afc 2013 programme et résumés
TRANSCRIPT

AFC 2013
Colloque de l’Association Française
de Cristallographie
Bordeaux 2 – 5 juillet
Programme et résumés

- 2 -

- 3 -
Chères et chers collègues, Nous vous souhaitons la bienvenue au Congrès AFC2013 de l'Association Française de Cristallographie, qui se tiendra dans la belle ville de Bordeaux qui avait déjà accueilli le Congrès de l'IUCr en 1990. Le Comité Scientifique a organisé un programme riche et varié de conférences plénières, de sessions thématiques ou interdisciplinaires et de rencontres autour de posters. Les scientifiques de nombreuses disciplines utilisant la cristallographie ou la cristallogenèse dans leurs recherches pourront y découvrir les résultats les plus récents et partager leurs points de vue. L'AFC a pour tradition de mettre à l'honneur les jeunes chercheurs, qu'elle soutient par des prix et des bourses. L'AFC2013 sera l'occasion de la remise du prix de Thèse à nos lauréats 2012, qui y présenteront leurs travaux. Un jury sélectionnera également les meilleurs posters, à qui l'AFC remettra un prix. A noter cette année une initiative inédite, "Meet the experts", où doctorants et jeunes chercheurs sont invités à venir rencontrer des chercheurs confirmés et leur poser toutes les questions qui leur tiennent à cœur. Nous comptons sur votre présence à l'Assemblée Générale, un événement majeur de la vie de notre Association. Le Conseil d'Administration vous y présentera le bilan de sa mandature qui s'achève et échangera avec vous sur les perspectives futures, en particulier les préparatifs de l'Année Internationale de la Cristallographie en 2014. Le Comité d'Organisation a enfin déployé tous ses efforts pour compléter ce programme par des soirées conviviales, qui devraient achever, nous l'espérons, de faire de l'AFC2013 à Bordeaux un "Grand Cru" à tous les égards. Au plaisir de vous y retrouver en personne, Jacqueline Cherfils, Présidente du Comité Scientifique Alain Dautant, Président du Comité d'Organisation

- 4 -
Remerciements
Le comité local d’organisation tient à remercier tout spécialement toutes celles et ceux sans qui ce congrès n’aurait pas été possible : Martine Galian, Catherine Alves-Magalhaes Isabelle Nicolas, (Service de Gestion de l’IBGC), Lydie Goutière (Services généraux de l’IBGC), Axel Catouillart et Jean Baptiste Maurange (Service informatique de l’IBGC), Annie Nadeau et Guillaume Moissonnié (Cellule Colloque à la DR 15 du CNRS), Jean-Louis Hodeau (Grenoble) qui vous a proposé l’exposition dans le Hall de l’ENSCBP, Mireille Frimigacci (Responsable HSE, ENSCBP), Marie Hénault (Responsable Cellule Congrès de l’Université Bordeaux Segalen), l’AFC et le conseil scientifique de l’AFC 2013, les chairs et les co-chairs pour l’organisation des sessions, les conférenciers qui ont bien voulu accepter l’invitation, les organismes qui nous ont soutenus, les partenaires, et tous les participants au congrès de l’AFC 2013.

- 5 -
Le Comité Scientifique
• Jacqueline Cherfils, présidente (LEBS, Gif, AFC) • René Guinebretière, vice-président (SPCTS, Limoges, AFC) • Nathalie Audebrand (ESI, Rennes) • Yves Bourne (AFMB, Marseille) • Alain Dautant (IBGC, Bordeaux) • Denis Gratias (ONERA, Paris) • Ivan Huc (IECB, Bordeaux) • Jean-Paul Itié (Soleil, AFC) • Mario Maglione (ICMCB, Bordeaux) • Claudine Mayer (Pasteur, Paris, AFC) • Olivier Perez (ENSI, Caen, AFC)

- 6 -
Le Comité Local d’Organisation
• Alain Dautant, président (IBGC, CNRS) • Pierre Dechambenoit (CRPP, UB1) • Stephan Dubernet (IRAMAT-CRP2A, UB3) • Sébastien Fribourg (IECB-ARNA, INSERM) • Bernard Gallois (CBMN, CNRS) • Philippe Guionneau (ICMCB, UB1) • Elizabeth Hillard (CRPP, CNRS) • Ivan Huc (IECB-CBMN, CNRS) • Brice Kauffmann (IECB, CNRS) • Aline Lacoudre (ISM, UB1) • Eric Lebraud (ICMCB, CNRS) • Stéphane Massip (Faculté de Pharmacie, UBS) • Denise Mondieg (LOMA, UB1) • Stanislas Pechev (ICMCB, CNRS) • Thierry Toupance (ISM, UB1)

- 7 -

Organismes soutenant le congrès
- 8 -
L’Association Bordelaise de Cristallographie www.abc.u-bordeaux1.fr La Région Aquitaine region.aquitaine.fr AVIESAN www.aviesan.fr L’Université de Bordeaux 1 www.u-bordeaux1.fr L’Université Bordeaux Segalen www.univ-bordeauxsegalen.fr CNRS – Délégation régionale Aquitaine www.cnrs.fr Le CROUS de Bordeaux www.crous-bordeaux.fr La CUB - Communauté Urbaine de Bordeaux www.lacub.fr Le Conseil Général de Gironde - CG33 www.gironde.fr L’IBGC www.ibgc.cnrs.fr L’INSERM www.inserm.fr L’IPB – ENSCBP www.enscbp.fr L’Office de Tourisme de Bordeaux - OTB www.bordeaux-tourisme.com TransBioMed www.transbiomed.u-bordeaux2.fr

Organismes soutenant le congrès
- 9 -

Partenaires
- 10 -
www.sourcedesabatilles.com
genomics.agilent.com
www.bruker.com
www.dectris.com
www.dutscher.com
www.dunnlab.de
www.elexience.fr
www.exploranova.com
www.inel.fr www.moleculardimensions.com

Partenaires
- 11 -
www.mitegen.com
www.natx-ray.com
www.oxcryo.com
www.panalytical.com
www.rigaku.com www.stoe.com
www.xenocs.com

Journal des sessions
- 12 -
Mardi 2 juillet 1A Matériaux fonctionnels (1) 15h00 – 16h30 A MARIO MAGLIONE et CHRISTINE MARTIN 1B Surfaces et interfaces 15h00 – 16h30 B GILLES RENAUD et DAVID BABONNEAU 1C Nouvelles structures en biologie 15h00 – 16h30 C MIRJAM CZJZEK et LIONEL MOUREY 2A Chimie supramoléculaire et Ingénierie moléculaire 17h00 – 18h30 A NATHALIE GUILLOU et IVAN HUC 2B Cristallographie pour les nanosciences 17h00 – 18h30 B RENE GUINEBRETIERE et VALERIE DEMANGE 2C Enzymologie et dynamique 17h00 – 18h30 C HERMAN VAN TILBEURGH et GERLIND SULZENBACHER
Mercredi 3 juillet 3A Croissance cristalline (GFCC) 10h30 – 12h00 A ALAIN IBANEZ et FRANÇOISE BONNETE 3B Cristallographie résolue en temps 10h30 – 12h00 B MARYLISE BURON et SYLVAIN RAVY 3C La cristallographie sous conditions extrêmes 10h30 – 12h00 C ERIC GIRARD et JEAN-PAUL ITIE 4A Matériaux fonctionnels (2) 13h30 – 15h00 A MARIO MAGLIONE et CHRISTINE MARTIN 4B Apériodicité, Structure modulée 13h30 – 15h00 B BERTRAND TOUDIC et DENIS GRATIAS 4C Biologie structurale des génomes 13h30 – 14h40 C CLAUDINE MAYEr et ANNE-CATHERINE DOCK-BREGEON 4D Formation ReNaFoBis 14h40 – 15h00 C JEAN CAVARELLI
Jeudi 4 juillet 5A Approches multitechniques en cristallochimie 10h30 – 12h00 A
NATHALIE AUDEBRAND et PASCAL ROUSSEL 5B Densité électronique, modélisation 10h30 – 12h00 B
PHILIPPE RABILLER et MARIE-BERNADETTE LEPETIT 5C Cristallographie biologique et Santé 10h30 – 12h00 C
YVES BOURNE et MAGALI MATHIEU 6A/B Méthodes émergentes en cristallographie 13h30 – 15h15 A
OLIVIER PEREZ ET DAVID LE BOLLOC'H 6C Biologie Structurale Intégrative et gros assemblages 13h30 – 15h15 C
DINO MORAS et JACQUELINE CHERFILS Vendredi 5 juillet
7A Cristallographie in situ, in operando 9h00 – 10h30 A ERIK ELKAÏM ET BEATRICE GILLON
7B Texture, Microstructure, déformation 9h00 – 10h30 B ALAIN LODINI ET DANIEL CHATEIGNER
7C GI Synchrotron et plateformes en biologie structurale 9h00 – 10h30 C ANDREW THOMPSONet MICHEL KOCHOYAN
8A Chiralité, polymorphisme 11h00 – 12h30 A GERARD COQUEREL et CHRISTIAN JARRY
8B De l'ordre local à l'ordre à longue distance 11h00 – 12h30 B PIERRE-ANTOINE ALBOUY; ET PAULINE MARTINETTO
8C Défis en cristallographie biologique - Success stories. 11h00 – 12h30 C PHILIPPE DUMAS ET JULIE MENETREY

- 13 -
Programme

- 14 -

Programme
- 15 -
Mardi 2 juillet
10h00 – 14h00 Accueil et enregistrement des participants Hall de l’ENSCPB
☺☺☺☺☺☺ Repas ☺☺☺☺☺☺
12h00 – 13h30 Déjeuner dans le hall de l’ENSCBP 13h30 – 14h00 Discours inauguraux et de bienvenue
ALAIN DAUTANT ; Président du comité local d’organisation JACQUELINE CHERFILS ; Présidente de l’Association Française de Cristallographie
* * * * * * * * * * Conférence plénière I * * * * * * * * *
14h00 – 15h00 Amphi C
Chair : RENE GUINEBRETIERE, vice président de l’AFC IVAN HUC ; CBMN & IECB, Bordeaux
SP1 - Les foldamères : une expansion de l'espace chimique basée sur l'analyse des structures.
= = = Session 1A Matériaux fonctionnels (1)= = =
15h00 – 16h30 Amphi A
Chair : MARIO MAGLIONE; ICMCB, Bordeaux Co-chair : CHRISTINE MARTIN; ENSI, Caen 15h00 CHRISTOPHE PAYEN; Institut des Matériaux Jean Rouxel, Nantes
O1 - Relations structure-magnétisme dans le composé magnéto-électrique MnWO4.
15h30 JULIEN ROBIN; Institut des Sciences Chimiques Université de Rennes 1 O2 - Synthèse et structures cristallines de nouveaux polymères de coordination chiraux à base de fluorène pour la séparation et la catalyse énantiosélective.
16h00 – 16h30 JEROME ROUQUETTE; CNRS Université de Montpellier II O3 - Origine de la fatigue ferroélectrique dans le zirconate-titanate de Plomb PZT.
= = = Session 1B Surfaces et interfaces = = = 15h00 – 16h30 Amphi B
Chair : GILLES RENAUD; ESRF, Grenoble Co-Chair : DAVID BABONNEAU; Université de Poitiers 15h00 YVONNE SOLDO-OLIVIER; LEPMI, Grenoble
O4 - SXRD in situ couplée à l’électrochimie : étude de l’électro-insertion d’hydrogène dans des nanofilms de Pd.
15h30 ALESSANDRO COATI; Synchrotron SOLEIL, Gif-sur-Yvette O5 - Surfaces vicinales : des nano-gabarits pour croissance épitaxiale.

Programme
- 16 -
16h00 GABIN GBABODE; Université de Rouen O6 - Mise en évidence de phases induites par le substrat pour des mésogènes π-conjugués.
16h15 – 16h30 RENE GUINEBRETIERE; ENS de Céramique Industrielle, Université de Limoges
O7 - Etude de surfaces vicinales ordonnées par diffusion centrale des rayons X sous incidence rasante. Nouveaux développements sur la ligne BM02 à l’ESRF.
= = = Session 1C Nouvelles structures en biologie = = =
15h00 – 16h30 Amphi C
Chair : MIRJAM CZJZEK; Station Biologique, Roscoff Co-chair : LIONEL MOUREY; IBPS, Toulouse 15h00 MARIE-CHRISTINE VANEY; Institut Pasteur, Unité de Virologie Structurale Paris
O8 - Structure de l'ectodomaine de la glycoprotéine d'enveloppe E1, dans sa forme post-fusion, du virus de la Rubéole.
15h30 SEBASTIEN FRIBOURG; Institut Européen de Chimie et Biologie, Bordeaux O9 - Bases structurales de l'initiation de la transcription de l'ARN polymerase III.
16h00 CORALIE BOMPARD; Université Lille 1 O10 - Analyse structurale par SAXS de SopB, un effecteur de type III de Salmonella en complexe avec sa chaperonne spécifique SigE.
16h15 – 16h30 PASCAL ARNOUX ; CEA Cadarache O11 - Bases structurales et fonctionnelles de la biominéralisation de la magnétite : importance des domaines magnétochromes.
☺☺☺☺☺☺ Tout le monde rencontre tout le monde ☺☺☺☺☺☺
16h30 – 17h00 Pause café sponsorisée par l’ABC – Rencontre avec les partenaires industriels Salle ChemInnov.
= = = Session 2A Chimie supramoléculaire et Ingénierie moléculaire = = =
17h00 – 18h30 Amphi A
Chair : NATHALIE GUILLOU; Institut Lavoisier, Versailles St-Quentin-en-Yvelines Co-chair : IVAN HUC; CBMN & IECB, Bordeaux 17h00 NICOLAS MERCIER; MOLTECH-Anjou, Université d'Angers
O12 - Analyse structurale d’hybrides organique-inorganique et relations structure-propriétés : l’importance de l’interface et des liaisons faibles.
17h30 JONATHAN NITSCHKE; Cambridge University O13 - Crystallography as a critical resource in metal-organic self assembly.
18h00 MARC FOURMIGUE; Université de Rennes 1 O14 - La liaison halogène pour l'élaboration d'architectures complexes: contribution électrostatique et réseaux d'anions (2D, 3D).
18h15 – 18h30 CLEMENT FALAISE; UCCS O15 - Les carboxylates d’uranium tétravalents : précurseurs du combustible nucléaire.

Programme
- 17 -
= = = Session 2B Cristallographie pour les nanosciences = = =
17h00 – 18h30 Amphi B
Chair : RENE GUINEBRETIERE; Université de Limoges Co-chair : VALERIE DEMANGE; Institut des Sciences Chimiques de Rennes, Rennes 1 17h00 ANNICK LOISEAU; Laboratoire d'Etude des Microstructures ONERA – CNRS
O16 - Structure des nanostructures de nitrure de bore: quel impact sur leurs propriétés de luminescence ?
17h30 ETIENNE SNOECK; CEMES, Toulouse O17 - Imagerie des champs de déformations par holographie électronique.
18h00 PASCALE LAUNOIS, CRM2, Université Paris Sud O18 - Structuration de l’eau pendant le remplissage des nanotubes de carbone.
18h15 – 18h30 DOMINIK SCHANIEL, CRM2, Université Nancy 1 O19 - Structure et dynamique de molécules isolées dans des matrices poreuses.
= = = Session 2C Enzymologie et dynamique.= = =
17h00 – 18h30 Amphi C
Chair : HERMAN VAN TILBEURGH; IBBMC, Université Paris-Sud, Orsay Co-chair : GERLIND SULZENBACHER; AFMB, Marseille 17h00 AUDE ECHALIER-GLAZER; CBS, Montpellier
O20 - Activation et activité du complexe COP9 signalosome.
17h30 MURIEL GONDRY; CEA, Saclay O21 - Les synthases de cyclodipeptides (CDPS), des homologues d’aminoacyl-ARNt synthétases impliquées dans la synthèse peptidique non ribosomale.
18h00 JULIEN HIBLOT; Faculté de Médecine Marseille O22 - Promiscuité enzymatique lactonase/phosphotriestérase : implications évolutives et biotechnologiques.
18h15 – 18h30 VALERIE CAMPANACCI; LEBS Gif-sur-Yvette O23 - Aperçu structural du mécanisme enzymatique d’AnkX, une protéine de Legionella pneumophila modifiant la petite protéine G Rab.
☺☺☺☺☺☺ Tout le monde rencontre tout le monde ☺☺☺☺☺☺
18h30 – 20h30 Soirée d’accueil dans le hall de l’ENSCBP

Programme
- 18 -

Programme
- 19 -
Mercredi 3 juillet
* * * * * * * * * * Conférence plénière II * * * * * * * * *
9h00-10h00 Amphi C
Chair : JACQUELINE CHERFILS, présidente de l’AFC 9h00-10h00 Amphi C - RAY STEVENS, Department of Molecular Biology, Scripps, USA
SP2 - The Amazing Diversity of the Human GPCR Superfamily.
☺☺☺☺☺☺ Tout le monde rencontre tout le monde ☺☺☺☺☺☺
10h00 – 10h30 Pause café – Rencontre avec les partenaires industriels Salle ChemInnov.
= = = Session 3A Croissance cristalline (GFCC) = = = 10h30 – 12h00 Amphi A
Chair : ALAIN IBANEZ; Institut Néel, Grenoble Co-chair : FRANÇOISE BONNETE, Université d'Avignon et des Pays de Vaucluse 10h30 VALENTIN GORDELIY; IBS Grenoble
O24 - Cristallisation des protéines membranaires in cubo.
11h00 PASCAL LOISEAU; Chimie Paris Tech / ENSCP Paris O25 - Croissance, Structure et Optique Non Linéaire Quadratique.
11h30 MARJORIE ALBINO; ICMCB Bordeaux O26 - Etude structurale par diffraction des rayons X sur monocristaux relaxeurs de Ba2LnFeNb4O15 (Ln=La, Pr, Nd, Sm et Eu).
11h45 – 12h00 PIERRE GRAS; ENSIACET Toulouse O27 - Synthèse, caractérisation et croissance cristalline de cristaux de pyrophosphate de calcium d’intérêt biologique.
= = = Session 3B Cristallographie résolue en temps = = =
10h30 – 12h00 Amphi B
Chair : MARYLISE BURON; Université Rennes 1 Co-chair : SYLVAIN RAVY; CRISTAL, Synchrotron SOLEIL 10h30 CAROLINE CURFS; ESRF, Grenoble
O28 -Etudes par Diffraction Résolue en Temps de Phénomènes Irréversibles: Application aux Synthèses Auto-Propagées.
11h00 PAUL BEAUD; SLS (Swiss Light Source) O29 - Ultrafast structural dynamics in charge and orbitally ordered manganites under non-equilibrium conditions.

Programme
- 20 -
11h30 – 12h00 CLAIRE LAULHE; Synchrotron-soleil O30 - Diffraction pompe-sonde résolue en temps sur la ligne de lumière CRISTAL à SOLEIL.
= = = Session 3C La cristallographie sous conditions extrêmes = = = 10h30 – 12h00 Amphi C
Chair : ERIC GIRARD; IBS, Grenoble Co-chair : JEAN-PAUL ITIE; Synchrotron SOLEIL 10h30 JULIEN HAINES; Institut Charles Gerhardt, Montpellier
O31 - Diffraction des rayons X sur monocristaux sous pression : application à l’étude de structures complexes.
11h00 LOUISE LASSALLE; IBS Grenoble O32 - Molecular basis of piezophilic adaptation.
11h20 NATHALIE COLLOC'H; CNRS CEA Université de Caen, Centre Cyceron O33 - Structure cristallographique de la neuroglobine sous pression.
11h40 – 12h00 PHILIPPE GUIONNEAU; ICMCB Bordeaux O34 - Haute pression et cristallographie des matériaux moléculaires à conversion de
spin.
☺☺☺☺☺☺ Repas ☺☺☺☺☺☺ 12h00-13h30 Déjeuner dans le hall de l’ENSCBP
= = = Session 4A Matériaux fonctionnels (2)= = = 13h30 – 15h00 Amphi A
Chair: MARIO MAGLIONE; ICMCB, Bordeaux Co-chair: CHRISTINE MARTIN; ENSI Caen 13h30 MATHIEU ALLIX; CEMHTI, Orléans
O35 -Céramiques transparentes par cristallisation complète du verre.
14h00 JEAN-MICHEL RUEFF; ENSI Caen O36 -Matériaux hybrides à base d’argent et d’acides phosphoniques et carboxyliques : exploration de nouvelles architectures à propriétés bactéricides.
14h20 LIANG TAO; Université de Picardie O37 - Crystal and Magnetic Structure of LiMBO3 (M=Fe, Co, Mn).
14h40 – 15h00 ARNAUD GROSJEAN; ICMCB Bordeaux O38 -Matériaux polymériques à conversion de spin de type [Fe(R-trz)3]•Xn: Comportement structural et microstructural.

Programme
- 21 -
= = = Session 4B Apériodicité, Structure modulée = = = 13h30 – 15h00 Amphi B
Chair : BERTRAND TOUDIC; GMCM, Rennes Co-chair : DENIS GRATIAS; ONERA, Chatillon 13h30 BERTRAND DEVOUARD; Observatoire de Physique du Globe, Clermont-Ferrand
O39 -Structures naturelles apériodiques et multimacles icosaédriques. (30 min)
14h00 MARIANNE QUIQUANDON; LEM CNRS/ONERA, Chatillon O40 - Etude de la filiation structurale entre les phases icosaédriques F (Faces Centrées) et P (Primitives).
14h20 SYLVAIN RAVY; Synchrotron SOLEIL O41 - L'homométrie à la lumière des faisceaux cohérents.
14h40 – 15h00 WERNER PAULUS; Institut Charles Gerhard, Université de Montpellier 2 O42 - L’impact des corrélations structurales à grande échelle pour la réactivité des solides : l'exemple des oxydes AO(ABO3)n Ruddlesden-Popper.
= = Session 4C Biologie structurale des génomes (interactions protéines/ADN, ARN)= =
13h30 – 14h30 Amphi C
Chair : CLAUDINE MAYER; Institut Pasteur, Université Paris 7, Paris Co-Chair : ANNE-CATHERINE DOCK-BREGEON; ENS, Paris 13h30 MARIE-HELENE LE DU; Laboratoire de Biologie Structurale et Radiologie, CEA, CNRS, Université Paris Sud, Gif-sur-Yvette
O43 -Vers l'architecture du télomère de la levure bourgeonnante par biologie structurale intégrative.
14h00 VALERIE LAMOUR; IGBMC, Illkirch O44 - Nouvelles informations sur le mécanisme de surenroulement de l'ADN obtenues par cryo-microscopie électronique d'une ADN gyrase complète.
14h20 – 14h40 JOANNA TIMMINS; IBS, Grenoble O45 - Etudes structurales de la recombinaison homologue chez Deinococcus
radiodurans.
= = = 4D Formation ReNaFoBis = = = 14h40 – 15h00 Amphi C
Chair : JEAN CAVARELLI; IGBMC, Strasbourg 14h40 – 15h00 CLAUDINE MAYER; Institut Pasteur, Université Paris 7
O46 - ReNaFoBis : Réseau National de Formation Doctorale en Biologie Structurale Intégrative

Programme
- 22 -
* * * * * * * * * * Hommage à Roger Fourme * * * * * * * * * 15h00 – 15h30 Amphi C
15h00 – 15h30 ERIC GIRARD; IBS, Grenoble
SP3 - Un hommage à Roger Fourme
☺☺☺☺☺☺ Tout le monde rencontre tout le monde ☺☺☺☺☺☺ 15h30 – 16h00 Pause café – Rencontre avec les partenaires industriels Salle ChemInnov. 16h00 – 17h00 Première séance Poster 17h00 – 18h00 Transfert en TRAMWAY Ligne B
(Direction Bassins à flot ou Claveau Station Victoire)
= = = = = = = = = = Conférence grand public = = = = = = = = = Amphitéatre Denigès
Université Bordeaux Segalen Place de la Victoire Bordeaux
18h00 : Photo de groupe sur le parvis de l’Université de Bordeaux Segalen – Site Victoire 18h15 Accueil par MANUEL TUNON DE LARA ; Président de l’Université Bordeaux Segalen
18h25 AN-PANG TSAI, Tohoku University, Japan SP4 - Quasicrystals : Structures, properties and applications
19h25 – 20h00 Réception dans l’atrium de l’Université offert par l’Université Bordeaux Segalen

Programme
- 23 -
Jeudi 4 juillet
* * * * * * * * * * Conférence plénière III * * * * * * * * * 9h00-10h00 Amphi C
Chair : PASCAL ROUSSEL, vice-président de l’AFC MARC DE BOISSIEU, SIMAP, Grenoble
SP5 - Les quasicristaux : structure atomique et dynamique.
☺☺☺☺☺☺ Tout le monde rencontre tout le monde ☺☺☺☺☺☺
10h00 – 10h30 Pause café – Rencontre avec les partenaires industriels Salle ChemInnov.
= = = Session 5A Approches multitechniques en cristallochimie = = = 10h30 – 12h00 Amphi A
Chair : NATHALIE AUDEBRAND; Université Rennes 1 Co-chair : PASCAL ROUSSEL; ENSC, Lille 10h30 PHILIPPE BOULLAY; CRISMAT, Caen
O47 - Combinaison des données de diffraction des électrons et de diffraction des rayons X poudre: que peut-on en attendre?
11h00 FLORENT BOUCHER; Institut des Matériaux Jean Rouxel, Nantes O48 - Apport des calculs ab initio à la résolution des structures cristallines : intérêt de la modélisation en spectrométrie RMN et Mössbauer.
11h30 VALERIE DEMANGE; Université de Rennes 1 O49 - Nanorods de niobates et niobo-tantalates de potassium épitaxiés sur substrats monocristallins : étude par diffraction des rayons X, diffraction des électrons en précession et tomodiffraction des électrons.
11h45 – 12h00 LUCY COOPER; Institut Lavoisier, CNRS Université de Versailles St-Quentin-en-Yvelines
O50 - Solides hybrides à base de ligands d’origine naturelle : résolution structurale via une approche multitechnique « diffraction / RMN ».
= = = Session 5B Densité électronique, modélisation = = =
10h30 – 12h00 Amphi B
Chair : PHILIPPE RABILLER; Université de Rennes 1, Rennes Co-Chair : MARIE-BERNADETTE LEPETIT; Institut Néel, Grenoble 10h30 NOUR EDDINE GHERMANI; Laboratoire Structures, Propriétés de Modélisation des Solides, Ecole centrale de Paris
O51 - Propriétés électrostatiques de molécules d'intérêt pharmaceutique.
11h00 ALESSANDRO ERBA; Dipartimento di Chimica, Université de Turin, Italie O52 - Ab initio description of nuclear motion and dispersion effects on the electron density matrix of crystals.

Programme
- 24 -
11h30 ENRIQUE ESPINOSA; CRM2, Université de Lorraine O53 - Les liaisons halogène et chalcogène pour l'ingénierie cristalline et la chimie supramoléculaire.
11h45 – 12h00 SAMIR BENTATA; Faculté des Sciences et de la Technologie, Mostaganem, Algérie
O54 - Ab-initio Study of structural, Electronic and Magnetic Properties of CdTe Doped Transition Metal Co.
= = = Session 5C Cristallographie biologique et Santé = = =
10h30 – 12h00 Amphi C
Chair : YVES BOURNE; AFMB, Marseille Co-chair : MAGALI MATHIEU, Sanofi, Chilly-Mazarin 10h30 WILLIAM BOURGUET; CBS, Montpellier
O55 - Mécanismes de reconnaissance et d'activation des récepteurs hormonaux par les perturbateurs endocriniens environnementaux.
11h00 THOMAS BERTRAND; Sanofi, Vitry sur Seine O56 -Une approche structurale pour l’inhibition sélective de PI3Kbeta.
11h20 VALERIE GUILLET; IPBS, Toulouse O57 - Structure cristallographique de la protéine FadD32 de Mycobacterium marinum, une cible potentielle pour le développement de nouveaux antituberculeux.
11h40 – 12h00 PASCALE MARCHOT; AFMB, Marseille O58 - Toward closing the synaptic gap: a molecular approach to explore neuronal connectivity deficiencies associated with autism.
☺☺☺☺☺☺ Repas ☺☺☺☺☺☺
12h00-13h30 Déjeuner dans le hall de l’ENSCBP
= = =Session 6A/B Méthodes émergentes en cristallographie = = = 13h30 – 15h15 Amphi A
Chair : OLIVIER PEREZ; ENSI Caen Co-chair : DAVID LE BOLLOC'H; LPS, Orsay 13h30 PIERRE BORDET; Institut Néel, Grenoble
O59 - Application de la Fonction de Distribution de Paires à l'étude de matériaux nano-cristallins ou mal ordonnés.
14h00 JAN LÜNING; Université Pierre et Marie Curie, Paris O60 - Nouvelles possibilités expérimentales en physique introduites par les XFELs.
14h30 BERTRAND TOUDIC; GMCM, Rennes O61 - Méthodes rapides pour analyser les désordres structuraux statiques et dynamiques dans l’immense espace réciproque.

Programme
- 25 -
14h55 VINCENT JACQUES; LPS Université Paris Sud O62 - Défauts de phase dans les cristaux électroniques étudiés par diffraction cohérente des rayons X.
15h15 – 15h30 NICOLETA GALATANU; Xenocs O63 - La société Xenocs
= = = Session 6C Biologie Structurale Intégrative et gros assemblages = = =
13h30 – 15h15 Amphi C
Chair : DINO MORAS ; IGBMC, Illkirch Co chair : JACQUELINE CHERFILS; LEBS, Gif-sur-Yvette 13h30 YVES MECHULAM; Ecole Polytechnique Palaiseau
O64 - Démarrage de la traduction chez les eucaryotes et les archées : le facteur e/aIF2.
14h00 NICOLAS GARREAU DE LOUBRESSE; Equipe Marat Yusupov, IGBMC Strasbourg O65 - Crystal structure of the eukaryotic 80S ribosome.
14h30 ZEINEB FOURATI-KAMMOUN; Ecole Polytechnique, Palaiseau et IBBMC, Orsay O66 - Etude structurale et fonctionnelle de la protéine Pat1 de Saccharomyces cerevisiae.
14h50 LUDOVIC SAUGUET; Institut Pasteur, Paris O67 - Détermination des bases structurales du mécanisme de perméation des ions chez les récepteurs-canaux pentamériques de la famille Cys-loop.
15h10 – 15h30 MARC RUFF; IGBMC, Illkirch O68 - Etudes structurales et fonctionnelles du complexe de pré-intégration du VIH-1.
☺☺☺☺☺☺ Tout le monde rencontre tout le monde ☺☺☺☺☺☺
☺☺☺☺☺☺ Meet the experts ☺☺☺☺☺☺
15h30 – 16h00 Amphis A/B Jeunes chercheurs, venez rencontrer et poser vos questions à des chercheurs et éditeurs de l'AFC:
Comment choisir le "bon" journal ? Comment rédiger une demande de financement ? Etc…
15h30 – 16h00 Pause café – Rencontre avec les partenaires industriels Salle ChemInnov. 16h00 – 17h00 Seconde séance Poster
= = = Assemblée Générale de l’AFC = = = 17h00 – 18h00 Amphi C
= = = Prix de thèse 2012 de l’AFC = = =
18h00 – 19h00 Amphi C Conférences des trois Lauréats des prix de l'AFC 2012. Chair : ENRIQUE ESPINOSA ; Nancy

Programme
- 26 -
Chair : JACQUELINE CHERFILS; Présidente de l’AFC Co-Chair : CLAUDINE MAYER; Vice-présidente de l’AFC Co-Chair : RENE GUINEBRETIERE ; Vice-président de l’AFC Co-Chair : PASCAL ROUSSEL; Vice-président de l’AFC 18h00 (GT Bio) - TRISTAN WAGNER ; Institut Pasteur, Paris
O69 - Contrôle du métabolisme central chez les mycobactéries par la régulation allostérique de l´alpha-kétoglutarate déshydrogénase.
18h20 (GT Chimie) FLORIAN MOREAU; Laboratoire des Sciences Chimiques, Rennes O70 - Cristallochimie de nouveaux polymères de coordination à noyau spirobifluorène ou tétraphénylméthane : du design du ligand à la topologie et aux propriétés du solide hybrides.
18h40 – 19h00 (GT Physique) ERWAN PAINEAU; Institut National Polytechnique Lorraine, Nancy
O71 - Transitions de phases cristal-liquides et comportement sous champs de suspensions colloïdales d'argile naturelle.
19h00 – 20h00 Transfert en TRAMWAY Ligne B (direction Bassins à flot ou Claveau, Station CAPC)
☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺ ☺☺☺☺☺☺ ☺☺☺☺☺☺
☺☺☺☺☺☺ 20h00 – 23h00 ☺☺☺☺☺☺ ☺☺☺☺☺☺ Cité Mondiale du vin ☺☺☺☺☺☺
☺☺☺☺☺☺ Dîner de Gala ☺☺☺☺☺☺ ☺☺☺☺☺☺ Remise des prix des posters ☺☺☺☺☺☺ ☺☺☺☺☺☺ ☺☺☺☺☺☺ ☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺☺

Programme
- 27 -
Vendredi 5 juillet
= = = Session 7A Cristallographie in situ, in operando = = = 9h00 – 10h20 Amphi A
Chair: ERIK ELKAÏM; Synchrotron SOLEIL Co-chair: BEATRICE GILLON; Laboratoire Léon Brillouin, CEA-CNRS, Saclay 9h00 MONICA CERETTI; Institut Charles Gerhardt, Montpellier
O72 - Structural complexity in (Re)2NiO4+δ : electrochemically controlled oxygen intercalation reactions, explored by neutron and synchrotron scattering methods under operando conditions.
9h30 LAURENCE CROGUENNEC; ICMCB, Bordeaux O73 - Apport des études menées operando par diffraction et absorption des rayons X à la compréhension des mécanismes mis en jeu au sein des matériaux d'électrode pour batteries Li-ion.
10h00 – 10h20 OMAR KAMMOUN; Institut des Sciences Chimiques de Rennes O74 - Structures lamellaires supramoléculaires de sulfates hybrides : du design à la stabilité thermique et aux transitions de phases.
= = = Session 7B Texture, Microstructure, déformation = = =
9h00 – 10h30 Amphi B
Chair : ALAIN LODINI; LACM, Reims Co-Chair : DANIEL CHATEIGNER; ENSI Caen 9h00 CHRISTOPHE WIERZBANOWSKI; Université de Cracovie
O75 - Study of texture development in asymmetrically rolled titanium. Experimental study and calculations.
9h30 VINCENT KLOSEK; CEA - LLB – Saclay O76 - Evolutions microstructurales et hétérogénéités de déformation dans les aciers ODS Fe-14Cr1W.
10h00 – 10h30 HÉLÈNE ROTELLA, ENSI Caen O77 - Résolution et affinement de structure d’un film mince épitaxié de LaVO3.
= = = Session 7C Grands Instruments Synchrotron et plateformes en biologie structurale - (Développements, XFEL, nouveautés) = = =
9h00 – 10h30 Amphi C
Chair : ANDREW THOMPSON; Synchrotron SOLEIL Co-Chair : MICHEL KOCHOYAN; CBS, Montpellier 9h00 JEAN DAILLANT; DSM/IRAMIS/SIS2M/LIONS CEA
O78 - La cristallographie à SOLEIL.

Programme
- 28 -
9h30 Marie-EMMANUELLE COUPRIE; Synchrotron SOLEIL O79 - FELs dans le monde, leurs propriétés et avantages cf synchrotrons, et l'avenir des projets en France.
9h50 JAN LÜNING; Université Pierre et Marie Curie, Paris O80 - Nouvelles possibilités expérimentales en cristallographie introduites par les XFELs.
10h10 – 10h30 HASSAN BELRHALI; EMBL Grenoble O81 - BM14-2: une ligne MX MAD à l’ESRF avec de nouveaux outils pour exposer des cristaux biologiques à température ambiante.
☺☺☺☺☺☺ Tout le monde rencontre tout le monde ☺☺☺☺☺☺
10h30 – 11h00 Pause café – Rencontre avec les partenaires industriels Salle ChemInnov.
= = = Session 8A Chiralité, polymorphisme = = = 11h00 – 12h30 Amphi A
Chair : GERARD COQUEREL; TSHO, Rouen Co-Chair : CHRISTIAN JARRY; Pharmacochimie, Université Bordeaux Segalen 11h00 PHILIPPE ESPEAU; Laboratoire de Chimie Physique, Université Paris Descartes
O82 - Formulations Pharmaceutiques et Polymorphisme
11h30 HOWARD D. FLACK; Genève, Suisse O83 - Configuration absolue et structure absolue: notions de base et évaluation.
12h00 – 12h30 PATRICK ROSA; ICMCB Bordeaux O84 - Composés multifonctionnels : transition de spin et chiralité.
= = = Session 8B De l'ordre local à l'ordre à longue distance = = =
11h00 – 12h30 Amphi B
Chair : PIERRE-ANTOINE ALBOUY; LPS, Orsay Co-chair : PAULINE MARTINETTO; Institut Néel, Grenoble 11h00 DOMINIQUE THIAUDIERE; Synchrotron SOLEIL
O85 - Apport du synchrotron pour des expériences combinées DRX et XAS.
11h30 MICHELA BRUNELLI; ILL, Grenoble O86 - Analyse par PDF (rayons X et neutrons) à l'échelle nanométrique des inhomogénéités dans des oxydes de cérium dopés au rhénium.
12h00 – 12h30 CELINE MARIETTE; Institut de Physique de Rennes O87 - Désordre "quasi-liquide" de chaines d'alcane sous-confinement subnanométrique supramoléculaire.

Programme
- 29 -
= = = 8C Défis en cristallographie biologique - Success stories.= = = 11h00 – 12h30 Amphi C
Chair : Philippe Dumas; IBMC, Strasbourg Co-chair : Julie Ménétrey; LEBS, Gif-sur-Yvette 11h00 ANNE HOUDUSSE; Institut Curie
O88 - La myosine VI, un nanomoteur à contre-sensMyosin VI.
11h25 JEAN-BAPTISTE CHARBONNIER; IBiTec, CEA Saclay O89 - Control of the DNA mismatch repair and the meiosis recombination by eukaryotic MutL homologs.
11h50 MIKAEL ELIAS; Weizmann Institute of Science, Rehovot – Israel O90 - La survie en milieu riche en arséniate : le mécanisme moléculaire de discrimination du phosphate.
12h10 – 12h30 JACQUELINE CHERFILS; LEBS, Gif-sur-Yvette O91 - Integrated conformational and lipid-sensing regulation of ArfGEFs.
☺☺☺☺☺☺ Repas ☺☺☺☺☺☺
12h30-14h00 Déjeuner dans le hall de l’ENSCBP
☺☺☺☺☺☺ Clôture de l’AFC 2013☺☺☺☺☺☺
Au revoir et
à bientôt

Programme
- 30 -

- 31 -
Résumés des conférences plénières
SP1-5 et orales O1-91

- 32 -

Session plénière I – Mardi 2 juillet 14h00-15h00
- 33 -
SP1 - Les foldamères: une expansion de l'espace chimique basée sur l'analyse des structures
Ivan Huc
Université de Bordeaux – CNRS – IPB UMR5248 (CBMN), Institut Européen de Chimie et Biologie, 2 rue Robert Escarpit, 33600 Pessac, France. [email protected]
Nous avons entrepris un programme de recherche dans le domaine des foldamères – des architectures moléculaires artificielles repliées – dont le succès a reposé très largement sur l'analyse des structures cristallographiques. Spécifiquement, nous avons développé des foldamères hélicoïdaux dérivés d'acides aminés aromatiques.[1] Certains de ces objets repliés présentent une stabilité conformationnelle sans précédent,[2] et constituent des briques élémentaires bien définies pour l'élaboration de structures repliées artificielles de la taille de petites protéines (Fig. 1).[3] Ils possèdent fréquemment une forte propension à s'assembler en hélices double, triple voire quadruple.[4] Des cavités peuvent être conçues à l'intérieur de ces hélices qui leur confèrent des propriétés de récepteurs artificiels[5] et de moteurs moléculaires.[6] Des analogues solubles dans l'eau de ces foldamères se montrent prometteurs pour la reconnaissance d'acides nucléiques et de protéines.[7]
Figure 1. A gauche et au centre: structure dans le cristal d'un grand foldamère constitué de deux hélices de sens d'hélicité opposés orientées à 90°. A droite: la structure cristalline d'une petite protéine est montrée à la même échelle pour en comparer la taille.
[1] G. Guichard, I. Huc, Chem. Commun. 2011, 47, 5933. [2] H. Jiang, J.-M. Léger, I. Huc, J. Am. Chem. Soc. 2003, 125, 3448; N. Delsuc, T. Kawanami, J. Lefeuvre, A. Shundo, H. Ihara, M. Takafuji, I. Huc ChemPhysChem 2008, 9, 1882. [3] N. Delsuc, J.-M. Léger, S. Massip, I. Huc Angew. Chem. Int. Ed. 2007, 46, 214; N. Delsuc, S. Massip, J.-M. Léger, B. Kauffmann, I. Huc, J. Am. Chem. Soc. 2011, 133, 3165. [4] Q. Gan, C. Bao, B. Kauffmann, A. Grélard, J. Xiang, S. Liu, I. Huc, H. Jiang, Angew. Chem. Int. Ed. 2008, 47, 1715; D. Haldar, H. Jiang, J.-M. Léger, I. Huc, Angew. Chem. Int. Ed. 2006, 45, 5483. [5] Y. Ferrand, A. M. Kendhale, B. Kauffmann, A. Grélard, C. Marie, V. Blot, M. Pipelier, D. Dubreuil, I. Huc, J. Am. Chem. Soc. 2010, 132, 7858. [6] Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang, I. Huc, Science 2011, 331, 1172. [7] L. Delaurière, Z. Dong, K. Laxmi-Reddy, F. Godde, J.-J. Toulmé, I. Huc, Angew. Chem. Int. Ed. 2012, 51, 473.

1a - Matériaux fonctionnels (1) – Mardi 2 juillet 15h00-16h30
- 34 -
O1 - Relations structure-magnétisme dans le composé magnéto-électrique MnWO4
Christophe Payen1, Lynda Meddar1, Michael Josse2, Pascaline Patureau1, Philippe
Deniard1, Mario Maglione2, Françoise Damay3, Gilles André3 1Institut des Matériaux Jean Rouxel, Université de Nantes - CNRS, 2CNRS, ICMCB, Bordeaux, 2LLB, CEA,CNRS
MnWO4 est un composé magnétoélectrique de type II bien étudié depuis quelques années [1]. Cet oxyde de Mn2+ (spin S=5/2) présente une structure cristalline simple (hubnerite ou wolframite) avec des chaînes zigzag issues du partage d’arêtes d’octaèdres MnO6 distordus [2]. Trois phases magnétiques sont présentes à basse température [3]. Une de ces phases (AF2, entre T1 = 7.5 K et T2 = 12.5 K) associe une structure magnétique hélicoïdale incommensurable et une polarisation ferroélectrique spontanée dans une direction perpendiculaire aux chaînes « MnO4 » (phase « multiferroïque »). La succession d'états magnétiques à basse température est la conséquence d'une compétition entre interactions magnétiques et anisotropie magnétique locale. Il est possible de réaliser des substitutions chimiques du Mn2+ ou du W6+ par d’autres métaux de transition, ce qui ouvre des possibilités de modulation des propriétés.
Nous présenterons différents travaux réalisés pour comprendre les propriétés magnétiques du composé MnWO4 et celles des solutions solides issues des différentes substitutions chimiques. L’état « multiferroïque » peut être déstabilisé ou stabilisé selon le type de substitution, les substitutions impliquant des modifications des interactions magnétiques et de l’anisotropie magnétique qui modulent les propriétés magnétiques. [1] A.H. Arkenbout, Phys. Rev. B, 74, 184431 (2006) ; K. Taniguchi et al., Phys. Rev. Lett. 97, 097203 (2006) ; O. Heyer et al., J. Phys.: Condens. Matter 18, L471 (2006). [2] H. Weitzel, Z. Kristallogr. Kristallgeom. Kristallphys. Kristallchem., 144, 238 (1976) ; J. Macavei, H. Schulz, H., Z. Kristallogr. 207, 193 (1993). [3] G. Lautenschläger et al., Phys. Rev. B, 48, 6087 (1993).

1a - Matériaux fonctionnels (1) – Mardi 2 juillet 15h00-16h30
- 35 -
O2 - Synthèse et structures cristallines de nouveaux polymères de coordination chiraux à base de fluorène pour la séparation et la catalyse énantiosélective
Julien Robin 1, Nathalie Audebrand 1, Cyril Poriel 2, Carmelo Prestipino 1
1Equipe Chimie du Solide et Matériaux, 2Equipe Matière Condensée et Systèmes Electroactifs. Institut des Sciences chimiques de Rennes, UMR CNRS 6226, Université de Rennes 1.
Les recherches sur les Metal Organic Frameworks (MOFs), ou polymères de coordination, se sont largement développées ces vingt dernières années [1] et les applications potentielles sont nombreuses notamment dans le domaine de l’environnement (stockage et séparation de gaz, dépollution des eaux…). Les MOFs chiraux sont de plus en plus étudiés compte tenu de leurs propriétés en catalyse hétérogène et séparation énantiosélective [2,3]. Nous avons synthétisé trois ligands originaux à cœur fluorène sur lesquels ont été greffées deux chaînes chirales (Figure 1).
Figure 1 : Ligands chiraux à cœur fluorène synthétisés
La réactivité de ces ligands avec des sels métalliques (Zn, Cu, Cd, Y, La) a été étudiée avec différentes conditions de synthèses solvothermales. Les structures cristallines des MOFs synthétisés ont été déterminées par diffraction des rayons X (DRX) par le monocristal. La variation de la taille, de la géométrie et de la connectivité du ligand a permis de contrôler l’interpénétration, la topologie, la dimensionnalité et la porosité des MOFs, comme l’illustre la Figure 2.
Figure 2 : Série de MOFs au cuivre : a) [Cu L1H2O] porosité potentielle de 15% ; b) [Cu2 (L2)2
(DMF) (H2O)] porosité potentielle de 35% ; c) [Cu4 (L3)2 DMF (H2O)3] porosité potentielle de 60%.
La réactivité de ces MOFs a été étudiée en particulier par DRX par les poudres in situ avec des diffractomètres de laboratoire et le rayonnement synchrotron. [1] L. R. MacGillvray, “Metal-Organic Frameworks: Design and applications”, John Wiley & Sons, 2010. [2] Y. Minyoung, S. Renganathan, and K. Kimoon, Chem. Rev, 2012, 112, 1196-1231. [3] L. Jian-Rong, J. Sculley, and H. C. Zhou, 2012, Chem. Rev, 112, 869-932.

1a - Matériaux fonctionnels (1) – Mardi 2 juillet 15h00-16h30
- 36 -
O3 - Origine de la fatigue Ferroélectrique dans le zirconate-titanate de Plomb PZT
J. Rouquette,1 M. Hinterstein,2 J. Haines,1 Ph. Papet,1 M. Knap,3 J. Glaum,4 H. Fuess5 1Institut Charles Gerhardt UMR CNRS 5253 Equipe C2M, Montpellier cedex 5, France. 2Institut für Werkstffwissenschaften, Technische Universität Dresden, Germany 3CELLS-ALBA, Barcelona, Spain 4School of Materials Science and Engineering, Australian 5Institute for Materials Science, Technische Universität Darmstadt, Germany . E-mail: [email protected]
Les Ferroélectriques présentent un fort potentiel pour des applications microélectroniques. Dans les mémoires ferroélectriques non volatiles (FeRAM), l’information est stockée et lue en renversant la direction de la polarisation électrique. La dégradation du basculement de polarisation, i.e. fatigue ferroélectrique, est une contrainte technique à la commercialisation des mémoires ferroélectriques ; la quantité de charges basculées sous champs diminue lors de cycles (de l’ordre de 107) répétés. Malgré de nombreuses études théoriques et expérimentales, l’origine de la fatigue n’est pas connue. Dans la littérature la fatigue serait causée par la formation d’une couche à l’interface entre l’électrode métallique et le matériau ferroélectrique, des déformations résultantes de domaines à 90°, de l’électro-migration de lacunes d’oxygène formant des défauts étendus pouvant bloquer des domaines, de mécanismes d’inhibition de nucléation d’interface induit par injection de charge, de fissures micro- et macro-scopiques, …, mais à notre connaissance l’origine structurale de la fatigue n’a jamais été proposée.
Dans cette étude, la fatigue ferroélectrique de céramiques commerciales de PZT est étudiée par diffraction X synchrotron en fonction du champ électrique appliqué. Utilisant la méthodologie rapportée récemment [1], nous avons caractérisé l’origine structurale de la fatigue par une dégradation de la transformation quadratique-monoclinique sous champ, qui traduit la réduction de l’efficacité piézoélectrique. La perte de polarisation rémanente peut aussi être détectée par une anomalie moins intense et plus diffuse du déplacement quadratique moyen du plomb Biso(Pb) sous champ pour l’échantillon fatigué (107 cycles) qui traduit le basculement de polarisation. Nous avons aussi pu caractériser la cinétique de la réaction induite sous champ, de l’ordre de la milliseconde, qui conforte sans aucun doute notre interprétation sur l’origine structurale de la fatigue ferroélectrique. [1] M. Hinterstein, J. Rouquette*, J. Haines, P. Papet, M. Knapp, J. Glaum, H. Fuess, Structural Description of the Macroscopic Piezo- and Ferroelectric Properties of Lead Zirconate Titanate, Phys. Rev. Lett., 107 (2011).

1b - Surfaces et Interfaces – Mardi 2 juillet 15h00-16h30
- 37 -
O4 - SXRD in situ couplée à l’électrochimie : étude de l’électro-insertion d’hydrogène dans des nanofilms de Pd
Yvonne Soldo-Olivier1, Eric Sibert1, W. Liang1, M. De Santis2 1Laboratoire d’Electrochimie et de Physico Chimie des Matériaux et des Interfaces, CNRS-Grenoble INP-UJF-UdS, 1130 rue de la Piscine, 30402 St . Martin d’Hères, France 2 Institut Néel, CNRS-UJF, 25 av. des Martyrs, 38042 Grenoble, France
Le palladium présente non seulement des propriétés catalytiques remarquables vis-à-vis de la dissociation d'hydrogène, mais il est aussi caractérisé par une grande cinétique d’insertion/désinsertion de cet élément. Par rapport au Pd massif, la taille nanométrique des films ultra-minces est de nature à induire de profondes modifications sur les propriétés thermodynamiques. C'est le cas pour les nanoparticules de Pd, qui présentent une solubilité de l'hydrogène réduite [1-4].
Afin d'obtenir une compréhension approfondie des mécanismes régissant l'insertion d'hydrogène dans les films ultra-minces de Pd, nous avons étudié l'influence de la taille nanométrique et du substrat monocristallin sur les isothermes d’insertion électrochimique de l’hydrogène. Le comportement original des différentes paramètres thermodynamiques, tel le taux d'insertion maximale d'hydrogène, a été observé pour les systèmes Pd/Pt(111) et Pd/Au(111).
Nous avons réalisé des mesures de diffraction de surface des rayons X (SXRD) in situ en milieu électrochimique. Pour ceci, nous avons conçu une cellule électrochimique adaptée à la diffraction de surface et venant s’insérer dans le diffractomètre de la ligne française CRG-D2AM à l’ESRF (Grenoble, France). Ces mesures nous ont permis d’obtenir la structure microscopique des films de Pd avant et après hydruration. L’existence de zones au sein des films de Pd avec des paramètres cristallographiques différents a été mise en évidence.
Nous avons ainsi été en mesure de donner une description détaillée de la relation forte entre la structure des films à l'échelle atomique et le comportement des isothermes d’insertion en fonction du substrat et de l’épaisseur du film [5,6].
[1] A. Pundt, M. Suleiman, C. Bähtz, M.T. Reetz, R. Kirchheim, N.M. Jisrawi, Mat. Sci. Engi. 2004, B108, 19. [2] A. Pundt, Adv. Engi. Mat. 2004, 6, 11. [3] C. Lebouin, Y. Soldo, S.A. Grigoriev, M. Guymont, P. Millet, Int. J. Hydrogen Energ., 2013, 38(2), 966. [4] C. Lebouin, Y. Soldo-Olivier, E. Sibert, P. Millet, M. Maret, R. Faure J. Electroanal. Chem., 2009, 626, 59. [5] C. Lebouin, Y. Soldo-Olivier, E. Sibert, M. De Santis, F. Maillard, R. Faure, Langmuir 2009, 25(8), 4251. [6] Y. Soldo-Olivier, M. C. Lafouresse, M. De Santis, C. Lebouin, M. de Boissieu, E. Sibert, J. Phys. Chem. C, 2011, 115(24), 12041.

1b - Surfaces et Interfaces – Mardi 2 juillet 15h00-16h30
- 38 -
O5 - Surfaces vicinales : des nano-gabarits pour croissance épitaxiale Alessandro Coati1, Yves Garreau1,2 1Synchrotron SOLEIL, L’Orme des Merisiers, Saint Aubin, B.P. 48, 91192 Gif sur Yvette Cedex, France, 2Matériaux et Phénomènes Quantiques (MPQ), Bâtiment Condorcet, Case courrier 7021, 10 rue Alice Domon et Léonie Duquet, 75205 Paris Cedex 13
Les surfaces vicinales métalliques sont des bons gabarits pour la croissance de
nanostructure. En effet, elles présentent une succession régulière de terrasses séparées par des marches et constituent elles mêmes de nanostructures dont on peut contrôler les dimensions (largeur de terrasses, période) en choisissant l’angle de coupe par rapport à une surface nominale. Suite à un recuit thermique ou à l’adsorption d’une espèce chimique, ou encore à un dépôt d’un autre matériau, les surfaces vicinales peuvent se réorganiser et donner naissance à des nouvelles structures périodiques, comme par exemple des facettes, qui peuvent être contrôlées par les paramètres de recuit, d’adsorption ou de dépôt.
Nous nous sommes intéressés à l’organisation de surfaces vicinales métalliques et à la modification de leurs morphologies suite au dépôt sub-monocouche d’une espèce chimique en utilisant la diffraction de rayons X en incidence rasante (GIXD) et la microscopie à effet tunnel (STM).
Pour des dépôts dépassant la monocouche, nous avons pu observer des phénomènes intéressants d’épitaxie sur les surfaces métalliques vicinales étudiées. Par exemple, le dépôt d’Ag sur une surface vicinale de Cu ou de Ni (espèces présentant une forte non miscibilité en volume), une croissance épitaxiale à lieu, avec une interface abrupte entre le substrat et la couche déposée. Nous avons pu mettre en évidence que l’interface entre les deux métaux est constituée de deux surfaces vicinales qui s’emboitent parfaitement. Nous obtenons ainsi une croissance avec une couche bien cristalline et sans défauts, présentant une surface bidimensionnelle, même en présence de matériaux qui présentent au départ des paramètres réticulaires très différents.
L’action d’une surface vicinale dans l’épitaxie d’une couche peut avoir aussi d’autres effets étonnants : c’est le cas de la croissance d’une couche de Co sur une surface vicinale d’Au. Dans ce cas le Co, qui présente naturellement une structure fcc, est contraint à croitre avec une structure fcc, héritée du substrat d’Au.
Dans cet exposé on pourra mettre en évidence ces phénomènes très particuliers liés à la croissance sur substrats vicinaux.

1b - Surfaces et Interfaces – Mardi 2 juillet 15h00-16h30
- 39 -
O6 - Mise en évidence de phases induites par le substrat pour des mésogènes π-conjugués
Gabin Gbabode1,4, Oliver Werzer2, Armin Moser2, Roland Resel2, Yves Geerts3, Johann
de Silva4, Michele Sferrazza4 1Sciences et Méthodes Séparatives, EA 3233, Université de Rouen, 76821 Mont-Saint-Aignan, France, 2Institute of Solid State Physics, Graz University of Technology, Petersgasse 16, 810 Graz, Autriche, 3Laboratoire de Chimie des Polymères, Faculté des Sciences, Université libre de Bruxelles (ULB), Boulevard du Triomphe, 1050, Bruxelles, Belgique, 4Département de Physique, Faculté des Sciences, Université libre de Bruxelles, Boulevard du Triomphe, 1050 Bruxelles, Belgique.
L’électronique organique est un domaine de recherche très attractif qui est basé sur l’utilisation de molécules semi-conductrices organiques pour la fabrication de composants électroniques comme les transistors à effet de champ organiques (ou OFETs). Il est connu, pour ces derniers, que le transport de charges s’effectue dans les premières couches moléculaires du semi-conducteur organique (OSC) à l’interface entre celui-ci et le diélectrique [1]. Il est ainsi crucial de pouvoir connaître l’organisation structurale des OSCs proche du substrat sur lequel ils sont déposés car elle est directement reliée à leurs propriétés de transport de charges. Nous présenterons deux exemples de mésogènes π-conjugués déposés en film minces (épaisseur inférieure à 100 nm) par la méthode de spin-coating sur des wafers de silicium conventionnels. Nous avons mis en évidence pour chacun d’eux la présence d’une organisation structurale proche du substrat (phase induite par le substrat) différente de celle observée dans le reste du film (phase « bulk »), notamment grâce à des analyses structurales combinant principalement diffraction des rayons X spéculaire et en incidence rasante.
Le premier composé, le ,-dioctylterthiophène, présente un comportement thermotrope assez complexe avec des transitions de phases à 337 K (cristalline - smectique G), 344 K (smectique G – smectique F), 358 K (smectique F – smectique C) et fond à 363 K. La présence de deux phases cristallines en films minces à température ambiante a pu être prouvée dont la proportion relative dépend des conditions de fabrication du film. Pour le deuxième, un dérivé de phthalocyanine, nous avons pu montrer l’existence d’une phase induite par le substrat sur une épaisseur d’environ 30 nm. De plus, de manière tout à fait originale, cette phase possède une organisation structurale tridimensionnelle alors que la phase « bulk » est une mésophase colonnaire bidimensionnelle typique [2]. [1] Dinelli et al., Phys. Rev. Lett. 2004, 92, 116802. [2] Gbabode et al., Adv. Mater. 2012, 24, 658-662.

1b - Surfaces et Interfaces – Mardi 2 juillet 15h00-16h30
- 40 -
O7 - Etude de surfaces vicinales ordonnées par diffusion centrale des rayons X sous incidence rasante. Nouveaux développements sur la ligne BM02 à l’ESRF
Caroline Matringe1, Elsa Thune1, René Guinebretière1
, David Babonneau2, Nathalie Boudet3, Nils Blanc3, Mireille Maret4 1Science des Procédés Céramiques et de Traitements de Surface, SPCTS, UMR CNRS 7315,
ENSCI, Centre Européen de la Céramique (CEC), 12 rue Atlantis, 87068 Limoges Cedex 2Institut PPRIME, UPR CNRS 3346, Université de Poitiers, SP2MI, Téléport 2, Boulevard
Marie et Pierre Curie, BP 30179, 86962 Futuroscope Chasseneuil Cedex 3Institut Néel, UPR CNRS 2940, 25 rue des Martyrs, BP 166, 38042 Grenoble cedex 9 4Science et Ingénierie des Matériaux et Procédés, SIMAP UMR 5266 CNRS, 1130 rue de la Piscine, BP 75, 38402 St Martin d’Hères.
Les surfaces vicinales sont des substrats utilisés pour faire croître des nanostructures auto-organisées. D’une manière générale, après découpe des monocristaux ces surfaces présentent des marches de dimension latérale et de hauteur variables. Des traitements thermiques judicieusement choisis conduisent, par simple minimisation de l’entropie, à la formation de réseaux de marches périodiques qui peuvent a priori être mono ou bidimensionnels. Les dimensions caractéristiques des nanostructures élaborées sur ces surfaces gabarits sont directement dépendantes de la période des réseaux formés et de la hauteur des marches qui les constituent. La mesure de l’évolution de la période ainsi que du profil des marches constitue donc une étape clef de l’élaboration de ces nanostructures par auto-organisation.
La diffusion centrale des rayons X sous incidence rasante est une méthode de choix pour déterminer à la fois le profil et les dimensions caractéristiques des réseaux de marches. L’orientation relative entre l’échantillon, et plus particulièrement les bords de marche, et le faisceau incident de rayons X doit toutefois être contrôlée de façon très précise. De cette précision dépendra l’aptitude à déterminer notamment le profil des marches. Les bords de marche étant des directions cristallographiques, l’orientation azimutale de l’échantillon, qui est un monocristal, peut être déterminée par diffraction. Nous montrerons dans cette communication, comment l’association du nouveau goniomètre implanté sur la ligne BM02 à l’ESRF et du banc de diffusion centrale présent sur cette même ligne nous a permis d’étudier de façon complète des réseaux mono ou bidimensionnel formés par traitements thermiques à très haute température de surfaces de saphir. La pertinence de l’approche proposée sera illustrée notamment par le très bon accord entre des observations locales dans l’espace réel menées par microscopie à force atomique et la cartographie bidimensionnelle du réseau réciproque au voisinage du centre de ce réseau.

1c - Nouvelles structures en biologie – Mardi 2 juillet 15h00-16h30
- 41 -
O8 - Structure de l'ectodomaine de la glycoprotéine d'enveloppe E1, dans sa forme post-fusion, du virus de la Rubéole. Marie-Christine Vaney, Rebecca Phillips, Alejandra Tortorici, Rana Al-Kurdi, Giovanna Barba-Spaeth, Thomas Krey, Félix A. Rey Unité de Recherche de Virologie Structurale, Département de Virologie, Institut Pasteur, CNRS UMR 3569,28 rue du Dr. Roux, 75015 Paris, France
Le virus de la rubéole (RV) est l’agent responsable de cette maladie généralement bénigne touchant les enfants, mais qui peut provoquer de graves malformations congénitales au foetus lors d'une infection in utero. RV est le seul membre du genre des Rubivirus appartenant à la famille des Togaviridae, qui inclut aussi le genre Alphavirus dont le virus chikungunya fait partie. Malgré son importance médicale, l'organisation structurale de RV reste peu connue à l'heure actuelle. Les glycoprotéines E1 et E2 sont les protéines d'enveloppe de RV. Nous avons déterminé la structure de la forme post-fusion de l'ectodomain de E1 (E1e) à 1.8Å de résolution [1]. E1 est l'antigène principal et la seule cible des anticorps neutralisants contre RV, et l'épitope d'un de ces anticorps neutralisants a été localisé sur la structure. E1 est aussi impliquée dans l'entrée du virus dans les cellules cibles par sa liaison à des récepteurs cellulaires de surface, dont l'un d'entre eux a été récemment identifié [2], ainsi que dans la fusion membranaire. La structure du trimère de E1e révèle deux boucles de fusion et un site métallique par protomère, ainsi qu'une très large surface de fusion (~8000 Å2). Celle-ci pourrait servir à induire une pression importante lors de l'insertion membranaire du virus, de façon à catalyser plus efficacement la fusion des membranes virale et cellulaire. Nous avons aussi montré que les propriétés de liaison de E1e aux lipides sont différentes en fonction de la nature de l'ion (Na+ ou Ca2+) et du pH. La présence de métal dans E1e évoque les protéines cellulaires de la famille "TIM" [3], lesquelles reconnaissent spécifiquement, via un métal, les têtes lipidiques des phosphatidylsérines à la surface plasmatique des cellules en apoptose. Les caractéristiques structurales de RV E1e et sa comparaison avec les autres protéines de fusion de classe II, qui englobe les protéines d'enveloppe des alphavirus et flavivirus, seront présentées. [1] DuBois RM, Vaney MC, Tortorici MA, Kurdi RA, Barba-Spaeth G, Krey T, Rey FA. Functional and evolutionary insight from the crystal structure of rubella virus protein E1. Nature. 2013, 493(7433):552-556. [2] Cong H, Jiang Y, Tien P. Identification of the Myelin Oligodendrocyte Glycoprotein as a Cellular Receptor for Rubella Virus. J. Virology. 2011, 85(21):11038–11047. [3] Santiago C, Ballesteros A, Martinez-Munoz L, Mellado M, Kaplan GG, Freeman GJ, Casasnovas JM. Structures of T cell immunoglobulin mucin receptors 1 and 2 reveal mechanisms for regulation of immune responses by the TIM receptor family. Immunity. 2007, 26:299–310.

