alla mia famiglia “malaussène” zanette.pdf · alla mia famiglia “malaussène ... stessa...
TRANSCRIPT

Alla mia famiglia “Malaussène”

INDICE

Indice
CARATTERIZZAZIONE DEI MECCANISMI NEUROPROTETTIVI DELLA GUANOSINA 1. Introduzione
1.1 Il sistema purinergico 1
1.1.1 Sintesi dei nucleotidi purinici
1.1.2 Metabolismo intracellulare
1.1.3 Metabolismo extracellulare
1.1.4 Rilascio dei nucleotidi e nucleosidi purinici dai neuroni
1.1.5 Rilascio dei nucleotidi e nucleosidi purinici dalla glia
1.2 Trasportatori dei nucleosidi 12
1.2.1 I Trasportatori Nucleosidici Concentrativi (CNT)
1.2.2 I Trasportatori Nucleosidici Equilibrativi (ENT)
1.2.3 La regolazione cellulare dei carrier nucleosidici
1.3 Sistemi recettoriali 19
1.3.1 Purine a basa adeninica
1.3.2 Purine a base guaninica
1.4 Effetti delle purine nel sistema nervoso centrale e periferico 20
1.4.1 Neuroprotezione
1.4.2 Effetti protettivi dell’adenosina
1.4.3 Effetti protettivi della guanosina
1.4.4 Effetto proliferativo e apoptotico delle purine
1.4.5 Effetti del derivato purinico AIT-082
1.5 Differenziazione 30
1.5.1 Effetto differenziante dell’adenosina
1.5.2 Effetto differenziante della guanosina
1.5.3 Differenziazione e citoscheletro
1.6 Linea cellulare SH-SY5Y 35
2. Scopo della Ricerca 37
3. Materiali e Metodi
3.1 Composti chimici utilizzati 40
3.2 Coltura cellulare 41
3.3 Test della Sulforodamina B (SRB) 41
3.4 Test di vitalità (MTT test) 42
3.5 Valutazione della neuritogenesi 42

Indice
3.6 Dosaggio proteico 43
3.7 Esperimenti di uptake su cellule di neuroblastoma umano SH-SY5Y 44
3.8 Esperimenti di Radioligand Binding 45
3.8.1 Esperimenti di saturazione su cellule intatte
3.8.2 Preparazione di membrane da cellule SH-SY5Y
3.8.3 Esperimenti di saturazione su membrane di cellule SH-SY5Y
3.8.4 Esperimenti di competizione su membrane di cellule SH-SY5Y
3.8.5 Analisi dei dati
3.9 Tecniche di immunocitochimica 48
3.9.1 Immunocitochimica con falloidina
3.10 Tecniche di Western Blot 50
3.10.1 Preparati dei lisati cellulari
3.10.2 Elettroforesi delle proteine in gel di acrilamide con SDS (SDS-PAGE)
3.10.3 Western Blot
3.11 Determinazione dei livelli di AMP ciclico 53
3.11.1 Stimolazione della sintesi di AMP ciclico
3.11.2 Dosaggio dell’AMP ciclico
3.12 Esperimenti di Calcium Videoimaging 56
3.13 Fosforilazione di ERK1/2 57
3.14 Tecniche di manipolazione dell’RNA e del cDNA 59
3.14.1 Estrazione di RNA totale da cellule
3.14.2 Reazione di trascrizione inversa
3.14.3 Reazione di polimerizzazione
3.14.4 Elettroforesi in gel di agarosio
4. Risultati
4.1 Effetto della guanosina su cellule di neuroblastoma SH-SY5Y 62
poste in condizioni di deprivazione da siero
4.1.1 Valutazione visiva dell’effetto protettivo della guanosina sulle
cellule SH-SY5Y
4.2 Test di vitalità cellulare (MTT test) 63
4.3 Effetto della guanosina sulla via delle MAPk in cellule SH-SY5Y in starvation 65
4.4 Effetto della guanosina sui livelli di AMP ciclico nelle cellule SH-SY5Y 66 poste in deprivazione da siero
4.5 Studio del trasporto della guanosina da parte di trasportatori equilibrativi dei 68 nucleosidi nelle cellule SH-SY5Y

Indice
4.5.1 Determinazione dell’espressione dei trasportatori equilibrativi dei nucleosidi
4.5.2 Uptake di [3H]Adenosina e [
3H]Guanosina
4.5.3 Verifica dell’asenza di carrier di tipo concentrativo
4.5.4 Esperimenti di competizione con adenosina e guanosina verso [3H]NBTI
4.6 Espressione di purine nucleoside phosphorylase (PNP) nelle cellule SH-SY5Y 73
4.7 Esperimenti di differenziazione 74
4.7.1 Metodo visivo
4.7.2 Esperimenti di immunocitochimica: effetto della guanosina sul citoscheletro
4.7.3 Esperimenti di immunocitochimica: effetto della guanosina su marker
neuronali
4.8 Effetto antiproliferativo della guanosina (test della SRB) 86
4.9 Effetto della guanosina sui livelli intracellulari di AMP ciclico 92 nelle cellule SH-SY5Y
4.9.1 Effetto degli inibitori delle fosfodiesterasi rolipram e milrinone
sull’accumulo di AMP ciclico
4.9.2 Modulazione della sintesi di AMP ciclico su cellule SH-SY5Y
differenziate con guanosina o acido retinoico
- differenziazione con guanosina
- differenziazione con acido retinoico
- differenziazione con guanosina (stimolazione con forskolina)
- differenziazione con acido retinoico (stimolazione con forskolina)
4.10 Determinazione dei livelli intracellulari di calcio 100
4.11 Attivazione della via delle MAP chinasi 101
4.12 Attivazione della via di PI3 chinasi 102
5. Discussione 103 6. Bibliografia 114

Indice
CARATTERIZZAZIONE DEI MECCANISMI NEUROPROTETTIVI DEI LIGANDI DEI RECETTORI SIGMA 1. INTRODUZIONE 1.1 Identificazione dei recettori sigma 122
1.2 Classificazione dei recettori sigma 123
1.3 Distribuzione anatomica 125
1.4 Il sottotipo recettoriale sigma-1 127
1.4.1 Ruoli fisiologici del recettore sigma-1
1.5 Il sottotipo recettoriale sigma-2 129
1.6 Ligandi endogeni dei recettori sigma 130
1.7 Recettori sigma e trasduzione del segnale 132
1.8 Recettori sigma come modulatori 134
1.8.1 Modulazione dei canali per il k+
1.8.2 Modulazione delle correnti di ca2+
1.8.3 Modulazione del glutammato
1.8.4 Modulazione allosterica mediata dalla fenitoina
1.9. Ruolo dei recettori sigma nella neuroprotezione 137
1.9.1 Studi in vivo
1.9.2 Studi in vitro
1.10 Applicazioni terapeutiche di ligandi sigma-1 141
1.10.1 Schizofrenia
1.10.2 Memoria e deficit di attenzione
1.10.3 Depressione e ansia
1.10.4 Abuso di sostanze stupefacenti
1.11 Tecnica del Radioligand Binding 146
2. SCOPO DELLA TESI 148
3. MATERIALI E METODI
3.1 Esperimenti di radioligand binding 150
3.1.1 Preparazione delle membrane di fegato di ratto
3.1.2 Esperimenti di competizione su membrane di fegato di
ratto verso [3H](+)pentazocina
3.1.3 Esperimenti di competizione su membrane di fegato d

Indice
i ratto verso [3H]DTG
3.1.4 Calcolo del valore di IC50 e Ki
3.1.5 Esperimenti di competizione su membrane di fegato di ratto verso
[3H](+)Pentazocina in presenza di fenitoina
3.1.6 Esperimenti di saturazione con [3H](+)Pentazocina e [
3H]DTG sulle cellule
di neuroblastoma umano SH-SY5Y
3.1.7 Esperimenti di competizione con (+)Pentazocina e Aloperidolo, su cellule di
neuroblastoma umano SH-SY5Y
3.2. Esperimenti di differenziazione 157
4. RISULTATI
4.1 Esperimenti di competizione su membrane 158
di fegato di ratto verso [3H](+)pentazocina e [3H]DTG
4.2 Esperimenti di competizione su membrane 163
di fegato di ratto verso [3H](+)pentazocina
in presenza di fenitoina
4.3 Esperimenti di saturazione con [3H](+)pentazocina 165
e [3H]DTG su cellule di neuroblastoma umano SH-SY5Y
4.4 Esperimenti di competizione con (+)-pentazocina 166
e aloperidolo su cellule di neuroblastoma umano SH-SY5Y
4.5 Esperimenti di inibizione della proliferazione (test della SRB 169
4.6 Studio dei cambiamenti morfologici indotti 172
da ligandi sigma su cellule di neuroblastoma umano SH-SY5Y
5. DISCUSSIONE 174
6. BIBLIOGRAFIA 181

CARATTERIZZAZIONE DEI
MECCANISMI NEUROPROTETTIVI
DELLA GUANOSINA

1. Introduzione

Introduzione
1
1.1 Il Sistema Purinergico
Il termine purina, coniato nel 1884 dal chimico tedesco Emil Fischer (1852-1919),
viene attualmente usato per indicare tutte le molecole eterocicliche aromatiche composte
da un anello purinico, a sua volta formato da un anello pirimidinico fuso con un imidazolo,
sostituito con diversi gruppi funzionali. Nel 1898 Fischer riuscì a sintetizzare questa
molecola e a dimostrare che tutte le molecole oggi note come purine appartengono alla
stessa famiglia di composti chimici. Le nucleobasi, in generale, sono molecole organiche
N-eterocicliche aromatiche che possono essere di due tipi: pirimidiniche se presentano un
anello pirimidinico (uracile, timina e citosina) o puriniche, come nel caso di adenina e
guanina, con un anello purinico sostituito con un gruppo amminico nella prima, e con un
gruppo amminico ed uno carbonilico nella seconda. Ipoxantina e xantina sono altre due
comuni basi puriniche (Henderson, 1972; Brown e Foote, 1998).
(A) (G)
Figura 1: Struttura chimica delle basi puriniche adenina (A) e guanina (G).
Introduzione
2
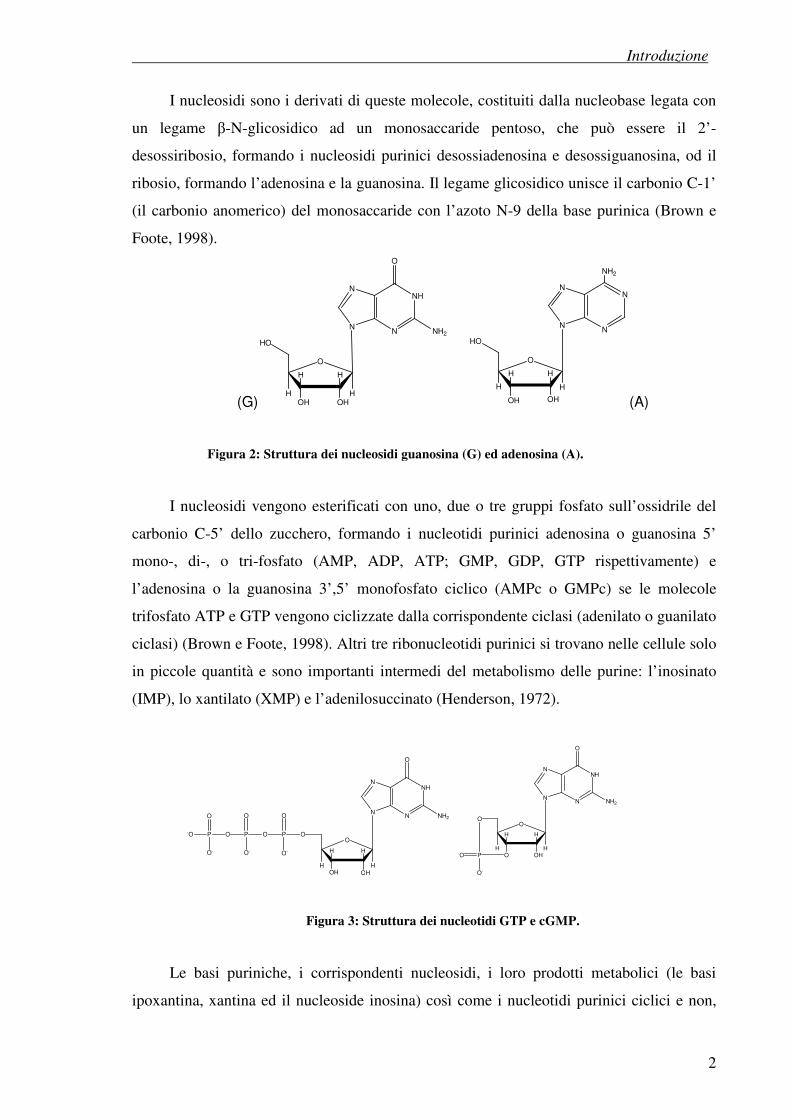
I nucleosidi sono i derivati di queste molecole, costituiti dalla nucleobase legata con
un legame β-N-glicosidico ad un monosaccaride pentoso, che può essere il 2’-
desossiribosio, formando i nucleosidi purinici desossiadenosina e desossiguanosina, od il
ribosio, formando l’adenosina e la guanosina. Il legame glicosidico unisce il carbonio C-1’
(il carbonio anomerico) del monosaccaride con l’azoto N-9 della base purinica (Brown e
Foote, 1998).
(G)
NH
N
N
O
NH2N
O
OHOH
HH
HH
HO
N
NN
N
NH2
O
OHOH
HH
HH
HO
(A)
Figura 2: Struttura dei nucleosidi guanosina (G) ed adenosina (A).
I nucleosidi vengono esterificati con uno, due o tre gruppi fosfato sull’ossidrile del
carbonio C-5’ dello zucchero, formando i nucleotidi purinici adenosina o guanosina 5’
mono-, di-, o tri-fosfato (AMP, ADP, ATP; GMP, GDP, GTP rispettivamente) e
l’adenosina o la guanosina 3’,5’ monofosfato ciclico (AMPc o GMPc) se le molecole
trifosfato ATP e GTP vengono ciclizzate dalla corrispondente ciclasi (adenilato o guanilato
ciclasi) (Brown e Foote, 1998). Altri tre ribonucleotidi purinici si trovano nelle cellule solo
in piccole quantità e sono importanti intermedi del metabolismo delle purine: l’inosinato
(IMP), lo xantilato (XMP) e l’adenilosuccinato (Henderson, 1972).
NH
N
N
O
NH2N
O
OH
HH
HHOH
OPO
O
O-
P
O
O-
OP-O
O-
O
NH
N
N
O
NH2N
O
OHO
HH
HHP
O-
O
O
Figura 3: Struttura dei nucleotidi GTP e cGMP.
Le basi puriniche, i corrispondenti nucleosidi, i loro prodotti metabolici (le basi
ipoxantina, xantina ed il nucleoside inosina) così come i nucleotidi purinici ciclici e non,

Introduzione
3
sono tutte molecole ubiquitariamente presenti nelle cellule animali e vegetali (Rathbone et
al., 1999).
Oltre a svolgere importanti ruoli biologici, le basi puriniche e pirimidiniche, sono i
costituenti del DNA e dell’RNA, mentre i nucleotidi e i nucleosidi purinici partecipano a
numerose reazioni biochimiche e sono coinvolti, soprattutto l’ATP, nel trasporto di energia
all’interno della cellula.
Inoltre, i nucleotidi ciclici adenosina 3’, 5’-monofosfato ciclico (AMPc) e guanosina
3’, 5’-monofosfato ciclico (GMPc) agiscono come importanti secondi messaggeri
intracellulari durante la trasduzione del segnale (Rathbone et al., 1998; Rathbone et al.,
1999; Schmidt et al., 2007).
Le purine a base adeninica rivestono diversi ruoli biologici e sono coinvolte in modo
particolare nel metabolismo energetico mentre le purine a base guaninica sono state
studiate principalmente come modulatori di processi intracellulari, specialmente nei
confronti dell’attività delle proteine G nella trasduzione del segnale, ma è stato anche
dimostrato che queste possiedono effetti extracellulari che includono una modulazione
dell’attività glutammatergica in vitro e in vivo, effetti comportamentali ed effetti trofici
sulle cellule neuronali (Schmidt et al., 2007).
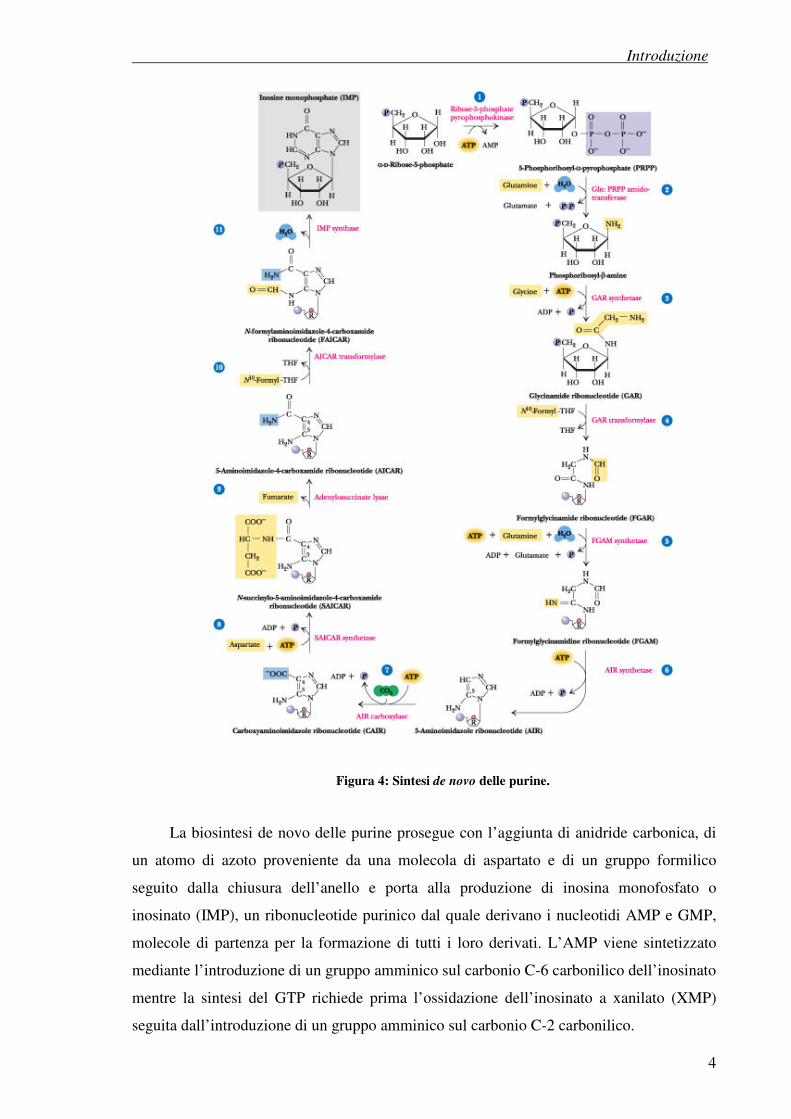
1.1.1 Sintesi dei nucleotidi purinici
I nucleotidi purinici vengono prodotti dalle cellule seguendo due diverse vie
biosintetiche. La prima è una sintesi de novo che utilizza semplici materiali di partenza
come amminoacidi e bicarbonato e, a differenza delle pirimidine, sintetizza le basi
puriniche già legate alla molecola di ribosio.
La prima reazione nella sintesi de novo delle purine consiste nella formazione della
5-fosforibosilammina, una molecola proveniente dalla reazione tra l’amminoacido
glutammina e il 5-fosforibosil-1-pirofosfato (PRPP), un donatore di gruppi fosfato
sintetizzato dalla reazione di un gruppo pirofosforico dell’ATP con una molecola di
ribosio-5-fosfato.
Successivamente, mediante l’aggiunta della glicina seguita da reazioni di
formilazione, amminazione e chiusura dell’anello, si giunge alla sintesi del ribonucleotide
5-aminoimidazolo, un intermedio contenente nella sua struttura lo scheletro dell’anello
purinico.
Introduzione
4
Figura 4: Sintesi de novo delle purine.
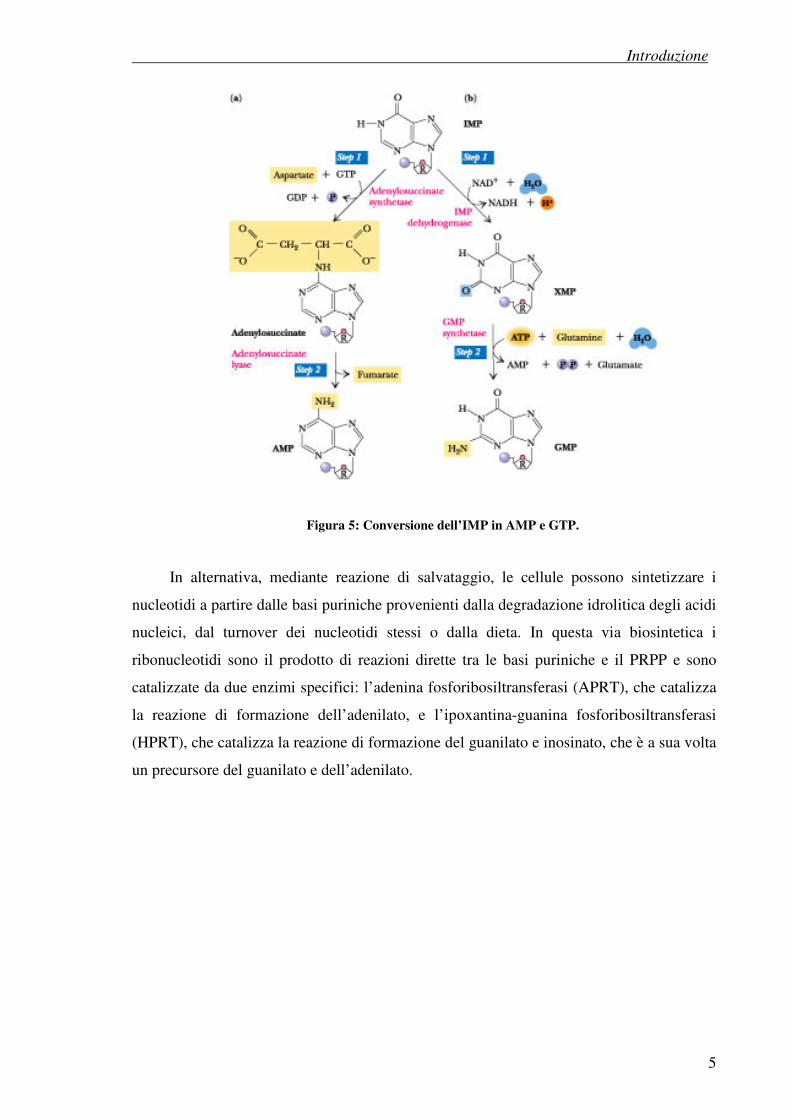
La biosintesi de novo delle purine prosegue con l’aggiunta di anidride carbonica, di
un atomo di azoto proveniente da una molecola di aspartato e di un gruppo formilico
seguito dalla chiusura dell’anello e porta alla produzione di inosina monofosfato o
inosinato (IMP), un ribonucleotide purinico dal quale derivano i nucleotidi AMP e GMP,
molecole di partenza per la formazione di tutti i loro derivati. L’AMP viene sintetizzato
mediante l’introduzione di un gruppo amminico sul carbonio C-6 carbonilico dell’inosinato
mentre la sintesi del GTP richiede prima l’ossidazione dell’inosinato a xanilato (XMP)
seguita dall’introduzione di un gruppo amminico sul carbonio C-2 carbonilico.
Introduzione
5
Figura 5: Conversione dell’IMP in AMP e GTP.
In alternativa, mediante reazione di salvataggio, le cellule possono sintetizzare i
nucleotidi a partire dalle basi puriniche provenienti dalla degradazione idrolitica degli acidi
nucleici, dal turnover dei nucleotidi stessi o dalla dieta. In questa via biosintetica i
ribonucleotidi sono il prodotto di reazioni dirette tra le basi puriniche e il PRPP e sono
catalizzate da due enzimi specifici: l’adenina fosforibosiltransferasi (APRT), che catalizza
la reazione di formazione dell’adenilato, e l’ipoxantina-guanina fosforibosiltransferasi
(HPRT), che catalizza la reazione di formazione del guanilato e inosinato, che è a sua volta
un precursore del guanilato e dell’adenilato.

Introduzione
6
Figura 6: Via di salvataggio delle purine.
Le vie di salvataggio delle purine sono molto importanti perché consentono alla
cellula di risparmiare energia e un loro difetto provoca delle importanti conseguenze.
Mutazioni dei geni che codificano per gli enzimi coinvolti in queste vie biosintetiche,
infatti, possono provocare una riduzione della concentrazione dei nucleotidi e un notevole
aumento degli intermedi di reazione. La sindrome di Lesch-Nyhan è dovuta ad un errore
congenito che provoca la totale assenza dell’enzima ipoxantina-guanina
fosforibosiltransferasi e causa, nei pazienti affetti da tale sindrome, un comportamento
autolesionista, deficienza mentale e spasticità. A livello biochimico, la mancanza di questo
enzima determina un’elevata concentrazione di PRPP, un marcato incremento della sintesi
de novo delle purine e una sovrapproduzione di urato che successivamente può portare ad
attacchi di gotta. La relazione tra l’assenza dell’enzima transferasi e gli effetti neurologici
non è ancora stata chiarita ma potrebbe essere correlata alla presenza nel cervello di cellule
specifiche che dipendono dalla via di salvataggio per la sintesi dell’IMP e del GMP oppure
da un danno provocato dall’alta concentrazione degli intermedi di reazione (Berg et al.,
2002).
1.1.2 Metabolismo intracellulare
In condizioni fisiologiche gli enzimi coinvolti nel metabolismo delle purine
mantengono all’interno della cellula un corretto equilibrio tra i nucleosidi, i nucleotidi e i
loro deossi-derivati, determinando così la quantità di purine disponibile per un eventuale
rilascio in seguito ad uno stimolo appropriato (Ciccarelli et al., 2001).
Introduzione
7
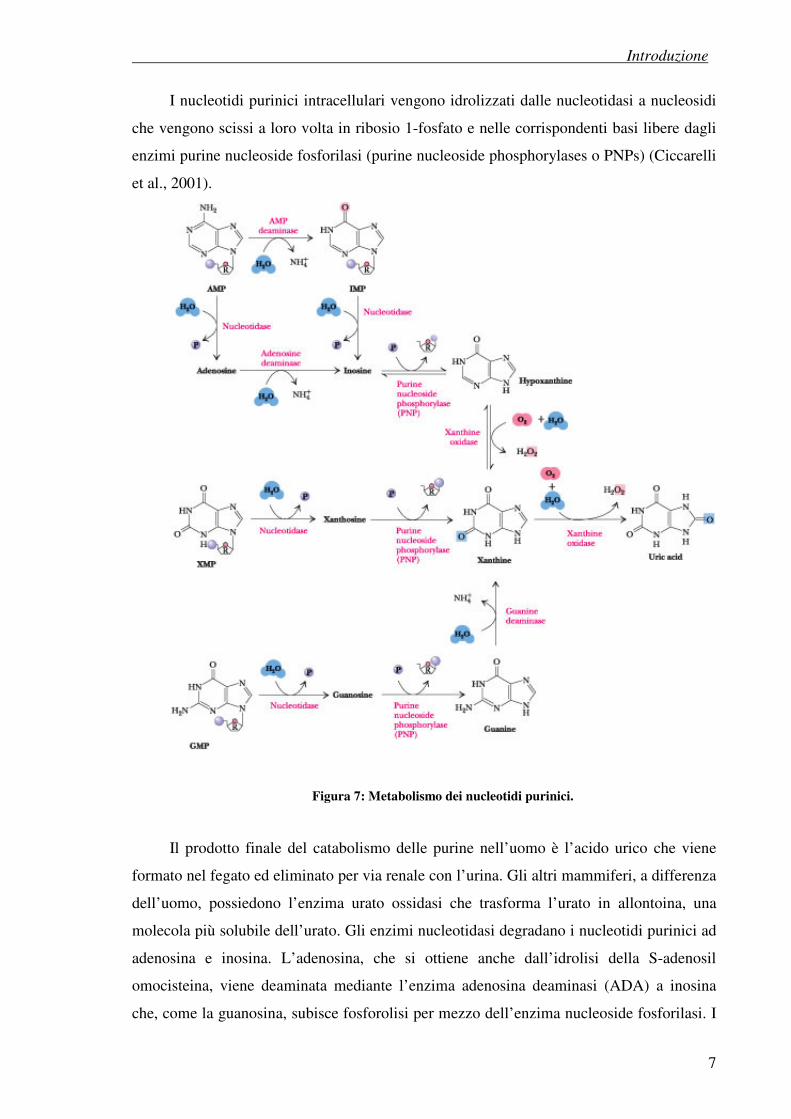
I nucleotidi purinici intracellulari vengono idrolizzati dalle nucleotidasi a nucleosidi
che vengono scissi a loro volta in ribosio 1-fosfato e nelle corrispondenti basi libere dagli
enzimi purine nucleoside fosforilasi (purine nucleoside phosphorylases o PNPs) (Ciccarelli
et al., 2001).
Figura 7: Metabolismo dei nucleotidi purinici.
Il prodotto finale del catabolismo delle purine nell’uomo è l’acido urico che viene
formato nel fegato ed eliminato per via renale con l’urina. Gli altri mammiferi, a differenza
dell’uomo, possiedono l’enzima urato ossidasi che trasforma l’urato in allontoina, una
molecola più solubile dell’urato. Gli enzimi nucleotidasi degradano i nucleotidi purinici ad
adenosina e inosina. L’adenosina, che si ottiene anche dall’idrolisi della S-adenosil
omocisteina, viene deaminata mediante l’enzima adenosina deaminasi (ADA) a inosina
che, come la guanosina, subisce fosforolisi per mezzo dell’enzima nucleoside fosforilasi. I

Introduzione
8
prodotti di tale reazione sono le basi libere ipoxantina e guanina e il ribosio 1-fosfato. Le
basi libere vengono quindi ossidate a xantina e successivamente ad acido urico per mezzo
degli enzimi xantina ossidasi e guanasi (guanina deaminasi), rispettivamente.
Negli astrociti la via metabolica principale per il metabolismo dei nucleosidi è la
sintesi diretta dei nucleotidi AMP e GMP catalizzata dagli enzimi adenosina e guanosina
chinasi. Nell’uomo il catabolismo dell’AMP è dovuto all’enzima adenilato deaminasi che
deamina l’AMP a IMP in quanto gli enzimi nucleotidasi hanno una bassa affinità per
questo nucleotide (Ciccarelli et al., 2001).
1.1.3 Metabolismo extracellulare
Una volta rilasciati all’esterno della cellula, i nucleotidi e i nucleosidi purinici
vengono sottoposti all’azione di numerosi enzimi localizzati sulla superficie cellulare.
Infatti, oltre alle nucleotidasi intracellulari che si trovano nelle cellule della glia e nei
neuroni, sono state identificate delle ecto-nucleotidasi specifiche che regolano i livelli dei
nucleotidi e nucleosidi purinici all’esterno della cellula (Ciccarelli et al., 2001; Rathbone et
al., 1999).
La principale famiglia di ectoenzimi presente sulla membrana cellulare è quella delle
ecto-nucleoside trifosfatasi (E-NTPase) a cui appartengono gli enzimi ecto-ATPasi che
trasformano l’ATP in ADP, ecto-ATP difosfoidrolasi (chiamate anche ecto-apirasi) che
idrolizzano sia l’ATP che l’ADP ad AMP e ecto-5’-nucleotidasi che catalizzano l’idrolisi
del nucleoside 5’-monofosfato in nucleoside. La selettività di questi enzimi per i nucleotidi
a base adeninica non è assoluta in quanto idrolizzano tutti i nucleotidi purinici e
pirimidinici.
L’idrolisi extracellulare dell’ATP è simile nei neuroni, nelle cellule simil-neuronali
(come le cellule PC12), nelle giunzioni neuromuscolari, negli astrociti in coltura e nelle
cellule non neuronali come quelle dell’endotelio vascolare.
Anche i nucleosidi vengono metabolizzati all’esterno della cellula. L’adenosina
deaminasi (ADA), un enzima principalmente citosolico che controlla i livelli intracellulari
di adenosina prevenendo eventuali effetti citotossici nel sistema nervoso centrale e nei
tessuti periferici, è espressa anche sulla superficie esterna delle cellule nel SNC e la sua
attività nelle cellule della glia è nove volte maggiore rispetto ai neuroni. A differenza
dell’adenina, la guanina derivante dal metabolismo intra- ed extracellulare non viene
riutilizzata in grande quantità per formare nucleosidi e nucleotidi perché, nel cervello,

Introduzione
9
l’attività della guanasi è più alta dell’attività della fosforibosiltransferasi (HPRT), l’enzima
che catalizza la sintesi dei nucleotidi nella via di salvataggio (Ciccarelli et al., 2001;
Rathbone et al., 1999).
Figura 8: Metabolismo extracellulare nucleotidi/nucleosidi.
1.1.4 Rilascio dei nucleotidi e nucleosidi purinici dai neuroni
Le purine a base adeninica e guaninica e i loro metaboliti sono presenti nel fluido
cerebrospinale umano e animale (Schmidt et al., 2007). La concentrazione extracellulare
delle purine è regolata da diversi fattori tra cui la quantità rilasciata, il volume
extracellulare locale, i meccanismi di uptake che le internalizzano e la presenza di enzimi
extracellulari che le metabolizzano.
Le fonti di rilascio delle purine nel sistema nervoso centrale sono i neuroni, le cellule
della glia e microglia, le cellule endoteliali e quelle ematiche (Rathbone et al., 1999).
Il rilascio neuronale dei nucleotidi purinici avviene attraverso un processo
esocitotico.
L’ATP e il GTP sono co-localizzati nelle vescicole sinaptiche dei neuroni
adrenergici, colinergici e purinergici che vengono rilasciate in seguito a depolarizzazione.
L’ATP viene liberato anche insieme a diversi neurotrasmettitori dai terminali dei neuroni
centrali e periferici in maniera Na+ e Ca+2 dipendente.
I nucleosidi purinici, invece, raggiungono lo spazio extracellulare mediante diversi
meccanismi. Uno è costituito dal trasportatore bidirezionale dei nucleosidi, presente sulle
cellule neuronali e gliali, che trasporta i nucleosidi secondo gradiente di concentrazione. La
presenza dei nucleotidi nello spazio extracellulare può inoltre essere il risultato di un

Introduzione
10
rilascio diretto da parte delle cellule danneggiate oppure del metabolismo dei nucleotidi
extracellulari da parte delle ecto-nucleotidasi, gli enzimi che li idrolizzano.
Nei neuroni che rilasciano adenosina come neurotrasmettitore nel cervello si può
osservare, mediante immunoreattività specifica, un’intensa colorazione del citoplasma del
pericario e nelle fibre delle cellule neuronali in molte regioni del cervello di ratto e in
specifiche sottopopolazioni di cellule retiniche di coniglio, topo e scoiattolo. Numerosi dati
dimostrano che il rilascio di adenosina da fettine di cervello oppure di sinaptosomi viene
innescato da diversi fenomeni come depolarizzazione da KCl o altri agenti depolarizzanti,
da aminoacidi eccitatori e da stimolazione elettrica.
A differenza del rilascio dei nucleotidi a base adeninica che avviene mediante
vescicole dai terminali nervosi, l’adenosina viene anche rilasciata lungo l’assone del nervo
vago dei mammiferi e del nervo ottico in modo non vescicolare. In condizioni fisiologiche,
la concentrazione extracellulare di adenosina nel sistema nervoso centrale è pari a 1 µM,
valore che cresce drammaticamente in condizioni di ipossia, ischemia, o convulsioni
(Rathbone et al., 1999).
Il rilascio delle purine a base adeninica è regolato dai recettori purinergici:
l’attivazione dei recettori A1 inibisce il rilascio delle purine mentre l’attivazione dei
recettori A2 e P2Y lo stimola.
Per quanto riguarda l’altro nucleoside purinico, la guanosina, evidenze indirette
indicano che anche questa molecola può essere rilasciata dai neuroni in seguito a
depolarizzazione e che il rilascio delle purine a base guaninica è maggiore rispetto a quello
delle loro controparti adeniniche. Come avviene per l’adenosina, anche una parte della
guanosina extracellulare deriva in una certa misura dal metabolismo del corrispondente
nucleotide. Infatti, l’inibizione dell’enzima ecto-5’-nucleotidasi, che idrolizza i nucleotidi
purinici a nucleosidi, causa una diminuzione dell’accumulo di guanosina extracellulare.
Per quanto riguarda la biodisponibilità, la somministrazione orale di guanosina causa
un raddoppio della concentrazione nel fluido cerebrospinale degli animali trattati rispetto al
gruppo di controllo, mentre la somministrazione intraperitoneale di GMP in dosi
anticonvulsivanti triplica i livelli di guanosina nel fluido cerebrospinale dei ratti senza
interferire con i livelli di GMP (Ciccarelli et al., 2001; Schmidt et al., 2007).

Introduzione
11
1.1.5 Rilascio dei nucleotidi e nucleosidi purinici dalla glia
Le cellule della glia, come i neuroni, rilasciano nucleotidi e nucleosidi purinici a
riposo e sotto stimolazione ma le fonti intracellulari delle purine e il meccanismo del loro
rilascio sono molto differenti tra neuroni e cellule gliali (Rathbone et al., 1999). Le cellule
della glia, insieme ai neuroni, costituiscono il sistema nervoso. Hanno funzione nutritiva e
di sostegno per i neuroni e assicurano l’isolamento dei tessuti nervosi e la protezione da
corpi estranei in caso di lesioni. Alcune cellule della glia regolano l’ambiente interno del
cervello e in particolare i fluidi che circondano i neuroni e le loro sinapsi provvedendo al
nutrimento delle cellule nervose. Altri tipi di cellule della glia producono molecole in
grado di influenzare la crescita degli assoni. Studi recenti hanno identificato un loro ruolo
attivo nella trasmissione degli impulsi nervosi. La riproduzione delle cellule della glia
avviene molto frequentemente per mitosi contrariamente ai neuroni per i quali il fenomeno
è molto raro.
La maggior parte delle purine cerebrali deriva dagli astrociti, un tipo di cellule della
glia coinvolte in numerose funzioni del cervello e che partecipano allo sviluppo neuronale,
all’attività sinaptica, al controllo omeostatico dell’ambiente extracellulare e anche ai
processi correlati agli insulti del cervello arrestando e riparando eventuali danni. Inoltre gli
astrociti, come i neuroni, sono responsabili del metabolismo e dell’uptake dell’adenosina e
della guanosina (Schmidt et al., 2007).
Il rilascio delle purine dalle cellule della glia non avviene attraverso un meccanismo
esocitotico come per i neuroni ma sfrutta i trasportatori transmembranari bidirezionali dei
nucleosidi e le proteine trasportatrici della famiglia chiamata ATP binding cassette (ABC).
Inoltre anche cambiamenti dell’osmolarità intracellulare e della tensione delle membrane,
come accade in condizioni patologiche, causano un rilascio non vescicolare di aminoacidi
neuroattivi e di purine.
Oltre al ben conosciuto rilascio di purine a base adeninica, le cellule della glia come i
neuroni rilasciano anche purine a base guaninica.
Il rilascio dei nucleotidi guaninici e della guanosina da parte degli astrociti avviene in
condizioni basali e subisce un forte incremento durante uno stress ipossico o ipoglicemico.
Anche i livelli extracellulari di adenosina aumentano in condizioni di ipossia o ischemia
cerebrale ma le concentrazioni di guanosina restano alte per diversi giorni dopo il danno.
Probabilmente solo una piccola parte della guanosina rilasciata rientra all’interno della
cellula per mezzo dei trasportatori mentre la maggior parte tende ad accumularsi nello

Introduzione
12
spazio extracellulare. Molti studi dimostrano che le cellule della glia sono la principale
fonte di guanosina sia in condizioni fisiologiche che in condizioni patologiche (Ciccarelli
et al., 2001; Rathbone et al., 1999).
1.2 I trasportatori dei nucleosidi
Non tutti i tipi cellulari sono in grado di sintetizzare da sé le molecole nucleosidiche,
come, nel caso dei mammiferi, gli enterociti, gli eritrociti, i leucociti, certe cellule cerebrali
e quelle del midollo spinale (Cabrita et al., 2002). In tutti questi casi è necessario un
sistema di recupero per le purine ed i nucleosidi presenti nell’ambiente extracellulare come
nutrienti o per loro neosintesi da parte di altre cellule. Data la natura idrofila di queste
molecole, i trasportatori sono indispensabili anche per la loro liberazione nell’ambiente
esterno alla cellula, e sono quindi un fattore determinante per la farmacocinetica, la
distribuzione e l’attività biologica di nucleobasi, nucleosidi, e loro analoghi ad uso clinico
(come, ad esempio, 3’-azido-3’-deossitimidina o AZT, Ribavirina, Vidarabina o Ara-A, 5-
fluorouracile) (Griffith e Jarvis, 1996).
I carrier per i nucleosidi (Nucleoside Transporters, NT) sono stati identificati e
classificati in cinque superfamiglie, due delle quali si trovano anche nei mammiferi come
proteine integrali di membrana; e nel tessuto nervoso in particolare ne sono provviste sia le
cellule di glia sia i neuroni (Cabrita et al., 2002).
Le due famiglie di trasportatori per le cellule dei mammiferi sono state distinte in
base alla diversa struttura molecolare, alle caratteristiche farmacologiche e a quelle
funzionali, che comprendono il meccanismo di trasporto, attivo-concentrativo o passivo-
equilibrativo; la sensibilità ad inibitori, e la selettività di substrato. Secondo la
classificazione funzionale, distinguiamo i trasportatori nucleosidici concentrativi (CNT)
dai trasportatori nucleosidici equilibrativi (ENT).
1.1.1 I Trasportatori Nucleosidici Concentrativi (CNT)
Localizzati in numerosi organi, tessuti e cellule specializzate di mammiferi (plesso
coroideo, macrofagi, splenociti, reni, fegato, cervello, epitelio dell’intestino tenue), questi
carrier lavorano secondo il gradiente transmembranario del sodio ([Na+]extracellulare=140
mM vs [Na+]intracellulare=5-10 mM) generato dalla Na+/K+ ATPasi (Wang et al., 1997),
attuando un cotrasporto di tipo simporto diretto verso l’interno della cellula e con un

Introduzione
13
rapporto sodio-nucleoside pari a 1:1 per quasi tutti i sottotipi. Il sistema è selettivo per
questo ione: sono pochi gli studi che hanno riscontrato in un altro catione la capacità di
regolare il trasporto concentrativo: negli epatociti di ratto, dove il litio sembra essere
efficace quanto il sodio nello stimolare l’entrata dell’adenosina, e nelle cellule renali con
orletto a spazzola di ratto, nelle quali il trasporto nucleosidico è regolato dal potassio
(Griffith e Jarvis, 1996).
Figura 9: Struttura molecolare della famiglia di trasportatori concentrativi. Il trasportatore concentrativo rCNT1, espresso nei tessuti di ratto, è stato il primo membro di questa famiglia ad essere scoperto. Questo carrier è costituito da 13 domini transmembranari con un esteso terminale carbossilico extracellulare. Si ipotizza che anche tutte le altre proteine della famiglia CNT dei mammiferi presentino una struttura simile (Cabrita et al., 2002) (figura tratta da Baldwin et al., 1999).
I trasportatori CNT sono attualmente distinti in cinque sottoclassi in base alla loro
sensibilità ad inibitori (cs sensibili e ci insensibili), selettività di substrato o secondo
l’ordine numerico di caratterizzazione (N1-N5):
N1, CNT2 o cif (concentrativi, inibitore-insensibili, per la formicina B)
I carrier N1 sono selettivi per i nucleosidi purinici, ma accettano come substrato anche la
formicina B, isomero citotossico dell’inosina scarsamente metabolizzato, l’uridina e la
deossiuridina. Sono trasportatori ubiquitariamente distribuiti: li troviamo a livello
intestinale, renale, epatico, pancreatico, cardiaco, polmonare, placentare, cerebrale, nella
muscolatura scheletrica ed in molte linee cellulari di mammifero (ratto, coniglio, uomo)
(Wang et al., 1997; Cabrita et al., 2002).

Introduzione
14
N2, CNT1 o cit (concentrativi, inibitore-insensibili, per la timidina)
Questo secondo sottotipo comprende carrier specifici per le pirimidine, che accettano come
substrato anche l’adenosina, e, analogamente ai precedenti, sono ampiamente diffusi: li
troviamo nel rene, nel fegato, nell’intestino, nella placenta e nel cervello.
N3, CNT3 o cib (concentrativi, inibitore-insensibili, ad ampia selettività di substrato)
I carrier N3 trasportano entrambi i tipi di nucleosidi e sono gli unici a legare due ioni sodio
per la traslocazione di una molecola di nucleoside (rapporto stechiometrico pari a 2:1).
Sono stati identificati nel midollo spinale, nell’intestino, nel pancreas, nella trachea, nella
ghiandola mammaria, nella prostata, nei testicoli, nel fegato, nei polmoni, nei reni, nella
placenta, nelle linee cellulari HL-60 (Cabrita et al., 2002) e Caco-2 (Griffith e Jarvis,
1996).
N4 o cit
I trasportatori di tipo N4 sono analoghi agli N2 per selettività di substrato, accettano però
anche la guanosina. Sono stati caratterizzati a livello renale nelle cellule con orletto a
spazzola nell’uomo. Si può notare come tutte queste prime quattro sottoclassi di carrier
concentrativi condividano adenosina ed uridina come substrati (Thorn e Jarvis, 1996).
L’affinità di questi sottotipi di trasportatori CNT per il loro substrato è alta (Km=1-40 µM
a 20°C in presenza di una concentrazione di sodio extracellulare pari a 100 mM) (Griffith e
Jarvis, 1996), e sono insensibili all’esposizione agli inibitori nitrobenziltioinosina (NBTI o
NBMPR) e dipiridamolo fino ad una concentrazione pari a 10 µM (Flanagan e Meckling-
Gill, 1997).
N5 o cs, carrier concentrativi inibitore-sensibili
Il sottotipo N5 è stato identificato nelle cellule di leucemia umana, trasporta la formicina B
ed è molto sensibile anche a basse concentrazioni (<10 nM) degli inibitori NBTI e
dipiridamolo (Griffith e Jarvis, 1996).
Cgs, trasportatore concentrativo sodio-dipendente, specifico per la guanosina,
Nitrobenziltioinosina-sensibile
Questo sesto sottotipo di trasportatori concentrativi è stato caratterizzato negli ultimi dieci
anni nella linea cellulare di leucemia promielocitica umana acuta (NB4) e di leucemia
linfocitica murinica acuta (L1210). Il rapporto stechiometrico Na+:nucleoside è anche in
questo caso 1:1, ed è selettivo per questo catione. Il sistema cgs trasporta nucleosidi,

Introduzione
15
nucleobasi ed analoghi, ma è altamente selettivo per la guanosina, il cui uptake può essere
ridotto non oltre il 50% in presenza di nucleosidi competitivi a concentrazioni dieci volte
più elevate. In presenza dell’inibitore NBTI 1 µM o del dipiridamolo 20 µM si arriva ad
una riduzione dell’attività anche del 90% (Flanagan e Meckling-Gill, 1997; Flanagan et al.,
2003).
1.2.2 I Trasportatori Nucleosidici Equilibrativi (ENT)
Questi carrier sono ampiamente distribuiti nelle cellule e nei tessuti dei mammiferi e
lavorano secondo il gradiente di concentrazione del nucleoside tra i compartimenti intra-
ed extra-cellulare, attuando una diffusione facilitata bidirezionale simmetrica, senza
consumo di energia (Thorn e Jarvis, 1996).
Figura 10: Struttura molecolare della famiglia di trasportatori equilibrativi. I membri della famiglia ENT sono costituiti da 11 α-eliche transmembranarie, con il terminale amminico citoplasmatico, il terminale carbossilico extracellulare ed un grande anello citoplasmatico che collega il sesto dominio transmembranario al settimo (Hammond et al., 2004).
Questa categoria di trasportatori, grazie alla bidirezionalità con cui operano,
rappresenta il mezzo principale per il rilascio dei suoi substrati nell’ambiente
extracellulare, anche se in condizioni di alterato metabolismo o comunque quando i
gradienti ionici vengono massicciamente perturbati (come nel caso di infarto), anche il
sistema concentrativo è sfruttato per rilasciare i nucleosidi dalle cellule (Rathbone et al.,
1999).

Introduzione
16
Si tratta di trasportatori per i nucleosidi ad ampia selettività di substrato, legano
quindi sia le molecole puriniche che quelle pirimidiniche con un’affinità bassa o media
(Km~20-400 µM) (Thorn e Jarvis, 1996) e sono distinti in due classi con proprietà
farmacocinetiche simili, che differiscono marcatamente per la sensibilità verso la
nitrobenziltioinosina (NBTI) (Griffith e Jarvis, 1996), e, nel caso delle due isoforme
umane, anche per la sensibilità verso l’inibizione da dipiridamolo e dilazep (l’ hENT1 è tra
le 100 e le 1000 volte più sensibile dell’ hENT2) (Baldwin et al., 2005).
Distinguiamo quindi:
ENT1 o es, carrier equilibrativi sensibili
Trasportatori potentemente inibiti già a basse concentrazioni dall’inibitore
nitrobenziltioinosina, nel range nM (≤1-10 nM), a causa dell’interazione reversibile di
questa molecola con siti di binding ad alta affinità (Kd=0.1-10 nM). Questa sottoclasse
mostra un’affinità variabile nei confronti dei substrati nei diversi tipi cellulari, il che fa
pensare all’esistenza di varie isoforme. Ad esempio, l’affinità può variare anche sulla base
della configurazione dell’enantiomero del nucleoside e della presenza o meno di
determinanti strutturali, come il gruppo polare ossidrilico in posizione 2’, o 5’ e soprattutto
in posizione 3’: la loro assenza riduce l’affinità in termini di Km e in molti tipi di cellule
alcuni deossi-nucleosidi passano la membrana per diffusione semplice anziché tramite
proteina carrier, probabilmente perché viene a mancare un legame a idrogeno col
trasportatore (Griffith e Jarvis, 1996).
ENT2 o ei, carrier equilibrativi insensibili
Questa sottoclasse di trasportatori equilibrativi presenta tre caratteristiche distintive dalla
precedente: è meno sensibile alla presenza dell’inibitore NBTI e mantiene un’attività
massima fino all’esposizione a concentrazioni del range µM (1-10 µM) della molecola, per
l’assenza di siti ad alta affinità per la molecola. Gli ENT2 trasportano anche le nucleobasi
puriniche e pirimidiniche (Cabrita et al., 2002; Baldwin et al., 2005) e presentano in genere
un’affinità minore per il substrato, anche di sedici volte, come è stato riscontrato per la
guanosina nelle cellule di tumore ascitico di Ehrlic (Griffith e Jarvis, 1996).

Introduzione
17
Studi su membrane prelevate da diverse regioni di cervello umano hanno mostrato
che ENT1 ed ENT2 hanno distinte distribuzioni cerebrali e che il pattern degli ENT1
dell’uomo (hENT1) è correlato a quello dei recettori adenosinici A1.
Sono pochi gli esempi di cellule o tessuti che possiedono solo un tipo di trasportatori
nucleosidici ENT, e ne è esempio la linea cellulare linfoblastoide CCRF-CEM derivata da
una leucemia umana, dotata di carrier es. La maggior parte delle linee cellulari studiate
fino ad ora esprime invece entrambi i sottotipi di trasportatori equilibrativi
contemporaneamente ed in diverse proporzioni, ma quelli più numerosi sono di gran lunga
gli es: la loro attività copre anche l’80% di quella totale.
Nel 2001 sono state scoperte sequenze di DNA per un terzo membro della famiglia
di trasportatori equilibrativi ENT, sia nell’uomo sia nel topo (hENT3, mENT3), ma non ne
sono ancora state descritte le proprietà funzionali. Si ipotizza però, data la presenza di
un’estesa regione ammino-terminale idrofila, che funzioni da trasportatore intracellulare
(Cabrita et al., 2002; Baldwin et al., 2005).
Recentemente è stato anche identificato e caratterizzato il membro ENT4: si tratta di
un trasportatore di membrana ampiamente distribuito nei tessuti umani, ed in particolare
nel cuore, nel cervello e nella muscolatura scheletrica; e sembra essere un carrier
insensibile alla nitrobenziltioinosina e fortemente pH-dipendente. Attualmente è
considerato un trasportatore di ioni organici e di monoammine piuttosto che dei nucleosidi,
sebbene sia stata dimostrata per l’omologo umano (hENT4) e del topo (mENT4) la
capacità di trasportare adenosina a valori di pH acidi (6.0 e 5.5 rispettivamente) (Barnes et
al., 2006; Baldwin et al., 2005).
1.2.3 La regolazione cellulare dei carrier nucleosidici
L’attività e l’espressione dei diversi trasportatori per i nucleosidi cambia in uno
stesso tipo di cellula in risposta a diversi fattori, tra cui stimoli collegati alla crescita, la
differenziazione e la progressione lungo il ciclo cellulare. Ad esempio nei macrofagi-S1
quiescenti la maggior parte dell’uptake avviene via ei, ma in seguito a trattamento con il
colony-stimulating factor 1 (CSF1) l’entità del trasporto triplica grazie all’aumentata
espressione degli es (Griffith e Jarvis, 1996). Nelle cellule epiteliali HeLa sincronizzate,
linea cellulare che esprime solo trasportatori ENT (Coe et al., 2002), l’attività es ed i siti di
legame per l’inibitore NBTI restano costanti relativamente alle dimensioni cellulari nel
progredire attraverso il ciclo.

Introduzione
18
La differenziazione in senso neutrofilo o monocitico della linea cellulare di leucemia
umana HL-60, inducibile per esposizione agli esteri del forbolo, al dimetilsulfossido
(DMSO) o all’acido retinoico, porta all’aumento del numero dei trasportatori concentrativi
a discapito della classe equilibrativa (Cabrita et al., 2002; Flanagan e Meckling-Gill, 1997),
e questo accade in molte linee cellulari (Coe et al., 2002). In generale la differenziazione
cambia sia la tipologia di trasporto predominante, come nell’esempio precedente, sia
l’attività stessa del trasportatore: i reticolociti immaturi di pecora adulta hanno alta attività
es, persa in seguito con la maturazione ad eritrociti (Griffith e Jarvis, 1996).
Gli altri fattori che modulano attività ed espressione dei carrier possono essere la
trasformazione neoplastica delle cellule (il parenchima epatico di ratto esprime un’attività
di trasporto Na+-dipendente (CNT) che le cellule d’epatoma sembrano aver perso a favore
del sistema equilibrativo), l’attivazione diretta delle protein chinasi PKA e PKC in modo
diretto od attraverso i recettori di membrana nelle colture di cellule cromaffini,
l’esposizione a glucocorticoidi, steroidi sessuali, acido retinoico, ormone tiroideo T3 e
all’etanolo, che inibisce il trasporto dell’adenosina via es (Griffith e Jarvis, 1996; Coe et
al., 2002).
Non sono stati ancora identificati tipi cellulari che esprimano solo trasportatori di
tipo concentrativo (Cabrita et al., 2002). Nelle cellule e nei tessuti umani il trasportatore
più ampiamente espresso e distribuito è l’ENT1; al contrario i carrier CNT ed ENT2 sono i
più specifici per i tessuti, i primi soprattutto a livello dei sistemi gastro-intestinale, epatico
e renale, i secondi espressi prevalentemente nel tessuto muscolare scheletrico (Coe et al.,
2002).
Molti tipi di cellule esprimono più sottotipi insieme, così che uno stesso nucleoside
può essere substrato di più di un meccanismo in una cellula; questo ha reso più difficoltoso
l’identificazione definitiva della selettività specifica di ogni sottoclasse. Grazie agli studi
sui carrier clonati ed espressi isolatamente si è potuti risalire ad informazioni più
dettagliate (Wang et al., 1997).

Introduzione
19
1.3 Sistemi Recettoriali
1.3.1 Purine a base adeninica
Gli effetti delle purine a base adeninica sono mediati da diversi tipi di recettori
localizzati sulla superficie cellulare.
Questi recettori, che possiedono caratteristiche farmacologiche ben definite e che
sono stati clonati, si dividono in recettori P1 che rispondono all’adenosina e recettori P2
che rispondono all’ATP e all’uracile trifosfato (UTP).
I recettori P1 sono accoppiati a proteine G e si suddividono in 4 sottotipi: A1, A2A,
A2B e A3. L’attivazione dei recettori A2A e A2B, accoppiati a proteine Gs, stimola
l’adenilato ciclasi provocando un aumento della concentrazione dell’AMP ciclico mentre
l’attivazione dei recettori A1 e A3, accoppiati a proteine Gi, inibisce l’adenilato ciclasi
portando a una diminuzione della concentrazione dell’AMP ciclico. Inoltre l’attivazione
dei recettori A1 e A3 provoca un aumento dei livelli di calcio intracellulare attraverso una
via che coinvolge l’attivazione della fosfolipasi C.
I recettori P2, invece, si dividono in recettori P2X che sono dei canali ionici e in
recettori metabotropici P2Y accoppiati a proteine G (Canals et al., 2005; Rathbone et al.,
1998; Schmidt et al., 2007).
È ampiamente accettato che i recettori A1 e A2A siano coinvolti nel ruolo
neuroprotettivo dell’adenosina in condizioni di ischemia cerebrale e ci sono evidenze che
indicano che l’adenosina, attivando i recettori A1 e A2A, possa influire sullo sviluppo del
sistema nervoso. L’espressione di questi due tipi recettoriali, infatti, avviene nel tardo
periodo prenatale e i loro livelli aumentano dopo la nascita.
Inoltre, dato che l’espressione dei recettori A1 nel cervello avviene durante la
seconda metà del periodo di gestazione in concomitanza con la differenziazione e la
migrazione neuronale, è stato possibile ipotizzare un coinvolgimento di tali recettori in
questi fenomeni. In modo simile, sono stati osservati dei cambiamenti nell’espressione dei
recettori A2A durante lo sviluppo neuronale e ciò suggerisce un possibile coinvolgimento di
questi recettori nella differenziazione neuronale (Canals et al., 2005).

Introduzione
20
1.3.2 Purine a base guaninica
Diversamente dalle purine a base adeninica, la guanosina e il GTP non possiedono
un’alta affinità per i recettori P1 e P2 adenosinici e ciò suggerisce l’esistenza di recettori o
siti di legame specifici per queste molecole (Schmidt et al., 2007).
Sebbene alcuni effetti delle purine a base guaninica siano conseguenti al loro uptake
e quindi avvengano all’interno della cellula, molti degli effetti trofici delle purine a base
guaninica non vengono influenzati dalla presenza dell’NBTI (nitrobenziltioinosina), un
inibitore dei trasportatori nucleosidici.
Inoltre, sebbene la guanosina stimoli il rilascio delle purine a base adeninica dagli
astrociti, molti degli effetti delle purine a base guaninica persistono in presenza degli
antagonisti dei recettori P1 e P2 e ciò indica che molti degli effetti della guanosina esogena
non coinvolgono i recettori adenosinici. Una possibile ipotesi alternativa comprende
l’esistenza di recettori specifici per le purine a base guaninica. Le cellule PC12 e gli
astrociti in coltura, ad esempio, esprimono dei siti di legame per il GTP con caratteristiche
simili a quelle dei recettori di membrana (Oleskovicz et al., 2008; Traversa et al., 2002;
Traversa et al., 2003).
Per quanto riguarda la guanosina, è stato trovato uno specifico sito di legame sulle
membrane del cervello di ratto al quale la molecola si associa molto rapidamente in modo
reversibile e saturabile. La curva di saturazione indica la presenza di un singolo sito di
legame ad alta affinità. Sebbene il meccanismo di trasduzione del segnale sia ancora
sconosciuto, è stato suggerito che questi recettori siano accoppiati a proteine G e
coinvolgano nucleotidi ciclici o attivino la cascata delle MAP chinasi (mitogen activated
protein kinase o MAPK) sulla base della capacità della guanosina di aumentare la sintesi di
fattori trofici negli astrociti associata ad un incremento della fosforilazione di ERK1 e
ERK2 (extracellular signal-regulated kinase o ERK 1/2), effetto bloccato dal pre-
trattamento con la tossina della pertosse (Traversa et al., 2002; Traversa et al., 2003).
1.4 Effetti delle purine nel Sistema Nervoso Centrale e Periferico
Oltre ad essere considerate neurotrasmettitori e modulatori del sistema nervoso
centrale, periferico ed enterico, le purine extracellulari svolgono anche un ruolo trofico
nello sviluppo e nel mantenimento del sistema nervoso e delle sue risposte a malattie e
danni. I nucleosidi e i nucleotidi purinici agiscono come agenti trofici sulle cellule del

Introduzione
21
sistema nervoso attraverso diversi meccanismi: 1. inducendo cambiamenti funzionali nelle
cellule gliali che modulano la differenziazione neuronale; 2. stimolando la sintesi e il
rilascio di fattori trofici dalle cellule neuronali e non; 3. migliorando gli effetti dei fattori di
crescita sulle loro cellule bersaglio; 4. interagendo direttamente con i neuroni o precursori
neuronali inducendo neuritogenesi e differenziazione e aumentando la sopravvivenza
neuronale (Rathbone et al., 1999).
1.4.1 Neuroprotezione
In patologie del sistema nervoso centrale come convulsioni, traumi, ischemia e
ipossia, i neuroni e gli astrociti rilasciano elevate quantità di nucleotidi e nucleosidi
purinici che, accumulandosi a livello extracellulare, innescano degli eventi riparativi che
comprendono astrogliosi, attivazione delle cellule della microglia e rigenerazione degli
assoni danneggiati. Generalmente i nucleotidi promuovono la proliferazione degli astrociti
in modo da formare una cicatrice nell’area danneggiata mentre i nucleosidi stimolano la
produzione di fattori trofici che favoriscono il recupero neuronale.
Oltre ad agire direttamente sulle cellule danneggiate, inoltre, le purine possono
svolgere il loro ruolo indirettamente stimolando uno o più tipi di cellule a produrre e
rilasciare dei fattori trofici e aumentando gli effetti di questi fattori sulle cellule bersaglio
(Ciccarelli et al., 2001; Rathbone et al., 1999).
1.4.2 Effetti protettivi dell’adenosina
L’adenosina è una molecola ubiquitaria distribuita in tutti i tessuti dove modula
importanti processi fisiologici e svolge un ruolo cardine nel controllo della
neurodegenerazione del sistema nervoso centrale (SNC). Nel fluido extracellulare è
normalmente presente una concentrazione nanomolare di adenosina che aumenta in modo
significativo (fino a raggiungere concentrazioni micromolari) in condizioni ischemiche,
ipossiche, in presenza di attacchi epilettici o durante un’aumentata attività nervosa,
prevenendone i danni.
In queste condizioni, gli agenti che aumentano i livelli dell’adenosina endogena
inibendo la sua degradazione (inibitori dell’adenosina deaminasi e dell’adenosina chinasi)
o bloccando il suo trasporto all’interno della cellula, offrono protezione contro i danni
neuronali.

Introduzione
22
Figura 11: Principali meccanismi di modulazione della trasmissione adenosinica coinvolti nella neuroprotezione (da Wardas, 2002).
Diverse evidenze indicano il coinvolgimento dei recettori A1 e A2A nei meccanismi
neuroprotettivi e dati recenti hanno dimostrato che è possibile ottenere lo stesso effetto
protettivo o stimolando i recettori A1 o bloccando i recettori A2A.
Studi condotti su fettine di cervello indicano che l’adenosina e gli agonisti A1
possono attenuare la degenerazione e la perdita di cellule neuronali provocate da
ipossiemia o ipoglicemia. È stato dimostrato che queste molecole attenuano il danno
neuronale sia in vivo che in vitro in condizioni ischemiche o eccitotossiche mentre gli
antagonisti A1 lo aumentano.
Sembra comunque che negli esperimenti in vivo diversi fattori, non necessariamente
a livello cellulare, contribuiscano all’effetto protettivo dell’adenosina. Questo nucleoside,
infatti, svolge un ruolo cruciale nel controllo del circolo cerebrale e, provocando la
vasodilatazione delle arterie cerebrali, può ridurre le conseguenze negative di uno stato
ischemico. All’effetto neuroprotettivo contribuisce probabilmente anche la capacità
dell’adenosina e dei suoi analoghi di prevenire l’attivazione e l’adesione dei leucociti sulle
cellule endoteliali dei vasi sanguigni e di ridurre la temperatura corporea.

