centre de référence des maladies lysosomales … · bichat médecine interne o.lidove...
TRANSCRIPT

Centre de Référence des Maladies LysosomalesReferal Center for Lysosomal Diseases
Hôpitaux Universitaires Nord Val de Seine, Hôpital Beaujon
Trousseau Neuropédiatrie T. Billette, B. Heron, C. Mignot Bichat Médecine Interne O.LidovePitié-Salpétrière Neurology N. Baumann
F. Sedel (A. Lyon-Caen)C. Mignot
Cochin Génétique C. Caillaud, G. Viot
Beaujon Médicine Interne N. Belmatoug (coordinateur) Infirmière S. Mamine Assistante de Recherche clinique M. Bengherbia
Secrétaire S. Zebiche
Inserm Epidémiologie J. Stirnemann, Genève (Suisse) F. Mentre, Hôpîtal Bichat D. Hamroun, Montpellier

Les Muco Poly SaccharidosesNadia BelmatougBénédicte Héron
- Macromolécules formant la matrice extracellulaire
- Polysaccharides : Unités répétées de disaccharides N -acetyl-hexosamine

LYSOSOME
- 1949 C. de Duve (Nobel Price 1979)- sacs contenant de nombreux enzymes

LYSOSOME
•••• Organites intracellulaires•••• Saccules 0.5 microns •••• Présents dans toutes les cellules de mammifères (sauf GR)
•••• Équipement enzymatique riche (hydrolases lysosomales)•••• Digestion de composés, macromolécules d’organites intracellulaires (mitochondries etc.)
•••• Etape finale de dégradation des constituants intracellulaires ou autophagie

LE LYSOSOME
•••• Structure dynamique non figées
•••• Fusion avec d’autres membranes
•••• Contient des protéines de type pompes à protons ent rée d’ion H+ dans la lumière des lysosomes assurent un pH acide (pH : 5)acide (pH : 5)
•••• Confine l’action des hydrolases lysosomales à ce compartiment
•••• Empêche l’action des enzymes dans le cytoplasme
Contient certaines protéines spécifiques- Lysosome-Associated Membrane Proteins : LAMPs, LAMP1 , LAMP2- Lysosomal Integral Membrane Proteins : LIMPS

LES MALADIES LYSOSOMALES
•••• Environ 50 maladies, maladies rares
•••• Transmission autosomique récessive Sauf : Fabry, Hunter , Danon : X
•••• Accumulation de molécules non dégradées /non •••• Accumulation de molécules non dégradées /non dégradables dans les lysosomes
•••• Déficit mono-enzymatique +++ ou protéine activatri ce
•••• Anomalies des protéines membranaires
•••• Déficit multi-enzymatique (MLII et III, Austin)

•••• Maladies avec signes viscéraux et/ou dysmorphiques
- Maladies de surcharge
- Organomégalie- Infiltration des organes - Maladies à expression musculaire, neurologique
LES MALADIES LYSOSOMALES
- Maladies à expression musculaire, neurologique osseuse etc.•••• Altération de la fonction des organes •••• Altération des fonctions cellulaires •••• Maladies avec perte des acquisitions psychomotrices « préalablement acquises »
•••• Maladies avec « accidents aigus révélateurs »

LES MALADIES LYSOSOMALES
LIPIDOSES• Maladie d’Austin• Maladie de Fabry• Maladie de Farber • Maladie de Gaucher• Maladie de Landing (GM1)• Maladie de Tay-Sachs et Sandhoff (GM2)• Maladie de Krabbe • Leucodystrophie métachromatique• Maladies de Niemann-Pick type A/B• Maladie de Niemann -Pick C
MUCOPOLYSACCHARIDOSES• Maladie de Hurler/Scheie (MPS IH/S)• Maladie de Hunter (MPS II)• Maladie de Sanfilippo (MPS III A, B, C, D)• Maladie de Morquio A (MPS IVA )• Maladie de Maroteaux-Lamy (MPS VI)• Maladie de Sly (MPS VII)• Mucopolysaccharidose de type IX
GLYCOPROTEINOSES• Aspartylglucosaminurie• Maladie de Niemann -Pick C
• Maladie de Wolman
CEROIDE-LIPOFUSCHINOSES
DEFICIT TRANSPORTEURS• Cystinose• Maladie de Danon • Maladie de Salla
GLYCOGENOSE• Maladie de Pompe (Glycogénose type 2)
• Aspartylglucosaminurie• Fucosidose� αααα mannosidose� ββββ mannosidose• Sialidose et galactosialidose• Maladie de Schindler et Kanzaki• Mucolipidoses type II et III• Mucolipidose de type IV
AUTRES PATHOLOGIES• Pycnodysostose• Chediak-Higashi• Papillon-Lefèvre