1c - Nouvelles structures en biologie – Mardi 2 juillet 15h00-16h30
- 42 -
O9 - Bases structurales de l'initiation de la transcription de l'ARN polymerase III Stéphane Lefèvre 1,2, Hélène Dumay-Odelot 1,2, Leyla El Ayoubi 1,2, Noël Pinaud 1,2, Martin Teichmann 1,2, Sébastien Fribourg 1,2 1 Univ. Bordeaux, IECB, F-33607 Pessac, France, 2 INSERM, U869, Laboratoire ARNA, F-33000, Bordeaux, France,
L’ARN polymérase III humaine (hPol III) transcrit les gènes codants pour les ARN de transfert et l’ARN ribosomal 5S, ainsi que ceretains ARN non codants impliqués dans la régulation de la transcription et dans l’épissage tels que 7SK, U6 snRNA. hPol III est la plus complexe des trois ARN polymérases nucléaires avec 17 sous-unités. Parmi ces 17 sous-unités seulement 5 sont spécifiques de hPol III et sont réparties en deux sous-complexes stables de 2 (RPC4 et RPC5) et 3 sous-unités (RPC62, RPC39, RPC32) respectivement.
Le complexe ternaire formé par RPC62, RPC39 et RPC32 est impliqué dans
l’initiation spécifique de la transcription. Par ailleurs la sous-unité RPC32 est présente dans le
génome sous deux isoformes codées par deux gènes distincts [1]. RPC32 est la sous-unité
constitutive présente dans chaque type cellulaire testé à ce jour, alors que RPC32 la première historiquement identifiée, a une expression hautement régulée et n’est retrouvée que dans les cellules souches embryonnaires et les cellules en cours de transformation [1]. Dans le but de comprendre les bases moléculaires de la fonction de ce complexe ternaire et l’implication pour l’holoenzyme de la présence de l’une ou l’autre isoforme de RPC32, nous avons débuté une étude structure-fonction de ce complexe.
En résolvant la structure de la sous-unité humaine RPC62 nous avons identifié des
domaines classiques de liaison à l’ADN et une homologie forte avec TFIIE un facteur de transcription Pol II [2]. L’étude fonctionnelle in vitro associée a permis l’identification des domaines nécessaires à la formation du complexe et démontre une spécificité de RPC62 et RPC39 pour l’ADN simple ou double brin respectivement. Cette première étape dans la compréhension du rôle du complexe dans l’initiation de la transcription permet de proposer un modèle fonctionnel du complexe ternaire et démontre une homologie étendue entre les système de transcription Pol I/II et III. [1] Haurie et al. (2010) Proc. Nat. Acad. Sci. USA, 107 (9), 4176-81. [2] Lefèvre et al. (2011) Nat. Struct. Mol. Biol., 18(3), 352-8.

1c - Nouvelles structures en biologie – Mardi 2 juillet 15h00-16h30
- 43 -
O10 - Analyse structurale par SAXS de SopB, un effecteur de type III de Salmonella en complexe avec sa chaperonne spécifique SigE. Pierre Roblin1, Pierre Lebrun2, Prakash Rucktooa3, Fréderique Dewitte3, Zoé Lens3, Vincent Villeret3 & Coralie Bompard3.
1Synchrotron SOLEIL, L’orme des merisiers, Saint Aubin, BP 48, 91192 Gif sur Yvette Cedex, France 2Structural Biology, Brussels , Vlaams Instituut voor Biotechnologie (VIB) Vrije Universiteit Brussel (VUB) , Pleinlaan 2, 1050 Brussels, Belgium 3Interdisciplinary Research Institute, CNRS -USR 3078, Universite Lille Nord de France, 50 Avenue de Halley, 59658 Villeneuve d’Ascq, France
SopB/SigD est une inositol phosphatase multi-domaines impliquée dans l’entéro-
pathogénicité de Salmonella enterica. Ce facteur de virulence est injecté dans les cellules eucaryotes par le système de sécrétion de type III (TTSS) provoquant une réorganisation du cytosquelette actine qui aboutit à l’internalisation de la bactérie. Avant sa sécrétion, la protéine est maintenue dans le cytoplasme des bactéries par interaction avec une chaperonne spécifique SigE dont la structure atomique est connue. Nous avons réalisé l’étude du complexe SopB/SigE par diffusion des rayons-X aux petits angles (SAXS) afin d’avancer dans la connaissance des mécanismes de sécrétion de type III et notamment dans la reconnaissance des effecteurs par l’appareil de sécrétion lui-même. Le SAXS combiné à d’autres approches biochimique, biophysiques et de modélisation moléculaire s’est avéré être la technique la plus appropriée pour l’étude de ce complexe qui possède plusieurs régions désordonnées. L’interaction entre SopB et SigE a pu être caractérisée par l’analyse structurale de plusieurs constructions de l’effecteur co-exprimées avec la chaperonne. La structure a basse résolution du complexe obtenue par SAXS a abouti à la mise en évidence d’une structure quaternaire originale permettant de mieux comprendre les mécanismes de reconnaissance des complexes effecteur/chaperonne de type III et les composant du TTSS et a permis de proposer un mécanisme de sécrétion pour ce facteur de virulence.

1c - Nouvelles structures en biologie – Mardi 2 juillet 15h00-16h30
- 44 -
O11 - Bases structurales et fonctionnelles de la biominéralisation de la magnétite : importance des domaines magnétochromes.
Marina I Siponen1, Pierre Legrand2, Marc Widdrat3, Stéphanie R. Jones4, Michelle C.Y. Chang4, Damien Faivre3, David Pignol1, Pascal Arnoux1
1Laboratoire de Bioénergétique Cellulaire, CEA/DSV/IBEB, UMR 7265 CNRS/CEA, Université Aix-Marseille, Saint-Paul-lez-Durance, F-13180, France 2French National Synchrotron facility-SOLEIL, GIF-sur-Yvette, France 3Department of Biomaterials, Max Planck Institute of Colloids and Interfaces, Science Park Golm, 14424 Potsdam, Germany 4Departments of Chemistry and Molecular and Cell Biology, University of California, Berkeley, Berkeley, CA 94720-1460, United States
Les bactéries magnétotactiques s’alignent le long des lignes de champ magnétique
terrestre grâce à la mise en place d’un organite appelé le magnétosome : un cristal de magnétite [Fe(II)Fe(III)2O4] ou bien de greigite [Fe(II)Fe(III)2S4] entouré d’une vésicule protéo-lipidique. Alors que les besoins en Fer(II) et Fe(III) sont évidents, rien n’est connu concernant les mécanismes biologiques de contrôle de ce ratio Fe(II)/Fe(III). Nous avons proposé l’intervention d’un domaine contenant un motif CXXCH (typique des cytochromes c) qui est spécifique des MTB et qui se retrouve en tandem dans plusieurs protéines associées au magnétosome (MamE, P, T et X) et nous avons proposé le nom de magnétochrome pour définir ce domaine [1].
Ici, nous présentons la structure de MamP qui se caractérise par l’association d’un
domaine PDZ avec deux domaines magnétochrome [2]. Le domain PDZ joue un rôle central dans l’oligomérisation de MamP via la reconnaissance d’un peptide provenant de sa propre séquence (auto-inhibition). Quant aux deux domaines magnétochrome ils définissent une nouvelle classe de cytochrome de type c extrêmement compacte et caractérisée par une forte exposition de l’hème au solvant. L’arrangement relatif des domaines PDZ et magnétochromes dans le dimère de MamP contribue à la création d’une poche acide que l’on peut décrire comme un creuset entouré de quatre hèmes. Des expériences de cinétique, de co-cristallisation de MamP avec du Fe(II) ainsi que des expériences de production de magnétite in vitro en présence de MamP indiquent que MamP est une fer oxidase qui contribue à la formation et à la solubilisation d’espèces de Fe(III) dans le creuset. Ces espèces seraient par la suite nécessaires à la croissance des cristaux de magnétites ou de greigite. Ces résultats montrent les mécanismes moléculaires de la gestion du fer prenant place dans le magnétosome et soulignent l’importance des domaines magnétochromes dans ce processus. [1] Siponen, et al., Biochem Soc Trans. 2013, 40, 1319-23 [2] Siponen, et al., Soumis

2a - Chimie supramoléculaire et ingénierie moléculaire Mardi 2 juillet 17h00-18h30
- 45 -
O12 - Analyse structurale d’hybrides organique-inorganique et relations structure-propriétés : l’importance de l’interface et des liaisons faibles Nicolas Mercier MOLTECH-Anjou UMR CNRS 6200- Université d’Angers, 2Bd Lavoisier, 49045 Angers
Dans cette présentation, nous analyserons des structures cristallines (« retro-crystal-engineering analysis») de composés de type hybride organique-inorganique en relation avec leurs propriétés. Beaucoup d’exemples seront pris parmi des hybrides constitués d’anions halogénométallate et de cations organiques [1]. On attachera une importance particulière à l’interface organique-inorganique qui est le siège d’interactions faibles et on montrera l’impact de celles-ci sur les propriétés des matériaux. Ainsi, nous verrons comment la combinaison de liaisons halogène et liaisons hydrogène permet la réduction du gap BC-BV de perovskites hybrides [2], comment un changement de liaison hydrogène avec la température, corrélé à un changement conformationnel du cation cystaminium, a un impact drastique sur les propriétés optiques (doublage de fréquence) des matériaux [3], comment le methylviologène permet la formation (« effet template ») de chaines polaires d’octaèdres trans-connectés dans une nouvelle famille de ferroélectriques hybrides [4], ou comment les interactions N+…Cl ont un effet sur les propriétés de transfert de charge photo-induit de chlorometallates de viologène [5]. D’autres exemples seront empruntés à la chimie de coordination, en particulier à des complexes ou des polymères de coordination possédant des propriétés de photoluminescence [6].
[1] N. Mercier et al., CrystEngComm 2009, 11, 720-734. [2] S. Sourisseau, N. Mercier et al., Chem. Mater. 2007, 19, 600-607. [3] N. Mercier et al., Angew. Chemie 2006, 45, 2100-2103 ; W. Bi, N. Mercier et al., Adv. Mater. 2008, 20, 1013-1017 [4] N. Leblanc, N. Mercier et al., J. Am. Chem. Soc. 2011, 133, 14924-14927. [5] N. Mercier, Eur. J. Inorg. Chem. 2013, 19-31. [6] O. Toma, N. Mercier et al., CrystEngComm 2012, 14, 7844-7847 ; O. Toma, N. Mercier et al., Eur. J. Inorg. Chem. 2013, 1113-1117.

2a - Chimie supramoléculaire et ingénierie moléculaire Mardi 2 juillet 17h00-18h30
- 46 -
O13 - Crystallography as a critical resource in metal-organic self assembly Jonathan Nitschke Cambridge University, United Kingdom
Designing complex materials for new devices through the art of chemical synthesis brings challenges and opportunities; single-crystal X-ray diffraction is a key front-line technique in the structural characteration of new, complex self-assembled materials.This talk will focus upon the design of self-assembly processes that can bring together simple, organic molecules and first-row transition-metal ions into complex, functional structures, and how these structures may be characterised using state-of-the-art crystallographic techniques developed by our partners at Global Phasing Ltd. Examples include an octa-Fe(II) cubic cage that is capable of selectively binding to higher fullerenes,[1] a tetra-Fe(II) tetrahedral cage that is capable of rendering air-stable white phosphorus (P4), which is ordinarily pyrophoric,[2] and a deca-Co(II) pentagonal prism, shown at right, which forms part of a chemical network that behaves differently under the influences of different chemical signals.[3] This prism is formed through the action of hexafluorophosphate template ions, and it binds tightly to chloride once formed. [1] W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg, J. R. Nitschke, Angew. Chem. Int. Ed. 2011, 50, 3479. [2] P. Mal, B. Breiner, K. Rissanen, J. R. Nitschke, Science 2009, 324, 1697. [3] I. A. Riddell, M. M. J. Smulders, J. K. Clegg, Y. R. Hristova, B. Breiner, J. D. Thoburn, J. R. Nitschke, Nature Chem. 2012, 4, 751.

2a - Chimie supramoléculaire et ingénierie moléculaire Mardi 2 juillet 17h00-18h30
- 47 -
O14 - La liaison halogène pour l'élaboration d'architectures complexes: contribution électrostatique et réseaux d'anions (2D, 3D) Marc Fourmigué, Julien Lieffrig, Olivier Jeannin Institut des Sciences Chimiques de Rennes, Université Rennes 1, UMR CNRS 6226, Campus de Beaulieu, 35042 Rennes (France)
La liaison halogène est basée sur une distribution électronique anisotrope autour d’atomes d’halogène organiques, conduisant à une déformation et une lacune de densité électronique le long de la liaison C–Hal. Cette polarisation est à l’origine d’interactions Hal•••Hal directes mais aussi d’interactions C–Hal•••NC– avec des nitriles ou tout autre base de Lewis, qui peuvent être aussi fortes et plus directionnelles qu'une liaison hydrogène normale [1,2].
La contribution électrostatique à cette interaction est démontrée ici dans une série de complexes à transfert de charge formulés (EDT-TTFI2)2(TCNQFn) avec n = 0–2 dans lequel la charge des partenaires électro-actifs est modulable (et modulée) en fonction de n et/ou de la température, dans des composés présentant une conversion neutre-ionique [3,4].
Par ailleurs, la mise en œuvre d'anions halogénures (Cl–, Br–, I–) ou pseudo-halogénures (SCN–) [5] comme accepteurs de liaisons halogène permet la construction de structures polymériques avec des molécules iodées ditopiques, mais aussi des structures poreuses robustes avec des molécules iodées tritopiques, comme des structures 2D en nid d'abeille [6], evt. interpénétrées, mais aussi des structures 3D cubiques de type pyrite [7]:
[1] R. Rogers, Cryst. Growth Des. 2011, 11, 4721. [2] M. Fourmigué, Curr. Op. Solid State Mater. Sc. 2009, 13, 36. [3] J. Lieffrig, O. Jeannin, T. Guizouarn, P. Auban, M. Fourmigué, Cryst. Growth Design 2012, 12, 4248 [4] J. Lieffrig, O. Jeannin, S. Dahaoui, E. Espinosa, P. Auban-Senzier, M. Fourmigué et al. , en préparation [5] P. Cauliez, V. Polo, T. Roisnel, R. Llusar, M. Fourmigué, CrystEngComm 2010, 12, 558 [6] S. Triguero, R. Llusar, V. Polo, M. Fourmigué, Cryst Growth Design 2008, 8, 2241 [7] J. Lieffrig, O. Jeannin, M. Fourmigué, J. Am. Chem. Soc. 2013, sous presse, DOI: 10.1021/ja400740v
C Br+
-
N C-
EDT-TTFI2TCNQFn, -1 < < 0
EDT-TTFI2S
S
S
S
S
S
I
I
NC
NC
CN
CN
F
F
S
S
S
S
S
S
I
I

2a - Chimie supramoléculaire et ingénierie moléculaire Mardi 2 juillet 17h00-18h30
- 48 -
O15 - Les carboxylates d’uranium tétravalents : précurseurs du combustible nucléaire
Clément Falaise1, Christophe Volkringer1, Natacha Henry1, Thierry Loiseau1
1 Unité de Catalyse et Chimie du Solide (UCCS) – UMR CNRS 8181, Université de Lille, USTL-ENSCL, BatC7, BP 90108, 59652 Villeneuve d’Ascq
Depuis quelques années la chimie de l’uranium connait un regain d’intérêt, notamment
grâce à sa place dans l’industrie énergétique mondiale. La maitrise de la technologie nucléaire implique une parfaite compréhension de la chimie associé à l’uranium, que ce soit pour la fabrication du combustible nucléaire (oxyde d’uranium) ou dans les méthodes de séparation.
Aujourd’hui la chimie de l’uranium hexavalent (UVI) est largement étudiée, celle des degrés d’oxydation inférieurs (UV, UIV, UIII) reste encore assez peu explorée. Ce constat est surprenant, si l’on considère que l’uranium tétravalent est impliqué dans certaines étapes de fabrication du combustible nucléaire conduisant à la formation de la céramique UO2 utilisée comme combustible dans les centrales nucléaires actuelles. Ce paradoxe s’explique simplement par la difficulté d’isoler et de caractériser des espèces chimiques présentant de faibles degrés d’oxydation dans les conditions atmosphériques ambiantes.
Cette communication sera centrée sur la cristallochimie de nouveaux composés à base d’uranium tétravalents (UIV) et leurs propriétés thermiques. Des méthodes de synthèses impliquant une maitrise de la cristallisation et de l’hydrolyse seront présentées. Ces stratégies de synthèse ont donné lieu à des chimies de coordination inédites de l’uranium, associant notamment clusters inorganiques polynucléaire et ligands organiques. La présentation s’articulera principalement autour des topologies associant UIV et de ligands carboxylates :
- dans un premier temps le système UIV/dicarboxylates linéaires sera exposé. Ce système très riche d’un point de vue cristallochimique permet l’obtention d’une famille de matériaux microporeux (U6O4(OH)4(H2O)6(OOC-X-COO)6) dont les dimensions des cavités sont modulables [1].
- l’étude du système UIV/benzoate a également été effectuée. Ainsi nous avons réussi à isoler des agrégats nanométriques composés de 38 atomes d’uranium [2]. La formation de ces assemblages polymétalliques ainsi que les difficultés liées à la résolution structurale seront abordées.
[1] Falaise et al., Chemistry- A european journal. 2013, 19, 5324–533. [2] Falaise et al., submitted to Angew. Chem. Int. Ed.

2b - Cristallographie pour les nanosciences – Mardi 2 juillet 17h00-18h30
- 49 -
O16 - Structure of BN nanostructures: which impact on their luminescent properties? J. Loayza1,2, J. Barjon2, A. Pierret1,3, S. Moldovan4, O. Ersen4, F. Ducastelle1 and A. Loiseau1 1LEM, ONERA-CNRS, 29 avenue de la Division Leclerc, Châtillon, France 2Group ‘Nanophysique et Semi-conducteurs’, CEA/INAC –CNRS-UJF, 17 rue des Martyrs, Grenoble, France 3GEMAC, Université Versailles St Quentin – CNRS, 45 avenue des Etats Unis, Versailles, France 4LPA, ENS-CNRS, 24 rue Lhomond, Paris, France 4IPCMS, CNRS – Université de Strasbourg, 23 rue du Loess, Strasbourg, France [email protected]
Hexagonal boron nitride (h-BN) is a wide band gap semiconductor (~ 6.5 eV), which can be synthesized, as graphite, its carbon analog, as bulk crystallites, nanotubes and layers. These structures meet a growing interest for deep UV LED and graphene engineering [1]. For instance electron mobility of graphene has been shown to be preserved when graphene is supported by a h-BN film.
Until recently, properties of h-BN materials were poorly known due to both the scarcity of crystals and suitable investigation tools. This situation has changed thanks, first, to the development of dedicated photoluminescence (PL) and cathodoluminescence (CL) experiments running at 4K and adapted to the detection in the far UV range [2, 3, 4], and second to the avaibility of high quality single crystals [5]. Thanks to these tools, h-BN has been shown to display original optical properties, governed, in the energy range 5.5 – 6 eV, by strong excitonic effects [2, 3, 6], which has been confirmed by reliable theoretical calculations [7, 8]. Furthermore, experimental investigations combining cathodo-luminescence measurements and transmission electron microscopy (TEM) observations have revealed that excitonic luminescence is highly sensitive to their environment and are easily perturbed by structural defects such as dislocations [4].
In this talk, we will examine the interplay between structure, defects and luminescence properties of BN layers and BN nanotubes and how these properties can be further exploited for the characterization of these nanostructures. We carry out optical and structural characterizations of this material by combining PL, CLmeasurements at 4K in the UV range (up to 7eV) and TEM analyses using HRTEM, diffraction contrast imaging and electron tomography. Thin layers have been obtained by mechanically exfoliating small crystallites. Exfoliated flakes were reported first on SiO2 substrates for AFM thickness measurements, as described in [9] and second on TEM grids. Nanotubes are multi wall nanotubes made of typically 10 to 20 layers provided by the team of D. Golberg (NIMS, Japan).

2b - Cristallographie pour les nanosciences – Mardi 2 juillet 17h00-18h30
- 50 -
O16 - Structure of BN nanostructures: which impact on their luminescent properties?
We will show that, whatever the structure, bulk or nanoscale, excitonic luminescence
consists of two series of lines called S and D. S excitons are found to be self-trapped, due to a Jahn-Teller effect [3]. Thanks to the imaging capability of the CL, emission, related to D lines, is found to be localized on defects, identified by TEM as grain boundaries. In defect free areas of thin layers, D lines completely vanish and S lines only are observed. D/S ratio can therefore be used as a qualification parameter of the defect densities present in the layers [10]. Concerning nanotubes, we will show that exciton trapping on structural defects gives rise to a spectacular localization of the luminescence caused by a complex structure of these tubes that we have analysed by combining electron diffraction, high resolution imaging and electron tomography. [1] C.R. Dean et al. Nature Nanotechnology 5 (2010) 722 [2] P. Jaffrennou el al., Phys. Rev. B 77 (2008) 235422 [3] K. Watanabe et al., Phys. Rev. B 79 (2009) 193104 [4] P. Jaffrennou el al., J. Appl. Phys. 102 (2007) 116102 [5] Y. Kubota et al.. Science 317 (2007) 932 [6] L. Museur et al., Phys. Stat. Sol. rrl, 5 (2011) 414 [7] B. Arnaud, et al. Phys. Rev. Lett. 96 (2006) 026402 [8] C.-H. Park et al., Phys. Rev Lett. 96 126105 (2006) [9] G. F. Schneider, et al, Nano. Lett. 10 (2010)1912 [10] J. Loayza et al, Nanoletters, submitted (2013)

2b - Cristallographie pour les nanosciences – Mardi 2 juillet 17h00-18h30
- 51 -
O17 - Imagerie des champs de déformations par holographie électronique Etienne Snoeck, Christophe Gatel, Florent Houdellier, Nikolay Cherkashin, Martin Hytch CEMES-CNRS 29 rue Jeanne Marvig, 31055 Toulouse Cedex
Dans une expérience de Microscopie Electronique à Transmission (MET) conventionnelle ou de spectroscopie seules la distribution d’intensité et la distribution en énergie du faisceau d’électron sont mesurées alors que l’information contenue dans la phase des faisceaux transmis et diffractés est perdue. Le déphasage du faisceau d’électron porte des informations résultant de l’interaction entre l’onde électronique et les champs magnétiques et électrostatiques. La mesure de ce déphasage permet d’évaluer ces champs [1]. Nous avons également montré que la phase des faisceaux diffractés permet aussi de sonder les champs de déformation locaux [2, 3]. Les techniques d’interférométrie électronique menées dans un microscope électronique à transmission permettent de mesurer ces phases et de cartographier ces champs avec une résolution spatiale de l’ordre de celle du microscope. Je détaillerai la technique d’holographie en champ sombre qui permet de mesurer la phase géométrique des faisceaux diffractés et par là, de cartographier les champs de déformation. J’illustrerai les potentialités de cette méthode en étudiant l’état de déformation de divers système contraints.
[1] Electron holography of nanostructured materials R.E. Dunin-Borkowski, M.R. McCartney and D.J. Smith. Chapter in Volume 3 of the "Encyclopaedia of Nanoscience [2] International patent PCT N° PCT/FR2008/001302 - M.J. Hÿtch, E. Snoeck, F. Houdellier, F. Hüe. [3] Nanoscale holographic interferometry for strain measurements in electronic devices M. Hÿtch, F. Houdellier, F. Hüe and E. Snoeck, Nature, 453, (June 2008)
2b - Cristallographie pour les nanosciences – Mardi 2 juillet 17h00-18h30
- 52 -
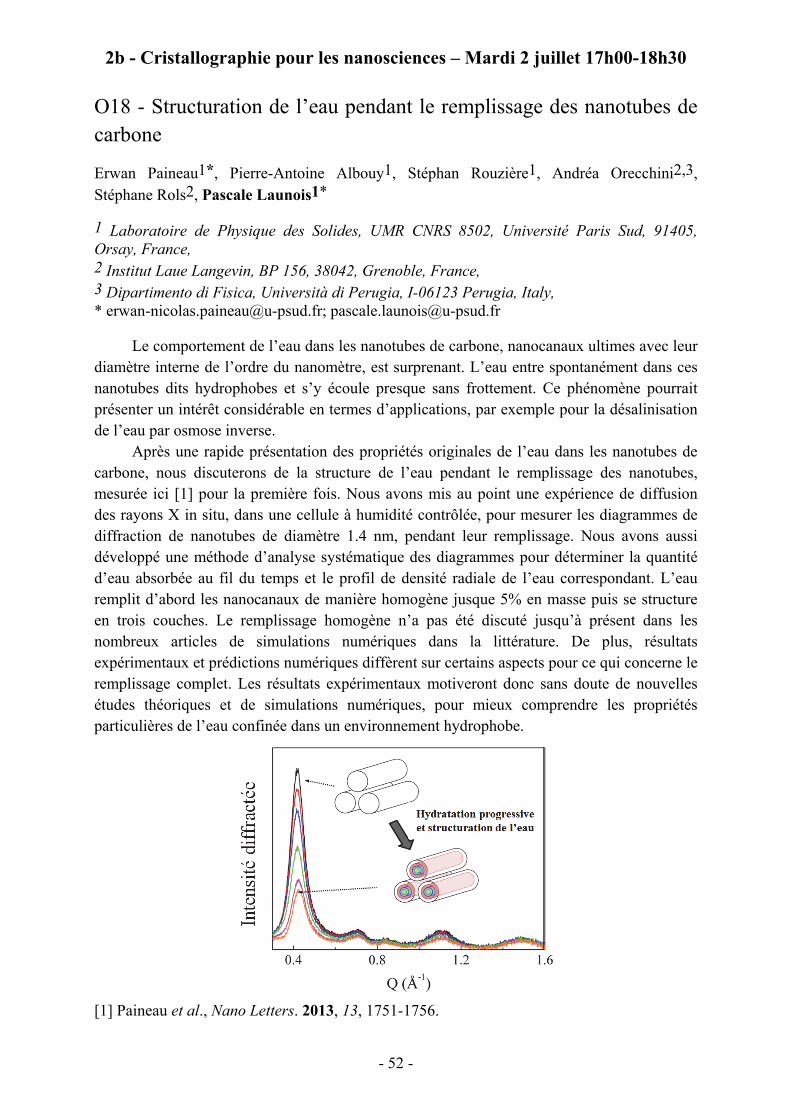
O18 - Structuration de l’eau pendant le remplissage des nanotubes de carbone
Erwan Paineau1*, Pierre-Antoine Albouy1, Stéphan Rouzière1, Andréa Orecchini2,3, Stéphane Rols2, Pascale Launois1*
1 Laboratoire de Physique des Solides, UMR CNRS 8502, Université Paris Sud, 91405, Orsay, France, 2 Institut Laue Langevin, BP 156, 38042, Grenoble, France, 3 Dipartimento di Fisica, Università di Perugia, I-06123 Perugia, Italy, * [email protected]; [email protected]
Le comportement de l’eau dans les nanotubes de carbone, nanocanaux ultimes avec leur diamètre interne de l’ordre du nanomètre, est surprenant. L’eau entre spontanément dans ces nanotubes dits hydrophobes et s’y écoule presque sans frottement. Ce phénomène pourrait présenter un intérêt considérable en termes d’applications, par exemple pour la désalinisation de l’eau par osmose inverse.
Après une rapide présentation des propriétés originales de l’eau dans les nanotubes de carbone, nous discuterons de la structure de l’eau pendant le remplissage des nanotubes, mesurée ici [1] pour la première fois. Nous avons mis au point une expérience de diffusion des rayons X in situ, dans une cellule à humidité contrôlée, pour mesurer les diagrammes de diffraction de nanotubes de diamètre 1.4 nm, pendant leur remplissage. Nous avons aussi développé une méthode d’analyse systématique des diagrammes pour déterminer la quantité d’eau absorbée au fil du temps et le profil de densité radiale de l’eau correspondant. L’eau remplit d’abord les nanocanaux de manière homogène jusque 5% en masse puis se structure en trois couches. Le remplissage homogène n’a pas été discuté jusqu’à présent dans les nombreux articles de simulations numériques dans la littérature. De plus, résultats expérimentaux et prédictions numériques diffèrent sur certains aspects pour ce qui concerne le remplissage complet. Les résultats expérimentaux motiveront donc sans doute de nouvelles études théoriques et de simulations numériques, pour mieux comprendre les propriétés particulières de l’eau confinée dans un environnement hydrophobe.
[1] Paineau et al., Nano Letters. 2013, 13, 1751-1756.

2b - Cristallographie pour les nanosciences – Mardi 2 juillet 17h00-18h30
- 53 -
O19 - Structure et dynamique de molécules isolées dans des matrices poreuses
Dominik Schaniel1, Kuan-Ying Hsieh1, El-Eulmi Bendeif1, Axel Gansmuller1, Sébastien
Pillet1, Theo Woike2 1Universite de Lorraine, CRM2, UMR 7036, Vandoeuvre- les-Nancy, 2Institut für Strukturphysik, TU Dresden
L’inclusion de molécules fonctionnelles dans des matrices nanostructurées est un domaine de recherche très actif [1], en vertu des applications potentielles dans plusieurs domaines, p.ex. optique, catalyse, médecine [1-4]. Dans le cas d’une fonctionnalité optique comme la photocommutation du ligand NO dans Na2[Fe(CN)5NO]2H2O (SNP) leurs conférant des propriétés photoréfractives intéressantes [5], ces capacités devraient être conservées après encapsulation dans la matrice. Il existe donc un intérêt fondamental pour déterminer la structure de molécules encapsulées dans des matrices afin de vérifier l’intégrité structurale des complexes fonctionnels et d’étudier les interactions hôte-matrices.
Nous avons réalisé des nanohybrides contenant des molécules isolées spécifiques du
complexe SNP en imprégnant des matrices poreuses de SiO2 (taille de pore entre 1-2 nm) par une solution de SNP. La structure de ces nanohybrides a été étudiée par diffusion totale et l’analyse de la fonction de distribution de paires (PDF), et par résonance magnétique nucléaire (RMN). L’analyse de la PDF différentielle permet d’identifier les complexes de SNP à l’intérieur des pores, sous forme de molécules isolées ou de clusters moléculaires, et de déterminer l’arrangement anion-cation et la taille du cluster. L’analyse RMN révèle une certaine dynamique des molécules qui dépend du taux d’hydratation de la matrice.
En conclusion, l’analyse PDF des données de diffusion totale combinée à des mesures
RMN du solide permet de déterminer la structure et la dynamique des molécules isolées dans les pores d’un matériau amorphe. [1] Sanchez et al., Adv. Mater. 2003, 15, 1669 ; Vinu et al., J. Nanosci. Nanotec. 2005, 5, 347. [2] Deniz et al., Chem. Eur. J. 2012, 18, 15782. [3] Blecher et al., Nanomedicine : Nanotechnology, Biology, and Medicine 2012, 8, 1364. [4] T.-W. Sung, Y.-L. Lo, Sensors and Actuators B : Chemical 2012, 173, 406. [5] Schaniel et al., Adv. Mater. 2007, 19, 723.

2c - Enzymologie et dynamique – Mardi 2 juillet 17h00-18h30
- 54 -
O20 - The COP9 signalosome: Activity and regulation M. Birol, F. Hoh, Y. Yang, A. Padilla, C. Dumas & A. Echalier * Centre de Biochimie Structurale. CNRS, INSERM, UM1, UM2. 29 rue de Navacelles. 34090 Montpellier cedex * [email protected] The Cop9 (Constitutive photomorphogenesis 9) signalosome (CSN) is a large multiprotein complex that, through its role in the ubiquitin-proteasome pathway, is implicated in various cellular functions spanning from cell cycle to circadian rhythm and reaching the immunity of organisms. To date the CSN complex has mostly been studied for its implication in the regulation of the E3-cullin RING ubiquitin ligases (CRLs). The isopeptidase catalytic activity of the CSN complex is carried by the subunit 5, CSN5 (also known as Jab1). CSN5 is involved in the deneddylation of CRLs by performing the hydrolysis of the isopeptide bond between an ubiquitin-like molecule, Nedd8 (cullin-neural precursor cell expressed developmentally downregulated gene 8) and the CRL cullin scaffolding subunit. CSN5 when incorporated within the CSN complex displays isopeptidase activity, however it is intrinsically inactive in a stand-alone form. In light of our work, the determination of the CSN5 crystal structure together with biochemical and in silico investigations contributed to understand the molecular regulation of CSN5 activity as well as to identify a potential molecular trigger that is involved in the transition of an inactive CSN5 to an active isopeptidase enzyme. A single point mutation located in a flexible region of the active site of CSN5, known as the Ins-1 segment, was used to obtain a constitutively active variant of CSN5 [1]. We will present here how the activity of the CSN catalytic subunit is regulated. [1] Echalier et al. (2013). PNAS. 110(4), 1273-1278.

2c - Enzymologie et dynamique – Mardi 2 juillet 17h00-18h30
- 55 -
O21 - Les synthases de cyclodipeptides, des homologues d’aminoacyl-ARNt synthétases impliquées dans la synthèse peptidique non ribosomale Mireille Moutiez1, Jérôme Seguin1, Pascal Belin1, Jean-Luc Pernodet2, Muriel Gondry1
1CEA, iBiTec-S/SIMOPRO, 91191 Gif-sur-Yvette, 2Univ. Paris-Sud 11, CNRS, UMR8621, 91405 Orsay
Nous avons identifié une nouvelle famille d’enzymes, les synthases de cyclodipeptides (CDPS), qui sont impliquées dans la biosynthèse d’une classe de peptides non ribosomaux, les cyclodipeptides [1,2]. Les cyclodipeptides et leurs dérivés, les dicétopipérazines (DKP), sont extrêmement répandus chez les microorganismes et se caractérisent par une grande variété de structures chimiques et une large gamme d’activités pharmacologiques. Les CDPSs utilisent des ARNt aminoacylés (aa-ARNt) comme substrats pour synthétiser les liaisons peptidiques de différents cyclodipeptides ; elles détournent donc à leur profit des composés qui sont normalement dévolus à la synthèse protéique ribosomale. La résolution des structures cristallographiques de CDPS [3-5] a permis de montrer que ces enzymes présentent une homologie structurale importante avec les domaines catalytiques des aminoacyl-ARNt synthétases (aaRS), les enzymes qui catalysent l’activation et le transfert des acides aminés sur les ARNt pour former des aa-ARNt (Figure 1).
substrats cyclodipeptides
NH
HNO
O
aa
aa
aaRS aaRS
substrats Produit
aa
CDPS CDPS
Figure 1 : une similitude structurale entre deux familles d’enzymes, les aaRS et les CDPS
Les CDPS possèdent une poche qui accommode la partie aminoacyle d’un aa-ARNt et contient les résidus catalytiques. Elles utilisent un mécanisme catalytique de type ping-pong qui transite par la formation d’un intermédiaire acyl-enzyme provenant du transfert de la partie aminoacyle d’un premier aa-ARNt vers une sérine catalytique [3]. Les étapes subséquentes de la catalyse et les bases moléculaires des interactions entre les CDPS et leurs substrats sont actuellement en cours d’étude au laboratoire.

2c - Enzymologie et dynamique – Mardi 2 juillet 17h00-18h30
- 56 -
O21 - Les synthases de cyclodipeptides, des homologues d’aminoacyl-ARNt synthétases impliquées dans la synthèse peptidique non ribosomale Dans les voies de biosynthèse de DKP, les CDPS sont associées à des enzymes de modification des cyclodipeptides produits. Par exemple, la voie de biosynthèse isolée chez M. tuberculosis est constituée d’une CDPS produisant du cyclo(L-Tyr-L-Tyr) qui est utilisé comme substrat par le cytochrome P450 CYP121 pour former une DKP dont les chaînes latérales sont pontées par une liaison C-C intramoléculaire [1,2]. Notre objectif est d’utiliser les connaissances acquises pour effectuer une ingénierie tant génétique que protéique des voies de biosynthèse des DKP [6] afin de créer de nouvelles DKP aux propriétés biologiques et pharmacologiques optimisées. [1] Gondry et al., Nat Chem Biol. 2009, 5, 414-420. [2] Belin et al., Proc Natl Acad Sci U S A. 2009, 106, 7426-7431. [3] Sauguet et al., Nucleic Acids Res. 2011, 39, 4475-4489. [4] Vetting et al., Nat Chem Biol. 2010, 6, 797-799. [5] Bonnefond et al., Proc Natl Acad Sci U S A. 2011, 108, 3912-3917. [6] Belin et al., Nat. Prod. Rep. 2012, 29, 961-979.

2c - Enzymologie et dynamique – Mardi 2 juillet 17h00-18h30
- 57 -
O22 - Promiscuité enzymatique lactonase/phosphotriestérase : implications évolutives et biotechnologiques.
Julien Hiblot1, Guillaume Gotthard1, Jérémy Robin1, Patrick Masson2, Mikael Elias3 & Eric Chabrière1 1 Unité de Recherche sur les Maladies Infectieuses et Tropicales Emergentes, IRD/CNRS, Université de la Méditerranée (Aix-Marseille II), 13 385 Marseille, France, 2 IRBA-CRSSA, Département de Toxicologie. 38702 La Tronche Cedex, France, 3 Weizmann Institute of Science, Biological Chemistry, Rehovot, Israel.
La promiscuité enzymatique est la capacité fortuite d’une enzyme à prendre en charge un substrat non naturel, i.e. une fonction non soumise à pression de sélection [1]. Il s’agit de l’un des moteurs prépondérant de l’évolution des fonctions enzymatiques. Récemment, un lien phylogénétique fut mis en évidence entre les Phosphotriestérases bactériennes (PTEs) et les « Phosphotriesterases-Like Lactonase » (PLLs) [2]. Les PTEs auraient divergé des PLLs suite à l’utilisation massive d’insecticides organophosphorés (OPs) depuis 50 ans.
En effet, les PTEs présentent la capacité d’hydrolyser très efficacement certains OPs neurotoxiques [3] ouvrant des perspectives d’utilisation dans la bio-décontamination de ces composés. Les PLLs, quant à elles, sont des lactonases hydrolysant les AHLs, molécules impliquées dans la communication bactérienne (ou quorum sensing ; QS) [4] et ouvrant des perspectives d’utilisation dans l’inhibition de la virulence bactérienne (par quorum quenching ; QQ) [5]. La promiscuité entre ces deux activités enzymatiques proviendrait de la ressemblance géométrique entre l’état de transition d’hydrolyse des AHLs et celle des substrats OPs (tétraédriques) [4,6].
Afin de développer des produits d’intérêt biotechnologique, nous nous sommes inspiré de ce lien évolutif afin d’améliorer la PLL hyperthermostable SsoPox [7-11]. Nous avons utilisé une banque de données de mutations lors de protocoles d’évolution in vitro afin de redessiner (in silico) le site actif de SsoPox. Suite à son amélioration, l’analyse structurale de variants sélectionnés a révélé, notamment, une flexibilisation locale, principalement responsable de l’amélioration. Cette observation soutient le modèle de polarité/modularité corps-site actif des topologies présentant un haut potentiel évolutif [12].
[1] Khersonsky & Tawfik, Annu Rev Biochem. 2010, 79, 471-505. [2] Afriat-Jurnou et al., Biochemistry. 2012, 51, 6047-55; Afriat et al. Biochemistry. 2006, 45, 13677-13686; Elias & Tawfik, J Biol Chem. 2012, 287, 11-20. [3] Raushel. Nature. 2011, 469, 310-311. [4] Popat et al., Br Med Bull. 2008, 87, 63-75. [5] Amara et al., Chem Rev. 2011, 111, 195-208. [6] Ben-David et al., J Mol Biol. 2012, 418, 181-196. [7] Gotthard et al., Acta Crystallogr Sect F. 2013, 69, 73-76. [8] Hiblot et al., Sci Rep. 2012, 2, 779. [9] Hiblot et al., PLoS One. 2012, 7, e47028. [10] Del Vecchio et al., Extremophiles. 2009, 13, 461-470. [11] Elias et al., J Mol Biol. 2008, 379, 1017-1028. [12] Dellus-Gur et al., J Mol Biol. 2013, accepted.

2c - Enzymologie et dynamique – Mardi 2 juillet 17h00-18h30
- 58 -
O23 - Aperçu structural du mécanisme enzymatique d’AnkX, une protéine de Legionella pneumophila modifiant la petite protéine G Rab Valérie Campanacci1, Shaeri Mukherjee2, Craig R. Roy2, Jacqueline Cherfils1 1 Laboratoire d’Enzymologie et Biochimie Structurales, Centre de Recherche de Gif, CNRS, Gif-sur-Yvette, France, 2 Department of Microbial Pathogenesis, Yale University School of Medicine, New Haven, CT, USA
Legionella pneumophila est la bactérie responsable d’une pneumonie sévère appelée
maladie du Légionnaire. La bactérie infecte les macrophages alvéolaires et se développe en créant une vacuole spécialisée, la LCV (legionella-containing vacuole), similaire d’un point de vue morphologique au réticulum endoplasmique de son hôte [1]. Afin d’assurer la biogenèse et la survie de cet organelle de réplication, L. pneumophila injecte un arsenal de protéines appelées effecteurs dans le cytosol de la cellule infectée via un système de sécrétion de type IV (Dot/Icm). Ces effecteurs vont pirater les machineries du trafic cellulaire et ainsi permettre au pathogène d’échapper aux défenses de l’hôte [2].
AnkX, un des effecteurs sécrétés par L. pneumophila, est une protéine comprenant un
domaine FIC et des séquences ankyrines répétées [3]. Les domaines FIC sont caractérisés par un motif conservé HPFx[D/E]GN[G/K]R. Alors que la plupart des domaines FIC bactériens permettent le transfert d’AMP à partir d’ATP sur des protéines cibles, AnkX utilise la cytidine diphosphate-choline (CDP-choline) comme substrat pour attacher de manière covalente un groupement phosphocholine sur la petite protéine G Rab [4]. Cette modification de Rab altère son interaction avec ses partenaires cellulaires.
Nous avons résolu les structures cristallographiques de AnkX (résidus 1 à 484) sous forme apo, liée à son substrat la CDP-choline (en utilisant une forme inactive de l’enzyme), et liée aux produits de la réaction (CMP et phosphocholine) [5]. Ces structures révèlent pourquoi AnkX transfère la phosphocholine et non pas le nucléotide sur Rab, et comment les séquences ankyrines répétées contraignent le domaine FIC. Nos résultats généralisent un mécanisme de phosphoryl-transférase commun à toutes les enzymes à domaine FIC.
[1] Tilney et al., J. Cell Sci. 2001, 114, 4637. [2] Hubber et al., Annu. Rev. Cell Dev. Biol. 2010, 26, 261–83. [3] Pan et al., Science. 2008, 320, 1651–1654. [4] Mukherjee et al., Nature, 2011, 477, 103-106. [5] Campanacci et al., EMBO J. 2013 Apr 9. doi: 10.1038/emboj.2013.82. Remerciements : Nous remercions chaleureusement toute l’équipe de la ligne Proxima-1 du synchrotron Soleil, et tout particulièrement Beatriz Guimaraes, pour leur aide, conseils et disponibilité lors des collectes. Ce travail a bénéficié des infrastructures de la plateforme de cristallisation LEBS/IMAGIF.

Séance plénière II – Mercredi 3 juillet 9h00-10h00
- 59 -
SP2 - The Amazing Diversity of the Human GPCR Superfamily Ray Stevens The Scripps Research Institute, La Jolla CA, USA
GPCRs constitute the largest protein families in the human genome and play essential roles in normal cell processes, most notably in cell signaling. The human GPCR family contains more than 800 members and recognizes thousands of different ligands and activates a number of signaling pathways through interactions with a small number of binding partners. GPCRs have also been implicated in numerous human diseases, and represent more than 30-40% of drug targets. Delivering GPCR structures in close collaboration with the community on specific receptor systems is of immense value to the basic science community interested in cell signaling and molecular recognition, as well as the applied science community interested in drug discovery. This work is being followed up with additional biophysical characterization including NMR spectroscopy and community wide assessments with computational biology groups throughout the world. Crystal structures are now available for rhodopsin, adrenergic, and adenosine receptors in both inactive and activated forms, as well as for chemokine, dopamine, histamine, lipid, and all four opioid receptors in inactive conformations. A review of the common structural features seen in these receptors and the scope of structural diversity of GPCRs at different levels of homology provides insight into our growing understanding of the biology of GPCR action and their impact on drug discovery. Given the current set of GPCR structural data, a distinct modularity is now being observed between the extracellular (ligand-binding) and intracellular (signaling) regions. The rapidly expanding repertoire of GPCR structures provides a solid framework for experimental and molecular modeling studies, and helps to chart a roadmap for comprehensive structural coverage of the whole superfamily and an understanding of GPCR biological and therapeutic mechanisms. With the rapid accumulation of this data, one can now start to investigate GPCR evolution, and expanding this understanding to human evolution and cognition.
This work was supported by NIGMS PSI:Biology for GPCR structure processing (U54GM094618) and the NIH Roadmap Initiative (JCIMPT) for technology development (GM073197).

3a - Croissance cristalline (GFCC) – Mercredi 3 juillet 10h30-12h00
- 60 -
O24 - Membrane Protein Crystallization in Cubic Phases Valentin Gordeliy 1,2 1Institute of Structural Biology J.P. Ebel, CEA-CNRS-UJF UMR 5075, Grenoble F-38027, France 2Institute of Complex System-6, Research Centre Juelich, Juelich 52425, Germany
An overview of in meso approaches to membrane protein crystallization will be given and new developments in the field under discussion will be described. Recent progress in the corresponding high throughput instrumentation will also be in a focus of the presentation. In addition, a brief overview of membrane protein crystallization platform at the Institute of Structural Biology J.P. Ebel, Grenoble will be presented. A critical analysis of the perspectives of this approach will be done.

3a - Croissance cristalline (GFCC) – Mercredi 3 juillet 10h30-12h00
- 61 -
O25 - Croissance, Structure et Optique Non Linéaire Quadratique
Pascal Loiseau1, Gérard Aka1, Simon Ilas1, Federico Khaled1
1Laboratoire de Chimie de la Matière Condensée de Paris – ENSCP (Chimie-Paristech), UMR CNRS 7574, 11 rue Pierre et Marie Curie, 75231 Paris Cedex 05
Parmi tous les milieux qui peuvent conduire à la génération d’un rayonnement laser, les lasers solides sont souvent privilégiés pour des raisons de miniaturisation, de maintenance et de fiabilité. Cependant, alors que les applications des lasers sont fortement conditionnées par leurs longueurs d’onde d’émission, toutes ces longueurs d’onde ne sont pas facilement accessibles. En effet, les lasers solides les mieux maîtrisés pour de hautes performances émettent vers 1 µm (cas du cristal de Y3Al5O12 (YAG) dopé Nd3+ par exemple). Pour obtenir des longueurs d’onde plus courtes, la solution consiste alors à disposer derrière le cristal laser des cristaux qualifiés de non linéaires : ils ont la particularité de pouvoir modifier la fréquence des rayonnements laser qui les traversent. Il est alors possible d’envisager des dispositifs laser solides émettant dans le visible, voire même dans l’ultraviolet par des processus de somme de fréquence.
Les processus non linéaire quadratique de conversion de fréquence ne sont efficaces que dans des milieux biréfringents, non centrosymétriques, transparents dans le domaine optique d’intérêt, et dont la polarisabilité est significative. L’ensemble de ces critères peut s’avérer extrêmement sélectif et difficile à combiner, d’autant plus que la longueur d’onde laser visée est courte. Néanmoins, il s’avère que les composés à base de groupements borate présentent les meilleures propriétés pour les processus de conversion de fréquence dans le visible et l’ultraviolet.
Au-delà des ces propriétés intrinsèques, le cristal optique non linéaire idéal doit aussi présenter de bonnes propriétés d’usage et la faculté d’être élaboré avec la meilleure qualité cristalline et optique possible. L’ensemble de ces aspects sera abordé pendant la conférence au travers de l’expérience acquise au LCMCP dans ce domaine depuis plus de 20 ans.

3a - Croissance cristalline (GFCC) – Mercredi 3 juillet 10h30-12h00
- 62 -
O26 - Etude structurale par diffraction des rayons X sur monocristaux relaxeurs de Ba2LnFeNb4O15 (Ln=La, Pr, Nd, Sm et Eu) Marjorie Albino1,2, Philippe Veber1,2, Stanislav Pechev1,2, Matias Velázquez1,2, Mario Maglione1,2, Michaël Josse1,2
1 CNRS, ICMCB, UPR 9048, F-33600 Pessac, France 2 Univ. Bordeaux, ICMCB, UPR 9048, F-33600 Pessac, France
Les composés de formulation Ba2LnFeNb4O15 avec Ln=La, Pr, Nd, Sm, Eu
(BLnFNO) cristallisants dans la structure cristalline Tetragonal Tungsten Bronze (TTB) ont fait l’objet d’études sur céramiques au sein de l’ICMCB [1]. Les cations Nb5+ positionnés en site octaédrique, connus comme ions ferroélectriquement actifs, apportent à cette famille de matériaux des propriétés diélectriques intéressantes. La structure cristalline étudiée par diffraction des rayons X et neutronique sur poudre suggère une modulation de la structure TTB.
Pour étudier les comportements diélectriques et structuraux intrinsèques à la matrice TTB, l’élaboration de cristaux est indispensable. La technique de croissance cristalline par la méthode des flux, utilisant le borate de lithium LiBO2 en tant que solvant, a permis l’obtention de cristaux BLnFNO de taille millimétrique [2], malgré la volatilisation du solvant. Par la suite, d’autres essais ont été entrepris dans une enceinte en platine scellée sous vide primaire afin d’éviter l’évaporation du solvant lors de la croissance. Après optimisation du procédé de cristallogenèse [3], des cristaux de BLnFNO de taille sub-centimétrique voire centimétrique ont ainsi été formés et utilisés pour des mesures diélectriques.
Les études structurales sur monocristaux par DRX réalisées à température ambiante mettent en évidence, et ce, quelle que soit la nature de la terre rare insérée en site carré, une structure modulée incommensurable bidimensionnelle (3+2D) caractérisée par les vecteurs de modulation (α, ±α, ½). La résolution des structures cristallines en 3D sur les monocristaux de BLnFNO conduit à des facteurs de reliabilités satisfaisants en symétrie quadratique dans le groupe d’espace P4/mbm. Le remplissage partiel du site carré par les cations Ln3+, avec un taux de lacunes pouvant atteindre jusqu'à ~40% dans le cas de Ln=Sm ou Eu, induit une distorsion des octaèdres MO6 (M=Fe, Nb) environnants. L’évolution des distances M-O ainsi que des angles O-M-O ou encore M-O-M fera l’objet d’une discussion détaillée lors de cette présentation orale, afin d’évaluer les relations structure-propriétés dans ces matériaux. [1] M. Josse et al., Solid State Sci., 2009, 11(6), 1118-1123 [2] E. Castel & al., J. Cryst. Growth, 2012, 340, 156-165 [3]M. Albino et al., accepted in Eur. J. Inorg. Chem., doi: 10.1002/ejic.201300008

3a - Croissance cristalline (GFCC) – Mercredi 3 juillet 10h30-12h00
- 63 -
O27 - Synthèse, caractérisation et croissance cristalline de cristaux de pyrophosphate de calcium d’intérêt biologique. Pierre Gras,1 Christian Rey,1 Sébastien Teychené,2 Christian Bonhomme,3 Danielle Laurencin,4 Béatrice Biscans,2 Stéphanie Sarda5 et Christèle Combes1 1 CIRIMAT, ENSIACET, UMR 5085, INPT-CNRS-UPS, Université de Toulouse, Toulouse, France. 2 Laboratoire de Génie Chimique, ENSIACET, INPT-CNRS-UPS, Université de Toulouse, Toulouse, France. 3 Laboratoire de Chimie de la Matière Condensée de Paris, Collège de France, UMR 7574, UMPC Paris 6, Paris, France. 4 Institut Charles Gerhardt de Montpellier, UMR 5253, Montpellier, France. 5 CIRIMAT, Université Paul Sabatier, UMR 5085, INPT-CNRS-UPS, Université de Toulouse, Toulouse, France.
Les pyrophosphates de calcium hydratés (CPP : Ca2P2O7·nH2O) sont impliqués dans de nombreuses formes d’arthrite dont l’arthrose, la plus répandue des maladies articulaires. Deux types de CPP ont été identifiés in vivo dans les articulations de patients arthritiques : des cristaux de pyrophosphates de calcium dihydraté (CPPD : Ca2P2O7·2H2O) monoclinique (m-CPPD) et triclinique (t-CPPD), possédant chacun un fort potentiel inflammatoire [1]. Outre ces deux formes, plusieurs phases de CPP ont été synthétisées in vitro dont un composé tetrahydraté monoclinique m-CPPT β (CPPT: Ca2P2O7·4H2O) et une phase amorphe a-CPP identifiés comme précurseurs in vitro des phases pathologiques [2].
L’objectif de ce travail est de mettre au point une méthode de synthèse simple et rapide de plusieurs phases de CPP pures puis de les caractériser finement. Les composés ont été préparés par double décomposition de sels de pyrophosphate et de calcium en milieu aqueux tamponné à différentes températures. Quatre phases pures ont été synthétisées, m-CPPD, t-CPPD, m-CPPT β et a-CPP, et caractérisées notamment par DRX sur poudre, spectroscopies FTIR et Raman, et RMN du solide 31P et 43Ca. Dans un second temps, plusieurs conditions de croissance cristalline en gel ont été utilisées afin d’obtenir des cristaux de qualité suffisante, notamment m-CPPD dont la structure reste non résolue.
Les outils analytiques complémentaires ont permis d’étudier certaines propriétés structurales des phases de CPP ainsi que leur évolution en solution. Ces résultats ont contribué à l’identification de cristaux de CPP in situ dans le liquide synovial de patients souffrant d’arthrite. Des résultats préliminaires sur la croissance de cristaux en gel seront présentés. Le but final de cette étude serait de relier la morphologie et la chimie de surface des cristaux de CPP aux propriétés inflammatoires observées afin d’aider à la mise au point de nouveaux traitements visant contrôler voire inhiber leur formation in vivo. [1] M. Roch-Arveiller, R. Legros, B. Chanaud, O. Muntaner, S. Strzalko, A. Thuret, D. A. Willoughby et J. P. Giroud, Biomed. Pharmacother., 1990, 44, 467–474. [2] E. H. Brown, J. R. Lehr, J. P. Smith et A. W. Frazier, Agricultural and Food Chemistry, 1963, 11, 214–222.

3b - Cristallographie résolue en temps – Mercredi 3 juillet 10h30-12h00
- 64 -
O28 - Etudes par Diffraction Résolue en Temps de Phénomènes Irréversibles: Application aux Synthèses Auto-Propagées.
Caroline Curfs1
1ESRF, 6 rue J. Horowitz, 38046 Grenoble Cx
Irreversible phenomena can be challenging to study, especially when the succession of events is extremely fast. Thus, in order to avoid missing or overlapping information, the phenomenon has to be probed in the most continuous and fast way. When polycrystalline materials are involved, a possible technique is Time-Resolved X-Ray Powder Diffraction (TRXRD).
Combustion synthesis [1] is a good example of a fast and irreversible phenomenon. Indeed it can take only few seconds to obtain the final products and the lifetime of some intermediate phases can be as short as a couple of hundreds of milliseconds. This synthesis is based on the exothermic properties of the chemical reaction to synthesize a whole sample. Two combustion modes exist depending whether the synthesis occurs in the form of a heat wave travelling through the sample (Self-propagating High-temperature Synthesis SHS) or if it occurs simultaneously in the whole sample (Thermal Explosion Synthesis TES). Because these reactions can be extremely fast (less than 1 second to obtain the final products), the composition and physico-chemical properties of the final products are difficult to predict. Thus, in order to better control this process, it is necessary to determine the synthesis mechanisms. The crystallographic changes occurring during the synthesis are then probed by TRXRD without interfering with the propagation of the wave. TRXRD is also the only technique which can be used to study the mechanisms during the TES.
In this study, several samples of the same composition (equiatomic composition of Al-Ni-Ti-C) and density (55%) have been synthesised by Combustion Synthesis using either the SHS or the TES modes. For both modes, TRXRD experiments have been performed to determine the synthesis mechanisms. A comparison between the different synthesis mechanisms involved in the SHS and TES modes will be presented.
[1] Merzhanov, in Combustion and Plasma Synthesis of High-Temperature Materials, VCH, New York, p.1, 1990.

3b - Cristallographie résolue en temps – Mercredi 3 juillet 10h30-12h00
- 65 -
O29 - Ultrafast structural dynamics in charge and orbitally ordered manganites under non-equilibrium conditions Paul Beaud1
1 Light Source, Paul Scherrer Institut, CH-5232 Villigen PSI, Switzerland
A key challenge in condensed matter is the understanding of the dynamic interplay among valence charge distribution, orbital order, magnetism and distortions of the atomic lattice which give rise to complex phase diagrams. Material properties such as superconductivity, metal-insulator transitions, colossal magnetoresistance (CMR) and multiferroicity are observed. Very short, intense pulses of light have the ability to excite matter far from equilibrium on extremely short time scales. The ensuing real-time dynamics have the potential to unravel the complex interplay of forces that ultimately determine the properties of these materials. Femtosecond x-ray diffraction is the only method capable of directly measuring the dynamics of the long range order of both, the atomic lattice and the electronic subsystems.
In my talk I will first introduce the time-resolved x-ray diffraction techniques we
developed at the FEMTO slicing source [1] at the Swiss Light Source during its first years of operation by addressing fundamental questions on simple solid systems [2,3]. More recently we have shifted our research interests towards structural dynamics of strongly correlated systems occuring under highly non-equilibrium conditions. As an example, our progressing studies of the dynamics coupled to the melting of charge and orbital order in CMR manganites [4,5] will be discussed in more detail. [1] P. Beaud et al., Phys. Rev. Lett. 99 174801 (2007). [2] S. L. Johnson et al., Phys. Rev. Lett. 100 155501 (2008). [3] S. L. Johnson et al., Phys. Rev. Lett. 102 175503 (2009). [4] P. Beaud et al., Phys. Rev. Lett. 103 155702 (2009). [5] A. Caviezel et al., Phys. Rev. B 87 205104 (2013).

3b - Cristallographie résolue en temps – Mercredi 3 juillet 10h30-12h00
- 66 -
O30 - Diffraction pompe-sonde résolue en temps sur la ligne de lumière CRISTAL à SOLEIL
C. Laulhé1,2, S. Ravy2, S. Hustache2, Ph. Hollander2 and J.-P. Ricaud2 1Université Paris-Sud, 91405 Orsay Cedex, France, 2Synchrotron SOLEIL, Gif-sur-Yvette, France
Le synchrotron SOLEIL développe des expériences pompe-sonde s’appuyant à la fois sur des lasers à impulsions ultracourtes et sur le faisceau de rayons X pulsé produit dans l’anneau de stockage. La résolution temporelle de ces expériences est limitée en pratique par la durée des impulsions de rayons X, typiquement 70 ps FWHM dans les modes de fonctionnement standards de l’anneau. Afin d’étudier les dynamiques photoinduites ultrarapides, un mode “low-α” [1-2] a été mis en place, pouvant donner des impulsions X de 10 ps FWHM. Une résolution ultime de ~ 100 fs est attendue à l’horizon 2014 au synchrotron SOLEIL, dans le cadre du projet femto-slicing [3]. CRISTAL est une ligne de diffraction sur onduleur qui depuis avril 2012 utilise la structure temporelle du rayonnement synchrotron pour réaliser des expériences de type pompe-sonde. L’échantillon est d’abord excité par une impulsion laser infrarouge de 40 fs FWHM (« pompe »). Les changements structuraux photoinduits sont ensuite étudiés dans le domaine temporel, en mesurant la diffraction d’une
impulsion de rayons X monochromatiques au temps t après excitation (« sonde »). Les expériences se déroulent sur le diffractomètre 6-cercles de la ligne, qui offre une grande variété d’environnements échantillon, et une géométrie de détection très souple.
Les impulsions laser et X sont produites à des fréquences de l’ordre du kHz et du MHz, respectivement. Pour faire une expérience pompe-sonde, il faut donc sélectionner le signal de diffraction provenant d’une seule impulsion X après chaque excitation laser. Dans les modes d’opération standards à SOLEIL (hybride, 8 paquets, 1 paquet), l’impulsion à sélectionner est séparée de ses voisines de 147 ns. Sur CRISTAL, nous sélectionnons cette impulsion via une fenêtre d’activation de 90 ns appliquée au détecteur à pixels XPAD3.2 [4], de dimensions 7 x 12 cm² (taille de pixel : 150 µm).
Dans ma présentation, je décrirai l’installation de diffraction pompe-sonde résolue en temps sur la ligne CRISTAL. Je montrerai ensuite des exemples de mesure dans les modes standard (résolution 70 ps) et “low-α” (résolution 10 ps) [5-6]. Je terminerai en présentant les dernières avancées du projet femto-slicing à SOLEIL, qui vise à produire des impulsions de rayons X de 100 fs d’ici à 2014.
[1] P. Brunelle, F. Briquez, A. Loulergue et al., Proceedings of IPAC 2011, (2011) 2124. [2] M.-A. Tordeux, J. Barros, A. Bence et al., Proceedings of IPAC 2012, (2012) 1608. [3] A. Nadji, F. Briquez, M.-E. Couprie et al., Proceedings of IPAC 2010 (2010) 2499. [4] P. Pangaud, S. Basolo, N. Boudet et al., Nucl. Instrum. Methods Phys. Res. A 591, 159 (2008). [5] C. Laulhé, S. Ravy, P. Fertey et al., Acta Physica Polonica A 121, 332 (2012). [6] C. Laulhé, M. Cammarata, M. Servol et al., to be published in Eur. Phys. J. Special Topics.