Introduzione
23
La somministrazione cronica degli antagonisti A1 per 2-3 settimane prima di un
insulto ischemico riduce il danno neuronale e questo effetto protettivo potrebbe essere
determinato da una up-regulation dei recettori A1.
Dopo un attacco ipossico o ischemico si ha un rilascio immediato di amminoacidi
eccitatori che inducono depolarizzazione della membrana e un eccessivo aumento dei
livelli di calcio intracellulari, innescando una serie di processi che provocano la morte
cellulare. Contemporaneamente, il rilascio di glutammato aumenta la liberazione di
adenosina endogena che, agendo a livello pre e postsinaptico, svolge la sua funzione
neuroprotettiva nei confronti della neurotossicità indotta dagli amminoacidi eccitatori.
Attivando i recettori A1 presinaptici, l’adenosina potrebbe ridurre l’influsso del
calcio attraverso i canali voltaggio-dipendenti e quindi impedire un ulteriore rilascio del
glutammato. In questo modo si ha anche una diminuzione dell’attivazione dei recettori
NMDA (N-metil-D-aspartato) e di conseguenza viene ostacolato anche l’influsso di calcio
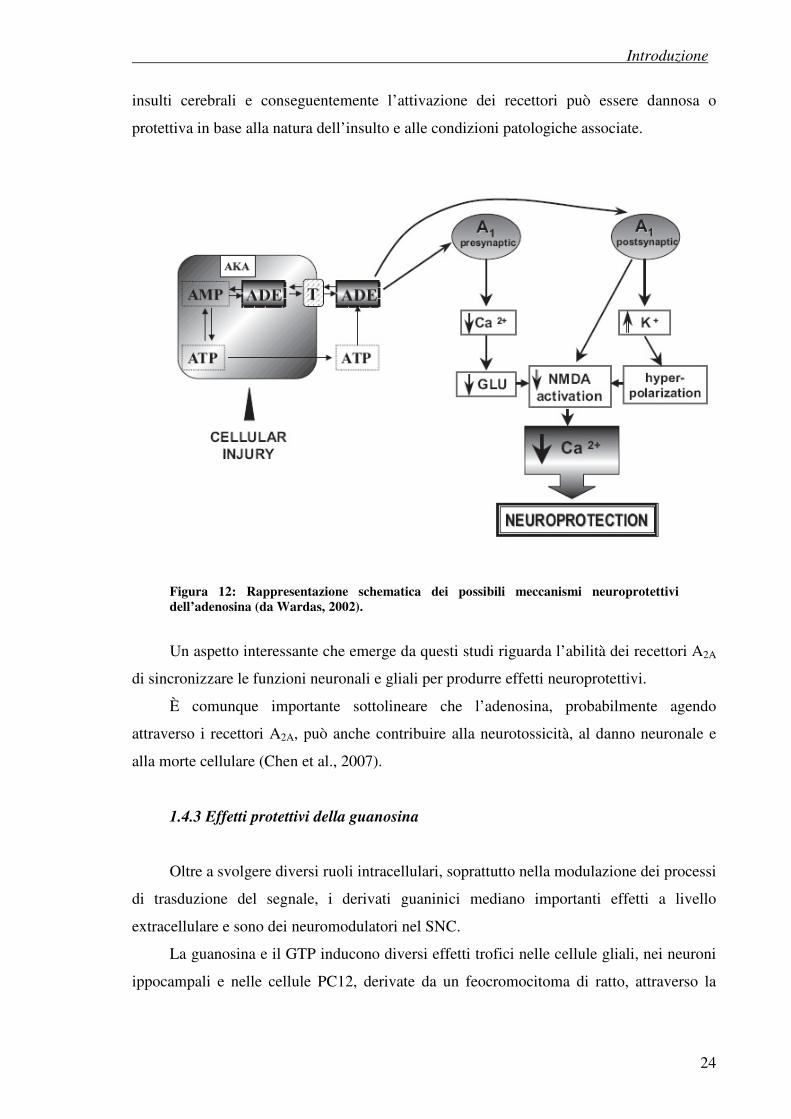
nei neuroni NMDA-mediato (Figura 12).
A livello postsinaptico, il legame dell’adenosina ai recettori A1 causa l’attivazione
dei canali del potassio e quindi un aumento dell’efflusso dello ione. Ciò porta a una
iperpolarizzazione dei neuroni postsinaptici e ad una inibizione dell’apertura dei canali del
calcio voltaggo-dipendenti. Inoltre, ostacolando la depolarizzazione della membrana,
l’adenosina innalza la soglia del potenziale di apertura dei canali NMDA.
Riassumendo, l’adenosina agisce come neuroprotettore stabilizzando il potenziale di
membrana e mantenendo l’omeostasi del calcio nei neuroni postsinaptici (Figura 12).
Meno conosciuto è il ruolo esercitato dai recettori A2A nella neuroprotezione. Alcuni
studi hanno evidenziato le proprietà protettive degli antagonisti di questi recettori anche se
il meccanismo d’azione non è stato chiarito. Nei topi knock-out per i recettori A2A
sottoposti a ischemia, l’infarto cerebrale e i deficit neurologici apparivano attenuati rispetto
ai topi non modificati (Wardas, 2002).
Gli antagonisti, e in alcuni casi anche gli agonisti, dei recettori A2A svolgono azione
protettiva contro diversi tipi di insulti cerebrali come ischemia, emorragia, eccitotossicità,
tossicità mitocondriale e insulti immunologici. È stato ipotizzato che questi recettori siano
in grado di modulare il rilascio del glutammato e la neuroinfiammazione contribuendo così
alla neuroprotezione durante gli insulti cerebrali. L’apparente paradosso della
neuroprotezione indotta sia dall’attivazione, sia dall’inattivazione, di questi recettori
riflette la complessità delle azioni dei recettori A2A espressi su vari elementi cellulari, vale
a dire neuroni, glia e vascolatura, ognuno dei quali può mediare effetti diversi in corso di
Introduzione
24
insulti cerebrali e conseguentemente l’attivazione dei recettori può essere dannosa o
protettiva in base alla natura dell’insulto e alle condizioni patologiche associate.
Figura 12: Rappresentazione schematica dei possibili meccanismi neuroprotettivi dell’adenosina (da Wardas, 2002).
Un aspetto interessante che emerge da questi studi riguarda l’abilità dei recettori A2A
di sincronizzare le funzioni neuronali e gliali per produrre effetti neuroprotettivi.
È comunque importante sottolineare che l’adenosina, probabilmente agendo
attraverso i recettori A2A, può anche contribuire alla neurotossicità, al danno neuronale e
alla morte cellulare (Chen et al., 2007).
1.4.3 Effetti protettivi della guanosina
Oltre a svolgere diversi ruoli intracellulari, soprattutto nella modulazione dei processi
di trasduzione del segnale, i derivati guaninici mediano importanti effetti a livello
extracellulare e sono dei neuromodulatori nel SNC.
La guanosina e il GTP inducono diversi effetti trofici nelle cellule gliali, nei neuroni
ippocampali e nelle cellule PC12, derivate da un feocromocitoma di ratto, attraverso la

Introduzione
25
promozione della proliferazione e della differenziazione cellulare e attraverso
l’arborizzazione e la crescita dei neuriti (Oleskovicz et al., 2008).
Diversi studi indicano che la guanosina può essere considerata un composto
neuroprotettivo endogeno ed è stato dimostrato che i derivati guaninici vengono rilasciati
dagli astrociti in condizioni di ipossia o ipoglicemia e che i loro livelli extracellulari
restano alti per un lungo periodo dopo l’insulto, aumentando progressivamente anche come
risultato dell’idrolisi dei nucleotidi.
E’ stato dimostrato che in fettine di ippocampo esposte a deprivazione di ossigeno e
glucosio per 15 minuti, seguite da due ore di riperfusione, l’aggiunta di guanosina e GMP
al mezzo di incubazione determina un recupero della vitalità cellulare. Inoltre, il mezzo
prelevato da colture di astrociti trattati con guanosina si è dimostrato in grado di prevenire
la neurotossicità indotta dall’NMDA (Oleskovicz et al., 2008; Schmidt et al., 2007).
In accordo con gli studi in vitro, studi in vivo hanno dimostrato che la
somministrazione di GMP o guanosina previene la comparsa di convulsioni e la morte
cellulare mediate dall’NMDA nel tessuto striato nei ratti.
Uno dei ruoli extracellulari principali dei derivati guaninici è il loro effetto
modulatorio sul sistema glutammatergico. Essi determinano una riduzione del legame del
glutammato e dei suoi analoghi ai loro recettori, con conseguente diminuzione della
neurotossicità indotta dall’amminoacido.
È stato inoltre dimostrato che la guanosina è in grado di modulare l’attività dei
trasportatori del glutammato aumentando l’uptake dell’amminoacido negli astrociti e
diminuendone l’accumulo nelle vescicole sinaptiche (Oleskovicz et al., 2008).
In condizioni fisiologiche, l’effetto della guanosina sull’uptake del glutammato in
fettine cerebrali sembra essere dipendente dall’età (maggiore negli animali giovani) e dalla
struttura coinvolta (maggiore nella corteccia). Viceversa, in condizioni eccitotossiche il
ruolo della guanosina nello stimolare l’uptake del glutammato da parte degli astrociti
sembra costituire un meccanismo protettivo generalizzato (Schmidt et al., 2007; Schmidt et
al., 2009).
La deprivazione di ossigeno e glucosio nelle cellule cerebrali riproduce una serie di
stati patologici e provoca un incremento della concentrazione extracellulare di glutammato
che passa da concentrazioni micromolari a concentrazioni millimolari. L’influsso del
calcio causato da un’eccessiva attivazione dei recettori del glutammato scatena una serie di
processi intracellulari tra cui proteolisi, perossidazione lipidica e generazione di specie
reattive dell’ossigeno che causano la morte cellulare.

Introduzione
26
I trasportatori degli amminoacidi ad alta affinità sodio-dipendenti provvedono
all’eliminazione del glutammato extracellulare dallo spazio sinaptico in un modo che
sembra dipendente dallo stato redox della cellula. È stato comunque dimostrato che
l’effetto neuroprotettivo della guanosina non è direttamente correlato ai recettori e ai
trasportatori del glutammato e che non sono neppure coinvolti i recettori adenosinici.
L’effetto della guanosina, infatti, non cambia se le fettine cerebrali vengono
precedentemente esposte al dipiridamolo, un inibitore non selettivo dei trasportatori dei
nucleosidi. Questo dato, inoltre, potrebbe suggerire l’esistenza di un sito d’azione
extracellulare della guanosina.
Dato che l’eccessivo rilascio di glutammato provoca un aumento dei livelli
intracellulari di calcio che porta a morte cellulare, è stata ipotizzata una correlazione tra
l’omeostasi del calcio e l’effetto protettivo della guanosina. L’incubazione di fettine di
ippocampo in un mezzo privo di calcio abolisce difatti l’effetto protettivo della guanosina,
sottolineando la dipendenza dell’effetto della purina dai livelli extracellulari di calcio.
Alcuni studi hanno dimostrato che in astrociti in coltura trattati con guanosina
aumenta l’espressione dei canali del potassio e questo potrebbe contribuire alla
neuroprotezione con un meccanismo non ancora conosciuto, poichè bloccando i canali del
potassio, l’effetto neuroprotettivo della guanosina viene abolito.
La guanosina, dunque, sembrerebbe agire mediante l’attivazione dei canali del
potassio e il suo effetto dipenderebbe dal calcio extracellulare.
Il coinvolgimento del calcio intracellulare nell’effetto della guanosina non è ancora
stato chiarito ma l’entrata dello ione attraverso i canali voltaggio-dipendenti oppure la
mobilitazione delle riserve intracellulari può attivare i canali del potassio calcio-dipendenti
favorendo la neuroprotezione indotta dalla guanosina.
Tenendo in considerazione la mobilitazione del calcio intracellulare durante un
attacco ischemico e il possibile coinvolgimento del calcio nell’effetto della guanosina,
sono state studiate le possibili vie di trasduzione del segnale che coinvolgono il calcio.
Sorprendentemente l’inibizione della proteina chinasi II calmodulina-dipendente (CaMKII)
non influenza l’effetto della guanosina che viene però abolito usando inibitori selettivi
della PKC, PKA, MEK (MAPK/ERK) e PI3-K.
Traversa e collaboratori (2002) hanno dimostrato che l’attivazione della cascata delle
MAPK da parte della guanosina viene bloccata dal pretrattamento delle cellule con la
tossina della pertosse ma non con gli antagonisti dei recettori adeninici. Questo suggerisce

Introduzione
27
che la guanosina agisca mediante recettori accoppiati a proteine G distinti da quelli attivati
dai derivati adeninici.
La guanosina potrebbe dunque agire attivando un recettore oppure un canale del
potassio calcio-dipendente e promuovendo l’attivazione della PKC, PKA, MEK oppure
PI3-K (Oleskovicz et al., 2008).
In vivo la guanosina e il GMP sono capaci di modulare i processi mnemonici dopo
somministrazione acuta o cronica per via intraperitoneale o orale, un effetto non prevenuto
dagli antagonisti dei recettori adenosinici (Schmidt et al., 2007).
La somministrazione di guanosina (8 mg/kg) per via intraperitoneale in ratti con
occlusione dell’arteria cerebrale prolunga la sopravvivenza dell’animale e diminuisce i
difetti neurologici e il danno tissutale derivanti dall’occlusione.
Inoltre la guanosina protegge i neuroni dagli effetti della deprivazione da ossigeno e
glucosio anche se somministrata 5 ore dopo lo stimolo (Chang et al., 2007).
1.4.4 Effetto proliferativo e apoptotico delle purine
In molte malattie del sistema nervoso centrale e in seguito ad un insulto cerebrale, le
cellule dell’astroglia vanno incontro ad un processo chiamato “astrogliosi reattiva”. In
questo processo, le cellule dell’astroglia vanno incontro a proliferazione, iperplasia,
ipertrofia e ramificazione dei processi fino ad assumere una forma stellata. In
concomitanza con questi fenomeni si osserva anche la sintesi e la liberazione di fattori di
crescita, neurotrofine e pleiotrofine (Rathbone et al., 1999).
Le purine a base adeninica inducono diversi effetti negli astrociti e in generale i
nucleotidi ne stimolano la proliferazione e la differenziazione mentre i nucleosidi
inibiscono la proliferazione cellulare e inducono la morte per apoptosi.
L’effetto proliferativo dell’ATP è mediato dall’attivazione dei recettori
metabotropici P2Y con conseguente stimolazione della via delle MAPK che controlla la
proliferazione cellulare e la differenziazione. La stimolazione dei recettori P2Y negli
astrociti mobilita anche il calcio intracellulare e attiva le proteine chinasi calcio-dipendenti.
Gli effetti dell’adenosina sulla proliferazione cellulare sono meno definiti. In colture
di astrociti di pulcino l’adenosina stimola la proliferazione cellulare, un effetto non
influenzato dall’NBTI, un inibitore dei trasportatori nucleosidici, ma inibito dal DMPX, un
antagonista dei recettori A2. Negli astrociti di ratto, inoltre, la stimolazione dei recettori A2

Introduzione
28
aumenta la proliferazione cellulare mentre la stimolazione dei recettori A1 ne causa
l’inibizione.
È stato dimostrato che anche la guanosina e le purine a base guaninica stimolano la
proliferazione di molti tipi di cellule in modo tempo-dipendente e che l’effetto mitogeno
causato dal GTP è indipendente da quello causato dalla guanosina. Inoltre, dato che
l’effetto proliferativo indotto dalla guanosina negli astrociti di cervello di pulcino è
antagonizzato da antagonisti dei recettori A2 e dato che l’effetto proliferativo indotto dal
GTP viene ridotto dalla suramina, un antagonista dei recettori P2, è possibile che l’effetto
proliferativo di almeno un componente delle purine a base guaninica sia secondario a un
aumento dei livelli extracellulari delle purine a base adeninica.
In colture di astrociti di ratto il GTP esogeno incrementa i livelli extracellulari di
nucleotidi adeninici, un effetto in parte attribuibile al blocco degli enzimi nucleotidasi,
mentre l’aggiunta della guanosina esogena stimola la produzione di nucleotidi adeninici e,
in misura minore, il rilascio di adenosina e inosina. Dato che non si osserva un
contemporaneo incremento dei livelli di ipoxantina extracellulare, si può ipotizzare
un’inibizione dell’attività dell’adenosina deaminasi extracellulare da parte della guanosina
oppure la presenza di un enzima purine nucleoside fosforilasi (PNP)-simile collocato sulla
membrana cellulare. L’incremento dei livelli di adenosina indotto dalla guanosina non è
sufficiente a causare apoptosi.
I nucleotidi purinici innescano il fenomeno dell’astrogliosi come primo tentativo di
limitare la perdita di tessuto neuronale dopo un insulto e per offrire un supporto per la
rigenerazione assonale. Il ruolo dell’adenosina nel regolare la proliferazione cellulare è
cruciale. E’ stato riportato che la 2-cloro-adenosina, un analogo metabolicamente stabile
dell’adenosina, induce apoptosi negli astrociti in coltura, un effetto che sembra essere
mediato dai recettori A3 e che non sembra essere influenzato dagli antagonisti dei recettori
A1 e A2.
In condizioni patologiche, il ruolo pro-apoptotico dell’adenosina è coinvolto nel
rimodellamento delle cicatrici cerebrali dopo un trauma o dopo ischemia. In questo modo
vengono eliminate cellule inutili per lo sviluppo cerebrale o danneggiate e si impedisce,
così, il rilascio di sostanze neurotossiche o infiammatorie. Inoltre viene limitata l’eccessiva
proliferazione degli astrociti innescata dai nucleotidi purinici.
Contrariamente all’adenosina, la guanosina non causa apoptosi quando viene
aggiunta in colture di astrociti, ma l’aggiunta di un inibitore dell’ADA provoca la
comparsa di un moderato numero di cellule apoptotiche. Ciò indica che la guanosina

Introduzione
29
provoca il rilascio di adenosina che, se viene degradata, può indurre apoptosi (Ciccarelli et
al., 2001; Rathbone et al., 1999).
La guanosina svolge inoltre un ruolo anti-apoptotico concentrazione-dipendente che
protegge gli astrociti dall’apoptosi indotta dalla staurosporina, un inibitore delle proteine
chinasi. Questo effetto non viene influenzato dagli inibitori dei trasportatori nucleosidici,
dagli antagonisti dei recettori A1 e A2 o dagli antagonisti dei recettori P2X e P2Y. Al
contrario, il pre-trattamento degli astrociti con la tossina della pertosse, che disaccoppia le
proteine Gi dai loro recettori, abolisce l’effetto antiapoptotico della guanosina che viene
inoltre ridotto dal pre-trattamento degli astrociti con gli inibitori della fosfoinoside-3-
chinasi (PI3K) o della via delle MAPK (Di Iorio et al., 2004).
Infine, è stato dimostrato che la guanosina protegge le cellule di neuroblastoma
umano SH-SY5Y dall’apoptosi indotta dalla proteina neurotossica β-amiloide, un effetto
mediato dall’attivazione delle vie PI3K/Akt/PKB e MAPK (Pettifer et al., 2004).
1.4.5 Effetti del derivato purinico AIT-082
AIT-082 (4-[[3-(1,6-dihydro-6-oxo-9-purin-9-yl)-1-oxopropyl]amino]benzoic acid) è
un analogo stabile della purina ipoxantina, che ha dimostrato di indurre effetti in parte
analoghi, ma non totalmente sovrapponibili, a quelli indotti dalla guanosina sia in vitro che
in vivo.
AIT-082 ha suscitato inizialmente interesse per la sua capacità di aumentare la
memoria in topi in cui era stato indotto un deficit medio di memoria senza indurre effetti
avversi (Glasky et al., 1994).
Studi in vitro hanno dimostrato la capacità di AIT-082 di potenziare l’effetto
differenziante di NGF sulle cellule PC12 (Middlemiss et al., 1995) e di stimolare il rilascio
di adenosina e neurotrofine dagli astrociti (Glasky, 1996; Rathbone, 1999).
In vivo, AIT-082 è anche in grado di proteggere i neuroni ippocampali di ratto dalla
tossicità a lungo termine da kainato riducendo la perdita di neuroni e la diminuzione di
attività di GAD (glutamic acid decarboxylase) (Di Iorio et al., 2001). E’ stato accertato che
AIT-082 è in grado di passare la barriera ematoencefalica con un meccanismo non
saturabile mentre viene portato fuori tramite un meccanismo attivo e saturabile (Taylor et
al., 2000).
Uno studio su topi dimostra che AIT-082 è in grado di ridurre il tremore indotto da
arecolina e oxotremorina diminuendone l’intensità e la durata (Nannan et al., 2007).

Introduzione
30
Questo composto sembra essere un buon candidato per la cura di malattie
neurodegenerative quali il morbo di Alzheimer ed in questo momento è in fase di
sperimentazione clinica.
1.5 Differenziazione
La “differenziazione cellulare” è definita come un processo complesso in cui una
cellula acquisisce o mostra un nuovo fenotipo stabile senza cambiare il proprio genotipo
(Ham et al., 1980). La “differenziazione neuronale” è un tipo di differenziazione cellulare
in cui le cellule acquisiscono caratteristiche tipiche delle cellule neuronali come
l’eccitabilità elettrica, la presenza di assoni e dendriti e l’espressione di proteine neuronali
specifiche. Nello sviluppo embrionale, gruppi di cellule indifferenziate vanno incontro a
differenziazione attraverso diversi passaggi, così come i neuroni ad esempio originano dai
neuroblasti che a loro volta derivano dalla cresta neurale. La differenziazione neuronale è
un evento fondamentale per lo sviluppo del sistema nervoso e per la rigenerazione del
tessuto nervoso danneggiato (Buttiglione et al., 2007) che coinvolge diverse vie di
trasduzione del segnale (Sànchez et al., 2004) ed è regolata da interazioni cellula-cellula e
cellula-substrato. Inoltre partecipano a questo fenomeno una gran quantità di molecole
solubili tra cui ormoni, fattori di crescita, citochine, fattori trofici e morfogenici (Lòpez-
Carballo et al., 2002).
Una delle caratteristiche più importanti delle cellule differenziate è l’arresto della
crescita. Ad esempio, quando le cellule PC12 vengono differenziate con NGF cessano di
proliferare ed estendono neuriti.
L’acido retinoico, la forma biologicamente attiva della vitamina A, svolge un ruolo
importante nello sviluppo embrionale e nella generazione di diversi organi e sistemi
(Lòpez-Carballo et al., 2002) e induce differenziazione neuronale mediante interazione con
specifici recettori nucleari (RA e RXR-α, β, γ) che fungono da regolatori trascrizionali
(Gangadharan et al., 2002).
Nelle cellule di neuroblastoma SH-SY5Y l’acido retinoico induce inibizione della
crescita e differenziazione che si manifesta con la crescita di neuriti (Kim et al., 2000) e
con la conversione morfologica delle cellule in un fenotipo simil-neuronale (Ammer e
Schultz, 1994).
Uno dei bersagli degli eventi regolatori associati alla differenziazione delle cellule
SH-SY5Y indotta da acido retinoico è rappresentato dalle proteine G. E’ stato infatti

Introduzione
31
dimostrato che sei giorni di trattamento con acido retinoico 10 µM aumentano in maniera
significativa i livelli di proteine G inibitorie (Giα1 e Giα2) mentre riducono i livelli delle
proteine Gsα (Ammer e Schultz, 1994).
Uno studio svolto su cellule SH-SY5Y dimostra che il trattamento a lungo termine
con acido retinoico porta ad un notevole aumento dei livelli di AMPc. Questo sembra
essere dovuto ad un incremento dell’attività dell’adenilato ciclasi indotta dai recettori delle
prostaglandine E1 che vanno incontro ad up-regulation. La riduzione delle proteine Gsα
potrebbe servire a bilanciare l’eccessiva produzione di AMPc (Ammer e Schultz, 1994).
Le cellule di neuroblastoma rispondono all’AMPc formando estensioni simili a
neuriti ed è stato dimostrato che le sostanze che aumentano i livelli intracellulari di AMPc
inducono differenziazione neuronale (Sànchez et al., 2004).
Uno studio recente dimostra come sulle cellule SH-SY5Y la differenziazione con
acido retinoico provoca una down-regulation e una ri-localizzazione dei geni inibitenti la
differenziazione ID1, ID2 e ID3. Sempre in questo studio è stata dimostrata la capacità
dell’acido retinoico di fosforilare Akt sulla Ser-473 già dopo 30 minuti di stimolazione. La
stimolazione della via PI3K/Akt sembra essere importante ma non sufficiente ad indurre
differenziazione (Lopez-Carballo et al., 2002).
1.5.1 Effetto differenziante dell’adenosina
Come detto precedentemente, l’adenosina esplica il suo ruolo neuroprotettivo in
condizioni di ischemia e contribuisce allo sviluppo del sistema nervoso mediante i recettori
A1 e A2A. L’attivazione dei recettori A1 e A2A nella linea cellulare SH-SY5Y e nelle colture
primarie dei neuroni striatali di ratto promuove la neuritogenesi e la differenziazione
mediante l’attivazione della via delle MAPK e l’attivazione della proteina chinasi C
(PKC), anche se in modo indipendente l’una dall’altra.
Nelle cellule SH-SY5Y i recettori adenosinici mediano la fosforilazione di ERK-1/2
attraverso la classica via Ras/Raf/MEK/ERK ma l’attivazione di questa via da parte dei
recettori A1 è indipendente dalla PKA mentre è mediata dalla PKA nel caso dei recettori
A2A, che sono accoppiati positivamente all’adenilato ciclasi. Gli agonisti dei recettori A2A,
infatti, aumentano le concentrazioni intracellulari di AMPc che di conseguenza attiva la
PKA. Inoltre, in queste cellule la fosforilazione di ERK-1/2 è molto sensibile all’inibizione
della PKA e ciò suggerisce che l’attivazione delle MAPK è mediata dai recettori A2A.

Introduzione
32
L’attivazione di ERK-1/2 potrebbe non essere, però, l’unica via coinvolta nella
differenziazione morfologica delle cellule SH-SY5Y. L’attivazione della PKC, infatti, è
necessaria per indurre neuritogenesi dopo la stimolazione dei recettori adenosinici.
È stato dimostrato che l’attivazione delle diverse isoforme della PKC da parte degli
esteri del forbolo induce crescita dei neuriti in questa linea cellulare.
Dato che gli inibitori di ERK e della PKC sono in grado di annullare l’effetto
neuritogenico solo se usati in combinazione, è possibile ipotizzare che queste due vie siano
indipendenti l’una dall’altra ma che portino, mediante l’attivazione dei recettori
adenosinici, a un risultato simile.
L’effetto neuritogenico ottenuto dalla stimolazione contemporanea dei recettori A1 e
A2A è simile a quello ottenuto stimolando uno solo dei due recettori e questa mancanza di
sinergismo può essere dovuta alle loro reciproche interazioni antagonistiche.
L’adenosina endogena ha maggior affinità per i recettori A1 rispetto ai recettori A2A e
ciò suggerisce che in condizioni basali predominino le funzioni mediate dai recettori A1
mentre in condizioni ipossiche o dopo depolarizzazione, quando si verifica un aumentato
rilascio di adenosina, acquisterebbe importanza il ruolo dei recettori A2A. Le interazioni
antagonistiche tra i due tipi recettoriali fanno sì che basse e alte concentrazioni di
adenosina possano stimolare rispettivamente i recettori A1 e A2A.
Dato che la stimolazione di entrambi i tipi recettoriali produce lo stesso effetto
differenziante, sia alte che basse concentrazioni di adenosina possono indurre
neuritogenesi.
I difetti della crescita dei neuriti sono stati associati alla malattia di Alzheimer e le
mutazioni geniche che coinvolgono la differenziazione neuronale causano diverse malattie
come ritardo mentale o eteropatie che portano a morte prematura. Per questi motivi i
recettori adenosinici potrebbero essere considerati un target per la cura di queste malattie.
(Canals et al., 2005).
1.5.2 Effetto differenziante della guanosina
È ampiamente riconosciuto che le purine extracellulari sono importanti molecole ad
azione trofica che influenzano la crescita, la differenziazione ma anche la morte cellulare.
La guanosina e il GTP extracellulari promuovono la crescita dei neuriti nelle cellule
PC12 e potenziano l’effetto neuritogenico del fattore di crescita neuronale (nerve growth

Introduzione
33
factor, NGF) con un meccanismo separato ma complementare. È stato dimostrato che
l’effetto della guanosina è sia AMPc-dipendente che AMPc-indipendente.
Nell’azione AMPc-indipendente della guanosina sembra che sia coinvolta la
guanilato ciclasi la cui attivazione incrementa i livelli di GMPc. È stato dimostrato, infatti,
che i composti che aumentano i livelli di questo nucleotide ciclico, come l’ossido nitrico o
analoghi del GMPc che riescono a permeare la membrana cellulare, possono favorire la
neuritogenesi nelle cellule PC12 e in altri sistemi cellulari. Tuttavia non è noto il
meccanismo d’azione del GMPc che potrebbe agire regolando le fosfodiesterasi, attivando
la PKC o regolando canali ionici.
Diversamente dalla guanosina, il meccanismo d’azione mediante il quale il GTP
extracellulare migliora la crescita dei neuriti non coinvolge né l’AMPc né il GMPc.
Il GTP extracellulare innalza i livelli del Ca2+ intracellulare nelle cellule PC12
mediante un aumento dell’influsso dello ione attraverso i canali di tipo L e incrementando
il suo rilascio dalle riserve intracellulari, incremento direttamente correlato alla
neuritogenesi.
L’esposizione delle cellule PC12 a GTP determina attivazione della PKC che può
essere il risultato della stimolazione della fosfolipasi C che genera IP3 e diacilglicerolo. Il
diacilglicerolo stimola direttamente la PKC mentre l’IP3 libera il calcio dalle riserve che a
sua volta va ad attivare la proteina chinasi.
L’attivazione della PKC non è però sufficiente ad indurre l’effetto neuritogenico ma
è sinergica con l’attivazione della cascata delle MAPK.
In qualunque modo venga stimolato, l’incremento dei livelli del Ca2+ sembra essere il
meccanismo principale mediante il quale il GTP aumenta la neuritogenesi indotta
dall’NGF (Gysbers et al., 2000).
Quindi, sia la guanosina (mediante la via dell’AMPc) che il GTP (mediante la via del
calcio) attivano la via delle MAPK e potenziano in modo simile la neuritogenesi indotta
dall’NGF. Se l’effetto del GTP fosse dovuto alla sua conversione a guanosina da parte
delle ectonucleotidasi, anche il GDP e il GMP dovrebbero indurre neuritogenesi, ma ciò
non sembra essere il caso (Rathbone et al.,1999).
E’ stato suggerito che il GTP potrebbe indurre differenziazione legandosi ad un sito
di legame presente sulle cellule PC12, probabilmente associato ai recettori del glutammato
(Gysbers et al., 2000).

Introduzione
34
1.5.3 Differenziazione e citoscheletro
Le malattie neurodegenerative sono caratterizzate da patologie assonali e neuronali e
la rigenerazione cellulare richiede estensione dei neuriti e orientazione cellulare. In queste
condizioni, i neuroni non connessi correttamente con i propri bersagli vengono rimossi per
apoptosi (Arnon e Aharoni, 2007; Hong Yang et al., 2005).
Il citoscheletro gioca un ruolo fondamentale nel mantenimento della forma altamente
asimmetrica e nella polarità strutturale dei neuroni che sono caratteristiche fondamentali
della fisiologia neuronale. Fondamentalmente, il citocheletro è costituito da tre diversi tipi
di proteine: i filamenti di actina, i filamenti intermedi come le keratine o i neurofilamenti e
i microtubuli. I microtubuli sono bastoncelli cavi di varia lunghezza che si presentano
isolati o in fasci poco serrati nel citoplasma di quasi tutte le cellule eucariotiche. Essi sono
anche i principali componenti strutturali delle ciglia e dei flagelli, dove contribuiscono a
produrre la forza del battito nel movimento (Maccioni et al., 1995). Nella cellula, ad ogni
cambiamento di forma il citoscheletro si riplasma, e i microtubuli del citoplasma si trovano
continuamente ad essere dissociati. La loro natura dinamica viene utilizzata a pieno quando
viene modificata la forma della cellula: un esempio ne e' la mitosi. In questo processo la
dissociazione del microtubulo è accompagnata da quella dei microfilamenti e dallo
smembramento dell’organizzazione dei filamenti intermedi, e quindi da un
rimaneggiamento globale del citoscheletro: come risultato la cellula perde la sua forma
interfasica e diventa sferica. I microtubuli si formano per associazione in quantità uguali di
due tipi di molecole tubuliniche, dette α-tubulina e β-tubulina, avente ciascuna un peso
molecolare di 50000 Dalton. L'alfa-tubulina si lega alla beta-tubulina per formare un
dimero e i dimeri si legano l'uno all'altro per formare dei protofilamenti: a loro volta i
dimeri di un protofilamento si legano ai dimeri adiacenti per formare un tubulo completo.
Questo processo di polimerizzazione viene facilitato da proteine accessorie che si
suddividono in due classi principali: le proteine associate ai microtubuli o MAP
(microtubule-associated protein) che hanno un peso molecolare tra 200000 e 300000
Dalton, e le proteine tau che hanno un peso molecolare che va da 60000 a 70000 Dalton.
Le MAP sono formate da due componenti: una si lega ai microtubuli mentre l'altra si
estende verso l'esterno e può contribuire a legare trasversalmente i microtubuli alle altre
strutture cellulari.
Nel morbo di Alzheimer’s, le placche amiloidi e i grovigli neurofibrillari
costituiscono due delle maggiori alterazioni neuropatologiche presenti nel cervello. I

Introduzione
35
grovigli neurofibrillari sono costituiti da un accumulo di proteina associata ai microtubuli
tau, che a causa di una iperfosforilazione si ammassa e non svolge più la sua normale
funzione di sostegno ai microtubuli (Benitez-King et al., 2004).
Un altro esempio di anormalità del citoscheletro presente in una patologia
neurodegenerativa sono i corpi di Lewis, considerati markers citopatologici nella malattia
di Parkinson. I corpi di Lewis sono costituiti da tubulina, MAP1 e MAP2. Inoltre, la
perdita di dendriti e la distribuzione irregolare dei processi neuronali si ritrova in aree del
cervello di pazienti schizofrenici. Numerose evidenze quindi suggeriscono che le malattie
neurodegenrative e le malattie psichiatriche sono associate ad alterazioni del citoscheletro
nei neuroni che, a loro volta, fanno perdere la connettività sinaptica e l’abilità di
trasmettere informazioni assonali al dominio somatodendritico (Benitez-King et al., 2004) .
La presenza di neuriti è uno dei principali ed essenziali markers di differenziazione
neuronale e per questa ragione la valutazione dell’estensione dei neuriti è uno dei metodi
più comunemente usato per valutare la differenziazione neuronale.
1.6 Linea cellulare SH-SY5Y
Le cellule di neuroblastoma umano SH-SY5Y sono un subclone neuroblastico
estensivamente caratterizzato della linea cellulare SK-N-SH (Nakagawa-Yagi et al., 1991;
Seidenfaden et al., 2006) ricavata nel 1970 dal tessuto di un neuroblastoma metastatico di
una bambina di quattro anni (Biedler et al., 1973). I neuroblastoma sono tumori ad
insorgenza in età pediatrica che originano da neuroblasti immaturi durante lo sviluppo del
sistema nervoso periferico (Olsson et al., 2000).
Le cellule della linea SH-SY5Y sono una popolazione cellulare dalla morfologia
simil-neuroblastica, con un corpo cellulare a forma di goccia (Jalava et al., 1990) e piccoli
neuriti, che crescono per buona parte come monostrato formando piccoli aggregati o
clusters (Biedler et al., 1973; 1978), mentre una parte minore si trova in sospensione e
rappresenta le cellule in mitosi.
Queste cellule costituiscono un modello ben caratterizzato per lo studio in vitro della
differenziazione in un fenotipo neuronale-simile inducibile da vari agenti differenzianti
esogeni, come l’acido retinoico, gli esteri del forbolo, il fattore di crescita dei nervi (NGF),
e l’analogo membrana-permeabile del cAMP, il dibutiril-cAMP (Seidenfaden et al., 2006;
Peterfreund et al., 1997; Ammer e Schulz, 1994). La differenziazione di queste cellule è

Introduzione
36
associata all’estensione di lunghi neuriti e può essere quantificata tramite analisi
morfologica della neuritogenesi (Jalava et al., 1990, Seidenfaden et al., 2006).
La linea SH-SY5Y è molto usata anche come modello per lo studio della morte
cellulare neuronale indotta da stress ossidativi, associato a diverse malattie
neurodegenerative croniche come il morbo di Parkinson, di Alzheimer e di Huntington
(Ruffels et al., 2004). Sono utilizzate anche come modello per la neurotrasmissione
simpatica e quella dopaminergica nello studio del morbo di Parkinson (Ault e Werling,
2000).
Nelle membrane cellulari sono state isolate proteine GI/GO inattivabili
specificamente dalla tossina PTX per ADP-ribosilazione (Nakagawa-Yagi et al., 1991).
Esprimono i recettori adenosinici A1 ed A2A e, in presenza di inibitori delle fosfodiesterasi,
i livelli del cAMP aumentano anche in assenza di una stimolazione dell’adenilato ciclasi
recettore-mediata, evidenza che suggerisce che i livelli basali del nucleotide ciclico siano
soggetti ad un alto turnover (Canals et al., 2005; Morgan et al., 1993).
Altre caratteristiche della linea cellulare comprendono la capacità di convertire il
glutammato in GABA e la colina in acetilcolina, e la presenza di attività dopamina-β-
idrossilasica (Biedler et al., 1978). Esprimono i recettori oppioidi µ e δ, in particolare i δ2
(Toll et al., 1997), ed i recettori α2 adrenergici (Ammer e Schulz, 1994). Sono anche
considerate un modello appropriato per lo studio dei recettori σ (Ault e Werling, 2000).
Le cellule esprimono inoltre i canali per il calcio, sia di tipo N sia di tipo L, i
recettori muscarinici (M3) e nicotinici, i recettori per il neuropeptide Y (recettori Y2) (Ault
e Werling, 2000; Svensson, 2000).

2. Scopo della Ricerca

Scopo della Ricerca
37
L’interesse della comunità scientifica nei confronti dei derivati purinici come
possibili neurotrasmettitori è stato sollevato nel 1972 quando G. Burnstock pubblicò una
review in cui raccoglieva le molteplici evidenze sperimentali a favore del ruolo svolto
dall’ATP quale principale molecola-segnale di un sistema non-adrenergico non-colinergico
(NANC), presente almeno a livello gastro-enterico, e da lui denominato sistema
purinergico (Burnstock G., 1972). Da allora, il moltiplicarsi degli studi ha permesso non
solo di confermare l’ATP quale trasmettitore di un sistema NANC, ma anche di definire la
presenza di due distinti sistemi recettoriali, uno per il nucleotide adeninico ATP, l’altro per
il suo derivato nucleotidico adenosina. Ambedue i sistemi sono diffusi a livello del sistema
nervoso sia centrale, sia periferico, influenzando così l’attività dell’organismo in toto.
Tra i molteplici effetti esercitati a livello del sistema nervoso centrale, quelli di più
recente identificazione riguardano la capacità di ATP e adenosina di svolgere attività a
lungo termine, quali neuroprotezione o neurolesività, stimolazione o inibizione della
proliferazione, effetti trofici o di riduzione della sopravvivenza cellulare.
Rispetto ai derivati purinici su base adeninica, quelli a base guaninica come il GTP e
la guanosina sono stati oggetto di un numero di studi di molto inferiore, forse a causa della
difficoltà di identificazione di un sistema recettoriale specifico e della conseguente
mancata caratterizzazione farmacologica dello stesso. Ciononostante, svariate recenti
evidenze sperimentali indicano che la guanosina è in grado di esercitare, in modo sia
diretto, sia indiretto, funzioni trofiche ed anti-apoptotiche su astrociti (Rathbone et al.,
1998; Di Iorio et al., 2004), funzioni neuroprotettive su fettine di ippocampo o in modelli
di ischemia cerebrale (Oleskovicz et al., 2008; Chang et al., 2007) ed effetti

Scopo della Ricerca
38
comportamentali, quale un effetto amnesico (Saute et al., 2006) e anticonvulsivante
(Soares et al., 2004). Ed è sulla base di questo crescente numero di studi che si basa la
recente proposta dell’esistenza di un sistema purinergico su base guaninica,
particolarmente attivo a livello del sistema nervoso centrale, che potrebbe costituire un
ulteriore bersaglio per la modulazione farmacologica di svariati processi neuronali
(Schmidt et al., 2007).
L’interesse in questo studio deriva principalmente dalla scarsità di dati riguardanti
l’effetto della guanosina sui neuroni in generale, rispetto a quelli ottenuti da studi eseguiti
impiegando cellule gliali, ed è per questo che la scelta della linea cellulare da utilizzare è
ricaduta su un neuroblastoma umano: le cellule SH-SY5Y. Questa linea cellulare
ampliamente usata costituisce un modello adatto a questo studio in quanto esprime diversi
recettori quali i recettori adenosinici di tipo A1 e di tipo A2A (Canals et al., 2005; Morgan et
al., 1993) e i recettori muscarinici (Lambert et al., 1989); presenta un turnover dell’ AMPc
intracellulare e movimenti del calcio intracellulare adeguatamente elevati e, in presenza di
agenti differenzianti come l’acido retinoico, va incontro a cambiamenti morfologici con
comparsa di un aspetto simil-neuronale dovuto alla crescita di numerosi neuriti (Ammer e
Schulz., 1994). Inoltre, è stato dimostrato che la linea risponde all’esposizione ad agonisti
dei recettori adenosinici con un processo di differenziazione simile a quello indotto
dall’acido retinoico (Canals et al., 2005) e all’esposizione a guanosina con una riduzione
dell’apoptosi indotta da β-amiloide (Pettifer et al., 2004).
Il primo scopo di questo studio è quello di indagare la capacità della guanosina di
proteggere le cellule poste in condizioni di stress, come ad esempio la deprivazione da
siero, e i principali meccanismi alla base di questo effetto.
In secondo luogo ci siamo posti l’obiettivo di investigare la presenza di meccanismi
di trasporto transmembrana per la guanosina e la loro successiva caratterizzazione. Questo
studio è utile non solo per valutare l’entità dell’uptake di per se, ma serve anche da
premessa metodologica per la ricerca di siti di legame specifici per la guanosina.
Infine lo scopo di questa tesi è quello di valutare l’effetto neuritogenico della
guanosina in questa linea cellulare basandoci su dati di letteratura che dimostrano la
capacità della guanosina di favorire la crescita dei neuriti indotta dal Fattore di Crescita
neuronale (NGF) nelle cellule PC12, derivate da un feocromocitoma di ratto (Gysbers et
al., 2000); e la differenziazione di neuroni di ippocampo in vitro (Rathbone et al., 1999;
Traversa et al., 2003) con induzione della neuritogenesi e aumento della sintesi e del
rilascio da parte degli astrociti di fattori trofici, come NGF e la proteina neurotrofica

Scopo della Ricerca
39
S100β. Per quanto riguarda lo studio sulla differenziazione indotta da guanosina, in questo
studio verrà valutato il coinvolgimento del sistema adenosinico, l’effetto anti-proliferativo
della guanosina e i possibili meccanismi d’azione alla base degli effetti della guanosina in
questa linea cellulare.

3. Materiali e Metodi

Materiali e Metodi
40
3.1 Composti chimici utilizzati
Backer: Acido cloridrico (HCl) 37 %, Potassio cloruro (KCl).
Boehringer: Hepes.
Carlo Erba: Sodio cloruro (NaCl).
Euro€lone: Dulbecco’s Modified Eagle’s Medium High Glucose (DME/HIGH),
Dulbecco’s Phosphate Buffered Saline (D-PBS), Trypsin 0.05 % con EDTA 0.02 % in
PBS, Amphotericin B (Fungizone) 250 µg/ml, Penicillin Streptomycin solution 100x, L-
Glutamine solution 100x, Fetal bovine serum (FBS).
Merck: Sodio idrossido (NaOH), Fosfato di potassio monobasico (KH2PO4), Fosfato di
sodio bibasico diidrato (Na2HPO4·2H2O).
RBI (Research Biochemicals Inc.): 3,7-dimetil-1-propargylxantine (DMPX), 8-
cyclopentyl-1,3-dipropylxanthine (DPCPX).
SIGMA: Trypan blue, guanosina, Adenosine deaminasi (ADA), forskolina, rolipram,
nitrobenziltioinosina (NBTI), acido retinoico, adenosina-3’,5’-monofosfato ciclico
(cAMP), dimetil sulfossido biotechnology performance certified (DMSO), TRIZMA®
BASE, acido etilendiamino tetracetico (EDTA), Giemsa stain stock solution, May-
Grünwald stain solution, Phosphate Buffered Saline (PBS).

Materiali e Metodi
41
3.2 Coltura cellulare
Le cellule di neuroblastoma umano della linea cellulare SH-SY5Y vengono
mantenute in bottiglie aventi superficie uguale a 75 cm2 e tappo con filtro sterilizzante
(Corning®), nelle quali sono seminate ad una densità di 1,5x106 in 25 ml di terreno di
coltura costituito dal mezzo Dulbecco’s Modified Eagle’s Medium High Glucose
(DME/HIGH) contenente il 10 % v/v di siero fetale bovino (FBS), inattivato per
riscaldamento a 60°C per un ora, l’1 % v/v degli antibiotici Amfotericina B (1,35x10-3
mM), L-glutammina (2 mM) e una soluzione di penicillina-streptomicina (rispettivamente
100 UI/ml e 100 µg/ml). Le cellule sono mantenute in termostato umidificato alla
temperatura di 37°C in un’atmosfera composta per il 5 % da CO2 e il 95 % da aria. Il
passaggio delle cellule viene effettuato settimanalmente al raggiungimento di una
confluenza dell’80%: le cellule vengono lavate una o due volte con 5 ml di una soluzione
salina D-PBS (Dulbecco’s Phosphate Buffered Saline) sterile e staccate dalla bottiglia con
2 ml una soluzione contenente tripsina 0,05 % ed EDTA 0,02 % in PBS. Dopo 10 minuti in
incubatore a 37°C, la tripsina viene inattivata per aggiunta di 10 ml di terreno completo
DME/HIGH al 10 % FBS. Dopo accurata risospensione delle cellule contenute nella
bottiglia, si procede trasferendo le stesse in una provetta da 15 ml e centrifugando a 1850
rpm (547xg) per 5 minuti a temperatura ambiente.
3.3 Test della Sulforodamina B (SRB)
Il test della Sulforodamina B è un test colorimetrico che evidenzia il comportamento
proliferativo delle cellule sottoposte all’azione della sostanza che si vuole testare. Poichè la
Sulforodamina B è un colorante in grado di legarsi alle proteine cellulari, la quantità del
colorante stesso, rivelata dalla lettura con lo spettrofotometro, è direttamente proporzionale
al numero di cellule vive in ogni pozzetto.
Le cellule vengono seminate in piastre da 96 pozzetti, alla densità di 2x103-10x104
cellule per pozzetto, a seconda dei tempi di esposizione alle sostanze in esame. Il giorno
della valutazione, dopo aver controllato al microscopio le condizioni delle cellule, si
centrifuga la piastra a 1710 rpm (381 xg) per 5 minuti a temperatura ambiente (SIGMA
3K30-Celbio).
La piastra viene svuotata per rovesciamento, lavata con 100 µl di CMF-PBS e
fissata in 50 µL di TCA (acido tricloroacetico). Dopo aver lasciato la piastra a 4°C per un

Materiali e Metodi
42
ora, si procede con la colorazione: si svuota la piastra e si lava con 100 µl di acqua
bidistillata per rimuovere i residui di TCA, quindi si aggiungono 100 µl per pozzetto di una
soluzione di Sulforodamina B (SRB) allo 0,4 % in Acido Acetico all’1 % che viene
lasciata a temperatura ambiente per 30 minuti. Si lava quindi ogni pozzetto con 100 µl di
Acido Acetico all’1 % fino alla rimozione del colorante non legato. Quindi si procede con
la solubilizzazione del colorante con 200 µl per pozzetto di una soluzione di TRIS 10 mM.
La lettura viene effettuata a 570 nm mediante lo spettrofotometro Microplate Autoreader
(Bio-Tek Instruments).
3.4 Test di vitalità (MTT test)
Per valutare l’attività mitocondriale è stato impiegato il test dei sali di tetrazolio che
è un test colorimetrico in grado di stimare il numero delle cellule aventi ancora attività
mitocondriale, e quindi la vitalità delle cellule. Questo test si basa su un indicatore
metabolico, il sale solubile di tetrazolo che, nelle cellule vitali, è ridotto nel mitocondrio ad
opera di enzimi deidrogenasi attivi, a formare un cristallo insolubile in acqua di color viola.
Dopo il tempo desiderato di trattamento vengono quindi aggiunti 20 µl di
soluzione MTT (5 mg/ml) in ogni pozzetto e in seguito a 2 ore di incubazione a 37°C al 5
% di CO2, viene eliminata la soluzione dei sali di tetrazolio per rovesciamento e ai pozzetti
sono aggiunti 200 µl di DMSO per solubilizzare i precipitati di formazano. La lettura con
lo spettrofotometro (lettore di piastra da 96 pozzetti, Microplate Autoreader, Bio-Tek
Instruments) è stata effettuata alla lunghezza d’onda doppia di 540 e 630 nm.
3.5 Valutazione della neuritogenesi
Per valutare la potenziale azione differenziante della Guanosina (GUO) e i possibili
meccanismi coinvolti, sono stati eseguiti esperimenti di differenziazione il cui esito è stato
valutato visivamente.
Le cellule vengono seminate in piastre da sei pozzetti e trattate immediatamente con i
composti in esame e con l’acido retinoico come controllo positivo interno. Trascorso il
tempo stabilito, le cellule vengono fissate in metanolo per un minuto e colorate secondo il
metodo May-Grunwald Stain: si ricoprono i pozzetti con il colorante May-Grunwald per 5
minuti e successivamente, senza eliminare il colorante, si aggiunge goccia a goccia il PBS
fino a riempire il pozzetto e si lascia agire per 7 minuti. Quindi si svuota la piastra per

Materiali e Metodi
43
rovesciamento, si effettua un lavaggio con altro PBS per 7 minuti, in modo da eliminare il
colorante non legato e si procede eliminando il PBS e ricoprendo il pozzetto con il
colorante Giemsa diluito in PBS e lo si lascia agire per 7 minuti, trascorsi i quali si
effettuano abbondanti risciacqui con acqua bidistillata.
Le piastre vengono osservate con un microscopio Leica Leitz Wetzlar con
ingrandimento 40x collegato a un computer. Le immagini di almeno quattro campi scelti a
caso sono state acquisite mediante il programma Leica IM (V.4, Leica Microsystem
Digital Imaging, Cambridge, U.K.).
La determinazione della percentuale di cellule differenziate si effettua rapportando il
numero di prolungamenti con lunghezza almeno doppia rispetto al corpo cellulare al
numero di cellule totali. Sulle stesse immagini, utilizzando il programma “Scion Image” è
stato possibile misurare la lunghezza effettiva dei neuriti utilizzando una scala
micrometrica.
3.6 Dosaggio proteico
La quantificazione delle proteine presenti nei campioni delle membrane cellulari per
il Radioligand Binding e dei lisati cellulari per il Western Blot è stata eseguita utilizzando
il kit BioRadDC Protein Assay, basato sul metodo di Lowry et al.
Il saggio è condotto su una piastra da 96 pozzetti, in cui i campioni vengono
distribuiti in quantità di 2 e 3 µl per pozzetto e portati al volume finale di 10 µl con acqua
bidistillata o lysis buffer.
Per valutare la concentrazione proteica incognita nei campioni si è costruita una
curva di taratura, utilizzando concentrazioni crescenti di Siero Albumina di Bue (BSA),
sciolta in acqua distillata alla concentrazione di 2 µg/µl. Tutte le prove, compresa la curva
di taratura, sono allestite in duplicato.
Alle soluzioni precedentemente preparate sono stati aggiunti 25 µl per pozzetto di
reagente A, una soluzione acida di tartrato di rame, per 5 minuti a temperatura ambiente.
Sono stati quindi aggiunti 200 µl per pozzetto di reagente B, il reattivo di Folin, che
reagisce con le proteine, per 5 minuti a temperatura ambiente.
La presenza di proteine viene indicata dallo sviluppo di un colore violetto e
l’intensità della colorazione, misurata spettrofometricamente, è proporzionale alla
concentrazione proteica. L’assorbanza viene rilevata alla lunghezza d’onda di 630 nm con
un lettore per piastre Microplate Reader (Bio -Tek Instrument).

Materiali e Metodi
44
La retta di taratura, ottenuta con concentrazioni note di BSA (standard), ha
consentito di estrapolare, dai dati di assorbanza dei campioni, le relative concentrazioni di
proteine, basandosi sulla legge di Lambert-Beer, che stabilisce una proporzionalità diretta
tra assorbanza e concentrazione proteica di una soluzione, e che graficamente assume un
andamento rettilineo. Le concentrazioni finali sono espresse in mg/ml e vengono calcolate
come rapporto tra l’assorbanza del campione e l’assorbanza della retta standard attraverso
programma di calcolo Excel.
3.7 Esperimenti di uptake su cellule di neuroblastoma umano SH-
SY5Y
Sulla linea cellulare SH-SY5Y sono stati svolti due tipi di esperimenti per lo studio
del trasporto della [3H]-guanosina (Guanosine [8-3H], ICN Pharmaceutical, attività
specifica 20 Ci/mmol) e della [3H]-adenosina (PerkinElmer, Boston, MA, USA, attività
specifica 35.9 Ci/mmol). Per questi esperimenti, le cellule vengono seminate 48 h prima
dell’esperimento in piastre da 24 pozzetti (FALCON®) ad una densità di 2.5 x 105 cellule
in 1 ml di terreno completo per pozzetto e mantenute in termostato a 37°C. Il giorno
dell’esperimento vengono allestite le soluzioni di prestimolazione e di stimolazione,
costituite da [3H]-adenosina o [3H]-guanosina ad un’unica concentrazione, 1 µM e 100 µM
rispettivamente, in PBS, oppure dal tampone Locke preparato con o senza NaCl 140 mM,
pH 7.4, contenente [3H]-adenosina 1 µM con e senza NBTI non marcato (10 µM) o
guanosina 100 µM. Quindi si procede secondo il seguente protocollo: si effettua un
risciacquo del pozzetto con il tampone PBS (200 µl) a 4 °C e successivamente una
preincubazione della durata di 10 minuti, allo scadere dei quali si aspira la soluzione per
sostituirla con 200 µl di una soluzione di stimolazione come descritto. Si lascia incubare il
tempo desiderato a temperatura ambiente (25 °C). Quindi si aspira nuovamente il
surnatante e si procede con due lavaggi consecutivi per pozzetto con 200 µl di tampone
PBS, 4 °C, e si aggiungono 250 µl per pozzetto di una sospensione di NaOH 1 M + 1 % di
sodio dodecil solfato (SDS). Dopo pochi minuti, si trasferiscono 200 µl della sospensione
ottenuta da ogni pozzetto in altrettanti pozzetti di una piastra flessibile (Wallac) che viene
inserita in un apposito supporto. Si aggiungono 250 µl di liquido di scintillazione
(Optiphase ‘SuperMix’), e si sigilla la piastra con un film di plastica. Due pozzetti vengono
utilizzati per effettuare le conte di 50 µl del nucleotide triziato utilizzato (standard) in 250
µl di NaOH 1 M + 1 % SDS e di liquido di scintillazione. La lettura è effettuata dopo aver

Materiali e Metodi
45
incubato la piastra flessibile al buio overnight. La concentrazione proteica è determinata
impiegando le soluzioni della Bio-Rad (DC Protein Assay). L’analisi dei dati è stata svolta
utilizzando il programma Excel in modo analogo a quanto riportato per gli esperimenti di
radioligand binding. I valori in CPM (conte per minuto) sono trasformati in DPM
(disintegrazioni per minuto) sulla base dell’efficienza di lettura dello standard e quindi
rapportati all’attività specifica per calcolare le picomoli di [3H]-adenosina e di [3H]-
guanosina trasportate al secondo in ogni campione, che sono state riportate come media ±
deviazione standard, oppure le picomoli di [3H]-adenosina trasportate sono state rapportate
ai mg di proteine presenti nel pozzetto e riportate come media ± errore standard della
media.
3.8 Esperimenti di Radioligand Binding
Sono stati svolti esperimenti per lo studio dei trasportatori nucleosidici equilibrativi
utilizzando la [3H]-nitrobenziltioinosina ([3H]-NBTI, PerkinElmer, Boston, MA, USA,
attività specifica 30.0 Ci/mmol). Gli esperimenti di saturazione sono stati condotti sia su
cellule intatte che su membrane di cellule SH-SY5Y mentre gli esperimenti di
competizione sulle sole membrane.
3.8.1 Esperimenti di saturazione su cellule intatte SH-SY5Y
Le cellule vengono seminate in piastre da 24 pozzetti come descritto per gli
esperimenti di uptake e il giorno dell’esperimento vengono allestite le soluzioni costituite
da [3H]-NBTI a diverse concentrazioni (0.1 nM, 0.3 nM, 1 nM, 3 nM) disciolto in tampone
PBS con o senza NBTI non marcato alla concentrazione di 1 µM. Quindi si procede
secondo il seguente protocollo: si aspira con una pompa a vuoto il mezzo di coltura da ogni
pozzetto, si effettua un risciacquo con il tampone PBS (200 µl) e lo si sostituisce con 200
µl della soluzione contenente NBTI marcato, procedendo analogamente sui pozzetti
successivi ad intervalli di dieci secondi l’uno dall’altro. Le piastre vengono lasciate
incubare per 1 h a partire dal primo pozzetto trattato, a temperatura ambiente (25 °C). Allo
scadere del tempo stabilito, si aspira nuovamente il surnatante e si procede con due lavaggi
consecutivi per pozzetto, analogamente al primo risciacquo (200 µl di PBS freddo), e si
aggiungono 250 µl per pozzetto di una sospensione di NaOH 1 M + 1 % di sodio dodecil
solfato (SDS) per disgregare le membrane cellulari e facilitare il distacco delle cellule dal

Materiali e Metodi
46
fondo del pozzetto. Dopo pochi minuti, vengono trasferiti 200 µl della sospensione
ottenuta da ogni pozzetto nei corrispettivi pozzetti di una piastra flessibile (Wallac) da 24
pozzetti che viene inserita in un apposito supporto. Si aggiungono 250 µl di liquido di
scintillazione (Optiphase ‘SuperMix’), si sigilla la piastra con film di plastica (sealing tape,
PerkinElmer) e si introduce nel contatore a scintillazione liquida (1450 Microbeta Trilux,
Wallac) per la lettura dopo 720 minuti. In due pozzetti si effettua la conta di un volume
noto di [3H]-NBTI 6 nM (25 µl) nello stesso volume di NaOH 1 M + 1 % SDS e di liquido
di scintillazione utilizzati per la lettura dei campioni. Questa lettura permette di risalire
all’efficienza della conta.
3.8.2 Preparazione di membrane da cellule SH-SY5Y
Per quanto riguarda il legame di [3H]-NBTI su membrane, le membrane di cellule
SH-SY5Y sono preparate secondo il seguente procedimento: dieci flask da 75 cm2
(contenenti circa 300000000 di cellule) vengono lavate due volte con 5 ml di PBS e poi
staccate in 10 ml di buffer di lisi ghiacciato (HEPES 5 mM, EDTA 2 mM, pH 8.15,
fluoruro di fenilmetansulfonile o PMSF 1 mM, Leupeptina 0,1 mM) mediante uno scraper.
Le cellule vengono quindi centrifugate per 10 minuti a 4 °C a 12000 xg (SIGMA
3K30-Celbio) e il pellet viene risospeso in 10 ml di PBS 8 mM pH 7.4. La sospensione
viene quindi potterizzata con un potter di vetro (Thomas Company, Philadelphia) per una
trentina di volte, centrifugata per 3 volte a 48000 xg per 20 minuti risospendendo il pellet
di volta in volta in PBS fresco. Dopo la terza centrifugazione le membrane vengono
risospese in 4 ml di PBS e su questa sospensione viene effettuato il dosaggio delle proteine
secondo il metodo di Lowry et al. (1951).
3.8.3 Esperimenti di saturazione su membrane di cellule SH-SY5Y
In una piastra per filtrazione da 96 pozzetti ( MultiScreen Vacuum Manifold,
Millipore) precedentemente bagnata con 200 µl di tampone Tris-HCl 50 mM, pH 7.4,
vengono aggiunte concentrazioni crescenti di [3H]-NBTI (0.01-10 nM), NBTI freddo alla
concentrazione di 1 µM per la determinazione del legame aspecifico, circa 50 µg di
proteine di membrane cellulari e una quantità di tampone adeguata per raggiungere in ogni
pozzetto il volume finale di 250 µl. La piastra viene lasciata ad incubare per 1 ora a 4 °C,
tempo necessario per raggiungere l’equilibrio. La piastra viene quindi filtrata con pompa a

Materiali e Metodi
47
vuoto per eliminare l’[3H]-NBTI non legato e lavata con 200 µl di tampone Tris-HCl 50
mM freddo. A ciascun pozzetto vengono aggiunti 25 µl di liquido di scintillazione
(Optiphase “Supermix”). La piastra viene lasciata al buio overnight e letta tramite il
contatore a scintillazione liquida (Microbeta Trilux, Wallac). I dati sono espressi come
media di pmol di [3H]-NBTI legato rispetto ai mg di proteine ± errore standard.
3.8.4 Esperimenti di competizione su membrane di cellule SH-SY5Y
Per gli esperimenti di competizione è stato utilizzato come ligando marcato [3H]-
NBTI alla concentrazione fissa di 0.5 nM, concentrazioni crescenti di guanosina (0.3 –
10000 µM) o adenosina (0.1 – 1000 µM) e NBTI freddo alla concentrazione di 1 µM per la
determinazione del valore di aspecifico. I risultati sono riportati come picomoli di [3H]-
NBTI legato rispetto ai mg di proteine. I dati sono espressi come media ± deviazione
standard.
3.8.5 Analisi dei dati
L’analisi dei dati è stata svolta utilizzando il programma Excel. Dalle conte per
minuto (cpm) ottenute dallo standard si ricava l’efficienza di conteggio della piastra come
rapporto tra le cpm di una quantità nota di radioattivo letta e le disintegrazioni per minuto
(dpm) teoriche, ottenute moltiplicando la quantità di triziato espressa in nCi per il numero
di disintegrazioni per minuto corrispondenti ad 1 nCi (ca. 2220). Le disintegrazioni per
minuto sono rapportate a loro volta alle picomoli note di standard utilizzate, per ricavare il
valore dell’attività specifica. Le cpm di ogni campione sono quindi rapportate
all’efficienza di lettura della piastra ricavando così le dpm corrispondenti. I loro valori
rapportati all’attività specifica calcolata danno le picomoli di triziato di ogni singolo
pozzetto, che sono state riportate come media ± deviazione standard delle femtomoli di
[3H]-NBTI legate per pozzetto. I valori di Kd, Bmax e IC50 sono stati ottenuti con il
programma SigmaPlot 2001.

Materiali e Metodi
48
3.9 Tecniche di immunocitochimica
L’immunocitochimica è una tecnica che consente l’identificazione e la localizzazione
cellulare di proteine, grazie all’utilizzo di anticorpi specifici, riconosciuti a loro volta da
anticorpi secondari coniugati a molecole fluorescenti, mediante un microscopio ad epi-
fluorescenza.
Le cellule SH-SY5Y sono state seminate in piastre da 24 o da 6 pozzetti, in cui era
stato precedentemente inserito un vetrino sterile da 13 mm (Zeus, Vetrotecnica, Padova,
Italy) o rettangolare da 21x26 mm (Micro Standard Cover Glass, Matsunami Glass
Industries, LTD., Japan), alla concentrazione di 10-25x105 cellule per pozzetto per
trattamenti da 24-96 h. Le cellule sono state trattate subito dopo la semina e sempre almeno
in doppio.
Dopo 24, 48, 72 e 96 h le cellule sono state fissate con PFA in PBS al 2 %
(massa:vol) per 30 minuti. Le cellule sono state poi lavate per 3 volte con PBS per 5-10
minuti sotto agitazione, e successivamente permeabilizzate con Triton X-100 allo 0.2 %
(vol:vol) per 5 minuti sotto agitazione. Per l’identificazione di NeuN i vetrini sono stati
invece immersi in un tampone citrato 10 mM a pH 6 e contenente Triton X-100 allo 0.3 %
e scaldati in microonde fino al bollire della soluzione.
Dopo ulteriori lavaggi in PBS, si è proceduto al bloccaggio dei siti aspecifici
utilizzando una soluzione di blocco costituita da BSA al 4 % e siero di capra al 5 %
(Normal Goat Serum) in PBS. L’incubazione è durata 1 ora a 37°C in camera umida.
Le cellule sono state dunque incubate con le soluzioni degli anticorpi primari diluiti
nella soluzione di blocco:
• Anticorpo monoclonale anti-β-Tubulina III (Sigma-Aldrich, MI, Italy) dil 1:250
• Anticorpo monoclonale anti-MAP2 (Sigma-Aldrich, MI, Italy) dil 1:250
• Anticorpomonoclonale anti-NeuN (Sigma-Aldrich, MI, Italy ) dil 1:250
L’incubazione con l’anticorpo primario è stata effettuata overnight a 4°C sempre in
camera umida.
Il giorno seguente i vetrini, dopo 3 lavaggi in PBS, sono stati incubati per 1 ora a
37°C con l‘anticorpo secondario coniugato con fluoresceina isotiocianato (Goat anti-
Mouse-FITC conjugated, Sigma-Aldrich, Milano, Italy) diluito 1:300 nella soluzione di
blocco.

Materiali e Metodi
49
Dopo ulteriori lavaggi in PBS, si è proceduto alla colorazione dei nuclei incubando le
cellule per 7 minuti al buio con una soluzione di DAPI (4’,6-diamino-2-fenilindolo
cloridrato, Sigma Aldrich) alla concentrazione di 1 µg/ml in PBS.
Infine è stata eseguita una disidratazione alcolica eseguendo lavaggi di 5 minuti
ciascuno con soluzioni di etanolo in acqua a concentrazione crescente (70, 90, 100%)
(vol/vol).
Per il montaggio è stato utilizzato come agente antiquencing il Glicerolo-DABCO (4-
diazabicyclo[2.2.2]octane) (3 % DABCO stock + 0.02 % sodio azide + 10 % Tris HCl 200
mM, pH 8).
Le immagini sono state quindi acquisite con il microscopio a epifluorescenza Eclipse
E800- NIKON, collegato a fotocamera DMX 1200, mediante il programma ACT-1. Sono
stati acquisiti almeno quattro campi scelti a caso per ciascun trattamento e gli esperimenti
sono stati condotti in triplicato.
3.9.1 Immunocitochimica con falloidina
La falloidina è una tossina proveniente dalla pianta Amanita Phalloides in grado di
legarsi all’interfaccia dei filamenti di F-actina prevenendone la depolarizzazione.
Se coniugata a dei fluorofori risulta visibile al microscopio a fluorescenza. Abbiamo
quindi effettuato una serie di esperimenti di tempo-dipendenza per cercare di
caratterizzare l’effetto differenziante di guanosina fino a sette giorni di trattamento
utilizzando falloidina coniugata a tetrametil rodamina isotiocianato, TRITC (Phalloidin
TRITC labeled mixed isomers, Sigma-Aldrich, Milano, Italy).
Le cellule vengono seminate in piastre da 6 pozzetti contenenti dei vetrini
rettangolari o in piastre da 24 contenenti vetrini rotondi sterili alla concentrazione di
100000-250000 cellule per pozzetto per i trattamenti da 24-96 h e alla concentrazione di
20000 cellule per pozzetto per i trattamenti da 7 giorni. Trascorso il tempo desiderato, le
cellule sono state fissate in PFA al 2 % in PBS, lavate 3 volte per 5 minuti in PBS, trattate
con Glicina allo 0.1 % in PBS per 5 minuti sotto agitazione e permeabilizzate con Triton
X-100 allo 0.2 % per 5 minuti. Le cellule sono state quindi incubate al buio con falloidina
(1 µg/ml in PBS) per 10-15 min e con DAPI (1 µg/ml in PBS) per 7 min. Tra un
trattamento e l’altro sono sempre stati effettuati 3 lavaggi da 5 minuti in PBS. Si procede
come descritto sopra con la disidratazione con alcoli e si montano i vetrini con glicerolo-
DABCO su vetrini portaoggetto.

Materiali e Metodi
50
3.10 Tecniche di Western Blot
L’attivazione della via delle MAP chinasi è stata studiata mediante l’utilizzo della
tecnica del western blot e nello specifico le proteine visualizzate sono state Akt-1, p-Akt-1
e l’actina come controllo interno.
3.10.1 Preparazione dei lisati cellulari
Le cellule sono state staccate in PBS mediante uno scraper, raccolte in una provetta
da 15 ml e centrifugate a 397 xg per 5 minuti. Successivamente, è stato eliminato il
sovranatante, il pellet è stato risospeso in 50 µl di Lysis Buffer con inibitori di fosfatasi
(NaF 1 mM; sodio ortovandato 1 mM) e inibitori di proteasi (Protease inhibitor cockatil,
Sigma-Aldrich) e trasferito in un microtubo da 1.5 ml. Il microtubo è stato centrifugato a
10000 xg per 10 minuti a 4°C (SIGMA 4K15C-Bio.Tec.). Sul lisato cellulare così ottenuto
è stato effettuato il dosaggio delle proteine utilizzando il metodo Lowry già descritto. I
campioni sono stati mantenuti a -20 °C. Il giorno dell’esperimento, al volume del
campione contenete la quantità di proteina stabilita, è stato aggiunto un volume equivalente
di tampone di caricamento costituito da Tris HCl pH 6.8, 1 M, 0.001 % di blu-
bromofenolo, glicerolo al 10 %, SDS al 10 % per denaturare le proteine, e β-
mercaptoetanolo al 5 % per ridurre i ponti disolfuro.
3.10.2 Elettroforesi delle proteine in gel di poliacrilamide con SDS (SDS-PAGE)
Questa metodica consente di separare proteine di diverso peso molecolare mediante
la loro migrazione attraverso le maglie di un gel di poliacrilamide sotto l’azione di un
campo elettrico. Ciò è consentito dalla presenza del sodiododecilsolfato (SDS), un
detergente anionico capace di complessarsi alle proteine e di conferire loro una carica netta
negativa costante per unità di massa. Ne consegue che esse vengono separate sulla base del
loro peso molecolare.
E’ stata effettuata una separazione proteica mediante elettroforesi in verticale su gel di
poliacrilamide, secondo il metodo di Laemmli (Laemmli et al., 1970).
I campioni, contenenti uguali quantità di proteine del lisato cellulare (70 µg) e
sospesi in buffer di caricamento, vengono caricati su gel di poliacrilamide preparati la sera
prima o su gel precast (PAGEr® Precast Gels, Lonza Rockland, U.S.A.). Il gel di

Materiali e Metodi
51
poliacrilamide è un gel dello spessore di circa 1 mm formato da un gradiente discontinuo
di due gel a pH e concentrazioni di acrilamide diversi: un gel superiore di impaccamento
(stacking gel) e uno inferiore di corsa (running gel), dove avviene la separazione delle
proteine. Lo stacking gel è stato preparato al 4 % mentre il running gel al 10 % di
acrilamide.
Come marcatori dei pesi molecolari sono stati utilizzati il ColorBurstTM Wide Molecular
Weight Range, (Sigma-Aldrich, Milano, Italy).
La corsa elettroforetica è avvenuta in una camera per elttroforesi (PAGErTM Minigel
Chamber, Lonza Rockland, U.S.A.) riempita con il tampone di corsa costituito da 192 mM
glicina, 25 mM Tris e 0.1 % SDS. Alla camera elettroforetica è stato applicato un
amperaggio di 10 mA per ogni gel per 30 minuti, al fine di favorire l’impaccamento delle
bande nello stacking gel; e successivamente, una corrente di 20 mA per 1 ora e 30 minuti,
allo scopo di separare le proteine nel running gel.
Figura 13: PAGErTM Minigel Chamber, Lonza Rockland, U.S.A.
3.10.3 Western Blot
Il metodo consiste nell’elettrotrasferimento di proteine dalla matrice del gel di
acrilamide, in cui sono state fatte migrare, ad una membrana di nitrocellulosa. Una volta
fissate nella membrana, le proteine possono poi essere analizzate per mezzo di anticorpi
specifici diretti contro epitopi antigenici esposti dalle proteine stesse.

