chimie analytique appliquée à l'environnement
TRANSCRIPT

Chimie analytique appliquée à l’environnement
Karine Desboeufs
Prof Université Paris Diderot, LISA

Organisation du cours
UE répartie entre :
12h de cours
Cours disponible sur portail du master
et sur http://www.lisa.univ-paris12.fr/~desboeufs/
Pour vous évaluer, 1 QCM à chaque cours
En cas de désespoir: [email protected]
20h de TP (4 séances de 5h)
Notation: 80% pour l’examen final et 20 % pour les TP

Qu’est ce que la chimie analytique appliquée à l’environnement?
Définition
La chimie analytique est la science qui permet d'acquérir des informations concernant un matériau ou un échantillon et d'en tirer une composition chimique (élémentaire/moléculaire) à l'aide de méthodes scientifiques
Application à l’environnement:
Chimie analytique appliquée aux matrices environnementales permet de mesurer des milliers de substances jusqu’alors inconnues afin d’évaluer leurs risques environnementaux.

Qu’est ce que la chimie analytique appliquée à l’environnement?
Application à l’environnement: Toutefois pour évaluer un risque, il faut pouvoir comparer les
valeurs trouvées. On a donc eu besoin d’intégrer la notion d’incertitude de la mesure pour comparer des données provenant de laboratoires différents, de méthodologies différentes, de pays différents…
Utilisation d’outils analytiques adaptés pour fournir des valeursfiables de composition chimique qui permettront le suivi de la qualité de l’environnement, une évaluation réelle des impacts et par la suite d’amener à des prises de décisions sereines et acceptées.
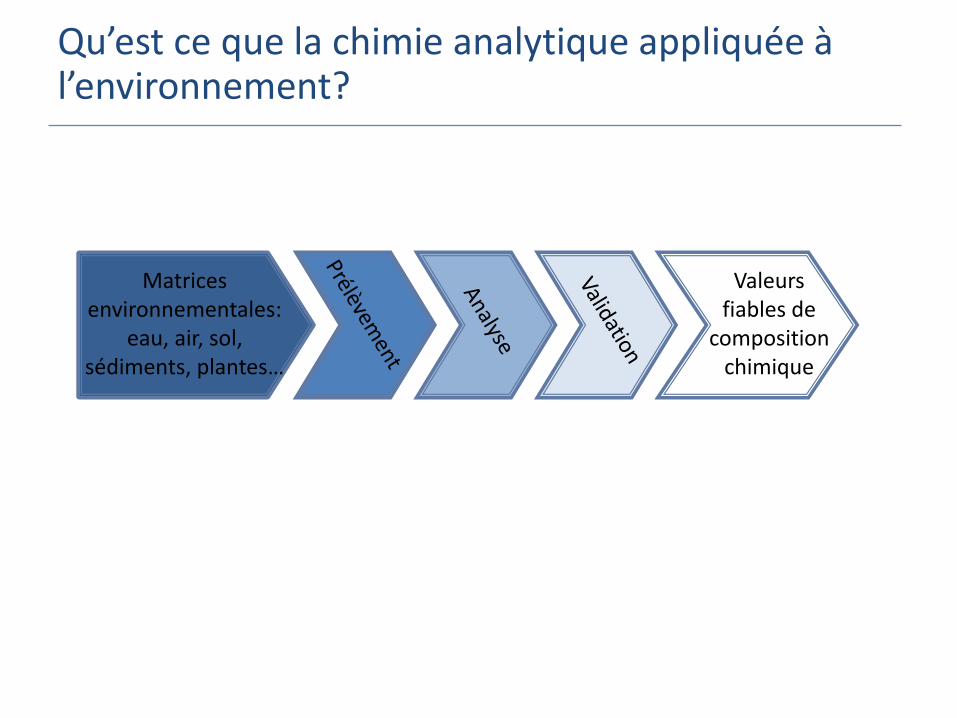
Matrices environnementales:
eau, air, sol, sédiments, plantes…
Valeurs fiables de
composition chimique
Qu’est ce que la chimie analytique appliquée à l’environnement?
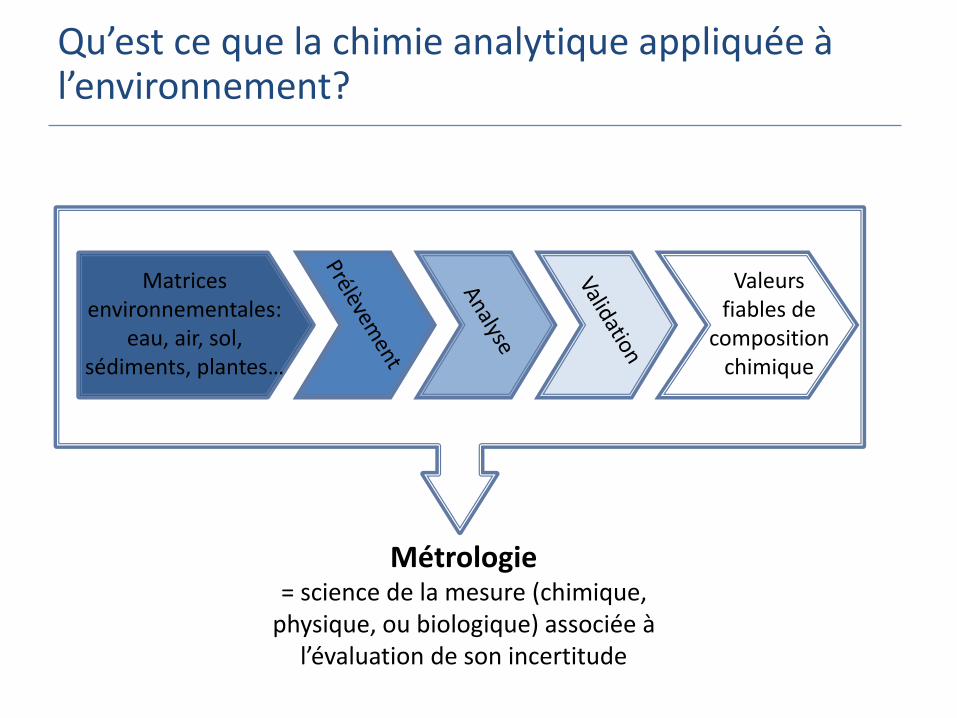
Matrices environnementales:
eau, air, sol, sédiments, plantes…
Valeurs fiables de
composition chimique
Métrologie = science de la mesure (chimique,
physique, ou biologique) associée à l’évaluation de son incertitude
Qu’est ce que la chimie analytique appliquée à l’environnement?
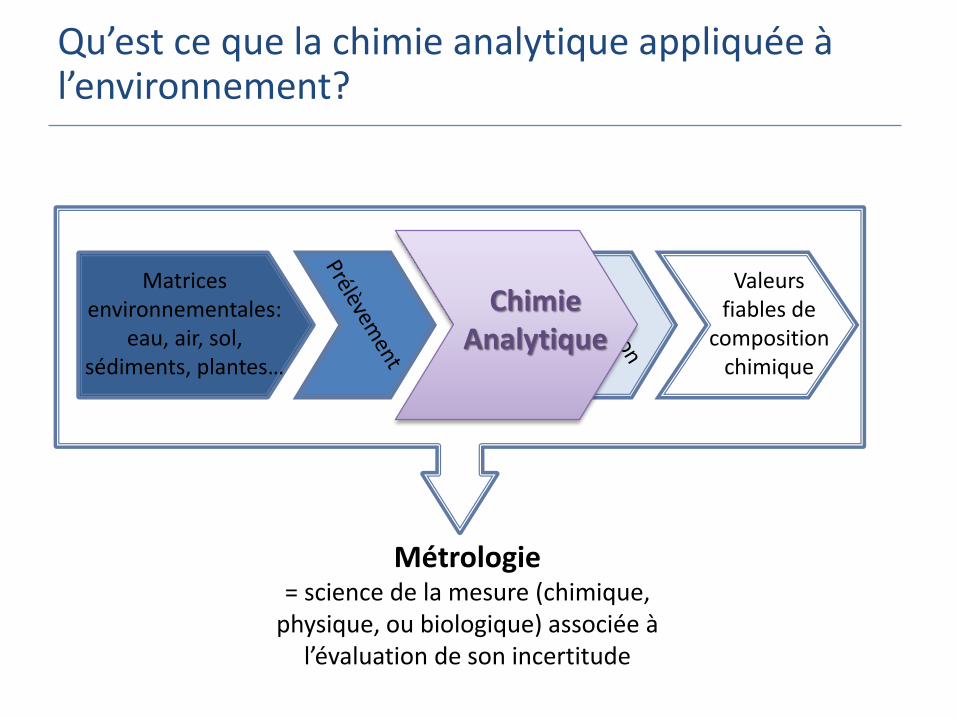
Matrices environnementales:
eau, air, sol, sédiments, plantes…
Valeurs fiables de
composition chimique
Métrologie = science de la mesure (chimique,
physique, ou biologique) associée à l’évaluation de son incertitude
Chimie Analytique
Qu’est ce que la chimie analytique appliquée à l’environnement?

Matrices environnementales:
eau, air, sol, sédiments, plantes…
Valeurs fiables de
composition chimique
Métrologie = science de la mesure (chimique,
physique, ou biologique) associée à l’évaluation de son incertitude
Chimie Analytique
UE Métrologie 2nd semestre
(B. Picquet-Varrault)+
Stage de terrain
Qu’est ce que la chimie analytique appliquée à l’environnement?

Plan du cours
1. Généralités sur l’analyse chimique
2. Préparation de l’échantillon
3. Analyse des métaux
4. Analyse des polluants inorganiques solubles
5. Analyse des polluants organiques
6. Assurance qualité d’une analyse

1. Généralités sur l’analyse chimique: Vocabulaire
Echantillon (sample) = une fraction de l’objet à analyser
Analyte(s) (analyte) = espèce(s) à doser Solutés (solute)= analytes dissous dans un échantillon liquide
Matrice (matrix)= reste des composés présents dans l’échantillon Solvant (solvant)= matrice liquide Matrice aqueuse (aqueous matrix)= quand le solvant est de l’eau Matrice organique (organic matrix)= quand le solvant ou le milieu
sont organiques (huile, feuille,..)
Organique vs inorganique? Organique = qui contient du carbone et de l’hydrogène (H-C) Inorganique = le reste (N, S, …)
Minérale = qui ne contient pas de carbone

1. Généralités sur l’analyse chimique: Pourquoi mesurer
les espèces chimiques dans les différents milieux?
Suivis des espèces réglementées (= polluants) pour leur impact sanitaire ou climatique
Méthodes souvent automatisées
Procédures normalisées
Norme ISO 17381 (2003) = Choix et application des méthodes utilisant des kits prêts à l'emploi en analyse de l'eau
Norme NF EN ISO 17294-2 (2005) = Analyse des métaux lourds dans les boues et les sédiments par ICP-MS.
Suivis de composés traceurs de sources
Suivis d’exposition particulière en cas d’épidémie.
Suivi d’espèces réactives pour comprendre la chimie qui a lieu dans les différents milieux
Méthodes spécifiques et généralement « maison »

Quelles sont les espèces à analyser = espèces réglementées?
Les espèces inorganiques: Les composés « solubles »: nitrates (nitrates), sulfates (sulphates),
chlorures (chlorides), fluorures (fluorides)… Les métaux et métaux lourds (heavy metals) Al, Cd, Cr, Cu, Fe, Pb,
Mo, Ni…
Les espèces organiques: Les hydrocarbures (hydrocarbons): benzène, dichloroéthane, …
Les carbonylés (carbonyl compounds): formaldéhyde, acétone…
Les HAP: Hydrocarbures Aromatiques Polycycliques (PolycyclicAromatic Hydrocarbons)
Les pesticides/herbicides (Pesticide/herbicide)(glyphosate, triazine..) Les espèces organohalogénées (comme organochlorées (organochlorines):
épichlorhydrine, acrylamide, dioxines…)

1.Généralités sur l’analyse chimique: Les différentes étapes d’une analyse
Samplepreparation
MeasurementData
treatment
Mise en condition de l’échantillon pour faire
l’analyse
Etalonnage (calibration) = relation entre le signal mesuré et la concentration recherchée
Obtention d’un signal proportionnel à la teneur en analyte
Obtention d’une concentration fiable en analyte
Différentes techniques de mesures:•Chromatographiques•Spectrométriques•Electrochimiques
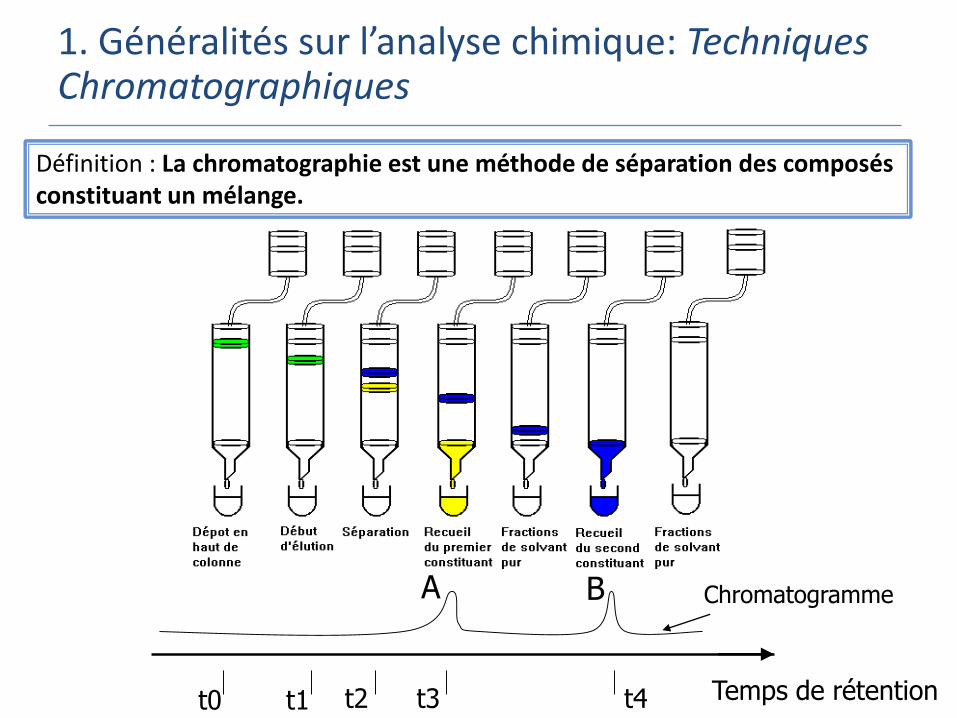
Définition : La chromatographie est une méthode de séparation des composés constituant un mélange.
A B
Temps de rétentiont0 t1 t2 t3 t4
Chromatogramme
1. Généralités sur l’analyse chimique: Techniques Chromatographiques

1906 Le chimiste Tswett sépare des pigments végétaux colorés sur une colonne remplie de carbonate de calcium. Les pigments étaient entraînés avec de l’éther de pétrole .
Formation de bande de couleurs différentes sur la colonne (vert, orange, jaune,…)
Technique devient CHROMATOGRAPHIE(écriture des couleurs)
1940 Martin et Synge : Pratique et théorie de la chromatographie(Prix Nobel en 1952)
1952 Chromatographie en Phase Gazeuse (CPG ou GC)
1968 Chromatographie Liquide Haute Performance (CLHP ou HPLC)
Historique
1. Généralités sur l’analyse chimique: Techniques Chromatographiques
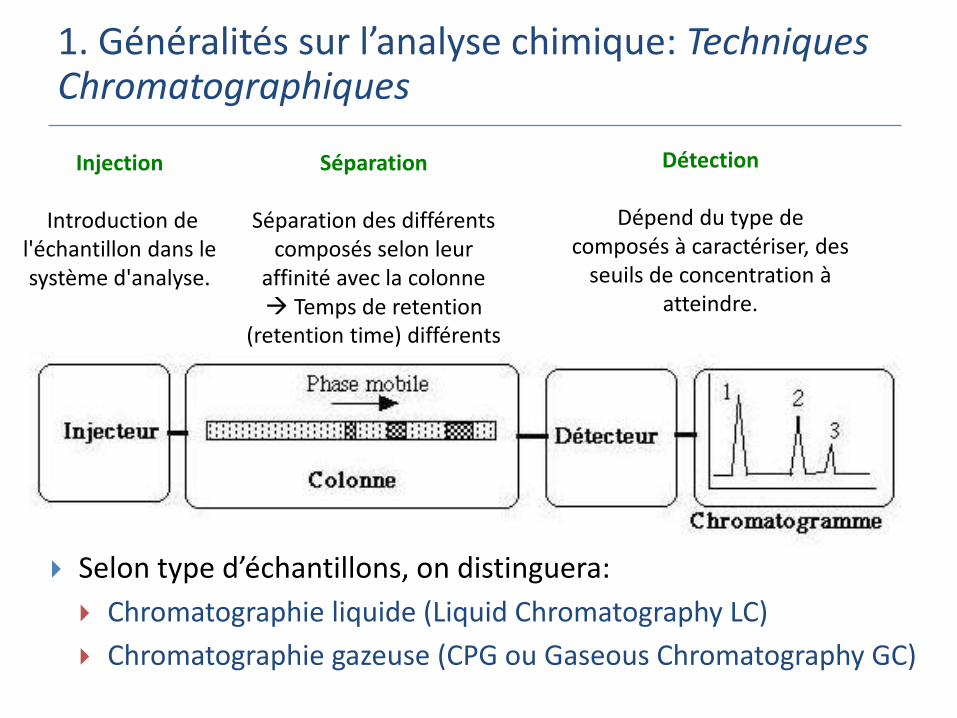
Séparation
Séparation des différents composés selon leur
affinité avec la colonne Temps de retention
(retention time) différents
Injection
Introduction de l'échantillon dans le système d'analyse.
Détection
Dépend du type de composés à caractériser, des
seuils de concentration à atteindre.
1. Généralités sur l’analyse chimique: Techniques Chromatographiques
Selon type d’échantillons, on distinguera:
Chromatographie liquide (Liquid Chromatography LC)
Chromatographie gazeuse (CPG ou Gaseous Chromatography GC)
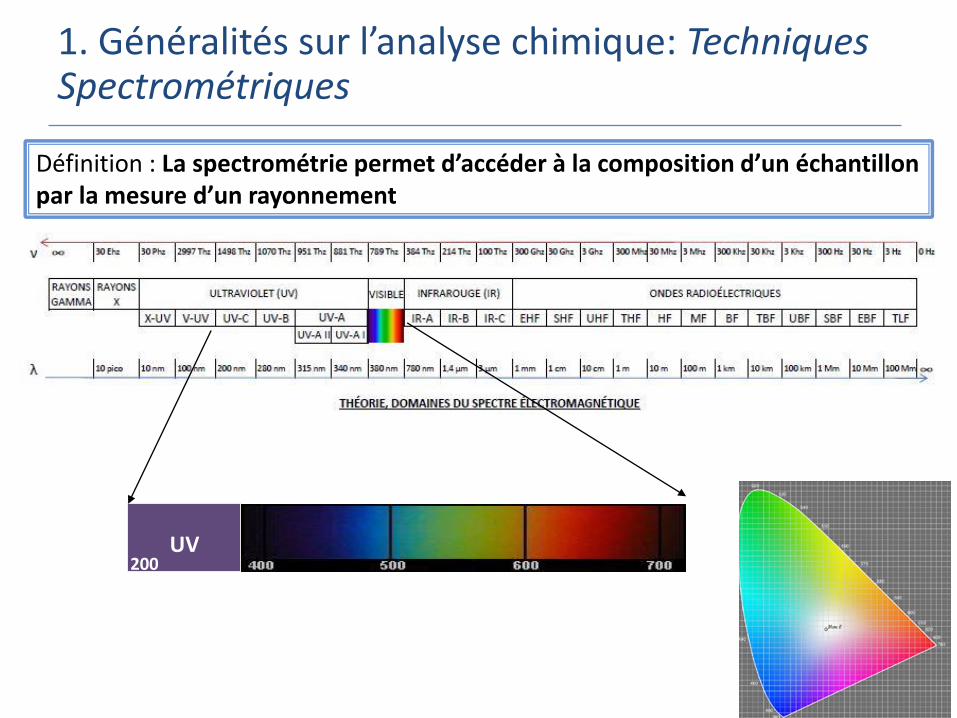
Définition : La spectrométrie permet d’accéder à la composition d’un échantillon par la mesure d’un rayonnement
1. Généralités sur l’analyse chimique: Techniques Spectrométriques
UV200