3c - Cristallo sous conditions extrêmes – Mercredi 3 juillet 10h30-12h00
- 67 -
O31 - Diffraction des rayons X sur monocristaux sous pression : application à l’étude de structures complexes
Julien Haines1, Jérôme Rouquette
1, Arie van der Lee
2
1Institut Charles Gerhardt Montpellier, UMR 5253 CNRS-Université Montpellier 2, France,
2Institut Européen des Membranes, UMR CNRS 5635, Université Montpellier 2, France
Grâce à la qualité des données, la diffraction de rayons X sur monocristal sous pression
permet de suivre la structure cristallographique fine de matériaux complexes. Les spécificités de la technique de diffraction des rayons X sur monocristal en cellule à enclume de diamants seront présentées. Des résultats obtenus sur source de laboratoire et au synchrotron seront comparés. Nous présenterons des exemples des études sous pression sur des matériaux à charpente ouverte comme les zéolithes et les cyanures de métaux de transition [1] présentant des structures particulièrement complexes (très grandes mailles, transitions de phases, désordre, insertion d’espèces). [1] Cairns et al., Nature Mater. 2013, 12, 212-216.

3c - Cristallo sous conditions extrêmes – Mercredi 3 juillet 10h30-12h00
- 68 -
O32 - Molecular basis of piezophilic adaptation
Louise Lassalle1, Marie Trovaslet2, Anne-claire Dhaussy3, Dominique Madern1, Bruno Franzetti1, Eric Girard1 1 IBS (UMR 5075 CEA-CNRS-UJF-PSB), 41 rue Jules Horowitz, 38027 Grenoble Cedex, France
2 Institut de Recherches Biomédicales des Armées, antenne de La Tronche, France 3 CRISMAT, ENSICAEN, 6 Boulevard du Maréchal Juin, 14000 Caen, France
Notre planète vit majoritairement sous pression. En effet, plus de 60 % de la biosphère sont caractérisés par des pressions supérieures à 100 bars. Ces pressions peuvent atteindre 1000 bars dans les fosses sous-marines les plus profondes. La plupart des micro-organismes y sont toutefois représentés. La découverte récente de Pyrococcus yayanosii CH1 [1], premier hyperthermo-piezophile strict, a relancé la question de l’adaptation aux fortes pressions. En effet, cette nouvelle archée n’est capable de croître que sous hautes pressions (optimum de croissance : 98 °C et 520 bars) et supporte des pressions pouvant atteindre 1500 bars.
Je présenterai l’approche choisie basée sur une étude comparative de deux protéines modèles ainsi que les résultats préliminaires obtenus. L’effet de la pression sur les malate déshydrogénases (MalDH) et les glyoxylate réductases (GR) est étudié aussi bien d’un point de vue structurale qu’enzymatique. Le suivi de l’activité enzymatique est effectué grâce à un spectrophotomètre couplé à une enceinte pression disponible au sein de la plateforme Haute Pression de l’IBS. L’étude structurale est effectuée par cristallographie des rayons X grâce à une cellule à enclume diamant [2].
Les résultats préliminaires indiquent une différence de comportement entre les enzymes issues d’organismes isolés à la surface et celles issues d’organismes des profondeurs. Cette différence suggère que les enzymes issues d’organismes des fonds marins sont adaptées pour fonctionner de manière optimale sous hautes pressions. [1] Xiang Zeng, et al.,. The ISME journal. 2009, 3 :873-876. [2] Fourme et al., Curr Opin Struct Biol. 2012, 22(5):636-42

3c - Cristallo sous conditions extrêmes – Mercredi 3 juillet 10h30-12h00
- 69 -
O33 - Structure cristallographique de la neuroglobine sous pression
Nathalie Colloc’h1, Eric Girard2, Roger Fourme3, Anne-Claire Dhaussy4, Thierry Prangé5,
Beatrice Vallone6 1ISICT UMR 6301 CNRS – CEA – Université de Caen, centre Cyceron, Caen,, 2IBS, UMR 5075 CEA-CNRS-UJF, Grenoble, 3Synchrotron Soleil, Gif-sur-Yvette, 4CRISTMAT, ENSICAEN, Caen, 5LCRB UMR 8015 CNRS – Université Paris Descartes, 6Department of Biochemical Sciences, University of Rome La Sapienza, Rome, Italie
La neuroglobine (Ngb) est une globine impliquée dans la protection des neurones contre l’hypoxie et surexprimée dans les glioblastomes qui sont des tumeurs cérébrales très agressives. La Ngb participerait aux mécanismes de défense de ce type de cancer pour survivre dans des environnements hypoxiques. La Ngb est également surexprimée dans le cerveau des mammifères marins vivant en profondeur, donc dans un environnement sous pression et hypoxique.
La structure de la Ngb murine a été déterminée dans les formes deoxy et liée au monoxyde de carbone, révélant la présence d’une large cavité interne impliquée dans la transition structurale entre les deux formes [1,2]. La pression permettant de piéger des sous-états conformationnels de haute énergie [3], nous avons donc déterminer la structure de la Ngb sous haute pression hydrostatique pour espérer atteindre des sous-états impliqués dans son fonctionnement.
Les données de diffraction ont été enregistrées sur la ligne CRISTAL du synchrotron SOLIEL sur des cristaux de Ngb murine placés dans une cellule à enclume diamant soumis à une pression élevée. L’analyse de la courbe de compressibilité a clairement révélé qu’il existait une transition aux alentours de 300 MPa. Nous avons enregistré trois jeux complets de diffraction, l’un à pression ambiante, le deuxième avant la transition (270 MPa) et le troisième proche de la transition (310 MPa). La résolution des différents jeux de donnes est de 2 Å, ce qui permet d’analyser les différences.
La différence la plus notable est la grande diminution du volume de la cavité interne dans les structures sous pression, avec une rupture de la connexion entre l’hème et l’arrière de la cavité, qui serait un site réservoir pour l’oxygène. Il semble que les structures sous pression correspondent à un intermédiaire entre la structure deoxy et la structure en présence de monoxyde de carbone. [1] Vallone et al., Proteins, 2004, 56, 85-92. [2] Vallone et al., Proc. Natl. Acad. Sci. USA, 2004, 101, 17351-17356. [3] Fourme et al., Ann. Rev. Biophys., 2009, 38, 53-171.

3c - Cristallo sous conditions extrêmes – Mercredi 3 juillet 10h30-12h00
- 70 -
O34 - Haute Pression et Cristallographie des Matériaux Moléculaires à Conversion de Spin.
Philippe Guionneau1, Patrick Rosa1, Helena J. Shepherd2, Samir Matar1, Vincent
Legrand3, Marie-Hélène Lemée-Cailleau3, Stanislav Pechev1, Alain Marbeuf4, P. Négrier4,
Laure Vendier2, Gabor Molnar2, Jean-François Létard1 1 Univ. de Bordeaux, ICMCB 2 Univ.de Toulouse, LCC 3 ILL, Grenoble 4 Univ. de Bordeaux, LOMA
Le comportement sous haute pression des matériaux moléculaires à conversion de
spin apparaît complexe et même surprenant à bien des égards [1]. En effet, si l’influence de l’application d’une pression sur une conversion de spin semble bien comprise et anticipée à l’échelle de la molécule, il n’en est rien à l’échelle du cristal. D’ailleurs, la plupart, voire la totalité, des propriétés de conversion de spin sous pression qui apparaissent non conformes aux prédictions, atypiques ou inédites sont généralement attribuées à une origine structurale. Encore faut-il le prouver et comprendre les phénomènes mis en jeu, afin, en retour, d’utiliser les relations structure-propriétés dans le développement de ces matériaux piézo-commutables.
Nous montrerons comment l’investigation cristallographique en conditions extrêmes
a récemment permis de déterminer les diagrammes de phase (pression-température-lumière) de complexes moléculaires à conversion de spin. L’accent sera porté sur la complémentarité des approches d’investigation structurale sous pression in situ utilisées : diffraction X sur monocristal [2, 3], diffraction des neutrons sur poudre [4] y compris avec couplage pression-température [5] et approches théoriques [6].
[1] P.Guionneau and E. Collet in Spin-Crossover Materials: Properties and Applications, John Wiley & Sons Ltd, Oxford UK, (2013), Chapter 20, pp 507 -526.
[2] H.J. Shepherd et al. Angew. Chem. Int. Ed. 51, 3910-3914 (2012). [3] H.J. Shepherd et al. Phys. Chem. Chem. Phys., 14, 5265-5271 (2012). [4] V. Legrand et al. J. Appl. Cryst, 47, 637-640 (2008). [5] V. Legrand et al. soumis, (2013). [6] A. Marbeuf et al. Chemical Physics, 420, 25–34 (2013).
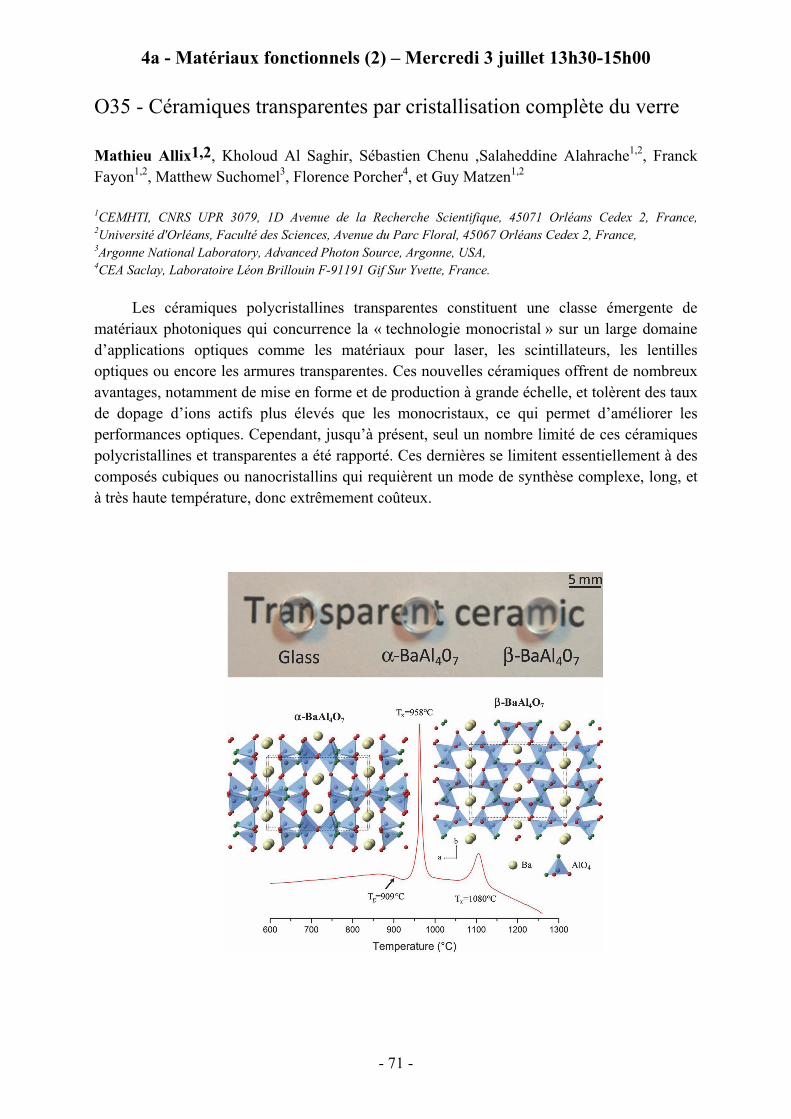
4a - Matériaux fonctionnels (2) – Mercredi 3 juillet 13h30-15h00
- 71 -
O35 - Céramiques transparentes par cristallisation complète du verre
Mathieu Allix1,2, Kholoud Al Saghir, Sébastien Chenu ,Salaheddine Alahrache1,2, Franck Fayon1,2, Matthew Suchomel3, Florence Porcher4, et Guy Matzen1,2 1CEMHTI, CNRS UPR 3079, 1D Avenue de la Recherche Scientifique, 45071 Orléans Cedex 2, France, 2Université d'Orléans, Faculté des Sciences, Avenue du Parc Floral, 45067 Orléans Cedex 2, France, 3Argonne National Laboratory, Advanced Photon Source, Argonne, USA, 4CEA Saclay, Laboratoire Léon Brillouin F-91191 Gif Sur Yvette, France.
Les céramiques polycristallines transparentes constituent une classe émergente de
matériaux photoniques qui concurrence la « technologie monocristal » sur un large domaine d’applications optiques comme les matériaux pour laser, les scintillateurs, les lentilles optiques ou encore les armures transparentes. Ces nouvelles céramiques offrent de nombreux avantages, notamment de mise en forme et de production à grande échelle, et tolèrent des taux de dopage d’ions actifs plus élevés que les monocristaux, ce qui permet d’améliorer les performances optiques. Cependant, jusqu’à présent, seul un nombre limité de ces céramiques polycristallines et transparentes a été rapporté. Ces dernières se limitent essentiellement à des composés cubiques ou nanocristallins qui requièrent un mode de synthèse complexe, long, et à très haute température, donc extrêmement coûteux.

4a - Matériaux fonctionnels (2) – Mercredi 3 juillet 13h30-15h00
- 72 -
O35 - Céramiques transparentes par cristallisation complète du verre Nos récents travaux démontrent la possibilité d’obtenir simplement des céramiques
polycristallines transparentes par cristallisation complète d’un verre de même composition, malgré des cristaux de taille micrométrique et de symétrie non cubique (donc non isotrope). De plus, la cristallisation à partir du verre permet de synthétiser de nouveaux matériaux grâce aux températures de cristallisation relativement basses. Ceci est démontré dans le cas d’une nouvelle composition, BaAl4O7, possédant deux polymorphes orthorhombiques et montrant une haute transparence dans les domaines visible et proche infrarouge [1,2]. Les structures cristallographiques de ces composés ont été déterminées ab initio à partir de données de diffraction de poudre. A partir des structures ainsi établies, les indices de réfraction ont été calculés, permettant de remonter à la biréfringence des matériaux et de discuter les propriétés de transparence en fonction des structures cristallines établies et des microstructures observées. Dernièrement, ce même procédé de synthèse a été appliqué sur de nouvelles compositions d’aluminosilicates de strontium, permettant de mettre en évidence l’existence de nouvelles solutions solides qui ont été élaborées sous forme de matériaux polycristallins hautement transparents. [1] M. Allix, Highly Transparent BaAl4O7 Polycrystalline Ceramic Obtained by Full Crystallization from Glass, Advanced Mater., 24 5570 (2012) [2] M. Allix, Verres, vitrocéramique et céramiques transparentes d’aluminates, Brevet (dépôt en France n°1161025 décembre 2011 – Extension internationale mars 2012). [3] S. Alahraché, Perfectly transparent Sr3Al2O6 polycristalline ceramic elaborated from glass crystallization, Chemistry of Materials, submitted.

4a - Matériaux fonctionnels (2) – Mercredi 3 juillet 13h30-15h00
- 73 -
O36 - Matériaux hybrides à base d’argent et d’acides phosphoniques et carboxyliques : exploration de nouvelles architectures à propriétés bactéricides. Jean-Michel Rueff1, Olivier Perez1, Marion Galmiche1, Hélène Couthon-Gourvès2, Mathieu Berchel2, Paul-Alain Jaffrès2, Gary Hix3
1 CRISMAT, CNRS UMR 6508, ENSICAEN-Caen; 2 Université de Brest, CNRS, UMR 6521, CEMCA; 3 Nottingham Trent University – UK.
Les sels, métaux ou nanoparticules à base d’argent présentent des propriétés bactéricides, dues aux ions argent qu’ils libèrent, exploitées dans le domaine médical (pansements, instruments chirurgicaux). Nos récents travaux ont montré qu’il était possible de synthétiser de nouveaux matériaux hybrides à base d’argent capables de relarguer des ions Ag+ en solution aqueuse conduisant alors à un effet bactéricide sur six souches bactériennes et tout particulièrement sur les bactéries Gram négatives (A. aeruginosa, E. coli)[1]. Or, la dimensionnalité (1D, 2D ou 3D) et la structure d’un matériau hybride, pour un métal donné, est aisément contrôlables via la nature des briques organiques ou/et le milieu réactionnel (pH, température). Dès lors notre objectif fut d’explorer l’influence de la dimensionnalité et la structure du matériau final sur le relargage d’argent. A cette fin, nous avons réalisé des hybrides à l’argent présentant des architectures originales à partir de molécules homo[2] ou hétéropolyfonctionelles [3] possédant des groupes réactifs du type acide phosphonique ou acides phosphoniques et acides carboxyliques liés à un squelette rigide ; différentes conditions expérimentales furent appliquées (pH, température). La théorie HSAB (Hard and Soft Acids and Bases theory) a été appliquée afin d’orienter nos synthèses et obtenir des matériaux à charpentes optimisées pour contrôler le relargage d’argent. Cette stratégie a permis d’obtenir plusieurs matériaux dont les structures ont été caractérisées par diffraction de rayons-X sur monocristal. La synthèse, la structure et la stabilité de matériaux hybrides polyfonctionnels à base d’argent ainsi que la quantification du relargage de sel d’argent en solution aqueuse seront présentés. [1] Berchel, M.; Le Gall, T.; Denis, C.; Le Hir, S.; Quentel, F.; Elléouet, C.; Montier, T.; Rueff, J.M.; Salaün, J.Y.; Haelters, J.P.; Lehn, P.; Hix, G.B.; Jaffrès, P.A. New J. Chem. 35 (2011) 1000. [2] Rueff, J.M.; Perez, O.; Pautrat, A.; Barrier, N.; Hix, G.; Hernot, S.; Couthon-Gourvès, H.; Jaffrès, P.A. Inorg. Chem. (2012), 51, 10251-10261. [3] A) Rueff, J.M.; Caignaert, V.; Chausson, S.; Leclaire, A.; Simon, C.; Perez, O.; le Pluart, L.; Jaffrès, P.A. Eur. J. Inorg. Chem. (2008) 4117. b) Rueff, J.M.; Perez, O.; Simon, C; Couthon-Gourvès, H.; Lorilleux, C.; Jaffrès, P.A. Cryst. Growth Des. 9 (2009) 4262.

4a - Matériaux fonctionnels (2) – Mercredi 3 juillet 13h30-15h00
- 74 -
O37 - Crystal and Magnetic Structure of LiMBO3 (M=Fe, Co, Mn)
Liang Tao1, Gwenaëlle Rousse
2, Christian Masquelier
1
1 LRCS, CNRS UMR7314, Université de Picardie Jules Verne, 33 rue Saint Leu, 80039
Amiens, France 1, 2 IMPMC UMR 7590 CNRS-Université Pierre et Marie Curie, 4 Place Jussieu, 75252 Paris
Cedex 05, France 2
LiMBO3 (M = Fe, Co, Mn) compositions, especially LiFeBO3, have been spotted as potential interesting cathode materials for Li-ion battery [1-6] due to the light and small triangular oxyanion (BO3)
3- which would allow in theory for LiFeBO3 to reach a capacity of 220mAh/g for the extraction of Li+ at ~2.8 V vs. Li.
The crystallographic unit cells of LiMBO3 (M = Fe, Mn, Co) are usually described in monoclinic C2/c symmetry except for one polymorph of LiMnBO3 described in the hexagonal (P-6) space group. Up to now, three structural models have been proposed for the monoclinic form of LiFeBO3. Pure LiFeBO3 was prepared and analysed by state of the art synchrotron X-Ray diffraction and PDF data, confronted with 57Fe Mossbauer, Neutron Powder Diffraction (NPD) and 6Li-NMR. All the results [7] suggested a new model for LiFeBO3: Li has two partially occupied sites and Fe atom either vibrates along the long axis of ellipsoid[001], or randomly distribute inside the anisotropic thermal motion ellipsoid of Fe atom. Similar model was tested against NPD data of LiCoBO3 and LiMnBO3 recorded at D2B ILL Grenoble.
Both temperature dependent magnetic susceptibility measurements and low temperature NPD (D20 ILL Grenoble) experiments were carried out. The results indicate the formation of an intermediate incommensurately ordered phase on cooling, followed by formation of commensurate long-range antiferromagnetic order below TN2 = 10K for LiMnBO3, TN2 = 25 K for LiFeBO3, and TN2 = 11 K for LiCoBO3. [1] V. Legagneur et al., Solid States Ionics, 2001, 139 (1-2), 37. [2] Y.Z. Dong et al., Electrochimica Acta, 2008, 53 (5), 2339 [3] A. Yamada et al., Advanced Materials, 2010, 22 (32), 3583. [4] A.Yamada et al., J. Mater. Chem., 2011, 21 (29), 10690 [5] J. C. Kim et al., J. Electrochem. Soc., 2011, 158 (3), A309 [6] Y. Janssen et al., J. Am. Chem. Soc., 2012, 134, 12516 [7] L. Tao, B. C. Melot, G. Rousse, D. Hanzel, G. Mali, J. R. Neilson, T. M. McQueen, R. Dominko, S. Levasseur, C. Masquelier; submitted, March 2013

4a - Matériaux fonctionnels (2) – Mercredi 3 juillet 13h30-15h00
- 75 -
O38 - Matériaux polymériques à conversion de spin de type [Fe(R-trz)3]·Xn: Comportement structural et microstructural
Arnaud Grosjean1, Nathalie Daro1, Cindy Mauriac1, Céline Etrillard1, Brice Kauffmann2, Philippe Négrier3, Denise Mondieig3, Jean-François Létard1 et Philippe Guionneau1. 1 CNRS, Univ. Bordeaux, ICMCB, UPR 9048, 87 avenue du Dr A. Schweitzer, F-33608 Pessac 2 European Institute of Chemistry and Biology, 2 Rue Robert Escarpit, 33607 Pessac Cedex 3 CNRS, Univ. Bordeaux, LOMA, UMR 5798, 351 cours de la libération, F-33400 Talence E-mail: [email protected] ; [email protected]
Les métaux de transition allant de 3d4 à 3d7 sont connus pour former des complexes pouvant avoir deux configurations électroniques différentes, appelés états Bas Spin et Haut Spin, en fonction de leurs environnements cristallins. Dans certains cas ces composés peuvent passer d’un état à un autre en fonction de conditions extérieures comme la température, la pression, une irradiation lumineuse, l’application d’un champ magnétique, ou encore sous l’effet d’une désolvatation : c’est le phénomène de la conversion de spin. Il a déjà été démontré que les relations structure-propriétés ont un rôle crucial dans la compréhension et le développement de ce type de matériaux [1].
Parmi tous les matériaux à conversion de spin, la famille des complexes polymériques [Fe(Rtrz)3]Xn possède les caractéristiques au plus proche des pré-requis pour des applications industrielles [2]. En effet, ils peuvent présenter une transition de spin abrupte avec large hystérèse autour des conditions ambiantes de température et de pression. Cependant, bien que cette famille de composés soit connue depuis plus de vingt ans son comportement reste encore mal compris et, notamment, les propriétés structurales restaient à déterminer. En effet, jusqu’à présent, aucune structure cristalline n’avait été obtenue. Au cours de ce travail, un premier résultat basé sur des investigations par diffraction X sur monocristal [3] a permis de décrire de façon fiable les propriétés structurales d’un complexe de cette famille, confirmant d’ailleurs l’hypothèse de structure polymérique 1D admise jusqu’ici sur la base d’investigation sur poudre [4, 5]. Ce premier résultat a permis d’ouvrir la voie à de nombreuses études cristallographiques complètes sur les différents aspects du comportement structural de ces matériaux autour de la transition de spin. A ce jour, une véritable banque de données structurales sur les complexes [Fe(Rtrz)3]Xn est en cours de construction et permet déjà d’envisager des relations structures-propriétés [6]. [1] P. Guionneau et al., Top. Curr. Chem. 234, 97-128 (2004) ; Malcolm A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 [2] A. Bousseksou et al., Chem. Soc. Rev., 2011, 40, 3313–3335 ; G. Aromi et al., Coordination Chemistry Reviews 255 (2011) 485–546 [3] A. Grosjean et al., Chem. Commun., 2011, 47, 12382–12384 [4] A. Urakawa et al., J. Phys. Chem. C 2011, 115, 1323–1329 [5] A. Michalowicz et al., Chem. Mater. 1995, 7, 1833-1842 [6] A. Grosjean et al., Eur. J. Inorg. Chem. 2013 DOI: 10.1002/ejic.201201121

4b - Apériodicité, structure modulée – Mercredi 3 juillet 13h30-15h00
- 76 -
O39 - Structures naturelles apériodiques et multimacles icosaédriques
Bertrand Devouard1
1Aix-Marseille Université - CEREGE, BP80, 13545 Aix-en Provence Cedex 04
Cette contribution présente un florilège de minéraux ayant des structures apériodiques,
du tube à l'icosaèdre. Les principaux exemples sont des composés lamellaires, phyllosilicates, sulfosels ou des composés mixtes hydroxyde-sulfure (e.g., tochilinite) associant deux types de feuillets incommensurables. Ces structures apériodiques présentent des similarités avec les nanostructures de carbone, mais possèdent souvent un plus grnd degré d'organisation du fait de forces de liaison inter-feuillets plus importantes.
Les serpentines, de composition idéale Mg3Si2O5, présentent des structures "plates" (lizardite), tubulaires (chrysotile, e.g. [1]), côniques (certains "chrysotiles" synthétiques, e.g. [2]), modulées (antigorite), mais aussi des structures cylindriques polygonisées (serpentines polygonales [3, 4]) ou polyhédriques [5]. Les serpentines polygonales sont formées de 15 ou 30 secteurs de types lizardite et présentent des symétries globales d'ordre 5 [3, 4].
Les serpentines polygonales ont été décrites comme des macles multiples de lizardite [6]. De fait, des multi-macles "parfaites" de symétrie 5 sont possibles, telle la pentagonite [7] qui présente des macles cycliques à cinq individus. Le composé synthétique B60, quand à lui, peut former de spectaculaires macles multiples à symétrie icosaédrique [8]. Si de telles multi-macles icosaédriques ne sont pas (encore?) connues à l'état naturel, on notera qu'un minéral quasicristallin a été décrit récemment [9].
Dans le cas des serpentines polygonales, toutefois, il a été démontré par diffraction électronique et microscopie électronique à transmission que les différents secteurs sont délimités par des jonctions courbes qui sont des murs de dislocations partielles et qui génèrent un kaléidoscope de polytypes d'un secteur à l'autre [3, 4].
Certains sulfosels lamellaires synthétiques [e.g., 10] sont connus pour former des structures enroulées ou modulées semblables aux serpentines. La cylindrite et la franckéite sont deux sulfosels naturels de Pb, Sb, Sn, Fe qui présentent des structures cylindriques (centimétriques!) et modulées, respectivement [11]. Enfin, la tubulite, Ag2Pb22Sb20S53 [12], forme des tubes creux millimétriques qui sont une merveilleuse illustration de la complexité de ces structures minérales apériodiques.
[1] Devouard & Baronnet, Eur. J. Miner, 1995, 7, 835-846. [2] Amelinckx et al., Acta Cryst. A, 1996, 52, 850-878. [3] Baronnet et al., Phys. Chem. Minerals, 1994, 21, 330-343. [4] Baronnet & Devouard, Can. Mineralogist, 2005, 43, 513-542. [5] Baronnet et al., Amer. Mineralogist, 2007, 92, 687-690. [6] Chisholm, J. Phys. D, 1991, 24, 199-202. [7] Evans, Amer. Mineralogist, 1973, 58, 412-424. [8] Hubert et al., Nature, 1998, 391, 376-378. [9] Bindi et al., Amer. Mineralogist, 2011, 96, 928–931. [10] Bernaerts, 1997, PhD thesis, Univ. Antwerpen, Belgium. [11] Williams and Hyde, Phys. Chem. Minerals, 1988, 15, 521-544. [12] Möelo et al., Min. Mag. 2012, 76, 807-817.

4b - Apériodicité, structure modulée – Mercredi 3 juillet 13h30-15h00
- 77 -
O40 - Etude de la filiation structurale entre les phases icosaédriques F (Faces Centrées) et P (Primitives)
Marianne Quiquandon1 et Denis Gratias
2
1,2
LEM CNRS/ONERA ; 29 avenue de la division Leclerc, 92322 CHATILLON CEDEX Nous décrivons comment l’on peut passer d’une structure atomique quasicristalline
icosaédrique de type F (Faces Centrées), comme celle proposée par M. Quiquandon and D. Gratias [1], vers celle de type P (Primitive), proposée par C. Pay Gomez and S. Lidin [2]. L’une et l’autre peuvent, en effet, être obtenues avec des surfaces atomiques construites à partir du même polyèdre élémentaire proposé dès 1986 par C. L. Henley [3] et se déduisent l’une de l’autre par simple mise en ordre chimique de ces surfaces de façon analogue aux transformations ordre-désordre qu’on rencontre dans les alliages ordonnés simples.
Notre précédent modèle de structure atomique de type F [1] s’appuie à l’origine sur un
petit triacontaèdre avec des surfaces atomiques différentes entre les deux sites n et n’ de l’hyperespace adéquatement tronqués pour éviter la génération d’atomes à trop proche distance pour qu’il n’y ait pas de recrouvrement entre les deux surfaces. Dans le cas d’une structure P, les deux sites n et n’ doivent être occupés par une même surface atomique construite avec le polyèdre de Henley [3] qui satisfait à la fois le non recouvrement des surfaces atomiques aux courtes distances et la condition de fermeture. Reformulant en retour notre modèle de structure F en utilisant des surfaces atomiques construites à partir de ce polyèdre [3], nous montrons comment on peut alors passer de P à F par simple décoration chimique des cellules engendrées par la décomposition des surfaces atomiques des atomes proches. [1] M. Quiquandon and D. Gratias, Phys. Rev. B 74, 214205 (2006). [2] C. Pay Gomez and S. Lidin, Phys. Rev. B 68, 024203 (2003). [3] C. L Henley, Phys. Rev. B 34, 797 (1986).

4b - Apériodicité, structure modulée – Mercredi 3 juillet 13h30-15h00
- 78 -
O41 - L’homométrie à la lumière des faisceaux cohérents
Sylvain Ravy1, 1Synchrotron-SOLEIL, L’Orme des merisiers, Saint Aubin BP48, 911912 Gif-sur-Yvette Cedex, France
Introduite par Arthur L. Patterson en 1939 [1], l’homométrie décrit des structures cristallines ayant les mêmes distances interatomiques et donc des réflexions de Bragg de même intensité. Étendue à la diffusion diffuse, elle décrit des systèmes désordonnés qui ont statistiquement la même fonction de corrélation de paires. De telles structures apparaissent donc indistinguables par diffraction.
Depuis une quinzaine d’année, l’utilisation de faisceaux cohérents a permis de mesurer des diagrammes de diffraction qui présentent des franges d’interférence ou des tavelures (speckles) moyennées par les techniques classiques. Nous montrerons que la diffusion cohérente permet de résoudre en théorie certains cas d’homométrie de Bragg, ce qui est connu, mais qu’elle ouvre des voies pour traiter les cas d’homométrie de diffusion diffuse, ce qui l’est moins [3].
Grâce à suite de Rudin-Shapiro [2] : 1, 1, 1, -1, 1, 1, -1, 1, 1, 1, 1, -1, -1, -1, 1, -1, 1, 1, 1, -1, 1, 1, -1, 1, -1, -1, -1, 1, 1, 1, -1, 1, 1, 1, 1, -1, 1, 1, -1, 1…
dont la diffusion diffuse est similaire à celle d’un suite aléatoire alors qu’elle est déterministe, il est possible de manipuler indépendamment les fonctions de corrélation de quadruplets des fonctions de corrélation de paires. On peut ainsi montrer que l’analyse statistique des tavelures des diagrammes de diffraction cohérente permet de distinguer des ordres qui concernent des fonctions de corrélation d’ordre supérieur à 2.
La diffraction pourrait-elle, enfin, explorer au-delà des corrélations de paires ?
[1] A.L. Patterson, Nature (London) 143, 939 (1939); Phys. Rev. 65, 195 (1944) [2] http://oeis.org/A020985 [3] Sylvain Ravy, Submitted, (http://arxiv.org/abs/1303.3792)

4b - Apériodicité, structure modulée – Mercredi 3 juillet 13h30-15h00
- 79 -
O42 - L’impact des corrélations structurales à grande échelle pour la réactivité des solides : l’exemple des oxydes AO(ABO3)n Ruddlesden-Popper W. Paulus1, M. Ceretti1, O. Wahyudi2, A. Perrichon1, A. Villesuzanne3, D. Chernychov4, A. Bosak4
1 Université de Montpellier 2, Institut Charles Gerhard, UMR 5253, Montpellier 2 Université de Rennes 1, Instiut de Chimie UMR 6226, Rennes 3 ICMCB, Bordeaux 4 ESRF, Grenoble
Les oxydes des métaux de transition du type K2NiF4 montrent une haute mobilité de l’ion oxygène dès la température ambiante. Pour cette raison ils sont actuellement considérés comme membrane d’oxygène dans des futures piles à combustible type SOFC. Certains composés comme le (Pr/Nd)2NiO4+δ montrent des propriétés de conductivité ionique de l’oxygène très prometteuses déjà à température modérées et peuvent intercaler l’oxygène sur
des sites interstitiels qui peut aller jusqu’à =0.25 pour (Pr/Nd)2NiO4+δ. Par diffraction neutronique combinée avec des calculs de la DFT, nous avons montré
une délocalisation dynamique de l’oxygène apical à faible potentiel. Cette délocalisation est à l’origine de la formation des super-réseaux, impliquant des fortes corrélations entre ordre structural des atomes de oxygène apicaux et interstitiels, associé à des ordres de charge et/ou orbital du métal de transition. L’analyse de ces interactions est importante pour mieux comprendre leur réactivité chimique et notamment leur haute mobilité ionique de l’oxygène à basse température.
Nous allons focaliser sur le cas complexe du système Pr2NiO4+δ, qui montre à part d’une très forte délocalisation dynamique de l’oxygène apical, des corrélations structuraux au delà de 300Å pour la phase PrNiO4.25.
Nous discutons l’enjeu compétitif entre corrélations et instabilités structurales, provocant une dynamique de réseau modifiée via la création des modes mous, qui in fine peuvent promouvoir la mobilité de l’oxygène dès la température ambiante. [1] From T to T’-La2CuO4 via Oxygen Vacancy Ordered La2CuO3.5, M. Ikbel Houchati, M. Ceretti, C. Ritter and W. Paulus, Chem. Mater. 2012, 24, 3811-3815 [2] Lattice Dynamics to Trigger Low Temperature Oxygen Mobility in Solid Oxide Ion Conductors, W.Paulus, H. Schober, S. Eibl, M. Johnson, T. Berthier, O. Hernandez, M. Ceretti, M. Plazanet, K. Conder, C. Lamberti, J. Am. Chem. Soc. (2008) 130 (47) 16080-85 [3] On the role of lattice dynamics on low-temperature oxygen mobility in solid oxides: a neutron diffraction and first-principles investigation of La2CuO4+δ, Villesuzanne Antoine, Paulus Werner, Cousson, Alain, Hosoya Shoichi, Le Dreau Loic, Hernandez Olivier, Prestipino Carmelo, Houchati Mohamed Ikbel, Schefer Juerg, J Solid State Electrochemistry Volume: 15 Issue: 2 Pages: 357-366 (2011)

4c - Biologie structurale des génomes (interactions protéines/ADN, ARN Mercredi 3 juillet 13h30-14h40
- 80 -
O43 - Vers l’architecture du télomère de la levure bourgeonnante par biologie structurale intégrative. Yann-Vaï Le Bihan1, Béatrice Matot1, Olivier Piétrement2, Rachel Lescasse3, Javier Pérez4, Marie-Josèphe Giraud-Panis5, Patrick Weber6, Bianca Sclavi7, Sophie Zinn-Justin1, Eric Le Cam4, Eric Gilson5, Simona Miron1, and Marie-Hélène Le Du1. 1 Lab Biologie Structurale et Radiobiologie, CEA, CNRS et Univ Paris Sud, Gif-sur-Yvette. 2 Maintenance des Génomes, IGR, CNRS et Univ. Paris Sud, Villejuif. 3 Lab Télomère et Réparation du Chromosome, CEA, Fontenay-aux-roses. 4 Synchrotron SOLEIL, Gif-sur-Yvette. 5 IRCAN, Univ Nice CNRS INSERM, Nice. 6 PF6, Institut Pasteur CNRS, Paris. 7 LBPA, CNRS, ENS Cachan.
Les télomères constituent l’extrémité non codante des chromosomes linéaires chez les eucaryotes. Une multitude de données atteste de leur implication dans le cancer et le vieillissement. Leur raccourcissement progressif lors de la réplication de l'ADN conduit à la sénescence cellulaire, ce qui est considéré comme une barrière anti-tumorale et participe au vieillissement. Inversement, leur allongement par l'enzyme télomérase et la voie ALT est requis pour l’initiation et le maintien des cancers. Les mécanismes biologiques de maintien des télomères sont bien établis, mais manquent dramatiquement de données structurales. Pratiquement rien n'est connu sur le repliement approprié et la dynamique des assemblages macromoléculaires télomériques. Par conséquent, une question clé de la biologie moderne des télomères est de relier les fonctions associées aux télomères à la structure des complexes nucléoprotéiques qui constituent les extrémités des chromosomes.
Chez la levure, Rap1 lie étroitement l’ADN double-brin avec une fréquence moyenne de une protéine indépendante tous les 18 paires de base. Son interaction avec l'ADN induit une hypersensibilité au permanganate de potassium que nous avons attribuée à un résidu d’arginine conservé, et non pas à une fusion ou une altération locale de l'ADN (Le Bihan et al., 2013). Rap1 recrute des partenaires protéiques fonctionnels essentiels et spécifiques pour la régulation négative de l'allongement des télomères, pour la répression de la transcription, ou pour l'inhibition de la voie de réparation par jonction des extrémités non homologues (NHEJ). Rap1 est organisée en domaines globulaires connectés par de longues régions non structurées, ce qui suggère que ses fonctions multiples sont associées à un comportement dynamique favorisant la formation de structures quaternaires spécifiques. Nous avons utilisé une approche intégrée de méthodes complémentaires de biologie structurale (cristallographie, RMN, SAXS) pour construire les architectures de Rap1 et du complexe Rap1/DNA. Nos résultats révèlent un réarrangement structural de Rap1 lors de la fixation à l’ADN qui implique notamment une réorientation du domaine C-terminal par rapport au domaine Myb. Une structure cristallographique du domaine Myb en complexe avec un ADN double-brin permet d’identifier une boucle essentielle s’enroulant autour de l’ADN, qui contraint l’orientation du domaine C-terminal et affecte l’affinité de Rap1 pour l’ADN (Matot et al., 2012). Enfin, la combinaison des informations structurales à l’analyse de la liaison de Rap1 aux répétitions télomériques par AFM, nous permet de proposer un modèle d’assemblage de Rap1 aux télomères.

4c - Biologie structurale des génomes (interactions protéines/ADN, ARN Mercredi 3 juillet 13h30-14h40
- 81 -
O44 - Nouvelles informations sur le mécanisme de surenroulement de l'ADN obtenues par cryo-microscopie électronique d'une ADN gyrase complète. Julie Papillon1, Jean-François Ménétret1, Claire Batisse1, Reynald Hélye2, Patrick Schultz1, Noëlle Potier2, Valérie Lamour1,3 1 IGBMC, UMR7104 CNRS-UdS, U964 Inserm, Illkirch, France 2 Institut de Chimie de Strasbourg, UMR7177 CNRS-UdS, Strasbourg, France 3 Hôpitaux Universitaires de Strasbourg, Strasbourg, France
Les ADN topoisomérases de type 2 (Topo2A) remodèlent la topologie de l’ADN lors de la réplication, la transcription et la ségrégation des chromosomes. Ces enzymes modulaires catalysent le transport d’un ADN double brin à travers la coupure d’un autre duplex par un mécanisme d’ouverture successive de «portes» le long de son interface hétérodimérique. L’ADN gyrase bactérienne, une cible avérée pour les antibiotiques, est la seule Topo2A capable d’introduire des supertours négatifs dans l’ADN nécessitant la fixation de longs segments contigus d’ADN. Les données structurales morcelées de ces complexes multi-conformationnels laissent aujourd’hui de nombreuses questions mécanistiques en suspens et ne permettent pas toujours d’identifier les états conformationnels visés par les antibiotiques. Nous avons déterminé par cryo-microscopie électronique l’architecture complète de l’ADN gyrase de T. Thermophilus et son complexe avec un ADN long de 155pb bloqué par l’antibiotique ciprofloxacine, après caractérisation de ses complexes nucléoprotéiques de plus de 320kDa par spectrométrie de masse supramoléculaire. Ces premières données structurales sur l’ADN gyrase entière révèlent l’organisation quaternaire de ses domaines et le trajet de l’ADN autour de l’enzyme menant à la formation de surenroulement négatif. La conformation de l’enzyme en présence d’ADN et de la ciprofloxacine suggère que l’antibiotique bloque le transport de l’ADN dans les premières étapes du cycle catalytique. Cette étude montre également l’implication du domaine ATPase dans la capture et la stabilisation des croisements d’ADN avant translocation, un mécanisme qui peut être étendu à toutes les topoisomérases de type 2.

4c - Biologie structurale des génomes (interactions protéines/ADN, ARN Mercredi 3 juillet 13h30-14h40
- 82 -
O45 - Études structurales de la recombinaison homologue chez Deinococcus radiodurans Simone Pellegrino1, Jens Radzimanowski1, François Dehez2, Daniele de Sanctis1, Sean McSweeney1 and Joanna Timmins1,3 1European Synschrotron Radiation Facility, Groupe de Biologie Structurale, Grenoble,
2Université de Lorraine/CNRS, Nancy, 3Institut de Biologie Structurale, Groupe Infection
virale et Cancer, Grenoble.
Chez Deinococcus radiodurans, un organisme présentant une très forte résistance aux rayonnements ionisants, la principale voie impliquée dans la réparation des cassures double-brin est la voie de recombinaison homologue faisant intervenir une série de protéine Rec ayant pour objectif de charger la recombinase RecA sur l’ADN afin de catalyser l’échange de brins d’ADN. Nous avons utilisé des méthodes de biologie structurale et de biochimie in vitro pour caractériser la structure et les activités de plusieurs de ces protéines et notamment la protéine RecN et le complexe hétéro-héxamérique RecO-RecR. RecN joue un rôle essentiel dans les premières étapes de reconnaissance de cassures double-brin dans l’ADN, alors que le complexe RecOR reconnaît les jonctions simple brin-double brin et facilite le chargement de RecA sur l’ADN. Nous avons déterminé les structures cristallines de trois fragments de RecN et, avec l’aide d’une enveloppe de la protéine entière déterminée par SAXS (small-angle X-ray scattering), nous avons pu reconstituée une structure quasi atomique de cette protéine très allongée (300 Å de long) appartenant à la famille des SMC (Structural Maintenance of Chromosomes). Nous avons également étudié le rôle de la fixation et de l’hydrolyse de l’ATP dans l’activité et l’oligomérisation de RecN et identifié les régions de RecN impliquées dans l’interaction avec l’ADN. Nous avons utilisé une approche similaire pour RecO-RecR : nous avons déterminé deux structures cristallines du complexe révélant un changement de conformation permettant la fixation de l’ADN au centre de ce complexe. Cette hypothèse a été confirmé par l’étude du complexe par SANS (small-angle neutron scattering), et les résidus impliqués dans la fixation de l’ADN simple brin ont été identifiés par l’utilisation de simulations de dynamique moléculaire et de mutagénèse dirigée. Ces travaux nous permettent de mieux comprendre le rôle et le mécanisme d’action de ces protéines dans la voie de recombinaison homologue.

4d - ReNaFoBis – Mercredi 3 juillet 14h40-15h00
- 83 -
O46 - Réseau National de Formation Doctorale en Biologie Structurale Intégrative
Claudine Mayer
Institut Pasteur, Université Paris
Les défis actuels de la biologie structurale nécessitent de former des étudiants ayant une culture biologique de plus en plus vaste en complément d’une formation initiale suffisante en biologie, physique, chimie, et mathématiques. En effet, la compréhension des systèmes biologiques impose d’intégrer des méthodes et des outils aux interfaces de plusieurs disciplines. Inclure un tel ensemble de connaissances dans un cursus Licence-Master est incompatible avec son format actuel, et conduirait à une formation probablement hors de portée de la majorité des étudiants en biologie à l’université. Depuis de nombreuses années, la communauté française de biologie structurale a eu à faire face à de telles difficultés, les défis à relever ne pouvant le plus souvent pas être abordés au sein d’une unique université.
Il est aujourd’hui plus que nécessaire de développer des collaborations et des échanges entre les universités françaises dans le cadre de l'enseignement de la biologie structurale intégrative afin d’assurer la formation et le recrutement de futurs chercheurs dans un niveau d’excellence international. Dans ce contexte, plusieurs représentants des équipes de recherche et/ou responsables de formation de cinq universités françaises (Grenoble, Marseille, Montpellier, Paris et Strasbourg) se sont associés pour mettre en place le Réseau National de Formation en Biologie Structurale Intégrée, RéNaFoBis. Soutenue par l’ITMO Bases Moléculaires et Structurales du Vivant, cette initiative s’appuie sur des collaborations définies au sein de la recherche nationale (projet FRISBI) et européenne (projet INSTRUCT). Nous présenterons, au cours de cet exposé, les objectifs et les actions en cours de RéNaFoBiS.

Roger Fourme – Mercredi 3 juillet 15h00-15h30
- 84 -
SP3 - Un hommage à Roger Fourme… Eric Girard1
1 Institut de Biologie Structurale J.-P. Ebel, UMR5075 CEA-CNRS-UJF-PSB, Grenoble, France.
Au cours de cette présentation, je reprendrai les grandes lignes de la carrière scientifique de Roger Fourme (1942-2012).
En particulier, je me focaliserai sur le dernier domaine de recherche que Roger avait initié en 2000 : les études sur les effets des hautes pressions hydrostatiques sur les macromolécules biologiques [1,2]. [1] R. Fourme, E. Girard, R. Kahn, A.-C. Dhaussy, I. Ascone, Annu. Rev. Biophys. 2009, 38, 153–171. [2] R. Fourme, E. Girard, K. Akasaka, Curr Opin Struct Biol 2012, 22, 636–642.

Conférence Grand Public à l’Université Bordeaux Segalen Mercredi 3 juillet 18h25-19h25
- 85 -
SP4 - Quasicrystals: Structures, properties and applications A.P. Tsai Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, Sendai 980-8577, Japan
Quasi-crystal (Qc) is no longer a unique structure matter since it has been confirmed as an equilibrium phase in a large number of alloys. It is clear that stability of stable Qcs can be understood within the framework of Hume-Rothery rules. Even more interesting, it is found that stable Qcs are a strict electron compound, which only formed for alloys with sharp valence electron concentration (e/a: electron-atom ratio). Actually, most stable Qcs were discovered on the basis of the e/a criterion. With these stable Qcs, structural analysis is allowed to study on a single grain sample.
Although few structural models have been proposed, they still suffered from considerable uncertainty due to topological and chemical disorder. Recently, understanding in structure has been highly improved in a binary Cd-Yb Qc. Furthermore, observed surface structures of quasicrystals by STM have been well interpreted by bulk structural model.
This talk will mainly deal with three topics; the first one is the Hume-Rothery rules for
stable Qcs, the second one is regarding the structure of a binary Cd-Yb Qc and the third one is the surface structures in relation to bulk structural models for Qcs.

Séance plénière III – Jeudi 4 juillet 9h00-10h00
- 86 -
SP5 - Les quasicristaux : structure atomique et dynamique.
Marc de Boissieu SIMaP, CNRS, Université de Grenoble, Saint Martin d’Hères, France
La découverte des quasicristaux par Dan Shechtman a été une révolution en cristallographie qui a profondément modifiée notre compréhension des matériaux ordonnés. En effet les quasicristaux sont des matériaux ordonnés à grande distance (comme le montre leur diagramme de diffraction avec des pics de Bragg) mais non périodiques. Après une introduction sur les quasicristaux, je présenterai les résultats récents obtenus dans le système binaire CdYb, pour lequel la structure atomique de cette phase icosaédrique est maintenant bien comprise. Est ce que l’ordre quasipériodique à grande distance induit de nouvelles propriétés physiques ? J’aborderai cette question en présentant des résultats sur la dynamique de ces matériaux : la dynamique de réseau et les phonons d’une part, et les phasons d’autre part, qui sont des fluctuations caractéristiques de l’ordre quasipériodique. Finalement j’aborderai la question des mécanismes qui peuvent stabiliser ces structures ordonnées et non périodiques.

5a - Approches multitechniques en cristallochimie – Jeudi 4 juillet 10h30-12h00
- 87 -
O47 - Combinaison des données de diffraction des électrons et de diffraction des rayons X poudre: que peut-on en attendre?
P. Boullay1, N. Barrier1, S. Malo1, O. Perez1 and L. Palatinus2
1CRISMAT, CNRS UMR 6508, 6 Bd du Maréchal JUIN 14050 CAEN Cedex, France 2Institute of Physics of the AS CR, v.v.i. Na Slovance 2, 182 21 Prague, Czechia.
Déterminer la structure de matériaux complexes et/ou de taille nanométrique est un enjeu important en Science des Matériaux, essentiel pour définir une stratégie d’élaboration de matériaux et connaître l’influence des mécanismes structuraux sur les propriétés. L’heureux possesseur d’un monocristal de bonne qualité n’aura vraisemblablement pas à se soucier d’une «approche multitechnique» pour déterminer la structure du composé nouvellement synthétisé. De même, un composé pulvérulent monophasé de difficulté structurale moyenne ne devrait guère poser de problème aux outils de résolution de structures applicables aux diagrammes de diffraction de poudres. Pour certains matériaux de complexité structurale élevée (maille de très grand volume, phases apériodiques, présence de désordre, …) et/ou de faible volume diffractant (films minces, nanoparticules, …), une approche multitechnique prend, en revanche, tout son sens.
La microscopie électronique en transmission a depuis longtemps fait ses preuves en tant que technique complémentaire de la diffraction des rayons X sur poudre. Souvent première étape vers la résolution structurale, elle permet d’apporter des informations sur les paramètres de maille, la symétrie et/ou la présence de motifs structuraux (voir [1-3] pour quelques exemples). Loin de se limiter à un simple aspect qualitatif, l’émergence de la technique de précession des électrons [4] en mode dit « tomographique » [5] combiné à la méthode de résolution par charge-flipping [6] permet d’envisager une utilisation plus quantitative des données de diffraction des électrons aboutissant à un premier modèle structural pouvant être affiné à l’aide du diagramme de diffraction de poudres. Dans le cadre de cet exposé, nous présenterons un aperçu de ce qui peut être obtenu par précession des électrons dans le cas de la résolution de structures modulées incommensurables avec
notamment l’exemple des composés -CaTe2O5 [7] et Bi5Nb3O15 [8]. Nous verrons que la combinaison des données de diffraction des rayons X, des neutrons et des électrons peut être réalisée (JANA2006 [9]) et nous en discuterons l’intérêt dans le cadre d’un affinement structural. [1] P. Boullay et al, J. Solid State Chem. 1997, 132, 239-248. [2] M. Huvé et al, Chem. Mater. 2004, 16, 2628–2638. [3] L.B. McCusker et al, Z. Kristallogr. 2013, 228, 1-10. [4] R. Vincent et al, Ultramicroscopy 1994, 53, 271-282. [5] U. Kolb et al, Ultramicroscopy 2007, 107, 507. [6] G. Oszlányi et al, Acta Cryst. A 2004, 60, 134-141. [7] N. Barrier, Habilitation à Diriger des Recherches 2012, Université de Caen-Basse Normandie. [8] P. Boullay et al, Inorg. Chem. 2013, 52, 6127–6135. [9] V. Petricek et al, Institute of Physics, Praha, Czech Republic, 2006.

5a - Approches multitechniques en cristallochimie – Jeudi 4 juillet 10h30-12h00
- 88 -
O48 - Apport des calculs ab initio à la résolution des structures cristallines : intérêt de la modélisation en spectrométrie RMN et Mössbauer Florent Boucher* Institut des Matériaux Jean Rouxel (IMN), Université de Nantes, CNRS, 2 rue de la Houssinière, BP 32229, 44322 Nantes Cedex *[email protected]
Concernant la caractérisation des matériaux par diffraction des rayons X, nous
observons depuis plusieurs années un engouement marqué pour les études sur poudres délaissant de fait l’intérêt porté aux approches sur mono cristaux. Cette mutation dans le domaine des techniques de diffraction, essentiellement guidée par une évolution des problématiques matériaux : étude in situ, matériaux polycristallins ou nanostructurés, matériaux d’électrodes, n’est pas sans conséquences sur la qualité des données que l’on peut en extraire. Bien que les diffractomètres et les détecteurs soient de plus en plus performants, les informations structurales issues des études de diffraction sur poudre sont par nature entachées d’une plus grande imprécision. Le cristallochimiste ou le chimiste des matériaux doit en être conscient lorsqu’il cherche à interpréter telle ou telle propriété à partir de l’analyse des longueurs de liaisons ou de l’étude des environnements atomiques.
Cette difficulté peut en partie être contournée en combinant plusieurs techniques de
caractérisations telles que la diffraction électronique ou neutronique ou des techniques de sondes locales telles que la RMN, le Mössbauer ou la spectrométrie d’absorption des rayons X. Dans cet exposé, nous montrerons comment les calculs de structure électronique ab initio (basés sur la théorie de la fonctionnelle de la densité, DFT) sont également complémentaires de toutes ces approches. Les optimisations de géométrie permettent par exemple d’améliorer la description des distances lorsque qu’existent, au sein d’une même structure, des contrastes marqués entre atomes lourds et légers ou que des atomes d’hydrogène doivent être positionnés. L’accès aux composantes du tenseur de gradient de champs électrique autorise en parallèle une corrélation directe aux données de la RMN (Cq) et du Mössbauer (éclatement quadripolaire). Finalement, le calcul des constantes d’écran ou des densités de charge au noyau permet la comparaison avec les valeurs de déplacement chimique (RMN) ou de déplacement isomérique (Mössbauer). Nous illustrerons cela au travers de quelques exemples issus d’études récentes.

5a - Approches multitechniques en cristallochimie – Jeudi 4 juillet 10h30-12h00
- 89 -
O49 - Nanorods de niobates et niobo-tantalates de potassium épitaxiés sur substrats monocristallins : étude par diffraction des rayons X, diffraction des électrons en précession et tomodiffraction des électrons Valérie Demange1, Vincent Dorcet1, Lukas Palatinus2, Philippe Boullay3, Anne Waroquet1, Noha Hakmeh1, Quentin Simon4, Maryline Guilloux-Viry1
1 Institut des Sciences Chimiques de Rennes, UMR 6226 CNRS/Université de Rennes 263,
avenue Général Leclerc, Campus de Beaulieu, 35042 Rennes, France, 2
Institute of Physics, Department of Structure Analysis, Cukrovarnicka 10, 16253 Praha 6, Czech Republic, 3
Laboratoire de Cristallographie et Sciences des Matériaux, CRISMAT – UMR 6508 CNRS / ENSICAEN, 6, boulevard du Maréchal Juin, 14050 Caen Cedex 4, France, 4
Institut de Chimie de la Matière Condensée de Bordeaux, ICMCB – UPR 9048 CNRS, 87, Avenue du Docteur Schweitzer, 33608 PESSAC Cedex, France.
Notre étude porte sur la synthèse par ablation laser pulsé, sur des substrats monocristallins de (100)SrTiO3 et de saphir R, de niobates et niobo-tantalates de potassium et la caractérisation de leur structure et microstructure. L'augmentation du taux de potassium relativement à celui du niobium, ou niobium et tantale, permet d'obtenir successivement les phases référencées dans le diagramme de phases KNbO3-Nb2O5 (i.e. K(Nb,Ta)3O8, K3(Nb,Ta)7O19, K4(Nb,Ta)6O19, K6(Nb,Ta)10O30, K(Nb,Ta)O3) [1]. Les composés anisotropes présentent pour la plupart une croissance sous forme de nanorods épitaxiés. Parmi eux, nous avons étudié plus spécifiquement les composés de structure-type bronze de tungstène quadratique dont la structure a été résolue par diffraction électronique en précession et tomodiffraction des électrons, techniques couplées à la microscopie électronique en transmission. Les relations d'épitaxie de ces nanorods ont été déterminées par diffraction des rayons X sur un diffractomètre 4-cercle.
Structure après affinement d'un nanorod K5.96Nb10.56O29.2
à partir des données de diffraction électronique en précession. [1] A. Reisman, F. Holtzberg. J. Am. Chem. Soc. 1955, 77, 2115-1955.

5a - Approches multitechniques en cristallochimie – Jeudi 4 juillet 10h30-12h00
- 90 -
O50 - Solides hybrides à base de ligands d’origine naturelle : résolution structurale via une approche multitechnique « diffraction / RMN » Lucy Cooper1, Thomas Devic1, Nathalie Guillou,1 Charlotte Martineau,1 Christian Serre1
1 Institut Lavoisier, UMR CNRS 8180, Université de Versailles St-Quentin-en-Yvelines, 45 avenue des États-Unis, 78035, Versailles, France
Les « Metal Organic Frameworks » (MOFs) sont des polymères hybrides cristallisés poreux formés de ligands organiques et de briques inorganiques interagissant par liaisons fortes [1]. Ils sont souvent obtenus à partir de poly-carboxylates et de cations M(II) ; ce système conduit à des cristaux, généralement peu stables à l’hydrolyse. A l’inverse, les cations plus chargés (M(III), M(IV)) et les ligands plus basiques, conduisent souvent à des matériaux plus stables, mais moins bien cristallisés. Dans cette étude, nous nous sommes intéressés à la réactivité du système Zr(IV) / acide gallique, un carboxyphénolate d’origine naturelle. La complexité des solides obtenus (incertitude sur l’état de protonation des ligands et diversité des polyèdres de coordination) rend difficile le processus de détermination structurale uniquement à partir de la diffraction sur poudre. Nous avons donc choisi une stratégie alternative, combinant les deux techniques complémentaires que sont la diffraction et la Résonance Magnétique Nucléaire (RMN) à l’état solide.
Les données RMN 1H et 13C, en renseignant sur la nature, les modes de coordination et la multiplicité des ligands organiques, ont permis d’amorcer la résolution structurale par diffraction de deux des nombreuses phases pulvérulentes identifiées dans ce système. La première structure, obtenue en présence d’excès d’acide salicylique, Zr[C6H2O3CO2][C6H4OCO2][HCON(CH3)2], peut se décrire à partir de chaînes d’antiprismes à base carrée connectés par arêtes. Dans le second composé synthétisé à plus haute température, l’acide gallique perd sa fonction carboxylique, pour former Zr[C6H3O3]2.N(CH3).
La synthèse, la structure, les caractérisations physicochimiques et l’approche conjointe « diffraction sur poudre / RMN du solide » utilisée seront présentées.
Fig. 1 : Structures des deux composés obtenus avec (a) et sans acide salicylique (b).
[1] G. Férey, Chem. Soc. Rev, 2008, 37, 191.

5b - Densité électronique, modélisation – Jeudi 4 juillet 10h30-12h00
- 91 -
O51 - Propriétés électrostatiques de molécules d’intérêt pharmaceutique Nour Eddine Ghermani,1,2 Nouha El Hassan,1 Aziza Ikni,1 Xiaoxuan Shi,1 Jean-Michel Gillet1 et Anne Spasojevic-de Biré1 1Laboratoire Structures, Propriétés et Modélisation des Solides (SPMS) UMR CNRS 8580, Ecole Centrale Paris, 1, Grande Voie des Vignes, 92295 Châtenay-Malabry, France. 2Institut Galien Paris Sud, UMR CNRS 8612, Université Paris Sud, Faculté de Pharmacie, 5, rue Jean-Baptiste Clément, 92296 Châtenay-Malabry, France.
Les interactions moléculaires sont régies par les propriétés électrostatiques tells que les charges et les moments atomiques, le potentiel et le champ électrostatique. Ces propriétés sont dérivées de la distribution électronique de la molécule isolée ou à l’intérieur d’un cristal. Deux méthodes existent pour la détermination de la distribution électronique : une méthode expérimentale basée sur la diffraction X haute résolution et une méthode théorique basée sur les calculs quantiques Hartree-Fock ou DFT (fonctionnelle de densité). Ces deux méthodes donnent des résultats très comparables. Depuis plus de dix ans, nous avons appliqué ces méthodes à différentes molécules pharmaceutiques qui ont dans le domaine de la recherche et de la santé en général un intérêt certains.
Dans cette présentation orale, nous commencerons par donner les bases de calculs des
propriétés électrostatiques avant de présenter nos résultats sur les molécules pharmaceutiques que nous avons étudiées.

5b - Densité électronique, modélisation – Jeudi 4 juillet 10h30-12h00
- 92 -
O52 - Ab initio Description of Nuclear Motion and Electron Correlation Effects on the Density Matrix of Crystals Alessandro Erba,a,* Cesare Pisani,a Matteo Ferrabone a and Roberto Dovesi a
a Dipartimento di Chimica, Università di Torino, via Giuria 5 10125, Torino, Italy * E-mail: [email protected] A large number of effects has to be accurately taken into account when comparing theoretical predictions with experimental determinations of the electron charge distribution of crystals. Within the framework of ab initio simulations, in this contribution we address two of them: thermal nuclear motion and dynamic electron correlation. Two schemes for the inclusion of nuclear motion effects are presented: (i) a fully ab initio technique for the determination of atomic anisotropic displacement parameters (ADP) of crystalline materials within the harmonic approximation to the lattice potential [1]; (ii) a harmonic Monte Carlo ab initio approach for the description of nuclear motion effects on the electronic density matrix of crystalline materials [2]: in the frame of the Born-Oppenheimer approximation, nuclear motions in crystals can be simulated rather accurately using a harmonic model. In turn, the electronic first-order density matrix can be expressed as the statistically weighted average over all its determinations each resulting from an instantaneous nuclear configuration. As regards electron correlation, the CRYSCOR program implements the MP2 method for crystals, reformulated according to the so-called local approach (namely LMP2) [3]. The program provides both the LMP2 correction to the Hartree-Fock energy and one-electron Density Matrix (DM) [4]. As a consequence, the correlation correction at LMP2 level to several DM-related quantities such as electron charge and momentum densities, Compton profiles, static structure factors, etc. can be evaluated. Such schemes have been developed within the formalism of all-electron atom-centered basis sets, one-electron Hamiltonians (Hartree-Fock, Density-functionaltheory, hybrids) and periodic boundary conditions, and implemented in the CRYSTAL and CRYSCOR programs for solid state quantum chemistry. [1] A. Erba, M. Ferrabone, R. Orlando and R. Dovesi, J. Comput. Chem., 34, 346 (2013). [2] C. Pisani, A. Erba, M. Ferrabone and R. Dovesi, J. Chem. Phys., 137, 044114 (2012). [3] C. Pisani, M. Schütz, S. Casassa, D. Usvyat, L. Maschio, M. Lorenz and A. Erba, Phys. Chem. Chem. Phys., 14, 7615 (2012) [4] D. Usvyat and M. Schütz, J. Phys.: Conf. Ser., Honorary issue Pisani, 117, 012027 (2008).

