bastien burat | thèse de doctorat | université de limoges...
TRANSCRIPT

Université de Limoges École Doctorale Bio-Santé (ED 524) Unité Mixte de Recherche INSERM S850 – Pharmacologie des Im-munosuppresseurs et de la Transplantation
Thèse pour obtenir le grade de
Docteur de l’Université de Limoges
Discipline / Spécialité : Pharmacologie et Sciences du Médicament
Présentée et soutenue par
Bastien Burat
Le 19 décembre 2017
Apports de la protéomique quantitative différentielle haut-débit à
l’étude des mécanismes de modification du cytosquelette de cel-
lules tubulaires proximales induits par les Inhibiteurs de la Calci-
neurine.
Thèse dirigée par le Professeur Marie ESSIG
JURY :
Rapporteurs
Professeur Laurent JUILLARD
Docteur Joost-Peter SCHANSTRA
Examinateurs
Docteur Chantal BARIN-LE GUELLEC
Docteur Nicolas PALLET (président du jury)
Directrice de thèse
Professeur Marie ESSIG
Thèse de doctorat

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 2

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 3
Remerciements
Aux membres du jury, Messieurs Laurent Juillard et Joost-Peter Schanstra en leur
qualité de rapporteurs ainsi que Madame Chantal Barin-Le Guellec et Monsieur Nicolas Pal-
let en leurs qualité d’examinateurs,
C’est un honneur de vous compter parmi les membres de ce jury. Merci à chacun
d’entre vous pour le temps consacré à la lecture critique de mes travaux.
A Marie Essig,
Merci de m’avoir accordé ta confiance dès le début de cette aventure. Merci d’avoir
prêté une oreille attentive et compatissante à mes doutes et d’avoir su prodiguer les meil-
leurs conseils aux moments où j’en avais le plus besoin. Je suis fier du chemin parcouru,
scientifiquement et humainement, grâce à toi.
A Julien Gonzalez
C’est par toi que tout a commencé. Merci de m’avoir donné ma chance, de m’avoir
soutenu et de m’avoir tant appris.
Aux membres de l’UMR INSERM 850,
Merci à tous pour votre accueil, et votre contribution, quelque qu’elle soit, à ces tra-
vaux de thèse.
Merci à Hélène Arnion. Tu as mis autant d’énergie et de ressources à m’aider que
moi à t’embêter, ce qui n’est pas peu dire… Merci d’avoir été une collègue dévouée et une
amie patiente.
Merci à François-Ludovic Sauvage. L’optimisation de l’iTRAQ doit beaucoup à ton
expertise technique et à ta capacité à résoudre tous mes problèmes en un temps record.
Merci à James Javellaud. Nos discussions vont beaucoup me manquer. Chevalier,
iTRAQman te salue.
Merci à Khadija Baali. Pour ta gentillesse et ton amitié autant que pour le bon souve-
nir d’avoir travaillé à tes côtés.
Merci à Zora El-Ouafi et Quentin Faucher, stagiaires 4 étoiles. Ce fût un plaisir et un
honneur de vous initier à la recherche et de vous accompagner au cours de vos projets res-
pectifs qui s’articulent avec une importance majeure dans ces travaux de thèse.
Merci à Benjamin Chantemargue et Danko Stamenic, braves compagnons
d’infortune.
Merci à Patrick Trouillas d’avoir apporté un œil neuf et pertinent à nos questionne-
ments scientifiques.
Merci à Florent Di Meo. Pour tes conseils, ton recul et ton indulgence.
Merci à Jean-Hervé Comte et Patricia Festa pour leur présence et leur aide au quoti-
dien.
Au magasinier, à ses parents et à son petit frère…

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 4
Résumé
En transplantation d’organe solide, les Inhibiteurs de la Calcineurine, Cyclosporine A
et Tacrolimus, ont permis une amélioration significative de la survie à court terme du greffon
en prévenant le rejet d’allogreffe. Cette évolution positive est contrebalancée par la toxicité
chronique, notamment rénale, de ces immunosuppresseurs, susceptible de contribuer au
développement complexe et multifactoriel de la dysfonction chronique du greffon, facteur
pronostique majeur d’une insuffisance rénale terminale (perte définitive de la fonction ré-
nale).
Bien que très largement étudiés, les mécanismes physiopathologiques impliqués
dans les effets néphrotoxiques des Inhibiteurs de la Calcineurine sont encore en grande par-
tie mal compris. L’objectif principal de ce travail a été de combiner deux approches complé-
mentaires, une ciblée l’autre non, afin de mettre en lumière des aspects encore non décrits
de la physiopathologie des ICN sur des cellules tubulaires proximales.
La première approche repose sur l’application des outils de protéomique quantitative
« shotgun » à l’analyse, sans a priori, haut-débit, à grande échelle, des variations
d’expression protéique au sein d’un modèle de cellules tubulaires proximales en culture. La
technologie choisie est le marquage iTRAQ (« isobaric Tags for Relative & Absolute Quanti-
tation ») qui consiste en un marquage chimique par quatre tags isobares, indiscernables en
mode MS, caractérisés individuellement en mode MS/MS. L’optimisation de la technologie
iTRAQ a permis la mise au point d’un protocole, couvrant les aspects successifs de prépara-
tion et de passage d’échantillon, d’analyse en masse et de retraitement bio-informatique,
compatible avec une analyse fiable et robuste par chromatographie liquide haute-
performance couplée à un spectromètre de masse haute résolution. En particulier, ce travail
a abouti au développement d’un algorithme permettant le tri des données peptidiques et le
calcul des ratios iTRAQ protéiques sur la base de la variabilité biologique.
La seconde approche applique de manière ciblée les outils classiques de biologie
moléculaire à l’étude du cytosquelette d’Actine de cellules tubulaires proximales en culture.
La combinaison de ces deux stratégies complémentaires a permis de mettre en lu-
mière un rôle inédit du cytosquelette d’Actine dans les effets physiopathologiques de la Cy-
closporine A sur des cellules tubulaires proximales en apportant des éléments en faveur d’un
mécanisme reposant sur une régulation originale de la dynamique intracellulaire de l’Actine.
Mots-clés : Cyclosporine A, Tacrolimus, cellules tubulaires proximales, protéomique
quantitative haut-débit, iTRAQ, LC-MS/MS, CiR-C, cytosquelette d’Actine, Cofiline

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 5
Droits d’auteurs
Cette création est mise à disposition selon le Contrat :
« Attribution-Pas d'Utilisation Commerciale-Pas de modification 3.0 France »
disponible en ligne : http://creativecommons.org/licenses/by-nc-nd/3.0/fr/

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 6
Table des abréviations
ABP : Actin-Binding Protein
ADP : Adénosine Diphosphate
Arp : Actin-related protein
AT (TA) : Atrophie Tubulaire
ATP : Adénosine Triphosphate
CaN : Calcineurine
CsA : Cyclosporine A
CFL : Cofiline
(mono)CFL : Cofiline monomérique
(di)CFL : Cofiline dimérique
(tetra)CFL : Cofiline tétramérique
CRAC : Calcium-release activated channels
CypA : Cyclophiline A
CypB : Cyclophiline B
CypD: Cyclophiline D
CiR-C : Customizable iTRAQ Ratios Calculator
CTGF : Connective Tissue Growth Factor
DAG : DiAcylGlycerol
ECA : Enzyme de Conversion de l’Angiotensine
EGF : Epidermal Growth Factor
ERAD : ER-Associated Degradation
ERO (ROS) : Espèces Réactives de l’Oxygène
FDA: Food & Drug Administration
FI (IF) : Fibrose Interstitielle
FKBP12 : 12 kDa FK506-Binding Protein
H+/K+ ATPase : pompe Proton-Potassium ATPase
HK-2 : Human Kidney 2
ICAT : Isotope-Coded Affinity Tags
ICN : Inhibiteurs de la CalciNeurine
IP3 : inositol-1.4.5-triphosphate
iTRAQ : isobaric Tags for Relative and Absolute Quantitation
LC : Liquid Chromatography
LFQ : Label-Free Quantification
LLC PK-1 : Lilly Laboratories Cells Porcine Kidney 1
LZ : Leucine Zipper
MEC (ECM) : Matrice ExtraCellulaire
MMF : Mycophénolate Mofétil

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 7
MRTF : Myocardin-Related Transcription Factors
MS/MS : spectrométrie de masse en tandem
Na+/K+ ATPase : pompe Sodium/Potassium ATPase
NFAT : Nuclear Factor of Activated T cells
NHS : N-hydroxysuccinimide
NKCC2 : cotransporteur Sodium/Potassium/Chlorure 2
NO : monoxyde d’azote
O2- : ion superoxyde
PIP2 : phosphatidyl inositol-4,5-diphosphate
PPIase : Peptidyl Prolyl cis/trans Isomérase
RE (ER) : Réticulum Endoplasmique
RPTEC : Renal Primary Tubular Epithelial Cells
SILAC : Stable Isotope Labeling for Amino acids in Cell culture
SRAA : Système Rénine-Angiotensine-Aldostérone
SRF : Serum Response Factor
TAC/FK506 : Tacrolimus
TCF : Ternary Complex Factors
TCR : T-Cell Receptor
TEM (EMT) : Transition Épithélio-Mésenchymateuse
TGF : Transforming Growth Factor
TNF : Tumor Necrosis Factor
UPR : Unfolded Protein Response

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 8
Table des matières
Introduction générale ........................................................................................................... 11
Chapitre I. Transplantation, immunosuppression et néphrotoxicité des Inhibiteurs de la
Calcineurine ......................................................................................................................... 13
I.1. Etat des lieux en transplantation d’organe solide ........................................................ 13
I.2. Les Inhibiteurs de la Calcineurine : Cyclosporine A et Tacrolimus .............................. 14
I.3. Mécanisme d’action pharmacologique des Inhibiteurs de la Calcineurine................... 17
I.4. Manifestations histologiques et fonctionnelles de la néphrotoxicité des Inhibiteurs de la
Calcineurine (17,18) ......................................................................................................... 19
I.4.1. Néphrotoxicité aigüe ............................................................................................ 19
I.4.2. Néphrotoxicité chronique (48–51) ........................................................................ 20
I.5. Mécanismes physiopathologiques cellulaires et moléculaires des cellules tubulaires
exposées aux Inhibiteurs de la Calcineurine..................................................................... 21
I.5.1. Perturbation du cycle cellulaire : sénescence et apoptose ................................... 21
I.5.2. Stress du réticulum endoplasmique ..................................................................... 23
I.5.3. Stress oxydant ..................................................................................................... 24
I.5.4. Perturbation de l’homéostasie électrochimique .................................................... 25
I.5.5. Transition épithélio-mésenchymateuse ................................................................ 26
I.5.6. Réorganisation du cytosquelette d’Actine induite par la Cyclosporine A ............... 28
Chapitre II. Approches protéomiques quantitatives différentielles à haut-débit « bottom-
up/shotgun » ........................................................................................................................ 29
II.1. Technologies de marquage isotopique stable ............................................................ 29
II.1.1. Marquage isotopique : technologie « Isotope-Coded Affinity Tags » ou ICAT ..... 29
II.1.2. Marquage métabolique : technologie « Stable Isotope Labeling by Amino Acids in
Cell Culture » ou SILAC ................................................................................................ 32
II.1.3. Marquage isobare : « isobaric Tags for Relative and Absolute Quantitation »
(iTRAQ) ........................................................................................................................ 34
II.2. Quantification « label-free » (LFQ)(121) .................................................................... 37
II.3. Apports et limites de la technologie iTRAQ par rapport aux technologies ICAT, SILAC
et LFQ .............................................................................................................................. 39
II.4. Etude protéomique haut-débit de cellules tubulaires proximales exposées aux ICN par
protéomique Shotgun-iTRAQ ........................................................................................... 41
II.4.1. Hypothèse de travail et objectifs ......................................................................... 41
II.4.2. Développement et optimisation d’une stratégie d’analyse intégrée des données
MS/MS pour l’étude protéomique quantitative par la technologie iTRAQ ; Application aux
cellules rénales tubulaires proximales exposées aux ICN ............................................. 43
II.4.2.1 Article 1. ........................................................................................................ 43
II.4.2.2 Seuils des variations biologiquement significatives ........................................ 86
II.4.2.3 Cartographie dynamique d’un sous-protéome d’intérêt : les protéines
associées à l’Actine ................................................................................................... 87
Chapitre III. Organisation du cytosquelette d’Actine des cellules épithéliales tubulaires
proximales ........................................................................................................................... 89
III.1. Organisation générale du cytosquelette d’Actine (130) ............................................. 89
III.2. La famille ADF/Cofiline (136,137) ............................................................................. 92
III.2.1. Fonctions biologiques de la Cofiline ................................................................... 92
III.2.1.1 Renouvellement du cytosquelette d’Actine ................................................... 92
III.2.1.2 Transport de l’Actine et transcription............................................................. 92

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 9
III.2.1.3 Réponse au stress........................................................................................ 93
III.2.1.4 Apoptose ...................................................................................................... 93
III.2.2. Mécanismes de régulation de la Cofiline (Figure 14) (159) ................................ 95
III.2.2.1 Phosphorylation / Déphosphorylation (Figure 13) ......................................... 95
III.2.2.2 Liaison à PIP2 et sensibilité au pH cellulaire ................................................. 97
III.2.2.3 Sensibilité au stress oxydatif ........................................................................ 98
III.2.2.4 Etat d’oligomérisation de la Cofiline .............................................................. 99
III.2.2.5 Concentration totale et rapport Cofiline / Actine .......................................... 100
III.3. Régulation de l’expression génique par la dynamique du cytosquelette d’Actine .... 102
III.3.1. Implication de l’Actine nucléaire dans la transcription génique (140,189,190) .. 102
III.3.2. Cas particulier de la régulation des facteurs de transcription MRTF et SRF
(191,192) .................................................................................................................... 103
III.3.2.1 Mécanistique de l’axe RhoGTPases-Actine-SRF ........................................ 103
III.3.2.2 Gènes cibles de MRTF-SRF ....................................................................... 104
III.4. Signalisation CsA-dépendante de l’axe Actine-MRTF-SRF de cellules tubulaires
proximales ...................................................................................................................... 107
III.4.1. Hypothèses de travail et objectifs ..................................................................... 107
III.4.2. Etude des modifications Cofiline-dépendantes de l’axe Actine-MRTF-SRF de
cellules tubulaires proximales induites par la Cyclosporine A...................................... 108
Chapitre IV. Discussion, conclusion et perspectives .......................................................... 133
IV.1. Discussion et conclusion ........................................................................................ 133
IV.1.1. Evolution de la stratégie de protéomique quantitative ...................................... 133
IV.1.2. Problématique des outils de quantification ....................................................... 134
IV.1.3. Evolution de l’algorithme CiR-C ....................................................................... 136
IV.1.4. Interprétation interactomique et évolution vers la biologie des systèmes ......... 137
IV.1.5. Détermination des variations biologiques significatives.................................... 138
IV.1.6. Résultats préliminaires : application de la technologie iTRAQ à l’étude du
protéome de cellules tubulaires proximales exposées aux ICN ................................... 139
IV.2. Perspectives .......................................................................................................... 145
Références bibliographiques .............................................................................................. 146
Annexes............................................................................................................................. 160

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 10
Table des figures et tableaux
Figure 1. Structures chimiques des Inhibiteurs de la Calcineurine ....................................... 16
Figure 2. Voie d’activation des TCD4+ et mode d’action pharmacologique des ICN. .............. 18
Figure 3. Effets différentiels des ICN sur l’organisation du cytosquelette d’Actine des TCP. 28
Figure 4. Principe de l’ICAT. ................................................................................................ 31
Figure 5. Principe du SILAC. ................................................................................................ 33
Figure 6 1. Principe de l’iTRAQ. ........................................................................................... 35
Figure 7. Principes de la LFQ. ............................................................................................. 38
Figure 8. Dose-réponse de l’activité de transcription du facteur de transcription NFAT de LLC
PK-1 exposées 24 h aux ICN. .............................................................................................. 42
Figure 9. Distribution gaussienne du nombre de protéines en fonction de la valeur des
quantifications iTRAQ RSPI. ................................................................................................ 86
Figure 10. Réseau STRING d’intéractions des protéines associées à l’Actine, identifiées et
quantifiées au cours des travaux rapportés dans l’Article 1. ................................................. 87
Figure 11. Représentation en heat-map des variations d’abondance relative des protéines
associées à l’Actine, identifiées et quantifiées au cours des travaux rapportés dans l’Article 1.
............................................................................................................................................ 88
Figure 12. Organisation générale du cytosquelette d’Actine. ................................................ 91
Figure 13. Voies de signalisation impliquées dans la régulation par phosphorylation-
déphosphorylation de la CFL (d’après (159)). ...................................................................... 96
Figure 14. Mécanismes de régulation de la CFL. ................................................................. 98
Figure 15. Représentation en heat-map de la quantification iTRAQ du sous-protéome
d’intérêt de LLC-PK-1 traitées 24 h par les ICN (iTRAQ-nanoLC-QqTOF, ProteinPilot-
Paragon-CiR-C). ................................................................................................................ 134
Figure 16. Réseau STRING des 130 protéines identifiées et quantifiées au cours de l’analyse
du protéome de cellules LLC PK-1 exposées 24 h aux ICN (iTRAQ-nanoLC-QqTOF, Mascot-
jTRAQx-CiR-C). ................................................................................................................. 140
Figure 17. Représentation en heat-map de la quantification iTRAQ (RSPI) du sous-protéome
d’intérêt de LLC-PK-1 traitées 24 h par les ICN (iTRAQ-nanoLC-QqTOF, Mascot-jTRAQx-
CiR-C). .............................................................................................................................. 143
Figure 18. Réseau STRING des 70 protéines dont l’expression est perturbée par l’exposition
aux ICN (iTRAQ-nanoLC-QqTOF, Mascot-jTRAQx-CiR-C). ............................................... 144
Tableau 1. Liste de gènes cibles de SRF validés expérimentalement (d’après (200)). ....... 105
Tableau 2. Profils de variations d’expression protéique du sous-protéome d’intérêt (n=70) de
LLC PK-1 traitées 24 h par les ICN (iTRAQ-nanoLC-QqTOF, Mascot-jTRAQx-CiR-C). ..... 141
Tableau 3. Liste des protéines identifiées dont l’expression n’est pas impactée par les ICN
(iTRAQ-nanoLC-QqTOF, Mascot-jTRAQx-CiR-C). ............................................................ 142

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 11
Introduction générale
La situation actuelle en transplantation d’organe solide est un enjeu majeur de santé
publique. La demande croissante associée à une offre limitée de greffons pousse à
l’optimisation des protocoles de prélèvement, de conservation, d’implantation et
d’immunosuppression dans le but d’augmenter la durée de vie des organes effectivement
transplantés.
L’introduction des médicaments immunosuppresseurs Inhibiteurs de la Calcineurine
aux protocoles d’immunosuppression a permis une prévention efficace du risque de rejet
aigu d’allogreffe et une amélioration significative de la survie à court terme du greffon. Mal-
gré l’apparition de nouvelles classes de traitements immunosuppresseurs, les ICN sont en-
core massivement utilisés et restent la thérapeutique de choix.
Cependant, l’utilisation clinique des ICN pose le problème de la survie à long terme
du greffon et du patient transplanté. En effet, l’exposition aux ICN est associée à une né-
phrotoxicité aigüe, caractérisée par des atteintes vasculaires réversibles et par une néphro-
toxicité chronique marquée par le développement d’atteintes rénales irréversibles générali-
sées, tubulo-interstitielles (fibrose interstitielle et atrophie tubulaire) et glomérulaires (glomé-
ruloscléroses) conduisant à une dysfonction chronique du greffon et à une insuffisance ré-
nale chronique puis terminale.
Alors que les mécanismes immunologiques impliqués dans l’action pharmacologique
des ICN sont parfaitement décrits, les mécanismes physiopathologiques impliqués dans les
effets néphrotoxiques sont en grande partie non élucidés à ce jour.
Au cours de ce travail de thèse, l’étude des mécanismes physiopathologiques a été
envisagée selon deux stratégies complémentaires appliquées à des cellules tubulaires
proximales en culture exposées à des concentrations cliniquement compatibles d’ICN.
La première stratégie, sans a priori, a reposé sur l’approche de protéomique quantita-
tive à haut débit iTRAQ (pour « isobaric Tags for Relative & Absolute Quantitation »), adap-
tée à l’analyse multiplexée des niveaux d’expression protéique au sein d’échantillons de na-
ture variée, par chromatographie liquide haute performance couplée à la spectrométrie de
masse en tandem.
La technologie iTRAQ a été optimisée pour son application à l’étude du protéome de
cellules tubulaires proximales en culture afin d’observer, à l’échelle cellulaire et de manière
simultanée, les variations d’expression protéique induites par l’exposition prolongée (24 h)
aux ICN.
La seconde stratégie, avec a priori, a consisté en l’application des techniques clas-
siques de biologie moléculaire à l’étude du cytosquelette d’Actine de cellules tubulaires

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 12
proximales en culture, en tant qu’acteur principal d’un nouveau mécanisme physiopatholo-
gique de la CsA.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 13
Chapitre I. Transplantation, immunosuppression et néphrotoxicité des Inhibi-
teurs de la Calcineurine
I.1. Etat des lieux en transplantation d’organe solide
La transplantation d’organes constitue la thérapeutique de choix des pathologies res-
ponsables de la perte définitive de fonction d’un organe vital. Selon le rapport médical et
scientifique de l’Agence de la Biomédecine, au cours de l’année 2015 en France, 5746 nou-
veaux patients ont été transplantés (+ 22.9 % sur la période 2006-2015) pour une population
totale 57 171 sujets possédant un greffon fonctionnel au 31 décembre. La transplantation
rénale représente la majorité des greffes (incidence / prévalence de 61 %). Tous organes
confondus, le nombre de patients en attente de transplantation a doublé sur la période 2006-
2015 pour atteindre 14 500 inscrits sur liste d’attente au 1er janvier 2016. La situation en
transplantation est donc la suivante : il existe une forte demande révélant une pénurie de
greffons. Cet enjeu majeur de santé publique nécessite l’augmentation du nombre de prélè-
vements et l’optimisation de la durée de vie des organes transplantés. Ce dernier objectif est
compliqué par l’augmentation de l’âge des donneurs (+ 7 ans entre 2006 et 2015) et par
leurs antécédents avec une qualité moindre des organes prélevés.
Les étapes de prélèvement, de conservation intermédiaire et d’implantation condi-
tionnent le succès à court et à long terme de la transplantation. La période pendant laquelle
le greffon n’est plus vascularisé est appelée ischémie froide. En France, en 2013, elle est en
moyenne de 17,1 heures en transplantation rénale. Les phénomènes d’ischémie-reperfusion,
d’autant plus importants lorsque l’ischémie froide est prolongée, favorisent l’apparition de
lésions cellulaires qui participent à l’altération du greffon à long terme.
Au final, l’organe est implanté au sein d’un organisme qui le considère comme un
élément étranger à éliminer. La meilleure histocompatibilité possible entre donneur et rece-
veur est indispensable mais reste le plus souvent incomplète. Sans prise en charge, le rejet
aigu d’allogreffe par le système immunitaire de l’hôte est inéluctable et conduit à l’arrêt, pré-
coce, du fonctionnement du greffon. Un traitement immunosuppresseur est nécessaire pour
inhiber la réponse immunitaire à médiation cellulaire et humorale et éviter ainsi les phéno-
mènes de rejet.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 14
I.2. Les Inhibiteurs de la Calcineurine : Cyclosporine A et Tacrolimus
Les Inhibiteurs de la Calcineurine (ICN) sont les chefs de file des traitements immu-
nosuppresseurs à la base des protocoles cliniques post-transplantation. Selon les rapports
annuels Organ Procurement and Transplantation Network / Scientific Registry of Transplant
Recipients 2014 et 2015, un traitement immunosuppresseur intégrant un ICN est reçu par
plus de 95 % des transplantés rénaux aux Etats-Unis dont 93 % reçoivent un régime immu-
nosuppresseur à base de Tacrolimus contre 2 % recevant un régime à base de Cyclosporine
A (1,2).
La Cyclosporine A (CsA) a été isolée en 1970, au cours d’une de campagne de re-
cherche de nouvelles molécules antibiotiques ou antifongiques menée par le Département
de Microbiologie du laboratoire pharmaceutique Sandoz, à partir d’une souche norvégienne
de Tolypocladium inflatum, un champignon du sol. La CsA appartient à une superfamille de
métabolites fongiques aux propriétés insecticides et fongicides, polypeptides non riboso-
maux cycliques de 11 acides aminés synthétisés (Figure 1) par un polypeptide multienzyma-
tique, la Cyclosporine synthétase.
Dès 1976, Borel et coll. décrivent l’action immunosuppressive de la CsA, en obser-
vant notamment une augmentation de la survie et une diminution du rejet significatives au
cours d’expériences d’allogreffe de peau chez la Souris et le Rat (3).
L’incorporation de la CsA aux protocoles cliniques d’immunosuppression en trans-
plantation d’organe solide est expérimentée à la fin des années 1970. En 1979, Calne et coll.
rapportent l’efficacité de l’immunosuppression par la CsA, chez 34 patients transplantés ré-
naux, hépatiques ou pancréatiques (4,5). La Food & Drug Administration (FDA) américaine
autorise l’utilisation clinique de la CsA en tant qu’immunosuppresseur pour le traitement du
rejet aigu d’allogreffe ou de maladies auto-immunes en 1983.
Le Tacrolimus (pour Tsukuba mACROLide IMmUnoSuppressant) ou FK-506 (TAC),
découvert en 1984 au cours d’une campagne de recherche de nouvelles molécules immuno-
suppressives et chimiothérapeutiques menée par le laboratoire pharmaceutique Fujisawa,
est un macrolide lactone neutre (Figure 1) aux propriétés antimicrobiennes, synthétisé par la
bactérie du sol Streptomyces tsukubaensis.
En 1987, Kino et coll. s’intéressent aux propriétés immunosuppressives de TAC, chez
la Souris, et concluent que le TAC présente un potentiel immunosuppresseur supérieur à la
CsA, à concentrations équivalentes (6).
L’utilisation du TAC dans les protocoles d’immunosuppression post-transplantation
d’organe solide est envisagée dès la fin des années 1980. En 1989, Starzl et coll. ont re-
cours à TAC chez 14 patients transplantés hépatiques et rapportent une efficacité supérieure

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 15
à la CsA (7). La FDA autorise l’indication clinique de TAC en tant qu’immunosuppresseur
pour le traitement du rejet aigu d’allogreffe en transplantation, en 1994.
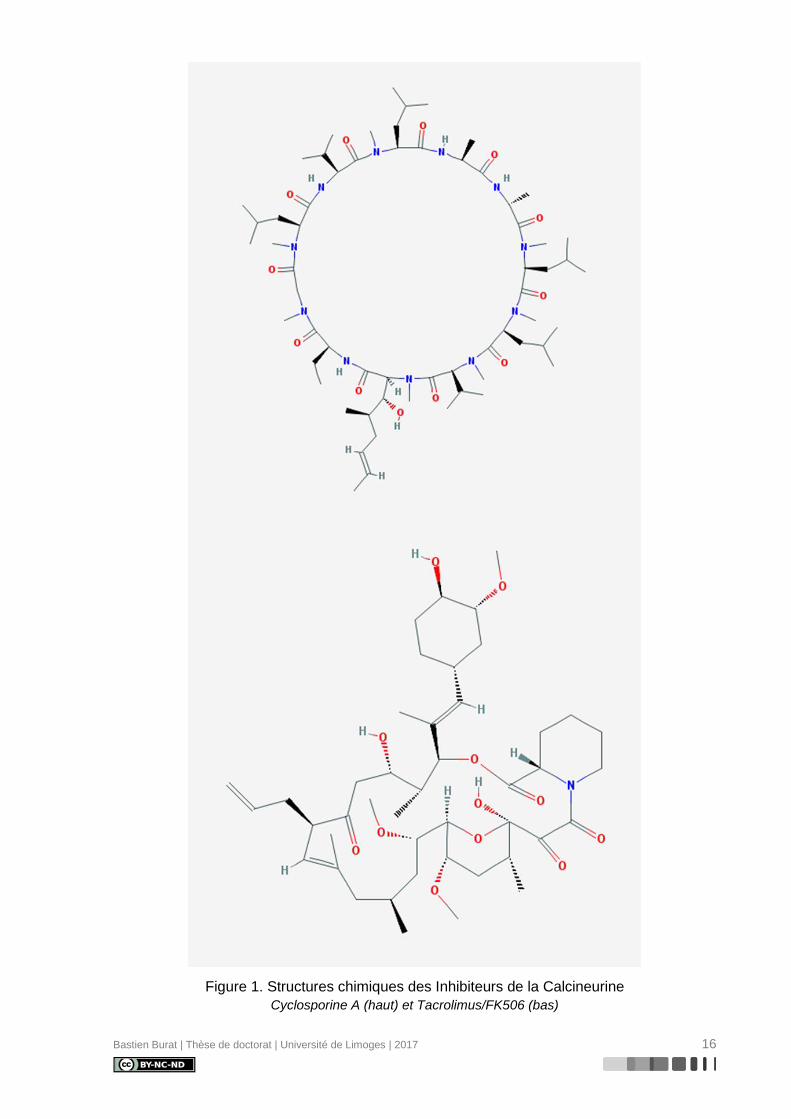
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 16
Figure 1. Structures chimiques des Inhibiteurs de la Calcineurine
Cyclosporine A (haut) et Tacrolimus/FK506 (bas)

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 17
I.3. Mécanisme d’action pharmacologique des Inhibiteurs de la Calcineurine
Bien que d’origine et de nature chimique différente, les ICN présentent un mode
d’action pharmacologique identique en ciblant la Calcineurine au sein de la voie Calcium-
dépendante d’activation des lymphocytes T.
Pour rappel, la reconnaissance par le récepteur des cellules T (TCR) d’un peptide an-
tigénique présenté par les molécules du Complexe Majeur d’Histocompatibilité d’une cellule
présentatrice d’antigène a pour conséquence l’activation de la phospholipase C-γ, respon-
sable de l’hydrolyse du phosphatidyl inositol-4,5-diphosphate (PIP2) membranaire en inosi-
tol-1.4.5-triphosphate (IP3) et diacylglycérol (DAG). L’interaction d’IP3 avec des récepteurs
IP3R intracellulaires entraîne un relargage des réserves calciques du réticulum endoplas-
mique. Cet efflux stimule les canaux CRAC (Calcium release-activated channels) de la
membrane plasmique. L’influx total obtenu active la protéine calmoduline qui recrute la Cal-
cineurine (CaN). Cette protéine est une Sérine-Thréonine phosphatase dépendante du cal-
cium, constituée de deux sous-unités A et B permettant la déphosphorylation – entre autres
cibles – du facteur de transcription NFAT (sur les résidus Sérine du NFAT regulatory do-
main). Cette déphosphorylation est nécessaire à la translocation vers le noyau et à la trans-
cription de gènes cibles dont la transcription du gène de l’interleukine IL-2 (8,9). L’IL-2 est
une cytokine essentielle à la survie, à la croissance et à la prolifération des lymphocytes T,
grâce à une action autocrine et paracrine sur le récepteur membranaire IL-2R aboutissant à
l’activation de la voie de la Sérine/Thréonine kinase mTOR, via la cascade de signalisation
impliquant les protéines JAK/STAT et PI3K/Akt (10). Les lymphocytes T occupent une place
centrale dans la mobilisation de la réponse immunitaire à médiation cellulaire et par consé-
quent dans le rejet aigu d’allogreffe (Figure 2).
L’action pharmacologique des ICN s’effectue après liaison à des protéines cytoplas-
miques appelées immunophilines (11). Les complexes ICN-immunophiline inhibent l’activité
phosphatase de la CaN (12,13) en interagissant avec une région formée par le domaine ca-
talytique de la sous-unité A, le domaine de liaison à la sous-unité B et la sous-unité B
(14,15). L’absence de déphosphorylation de NFAT conduit à sa séquestration cytoplasmique
et à l’inhibition de l’activation lymphocytaire T par la voie dépendante du Calcium (16).
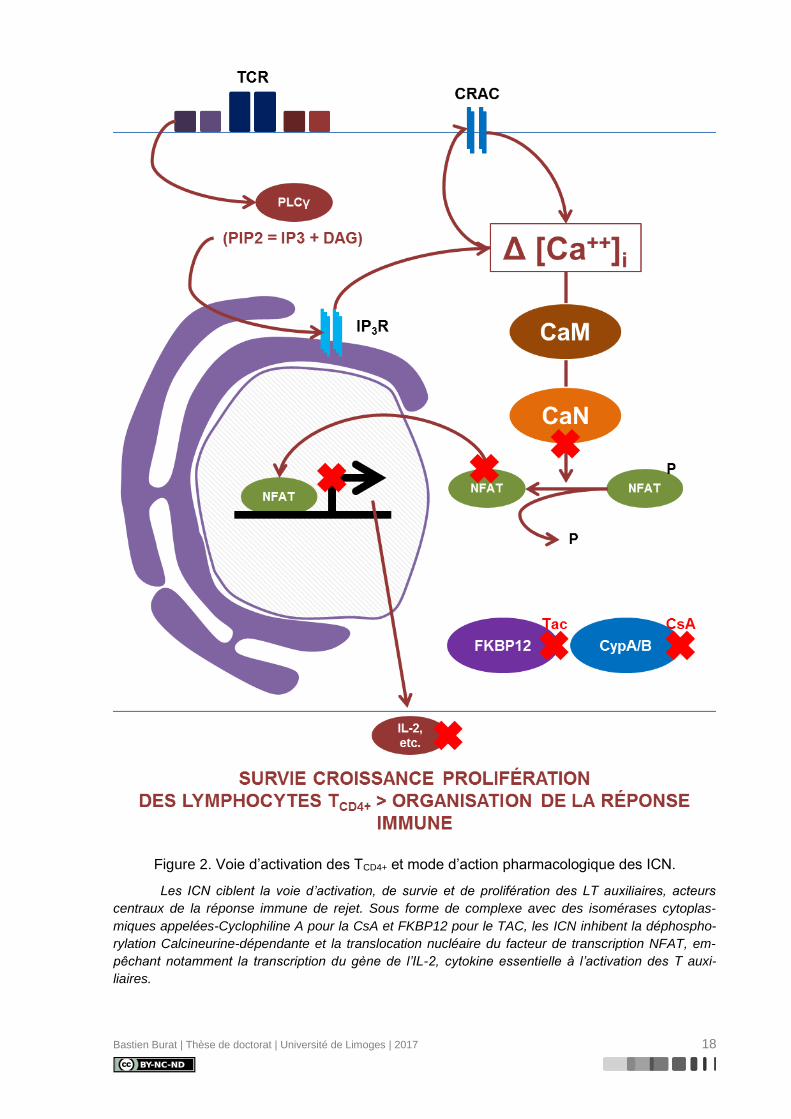
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 18
Figure 2. Voie d’activation des TCD4+ et mode d’action pharmacologique des ICN.
Les ICN ciblent la voie d’activation, de survie et de prolifération des LT auxiliaires, acteurs
centraux de la réponse immune de rejet. Sous forme de complexe avec des isomérases cytoplas-
miques appelées-Cyclophiline A pour la CsA et FKBP12 pour le TAC, les ICN inhibent la déphospho-
rylation Calcineurine-dépendante et la translocation nucléaire du facteur de transcription NFAT, em-
pêchant notamment la transcription du gène de l’IL-2, cytokine essentielle à l’activation des T auxi-
liaires.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 19
I.4. Manifestations histologiques et fonctionnelles de la néphrotoxicité des Inhibiteurs
de la Calcineurine (17,18)
Dès la première description de l’action immunologique de la CsA à la fin des années
1980, Calne et coll. ont rapporté le développement précoce d’une insuffisance rénale au sein
de la cohorte de patients transplantés rénaux traités par l’ICN (4).
L’observation d’atteintes rénales similaires chez des patients transplantés cardiaques
recevant un régime d’immunosuppression à base de CsA a confirmé l’implication de cette
molécule dans l’apparition d’effets secondaires néphrotoxiques (19).
I.4.1. Néphrotoxicité aigüe
Les manifestations aigües de la néphrotoxicité des ICN sont essentiellement vascu-
laires par altération des propriétés hémodynamiques de la micro-vascularisation rénale
(20,21). Des perturbations du système rénine-angiotensine-aldostérone consécutives à
l’augmentation de la concentration en facteurs vasoconstricteurs associée à la diminution de
la concentration en facteurs vasodilatateurs induisent une vasoconstriction des artérioles
afférentes.
La comparaison de l’évolution entre les concentrations plasmatiques et urinaires de
facteurs vasoconstricteurs tels que l’Endothéline et le Thromboxane, deux ans après une
greffe hépatique, montrent que les ICN stimulent leur expression et leur sécrétion par les
cellules épithéliales rénales (22) comme cela a déjà pu être observé in vitro, avec la lignée
tubulaire proximale porcine LLC PK-1 (23) et in vivo, chez le Rat (24).
La promotion de facteurs vasoconstricteurs s’accompagne d’une diminution de la
concentration ou de la production de facteurs vasodilatateurs tels que la Prostacycline (22)
ou le monoxyde d’azote (NO) (25). En particulier, dans un contexte favorable
d’ischémie/reperfusion et d’hypoxie, les ICN augmente la production de radicaux libres et de
dérivés réactifs de l’Oxygène par les cellules rénales (26). Par exemple, la production
d’anion superoxyde O2- par les cellules endothéliales (27) impacte la disponibilité de NO par
la formation de l’ion péroxynitrite (NO + O2- ONOO-) (28).
Cette perturbation des facteurs vasoconstricteurs et vasodilatateurs participe à
l’activation du système rénine-angiotensine-aldostérone (SRAA). Le SRAA est un méca-
nisme de régulation des homéostasies cardiovasculaire, hydrosodée et de la fonction rénale
reposant sur l’action endocrine de l’angiotensine II sur les cellules endothéliales artériolaires
et les cellules musculaires lisses associées (vasoconstriction), les cellules épithéliales tubu-
laires (réabsorption, excrétion), les surrénales (synthèse d’aldostérone) et le système ner-
veux central (synthèse d’ADH par l’hypophyse, système sympathique). La synthèse
d’angiotensine II, à partir de son précurseur l’angiotensinogène, est sous le contrôle de deux
enzymes, la rénine et l’enzyme de conversion de l’angiotensine (ECA) respectivement sécré-
tées par le rein et les poumons. Les ICN augmente la production de rénine par les cellules

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 20
de l’appareil juxtaglomérulaire (29) tout en potentialisant l’effet vasoconstricteur de
l’angiotensine II sur les cellules musculaires lisses de la micro-vascularisation (30)
Aux atteintes vasculaires s’ajoutent des atteintes tubulaires non spécifiques. La né-
phrotoxicité aigüe des ICN peut notamment être associée à l’apparition de vacuoles isomé-
triques et de corps d’inclusion dans le cytoplasme des cellules épithéliales tubulaires. Ces
manifestations correspondent, respectivement, à un accroissement de la taille du réticulum
endoplasmique et des vésicules lysosomales, d’une part, et à un accroissement de la taille
des mitochondries et des autolysosomes, d’autre part (31–38). Mais leur développement
n’est pas exclusivement consécutif à une exposition aux ICN (39–43) et ne peut servir de
facteur pronostique à l’évolution vers une toxicité rénale chronique (44,45).
Les manifestations histologiques de la néphrotoxicité aigüe sont majoritairement ré-
versibles. Une amélioration de la fonction rénale peut être observée plusieurs mois après la
greffe en cas d’ajustement/diminution de dose ou de conversion vers un autre immunosup-
presseur (46,47).
I.4.2. Néphrotoxicité chronique (48–51)
Les manifestations chroniques de la néphrotoxicité des ICN correspondent à une ag-
gravation des atteintes vasculaires et une progression vers les structures glomérulaires et
tubulo-interstitielles
À long terme, les perturbations hémodynamiques sont amplifiées, de manière irréver-
sible, par le rétrécissement de la lumière des vaisseaux à la faveur de dépôts hyalins dans la
tunica media et à la surface extérieure de la tunica adventitia des artérioles afférentes (35).
Les ICN provoquent la formation de granules éosinophiles dans le cytoplasme des cellules
musculaires lisses tapissant les portions les plus distales de l’endothélium des artères inter-
lobulaires. Cette transformation des cellules musculaires lisses s’accompagne de leur hyper-
trophie et de leur vacuolisation et aboutit à leur remplacement par des dépôts hyalins locali-
sés (agrégats protéiques et lipidiques éosinophiles) (52–54).
Cette artériolopathie chronique a notamment pour conséquence une ischémie du
glomérule participant au développement de la glomérulosclérose induite par les ICN.
L’exposition aux ICN conduit également à l’apparition de glomérules atubulaires, des
glomérules vascularisés mais totalement déconnectés du tubule proximal adjacent (55), évo-
luant vers des phénotypes fibrosants (fibrose périglomérulaire) ou kystiques (56).
D’un point de vue histologique, les atteintes tubulaires chroniques se résument en
partie au développement des phénomènes d’atrophie tubulaire (AT) et fibrose interstitielle
(FI) résultant de plusieurs mécanismes physiopathologiques au sein des cellules épithéliales
tubulaires.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 21
I.5. Mécanismes physiopathologiques cellulaires et moléculaires des cellules tubu-
laires exposées aux Inhibiteurs de la Calcineurine
I.5.1. Perturbation du cycle cellulaire : sénescence et apoptose
Le cycle cellulaire organise la vie de la cellule de la réplication de son matériel géné-
tique à sa mitose en deux cellules-filles, selon des phases successives rythmées par des
points de vérification, garants de l’intégrité cellulaire.
Depuis la découverte de la CsA à la fin des années 1970, de nombreuses études, in
vivo comme in vitro, ont mis en lumière une perturbation directe du cycle cellulaire par les
ICN (57–63). Les ICN étant des molécules immunosuppressives, les premières observations
concernaient une inhibition de la prolifération et de la croissance cellulaires de cellules im-
munitaires (64–67) avant de s’étendre à d’autres types cellulaires notamment les cellules
rénales. Ainsi, in vitro, CsA et Tac induisent une inhibition de la prolifération cellulaire de cel-
lules tubulaires proximales humaines en culture, dépendante de la concentration et du temps
d’exposition (68,69). La CsA provoque un arrêt du cycle cellulaire en phase G0/G1 ou G2/M
de cellules tubulaires proximales porcines LLC PK-1, d’une manière dépendante de la con-
centration. Cet arrêt semble consécutif à des dommages à l’ADN amenant à une accumula-
tion de la protéine p53 (70). Des travaux ultérieurs ont précisé la nature de la perturbation du
cycle cellulaire. Des cellules tubulaires proximales exposées à la CsA déclenchent des mé-
canismes associés à l’entrée en sénescence : l’exposition à l’ICN crée des conditions favo-
rables à un raccourcissement des télomères qui active en cascade la protéine p53 et une
protéine inhibitrice du cycle cellulaire, l’inhibiteur de kinase dépendante des cyclines
CDKN1A ou p21 (71).
L’arrêt du cycle cellulaire et l’’entrée en sénescence est la première alternative quand
les conditions requises pour progresser dans le cycle ne sont pas réunies. La seconde alter-
native est l’entrée en apoptose qui déclenche les mécanismes d’une mort cellulaire contrôlée
(72). Les observations cliniques ont rapporté une association entre CsA et apoptose in vivo
(73). In vitro, la CsA induit apoptose et nécrose de cellules tubulaires proximales en fonction
du temps et de la concentration (74,75). Bien que la CsA augmente l’expression du récep-
teur de mort Fas, la voie extrinsèque ne semble pas impliquée puisque son inhibition au ni-
veau de FasL ou de la caspase 8 n’empêche pas les effets de la CsA. Ainsi, les mécanismes
en jeu appartiennent à la voie intrinsèque, mitochondriale. Indépendamment des caspases,
la CsA favorise la translocation de la protéine pro-apoptotique Bax à la mitochondrie qui dé-
clenche la libération des médiateurs pro-apoptotiques Cytochrome c et Smac/Diablo, entrai-
nant l’activation en cascade des caspases finales 9 et 3 (76).
Les effets pro-apoptotiques de la CsA peuvent être expliqués par l’inhibition de
l’interaction entre la Cyclophiline D mitochondriale (CypD) et le facteur anti-apoptotique Bcl-
2. En effet, CypD interagit avec Bcl-2 et potentialise ses propriétés anti-apoptotiques, telle

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 22
que l’inhibition de la libération cytosolique de CytC, de manière CsA-dépendante. CsA, tout
comme le knockdown de CypD, sensibilise les cellules à l’apoptose en empêchant la liaison
de CypD à Bcl-2 (77).
En revanche, les effets pro-nécrotiques de la CsA ne peuvent être expliqués par son
interaction avec CypD. En effet, CypD a un rôle majeur dans l’ouverture du pore de transition
de perméabilité mitochondrial au cours de la mort cellulaire de type nécrotique induite par un
stress oxydant ou un choc calcique. Des cellules provenant de souris Ppif-/- (KO du gène de
CypD) sont insensibles à l’exposition aux ERO et à une surcharge de Ca++ tout comme
leurs organes ne subissent pas de dommages d’ischémie/reperfusion. En revanche, des
cellules Ppif-/- sont toujours sensibles à l’apoptose (78,79).
Les études portant sur les modalités de conditionnement par la CsA dans un contexte
d’ischémie/reperfusion ont rapporté des effets protecteurs, anti-nécrotiques de la CsA par
inhibition de la CypD et de l’ouverture du pore de transition mitochondrial (80,81).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 23
I.5.2. Stress du réticulum endoplasmique
Les protéines destinées à être sécrétées ou implantées dans les membranes plas-
miques et des organites sont synthétisées dans la lumière du réticulum endoplasmique (RE)
où les conditions physico-chimiques sont optimales pour le bon repliement des structures
tertiaire et quaternaire par les protéines chaperonnes, la maturation et les modifications post-
traductionnelles. Les protéines matures sont soumises à un contrôle de la qualité du replie-
ment qui détermine leur orientation vers l’appareil de Golgi en vue de leur sécré-
tion/implantation ou vers le cytoplasme en vue de leur dégradation par le protéasome (pro-
cessus ER-associated degradation, ERAD). L’accumulation de protéines mal repliées dé-
clenche un stress du RE et l’Unfolded Protein Response (UPR), mécanisme d’adaptation et
de réparation comprenant l’arrêt de la biosynthèse protéique classique (afin d’endiguer
l’afflux de protéines immatures au RE), la biosynthèse de protéines chaperonnes supplé-
mentaires (pour renforcer les capacités de repliement), la dégradation des protéines mal
repliées par ERAD et, le cas échéant, la mort cellulaire par apoptose.
In vitro, des cellules épithéliales rénales humaines en culture primaire exposées à
des concentrations cliniques de CsA développent un stress du réticulum endoplasmique et
de la synthèse protéique caractérisés par la surexpression de marqueurs de l’Unfolded Pro-
tein Response (UPR). Le stress du RE déclenche des modifications phénotypiques des cel-
lules épithéliales rénales, notamment, le développement important et la dilatation du RE et
conduit à la mort cellulaire par apoptose (82–85) ou à la survie par autophagie (86).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 24
I.5.3. Stress oxydant
L’hypoxie locale engendrée par l’hypoperfusion ou l’ischémie résultant des perturba-
tions vasculaires et hémodynamiques induites par l’exposition chronique aux ICN est sus-
ceptible d’installer un environnement favorable à la formation de radicaux libres et d’espèces
réactives de l’Oxygène (ERO) et à l’apparition d’un stress oxydatif au sein des cellules tubu-
laires proximales (87).
In vitro, la CsA est capable d’activer la NADPH oxydase et d’augmenter la production
d’anion superoxyde O2- et de péroxynitrite ONOO- de cellules tubulaires proximales LLC PK-
1 (88). Par l’utilisation d’antioxydants tels que le resvératrol et la vitamine E, Galletti et coll.
ont été capable de prévenir la cytotoxicité associée à l’accumulation intracellulaire CsA-
dépendante de ERO de cellules tubulaires proximales immortalisées de Rat (89). In vivo,
l’administration de l’antioxydant N-acétylcystéine (NAC) combinée à la CsA empêche
l’apparition de marqueurs histologiques de néphrotoxicité chez des Rats traités (90).
Ces travaux mettent en évidence l’importance du stress oxydatif par la production
d’espèces réactives de l’Oxygène dans les mécanismes physiopathologiques de la néphro-
toxicité chroniques des ICN.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 25
I.5.4. Perturbation de l’homéostasie électrochimique
Les atteintes tubulaires chroniques des ICN se manifestent chez les patients traités
par des perturbations de l’homéostasie des électrolytes (91) telles que des hyperkaliémies et
hyponatrémies (92–94), des hypomagnésémies (95,96), des acidoses métaboliques hyper-
chlorémiques (97) et des hyperuricémies.
Ces perturbations de l’homéostasie électrochimique peuvent être expliquées en pre-
mier lieu par des variations ICN-dépendantes de l’activité ou de l’expression de transporteurs
membranaires tubulaires responsables des échanges, de la sécrétion ou de la réabsorption
des électrolytes.
En particulier, le développement d’’hyperkaliémies et d’hyponatrémies a été rattaché
aux effets potentiels des ICN sur la pompe Sodium/Potassium ATPase (Na+/K+ ATPase), le
cotransporteur Sodium/Potassium/Chlorure 2 (NKCC2), la pompe Proton-Potassium ATPase
(H+/K+ ATPase) ou les canaux potassiques activés par le Calcium.
La pompe Na+/K+ ATPase est exprimée à la membrane basale des cellules tubu-
laires et échange deux Na+ contre trois K+. Cet échange électrogénique est la base du gra-
dient de concentration du Sodium permettant la réabsorption transcellulaire de la plupart des
électrolytes.
La CsA inhibe spontanément mais de manière partielle (- 20 %, p < 0.05) l’activité de
la Na+/K+ ATPase humaine, purifiée à partir de préparations de Rein (98). In vitro comme in
vivo ,l’inhibition de la Na+/K+ ATPase s’accompagne d’une sous-expression de l’ARNm de la
sous-unité alpha 1 (99,100). Au niveau rénal, la CsA et le TAC induisent une inhibition diffé-
rentielle de l’activité de la pompe Na+/K+ ATPase, caractéristique du segment du système
tubulaire considéré. Les portions médullaires ascendantes de l’anse de Henlé et les portions
corticales et médullaires externes du tube collecteur sont les segments les plus sensibles à
la CsA (101,102).
Le cotransporteur NKCC2 est exprimé à la membrane apicale des cellules tubulaires
de l’anse de Henlé afin de réabsorber les ions Na +, Cl- et K+.
A l’instar de la Na+/K+ ATPase , le NKCC2 est inhibé par la CsA (103–105).
Au contraire, la CsA est capable d’induire, in vitro, une fermeture des canaux potas-
siques exprimés à la membrane apicale de cellules de tubules collecteurs issues de cultures
primaires (106).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 26
I.5.5. Transition épithélio-mésenchymateuse
La transition épithélio-mésenchymateuse (TEM) est un mécanisme physiologique mis
en jeu lors du développement embryonnaire, lors de la cicatrisation ou de la dispersion des
métastases et constitue un aspect hypothétique du développement de la fibrose interstitielle,
caractéristique de la néphrotoxicité chronique des ICN et de la progression vers
l’insuffisance rénale terminale.
D’un point de vue histologique, la toxicité des ICN est caractérisée par le phénomène
de fibrose interstitielle et atrophie tubulaire FI/AT affectant la cohérence mécanique et fonc-
tionnelle des tubules et de l’interstitium (interface séparant les tubules de la micro-
vascularisation parallèle). Consécutivement à un stress, les cellules épithéliales tubulaires
rénales libèrent des cytokines pro-inflammatoires et chimiokines (dont la chimiokine CCL2)
recrutant majoritairement des macrophages, distingués en type 1 (M1) et 2 (M2). Les M1
activés par le TNFα, sécrètent notamment l’interleukine 1-β (IL1-β) et des espèces réactives
de l’oxygène, délétères. Les macrophages de type M2, bien qu’activés par le TGF-β, produi-
sent des cytokines anti-inflammatoires et tendent à protéger le rein. Le développement local
de l’inflammation favorise l’émergence d’un type cellulaire d’ordinaire associé au contexte de
cicatrisation, les myofibroblastes. Sous l’influence du TGF-β et du CTGF, leurs effectifs
augmentent à partir de plusieurs réservoirs, les fibroblastes interstitiels, les péricytes ou les
cellules épithéliales suivant, pour ces dernières, un processus de transition épithélio-
mésenchymateuse.
La TEM est caractérisée par une perte du phénotype épithélial (polarité cellulaire et
adhésion intercellulaire impliquant l’E-cadhérine) et l’acquisition du phénotype mésenchyma-
teux (réorganisation du cytosquelette d’Actine, expression de novo de l’α-SMA (« α-smooth
muscle actin »)). Les myofibroblastes ont pour fonction essentielle la mise en place et
l’entretien d’une matrice extracellulaire. Ainsi, des protéines constitutives de cette MEC tels
la fibronectine ou les divers types de collagènes sont synthétisées tandis que la dégradation
de cette MEC par les métalloprotéases matricielles (MMPs) est notamment entravée par les
TIMP (tissue inhibitors of metalloproteases). La progression de la fibrose est d’autant plus
rapide que les myofibroblastes sont eux-mêmes capables de produire TGF-β et CTGF (107–
109).
In vivo, une étude menée sur 108 patients transplantés rénaux recevant un régime
d’immunosuppression CsA-MMF-Prednisone a mis en avant la corrélation entre observation
d’une TEM précoce et développement d’une néphrotoxicité CsA-dépendante à long terme
(110). La comparaison de deux cohortes après ajustement du régime d’immunosuppression
(retrait de CsA ou retrait du MMF) a montré une progression différentielle de biomarqueurs
protéiques spécifiques de la TEM corrélée à une altération différentielle de la fonction rénale
(mesurée par le débit de filtration glomérulaire).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 27
In vitro, deux études menées parallèlement sur des cultures primaires (RPTEC) et
une lignée immortalisée (HK-2) de cellules épithéliales tubulaires proximales humaines ont
démontré la capacité de la CsA d’induire la transition épithélio-mésenchymateuse à partir de
cellules tubulaires proximales exposées à une gamme de concentrations proches des con-
centrations observées en clinique (111,112).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 28
I.5.6. Réorganisation du cytosquelette d’Actine induite par la Cyclosporine A
L’étude de l’organisation du cytosquelette des cellules rénales exposées aux ICN est
une piste récente et encore peu développée dans l’investigation des mécanismes de néphro-
toxicité des ICN. Des travaux sur des modèles de podocytes en culture ont montré que la
CsA induisait une stabilisation du cytosquelette d’Actine structurant les pédicelles. Cette sta-
bilisation est consécutive à l’inhibition pharmacologique de l’activité phosphatase de la CaN.
Au sein des podocytes, la CaN déphosphoryle la Synaptopodine, une Actin-binding protein
essentielle pour la mise en place et le maintien des pédicelles. La Synaptopodine est alors
ciblée par la protéolyse dépendante de la Cathepsine L. En inhibant la CaN, CsA empêche
la dégradation protéique de la Synaptopodine qui participe alors à l’organisation physiolo-
gique du cytosquelette d’Actine des pédicelles via, notamment, son interaction avec 14-3-3
et l’activation de la voie des petites GTPases Rho (113). Dans le même temps, indépen-
damment des effets sur la Synaptopodine, CsA induit la surexpression protéique de l’Actin-
binding protein Cofiline, sous sa forme déphosphorylée (114).
La caractérisation du cytosquelette d’Actine de cellules de la lignée tubulaire proxi-
male porcine LLC PK-1 après exposition aux ICN à doses pharmacologiques, révèle une
réorganisation différentielle où la CsA, et non le TAC, semble induire la déstructuration du
cytosquelette d’Actine en périphérie de la cellule (Figure 3), indépendamment de l’inhibition
de la Calcineurine, ce qui entraîne une perte réversible de sa morphologie physiologique
(115). Le mécanisme de modification du cytosquelette d’Actine et ses conséquences sur le
phénotype des cellules tubulaires proximales étaient inconnus au début de ces travaux de
thèse.
Figure 3. Effets différentiels des ICN sur l’organisation du cytosquelette d’Actine des TCP.
(a) Contrôle, (b) CsA 5 µM, 24 h, (c) Tac 0,05 µM, 24 h.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 29
Chapitre II. Approches protéomiques quantitatives différentielles à haut-débit
« bottom-up/shotgun »
Les observations réalisées à l’aide d’approches expérimentales ciblées ne décrivent
que partiellement la toxicité des ICN et ne permettent pas de comprendre les mécanismes
physiopathologiques en jeu, dans leur ensemble.
Alors que les mécanismes de néphrotoxicité des ICN semblent encore largement non
élucidés, la première problématique de ces travaux de thèse pose la question des modalités
expérimentales d’une stratégie non ciblée, sans a priori, pour valider mais surtout compléter
les connaissances actuelles par la découverte de nouveaux mécanismes physiopatholo-
giques impliqués dans la toxicité tubulaire des ICN.
Les approches omiques basées sur une analyse haut-débit de systèmes biologiques
sont spécifiquement adaptées à ce dessein. En particulier, les approches protéomiques
quantitatives bottom-up/shotgun sont des méthodes analytiques reposant sur la digestion
protéolytique de l’échantillon protéique, la séparation des peptides par chromatographie li-
quide et l’analyse de l’échantillon digéré par spectrométrie de masse. Les technologies pré-
sentées ici complètent les aspects purement qualitatifs de la protéomique shotgun par une
quantification protéique relative selon une gamme variable de performances en termes de
précision, de justesse, de reproductibilité, de polyvalence d’application, de couverture de
protéome et de séquence, qui surpassent les approches de protéomique quantitative basée
sur une séparation électrophorétique (116,117).
II.1. Technologies de marquage isotopique stable
II.1.1. Marquage isotopique : technologie « Isotope-Coded Affinity Tags » ou ICAT
Décrite en 1999 par Gygi et coll. (118), la technologie ICAT (Figure 4) fut la première
technologie de protéomique quantitative développée sur la base d’une séparation chromato-
graphique de peptides marqués et d’une analyse par spectrométrie de masse en tandem. Le
marquage chimique consiste en la formation d’une liaison covalente par alkylation entre la
fonction thiol d’un résidu cystéine et un tag composé de trois groupes fonctionnels distincts :
- un groupe réactif alkylant (iodoacétamide) pour la liaison covalente du tag au groupe
thiol (-SH) du résidu cystéine
- un groupe de liaison incorporant huit atomes d’un isotope lourd ou léger de
l’hydrogène (couple 1H / 2H) ou du carbone (couple 12C / 13C)
- un groupe d’affinité (biotine) pour l’isolation des peptides marqués par chromatogra-
phie d’affinité avec l’avidine
L’étape de marquage est précoce au cours de la préparation des échantillons protéiques,
succédant directement à l’exposition aux deux conditions expérimentales étudiées. Chacun

