rubrique 5. paramètres et méthodes -...
TRANSCRIPT

ETUDE DE FAISABILITE DE LA MISE EN PLACE
D'UN RESEAU NATIONAL D'OBSERVATION DE LA
QUALITE DES EAUX LAGUNAIRES ET MARINES
EN COTE D'IVOIRE (R.N.O.C.I.)
Rubrique V
1PARAMETRES ET METHODES lpar
J.M. CHANTRAINE*, Ph. DUFOUR** et J.L. PESCHET***
1 - Introduction v,4
2 - Recommandations générales V,6
3 - Qualité de l'eau V,6
4 Micropolluants organiques et minéraux V,16
5 - Biologie V,17
6 - Minéralogie V,22
7 - Hydrocarbures et macrodéchets sur le littoral V,22
8 - Annexes V,25
* Chercheur ORSTOM - Centre de Recherches OcéanographiquesB.P. V 18 - Abidjan - Côte d'Ivoire
** Chercheur ORSTOM - Institut de LimnologieAvenue de Corzent - 74203 Thonon les Bains - France
*** Ingénieur biochimiste - Ministère de l'EnvironnementB.P. V_2_~4 - AQidi~_~_:.C~e d'Ivoir~. _

- V,2 -
PARAHETRES ET METHODES
1 - INTRODUCTION
2 - RECOMMANDATIONS GENERALES
3 - QUALITE DE L'EAU
3.1 - Température
3.2 - Salinité
3.3 - pH
3.4 - Oxygène dissous
3.5 - Turbidité
3.6 - Matières en suspension
3.7 - Sels nutritifs
3.7.1. - Prélèvement, conservation, dosage
3.7.2. - Azote ammoniacal
3.7.3. - Azote nitreux
3.7.4. - Azote nitrique
3.7.5. - Phosphate minéral dissous
3.7.6. - Silice dissoute
3.7.7. - Sulfates·
3.8 - Sulfures
3.9 Carbone organique particulaire
3.10- Demande· chimique en oxygène
3.11- Oxydabilité par le permanganate à chaud en milieu alcalin
3.12- Demande biochimique en oxygène
3.13- Auto-consommation en oxygène en 48 h
3.14- Essai de putrescibilité
4 - MICROPOLLUANTS OR~NIQUES ET MINERAUX
4.1 - Détergents anioniques
4.2 - Hydrocarbures dans l'eau de mer et les sédiments marins
4.3 - Organochlorés
v,4
V,6
v,6V,7
V,7
v,7V,8
v,9v,9V,10
V,10
V,10
V,10
V,10
V,10
V,10
V,l!
V,l!
V,13
V,13
V,14
V,14
V,15
V,16
V,16
V,16
V,16
V,16
4.4 - Micropolluants minéraux : Ag, Cd, Cu, Zn, Co, Cr, Fe, Mn,Ni, Pb, Hg V,17
4.4.1 - Traces métalliques dans l'eau V,17
4.4.2 - Mercure dans l'eau et la matière vivante V,17
4.4.3 - Traces métalliques dans les sédiments
4.5 - Autres
V,17
V,17

5 - BIOLOGIE
5.1 - Chlorophylle "a" et phéopigments
5.2 - Zooplancton
5.3 - Surveillance de l'ichtyofaune
5.4 Bactéries et virus
5.4.1 - Numération des coliformes totaux et d'EscherichiaColi en milieu liquide
5.4.2 - Numération des streptocoques et des streptocoquesfécaux en milieu liquide
5.4.3 - Analyse bactériologique des coquillages
5.4.4-- Clostridium perfringens
5.4.5 - Germes pathogènes
6 - MINERALOGIE
7 - HYDROCARBURES ET MACRODECHETS SUR LE LITTORAL
8 - ANNEXES
8.1 - Choix d'une technique de mesure des hydrocarbures
8.2 - Dosage des hydrocarbures dans l'eau de mer et les sédimentsmarins
8.3 - Dosage des composés organochlorés et polychlorobiphénylesdans l'eau de mer, les sédiments et les organismes marins
8.4 - Protocole de minéralisation du sédiment pour l'analysedes métaux absorbés
8.5 - Proposition pour la surveillance écologique de l'ichtyofaunede la lagune Ebrié
8.6 - Numération des streptocoques et des streptocoques fécaux enmilieu liquide •
V,17
V,18
V,19
V,19
V,19
V,20
V,21
V,21
V,22
V,22
V,22
V,22