5b - Densité électronique, modélisation – Jeudi 4 juillet 10h30-12h00
- 93 -
O53 - Les liaisons halogène et chalcogène pour l’ingénierie cristalline et la chimie supramoléculaire
M. Brezgunova1, E. Aubert1, S. Dahaoui1, T.T.T. Bui,1 J. Lieffrig,2 S. Lebègue,1 C. Jelsch,1 P.
Fertey,3 C. Lecomte,1 J.G. Ángyán,1 M. Fourmigué,2 Enrique Espinosa1 1Laboratoire de Cristallographie, Résonance Magnétique et Modélisations (CRM2), UMR
CNRS 7036, Université de Lorraine, BP 70239, Bd. des Aiguillettes, 54506 Vandoeuvre-lès-Nancy, France. 2Institut des Sciences Chimiques de Rennes Université Rennes 1, UMR CNRS 6226 Campus
de Beaulieu, 35042 Rennes, France. 3Synchrotron Soleil, L’Orme des Merisiers Saint-Aubin, BP 48, 91192 Gif-sur-Yvette, France.
E-mail : [email protected]
La liaison halogène est une interaction attractive et directionnelle. Elle se forme entre un atome d’halogène, lié à un atome de carbone, et un autre atome d’halogène ou une base de Lewis (C–Hal…X, X = Hal, B). L’origine de cette interaction faible a été associée à l’anisotropie de la distribution électronique autour du noyau de l’halogène. Cette anisotropie conduit à des régions électrophiles (δ+) selon la direction C–Hal et à des régions nucléophiles (δ-) dans le plan perpendiculaire à cette direction. L’intensité et les aspects directionnels de la liaison halogène peuvent être aussi efficaces que ceux de la liaison hydrogène pour guider l’assemblage moléculaire. Malgré le fait que cette interaction puisse être mise à profit dans les domaines de l’ingénierie cristalline et de la chimie supramoléculaire, la caractérisation et la modélisation expérimentale de la liaison halogène ont été très peu explorées. Notre groupe a étudié la structure et la densité électronique cristalline des composés hexachlorobenzène (C6Cl6), hexabromobenzène (C6Br6), pentachlorophénol (C6Cl5OH) et pentabromophénol (C6Br5OH), ce qui a permis de mettre en évidence les régions électrophile/nucléophile autour des noyaux des atomes de chlore et de brome et les interactions que celles-ci réalisent.[1,2] Par ailleurs, la liaison chalcogène présente de nombreuses similitudes avec la liaison halogène. En effet, elle est également contrôlée par des interactions électrostatiques du type électrophile-nucléophile entre régions atomiques δ+ et δ-. Avec l’étude du composé C8H4O2Se nous avons prouvé le fort caractère directionnel de la liaison chalcogène Chal…X (X = Chal, Hal, B).[3]
Afin de les utiliser dans les domaines de l’ingénierie cristalline et de la chimie supramoléculaire, l’intensité électrostatique et l’énergie des liaisons halogène et chalcogène ont été caractérisées et comparées à celles des liaisons hydrogène dans les mêmes structures cristallines.[2,3] [1] (a) T. T. T. Bui, S. Dahaoui, C. Lecomte, G. R. Desiraju, E. Espinosa, Angew. Chem. Int. Ed., 2009, 48, 3838-3841; (b) E. Aubert, S. Lebègue, M. Marsman, T.T.T. Bui, C. Jelsch, S. Dahaoui, E. Espinosa and J.G. Ángyán, J. Phys. Chem. A, 2011, 115, 14484-14494. [2] M. E. Brezgunova, E. Aubert, S. Dahaoui, P. Fertey, S. Lebègue, C. Jelsch, J. G. Ángyán, E. Espinosa, Cryst. Growth & Des., 2012, 12, 5373-5386. [3] M. E. Brezgunova, J. Lieffrig, E. Aubert, S. Dahaoui, P. Fertey, S. Lebègue, J. G. Ángyán, M. Fourmigué, E. Espinosa, Cryst. Growth & Des., 2013 (submitted).

5b - Densité électronique, modélisation – Jeudi 4 juillet 10h30-12h00
- 94 -
O54 - Ab-initio Study of structural, Electronic and Magnetic
Properties of CdTe Doped Transition Metal Co
Samir Bentata*, Ali Zitouni, Bouabdellah Bouadjemi and Wissam Benstaali
Laboratoire de Technologie et des Propriétés du Solide Faculty of Sciences and Technology, BP227 Abdelhamid Ibn Badis University, Mostaganem (27000) Algeria *E-mail: [email protected]
The full potential linear augmented plane wave (FPLAPW) based on density-functional theory (DFT) is employed to study the structural, electronic and magnetic properties of transition metal Co doped CdTe. The 3d transition element Co is used as a dopant in order to induce spin polarization. We have analyzed the structural parameters, charge and spin densities, total and partial densities of states within the generalized gradient approximation (GGA). The results show a half-metallic ferromagnetic character with an important magnetic moment. The results obtained, make the CoxZn1-xTe a promising candidate for application in spintronics.
Keywords: magnetic moment, half-metallic, spin-up/spin-down, GGA.

5c - Cristallographie biologique et santé – Jeudi 4 juillet 10h30-12h00
- 95 -
O55 - Mécanismes de reconnaissance et d'activation des récepteurs hormonaux par les perturbateurs endocriniens environnementaux William Bourguet Centre de Biochimie Structurale, Inserm U1054-CNRS U5048-UM1, 34090 Montpellier, France
Nous sommes quotidiennement exposés à des milliers de polluants environnementaux dont certains peuvent agir comme des perturbateurs endocriniens. Le terme de “perturbation endocrinienne” fait référence à la capacité de certaines substances exogènes à moduler le système hormonal de manière inappropriée et à provoquer des effets délétères sur les organismes vivants tels que des troubles de la reproduction, du comportement, du métabolisme ou encore des cancers. Les perturbateurs endocriniens sont le plus souvent des composés de synthèse produits par l’industrie mais on en trouve également à l’état naturel, notamment dans les plantes et les champignons. Ils ont des structures chimiques généralement très éloignées de celles des hormones naturelles qui rendent la compréhension de leur liaison aux récepteurs nucléaires hormonaux particulièrement difficile.
Dans ce contexte, nous développons un projet original visant à étudier de manière
systématique les modes de reconnaissance et d’activation utilisés par les grandes familles de polluants environnementaux (phtalates, parabènes, benzophénones, pesticides, alkylphenols, organoétains, etc.) lors de l’interaction avec leurs récepteurs hormonaux cibles. Lors de cet exposé, je présenterai plus particulièrement les cas du bisphénol A (BPA) [1], des organoétains [2] et du phtalate MEHP [3] afin d’illustrer la diversité des mécanismes mis en œuvre. J’évoquerai également le développement d’un outil bioinformatique qui utilise ces structures cristallographiques expérimentales pour faciliter la modélisation de l’interaction des molécules environnementales avec les récepteurs nucléaires et donc la prédiction de leur activité hormonale.
Une application directe de ces résultats dans le domaine de l’environnement est de
pouvoir proposer des modifications chimiques des composés existants afin de dissocier leurs propriétés industrielles de leur activité hormonale résiduelle. Ces recherches peuvent avoir également des retombées dans le domaine de la santé car elles permettent de révéler des modes de liaison insoupçonnés pouvant inspirer la conception de nouvelles molécules thérapeutiques. [1] Delfosse et al., Proc Natl Acad Sci U S A. 2012, 109, 14930-5. [2] le Maire et al., EMBO Rep. 2009, 10, 367-73. [3] Résultats non publiés.

5c - Cristallographie biologique et santé – Jeudi 4 juillet 10h30-12h00
- 96 -
O56 - Une approche structurale pour l’inhibition sélective de PI3Kbeta
Thomas Bertrand1, Jean-Pierre Marquette1, Andreas Karlsson1, Nadine Michot2, Angela
Virone-Oddos3, Victor Certal3, Frank Halley3 1Structure Design and Informatics,
2Protein Production,
3Oncology Drug Discovery
Sanofi, 13 Quai Jules Guesde, 94403 Vitry-sur-Seine France.
La voie de signalisation PI3K/mTOR est impliquée dans la prolifération, la survie des cellules tumorales et les métastases. L’identification de mutations somatiques activatrices du gène PIK3CA ainsi que la fréquence très élevée de la perte du suppresseur de tumeur PTEN (Phosphatase and TENsin homologue), activant cette voie dans les tumeurs humaines, ont conduit au développement de nouvelles thérapies anticancéreuses ciblant cette voie. Dans le cas de la perte de PTEN, l’activation anormale de cette voie de signalisation est due à l’isoforme PI3Kbeta. La plupart des inhibiteurs de PI3K en développement clinique étant des molécules pan-PI3K, il serait utile de découvrir des inhibiteurs sélectifs de l’isoforme PI3Kbeta pour le traitement des cancers humains présentant une déficience en PTEN. Les structures de PI3Kbeta et d’autres isoformes en complexe avec différents inhibiteurs ont été résolues par cristallographie des rayons X. Elles ont permis de mieux comprendre la relation structure-activité de la série chimique du candidat actuellement en phase clinique ainsi que sa sélectivité1. [1] Certal et al., J. Med. Chem. 2012, 55(10), 4788-4805.

5c - Cristallographie biologique et santé – Jeudi 4 juillet 10h30-12h00
- 97 -
O57 - Structure cristallographique de la protéine FadD32 de Mycobacterium marinum, une cible potentielle pour le développement de nouveaux antituberculeux
Valérie Guillet1, Ségolène Galandrin
2, Mamadou Daffé
2, Hédia Marrakchi
2 et Lionel
Mourey1
1Groupe de Biophysique Structurale, IPBS/CNRS,
2Enveloppes mycobactériennes : Structure, biosynthèse et fonctions, IPBS/CNRS – Université
de Toulouse, F-31077 Toulouse, France
La découverte de nouveaux antituberculeux est nécessaire pour permettre un traitement efficace de la maladie quel que soit le stade de l’infection. Un des axes pour la découverte de nouvelles molécules est de cibler la biosynthèse de composants de l’enveloppe Mycobacterium tuberculosis, parmi lesquels les acides mycoliques. Ces lipides sont des composants majeurs et spécifiques de l’enveloppe mycobactérienne et essentiels à la survie.
L’étape clef de la formation des acides mycoliques est la condensation de deux chaînes d’acides gras. Cette étape est contrôlée par l’opéron fadD32-pks13-accD4, essentiel à la croissance du bacille tuberculeux [1]. FadD32 est une fatty acyl-AMP ligase (FAAL) qui active un acide gras en C50-C60 (conversion des acides gras en acyl-adénylates [2]) avant condensation avec un acide gras en C22-C26 par l’enzyme Pks13 [3]. La protéine FadD32 est conservée dans tous les génomes des espèces mycobactériennes séquencées et représentent une cible très prometteuse. L’activité enzymatique in vitro de FadD32 de M. tuberculosis étant relativement faible, nous avons élargi cette étude à d’autres enzymes orthologues complexées à des analogues de substrats ou inhibiteurs. Des cristaux de FadD32 de M. marinum et M. smegmatis ont été obtenus et les structures résolues en complexe avec un inhibiteur analogue de substrat [4].
Ces structures sont essentielles pour comprendre les disparités de spécificité de substrat et d’activité enzymatique entre les différentes FadD32 et au sein de la famille des FAAL, ouvrant ainsi de nouvelles perspectives en drug design. Outre la résolution de la structure, nous avons également développé un test de criblage haut-débit (HTS). L’identification de ligands potentiels et la validation de ces composés a été réalisée par mesure de la modulation de la stabilité thermique. Ce criblage a permis l’identification de nouvelles classes d’inhibiteurs [5]. [1] Portevin D. et al., J. Biol. Chem. 2005, 280, 8862-8874. [2] Léger M. et al., Chem. Biol. 2009, 16 (5): 510−519. [3] Gavalda S. et al., J. Biol. Chem. 2009, 284 (29): 19255−19264. [4] Guillet V. et al., manuscrit en préparation [5] Galandrin S. et al., J. Biomol. Screen. 2013, 18(5):576-87.

5c - Cristallographie biologique et santé – Jeudi 4 juillet 10h30-12h00
- 98 -
O58 - Toward closing the synaptic gap: a molecular approach to explore neuronal connectivity deficiencies associated with autism Pascale Marchot1,2, Philippe Leone1, Igor P Fabrichny1,2, Davide Comoletti3, Meghan T Miller3, Géraldine Ferracci4, Simon U Garcia3, Sandrine Conrod2, Palmer Taylor3, Yves Bourne1 1 Architecture et Fonction des Macromolécules Biologiques (AFMB), CNRS/AMU, Marseille, France. 2 Biochimie des Interactions Cellulaires et Moléculaires (BIMC), CNRS/AMU, and 4Centre d’Analyse Protéomique de Marseille (CAPM), Faculté de Médecine Secteur Nord, Marseille, France. 3 Dept. of Pharmacology, Skaggs School of Pharmacy and Pharmaceutical Sciences (SSPPS), UCSD, La Jolla, CA.
Neuronal circuits are maintained by homeostatic mechanisms controlling synapse maturation and signaling. The neuroligins are postsynaptic cell adhesion proteins whose transient associations with presynaptic alpha- and beta-neurexins participate in synaptogenesis and may regulate the fine balance between excitation and inhibition. This heterophilic interaction is regulated by gene selection, alternative mRNA splicing and post-translational modifications. Mutations in the neuroligin and neurexin genes appear to be associated with autism and mental retardation.
Structural analyses of neuroligin-4 [1] and its complex with a beta-neurexin partner [2] led to position the neuroligin structural defects associated with autism-linked mutations and precluding either proper folding and trafficking to the cell membrane, or recognition of the neurexin partner. These analyses also showed that whereas neuroligin-1 (found at glutamatergic and central cholinergic synapses) and neuroligin-2 (found at gabaergic and glycinergic synapses) are preset conformationally to respectively bind and not bind neurexin, conformational reshaping at the surface of neuroligin-4 (found at glycinergic synapses), controled by environmental variations, is required for accommodating neurexin, thus adding another mean for regulation of the neurexin-neuroligin interaction.
This work illustrates how structural biology, associated with molecular biology and biochemistry approaches, can contribute to dissect the organization and functioning of inter-cellular interactions crucial for neuronal activity. [1] Fabrichny, Leone, Sulzenbacher, Comoletti, Miller, Taylor, Bourne, Marchot (2007) Structural analysis of the synaptic protein neuroligin and its -neurexin complex: determinants for folding and cell adhesion. Neuron 56, 979-91. [2] Leone, Comoletti, Ferracci, Conrod, Garcia, Taylor, Bourne, Marchot (2010) Structural insights into the exquisite selectivity of neurexin/neuroligin synaptic interactions. EMBO J 29, 2461-71. Work supported by the SPINE2-Complexes Consortium (to YB, PM and PL); the CNRS Direction des Relations Internationales – Sciences de la Vie (to PM and YB); the CNRS and Fondation pour la Recherche Médicale (to IF); the Autism Speaks and the Cure Autism Now Pilot Study (to DC), and NIH grants (to PT and MM).

6a - Méthodes émergentes en cristallographie – Jeudi 4 juillet 13h30-15h30
- 99 -
O59 - Application de la Fonction de Distribution de Paires à l'étude de matériaux nano-cristallins ou mal ordonnés Pierre Bordet Institut Néel, CNRS-UJF, Grenoble
Depuis quelques années, l’analyse de la diffusion totale par la fonction de distribution
de paires (PDF) connait un renouveau important [1], motivé, d’une part, par le développement de nouvelles sources de haute énergie et intensité alliées à des méthodes d’analyse robustes et d’autre part par l’émergence de thématiques matériaux nécessitant la prise en compte de la structure locale. Ainsi, l’analyse PDF a-t-elle été appliquée avec succès à des problèmes très variés allant de l’étude de la transition Jahn-Teller dans les manganites magnéto-résistives [2] à celle d’inclusions nanocristallines dans des minéraux à base de titane [3].
Cependant, l’utilisation de la PDF a été jusqu’ici cantonnée pour l’essentiel au domaine des composés minéraux de structures relativement simples. Nous voulons ici montrer à travers deux exemples que la PDF peut être un outil précieux dans le cadre d’études structurales de matériaux plus complexes
Le premier de ces exemples portera sur l’apport de la PDF dans l’étude de composés moléculaires ou pharmaceutiques. Le deuxième, sur l’étude du verdissement progressif de pigments de bleu de Prusse anciens. Dans les deux cas, on précisera l’apport de l’analyse PDF ainsi que les difficultés nouvelles qui devront être prises en compte pour un application optimale de la méthode à ce type de composés [1] EGAMI T., BILLINGE S. J. L. (2003) – Underneath the Bragg Peaks: Structural analysis of complex materials, Pergamon, Oxford, England. [2] BOZIN ET AL. (2007) – Phys. Rev; Lett. 98, 137203. [3] GREY I.E., et al. (2010) American Mineralogist, 95, 161

6a - Méthodes émergentes en cristallographie – Jeudi 4 juillet 13h30-15h30
- 100 -
O60 - Nouvelles possibilités expérimentales en physique introduites par les XFELs Jan Lüning Université Pierre et Marie Curie, Paris
Le rayonnement des nouveaux lasers à électrons libres émettant des rayons X (XFEL,
pour X-ray Free Electron Laser) permet de réaliser des expériences entièrement nouvelles grâce aux propriétés uniques de leur rayonnement : des impulsions ultra-brèves (domaine de la femtoseconde) et ultra-intenses avec un très haut degré de cohérence. Ceci crée des nouvelles possibilités pour l'étude expérimentale dans une grande diversité de domaines scientifiques, aller de la physique à la chimie, la biologie et autres.
Pour leur exploitation, un grand nombre de nouvelles techniques expérimentales et d'instrumentations originaux ont été conçus pendant les quelques années depuis le XFEL FLASH en Hambourg est devenu une source ouverte aux utilisateurs. Entre temps, il y a quatre XFELs qui servent une communauté d'utilisateurs en pleine croissance : les XUV-FELs FLASH et FERMI (Italie) et les XFELs LCLS (Etats-Unis) et SACLA (Japon).
Cette présentation commencera par un résumé de ce développement récent en mettant
l'accent sur les capacités et les défis expérimentaux qui dérivent des propriétés uniques de ces sources.
Après une présentation plus générale de l'état de l'art des techniques de diffusion aux XFELs, quelques exemples récents concernant la physique de la matière condensée sont discutés en plus de détails.

6a - Méthodes émergentes en cristallographie – Jeudi 4 juillet 13h30-15h30
- 101 -
O61 - Méthodes rapides pour analyser les désordres structuraux statiques et dynamiques dans l’immense espace réciproque. Laurent Guérin1, Bertrand Toudic1, Céline Mariette1, Philippe Rabiller1, Alexei Bosak2, Danièle de Sanctis2, Jacques Ollivier3 1Institut de Physique de Rennes, 2ESRF, Grenoble, 3ILL, Grenoble
Une tendance constante de la cristallographie est d’aller vers l’étude de systèmes de plus en plus complexes. Ceci se caractérise par des mailles de plus en plus grandes, voire infinies pour des cristaux apériodiques qui possèdent un ordre à grande distance mais sans la symétrie de translation. Aux structures moyennes sont associés des désordres, fluctuations de différents types qui induisent la réduction de l’intensité des pics de Bragg et simultanément l’apparition de la diffusion diffuse dans l’espace réciproque. Un exemple fameux est le Thermal Diffuse Scattering (T.D.S.), signature des branches de phonon dont l’intensité croît comme le module du vecteur réciproque au carré. Bien sûr de nombreux autres sources de désordre existent en lien, comme le T.D.S., avec des fluctuations collectives (modes de phason si matériaux apériodiques, phénomènes prétransitionnels critiques de type mode mou ou ralentissement critique des fluctuations autour d’instabilités structurales) ou des fluctuations locales d’arrangement structural ou de composition. D’un point de vue structural, le challenge est évidemment de résoudre la structure en affinant simultanément la diffusion diffuse et les pics de Bragg. Cette méthode dite 3D-PDF ne fera pas l’objet de cette présentation et nous nous focaliserons ici sur le caractère potentiellement dynamique du désordre présent dans ces matériaux. Cette information en énergie est essentielle pour déterminer l’origine du désordre et le caractériser. L’étude couplée de la diffraction X et des X inélastiques permet une analyse rapide et complète grâce aux développements récents de détecteurs très haute résolution à bruit nul et aux spectromètres à rétrodiffusion sur sources synchrotrons. Si nécessaire, des mesures par diffusion neutronique en temps de vol peuvent compléter ces informations pour les dynamiques les plus lentes. Cette approche expérimentale sera illustrée par une étude du désordre dans un cristal composite apériodique organique.

6a - Méthodes émergentes en cristallographie – Jeudi 4 juillet 13h30-15h30
- 102 -
O62 - Défauts de phase dans les cristaux électroniques étudiés par diffraction cohérente des rayons X Vincent Jacques LPS, Université Paris Sud
La diffraction cohérente des rayons X est une technique très sensible à la présence de
défauts de phase dans les cristaux. Une dislocation de réseau est un exemple typique de défaut de phase cristallin qui crée un effet d’interférence visible en diffraction cohérente. Cet effet a été démontré expérimentalement sur des boucles de dislocation isolées dans le silicium [1]. Au-delà des dislocations de réseau, les défauts de phase se rencontrent dans tout type de structure, et en particulier dans les cristaux électroniques, qui présentent des modulations statiques de la densité de charge appelées Ondes de Densité de Charge (ODC). Ces structures électroniques sont des cristaux à part entière qui se forment dans une structure atomique sous-jacente par un mécanisme de distorsion périodique de réseau appelée distorsion de Peierls. Dans certains systèmes, les ODC sont incommensurables et elles ont la possibilité de glisser lorsqu’elles sont soumises à l’action d’une force extérieure telle que celle engendrée par l’application d’un courant électrique. Nous montrerons comment les ODC des systèmes quasi-unidimensionnels K0.3MoO3 et NbSe3 se comportent sous courant, et notamment comment la diffraction cohérente des rayons X a permis de mettre en évidence les différents régimes de glissement de l’ODC dans NbSe3 [2] et un réseau de solitons dans K0.3MoO3 [3,4]. [1] V.L.R. Jacques et al., PRL 106, 065502 (2011) [2] E. Pinsolle et al., PRL 109, 256402 (2012) [3] D. Le Bolloc’h et al., PRL 100, 096403 (2008) [4] V.L.R. Jacques et al., PRB 85, 035113 (2012)

6b - Biologie structurale intégrative et gros assemblages Jeudi 4 juillet 13h30-15h30
- 103 -
O64 - Démarrage de la traduction chez les eucaryotes et les archées: le facteur e/aIF2 Yves Mechulam, Pierre-Damien Coureux, Etienne Dubiez, Auriane Monestier, Marie Naveau, Christine Lazennec-Schurdevin, Michel Panvert et Emmanuelle Schmitt Laboratoire de Biochimie, Unité mixte de Recherche 7654, Ecole Polytechnique, Centre National de la Recherche Scientifique, F-91128 Palaiseau cedex.
Chez tous les êtres vivants, le démarrage de la traduction implique le recrutement d’un ARNt initiateur spécialisé, systématiquement aminoacylé par une méthionine. Cet ARNt joue un rôle clé dans l’identification du codon de démarrage sur l’ARNm. Malgré l’universalité de ce processus, les mécanismes de sélection et d’utilisation de cet ARNt différent suivant le domaine, bactérien, eucaryote ou archéen. Chez les eucaryotes et les archées, le facteur hétérotrimérique de démarrage de la traduction 2 (e/aIF2) forme un complexe ternaire avec le GTP et l'ARN de transfert initiateur, puis vient se lier à la petite sous-unité du ribosome. L’appariement entre l’anticodon de l’ARNt et le codon de démarrage sur l’ARNm engendre une cascade d’évènements qui aboutit au départ des facteurs de démarrage et au positionnement correct du ribosome pour une traduction fidèle.
Nous utilisons un ensemble de méthodes biochimiques et structurales pour décrire la formation du complexe de démarrage archéen, et le rôle des facteurs dans la spécificité du système. Nous décrirons plus particulièrement les résultats obtenus concernant la liaison de l’ARNt initiateur au facteur aIF2, ainsi que le rôle du facteur aIF1 dans la formation du complexe de démarrage. De manière surprenante, malgré une similitude structurale considérable entre la sous unité centrale d'e/aIF2 et le facteur d'allongement de la traduction EF-Tu, ces deux protéines G de l'appareil de traduction utilisent des stratégies de liaison de l'ARNt radicalement différentes [1]. Ces résultats seront également discutés dans le contexte du démarrage de la traduction chez les eucaryotes [2]. [1] Schmitt et al., Nat. Struct. Mol. Biol. 2012, 19, 450-454. [2] Naveau et al., Nucleic Acids Res. 2013, 41, 1047-1057.

6b - Biologie structurale intégrative et gros assemblages Jeudi 4 juillet 13h30-15h30
- 104 -
O65 - Crystal structure of the eukaryotic 80S ribosome
Nicolas Garreau de Loubresse and Marat Yusupov Institut de Génetique et de Biologie Moléculaire et Cellulaire, UMR7104 IGBMC, U964 INSERM, Département de Biologie Structurale Intégrative, Illkirch, France.
Ribosomes translate genetic information encoded by messenger RNA into proteins. Many aspects of translation and its regulation are specific to eukaryotes, whose ribosomes are much larger and intricate than their bacterial counterparts. We report the crystal structure of the 80S ribosome from the yeast Saccharomyces cerevisiae at a resolution of 3.0 angstroms. This atomic model reveals the precise architecture of eukaryote-specific elements and their interaction with the universally conserved core. Together, it forms the structural and experimental framework to explore the eukaryotic translation apparatus as well as small molecules of therapeutic interest against infectious diseases, genetic disorders and cancers. Emphasis will be placed on methods development and recent advances to investigate small molecules on the yeast ribosome.

6b - Biologie structurale intégrative et gros assemblages Jeudi 4 juillet 13h30-15h30
- 105 -
O66 - Etude structurale et fonctionnelle de la protéine Pat1 de Saccharomyces cerevisiae
Zeineb Fourati-Kammoun1,2
, Olga Kolesnikova3, Jenny Keller
2, Noureddine Lazar
2,
Anthony Doizy2, Herman van Tilbeurgh
2, Bertrand Séraphin
3 et Marc Graille
1,2 1Laboratoire de Biochimie, Ecole Polytechnique, Route de Saclay, Palaiseau,
2Institut de Biochimie et Biophysique Moléculaire et Cellulaire, Université Paris sud, Orsay
3Equipe Labellisée La Ligue, IGBMC (Institut de Génétique et de Biologie Moléculaire et
Cellulaire), Illkirch F-67400, France; CNRS, UMR7104, Illkirch F-67404, France; Inserm, U964, Illkirch F-67400, France; and Université de Strasbourg, Strasbourg F-67000, France
La quantité d’un ARNm eucaryote dans la cellule est finement régulée par l’équilibre entre son niveau de transcription et sa vitesse de dégradation. Chez la levure S. cerevisiae, la dégradation des ARNm se fait essentiellement par la voie 5’-3’, tributaire d’une étape de « decapping ». Cette étape, catalysée par l’enzyme Dcp2, vise à détruire la coiffe stabilisatrice en 5’ des ARNm pour permettre à l’exonucléase Xrn1 de dégrader l’ARNm dans le sens 5’-3’. L’activité catalytique intrinsèque de Dcp2 est très faible et requiert l’intervention d’un ensemble de facteurs formant le complexe activateur du « decapping ». La protéine Pat1 constitue un élément central de ce complexe [1]. En effet, Pat1 active directement la catalyse par Dcp2 à travers son domaine C terminal, et utilise ce domaine pour fixer Dcp2 et certains autres activateurs du « decapping » [2, 3].
La structure cristallographique du domaine C terminal de Pat1 de S. cerevisiae ainsi que la caractérisation de son interaction avec l’ARN seront présentés. La structure de ce domaine révèle une particularité par rapport à son orthologue humain [4]. Il s’agit d’une extension C terminale caractéristique des levures, dont la délétion induit un phénotype thermosensible in vivo, suggérant qu’il s’agit d’un domaine fonctionnellement important. En considérant qu’il existe deux paralogues Pat1 chez l’homme (Pat1a et Pat1b) dont un seul est impliqué dans le « decapping », ce résultat stipule que Pat1 ait évolué en séparant deux domaines fonctionnellement importants chez les eucaryotes supérieurs. [1] Bonnerot C, Boeck R, Lapeyre B (2000) The two proteins Pat1p (Mrt1p) and Spb8p interact in vivo, are required for mRNA decay, and are functionally linked to Pab1p. Mol Cell Biol 20: 5939-5946 [2] Nissan T, Rajyaguru P, She M, Song H, Parker R (2010) Decapping activators in Saccharomyces cerevisiae act by multiple mechanisms. Mol Cell 39: 773-783 [3] Pilkington GR, Parker R (2008) Pat1 contains distinct functional domains that promote P-body assembly and activation of decapping. Mol Cell Biol 28: 1298-1312 [4] Braun JE, Tritschler F, Haas G, Igreja C, Truffault V, Weichenrieder O, Izaurralde E (2010) The C-terminal alpha-alpha superhelix of Pat is required for mRNA decapping in metazoa. EMBO J 29: 2368-2380

6b - Biologie structurale intégrative et gros assemblages Jeudi 4 juillet 13h30-15h30
- 106 -
O67 - Détermination des bases structurales du mécanisme de perméation des ions chez les récepteurs-canaux pentamériques de la famille Cys-loop
Ludovic Sauguet1, Samuel Murail
2, Catherine Van Renterghem
3, Laurie Malherbe
1, Frédéric
Poitevin1, Andy Thompson4, Pierre-Jean Corringer
3, Marc Baaden
2 et Marc Delarue
1
1Unité de Dynamique Structurale des Macromolécules, Institut Pasteur, Paris
2IBPC, Paris,
3Unité Récepteurs-Canaux, Institut Pasteur, Paris,
4Synchrotron SOLEIL, Gif-sur-Yvette
Chez les vertébrés, les récepteurs-canaux de la famille Cys-loop (RCPs) assurent la transmission chimio-électrique du signal électrique nerveux par l’intermédiaire de transitions allostériques qui couplent la liaison d’un agoniste avec l’ouverture ou la fermeture du canal
1. Chez l’homme, ces protéines membranaires pentamériques comprennent les récepteurs nicotiniques, GABA, glycine et sérotonine, qui sont la cible de nombreuses classes de molécules thérapeutiques. Les mécanismes de perméation des ions chez les RCPs demeurent
néanmoins peu documentés. La détermination de la structure de GLIC 2, un homologue
procaryote de la famille, dans une conformation ouverte perméante aux ions 3-4, constitue un bon modèle structural permettant d’étudier les mécanismes de perméation chez les RCPs. En améliorant récemment la structure de GLIC à 2.4 Å de résolution, la plus haute résolution atteinte pour des RCPs, nous avons pu pour la première fois décrire expérimentalement la géométrie d’hydratation dans le pore de ces récepteurs et notamment la
présence de deux pentagones d’eau 5. A partir de co-cristaux obtenus en présence de diffuseurs anomaux (Br-, Cs+ et Rb+), nous avons cartographié les sites de fixation des ions monovalents positifs et négatifs dans la structure de GLIC et notamment dans le pore transmembranaire. Cette étude structurale a été complétée par des simulations de dynamique moléculaire et des expériences d’électrophysiologie sur canal-unique qui ont permis de
proposer un mécanisme pour la perméation des ions 5.
[1] Corringer P.J., Poitevin F., Prevost M.S., Sauguet L. et al. Structure. 2012, 20 (6) 941-56 [2] Bocquet N. et al. Nature 2009, 457 (7225) 111-4 [3] Prevost M., Sauguet L. et al. Nature Struct. Mol. Biol. 2012, 19 (6) 642-9 [4] Sauguet L. et al. Nature communications. 2013, 16 (4) 1697 [5] Sauguet L. et al. EMBO J. 2013, 32 (5) 728-41

6b - Biologie structurale intégrative et gros assemblages Jeudi 4 juillet 13h30-15h30
- 107 -
O68 - Études structurales et fonctionnelles du complexe de pré-intégration du VIH-1 Nicolas Levy1, Sylvia Eiler1, Aurélie Schaetzel1, Corinne Crucifix1, Karine Pradeau-Aubtreton1, Vincent Parissi2, Stéphane Emiliani3, Yves Mély4, Patrick Schultz1, Marc Ruff1 1IGBMC, 1 rue Laurent Fries, 67404 Illkirch,
2Laboratoire de Microbiologie Fondamentale et Pathogénicité, Université de Bordeaux 2,
Bordeaux, 3Institut Cochin, Université Paris Descartes, Paris,
4Laboratoire de Biophotonique et Pharmacologie. UMR 7213 CNRS, UDS, Faculté de
Pharmacie, Illkirch
Dans les étapes précoces de la réplication du VIH-1, le génome viral est converti en ADN qui va interagir avec des protéines cellulaires et virales pour former le complexe de pré-intégration (PIC). L’intégrase (IN) est une protéine essentielle du PIC et intervient dans plusieurs étapes de la réplication du virus, comme la décapsidation, la réverse transcription, l’import nucléaire, le ciblage de la chromatine et l’intégration. L’intégrase va recruter des protéines de la cellule hôte pour effectuer ces diverses fonctions. Les mécanismes moléculaires et la dynamique de ces processus ainsi que le rôle des cofacteurs cellulaires restent largement inconnus. Les intégrases rétrovirales sont des protéines montrant une grande flexibilité inter domaine. Comme pour d’autres protéines désordonnées, cette flexibilité intrinsèque confère à l’intégrase la capacité d’interagir avec des partenaires multiples. Nous assumons que les différentes fonctions du PIC dans l’infection virale reflètent les différentes conformations de ses composant protéiques qui peuvent varier avec les protéines partenaires et les modifications post traductionnelles. Nous avons démontré que la faible solubilité et la flexibilité inter domaine peuvent être contournées par la formation de complexes stables et spécifiques avec des substrats comme des cofacteurs protéiques et par des modifications post traductionnelles. Nous avons reconstitués in vitro des complexes solubles et stables autour de l’intégrase qui est présente à toutes les étapes de la transcription inverse à l’intégration. Les structures des complexes IN/LEDGF/DNA [1] et IN/LEDGF/INI1/DNA [2] ont été résolues par cryo microscopie électronique. Ces structures combinées aux essais fonctionnels ont donnés des informations importantes pour les rôles de LEDGF et INI1 dans l’infection virale. Un autre sous complexe du PIC a été caractérisé au laboratoire, le complexe IN/transportine-SR2/VBP1 impliqué dans le transfert nucléaire du PIC. Des résultats préliminaires sur la structure de ce complexe seront présentés. [1] Michel et al, EMBO J.. 2009, 28, 980-991. [2] Maillot et al, PLoS ONE, 2013, 8(4): e60734. doi:10.1371/journal.pone.0060734

Lauréats des prix de thèse de l’AFC 2012 – Jeudi 4 juillet 18h00-19h00
- 108 -
(GT-BIO)
O69 - Contrôle du métabolisme central chez les mycobactéries par la régulation allostérique de l´alpha-kétoglutarate déshydrogénase. Tristan Wagner Institut Pasteur, Paris
L’α-kétoglutarate déshydrogénase (KDH) est un complexe multienzymatique constitué de trois composants (E1o, E2o, E3) situé au nœud métabolique entre le cycle de Krebs et l’assimilation de l’azote. Vu son rôle clef, ce complexe nécessite une régulation fine pour coordonner le flux de carbone ainsi que la production d’énergie par le cycle de Krebs. Les Corynebacterineae ont une situation particulière car E2o est absent, son domaine catalytique est fusionné à E1o. Ce nouveau polypeptide (appelé KGD pour E1o+E2o) possède un système unique de contrôle par GarA, une protéine à domaine en tête de fourche (ce domaine appelé FHA reconnait les thréonines phosphorylées). GarA est une sorte d’ « interrupteur moléculaire » qui régule la synthèse du glutamate et est contrôlé lui-même par phosphorylation. Dans l’état non phosphorylé, GarA lie E1o, le cœur catalytique du complexe KDH, en inhibant son activité.
Le but de cette thèse est de déchiffrer le mécanisme catalytique de E1o, ainsi que les bases moléculaires de l’inhibition par GarA. Nous avons résolu la structure cristalline de KGD de Mycobacterium smegmatis en complexe avec son cofacteur thiamine diphosphate. Nous montrons pour la première fois que KGD effectue d’importants réarrangements conformationnels pendant le cycle catalytique, le principal étant une translocation d’un tour
d’une hélice αexterne (αE). GarA agit comme inhibiteur en liant cette hélice par son domaine FHA, bloquant ainsi sa translocation. L’interaction KGD-GarA n’implique aucun résidu phospho-thréonine mais un aspartate mimant la charge phosphate sur l’hélice αE, un comportement sans précédents pour une protéine à domaine FHA. Le domaine additionnel de type E2o interagit également avec la même hélice αE, générant un effet d’inhibition synergique avec GarA. Cet effet peut néanmoins être contrecarré par l’action de l’acétyl-CoA, un activateur allostérique qui se lie à un site distinct. L’action de l’acétyl-CoA, jusqu’à présent unique pour l’enzyme mycobactérienne, est due à des changements subtils de certains résidus clefs pour le réarrangement conformationnel catalytique.

Lauréats des prix de thèse de l’AFC 2012 – Jeudi 4 juillet 18h00-19h00
- 109 -
Structure de l´organisation hexamérique de KGD composé de E1o (en vert) et du domaine E2o (en bleu). Le domaine FHA de GarA est représenté en surface (violet) alors que l´emplacement du site actif de E1o est localisé par le cofacteur thiamine pyrophosphate (rouge).

Lauréats des prix de thèse de l’AFC 2012 – Jeudi 4 juillet 18h00-19h00
- 110 -
(GT CHIMIE) O70 - Cristallochimie de nouveaux polymères de coordination à noyau spirobifluorène ou tétraphénylméthane : du design du ligand à la topologie et aux propriétés du solide hybrides. Florian Moreau
Laboratoire des Sciences Chimiques, Rennes
Ce travail concerne la cristallochimie de nouveaux polymères de coordination. Afin de
diriger la topologie des réseaux formés et d'obtenir des polymères de coordination poreux, nous avons proposé le design de ligands tétracarboxylates comportant deux types de noyaux de géométries fixées : le noyau tétraphénylméthane de symétrie Td et le noyau spirobifluorène de symétrie D2d. des espaceurs phényles ont été introduits dans une stratégie de chimie d'échelle et la position de la fonction coordinante sur l'espaceur a été modulée afin de permettre une flexibilité conformationnelle des ligands. La réactivité de ces ligands a été explorée vis-à-vis de sels de zinc, de cuivre et de lanthane. Les structures des polymères de coordination ont été déterminées par diffraction des rayons x par le monocristal. La stabilité thermique, la réactivité chimique et les propriétés d'adsorption de gaz ont été investiguées. Le premier chapitre de ce mémoire est consacré à l'étude bibliographique des polymères de coordination poreux. Le deuxième chapitre expose la stratégie de synthèse organique des ligands visés. Les trois chapitres suivants regroupent selon leurs caractéristiques structurales les polymères de coordination synthétisés. Un éclairage particulier est porté sur les topologies et le phénomène d'interpénétration des réseaux ainsi que sur l'influence de la flexibilité des ligands. Un dernier chapitre est consacré aux polymères de coordination 2D à noyau spirobifluorène. Enfin, un récapitulatif des traits marquants des matériaux synthétisés clôture ce mémoire en abordant les perspectives offertes par ce travail.

Lauréats des prix de thèse de l’AFC 2012 – Jeudi 4 juillet 18h00-19h00
- 111 -
(GT PHYSIQUE)
O71 - Transitions de phases dans les argiles. Influence de la minéralogie et de la morphologie. Comportement sous écoulement et sous champs. Erwan Paineau
Institut National Polytechnique Lorraine, Nancy
L’objectif de ce travail est d’étudier les transitions de phases sol-gel et isotrope-nématique dans des suspensions de smectites dioctaédriques en fonction de la morphologie et de la nature minéralogique des argiles. Bien que tous les systèmes étudiés présentent une transition sol-gel à de faibles fraction volumique, la transition cristal-liquide isotrope-nématique n’a pu être identifiée que dans le cas de suspensions de smectites ayant un déficit de charge tétraédrique. L’effet de la localisation de la charge sur le comportement colloïdal a été déterminée à l’aide de la diffusion des rayons X aux petits angles (SAXS) et par des mesures rhéologiques. La nature des interactions électrostatiques dans ces suspensions est purement répulsive et rejette l’idée d’une structure tridimensionnelle de type « château de carte ». Cependant, les smectites ayant un déficit de charge tétraédrique sont plus répulsives et ont des propriétés viscoélastiques plus faibles que celles ayant un déficit octaédrique. Il a également été montré que la dépendance en taille de particules de la position de la transition sol-gel était liée à une statistique de piégeage hydrodynamique des plaquettes d’argile. Finalement, l’application de champs externes (électrique et magnétique) a permis d’obtenir l’alignement de la phase nématique tandis que dans la phase isotrope, le champ électrique induit un ordre antinématique parfait. Afin de préserver l’ordre induit, ces suspensions ont été polymérisées sous champ permettant l’obtention de nanocomposites orientées et structurés.

7a - Cristallographie in situ, in operando – Vendredi 5 juillet 9h00-10h30
- 112 -
O72 - Complexité structurale de (RE)2NiO4+ explorée par diffraction in situ des neutrons et du rayonnement synchrotron pendant une réaction d'intercalation de l’oxygène contrôlée électrochimiquement
M Ceretti1, O. Wahyudi2,3, J. Stern1, A. Villesuzanne3, J.M. Bassat3, G. André4, F. Porcher4, W. Paulus1
1Université de Montpellier 2, Institut Charles Gerhardt, UMR CNRS 5253, Montpellier, 2Université de Rennes 1, Rennes, 3CNRS, Université de Bordeaux, ICMCB, 87 Av. Dr. A. Schweitzer, Pessac, 4Laboratoire Léon Brillouin, CEA-CNRS, CE Saclay, 91191 Gif sur Yvette
Les conducteurs d'ions oxygène à basse température sont des matériaux d'intérêt majeur
pour une série d'applications dans le domaine des conducteurs ioniques. Nd2NiO4+ et
Pr2NiO4+ appartiennent à ces oxydes non stœchiométriques, avec une structure type K2NiF4, qui montrent une mobilité d'ions d'oxygène dès la température ambiante. Pour cette raison, ils figurent parmi les candidats les plus prometteurs pour les membranes d'oxygène, les capteurs ou les catalyseurs, opérant à température modérée. Une des caractéristiques les plus intéressantes est la possibilité d'accueillir une quantité importante d'atomes d'oxygène
supplémentaires sur les sites interstitiels, la région non stœchiométrique étant 0 < <0,26 [1]. Dans cette étude, nous avons suivi par diffraction in situ des neutrons sur poudre et par
diffraction du rayonnement synchrotron sur monocristal, l’évolution structurale en fonction de
la stœchiométrie en oxygène de (Nd,Pr)2NiO4+ à température ambiante, pendant une réaction électrochimique. La réaction a été effectuée dans des cellules spécialement conçues [2-3]. Nous rapportons ici le riche diagramme de phase en fonction du transfert de charge, allant d'une phase orthorhombique incommensurable pour (Nd,Pr)2NiO4.25, passant par une phase ordonnée intermédiaire quadratique, jusqu’à la phase stœchiométrique AF (Nd,Pr)2NiO4.0. La réaction de Nd2NiO4.0 s'est révélée être partialement réversible en s’arrêtant à une
stœchiométrie =0.10. Une étude structurale de cette phase a révélé une structure P42/ncm. Les calculs préliminaires de dynamique de réseau (algorithme DFT ab initio) indiquent que cette réversibilité limitée est lié aux contraintes de structure et en particulier à la valeur du paramètre c, accompagné d'un facteur de Debye-Waller réduit pour les atomes d'oxygène
apicaux. Pr2NiO4+ présente un mécanisme d'intercalation différente, et la phase réduite peut
être chargé jusqu'à sa stœchiométrie initiale =0.25. Il s'agit d'une différence importante qui nous pensons être liée au rayon ionique légèrement plus élevé de Pr3+ par rapport à Nd3+ et associée à une dynamique du réseau modifiée [4-5].
[1] Y. Toyosumi et al. J. Alloys and Compounds 408‐412 (2006) 1200‐1204. [2] W. Paulus, et al, JACS (2008) [3] R. Le Toquin, et al, J. Am. Chem. Soc. 128 (2006) 13161‐13174 [4] A Villesuzanne et al., J. Solid State Electrochem. 15, 2 (2012), 357 [5] O. Wayudi, PhD Thesis, University Rennes 1, dec. 2011

7a - Cristallographie in situ, in operando – Vendredi 5 juillet 9h00-10h30
- 113 -
O73 - L’apport des études menées operando par diffraction et absorption des rayons X à la compréhension des mécanismes mis en jeu au sein des matériaux d’électrode pour batteries Li-ion.
Laurence Croguennec1, Jean-Marcel Ateba Mba1,2, Matteo Bianchini1,2,3, Hideyuki Koga1,
Emmanuelle Suard3, Stéphanie Belin4, Claude Delmas1 et Christian Masquelier2
1 ICMCB-CNRS, Université de Bordeaux, IPB-ENSCBP, 33600 Pessac, France, 2 LRCS-CNRS, Université de Picardie Jules Vernes, 80039 Amiens Cedex 1, France, 3 Institut Laue-Langevin, 38000 Grenoble, France, 4 Synchrotron Soleil, 91192 Gif-sur-Yvette, France
Ces vingt dernières années les efforts de recherche menées dans le domaine des batteries Li-ion ont été largement orientés vers la recherche de nouveaux matériaux d’électrode et de nouveaux électrolytes, avec l’objectif d’accroitre la sécurité, la durée de vie et les densités d’énergie et de puissance de ces systèmes électrochimiques [1]. Il s’est avéré essentiel de suivre les évolutions structurales et les processus redox mis en jeu au cours des réactions de désintercalation et de réintercalation des ions Li+ des matériaux d’électrodes, pour mieux prédire la réversibilité de ces batteries Li-ion. Des méthodes de caractérisation ont été rapidement développées pour suivre in situ ces réactions dynamiques au cours du fonctionnement de la batterie, notamment par diffraction des rayons X (de laboratoire ou Synchrotron) [2-3] ou par spectroscopie d’absorption des rayons X [3-4]. Ces études menées operando apportent une information globale sur la nature des réactions mises en jeu, dans des conditions proches des conditions réelles d’utilisation d’une batterie. Les caractérisations menées ex situ sur des compositions clés restent néanmoins essentielles car complémentaires : elles permettent en effet une détermination plus fine de leur structure atomique et électronique. Nous illustrerons à travers quelques exemples l’apport de ces caractérisations, réalisées operando, pour la compréhension de systèmes de choix : ici, des matériaux d’électrodes positives de type polyanioniques (LiFePO4 et LiVPO4F) et oxydes lamellaires surlithiés (Li1.20Ni0.13Co0.13Mn0.54O2) [3-6]. Nous présenterons également pour la première fois la cellule électrochimique que nous avons développée pour réaliser de la diffraction des neutrons in situ, de résolution suffisante pour réaliser une détermination structurale par la méthode de Rietveld. Remerciements : Les auteurs remercient l’Institut de Recherche Européen Alistore-ERI, le Réseau Français sur le Stockage Electrochimique de l’Energie (RS2E), l’industriel Toyota Motor Europe, l’Institut Laue-Langevin (ILL-Grenoble), le Synchrotron SOLEIL et la Région Aquitaine pour leur soutien aux recherches présentées. [1] Goodenough et al., Chem. Mater. 2010, 22, 587-603. [2] Morcrette et al., Electrochimica Acta 2002, 47(19), 3137-3149. [3] Leriche et al., J. Electrochem. Soc. 2010, 157(5), A606-A610. [4] Ouvrard et al., J. Power Sources 2013, 229, 16-21. [5] Ateba Mba et al., J. Electrochem. Soc. 2012, 159(8), A1171-A1175. [6] Koga et al., J. Phys. Chem. C, soumise. [7] Bianchini et al., J. Electrochem. Soc., soumise.
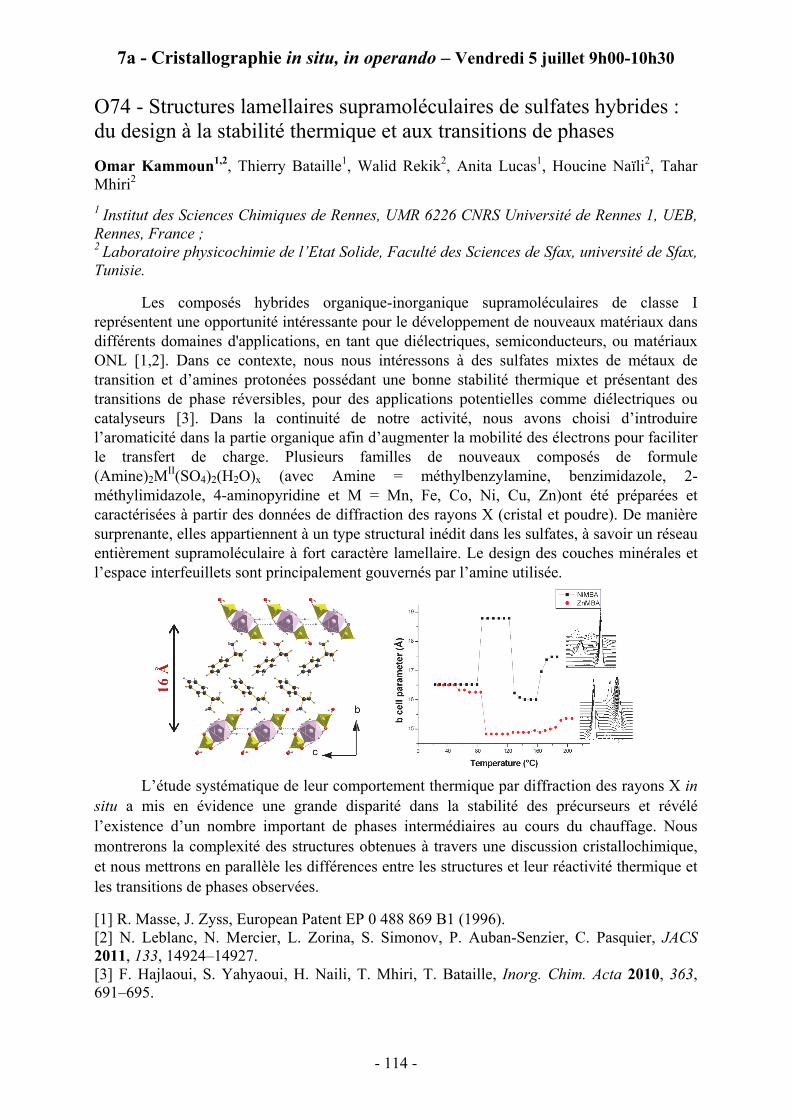
7a - Cristallographie in situ, in operando – Vendredi 5 juillet 9h00-10h30
- 114 -
O74 - Structures lamellaires supramoléculaires de sulfates hybrides : du design à la stabilité thermique et aux transitions de phases
Omar Kammoun1,2, Thierry Bataille1, Walid Rekik2, Anita Lucas1, Houcine Naïli2, Tahar Mhiri2 1 Institut des Sciences Chimiques de Rennes, UMR 6226 CNRS Université de Rennes 1, UEB, Rennes, France ; 2 Laboratoire physicochimie de l’Etat Solide, Faculté des Sciences de Sfax, université de Sfax, Tunisie.
Les composés hybrides organique-inorganique supramoléculaires de classe I représentent une opportunité intéressante pour le développement de nouveaux matériaux dans différents domaines d'applications, en tant que diélectriques, semiconducteurs, ou matériaux ONL [1,2]. Dans ce contexte, nous nous intéressons à des sulfates mixtes de métaux de transition et d’amines protonées possédant une bonne stabilité thermique et présentant des transitions de phase réversibles, pour des applications potentielles comme diélectriques ou catalyseurs [3]. Dans la continuité de notre activité, nous avons choisi d’introduire l’aromaticité dans la partie organique afin d’augmenter la mobilité des électrons pour faciliter le transfert de charge. Plusieurs familles de nouveaux composés de formule (Amine)2M
II(SO4)2(H2O)x (avec Amine = méthylbenzylamine, benzimidazole, 2-méthylimidazole, 4-aminopyridine et M = Mn, Fe, Co, Ni, Cu, Zn)ont été préparées et caractérisées à partir des données de diffraction des rayons X (cristal et poudre). De manière surprenante, elles appartiennent à un type structural inédit dans les sulfates, à savoir un réseau entièrement supramoléculaire à fort caractère lamellaire. Le design des couches minérales et l’espace interfeuillets sont principalement gouvernés par l’amine utilisée.
L’étude systématique de leur comportement thermique par diffraction des rayons X in
situ a mis en évidence une grande disparité dans la stabilité des précurseurs et révélé l’existence d’un nombre important de phases intermédiaires au cours du chauffage. Nous montrerons la complexité des structures obtenues à travers une discussion cristallochimique, et nous mettrons en parallèle les différences entre les structures et leur réactivité thermique et les transitions de phases observées.
[1] R. Masse, J. Zyss, European Patent EP 0 488 869 B1 (1996). [2] N. Leblanc, N. Mercier, L. Zorina, S. Simonov, P. Auban-Senzier, C. Pasquier, JACS 2011, 133, 14924–14927. [3] F. Hajlaoui, S. Yahyaoui, H. Naili, T. Mhiri, T. Bataille, Inorg. Chim. Acta 2010, 363, 691–695.

7b - Texture, Microstructure, déformation – Vendredi 5 juillet 9h00-10h30
O75 - Study of texture development in asymmetrically rolled titanium. Experimental study and calculations. M. Wronski1, K. Wierzbanowski1, S. Wronski1, B. Bacroix2, A. Lodini3 , P. Lipinski4
1 AGH University of Science and Technology, Faculty of Physics and Applied Computer Science, al. Mickiewicza 30, 30-059 Kraków, Poland, 2 LSPM-CNRS, Université Paris XIII, 99, av. J.B. Clement, 93 430 Villetaneuse, France 3 LISM, UFR SEN, Université de Reims Champagne Ardenne, Moulin de la Housse BP1039, 51687 Reims, France 4 LaBPS, ENIM, 1, Route d’Ars Laquenexy CS 65820, 57078 Metz Cedex 3, France
The goal of this work was to study the asymmetric rolling process using Finite Element (FEM) method coupled with the crystallographic deformation model of polycrystalline material. The Leffers-Wierzbanowski (LW) model was selected to be coupled with Finite Elements Method (FEM). The implementation of LW model into FEM enabled a study of heterogeneous plastic deformation process (with elastic interactions), like asymmetric rolling, taking into account its crystallographic nature. The experimental textures of asymmetrically rolled titanium were determined using Electron Back Scattering Diffraction (EBSD) technique. The observed trends of texture formation were confirmed by our calculations.
The studied asymmetric rolling process was realized using two identical rolls, driven
by independent motors, rotating with different angular velocities 1 and 2. This ensured a high range of rolling asymmetry. During asymmetric rolling a strong stress component is induced in the material. Our aim was to examine the influence of this shear stress on the microstructure and crystallographic texture.
It should be mentioned that asymmetric rolling offers numerous possibilities of material modification, it can provide large volumes of deformed material and is easy to implement on existing industrial rolling mills.

7b - Texture, Microstructure, déformation – Vendredi 5 juillet 9h00-10h30
- 116 -
O76 - Evolutions microstructurales et hétérogénéités de déformation dans les aciers ODS Fe-14Cr1W
S. Zhong1, V. Klosek1, Y. de Carlan2, V. Ji3, M.H. Mathon1
1 CEA, IRAMIS, Laboratoire Léon Brillouin (CEA – CNRS), F-91191 Gif-sur-Yvette cedex 2 CEA, DEN, SRMA, F-91191 Gif-sur-Yvette cedex 3 ICMMO / LEMHE, Univ. Paris Sud XI, F-91405 Orsay cedex
Les aciers ODS (Oxide Dispersion Strengthening) sont envisagés comme matériaux de
structure pour les réacteurs de future génération à neutrons rapides (RNR) ou à fusion. Le renforcement de ces matériaux par une dispersion fine d’oxydes de type Y2O3 a ouvert de nombreuses perspectives d’applications de ces aciers ODS. Ces systèmes sont élaborés par mécanosynthèse (mechanical alloying). Il est acquis que ce procédé permet d’obtenir ces matériaux nanostructurés avec mise en solution des atomes d’yttrium et d’oxygène introduits sous forme de nanocomposés durant le co-broyage, ce qui permet ensuite de contrôler la phase de précipitation des particules durcissantes. Les propriétés mécaniques étant étroitement dépendantes de la dispersion d’oxydes, la maitrise de la précipitation est un enjeu important dans le développement de ces aciers.
L’objectif de ce travail a été, d’abord, de préciser les cinétiques de précipitation ainsi
que les conditions de recristallisation entre 850° et 1450°C dans un matériau choisi comme référence (Fe-14Cr-1W-0,3Ti-0,3Y2O3) ainsi que dans 4 nuances présentant des teneurs en Ti, Y et O différentes, afin de mettre en évidence l’effet de la composition chimique sur le comportement de ce type d’alliage sous traitement thermique. La microstructure au sens large du terme (taille des grains, dispersion des oxydes, textures cristallographiques) a été caractérisée par diffraction et diffusion des neutrons ainsi que par microscopie électronique.
Dans un deuxième temps, l’impact de ces évolutions microstructurales sur les
propriétés mécaniques a été étudié. Une partie du travail a notamment été focalisée sur l’analyse sous sollicitation uni-axiale des hétérogénéités de déformation des grains en fonction de leur orientation cristallographique. Cette étude, réalisée sur plusieurs nuances avec différentes distributions de tailles de nanoparticules, s’est appuyée sur la technique de diffraction des neutrons pour l’analyse des déformations et des textures cristallographiques. Les comportements observés ont été confrontés à une modélisation micromécanique.

7b - Texture, Microstructure, déformation – Vendredi 5 juillet 9h00-10h30
- 117 -
O77 - Résolution et affinement de structure d’un film mince épitaxié de LaVO3.
Hélène Rotella1, Pascal Roussel2, Magali Morales3, Henni Ouerdane1, Philippe Boullay1, Olivier Copie1, Adrian David1, Daniel Chateigner1, Lucas Lutterotti4, Wilfrid Prellier1. 1 Laboratoire CRISMAT, CNRS UMR 6508, ENSICAEN et Université de Caen Basse-Normandie, 6 bd Maréchal Juin, 14050 Caen Cedex 4, France. 2 Laboratoire UCCS, CNRS UMR 8181, ENSCL, Cité Scientifique, Bâtiment C7 - BP 90108, 59652 Villeneuve d'Ascq Cedex, France. 3 Laboratoire CIMAP, CNRS UMR 6252, ENSICAEN et Université de Caen Basse-Normandie, 6 bd Maréchal Juin, 14050 Caen Cedex 4, France. 4 Dipartimento di Ingegneria dei Materiali, Univ. di Trento, I-38050 Trento, Italy E-mail: [email protected]
L’étude des pérovskites ABO3 synthétisées sous forme de films minces suscite depuis de nombreuses années un grand intérêt tant ils présentent un large spectre de propriétés fonctionnelles, telles que la ferroélectricité, la supraconductivité, ou encore les transitions métal-isolant. Ces propriétés, souvent liées à la structure de ces matériaux, nécessitent une détermination de plus en plus précise des propriétés structurales. La connaissance des angles B-O-B permet par exemple de comprendre les recouvrements orbitalaires responsables du type de transport des électrons au sein du matériau ou encore interviennent dans les interactions d’échange entre les spins des électrons.
Ce travail présente la résolution et l’affinement de la structure d’un film mince de LaVO3 réalisée par diffraction de rayons X en haute résolution et s’inscrit dans cette optique. L’orthovanadate de lanthane présente, sous forme massive, une structure orthorhombique Pnma commune dans les pérovskites, qui se déforme facilement sous la contrainte à travers les rotations d’octaèdres d’oxygènes. Le film mince ainsi déposé sur un substrat de SrTiO3 orienté (001), s’adapte pour compenser la contrainte qui lui est imposée [1]. Bien que le principe de cette étude soit de considérer le film comme un monocristal, on ne peut se soustraire aux contraintes de mesures imposées par la géométrie de l’échantillon. Sa taille est en effet un inconvénient majeur (l’épaisseur du film étant d’un peu moins d’une centaine de nanomètres) qui conduit à une intensité diffractée fortement diminuée. A cela, s’ajoute l’orientation préférentielle du cristal induite par la croissance épitaxiale du film sur le substrat monocristallin. En conséquence, déterminer la structure précise d’un film mince devient un challenge intéressant.
Dans cet exposé, je montrerai, en m’appuyant sur les développements récents de la diffraction des rayons X, qu’il devient possible d’accéder à un grand nombre de réflexions et, après une analyse fine, d’accéder à la structure complète d’un film mince d’oxyde.
[1] Rotella et al., Phys. Rev. B, 2012, 85, 184101

7c - Grands Instruments Synchrotron et plateformes en biologie structurale - (Développements, XFEL, nouveautés) – Vendredi 5 juillet 9h00-10h30
- 118 -
O78 - La cristallographie à SOLEIL Jean Daillant DSM/IRAMIS/SIS2M/LIONS CEA
L’année 2014 a été choisie par l’UNESCO pour être l’année internationale de la cristallographie. Dans ce cadre, je dresserai un panorama de la cristallographie à SOLEIL en insistant sur les axes stratégiques que nous avons définis :
Biocristallographie, résolution temporelle, utilisation de la cohérence, conditions extrêmes, mesures in operando et couplage de techniques.
Ces différents points seront illustrés par des exemples récents.