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 30
des deux échantillons est alors incubé soit en présence des tags légers, soit en présence
des tags lourds, avant d’être combinés en un mélange équimolaire et digérés en peptides
trypsiques. Les peptides sont alors isolés par chromatographie d’affinité sur colonne
d’avidine, puis séparés et analysés par LC-MS/MS.
Au cours de l’expérience MS, des peptides de séquence identique marqués par deux
tags de composition isotopique différente apparaissent comme deux signaux MS correspon-
dant à deux ions précurseurs de masse différente, en raison d’un décalage de masse dû à
l’enrichissement différentiel en isotopes légers ou lourds. La quantification relative est alors
possible en comparant les intensités des signaux obtenus pour l’ion précurseur léger et l’ion
précurseur lourd.
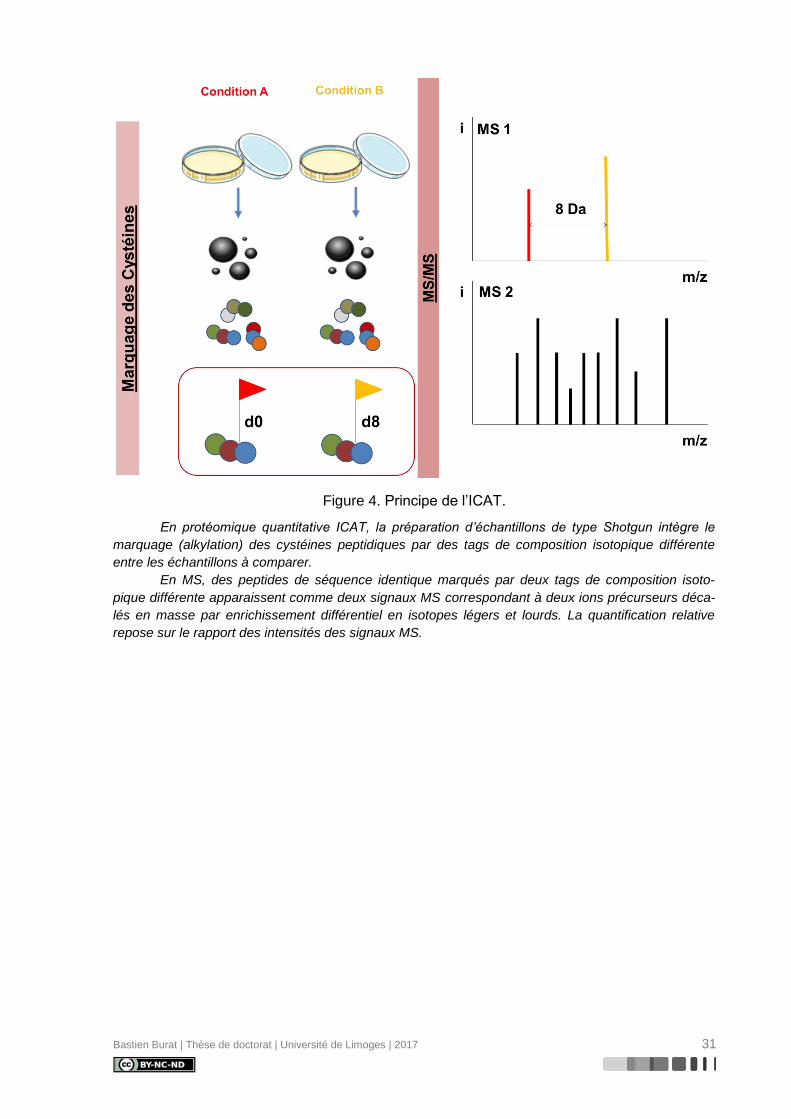
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 31
Figure 4. Principe de l’ICAT.
En protéomique quantitative ICAT, la préparation d’échantillons de type Shotgun intègre le
marquage (alkylation) des cystéines peptidiques par des tags de composition isotopique différente
entre les échantillons à comparer.
En MS, des peptides de séquence identique marqués par deux tags de composition isoto-
pique différente apparaissent comme deux signaux MS correspondant à deux ions précurseurs déca-
lés en masse par enrichissement différentiel en isotopes légers et lourds. La quantification relative
repose sur le rapport des intensités des signaux MS.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 32
II.1.2. Marquage métabolique : technologie « Stable Isotope Labeling by Amino Acids
in Cell Culture » ou SILAC
Décrite en 2002 par Ong et coll. (119), la technologie SILAC (Figure 5) fait évoluer le
marquage isotopique vers un marquage métabolique, reposant sur l’incorporation différen-
tielle d’analogues enrichis d’acides aminés essentiels au cours de la biosynthèse protéique
de cellules en culture in vitro, de tissus ou d’organes in vivo.
Le marquage SILAC ne fait donc pas intervenir de tags à proprement parler mais un
approvisionnement différentiel des cellules en croissance en acides aminés essentiels (non
synthétisés par l’organisme modèle) enrichis en isotopes lourds (2H, 13C, 15N) selon les deux
conditions expérimentales distinctes étudiées. Pour cela, les cellules sont cultivées dans des
conditions de déplétion en acides aminés essentiels (milieu déplété et sérum dialysé) aux-
quelles sont incorporés soit l’acide aminé léger, soit l’acide aminé lourd.
Comme pour l’ICAT, en MS, des peptides de séquence identique marqués par des
acides aminés de composition isotopique différente apparaissent sous la forme d’une paire
de signaux MS décalés en masse correspondant à deux ions précurseurs enrichis différem-
ment en isotopes légers ou lourds. La quantification relative résulte de la comparaison des
intensités des signaux obtenus pour l’ion précurseur léger et l’ion précurseur léger.
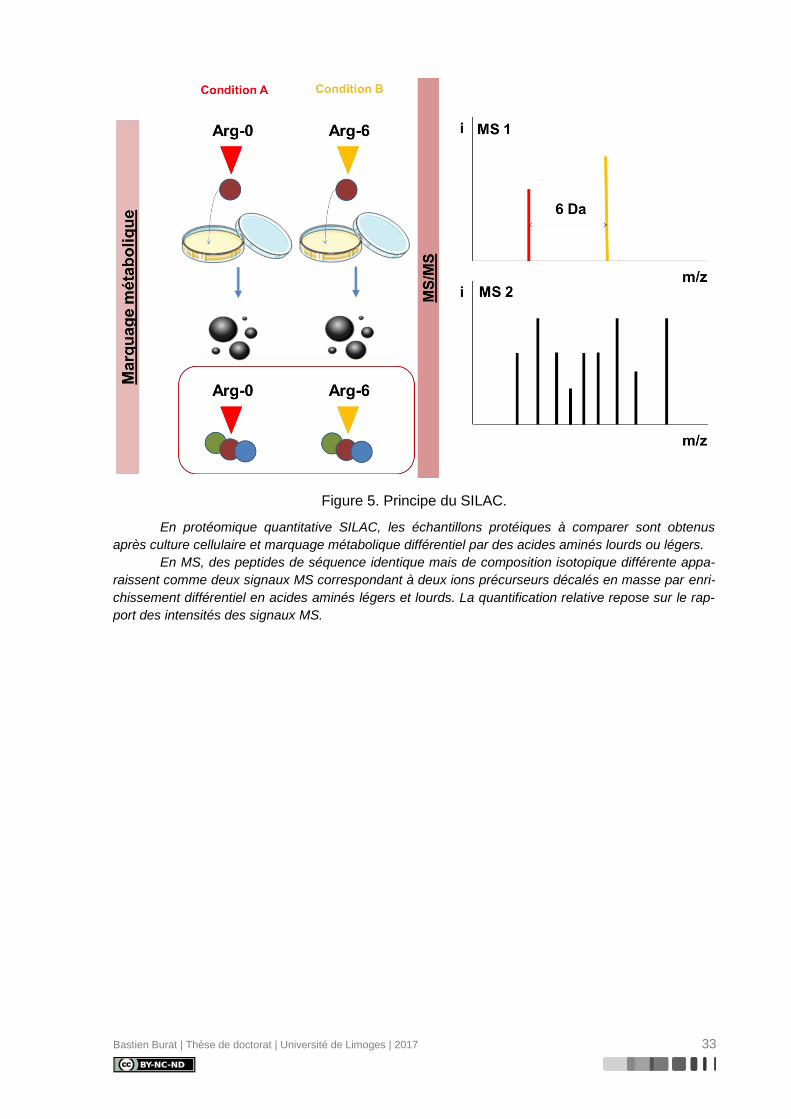
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 33
Figure 5. Principe du SILAC.
En protéomique quantitative SILAC, les échantillons protéiques à comparer sont obtenus
après culture cellulaire et marquage métabolique différentiel par des acides aminés lourds ou légers.
En MS, des peptides de séquence identique mais de composition isotopique différente appa-
raissent comme deux signaux MS correspondant à deux ions précurseurs décalés en masse par enri-
chissement différentiel en acides aminés légers et lourds. La quantification relative repose sur le rap-
port des intensités des signaux MS.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 34
II.1.3. Marquage isobare : « isobaric Tags for Relative and Absolute Quantitation »
(iTRAQ)
La technologie iTRAQ (Figure 6) a été décrite dès 2004 par Ross et coll. (120) dans
l’optique de multiplexer l’analyse de protéomique quantitative par spectrométrie de masse en
tandem en permettant la comparaison de profils d’expression protéique provenant de 2 à 8
conditions expérimentales distinctes.
Le marquage iTRAQ consiste en la formation d’une liaison covalente et d’une fonc-
tion amide (similaire à une liaison de type peptidique) entre l’amine N-terminale d’un peptide
(ou l’amine ε d’un résidu Lysine) et l’ester de N-hydroxysuccinimide d’un tag isobare compo-
sé de trois groupes fonctionnels :
- un groupe réactif amine-spécifique, (amine specific peptide reactive group), ester de
NHS responsable de la liaison au peptide
- un groupe d’équilibre (balance group) de 28, 29, 30 ou 31 Da, selon la substitution
progressive d’isotopes légers de carbone (12C) et d’oxygène (16O) par leurs équiva-
lents lourds (13C et 18O) au sein d’un groupe carbonyle. Ce groupe est chargé de
compenser la variation de masse d’ :
- un groupe rapporteur (reporter group), dont la masse est de 114,1, 115,1, 116,1 ou
117,1 Da selon la substitution progressive d’isotopes légers de carbone (12C) et
d’azote (14N) par leurs équivalents lourds (13C et 15N) au sein d’une N-
méthylpiperazine, pour une masse totale constante « groupe rapporteur – groupe
d’équilibre » de 145,1 Da
Une déclinaison 8-plex complète la déclinaison 4-plex décrite ci-dessus par l’ajout de 4 tags
isobares intégrant des groupes rapporteurs de masse 113, 118, 119 et 121 Da.
Au cours de la préparation d’échantillon, le tag est introduit après digestion trypsique
des protéines. Les peptides trypsiques marqués sont mélangés en proportions équivalentes
entre les différentes conditions expérimentales étudiées (1 :1 :1 :1) avant d’être analysés par
LC-MS/MS.
L’expérience MS caractérise l’ensemble du mélange peptidique, indépendamment de
l’origine du peptide. Des peptides identiques marqués par quatre tags isobares différents
apparaissent sous un seul signal MS correspondant à un ion précurseur unique en raison
d’un rapport masse/charge m/z identique et unique.
En MS/MS, la CID fragmente les peptides au niveau des liaisons amide peptidiques,
peptide-tag et groupe d’équilibre-groupe rapporteur. A chaque ion précurseur en MS, sont
alors associés quatre ions groupe rapporteur de masses distinctes (114,1, 115,1, 116,1 et
117,1 Da), spécifiques d’une condition expérimentale initiale et un ensemble d’ions frag-
ments permettant le séquençage du peptide. La quantification relative est alors possible en

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 35
comparant les signaux MS/MS, proportionnels au nombre d’ions rapporteurs détectés et à la
concentration condition-dépendante d’ions précurseurs (ou peptides trypsiques), pour
chaque ion rapporteur et chaque condition expérimentale initiale.
Deux stratégies de quantification majeures existent. La première consiste en la com-
paraison des aires des pics MS/MS (intégration trapézoïdale) relatifs aux ions rapporteurs de
masse. La seconde consiste en la comparaison des sommes des intensités des pics MS/MS
des ions rapporteurs de masse.
Figure 6 1. Principe de l’iTRAQ.
En protéomique quantitative iTRAQ, la préparation d’échantillons de type Shotgun intègre le
marquage des groupements amines peptidiques par des tags isobares de composition isotopique
différente entre les échantillons à comparer.
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 36
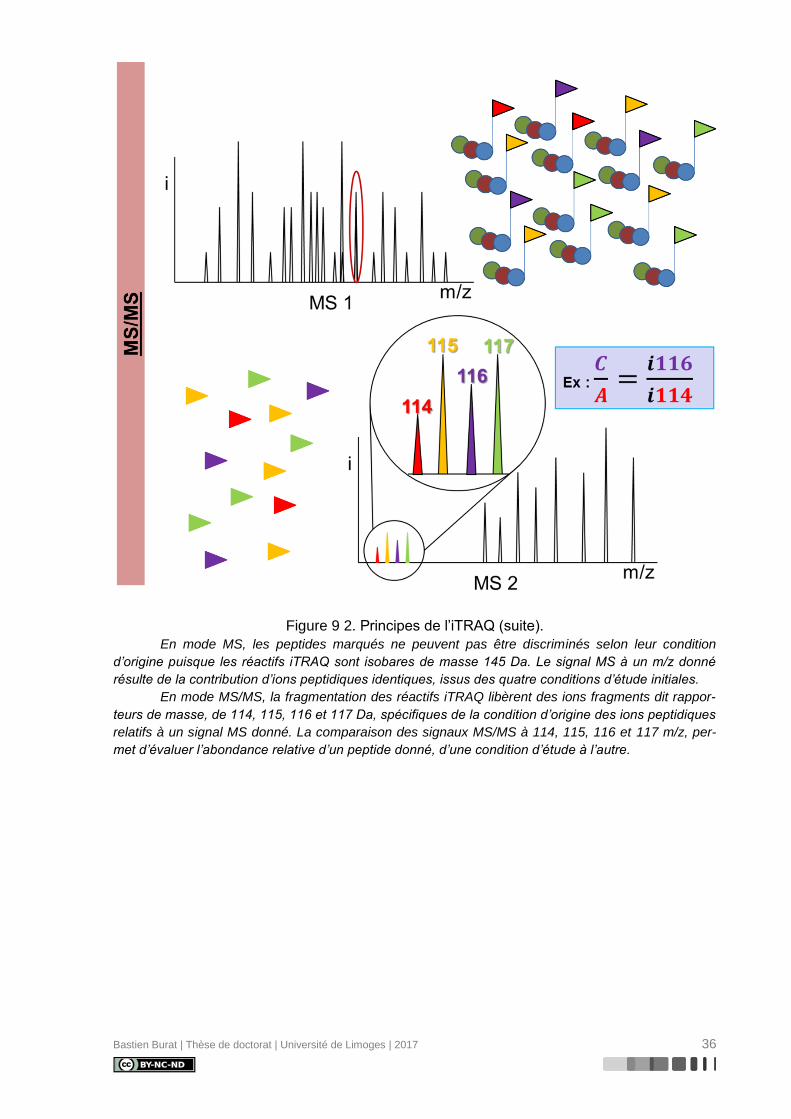
Figure 9 2. Principes de l’iTRAQ (suite).
En mode MS, les peptides marqués ne peuvent pas être discriminés selon leur condition
d’origine puisque les réactifs iTRAQ sont isobares de masse 145 Da. Le signal MS à un m/z donné
résulte de la contribution d’ions peptidiques identiques, issus des quatre conditions d’étude initiales.
En mode MS/MS, la fragmentation des réactifs iTRAQ libèrent des ions fragments dit rappor-
teurs de masse, de 114, 115, 116 et 117 Da, spécifiques de la condition d’origine des ions peptidiques
relatifs à un signal MS donné. La comparaison des signaux MS/MS à 114, 115, 116 et 117 m/z, per-
met d’évaluer l’abondance relative d’un peptide donné, d’une condition d’étude à l’autre.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 37
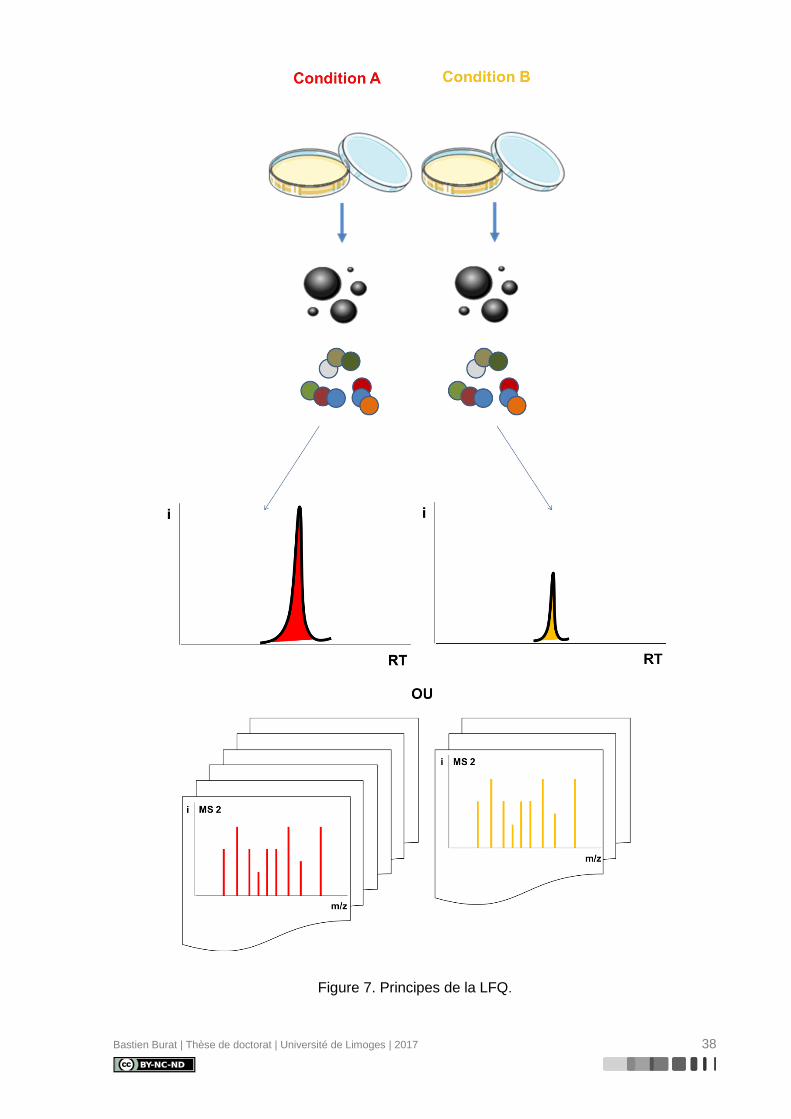
II.2. Quantification « label-free » (LFQ)(121)
Bien que devenus des techniques de référence en protéomique quantitative, les ap-
proches basées sur un marquage isotopique stable complexifient la préparation
d’échantillons, rallongent le temps d’analyse et augmentent les coûts expérimentaux. Ainsi,
des approches label-free (Figure 7) ont été développées pour permettre une analyse
d’échantillons simple et rapide.
Contrairement aux approches de marquage isotopique stable, où les échantillons à
comparer sont préparés, marqués, mélangés après marquage et analysés par LC-MS/MS en
une seule fois, les approches label-free reposent sur la comparaison d’analyses LC-MS/MS
d’échantillons préparés et analysés séparément.
Deux méthodes de quantification relative ont été développées :
• la comparaison des intensités des signaux MS des ions précurseurs après
l’extraction des chromatogrammes d’ions et l’intégration des aires de pics MS
sur le temps de rétention chromatographique, pour chaque peptide identifié
• la comparaison du nombre de spectres MS/MS relatifs aux ions fragments,
pour chaque peptide identifié (spectral counting)
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 38
Figure 7. Principes de la LFQ.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 39
II.3. Apports et limites de la technologie iTRAQ par rapport aux technologies ICAT,
SILAC et LFQ
La possibilité de multiplexer l’analyse protéomique représente l’évolution majeure ap-
portée par la technologie iTRAQ. Auparavant, les études menées à l’aide des technologies
ICAT et SILAC devaient se limiter à la comparaison de deux conditions expérimentales dis-
tinctes puisque ces marquages protéiques reposent sur un principe binaire d’enrichissement
en isotopes légers et lourds.
Plusieurs études comparatives ont permis d’établir une hiérarchie des techniques de
protéomique « shotgun »(122,123).
Comparé à l’ICAT, l’iTRAQ offre une meilleure sensibilité, une meilleure couverture
du protéome d’intérêt tout en n’étant pas limité par le marquage biaisé des seules protéines
possédant un résidu Cystéine (avantage partagé par le SILAC et la LFQ) (116).
À sensibilité équivalente, l’iTRAQ assure une meilleure couverture que le SILAC tout
en bénéficiant d’une gamme plus large d’applications (le recours au SILAC est restreint à
des cellules en culture) et d’un temps de préparation d’échantillons plus court. Toutefois, le
SILAC a l’avantage d’un marquage extrêmement stable (incorporation du marquage au
cours de la synthèse protéique) et virtuellement total (six à huit doublements cellulaires pour
marquer l’ensemble des protéines) (117,124).
Bien que la LFQ bénéficie d’une couverture supérieure, d’une gamme dynamique
plus large et de temps de préparation et d’analyse significativement plus courts (pas d’étape
de marquage), l’iTRAQ, et de manière générale l’ensemble des techniques reposant sur un
marquage isotopique stable, surpasse ses performances par une meilleure précision, une
plus grande justesse de mesure et une plus grande reproductibilité (125,126).
La technologie iTRAQ possède une variabilité (dispersion des valeurs expérimentales
par rapport à une valeur théorique) à trois composantes : une variabilité technique, une va-
riabilité expérimentale et une variabilité biologique (127).
La variabilité technique traduit la dispersion résultant de l’analyse d’un échantillon
donnée dans des conditions données et peut être compensée par la répétition au sein de
réplicats techniques. Dans le cas de l’iTRAQ, il s’agit majoritairement de la variabilité intro-
duite lors de la préparation et du marquage des échantillons. La variabilité technique com-
prend l’erreur relative au marquage par deux tags différents et est évaluée à environ 11-12
% de variation (128).
La variabilité expérimentale traduit la dispersion observée entre deux expériences
identiques mais indépendantes et peut être résumée à l’écart observé entre deux marquages
iTRAQ par deux jeux de tags différents pour un même échantillon. La variabilité expérimen-

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 40
tale s’élève à environ 23 % de variation et peut être appréhendée par des réplicats expéri-
mentaux et la répétition de marquages.
La variabilité biologique résulte des variations aléatoires propres à tout système bio-
logique vivant. Il s’agit de la source de variabilité la plus importante, à hauteur de 25 % de
variation.
Cette variabilité multifactorielle conditionne la reproductibilité et la précision des quan-
tifications protéiques et s’ajoute aux limites intrinsèques à la technologie iTRAQ telles que
décrites par Ow et coll. en 2009 (129). Le marquage iTRAQ permet de visualiser des varia-
tions d’expression protéique dans la limite de deux ordres de grandeur en raison d’un écra-
sement des ratios et d’une sous-estimation systématique résultant de :
- la présence d’« impuretés » au sein des différents tags, des contaminations croisées,
dues aux abondances relatives des isotopes constitutifs, amenant à des contributions
croisées M-1/M+1 lors en MS/MS. Ce problème est résolu par l’application de fac-
teurs de correction isotopique correspondant aux pourcentages de chaque tag con-
taminant pour un tag donné
- la contribution du bruit de fond en MS/MS, lorsque l’intensité de l’ion rapporteur de
masse est faible, contribution d’autant plus importante que l’échantillon est complexe
ou que la masse du tag considéré est élevée. Cette contamination peut être partiel-
lement contournée en se focalisant sur les signaux MS/MS de plus haute intensité
tout en gardant à l’esprit que plus l’ion rapporteur de masse a une masse élevée plus
son signal a une intensité faible

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 41
II.4. Etude protéomique haut-débit de cellules tubulaires proximales exposées aux ICN
par protéomique Shotgun-iTRAQ
II.4.1. Hypothèse de travail et objectifs
Après avoir réalisé une synthèse de la littérature traitant des mécanismes physiopa-
thologiques participant à la néphrotoxicité des ICN, dans un premier temps, et des ap-
proches de protéomique quantitative haut débit, dans un second temps, l’hypothèse formu-
lée est la suivante : une cartographie dynamique du protéome de cellules tubulaires proxi-
males exposées aux ICN, obtenue sans a priori, par protéomique quantitative haut-débit
Shotgun-iTRAQ devrait permettre la découverte de nouveaux mécanismes physiopatholo-
giques impliqués dans la toxicité tubulaire des ICN.
Les objectifs de travail étaient donc :
1. d’optimiser la technologie iTRAQ aux étapes successives de préparation,
d’analyse et de retraitement bio-informatique
2. d’appliquer la technologie iTRAQ optimisée à l’étude du protéome de cellules
tubulaires proximales afin d’observer et classer les variations d’expression
protéique consécutives à une exposition prolongée à des concentrations cli-
niques d’ICN puis déterminer les mécanismes cellulaires et les voies de signa-
lisation impactés
Le premier article détaille le développement d’une nouvelle méthodologie de traite-
ment des données MS/MS, de l’identification protéique à la quantification relative, dans le
cadre de l’application d’un protocole optimisé de protéomique quantitative haut-débit iTRAQ
à l’étude des perturbations du protéome de cellules tubulaires proximales porcines LLC PK-1
analysées par chromatographie liquide haute performance (nanoHPLC) en ligne avec un
spectromètre de masse haute résolution (QqTOF). Cet article est en révision pour publica-
tion dans la revue Proteomics.
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 42
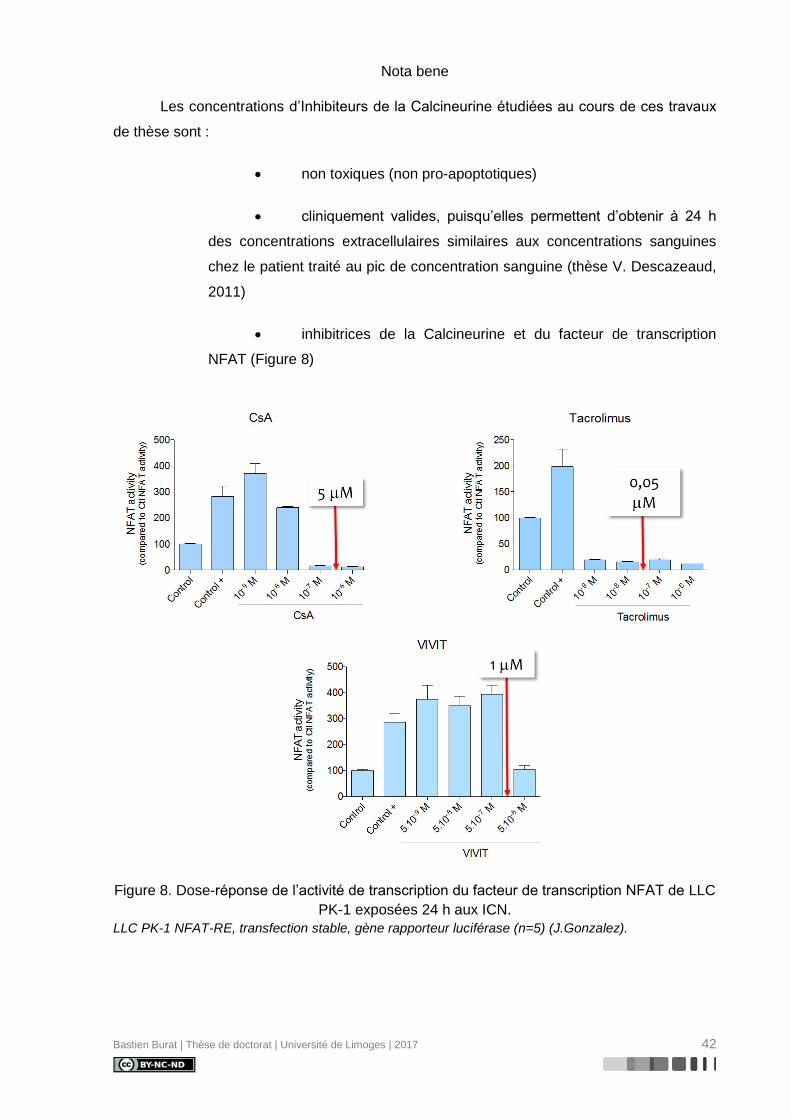
Nota bene
Les concentrations d’Inhibiteurs de la Calcineurine étudiées au cours de ces travaux
de thèse sont :
• non toxiques (non pro-apoptotiques)
• cliniquement valides, puisqu’elles permettent d’obtenir à 24 h
des concentrations extracellulaires similaires aux concentrations sanguines
chez le patient traité au pic de concentration sanguine (thèse V. Descazeaud,
2011)
• inhibitrices de la Calcineurine et du facteur de transcription
NFAT (Figure 8)
Figure 8. Dose-réponse de l’activité de transcription du facteur de transcription NFAT de LLC
PK-1 exposées 24 h aux ICN.
LLC PK-1 NFAT-RE, transfection stable, gène rapporteur luciférase (n=5) (J.Gonzalez).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 43
II.4.2. Développement et optimisation d’une stratégie d’analyse intégrée des données
MS/MS pour l’étude protéomique quantitative par la technologie iTRAQ ; Application
aux cellules rénales tubulaires proximales exposées aux ICN
II.4.2.1 Article 1.
Sum of peak intensities may improve upon peak area integration in a more performant
iTRAQ data analysis pipeline when using a TripleTOF 5600+ platform
BASTIEN BURAT*, JULIEN GONZALEZ, FRANÇOIS-LUDOVIC SAUVAGE, HÉLÈNE AR-
NION, EMILIE PINAULT, PIERRE MARQUET, MARIE ESSIG
En révision pour publication au journal Proteomics.

For Peer Review
Sum of peak intensities may improve upon peak area
integration in a more performant iTRAQ data analysis pipeline when using a TripleTOF 5600+ platform.
Journal: PROTEOMICS
Manuscript ID pmic.201700372.R1
Wiley - Manuscript type: Research Article
Date Submitted by the Author: n/a
Complete List of Authors: Burat, Bastien; Universite de Limoges Faculte de Medecine, UMR INSERM S-850 Pharmacology of Immunosuppressive drugs and Transplantation Gonzalez, Julien; Universite de Limoges Faculte de Medecine, UMR INSERM S-850 Pharmacology of Immunosuppressive drugs and Transplantation Sauvage, François-Ludovic; Universite de Limoges Faculte de Medecine, UMR INSERM S-850 Pharmacology of Immunosuppressive drugs and Transplantation Arnion, Hélène; Universite de Limoges Faculte de Medecine, UMR INSERM S-850 Pharmacology of Immunosuppressive drugs and Transplantation Pinault, Emilie; Universite de Limoges, BISCEm, FR CNRS 3503 GEIST Marquet, Pierre; Universite de Limoges Faculte de Medecine, UMR INSERM S-850 Pharmacology of Immunosuppressive drugs and Transplantation; Centre Hospitalier Universitaire de Limoges, Department of Pharmacology and Toxicology Essig, Marie; Universite de Limoges Faculte de Medecine, UMR INSERM S-850 Pharmacology of Immunosuppressive drugs and Transplantation; Centre Hospitalier Universitaire de Limoges, Department of Nephrology, Dialysis, Transplantation
Keywords: iTRAQ, peak intensities, peak areas
Wiley - VCH
PROTEOMICS

For Peer Review
1
Sum of peak intensities may improve upon peak area
integration in a more performant iTRAQ data analy-
sis pipeline when using a TripleTOF 5600+ plat-
form.
Bastien Burat1*, Julien Gonzalez
1, François-Ludovic Sauvage
1, Hélène Arnion
1, Emilie Pinault
4,
Pierre Marquet12, Marie Essig
13*
1INSERM, UMR S-850, University of Limoges, Limoges, France
2CHU Limoges, Department of Pharmacology and Toxicology, Limoges, France
3CHU Limoges, Department of Nephrology, Dialysis, Transplantation, Limoges, France
4BISCEm, FR CNRS 3503 GEIST, University of Limoges, Limoges, France
Correspondence: Bastien Burat, Tel.: +33 5 19 56 42 49; E-mail: [email protected].
Marie Essig Tel.: +33 555 43 58 95; Fax: +33 555 43 59 36; E-mail: [email protected].
UMR INSERM S-850, Pharmacology of Immunosuppressive drugs and Transplantation, Faculty
of Medicine and Pharmacy, University of Limoges, 2, rue du Dr Marcland 87025 Limoges
CEDEX, France.
Page 1 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
2
ABBREVIATIONS
CNI: CalciNeurin Inhibitors
CsA: Cyclosporine A
CiR-C: Customizable iTRAQ Ratios Calculator
ICAT: Isotope-Coded Affinity Tags
iTRAQ: isobaric Tags for Relative and Absolute Quantitation
MS/MS: tandem mass spectrometry
nano-HPLC: nano-scale high-performance liquid chromatography
NFAT: Nuclear Factor of Activated T cells
RPA: Ratios of Peak Areas
RSPI: Ratios of the Sums of Peak Intensities
SILAC: Stable Isotope Labeling of Amino acids in Cell culture
Tac: Tacrolimus
Keywords: iTRAQ, peak intensities, peak areas
6681 words
Page 2 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
3
ABSTRACT
In the field of quantitative proteomics, the Isobaric Tags for Relative & Absolute Quanti-
tation (iTRAQ) technology has demonstrated efficacy for proteome monitoring despite its lack of
a consensus for data handling. In this study, after peptide and protein identification, we compared
the widespread quantitation method based on the calculation of MS/MS reporter ion peaks areas
ratios (ProteinPilot) to the alternative method based on the calculation of ratios of the sum of
peak intensities (jTRAQx (Quant)) and we processed output data with the in-house Customizable
iTRAQ Ratios Calculator (CiR-C) algorithm. Quantitation based on peak area ratios displayed no
significant linear correlation with Western blot quantitation. In contrast, quantitation based on the
sum of peak intensities displayed a significant linear association with Western blot quantitation
(non-zero slope; Pearson correlation coefficient test, r = 0.2962, p = 0.0099 **) with an average
bias of 0.08747 ± 0.5004 and 95% Limits of Agreement from - 0.8932 to 1.068. We proposed the
Mascot-jTRAQx-CiR-C strategy as a simple yet powerful data processing adjunct to the iTRAQ
technology.
Page 3 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
4
SIGNIFICANCE OF THE STUDY
In this work, an in-house algorithm named Customizable iTRAQ Ratio Calculator (CiR-
C) was implemented to process large datasets and compute final quantitation (median, weighted
mean and standard deviation) for iTRAQ-based shotgun proteomics. This algorithm was used to
retreat datasets in the comparison between two workflows based on the two strategies of MS/MS
signal integration (ratios of peak areas (RPA) versus ratios of the sum of peak intensities (RSPI))
for iTRAQ quantitation in the perspective of the proteome monitoring of tubular proximal cell
lysates. RSPI was confirmed to be the best-suited strategy when using high-resolution MS plat-
forms. The RSPI-based iTRAQ workflow happened to allow reliable and robust protein expres-
sion measurement. CiR-C proved to be a promising, simple and powerful, adjunct to iTRAQ data
processing.
Page 4 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
5
INTRODUCTION
The recent and already widespread large-scale omics technologies enabled the discovery
of unexpected mechanisms in the field of pathophysiology. These techniques investigate DNA
(genomics, epigenetics), mRNAs or microRNAs (transcriptomics), proteins (proteomics), lipids
and small molecules (metabolomics). When employed in parallel onto these different targets,
large-scale omics techniques help seize the many layers of cell responses to pathophysiological
stimuli or to drugs, e.g., regulation and transcription of genes, handling of transcripts and transla-
tion into proteins. They are major tools to comprehensively explore all intracellular pathways
beyond the first known drug targets. This enables a better understanding of cellular side effects of
drugs.
A number of MS-based high-throughput proteomics or “shotgun proteomics” technologies
are compatible with relative protein quantitation and offer variable performances in terms of pro-
teome and sequence coverage, precision, accuracy and reproducibility of quantitation or versatili-
ty of sample application [1]. Using Label-free Quantification (LFQ), either protein abundance
correlates with the measure of peptide precursor ion MS signal intensities or is obtained from the
counting of peptide fragment ion MS/MS spectra (spectral counting) [2]. Isotope-coded Affinity
Tags (ICAT) was the first labeling technique, which was based on biased protein labeling through
tagging of the non-universal residue cysteine with heavy or light tags [3]. Stable Isotope Labeling
of Amino Acids in Cell Culture (SILAC) is a metabolic labeling technique based on the introduc-
tion of heavy or light amino acids during protein biosynthesis [4], hence a high-level reliability
and robustness in terms of labeling stability, precision and accuracy [5]. Isobaric Tags for Rela-
tive & Absolute Quantitation (iTRAQ) is a chemical labeling technique that has been developed
to multiplex comparison between protein sets issued from different biological samples, as ob-
Page 5 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
6
tained from a single tandem mass spectrometry run. In iTRAQ, digested peptides, from up to
eight different conditions, are labeled by isobaric tags. This allows characterizing peptides, in a
condition-independent way, using the first mass spectrometry filter (MS mode) and measuring
their relative abundance between the different conditions using the second stage of the tandem
mass spectrometer (MS/MS mode) [6], upon the detection of the condition-specific reporter ions
of distinct masses in the low m/z region of the MS/MS spectrum.
Although extended multiplexing is the major advantage of iTRAQ, its use benefits from:
high reproducibility, precision and accuracy compared to LFQ (like all stable isotope labeling
versus LFQ), to the cost of a wider dynamic range, better proteome coverage and faster sample
preparation and analysis [7,8]; better sensitivity and proteome coverage compared to ICAT [9];
wide sample applicability, faster sample processing and better proteome coverage compared to
SILAC [10]; as well as a valuable ‘toolbox’ that has been built over the past decade thanks to the
literature addressing technique drawbacks and methodological solutions to overcome them [11].
The best method to quantify the isobaric tags, hence the relative peptide abundances, is
still under scrutiny. The commercially available software used the ratios of peak areas (RPA)
based on the initial description stating that the abundance of a collision-released mass reporter
ion appeared to be proportional to the trapezoidal integration of peaks at the theoretical mass-to-
charge ratio (m/z). Alternatively, the ratios of sum of peak intensities (RPSI) was shown to result
in higher sensitivity and more reliable quantitation [12–14]. In this case, the abundance of a mass
reporter ion is directly related to its ion counts – height of the peaks – at the theoretical m/z. To
the best of our knowledge, performances of the two methodologies (RPA and RSPI) have never
been studied and compared in the light of classical molecular biology approaches for protein
quantitation (e.g. Western blot).
Page 6 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
7
In iTRAQ, a correct peptide and protein quantitation needs a correct interpretation of
MS/MS spectra, which depends on a trustworthy peptide and protein identification. The identifi-
cation and quantitation can invariably be performed by commercial built-in algorithms integrated
to companion software packages; they fully retreat data generated by the manufacturer’s mass
spectrometers (e.g. Paragon - ProteinPilot for ABSciex mass spectrometers). Alternatively, anal-
yses may be split into separate stages through a composite suite of commercial or free algorithms
integrated to manufacturer-independent software (e.g. Mascot in Mascot Server, jTRAQx). It is
noteworthy that, although tools to compute RPA are widely available, the computation of the
ratios of RSPI is not supported by any available companion software, and so, it requires an alter-
native suite.
The first aim of our work was to compare these two strategies of quantitation (RPA and
RSPI) to the classical Western blot technique, commonly used as a non-MS validation technique
for iTRAQ-based quantitation. The second aim was to further develop an all-in-one protocol,
from sample preparation to result reporting, based on the best strategy of quantitation followed by
in-house data processing. This study was carried out on the respective effects of the CalciNeurin
inhibitors (CNI) Cyclosporine A (CsA) and Tacrolimus (Tac, a.k.a. FK506) on renal proximal
tubular cells, used as a study model. iTRAQ was combined to nano-scale liquid chromatography
online with Q-Q-TOF tandem mass spectrometry on proximal tubular cells to investigate whether
CsA and Tac nephrotoxicity results from the inhibition of calcineurin or from the modulation of
other intracellular pathways targeted by immunophilins.
In this work, we validated the RSPI methodology and we built an automated data pro-
cessing algorithm called Customizable iTRAQ Ratio Calculator (CiR-C) to refine critical pa-
rameters related to peptide confidence and selection. The composite suite made up of the Mascot
Page 7 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
8
algorithm, for peptide and protein identification, end-to-end with the jTRAQx software for com-
putation of RSPI at the peptide level and the CiR-C algorithm for data integration and definitive
protein quantitation turned out to be a successful combination. It has provided a great improve-
ment compared to already available solutions for iTRAQ-based high-throughput quantitative pro-
teomics and multiplexed analysis of biological systems.
Page 8 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
9
EXPERIMENTAL METHODS
MATERIAL AND CHEMICALS
Dulbecco’s Modified Eagle’s Medium-Ham’s F12 (1:1, #31331, Gibco), Fetal Bovine Se-
rum (#10500), 1 M HEPES (#15630), 7.5 % Sodium bicarbonate (#25080), 10,000 UI.mL-1 Peni-
cillin / Streptomycin (#15140), Dulbecco’s Phosphate Buffer Saline (#14190) were purchased
from Gibco. Sodium selenite (S5261), CsA (#30024), Tac (F4679), insulin (I4011), triiodothyro-
nine (T6397) and dexamethasone (D4902) were purchased from Sigma-Aldrich. Primary antibod-
ies against porcine Cyclophilin A (ab41984, 1:1,000) was purchased from Abcam, anti-β-Actin
(MA1-91399, 1:10,000), anti-Na+/K+ ATPase alpha subunit 1 (MA3-929, 1:2,000), anti-Cofilin-
1 (PA1-24931, 1:10,000) and anti-Galectin-1 (#437400, 1:500) were purchased from Ther-
moFisher. Secondary antibodies were purchased from Sigma-Aldrich (Anti-Mouse IgG (whole
molecule)–Peroxidase antibody produced in rabbit, A9044, 1:10,000; Anti-Rabbit IgG (whole
molecule)–Peroxidase antibody produced in goat, A9169, 1:10,000) and ThermoFisher (F(ab')2-
Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP, A24531, 1:10,000).
CELL CULTURE CONDITIONS & DRUG EXPOSURE
LLC PK-1 (Lilly Laboratories Porcine Kidney-1) porcine proximal tubule cells (ATCC-
CL-101, ATCC, Manassas, VA) were expanded in 75 cm² flasks at 37 °C with 5 % CO2 and
passed once confluence was reached. Culture medium consisted in a 1:1 Dulbecco’s Modified
Eagle’s Medium-Ham’s F12 mix supplemented with 5 % FBS, 15 mM HEPES, 0.1 % Sodium
bicarbonate, 100 UI.mL-1 Penicillin / Streptomycin and 50 nM Sodium selenite. LLC PK-1 cells
were cultured between passage 7 and passage 25.
Page 9 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
10
LLC PK-1 were seeded in four 60 mm Petri dishes (one per condition) and expanded up
to sub-confluence in the routine cell culture medium.
Seeded LLC PK-1 sustained serum starvation and were fed with hormonally-
defined (25 µg.mL-1 insulin, 11 µg.mL-1 transferrin, 50 nM triiodothyronine, 0.1 µM dexame-
thasone, 0.1 µg.mL-1 desmopressin) fresh medium to engage epithelial differentiation, for 24
hours.
Differentiated LLC PK-1 cells were exposed for 24 hours to four different conditions:
Control (vehicle: 96 % Ethanol), 5 µM CsA, 0.05 µM Tac or 1 µM VIVIT (a specific NFAT in-
hibitor [15]).
PROTEIN EXTRACTION, SAMPLE PREPARATION, ITRAQ LABELLING AND ISOE-
LECTRIC FOCUSING
After 24 h drug exposure, LLC PK-1 cells were washed twice with Dulbecco’s Phosphate
Buffer Saline and lysed by scrapping in a custom RIPA lysis buffer (150 mM NaCl, 50 mM
TRIS-HCl, 0.1 % NP-40, 0.1 % SDS, 1 mM EDTA in ultrapure H2O, supplemented with an anti-
protease / anti-phosphatase mix). Cell lysates were incubated on ice for 30 min and centrifuged
for 15 min at 21000 g. Supernatants were stored until protein concentration was measured using
the Bradford colorimetric method and iTRAQ labeling. Twenty-five micrograms of proteins were
precipitated by - 20 °C cold acetone. After acetone evaporation, the precipitates were solubilized
in 25 mM ammonium bicarbonate then were incubated with 50 mM dithiothreitol for 40 min at
60 °C, to reduce disulfide bonds, 100 mM iodoacetamide in the dark for 40 min at room tempera-
ture, to alkylate/block cysteine residues and eventually were digested for 24 h at 37 °C with
mass-spectrometry grade trypsin (V5280, Promega) at a 1: 50 enzyme: substrate ratio.
Page 10 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
11
After digestion, samples were incubated with iTRAQ tags (iTRAQ Reagents Multiplex
kits, 4-plex, #4352135, Sigma-Aldrich) – one tag per drug exposure condition and five different
tag/condition associations over five experiments – for 1 h at room temperature. Labeled samples
were mixed and separated into 12 fractions by isoelectric focusing (OFFGEL 3100 Fractionator,
Agilent Technologies, Santa Clara, CA) for 24 h at increasing voltage and steady intensity of 50
µA in a 3-10 pH IPG strip. Fractions were retrieved for further MS analysis after the IPG strip
was incubated in a 1:1 acetonitrile (ACN): water, 0.1 % formic acid (FA) wash solution for 15
min at room temperature (Scheme 1A).
NANO-LC PEPTIDE SEPARATION AND Q-Q-TOF MASS SPECTROMETRY
IEF fractions were analyzed by nano-LC MS/MS using a nano-chromatography liquid Ul-
timate 3000 system (LC Packings DIONEX, Sunnyvale, CA) coupled to a Triple TOF 5600+
mass spectrometer (ABSciex, Toronto, Canada). For each sample, 5 µL were injected into a pre-
column (C18 PepmapTM 300 µm ID x 5 mm, LC Packings DIONEX) using the loading unit. Af-
ter desalting for 3 min with loading solvent (2 % ACN, 0.05 % trifluoroacetic acid (TFA)), the
pre-column was switched online with the analytical column (C18 PepmapTM 75 µm ID x
150 mm, LC Packings DIONEX) pre-equilibrated with 95 % solvent A (ACN 5 % - FA 0.1 %).
Peptides were eluted from the pre-column into the analytical column and then into the mass spec-
trometer by a linear gradient from 5 % to 25 % in 70 min, then to 95 % of solvent B (98 % ACN,
0.1 % FA) over 120 min at a flow rate of 200 nL/min.
Data acquisition was carried out by IDA (Information-Dependent Acquisition) mode of
Analyst 1.7 TF software (ABSciex). The data from MS and MS/MS were continuously recorded
with a cyclic duration of 3 s. For each MS scan, up to 50 precursors were selected for fragmenta-
Page 11 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
12
tion based on their intensity (greater than 20,000 cps), their charge state (2+, 3+) and if the pre-
cursor had already been selected for fragmentation (dynamic exclusion). The collision energies
were automatically adjusted according to charge state, ionic mass of selected precursors and
iTRAQ labelling.
MASS SPECTROMETRY DATA PROCESSING AND RELATIVE PROTEIN IDENTIFICA-
TION / QUANTIFICATION
QUANTITATION METHOD BASED ON RSPI
MS and MS/MS data for five independent experiments (biological replicates) (*.wiff, 1
per fraction, 12 files per experiment) were submitted to Mascot Server 2.2.03 via Pro-
teinPilot (version 5.0, ABSciex) for protein identification, and searched against two complemen-
tary Sus scrofa databases: a Swiss-Prot database (2015_10 release) and a TrEMBL data-
base (2015_10 release). Carbamidomethyl (C) was defined as a fixed modification. Oxidation
(O), iTRAQ4plex (K), iTRAQ4plex (Y), iTRAQ4plex (N-term) were defined as variable modifi-
cations. MS/MS fragment mass tolerance was set at 0.3 Da. Precursor mass tolerance was set at
0.2 Da.
Mascot raw data files (*.dat, 1 per experiment) were saved for further isobaric tags-based
peptide and protein quantitation with the Java implementation of the Quant algorithm,
jTRAQx (version 1.13, [16]). Reporter mass tolerance was set at 0.05 Da while iTRAQ correc-
tion factors were implemented as provided by ABSciex. This tool generated one .jpf file (tab-
delimited text file) for each series.
Page 12 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
13
QUANTITATION METHOD BASED ON RPA
MS and MS/MS data for five independent experiments (biological replicates) (*.wiff, 1
per fraction, 12 files per experiment) were submitted to protein identification using the Paragon
algorithm as implemented in the ProteinPilot software (version 5.0, AB SCIEX) and searched
against two complementary Sus scrofa databases: a Swiss-Prot database (2015_10 release, 1422
entries) and a TrEMBL database (2015_10 release, 47465 entries). Quantitation was conducted
with or without auto bias correction, an available option implemented in the ProteinPilot software
to normalize uneven protein across the multiplex samples, improving further quantitation. Mass
tolerances and identification parameters were automatically set and optimized for the ABSciex
5600+ TripleTOF™-generated MS/MS data (MS/MS Fragment mass tolerance was set at 0.1 Da.
Precursor mass tolerance was set at 0.05 Da).
The *.group results files (1 per experiment) were exported as Peptide Summaries.
WESTERN BLOT
Western blots were performed on total cell lysates from 5 independent experiments (bio-
logical replicates), prepared in custom RIPA buffer (see above). Forty micrograms of proteins per
exposure condition were separated by electrophoresis under reducing and denaturing conditions
on a NuPAGE® Novex® Bis-Tris pre-cast gel (NP0341, ThermoFisher) in 1X NuPAGE™
MOPS SDS running buffer (NP0001, ThermoFisher) and transferred to a nitrocellulose (NC)
membrane (NP23001, ThermoFisher) using the iBLOT 2 Dry Blotting system (IB21001, Ther-
moFisher). Membranes were blocked for 1 h at room temperature under agitation with TBS-
Tween buffer (10 mM Tris 7.6, 150 mM NaCl, 0.1 % Tween-20) complemented with 5 % (W/V)
non-fat milk powder to obtain BLOTTO buffer. Primary antibody incubation was done in
Page 13 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
14
BLOTTO for 1 h at room temperature. After three 5-min washes in TBS-T, secondary antibody
incubation was performed in BLOTTO for 1 h at room temperature then washed again. Mem-
branes were incubated in a 1: 1 mix of SuperSignal™ West Pico PLUS Chemiluminescent Sub-
strate kit (#34577, ThermoFisher) and analyzed by the ChemiDoc Imaging system (Bio-Rad) for
chemiluminescent signal detection and acquisition. Quantitation was computed via the ImageLab
software (Bio-Rad).
STATISTICAL ANALYSIS
Western blot quantitation was compared to RSPI- and RPA-based quantitation using the
Bland-Altman comparison method [17,18] and Pearson correlation coefficient test as implement-
ed in the GraphPad Prism software (version 5.04).
DATA AVAILABILITY
The MS proteomics data have been deposited to the ProteomeXchange Consortium
(http://proteomecentral.proteomexchange.org) [19] via the PRIDE partner repository [20] with
the data set identifier PXD007891 (username: [email protected], password: TPSGICw9).
Page 14 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
15
RESULTS
COMPARISON BETWEEN THE DATA PROCESSING OUTPUTS: MASCOT – jTRAQx
VERSUS PARAGON – PROTEINPILOT
Multiplex analysis of CNI-exposed partial proteomes was conducted and optimized in
vitro using the epithelial tubular proximal cell line LLC PK-1 serving as a model. Five series of
samples were prepared and processed according to the optimized custom-made iTRAQ proto-
cols (Scheme 1A). The samples labeling strategy is summarized in Table 1.
The RPA strategy resulted in Paragon identifying 9788 ± 3270 peptides related to 1100 ±
132 proteins per series. After identification and quantitation by the Paragon – ProteinPilot suite,
the CiR-C algorithm included 4105 peptides (114 proteins) split into 35 Swiss-Prot entries and 79
TrEMBL entries.
The RSPI strategy resulted in Mascot Server identifying 14291 ± 4582 trypsin-digested
peptides related to 1160 ± 115 proteins per series. After identification by the Mascot – jTRAQx
suite, the CiR-C algorithm included 32169 peptides (370 proteins) split into 131 Swiss-Prot and
239 TrEMBL entries.
The CiR-C shell script excluded irrelevant data according to criteria summed up in Table
2: i) identification confidence: peptides are retained if the probability that the observed positive
match is a random match is below 5 % (p < 0.05, Mascot score > 30; Paragon confidence score >
95); ii) quantification confidence: peptides are retained if all iTRAQ ratios have been successful-
ly calculated i.e., peptides with 0.0 ratios or uncalculated ratios (null ratios) are discarded; iii)
peptides related to ‘Fragment’- and ‘REVERSED’-annotated proteins are discarded. After irrele-
vant data removal, CiR-C drew up an exhaustive catalogue of identified peptide sequences with
their associated Swiss-Prot or TrEMBL accession IDs. Peptides were assigned to a frequency
Page 15 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
16
index of positive matches (identification in {1;2;3;4;5} out of 5 independent experiments (biolog-
ical replicates)) and CiR-C drew a second catalogue of peptides with the highest frequency index
(n=5). Protein ratios were calculated as both overall and series-specific median values of peptide
ratios associated with a given accession ID and frequency index.
CiR-C discarded 7597 ± 3291 (75.4 % ± 9.0) peptides quantified by RPA and 4377 ±
2367 (29.4 % ± 5.5) peptides quantified by the RSPI (Table 2).
Page 16 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
17
COMPARISON BETWEEN THE TWO QUANTITATION STRATEGIES
The results of the two quantitation strategies (RPA from the Paragon – ProteinPilot – CiR-
C data processing and RSPI from the Mascot Server – jTRAQx – CiR-C data processing, Scheme
1B) were compared to the Western blot analysis of five proteins picked from the 370-protein final
list (Figure 1A). The selection comprised the cytoskeleton-structuring β-Actin, cytoplasmic Co-
filin-1, alpha 1 subunit of membrane-attached Na+-K
+ ATPase, cytosolic Cyclosporine-
complexing Cyclophilin A and Galectin-1, i.e., a panel of both spatially scattered and differently
expressed proteins (Figure 1B).
iTRAQ quantitation using the commercial method based on RPA displayed no significant
linear correlation with Western blot quantitation (zero-slope; Pearson correlation coefficient r =
-0.07443, p = 0.5257) (Figure 2A). Furthermore, the average difference between Paragon – Pro-
teinPilot – CiR-C and Western blot methods was -0.2747 ± 0.9610 (approx. 30 % of a given
iTRAQ ratio) while 95% Limits of Agreement were -2.163 and 1.609 (Figure 2B). Auto bias cor-
rection did improve neither accuracy nor precision as it worsened ratio compression and non-
correlation to Western blot (zero-slope; Pearson correlation coefficient r = 0.07245, p = 0.5368)
and the average bias increased (-0.3837 ± 0.7308) even though 95% Limits of Agreement were
tighter (-1.816 and 1.049) (data not shown). Moreover, the RPA method resulted in several mis-
matches where Western Blot couldn’t be compared to iTRAQ because of missing iTRAQ quanti-
tation (iTRAQ ratio = 0).
Concerning the quantitative results obtained from the Mascot Server – jTRAQx – CiR-C
strategy based on RSPI, a statistically-significant linear association with Western blot quantita-
tion was observed (non-zero slope; Pearson correlation coefficient r = 0.2962, p = 0.0099 **)
Page 17 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
18
(Figure 3A). The average difference was 0.08747 ± 0.5004 (approx. 9 % of a given ratio). The
95% Limits of Agreement were closer, i.e., -0.8932 and 1.068 (Figure 3B).
Page 18 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
19
REFINMENT OF THE CiR-C ALGORITHM
The CiR-C script uses two thresholds for peptide selection: a peptide significance thresh-
old (given by the Mascot score) and a peptide frequency threshold (defined as the minimum
number of experiments in which a given peptide must be identified and quantified). To establish
the best compromise between reliability of iTRAQ quantitation and time/cost efficiency, we ex-
plored how threshold tuning may affect (strengthen or weaken) CiR-C results.
The RSPI calculated after tuning the first threshold to either a permissive (Mascot score >
20) or a stringent (Mascot score > 40) cut-off value were compared to Western blot standards
(Figure 4). When opening to more peptides (lower Mascot score) the linear correlation was lost
(zero slope; Pearson correlation coefficient test, r = 0.06231, p = 0.5954) (Figure 4A); doing so,
the mean difference compared to Western blot was slightly reduced (-0.06338 ± 0.5367, 95%
Limits of Agreement 0.9886 and -1.115) (Figure 4B). When restricting to less peptides (higher
Mascot score) the linear correlation was not improved (non-zero slope; Pearson correlation coef-
ficient r = 0.2737, p = 0.0175 *) (Figure 4C); again, the difference compared to Western blot was
slightly reduced (-0.07496 ± 0.5098, 95% Limits of Agreement 0.9243 and -1.074) (Figure 4D).
The RSPI computed after adjustment of peptide frequency threshold to lower levels were
plotted against Western blot results (Figure 5). Using peptides from four out of five independent
experiments (biological replicates) resulted in a significant improvement of correlation (non-zero
slope; Pearson correlation coefficient r = 0.3466, p = 0.0067 **) (Figure 5A) and a greater differ-
ence compared to Western blot (-0.05458 ± 0.5186, 95% Limits of Agreement 0.9618 and -
1.071) (Figure 5B). Conversely, using peptides from only three out of five independent experi-
ments (biological replicates) led to a loss of correlation (zero slope; Pearson correlation coeffi-
Page 19 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
20
cient r = 0.06591, p = 0.6671) (Figure 5C) and a greater difference compared to Western blot (-
0.09232 ± 0.6069, 95% Limits of Agreement 1.097 and -1.282) (Figure 5D).
Page 20 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
21
DISCUSSION – CONCLUSION
Since its first description by Ross et al. in 2004, iTRAQ has been widely used for multi-
plexed analysis of proteomes. The advantages and drawbacks of iTRAQ have been widely ad-
dressed in the literature, following the many developments of technical optimizations, analysis
strategies and tools to improve quantitation precision and accuracy [21–24]. However, there is
still no consensual technique in sample preparation and analysis or data processing.
iTRAQ results in rich and complex MS/MS datasets which require thorough processing
and solid statistics to reach relevant conclusions. In this respect, the choice of a tag ratio calcula-
tion method is crucial. Two potential strategies have been proposed: either RPA or RSPI. The
two methods are not equivalent since peak area measurement suffers from a major bias originated
from the way reporter ion signals are processed, as described by Boehm et al. [13]. It is worth
noting that peak intensities are proportional to reporter mass ion counts, whereas peak areas are
not (Scheme 2). Therefore, the RSPI is more likely to provide a reliable rendering of the actual
ion count detected by mass spectrometer. Numerous studies reported the necessity of RPSI-based
quantitation workflows to obtain robust, precise, accurate and sensitive when using high resolu-
tion MS platforms (Orbitraps and TripleTOF 5600) [25–27] We confirmed the superiority of
RSPI since the commercially available RPA strategy failed to report significant biological varia-
tions assessed by Western Blot analysis. In contrast, with the lowest bias and best correlation, the
RSPI from Mascot – jTRAQx – CiR-C data processing strategy provided the most reliable set of
quantitation ratios when compared to Western Blot performance. As compared to the Western
blot ‘quality control’, the median RPA showed dramatic differences leading to a global misrepre-
sentation of changes in protein expression. Conversely, the median RSPI for the set of proteins of
Page 21 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
22
interest were well correlated and showed moderate and unbiased differences compared to West-
ern Blot.
Western blot was chosen as a non-MS-based quality control for iTRAQ because the tech-
nique has already been successfully used to validate iTRAQ. Either alone [28,29], in parallel with
other classical molecular biology techniques such as RT-qPCR [30,31], or, in parallel with MS-
based targeted methods like selected reaction monitoring/multiple reaction monitoring
(SRM/MRM) [32,33], Western blot always managed to validate and confirm results from both
iTRAQ and other, more performant, validation techniques. Most importantly, the technique was
the first to assess performance issues when discrepancies between Western blot and iTRAQ re-
sults highlight ratio compression and underestimation [34]. The observed difference between
iTRAQ ratios and Western blot ratios may be explained by the intrinsic ratio compression due to
background noise at the low m/z end, by co-elution of peptides with close m/z, and to a lesser
extent by tag purity and inter-contamination [35]. Even if correction factors are provided to take
tag ‘impurity’ into account, the background mass spectrometry noise brings the ratio towards
unity. However, the custom-made strategy using the RSPI appeared to minimize these differences
with respect to the commercially available strategy.
Differences between iTRAQ and Western blot may also be explained by the technical
evolution of LC-MS-based quantitative proteomics when compared to antibody-based approach-
es. On one hand, Western blot remains a qualitative and semi-quantitative technique with an in-
herent variability in analytical sensitivity, specificity and reproducibility, especially because of
the multifactorial (e.g. variability of antibodies specificity, non-linearity of chemiluminescent
reaction) non-linearity between protein abundance and signal intensity [36,37]. On the other
hand, the use of high-performance liquid chromatography (such as nanoLC) online with high-
Page 22 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
23
resolution mass spectrometers (such as TripleTOF 5600+ QqTOF mass spectrometer) has outper-
formed Western blot with greater sensitivity, selectivity, specificity and reproducibility, wider
linear dynamic range, increased accuracy and precision, high-throughput of in-depth information
[38–42].
A major point of iTRAQ data processing is the upstream tolerance for peptide characteri-
zation and identification. More peptides were identified with Mascot Server than with Paragon,
mainly because mass tolerances were more stringent with the latter, due to the use of non-
customizable manufacturer’s parameters optimized for data generated by TripleTOF 5600+ mass
spectrometers. Mass tolerances with Mascot Server were chosen to be sufficiently permissive to
provide enough data for further selection. The choice of stringent parameters for identification is
in complete agreement with the RPA quantitation strategy, as implemented in ProteinPilot, where
peak width is essential to reliable quantitation. Conversely, our approach postpones the applica-
tion of stringent conditions to after quantitation.
Another major point of iTRAQ data processing is the downstream management of irrele-
vant data. It can be done manually but it is time-consuming and error-prone due to the huge
amount of information at the peptide level as well as the number of replicates. Just like with eve-
ry high-throughput approach, the analysis of iTRAQ results needs to be automated. The CiR-C
algorithm was designed to be the simplest in terms of data elimination and transformation. Likely
to exacerbate inherent issues – such as ratio-compressing variance-stabilizing normalization –
heavy data transformation was not used after jTRAQx or ProteinPilot data processing. The bio-
logical significance of the results arose from: i) a focus on peptide selection thanks to the proba-
bilistic Mascot score or its Paragon counterpart (Confidence score); ii) the restriction to selected
peptides based on their occurrence among multiple biological replicates; iii) the calculation of
Page 23 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
24
median and weighted mean ratios for each set of peptides obtained from a given protein. As men-
tioned above, upstream data processing was willingly permissive to sustain this statistical ap-
proach. It resulted in less loss of information and good data fitting. Paradoxically, stringent iden-
tification criteria also resulted in increased output, including a large amount of irrelevant data:
when parsing peptide summaries, it appeared that irrelevant data were essentially null ratios, i.e.,
the technical impossibility to provide peptide quantitation from the MS2 mass spectra.
The confidence threshold for the Mascot score was a compromise between the generation
of aberrant information and the loss of information. This threshold highly depends on the number
of peptides characterized by mass spectrometry. The probability of a random match was first set
to 1 out of 1000 (s = 30, p = 0.001) and the test of scores 10-fold apart from our initial choice (s =
20, p = 0.01) confirmed how close it is from a potential optimum for the size of our datasets. In-
deed, this threshold highly depends on the number of peptides characterized in MS/MS.
The occurrence threshold was set to the largest possible values – the more peptides, the
better – hence the restriction to ubiquitous peptides, i.e., peptides retrieved from all five biologi-
cal replicates. The main source of variations when using iTRAQ is of biological origin (± 25 %)
[43]. The number of biological replicates was set to five independent experiments (biological
replicates), corresponding to the use of a complete given set of iTRAQ tags. However, as a matter
of time and cost efficiency, the possibility to reduce the number of independent experiments (bio-
logical replicates) had to be addressed. As expected, considering biological variations are the
most impactful source of variability of iTRAQ, we showed that using four independent replicates
is the lowest limit to obtain reliable results, five replicates appearing optimal under our experi-
mental conditions.
Page 24 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
25
In summary, this work demonstrated that RSPI outperform the commercially available
RPA in quantifying biological modifications using iTRAQ. Furthermore, we propose a Mascot –
jTRAQx – CiR-C strategy as a simple yet powerful answer to the need for an all-inclusive suite
for iTRAQ data processing.
Page 25 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
26
ACKNOWLEDGEMENTS
CONFLICT OF INTEREST STATEMENT
The authors have declared no conflict of interest
Page 26 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
27
REFERENCES
[1] Li, Z., Adams, R.M., Chourey, K., Hurst, G.B., et al., Systematic Comparison of Label-Free, Metabolic Labeling, and Isobaric Chemical Labeling for Quantitative Proteomics on LTQ Orbitrap Velos. J. Proteome Res. 2012, 11, 1582–1590.
[2] Bantscheff, M., Schirle, M., Sweetman, G., Rick, J., Kuster, B., Quantitative mass spec-trometry in proteomics: a critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031.
[3] Gygi, S.P., Rist, B., Gerber, S.A., Turecek, F., et al., Quantitative analysis of complex pro-tein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999.
[4] Ong, S.-E., Blagoev, B., Kratchmarova, I., Kristensen, D.B., et al., Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression pro-teomics. Mol. Cell. Proteomics 2002, 1, 376–386.
[5] Chen, X., Wei, S., Ji, Y., Guo, X., Yang, F., Quantitative proteomics using SILAC: Princi-ples, applications, and developments. PROTEOMICS 2015, 15, 3175–3192.
[6] Ross, P.L., Huang, Y.N., Marchese, J.N., Williamson, B., et al., Multiplexed Protein Quanti-tation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169.
[7] Patel, V.J., Thalassinos, K., Slade, S.E., Connolly, J.B., et al., A Comparison of Labeling and Label-Free Mass Spectrometry-Based Proteomics Approaches. J. Proteome Res. 2009, 8, 3752–3759.
[8] Wang, H., Alvarez, S., Hicks, L.M., Comprehensive Comparison of iTRAQ and Label-free LC-Based Quantitative Proteomics Approaches Using Two Chlamydomonas reinhardtii Strains of Interest for Biofuels Engineering. J. Proteome Res. 2012, 11, 487–501.
[9] Wu, W.W., Wang, G., Baek, S.J., Shen, R.-F., Comparative Study of Three Proteomic Quantitative Methods, DIGE, cICAT, and iTRAQ, Using 2D Gel- or LC−MALDI TOF/TOF. J. Proteome Res. 2006, 5, 651–658.
[10] Pütz, S.M., Boehm, A.M., Stiewe, T., Sickmann, A., iTRAQ Analysis of a Cell Culture Model for Malignant Transformation, Including Comparison with 2D-PAGE and SILAC. J. Proteome Res. 2012, 11, 2140–2153.
[11] Evans, C., Noirel, J., Ow, S.Y., Salim, M., et al., An insight into iTRAQ: where do we stand now? Anal. Bioanal. Chem. 2012, 404, 1011–1027.
[12] Lin, W.-T., Hung, W.-N., Yian, Y.-H., Wu, K.-P., et al., Multi-Q: A Fully Automated Tool for Multiplexed Protein Quantitation. J. Proteome Res. 2006, 5, 2328–2338.
[13] Boehm, A.M., Pütz, S., Altenhöfer, D., Sickmann, A., Falk, M., Precise protein quantifica-tion based on peptide quantification using iTRAQTM. BMC Bioinformatics 2007, 8, 214.
[14] Carrillo, B., Yanofsky, C., Laboissiere, S., Nadon, R., Kearney, R.E., Methods for combin-ing peptide intensities to estimate relative protein abundance. Bioinformatics 2010, 26, 98–103.
[15] Aramburu, J., Yaffe, M.B., López-Rodrı́guez, C., Cantley, L.C., et al., Affinity-Driven Pep-tide Selection of an NFAT Inhibitor More Selective Than Cyclosporin A. Science 1999, 285, 2129.
[16] Muth, T., Keller, D., Puetz, S.M., Martens, L., et al., jTraqX: A free, platform independent tool for isobaric tag quantitation at the protein level. PROTEOMICS 2010, 10, 1223–1225.
[17] Altman, D.G., Bland, J.M., Measurement in medicine: the analysis of method comparison studies. The statistician 1983, 307–317.
Page 27 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
28
[18] Bland, J.M., Altman, D., Statistical methods for assessing agreement between two methods of clinical measurement. The lancet 1986, 327, 307–310.
[19] Deutsch, E.W., Csordas, A., Sun, Z., Jarnuczak, A., et al., The ProteomeXchange consorti-um in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45, D1100–D1106.
[20] Vizcaíno, J.A., Csordas, A., del-Toro, N., Dianes, J.A., et al., 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456.
[21] Karp, N.A., Huber, W., Sadowski, P.G., Charles, P.D., et al., Addressing accuracy and pre-cision issues in iTRAQ quantitation. Mol. Cell. Proteomics 2010, 9, 1885–1897.
[22] Mahoney, D.W., Therneau, T.M., Heppelmann, C.J., Higgins, L., et al., Relative Quantifica-tion: Characterization of Bias, Variability and Fold Changes in Mass Spectrometry Data from iTRAQ-Labeled Peptides. J. Proteome Res. 2011, 10, 4325–4333.
[23] Hultin-Rosenberg, L., Forshed, J., Branca, R.M.M., Lehtio, J., Johansson, H.J., Defining, Comparing, and Improving iTRAQ Quantification in Mass Spectrometry Proteomics Data. Mol. Cell. Proteomics 2013, 12, 2021–2031.
[24] Pascovici, D., Song, X., Solomon, P.S., Winterberg, B., et al., Combining Protein Ratio p -Values as a Pragmatic Approach to the Analysis of Multirun iTRAQ Experiments. J. Prote-ome Res. 2015, 14, 738–746.
[25] Bantscheff, M., Boesche, M., Eberhard, D., Matthieson, T., et al., Robust and Sensitive iTRAQ Quantification on an LTQ Orbitrap Mass Spectrometer. Mol. Cell. Proteomics 2008, 7, 1702–1713.
[26] Köcher, T., Pichler, P., Schutzbier, M., Stingl, C., et al., High Precision Quantitative Prote-omics Using iTRAQ on an LTQ Orbitrap: A New Mass Spectrometric Method Combining the Benefits of All. J. Proteome Res. 2009, 8, 4743–4752.
[27] Jones, K.A., Kim, P.D., Patel, B.B., Kelsen, S.G., et al., Immunodepletion Plasma Prote-omics by TripleTOF 5600 and Orbitrap Elite/LTQ-Orbitrap Velos/Q Exactive Mass Spec-trometers. J. Proteome Res. 2013, 12, 4351–4365.
[28] Chang, H., Jiang, S.-F., Dang, K., Wang, H.-P., et al., iTRAQ-based proteomic analysis of myofibrillar contents and relevant synthesis and proteolytic proteins in soleus muscle of hi-bernating Daurian ground squirrels ( Spermophilus dauricus ). Proteome Sci. 2016, 14, 16.
[29] Hou, Q., Tan, H.T., Lim, K.H., Lim, T.K., et al., Identification and Functional Validation of Caldesmon as a Potential Gastric Cancer Metastasis-associated Protein. J. Proteome Res. 2013, 12, 980–990.
[30] Qin, L., Liu, X., Liu, S., Liu, Y., et al., Differentially expressed proteins underlying child-hood cortical dysplasia with epilepsy identified by iTRAQ proteomic profiling. PLOS ONE 2017, 12, e0172214.
[31] Kaur, P., Rizk, N.M., Ibrahim, S., Younes, N., et al., iTRAQ-Based Quantitative Protein Expression Profiling and MRM Verification of Markers in Type 2 Diabetes. J. Proteome Res. 2012, 11, 5527–5539.
[32] Légaré, C., Droit, A., Fournier, F., Bourassa, S., et al., Investigation of Male Infertility Us-ing Quantitative Comparative Proteomics. J. Proteome Res. 2014, 13, 5403–5414.
[33] Narumi, R., Murakami, T., Kuga, T., Adachi, J., et al., A Strategy for Large-Scale Phospho-proteomics and SRM-Based Validation of Human Breast Cancer Tissue Samples. J. Prote-ome Res. 2012, 11, 5311–5322.
[34] DeSouza, L.V., Romaschin, A.D., Colgan, T.J., Siu, K.W.M., Absolute Quantification of Potential Cancer Markers in Clinical Tissue Homogenates Using Multiple Reaction Moni-
Page 28 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
29
toring on a Hybrid Triple Quadrupole/Linear Ion Trap Tandem Mass Spectrometer. Anal. Chem. 2009, 81, 3462–3470.
[35] Ow, S.Y., Salim, M., Noirel, J., Evans, C., et al., iTRAQ Underestimation in Simple and Complex Mixtures: “The Good, the Bad and the Ugly.” J. Proteome Res. 2009, 8, 5347–5355.
[36] Charette, S.J., Lambert, H., Nadeau, P.J., Landry, J., Protein quantification by chemilumi-nescent Western blotting: Elimination of the antibody factor by dilution series and calibra-tion curve. J. Immunol. Methods 2010, 353, 148–150.
[37] Heidebrecht, F., Heidebrecht, A., Schulz, I., Behrens, S.-E., Bader, A., Improved semiquan-titative Western blot technique with increased quantification range. J. Immunol. Methods 2009, 345, 40–48.
[38] Barnidge, D.R., Dratz, E.A., Martin, T., Bonilla, L.E., et al., Absolute Quantification of the G Protein-Coupled Receptor Rhodopsin by LC/MS/MS Using Proteolysis Product Peptides and Synthetic Peptide Standards. Anal. Chem. 2003, 75, 445–451.
[39] Li, N., Nemirovskiy, O.V., Zhang, Y., Yuan, H., et al., Absolute quantification of multidrug resistance-associated protein 2 (MRP2/ABCC2) using liquid chromatography tandem mass spectrometry. Anal. Biochem. 2008, 380, 211–222.
[40] Atrih, A., Turnock, D., Sellar, G., Thompson, A., et al., Stoichiometric Quantification of Akt Phosphorylation Using LC-MS/MS. J. Proteome Res. 2010, 9, 743–751.
[41] Yang, T., Xu, F., Xu, J., Fang, D., et al., Comparison of liquid chromatography–tandem mass spectrometry-based targeted proteomics and conventional analytical methods for the determination of P-glycoprotein in human breast cancer cells. J. Chromatogr. B 2013, 936, 18–24.
[42] Zhang, W., Zhong, T., Chen, Y., LC-MS/MS-based targeted proteomics quantitatively de-tects the interaction between p53 and MDM2 in breast cancer. J. Proteomics 2017, 152, 172–180.
[43] Gan, C.S., Chong, P.K., Pham, T.K., Wright, P.C., Technical, Experimental, and Biological Variations in Isobaric Tags for Relative and Absolute Quantitation (iTRAQ). J. Proteome Res. 2007, 6, 821–827.
Page 29 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
30
Scheme 1. Introduction to iTRAQ technology. (A) Protocol walk-through. (B) Theoretical com-
parison between strategies of MS/MS peak integration, RPA and RSPI.
Figure 1. (A) Representative Western Blot of β-Actin, Cyclophilin A, Cofilin-1, Na+ / K+
ATPase subunit α1, and Galectin-1 after 24h exposure to : a. Vehicle, b. CsA 5 µM, c. Tac 0.05
µM, d. VIVIT 1 µM. (B) Scatter plots of Western blot ratios and relative protein abundance of β-
Actin, Cyclophilin A, Cofilin-1, Na+ / K+ ATPase subunit α1, normalized to Galectin-1, ex-
pressed as mean ± S.E.M.
Figure 2. Linear regression plus Pearson correlation coefficient test (A) and Bland-Altman com-
parison plot (B) to assess correlation and agreement between iTRAQ based on RPA and Western
blot protein quantitation along five independent experiments (biological replicates).
Figure 3. Linear regression plus Pearson correlation coefficient test (A) and Bland-Altman com-
parison plot (B) to assess correlation and agreement between iTRAQ based on RSPI and Western
blot protein quantitation along five independent experiments (biological replicates).
Figure 4. Linear regression plus Pearson correlation coefficient test (A,C) and Bland-Altman
comparison (B,D) to assess the correlation and agreement between RSPI and Western blot ratios
in five independent experiments (biological replicates) after adjustment to more (A,B) or less
(C,D) permissive Mascot score.
Figure 5. Linear regression plus Pearson correlation coefficient test (A,C) and Bland-Altman
comparison plot (B,D) to assess correlation and agreement between RSPI and Western blot ratios
along 4 (A,B) or 3 (C,D) independent experiments (biological replicates).
Scheme 2. RSPI fit better than RPA to report tag signature ion counts.
Page 30 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
31
Table 1. Summary of experimental design. Proteins from five independent replicates of four dis-
tinct experimental conditions were extracted from LLC PK-1 cells. Peptides were labeled with
iTRAQ 4-plex reagents and analyzed by nano-HPLC MS/MS. 4-plex distribution rotated between
independent experiments (biological replicates) to circumvent tag-related bias.
Page 31 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
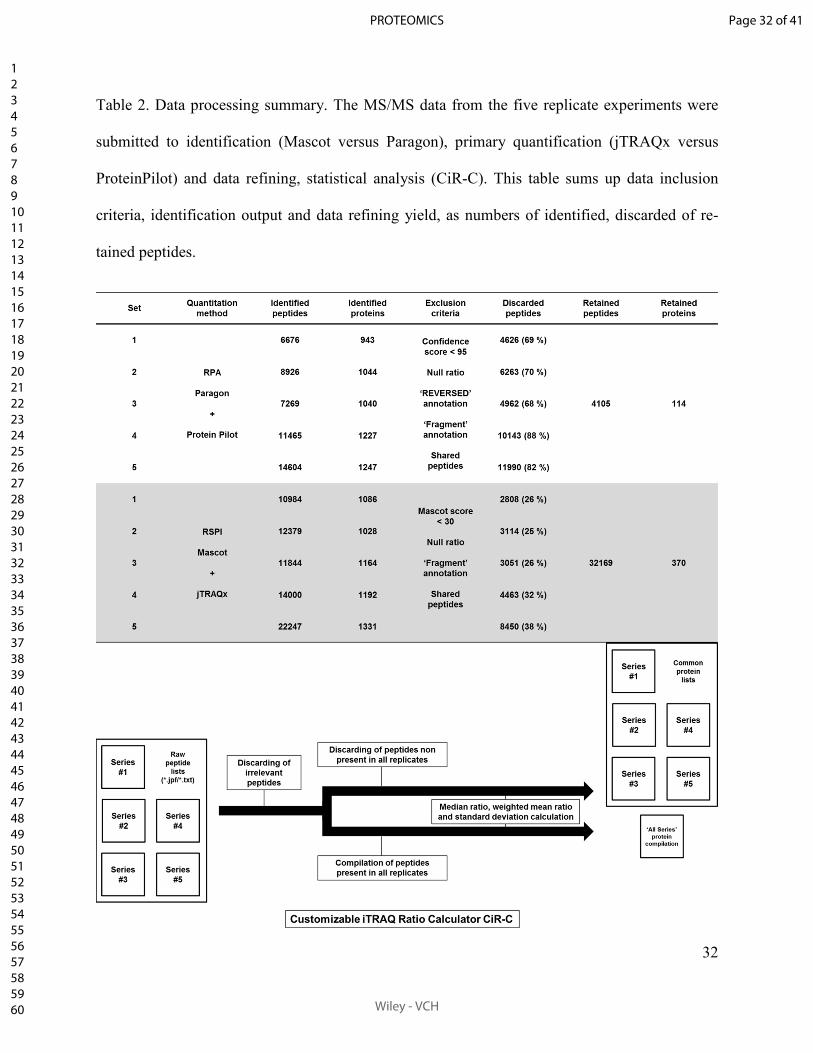
For Peer Review
32
Table 2. Data processing summary. The MS/MS data from the five replicate experiments were
submitted to identification (Mascot versus Paragon), primary quantification (jTRAQx versus
ProteinPilot) and data refining, statistical analysis (CiR-C). This table sums up data inclusion
criteria, identification output and data refining yield, as numbers of identified, discarded of re-
tained peptides.
Page 32 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
Scheme 1. Introduction to iTRAQ technology. (A) Protocol walk-through. (B) Theoretical com-parison between strategies of MS/MS peak integration, RPA and RSPI.
182x177mm (300 x 300 DPI)
Page 33 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
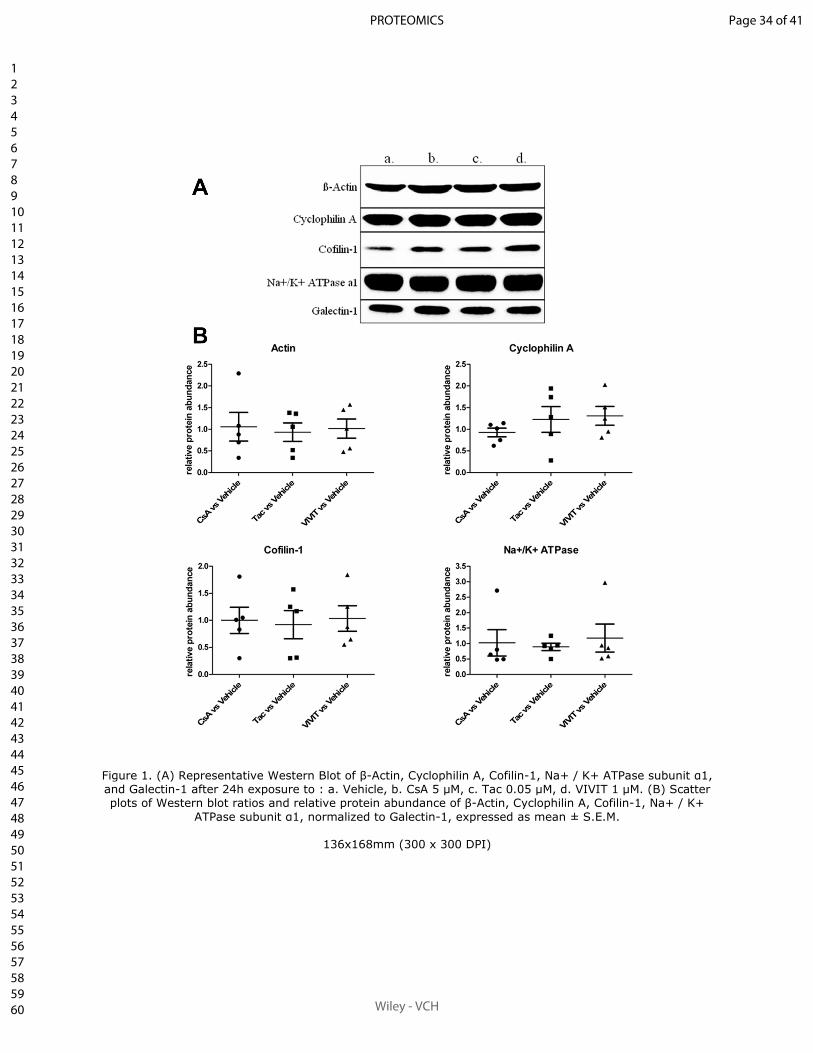
For Peer Review
Figure 1. (A) Representative Western Blot of β-Actin, Cyclophilin A, Cofilin-1, Na+ / K+ ATPase subunit α1, and Galectin-1 after 24h exposure to : a. Vehicle, b. CsA 5 µM, c. Tac 0.05 µM, d. VIVIT 1 µM. (B) Scatter plots of Western blot ratios and relative protein abundance of β-Actin, Cyclophilin A, Cofilin-1, Na+ / K+
ATPase subunit α1, normalized to Galectin-1, expressed as mean ± S.E.M.
136x168mm (300 x 300 DPI)
Page 34 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
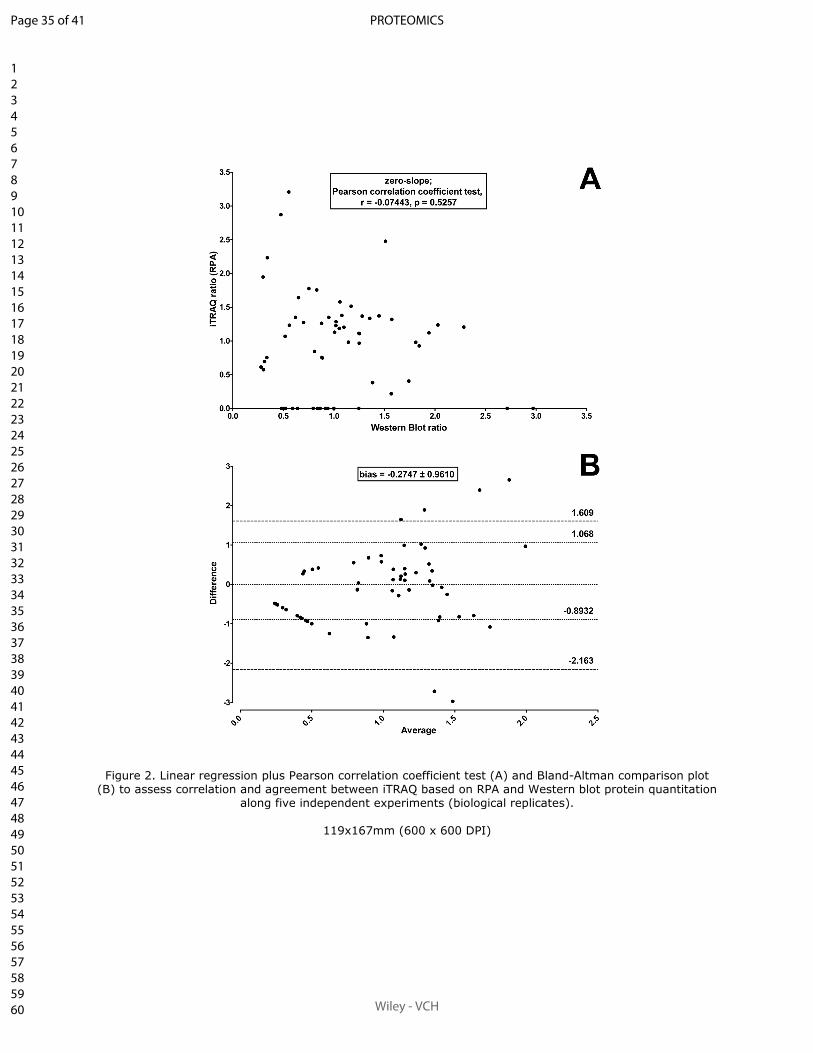
For Peer Review
Figure 2. Linear regression plus Pearson correlation coefficient test (A) and Bland-Altman comparison plot (B) to assess correlation and agreement between iTRAQ based on RPA and Western blot protein quantitation
along five independent experiments (biological replicates).
119x167mm (600 x 600 DPI)
Page 35 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
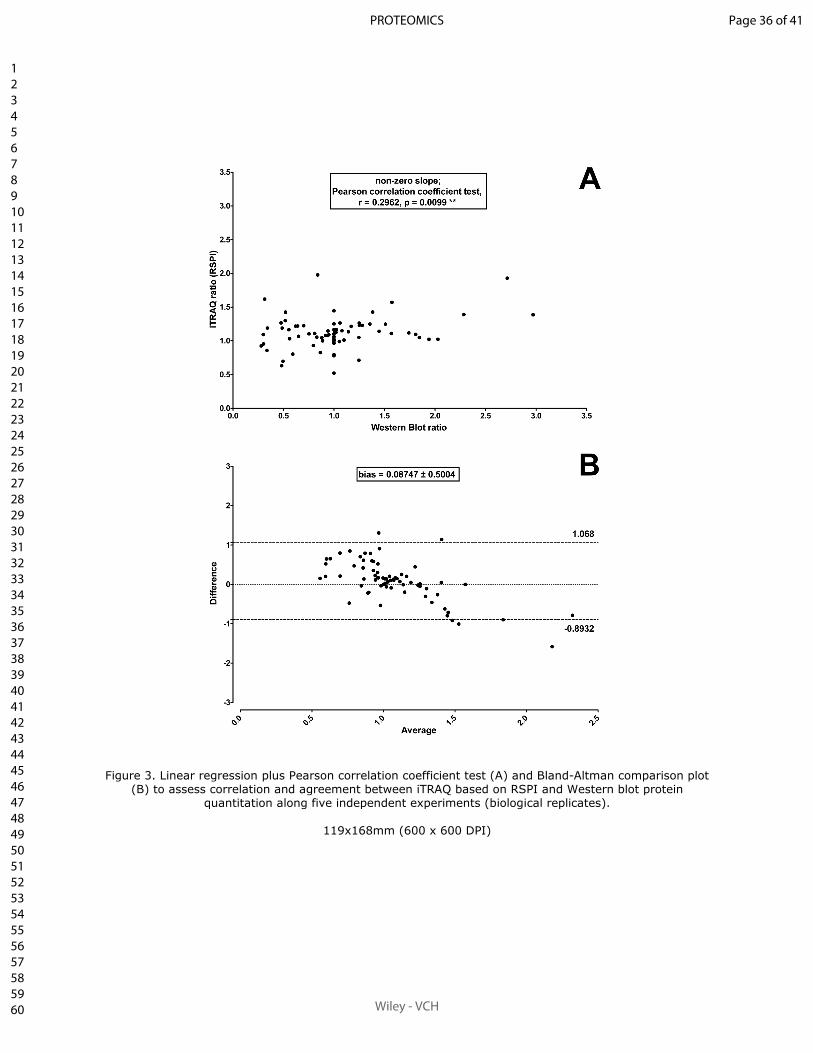
For Peer Review
Figure 3. Linear regression plus Pearson correlation coefficient test (A) and Bland-Altman comparison plot (B) to assess correlation and agreement between iTRAQ based on RSPI and Western blot protein
quantitation along five independent experiments (biological replicates).
119x168mm (600 x 600 DPI)
Page 36 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
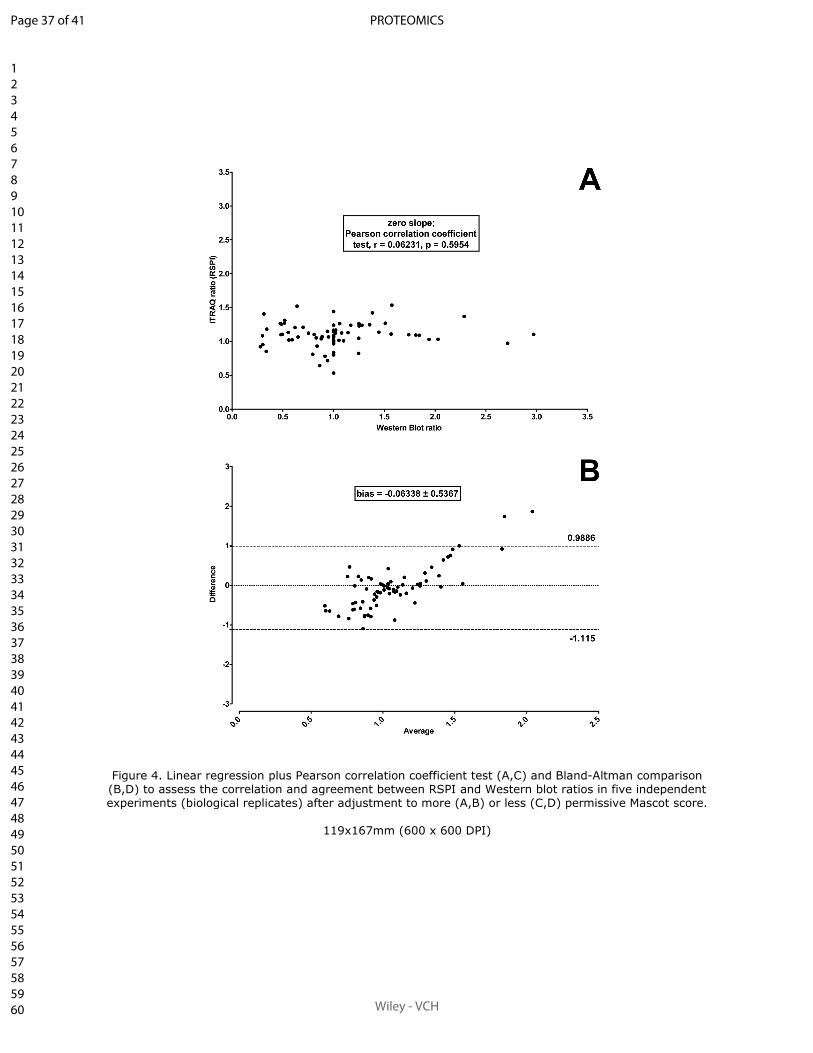
For Peer Review
Figure 4. Linear regression plus Pearson correlation coefficient test (A,C) and Bland-Altman comparison (B,D) to assess the correlation and agreement between RSPI and Western blot ratios in five independent experiments (biological replicates) after adjustment to more (A,B) or less (C,D) permissive Mascot score.
119x167mm (600 x 600 DPI)
Page 37 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
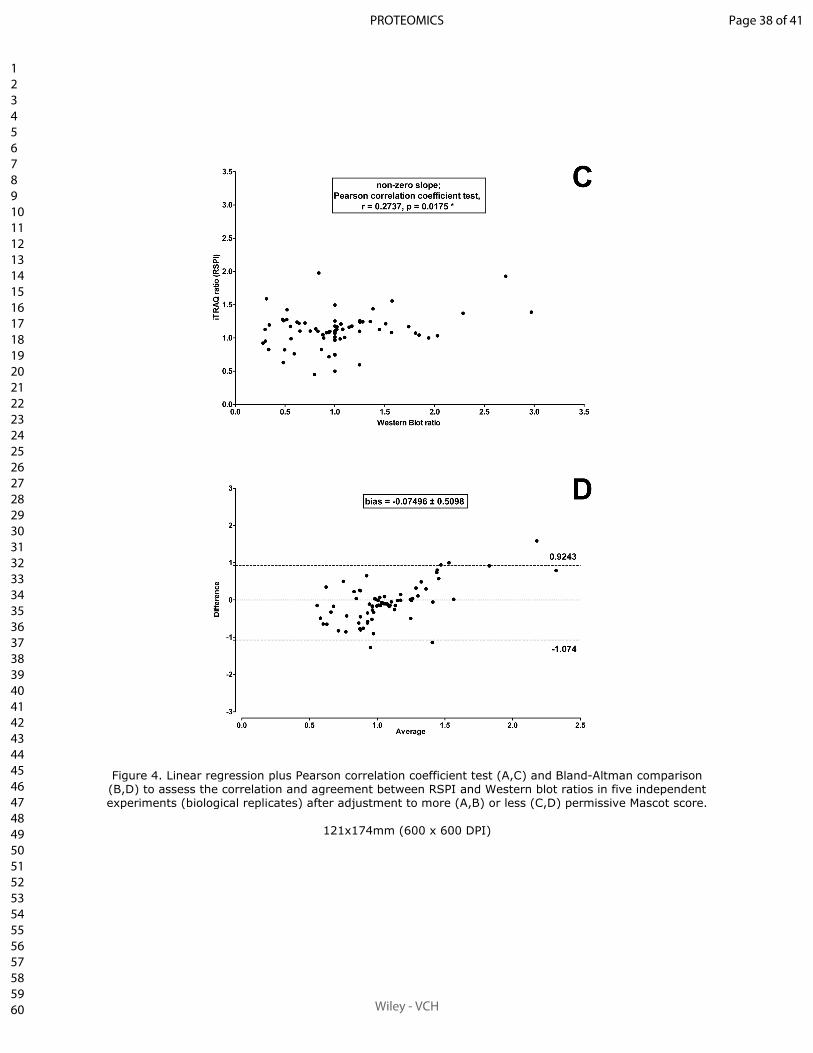
For Peer Review
Figure 4. Linear regression plus Pearson correlation coefficient test (A,C) and Bland-Altman comparison (B,D) to assess the correlation and agreement between RSPI and Western blot ratios in five independent experiments (biological replicates) after adjustment to more (A,B) or less (C,D) permissive Mascot score.
121x174mm (600 x 600 DPI)
Page 38 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
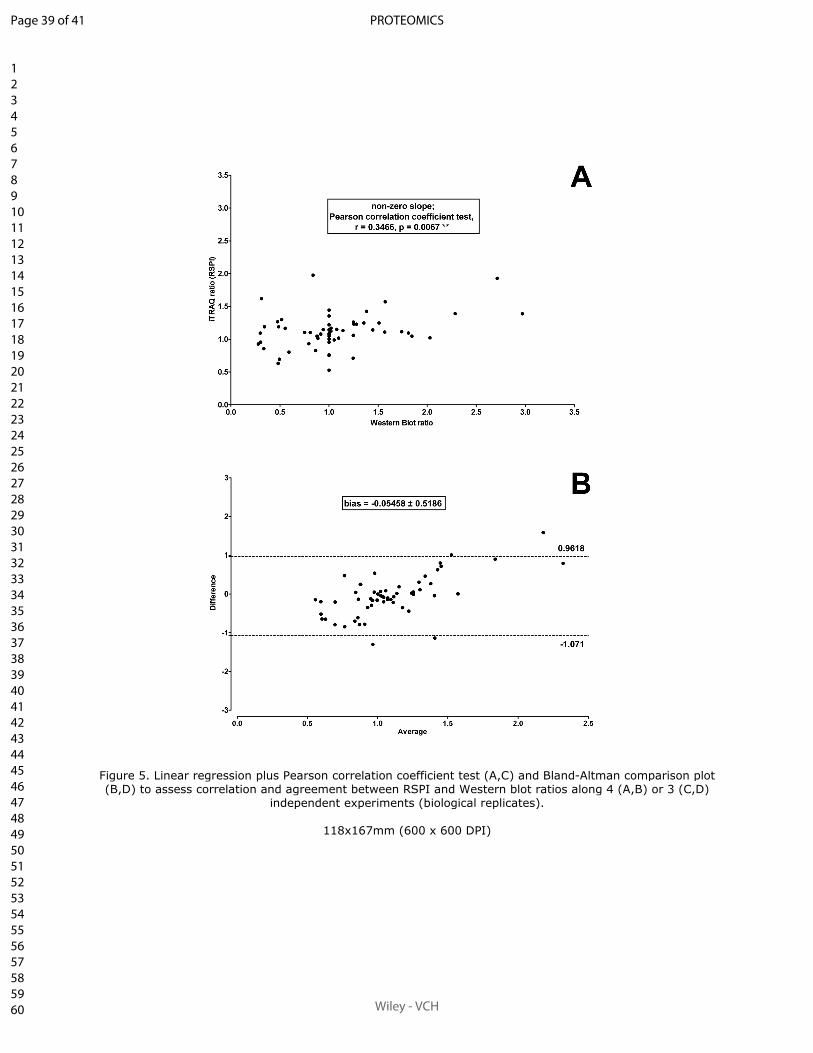
For Peer Review
Figure 5. Linear regression plus Pearson correlation coefficient test (A,C) and Bland-Altman comparison plot (B,D) to assess correlation and agreement between RSPI and Western blot ratios along 4 (A,B) or 3 (C,D)
independent experiments (biological replicates).
118x167mm (600 x 600 DPI)
Page 39 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
Figure 5. Linear regression plus Pearson correlation coefficient test (A,C) and Bland-Altman comparison plot (B,D) to assess correlation and agreement between RSPI and Western blot ratios along 4 (A,B) or 3 (C,D)
independent experiments (biological replicates).
118x166mm (600 x 600 DPI)
Page 40 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960