- V,4 -
1 - INTRODUCTION
La présente rubrique se propose:
1°/ de dresser la liste des paramètres qui peuvent ~tre nécessaires
ou utiles de mesurer à un moment ou à un autre dans le dadre du
R. N. O. C. r.
2°/ de décrire ou citer les méthodes appropriées de prélèvement, de
conditionnement, de stockage et d'analyse.
L'objectif primordial est de fournir une méthodologie aux futurs
exécutants du R.N.O.C.I. Il ne peut s'agir d'un état définitif. En effet
la méthodologie peut Atre sujette à des modifications liées au progrès
des techli'fq1i'Eis~':Î""dlimatériel d'une part, èt,i;jdT~û=tre part, à l'évolution
du milieu étudié, aux objectifs de l'étude et aussi à l'examen des pre
miers résultats.
La liste des paramètres considérés est issue de celles du R.N.O.
et du R.N.C.* français. Elle a été amoindrie ou élargie en fonction du
contexte de l'étude: milieu marin et lagunaire tropical.
Les protocoles d'analyses ont également été empruntés à ces m3mes
réseaux lorsqu'ils semblaient adaptés aux conditions des milieux étudiés.
On notera parfois des remarques formulées de par la spécificité des
milieux. Ces méthodes sont soit les plus performantes et les plus fiables
à l'heure actuelle, soit les plus accessibles, compte tenu du matériel
disponible en Cate d'Ivoire. Enfin, ces méthodes ne sont pas développées
ici lorsqu'elles sont présentées dans des ouvrages édités et facilement
accessibles (liste ci-après) alors qu'elles sont décrites si elles ont
été adaptées ou extraites d'ouvrages difficilement accessibles. Il y aura
m3me parfois plusieurs méthodes valables proposées dont le choix pourra
par exemple être dicté par des considérations matérielles.
* Réseau National de Contrôle de la qualité des eaux littorales. C'est
une structure complémentaire au R.N.O.

- V,5 -
Les paramètres sont regroupés en quatre classes:
- qualité de l'eau
- micropolluants organiques et minéraux (dans l'eau, la matière vivante,
les matières en suspension et les sédiments)
- biologie (sur l'eau et la matière vivante)
minéralogie (sur les matières en suspension et les sédiments).
Il va de soi que parallèlement à la collecte des échantillons
et aux analyses effectuées in si tu, devront 3tre réalisées un minimum
de mesures météorologiques (vents, température de l'air, précipitations ••
et hydrographiques (marées, nivea.u de l'ea.u, dérive superficielle, cou
rants, vagues ~ •• ) lorsqu'on ne peut se les..c:ÎJ?~:\r.i"é'i~/j;à.Ü:prètf'.:'aé'â,,'gê'i'V'ides
nationaux compétents (ASECNA, Port ••• ).
Liste des références citées
l
II
III -
IV -
Manuel des méthodes de prélèvements et dt~yses.1:
caractéristiques physico-chimiques et hydrobiologiques.
Réseau National d'Observation de la qualité du milieu marin.
Manuel des méthodes de prélèvements et d t analyses.2:
micropolluants orga.niques et minéraux. Réseau National d'Obse~
vation de la qualité du milieu marin.
MARCHA.ND(M.) et MARTIN(J-L.), 1983 - Etude de faisabilité du
R.N.O. eate d'Ivoire~ polluants chimiques. Centre National
pour l'EXplOitation des Océans.
Manuel des méthodes, 1983 - Réseau National d'Observation de
la qualité du milieu marin. Document provisoire. Centre Natio
nal pour ItExploitation des Océans.
V - Nonnes AFNOR - Tour Europe Cedex 7, 92080 PARIS.
VI - STRICKLAND (J.D.H.) and PARSONS (T.R.), 1968 - A practical
handbook of seawater analysis. Bulletin 167.Fisheries Research
Board of Canada, Ottawa.