TRAITMENT DES MALADIES LYSOSOMALES
Maladie DCI Nom AMM Laboratoire
Gaucher imiglucerasevelaglucerase
CerezymeV-Priv
19942010
GenzymeShire
Fabry agalsidase α
agalsidase β
ReplagalFabrazyme
2002 ShireGenzyme
MPS I iaronidase Aldurazyme 2003 Genzyme
MPS II idursulfase Elaprase 2007 Shire
MPS VI galsulfase Naglazyme 2006 BioMarin
Pompe alglucosidase Myozyme 2006 Genzyme
Miglustat Inhibiteur de substrat : Gaucher, NPC

- 6 ansRaideur d’un doigt depuis 3 ans
- 20 ansOpacités cornéennesRaideur doigts, épaules (diagnostic de PR)
- 31 ansDysplasie du col fémoral et acetabulumPied en équin varusDysostoses multiples Dysfonction cardiaque progressive , Dysfonction cardiaque progressive , Valvulopathie
- 53 ansChirurgie cardiaqueOpacitées cornéennes (grefe de cornée)Atrophie rétinienne, cécitéHypoacousieVertigesDiagnostic de MPS I par un neurologueSyndrome de Scheie(MPS I, forme atténuée )

Accumulation de Glycosaminoglycanes
Dermatane-S + Héparane-S MPS I, II (Hurler-Sheie, Hunter)
Héparane -S MPS III (San filippo )
Muco Poly Saccharidoses
Héparane -S MPS III (San filippo )
Kératane-S MPS IV (Morquio)
Dermatane-S MPS VI (Maroteaux Lamy)
Chondroïtine-S A et C MPS VII (Sly)

Enzyme déficitaire sans les MPS
MPS type Nom Enzyme décicitaire
MPS IHMPS ISMPS IH/S
Hurler ScheieHurler–Scheie
α-L-iduronidaseα-L-iduronidaseα-L-iduronidase
MPS II Hunter Iduronate-2-sulfatase
MPS III A MPS III B
Sanfilippo heparan N-sulfataseα-N-acetylglucosaminidase
MPS, mucopolysaccharidosis.1. Neufeld E & Muenzer J. The mucopolysaccharidoses. In: The Metabolic and Molecular Bases of Inherited Disease 2001:3421–52.
MPS III BMPS III CMPS III D
α-N-acetylglucosaminidaseacetyl-CoA: α-glucosaminide acetyltransferaseN-acetylglucosamine-6-sulfatase
MPS IV AMPS IV B
Morquio N-acetylgalactosamine-6-sulfataseβ-galactosidase
MPS VI Maroteaux–Lamy N-acetylgalactosamine-4-sulfatase
MPS VII Sly β-glucuronidase
MPS IX Hyaluronidase(Natowicz) syndrome
hyaluronidase

GAG accumulation
GAG
Récepteur du M6P
Vésicule de Transport
Noyau
Lysosomes
GAG
I2S, iduronate-2-sulfatase; M6P, mannose-6-phosphate; GAG, glycosaminoglycan; ER, endoplasmic reticulum.1. Alberts B, et al. Molecular Biology of the Cell. 4th ed, 2002.
Noyau
Golgi
RE
Site de clivage Site de clivage GAGs are normally
degraded in thelysosomes by
enzymes, including I2S
En l’absence I2S, la voie de
dégradation de GAG
est bloqué GAGs
s’accumulent dans les
lysosomes13