1. Généralités sur l’analyse chimique: Techniques Spectrométriques
Pour un rayonnement électromagnétique, on mesure:
L’énergie absorbée par un atome ou une molécule:
Spectrométrie UV-Visible
Spectrométrie d’absorption atomique
Spectrométrie à résonance magnétique (RMN)
L’énergie émise par un atome ou une molécule
Spectrométrie d’émission atomique
Spectrométrie de fluorescence
Spectrométrie de luminescence
Pour des ions ou des molécules ionisées, on mesure:
Leur rapport masse sur charge
Spectrométrie de masse

1. Généralités sur l’analyse chimique: Techniques Electrochimiques
Définition : Les méthodes électrochimiques permettent d’accéder à lacomposition d’un échantillon à partir de réactions d’oxydoréductions
Lors d’une réaction de dosage oxydo-reducteur, on mesure soit :
Les charges échangées Coulométrie
Le potentiel d’un couple redox Potentiométrie (pHmétrie)
L’intensité de la réaction d’oxydo-reductionVoltampérométrie/Polarographie
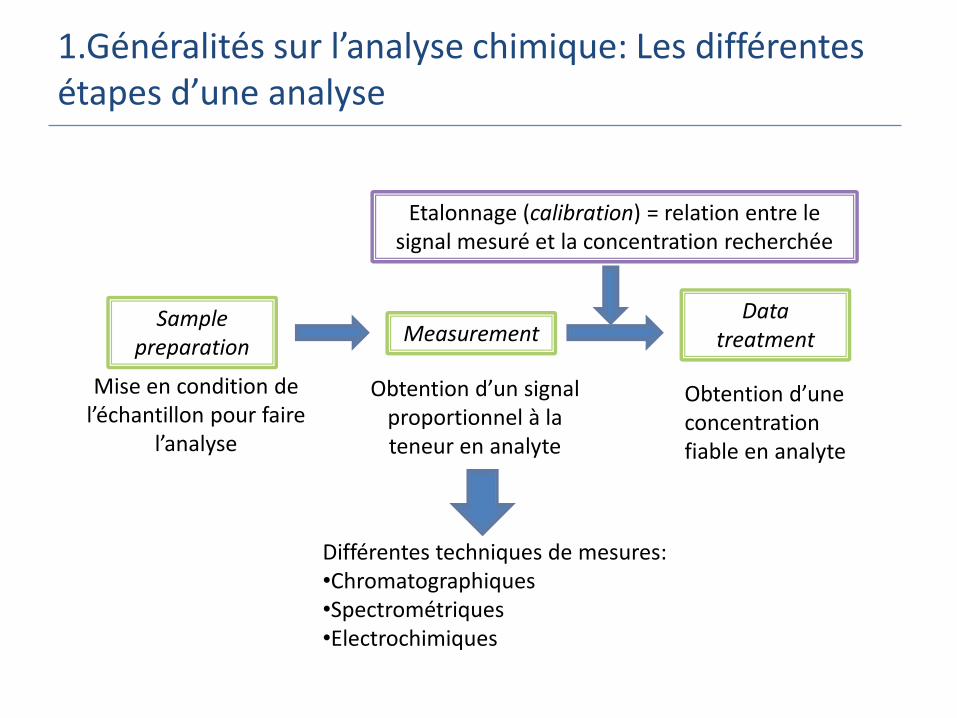
1.Généralités sur l’analyse chimique: Les différentes étapes d’une analyse
Samplepreparation
MeasurementData
treatment
Mise en condition de l’échantillon pour faire
l’analyse
Etalonnage (calibration) = relation entre le signal mesuré et la concentration recherchée
Obtention d’un signal proportionnel à la teneur en analyte
Obtention d’une concentration fiable en analyte
Différentes techniques de mesures:•Chromatographiques•Spectrométriques•Electrochimiques

Plan du cours
1. Généralités sur l’analyse chimique
2. Préparation de l’échantillon
3. Analyse des métaux
4. Analyse des polluants inorganiques solubles
5. Analyse des polluants organiques
6. Assurance qualité d’une analyse

2.Préparation de l’échantillon
Afin de mettre l’analyte à une concentration adaptée à la méthode d’analyse: Dilution Pré-concentration
Afin de l’isoler des autres constituants (Purification): Précipitation Extraction Filtration/ultrafiltration/dialyse
Afin de le changer de matrice: Extraction Mise en solution/Minéralisation
Afin de le rendre détectable (émetteur d’un signal) par la méthode d’analyse: Complexation/Indicateurs colorés Dérivatisation

2.Préparation de l’échantillon
Afin de mettre l’analyte à une concentration adaptée à la méthode d’analyse: Dilution Pré-concentration
Afin de l’isoler des autres constituants (Purification): Précipitation Extraction Filtration/ultrafiltration/dialyse
Afin de le changer de matrice: Extraction Mise en solution/ Minéralisation
Afin de le rendre détectable (émetteur d’un signal) par la méthode d’analyse: Complexation/Indicateurs colorés Dérivatisation

2.Préparation de l’échantillon
Afin de mettre l’analyte à une concentration adaptée à la méthode d’analyse: Dilution Pré-concentration
Afin de l’isoler des autres constituants (Purification): Précipitation Extraction Filtration/ultrafiltration/dialyse
Afin de le changer de matrice: Extraction Mise en solution/ Minéralisation
Afin de le rendre détectable (émetteur d’un signal) par la méthode d’analyse: Complexation/Indicateurs colorés Dérivatisation
2.1. Cas des phase solides• Mise en solution• Extraction
2.2 Cas des phases liquides • Extraction SPE / pré-concentration• Dérivatisation

2.1. Mise en solution (Decomposition into soluble forms)
But: Solubiliser et homogénéiser les espèces à doser à partir d’une matrice solide
Principe: Méthode basée
soit sur la destruction totale de la matrice de solide (total digestion)
soit sur l’extraction des analytes dans une phase liquide à partir de la matrice solide (extraction).
Applications:
Analyse des métaux dans des échantillons solides (boues, sols, aérosols): par des méthodes en voie liquide (ICP)
Analyse des composés organiques (HAP, pesticides, herbicides) dans des échantillons solides (boues, sols, aérosols) par des méthodes en voie liquide (LC)

2.1. Mise en solution: Destruction de la matrice
Pour analyse élémentaire
Si échantillons totalement minéraux :
Mise en solution = dissolution de la matrice minérale
Si échantillons organiques ou mixtes (Cas des échantillons environnementaux):
Minéralisation = décomposition de la matière organique en matière minérale
Mise en solution
Les deux étapes se font simultanément ou pas

2.1. Mise en solution: Minéralisation
Deux voies de minéralisation Par voie sèche:
Par calcination
Par voie humide: Par oxydation/digestion à chaud
Systèmes de chauffage: Répartition homogène de la température
Plaque-chauffantes
Micro-ondes Contrôle de la température
Contrôle de la pression

2.1. Mise en solution: Minéralisation
Protocole Calcination (Ashing) en plusieurs étapes:
1. Séchage (103-105°C): pour ôter l’eau
2. Pesée du résidu à sec
3. Chauffage pour atteindre 450-500°C sur quelques heures
Récupération de cendres (Eléments minéraux sous forme de carbonates ou d'oxydes)
Mise en solution des cendres par dissolution dans acide nitrique ou chlorhydrique
Points critiques: Destruction complète de la matière organique mais perte par
volatilisation (Hg, As, Se, P)
Par voie sèche

2.1. Mise en solution: Minéralisation
Protocole
Oxydation/digestion de l’échantillon
1. Séchage (103-105°C): pour ôter l’eau
2. Pesée du résidu à sec
3. Mise en contact échantillon/réactifs + chauffage
4. Evaporation des réactifs
5. Reprise dans solvant adapté pour l’analyse
Multitude de modes opératoires (types de réactifs, succession ou mélange des réactifs, température de chauffage, temps de contact..)
Problème: La minéralisation est difficilement complète!
Par voie humide

2.1. Mise en solution: Minéralisation
Les principaux réactifs:
Pour la minéralisation:
HClO4, H2O2: oxydants
H2SO4 /HNO3: acides qui accentuent l'effet des oxydants
Pour la mise en solution de la matrice minéralisée:
HF: permet la destruction des matrices alumino-silicatées
Eau régale (Aqua regia: 3 HCl/ 1 HNO3): méthode normalisée
Les principaux contenants:
Borosilicates, quartz ne peuvent être utilisés avec HF
PTFE peut être utilisé pour des acides à bas points d'ébullition car il fond si t°> 250°C
Téflon ne peut être utilisé avec H2SO4, car il le corrode et fond avant son point d'ébullition
Par voie humide
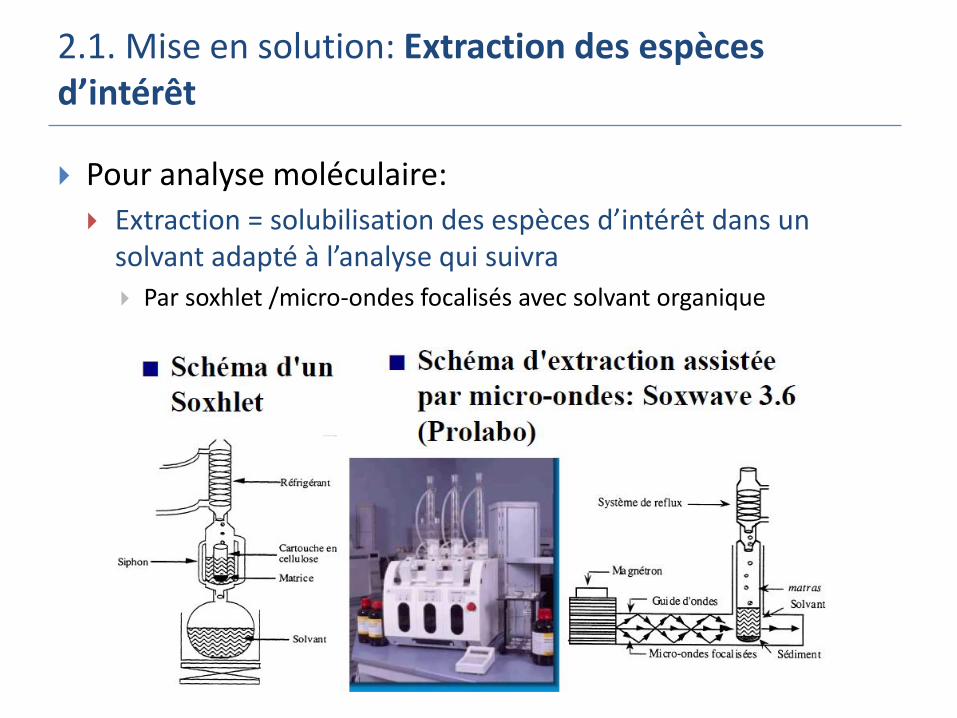
2.1. Mise en solution: Extraction des espèces d’intérêt
Pour analyse moléculaire:
Extraction = solubilisation des espèces d’intérêt dans un solvant adapté à l’analyse qui suivra
Par soxhlet /micro-ondes focalisés avec solvant organique

2.1. Mise en solution: Extraction des espèces d’intérêt
Pour analyse moléculaire:
Extraction = solubilisation des espèces d’intérêt dans un solvant adapté à l’analyse qui suivra
Par soxhlet /micro-ondes folcalisés avec solvant organique
Filtration: pour éliminer la phase solide restante
Concentration: évaporation du solvant d’extraction pour concentrer les analytes dans un plus petit volume

2.1. Mise en solution: En pratique
Méthodes riches en manipulations qui nécessitent des contrôles liées aux:
Risque de contamination :
N’utiliser que des réactifs purs
Réduire la quantité de réactifs
Simplifier les manipulations
Faire des blancs de manipulations
Risque de perte (volatilisation) ou de solubilisation ou d’extraction incomplètes
Vérifier le rendement de mise en solution à l'aide d'échantillons référence

2.Préparation de l’échantillon
Afin de mettre l’analyte à une concentration adaptée à la méthode d’analyse: Dilution Pré-concentration
Afin de l’isoler des autres constituants (Purification): Précipitation Extraction Filtration/ultrafiltration/dialyse
Afin de le changer de matrice: Extraction Mise en solution/ Minéralisation
Afin de le rendre détectable (émetteur d’un signal) par la méthode d’analyse: Complexation/Indicateurs colorés Dérivatisation
2.1. Cas des phase solides• Mise en solution totale• Extraction
2.2 Cas des phases liquides • Extraction SPE / pré-concentration• Derivatisation

2.2. Méthode d’extraction sur phase solide = SPE (Solid Phase Extraction)
But: concentrer ou isoler d’une matrice liquide (ou gazeuse) le composé qu’on veut analyser
Principe: méthode basée sur l’adsorption sur une phase solide (adsorbant, adsorbent) des composés d’intérêt présents dans un échantillon liquide (ou gazeux)
Applications:
Analyse des herbicides/pesticides/hydrocarbures/HAP dans les eaux de surface: pour les concentrer avant analyse en HPLC ou LC-MS
Analyse des métaux/polluants organiques dans l’eau de mer: pour les isoler de la matrice « salée » (effet de matrice) avant analyse….
Analyse des composés organiques volatiles dans l’air : pour les capturer et les concentrer avant analyse
Schéma d’une cartouche SPE (cartridge)
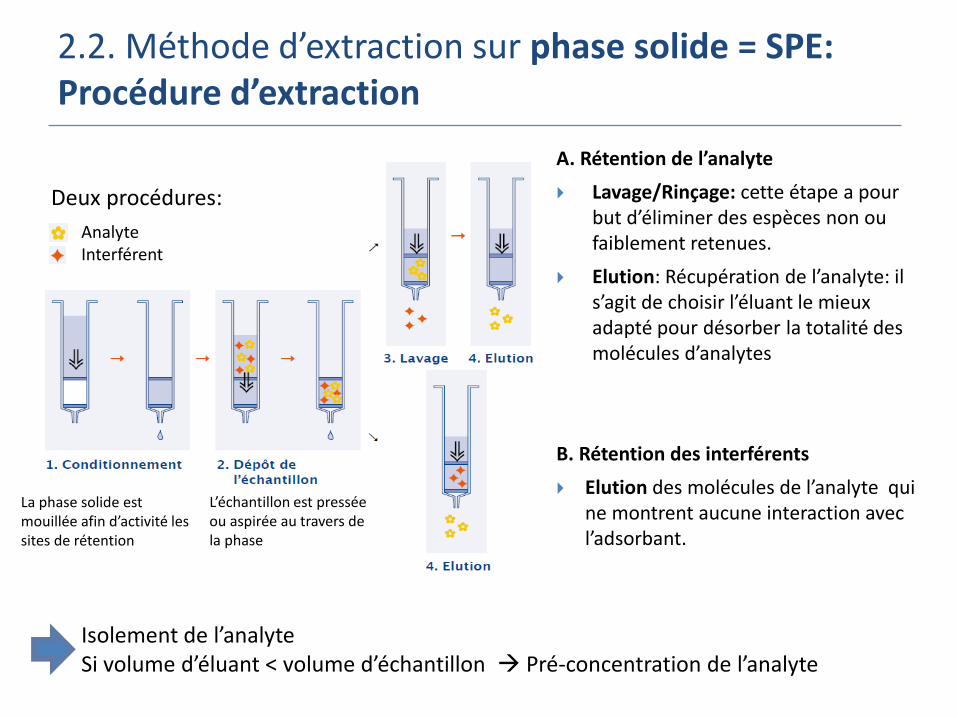
2.2. Méthode d’extraction sur phase solide = SPE:Procédure d’extraction
A. Rétention de l’analyte
Lavage/Rinçage: cette étape a pour but d’éliminer des espèces non ou faiblement retenues.
Elution: Récupération de l’analyte: il s’agit de choisir l’éluant le mieux adapté pour désorber la totalité des molécules d’analytes
B. Rétention des interférents
Elution des molécules de l’analyte qui ne montrent aucune interaction avec l’adsorbant.
Isolement de l’analyte Si volume d’éluant < volume d’échantillon Pré-concentration de l’analyte
La phase solide est mouillée afin d’activité les sites de rétention
L’échantillon est pressée ou aspirée au travers de la phase
Deux procédures:
AnalyteInterférent

2.2. Méthode d’extraction sur phase solide = SPE: Les adsorbants
Le choix de l’adsorbant permet de définir une sélectivité spécifique aux composés d’intérêt ainsi qu’une capacité de charge suffisante à l’entière adsorption de ceux-ci.
Les adsorbants sont souvent des gels avec une granulométrie entre 40 et 100 µm pour permettre la percolation.
Deux grandes familles : les silices vierges ou greffées (85% des cas): très sélectives mais faible
capacité de charge (faible surface spécifique), pH compris entre 2 et 7,5
les polymères: faiblement sélectifs mais très stables chimiquement, pH
compris entre 1 et 14 et capacité de charge bien supérieure aux silicesAmberlite (XAD)
(Carbone graphite)
Perles de gel de silice
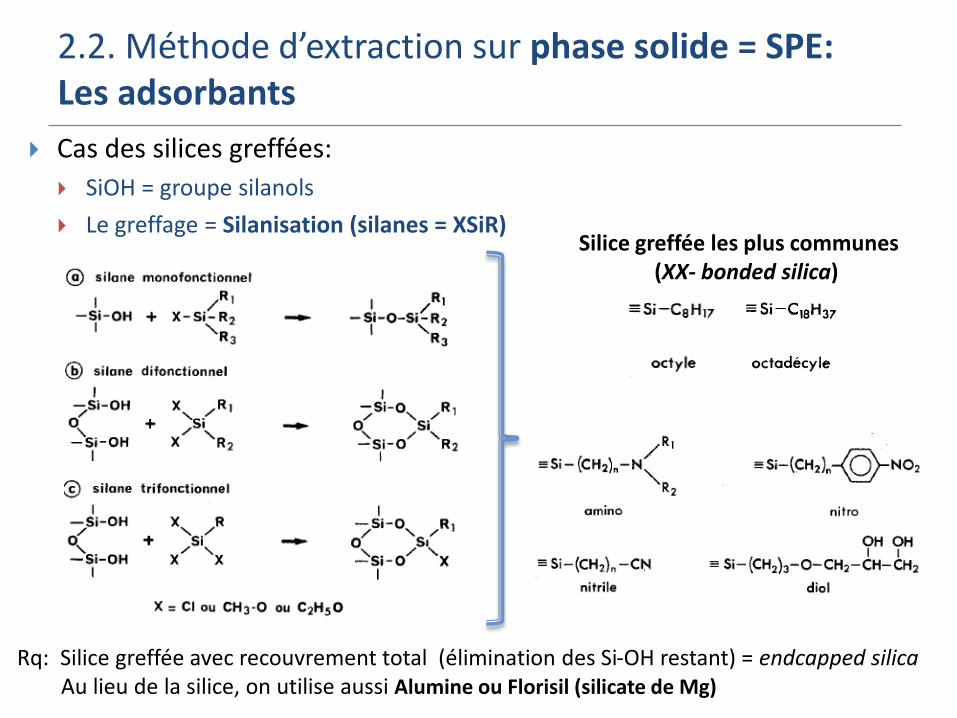
2.2. Méthode d’extraction sur phase solide = SPE: Les adsorbants
Cas des silices greffées:
SiOH = groupe silanols
Le greffage = Silanisation (silanes = XSiR)Silice greffée les plus communes
(XX- bonded silica)
Rq: Silice greffée avec recouvrement total (élimination des Si-OH restant) = endcapped silicaAu lieu de la silice, on utilise aussi Alumine ou Florisil (silicate de Mg)