Materiali e Metodi
52
Il trasferimento delle proteine dal gel alla membrana di nitrocellulosa è stato
effettuato mediante un sistema continuo assemblato in un apposito blottatore (YRDIMES,
wealtech corp, Lakeside, GA,USA).
E stato allestito un sandwich costituito, nell’ordine dall’anodo al catodo, da 3 fogli di
carta da filtro (Blotting Paper, Sigma-Aldrich), dalla membrana di nitrocellulosa (Trans-
Blot® Transfer Medium, BioRad), dal gel di poliacrilamide e da altri 3 fogli di carta da
filtro. I fogli di carta da filtro sono stati precedentemente immersi per qualche minuto nel
tampone di trasferimento (Tris 49.6 mM, glicina 384 mM, 20 % metanolo e 0,01 % SDS),
mentre la membrana di nitrocellulosa è stata pre-idratata in acqua bidistillata.
La procedura di blottaggio viene eseguita applicando una corrente di 300 mA per 1
ora e mezza. Il trasferimento delle proteine dal gel è stato a volte verificato colorando la
membrana di nitrocellulosa con Rosso Ponceau (0.1 % Ponceau S (peso/vol) in 5 % acido
acetico (vol/vol) (Sigma-Aldrich, Milano, Italy), preparato per una colorazione rapida che
si ottiene immergendo la membrana per pochi minuti nella soluzione e poi sciacquandola
più volte con acqua bidistillata. Per lo studio di P-Akt1 non è stato utilizzato il Rosso
Ponceau poiché l’acido acetico in cui è disciolto potrebbe defosforilare le proteine presenti
sulla membrana.
La membrana è stata quindi tagliata in base alla corsa delle proteine, identificabili
grazie ai marcatori di peso molecolare, e conservata nella soluzione di blocco (5% di latte
scremato in polvere (Euroclone, Pavia, Italia) e 0.05 % Tween-20 (poliossimetilene-
sorbitanmonolaurato, Sigma-Aldrich, Milano, Italy) in TBS (12.5 mM Tris HCl e 125 mM
NaCl a pH 7.4 in acqua bidistillata), a 4 °C overnight.
Il giorno successivo la membrana è stata incubata con gli anticorpi primari:
• Anticorpo monoclonale anti-Akt1 (Santa Cruz Tebu-Bio) (dil 1:1000)
• Anticorpo monoclonale anti- phospho-Akt1 (Santa Cruz Tebu-Bio) (dil 1:1000)
• Anticorpo anti-actina (Sigma) (1:30000)
L’incubazione degli anticorpi primari è condotta o a 37°C per un’ora o overnight a 4
°C quando il blocco viene effettuato a temperatura ambiente per 1 ora. Dopo ogni
incubazione, una serie di lavaggi permette di rimuovere l’anticorpo legatosi alla membrana
in modo aspecifico.
Le membrane sono incubate quindi con l’anticorpo secondario per 60 minuti sempre
a 37°C (IgG Anti-mouse coniugato con fosfatasi alcalina o perossidasi) diluito da 1:2000 a
1:50000.

Materiali e Metodi
53
I complessi antigene-anticorpo sono analizzati mediante una reazione di
chemioluminescenza. Questa reazione avviene in condizione di semibuio bagnando la
membrana con una soluzione di Chemioluminescent AP Substrate (ImmobilonTM Western,
Millipore).
I prodotti chemioluminescenti di questa reazione vengono visualizzati tramite
autoradiografia su lastre CL-X POSURETM FILM (Thermo Scientific).
3.11 Determinazione dei livelli di AMP ciclico
L’accumulo di AMP ciclico è stato valutato in seguito a stimolazione con diverse
concentrazioni di guanosina e a tempi diversi su cellule SH-SY5Y indifferenziate o
differenziate sia con guanosina che con acido retinoico. Le cellule SH-SY5Y vengono
seminate 48 ore prima dell’esperimento in piastre da 24 pozzetti (Falcon®) ad una densità
pari a 2.5x105 cellule in un millilitro di terreno completo per pozzetto.
In una serie di esperimenti le cellule sono state dunque differenziate per 48 ore con
guanosina o acido retinoico.
3.11.1 Stimolazione della sintesi di AMP ciclico
L’esperimento consiste nel sottoporre le cellule ad una pre-stimolazione e a una
stimolazione, entrambe della durata di 10 minuti, con soluzioni contenenti le sostanze che
si vogliono testare. Le soluzioni di prestimolazione sono costituite dal terreno completo ma
privo di siero (DME/HIGH 0 % FBS), le soluzioni per la stimolazione contengono le
sostanze in esame. Due dei 24 pozzetti di ogni piastra non sono stati trattati, per procedere
alla tripsinizzazione e conta delle cellule in camera di Burker.
Trascorso il tempo stabilito, si sostituisce la soluzione di prestimolazione con quella
di stimolazione e si lascia la piastra in incubatore a 37°C per 10 minuti. Nei pozzetti adibiti
alla conta cellulare si introducono 200 µl di tripsina e, dopo il distacco delle cellule, si
aggiungono 800 µl di terreno.
La stimolazione viene bloccata mediante l’aggiunta in ogni pozzetto di 250 µl di HCl
0.084 N a 4°C e si neutralizza la soluzione con aggiunta di 250 µl di TRIS 0,1 M a 4°C. Si
procede con la disgregazione delle cellule mediante sonicazione con ultrasuoni per circa 5
secondi per pozzetto.
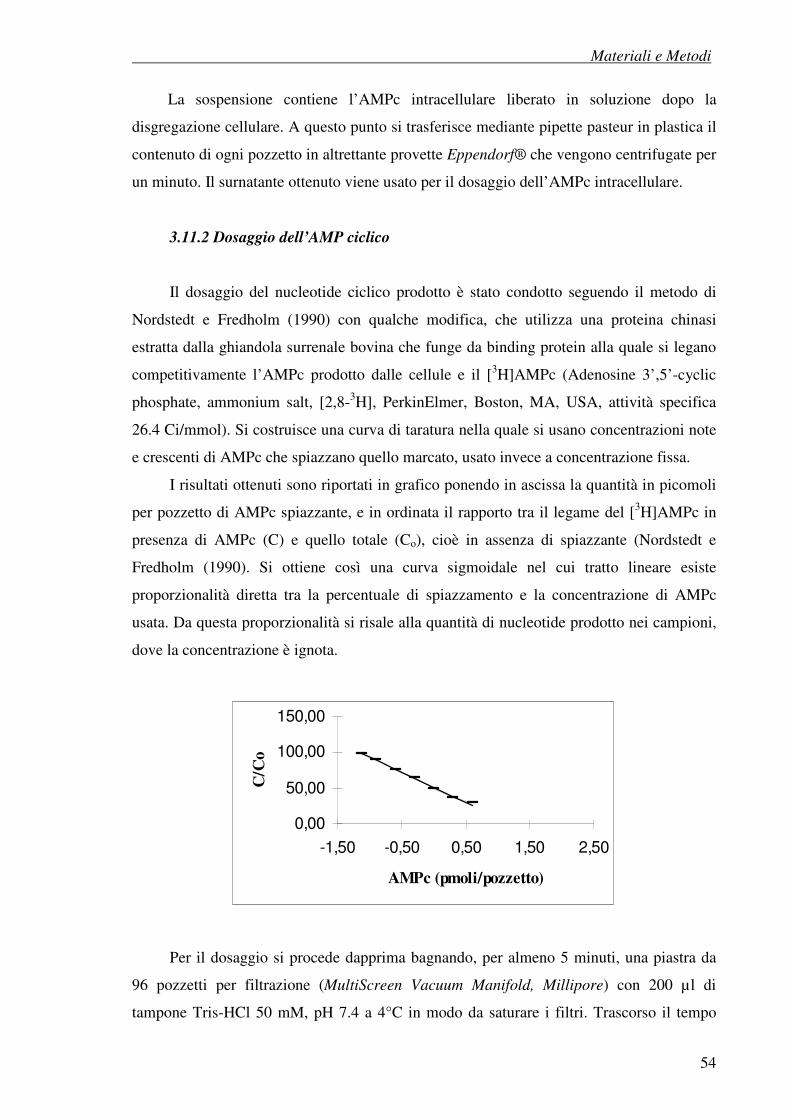
Materiali e Metodi
54
La sospensione contiene l’AMPc intracellulare liberato in soluzione dopo la
disgregazione cellulare. A questo punto si trasferisce mediante pipette pasteur in plastica il
contenuto di ogni pozzetto in altrettante provette Eppendorf® che vengono centrifugate per
un minuto. Il surnatante ottenuto viene usato per il dosaggio dell’AMPc intracellulare.
3.11.2 Dosaggio dell’AMP ciclico
Il dosaggio del nucleotide ciclico prodotto è stato condotto seguendo il metodo di
Nordstedt e Fredholm (1990) con qualche modifica, che utilizza una proteina chinasi
estratta dalla ghiandola surrenale bovina che funge da binding protein alla quale si legano
competitivamente l’AMPc prodotto dalle cellule e il [3H]AMPc (Adenosine 3’,5’-cyclic
phosphate, ammonium salt, [2,8-3H], PerkinElmer, Boston, MA, USA, attività specifica
26.4 Ci/mmol). Si costruisce una curva di taratura nella quale si usano concentrazioni note
e crescenti di AMPc che spiazzano quello marcato, usato invece a concentrazione fissa.
I risultati ottenuti sono riportati in grafico ponendo in ascissa la quantità in picomoli
per pozzetto di AMPc spiazzante, e in ordinata il rapporto tra il legame del [3H]AMPc in
presenza di AMPc (C) e quello totale (Co), cioè in assenza di spiazzante (Nordstedt e
Fredholm (1990). Si ottiene così una curva sigmoidale nel cui tratto lineare esiste
proporzionalità diretta tra la percentuale di spiazzamento e la concentrazione di AMPc
usata. Da questa proporzionalità si risale alla quantità di nucleotide prodotto nei campioni,
dove la concentrazione è ignota.
0,00
50,00
100,00
150,00
-1,50 -0,50 0,50 1,50 2,50
AMPc (pmoli/pozzetto)
C/C
o
Per il dosaggio si procede dapprima bagnando, per almeno 5 minuti, una piastra da
96 pozzetti per filtrazione (MultiScreen Vacuum Manifold, Millipore) con 200 µl di
tampone Tris-HCl 50 mM, pH 7.4 a 4°C in modo da saturare i filtri. Trascorso il tempo

Materiali e Metodi
55
necessario, si aspira il tampone in eccesso mediante pompa a vuoto e si asciuga la
superficie della piastra con carta assorbente.
A questo punto si procede caricando la piastra con la curva di taratura da 0.125 a 4
picomoli finali e con i campioni a concentrazione ignota. I campioni vengono diluiti nei
pozzetti con lo specifico buffer in modo tale che l’entità dello spiazzamento possa essere
rapportabile alla parte lineare della curva di taratura.
In ogni pozzetto si aggiungono 150 µl di una soluzione di proteina legante l’AMPc,
la binding protein, e 50 µl di [3H]AMPc 2nM in modo da avere un volume finale di 250 µl
in ogni pozzetto. Le piastre vengono lasciate ad incubare per due ore e mezza a 4 °C e
quindi filtrate con pompa a vuoto per eliminare il nucleotide ciclico non legato alla binding
protein, e lavata con 200 µl di tampone Tris-HCl mM freddo. A ciascun pozzetto vengono
aggiunti 25 µl di liquido di scintillazione Optiphase “Supermix”. La piastra viene lasciata
al buio overnight e letta tramite il contatore a scintillazione liquida (Microbeta Trilux,
Wallac).
Figura 14: Immagini delle piastre da 96 pozzetti per filtrazione (MultiScreen Vacuum Manifold, Millipore) e dell’apposito supporto per lo scintillatore.
I dati ottenuti dal contatore a scintillazione liquida, come conta per minuto, vengono
analizzati mediante un metodo di fitting o regressione non lineare e vengono interpolati
nella porzione lineare della curva di taratura permettendo così di risalire alla quantità di
nucleotide ciclico prodotto per milione di cellule. I dati vengono espressi come media ±
errore standard.

Materiali e Metodi
56
3.12 Esperimenti di Calcium Videoimaging
Gli esperimenti di videoimaging sono effettuati su cellule seminate su vetrini da 13
mm. Fura-2 pentacetoxymetilestere (fura-2 AM) è usato come indicatore di Ca2+. Le cellule
vengono caricate incubandole per 30 min a temperatura ambiente in soluzione salina
fisiologica (NES, in mM: 2.8 KCl, 140 NaCl, 2 CaCl2, 10 Hepes, 2 MgCl2, 10 glucosio, pH
7.3) contenente 10 mg / ml di BSA e fura-2 AM 5 µM. Il sistema di microscopia digitale
in fluorescenza è costruito attorno ad un microscopio invertito (Zeiss Axiovert 135,
Oberkochen, Germany) equipaggiato con una fotocamera CCD (Hamamatsu Photonics,
Hamamatsu, Japan). Le cellule caricate sono alternativamente eccitate a 340 e 380 nm
usando un microfluorimetro modificato a doppia lunghezza (Jasco CAM-230, Tokyo,
Japan). Le immagini fluorescenti sono state acquisite con la fotocamera CCD.
L’aquisizione dell’immagine viene effettuata ogni 4 secondi. Il calcolo del rapporto e
i grafici temporali del segnale di fluorescenza sono stati calcolati off line. La variazione
dell’intensità di fluorescenza è stata calcolata dal rapporto tra le immagini nelle aree di
interesse. Una camera microincubatore a temperatura controllata (Medical System
Corporation, Greenvale, NY, USA) mantiene la temperature a 37°C durante gli esperimenti
di videoimaging. I composti sono stati gentilmente applicati sulle cellule caricando un
appropriato volume (100x) di una soluzione concentrata in una siringa da 2 mL collegata
alla camera del microincubatore tramite un piccolo tubo.

Materiali e Metodi
57
3.13 Fosforilazione di ERK1/2
La fosforilazione di ERK1/2 è stata valutata mediante l’utilizzo di un kit ELISA
(CASE™ Kit-Cellular Activation of Signaling ELISA). Questo kit è in grado di
quantificare la quantità di proteine ERK1/2 attivate (fosforilate) rispetto alle proteine totali.
La fosforilazione di ERK avviene sia a livello della serina 202 che della tirosina 204. I dati
sono espressi come rapporto tra proteine fosforilate e non fosforilate e normalizzate per il
numero di cellule.
Sono seminate in piastre da 96 pozzetti in terreno completo, 5-15x104 cellule per
pozzetto, e per gli esperimenti in starvation, dopo 24 h dalla semina il terreno completo è
stato sostituito con terreno allo 0 % di siero.
Dopo 48 h totali di coltura le cellule sono state stimolate a tempi crescenti (0-10
minuti) con concentrazioni diverse di Guanosina (30, 100 e 300 µM) o solo con guanosina
100 µM per le cellule mantenute al 10 % FBS. Successivamente si esegue
pedissequamente il procedimento previsto dal kit.
Le cellule sono fissate con 200 µl di Cell Fixing Buffer per 20 minuti, lavate 2 volte
con 200 µl di Washing Buffer per 5 minuti sotto agitazione e incubate con 100 µl di
Quencing Buffer per 20 minuti a temperatura ambiente. Dopo un ulteriore lavaggio da 5
minuti, vengono aggiunti 100 µl di Antigen Retrieval Buffer e la piastra viene scaldata in
microonde a 440 Watts per 3 minuti. Una volta raffreddata la piastra e dopo aver lavato
con 200 µl di Washing Buffer, si lascia in incubazione per 1 ora a temperatura ambiente
con 100 µl di Blocking Buffer. Dopo aver lavato nuovamente la piastra con 200 µl di
Washing Buffer, si aggiungono 50 µl di anticorpo primario diluito 1:100 (Anti-phospho-
ERK1/2 o Anti-Pan-ERK1/2) e si lascia incubare per 1 ora a temperatura ambiente. In una
fila di pozzetti vengono aggiunti solo 50 µl di Buffer. Dopo aver lavato 2 volte per
eliminare l’eccesso di anticorpo primario, si aggiungono 100 µl di anticorpo secondario
diluito 1:16 in tutti i pozzetti e si lascia incubare sempre per 1 ora a temperatura ambiente.
Finita l’incubazione, i pozzetti vengono lavati 2 volte con 200 µl di Washing Buffer, e 1
volta con 200 µl di PBS sempre sotto agitazione.
Lo sviluppo procede con l’aggiunta di 100 µl di Developing Solution e dopo circa 8
minuti con l’aggiunta di altrettanti µl di Stop Solution che fa virare il colore da blu a giallo.
Dopo 5 minuti si effettua la lettura della piastra a 450 nm.
Si finisce il saggio con la quantificazione del numero di cellule: si svuota la piastra,
si lava prima con 200 µl di Washing Buffer e poi con 200 µl di acqua bidistillata e si
Materiali e Metodi
58
colorano le cellule con 100 µl di Cell Staining Buffer per 30 minuti a temperatura
ambiente. Alla fine dell’incubazione i pozzetti vengono lavati più volte con acqua
bidistillata per eliminare l’eccesso di colorante. Infine la piastra viene messa ad incubare
sotto agitazione per 1 ora a temperatura ambiente con 100 µl di SDS all’1 %. La lettura
viene effettuata a 595 nm.
I valori di assorbanza ottenuti con gli anticorpi specifici vengono normalizzati con i
valori di assorbanza relativi al numero di cellule e infine i valori di assorbanza relativa di
ERK1/2 fosforilato vengono normalizzati rispetto ai valori ottenuti per ERK1/2 totali.
Figura 15: Esempio schematico della preparazione di una piastra per l’esecuzione del CASE kit ELISA (Cell Signaling).

Materiali e Metodi
59
3.14 Tecniche di manipolazione dell’RNA e del cDNA
Questi esperimenti sono stati effettuati per valutare l’espressione genica della
proteina Purine Nucleoside Phosphorylase (PNP) nelle cellule SH-SY5Y.
3.14.1 Estrazione di RNA totale da cellule
Per estrarre l’RNA da circa 2.5x106 cellule SH-SY5Y è stato utilizzato il kit SV
Total RNA Isolation System (Promega, U.S.A.). Al pellet cellulare sono aggiunti 175 µl di
SV RNA Lysis Buffer, successivamente, 350 µl di SV Dilution Buffer. Il preparato viene
prima incubato in un bagno termostatato a 70 °C per 3 minuti, e poi centrifugato a 13000
xg per 10 minuti. Il surnatante è stato recuperato, addizionato di 200 µl di etanolo al 95 %
(Fluka) e trasferito in una colonnina provvista di una membrana di silicio (Spin Basket
Assembly, Promega, U.S.A.). La colonnina viene centrifugata a 14.000 xg per 1 minuto,
lavata con 600 µl di SV RNA Wash Solution e bloccata con l’aggiunta di 200 µl di SV
DNAse Stop Solution. La colonnina viene quindi lavata per 2 volte con SV RNA Wash
Solution e l’RNA legato alla membrana viene eluito per centrifugazione a 14000 xg per 1
minuto con 100 µl di Nuclease-Free Water, e conservato a -20 °C fino all’utilizzo.
3.14.2 Reazione di trascrizione inversa
L’RNA può essere convertito in una copia di cDNA a doppio filamento mediante
l’utilizzo di un enzima chiamato “trascrittasi inversa”.
Il cDNA sintetizzato può quindi essere utilizzato come stampo per l’amplificazione della
sequenza di interesse mediante una classica reazione di polimerizzazione a catena (PCR),
che viene di conseguenza chiamata RT-PCR (Reverse Trascriptase-PCR o PCR dopo
trascrizione inversa).
La procedura avviene in questo modo: a 10 µl di RNA vengono aggiunti 1 µl di
Rnadom Primers (Promega) e 1 µl di dNTPs 10 mM (deoxy-Nucleotide Tri-Phosphate)
(Promega). La soluzione viene incubata a 65°C per 5 minuti e poi posta a raffreddare in
ghiaccio. Ad essa vengono aggiunti 4 µl di First-Strand Buffer (Invitrogen), 2 µl di DTT
(Dithiothreitol) (Invitrogen) 0.1 M e 1 µl di acqua bi distillata filtrata. L’incubazione viene
eseguita a 42°C per 2 minuti. Infine, viene aggiunto 1 µl di SuperScriptII (200 U)
(Invitrogen), la polimerasi che catalizza la formazione dei filamenti complementari. La

Materiali e Metodi
60
retro trascrizione avviene mediante un’incubazione per 50 minuti a 42°C e poi per 15
minuti a 70°C. I campioni vengono poi conservati a 4°C.
3.14.3 Reazione di polimerizzazione a catena (PCR)
La metodica consiste nella selettiva amplificazione di una specifica sequenza di
DNA bersaglio utilizzando una coppia di primers complementari alle regioni che
fiancheggiano la sequenza di interesse. Si tratta di una reazione a catena in più cicli, in
ciascuno dei quali fungono da stampo per la sintesi i filamenti di DNA sintetizzati nel ciclo
precedente. Il programma di PCR prevede una prima denaturazione a 94°C per 10 minuti e
43 cicli caratterizzati ciascuno da tre passaggi: una denaturazione a 94 °C per 15 secondi,
una fase di annealing ovvero di appaiamento dei primers alle regioni complementari a 61
°C per 1 minuto e una fase di elongation ovvero di sintesi del DNA a 72 °C per 30 secondi.
Terminati i cicli previsti si procede all’elongation finale a 72°C per 15 minuti.
Nelle reazioni svolte per la valutazione dell’espressione genica dell’enzima Purine
Nucleoside Phosphorylase (PNP) sono stati preparati 10 µl totali di mix di reazione
contenenti 1 µl di cDNA, 1 µl di Primer Reverse, 1 µl di Primer Forward, 1 µl di buffer
Red Taq polimerasi, 1 µl di dNTPs 2.5 mM, 0.5 µl di Red Taq polimerasi (contenente già
MgCl2) (Sigma-Aldrich, Milano, Italy) e 4.5 µl di acqua bidistillata filtrata, ottenendo un
volume finale di 10 µl totali.
Entrambi i primer sono stati diluiti subito prima dell’uso e hanno la seguente sequenza:
FORWARD PRIMER: 5’- AAT GGA GAA CGG ATA CAC CTA TG -3’
REVERSE PRIMER: 5’- ATC AAC TGG CTT TGT CAG GGA G -3’
Schema 1: ciclo di PCR.
94°C 94°C 72°C 72°C
61°C 4°C 10’ 15’’
1’
30’’ 15’
∞ _
43 cicli

Materiali e Metodi
61
3.14.4 Elettroforesi in gel di agarosio
La metodica consiste nell’indurre la migrazione di molecole di acido nucleico
attraverso le maglie di un gel di agarosio in presenza di un campo elettrico. Poiché tutti gli
acidi nucleici hanno composizione chimica molto simile e un elevato rapporto
carica/massa a pH fisiologico, la loro mobilità in agarosio dipende soprattutto dalla
lunghezza e dalla conformazione.
Si sciolgono 3 g di agarosio (Sigma-Aldrich, Milano, Italy) ad una temperatura
superiore a 90 °C in tampone TBE 1X (0,089M Acido Borico, 0.0025 M EDTA) e si
aggiunge il colorante Gel RedTM 10000x (Botium, Inc.). La soluzione di agarosio viene
versata in una celletta elettroforetica orizzontale, munita di spaziatori e pettine, e lasciata
gelificare. Versato il TBE 1X e caricati nei pozzetti i campioni di DNA, si applica un
campo elettrico di 170 V. Il DNA, carico negaticamente, migra in direzione del polo
positivo.
Il colorante Gel RedTM emette fluorescenza quando esposto a luce ultravioletta solo
se è intercalato tra le basi di acidi nucleici, pertanto i frammenti di DNA separati tramite
elettroforesi vengono visualizzati grazie a un transilluminatore a luce UV (UV corti, circa
300 nm). Per verificare la lunghezza dei frammenti amplificati i campioni sono stati
caricati assieme ad un marcatore di peso molecolare (100 bp Low Ladder, Sigma-Aldrich).

4. Risultati

Risultati
62
4.1 Effetto della guanosina su cellule di neuroblastoma SH-SY5Y poste
in condizioni di deprivazione da siero
E’ stato dimostrato che la guanosina è in grado di aumentare la sopravvivenza di
linee cellulari non neuronali come gli astrociti o la glia, poste in condizioni di stress come
l’ipossia o sottoposte a tossicità da MPP+. Questo studio è volto a valutare l’effetto
citoprotettivo della guanosina sulla linea cellulare neuronale SH-SY5Y, posta in una
condizione di stress come la deprivazione da siero (starvation).
4.1.1 Valutazione visiva dell’effetto protettivo della guanosina sulle cellule SH-
SY5Y
Le cellule sono state seminate in terreno completo al 10 % di siero e lasciate aderire
per 24h. Successivamente sono state poste in condizioni di deprivazione da siero (0% siero
FBS) per altre 24 ora in presenza o in assenza di guanosina 75 µM. Le cellule poste in
condizioni di stravation tendono a cambiare morfologia rispetto ai controlli al 10 % FBS.
Il trattamento con guanosina 75 µM è in grado di riportare le cellule ad una morfologia più
simile a quella dei controlli rispetto a quella delle cellule in starvation (Figura 16).
starvation 24 h (0 % FBS) controlli (10 % FBS) starvation + GUO 75 µM
Figura 16: Effetto della guanosina sulle cellule SH-SY5Y poste in starvation per 24 ore. Le immagini sono state acquisite utilizzando il microscopio Leica Leitz Wetzlar obiettivo 40x collegato ad un computer dotato del programma Leica IM (W.4, Leica Microsystem Digital Imaging, Cambrige UK).
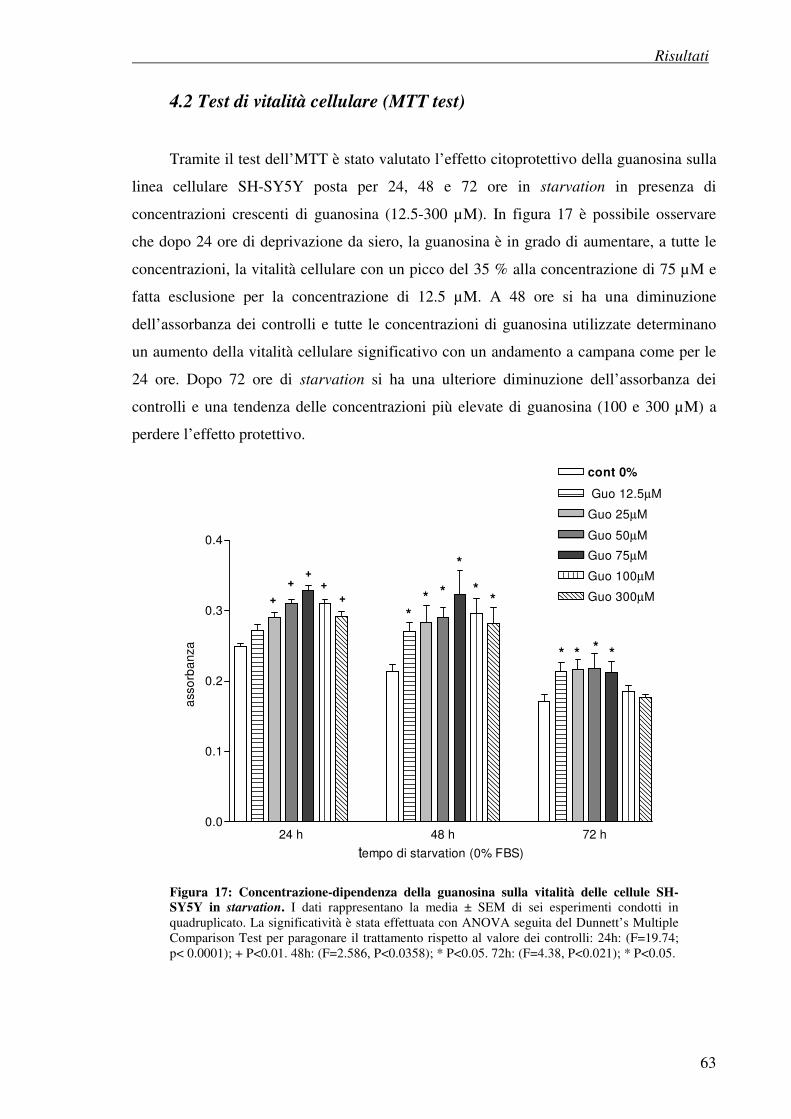
Risultati
63
4.2 Test di vitalità cellulare (MTT test)
Tramite il test dell’MTT è stato valutato l’effetto citoprotettivo della guanosina sulla
linea cellulare SH-SY5Y posta per 24, 48 e 72 ore in starvation in presenza di
concentrazioni crescenti di guanosina (12.5-300 µM). In figura 17 è possibile osservare
che dopo 24 ore di deprivazione da siero, la guanosina è in grado di aumentare, a tutte le
concentrazioni, la vitalità cellulare con un picco del 35 % alla concentrazione di 75 µM e
fatta esclusione per la concentrazione di 12.5 µM. A 48 ore si ha una diminuzione
dell’assorbanza dei controlli e tutte le concentrazioni di guanosina utilizzate determinano
un aumento della vitalità cellulare significativo con un andamento a campana come per le
24 ore. Dopo 72 ore di starvation si ha una ulteriore diminuzione dell’assorbanza dei
controlli e una tendenza delle concentrazioni più elevate di guanosina (100 e 300 µM) a
perdere l’effetto protettivo.
24 h 48 h 72 h0.0
0.1
0.2
0.3
0.4
cont 0%
Guo 12.5µM
Guo 25µM
Guo 50µM
Guo 75µM
Guo 100µM
Guo 300µM+
** *
*
**
* * * *
+
++
+
tempo di starvation (0% FBS)
asso
rba
nza
Figura 17: Concentrazione-dipendenza della guanosina sulla vitalità delle cellule SH-SY5Y in starvation. I dati rappresentano la media ± SEM di sei esperimenti condotti in quadruplicato. La significatività è stata effettuata con ANOVA seguita del Dunnett’s Multiple Comparison Test per paragonare il trattamento rispetto al valore dei controlli: 24h: (F=19.74; p< 0.0001); + P<0.01. 48h: (F=2.586, P<0.0358); * P<0.05. 72h: (F=4.38, P<0.021); * P<0.05.
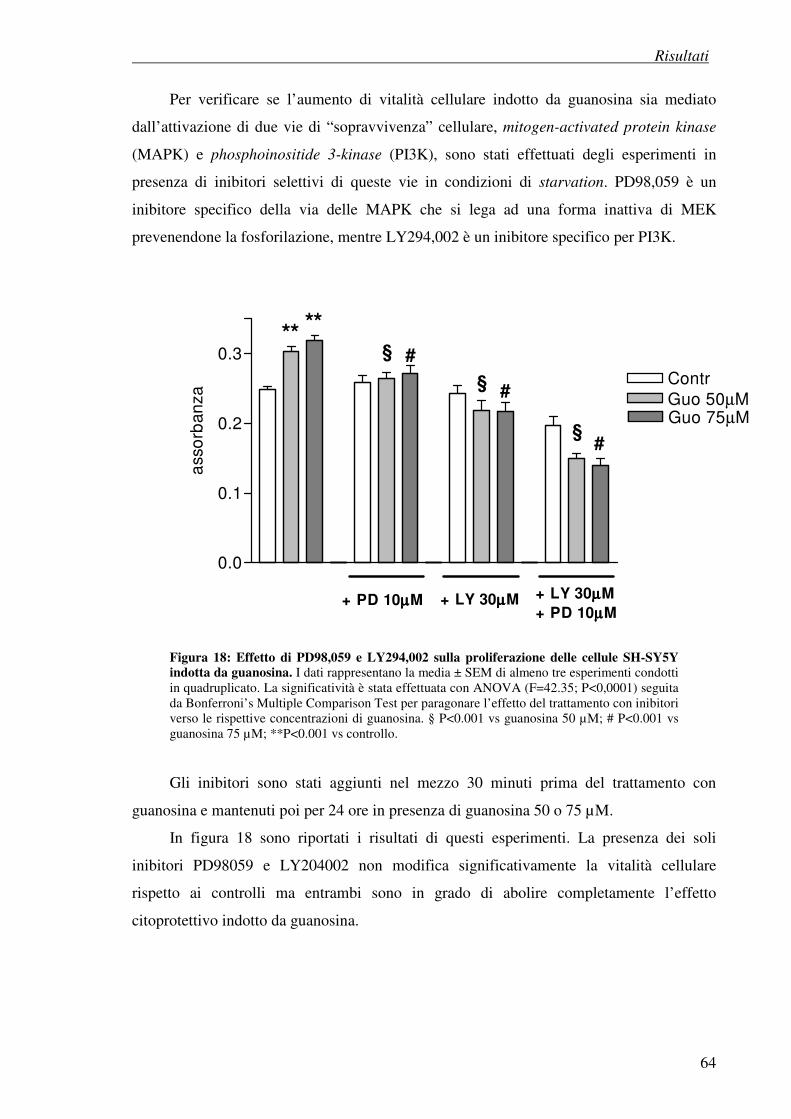
Risultati
64
Per verificare se l’aumento di vitalità cellulare indotto da guanosina sia mediato
dall’attivazione di due vie di “sopravvivenza” cellulare, mitogen-activated protein kinase
(MAPK) e phosphoinositide 3-kinase (PI3K), sono stati effettuati degli esperimenti in
presenza di inibitori selettivi di queste vie in condizioni di starvation. PD98,059 è un
inibitore specifico della via delle MAPK che si lega ad una forma inattiva di MEK
prevenendone la fosforilazione, mentre LY294,002 è un inibitore specifico per PI3K.
0.0
0.1
0.2
0.3
+ LY 30µµµµM
Contr
Guo 50µMGuo 75µM
+ LY 30µµµµM
+ PD 10µµµµM+ PD 10µµµµM
#§** **
#
#
§
§
asso
rba
nza
Figura 18: Effetto di PD98,059 e LY294,002 sulla proliferazione delle cellule SH-SY5Y indotta da guanosina. I dati rappresentano la media ± SEM di almeno tre esperimenti condotti in quadruplicato. La significatività è stata effettuata con ANOVA (F=42.35; P<0,0001) seguita da Bonferroni’s Multiple Comparison Test per paragonare l’effetto del trattamento con inibitori verso le rispettive concentrazioni di guanosina. § P<0.001 vs guanosina 50 µM; # P<0.001 vs guanosina 75 µM; **P<0.001 vs controllo.
Gli inibitori sono stati aggiunti nel mezzo 30 minuti prima del trattamento con
guanosina e mantenuti poi per 24 ore in presenza di guanosina 50 o 75 µM.
In figura 18 sono riportati i risultati di questi esperimenti. La presenza dei soli
inibitori PD98059 e LY204002 non modifica significativamente la vitalità cellulare
rispetto ai controlli ma entrambi sono in grado di abolire completamente l’effetto
citoprotettivo indotto da guanosina.
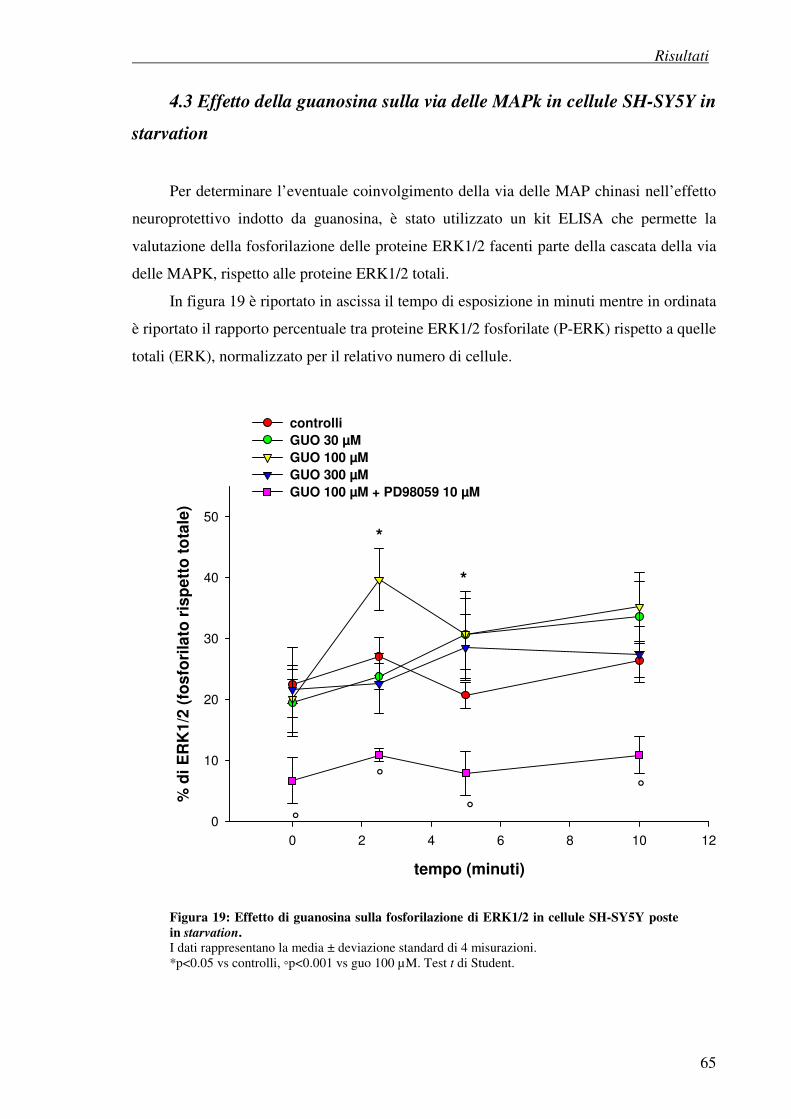
Risultati
65
4.3 Effetto della guanosina sulla via delle MAPk in cellule SH-SY5Y in
starvation
Per determinare l’eventuale coinvolgimento della via delle MAP chinasi nell’effetto
neuroprotettivo indotto da guanosina, è stato utilizzato un kit ELISA che permette la
valutazione della fosforilazione delle proteine ERK1/2 facenti parte della cascata della via
delle MAPK, rispetto alle proteine ERK1/2 totali.
In figura 19 è riportato in ascissa il tempo di esposizione in minuti mentre in ordinata
è riportato il rapporto percentuale tra proteine ERK1/2 fosforilate (P-ERK) rispetto a quelle
totali (ERK), normalizzato per il relativo numero di cellule.
tempo (minuti)
0 2 4 6 8 10 12
% d
i E
RK
1/2
(fo
sfo
rila
to r
isp
ett
o t
ota
le)
0
10
20
30
40
50
controlli
GUO 30 µM
GUO 100 µM
GUO 300 µM
GUO 100 µM + PD98059 10 µM
*
*
°
°°
°
Figura 19: Effetto di guanosina sulla fosforilazione di ERK1/2 in cellule SH-SY5Y poste in starvation. I dati rappresentano la media ± deviazione standard di 4 misurazioni. *p<0.05 vs controlli, ◦p<0.001 vs guo 100 µM. Test t di Student.
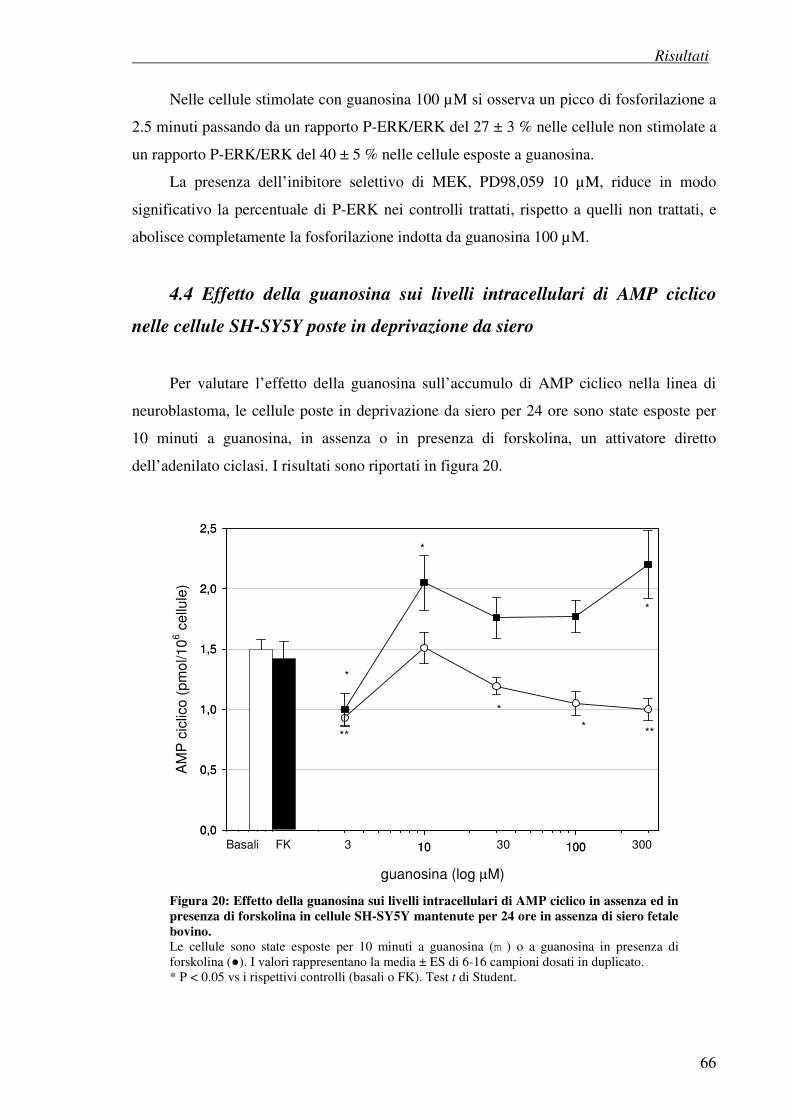
Risultati
66
Nelle cellule stimolate con guanosina 100 µM si osserva un picco di fosforilazione a
2.5 minuti passando da un rapporto P-ERK/ERK del 27 ± 3 % nelle cellule non stimolate a
un rapporto P-ERK/ERK del 40 ± 5 % nelle cellule esposte a guanosina.
La presenza dell’inibitore selettivo di MEK, PD98,059 10 µM, riduce in modo
significativo la percentuale di P-ERK nei controlli trattati, rispetto a quelli non trattati, e
abolisce completamente la fosforilazione indotta da guanosina 100 µM.
4.4 Effetto della guanosina sui livelli intracellulari di AMP ciclico
nelle cellule SH-SY5Y poste in deprivazione da siero
Per valutare l’effetto della guanosina sull’accumulo di AMP ciclico nella linea di
neuroblastoma, le cellule poste in deprivazione da siero per 24 ore sono state esposte per
10 minuti a guanosina, in assenza o in presenza di forskolina, un attivatore diretto
dell’adenilato ciclasi. I risultati sono riportati in figura 20.
guanosina (log µM)
1 10 100
0,0
0,5
1,0
1,5
2,0
2,5
1 10 100
AM
P c
iclic
o (
pm
ol/10
6 c
ellu
le)
0,0
0,5
1,0
1,5
2,0
2,5
3 30Basali FK
**
*
***
*
*
*
300
Figura 20: Effetto della guanosina sui livelli intracellulari di AMP ciclico in assenza ed in presenza di forskolina in cellule SH-SY5Y mantenute per 24 ore in assenza di siero fetale bovino. Le cellule sono state esposte per 10 minuti a guanosina (m ) o a guanosina in presenza di forskolina (●). I valori rappresentano la media ± ES di 6-16 campioni dosati in duplicato. * P < 0.05 vs i rispettivi controlli (basali o FK). Test t di Student.

Risultati
67
In condizioni di starvation, l’esposizione delle cellule a concentrazioni crescenti di
guanosina determina una variazione della produzione di AMP ciclico che presenta un
andamento non lineare, con una riduzione statisticamente significativa (p<0.001) della
produzione del nucleotide ciclico, rispetto al valore basale, del 38 ± 5 % alla
concentrazione di 3 µM. Alla concentrazione di 10 µM si osserva un ritorno ai livelli basali
seguito da una progressiva riduzione, fino a -33 ± 4 %, alla concentrazione di 300 µM
(p<0.001).
L’esposizione delle cellule a sola forskolina (3 µM) non determina un aumento
significativo dei livelli di AMP ciclico intracellulare rispetto ai valori basali (1.42 ± 0.14
pmol/106 cellule).
L’aggiunta di guanosina causa nuovamente una riduzione dei livelli di AMPc,
rispetto alla sola forskolina, alla concentrazione 3 µM (1.00 ± 0.13 pmol/106 cellule,
p<0.001), seguita da un aumento a partire dalla concentrazione di 10 µM, con un valore
pari a 2.05 ± 0.23 pmol/106 cellule, e dal mantenimento a plateau fino alla concentrazione
di 300 µM (2.20 ± 0.28 pmol/106 cellule, p<0.01).
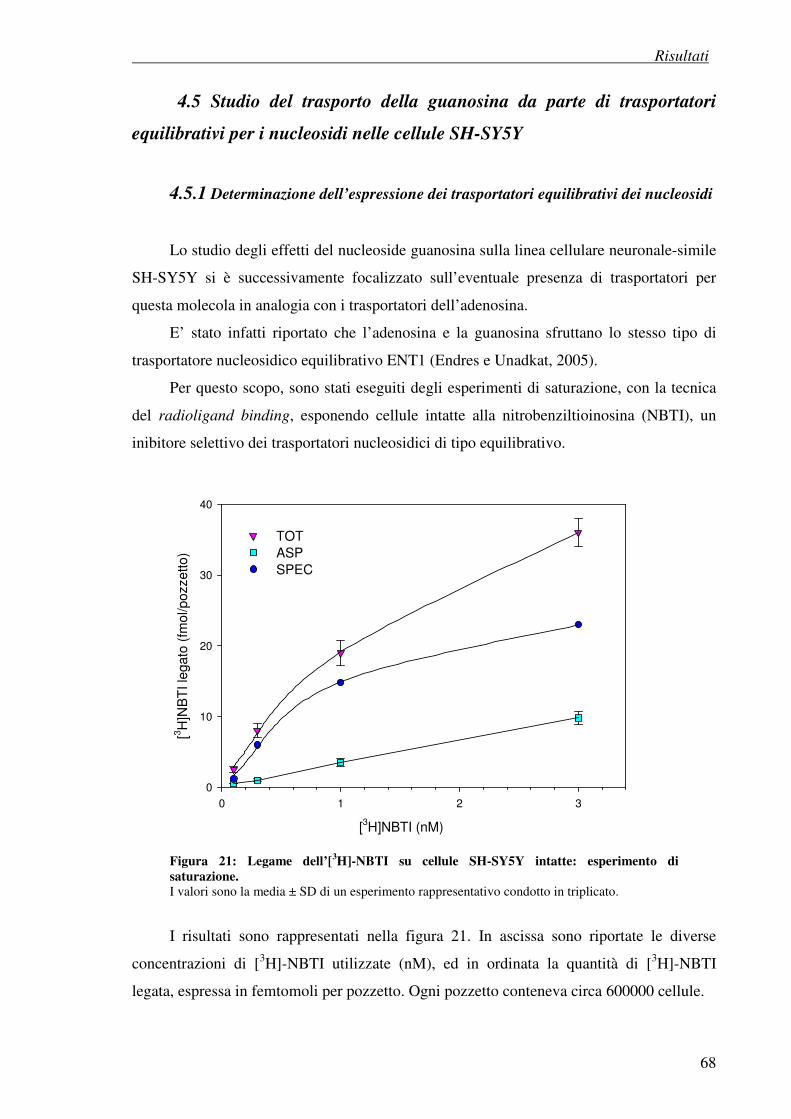
Risultati
68
4.5 Studio del trasporto della guanosina da parte di trasportatori
equilibrativi per i nucleosidi nelle cellule SH-SY5Y
4.5.1 Determinazione dell’espressione dei trasportatori equilibrativi dei nucleosidi
Lo studio degli effetti del nucleoside guanosina sulla linea cellulare neuronale-simile
SH-SY5Y si è successivamente focalizzato sull’eventuale presenza di trasportatori per
questa molecola in analogia con i trasportatori dell’adenosina.
E’ stato infatti riportato che l’adenosina e la guanosina sfruttano lo stesso tipo di
trasportatore nucleosidico equilibrativo ENT1 (Endres e Unadkat, 2005).
Per questo scopo, sono stati eseguiti degli esperimenti di saturazione, con la tecnica
del radioligand binding, esponendo cellule intatte alla nitrobenziltioinosina (NBTI), un
inibitore selettivo dei trasportatori nucleosidici di tipo equilibrativo.
[3H]NBTI (nM)
0 1 2 3
[3H
]NB
TI le
ga
to (
fmo
l/p
ozze
tto
)
0
10
20
30
40
TOT
ASP
SPEC
Figura 21: Legame dell’[3H]-NBTI su cellule SH-SY5Y intatte: esperimento di saturazione. I valori sono la media ± SD di un esperimento rappresentativo condotto in triplicato.
I risultati sono rappresentati nella figura 21. In ascissa sono riportate le diverse
concentrazioni di [3H]-NBTI utilizzate (nM), ed in ordinata la quantità di [3H]-NBTI
legata, espressa in femtomoli per pozzetto. Ogni pozzetto conteneva circa 600000 cellule.
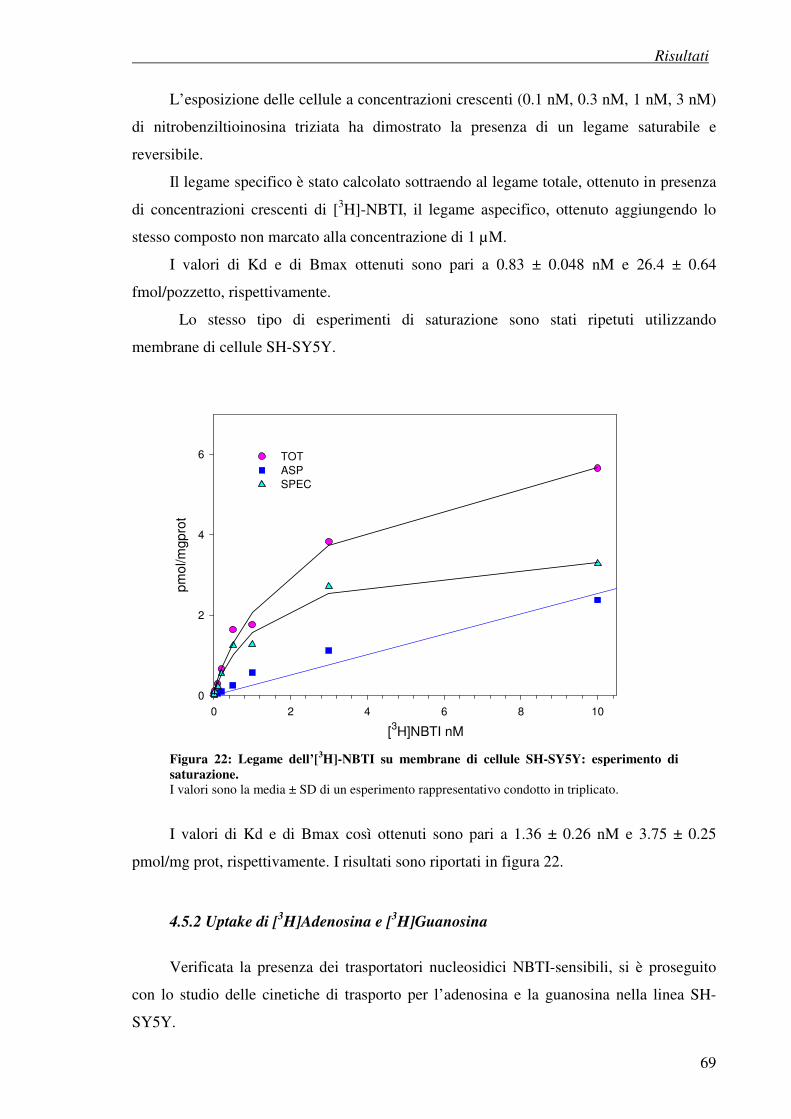
Risultati
69
L’esposizione delle cellule a concentrazioni crescenti (0.1 nM, 0.3 nM, 1 nM, 3 nM)
di nitrobenziltioinosina triziata ha dimostrato la presenza di un legame saturabile e
reversibile.
Il legame specifico è stato calcolato sottraendo al legame totale, ottenuto in presenza
di concentrazioni crescenti di [3H]-NBTI, il legame aspecifico, ottenuto aggiungendo lo
stesso composto non marcato alla concentrazione di 1 µM.
I valori di Kd e di Bmax ottenuti sono pari a 0.83 ± 0.048 nM e 26.4 ± 0.64
fmol/pozzetto, rispettivamente.
Lo stesso tipo di esperimenti di saturazione sono stati ripetuti utilizzando
membrane di cellule SH-SY5Y.
[3H]NBTI nM
0 2 4 6 8 10
pm
ol/m
gp
rot
0
2
4
6 TOT
ASP
SPEC
Figura 22: Legame dell’[3H]-NBTI su membrane di cellule SH-SY5Y: esperimento di saturazione. I valori sono la media ± SD di un esperimento rappresentativo condotto in triplicato.
I valori di Kd e di Bmax così ottenuti sono pari a 1.36 ± 0.26 nM e 3.75 ± 0.25
pmol/mg prot, rispettivamente. I risultati sono riportati in figura 22.
4.5.2 Uptake di [3H]Adenosina e [
3H]Guanosina
Verificata la presenza dei trasportatori nucleosidici NBTI-sensibili, si è proseguito
con lo studio delle cinetiche di trasporto per l’adenosina e la guanosina nella linea SH-
SY5Y.
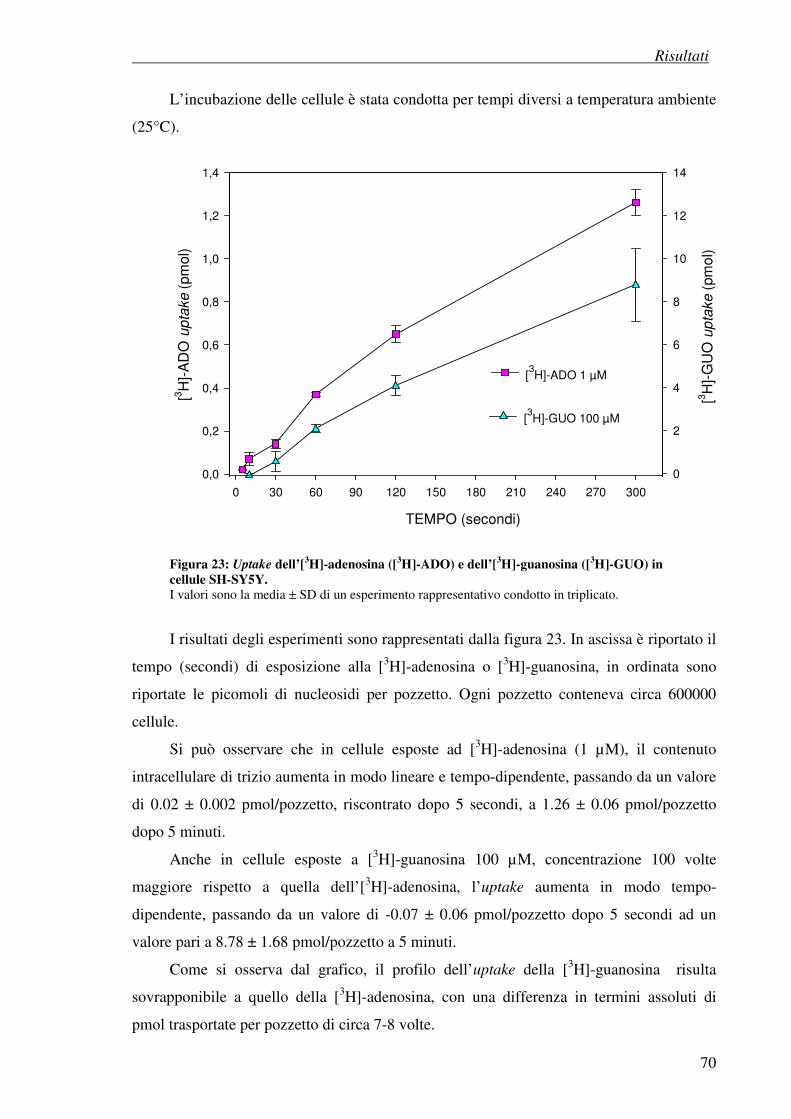
Risultati
70
L’incubazione delle cellule è stata condotta per tempi diversi a temperatura ambiente
(25°C).
[3H
]-G
UO
up
take (
pm
ol)
0
2
4
6
8
10
12
14
[3H]-GUO 100 µM
0 30 60 90 120 150 180 210 240 270 300
[3H
]-A
DO
up
take (
pm
ol)
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
[3H]-ADO 1 µM
TEMPO (secondi)
Figura 23: Uptake dell’[3H]-adenosina ([3H]-ADO) e dell’[3H]-guanosina ([3H]-GUO) in cellule SH-SY5Y. I valori sono la media ± SD di un esperimento rappresentativo condotto in triplicato.
I risultati degli esperimenti sono rappresentati dalla figura 23. In ascissa è riportato il
tempo (secondi) di esposizione alla [3H]-adenosina o [3H]-guanosina, in ordinata sono
riportate le picomoli di nucleosidi per pozzetto. Ogni pozzetto conteneva circa 600000
cellule.
Si può osservare che in cellule esposte ad [3H]-adenosina (1 µM), il contenuto
intracellulare di trizio aumenta in modo lineare e tempo-dipendente, passando da un valore
di 0.02 ± 0.002 pmol/pozzetto, riscontrato dopo 5 secondi, a 1.26 ± 0.06 pmol/pozzetto
dopo 5 minuti.
Anche in cellule esposte a [3H]-guanosina 100 µM, concentrazione 100 volte
maggiore rispetto a quella dell’[3H]-adenosina, l’uptake aumenta in modo tempo-
dipendente, passando da un valore di -0.07 ± 0.06 pmol/pozzetto dopo 5 secondi ad un
valore pari a 8.78 ± 1.68 pmol/pozzetto a 5 minuti.
Come si osserva dal grafico, il profilo dell’uptake della [3H]-guanosina risulta
sovrapponibile a quello della [3H]-adenosina, con una differenza in termini assoluti di
pmol trasportate per pozzetto di circa 7-8 volte.
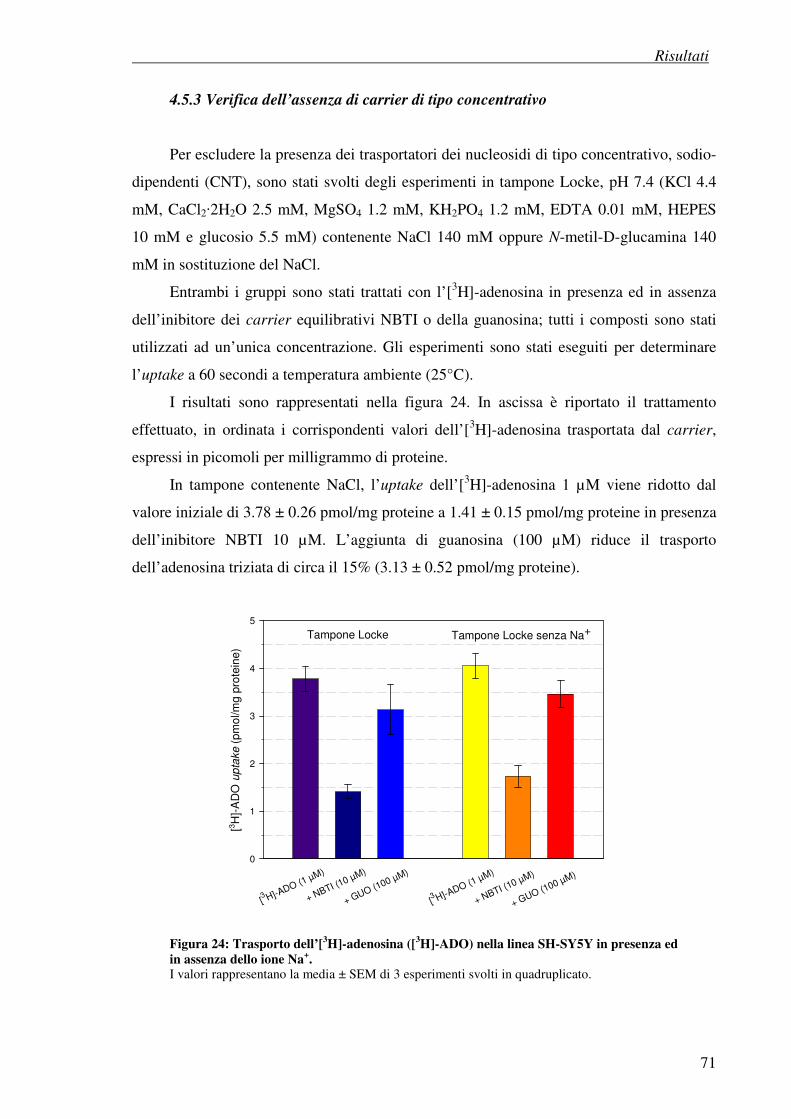
Risultati
71
4.5.3 Verifica dell’assenza di carrier di tipo concentrativo
Per escludere la presenza dei trasportatori dei nucleosidi di tipo concentrativo, sodio-
dipendenti (CNT), sono stati svolti degli esperimenti in tampone Locke, pH 7.4 (KCl 4.4
mM, CaCl2·2H2O 2.5 mM, MgSO4 1.2 mM, KH2PO4 1.2 mM, EDTA 0.01 mM, HEPES
10 mM e glucosio 5.5 mM) contenente NaCl 140 mM oppure N-metil-D-glucamina 140
mM in sostituzione del NaCl.
Entrambi i gruppi sono stati trattati con l’[3H]-adenosina in presenza ed in assenza
dell’inibitore dei carrier equilibrativi NBTI o della guanosina; tutti i composti sono stati
utilizzati ad un’unica concentrazione. Gli esperimenti sono stati eseguiti per determinare
l’uptake a 60 secondi a temperatura ambiente (25°C).
I risultati sono rappresentati nella figura 24. In ascissa è riportato il trattamento
effettuato, in ordinata i corrispondenti valori dell’[3H]-adenosina trasportata dal carrier,
espressi in picomoli per milligrammo di proteine.
In tampone contenente NaCl, l’uptake dell’[3H]-adenosina 1 µM viene ridotto dal
valore iniziale di 3.78 ± 0.26 pmol/mg proteine a 1.41 ± 0.15 pmol/mg proteine in presenza
dell’inibitore NBTI 10 µM. L’aggiunta di guanosina (100 µM) riduce il trasporto
dell’adenosina triziata di circa il 15% (3.13 ± 0.52 pmol/mg proteine).
[3H
]-A
DO
up
take
(p
mo
l/m
g p
rote
ine)
0
1
2
3
4
5
[3 H]-A
DO (1 µM)
+ NBTI (10 µM)
+ GUO (100 µM)
Tampone Locke Tampone Locke senza Na+
[3 H]-A
DO (1 µM)
+ NBTI (10 µM)
+ GUO (100 µM)
Figura 24: Trasporto dell’[3H]-adenosina ([3H]-ADO) nella linea SH-SY5Y in presenza ed in assenza dello ione Na+. I valori rappresentano la media ± SEM di 3 esperimenti svolti in quadruplicato.
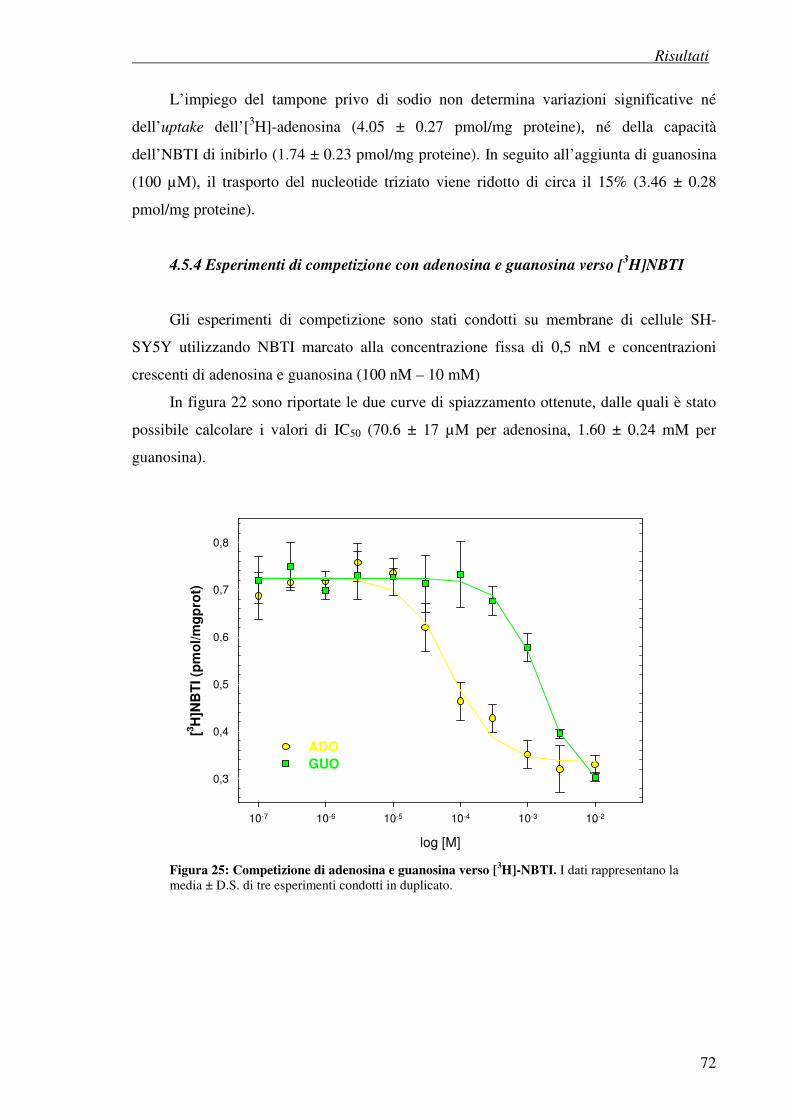
Risultati
72
L’impiego del tampone privo di sodio non determina variazioni significative né
dell’uptake dell’[3H]-adenosina (4.05 ± 0.27 pmol/mg proteine), né della capacità
dell’NBTI di inibirlo (1.74 ± 0.23 pmol/mg proteine). In seguito all’aggiunta di guanosina
(100 µM), il trasporto del nucleotide triziato viene ridotto di circa il 15% (3.46 ± 0.28
pmol/mg proteine).
4.5.4 Esperimenti di competizione con adenosina e guanosina verso [3H]NBTI
Gli esperimenti di competizione sono stati condotti su membrane di cellule SH-
SY5Y utilizzando NBTI marcato alla concentrazione fissa di 0,5 nM e concentrazioni
crescenti di adenosina e guanosina (100 nM – 10 mM)
In figura 22 sono riportate le due curve di spiazzamento ottenute, dalle quali è stato
possibile calcolare i valori di IC50 (70.6 ± 17 µM per adenosina, 1.60 ± 0.24 mM per
guanosina).
log [M]
10-7 10-6 10-5 10-4 10-3 10-2
[3H
]NB
TI (p
mo
l/m
gp
rot)
0,3
0,4
0,5
0,6
0,7
0,8
ADO
GUO
Figura 25: Competizione di adenosina e guanosina verso [3H]-NBTI. I dati rappresentano la media ± D.S. di tre esperimenti condotti in duplicato.
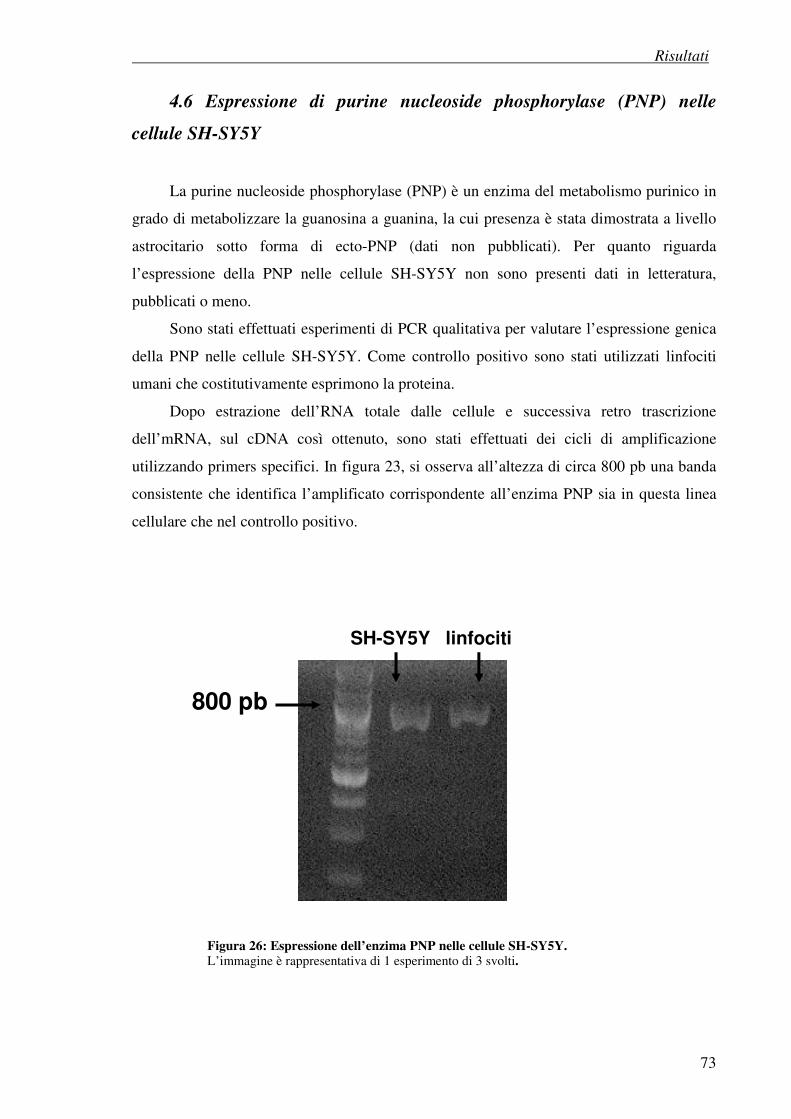
Risultati
73
4.6 Espressione di purine nucleoside phosphorylase (PNP) nelle
cellule SH-SY5Y
La purine nucleoside phosphorylase (PNP) è un enzima del metabolismo purinico in
grado di metabolizzare la guanosina a guanina, la cui presenza è stata dimostrata a livello
astrocitario sotto forma di ecto-PNP (dati non pubblicati). Per quanto riguarda
l’espressione della PNP nelle cellule SH-SY5Y non sono presenti dati in letteratura,
pubblicati o meno.
Sono stati effettuati esperimenti di PCR qualitativa per valutare l’espressione genica
della PNP nelle cellule SH-SY5Y. Come controllo positivo sono stati utilizzati linfociti
umani che costitutivamente esprimono la proteina.
Dopo estrazione dell’RNA totale dalle cellule e successiva retro trascrizione
dell’mRNA, sul cDNA così ottenuto, sono stati effettuati dei cicli di amplificazione
utilizzando primers specifici. In figura 23, si osserva all’altezza di circa 800 pb una banda
consistente che identifica l’amplificato corrispondente all’enzima PNP sia in questa linea
cellulare che nel controllo positivo.
Figura 26: Espressione dell’enzima PNP nelle cellule SH-SY5Y. L’immagine è rappresentativa di 1 esperimento di 3 svolti.
800 pb
SH-SY5Y linfociti