Manifestations cliniques des MPSMultisystémiques
Consultation Multi Disciplinaire Passage Enfants-Adulte
Trousseau-Beaujon- Pitié SalpétrièreBénédicte Héron , Frédéric Sedel, Nadia Belmatoug
MPSIncidence 1/100 000

Neuro-comportemental Ostéo-articulaire Cutanéo-articulaire
MucoPolySaccharidoses
•••• Dysmorphie visage•••• Macrocéphalie•••• Macrocéphalie
•••• Avance staturopondérale initiale •••• Dysostoses multiples
•••• Atteintes viscérales

Neuro-comportemental Ostéo-articulaire Cutanéo-articu laire
MucoPolySaccharidoses
San Filippo IIIHunter IIHunter II
Hürler – Scheie ISly VIIMaroteaux -Lamy VI
Morquio IVMPS IX

Hürler-Scheie, Hunter, SlySurcharge lysosomale progressiveAtteinte multisystémique évolutive
Ophtalmologiques - mégacornée- dépots cornéens- glaucome - rétinopathie, œdème papillaire, atrophie optique
Cardiaques - valvulopathie (osler++)Cardiaques - valvulopathie (osler++)- cardiomyopathie- coronaropathie- HTA, HTAP
Respiratoires - encombrement chronique et surinfections- infiltration pharyngo-laryngo-trachéo-bronchique - pneumopathie interstitielle - syndrome respiratoire obstructif et restrictif
Stomatologiques - retard de poussée dentaire, dents pointues - émail anormal, hypertrophie gingivale, kystes

Hürler-Scheie, Hunter, SlySurcharge lysosomale progressiveAtteinte multisystémique évolutive
Abdomino-digestive - hernies inguinales, ombilicale- hépato-splénomégalie - diarrhée, constipation- troubles de déglutition
ORL auditive - OSM, surdité ORL auditive - OSM, surdité
Ostéo-articulaire - cyphose thoraco-lombaire, genu valgum - dysostoses multiples, enraidissement articulaire- cassure staturopondérale puis petite taille
Neurologique - retard /régression psychomotrice- agitation, troubles du sommeil- syndrome canal carpien- compression médullaire- hydrocéphalie, atrophie cérébrale- épilepsie, tétraparésie spastique

Hürler-Scheie, Hunter, SlyFormes sévères Hernies, viscéromégalieOtite SM, chirurgie ORLInfections respiratoires Cyphose thoracolombaireDysostoses multiples
Formes atténuéesPolyarthropathieValvulopathieCompression nerveuse(Médullaire, canal carpien)
Opacités cornéennes, glaucome
Dysostoses multiplesDysmorphie
RégressionpsychomotriceEvolution rapideAtteinte multisystémique
glaucomeSurditéDiarrhée chronique
Intelligence normaleEvolution lente, insidieuseAtteinte polymorphe

Signes de début- Anasarque foeto-placentaire-Tache mongoloïde - Hernies, viscéromégalie
Infections ORL et respiratoires- Macrocéphalie, Avance staturale- Traits épaissis, Hirsutisme
Cyphose thoraco-lombaire- Enraidissement articulaire

Cas clinique 1 : Maladie de Hürler
4 ans •••• Limitations articulaires multiples•••• Traits du visage épais et macroglossie, •••• Hépatomégalie •••• Otite séreuse bilatérale •••• Otite séreuse bilatérale •••• Hernies inguinales : chirurgie à 3 mois puis à 7ans•••• Hernie ombilicale•••• Dysplasie valve mitrale•••• Dysostoses multiples•••• Mégacornée