2.2. Méthode d’extraction sur phase solide = SPE: Les adsorbants
Le choix de l’adsorbant permet de définir une sélectivité spécifique aux composés d’intérêt ainsi qu’une capacité de charge suffisante à l’entière adsorption de ceux-ci.
Les adsorbants sont souvent des gels avec une granulométrie entre 40 et 100 µm pour permettre la percolation.
Deux grandes familles : les silices vierges ou greffées (85% des cas): très sélectives mais faible
capacité de charge (faible surface spécifique), pH compris entre 2 et 7,5
les polymères: faiblement sélectifs mais très stables chimiquement, pH
compris entre 1 et 14 et capacité de charge bien supérieure aux silicesAmberlite (XAD)
(Carbone graphite)
Perles de gel de silice
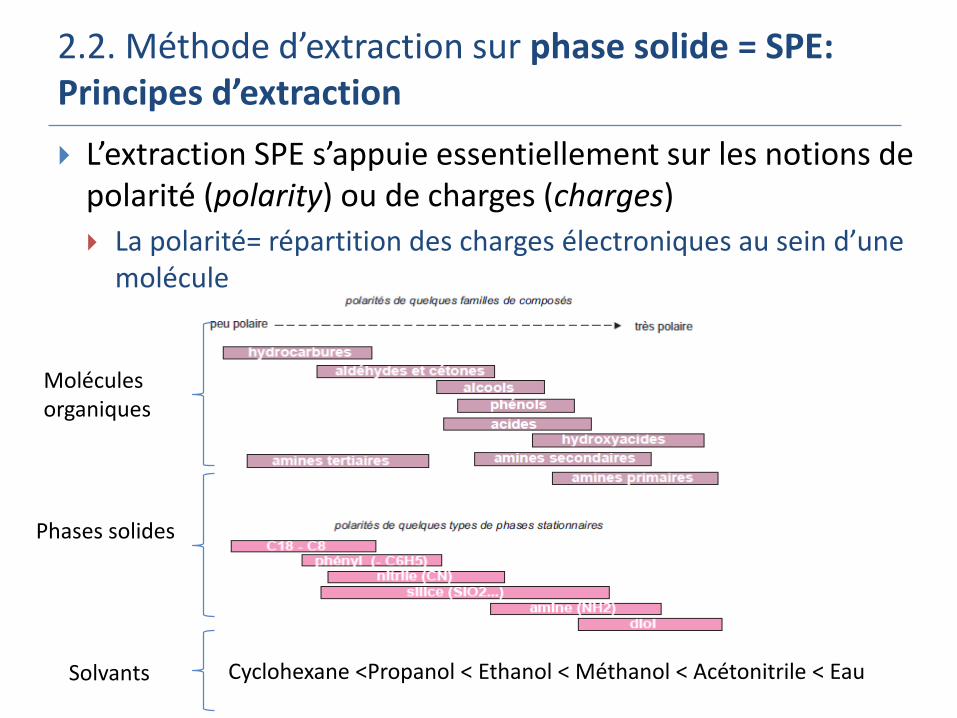
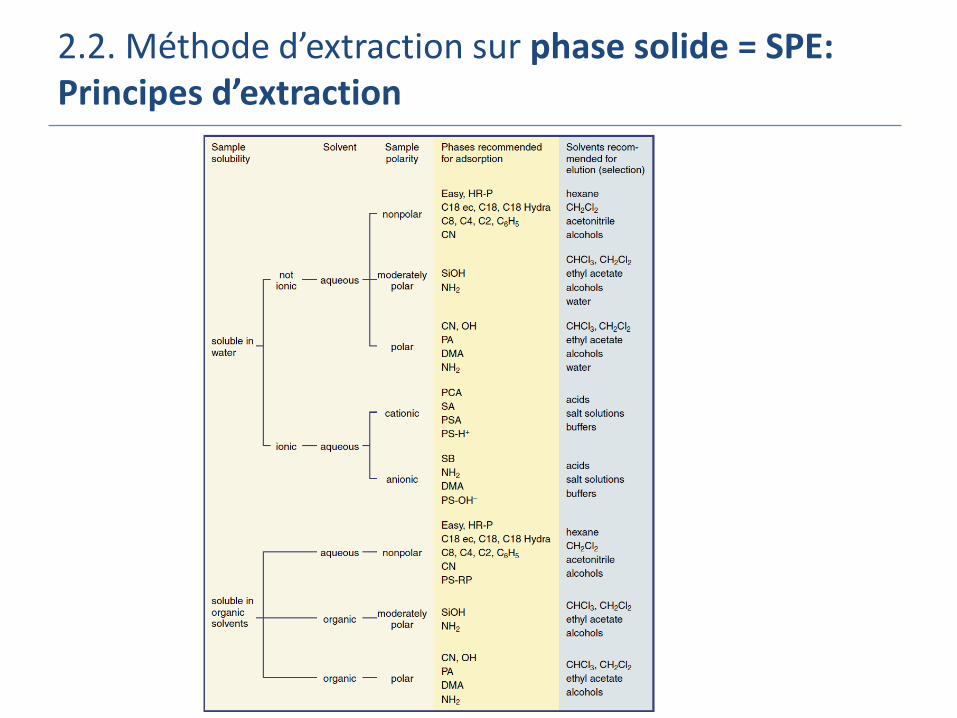
2.2. Méthode d’extraction sur phase solide = SPE: Principes d’extraction
L’extraction SPE s’appuie essentiellement sur les notions de polarité (polarity) ou de charges (charges)
La polarité= répartition des charges électroniques au sein d’une molécule
Molécules organiques
Phases solides
Cyclohexane <Propanol < Ethanol < Méthanol < Acétonitrile < Eau Solvants

2.2. Méthode d’extraction sur phase solide = SPE: Principes d’extraction
3 mécanismes d’extraction:
les molécules polaires s’attirent et inversement! Phase inversée (Reversed phase): extraction d’analytes peu polaires ou
apolaires (hydrocarbures, HAP, pesticide peu polaires ..) Echantillon liquide polaire = typiquement matrice aqueuse
Phase solide = phase non polaire (C18, C8, XAD)
Eluant = solvant non polaire ou moins polaire que l’eau

2.2. Méthode d’extraction sur phase solide = SPE: Principes d’extraction
Phase inverse

2.2. Méthode d’extraction sur phase solide = SPE: Principes d’extraction
3 mécanismes d’extraction:
les molécules polaires s’attirent et inversement! Phase inversée (Reversed phase): extraction d’analytes peu
polaires ou apolaires (hydrocarbures, HAP, pesticide peu polaires ..) Echantillon liquide polaire = typiquement matrice aqueuse
Phase solide = phase non polaire (C18, C8, XAD)
Eluant = solvant non polaire ou moins polaire que l’eau
Phase normale: extraction d’analytes polaires (pesticides polaires) Echantillon liquide mi ou non polaire (huile, hexane,…)
Phase solide = phase polaire (Si greffée –CN, -NH2 et –diol, silice vierge, alumine, florisil)
Eluant = solvant polaire ou plus polaire que la matrice
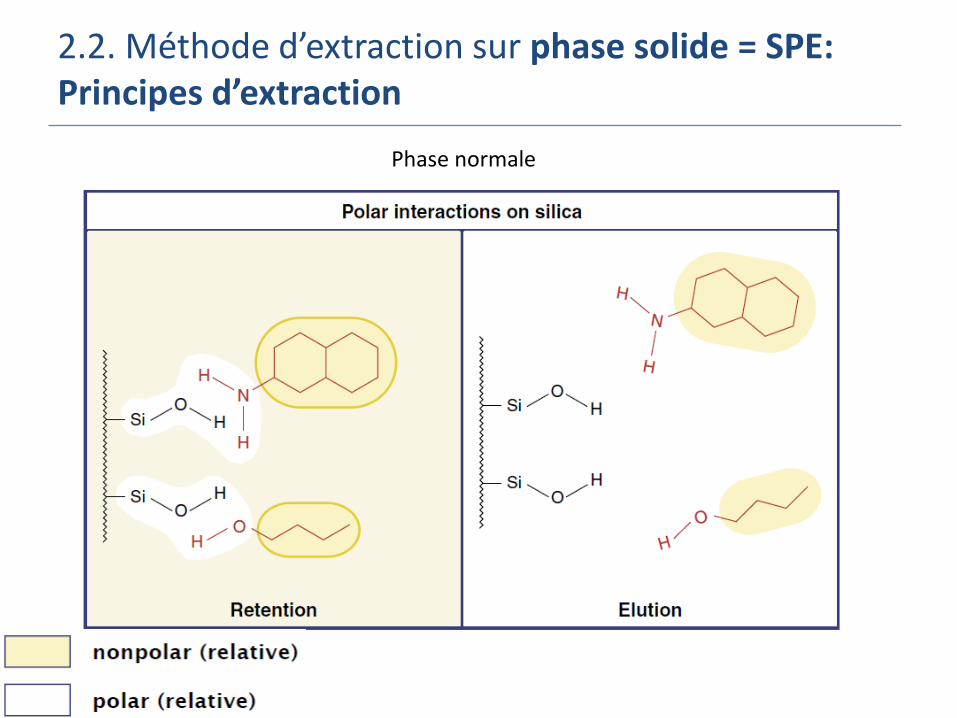
2.2. Méthode d’extraction sur phase solide = SPE: Principes d’extraction
Phase normale

2.2. Méthode d’extraction sur phase solide = SPE: Principes d’extraction
3 mécanismes d’extraction:
les molécules polaires s’attirent et inversement! Phase inversée (Reversed phase): extraction d’analytes peu polaires ou
apolaires (hydrocarbures, HAP, pesticide peu polaires ..) Echantillon liquide polaire = typiquement matrice aqueuse
Phase solide = phase non polaire (C18, C8, XAD)
Eluant = solvant non polaire ou moins polaire que l’eau
Phase normale: extraction d’analytes polaires (pesticides polaires) Echantillon liquide mi ou non polaire (huile, hexane,…)
Phase solide = phase polaire (Si greffée –CN, -NH2 et –diol, silice vierge, alumine, florisil)
Eluant = solvant polaire ou plus polaire que la matrice
Echange d’ion: extraction de composés chargés (métaux, acides/bases) Echantillon liquide aqueux ou organique
Phase solide chargée (Si greffée –SO3- and –N+(CH3)3 (SAX))
Eluant = eau à différents pH ou à forte force ionique
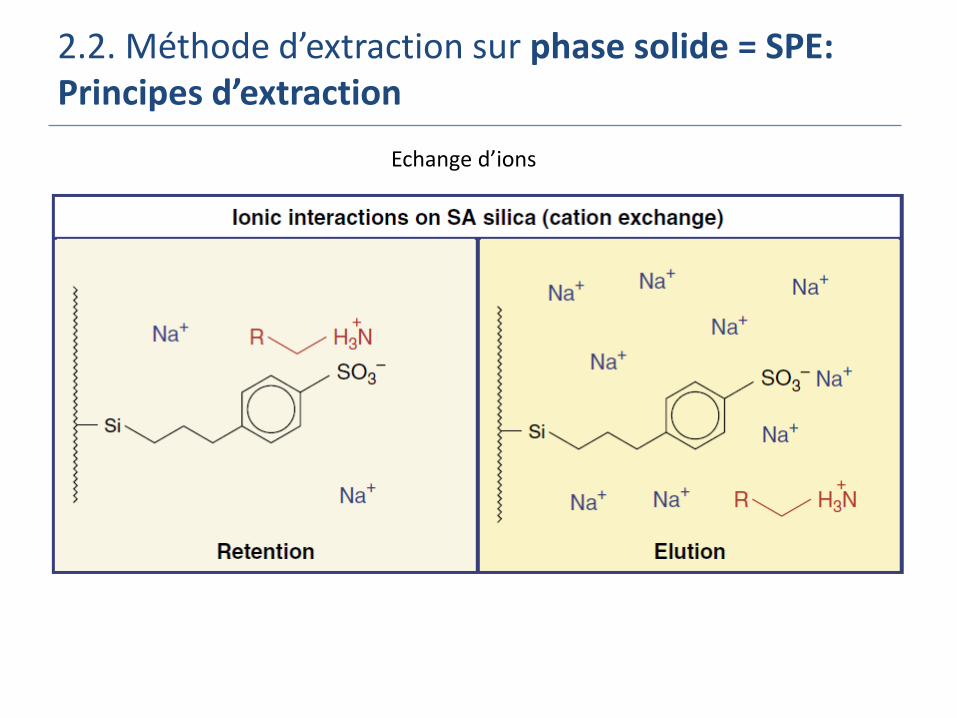
2.2. Méthode d’extraction sur phase solide = SPE: Principes d’extraction
Echange d’ions
2.2. Méthode d’extraction sur phase solide = SPE: Principes d’extraction

2.2. Méthode d’extraction sur phase solide = SPE:En pratique
Les points clé d’une bonne extraction
Vérifier le volume de percée de la résine (ou volume de fin de fixation ) = volume au-delà duquel une partie des analytes n’est plus retenue sur la cartouche
Utiliser des modules d’extraction en cas de gros volumes d’échantillons
Le débit de rétention et d’élution
Estimer le rendement d’extraction
Extraction peut se faire:
Hors ligne
En ligne = couplage SPE-LC, SPE-GC…

2.2. Dérivatisation But: rendre détectable dans une matrice liquide (gazeuse) le composé
qu’on veut analyser
Principe: méthode basée sur la complexation de l’espèce à analyser de façon à la rendre absorbante d’un rayonnement
Applications:
Analyse des espèces carbonylés par HPLC dans les matrices aqueuses ou après extraction en phase liquide

Plan du cours
1. Généralités sur l’analyse chimique
2. Préparation de l’échantillon
3. Analyse des métaux
4. Analyse des polluants inorganiques
5. Analyse des polluants organiques
6. Assurance qualité d’une analyse

3. Analyse des métaux
Les métaux lourds (heavy metals) ou éléments traces métalliques (ETM / trace metals) = métaux présents à l’état de trace (Ag, Cd, Cr, Cu, Pb, Hg, Ni..) +
éléments non métalliques (As, Se..), qui sont classés comme substances dangereuses
Souvent oligo-éléments mais toxiques à fortes doses (Cu, Ni,..) ou seulement toxiques (Pb, Cd, Sb..) Toxicité dépend de leur forme chimique :
Chrome hexavalent : Cr6+, CrO42-, Cr2O7
2-, CrO3, ...
Mercure: Hg0, Hg2Cl2, (CH3)2Hg…
Fraction labile = Fraction biodisponible
Ubiquistes dans les différents milieux: sol, eau, air (particules) Problématique d’accumulation
Sources: combustion charbon/pétrole/usine d’incinération + industries minières

3. Analyse des métaux:
But: Analyses des métaux et métaux lourds
Techniques applicables aux phases liquides et solides (après mise en solution):
Spectrométrie atomique d’absorption
Spectrométrie d’émission
Spectrométrie de masse
Techniques applicables aux phases solides:
Spectrométrie de rayons X
Couplage avec la technologie plasma: ICP-AES et ICP-MS

3.1. Analyses élémentaires: Les méthodes ICP
ICP: Inductively Coupled Plasma Techniques basées sur la technologie Plasma But: Faire passer toutes les molécules sous forme élémentaire Principe: technologie couplée avec des méthodes spectrométriques
ICP-AES: ICP-Atomic Emission Spectrometry Couplage avec Spectrométrie d’Emission Atomique
ICP-MS: ICP-Mass Spectrometry Couplage avec Spectrométrie de Masse
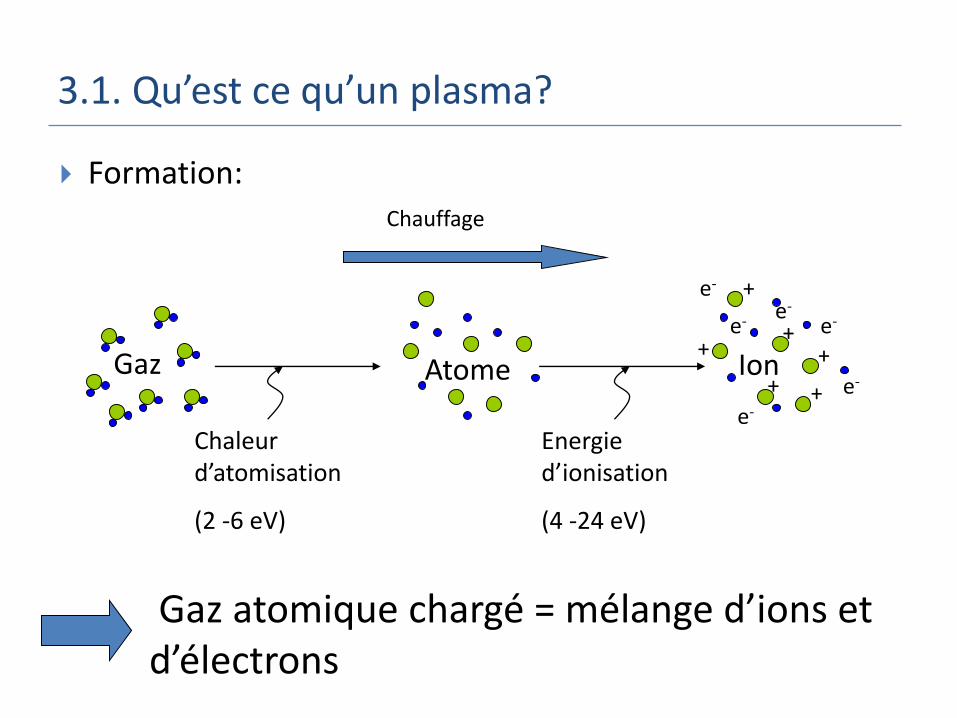
3.1. Qu’est ce qu’un plasma?
Formation:
+e-
Gaz atomique chargé = mélange d’ions et d’électrons
IonGaz Atome
Chaleur d’atomisation
(2 -6 eV)
Energie d’ionisation
(4 -24 eV)
+
+ +
++e-
e-
e-
e-e-
Chauffage

3.1. Energie d’ionisation (eV)
4.34 6.82 7.90 7.72 9.78 13.99
15.75
10.36Les gaz rares sont les plus difficilement ionisables

3.1. En pratique…
Plasma existe sur une large gamme de température ou de densité
Température d’initiation dépend de la substance à ioniser: 4 000 K pour les éléments facilement ionisables (Cs)
20 000K pour les éléments difficilement ionisables (He, gaz rares..)
En pratique, plasmas synthétiques utilisent autres techniques que le chauffage (décharge électrique, laser…)


3.1. En pratique…
Plasma existe sur une large gamme de température ou de densité
Température d’initiation dépend de la substance à ioniser: 4 000 K pour les éléments facilement ionisables (Cs)
20 000K pour les éléments difficilement ionisables (He, gaz rares..)
En pratique, plasmas synthétiques utilisent autres techniques que le chauffage (décharge électrique, laser…)

3.1. Application en Chimie Analytique
Source très chaude Température plus élevée que celle produite par les
flammes ou les décharges électriques (arc/étincelle), donc plus efficace
Méthode Température (°C)
Flamme 1700-3150
Electrothermie 2200-3000
Plasma 6500-10000
Arc électrique 4000-5000

3.1. Application en Chimie Analytique
Source très chaude Température plus élevée que celle produite par les flammes
ou les décharges électriques (arc/étincelle), donc plus efficace
Donc très énergétique: on y atteint facilement les chaleurs d’atomisation des
molécules et les énergie d’ionisation et d’excitation des atomes
Utiliser pour la détermination de la composition chimique élémentaire