Risultati
74
4.7 Esperimenti di differenziazione
4.7.1 Metodo visivo
Lo studio dell’azione differenziante della guanosina è iniziato con la valutazione
dell’effetto del nucleoside sulla morfologia delle cellule SH-SY5Y.
A questo scopo le cellule sono state trattate con concentrazioni crescenti di
guanosina (10, 75 e 150 µM) e con acido retinoico (10 µM), composto di riferimento per
un’eventuale attività differenziante. Inoltre in ogni esperimento è stato incluso un gruppo
di controllo contenente cellule non trattate. Le cellule sono state mantenute in siero fetale
bovino al 10 % e sono state sottoposte a 24 e 72 ore di trattamento, effettuato
contemporaneamente alla semina.
Figura 27: Differenziazione delle cellule SH-SY5Y dopo trattamento con guanosina e acido retinoico per 24 ore. Le cellule sono state trattate per 24 ore con acido retinoico 10 µM (B) e con guanosina 10 µM (C), 75 µM (D) e 150 µM (E). In ogni esperimento è stato incluso un gruppo di controllo contenente cellule non trattate (A). Le immagini sono state acquisite utilizzando il microscopio Leica Leitz Wetzlar obiettivo 40x collegato ad un computer dotato del programma Leica IM (W.4, Leica Microsystem Digital Imaging, Cambrige UK).
Come riportato in figura 27, l’esposizione delle cellule ad acido retinoico 10 µM per
24 ore (B) ha indotto, rispetto ai controlli (A), la comparsa di spessi prolungamenti che si
estendono dal corpo cellulare e che possiedono una lunghezza maggiore rispetto allo
A. Controlli
B. Acido Retinoico 10 µM
C. Guo 10 µM
D. Guo 75 µM
E. Guo 150 µM
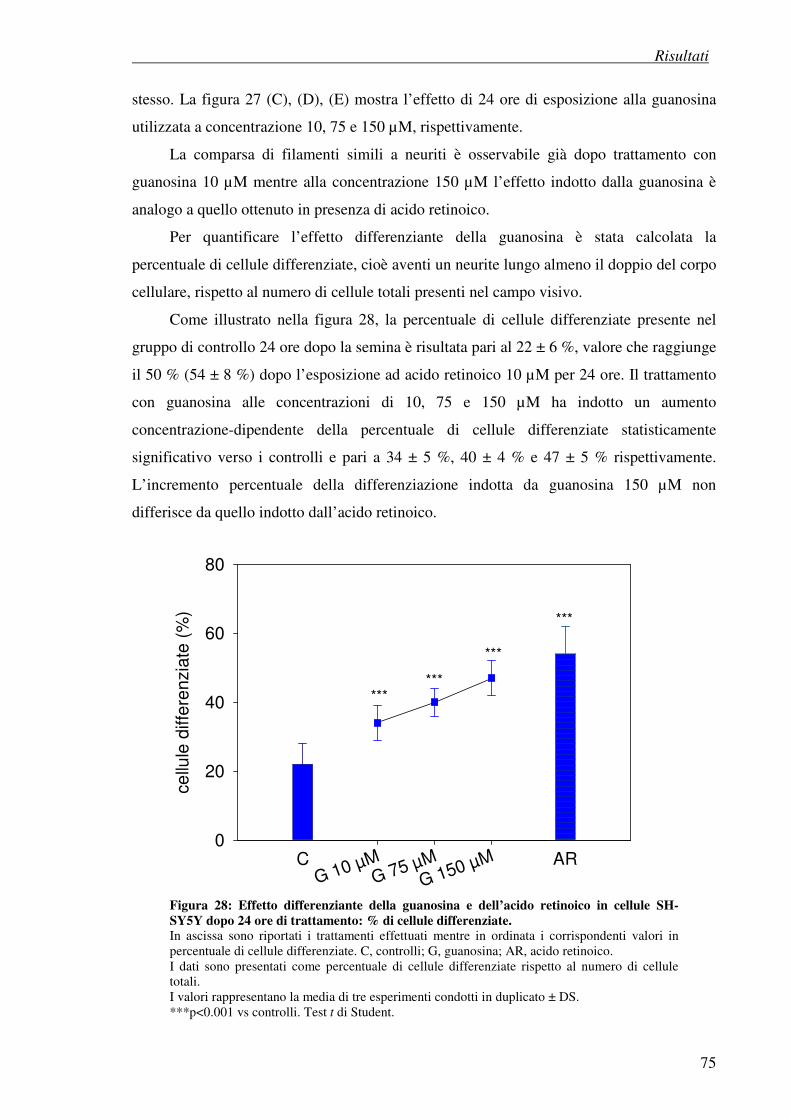
Risultati
75
stesso. La figura 27 (C), (D), (E) mostra l’effetto di 24 ore di esposizione alla guanosina
utilizzata a concentrazione 10, 75 e 150 µM, rispettivamente.
La comparsa di filamenti simili a neuriti è osservabile già dopo trattamento con
guanosina 10 µM mentre alla concentrazione 150 µM l’effetto indotto dalla guanosina è
analogo a quello ottenuto in presenza di acido retinoico.
Per quantificare l’effetto differenziante della guanosina è stata calcolata la
percentuale di cellule differenziate, cioè aventi un neurite lungo almeno il doppio del corpo
cellulare, rispetto al numero di cellule totali presenti nel campo visivo.
Come illustrato nella figura 28, la percentuale di cellule differenziate presente nel
gruppo di controllo 24 ore dopo la semina è risultata pari al 22 ± 6 %, valore che raggiunge
il 50 % (54 ± 8 %) dopo l’esposizione ad acido retinoico 10 µM per 24 ore. Il trattamento
con guanosina alle concentrazioni di 10, 75 e 150 µM ha indotto un aumento
concentrazione-dipendente della percentuale di cellule differenziate statisticamente
significativo verso i controlli e pari a 34 ± 5 %, 40 ± 4 % e 47 ± 5 % rispettivamente.
L’incremento percentuale della differenziazione indotta da guanosina 150 µM non
differisce da quello indotto dall’acido retinoico.
C AR
ce
llule
diffe
renzia
te (
%)
0
20
40
60
80
G 10 µMG 75 µM
G 150 µM
******
***
***
Figura 28: Effetto differenziante della guanosina e dell’acido retinoico in cellule SH-SY5Y dopo 24 ore di trattamento: % di cellule differenziate. In ascissa sono riportati i trattamenti effettuati mentre in ordinata i corrispondenti valori in percentuale di cellule differenziate. C, controlli; G, guanosina; AR, acido retinoico. I dati sono presentati come percentuale di cellule differenziate rispetto al numero di cellule totali. I valori rappresentano la media di tre esperimenti condotti in duplicato ± DS. ***p<0.001 vs controlli. Test t di Student.
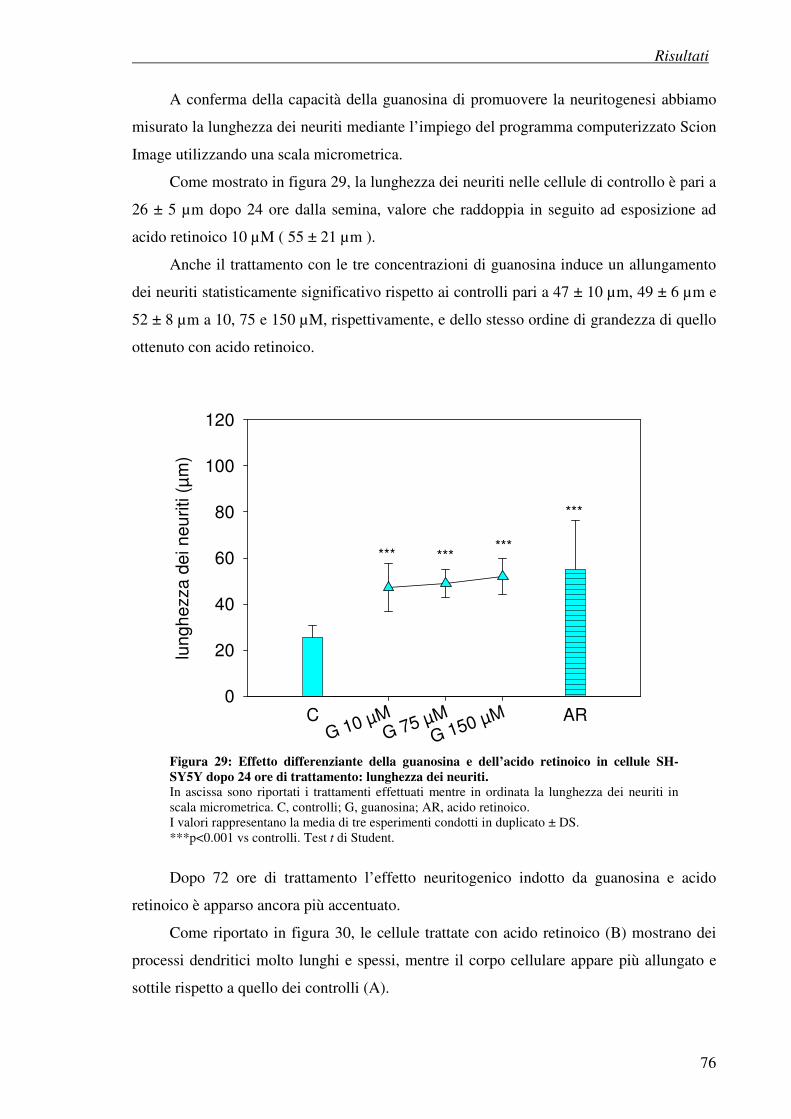
Risultati
76
A conferma della capacità della guanosina di promuovere la neuritogenesi abbiamo
misurato la lunghezza dei neuriti mediante l’impiego del programma computerizzato Scion
Image utilizzando una scala micrometrica.
Come mostrato in figura 29, la lunghezza dei neuriti nelle cellule di controllo è pari a
26 ± 5 µm dopo 24 ore dalla semina, valore che raddoppia in seguito ad esposizione ad
acido retinoico 10 µM ( 55 ± 21 µm ).
Anche il trattamento con le tre concentrazioni di guanosina induce un allungamento
dei neuriti statisticamente significativo rispetto ai controlli pari a 47 ± 10 µm, 49 ± 6 µm e
52 ± 8 µm a 10, 75 e 150 µM, rispettivamente, e dello stesso ordine di grandezza di quello
ottenuto con acido retinoico.
G 10 µMG 75 µM
G 150 µM
******
***
***
C AR
lun
gh
ezza d
ei n
eu
riti (
µm
)
0
20
40
60
80
100
120
Figura 29: Effetto differenziante della guanosina e dell’acido retinoico in cellule SH-SY5Y dopo 24 ore di trattamento: lunghezza dei neuriti. In ascissa sono riportati i trattamenti effettuati mentre in ordinata la lunghezza dei neuriti in scala micrometrica. C, controlli; G, guanosina; AR, acido retinoico. I valori rappresentano la media di tre esperimenti condotti in duplicato ± DS. ***p<0.001 vs controlli. Test t di Student.
Dopo 72 ore di trattamento l’effetto neuritogenico indotto da guanosina e acido
retinoico è apparso ancora più accentuato.
Come riportato in figura 30, le cellule trattate con acido retinoico (B) mostrano dei
processi dendritici molto lunghi e spessi, mentre il corpo cellulare appare più allungato e
sottile rispetto a quello dei controlli (A).

Risultati
77
Le cellule esposte all’azione della guanosina (C, D, E) mostrano dei prolungamenti
più sottili ma in questo caso il corpo cellulare sembra essere più tondeggiante e le cellule
maggiormente adese alla superficie della piastra tanto da apparire schiacciate. Questo
effetto è maggiormente evidente nelle cellule trattate con guanosina 150 µM (E) dove
inoltre i neuriti tendono a formare una fitta rete che collega i corpi cellulari.
A. Controlli
B. Acido Retinoico 10 µM
C. Guanosina 10 µM
D. Guanosina 75 µM
E. Guanosina 150 µM
Figura 30: Differenziamento delle cellule SH-SY5Y per trattamento con guanosina e acido retinoico per 72 ore. Le cellule sono state trattate per 72 ore con acido retinoico 10 µM (B) e con guanosina 10 µM (C), 75 µM (D) e 150 µM (E). In ogni esperimento è stato incluso un gruppo di controllo contenente cellule non trattate (A). Le immagini sono state acquisite utilizzando il microscopio Leica Leitz Wetzlar obiettivo 40x collegato ad un computer dotato del programma Leica IM (W.4, Leica Microsystem Digital Imaging, Cambrige UK).
Come illustrato nella figura 31 la percentuale di cellule differenziate presente nei
campi delle cellule di controllo a 72 ore dalla semina è pari a 27 ± 4 % e raggiunge il 67 ±
6 % dopo trattamento con acido retinoico 10 µM.
L’esposizione alla guanosina 10, 75 e 150 µM per 72 ore incrementa la percentuale
di cellule differenziate del 42 ± 5 %, 45 ± 3 % e 56 ± 5 % rispettivamente, rispetto ai valori
basali.
A differenza dell’esposizione per 24 ore, l’incremento percentuale della
differenziazione indotta da guanosina 150 µM dopo 72 ore è inferiore e statisticamente
diverso da quello indotto dall’acido retinoico.
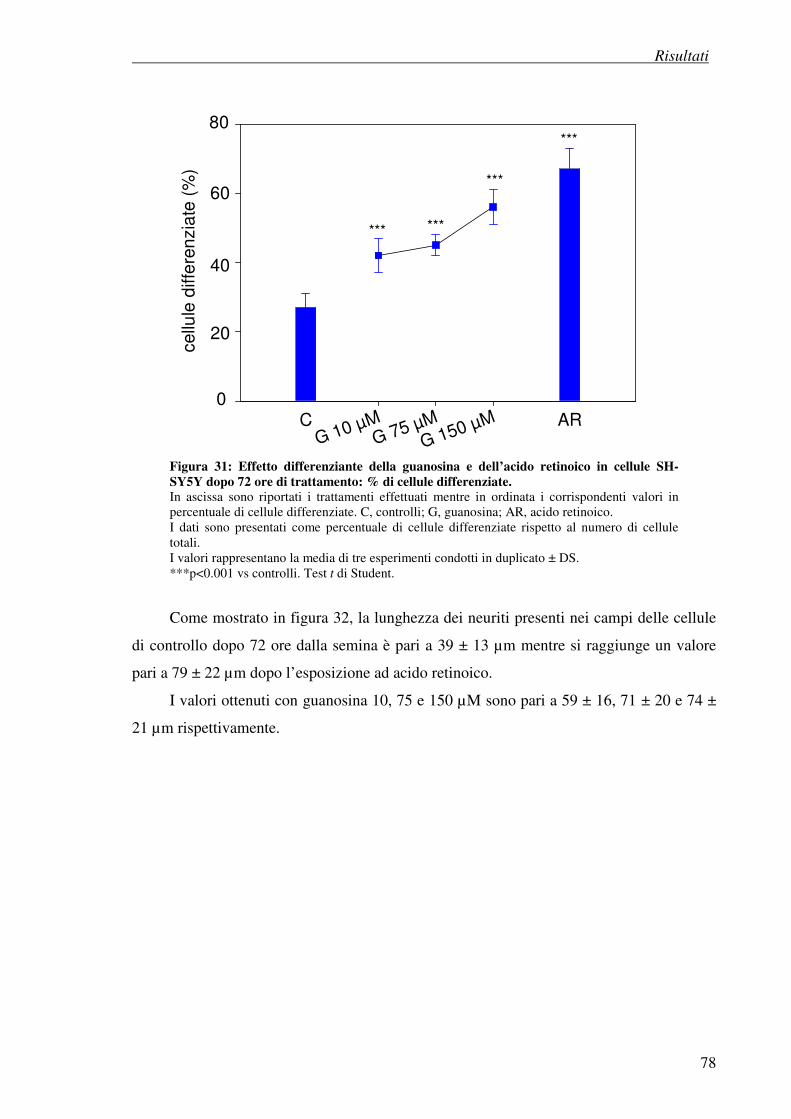
Risultati
78
C AR
cellu
le d
iffe
ren
zia
te (
%)
G 10 µMG 75 µM
G 150 µM
0
20
40
60
80
*** ***
***
***
Figura 31: Effetto differenziante della guanosina e dell’acido retinoico in cellule SH-SY5Y dopo 72 ore di trattamento: % di cellule differenziate. In ascissa sono riportati i trattamenti effettuati mentre in ordinata i corrispondenti valori in percentuale di cellule differenziate. C, controlli; G, guanosina; AR, acido retinoico. I dati sono presentati come percentuale di cellule differenziate rispetto al numero di cellule totali. I valori rappresentano la media di tre esperimenti condotti in duplicato ± DS. ***p<0.001 vs controlli. Test t di Student.
Come mostrato in figura 32, la lunghezza dei neuriti presenti nei campi delle cellule
di controllo dopo 72 ore dalla semina è pari a 39 ± 13 µm mentre si raggiunge un valore
pari a 79 ± 22 µm dopo l’esposizione ad acido retinoico.
I valori ottenuti con guanosina 10, 75 e 150 µM sono pari a 59 ± 16, 71 ± 20 e 74 ±
21 µm rispettivamente.
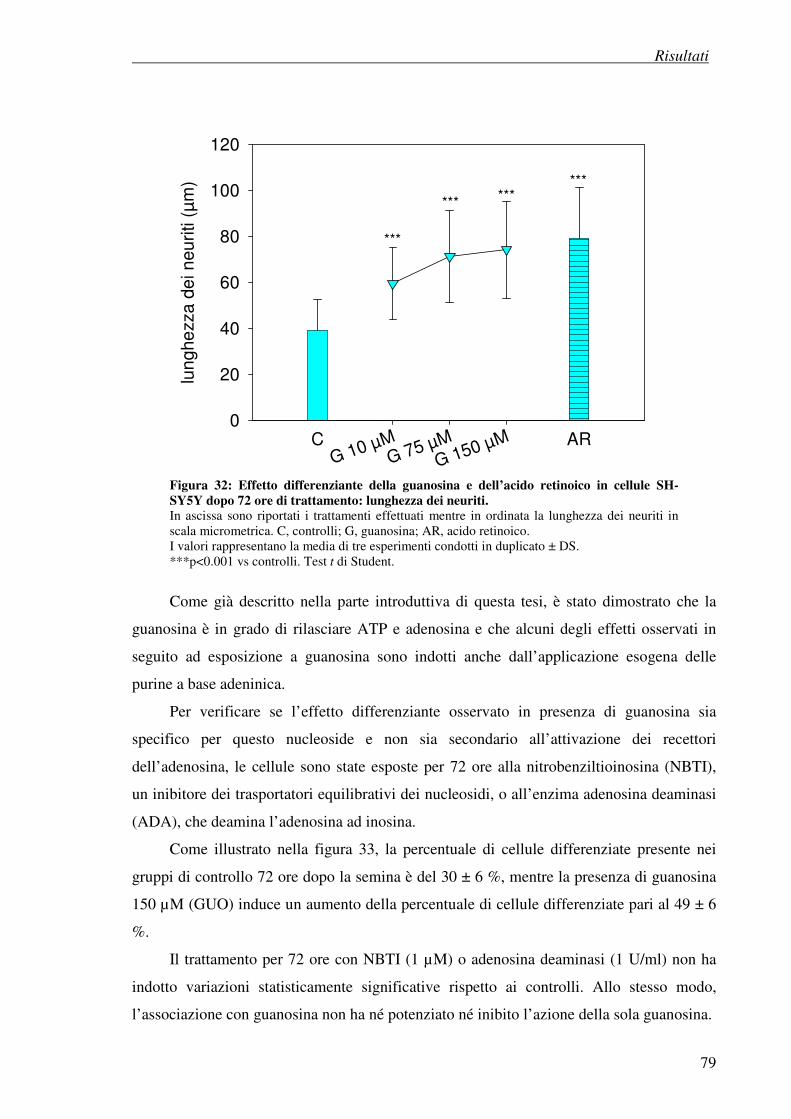
Risultati
79
C AR
lun
ghe
zza
de
i n
euri
ti (
µm
)
0
20
40
60
80
100
120
G 10 µMG 75 µM
G 150 µM
***
*** ******
Figura 32: Effetto differenziante della guanosina e dell’acido retinoico in cellule SH-SY5Y dopo 72 ore di trattamento: lunghezza dei neuriti. In ascissa sono riportati i trattamenti effettuati mentre in ordinata la lunghezza dei neuriti in scala micrometrica. C, controlli; G, guanosina; AR, acido retinoico. I valori rappresentano la media di tre esperimenti condotti in duplicato ± DS. ***p<0.001 vs controlli. Test t di Student.
Come già descritto nella parte introduttiva di questa tesi, è stato dimostrato che la
guanosina è in grado di rilasciare ATP e adenosina e che alcuni degli effetti osservati in
seguito ad esposizione a guanosina sono indotti anche dall’applicazione esogena delle
purine a base adeninica.
Per verificare se l’effetto differenziante osservato in presenza di guanosina sia
specifico per questo nucleoside e non sia secondario all’attivazione dei recettori
dell’adenosina, le cellule sono state esposte per 72 ore alla nitrobenziltioinosina (NBTI),
un inibitore dei trasportatori equilibrativi dei nucleosidi, o all’enzima adenosina deaminasi
(ADA), che deamina l’adenosina ad inosina.
Come illustrato nella figura 33, la percentuale di cellule differenziate presente nei
gruppi di controllo 72 ore dopo la semina è del 30 ± 6 %, mentre la presenza di guanosina
150 µM (GUO) induce un aumento della percentuale di cellule differenziate pari al 49 ± 6
%.
Il trattamento per 72 ore con NBTI (1 µM) o adenosina deaminasi (1 U/ml) non ha
indotto variazioni statisticamente significative rispetto ai controlli. Allo stesso modo,
l’associazione con guanosina non ha né potenziato né inibito l’azione della sola guanosina.
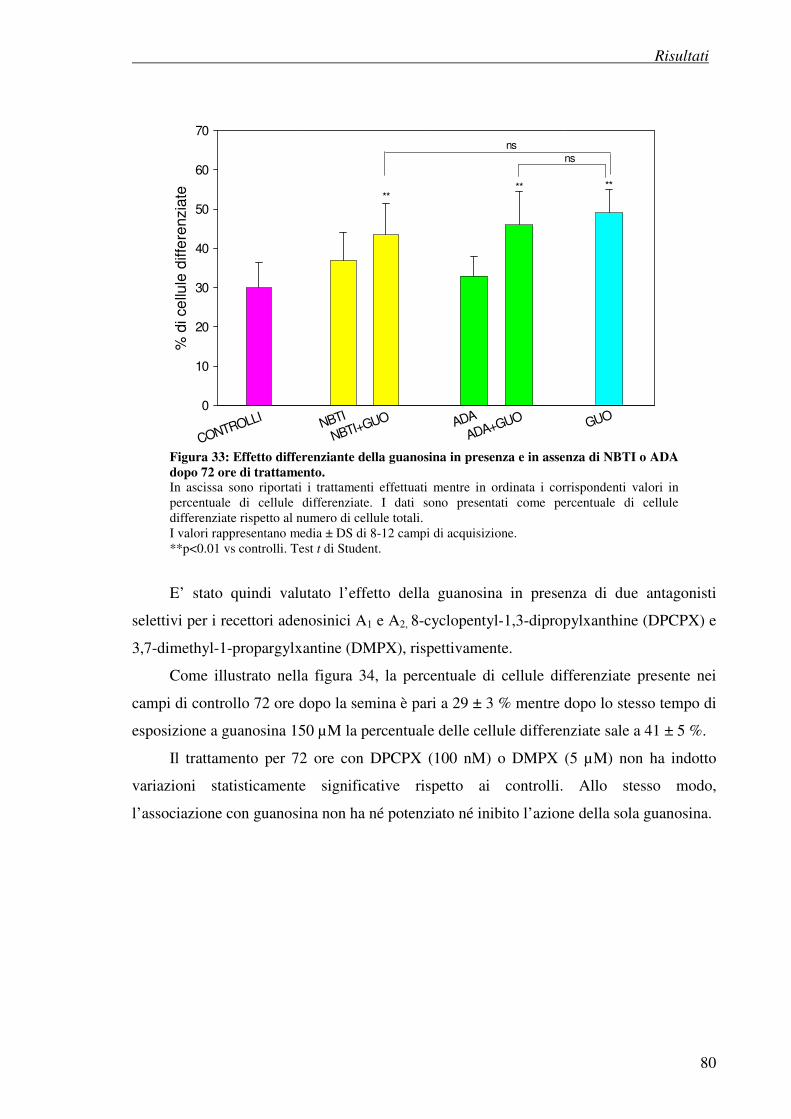
Risultati
80
CONTROLLI
% d
i ce
llule
diffe
renzia
te
0
10
20
30
40
50
60
70
NBTI
NBTI+GUO ADA
ADA+GUO GUO
**** **
nsns
Figura 33: Effetto differenziante della guanosina in presenza e in assenza di NBTI o ADA dopo 72 ore di trattamento. In ascissa sono riportati i trattamenti effettuati mentre in ordinata i corrispondenti valori in percentuale di cellule differenziate. I dati sono presentati come percentuale di cellule differenziate rispetto al numero di cellule totali. I valori rappresentano media ± DS di 8-12 campi di acquisizione. **p<0.01 vs controlli. Test t di Student.
E’ stato quindi valutato l’effetto della guanosina in presenza di due antagonisti
selettivi per i recettori adenosinici A1 e A2, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) e
3,7-dimethyl-1-propargylxantine (DMPX), rispettivamente.
Come illustrato nella figura 34, la percentuale di cellule differenziate presente nei
campi di controllo 72 ore dopo la semina è pari a 29 ± 3 % mentre dopo lo stesso tempo di
esposizione a guanosina 150 µM la percentuale delle cellule differenziate sale a 41 ± 5 %.
Il trattamento per 72 ore con DPCPX (100 nM) o DMPX (5 µM) non ha indotto
variazioni statisticamente significative rispetto ai controlli. Allo stesso modo,
l’associazione con guanosina non ha né potenziato né inibito l’azione della sola guanosina.
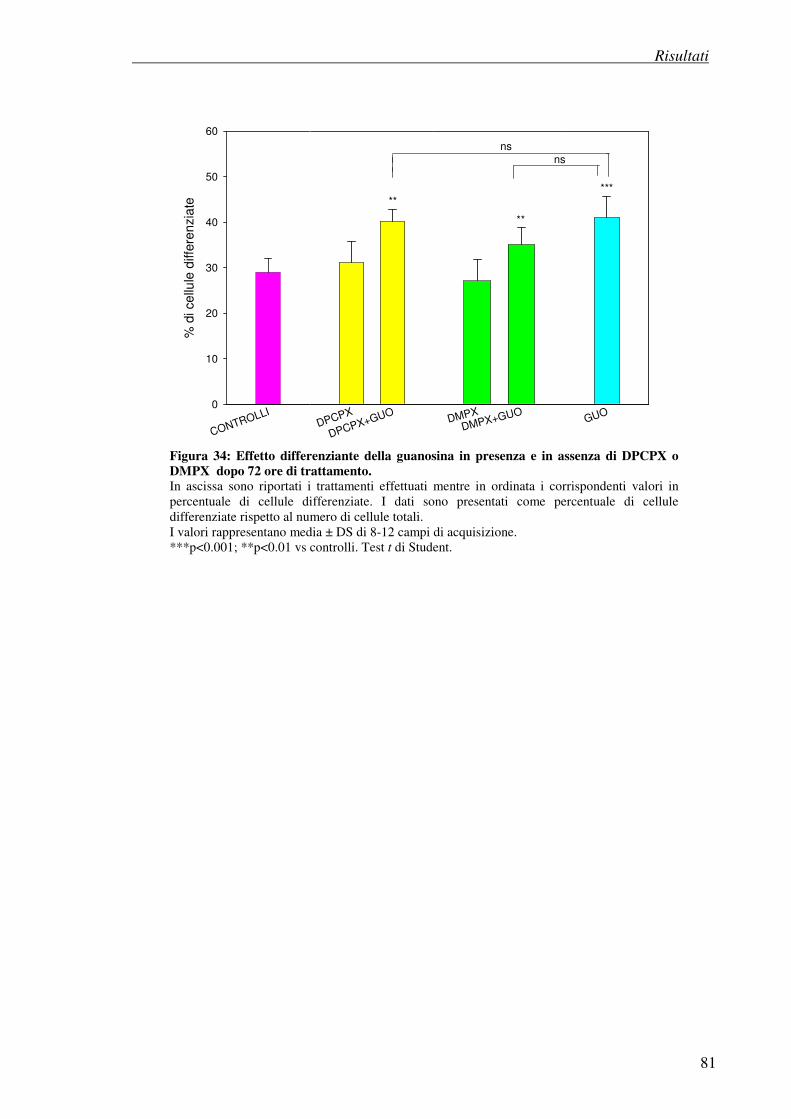
Risultati
81
CONTROLLI
% d
i ce
llule
diffe
ren
zia
te
0
10
20
30
40
50
60
DPCPX
DPCPX+GUODMPX
DMPX+GUOGUO
**
**
***
ns
ns
Figura 34: Effetto differenziante della guanosina in presenza e in assenza di DPCPX o DMPX dopo 72 ore di trattamento. In ascissa sono riportati i trattamenti effettuati mentre in ordinata i corrispondenti valori in percentuale di cellule differenziate. I dati sono presentati come percentuale di cellule differenziate rispetto al numero di cellule totali. I valori rappresentano media ± DS di 8-12 campi di acquisizione. ***p<0.001; **p<0.01 vs controlli. Test t di Student.

Risultati
82
4.7.2 Esperimenti di immunocitochimica: effetto della guanosina sul citoscheletro
La falloidina, come descritto con maggiori dettagli nel capitolo Materiali e Metodi
(pag. 49), è una sostanza in grado di legarsi ai filamenti di actina. Per caratterizzare meglio
l’effetto differenziante della guanosina sulla linea cellulare SH-SY5Y, le cellule sono state
trattate con guanosina 150 µM per 24, 48, 72, 96 h e 7 giorni e quindi i filamenti di actina
sono stati evidenziati con falloidina.
controlli guanosina acido retinoico
24 h
48 h
72 h
96 h
7 gg
Figura 35: Espressione di falloidina in seguito a trattamento con guanosina o acido retinoico. Le immagini sono rappresentative di uno di 3 esperimenti condotti in duplicato.
Nelle immagini riportate in figura 35, si osserva che le cellule SH-SY5Y
mantengono un aspetto simil-neuronale in seguito a trattamento con guanosina e acido
retinoico fino a 7 giorni di trattamento.
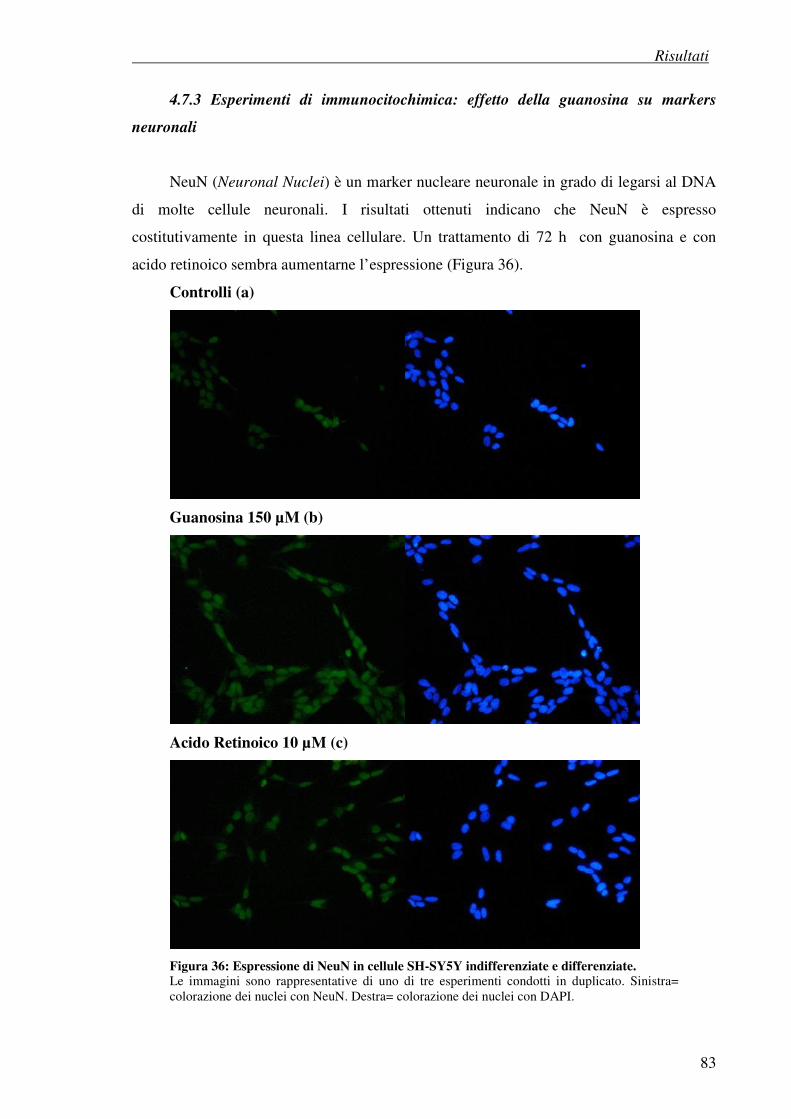
Risultati
83
4.7.3 Esperimenti di immunocitochimica: effetto della guanosina su markers
neuronali
NeuN (Neuronal Nuclei) è un marker nucleare neuronale in grado di legarsi al DNA
di molte cellule neuronali. I risultati ottenuti indicano che NeuN è espresso
costitutivamente in questa linea cellulare. Un trattamento di 72 h con guanosina e con
acido retinoico sembra aumentarne l’espressione (Figura 36).
Controlli (a)
Guanosina 150 µM (b)
Acido Retinoico 10 µM (c)
Figura 36: Espressione di NeuN in cellule SH-SY5Y indifferenziate e differenziate. Le immagini sono rappresentative di uno di tre esperimenti condotti in duplicato. Sinistra= colorazione dei nuclei con NeuN. Destra= colorazione dei nuclei con DAPI.
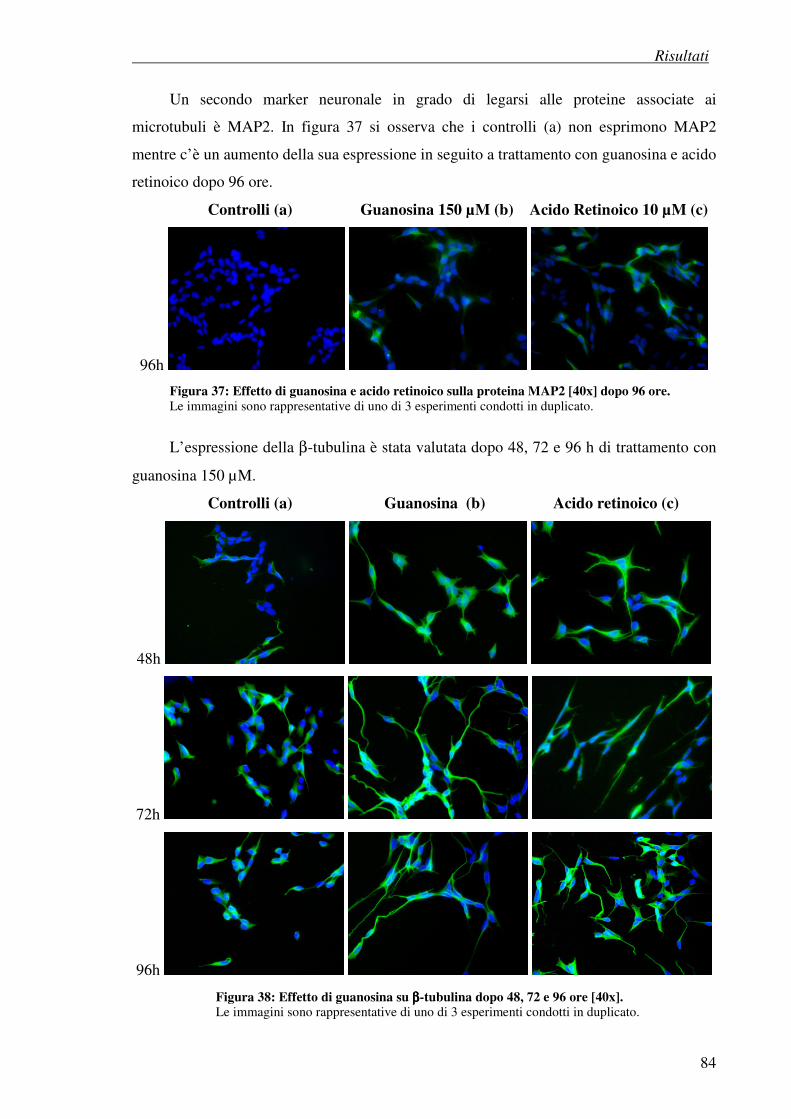
Risultati
84
Un secondo marker neuronale in grado di legarsi alle proteine associate ai
microtubuli è MAP2. In figura 37 si osserva che i controlli (a) non esprimono MAP2
mentre c’è un aumento della sua espressione in seguito a trattamento con guanosina e acido
retinoico dopo 96 ore.
Controlli (a) Guanosina 150 µM (b) Acido Retinoico 10 µM (c)
96h
Figura 37: Effetto di guanosina e acido retinoico sulla proteina MAP2 [40x] dopo 96 ore. Le immagini sono rappresentative di uno di 3 esperimenti condotti in duplicato.
L’espressione della β-tubulina è stata valutata dopo 48, 72 e 96 h di trattamento con
guanosina 150 µM.
Controlli (a) Guanosina (b) Acido retinoico (c)
48h
72h
96h
Figura 38: Effetto di guanosina su ββββ-tubulina dopo 48, 72 e 96 ore [40x]. Le immagini sono rappresentative di uno di 3 esperimenti condotti in duplicato.
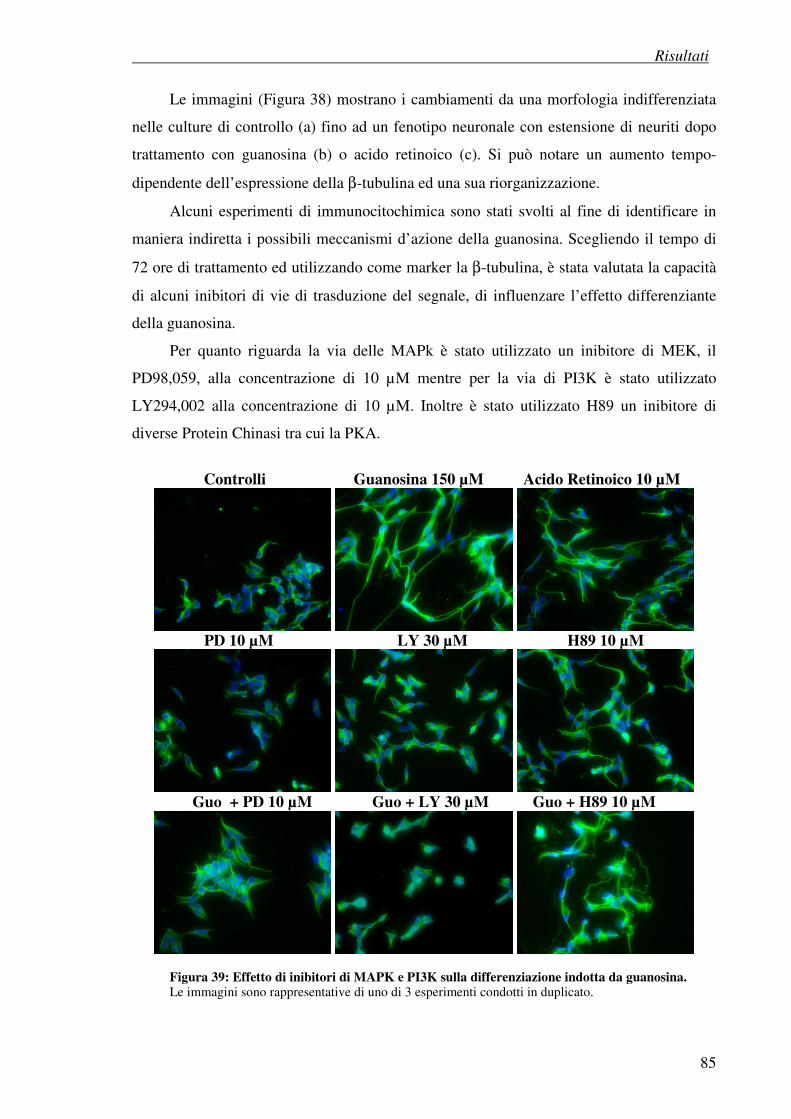
Risultati
85
Le immagini (Figura 38) mostrano i cambiamenti da una morfologia indifferenziata
nelle culture di controllo (a) fino ad un fenotipo neuronale con estensione di neuriti dopo
trattamento con guanosina (b) o acido retinoico (c). Si può notare un aumento tempo-
dipendente dell’espressione della β-tubulina ed una sua riorganizzazione.
Alcuni esperimenti di immunocitochimica sono stati svolti al fine di identificare in
maniera indiretta i possibili meccanismi d’azione della guanosina. Scegliendo il tempo di
72 ore di trattamento ed utilizzando come marker la β-tubulina, è stata valutata la capacità
di alcuni inibitori di vie di trasduzione del segnale, di influenzare l’effetto differenziante
della guanosina.
Per quanto riguarda la via delle MAPk è stato utilizzato un inibitore di MEK, il
PD98,059, alla concentrazione di 10 µM mentre per la via di PI3K è stato utilizzato
LY294,002 alla concentrazione di 10 µM. Inoltre è stato utilizzato H89 un inibitore di
diverse Protein Chinasi tra cui la PKA.
Controlli Guanosina 150 µM Acido Retinoico 10 µM
PD 10 µM LY 30 µM H89 10 µM
Guo + PD 10 µM Guo + LY 30 µM Guo + H89 10 µM
Figura 39: Effetto di inibitori di MAPK e PI3K sulla differenziazione indotta da guanosina. Le immagini sono rappresentative di uno di 3 esperimenti condotti in duplicato.

Risultati
86
La presenza degli inibitori di MAPK e PI3K, che di per sé non modificano la
percentuale di cellule differenziate rispetto ai controlli, ha completamente abolito l’effetto
neuritogenico della guanosina (Figura 39). Mentre la presenza dell’inibitore di PKA ha
avuto un effetto sinergico con la guanosina ed ha modificato significativamente la
morfologia cellulare dopo 72 ore di trattamento.
4.8 Effetto antiproliferativo della guanosina (test della SRB)
Poiché differenziazione e proliferazione sono fenomeni mutuamente esclusivi, è stata
valutata la capacità della guanosina di ridurre la proliferazione cellulare. E’ stato usato
come controllo positivo sempre l’acido retinoico di cui è nota la capacità di ridurre la
proliferazione in questa linea cellulare in concomitanza con l’effetto differenziante
(Encinas et al., 2000).
Gli esperimenti sono stati condotti esponendo le cellule a concentrazioni crescenti di
guanosina e acido retinoico per 48 ore o 7 giorni di trattamento.
Come illustrato in figura 40, dopo 48 ore di esposizione a concentrazioni crescenti di
guanosina (da 19 nM a 3 mM) si osserva una riduzione della proliferazione cellulare a
partire da 37 µM. Alle concentrazioni più elevate, il valore di assorbanza delle cellule
trattate alla concentrazione più alta di guanosina (0.331 ± 0.05 D.O.) non è
significativamente diverso dal valore di assorbanza delle cellule presenti al momento della
semina (0.268 ± 0.06 D.O.).
Come riportato in figura 41, l’esposizione delle cellule per 7 giorni alle stesse
concentrazioni di guanosina provoca una diminuzione della proliferazione cellulare
concentrazione-dipendente, per la quale è stato possibile calcolare il valore di EC50 che è
risultato pari a 172 ± 113 µM.
Le Figure 42 e 43 riportano l’effetto dell’esposizione delle cellule a concentrazioni
crescenti di acido retinoico (19 nM-3.33 µM) per 48 ore e (0.7 nM-3.33 µM) per 7 giorni.
Non è stato possibile calcolare il valore di EC50 dopo 48 ore di trattamento, anche nel
caso dell’acido retinoico mentre dopo 7 giorni di esposizione, l’andamento concentrazione-
dipendente ha permesso di calcolare il valore di EC50 pari a 0.20 ± 0.15 µM.
Ulteriori esperimenti di proliferazioni sono stati condotti per valutare l’effetto di
alcuni analoghi della guanosina su questa linea cellulare. In figura 44 e 45 sono riportati i
valori di assorbanza ottenuti in seguito ad esposizione delle cellule SH-SY5Y a

Risultati
87
concentrazioni crescenti di guanina (20 nM-3.3 mM) dopo 48 ore e 7 giorni,
rispettivamente.
Non è stato possibile calcolare il valore di EC50 dopo 48 ore di trattamento, anche nel
caso della guanina mentre dopo 7 giorni di esposizione, l’andamento concentrazione-
dipendente ha permesso di calcolare il valore di EC50 pari a 190 ± 17 µM.
Le Figure 46 e 47 riportano l’effetto dell’esposizione delle cellule a concentrazioni
crescenti di GTP (19 nM-3.33 µM) per 48 ore e per 7 giorni. Ad entrambi i tempi di
esposizione, solo la concentrazione più alta di GTP è stata in grado di ridurre in maniera
significativa la proliferazione cellulare.
Nelle figure 48 e 49 è riportato l’effetto di concentrazioni crescenti di un analogo
della guanosina, il composto AIT-082, dopo 48 ore e 7 giorni di trattamento.
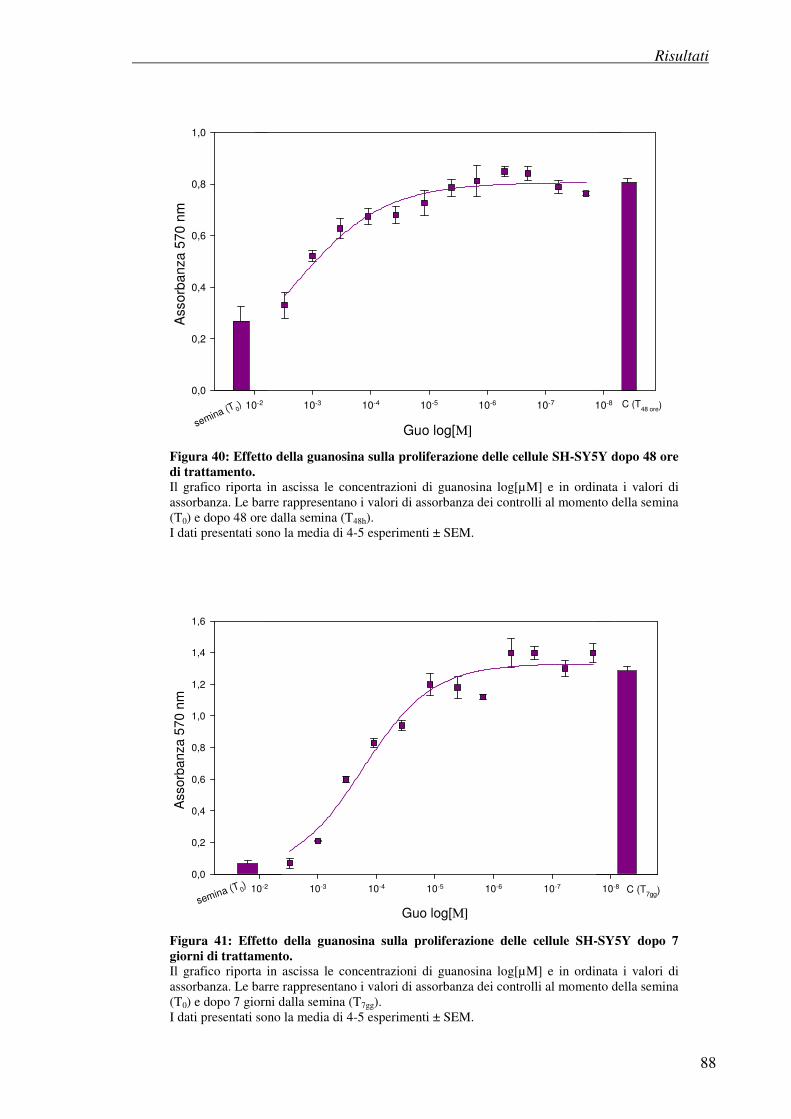
Risultati
88
semina (T 0) C (T
48 ore)
Asso
rba
nza
57
0 n
m
0,0
0,2
0,4
0,6
0,8
1,0
Guo log[Μ]
10-810-710-610-510-410-310-2
Figura 40: Effetto della guanosina sulla proliferazione delle cellule SH-SY5Y dopo 48 ore di trattamento. Il grafico riporta in ascissa le concentrazioni di guanosina log[µM] e in ordinata i valori di assorbanza. Le barre rappresentano i valori di assorbanza dei controlli al momento della semina (T0) e dopo 48 ore dalla semina (T48h). I dati presentati sono la media di 4-5 esperimenti ± SEM.
semina (T 0) C (T
7gg)
Guo log[Μ]
10-810-710-610-510-410-310-2
Assorb
anza 5
70 n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
Figura 41: Effetto della guanosina sulla proliferazione delle cellule SH-SY5Y dopo 7 giorni di trattamento. Il grafico riporta in ascissa le concentrazioni di guanosina log[µM] e in ordinata i valori di assorbanza. Le barre rappresentano i valori di assorbanza dei controlli al momento della semina (T0) e dopo 7 giorni dalla semina (T7gg). I dati presentati sono la media di 4-5 esperimenti ± SEM.
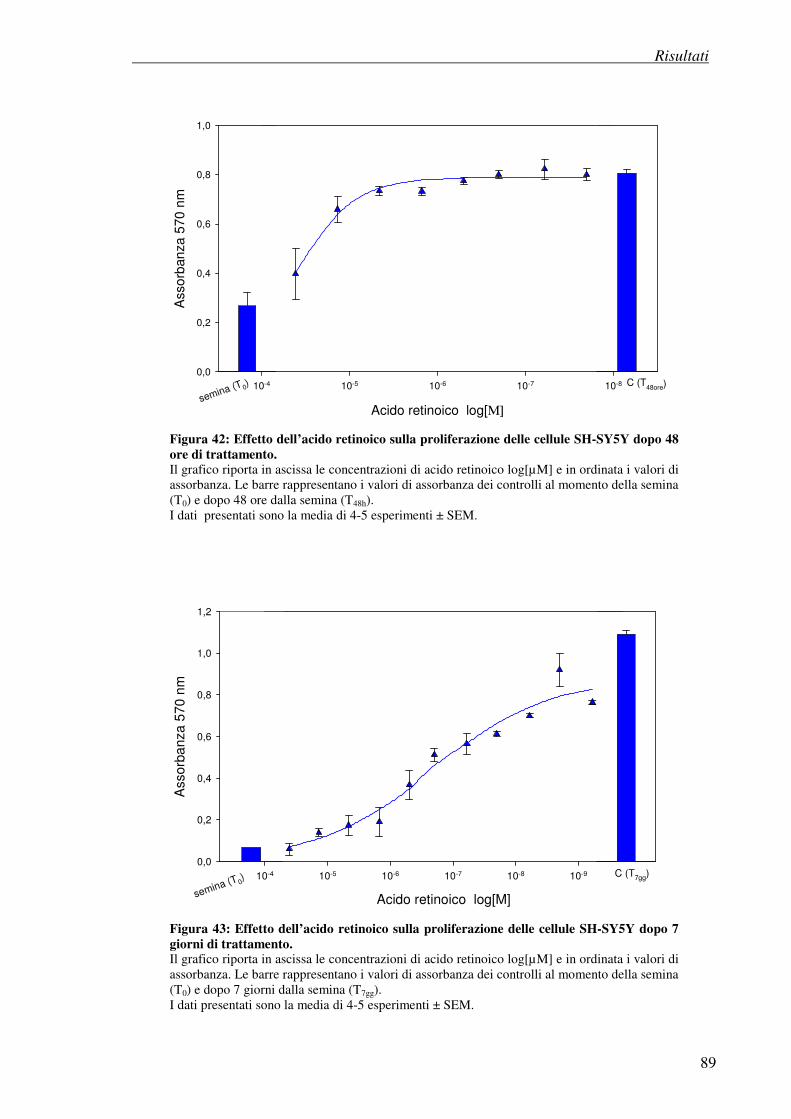
Risultati
89
semina (T 0) C (T
48ore)
Acido retinoico log[Μ]
10-810-710-610-510-4
Asso
rban
za
570
nm
0,0
0,2
0,4
0,6
0,8
1,0
Figura 42: Effetto dell’acido retinoico sulla proliferazione delle cellule SH-SY5Y dopo 48 ore di trattamento. Il grafico riporta in ascissa le concentrazioni di acido retinoico log[µM] e in ordinata i valori di assorbanza. Le barre rappresentano i valori di assorbanza dei controlli al momento della semina (T0) e dopo 48 ore dalla semina (T48h). I dati presentati sono la media di 4-5 esperimenti ± SEM.
C (T7gg
)
semina (T 0)
Acido retinoico log[M]
10-910-810-710-610-510-4
Assorb
anza
570
nm
0,0
0,2
0,4
0,6
0,8
1,0
1,2
Figura 43: Effetto dell’acido retinoico sulla proliferazione delle cellule SH-SY5Y dopo 7 giorni di trattamento. Il grafico riporta in ascissa le concentrazioni di acido retinoico log[µM] e in ordinata i valori di assorbanza. Le barre rappresentano i valori di assorbanza dei controlli al momento della semina (T0) e dopo 7 giorni dalla semina (T7gg). I dati presentati sono la media di 4-5 esperimenti ± SEM.
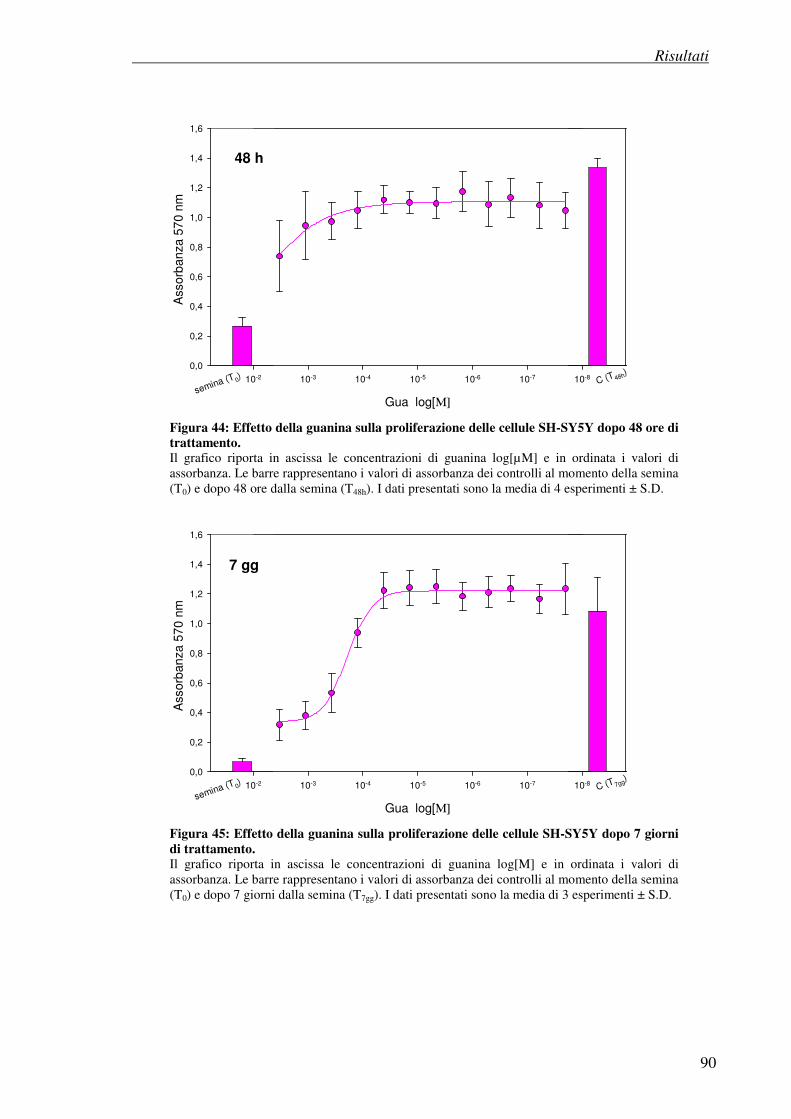
Risultati
90
48hIC50 GUA 190 ± 17 µM
semina (T 0)
C (T 48h)
Gua log[Μ]
10-810-710-610-510-410-310-2
Assorb
an
za 5
70 n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
48 h
Figura 44: Effetto della guanina sulla proliferazione delle cellule SH-SY5Y dopo 48 ore di trattamento. Il grafico riporta in ascissa le concentrazioni di guanina log[µM] e in ordinata i valori di assorbanza. Le barre rappresentano i valori di assorbanza dei controlli al momento della semina (T0) e dopo 48 ore dalla semina (T48h). I dati presentati sono la media di 4 esperimenti ± S.D.
semina (T 0)
C (T 7gg)
Gua log[Μ]
10-810-710-610-510-410-310-2
Assorb
an
za 5
70 n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
7 gg
Figura 45: Effetto della guanina sulla proliferazione delle cellule SH-SY5Y dopo 7 giorni di trattamento. Il grafico riporta in ascissa le concentrazioni di guanina log[M] e in ordinata i valori di assorbanza. Le barre rappresentano i valori di assorbanza dei controlli al momento della semina (T0) e dopo 7 giorni dalla semina (T7gg). I dati presentati sono la media di 3 esperimenti ± S.D.
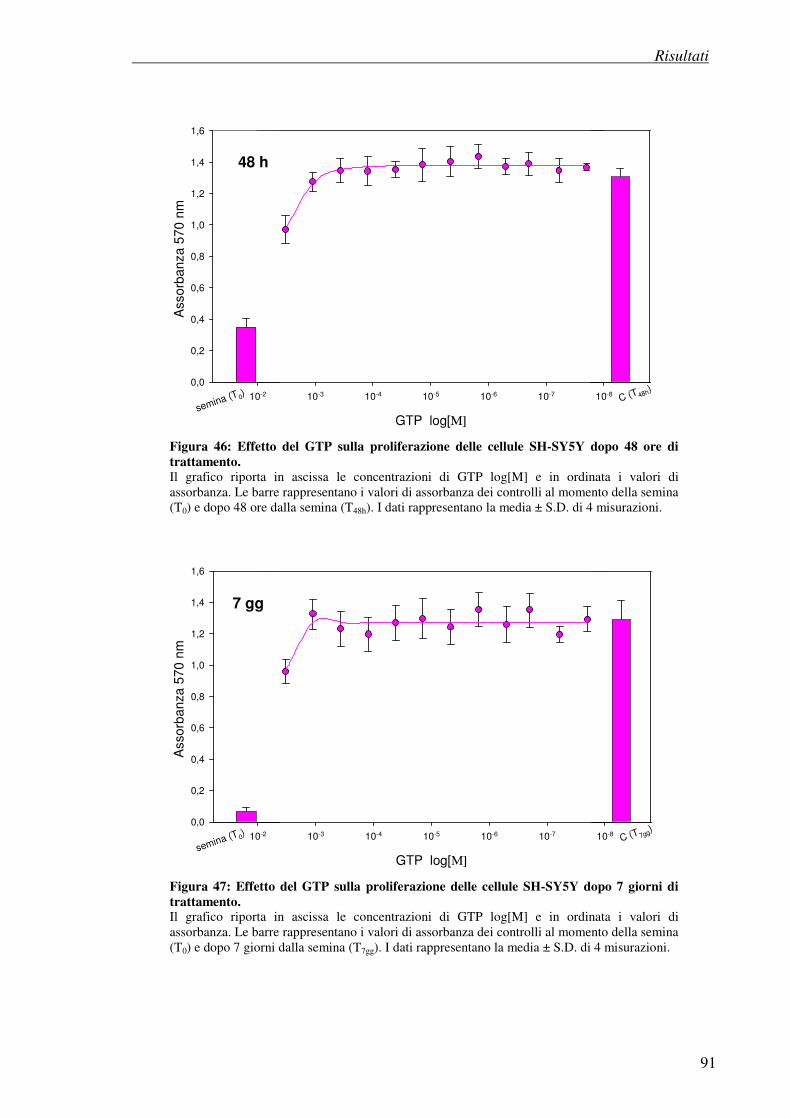
Risultati
91
48hIC50 GUA 190 ± 17 µM
semina (T 0)
C (T48h)
GTP log[Μ]
10-810-710-610-510-410-310-2
Asso
rba
nza
57
0 n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
48 h
Figura 46: Effetto del GTP sulla proliferazione delle cellule SH-SY5Y dopo 48 ore di trattamento. Il grafico riporta in ascissa le concentrazioni di GTP log[M] e in ordinata i valori di assorbanza. Le barre rappresentano i valori di assorbanza dei controlli al momento della semina (T0) e dopo 48 ore dalla semina (T48h). I dati rappresentano la media ± S.D. di 4 misurazioni.
semina (T 0)
C (T 7gg)
GTP log[Μ]
10-810-710-610-510-410-310-2
Asso
rba
nza
57
0 n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
7 gg
Figura 47: Effetto del GTP sulla proliferazione delle cellule SH-SY5Y dopo 7 giorni di trattamento. Il grafico riporta in ascissa le concentrazioni di GTP log[M] e in ordinata i valori di assorbanza. Le barre rappresentano i valori di assorbanza dei controlli al momento della semina (T0) e dopo 7 giorni dalla semina (T7gg). I dati rappresentano la media ± S.D. di 4 misurazioni.
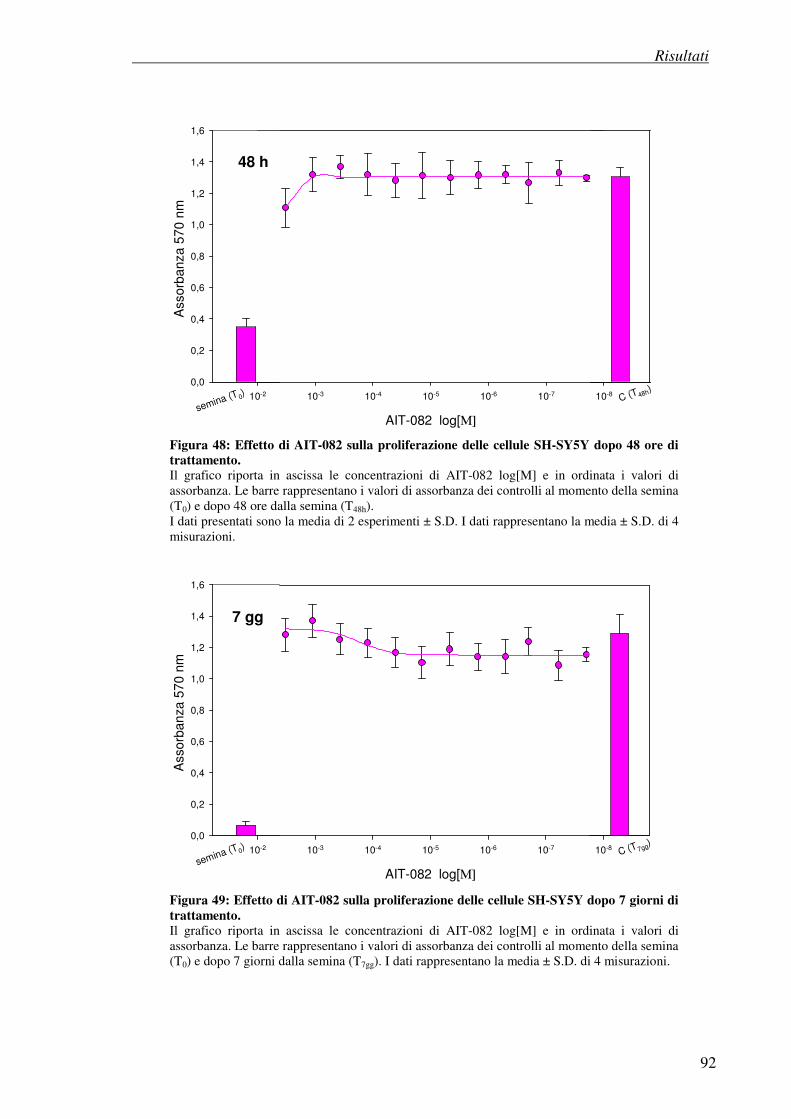
Risultati
92
48hIC50 GUA 190 ± 17 µM
semina (T 0)
C (T48h)
AIT-082 log[Μ]
10-810-710-610-510-410-310-2
Asso
rba
nza
57
0 n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
48 h
Figura 48: Effetto di AIT-082 sulla proliferazione delle cellule SH-SY5Y dopo 48 ore di trattamento. Il grafico riporta in ascissa le concentrazioni di AIT-082 log[M] e in ordinata i valori di assorbanza. Le barre rappresentano i valori di assorbanza dei controlli al momento della semina (T0) e dopo 48 ore dalla semina (T48h). I dati presentati sono la media di 2 esperimenti ± S.D. I dati rappresentano la media ± S.D. di 4 misurazioni.
semina (T 0)
C (T 7gg)
AIT-082 log[Μ]
10-810-710-610-510-410-310-2
Asso
rba
nza
57
0 n
m
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
7 gg
Figura 49: Effetto di AIT-082 sulla proliferazione delle cellule SH-SY5Y dopo 7 giorni di trattamento. Il grafico riporta in ascissa le concentrazioni di AIT-082 log[M] e in ordinata i valori di assorbanza. Le barre rappresentano i valori di assorbanza dei controlli al momento della semina (T0) e dopo 7 giorni dalla semina (T7gg). I dati rappresentano la media ± S.D. di 4 misurazioni.
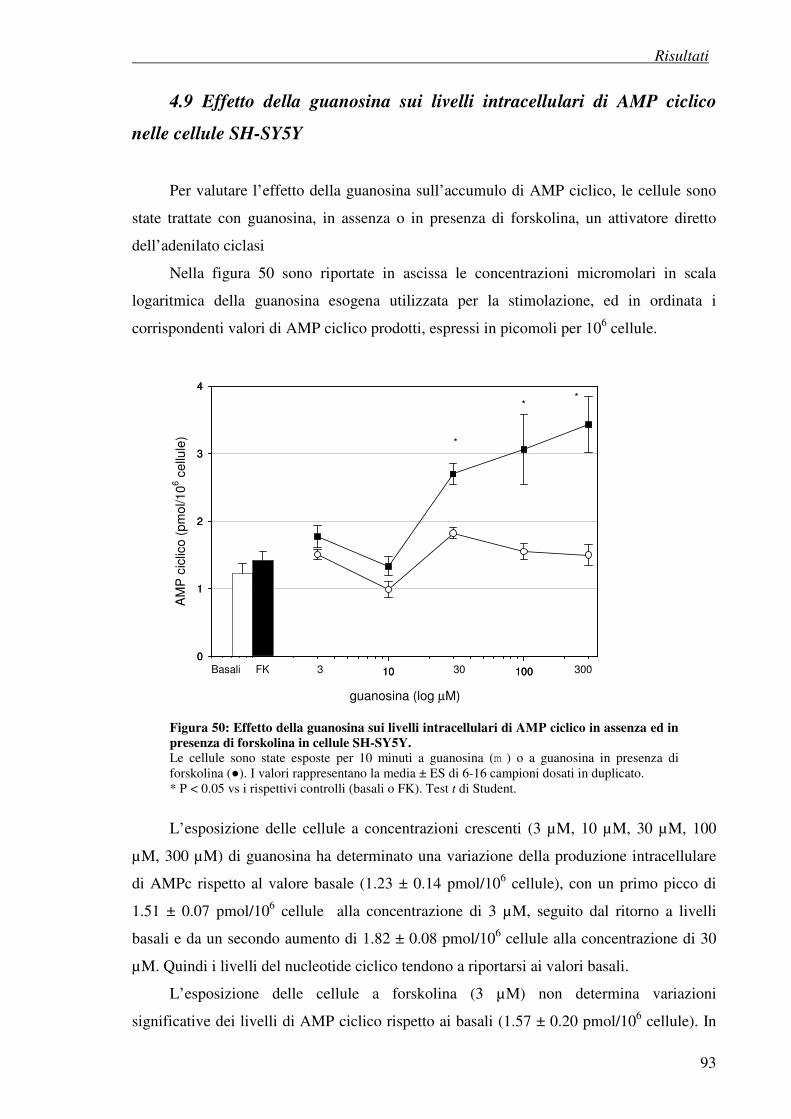
Risultati
93
4.9 Effetto della guanosina sui livelli intracellulari di AMP ciclico
nelle cellule SH-SY5Y
Per valutare l’effetto della guanosina sull’accumulo di AMP ciclico, le cellule sono
state trattate con guanosina, in assenza o in presenza di forskolina, un attivatore diretto
dell’adenilato ciclasi
Nella figura 50 sono riportate in ascissa le concentrazioni micromolari in scala
logaritmica della guanosina esogena utilizzata per la stimolazione, ed in ordinata i
corrispondenti valori di AMP ciclico prodotti, espressi in picomoli per 106 cellule.
guanosina (log µM)
1 10 100
0
1
2
3
4
1 10 100
AM
P c
iclic
o (
pm
ol/1
06 c
ellu
le)
0
1
2
3
4
3 30Basali FK
*
**
300
Figura 50: Effetto della guanosina sui livelli intracellulari di AMP ciclico in assenza ed in presenza di forskolina in cellule SH-SY5Y. Le cellule sono state esposte per 10 minuti a guanosina (m ) o a guanosina in presenza di forskolina (●). I valori rappresentano la media ± ES di 6-16 campioni dosati in duplicato. * P < 0.05 vs i rispettivi controlli (basali o FK). Test t di Student.
L’esposizione delle cellule a concentrazioni crescenti (3 µM, 10 µM, 30 µM, 100
µM, 300 µM) di guanosina ha determinato una variazione della produzione intracellulare
di AMPc rispetto al valore basale (1.23 ± 0.14 pmol/106 cellule), con un primo picco di
1.51 ± 0.07 pmol/106 cellule alla concentrazione di 3 µM, seguito dal ritorno a livelli
basali e da un secondo aumento di 1.82 ± 0.08 pmol/106 cellule alla concentrazione di 30
µM. Quindi i livelli del nucleotide ciclico tendono a riportarsi ai valori basali.
L’esposizione delle cellule a forskolina (3 µM) non determina variazioni
significative dei livelli di AMP ciclico rispetto ai basali (1.57 ± 0.20 pmol/106 cellule). In