7c - Grands Instruments Synchrotron et plateformes en biologie structurale - (Développements, XFEL, nouveautés) – Vendredi 5 juillet 9h00-10h30
- 119 -
O79 - X-ray Free Electron Laser sources Marie-Emmanuelle Couprie Synchrotron SOLEIL
Since the first laser discovery in 1960 and the first Free Electron Laser (FEL) in 1977, Free Electron Lasers provide intense coherent fs pulses in the X-ray range for multidisciplinary investigations of matter. After introducing the concepts, and the various configurations, the panorama of the presently available facilities around the world will be described. The different properties of these sources will be presented. New recent trends will be givcn, such as two-color operation. Further prospects including multi- Free Electron Laser beamlines on superconducting linear accelerators, laser wakefield accelerators based Free Electrons Lasers will be open. Future advanced FELs will be discussed as well, in particular with the case of the LUNEX5 (free electron Laser Using a New accelerator for the Exploitation of X-ray radiation of 5th generation) project in France, which aims at investigating the production of short, intense, and coherent pulses in the soft X-ray region, with a 400 MeV superconducting linear accelerator and a laser wakefield accelerator (LWFA), feeding a single Free Electron Laser line with seeding with High order Harmonic in Gas and Echo Enable Harmonic Generation.

7c - Grands Instruments Synchrotron et plateformes en biologie structurale - (Développements, XFEL, nouveautés) – Vendredi 5 juillet 9h00-10h30
- 120 -
O80 - Nouvelles possibilités expérimentales en cristallographie introduites par les XFELs Jan Lüning Université Pierre et Marie Curie, Paris
Le rayonnement des nouveaux lasers à électrons libres émettant des rayons X (XFEL, pour X-ray Free Electron Laser) permet de réaliser des expériences entièrement nouvelles grâce aux propriétés uniques de leur rayonnement : des impulsions ultra-brèves (domaine de la femtoseconde) et ultra-intenses avec un très haut degré de cohérence. Ceci crée des nouvelles possibilités pour l'étude expérimentale dans une grande diversité de domaines scientifiques, aller de la physique à la chimie, la biologie et autres.
Pour leur exploitation, un grand nombre de nouvelles techniques expérimentales et
d'instrumentations originaux ont été conçus pendant les quelques années depuis le XFEL FLASH en Hambourg est devenu une source ouverte aux utilisateurs. Entre temps, il y a quatre XFELs qui servent une communauté d'utilisateurs en pleine croissance : les XUV-FELs FLASH et FERMI (Italie) et les XFELs LCLS (États-Unis) et SACLA (Japon).
Cette présentation commencera par un résumé de ce développement récent en mettant
l'accent sur les capacités et les défis expérimentaux qui dérivent des propriétés uniques de ces sources. Après une présentation plus générale de l'état de l’art des techniques de diffusion aux XFELs, quelques exemples récents concernant l'étude de la structure des nano-structures biologiques sont discutés en plus de détails.

7c - Grands Instruments Synchrotron et plateformes en biologie structurale - (Développements, XFEL, nouveautés) – Vendredi 5 juillet 9h00-10h30
- 121 -
O81 - BM14-2: une ligne MX MAD à l’ESRF avec de nouveaux outils pour exposer des cristaux biologiques à température ambiante.
Belrhali H1, Nanao M, Manjasetty B1, Zander U, Hoffmann G, Márquez JA1, Felissaz F, Gobbo A, Papp G, Cipriani F1, Nasri JA, Geoffroy L, Elhorga V, Lavault B, Siebrecht R2 1EMBL Grenoble Outstation,
2Arina.x
BM14 est la station expérimentale située sur l’aimant de courbure 14 de l’ESRF. Elle est dédiée aux expériences de cristallographie des macromolécules biologiques et notamment celles qui utilisent la méthode expérimentale MAD (Multiple Anomalous Diffraction).
BM14 a bénéficié en 2011 et 2012 d’un renouvellement complet de ses éléments optiques, augmentant la brillance de son faisceau d’un facteur 3 à 4 selon l’énergie utilisée (de 7 à 17 keV). Il en résulte pour l’utilisateur un temps moyen d’exposition de 5 à 7 secondes par image.
En collaboration avec les équipes de Florent Cipriani et de Jose-Antonio Marquez de l’EMBL, nous avons récemment développé une nouvelle tête goniométrique pour le microdiffractomètre de la ligne (MDII) qui permet de manipuler les plaques de cristallisation de type « Crystal Direct [2]» et ainsi offrir aux utilisateurs la possibilité d’exposer leur échantillons in situ, c'est-à-dire encore dans leur milieu de cristallisation, aux rayons X de la ligne de lumière. Ces plaques ont été optimisées pour ne diffuser que très faiblement sous les rayons-X minimisant le bruit accumulé sur les clichés de diffraction.
Nous présenterons les expériences initiales de collectes de données et in situ ainsi que leur traitement automatisé et les perspectives qu’offre ce nouvel instrument sur la ligne.
L’équipe de BM14 a également développé une collaboration avec l’entreprise Arinax un changeur de buse pour optimiser l’utilisation de la déshydratation contrôlée des échantillons à température ambiante [3]. Grâce à ce système, l’utilisateur peut bascule d’une expérience de déshydration à une expérience de collecte de données cristallographiques sous flux d’azote cryogénique à distance. Nous avons pu vérifier que le séchage de cristaux tests permet leur congélation sans ajout de cryo-protectant [4].
Nous nous proposons d’exposer les principes de ces développements et leur modus operandi ainsi que les premiers résultats enregistrés qui semblent prometteur.
[1] A. Thompson et al 1993-1994 (EMBL Report 1993-1994) [2] CrystalDirect: a new method for automated crystal harvesting based on laser-induced photoablation of thin films F. Cipriani, M. Röwer, C. Landret, U. Zander, F. Felisaz and J. A. Márquez Acta Cryst. (2012). D68, 1393-1399. [ doi:10.1107/S0907444912031459 ] [3] Improvement of the diffraction quality of macromolecular crystals at European synchrotrons using the HC1 dehydration control device U. Mueller, M. S. Weiss, J. Sanchez-Weatherby, T. L.-M. Sorensen, M. Thunnissen, T. Ursby, A. Gobbo, S. R. Russi, M. W. Bowler and F. Cipriani. Acta Cryst A. (2012). A68, s14 [4] Direct cryocooling of naked crystals: are cryoprotection agents always necessary? E. Pellegrini, D. Piano and M. W. Bowler. Acta Cryst. (2011). D67, 902-906

8a - Chiralité, polymorphisme – Vendredi 5 juillet 11h00-12h30
- 122 -
O82 - Formulations Pharmaceutiques et Polymorphisme Philippe Espeau1, Yohann Corvis1, Philippe Négrier2, Stéphane Massip3, Jean-Michel Léger3 1 EA 4066 « Physico-Chimie Industrielle du Médicament », Faculté des Sciences Pharmaceutiques et Biologiques, 4 avenue de l’Observatoire, F-75006 Paris. 2 LOMA, UMR CNRS 5798, Université Bordeaux, F-33400 Talence. 3 Pharmacochimie, FRE CNRS 3396, Université Bordeaux 2, 33000 Bordeaux.
Le polymorphisme revêt un grand intérêt en pharmacie, et plus particulièrement dans l’industrie pharmaceutique, pour des raisons d’activité thérapeutique mais aussi de brevetabilité. Un polymorphisme mal caractérisé ou méconnu peut entraîner des conséquences dramatiques tant d’un point de vue sanitaire (inefficacité, surdosage) que d’un point de vue financier. C’est pour la première raison que la recherche et la caractérisation des polymorphes pour une substance active donnée sont devenues obligatoires depuis une quinzaine d’années.
En effet, pour une substance active donnée, l’activité thérapeutique peut varier d’un
polymorphe à un autre car sa « biodisponibilité » n’est pas la même. La forme solide retenue sera celle qui répond à des exigences de stabilité physique et chimique, pharmacocinétiques, et donc d’activité thérapeutique. Si généralement, le choix se porte sur la forme solide la plus stable dans des conditions ordinaires de température et de pression, dans certains cas, une forme métastable peut lui être préférée pour des raisons de biodisponibilité ou de contournement de brevet. Tout ceci nécessite de procéder, en amont, à un criblage des polymorphes pour ensuite établir leur stabilité relative. Grâce à la calorimétrie et à la diffraction des rayons X, des règles simples de hiérarchisation peuvent être établies [1].
Le polymorphisme cristallin, qu’il soit lié à la substance active [2] ou aux excipients [3],
sera donc abordé sous l’angle thermodynamique et cristallographique. Son impact, en termes d’activité thérapeutique, de formulation galénique, mais également de brevet, sera discuté aux travers d’exemples. [1] H.G. Brittain, in « Polymorphism in pharmaceutical solids », Ed.Informa Healthcare, 2009. [2] P. Espeau et al., J. Pharm. Sci. 2005, 94(3), 524-39. [3] Y. Corvis et al., CrystEngComm. 2012, 14, 7055-64.

8a - Chiralité, polymorphisme – Vendredi 5 juillet 11h00-12h30
- 123 -
O83 - Configuration absolue et structure absolue: notions de base et évaluation
Howard D. Flack1 1Chimie minérale, analytique et appliquée, Université de Genève, Suisse
On traitera les questions de symétrie d’importance dans la compréhension et la détermination de la configuration absolue et la structure absolue. Les définitions de chiralité, énantiomorphe, énantiomère et racémate vont être exposées. On va considérer la formation de structures cristallines chirales et achirales à partir de molécules chirales et achirales. Le phénomène de maclage par inversion sera traité par les exemples du quartz et du hexahelicène. Par la suite, c’est la symétrie de l’image de diffraction en présence de la diffusion resonante qui va occuper notre attention. La modélisation de tout cristal non-centrosymétrique comme maclé par inversion en suit naturellement. Pour terminer la partie notions de base, il convient d’en déduire les règles concernant la détermination de la configuration absolue d’une molécule chirale à partir des mesures de diffraction.
Les travaux exposés sous le thème évaluation ont débuté en 2007. Ils concernent uniquement les structures cristallines non-centrosymétriques. On utilise la moyenne (A) et la différence (D) d’intensité des opposés de Friedel i.e. A(hkl) = ½[|F(hkl)|2 + |F(h̅k̅l ̅)|2], D(hkl) = |F(hkl)|2 - |F(h̅k̅l ̅)|2. Par un calcul des valeurs attendues de A et de |D|2 on arrive à Friedifstat qui quantifie a priori la grandeur du signale de la diffusion resonante d’un cristal à partir de sa composition chimique et la longueur d’onde de la radiation. On peut aussi calculer les valeurs Friedifobs et Friedifmodel, et les comparer avec Friedifstat, ce qui donne les indications intéressantes concernant la présence d’un centre de symétrie dans le cristal. Les graphiques de Aobs contre Amodel et de Dobs contre Dmodel permettent de visualiser l’accord entre paramètres. On observe souvent, même trop souvent, que les valeurs de Dobs sont dominées par les incertitudes aléatoires et les erreurs systématiques. L’utilisation de valeurs comme Rint ou Rmerge permet, dans les bons cas, de déterminer le groupe ponctuel du cristal. [1] Parsons, S., Pattison, P. & Flack, H.D., Acta Cryst. A. 2012, 68, 736-749. [2] Flack, H.D., Sadki, M., Thompson, A.L. & Watkin, D.J., Acta Cryst. A. 2011, 67, 21-34.

8a - Chiralité, polymorphisme – Vendredi 5 juillet 11h00-12h30
- 124 -
O84 - Composés multifonctionnels : transition de spin et chiralité Patrick Rosa, Ahmad Naim, Elen Duverger-Nedellec, Philippe Guionneau CNRS, Université de Bordeaux, ICMCB, 87 avenue du Dr. A. Schweitzer, Pessac, F-33608, France [email protected]
La synthèse de nouveaux composés à transition de spin est intéressante tant d'un point de vue fondamental, pour la compréhension physique des transitions de phase impliquées, que pour les applications technologiques éventuelles de ces composés. L'obtention de matériaux magnétiques non-centrosymétriques constitue un objet d'étude intéressant en optique non-linéaire (effet potentiel des électrons non-appariés), en cristallographie (mise en évidence récente d’un arrangement incommensurable d’états de spin) [1].
Pour obtenir des matériaux moléculaires à transition de spin non-centrosymétriques,
nous développons actuellement plusieurs stratégies au sein du groupe de Sciences Moléculaires de l'ICMCB. Ces stratégies passent par l'introduction de la chiralité par les deux composantes variables du matériau, les ligands du métal de transition (typiquement le Fe(II) ou le Co(II)) et les éventuels contre-anions. Nous exposerons les résultats récents que nous avons obtenus avec cette dernière approche, en détaillant l'utilisation de plusieurs familles d'anions chiraux, utilisées précédemment principalement en HPLC chirale et/ou différentiation énantiosélective en RMN, rarement pour la synthèse de matériaux moléculaires, mais jamais dans le contexte de la transition de spin: des tris(dioxolènes) de phosphate(V) (famille TRISPHAT développée par le Prof. Lacour de l'Université de Genève)[2] ou d’arsenic(V)[3], des adduits de (+)- ou (-)-tartrate avec As(III) ou Sb(III).[4] Nous présenterons les structures de nouveaux anions obtenus, ainsi que divers complexes de Fe(II) cristallisés en présence de ces anions. Nous montrerons que l'analyse des interactions chirales en phase solide peut présenter des résultats inversés par rapport aux résultats en solution (RMN, dichroïsme circulaire), et que celles-ci dépendent fortement, comme le phénomène de transition de spin lui-même, d’interactions intermoléculaires non-covalentes, en particulier les liaisons hydrogène. [1] E. Collet et collab., Phys. Rev. Lett., 2012, 109, 257206 1-5. [2]. J. J. Jodry et collab., Inorg. Chem., 2004, 43, 3329. [3] T. Ito et collab. Inorg. Nucl. Chem. Lett., 1971, 7, 1097-1102. B. A. Borgias, Inorg. Chem., 1986, 25, 1057-1060. [4] A. Zalkin, Inorg. Chem., 1973, 12, 1641-1646.

8b - De l'ordre local à l'ordre à longue distance – Vendredi 5 juillet 11h00-12h30
- 125 -
O85 - Apport du synchrotron pour des expériences combinées de diffraction des rayons X et de spectroscopie d’absorption des rayons X Dominique Thiaudière Synchrotron SOLEIL, l’orme des merisiers, Saint-Aubin. 91192 Gif sur Yvette
La diffraction des rayons X (XRD) et la spectroscopie d’absorption des rayons X (XAS) sont deux techniques d’analyse indispensables pour obtenir des informations sur la structure de matériaux. Puisque le rayonnement synchrotron permet de choisir aisément l’énergie du faisceau de photons, il est un outil incontournable pour des mesures XAS. En ce qui concerne la diffraction des rayons X, en plus du choix de l’énergie, les flux importants de photons permettent de réduire considérablement les temps d’enregistrement de diagrammes ou de clichés de diffraction. On peut aussi envisager d’étudier des matériaux dont les quantités de matière diffractantes sont très faibles. Enfin, les tailles de faisceau pouvant être de plus en plus réduites, les mesures XRD (mais aussi les mesures XAS) peuvent être menées localement en vue de cartographier des matériaux.
Les informations pouvant être recueillies par ces deux techniques sont dans la plupart des cas complémentaires, il n’est donc pas rare de devoir faire des mesures combinées. Celles-ci peuvent être dissociées dès lors que les conditions expérimentales n’ont pas pour conséquence une irréversibilité des propriétés des matériaux à étudier. Pour des études de transformation in-situ, ces mesures combinées en quasi-simultané deviennent importantes si on souhaite garantir que les deux expériences sont effectuées sur la même zone de l’échantillon et dans des conditions physico-chimiques (température, pression, sollicitation mécanique, processus réactionnels, etc) totalement identiques, ce qui est très important pour établir des corrélations entre les informations complémentaires données par les deux types de mesures.
Mon exposé débutera par un rappel des techniques d’analyse couramment utilisées sur les installations synchrotrons. J’expliquerai l’intérêt de combiner des mesures XRD et XAS pour obtenir des informations à courte, moyenne et grande distance. Je donnerai les conditions expérimentales optimales pour réussir de telles mesures. Enfin, je présenterai différentes études qui ont nécessitées des mesures combinées en quasi-simultané.

8b - De l'ordre local à l'ordre à longue distance – Vendredi 5 juillet 11h00-12h30
- 126 -
O86 - Structure d'oxydes fonctionnels: le rôle de l'échelle mésoscopique Michela Brunelli Institut Laue-Langevin, Grenoble, France.
Les oxydes fonctionnels présentent des propriétés physiques complexes et fascinantes liés à leur structure. La présence de fortes corrélations électroniques et/ou d’une importante concentration en défauts affectent leur structure à différentes échelles. En particulier, l’importance des inhomogénéités à une échelle nanométrique (et parfois à plus grande échelle) devient de plus en plus évidente.
Je présenterai dans cette contribution quelques résultats récents obtenus à l’ESRF et à l’ILL en collaboration avec le département de chimie de l’Université de Milan I. Nous avons effectués des mesures de diffraction X et neutronique sur des poudres d’oxydes à fort défaut de concentration de type Ce1-xTRxO2-x/2 où TR est une terre rare (ces oxydes servent d’électrolytes dans des piles à combustible). Les données ont été analysées par PDF afin de remonter aux inhomogénéités nanométriques. Grâce à l’extrême souplesse de cette technique, il est possible de se concentrer sur différentes échelles de longueur à partir des seules données de diffraction de poudre.
A partir des quelques exemples concernant ce type de matériau, je voudrais souligner la complémentarité entre les informations obtenues en utilisant les neutrons ou les rayons X, une analyse dans l’espace direct ou réciproque.
[1] E. Dagotto, Science 309 (2005) 257-262. [2] M. Coduri et al., Phys. Chem. Chem. Phys. 15 (2013), 8495 – 8505. [3] M. Scavini et al., Chem. Mater., 24 (2012) 1338-1345. [4] M. Coduri et al., Z. Kristallogr. 227 (2012) 272-79.

8b - De l'ordre local à l'ordre à longue distance – Vendredi 5 juillet 11h00-12h30
- 127 -
O87 - Désordre "quasi-liquide" de chaines d'alcane sous-confinement subnanométrique supramoléculaire.
Céline Mariette1, Bertrand Toudic
1, Philippe Rabiller
1, Laurent Guérin
1, Alexei Bosak
2
1Institut de Physique de Rennes,
2ESRF, Grenoble
Il existe une très vaste littérature concernant les phases uni-dimensionnelles de type "liquide" généralement sous confinement dans des matrices hôtes [1, 2]. Ce travail a été généralisé dans le cadre du confinement de molécules linéaires d'alcanes (CnH2n+2) dans une matrice hôte d'urée (CO(NH2)2). Ces composés d'inclusion sont connus pour être une famille prototype de matériaux apériodiques lorsque la molécule invitée est suffisamment longue (n supérieur à 13) [3]. Nous montrerons ici que des signatures spécifiques existent dans le cas des molécules invitées plus courtes, le cristal étant stable au delà de n=7. Le désordre translationnel y apparait alors important, pouvant autoriser de véritables glissements de ces molécules confinées. Cette ordre purement local dans la phase haute température "liquide quasi-unidimensionnel" conduit à froid à un ordre à longue distance soit commensurable avec le réseau hôte (n=8 et n=11) soit fabriquant un composite incommensurable intermodulé (n=7 [4] ou n=12). L'ordre local dans la phase "quasi-liquide" se traduit par une image de diffusion du sous-réseau invité particulièrement structurée. L'extrême richesse de ces informations en font des matériaux adaptés pour des études 3D-PDF.
FIGURE: Plans de diffusion associés à la périodicité de la molécule confinée d'alcane dans la matrice hôte d'urée, mesurés par rayonnement synchrotron sur la ligne de lumière ID23 (ESRF): à gauche, dans le n-nonane/urée et à droite, dans le n-dodécane/urée. [1] Heilmann et al., Phys. Rev. B, 1979, 20, 2, 751. [2] Albouy et al., Phys. Rev. B, 1987, 35, 173. [3] Toudic et al., Science, 2008, 319, 69. [4] Mariette et al., J. Chem. Phys. 2012, 136, 104507.

8c - Défis en cristallographie biologique – Success stories Vendredi 5 juillet 11h00-12h30
- 128 -
O88 - La myosine VI, un nanomoteur à contre-sens.
Anne Houdusse1, Amel Bahloul1, Julie Ménétrey1, Paola Llinas1, Tatiana Isabet1, Olena
Pylypenko1, Hannah Benisty1, Virginie Ropars1, Serena Sirigu1, Carlos Kikuti1, Helena
Sirkia1, H. Lee Sweeney2. 1Structural Motility, Institut Curie UMR 144, Paris, France 2University Pennsylvania, Philadelphia, USA
La myosine VI est le seul moteur de la superfamille capable de diriger la production de force dans une direction opposée à celle des autres le long des filaments d’actine. De nombreuses études structurales couplées à des expériences de molécule unique ont permis de déterminer les éléments structuraux et le mécanisme particulier de ce nanomoteur. Alors qu’il ne fait plus de doute que les réarrangements du domaine moteur sont similaires à ceux des autres myosines, la procession du moteur le long du filament d’actine requiert un découplage de son bras de levier. Ce qui rend le mécanisme de cette myosine plus proche de celui déjà décrit pour le mouvement de kinésines. Les défis restant à relever pour ce moteur consistent à décrire comment il s’assemble en dimère lors de la reconnaissance de partenaires et comment cela définit le rôle cellulaire qu’il peut jouer en transportant ou en ancrant ses partenaires au cytosquelette d’actine. [1] Ménétrey J*, Bahloul A*, et al. Nature (London) 435, 779-85, 2005. [2] Ménétrey J*, Llinas P*, et al. The structural basis for the large powerstroke of myosin VI. Cell 131:300-308, 2007. [3] Mukherjea M*, Llinas P*, et al. Myosin VI dimerization triggers an unfolding of a 3-helix bundle in order to extend its reach. Molecular Cell, 35:305-15, 2009. [4] Sweeney HL and Houdusse A, Myosin VI rewrites the rules for myosin motors, Cell, 141:573-82, 2010. [5] Ménétrey J*, Isabet T*, et al. Processive steps in the reverse direction require uncoupling of the lead head lever arm of myosin VI. Mol Cell. 48,75-86, 2012.

8c - Défis en cristallographie biologique – Success stories Vendredi 5 juillet 11h00-12h30
- 129 -
O89 - Control of the DNA mismatch repair and the meiosis recombination by eukaryotic MutL homologs. JB Charbonnier1, E Gueneau1, C Tellier-Lebegue1, P Legrand2, J Marquèz3, C Dherin4, S Boiteux4 1 UMR 8221, CEA, CNRS, Univ Paris Sud, iBiTec-S, Gif-s-Yvette 2 Synchrotron SOLEIL, Proxima 1, St Aubin 3 HTX platform, EMBL, Grenoble 4 UPR 4301, CNRS, Centre Biophysique Moléculaire, Orléans [email protected]
The DNA mismatch-repair (MMR) system corrects replication errors and is thus
essential for maintaining genome integrity. MMR factors are also involved in other DNA-metabolic processes including a correct meiotic recombination. These functions depend on MutL homolog heterodimers with Mlh1 as a common subunit. In human, MLH1 mutations underlie half of hereditary nonpolyposis colorectal cancers (HNPCC). We solved the crystal structures of MutLa (Mlh1/Pms1 heterodimer) C-terminal domain (CTD) from S. cerevisiae, alone and in complex with fragments derived from Mlh1 partners [1]. These structures reveal structural rearrangements and additional domains compared to the bacterial MutL counterparts and in particular they highlight a role of Mlh1 subunit in Pms1 endonuclease site. We determined the structures of the ternary complexes between MutLa-CTD and Exo1 or Ntg2 fragments reveal the binding mode of the MIP-box motif shared by several Mlh1 partners [2, 3, 4]. Finally, the structures provide a rationale for the deleterious impact of mutations in HNPCC. [1] Gueneau, E et al (2013) Nat Struct Mol Biol [2] Gellon, L et al (2002) J Biol Chem [3] Dherin, C, et al (2009) Mol Cell Biol [4] Zakharyevich, K et al (2010) Mol Cell

8c - Défis en cristallographie biologique – Success stories Vendredi 5 juillet 11h00-12h30
- 130 -
O90 - La survie en milieu riche en arséniate : le mécanisme moléculaire de discrimination du phosphate
Mikael Elias1, Alon Wellner1, Korina Goldin-Azulay1, Eric Chabriere2, Julia A. Vorholt3,
Tobias J. Erb3 & Dan S. Tawfik1 1Department of Biological Chemistry, Weizmann Institute of Science, Rehovot 76100, Israel. 2URMITE CNRS-Université de la Méditerranée, Marseille, France 3Institute of Microbiology, ETH Zurich, Switzerland
L’arséniate et le phosphate sont abondants sur notre planète et sont des espèces proches sur les plans physico-chimiques : pKa quasi identiques, charges similaires sur les atomes d’oxygène, et une taille qui ne diffère que de 4%. Le phosphate est pourtant indispensable à la vie (étant notamment un constituant essentiel de l’ADN) alors que l’arséniate est extrêmement toxique. Cette similarité a suscité l’idée que l’arséniate pourrait substituer le phosphate dans certaines niches [1]. Mais qu’il soit utilisé ou exclu, discriminer l’arséniate du phosphate est une tâche très complexe : toutes les enzymes qui utilisent le phosphate sont incapables de discriminer ces deux anions. Ceci constitue par ailleurs une des raisons principales de la très grande toxicité de l’arséniate. Les protéines sont-elles capables de discriminer ces deux ions ? Et si oui, par quel mécanisme ?
Nous nous sommes intéressés en particulier au système d’import cellulaire de
phosphate, qui est soumis à ce challenge dans les environnements riches en arséniate. Plus particulièrement, nous avons examiné les Phosphate-Binding Protein périplasmiques (PBPs) associées au transporteur ABC qui permettent l’import de phosphate. Nous avons pu montrer que toutes ces protéines sont capables de discriminer efficacement le phosphate de l’arséniate (>500 fois) [2]. Nous avons résolu la structure de l’une de ces PBPs, en présence de phosphate et d’arséniate, à des résolutions sub-atomiques (0.88-0.98Å). Les anions sont fixés et immobilisés par une constellation de liaisons hydrogène (12) avec la protéine et des interactions répulsives. Ce mode de fixation provoque la distorsion d’une liaison hydrogène courte, unique (Low barrier H-bond) par l’ion arséniate, 4% plus gros que le phosphate [2]. Ces déterminants structuraux permettent au système d’import de phosphate de fixer sélectivement le phosphate par rapport à l’arséniate, et ce même dans des environnements riches en arséniate tels que le Lac Mono (californie, USA).
[1] Wolfe-Simon et al., Science. 2011, 332, 1163-6. [2] Elias et al., Nature. 2012, 491, 134-7.

8c - Défis en cristallographie biologique – Success stories Vendredi 5 juillet 11h00-12h30
- 131 -
O91 - Integrated conformational and lipid-sensing regulation of ArfGEFs
Jacqueline Cherfils, Kaheina Aizel, Valérie Biou, Jorge Navaza, Mahel Zeghouf
Laboratoire d'Enzymologie et Biochimie Structurales, CNRS, Gif-sur-Yvette, France
The structural and biochemical mechanisms whereby GEFs coordinate their subcellular targeting to their nucleotide exchange activity remain poorly understood. I will present the crystal structure of an ArfGEF in a complex that mimics a membrane-bound intermediate combined with the analysis of its exchange activity reconstituted on membranes. This uncovered a novel regulatory mechanism mediated by an atypical PH domain, which restricts the activity of the GEF to specific membranes and potentiates its efficiency by about 2000-fold by concurrent optimization of membrane recruitment and nucleotide exchange.
- 132 -

- 133 -
Résumés des POSTERS
P1-70

- 134 -

Session Posters 1a/4a Matériaux fonctionnels (1 et 2)
- 135 -
P1 - Etude de différentes structures des conducteurs protoniques à base de sulfate, de phosphate et de sélénate avec le benzylamine Asma BEN RACHED1, Zakaria ELAOUD1, Arnaud GROSJEAN2, Philippe GUIONNEAU2, et Tahar MHIRI1 1Laboratoire de l’Etat Solide, Faculté des Sciences de Sfax, BP1171 Sfax Tunisie, 2CNRS, Univ. Bordeaux, ICMCB, UPR 9048, 87 avenue du Dr A. Schweitzer, F-33608 Pessac
La recherche et la caractérisation de nouveaux matériaux hybrides de type MHnXO4-n (M= ligand organique, X=S, Se, P, As …) constituent un thème de recherche très prometteur en cristallochimie. Ces matériaux présentent un intérêt considérable parce qu’ils rassemblent les propriétés physiques des éléments organiques et inorganiques [1,2]. Les applications de ces matériaux couvrent des domaines aussi variés que la bioélectronique ou l’optique par exemple [3,4]. Notre démarche est basée sur une synthèse systématique de composés cristallisés suivie de la détermination de leurs structures cristallines et de la nature des liaisons chimiques qui sont donc la clé de la compréhension du comportement physique de ces matériaux.
Dans cette optique nous poursuivons nos recherches sur les synthèses de benzylamine avec différents acides comme les acides sulfurique, phosphorique, phosphoreux et sélénique.
L’étude cristallographique par diffraction X a montré que ces matériaux ne sont pas isotypes bien que basés sur le même amine (M=C7 H9 N1). Le sulfate organique (X=S) cristallise dans le système monoclinique de groupe d’espace P21/c alors que le sélénate (X=Se) et le phosphate (X=P) organiques cristallisent dans le système triclinique de groupe d’espace P-1 tandis que le composé à base de l’acide phosphoreux cristallise dans le système orthorhombique de groupe d’espace Pbca. L’examen de ces structures montre une alternance de plans contenant des chaînes organiques [C6H5CH2NH3
+] et de plans formés de chaînes de tétraèdres [HSO4]
- ,[SeO3]2-, [HPO3]
- et [PO4]2-. Les plans inorganiques et organiques sont liés
entre eux par des liaisons hydrogène du type N–H…O.et O_H…O. Les liaisons inter-chaînes sont assurées par l’établissement d’interactions de type van der Waals. Toutes ces liaisons assurent la cohésion et la stabilité de l’édifice cristallin et forment ainsi un réseau tridimensionnel. [1] F.Z. Arrakhiz. Materials and Design.2013, 50, 376–381. [2] X. Yu. Dyes and Pigments . 2013, 98, 479-485. [3] A.K.Tatikonda. Biosensors and Bioelectronics .2013, 45, 201–205. [4] M.H. Ibrahim. Optik .2013, 124 , 1532– 1535.

Session Posters 1a/4a Matériaux fonctionnels (1 et 2)
- 136 -
P2 - Relation structure propriétés dans la solution solide (Ba1-xSrx)2NdFeNb4O15 Flora Molinari1,2, Eric Lebraud1,2, Rodolphe Decourt1,2, Dominique Michau1,2, Michaël Josse1,2
1 CNRS, ICMCB, UPR 9048, F-33600 PESSAC, FRANCE 2 UNIV. BORDEAUX, ICMCB, UPR 9048, F-33600 PESSAC, FRANCE
Des études céramiques sur des composés Ba2LnFeNb4O15 (Ln = La, Pr, Nd, Sm, Eu, Gd), de structure type « bronze quadratique de tungstène » (TTB), ont révélées des comportements diélectriques originaux : relaxeur pour Ln=La et Pr et ferroélectrique pour Ln=Nd, Sm, Eu [1]. L’étude des propriétés diélectriques dans la solution solide Ba2PrxNd1-xFeNb4O15, a permis de mettre en évidence pour certaines compositions (0,2 ≤ x ≤ 0,8) une transition relaxeur-ferroélectrique-paraélectrique lors du réchauffement [2]. Des comportements similaires (usuellement dénommés « crossover ») ont été obtenus dans d'autres solutions solides basées sur des mécanismes de substitution différents (par exemple, substitution Nb-Ta).
Dans ce poster, nous discuterons des résultats obtenus sur des céramiques au sein d’une nouvelle solution solide (Ba1-xSrx)2NdFeNb4O15, basée elle aussi sur le niobate ferroélectrique Ba2NdFeNb4O15. Les analyses MEB et les propriétés diélectriques, ainsi que les études structurales sur poudre par Diffraction des Rayons X réalisées à l’ambiante dans le groupe d’espace P4/mbm menées dans cette solution solide seront alors présentés.
[1] M. Josse et al., Solid State Sci., 2009, 11(6), 1118-1123 [2] E. Castel et al., J. Phys.: Condens. Matter 21 (2009) 452201

Session Posters 1a/4a Matériaux fonctionnels (1 et 2)
- 137 -
P3 - Elaboration, structure et caractérisation spectroscopique du phosphate NaxCayAlzTit(PO4)3
A. Nazih1, S. Pechev2, S. Krimi3, M. Velazquez2, M. Couzi4, A. El Jazouli1
1LCMS, URAC17, Faculté des Sciences Ben M'Sik-UH2M, Casablanca, Maroc. 2ICMCB-CNRS, Université de Bordeaux1, France. 3LPCMI, Faculté des Sciences Ain Chock – UH2C, Casablanca, Maroc. 4ISM-CNRS, Université de Bordeaux1, France.
Les matériaux phosphatés vitreux ou cristallisés sont très étudiés en raison de leur importance dans divers domaines : batteries, optique non linéaire, conducteurs ioniques, biomatériaux, lasers, etc. Nous avons montré précédemment que les phosphates cristallisés Na5-2xCaxTi(PO4)3 (0 < x <1) appartiennent à la famille Nasicon (Na super ionic conductors) et cristallisent dans le groupe d’espace non-centrosymétrique R32 [1-3]. Les essais de substitution, dans Na3CaTi(PO4)3 (x = 1), de l'aluminium au titane, nous a permis d’isoler un nouveau phosphate de formule NaxCayAlzTit(PO4)3. Nous présentons ici sa méthode de préparation, sa structure cristalline et sa caractérisation par Raman, infrarouge et absorption optique.
Le phosphate NaxCayAlzTit(PO4) a été préparé, sous forme de monocristaux et de poudre polycristalline, à partir de Na2CO3, CaCO3, Al2O3, TiO2 et (NH4)2HPO4. Les monocristaux ont été obtenus par fusion suivie d’un refroidissement lent. La poudre a été obtenue par réaction à l’état solide. La structure, résolue par diffraction des rayons X sur monocristal, appartient au type structural Nasicon, groupe d’espace R32. Les paramètres cristallins de la maille hexagonale équivalente sont : ah = 8,9976(1) Å et ch= 21,8312(3) Å. La structure est formée par un réseau 3D d’octaèdres AO6 (A=Na/Ca, Al/Ti) et de tétraèdres PO4 liés par les sommets. Les sites interstitiels, usuellement appelés M1 et M2, sont occupés par le sodium.
L’étude du phosphate NaxCayAlzTit(PO4) par spectroscopie vibrationnelle Raman et infrarouge et par absorption optique sera également présentée.
[1] S. Krimi, A. El Jazouli, L. Rabardel, M. Couzi, I. Mansouri and G. Le Flem. J. Solid State Chem., 1993, 102, 400-407. [2] S. Krimi, I. Mansouri, A. El Jazouli, J. P. Chaminade, P. Gravereau and G. Le Flem. J. Solid State Chem., 1993, 105, 561-566. [3] S. Krimi, A. El Jazouli, A. Lachgar, L. Rabardel, D. de Waal, J. R. Ramos-Barrado. Ann. Chim. Sci. Mat., 2000, 25 Supp. 1, S75-78. Remerciements : Nous remercions le CNRST/Maroc, le CNRS/France (Accord n° 24494) et le comité franco-marocain Volubilis (Action Intégrée n° MA/10/229), pour leur soutien financier.

Session Posters 1a/4a Matériaux fonctionnels (1 et 2)
- 138 -
P4 - Diffraction X et cristaux à conversion de spin : approches inédites et résultats nouveaux.
Sabine Lakhloufi1,2
, Philippe Guionneau2, Marie-Hélène Lemée-Cailleau
1
1Institut Laue-Langevin, 6 rue Jules Horowitz, BP 156, Grenoble, F-38042, France,
2CNRS, Université de Bordeaux, ICMCB, 87 avenue du Dr A. Schweitzer, Pessac, F-33608,
France
La diffraction des rayons X (DRX) est un outil privilégié pour l’étude fine de la matière, depuis l’échelle atomique jusqu’à celle de l’arrangement complet macroscopique. Les développements techniques récents de cette méthode, en matière de source de rayonnement, de détection et d’analyse de données, nous ont permis de développer deux approches innovantes pour l’étude des transitions de phase, offrant dans le cas de cristaux à conversion de spin une description du mécanisme de conversion depuis l’échelle de la première sphère de coordination de l’ion métallique jusqu’à celle, macroscopique, du cristal.
Une étude multi-structurale multi-température du complexe à conversion thermique graduelle [Fe(PM-AzA)2(NCS)2] (Fe-AzA), comprenant plus de 40 structures acquises par DRX entre 100K et 300K a donné lieu à l’élaboration de films de la conversion de spin, permettant de visualiser quasiment en continu toutes les modifications structurales induites par la commutation. Allié à l’étude multi-température du complexe isostructural à base de ZnII qui ne transite pas, ce procédé a permis de décorréler les effets thermiques de ceux de la conversion de spin, et d’identifier et quantifier précisément les conséquences propres de cette dernière, depuis l’échelle atomique à celle de la maille cristalline.
L’étude aux échelles supérieures, mésoscopique et macroscopique, a nécessité un protocole expérimental pionnier mis en œuvre au laboratoire. Basé sur l’évolution des forme et taille des taches de Bragg des clichés de DRX, il a permis le suivi de l’évolution de la qualité cristalline des matériaux pendant et au fil des conversions de spin. Appliqué aux cristaux de Fe-AzA, il a mis à jour une fatigabilité initiale en lien avec le mécanisme de conversion de spin, vieillissement inattendu dans un complexe à conversion graduelle [1].
Les avancées réalisées à l’aide de ces deux techniques concernent aussi bien le niveau fondamental, apportant des informations de base sur le phénomène de conversion de spin, que le niveau expérimental, ouvrant de nouvelles voies d’investigation, en allant jusqu’à des éléments intéressants le domaine de l’application industrielle. [1] Guionneau et al., Chem. Phys. Lett. 2012, 542, 52-55.

Session Posters 1b Surfaces et interfaces
- 139 -
P5 - Etude de la transition de phase cubique/quadratique dans des nanofils auto-organisés de FePt
David Babonneau1, Mathieu Garel
1, Frédéric Pailloux
1, Alexandre Boulle
2, Alessandro
Coati3, Yves Garreau
3,4
1Institut PPRIME, UPR CNRS 3346, Université de Poitiers, SP2MI, 11 Bvd Marie et Pierre
Curie, BP 30179, 86962 Futuroscope Chasseneuil Cedex 2Science des Procédés Céramiques et de Traitements de Surface, SPCTS, UMR CNRS 7315,
ENSCI, Centre Européen de la Céramique (CEC), 12 rue Atlantis, 87068 Limoges Cedex 3Synchrotron SOLEIL, L'Orme des merisiers, BP 48, 91192 Gif-sur-Yvette Cedex, France
4Université Paris Diderot, Sorbonne-Paris-Cité, MPQ, UMR 7162 CNRS, Bâtiment
Condorcet, Case 7021, 75205 Paris Cedex 13, France
Les nanofils ferromagnétiques présentent des propriétés physiques originales qui découlent de leurs dimensions sub-microniques et de leur géométrie spécifique. Ils offrent des perspectives nouvelles pour des applications, notamment dans le domaine du stockage de l’information, de l’électronique de spin et des dispositifs hyperfréquences. Dans cette étude, nous nous intéressons à des nanofils d’alliage FePt dont l’organisation, la morphologie, la composition et les transitions de phases sont autant de paramètres clefs gouvernant leurs propriétés physico-chimiques.
Les nanofils sont élaborés sur des surfaces d’alumine amorphe (Al2O3) préalablement nanostructurées par pulvérisation ionique en incidence oblique conduisant à l’obtention de rides périodiques unidirectionnelles [1,2]. Un co-dépôt de FePt est ensuite effectué en incidence rasante (5°) et à température ambiante par pulvérisation ionique. Une couche d’Al2O3 est finalement déposée pour recouvrir et enrober les nanofils, puis des traitements thermiques ex situ à 600°C sous vide sont effectués. L’organisation et la morphologie des nanofils ont été analysées avant et après traitement par GISAXS et HAADF-STEM, et les phases en présence ont été identifiées par diffraction des rayons X en incidence rasante et par diffraction électronique en aire sélectionnée filtrée en énergie. Nos observations montrent que la microstructure résultant de la croissance à température ambiante est assez bien préservée lors des traitements thermiques, qui entraînent par ailleurs une transition de phase cubique (désordonnée)/quadratique (ordonnée).
Afin de préciser la cinétique de mise en ordre chimique, des traitements thermiques in situ ont également été réalisés dans un microscope électronique. En utilisant un dispositif de détection des très faibles intensités diffractées et en procédant à un post-traitement des clichés obtenus, la détection des premiers stades de mise en ordre a ainsi pu être mise en évidence.
[1] Camelio et al., Phys. Rev. B 2009, 80, 155434. [2] Babonneau et al., Phys. Rev. B 2012, 85, 235415.

Session Posters 1c Nouvelles structures en biologie
- 140 -
P6 - One protein, many assemblies. Membrane fusion by a single viral glycoprotein involves formation of radically different pH-dependent oligomers. Eduard Baquero1*, Aurélie A. Albertini1*, Malika Ouldali1, Linda Buonocore2, John K. Rose2, Jean Lepault1+, Yves Gaudin1+, Stéphane Bressanelli1+ 1Centre de Recherche de Gif, Laboratoire de Virologie Moléculaire et Structurale, CNRS
(UPR 3296), Avenue de la Terrasse, 91198, Gif sur Yvette Cedex, France, 2Yale University School of Medicine, 310 Cedar St., New Haven, CT 06510, USA
Enveloped viruses enter cells through a membrane fusion reaction driven by
conformational changes of viral fusion glycoproteins. For some of these fusion glycoproteins, crystal structures have provided atomic, static pictures of pre- and post-fusion conformations, but intermediates in the transition have remained elusive [1]. Thus, in different pH conditions the vesiculovirus (family Rhabdoviridae) fusion protein G may form two kinds of symmetric, trimeric spikes. Each trimer is made of a different folding state of G [2] [3].
We report here a crystal structure in which G forms a flat heterotetramer with twofold symmetry. The tetramer is made up of two different refolding intermediates between the pre- and post-fusion states. The tetramer interfaces feature pH-dependent molecular switches and residues where mutants were previously reported to change the phenotype for rhabdovirus fusion.
These results extend the noted convergence of fusion proteins of so-called class III (e.g. Rhabdoviruses) and class II (e.g. flaviviruses) in a completely unexpected direction. In agreement with electron microscopy and tomography results of the states of G at the viral surface, we propose that the different assemblies of G act at different stages of fusion. This proposed division of labor has profound consequences on virally-catalyzed membrane fusion. [1] E. Baquero, A.A. Albertini, P. Vachette, J. Lepault, S. Bressanelli, Y. Gaudin, Curr. Opin. Virol. 2013. [2] S. Roche, S. Bressanelli, F.A. Rey, Y. Gaudin, Science 2006, 313, 187. [3] S. Roche, F.A. Rey, Y. Gaudin, S. Bressanelli, Science 2007, 315, 843.

Session Posters 1c Nouvelles structures en biologie
- 141 -
P7 - La protéine périplasmique AccA d’Agrobacterium tumefaciens : Etude de la relation Structure fonction El Sahili Abbas
1, Denis Faure
2, Solange Moréra1
1LEBS UPR CNRS 3082, 2ISV UPR CNRS 2355
Agrobacterium tumefaciens est une bactérie pathogène du sol qui cause, chez les
plantes, la maladie de la galle du collet se caractérisant par la formation de tumeurs colonisées par la bactérie. Ce mécanisme d’infection est bien connu et se compose de 4 étapes:
1- La blessure de la plante active chez A. tumefaciens le plasmide de virulence Ti (pTi) 2- Un fragment de ce pTi, le T-DNA est transféré dans le génome de la cellule
végétale 3- La plante sécrète alors des hormones de croissances végétales à l’origine des
tumeurs et de l’Agrocinopine qui sert de source d’énergie à la bactérie 4- L’Agrocinopine active également des mécanismes de quorum sensing chez la
bactérie qui conduisent à la propagation du pTi chez des bactéries non pathogènes les rendant pathogènes.
L’Agrocinopine est importée dans la bactérie par le système Acc, composé d’un transporteur ABC et d’une protéine périplasmique AccA qui reconnait l’Agrocinopine.
Cependant, des études ont montré qu’AccA était également la voie d’entrée de l’Agrocine 84, qui est un agent phytosanitaire qui tue A. tumefaciens [1].
Il s’agit donc de comprendre la spécificité de l’interaction d’AccA avec l’Agrocinopine et l’Agrocine 84 par une étude structure fonction.
Je présenterai mes premiers résultats de doctorat (débuté en Octobre 2012) sur cette étude à travers des structures cristallines obtenues et des mesures de microcalorimétrie. [1] Kim, H. & Farrand, S. K. (1997). Journal of Bacteriology. 179, 7559–7572.

Session Posters 1c Nouvelles structures en biologie
- 142 -
P8 - Le criblage des conditions de cristallisation des protéines est sous-échantillonné. Fabrice Gorrec MRC Laboratory of Molecular Biology, Cambridge, UK
Les conditions de cristallisation qui ont permis d'obtenir les structures de cristaux de protéines au MRC Laboratory of Molecular Biology (MRC-LMB, Cambridge, Angleterre) ont été analysées [1]. Plus un agent cristallisant est employé pour formuler les conditions de nos kits de criblages, plus fréquemment il apparait dans les formulations de conditions pour nos structures publiées. Cette analyse montre que, malgré la grande variété des agents employés [2], ils ont tous le même impact sur nos chances de succès d'obtenir les structures à partir de cristaux. Et surtout, l'analyse suggère que même si nous employons de nombreux kits de criblage, ce criblage est sous-échantillonné. [1] Gorrec, J. Appl. Cryst. 2013, in press. [2] Stock et al., Prog. Biophys. Mol. Biol. 2005, 88, 311-327.

Session Posters 2a Chimie supramoléculaire et ingénierie moléculaire
- 143 -
P9 - Service de Radiocristallographie de l’Institut de Chimie de Strasbourg Corinne Bailly, Lydia Brelot Service de Radiocristallographie, Institut de Chimie de Strasbourg (UMR 7177), 1 rue Blaise Pascal, B.P.296 R8, 67008 Strasbourg CEDEX, France
Nous vous présentons ici le Service de Radiocristallographie de l’Institut de Chimie de Strasbourg. Ce service assure la détermination de structures moléculaires et/ou supramoléculaires par diffraction des rayons X sur monocristaux de tous composés, produits naturels ou produits de synthèse.
Le service dispose de deux diffractomètres : un diffractomètre 4-cercles CCD Bruker
Kappa APEX II DUO IµS équipé de deux sources (tube scellé au molybdène et micro-source au cuivre) ainsi qu’un diffractomètre 4-cercles Nonius Kappa CCD équipé d’un tube scellé au molybdène. Ces deux diffractomètres sont chacun équipés d’un système cryogénique Oxford Cryosystems pour les enregistrements à basse température.
Le service dispose également d’un stéréomicroscope LEICA M125 à lumière polarisée, équipé d’une caméra permettant de prendre en photo les cristaux.
Le service étant ouvert à toute la communauté strasbourgeoise ainsi qu’à tout
laboratoire extérieur, la nature des échantillons étudiés est très variée : composés supramoléculaires (grilles de mercure [1]), porphyrines [2], composés organométalliques (complexes de titane [3]), composés organiques [4], etc… La microsource au cuivre permet également de déterminer la configuration absolue de la structure d’un composé énantiomériquement pur.
[1] J.Ramirez, A.-M.Stadler, Z-Anorg.Allg.Chem. 2009, 635, 1348-1351. [2] J.Taesch, T.T.Dang, V.Heitz, Tetrahedron Lett. 2012, 53, 333-337. [3] C.Romain, L.Brelot, S.Bellemin-Laponnaz, S.Dagorne, Organometallics. 2010, 29, 1191-1198. [4] B.Manteau, P.Genix, L.Brelot, J.-P.Vors, S.Pazenock, F.Giornal, C.Leuenberger, F.R.Leroux, Eur.J.Org.Chem. 2010, 6043-6066.

Session Posters 2a Chimie supramoléculaire et ingénierie moléculaire
- 144 -
P10 - Résolution d’une macle lors de la détermination structurale par diffraction X d’un complexe de cuivre
Carine Duhayon, Jean-Pierre Costes
CNRS, LCC (Laboratoire de Chimie de Coordination), 205 route de Narbonne, BP 44099, F-31077 Toulouse Cedex 4.
Le ligand 2-hydroxy-N'-[(2-hydroxy-3-methoxyphenyl)methylidene]benzohydrazide possède trois fonctions déprotonables et deux sites de coordination devant permettre de ce fait la formation de complexes hétérométalliques, soit par synthèse directe, soit par synthèse multi-étapes. Afin de mieux comprendre l’affinité des ions métalliques vis-à-vis des sites possibles de coordination, nous avons d’abord envisagé la complexation d’un seul centre métallique, en l’occurrence l’ion cuivre(II) en présence d’un excès de pipéridine qui joue le rôle d’agent déprotonant mais qui peut aussi se complexer au cuivre.
Lors de la détermination structurale par diffraction des rayons X sur les monocristaux obtenus, nous avons été confrontés à la présence d’une macle par pseudo-mérihédrie. Nous montrerons comment les outils logiciels (en particulier Crystals[1], Platon[2] et Rotax[3]) mis à la disposition du cristallographe ont permis de trouver la bonne maille cristalline et la loi de macle, le traitement final aboutissant à la résolution de cette structure.
Figure. Vue de la molécule obtenue
Le résultat obtenu confirme la formation d’un complexe mononucléaire neutre. L’ion cuivre se trouve dans un environnement plan carré, lié à deux atomes d’oxygène et un atome d’azote du ligand, la quatrième position étant occupée par l’atome d’azote de la pipéridine. Jusqu’à présent les ligands similaires à celui utilisé ici conduisent à des complexes tétranucléaires,[4] avec les quatre centres métalliques positionnés aux sommets d’un carré. L’utilisation de la pipéridine permet donc d’obtenir un complexe simple qui pourra servir à la formation ultérieure d’entités hétérométalliques.
[1] Betteridge, P.W., Carruthers, J.R., Cooper, R.I., Prout, K. & Watkin, D.J. 2003, J. Appl. Cryst. 36, 1487. [2] A.L.Spek, Acta Cryst. 2009, D 65, 148-155 [3] R. I. Cooper, R. O. Gould, S. Parsons and D. J. Watkin, 2002, J. Appl. Cryst., 35, 168-174 [4] Y. S. Moroz, K. Kulon, M. Haukka, E. Gumienna-Kontecka, H. Kozlowski, F. Meyer, I. O. Fritsky, Inorg. Chem. 2008, 47, 5656.

Session Posters 2a Chimie supramoléculaire et ingénierie moléculaire
- 145 -
P11 - Structure Elucidation of Host-Guest Complexes of Tartaric and Malic Acid by Quasi-Racemic Crystallography
Guillaume Lautrette,1 Brice Kauffmann,1 Yann Ferrand,1 Christophe Aube,2 Nagula Chandramouli,1 Didier Dubreuil,2 Ivan Huc1 1 Université de Bordeaux, CBMN, UMR5248 Institut Européen de Chimie et Biologie 2 rue Escarpit, 33600 Pessac, France and CNRS, CBMN, UMR5248 2 Université de Nantes, CEISAM, UMR6230 Faculté des Sciences et des Techniques 2 rue de la Houssinière, BP 92208 44322 Nantes Cedex 3, France and CNRS, CEISAM, UMR6230
The propensity of racemic solutions of organic molecules to frequently produce racemic crystals and to only rarely resolve into crystals containing exclusively one enantiomer (conglomerates) has been known for long. Over a decade of foldamer chemistry has provided strong empirical evidence that this observation also holds true in the case of helical aromatic foldamers. Racemic crystallography has been used to solve the structure of some proteins that can be produced by chemical synthesis and which were shown to crystallize more readily as a racemic pair than as a single enantiomer.[1] As an extension to racemic crystallography, quasi-racemates are sometimes found to co-crystallize, as was originally described by Pasteur for malate and tartrate salts.[2] A quasi-racemic crystal comprise a pair of molecules the structures of which are almost, but not exactly, mirror images.[3]
In this poster, we introduce the use of quasi-racemic crystallography as a method to elucidate the structures of host-guest foldamers complexes of tartaric and malic acid. We took advantage of the high propensity of enantiomeric P and M helices to co-crystallize, and demonstrated that co-crystals still form even when the guests introduced in the cavities are not mirror images. [1] (a) C. Toniolo, C. Peggion, M. Crisma, F. Formaggio, X. Shui, D.S. Eggleston, Nat. Struct. Biol. 1994, 1, 908–914; (b) B. L. Pentelute, Z. P. Gates, V. Tereshko, J. L. Dashnau, J. M. Vanderkooi, A. Kossiakoff, S. B. H. Kent, J. Am. Chem. Soc. 2008, 130, 9695– 9701. [2] (a) L. Pasteur, Ann. Chim. Phys. 1853, 28, 437; (b) K. A. Wheeler, R. C. Grove, R. E. Davis, W. S. Kassel, Angew. Chem. 2008, 120, 84–87; Angew. Chem. Int. Ed. 2008, 47, 78–81. [3] (a) D. E. Mortenson, K. A. Satyshur, I. A. Guzei, K. T. Forest, S. H. Gellman, J. Am. Chem. Soc. 2012, 134, 2473; (b) K. Mandal, B. L. Pentelute, D. Bang, Z. P. Gates, V. Yu. Torbeev, S. B. H. Kent, Angew. Chem. 2012, 124, 1510–1515; Angew. Chem. Int. Ed. 2012, 51, 1481.

Session Posters 2a Chimie supramoléculaire et ingénierie moléculaire
- 146 -
P12 - Synthèse et caractérisation nouveaux carboxylates hétérométallique impliquant le cation uranyle Natacha Henry, Ionut Mihalcea, Clément Falaise, Christophe Volkringer, Thierry Loiseau
Unité de Catalyse et Chimie du Solide (UCCS) – UMR CNRS 8181, Université de Lille, USTL-ENSCL, BatC7, BP 90108, 59652 Villeneuve d’Ascq
La chimie des actinides a connu un net regain d’intérêt ces dernières années, qui devrait s’accentuer avec le renouvellement du parc électronucléaire et notamment vers le développement d’un nucléaire durable. Dans ce cadre, la maîtrise des degrés d’oxydation des actinides au sein de ces composés constitue un objectif important afin de mieux relier les propriétés du solide aux caractéristiques redox originales de ces éléments. Des systèmes comportant l’uranium au degré d’oxydation +6 et un lanthanide (permettant de simuler le comportement du plutonium ou des actinides mineurs) ou des métaux de transition (présents dans le cycle du combustible) sont donc privilégiés, en leur associant différents anions organiques (carboxylates).
Les synthèses et les structures d’hétérométallique de carboxylates formés d'uranyle associé à un lanthanide trivalent (Ce, Nd) ou un métal de transition divalent (Cu, Zn) seront présentées. Dans le cas de lanthanide-uranyle mixte, nous discuterons d'une configuration unique d’interaction cation-cation avec UVI=O-LnIII (Ln = Ce, Nd) (figure a), ainsi que de leur dégradation thermique en oxydes mixtes (U,Ln)O2 dans le perspective de produire de nouveaux types de combustibles nucléaires.[1,2]
La synthèse de composés mixtes contenant cuivre ou zinc[3,4] mène à une série dont certains ont un comportement de déshydratation inattendu (figure b).
[1] C. Volkringer, N. Henry, S. Grandjean, T. Loiseau, J. Am. Chem. Soc. 2012, 134, 1275. [2] I. Mihalcea, C. Volkringer, N. Henry, T. Loiseau, Inorg. Chem. 2012, 51, 9610. [3] J. Olchowka, C. Falaise, C. Volkringer, N. Henry, T. Loiseau, Chem. Eur. J. 2013, 19, 2012. [4] J. Olchowka, C. Volkringer, N. Henry, T. Loiseau, Eur. J. Inorg. Chem. 2013 2109.

Session Posters 2a Chimie supramoléculaire et ingénierie moléculaire
- 147 -
P13 - Matériaux hybrides organiques inorganiques : synthèse et
caractérisation
Insaf ABDI et Amor BENALI
Unité de Recherche synthèse et structure de nanomatériaux, Université de Carthage, Faculté des Sciences de Bizerte 7021, Jarzoune, Bizerte, Tunisie [email protected]
Les matériaux hybrides organique-inorganique sont généralement élaborés par voie hydro(solvo)thermale en condition sub-critique par voie classique en utilisant des autoclave type Parr ou par chauffage micro-ondes. Deux types de composés hybrides sont obtenus : les composés hybrides type I où les interactions entre les deux sous-réseaux sont faibles (liaisons hydrogène, liaison de type Van der Waals et les composés hybrides type II ou MOF (Metal Organic Framework), où des liaisons fortes (covalentes) s’établissent entre les sous-réseaux inorganique organique. Compte tenu de leurs porosité élevée, ces composés peuvent avoir des applications dans plusieurs domaines tel que : chimie du pétrole, catalyse, stockage-séparation de gaz (H2, CO2, CH4,...).
Dans la littérature peu de travaux concernent les fluorures hybrides. L’équipe
allemande, Kemnitz et al., a étudié jusqu’à la fin des années 90, différents systèmes chimiques impliquant des cations métalliques tels que Al3+, Mn2+ ou Fe3+. Durant cette même période, Gerasimenko et al., ont synthétisé de nombreux fluorures à base de zirconium en particulier un fluorozirconate de guanidinium tridmensionnel [H3O]·[Hgua]5.[ZrF5]6 [1]. Ce dernier présente des cavités de diamètre proche de 5 Å. Quelques années plus tard, Lightfoot et al. ont élaboré des fluorures de vanadium [2] ou d’yttrium hybrides [3] présentant des structures tridimensionnelles.
C’est dans ce contexte que nous nous sommes intéressés à l’investigation des systèmes
FeF3 – amine - HF – éthanol/H2O par synthèse hydrothermale classique. Une nouvelle phase a été mise en évidence de formulation [C2H7N4]2 FeF6 (F) H2O. Sa structure monoclinique a été déterminée sur monocristal dans le groupe d’espace Pc. Elle peut être décrite en termes d’octaèdres FeF6 isolés et de cations organiques [C3H7N4]
2+. La caractérisation du fluoroferrate hybride par spectrométrie Mössbauer indique un environnement octaédrique et une valence trois pour les cations Fe3+.
Mots clés : hybdride, hydro(solvo)thermale, DRX
[1] A.V. Gerasimenko, B.V. Bukvetskii, V.B. Logvinova, R.L. Davidovich, Koord. Khim., 22 (1996) 584. [2] D.W. Aldous, N.F. Stephens, P. Lightfoot, Inorg. Chem., 46 (2007) 3996. [3] N.F. Stephens, Philip Lightfoot, J. Solid State Chem., 180 (2007) 259.

Session Posters 2a Chimie supramoléculaire et ingénierie moléculaire
- 148 -
P14 - Evidence of the unprecedented conversion of intermolecular proton- to water-bridging two phosphoryl metal complexes Rémy Sylvain,1 Laure Vendier,1 Christian Bijani,1 Antonio Santoro,2 Fausto Puntoriero,2 Sebastiano Campagna,2 Pierre Sutra1 and Alain Igau,1 1Laboratoire de Chimie de Coordination, UPR-CNRS 8241, 205 Route de Narbonne, 31077 Toulouse cedex 4, France, 2Dipartimento di Scienze Chimishe, University of Messina, Via Sperone 31, I-98166 Vill. S.Agata, Messina, Italy
We evidenced the conversion of an unprecedented intermolecular bridged-proton bimetallic P-metallated phosphoryl complex [{[Ru](Ph 2 P=O)} 2 H][PF 6] 3 to its corresponding H-bonded water complex [{[Ru](Ph 2 P=O)} 2 (H 2 O)][PF 6] 2, a rare motif. Surprisingly and to the best of our knowledge, hydrogen-bonded dimers of P-metallated phosphoryl complexes {[L n M](R 2 P=O) 2 }H] have not yet been described in the literature [1-3]. Moreover, the H-bonded water bridging intermolecularly two P=O unit is a rare motif [4-6].
Their synthesis, NMR data, X-ray crystal structures, electrochemical and photo physical properties were performed.
We have demonstrated that the P=O group in the polypyridine ruthenium(II) complex {[Ru](Ph 2 PO)]} 1 is a good hydrogen-bond acceptor to form quantitatively the intermolecular proton-bridged [{[Ru](Ph 2 P=O)} 2 H]3+2 and water-bridged [{[Ru](Ph 2 P=O)} 2 -(H 2 O)]2+3 dimer complexes which have been successfully characterized in solution and their structure determined by X-ray crystallography. We have demonstrated that the unprecedented intermolecular H-bonded water bimetallic complex 3 is formed through the formation of the proton-bridged complex 2. Our studies emphasize the ability of P-metallated P=O unit to act as a proton acceptor. Hydrogen bonding belongs to the panel of the interactions that hold groups of molecules together. Therefore, the properties of P=O unit to form hydrogen bonding pave the way for future investigations in many areas. We are therefore currently exploring the potential of bridged hydrogen-bonded organic entities to P-metallated phosphoryl complexes in crystal engineering.
[1] Dupont et al., Acta Cryst. D. 2013, 04, 3-6 ; B. Kurscheid, H.-G. 30 Stammler, B. Neumann and B. Hoge, Z. Anorg. Allg. Chem., 2012, 638, (3-4), 534. [2] T. Achard, L. Giordano, A. Tenaglia, Y. Gimbert and G. Buono Organometallics, 2010, 29, 3936 [3] A. Christiansen, D. Selent, A. Spannenberg, W. Baumann, R. Franke and A. Börner, Organometallics, 2010, 29, 3139. [4] C. Givelet, B. Tinant, L. Van Meervelt, T. Buffeteau, N. Marchand-Geneste and B. Bibal, J. Org. Chem., 2009, 74, 652 [5] G. Guerrero, P. H. Mutin, F. Dahan, and A. Vioux J. Organomet. 40 Chem., 2002, 649, 113. [6] P. Calcagno, B. M. Kariuki, S. J. Kitchin, J. M. A. Robinson, D. Philp, and K. D. M. Harris, Chem. Eur. J., 2000, 6, 2338.