VII-
VIII -
IX -
- V,6 -
MAZIERES (J.), 1981 - Méthodes usuelles d'analyse bactériolo
gique pour le contrôle sanitaire courant des eaux de mer et
des coquillages. Institut Scientifique et Technique des P3ches
Maritimes, Nantes.
BRISOU (J.F.) et DENIS (F.A.), 1980 - Techniques de surveillance
de l'environnement maritime. Masson ed., Paris, 206 p.
O. M.S" 1977 - Directives applicables à la surveillance sanitaire
de la qualité des eaux littorales. Bureau régional de l'Europe,
Copenhague, 172 p.
2 - RECOMMANDATIO,S GENERALES~. l. .
Avant de passer en revue les méthodes préconisées pour la déter
mination des trois classes de paramètres, il est préférable de prendre
connaissance, dans un chapitre préalable, du dénominateur commun des pro
tocoles constitué de recommandations générales ~ant trait à:
l'équipement du navire: portique, treuil, câble, lest, poulie compteuse ••
le matériel de prélèvement: bouteilles à clapets avec supports, messa
gers, récipients •••
des accessoires spéciaux: congélateur ou glacière, matériel de préfil
tration, distributeurs de réactifs •••
le fla.connage selon le pa.ramètre: matéria.u (verre, plastique, polyéthy
lène), système de fermeture, volume •••
Ces recommandations ont été formulées par A.AMINOT (Référence l,
Chapi tre 1).
3 - QUALITE DE L'EAU
Tous les paramètres de qualité de l'eau peuvent ~tre considérés
comme indicateurs de pollution lorsqu'ils dépassent des valeurs moyennes

- V,7 -
limites à définir pour ohaque milieu.
Exemples: - élévation de température par rejet d'usine thermique,
conoentrations excessives en phosphate du fait des rejets
d'eaux domestiques,
- élévation ou baisse anormale du p.li du fait de rejets indus-
triels,
3.1 - ,Température
eto•••
Référence IV, Chapitre; 2.
3.2 - Salinité
La salinité est un paramètre oonservatif qui sert de traoeur de
masse d'eau et intervient dans certains calculs d'hydro~amique. Seul le
premier usage est utile dans le cadre du R.N.O. Dans ces oonditions la
précision néoessaire est modeste: 0,05 g/l en mer et 0,5g/l en lagunes~
Dans ces oonditions, la salinité peut @tre mesurée au salinomè
tre à induction en mer, au salinomètre optique en lagune.
Le salinomètre optique est un réfractomètre qui donne une leoture
directe et immédiate. Son coût est de l'ordre de 100 000 F CFA.
Pour la méthode au salinomètre à induction on adoptera le proto
oole développé dans la référence IV, Chapitre 3.
3.3 - pH
La méthode éleotrochimique aveo électrodes de verre est la plus
pratique et suffisamment préoise dans le oadre d'un réseau de surveil
lanoe de la pollution (Référence IV, Chapitre 4).
On notera que la valeur du pH est dépendante de l'aotivité bio
logique, en partioulier de la photosynthèse.
Dans un milieu bien tamponné comme l'océan, les variations nyc
th&~es du pH son~ peu importantes. Dans un milieu moins bien tamponné