For Peer Review
Scheme 2. RSPI fit better than RPA to report tag signature ion counts.
177x60mm (300 x 300 DPI)
Page 41 of 41
Wiley - VCH
PROTEOMICS
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354555657585960
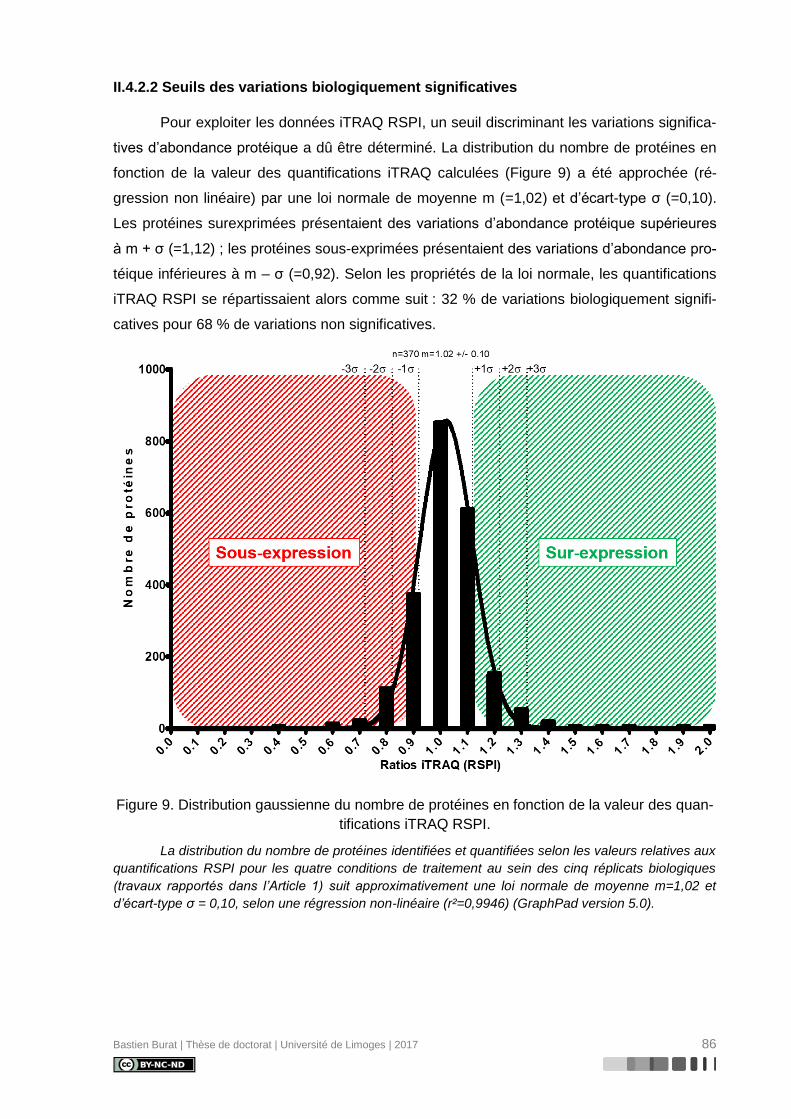
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 86
II.4.2.2 Seuils des variations biologiquement significatives
Pour exploiter les données iTRAQ RSPI, un seuil discriminant les variations significa-
tives d’abondance protéique a dû être déterminé. La distribution du nombre de protéines en
fonction de la valeur des quantifications iTRAQ calculées (Figure 9) a été approchée (ré-
gression non linéaire) par une loi normale de moyenne m (=1,02) et d’écart-type σ (=0,10).
Les protéines surexprimées présentaient des variations d’abondance protéique supérieures
à m + σ (=1,12) ; les protéines sous-exprimées présentaient des variations d’abondance pro-
téique inférieures à m – σ (=0,92). Selon les propriétés de la loi normale, les quantifications
iTRAQ RSPI se répartissaient alors comme suit : 32 % de variations biologiquement signifi-
catives pour 68 % de variations non significatives.
Figure 9. Distribution gaussienne du nombre de protéines en fonction de la valeur des quan-
tifications iTRAQ RSPI.
La distribution du nombre de protéines identifiées et quantifiées selon les valeurs relatives aux
quantifications RSPI pour les quatre conditions de traitement au sein des cinq réplicats biologiques
(travaux rapportés dans l’Article 1) suit approximativement une loi normale de moyenne m=1,02 et
d’écart-type σ = 0,10, selon une régression non-linéaire (r²=0,9946) (GraphPad version 5.0).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 87
II.4.2.3 Cartographie dynamique d’un sous-protéome d’intérêt : les protéines asso-
ciées à l’Actine
Parmi les 130 protéines de la compilation finale identifiées au sein de Swiss-Prot (an-
notation et vérification manuelles), 70 présentaient une variation significative d’abondance
relative. L’outil en ligne du système de classification PANTHER (version 12.0, Sus scrofa,
PANTHER Protein Class annotation data set) montrait une sur-représentation des protéines
cytosquelettiques de la famille de l’Actine (Actin family cytoskeletal proteins PC00041) lors
du test de sur-représentation PANTHER (PANTHER Overrepresentation test, release date
20170413). La visualisation de réseau d’interactions protéiques telle que permise par l’outil
en ligne STRING (version 13.5) associait ses protéines au sein d’un super-nœud de pro-
téines impliquées dans la dynamique du cytosquelette d’Actine (Figure 10 et 18).
Figure 10. Réseau STRING d’intéractions des protéines associées à l’Actine, identifiées et
quantifiées au cours des travaux rapportés dans l’Article 1.
Actin family cytoskeletal proteins PC00041, Sus scrofa, STRING version 13.5

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 88
Le réseau de protéines associées à la dynamique du cytosquelette d’Actine présen-
tait un profil d’expression propre à chaque ICN (Figure 11). D’un point de vue phénotypique,
cela se traduisait par une réorganisation CsA-spécifique du cytosquelette d’Actine de LLC
PK-1 (Figure 3).
Figure 11. Représentation en heat-map des variations d’abondance relative des protéines
associées à l’Actine, identifiées et quantifiées au cours des travaux rapportés dans l’Article 1.
Actin family cytoskeletal proteins PC00041, Sus scrofa, Euclidian distance hierarchical clustering
GENE-E version 3.0.204.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 89
Chapitre III. Organisation du cytosquelette d’Actine des cellules épithéliales
tubulaires proximales
À partir des données d’identification Mascot, la visualisation de réseaux STRING
d’interactions protéiques montrait une sur-représentation de protéines impliquées dans la
dynamique du cytosquelette d’Actine.
Le réseau de protéines associées à la dynamique du cytosquelette d’Actine présen-
tait un profil d’expression ICN-spécifique expliquant le phénotype de réorganisation CsA-
spécifique du cytosquelette d’Actine des LLC PK-1.
À la lumière de ces résultats préliminaires, ce travail de thèse a été poursuivi par
l’étude, plus spécifique, des effets des ICN sur la dynamique du cytosquelette d’Actine de
cellules tubulaires proximales.
Après une présentation des données de la littérature sur l’organisation et la régulation
du cytosquelette d’actine, ce chapitre décrira les résultats de l’étude de l’effet de la CsA sur
l’axe Actine-MRTF-SRF des cellules tubulaires proximales rénales.
III.1. Organisation générale du cytosquelette d’Actine (130)
La dénomination Actine est une terminologie générique désignant une famille de pro-
téines hautement conservées participant notamment à la cohésion mécanique et aux mou-
vements à l’échelle nucléaire, cytoplasmique, cellulaire et intercellulaire. En conditions phy-
siologiques, les protéines de cette famille sont remarquables par leur capacité à homopoly-
mériser spontanément en des structures dynamiques et polaires appelées microfilaments
d’Actine F (filamentaire) par l’assemblage ATP-dépendant de sous unités monomériques
libres de 42 kDa appelées Actine G (globulaire). Les microfilaments d’Actine F sont arrangés
en hélices de 5 à 9 nm de diamètre pour une longueur de chevauchement variant entre 385
et 358 Å et une torsion variable de 2,154 à 2,167 monomères/tour, composées d’Actine G
associées à l’ATP (ATP-Actine G) et soumis à un renouvellement permanent selon une lo-
gique de tapis roulant (Actin treadmilling). Ce renouvellement induit la polarisation du microfi-
lament entre une extrémité + dite barbelée (barbed end), à croissance rapide par incorpora-
tion d’ATP-Actine G, et une extrémité - dite pointue (pointed end), à croissance lente, par
hydrolyse de l’ATP (ADP-Pi Actine F) et dissociation d’ADP-Actine G, moins affine pour la
liaison au microfilament. Le renouvellement est entretenu par la régénération ATP-
dépendante d’ATP-Actine G (131).
Au sein de la cellule, l’organisation du cytosquelette d’Actine (Figure 12) incombe à
une superfamille de plus de 250 protéines (isoformes incluses), dont 162 distinctes (chez
l’Homme), liant l’Actine (Actin-binding proteins ou ABP) (132,133) chargées de maintenir un
réservoir d’Actine G prêt à la polymérisation, de surveiller et réguler le degré de polymérisa-
tion de l’Actine en bloquant les extrémités des microfilaments, en découpant les microfila-

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 90
ments existants ou, à l’inverse, en initiant la synthèse de nouveaux microfilaments ou la for-
mation et l’élongation de nouveaux embranchements. Ainsi, les ABP ont également pour rôle
de structurer le cytosquelette en faisceaux et réseaux tridimensionnels d’Actine F et de régu-
ler la compétition pour leur approvisionnement en Actine G, selon les besoins cellulaires.
La dépolymérisation est le mécanisme le plus important dans la dynamique du cy-
tosquelette d’Actine puisque c’est ainsi que le réservoir d’Actine G polymérisable peut être
régénéré le plus rapidement (principe des vases communicants), la quantité totale d’Actine
dans la cellule étant constante (134). La dépolymérisation est à la fois initiatrice et motrice de
la dynamique du cytosquelette d’Actine.
Les ABPs de la superfamille de la Gelsoline et de la famille de la Cofiline sont res-
ponsables de la dépolymérisation des microfilaments (132,135) :
• en sectionnant les microfilaments
• en induisant des contraintes mécaniques (torsions) au microfilament
• en favorisant la dissociation d’ADP-Actine G à l’extrémité - pointue
• en empêchant la réincorporation immédiate des monomères d’Actine G libé-
rées :
▪ par le blocage direct des extrémités + barbelées via les pro-
téines de coiffe
▪ par la séquestration des monomères d’Actine G
▪ par l’inhibition de l’échange de l’ADP-ATP lié à l’Actine.
La gestion du réservoir de monomères d’Actine G ainsi constitué est principalement
dévolu à l’ABP Profiline qui assure notamment la mobilisation des monomères en promou-
vant le remplacement du nucléotide lié à l’Actine (ADP > ATP] et en inhibant l’hydrolyse de
l’ATP. La Profiline est ensuite responsable de la répartition des monomères mobilisés entre
les différents réseaux et faisceaux de microfilaments grâce à un transport direct aux extrémi-
tés + barbelées associé à l’inhibition différentielle de l’incorporation des monomères aux ma-
crostructures existantes. En compétition avec la Profiline pour la liaison aux monomères
libres, les Thymosines, en particulier la Thymosine β4, jouent également un rôle de gestion
du réservoir, empêchant la mobilisation des monomères en vue de la polymérisation par sé-
questration de l’ATP-Actine G et inhibition de l’échange de nucléotide (ADP > ATP).
Les microfilaments polymérisés sont organisés en macrostructures adaptées à leurs
localisations cellulaires et à leurs missions ; chaque structure possède une architecture et
des propriétés mécaniques propres résultant d’une organisation spatiale particulière gérée
par des ABP spécifiques.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 91
Le complexe protéique Arp2/3, composé des protéines Arp2 et Arp3 apparentées en
séquence et en structure à l’Actine (Actin-related protein) et de sept sous-unités Arc, permet
la nucléation de nouveaux embranchements pour l’organisation de réseaux tridimensionnels
de microfilaments pouvant servir de points d’ancrage et d’appui, tels que les lamellipodes ou
les vésicules d’endocytose. Arp2/3 se lie à un premier microfilament d’Actine et sert
d’ancrage à l’extrémité – d’un second microfilament, selon une déviation de 70°. La nucléa-
tion par Arp2/3 est sous le contrôle des promoteurs de nucléation tel que WASP.
En compétition avec les protéines de coiffe pour la liaison aux extrémités +, les For-
mines provoquent la nucléation et l’élongation de faisceaux de microfilaments parallèles,
dépourvus de tout embranchement, tels que retrouvés au sein des filopodes, des adhé-
rences focales ou de l’anneau contractile lors de la cytodiérèse. Pour cela, des homodimères
enserrent l’extrémité + des microfilaments (domaines formin-homology FH2) et stabilisent les
dimères d’Actine G recrutés par la Profiline. La Profiline est recrutée via son affinité pour des
domaines riches en Proline (domaines FH1). Les tétramères ENA/VASP sont responsables
de la formation de structures similaires mais ne sont pas capables de nucléer des microfila-
ments.
Les protéines de coiffe ainsi que les Myosines et Tropomyosines stabilisent et protè-
gent les microfilaments et macrostructures existantes.
Figure 12. Organisation générale du cytosquelette d’Actine.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 92
III.2. La famille ADF/Cofiline (136,137)
La Cofiline-1 (CFL1/CFL) est le représentant ubiquitaire de la famille des facteurs de
dépolymérisation de l’Actine (actin depolymerizing factors, ADF). Exprimés chez tous les
Eucaryotes, les ADF/Cofiline regroupent également, chez les Mammifères, la Destrine
(DSTN, épithéliale, ou ADF) et la Cofiline-2 (CFL2, musculaire). La CFL est une protéine de
faible poids moléculaire (17 kDa) dont les fonctions biologiques résultent majoritairement,
mais pas exclusivement, de sa haute affinité pour l’Actine (138).
III.2.1. Fonctions biologiques de la Cofiline
III.2.1.1 Renouvellement du cytosquelette d’Actine
Les ADF/Cofiline au sens large sont connus depuis le début des années 80 pour leur
capacité à dépolymériser les microfilaments d’Actine. La CFL favorise le relargage de Pi des
ADP-Pi Actine G constituant les segments les plus anciens à l’extrémité – des microfila-
ments, permet la dissociation d’ADP-Actine, et ainsi, la régénération du réservoir et le renou-
vellement perpétuel du microfilament. De plus, la CFL fragilise les microfilaments en modi-
fiant leurs propriétés physico-chimiques et structurales, provoquant notamment une modifi-
cation de la torsion de l’hélice (2,218 unités/tour, + 5°, longueur de chevauchement : 269 Å
(139)). Au sein de la cellule, ces mécanismes sont corrélés à l’action de la Profiline et
s’inscrivent dans le treadmilling des microfilaments ainsi que dans la gestion des réseaux et
faisceaux ; la dépolymérisation à proprement parler n’intervient que si la vitesse de dissocia-
tion à l’extrémité - devient significativement supérieure à la vitesse d’incorporation à
l’extrémité + comme c’est le cas lorsque l’action de la CFL est coordonnée au blocage des
extrémités + par des protéines de coiffe.
III.2.1.2 Transport de l’Actine et transcription
Au sein de la cellule eucaryote, l’Actine totale est répartie entre le cytoplasme et le
noyau où monomères d’Actine-G et microfilaments d’Actine-F jouent un rôle prépondérant
dans la transcription génique (140). Cependant, la structure primaire de l’Actine ne comporte
ni séquence de localisation nucléaire (nuclear localization sequence, NLS), ni séquence
d’export nucléaire (nuclear export sequence, NES) permettant la prise en charge et le trans-
port nucléo-cytoplasmique par les protéines de la famille des Karyophérines, Importines et
Exportines. En revanche, la CFL possède un NLS bipartite (21-34) dépendant de l’Importine
α/β et un NES (11-20) dépendant de l’Exportine-1 (141). Ainsi, l’Actine « emprunte » le NLS
de la CFL afin d’être transférée dans le noyau par l’Importine-9 (142).
Mais la CFL ne se limite pas à l’import/export d’Actine puisque la CFL nucléaire est
un constituant nécessaire du complexe d’élongation de la transcription établi autour de l’ARN
Polymérase II. In vivo, un complexe CFL-Actine-ARN Pol II a été caractérisé au cours
d’expériences de cross-linking et de co-immunoprécipitation alors que des expériences de

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 93
gene silencing anti-CFL ont démontré l’importance de CFL pour la synthèse de nouveaux
ARNm. La CFL est principalement associée aux régions codantes du gène où le complexe
formé par l’Actine et l’ARN Pol II au cours de la phase d’élongation est strictement CFL-
dépendant (143).
III.2.1.3 Réponse au stress
Dans des conditions de stress cellulaire majeur tels qu’une pénurie énergétique et un
ralentissement du métabolisme par déplétion d’ATP, un choc thermique ou un choc osmo-
tique, la CFL est massivement mobilisée au noyau (à la faveur de son domaine NLS ) où son
accumulation extrême conduit à une saturation totale des microfilaments d’Actine nucléaires
résultant en la formation de structures appelées « bâtonnets » (« rods ») de Cofiline-Actine
(141).
Ce phénomène a d’abord été observé au sein de cultures de fibroblastes et de cul-
tures primaires de myocytes de Poulet exposées à un choc osmotique, un choc thermique
ou à une concentration élevée de DMSO (144–147).
La formation de bâtonnets de CFL-Actine peut être considérée comme une mesure
d’urgence destinée à économiser l’énergie dépensée, notamment, dans la dynamique du
cytosquelette d’Actine. Ces structures atypiques ont d’abord été observées dans les neurites
de neurones stressés où elles monopolisent le pool de CFL et permettent un ralentissement
à court terme de la consommation d’ATP (148). Quoique majoritairement décrit comme un
mécanisme nucléaire, l’observation de bâtonnets de CFL-Actine cytoplasmiques consécuti-
vement à un stress cellulaire de déplétion en ATP abonde dans le sens de l’hypothèse de la
sauvegarde énergétique (149,150). Au sein de cellules tubulaires proximales telles que LLC
PK-1, ce mécanisme peut être expliquée à la lumière de la relation CFL-Na+/K+ ATPase où
la CFL joue le rôle d’alimentateur énergétique en transférant la triose phosphate isomérase
(cf. III.2.2.1.)
L’étude de la formation de bâtonnets de CFL-Actine se concentre depuis de nom-
breuses années sur leur implication dans le développement de maladies neurodégénératives
(151).
III.2.1.4 Apoptose
L’analyse protéomique comparative de la mitochondrie et du cytosol de cellules ex-
posées à la Staurosporine (un inhibiteur aspécifique des kinases, inducteur rapide de
l’apoptose par la voie intrinsèque, dépendante des caspases) a démontré que la CFL est
une des protéines transloquées le plus précocement à la mitochondrie au cours de la phase
d’initiation de l’apoptose, en amont de la transition de perméabilité mitochondriale, de la libé-
ration de Cytochrome C dans le cytosol et de l’activation des Caspases effectrices. Des ex-
périences de gene silencing (siRNA anti-CFL) et de mutagénèse dirigée ont établi que la

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 94
translocation mitochondriale de la CFL sous sa forme déphosphorylée est essentielle à
l’initiation de la mort cellulaire par la voie intrinsèque. Le domaine de liaison à l’Actine est
alors nécessaire pour l’activité pro-apoptotique (152).
Des mécanismes similaires ont été observés au cours d’études in vitro ultérieures
permettant de préciser les conditions requises à la relocalisation pro-apoptotique de la CFL.
Ainsi, l’apoptose induite par le TGF-beta au sein de cellules de Carcinome de la Prostate
implique la translocation des CFL cytoplasmique et nucléaire vers la mitochondrie (153).
L’exposition de cellules humaines de Leucémie et de Cancer du Sein à des dérivés isothio-
cyanates déclenche une mort cellulaire passant par la dépolymérisation du cytosquelette
d’Actine et la translocation de CFL liée à l’Actine-G après activation de la voie de signalisa-
tion ROCK1/PTEN/PI3K (154,155). Dans le contexte de l’apoptose déclenchée par un stress
oxydant (156), la translocation de la CFL sous une forme déphosphorylée, non liée à l’Actine
et oxydée au niveau de ses résidus Cystéine est requise pour induire la transition de per-
méabilité mitochondriale (157,158).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 95
III.2.2. Mécanismes de régulation de la Cofiline (Figure 14) (159)
III.2.2.1 Phosphorylation / Déphosphorylation (Figure 13)
La stratégie principale de régulation de la CFL consiste à empêcher la liaison de
l’ABP à l’Actine. La structure primaire de la CFL comporte un résidu Sérine en position 3
dont la phosphorylation inhibe l’interaction CFL-Actine (160–162). La phosphorylation de
Ser3 est assurée par les kinases LIM, effectrices finales de la voie des petites GTPases
Rho, RhoA, Rac et Cdc42 et de leur kinases associées ROCK, PAK et MRCK (163–171).
La déphosphorylation de Ser3 est catalysée spécifiquement par les phosphatases
Slingshot et Chronophine ou de manière plus générique par les Sérine/Thréonine phospha-
tases PP1 et PP2A (172).
Toutefois, des travaux portant sur la nature de la relation entre état de phosphoryla-
tion et activité de la CFL ont remis en cause ce modèle binaire, soulignant le caractère mul-
tiple des mécanismes de régulation de la CFL. Après stimulation du chimiotactisme de cel-
lules métastatiques d’adénocarcinomes mammaires de Rat (lignée MTLn3) par l’Epidermal
Growth Factor (EGF), Song et coll. ont mesuré en parallèle l’activité de la CFL et son état de
phosphorylation tout en modulant la régulation par Slingshot et LIMK. Un découplage total
(temporel et spatial) entre activité et phosphorylation est observé puisque la CFL conserve
une activité de sectionnement et un rôle dans la réorganisation du cytosquelette de ces cel-
lules identique, que sa forme phosphorylée pCFL soit favorisée (inhibition de Slingshot par
Na3VO4) ou non (inhibition de ROCK par Y-27632 ou siRNA anti-LIMK) (173,174).
Déphosphorylation par inhibition de la pompe Sodium/Potassium ATPase
Dans leur recherche de nouveaux partenaires protéiques et nouveaux mécanismes
de régulation de la pompe Sodium-Potassium ATPase, Lee et coll. ont décrit l’interaction
entre la CFL et la Na+/K+ ATPase, soulignant également l’importance de la CFL dans
l’activité de la pompe. Des expériences de double hybride et de co-immunoprécipitation ont
démontré l’association directe entre la région N-terminale de la CFL et la région de la grande
boucle intracytoplasmique formée par la sous-unité alpha de la Na+/K+ ATPase (indépen-
damment de l’isoforme considérée) (175,176).
Par cette association, la CFL joue notamment un rôle d’alimentateur énergétique
grâce à l’interaction RhoGTPase-dépendante de sa forme phosphorylée (p-CFL) avec la
triose-phosphate isomérase (TPI). Le complexe pCFL-TPI est transloqué à la membrane
plasmique où son action enzymatique glycolytique permet l’approvisionnement de la Na+/K+
ATPase en ATP (177).
La CFL relie la voie dépendante du Calcium et la voie des petites GTPases Rho à la
transduction énergétique et aux échanges ioniques au sein de la cellule. Réciproquement, la
CFL peut relier un déséquilibre ionique ou une perturbation métabolique à la dynamique du
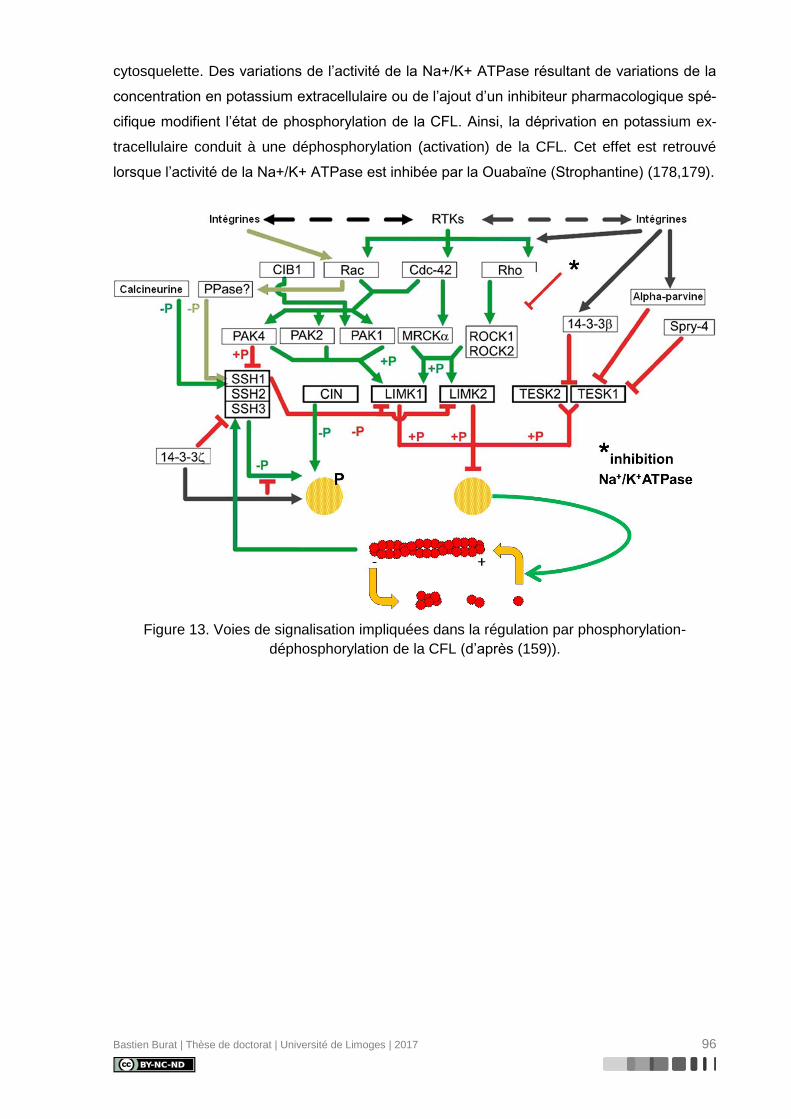
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 96
cytosquelette. Des variations de l’activité de la Na+/K+ ATPase résultant de variations de la
concentration en potassium extracellulaire ou de l’ajout d’un inhibiteur pharmacologique spé-
cifique modifient l’état de phosphorylation de la CFL. Ainsi, la déprivation en potassium ex-
tracellulaire conduit à une déphosphorylation (activation) de la CFL. Cet effet est retrouvé
lorsque l’activité de la Na+/K+ ATPase est inhibée par la Ouabaïne (Strophantine) (178,179).
Figure 13. Voies de signalisation impliquées dans la régulation par phosphorylation-
déphosphorylation de la CFL (d’après (159)).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 97
III.2.2.2 Liaison à PIP2 et sensibilité au pH cellulaire
La superposition de régions de la structure tertiaire de la CFL impliquées dans les in-
teractions Actine-CFL et PIP2-CFL confère à PIP2 un rôle dans la modulation de l’activité de
la CFL par une inhibition compétitive de la liaison à l’Actine et de l’activité de dépolymérisa-
tion du microfilament (180,181). À la membrane, au sein des réseaux périphériques d’Actine,
la CFL est sensible à la densité de PIP2 membranaires et forme des regroupements (« clus-
ters » ) de plusieurs PIP2 par des interactions électrostatiques entre les résidus positivement
chargés K95, K96, K112, K114, K125, K126, K127 et les têtes polaires phospholipidiques
(182). Ce mécanisme permet une régulation locale de la CFL, indépendante de la régulation
générale par LIMK mais dépendante du pH périphérique et notamment de l’efflux de H+ par
l’échangeur Na+-H+ NHE1 (173,183). Ainsi, PIP2 participe à la sensibilité intrinsèque de la
CFL au pH cellulaire. En effet, la CFL, seule, sectionne et dépolymérise les microfilaments
d’Actine de manière optimale à pH basique (pH 8) alors que son activité décline et favorise la
polymérisation et l’élongation du microfilament quand le pH devient plus acide (pH 6.5)
(184).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 98
III.2.2.3 Sensibilité au stress oxydatif
Comme décrit précédemment, l’oxydation des résidus Cystéine (Cys39, Cys80,
Cys139 et Cys147) de la CFL est nécessaire à sa translocation à la mitochondrie et à sa
fonction pro-apoptotique lors de l’apoptose consécutive à un stress oxydant (157,158). Dans
ces conditions, la CFL est incapable de dépolymériser l’Actine ce qui peut être expliqué par
deux hypothèses. La première hypothèse repose sur la capacité de la CFL oxydée à former
des homodimères par la formation de ponts disulfure intermoléculaires. Ces dimères, obte-
nus uniquement in vitro en présence de de glutathione oxydé, permettent la promotion des
faisceaux d’Actine-F (185). La seconde hypothèse repose sur la capacité de la CFL oxydée
à altérer sa structure tertiaire et former des ponts disulfures intramoléculaires entre les rési-
dus Cys39 et Cys80, et, Cys139 et Cys147. Cette forme particulière de CFL, observée au
sein de lymphocytes T ciblés par des granulocytes et de lymphomes traités par l’oxydant des
neutrophiles, la Taurine Chloramine, perd son affinité pour l’Actine (186).
Figure 14. Mécanismes de régulation de la CFL.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 99
III.2.2.4 Etat d’oligomérisation de la Cofiline
Au cours de travaux visant à caractériser in vitro, en solution, les interactions entre
Actine-F et CFL par cross-linking, Pfannstiel et coll. ont observé la formation d’homo-
oligomères de CFL à la faveur de ponts disulfure entre les Cystéines en position 39 (Cys39)
et 147 (Cys147) de la structure primaire. Ce phénomène est d’autant plus favorisé que la
concentration de CFL ou de PIP2 est importante et est sensible au pH (les oligomères sont
plus facilement observés à pH 8 qu’à pH 6,5) (185). Le phénomène d’oligomérisation spon-
tanée de la CFL a été confirmé par des expériences identiques menées in vivo (187).
Le degré d’oligomérisation de la CFL dépend de l’état général de phosphorylation du
réservoir intracellulaire de CFL : les oligomères de CFL (dimères diCFL et tétramères té-
traCFL) résultent de l’association entre protomères non phosphorylés. Plus la phosphoryla-
tion du réservoir de CFL est importante, plus la concentration de monoCFL non phosphory-
lée est faible et, par conséquent, plus la probabilité de rencontre de deux (ou quatre) mo-
noCFL non phosphorylées est faible.
L’activité de la CFL vis-à-vis des microfilaments d’Actine-F est fonction de son état
d’oligomérisation ; monoCFL est responsable du sectionnement et de la dépolymérisation
des microfilaments d’Actine-F alors que tetraCFL et diCFL sont incapables de sectionner
l’Actine-F et participent paradoxalement à la mobilisation de monomères et à la nucléation
de nouveaux microfilaments.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 100
III.2.2.5 Concentration totale et rapport Cofiline / Actine
L’état d’oligomérisation de la CFL est corrélé à sa concentration intracellulaire. Plus la
concentration intracellulaire de CFL est élevée, plus la probabilité de rencontre des proto-
mères conduisant à la formation d’un oligomère de haut poids moléculaire est grande. Inver-
sement, plus la concentration intracellulaire de CFL est faible, plus cette probabilité est
faible.
La concentration intracellulaire de CFL disponible est modulée artificiellement par
phosphorylation/déphosphorylation. La forme oligomérisable et active vis-à-vis de l’Actine est
la forme déphosphorylée.
La concentration intracellulaire de CFL et son rapport avec la concentration intracellu-
laire totale d’Actine ont été décrits comme des paramètres déterminants dans l’orientation de
la CFL vers une activité de sectionnement et de dépolymérisation, une activité d’entretien et
de renouvellement de microfilaments existants, ou une activité de nucléation de nouveaux
microfilaments (184,188).
In vitro, l’activité de sectionnement par la CFL est optimale pour une concentration de
10 nM (56 x 10-6 évènements.µm-1 Actine.s-1 pour CFL) et est réalisé de manière isolée par
des clusters de petite taille (une dizaine de CFL. Alors, la probabilité d’un évènement de sec-
tionnement augmente avec la longueur du microfilament, et diminue quand la densité de
CFL par rapport au microfilament augmente.
En parallèle, CFL accélère la dépolymérisation des microfilaments sectionnés à leur
extrémité pointue (5 fois le taux de dissociation de l’Actine seule) de manière concentration-
dépendante.
À hautes concentrations (1 µM) et à densités élevées (à partir 49 % de saturation des
microfilaments), la CFL n’entraîne plus de sectionnement de l’Actine-F et favorise la nucléa-
tion de microfilaments de novo de manière concentration-dépendante (selon les concentra-
tions d’Actine-G et de CFL).
Ce mode de régulation est bien adapté à l’organisation spatiale et temporelle d’une
macrostructure dynamique telle que le cytosquelette d’Actine. Physiologiquement, la CFL
n’est pas restreinte à la déstructuration des microfilaments d’Actine-F et à l’entretien du ré-
servoir d’Actine G mobilisables, grâce à cette régulation plus fine de son activité, reposant
localement sur sa concentration et son niveau d’occupation du microfilament. Ainsi, à la fa-
veur de gradients intracellulaires plus ou moins localisés, la CFL peut orienter la dynamique
et l’architecture du cytosquelette entre des régions de basses concentrations, où les microfi-
laments existants, faiblement associés à la CFL, sont soumis à des forces de torsion sup-
plémentaires, sectionnés et dépolymérisés, et des régions à concentrations intermédiaires