Session Posters 2c Enzymologie et dynamique
- 149 -
P15 - Caractérisations biochimique et structurale des glutathion transférases du basidiomycète Phanerochaete chrysosporium Claude Didierjean1, Pascalita Prosper1, Thomas Roret1, Mélanie Morel2, Eric Gelhaye2, Frédérique Favier1 1CRM2 UMR UHP-CNRS 7036, faculté des sciences et technologies, Université de Lorraine, BP 70239 54506 Vandoeuvre-lès-Nancy, France. [email protected] 2IAM, UMR UHP-INRA 1136, faculté des sciences et technologies, Université de Lorraine, BP 70239 54506, Vandoeuvre-lès-Nancy, France.
Les interactions entre les champignons et les plantes constituent un des exemples les plus intéressants pour l’écologie. D’un côté, les champignons symbiotiques (tels que les mycorhiziens) ont la capacité d’amplifier la production des matières premières végétales et d’un autre côté les pathogènes peuvent engendrer des conséquences dramatiques sur les cultures. Les séquençages récents de nombreux génomes de champignons ont l’ambition d’apporter de nouvelles informations pour comprendre ces interactions en identifiant de nouvelles enzymes fongiques impliquées, par exemple, dans les processus de dégradation du bois1. Notre analyse s’est focalisée sur les glutathion transférases (GST) qui constituent une vaste superfamille d’enzymes classées comme des enzymes du métabolisme secondaire. Dans ce projet, nous tentons de caractériser toutes les GST présentes dans le champignon saprophyte de la pourriture blanche Phanerochaete chrysosporium en étudiant leurs spécificités de substrat et leurs relations structure-fonction. Notre analyse phylogénétique des GST fongiques a permis de montrer que les champignons saprophytes possèdent en général un plus grand nombre de séquences codantes pour les GST (>25) que les autres champignons2. Par exemple, la levure Saccharomyces cerevisiae en possède 11 alors que P. chrysosporium en possède 27. Ainsi, nous avons pu classer les GST fongiques en cinq classes : Omega, Ure2p, GTT1, GTT2 et GSTFuA dont plusieurs sont spécifiques aux champignons. Nos études biochimiques et structurales récentes ont mis en évidence de nouvelles classes structurales de GSTs avec des propriétés parfois non catalytiques mais plutôt de transport3. Ces propriétés seront présentées et les structures cristallographiques seront resituées dans la superfamille structurale des GST. [1] Eastwood et al., Science. 2011, 333, 762-765 [2] M. Morel et al., Cell. Mol. Life Sc. 2009, 66, 3711-3725. [3] Meux et al., J. Biol. Chem. 2011, 286, 9162-9173 ; Mathieu et al., J. Biol. Chem. 2012, 287, 39001-39011 ; Meux et al., FEBS Letters, 2012, 586, 3944-3950 ; Thuillier et al., 2013, FEBS Letters, accepté.

Session Posters 2c Enzymologie et dynamique
- 150 -
P16 - Étude structurale et fonctionnelle de la pseudouridine synthase PUS1 Tiphaine Huet1, Jeffrey Patton2 et Stéphane Thore1
1 Department of Molecular Biology, University of Geneva, Sciences III, Quai Ernest Ansermet 30, 1211 Geneva, Switzerland. 2 Department of Pathology, Microbiology, and Immunology, University of South Carolina, School of Medicine, 6439 Garners Ferry Road, Columbia, SC 29208, United States.
Considéré comme le “cinquième” nucléotide, la pseudouridine est le nucléotide modifié le plus abondant. Elle résulte de l’action des enzymes pseudouridine synthases. Ces enzymes sont responsables de la modification des ARNs non-codant, tels que les ARNt, RNAr [1]. Récemment, il a été montré que la pseudouridine synthase PUS1 modulait les réponses des récepteurs nucléaires de class I et II en impliquant l’ARN non-codant SRA (Steroid receptor RNA Activator) [2]. Le SRA intervient dans la signalisation du récepteur de l’acide rétinoïque (RARγ) et présente plusieurs pseudouridines, suggérant une nouvelle classe de substrats pour l’enzyme PUS1 [3]. Ces résultats indiquent un nouveau niveau de régulation de la transcription induite par les récepteurs nucléaires.
Nous présentons ici les modèles atomiques, obtenus par cristallisation, des enzymes PUS1 sauvage et de sa forme inactive mutée D146A. Les constantes d’affinité et les activités ont été caractérisées pour de ces deux formes de PUS1 en complexe avec plusieurs substrats SRA. Ces résultats révèlent les bases moléculaires de la reconnaissance du SRA par l’enzyme PUS1.
[1] Patton, J.R. Multiple pseudouridine synthase activities for small nuclear RNAs. Biochem J (1993) 290: 595-600. [2] Zhao, X., Patton, J.R., Davis, S.L., Florence, B., Ames, S.J. and Spanjaard, R.A. Regulation of nuclear receptor activity by a pseudouridine synthase through post-transcriptional modification of steroid receptor RNA activator. Mol Cell (2004) 15: 549-558. [3] Hao, X., Patton, J.R., Ghosh, S.K., Fischel-Ghodsian, N., Shen, L. and Spanjaard, R.A. Pus3p- and Pus1p-dependent pseudouridylation of steroid receptor RNA activator controls a functional switch that regulates nuclear receptor signaling. Mol Endocrinol (2007) 21: 686-699.

Session Posters 2c Enzymologie et dynamique
- 151 -
P17 - Etudes structurales et fonctionnelles de deux laminarinases de la bactérie marine Zobellia galactanivorans Aurore Labourel1,2, Murielle Jam1,2, Alexandra Jeudy1,2, Jan-Hendrik Hehemann1,2,
Mirjam Czjzek1,2 et Gurvan Michel1,2
1UPMC Université Paris 6 et 2CNRS, UMR 7139 Végétaux Marins et Biomolécules, Station Biologique de Roscoff, F-29682 Roscoff, Bretagne, France
Les algues brunes sont des organismes photosynthétiques marins ayant un
métabolisme du stockage du carbone particulier. Le D-fructose-6-phosphate issue de la photosynthèse n’est pas converti en saccharose et en amidon comme chez les plantes terrestres, mais en D-mannitol et en laminarine, un beta-1,3-glucane vacuolaire portant des branchements occasionnels en beta-1,6 [1]. La laminarine peut être utilisée comme source de carbone par des bactéries marines hétérotrophes. Tel est le cas de la flavobactérie Zobellia galactanivorans isolée de l’algue rouge Delesseria sanguinea [2] et véritable modèle émergent pour la bioconversion des polysaccharides d’algues [3-6]. L’analyse de son génome séquencé révèle la présence de 5 laminarinases putatives (4 GH16 et 1 GH64) présentant des architectures modulaires variées. Afin de confirmer les prédictions, les modules catalytiques des laminarinases A et C (LamA_GH16 et LamC_GH16, 38% d’identité de séquence), ainsi que le CBM6 de LamC (LamC_CBM6) ont été clonés et surexprimés dans E. coli. LamA_GH16 et LamC_GH16 sont toutes les deux actives sur la laminarine extraite de l’algue brune Laminaria digitata et sur le (1,3:1,4)-β-D-glucane de l’orge. Leurs paramètres cinétiques ont ainsi été déterminés sur chacun des substrats. Nous avons également pu montrer par gel retard que LamC_CBM6 était capable de lier la laminarine. Les structures cristallographiques de LamA_GH16, LamC_GH16 et LamC_CBM6 ont été résolues par remplacement moléculaire à 1.5 Å, 1.6 Å et 1.4 Å de résolution respectivement, et expliquent la spécificité de LamA_GH16 pour la laminarine. Finalement des mutants inactifs de LamA_GH16 et de LamC_GH16 ont été obtenus par mutagénèse dirigée afin d’obtenir la structure des complexes enzyme-substrat. LamA_GH16 a été cristallisée avec des tétrasaccharides purifiés de laminarine et un trisaccharide de (1,3:1,4)-β-D-glucane dans la gorge catalytique. LamC_GH16 a pu être cristallisée avec un hexasaccharide de synthèse contenant deux liaisons thiols. Les mécanismes moléculaires de reconnaissance de la laminarine par ces GHs et CBM marins seront finalement discutés. [1] Michel et al., New Phytologist.2010, 188, 67-81. [2] Barbeyron et al., Int J Syst Evol Microbiol, 2001, 51: 985-997. [3] Michel et al., Applied Microbiology and Biotechnology, 2006, 71, 23-33. [4] Hehemann et al., Nature, 2010, 464, 908-912. [5] Rebuffet et al., Environmental microbiology, 2011, 13,1253-1270. [6] Thomas et al., Journal of Microbiological Methods, 2011, 84, 61-66.

Session Posters 2c Enzymologie et dynamique
- 152 -
P18 - A quoi sert une chlorophylle qui ne capte pas la lumière ? Audrey Le Bas
1, Silvia Zambolin
1, Fabrice Rappaport
2, Daniel Picot
1, Francesca Zito
1
1Laboratoire de Biologie Physico-Chimique des Protéines Membranaires, UMR 7099,
2Laboratoire de Physiologie Membranaire et Moléculaire du Chloroplaste, UMR 7141,
Institut de Biologie Physico-Chimique IBPC, 13 rue Pierre et Marie Curie, 75005 Paris
La structure cristallographique du complexe membranaire cytochrome b6f de la photosynthèse oxygénique a été résolue en présence [1] et en absence d'un inhibiteur (TDS). Des changements structuraux de faible amplitude, à l'exception de la réorganisation d'un domaine, et liés au turn-over de la protéine sont mis en évidence dans le b6f, principalement autour de la molécule de chlorophylle. De tels mouvements sont aussi retrouvés dans l'homologue mitochrondrial, le cytochrome bc1.
Le b6f, bien que très similaire au bc1, possède des caractéristiques propres comme une
molécule de chlorophylle a de fonction inconnue, étudiée par une approche de mutagénèse dirigée chez l'algue verte unicellulaire Chlamydomonas reinhardtii. L'alanine 140 de la sous-unité IV, située dans le site natif de la chlorophylle, a été mutée en phénylalanine dans le but de modifier la fixation et/ou la stabilité de ce pigment. La structure cristallographique du mutant A140F a montré, pour la première fois, un complexe b6f sans molécule de chlorophylle. Cette perte est accompagnée d'une diminution du turn-over de la protéine, suggérant un rôle fonctionnel pour ce pigment.
En effet, les mêmes changements structuraux observés dans la souche de référence sont
retrouvés dans ce mutant avec une plus forte amplitude. Le mutant A140F peut ainsi apporter une meilleure compréhension des mécanismes de transfert d'électrons et de leur contrôle, domaine très controversé dans l'étude des cytochromes de type bc1 et b6f.
[1] Stroebel et al., Nature. 2003, 426(6965), 413-418.

Session Posters 2c Enzymologie et dynamique
- 153 -
P19 - Etudes structurales de la « Quinone Envelope Oxidoreductase homolog » d'Arabidopsis thaliana. Sarah Mas-y-mas1, Cécile Giustini2, Lucas Moyet2, Daniel Salvi2, Jean-Luc Ferrer2, Norbert Rolland2, Gilles Curien2 et David Cobessi1 1Institut de Biologie Structurale 2Laboratoire de Physiologie Cellulaire et Végétale, CEA, CNRS, Université Joseph Fourier, Grenoble
La ceQORH (Chloroplast envelope Quinone Oxidoreductase homolog) est une enzyme chloroplastique qui participe à la détoxification des plantes en agissant sur des longues chaînes carbonées toxiques issues du stress oxydatif. Cette protéine est codée par un gène nucléaire et est importée dans le chloroplaste par une voie alternative au système d'import principal TOC et TIC [1], voie d’import qui est encore inconnue. De manière générale, les protéines chloroplastiques possèdent un peptide signal situé à l'extrémité N-terminale qui empêche leur repliement jusqu’à ce que ce peptide signal soit clivé au cours du transport au travers des membranes. Cependant, pour la ceQORH, cette séquence est interne (résidu 60 à 100), et n'est pas clivée [2].
Nous avons cloné et purifié la ceQORH d'Arabidopsis thaliana. Seuls les complexes
de la ceQORH avec le NADP et le 13-Oxo-9,11,15-octadecatrienoic (13-KOT), ou de la ceQORH avec le NADPH cristallisent. La structure de la ceQORH en complexe avec le NADP et le 13-KOT a été déterminée à 2,8 Å de résolution en utilisant le remplacement moléculaire. L’unité asymétrique est composée de deux tétramères. À la différence des quinones oxydo-réductases de structure connue, les monomères de la ceQORH ne s'associent pas sous forme d'un dimère fonctionnel dû à l'orientation d'une hélice α empêchant la dimérisation de l'enzyme. La surface enfouie de chaque monomère dans le tétramère est très faible. Le 13-KOT est stabilisé par trois molécules de ceQORH. Le nicotinamide du NADP est situé à une distance du 13-KOT qui est compatible pour la réduction d'une de ses doubles liaisons C-C. Le peptide signal est partiellement enfoui dans la structure. Afin de déterminer l'état d'oligomérisation des différents complexes de la ceQORH avec ses cofacteurs et ligands en solution, des études d'ultracentrifugation analytique seront entreprises.
[1] Miras et al., J. Biol. Chem., 2007, 282, 29482-29492. [2] Miras et al., J. Biol. Chem., 2002, 227, 47770-47778.

Session Posters 2c Enzymologie et dynamique
- 154 -
P20 - Bases structurales de l'activité oxydase de la flavoprotéine AnaB impliquée dans la biosynthèse de la cyanotoxine anatoxine-a.
Karine Moncoq1, Leslie Regad2, Stéphane Mann3, Annick Méjean3, Olivier Ploux3 1CNRS/Université Paris-Diderot 7, UMR7099, IBPC, Paris, 2INSERM, UMR-S973, MTi, Paris, 3CNRS, UMR7223, Chimie ParisTech, ENSCP, Paris.
Les cyanobactéries sont des procaryotes photosynthétiques qui colonisent la plus part des environnements aquatiques ou terrestres. Ces micro-organismes produisent de nombreux métabolites secondaires dont des toxines pour les animaux et l'homme. Parmi ces toxines, l'anatoxine-a est une neurotoxine extrêmement puissante, agoniste du récepteur nicotinique de l'acétylcholine. La voie de biosynthèse de cette toxine a été récemment identifiée et une des enzymes clé de cette biosynthèse, AnaB, a été caractérisée [1].
Cette flavoprotéine catalyse l'oxydation du substrat prolyl-AnaD (proline chargée sur AnaD, protéine de transport d'acyles (ACP)) en déhydroprolyl-AnaD (1-pyrroline-5-carboxyl-AnaD, P5C) en utilisant l'oxygène comme deuxième substrat.
Voie de biosynthèse de l'anatoxine-a
AnaB présente donc une activité oxydase bien qu'elle appartienne à la famille des acyl-CoA déshydrogénases (ACADs) qui, à la place de l'oxygène, utilise des protéines de transport d'électrons comme accepteur d'électrons. Afin de comprendre la réactivité à l'oxygène de cette enzyme peu commune, sa structure cristallographique a été résolue à 2.8 Å. AnaB est un homotétramère qui présente un repliement très proche des ACADs avec une poche de fixation au FAD similaire et la base du site actif conservée. Si la similarité d'AnaB avec les ACADs est frappante, la structure révèle plusieurs différences importantes : i) une plus grande accessibilité du FAD au solvant, ii) la présence d'un canal d'eau proche du FAD pouvant constituer un chemin d'entrée potentiel de l'oxygène, et iii) la présence d'une charge positive dans l'environnement du site actif. L'ensemble de ces données fournissent un aperçu structural et moléculaire sur le mécanisme d'action d'AnaB et en particulier sa réactivité à l'oxygène.
[1] Mann et al., Biochemistry 2011, 50, 7184-7197.

Session Posters 2c Enzymologie et dynamique
- 155 -
P21 - Caractérisation structurale et fonctionnelle d’amylosaccharases Frédéric Guérin1,2, Sophie Barbe2, Sandra Pizzut-Serrin2, Gabrielle Véronèse2, David Guieysse2, Valérie Guillet1, Pierre Monsan2, Lionel Mourey1, Magali Remaud-Siméon2, Isabelle André2 et Samuel Tranier1
1Institut de Pharmacologie et de Biologie Structurale, CNRS-Université de Toulouse, F-31077 Toulouse, France, 2Université de Toulouse ; INSA, UPS, INP, LISBP, F-31077 Toulouse, France ; CNRS, UMR 5504, F-31400 Toulouse, France ; INRA, UMR 792, F-31400 Toulouse, France
Les amylosaccharases sont des α-transglucosidases de la famille GH13 [1] utilisant le saccharose comme donneur d’unité glucosyle pour synthétiser des produits tels que l’amylose ou des isomères du saccharose (turanose, tréhalulose). Ces enzymes présentent également un grand intérêt pour la synthèse de glucoconjugués en mettant à profit leur promiscuité vis-à-vis d’accepteurs exogènes [2]. La seule structure d’amylosaccharase disponible était celle de l'amylosaccharase de Neisseria polysaccharea (ASNp) [3]. Par comparaison avec l’ASNp, l’ASDg, une amylosaccharase homologue de Deinococcus geothermalis, présente des différences de profil de produits et de caractéristiques biochimiques [4].
Les structures de l’ASDg, ainsi que celles de l’ASDg et de l’ASNp en complexe avec le
turanose ont été déterminées [5]. L’ASDg révèle une organisation homodimérique inédite pour les amylosaccharases pouvant expliquer sa thermostabilité accrue. Les interactions observées au sein du complexe ASNp-turanose contraignent la sous-unité fructosyle à adopter une conformation exclusivement ouverte, suggérant une orientation sélective du fructose accepteur dans le site actif et expliquant ainsi la formation préférentielle de turanose par cette enzyme. L’ASDg lie majoritairement le tautomère α-furanoyl du fructose au travers d’un réseau moins dense d’interactions. Cette topologie du sous-site +1, moins contrainte que dans l’ASNp, peut autoriser d’autres modes de liaison du fructose conduisant à la formation de tréhalulose en quantité plus importante.
Parallèlement, nous avons déterminé la structure cristallographique de trois doubles
mutants présentant une amélioration spectaculaire de spécificité à la fois vis-à-vis du saccharose et de l’accepteur α-allyl-N-acetyl-2-désoxy-α-D-glucopyranoside par rapport au type sauvage. L’analyse des structures et des simulations de dynamique moléculaire montrent une rigidité locale du sous-site -1 couplée à une flexibilité des boucles impliquées dans la topologie du site actif [6].
Tout ceci montre l'importance de la conformation locale des résidus catalytiques mais
aussi de la dynamique de ces protéines au cours de la catalyse.
[1] Henrissat et al, Curr. Opin. Struct. Biol. 1997, 7, 637-44. [2] André et al, Top. Curr. Chem. 2010, 294, 25-48. [3] Skov et al, J. Biol. Chem. 2001, 276, 25273-78. [4] Emond et al, FEMS Microbiol. Lett. 2008, 285, 25-32. [5] Guérin et al, J. Biol. Chem. 2012, 287, 6642-54. [6] Champion et al, J Am Chem Soc. 2012, 134, 18677-88.

Session Posters 3a Croissance cristalline (GFCC)
- 156 -
P22 - PCC-Maltoside : un nouveau tensio-actif pour la cristallisation des protéines membranaires
Laurie-Anne Barret1,2, Barrot-Ivolot Cherone1, Raynal Simon1, Colette Jungas2, Ange Polidori1, Françoise Bonneté1 1 Equipe Chimie Bioorganique et des Systèmes Amphiphiles, IBMM UMR 5247, 33 rue L. Pasteur, Université d’Avignon et des Pays de Vaucluse, 84000 Avignon, France 2 Laboratoire de Bioénergétique Cellulaire, Institut de Biologie Environnementale et de Biotechnologie, CEA Cadarache, 13108 Saint-Paul-lez-Durance, France
La cristallisation des protéines membranaires (PMs) représente un défi de la biologie structurale. En effet, la résolution structurale de ces protéines à l'échelle atomique se heurte à une cristallisation plus compliquée que celle des protéines solubles du fait du caractère amphiphile des PMs, qui nécessitent l'emploi de détergents (molécules amphiphiles, lipophiles) pour leur solubilisation, leur purification et leur manipulation. Néanmoins l’utilisation de ces tensio-actifs détergents (DDM, OG) peut parfois déstabiliser la protéine membranaire et amener à l’obtention de cristaux de faible pouvoir diffractant.
L’utilisation de tensio-actifs à chaine hydrophobe plus rigide [1] ou hémifluorée lipophobe, moins déstabilisante pour les PMs, peut être une alternative intéressante par rapport aux détergents classiques hydrocarbonés.
PCC--malt FH--malt
Dans ce projet, nous avons étudié le rôle de la nature de la chaîne hydrophobe (rigide
cyclique ou fluorée) sur la stabilité et la cristallisation du complexe photosynthétique RC-LH1-pufX de Rhodobacter blasticus. Nous avons abordé la cristallisation par l’étude des forces d'interaction (via la mesure du second coefficient du viriel) entre micelles de tensio-actifs en utilisant des techniques de diffusion des rayonnements. Cette approche déjà utilisée avec succès [2-4], nous a permis de corréler diagrammes de phases des tensio-actifs et diagrammes de phases des complexes membranaires [5], nécessaire au contrôle de la croissance cristalline et à l’amélioration des cristaux de protéines. [1] Hovers et al. Molecular Membrane Biology (2011) 28(3): 171-81 [2] Bonneté et al, J. Cryst Growth (1999) 196: 403-414. [3] Loll et al, J. Cryst. Growth (2001) 232: 432-438 [4] Loll et al, Cryst Growth Des. (2002) 2: 533-539 [5] Barret et al, J Phys Chem B (2013) accepted

Session Posters 3a Croissance cristalline (GFCC)
- 157 -
P23 - Réseau National des Technologies et Procédés de la Croissance Cristalline
Françoise Bonneté1, Pascal Lejay2,*
1 Equipe Chimie Bioorganique et des Systèmes Amphiphiles, IBMM UMR 5247, 33 rue L. Pasteur, Université d’Avignon et des Pays de Vaucluse, 84000 Avignon, France 2 Institut NEEL, 25 avenue des Martyrs, BP 166, 38042 Grenoble cedex 9 CNRS - Mission Ressources et Compétences Technologiques http://cristech.cnrs.fr * Responsable du Réseau Cristech
Le réseau CRISTECH, créé en 2005, est un réseau technologique issu de la Mission Ressources et Compétences Technologiques (MRCT) du CNRS. A travers différents organismes de recherche (CNRS, INSERM, Universités, Ecoles et CEA) et des Industriels, le réseau Cristech est ouvert aux chercheurs, ingénieurs et techniciens impliqués dans l’élaboration, la mise en forme et l’étude de matériaux monocristallins.
Les technologies couvertes par le réseau sont variées, car elles dépendent fortement des composés/matériaux considérés et de la thématique scientifique : cristaux pour la biologie, nouveaux matériaux, cristaux pour l’optique, métallurgie semi-conducteurs, etc. Les méthodes de préparation vont de conditions douces (pression ambiante, basse température) à des conditions extrêmes (haute température, haute pression). De la même façon, les technologies dépendent fortement de la taille recherchée pour les monocristaux : d’une centaine de microns pour les nouveaux matériaux ou les cristaux de protéines, à plusieurs kilogrammes pour des applications telles que le silicium photovoltaïque et le saphir. Les objectifs du Réseau Cristech pour la communauté sont multiples : Etablir un état des lieux et créer un référentiel complet et actualisable de la croissance cristalline en France, Représenter l’ensemble des thèmes et des technologies de cristallogenèse, Sauvegarder un savoir faire et le faire évoluer, Fédérer des compétences et aider aux collaborations entre les équipes et les laboratoires, Former une entité connue et visible des autres communautés scientifiques susceptibles d’interagir, Mener des actions nationales de formation.

Session Posters 3a Croissance cristalline (GFCC)
- 158 -
P24 - Crystal structures of E. coli outer-membrane porin OmpF in two different C2 space-groups, with and without tNCS. Vincent Chaptal1, Arnaud Kilburg1, Ben Wiseman2, Jonathan Sarwan2, Gina Catalina Reyes-Mejia2, Jean-Michel Jault2 and Pierre Falson1 1 Drug Resistance Mechanism and Modulation, CNRS UMR5086 BMSSI, Univ. Lyon-1, IBCP, 7 passage du vercors, Lyon, 69007, France, 2 Institut de Biologie Structurale, UMR 5075, Université Joseph Fourier/CEA/CNRS, 41 rue jules Horowitz, Grenoble, F-38027 cedex-1, France
OmpF est la protéine la plus abondante de la membrane externe de E. coli. OmpF
appartient à la famille des porines générales qui n’ont pas de spécificité particulière pour leurs substrats, mais permet la diffusion de molécules à travers son pore ayant une limite de taille de 600 Da [1]. Nous avons cristallisé OmpF comme contaminant d’une purification d’un transporteur ABC de B. subtilis, BmrA, exprimé dans E. coli C41(DE3) [2]. OmpF a été co-purifiée sur chromatographie d’échange d’ions, colonne d’affinité au Nickel et une chromatographie d’exclusion de taille. L’identification de OmpF dans les cristaux n’a été possible qu’après une optimisation extensive de ces derniers, permettant la séparation de plusieurs gros cristaux, leurs dépôts sur gel d’acrylamide en conditions dénaturantes, et identification par spectrométrie de masse. Cette optimisation de la taille des cristaux nous a permis de collecter de façon concomitante des données de diffraction jusqu’à 3.5Å et 3.8Å de résolution pour deux formes cristallines distinctes. Ces deux formes appartiennent au groupe d’espace C2, qui n’a jamais été observée auparavant dans les structures de OmpF déjà déposées dans la PDB. Malgré des paramètres de maille très similaires pour ces deux formes cristallines, l’empilement est différent et se traduit par une symétrie non-cristallographique translationnelle (tNCS) pour la forme diffractant à 3.8Å.
Cette cristallisation d’un contaminant d’une purification de protéine membranaire nous
a amené à revoir notre stratégie de purification et à identifier plusieurs autres contaminants, spécifiques de certains détergents. Les enseignements tirés de ces expériences apportent de nouveaux éléments pour la purification des protéines membranaires. [1] Koebnik, R., K.P. Locher, and P. Van Gelder, Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol Microbiol, 2000. 37(2): p. 239-53. [2] Steinfels, E., et al., Characterization of YvcC (BmrA), a multidrug ABC transporter constitutively expressed in Bacillus subtilis. Biochemistry, 2004. 43(23): p. 7491-502.

Session Posters 3a Croissance cristalline (GFCC)
- 159 -
P25 - Des contaminants cachés dans la purification du transporteur ABC BmrA de Bacillus subtilis Arnaud Kilburg1, Vincent Chaptal1, Benjamin Wiseman2, Jonathan Sarwan2, Gina Catalina Reyes-Mejia2, Jean-Michel Jault2 and Pierre Falson1. 1 Drug Resistance Mechanism and Modulation, CNRS UMR5086 BMSSI, Univ. Lyon-1, IBCP, 7 passage du vercors, Lyon, 69007, France, 2 Institut de Biologie Structurale, UMR 5075, Université Joseph Fourier/CEA/CNRS, 41 rue jules Horowitz, Grenoble, F-38027 cedex-1, France
BmrA (« Bacillus Multidrug Resistance ATP ») est une pompe d’efflux membranaire – transporteur ABC – qui utilise l’énergie issue de l’hydrolyse de l’ATP pour transporter une grande diversité de molécules vers l’extérieur des cellules. Les recherches actuelles se concentrent sur l’obtention de données structurales en complexe avec des substrats transportés afin de comprendre leur fonction d’efflux. A ce jour, la structure de trois exportateurs ABC bactériens et celle de la P-gp de souris ont été publiés [1]. Aucune n’a été résolue en présence de substrats transportés. Notre objectif est de résoudre la structure 3D de BmrA en complexe avec des nucléotides, des substrats et/ou des modulateurs. BmrA a été purifiée avec six détergents différents sur chromatographie échangeuse d’ions, colonne d’affinité au nickel et chromatographie d’exclusion de taille. Avec 75% d’extraction, la foscholine 12 (FC12) est le détergent le plus efficace pour solubiliser BmrA. Sa purification a conduit à la cristallisation d’un contaminant, OmpF, une porine de la membrane externe d’E.coli. Nous avons résolu sa structure à 3.5 Å de résolution. Cette protéine possède un profil de migration sur gel d’acrylamide en conditions dénaturantes identique à celui de BmrA rendant son identification difficile. L’analyse par spectrométrie de masse a révélé la présence d’OmpF uniquement dans les échantillons purifiés en FC12. Une autre protéine, AcrB (acriflavine resistance protein B) a été détectée dans les échantillons purifiés avec des détergents maltosides mais était absente des purifications en FC12 et LDAO. OmpF et AcrB sont deux contaminants fréquemment rencontrés dans la purification des protéines membranaires et ont déjà été cristallisés à plusieurs reprises [2], [3]. Nous sommes parvenus à éliminer OmpF en optimisant les conditions d’extractions en FC12 et AcrB en centrifugeant la fraction membranaire avant solubilisation. Nos résultats apportent de nouveaux éléments dans la stratégie d’élimination d’OmpF et AcrB pour la purification des protéines membranaires. [1] Aller SG, Yu J, Ward A, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science (2009) 323:1718-1722. [2] Kefala G, Chihoon A, Krupa M, et al. Structures of the OmpF porin crystallized in the presence of foscholine 12. Protein science (2010) 19 :1117-1125. [3] Psakis G, Polaczek J et Essen L. Obstinate contaminants in a picogram scale. One more bottleneck in the membrane protein structure pipeline. Journal of structural biology (2009) 166 :107-111.

Session Posters 3a Croissance cristalline (GFCC)
- 160 -
P26 - Étude de la croissance cristalline de l’aragonite (CaCO3) en présence d’ions phosphates S. Tadier1, P.G. Koutsoukos2, C. Rey1, C. Combes1
1 Université de Toulouse, CIRIMAT, UPS-INPT-CNRS, ENSIACET, 4, allée Emile Monso, BP
44362, 31030 Toulouse Cedex 4, France 2 Department of Chemical Engineering and FORTH-ICEHT, University of Patras, GR 26 500,
Patras, Greece
Le carbonate de calcium (CaCO3) est un composé abondant, notamment dans les calcifications d’origine biologique. L’étude de sa germination et de sa croissance cristalline, en présence ou non d’ions phosphates (PO4
3-) qui pourraient les contrôler, est donc particulièrement intéressante. A notre connaissance, aucune étude n’a été réalisée sur l’aragonite, l’un des quatre polymorphes non hydratés du CaCO3, et constituant final d’un ciment pour comblement osseux mis au point au CIRIMAT. Afin de mieux appréhender la prise de ce ciment in-vivo (donc en présence de PO4
3-), nous avons étudié leur influence sur la cristallisation de l’aragonite.
La croissance cristalline d’aragonite à partir d’une solution sursaturée en CaCO3 a été
mise au point par la méthode de croissance cristalline à composition constante : les mécanismes de cristallisation de l’aragonite à partir de germes d’aragonite (synthétisés par précipitation à 100°C) ont été étudiés en conditions contrôlées (pH= 7,8, T = 37°C, force ionique et sursaturation constantes), en présence ou non d’ions PO4
3- (0,25 µmol.L-1 - 1 mmol.L-1).
Nous montrons qu’en l’absence de phosphates, la croissance des germes d’aragonite est contrôlée soit par le transport de masse au sein de la solution vers la surface du cristal, soit par la diffusion de surface. En présence d’ions PO4
3-, trois zones de comportements ont été mises en évidence : à partir de 0,5 µmol.L-1, les ions PO4
3- diminuent la vitesse de cristallisation de l’aragonite et inhibent même toute croissance pour une concentration de 7,5 µmol.L-1. A partir de 8 µmol.L-1, il semblerait que les germes introduits se dissolvent en début d’expérience. Au-delà de 1 mmole.L-1, le précipité est composé de phosphate de calcium mal cristallisé et non plus de CaCO3. Cette étude fondamentale menée in-vitro contribue à une meilleure connaissance de la réaction de prise in-vivo du ciment biomédical à base de CaCO3.

Session Posters 3a Croissance cristalline (GFCC)
- 161 -
P27 - Croissance hydrothermale de monocristaux isotypes du quartz, relations structure- propriétés Vincent Ranieri, Manhal Souleiman, Gopal Bhalerao, Julien Haines et Olivier Cambon Institut Charles Gerhardt Montpellier, UMR-CNRS-UM2-ENSCM-UM1 5253, Université Montpellier 2, Equipe C2M, Place E. Bataillon, 34095 Montpellier, Cedex 5, France
Parmi les matériaux piézoélectriques, le quartz-α est encore le plus utilisé. Cependant ses propriétés physiques atteignent leurs limites (transition α ↔ β à 846 K, coefficient de couplage électromécanique k =8,5%) pour les applications émergentes. Des relations structure-propriétés liant à la fois les propriétés physiques et la stabilité thermique à la distorsion structurale ont été établies pour les matériaux isotypes du quartz [1]. GeO2 et GaAsO4 sont les matériaux les plus prometteurs de cette famille.
Nous présenterons les travaux effectués sur la phase pure GaAsO4 et sur une solution solide Si1-xGexO2.
Les monocristaux de GaAsO4 ont été obtenus par épitaxie sur des germes X (1-20) de GaPO4 et de GaAsO4 en conditions hydrothermales (P≤2MPa). Les mesures confirment ses remarquables performances avec une stabilité thermique atteignant 1273 K et un coefficient de couplage le plus important des isotypes du quartz (k = 21%) [2,3]. De plus, sa structure non centro-symétrique et sa composition chimique lui confère des propriétés optiques non linéaires. Les premières mesures donnent d11=3.03 pm/V (χ(2)=6.06 pm/V).
Des solutions solides Si1-xGexO2 (0<x<0,24) ont été obtenues par épitaxie sur germe Z (001) de quartz par voie hydrothermale (100 MPa≤ P ≤200MPa) [4]. Les mesures par DRX et par spectroscopie Raman montrent que la distorsion structurale et la température de transition α ↔ β augmentent avec x. De plus, l’étude des modes de vibration Raman impliqués dans la transition α ↔ β montre que le désordre dynamique est réduit avec la substitution de germanium [5]. Ce résultat est important pour les applications car le désordre dynamique est responsable des pertes des propriétés piézoélectriques du quartz et limite son utilisation à 200°C [6].
[1] Haines J., Cambon, O., Keen D. A. Physica B., 2004, 350, 1-3, E979-E981. [2] Cambon, O., Haines, J., Fraysse, G., Détaint, J., Capelle, B., Van der Lee, A. J. Appl. Phys., 2005, 97, 074110. [3] Cambon, O., Bhalerao, G. M., Bourgogne, D., Haines, J., Hermet, P., Keen, D. A., Tucker, M. G. J. Am. Chem. Soc, 2011, 20, 133, pp 8048–8056. [4] Ranieri, V., Darracq, S., Cambon, M., Haines, J., Cambon, O., Largeteau, A., Demazeau, G. Inorganic chemistry, 2011, 50 (10), pp 4632–4639. [5] Ranieri, V., Bourgogne, D., Darracq, S., Cambon, M., Haines, J., Cambon, O., Le Parc, R., Levelut, C., Largeteau, A., Demazeau, G. Phys. Rev. B, 2009, 79, 224304. [6] Haines J., Cambon, O., Keen D. A., Tucker M. G., and Dove M. T. Appl. Phys. Lett., 2002, 81, 2968.

Session Posters 3a La cristallographie sous conditions extrêmes (GFCC)
- 162 -
P28 - Premières expériences Hautes Pressions en presses Gros Volumes (Paris-Edimbourg et multi-enclumes) de diffraction X en dispersion d’énergie sur la ligne PSICHÉ de SOLEIL Nicolas Guignot1, Jean-Paul Itié1, Pierrick Zerbino1, Kirill Chernidchenko1,2, Yann Le Godec2, Alain Prat3, Geeth Manthilake4, Julien Chantel4, Franck Pointud4 et Denis Andrault4 1 Synchrotron SOLEIL, 2 IMPMC, Université Pierre et Marie Curie, 3 Institut Néel, 4 LMV, Université Blaise Pascal
Deux types de presses "gros volume" ont été installés et testés sur la ligne PSICHÉ de SOLEIL. La diffraction X in situ a permis de suivre la densité et les transformations de phases grâce à la détection combinée en dispersion d’énergie et dispersion angulaire (CAESAR [1]) Dans cette présentation nous montrerons l’intérêt de ces systèmes pour l’étude des matériaux en conditions extrêmes de pression et de température. Dans une première expérience réalisée en cellule Paris-Edimbourg, nous avons pu observer la croissance de nanoparticules de MoB4 à haute pression et haute température. Dans une seconde expérience nous avons aligné une nouvelle presse équipée d'un module original de type DIA-100, et pouvant délivrer 1200 tonnes de poussée. Nous avons mesuré la compression d'un mélange de MgO et de KBr jusqu’à 21.5 GPa. Quelques améliorations restent à apporter à ces dispositifs avant de les mettre à la disposition des utilisateurs. A terme, leur complémentarité permettra de réaliser une grande diversité d'expériences jusqu'à plus de 25 GPa et 2000°C.
[1] Wang et al. (2004) Journal of Applied Crystallography, 37, 947-956.

Session Posters 4b Apériodicité, structure modulée
- 163 -
P29 - Structural and thermal investigations of a Tunisian natural phosphate rock
Béchir Badraoui1, Hassen Bachouâ1, Masseoud Othmani1, Yannick Coppel2, Nabil Fatteh3, Mongi Debbabi1
1 Laboratoire de Physico-Chimie des Matériaux, Université de Monastir, Faculté des Sciences, 5019 Monastir, Tunisia 2 CNRS, LCC (Laboratoire de Chimie de Coordination), 205 route de Narbonne, BP 44099, F-31077 Toulouse Cedex 4, France 3 Centre de recherches CPG Métlaoui 2134 Gafsa, Tunisia E-mail : [email protected]
Tunisian natural phosphate rock from M’dhilla deposits are characterized by different methods. The principal constituent of the phosphate rock sample is the fluoroapatitic phase to which other clay and siliceous mineral; sulfates, carbonates and organic matter are associated.
The crystallographic structure of natural phosphate was examined by powder X-Ray diffraction (XRD) at different temperatures of calcinations. The XRD diagrams show a significant structure change with increasing of the temperature to 1100°C. The MAS-NMR technique was used to study the carbon, phosphorus, silicon and fluoride environment.
The result confirms the single phosphate apatitic phase; the NMR study also indicates the presence of a small amount of organic compounds which was detected by chemical analysis (only 0.6%), IR spectroscopy and thermogravimetric analysis.

Session Posters 4b Apériodicité, structure modulée
- 164 -
P30 - Structure cristalline de composés approximants dans les systèmes (Ce, Nd, Sm, Gd)-Au-Sn Pascal Boulet, Marie-Cécile De Weerd, Jean-Marie Dubois Institut Jean Lamour, UMR CNRS 7198, Université de Lorraine, Parc de Saurupt, 54011 Nancy Cedex
Depuis la découverte de composés quasicristallins stables dans les systèmes binaires Cd5.7Yb [1] et Cd5.7Ca [2], un intérêt croissant s’est porté sur ces structures quasicristallines et leurs structures approximantes. Ces différentes phases de type Tsai sont formées de plusieurs enveloppes concentriques : un tétraèdre partiellement occupé, suivi d’un dodécaèdre, puis un icosaèdre, un icosidodécaèdre, et enfin un triacontaèdre plus ou moins déficitaire. En fait, toutes ces phases coexistent dans un domaine de composition très restreint et évalué par exemple à moins de 2% atomique dans les systèmes binaires comme Cd-Yb.
Par ailleurs, des structures similaires ont été obtenues dans des systèmes ternaires de
type TR-Au-Sn [5,6], avec TR = Ce, Pr, Eu, Gd, Dy et Tb. Les déterminations expérimentales de des diagrammes ternaires à base de Cérium [7] ou de Praséodyme[8] ont révélé des domaines de compositions légèrement plus élevés concernant ce type de phases, dû à un échange sur certains sites cristallographiques d’or et d’étain. En fait, une étude précise du diagramme ternaire Ce-Au-Sn[7] a montré un domaine d’homogénéité donnant une formule type Ce3Au15±xSn 3±y, avec 0<x<2 and 0<y<1.
Dans ce papier, nous décrirons les structures obtenues à partir de données
monocristallines de 4 structures isostructurales adoptant la structure approximante 1/1 de type Tsai dans les systèmes ternaires TR-Au-Sn avec TR= cérium, néodyme, samarium et gadolinium.
[1] A.P. Tsai, J. Q. Guo, E. Abe, H. Takakura and T.J. Sato, Nature 408 (2000) 537. [2] J. Q. Guo, E. Abe and A.P. Tsai, Phys. Rev. B 62 (2000) R14605. [3] A. Palenzona, J. Less Common Met. 25 (1971) 367. [4] C.P. Gomez and S. Lidin, Angew. Chem., Int. Ed. 40 (2001) 4037. [5] S. Kenzari, V. Demange, P. Boulet, M.C. de Weerd, J. Ledieu, J.M. Dubois and V. Fournée, J. Phys. Condens Matter 20 (2008) 095218. [6] Y. Morita and A.P. Tsai, Jap. J. App. Phys. 47 (2008) 7975 [7] P. Boulet, D. Mazzone, H. Noel, P. Rogl and R. Ferro, J. Alloys and Compd. 317/318 (2001) 350. [8] D. Mazzone, R. Marazza, P. Riani, G. Zanicchi, G. Cacciamani, M.L. Fornasini and P. Manfrinetti, Calphad 33 (2009) 31.

Session Posters 4b Apériodicité, structure modulée
- 165 -
P31 - Zn(O3P-C3H5) : Où la nature est plus propre à induire l’ordre que le chimiste ! Marion Galmiche1,2, Olivier Perez1, Jean-Michel Rueff1, Bernard Nysten2, Paul-Alain Jaffrès3 1 CRISMAT, CNRS UMR 6508, ENSICAEN-Caen, 2 Université Catholique de Louvain IMCN/BSMA - Belgique, 3 Université de Brest, CNRS, UMR 6521, CEMCA
Dans l’optique d’obtenir des matériaux hybrides présentant des propriétés de conduction électronique, des synthèses par voie hydrothermale ont été réalisées à partir de l’acide de 2-thiophen-2-ylphosphonic et d’acétate de zinc. Des mono cristaux ont été obtenus et étudiés par diffraction des rayons X sur un diffractomètre 4-cercles (Nonius) équipé d’une caméra CCD (APEX2) et d’une source micro foyer Mo (INCOATEC). Le réseau réciproque associé à ce matériau est caractérisé par la présence de réflexions intenses, s’indexant dans la
maille a=4.8110(4)Å, b=27.193(3)Å, c=5.6571(4) Å et ===90°, et de réflexions de faibles intensité en position irrationnelle avec le réseau précédemment défini. L’indexation de ces dernières nécessite l’introduction du vecteur de modulation q= -0.034 a* + 0.216 b*, incompatible avec une symétrie orthorhombique. Le composé synthétisé a donc une structure modulée incommensurable de symétrie monoclinique. La résolution structurale a été réalisée
à l’aide du formalisme des superespaces[1,2] dans le groupe de Xn(103)0 avec comme vecteur de centrage X(0,½,0,½). La détermination de la structure modulée met en évidence que i) la molécule organique utilisée lors de la synthèse s’est brisée : le cycle aromatique thiophène a laissé la place à une chaine linéaire à trois atomes de carbone comportant une double liaison C=C compatible avec l’une des deux structures suivantes : Zn(O3P-CH=CH-
CH3)H2O ou Zn(O3P-CH2-CH=CH3)H2O ii) la modulation conduit à la mise en ordre des différents régio-isomères au sein du matériau.
Nous avons cherché à reproduire la synthèse de ce matériau par voie hydrothermale à
partir de la molécule organique commerciale, l’acide allyl phosphonique (H2O3P-CH2-CH=CH2), et de l’acétate de zinc. Les cristaux obtenus présentent les mêmes paramètres de maille. Toutefois, aucune modulation n’est observée et la résolution structurale révèle un important désordre affectant les chaines carbonées. Ce désordre correspond à une moyenne des conformations des chaines révélées par l’étude de la phase modulée. [1] P.M. De Wolff. Acta Crystallogr. A, 30 :777, 1974. [2] P.M. De Wolff, T. Jansen, and A. Janner. Acta Crystallogr. A, 37 :625, 1981.

Session Posters 4b Apériodicité, structure modulée
- 166 -
P32 - Structural studies of tetragonal tungsten bronze (TTB) crystals and ceramics by XRD, neutron and electron diffraction Pierre Heijboer1, Michaël Josse1, Matias Velazquez1, Florence Porcher2, François Weill1, Mario Maglione1 1
ICMCB, Université Bordeaux 1, CNRS, 87 Avenue du Docteur Albert Schweitzer, 33600 Pessac 2 LLB, UMR12 CEA-CNRS, Bât. 563 CEA Saclay, 91191 Gif sur Yvette
The tetragonal tungsten bronze structure (TTB) has been first reported in 1949 [1].
They observed that such materials have both ferroelectric and ferromagnetic properties at room temperature. This recently came back through the investigation of Ba2LnFeNb4O15 (where Ln = La, Pr, Nd, Sm, Eu, Gd) ceramics and crystals, which revealed their composite multiferroic nature.
Following the recent studies on ceramics from this family [2, 3, 4] we investigated their crystal growth by the optical vertical floating zone. By using this last technique, we grew cm-sized single-crystal sections of the Ba2LaFeNb4O15 compound, to investigate more precisely the crystal structure by XRD, neutron and electronic diffraction. We confirmed the existence of a modulated structure (incommensurate or not, depending on the composition) and the average structure as tetragonal (P4/mbm).
In order to investigate the effect of substitution (lanthanum or neodymium by cerium)
on the structure and physical properties, we also synthesized new adjusted compositions TTB in ceramics. their characterization by XRD and neutron diffraction in temperature support a P4/mbm space group. We recently performed electron diffraction in temperature on those ceramics, and they also exhibit this modulation. Furthermore, some XPS experiments on both ceramics and crystals are giving us more information on the lattice disorder, including the octahedral sites both filled by iron and niobium. We added physical properties investigations, including dielectric measurements, that revealed particular and interesting relaxor-ferroelectric-paraelectric transitions by increasing the temperature in some of the ceramics.
The aim of our studies is to link the composition, the structure (including the
incommensurate modulation) and the physical properties of such advanced materials. [1] Magneli et al., Ark. Kemi 1, 1949, 213. [2] Roulland et al., Solid State Sciences 2009, 11, 1709–1716. [3] Graetsch et al., Acta Cryst. B. 2012, 68, 101-106. [4] Albino et al., accepted in Eur. J. Inorg. Chem., doi: 10.1002/ejic.201300008

Session Posters 4b Apériodicité, structure modulée
- 167 -
P33 - Tetrahedron ordering in the Cd6Tb and Zn6Sc 1/1 quasicrystalline approximants: Experiment and Simulation
D. Liu1, G. Beutier1, H. Euchner2, T. Yamada3, M. Dupraz1, M. de Boissieu1 1 Sciences et Ingénierie des Matériaux et Procédés, INP Grenoble CNRS UJF, Grenoble, France 2 Institutfu ̈rTheoretische und AngewandtePhysik, Universitt Stuttgart, Pfaffenwaldring 57, 70550 Stuttgart, Germany 3 IMRAM, Tohoku University, Sendai, Japan
[email protected] Keywords: quasicrystal approximant, phase transition, domain
The discovery of binary quasicrystal Cd5.7Yb and Cd5.7Ca breaks a path to deeply investigate structure and physical properties of quasicrystal [1, 2]. For nowadays, the technique determining atomic structure of binary quasicrystal has been developed maturely [3], however, study quasicrystal directly is still a challenging work. As counterparts of icosahedral quasicrystal, Cd6R (R=rare elements) as well as Zn6Sc 1/1 approximants, are expected to play a key role to understand different properties of quasicrystal.
Other than quasicrystals, most of the cubic approximants have been evidenced an order-disorder phase transition from space group Im-3 to Cc or C2/c, and the ordering of innermost Cd4/Zn4 tetrahedron in Tsai-type cluster is indicated as the trigger factor [4, 5, 6, 7]. According to symmetric operation, 6 domains with different orientations are obtained in the low temperature phase [8].
We will present high resolution x-ray diffraction and diffuse scattering in situ measurements using synchrotron data. And this will be compared with atomic scale molecular dynamic simulation. The structural phase transition is observed at ~190K and 159K for Cd6Tb and Zn6Sc respectively. Diffuse scattering with respect of temperature is discussed both experimentally and simulatively to Zn6Sc. A deep measurement is carried out with some fundamental reflections below Tc, and the 3-D peak splitting contributed by different domains is visualized using a new method.
[1] A.P. Tsai, J.Q. Guo, E. Abe, H. Takakura and T. J. Sato, Nature 408 (2000), 537. [2] J.Q. Guo, E. Abe and A.P. Tsai, Phys. Rev. B 62 (2000), R14605. [3] H. Takakura, C.P. Gómez, A. Yamamoto, M. De Boissieu and A.P. Tsai, Nature Materials 6 (2007), 58. [4] R. Tamura, Y. Murao, S. Takeuchi, M. Ichihara, M. Isobe and Y. Ueda, J. J. Appl. Phys. 41 (2002), L524. [5] R. Tamura, K. Edagawa, C. Aoki, S. Takeuchi and K. Suzuki, Phys. Rev. B 68 (2003), 174105. [6] R. Tamura, K. Edagawa, Y. Murao, S. Takeuchi, K. Suzuki, M. Ichihara, M. Isobe and Y. Ueda, J. Non-Cryst. Solids 334-335 (2004), 173. [7] R. Tamura, K. Nishimoto, S. Takeuchi, K. Edagawa, M. Isobe and Y. Ueda. Phys. Rev. B 71 (2005), 092203. [8] T. Yamada, H. Euchner, C.P. Gómez, H. Takakura, R. Tamura and M. de Boissieu, J. Phys.: Condens. Matter 25 (2013), 205405.

Session Posters 4b Apériodicité, structure modulée
- 168 -
P34 - Transitions de phases vers des états à ondes de densité de charges dans les bronzes phosphates de tungsten (PO2)4(WO3)2m
Olivier Pérez1, Kamil Kolincio1, Laurent Guérin2, Pierre Fertey3
1 Ensicaen, 6 Bd du Maréchal Juin, 14050 Cedex 04, France 2 IPR, Université de Rennes 1, Campus de Beaulieu, Bâtiment 11A, 35042 RENNES 3 Synchrotron SOLEIL, L’Orme des Merisiers - BP 48, 91190 SAINT-AUBIN
Les bronzes phosphate de tungstène [1] à tunnels pentagonaux (MPTBp), de formule chimique (PO2)4(WO3)2m avec m variant de 4 à 14, peuvent être décrits comme l’intercroissance régulière de tranches de tétraèdres PO4 et de tranches d’octaèdres WO6 liés par les sommets et d’épaisseur proportionnelle à m. Cette famille de composés dont la dimensionnalité peut être gouvernée par le paramètre m, fournit un système modèle pour l’étude des conducteurs quasi 2D et des instabilités électroniques qui les caractérisent . Les MPTBp présentent ainsi jusqu’à trois transitions de phase successives de type Peierls en fonction de la température vers des états à ondes de densité de charges (ODC). Ces états sont associés à l’apparition de réflexions satellites de faible intensité et donc à des modulations structurales commensurables ou incommensurables. L’étude de la surface de Fermi permet de définir trois possibles vecteurs de modulation via l’hypothèse de mécanisme d’emboîtement caché [2,3]. Notons que des comportements assez différents sont observés pour les MPTBp en fonction de la parité de m (symétries différentes pour les termes paires ou impaires) ou de la valeur de m, i.e. de la dimensionnalité (transition en dessous de la température ambiante pour m≤ 8 et au-dessus pour m ≥ 9).
Afin d’obtenir des informations sur les mécanismes régissant ces instabilités électroniques, i.e. d’expliquer les différentes transitions de phases observées, une analyse précise des propriétés de transport et des diagrammes de diffraction de différents membres de la famille des MPTBp au-dessus et au-dessous de la température de chaque transition a été réalisée. Notre choix s’est orienté sur des membres présentant des différences de dimensionnalité marquées (P4W12O44 (m=6) ou P4W20O68 (m=10)), des parités différentes pour m (P4W18O62 (m=9) ou P4W20O68 (m=10)) ou des valeurs inusuelles de m (P4W23O77 (m=11.5)). L’étude structurale des phases modulées en utilisant le formalisme des superespaces est proposée.
[1] P. Roussel, O. Pérez and Ph. Labbé, Acta Cryst. B57, 603-632 (2001) [2] Foury, P. & Pouget, J. P. (1993). Int. J. Mod. Phys. B7, 3973-4003. [3] E. Wang, M. Greenblatt, E.I. Rachidi, E. Canadell, & M.H. Whangbo, J.S.S.C. 80, 266- 275 (1989).

Session Posters 4b Apériodicité, structure modulée
- 169 -
P35 - Structure modulée commensurable d’un nouveau polymorphe β-CaTeO3(H2O)
M. Poupon, O. Pérez, N. Barrier, J.M. Rueff, H. Latappy
Laboratoire CRISMAT, UMR 6508 CNRS ENSICAEN, 6 bd Maréchal Juin 14050 CAEN Cedex 4 France.
Ces dernières années, nous avons montré que la synthèse de phases hydratées ATeO3(H2O) (A = Ca, Sr, Ba) par voie hydrothermale, suivie ou non d’une déshydratation, était une méthode de synthèse originale pour obtenir de nouvelles phases polymorphes [1,2] ; ces phases peuvent présenter des propriétés de ferroélectricité ou d’optique non-linéaire [3,4]. Récemment nous avons pu obtenir, par voie hydrothermale un nouveau polymorphe β-CaTeO3(H2O). Des monocristaux de cette phase ont pu être isolés et des données de diffraction ont été mesurées sur un diffractomètre 4-cercles équipé d’une caméra CCD (Apex2) et d’une source microfocus Mo (INCOATEC). β-CaTeO3(H2O) présente une symétrie monoclinique et une maille cristalline a=13.60Å, b=3.74Å, c =9.40Å et β=124.53°. Une première étude réalisée dans le groupe d’espace C2/m, révèle l’existence d’un désordre de position affectant une partie des atomes d’oxygène qui interdit toute analyse fine de l’environnement de l’atome de tellure. Les plans de diffraction reconstruits à partir des images expérimentales présentent des réflexions supplémentaires noyées dans des lignes de diffusion diffuse ; le précédent résultat d’affinement ne correspondrait donc qu’à une structure moyenne. Bien que ces réflexions supplémentaires soient localisées en position rationnelle dans le réseau précédemment défini, nous avons choisi d’utiliser le formalisme des superespaces en (3+1)d ; cette approche permet d’interpréter la structure réelle à partir de notre structure moyenne et d’une modulation de certains paramètres atomiques tout en réduisant les corrélations entre atomes grâce aux symétries (3+1)d. Le groupe de superespace
choisi est X2/n(α0γ)0s avec X(0,½,0,½) et vecteur de modulation . L’analyse des densités électroniques à (3+1) dimensions a révélé une mise en ordre des atomes d’oxygène précédemment trouvés désordonnés. Cette mise en ordre conduit pour les atomes de tellure à une configuration tétraédrique [TeO3E], E désignant la paire d’électrons libres du tellure. Une hypothèse sur l’existence de diffusion diffuse est finalement proposée. [1] Rai R., Sharma S. & Choudhary R.N.P., J. Mat. Sci. Lett., 2002, 21, 297. [2] Yamada T & Iwasaki H., J. Appl. Phys., 1973, 44, 3934. [3] Stöger B., Weil M., Baran E.J., González-Baró A. C., Malo S., Rueff J. M., Petit S.,
Lepetit M. B. Raveau B. & Barrier N., DaltonTrans., 2011, 40, 5538. [4] N. Barrier, Habilitation à Diriger des Recherches, Université de Caen, 2012.

Session Posters 4b Apériodicité, structure modulée
- 170 -
P36 - Transitions de phase dans les composés d'inclusion d'urée/alcane. Philippe Rabiller1, Céline Mariette1, Bertrand Toudic1, Laurent Guérin1, Mark D. Hollingsworth2 1Institut de Physique de Rennes, 2 Kansas State University, Manhattan, USA
Les cristaux apériodiques possèdent la propriété d’avoir un ordre à grande distance mais sans la symétrie de translation. Ces cristaux se décrivent dans des superespaces cristallographiques de dimension supérieure à trois. Dans cette affiche, nous nous intéressons plus particulièrement aux brisures de symétrie présentes dans de tels espaces cristallographiques en considérant la famille prototype hôte/invité d'urée/alcane. Des études par diffraction de rayons X sur sources synchrotron révèlent de multiples solutions structurales impliquant des changements ou non de la dimension du groupe de superespace. Un cas particulièrement intéressant est illustré par les molécules invitées les plus courtes. Cette cristaux présentent une phase de haute symétrie de dimension 3 avec confinement d'une phase "quasi-liquide" des alcanes. A basse température, la mise en ordre structural conduit à la formation de composites apériodiques intermodulés. Un tel exemple monoclinique est ainsi obtenu pour l'heptane [1].
FIG: (a) Représentation schématique de la structure du n-heptane/urée. (b) Projection suivant la direction des canaux (c) Représentation bidimensionnelle du composé définissant les paramètres hôte ch, invité cg et le déphasage entre les molécules invitées de canal à canal g. [1] Mariette et al., J. Chem. Phys. 2012, 136, 104507.

Session Posters 5a Approches multitechniques en cristallochimie
- 171 -
P37 - Résolution structurale par diffraction RX d’azo composés A. Benosmane1, A. Mili1, A. Bouchoul1, L. Ouahab2
1Unité de recherche de chimie de l’environnement et Moléculaire structurale. Faculté des Sciences Exactes, Université Mentouri, Constantine Algérie. 2LCSIM/UMR 6511 CNRS, Université de Rennes I. France. E-mail : [email protected]
Les azo-composés ont été préparés sous forme de poudre de couleur rouge par la méthode de synthèse classique des colorants azoïques (réaction de diazotation d’amine aromatique primaire suivie d’une copulation sur le β-naphtol).
R= H, Cl, SO2-NH2, O-CH3
Des cristaux rouges sous forme d’aiguilles, stables à l’air, ont été obtenus par recristallisation dans un mélange composé de deux solvants (l’éthanol et Acétone). Les intensités sont collectées par un diffractomètre automatique à quatre cercles munis d’un détecteur bidimensionnel Kappa CCD.
Les structures moléculaires analysées correspondant au monomère ou dimère, se présentent sous la forme quinonehydrazone qui constitue en effet le tautomère thermodynamiquement favorisé en raison de son énergie plus basse. Farrugia, L. J. (1997). J. Appl. Cryst. 30, 565. Lee, S. H., Kim, J. Y., Ko, J., Lee, J. Y. & Kim, J. S. (2004). J. Org. Chem. 69, 2902–2905. Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. Oueslati, F., Dumazet-Bonnamour, I. & Lamartine, R. (2004). New J. Chem.28, 1575–1578. Spek, A. L. (2009). Acta Cryst. D65, 148–155. Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor,R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19. Chen, Z. H., Morimoto, H., Matsunaga, S. & Shibasaki, M. (2008). J. Am. Chem. Soc. 130, 2170–2171. Dao, V.-T., Gaspard, C., Mayer, M., Werner, G. H., Nguyen, S. N. & Michelot, R. J. (2000). Eur. J. Med. Chem. 35, 805–813.

Session Posters 5b Densité électronique, modélisation
- 172 -
P38 - Electronic and Magnetic Properties of the PrMnO3 Perovskite: A comparative study of LSDA+U and GGA+U Zoubir Aziz*, Bouabdellah Bouadjemi and Samir Bentata Laboratoire de Technologie et des Propriétés du Solide Faculty of Sciences and Technology, BP227 Abdelhamid Ibn Badis University, Mostaganem (27000) Algeria *E-mail: [email protected]
The electronic and magnetic properties of the cubic PrMnO3 perovskite structure are studied using the full potential linear augmented plane wave (FPLAPW) based on density-functional theory (DFT) in the local spin density approximation (LSDA) and the generalized gradient approximation (GGA) with the on-site Hubbard Ueff parameter (LSDA + U and GGA+U). We have analyzed the structural parameters, densities of states, partial densities of states and band structures. We have shown that this component is half-metallic and ferromagnetic with an important magnetic moment.
Keywords: magnetic moment, half-metallic, spin-up/spin-down, GGA+U.

Session Posters 5b Densité électronique, modélisation
- 173 -
P39 - Etude de la distribution de la densité de charge dans le composé 4-Methoxy Benzene Carbothio Amide (C8H9NOS) Younes Magrous, Abdelkader Chouiah and Fodil Hamzaoui Laboratoire Structure et Elaboration des Matériaux Moléculaires Faculté des Sciences & de la Technologie - Université de Mostaganem - Algérie
Le présent travail concerne la détermination de la distribution de la densité électronique
du composé de formule chimique C8H9NOS à partir d’un spectre de diffraction des rayons X haute résolution sur monocristal [1]. Le Composé investi est connu pour ses remarquables applications en optiques non linéaire.
Nous avons utilisé le formalisme de Blessing pour la réduction et le traitement des
données brutes. L’affinement de la structure a été effectué en utilisant le programme XD basé sur le model multipolaire de Hansen-Coppens [2] tenant compte l’asphéricité des électrons de valence. Les résultats obtenus témoignent de la qualité du spectre de diffraction d’une part et de l’efficacité du model utilisé. Nous avons exploré systématiquement les différentes sections planes de la molécule. Les cartes de densité obtenues seront présentées lors de la rencontre.
Carte de densité dynamique dans le plan du
cycle benzénique
Carte de densité dynamique
autour de l'atome de Liaison C-S [1] Dupont et al., Acta Cryst. D. 2013, 04, 3-6. [2] Hansen, N. K. & Coppens, P. (1978). Acta Cryst. A34, 909.