3.1. Application en Chimie Analytique
Le plasma permet ainsi de:
casser les liaisons moléculaires
produire des atomes et des ions libres
d'exciter ces atomes/ions
Source d’ions Spectrométrie de Masse
Source de photons Spectrométrie d’émission atomique
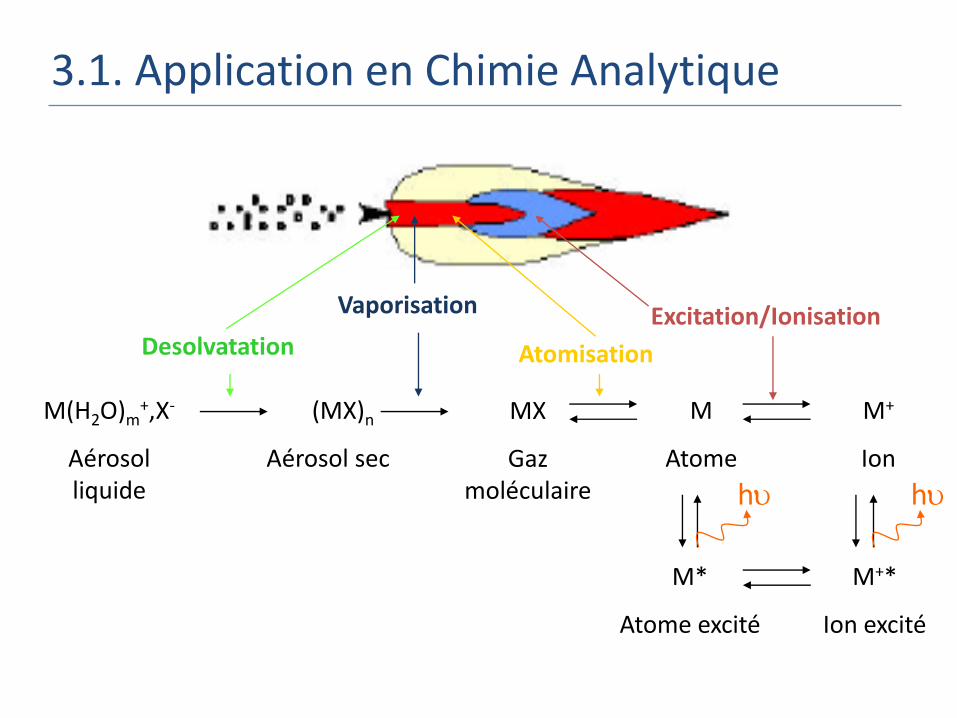
3.1. Application en Chimie Analytique
M(H2O)m+,X-
Aérosol liquide
(MX)n
Aérosol sec
M
Atome
M+
Ion
MX
Gaz moléculaire
Desolvatation
Vaporisation
Atomisation
Excitation/Ionisation
M*
Atome excité
M+*
Ion excité
h h

3.1. En pratique : 1.Gaz utilisé?
L’argon est le gaz le plus usité pour les plasmas en chimie analytique car
Potentiel d ’ionisation ~16 eV : on utilise gaz rare car les plus difficilement ionisables donc plasma avec une température très élévée
Températures de 10000 K
Ar gaz rare le plus courant sur la terre… He : OK, mais cher
Gaz rare monoatomique : spectre simple, ne se recombine pas avec les espèces chimiques d'une façon stable
Inconvénient : mauvaise conductibilité thermique donc peu stable Necéssité de le perturber le moins possible si on veut avoir des conditions
répétables, notamment en température

3.1. En pratique : 1.Gaz utilisé?
4.34 6.82 7.90 7.72 9.78 13.99
15.75
10.36Energie d’ionisation (eV)

Potentiel d’ionisation d’un plasma d’argon
0 10 20 30 40 50 60 70 80 900
20
40
60
80
100
Nombre atomique
Effi
caci
té d
'ion
isat
ion
(%
)
Li
Be
B
C
BaMg
Al
Si
P
S
Cl
K
Ca
Sc V
Ti
Cr
Mn
Zn
Ga
GeCu
Fe
Ni
Co
As
Se
Br
Rb
Sr
Y
Zr
Nb
Mo
Tc
Ru
Rh
Pd
Ag
Cd
In
Sn
Sb
Te
I
Xe
CsNa Lanthanoides Hf
Ta
W
Re
Os
Ir
Pt
Au
Hg
Tl Pb
Bi
Po
Rn
Ra
Ac
N FKrHe
ONe Ar
Electron temperature : 6,680K
Electron density : 1.4714 x 10 cm-1

Elements peu ou pas ionisables en plasma d’Ar

3.1. En pratique : 1.Gaz utilisé?
L’argon est le gaz le plus usité pour les plasmas en chimie analytique car
Potentiel d ’ionisation ~16 eV : on utilise gaz rare car les plus difficilement ionisables donc plasma avec une température très élévée
Températures de 10000 K
Ar gaz rare le plus courant sur la terre… He : OK, mais cher
Gaz rare monoatomique : spectre simple, ne se recombine pas avec les espèces chimiques d'une façon stable
Inconvénient : mauvaise conductibilité thermique donc peu stable Necéssité de le perturber le moins possible si on veut avoir des conditions
répétables, notamment en température
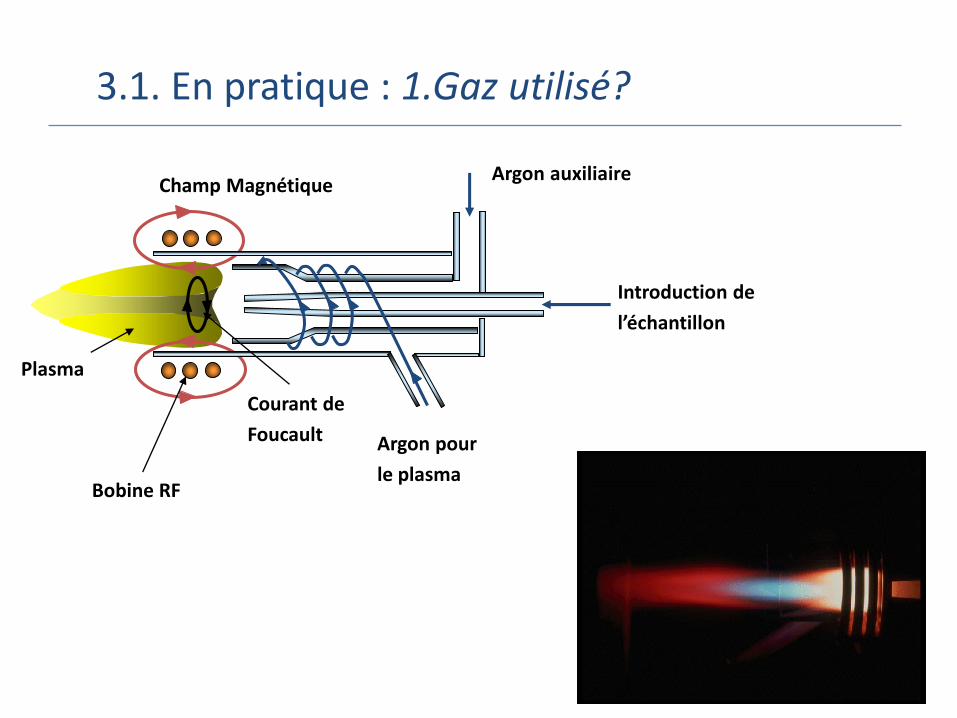
3.1. En pratique : 1.Gaz utilisé?
Argon pour
le plasma
Introduction de
l’échantillon
Argon auxiliaire
Bobine RF
Champ Magnétique
Courant de
Foucault
Plasma

3.1. En pratique : 2. Introduction de l’échantillon vers le plasma
Échantillon : Liquide
Echantillon liquide, nécessité de mise en solution = minéralisation

3.1. En pratique : 2. Introduction de l’échantillon vers le plasma
Échantillon : Liquide
Echantillon liquide, nécessité de mise en solution = minéralisation….puis:
Étape 1 : Génération de l’aérosol = Nébulisation
Étape 2 : Introduction de l’aérosol dans le plasma = Sélection des aérosols
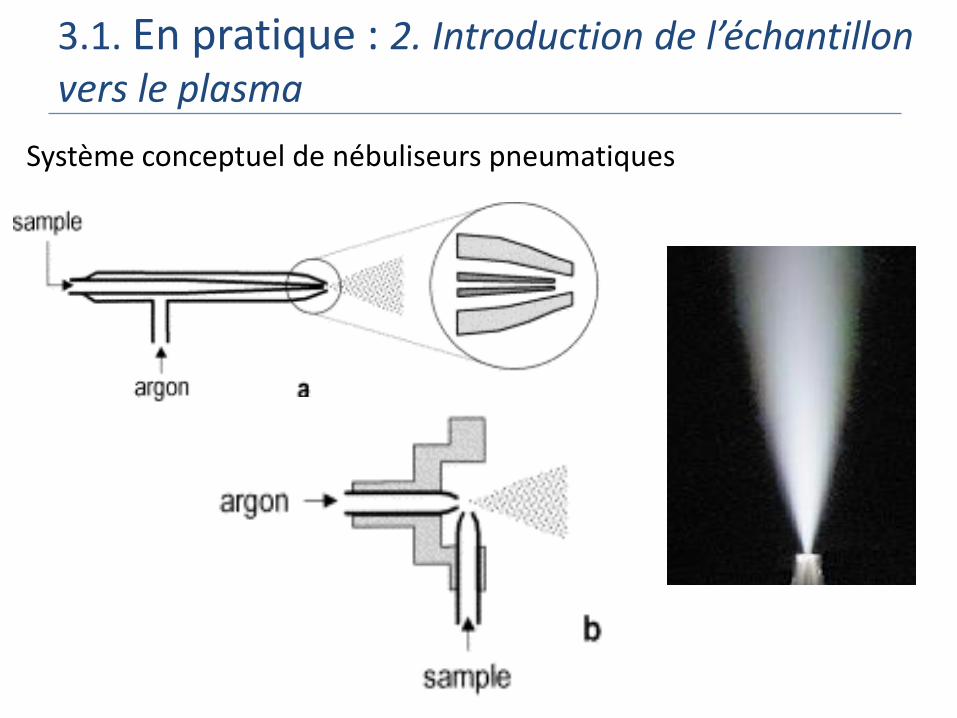
3.1. En pratique : 2. Introduction de l’échantillon vers le plasma
Système conceptuel de nébuliseurs pneumatiques

3.1. En pratique : 2. Introduction de l’échantillon vers le plasma
Échantillon : Liquide
Echantillon liquide, nécessité de mise en solution = minéralisation….puis:
Étape 1 : Génération de l’aérosol = Nébulisation
Étape 2 : Introduction de l’aérosol dans le plasma = Sélection des aérosols = Chambre de conditionnement

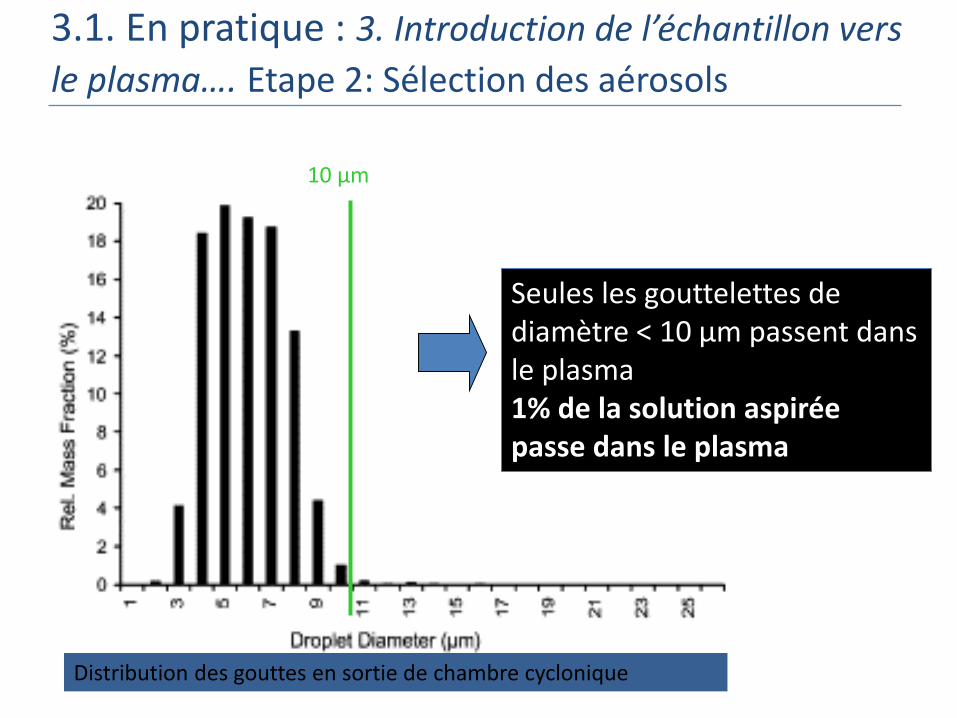
Distribution des gouttes en sortie de nébuliseur Meinhard
10 µm
Distribution des gouttes en sortie de chambre cyclonique
Seules les gouttelettes de diamètre < 10 μm passent dans le plasma1% de la solution aspirée passe dans le plasma
3.1. En pratique : 3. Introduction de l’échantillon vers
le plasma…. Etape 2: Sélection des aérosols
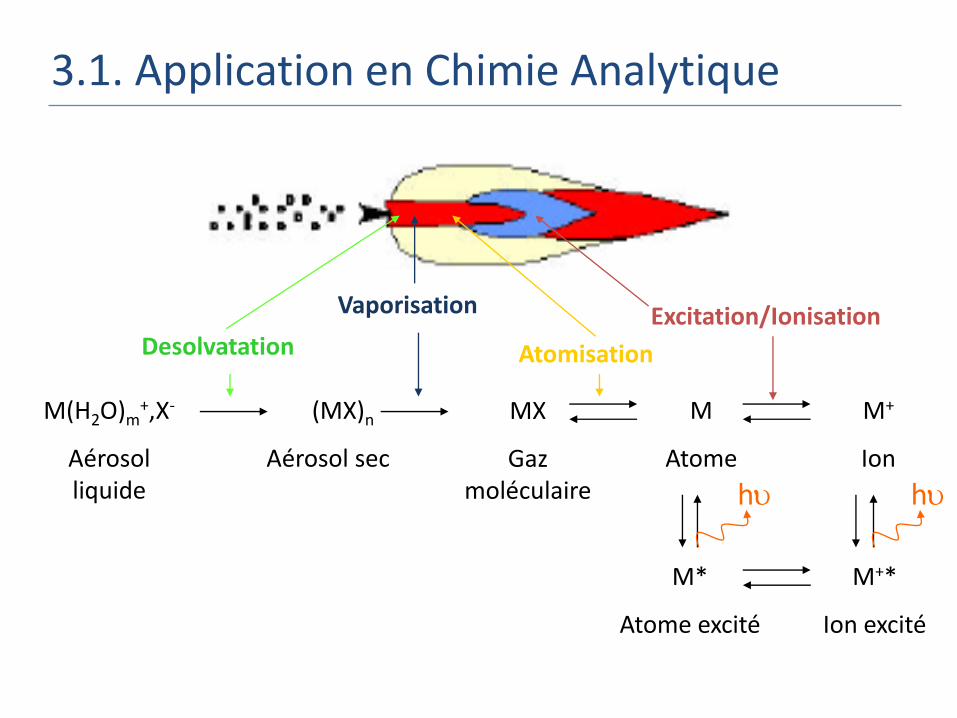
3.1. Application en Chimie Analytique
M(H2O)m+,X-
Aérosol liquide
(MX)n
Aérosol sec
M
Atome
M+
Ion
MX
Gaz moléculaire
Desolvatation
Vaporisation
Atomisation
Excitation/Ionisation
M*
Atome excité
M+*
Ion excité
h h

3.1. En pratique : 4. Connexion avec la spectrométrie
Excitation/IonisationTransfert de charge
Atomes et ions excités Ions
MSAES

A. ICP-AESInductively Coupled Plasma-Atomic Emission Spectrometry

3.1.A ICP-AES:
ICP-AES ou encore OES, pour Optical Emission, car les raies analysées sont souvent des raies ioniques et pas seulement atomiques.
ICP = Source de photons
AES = Compteur de photons

3.1.A ICP-AES: Emission atomique: Principe
Le retour à un état moins énergétique s ’accompagne d ’une émission de photons
Une longueur d’onde unique est associée à chaque transition et est donc typique d’un élément donné:
En – Ep = h. = hc/λ
h : cte de Planck (6.63 x 10-34 J.s), c : célérité de la lumière (3 x 108 m/s)
: fréquence (Hz), λ : longueur d ’onde (m)

3.1.A ICP-AES: Emission atomique: Principe
Lors de l’excitation, plusieurs transitions électroniques possibles:
Les longueurs d’ondes faibles (UV) correspondent aux transitions les plus énergétiques
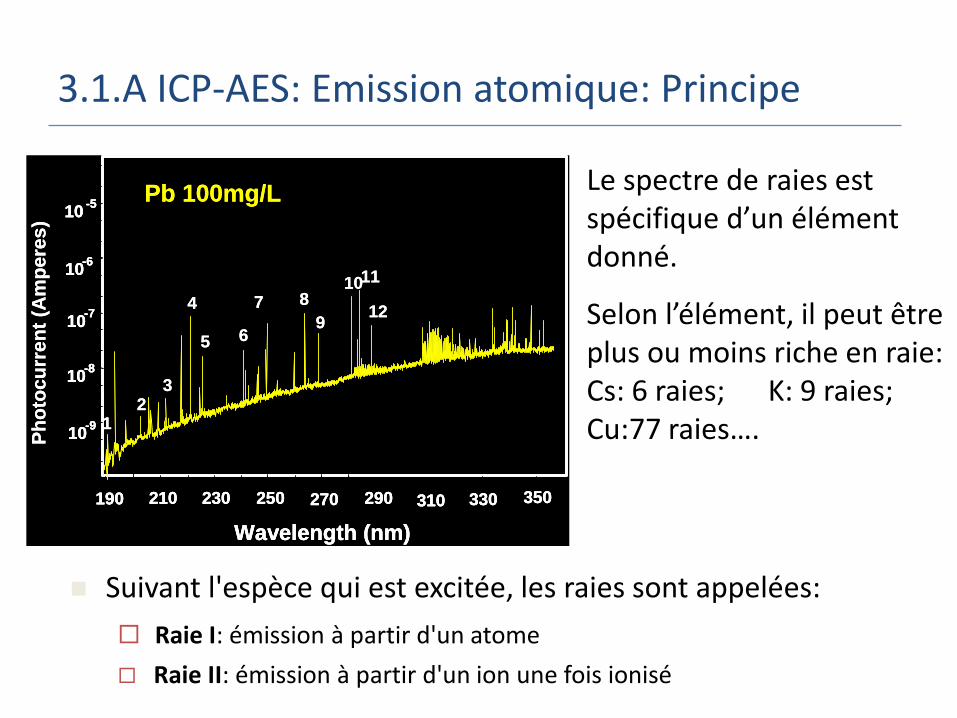
3.1.A ICP-AES: Emission atomique: Principe
Ph
oto
cu
rre
nt
(Am
pe
res
)
-9 10
-8 10
-7 10
-6 10
-5 10
Wavelength (nm)
190 270 310 330230 250 290210 350
Pb 100mg/L
12
3
4
5 6
7 8
912
1110
Ph
oto
cu
rre
nt
(Am
pe
res
)
-9 10
-8 10
-7 10
-6 10
-5 10
-9 10
-8 10
-7 10
-6 10
-5 10
Wavelength (nm)
190 270 310 330230 250 290210 350
Wavelength (nm)
190 270 310 330230 250 290210 350
Pb 100mg/L
12
3
4
5 6
7 8
912
1110
Le spectre de raies est spécifique d’un élément donné.
Selon l’élément, il peut être plus ou moins riche en raie: Cs: 6 raies; K: 9 raies; Cu:77 raies….
Suivant l'espèce qui est excitée, les raies sont appelées:
Raie I: émission à partir d'un atome
Raie II: émission à partir d'un ion une fois ionisé

3.1.A ICP-AES: Emission atomique: Principe
Intensité de la raie est :
• spécifique de la longueur d’onde
• proportionnelle à la concentration d’un élément donné…
0
20
40
60
80
100
0 0 .2 0 .4 0 .6 0 .8 1
Concentration (c )
Sig
na
l (S
)
m2
m1
Sbl
267.531267.531 267.779267.779
648648
2k2k
Nb
co
up
s
Cu 224.700
Cu 324.754
Les raies ioniques (II) sont les plus utilisées pour les faibles concentrations car ce sont les plus sensibles…
Cu 224.700
Cu 324.754Cu 224 Cu 324