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 101
ou fortes, où les microfilaments existants sont saturés, stabilisés et où de nouveaux microfi-
laments peuvent être nucléés de novo.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 102
III.3. Régulation de l’expression génique par la dynamique du cytosquelette d’Actine
III.3.1. Implication de l’Actine nucléaire dans la transcription génique (140,189,190)
Le cytosquelette d’Actine structure l’ensemble de la cellule par un équilibre dyna-
mique entre des réservoirs cytoplasmiques et nucléaires. L’accessibilité moindre de l’Actine
nucléaire rend son étude plus difficile. Néanmoins, de nombreuses études amènent à penser
que, comme dans le cytoplasme, l’Actine ne se limite pas à un rôle passif de structure et que
l’Actine nucléaire s’organise selon des mécanismes similaires à son pendant cytoplasmique,
au rythme de polymérisations et dépolymérisations sous le contrôle d’ABP nucléaires.
L’Actine nucléaire est notamment impliquée dans les étapes successives du proces-
sus de transcription :
• au sein des complexes de pré-initiation et d’initiation de la transcription par les
ARN Polymérases. L’isoforme β d’Actine-G interagit avec les sous-unités
RPABC2 et RPABC3, communes à toutes les ARN Polymérases eucaryotes I,
II et III.
• au cours de la phase d’élongation. L’Actine-G recrute le facteur d’élongation
P-TEFb, responsable de la phosphorylation du domaine C-terminal de l’ARN
Polymérase Il, nécessaire à la sortie du complexe d’initiation. Via son interac-
tion avec les hnRNP (heterogenous nuclear ribonucleoproteins), l’Actine-G lie
les ARN en cours de transcription (pre-mRNP/mRNP) et recrute des histone
acétyltransférases (telle PCAF qui acétyle H3K9) qui assurent la transition
vers une configuration plus permissive de la chromatine.
• au sein des complexes de remodelage de la chromatine BAF et INO80.
L’isoforme β d’Actine-G est essentielle pour l’activité ATPase de ces com-
plexes ainsi que pour l’interaction avec la chromatine et les nucléosomes.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 103
III.3.2. Cas particulier de la régulation des facteurs de transcription MRTF et SRF
(191,192)
Le caractère hautement dynamique du cytosquelette d’Actine lui confère un rôle de
premier plan dans l’adaptation mécanique à son environnement. Actuellement, six familles
de protéines membranaires sont décrites comme étant capables d’initier la transduction d’un
signal intracellulaire transmis au cytosquelette d’Actine en réponse à un stimulus extracellu-
laire :
- les protéines de la famille des Intégrines. Impliquées dans l’ancrage cellulaire à
une matrice extracellulaire (MEC) au niveau des contacts focaux,
- les protéines de la famille des Cadhérines telles que les Cadhérines épithéliales
(E-Cadhérines). Impliquées dans les jonctions intercellulaires au sein des jonc-
tions d’adhérence,
- les récepteurs aux ligands de la superfamille des TGF-beta,
- les récepteurs à Tyrosine kinase,
- les récepteurs couplés aux petites protéines G,
- le récepteur Frizzled.
Hors interaction mécanique directe avec les microfilaments d’Actine, ces récepteurs
membranaires déclenchent des cascades de signalisation convergeant invariablement vers
la voie des RhoGEFs, Rho GTPases et kinases associées. La voie des RhoGTPases consti-
tue la voie de régulation majeure des Actin-binding proteins (ABP), responsables de
l’organisation et de la dynamique du cytosquelette d’Actine.
III.3.2.1 Mécanistique de l’axe RhoGTPases-Actine-SRF
Au-delà de l’adaptation mécanique, l’axe RhoGTPases-Actine est un levier important
de l’adaptation générale de la cellule à son environnement par sa régulation de la transcrip-
tion génique dépendante du facteur de transcription Serum Response Factor (SRF) associé
à son cofacteur Myocardin-Related Transcription Factor (MRTF) (164,193–196)
La stimulation par des mitogènes active les petites GTPases Rho, RhoA, d’une part,
et Rac1 et Cdc42, d’autre part, qui potentialisent la transduction du signal, en parallèle et de
manière indépendante (193). Une cascade de phosphorylation active consécutivement les
kinases associées à RhoA, Rho-associated protein kinases (ROCK), et les kinases en aval
de Rac1 et Cdc42, p21-activated kinases (PAK) et Myotonic dystrophy-related Cdc42-
associated kinases (MRCK) puis les LIM domain kinases (LIMK). Les LIMK activées phos-
phorylent la Cofiline conduisant à une stabilisation des microfilaments et une diminution de la
quantité d’Actine-G cytoplasmique (164,194,195). L’activité des LIMK sur la dynamique de
renouvellement du cytosquelette, la stabilisation de l’Actine-F et les niveaux d’Actine-G cyto-

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 104
plasmique est contrebalancée par les phosphatases Slingshot sous contrôle de la voie Cal-
cium-dépendante et de la Calcineurine (172). Les phosphatases Slingshot activées déphos-
phorylent la Cofiline qui est alors responsable de la dépolymérisation des microfilaments et
de l’accumulation cytoplasmique d’Actine G.
La translocation nucléaire du cofacteur MRTF résulte de la dissociation du complexe
cytoplasmique MRTF-Actine-G par déplétion d’Actine-G. L’Actine-G est capable de se fixer
aux motifs RPEL de MRTF conduisant à sa séquestration cytoplasmique (196).
Une fois le cofacteur MRTF transféré dans le noyau, la formation du complexe
MRTF-SRF nécessite l’homodimérisation de MRTF, via ses domaines LZ (Leucine zipper),
et l’interaction entre le domaine MADS de SRF et les régions B1 (basique) et Q (riche en
Glutamate).
Par cet axe, l’activité de transcription par SRF est modulable par des drogues du cy-
tosquelette. Ainsi, le Jasplakinolide, un stimulateur de la polymérisation de l’Actine, active
SRF tout comme le Swinholide A, responsable de la séquestration de dimères d’Actine-G.
En revanche, les Latrunculines A et B ou la Cytochalasine D, molécules inhibitrices de la
polymérisation et activatrices de la dépolymérisation, inhibent SRF.
III.3.2.2 Gènes cibles de MRTF-SRF
Le complexe MRTF-SRF active la transcription de gènes cibles (197,198) sous con-
trôle de promoteurs intégrant un élément de réponse au sérum (Serum Response Element,
SRE) composé d’un ou plusieurs exemplaires d’une séquence consensus
CC[A/T]2A[A/T]3GG appelé boîte CArG. La transcription du CArGome (Tableau 1) est dé-
pendante de la formation de complexes liant SRF avec des cofacteurs de transcription spéci-
fiques d’un tissu, d’un type cellulaire et/ou d’une voie de signalisation en amont. Les
membres de la famille des MRTF en aval de l’axe RhoGTPases-Actine et les membres de la
famille des protéines TCF (Ternary Complex Factors) à domaine Ets, en aval de la voie des
MAPK (199) interagissent avec SRF de manière mutuellement exclusive.
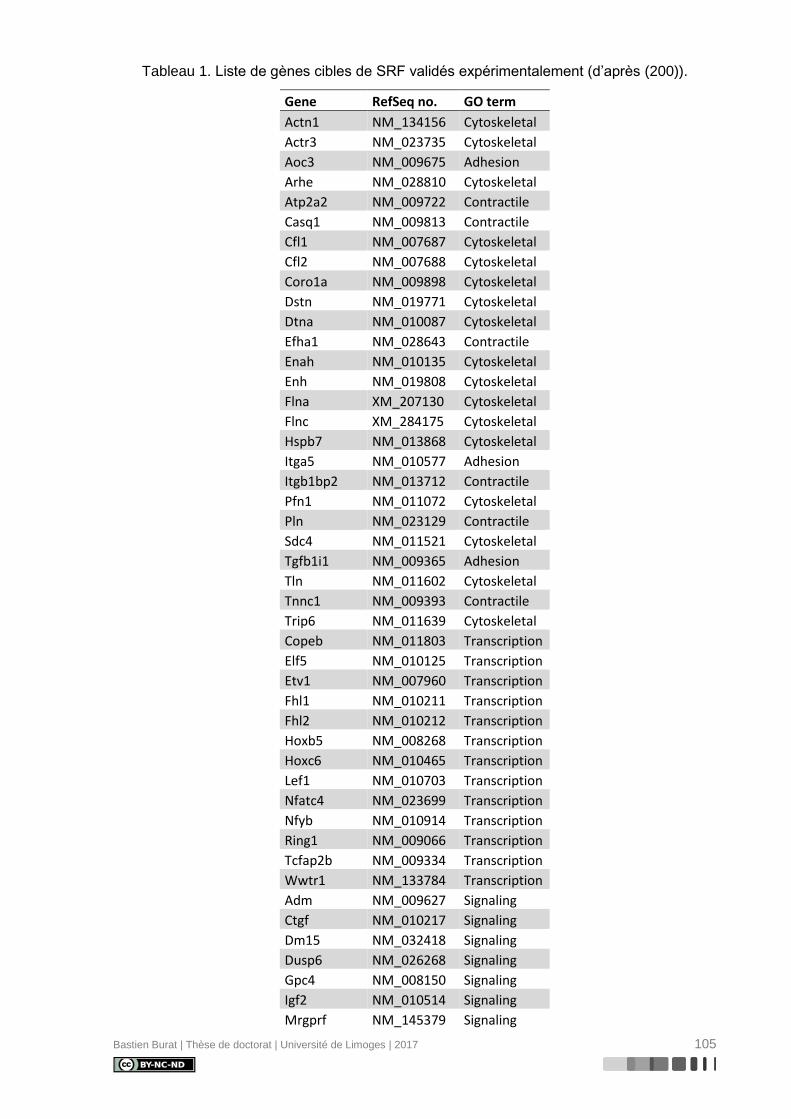
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 105
Tableau 1. Liste de gènes cibles de SRF validés expérimentalement (d’après (200)).
Gene RefSeq no. GO term
Actn1 NM_134156 Cytoskeletal
Actr3 NM_023735 Cytoskeletal
Aoc3 NM_009675 Adhesion
Arhe NM_028810 Cytoskeletal
Atp2a2 NM_009722 Contractile
Casq1 NM_009813 Contractile
Cfl1 NM_007687 Cytoskeletal
Cfl2 NM_007688 Cytoskeletal
Coro1a NM_009898 Cytoskeletal
Dstn NM_019771 Cytoskeletal
Dtna NM_010087 Cytoskeletal
Efha1 NM_028643 Contractile
Enah NM_010135 Cytoskeletal
Enh NM_019808 Cytoskeletal
Flna XM_207130 Cytoskeletal
Flnc XM_284175 Cytoskeletal
Hspb7 NM_013868 Cytoskeletal
Itga5 NM_010577 Adhesion
Itgb1bp2 NM_013712 Contractile
Pfn1 NM_011072 Cytoskeletal
Pln NM_023129 Contractile
Sdc4 NM_011521 Cytoskeletal
Tgfb1i1 NM_009365 Adhesion
Tln NM_011602 Cytoskeletal
Tnnc1 NM_009393 Contractile
Trip6 NM_011639 Cytoskeletal
Copeb NM_011803 Transcription
Elf5 NM_010125 Transcription
Etv1 NM_007960 Transcription
Fhl1 NM_010211 Transcription
Fhl2 NM_010212 Transcription
Hoxb5 NM_008268 Transcription
Hoxc6 NM_010465 Transcription
Lef1 NM_010703 Transcription
Nfatc4 NM_023699 Transcription
Nfyb NM_010914 Transcription
Ring1 NM_009066 Transcription
Tcfap2b NM_009334 Transcription
Wwtr1 NM_133784 Transcription
Adm NM_009627 Signaling
Ctgf NM_010217 Signaling
Dm15 NM_032418 Signaling
Dusp6 NM_026268 Signaling
Gpc4 NM_008150 Signaling
Igf2 NM_010514 Signaling
Mrgprf NM_145379 Signaling

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 106
P2rx1 NM_008771 Signaling
Rrad NM_019662 Signaling
Tspan13 NM_025359 Signaling
Bin1 NM_009668 Transport
Dnajb1 NM_018808 Transport
Mrvil NM_010826 Transport
Car3 NM_007606 Metabolism
Mrrf NM_026422 Metabolism
Urod NM_009478 Metabolism
Galnt3 NM_015736 Transferase
D14Ertd231e NM_153414 Unknown
Impact NM_008378 Unknown
Lzf NM_133185 Unknown
Shkbp1 NM_138676 Unknown

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 107
III.4. Signalisation CsA-dépendante de l’axe Actine-MRTF-SRF de cellules tubulaires
proximales
III.4.1. Hypothèses de travail et objectifs
Les résultats de l’analyse protéomique iTRAQ suggérait un effet spécifique de la CsA
sur le remodelage du cytosquelette par le biais d’une nouvelle dynamique de l’Actine. Le but
de ce deuxième travail a donc été d’identifier, à l’aide des approches classiques de biologie
moléculaire, les mécanismes en jeu, en particulier l’implication éventuelle des RhoGTPases,
la nature des ABP impliquées dans la régulation de l’Actine et enfin les conséquences en
aval sur la modulation éventuelle de l’axe MRTF/SRF. Ce travail fait l’objet d’un article qui
sera prochainement soumis à publication.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 108
III.4.2. Etude des modifications Cofiline-dépendantes de l’axe Actine-MRTF-SRF de
cellules tubulaires proximales induites par la Cyclosporine A
Article 2.
Cyclosporine A inhibits MRTF-SRF signaling through Na+/K+ ATPase inhibition & Ac-
tin remodeling
BASTIEN BURAT*, QUENTIN FAUCHER, HELENE ARNION, PIERRE MARQUET, MARIE
ESSIG

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 109
Introduction
The development of immunosuppressive regimens based on the Calcineurin Inhibi-
tors (CNI), Cyclosporine A (CsA) and Tacrolimus (Tac), represented a breakthrough in the
prevention of allograft rejection in solid organ transplantation (4,5,7). Although CNI are now
widely used in clinical protocols of immunosuppression (1,2) and have significantly improved
short-term graft and patient survival, long-term exposure is associated with major limiting
deleterious side effects such as nephrotoxicity, leading to end-stage renal disease (19).
CNI nephrotoxicity evolves from acute, reversible vascular and hemodynamic im-
pairments to chronic irreversible and generalized renal injuries (17,18). In particular, chronic
CNI nephrotoxicity results in the interstitial fibrosis and tubular atrophy (IF/TA) of proximal
tubules, whose mechanisms remain largely unsolved. Up to now, CNI are known to : disrupt
cell cycle and engage cell death (70,71,73–76); induce ER stress and UPR (82–86); promote
oxidative stress (87,88); impact ion homeostasis (91), e.g. by inhibition of the Na+/K+ AT-
Pase pump (98–102); or engage epithelial-mesenchymal transition (EMT) (110–112).
In the kidney, the study of podocytes exposed to CsA revealed that the dynamic of
the Actin cytoskeleton was sensitive to CsA. Indeed, CsA exerted the stabilization of the Ac-
tin cytoskeleton of foot processes, explaining the anti-proteinuric effect of CsA observed in
nephrosis. This mechanism was demonstrated to be Calcineurin-dependent with the inhibi-
tion of the dephosphorylation and consequent proteolysis of Synaptopodin, an Actin-binding
protein (ABP) whose activity is essential for podocyte structure-function (113). Besides, CsA
induced the overexpression of Cofilin and its regulation in a non-phosphorylated state, inde-
pendently from the effects on Synaptopodin (114).
In proximal tubular cells (PTC), we also demonstrated that the Actin cytoskeleton
could be modified by CsA (115). Indeed, independently from the inhibition of NFAT, CsA trig-
gered a reversible disorganization of Actin scaffolding at the periphery of the cell. Whether a
mechanism involving the CsA-related regulation of CFL was responsible for the effects of
CsA on PTC remained to be addressed.
CFL was initially described as an Actin-depolymerizing factor involved in Actin tread-
milling. CFL modifies the physical-chemical properties of the microfilaments, promotes Pi
release and ADP-Actin-G dissociation from older segments to replenish the pool of G-Actin
and to supply enough material for microfilament renewal. Since then, the scope of CFL activi-
ty was expanded in the light of the multiple mechanisms of CFL regulation. Up to date, CFL
activity is known to be sensitive to F-Actin saturation, phosphorylation-dephosphorylation of
its Ser3 residue, PIP2 interaction, intracellular pH and oxidative stress (159).
Since direct interaction with F-Actin microfilaments is necessary for the Actin-related
activity CFL, the degree of saturation of the microfilament (or the density of CFL near the
microfilament) play a major role in the regulation of CFL. Saturation is correlated to global

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 110
CFL concentration, CFL: Actin ratio and CFL oligomerization. At high saturation and CFL
density, F-actin filaments are severed and prone to the nucleation and polymerization of new
branches. Conversely, at low saturation and CFL density, F-actin filaments are severed and
completely depolymerized into G-Actin. Low CFL concentrations are related to low satura-
tion, and so, the higher the concentration, the higher the saturation of microfilaments
(184,188). Besides, in vivo and in vitro, CFL may exist as monomers, dimers and tetramers
with higher-order oligomers related to higher CFL concentration and F-Actin saturation
(185,187).
The switches between phosphorylated and dephosphorylated status are an-other lev-
el of regulation between active and inactive CFL. Phosphorylation of the Ser3 residue pre-
vents CFL-Actin interaction while dephosphorylation rescues CFL-Actin interaction and ena-
bles Actin-related CFL activity. Phosphorylation of Ser3 is only catalysed by LIM kinases,
downstream small RhoGTPases and their associated kinases (195). Dephosphorylation of
CFL Ser3 is catalysed by Slingshot phosphatases (172), which itself is activated by Calcineu-
rin.
Outside Actin-related functions, CFL is known to interact with Na+/K+ ATPase to pro-
vide energetic fuel to the pump. CFL complexes triose phosphate isomerase and interacts
with the alpha subunit of Na+/K+ ATPase to locally provide ATP. When Na+/K+ ATPase is
inhibited (by potassium depletion or the action of a pharmacological inhibitor), a feedback
mechanism disrupts energetic supply by CFL dephosphorylation (177–179).
The regulation of CFL orientates Actin dynamics towards the polymerization of G-
Actin monomers into F-Actin microfilaments, or, conversely, promotes the depolymerization
of F-actin into G-Actin. In case of a cytoplasmic accumulation of G-Actin, G-Actin traps Myo-
cardin-Related Transcription Factor (MRTF), a co-factor of the Serum Response Factor
(SRF) transcription factor. Complexes of MRTF-G-Actin are restricted to the cytoplasm.
Gene transcription by MRTF-SRF complexes depends on the nuclear import of MRTF hence
the cytoplasmic accumulation of G-Actin leads to SRF transcription inhibition (196). In that
way, the Actin dynamic was described as a potent intracellular fulcrum for cell adaptation to
its environment, connecting upstream RhoGTPases to downstream MRTF-SRF pathway
(164,193,194).
In this study, we aimed at the deciphering of the intracellular mechanisms of CsA-
induced reorganization of the Actin cytoskeleton of PTC and its downstream consequences.
We showed that CsA-induced reorganization of Actin cytoskeleton is associated with the de-
polymerization of F-Actin structures and the cytoplasmic accumulation of G-Actin which leads
to the inhibition of MRTF-SRF transcription activity. This mechanism involves changes in
CFL concentration and oligomerization state as well as interactions with CFL phosphorylation

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 111
site. However, the modifications were independent from the activity of LIM kinases but in-
volved the interaction between the Na+/K+ ATPase and CFL.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 112
Results
In proximal tubular cells, CsA, but not TAC, elicited a strong reorganization of Actin
cytoskeleton.
In fluorescence microscopy, cell monolayers of proximal tubular LLC PK-1 show-
cased a puzzled pattern as phalloidin-labelled F-Actin cytoskeleton organized cell mem-
branes into intercellular convolutions (Figure 1 A, panel a). These peculiar structuring of
plasma membranes were reminiscent of the way basolateral membranes connect inside
proximal tubular epithelia, in vivo.
Upon 24-hour CsA exposure, LLC PK-1 sustained a significant reorganization of cor-
tical Actin cytoskeleton (Figure 1 A, panel b) related to a significant loss of F-Actin-based
structures (Figure 1 B, -0.46 %, p<0.01**). CsA effects seemed to be related to the inhibition
of the peptidylprolyl cis-trans isomerase activity of its target immunophilin Cyclophilin A
(CypA) since a specific pharmacological inhibitor of CypA (CAI), without any immunological
effect, elicited similar Actin modifications (Figure 1 A panel e, Figure 1 B, -0.51 %, p<0.01**).
However, CsA effects seemed independent of Calcineurin inhibition since neither Tac, VIVIT,
a specific inhibitor of NFAT dephosphorylation by Calcineurin, nor GPI 1046, a pharmacolog-
ical inhibitor of FKBP12, modified Actin organization (Figure 1A panels c, d and f, Figure 1B).
In conclusion, these observations suggested that CsA triggered the deep remodelling
of proximal tubular Actin cytoskeleton and the depolymerization of F-Actin, in a Cyclophilin-
dependent way, independently from Calcineurin inhibition.
Actin depolymerization led to a decrease in SRF-MRTF transcription activity in LLC
PK-1 upon CsA exposure
Profiles of drug-related Actin dynamics strictly superimposed to profiles of drug-
related SRF transcription activity. Upon 24-hour CsA exposure, SRF transcription activity
was significantly reduced (Figure 2 A, -48.5 % of control activity, p<0.001***). Likewise, CAI
elicited a concentration-dependent negative regulation of SRF (Figure 2 C). On the contrary,
neither Tac, VIVIT (Figure 2 B) nor GPI 1046 (Figure 2 D) reduced SRF transcription activity
which was slightly increased upon Tac and VIVIT exposure.
In conclusion, CsA downregulated the transcription of SRF-MRTF target genes, in a
Cyclophilin-dependent way, independently from Calcineurin inhibition.
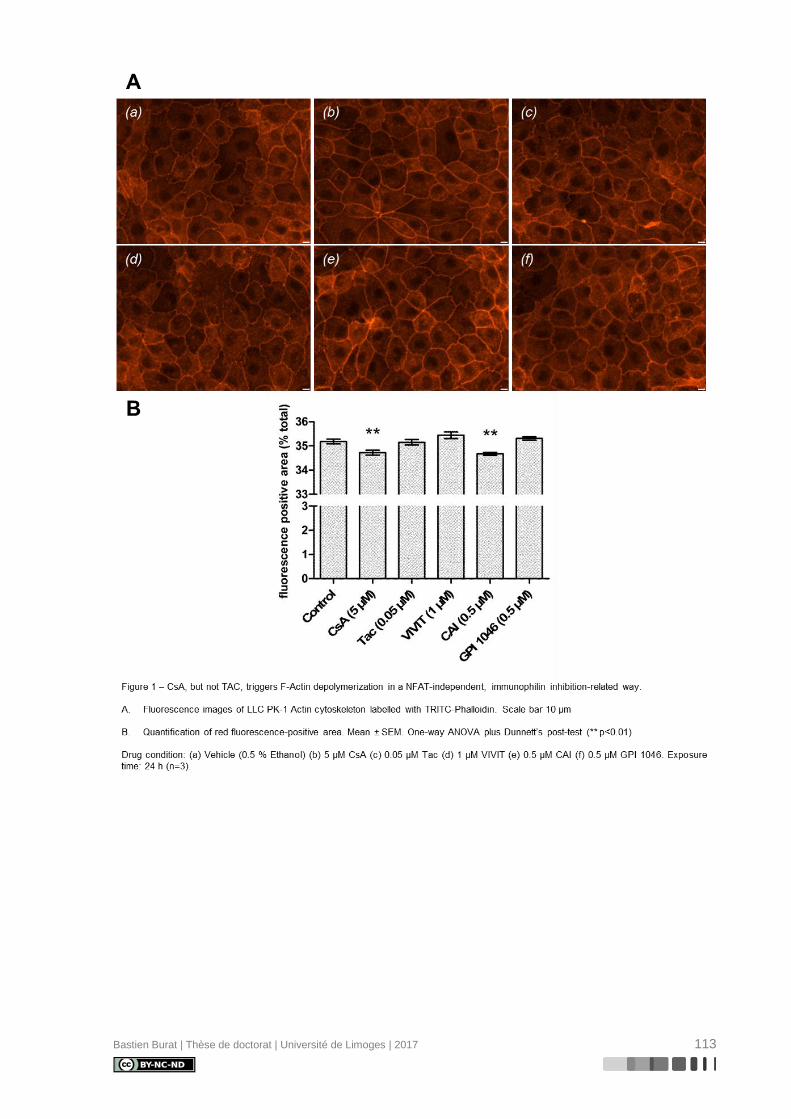
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 113
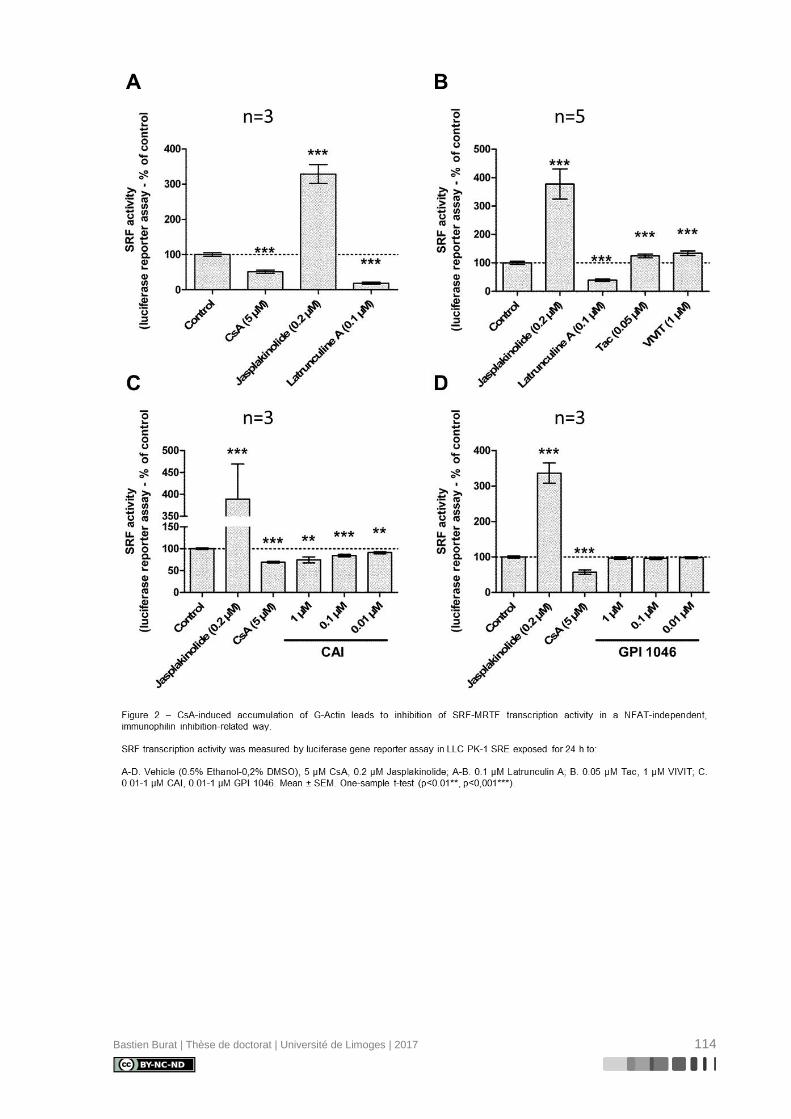
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 114

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 115
CsA regulates Actin-binding protein Cofilin through changes in Cofilin-Actin ratio and
oligomerization state
Differential quantitative proteomic analysis of identified Actin family cytoskeletal pro-
teins (PANTHER Protein Class PC00041) showed that CsA decreased CFL: Actin ratio since
Actin was overexpressed while CFL levels remained steady whereas TAC induced equiva-
lent overexpression for both Actin and CFL (Figure EV1). A similar pattern of expression was
observed for F-Actin capping protein subunits α2 and β.
Western blot analysis of cross-linked CFL oligomers (Figure 3 A) indicated that CsA
induced a significant decrease in dimers and tetramers (RdiCFL = 0.40 ± 0.09; RtetraCFL =
0.38 ± 0.16) while monomers remain steady (RmonoCFL = 0.90 ± 0.06) (Figure 3 B).
In conclusion, CsA elicited a shift in the balance between oligomeric forms of CFL.
CsA decreased the protein abundance of CFL dimers and tetramers.
CsA effects seemed partly independent from phosphorylation/dephosphorylation of
Cofilin
The S3-R peptide, an analogue of the CFL phosphorylation site and a non-specific
inhibitor of Ser3-targeting proteins, didn’t impact the organization of the Actin cytoskeleton
(Figure 4 A, panel b) but it is associated with a slight yet significant increase in F-Actin levels
(Figure 4 B, +0.36 %, p<0.05*). When in a co-treatment with CsA, S3-R nullified CsA Actin
remodelling (Figure 4 A, panel e, Figure 4 B). Conversely, the addition of a specific pharma-
cological inhibitor of LIMK, LIMKi3, alone or associated with CsA triggered CsA-like pheno-
type of Actin dynamics (Figure 4 A, panel c & f) and significant decreases in F-Actin levels
(Figure 4 B, LIMKi3 alone: -1.62 %, p<0.001***; LIMKi3 + CsA: -1.22 %, p<0.001***).
S3-R alone didn’t impact the MRTF-SRF transcription activity (Figure 5 A). When in a
co-treatment with CsA, S3-R significantly decreased the CsA-induced inhibition of MRTF-
SRF transcription activity (CsA: -33.1 %; S3-R + CsA: -24.5 %; +8.6 %, p<0.05*). LIMKi3
alone induced an inhibition of SRF-MRTF transcription activity (Figure 5 B, -52.3 %,
p<0.001***) like what was observed with CsA alone (-35.9 %, p<0.001***). Furthermore,
LIMKi3 reinforced the CsA-induced inhibition of SRF transcription activity (-61.5 %,
p<0.001***).
The pharmacological inhibitors of the RhoGTPases pathway, Y27632 (ROCK inhibi-
tor) and EHT1864 (Rac1 inhibitor) elicited the same effects as LIMKi3 at both cytoskeletal
(Figure EV2 A) and transcriptional (Figure EV2 B) levels.
In conclusion, CsA effects involved the phosphorylation site of CFL as a target for
dephosphorylation. Inhibitors of the RhoGTPases pathway triggered CsA-like effects.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 116

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 117

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 118

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 119
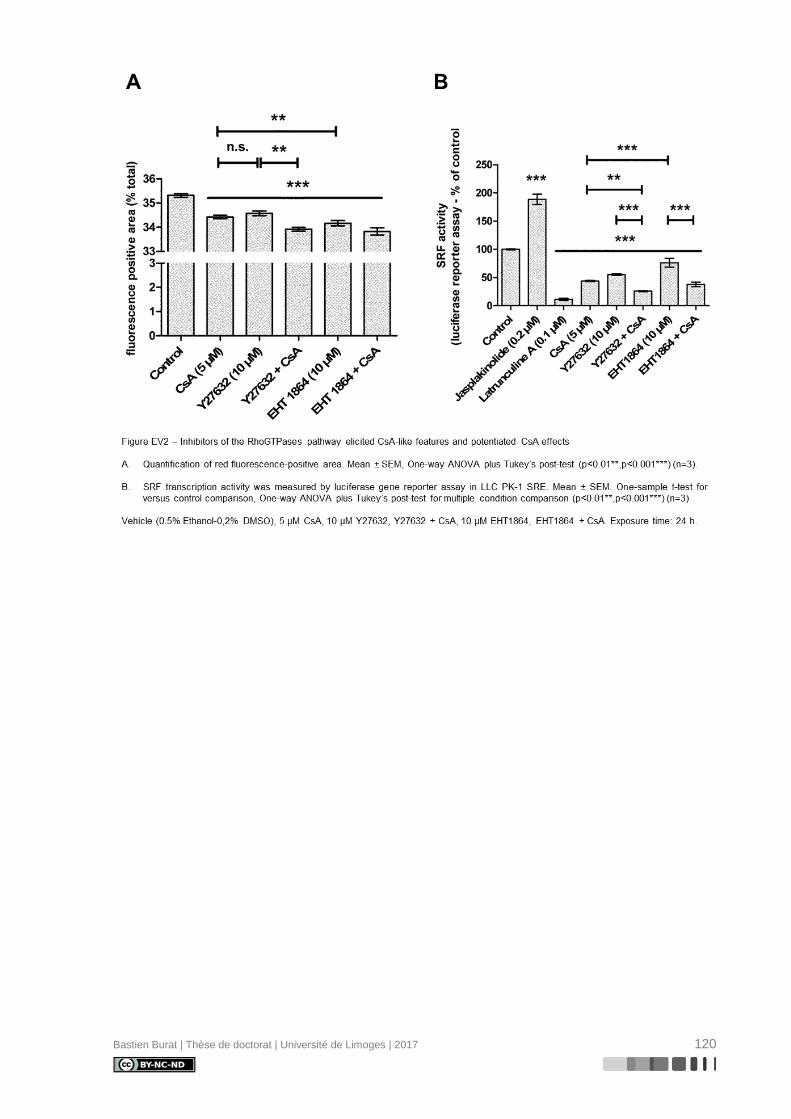
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 120

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 121
CsA inhibited Na+/K+ ATPase activity in tubular proximal cells and Ouabain-induced
inhibition of Na+/K+ ATPase mimicked Actin reorganization and inhibition of MRTF-SRF
transcription activity observed upon CsA exposure
A significant decrease in Na+/K+ ATPase activity (-23.1 % of control, p<0.01**) was
observed in LLC PK-1 exposed for 24 h to CsA (Figure 6 A) whereas Tac treatment had no
effect.
LLC PK-1 exposed for 24 h to 100 nM Ouabain featured CsA-like Actin reorganization
(Figure 6B panel c) with a significantly greater loss of F-Actin (Figure 6 c, CsA: -0.76 %;
OUA: -1.35 %, p<0.001***). Moreover, Ouabain co-treatment with CsA potentiated CsA-
induced cell stiffening (Figure 6B panel d) and F-Actin decrease (Figure 6 C, -2.26 %,
p<0.001***). Likewise, Ouabain exposure led to the inhibition of MRTF-SRF transcription
activity (Figure 6 D, -26.6 %). Ouabain co-treatment with CsA reinforced CsA effects (CsA: -
47.0 %; OUA + CsA: -62.0 %, p<0.001***).
In conclusion, CsA targeted Na+/K+ ATPase and inhibited its activity. Na+/K+
ATPase inhibitor Ouabain elicited Actin reorganization the same way CsA did. Ouabain-
related Actin dynamics induced the partial inhibition of MRTF-SRF transcription activity. The
association of Ouabain with CsA tended to potentiate CsA effects on tubular proximal mor-
phology and Actin dynamics.
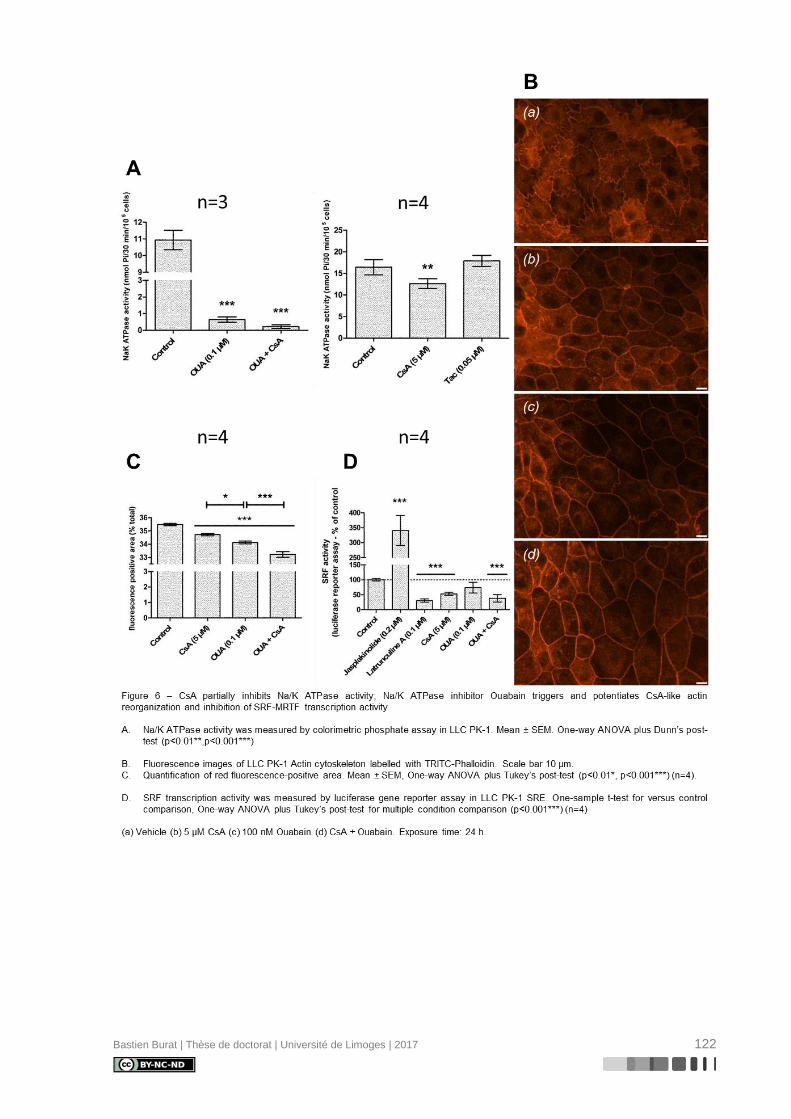
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 122

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 123
Discussion
In the light of early observations of CsA effects on the Actin cytoskeleton of podo-
cytes, the study of the Actin dynamic in PTC appeared of utmost interest in the elucidation of
pathophysiological mechanisms of tubular atrophy upon CNI exposure.
In this work, we demonstrated that CsA-induced remodelling of the Actin cytoskeleton
of PTC was associated with a modification of the F-Actin: G-Actin ratio through an original
regulation of CFL: Indeed, CsA triggered decreases in the total CFL: Actin ratio and the lev-
els of CFL dimers and tetramers, independently from the phosphorylation by LIM kinases but
most likely related to ta CsA-induced inhibition of Na+/K+ ATPase.
Studies on podocytes reported that CsA had anti-proteinuric effects via the stabiliza-
tion of the Actin cytoskeleton structuring the foot processes. This stabilization resulted from
the overexpression and dephosphorylation of Cofilin. So, although the study didn’t focus on
CFL oligomerization, the CFL: Actin ratio was in favour of CFL and the CFL concentration
was higher, therefore the activity of CFL was regulated to promote Actin polymerization and
stabilize microfilaments. What we observed in PTC was a destabilization of the Actin cyto-
skeleton structuring the intercellular convolutions. This indicated a different regulation of CFL
activity where CFL would have to exert low saturation of F-Actin microfilaments (lower CFL:
Actin ratio; lower levels of CFL oligomers) and to be active for Actin-related functions i.e.
dephosphorylated.
CsA had already been reported to elicit the remodelling of the Actin cytoskeleton of
PTC (201). In this study, CsA (4.2 µM, 24 h) activated RhoA and the phosphorylation cas-
cade, which led to CFL phosphorylation by LIMK. CFL inhibition results in the formation of
stress fibres and the tightening of tight junctions. Our findings are just the opposite of these
previous results and are incompatible with the mechanism they described. Indeed, all the
pharmacological inhibitors of the RhoGTPases pathway we used elicited CsA-like effects i.e.
the loss of proximal tubular morphology and the inhibition of MRTF-SRF activity. These dis-
crepancies might result from differences in cell culture conditions as, in the study by Martin-
Martin et al, control LLC PK-1 seemed non-confluent, less differentiated, with non-convoluted
membranes and a significant amount of stress fibres. Although the cell culture protocol ap-
peared to be similar, the serum deprivation prior to drug exposure differed as it is not sup-
plemented with hormones allowing advanced epithelial differentiation of LLC PK-1 cells at
the morphological and cytoskeletal levels. Our present model seemed closer to the in vivo
aspect of PTC than the one Martin-Martin et al developed.
CsA effects on TCP were reminiscent of OUA effects on HeLa cells (178). OUA ex-
erted a similar reorganization of the Actin cytoskeleton by dephosphorylating CFL via a
mechanism where the inhibition of Na+/K+ ATPase activates the Ras/Raf/MEK cascade, in a
Src-EGFR-dependent way, leading to the inhibition of LIMK downstream the small RhoG-

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 124
TPases pathway. OUA effects were reproduced by Y27632 just like CsA effects were mim-
icked by all the RhoGTPases inhibitors we used.
Earlier findings about the mechanism of Na+/K+ ATPase energetic supply by pCFL-
TPI complexes (177) reported that the overexpression of constitutively active CFL (Ser3 was
replaced by Ala) was enough to cut endogenous CFL out of phosphoryla-
tion/dephosphorylation-based regulation which made TPI localization insensitive to the acti-
vation/inhibition of the RhoGTPases pathway. Likewise, the addition of S3-R was enough to
partially exclude endogenous CFL from CsA-induced regulation.
Dephosphorylation of CFL by Slingshot phosphatases seemed unlikely since Sling-
shot phosphatases are activated by Calcineurin. CsA effects on CFL were unlikely to be re-
lated to Calcineurin since Tac and VIVIT didn’t elicit CsA-like effects on the Actin cytoskele-
ton or the MRTF-SRF transcription activity.
Numerous studies have already reported the inhibition of Na+/K+ ATPase by CsA, in
vitro and in vivo. However, the observations were limited to the consequences in ion
transport (98,99,101,102,104,202–204). Although this work isn’t the first one to study the
relationship between the Na+/K+ ATPase pump and the Actin cytoskeleton, the partial inhibi-
tion (about 25%) of proximal tubular Na+/K+ ATPase by CsA is, to date, the first observation
of a direct correlation between variations in the pump activity and cytoskeletal remodelling.
Besides, the potentiation of the CsA-induced depolymerization of F-Actin by OUA is in favour
of a Na+/K+ ATPase -related mechanism.
How CsA inhibits Na+/K+ ATPase has yet to be elucidated. CsA is known to induce
over-expression of endothelin, which is a well-known inhibitor of Na+/K+ ATPase activity
(23,205). Furthermore, CsA can downregulate Na+/K+ ATPase activity through the inhibition
of Cyclophilin B. The PPIase was described as a crucial partner for the structure and activity
of Na+/K+ ATPase catalytic subunit (206). Besides, the proteome monitoring of HEK cells by
SILAC-LC-MS/MS reported the decrease in CypB protein abundance upon CsA exposure
(207). These findings, together with the observations of CsA-like effects upon CAI exposure,
support the implication of Cyps in the inhibition of Na+/K+ ATPase by CsA. To what extent
the Cyps are implicated has yet to be elucidated.
We propose a model (Figure 7) where: the inhibition of Na+/K+ ATPase by CsA leads
to the dephosphorylation of CFL, independently from the classical regulation pathways; CsA
decreases the CFL: Actin ratio and the protein abundance of F-Actin-stabilizing and polymer-
ization-promoting CFL oligomers; active, minority, monomeric CFL depolymerizes low-
saturated F-Actin microfilaments and replenish G-Actin pools; the accumulation of G-Actin
leads to the cytoplasmic trapping of the SRF co-factor MRTF; the low nuclear abundance of
MRTF results in the low abundance of transcription-ready MRTF-SRF complexes and the

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 125
decrease in MRTF-SRF transcription activity; overall, CsA inhibits gene transcription by the
MRTF-SRF complex via drug-specific, G-Actin-prone Actin-dynamics.
The CsA-induced inhibition of MRTF-SRF gene transcription might have feedback
consequences on Actin dynamics since ACTB, Cfl1 and other ABP-coding genes are identi-
fied genes of the CArGome (the ensemble of MRTF-SRF genes whose promoters contain
CArG boxes) and the activity of the MRTF-SRF complex was shown to be required for the
homeostasis of Actin levels and the organization of the Actin cytoskeleton (200).
As a matter of facts, cellular consequences of MRTF-SRF inhibition might be large-
scale perturbations since MRTF-SRF regulates the transcription of genes coding for
transport and adhesion proteins, enzymes of the lipid and glucose metabolisms, transcription
factors, growth factors, growth factor receptors, etc.
For instance, past studies clearly elucidated the role of MRTF-SRF inhibition in multi-
factorial biological processes like proapoptotic mechanisms (208,209) and epithelial-
mesenchymal transition (210,211), which have been widely described as CsA-related patho-
physiological mechanisms.
Hence why the proposed model might be of utmost interest to explain and link to-
gether known in vitro effects of CsA in the light of an original regulation of Actin dynamics
upon CsA exposure.
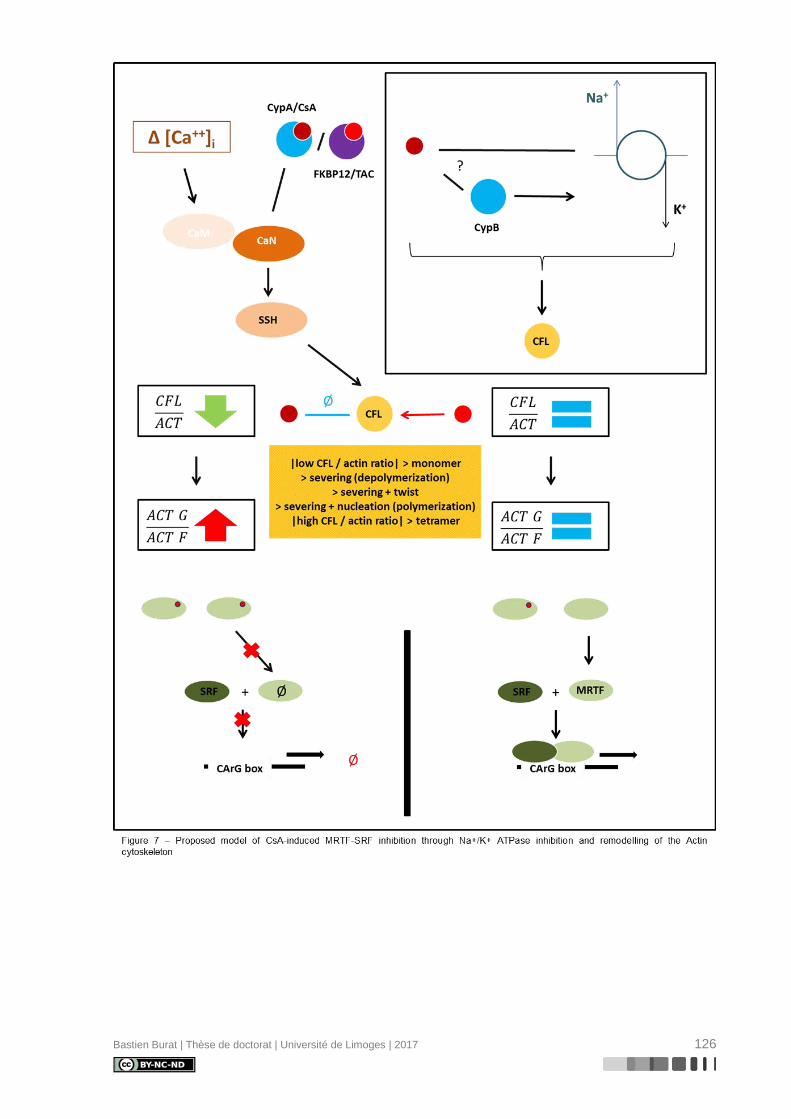
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 126

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 127
Materials and methods
Chemicals
Dulbecco’s Modified Eagle’s Medium-Ham’s F12 (1:1, #31331, Gibco), Fetal Bovine
Serum (#10500), 1 M HEPES (#15630), 7.5 % Sodium bicarbonate (#25080), 10,000 UI.mL-
1 Penicillin / Streptomycin (#15140), Dulbecco’s Phosphate Buffer Saline (#14190) were pur-
chased from Gibco. Sodium selenite (S5261), CsA (#30024), Tac (F4679), Jasplakinolide,
Latrunculin A, ROCK inhibitor Y27632, Rac1 inhibitor EHT 1864, Na+/K+ ATPase inhibitor
Ouabain, insulin (I4011), triiodothyronine (T6397) and dexamethasone (D4902) were pur-
chased from Sigma-Aldrich. Cyclophilin A inhibitor was purchased from Millipore. LIMK1/2
inhibitor LIMKi3 was provided by Tocris.
S3-R peptide (MASGVMVSDDVVKVFNRRRRRRRR), an analogue of the Cofilin
phosphorylation site, was synthetized by ProteoGenix (Schiltigheim, France).
Cell culture conditions
LLC PK-1 (Lilly Laboratories Porcine Kidney-1) porcine proximal tubule cells (ATCC-
CL-101, ATCC, Manassas, VA) were expanded in 75 cm² flasks at 37 °C with 5 % CO2 and
passed once confluence was reached. Culture medium consisted in a 1:1 Dulbecco’s Modi-
fied Eagle’s Medium-Ham’s F12 mix supplemented with 5 % FBS, 15 mM HEPES, 0.1 %
Sodium bicarbonate, 100 UI.mL-1 Penicillin / Streptomycin and 50 nM Sodium selenite. LLC
PK-1 cells were cultured between passage 7 and passage 30.
Seeded LLC PK-1 sustained serum starvation and were fed with hormonally-defined
(25 µg.mL-1 insulin, 11 µg.mL-1 transferrin, 50 nM triiodothyronine, 0.1 µM dexamethasone,
0.1 µg.mL-1 desmopressin) fresh medium to engage epithelial differentiation, for 24 hours.
Hormonally-defined LLC PK-1 cells sustained cell transfection or were exposed for 24
hours to: vehicle mix (Ethanol, DMSO), 5 µM CsA, 0.05 µM Tac, 1 µM VIVIT (a specific
NFAT inhibitor), 0.5 µM Cyclophilin A inhibitor, 200 nM Jasplakinolide, 100 nM Latrunculin
A,10 µM ROCK inhibitor Y27632, Rac1 inhibitor EHT 1864, CDC42 inhibitor ML141, Arp2/3
inhibitor CK666, formins inhibitor SMIFH2 and LIMK inhibitor LIMKi3, 100 nM Na+/K+
ATPase inhibitor Ouabain or 100 µg.L-1 S3-R.
Actin cytoskeleton characterization
Immunocytochemistry
Serum-starved, hormonally-defined, treated LLC PK-1 cells seeded on glass cover
slips in 6-well plates were washed twice for 3 min in PBS. Then, cells were fixed in 4 % para-
formaldehyde-PBS for 10 min, at room temperature. Cells were washed once again in PBS
for 3 min then permeabilised with 0.1 % Triton/X-100-PBS for 5 min, at room temperature.
Permeabilised cells were washed three times for 3 min in PBS before incubation in Phal-

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 128
loidin-TRITC-PBS for 30 min, at 37 °C, in the dark. Eventually, cover slips were washed
three times for 3 min in PBS then mounted on glass microscope slides using ProLong® Gold
Antifade Reagent as a mounting medium and a DAPI nuclei staining. Florescence labelling
was visualized using a Leica DM-RX microscope.
Image analysis
For each exposure condition, ten photographs (*.tiff) were randomly taken to report
from an independent experiment. Image analysis was conducted using the ImageJ software
(v.1.48). Early image processing consisted in a restriction to the green channel of the RGB
picture. The implemented Auto Local Threshold tool further selected the fluorescence-
positive pixels (Niblack thresholding method, radius = 15). Thus, for each photograph, a
semi-quantitative ratio was calculated as a percentage of the total image area.
All above-mentioned steps were automatized thanks to the macro editing tool of the
ImageJ software.
Statistical analysis of fluorescence area data was performed using the one-way anal-
ysis of variance (1-way ANOVA) test followed by the Bonferroni’s Multiple Comparison post-
test with a significance threshold at p<0.05, as implemented in GraphPad Prism (v. 5.04).
Luciferase Gene Reporter Assay for SRF/SRE Activity
Stable cell transfection & clone selection
Volumes equivalent to 0 µg, 2 µg or 4 µg of plasmid DNA () and transfection reagent
lipofectamine (1:25) were separately diluted in FBS-free, antibiotic-free routine medium.
Then, DNA and lipofectamine preparations were blended and put at rest for 15 min, at room
temperature. Serum-starved, hormonally-defined LLC PK-1 cells in 6-well plates were incu-
bated with DNA-lipofectamine mixtures for 24 h, at 37 °C.
Transfected LLC PK-1 cells (LLC PK-1 SRE) were incubated in routine medium for 72
h before antibiotic selection to allow proper plasmid integration. Then, cells were re-
suspended in 0.05 % Trypsin-EDTA, 10 min at 37 °C. Cell suspensions were diluted 10 times
and re-seeded in routine medium completed with 1 % Hygromycin B (50 mg.mL-1) refreshed
every 48 h until colonies appear. Colonies were harvested thanks to trypsin-soaked paper
disks () and re-seeded to start routine culture in antibiotic-supplemented growth medium.
Luciferase gene reporter assay/Bioluminescence assay
Serum-starved, hormonally-defined, treated LLC PK-1 SRE were re-suspended me-
chanically in 1X lysis buffer (Cell Culture Lysis 5X Reagent, E153A, Promega). Cell lysates
were distributed in technical duplicates in a white 96-well plate (#3912, Costar). Biolumines-
cence signals were detected by the Enspire Multimode Reader (Perkin-Elmer).
Oligomer cross-linking and Western blot

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 129
Serum-starved, hormonally-defined, treated LLC PK-1 cells in 60 mm Petri dishes
were incubated in hormonally-defined medium – 1 % formaldehyde (cross-linking range 2.9
Å) for 15 min under agitation. Formaldehyde cross-linking was quenched by the addition of 1
M glycine (final concentration 125 mM). LLC PK-1 were washed twice with Dulbecco’s Phos-
phate Buffer Saline and lysed by scrapping in a custom RIPA lysis buffer (150 mM NaCl, 50
mM TRIS-HCl, 0.1 % NP-40, 0.1 % SDS, 1 mM EDTA in ultrapure H2O, supplemented with
a 1:100 anti-protease / anti-phosphatase mix). Cell lysates were incubated on ice for 30 min
and centrifuged for 15 min at 21000 g. Supernatants were stored until protein concentration
was measured using the Bradford colorimetric method. Forty micrograms of proteins per ex-
posure condition were separated by electrophoresis under reducing and denaturing condi-
tions on a NuPAGE® Novex® Bis-Tris pre-cast gel (NP0341, ThermoFisher) in 1X Nu-
PAGE™ MOPS SDS running buffer (NP0001, ThermoFisher) and transferred to a nitrocellu-
lose (NC) membrane (NP23001, ThermoFisher) using the iBLOT 2 Dry Blotting system
(IB21001, ThermoFisher). Membranes were blocked for 1 h at room temperature under agi-
tation with TBS-Tween buffer (10 mM Tris 7.6, 150 mM NaCl, 0.1 % Tween-20) comple-
mented with 5 % (W/V) non-fat milk powder to obtain BLOTTO buffer. Primary antibody incu-
bation was done in BLOTTO for 1 h at room temperature. After three 5-min washes in TBS-
T, secondary antibody incubation was performed in BLOTTO for 1 h at room temperature
then washed again. Membranes were incubated in a 1: 1 mix of SuperSignal™ West Pico
PLUS Chemiluminescent Substrate kit (#34577, ThermoFisher) and analyzed by the Chemi-
Doc Imaging system (Bio-Rad) for chemiluminescent signal detection and acquisition. Quan-
titation was computed via the ImageLab software (Bio-Rad)
Na+/K+ ATPase activity/Phosphate release assay
Serum-starved, hormonally-defined, treated LLC PK-1 cells in 24-well plates were
permeabilised by osmotic shock in ultrapure water (150 µL) and fast freeze in liquid nitrogen
(10 s). Cells were thawed (200 µL) in a buffer with a composition of 200 mM sodium chloride,
80 mM histidine, 20 mM potassium chloride, 6 mM magnesium chloride, 2 mm EGTA, 2
µg.mL-1 Alamethicin, 30 µM Digitonin. For each drug exposure condition, a volume of 30 mM
Ouabain (final concentration 1 mM) or ultrapure water was added to every other well. Cells
were either incubated 30 min at 37 °C. A volume of 30 mM ATP (final concentration 1 mM)
was added to each well. Cells were incubated 30 min more at 37 °C. On ice, trichloroacetic
acid: water (1:1) terminated ATP hydrolysis reactions. Plates were centrifuged at 3000 rpm
for 10 min at room temperature. Supernatants were diluted 50 times and distributed (50 µL)
in technical triplicates to a 96-well plate.
Concentration of free phosphate released from ATP hydrolysis was measured using
the BIOMOL® Green kit (Enzo). NaK ATPase activity (expressed in nmol of released phos-

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 130
phate/30min/105 cells) was calculated as the difference in free phosphate concentration in
the presence and absence of Ouabain.
iTRAQ shotgun proteomics
Protein extraction, sample preparation, iTRAQ labelling and isoelectric focus-
ing
After 24 h drug exposure, LLC PK-1 cells were washed twice with Dulbecco’s Phos-
phate Buffer Saline and lysed by scrapping in a custom RIPA lysis buffer (150 mM NaCl, 50
mM TRIS-HCl, 0.1 % NP-40, 0.1 % SDS, 1 mM EDTA in ultrapure H2O, supplemented with
an anti-protease / anti-phosphatase mix). Cell lysates were incubated on ice for 30 min and
centrifuged for 15 min at 21000 g. Supernatants were stored until protein concentration was
measured using the Bradford colorimetric method and iTRAQ labeling. Twenty-five mi-
crograms of proteins were precipitated by - 20 °C cold acetone. After acetone evaporation,
the precipitates were solubilized in 25 mM ammonium bicarbonate then were incubated with
50 mM dithiothreitol for 40 min at 60 °C, to reduce disulfide bonds, 100 mM iodoacetamide in
the dark for 40 min at room temperature, to alkylate/block cysteine residues and eventually
were digested for 24 h at 37 °C with mass-spectrometry grade trypsin (V5280, Promega) at a
1: 50 enzyme: substrate ratio.
After digestion, samples were incubated with iTRAQ tags (iTRAQ Reagents Multi-plex
kits, 4-plex, #4352135, Sigma-Aldrich) – one tag per drug exposure condition and five differ-
ent tag/condition associations over five experiments – for 1 h at room temperature. Labeled
samples were mixed and separated into 12 fractions by isoelectric focusing (OFFGEL 3100
Fractionator, Agilent Technologies, Santa Clara, CA) for 24 h at increasing voltage and
steady intensity of 50 µA in a 3-10 pH IPG strip. Fractions were retrieved for further MS anal-
ysis after the IPG strip was incubated in a 1:1 acetonitrile (ACN): water, 0.1 % formic acid
(FA) wash solution for 15 min at room temperature.
nano-LC peptide separation and Q-Q-TOF mass spectrometry
IEF fractions were analyzed by nano-LC MS/MS using a nano-chromatography liquid
Ultimate 3000 system (LC Packings DIONEX, Sunnyvale, CA) coupled to a Triple TOF
5600+ mass spectrometer (ABSciex, Toronto, Canada). For each sample, 5 µL were injected
into a pre-column (C18 Pepmap™ 300 µm ID x 5 mm, LC Packings DIONEX) using the load-
ing unit. After desalting for 3 min with loading solvent (2 % ACN, 0.05 % trifluoroacetic acid
(TFA)), the pre-column was switched online with the analytical column (C18 Pepmap™ 75
µm ID x 150 mm, LC Packings DIONEX) pre-equilibrated with 95 % solvent A (ACN 5 % - FA
0.1 %). Peptides were eluted from the pre-column into the analytical column and then into
the mass spectrometer by a linear gradient from 5 % to 25 % in 70 min, then to 95 % of sol-
vent B (98 % ACN, 0.1 % FA) over 120 min at a flow rate of 200 nL/min.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 131
Data acquisition was carried out by IDA (Information-Dependent Acquisition) mode of
Analyst 1.7 TF software (ABSciex). The data from MS and MS/MS were continuously rec-
orded with a cyclic duration of 3 s. For each MS scan, up to 50 precursors were selected for
fragmentation based on their intensity (greater than 20,000 cps), their charge state (2+, 3+)
and if the precursor had already been selected for fragmentation (dynamic exclusion). The
collision energies were automatically adjusted according to charge state, ionic mass of se-
lected precursors and iTRAQ labelling.
Mass spectrometry data processing and relative protein identification / quanti-
fication
MS and MS/MS data for five independent experiments (*.wiff, 1 per fraction, 12 files
per experiment) were submitted to Mascot Server 2.2.03 via ProteinPilot (version 5.0, AB-
Sciex) for protein identification, and searched against two complementary Sus scrofa data-
bases: a Swiss-Prot database (2015_10 release) and a TrEMBL database (2015_10 re-
lease). Carbamidomethyl (C) was defined as a fixed modification. Oxidation (O), iTRAQ4plex
(K), iTRAQ4plex (Y), iTRAQ4plex (N-term) were defined as variable modifications. MS/MS
fragment mass tolerance was set at 0.3 Da. Precursor mass tolerance was set at 0.2 Da.
Mascot raw data files (*.dat, 1 per experiment) were saved for further isobaric tags-
based peptide and protein quantitation with the Java implementation of the Quant algorithm,
jTRAQx (version 1.13, (212)). Reporter mass tolerance was set at 0.05 Da while iTRAQ cor-
rection factors were implemented as provided by ABSciex. This tool generated one *.jpf file
(tab-delimited text file) for each series.
*.jpf were submitted to the CiR-C (Customizable iTRAQ Ratio Calculator) algorithm
which excluded irrelevant data according to: i) identification confidence: peptides are retained
if the probability that the observed positive match is a random match is below 5 % (p < 0.05,
Mascot score > 30); ii) quantification confidence: peptides are retained if all iTRAQ ratios
have been successfully calculated i.e., peptides with 0.0 ratios or uncalculated ratios (null
ratios) are discarded; iii) peptides related to ‘Fragment’- and ‘REVERSED’-annotated pro-
teins are discarded. After irrelevant data removal, CiR-C drew up an exhaustive catalogue of
identified peptide sequences with their associated Swiss-Prot or TrEMBL accession IDs.
Peptides were assigned to a frequency index of positive matches (identification in {1;2;3;4;5}
out of 5 independent experiments) and CiR-C drew a second catalogue of peptides with the
highest frequency index (n=5). Protein ratios were calculated as both overall and series-
specific median values of peptide ratios associated with a given accession ID and frequency
index.
The protein list analysis was performed by submitting Swiss-Prot accession IDs to the
online tool powered by the PANTHER Classification system. The PANTHER Overrepresen-
tation test (release date 20170413) parsed the PANTHER database (version 12.0, released