Risultati
94
presenza di concentrazioni crescenti di guanosina (3 µM, 10 µM, 30 µM, 100 e 300 µM) si
osserva un aumento significativo dell’accumulo di AMPc intracellulare, rispetto ai valori
ottenuti in presenza di sola forskolina, a partire dalla concentrazione di 30 µM (2.70 ± 0.15
pmol/106 cellule) fino ad un massimo di 3.43 ± 0.41 pmol/106 cellule alla concentrazione
di guanosina 300 µM.
4.9.1 Effetto degli inibitori delle fosfodiesterasi rolipram e milrinone
sull’accumulo di AMP ciclico
L’effetto di concentrazioni equimolecolari (100 µM) di due inibitori delle
fosfodiesterasi, rolipram (PDE4) e milrinone (PDE3), sull’accumulo di AMP ciclico in
condizioni basali e in presenza di forkolina è riportato nella Tabella 1.
In condizioni basali, la presenza di 100 µM rolipram causa un aumento di circa 5
volte dei livelli di AMP ciclico (da 1.37 ± 0.10 a 6.91 ± 2.00 pmol/106 cellule, P<0.01),
mentre il milrinone non determina variazioni significative (1.26 ± 0.11 pmol/106 cellule).
In presenza di forskolina l’efficacia del rolipram aumenta, determinando un aumento dei
livelli del nucleotide ciclico di circa 37 volte (da 1.61 ± 0.06 a 61.3 ± 28 pmol/106 cellule,
P<0.01), mentre il milrinone determina un piccolo ma significativo aumento (P<0.05).
Tabella 1. Effetto degli inibitori delle fosfodiesterasi rolipram (PDE4) e milrinone (PDE3) sui livelli di AMPciclico in condizioni basali e in presenza di forskolina.
AMP ciclico
(pmol/106 cellule)
Basali 1.37±0.10
Rolipram 100 µM 6.91±2.00 P<0.01
Milrinone 100 µM
1.26±0.11 n.s
Forskolina 3 µM 1.61±0.06
Rolipram 100 µM 61.3±28 P<0.01
Milrinone 100 µM 2.21±0.15 P<0.05
I dati sono la media ±SEM di 3 esperimenti separati condotti in triplicato. ANOVA t-test seguito dal Tukey’s comparison test. n.s., non significativo

Risultati
95
4.9.2 Modulazione della sintesi di AMP ciclico su cellule SH-SY5Y differenziate
con guanosina o acido retinoico
Poiché la differenziazione è un fenomeno che porta ad una vasta riorganizzazione
cellulare, si è voluto valutare un’eventuale corrispondenza tra la differenziazione cellulare
e la produzione di AMP ciclico mediante esperimenti volti a quantificare la sintesi del
nucleotide ciclico intracellulare sia in cellule non-differenziate (ND), sia in cellule
differenziate per 48 ore con guanosina 150 µM (DIFF/GUO) o acido retinoico 10 µM
(DIFF/AR).
Gli esperimenti sono stati condotti su cellule differenziate con guanosina o con acido
retinoico, in assenza e in presenza di forskolina. Inoltre, in entrambe le condizioni è stato
valutato l’effetto dell’inibitore delle fosfodiesterasi-4 (PDE-4) rolipram.
- Differenziazione con guanosina
Nella figura 51 sono riportati i valori di AMPc espressi in pmoli/106cellule, ottenuti
stimolando per 10 minuti le cellule ND o DIFF/GUO con guanosina 100 µM in assenza o
presenza di rolipram 10 µM. Nelle cellule DIFF/GUO, i livelli basali di AMPc (2.19 ±
0.69) risultano significativamente aumentati (p<0.05) rispetto a quelli delle cellule ND
(1.35 ± 0.33). L’esposizione a guanosina 100 µM per 10 minuti non causa alcuna
variazione significativa dei livelli di AMPc rispetto ai valori basali relativi alle due
condizioni sperimentali (ND o DIFF/GUO). La presenza dell’inibitore delle PDE-4
rolipram determina un significativo incremento dei valori di AMPc, sia verso i basali ND
(p<0.001), sia verso i basali DIFF/GUO (p<0.05) (3.32 ± 1.04 e 4.11 ± 1.88,
rispettivamente).
L’associazione guanosina/rolipram produce nelle cellule differenziate un incremento
dei livelli di AMPc 10 volte maggiore rispetto a quanto osservato nelle cellule ND (29.17 ±
12.28 vs 3.88 ± 2.34; p<0.001).
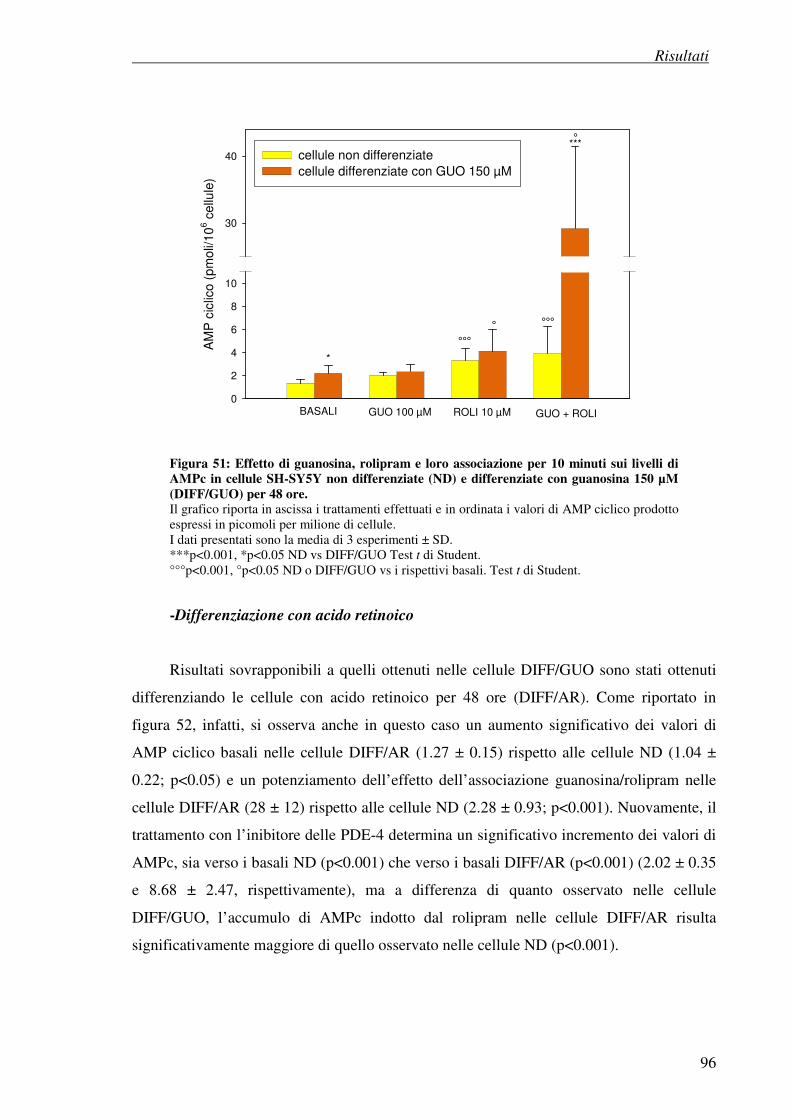
Risultati
96
AM
P c
iclic
o (
pm
oli/
10
6 c
ellu
le)
0
2
4
6
8
10
30
40
*
***
BASALI GUO 100 µM ROLI 10 µM GUO + ROLI
cellule non differenziate
cellule differenziate con GUO 150 µM
°°°
° °°°
°
Figura 51: Effetto di guanosina, rolipram e loro associazione per 10 minuti sui livelli di AMPc in cellule SH-SY5Y non differenziate (ND) e differenziate con guanosina 150 µM (DIFF/GUO) per 48 ore. Il grafico riporta in ascissa i trattamenti effettuati e in ordinata i valori di AMP ciclico prodotto espressi in picomoli per milione di cellule. I dati presentati sono la media di 3 esperimenti ± SD. ***p<0.001, *p<0.05 ND vs DIFF/GUO Test t di Student. °°°p<0.001, °p<0.05 ND o DIFF/GUO vs i rispettivi basali. Test t di Student.
-Differenziazione con acido retinoico
Risultati sovrapponibili a quelli ottenuti nelle cellule DIFF/GUO sono stati ottenuti
differenziando le cellule con acido retinoico per 48 ore (DIFF/AR). Come riportato in
figura 52, infatti, si osserva anche in questo caso un aumento significativo dei valori di
AMP ciclico basali nelle cellule DIFF/AR (1.27 ± 0.15) rispetto alle cellule ND (1.04 ±
0.22; p<0.05) e un potenziamento dell’effetto dell’associazione guanosina/rolipram nelle
cellule DIFF/AR (28 ± 12) rispetto alle cellule ND (2.28 ± 0.93; p<0.001). Nuovamente, il
trattamento con l’inibitore delle PDE-4 determina un significativo incremento dei valori di
AMPc, sia verso i basali ND (p<0.001) che verso i basali DIFF/AR (p<0.001) (2.02 ± 0.35
e 8.68 ± 2.47, rispettivamente), ma a differenza di quanto osservato nelle cellule
DIFF/GUO, l’accumulo di AMPc indotto dal rolipram nelle cellule DIFF/AR risulta
significativamente maggiore di quello osservato nelle cellule ND (p<0.001).
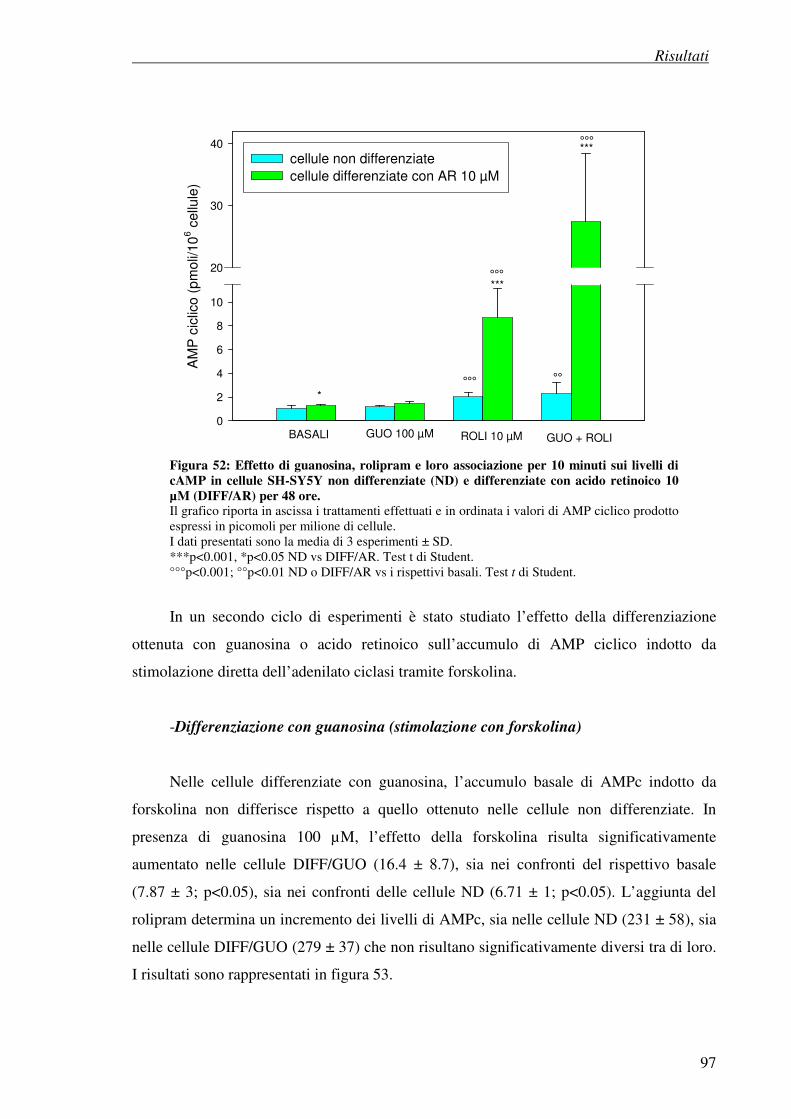
Risultati
97
BASALI GUO 100 µM ROLI 10 µM GUO + ROLI
AM
P c
iclic
o (
pm
oli/
10
6 c
ellu
le)
0
2
4
6
8
10
20
30
40
*
***
***
°°
°°°
cellule non differenziate
cellule differenziate con AR 10 µM
°°°
°°°
Figura 52: Effetto di guanosina, rolipram e loro associazione per 10 minuti sui livelli di cAMP in cellule SH-SY5Y non differenziate (ND) e differenziate con acido retinoico 10 µM (DIFF/AR) per 48 ore. Il grafico riporta in ascissa i trattamenti effettuati e in ordinata i valori di AMP ciclico prodotto espressi in picomoli per milione di cellule. I dati presentati sono la media di 3 esperimenti ± SD. ***p<0.001, *p<0.05 ND vs DIFF/AR. Test t di Student. °°°p<0.001; °°p<0.01 ND o DIFF/AR vs i rispettivi basali. Test t di Student.
In un secondo ciclo di esperimenti è stato studiato l’effetto della differenziazione
ottenuta con guanosina o acido retinoico sull’accumulo di AMP ciclico indotto da
stimolazione diretta dell’adenilato ciclasi tramite forskolina.
-Differenziazione con guanosina (stimolazione con forskolina)
Nelle cellule differenziate con guanosina, l’accumulo basale di AMPc indotto da
forskolina non differisce rispetto a quello ottenuto nelle cellule non differenziate. In
presenza di guanosina 100 µM, l’effetto della forskolina risulta significativamente
aumentato nelle cellule DIFF/GUO (16.4 ± 8.7), sia nei confronti del rispettivo basale
(7.87 ± 3; p<0.05), sia nei confronti delle cellule ND (6.71 ± 1; p<0.05). L’aggiunta del
rolipram determina un incremento dei livelli di AMPc, sia nelle cellule ND (231 ± 58), sia
nelle cellule DIFF/GUO (279 ± 37) che non risultano significativamente diversi tra di loro.
I risultati sono rappresentati in figura 53.
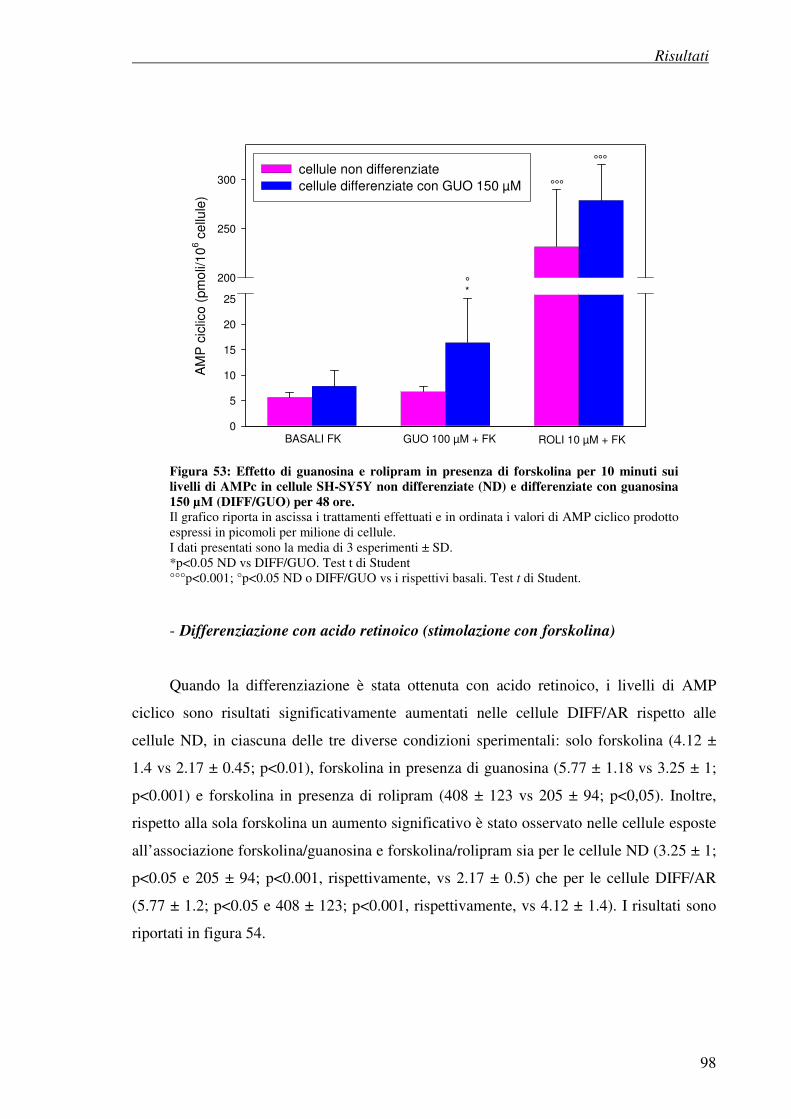
Risultati
98
AM
P c
iclic
o (
pm
oli/
10
6 c
ellu
le)
0
5
10
15
20
25
200
250
300
*
BASALI FK GUO 100 µM + FK ROLI 10 µM + FK
°
°°°
°°°
cellule non differenziate
cellule differenziate con GUO 150 µM
Figura 53: Effetto di guanosina e rolipram in presenza di forskolina per 10 minuti sui livelli di AMPc in cellule SH-SY5Y non differenziate (ND) e differenziate con guanosina 150 µM (DIFF/GUO) per 48 ore. Il grafico riporta in ascissa i trattamenti effettuati e in ordinata i valori di AMP ciclico prodotto espressi in picomoli per milione di cellule. I dati presentati sono la media di 3 esperimenti ± SD. *p<0.05 ND vs DIFF/GUO. Test t di Student °°°p<0.001; °p<0.05 ND o DIFF/GUO vs i rispettivi basali. Test t di Student.
- Differenziazione con acido retinoico (stimolazione con forskolina)
Quando la differenziazione è stata ottenuta con acido retinoico, i livelli di AMP
ciclico sono risultati significativamente aumentati nelle cellule DIFF/AR rispetto alle
cellule ND, in ciascuna delle tre diverse condizioni sperimentali: solo forskolina (4.12 ±
1.4 vs 2.17 ± 0.45; p<0.01), forskolina in presenza di guanosina (5.77 ± 1.18 vs 3.25 ± 1;
p<0.001) e forskolina in presenza di rolipram (408 ± 123 vs 205 ± 94; p<0,05). Inoltre,
rispetto alla sola forskolina un aumento significativo è stato osservato nelle cellule esposte
all’associazione forskolina/guanosina e forskolina/rolipram sia per le cellule ND (3.25 ± 1;
p<0.05 e 205 ± 94; p<0.001, rispettivamente, vs 2.17 ± 0.5) che per le cellule DIFF/AR
(5.77 ± 1.2; p<0.05 e 408 ± 123; p<0.001, rispettivamente, vs 4.12 ± 1.4). I risultati sono
riportati in figura 54.
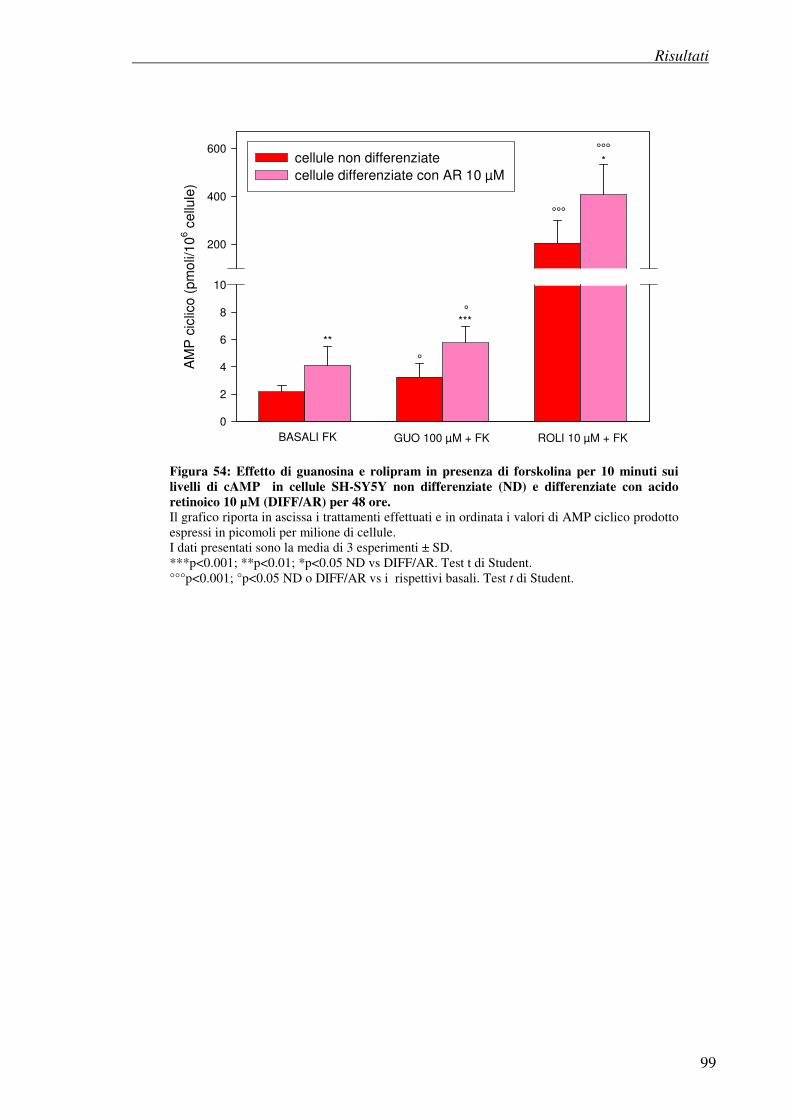
Risultati
99
AM
P c
iclic
o (
pm
oli/
10
6 c
ellu
le)
0
2
4
6
8
10
200
400
600
**
***
*
BASALI FK GUO 100 µM + FK ROLI 10 µM + FK
°
°
°°°
°°°cellule non differenziate
cellule differenziate con AR 10 µM
Figura 54: Effetto di guanosina e rolipram in presenza di forskolina per 10 minuti sui livelli di cAMP in cellule SH-SY5Y non differenziate (ND) e differenziate con acido retinoico 10 µM (DIFF/AR) per 48 ore. Il grafico riporta in ascissa i trattamenti effettuati e in ordinata i valori di AMP ciclico prodotto espressi in picomoli per milione di cellule. I dati presentati sono la media di 3 esperimenti ± SD. ***p<0.001; **p<0.01; *p<0.05 ND vs DIFF/AR. Test t di Student. °°°p<0.001; °p<0.05 ND o DIFF/AR vs i rispettivi basali. Test t di Student.

Risultati
100
4.10 Determinazione dei livelli intracellulari di calcio
E’ stata effettuata una serie di esperimenti, condotti in collaborazione con la Prof.ssa
P. Lorenzon del nostro Dipartimento, volti a valutare la capacità della guanosina di
modulare i livelli di calcio intracellulare.
In figura 55, è riportato in ascissa la concentrazione di guanosina più alta utilizzata e
i controlli stimolati con sola fisiologica. In ordinata invece vediamo riportata la percentuale
di cellule che hanno dato liberazione di calcio nei 15 minuti di osservazione. Il 100% delle
cellule osservate ha risposto positivamente alla stimolazione con carbacolo (dati non
mostrati). Vediamo come la guanosina invece, fino alla concentrazione di 1 mM, non abbia
aumentato in maniera statisticamente significativa la concentrazione di calcio
intracellulare.
C GUO 1 mM
ce
llu
le c
on
att
ivazio
ne
di C
a+
2 (
%)
0
10
20
30
40
50
Figura 55: Aumento dei livelli di calcio in seguito a stimolazione con guanosina 1 mM. I dati sono espressi come % di cellule attive su cellule totali e sono state osservate circa 400 cellule.
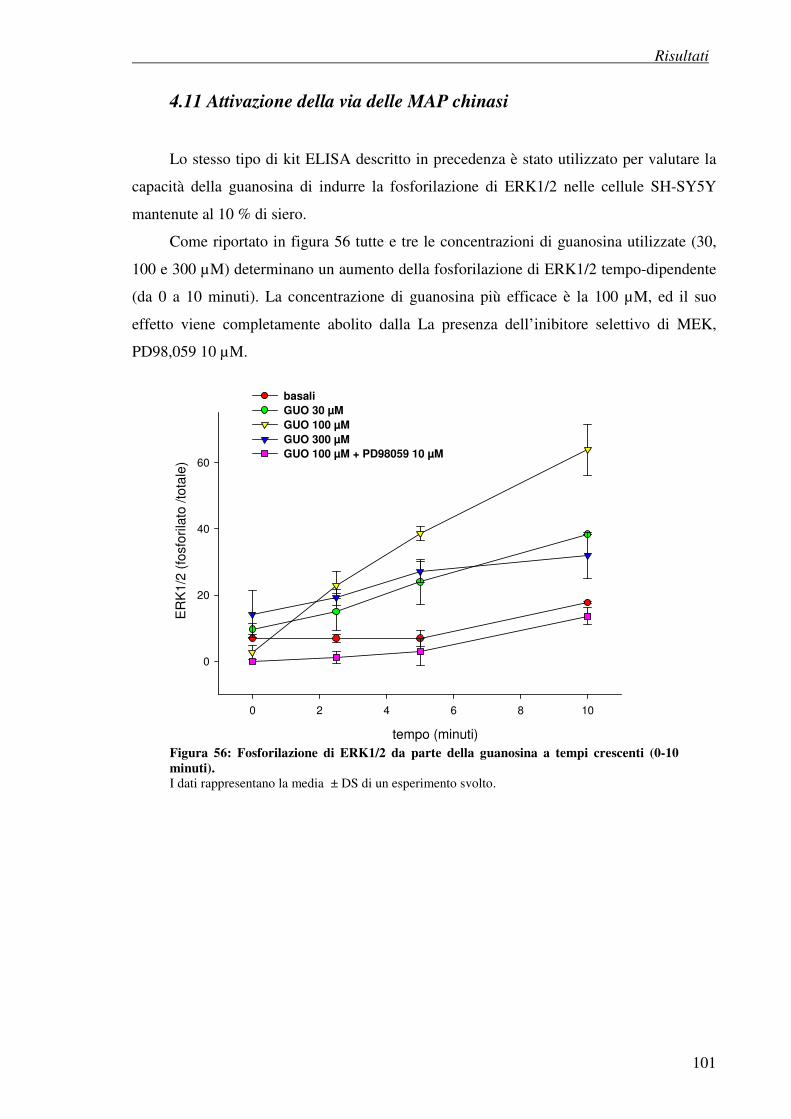
Risultati
101
4.11 Attivazione della via delle MAP chinasi
Lo stesso tipo di kit ELISA descritto in precedenza è stato utilizzato per valutare la
capacità della guanosina di indurre la fosforilazione di ERK1/2 nelle cellule SH-SY5Y
mantenute al 10 % di siero.
Come riportato in figura 56 tutte e tre le concentrazioni di guanosina utilizzate (30,
100 e 300 µM) determinano un aumento della fosforilazione di ERK1/2 tempo-dipendente
(da 0 a 10 minuti). La concentrazione di guanosina più efficace è la 100 µM, ed il suo
effetto viene completamente abolito dalla La presenza dell’inibitore selettivo di MEK,
PD98,059 10 µM.
tempo (minuti)
0 2 4 6 8 10
ER
K1
/2 (
fosfo
rila
to /to
tale
)
0
20
40
60
basali
GUO 30 µM
GUO 100 µM
GUO 300 µM
GUO 100 µM + PD98059 10 µM
Figura 56: Fosforilazione di ERK1/2 da parte della guanosina a tempi crescenti (0-10 minuti). I dati rappresentano la media ± DS di un esperimento svolto.

Risultati
102
4.12 Attivazione della via di PI3K
Gli studi sull’attivazione della via di PI3K durante gli eventi differenzianti indotti da
acido retinoico e guanosina sono stati condotti con la tecnica del Western blot e sono
ancora in via di svolgimento. Dati preliminari indicano che il trattamento di 1 ora con
acido retinoico aumenta la fosforilazione di Akt rispetto ai controlli (n=3). Per quanto
riguarda la guanosina, la conferma dell’attivazione di questa via richiede ulteriori
esperimenti, in particolare per la caratterizzazione della tempo-dipendenza.
Figura 57: Fosforilazione di Akt da parte dell’acido retinoico. L’immagine è rappresentativa di uno di tre esperimenti.
Controlli Guo 1h A.R. 1h
60 KDa P-Akt
actina 40 KDa

5. Discussione

Discussione
103
Da questo studio sono emerse diverse evidenze sulla capacità della guanosina di
esercitare un effetto protettivo su cellule derivate da un neuroblastoma umano, la linea
cellulare SH-SY5Y. Questo effetto si manifesta con la capacità della guanosina, da una
parte, di opporsi ai danni indotti da deprivazione da siero, e dall’altra di indurre
differenziazione in cellule mantenute in condizioni di coltura ottimali.
Guanosina e deprivazione da siero
Nelle cellule SH-SY5Y sottoposte a deprivazione da siero, l’esposizione a guanosina
è in grado di prevenire la comparsa di alterazioni morfologiche, indice di sofferenza
cellulare, osservate nelle cellule sottoposte a stress da starvation. Questo effetto si
accompagna ad un aumento della proliferazione cellulare, come evidenziato con il test
dell’MTT. Infatti dopo 24, 48 e 72 ore di starvation, la presenza di concentrazioni
crescenti di guanosina esercita un’azione protettiva, con un aumento della vitalità cellulare
che raggiunge un massimo alle concentrazioni di 75 e 100 µM (+35-40 %). E’ da notare
che l’andamento dell’effetto rispetto alle concentrazioni assume un aspetto “a campana” e
non quello sigmoidale, classico delle curve concentrazione-risposta. L’ipotesi che può
essere avanzata è che la guanosina eserciti la propria azione interagendo con bersagli
diversi, o ad attività contrastante tra di loro, al crescere della concentrazione,
comportandosi quindi più da modulatore dell’attività cellulare che da molecola-segnale.
La capacità della guanosina di indurre proliferazione delle cellule SH-SY5Y
sottoposte a stress da carenza da siero è stata confermata da studi di citofluorimetria
condotti in collaborazione con la Dr.ssa Pacor del Dipartimento di Scienze della Vita

Discussione
104
dell’Università di Trieste. In questi esperimenti la guanosina si è dimostrata in grado di
aumentare la percentuale di cellule in fase di sintesi e di ridurre la percentuale di cellule in
fase G0-G1. Sugli stessi campioni su cui è stata effettuata l’analisi del ciclo cellulare, è
stata fatta la conta delle cellule che ha confermato un aumento del numero delle stesse.
In studi condotti su astrociti primari di ratto in coltura, è stato dimostrato che l’azione
proliferativa della guanosina è dipendente dalla capacità del nucleoside di stimolare la
sintesi ed il rilascio di fattori trofici, mediata dall’attivazione delle mitogen-activated
protein kinase (MAPK) (Di Iorio et al., 2001). Inoltre è stato dimostrato che la guanosina è
anche in grado di attivare la via della phosphoinositide 3-kinase (PI3K) che, a sua volta,
innesca le vie di sopravvivenza cellulare (Di Iorio et al., 2004). La capacità degli inibitori
specifici delle vie delle MAPK e di PI3K di abolire completamente l’effetto proliferativo
della guanosina, fornisce una prova indiretta del coinvolgimento di queste vie di
trasduzione del segnale nell’azione della guanosina anche a livello neuronale.
Il coinvolgimento della via di MAPK è stato ulteriormente indagato misurando
l’entità di fosforilazione di ERK1/2 in cellule mantenute in starvation per 24 ore. In questo
caso, la concentrazione di guanosina 100 µM è risultata essere la sola efficace, con un
incremento significativo dopo 2,5-5 minuti di esposizione, effetto totalmente abolito dalla
presenza dell’inibitore PD98059. La presenza dell’inibitore ha inoltre ridotto in modo
significativo la percentuale di ERK fosforilato non solo nelle cellule esposte a guanosina,
ma anche nelle cellule utilizzate come controlli, suggerendo che nelle cellule SH-SY5Y la
deprivazione da siero porti ad attivazione delle vie di sopravvivenza cellulare mediate da
ERK1/2, al pari di quanto osservato in cellule di carcinoma del colon umano (Jung et al.,
1999). Sull’attività di questa via, quindi, la guanosina sembra esercitare un effetto
facilitatorio.
Un’altra via la cui attivazione è correlata ad effetti neuroprotettivi è quella
dell’adenilato ciclasi. Infatti, l’AMP ciclico è un potente attivatore di CREB (cyclic AMP
response element binding protein), un fattore di trascrizione che svolge un ruolo
importante nella sopravvivenza neuronale. Ad esempio, è stato dimostrato che nella linea
SH-SY5Y il nucleotide ciclico è in grado di proteggere le cellule da stress ossidativo
(Chalovich, 2006). Sono stati quindi eseguiti esperimenti volti a caratterizzare l’effetto
della guanosina sui livelli intracellulari di AMP ciclico.
In condizioni di coltura standard l’esposizione delle cellule SH-SY5Y a
concentrazioni crescenti di guanosina (da 3 a 300 µM) non ha determinato variazioni
significative dei livelli di AMP ciclico. Viceversa, in condizioni di starvation si è osservata

Discussione
105
una lieve ma significativa riduzione, con un andamento oscillante. Questi cambiamenti
nella risposta al nucleoside possono essere conseguenza degli importanti cambiamenti che
si instaurano a livello cellulare durante la deprivazione da siero. Numerosi sono i dati
presenti in letteratura che riportano variazioni dell’espressione di proteine recettoriali,
effettrici e/o regolatorie in seguito ad esposizione a condizioni stressanti. Ad esempio,
nelle cellule SH-SY5Y uno stress metabolico è in grado di variare l’espressione di proteine
regolatrici dell’attività di proteine G, in particolare di RGS2 e RGS4 (Song e Jope, 2006).
Anche l’attività delle adenilato ciclasi (AC) è regolata da una classe di proteine di recente
identificazione, denominate AGS, in grado di influenzare lo stato di attivazione delle
proteine G in modo recettore-indipendente (Lanier, 2004). Una variazione di espressione di
queste proteine in condizioni di stress è stata recentemente dimostrata nel cuore ischemico
(Sato et al., 2006). La diversa risposta alla guanosina in condizioni di starvation potrebbe
quindi essere conseguenza della variazione dei livelli di proteine regolatrici RGS e/o
AGS1.
La condizione di starvation, viceversa, non ha influito sulla capacità della guanosina
di potenziare l’accumulo di AMP ciclico indotto da forskolina. Un simile potenziamento
dell’attività del diterpene è stato descritto, sempre nelle cellule SH-SY5Y, da parte del
carbacolo, che di per sé induce al pari della guanosina solo un leggero aumento dei livelli
di cAMP (Nakagawa-Yagi et al., 1991). Tale potenziamento è stato attribuito
all’attivazione di chinasi calcio-calmodulina dipendenti. Allo stesso modo, anche la
stimolazione dell’adenilato ciclasi da parte della melatonina nelle cellule SH-SY5Y è
risultata dipendente dal calcio intracellulare (Schuster et al., 2005). Nel caso della
guanosina, esperimenti di calcium imaging hanno messo in evidenza solo leggere e non
significative fluttuazioni dei livelli di Ca2+ intracellulare in seguito ad esposizione al
nucleoside e ciò rende necessaria un’ipotesi diversa. Questa potrebbe risiedere nella
capacità di recettori accoppiati a proteine GI/O, di stimolare l’attività di adenilato ciclasi
tramite il dimero βγ. L’incertezza dell’esistenza o meno di un recettore specifico per la
guanosina rende estremamente speculativa questa discussione. E’ però importante
ricordare che un sito di legame specifico per l’[3H]-guanosina è stato recentemente
caratterizzato in membrane di cervello di ratto (Traversa et al., 2002) ed in colture primarie
di astrociti di ratto (Traversa et al., 2003) che è risultato sensibile alla tossina della
pertosse.
Questi risultati, nell’insieme, confermano che la guanosina è in grado di aumentare la
vitalità e la proliferazione sulle cellule SH-SY5Y poste in condizioni di deprivazione da

Discussione
106
siero e suggeriscono che questa possa agire come molecola endogena potenzialmente
partecipe alla regolazione del delicato bilancio tra fattori che promuovono danno cellulare
e fattori ad attività protettiva. Il meccanismo tramite cui il nucleoside esercita tale azione
resta ancora da definire, ma sembra essere correlato ad una breve attivazione delle via di
MAPK.
Guanosina e trasportatori
Al pari dell’adenosina, la guanosina è presente a livello extracellulare e la loro
concentrazione aumenta dopo rilascio da cellule gliali e neuronali in condizioni
patologiche (Rathbone et. Al, 1999). In generale, i livelli extracellulari delle purine sono
governati da sistemi di trasporto specifici che funzionano secondo gradiente (equilibrativi)
o con meccanismo concentrativo, che quindi costituiscono degli importanti regolatori della
durata e dell’intensità degli effetti mediati dalle purine stesse. La seconda parte di questo
studio è stata quindi rivolta alla determinazione dell’espressione nella linea cellulare SH-
SY5Y, di trasportatori per i nucleosidi adenosina e guanosina ed alla loro caratterizzazione.
Tramite esperimenti di binding è stata dimostrata la capacità del bloccante dei
trasportatori equilibrativi, NBTI, di legarsi in modo saturabile e reversibile sia a cellule
SH-SY5Y intatte, sia a preparazioni di membrana. Questa è, a nostra conoscenza, la prima
evidenza della presenza di carrier NBTI-sensibili nelle cellule di neuroblastoma umano. Il
valore di Kd ottenuto (0.83 ± 0.048 nM) è inoltre ampiamente in accordo con dati riportati
in letteratura, ad esempio, su cellule cromaffini (0.56 ± 0.2 nM) (Delicado et al., 1990).
Sono invece risultati assenti i trasportatori concentrativi, in quanto non si sono rilevate
differenze significative nell’uptake dell’adenosina triziata in assenza di ioni sodio. In
ambedue le condizioni (in presenza e in assenza di sodio), l’uptake dell’adenosina è
risultato sensibile al blocco da NBTI, come atteso, mentre la contemporanea presenza di
guanosina 100 µM ha determinato solo una tendenza alla riduzione dell’uptake, che non ha
raggiunto la significatività. Questo risultato, comunque, non implica che i due composti
non utilizzino lo stesso trasportatore per accedere all’interno della cellula. Infatti, con
esperimenti di competizione è stato dimostrato che, oltre all’adenosina, anche la guanosina
è in grado di determinare uno spiazzamento concentrazione-dipendente dell’NBTI
marcato, ma con un’affinità 25 volte inferiore (IC50 di circa 1.5 mM per la guanosina
rispetto ad una IC50 di circa 70 µM per l’adenosina). Quindi, la mancata competizione tra
adenosina e guanosina osservata negli esperimenti volti a definire la presenza o meno dei
trasportatori concentrativi è riferibile piuttosto alla bassa concentrazione di guanosina

Discussione
107
usata. La capacità delle due purine di utilizzare gli stessi trasportatori, seppure con diversa
efficienza, è stata riportata utilizzando ceppi di lievito mutati per i trasportatori hENT1
(Endres e Unadkat, 2005).
Le cinetiche di uptake dell’adenosina e della guanosina sono risultate ampiamente
sovrapponibili, con un incremento lineare fino a 300 secondi di esposizione ai substrati
marcati. Questo risultato indica che viene mantenuto il gradiente che favorisce il
movimento dei nucleosidi dall’esterno verso l’interno della cellula, anche dopo tempi
relativamente lunghi, e che il mantenimento del gradiente è possibilmente legato
all’utilizzo dei substrati a livello intracellulare.
E’ importate ricordare che sia i neuroni che le cellule gliali rilasciano purine in
condizioni fisiologiche e patologiche. In stato di riposo, la guanosina viene rilasciata in
quantità pari a 1 volta e mezza rispetto all’adenosina, mentre in condizioni di stress, come
accade nell’ipoglicemia o nell’ipossia cerebrale, ne aumenta ulteriormente il rilascio,
arrivando ad una quantità dieci volte superiore a quella dell’adenosina (Rathbone et al.,
1999).
Queste considerazioni sono alla base della scelta di concentrazioni diverse (1 µM per
ADO e 100 µM per GUO) per lo studio dell’uptake. Può essere interessante ricordare che
anche l’espressione dei trasportatori equilibrativi viene modulata dalla disponibilità di
ossigeno (Eltzschig et al., 2005).
I risultati sopra riportati presentano anche un risvolto pratico per quanto riguarda
l’identificazione di putativi recettori selettivi per la guanosina. La cinetica lenta dell’uptake
e, soprattutto la bassa affinità del trasporto, permettono di impiegare anche cellule intatte
per studi di binding, in quanto il contributo delle proteine trasportatrici nei confronti dei
siti recettoriali risulta trascurabile sulla base delle concentrazioni molto basse (nanomolari)
impiegate negli esperimenti di binding.
Guanosina e differenziazione
Durante lo svolgimento degli studi condotti sulle cellule SH-SY5Y sottoposte a
stress da deprivazione da siero, è stata osservata nelle cellule di controllo, mantenute in
condizioni ottimali di coltura, la capacità della guanosina di determinare cambiamenti
morfologici interpretabili come attivazione di processi di differenziazione neuronale.
Questo aspetto è stato quindi approfondito impiegando come controllo positivo l’acido
retinoico, di cui è nota la capacità di indurre differenziazione anche nelle cellule SH-SY5Y
(Ammer e Schultz, 1994).

Discussione
108
L’effetto differenziante è stato quantizzato sia come percentuale di cellule
differenziate rispetto alle cellule totali, sia come lunghezza dei neuriti in µm con l’impiego
di un programma computerizzato. La capacità della guanosina di indurre differenziazione è
risultata essere in larga parte sovrapponibile a quella del composto di riferimento, l’acido
retinoico. In ambedue i casi, infatti, si è osservato un aumento significativo del numero
delle cellule differenziate, in rapporto al numero di cellule totali, e della lunghezza dei
neuriti. L’effetto differenziante ha insorgenza precoce, essendo evidente già dopo 24 ore di
trattamento e, nel caso dell’esposizione al nucleoside, è risultato concentrazione-
dipendente rispetto alle tre concentrazioni utilizzate (10, 75 e 150 µM). Dopo 72 ore di
trattamento, la lunghezza dei neuriti delle cellule considerate come differenziate, cioè
aventi almeno un prolungamento di lunghezza pari o superiore a due volte il corpo
cellulare, era simile nelle cellule differenziate in presenza della concentrazione più alta di
guanosina (150 µM) e in quelle differenziate con l’acido retinoico (10 µM).
E’ stato riportato che la guanosina è in grado di stimolare gli astrociti a rilasciare
adenosina (Ciccarelli et al., 2001), che a sua volta contribuisce all’instaurarsi dei processi
riparativi a livello neuronale per attivazione dei recettori A1 e A2A. E’ stato dimostrato che
questi stessi recettori sono in grado di indurre differenziazione delle cellule SH-SY5Y
(Canals et al., 2005). Per determinare se l’effetto differenziante della guanosina fosse una
conseguenza del rilascio di adenosina indotto da guanosina, e quindi dell’attivazione del
sistema adenosinico, abbiamo utilizzato due diversi approcci. Nel primo caso è stato
valutato l’effetto dell’adenosina endogena tramite blocco del trasportatore equilibrativo
NBTI-sensibile, cui segue l’accumulo di adenosina endogena a livello extracellulare, e
tramite rimozione dell’adenosina endogena per l’aggiunta dell’enzima adenosina
deaminasi, che degrada l’adenosina ad inosina. Quest’ultimo composto è privo di affinità
nei confronti dei recettori A1 e A2A. Sulla base della conta delle cellule differenziate è
risultato che l’effetto differenziante indotto dalla guanosina 150 µM non ha subito
modificazioni significative. Nel secondo tipo di approccio è stato valutato direttamente il
coinvolgimento dei recettori adenosinici A1 e A2A, impiegando due antagonisti selettivi,
rispettivamente DPCPX e DMPX. Anche questa serie di esperimenti ha confermato il
mancato coinvolgimento dei recettori P1 nell’effetto differenziante della guanosina.
Per caratterizzare meglio, da un punto di visto molecolare, l’effetto differenziante
della guanosina, abbiamo effettuato degli studi di immunocitochimica utilizzando la
concentrazione di guanosina 150 µM che aveva dato i risultati paragonabili a quelli indotti
dall’acido retinoico e andando a valutare la variazione di espressione di alcuni markers

Discussione
109
tipicamente neuronali come β-tubulina e MAP2. L’espressione e la riorganizzazione della
β-tubulina è già evidente dopo 48 ore di esposizione a guanosina e aumenta in maniera
tempo-dipendente a 72 e 96 ore. L’espressione della proteina associata ai microtubuli
MAP2, che generalmente si manifesta meno precocemente rispetto alla β-tubulina,
aumenta in maniera evidente rispetto ai controlli, nei quali l’espressione è molto bassa,
dopo 96 ore di trattamento con guanosina.
Come controllo negativo interno è stato infine utilizzato un anticorpo verso una
proteina nucleare neuronale specifica denominata NeuN (Neuronal Nuclei), impiegata per
l’identificazione del fenotipo neuronale. Nel confronto tra la colorazione dei nuclei
evidenziati con DAPI e con NeuN si è riscontrata un’ampia sovrapposizione, sia nelle
cellule di controllo, sia in quelle differenziate. Nelle cellule differenziate, inoltre, il livello
di espressione sembra aumentato, rispetto ai controlli, in accordo con uno stato di
differenziazione più avanzato (Mullen et al, 1992).
Oltre alla comparsa di marker tipici di cellule neuronali, la differenziazione si
accompagna a cambiamenti morfologici dovuti a riorganizzazione delle proteine del
citoscheletro. I cambiamenti della disposizione dei filamenti di actina sono stati identificati
utilizzando il composto falloidina, che ha permesso di evidenziare come la differenziazione
indotta da guanosina sia un processo che si protrae almeno fino a 7 giorni di trattamento.
Dopo un tempo così prolungato, l’intreccio e l’estensione dei neuriti è tale da non
permettere la valutazione visiva del numero di cellule differenziate o della lunghezza delle
estensioni.
Il meccanismo con cui la guanosina induce differenziazione non è ancora chiaro, ma
a questo proposito possono essere fatte alcune considerazioni. E’ noto che l’acido
retinoico, di cui è conosciuta la capacità di influenzare lo sviluppo, il mantenimento e la
rigenerazione neuronale, esercita i suoi effetti per interazione con specifici recettori
appartenenti alla superfamiglia dei recettori intranucleari, fattori di trascrizione in grado di
modulare l’espressione di oltre 500 geni (Maden, 2007). E’ stato anche dimostrato che
nelle cellule SH-SY5Y la differenziazione indotta dall’acido retinoico è preceduta
dall’attivazione di due vie importanti per la sopravvivenza cellulare, la via delle MAP
chinasi (Singh et al., 2003) e la via di PI3K/Akt/PKB (Lopez-Carballo et al., 2002). Nel
caso della guanosina, la sua capacità di attivare le stesse vie è stata dimostrata sia nelle
cellule SH-SY5Y (Pettifer et al., 2007; Zanette, 2007), sia in astrociti (Di Iorio et al.,
2004), e i dati presentati in questa tesi dimostrano la capacità degli inibitori delle vie
MAKP e PI3K di abolire l’espressione della β-tubulina, e quindi la differenziazione indotta

Discussione
110
da guanosina, dopo 72 ore di esposizione. Inoltre, i presenti risultati indicano che la
guanosina è in grado di aumentare la fosforilazione di ERK1/2 e, possibilmente, anche di
Akt. Nonostante tutto, questi dati non costituiscono una dimostrazione inequivocabile della
presenza di un recettore specifico per il nucleoside guaninico nelle stesse cellule e non
danno indicazioni sul meccanismo tramite cui vengono innescate queste vie di
sopravvivenza. In ogni caso, la possibilità che l’effetto sia mediato da un recettore
intracellulare, come nel caso dell’acido retinoico, appare remota, considerato che questo
tipo di meccanismo è in generale circoscritto a molecole lipofile, in grado di permeare la
membrana plasmatica. Per la sua struttura idrofilica, invece, la guanosina, al pari
dell’adenosina, passa dall’ambiente extracellulare a quello intracellulare tramite proteine di
membrana di tipo equilibrativo, che operano il trasporto dei nucleosidi secondo gradiente.
Per quanto riguarda la guanosina, essa è presente a livello extracellulare a concentrazioni
superiori, rispetto all’adenosina, e la sua affinità verso i trasportatori è di almeno due ordini
inferiore. Queste diversità, se da un lato rafforzano l’ipotesi di un meccanismo d’azione
extracellulare per la guanosina, sottolineano anche la diversità di ruolo tra le due molecole:
l’adenosina quale molecola sentinella per stati di emergenza quali l’ipossia, la guanosina
come modulatore trofico.
Un dato interessante è emerso con l’impiego del composto H89, considerato un
inibitore delle protein-chinasi AMP ciclico-dipendenti. Già di per sé, H89 ha indotto un
aumento dell’espressione della β-tubulina, paragonabile a quella prodotta della guanosina
stessa. Questo effetto può essere attribuito alla scarsa selettività del composto, di cui è stata
dimostrata la capcità di inibire almeno 6 diversi tipi di chinasi con la stessa affinità, se non
superiore, con cui inibisce la PKA. Ad esempio H89 è in grado di inibire le RhoA,
RhoGTPasi in grado di modificare il citoscheletro (Lochner e Moolman, 2006).
La valutazione dell’effetto differenziante in cellule simil-neuronali viene solitamente
effettuata come riportato nella presente tesi, vale a dire con la conta visiva delle cellule la
cui morfologia è caratterizzata dalla presenza di processi dendritici e con la misurazione
della lunghezza degli stessi. Inoltre, con tecniche di immunoistochimica è possibile
evidenziare la presenza di specifici marker neuronali. Nessuno di questi metodi, però,
permette di quantificare la potenza dell’effetto differenziante poiché la gradualità della
risposta rispetto alla concentrazione usata è difficile, se non impossibile, da misurare.
Abbiamo quindi fatto ricorso alla correlazione inversa esistente tra differenziazione e
proliferazione, due fenomeni che si escludono a vicenda. Infatti, durante la
differenziazione le cellule vanno incontro ad una profonda riorganizzazione che comporta,

Discussione
111
oltre a cambiamenti morfologici, un diverso assetto di proteine strutturali, delle vie di
trasduzione del segnale e delle interazioni tra cellule, nonché e principalmente il ciclo
cellulare, che viene bloccato in fase G1 (Encinas et al., 2000). Di conseguenza, cellule di
origine neuronale indotte a differenziarsi cessano di proliferare. Abbiamo quindi impiegato
un test colorimetrico per il quale il valore di assorbanza è indice diretto della massa
cellulare, il test della sulforodamina B, che permette quindi di apprezzare la relazione
esistente tra concentrazione del composto differenziante e l’effetto sulla differenziazione
stessa delle cellule, cioè della biomassa. Con questo metodo, dopo 48 ore di esposizione a
guanosina e acido retinoico, una riduzione della proliferazione è evidente solo alle
concentrazioni più alte utilizzate, un risultato comunque congruo con il tempo di raddoppio
della linea cellulare, che è di 48 ore. Dopo esposizione agli agenti differenzianti per 7
giorni, si è potuta invece osservare una riduzione della proliferazione che è risultata
graduale e dipendente dalla concentrazione dell’agente differenziante. In queste
condizioni, quindi, è stato possibile determinare i valori di IC50. che sono risultati pari a
172 ± 113 µM per la guanosina e 0.20 ± 0.15 µM per l’acido retinoico. E’ da sottolineare
l’assenza di effetti tossici dovuti all’esposizione delle cellule alle concentrazioni più
elevate dei due composti in esame, dimostrato dal fatto che i valori di assorbanza non sono
mai risultati inferiori a quelli dei controlli al momento della semina.
Utilizzando il test della sulforodamina B è stata valutata anche la capacità di derivati
della guanosina come guanina e GTP, e di una analogo di sintesi, l’AIT-082, di mimarne
l’effetto.
Né GTP, né il composto AIT-082 sono stati in grado di ridurre la proliferazione
cellulare fatta eccezione per la concentrazione più alta di GTP. Per quanto riguarda invece
la guanina, il suo effetto antiproliferativo è risultato tempo- e concentrazione-dipendente
con un valore di IC50 a sette giorni di trattamento, pari a 190 ± 17 µM, valore
sovrapponibile a quello ottenuto in seguito ad esposizione a guanosina. Sulla base di
questo risultato si è tentati di ipotizzare che almeno in parte l’effetto differenziante indotto
dalla guanosina possa essere dovuto al suo metabolismo a guanina da parte di una ecto-
PNP, la cui presenza nelle cellule SH-SY5Y resta ancora da dimostrare. In ogni caso,
l’evidenza dell’espressione di PNP, ottenuta con la tecnica PCR, costituisce un’indicazione
iniziale. Questi dati sono comunque in accordo con quanto riportato da Watanabe et at.,
che hanno osservato un effetto neuroprotettivo di guanosina e guanina su neuroni
cerebellari della stessa entità e a concentrazioni equimolecolari (Watanabe et al, 2003).
Oltre all’acido retinoico, altri agenti in grado di indurre differenziazione

Discussione
112
comprendono il forbolo miristato acetato, attivatore della protein chinasi C, e il dibutirril
AMPc, analogo del AMPc in grado di attraversare le membrane. Nelle cellule SH-SY5Y,
un aumento dei livelli di AMP ciclico è stato spesso messo in relazione alla comparsa di
differenziazione (Sanchez et al., 2004; Monaghan et al., 2008) ed è stato riportato che nelle
cellule SH-SY5Y differenziate con acido retinoico l’attività basale dell’adenilato ciclasi e i
livelli di AMP ciclico erano superiori, rispetto alle cellule non differenziate (Ammer e
Schultz, 1994). Per questo motivo, abbiamo indagato se simili effetti sul nucleotide ciclico
potessero essere indotti anche dalla guanosina, in condizioni in cui esercitava un effetto
differenziante. I risultati ottenuti confermano che nelle cellule SH-SY5Y differenziate, sia i
livelli basali di AMP ciclico, sia quelli stimolati da forskolina, sono statisticamente più
elevati, rispetto alle cellule non-differenziate, e che ciò avviene indipendentemente
dall’agente differenziante utilizzato Questo suggerisce l’esistenza di un bersaglio,
presumibilmente post-recettoriale, comune alla guanosina e all’acido retinoico, capace di
influenzare la via di trasduzione del segnale adenilato ciclasi-AMPc, apparentemente
aumentando la sensibilità del sistema.
Dati riportati in letteratura indicano che nelle cellule SH-SY5Y, le PDE di tipo IV
cAMP-specifiche costituiscono un importante elemento nella regolazione dei livelli di
AMP ciclico (Canals et al., 2005; Mogan et al., 1993). In accordo con questi dati, la
presenza dell’inibitore delle fosfodiesterasi di tipo IV, rolipram, ha determinato un
aumento significativo dei livelli basali di AMP ciclico, indice che nelle cellule SH-SY5Y il
nucleotide ciclico è sottoposto ad un rapido turnover almeno in parte dovuto all’attività di
PDE di tipo IV. Una elevata attività fosfodiesterasica spiegherebbe anche lo scarso effetto
della forskolina sui livelli di cAMP osservato in queste cellule. Il dato più interessante
riguarda comunque e soprattutto la capacità del rolipram di potenziare, con incrementi di
circa 10 volte dei livelli di AMP ciclico, l’effetto della guanosina nelle cellule
differenziate. Infatti, recentemente una sempre più crescente attenzione è stata rivolta al
possibile impiego di inibitori delle PDE-IV nel trattamento di malattie psichiatriche e
neurogedenerative (Halene e Siegel). Composti endogeni come la guanosina potrebbero
essere elementi determinanti nell’efficacia clinica di questi composti.
In conclusione, sulla base dei risultati riportati in questa tesi è possibile suggerire che
la guanosina potrebbe essere impiegata come agente terapeutico per indurre rigenerazione
neuronale in patologie neurodegenerative quali il morbo di Alzheimer’s, il morbo di
Parkinson’s e nella malattia degenerativa del motoneurone. In questa ottica è importante

Discussione
113
sottolineare che uno studio recente ha dimostrato la capacità della guanosina somministrata
per via sistemica di indurre rimielinizzazione della spina dorsale dopo parziale lesione
della stessa nei ratti (Jiang et al., 2008). La capacità inoltre della guanosina di indurre
differenziazione apre un nuovo capitolo riguardante potenziali agenti neuroprotettivi e
aggiunge nuovo slancio alla ricerca del meccanismo d’azione molecolare della guanosina

6. BIBLIOGRAFIA

Bibliografia
114
Ammer H. e Schulz R. Retinoic acid-induced differentiation of human neuroblastoma SH-SY5Y cells is associated with changes in the abundance of G proteins. J Neurochem 1994; 62: 1310-1318 Arnon R. e Aharoni R. Neurogenesis and neuroprotection in the CNS – fundamental elements in the effect of glatiramer acetate on treatment of autoimmune neurological disorders. Mol Neurobiol 2007; 36: 245-253 Ault D.T. e Werling L.L. SHSY-5Y cells as a model for sigma receptor regulation of potassium-stimulated dopamine release. Brain Res 2000; 877: 354-360 Baldwin S.A., Mackey R.J., Cass C.E. e Young J.D. Nucleoside transporters: molecular biology and implications for therapeutic development. Mol Med Today 1999; 5: 216-224 Baldwin S.A.,Yao S.Y.M., Hyde R.J., Ng A.M.L., Foppolo S., Barnes K., Ritzel M.W.L., Cass C.E. e Young J.D. Functional characterization of novel human and mouse equilibrative nucleoside transporters (hENT3 and mENT3) located in intracellular membranes. J Biol Chem 2005; 280: 15880-15887 Barnes K., Dobrzynski H., Foppolo S., Beal P.R., Tallez J., Claycomb W.C., Cass C.E., Young J.D., Billeter-Clark R., Boyett M.R. e Baldwin S.A. The distribution and functional characterization of ENT4, a bifunctional nucleoside and organic cation transporter. Purinergic Signalling 2006; 2: 311 Benitez-King G., Ramirez-Rodriguez G., Ortiz L. e Meza I. The neuronal cytoskeleton as a potential therapeutical target in neurodegenerative diseases and schizophrenia. Curr Drug Targets CNS Neurol Disord 2004; 3(6): 515-533 Berg J.M., Tymoczko J.L. e Stryer L. Biochemistry. W.H. Freeman and company 2005; Part III, cap.25 http: / / www.ncbi.nlm.gov Biedler J.L., Helson L. e Spengler B.A. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973; 33: 2643-2652 Biedler J.L., Roffler-Tarlov S., Schachner M. e Freedman L.S. Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. Cancer Res 1978; 38: 3751-3757 Brown W.H. e Foote C.S. Organic chemistry. EdiSES 1998; 1095-1098 Burnstock G. Purinergic Signalling. Br J Pharmacol 2006; 147:172-182

Bibliografia
115
Buttiglione M., Vitiello F., Sardella E., Petrone L., Nardulli M., Favia P., d’Agostino R. e Gristina R. Behaviour of SH-SY5Y neuroblastoma cell line grown in different media and on different chemically modified substrates. Biomaterials 2007; 28: 2932-2945 Cabrita M.A., Baldwin S.A., Young J.D., e Cass C.E. Molecular biology and regulation of nucleoside and nucleobase transporter proteins in eukaryotes and prokaryotes. Biochem Cell Biol 2002; 80: 623-638 Canals M., Angulo E., Casadò V., Canela E.I., Mallol J., Viñals F., Staines W., Tinner B., Hillion J., Agnati L., Fuxe K., Ferré S., Lluis C. e Franco R. Molecular mechanisms involved in the adenosine A1 and A2 receptor-induced neuronal differentiation in neuroblastoma cells and striatal primary cultures. J Neurochem 2005; 95: 337-348 Chalovich E.M., Zhu J.H., Caltagarone J., Bowser R. e Chu C.T. Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J Biol Chem 2006; 281(26): 17870-17881 Chang R., Algird A., Bau C., Rathbone M.P. e Jiang S. Neuroprotective effects of guanosine on stroke models in vitro and in vivo. Neurosci Lett 2008; 431 : 101-105 Chen J., Sonsalla P.K., Pedata F., Melani A., Domenici M.R., Popoli P., Geiger J., Lopes L.V. e de Medonça A. Adenosine A2A receptors and brain injury: broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Prog Neurobiol 2007; 83: 310-331 Ciccarelli R., Ballerini P., Sabatino G., Rathbone M.P., D’Onofrio M., Caciagli F. e Di Iorio P. Involvement of astrocytes in purine-mediated reparative processes in the brain. Int J Dev Neurosci 2001; 19: 395-414 Coe I., Zhang Y., McKenzie T. e Naydenova Z. PKC regulation of the human equilibrative nucleoside transporter, hENT1. FEBS Lett 2002; 517: 201-205 Delicado E.G., Rodrigues A., Sen R.P., Sebastiao A.M., Ribeiro J.A. e Miras-Portugal M.T. Effect of 5’-(N-Ethylcarboxamido)adenosine on adenosine transport in cutured chromaffin cells. J Neurochem 1990; 54: 1941-1946 Di Iorio P., Virgilio A., Giuliani P., Ballerini P., Vianale G., Middlemiss P.J., Rathbone M.P. e Ciccarelli R. AIT-082 is neuroprotective against kainate-induced neuronal injury in rats. Exp Neurol 2001; 169(2): 392-399