• Avant ERT puis• 3 mois d’aldurazyme
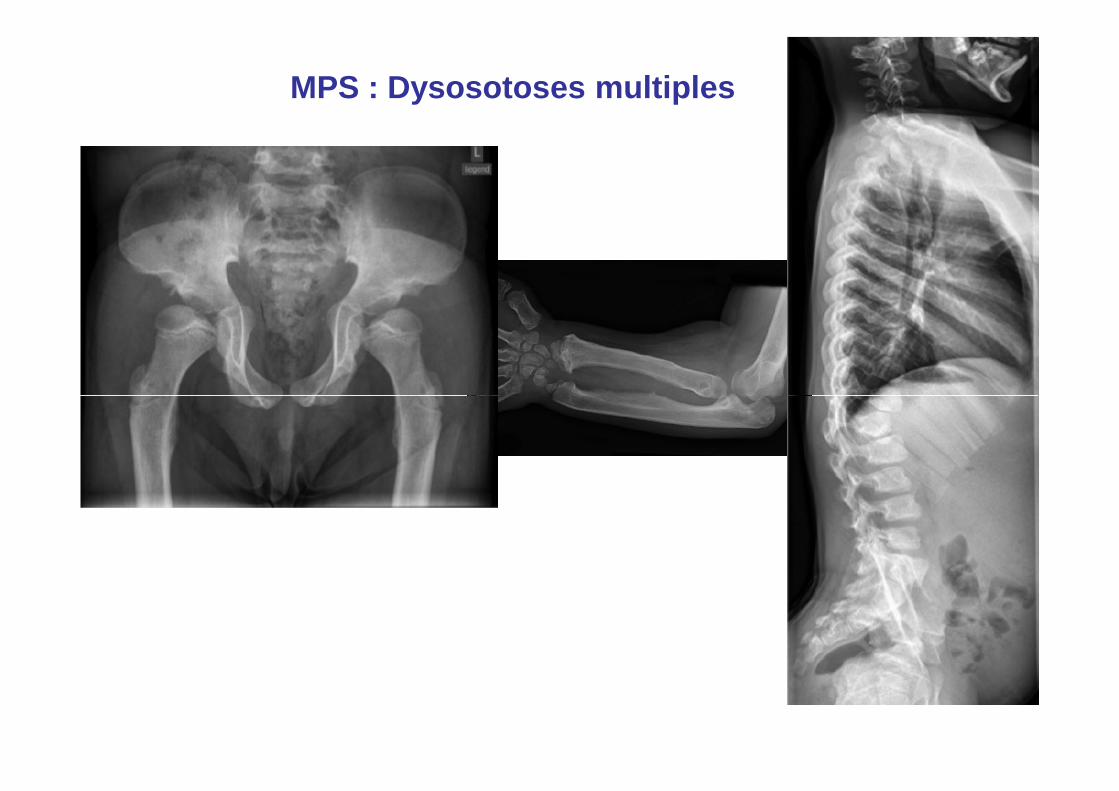
MPS : Dysosotoses multiples


• Dysmorphie faciale modérée• Doigts trapus et courts, • Raccourcissement des
tendons • Raideur articulaire
progressive sans signe
MPS 1 : Hürler/Sheie 1/100000 naissances
progressive sans signe inflammatoire, ou érosions
• Syndrome du canal carpien, bilatéral avant 35 ans

Mains en griffe • Doigts trapus et courts • Raccourcissement des
tendons • Raideur articulaire
progressive sans signe
MPS 1 : Hürler
progressive sans signe inflammatoireSyndrome du canal
carpien

MPS 1 : Hürler• Valvulopathie 87.9 %• Raideur articulaire 87.7 %• Opacités cornéennes 81.8 %• SCC 66.7 %• ATCD de hernie 65.1 %• Dysmorphie faciale 51.6 %• Dysostose multiple 49.2 %

incidence 1 in 162,000 live male births3
MPS II atténuéesHunter
Incidence 1/162 000

Hunter : MPS II Forme atténuée Forme sévère
• Troubles cognitifs • Atteinte viscérale
1. Neufeld EF and Muenzer J. The mucopolysaccharidoses. In: Scriver et al. The Metabolic and Molecular Bases of Inherited Disease. 2001:3421–52. Photograph (left) reproduced with permission from the authors and McGraw-Hill.
• Pas de trouble cognitif• Atteinte viscérale isolée

Hunter• Raideur articulaire• Mains en griffe • Douleurs • Parfois révélateurs
• Contractures articulaires• Limitation articulaire • Syndrome du canal carpien• Pas de signe inflammatoire
1. Link B et al. Orthopedic Reviews 2010;2:56–64.

Hunter• Cou court • Os épais de forme anormale• Déformations du rachis
- Cyphose- Cyphose- Déformation des vertèbres- Compression médullaire
- Spondylolysthésis - Compression médullaire
• Petite taille 1. Link B et al. Orthopedic Reviews 2010;2:56–64.