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 132
on 2017-07-10) using the Sus scrofa reference list and the PANTHER Protein Class annota-
tion data set. Only p<0.05 items were retained and considered significantly over-represented.
Biological significance criteria
Frequency distribution of the iTRAQ peak intensities ratios could be approximated as
a Gaussian distribution: iTRAQ peak intensities ratios frequency distribution significantly fit-
ted implemented Gaussian non-linear regression (n = 370, R² = 0.9946, mean = 1.02 ± 0.10).
One of the Gaussian distribution properties is: I) 66% of the values lie in a 1 S.D. range
around the mean, II) 95 % of the values lie in a 2 S.D. range around the mean, III) 99 % of
the values lie in a 3 S.D. range around the mean. Applying the “mean ± n S.D.” property to
the set of iTRAQ peak intensity ratios, the upper thresholds for significant upregulations upon
drug exposure would be “1.02 – 0.10 = 0.92”, “1.02 – 2 x 0.10 = 0.82” and “1.02 – 3 x 0.10 =
0.72” while the lower thresholds for significant downregulations upon drug exposure would
be “1.02 + 0.10 = 1.12”, “1.02 + 2 x 0.10 = 1.22” and “1.02 + 3 x 0.10 = 1.32”. Thus, proteins
were classified thanks to their distance from the unity: proteins with median ratios between
0.92 and 1.12 were annotated as non-impacted proteins; up-regulated proteins with quantita-
tive ratios beyond 1.12 were ranked according to three tiers of significant fold increase: +σ
(1.12 ≤ r < 1.22), +2σ (1.22 ≤ r < 1.32) or +3σ (1.32 ≤ r). The same way down-regulated pro-
teins with quantitative ratios below 0.82 were ranked according to three tiers of significant
fold decrease: -σ (0.92 ≥ r > 0.82), -2σ (0.82 ≥ r > 0.72) or -3σ (0.72 ≥ r).
Hierarchical clustering & heat-map representation
Heat map representation and hierarchical clustering were generated using the Euclid-
ian distance calculation as provided by the GENE-E software (version 3.0.204).
Data availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium
(http://proteomecentral.proteomexchange.org) (213) via the PRIDE partner repository (214)
with the data set identifier PXD007891 (username: [email protected], password:
TPSGICw9).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 133
Chapitre IV. Discussion, conclusion et perspectives
IV.1. Discussion et conclusion
IV.1.1. Evolution de la stratégie de protéomique quantitative
Ces travaux de thèse s’inscrivent dans une thématique du laboratoire visant à appré-
hender le caractère multifactoriel de la néphrotoxicité des ICN par les approches non ciblées
de protéomique quantitative haut-débit, alors que les approches classiques de biologie mo-
léculaire, ciblées, ont montré leurs limites par l’impossibilité d’investigations à grande
échelle. Cette stratégie repose sur les performances des techniques de protéomique dite
« shotgun », couplant la séparation d’échantillons peptidiques par chromatographie liquide à
l’analyse par spectrométrie de masse.
Les premières études menées ont été le fruit de l’optimisation de la technologie SI-
LAC en vue de son application à l’analyse par nanoHPLC-MALDI-TOF/TOF des lysats cellu-
laires et des surnageants de culture de cellules HEK 293 après une exposition de 24 h à la
Cyclosporine A et au Tacrolimus. Pour la première fois, l’évolution du protéome de cellules
rénales soumises à des concentrations cliniques d’ICN a pu être observée, confirmant des
mécanismes physiopathologiques précédemment décrits tout en mettant en évidence des
profils de toxicité spécifiques à la CsA et au Tac ainsi que des mécanismes encore jamais
observés.
Afin d’approfondir les résultats prometteurs obtenus avec la technologie SILAC, la
stratégie a évolué vers l’optimisation de la technologie iTRAQ, présentant entre autres
l’avantage d’une analyse multiplexée. Cette transition s’est accompagnée d’une évolution du
modèle d’étude et de la plateforme d’analyse :
• les cellules rénales embryonnaires humaines HEK 293 ont été remplacées par
les cellules tubulaires proximales porcines LLC PK-1 plus adaptées à une
étude fonctionnelle grâce à leur degré de différentiation avancé
• l’analyse par nanoHPLC-MALDI-TOF/TOF a été mise à jour vers une analyse
nano-HPLC-QqTOF grâce à l’acquisition d’un spectromètre de masse Tri-
pleTOF 5600+ de haute résolution
La technologie iTRAQ a été optimisée puis appliquée à l’analyse du protéome de ly-
sats cellulaires extraits de cellules LLC PK-1 exposées 24 h à la CsA, au Tac et à un inhibi-
teur spécifique de l’interaction CaN-NFAT, le peptide VIVIT.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 134
IV.1.2. Problématique des outils de quantification
Le traitement bio-informatique des données MS/MS a d’abord été confié au logiciel
ProteinPilot qui intègre à la fois l’algorithme d’identification Paragon et l’algorithme de quanti-
fication iTRAQ basé sur la RPA. Bien que les performances d’identification de Paragon se
soient révélées satisfaisantes, ProteinPilot s’est montré particulièrement inadapté à la quanti-
fication iTRAQ à partir des données MS/MS générées par le TripleTOF 5600+, en introdui-
sant un biais systématique (Figure 15).
Figure 15. Représentation en heat-map de la quantification iTRAQ du sous-protéome
d’intérêt de LLC-PK-1 traitées 24 h par les ICN (iTRAQ-nanoLC-QqTOF, ProteinPilot-
Paragon-CiR-C).
Au contraire des utilisateurs de spectromètres de masse haute résolution de type Or-
bitrap (ThermoFisher Scientific) qui bénéficient d’un logiciel intégrant la quantification RSPI,
les utilisateurs de spectromètres de masse AB SCIEX doivent opter pour des outils alterna-
tifs (majoritairement open source) et fractionner le traitement bio-informatique en fonction
des capacités et des fonctionnalités de ces logiciels.
Au cours de ces travaux, le logiciel de quantification Quant a été choisi, parmi les dif-
férentes options disponibles, en raison de l’intégration de la quantification RSPI, de sa prise
en main et de son utilisation particulièrement simples, rapides et intuitives au travers de son
implémentation Java, jTRAQx.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 135
Malgré le fait que jTRAQx intègre des options de correction isotopique et de prise en
charge des peptides partagés, les données sortantes ne sont pas exploitables en l’état. La
quantification protéique ne peut pas être extrapolée à partir de la quantification peptidique en
raison de la persistance de données susceptibles d’introduire un biais dans la quantification
finale telles que des peptides identifiés avec une faible confiance ou des quantifications pep-
tidiques manquantes.
jTRAQx permet d’exclure manuellement des peptides des calculs de quantification
protéique mais, en raison de la taille des jeux de données, ce processus aurait été trop long
et sujet à erreur. Ainsi, le tri devait être informatisé et automatisé.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 136
IV.1.3. Evolution de l’algorithme CiR-C
L’algorithme CiR-C a d’abord été développé pour compléter jTRAQx et exclure les
données peptidiques aberrantes. Pour cette raison, le langage de l’interpréteur en ligne de
commande Bash (shell UnIx/GNU) a été choisi comme langage de programmation car il est
particulièrement adapté à la manipulation efficace de fichiers texte, de chaine de caractères,
de lignes et de colonnes par des scripts courts.
CiR-C est ensuite devenu un algorithme d’analyse et de quantification à part entière
avec les ajouts successifs :
• du classement des peptides selon leur fréquence afin de déterminer les pep-
tides identifiés et quantifiés dans l’ensemble des expériences indépendantes
(peptides de référence)
• du calcul d’un rapport médian, d’un rapport moyen pondéré et d’un écart-type
pour chaque protéine identifiée à partir de peptides de référence
• de la restriction des listes relatives à chaque expérience indépendante aux
seuls peptides de référence. Cette étape a été réalisée en prévision de la va-
lidation de la technique par Western blot et de la comparaison des quantifica-
tions relatives iTRAQ et WB par la méthode de Bland et Altman.
La dernière version développée avait pour objectif de préparer la transition vers les
outils d’interactomique en synchronisant, pour une protéine donnée, les données de quantifi-
cation iTRAQ avec les informations disponibles au sein de la base de données UniProt, en
particulier les informations de Gene-Ontology relatives aux fonctions biologiques. Le but était
de regrouper les protéines par fonction biologique afin d’observer les variations d’expression
sur un jeu restreint de protéines liées tout en identifiant les processus biologiques les plus
représentés susceptibles d’être également les plus impactés.
Classer selon les fonctions biologiques génère trop de redondances. Par exemple,
les protéines impliquées dans la dynamique du cytosquelette d’Actine ont été classées et
réparties en 22 fonctions biologiques au lieu d’une seule
L’amélioration future de CiR-C pour tendre vers la vision holistique de la biologie in-
tégrative repose sur trois points essentiels :
• définir une précision maximale dans la définition des fonctions biologiques
synchronisées afin de ne pas diluer l’information
• étendre la synchronisation à d’autres bases de données
• développer un outil de visualisation des résultats

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 137
IV.1.4. Interprétation interactomique et évolution vers la biologie des systèmes
La protéomique quantitative iTRAQ se définit comme un criblage à haut-débit à
l’échelle du protéome entier. Parallèlement aux travaux visant à doter CiR-C de fonctionnali-
tés interactomiques, la liste de protéines identifiées a été analysée grâce à des outils bio-
informatiques déjà existants : DAVID, STRING et le système de classification PANTHER.
Ces outils en ligne travaillent à partir d’une simple liste d’identifiants d’accession Uni-
Prot. Aucun ne possède une panoplie exhaustive de fonctionnalités, aussi il est impératif de
les utiliser de manière complémentaire pour en tirer l’analyse la plus complète possible.
DAVID et PANTHER reposent sur des algorithmes capables de classer les protéines
de manière statistique selon
STRING permet la visualisation 2D des réseaux d’interaction moléculaire et peut être
comparé à une version simplifiée de Cytoscape. Les réseaux générés sont structurés en
nœuds (protéines) et liens (interactions).
La principale limite de ces outils est l’absence de représentation des données de
quantification. Il est possible d’avoir une vision intégrée des mécanismes et voies de signali-
sation impactés mais il est impossible de conclure quant à la manière dont ils sont impactés.
L’utilisateur dispose de cartes figées hors de tout contexte de variation d’abondance pro-
téique.
A l’inverse, des logiciels tels que GENE-E sont capables de générer des heat-maps
résumant les variations d’expression d’une liste de protéines donnée en dehors de tout con-
texte de fonction et interaction biologiques.
L’enjeu est donc d’utiliser ces outils de manière complémentaire afin d’aboutir à une
cartographie dynamique du protéome d’intérêt

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 138
IV.1.5. Détermination des variations biologiques significatives
L’analyse des variations d’abondance protéique nécessite la définition de valeurs
seuils qui permettent de discriminer les écarts significatifs par rapport à la condition de réfé-
rence des fluctuations non significatives. Ces valeurs seuils peuvent être fixées arbitraire-
ment selon une estimation subjective de la qualité du jeu de données. La stratégie choisie
s’est appuyée sur la recherche d’un critère objectif défini par une approche statistique adap-
tées de la stratégie déjà appliquée dans le laboratoire pour la détermination des valeurs
seuils en quantification SILAC. La distribution du nombre de protéines en fonction de la va-
leur des ratios iTRAQ peut être approchée par une loi normale de moyenne m (≈1) et
d’écart-type σ (par régression non linéaire). Les propriétés de la loi normale énoncent que
les valeurs sont réparties comme suit :
• 68 % dans l'intervalle [m − σ ; m + σ]
• 95 % dans l'intervalle [m − 2 σ ; m + 2 σ]
• 99,7 % dans l'intervalle [m − 3 σ ; m + 3 σ]
Sous réserve que la loi normale soit approximativement centrée sur 1, les protéines
dont les abondances relatives correspondent à des rapports iTRAQ supérieurs à m + σ ou
inférieurs à m − σ sont considérées comme étant significativement surexprimées ou sous-
exprimées.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 139
IV.1.6. Résultats préliminaires : application de la technologie iTRAQ à l’étude du pro-
téome de cellules tubulaires proximales exposées aux ICN
L’application de la technologie iTRAQ à l’analyse du protéome de lysats cellulaires
extraits de cellules LLC PK-1 exposées 24 h à la CsA, au Tac et au VIVIT a permis de quan-
tifier simultanément les abondances protéiques relatives de 130 protéines (Tableaux 2 et 3).
Les résultats interactomiques préliminaires indiquent que les ICN perturbent majori-
tairement l’expression de protéines impliquées dans la transcription, la traduction, la matura-
tion et les modifications post-traductionnelles, l’homéostasie électrochimique, le cycle cellu-
laire, l’autophagie, l’apoptose, les métabolismes glucidique, lipidique, des nucléotides et des
acides aminés, le stress du réticulum endoplasmique, le stress oxydant et l’organisation du
cytosquelette d’Actine (Figures 16 et 18).
L’analyse des ratios iTRAQ a permis de classer les protéines selon 8 profils
d’expression (Tableau 2, Figure 17) :
• Sans effet
• ICN-VIVIT
• CsA-Tac
• CsA seule
• Tac seul
• CsA-VIVIT
• Tac-VIVIT
• VIVIT seul
La confrontation des résultats doit permettre d’établir une première cartographie dy-
namique du protéome des cellules tubulaires proximales exposées aux ICN.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 140
Figure 16. Réseau STRING des 130 protéines identifiées et quantifiées au cours de l’analyse
du protéome de cellules LLC PK-1 exposées 24 h aux ICN (iTRAQ-nanoLC-QqTOF, Mascot-
jTRAQx-CiR-C).
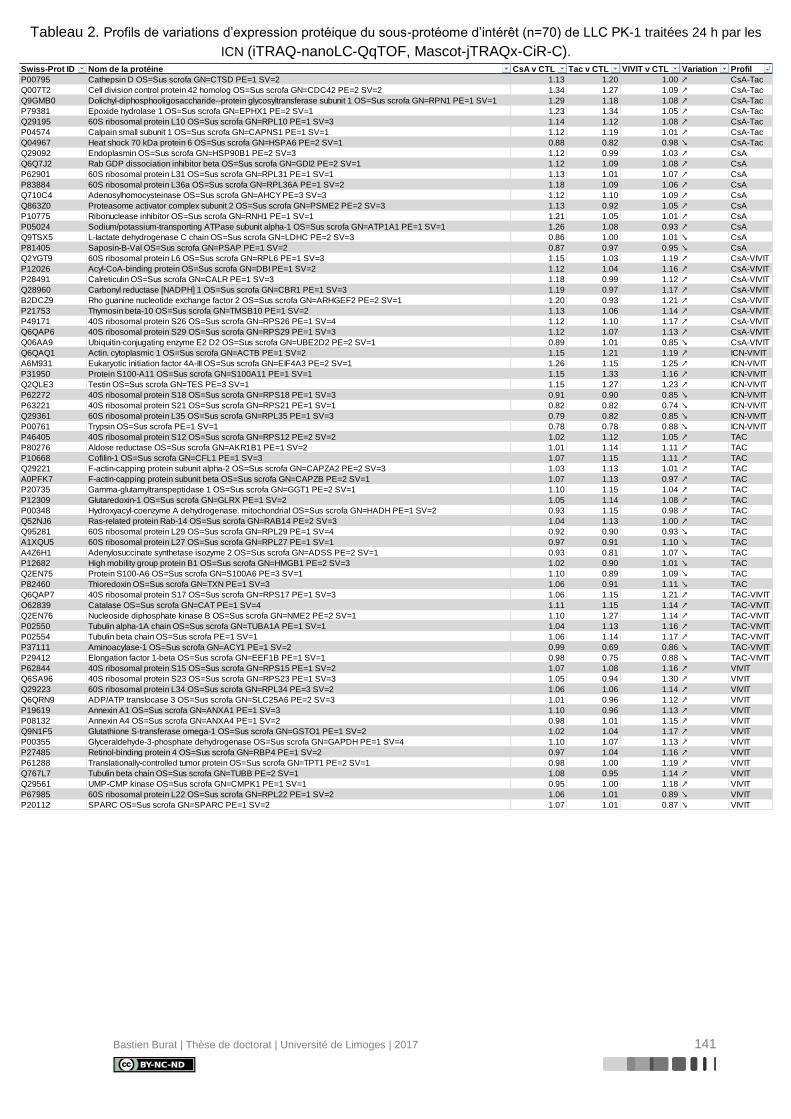
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 141
Swiss-Prot ID Nom de la protéine CsA v CTL Tac v CTL VIVIT v CTL Variation Profil
P00795 Cathepsin D OS=Sus scrofa GN=CTSD PE=1 SV=2 1.13 1.20 1.00 ↗ CsA-Tac
Q007T2 Cell division control protein 42 homolog OS=Sus scrofa GN=CDC42 PE=2 SV=2 1.34 1.27 1.09 ↗ CsA-Tac
Q9GMB0 Dolichyl-diphosphooligosaccharide--protein glycosyltransferase subunit 1 OS=Sus scrofa GN=RPN1 PE=1 SV=1 1.29 1.18 1.08 ↗ CsA-Tac
P79381 Epoxide hydrolase 1 OS=Sus scrofa GN=EPHX1 PE=2 SV=1 1.23 1.34 1.05 ↗ CsA-Tac
Q29195 60S ribosomal protein L10 OS=Sus scrofa GN=RPL10 PE=1 SV=3 1.14 1.12 1.08 ↗ CsA-Tac
P04574 Calpain small subunit 1 OS=Sus scrofa GN=CAPNS1 PE=1 SV=1 1.12 1.19 1.01 ↗ CsA-Tac
Q04967 Heat shock 70 kDa protein 6 OS=Sus scrofa GN=HSPA6 PE=2 SV=1 0.88 0.82 0.98 ↘ CsA-Tac
Q29092 Endoplasmin OS=Sus scrofa GN=HSP90B1 PE=2 SV=3 1.12 0.99 1.03 ↗ CsA
Q6Q7J2 Rab GDP dissociation inhibitor beta OS=Sus scrofa GN=GDI2 PE=2 SV=1 1.12 1.09 1.08 ↗ CsA
P62901 60S ribosomal protein L31 OS=Sus scrofa GN=RPL31 PE=1 SV=1 1.13 1.01 1.07 ↗ CsA
P83884 60S ribosomal protein L36a OS=Sus scrofa GN=RPL36A PE=1 SV=2 1.18 1.09 1.06 ↗ CsA
Q710C4 Adenosylhomocysteinase OS=Sus scrofa GN=AHCY PE=3 SV=3 1.12 1.10 1.09 ↗ CsA
Q863Z0 Proteasome activator complex subunit 2 OS=Sus scrofa GN=PSME2 PE=2 SV=3 1.13 0.92 1.05 ↗ CsA
P10775 Ribonuclease inhibitor OS=Sus scrofa GN=RNH1 PE=1 SV=1 1.21 1.05 1.01 ↗ CsA
P05024 Sodium/potassium-transporting ATPase subunit alpha-1 OS=Sus scrofa GN=ATP1A1 PE=1 SV=1 1.26 1.08 0.93 ↗ CsA
Q9TSX5 L-lactate dehydrogenase C chain OS=Sus scrofa GN=LDHC PE=2 SV=3 0.86 1.00 1.01 ↘ CsA
P81405 Saposin-B-Val OS=Sus scrofa GN=PSAP PE=1 SV=2 0.87 0.97 0.95 ↘ CsA
Q2YGT9 60S ribosomal protein L6 OS=Sus scrofa GN=RPL6 PE=1 SV=3 1.15 1.03 1.19 ↗ CsA-VIVIT
P12026 Acyl-CoA-binding protein OS=Sus scrofa GN=DBI PE=1 SV=2 1.12 1.04 1.16 ↗ CsA-VIVIT
P28491 Calreticulin OS=Sus scrofa GN=CALR PE=1 SV=3 1.18 0.99 1.12 ↗ CsA-VIVIT
Q28960 Carbonyl reductase [NADPH] 1 OS=Sus scrofa GN=CBR1 PE=1 SV=3 1.19 0.97 1.17 ↗ CsA-VIVIT
B2DCZ9 Rho guanine nucleotide exchange factor 2 OS=Sus scrofa GN=ARHGEF2 PE=2 SV=1 1.20 0.93 1.21 ↗ CsA-VIVIT
P21753 Thymosin beta-10 OS=Sus scrofa GN=TMSB10 PE=1 SV=2 1.13 1.06 1.14 ↗ CsA-VIVIT
P49171 40S ribosomal protein S26 OS=Sus scrofa GN=RPS26 PE=1 SV=4 1.12 1.10 1.17 ↗ CsA-VIVIT
Q6QAP6 40S ribosomal protein S29 OS=Sus scrofa GN=RPS29 PE=1 SV=3 1.12 1.07 1.13 ↗ CsA-VIVIT
Q06AA9 Ubiquitin-conjugating enzyme E2 D2 OS=Sus scrofa GN=UBE2D2 PE=2 SV=1 0.89 1.01 0.85 ↘ CsA-VIVIT
Q6QAQ1 Actin. cytoplasmic 1 OS=Sus scrofa GN=ACTB PE=1 SV=2 1.15 1.21 1.19 ↗ ICN-VIVIT
A6M931 Eukaryotic initiation factor 4A-III OS=Sus scrofa GN=EIF4A3 PE=2 SV=1 1.26 1.15 1.25 ↗ ICN-VIVIT
P31950 Protein S100-A11 OS=Sus scrofa GN=S100A11 PE=1 SV=1 1.15 1.33 1.16 ↗ ICN-VIVIT
Q2QLE3 Testin OS=Sus scrofa GN=TES PE=3 SV=1 1.15 1.27 1.23 ↗ ICN-VIVIT
P62272 40S ribosomal protein S18 OS=Sus scrofa GN=RPS18 PE=1 SV=3 0.91 0.90 0.85 ↘ ICN-VIVIT
P63221 40S ribosomal protein S21 OS=Sus scrofa GN=RPS21 PE=1 SV=1 0.82 0.82 0.74 ↘ ICN-VIVIT
Q29361 60S ribosomal protein L35 OS=Sus scrofa GN=RPL35 PE=1 SV=3 0.79 0.82 0.85 ↘ ICN-VIVIT
P00761 Trypsin OS=Sus scrofa PE=1 SV=1 0.78 0.78 0.88 ↘ ICN-VIVIT
P46405 40S ribosomal protein S12 OS=Sus scrofa GN=RPS12 PE=2 SV=2 1.02 1.12 1.05 ↗ TAC
P80276 Aldose reductase OS=Sus scrofa GN=AKR1B1 PE=1 SV=2 1.01 1.14 1.11 ↗ TAC
P10668 Cofilin-1 OS=Sus scrofa GN=CFL1 PE=1 SV=3 1.07 1.15 1.11 ↗ TAC
Q29221 F-actin-capping protein subunit alpha-2 OS=Sus scrofa GN=CAPZA2 PE=2 SV=3 1.03 1.13 1.01 ↗ TAC
A0PFK7 F-actin-capping protein subunit beta OS=Sus scrofa GN=CAPZB PE=2 SV=1 1.07 1.13 0.97 ↗ TAC
P20735 Gamma-glutamyltranspeptidase 1 OS=Sus scrofa GN=GGT1 PE=2 SV=1 1.10 1.15 1.04 ↗ TAC
P12309 Glutaredoxin-1 OS=Sus scrofa GN=GLRX PE=1 SV=2 1.05 1.14 1.08 ↗ TAC
P00348 Hydroxyacyl-coenzyme A dehydrogenase. mitochondrial OS=Sus scrofa GN=HADH PE=1 SV=2 0.93 1.15 0.98 ↗ TAC
Q52NJ6 Ras-related protein Rab-14 OS=Sus scrofa GN=RAB14 PE=2 SV=3 1.04 1.13 1.00 ↗ TAC
Q95281 60S ribosomal protein L29 OS=Sus scrofa GN=RPL29 PE=1 SV=4 0.92 0.90 0.93 ↘ TAC
A1XQU5 60S ribosomal protein L27 OS=Sus scrofa GN=RPL27 PE=1 SV=1 0.97 0.91 1.10 ↘ TAC
A4Z6H1 Adenylosuccinate synthetase isozyme 2 OS=Sus scrofa GN=ADSS PE=2 SV=1 0.93 0.81 1.07 ↘ TAC
P12682 High mobility group protein B1 OS=Sus scrofa GN=HMGB1 PE=2 SV=3 1.02 0.90 1.01 ↘ TAC
Q2EN75 Protein S100-A6 OS=Sus scrofa GN=S100A6 PE=3 SV=1 1.10 0.89 1.09 ↘ TAC
P82460 Thioredoxin OS=Sus scrofa GN=TXN PE=1 SV=3 1.06 0.91 1.11 ↘ TAC
Q6QAP7 40S ribosomal protein S17 OS=Sus scrofa GN=RPS17 PE=1 SV=3 1.06 1.15 1.21 ↗ TAC-VIVIT
O62839 Catalase OS=Sus scrofa GN=CAT PE=1 SV=4 1.11 1.15 1.14 ↗ TAC-VIVIT
Q2EN76 Nucleoside diphosphate kinase B OS=Sus scrofa GN=NME2 PE=2 SV=1 1.10 1.27 1.14 ↗ TAC-VIVIT
P02550 Tubulin alpha-1A chain OS=Sus scrofa GN=TUBA1A PE=1 SV=1 1.04 1.13 1.16 ↗ TAC-VIVIT
P02554 Tubulin beta chain OS=Sus scrofa PE=1 SV=1 1.06 1.14 1.17 ↗ TAC-VIVIT
P37111 Aminoacylase-1 OS=Sus scrofa GN=ACY1 PE=1 SV=2 0.99 0.69 0.86 ↘ TAC-VIVIT
P29412 Elongation factor 1-beta OS=Sus scrofa GN=EEF1B PE=1 SV=1 0.98 0.75 0.88 ↘ TAC-VIVIT
P62844 40S ribosomal protein S15 OS=Sus scrofa GN=RPS15 PE=1 SV=2 1.07 1.08 1.16 ↗ VIVIT
Q6SA96 40S ribosomal protein S23 OS=Sus scrofa GN=RPS23 PE=1 SV=3 1.05 0.94 1.30 ↗ VIVIT
Q29223 60S ribosomal protein L34 OS=Sus scrofa GN=RPL34 PE=3 SV=2 1.06 1.06 1.14 ↗ VIVIT
Q6QRN9 ADP/ATP translocase 3 OS=Sus scrofa GN=SLC25A6 PE=2 SV=3 1.01 0.96 1.12 ↗ VIVIT
P19619 Annexin A1 OS=Sus scrofa GN=ANXA1 PE=1 SV=3 1.10 0.96 1.13 ↗ VIVIT
P08132 Annexin A4 OS=Sus scrofa GN=ANXA4 PE=1 SV=2 0.98 1.01 1.15 ↗ VIVIT
Q9N1F5 Glutathione S-transferase omega-1 OS=Sus scrofa GN=GSTO1 PE=1 SV=2 1.02 1.04 1.17 ↗ VIVIT
P00355 Glyceraldehyde-3-phosphate dehydrogenase OS=Sus scrofa GN=GAPDH PE=1 SV=4 1.10 1.07 1.13 ↗ VIVIT
P27485 Retinol-binding protein 4 OS=Sus scrofa GN=RBP4 PE=1 SV=2 0.97 1.04 1.16 ↗ VIVIT
P61288 Translationally-controlled tumor protein OS=Sus scrofa GN=TPT1 PE=2 SV=1 0.98 1.00 1.19 ↗ VIVIT
Q767L7 Tubulin beta chain OS=Sus scrofa GN=TUBB PE=2 SV=1 1.08 0.95 1.14 ↗ VIVIT
Q29561 UMP-CMP kinase OS=Sus scrofa GN=CMPK1 PE=1 SV=1 0.95 1.00 1.18 ↗ VIVIT
P67985 60S ribosomal protein L22 OS=Sus scrofa GN=RPL22 PE=1 SV=2 1.06 1.01 0.89 ↘ VIVIT
P20112 SPARC OS=Sus scrofa GN=SPARC PE=1 SV=2 1.07 1.01 0.87 ↘ VIVIT
Tableau 2. Profils de variations d’expression protéique du sous-protéome d’intérêt (n=70) de LLC PK-1 traitées 24 h par les
ICN (iTRAQ-nanoLC-QqTOF, Mascot-jTRAQx-CiR-C).
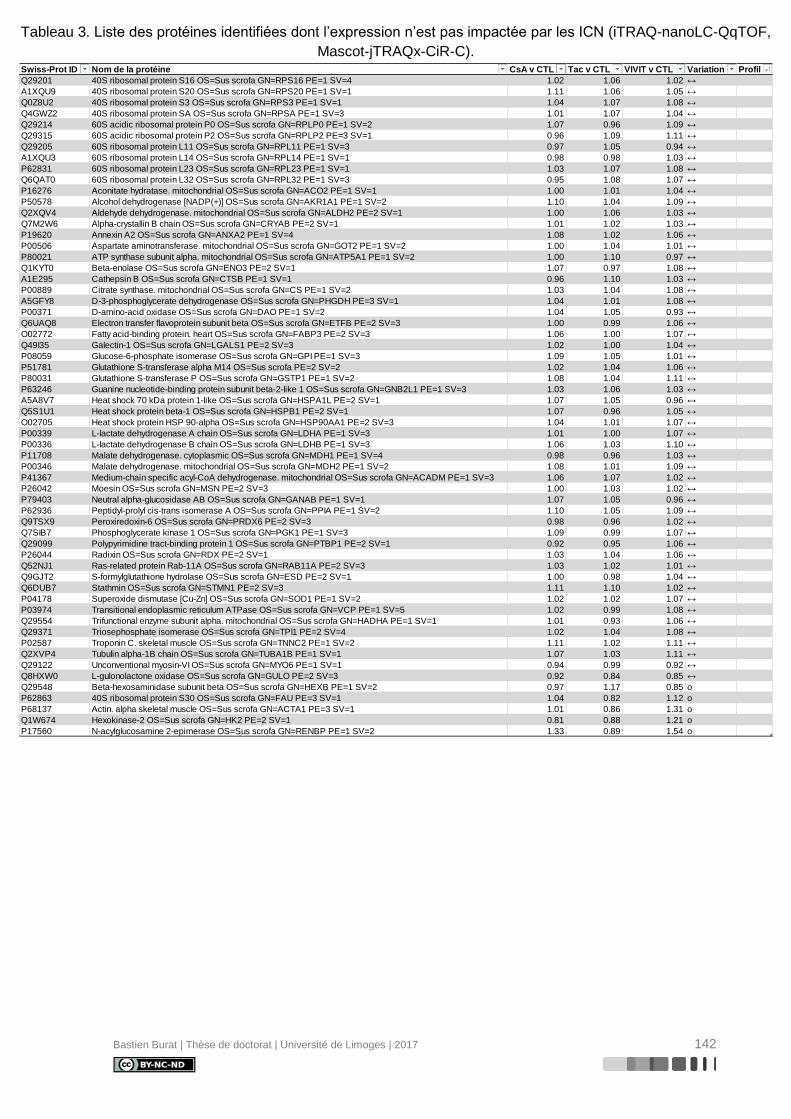
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 142
Swiss-Prot ID Nom de la protéine CsA v CTL Tac v CTL VIVIT v CTL Variation Profil
Q29201 40S ribosomal protein S16 OS=Sus scrofa GN=RPS16 PE=1 SV=4 1.02 1.06 1.02 ↔
A1XQU9 40S ribosomal protein S20 OS=Sus scrofa GN=RPS20 PE=1 SV=1 1.11 1.06 1.05 ↔
Q0Z8U2 40S ribosomal protein S3 OS=Sus scrofa GN=RPS3 PE=1 SV=1 1.04 1.07 1.08 ↔
Q4GWZ2 40S ribosomal protein SA OS=Sus scrofa GN=RPSA PE=1 SV=3 1.01 1.07 1.04 ↔
Q29214 60S acidic ribosomal protein P0 OS=Sus scrofa GN=RPLP0 PE=1 SV=2 1.07 0.96 1.09 ↔
Q29315 60S acidic ribosomal protein P2 OS=Sus scrofa GN=RPLP2 PE=3 SV=1 0.96 1.09 1.11 ↔
Q29205 60S ribosomal protein L11 OS=Sus scrofa GN=RPL11 PE=1 SV=3 0.97 1.05 0.94 ↔
A1XQU3 60S ribosomal protein L14 OS=Sus scrofa GN=RPL14 PE=1 SV=1 0.98 0.98 1.03 ↔
P62831 60S ribosomal protein L23 OS=Sus scrofa GN=RPL23 PE=1 SV=1 1.03 1.07 1.08 ↔
Q6QAT0 60S ribosomal protein L32 OS=Sus scrofa GN=RPL32 PE=1 SV=3 0.95 1.08 1.07 ↔
P16276 Aconitate hydratase. mitochondrial OS=Sus scrofa GN=ACO2 PE=1 SV=1 1.00 1.01 1.04 ↔
P50578 Alcohol dehydrogenase [NADP(+)] OS=Sus scrofa GN=AKR1A1 PE=1 SV=2 1.10 1.04 1.09 ↔
Q2XQV4 Aldehyde dehydrogenase. mitochondrial OS=Sus scrofa GN=ALDH2 PE=2 SV=1 1.00 1.06 1.03 ↔
Q7M2W6 Alpha-crystallin B chain OS=Sus scrofa GN=CRYAB PE=2 SV=1 1.01 1.02 1.03 ↔
P19620 Annexin A2 OS=Sus scrofa GN=ANXA2 PE=1 SV=4 1.08 1.02 1.06 ↔
P00506 Aspartate aminotransferase. mitochondrial OS=Sus scrofa GN=GOT2 PE=1 SV=2 1.00 1.04 1.01 ↔
P80021 ATP synthase subunit alpha. mitochondrial OS=Sus scrofa GN=ATP5A1 PE=1 SV=2 1.00 1.10 0.97 ↔
Q1KYT0 Beta-enolase OS=Sus scrofa GN=ENO3 PE=2 SV=1 1.07 0.97 1.08 ↔
A1E295 Cathepsin B OS=Sus scrofa GN=CTSB PE=1 SV=1 0.96 1.10 1.03 ↔
P00889 Citrate synthase. mitochondrial OS=Sus scrofa GN=CS PE=1 SV=2 1.03 1.04 1.08 ↔
A5GFY8 D-3-phosphoglycerate dehydrogenase OS=Sus scrofa GN=PHGDH PE=3 SV=1 1.04 1.01 1.08 ↔
P00371 D-amino-acid oxidase OS=Sus scrofa GN=DAO PE=1 SV=2 1.04 1.05 0.93 ↔
Q6UAQ8 Electron transfer flavoprotein subunit beta OS=Sus scrofa GN=ETFB PE=2 SV=3 1.00 0.99 1.06 ↔
O02772 Fatty acid-binding protein. heart OS=Sus scrofa GN=FABP3 PE=2 SV=3 1.06 1.00 1.07 ↔
Q49I35 Galectin-1 OS=Sus scrofa GN=LGALS1 PE=2 SV=3 1.02 1.00 1.04 ↔
P08059 Glucose-6-phosphate isomerase OS=Sus scrofa GN=GPI PE=1 SV=3 1.09 1.05 1.01 ↔
P51781 Glutathione S-transferase alpha M14 OS=Sus scrofa PE=2 SV=2 1.02 1.04 1.06 ↔
P80031 Glutathione S-transferase P OS=Sus scrofa GN=GSTP1 PE=1 SV=2 1.08 1.04 1.11 ↔
P63246 Guanine nucleotide-binding protein subunit beta-2-like 1 OS=Sus scrofa GN=GNB2L1 PE=1 SV=3 1.03 1.06 1.03 ↔
A5A8V7 Heat shock 70 kDa protein 1-like OS=Sus scrofa GN=HSPA1L PE=2 SV=1 1.07 1.05 0.96 ↔
Q5S1U1 Heat shock protein beta-1 OS=Sus scrofa GN=HSPB1 PE=2 SV=1 1.07 0.96 1.05 ↔
O02705 Heat shock protein HSP 90-alpha OS=Sus scrofa GN=HSP90AA1 PE=2 SV=3 1.04 1.01 1.07 ↔
P00339 L-lactate dehydrogenase A chain OS=Sus scrofa GN=LDHA PE=1 SV=3 1.01 1.00 1.07 ↔
P00336 L-lactate dehydrogenase B chain OS=Sus scrofa GN=LDHB PE=1 SV=3 1.06 1.03 1.10 ↔
P11708 Malate dehydrogenase. cytoplasmic OS=Sus scrofa GN=MDH1 PE=1 SV=4 0.98 0.96 1.03 ↔
P00346 Malate dehydrogenase. mitochondrial OS=Sus scrofa GN=MDH2 PE=1 SV=2 1.08 1.01 1.09 ↔
P41367 Medium-chain specific acyl-CoA dehydrogenase. mitochondrial OS=Sus scrofa GN=ACADM PE=1 SV=3 1.06 1.07 1.02 ↔
P26042 Moesin OS=Sus scrofa GN=MSN PE=2 SV=3 1.00 1.03 1.02 ↔
P79403 Neutral alpha-glucosidase AB OS=Sus scrofa GN=GANAB PE=1 SV=1 1.07 1.05 0.96 ↔
P62936 Peptidyl-prolyl cis-trans isomerase A OS=Sus scrofa GN=PPIA PE=1 SV=2 1.10 1.05 1.09 ↔
Q9TSX9 Peroxiredoxin-6 OS=Sus scrofa GN=PRDX6 PE=2 SV=3 0.98 0.96 1.02 ↔
Q7SIB7 Phosphoglycerate kinase 1 OS=Sus scrofa GN=PGK1 PE=1 SV=3 1.09 0.99 1.07 ↔
Q29099 Polypyrimidine tract-binding protein 1 OS=Sus scrofa GN=PTBP1 PE=2 SV=1 0.92 0.95 1.06 ↔
P26044 Radixin OS=Sus scrofa GN=RDX PE=2 SV=1 1.03 1.04 1.06 ↔
Q52NJ1 Ras-related protein Rab-11A OS=Sus scrofa GN=RAB11A PE=2 SV=3 1.03 1.02 1.01 ↔
Q9GJT2 S-formylglutathione hydrolase OS=Sus scrofa GN=ESD PE=2 SV=1 1.00 0.98 1.04 ↔
Q6DUB7 Stathmin OS=Sus scrofa GN=STMN1 PE=2 SV=3 1.11 1.10 1.02 ↔
P04178 Superoxide dismutase [Cu-Zn] OS=Sus scrofa GN=SOD1 PE=1 SV=2 1.02 1.02 1.07 ↔
P03974 Transitional endoplasmic reticulum ATPase OS=Sus scrofa GN=VCP PE=1 SV=5 1.02 0.99 1.08 ↔
Q29554 Trifunctional enzyme subunit alpha. mitochondrial OS=Sus scrofa GN=HADHA PE=1 SV=1 1.01 0.93 1.06 ↔
Q29371 Triosephosphate isomerase OS=Sus scrofa GN=TPI1 PE=2 SV=4 1.02 1.04 1.08 ↔
P02587 Troponin C. skeletal muscle OS=Sus scrofa GN=TNNC2 PE=1 SV=2 1.11 1.02 1.11 ↔
Q2XVP4 Tubulin alpha-1B chain OS=Sus scrofa GN=TUBA1B PE=1 SV=1 1.07 1.03 1.11 ↔
Q29122 Unconventional myosin-VI OS=Sus scrofa GN=MYO6 PE=1 SV=1 0.94 0.99 0.92 ↔
Q8HXW0 L-gulonolactone oxidase OS=Sus scrofa GN=GULO PE=2 SV=3 0.92 0.84 0.85 ↔
Q29548 Beta-hexosaminidase subunit beta OS=Sus scrofa GN=HEXB PE=1 SV=2 0.97 1.17 0.85 o
P62863 40S ribosomal protein S30 OS=Sus scrofa GN=FAU PE=3 SV=1 1.04 0.82 1.12 o
P68137 Actin. alpha skeletal muscle OS=Sus scrofa GN=ACTA1 PE=3 SV=1 1.01 0.86 1.31 o
Q1W674 Hexokinase-2 OS=Sus scrofa GN=HK2 PE=2 SV=1 0.81 0.88 1.21 o
P17560 N-acylglucosamine 2-epimerase OS=Sus scrofa GN=RENBP PE=1 SV=2 1.33 0.89 1.54 o
Tableau 3. Liste des protéines identifiées dont l’expression n’est pas impactée par les ICN (iTRAQ-nanoLC-QqTOF,
Mascot-jTRAQx-CiR-C).
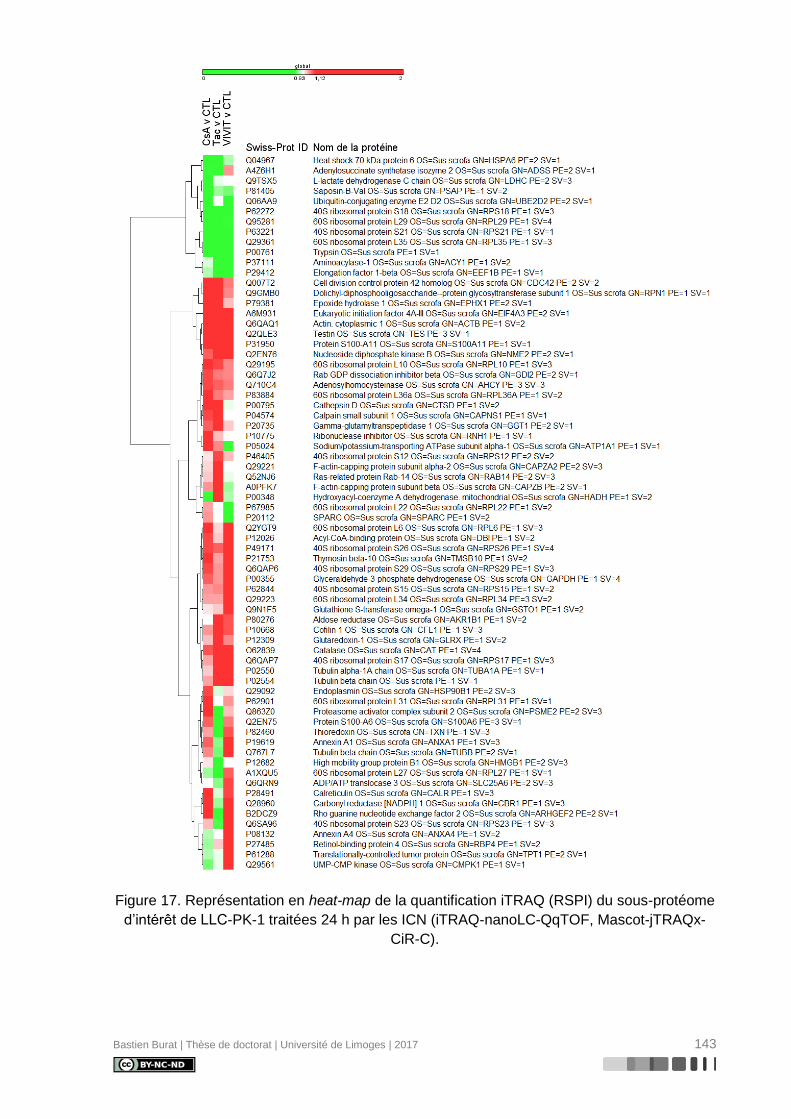
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 143
Figure 17. Représentation en heat-map de la quantification iTRAQ (RSPI) du sous-protéome
d’intérêt de LLC-PK-1 traitées 24 h par les ICN (iTRAQ-nanoLC-QqTOF, Mascot-jTRAQx-
CiR-C).
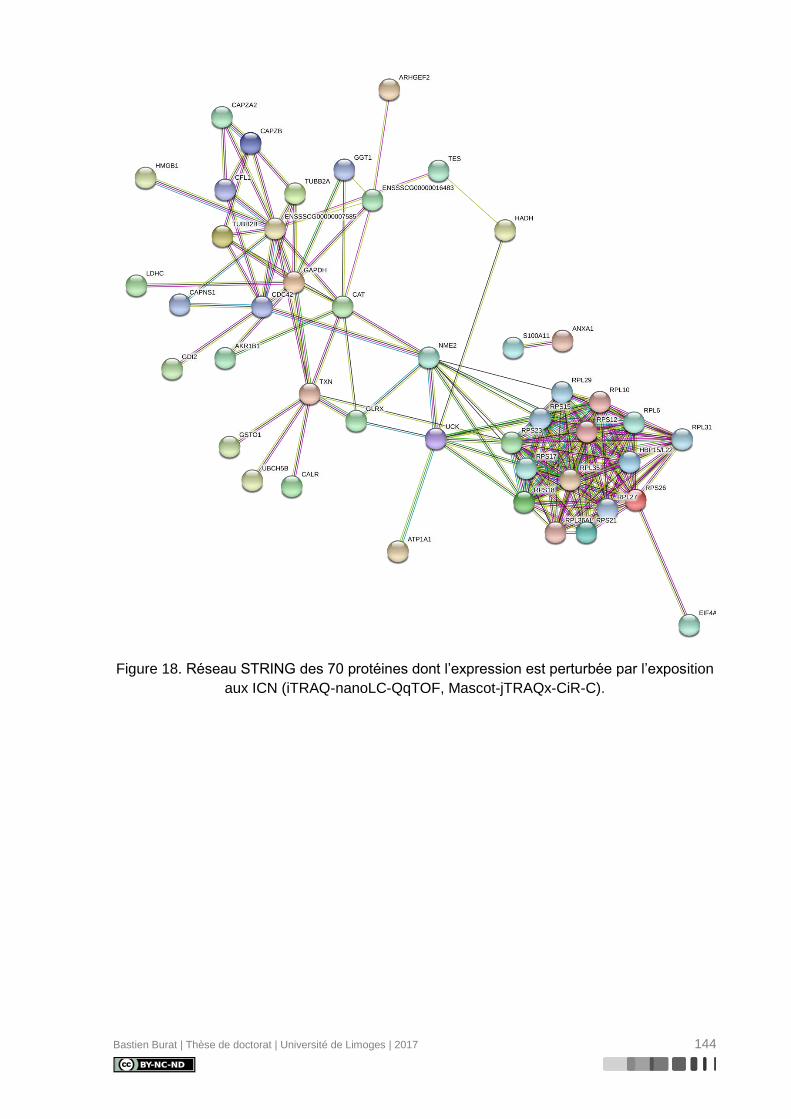
Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 144
Figure 18. Réseau STRING des 70 protéines dont l’expression est perturbée par l’exposition
aux ICN (iTRAQ-nanoLC-QqTOF, Mascot-jTRAQx-CiR-C).