Bibliografia
116
Di Iorio P., Ballerini P., Traversa U., Nicoletti F., D’Alimonte I., Kleywegt S., Werstiuk E.S., Rathbone M.P., Caciagli F. e Ciccarelli R. The antiapoptotic effect of guanosine is mediate by the activation of the PI3-kinase/AKT/PKB pathway in cultured rat astrocytes. Glia 2004; 46: 356-368 Eltzschig H.K., Abdulla P., Hoffman E., Hamilton K.E., Daniels D., Schonfeld C., Loffler M., Reyes G., Duszenko M., Karhausen J., Robinson A., Westerman K.A., Coe I. Colgan S. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med 2005; 202: 1493-1505 Encinas M., Iglesias M., Liu Y., Wang H., Muhaisen A., Ceña V., Gallego C. e Comella J.X. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J Neurochem 2000; 75 (3): 991-1002 Endres C.J. e Unadkat J.D. Residues Met89 and Ser160 in the human equilibrative nucleoside transporter 1 affect its affinity for adenosine, guanosine, S
6-(4-Nitrobenzyl)-mercaptopurine riboside, and
dipyridamole. Mol Pharmacol 2005; 67: 837-844 Feng Z. e Porter A.G. NF-κB/Rel proteins are required for neuronal differentiation of SH-SY5Y neuroblastoma cells. J Biol Chem 1999; 274 (43): 30341-30344 Flanagan S.A. e Meckling-Gill K.A. Characterization of a novel Na
+-dependent, guanosine-specific, nitrobenzylthioinosine-
sensitive transporter in acute promyelocytic leukemia cells. J Biol Chem 1997; 272: 18026-18032 Flanagan S.A., Gandhi V., Secrist III J.A. e Meckling K.A. The novel nucleoside transport system exhibited by NB4 cells, csg, transports deoxyguanosine analogues, including ara-G. Biochem Pharmacol 2003; 66: 733-737 Gangadharan S., Henri H., Heikki R. e Gerald M. Receptor for advanced glycation end products plays more important role in cellular survival than in neurite outgrowth during retinoic acid-induced differentiation of neuroblastoma cells. J Biol Chem 2002; 277 : 6888-6897 Glasky A.J., Ritzman R.F., Prisecaru J., Santos S., Kati K., Rathbone M.P., Middlemiss Y.J. e Crocker C.E. Elevation of brain mRNA for neurotrophins by oral AIT-082 in mice. Abstr Soc Neurosci 1996; 22, 751 Glasky A.J., Melchior C.L., Pirzadeh B., Heydari N e Ritzman R.F. Effect of AIT-082, a purine analog, on working memory in normal and aged mice. Pharmacol Biochem Behav 1994; 47(2): 325-329

Bibliografia
117
Griffith D.A. e Jarvis S.M. Nucleoside and nucleobase transport systems of mammalian cells. Biochem Biophys Acta 1996; 1286 : 153-181 Gysbers J.W., Guarnieri S., Mariggiò M.A., Pietrangel T., Fanò G. e Rathbone M.P. Extracellular GTP enhances nerve growth factor-induced neurite outgrowth via increases in intracellular calcium. Neurosci 2000; 96 : 817-824 Hartley C.L., Johnston H.B., Nicol S., Chan K.M., Baines A.J., Anderton B.H. e Thomas S.M. Phenotypic morphology and the expression of cytoskeletal markers during long-term differentiation of human SH-SY5Y neuroblastoma cells. Toxicol in Vitro 1996; 10: 539-550 Ham R.G. e Veomett M.J. Mechanisms of development. C.V. Mosby, St Louis, USA, 1980 Hammond J.R., Stolk M., Archer R.G.E. e McConnel K. Pharmacological analysis and molecular cloning of the canine equilibrative nucleoside transporter 1. Eur J Pharmacol 2004 ; 491: 9-19 Halene T.B. e Siegel S.J. PDE inhibitors in psychiatry-future options for dementia, depression and schizophrenia? Drug Discov Today 2007; 12: 870-878 Henderson J.F. Regulation of purine biosynthesis. American Chemical Society, Washington, D.C. Martin DW Jr 1972; 1-191 Hong Yang I., Co C.C. e Ho C. Alteration of human neuroblastoma cell morphology and neurite extension with micropatterns. Biomaterials 2005; 26: 6599-6609 Jalava A.M., Heikkilä J., Åkerlind G., Pettitt G.R. e Åkerman K.E.O. Effects of bryostatins 1 and 2 on morphological and functional differentiation of SH-SY5Y human neuroblastoma cells. Cancer Res 1990; 50: 3422-3428 Jiang S., Fischione G., Guiliani P., Romano S., Caciagli F. e Di Iorio P. Metabolism and distribution of guansine given intraperitoneally: implication for spinal cord injury. Nucleotides Nucleosides Nucleic Acids 2008; 27(6): 673-680. Jung Y.D., Nakano K., Liu W., Gallick G.E. e Ellis L.M. Extracellular signal-regulated kinase activation is required for up-regulation of Vascular Endothelial Growth Factor by serum starvation in human colon carcinoma cells. Cancer Res 1999; 59: 4804:4807 Kim S.N., Kim S.G., Park S.D., Cho-Chung Y.S. e Hong S.H. Participation of Tipe II protein kinase A in the retinoic acid-induced growth inhibition of SH-SY5Y human neuroblastoma cells. J Cell Physiol 2000; 182: 421-428

Bibliografia
118
Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227(5259): 680-685 Lambert D.G., Ghataorre A.S. e Nahorski S.R. Muscarinic receptor binding characteristics of a human neuroblastoma SK-N-SH and its clones SH-SY5Y and SH-EP1. Eur J Pharmacol 1989; 165: 71-77 Lanier S.A. AGS proteins, GPR motifs and the signals processed by heterotrimeric G proteins. Biol. Cell 2004; 96: 369-372 Lochner A. e Moolman J.A. The many faces of H89: a review. Cardiovasc Drug Rev 2006; 24: 261-27 Lòpez-Caballo G., Moreno L., Masià S., Pérez P. e Barettino D. Activation of the phosphatidylinositol 3-kinase/Akt signaling pathway by retinoic acid is required for neuronal differentiation of SH-SY5Y human neuroblastoma cells. J Biol Chem 2002; 277 : 25297-25304 Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ Protein measurement with the folin phenol reagent. J Biol Chem, 1951; 93: 265-275 Maccioni R.G. e Cambiazo V. Role of microtubul-associated proteins in the control of microtubul assembly. Physiol Rev 1995; 75(4): 835-864 Maden M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci 2007; 8(10):755-65 Middlemiss P.J., Glasky A.J., Rathbone M.P., Werstuik E., Hindley S. e Gysbers J. AIT-082, a unique purine derivative, enhances nerve growth factor mediated neurite outgrowth from PC12 Neurosci Lett 1995; 199(2): 131-134 Monaghan F.K., pou C., Mackenzie C.J., Plevin R. e Lutz E.M. Neurotrophic actions of PACAP-38 and LIF on Human neuroblastoma SH-SY5Y cells. J Mol Neurosci 2008; 36(1-3): 45-56 Morgan A.J., Murray K.J. e ChallissR.A.J. Comparison of the effect of isobutylmethylxanthine and phosphodiesterase-selective inhibitors on cAMP levels in SH-SY5Y neuroblastoma cells. Biochem Pharmacol 1993; 45: 2373-2380 Mullen R.J., Charles R.B. e Smith A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Development 1992; 116: 201-211 Nakagawa-Yagi Y., Saito Y., Takada Y. e Takayama M. Carbachol enhances forskolin-stimulated cyclic AMP accumulation via activation of calmodulin system in human neuroblastoma SH-SY5Y cells. Biochem Biophys Res Comm 1991; 178: 116-123

Bibliografia
119
Nannan G., Runmei Y., Fusheng L., Shoulan Z. e Guangqing L. Effects of AIT-082, a purine derivative, on tremor induced by arecoline or oxotremorine in mice. Pharmacology 2007; 80(1): 21-26 Nordstedt C. e Fredholm B.B. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem 1990; 189: 231-234 Oleskovicz S. P.B., Martins W.C., Leal R.B., Tasca C.I. Mechanism of guanosina-induced neuroprotection in rat hippocampal slices submitted to oxygen-glucose deprivation. Neurochem Int 2008; 52: 411-418 Olsson A.-K., Vadhammar K. e Nånberg Activation and protein kinase C-dependent nuclear accumulation of ERK in differentiating human neuroblastoma cells. Exp Cell Res 2000; 256: 454-467 Peterfreund R.A., Gies E.K. e Fink J.S. Protein kinase C regulates adenosine A2A receptor mRNA expression in SH-SY5Y cells. Eur J Pharmacol 1997; 336: 71-80 Pettifer K.M., Kleywegt S., Bau C.J., Ramsbottom J.D., Vertes E., Ciccarelli R., Caciagli F., Werstiuk E.S. e Rathbone M.P. Guanosine protects SH-SY5Y cells against β-amyloid-induced apoptosis. Dev Neurosci 2004; 15 : 833-836 Rathbone M.P., Middlemiss P., Andrew C., Caciagli F., Ciccarelli R., Di Iorio P. e Huang R. The trophic effects of purines and purinergic signaling in pathologic reactions of astrocytes. Alzheimer Dis Assoc Disord 1998; 12 (2): S36-S45 Rathbone M.P., Middlemiss P.J., Gysbers J.W., Herman M. A.R., Reed J.K., Ciccarelli R., Di Iorio P. e Caciagli F. Trophic effects of purines in neurons and glial cells. Prog Neurobiol 1999; 59: 663-690 Ruffels J., Griffin M. e Dickenson J.M. Activation of ERK 1/2, JNK and PKB by hydrogen peroxide in human SH-SY5Y neuroblastoma cells: role of ERK 1/2 in H2O2-induced cell death. Eur J Pharmacol 2004; 483: 163-173 SánchezS., Jiménez C., Carrera A.C., Diaz-Nido J., Avila J. E Wandosell F. A cAMP-activated pathway, including PKA and PI3K, regulates neuronal differentiation. Neurochem Int 2004; 44: 231-242 Sato M., Cismowski M.J., Toyota E., Smrcka A.V., Lucchesi P., Chilian W.M. Identification of a receptor-independent activator of G protein signaling (AGS8) in ischemic heart and its interaction with Gβγ. Proc Natl Acad Sci USA 2006; 103: 797-802 Schmidt A.P., Lara D.R. e Souza D.O. Proposal of guanine-based purinergic system in the mammalian central nervous system. Pharmacol Therc 2007; 116 (3): 401-416

Bibliografia
120
Schmidt A.P., Tort A.B.L., Silveira P.P., Bohmer A.E., Hansel G., Knorr L., Schallenber C., Dalmaz C., Elisabetsky E., Crestana R.H., Lara D.R. e Souza D.O. The NMDA antagonist MK-801 induces hyperalgesia and increases CSF excitatory amino acids in rats: Reversal by guanosine. Pharmacol Biochem Behav 2009; 91: 549-553 Schuster C., Williams L.M., Morris A., Morgan P.J. e Barrett P. The human MT1 melatonin receptor stimulates cAMP production in the human neuroblastoma cell line SH-SY5Y cells via a calcium-calmodulin signal transduction pathway. J Neuroendocrinol 2005, 17: 170-178 Seidenfaden R., Krauter A. e Hildebrandt H. The neural cell adhesion molecule NCAM regulates neuritogenesis by multiple mechanisms of interaction. Neurochem Int 2006; 49(1): 1-11 Singh U.S., Pan J., Kao Y., Joshi S., Young J.L. e Baker K.M. Tissue transglutaminase activation of RhoA and MAP kinase pathways during retinoic acid-induced neyronal differentiation of SH-SY5Y cells. J Biol Chem 2003; 278 (1): 391-399 Song L. e Jope R.S. Cellular stress increases RGS2 mRNA and decreases RGS4 mRNA levels in SH-SY5Y cells. Neurosci Lett 2006; 402: 205-209 Thorn J.A. e Jarvis S.M. Adenosine transporters. Gen Pharmacol 1996; 27: 613-620 Taylor E.M., Yan R., Hauptmann N., Maher T.J., Djahandideh D. e Glasky A.J. AIT-082, a cognitive enhancer, is transported into brain by a nonsaturable influx mechanism and out of brain by a saturable efflux mechanism. J Pharmacol Exp Ther 2000; 293(3): 813-821 Thorn J.A. e Jarvis S.M. Adenosine transporters. Gen Pharmacol 1996; 27: 613-620 Toll L., Polgar W.E., Auh J.S. Characterization of the delta-opioid receptor found in SH-SY5Y neuroblastoma cells. Eur J Pharmacol 1997; 323: 261-267 Traversa U., Bombi G., Di Iorio P., Ciccarelli R., Werstiuk E.S. e Rathbone M.P. Specific [
3H]-guanosine binding sites in rat brain membranes.
Br J Pharmacol 2002; 135: 969-976 Traversa U., Bombi G., Camaioni E., Macchiarulo A., Costantino G., Palmieri C., Caciagli F. e Pellicciari R. Rat brain guanosine binding site: biological studies and pseudo-receptor construction. Bioor Med Chem 2003; 11: 5417-5425 Svensson A.L. Tacrine interacts with different sites on nicotinic receptor subtypes in SH-SY5Y neuroblastoma and M10 cells. Behav Brain Res 2000; 113: 193-197

Bibliografia
121
Wang J., Schaner M.E., Thomassen S., Su S.-F., Piquette-Miller M., e Giacomini K.M. Functional and molecular characteristics of Na
+-dependent nucleoside transporters.
Pharmacol Res 1997; 14: 1524-1531 Wardas J. Neuroprotective role of adenosine in the CNS. Pol J Pharmacol 2002; 54: 313-326 Watanabe S., Yoshimi Yoji e Ikekita M. Neuroprotective effect of adenine on purkinje cell survival in rat cerebellar primary cultures. J Neurosci Research 2003; 74: 754-759 Zanette C. Neuroprotective effect of guanosine in human neuroblastoma SH-SY5Y cells. XI Seminario per Dottorandi in Farmacologia e Scienze Affini - Siena, Certosa di Pontignano 24-27 Settembre 2007 http://library.med.utah.edu www.ncbi.nlm.nih.gov

CARATTERIZZAZIONE DEI
MECCANISMI NEUROPROTETTIVI
DEI LIGANDI DEI RECETTORI
SIGMA

1. INTRODUZIONE

INTRODUZIONE
122
1.1 Identificazione dei recettori sigma
I recettori sigma sono stati identificati da Martin e collaboratori nel 1976, sulla base
delle azioni del composto N-allilnormetazocina (SKF-10,047) e di altri benzomorfani ad
esso correlati (Matzumoto et al., 2003). Questi ligandi oppioidi inducevano una serie di
comportamenti, come delirio nel cane ed effetti psicotomimetici nell’uomo, che non erano
attribuibili all’interazione con i classici recettori oppioidi di tipo κ e µ. Quindi siti furono
inizialmente classificati come sottotipi della famiglia dei recettori oppiacei e denominati
recettori sigma, ma studi successivi dimostrarono che essi rappresentano recettori
indipendenti i cui ligandi endogeni non sono ancora a tutt’oggi ben identificati.
Numerose sono le caratteristiche farmacologiche che servono a differenziarli dai
recettori degli oppioidi. Ad esempio, i recettori degli oppioidi sono enantioselettivi per gli
isomeri (-) dei farmaci narcotici derivati dell'oppio e dei loro congeneri, mentre i recettori
sigma sono enantioselettivi per gli isomeri (+). L’isomero (-) del SKF-10,047, prototipo
dei ligandi sigma, presenta maggiore affinità per i recettori oppioidi di tipo κ ed è
responsabile di azioni che sono antagonizzate dal naloxone. L’isomero (+)SKF-10,047
invece si lega a due siti: con moderata affinità al sito della fenciclidina (PCP) all’interno
del canale ionico del recettore del glutammato del tipo N-metil-D-aspartato (NMDA), e
con alta affinità ad un altro sito, appunto designato recettore sigma (Matzumoto et al.
2003).
La selettività enantiomerica del recettore sigma per i (+)-benzomorfani e il fatto che
gli effetti di tipo comportamentale sopra indicati non venissero bloccati né in vivo né in

INTRODUZIONE
123
vitro da naloxone e naltrexone, hanno portato all’esclusione di questi recettori dalla
famiglia dei recettori oppioidi.
Con l’impiego di radioligandi piu selettivi si è poi osservato che il sito cui si lega la
fenciclidina e il sito denominato “sigma” non appartenevano allo stesso complesso
recettoriale. In aggiunta i recettori sigma possono essere differenziati dai siti della
fenciclidina anche in base alla differente distribuzione anatomica nel sistema nervoso
centrale (SNC) (Skuza, 2003).
In definitiva, i recettori sigma sono stati recentemente riclassificati come recettori
unici e distinti dai siti di legame degli oppioidi e della fenciclidina (Matzumoto et al.,
2003).
Inizialmente i recettori sigma sono stati associati a vari disturbi del sistema nervoso
centrale, come la schizofrenia o i disordini motori. Oggi, dopo un gran numero di studi
sull’importanza dei recettori sigma nel sistema nervoso centrale, è ampiamente
riconosciuto che tali recettori rappresentano una nuova strada nel possibile trattamento
farmacologico di queste ed altre patologie del SNC.
1.2 Classificazione dei recettori sigma
Studi di binding hanno meglio caratterizzato i recettori sigma ed è emerso che questi
recettori si distinguono da tutti gli altri recettori di neurotrasmettitori noti; è stato possibile
definirli come siti di legame non-oppioidi, con alta affinità per la 1,3-di-O-
tolilmetilguanidina (DTG), la (+)-3-(3-idrossifenil)-N-(1-propil)piperidina [(+)-3-PPP] e
l’aloperidolo (Guitart et al., 2004).
Studi biochimici e farmacologici hanno evidenziato l’esistenza di due sottotipi
recettoriali, oggi denominati sigma-1 e sigma-2. Questi sottotipi recettoriali si
differenziano innanzitutto per il profilo farmacologico nei confronti di alcune molecole
specifiche, in quanto i siti sigma-1 presentano una alta affinità (nM) e stereoselettività per
gli isomeri (+) della pentazocina, dell’ SKF-10,047, e di altri benzomorfani mentre i
recettori sigma-2 esibiscono per gli stessi bassa affinità (µM). Entrambi i sottotipi hanno
affinità da bassa a moderata per gli isomeri (-) di questi composti. Pertanto si nota una
diversa stereoselettività tra questi due siti: gli isomeri (+) risultano più potenti verso i siti
sigma-1 mentre una situazione opposta si osserva verso i siti sigma-2.
Altri composti che permettono di distinguere questi siti sono il carbapentano, il
dextrometorfano e la fenitoina. I primi due sono ligandi piuttosto selettivi per il recettore
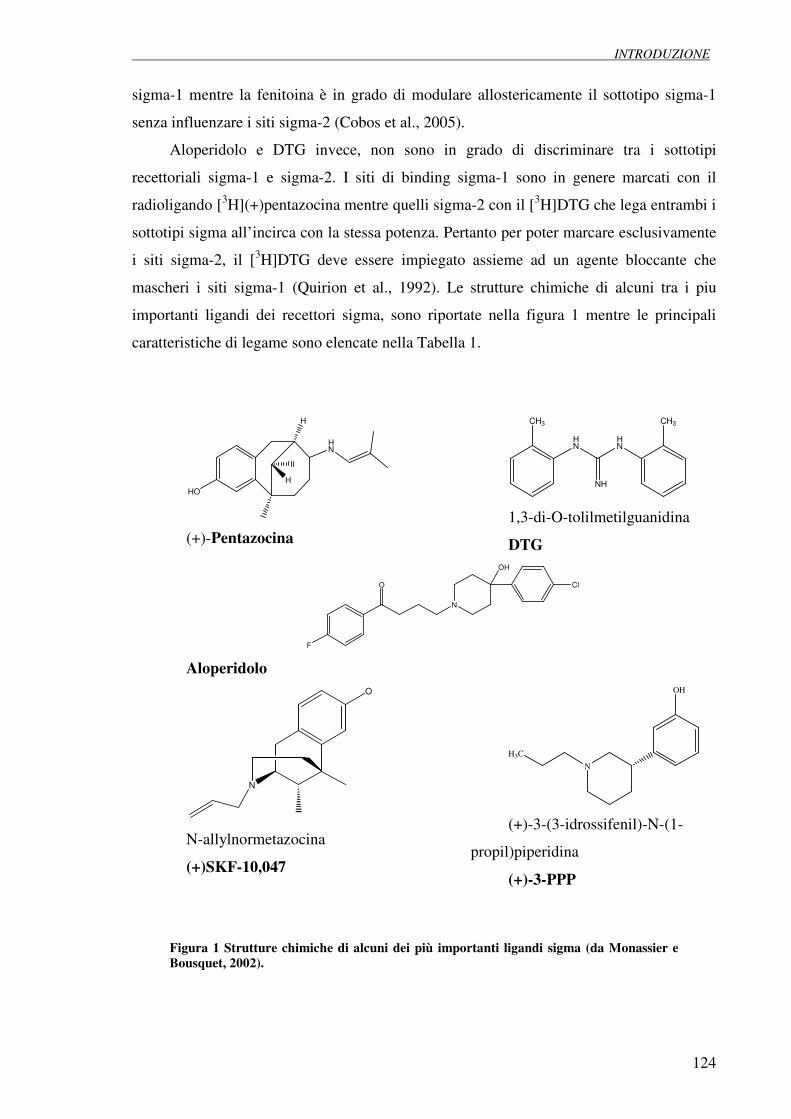
INTRODUZIONE
124
sigma-1 mentre la fenitoina è in grado di modulare allostericamente il sottotipo sigma-1
senza influenzare i siti sigma-2 (Cobos et al., 2005).
Aloperidolo e DTG invece, non sono in grado di discriminare tra i sottotipi
recettoriali sigma-1 e sigma-2. I siti di binding sigma-1 sono in genere marcati con il
radioligando [3H](+)pentazocina mentre quelli sigma-2 con il [3H]DTG che lega entrambi i
sottotipi sigma all’incirca con la stessa potenza. Pertanto per poter marcare esclusivamente
i siti sigma-2, il [3H]DTG deve essere impiegato assieme ad un agente bloccante che
mascheri i siti sigma-1 (Quirion et al., 1992). Le strutture chimiche di alcuni tra i piu
importanti ligandi dei recettori sigma, sono riportate nella figura 1 mentre le principali
caratteristiche di legame sono elencate nella Tabella 1.
HN
H
H
HO
(+)-Pentazocina
HN
HN
CH3CH3
NH
1,3-di-O-tolilmetilguanidina
DTG
O
F
N
OH
Cl
Aloperidolo O
N
N-allylnormetazocina
(+)SKF-10,047
OH
N
H3C
(+)-3-(3-idrossifenil)-N-(1-
propil)piperidina
(+)-3-PPP
Figura 1 Strutture chimiche di alcuni dei più importanti ligandi sigma (da Monassier e Bousquet, 2002).
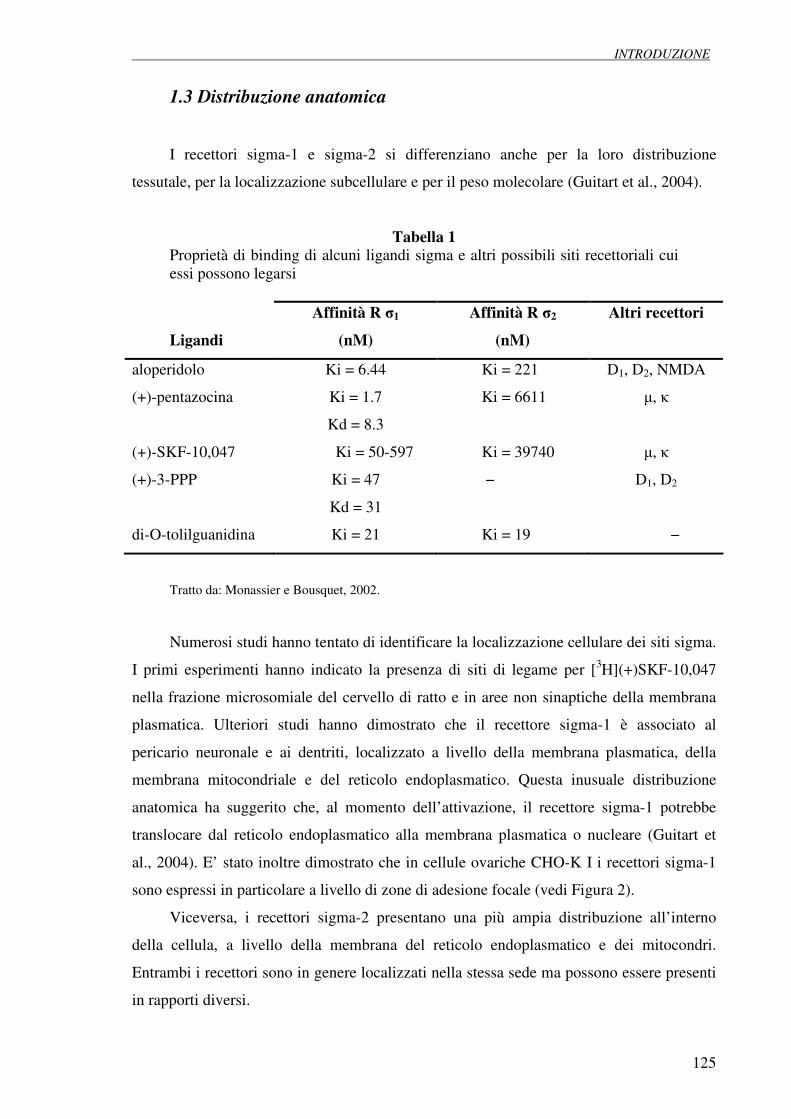
INTRODUZIONE
125
1.3 Distribuzione anatomica
I recettori sigma-1 e sigma-2 si differenziano anche per la loro distribuzione
tessutale, per la localizzazione subcellulare e per il peso molecolare (Guitart et al., 2004).
Tabella 1 Proprietà di binding di alcuni ligandi sigma e altri possibili siti recettoriali cui essi possono legarsi
Ligandi
Affinità R σ1
(nM)
Affinità R σ2
(nM)
Altri recettori
aloperidolo Ki = 6.44 Ki = 221 D1, D2, NMDA
(+)-pentazocina Ki = 1.7
Kd = 8.3
Ki = 6611 µ, κ
(+)-SKF-10,047 Ki = 50-597 Ki = 39740 µ, κ
(+)-3-PPP Ki = 47
Kd = 31
− D1, D2
di-O-tolilguanidina Ki = 21 Ki = 19 −
Tratto da: Monassier e Bousquet, 2002.
Numerosi studi hanno tentato di identificare la localizzazione cellulare dei siti sigma.
I primi esperimenti hanno indicato la presenza di siti di legame per [3H](+)SKF-10,047
nella frazione microsomiale del cervello di ratto e in aree non sinaptiche della membrana
plasmatica. Ulteriori studi hanno dimostrato che il recettore sigma-1 è associato al
pericario neuronale e ai dentriti, localizzato a livello della membrana plasmatica, della
membrana mitocondriale e del reticolo endoplasmatico. Questa inusuale distribuzione
anatomica ha suggerito che, al momento dell’attivazione, il recettore sigma-1 potrebbe
translocare dal reticolo endoplasmatico alla membrana plasmatica o nucleare (Guitart et
al., 2004). E’ stato inoltre dimostrato che in cellule ovariche CHO-K I i recettori sigma-1
sono espressi in particolare a livello di zone di adesione focale (vedi Figura 2).
Viceversa, i recettori sigma-2 presentano una più ampia distribuzione all’interno
della cellula, a livello della membrana del reticolo endoplasmatico e dei mitocondri.
Entrambi i recettori sono in genere localizzati nella stessa sede ma possono essere presenti
in rapporti diversi.
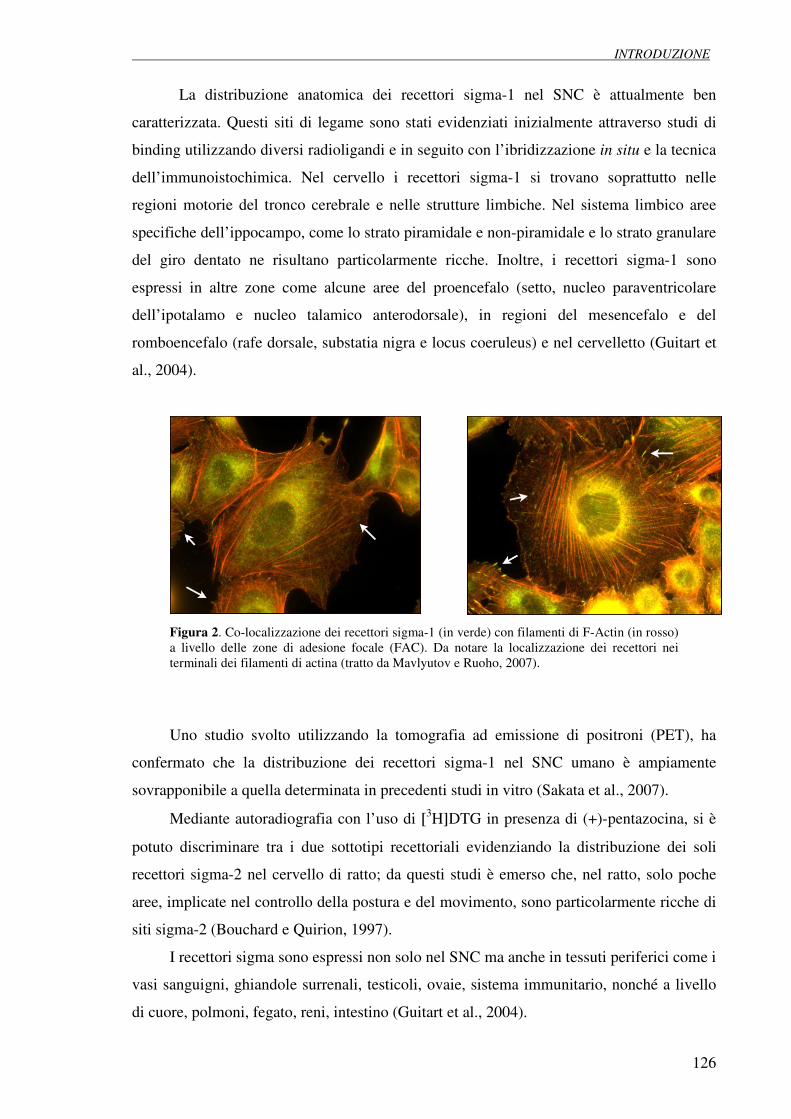
INTRODUZIONE
126
La distribuzione anatomica dei recettori sigma-1 nel SNC è attualmente ben
caratterizzata. Questi siti di legame sono stati evidenziati inizialmente attraverso studi di
binding utilizzando diversi radioligandi e in seguito con l’ibridizzazione in situ e la tecnica
dell’immunoistochimica. Nel cervello i recettori sigma-1 si trovano soprattutto nelle
regioni motorie del tronco cerebrale e nelle strutture limbiche. Nel sistema limbico aree
specifiche dell’ippocampo, come lo strato piramidale e non-piramidale e lo strato granulare
del giro dentato ne risultano particolarmente ricche. Inoltre, i recettori sigma-1 sono
espressi in altre zone come alcune aree del proencefalo (setto, nucleo paraventricolare
dell’ipotalamo e nucleo talamico anterodorsale), in regioni del mesencefalo e del
romboencefalo (rafe dorsale, substatia nigra e locus coeruleus) e nel cervelletto (Guitart et
al., 2004).
Figura 2. Co-localizzazione dei recettori sigma-1 (in verde) con filamenti di F-Actin (in rosso) a livello delle zone di adesione focale (FAC). Da notare la localizzazione dei recettori nei terminali dei filamenti di actina (tratto da Mavlyutov e Ruoho, 2007).
Uno studio svolto utilizzando la tomografia ad emissione di positroni (PET), ha
confermato che la distribuzione dei recettori sigma-1 nel SNC umano è ampiamente
sovrapponibile a quella determinata in precedenti studi in vitro (Sakata et al., 2007).
Mediante autoradiografia con l’uso di [3H]DTG in presenza di (+)-pentazocina, si è
potuto discriminare tra i due sottotipi recettoriali evidenziando la distribuzione dei soli
recettori sigma-2 nel cervello di ratto; da questi studi è emerso che, nel ratto, solo poche
aree, implicate nel controllo della postura e del movimento, sono particolarmente ricche di
siti sigma-2 (Bouchard e Quirion, 1997).
I recettori sigma sono espressi non solo nel SNC ma anche in tessuti periferici come i
vasi sanguigni, ghiandole surrenali, testicoli, ovaie, sistema immunitario, nonché a livello
di cuore, polmoni, fegato, reni, intestino (Guitart et al., 2004).

INTRODUZIONE
127
1.4 Il sottotipo recettoriale sigma-1
Il recettore sigma-1 è il sottotipo più studiato e caratterizzato. E’ stato clonato a
partire da fegato di porcellino d’india, cellule di coriocarcinoma placentare umano,
cervello umano, cervello di ratto e di topo.
Il gene per il recettore sigma è localizzato sul cromosoma 9 sulla banda p13, regione
nota per essere associata con diversi disordini psichiatrici, ed è lungo circa 7kbp. E’
costituito da quattro esoni e tre introni. L’esone 3 è il più corto (93 bp) metre l’esone 4 è il
più lungo (1.132bp). L’esone 2 codifica per il dominio transmembrana presente nel
recettore. Per quanto riguarda gli introni, l’introne 3 è il più lungo (1.250 bp).
L’unica somiglianza è stata trovata con un enzima dell’Aradopsis thaliana: questo
enzima di lievito, coinvolto nella sintesi dell’ergosterolo, è una C8-C7 isomerasi e sembra
essere strutturalmente correlato con il recettore sigma-1 mentre l’omologo umano
dell’enzima di lievito non presenta alcuna similitudine strutturale. Inoltre si è visto che
ligandi sigma come l’aloperidolo inibiscono la biosintesi dello sterolo nel lievito e ciò
dimostra la relazione farmacologica che esiste tra i siti di legame sigma e l’attività della
C8-C7 sterolo isomerasi. Di contro, i recettori sigma non possiedono attività isomerasica
(Moebius et al., 1997; Guitart et al., 2004).
Studi su una variante del recettore sigma-1 carente dell’esone 3 hanno evidenziato
che quest’ultimo perde la capacità funzionale di legare i suoi ligandi. E’ stato quindi
individuato attorno a questa regione il sito attivo del recettore con i suoi amminoacidi
critici. Il recettore umano sigma-1 è stato mutato singolarmente per ciascuno degli
amminoacidi espressi in questa regione ed è stata valutata l’influenza di ciascuna
mutazione sulla capacità di legame. Questi studi hanno evidenziato la presenza di due
amminoacidi anionici fondamentali che sono D126 ed E172. Sebbene la funzione legante
sia abolita da entrambe queste due mutazioni, l’espressione dei mutanti a livello proteico è
normale. E’ comunque possibile che queste mutazioni portino al mal ripiegamento della
proteina o ad una sua sbagliata localizzazione.
In tutto la proteina è costituita da 223 amminoacidi e sembrava includesse un unico
dominio transmembrana mentre da studi più recenti si è visto essere costituita da due
domini trasmembrana, risultanti in un loop extracellulare di circa 50 amminoacidi e in un
C-terminale intracellulare di circa 125 amminoacidi. In accordo con questo modello, l’N-
terminale è molto corto e sempre localizzato nella porzione intracellulare. Alla fine di
questa sequenza, in prossimità di un gruppo anionico, si trovano due residui di arginina (R-

INTRODUZIONE
128
R) che fungono da segnale per l’inserimento della proteina recettoriale nel reticolo
endoplasmatico (Figura 3). Una regione idrofobica potrebbe essere importante per il
binding di (+)-pentazocina (Yamamoto et al., 1999). Non è ancora chiaro se questo sito di
legame sia condiviso o meno con neurosteroidi.
La sequenza amminoacidica non corrisponde a quella dei recettori accoppiati alle
proteine G e ciò fa cadere l’ipotesi che vede i recettori sigma appartenenti a questa classe.
Inoltre, la conferma che i recettori sigma non appartengono alla classe dei recettori
accoppiati a proteine G è stata ottenuta da Hong e Werling (2000). Una seconda regione
idrofobica sembra essere il sito di legame per la (+)-pentazocina ma dev’essere ancora
chiarito se questo sito è condiviso anche dai neurosteroidi.
Figura 3. Modello strutturale del recettore sigma-1. E’ una proteina formata da 223 aminoacidi, comprendente due domini transmembrana idrofobici e due porzioni intracellulari C- e N-terminali. In prossimità del gruppo –NH2 ci sono due residui di arginina (R-R) importanti per l’inserimento della proteina nel reticolo endoplasmatico (da Duncan e Wang, 2005).
1.4.1 Ruoli fisiologici del recettore sigma-1
La clonazione della proteina recettoriale sigma-1 ha permesso di progredire nello
studio delle funzioni biochimiche e fisiologiche di questi siti di binding.
Sono state descritte numerose funzioni fisiologiche mediate dai recettori sigma-1,
quali:

INTRODUZIONE
129
• Modulazione della sintesi e del rilascio dei neurotrasmettitori acetilcolina e
dopamina
• Effetti neuromodulatori su sistemi glutammatergici di tipo NMDA
• Modulazione del turnover dei fosfoinositidi indotti dall’attivazione dei
recettori muscarinici
• Miglioramento dell’apprendimento e della memoria in modelli animali di
amnesia
• Attività neuroprotettiva e anti-amnesica
• Modulazione dell’analgesia indotta dagli oppioidi
• Alterazione dell’attivita locomotoria e della tossicità indotta da cocaina
Il recettore sigma-1 risulta quindi coinvolto in diverse patologie a carico del SNC
come la schizofrenia, la demenza, la depressione e in disordini a carico del sistema nervoso
periferico.
I meccanismi molecolari coinvolti in questi effetti non sono stati ancora identificati,
ma diversi studi indicano un possibile ruolo del recettore nella modulazione della
mobilizzazione del Ca2+ intracellulare, forse a seguito della formazione di un complesso
recettore sigma-1-ankirina-recettore dell’inositolo 1,4,5-trifosfato (IP3) (Hayashi e Su,
2000).
1.5 Il sottotipo recettoriale sigma-2
Il sottotipo recettoriale sigma-2 non è stato ancora caratterizzato come il sigma-1.
Inizialmente identificato in cellule di feocromocitoma di ratto PC12; ha un peso
molecolare di circa 18-21 KDa e bassa affinità e stereoselettività per la (+)-N-
allilnormetazocina. La proteina non è stata ancora clonata e questo spiega la relativa
scarsità di informazioni attualmente disponibili (Guitart et al., 2004).
A livello del SNC i recettori sigma-2 sono espressi a livello del cervelletto, del locus
niger e del nucleo rosso, zone cerebrali coinvolte nel controllo motorio. Questa
distribuzione è in accordo con la comparsa di tipici effetti collaterali extrapiramidali a
seguito del trattamento con farmaci neurolettici, come l’aloperidolo, capaci di legarsi ai
recettori sigma-2. A livello periferico, siti sigma-2 sono presenti nel fegato, nei reni e in
altri tessuti endocrini.

INTRODUZIONE
130
La caratterizzazione dei recettori sigma-2 è risultata più difficile a causa della
mancanza di ligandi selettivi e ad alta affinità. Recentemente sono stati identificati alcuni
composti che presentano una notevole selettività sigma-2, come l’alcaloide ibogaina, la
cicloesilpiperazina (PB-28) (Colabufo et al., 2004), i fenilmorfani, (+)-1R,5R-(E)-8-
Benziliden-5-(3-idrossifenil)-2-metilmorfan-7-one (CB-64D) e il (+)-1R,5R-(E)-8-(3,4-
diclorobenziliden)-5-(3-idrossifenil)-2-metilmorfan-7-one (CB-184), che hanno dimostrato,
rispettivamente, un’affinità per il recettore sigma-2 185 volte e 554 volte maggiore rispetto
a quella per il recettore sigma-1 (Guitart et al., 2004).
Un’elevata espressione del sottotipo recettoriale sigma-2 è stata inoltre riscontrata in
diverse linee cellulari tumorali neuronali e non, come in tumori della mammella, dei
polmoni e della prostata e in melanomi. Questa iperespressione, ha suggerito un importante
ruolo di questi siti recettoriali nella proliferazione cellulare. Proprio in questo contesto è
già stato dimostrato sia in vivo che in vitro l’effetto citotossico e antiproliferativo di ligandi
sigma-2 selettivi come ibogaina, CB-64D e PB-28.
Ligandi sigma-2 inducono morte cellulare in vari tipi di cellule attraverso apoptosi,
regolando i livelli intracellulari di Ca+2 e alterandone l’omeostasi (Guitart et al., 2004). Il
ruolo svolto dai recettori sigma-2 nell’induzione dell’apoptosi individua in potenti e
selettivi ligandi sigma-2, strumenti diagnostici e farmaci utili nella terapia dei tumori. Ad
esempio, attraverso la PET (tomografia ad emissione di positroni) si ottengono immagini
sulla distribuzione in vivo dei recettori sigma nelle cellule tumorali. Si tratta di un indagine
non invasiva in grado di evidenziare tumori ed eventuali metastasi attraverso marcatura
diretta delle cellule neoplastiche con ligandi sigma-2 selettivi contenti isotopi emettenti
positroni (Hashimoto e Ishiwata, 2006).
1.6 Ligandi endogeni dei recettori sigma
Nonostante negli ultimi anni le nostre conoscenze riguardo i recettori sigma siano
aumentate, non è ancora stato possibile identificare un loro ligando endogeno, cosa che
permetterebbe di migliorare il grado di conoscenza sul ruolo degli stessi recettori nella
regolazione delle funzioni fisiologiche e patofisiologiche del cervello. Numerosi peptidi
noti e altri neurotrasmettitori si sono dimostrati chiaramente inefficaci nello spiazzare
ligandi selettivi dai siti sigma. Tra i neurotrasmettitori che non interagiscono con il
recettore sigma si possono citare, ad esempio, le monoammine serotonina, noradrenalina,

INTRODUZIONE
131
dopamina ed istamina; aminoacidi come glutammato, glicina, aspartato e cisteina, e peptidi
quali endorfine, dinorfine, encefaline e sostanza P.
Il neuropeptide Y (NPY) merita una particolare attenzione, dal momento che è stata
ripetutamente ipotizzata la sua natura di ligando endogeno per i recettori sigma.
Esperimenti in vivo hanno dimostrato che il NPY e i ligandi sigma possono modulare
l’attività neuronale NMDA-dipendente nell’ippocampo ed il rilascio di [3H]dopamina
stimolato dall’NMDA da sezioni corticali e striatali di ratto (Guitart et al., 2004). E’ stato
quindi ipotizzato che NPY e ligandi del recettore sigma-1 potessero avere un recettore
comune accoppiato a proteine G, e che l’opposto effetto di modulazione fosse dovuto a
diversa attività intrinseca, con i ligandi sigma in qualità di agonisti inversi (Ault e Werling,
1997). Studi recenti hanno però dimostrato tramite autoradiografia che concentrazioni
micromolari di agonisti o antagonisti del recettore sigma-1 non alterano il binding del
[35S]GTPγS indotto da concentrazioni nanomolari di NPY nel cervello di ratto, indicando
che il NPY non costituisce un ligando endogeno per i recettori sigma-1 (Hong e Werling,
2001). Inoltre, questo studio pone ulteriormente in dubbio che il meccanismo di
trasduzione del segnale dei recettori sigma si basi sull’interazione con un recettore in grado
di attivare proteine G dopo interazione con un ligando.
Gli ormoni steroidei, sia quelli sintetizzati nelle ghiandole periferiche, sia i
neurosteroidi, sono stati considerati come possibili ligandi endogeni dei recettori sigma-1,
e tale interazione è stata inizialmente suggerita da esperimenti di binding in vitro nel
cervello e nella milza di porcellino d’india. Su e collaboratori, nel 1988, avevano
dimostrato come il progesterone fosse in grado di inibire il binding del [3H](+)SKF-10,047
nel cervello di porcellino d’india e del [3H]-aloperidolo nella milza, con valori di Ki
dell’ordine nanomolare. E’ stato in seguito dimostrato che altri steroidi, come il
testosterone, il pregnenolone solfato o il deossicorticosterone, inibiscono il binding ai siti
sigma-1 con valori di Ki in un range micromolare. Altri gruppi hanno poi dimostrato che il
progesterone è effettivamente in grado di inibire potentemente il legame di ligandi sigma-1
selettivi radiomarcati in diverse preparazioni, come il cervello di ratto, la membrana
plasmatica degli splenociti e i microsomi epatici. Inoltre, molti ligandi sigma-1 tra cui
aloperidolo, carbapentano, DTG, (+)-3-PPP e rimcazolo, sono in grado di inibire il binding
del [3H]-progesterone in preparazioni di fegato di ratto.
Altri esperimenti hanno confermato come la somministrazione di neurosteroidi porti
ad una inibizione dose-dipendente del legame [3H](+)SKF-10,047 ai recettori sigma-1. La
modulazione dei livelli endogeni di steroidi è in grado di aumentare il binding del

INTRODUZIONE
132
[3H](+)SKF-10,047 in vivo, mentre l’uso di agenti farmacologici che aumentano la
concentrazione di progesterone, provoca una riduzione del binding in vivo del [3H](+)SKF-
10,047.
Risulta chiaro che gli steroidi neuroattivi possono interagire direttamente con il
recettore sigma-1. Il progesterone sembra essere l’inibitore più potente dal momento che il
suo valore di Ki, pari a 300 nM (Collier et al., 2007) è compatibile con un’interazione in
vivo. L’interazione dei neurosteroidi con il recettore sigma si verifica attraverso un
meccanismo simile a quello con cui i neurosteroidi interagiscono con il recettore NMDA o
GABAA (Guitart et al., 2004; Monnet e Maurice, 2006). Questi recettori ionotropici
mediano le azioni neuromodulatorie non genomiche degli steroidi e dei loro metaboliti, i
quali si legano ad un sito specifico nel canale per il Ca2+ (recettore NMDA) o nel canale
anionico per il Cl- (recettore GABAA). Il meccanismo d’azione dipende dalle
concentrazioni degli steroidi: basse concentrazioni di steroidi (nM) sono sufficienti ad
attivare i recettori steroidei intracellulari e a modulare il recettore GABAA con
meccanismo allosterico positivo, mentre elevate concentrazioni (µM) sono necessarie per
la modulazione negativa di questi recettori.
1.7 Recettori sigma e trasduzione del segnale
I primi studi di binding con radioligandi hanno dimostrato che i parametri del legame
di alcuni ligandi sigma, come il (+)SKF-10,047 e la (+)-3-PPP, venivano alterati dalla
guanosina trifosfato (GTP), il che suggeriva che il recettore sigma fosse accoppiato a un
meccanismo di trasduzione del segnale che vedeva coinvolte le proteine G. Più di recente,
è emerso che alcuni ligandi del recettore sigma stimolano l’attività della proteina G in
membrane sinaptiche estratte dalla corteccia prefrontale di topo e che il binding della
[35S]GTPγS nella milza di cavia è stimolato dalla (+)-pentazocina. Inoltre, molti studi
hanno dimostrato che l’affinità di legame di certi ligandi sigma al recettore è nettamente
diminuita in membrane trattate con la tossina della pertosse o con la tossina del colera.
Questi studi quindi suggerirono che alcuni effetti dei recettori sigma fossero mediati per lo
meno indirettamente dalle proteine G.
Nonostante numerose evidenze sostengano che i siti sigma siano accoppiati a
proteine G, altri riscontri escludono questo tipo di accoppiamento oppure ne propongono
caratteristiche di natura peculiare. E’ stato stabilito, ad esempio, che i parametri di legame
dei radioligandi sigma-1 agonisti come [3H](+)pentazocina e [3H]aloperidolo non vengono

INTRODUZIONE
133
alterati in risposta al trattamento con GTPγS in preparazioni di membrana derivanti da
cervelletto di ratto e di cavia (Guitart et al., 2004).
Anche studi elettrofisiologici hanno dato risultati contrastanti. Per esempio, l’effetto
della (+)-pentazocina sulle risposte indotte dal NMDA nei neuroni ippocampali non viene
alterato da un pretrattamento con la tossina della pertosse, mentre lo stesso composto è
capace di modificare le correnti del potassio nelle cellule melanotropiche dell’ipofisi di
rana dopo pretrattamento con la tossina del colera (Hong e Werling, 2000).
Questi ed altri risultati sperimentali evidenziano che non è stato ancora raggiunto un
accordo sul coinvolgimento delle proteine G nella mediazione della transduzione del
segnale nei recettori sigma. Bowen e i suoi collaboratori hanno dimostrato come i ligandi
sigma possano modulare la stimolazione del turnover del fosfoinositolo. Nei miociti
cardiaci di ratto, i ligandi sigma si sono rivelati in grado di influenzare la contrazione
cellulare e il movimento del calcio. In queste cellule infatti, un agonista sigma-1 come il
1S,2R-(−)-cis -N- [2-(3,4-dichloro phenyl) ethyl] -N-methyl-2-(1-pyrrolidinyl) cyclo
hexylamina (BD-737) determina un rapido aumento della produzione di inositolo trifosfato
(IP3), mentre il [2-(3,4-Dichlorophenyl)ethyl]-N-methyl-2-(dimethylamino)ethylamine
dihydrobromide] (BD-104), un presunto antagonista sigma-1, provoca un marcato aumento
dei livelli di IP3. Pertanto si è ipotizzato che il BD-1047 non agisca come antagonista vero
in miociti cardiaci. Tuttavia, in linee cellulari neuronali ed in sinaptosomi cerebrali, ligandi
sigma-1 (agonisti e antagonisti) non aumentano i livelli di IP3. Sulla base di questi dati, si è
ipotizzato che differenti tipi cellulari rispondano in modo diverso all’attivazione del
recettore sigma-1.
Recentemente è stato dimostrato che il recettore sigma-2 è in grado di regolare il
rilascio di Ca2+ dalle riserve del reticolo endoplasmatico. In questo studio si evidenzia che i
ligandi sigma-2 portano ad un incremento della concentrazione di Ca2+ in due momenti
distinti, uno immediato e transitorio, probabilmente dovuto al rilascio da parte del reticolo
endoplasmatico, l’altro invece più consistente, proveniente verosimilmente dalle riserve
mitocondriali. E’ stato anche ipotizzato che l’attivazione dei recettori sigma-2 determini
l’inizio di un processo Ca2+-dipendente nelle cellule PC12, che potrebbe regolare il rilascio
di dopamina, stimolato dall’amfetamina, attraverso la via Ca2+/calmodulina chinasi II
(Guitart et al., 2004).
Tutti questi dati suggeriscono chiaramente come i classici sistemi di secondi
messaggeri giochino un ruolo importante nell’attività dei recettori sigma, benché questo
probabilmente avvenga attraverso un meccanismo diverso da quelli tipici e ben conosciuti.

INTRODUZIONE
134
Molte sono ancora le domande che restano aperte. Bisognerà infatti verificare se
esistano diversi tipi di recettori sigma-1 nei diversi tipi di cellule: se la proteina sigma-1,
recentemente clonata sia solo una componente di un complesso recettoriale a più subunità,
e infine se il recettore sigma sia in realtà solo una porzione di un più ampio canale ionico
(Guitart et al., 2004).
1.8 Recettori sigma come modulatori
1.8.1 Modulazione dei canali per il K+
Per tentare di delineare un meccanismo di traduzione del segnale che caratterizzi i
recettori sigma, occorre considerare anche l’interazione tra questi recettori e i canali ionici.
I recettori sigma sono in grado di modulare canali ionici in molti tipi di cellule. Per
esempio ligandi sigma come (+)-pentazocina e (+)SKF-10,047 possono variare le correnti
di potassio indipendentemente dalla presenza dei nucleotidi guaninici GTPγS o di GDPβS,
suggerendo che nella mediazione di tali risposte le proteine G non svolgano alcun ruolo.
E’ stato dimostrato che i recettori sigma sono in grado di modulare i canali del K+ a
controllo di potenziale, in modo diverso a seconda della presenza o dell’assenza del
ligando sigma esogeno. E’ stato quindi suggerito che i recettori sigma di per sé possono
costituire delle subunità ausiliarie per i canali del K+ voltaggio dipendenti (Kv 1.4, KV 1.5)
fornendo un diverso meccanismo di trasduzione del segnale dipendente dall’interazione
proteina-proteina (Aydar et al., 2002).
1.8.2 Modulazione delle correnti di Ca2+
I recettori sigma sembrano essere coinvolti nella regolazione dell’omeostasi del Ca2+.
E’stata ipotizzata la formazione di un complesso ternario formato dal recettore sigma-1,
dall’ankirina e dal recettore dell’IP3 (Hayashi e Su, 2003). In accordo con questo modello,
agonisti quali (+)-pentazocina o neurosteroidi, interagendo con il loro sito di binding,
fanno sì che il complesso recettore sigma-1/ankirina si dissoci dal recettore dell’IP3 e che
traslochi sulla membrana nucleare o plasmatica. La dissociazione causerebbe un
incremento del legame dell’IP3, il quale a sua volta determinerebbe un rilascio di Ca2+ dai
depositi intracellulari. Viceversa, antagonisti del recettore sigma-1, provocherebbero la

INTRODUZIONE
135
dissociazione dello stesso dall’ankirina, la quale rimarrebbe associata al recettore dell’IP3,
sul reticolo endoplasmatico.
In esperimenti volti a studiare meccanismi neuroprotettivi è stata osservata la
capacità di ligandi sigma di regolare i canali per il calcio voltaggio-dipendenti in colture di
neuroni corticali di ratto: in particolare composti agonisti sembrano deprimere velocemente
le correnti di calcio (Guitart et al., 2004).
Inoltre è stato dimostrato che agonisti dei recettori sigma-1 come (+)-pentazocina e
BD-737 riducono le concentrazioni intracellulari di Ca2+ indotte da muscarina in cellule di
neuroblastoma umano SH-SY5Y. Questo effetto inibitorio però è probabilmente dovuto al
blocco dei recettori muscarinici da parte dei suddetti ligandi piuttosto che all’attivazione
dei recettori sigma-1 (Hong e Werling, 2002).
1.8.3 Modulazione del glutammato
I recettori sigma possono regolare la trasmissione glutammatergica attraverso la
modulazione del complesso recettoriale NMDA. Somministrazioni microiontoforetiche o
endovenose di basse dosi di ligandi sigma-1 come la (+)-pentazocina potenziano
l’eccitabilità neuronale indotta dall’attivazione dei recettori NMDA nei neuroni piramidali
dell’area CA3 dell’ippocampo di ratto.
Questo effetto non si verifica in risposta ad antagonisti sigma-1 come aloperidolo o
N,N-dipropil-2-[4-metossi-3-(2-feniletossi)-fenil]etilamina (NE-100) che, al contrario,
sopprimono il potenziamento prodotto dagli agonisti sigma.
Analogamente ai ligandi sigma-1, anche gli agonisti sigma-2 potenziano le risposte
NMDA-indotte (Skuza, 2003). Ad esempio la somministrazione di ligandi ad alta affinità
per il recettore sigma-2 aumenta in maniera dose-dipendente la risposta NMDA-indotta
nella regione CA3 dell’ippocampo di ratto, mentre l’ibogaina, un ligando con moderata
affinità per il recettore sigma-2, incrementa debolmente la stessa risposta. Per spiegare tali
differenze è stata ipotizzata l’esistenza di più di un sottotipo recettoriale sigma-2.
Anche gli steroidi neuroattivi possono modulare le risposte NMDA-mediate. In
particolare, il deidroepiandrosterone (DHEA) potenzia l’attività elettrica NMDA-evocata
nei neuroni piramidali nell’area CA3 dell’ippocampo, al pari degli agonisti sigma-1, e tale
effetto più venire antagonizzato dall’aloperidolo o dal NE-100, nonché dal progesterone
(Skuza, 2003). Uno schema riassuntivo del ruolo dei recettori sigma-1 nei confronti
dell’omeostasi del calcio e dei recettori ionotropici del glutammato NMDA è riportato in
figura 4.

INTRODUZIONE
136
Figura 4: Modello del ruolo del recettore sigma-1 nella trasmissione glutammatergica. I recettori sigma-1 presenti sul reticolo endoplasmatico regolano i livelli di Ca2+ attraverso il recettore dell’IP3 mediante l’associazione del recettore con proteine del citoscheletro. Tramite questa modulazione, i recettori sigma-1 influenzano svariati processi fisiologici a livello della membrana plasmatica (da Hashimoto e Ishiwata, 2006).
1.8.4 Modulazione allosterica mediata dalla fenitoina
Per quanto riguarda i recettori sigma-1, tra i metodi utilizzati per discriminare
composti ad attività agonista da quelli ad attività antagonista, l’unico test che impiega una
metodica in vitro si basa sulla modulazione allosterica della fenitoina verso recettori
sigma-1 riportata da Cobos e collaboratori (2005) in membrane di cervello di porcellino
d’india. La modulazione allosterica si verifica quando il legame di un ligando o di un
substrato facilita l'interazione di altre molecole di ligando o substrato con altri siti di
legame sul medesimo complesso recettoriale o enzimatico determinando una variazione
conformazionale della subunità cui è legato. Questo legame modifica i restanti siti attivi
accrescendo la loro affinità per il ligando stesso.
E’ stato inizialmente riportato che la presenza di fenitoina (DPH) aumenta
preferenzialmente il binding di radioligandi sigma-1 come [3H]-destrometorfano,
[3H](+)SKF-10,047 (Musacchio et al., 1988) e [3H](+)pentazocina (Cobos et al., 2005). In

INTRODUZIONE
137
seguito altri studi misero in evidenza che l’aggiunta di questo composto in esperimenti di
competizione con la tecnica del radioligand binding è in grado di variare l’affinità di un
agonista come la (+)-pentazocina verso il recettore e di conseguenza di spostare verso
sinistra la curva di spiazzamento, mentre non ha alcun effetto su composti antagonisti
come aloperidolo e NE-100 (Cobos et al., 2006). Questo effetto è stato spiegato sulla base
del modello del recettore a due stati, in base al quale un modulatore allosterico positivo
stabilizza la forma attiva del recettore, a spese della forma inattiva. In questo modo
aumenterebbe il binding di composti agonisti, mentre si ridurrebbe il legame di agonisti
inversi (Cobos et al, 2006).
1.9 Ruolo dei recettori sigma nella neuroprotezione
La possibilità di progettare farmaci per il trattamento delle diverse patologie del SNC
è legata allo sviluppo delle conoscenze del ruolo svolto dai diversi sistemi recettoriali e dai
rispettivi neurotrasmettitori e neuromodulatori coinvolti. Infatti, l’insorgenza delle
patologie stesse potrebbe essere ritardata sia stimolando l’attivazione dei sistemi
neuroprotettivi, sia inibendo quella dei sistemi neurolesivi.
E’ stato dimostrato che il recettore sigma-1 è in grado di modulare l’attività di diversi
sistemi recettoriali coinvolti nelle patologie neurodegenerative.
I meccanismi tramite cui i ligandi sigma esercitano un effetto protettivo in modelli
animali sembrano essere diversi, e comprendono (Maurice et al., 2006; Goyagi et al.,
2001):
• inibizione del rilascio di glutammato indotto da ischemia
• riduzione dei livelli intracellulari di Ca2+ indotto dall’attivazione dei recettori
NMDA post-sinaptici
• riduzione della produzione di ossido nitrico (a livello neuronale)
• riduzione della risposta indotta dall’attivazione dei recettori NMDA
• riduzione della trasmissione dopaminergica
• prevenzione della diffusione della depressione corticale
1.9.1 Studi in vivo
Numerosi studi condotti in vivo hanno dimostrato la capacità di composti ad attività
agonista sigma-1 di esercitare effetti benefici a livello del SNC. Ad esempio, agonisti come
dimemorfano e 2-(4-morpholino)ethyl 1-phenylcyclohexane-1-carboxylate hydrochloride

INTRODUZIONE
138
(PRE-084), somministrati durante riperfusione post-ischemica indotta in ratti, hanno
dimostrato di diminuire i livelli di glutammato e di sopprimere gli stadi iniziali dello stato
infiammatorio, riducendo danno tessutale e morte cellulare (Matsuno, 1999; Shen et al.,
2008). Similmente, il composto agonista (4-phenyl-1-(4-phenylbutyl)piperidine (PPBP) si
è dimostrato in grado di attenuare la produzione di ossido nitrico in ratti e topi in cui il
danno ischemico focale è stato indotto da occlusione transitoria di un arteria cerebrale
(Goyagi et al., 2001).
In modelli di amnesia indotta da antagonisti colinergici o da antagonisti non-
competitivi del recettore NMDA, agonisti sigma-1 come (+)-pentazocina e PRE-084 hanno
mostrato proprietà anti-amnesiche, attenuando lo “spatial learning-impairement” e il
“memory impairement” associati all’invecchiamento (Maurice, 2002). Allo stesso modo,
diversi ligandi sigma-1 si sono altresì mostrati capaci di alleviare il deficit di
apprendimento e di memoria in topi trattati con il frammento attivo 25-35 della proteina β-
amiloide, modello di studio per il morbo di Alzheimer (Marrazzo et al, 2005). Anche
alcuni prototipi di agonisti come la igmesina nonché il PRE-084 si sono dimostrati efficaci
in test in vivo volti a valutare l’entità del deficit di memoria e apprendimento nel “water
maze test” in ratti giovani (Meunier et al., 2004; Maurice, 2001).
Dato che i recettori sigma-1 sono localizzati a livello di ipotalamo e amigdala, zone
collegate alla regolazione di diversi stati emozionali quali l’ansia e l’aggressività, si è
studiato il potenziale effetto di ligandi agonisti come (+)SKF 10,047, in modelli animali
quali quello dell’ isolamento forzato, registrando una significativa attenuazione del
comportamento aggressivo (Beltran et al., 2006). Anche il comportamento ossessivo-
compulsivo viene ridotto dopo somministrazione degli agonisti sigma-1 (+)SKF-10,047 e
PRE-084, come dimostrato impiegando il “marble burying test” (Egashira et al., 2007).
Inoltre, altri composti hanno dimostrato, nei modelli animali di depressione quali il “forced
swimming test” e il “tail suspension test”, di ridurre il tempo d’immobilità e di attenuare
altri comportamenti indotti da stress e/o paura, suggerendo una loro potenziale utilità nel
trattamento di stati depressivi e ansiosi (Hashimoto e Ishiwata, 2006).
L’efficacia dei ligandi agonisti nei modelli sopra citati è stata generalmente avvallata
dalla capacità di composti ad attività antagonista di prevenire o di antagonizzare l’effetto
comportamentale degli agonisti. Ciò nonostante, alcuni ligandi sigma-1 ad attività
antagonista sono risultati neuroprotettivi in modelli di ischemia cerebrale. Ad esempio, è
emerso da diversi studi che l’aloperidolo, potente antipsicotico e antagonista dei recettori
sigma-1, a basse concentrazioni ha la capacità di diminuire lesioni ischemiche indotte da
occlusione transitoria di una arteria cerebrale (Schetz et al., 2007).

INTRODUZIONE
139
Infine, è stato dimostrato che, al pari dell’aloperidolo, il composto BD-1047 esercita
attività anti-psicotica, valutata come la capacità di ridurre l’iperattività indotta da
amfetamina e il comportamento di arrampicamento indotto dall’apomorfina (Skuza e
Rògoz, 2006). Lo stesso composto BD-1047 è risultato in grado di ridurre le sensazioni
dolorifiche causate dal test della formalina, a seguito del blocco selettivo dei recettori
sigma-1 (Kim et al., 2006).
1.9.2 Studi in vitro
La capacità degli agonisti selettivi dei recettori sigma-1 di esercitare effetti
neuroprotettivi in vivo è stata confermata da numerosissimi studi condotti in vitro, in cui
l’insulto in grado di danneggiare cellule di origine neuronale è stato causato con diversi
paradigmi come (Maurice et al., 2006):
• alte dosi di glutammato o NMDA
• condizioni ipossiche o da deprivazione di glucosio
• stress ossidativo
• applicazione della proteina β-amiliode
• induzione di apoptosi
• ischemia
E’ stato ipotizzato che gli steroidi possano essere ligandi endogeni dei recettori
sigma-1 e che i recettori sigma possano avere un ruolo nel metabolismo dei neurosteroidi.
A conferma di ciò è stato dimostrato che steroidi endogeni neuroattivi, quali il
pregnenolone e il deidroepiandrosterone, interagiscono effettivamente con la proteina
sigma-1 attivandola e mostrando in vitro attività neuroprotettiva contro i danni provocati
da stress ossidativo implicati nella patogenesi del morbo di Alzheimer (Maurice et al.,
2006).
Prendendo in considerazione il prototipo ad attività agonista PPBP, in grado di
attenuare in vivo i danni da ischemia cerebrale focale, è stata dimostrata la sua capacità di
proteggere in vitro colture primarie di neuroni esposte all’azione eccitotossica del
glutammato o alla deprivazione di ossigeno e/o glucosio (Goyagi et al., 2001). Lo stesso
composto è risultato in grado di esercitare un effetto antiapoptotico, favorendo
l’espressione del gene bcl-2, effetto antagonizzato dal rimcazolo, un antagonista selettivo
dei recettori sigma-1 (Yang et al., 2007 ).