Hunter : 23 ansAprès 12 mois d’Elaprase
32

MPS I ET II atténuées
35 – 40 ans Diarrhée chronique
EnfanceAmygdalectomieRaideurs articulaires progressives « Polyarthrite chronique»20 ansDyspnée d’effort« Polyarthrite rhumatoïde »
Diarrhée chronique HépatomégalieSurdité progressiveValvuloplastie aortique et mitraleCompression médullaire : LaminectomieOpacités cornéennes : Greffe de cornée

Maroteaux -Lamy
- 2 formes évolutives (rapide/lente)- Pas d’atteinte intellectuelle- Valvulopathie constante
- Atteinte ostéo-articulaire +++Compression médullaire : occipito cervicale +++- Traitement : Naglazymz

Morquio A
- Pas d’atteinte intellectuelle
- Atteinte ostéo-articulaire avec déformations
- hyperlaxité- Compression médullaire - Luxation atloïdo-axoïdienne +++ sténose cervicale
- Atteinte sensorielle tardive

DIAGNOSTIC
Suspicion clinique
NFS : cellules de surchargeMucopolysaccharides urinaires (GAG)
Activité enzymatique leucocytaireEtude du gène (mutations)
Diagnostic anténatal pour les futurs grossesses

GAG urinaires•••• Orientation diagnostique
Dermatane-S + Héparane-S MPS I, II, (VII)Héparane-S MPS IIIKératane-S MPS IVDermatane -S MPS VIDermatane -S MPS VIChondroïtine-S A et C MPS VII
•••• Peuvent être normaux Formes atténuéesAdultes

Traitements symptomatiques– Kinésithérapie, orthophonie– Ergothérapie, psychomotricité– Appareillages, installations– Chirurgie viscérale, ORL, orthopédique,
cardiaque, ophtalmologique, neurochirurgie
• difficultés d’intubation • difficultés d’abord veineux
si possible regrouper les interventions +++
– Traitements médicamenteux– Ventilation, nutrition

MPS I (MPS II, III, VI, VII)Traitements spécifiques
Transplantation de cellules souches hématopoïétique s
- Greffe de moelle osseuse 1980- Sang de cordon- Mortalité/morbidité 20%- Echec, GVH, hémorragie pulmonaire, décès - Echec, GVH, hémorragie pulmonaire, décès - Tissus difficiles à atteindre
- Os +++- Cerveau stabilisation neuro -cognitive
(Hürler)- Œil : greffe de cornée
Indications : MPS I sévère < 2ans

MPS I, II et VITraitement enzymothérapie substitutive
Enzyme recombinante
• MPS I : ALDURAZYME (AMM 2003)100 à 200 UI/kg/sem
• MPS VI : NAGLAZYME (AMM 2006)1 mg/kg/sem
• MPS II : ELAPRASE (AMM 2007)0,5 mg/kg/sem
• Perfusion IV hebdomadaire de 3 à 4 heures
• En hospitalisationEffets indésirables : réactions liées à la perfusi on (allergiques)attention si atteinte des voies respiratoires

•••• Traitement MPS I : Aldurazyme
- Perfusion hebdomadaire, IV lente
- Posologie = 100 U/kg de poids corporel
- Vitesse de perfusion initiale de 2 U/kg/h - Vitesse de perfusion initiale de 2 U/kg/h augmentée /15 mn, jusqu’à 43 U/kg/h.
- Chambre implantable parfois nécessaire

ALDURAZYMEEvaluation à 5 ans
AMELIORATIONTolérance à l’effort Mobilité globale Force musculaireMobilité articulaire
STABILISATIONAtteinte myocardique et valvulaireTroubles oculaires Acuité visuelle
Mobilité articulaireFonction respiratoireSommeilTaille foie et rateTroubles digestifsSouplesse peau et
cheveux
Acuité visuelleAuditionCroissance staturo-
pondéraleFonction cognitive ?