- V,B -
et plus riche biologiquement comme les lagunes, les variations nycthémé
rales du pH peuvent devenir importantes et largement excéder les varia
tions spatiales et à grande échelle de temps, d'où l'intér3t d'effectuer
les mesures de pH toujours à la m~me heure, ou si impossible, de tenir
compte des variations nycthémérales.
3.4 - Oxygène dissous
L'o~gène moléculaire dissous est un paramètre très important
des écosystèmes aquatiques qui influence la majorité des processus biolo
giques et chimiques qui s'y déroulent.
La concentratioI1",~~~,)p~J,;\~ne)9+s~Ql.lS".~l'Jt" l~.rés:ill1ïante .de.s·f'acteurs
physiques, chimiques et biologiques suivants:
- échanges à l'interface eau-atmosphère,
- diffusion et mélange au sein de la masse d'eau,
- consommation par les réactions d'oxydation chimique,
consommation par les phénomènes de photo-oxydation,
respiration des organismes aquatiques, ce qui inclut au sens
large la dégl~dation bactérienne des matières organiques, la
nitrification, la sulfo-oxydation•••
production par la photosynthèse.
Le pourcentage d'oxygène dans l'eau par rapport à son niveau
d'équilibre avec l'atmosphère sus-jacente (100% de saturation) est un
élément à considérer, aussi important que sa concentration.
En lagune Ebrié, dans certains secteurs eutrophisés par des
apports organiques exogènes, le pourcentage de saturation peut dépasser
200% en fin d'après-midi en surface et ~tre nul en fin de nuit en surface
ou en pennanence en profondeur. Ces valeurs extr~mes et ces fortes varia
tions ne sont pas sans influencer la vie aquatique. Les milieux totalement
désoxygénés peuvent aussi ~tre une nuisance olfactive pour les riverains
(dégagement d'hydrogène sulfuré).

- V,9 -
Protocoles d'échantillonnage, de conservation, d'analyse et de
calcul (Référence IV, Chapitre 5).
La méthode d'analyse préconisée est celle de WINKLER améliorée.
Notons ici que le développement des électrodes à oxygène permet
des mesures rapides et qui, utilisées dans de bonnes conditions, sont
aussi sensibles et précises que les mesures chimiques. Le maniement et
surtou.t l'étalonnage des sondes à oxygène est cependant délicat et destiné
à un personnel hautement qualifié. Ce qui les rend inutilisables dans le
cadre d'un réseau de routine où la priorité doit être accordée à la simpli
cité de mise en oeuvre et à la fiabilité.
Notons enfin que la concentration en oxygène est coDiÎi1é~ i~'~aieu~ ,',
du pH très dépendante de l'activité biologique, en particulier des
rythmes nycthéméraux de la photosynthèse. D'où les mêmes remarques concer
nant l'heure des prélèvements que celles formulées au paragraphe précédent.
3.5 - Turbidi té
Référence IV, Chapitre 13.
~~if!~~.!~!~e:Une simple mesure de la profondeur de disparition du
disque de Secchi (disque métallique blanc ou à 4 secteurs noirs
et blancs S ,de 25 à 30 om de diamètre) peut suffire pour
cette mesure. Elle est plus adaptée à des eaux: lagunaires où elle
peut varier d'un facteur 10, et elle est beaucoup plus simple
bien quI elle présente 1'inconvénient d'Itre subjeotive puisqu'elle
dépend de l'observateur et aussi de Ilheure de la journée (éviter
de faire cette mesure à des moments trop proches du lever et du
coucher de saleiV. Elle présente l'inconvénient de nI intégrer la
turbidité qu'entre la surface et la profondeur de disparition du
disque de Secchi, inconvénient limité en lagune par la faible
profondeur et la faible hétérogénéité verticale.
3.6 - Mati~res en suspension (M.E.S.)
C'est la méthode par filtration sur filtre en fibre de verre qui
a été choisie pour son caractère rapide et universel (Référence V, NF,
T 90-105).

... V,lO ...
3.7 - Sels nutritifs
Les paragraphes 3.7.1 à 3.7.7. concernent les sels nutritifs du p~
toplancton. Ils se présentent sous plusieurs formes minérales en solution.
Pour l'azote on distingue selon le degré d'o~dation: nitrates
(N03-), nitrites(N02-) et azote ammoniacal (RH3 et surtout NH4+).
Pour le phosphore on utilise le terme de phosphate qui englobe en
fait toutes les formes d'orthophosphates présentes.
Le terme de silice englobe les formes Si(OH)4 et Si 0(OH)3-.
En mer aussi bien qu'en lagune les concentrations peuvent dans
certaines conditions devenir inférieures à la limite de détection, d'où des
précautions nécessaires de prélèvement, conservation et dosages.
3.7.1. - Prélèvement, conservation, dosages (généralités).
Référence IV, Chapitre 6 -
3.7.2. - Azote ammoniacal.
Référence IV, Chapitre 7 -
3.7.3. - Azote nitreux.
Référence IV, Chapitre 8 -
3.7.4. - Azote nitrique.
Référence IV, Chapitre 9 -
3.7.5.- Phosphore minéral dissous.
Référence IV, Chapitre 10 -
3.7.6.- Silice dissoute.
Référence I, Chapitre 3-E.
En lagune, l'inté~t de ce paramètre est limité. D'une part, la
silice n'y est jamais un âlément nutritif limitant. D'autre part, sa