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 145
IV.2. Perspectives
Le marquage iTRAQ étant réalisé au cours de la préparation d’échantillons, le do-
maine d’applications comprend virtuellement des échantillons de toute nature et de tout ori-
gine, pour des études protéomiques quantitatives in vitro, in vivo ou ex vivo, sous réserve
que l’extraction, la préparation et la digestion protéique soient efficaces.
En cela, l’iTRAQ n’est surpassé que par les techniques de LFQ.
La technologie iTRAQ a été optimisée pour une application à des cellules tubulaires
proximales, adhérentes, en culture. La transposition à l’analyse de cultures primaires de cel-
lules rénales exposées aux ICN in vitro ou in vivo, à l’analyse des prélèvements tissulaires
chez des animaux traités de manière chronique ou à l’analyse de biopsies de patients trans-
plantés ne nécessiterait pas d’ajustement majeur du protocole existant.
Par ailleurs, le protocole appliqué aux cellules en culture a été utilisé avec succès
pour l’analyse du protéome urinaire de patients transplantés rénaux (cf. Annexe 2.), sans
ajustement. De futures applications pourraient donc également consister en l’analyse des
surnageants de culture de cellules exposées aux ICN afin d’en étudier le sécrétome.
L’outil iTRAQ apparait donc comme particulièrement utile pour approfondir les con-
naissances sur les effets rénaux des ICN, dans une stratégie globale de mise au point de
nouveaux immunosuppresseurs dénués d’effets néphrotoxiques.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 146
Références bibliographiques
1. Hart A, Smith JM, Skeans MA, Gustafson SK, Stewart DE, Cherikh WS, et al. Kidney. Am J Transplant. janv 2016;16(S2):11‑46.
2. Hart A, Smith JM, Skeans MA, Gustafson SK, Stewart DE, Cherikh WS, et al. OPTN/SRTR 2015 Annual Data Report: Kidney. Am J Transplant. janv 2017;17:21‑116.
3. Borel JF, Feurer C, Gubler HU, Stähelin H. Biological effects of cyclosporin A: a new antilymphocytic agent. Agents Actions. juill 1976;6(4):468‑75.
4. Calne R., Thiru S, Mcmaster P, Craddock G., White DJ., Evans D., et al. CYCLOSPOR-IN A IN PATIENTS RECEIVING RENAL ALLOGRAFTS FROM CADAVER DONORS. The Lancet. déc 1978;312(8104):1323‑7.
5. Calne RY, Rolles K, Thiru S, Mcmaster P, Craddock GN, Aziz S, et al. CYCLOSPORIN A INITIALLY AS THE ONLY IMMUNOSUPPRESSANT IN 34 RECIPIENTS OF CADAVER-IC ORGANS: 32 KIDNEYS, 2 PANCREASES, AND 2 LIVERS. The Lancet. nov 1979;314(8151):1033‑6.
6. Kino T, Hatanaka H, Miyata S, Inamura N, Nishiyama M, Yajima T, et al. FK-506, a novel immunosuppressant isolated from a Streptomyces. II. Immunosuppressive effect of FK-506 in vitro. J Antibiot (Tokyo). 1987;40(9):1256‑65.
7. Starzl T, Fung J, Venkataramman R, Todo S, Demetris A, Jain A. FK 506 FOR LIVER, KIDNEY, AND PANCREAS TRANSPLANTATION. The Lancet. oct 1989;334(8670):1000‑4.
8. Hogan PG. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 15 sept 2003;17(18):2205‑32.
9. Li H, Rao A, Hogan PG. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. févr 2011;21(2):91‑103.
10. Malek TR, Castro I. Interleukin-2 Receptor Signaling: At the Interface between Tolerance and Immunity. Immunity. août 2010;33(2):153‑65.
11. Schreiber S. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science. 18 janv 1991;251(4991):283‑7.
12. Liu J, Farmer JD, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. août 1991;66(4):807‑15.
13. Liu J, Albers MW, Wandless TJ, Luan S, Alberg DG, Belshaw PJ, et al. Inhibition of T cell signaling by immunophilin-ligand complexes correlates with loss of calcineurin phospha-tase activity. Biochemistry (Mosc). 28 avr 1992;31(16):3896‑901.
14. Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, Fitzgibbon MJ, et al. X-ray struc-ture of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell. août 1995;82(3):507‑22.
15. Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, et al. Crys-tal structures of human calcineurin and the human FKBP12–FK506–calcineurin complex. Nature. déc 1995;378(6557):641‑4.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 147
16. McCaffrey PG, Perrino BA, Soderling TR, Rao A. NF-ATp, a T lymphocyte DNA-binding protein that is a target for calcineurin and immunosuppressive drugs. J Biol Chem. 15 févr 1993;268(5):3747‑52.
17. Gaston RS. Chronic Calcineurin Inhibitor Nephrotoxicity: Reflections on an Evolving Par-adigm. Clin J Am Soc Nephrol. 1 déc 2009;4(12):2029‑34.
18. Naesens M, Kuypers DRJ, Sarwal M. Calcineurin Inhibitor Nephrotoxicity. Clin J Am Soc Nephrol [Internet]. 18 févr 2009 [cité 31 mars 2017]; Disponible sur: http://cjasn.asnjournals.org/cgi/doi/10.2215/CJN.04800908
19. Myers BD, Ross J, Newton L, Luetscher J, Perlroth M. Cyclosporine-Associated Chronic Nephropathy. N Engl J Med. 13 sept 1984;311(11):699‑705.
20. Murray BM, Paller MS, Ferris TF. Effect of cyclosporine administration on renal hemody-namics in conscious rats. Kidney Int. nov 1985;28(5):767‑74.
21. English J, Evan A, Houghton DC, Bennett WM. Cyclosporine-induced acute renal dys-function in the rat. Evidence of arteriolar vasoconstriction with preservation of tubular function. Transplantation. juill 1987;44(1):135‑41.
22. Textor SC, Burnett JC, Romero JC, Canzanello VJ, Taler SJ, Wiesner R, et al. Urinary endothelin and renal vasoconstriction with cyclosporine or FK506 after liver transplanta-tion. Kidney Int. mai 1995;47(5):1426‑33.
23. Nakahama H. Stimulatory effect of cyclosporine A on endothelin secretion by a cultured renal epithelial cell line, LLC-PK1 cells. Eur J Pharmacol. mai 1990;180(1):191‑2.
24. Ramírez C, Olmo A, O’Valle F, Masseroli M, Aguilar M, Gómez-Morales M, et al. Role of intrarenal endothelin 1, endothelin 3, and angiotensin II expression in chronic cyclosporin A nephrotoxicity in rats. Exp Nephrol. juin 2000;8(3):161‑72.
25. HORTELANO S, CASTILLA M, TORRES AM, TEJEDOR A, BOSCÁ L. Potentiation by Nitric Oxide of Cyclosporin A and FK506-Induced Apoptosis in Renal Proximal Tubule Cells. J Am Soc Nephrol. 1 déc 2000;11(12):2315‑23.
26. Zhong Z, Arteel GE, Connor HD, Yin M, Frankenberg MV, Stachlewitz RF, et al. Cyclo-sporin A increases hypoxia and free radical production in rat kidneys: prevention by die-tary glycine. Am J Physiol - Ren Physiol. 1 oct 1998;275(4):F595.
27. Diederich D, Skopec J, Diederich A, Dai FX. Cyclosporine produces endothelial dysfunc-tion by increased production of superoxide. Hypertension. 1 juin 1994;23(6_Pt_2):957‑61.
28. Navarro-Antolín J, López-Muñoz MJ, Klatt P, Soria J, Michel T, Lamas S. Formation of peroxynitrite in vascular endothelial cells exposed to cyclosporine A. FASEB J [Internet]. 20 mars 2001; Disponible sur: http://www.fasebj.org/content/early/2001/05/02/fj.00-0636fje.short
29. Kurtz A, Bruna RD, Kühn K. Cyclosporine A enhances renin secretion and production in isolated juxtaglomerular cells. Kidney Int. mai 1988;33(5):947‑53.
30. Lassila M. Interaction of Cyclosporine A and the Renin-Angiotensin System New Per-spectives. Curr Drug Metab. 1 févr 2002;3(1):61‑71.
31. Mihatsch MJ, Thiel G, Basler V, Ryffel B, Landmann J, von Overbeck J, et al. Morpholog-ical patterns in cyclosporine-treated renal transplant recipients. Transplant Proc. août 1985;17(4 Suppl 1):101‑16.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 148
32. Mihatsch MJ, Thiel G, Ryffel B. Morphology of ciclosporin nephropathy. Prog Allergy. 1986;38:447‑65.
33. Mihatsch MJ, Ryffel B, Hermle M, Brunner FP, Thiel G. Morphology of cyclosporine ne-phrotoxicity in the rat. Clin Nephrol. 1986;25 Suppl 1:S2-8.
34. Mihatsch MJ, Thiel G, Ryffel B. Brief review of the morphology of ciclosporin nephropa-thy. Contrib Nephrol. 1986;51:156‑61.
35. Mihatsch MJ, Kyo M, Morozumi K, Yamaguchi Y, Nickeleit V, Ryffel B. The side-effects of ciclosporine-A and tacrolimus. Clin Nephrol. juin 1998;49(6):356‑63.
36. Randhawa PS, Shapiro R, Jordan ML, Starzl TE, Demetris AJ. The histopathological changes associated with allograft rejection and drug toxicity in renal transplant recipients maintained on FK506. Clinical significance and comparison with cyclosporine. Am J Surg Pathol. janv 1993;17(1):60‑8.
37. Kim JY, Suh KS. Light microscopic and electron microscopic features of cyclosporine nephrotoxicity in rats. J Korean Med Sci. 1995;10(5):352.
38. Morozumi K, Takeda A, Uchida K, Mihatsch M. Cyclosporine nephrotoxicity: how does it affect renal allograft function and transplant morphology? Transplant Proc. mars 2004;36(2):S251‑6.
39. Heyman SN, Brezis M, Reubinoff CA, Greenfeld Z, Lechene C, Epstein FH, et al. Acute renal failure with selective medullary injury in the rat. J Clin Invest. 1 août 1988;82(2):401‑12.
40. Mihatsch MJ, Basler V, Curschellas E, Kyo M, Duerig M, Huser B, et al. Giant Mitochon-dria in “Zero-hour” Transplant Biopsies. Ultrastruct Pathol. 10 juill 2009;16:277‑82.
41. Lee HT. A1 adenosine receptor knockout mice are protected against acute radiocontrast nephropathy in vivo. AJP Ren Physiol. 1 juin 2006;290(6):F1367‑75.
42. Haas M, Sonnenday CJ, Cicone JS, Rabb H, Montgomery RA. Isometric Tubular Epithe-lial Vacuolization in Renal Allograft Biopsy Specimens of Patients Receiving Low-Dose Intravenous Immunoglobulin for a Positive Crossmatch: Transplantation. août 2004;78(4):549‑56.
43. Bollee G, Anglicheau D, Loupy A, Zuber J, Patey N, Gregor DM, et al. High-Dosage In-travenous Immunoglobulin-Associated Macrovacuoles Are Associated with Chronic Tu-bulointerstitial Lesion Worsening in Renal Transplant Recipients. Clin J Am Soc Nephrol. 30 juill 2008;3(5):1461‑8.
44. Naesens M, Lerut E, Damme BV, Vanrenterghem Y, Kuypers DRJ. Tacrolimus Exposure and Evolution of Renal Allograft Histology in the First Year After Transplantation. Am J Transplant. sept 2007;7(9):2114‑23.
45. Naesens M, Kambham N, Concepcion W, Salvatierra O, Sarwal M. The Evolution of Nonimmune Histological Injury and Its Clinical Relevance in Adult-Sized Kidney Grafts in Pediatric Recipients. Am J Transplant. nov 2007;7(11):2504‑14.
46. Klintmalm GB., Iwatsuki S, Starzl T. NEPHROTOXICITY OF CYCLOSPORIN A IN LIV-ER AND KIDNEY TRANSPLANT PATIENTS. The Lancet. févr 1981;317(8218):470‑1.
47. Morris PJ, French ME, Dunnill MS, Hunnisett AG, Ting A, Thompson JF, et al. A con-trolled trial of cyclosporine in renal transplantation with conversion to azathioprine and prednisolone after three months. Transplantation. sept 1983;36(3):273‑7.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 149
48. Nankivell BJ, Borrows RJ, Fung CL-S, O’Connell PJ, Allen RDM, Chapman JR. The Nat-ural History of Chronic Allograft Nephropathy. N Engl J Med. 11 déc 2003;349(24):2326‑33.
49. Nankivell BJ, Borrows RJ, Fung CL-S, OʼConnell PJ, Chapman JR, Allen RDM. Cal-cineurin Inhibitor Nephrotoxicity: Longitudinal Assessment by Protocol Histology: Trans-plantation. août 2004;78(4):557‑65.
50. Nankivell BJ, Borrows RJ, Fung CLS, O???Connell PJ, Chapman JR, Allen RDM. Delta Analysis of Posttransplantation Tubulointerstitial Damage: Transplantation. août 2004;78(3):434‑41.
51. Nankivell BJ, Borrows RJ, Fung CLS, O???Connell PJ, Allen RDM, Chapman JR. Evolu-tion and Pathophysiology of Renal-Transplant Glomerulosclerosis: Transplantation. août 2004;78(3):461‑8.
52. Young BA, Burdmann EA, Johnson RJ, Andoh T, Bennett WM, Couser WG, et al. Cyclo-sporine A induced arteriolopathy in a rat model of chronic cyclosporine nephropathy. Kidney Int. août 1995;48(2):431‑8.
53. Young BA, Burdmann EA, Johnson RJ, Alpers CE, Giachelli CM, Eng E, et al. Cellular proliferation and macrophage influx precede interstitial fibrosis in cyclosporine nephrotox-icity. Kidney Int. août 1995;48(2):439‑48.
54. Horike K, Takeda A, Yamaguchi Y, Ogiyama Y, Yamauchi Y, Murata M, et al. Is arteriolar vacuolization a predictor of calcineurin inhibitor nephrotoxicity?: Arteriolar vacuolization as calcineurin inhibitor nephrotoxicity. Clin Transplant. juill 2011;25:23‑7.
55. Gandhi M, Olson JL, Meyer TW. Contribution of tubular injury to loss of remnant kidney function. Kidney Int. oct 1998;54(4):1157‑65.
56. Gibson IW, Downie TT, More IAR, Lindop GBM. ATUBULAR GLOMERULI AND GLO-MERULAR CYSTS—A POSSIBLE PATHWAY FOR NEPHRON LOSS IN THE HUMAN KIDNEY? J Pathol. août 1996;179(4):421‑6.
57. Martin M, Krichbaum M, Kaever V, Goppelt-Strübe M, Resch K. Cyclosporin A suppress-es proliferation of renal mesangial cells in culture. Biochem Pharmacol. 15 mars 1988;37(6):1083‑8.
58. Kim YI, Calne RY, Nagasue N. Cyclosporin A stimulates proliferation of the liver cells after partial hepatectomy in rats. Surg Gynecol Obstet. avr 1988;166(4):317‑22.
59. Jonasson L, Holm J, Hansson GK. Cyclosporin A inhibits smooth muscle proliferation in the vascular response to injury. Proc Natl Acad Sci U S A. avr 1988;85(7):2303‑6.
60. Urabe A, Kanitakis J, Viac J, Thivolet J. Cyclosporin a inhibits directly in vivo keratinocyte proliferation of living human skin. J Invest Dermatol. mai 1989;92(5):755‑7.
61. Healey M, Rosenberg L, Clas D, Duguid WP. Inhibition of pancreatic islet cell differentia-tion and proliferation by cyclosporine A. Transplant Proc. avr 1990;22(2):861‑2.
62. Karashima T, Hachisuka H, Sasai Y. FK506 and cyclosporin A inhibit growth factor-stimulated human keratinocyte proliferation by blocking cells in the G0G1 phases of the cell cycle. J Dermatol Sci. sept 1996;12(3):246‑54.
63. Khanna AK, Hosenpud JD. Cyclosporine induces the expression of the cyclin inhibitor p21. Transplantation. 15 mai 1999;67(9):1262‑8.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 150
64. Pawelec G, Wernet P. Cyclosporin a inhibits interleukin 2-dependent growth of alloacti-vated cloned human T-lymphocytes. Int J Immunopharmacol. janv 1983;5(4):315‑21.
65. Muraguchi A. Selective suppression of an early step in human B cell activation by cyclo-sporin A. J Exp Med. 1 sept 1983;158(3):690‑702.
66. Kermani-Arab V, Salehmoghaddam S, Danovitch G, Hirji K, Rezai A. Mediation of the antiproliferative effect of cyclosporine on human lymphocytes by blockade of interleukin 2 biosynthesis. Transplantation. avr 1985;39(4):439‑42.
67. Hannam-Harris AC, Taylor DS, Nowell PC. Cyclosporin A directly inhibits human B-cell proliferation by more than a single mechanism. J Leukoc Biol. 1 août 1985;38(2):231‑9.
68. Blaehr H, Friis S. Acute Inhibition of Human Renal Tubular Cell Growth by Cyclosporin A. Pharmacol Toxicol. févr 1990;66(2):115‑20.
69. Blaehr H, Andersen CB, Ladefoged J. Acute effects of FK506 and cyclosporine A on cul-tured human proximal tubular cells. Eur J Pharmacol. 1 avr 1993;228(5‑6):283‑8.
70. Lally C, Healy E, Ryan MP. Cyclosporine A-induced cell cycle arrest and cell death in renal epithelial cells. Kidney Int. oct 1999;56(4):1254‑7.
71. Jennings P, Koppelstaetter C, Aydin S, Abberger T, Wolf AM, Mayer G, et al. Cyclospor-ine A induces senescence in renal tubular epithelial cells. AJP Ren Physiol. 18 juill 2007;293(3):F831‑8.
72. Elmore S. Apoptosis: A Review of Programmed Cell Death. Toxicol Pathol. juin 2007;35(4):495‑516.
73. Ito H, Kasagi N, Shomori K, Osaki M, Adachi H. Apoptosis in the human allografted kid-ney. Analysis by terminal deoxynucleotidyl transferase-mediated DUTP-botin nick end labeling. Transplantation. 27 oct 1995;60(8):794‑8.
74. Healy E, Dempsey M, Lally C, Ryan MP. Apoptosis and necrosis: Mechanisms of cell death induced by cyclosporine A in a renal proximal tubular cell line. Kidney Int. 1998;54(6):1955‑66.
75. Ortiz A, Lorz C, Catalán M, Ortiz A, Coca S, Egido J. Cyclosporine A induces apoptosis in murine tubular epithelial cells: role of caspases. Kidney Int Suppl. déc 1998;68:S25-29.
76. Justo P. Intracellular Mechanisms of Cyclosporin A-Induced Tubular Cell Apoptosis. J Am Soc Nephrol. 1 déc 2003;14(12):3072‑80.
77. Eliseev RA, Malecki J, Lester T, Zhang Y, Humphrey J, Gunter TE. Cyclophilin D Inter-acts with Bcl2 and Exerts an Anti-apoptotic Effect. J Biol Chem. 10 avr 2009;284(15):9692‑9.
78. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 31 mars 2005;434(7033):658‑62.
79. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclo-philin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 31 mars 2005;434(7033):652‑8.
80. Lemoine S, Pillot B, Rognant N, Augeul L, Rayberin M, Varennes A, et al. Postcondition-ing With Cyclosporine A Reduces Early Renal Dysfunction by Inhibiting Mitochondrial Permeability Transition: Transplantation. avr 2015;99(4):717‑23.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 151
81. Lemoine S, Pillot B, Augeul L, Rabeyrin M, Varennes A, Normand G, et al. Dose and timing of injections for effective cyclosporine A pretreatment before renal ischemia reper-fusion in mice. Gallyas F, éditeur. PLOS ONE. 10 août 2017;12(8):e0182358.
82. Pallet N, Bouvier N, Bendjallabah A, Rabant M, Flinois JP, Hertig A, et al. Cyclosporine-Induced Endoplasmic Reticulum Stress Triggers Tubular Phenotypic Changes and Death. Am J Transplant. nov 2008;8(11):2283‑96.
83. Pallet N, Rabant M, Xu-Dubois Y-C, LeCorre D, Mucchielli M-H, Imbeaud S, et al. Re-sponse of human renal tubular cells to cyclosporine and sirolimus: A toxicogenomic study. Toxicol Appl Pharmacol. 1 juin 2008;229(2):184‑96.
84. Han SW, Li C, Ahn KO, Lim SW, Song HG, Jang YS, et al. Prolonged Endoplasmic Re-ticulum Stress Induces Apoptotic Cell Death in an Experimental Model of Chronic Cyclo-sporine Nephropathy. Am J Nephrol. 2008;28(5):707‑14.
85. Hama T, Nakanishi K, Mukaiyama H, Shima Y, Togawa H, Sako M, et al. Endoplasmic reticulum stress with low-dose cyclosporine in frequently relapsing nephrotic syndrome. Pediatr Nephrol. juin 2013;28(6):903‑9.
86. Pallet N, Bouvier N, Legendre C, Gilleron J, Codogno P, Beaune P, et al. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy. août 2008;4(6):783‑91.
87. Djamali A. Oxidative stress as a common pathway to chronic tubulointerstitial injury in kidney allografts. AJP Ren Physiol. 9 mai 2007;293(2):F445‑55.
88. Vetter M, Chen Z-J, Chang G-D, Che D, Liu S, Chang C-H. Cyclosporin A Disrupts Brad-ykinin Signaling Through Superoxide. Hypertension. 1 mai 2003;41(5):1136‑42.
89. Galletti P, Di Gennaro CI, Migliardi V, Indaco S, Della Ragione F, Manna C, et al. Diverse effects of natural antioxidants on cyclosporin cytotoxicity in rat renal tubular cells. Neph-rol Dial Transplant. 1 août 2005;20(8):1551‑8.
90. Tariq M, Morais C, Sobki S, Al Sulaiman M, Al Khader A. N-acetylcysteine attenuates cyclosporin-induced nephrotoxicity in rats. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc. avr 1999;14(4):923‑9.
91. Heering P, Grabensee B. Influence of ciclosporin A on renal tubular function after kidney transplantation. Nephron. 1991;59(1):66‑70.
92. Higgins R, Ramaiyan K, Dasgupta T, Kanji H, Fletcher S, Lam F, et al. Hyponatraemia and hyperkalaemia are more frequent in renal transplant recipients treated with tacroli-mus than with cyclosporin. Further evidence for differences between cyclosporin and tac-rolimus nephrotoxicities. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc. févr 2004;19(2):444‑50.
93. Caliskan Y, Kalayoglu-Besisik S, Sargin D, Ecder T. Cyclosporine-associated hyper-kalemia: report of four allogeneic blood stem-cell transplant cases: Transplantation. avr 2003;1069‑72.
94. Takami A, Asakura H, Takamatsu H, Yamazaki H, Arahata M, Hayashi T, et al. Isolated hyperkalemia associated with cyclosporine administration in allogeneic stem cell trans-plantation for renal cell carcinoma. Int J Hematol. févr 2005;81(2):159‑61.
95. Miura K, Nakatani T, Asai T, Yamanaka S, Tamada S, Tashiro K, et al. Role of hypo-magnesemia in chronic cyclosporine nephropathy. Transplantation. 73(3):340‑7.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 152
96. Holzmacher R, Kendziorski C, Michael Hofman R, Jaffery J, Becker B, Djamali A. Low serum magnesium is associated with decreased graft survival in patients with chronic cy-closporin nephrotoxicity. Nephrol Dial Transplant. 1 juill 2005;20(7):1456‑62.
97. Stahl RA, Kanz L, Maier B, Schollmeyer P. Hyperchloremic metabolic acidosis with high serum potassium in renal transplant recipients: a cyclosporine A associated side effect. Clin Nephrol. mai 1986;25(5):245‑8.
98. Younes-Ibrahim M, Barnese M, Burth P, Castro-Faria MV. Inhibition of Purified Human Kidney Na + ,K + -ATPase by Cyclosporine A. Ann N Y Acad Sci. avr 2003;986(1):633‑5.
99. Ferrer-Martínez A, Felipe A, Barceló P, Casado FJ, Ballarín J, Pastor-Anglada M. Effects of cyclosporine A on Na,K-ATPase expression in the renal epithelial cell line NBL-1. Kid-ney Int. nov 1996;50(5):1483‑9.
100. Lim SW, Ahn KO, Sheen MR, Jeon US, Kim J, Yang CW, et al. Downregulation of Renal Sodium Transporters and Tonicity-Responsive Enhancer Binding Protein by Long-Term Treatment with Cyclosporin A. J Am Soc Nephrol. 17 janv 2007;18(2):421‑9.
101. Tumlin JA, Sands JM. Nephron segment-specific inhibition of Na+/K+-ATPase activity by cyclosporin A. Kidney Int. janv 1993;43(1):246‑51.
102. Lea JP, Sands JM, McMahon SJ, Tumlin JA. Evidence that the inhibition of Na+/K+-ATPase activity by FK506 involves calcineurin. Kidney Int. sept 1994;46(3):647‑52.
103. Aker S, Heering P, Kinne-Saffran E, Deppe C, Grabensee B, Kinne RK. Different ef-fects of cyclosporine a and FK506 on potassium transport systems in MDCK cells. Exp Nephrol. 2001;9(5):332‑40.
104. Deppe CE, Heering PJ, Tinel H, Kinne-Saffran E, Grabensee B, Kinne RK. Effect of cyclosporine A on Na+/K(+)-ATPase, Na+/K+/2Cl- cotransporter, and H+/K(+)-ATPase in MDCK cells and two subtypes, C7 and C11. Exp Nephrol. déc 1997;5(6):471‑80.
105. Esteva-Font C, Ars E, Guillen-Gomez E, Campistol JM, Sanz L, Jimenez W, et al. Ciclosporin-induced hypertension is associated with increased sodium transporter of the loop of Henle (NKCC2). Nephrol Dial Transplant. 7 juill 2007;22(10):2810‑6.
106. Ling BN, Eaton DC. Cyclosporin A inhibits apical secretory K+ channels in rabbit cor-tical collecting tubule principal cells. Kidney Int. nov 1993;44(5):974‑84.
107. Zeisberg M, Neilson EG. Mechanisms of Tubulointerstitial Fibrosis. J Am Soc Neph-rol. 1 nov 2010;21(11):1819‑34.
108. Klein J, Miravete M, Buffin-Meyer B, Schanstra JP, Bascands J-L. La fibrose tubulo-interstitielle rénale: Menace fantôme ou dernière croisade ? médecine/sciences. janv 2011;27(1):55‑61.
109. Lovisa S, Zeisberg M, Kalluri R. Partial Epithelial-to-Mesenchymal Transition and Other New Mechanisms of Kidney Fibrosis. Trends Endocrinol Metab. oct 2016;27(10):681‑95.
110. Hazzan M, Hertig A, Buob D, Copin M-C, Noel C, Rondeau E, et al. Epithelial-to-Mesenchymal Transition Predicts Cyclosporine Nephrotoxicity in Renal Transplant Re-cipients. J Am Soc Nephrol. 1 juill 2011;22(7):1375‑81.
111. McMorrow T, Gaffney MM, Slattery C, Campbell E, Ryan MP. Cyclosporine A induced epithelial-mesenchymal transition in human renal proximal tubular epithelial cells. Neph-rol Dial Transplant. 1 oct 2005;20(10):2215‑25.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 153
112. Slattery C, Campbell E, McMorrow T, Ryan MP. Cyclosporine A-Induced Renal Fibro-sis. Am J Pathol. août 2005;167(2):395‑407.
113. Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cy-closporine A. Nat Med. sept 2008;14(9):931‑8.
114. Li X, Zhang X, Li X, Wang X, Wang S, Ding J. Cyclosporine A protects podocytes via stabilization of cofilin-1 expression in the unphosphorylated state. Exp Biol Med. août 2014;239(8):922‑36.
115. Descazeaud V, Mestre E, Marquet P, Essig M. Calcineurin regulation of cytoskeleton organization: a new paradigm to analyse the effects of calcineurin inhibitors on the kid-ney. J Cell Mol Med. févr 2012;16(2):218‑27.
116. Wu WW, Wang G, Baek SJ, Shen R-F. Comparative Study of Three Proteomic Quan-titative Methods, DIGE, cICAT, and iTRAQ, Using 2D Gel- or LC−MALDI TOF/TOF. J Proteome Res. mars 2006;5(3):651‑8.
117. Pütz SM, Boehm AM, Stiewe T, Sickmann A. iTRAQ Analysis of a Cell Culture Model for Malignant Transformation, Including Comparison with 2D-PAGE and SILAC. J Prote-ome Res. 6 avr 2012;11(4):2140‑53.
118. Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. oct 1999;17(10):994‑9.
119. Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Sta-ble Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol Cell Proteomics. mai 2002;1(5):376‑86.
120. Ross PL. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol Cell Proteomics. 17 sept 2004;3(12):1154‑69.
121. Zhu W, Smith JW, Huang C-M. Mass spectrometry-based label-free quantitative pro-teomics. J Biomed Biotechnol. 2010;2010:840518.
122. Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spec-trometry in proteomics: a critical review. Anal Bioanal Chem. 1 oct 2007;389(4):1017‑31.
123. Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem. 1 sept 2012;404(4):939‑65.
124. Chen X, Wei S, Ji Y, Guo X, Yang F. Quantitative proteomics using SILAC: Princi-ples, applications, and developments. PROTEOMICS. sept 2015;15(18):3175‑92.
125. Patel VJ, Thalassinos K, Slade SE, Connolly JB, Crombie A, Murrell JC, et al. A Comparison of Labeling and Label-Free Mass Spectrometry-Based Proteomics Ap-proaches. J Proteome Res. 6 juill 2009;8(7):3752‑9.
126. Wang H, Alvarez S, Hicks LM. Comprehensive Comparison of iTRAQ and Label-free LC-Based Quantitative Proteomics Approaches Using Two Chlamydomonas reinhardtii Strains of Interest for Biofuels Engineering. J Proteome Res. janv 2012;11(1):487‑501.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 154
127. Gan CS, Chong PK, Pham TK, Wright PC. Technical, Experimental, and Biological Variations in Isobaric Tags for Relative and Absolute Quantitation (iTRAQ). J Proteome Res. févr 2007;6(2):821‑7.
128. Redding AM, Mukhopadhyay A, Joyner DC, Hazen TC, Keasling JD. Study of nitrate stress in Desulfovibrio vulgaris Hildenborough using iTRAQ proteomics. Brief Funct Ge-nomic Proteomic. juin 2006;5(2):133‑43.
129. Ow SY, Salim M, Noirel J, Evans C, Rehman I, Wright PC. iTRAQ Underestimation in Simple and Complex Mixtures: “The Good, the Bad and the Ugly”. J Proteome Res. 6 nov 2009;8(11):5347‑55.
130. Suarez C, Kovar DR. Internetwork competition for monomers governs actin cytoskele-ton organization. Nat Rev Mol Cell Biol. 14 sept 2016;17(12):799‑810.
131. Pollard TD, Cooper JA. Actin, a Central Player in Cell Shape and Movement. Sci-ence. 27 nov 2009;326(5957):1208‑12.
132. Dos Remedios CG, Chhabra D, Kekic M, Dedova IV, Tsubakihara M, Berry DA, et al. Actin Binding Proteins: Regulation of Cytoskeletal Microfilaments. Physiol Rev. 1 avr 2003;83(2):433‑73.
133. Paavilainen VO, Bertling E, Falck S, Lappalainen P. Regulation of cytoskeletal dy-namics by actin-monomer-binding proteins. Trends Cell Biol. juill 2004;14(7):386‑94.
134. Leavitt J, Ng SY, Aebi U, Varma M, Latter G, Burbeck S, et al. Expression of trans-fected mutant beta-actin genes: alterations of cell morphology and evidence for auto-regulation in actin pools. Mol Cell Biol. juill 1987;7(7):2457‑66.
135. Pollard TD. Actin and Actin-Binding Proteins. Cold Spring Harb Perspect Biol. août 2016;8(8):a018226.
136. Bernstein BW, Bamburg JR. ADF/Cofilin: a functional node in cell biology. Trends Cell Biol. avr 2010;20(4):187‑95.
137. Kanellos G, Frame MC. Cellular functions of the ADF/cofilin family at a glance. J Cell Sci. 1 sept 2016;129(17):3211‑8.
138. Bamburg JR, Harris HE, Weeds AG. Partial purification and characterization of an actin depolymerizing factor from brain. FEBS Lett. 17 nov 1980;121(1):178‑82.
139. McGough A, Pope B, Chiu W, Weeds A. Cofilin Changes the Twist of F-Actin: Impli-cations for Actin Filament Dynamics and Cellular Function. J Cell Biol. 25 août 1997;138(4):771‑81.
140. Miyamoto K, Gurdon JB. Transcriptional regulation and nuclear reprogramming: roles of nuclear actin and actin-binding proteins. Cell Mol Life Sci. sept 2013;70(18):3289‑302.
141. Munsie LN, Desmond CR, Truant R. Cofilin nuclear–cytoplasmic shuttling affects co-filin–actin rod formation during stress. J Cell Sci. 1 sept 2012;125(17):3977‑88.
142. Dopie J, Skarp K-P, Kaisa Rajakyla E, Tanhuanpaa K, Vartiainen MK. Active mainte-nance of nuclear actin by importin 9 supports transcription. Proc Natl Acad Sci. 28 févr 2012;109(9):E544‑52.
143. Obrdlik A, Percipalle P. The F-actin severing protein cofilin-1 is required for RNA pol-ymerase II transcription elongation. Nucl Austin Tex. févr 2011;2(1):72‑9.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 155
144. Nishida E, Iida K, Yonezawa N, Koyasu S, Yahara I, Sakai H. Cofilin is a component of intranuclear and cytoplasmic actin rods induced in cultured cells. Proc Natl Acad Sci U S A. août 1987;84(15):5262‑6.
145. Ohta Y, Nishida E, Sakai H, Miyamoto E. Dephosphorylation of cofilin accompanies heat shock-induced nuclear accumulation of cofilin. J Biol Chem. 25 sept 1989;264(27):16143‑8.
146. Ono S, Abe H, Nagaoka R, Obinata T. Colocalization of ADF and cofilin in intranucle-ar actin rods of cultured muscle cells. J Muscle Res Cell Motil. avr 1993;14(2):195‑204.
147. Abe H, Nagaoka R, Obinata T. Cytoplasmic Localization and Nuclear Transport of Cofilin in Cultured Myotubes. Exp Cell Res. mai 1993;206(1):1‑10.
148. Bernstein BW, Chen H, Boyle JA, Bamburg JR. Formation of actin-ADF/cofilin rods transiently retards decline of mitochondrial potential and ATP in stressed neurons. AJP Cell Physiol. 1 nov 2006;291(5):C828‑39.
149. Ashworth SL, Southgate EL, Sandoval RM, Meberg PJ, Bamburg JR, Molitoris BA. ADF/cofilin mediates actin cytoskeletal alterations in LLC-PK cells during ATP depletion. Am J Physiol - Ren Physiol. 1 avr 2003;284(4):F852‑62.
150. Minamide LS, Maiti S, Boyle JA, Davis RC, Coppinger JA, Bao Y, et al. Isolation and Characterization of Cytoplasmic Cofilin-Actin Rods. J Biol Chem. 19 févr 2010;285(8):5450‑60.
151. Munsie LN, Truant R. The role of the cofilin-actin rod stress response in neurodegen-erative diseases uncovers potential new drug targets. BioArchitecture. nov 2012;2(6):204‑8.
152. Chua BT, Volbracht C, Tan KO, Li R, Yu VC, Li P. Mitochondrial translocation of cofil-in is an early step in apoptosis induction. Nat Cell Biol. déc 2003;5(12):1083‑9.
153. Zhu B, Fukada K, Zhu H, Kyprianou N. Prohibitin and Cofilin Are Intracellular Effec-tors of Transforming Growth Factor β Signaling in Human Prostate Cancer Cells. Cancer Res. 1 sept 2006;66(17):8640‑7.
154. Li G, Cheng Q, Liu L, Zhou T, Shan C, Hu X, et al. Mitochondrial translocation of cofil-in is required for allyl isothiocyanate-mediated cell death via ROCK1/PTEN/PI3K signal-ing pathway. Cell Commun Signal. 2013;11(1):50.
155. Li G, Zhou J, Budhraja A, Hu X, Chen Y, Cheng Q, et al. Mitochondrial translocation and interaction of cofilin and Drp1 are required for erucin-induced mitochondrial fission and apoptosis. Oncotarget. 30 janv 2015;6(3):1834‑49.
156. Lee C-K, Park H-J, So HH, Kim HJ, Lee KS, Choi WS, et al. Proteomic profiling and identification of cofilin responding to oxidative stress in vascular smooth muscle. PRO-TEOMICS. déc 2006;6(24):6455‑75.
157. Klamt F, Zdanov S, Levine RL, Pariser A, Zhang Y, Zhang B, et al. Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat Cell Biol. oct 2009;11(10):1241‑6.
158. Zdanov S, Klamt F, Shacter E. Importance of cofilin oxidation for oxidant-induced apoptosis. Cell Cycle. mai 2010;9(9):1675‑7.
159. Vantroys M, Huyck L, Leyman S, Dhaese S, Vandekerkhove J, Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol. 4 sept 2008;87(8‑9):649‑67.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 156
160. Agnew BJ, Minamide LS, Bamburg JR. Reactivation of Phosphorylated Actin Depol-ymerizing Factor and Identification of the Regulatory Site. J Biol Chem. 21 juill 1995;270(29):17582‑7.
161. Moriyama K, Iida K, Yahara I. Phosphorylation of Ser-3 of cofilin regulates its essen-tial function on actin. Genes Cells. janv 1996;1(1):73‑86.
162. Nagaoka R, Abe H, Obinata T. Site-directed mutagenesis of the phosphorylation site of cofilin: its role in cofilin-actin interaction and cytoplasmic localization. Cell Motil Cyto-skeleton. 1996;35(3):200‑9.
163. Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, et al. Cofilin phos-phorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 25 juin 1998;393(6687):809‑12.
164. Maekawa M. Signaling from Rho to the Actin Cytoskeleton Through Protein Kinases ROCK and LIM-kinase. Science. 6 août 1999;285(5429):895‑8.
165. Ohashi K, Hosoya T, Takahashi K, Hing H, Mizuno K. A Drosophila Homolog of LIM-Kinase Phosphorylates Cofilin and Induces Actin Cytoskeletal Reorganization. Biochem Biophys Res Commun. oct 2000;276(3):1178‑85.
166. Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K. Rho-associated Kinase ROCK Activates LIM-kinase 1 by Phosphorylation at Threonine 508 within the Ac-tivation Loop. J Biol Chem. 4 févr 2000;275(5):3577‑82.
167. Dan C, Kelly A, Bernard O, Minden A. Cytoskeletal Changes Regulated by the PAK4 Serine/Threonine Kinase Are Mediated by LIM Kinase 1 and Cofilin. J Biol Chem. 24 août 2001;276(34):32115‑21.
168. Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. sept 1999;1(5):253‑9.
169. Edwards DC, Gill GN. Structural Features of LIM Kinase That Control Effects on the Actin Cytoskeleton. J Biol Chem. 16 avr 1999;274(16):11352‑61.
170. Sumi T, Matsumoto K, Shibuya A, Nakamura T. Activation of LIM Kinases by Myoton-ic Dystrophy Kinase-related Cdc42-binding Kinase α. J Biol Chem. 22 juin 2001;276(25):23092‑6.
171. Sumi T, Matsumoto K, Nakamura T. Specific Activation of LIM kinase 2 via Phos-phorylation of Threonine 505 by ROCK, a Rho-dependent Protein Kinase. J Biol Chem. 5 janv 2001;276(1):670‑6.
172. Huang TY, DerMardirossian C, Bokoch GM. Cofilin phosphatases and regulation of actin dynamics. Curr Opin Cell Biol. févr 2006;18(1):26‑31.
173. Song X, Chen X, Yamaguchi H, Mouneimne G, Condeelis JS, Eddy RJ. Initiation of cofilin activity in response to EGF is uncoupled from cofilin phosphorylation and dephosphorylation in carcinoma cells. J Cell Sci. 15 juill 2006;119(14):2871‑81.
174. Hosoda A, Sato N, Nagaoka R, Abe H, Obinata T. Activity of cofilin can be regulated by a mechanism other than phosphorylation/dephosphorylation in muscle cells in culture. J Muscle Res Cell Motil. 18 sept 2007;28(2‑3):183‑94.
175. Lee K, Jung J, Kim M, Guidotti G. Interaction of the α subunit of Na,K-ATPase with cofilin. Biochem J. 15 janv 2001;353(2):377‑85.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 157
176. Kim M, Jung J, Park C-S, Lee K. Identification of the cofilin-binding sites in the large cytoplasmic domain of Na,K-ATPase. Biochimie. oct 2002;84(10):1021‑9.
177. Jung J, Yoon T, Choi EC, Lee K. Interaction of Cofilin with Triose-phosphate Isomer-ase Contributes Glycolytic Fuel for Na,K-ATPase via Rho-mediated Signaling Pathway. J Biol Chem. 13 déc 2002;277(50):48931‑7.
178. Jung J, Kim M, Choi S, Kim M-J, Suh J, Choi EC, et al. Molecular mechanism of cofil-in dephosphorylation by ouabain. Cell Signal. nov 2006;18(11):2033‑40.
179. Jung J, Park H, Kim M, Kim M-J, Choi EC, Lee K. Extracellular potassium deprivation reversibly dephosphorylates cofilin. Biochem Biophys Res Commun. juill 2006;345(4):1393‑7.
180. Yonezawa N, Nishida E, Iida K, Yahara I, Sakai H. Inhibition of the interactions of cofilin, destrin, and deoxyribonuclease I with actin by phosphoinositides. J Biol Chem. 25 mai 1990;265(15):8382‑6.
181. Yonezawa N, Homma Y, Yahara I, Sakai H, Nishida E. A short sequence responsible for both phosphoinositide binding and actin binding activities of cofilin. J Biol Chem. 15 sept 1991;266(26):17218‑21.
182. Zhao H, Hakala M, Lappalainen P. ADF/Cofilin Binds Phosphoinositides in a Multiva-lent Manner to Act as a PIP2-Density Sensor. Biophys J. mai 2010;98(10):2327‑36.
183. Frantz C, Barreiro G, Dominguez L, Chen X, Eddy R, Condeelis J, et al. Cofilin is a pH sensor for actin free barbed end formation: role of phosphoinositide binding. J Cell Biol. 1 déc 2008;183(5):865‑79.
184. Yeoh S, Pope B, Mannherz HG, Weeds A. Determining the differences in actin bind-ing by human ADF and cofilin1. J Mol Biol. 25 janv 2002;315(4):911‑25.
185. Pfannstiel J, Cyrklaff M, Habermann A, Stoeva S, Griffiths G, Shoeman R, et al. Hu-man Cofilin Forms Oligomers Exhibiting Actin Bundling Activity. J Biol Chem. 28 déc 2001;276(52):49476‑84.
186. Klemke M, Wabnitz GH, Funke F, Funk B, Kirchgessner H, Samstag Y. Oxidation of Cofilin Mediates T Cell Hyporesponsiveness under Oxidative Stress Conditions. Immuni-ty. sept 2008;29(3):404‑13.
187. Goyal P, Pandey D, Brünnert D, Hammer E, Zygmunt M, Siess W. Cofilin Oligomer Formation Occurs In Vivo and Is Regulated by Cofilin Phosphorylation. Manser E, éditeur. PLoS ONE. 8 août 2013;8(8):e71769.
188. Andrianantoandro E, Pollard TD. Mechanism of Actin Filament Turnover by Severing and Nucleation at Different Concentrations of ADF/Cofilin. Mol Cell. oct 2006;24(1):13‑23.
189. Percipalle P. Co-transcriptional nuclear actin dynamics. Nucleus. janv 2013;4(1):43‑52.
190. Rajakylä EK, Vartiainen MK. Rho, nuclear actin, and actin-binding proteins in the reg-ulation of transcription and gene expression. Small GTPases. janv 2014;5(1):e27539.
191. Posern G, Treisman R. Actin’ together: serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol. nov 2006;16(11):588‑96.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 158
192. Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. mai 2010;11(5):353‑65.
193. Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Racl , and CDC42Hsregulate transcriptional activation by SRF. Cell. juin 1995;81(7):1159‑70.
194. Sotiropoulos A, Gineitis D, Copeland J, Treisman R. Signal-Regulated Activation of Serum Response Factor Is Mediated by Changes in Actin Dynamics. Cell. juill 1999;98(2):159‑69.
195. Geneste O, Copeland JW, Treisman R. LIM kinase and Diaphanous cooperate to regulate serum response factor and actin dynamics. J Cell Biol. 28 mai 2002;157(5):831‑8.
196. Miralles F, Posern G, Zaromytidou A-I, Treisman R. Actin Dynamics Control SRF Ac-tivity by Regulation of Its Coactivator MAL. Cell. mai 2003;113(3):329‑42.
197. Wang D-Z, Li S, Hockemeyer D, Sutherland L, Wang Z, Schratt G, et al. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc Natl Acad Sci. 12 nov 2002;99(23):14855‑60.
198. Cen B, Selvaraj A, Burgess RC, Hitzler JK, Ma Z, Morris SW, et al. Megakaryoblastic Leukemia 1, a Potent Transcriptional Coactivator for Serum Response Factor (SRF), Is Required for Serum Induction of SRF Target Genes. Mol Cell Biol. 15 sept 2003;23(18):6597‑608.
199. Treisman R. Ternary complex factors: growth factor regulated transcriptional activa-tors. Curr Opin Genet Dev. févr 1994;4(1):96‑101.
200. Sun Q. Defining the mammalian CArGome. Genome Res. 19 déc 2005;16(2):197‑207.
201. Martin-Martin N, Dan Q, Amoozadeh Y, Waheed F, McMorrow T, Ryan MP, et al. RhoA and Rho kinase mediate cyclosporine A and sirolimus-induced barrier tightening in renal proximal tubular cells. Int J Biochem Cell Biol. janv 2012;44(1):178‑88.
202. Ihara H, Hosokawa S, Ogino T, Arima M, Ikoma F. Activation of K+ channel and inhi-bition of Na(+)-K+ ATPase of human erythrocytes by cyclosporine: possible role in hy-perpotassemia in kidney transplant recipients. Transplant Proc. août 1990;22(4):1736‑9.
203. Marakhova II, Vereninov AA, Vinogradova TA, Toropova FV. Cyclosporin A inhibits long-term activation of Na+,K+ pump in phytohemagglutinin-stimulated human lympho-cytes. Membr Cell Biol. 1998;12(3):363‑74.
204. Marakhova I., Ivanova A., Toropova F., Vereninov A., Vinogradova T. Functional ex-pression of the Na/K pump is controlled via a cyclosporin A-sensitive signalling pathway in activated human lymphocytes. FEBS Lett. 6 août 1999;456(2):285‑9.
205. Zeidel ML, Brady HR, Kone BC, Gullans SR, Brenner BM. Endothelin, a peptide in-hibitor of Na(+)-K(+)-ATPase in intact renaltubular epithelial cells. Am J Physiol - Cell Physiol. 1 déc 1989;257(6):C1101‑7.
206. Suñé G, Sarró E, Puigmulé M, López-Hellín J, Zufferey M, Pertel T, et al. Cyclophilin B Interacts with Sodium-Potassium ATPase and Is Required for Pump Activity in Proxi-mal Tubule Cells of the Kidney. Câmara NOS, éditeur. PLoS ONE. 10 nov 2010;5(11):e13930.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 159
207. Lamoureux F, Mestre E, Essig M, Sauvage FL, Marquet P, Gastinel LN. Quantitative proteomic analysis of cyclosporine-induced toxicity in a human kidney cell line and com-parison with tacrolimus. J Proteomics. déc 2011;75(2):677‑94.
208. Cao X-L, Hu X-M, Hu J-Q, Zheng W-X. Myocardin-related transcription factor-A pro-moting neuronal survival against apoptosis induced by hypoxia/ischemia. Brain Res. 18 avr 2011;1385:263‑74.
209. Sisson TH, Ajayi IO, Subbotina N, Dodi AE, Rodansky ES, Chibucos LN, et al. Inhibi-tion of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am J Pathol. avr 2015;185(4):969‑86.
210. Korol A, Taiyab A, West-Mays JA. RhoA/ROCK signaling regulates TGFβ-induced epithelial-mesenchymal transition of lens epithelial cells through MRTF-A. Mol Med Camb Mass. 29 sept 2016;22.
211. Gasparics Á, Sebe A. MRTFs- master regulators of EMT: MRTFs- Master Regulators of EMT. Dev Dyn. mars 2018;247(3):396‑404.
212. Muth T, Keller D, Puetz SM, Martens L, Sickmann A, Boehm AM. jTraqX: A free, plat-form independent tool for isobaric tag quantitation at the protein level. PROTEOMICS. 7 janv 2010;10(6):1223‑5.
213. Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics pub-lic data deposition. Nucleic Acids Res. 4 janv 2017;45(D1):D1100‑6.
214. Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 4 janv 2016;44(D1):D447‑56.

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 160
Annexes
Annexe 1. Algorithme CiR-C .......................................................................................... 161
Annexe 2. Application de la technologie iTRAQ à l’étude des modifications dans le temps
du protéome urinaire de patients greffés rénaux (mémoire M2, Zhour El-Ouafi) ............. 180

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 161
Annexe 1. Algorithme CiR-C

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
#!/bin/bash
echo -n > start.timedate >> start.time
# switch the file extension from .jpf to .txt
for file in *.jpf ; do
mv "$file" "${file%.jpf}.txt"
done
dos2unix -q *.txt # adapt .txt files generated with Windows to a UNIX distribution
# convert the delimiter, replace colons by points, fill protein name and protein ID fields, keep fields of interest only
for i in `ls serie*.txt` ; do
echo -ne "Formatting source files... ($i)\r"
sed -e "s/;/\t/g" -e "s/,/./g" $i > $i.tmpmv $i.tmp dot$i
awk -F '\t' '$2 != ""{p1=$2} NF==3{p2=$2} p1 && $2 == ""{$2=p1} p2 && NF==2{$0=$2 OFS p2 OFS $2} 1' OFS='\t' dot$i > fill$i
awk -F '\t' '$101 != ""{p1=$101} NF==3{p2=$2} p1 && $101 == ""{$101=p1} p2 && NF==2{$0=$101 OFS p2 OFS $2} 1' OFS='\t' fill$i > full$i
head -n 1 full$i | cut -d$'\t' -f2,4,6,10,12,14,16,18,20,22,24,26,28,30,32,101 >> tag$i
tail -n +2 full$i | cut -d$'\t' -f2,4,6,10,12,14,16,18,20,22,24,26,28,30,32,101 >> data$i
echo -n > tags.tmp
grep "$i" iTRAQtags.txt >> tags.tmp
# synchronize the tags and the related drug exposure conditions according to the 'iTRAQtags.txt' list
tag114=`awk '{print $2}' tags.tmp`tag115=`awk '{print $3}' tags.tmp`tag116=`awk '{print $4}' tags.tmp`tag117=`awk '{print $5}' tags.tmp`
sed -e "s/114/condition $tag114/g" -e "s/115/condition $tag115/g" -e "s/116/condition $tag116/g" -e "s/117/condition $tag117/g" -e "s/ProteinID/accession ID|gene name/g" -e"s/Peptide/associated peptide sequence/g" -e "s/Description/associated protein name/g" -e"s/Score/associated peptide score/g" tag$i > tags$i
# transpose, rank fields by alphabaetical order and retrotranspose
cat tags$i data$i >> tagged$i
awk -F $'\t' 'BEGIN {OFS = FS} {for (i=1; i<=NF; i++) a[i,NR]=$i
-1-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
max=(max<NF?NF:max)} END {for (i=1; i<=max; i++) {for (j=1; j<=NR; j++) printf "%s%s", a[i,j], (j==NR?RS:FS) } }' tagged$i >> r2c$i.tmp
mv r2c$i.tmp r2c$isort r2c$i > r2c$i.tmpmv r2c$i.tmp r2c$i
awk -F $'\t' 'BEGIN {OFS = FS} {for (i=1; i<=NF; i++) a[i,NR]=$i max=(max<NF?NF:max)} END {for (i=1; i<=max; i++) {for (j=1; j<=NR; j++) printf "%s%s", a[i,j], (j==NR?RS:FS) } }' r2c$i >> c2r$i
head -n 1 c2r$i >> _$i
# get rid of rows of irrelevant data (0.0 ratios, Mascot score below 30)
tail -n +2 c2r$i | grep -v "Fragment" | awk -F $'\t' 'BEGIN {OFS = FS} {if ( ($2 > 30) && ($3 != "") && ($5 != 0) && ($6 != 0) && ($7 != 0) && ($8 != 0) && ($9 != 0) && ($10 != 0) && ($11 != 0) && ($12 != 0) && ($13 != 0) && ($14 != 0) && ($15 != 0) && ($16 != 0) ) print $0}' >> _$i
done
# checkpoint for correct format
for i in `ls _serie*.txt` ; do
if [ `wc -l $i | awk '{print $1}'` == 1 ] ; then
mv $i empty$iecho "$i is empty."
exit
fi
done
clear
# create the output folder
mkdir output 2> /dev/null
rm output/* 2> /dev/null
# write an exhaustive list of peptides
echo -n > peptides.txt
-2-
E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
for i in `ls _serie*.txt` ; do
tail -n +2 $i | awk -F $'\t' 'BEGIN {OFS = FS} {print $1"\t"$3}' >> peptides.txt
done
sort peptides.txt | uniq > peptides.txt.tmpmv peptides.txt.tmp peptides.txtseqnumber=`wc -l peptides.txt | awk '{print $1}'`progress=0maxcount=0
# sort peptides based on the presence in x out of n series
while read line ; do
progress=$((progress + 1))pourcentprogress=`awk -v progress=$progress -v total=$seqnumber 'BEGIN
{pourcent=progress/total*100 ; printf "%3i",pourcent}'`echo -ne "Sorting peptides... ($pourcentprogress %)\r"echo -n > match.txtseq=`echo $line | awk '{print $NF}'`accession=`echo $line | awk '{for (i=1; i<NF; i++) print $i}'`count=0
for i in `ls _serie*.txt` ; do
match=`grep $'\t'"$seq"$'\t' $i | grep "$accession"$'\t'`
if [ "$match" != "" ] ; then
count=$((count + 1))
grep $'\t'"$seq"$'\t' $i | grep "$accession"$'\t' >> match.txt
fi
done
weight=`wc -l match.txt | awk '{print $1}'`
if [ `wc -l match.txt | awk '{print $1}'` == 0 ] ; then
echo "match.txt is empty."cp match.txt match.txt.saveecho $accession $seq
fi
awk -F $'\t' -v weight="$weight" 'BEGIN {OFS = FS}{
printf "%s\t", $1printf "%s\t", $4printf "%s\t", $3printf "%12.6f\t", $5printf "%12.6f\t", $6
printf "%12.6f\t", $7
-3-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
printf "%12.6f\t", $8printf "%12.6f\t", $9printf "%12.6f\t", $10printf "%12.6f\t", $11printf "%12.6f\t", $12printf "%12.6f\t", $13printf "%12.6f\t", $14printf "%12.6f\t", $15printf "%12.6f\t", $16printf "%12.6f\t", 1/weightprintf "\n"
}' match.txt >> $count.txt
if [ $count -ge $maxcount ] ; then
maxcount=$countecho $maxcount > maxcount.txt
fi
done < peptides.txt
clear
maxcount=`cat maxcount.txt`
# compute the weighted mean ratio, median ratio & standard deviation for each protein
for i in `seq 1 $maxcount` ; do
echo -ne "Calculating weighted mean ratios, median ratios and standard deviations (series mash-up) ... ($i/$maxcount file(s))\r"
awk -F $'\t' 'BEGIN {OFS = FS} {print $1}' $i.txt | while read accession ; dogrep "$accession" $i.txt | awk -F $'\t' 'BEGIN {OFS = FS}
{wsum4+=$4*$16
wsum5+=$5*$16 wsum6+=$6*$16 wsum7+=$7*$16 wsum8+=$8*$16 wsum9+=$9*$16 wsum10+=$10*$16 wsum11+=$11*$16 wsum12+=$12*$16 wsum13+=$13*$16 wsum14+=$14*$16 wsum15+=$15*$16 sum16+=$16 } END {
wm4=wsum4/sum16 wm5=wsum5/sum16 wm6=wsum6/sum16 wm7=wsum7/sum16 wm8=wsum8/sum16 wm9=wsum9/sum16
-4-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
wm10=wsum10/sum16 wm11=wsum11/sum16 wm12=wsum12/sum16 wm13=wsum13/sum16 wm14=wsum14/sum16 wm15=wsum15/sum16 printf "%12.6f\t", wm4 printf "%12.6f\t", wm5 printf "%12.6f\t", wm6 printf "%12.6f\t", wm7 printf "%12.6f\t", wm8 printf "%12.6f\t", wm9 printf "%12.6f\t", wm10 printf "%12.6f\t", wm11 printf "%12.6f\t", wm12 printf "%12.6f\t", wm13 printf "%12.6f\t", wm14 printf "%12.6f\t", wm15 printf "\n"
}' >> output/wm$i.txt.tmpdone
mv output/wm$i.txt.tmp output/wm$i.txtpaste $i.txt output/wm$i.txt >> output/wm$i.txt.tmpmv output/wm$i.txt.tmp output/wm$i.txt
awk -F $'\t' 'BEGIN {OFS = FS} {print $1}' output/wm$i.txt | while read accession ; dogrep "$accession" output/wm$i.txt | awk -F $'\t' 'BEGIN {OFS = FS}
{sqmd4+=$16*($4-$18)^2
sqmd5+=$16*($5-$19)^2 sqmd6+=$16*($6-$20)^2 sqmd7+=$16*($7-$21)^2 sqmd8+=$16*($8-$22)^2 sqmd9+=$16*($9-$23)^2 sqmd10+=$16*($10-$24)^2 sqmd11+=$16*($11-$25)^2 sqmd12+=$16*($12-$26)^2 sqmd13+=$16*($13-$27)^2 sqmd14+=$16*($14-$28)^2
sqmd15+=$16*($15-$29)^2sum16+=$16c4[i]=$4 # initialization of column arraysc5[i]=$5c6[i]=$6c7[i]=$7c8[i]=$8c9[i]=$9c10[i]=$10c11[i]=$11c12[i]=$12c13[i]=$13c14[i]=$14c15[i]=$15asort(c4) # from the lowest to the highest valuesasort(c5)asort(c6)
-5-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
asort(c7)asort(c8)
asort(c9) asort(c10) asort(c11)
asort(c12) asort(c13) asort(c14) asort(c15) } END {
sd4=sqrt(sqmd4/sum16)sd5=sqrt(sqmd5/sum16)sd6=sqrt(sqmd6/sum16)sd7=sqrt(sqmd7/sum16)sd8=sqrt(sqmd8/sum16)sd9=sqrt(sqmd9/sum16)sd10=sqrt(sqmd10/sum16)sd11=sqrt(sqmd11/sum16)sd12=sqrt(sqmd12/sum16)sd13=sqrt(sqmd13/sum16)sd14=sqrt(sqmd14/sum16)sd15=sqrt(sqmd15/sum16)x=int(NR/2)y=NR/2if (x < y) {printf "%s\t", $1printf "%s\t", $2printf "%12.6f\t", $18 # meanprintf "%12.6f\t", c4[x+1] # median
printf "%12.6f\t", sd4 # standard deviation printf "%12.6f\t", $19
printf "%12.6f\t", c5[x+1]printf "%12.6f\t", sd5
printf "%12.6f\t", $20printf "%12.6f\t", c6[x+1]printf "%12.6f\t", sd6printf "%12.6f\t", $21printf "%12.6f\t", c7[x+1]
printf "%12.6f\t", sd7 printf "%12.6f\t", $22
printf "%12.6f\t", c8[x+1] printf "%12.6f\t", sd8
printf "%12.6f\t", $23printf "%12.6f\t", c9[x+1]printf "%12.6f\t", sd9
printf "%12.6f\t", $24printf "%12.6f\t", c10[x+1]
printf "%12.6f\t", sd10 printf "%12.6f\t", $25
printf "%12.6f\t", c11[x+1]printf "%12.6f\t", sd11
printf "%12.6f\t", $26printf "%12.6f\t", c12[x+1]
printf "%12.6f\t", sd12 printf "%12.6f\t", $27
printf "%12.6f\t", c13[x+1]
-6-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
printf "%12.6f\t", sd13 printf "%12.6f\t", $28
printf "%12.6f\t", c14[x+1] printf "%12.6f\t", sd14
printf "%12.6f\t", $29printf "%12.6f\t", c15[x+1]printf "%12.6f\t", sd15} else if (x == y) {printf "%s\t", $1printf "%s\t", $2printf "%12.6f\t", $18printf "%12.6f\t", (c4[x]+c4[x+1])/2printf "%12.6f\t", sd4
printf "%12.6f\t", $19printf "%12.6f\t", (c5[x]+c5[x+1])/2printf "%12.6f\t", sd5
printf "%12.6f\t", $20printf "%12.6f\t", (c6[x]+c6[x+1])/2printf "%12.6f\t", sd6printf "%12.6f\t", $21printf "%12.6f\t", (c7[x]+c7[x+1])/2printf "%12.6f\t", sd7
printf "%12.6f\t", $22printf "%12.6f\t", (c8[x]+c8[x+1])/2printf "%12.6f\t", sd8
printf "%12.6f\t", $23printf "%12.6f\t", (c9[x]+c9[x+1])/2printf "%12.6f\t", sd9
printf "%12.6f\t", $24printf "%12.6f\t", (c10[x]+c10[x+1])/2printf "%12.6f\t", sd10
printf "%12.6f\t", $25printf "%12.6f\t", (c11[x]+c11[x+1])/2
printf "%12.6f\t", sd11 printf "%12.6f\t", $26
printf "%12.6f\t", (c12[x]+c12[x+1])/2printf "%12.6f\t", sd12
printf "%12.6f\t", $27printf "%12.6f\t", (c13[x]+c13[x+1])/2printf "%12.6f\t", sd13
printf "%12.6f\t", $28printf "%12.6f\t", (c14[x]+c14[x+1])/2printf "%12.6f\t", sd14printf "%12.6f\t", $29printf "%12.6f\t", (c15[x]+c15[x+1])/2printf "%12.6f\t", sd15}printf "\n"
}' >> output/wm$i.txt.tmpdone
mv output/wm$i.txt.tmp output/wm$i.txtsort output/wm$i.txt | uniq > output/wm$i.txt.tmpmv output/wm$i.txt.tmp output/wm$i.txt
head -n 1 _serie*.txt | head -n 2 | tail -n 1 | awk -F $'\t' 'BEGIN {OFS = FS}{
-7-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
printf "%s\t", $1printf "%s\t", $4printf "%s\t", $3printf "%s\t", $5printf "%s\t", $6printf "%s\t", $7printf "%s\t", $8printf "%s\t", $9printf "%s\t", $10printf "%s\t", $11printf "%s\t", $12 printf "%s\t", $13printf "%s\t", $14printf "%s\t", $15printf "%s\t", $16printf "\n"
}' >> output/nm$i.txt.tmp
cat $i.txt >> output/nm$i.txt.tmpmv output/nm$i.txt.tmp output/nm$i.txt
head -n 1 _serie*.txt | head -n 2 | tail -n 1 | awk -F $'\t' 'BEGIN {OFS = FS}{
printf "%s\t", $1printf "%s\t", $4printf "%s\t", "Mean " $5printf "%s\t", "Median " $5printf "%s\t", "SD " $5printf "%s\t", "Mean " $6printf "%s\t", "Median " $6printf "%s\t", "SD " $6 printf "%s\t", "Mean " $7printf "%s\t", "Median " $7printf "%s\t", "SD " $7 printf "%s\t", "Mean " $8printf "%s\t", "Median " $8printf "%s\t", "SD " $8 printf "%s\t", "Mean " $9printf "%s\t", "Median " $9printf "%s\t", "SD " $9 printf "%s\t", "Mean " $10printf "%s\t", "Median " $10printf "%s\t", "SD " $10 printf "%s\t", "Mean " $11printf "%s\t", "Median " $11printf "%s\t", "SD " $11 printf "%s\t", "Mean " $12 printf "%s\t", "Median " $12printf "%s\t", "SD " $12 printf "%s\t", "Mean " $13printf "%s\t", "Median " $13printf "%s\t", "SD " $13 printf "%s\t", "Mean " $14printf "%s\t", "Median " $14printf "%s\t", "SD " $14 printf "%s\t", "Mean " $15printf "%s\t", "Median " $15
-8-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
printf "%s\t", "SD " $15 printf "%s\t", "Mean " $16printf "%s\t", "Median " $16printf "%s\t", "SD " $16 printf "\n"
}' >> output/wm$i.txt.tmp
cat output/wm$i.txt >> output/wm$i.txt.tmpmv output/wm$i.txt.tmp output/wm$i.txt
done
# write a list of ubiquitous peptides
echo -n > major_peptides.txt
tail -n +2 output/nm$maxcount.txt | awk -F $'\t' 'BEGIN {OFS = FS} {print $1"\t"$3}' >>major_peptides.txt
sort major_peptides.txt | uniq > major_peptides.txt.tmpmv major_peptides.txt.tmp major_peptides.txtseqnumber=`wc -l major_peptides.txt | awk '{print $1}'`progress=0
for i in `ls _serie*.txt` ; do
mkdir output_ubi$i 2> /dev/null
done
clear
# list ubiquitous peptides, series by series
while read line ; do
progress=$((progress + 1))pourcentprogress=`awk -v progress=$progress -v total=$seqnumber 'BEGIN
{pourcent=progress/total*100 ; printf "%3i",pourcent}'`echo -ne "Listing ubiquitous peptides, series by series... ($pourcentprogress %)\r"seq=`echo $line | awk '{print $NF}'`accession=`echo $line | awk '{for (i=1; i<NF; i++) print $i}'`
for i in `ls _serie*.txt` ; do
echo -n > matchsolo.txt
match=`grep $'\t'"$seq"$'\t' $i | grep "$accession"$'\t'`
if [ "$match" != "" ] ; thengrep $'\t'"$seq"$'\t' $i | grep "$accession"$'\t' >> matchsolo.txt
weight=`wc -l matchsolo.txt | awk '{print $1}'`
if [ `wc -l matchsolo.txt | awk '{print $1}'` == 0 ] ; thenecho "matchsolo.txt is empty."cp matchsolo.txt matchsolo.txt.save
-9-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
echo $accession $seqfi
awk -F $'\t' -v weight=$weight 'BEGIN {OFS = FS}{
printf "%s\t", $1printf "%s\t", $4printf "%s\t", $3printf "%12.6f\t", $5printf "%12.6f\t", $6printf "%12.6f\t", $7printf "%12.6f\t", $8printf "%12.6f\t", $9printf "%12.6f\t", $10printf "%12.6f\t", $11printf "%12.6f\t", $12printf "%12.6f\t", $13printf "%12.6f\t", $14printf "%12.6f\t", $15printf "%12.6f\t", $16printf "%12.6f\t", 1/weight printf "\n"
}' matchsolo.txt >> _ubi$i
fi
done
done < major_peptides.txt
clear
progress=0
# compute the weighted mean ratio, median ratio & standard deviation for each protein
for i in `ls _ubi_serie*.txt` ; do
progress=$((progress + 1))echo -ne "Calculating weighted mean ratios, median ratios and standard deviations
(series by series)...($progress/$maxcount file(s))\r"
awk -F $'\t' 'BEGIN {OFS = FS} {print $1}' $i | while read accession ; dogrep "$accession" $i | awk -F $'\t' 'BEGIN {OFS = FS}
{wsum4+=$4*$16 # sum of weighted ratios
wsum5+=$5*$16 wsum6+=$6*$16 wsum7+=$7*$16 wsum8+=$8*$16 wsum9+=$9*$16 wsum10+=$10*$16 wsum11+=$11*$16 wsum12+=$12*$16 wsum13+=$13*$16 wsum14+=$14*$16 wsum15+=$15*$16
-10-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
sum16+=$16 } END { wm4=wsum4/sum16 # weighted mean wm5=wsum5/sum16 wm6=wsum6/sum16 wm7=wsum7/sum16 wm8=wsum8/sum16 wm9=wsum9/sum16 wm10=wsum10/sum16 wm11=wsum11/sum16 wm12=wsum12/sum16 wm13=wsum13/sum16 wm14=wsum14/sum16 wm15=wsum15/sum16 printf "%12.6f\t", wm4 printf "%12.6f\t", wm5 printf "%12.6f\t", wm6 printf "%12.6f\t", wm7 printf "%12.6f\t", wm8 printf "%12.6f\t", wm9 printf "%12.6f\t", wm10 printf "%12.6f\t", wm11 printf "%12.6f\t", wm12 printf "%12.6f\t", wm13 printf "%12.6f\t", wm14 printf "%12.6f\t", wm15 printf "\n" }' >> output$i/wm$i.tmp
done
mv output$i/wm$i.tmp output$i/wm$ipaste $i output$i/wm$i >> output$i/wm$i.tmpmv output$i/wm$i.tmp output$i/wm$i
awk -F $'\t' 'BEGIN {OFS = FS} {print $1}' output$i/wm$i | while read accession ; dogrep "$accession" output$i/wm$i | awk -F $'\t' 'BEGIN {OFS = FS}
{sqmd4+=$16*($4-$18)^2
sqmd5+=$16*($5-$19)^2 sqmd6+=$16*($6-$20)^2 sqmd7+=$16*($7-$21)^2 sqmd8+=$16*($8-$22)^2 sqmd9+=$16*($9-$23)^2 sqmd10+=$16*($10-$24)^2 sqmd11+=$16*($11-$25)^2 sqmd12+=$16*($12-$26)^2 sqmd13+=$16*($13-$27)^2 sqmd14+=$16*($14-$28)^2
sqmd15+=$16*($15-$29)^2sum16+=$16c4[i]=$4 # initialization of column arraysc5[i]=$5c6[i]=$6c7[i]=$7c8[i]=$8c9[i]=$9
-11-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
c10[i]=$10c11[i]=$11c12[i]=$12c13[i]=$13c14[i]=$14c15[i]=$15asort(c4) # from the lowest to the highest valuesasort(c5)asort(c6)asort(c7)asort(c8)
asort(c9) asort(c10) asort(c11)
asort(c12) asort(c13) asort(c14) asort(c15) } END {
sd4=sqrt(sqmd4/sum16)sd5=sqrt(sqmd5/sum16)sd6=sqrt(sqmd6/sum16)sd7=sqrt(sqmd7/sum16)sd8=sqrt(sqmd8/sum16)sd9=sqrt(sqmd9/sum16)sd10=sqrt(sqmd10/sum16)sd11=sqrt(sqmd11/sum16)sd12=sqrt(sqmd12/sum16)sd13=sqrt(sqmd13/sum16)sd14=sqrt(sqmd14/sum16)sd15=sqrt(sqmd15/sum16)x=int(NR/2)y=NR/2if (x < y) {printf "%s\t", $1printf "%s\t", $2printf "%12.6f\t", $18 # meanprintf "%12.6f\t", c4[x+1] # median
printf "%12.6f\t", sd4 # standard deviation printf "%12.6f\t", $19
printf "%12.6f\t", c5[x+1]printf "%12.6f\t", sd5
printf "%12.6f\t", $20printf "%12.6f\t", c6[x+1]printf "%12.6f\t", sd6printf "%12.6f\t", $21printf "%12.6f\t", c7[x+1]
printf "%12.6f\t", sd7 printf "%12.6f\t", $22
printf "%12.6f\t", c8[x+1] printf "%12.6f\t", sd8
printf "%12.6f\t", $23printf "%12.6f\t", c9[x+1]printf "%12.6f\t", sd9
printf "%12.6f\t", $24printf "%12.6f\t", c10[x+1]
-12-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
printf "%12.6f\t", sd10 printf "%12.6f\t", $25
printf "%12.6f\t", c11[x+1]printf "%12.6f\t", sd11
printf "%12.6f\t", $26printf "%12.6f\t", c12[x+1]
printf "%12.6f\t", sd12 printf "%12.6f\t", $27
printf "%12.6f\t", c13[x+1] printf "%12.6f\t", sd13
printf "%12.6f\t", $28printf "%12.6f\t", c14[x+1]
printf "%12.6f\t", sd14printf "%12.6f\t", $29printf "%12.6f\t", c15[x+1]printf "%12.6f\t", sd15} else if (x == y) {printf "%s\t", $1printf "%s\t", $2printf "%12.6f\t", $18printf "%12.6f\t", (c4[x]+c4[x+1])/2printf "%12.6f\t", sd4
printf "%12.6f\t", $19printf "%12.6f\t", (c5[x]+c5[x+1])/2printf "%12.6f\t", sd5
printf "%12.6f\t", $20printf "%12.6f\t", (c6[x]+c6[x+1])/2printf "%12.6f\t", sd6printf "%12.6f\t", $21printf "%12.6f\t", (c7[x]+c7[x+1])/2printf "%12.6f\t", sd7
printf "%12.6f\t", $22printf "%12.6f\t", (c8[x]+c8[x+1])/2printf "%12.6f\t", sd8
printf "%12.6f\t", $23printf "%12.6f\t", (c9[x]+c9[x+1])/2printf "%12.6f\t", sd9
printf "%12.6f\t", $24printf "%12.6f\t", (c10[x]+c10[x+1])/2printf "%12.6f\t", sd10
printf "%12.6f\t", $25printf "%12.6f\t", (c11[x]+c11[x+1])/2
printf "%12.6f\t", sd11 printf "%12.6f\t", $26
printf "%12.6f\t", (c12[x]+c12[x+1])/2printf "%12.6f\t", sd12
printf "%12.6f\t", $27printf "%12.6f\t", (c13[x]+c13[x+1])/2printf "%12.6f\t", sd13
printf "%12.6f\t", $28printf "%12.6f\t", (c14[x]+c14[x+1])/2printf "%12.6f\t", sd14printf "%12.6f\t", $29printf "%12.6f\t", (c15[x]+c15[x+1])/2printf "%12.6f\t", sd15}printf "\n"
-13-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
}' >> output$i/wm$i.tmpdone
mv output$i/wm$i.tmp output$i/wm$isort output$i/wm$i | uniq > output$i/wm$i.tmpmv output$i/wm$i.tmp output$i/wm$i
head -n 1 _serie*.txt | head -n 2 | tail -n 1 | awk -F $'\t' 'BEGIN {OFS = FS} { printf "%s\t", $1 printf "%s\t", $4
printf "%s\t", $3 printf "%s\t", $5 printf "%s\t", $6 printf "%s\t", $7 printf "%s\t", $8 printf "%s\t", $9 printf "%s\t", $10 printf "%s\t", $11 printf "%s\t", $12 printf "%s\t", $13 printf "%s\t", $14 printf "%s\t", $15 printf "%s\t", $16 printf "\n" }' >> output$i/nm$i.tmp
cat $i >> output$i/nm$i.tmpmv output$i/nm$i.tmp output$i/nm$i
head -n 1 _serie*.txt | head -n 2 | tail -n 1 | awk -F $'\t' 'BEGIN {OFS = FS}{
printf "%s\t", $1printf "%s\t", $4printf "%s\t", "Mean " $5printf "%s\t", "Median " $5printf "%s\t", "SD " $5printf "%s\t", "Mean " $6printf "%s\t", "Median " $6printf "%s\t", "SD " $6 printf "%s\t", "Mean " $7printf "%s\t", "Median " $7printf "%s\t", "SD " $7 printf "%s\t", "Mean " $8printf "%s\t", "Median " $8printf "%s\t", "SD " $8 printf "%s\t", "Mean " $9printf "%s\t", "Median " $9printf "%s\t", "SD " $9 printf "%s\t", "Mean " $10printf "%s\t", "Median " $10printf "%s\t", "SD " $10 printf "%s\t", "Mean " $11printf "%s\t", "Median " $11printf "%s\t", "SD " $11 printf "%s\t", "Mean " $12 printf "%s\t", "Median " $12
-14-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
printf "%s\t", "SD " $12 printf "%s\t", "Mean " $13printf "%s\t", "Median " $13printf "%s\t", "SD " $13 printf "%s\t", "Mean " $14printf "%s\t", "Median " $14printf "%s\t", "SD " $14 printf "%s\t", "Mean " $15printf "%s\t", "Median " $15printf "%s\t", "SD " $15 printf "%s\t", "Mean " $16printf "%s\t", "Median " $16printf "%s\t", "SD " $16 printf "\n"
}' >> output$i/wm$i.tmp
cat output$i/wm$i >> output$i/wm$i.tmpmv output$i/wm$i.tmp output$i/wm$i
done
# download a protein database for Sus scrofa from UniProt
dir=`pwd`
wget -O $dir/gene_ssa.txt"http://www.uniprot.org/uniprot/?query=reviewed%3Ayes%20AND%20organism%3A%22Sus%20scrofa%20(Pig)%20%5B9823%5D%22&sort=score&columns=id%2Centry%20name&format=tab"
wget -O $dir/base_hsa.txt"http://www.uniprot.org/uniprot/?query=&fil=organism%3A%22Homo%20sapiens%20(Human)%20%5B9606%5D%22%20AND%20reviewed%3Ayes&columns=id%2Centry%20name%2Cgo(biological%20process)&format=tab"
tail -n +2 gene_ssa.txt | sed -e "s/_/\t/g" | cut -f1,2 | sort -k2,2 -u -t$'\t' >>gene_ssa.txt.tmp
tail -n +2 base_hsa.txt | sed -e "s/_/\t/g" | cut -f2,4 | sort -k1,1 -u -t$'\t' >>base_hsa.txt.tmp
join -1 2 -2 1 -t$'\t' gene_ssa.txt.tmp base_hsa.txt.tmp >> base_ssa.txt
# adjust the database file to fit the script requirements
cut -f2,3 base_ssa.txt | sort | sed -e "s/; /\t/g" -e "s/, /_/g" -e "s/\//-/g" -e "s/ \[/_/g"-e "s/ /_/g" -e "s/:/_/g" -e "s/\]/_/g" > base_ssa.txt.tmp
# link the proteins to their GO biological functions
for i in `seq 1 $maxcount` ; do
cp base_ssa.txt.tmp base_ssa$i.txt
tail -n +2 output/wm$i.txt | sort | sed -e "s/|/\t/g" > output/wm$i.txt.tmpjoin output/wm$i.txt.tmp base_ssa$i.txt -t $'\t' >> output/datamean$i.txt
head -n 1 output/wm$i.txt | sed -e "s/|/\t/g" > output/headmean$i.txtcat output/headmean$i.txt output/datamean$i.txt > output/go$i.txt
-15-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
done
for i in `ls _ubi_serie*.txt` ; do
cp base_ssa.txt.tmp base_ssa$i
tail -n +2 output$i/wm$i | sort | sed -e "s/|/\t/g" > output$i/wm$i.tmpjoin output$i/wm$i.tmp base_ssa$i -t $'\t' >> output$i/datamean$i
head -n 1 output$i/wm$i | sed -e "s/|/\t/g" > output$i/headmean$icat output$i/headmean$i output$i/datamean$i > output$i/go$i
done
# create a unique list of biological functions
sed -e "s/|/\t/g" peptides.txt | cut -d$'\t' -f1 | sort | uniq > accessions.txt.tmpjoin accessions.txt.tmp base_ssa.txt.tmp -t $'\t' >> go_series.txt.tmp
awk -F $'\t' 'BEGIN {OFS = FS} {for (i=1; i<=NF; i++) a[i,NR]=$i max=(max<NF?NF:max)} END {for (i=1; i<=max; i++) {for (j=1; j<=NR; j++) printf "%s%s", a[i,j], (j==NR?RS:FS) } }' go_series.txt.tmp >> r2cgo_series.txt
nf=`wc -l r2cgo_series.txt | awk '{print $1}'`
for i in `seq 2 $nf` ; do
cut -d$'\t' -f$i go_series.txt.tmp >> go_list.txt.tmp
done
sed -e "/^$/d" go_list.txt.tmp | sort | uniq > go_list.txtkeynumber=`wc -l go_list.txt | awk '{print $1}'`progress=0
clear
# sort proteins by biological functions (one function = one folder)
while read line ; do
progress=$((progress + 1))pourcentprogress=`awk -v progress=$progress -v total=$keynumber 'BEGIN
{pourcent=progress/total*100 ; printf "%3i",pourcent}'`echo -ne "Linking proteins to biological functions... ($pourcentprogress %)\r"echo -n > matchgo.txtkeyword=`echo $line | awk -F $'\t' 'BEGIN {OFS = FS} {print $1}'`mkdir output_$keyword 2> /dev/null
for i in `seq 1 $maxcount` ; do
echo -n > matchgo.txt
-16-