ICP AES
3.1.A ICP-AES: Schéma-principe
2. Focalisation
3. Tri en λ
4. Détection
1. Interface ICP-AES
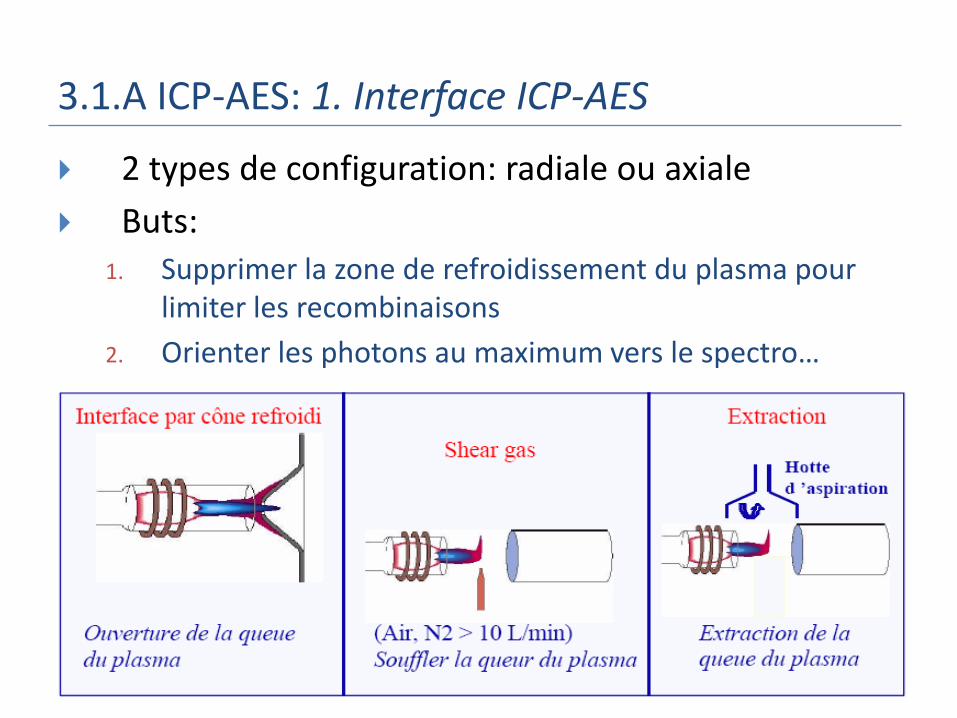
3.1.A ICP-AES: 1. Interface ICP-AES
2 types de configuration: radiale ou axiale
Buts: 1. Supprimer la zone de refroidissement du plasma pour
limiter les recombinaisons
2. Orienter les photons au maximum vers le spectro…

ICP AES
3.1.A ICP-AES: Schéma-principe
2. Focalisation
1. Interface ICP-AES
Lentille de collimation

ICP AES
3.1.A ICP-AES: Schéma-principe
2. Focalisation
3. Tri en λ
1. Interface ICP-AES

3.1.A ICP-AES: 3. Tri en λ = Systèmes dispersifs
But: Séparation des photons en fonction de leur longueur d’onde
2 types de systèmes dispersifs:
Le prisme
Indice de Réfraction du verre/quartz varie avec la longueur d'onde

3. Tri en λ = Systèmes dispersifs
But: Séparation des photons en fonction de leur longueur d’onde
2 types de systèmes dispersifs:
Le prisme
Le réseau de diffraction = surface optique permettant la dispersion de la lumière via une série de traits gravés
Ordre
1 2 3
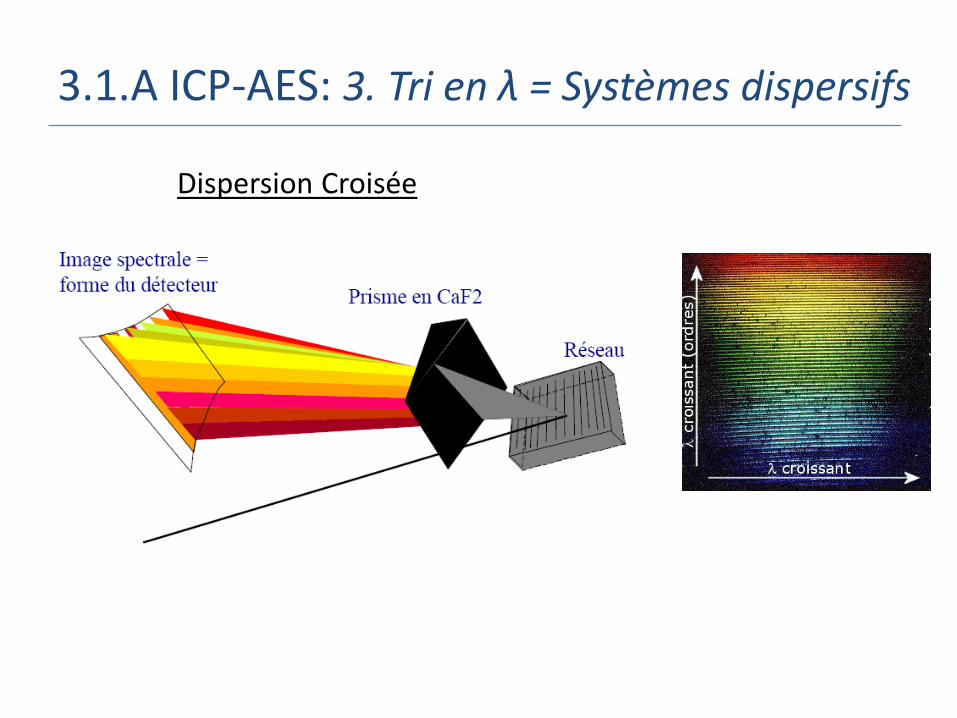
3.1.A ICP-AES: 3. Tri en λ = Systèmes dispersifs
Dispersion Croisée
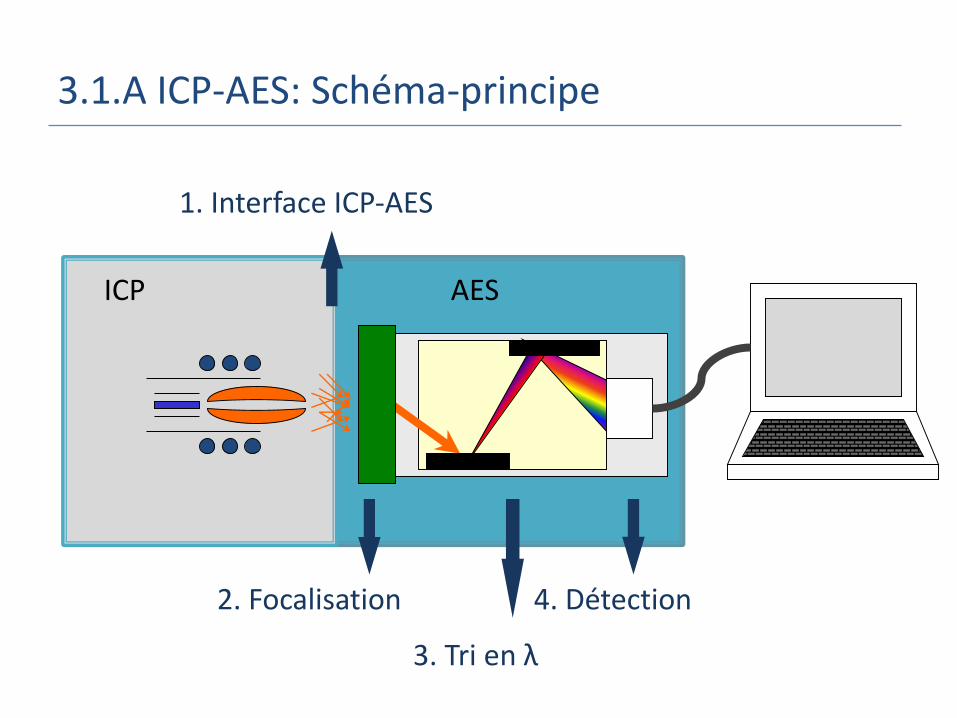
ICP AES
3.1.A ICP-AES: Schéma-principe
2. Focalisation
3. Tri en λ
4. Détection
1. Interface ICP-AES

3.1.A ICP-AES: 4. Détection:
Plaque comprenant des pixels photosensibles en silicium d'une taille allant de 10 à 30 µm, rangés en matrice ou en barrette, qui convertissent les photons incidents en électrons
But: Conversion des photons en courant électrique
Détecteur solide
CCD: Dispositif à Transfert de Charge
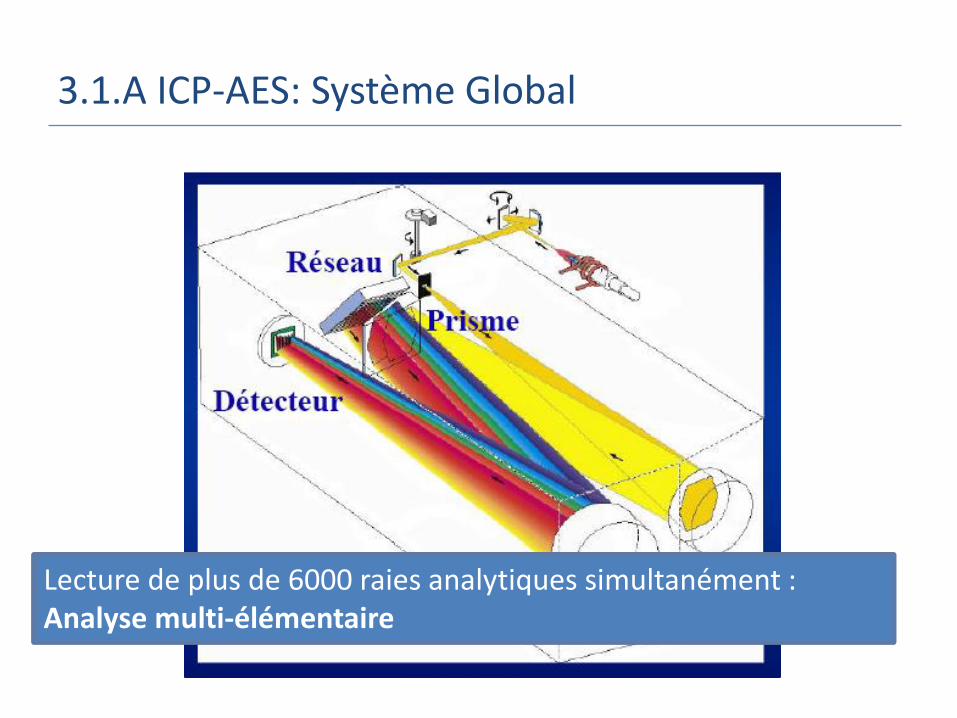
3.1.A ICP-AES: Système Global
Lecture de plus de 6000 raies analytiques simultanément : Analyse multi-élémentaire
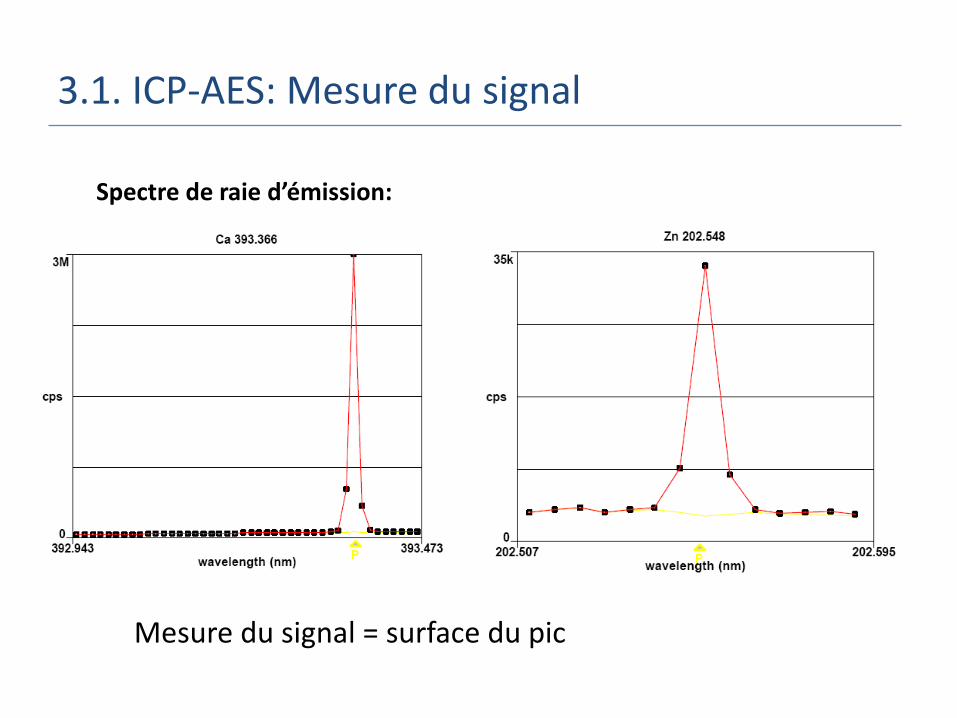
3.1. ICP-AES: Mesure du signal
Spectre de raie d’émission:
Mesure du signal = surface du pic
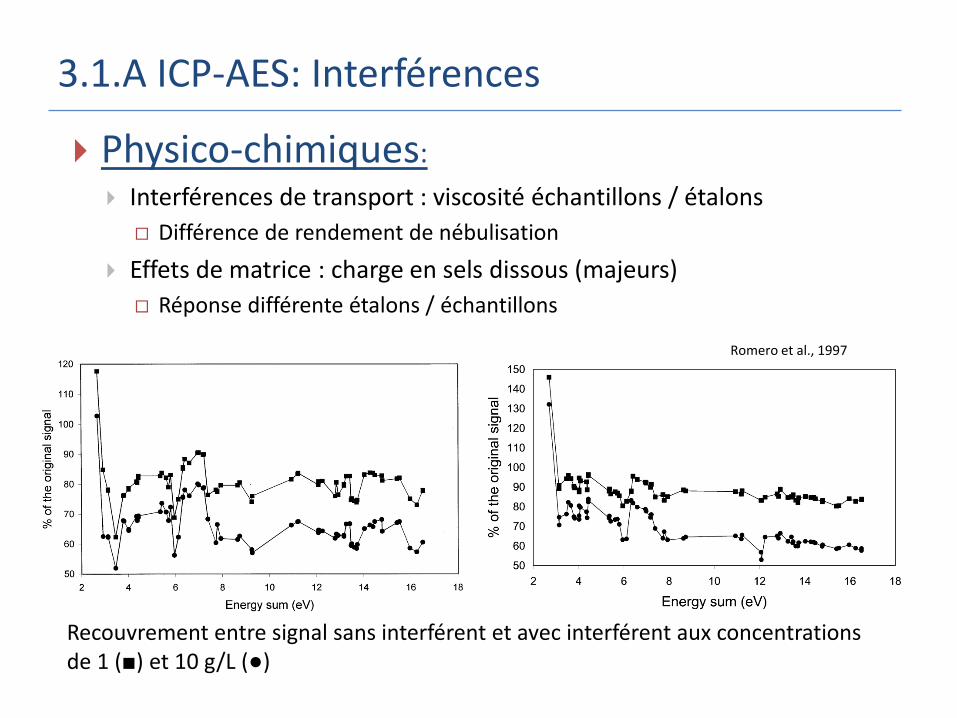
3.1.A ICP-AES: Interférences
Physico-chimiques:
Interférences de transport : viscosité échantillons / étalons
Différence de rendement de nébulisation
Effets de matrice : charge en sels dissous (majeurs)
Réponse différente étalons / échantillons
Recouvrement entre signal sans interférent et avec interférent aux concentrations de 1 (■) et 10 g/L (●)
Ajout de Na Ajout de Ca
Romero et al., 1997

3.1.A ICP-AES: Interférences
Physico-chimiques:
Interférences de transport : viscosité échantillons / étalons
Différence de rendement de nébulisation
Effets de matrice : charge en sels dissous (majeurs)
Réponse différente étalons / échantillons
Spectrales: Potentiellement les plus importantes : surdosages
Superposition de signaux

3.1.A ICP-AES: Interférence spectrale: chevauchement
310.163310.163 310.297310.297
2k2k
21k21k
Travail en hauteur de pics Travail à une autre longueur d'onde

B. ICP-MS
Inductively Coupled Plasma – Mass Spectrometry

3.1.B. ICP-MS:
ICP = source d’ions MS = Filtre: séparation des ions en fonctions de leur
charge et de leur masse avec un signal proportionnel au nombre d’ions
RQ: Le plasma produit aussi bien des ions positifs que négatifs, mais le système de séparation et de transport des cations et des anions étant différents, on ne peut pas analyser les deux en même temps en ICP-MS
A la différence de l'ICP-AES, ici récupération directe des ions à analyser!
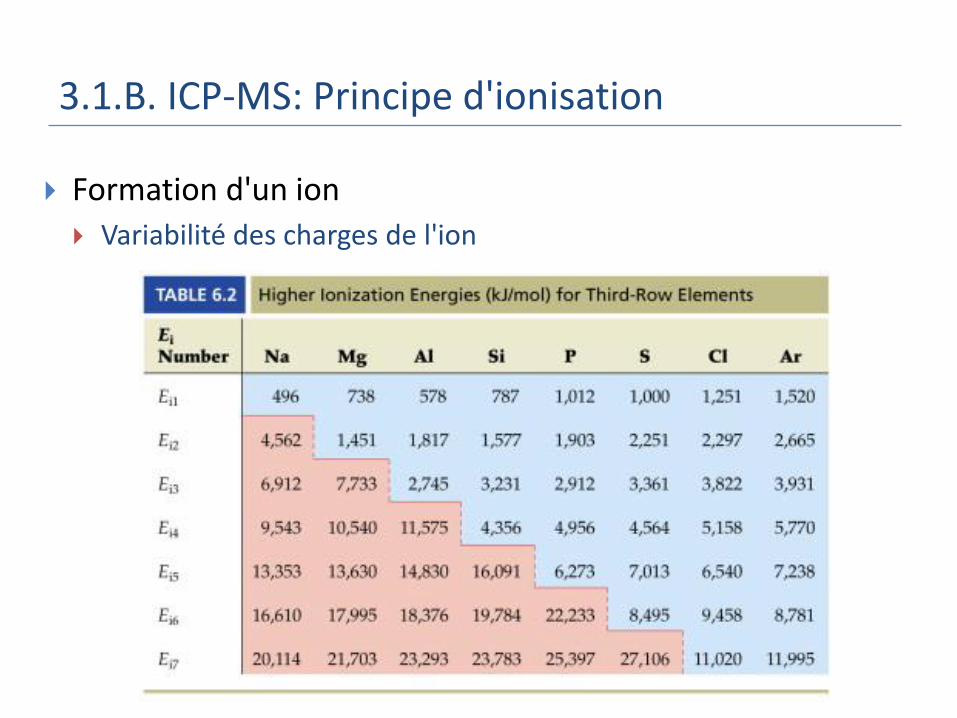
3.1.B. ICP-MS: Principe d'ionisation
Formation d'un ion
Variabilité des charges de l'ion