Session Posters 5c Cristallographie biologique et Santé
- 174 -
P40 - Mécanisme d’activation de Legionella RalF : étude biochimique et structurale Marcia Folly-Klan1, Eric Alix3, Danièle Stalder2, Lionel Duarte1, Anna Delprato1, Pampa Ray1, Mahel Zeghouf1, Bruno Antonny2, Valérie Campanacci1, Craig R. Roy3, Jacqueline Cherfils1 1 Laboratoire d’Enzymologie et Biochimie Structurales, Centre de Recherche de Gif, CNRS, Gif-sur-Yvette, France, 2 Institut de Pharmacologie Moléculaire et Cellulaire, Université de Nice-Sophia Antipolis et CNRS, 06560 Valbonne, France, 3 Department of Microbial Pathogenesis, Yale University School of Medicine, New Haven, CT, USA
Legionella pneumophila, l’agent responsable de la maladie du Légionnaire, est un
pathogène intracellulaire capable d’échapper à la réponse immunitaire de l’hôte et ainsi éviter sa destruction par les macrophages alvéolaires. Pour cela, la bactérie camoufle l’identité du phagosome pour éviter la voie lysosomale en créant une vacuole de réplication appelée LCV (Legionella-containing vacuole).
Dans ce but, L. pneumophila utilise un système de sécrétion de type IV appelé Dot/Icm
pour injecter différents effecteurs dans le cytosol de la cellule hôte. Un de ces effecteurs, RalF, va être responsable du recrutement de la petite protéine G Arf1 à la surface de la LCV [1, 2]. En effet, RalF possède un domaine (Sec7) apparenté aux facteurs d’échange cellulaires (ArfsGEFs) qui stimulent l’échange GDP/GTP nécessaire à l’activation de la petite protéine G Arf. La particularité de RalF réside dans la présence d’un domaine C-terminal, le capping domain, responsable de l’auto-inhibition de la protéine en solution [3].
Notre objectif est de décrypter les mécanismes fonctionnels de cette famille de protéines
afin de comprendre leur régulation et leur capacité à cibler certaines membranes. Ceci passe par une étude structurale par diffraction aux rayons X et par diffusion des rayons X aux petits angles (SAXS) combinée à une étude fonctionnelle de reconstitution de l’activité d’échange en présence ou non de membranes artificielles. Nos résultats montrent que le capping domain possède un « cluster » aromatique « senseur » de l’environnement lipidique. Le capping domain est ainsi capable d’interagir spécifiquement avec des lipides présents à la surface de la LCV permettant le passage à une conformation active de RalF dans laquelle le site actif du domaine Sec7 est libéré et catalyse la réaction d’échange de nucléotide de Arf1. [1] Nagai et al., Science, 2002, 295(5555), 679-682 [2] Ninio et al., TRENDS in Microbiology. 2007, 15(8), 372-380 [3] Amor et al., J. Biol. Chem., 2005, 280(2), 1392-1400

Session Posters 5c Cristallographie biologique et Santé
- 175 -
P41 - Etude structurale et fonctionnelle de la phosphatase alcaline LapA : un transporteur de phosphate chez Pseudomonas auruginosa PAO1 Ahmed Djeghader1, Guillaume Gotthard1, Daniel Gonzalez1, Julien Hiblot1, Mikael Elias2 et Eric Chabrière1 1Unité de recherche sur les maladies infectieuses tropicales émergentes (URMITE), CNRS-Université de la Méditerranée, Marseille, France 2Weizmann Institute of Science, Biological Chemistry, Rehovot, Israel
Le phosphore est un élément indispensable à la vie mais peu répandue dans la nature.
Son acquisition de l’environnement sous forme de phosphate inorganique (Pi) représente donc un enjeu majeur pour la survie et la croissance des microorganismes. Afin d’assurer son approvisionnement, les bactéries possèdent deux principaux systèmes complémentaires : le système Pit (Phosphate inorganic transport) à haute vélocité/faible affinité, et le système Pst (Phosphate specific transport) à haute affinité/faible vélocité [1]. Tandis que le système Pit est régulièrement utilisé par les bactéries dans les conditions normales, le système Pst est fortement sollicité en cas de carence en phosphate. En plus de ces deux systèmes, certaines bactéries possèdent un troisième système d’acquisition, fortement exprimé en conditions limitantes de phosphate.
Nous nous sommes intéressés à ce dernier système présent chez le pathogène Pseudomonas aeruginosa PAO1. Il s’agit en effet de la protéine LapA, une phosphatase alcaline à faible poids moléculaire [2], extracellulaire, et phylogénétiquement reliée à la famille des phosphate binding proteins (PBP). La structure cristallographique de cette protéine fut résolue à 0.87 Å de résolution. Elle présente un repliement de type venus-flytrap, similaire à celui des PBP avec des différences au niveau de la longueur des boucles exposées au solvant. Son activité phosphatase fut caractérisée envers le para-Nitro-Phenyl-Phosphate (pNPP). Nos résultats préliminaires montre que LapA présente une haute affinité pour ce substrat mais une très faible vélocité. Cela suggère un mode de fonctionnement et un rôle différent de celui des phosphatases alcalines classiques dans le système d’acquisition de phosphate. Des travaux de co-cristallisation avec des analogues de substrat et de mutagénèse dirigée sont actuellement en cours afin d’élucider les bases structuraux de cette activité. [1] Willsky et al., J Bacteriol. 1980, 144,356-365. [2] Tan et al., FEMS Microbiol Lett. 1993, 106, 281-286.

Session Posters 5c Cristallographie biologique et Santé
- 176 -
P42 - Solubilisation de protéines problématiques, et si la solution résidait dans la phylogénie ?
Daniel Gonzalez1, Julien Hiblot1, Guillaume Gotthard1, Ahmed Djeghader1, Mikael Elias2 et Eric Chabrière1 1 Unité de recherche sur les maladies infectieuses tropicales émergentes (URMITE), CNRS-Université de la Méditerranée, Marseille, France 2 Weizmann Institute of Science, Biological Chemistry, Rehovot, Israel
L’étude structurale, biochimique et mécanistique des protéines requière une protéine pure et en grand quantité. Cependant, de nombreuses protéines, problématiques mais à l’important rôle physiologique ne sont pas étudiées faute de pouvoir les obtenir en quantité/qualité suffisante; c’est notamment le cas des protéines hydrophobes. Bien que de multiples stratégies furent développées afin de surmonter ces limitations, elles s’avèrent souvent inefficaces et peuvent être complexes pour les plus pointues d’entre elles.
Dans le but de solubiliser une protéine problématique (HPBP), nous avons exploité une stratégie reposant sur la phylogénie: la résurrection ancestrale. En effet, connue pour générer des protéines ancestrales thermostables, nous avons posé l’hypothèse que cette méthode permettrait d’enrichir la protéine en mutations clés stabilisatrices; tout en conservant son activité physiologique. La Human Phosphate Binding Protein (HPBP) est une protéine humaine, issue du plasma humain. Cette lipoprotéine est associée la paraoxonase humaine (hPON1), présentant toutes deux une hydrophobicité prononcée et une grande instabilité [1,2]. D’ailleurs, la PON fut solubilisée par des approches méthodologiques lourdes en vue d’applications biotechnologiques [3].
En vue de solubiliser HPBP, nous avons réalisé un arbre phylogénétique de la famille des protéines DING, à laquelle appartient HPBP. Il a permis d’identifier 60 positions stabilisatrices potentielles dont 22 ont été sélectionnées par analyse structurale. Le gène codant ce variant fut synthétisé et la protéine exprimée de façon soluble contrairement à l’enzyme sauvage. Le variant conserve les multiples activités de HPBP dont celle de fixer le phosphate et d’inhiber le VIH, via l’étape de la transcription, une étape non ciblée par les thérapies actuelles [4,5]. Ainsi, nous proposons par cette étude une nouvelle méthodologie qui, par étude in sillico, permettant de solubiliser rapidement et relativement facilement certaines protéines complexes à étudier. Cette stratégie pourrait permettre de déverrouiller un verrou technique dans l’étude de certaines protéines hydrophobes.
[1] Dupont et al., Acta Cryst. D. 2013, 04, 3-6 ; Renault F, Chabriere E, Andrieu JP, Dublet B, Masson P, et al. (2006) J Chromatogr B Analyt Technol Biomed Life Sci 836: 15-21. [2] Rochu D, Renault F, Clery-Barraud C, Chabriere E, Masson P (2007) Biochim Biophys Acta 1774: 874-883. [3] Harel M, Aharoni A, Gaidukov L, Brumshtein B, Khersonsky O, et al. (2004) Nat Struct Mol Biol 11: 412-419. [4] Cherrier T, Elias M, Jeudy A, Gotthard G, Le Douce V, et al. Virol J 8: 352. [5] Morales R, Berna A, Carpentier P, Contreras-Martel C, Renault F, et al. (2006) Structure 14: 601-609.

Session Posters 5c Cristallographie biologique et Santé
- 177 -
P43 - Evolution convergente et divergente entre lactonases et phosphotriestérases Guillaume Gotthard1, Julien Hiblot1, Mikael Elias2 et Eric Chabrière1 1 Unité de recherche sur les maladies infectieuses tropicales émergentes (URMITE), CNRS-Université de la Méditerranée, Marseille, France 2 Weizmann Institute of Science, Biological Chemistry, Rehovot, Israel
Les organophosphorés (OPs) sont des composés synthétiques neurotoxiques introduits dans la nature à partir des années 50s. Certaines enzymes ont rapidement évolué (~ 30 ans) pour dégrader efficacement ces composés, approchant la perfection catalytique (BdPTE, kcat/KM ~ 108 M-1.s-1). L’émergence rapide d’une telle activité souleva la question de son origine puisque ces composés ne sont pas naturels. Récemment, la mise en évidence d’une promiscuité enzymatique phosphotriestérase chez des lactonases permit de proposer que les lactonases puissent être le point d’émergence des phosphotriestérases [1,2]. Ainsi, il est possible d’observer au sein de deux familles structurales distinctes (amidohydrolases et métallo-β-lactamases) des phénomènes d’évolution convergente et divergente illustrant parfaitement les relations de promiscuité entre ces deux activités.
SsoPox et SisLac sont des Phosphotriestérases-Like Lactonases (PLLs) hyperthermostables (Tm ~106°C) présentant des activités de promiscuité phosphotriestérase (kcat/KM ~ 103 M-1.s-1, à 25°C) [3]. Appartenant à la superfamille des amidohydrolases (topologie (α/β)8), elles présentent une importante homologie structurale avec la BdPTE. Cette dernière aurait divergé d’une lactonase appartenant probablement à la famille des PLLs [1]. Parallèlement aux PLLs, d’autres OP hydrolases (MPH et OPHC2) ayant également émergé au sein de la superfamille des métallo-β-lactamases (sandwich αβ/βα) sont présumées provenir de lactonases apparentées telles qu’AiiA.
Dans l’objectif de développer un bio-décontaminant efficace envers les OPs, nous avons utilisé les voies empruntées par la nature pour ingénieuriser une enzyme hyperthermostable (SsoPox) afin de rendre son cœur catalytique plus efficace envers les OPs. En effet, bien que la promiscuité d’activité phosphotriestérase des lactonases offre des possibilités d’évolution par mutagénèse aléatoire, criblage et sélection ; la nature offre des modèles déjà optimisés pouvant être mimés au sein d’enzyme préexistantes permettant ainsi de converger plus rapidement vers des catalyseurs efficaces et robustes. [1] Afriat-Jurnou et al., Biochemistry 51, 6047-6055 [2] Elias et al., The Journal of biological chemistry 287, 11-20 [3] Elias et al., Journal of molecular biology 379, 1017-1028

Session Posters 5c Cristallographie biologique et Santé
- 178 -
P44 - Bases structurales de l’antigénicité des complexes peptide-CMH Jean-Baptiste Reiser1, François Legoux2, Stéphanie Gras1, Eric Trudel1, Anne Chouquet1, Alexandra Léger2, Madalen Le Gorrec1, Paul Machillot1, Marc Bonneville2, Xavier Saulquin2, Dominique Housset1 1
Institut de Biologie Structurale, UMR 5075, CEA, CNRS, Université J. Fourier – Grenoble I, 41 rue J. Horowitz, 38027 Grenoble, 2
UMR 892 et Centre de Recherche en Cancérologie Nantes Angers, INSERM, Université de Nantes, 8 quai Moncousu, 44007 Nantes
Le projet que nous menons en collaboration avec l’équipe de M. Bonneville, a pour but de comprendre les bases structurales de la reconnaissance d’antigènes tumoraux, présentés par les molécules CMH, par les récepteurs du lymphocyte T (TCR). Nous cherchons à établir le lien entre les interactions intermoléculaires qu’un TCR peut établir avec les complexes peptide-CMH, observables grâce aux structures cristallographiques et quantifiables par différentes techniques biophysiques comme la résonance plasmonique de surface [1], et la capacité de ces mêmes complexes peptide-CMH à activer des lymphocytes T et à activer une réponse immune efficace. C’est un enjeu majeur pour le développement de l’immunothérapie anti-tumorale. En combinant études structurales de plusieurs complexes peptide-CMH et analyse de la fréquence des lymphocytes T spécifiques de ces complexes peptide-CMH dans le répertoire naïf de lymphocytes T [2], nous avons pu dégager un certain nombre de principes et montrer que plusieurs paramètres doivent être pris en compte. L’analyse de plusieurs TCR spécifiques d’un même antigène de mélanome, Melan-A [3], par SPR nous a montré très récemment une bonne corrélation entre affinité du TCR pour le peptide-CMH et l’avidité fonctionnelle pour les lymphocytes T qui portent ces TCR. De plus, cette approche va nous permettre de quantifier la contribution de chacune des chaînes α et β des TCR à l’interaction TCR-peptide-CMH, permettant de mieux comprendre les raisons de la prévalence d’une chaîne α particulière, TRAV12-2, dans le répertoire anti-Melana-A, et les bases de la différence d’affinité entre ces différents TCR. In fine, nos travaux visent à mieux comprendre toute la complexité de la réponse lymphocytaire T anti-tumorale, dans le but d’aider à développer des stratégies d’immunothérapie plus efficaces.
[1] Gras, S. et al. Structural bases for the affinity-driven selection of a public TCR against a dominant human cytomegalovirus epitope. J Immunol 183, 430-437 (2009). [2] Legoux, F. et al. Impact of TCR reactivity and HLA phenotype on naive CD8 T cell frequency in humans. J Immunol 184, 6731-6738, doi:jimmunol.1000295 [pii]10.4049/jimmunol.1000295 (2010). [3] Trautmann, L. et al. Dominant TCR V alpha usage by virus and tumor-reactive T cells with wide affinity ranges for their specific antigens. Eur J Immunol 32, 3181-3190 (2002).

Session Posters 5c Cristallographie biologique et Santé
- 179 -
P45 - Etude structurale de récepteurs membranaires bactériens impliqués dans le transport d'antibiotiques Lucile Moynie et James H Naismith Biomedical Sciences Research Complex, University of St Andrews, St Andrews KY16 9ST, Scotland
L'augmentation de la résistance aux antibiotiques des bactéries à Gram négatif telle que Pseudomonas aeruginosa est devenue un enjeu sanitaire majeur nécessitant le développement de nouveaux traitements [1]. Une des nouvelles stratégies développées consiste à utiliser les voies de transport du complexe sidérophore-fer à travers la paroi bactérienne afin de délivrer les antibiotiques de manière plus efficace. C'est le cas de BAL30072, un β-lactamine conjugué à un sidérophore développé par Basilea Pharmaceutica International qui possède une forte activité contre Pseudomonas aeruginosa (concentration minimal d'inhibition 8 mg/L) [2].
Récemment, deux transporteurs des complexes sidérophore-fer (PiuA et PirA) ont été identifiés comme étant impliqués dans le transport à travers la membrane externe de Pseudomonas aeruginosa de ces nouveaux antibiotiques [3,4].
Nous avons débuté l'étude structurale de ces protéines membranaires afin de comprendre les mécanismes de transport de cette nouvelle classe d'antibiotiques et d'améliorer leur efficacité. [1] Page and Heim, Cur Opin Pharmacol.. 2009, 09, 1-8. [2] Page et al., Antimicrob. Agents Chemother. 2010, 6, 2291-2302. [3] van delden et al., Antimicrob. Agents Chemother. 2013, 5, 2095-2101 [3] Mcpherson et al., Antimicrob. Agents Chemother. 2012, 12, 6334-6342

Session Posters 5c Cristallographie biologique et Santé
- 180 -
P46 - Caractérisations structurales d’inhibiteurs atypiques de la protéine kinase CK2 pour la conception de nouvelles molécules anti-cancéreuses
Jean-Baptiste Reiser1, Benoît Bestgen2,4, Thierry Lomberget2, Marc Le Borgne2, Mathias Engel3 Claude Cochet3 1Institut de Biologie Structurale Jean-Pierre Ebel (CEA-CNRS-UJF), 41 rue Jules Horowitz 38027 Grenoble Cedex, 2Université de Lyon, Université Lyon 1, Faculté de Pharmacie, ISPB, EA 4446 Biomolécules, Cancer et Chimiorésistances, 8 avenue Rockefeller 69373 Lyon cedex 8, France, 3Institut de Recherches en Technologie et Sciences pour le Vivant (CEA), 17 rue des Martyrs 30854 Grenoble Cedex 09 4Pharmaceutical and Medicinal Chemistry, Saarland University, Campus C2.3, D-66123 Saarbrücken, Germany.
CK2 est une protéine kinase qui fait partie d’une classe d’enzymes multifonctionnelles, constitutivement actives et exprimées de manière ubiquitaire. Sa surexpression et sa dérégulation ont été révélées dans de nombreux types de cellules cancéreuses, notamment les cellules leucémiques, de la prostate, du colon ou du sein. CK2 est aujourd’hui considérée comme une cible prometteuse pour le développement de nouvelles thérapies anticancéreuses spécifiques. Toutefois, peu des molécules inhibitrices identifiées à ce jour se sont avérées être de bons candidats pour le développement de molécules thérapeutiques ; essentiellement parce que beaucoup d’entre elles n’ont pas démontré leur efficacité dans des modèles cellulaires ou chez l’animal. Depuis plusieurs années, nos collaborateurs et notre équipe s’intéressent à la découverte et la caractérisation d’inhibiteurs originaux de cette protéine kinase en utilisant différentes approches de criblage, de biochimie, de biologie cellulaire et de biologie structurale. Ainsi, grâce à la stratégie employée, plusieurs nouvelles familles de molécules possédant une action inhibitrice à des degrés divers et spécifiques de CK2 ont été découvertes, dont certaines ont montré une efficacité in vivo sur la régression de xénogreffes de tumeurs humaines chimiorésistantes.
Dans un premier temps, nous nous sommes intéressé à plusieurs familles d’inhibiteurs ATP-compétitifs [1,2]. Les différentes structures de CK2 en complexe avec plusieurs molécules ont permis de décrire le mode d’interaction et les interactions clés nécessaires à l’inhibition et de rationaliser la sélectivité plus ou moins restreinte de ces composés pour CK2. Nous nous focalisons aujourd’hui sur des composés non-ATP-compétitifs atypiques qui sont capables de moduler l’activité kinase de façon allostérique. Ces nouveaux inhibiteurs ont la particularité d’être très spécifiques pour la kinase cible. Toutefois, le site de fixation et le mode d’inhibition restent encore peu connus. Nous présenterons les résultats préliminaires obtenus par SAXS sur un complexe entre le domaine catalytique de CK2 et un de ces nouveaux types d’inhibiteurs. [1] Prudent, R., et al., Antitumor activity of pyridocarbazole and benzopyridoindole derivatives that inhibit protein kinase CK2. Cancer Res. 2010, 70(23), 9865-74. [2] Lopez-Ramos, M., et al., New potent dual inhibitors of CK2 and Pim kinases: discovery and structural insights. FASEB J. 2010, 24(9), 3171-85.

Session Posters 5c Cristallographie biologique et Santé
- 181 -
P47 - Caractérisation structurale et fonctionnelle de la voie de conjugaison de FAT10 Prakash Rucktooa UMR 8576 CNRS – Université des Sciences et Technologies de Lille
Les modifications post-traductionnelles modulent la fonction des protéines, contribuant à la régulation fine de processus cellulaires. La fatylation est une de ces modifications, qui consiste en la conjugaison d’une protéine de type ubiquitine appelée FAT10 sur une protéine cible. Cette conjugaison se fait par l’intermédiaire de différentes enzymes agissant en cascade. Une enzyme d’activation (Uba6) active FAT10 avant de le transférer sur une enzyme de conjugaison (Ube2z) qui procède à la modification des protéines cibles avec l’action conjointe de E3-ligases.
Uba6 et Ube2z présentent toutefois la particularité de fonctionner à la fois lors de la fatylation et lors de l’ubiquitination. Cette double spécificité de substrat souligne la complexité de mécanismes de régulation intervenant dans la conjugaison de modificateurs de type ubiquitine sur des protéines cibles.
Nous utilisons des méthodes structurales, biophysiques et biochimiques afin de comprendre comment survient cette double spécificité de substrat pour Uba6 et Ube2z.

Session Posters 5c Cristallographie biologique et Santé
- 182 -
P48 - Inhibiteurs de Myosine du muscle lisse : une nouvelle méthode pour le traitement des maladies de hyper-contractilité des voies aériennes S. Sirigu1, J. Hartman2, G. Bergnes2, F. Malik2, A. Houdusse1 1 Motilité Structurale, Institut Curie, UMR 144, CNRS, Paris, France, 2 Recherche Préclinique et Développement, Cytokinetics, Inc., San Francisco, CA 94080, USA.
Les myosines sont des moteurs moléculaires essentiels pour de nombreuses fonctions cellulaires. La production de force est couplée à l’hydrolyse de l’ATP alors qu’un changement de conformation nommé powerstroke se produit sur le complexe acto-myosine au moment du relâchement des produits. Pendant ce cycle moteur, la myosine explore différents états conformationnels en fonction du nucléotide lié et de l’affinité pour l’actine qui en résulte. Plusieurs isoformes de myosines ont été impliquées dans des maladies humaines sévères et la recherche de drogues allostériques capables de moduler ces moteurs constitue une nouvelle approche thérapeutique pour ces déficits.
Notre collaborateur, Cytokinetics, Inc. (USA) a découvert des inhibiteurs sélectifs de
la Myosine II du muscle lisse qui empêchent l’hydrolyse de l’ATP 1, 2. Ce mécanisme d’inhibition augmente la population de moteurs moléculaires faiblement liés à l’actine et favorise la relaxation du muscle lisse. Le potentiel de cette drogue a été validé dans des modèles animaux de bronchoconstriction et représente une nouvelle voie thérapeutique pour le traitement des maladies de hyper-contractilité des voies aériennes.
Nous avons résolu la structure cristallographique de la Myosine II du muscle lisse liée à un de ces inhibiteurs de Cytokinetics. Notre structure décrit une poche inattendue dans laquelle la drogue se fixe. Cette poche se situe entre les deux connecteurs du moteur qui contrôlent l’élément mécanique de la myosine, le bras de levier. Cette région de la protéine subit des réarrangements structuraux qui déterminent la position exacte du bras de levier pendant le cycle de la myosine – et en particulier le powerstroke. Notre structure apporte des éléments nouveaux pour la compréhension des réarrangements allostériques du moteur et représente un outil important pour la réalisation de nouvelles générations d’inhibiteurs spécifiques de myosines.
1 Zhao et al., Pharmacol Exp Ther. 2011, 339(1):307-12 2 Ho et al., PLoS One. 2012, 7(5):e36302

Session Posters 5c Cristallographie biologique et Santé
- 183 -
P49 - Structural and functional studies of FNE, a bacterial adhesion protein of Streptococcus equi : a "Rebel" protein for crystallization Mounira Tiouajni, Dominique Durand, Agathe Urvoas, Marielle Lepiniec-Valerio, Karine Blondeau, Asma Guellouz, Marc Graille, Philippe Minard and Herman van Tilbeurgh Institut de Biochimie et Biophysique Moléculaire et Cellulaire (IBBMC), Université Paris Sud, UMR 8619, Orsay, France.
Streptococcus equi is a gram-positive bacteria responsible for diseases of the upper respiratory tract in horses that can be fatal such as strangles. Exceptionally, this bacteria is responsible for meningitis in humans. FNE is a protein involved in adhesion and virulence of Streptococcus equi by interacting with fibronectin in the extracellular matrix of the cell. Fibronectin is a long-glycoprotein (250 kDa) organized into functional domains that interact with different partners such as bacterial adhesion proteins. FNE Interacts with the Gelating Binding Domain of fibronectin (GBD), located near the N-terminus. We study the structural and functional aspects of this interaction as a model for bacterial adhesion to the GBD. We were unable to crystallize FNE or a truncated version lacking the disordered C-terminal peptide. We therefore developed artificial proteins that bind to FNE with the objective to create complexes of these proteins in complex with FNE and amenable to crystallization. We made use of a library coding for artificial protein constructed by repetition of a pattern designed HEAT from a thermophilic archaeal protein [1]. Three artificial proteins interacting with the FNE have been obtained by phage-display and the corresponding complexes with FNE were tested for crystallization. We will present this new innovative cristallogenesis technique and the structure of the complex FNE / artificial partner obtained at 1.83 Ǻ of resolution.
[1] Urvoas, Guellouz et al ; J Mol Biol. 2010, 404b307-27

Session Posters 6a Méthodes émergentes en cristallographie
- 184 -
P50 - Analyse structurale de noirs de carbone archéologiques par techniques combinées Sophie Cersoy1, Pauline Martinetto1, Pierre Bordet1, Jean-Louis Hodeau1, Elsa Van Eslande2, Philippe Walter2 1 Institut Néel, CNRS-UJF, Grenoble, 2LAMS, CNRS-UMPC, Paris
Les noirs dits « de carbone » ont été fréquemment utilisés depuis la Préhistoire en tant que pigments, cosmétiques ou encres. Ils regroupent en fait une très grande variété de produits obtenus par carbonisation d’un matériau d’origine animale, végétale ou minérale qu’on peut retrouver sous les termes de noir de charbon, noir de fumée, noir d’os…. Si les recettes de préparation sont relativement bien décrites, en particulier dans les ouvrages anciens [1], les différentes phases carbonées obtenues, souvent mal cristallisées, sont difficiles à identifier et à caractériser.
Des micro-prélèvements de cosmétiques et/ou encres noirs ont été effectués dans différents objets découverts pendant les fouilles de Pompéi. Leur composition, en lien avec leur usages et fonction, reste à déterminer [2]. Comprendre ces matériaux incite le développement de nouvelles méthodologies : combinaison diffraction des rayons X et tomographie, qui permet la reconstruction de coupes virtuelles structurales montrant la répartition des phases (cristallisées ou non) [3] et caractérisation de matériaux non/mal cristallisés par analyse de la fonction de distribution de paires (PDF) [4]. Des données de diffraction des rayons X ont été enregistrées sur la ligne BM2-CRG de l’ESRF en mode tomographie. Elles permettent d’identifier les phases cristallisées et de comparer les signaux de diffusion des différentes phases carbonées caractéristiques des échantillons. Des pigments noirs de référence, achetés chez différents fournisseurs de pigments, ont également été analysés pour aider à l’interprétation des échantillons anciens. Les résultats de diffraction des rayons X sont confrontés à ceux obtenus par d’autres techniques de caractérisation structurale comme la spectroscopie Raman, qui peut permettre de déterminer l’origine végétale, minérale ou animale des noirs de carbone étudiés et leur degré de désordre.
Ce projet participe à une série d’études menées sur les cosmétiques archéologiques, connus pour révéler des pratiques et savoir-faire anciens dans le domaine de la physico-chimie [5]. [1] DIOSCORIDE, De materia medica (I, 69); PLINE L'ANCIEN, Histoire naturelle, Livre XXXV.
[2] GAMBERINI et al., Vibrational Spectroscopy, 2008, 47, 82-90 ; CANEVALI et al., Anal. Bioanal. Chem., 2011, 401, (6), 1801-1818. [3] BLEUET P. et al. Nat. Mat., 2008, 7(6), 468-472 ; ALVAREZ-MURGA et al., J. Appl. Cryst. 2011, 44, 163-171 ; DE NOLF W. and JANSSENS K., Surf. Interface Anal., 2010, 42, 411-418. [4] EGAMI T., BILLINGE S. J. L., Underneath the Bragg Peaks: Structural analysis of complex materials, 2003, Pergamon, Oxford, England ; BORDET P., Collection de la Société Française de la Neutronique, 2008, 9, O. Isnard, Ed., EDP Sciences. [5] WALTER P. et al., Nature, 1999, 397, 483-484.

Session Posters 6a Méthodes émergentes en cristallographie
- 185 -
P51 - Cryscal
Thierry Roisnel1, Juan Rodríguez-Carvajal2, Javier González-Platas3 1 Centre de Diffractométrie X, Institut des Sciences Chimiques de Rennes, UMR6226 CNRS – Université de Rennes 1, Avenue du Général Leclerc, 35042 Rennes Cedex (France) 2 Institut Lauë Langevin, 6 rue Jules Horowitz, BP156, 38042 Grenoble Cedex 9 (France) 3 Departamento Fisica Fundamental II, Universidad de La Laguna, 38204 La Laguna, Tenerife (Spain)
Basé en grande partie sur l'utilisation de la librairie cristallographique modulaire CrysFML[1], Cryscal est une calculette cristallographique conçue pour réaliser de nombreuses opérations basiques en cristallographie / diffraction:
. lecture et analyse de fichiers d'intensités intégrées (.hkl); recherche de groupes d'espace
. transformations matricielles
. calculs de distances et angles interatomiques
. calcul de connectivité et de Bond Valence Sum
. calcul de facteurs de structure cristallographique (X, neutrons, e-)
. simulation de diagrammes de diffraction sur poudres (X, neutrons), avec prise en compte d'un élargissement des réflexions dues à une taille de particules non infinie
. calcul de coefficient d'absorption (X, neutrons)
. conversion de format de fichiers (.INS, .CIF, .CEL, .FST …) …
et également obtenir des informations cristallographiques (groupe d'espace, positions atomiques, …) ou atomiques (facteurs de diffusion, rayons ioniques, masses moléculaires, …) ou bien encore réaliser des rapports d'expérience de résolution structurale (.HTML, .PDF).
Les nombreuses potentialités offertes par Cryscal sont directement liées à l'utilisation de mots clés et des arguments associés, lesquels permettent de spécifier au programme les différentes tâches à accomplir. Ces instructions à exécuter peuvent être soit directement entrées au clavier (mode interactif), soit être lues dans un fichier d'entrée qui peut être passé comme argument à l'exécution du programme, ce qui permet aisément l'interfaçage par d'autres programmes (ex: WinGX, WinPLOTR ).
A noter que les informations structurales nécessaires à l'exécution de certaines tâches peuvent être soit introduites manuellement, soit être extraites directement de la lecture de fichiers de données structurales (formats CIF, .INS/.RES pour SHELXL, .PCR pour FullProf …).
Signalons enfin que Cryscal est évolutif en fonction des demandes des utilisateurs et qu'une version pour Windows est disponible et téléchargeable à l'adresse : www.cdifx.univ-rennes1.fr/cryscal.
[1] J. Rodríguez-Carvajal and J. González-Platas http://www.iucr.org/resources/commissions/crystallographic-computing/newsletters/1/crysfml

Session Posters 6b Biologie Structurale Intégrative et gros assemblages
- 186 -
P52 - Rotor architecture in the F1c10 sub-complex of the yeast F1F0-ATP synthase
Marie-France Giraud1,2, Patrick Paumard1,2, Corinne Sanchez1,2, Isabelle Larrieu1,2, Daniel Brèthes1,2, Alain Dautant1,2 1 Université de Bordeaux, IBGC, UMR 5095, F-33000 Bordeaux, France 2 CNRS, IBGC, UMR 5095, F-33000 Bordeaux, France
The yeast mitochondrial F1F0-ATP synthase is a hybrid molecular nanomotor. F1 is a chemical motor driven by ATP hydrolysis while F0 is an electrical motor driven by proton flow. The two stepping motors are mechanically coupled through a common rotary shaft. Two different crystal forms of the yeast F1c10 ATP-synthase sub-complex were obtained at a resolution of 3.43 Å (form I) and 6.5 Å (form II). Form I crystals were grown in the presence of Mg-ADP, dicyclohexylcarbodiimide (DCCD) and azide. The structure (yF1c10(I)) displayed new features compared with the unrefined yF1c10 [1] and the first yeast F1 structure (yF1) inhibited by adenylyl imidodiphosphate (AMPPNP) [2]. An ADP molecule was bound in both beta(DP) and beta(TP) catalytic sites. The alpha(DP)-beta(DP) pair was slightly open and resembled the novel conformation identified in yF1, whereas the alpha(TP)-beta(TP) pair was very closed and resembled more a DP pair. Hence, yF1c10(I) provided a model of a new Mg-ADP-inhibited state of the yeast F1 [3]. Like in the original yF1 [2] and yF1c10 [1] structures, the foot of the central stalk was rotated by ~40° with respect to the bovine F1 structure [4].
The form II crystal was obtained in the presence of AMPPNP, DCCD and azide.
Despite the low resolution of the dataset, the structure (yF1c10(II)) is informative on the enzyme. In form I crystals significant interactions of the F1-head with the c10-ring of a neighboring molecule affected the overall conformation of the rotor. This led to a tilt between the pseudo symmetry axis of the (alpha-beta)3 domain and the 10-fold symmetry axis of the c-ring near the F1-F0 rotor interface and resulted in an unbalanced machine. However, in the form II crystal, the packing has a weaker influence on the conformation of the rotor. The yeast yF1c10(II) and the bovine bF1c8 [5] structures share a common rotor architecture with the inertia center of the (alpha-beta)3 domain close to the rotor c-ring axis, thus providing a model of a more efficient motor. [1] Stock, D., Leslie, A.G., and Walker, J. E. (1999) Science 286, 1700-1705. [2] Kabaleeswaran, V., Puri, N., Walker, J. E., Leslie, A.G., and Mueller, D.M. (2006) EMBO J. 25, 5433-5442. [3] Dautant, A., Velours, J. and Giraud, M.-F. (2010) J. Biol. Chem. 285, 29502-29510. [4] Gibbons, C., Montgomery, M. G., Leslie, A. G., and Walker, J. E. (2000) Nat. Struct. Biol. 7, 1055-1061. [5] Watt, I.N., Montgomery, M.G., Runswick, M.J., Leslie, A.G., Walker J.E. (2010) Proc. Natl. Acad. Sci. USA. 107, 16823-16827.

Session Posters 6b Biologie Structurale Intégrative et gros assemblages
- 187 -
P53 - Structural insights into DNA recognition by Retinoid X Nuclear Receptor Judit Osz1, Alastair McEwan2, Pierre Poussin-Courtmontagne2, Catherine Birck2, Irwin Davidson3, Dino Moras1, Natacha Rochel1 1 Department of Integrative Structural Biology, 2 Structural Biology Platform, 3 Department of Functional Genomics and Cancer, Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Institut National de Santé et de Recherche Médicale (INSERM) U964 / Centre National de Recherche Scientifique (CNRS) UMR 7104 / Université de Strasbourg, 67404 Illkirch, France.
Retinoid X receptors (RXRs) are transcription factors with important function in embryonic development, metabolic processes, differentiation and apoptosis. A particular feature of RXRs is their ability to act as homodimer or as obligatory heterodimerisation partner of class II nuclear receptors. Functional RXR homodimers specifically regulate genes containing direct repeats (DR) of the half-site (A/G)G(G/T)TCA separated by 1 nucleotide (DR1). To unravel the mechanism of specificity of DNA binding by RXR, we have characterized the interaction between RXR and several DR1 from regulated genes using structural and biochemical approach. We identify the key interactions that lead to high and low affinity binding to DR1 sequence and show that the DNA sequence affects RXR homodimer structure. The solution structures of intact homodimeric RXR/DR1 show an extended asymmetrical arrangement with distinct hinge conformations.

Session Posters 7a Cristallographie in situ, in operando
- 188 -
P54 - Etude du photomagnétisme de composés moléculaires MnCuxZn(2-x) par diffraction de neutrons polarisés sous irradiation lumineuse in-situ. Béatrice Gillon1, Karl Ridier1, Grégory Chaboussant1, Nathalie Bridonneau2, Valérie Marvaud2 1Laboratoire Léon Brillouin (LLB), UMR12 CNRS-CEA, CEA Saclay, 91191 Gif-sur-Yvette (France), 2Institut Parisien de Chimie Moléculaire (IPCM), CNRS UMR7201, 75252 Paris (France)
La nature du processus microscopique qui pourrait expliquer les propriétés photomagnétiques des composés d’octacyanomolybdate de cuivre demeure en débat bien que les résultats de XMCD sur le complexe trinucléaire (MoCu2)-Meen [1] mettent en évidence un mécanisme de transition de spin (ST) de l’ion MoIV(BS) (S=0) vers un état haut spin MoIV(HS) (S=1). Toutefois, l’existence d’un mécanisme concomitant de transfert de charge métal-métal (MMCT) de l’ion MoIV vers l’ion CuII (déjà observé dans le complexe heptanucléaire photomagnétique MoCu6 par spectroscopie d’absorption X-ray [2]) ne peut pas être exclu comme le suggèrent les mesures SQUID effectuées récemment sur la famille de complexes (MoCuxZn2-x)-tren dans lesquels le Cu est substitué par le Zn non magnétique avec différentes concentrations 0<x<0,2.
Afin de mieux comprendre l’origine du photomagnétisme dans ces matériaux, nous utilisons la diffraction de neutrons polarisés (PND) associée à une irradiation in-situ par la lumière qui permet de caractériser les états photomagnétique à l’échelle atomique. Cette technique permet d’établir les cartes de densité de spin avant et après irradiation in-situ par un faisceau laser (405nm) afin de discriminer le rôle des atomes Mo and Cu dans le processus de photoexcitation. Les récentes investigations par PND sur un monocristal de (MoCu2)-Meen sur le diffractomètre 6T2 au LLB ont montré que des réflexions intenses apparaissent après irradiation lumineuse à des positions différentes de celles de l’état fondamental et disparaissent après relaxation thermique [3]. Ceci prouve l’existence d’un état métastable induit par la lumière possédant une structure différente du fondamental. Les résultats obtenus lors d’expériences similaires effectuées sur monocristal de (MoCu0,1Zn1,9)-tren sont présentés. [1] M.-A. Arrio, et al., J. Phys. Chem. C 2010, 114, 593-600 [2] J.M. Herrera, et al., Angew. Chem. Int. 2004, 43, 5468-5471 [3] A. Hammerschmied, Diplom Arbeit, TU Wien, 2010

Session Posters 7b Texture, microstructure, déformation
- 189 -
P55 - Analyse quantitative de diagrammes de diffraction électronique : nanoparticules et textures P. Boullay1 and L. Lutterotti2 1CRISMAT, CNRS UMR 6508, 6 Bd du Maréchal JUIN 14050 CAEN Cedex, France 2Department of Materials Engineering and Industrial Technologies, University of Trento, 38123 TRENTO, Italy
Les échantillons polycristallins composés de grains de taille nanométrique posent des difficultés quand à leur caractérisation structurale et microstructurale. L’objet de cette présentation sera de montrer comment la microscopie électronique en transmission (MET) peut être un outil de caractérisation permettant d’obtenir des résultats quantitatifs fiables et compléter avantageusement les informations obtenues par diffraction des rayons X. Parmi les différentes techniques disponibles dans un MET, les auteurs s’intéresseront plus particulièrement à l’analyse quantitative des diagrammes de diffraction de cercles classiquement obtenus pour une assemblée de nanoparticules. Ils montreront comment le programme MAUD [1] peut être utilisé pour extraire des informations telles que l’identification de la phase, les paramètres de maille, la texture, la taille et la forme des particules.
Typiquement, les données de diffraction électronique sont collectées sur un très faible volume diffractant en utilisant un faisceau parallèle illuminant une surface de 200µm2 à 1µm2 contenant un nombre suffisant de particules pour obtenir un diagramme de cercles. La durée d’enregistrement des données est de l’ordre de la seconde. Les intensités sont extraites en utilisant une routine ImageJ implémentée dans MAUD. Les informations structurales et microstructurales sont ensuite obtenues par la méthode de Rietveld de façon identique à ce qui est effectuée dans le cadre d’une analyse d’un diagramme de diffraction des rayons X sur poudre.
Les résultats obtenus sur quelques matériaux oxydes seront exposés et, lorsque disponibles, comparés à ceux obtenus par diffraction des rayons X. L’identification de la phase et l’analyse de la taille moyenne des domaines diffractant seront illustrées sur des nanoparticules de TiO2 [2] et de Mn3O4 [3]. Le cas d’une assemblée de nanoparticules présentant une forte texturation sera aussi abordé avec l’exemple d’un film de platine déposé sur silicium. Ces premiers résultats indiquent la pertinence de cette approche qui d’un point de vue technique est simple à mettre en œuvre. [1] L. Lutterotti, Nuclear Inst. and Methods in Physics Research 2010, B268, 334-340. [2] M.A. Reddy et al., ElectroChem. Com. 2006, 8, 1299-1303. [3] L. Sicard et al., J. Magn. Magn. Mater. 2010, 322, 2634-2640.

Session Posters 7c Grands Instruments Synchrotron et plateformes en biologie structurale - (Développements, XFEL, nouveautés)
- 190 -
P56 - PROXIMA 2A – Une nouvelle ligne de lumière micro-focus et accordable en énergie, dédiée à la bio-cristallographie Denis Duran1, Sebastien Le Couster1,2, Frederic Blache1, Aurélien Delmotte1, Roger Fourme1, Gavin Fox1, Rob Meijers1,3, Thierry Moreno1, Sandra Pierre-Joseph1, Jean-Pierre Samama1, Martin Savko1 & William Shepard1 1Synchrotron SOLEIL, l'Orme des Merisiers, Saint Aubin, BP 48, 91192 Gif-sur-Yvette, France 2Adresse actuelle: THALES, Domaine Corbeville Thomson, 91400 Orsay, France 3Adresse actuelle: EMBL c/o DESY, Building 25A, Notkestraße 85, 22603 Hamburg, Germany
PROXIMA 2A est une nouvelle ligne de lumière à Synchrotron SOLEIL, accordable en énergie, et dédiée à la micro-cristallographie des macromolécules biologiques. Elle vient d’ouvrir aux utilisateurs en mars 2013. Sa source est un puissant onduleur U24 sous vide. L’optique consiste en un monochromateur dit « channel-cut » en Si[111] refroidi avec l’azote liquide, un miroir pré-focalisant convexe et une paire de miroirs focalisants bimorphes en configuration Kirkpatrick-Baez. Ce schéma innovateur utilise un miroir convexe pour produire une source secondaire virtuelle afin de refocaliser le faisceau-X à 5 μm d’une large source horizontale. Les supports des éléments optiques sont étudiés pour minimiser les effets des vibrations transmis par l’environnement. Pour les dérives longues, la température dans les cabanes expérimentales est contrôlée à moins de 0.1°C près. Actuellement, en mode Kirkpatrick-Baez seul, le faisceau-X est focalisé à 10 μm × 5 μm (H×V FWHM) avec un flux de photons de 1×1013 – 4×1011 ph/s selon l’énergie (7 – 15 keV). La station expérimentale contient un micro-diffractomètre, un détecteur bidimensionnel, un cryostat, un robot pour transférer les échantillons et un détecteur de fluorescence-X pour les expériences MAD et SAD. Les utilisateurs gèrent leurs expériences par l’interface MXCuBE [1], qui permet le centrage de l’échantillon, le lancement des collectes, les enregistrements de spectres en énergie-X et les transferts des échantillons. Les données de diffraction-X enregistrées sont d’une excellente qualité, et les utilisateurs exploitent le micro-faisceau-X pour chercher la meilleure zone dans leurs cristaux. Un bilan des premiers résultats et les perspectives à venir seront présentés. [1] Gabadinho et al., J. Synchrotron Rad. 2010, 17, 700-707.

Session Posters 7c Grands Instruments Synchrotron et plateformes en biologie structurale - (Développements, XFEL, nouveautés)
- 191 -
P57 - Etude structurale de MglA, une GTPase régulatrice de la polarité bactérienne Paloma Fernández-Varela1, Armelle Vigouroux1, Mahel Zeghouf1, Lionel Duarte1, Mathilde Guzzo2, Tam Mignot2, Jacqueline Cherfils1 1Laboratoire d'Enzymologie et Biochimie Structurales (LEBS) Centre de Recherche de Gif – CNRS Bâtiment 34 - Avenue de la Terrasse 91190 Gif-sur-Yvette 2Laboratoire de Chimie Bactérienne, Institut de Microbiologie de la Méditerranée, Université Aix-Marseille, Marseille, France.
Comment la bactérie modèle Myxococcus xanthus contrôle-t-elle la direction de ses mouvements ? L’équipe de Tâm Mignot, au laboratoire de Chimie Bactérienne à Marseille, a démontré récemment que la polarité est établie par une protéine apparentée aux petites protéines G eukaryotes, MglA (1). La polarité du mouvement est déterminée par la localisation de MglA sous sa forme active, liée au GTP, au pôle avant de la bactérie, cependant qu’une GTPase activating protein, (GAP), MglB, l’inactive au pôle arrière. Occasionnellement, la bactérie s’arrête et repart en sens inverse : cette inversion est déclenchée par la relocalisation synchronisée de MglA et MglB aux pôles opposés. Ces travaux suggèrent pour la première fois que la régulation de la motilité par les petites GTPases, bien connue chez les eucaryotes, pourrait être bien plus largement conservée qu’il n’avait été envisagé jusqu’à ce jour.
Les bases structurales de ce mécanisme ont commencé à être étudiées chez une protéine homologue à MglA présente chez Thermus thermophilus, dont la fonction n’est pas connue (2). La forme inactive de MglATt (MglATt -GDP) adopte une conformation jamais encore observée chez les petites GTPases eucaryotes. Cette structure, ainsi que sa comparaison au complexe MglA-MglB, suggère que MglA pourrait subir un changement conformationnel de grande amplitude entre sa forme inactive (liée au GDP) et sa forme active (liée au GTP). Identifier la nature de ces réarrangements chez MglAMx est une étape importante dans la compréhension des mécanismes moléculaires de la motilité et de la polarité bactérienne. [1] Zhang Y et al. PLoS Genet 2012, 8(8):e1002872. [2] Miertzschke M et al. EMBO J. 2011, 30:4185-97.

Session Posters 8a Chiralité, polymorphisme
- 192 -
P58 - Towards the rational synthesis of Chiral Spin-Crossover compounds Ahmad Naim, Elen Duverger-Nedellec, Philippe Guionneau, Patrick Rosa CNRS, Université de Bordeaux, ICMCB, 87 Av. Doc. A. Schweitzer, 33608 Pessac, France
e-mail : [email protected]
Since the discovery of molecular chirality by Pasteur in 1849, and the helical structure of DNA by Wilkins, Watson and Crick in 1953, chirality has become a fundamental notion in Chemistry (e.g. enantio selective synthesis of drugs, NLO properties of chiral materials) and Biology (chirality of DNA, RNA, proteins, the origin of life). Chirality initially started dealing with the ‘simple’ case of quaternary carbons; with transition metals the situation got more complex due to the multiple and high valencies available to those atoms.
We have engaged towards the rational synthesis of new chiral spin-crossover materials starting from chiral enantiopure ligands and/or anions, or mixing with achiral ligands. We identified two potentially useful families of chiral anions; tartaric acid based anions ( antimonyl and arsenyl tartrate), which were widely used in the past in HPLC chiral separations, but only sparingly in the synthesis of molecular materials ; tris(dioxolene)-phosphate(V) and arsenate(V) anions. Lacour (U. Geneva) has synthesized tetrachlorocatechol-based phosphate chiral anions (TRISPHAT, BINPHAT,…) [1], with great synthetic versatility, and the same helicoidal chirality than hexacoordinated metal complexes. The more easily synthesized non-chlorinated analogue, the tris(catecholato)phosphate(V) anion (TRISCAT) was shown to epimerize quickly in solution [2]. Nevertheless the corresponding TRISCAS arsenate(V) was shown to be configurationally stable. We will present the results that we have achieved.
[1] a) J. Lacour, C. Ginglinger, F. Favarger, Tet. Lett. 1998, 39, 4825-4828. b) J. Lacour, S. Constant, V. Hebbe, Eur. J. Org. Chem. 2002, 3580-3588. c) F. Favarger, C. Goujon-Ginglinger, D. Monchaud, J. Lacour, J. Org. Chem. 2004, 69, 8521-8524. d) C. Pérollier, G. Bernardinelli, J. Lacour, Chirality 2008, 20, 313-324. e) S. Constant, J. Lacour, Top. Curr. Chem. 2005, 250, 1-41. [2] J. Cavezzan, G. Etemad-Moghadam, M. Koenig, A. Klaebe, Tet. Lett. 1979, 20, 795-798

Session Posters 8a Chiralité, polymorphisme
- 193 -
P59 - Caractérisation structurale de co-cristaux pharmaceutiques obtenus par cristallisation d’un mélange fondu.
Rachel Tabaroni1, Mathieu Marchivie1, Stéphane Massip1, Clémence Neurohr2 et Pascale Subra-Paternault2 1 Université Bordeaux Segalen, Pharmacochimie, CNRS FRE 3396, F-33000 Bordeaux, France 2 Université Bordeaux, CBMN-UMR5248, Allée Geoffroy St Hilaire, F-33600 Pessac, France
Les propriétés des matériaux à l’état solide sont largement dépendantes de l’arrangement tridimensionnel adopté par les molécules. Concernant les principes actifs (PA), il est bien connu que les propriétés comme la stabilité, la fluidité, l’hygroscopie ou la biodisponibilité, sont dépendantes de l’arrangement des molécules dans le solide. Ainsi, différentes stratégies ont été proposées pour modifier et contrôler de telles propriétés notamment par la formation de sels, de polymorphes, d’hydrates, de solvates et plus récemment de co-cristaux. Les co-cristaux sont des complexes moléculaires formés de deux dérivés différents (ou plus) cristallisant au sein de la même maille cristalline. Dans le domaine pharmaceutique, de tels systèmes sont préparés à partir d’un PA et d’une seconde molécule favorisant la formation du co-cristal appelé : « co-former ». Alors que le premier co-cristal fut découvert en 1844,[1] ce n’est que récemment que les co-cristaux sont apparus comme des composés prometteurs dans le développement du médicament afin de maîtriser la solubilité, réduire l’hygroscopie, contrôler le polymorphisme cristallin et bien d’autres paramètres.[2] Les techniques de croissance cristalline les plus couramment utilisées sont la recristallisation à partir de solvants, à partir d’un mélange fondu ou, de façon plus originale, par précipitation en milieu CO2 supercritique. Dans cette communication, nous décrivons un co-cristal formé à partir d’un mélange acétazolamide / nicotinamide par recristallisation classique et à partir d’un mélange équimolaire fondu. Si un hydrate de ce co-cristal était déjà décrit dans la littérature[3], le dérivé anhydre pressenti lors de ces travaux n’avait pas été caractérisé structuralement. La technique utilisée ici pour la formation de ce composé nous a permis de faire croître des cristaux de taille et de qualité suffisante pour une étude par diffraction des rayons X sur monocristaux. Les résultats confirment la stœchiométrie proposée et nous autorisent une comparaison détaillée de l’arrangement cristallin des deux pseudo-polymorphes.
[1] F. Wohler, Annalen Chem. Pharm., 1844, 51, 145-163; G. P. Stahly, Crystal Growth & Design, 2009, 9, n° 10, 4212-4229 [2] N. Rodriguez-Hornedo, Molecular Pharmaceutics, 2007, 4, n° 3, 299-300; N. A. Meanwell, Annual Reports in Medicinal Chemistry, 2008, 43, 373-404; N. Shan and M. J. Zaworotko, Drug discovery today, 2008, 13, n° 9-10, 440-446; D. J. Good and N. Rodriguez-Hornedo, Crystal Growth & Design, 2009, 9, n° 5, 2252-2264; N. Schultheiss and A. Newman, Cryst Growth Des, 2009, 9, n° 6, 2950-2967; N. Qiao, M. Li, W. Schlindwein, N. Malek, A. Davies and G. Trappitt, Int J Pharm, 2011, 419, n° 1-2, 1-11 [3] J. I. Arenas-Garcia, D. Herrera-Ruiz, K. Mondragon-Vasquez, H. Morales-Rojas and H. Hopfl, Crystal Growth & Design, 2010, 10, n° 8, 3732-3742

Session Posters 8a Chiralité, polymorphisme
- 194 -
P60 - Polymorphisme du 2-ChloroAdamantane Philippe Negrier1, Maria Barrio2, Josep Lluis. Tamarit2, Denise Mondieig1 1 Laboratoire Ondes et Matière d'Aquitaine, UMR 5798 au CNRS-Université Bordeaux I, 351, cours de la Libération, 33405 Talence Cedex, France. 2 Grup de Caracterització de Materials, Department de Física I Enginyeria Nuclear, ETSEIB, Diagonal 647, 08028 Barcelona, Universitat Politècnica de Catalunya, Catalonia
Les molécules organiques de forme pseudo-sphériques comme l'adamantane présentent souvent des transitions d'une phase solide ordonnée à une phase à désordre orientationel dite phase "plastique" [1-3]. La substitution en position 2 d'un atome d'hydrogène par le chlore réduit la symétrie (Cs) et conduit à un polymorphisme différent.[4-6]
Nous avons étudié le 2-ChloroAdamantane par diffraction des RX sur poudre en fonction de la température et par analyse enthalpique différentielle à pression atmosphérique. Quatre phases solides ont été identifiées dont trois phases avaient été mises en évidence par spectroscopie Raman et IR[7]. Nous présenterons les structures de ces phases résolues à partir de mesures par diffraction sur poudre à l'aide du diffractomètre CPS 120 INEL (en mode transmission et géométrie Debye-Scherrer et λCuKα1). [1] Disorder in Crystals, N. G. Parsonage and L.A.K. Staveley, Clarendon Press, Oxford, 1978. [2] J. P. Amoureux and M. Foulon, Acta Cryst. 1970, B43, 470. [3] J. P. Amoureux, M. Bee and J.C. Damien, Acta Cryst. 1980 ,B36, 2633 [4] T. Clark, T.M.O. Knox, H. Mackle and M.A. Mckervey, J. Chem.Soc. Faraday. Trans., 1977, 73, 1224. [5] A.B. Bazyleva, A.V. Blokin, G.J. Kabo, A.G. Kabo, Y.U. Paulechka, J. Chem. Thermodynamics, 2005, 37, 643. [6] M. Foulon and C. Gors, Acta Cryst. 1988, B44, 156. [7] M.R. Parolin, N.T. Kawaii, I. S. Butler and D.F. R. Gilson, Can. J. Chem. 1988, 66, 1973.

Session Posters 8a Chiralité, polymorphisme
- 195 -
P61 - Conformation et polymorphisme du 1,1,2,2-tetrachloroéthane (C2H2Cl4)
Ph. Negrier1, M. Barrio2, M. Zuriaga3, S.C. Pérez3, J.Ll. Tamarit2 and D. Mondieig1 1 Laboratoire Ondes et Matière d'Aquitaine, UMR 5798 au CNRS-Université Bordeaux I, 351, cours de la Libération, 33405 Talence Cedex, France. 2 Grup de Caracterització de Materials, Department de Física I Enginyeria Nuclear, ETSEIB, Diagonal 647, 08028 Barcelona, Universitat Politècnica de Catalunya, Catalonia 3 Facultad de Matemática, Astronomía y Física, Universidad Nacional de Córdoba and IFEG-CONICET, Ciudad Universitaria, X5016LAE Córdoba, Argentina
La molécule de 1,1,2,2-tetrachloroéthane adopte deux types de conformations: trans (antipériplanaire) et gauche (synclinale). La différence d'énergie entre les deux conformations est faible (<1 kcal mol-1)[1,2]. Ainsi, le polymorphisme observé sous différentes conditions de pression et de température est principalement lié aux interactions intermoléculaires.
Il a déjà été établi qu'à pression atmosphérique, la phase stable est orthorhombique (P212121,
Z=8, Z’=2) et présente uniquement des molécules en conformation gauche. A haute pression,
la structure d'une phase monoclinique (P21/c, Z=2, Z’=0.5) comportant deux molécules en
conformation trans a été déterminée.[3]
Ce travail présente une nouvelle phase solide dans laquelle 2 conformations gauche
coexistent. Cette phase métastable’, obtenue par recristallisation de la phase liquide
surfondue, se transforme de manière irréversible en phase stable lorsque l'on procède à une
élévation de température. Nous présenterons les relations thermodynamiques entre les
différentes phases condensées et les caractéristiques structurales de la phase ’. [1].J. E. Mark and C. Sutton, J. Am. Chem. Soc. 1972, 94, 1083. [2] (a) J. R. Thomas and W. D. Gwinn, J. Am. Chem. Soc. 1949, 71, 2785; (b) R. J. Abraham and K. Parry, Chem. Commun. 1969, 17, 963; (c) R. J. Abraham and K. Parry, J. Chem. Soc. B, 1970, 539. [3] M. Bujak, D. Bläser, A. Katrusiak, R. Boese, Chem. Commun. 2011, 47, 8769.

Session Posters
- 196 -
P62 - Reconnaissance de surfaces protéiques par des foldamères aromatiques. Jérémie Buratto1, Béatrice Langlois d’Estaintot1, Cinzia Colombo1,2, Lucile Fischer1,2, Bernard Gallois1 et Ivan Huc1,2
1 CBMN UMR 5248, Université Bordeaux1, Bât B14, Allée Geoffroy St Hilaire, 33600 Pessac France. 2 IECB, 2 Rue Robert Escarpit, 33607 Pessac France.
Les interactions protéine - protéine jouent un très grand rôle dans les processus biologiques, et représentent des cibles thérapeutiques de choix pour le traitement de maladies. Parmi les quelques 22 000 protéines humaines, 80% d’entre elles seraient actives sous forme de complexes. La conception rationelle de molécules antagonistes, susceptibles de reconnaître spécifiquement des surfaces protéiques étendues, permettrait d’inhiber ces interactions [1]. Les foldamères aromatiques présentent toutes les caractéristiques nécessaires pour accomplir un tel but [2] : i) structures stables mimant celles (hélices α, brins β) rencontrées dans les protéines, ii) fonctionnalisation importante, iii) obtention par synthèse sur support solide de molécules de grande taille, iv) cristallisation aisée.
Dans le cadre du contrat européen FOLDAPPI (FOLDamers Against Protein-Protein
Interaction, Action Marie Curie Industry-Academia Partnerships and Pathways, Programme FP7-People-2007), l’optimisation de tels foldamères s’appuie sur des outils structuraux (diffraction des rayons X et modélisation moléculaire). Les foldamères étudiés sont constitués de sous-unités quinolines. Ils adoptent une structure en hélice [2]. L’une des protéines cibles choisie pour démontrer l’interaction protéine - foldamère est l’anhydrase carbonique humaine (HCA II). Elle a été choisie car sa structure a été très étudiée et qu’elle offre la possibilité d’ancrer le foldamère à sa surface par l’intermédiaire d’un inhibiteur de forte affinité (Kd ~ 1 nM).
Nous présentons différents foldamères étudiés ainsi que leur faculté à adopter une hélicité donnée en présence de la protéine (expériences de CD induit).Après identification du meilleur candidat, un premier complexe a été cristallisé. Sa structure a été résolue à 3,2 Å. L’arrangement moléculaire révèle l’existence de dimères induits essentiellement par des interactions foldamère – foldamère. De nouvelles études (DLS, CD) en fonction de la nature et de la position des chaînes latérales du foldamère sont en cours afin de favoriser les interactions foldamère – protéine au sein d’un même complexe. [1] Cochran et al., Chem. Biol. 2000, 7(4), R85-94. [2] Gillies et al., J. Org. Chem. 2010, 46, 214.

Session Posters
- 197 -
P63 - Etude structurale d’une flavoprotéine fluorescente capable de générer une espèce réactive de l’oxygène Céline Lafaye1, David Von Stetten2, Marjolaine Noirclerc-Savoye1, Xiaokun Shu3, Antoine Royant1,2
1Institut de Biologie Structurale Jean-Pierre Ebel UMR5075-CNRS-CEA-UJF, 41 Rue Jules Horowitz, 38027 Grenoble, France. 2Structural Biology Group, European Synchrotron Radiation Facility, 6 Rue Jules Horowitz,
38043 Grenoble, France, 3Department of Pharmaceutical Chemistry, University of California San Francisco, California.
L’utilisation de la microscopie électronique en corrélation avec la microscopie de fluorescence permet la localisation protéique avec une très haute résolution spatiale. Un des challenges majeurs de cette technique corrélative reste le marquage spécifique des protéines. Cela nécessite le développement d’étiquettes fluorescentes, codées génétiquement pour une visualisation in situ au sein des cellules ou des tissus, et capables de générer une espèce réactive de l’oxygène, l’oxygène singulet. La flavoprotéine fluorescente miniSOG se révèle être un excellent candidat pour résoudre ce problème [1]. miniSOG (pour Singlet Oxygen Generator de taille minimale), est une petite (106 résidus) protéine fluorescente capable de générer efficacement de l’oxygène singulet après illumination par la lumière bleue. Dans les tissus fixés, l’oxygène singulet généré par photo illumination permet de polymériser localement la petite molécule diaminobenzidine en un précipité facilement visible par microscopie électronique [1]. Dans ce poster je présente l’avancement de nos études structurales de la protéine miniSOG destinée à comprendre ses propriétés de génération d’oxygène singulet et en proposer éventuellement une version améliorée. [1] Shu X, et al. PLoS Biol 2011, 9(4.): e1001041

Session Posters
- 198 -
P64 - Protection/diversion par C1q à l’interface hôte/pathogène
Christophe P. Moreau1,2
1Institut de Biologie Structurale, Grenoble 2Université Joseph Fourier, Grenoble
L’immunité innée constitue une première ligne de défense dont l’action est immédiate dès que les barrières physiques naturelles (muqueuses, épidermes) ont été franchies. Elle comprend un système majeur de défense nommé ‘complément’. Nous nous intéressons à la protéine multi-fonctionnelle C1q. Sa fonction dite ‘classique’ est l’activation de la cascade protéolytique du complément qui permet d’éliminer des complexes immuns, génère des signaux inflammatoires et instruit d’autres éléments de l’immunité. La seconde fonction est l’élimination rapide des cellules apoptotiques sans émission de signaux inflammatoires. Pour cela, C1q reconnaît différents ligands exposés spécifiquement en surface de ces cellules comme la phosphatidyl-sérine (PS, signal « eat me »), la calréticuline [1], ou la GAPDH [2].
De manière surprenante, des signaux similaires peuvent être retrouvés en surface de certains pathogènes. En effet, la CRT et la PS sont exprimées à la surface de Trypanosoma Cruzi, un parasite responsable de la maladie de Chagas. Ici, l’interaction C1q/calréticuline entraine une inactivation du complément et favorise la pénétration du parasite, suggérant une stratégie parasitaire de type ‘cheval de Troie’ [3]. En obtenant la structure de la calréticuline de ce parasite, seule ou en complexe avec les têtes globulaires de C1q, nous espérons découvrir des particularités permettant de développer des thérapies contre ces pathogènes en collaboration avec la faculté de médecine de Santiago au Chili.
Lorsque la GAPDH exprimée en surface des cellules apoptotiques humaines est reconnue par C1q ceci n’active pas le complément, tandis que son homologue à la surface des Pneumocoques conduit à l’activation du complément [2]. La résolution récente de la structure de la GAPDH de pneumocoque nous a donné des premiers indices pour expliquer ces différences, que nous souhaitons confirmer en obtenant la structure du complexe formé entre la GAPDH et C1q.
[1] Païdassi et al., J.Mol.Biol., 2011, 408(2), 277-90 [2] Terrasse et al., J.Mol.Biol., 2012, 287(51), 42620-33 [3] Ramirez et al., Mol. Immunol., 2012, 52(3-4), 133-40

Session Posters
- 199 -
P65 - Etude structurale et des propriétés magnétiques et électriques des pérovskites à doubles couches des manganates La2-xBixCaMn2O7 (avec x=1) M’hamed Oubla1
, Mohammed Lamire1*, Hassan Lassri2, El-Kebir Hlil3
1 Laboratoire de Physico-Chimie des Matériaux Inorganiques, Faculté des Sciences Aïn-Chock, Université Hassan II, B.P. 5366 Mâarif, Casablanca, Morocco, 2 Laboratoire de Physique des Matériaux, Micro-électronique, Automatique et Thermique, Faculté des Sciences Aïn-Chock, Université Hassan II, B.P. 5366 Mâarif, Casablanca, Morocco, 3 Institut Néel, CNRS - Université J. Fourier, BP 166, 38042 Grenoble, France. *Corresponding author email: [email protected]
L’oxyde pérovskite à couches LaCaBiMn2O7 a été préparé par la méthode conventionnelle de co-précipitation dans une solution aqueuse. Les études de diffraction des rayons X suggèrent que cette phase cristallise dans le groupe d’espace I4/mmm du système quadratique. Les propriétés magnétiques montrent que les interactions ferromagnétiques sont dominantes et que l’ion manganèse est présent au sein de cette phase dans des états de valence mixtes Mn3+ et Mn4+. La courbe de thermo-aimantation obtenue obéit à la loi de Bloch. La constante de rigidité de spin D et la valeur approximative de l’interaction d’échange JMn-Mn ont été estimées à partir des résultats expérimentaux. La courbe de variation de la résistivité électrique en fonction de la température montre que le composé présente une transition Metal-Isolant (M-I) au voisinage sa température de Curie (Tc).