ELAPRASEEvolution à 2 ans
Amélioration M1-M12M1 Etat général dynamisme activité quotidienneM2 Réduction HSM, GAG subnormauxM3 Amplitudes articulaires, croissanceM6 Test de marche de 6mn, fonction respiratoire
Stabilisation >M18La plupart des symptômes à partir de 18 mois
InconnusFormes de l’enfant < 5ansEvolution neurocognitive

MPS•••• Traitement précoce si Diagnostic précoce
– Diversité des signes cliniques
•••• Prise en charge pluridisciplinaire
•••• Traitements spécifiques•••• Traitements spécifiques– Enzymothérapie substitutive dès le diagnostic– Inefficacité sur lésions constituées osseuses,
cardiaques, neurologiques– Greffe pour maladie de Hürler <2ans
•••• Evaluation fonctionnelle et qualité de vie adaptée MPS

THERAPIES DES MALADIES LYSOSOMALESETS : inconvénients
- Faisabilité : perfusions itératives, PAC, domicile
- Risque : immuno -allergique
- Efficacité : perte d’efficacité / immunisation (faible) dépend de l’organe, de la maladie
du stade d’évolution- Coût

THERAPIES DES MALADIES LYSOSOMALESEnzymothérapie substitutives
ETS : l’efficacité dépend de différents facteurs
• Organes « bons répondeurs » : - organomégalie, moelle osseuse- tissus mous, appareil péri-articulaire
• « Répondeur tardif » : squelette
• « Non répondeurs » : cornée, valvulopathies (MPS)
• « Répondeur incertain » :précocité du traitement (MPS) ++++++

Perspectives thérapeutiques- Thérapies cellulaires
– Cellules souches hématopoiétiques / Sang de cordon
– CSH génétiquement modifiées
- Thérapie / réduction de substrat
– Génesteine– Miglustat– Eliglustat (essais cliniques)
- Enzymothérapie substitutive
– Spécifique– Problème
d’adressage (SNC, os, ...)
- Molécules chaperonnes– Stabilisation d’une protéine
mal conformée– Mutation – spécifique
- Thérapie génique intracérébrale
- Translecture des codons stop





Efficacité du traitement• Idursulfase treatment in 94 patients, for up to 3 ye ars, provided
sustained improvement in selected somatic parameters
• Clinical improvements in the 2-year extension phase included
– Increased walking capacity a
– Reduced liver and spleen volumes b– Reduced liver and spleen volumes– Reduced urinary GAG levels– Improved pulmonary function– Improved shoulder abduction and
flexion/extension and stabilization of mobility in other joints
GAG, glycosaminoglycan. aComponent of primary endpoint. bSecondary endpoints.
Muenzer J, et al. Genet Med. 2011;13:95–101
52

MALADIES LYSOSOMALES CONCLUSIONS
•••• Maladies génétiques, rares , invalidantes•••• Maladies souvent multi-systémiques
•••• Physiopathologie mal comprise
•••• Ciblage subcellulaire : comment entrer dans le •••• Ciblage subcellulaire : comment entrer dans le lysosome •••• Nombreuses sans traitement•••• Traitements symptomatiques +++ •••• Traiter avant les lésions irréversibles, fibrose •••• Amélioration de la connaissance d’autres maladies•••• Coût

Organisation du CETL
Comité d’Evaluation des Maladies LysosomalesCETG CETF CETMPS CETNP CETP
Comité d’Evaluation du Traitement de la Maladie de Gaucher
Comité d’Evaluation du Traitement de la Maladie de Fabry
Comité d’Evaluation du Traitement des MPS
Comité d’Evaluation du Traitement de la Maladie de Niemann Pick
Comité d’Evaluation du Traitement de la Pompe
Multidisciplinaire (Associations de patients (VML, APMF, San Filipo)
Orphanet (Ségolène Aymé)
Interlocuteur :
ANSM, Ministère de la santé, CNAMAssociation de patients : (VML, APMF, Glycogénose, San Filipo)

Pour en savoir plus , …
•••• Orphanet www.orphanet•••• CETL www.cetl.net•••• VML www.vml-asso.org•••• HAS PNDS•••• HAS PNDS•••• Association de patients Vaincre les Maladies Lysosomales•••• Le « Scriver » Textbook
Lysosomal disorders