3.1.B. ICP-MS: Principe d'ionisation = séparation m/z
Formation d'un ion
Variabilité des charges de l'ion
Variabilité de la masse de l'ion (notion d’isotope)
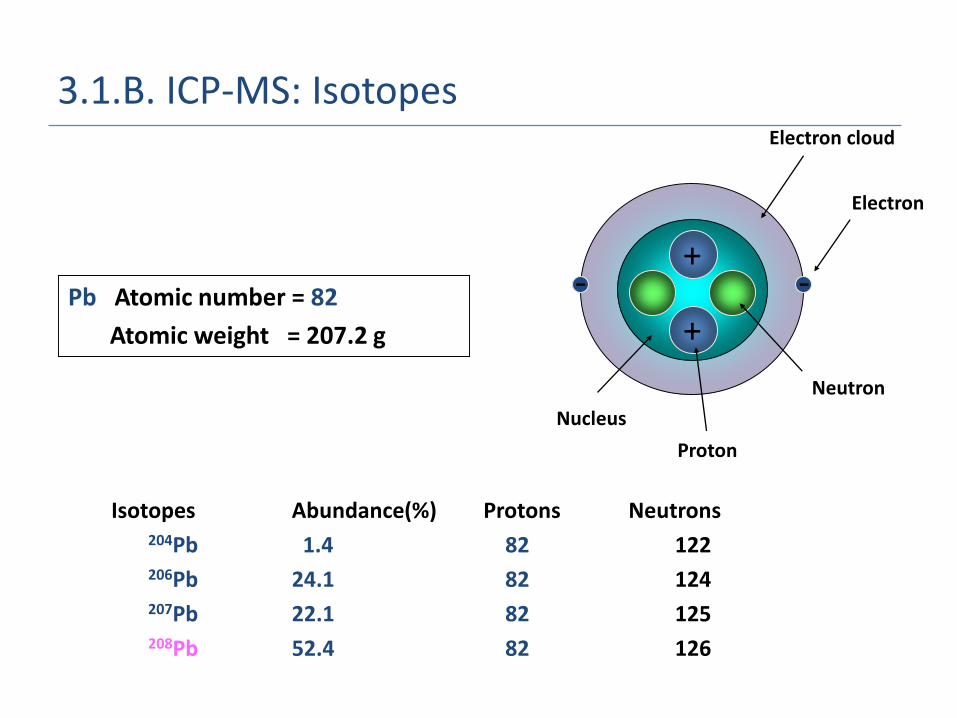
3.1.B. ICP-MS: Isotopes
Pb Atomic number = 82
Atomic weight = 207.2 g
Isotopes Abundance(%) Protons Neutrons 204Pb 1.4 82 122206Pb 24.1 82 124207Pb 22.1 82 125208Pb 52.4 82 126
+
-+
-
Nucleus
Proton
Electron
Neutron
Electron cloud
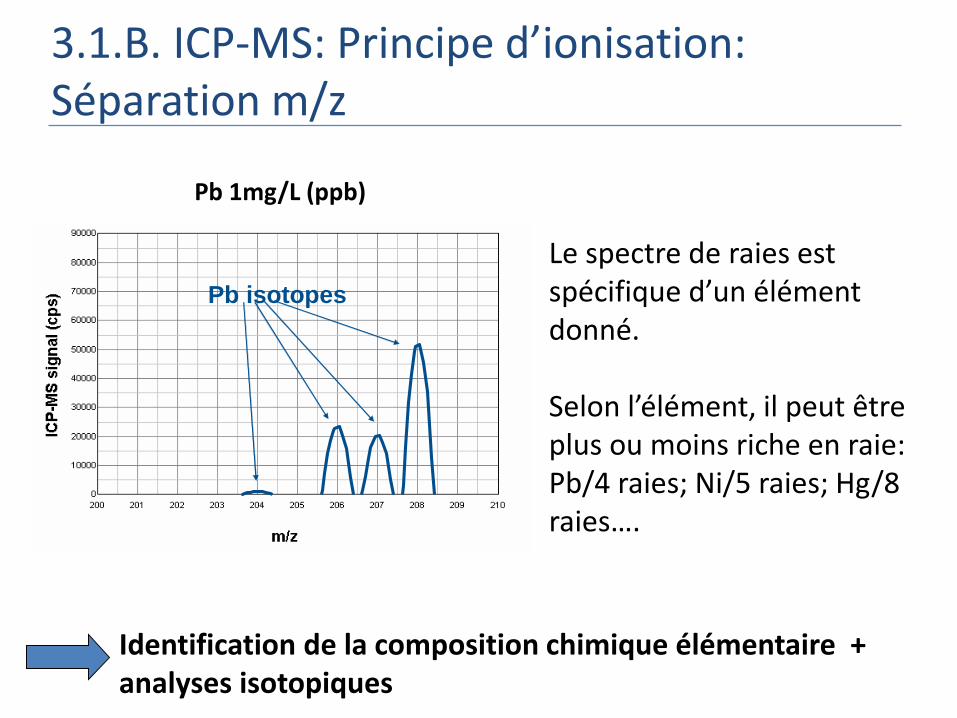
3.1.B. ICP-MS: Principe d’ionisation: Séparation m/z
Pb isotopesPb isotopes
Pb 1mg/L (ppb)
Identification de la composition chimique élémentaire + analyses isotopiques
Le spectre de raies est spécifique d’un élément donné.
Selon l’élément, il peut être plus ou moins riche en raie: Pb/4 raies; Ni/5 raies; Hg/8 raies….
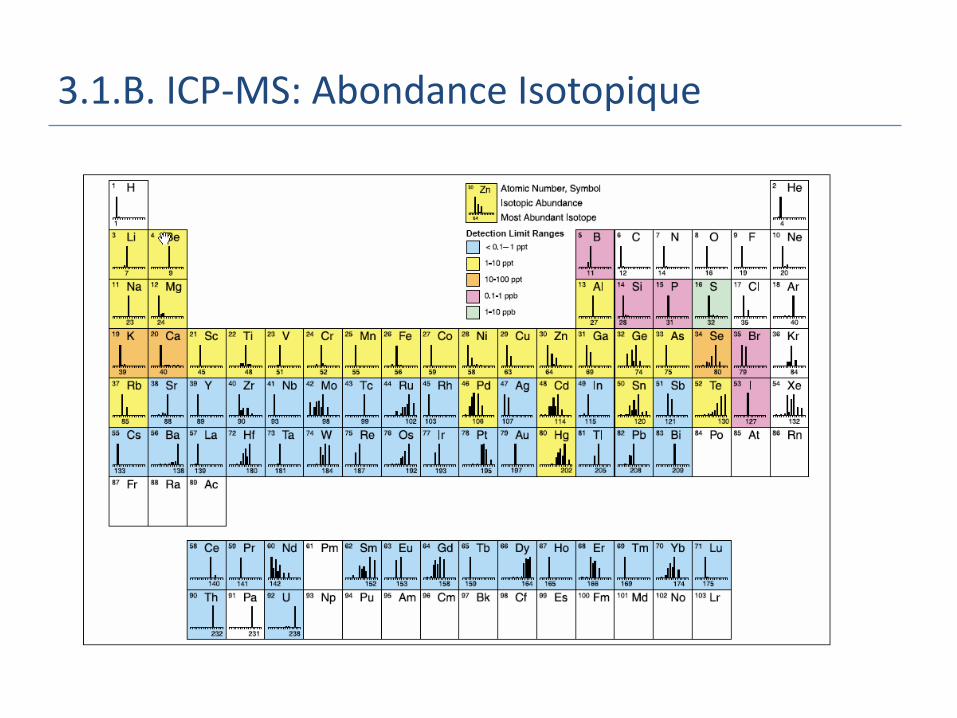
3.1.B. ICP-MS: Abondance Isotopique

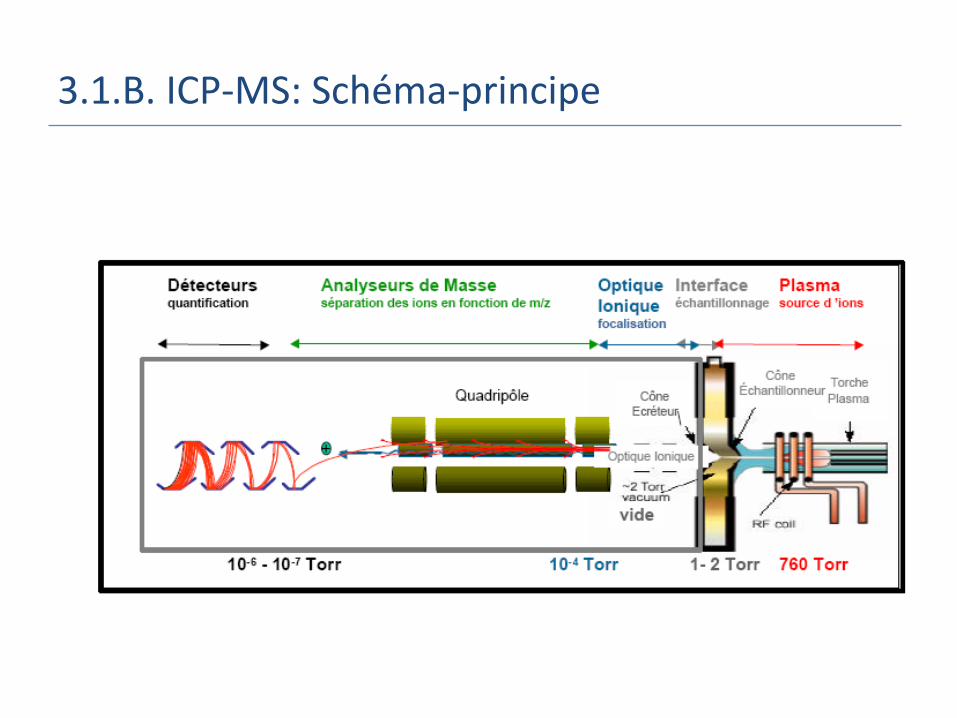
3.1.B. ICP-MS: Schéma-principe

3.1.B. ICP-MS: 1. Interface
Buts: Prélever les ions où ils sont formés, c’est-à-dire dans la
zone froide du plasma (ionisation + transfert de charge).
Passer de la pression atmosphérique à un vide compatible avec le spectromètre de masse.
Décroître la température de 6000 K à la température ambiante.
On utilise deux cônes: échantillonneur (sampler) puis écremeur/écorceur (skimmer) en Ni ou Pt car résistants aux hautes températures
3.1.B. ICP-MS: Schéma-principe

3.1.B. ICP-MS: 2. Optique ionique
Buts: Optimiser la trajectoire des ions pour une meilleure
focalisation dans l’analyseur de masse
Besoin d’arrêter les photons pour qu’ils n’atteignent pas le détecteur.
Utilisation d’optique ionique basée sur l’utilisation de lentille électrostatique
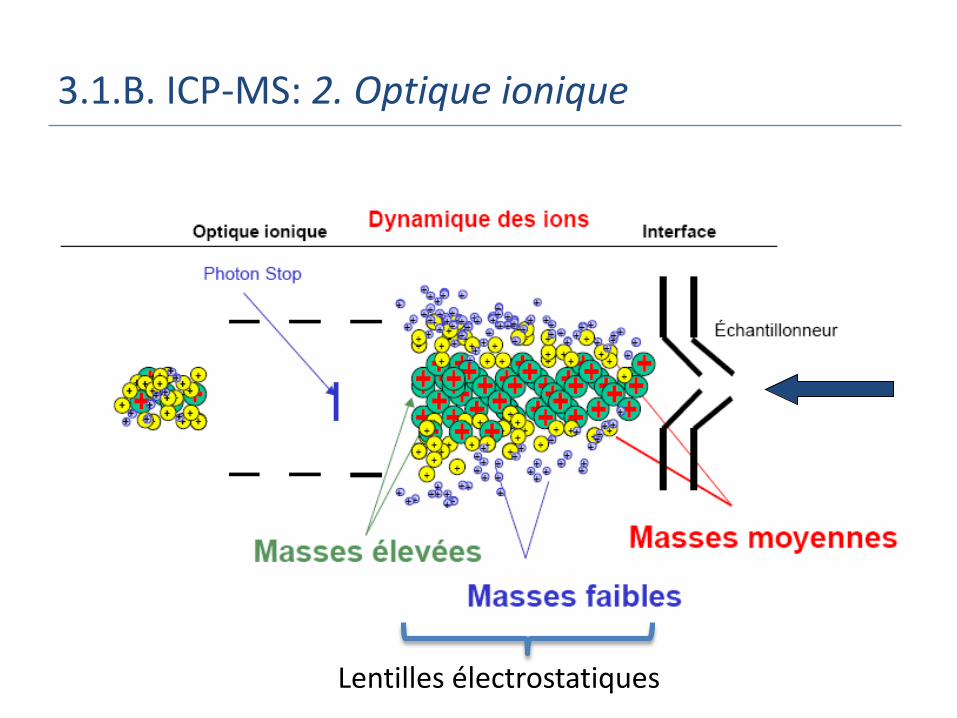
3.1.B. ICP-MS: 2. Optique ionique
Lentilles électrostatiques
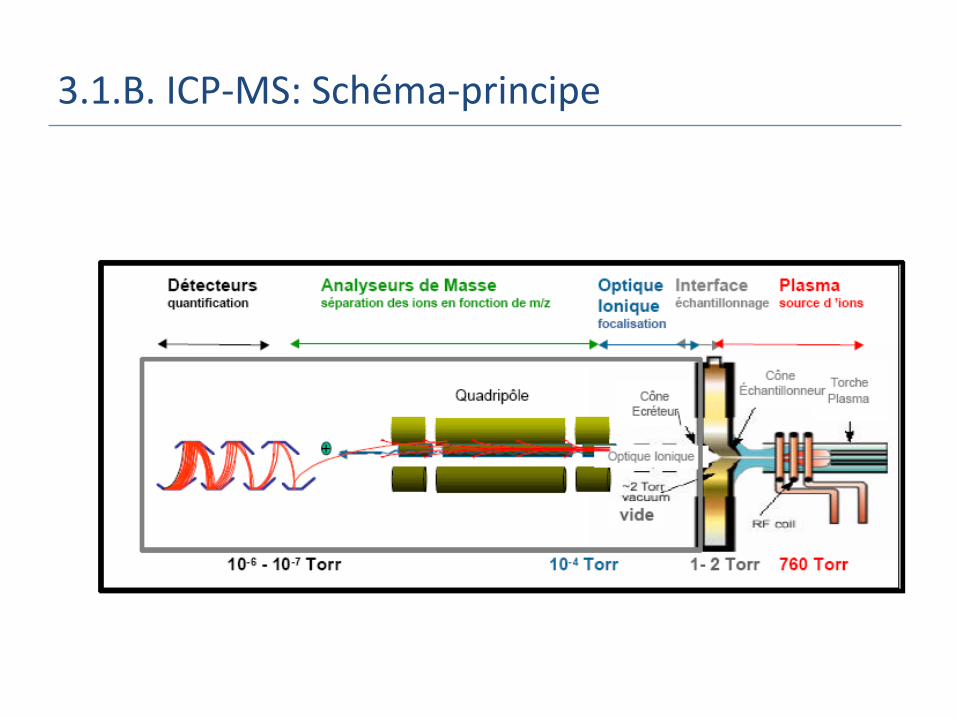
3.1.B. ICP-MS: Schéma-principe

3.1.B. ICP-MS: 3. Analyseur de Masse
Principaux Types:
Sélection par filtrage:
Filtre quadripolaire
Trappe à ions
Sélection dans le temps:
Temps de vol
Sélection dans l’espace:
Secteur (magnétique)
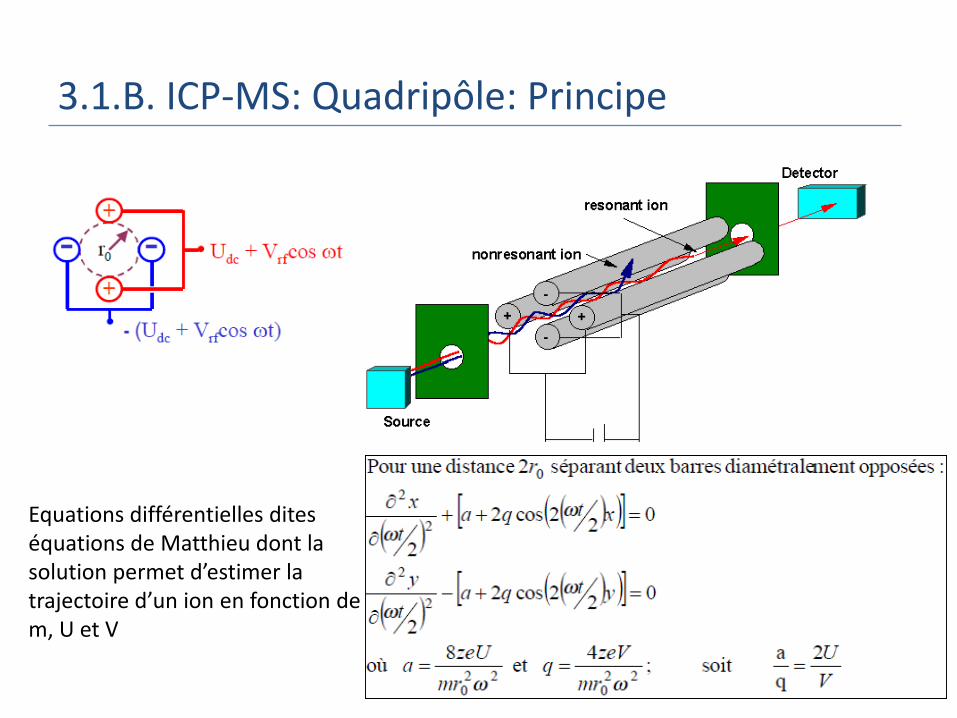
3.1.B. ICP-MS: Quadripôle: Principe
Equations différentielles dites équations de Matthieu dont la solution permet d’estimer la trajectoire d’un ion en fonction de m, U et V