E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
match=`grep "$keyword" output/go$i.txt`
if [ "$match" != "" ] ; thengrep "$keyword" output/go$i.txt >> matchgo.txt
if [ `wc -l matchgo.txt | awk '{print $1}'` == 0 ] ; thenecho "matchgo.txt is empty."cp matchgo.txt matchgo.txt.saveecho $keyword
fi
fi
awk -F $'\t' 'BEGIN {OFS = FS} {print $0}' matchgo.txt >> $keyword$i.txt
head -n 1 output/go$i.txt | head -n 2 | tail -n 1 | awk -F $'\t' 'BEGIN {OFS = FS}{
printf "%s\t", $0printf "\n"
}' >> output_$keyword/$keyword$i.txt.tmpcat $keyword$i.txt >> output_$keyword/$keyword$i.txt.tmpmv output_$keyword/$keyword$i.txt.tmp output_$keyword/$keyword$i.txt
done
echo -n > matchgo.txt
for i in `ls _ubi_serie*.txt` ; do
echo -n > matchgo.txtmatch=`grep "$keyword" output$i/go$i`
if [ "$match" != "" ] ; thengrep "$keyword" output$i/go$i >> matchgo.txt
if [ `wc -l matchgo.txt | awk '{print $1}'` == 0 ] ; thenecho "matchgo.txt is empty."cp matchgo.txt matchgo.txt.saveecho $keyword
fi
fi
awk -F $'\t' 'BEGIN {OFS = FS} {print $0}' matchgo.txt >> $keyword$i
head -n 1 output$i/go$i | head -n 2 | tail -n 1 | awk -F $'\t' 'BEGIN {OFS = FS}{
printf "%s\t", $0printf "\n"
}' >> output_$keyword/$keyword$i.tmpcat $keyword$i >> output_$keyword/$keyword$i.tmpmv output_$keyword/$keyword$i.tmp output_$keyword/$keyword$i
done
done < go_list.txt
-17-
E:\iTRAQ_m\isis_m\source\cir-c_x.sh lundi 30 octobre 2017 10:20
# cleaning
mv _serie*.txt outputmv peptides.txt output/mv major_peptides.txt output/mv go_list.txt output/mv output_*/ output/
rm -r *.tmp *.txt output/*.tmp output/data*.txt output/head*.txt output/output_ubi_*/*.tmpoutput/output_ubi_*/data*.txt output/output_ubi_*/head*.txt
echo -n > stop.timedate >> stop.time
echo -n > timespan.timecat start.time stop.time >> timespan.time
mv timespan.time output/rm -r *.time
# Bastien BURAT, 2015
-18-

Bastien Burat | Thèse de doctorat | Université de Limoges | 2017 180
Annexe 2. Application de la technologie iTRAQ à l’étude des modifications dans le
temps du protéome urinaire de patients greffés rénaux (mémoire M2, Zhour El-Ouafi)

MASTER II DE RECHERCHE
BIOLOGIE CELLULAIRE – PHYSIOLOGIE – PHYSIOPATHOLOGIE
APPLICATION DE LA TECHNOLOGIE iTRAQ A L’ÉTUDE DES
MODIFICATIONS DANS LE TEMPS DU PROTEOME URINAIRE DE
PATIENTS GREFFES RENAUX
Zhour EL OUAFI
29 SEPTEMBRE 2017
UMR INSERM-S850 – Pharmacologie des immunosuppresseurs et de la transplantation Limoges
Directeur de l’unité :
Professeur P. MARQUET
Responsables du stage :
Professeur M. ESSIG
B. BURAT

1
RESUME
Introduction : L’iTRAQ est une technique de protéomique quantitative différentielle validée. Les
urines sont un milieu d’étude complexe, riche en protéines et facilement accessible. L’objectif était
d’appliquer l’iTRAQ à l’exploration du protéome urinaire dans un schéma d’étude longitudinal per-
mettant l’analyse de ses variations dans le temps.
Matériels et méthodes: Un protocole iTRAQ adapté aux urines a été élaboré. Des urines de patients
ont été analysées dans un axe d’étude longitudinal, par iTRAQ couplée à la chromatographie liquide
haute performance (micro-LC), en ligne avec une spectrométrie de masse (QQTOF). Un traitement
bio-informatique permettait ensuite l’identification et la quantification relative des protéines entre
chaque condition chez chaque patient.
Résultats: Cinq patients, parmi les greffés rénaux du CHU de Limoges, ont été sélectionnés. Tous
atteints de polykystose rénale, traités par Tacrolimus, et sans épisode de rejet allo-immun. Quatre
échantillons d’urines par patients ont été recueillis, correspondant à quatre dates distinctes, réparties
sur la première année de greffe.1302 protéines ont été identifiées, dont 521 éléments uniques. Le
nombre de protéines identifiées par patient était similaire, autour de 260. La composition des pro-
téomes obtenus était variée, faite de protéines d’origine rénale et systémique. Une bonne reproducti-
bilité de la technique a été observée pour quatre des cinq patients testés, dont la distribution des ratios
(par rapport au temps M1) approchait une loi normale (moyenne ratios = 1.025, moyenne écarts-types
= 0.5325). L’analyse des ratios a permis de déceler plusieurs profils de variations significatives dans
le temps dont un groupe de 367 protéines non variantes, 43 en surexpression constante et 32 en sous-
expression constante.
Conclusion: L’iTRAQ semble être une technique adaptée à l’étude de variations temporelles du pro-
téome urinaire. Elle pourrait constituer un outil d’intérêt pour la compréhension de mécanismes tels
que la néphrotoxicité des anti-calcineurine en greffe rénale.
Mots clés :
Protéome urinaire, iTRAQ, néphrotoxicité des inhibiteurs de la Calcineurine

2
ABSTRACT
Introduction: iTRAQ is a validated technique of differential quantitative proteomics. Urine is a com-
plex, protein-rich and easily accessible medium. The objective was to apply the iTRAQ technique to
the analysis of time-dependent variations of the urinary proteome.
Materials: An iTRAQ protocol was optimized for urine analysis. Patients' urines were analyzed in a
longitudinal axis, using iTRAQ coupled with high-performance liquid chromatography (micro-LC),
on line with mass spectrometry (QQTOF). Then, bioinformatics allowed the identification and rela-
tive quantification of proteins between each condition for each patient.
Results: Five renal-transplanted patients of the Limoges University Hospital were selected. All af-
fected with polycystic kidney disease, treated with Tacrolimus, and without acute rejection. Four
urine samples per patient were collected, corresponding to four distinct dates over the first year of
transplantation.1302 proteins were identified including 521 unique elements. The number of proteins
identified per patient was similar, around 260. The proteome composition varied, made of proteins
from renal and systemic origins. A good reproducibility of the technique was observed for four of the
five patients tested, with ratio distribution (reported to time M1) approaching a normal distribution
(average ratios = 1.025, mean standard deviation = 0.5325). The ratio analysis revealed several sig-
nificant time-dependent patterns, including a group of 367 non-variant proteins, 43 with constant
over-expression and 32 with constant under-expression.
Conclusion: iTRAQ appears to be adapted to the study of temporal variations of the urinary prote-
ome. It could constitute an interesting tool for the understanding of mechanisms such as calcineurin
inhibitors nephrotoxicity.
Key words: Urine proteome, iTRAQ, Calcineurin inhibitors nephrotoxicity

3
SOMMAIRE
RESUME ............................................................................................................................................. 1
ABSTRACT ......................................................................................................................................... 2
SOMMAIRE ........................................................................................................................................ 3
LISTE DES ABREVIATIONS ............................................................................................................ 5
TABLE DES FIGURES....................................................................................................................... 6
TABLE DES TABLEAUX .................................................................................................................. 6
INTRODUCTION ............................................................................................................................... 7
1. Le protéome urinaire ................................................................................................................. 7
2. Protéomique quantitative différentielle: place de l’iTRAQ ...................................................... 8
2.a. ICAT et SILAC ...................................................................................................................... 8
2.b. iTRAQ ................................................................................................................................... 9
3. Utilisation de l’iTRAQ dans l’étude de la néphrotoxicité des inhibiteurs de la Calcineurine
(ICN) ................................................................................................................................................ 9
4. Objectif de travail .................................................................................................................... 11
MATÉRIELS ET MÉTHODES......................................................................................................... 12
1. Collection des échantillons ..................................................................................................... 12
2. Préparation des échantillons.................................................................................................... 12
2.a. Concentration des protéines urinaires .................................................................................. 12
2.b. Digestion trypsique .............................................................................................................. 13
2.c. Solid Phase Extraction (SPE) ............................................................................................... 13
3. iTRAQ et Spectrométrie de Masse ......................................................................................... 13
3.a. Marquage des échantillons ................................................................................................... 13
3.b. Focalisation isoélectrique (Agilent 3100 OFFGEL Fractionator Kit) ................................. 14
3.c. Micro-chromatographie liquide (micro-LC) ........................................................................ 14
3.d. Spectrométrie de masse ....................................................................................................... 14
3.e. Traitement bio-informatique des données de spectrométrie de masse et
identification/quantification relative des protéines basée sur les intensités de pics. .................. 15
RÉSULTATS ..................................................................................................................................... 16
1. Caractéristiques des patients : ................................................................................................. 16
2. Optimisation de la procédure d’extraction des protéines urinaires ......................................... 16
3. Analyse bio-informatique ....................................................................................................... 17
4. Etude des profils de variations significatives dans le temps. .................................................. 18

4
DISCUSSION .................................................................................................................................... 21
1. Optimisation du protocole ....................................................................................................... 21
2. Choix des patients ................................................................................................................... 22
3. Echec du patient CS ................................................................................................................ 23
4. Limites .................................................................................................................................... 23
5. Conclusion .............................................................................................................................. 23
REFERENCES BIBLIOGRAPHIQUES ........................................................................................... 24
ANNEXE ........................................................................................................................................... 27

5
LISTE DES ABREVIATIONS
ACN : Acétonitrile
ADN : Acide Désoxyribonucléique
ARNm : Acide Ribonucléique messager
BSA: Bovin Serum Albumin
CsA : Ciclosporine A
Da : Dalton
DTT : Dithiothreitol
GO : Gene Ontology
HEPES : Hydroxyethyl piperazine ethanesulfonic acid
IDA : Iodoacétamide
ICN : Inhibiteurs de la Calcineurine
Ig : Immunoglobuline
ICAT : Isotope Coded Affinity Tag
iTRAQ : Isobaric Tag for Relative and Absolute Quantitation
LC : Liquid Chromatogtraphy
LLC-PK1 : Lilly Laboratories Porcine Kidney-1
MMF : Micophenolate Mofetil
MOPS : Morpholinopropanesulfonic acid
MS : Spectrométrie de Masse
NFAT : Nuclear Factor of Activated T-cells
PKR : Polykystose Rénale
SILAC : Stable Isotope Labeling of Amino-acid in Cell Culture
SPE : Solid Phase Extraction
TAC : Tacrolimus
TFA : Trifluoro-acetic Acid
WB : Western Blot

6
TABLE DES FIGURES
Figure 1: Le réactif iTRAQ, d’après Zieske, J Exp Bot, 2006 ............................................................ 9
Figure 2 Principe de l’iTRAQ, d’après Masse-Spectrométrie.fr, 2016 .............................................. 9
Figure 3 Effets différentiels de la CsA et TAC sur le cytosquelette d’Actine de cellules LLC-PK1,
d’après Descazeaud, J Cell Mol Med, 2012....................................................................................... 10
Figure 4 Plaque résumée de la préparation des échantillons en iTRAQ sur urines,
d’après B Burat .................................................................................................................................. 15
Figure 5 Etude de la répartition des ratios pour chaque patient par une fonction de probabilité de
masse .................................................................................................................................................. 17
Figure 6 Diagramme de Venn comparant la composition des protéomes urinaire des différents pa-
tients ................................................................................................................................................... 17
Figure 7 Heat maps ........................................................................................................................... 18
Figure 8 Diagramme de Venn illustrant la répartition des protéines non variantes communes entre
les différents patients ......................................................................................................................... 18
Figure 9 Diagramme de Venn comparant les protéomes identifiés par iTRAQu vs Sigdel et al ..... 22
TABLE DES TABLEAUX
Tableau 1 Caractéristiques des patients ............................................................................................ 16
Tableau 2 Nombre de peptides et de protéines avant et après le tri effectué par CIR-C .................. 17
Tableau 3 Nombres de protéines par patient, selon trois profils de variation dans le temps : non va-
riantes, constamment sur-exprimées par rapport à M1 ou constamment sous-exprimées par rapport à
M1 Analyse quantitative et de la nature des protéines subissant des variations d’expressions dans le
temps. (a) Protéines sur-exprimées et (b) Protéines sous-exprimées ................................................. 18
Tableau 4 Analyse quantitative et de la nature des protéines subissant des variations d’expressions
dans le temps. ..................................................................................................................................... 19
Tableau 5 Protéines significativement enrichies selon le logiciel PANTHER ................................. 19

7
INTRODUCTION
Les progrès récents des technologies « omiques », permet aujourd’hui l’analyse à large échelle
et sans hypothèses a priori, des expressions d’ADN, d’ARN, de protéines ou encore de petites molé-
cules dérivées du métabolisme. Leur utilisation s’est démocratisée, permettant des avancées majeures
dans la compréhension de nombreux mécanismes physiologiques.
Une de leur application cible l’identification de nouveaux biomarqueurs dans les liquides bio-
logiques, dans le but in fine d’améliorer les performances diagnostiques et pronostiques en pratique
clinique. Des résultats ont été obtenus dans des thématiques variées comme le rejet de greffe (1), la
néphropathie a IgA (2) et l’oncologie médicale (rénale (3), prostatique (4), ovarienne (5)).
Ce travail est une étude préliminaire de l’utilisation de la technologie iTRAQ en protéomique
haut débit du protéome urinaire, afin d’en évaluer la variabilité temporelle, chez des patients trans-
plantés rénaux exposés aux Inhibiteurs de la Calcineurine (ICN).
1. Le protéome urinaire
La protéomique permet l’analyse qualitative et quantitative de l’ensemble du matériel pro-
téique d’un système biologique, définissant son protéome. Parmi les différents milieux biologiques
pouvant être exploités (sang, urines, tissus…), les urines constituent un fluide biologique de choix.
Le protéome urinaire se définit comme l’ensemble du matériel protéique excrété dans les
urines (en conditions physiologiques et pathologiques). Les mesures de protéinurie usuelles n’en dé-
tectent qu’une infime portion et sont uniquement quantitatives. L’excrétion urinaire dite physiolo-
gique représente moins de 150 mg de protéinurie par 24 heures. Ce protéome se compose d’un large
spectre de protéines et de peptides de bas poids moléculaire (polypeptides allant de 1000 à 20000 Da)
provenant d’une part de la circulation systémique après filtration glomérulaire (30 %) et d’autre part
de la sécrétion cellulaire rénale (glomérulaire et tubulaire), pour les 70 % restant (6). L’intérêt de son
étude réside dans le fait qu’il s’agit d’un milieu riche et facilement accessible. Son caractère dyna-
mique, susceptible de varier en composition et dans le temps offre théoriquement un reflet en temps
réel des évènements physio-pathologiques (maladies rénales ou systémiques, expositions à des thé-
rapeutiques néphrotoxiques…) en cours à un instant t (7).
L’étude initiale de Spahr et al. en 2001 (8) détectait par spectrométrie de masse (MS) 124
protéines urinaires. La majorité des spectres de MS étaient occupés par les 9 protéines les plus abon-
dantes dans le plasma (human serum albumin, serotransferrin, Ig kappa light chain c, Ig gamma
heavy chain c, uromodulin, apolipoprotein a-i, alpha-1 microglobulin, zinc-alpha-2 glycoprotein,
alpha-1-antitrypsin). Depuis cette époque, et grâce au développement des techniques de MS haute
résolution, plus de 2000 protéines ont été identifiées à ce jour dans des urines humaines saines (9).
Des facteurs de variations physiologiques supplémentaires viennent s’y additionner (âge, sexe, ré-
gime diététique, mode de vie…), susceptibles d’en influencer la composition. C’est de toute cette
dynamique dont il faut tenir compte pour espérer caractériser au mieux le protéome urinaire normal
ou pathologique, afin de dégager dans un second temps d’éventuels biomarqueurs de suivi.
D’un point de vue technique, il s’agit d’un fluide facilement accessible, pouvant être collecté
en grands volumes et de façon répétée, sans caractère invasif. Les autres milieux biologiques tels que
le sang où les tissus sont, quant à eux, limités par le caractère à risque des modalités de prélèvement.
Par exemple, les biopsies rénales exposent à des complications hémorragiques justifiant des mesures
de précaution (hospitalisation, asepsie, échographie, opérateur expérimenté, surveillance…) rendant
la procédure plus complexe, moins facile d’accès, avec un surcoût de prise en charge.
Enfin, ce milieu à l’avantage d’offrir une bonne stabilité protéique, contrairement notamment
aux prélèvements sanguins périphériques dans lesquels peut intervenir l’activation de protéases, dé-
gradant le matériel protéique et limitant ainsi la puissance de détection de la technique (10). A l’in-
verse, la traversée des voies urinaires basses et notamment la vessie peut constituer une source de
contamination potentielle.

8
2. Protéomique quantitative différentielle: place de l’iTRAQ
L’utilisation actuelle des techniques protéomiques est le plus souvent descriptive, évaluant la
situation à un instant t. Le caractère dynamique et complexe de la matrice urinaire rend son explora-
tion difficile par ces techniques. Elles ne permettent pas notamment de suivre l’évolution dans le
temps d’un biomarqueur pour un patient donné. Le recours aux techniques de protéomique différen-
tielle semble pertinent pour contourner cette difficulté.
En protéomique quantitative différentielle, 3 méthodes de marquage sont usuellement utili-
sées : ICAT (Isotope-Coded Affinity Tag), SILAC (Stable Isotope Labeling of Amino-acids in Cell
culture) et iTRAQ (Isobaric Tag for Relative and Absolute Quantitation).
2.a. ICAT et SILAC
Ces sont les deux stratégies les plus anciennes développées en protéomique quantitative dif-
férentielle basée sur la spectrométrie de masse. Elles offrent un marquage de qualité, stable et précis.
Cependant, certaines limites à leur utilisation subsistent.
L’ICAT repose sur un marquage chimique des protéines en greffant par alkylation un réactif
sur les fonctions thiols des résidus cystéines (11). Les tags utilisés existent sous deux formes stables :
lourde (8 atomes de 13C ou 2H) ou légère (8 atomes de 12C ou 1H). Ils sont donc chimiquement iden-
tiques, mais varient par leur composition isotopique. Deux conditions expérimentales sont compa-
rables par cette technique (marquage lourd et léger). Les échantillons tagués sont combinés puis di-
gérés par la trypsine. Lors de l’analyse de spectrométrie en tandem (LC-MS/MS), la quantification
différentielle de l’expression protéique est obtenue par la mesure du rapport « léger/lourd » pour
chaque peptide.
Une limite de l’ICAT est son manque de sensibilité dans la quantification, en raison d’un mar-
quage ciblant uniquement des protéines porteuses de groupements thiols. Cela peut induire une perte
d’information relative aux protéines ne contenant pas cette fonction (y compris en raison de modifi-
cations post-traductionnelles). Par ailleurs, lors de la phase d’élution, un même peptide, selon qu’il
est marqué par le tag lourd ou léger, pourra être libéré à deux temps différents (en raison du différen-
tiel de masse induit par le tag pour un même peptide). Enfin, le fragment biotine entrant dans la
composition du le tag peut induire des perturbations lors de l’interprétation du résultat en LC-MS/MS.
La SILAC utilise une technique de marquage métabolique des protéines. Elle est basée sur
l’incorporation d’acides aminés marqués lourds ou légers au cours de la synthèse protéique en culture
cellulaire (12). Elle nécessite de séparer d’emblée les cultures soumises aux conditions à comparer,
qui seront chacune cultivées en parallèle en présence soit de l’isotope lourd ou léger.
Cette stratégie offre un meilleur marquage que l’ICAT avec un taux d’incorporation du tag avoi-
sinant les 100%. Elle limite également les étapes de préparation chimique et de purification évitant
des biais de manipulations (13). En revanche, elle requiert des lignées cellulaires actives viables pour
assurer l’efficacité du processus de marquage, ce qui n’est pas toujours disponible pour l’ensemble
des échantillons expérimentaux. Surtout, il s’agit d’une technique expérimentale qui n’est pas utili-
sable pour le suivi de patients en routine.
Enfin, la principale faiblesse de ces deux techniques reste le nombre limité à deux conditions
d’études pouvant être comparées (14).

Figure 1 : Le réactif iTRAQ, d’après Zieske, J Exp Bot, 2006 (17)
Figure 2 : Principe iTRAQ, d’après Masse-Spectrométrie.fr, 2016.

9
2.b. iTRAQ
L’iTRAQ a été développée dans un second temps. Elle est basée sur un marquage chimique
par un tag isobarique spécifique de chaque condition (15).
Cette stratégie est aujourd’hui validée comme un outil de protéomique différentielle fiable.
Une supériorité en termes de sensibilité et couverture du protéome lui est même reconnue comparée
à l’ICAT (16). Comparée au SILAC, sa mise en œuvre ne nécessite pas de cultiver préalablement les
cellules dans des milieux différents. Mais l’intérêt majeur de l’iTRAQ est qu’il devient possible de
comparer jusqu’à 8 conditions différentes au cours d’une analyse unique de LC-MS/MS. Cette avan-
cée a été permise par le développement des caractéristiques des tags utilisés en iTRAQ. Ils se com-
posent de trois parties (Figure 1):
- le rapporteur de charge avec une masse allant de 114 à 117 Da, spécifique à chacun des 4
réactifs
- la balance neutre avec une masse allant de 28 à 31 Da, dont le but de maintenir une masse
totale de 145 Da commune aux 4 réactifs
- le groupement réactif peptidique, reconnaissant les groupements amines des peptides.
La combinaison entre le rapporteur et la balance fait toujours 145 Da, et la propriété physico-
chimique reste exactement la même entre les tags. La distinction du marquage réside dans la compo-
sition isotopique du tag en termes de nombre d’isotopes utilisé et leur position (13C, 18O…).
Dans cette méthode, les protéines sont réduites, alkylées et digérées par la trypsine avant d’être
marquées par un des quatre (à huit) réactifs iTRAQ. Les quatre échantillons sont ensuite mélangés de
manière équimolaire. Puis les peptides sont séparés par chromatographie et analysés en LC-MS/MS
(Figure 2).
La nature isobarique des tags utilisés permet, lors d’une 1ère MS, de caractériser chaque pep-
tide détecté selon sa masse et sa charge (ratio m/z) quelle que soit sa condition d’origine. Puis, la
seconde MS permet la quantification relative de chacun des peptides entre les différentes conditions,
grâce à la libération et la détection du tag.
A notre connaissance, l’usage réservé jusqu’ici à l’iTRAQ ainsi qu’aux techniques de protéo-
mique quantitatives antérieures était une comparaison transversale d’échantillons (sang, tissu, urines,
cultures cellulaires) soumis à des conditions variables. Cela a permis de comparer expérimentalement
l’impact de différentes thérapeutiques sur le protéome et de replacer cet effet à l’échelle d’une voie
de signalisation cellulaire. Cela a permis la compréhension de mécanismes physiopathologiques,
l’optimisation des thérapeutiques et l’identification de nouveaux biomarqueurs. Cependant cette ap-
plication reflète uniquement la situation à un instant t, sans offrir de visibilité chronologique. Elle ne
permet pas de ce fait, l’intégration et la mise en perspective dans le temps des résultats obtenus. Avec
la technologie iTRAQ, il devient pourtant possible d’analyser différents échantillons biologiques is-
sus d’un même patient mais prélevé à des temps différents, selon un axe d’étude longitudinal. Le
patient constitue donc son propre témoin et des variations temps-dépendantes physiologiques et/ou
pathologiques, pourraient être décelées. L’originalité de cette démarche apparait dans le fait que la
comparaison d’un protéome porte sur des conditions chronologiques et non plus sur des conditions
d’expositions différentes.
3. Utilisation de l’iTRAQ dans l’étude de la néphrotoxicité des inhibiteurs de la Calci-
neurine (ICN)
La technique iTRAQ semble tout particulièrement intéressante pour analyser la problématique
de la néphrotoxicité des immunosuppresseurs chez l’insuffisant rénal greffé. En effet, l’utilisation des
ICN, Ciclosporine (CsA) et Tacrolimus (TAC), en transplantation rénale a indéniablement révolu-
tionné le pronostic des patients greffés, permettant à celle-ci de devenir la stratégie de suppléance de
choix de l’insuffisance rénale chronique terminale (18). Cependant, les ICN restent grevés d’une né-

Figure 3 : Effets différentiels de la Ciclosporine A et du Tacrolimus sur le cytosquelette
d’actine de cellules LLC-PK1 (microscopie confocale, x100), d’après Descazeaud et al, J
Cell Mol Med, 2012 (24). Une modification globale de la structure cellulaire avec une
rigidification latérale sous membranaire du cytosquelette d’actine est observée in vitro dans des
cellules tubulaires proximales traitées par CsA.
Control CsA 5µM TAC 0.05 µM
Actin

10
phrotoxicité considérée comme inexorable, participant largement à la dysfonction chronique du gref-
fon et à terme à la perte du transplant (19), bien qu’elle soit encore à ce jour mal comprise. La stratégie
de prise en charge du retentissement toxique de ces molécules n’a que peu évolué depuis les débuts
de la greffe, il y a plus de 50 ans. Les marqueurs de routine actuels comprennent toujours la mesure
de la créatinine plasmatique, l’estimation du débit de filtration glomérulaire (DFG) où la recherche
d’une protéinurie, qui sont tous très peu spécifiques et sensibles (20). La référence en matière de
diagnostic reste l’analyse histologique de la ponction biopsie de greffon (21). Cet outil n’est pourtant
ni sans limites ni dénué de risques. En effet, il n’offre de visibilité que pour des lésions à des stades
déjà avancés, ne permettant pas leur dépistage précoce pour une prise en charge efficace. De plus, la
lecture histologique s’avère parfois peu discriminante entre différentes étiologies possibles pouvant
relever de traitements opposés. A cela s’ajoute une part de variabilité inter-observateurs et de biais
d’échantillonnage inhérent à la technique (seulement 0.02% de l’organe total analysé) affaiblissant
sa puissance diagnostique. Enfin, le caractère invasif du geste et ses complications potentielles ne
permettent pas son utilisation répétée dans un objectif de suivit/dépistage (22). Il n’existe d’ailleurs
que peu d’études dans la littérature explorant le retentissement histologique sur le greffon de l’usage
des ICN (23) et leur responsabilité dans la survenue de la dysfonction chronique du greffon. Cela
s’explique par le caractère progressif de l’installation de ces lésions toxiques, avec une longue période
d’évolution infraclinique et infrabiologique.
Face à cette problématique, l’exploitation d’échantillons urinaires pourrait permettre de con-
tourner la difficulté de mise en œuvre d’un suivi histologique longitudinal. L’étude de l’évolution du
protéome urinaire de patients exposés aux ICN pourrait fournir des informations cruciales, permettant
un diagnostic plus précoce et une meilleure compréhension des mécanismes qui sous-tendent cette
néphrotoxicité.
L’équipe de l’unité INSERM UMR-S850 a choisi d’aborder l’étude de cette néphrotoxicité
par une approche sans hypothèse a priori, utilisant la technologie de protéomique différentielle
iTRAQ, in vitro sur des cultures de cellules tubulaires proximales exposées aux ICN et ex vivo sur
des échantillons d’urines de patients issus de la cohorte des patients transplantés rénaux au CHU de
Limoges.
Les premiers résultats issus de cellules tubulaires proximales rénales porcines (LLC-PK1)
exposées aux ICN montraient d’importants remaniements du cytosquelette d’actine associés à une
perte du phénotype épithélial de ces cellules uniquement en condition d’exposition à la CsA. En effet,
les mêmes cellules, exposées à une autre molécule inhibitrice de la Calcineurine, le TAC, ou bien au
VIVIT (peptide inhibiteur sélectif de la déphosphorylation de NFAT) ne montraient aucune modifi-
cation structurelle (Figure 3).
Cependant, les voies de signalisations intracellulaires conduisant à cette réorganisation d’ac-
tine et l’impact de ces modifications du cytosquelette sur le phénotype global des cellules tubulaires
proximales n’étaient pas connus et l’approche protéomique différentielle a été choisie pour en avoir
une description la plus large possible.
Un premier travail s’est intéressé à l’étude des perturbations protéomiques induites par les
ICN dans les cellules embryonnaires rénales HEK-293 par application de la technique la SILAC (25).
Il apparaissait que la CsA et le TAC présentaient des profils d’expression protéique différents par
rapport à la situation contrôle, impactant des mécanismes moléculaires distincts. En effet, la CsA
induisait une surexpression des protéines impliquées dans le stress du réticulum endoplasmique (RE),
l’adressage des protéines, le métabolisme cellulaire, la fibrose rénale/processus pro inflammatoires
via la Basigin (CD147). En parallèle, elle provoquait une sous-expression de la cyclophiline B ainsi
que diverses protéines nucléaires (histones, ribonucléoprotéines = HNRNP) et traductionnelles. Le
TAC quant à lui ne modifiait pas l’expression des marqueurs de stress du RE ou des protéines chape-
ronnes, de même que celle des protéines du métabolisme des ARNm, de la traduction protéique, et
des cyclophilines. Pourtant, dans la littérature, ces deux molécules ICN avaient longtemps été consi-
dérées comme similaires. De nombreux effets observés sur la CsA (introduite de plus longue date,

11
avec un plus grand recul sur l’utilisation et les observations cliniques) avaient été généralisés comme
« effets ICN » et par extrapolation, attribués au TAC. Les résultats SILAC étaient en revanche con-
cordants avec plusieurs études qui avaient relaté un plus faible potentiel néphrotoxique avec le TAC,
traduisant des voies de toxicités différentes de celles de la CsA. Cliniquement, la créatinine plasma-
tique et la filtration glomérulaire étaient montrées comme meilleures sous TAC vs CsA (26-27), et
une amélioration significative de la fonction rénale était observée après switch de la CsA pour le TAC
en cas de dysfonction chronique du greffon où d’effets indésirables associés à la CsA (28). A l’échelle
in vitro, il semblait que le TAC avait à la fois un effet vasoconstricteur rénal (29-31) et un potentiel
fibrogénique plus faible que la CsA (32).
Cependant, ce travail mené avec la technologie SILAC ne comparait pas directement les deux
molécules entre elles. Par conséquent, cela ne permettait pas d’analyser ce qui relevait spécifiquement
de l’inhibition de la calcineurine par rapport aux autres voies intracellulaires potentiellement modi-
fiées par les ICN par le biais de leur interaction avec les immunophilines. La technologie iTRAQ a
donc été mise au point, initialement in vitro, pour répondre à cette problématique. Les premiers ré-
sultats montrant la puissance de cette approche, elle a été adaptée ex vivo pour le suivi longitudinal
des urines de patients.
4. Objectif de travail
L’objectif de ce travail était de mettre au point et de valider la pertinence de l’usage de la
technologie iTRAQ pour le suivi du protéome urinaire des patients transplantés rénaux.
L’objectif si cette stratégie était validée, était à terme de l’appliquer à une population de pa-
tients transplantés rénaux exposés aux différents ICN. La variabilité dans le temps du protéome uri-
naire et son association éventuelle avec des évènements clinico-biologiques où des modifications de
l’histologie rénale seraient ainsi analysés afin de dégager des mécanismes physiopathologiques spé-
cifiques.
Ce mémoire décrit l’étude préliminaire sur cinq patients transplantés rénaux, de mise au point
de la technique iTRAQ, pour le suivi du protéome urinaire.

12
MATÉRIELS ET MÉTHODES
1. Collection des échantillons
Ce travail a été mené sur la cohorte des transplantés rénaux du CHU de Limoges, à partir de
la collection d’urines conservée dans l’unité INSERM UMR S-850 dans le cadre du projet européen
BIOMARGIN et du projet local EPHEGREN.
Il s’agissait d’urines fraiches de milieu de jet, prélevées de façon autonome par le patient lui-
même lors des visites médicales, et secondairement conservées à - 80°C. Les prélèvements urinaires
correspondaient aux M1, M3, M6 et M12 (+/- 1 mois) post-transplantation. Pour chaque patient, ces
dates ont constitué les 4 conditions chronologiques étudiées.
2. Préparation des échantillons
2.a. Concentration des protéines urinaires
La concentration protéique de chaque échantillon a été déterminée par la méthode de Brad-
ford. L’échantillon était comparé en termes d’absorbance avec une gamme étalon préalablement éta-
blie à partir d’une solution mère de concentration connue (Bovine Serum Albumine, Sigma-Aldrich,
à 2 mg. mL-1) dont les différents points de concentration étaient obtenus par dilution au demi (1280
µg.ml-1 ; 640 µg.ml-1 ; 320 µg.ml-1 ; 160 µg.ml-1 ; 80 µg.ml-1 ; 40 µg.ml-1 ; 20 µg.ml-1 ; 10 µg.ml-
1 ; 5 µg.ml-1 ; 2.5 µg.ml-1 ; 1.25 µg.ml-1 ; 0.625 µg.ml-1 ; 0.3125 µg.ml-1 ; 0 µg.ml-1). Chaque point de
gamme et d’échantillon urinaire (correspondant aux urines prélevées à différentes dates pour chaque
patient) étaient déposés en triplicat, sur une plaque 96 puits, et dilués au centième par l’ajout de réactif
de Bradford (Quick Start TM Bradford Dye Reagent 1X, BIO-RAD. La plaque était ensuite lue au
spectrophotomètre (PerkinElmer, Enspire® Multimode Reader, protocole Bradford Assay/Custom) à
595 nm.
A partir de ce premier dosage, un volume d’urines correspondant à 150 µg de protéines était
prélevé et mis à précipiter dans l’acétone glaciale sur une nuit, à -20°C (minimum 12h, dans un vo-
lume d’acétone correspondant à six fois celui de l’échantillon). L’échantillon était ensuite centrifugé
à 5500 rpm, à 4°C, durant 5min, avant le retrait du surnageant à la pipette. Puis le culot était solubilisé
avec une solution de Bicarbonate d’Ammonium NH4CO3 à 100 mM. Une seconde gamme de BSA,
plus concentrée (2000 µg.ml-1 ; 1000 µg.ml-1 ; 500 µg.ml-1 ; 250 µg.ml-1 ; 125 µg.ml-1 ; 62.5 µg.ml-1 ;
31.25 µg.ml-1 ; 0 µg.ml-1), et réalisée dans du NH4CO3 à 100 mM, afin de se situer dans les mêmes
conditions chimiques que celles des échantillons à doser (solvants identiques et gamme adaptée aux
fortes concentrations des échantillons) était utilisée pour un second dosage post-précipitation. Une
fois la concentration finale déterminée, un volume d’échantillon correspondant à 25 µg de protéines
a été prélevé pour chaque condition chronologique chez chaque patient.

13
2.b. Digestion trypsique
Pour chaque volume d’échantillon correspondant à 25 µg de protéines, 1.5 µL de DTT (Di-
thiothreitol, Sigma-Aldrich, 50 mM) étaient ajoutés, suivit d’une incubation de 40 min à 60°C. Cette
étape permettait la réduction des ponts disulfures.
Puis, ont été ajoutés dans le mélange, 1.5 µL d’IDA (Iodoacétamide, Sigma-Aldrich, 100
Mm), et incubé durant 40 minutes à température ambiante, à l’obscurité afin d’obtenir une réaction
de carbamidométhylation des cystéines évitant la reformation de nouveaux ponts disulfures.
Le mélange était ensuite dilué par 54 µL d’eau afin de ne pas dénaturer la trypsine, avant
d’être incubé avec 0.5 µL de trypsine (Sequencing Grade Modified Trypsin, Porcine, Promega), à
37°C, permettant d’obtenir après 24h, la digestion protéique du mélange.
2.c. Solid Phase Extraction (SPE)
Cette étape permettait la concentration et la purification de l’échantillon. Au terme de la di-
gestion trypsique, le mélange était repris dans 10 µL d’acide formique pour stopper l’action de la
trypsine, puis centrifugé à 10000 rpm durant 5 minutes.
La SPE débutait avec le conditionnement des colonnes, par un passage premier de 2 mL mé-
thanol, suivit de 2 mL d’un mélange d’eau + acide formique 0.5%. L’échantillon était ensuite chargé
en totalité dans la colonne. Une étape de lavage par 2.6 mL de mélange d’eau 95% + méthanol 5% +
acide formique 0.5% permettait d’éliminer les composés non intéressants tels que l’urée, les sels.
Enfin l’élution avec 2.6 ml de méthanol pur permettait de décrocher et récupérer les composés pepti-
diques d’intérêt.
L’éluat était ensuite évaporé à sec au speedvacc, puis le culot à nouveau solubilisé dans 50 µL
d’eau 95% + méthanol 5% + acide formique 0.5%. Une dernière étape de centrifugation à 5000 Rpm
pendant 5 minutes était réalisée avant transfert en tube vial.
3. iTRAQ et Spectrométrie de Masse
3.a. Marquage des échantillons
Pour chaque patient, chaque condition (correspondant aux 4 temps post-greffe précédemment
cités) a été marquée par un tag spécifique identifiable par son groupe rapporteur de masse. Il a été
pris soin de ne pas attribuer les tags dans le même ordre chronologique entre les patients, pour s’af-
franchir d’un biais lié à un éventuel effet tag-spécifique.
Dans un premier temps, les tags iTRAQ ont étés repris par 70 µL d’éthanol absolu, puis un
tag spécifique (114117 kDa) a été attribué à chacune des 4 conditions chronologiques de chaque
patient. Le mélange a été homogénéisé par vortex soutenu et centrifugation de paillasse, puis incubé
1 heure à température ambiante.
L’ensemble des 4 échantillons tagués a ensuite été regroupé dans le même tube et homogé-
néisé (vortex soutenu + spin) pour finalement obtenir au total 5 tubes contenants chacun les 4 condi-
tions chronologiques taguées de chaque patient.

14
3.b. Focalisation isoélectrique (Agilent 3100 OFFGEL Fractionator Kit)
Chacun des 5 échantillon-patient, composé du mélange des 4 conditions temporelles, a été
fractionné par focalisation isoélectrique afin d’en faciliter l’analyse. Cette technique repose sur la
séparation des peptides en 12 fractions en exerçant un courant électrique responsable d’une migration
et d’une séparation des peptides en fonction de leur point isoélectrique propre (pHi).
L’échantillon a été préalablement conditionné en additionnant la totalité de son volume avec
1.44 mL de Peptide OFFGEL Stock Solution et un complément d’eau de manière à obtenir un volume
total de 1800 µL. En parallèle, une solution de réhydratation était élaborée en ajoutant 140 µL d’eau
dans 560 µL de Peptide OFFGEL Stock Solution.
Le montage du matériel de focalisation isoélectrique consistait à disposer dans une gouttière
composée de plusieurs couloirs en parallèle, une bande de gel par couloir (bandes pH 3-10), sur la-
quelle une rangée de 12 puits était ensuite déposée et enclenchée. Les bandes de gel étaient secondai-
rement réhydratées par 40 µL de solution de réhydratation par puit. Les gouttières étaient orientées
de telle sorte que l’anode, correspondant à l’électrode fixe, était à gauche et la cathode à droite. Le
montage était complété par la disposition de patins réhydratés au contact du gel aux deux extrémités
de chaque rangée. Puis, dans chaque rangée, 150 µL d’échantillon étaient placés dans chaque puit
avant de les sceller. Enfin, l’ajout d’huile minérale de chaque côté (400µL à l’anode et 1200µL à la
cathode) permettait d’isoler le circuit. La gouttière était ensuite enclenchée dans le réceptacle de l’ap-
pareil, et l’iso-électro-focalisation programmée pour une durée de 24 heures à voltage croissant et
Intensité constante de 50 µA.
3.c. Micro-chromatographie liquide (micro-LC)
Les 12 fractions obtenues pour chacun des 5 patients ont ensuite été récupérées dans 12 tubes
distincts de 1.5 mL, soit un total de 60 tubes. Une étape de lavage supplémentaire de chaque puit,
utilisant 150 µL d’un mélange composé à 50% d’ACN (Acétonitrile, Sigma-Aldrich), 50% d’eau et
0.1% de TFA (Acide Trifluoroacétique, Sigma-Aldrich) durant 15 min à température ambiante, était
systématiquement réalisée. Puis les lavages étaient récupérés et ajoutés à leur tube respectif. Les frac-
tions recueillies étaient ensuite soumises à évaporation et le volume résiduel (≤50µL) transféré dans
les tubes de micro-LC. L’ensemble des échantillons étaient conservés à -20°C avant la microLC et
l’analyse en MS/MS.
La séparation chromatographique des peptides a été effectuée sur un système Ultimate 300
microHPLC et une colonne C18 PepMap, en utilisant un gradient curvilinéaire du solvant B, de 120
min, (Acétonitrile 95% / Eau 5% / Acide formique 0.1%) dans le solvant A (Eau 98% / Acétonitrile
2% / Acide formique 0.1%), comme suit: jusqu'à 5% de B en 5 min, de 5% à 25% de B en 70 min,
de 25% à 95% de B en 12 min, maintenu à 95% de B pendant 10 min, de 95% à 5% en 3 min, maintenu
à 5% pendant 20 min.
3.d. Spectrométrie de masse
La spectrométrie de masse correspond à l’étape permettant de détecter, identifier et quantifier
des molécules d’intérêt par la mesure de leur masse. Pour ce faire, les peptides étudiés ont été chargés
dans un premier temps par leur passage dans une source d’ionisation de type electrospray. La transi-
tion se faisait ensuite par l’analyseur de type TOF (Time Of Flight) où les ions préalablement accélé-
rés par un champ électrique de valeur connue, étaient analysés en fonction du temps qu’ils mettaient
à parcourir une distance donnée jusqu’au détecteur permettant de les séparer selon leur rapport m/z
(masse/charge) ainsi déduit (conversion du courant ionique en courant électrique). Ces résultats
étaient représentés sous forme d’un spectre de masse.
Dans ce travail, cette étape se déclinait en 2 analyses de spectrométrie de masse successives
(TripleTOF 5600, ABSciex). Au cours de la première expérience (MS1) chaque peptide était carac-
térisé selon sa masse (m) et sa charge (z) définissant son rapport m/z spécifique, quel que soit sa

Figure 4: Plaque résumée de la préparation des échantillons, d’après B. Burat

15
condition d’origine (le tag ici ne jouait pas de rôle). La deuxième expérience MS2 se concentrait sur
un seul m/z. Les peptides qui lui étaient associés étaient fragmentés (liaisons peptidiques et tag-pep-
tides), ionisés et analysés par un algorithme qui permettait de remonter alors la séquence peptidique.
En parallèle, le nombre de groupes rapporteurs de masse libérés et détectés pour une séquence pepti-
dique donnée permettait de procéder à une quantification relative déterminant l’évolution de la quan-
tité de chaque peptide entre les différentes conditions (Figure 4).
3.e. Traitement bio-informatique des données de spectrométrie de masse et identifica-
tion/quantification relative des protéines basée sur les intensités de pics.
Les données MS/MS (*.wiff) pour les cinq séries de patients (12 fractions par expérience) ont
été soumises à l'identification de protéines en utilisant le logiciel Mascot Server 2.2.03 via Protein
Pilot (version 5.0, ABSciex) et recherchées dans deux bases de données complémentaires Homo Sa-
piens: SwissProt (version 2015_10) et TrEMBL (version 2015_10). Carbamidomethyl (C), Oxyda-
tion (O), iTRAQ4plex (K), iTRAQ4plex (Y), iTRAQ4plex (terme N) ont été définis en tant que va-
riables modifiables. Aucune modification définitive n'a été définie. Le seuil de tolérance en masse de
fragment a été fixée à 0,3 Da, et du précurseur à 0,2 Da.
Les fichiers de données brutes de Mascot (*.dat, 1 par expérience) ont été sauvegardés pour
des quantifications supplémentaires de peptides et de protéines, avec l'implémentation Java de l'algo-
rithme Quant, jTRAQx (version 1.13, 13). La tolérance en masse du rapporteur a été établie à 0,05
Da. Les facteurs de correction iTRAQ ont été mis en œuvre comme fourni par AB SCIEX. Cet outil
a généré un fichier *.jpf (fichier texte délimité par des tabulations) pour chaque série.
Les données ont été converties, formatées et triées grâce au script personnalisé CiR-C mis au
point dans le laboratoire (B Burat, J Proteome Res, soumis). Les peptides avec des ratios calculés
nuls, les peptides avec un score de Mascot <30 (indice de confiance traduisant la probabilité que le
signal peptide détecté soit due au hasard, à p=0.001, plus le score est élevé, plus cette probabilité est
faible) et les peptides provenant des protéines mal identifiées où annotées « fragmentées » ou « RE-
VERSED » (définissant l’identification de la séquence inverse d’un peptide) ont été éliminés.
Par la suite, la quantification relative de chaque protéine identifiée entre les différentes con-
ditions était effectuée à l’aide des ratios des tag iTRAQ, par rapport à la situation M1. En utilisant la
valeur de la moyenne de distribution (m) des ratios, on définissait le seuil de signification statistique
des variations d’expression obtenus pour chaque protéine, dans chaque condition :
- la sur-expression était définie par : [ratio > m + 1 SD]
- la sous-expression était définie par : [ratio < m – 1 SD]
Plusieurs groupes de protéines étaient individualisés selon leur niveau d’expression par rap-
port à M1: surexprimées, sous exprimées ou invariables entre les différentes conditions. L’interpré-
tation biologique fonctionnelle des données récupérées et classées était alors réalisée par recours à
plusieurs logiciels de traitement bio-informatique disponibles en ligne.
La base de données publique Gene Ontology (GO) donnait en termes biologiques standardi-
sés, les gènes et leurs produits dans 3 catégories : les processus biologiques, les fonctions molécu-
laires, les compartiments cellulaire dans lesquelles elles sont impliquées. Les voies d’enrichissement
de l’analyse fournies par GO étaient évaluées statistiquement (p-value pour que telle protéine soit
impliquée dans telle fonction), en suivant les instructions du logiciel PANTHER. Cet outil offrait la
possibilité d’une classification fonctionnelle des listings protéique selon les voies et processus biolo-
giques dans lesquelles elles étaient impliquées avec une précision jusqu’à un niveau moléculaire.
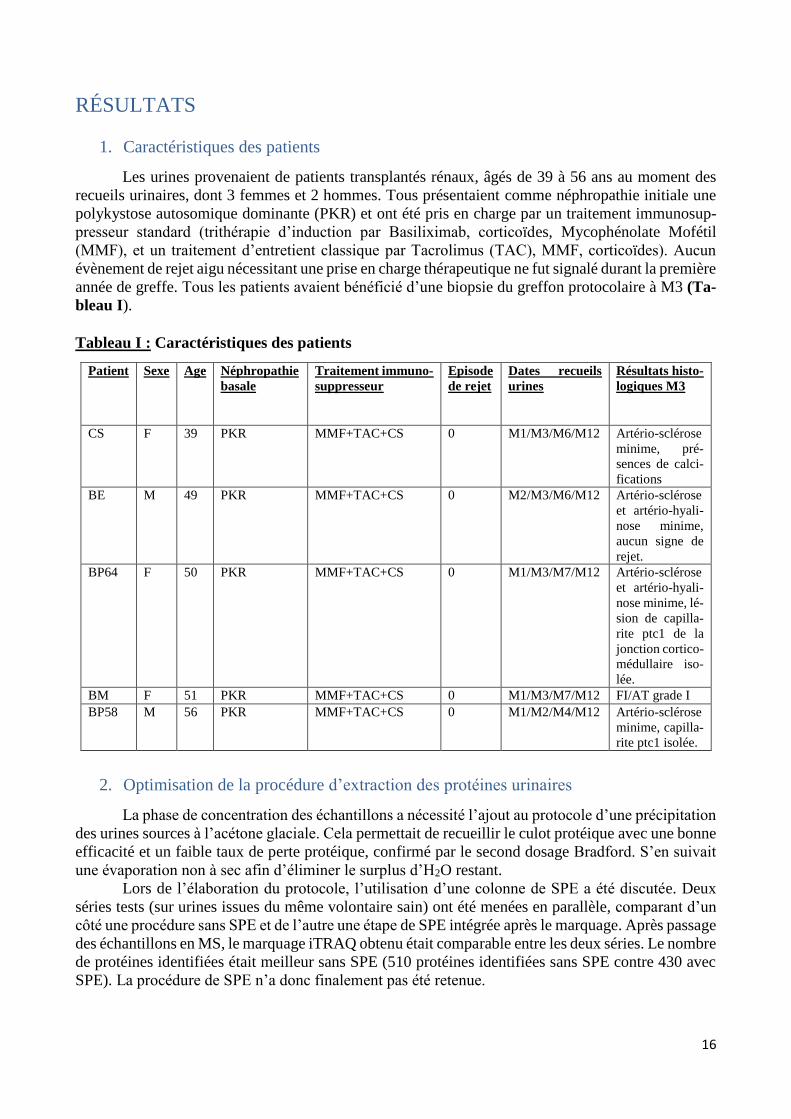
16
RÉSULTATS
1. Caractéristiques des patients
Les urines provenaient de patients transplantés rénaux, âgés de 39 à 56 ans au moment des
recueils urinaires, dont 3 femmes et 2 hommes. Tous présentaient comme néphropathie initiale une
polykystose autosomique dominante (PKR) et ont été pris en charge par un traitement immunosup-
presseur standard (trithérapie d’induction par Basiliximab, corticoïdes, Mycophénolate Mofétil
(MMF), et un traitement d’entretient classique par Tacrolimus (TAC), MMF, corticoïdes). Aucun
évènement de rejet aigu nécessitant une prise en charge thérapeutique ne fut signalé durant la première
année de greffe. Tous les patients avaient bénéficié d’une biopsie du greffon protocolaire à M3 (Ta-
bleau I).
Tableau I : Caractéristiques des patients
Patient Sexe Age Néphropathie
basale
Traitement immuno-
suppresseur
Episode
de rejet
Dates recueils
urines
Résultats histo-
logiques M3
CS F 39 PKR MMF+TAC+CS 0 M1/M3/M6/M12 Artério-sclérose
minime, pré-
sences de calci-
fications
BE M 49 PKR MMF+TAC+CS 0 M2/M3/M6/M12 Artério-sclérose
et artério-hyali-
nose minime,
aucun signe de
rejet.
BP64 F 50 PKR MMF+TAC+CS 0 M1/M3/M7/M12 Artério-sclérose
et artério-hyali-
nose minime, lé-
sion de capilla-
rite ptc1 de la
jonction cortico-
médullaire iso-
lée.
BM F 51 PKR MMF+TAC+CS 0 M1/M3/M7/M12 FI/AT grade I
BP58 M 56 PKR MMF+TAC+CS 0 M1/M2/M4/M12 Artério-sclérose
minime, capilla-
rite ptc1 isolée.
2. Optimisation de la procédure d’extraction des protéines urinaires
La phase de concentration des échantillons a nécessité l’ajout au protocole d’une précipitation
des urines sources à l’acétone glaciale. Cela permettait de recueillir le culot protéique avec une bonne
efficacité et un faible taux de perte protéique, confirmé par le second dosage Bradford. S’en suivait
une évaporation non à sec afin d’éliminer le surplus d’H2O restant.
Lors de l’élaboration du protocole, l’utilisation d’une colonne de SPE a été discutée. Deux
séries tests (sur urines issues du même volontaire sain) ont été menées en parallèle, comparant d’un
côté une procédure sans SPE et de l’autre une étape de SPE intégrée après le marquage. Après passage
des échantillons en MS, le marquage iTRAQ obtenu était comparable entre les deux séries. Le nombre
de protéines identifiées était meilleur sans SPE (510 protéines identifiées sans SPE contre 430 avec
SPE). La procédure de SPE n’a donc finalement pas été retenue.
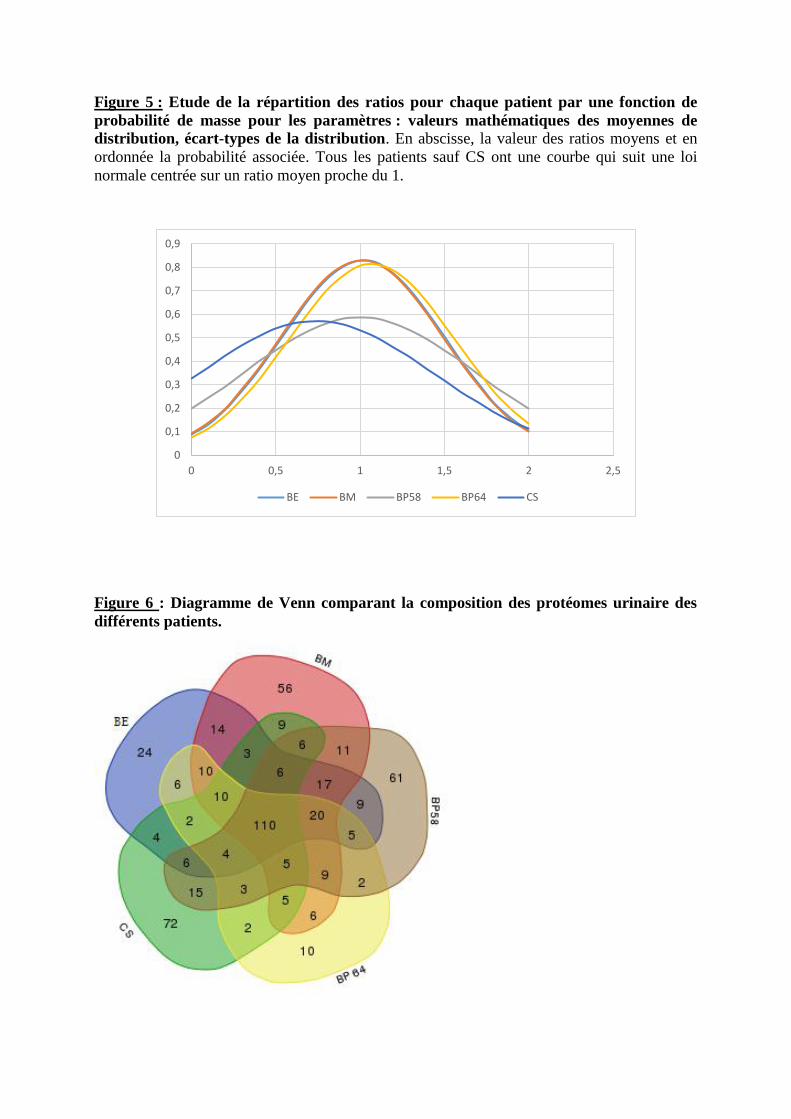
Figure 5 : Etude de la répartition des ratios pour chaque patient par une fonction de
probabilité de masse pour les paramètres : valeurs mathématiques des moyennes de
distribution, écart-types de la distribution. En abscisse, la valeur des ratios moyens et en
ordonnée la probabilité associée. Tous les patients sauf CS ont une courbe qui suit une loi
normale centrée sur un ratio moyen proche du 1.
Figure 6 : Diagramme de Venn comparant la composition des protéomes urinaire des
différents patients.
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
0 0,5 1 1,5 2 2,5
BE BM BP58 BP64 CS
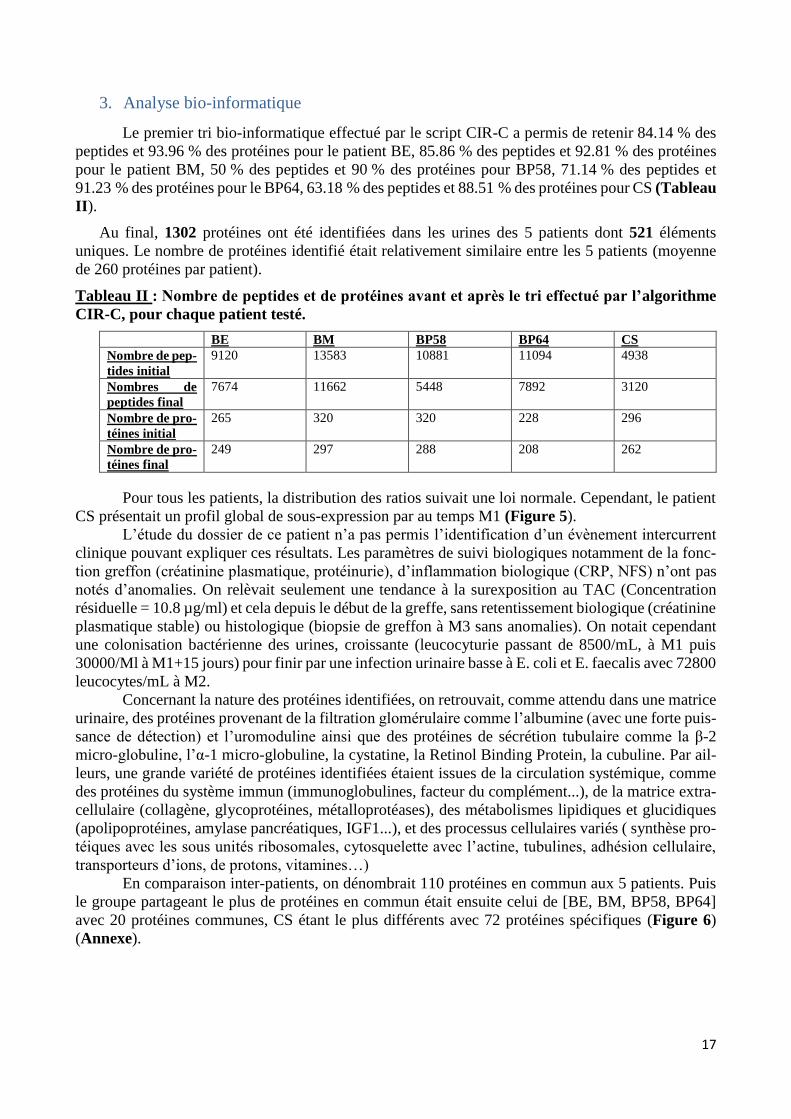
17
3. Analyse bio-informatique
Le premier tri bio-informatique effectué par le script CIR-C a permis de retenir 84.14 % des
peptides et 93.96 % des protéines pour le patient BE, 85.86 % des peptides et 92.81 % des protéines
pour le patient BM, 50 % des peptides et 90 % des protéines pour BP58, 71.14 % des peptides et
91.23 % des protéines pour le BP64, 63.18 % des peptides et 88.51 % des protéines pour CS (Tableau
II).
Au final, 1302 protéines ont été identifiées dans les urines des 5 patients dont 521 éléments
uniques. Le nombre de protéines identifié était relativement similaire entre les 5 patients (moyenne
de 260 protéines par patient).
Tableau II : Nombre de peptides et de protéines avant et après le tri effectué par l’algorithme
CIR-C, pour chaque patient testé.
BE BM BP58 BP64 CS
Nombre de pep-
tides initial
9120 13583 10881 11094 4938
Nombres de
peptides final
7674 11662 5448 7892 3120
Nombre de pro-
téines initial
265 320 320 228 296
Nombre de pro-
téines final
249 297 288 208 262
Pour tous les patients, la distribution des ratios suivait une loi normale. Cependant, le patient
CS présentait un profil global de sous-expression par au temps M1 (Figure 5). L’étude du dossier de ce patient n’a pas permis l’identification d’un évènement intercurrent
clinique pouvant expliquer ces résultats. Les paramètres de suivi biologiques notamment de la fonc-
tion greffon (créatinine plasmatique, protéinurie), d’inflammation biologique (CRP, NFS) n’ont pas
notés d’anomalies. On relèvait seulement une tendance à la surexposition au TAC (Concentration
résiduelle = 10.8 µg/ml) et cela depuis le début de la greffe, sans retentissement biologique (créatinine
plasmatique stable) ou histologique (biopsie de greffon à M3 sans anomalies). On notait cependant
une colonisation bactérienne des urines, croissante (leucocyturie passant de 8500/mL, à M1 puis
30000/Ml à M1+15 jours) pour finir par une infection urinaire basse à E. coli et E. faecalis avec 72800
leucocytes/mL à M2.
Concernant la nature des protéines identifiées, on retrouvait, comme attendu dans une matrice
urinaire, des protéines provenant de la filtration glomérulaire comme l’albumine (avec une forte puis-
sance de détection) et l’uromoduline ainsi que des protéines de sécrétion tubulaire comme la β-2
micro-globuline, l’α-1 micro-globuline, la cystatine, la Retinol Binding Protein, la cubuline. Par ail-
leurs, une grande variété de protéines identifiées étaient issues de la circulation systémique, comme
des protéines du système immun (immunoglobulines, facteur du complément...), de la matrice extra-
cellulaire (collagène, glycoprotéines, métalloprotéases), des métabolismes lipidiques et glucidiques
(apolipoprotéines, amylase pancréatiques, IGF1...), et des processus cellulaires variés ( synthèse pro-
téiques avec les sous unités ribosomales, cytosquelette avec l’actine, tubulines, adhésion cellulaire,
transporteurs d’ions, de protons, vitamines…)
En comparaison inter-patients, on dénombrait 110 protéines en commun aux 5 patients. Puis
le groupe partageant le plus de protéines en commun était ensuite celui de [BE, BM, BP58, BP64]
avec 20 protéines communes, CS étant le plus différents avec 72 protéines spécifiques (Figure 6)
(Annexe).