- V,l1 -
concentration est approximativement inversement proportionnelle à la
salinité.
3.7.7. - Sulfates.
En mer la concentration varie peu; en lagune elle peut ~tre
déduite de la salinité.
3.8 - Sulfures
Ils sont issus de la réduction des sulfates dans les eaux anoxi
que s, mais on peut aussi les doser en petite quan~é aux faibles concen
trations,.p.' o:xygè~~.:q~ sOAt .tqxiques pour les organismes aérobies et
abondants dans les eaux de fond de certaines zones de la lagune.
La méthode préconisée est celle au bleu de méthylène. Elle dose
les espèces H2S, HS- et S--
Prélèvement et conservation
H2S étant volatil et oxydable par l'oxygène, l'exposition à l'air
et les manipulations seront réduites au minimum. Dans une fiole jaugée de
100 ml contenant de l'acétate de zinc à 2%, on,verse l'eau à analyser juste
à la surface de la solution d'acétate, de 0,1 à 50 ml de manière à obtenir
une coloration convenable au moment de la colorimétrie. Cette opération fixe
les sulfures et permet de conserver l'échantillon à 4°C quelques jours au
maximum.
Réactifs
11 Acétate de zinc 2%. Dans une fiole jaugée de 1000 ml, verser
20g d'acétate de zinc, 1 ml d'acide acétique pur et compléter
à 1000 ml par de l'eau distillée;
~ Sulfate de diméthyl-p-phénylène diamine 0,2%. On verse 2g de
ce produit dans une fiole jaugée de 1000 ml contenant 200 ml
d'eau distillée. Puis on ajoute 200 ml d'H2S04 concentré, et l'on complète
jusqu'à 1000 ml avec de l'eau distillée.

- V, 12 -
11 Sulfate de fer et d'ammoniu~ 10~. A préparer extemporanément
5,7g de sulfate ferrique + 4,3g de sulfate d'ammonium + 2ml
de H2S04 concentré + H20 distillée (q.s.p. 100 ml).
N.B.- Pour les réactifs 2) et 3) l'eau distillée doit être fraîchementbouillie et refroidie.
Technique
Dans la fiole où le prélèvement a été fixé, on complète par de
l'eau distillée de manière à avoir environ 80ml. On ajoute a.lors 10ml de
la solution 2). On bouche la fiole immédiatement et on l'agite une fois.
On verse alors 0,5 ml de la solution 3) en ouvrant et refermant la fiole
très rapidement. On agit1t"'ê'1!"1j'R·'1"1:~iEfsé fêpè5s'èr/'fOnin.·dh'·c6'mplèteàÜ,,:ts par
de l'eau distillée jusqu'à 100 ml et on agite à nouveau.
Moins de 4 heures après, on mesure l'intensité de la coloration au
spectrocolorimètre à 670 nm (cuve de 1 cm). On compare le résultat à une
courbe d'étalonnage établie à partir de sulfure de sodium hydraté Na2S,9 H20
préalablement séché sur papier filtre.
Préoision
~ dans l'intervalle de confia.nce de 95%.
Sensibilité
0,15 pmole/l.
Référence
CLINE (J.D.), 1969 - Spectrophotometric determination of hydrogen
sulphide in natural waters. Limnology.and Oceanography,14 (3): 454-458.
N.B.- Si l'on peut se contenter d'un dosage grossier des sulfures, unesimple mesure du potentiel redox de l'eau suffit. H2S est présentau-dessous de - 250 mV et est approximativement proportionnel aupotentiel redox.