3.1.B. ICP-MS: Quadripôle
Trappe à ions ou piège à ions = Développement du filtre quadripolaire utilisant des électrodes à section hyperbolique
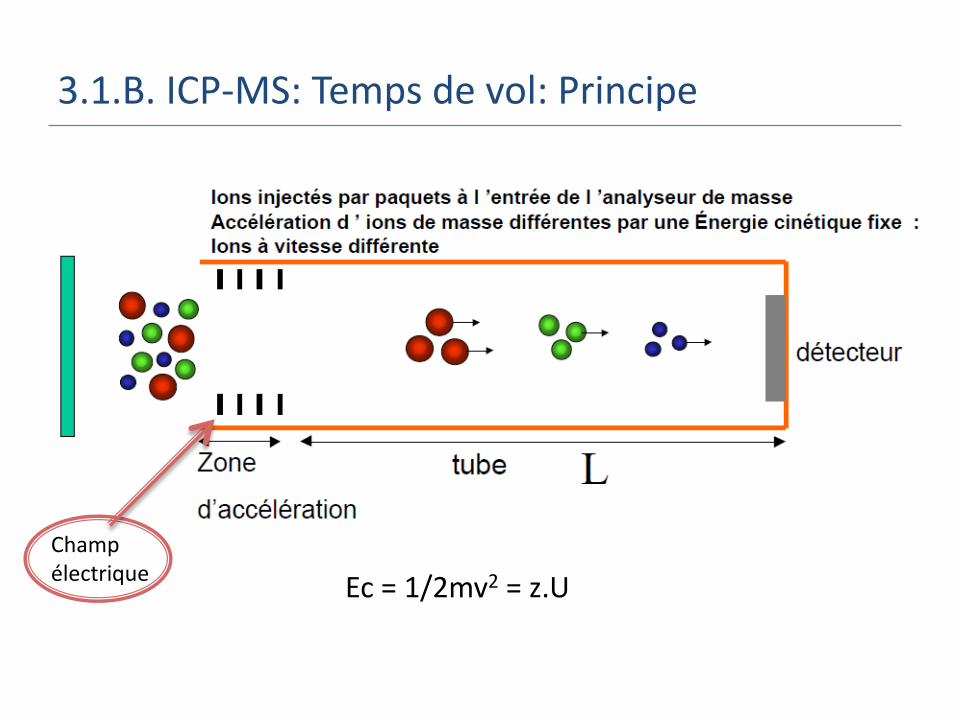
3.1.B. ICP-MS: Temps de vol: Principe
Ec = 1/2mv2 = z.U
Champ électrique
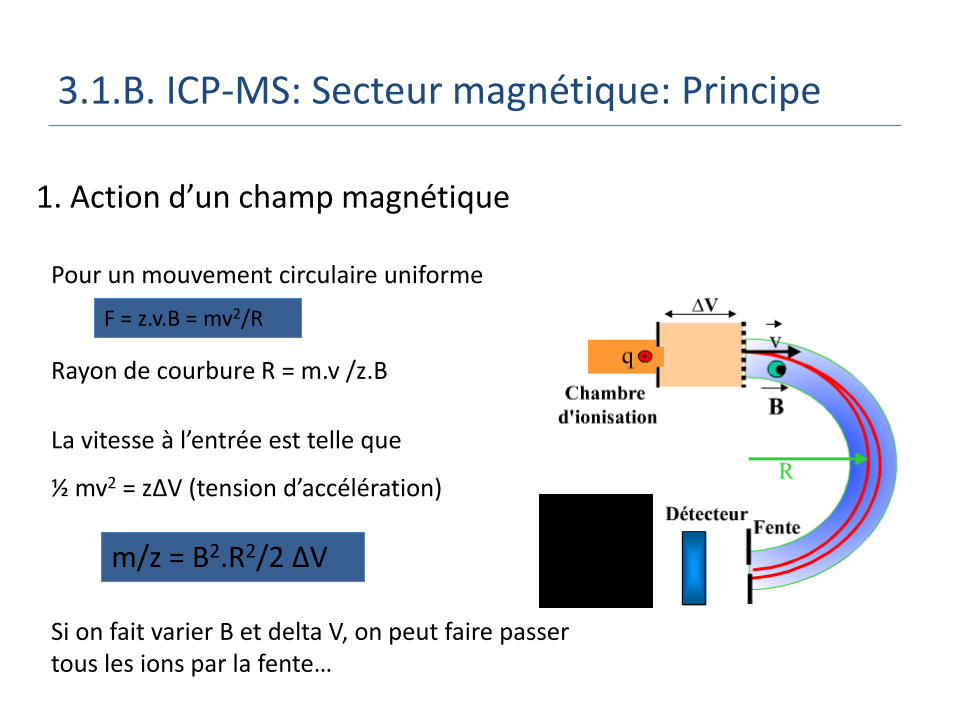
Pour un mouvement circulaire uniforme
Rayon de courbure R = m.v /z.B
La vitesse à l’entrée est telle que
½ mv2 = zΔV (tension d’accélération)
Si on fait varier B et delta V, on peut faire passer tous les ions par la fente…
1. Action d’un champ magnétique
F = z.v.B = mv2/R
m/z = B2.R2/2 ΔV
3.1.B. ICP-MS: Secteur magnétique: Principe
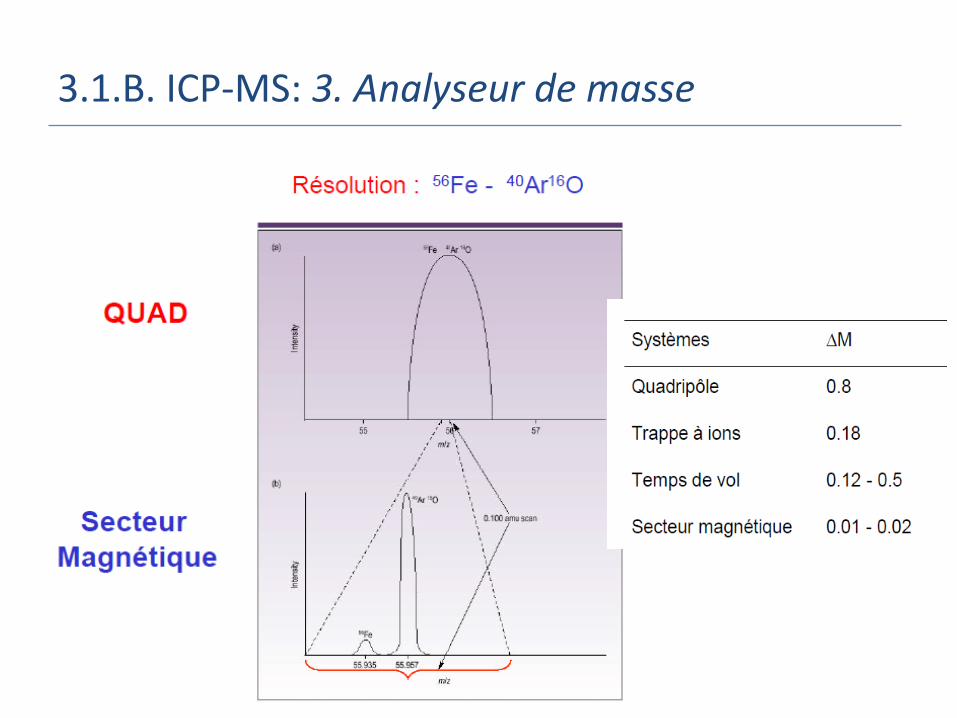
3.1.B. ICP-MS: 3. Analyseur de masse
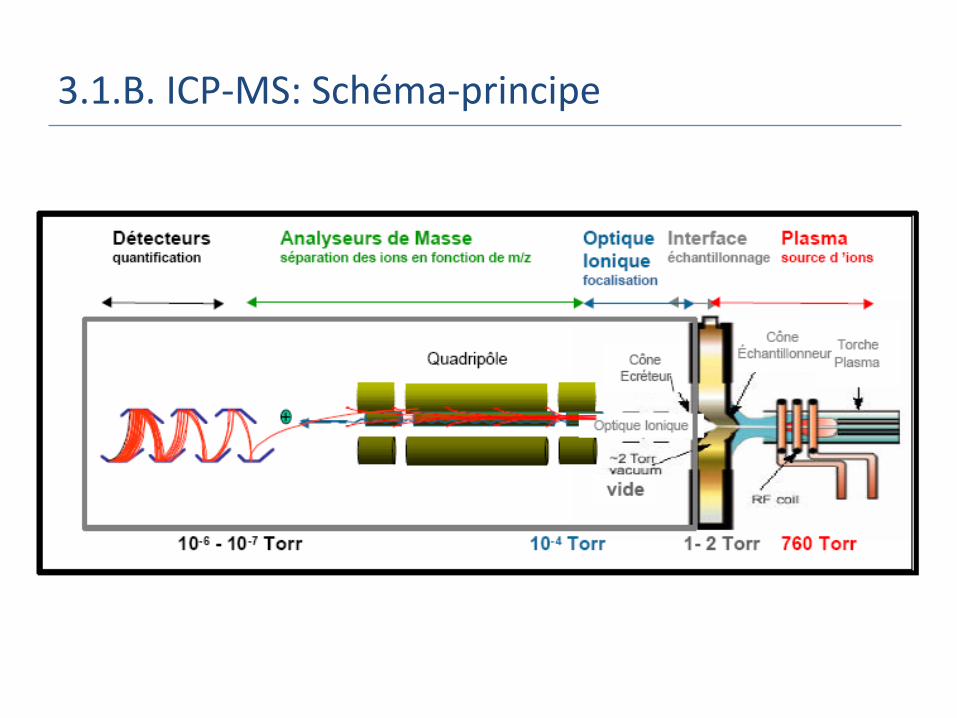
3.1.B. ICP-MS: Schéma-principe
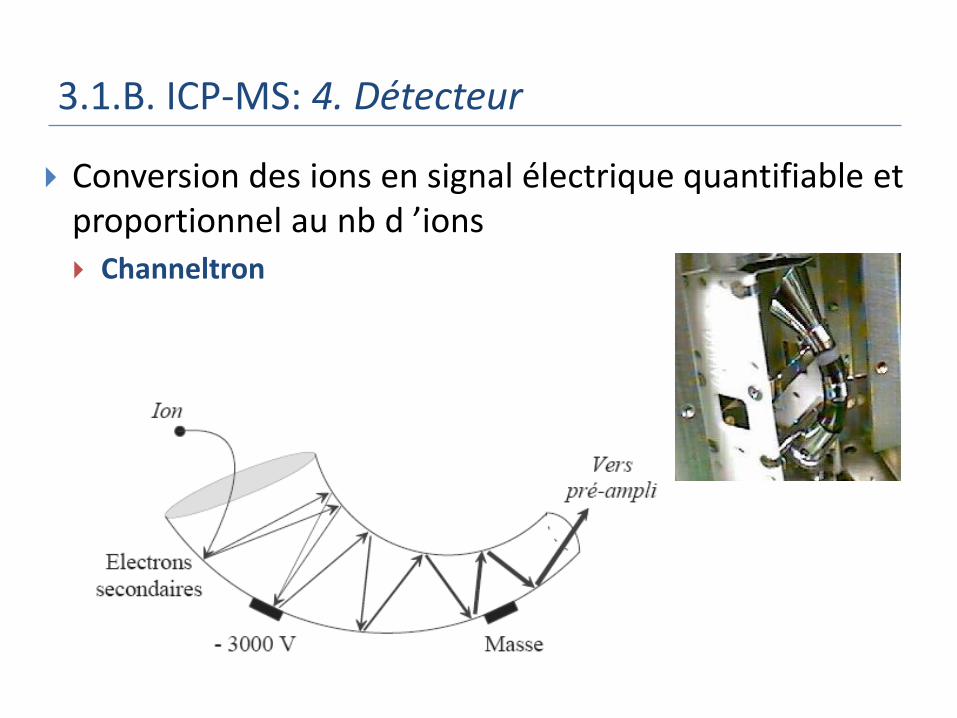
3.1.B. ICP-MS: 4. Détecteur
Conversion des ions en signal électrique quantifiable et proportionnel au nb d ’ions Channeltron
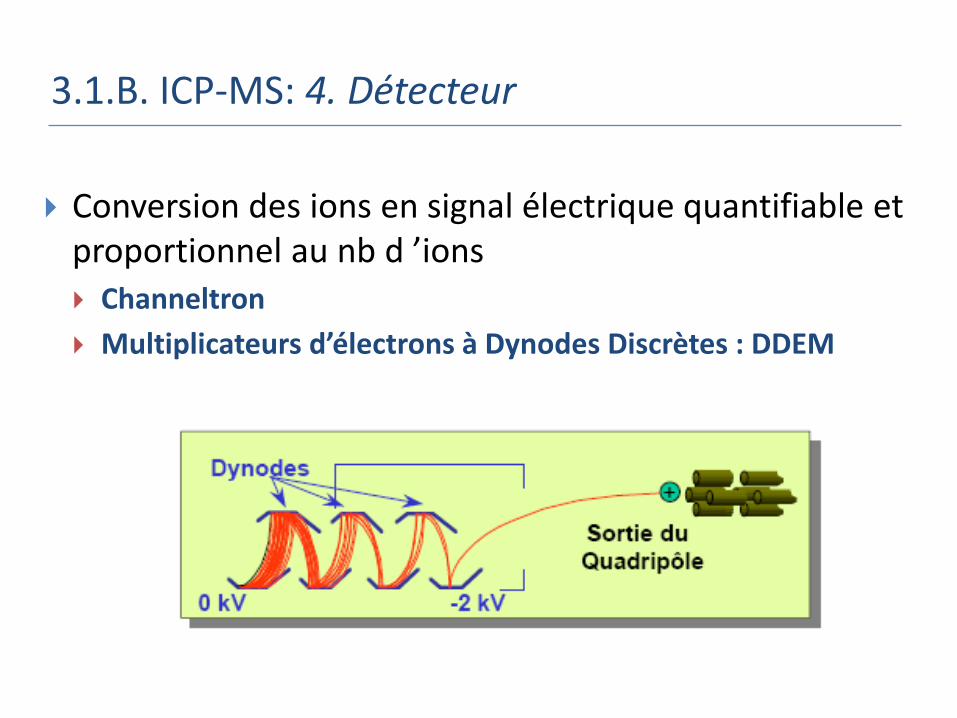
3.1.B. ICP-MS: 4. Détecteur
Conversion des ions en signal électrique quantifiable et proportionnel au nb d ’ions Channeltron
Multiplicateurs d’électrons à Dynodes Discrètes : DDEM

3.1.B. ICP-MS: Système global
Détection de plusieurs m/z quasi-simultanément : Analyse multi-élémentaire
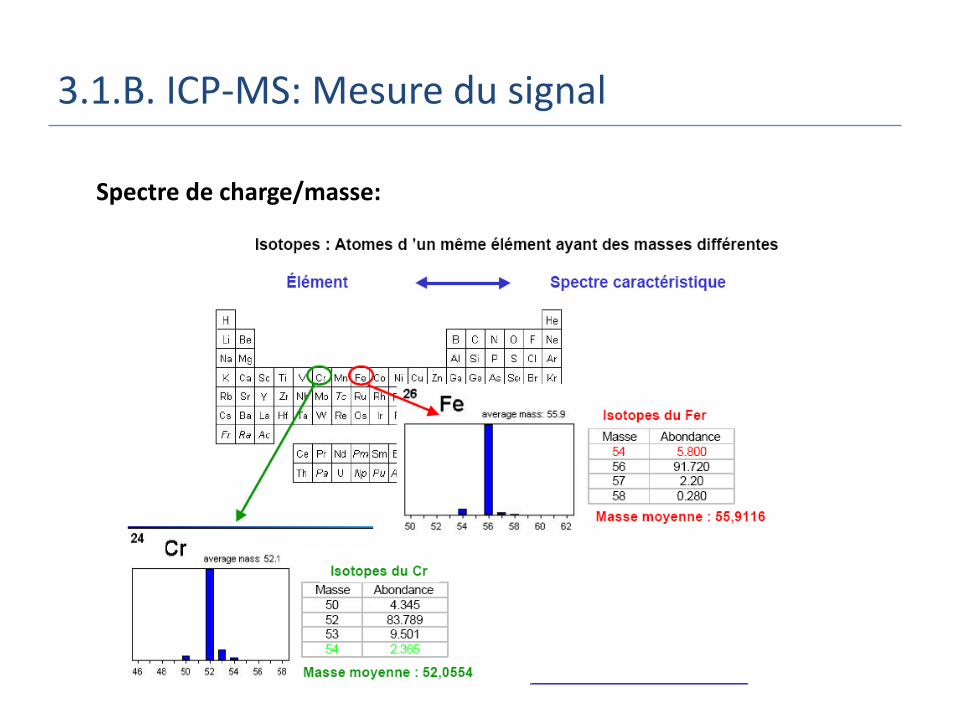
3.1.B. ICP-MS: Mesure du signal
Spectre de charge/masse:
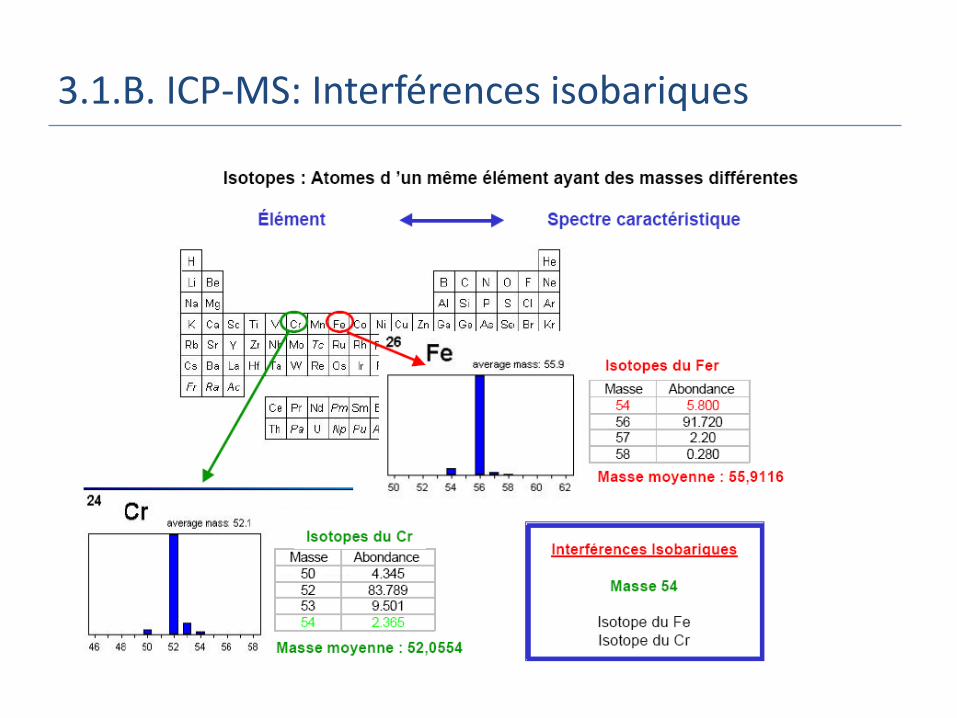
3.1.B. ICP-MS: Interférences isobariques

3.1.B. ICP-MS: Interférences
Interférences spectrales: Recouvrement isobarique des éléments: même
masse/charge Ions doublement chargés : à M/2
Ex: 136Ba2+ répondent au 68Zn+
Poly-atomiques, formés entre Ar et les éléments majeurs de la matrice (O, H, N)

3.1.B. ICP-MS: Interférences poly-atomiques

3.1.B. ICP-MS: Interférences poly-atomiques: comment s’en sortir?
Utilisation d’une cellule de collision/réaction:
Réactivité des espèces:
Réactions formant des espèces neutres
Cas: 40Ar16O+ et 56Fe+:
ArO+ + NH3 ArO + NH3+
avec une cinétique de 1.4 x10-9
Fe+ + NH3 Fe + NH3+
avec une cinétique de 9.1 x 10-12
Réactions formant des ions de masse différente
Cas 80Ar2+ et 80Se+
Ar2+ + CH4 Ar2H+ + CH3
Utilisation d’un gaz tampon (He) ou de collision (N2,H2)
Les ions polyatomiques interférants sont dissociés par collisions
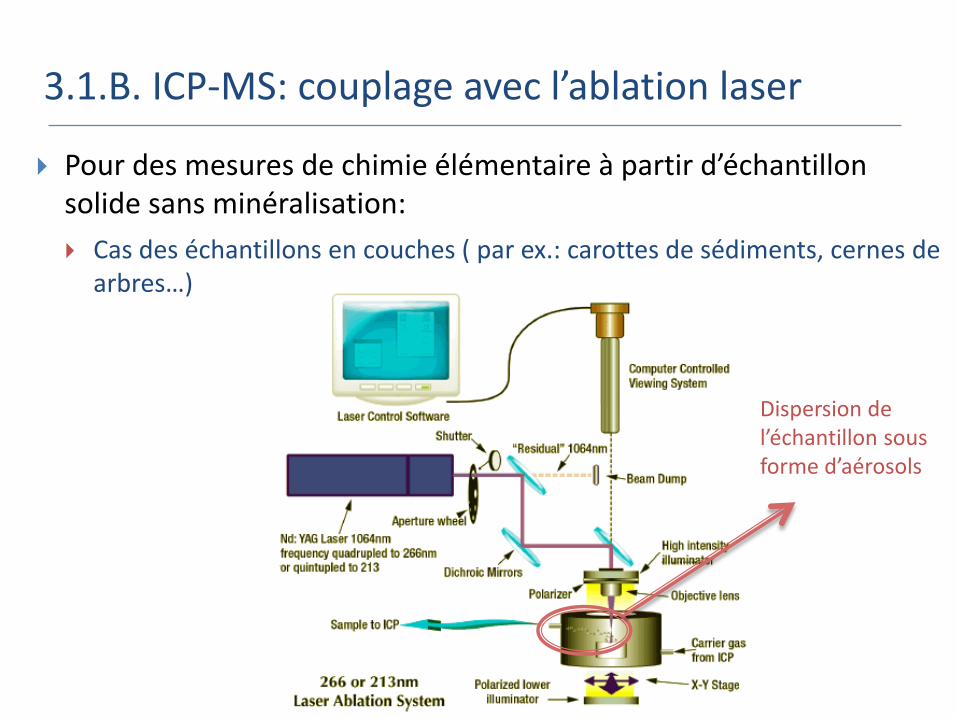
3.1.B. ICP-MS: couplage avec l’ablation laser
Pour des mesures de chimie élémentaire à partir d’échantillon solide sans minéralisation:
Cas des échantillons en couches ( par ex.: carottes de sédiments, cernes de arbres…)
Dispersion de l’échantillon sous forme d’aérosols