INTRODUZIONE
140
Altri studi hanno esaminato gli effetti tossici causati da iperomocisteinemia su cellule
gangliari di retina. L’omocisteina è un metabolita dell’amminoacido essenziale metionina
coinvolto in malattie come Alzheimer e morbo di Parkinson, in grado di attivare il
recettore per il glutammato NMDA, con conseguente danno neuronale derivante da un
aumentato flusso di calcio e attivazione della cascata apoptotica. Il suo effetto viene
bloccato in vitro da agonisti sigma-1 come la (+)-pentazocina, attraverso modulazione del
recettore NMDA (Martin et al., 2004).
Studi farmacologici hanno evidenziato che l’attivazione di recettori sigma-1 previene
l’aterazione dell’omeostasi del calcio intracellulare causata da deprivazione di sodio
azide/glucosio quale modello d’ischemia indotta in colture di neuroni corticali embrionali
di ratto. L’aumento di Ca2+ intracellulare causa danni irreversibili a livello della membrana
plasmatica attraverso attivazione di canali ionici calcio-sensibili, portando infine
all’attivazione di processi come proteolisi, lipolisi e produzione di specie reattive
dell’ossigeno. Ligandi recettoriali agonisti sigma-1 come DTG e (+)-pentazocina si sono
dimostrati in grado di bloccare diversi canali per il calcio e di deprimere l’ampiezza dei
transienti di Ca2+ indotti da ischemia (Katnik et al., 2006).
Possibili effetti neuroprotettivi sono stati studiati anche attraverso inibizione del
rilascio pre-sinaptico di amminoacidi eccitotossici come il glutammato in diversi tipi di
cellule. A questo scopo sono stati utilizzati ligandi sigma aventi diversa affinità per il sito
della PCP posto all’interno del recettore NMDA, sito che potrebbe essere selettivo per
ligandi sigma ad attività neuroprotettiva. Per questo studio sono state utilizzate colture
primarie neuronali di ratto, nelle quali è possibile indurre effetti neurotossici sia a livello
post-sinaptico, esponendo le cellule a basse concentrazioni di NMDA, sia a livello pre-
sinaptico, esponendo le cellule a condizioni ipossiche/ipoglicemiche. Questo studio,
condotto da Lockhart e collaboratori (1995), ha dimostrato che ligandi sigma come (+)-
pentazocina e (+)SKF-10,047 inibiscono gli effetti neurotossici seguenti ad eventi
postsinaptici, conseguenti al rilascio di aminoacidi eccitotossici, mentre non modificano gli
effetti neurotossici indotti da ipossia/ipoglicemia.
In conclusione, i recettori sigma sono in grado di modulare diversi sistemi di
neurotrasmettitori quali il sistema glutammatergico e dopaminergico e sono altresì
coinvolti in numerosi processi fisiologici (meccanismi neuroprotettivi) e patologici
(alterazioni motorie e di tipo psicotico). Le potenziali applicazioni terapeutiche dei ligandi
sigma-1 potrebbero essere le patologie neurodegenerative, i disordini neuropsichiatrici
come la schizofrenia, i disordini affettivi e comportamentali come la depressione, nonché

INTRODUZIONE
141
la demenza senile o la malattia di Alzheimer, soprattutto per alleviare i sintomi legati alla
depressione e alla perdita di memoria.
1.10 Applicazioni terapeutiche dei ligandi sigma
Nonostante sia necessario identificare con certezza i meccanismi d’azione e i ligandi
endogeni dei recettori sigma-1, numerose funzioni farmacologiche, biochimiche e anche
terapeutiche sono state proposte. Vista l’ampia distribuzione dei recettori sigma a livello
del SNC, i ligandi sigma possono venire utilizzati nel trattamento di diversi disturbi
neurologici di origine centrale come alterazione dell’umore, perdita della memoria e delle
percezioni sensoriali, effetti causati da abuso di droghe o discinesia tardiva. Inoltre,
considerando la distribuzione dei recettori sigma a livello periferico, questi ultimi possono
anche essere coinvolti nella modulazione dei movimenti gastrointestinali sotto condizioni
di stress, nella secrezione di bicarbonato a livello duodenale, nella contrazione dei vasi
deferenti, nella regolazione dei livelli plasmatici di ormoni come prolattina, corticosterone,
corticotropina (Guitart et al., 2004).
1.10.1 Schizofrenia
La schizofrenia è una patologia che coinvolge gli aspetti più misteriosi della mente
umana, come le emozioni e i processi cognitivi, e si dimostra di difficile indagine per la
ricerca.
Nella patogenesi della schizofrenia la teoria dopaminergica è stata per lungo tempo
la teoria predominante per tentare di spiegare le disfunzioni molecolari proprie della
patologia schizofrenica. Le numerose incongruenze tra meccanismo d’azione e l’efficacia
clinica dei farmaci dopaminergici hanno messo in evidenza le limitazioni della teoria
dopaminergica e hanno fatto ipotizzare il coinvolgimento di altri sistemi neurorecettoriali.
L’ipotesi alternativa implica alterazioni della funzione recettoriale glutammatergica di tipo
NMDA. E’ stato dimostrato che nel cervello di pazienti schizofrenici esistono alterazioni
strutturali e funzionali a carico della corteccia cerebrale nella quale l’amminoacido
glutammato è il principale neurotrasmettitore. Inoltre la fenciclidina (PCP), antagonista dei
recettori NMDA, è in grado di produrre psicosi simil-schizofrenica in soggetti sani con
comparsa di allucinazioni, deliri, disfunzioni cognitive, disturbi del pensiero.

INTRODUZIONE
142
Le prime evidenze a favore del coinvolgimento dei recettori sigma nella
patofisiologia della schizofrenia derivano dalla scoperta che ligandi sigma come (+)SKF-
10,047 esibiscono effetti psicotomimetici nell’uomo e che molti neurolettici hanno
un’elevata affinità per i recettori sigma. Queste scoperte lasciano supporre che i recettori
sigma possano giocare un ruolo determinante nell’azione antipsicotica. Studi post-mortem
su cervello umano hanno evidenziato la presenza di recettori sigma in strutture corticali e
limbiche, la cui densità risulta significativamente ridotta in individui affetti da
schizofrenia.
Nonostante siano state condotte numerose ricerche su nuovi composti antipsicotici
con alta affinità per il recettore sigma, fino ad oggi nessun ligando sigma selettivo è entrato
in commercio come farmaco neurolettico.
In base a numerosi test neurofarmacologici molti composti sono stati identificati
come potenziali antipsicotici, uno dei quali è il [α-(4-fluorofenil)-4-(5-fluoro-2-
pirimidinil)-1-piperazinbutanolo cloridrato] (BMY-14802). Questo composto ha alta
affinità per i recettori sigma e per quelli serotoninergici, 5-HT1A e 5-HT2 e bassa affinità
per i recettori della dopamina e di altri neurotrasmettitori. Il BMY-14802 è stato
identificato come possibile farmaco antipsicotico con potenza minore rispetto
all’aloperidolo, prototipo degli antipsicotici ma simile alla clozapina. Inoltre, il BMY-
14802 è in grado di bloccare il processo di sensibilizzazione alla metamfetamina e lo
sviluppo di sensibilizzazione alla cocaina, che rappresenta un modello farmacologico di
schizofrenia in quanto può generare una sindrome indistinguibile da un episodio acuto di
schizofrenia. Nonostante il profilo pre-clinico del BMY-14802 risultasse favorevole, in
grado di mediare gli effetti antipsicotici senza indurre effetti collaterali di tipo
extrapiramidale associati ai comuni neurolettici, trials clinici seguenti non hanno
evidenziato un significativo miglioramento dei sintomi psichiatrici.
NE-100 è un altro potenziale composto antipsicotico ad alta affinità per il recettore
sigma-1 e bassa affinità per i recettori sigma-2, della dopamina, della serotonina e della
fenciclidina. NE-100 antagonizza, nel ratto, gli effetti comportamentali della fenciclidina,
senza indurre catalessi. Inoltre, NE-100, così come altri antagonisti sigma-1, modula il
rilascio di dopamina mediato dal recettore NMDA e altera le correnti NMDA-mediate nei
neuroni dopaminergici. L’evidenza sperimentale suggerisce che i ligandi sigma sono in
grado di modulare sia la trasmissione dopaminergica che quella glutammatergica.
Oltre ai presunti effetti benefici sulla sintomatologia schizofrenica, è stato ipotizzato
che ligandi sigma contribuiscano al manifestarsi degli effetti collaterali di tipo motorio,
osservati dopo trattamento a lungo termine con farmaci antipsicotici. Gli studi condotti

INTRODUZIONE
143
negli ultimi anni sembrano confermare l’importanza dei recettori sigma nel controllo della
funzione motoria e nella regolazione delle reazioni distoniche (Guitart et al., 2004).
1.10.2 Memoria e Deficit di attenzione
In modelli di amnesia, gli agonisti sigma possono avere degli effetti benefici. Alcuni
ligandi sigma-1 come l’igmesina hanno dimostrato di prevenire o di far regredire l’amnesia
indotta dalla scopolamina in ratti e in topi. Questo risultato ha confermato l’ipotesi per cui
gli agonisti sigma-1 sono in grado di modulare le risposte comportamentali mediate dal
recettore colinergico di tipo nicotinico, aumentando il rilascio di acetilcolina sia
nell’ippocampo che nella corteccia frontale. In particolare l’agonista sigma-1 altamente
selettivo 1-(3,4-dimethoxyphenethyl)-4-(3-phenyl propyl)piperazine dihydrochloride (SA-
4503) è in grado di invertire l’amnesia indotta da scopolamina e di attenuare le difficoltà
dell’apprendimento negli animali che presentano delle disfunzioni colinergiche corticali.
Inoltre i ligandi sigma-1 mostrano effetti anti-amnesici nei disturbi dell’apprendimento
indotti dal blocco del recettore di tipo NMDA e possono potenziare molte risposte NMDA-
evocate in determinate regioni dell’ippocampo.
Anche gli steroidi neuroattivi possono avere un ruolo nella modulazione della
memoria da parte dei recettori sigma. Infatti il DHEA ed il progesterone hanno alta affinità
per il recettore sigma-1. Si è visto che pregnenolone, DHEA e loro esteri solfato
aumentano la capacità di fissazione della memoria nei topi con problemi di apprendimento,
mentre il progesterone è un antagonista e blocca gli effetti degli steroidi neuroattivi e di
alcuni agonisti sigma-1 (Guitart et al., 2004; Maurice et al., 2006).
1.10.3 Depressione e ansia
Recenti studi hanno dimostrato che farmaci antidepressivi come fluvoxamina,
fluoxetina, citalopram, sertralina e imipramina possiedono affinità da moderata ad alta per i
recettori sigma-1, suggerendo che i ligandi sigma possano esercitare azione antidepressiva.
Agonisti selettivi del recettore sigma-1 come (+)-pentazocina, (+)SKF-10,047 o SA-4503
riducono la durata dell’immobilità nel test del nuoto forzato (“forced swimming test”) e nel
test di sospensione della coda nel topo (“tail suspension test”), due modelli sperimentali
ampiamente utilizzati per testare l’attività antidepressiva di nuovi composti. Anche
l’agonista non selettivo 1-{3-[4-(3-chlorophenyl)-1-piperazinyl]propyl}-5-methoxy-3,4-
dihydro-2-quinolinone monomethanesulfonate (OPC-14523) e l’agonista selettivo sigma-1

INTRODUZIONE
144
igmesina mostrano un’attività di tipo antidepressivo nei modelli animali (Guitart et al.,
2004).
Recentemente è stato riportato che farmaci antidepressivi non inducono soltanto
alterazioni neurochimiche ma anche rimodellamenti strutturali come neo-formazione di
neuriti. E’ stato dimostrato che (+)-pentazocina, ligando sigma-1, imipramina e
fluvoxamina, farmaci antidepressivi aventi attività sui recettori sigma-1, possono
potenziare la neuritogenesi indotta dal fattore di crescita neuronale NGF nelle cellule di
feocromocitoma PC12, evidenza sperimentale di particolare interesse in quanto conferma
che l’attività terapeutica degli antidepressivi possa comprendere anche la neoformazione di
neuriti (Takebayashi et al., 2002; Takebayashi et al., 2004; Hashimoto et al., 2006).
I primi studi su topi knockout per il recettore sigma-1, non avevano evidenziato
particolari differenze tra topi mutati e wild-type tranne per quanto riguarda una
significativa diminuzione della risposta nell’ipermotilità nei topi knockout (Langa et al.,
2003).
Sabino e collaboratori hanno caratterizzato la risposta di topi knockout per il
recettore sigma-1 in due test comportamentali per lo stato ansioso (“light-dark transfer
test” e “elevated plus maze test”) e due per la valutazione dello stato depressivo (“forced
swim test” e “motor behavior test”). Da questi studi è emerso che i topi knockout
rispondono allo stesso modo dei topi wild-type per quanto riguarda i test comportamentali
per lo stato emozionale ansioso mentre si ha una diversa risposta nel “forced swim test” e
nel “motor behavior test”. Si ha infatti un aumento dell’immobilità e una contemporanea
diminuzione del nuoto nei topi knockout rispetto ai wild-type. Nessuna differenza è stata
notata invece per il climbing. Questi risultati portano alla conclusione che l’assenza di
recettori sigma-1 nel topo porta allo sviluppo di un fenotipo simil-depressivo e non simil-
ansiogeno (Sabino et al., 2009)
1.10.4 Abuso di sostanze stupefacenti
A tutt’oggi non sono state ancora individuate terapie farmacologiche efficaci per il
trattamento dell’abuso di cocaina, D-amfetamina e di altre sostanze stupefacenti. Il sistema
dopaminergico costituisce, almeno in parte, il substrato neuroanatomico della
tossicodipendenza. Infatti, il principale meccanismo d’azione della cocaina sembra essere
l’inibizione della ricaptazione delle amine simpaticomimetiche, in particolare della
dopamina, ma è stato anche dimostrato che la cocaina è in grado di legarsi ai recettori

INTRODUZIONE
145
sigma, con una preferenza dieci volte maggiore per il sottotipo sigma-1, rispetto al
sottotipo sigma-2.
Alcuni antagonisti selettivi del recettore sigma-1, come BMY-14802, BD-1047 e
BD-1063, sono in grado di ridurre gli effetti locomotori della cocaina. Un effetto simile è
stato osservato anche in topi trattati con oligodeossinucleotidi antisenso corrispondenti alla
sequenza sigma-1 recettoriale (Guitart et al., 2004). L’antagonismo funzionale dei recettori
sigma può essere ottenuto usando sia antagonisti farmacologici sia oligodeossinucleotidi
antisenso. Questi ultimi inibiscono la sintesi di nuove proteine recettoriali, causando la
deplezione dei recettori disponibili per il legame. E’ stato dimostrato che composti ad
attività antagonista sono in grado di ridurre le convulsioni indotte da cocaina in topi e di
attenuare la “place preference”, un modello animale usato per valutare i fenomeni di
gratificazione indotti dalla cocaina e da altre sostanze stupefacenti. Nel loro insieme, questi
dati suggeriscono che l’attivazione dei recettori sigma-1 da parte della cocaina potrebbe
essere importante per combattere gli effetti sia a breve, sia a lungo termine, della droga.
Tali dati inoltre indicano che i recettori sigma-1 rappresentano un bersaglio molto utile per
lo sviluppo di future strategie terapeutiche nel campo della tossicodipendenza.

INTRODUZIONE
146
1.11 Tecnica del Radioligand Binding
Una tecnica largamente impiegata per lo studio dei recettori è la tecnica di “binding
radiorecettoriale” (radioligand binding), che permette di ottenere informazioni dettagliate
su distribuzione e densità dei diversi recettori sia su tessuti animali, sia su linee cellulari
mammifere, e la loro caratterizzazione biochimica e farmacologica. Lo sviluppo e
l’utilizzo del “radioligand binding”, applicato per la prima volta negli anni ’70, ha un ruolo
di primaria importanza nella ricerca di base e clinica e in campo terapeutico per il suo
ruolo pilota nella scoperta di nuovi farmaci (Nicosia, 1999).
La tecnica di binding si basa sulla possibilità di misurare selettivamente e
quantitativamente la radioattività che si lega ai recettori. A questo scopo si utilizza un
ligando marcato, in genere trizio (3H), ad alta attività specifica, per assicurare stabilità ed
elevata selettività in modo che composti farmacologicamente simili a quello marcato
interferiscano con il legame di quest’ultimo al recettore. Inoltre, dato che la concentrazione
dei recettori è molto bassa, il radioligando deve essere un agonista o un antagonista ad
elevata affinità per il recettore.
Le principali tecniche di ”binding radiorecettoriale”, sono costituite da:
- esperimenti di saturazione: dove si utilizzano concentrazioni crescenti di ligando
marcato,
- esperimenti di competizione: esperimenti di spiazzamento, in cui tutti i campioni
contengono la stessa concentrazione di ligando marcato e concentrazioni crescenti di
composto in esame. Il vantaggio principale di questo procedimento consiste nella
possibilità di valutare l’affinità per un recettore di un farmaco non marcato, a condizione
che si abbia a disposizione un altro ligando radioattivo che si leghi allo stesso recettore.
L’esecuzione degli studi di binding comporta l’incubazione di una preparazione
(cellule o membrane) contenente il recettore e il ligando in esame. In condizioni di tempo,
temperatura e pH controllati determinati sperimentalmente, si raggiunge una situazione di
equilibrio in cui il radioattivo sarà:
a) libero in soluzione
b) legato al recettore specifico (legame specifico)
c) legato a siti non-specifici come lipidi, altre proteine o enzimi (legame non
specifico o aspecifico).
Per separare il complesso ligando-recettore si filtra il campione: le membrane (o
cellule) e quindi il recettore vengono trattenuti dal filtro, mentre il ligando rimasto libero

INTRODUZIONE
147
passa attraverso esso. Questo procedimento viene eseguito velocemente e con l’utilizzo di
tampone freddo in modo da non variare la concentrazione di complesso farmaco-recettore,
rimasta sul filtro. Si valuta infine la radioattività associata al filtro attraverso scintillazione
in fase liquida (Nicosia, 1999).
L’apparecchiatura fornisce letture in conte per minuto (CPM). Se si conosce l'attivita'
specifica della sostanza radioattiva (numero di DPM per mole), si può convertire la
quantità di radiattività legata in moli di ligando legate (e quindi il numero di recettori
legati).
Affinchè gli esperimenti abbiano un significato biologico, occorre che il legame
misurato sia saturabile e che raggiunga un plateau, nonostante l’aggiunta di concentrazioni
crescenti di radioattivo, indicando che tutti i recettori disponibili sono stati saturati. Dato
che i composti radioattivi si legano ai costituenti cellulari anche in modo non specifico (in
modo covalente), il legame specifico alle proteine recettoriali si ottiene per differenza tra il
legame totale e quello aspecifico. Per misurare la quota di legame aspecifico occorre
incubare il tessuto (o cellule) in presenza di composto marcato e di una quantità fissa a
concentrazione molto alta di legante freddo, non marcato. In questo modo il ligando freddo
spiazzerà il composto marcato da tutti i siti saturabili recettoriali, lasciando invariato il
legame ai siti aspecifici.
Riassumendo:
Legame totale: radioattività risultante dall'incubazione con radioattivo e membrane
Legame non-specifico: radioattività risultante dall'incubazione con radioattivo,
membrane, ed un eccesso di ligando non radioattivo.
Binding specifico = binding totale – binding non specifico
L’analisi quantitativa dei dati sperimentali di binding tiene conto solo della quota
legata in modo specifico (Racagni et al., 1986).

2. SCOPO DELLA TESI

SCOPO DELLA TESI
148
Durante gli ultimi anni, molti studi sono stati rivolti alla comprensione del sistema
dei recettori sigma, la cui identità separata da recettori di altri sistemi di neurotrasmettitori
è ormai largamente accettata. Il motivo di questi sforzi risiede nella convinzione, basata su
moltissime evidenze sperimentali, che questi recettori siano implicati in una miriade di
funzioni cellulari, processi biologici e patologie, tra cui ad esempio, la regolazione di
funzioni la cui alterazione è alla base di malattie neurodegenerative e neuropsichiatriche.
In particolare, queste attività sarebbero principalmente mediate dai recettori sigma-1,
mentre i recettori sigma-2 sarebbero principalmente implicati nella proliferazione di cellule
neoplastiche.
L’identificazione di nuovi siti recettoriali ha come conseguenza quella di
determinarne un profilo farmacologico, con l’identificazione di composti potenti e selettivi,
utili all’ulteriore approfondimento delle funzioni e dei ruoli dei recettori stessi. Di questi
composti, inoltre, è importante stabilirne l’efficacia, utile per l’identificazione delle
specifiche vie di trasduzione del segnale.
Lo scopo di questa tesi è stato quello di individuare, all’interno di una serie di
composti sintetizzati presso il Dipartimento di Scienze Farmaceutiche dell’Ateneo, quelli
dotati di maggior affinità e selettività verso i recettori sigma-1 e di stabilirne l’efficacia.
Per quanto riguarda la determinazione dell’affinità e selettività, questa è stata
effettuata con la tecnica del radioligand binding, che ha permesso di individuare tre nuovi
composti di potenziale interesse.
Per quanto riguarda l’identificazione dell’efficacia, un’analisi della letteratura ci ha
indirizzato verso la scelta di due metodiche in vitro: la prima legata alla modulazione
allosterica del legame di composti agonisti, e quindi eseguibile nuovamente con la tecnica

SCOPO DELLA TESI
149
del radioligand binding, la seconda basata sull’impiego di una linea di neuroblastoma
umano denominata SH-SY5Y. In questo caso, abbiamo pensato di sfruttare l’eventuale
capacità dei ligandi sigma di indurre differenziazione, misurabile con la riduzione della
proliferazione da una parte, e la stimolazione alla neuritogenesi dall’altra.
I risultati ottenuti suggeriscono che, tra i composti studiati, almeno uno presenta
un’affinità e una selettività che gli conferiscono una potenziale importanza nello sviluppo
di un profilo farmacologico dei recettori sigma. Inoltre, le tecniche impiegate per la
determinazione dell’efficacia sembrano essere adeguate allo scopo e potrebbero costituire
una valida alternativa alla metodica attualmente più avvalorata, il test in vivo del Morris
Water Maze (Tottori et al, 2002).

3. MATERIALI E METODI

MATERIALI E METODI
150
3.1 Esperimenti di Radioligand Binding Gli esperimenti di radioligand binding sono stati svolti per valutare l’affinità e
selettività di numerosi composti di neosintesi.
3.1.1 Preparazione delle membrane di fegato di ratto
Membrane contenenti recettori sigma, sono preparate a partire da fegato di ratto
secondo il metodo descritto da Matsumoto et al., (1995).
I ratti vengono uccisi con etere e decapitati, i fegati vengono posti su ghiaccio ed
eventualmente conservati a -20°C. In alternativa vengono utlizzati fegati prelevati da ratti
neonati (da 20 a 40 ratti) e trattati immediatamente dopo l’asportazione.
In ogni caso i fegati vengono pesati, sminuzzati con delle forbici e omogeneizzati in
10 volumi di una soluzione di saccarosio 0.32 M, pH 7.4 con l’aiuto di un potter (Thomas
Company, Philadelphia), alla temperatura di 4°C e centrifugati a 1000 xg (SIGMA 4K15C-
Bio.Tec.) per 10 minuti a 4°C.
Si ottiene un surnatante S1, conservato su ghiaccio, e un pellet P1 che viene
risospeso in 2 volumi di una soluzione fredda di saccarosio 0.32 M, pH 7.4 e ricentrifugato
come sopra. Il surnatante S2 viene unito al surnatante S1 precedentemente ottenuto, mentre
il pellet P2 viene eliminato. L’insieme dei surnatanti viene quindi centrifugato a 31000 xg
(SIGMA 3K30-Celbio) per 15 minuti a 4°C.
A questo punto si ottiene un pellet P3 che viene risospeso in 3 volumi di Tris-HCl
10 mM a pH 7.4 e la sospensione viene lasciata a incubare per 15-30 minuti a

MATERIALI E METODI
151
temperatura ambiente (25°C). Scaduto il tempo, la sospensione è centrifugata sempre a
31000 x g per 15 minuti a 4°C.
Il pellet finale viene infine risospeso in 1.53 volumi di Tris-HCl 10 mM a pH 7.4 e
viene effettuata un ulteriore omogeneizzazione con potter.
La sospensione viene ripartita in aliquote da 500 µl e conservate a -20°C.
La quantificazione delle proteine presenti nella sospensione di membrane usate
negli esperimenti di radioligand binding viene eseguita utilizzando il kit BioRad DC
Protein Assay, basato sul metodo di Lowry et al. (1951) già descritto nel capitolo Materiali
e Metodi a pag.43.
3.1.2 Esperimenti di competizione su membrane di fegato di ratto verso
[3H](+)Pentazocina (σσσσ1)
L’esperimento si basa sulla capacità di alcuni ligandi non marcati di competere con
il ligando triziato per lo stesso sito di legame, spiazzandolo dal recettore e permettendo di
misurare il grado di affinità del composto nei confronti del recettore.
A questo scopo, concentrazioni fisse di composto marcato sono poste in presenza di
concentrazioni crescenti di ligando non marcato per tempi definiti.
Vengono preparate le soluzioni costituite dal ligando preso in considerazione
(concentrazione iniziale 10 mM in DMSO) opportunamente disciolto in tampone Tris-HCl
50 mM, pH 8 e diluito in modo scalare, al fine di ottenere diverse concentrazioni (250 µl
pari a 0.1 – 10000 nM).
Le membrane vengono scongelate a temperatura ambiente, diluite in tampone Tris-
HCl 50 mM, pH 8 e risospese con un potter per ridurre la granulosità della preparazione.
L’incubazione di 2 ore a 37 ˚C inizia aggiungendo una quantità fissa di sospensione di
membrane (200 µl ≡ 250 µg di proteine → 1.25 mg/ml) e una concentrazione fissa di
[3H](+)PTZ (Pentazocina, [ring-1,3-3H], attività specifica 34.90 Ci/mmol) 1 nM finale in
presenza di concentrazioni crescenti dei composti in esame.
Il legame aspecifico di [3H](+)PTZ viene determinato in presenza di aloperidolo 10
µM finale in DMSO, concentrazione finale 1%.
A fianco della curva di concentrazioni dei composti si allestisce una curva con (+)-
pentazocina non marcata a tre concentrazioni fisse, come controllo interno.
Il volume finale pari a 500 µl, viene raggiunto, sia nelle provette relative al legame

MATERIALI E METODI
152
aspecifico, sia in quelle relative al legame totale, aggiungendo tampone Tris-HCl 50 mM,
pH 8. Le prove vengono condotte in duplicato.
Inoltre vengono preparati due standard, sempre in duplicato, costituiti da 50 µl di
[3H](+)PTZ 10 nM.
Il saggio viene terminato con filtrazione dei campioni sottovuoto. Si usano filtri in
microfibra di vetro Whatmann GF/B (Millipore) precedentemente bagnati con buffer Tris-
HCl contenente lo 0.5 % di PEI (polietilenimina) per 30 minuti.
La filtrazione viene affettuata con uno strumento (Millipore, MA, USA), costituito
da un supporto a tenuta per i filtri e da un attacco per la pompa a vuoto, il quale permette la
filtrazione di 12 campioni in successione. Dopo la filtrazione, ogni campione è risciacquato
due volte con 1 ml di buffer freddo.
Successivamente i filtri vengono asciugati sotto una lampada e inseriti nelle
apposite vials, in cui si aggiungono 3 ml di liquido di scintillazione Filter Count
(PerkinElmer) il quale permette di determinare la radioattività trattenuta sui filtri.
Le vials poste nei supporti vengono lasciate al buio nello spettrofotometro a
scintillazione liquida (Microbeta Trilux Wallac) e messe in conta dopo 12 ore.
I risultati ottenuti, espressi come conte per minuto CPM, vengono trasformati in
picomoli di (+)-pentazocina legati nel modo seguente: alle CPM relative al legame totale
viene sottratto il valore delle CPM relative al legame aspecifico in modo da ottenere la
quota di legame specifico. Le conte che derivano dalla lettura dello standard permettono di
risalire all’efficienza di conteggio ponendo in rapporto i valori delle CPM sperimentali e le
disintegrazioni per minuto (DPM) teoriche. Poiché 1 nCi corrisponde a 2220 DPM, le
DPM teoriche dello standard si ottengono moltiplicando la quantita di radioattivo teorico in
nCi per 2220 DPM.
Una volta nota l’efficienza di lettura è possibile trasformare i CPM relativi ad ogni
vials in DPM. Infine, conoscendo l’attività specifica dello standard (77478 DPM/pmol) si
possono determinare le pmoli legate per ogni vials.
Le pmoli vengono poi rapportate ai mg di proteine ottenute dal dosaggio proteico.

MATERIALI E METODI
153
3.1.3 Esperimenti di competizione su membrane di fegato di ratto verso [3H]DTG
(σσσσ2)
Gli esperimenti di competizione verso il sottotipo recettoriale σ2 vengono effettuati
analogamente a come descritto per gli esperimenti di competizione verso i recettori σ1 ma
utilizzando come composto marcato la [3H]-DTG ([5-3H]-1,3-Di-O-tolylguanidine, attività
specifica 56.0 Ci/mmol) 3 nM finale e una concentrazione di proteine pari a 188 µg per
campione. Inoltre, per mascherare i siti sigma-1, in ogni provetta si aggiungono 50 µl di
(+)-pentazocina non marcata alla concentrazione finale di 100 nM.
Dopo un’incubazione di 2 ore a temperatura ambiente, si procede con la filtrazione e
la conta come descritto sopra.
3.1.4 Calcolo del valore di IC50 e Ki
I risultati degli esperimenti di competizione permettono di calcolare il valore di IC50
e, da questo, il valore di Ki.
Il valore di IC50 o concentrazione inibente è la concentrazione di un competitore
necessaria a spiazzare il 50% del radioligando dal recettore. L'IC50 è perciò uno dei
parametri comunemente usati nella ricerca farmacologica per misurare la potenza di un
composto. Il valore è calcolato tramite regressione non lineare e può essere ottenuto con
l’aiuto di un programma computerizzato come il SigmaPlot (Jandel Scientific, Germany),
applicando la seguente equazione a 4 parametri:
y = min + (max-min) / 1 + 10 (log EC50-X)Hillslope
Dal valore di IC50 viene ricavato il valore di Ki secondo la formula di Cheng e Prusoff:
Ki = IC50 / 1 + [X]/KD
dove il valore di KD impiegato (costante di dissociazione all’equilibrio) rappresentante la
misura dell’affinità del radioligando verso il recettore all’equilibrio, è stato ottenuto da
esperimenti preliminari di saturazione.
[X] rappresenta la concentrazione di radioattivo impiegata.
La Ki esprime l'affinità del composto in esame per il recettore, cioè la
concentrazione del composto che all’equilibrio legherà il 50% dei siti disponibili in
assenza del radioligando o altri competitori.
La pendenza della parte rettilinea di una curva di competizione sigmoidale può
essere quantificata sulla base di un fattore, definito fattore di Hill (Hill slope).

MATERIALI E METODI
154
Questo fattore fornisce informazioni sulla natura dell’interazione ligando-recettore
e più precisamente è un indice della presenza di uno o più siti di interazione recettoriale.
Se la slope ha un valore uguale o vicino a 1, il ligando interagisce con una singola
popolazione recettoriale e la curva di competizione segue l’andamento previsto dalla legge
di azione di massa per un unico sito. Se invece la slope ha un valore che differisce
significativamente da 1, è probabile che il ligando interagisca con più sottotipi recettoriali
o che esista cooperatività positiva o negativa tra due o più siti.
3.1.5 Esperimenti di competizione su membrane di fegato di ratto verso
[3H](+)Pentazocina in presenza di fenitoina
Sono stati effettuati degli esperimenti di competizione prendendo in considerazione
due composti di riferimento: (+)-pentazocina e aloperidolo, rispettivamente agonista e
antagonista del recettore sigma-1. Gli esperimenti sono stati condotti seguendo il metodo
descritto per gli esperimenti di competizione verso i recettori sigma-1 incubando
concentrazioni fisse di radioligando [3H](+)PTZ con concentrazioni crescenti dei ligandi
non marcati in assenza e in presenza di fenitoina (DPH) 1 mM finale.
In questi esperimenti viene utilizzato il buffer a pH 7.4 per compensare l’aumento di pH
indotto dalla presenza del solvente in cui viene disciolta la fenitoina, l’NaOH, e in questo
modo in ogni provetta il pH finale non è superiore a 8.
In ogni provetta si aggiungono 50 µl di DPH (disciolta in NaOH 0.3M,
concentrazione iniziale di 25 mM) e parallelamente 50 µl di una soluzione di NaOH 0.12
M (12 mM finali) come controllo dell’influenza del solvente.
3.1.6 Esperimenti di saturazione con [3H](+)Pentazocina e [
3H]DTG sulle cellule di
neuroblastoma umano SH-SY5Y
Sono stati svolti esperimenti di saturazione volti a determinare, su cellule SH-SY5Y
intatte, la presenza di recettori σ1 e σ2 e i rispettivi valori di Kd e di Bmax.
Il valore di Kd o costante di dissociazione all’equilibrio è caratteristica di ogni
farmaco e del suo recettore; espressa come mol/L o M, equivale alla concentrazione di
farmaco necessaria ad occupare il 50 % dei recettori ed è quindi un indice di affinità. La
Kd è inversamente correlata all’affinità del ligando per il recettore, per cui ad un aumento
della Kd corrisponde una diminuizione dell’affinità.

MATERIALI E METODI
155
Il valore di Bmax, generalmente espresso come pmoli/mg di proteine presenti in un
dato tessuto, esprime il numero massimo di siti specifici per il ligando presenti nelle cellule
ed è coincidente con il numero di recettori.
Il legame specifico alle condizioni di equilibrio equivale alla frazione di recettori occupata
per il numero di recettori totale (Bmax) e dipende dalla concentrazione del radioligando
(L):
legame specifico = frazione di recettori occupata × [L] / Kd + [L]. Questa equazione descrive un’iperbole rettangolare dove il legame specifico è
ottenuto tramite regressione non lineare con il supporto del programma computerizzato
Sigma Plot (Jandel Scientific, Erkrath, Germany), applicando la seguente equazione:
y = Bmax × x / (Kd + x) Utilizzando il fit di questa iperbole rettangolare è possibile determinare l’affinità
del radioligando per il recettore e la densità dei siti recettoriali. Infatti, il Bmax corrisponde
al valore dell’asintoto a cui tende la curva, mentre la Kd è la concentrazione di
radioligando che satura il 50% di tutti i siti recettoriali presenti.
Le cellule vengono seminate 48 ore prima dell’esperimento in piastre da 24 pozzetti
(FALCON®) ad una densità di 2.5 x 105 cellule in 1 ml di terreno completo per pozzetto, e
mantenute in incubatore a 37°C. Il giorno dell’esperimento si preparano le soluzioni
costituite da [3H](+)PTZ o da [3H]DTG a diverse concentrazioni (1 nM, 3 nM, 10 nM, 30
nM, 50 nM) disciolti in tampone PBS (NaCl 137 mM, KCl 3 mM, Na2HPO4·2H2O 6.4
mM, KH2PO4 1.47 mM).
Quindi si procede secondo il seguente protocollo: si aspira sotto vuoto il mezzo di
coltura da ogni pozzetto, si effettua un risciacquo con il tampone PBS (250 µl) a 37 °C e lo
si sostituisce con le diverse concentrazioni di composto marcato per ottenere
concentrazioni crescenti, procedendo analogamente sui pozzetti successivi.
Per ogni concentrazione di radioligando, il valore del legame aspecifico è ottenuto
predisponendo delle prove in parallelo contenti aloperidolo in DMSO (25 µl, 10 µM
finale). Le prove vengono eseguite in triplicato. Il volume finale di 250 µl in ogni pozzetto,
viene raggiunto con tampone Tris-HCl 50 mM, pH 8.
Dopo un’incubazione di 120 minuti a 37 °C per la [3H](+)PTZ o a temperatura
ambiente per il [3H]DTG, si aspira il surnatante, si lava due volte ciascun pozzetto con
PBS a 4 °C per bloccare la reazione, e si aggiungono 250 µl per pozzetto di una
sospensione di NaOH 1 M contenente 1 % di sodio dodecil solfato (SDS) per disgregare le
membrane cellulari e facilitare il distacco delle cellule dal fondo del pozzetto.

MATERIALI E METODI
156
Successivamente, da ogni pozzetto vengono trasferiti 200 µl della sospensione nei
corrispettivi pozzetti di una piastra flessibile (Wallac) da 24 pozzetti che viene inserita in
un apposito supporto. Si aggiungono 250 µl di liquido di scintillazione Optiphase
‘SuperMix’, si sigilla la piastra con film di plastica (sealing tape, PerkinElmer) e la si
introduce nel contatore a scintillazione liquida (Microbeta Trilux Wallac, Turku, Finlandia)
per la lettura dopo 720 minuti. Inoltre si aggiungono 25 µl di [3H](+)PTZ 50 nM o di
[3H]DTG 30 nM, NaOH 1 M + 1% SDS e liquido di scintillazione, sempre in un volume
finale di 450 µl, per ottenere il valore dello standard.
Le CPM lette per ogni campione sono quindi corrette per l’efficienza di conteggio
della piastra ricavando così le DPM corrispondenti. I loro valori rapportati all’attività
specifica calcolata danno le picomoli di triziato di ogni singolo pozzetto, che sono riportate
come media ± deviazione standard.
3.1.7 Esperimenti di competizione con (+)-Pentazocina e Aloperidolo, su cellule di
neuroblastoma umano SH-SY5Y
L’esperimento si basa sulla capacità di alcuni ligandi non marcati di competere con
il ligando triziato per lo stesso sito di legame, spiazzandolo dal recettore e permette di
misurare il grado di affinità del composto nei confronti del recettore.
A questo scopo, concentrazioni fisse di composto marcato sono poste in presenza di
concentrazioni crescenti di ligando non marcato per tempi definiti.
Le cellule vengono seminate 48 ore prima dell’esperimento in piastre da 24 pozzetti
(FALCON®) ad una densità di 2.5 x 105 cellule in 1 ml di terreno completo per pozzetto, e
quindi mantenute in incubatore a 37°C.
Vengono preparate le soluzioni costituite dal ligando preso in considerazione a
diverse concentrazioni (125 µl pari a 0.0001, 0.001, 0.001, 1, 10, 100, 1000 nM)
opportunamente disciolto in tampone Tris-HCl 50 mM, pH 8. Le cellule vengono lavate
con PBS e l’incubazione inizia aggiungendo ad ogni pozzetto una determinata
concentrazione di ligando (125 µl) e una concentrazione fissa di [3H](+)PTZ 50 nM (15 µl,
3 nM finale) o di [3H]DTG 30 nM (15 µl, 3 nM finale) per ottenere un volume finale di
250 µl. Si allestiscono inoltre dei pozzetti, contenti solo le cellule, Tris-HCl e il
radioligando, che servono per determinare il legame totale. Tutte le prove vengono
condotte in triplicato.

MATERIALI E METODI
157
Le piastre sono riposte in termostato a 37°C per 2 ore e, trascorso il tempo stabilito,
si procede come descritto in precedenza negli esperimenti di saturazione: si aspira il
surnatante, si eseguono due lavaggi con PBS freddo e si aggiungono 250 µl per pozzetto di
una sospensione di NaOH 1 M + 1% di sodio dodecil solfato (SDS). Dopo pochi minuti, si
trasferiscono 200 µl della sospensione ottenuta da ogni pozzetto in altrettanti pozzetti di
una piastra flessibile (Wallac) che viene inserita in un apposito supporto. Si aggiungono
250 µl di liquido di scintillazione Optiphase ‘SuperMix’ (Wallac, Turku, Finlandia), e si
sigilla la piastra con film di plastica.
Due pozzetti vengono utilizzati per effettuare le conte di 15 µl del ligando triziato
utilizzato (standard) in un volume finale di 250 µl di NaOH 1 M + 1% SDS e di liquido di
scintillazione. La lettura si effettua dopo 12 ore.
3.2 Esperimenti di differenziazione
Per discriminare tra attività agonista o antagonista dei composti di riferimento e di
alcuni composti di neosintesi è stata valutata la capacità degli stessi di differenziare le
cellule SH-SY5Y. Questo approccio sfrutta la capacità degli agonisti sigma-1 di indurre
differenziazione e contemporaneamente ridurre la proliferazione cellulare. Questi
esperimenti sono stati condotti seguendo i protocolli descritti nel capitolo Materiali e
Metodi pag. 41 e 42.

4. RISULTATI

RISULTATI
158
4.1 Esperimenti di competizione su membrane di fegato di ratto verso
[3H](+)Pentazocina e [
3H]DTG
Tra i composti sintetizzati dal Dipartimento di Chimica Farmaceutica dell’Ateneo
di Trieste in collaborazione con l’Università degli Studi di Pavia, è iniziato uno screening
volto ad identificare i ligandi che presentano maggiore potenza e selettività nei confronti
dei recettori sigma. A questo scopo sono stati effettuati esperimenti di competizione
mediante la tecnica del Radioligand binding, impiegando due radioligandi utili per la
marcatura dei recettori sigma-1 e sigma-2. Lo screening ha interessato diverse serie di
composti per la maggior parte dei quali sono stati calcolati i valori di Ki.
Gli esperimenti di competizione sono stati condotti utilizzando i ligandi radioattivi
[3H](+)pentazocina e [3H]-di-O-tolilmetilguanidina (DTG) a concentrazione fissa (1 e 3
nM, rispettivamente) e concentrazioni crescenti dei ligandi non radioattivi, generalmente
pari a: 0.1, 0.3, 1, 3, 10, 30, 100, 300, 1000, 3000 e 10000 nM.
Preliminarmente, sono stati impiegati i tre composti di riferimento (+)-pentazocina,
DTG e aloperidolo, allo scopo di ottenere dei valori con cui confrontare i risultati dei
composti di neosintesi. La (+)-pentazocina è considerata un ligando ad alta affinità e
selettività per i recettori sigma-1. DTG e aloperidolo sono due isomeri (-) della
normetazocina che esibiscono affinità da moderata ad alta per entrambi i sottotipi
recettoriali sigma-1 e sigma-2, ma sono privi di selettività.
I risultati degli esperimenti di competizione dei composti di riferimento verso la
[3H](+)PTZ e il [3H]DTG sono riportati nella tabella sottostante come valori di IC50,
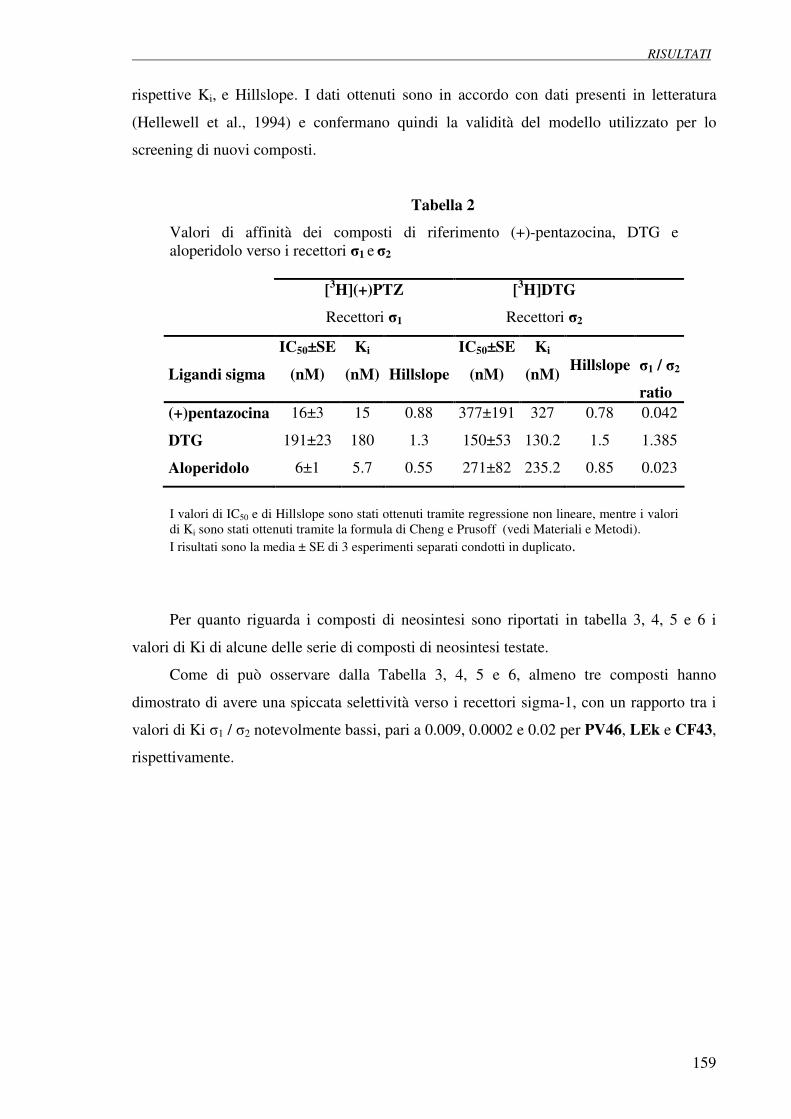
RISULTATI
159
rispettive Ki, e Hillslope. I dati ottenuti sono in accordo con dati presenti in letteratura
(Hellewell et al., 1994) e confermano quindi la validità del modello utilizzato per lo
screening di nuovi composti.
Tabella 2
Valori di affinità dei composti di riferimento (+)-pentazocina, DTG e aloperidolo verso i recettori σ1 e σ2
[3H](+)PTZ
Recettori σ1
[3H]DTG
Recettori σ2
Ligandi sigma
IC50±SE
(nM)
Ki
(nM)
Hillslope
IC50±SE
(nM)
Ki
(nM)
Hillslope
σ1 / σ2
ratio (+)pentazocina 16±3 15 0.88 377±191 327 0.78 0.042
DTG 191±23 180 1.3 150±53 130.2 1.5 1.385
Aloperidolo 6±1 5.7 0.55 271±82 235.2 0.85 0.023
I valori di IC50 e di Hillslope sono stati ottenuti tramite regressione non lineare, mentre i valori di Ki sono stati ottenuti tramite la formula di Cheng e Prusoff (vedi Materiali e Metodi). I risultati sono la media ± SE di 3 esperimenti separati condotti in duplicato.
Per quanto riguarda i composti di neosintesi sono riportati in tabella 3, 4, 5 e 6 i
valori di Ki di alcune delle serie di composti di neosintesi testate.
Come di può osservare dalla Tabella 3, 4, 5 e 6, almeno tre composti hanno
dimostrato di avere una spiccata selettività verso i recettori sigma-1, con un rapporto tra i
valori di Ki σ1 / σ2 notevolmente bassi, pari a 0.009, 0.0002 e 0.02 per PV46, LEk e CF43,
rispettivamente.
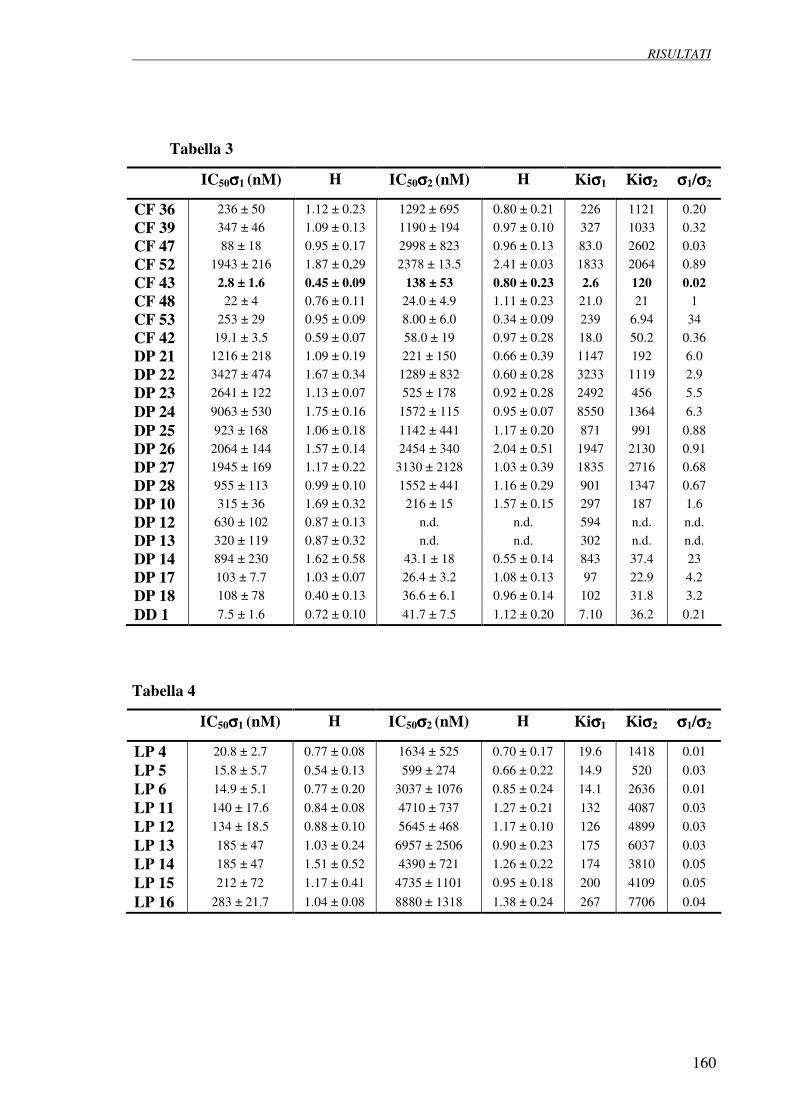
RISULTATI
160
Tabella 3
IC50σσσσ1 (nM) H IC50σσσσ2 (nM) H Kiσσσσ1 Kiσσσσ2 σσσσ1/σσσσ2
CF 36 236 ± 50 1.12 ± 0.23 1292 ± 695 0.80 ± 0.21 226 1121 0.20
CF 39 347 ± 46 1.09 ± 0.13 1190 ± 194 0.97 ± 0.10 327 1033 0.32
CF 47 88 ± 18 0.95 ± 0.17 2998 ± 823 0.96 ± 0.13 83.0 2602 0.03
CF 52 1943 ± 216 1.87 ± 0,29 2378 ± 13.5 2.41 ± 0.03 1833 2064 0.89
CF 43 2.8 ± 1.6 0.45 ± 0.09 138 ± 53 0.80 ± 0.23 2.6 120 0.02
CF 48 22 ± 4 0.76 ± 0.11 24.0 ± 4.9 1.11 ± 0.23 21.0 21 1
CF 53 253 ± 29 0.95 ± 0.09 8.00 ± 6.0 0.34 ± 0.09 239 6.94 34
CF 42 19.1 ± 3.5 0.59 ± 0.07 58.0 ± 19 0.97 ± 0.28 18.0 50.2 0.36
DP 21 1216 ± 218 1.09 ± 0.19 221 ± 150 0.66 ± 0.39 1147 192 6.0
DP 22 3427 ± 474 1.67 ± 0.34 1289 ± 832 0.60 ± 0.28 3233 1119 2.9
DP 23 2641 ± 122 1.13 ± 0.07 525 ± 178 0.92 ± 0.28 2492 456 5.5
DP 24 9063 ± 530 1.75 ± 0.16 1572 ± 115 0.95 ± 0.07 8550 1364 6.3
DP 25 923 ± 168 1.06 ± 0.18 1142 ± 441 1.17 ± 0.20 871 991 0.88
DP 26 2064 ± 144 1.57 ± 0.14 2454 ± 340 2.04 ± 0.51 1947 2130 0.91
DP 27 1945 ± 169 1.17 ± 0.22 3130 ± 2128 1.03 ± 0.39 1835 2716 0.68
DP 28 955 ± 113 0.99 ± 0.10 1552 ± 441 1.16 ± 0.29 901 1347 0.67
DP 10 315 ± 36 1.69 ± 0.32 216 ± 15 1.57 ± 0.15 297 187 1.6
DP 12 630 ± 102 0.87 ± 0.13 n.d. n.d. 594 n.d. n.d.
DP 13 320 ± 119 0.87 ± 0.32 n.d. n.d. 302 n.d. n.d.
DP 14 894 ± 230 1.62 ± 0.58 43.1 ± 18 0.55 ± 0.14 843 37.4 23
DP 17 103 ± 7.7 1.03 ± 0.07 26.4 ± 3.2 1.08 ± 0.13 97 22.9 4.2
DP 18 108 ± 78 0.40 ± 0.13 36.6 ± 6.1 0.96 ± 0.14 102 31.8 3.2
DD 1 7.5 ± 1.6 0.72 ± 0.10 41.7 ± 7.5 1.12 ± 0.20 7.10 36.2 0.21
Tabella 4
IC50σσσσ1 (nM) H IC50σσσσ2 (nM) H Kiσσσσ1 Kiσσσσ2 σσσσ1/σσσσ2
LP 4 20.8 ± 2.7 0.77 ± 0.08 1634 ± 525 0.70 ± 0.17 19.6 1418 0.01
LP 5 15.8 ± 5.7 0.54 ± 0.13 599 ± 274 0.66 ± 0.22 14.9 520 0.03
LP 6 14.9 ± 5.1 0.77 ± 0.20 3037 ± 1076 0.85 ± 0.24 14.1 2636 0.01
LP 11 140 ± 17.6 0.84 ± 0.08 4710 ± 737 1.27 ± 0.21 132 4087 0.03
LP 12 134 ± 18.5 0.88 ± 0.10 5645 ± 468 1.17 ± 0.10 126 4899 0.03
LP 13 185 ± 47 1.03 ± 0.24 6957 ± 2506 0.90 ± 0.23 175 6037 0.03
LP 14 185 ± 47 1.51 ± 0.52 4390 ± 721 1.26 ± 0.22 174 3810 0.05
LP 15 212 ± 72 1.17 ± 0.41 4735 ± 1101 0.95 ± 0.18 200 4109 0.05
LP 16 283 ± 21.7 1.04 ± 0.08 8880 ± 1318 1.38 ± 0.24 267 7706 0.04
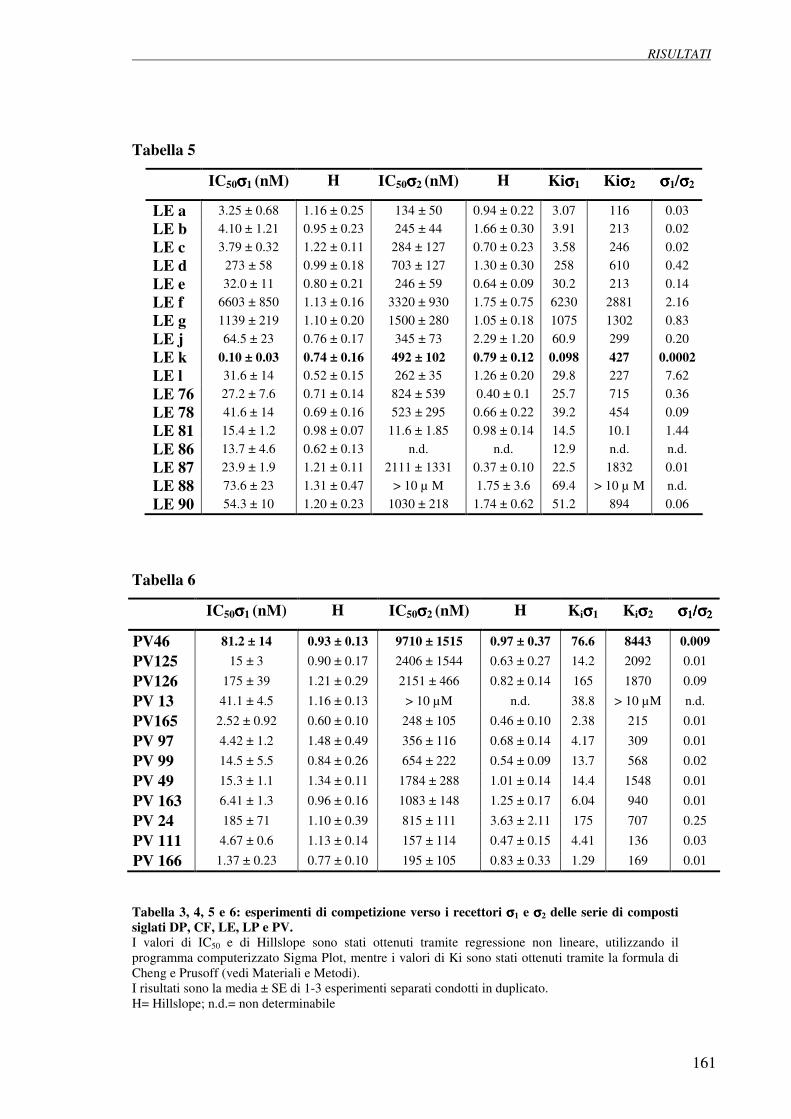
RISULTATI
161
Tabella 5
IC50σσσσ1 (nM) H IC50σσσσ2 (nM) H Kiσσσσ1 Kiσσσσ2 σσσσ1/σσσσ2
LE a 3.25 ± 0.68 1.16 ± 0.25 134 ± 50 0.94 ± 0.22 3.07 116 0.03
LE b 4.10 ± 1.21 0.95 ± 0.23 245 ± 44 1.66 ± 0.30 3.91 213 0.02
LE c 3.79 ± 0.32 1.22 ± 0.11 284 ± 127 0.70 ± 0.23 3.58 246 0.02
LE d 273 ± 58 0.99 ± 0.18 703 ± 127 1.30 ± 0.30 258 610 0.42
LE e 32.0 ± 11 0.80 ± 0.21 246 ± 59 0.64 ± 0.09 30.2 213 0.14
LE f 6603 ± 850 1.13 ± 0.16 3320 ± 930 1.75 ± 0.75 6230 2881 2.16
LE g 1139 ± 219 1.10 ± 0.20 1500 ± 280 1.05 ± 0.18 1075 1302 0.83
LE j 64.5 ± 23 0.76 ± 0.17 345 ± 73 2.29 ± 1.20 60.9 299 0.20
LE k 0.10 ± 0.03 0.74 ± 0.16 492 ± 102 0.79 ± 0.12 0.098 427 0.0002
LE l 31.6 ± 14 0.52 ± 0.15 262 ± 35 1.26 ± 0.20 29.8 227 7.62
LE 76 27.2 ± 7.6 0.71 ± 0.14 824 ± 539 0.40 ± 0.1 25.7 715 0.36
LE 78 41.6 ± 14 0.69 ± 0.16 523 ± 295 0.66 ± 0.22 39.2 454 0.09
LE 81 15.4 ± 1.2 0.98 ± 0.07 11.6 ± 1.85 0.98 ± 0.14 14.5 10.1 1.44
LE 86 13.7 ± 4.6 0.62 ± 0.13 n.d. n.d. 12.9 n.d. n.d.
LE 87 23.9 ± 1.9 1.21 ± 0.11 2111 ± 1331 0.37 ± 0.10 22.5 1832 0.01
LE 88 73.6 ± 23 1.31 ± 0.47 > 10 µ M 1.75 ± 3.6 69.4 > 10 µ M n.d.
LE 90 54.3 ± 10 1.20 ± 0.23 1030 ± 218 1.74 ± 0.62 51.2 894 0.06
Tabella 6
IC50σσσσ1 (nM) H IC50σσσσ2 (nM) H Kiσσσσ1 Kiσσσσ2 σσσσ1111/σ/σ/σ/σ2222
PV46 81.2 ± 14 0.93 ± 0.13 9710 ± 1515 0.97 ± 0.37 76.6 8443 0.009
PV125 15 ± 3 0.90 ± 0.17 2406 ± 1544 0.63 ± 0.27 14.2 2092 0.01
PV126 175 ± 39 1.21 ± 0.29 2151 ± 466 0.82 ± 0.14 165 1870 0.09
PV 13 41.1 ± 4.5 1.16 ± 0.13 > 10 µM n.d. 38.8 > 10 µM n.d.
PV165 2.52 ± 0.92 0.60 ± 0.10 248 ± 105 0.46 ± 0.10 2.38 215 0.01
PV 97 4.42 ± 1.2 1.48 ± 0.49 356 ± 116 0.68 ± 0.14 4.17 309 0.01
PV 99 14.5 ± 5.5 0.84 ± 0.26 654 ± 222 0.54 ± 0.09 13.7 568 0.02
PV 49 15.3 ± 1.1 1.34 ± 0.11 1784 ± 288 1.01 ± 0.14 14.4 1548 0.01
PV 163 6.41 ± 1.3 0.96 ± 0.16 1083 ± 148 1.25 ± 0.17 6.04 940 0.01
PV 24 185 ± 71 1.10 ± 0.39 815 ± 111 3.63 ± 2.11 175 707 0.25
PV 111 4.67 ± 0.6 1.13 ± 0.14 157 ± 114 0.47 ± 0.15 4.41 136 0.03
PV 166 1.37 ± 0.23 0.77 ± 0.10 195 ± 105 0.83 ± 0.33 1.29 169 0.01
Tabella 3, 4, 5 e 6: esperimenti di competizione verso i recettori σσσσ1 e σσσσ2 delle serie di composti siglati DP, CF, LE, LP e PV. I valori di IC50 e di Hillslope sono stati ottenuti tramite regressione non lineare, utilizzando il programma computerizzato Sigma Plot, mentre i valori di Ki sono stati ottenuti tramite la formula di Cheng e Prusoff (vedi Materiali e Metodi). I risultati sono la media ± SE di 1-3 esperimenti separati condotti in duplicato. H= Hillslope; n.d.= non determinabile
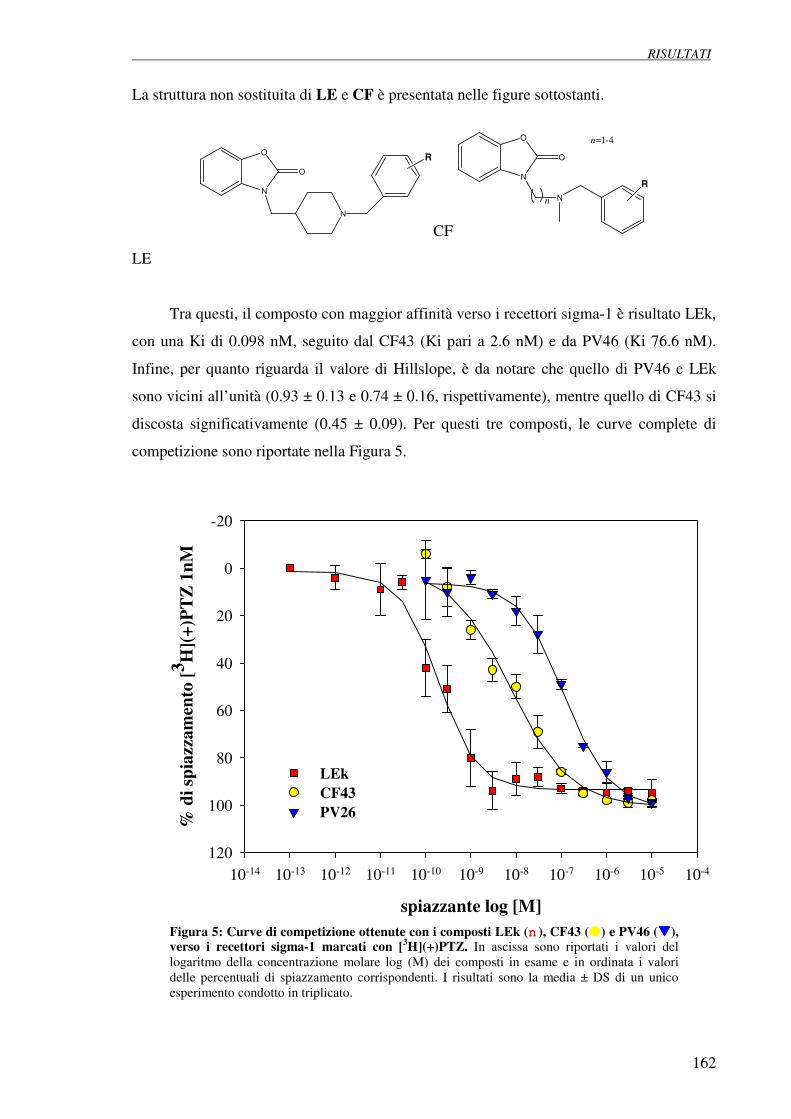
RISULTATI
162
La struttura non sostituita di LE e CF è presentata nelle figure sottostanti.
N
O
N
R
O
CF
N
O
N
R
O
n
n=1-4
LE
Tra questi, il composto con maggior affinità verso i recettori sigma-1 è risultato LEk,
con una Ki di 0.098 nM, seguito dal CF43 (Ki pari a 2.6 nM) e da PV46 (Ki 76.6 nM).
Infine, per quanto riguarda il valore di Hillslope, è da notare che quello di PV46 e LEk
sono vicini all’unità (0.93 ± 0.13 e 0.74 ± 0.16, rispettivamente), mentre quello di CF43 si
discosta significativamente (0.45 ± 0.09). Per questi tre composti, le curve complete di
competizione sono riportate nella Figura 5.
spiazzante log [M]
10-14 10-13 10-12 10-11 10-10 10-9 10-8 10-7 10-6 10-5 10-4
% d
i spi
azza
men
to [
3 H](
+)P
TZ
1nM
-20
0
20
40
60
80
100
120
LEkCF43 PV26
Figura 5: Curve di competizione ottenute con i composti LEk (nnnn ), CF43 (����) e PV46 (����), verso i recettori sigma-1 marcati con [3H](+)PTZ. In ascissa sono riportati i valori del logaritmo della concentrazione molare log (M) dei composti in esame e in ordinata i valori delle percentuali di spiazzamento corrispondenti. I risultati sono la media ± DS di un unico esperimento condotto in triplicato.