Figure 7 : Heat maps,
schématisant en couleur, pour
chaque protéine et chaque
patient, en rouge les protéine sur-
exprimées, en blanc celles se
trouvant dans la moyenne de
distribution +/- 1 écart type et en
bleu celle sous-exprimées,
toujours par rapport à M1.
Exemple du patient BM.
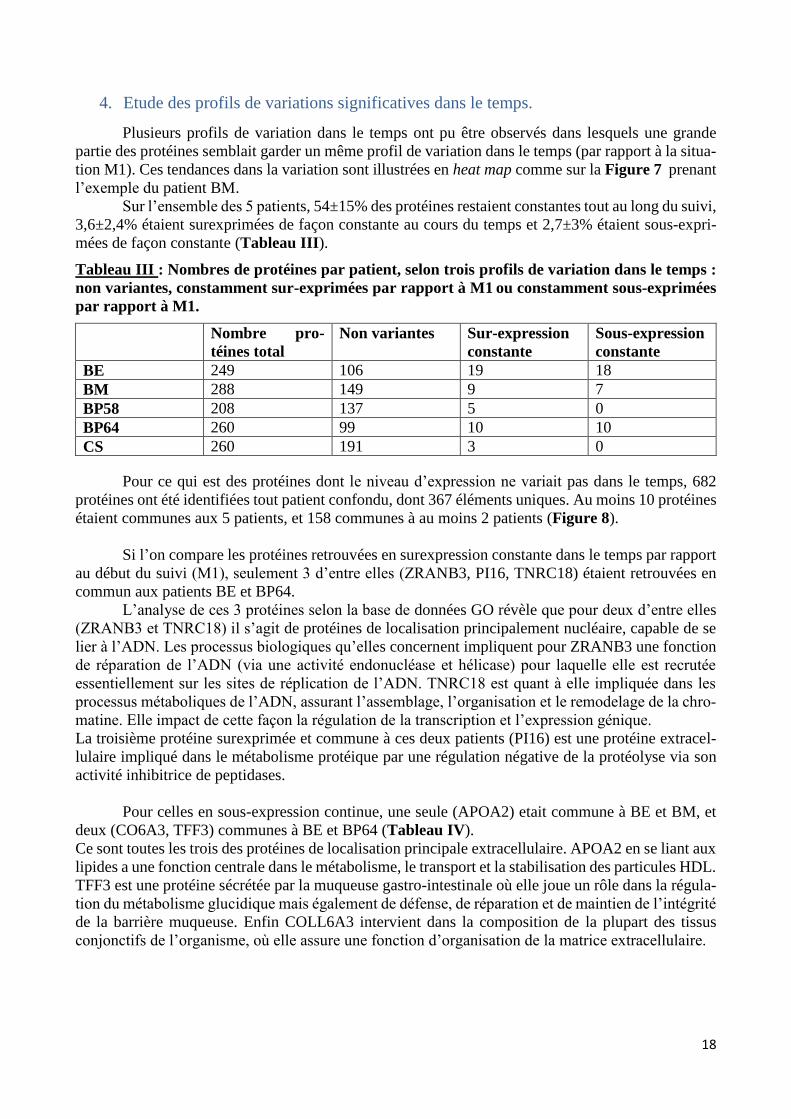
18
4. Etude des profils de variations significatives dans le temps.
Plusieurs profils de variation dans le temps ont pu être observés dans lesquels une grande
partie des protéines semblait garder un même profil de variation dans le temps (par rapport à la situa-
tion M1). Ces tendances dans la variation sont illustrées en heat map comme sur la Figure 7 prenant
l’exemple du patient BM.
Sur l’ensemble des 5 patients, 54±15% des protéines restaient constantes tout au long du suivi,
3,6±2,4% étaient surexprimées de façon constante au cours du temps et 2,7±3% étaient sous-expri-
mées de façon constante (Tableau III).
Tableau III : Nombres de protéines par patient, selon trois profils de variation dans le temps :
non variantes, constamment sur-exprimées par rapport à M1 ou constamment sous-exprimées
par rapport à M1.
Nombre pro-
téines total
Non variantes Sur-expression
constante
Sous-expression
constante
BE 249 106 19 18
BM 288 149 9 7
BP58 208 137 5 0
BP64 260 99 10 10
CS 260 191 3 0
Pour ce qui est des protéines dont le niveau d’expression ne variait pas dans le temps, 682
protéines ont été identifiées tout patient confondu, dont 367 éléments uniques. Au moins 10 protéines
étaient communes aux 5 patients, et 158 communes à au moins 2 patients (Figure 8).
Si l’on compare les protéines retrouvées en surexpression constante dans le temps par rapport
au début du suivi (M1), seulement 3 d’entre elles (ZRANB3, PI16, TNRC18) étaient retrouvées en
commun aux patients BE et BP64.
L’analyse de ces 3 protéines selon la base de données GO révèle que pour deux d’entre elles
(ZRANB3 et TNRC18) il s’agit de protéines de localisation principalement nucléaire, capable de se
lier à l’ADN. Les processus biologiques qu’elles concernent impliquent pour ZRANB3 une fonction
de réparation de l’ADN (via une activité endonucléase et hélicase) pour laquelle elle est recrutée
essentiellement sur les sites de réplication de l’ADN. TNRC18 est quant à elle impliquée dans les
processus métaboliques de l’ADN, assurant l’assemblage, l’organisation et le remodelage de la chro-
matine. Elle impact de cette façon la régulation de la transcription et l’expression génique.
La troisième protéine surexprimée et commune à ces deux patients (PI16) est une protéine extracel-
lulaire impliqué dans le métabolisme protéique par une régulation négative de la protéolyse via son
activité inhibitrice de peptidases.
Pour celles en sous-expression continue, une seule (APOA2) etait commune à BE et BM, et
deux (CO6A3, TFF3) communes à BE et BP64 (Tableau IV).
Ce sont toutes les trois des protéines de localisation principale extracellulaire. APOA2 en se liant aux
lipides a une fonction centrale dans le métabolisme, le transport et la stabilisation des particules HDL.
TFF3 est une protéine sécrétée par la muqueuse gastro-intestinale où elle joue un rôle dans la régula-
tion du métabolisme glucidique mais également de défense, de réparation et de maintien de l’intégrité
de la barrière muqueuse. Enfin COLL6A3 intervient dans la composition de la plupart des tissus
conjonctifs de l’organisme, où elle assure une fonction d’organisation de la matrice extracellulaire.
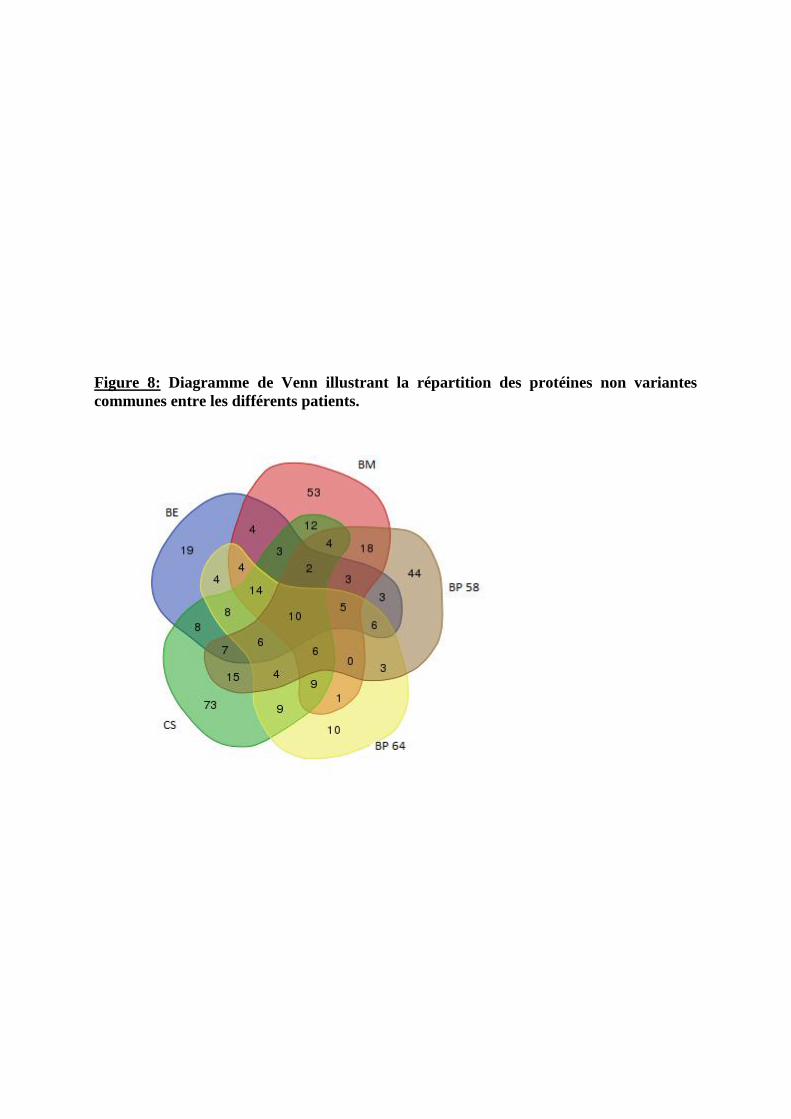
Figure 8: Diagramme de Venn illustrant la répartition des protéines non variantes
communes entre les différents patients.
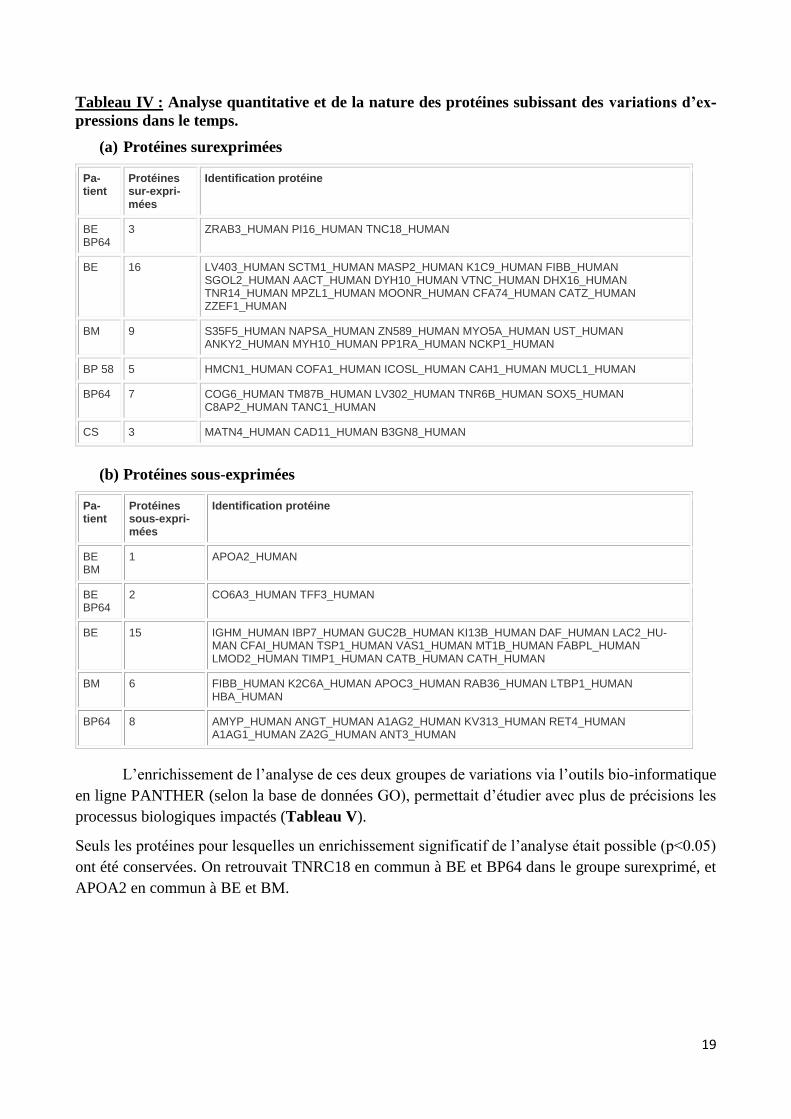
19
Tableau IV : Analyse quantitative et de la nature des protéines subissant des variations d’ex-
pressions dans le temps.
(a) Protéines surexprimées
Pa-tient
Protéines sur-expri-mées
Identification protéine
BE BP64
3 ZRAB3_HUMAN PI16_HUMAN TNC18_HUMAN
BE 16 LV403_HUMAN SCTM1_HUMAN MASP2_HUMAN K1C9_HUMAN FIBB_HUMAN SGOL2_HUMAN AACT_HUMAN DYH10_HUMAN VTNC_HUMAN DHX16_HUMAN TNR14_HUMAN MPZL1_HUMAN MOONR_HUMAN CFA74_HUMAN CATZ_HUMAN ZZEF1_HUMAN
BM 9 S35F5_HUMAN NAPSA_HUMAN ZN589_HUMAN MYO5A_HUMAN UST_HUMAN ANKY2_HUMAN MYH10_HUMAN PP1RA_HUMAN NCKP1_HUMAN
BP 58 5 HMCN1_HUMAN COFA1_HUMAN ICOSL_HUMAN CAH1_HUMAN MUCL1_HUMAN
BP64 7 COG6_HUMAN TM87B_HUMAN LV302_HUMAN TNR6B_HUMAN SOX5_HUMAN C8AP2_HUMAN TANC1_HUMAN
CS 3 MATN4_HUMAN CAD11_HUMAN B3GN8_HUMAN
(b) Protéines sous-exprimées
Pa-tient
Protéines sous-expri-mées
Identification protéine
BE BM
1 APOA2_HUMAN
BE BP64
2 CO6A3_HUMAN TFF3_HUMAN
BE 15 IGHM_HUMAN IBP7_HUMAN GUC2B_HUMAN KI13B_HUMAN DAF_HUMAN LAC2_HU-MAN CFAI_HUMAN TSP1_HUMAN VAS1_HUMAN MT1B_HUMAN FABPL_HUMAN LMOD2_HUMAN TIMP1_HUMAN CATB_HUMAN CATH_HUMAN
BM 6 FIBB_HUMAN K2C6A_HUMAN APOC3_HUMAN RAB36_HUMAN LTBP1_HUMAN HBA_HUMAN
BP64 8 AMYP_HUMAN ANGT_HUMAN A1AG2_HUMAN KV313_HUMAN RET4_HUMAN A1AG1_HUMAN ZA2G_HUMAN ANT3_HUMAN
L’enrichissement de l’analyse de ces deux groupes de variations via l’outils bio-informatique
en ligne PANTHER (selon la base de données GO), permettait d’étudier avec plus de précisions les
processus biologiques impactés (Tableau V).
Seuls les protéines pour lesquelles un enrichissement significatif de l’analyse était possible (p<0.05)
ont été conservées. On retrouvait TNRC18 en commun à BE et BP64 dans le groupe surexprimé, et
APOA2 en commun à BE et BM.
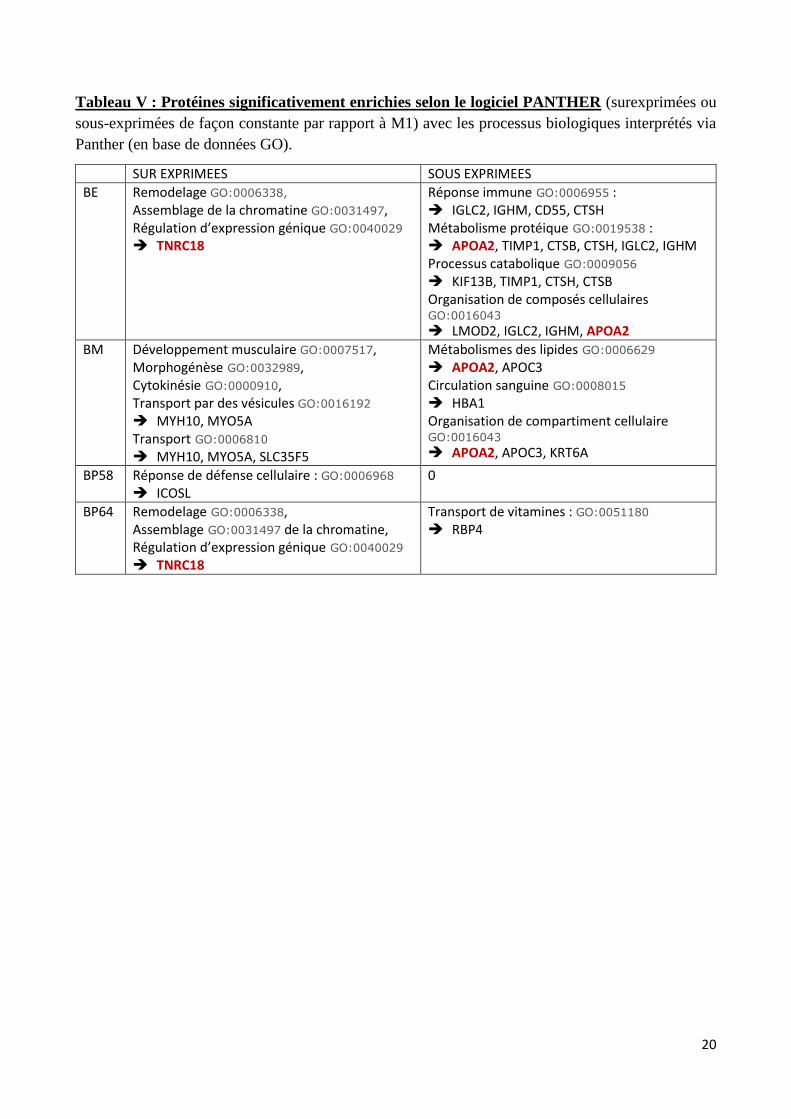
20
Tableau V : Protéines significativement enrichies selon le logiciel PANTHER (surexprimées ou
sous-exprimées de façon constante par rapport à M1) avec les processus biologiques interprétés via
Panther (en base de données GO).
SUR EXPRIMEES SOUS EXPRIMEES
BE Remodelage GO:0006338, Assemblage de la chromatine GO:0031497, Régulation d’expression génique GO:0040029 TNRC18
Réponse immune GO:0006955 : IGLC2, IGHM, CD55, CTSH Métabolisme protéique GO:0019538 : APOA2, TIMP1, CTSB, CTSH, IGLC2, IGHM Processus catabolique GO:0009056 KIF13B, TIMP1, CTSH, CTSB Organisation de composés cellulaires
GO:0016043 LMOD2, IGLC2, IGHM, APOA2
BM Développement musculaire GO:0007517, Morphogénèse GO:0032989, Cytokinésie GO:0000910, Transport par des vésicules GO:0016192 MYH10, MYO5A Transport GO:0006810 MYH10, MYO5A, SLC35F5
Métabolismes des lipides GO:0006629 APOA2, APOC3 Circulation sanguine GO:0008015 HBA1 Organisation de compartiment cellulaire
GO:0016043 APOA2, APOC3, KRT6A
BP58 Réponse de défense cellulaire : GO:0006968 ICOSL
0
BP64 Remodelage GO:0006338, Assemblage GO:0031497 de la chromatine, Régulation d’expression génique GO:0040029 TNRC18
Transport de vitamines : GO:0051180 RBP4

21
DISCUSSION
1. Optimisation du protocole
Au sein du laboratoire, la technologie iTRAQ a été initialement optimisée pour multiplexer
l’analyse de lysats cellulaires. La matrice urinaire constituant un milieu d’étude différent, une mise
au point du protocole a été nécessaire. Pour cela, nous avons eu recours au protocole urinaire utilisé
dans l’unité pour le projet européen en cours BIOMARGIN (identification de nouveaux biomarqueurs
en protéomique et peptidomique dans la thématique du rejet aigu en greffe rénale), en vue d’une
analyse en MS. Nous avons cherché à adapter le protocole « iTRAQ-cellules » en y mêlant la procé-
dure d’extraction des protéines urinaires telle qu’elle est réalisée dans BIOMARGIN ainsi que dans
de rares protocoles d’iTRAQ sur urines retrouvés dans la littérature (1 ; 33-34).
Dans les urines, les protéines sont en phase aqueuse et fortement diluées. Un préalable essen-
tiel fut donc la concentration des échantillons. L’estimation de la concentration protéique de chaque
échantillon constituait la première étape. Les patients sélectionnés ayant recouvré par la greffe une
fonction rénale, leur protéinurie était très faible, de rang physiologique. La gamme de BSA utilisée
pour le dosage par la technique de Bradford a dû être repensée et adaptée d’une part aux faibles
concentrations des urines pures, puis aux fortes concentrations des culots protéiques recueillis après
précipitation des urines. A noter également qu’après précipitation, la gamme a dû être réalisée dans
du NH4CO3 à 100 mM afin de se situer dans les mêmes conditions chimiques que celles dans lesquels
se trouvaient les échantillons à doser (solvant identique). Une mesure fiable et reproductible de la
concentration des échantillons pouvait ainsi être garantie.
Lors des manœuvres initiales d’élaboration du protocole, une étape de SPE était insérée juste
après la digestion trypsique de l’échantillon. Le but était de purifier au maximum le matériel protéique
obtenu en éliminant les résidus de sels, d’urée, d’impuretés pouvant interférer avec le marquage
iTRAQ. On augmentait ainsi en théorie la sensibilité de la technique (en augmentant la concentration
des composés d’intérêt) ainsi que sa reproductibilité et sa résolution puisqu’elle élimine une grande
partie des impuretés qui pourraient modifier la robustesse de l’analyse. Le premier test réalisé de cette
façon a été un échec avec une absence totale de liaisons tag-peptides malgré une détection satisfai-
sante des protéines en MS. Cette interférence dans l’établissement de la liaison du tag au peptide s’est
avérée être liée au pH de la solution de solubilisation du culot protéique après SPE. En effet, l’éluât
était évaporé avant d’être repris dans un mélange acide de méthanol et d’acide formique. Or le pH
optimal pour l’établissement de la liaison tag-peptide se situe en pH alcalin (pH=7-8). Par ailleurs,
l’expérience menée en parallèle comparant un échantillon préparé sans étape de SPE et un échantillon
ayant subi une SPE (solubilisé dans une solution tampon alcaline type NH4CO3, HEPES, MOPS,
n’interférant pas avec le marquage) a montré que le nombre de protéines détectées en MS est légère-
ment meilleur sans SPE (traduisant de possibles pertes protéiques en SPE) avec une couverture du
marquage équivalente. La phase de SPE a donc été supprimée du protocole. Son utilisation reste
possible à condition de contrôler le pH du milieu avant le marquage. On peut aussi discuter son utili-
sation en phase post-marquage afin d’éliminer l’éventuel surplus de tags par rapport aux peptides où
encore des résidus de glycérol (utilisé en isoéléctrofocalisation) pouvant gêner la lecture de l’échan-
tillon en MS.
Lors de la mise au point de l’iTRAQ sur cultures cellulaires, une validation de la technique a
été obtenue en parallèle par Western blot (WB) considérée comme le référentiel gold standard en
matière de quantification protéique (B Burat, J Proteome Res, soumis). Le projet actuel reprenant
finalement les mêmes conditions de réalisation (remplaçant uniquement le lysat cellulaire par un culot
protéique), l’étape de validation de la technique par WB n’a pas été jugé nécessaire.
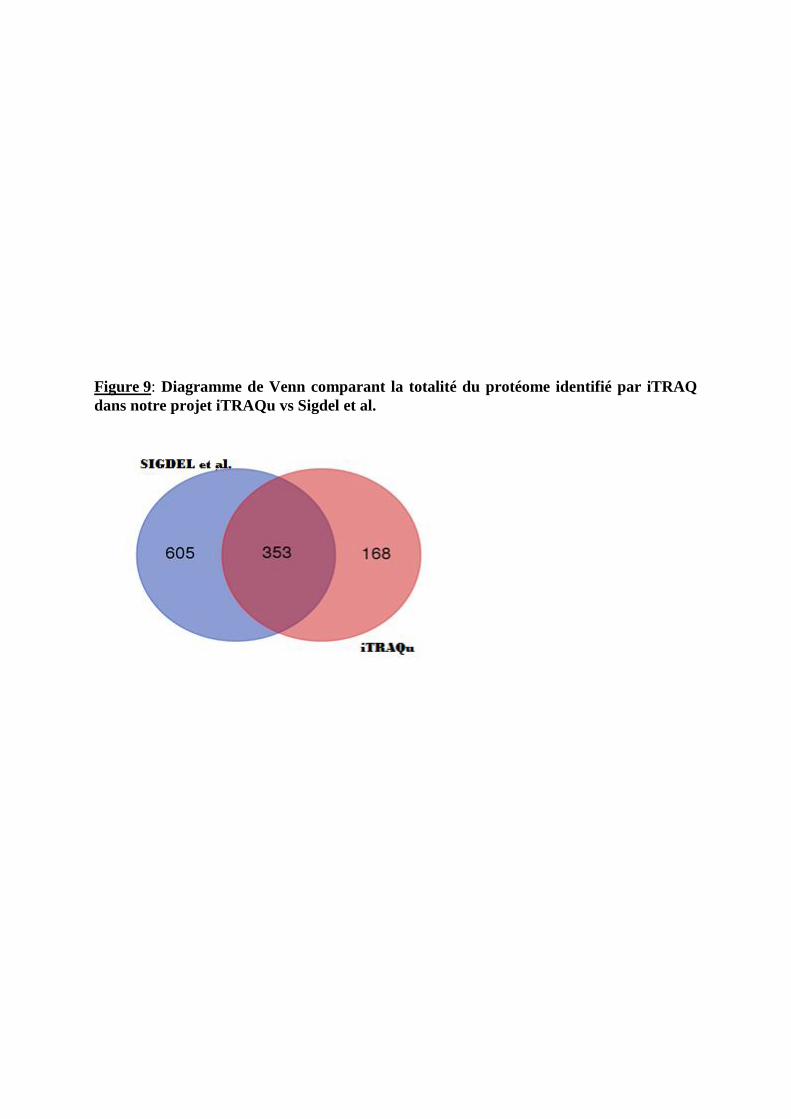
Figure 9: Diagramme de Venn comparant la totalité du protéome identifié par iTRAQ
dans notre projet iTRAQu vs Sigdel et al.

22
Notre travail permet de montrer que le suivi du protéome urinaire par technologie iTRAQ est
possible avec plusieurs points forts :
- une bonne sensibilité de détection, liée au couplage de la technique avec la MS (près de
260 protéines identifiées en moyenne par échantillon de 25µg de protéines)
- une bonne reproductibilité, tant en termes de nombre absolu de protéines identifiées (entre
200 et 300 éléments par échantillon), mais également sur la valeur des moyennes de dis-
tribution calculées à partir des ratios de médianes obtenus entre les conditions.
- une grande variété de protéines observées, malgré une forte représentation de certaines
protéines physiologiquement majoritaires dans le milieu urinaire (Albumine par exemple),
sans phénomène d’étouffement des capacités de détection des éléments minoritaires.
- une cohérence des protéines identifiées, puisqu’on retrouve comme attendu des protéines
issues de la filtration rénale et de la sécrétion tubulaire (Albumine, Uromoduline, Cysta-
tine…) ainsi que de la circulation systémique (Immunoglobulines, facteurs du Complé-
ment…) et non pas que des protéines issues du milieu vésical.
Dans la littérature, peu d’études ont utilisé l’iTRAQ en protéomique sur des urines (1 ; 33), et
aucune jusqu’ici dans une stratégie d’étude longitudinale à la recherche de variation dans le temps du
protéome urinaire.
Une analyse comparative de la composition protéique du protéome urinaire obtenu par
l’équipe de Sigdel en 2016 (33) et notre propre étude (Figure 9) retrouve un total de 521 éléments
unique dans notre projet (pool de 5 patients, à 4 temps différents, soit 20 échantillons de 25µg de
protéines brutes) contre 958 éléments uniques dans le travail de Sigdel et al. (pool de 108 échantil-
lons), soit un total de 1126 éléments uniques. Parmi eux, 168 protéines sont spécifiques à notre travail,
605 à celui de l’équipe de Sigdel et 353 sont communes aux deux.
Cette comparaison, réalisée à titre indicatif, possède l’avantage de croiser des données de pro-
téomique urinaire obtenues par technologie iTRAQ. En revanche, il est difficile d’approfondir cette
analyse en raison d’importantes différences dans la méthodologie appliquée entre les deux travaux.
En effet, le nombre d’échantillons et la population testée divergent (5 échantillons de greffés rénaux
sains dans notre projet vs 108 échantillons issus de greffés rénaux en rejet aigu, avec néphropathie à
BK virus où néphropathie chronique d’allogreffe chez Sigdel et al.) tout comme les conditions com-
parées (puisque nous comparions des temps différents et non des conditions cliniques pathologiques
différentes comme dans le travail de Sigdel et al.)
2. Choix des patients
Le choix d’une population de patients appartenant au groupe des malades greffés rénaux s’ex-
plique par la dynamique des travaux de recherche en cours au laboratoire, dont le but est l’étude de
la néphrotoxicité des traitements immunosuppresseurs et notamment les ICN. Il semblait donc justifié
de développer d’emblée la technique sur cette population cible, afin de répondre au mieux à ces ap-
plications futures.
Dans un souci d’homogénéisation des échantillons et de reproductibilité des résultats, les pa-
tients ont été sélectionnés avec une certaine comparabilité en termes d’âge, de néphropathie basale
(PKR), de traitements suivis (TAC), et de succès de la greffe a 1 an.
Enfin, le choix d’une population spécifique comme les greffés rénaux offre l’avantage d’une
accessibilité relativement aisée aux urines avec notamment des échantillons conservés dans le temps
dans le cadre du suivi médical, permettant une étude dans le temps comme il en était question dans
ce travail.

23
3. Echec du patient CS
Parmi les 5 patients étudiés, les résultats obtenus sur le patient CS se distinguent avec une
moyenne de distribution des ratios éloignée du 1. De plus, on observait un plus faible nombre de
protéines communes partagées avec les protéomes urinaires des autres patients.
Cette différence ne semblait pas être due à la technique. En effet, la détection montrait un
nombre de protéines semblable aux autres échantillons et un bon marquage par les tags, permettant
une quantification relative des protéines selon les différentes conditions chronologiques.
Le seul évènement intercurrent identifié dans l’histoire clinique du patient est celui d’une in-
fection urinaire basse concomitante à la réalisation des recueils urinaires. Une modification de la
composition du protéome par les germes présents et l’inflammation associée pourrait constituer une
hypothèse explicative aux différences observées dans les résultats de ce patient.
4. Limites
Un certain nombre de limites sont à soulever :
- Un faible nombre de patient a été recruté dans notre étude. Ce choix a été pris car ce
nombre suffisait pour la mise au point de la technique, sans consommer des échantillons
précieux.
- La reproductibilité intrinsèque de la technique, c’est-à-dire du coefficient de variabilité
intra-patient, n’a pas pu être testée en raison d’un manque de temps (suite à un problème
technique sur la chaine de micro-LC, la rendant indisponible pendant plusieurs mois). De
plus nous disposions d’une faible quantité d’échantillons résiduelle après réalisation des
tests.
- L’analyse statistique obtenue reste très préliminaire et ne permet pas de débuter une inter-
prétation poussée sur les protéines identifiées.
5. Conclusion
Ce travail confirme la possibilité de l’utilisation de la technologie iTRAQ dans l’étude du
protéome urinaire. Son application originale dans un schéma d’étude longitudinal suivant des condi-
tions chronologiques semble prometteuse dans l’appréciation des variabilités inter et intra indivi-
duelle des protéomes urinaires de patients dans le temps.
Dans le domaine plus spécifique de la greffe rénale et de la néphrotoxicité des ICN, l’iTRAQ
constitue un outil sérieux, permettant d’élargir la compréhension des mécanismes en jeu et l’identifi-
cation de nouveaux biomarqueurs. Ce travail constitue un préliminaire à la mise en application de
l’iTRAQ dans cette thématique au sein d’études de plus grande envergure.

24
REFERENCES BIBLIOGRAPHIQUES
(1) Sigdel TK, Salomonis N, Nicora CD, Ryu S, He J, Dinh V, et al. The Identification of Novel
Potential Injury Mechanisms and Candidate Biomarkers in Renal Allograft Rejection by Quan-
titative Proteomics. Mol & Cell Proteomics, 2014, 13 : 621‑631.
(2) Rocchetti MT, Centra M, Papale M, Bortone G, Palermo C, Centonze D, et al. Urine Protein
Profile of IgA Nephropathy Patients May Predict the Response to ACE-Inhibitor Therapy. Pro-
teomics, 2008, 8 : 206‑216.
(3) Frantzi M, Metzger J, Banks RE, Husi H, Klein J, Dakna M, et al. Discovery and validation of
urinary biomarkers for detection of renal cell carcinoma. J Proteomics, 2014, 98 :44-58.
(4) Davalieva K, Kiprijanovska S, Komina S, Petrusevska G, Chokrevska Zografska N, Polena-
kovic M. Proteomics Analysis of Urine Reveals Acute Phase Response Proteins as Candidate
Diagnostic Biomarkers for Prostate Cancer. Proteome Sci, 2015, 13 : 2.
(5) Smith CR., Batruch I, Bauça JM, Kosanam H, Ridley J, Bernardini MQ, et al. Deciphering the
Peptidome of Urine from Ovarian Cancer Patients and Healthy Controls. Clin Proteomics,
2014, 11 : 23.
(6) Bauça JM, Martínez-Morillo E, Diamandis EP. Peptidomics of Urine and Other Biofluids for
Cancer Diagnostics. Clin Chem, 2014, 60 : 1052‑61.
(7) Beasley-Green A. Urine Proteomics in the Era of Mass Spectrometry. Int Neurourol J, 2016,
20 : 70-75.
(8) Spahr CS, Davis MT, McGinley MD, Robinson JH, Bures EJ, Beierle J, et al. Towards Defining
the Urinary Proteome Using Liquid Chromatography-Tandem Mass Spectrometry. I. Profiling
an Unfractionated Tryptic Digest. Proteomics, 2001, 1 : 93‑107.
(9) Marimuthu A, O’Meally RN, Chaerkady R, Subbannayya Y, Nanjappa V, Kumar P, et al. A
Comprehensive Map of the Human Urinary Proteome. J Proteome Res, 2011, 10 : 2734‑43.
(10) Di Meo A, Batruch I, Yousef AG, Pasic MD, Diamandis EP, Yousef GM. An Integrated Pro-
teomic and Peptidomic Assessment of the Normal Human Urinome. Clin Chem Lab Med, 2017,
55 : 237‑47.
(11) Gygi, SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative Analysis of
Complex Protein Mixtures Using Isotope-Coded Affinity Tags. Nat Biotechnol, 1999, 17 :
994‑99.
(12) Ong S, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Stable Isotope
Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Ex-
pression Proteomics. Mol Cell Proteomics, 2002, 1 : 376‑86.
(13) Chen X, Wei S, Ji Y, Guo X, Yang F. Quantitative Proteomics Using SILAC: Principles, Ap-
plications, and Developments. Proteomics, 2015, 15 : 3175‑92.
(14) These lamoureux
(15) Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, et al. Multiplexed Pro-
tein Quantitation in Saccharomyces Cerevisiae Using Amine-Reactive Isobaric Tagging Rea-
gents. Mol Cell Proteomics, 2004, 3 : 1154‑69.
(16) Wells
(17) Zieske LR. A Perspective on the Use of ITRAQ Reagent Technology for Protein Complex and
Profiling Studies. J Exp Bot, 2006, 57 : 1501‑8.
(18) Wolfe RA, Ashby VB, Milford EL, Ojo AO, Ettenger RE, Agodoa LY, et al. Comparison of
Mortality in All Patients on Dialysis, Patients on Dialysis Awaiting Transplantation, and Re-
cipients of a First Cadaveric Transplant. N Engl J Med, 1999, 341 : 1725‑30.
(19) Naesens M, Kuypers D, Sarwal M. Calcineurin Inhibitor Nephrotoxicity. Clin J Am Soc
Nephrol, 2009, 4 : 481‑508.
(20) Anglicheau D, Naesens M, Essig M, Gwinner W, Marquet P. Establishing Biomarkers in Trans-
plant Medicine: A Critical Review of Current Approaches. Transplantation, 2016, 100 :

25
2024‑38.
(21) Henderson LK, Nankivell BJ, Chapman JR. Surveillance Protocol Kidney Transplant Biopsies
: Their Evolving Role in Clinical Practice. Am J Transplant, 2011, 11 : 1570‑75.
(22) Redfield RR, McCune KR, Rao A, Sadowski E, et al. Nature, Timing, and Severity of Compli-
cations from Ultrasound-Guided Percutaneous Renal Transplant Biopsy. Transplant Int, 2016,
29 : 167‑72.
(23) Nankivell BJ, Borrows RJ, Fung CLS, O’Connell PJ, Chapman JR, Allen RDM. Calcineurin
Inhibitor Nephrotoxicity: Longitudinal Assessment by Protocol Histology. Transplantation,
2004, 78 : 557‑65.
(24) Descazeaud V, Mestre E, Marquet P, Essig M. Calcineurin regulation of cytoskeleton organi-
zation: a new paradigm to analyse the effects of calcineurin inhibitors on the kidney. J Cell Mol
Med, 2012, 16 : 218‑27.
(25) Lamoureux F, Gastinel LN, Mestre E, Marquet P, Essig M. Mapping Cyclosporine-Induced
Changes in Protein Secretion by Renal Cells Using Stable Isotope Labeling with Amino Acids
in Cell Culture (SILAC). J Proteomics, 2012, 75 : 3674‑87.
(26) Jurewicz, AW. Tacrolimus versus Cyclosporin Immunosuppression: Long-Term Outcome in
Renal Transplantation. Nephrol Dial Transplant, 2003, 18 : i7-11.
(27) Kaplan B, Schold JD, Meier-Kriesche H. Long-Term Graft Survival with Neoral and Tacroli-
mus : A Paired Kidney Analysis. J Am Soc Nephrol, 2003, 14 : 2980‑84.
(28) Shihab FS, Waid TH, Conti DJ, Yang H, Holman MJ, Mulloy LC, et al. Conversion from Cy-
closporine to Tacrolimus in Patients at Risk for Chronic Renal Allograft Failure : 60-Month
Results of the CRAF Study. Transplantation, 2008, 85 : 1261‑69.
(29) Bagnis C, Deray G, Dubois M, Adabra Y, Jacquiaud C, Jaudon MC, et al. Comparative Acute
Nephrotoxicity of FK-506 and Ciclosporin in an Isolated in Situ Autoperfused Rat Kidney
Model. Am J Nephrol 1997, 17 : 17‑24.
(30) Gardiner SM., March JE, Kemp PA, Fallgren B, Bennett T. Regional Haemodynamic Effects
of Cyclosporine A, Tacrolimus and Sirolimus in Conscious Rats. Br J Pharmacol, 2004, 141 :
634‑43.
(31) Klein I, Abrahams A, Van Ede T, Hené RJ, Koomans HA, Ligtenberg G. Different Effects of
Tacrolimus and Cyclosporine on Renal Hemodynamics and Blood Pressure in Healthy Sub-
jects. Transplantation, 2002, 73 : 732‑36.
(32) Jain S, Bicknell GR, Nicholson ML. Tacrolimus Has Less Fibrogenic Potential than Cyclo-
sporin A in a Model of Renal Ischaemia-Reperfusion Injury. Br J Pharmacol, 2000, 87 :
1563‑68.
(33) Sigdel TK, Gao Y, He J, Wang A, Nicora CD, Fillmore TL, et al. Mining the Human Urine
Proteome for Monitoring Renal Transplant Injury. Kidney Int, 2016, 89 : 1244‑52.
(34) Sigdel TK., Nicora CD, Hsieh SC, Dai H, Qian WJ, Camp DG, et al. Optimization for Peptide
Sample Preparation for Urine Peptidomics. Clin Proteomics, 2014, 11 : 7.
(35) Clipstone NA, Crabtree GR. Identification of Calcineurin as a Key Signalling Enzyme in T-
Lymphocyte Activation. Nature, 1992, 357 : 695‑97.
(36) Becknell B, Greenbaum LA, Smoyer WE. A New “Tac” for Childhood Nephrotic Syndrome.
Kidney Int, 2012, 82 : 1049‑51.
(37) Pirsch JD, Miller J, Deierhoi MH, Vincenti F, Filo RS. A Comparison of Tacrolimus (FK506)
and Cyclosporine for Immunosuppression after Cadaveric Renal Transplantation. FK506 Kid-
ney Transplant Study Group. Transplantation, 1997, 63 : 977‑83.
(38) Margreiter R, European Tacrolimus vs Ciclosporin Microemulsion Renal Transplantation
Study Group. Efficacy and Safety of Tacrolimus Compared with Ciclosporin Microemulsion in
Renal Transplantation : A Randomised Multicentre Study. Lancet, 2002, 359 : 741‑46.
(39) Martins L, Ventura A, Branco A, Carvalho MJ, Henriques AC, Dias L, et al. Cyclosporine

26
versus Tacrolimus in Kidney Transplantation : Are There Differences in Nephrotoxi-
city? Transplant Proc, 2004, 36 : 877‑79.
(40) Lamoureux F, Mestre E, Essig M, Sauvage FL, Marquet P, Gastinel LN. Quantitative Proteomic
Analysis of Cyclosporine-Induced Toxicity in a Human Kidney Cell Line and Comparison with
Tacrolimus. J Proteomics, 2011, 75 : 677‑94.
(41) Justo P, Lorz C, Sanz A, Egido J, Ortiz A. Intracellular Mechanisms of Cyclosporin A-Induced
Tubular Cell Apoptosis. J Am Soc Nephrol, 2003, 14 : 3072‑80.
(42) Djamali A. Oxidative Stress as a Common Pathway to Chronic Tubulointerstitial Injury in Kid-
ney Allografts. Am J Physiol Renal Physiol, 2007, 293 : F445-455.
(43) Bravo-Cordero, Jose Javier, Marco A. Magalhaes O, Eddy RJ, Hodgson L, Condeelis J. Func-
tions of Cofilin in Cell Locomotion and Invasion. Nat Rev Mol Cell Biol, 2013, 14 : 405‑15.
(44) Kurtz A, Della Bruna R, Kühn K. Cyclosporine A Enhances Renin Secretion and Production in
Isolated Juxtaglomerular Cells. Kidney Int, 1988, 33 : 947‑53.
(45) Pichler RH, Franceschini N, Young BA, Hugo C, Andoh TF, Burdmann EA, et al. Pathogene-
sis of Cyclosporine Nephropathy: Roles of Angiotensin II and Osteopontin. J Am Soc Nephrol,
1995, 6 : 1186‑96.
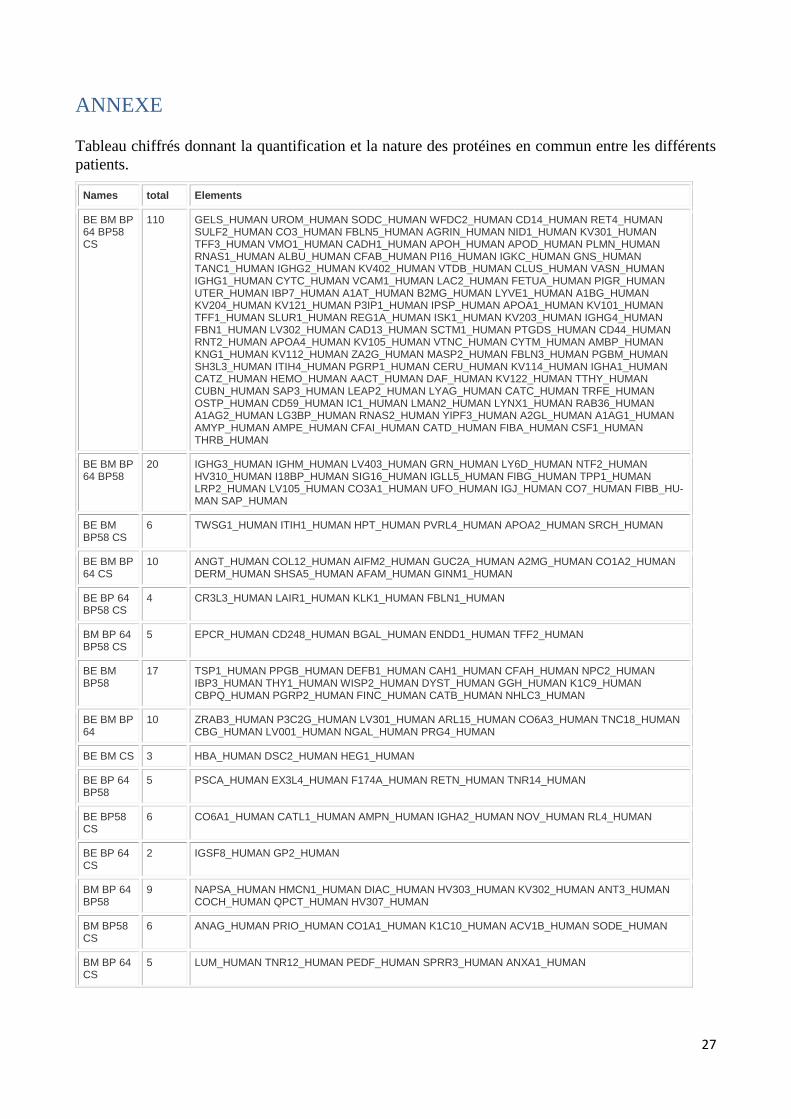
27
ANNEXE
Tableau chiffrés donnant la quantification et la nature des protéines en commun entre les différents
patients.
Names total Elements
BE BM BP 64 BP58 CS
110 GELS_HUMAN UROM_HUMAN SODC_HUMAN WFDC2_HUMAN CD14_HUMAN RET4_HUMAN SULF2_HUMAN CO3_HUMAN FBLN5_HUMAN AGRIN_HUMAN NID1_HUMAN KV301_HUMAN TFF3_HUMAN VMO1_HUMAN CADH1_HUMAN APOH_HUMAN APOD_HUMAN PLMN_HUMAN RNAS1_HUMAN ALBU_HUMAN CFAB_HUMAN PI16_HUMAN IGKC_HUMAN GNS_HUMAN TANC1_HUMAN IGHG2_HUMAN KV402_HUMAN VTDB_HUMAN CLUS_HUMAN VASN_HUMAN IGHG1_HUMAN CYTC_HUMAN VCAM1_HUMAN LAC2_HUMAN FETUA_HUMAN PIGR_HUMAN UTER_HUMAN IBP7_HUMAN A1AT_HUMAN B2MG_HUMAN LYVE1_HUMAN A1BG_HUMAN KV204_HUMAN KV121_HUMAN P3IP1_HUMAN IPSP_HUMAN APOA1_HUMAN KV101_HUMAN TFF1_HUMAN SLUR1_HUMAN REG1A_HUMAN ISK1_HUMAN KV203_HUMAN IGHG4_HUMAN FBN1_HUMAN LV302_HUMAN CAD13_HUMAN SCTM1_HUMAN PTGDS_HUMAN CD44_HUMAN RNT2_HUMAN APOA4_HUMAN KV105_HUMAN VTNC_HUMAN CYTM_HUMAN AMBP_HUMAN KNG1_HUMAN KV112_HUMAN ZA2G_HUMAN MASP2_HUMAN FBLN3_HUMAN PGBM_HUMAN SH3L3_HUMAN ITIH4_HUMAN PGRP1_HUMAN CERU_HUMAN KV114_HUMAN IGHA1_HUMAN CATZ_HUMAN HEMO_HUMAN AACT_HUMAN DAF_HUMAN KV122_HUMAN TTHY_HUMAN CUBN_HUMAN SAP3_HUMAN LEAP2_HUMAN LYAG_HUMAN CATC_HUMAN TRFE_HUMAN OSTP_HUMAN CD59_HUMAN IC1_HUMAN LMAN2_HUMAN LYNX1_HUMAN RAB36_HUMAN A1AG2_HUMAN LG3BP_HUMAN RNAS2_HUMAN YIPF3_HUMAN A2GL_HUMAN A1AG1_HUMAN AMYP_HUMAN AMPE_HUMAN CFAI_HUMAN CATD_HUMAN FIBA_HUMAN CSF1_HUMAN THRB_HUMAN
BE BM BP 64 BP58
20 IGHG3_HUMAN IGHM_HUMAN LV403_HUMAN GRN_HUMAN LY6D_HUMAN NTF2_HUMAN HV310_HUMAN I18BP_HUMAN SIG16_HUMAN IGLL5_HUMAN FIBG_HUMAN TPP1_HUMAN LRP2_HUMAN LV105_HUMAN CO3A1_HUMAN UFO_HUMAN IGJ_HUMAN CO7_HUMAN FIBB_HU-MAN SAP_HUMAN
BE BM BP58 CS
6 TWSG1_HUMAN ITIH1_HUMAN HPT_HUMAN PVRL4_HUMAN APOA2_HUMAN SRCH_HUMAN
BE BM BP 64 CS
10 ANGT_HUMAN COL12_HUMAN AIFM2_HUMAN GUC2A_HUMAN A2MG_HUMAN CO1A2_HUMAN DERM_HUMAN SHSA5_HUMAN AFAM_HUMAN GINM1_HUMAN
BE BP 64 BP58 CS
4 CR3L3_HUMAN LAIR1_HUMAN KLK1_HUMAN FBLN1_HUMAN
BM BP 64 BP58 CS
5 EPCR_HUMAN CD248_HUMAN BGAL_HUMAN ENDD1_HUMAN TFF2_HUMAN
BE BM BP58
17 TSP1_HUMAN PPGB_HUMAN DEFB1_HUMAN CAH1_HUMAN CFAH_HUMAN NPC2_HUMAN IBP3_HUMAN THY1_HUMAN WISP2_HUMAN DYST_HUMAN GGH_HUMAN K1C9_HUMAN CBPQ_HUMAN PGRP2_HUMAN FINC_HUMAN CATB_HUMAN NHLC3_HUMAN
BE BM BP 64
10 ZRAB3_HUMAN P3C2G_HUMAN LV301_HUMAN ARL15_HUMAN CO6A3_HUMAN TNC18_HUMAN CBG_HUMAN LV001_HUMAN NGAL_HUMAN PRG4_HUMAN
BE BM CS 3 HBA_HUMAN DSC2_HUMAN HEG1_HUMAN
BE BP 64 BP58
5 PSCA_HUMAN EX3L4_HUMAN F174A_HUMAN RETN_HUMAN TNR14_HUMAN
BE BP58 CS
6 CO6A1_HUMAN CATL1_HUMAN AMPN_HUMAN IGHA2_HUMAN NOV_HUMAN RL4_HUMAN
BE BP 64 CS
2 IGSF8_HUMAN GP2_HUMAN
BM BP 64 BP58
9 NAPSA_HUMAN HMCN1_HUMAN DIAC_HUMAN HV303_HUMAN KV302_HUMAN ANT3_HUMAN COCH_HUMAN QPCT_HUMAN HV307_HUMAN
BM BP58 CS
6 ANAG_HUMAN PRIO_HUMAN CO1A1_HUMAN K1C10_HUMAN ACV1B_HUMAN SODE_HUMAN
BM BP 64 CS
5 LUM_HUMAN TNR12_HUMAN PEDF_HUMAN SPRR3_HUMAN ANXA1_HUMAN
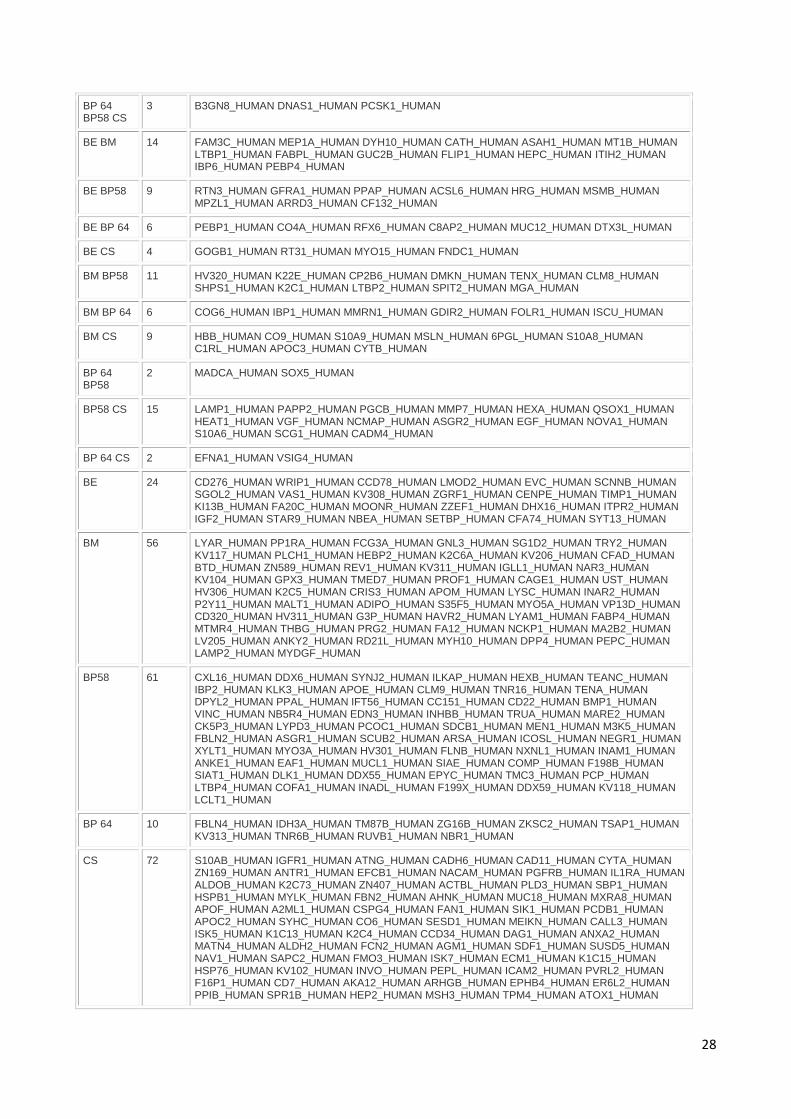
28
BP 64 BP58 CS
3 B3GN8_HUMAN DNAS1_HUMAN PCSK1_HUMAN
BE BM 14 FAM3C_HUMAN MEP1A_HUMAN DYH10_HUMAN CATH_HUMAN ASAH1_HUMAN MT1B_HUMAN LTBP1_HUMAN FABPL_HUMAN GUC2B_HUMAN FLIP1_HUMAN HEPC_HUMAN ITIH2_HUMAN IBP6_HUMAN PEBP4_HUMAN
BE BP58 9 RTN3_HUMAN GFRA1_HUMAN PPAP_HUMAN ACSL6_HUMAN HRG_HUMAN MSMB_HUMAN MPZL1_HUMAN ARRD3_HUMAN CF132_HUMAN
BE BP 64 6 PEBP1_HUMAN CO4A_HUMAN RFX6_HUMAN C8AP2_HUMAN MUC12_HUMAN DTX3L_HUMAN
BE CS 4 GOGB1_HUMAN RT31_HUMAN MYO15_HUMAN FNDC1_HUMAN
BM BP58 11 HV320_HUMAN K22E_HUMAN CP2B6_HUMAN DMKN_HUMAN TENX_HUMAN CLM8_HUMAN SHPS1_HUMAN K2C1_HUMAN LTBP2_HUMAN SPIT2_HUMAN MGA_HUMAN
BM BP 64 6 COG6_HUMAN IBP1_HUMAN MMRN1_HUMAN GDIR2_HUMAN FOLR1_HUMAN ISCU_HUMAN
BM CS 9 HBB_HUMAN CO9_HUMAN S10A9_HUMAN MSLN_HUMAN 6PGL_HUMAN S10A8_HUMAN C1RL_HUMAN APOC3_HUMAN CYTB_HUMAN
BP 64 BP58
2 MADCA_HUMAN SOX5_HUMAN
BP58 CS 15 LAMP1_HUMAN PAPP2_HUMAN PGCB_HUMAN MMP7_HUMAN HEXA_HUMAN QSOX1_HUMAN HEAT1_HUMAN VGF_HUMAN NCMAP_HUMAN ASGR2_HUMAN EGF_HUMAN NOVA1_HUMAN S10A6_HUMAN SCG1_HUMAN CADM4_HUMAN
BP 64 CS 2 EFNA1_HUMAN VSIG4_HUMAN
BE 24 CD276_HUMAN WRIP1_HUMAN CCD78_HUMAN LMOD2_HUMAN EVC_HUMAN SCNNB_HUMAN SGOL2_HUMAN VAS1_HUMAN KV308_HUMAN ZGRF1_HUMAN CENPE_HUMAN TIMP1_HUMAN KI13B_HUMAN FA20C_HUMAN MOONR_HUMAN ZZEF1_HUMAN DHX16_HUMAN ITPR2_HUMAN IGF2_HUMAN STAR9_HUMAN NBEA_HUMAN SETBP_HUMAN CFA74_HUMAN SYT13_HUMAN
BM 56 LYAR_HUMAN PP1RA_HUMAN FCG3A_HUMAN GNL3_HUMAN SG1D2_HUMAN TRY2_HUMAN KV117_HUMAN PLCH1_HUMAN HEBP2_HUMAN K2C6A_HUMAN KV206_HUMAN CFAD_HUMAN BTD_HUMAN ZN589_HUMAN REV1_HUMAN KV311_HUMAN IGLL1_HUMAN NAR3_HUMAN KV104_HUMAN GPX3_HUMAN TMED7_HUMAN PROF1_HUMAN CAGE1_HUMAN UST_HUMAN HV306_HUMAN K2C5_HUMAN CRIS3_HUMAN APOM_HUMAN LYSC_HUMAN INAR2_HUMAN P2Y11_HUMAN MALT1_HUMAN ADIPO_HUMAN S35F5_HUMAN MYO5A_HUMAN VP13D_HUMAN CD320_HUMAN HV311_HUMAN G3P_HUMAN HAVR2_HUMAN LYAM1_HUMAN FABP4_HUMAN MTMR4_HUMAN THBG_HUMAN PRG2_HUMAN FA12_HUMAN NCKP1_HUMAN MA2B2_HUMAN LV205_HUMAN ANKY2_HUMAN RD21L_HUMAN MYH10_HUMAN DPP4_HUMAN PEPC_HUMAN LAMP2_HUMAN MYDGF_HUMAN
BP58 61 CXL16_HUMAN DDX6_HUMAN SYNJ2_HUMAN ILKAP_HUMAN HEXB_HUMAN TEANC_HUMAN IBP2_HUMAN KLK3_HUMAN APOE_HUMAN CLM9_HUMAN TNR16_HUMAN TENA_HUMAN DPYL2_HUMAN PPAL_HUMAN IFT56_HUMAN CC151_HUMAN CD22_HUMAN BMP1_HUMAN VINC_HUMAN NB5R4_HUMAN EDN3_HUMAN INHBB_HUMAN TRUA_HUMAN MARE2_HUMAN CK5P3_HUMAN LYPD3_HUMAN PCOC1_HUMAN SDCB1_HUMAN MEN1_HUMAN M3K5_HUMAN FBLN2_HUMAN ASGR1_HUMAN SCUB2_HUMAN ARSA_HUMAN ICOSL_HUMAN NEGR1_HUMAN XYLT1_HUMAN MYO3A_HUMAN HV301_HUMAN FLNB_HUMAN NXNL1_HUMAN INAM1_HUMAN ANKE1_HUMAN EAF1_HUMAN MUCL1_HUMAN SIAE_HUMAN COMP_HUMAN F198B_HUMAN SIAT1_HUMAN DLK1_HUMAN DDX55_HUMAN EPYC_HUMAN TMC3_HUMAN PCP_HUMAN LTBP4_HUMAN COFA1_HUMAN INADL_HUMAN F199X_HUMAN DDX59_HUMAN KV118_HUMAN LCLT1_HUMAN
BP 64 10 FBLN4_HUMAN IDH3A_HUMAN TM87B_HUMAN ZG16B_HUMAN ZKSC2_HUMAN TSAP1_HUMAN KV313_HUMAN TNR6B_HUMAN RUVB1_HUMAN NBR1_HUMAN
CS 72 S10AB_HUMAN IGFR1_HUMAN ATNG_HUMAN CADH6_HUMAN CAD11_HUMAN CYTA_HUMAN ZN169_HUMAN ANTR1_HUMAN EFCB1_HUMAN NACAM_HUMAN PGFRB_HUMAN IL1RA_HUMAN ALDOB_HUMAN K2C73_HUMAN ZN407_HUMAN ACTBL_HUMAN PLD3_HUMAN SBP1_HUMAN HSPB1_HUMAN MYLK_HUMAN FBN2_HUMAN AHNK_HUMAN MUC18_HUMAN MXRA8_HUMAN APOF_HUMAN A2ML1_HUMAN CSPG4_HUMAN FAN1_HUMAN SIK1_HUMAN PCDB1_HUMAN APOC2_HUMAN SYHC_HUMAN CO6_HUMAN SESD1_HUMAN MEIKN_HUMAN CALL3_HUMAN ISK5_HUMAN K1C13_HUMAN K2C4_HUMAN CCD34_HUMAN DAG1_HUMAN ANXA2_HUMAN MATN4_HUMAN ALDH2_HUMAN FCN2_HUMAN AGM1_HUMAN SDF1_HUMAN SUSD5_HUMAN NAV1_HUMAN SAPC2_HUMAN FMO3_HUMAN ISK7_HUMAN ECM1_HUMAN K1C15_HUMAN HSP76_HUMAN KV102_HUMAN INVO_HUMAN PEPL_HUMAN ICAM2_HUMAN PVRL2_HUMAN F16P1_HUMAN CD7_HUMAN AKA12_HUMAN ARHGB_HUMAN EPHB4_HUMAN ER6L2_HUMAN PPIB_HUMAN SPR1B_HUMAN HEP2_HUMAN MSH3_HUMAN TPM4_HUMAN ATOX1_HUMAN

Apports de la protéomique quantitative différentielle haut-débit à l’étude des méca-nismes de modification du cytosquelette de cellules tubulaires proximales induits par les Inhibiteurs de la Calcineurine.
En transplantation d’organe solide, les Inhibiteurs de la Calcineurine (ICN), Cyclosporine A et
Tacrolimus, ont permis une amélioration significative de la survie à court terme du greffon en
prévenant le rejet d’allogreffe. Cette évolution positive est contrebalancée par une néphro-
toxicité susceptible de contribuer au développement complexe et multifactoriel de la dysfonc-
tion chronique du greffon, facteur pronostique majeur d’une insuffisance rénale terminale.
L’objectif principal de ce travail a été de combiner deux approches expérimentales complé-
mentaires, afin de mettre en lumière des aspects inédits de la physiopathologie des ICN. La
première approche repose sur l’application de la technique de protéomique quantitative
« shotgun » iTRAQ (« isobaric Tags for Relative & Absolute Quantitation ») à l’analyse non
ciblée du protéome de cellules tubulaires proximales. La seconde approche applique de ma-
nière ciblée les outils classiques de biologie moléculaire à l’étude du cytosquelette d’Actine
de cellules tubulaires proximales. La combinaison de ces deux stratégies complémentaires a
permis de mettre en lumière un rôle inédit du cytosquelette d’Actine dans les effets physiopa-
thologiques de la Cyclosporine A en apportant des éléments en faveur d’un mécanisme re-
posant sur une régulation originale de la dynamique intracellulaire de l’Actine.
Mots-clés : Cyclosporine A, Tacrolimus, cellules tubulaires proximales, protéomique quantita-tive haut-débit, iTRAQ, LC-MS/MS, CiR-C, cytosquelette d’Actine, Cofiline
Contributions of the differential high-throughput quantitative proteomic analysis of tubular proximal cells to the study of Calcineurin Inhibitors-induced modifications of the Actin cytoskeleton.
In solid organ transplantation, Calcineurin Inhibitors, Cyclosporine A and Tacrolimus, prevent
allograft rejection and ensure short-term allograft survival. However, CNI elicit nephrotoxic
side effects whose mechanisms remain widely unsolved and are thought to participate to the
multifactorial development of chronic kidney disease, leading to renal failure. The aim of this
work was to combine targeted and untargeted experimental strategies to better understand
CNI-induced physiopathological mechanisms. The first approach was based on the untarget-
ed monitoring of the proximal tubular proteome by the quantitative shotgun proteomic tech-
nique, iTRAQ (« isobaric Tags for Relative & Absolute Quantitation »). The second approach
consisted in the study of the Actin cytoskeleton of proximal tubular cells by classical molecu-
lar biology techniques. In the light of results from both approaches, this work reported that
the Actin cytoskeleton of proximal tubular cells may play a part in the pathophysiology of CsA
thanks to a mechanism based on an original regulation of the intracellular dynamics of Actin.
Keywords: Cyclosporine A, Tacrolimus, tubular proximal cells, high-throughput quantitative proteomics, iTRAQ, LC-MS/MS, CiR-C, Actin cytoskeleton, Cofilin