3.1.B. ICP-MS: couplage avec HPLC
ICP-AES et ICP-MS = méthodes basées
Sur l’atomisation des molécules technique élémentaire
Sur la mesure des photons ou des ions pas d’accès à l’état de valence
Méthodes d’analyse atomique pure, pas d’analyse de la spéciation (redox ou organique/inorganique)
Pour avoir accès à cette information, il faut séparer les différentes formes au préalable HPLC-ICP-MS
Mesure des formes redox des métaux (CrIII ou CrVI)
Mesure des formes organiques des métaux (As organique)

3.1.B. ICP-MS: couplage avec HPLC
HPLC: Colonne de séparation des espèces à considérer (séparation par polarité ou charges)
ICP-MS: détermination des teneurs en métaux dans chacune des espèces
Méthode la plus sensible car très faible teneur
Comme multi-élémentaire: possibilité de mesurer plusieurs formes métalliques simultanément dans un même échantillon CPS
Ex: Spéciation simultanée de la speciation de l’arsenic, du sélénium , de l’antimoine et du tellure
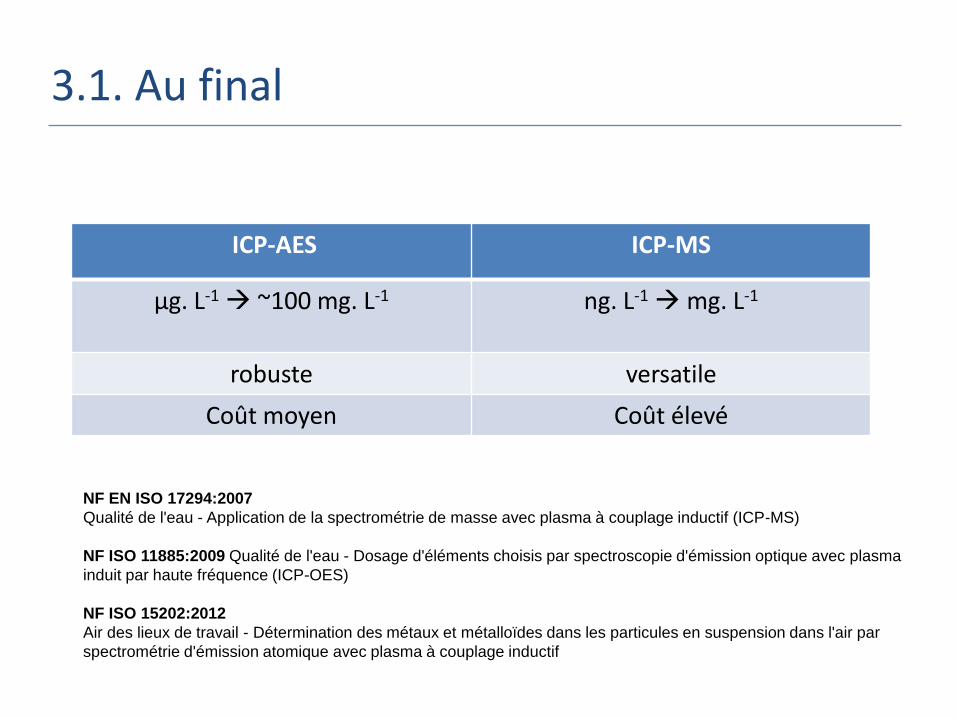
3.1. Au final
ICP-AES ICP-MS
µg. L-1 ~100 mg. L-1 ng. L-1
mg. L-1
robuste versatile
Coût moyen Coût élevé
NF EN ISO 17294:2007
Qualité de l'eau - Application de la spectrométrie de masse avec plasma à couplage inductif (ICP-MS)
NF ISO 11885:2009 Qualité de l'eau - Dosage d'éléments choisis par spectroscopie d'émission optique avec plasma
induit par haute fréquence (ICP-OES)
NF ISO 15202:2012
Air des lieux de travail - Détermination des métaux et métalloïdes dans les particules en suspension dans l'air par
spectrométrie d'émission atomique avec plasma à couplage inductif

3. Analyses élémentaires:
But: Analyses des métaux et métaux lourds
Techniques applicables aux phases liquides et solides (après mise en solution):
Spectrométrie atomique d’absorption
Spectrométrie d’émission
Spectrométrie de masse
Techniques applicables aux phases solides:
Spectrométrie de rayons X
Couplage avec la technologie plasma: ICP-AES et ICP-MS

3.2. Spectrométrie des rayons X
Rayons X?
Découverte le 8 novembre 1895 par Wilhelm Conrad RÖNTGEN (All.)
Publication le 22 décembre 1895 dans un article à l’académie dessciences avec comme illustration la « radiographie » de la main de safemme
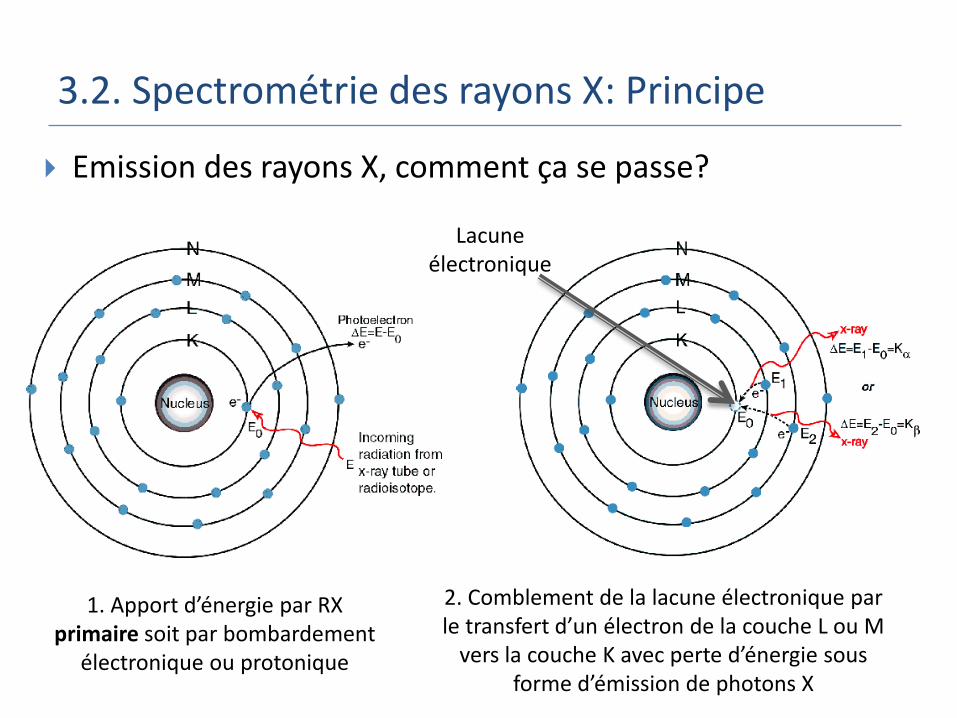
3.2. Spectrométrie des rayons X: Principe
Emission des rayons X, comment ça se passe?
1. Apport d’énergie par RX primaire soit par bombardement
électronique ou protonique
Lacune électronique
2. Comblement de la lacune électronique par le transfert d’un électron de la couche L ou M
vers la couche K avec perte d’énergie sous forme d’émission de photons X
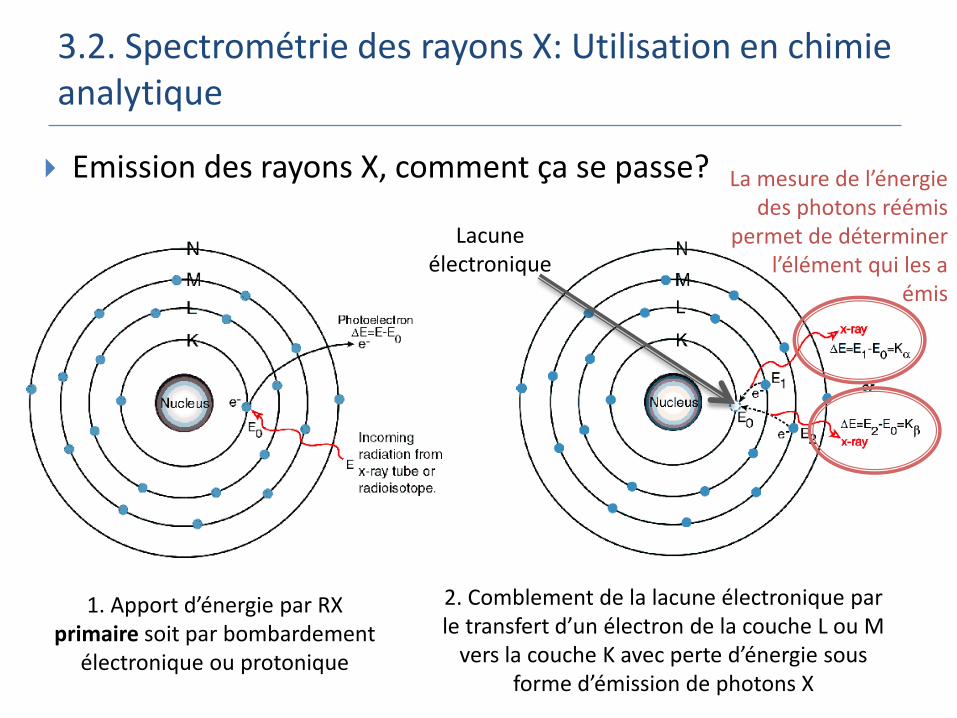
3.2. Spectrométrie des rayons X: Utilisation en chimie analytique
Emission des rayons X, comment ça se passe?
1. Apport d’énergie par RX primaire soit par bombardement
électronique ou protonique
Lacune électronique
2. Comblement de la lacune électronique par le transfert d’un électron de la couche L ou M
vers la couche K avec perte d’énergie sous forme d’émission de photons X
La mesure de l’énergie des photons réémis
permet de déterminer l’élément qui les a
émis

3.2. Spectrométrie des rayons X: Principe
Emission des rayons X, comment ça se passe?
3. La nouvelle lacune créée est comblée par le transfert d’électron des couches supérieures (M ou N) avec à
nouveau émission de photons X
Pour un électron d’arraché plusieurs transitions électroniques plusieurs photons X émis
Lacune électronique

3.2. Spectrométrie des rayons X: Utilisation en chimie analytique
Principe d’utilisation en chimie analytique est basée sur le comptage des photons X émis suite à l’arrachement des électrons des couches internes (K et L):
Chaque photon X a un niveau d’énergie propre à chaque élément
La quantité de photons émis est proportionnelle à la quantité de l’élément présent dans l’échantillon
Pour pouvoir étudier un échantillon, il nous faut donc :
générer des rayons X susceptibles d'exciter un atome et d'éjecter un électron,
Récupérer, trier et compter les photons émis

3.2. Spectrométrie des rayons X: Utilisation en chimie analytique
Pour pouvoir étudier un échantillon, il nous faut donc :
1. Générer des rayons X susceptibles d'exciter un atome et d'éjecter un électron,
Spectrométrie de fluorescence X (XRF) avec excitation par des photons X

3.2. A. La spectrométrie de fluorescence X
Condition d’excitation des électrons:
Il faut qu’il y ait possibilité d’avoir des transitions électronique entre K et L:
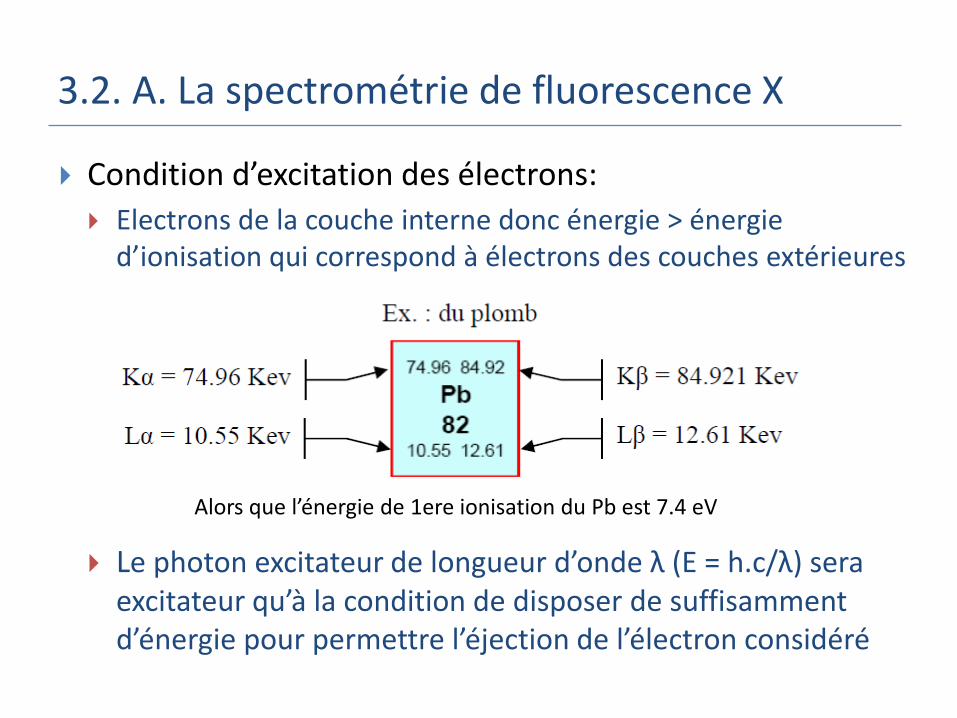
3.2. A. La spectrométrie de fluorescence X
Condition d’excitation des électrons:
Electrons de la couche interne donc énergie > énergie d’ionisation qui correspond à électrons des couches extérieures
Le photon excitateur de longueur d’onde λ (E = h.c/λ) sera excitateur qu’à la condition de disposer de suffisamment d’énergie pour permettre l’éjection de l’électron considéré
Alors que l’énergie de 1ere ionisation du Pb est 7.4 eV

3.2. A. La spectrométrie de fluorescence X
Pour produire les photons X primaires (ou excitateurs) Utilisation d’un tube de rayons X
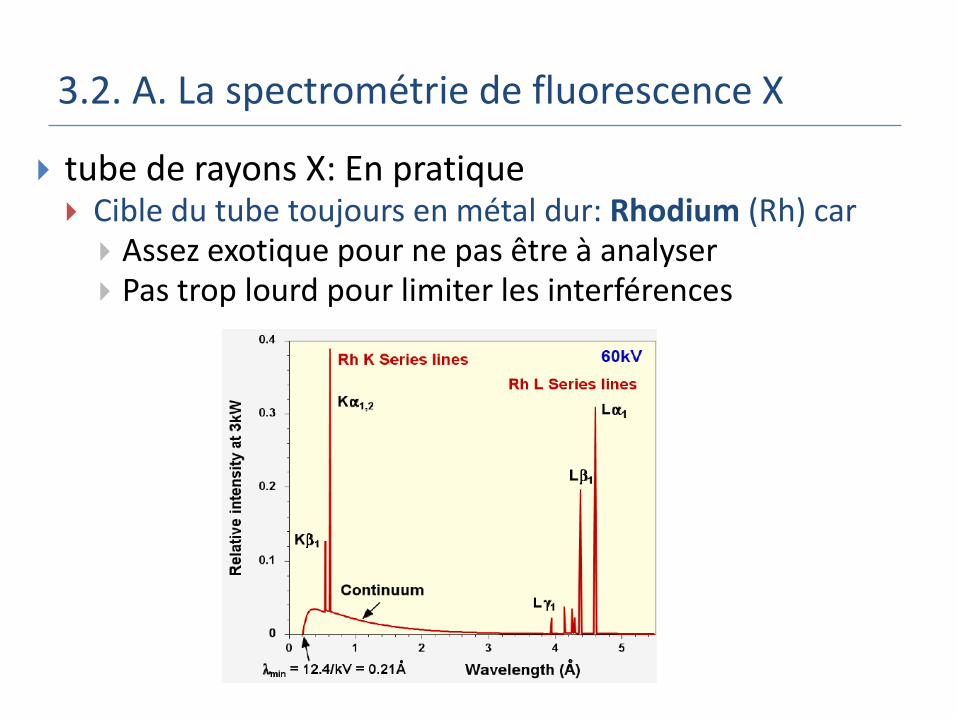
3.2. A. La spectrométrie de fluorescence X
tube de rayons X: En pratique Cible du tube toujours en métal dur: Rhodium (Rh) car
Assez exotique pour ne pas être à analyser Pas trop lourd pour limiter les interférences

3.2. A. La spectrométrie de fluorescence X
Les porte-échantillons: Echantillons solide ou liquide
Pour les liquides: nécessité de mettre un film: Attention de choisir un matériau peu absorbant

3.2. Spectrométrie des rayons X: Utilisation en chimie analytique
Pour pouvoir étudier un échantillon, il nous faut donc :
1. Générer des rayons X susceptibles d'exciter un atome et d'éjecter un électron,
Spectrométrie de fluorescence X (XRF) avec excitation par des photons X
2. Récupérer, trier et compter les photons émis
Système de dispersion en énergie
Système de dispersion en longueurs d’onde

3.2. A. La spectrométrie de fluorescence X
Les systèmes de comptages des photons
Deux possibilités pour les trier:
En fonction de leurs énergies système à dispersion d’énergie (EDXRF)
En fonction de leur longueur d’ondes (wavelength )système à dispersion de longueur d’ondes ( WDXRF)
Loi deBragg
Théorie des quanta
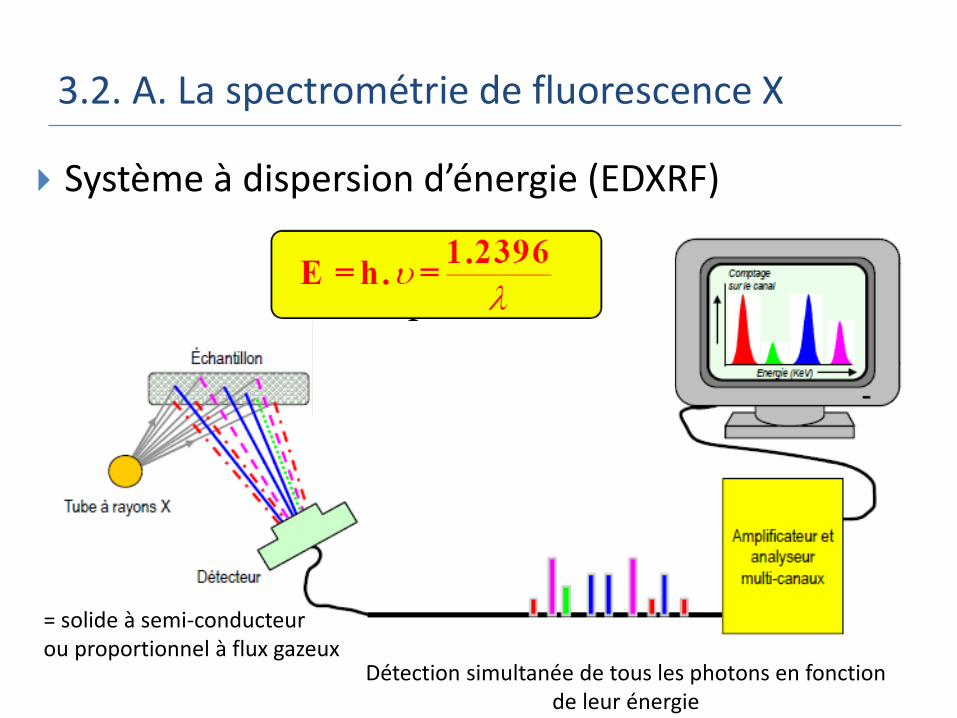
3.2. A. La spectrométrie de fluorescence X
Système à dispersion d’énergie (EDXRF)
Détection simultanée de tous les photons en fonction de leur énergie
= solide à semi-conducteur ou proportionnel à flux gazeux

3.2. A. La spectrométrie de fluorescence X
Système à dispersion d’énergie (EDXRF)
Peu sensible et peu résolu mais simultané

3.2. A. La spectrométrie de fluorescence X
Système à dispersion en longueurs d’ondes (WDXRF)
Détection séquentielle des longueurs d’ondes
ou Cristaux de Bragg

3.2. A. La spectrométrie de fluorescence X
Système à dispersion en longueurs d’ondes (WDXRF)
Plus sensible mais séquentiel

A suivre