Session Posters
- 200 -
P66 - Caractérisation structurale de la région centrale de la protéine pUL36 du virus de l’Herpès Simplex de type 1 Nathalie Scrima, Stéphane Bressanelli et Stéphane Roche Laboratoire de Virologie Moléculaire et Structurale, CNRS, Gif-sur Yvette
Les virus de la famille des herpèsviridae tels que le virus de l’herpès simplex de type 1 (HSV1) ont une structure caractéristique en quatre couches. Le génome viral est enfermé dans une capside icosaédrique elle-même entourée d’un « tégument » protéique et enfin d’une enveloppe lipidique. Le tégument se subdivise lui-même en deux sous-composants : le tégument interne regroupe les protéines interagissant fortement avec la capside tandis que les protéines de tégument externe se détachent aisément de la capside.
Deux des protéines du tégument interne, UL36 et UL37 forment un complexe de stœchiométrie inconnue. Elles remplissent des rôles multiples au cours du cycle viral. Elles sont ainsi impliquées dans le transport des capsides virales, dans l’injection du matériel génétique viral dans le noyau ou encore dans l’assemblage de nouvelles particules virales. Notamment, UL36 interagit avec la protéine UL25 localisée aux sommets de la capside ainsi qu’avec la protéine de tégument externe UL48, ce qui suggère qu’elle traverse le tégument de part en part. UL36 est une protéine de 337 kDa et à l’exception de son domaine N-terminal aucune information structurale n’est disponible, ce qui rend son étude délicate. Notamment, les domaines fonctionnels de la protéine restent inconnus.
Nous sommes parvenus à produire et à purifier divers fragments d’UL36 de HSV1 représentant au total un tiers de la protéine. Des expériences de gel-filtration indiquent que ces fragments sont tous très allongés et qu’ils sont probablement tous capables de former des oligomères, ce qui suggère qu’ils pourraient faire partie de la « tige » de la protéine traversant le tégument. Nous sommes parvenus à résoudre la structure cristallographique d’un de ces fragments. Celui-ci forme un dimère antiparallèle très allongé. La présence de contact cristallographique très étendu suggère qu’il pourrait former des oligomères plus instables d’ordres supérieurs. Les conséquences de ces données pour l’assemblage des particules virales seront discutées.

Session Posters
- 201 -
P67 - Evolution structurale en fonction de la température du composé LiNbO3
Charef TABTI, Abdelkader CHOUAIH & Fodil HAMZAOUI Laboratoire SEA2M, Département de Chimie Faculté des sciences et technologie, Université de Mostaganem BP. 188, Mostaganem 27000 , Algérie.
A comparative study was made by X-ray diffraction on a single crystal of lithium niobate (LiNbO3) at low temperature (120K) and room temperature (293K). LiNbO3 is a ferroelectric
compound particularly interesting for applications in nonlinear optic field. After a recording of very good quality of X-ray diffraction spectrum, we used Blessing formalism for the reduction and the processing raw data. Structure refinement was carried out by program SHELXL. The results of the refinement led to a reliability factor of about 6% to T = 293K and of 3% to T = 120K. The structure evolution study of lithium niobate according to the temperature, made it possible to highlight the compound stability in the invested temperature range. The found results show a light displacement (about 0,01Å) of oxygen atoms around the connection Li – Nb. Une étude comparative a été faite par diffraction des rayons X sur un monocristal de niobate de lithium (LiNbO3) à basse température (120K) et à température ambiante (293K) . Le LiNbO3 est un composé inorganique ferroélectrique particulièrement intéressant pour des applications dans le domaine de l’optique non linéaire. Après un enregistrement de très bonne qualité du spectre de diffraction X, nous avons utilisé le formalisme de Blessing pour la réduction et le traitement des données brutes. L’affinement de la structure a été réalisé par le programme SHELXL. Les résultats de l’affinement ont conduit à un facteur de reliabilité de l’ordre de 6% à T = 293K et de 3% à T = 120K. L’étude de l’évolution de la structure du niobate de lithium en fonction de la température, a permis de mettre en évidence la stabilité du composé dans le domaine de température investi. Les résultats trouvés montrent un léger déplacement (de l’ordre de 0,01Å) des atomes d’oxygène autour de la liaison Li – Nb.

Session Posters
- 202 -
P68 - Etude structurale de deux nouveaux composés de coordination supramoléculaires à base d’oxalate et de 2-picolylamine. Patrice Tsobnang Kenfack1,2, Siméon Ponou3, Emmanuel Wenger2, Claude Lecomte2, John. Ngolui Lambi3
1 Département de Chimie Inorganique, Université de Yaoundé I, Faculté de Sciences, BP 812, Yaoundé, Cameroun.; 2 CRM2, UMR 7036 CNRS, Université de Lorraine, Faculté des Sciences et Technologies, BP 70239, 54506 Vandoeuvre-lès-Nancy, France.; 3 Département de Chimie, Ecole Normale Supérieure de Yaoundé, BP 47 Yaoundé, Cameroun.
Le design et la synthèse des MOF « Metal Organic Framework » suscitent depuis
quelques années un intérêt considérable dans la chimie supramoléculaire et dans l’ingénierie cristalline en raison de leurs potentielles applications dans le magnétisme, la catalyse, l’optique non linéaire et la conductivité électrique [1,2]. Nous sommes intéressés par la rationalisation de la synthèse des composés de coordination hétérométalliques supramoléculaires basées sur les interactions intermoléculaires (liaisons hydrogènes, interaction ion –dipôle, interaction π-π stacking etc.) [3]. Nous présentons ainsi dans cette communication deux nouveaux composés du genre, à base des ligands oxalate et 2-aminométhylpyridine (amp) récemment obtenus: [Co(amp)3][Cr(C2O4)3].6H2O et [Cu2(amp)2Cl][Cr(C2O4)].H2O (Figure 1). Les interactions intermoléculaires très directionnelles mises en jeu dans la formation de ces composés ont été particulièrement analysées au cours des études structurales que nous avons réalisées par diffraction de rayons X sur monocristal. Ces complexes hétérométalliques présentent des caractéristiques intéressantes pour le magnétisme moléculaire.
Figure 1 : Unités asymétriques des composés Co(pico)3Cr(C2O4)3.6H2O (gauche) et
Cu2(pico)4Cl(Cr(C2O4)3.H2O (droite)
[1] Jean N., Corado D., Justin N., Claudio P., Eleuterio A., Siméon P. Transition Met chem. 2013, 38, 21-29. [2] Takanori O., Naoya K., Masakatsu S. Chem., Int. Ed. 2013, 52(24), 6196–6201. [3] Patrice T. K., Siméon P., Emmanuel W., Claude L., John. N. L. (en préparation) Nous remercions grandement l’UICr pour son Initiative Africaine et le Service de Coopération d’Action Culturelle (SCAC) de l’Ambassade de France au Cameroun pour le financement de ce travail.

- 203 -
P69 - Molecular Jewellery Aline Lacoudre et Noël Pinaud CESAMO, ISM, UMR5255, Université Bordeaux1, 351 cours de la Libération, 33405 Talence Cedex
La cristallogenèse est un art subtil et éphémère où la patience est reine. Cet Art présomptueux, qui consiste à assister Dame Nature dans la réalisation de structures architecturales moléculaires, nous livre parfois un spectacle oculaire à en faire pâlir ‘Van Cleef & Arpels’ !
Qu’ils diffractent ou non, incolores ou enluminés, maclés ou sublimement sculptés,
translucides ou opaques, les cristaux nous procurent un plaisir visuel que l’on transpose aussitôt en plaisir partagé.
Nous proposons au CESAMO de lier cet Art à celui de la photographie en réalisant
des clichés de tous les cristaux que nous ‘mettons au monde’ dans le service. Nous vous offrons ici un instant de flânerie, où formes, couleurs et lumière créent un poème graphique, afin de vous procurer le plus simple des plaisirs : le plaisir des yeux.

- 204 -
P70 - Les quasicristaux : chronologie d’une découverte
Marianne Quiquandon, Denis Gratias LEM CNRS/ONERA ; 29 avenue de la division Leclerc, 92322 CHATILLON CEDEX
Suite à l’attribution du prix Nobel de chimie 2011 à Daniel Shechtman pour la découverte des quasicristaux [1-2], nous présenterons le contexte scientifique de l’époque via l’historique des principaux articles sur le sujet [3-11] précurseurs de cette découverte. Nous verrons comment, malgré la tenace opposition d’une majorité de cristallographes américains, les concepts quasicristallins se sont rapidement développés pour rejoindre, en les complétant, ceux des phases incommensurables [12-16] et ont donné corps à un corpus maintenant bien structuré de la thématique Apériodicité au coeur du débat sur une redéfinition attendue de la notion de cristal. [1] D. Shechtman & I. Blech, Met. Trans. 16A, 1005 (1985). [2] D. Shechtman, I. Blech, D. Gratias & J. W. Cahn, Phys. Rev. Lett. 53 (20), 1951-1953 (1984). [3] E. Esclangon, Thèse à la faculté des sciences de Paris : Fonctions Quasi-Périodiques (1904). [4] H. Bohr, Acta Math. 45, 29 (1924); Acta Math. 46, 101 (1925); Acta Math. 47, 237 (1926). [5] A. S. Besicovitch, Almost periodic functions, Cambridge (1932). [6] Y. Meyer, lectures notes in mathematics n°117, Springer (1970); algebraic numbers and harmonic analysis, North Holland (1972). [7] R. Penrose Bull Inst Maths its Appl 10 n°7/8 266 (1974); ibid. Math. Intelligencer 2 32 (1979). [8] P. M. de Wolff Acta Cryst. A30, 777 (1974); Acta Cryst. A33, 493 (1977). [9] A. Janner & T. Janssen, Phys. Rev. B15, 643 (1977); Physica (Utrecht) 99A, 47 (1979). [10] M. Gardner Sci. Amer. 236, n°110 (1977). [11] A. L. Mackay, Sov. Phys. Crystallogr. 26 (5), 517 sept-oct (1981); ibid. Physica 114A, 609 North-Holland Publishing Co. (1982). [12] D. Levine & P.J. Steinhardt, Phys. Rev. Lett. 53 (26), 2477-2480 (1984). [13] M. Duneau & A. Katz, Phys. Rev. Lett. 54, 2688 (1985). [14] P. A. Kalugin, A. Y. Kitaev & L. S. Levitov, J. Phys. Lett. 46, L-601 (1985). [15] V. Elser, Phys. Rev. Lett. 54, 1730 (1985). [16] P. Bak, Phys. Rev. Lett. 56, 861 (1986).

- 205 -
P71 - Résolution de la structure cristalline du DL-menthol sur monocristal et criblage du polymorphisme de l’énantiomère et du composé racémique.
Yohann Corvis1, Philippe Négrier,2 Stéphane Massip,3 Mathieu Marchivie,3 Philippe Espeau1 1Laboratoire Physico-Chimie Industrielle du Médicament, EA 4066, Université Paris Descartes, Sorbonne Paris Cité, Faculté des Sciences Pharmaceutiques et Biologiques, 4 Avenue de l’Observatoire, 75 006 Paris, 2Univ. Bordeaux, LOMA, UMR 5798, 33 400 Talence, France, and CNRS, LOMA, UMR 5798, 33 400 Talence, France, 3Univ. Bordeaux, Pharmacochimie, FRE CNRS 3396, 33 000 Bordeaux, France, and CNRS, Pharmacochimie, FRE CNRS 3396, 33 000 Bordeaux, France.
Nos précédents travaux, qui ont montré qu’un dérivé amide pouvait induire un
polymorphisme de l’énantiomère lévogyre du menthol [1], nous ont conduit aux constats suivants : (i) la structure cristalline du DL-menthol, décrit comme étant un pseudo-racémate [2], était non résolue, (ii) depuis près d’un siècle, date à laquelle le polymorphisme du L-menthol a été mis en évidence [3], aucune étude quantitative des polymorphes de l’énantiomère n’a été réalisée, alors que la structure cristalline de sa forme stable a été résolue (système trigonal, P31 [4]). Le polymorphisme du DL-menthol est, quand à lui, non élucidé jusqu’à ce jour.
La structure cristalline du DL-menthol n’ayant pu être déterminée à partir des clichés
RX de poudres, la méthode de sublimation/condensation a été adaptée au laboratoire de manière à optimiser la croissance cristalline du composé racémique. Ainsi, nous avons pu résoudre la structure sur monocristal (système triclinique Pī, Z’ = 3). Des expériences d’analyse thermique et de diffraction des rayons X sur poudres ont été menées dans le but de caractériser les polymorphes du menthol, de déterminer leurs conditions de stabilité et d’accéder aux cinétiques de transition de phases. Les phases stables, à pression et température
ordinaires, ont été dénommées . Les résultats montrent que les polymorphes métastables
du L- et du DL-menthol cristallisent dans une maille triclinique P1. La transition → pour le L-menthol implique une modification structurale de sa maille (triclinique à trigonale), alors que pour le DL-menthol, cette transition ne s’accompagne d’aucun changement de système cristallin. Pour aller plus loin dans ces études, les interactions entre le L- et le D-menthol ont été appréhendées par détermination des diagrammes de phases stable et métastables.
La complémentarité des approches thermodynamique et cristallographique permet de
démontrer sans ambiguïté que le menthol racémique est un racémate alors que les polymorphes métastables correspondants sont des pseudo-racémates [5].
[1] Y. Corvis, P. Négrier, M. Lazerges, S. Massip, J.-M. Leger, P. Espeau, J. Phys. Chem. B 2010, 114, 5420-5426. [2] M. Kuhnert-Brandstaetter, R. Ulmer, L. Lanaghammer, Arch. Pharm. 1974, 307, 497-503. [3] F. E. Wright, J. Am. Chem. Soc 1917, 39, 1515-1524. [4] P. Bombicz, J. Buschmann, P. Luger, N. X. Dung, C. B. Nam, Z. Kristallogr. 1999, 214, 420-423. [5] Y. Corvis, P. Négrier, S. Massip, J.-M. Leger, P. Espeau, CrystEngComm 2012, 14, 7055-7064.

- 206 -
P72 - News in Open structure and property Databases
Daniel Chateigner1,*, Saulius Grazulis2, Justas Butkus2, Andrius Merkys2, Adriana Daškevič2, Olivier Pérez1, Giancarlo Pepponi3, Luca Lutterotti1,4, Miguel Quirós Olozábal5, Armel Le Bail6, Robert Downs7, Peter Moeck8, Alexandre F.T. Yokochi9 1 CRISMAT-ENSICAEN-CNRS, IUT Caen, Normandie Universités, Caen, France, 2 Inst. of Biotechnology, Vilnius, Lithuania, 3Fundazione Bruno Kessler, Trento, Italy, 4Dept. of Industrial Engineering, Univ. of Trento, Italy, 5Dept de Química Inorgánica, Univ. de Granada, Spain, 6Lab des Oxydes et Fluorures, CNRS and Univ. du Maine, Le Mans, France, 7Dept of Geosciences, Univ. of Arizona, Tucson, Arizona, USA, 8Portland State Univ., Dept of Physics, Portland, Oregon, USA, 9School of Chemical, Biological and Environmental Engineering, Oregon State Univ., Corvallis, Oregon, USA
The COD project (abbreviated from the "Crystallography Open Database", http://www.crystallography.net/) aims at collecting in a single open access database all organic, inorganic and metal organic structures [1]. It grew to more than 235000 cif files in the past 10 years. Since December 2007 the main database server is maintained and new software is developed in the Vilnius University Institute of Biotechnology. Structure file harvesting is now routinely operated and automated for a growing number of publishers' journals [2]. Registered users can deposit new data into the database, either form the previous publications or as personal communications. The deposition software performs rigorous checks of syntax and semantics, thus ensuring high quality depositions. The search engine acquired new capabilities, and COD records can be viewed on-line or downloaded. For massive data mining, COD permits downloads and updates of the whole database using Subversion, Rsync or HTTP protocols. The ease of access to COD data has spurred the use of this resource for software testing [3], teaching [4], and research [5].
For the powder diffraction community, COD is recently used for open Full-Pattern Search-Match (cod.iutcaen.unicaen.fr). It allows phase quantification from x-rays, neutron and electron powder patterns, with high or medium resolution instruments, provided the structures are already in COD. This tool is particularly suited for nanocrystalline powders in which severe line broadening appears precluding phase identification from only peak maxima. COD-derived databases are offered for software produced by many diffractometer vendors. In addition to COD, search match can be done against its sister database, PCOD, that contains structures predicted by the GRINSP program [6]. Recently launched the new TCOD database collects structures optimized from COD using first-principles calculations. The open nature of the COD permitted numerous mirrors around the globe [7] and specifically tailored COD database variants. Meanwhile, the Material Properties Open Database [8] collects tensor properties. This latter sister is on a demanding growth.
1. Gražulis, S.; Chateigner, D.; Downs, R. T.; Yokochi, A. F. T.; Quirós, M.; Lutterotti, L.; Manakova, E.; Butkus, J.; Moeck, P. & Le Bail, A. (2009). Crystallography Open Database -- an open-access collection of crystal structures, Journal of Applied Crystallography 42 : 726-729.

- 207 -
2. Gražulis, S.; Daškevič, A.; Merkys, A.; Chateigner, D.; Lutterotti, L.; Quirós, M.; Serebryanaya, N. R.; Moeck, P.; Downs, R. T. & Le Bail, A. (2012). Crystallography Open Database (COD): an open-access collection of crystal structures and platform for world-wide collaboration, Nucleic Acids Research 40 : D420-D427. 3. Grosse-Kunstleve, R. & Gildea, R. (2011). Computational Crystallography Initiative: COD stats, http://cci.lbl.gov/cod_stats/ (retrieved 2013.01.31). 4. Moeck, P. (2004). EDU-COD: Educational Subset of COD, http://nanocrystallography.research.pdx.edu/search/edu/ (retrieved 2013.01.31). 5. First, E. L. & Floudas, C. A. (2013). MOFomics: Computational pore characterization of metal–organic frameworks, Microporous and Mesoporous Materials 165 : 32-39. 9 L. Lutterotti, H. Pilliere, C. Fontugne, P. Boullay, D. Chateigner (2013), in preparation 6. Le Bail, A. (2005). Inorganic structure prediction with it GRINSP, Journal of Applied Crystallography 38 : 389-395. 7. Quirós-Olozábal, M. (2006). COD Mirror of Granada University, http://qiserver.ugr.es/cod/. Moeck, P. (2007). Crystallography Open Database Mirror, http://nanocrystallography.research.pdx.edu/search/codmirror/. Gražulis, S. (2007). COD Mirror in Vilnius, http://cod.ibt.lt/. Chateigner, D. (2010). Crystallography Open Database Mirror at ENSICAEN, http://cod.ensicaen.fr/. 8 G. Pepponi, S. Grazulis, D. Chateigner (2012). MPOD: a Material Property Open Database linked to structural information. Nuclear Instruments and Methods in Physics Research B 284, 2012, 10-14. http://www.materialproperties.org/

- 208 -
P73 - Structure de FemXwt en complexe avec un conjugué peptidyl-ARN.
Inés Li de la Sierra-Gallay,2 Matthieu Fonvielle,1 Maxime Lecerf,1 Delphine Patin,3 Dénia
Mellal,4 Didier Blanot,3 Herman van Tilbeurgh,2 Mélanie Ethève-Quelquejeu,4 Michel Arthur1
1Centre de Recherche des Cordeliers, LRMA, Equipe 12, Université Pierre et Marie Curie –Paris 6, UMR S 872, Paris, F-75006 France ; Université Paris Descartes, Sorbonne Paris Cité, UMR S 872, Paris F-75006 France ; INSERM, U872, Paris, F-75006 France 2Fonction et Architecture des Assemblages Macromoléculaires, Institut de Biochimie et de Biophysique Moléculaire et Cellulaire, Université Paris-Sud, UMR 8619, Orsay, F-91405, France; CNRS, UMR 8619, Orsay, F-91405, France 3Laboratoire des Enveloppes Bactériennes et Antibiotiques, Institut de Biochimie et de Biophysique Moléculaire et Cellulaire, Université Paris-Sud, UMR 8619, Orsay, F-91405, France; CNRS, UMR 8619, Orsay, F-91405, France 4Laboratoire de Chimie et de Biochimie Pharmacologiques et Toxicologiques, Université
Paris Descartes, UMR 8601, Paris, F-75005; CNRS UMR 8601, Paris, F-75005 France
Les aminoacyl transférases de la famille Fem interviennent dans la voie de synthèse peptidique non-ribosomale du peptidoglycane pariétal des bactéries, en utilisant des aminoacyl-ARNt en tant que donneurs de résidus d’acides aminés. Ces enzymes sont des cibles attractives pour le développement d’antibiotiques, car elles n'ont pas d’homologue dans les cellules de mammifères et interviennent dans des étapes essentielles de l'assemblage de la paroi de pathogènes résistants aux antibiotiques, comme Staphylococcus aureus, Streptococcus pneumoniae et Enterococcus faecalis. FemXWv de Weissella viridescens est une enzyme modèle pour la famille Fem. Elle intervient dans l'assemblage de la chaîne latérale du précurseur du peptidoglycane, plus particulièrement dans le transfert du résidu Ala de l'Ala-ARNt
Ala à la chaine latérale du résidu Lys en troisième position du précurseur du peptidoglycane UDP-MurNAc-pentapeptide.
La structure de FemXWv a été précédemment déterminée tant pour la forme apo que pour des complexes contenant l’UDP-MurNAc-pentapeptide [1] ou le produit de la réaction UDP-MurNAc-pentapeptide(Ala). La co-cristallisation avec l'ARN a échoué, vraisemblablement en raison de la basse affinité de FemXWv pour l'ARNt
Ala libre, l'instabilité de la liaison ester Ala-ARNt
Ala et l'ordre de fixation des substrats, c'est-à-dire le précurseur peptidoglycane suivi par Ala-ARNt
Ala, ce qui pourrait prévenir la formation d'un complexe binaire Fem/Ala-ARNt
Ala. Nous avons synthétisé un conjugué peptidyl-ARN stable qui mime les deux substrats de FemXWv. Ces molécules sont composées d'un analogue du précurseur du peptidoglycane lié de façon covalente à différentes portions du bras accepteur de l’ARNt
Ala. Deux de ces bi-substrats, présentant de très bonnes affinités pour FemX (IC50 de 0.05 à
2 nM) [2], ont été cristallisés avec FemXWv. Nous présentons ici la structure cristallographique à 1.7Å de résolution de l’un de ces complexes [3].
La structure du complexe et l'analyse des mutants a révélé le mécanisme par lequel FemXWv lie ses substrats pour la catalyse et la stabilisation de l'intermédiaire tétraédrique
[1] S. Biarrotte-Sorin et al., Structure 2004, 12, 257-267 [2] Fonvielle et al., Chem. Eur. J. 2013, 19, 1357-1363. [3] Fonvielle et al., Angew. Chem. 2013, 125, 1-5.

- 209 -
P74 - PknG mediated signalling pathways in Mycobacterium tuberculosis
Maria Natalia Lisa1, Nathalie Barilone1, Pedro Alzari1
1Unité de Microbiologie Structurale, Institut Pasteur, Paris, France.
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis and is still a major world health problem. During its life cycle Mtb goes through distinct replicative stages and is also capable of lying in a dormant state for several years. Then, Mtb bacilli must possess efficient signaling systems to sense the environment and to adapt the bacterial physiology accordingly. Structural and mechanistic information about these mechanisms will contribute to the development of new drugs against Mtb.
Mtb possess eukaryotic-like serine/threonine protein kinases that regulate metabolic processes and the interaction with the host [1-2]. Among them, protein kinase G (PknG) is essential for mycobacterial survival inside macrophages [3] and also controls the glutamate metabolism by regulating downstream partners [4]. Then, PknG is an attractive candidate for drug target against Mtb, and we are conducting structural and kinetic studies to better understand the molecular determinants of the enzyme activity. We will present several high resolution X-ray crystal structures obtained for PknG. In all of the cases a tilted and/or rotated orientation of helix alpha-C (a known hotspot for regulation) is found, which precludes the formation of a conserved and essential Lys-Glu ionic pair at the kinase active site [6]. Thus, we conclude that these structures represent inactive conformations of PknG. On the other hand, it has been shown that PknG autophosphorylates its N-terminal region, outside the catalytic core [4]. This is critical for the binding of the protein substrate GarA, which contains a C-terminal domain capable of recognizing pThr residues. Interestingly, while we have detected PknG activity in the presence of high concentrations of a 17mer peptide containing the GarA phosphorylation site [4], this activity disappears when deleting PknG N-terminal region. This indicates that phosphorylation outside PknG kinase core may be important for the kinase activity. [1] Cole ST et al., Nature 1998. [2] Sassetti CM et al., Mol Microbiol 2003. [3] Walburger A et al., Science 2004. [4] O'Hare HM et al., Mol Microbiol 2008. [5] Scherr N et al., PNAS 2007. [6] Huse M and Kuriyan J, Cell 2002.

- 210 -
P75 - Implication de la structure quaternaire dans la spécificité enzymatique d’une Glycoside Hydrolase de la famille GH5 Thibaud Frey1, Mickael Lafond2, Gerlind Sulzenbacher1, Jean-Guy Berrin2, Marie-Line Garron1
1 Architecture et Fonction des Macromolécules Biologique, UMR 7257 CNRS, Aix Marseille Université, Marseille, France 2 Biotechnologie des Champignons Filamenteux, UMR1163 INRA, Aix Marseille Université, Polytech Marseille, Marseille, France
Les cellulases sont des CAZymes (Carbohydrate Active Enzymes) impliquées dans de nombreux processus industriels comme la conversion de la biomasse en biocarburant. Largement étudiées, elles constituent la première famille de CAZyme décrite en 1989 sous le nom de « cellulase family A » [1]. La « cellulase family A », référencée maintenant comme Glycoside Hydrolase famille 5 (GH5), est une large famille de GH avec plus de 3800 séquences [2].
Même si les GH5 présentent une certaine prédisposition pour la dégradation des polysaccharides végétaux, il est encore complexe de prédire leur activité enzymatique puisque 20 activités ont été recensées au sein de cette famille. Afin de mieux comprendre la spécificité et l’évolution de cette grande famille, récemment une subdivision en sous familles a été réalisée [3]. Ce travail a permis d’identifier 51 sous familles dont 20 n’ont aucune caractérisation enzymatique et 38 n’ont aucun représentant structural.
La famille GH5_26 est une petite sous famille regroupant 23 séquences, pour laquelle deux activités glycosidiques, endo-β-1,4-glucanase (EC 3.2.1.4) et β-1,3/1,4-glucanase (EC 3.2.1.73), ont été reportées. Nos travaux ont portés sur la GH5_26 provenant de Saccharophagus degradans (GH5_26Sd). La caractérisation enzymatique montre que GH5_26Sd est une β-1,3/1,4-glucanase avec une activité cellulosique minoritaire. La GH5_26Sd adopte une structure quaternaire trimérique stable observable en solution et en cristallisation. La particularité de cette structure est qu’en plus de son domaine catalytique classique en TIM barrel, GH5_26Sd présente une extrémité N-terminale venant partiellement occuper le site actif du monomère contigu. Pour comprendre si cette occupation partielle du site actif pouvait influencer l’activité nous avons réalisé une caractérisation enzymatique complète de GH5_26Sd et d’un mutant tronqué. Nos résultats montrent que l’extrémité N-terminale influe effectivement sur la spécificité de substrat puisque la délétion de 16 acides aminés en N-ter confère à l’enzyme une activité cellulosique significative de type endo-β-1,4-glucanase. [1] Henrissat B. et al., Gene, 1989, 81(1):83-95. [2] www.cazy.org [3] Aspeborg H. et al., BMC Evol. Biol., 2012;12:186.

- 211 -
P76 - PknB: une phosphorylation en trans compatible avec une dimérisation ‘back to back’ G. André-Leroux*#, N. Barilone*, M.N. Lisa* et P. M. Alzari*
* Institut Pasteur, Unité de Microbiologie Structurale, 25 rue du Dr Roux, 75724 Paris Cedex 15, France # Inra, MIG, Domaine de Vilvert 78352 Jouy-en-Josas, France. [email protected]
La tuberculose, maladie infectieuse causée par Mycobacterium tuberculosis (Mtb), tue chaque année 2 millions de personnes. Bacille intracellulaire avec une stratégie d’infection redoutable, il alterne formes de vie réplicative et latente. Pour être virulent, le bacille a développé une signalisation cellulaire impliquant des kinases de type eucaryotes ou STPKs. PknB, STPK essentielle à la croissance, à la division cellulaires de la bactérie[1-2] et à l’infection des macrophages[3], montre un domaine kinase et une machinerie catalytique comparables aux homologues eucaryotes[4-5]. Ainsi, le mécanisme fonctionnel des STPKs suppose une activité optimale subordonnée à une activation de la boucle d’activation par autophosphorylation[5; 6] et à une homodimérisation via une interface ‘back-to-back’ extrêmement conservée[7-9]. Cette interface promeut l’autophosphorylation et active le domaine catalytique pour la phosphorylation de protéines hétérologues[8-10]; ensuite PknB monomérique est pleinement active[5-6; 8; 11]. Que l’autophosphorylation soit un mécanisme cis ou trans reste à éclaircir. Les travaux de Mieczkowski supportent un modèle d’assemblage ‘front-to-front’ compatible avec une transphosphorylation et montrent une seconde interface[11]. Nous discuterons nos résultats cristallographiques récents qui appuient fortement cette hypothèse de trans-autophosphorylation. [1] Sassetti et al., PNAS. 2003, 100, 12989-12994, [2] Fernandez et al., J Bacteriol. 2006,188, 7778-84. [3] Av-Gay et al., Infect Immun.1999, 67, 5676-5682. [4] Ortiz-Lombardía et al., J Biol Chem. 2003, 278, 13094-13100. [5] Young et al., Nat Struct Biol. 2003, 10, 168-174. [6] Boitel et al., Mol. Microbiol. 2003, 49, 1493-508. [7] Chenna et al., NAR. 2003, 31, 3497-3500. [8] Greestein et al., J. Biol. Chem. 2007, 282, 11427-11435. [9] Lombana et al., Structure. 2010, 18, 1667-1677. [10] Alber. Curr. Opin. Struct. Biol. 2009, 19, 650-657. [11] Mieczkowsky et al., EMBO J. 2008, 27, 3186-3197.

- 212 -
P77 - Confinement de liquides moléculaires dans des gels de silice et des verres bioactifs à porosité contrôlée. Rachida Dorbez-Sridi
1, Nouha Letaief, Nizar Bchellaoui, Hassane Oudadesse
2
1Laboratoire Physico-Chimie des Matériaux ; Faculté des Sciences de Monastir ; Université de Monastir. 2Unité Sciences Chimiques de Rennes ; UMR 6226 CNRS/ Université de Rennes 1.
Les propriétés des fluides moléculaires au voisinage d’une interface plane dépendent
presque uniquement de la nature chimique de la surface et de ses interactions avec le fluide moléculaire. À l’inverse, l’effet du confinement d’un fluide dans un matériau poreux est complexe et dépend non seulement de la nature chimique du matériau mais également de sa structure poreuse, caractérisée par la taille des pores et leur topologie.
Deux familles de matériaux monopolisent l’intérêt, à savoir la silice mésoporeuse et les verres bioactifs à base de silice ayant des caractéristiques contrôlées qui constituent des matériaux hôtes de confinement et qui peuvent être immergés dans des fluides complexes.
On s’est intéressé, en première étape, à l’étude, par diffusion de rayons X et de neutrons, de la structure, la cinétique ainsi que les transitions de phases de l’eau confinée dans deux échantillons de gels de silice ayant une porosité contrôlée l’un, non modifié, se comporte comme une matrice hydrophile et l’autre modifié présente une surface hydrophobe [1, 2].
En seconde étape, nous avons élaboré, par la méthode sol-gel, un verre ternaire mésoporeux dont la taille des pores est comprise entre 2 et 50 nm, composé de 85% SiO2
10% CaO et 5% P2O5. Ce verre, M85S, est synthétisé en se basant sur le protocole de synthèse du MCM-41 et a une porosité contrôlée dans le but de l’utiliser en tant que système de libération de principes actifs pour des applications concernent particulièrement le comblement de défauts osseux en chirurgies orthopédique et dentaire.
[1] R. Dorbez-Sridi, R. Cortes, E. Mayer et S. PIN. J. Chem Phys, 2002, 116(16), 7269. [2] J. Jelassi, T. Grosz, I. Bako, M.C. Bellissnt-funel, John Dore, H.L. Castricum and R. Sridi-Dorbez. J. Chem Phys 2011, 134, 064509.

- 213 -
P78 - Molecular basis of piezophilic adaptation
Louise Lassalle1, Marie Trovaslet2, Anne-claire Dhaussy3, Dominique Madern1, Bruno Franzetti1, Eric Girard1 1 IBS (UMR 5075 CEA-CNRS-UJF-PSB), 41 rue Jules Horowitz, 38027 Grenoble Cedex, France
2 Institut de Recherches Biomédicales des Armées, antenne de La Tronche, France 3 CRISMAT, ENSICAEN, 6 Boulevard du Maréchal Juin, 14000 Caen, France
Notre planète vit majoritairement sous pression. En effet, plus de 60 % de la biosphère sont caractérisés par des pressions supérieures à 100 bars. Ces pressions peuvent atteindre 1000 bars dans les fosses sous-marines les plus profondes. La plupart des micro-organismes y sont toutefois représentés. La découverte récente de Pyrococcus yayanosii CH1 [1], premier hyperthermo-piezophile strict, a relancé la question de l’adaptation aux fortes pressions. En effet, cette nouvelle archée n’est capable de croître que sous hautes pressions (optimum de croissance : 98 °C et 520 bars) et supporte des pressions pouvant atteindre 1500 bars.
Je présenterai l’approche choisie basée sur une étude comparative de deux protéines modèles ainsi que les résultats préliminaires obtenus. L’effet de la pression sur les malate déshydrogénases (MalDH) et les glyoxylate réductases (GR) est étudié aussi bien d’un point de vue structurale qu’enzymatique. Le suivi de l’activité enzymatique est effectué grâce à un spectrophotomètre couplé à une enceinte pression disponible au sein de la plateforme Haute Pression de l’IBS. L’étude structurale est effectuée par cristallographie des rayons X grâce à une cellule à enclume diamant [2].
Les résultats préliminaires indiquent une différence de comportement entre les enzymes issues d’organismes isolés à la surface et celles issues d’organismes des profondeurs. Cette différence suggère que les enzymes issues d’organismes des fonds marins sont adaptées pour fonctionner de manière optimale sous hautes pressions. [1] Xiang Zeng, et al.,. The ISME journal. 2009, 3 :873-876. [2] Fourme et al., Curr Opin Struct Biol. 2012, 22(5):636-42

- 214 -
P79 - Les Congrès de Cristallographie à Bordeaux
M. Hospital & J. Housty avec les souvenirs et les documents des anciens : M. Alléaume, Y. Barrans, N.B. Chanh, C. Hauw, P. Marsau, et des plus jeunes : A. Dautant, B. Gallois etc ... Laboratoire de Cristallographie, (1954 -1994) Bordeaux
Depuis les années 1950, l'Université de Bordeaux a toujours été active dans le domaine de la Cristallographie
en association avec le CNRS. Le laboratoire de Cristallographie (ERA 06 puis LA 144 du CNRS) a été membre de
l'A.F.C. depuis sa création en 1954 et a eu l'honneur et le plaisir d'organiser 3 Congrès :
• En 1962 le Congrès national de l'AFC (120 participants), • En 1973 le 1er Congrès européen de I'EUC (European Union of Crystallography) avec la présence de Dorothy
Hodgkin (prix Nobel de chimie 1964) (900 participants),
• En 1990 le XVème Congrès International de Cristallographie et Assemblée Générale de l'lUCr (International Union of
Crystallography) avec la présence de Dorothy Hodgkin, Herbert A. Hauptmann (prix Nobel de chimie 1985) et
Robert Huber (prix Nobel de chimie 1988) (2400 participants).
Les Anciens se réjouissent de voir le collectif bordelais de cristallographes recevoir leurs collègues français et étrangers et souhaitent à tous un bon Congrès et un agréable séjour.

- 215 -
Liste
des
participants

- 216 -

Liste des participants
- 217 -
Abdi Insaf P13 [email protected] Faculté des sciences de Bizerte Bousalem (Tunisie) Albino Marjorie O26 [email protected] ICMCB UPR9048 Bordeaux (France) Albouy Pierre-Antoine O18 [email protected] LPS UMR 8502 Paris (France) Allain Magali Chair [email protected] Plateforme PIAM Angers (France) Allix Mathieu O35 [email protected] CEMHTI UPR3079 Orléans (France) Arnoux Pascal O11 [email protected] CEA Cadarache (France) Audebrand Nathalie O2 [email protected] Institut des Sciences Chimiques UMR6226 Rennes (France) Aziz Zoubir P38 [email protected] Laboratoire de Technologie et des Propriétés du solide Mostaganem (Algérie) Babonneau David O7, P5 [email protected] PHYMAT CNRS Poitiers (France)
Badraoui Bechir P29 [email protected] IPEIM Monastir (Tunisie) Bailly Corinne P9 [email protected] Institut de Chimie de Strasbourg UMR7177 Strasbourg (France) Baptiste Benoit [email protected] IMPMC UMR 7590 Paris (France) Bataille Thierry O74 [email protected] Institut des Sciences Chimiques UMR6226 Rennes (France) Beaud Paul O29 [email protected] Paul Scherrer Institut Genève (Suisse) Belrhali Hassan O81 [email protected] EMBL Grenoble (France) Ben Rached Asma P1 [email protected] ICMCB UPR9048 Bordeaux (France) Benosmane Ali P37 [email protected] Université Hassiba Benbouali Chlef (Algérie) Bentata Samir O54, P38 [email protected] Université Abdelhamid Ibn Badis Mostaganem (Algérie)

Liste des participants
- 218 -
Berthier Eric Industriel [email protected] INEL Artenay (France) Bertrand Thomas O56 [email protected] SANOFI R&D Vitry-sur-Seine (France) Birck Catherine P53 [email protected] IGBMC UMR7104 U964 Strasbourg (France) Boissier Fanny [email protected] IECB Bordeaux (france) Bompard Coralie O10 [email protected] IRI USR3078 Lille (France) Bonneté Françoise P22, P23 [email protected] IBMM UMR5247 Avignon (France) Bordet Pierre O59, P50 [email protected] Institut Néel UPR2940 Grenoble (France) Bouchene Rafika [email protected] Université d’Oum El Bouaghi Oum El Bouaghi (Algérie) Boucher Florent O48 [email protected] IMN Jean Rouxel UMR6502 Nantes (France) Boulet Pascal P30 [email protected] Institut Jean Lamour UMR7198 Nancy (France)
Boullay Philippe O47, O49, P55 [email protected] CRISMAT UMR6508 Caen (France) Bourguet William O55 [email protected] CBS UMR5048 Montpellier (France) Bourne Yves O58 [email protected] AFMB UMR7257 Marseille (France) Brèthes Daniel P52 [email protected] IBGC UMR5095 Bordeaux (France) Bressanelli Stéphane P6, P66 [email protected] LVMS UPR3296 Gif-sur-Yvette (France) Brunelli Michela O86 [email protected] ILL Grenoble (France) Buratto Jérémie P62 [email protected] CBMN UMR5248 Bordeaux (France) Buron-Le Cointe Marylise Chair [email protected] Institut de Physique UMR6251 Rennes (France) Campanacci Valérie O23, P40 [email protected] LEBS UPR3082 Gif sur Yvette (France)

Liste des participants
- 219 -
Cattey Hélène [email protected] ICMUB UMR6302 Dijon (France) Cavagnino Andrea [email protected] IBBMC UMR8619 Orsay (France) Cavarelli Jean Chair [email protected] IGBMC UMR7104 U964 Strasbourg (France) Ceretti Monica O42, O72 [email protected] Institut Charles Gerhardt UMR5253 Montpellier (France) Chaptal Vincent P24, P25 [email protected] IBCP FR3302 Lyon (France) Charbonnier Jean-Baptiste O89 [email protected] CEA Saclay (France) Chateigner Daniel O77, P72 [email protected] CRISMAT UMR6508 Caen (France) Cherfils Jacqueline O23, O91, P40, P57 [email protected] LEBS UPR3082 Gif-sur-Yvette (France) Coati Alessandro O5, P5 [email protected] Synchrotron SOLEIL Saint-Aubin (France) Cochet Damien Industriel [email protected] Bruker AXS SAS Champs sur Marne (France)
Colloc'h Nathalie O33 [email protected] ISTCT UMR 6301 Caen (France) Combes Christèle O27, P26 [email protected] CIRIMAT, UPS-INPT-CNRS, ENSIACET Toulouse (France) Cooper Lucy O50 [email protected] ILV UMR8180 Versailles (France) Coquerel Gérard Chair [email protected] SMS EA3233 Rouen (France) Corvis Yohann O82, P71 [email protected] Faculté de Pharmacie Paris Descartes (France) Couprie Marie-Emmanuelle O79 [email protected] Saint-Aubin Synchrotron SOLEIL (France) Croguennec Laurence O73 [email protected] ICMCB UPR9048 Bordeaux (France) Curfs Caroline O28 [email protected] ESRF Grenoble (France) Czjzek Mirjam P17 [email protected] Station Biologique UMR7139 Roscoff (France) Daillant Jean O78 [email protected] Synchrotron SOLEIL Saint-Aubin (France)

Liste des participants
- 220 -
Dautant Alain P52 [email protected] IBGC CNRS Univ. Bdx Segalen Bordeaux (France) De Boissieu Marc SP5, P33 [email protected] SIMAP Grenoble (France) Dechambenoit Pierre CLO [email protected] CRPP UPR8641 Bordeaux (France) Demange Valérie O49 [email protected] Institut des Sciences Chimiques UMR6226 Rennes (France) Descamps Marc [email protected] UMET UMR8207 Lille (France) Devouard Bertrand O39 [email protected] CEREGE UMR7330 Aix-en-Provence (France) Didierjean Claude P15 [email protected] CRM2 UMR7036 Nancy (France) Djeghader Ahmed P41 [email protected] URMITE UMR6236 Marseille (France) Dock-Bregeon Anne-Catherine Chair [email protected] IBENS Paris (France) Dorbez-Sridi Rachida P77 [email protected] Faculté des Sciences Monastir (Tunisie)
Dorn Andy Industriel [email protected] Agilent Technologies XRD Cologne (Allemagne) Duhayon Carine P10 [email protected] LCC UPR8241 Toulouse (France) Dumas Philippe Chair [email protected] IBMC FRC1589 Strasbourg (France) Dupin Adrien [email protected] IECB Bordeaux (France) Echalier Aude O20 [email protected] CBS UMR5048 Montpellier (France) El Sahili Abbas P7 [email protected] LEBS UPR3082 Gif-sur-Yvette (France) Elias Mikael O22, O90, P41, P42, P43 [email protected] Weizmann Institute of Science Rehovot (Israel) Elkaim Erik Chair [email protected] Synchrotron SOLEIL Saint-Aubin (France) Erba Alessandro O52 [email protected] Université di Torino Torino (Italie) Espeau Philippe O82, P71 [email protected] Faculté de Pharmacie Paris Descartes (France)

Liste des participants
- 221 -
Espinosa Enrique O53 [email protected] CRM2 UMR7036 Nancy (France) Falaise Clément O15, P12 [email protected] UCCS UMR8181 Lille (France) Fauvet Gérard Industriel [email protected] Bruker AXS SAS Champs sur Marne (France) Favier Frédérique P15 [email protected] CRM2 UMR7036 Nancy (France) Fernandez-Varela Paloma P57 [email protected] LEBS UPR3082 Gif-sur-Yvette (France) Ferrand Yann P11 [email protected] CBMN UMR5248 Bordeaux (France) Ferrer Jean-Luc P19 [email protected] IBS UMR5075 Grenoble (France) Fetics Susan [email protected] LEBS UPR3082 Gif-sur-Yvette (France) Flack Evelyne [email protected] Université de Genève Genève (Suisse) Flack Howard O83 [email protected] Université de Genève Genève (Suisse)
Folly-Klan Marcia P40 [email protected] LEBS UPR3082 Gif-sur-Yvette (France) Fourati Kammoun Zeineb O66 [email protected] Ecole Polytechnique UMR7654 Palaiseau (France) Fourmigué Marc O14, O53 [email protected] Institut des Sciences Chimiques UMR6226 Rennes (france) Fribourg Sebastien O9 [email protected] IECB Bordeaux (France) Galatanu Nicoleta O63 [email protected] Xenocs Sassenage (France) Gallois Bernard P62 [email protected] CBMN UMR5248 Bordeaux (France) Galmiche Marion O36, P31 [email protected] CRISMAT UMR6508 Caen (France) Garreau de Loubresse Nicolas O65 [email protected] IGBMC UMR7104 U964 Strasbourg (France) Garron Marie-Line P75 [email protected] AFMB UMR7257 Marseille (France) Gbabode Gabin O6 [email protected] SMS EA3233 Rouen (France)

Liste des participants
- 222 -
Ghermani Nour Eddine O51 [email protected] LSPMS Ecole Centrale de Paris Paris (France) Gillon Béatrice P54 [email protected] CEA Saclay (France) Giorgi Michel [email protected] Fédération de Chimie FR 1739 Marseille (France) Girard Eric SP3, O32, O33, P78 [email protected] IBS UMR5075 Grenoble (France) Giraud Marie-France P52 [email protected] IBGC UMR5095 Bordeaux (France) Gondry Muriel O21 [email protected] CEA Saclay (France) Gonzalez Cécile [email protected] IBGC UMR5095 Bordeaux (France) Gonzalez Daniel P41, P42 [email protected] URMITE UMR6236 Marseille (France) Gorrec Fabrice P8 [email protected] MRC Laboratory of Molecular Biology, Cambridge (UK) Gordeliy Valentin O24 [email protected] IBS UMR5075 Grenoble (France)
Gotthard Guillaume O22, P41, P42, P43 [email protected] URMITE UMR6236 Marseille (France) Gras Pierre O27 [email protected] CIRIMAT UMR5085 Toulouse (France) Gratias Denis O40, P70 [email protected] LEM UMR104 Chatillon (France) Grosjean Arnaud O38, P1 [email protected] ICMCB UPR9048 Bordeaux (France) Guérin Laurent O87, P34, P36 [email protected] Institut de Physique UMR6251 Rennes (France) Gueguen-Chaignon Virginie [email protected] IBPC FR3302 Lyon (France) Guillet Valérie O57, P21 [email protected] IPBS UMR5089 Toulouse (France) Guillin Jacques Industriel [email protected] Bruker AXS SAS Champs sur Marne (France) Guillou Nathalie O50 [email protected] ILV UMR8180 Versailles (France) Guinebretière René O7 [email protected] SPCTS UMR7315 Limoges (France)

Liste des participants
- 223 -
Guionneau Philippe O34, O38, O84, P1, P4, P55 [email protected] ICMCB UPR9048 Bordeaux (France) Haines Julien O3, O31, P27 [email protected] Institut Charles Gerhardt UMR5253 Montpellier (France) Hamzaoui Fodil P39, P67 [email protected] SEA2M Université Abdelhamid Ibn Badis Mostaganem (Algérie) Hartmann Thomas Industriel [email protected] Stoe & Cie gmbh Darmstadt (Allemagne) Heijboer Pierre P32 [email protected] ICMCB UPR9048 Bordeaux (France) Henry Natacha O15, P12 [email protected] UCCS UMR8181 Lille (France) Hiblot Julien O22, P41, P42, P43 [email protected] URMITE UMR6236 Marseille (France) Hillard Elizabeth CLO [email protected] CRPP UPR8641 Bordeaux (France) Hobbs Jeanette Industriel [email protected] MolecularDimensions Newmarket (UK) Hodeau Jean-Louis P50 [email protected] Institut Néel UPR2940 Grenoble (France)
Hospital Michel O79 [email protected] Laboratoire de Cristallographie Université Bordeaux 1 Talence (France) Housty Jacques O79 [email protected] Laboratoire de Cristallographie Bdx 1 Cestas (France) Houdusse Anne O88, P48 [email protected] Institut Curie UMR144 Paris (France) Housset Dominique P44 [email protected] IBS UMR5075 Grenoble (France) Huc Ivan SP1, P11, P62 [email protected] CBMN UMR5248 Bordeaux (France) Huet Tiphaine P16 [email protected] Department of Molecular Biology Geneva (Switzerland) Ibanez Alain Chair [email protected] Institut Néel UPR2940 Voiron (France) Itié Jean-Paul P28 [email protected] Synchrotron SOLEIL Saint-Aubin (France) Jacques Vincent O62 [email protected] LPS UMR8502 Orsay (France) Jarry Christian Chair [email protected] Faculté de Pharmacie Bordeaux (France)

Liste des participants
- 224 -
Josse Michael O1, O26, P2, P32 [email protected] ICMCB UPR9048 Bordeaux (France) Kammoun Omar O74 [email protected] Institut des Sciences Chimiques UMR6226 Rennes (France) Karmazin-Brelot Lydia [email protected] Institut de Chimie de Strasbourg UMR7177 Strasbourg (France) Kauffmann Brice O38, P11 [email protected] IECB Bordeaux (France) Kenfack Tsobnang Patrice P68 [email protected] CRM2 UMR7036 Nancy (France) Khadri Amina [email protected] Université d’Oum El Bouaghi Oum El Bouaghi (Algérie) Kilburg Arnaud P24, P25 [email protected] IBCP FR3302 Lyon (France) Klosek Vincent O76 [email protected] CEA Saclay (France) Labesse Gilles [email protected] CBS UMR5048 Montpellier (France)
Labourel Aurore P17 [email protected] Station biologique UMR7139 Roscoff (France) Lacoudre Aline P69 [email protected] ISM UMR5255 Bordeaux (France) Lafaye Céline P63 [email protected] IBS UMR5075 Grenoble (France) Laguerre Michel [email protected] CBMN UMR5248 Bordeaux (France) Lakhloufi-Mathieu Sabine P4 [email protected] ICMCB UPR9048 Bordeaux (France) Lamour Valérie O44 [email protected] IGBMC UMR7104 Strasbourg (France) Larrieu Isabelle P52 [email protected] IBGC UMR5095 Bordeaux (France) Lassalle Louise O32, P78 [email protected] IBS UMR5075 Grenoble (France) Laulhé Claire O30 [email protected] Synchrotron SOLEIL Saint-Aubin (France)

Liste des participants
- 225 -
Launois Pascale O18 [email protected] LPS UMR8502 Orsay (France) Le Bas Audrey P18 [email protected] IBPC UMR7099 Paris (France) Le Bolloc'h David Chair [email protected] LPS UMR8502 Orsay (France) Le Dreau Loic Industriel [email protected] Bruker AXS SAS Champs sur Marne (France) Le Du Marie-Hélène O43 [email protected] CEA Saclay (France) Lebraud Eric P2 [email protected] ICMCB UPR9048 Bordeaux (France) Leger Jean-Michel O82 [email protected] Faculté de Pharmacie Bordeaux (France) Lemée-Cailleau Marie-hélène O34, P4 [email protected] ILL Grenoble (France) Leroux Gwenaëlle P76 [email protected] MIG Jouy en Josas (France) Li de la Sierra-Gallay Ines P73 [email protected] IBBMC UMR8619 Orsay (France)
Lisa Maria Natalia P74, 76 [email protected] Institut Pasteur Paris (France) Lisboa Johnny [email protected] IBBMC UMR8619 Orsay (France) Liu Dan P33 [email protected] SIMAP UMR5266 Grenoble (France) Lodini Alain O75 [email protected] LACM Reims (France) Loiseau Annick O16 [email protected] LEM UMR104 Chatillon (France) Loiseau Pascal O25 [email protected] LCMCP-ENSCP UMR7574 Paris (France) Lüning Jan O60, O80 [email protected] LCPMR UMR7614 Paris (France) Maglione Mario O1, O26, P32 [email protected] ICMCB Bordeaux (France) Marchivie Mathieu P59, P71 [email protected] Pharmacochimie Bordeaux (France) Marchot Pascale O58 [email protected] AFMB UMR7257 Marseille (France)

Liste des participants
- 226 -
Mariette Céline O61, O87, P36 [email protected] Institut de Physique UMR6251 Rennes (France) Martin Christine Chair [email protected] CRISMAT UMR6508 Caen (France) Martinetto Pauline P50 [email protected] Institut Néel UPR2940 Grenoble (France) Mas-y-mas Sarah P19 [email protected] IBS UMR5075 Grenoble (France) Massip Stéphane O82, P59, P71 [email protected] Pharmacochimie Bordeaux (France) Mathieu Magali Chair [email protected] Sanofi Vitry sur seine (France) Matringe Caroline O7 [email protected] SPCTS UMR7315 Limoges (France) Mayer Claudine O46 [email protected] Institut Pasteur Paris (France) Maziarz Steve Industriel [email protected] EXPLORA NOVA SARL Bordeaux (France) Mechulam Yves O64 [email protected] Ecole Polytechnique UMR7654 Palaiseau (France)
Menetrey Julie O88 [email protected] LEBS UPR3082 Gif-sur-Yvette (France) Mercier Nicolas O12 [email protected] MOLTECH UMR6200 Angers (France) Merzoug Meriem [email protected] Université d’Oum El Bouaghi Oum El Bouaghi (Algérie) Molinari Flora P2 [email protected] ICMCB UPR9048 Bordeaux (France) Moncoq Karine P20 [email protected] IBPC UMR7099 Paris (France) Mondieig Denise O38, P60, P61 [email protected] LOMA UMR5798 Bordeaux (France) Moras Dino P53 [email protected] CERBM GIE Strasbourg (France) Moreau Christophe P64 [email protected] IBS UMR5075 Grenoble (France) Moreau Florian O70 [email protected] Nottingham (UK) Mourey Lionel O57, P21 [email protected] IPBS UMR5089 Toulouse (France)

Liste des participants
- 227 -
Moynie Lucile P45 [email protected] Université de St Andrews St Andrews (Ecosse) Naim Ahmad O84, P58 [email protected] ICMCB UPR9048 Bordeaux (France) Négrier Philippe O34, O38, O82, P60, P61, P71 [email protected] LOMA UMR5798 Bordeaux (France) Nitschke Jonathan O13 [email protected] Cambridge (UK) Oubla M'hamed P65 [email protected] Faculté des Sciences Hassan II Ain Chock Casablanca (Maroc) Paineau Erwan O18, O70 [email protected] LPS UMR8502 Orsay (France) Paulus Werner O42, O72 [email protected] Institut Charles Gerhard Université Montpellier 2 Montpellier (France) Payen Christophe O1 [email protected] IMN Jean Rouxel UMR6502 Nantes (France) Pechev Stanislav O26, O34, P3 [email protected] ICMCB UPR9048 Bordeaux cedex (France)
Pedelacq Jean-Denis [email protected] IPBS UMR5089 Toulouse (France) Perez Olivier O36, O47, P31, P34, P35, P72 [email protected] CRISMAT UMR6508 Caen (France) Perez Philippe Industriel [email protected] ELEXIENCE Verrières-le-Buisson (France) Petit Pierre-Emmanuel [email protected] IMN Jean Rouxel UMR6502 Nantes (France) Poupon Morgane P35 [email protected] CRISMAT UMR6508 Caen (France) Philouze Christian [email protected] DCM UMR5250 Grenoble (France) Picot Daniel P18 [email protected] LBPCPM UMR7099 Paris (France) Pignol David O11 [email protected] CEA UMR7265 Cadarache (France) Pinaud Noël O9 [email protected] ISM UMR5255 Bordeaux (France)

Liste des participants
- 228 -
Poupon Morgane P35 [email protected] CRISMAT UMR6508 Caen (France) Prangé Thierry O33 [email protected] LCRB UMR 8015 Paris (France) Quiquandon Marianne O40, P70 [email protected] LEM UMR104 Chatillon (France) Rabiller Philippe O61, O87, P36 [email protected] Institut de Physique UMR6251 Rennes (France) Ranieri Vincent P27 [email protected] Institut Charles Gerhardt UMR5253 Montpellier (France) Ravy Sylvain O30, O41 [email protected] Synchrotron SOLEIL Saint Aubin (France) Reiser Jean-Baptiste P44, P46 [email protected] IBS UMR5075 Grenoble (France) Retailleau Pascal [email protected] ICSN UPR2301 Gif-sur-Yvette (France) Richard Philippe [email protected] ICMUB UMR6302 Dijon (France) Robin Julien O2 [email protected] Institut des Sciences Chimiques UMR6226 Rennes (France)
Rochel Natacha P53 [email protected] IGBMC UMR7104 Strasbourg (France) Rogues Pierrick Industriel [email protected] Natx-ray Grenoble (France) Roisnel Thierry P51 [email protected] Institut des Sciences Chimiques UMR6226 Rennes (France) Ropars Virginie O88 [email protected] Institut Curie UMR144 Paris (France) Roret Thomas P15 [email protected] CRM2 UMR7036 Nancy (France) Rosa Patrick O34, O84, P58 [email protected] ICMCB UPR9048 Bordeaux (France) Rotella Hélène O77 [email protected] CRISMAT UMR6508 Caen (France) Rouquette Jérome O3, O31 [email protected] Institut Charles Gerhardt UMR5253 Montpellier (France) Roussel Pascal O77 [email protected] UCCS UMR8181 Lille (France) Rousselin Yoann [email protected] ICMUB UMR6302 Dijon (France)

Liste des participants
- 229 -
Rucktooa Prakash O10, P47 [email protected] IRI UMR8576 Lille (France) Rueff Jean-Michel O36, P31, P35 [email protected] CRISMAT UMR6508 Caen (France) Ruff Marc O68 [email protected] IGBMC UMR7104 Strasbourg (France) Samama Jean-Pierre P56 [email protected] Synchrotron SOLEIL Saint Aubin (France) Sanchez Corinne P52 [email protected] IBGC UMR5095 Bordeaux (France) Sangiorgio Léa [email protected] Grenoble (France) Sauguet Ludovic O67 [email protected] Institut Pasteur Paris (France) Schaniel Dominik O19 [email protected] CRM2 UMR7036 Nancy (France) Seddiki Nadir [email protected] IBGC UMR5095 Bordeaux (France) Shepard William P56 [email protected] Synchrotron SOLEIL Saint Aubin (France)
Sirigu Serena P48 [email protected] Institut Curie UMR144 Paris (France) Sleator Olivia Industriel [email protected] Rigaku Europe Kemsing, Sevenoaks (UK) Snoeck Etienne O17 [email protected] CEMES UPR9011 Toulouse (France) Soldo-Olivier Yvonne O4 [email protected] LEPMI UMR5279 Grenoble (France) Specklin David [email protected] Institut de Chimie UMR 7177 Strasbourg (France) Sridi Taha Faculté des Sciences Monastir (Tunisie) Stevens Ray SP2 [email protected] Department of Molecular Biology, Scripps, USA La Jolla (U.S.A.) Sulzenbacher Gerlind P75 [email protected] AFMB UMR7257 Marseille (France) Tabti Charef P67 [email protected] Université Abdelhamid Ibn Badis Mostaganem (Algérie) Tao Liang O37 [email protected] LRCS CNRS UMR7314 Univ de Picardie Amiens (France)

Liste des participants
- 230 -
Thiaudière Dominique O85 [email protected] Synchrotron SOLEIL Saint Aubin (France) Thompson Andrew O67 [email protected] Synchrotron SOLEIL Saint Aubin (France) Timmins Joanna O45 [email protected] IBS UMR5075 Grenoble (France) Tola Fabienne [email protected] DCM UMR5250 Grenoble (France) Toudic Bertrand O61, O87, P36 [email protected] Institut de Physique UMR6251 Rennes (France) Tranier Samuel P21 [email protected] IPBS UMR5089 Toulouse (France) Trapani Stefano [email protected] CBS UMR5048 Montpellier (France) Tsai An-Pang SP4 [email protected] SIMAP Grenoble (France) Van der Lee Arie O31 [email protected] IEM UMR5635 Montpellier (France) Van Tilbeurgh Herman O66, P49, P73 [email protected] IBBMC UMR 8619 Orsay (France)
Vaney Marie-Christine O8 [email protected] Institut Pasteur Paris (France) Vendier Laure O34, P14 [email protected] LCC UPR8241 Aureville (France) Wagner Tristan O69 [email protected] Max Planck Institute Marburg (Allemagne) Wehenkel Annemarie [email protected] Institut Curie UMR3306 Paris (France) Welter Richard [email protected] Institut de Chimie UMR 7177 Strasbourg (France) Wierzbanowski Krzysztof O75 [email protected] Universite de Science et Technologie AGH Zabierzow (Pologne) Ingrid Gautier Industriel [email protected] PANalytical Le Plessis Robinson (France)

Liste des participants
- 231 -