumor necrosis factor recep-tor I. Arthritis Rheum 2005;52:531–8.
] Hayer S, Redlich K, Korb A, Hermann S, Smolen J, Schett G. Tenosy-novitis and osteoclast formation as the initial preclinical changes in amurine model of inflammatory arthritis. Arthritis Rheum 2007;56:79–88.