RISULTATI
163
4.2 Esperimenti di competizione su membrane di fegato di ratto verso
[3H](+)Pentazocina in presenza di fenitoina
NH
HN
O
O Figura 6: Struttura chimica della fenitoina
E’ stato riportato (Cobos et al., 2005) che l’aggiunta di fenitoina (DPH) nel mezzo
di incubazione in esperimenti di competizione con la tecnica del radioligand binding è in
grado di variare lo stato conformazionale del recettore sigma-1, determinando uno
spostamento verso sinistra della curva di competizione di composti ad attività agonista,
mentre risulta non avere effetto sugli antagonisti. Con questo presupposto, è stato
investigato l’eventuale effetto modulatorio della fenitoina sul legame di due dei composti
di riferimento nelle condizioni sperimentali da noi adottate. I ligandi scelti sono stati
pentazocina e aloperidolo, in quanto considerati, rispettivamente, agonista e antagonista
dei recettori sigma-1.
Gli esperimenti sono eseguiti incubando concentrazioni fisse del radioligando
[3H](+)PTZ (1 nM finale) con concentrazioni crescenti di ligandi non marcati (0.1 – 10000
nM), in assenza e in presenza di DPH (1 mM finale).
Nella tabella 7 sono riportati i valori di IC50 e Hillslope, mentre le curve complete
di competizione sono riportate nelle Figure 7 e 8 per quanto riguarda (+)-pentazocina e per
aloperidolo, rispettivamente.
Come si può osservare dalla Tabella 7, la presenza di DPH determina una riduzione
del valore di IC50 della (+)-pentazocina, che passa da 25.0 ± 2.4 a 18.6 ± 1.7 nM, mentre
non altera quello di aloperidolo (2.82 ± 0.57 nM e 4.80 ± 0.90 nM). Viceversa, i valori di
Hillslope non presentano variazioni significative.
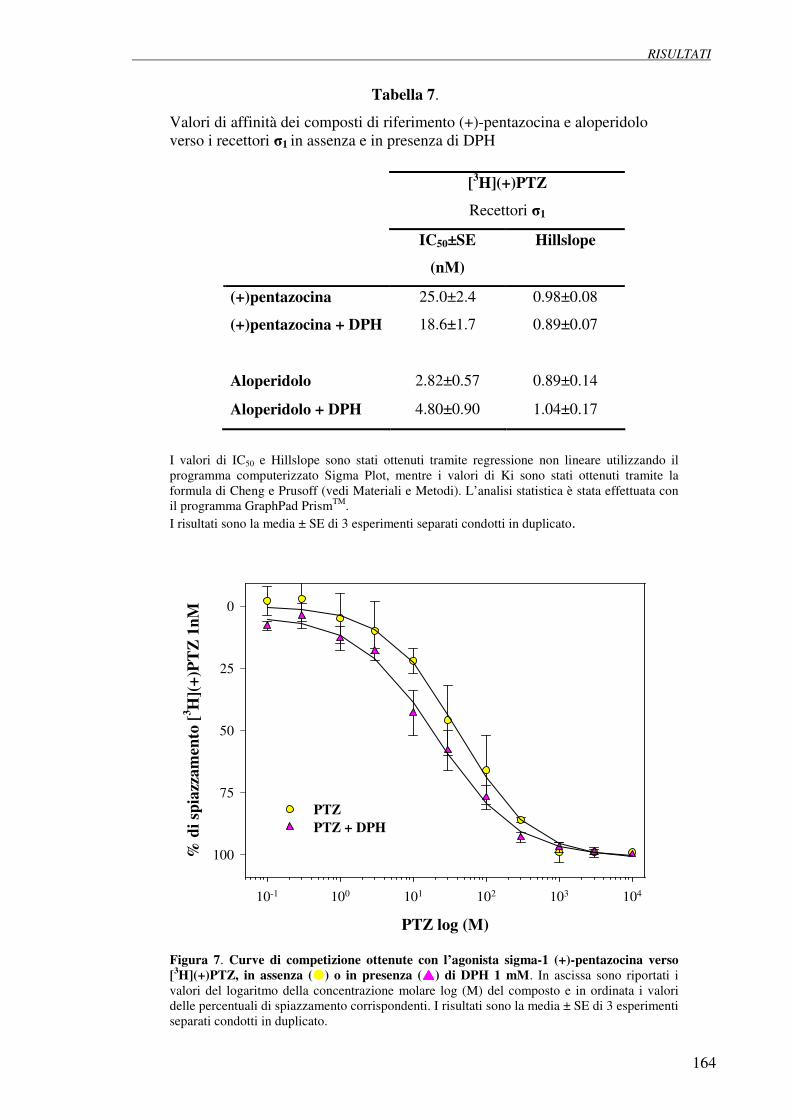
RISULTATI
164
Tabella 7.
Valori di affinità dei composti di riferimento (+)-pentazocina e aloperidolo verso i recettori σ1 in assenza e in presenza di DPH
I valori di IC50 e Hillslope sono stati ottenuti tramite regressione non lineare utilizzando il programma computerizzato Sigma Plot, mentre i valori di Ki sono stati ottenuti tramite la formula di Cheng e Prusoff (vedi Materiali e Metodi). L’analisi statistica è stata effettuata con il programma GraphPad PrismTM. I risultati sono la media ± SE di 3 esperimenti separati condotti in duplicato.
A
PTZ log (M)
10-1 100 101 102 103 104
% d
i spi
azza
men
to [
3 H](
+)P
TZ
1nM
0
25
50
75
100
PTZ PTZ + DPH
Figura 7. Curve di competizione ottenute con l’agonista sigma-1 (+)-pentazocina verso [3H](+)PTZ, in assenza (����) o in presenza (����) di DPH 1 mM. In ascissa sono riportati i valori del logaritmo della concentrazione molare log (M) del composto e in ordinata i valori delle percentuali di spiazzamento corrispondenti. I risultati sono la media ± SE di 3 esperimenti separati condotti in duplicato.
[3H](+)PTZ
Recettori σ1
IC50±SE
(nM)
Hillslope
(+)pentazocina 25.0±2.4 0.98±0.08
(+)pentazocina + DPH 18.6±1.7 0.89±0.07
Aloperidolo 2.82±0.57 0.89±0.14
Aloperidolo + DPH 4.80±0.90 1.04±0.17
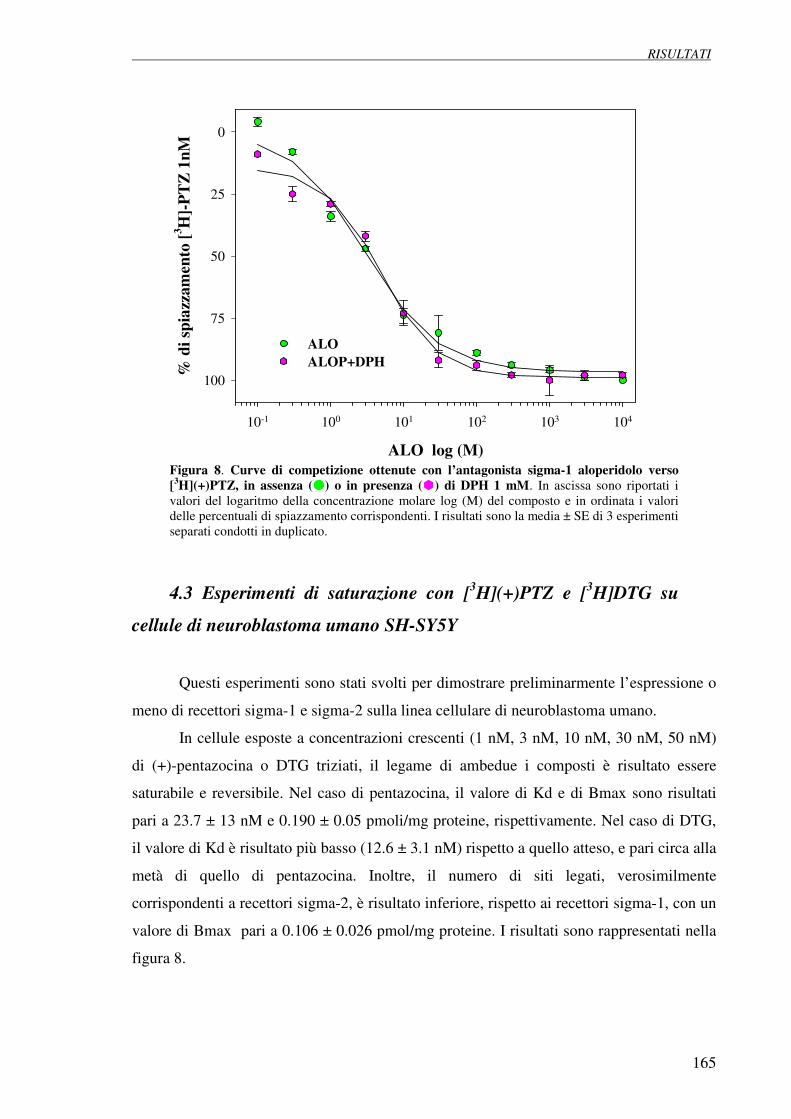
RISULTATI
165
ALO log (M)
10-1 100 101 102 103 104
% d
i spi
azza
men
to [
3 H]-
PT
Z 1
nM
0
25
50
75
100
ALO ALOP+DPH
Figura 8. Curve di competizione ottenute con l’antagonista sigma-1 aloperidolo verso [3H](+)PTZ, in assenza (����) o in presenza (����) di DPH 1 mM. In ascissa sono riportati i valori del logaritmo della concentrazione molare log (M) del composto e in ordinata i valori delle percentuali di spiazzamento corrispondenti. I risultati sono la media ± SE di 3 esperimenti separati condotti in duplicato.
4.3 Esperimenti di saturazione con [3H](+)PTZ e [
3H]DTG su
cellule di neuroblastoma umano SH-SY5Y
Questi esperimenti sono stati svolti per dimostrare preliminarmente l’espressione o
meno di recettori sigma-1 e sigma-2 sulla linea cellulare di neuroblastoma umano.
In cellule esposte a concentrazioni crescenti (1 nM, 3 nM, 10 nM, 30 nM, 50 nM)
di (+)-pentazocina o DTG triziati, il legame di ambedue i composti è risultato essere
saturabile e reversibile. Nel caso di pentazocina, il valore di Kd e di Bmax sono risultati
pari a 23.7 ± 13 nM e 0.190 ± 0.05 pmoli/mg proteine, rispettivamente. Nel caso di DTG,
il valore di Kd è risultato più basso (12.6 ± 3.1 nM) rispetto a quello atteso, e pari circa alla
metà di quello di pentazocina. Inoltre, il numero di siti legati, verosimilmente
corrispondenti a recettori sigma-2, è risultato inferiore, rispetto ai recettori sigma-1, con un
valore di Bmax pari a 0.106 ± 0.026 pmol/mg proteine. I risultati sono rappresentati nella
figura 8.
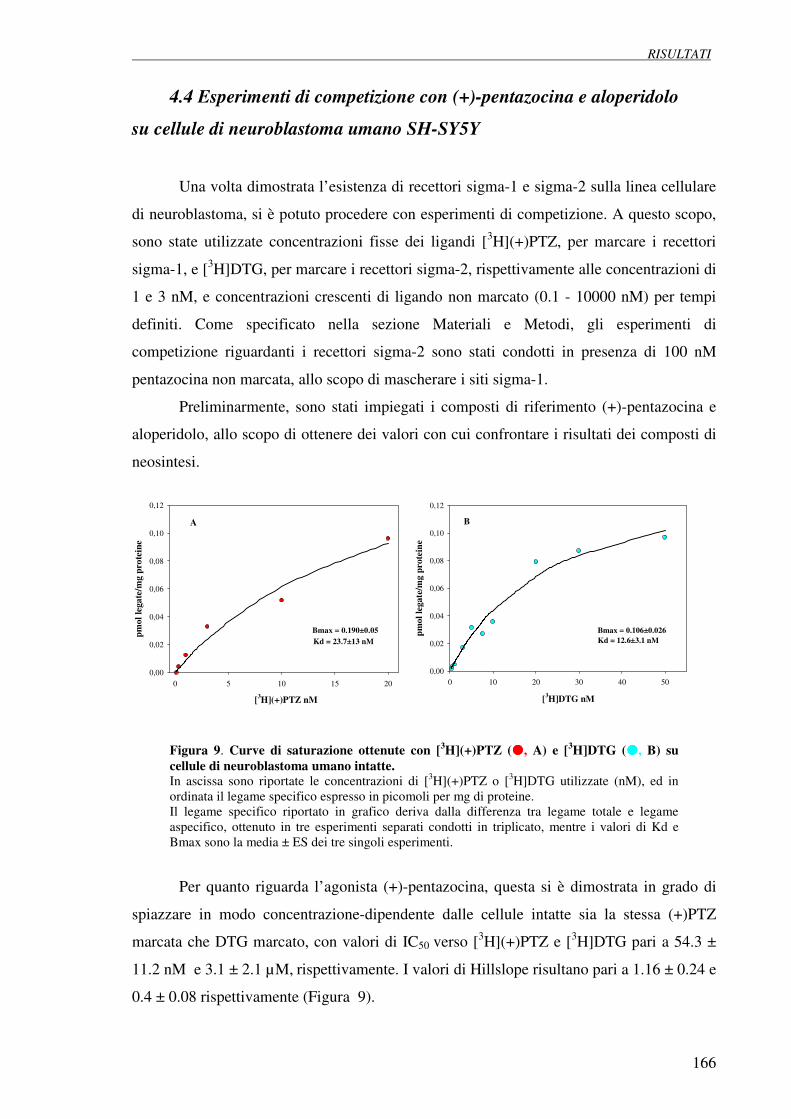
RISULTATI
166
4.4 Esperimenti di competizione con (+)-pentazocina e aloperidolo
su cellule di neuroblastoma umano SH-SY5Y
Una volta dimostrata l’esistenza di recettori sigma-1 e sigma-2 sulla linea cellulare
di neuroblastoma, si è potuto procedere con esperimenti di competizione. A questo scopo,
sono state utilizzate concentrazioni fisse dei ligandi [3H](+)PTZ, per marcare i recettori
sigma-1, e [3H]DTG, per marcare i recettori sigma-2, rispettivamente alle concentrazioni di
1 e 3 nM, e concentrazioni crescenti di ligando non marcato (0.1 - 10000 nM) per tempi
definiti. Come specificato nella sezione Materiali e Metodi, gli esperimenti di
competizione riguardanti i recettori sigma-2 sono stati condotti in presenza di 100 nM
pentazocina non marcata, allo scopo di mascherare i siti sigma-1.
Preliminarmente, sono stati impiegati i composti di riferimento (+)-pentazocina e
aloperidolo, allo scopo di ottenere dei valori con cui confrontare i risultati dei composti di
neosintesi.
0 5 10 15 20
pmol
lega
te/m
g pr
otei
ne
0,00
0,02
0,04
0,06
0,08
0,10
0,12
[3H](+)PTZ nM
Bmax = 0.190±0.05Kd = 23.7±13 nM
A
[3H]DTG nM
0 10 20 30 40 50
pmol
lega
te/m
g pr
otei
ne
0,00
0,02
0,04
0,06
0,08
0,10
0,12
Bmax = 0.106±0.026Kd = 12.6±3.1 nM
B
Figura 9. Curve di saturazione ottenute con [3H](+)PTZ (����, A) e [3H]DTG (����, B) su cellule di neuroblastoma umano intatte. In ascissa sono riportate le concentrazioni di [3H](+)PTZ o [3H]DTG utilizzate (nM), ed in ordinata il legame specifico espresso in picomoli per mg di proteine. Il legame specifico riportato in grafico deriva dalla differenza tra legame totale e legame aspecifico, ottenuto in tre esperimenti separati condotti in triplicato, mentre i valori di Kd e Bmax sono la media ± ES dei tre singoli esperimenti.
Per quanto riguarda l’agonista (+)-pentazocina, questa si è dimostrata in grado di
spiazzare in modo concentrazione-dipendente dalle cellule intatte sia la stessa (+)PTZ
marcata che DTG marcato, con valori di IC50 verso [3H](+)PTZ e [3H]DTG pari a 54.3 ±
11.2 nM e 3.1 ± 2.1 µM, rispettivamente. I valori di Hillslope risultano pari a 1.16 ± 0.24 e
0.4 ± 0.08 rispettivamente (Figura 9).
RISULTATI
167
Per quanto riguarda l’antagonista aloperidolo, i valori di IC50 verso [3H](+)PTZ e
[3H]DTG sono pari a 176 ± 300 nM e 58 ± 27 nM, rispettivamente, con valori di Hillslope
di 0.32 ± 0.2 e 0.5 ± 0.17.
Tabella 8.
Valori di affinità dei composti di riferimento (+)-pentazocina e aloperidolo verso i recettori σ1 e σ2 su cellule intatte di neuroblasoma umano
[3H](+)PTZ
Recettori σ1
[3H]DTG
Recettori σ2
Ligandi sigma
IC50±SE
(nM)
Hillslope IC50±SE
(nM)
Hillslope
(+)pentazocina 54.3±11.2 1.16±0.24 3100±2100 0.4±0.08
Aloperidolo 176±300 0.32±0.2 58±27 0.5±0.17
I valori di IC50 e di Hillslope sono ottenuti tramite regressione non lineare, utilizzando il programma computerizzato Sigma Plot. I risultati sono la media ± SE di 3 esperimenti separati condotti in duplicato.
A seguito, nelle figure 10 e 11 sono riportate le curve di competizione relative a (+)-
pentazocina e aloperidolo nei confronti di [3H](+)PTZ e [3H]DTG.
Per quanto riguarda l’antagonista DTG, le curve di competizione verso antrambi i
siti, sia sigma-1 che sigma-2, non presentavano un andamento sigmoidale e non è stato
quindi possibile ottenere un valore di IC50.
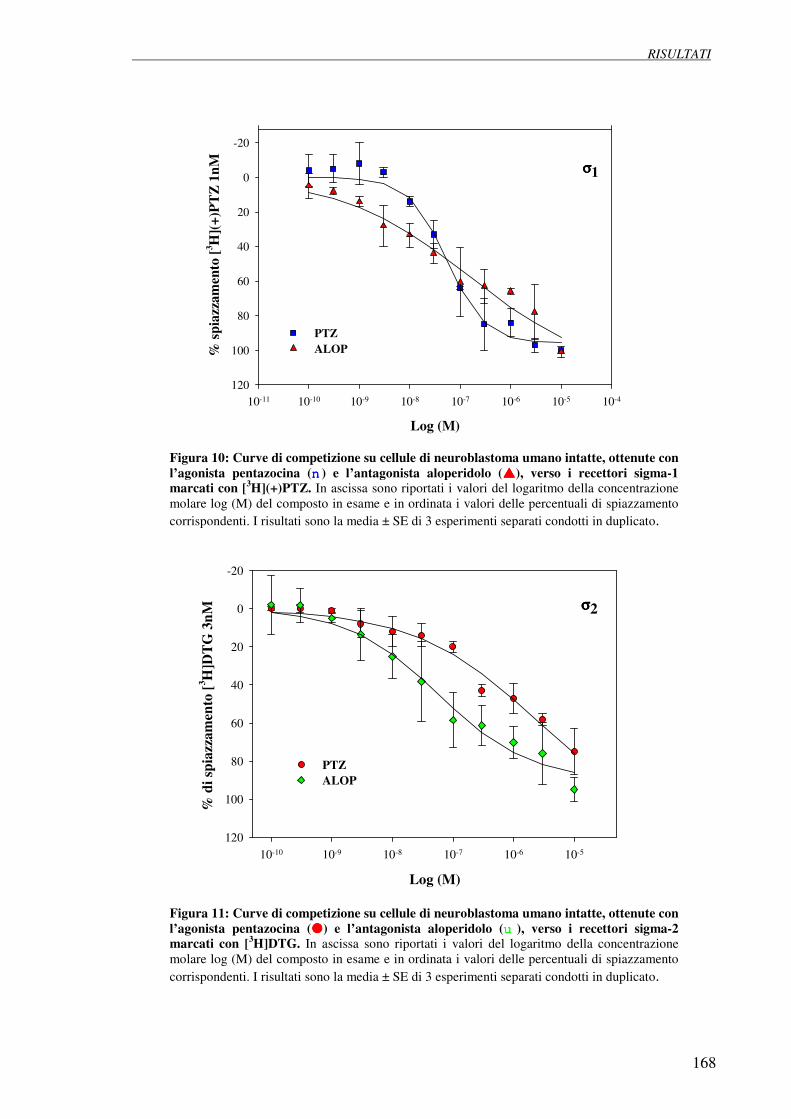
RISULTATI
168
Log (M)
10-11 10-10 10-9 10-8 10-7 10-6 10-5 10-4
% s
piaz
zam
ento
[3 H](
+)P
TZ
1nM
-20
0
20
40
60
80
100
120
PTZ ALOP
σσσσ1
Figura 10: Curve di competizione su cellule di neuroblastoma umano intatte, ottenute con l’agonista pentazocina (nnnn ) e l’antagonista aloperidolo (����), verso i recettori sigma-1 marcati con [3H](+)PTZ. In ascissa sono riportati i valori del logaritmo della concentrazione molare log (M) del composto in esame e in ordinata i valori delle percentuali di spiazzamento corrispondenti. I risultati sono la media ± SE di 3 esperimenti separati condotti in duplicato.
Log (M)
10-10 10-9 10-8 10-7 10-6 10-5
% d
i spi
azza
men
to [
3 H]D
TG
3nM
-20
0
20
40
60
80
100
120
PTZALOP
σσσσ2
Figura 11: Curve di competizione su cellule di neuroblastoma umano intatte, ottenute con l’agonista pentazocina (����) e l’antagonista aloperidolo (uuuu ), verso i recettori sigma-2 marcati con [3H]DTG. In ascissa sono riportati i valori del logaritmo della concentrazione molare log (M) del composto in esame e in ordinata i valori delle percentuali di spiazzamento corrispondenti. I risultati sono la media ± SE di 3 esperimenti separati condotti in duplicato.

RISULTATI
169
4.5 Esperimenti di inibizione della proliferazione (test della SRB)
Preliminarmente, è stato studiato l’effetto antiproliferativo dei tre composti di
riferimento (+)-pentazocina, DTG e aloperidolo, sulla linea cellulare SH-SY5Y di
neuroblastoma umano.
In questi esperimenti le cellule vengono trattate a tempi diversi, 48 e 72 ore,
utilizzando concentrazioni decrescenti dei composti in esame, opportunamente disciolti al
momento dell’esperimento in terreno di coltura completo. Allo scadere del tempo stabilito,
il mezzo viene rimosso e sulla stessa piastra viene eseguito il test della sulforodamina B.
Dai dati ottenuti è possibile calcolare i rispettivi valori di IC50, la concentrazione di ligando
capace di inibire la proliferazione cellulare del 50 % rispetto ai controlli.
Tra i composti di riferimento solo l’agonista sigma-1 (+)-pentazocina è in grado di
indurre un effetto antiproliferativo concentrazione- e tempo-dipendente. A 48 ore il valore
di IC50 risulta pari a 70 ± 6.5 µM con una Hillslope di 1.5 ± 0.18. Dopo 72 ore di
trattamento il valore di IC50 risulta inferiore e pari a 44 ± 4 µM con una Hillslope di 2.2 ±
0.5.
Al contrario, gli antagonisti aloperidolo e DTG non stati in grado di influenzare la
proliferazione cellulare in maniera concentrazione- e tempo-dipendente. La riduzione di
assorbanza osservata alle concentrazioni più alte può essere ascritta sia ad un effetto
tossico del solvente utilizzato (DMSO), in quanto una riduzione dell’assorbanza di entità in
parte sovrapponibile è stata ottenuta trattatando le cellule con equivalenti percentuali di
solo solvente (dati non mostrati), sia ad un effetto tossico del composto che si evidenzia
alle concentrazioni più elevate, sia alla combinazione dei due fattori (Figura 12).
Per quanto riguarda i composti di neosintesi, nessuno ha determinato una riduzione
della proliferazione con andamento sigmoidale, simile a quello ottenuto con (+)-
pentazocina, con la possibile sola eccezione del composto LEk, e solo dopo 72 ore di
esposizione. In questo caso, il valore di IC50 calcolato è pari a 1000 ± 626 nM. Inoltre, alla
concentrazione più elevata utilizzata, pari a 3.3 mM, il composto PV46 ha determinato una
notevole riduzione dei valori di assorbanza, sia dopo 48, sia dopo 72 ore di trattamento
(Figura 13).
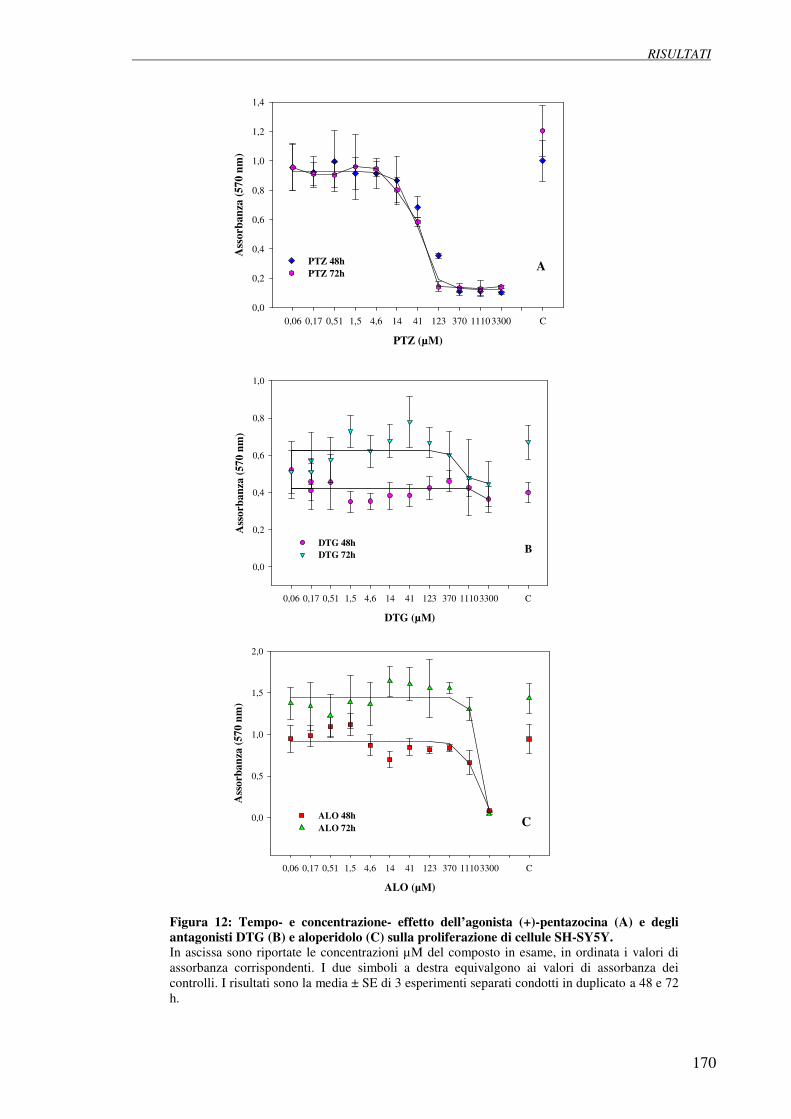
RISULTATI
170
PTZ (µM)
C3300111037012341144,61,50,510,170,06
Ass
orba
nza
(570
nm
)
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
PTZ 48h PTZ 72h
A
DTG (µM)
C3300111037012341144,61,50,510,170,06
Ass
orba
nza
(570
nm
)
0,0
0,2
0,4
0,6
0,8
1,0
DTG 48hDTG 72h B
ALO (µM)
C3300111037012341144,61,50,510,170,06
Ass
orba
nza
(570
nm
)
0,0
0,5
1,0
1,5
2,0
ALO 48h ALO 72h C
Figura 12: Tempo- e concentrazione- effetto dell’agonista (+)-pentazocina (A) e degli antagonisti DTG (B) e aloperidolo (C) sulla proliferazione di cellule SH-SY5Y. In ascissa sono riportate le concentrazioni µM del composto in esame, in ordinata i valori di assorbanza corrispondenti. I due simboli a destra equivalgono ai valori di assorbanza dei controlli. I risultati sono la media ± SE di 3 esperimenti separati condotti in duplicato a 48 e 72 h.
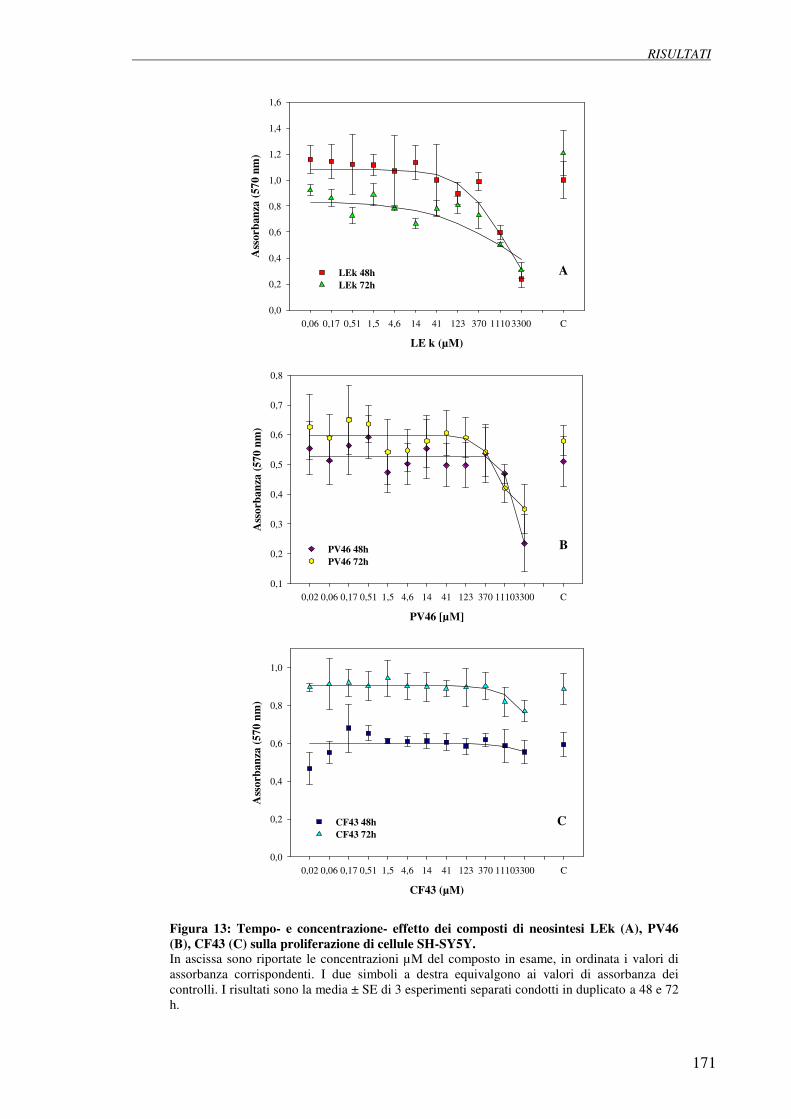
RISULTATI
171
LE k (µM)
C3300111037012341144,61,50,510,170,06
Ass
orba
nza
(570
nm
)
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
LEk 48h LEk 72h
A
PV46 [µM]
C3300111037012341144,61,50,510,170,060,02
Ass
orba
nza
(570
nm
)
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
PV46 48h PV46 72h
B
CF43 (µM)
C3300111037012341144,61,50,510,170,060,02
Ass
orba
nza
(570
nm
)
0,0
0,2
0,4
0,6
0,8
1,0
CF43 48h CF43 72h
C
Figura 13: Tempo- e concentrazione- effetto dei composti di neosintesi LEk (A), PV46 (B), CF43 (C) sulla proliferazione di cellule SH-SY5Y. In ascissa sono riportate le concentrazioni µM del composto in esame, in ordinata i valori di assorbanza corrispondenti. I due simboli a destra equivalgono ai valori di assorbanza dei controlli. I risultati sono la media ± SE di 3 esperimenti separati condotti in duplicato a 48 e 72 h.

RISULTATI
172
4.6 Studio dei cambiamenti morfologici indotti da ligandi sigma su
cellule di neuroblastoma umano SH-SY5Y
Il presente studio ha infine rivolto l’attenzione al possibile effetto sulla morfologia
delle cellule SH-SY5Y da parte dei composti di riferimento (+)-pentazocina, DTG,
aloperidolo, e di alcuni dei composti di neo-sintesi, LEk, PV46 e CF43. Per questo scopo,
le cellule sono state mantenute in mezzo arricchito con il 10% di siero fetale bovino per 24
ore dopo la semina e quindi trattate con concentrazioni pari a 10 µM dei composti in
esame. Dopo 48 o 72 ore di trattamento, le cellule sono state fissate e osservate al
microscopio ottico. In ogni esperimento è stato incluso un gruppo di controllo, costituito da
cellule trattate con solo mezzo o trattate con mezzo contenente il solvente in cui sono
disciolti i composti (DMSO 0,1% finale).
Come riportato nella figura 13, rispetto ai controlli, l’esposizione delle cellule di
neuroblastoma umano alla (+)-pentazocina 10 µM ha indotto, dopo 48 ore di trattamento,
un effetto neuritogenico, evidenziato dalla comparsa di spessi prolungamenti che si
estendono dal corpo cellulare. La percentuale di cellule differenziate, rispetto ai controlli
(10.1 ± 3 %), è stata calcolata pari a 23.0 ± 5.1 %, differenza statisticamente significativa
(p<0.001).
DTG (21.8 ± 5.2 % di cellule differenziate) e aloperidolo (15.2 ± 4.6 % di cellule
differenziate) non hanno causato un effetto differenziante statisticamente significativo
rispetto ai controlli trattati con DMSO 0.1 % (16.9 ± 3.5 %). Il DMSO sembra avere di per
sé un effetto differenziante, portando ad un incremento della percentuale di cellule
differenziate di circa il 7 %, rispetto ai controlli trattati con solo mezzo (p<0.001).
Per quanto riguarda gli altri ligandi, solo LEk ha dimostrato di avere un
significativo effetto neuritogenico rispetto ai controlli, con una media di 31 ± 7 % di
cellule differenziate (p<0.001). Viceversa, al pari dei composti di riferimento DTG e
aloperidolo, i composti di neosintesi PV46 e CF43 non hanno provocato alcuna evidente
modifica, con percentuali di differenziazione pari a 19.8 ± 5.7 % e 11.2 ± 9.1 %
rispettivamente.
Prolungando l’esposizione delle cellule a 72 ore non si sono evidenziati ulteriori
cambiamenti nella capacità dei composti in esame di indurre neuritogenesi. Nuovamente, i
soli composti efficaci sono risultati (+)-pentazocina, con una percentuale di cellule
differenziate, rispetto ai controlli (6 ± 2.4 %) pari a 18.8 ± 5.7 % (p<0.001) e LEk, con una
percentuale di cellule differenziate pari a 19.2 ± 4.5 % (p<0.001). Nel caso di LEk, il
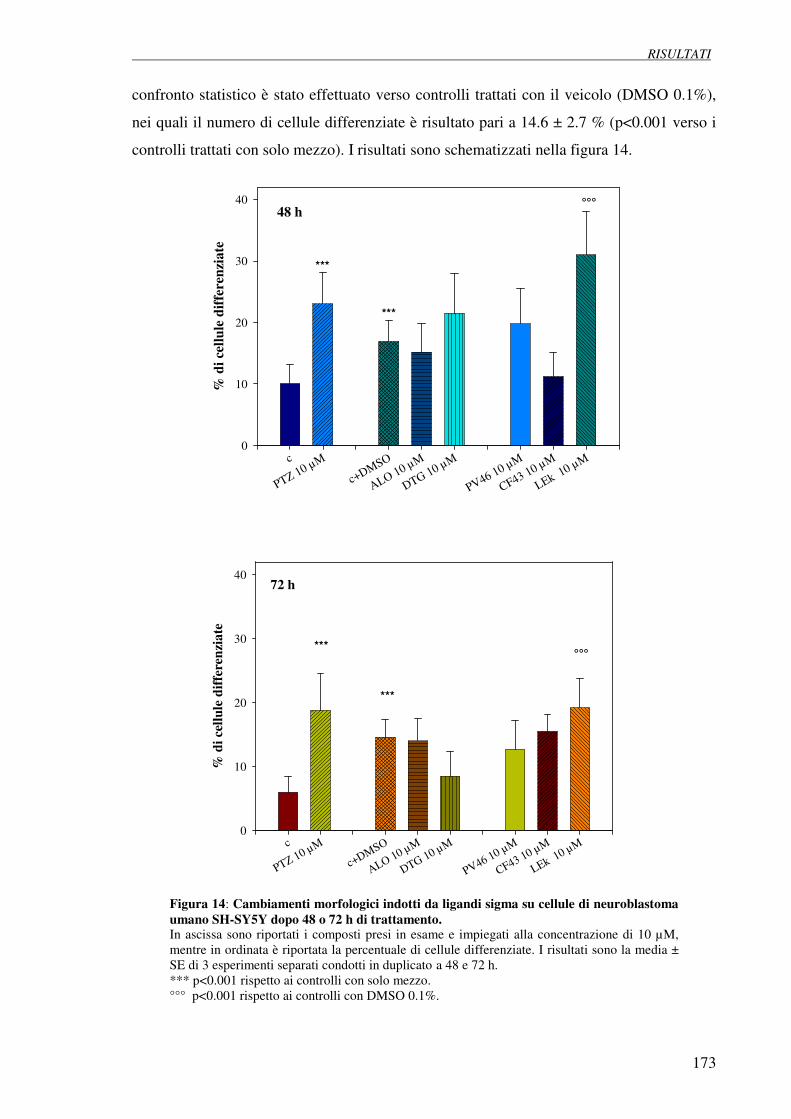
RISULTATI
173
confronto statistico è stato effettuato verso controlli trattati con il veicolo (DMSO 0.1%),
nei quali il numero di cellule differenziate è risultato pari a 14.6 ± 2.7 % (p<0.001 verso i
controlli trattati con solo mezzo). I risultati sono schematizzati nella figura 14.
48 h
c
PTZ 10 µM
c+DMSO
ALO 10 µM
DTG 10 µM
PV46 10 µM
CF43 10 µM
LEk 10 µM
% d
i cel
lule
dif
fere
nzia
te
0
10
20
30
40
***
°°°
***
72 h
c
PTZ 10 µM
c+DMSO
ALO 10 µM
DTG 10 µM
PV46 10 µM
CF43 10 µM
LEk 10 µM
% d
i cel
lule
dif
fere
nzia
te
0
10
20
30
40
***
***
°°°
Figura 14: Cambiamenti morfologici indotti da ligandi sigma su cellule di neuroblastoma umano SH-SY5Y dopo 48 o 72 h di trattamento. In ascissa sono riportati i composti presi in esame e impiegati alla concentrazione di 10 µM, mentre in ordinata è riportata la percentuale di cellule differenziate. I risultati sono la media ± SE di 3 esperimenti separati condotti in duplicato a 48 e 72 h. *** p<0.001 rispetto ai controlli con solo mezzo. °°° p<0.001 rispetto ai controlli con DMSO 0.1%.

5. DISCUSSIONE

DISCUSSIONE
174
I risultati presentati in questa tesi possono essere così riassunti: innanzitutto, lo
screening di nuovi composti con possibile affinità verso i recettori sigma, sintetizzati
presso il Dipartimento di Scienze Farmaceutiche dell’Ateneo, ha permesso di individuare,
tramite la tecnica del radioligand binding, tre composti dotati di alta affinità e selettività
verso i recettori sigma-1. La stessa tecnica, pur potenzialmente adatta alla discriminazione
di composti ad attività agonista, rispetto a quelli ad attività antagonista, grazie alla
modulazione di tipo allosterico esercitata dalla fenitoina, è risultata poco sensibile e
indaginosa. Con la dimostrazione della presenza di recettori sigma-1 e sigma-2 sulle
cellule di neuroblastoma umano SH-SY5Y è stato possibile correlare la capacità della
pentazocina di ridurre la proliferazione cellulare e di indurre differenziazione, alla sua
capacità di interazione con i recettori sigma-1. Infine, tra i diversi composti di neosintesi,
quello dotato di maggior potenza e selettività, LEk, è risultato in grado di mimare l’effetto
della pentazocina sulla proliferazione e differenziazione cellulare. Questo suggerisce un
suo possibile e prezioso impiego come agonista altamente selettivo e ad alta affinità per lo
studio dei recettori sigma-1 e dei potenziali impieghi terapeutici ad essi correlati.
Nel dettaglio, per quanto riguarda lo screening dei composti di neosintesi, questo è
stato preceduto dalla verifica delle appropriate condizioni sperimentali, eseguita
impiegando tre composti di riferimento: pentazocina, DTG ed aloperidolo. La prima è
considerata un ligando selettivo dei recettori sigma-1, e come tale la sua forma marcata con
trizio è largamente impiegata per lo studio, tramite la tecnica del radioligand binding, del
sottotipo recettoriale sigma-1. Inoltre, la sua attività intrinseca sembra collocarla nel
novero dei composti ad attività agonista verso gli stessi recettori. Per questo composto, i
valori di Ki ottenuti nei confronti dei siti sigma-1 e sigma-2 (15 e 327 nM,

DISCUSSIONE
175
rispettivamente), sono in linea con dati riportati in letteratura (Hellewell et al, 1994), con
un rapporto σ1/σ2 di 0,042.
Al contrario della pentazocina, il DTG è dotato di scarsa affinità e selettività nei
confronti di ambedue i sottotipi recettoriali. Ciononostante, è un radioligando
comunemente utilizzato per identificare i recettori sigma-2, previo mascheramento dei siti
sigma-1 con pentazocina o altro ligando sigma-1 selettivo. Per questo composto, i valori di
Ki ottenuti nei confronti dei siti sigma-1 e sigma-2 sono stati di 180 e 130 nM,
rispettivamente, con un rapporto σ1/σ2 di 1.4, a conferma della mancata selettività. In
modo consistente, anche i valori di Hillslope sono risultati maggiori dell’unità, indicando
l’interazione con più di un sito di riconoscimento.
L’aloperidolo, infine, possiede alta affinità per i recettori sigma-1, verso i quali
esplica attività antagonista, e bassa affinità verso i recettori sigma-2. Anche in questo caso,
i valori di Ki ottenuti sono dell’ordine di grandezza di quelli riportati in letteratura, essendo
pari a 5.7 e 235 nM per i siti sigma-1 e sigma-2, rispettivamente (Hellwell et al., 1994), cui
consegue un rapporto σ1/σ2 di 0.023.
La congruità dei risultati ottenuti per i composti di riferimento con i dati di
letteratura ci permette di affermare che alcuni tra i numerosi composti di neosintesi di cui è
stato effettuato lo screening presentano valori di affinità e selettività tali da farne composti
potenzialmente utili per lo studio del sistema recettoriale sigma. Il composto con sigla
PV46, pur non presentando, rispetto a pentazocina o aloperidolo, un valore di Ki per i siti
sigma-1 particolarmente basso (77 nM), risulta essere circa 5- e 3- volte più selettivo della
pentazocina e dell’aloperidolo, rispettivamente. I valori di Hillslope sono inoltre risultati
molto vicini all’unità, sia per quanto riguarda i siti sigma-1, sia per quanto riguarda i siti
sigma-2, indicando una interazione con un singolo sito e a conferma che le costanti di
affinità verso ambedue i recettori sono sufficientemente distanziate da conferire una
spiccata selettività.
Nella ricerca di composti ad alta affinità e selettività verso i recettori sigma-1,
risultati ancora più incoraggianti sono stati ottenuti con la determinazione dei parametri di
binding di uno dei composti della serie LE, denominato LEk. Questo composto è risultato
circa 150- e 60- volte più affine della pentazocina e dell’aloperidolo, rispettivamente, nei
confronti dei recettori sigma-1, con una Ki di circa 0.1 nM. Il rapporto σ1/σ2 è risultato
essere di 0.0002, indicando una selettività di 200 volte maggiore della pentazocina e 125
volte superiore a quella dell’aloperidolo. In senso assoluto, l’affinità verso i siti sigma-1 è
quindi 5000 volte maggiore rispetto ai siti sigma-2. Come nel caso del composto PV46, i

DISCUSSIONE
176
valori di Hillslope vicino all'unità sono indice di un legame ad un singolo sito secondo la
legge dell’azione di massa.
Tra i composti della serie CF, quello con maggior affinità e selettività verso i siti
sigma-1 è risultato essere il CF43, con caratteristiche sovrapponibili a quelle
dell’aloperidolo. Infatti, nonostante i valori di Ki verso i siti sigma-1 (2.6 nM) e sigma-2
(120 nM) risultino dimezzati rispetto a quelli dell’aloperidolo (5.7 e 235 nM,
rispettivamente), il rapporto σ1/σ2 è, per ambedue i composti, di 0.02. Inoltre, ambedue i
ligandi presentano una Hillslope verso i siti sigma-1 di circa 0.5, un dato che suggerisce
una forma di cooperatività negativa o la presenza di ulteriori siti di riconoscimento che, nel
caso dell’aloperidolo potrebbero essere recettori di altri neurotrasmettitori, quali i recettori
dopaminergici verso i quali presenta affinità nell’ordine del nanomolare (Costantino et al.,
2005). Questa ipotesi potrebbe essere estesa al composto CF43: non è infrequente infatti
che ligandi sigma presentino un grado non irrilevante di affinità nei confronti di più di un
tipo di recettore di altri neurotrasmettitori, quali i recettori degli oppioidi, muscarinici,
serotoninergici, adrenergici o dopaminergici (Costantino et al., 2005).
Una volta determinato il grado di selettività e di affinità di un nuovo ligando verso
uno specifico recettore, è importante determinarne l’attività intrinseca. Nel caso dei
recettori sigma, Cobos et al. (2005) hanno riportato un metodo che, tramite la tecnica del
radioligand binding, evidenzia la capacità della fenitoina di modulare in modo allosterico
l’interazione dei ligandi sigma-1, ma non dei ligandi sigma-2, con il proprio recettore. Nel
caso di composti ad attività agonista, la presenza della fenitoina ne determinerebbe uno
spostamento della curva di competizione verso sinistra, mentre la capacità di spiazzamento
di composti ad attività antagonista resterebbe inalterata. Abbiamo quindi condotto una
serie di esperimenti utilizzando la pentazocina come composto di riferimento ad attività
agonista, l’aloperidolo come composto antagonista di riferimento, e la pentazocina triziata
per marcare i siti sigma-1. I risultati ottenuti sono in linea con quelli di letteratura, in
quanto ci permettono di confermare la capacità della fenitoina di modulare il legame della
pentazocina, il cui valore di IC50 si è ridotto in modo significativo da 25 a 19 nM, ma non
quello dell’antagonista aloperidolo, il cui valore di IC50 non ha subito alcun cambiamento.
Rispetto a quanto riportato da Cobos et al, però, lo spostamento della curva dell’agonista
era molto poco evidente, ed evidenziabile solo dopo un’analisi statistica dedicata. E’
possibile che la scarsa sensibilità del metodo da noi ottenuta derivi dal fatto di avere usato
come competitore la stessa molecola usata come marcatore i siti sigma-1, la pentazocina, il
cui legame al recettore è soggetto anch’esso a modulazione allosterica. In questo caso,
sarebbe necessario disporre di un ligando sigma-1 a comprovata attività agonista diverso

DISCUSSIONE
177
dal ligando marcato, come destrometorfano o SKF-10,047, da impiegare come composto
spiazzante di riferimento. Esiste infine la possibilità che alla base del diverso risultato ci
sia il diverso tessuto impiegato, nella fattispecie cervello di cavia (Cobos et al., 2005) e
fegato di ratto (presente studio). Il discreto accordo tra i parametri di binding dei composti
di riferimento da noi ottenuti impiegando fegato di ratto con quelli in letteratura, riferiti sia
a cervello di cavia, sia a fegato di ratto, rende questa ipotesi poco probabile. In ogni caso,
la mancanza di un composto di riferimento non ci ha permesso di impiegare questo tipo di
approccio per la determinazione dell’efficacia dei nuovi composti.
Considerata la scarsa praticabilità degli esperimenti di modulazione del binding da
parte della fenitoina, allo scopo di determinare l’attività intrinseca dei composti di
neosintesi si è cercato di identificare, tra i metodi in vitro descritti in letteratura, quelli più
sensibili, meno costosi e più adatti ad uno studio di screening. Dopo un’attenta analisi è
però emerso un certo grado di confusione. Ad esempio, un effetto neuroprotettivo è stato
imputato da alcuni autori a composti riferiti quali agonisti sigma-1 (Nakazawa et al, 1998;
Yang et al, 2007), da altri autori a composti ritenuti antagonisti degli stessi recettori
(Schetz et al, 2007). In questi studi, la neuroprotezione era valutata sulla base della
capacità di ligandi sigma di proteggere colture neuronali da stress indotto da privazione di
ossigeno/glucosio. Considerato che ligandi endogeni a comprovato effetto neuroprotettivo
come l’adenosina sono in grado di indurre differenziazione, e che questo fenomeno si
accompagna alla inibizione della proliferazione, abbiamo deciso di verificare se la linea
cellulare SH-SY5Y, in cui la stimolazione dei recettori adenosinici A1 e A2 determina
differenziazione (Canals et al., 2005), esprimesse siti sigma-1 e -2, e questo allo scopo di
verificare in seguito la comparsa o meno di un effetto differenziante mediato da ligandi
considerati prevalentemente sigma-1 selettivi. In caso positivo, l’attività differenziante
poteva essere verificata con due approcci diversi e complementari: la verifica della
riduzione della proliferazione da una parte e della comparsa di neuritogenesi dall’altra.
Per accertare la presenza di recettori sigma nella linea cellulare di neuroblastoma
umano SH-SY5Y sono stati eseguiti esperimenti di saturazione impiegando nuovamente
[3H]pentazocina e [3H]DTG per marcare, rispettivamente, i siti sigma-1 e sigma-2. Gli
esperimenti, inoltre, sono stati eseguiti su cellule intatte, e non su membrane, e questo allo
scopo di ottenere dei parametri di binding dei composti di riferimento da poter usare non
solo come termine di confronto verso i composti di neosintesi, ma specialmente per poter
correlare in modo diretto questi valori con quelli eventualmente ottenuti negli esperimenti
di tipo funzionale (inibizione della proliferazione).

DISCUSSIONE
178
Per quanto riguarda gli esperimenti di saturazione, i risultati ottenuti indicano che i
due ligandi presentano all’incirca lo stesso grado di affinità, con valori di Kd pari a 23.7 e
12.6 nM, rispettivamente, per [3H]pentazocina e [3H]DTG. Questi valori sono ampiamente
in accordo con quelli ottenuti da Colabufo et al. (2004) impiegando membrane di cellule di
neuroblastoma SK-N-SH. Questa linea costituisce la cultura da cui, per clonazione, è stata
ottenuta quella da noi usata, e se ne differenzia per la presenza di una doppia popolazione
cellulare, una simil-neuronale ed una simil-epiteliale, ed è quest’ultima popolazione ad
essere assente nelle cellule SH-SY5Y. Al contrario dei valori di Kd, i valori di Bmax sono
risultati di molto inferiori, rispetto alle cellule SK-N-SH (525-656 pmoli/mg proteine), con
una espressione maggiore di quasi il 100% di siti sigma-1 (0.190 ± 0.05 pmoli/mg
proteine), rispetto ai siti sigma-2 (0.106 ± 0.026 pmoli/mg proteine). E’ possibile che
questa differenza sia dovuta, più che alla diversità della linea cellulare, all’aver impiegato
cellule intatte al posto di membrane: in questo modo, infatti, possono essere evidenziati
solo i recettori espressi a livello di membrana, escludendo quelli situati a livello del
citoplasma. Per quanto riguarda gli esperimenti di competizione, i valori di IC50 per la
pentazocina sono risultati più elevati di circa 3 e 10 volte, rispettivamente, verso i siti
sigma-1 e sigma-2, se confrontati con quelli ottenuti negli esperimenti eseguiti su
membrane. Questo risultato non è inaspettato e deriva dall’aver usato cellule intatte, in cui
lo stato conformazionale del recettore è presumibilmente a minore affinità, rispetto a
quello assunto in membrane isolate. Inaspettato è stato invece il risultato ottenuto con DTG
e aloperidolo, per i quali le curve di competizione verso i recettori sigma-1 hanno assunto
un aspetto rettilineo o quasi, tanto da impedire il calcolo del valore di IC50 nel caso del
DTG, e di calcolare valori elevati, e probabilmente non attendibili, per l’aloperidolo, con
riduzione di affinità, valutata come IC50, verso i siti sigma-1 da 6 nM ottenuto su
membrane a 176 nM nelle cellule intatte, con valore di Hillslope di 0.32. Questi risultati
potrebbero essere dovuti alla lipofilicità di questi composti, rispetto alla pentazocina, con
conseguente “intrappolamento” degli stessi all’interno delle cellule, lontano dal sito di
riconoscimento espresso sulla membrana. Inoltre, per quanto riguarda l’aloperidolo,
un’ulteriore spiegazione potrebbe derivare dalla dimostrazione che nelle cellule SH-SY5Y
questo composto va incontro a metabolismo, con formazione di un derivato ridotto in
grado di bloccare selettivamente e in maniera irreversibile i recettori sigma-1 (Cobos et al.,
2007).
I risultati ottenuti negli esperimenti di competizione hanno trovato conferma negli
esperimenti di inibizione della proliferazione, nei quali la sola pentazocina, tra i composti
di riferimento, è risultata in grado di ridurre, in modo concentrazione dipendente, la

DISCUSSIONE
179
biomassa. Tale effetto è risultato anche tempo-dipendente, con una riduzione del valore di
IC50 da 70 µM, dopo 48 ore di trattamento, a 40 µM, dopo 72 ore di esposizione. Il
mancato effetto di DTG e aloperidolo, almeno fino alla concentrazione di 1 mM, potrebbe
essere ascritto sia a fattori farmacocinetici, come sopra suggerito, sia a fattori
farmacodinamici, cioè alla loro presunta attività antagonista. Nel caso della validità della
seconda ipotesi, si potrebbe suggerire che, tra i composti di neosintesi, solo LEk sia dotato
di attività intrinseca, in quanto solo per questo composto è stato possibile calcolare un
valore di IC50, e comunque solo dopo 72 ore di trattamento. Viceversa, sia PV46, sia CF43
si sono dimostrati inefficaci, nuovamente almeno fino alla concentrazione di 1 mM, e in tal
senso potrebbero essere annoverati tra ligandi sigma-1 ad attività antagonista. La riduzione
di assorbanza osservata alle concentrazioni più elevate potrebbero essere ascritte ad un
effetto tossico, come nel caso di aloperidolo e PV46, o all’effetto differenziante del
DMSO. Questo solvente è stato impiegato per solubilizzare tutti i composti, con
l’eccezione della pentazocina, e la sua capacità di differenziante è ampiamente
documentata (Oh et al., 2005).
Verificata la capacità della pentazocina e del composto LEk di ridurre la
proliferazione cellulare, lo studio è proseguito con la determinazione della eventuale
capacità degli stessi di indurre differenziazione. E’ noto che infatti proliferazione e
differenziazione sono due fenomeni che si escludono a vicenda, e che la differenziazione
può essere efficacemente valutata visivamente, osservando il numero di cellule che
presentano dei prolungamenti di lunghezza pari almeno il doppio di quella del corpo
cellulare. Con questo metodo abbiamo potuto confermare le proprietà differenzianti di
pentazocina e LEk, per i quali la percentuale di cellule differenziate è risultata
significativamente aumentata rispetto ai rispettivi controlli, con percentuali pari a 23% e
31%, rispettivamente, dopo 48 ore di contatto. Anche in questo caso, né i due composti di
riferimento DTG e aloperidolo, né i composti di neosintesi PV46 e CF43, hanno
determinato variazioni significative della lunghezza delle estensioni neuritiche, e questo
rafforza l’ipotesi che si tratti di composti privi di attività intrinseca.
In conclusione, i risultati presentati in questa tesi suggeriscono che il composto
LEk, selezionato all’interno di una ampia serie di composti di neosintesi per la sua
elevatissima potenza e selettività, possa agire come agonista dei recettori sigma-1. Questo
riveste un certa importanza considerato che, nonostante non esistano più dubbi riguardo al
fatto che i recettori sigma costituiscano una classe separata, rispetto ad altri sistemi
recettoriali, non sono ancora noti né il ligando endogeno, né le vie di trasduzione del
segnale. Di conseguenza lo sviluppo di agonisti ed antagonisti potenti e selettivi può

DISCUSSIONE
180
contribuire notevolmente ad una miglior comprensione della funzione e del ruolo dei
recettori sigma in condizioni fisiologiche e in svariate patologie.

6. BIBLIOGRAFIA

BIBLIOGRAFIA
181
Ault D.T., Werling L.L. Differential modulation of NMDA-stimulated [
3H]dopamine release from rat striatum by
neuropeptide Y and sigma receptor ligands.
Brain Res 1997; 760: 210-217 Ault D.T., Werling L.L. SH-SY5Y cells as a model for sigma receptor regulation of potassium-stimulated dopamine
release.
Brain Res 2000; 877: 354-360 Aydar E., Palmer C.P., Klyachko V.A., Jackson M.B. The sigma receptor as a ligand-regulated auxiliary potassium channel subunit.
Neuron 2002; 34: 399-410 Beltran D., Cavas M., Navarro J.F. Effects of (+)SKF 10047, a sigma-1 selective agonist, on isolation-induced aggression in
male mice.
Methods Find Exp Clin Pharmacol 2006; 28: 601-4 Bouchard P. Quirion R. [
3H]1,3-di(2-tolyl)guanidine and [
3H](+)pentazocine binding sites in the rat brain:
autoradiographic visualization of the putative sigma-1 and sigma-2 receptor subtypes.
Neuroscience 1997; 76: 467-77 Canals M., Angulo E., Casadó V., Canela E.I., Mallol J., Viñals F., Staines W., Tinner B., Hillion J., Agnati L. Fuxe K., Ferré S., Lluis C., Franco R. Molecular mechanisms involved in the adenosine A1 and A2A receptor-induced neuronal
differentiation in neuroblastoma cells and striatal primary cultures.
J Neurochem 2005; 92:337-348 Cobos E.J., Lucena G., Baeyens J.M., Del Pozo E. Phenytoin differentially modulates the affinity of agonist and antagonist ligands for σ1
receptors af guinea pig brain.
Synapse 2005; 55: 192-195 Cobos E.J., Lucena G., Baeyens J.M., Del Pozo E. Differences in the allosteric modulation by phenytoin of the binding properties of the σ1
ligands [3H](+)-Pentazocine and [
3H]NE-100.
Synapse 2006 59: 152-161 Cobos E.J., Del Pozo E., Baeyens J.M. Irreversible blockade of sigma-1 receptors by haloperidol and its metabolites in guinea pig
brain and SH-SY5Y human neuroblastoma cells.
J Neurochem 2007; 102: 812-825 Colabufo N.A., Brardi F., Contino M., Niso M., Abate C., Perrone R., Tortorella V. Antiproliferative and cytotoxic effects of some sigma-2 agonist and sigma-1 antagonist in
tumor cell lines.
Naunyn-Schmiedeberg’s Arch Pharmacol 2004; 370: 106-113

BIBLIOGRAFIA
182
Collier T.L., Waterhouse R.N., Kassiou M. Imaging sigma receptors: application in drug development.
Curr Pharm Des 2007; 13: 51-72 Costantino L., Gandolfi F., Sorbi C., Franchini S., Prezzavento O., Vittorio F., Ronsisvalle G., Leonardi A., Poggesi E., Brasili L. Synthesis and structure-activity relationships of 1-aralkyl-4-benzylpiperidine and 1-
aralkyl-4-benzylpiperazine derivatives as potent sigma ligands.
J Med Chem 2005; 48: 266-73 DeCostner M.A., Klette K.L., Knight E., Tortella F.C. σ Receptor-mediated neuroprotection against glutamate toxicity in primari rat neuronal
culture.
Brain Res 1995; 671: 45-53 Duncan G., Wang L. Focus on molecules: the sigma-1 receptor.
Exp Eye Res 2005; 81: 121-122 Egashira N., Harada S., Okuno R., Matsushita M., Nishimura R., Mishima K., Iwasaki K., Orito K., Fujiwara M. Involvement of the sigma1 receptor in inhibiting activity of fluvoxamine on marble-burying
behavior: comparison with paroxetine.
Eur J Pharmacol 2007; 563: 149-54 Goyagi T., Goto S., Bhardwaj A., Dawson V.L., Hurn P.D., Kirsch J.R. Neuroprotective effect of sigma(1)-receptor ligand 4-phenyl-1-(4-phenylbutyl) piperidine
(PPBP) is linked to reduced neuronal nitric oxide production.
Stroke 2001; 32: 1613-1620 Guitart X., Codony X., Monroy X. Sigma receptors: biology and therapeutic potential.
Psychopharmacology 2004; 174: 301-319 Hashimoto K., Ishiwata K. Sigma receptors ligands: possible application as therapeutic drugs and as
radiopharmaceuticals.
Curr Pharm Des 2006; 12: 3857-3876 Hayashi T., Su T.P. Regulating ankryn dynamics: roles of sigma-1 receptors.
Proc Natl Acad Sci USA 2000; 98: 491-496 Hellewell S.B., Bruce A., Feinstein G., Orringer J., Williams W., Bowen W.D. Rat liver and kidney contain high densities of sigma-1 and sigma-2 receptors:
characterization by ligand binding and phtoaffinity labelling.
Eur J Pharmacol 1994; 268: 9-18 Hong W., Werling L.L. Evidence that the sigma-1 receptor is not directly coupled to G proteins.
Eur J Pharmacol 2000; 408: 117-125

BIBLIOGRAFIA
183
Hong W., Werling L.L. Lack of effects by sigma ligands on neuropeptide Y-induced G-protein activation in rat
hippocampus and cerebellum.
Brain Res 2001; 901: 208-218 Hong W., Werling L.L. Binding of sigma receptor ligands and their effects on muscarine-induced Ca
2+ changes in
SH-SY5Y cells.
Eur J Pharmacol 2002; 436: 35-45 Katnik C., Guerrero W.R., Pennypacker K.R., Herrera Y., Cuevas J. Sigma-1 receptor activation prevents intracellular calcium dysregulation in cortical
neurons during in vitro ischemia.
J Pharmacol Exp Ther 2006; 319: 1355-1365 Kim H.W., Kwon Y.B., Roh D.H., Yoon S.Y., Han H.J., Kim K.W., Beitz A.J., Lee J.H. Intrathecal treatment with sigma1 receptor antagonists reduces formalin-induced
phosphorylation of NMDA receptor subunit 1 and the second phase of formalin test in
mice.
Br J Pharmacol 2006;148 :490-498 Langa F., Codony X., Tovar V., Lavado A., Gimenez E., Cozar P., Cantero M., Dordal A., Hernandez E., Perez R., Monroy X., Zamanillo D., Guitart X. e Montoliu L. Generation and phenotypic analysis of sigma receptor type I (sigma 1) knockout mice.
Eur J Neurosci 2003; 18(8): 2188-2196 Lockhart B.P., Soulard P., Benicourt C., Privat A., Junien J.L. Distinct neuroprotective profiles for sigma ligands against N-methyl-D-aspartate (NMDA),
and hypoxia-mediated neurotoxicity in neuronal culture toxicity studies.
Brain Res 1995; 675: 110-120 Lowry O.H., Rosenbrough N.J., Farr A.L., Randall R.J. Protein measurement with the folin phenol reagent.
J Biol Chem 1951; 93: 265-275 Marrazzo A., Caraci F., Salinaro E.T., Su T.P., Copani A., Ronsisvalle G. Neuroprotective effects of sigma-1 receptor agonists against beta-amyloid-induced
toxicity.
Neuroreport 2005; 16: 1223-1226 Martin Moore P., Ola M.S., Agarwal N., Ganapathy V., Smith S.B. The sigma receptor ligand (+)-pentazocine prevents apoptotical retinal ganglion cell
death induced in vitro by homocysteine and glutamate.
Mol Brain Res 2004; 123: 66-75 Matsumoto R.R., Liu Y., Lerner M., Howard E.W., Brackett D.J. Sigma receptors: potential medications development target for anti-cocaine agents.
Eur J Pharmacol 2003; 469: 1-12

BIBLIOGRAFIA
184
Matsuno K. Anti-amnesic effects of sigma-receptors agonists.
Folia Pharmacol Jpn [abstract] 1999; 114: 25-33 Maurice T. Beneficial effect of the sigma-1 receptor agonist PRE-084 against the spatial learning
deficits in aged rats.
Eur J Pharmacol 2001; 431: 223-227 Maurice T., Urani A., Phan V.L., Romieu P. The interaction between neuroactive steroids and the sigma-1 receptor function:
behavioral consequences and therapeutic opportunities.
Brain Res Rev 2001; 37: 116-132 Maurice T. Improving Alzheimer’s desease related cognitive deficits with sigma-1 receptor agonists.
Drug News Perspect 2002; 15: 617-625 Maurice T., Grègorie C., Espallergues J. Neuro(active)steroids actions at the neuromodulatory sigma-1 receptor: biochemical and
physiological evidences, consequences in neuroprotection.
Pharmacol Biochem Behav 2006; 84: 581-597 Mavlyutov T.A., Ruoho A.E. Ligand-dependent localization and intracellular stability of sigma-1 receptors in CHO-Kl
cells.
J Mol Signal 2007; 2: 1-8 Meunier J., Guè M., Rècasens M., Maurice T. Attenuation by a sigma-1 receptor agonist of the learning and memory deficits induced by
a prenatal restraint stress in juvenile rats.
Br J Pharmacol 2004; 142: 689-700 Moebius F.F., Reiter R.J., Hanner M., Glossmann H. High affinity of sigma 1-binding sites for sterol isomerization inhibitors: evidence for a
pharmacological relationship with the yeast sterol C8-C7 isomerase.
Br J Pharmacol 1997; 121: 1-6 Monassier L., Bousquet P. Sigma receptors: from discovery to highlights of their implications in the cardiovascular
system.
Fundam Clin Pharmacol 2002; 16: 1-8 Monnet F.P., Maurice T. The sigma-1 protein as a target for non-genomic effects of neuroactivesteroids: molecular,
physiological and behavioural aspects.
J Pharmacol Sci 2006; 100: 93-118 Musacchio JM, Klein M, Paturzo JJ Effects of dextromethorphan site ligands and allosteric modifiers on the binding of (+)-
[3H]3-(-3-hydroxyphenyl)-N-(1-propyl)piperidine.
Mol Pharmacol 1989; 35: 1-5

BIBLIOGRAFIA
185
Nakazawa M., Matsuno K., Mita S. Activation of sigma1 receptor subtype leads to neuroprotection in the rat primary neuronal
cultures.
Neurochem Int 1998; 32 :337-343 Nicosia S. Iterazioni farmaco-recettore e risposta quantitative ai farmaci.
In: Farmacologia generale e molecolare. Clementi F, Fumagalli G, UTET, 1999; pp. 43-61 Oh J., Karlmark K.R., Shin J., Pollak A., Freilinger A., Hengstschläger M., Lubec G. Differentiation of neuroblastoma cell line N1E-115 involves several signalling cascades.
Neurochem Res 2005; 30: 333-348 Quirion R., Bowen W.D., Itzhak Y., Julien J.L., Musacchio J.M., Rothman R.B., Su T.P., Tam S.W., Taylor D.P. A proposal for the classification of sigma binding site.
Trends Pharmacol Sci 1992; 13: 85-86 Racagni G., Cantalupi S., Fumagalli R. Misura della cinetica recettoriale. In: farmacologia generale e applicata.
Masson, Masson Italia Editori, Milano 1986, pp. 313-314 Sabino V., Cottone P., Parylak S.L., Steardo L. e Zorrilla E.P. Sigma-1 receptor knockout mice display a depressive-like phenotype.
Behav Brain Res 2009 (doi:10.1016/j.bbr.2008.11.036) Sakata M., Kimura Y., Naganawa M., Oda K., Ishii K., Chihara K., Ishiwata K. Mapping of human cerebral sigma1 receptors using positron emission tomography and
[11
C]SA4503.
Neuroimage 2007; 35: 1-8 Schetz J.A., Perez E., Liu R., Chen S., Lee I., Simpkins J.W. A prototypical sigma-1 receptor antagonist protects against brain ischemia.
Brain. Res 2007; 1181: 1-9 Senda T., Matsuno K., Kobayashi T., Mita S. Reduction of the scopolamine-induced impaiment of passive-avoidance performance by
sigma receptor agonist in mice.
Physiol Behav 1997; 61: 257-264 Shen Y.C. Wang Y.H., Chou Y.C., Liou K.T., Yen J.C., Wang W.Y., Liao J.F.J. Dimemorfan protects rats against ischemic stroke through activation of sigma-1 receptor-
mediated mechanisms by decreasing glutamate accumulation.
J Neurochem 2008; 104: 558-72 Skehan P., Storeng R., Scudiero D., Monks A., MacMahon J., Viostica D., Warren J.T., Bokesch H., Kenney S., Boyd M.R. New colorimetric cytotoxicity assay for anticancer-drug screening.
J Natl Cancer Inst 1990; 82: 1107-1112

BIBLIOGRAFIA
186
Skuza G. Potential antidepressant activity of sigma ligand. Pol J Pharmacol 2003; 55: 923–934 Skuza G., Rogoz Z. Effect of BD 1047, a sigma1 receptor antagonist, in the animal models predictive of
antipsychotic activity.
Pharmacol Rep 2006; 58: 626-35 Skuza G., Rogoz Z. Antidepressant-like effect of combined treatment with selective sigma receptor agonists
and a 5-HT(1A) receptor agonist in the forced swimming test in rats.
Pharmacol Rep,2007; 59: 773-7 Su T.P., London E.D., Jaffe J.H. Steroid binding at sigma receptors suggests a link between endocrine, nervous, and
immune systems.
Science 1988; 240: 219-21 Su T.P., Hayashi T. Understanding the molecular mechanism of sigma-1 receptors: towards a hypothesis that
sigma-1 receptors are intracellular amplifiers for signal transduction.
Curr Med Chem 2003; 10: 2073-2080 Takebayashi M., Hayashi T., Su T.P. Nerve growth factor-induced neurite sprouting in PC12 cells involves sigma-1 receptors:
implications for antidepressants.
J Pharmacol Exp Ther 2002; 303:1227-37 Takebayashi M., Hayashi T., Su T.P. Sigma-1 receptors potentiate epidermal growth factor signaling towards neuritogenesis in
PC12 cells: potential relation to lipid raft reconstitution.
Synapse 2004; 53: 90-103 Tottori K., Nakai M., Uwahodo Y., Miwa T., Yamada S., Oshiro Y., Kikuchi T., Altar C.A. Attenuation of scopolamine-induced and age-associated memory impairments by the sigma
and 5-hydroxytryptamine(1A) receptor agonist OPC-14523 (1-[3-[4-(3-chlorophenyl)-1-
piperazinyl]propyl]-5-methoxy-3,4-dihydro-2[1H]-quinolinone monomethanesulfonate).
J Pharmacol Exp Ther 2002; 301: 249-257 Yamamoto H., Miura R., Yamamoto T., Shinohara K., Watanabe M., Okuyama S., Nakazato A., Nukada T. Amino acid residues in the transmembrane domain of the type 1 sigma receptor critical for
ligand binding.
FEBS Lett 1999; 445: 19-22 Yang S., Bhardwaj A., Cheng J., Alkyed N.J., Hurn P.D., Kirsch J. Sigma receptor agonists provide neuroprotection in vitro by preserving bcl-2.
Anesth Analg 2007; 104: 1179-1184

I miei più sentiti ringraziamenti vanno alla Dott.ssa Chiara Florio. In questi tre anni mi ha
seguito con instancabile pazienza, mi ha permesso di crescere, di nutrire entusiasmo per il mio
lavoro e di realizzare questo progetto. Rappresenta per me un esempio di correttezza e umanità.
Ringrazio il Prof. Ugo Traversa, la Dott.ssa Sabrina Pacor e la Prof. ssa Paola Lorenzon
perché con la loro disponibilità hanno contribuito alla completezza di questo lavoro.
La collaborazione con la Prof.ssa Maria Grazia Mamolo e il Prof. Luciano Vio origina da
una stima e un affetto nati ancora durante gli anni della tesi di laurea… saranno sempre un punto
di riferimento!
Doverosi però sono i ringraziamenti verso tutto il Dipartimento di Scienze della Vita perché
in questi anni ho trovato sempre persone pronte ad aiutarmi scientificamente e moralmente.
… non avrei potuto farcela senza il mio gruppo del “Castelletto” che ha reso indimenticabili
questi anni e ha colorato tutte le mie giornate lavorative anche nei momenti più difficili.
Grazie Valeria, Sara, Marco, Alessandra, Chiara, Giulia, Ilaria, Elena, Egle, Alberto….
…per tutto quello che avete fatto per me!