- V, 13 -
Les paramètres qui suivent: earbone organique partioulaire (c.a.p.),oarbone organique total (C.a.T.), demande chimique en oxygène (D.C.a.),
demande biochimique en oxygène (D.B.a.), putrescibilité sont des indica
teurs de la teneur en matière organique globale du milieu et de son poten
tiel de dégradabilité.
3.9 - Carbone organique particulaire {c.a.p.)
Le carbone est l'élément de base de la matière organique. Chez les
3tres vivants il représente environ 50% de leur poids sec.
Le carbone organique total se répartit en deux fractions séparêes
arbitrairement par filtration.
Le carbone organique particulaire (c.a.p.) est la fraction retenue
sur un filtre de porosité 0,45 J'lm.
Le carbone organique dissous est la fraction qui passe au travers
de ce filtre.
c.a.p. Méthode recommandée: chimique
oxydation par le mélange sulfochromique(Référence IV, Chapitre 17).
Des méthodes plus précises sont basées sur la combustion de la
matière organique dans l'oxygène suivie d'un dosage du gaz carbonique.
Elles nécessitent un matériel sophistiqué: ana.lyseur CliN, analyseur infra
rouge actuellement non disponiblooà Abidjan.
Méthodes alternatives en cas d'acquisition de ce matériel-
Combustion dans l'oxygène (fortes concentra.tions)(Référence VI, Chapitre 4,4, II).
Combustion dans l'oxygène (faibles concentrations) .(Référence VI, Chapitre 4,4, III).
3.10 - Demande chimique en oxygène (D.C.a.)
Cette analyse normalisée sous le numéro AFNOR NF T 90 - 101
(Référence V) est couramment utilisée pour déterminer la pollution. Elle

- V, 14 -
consiste à oxyder énergiquement, en présence d'un catalyseur, et de façon
totale (mélange sulfo-chromique à chaud pendant 2 heures ) la matière organique.
C'est donc le reflet de la quantité totale de matières oxydables, biodégrada
bles ou non. Elle s'exprime en milhgrammes d'oxygène par litre.
Malheureusement, son emploi est limité à des échantillons dont la
teneur en chlorures est inférieure à 3 g/l. En effet, les chlorures réagissent
eux aussi avec le bichromate.
Quelques essais effectués en laboratoire ont montré que les chlorures
augmentaient la D.C.O., mais jamais de façon similaire, malgré une complexa
tion des ions chlorures par le sulfate de mercure dans un rapport 1 à 10 en
poids. Etant donné que la concentration des chlorures varie de 0 à 20 gll au
cours de l'année dans le milieu lagunaire, il serait impossible d'exploiter
un résultat de D.C.O., l'augmentation de cette dernière pouvant être aussi
bien due aux chlorures qu'à la pollution.
Cette analyse n'a donc pas à être prise en compte dans le R.N.O.C.I.
3.11 - Oxydabilité par le permanganate à chaud en milieu alcalin
Cette analyse normahsée sous le numéro AFNOR T 90 - 018 (Référence V:
est couramment uti lisée pour le dosage des matières organiques oxydables essen
tiellement dans les eaux d'alimentation. C'est une méthode conventionnelle
qu'il est important de suivre rigoureusement pour assurer la constance des
résultats.
Cette réaction est perturbée par la présence des chlorures, des
sulfures, du fer ferreux, de l'azote ammoniacal et nitreux et de matières
en suspension.
Cette analyse n'a donc pas à âtre prise en compte dans le R.N.O.C.I.
3.12 - Demande biochimique en oxygène (D.B.O.)
Cette analyse normalisée sous le numéro AFNOR T 90 - 103 (Référence V:
est cour~~ent utilisée pour déterminer la quantité de matières biodégradables
présentes ~ans une eau. Dans les eaux très polluées il est nécessaire de rajou
ter des quantités connues d'oxygène à l'échantillon, ou d'effectuer une gamme
de dilutions. Dans la plupart des eaux naturelles on peut utiliser l'échantil
lon brut 5 jours plus tard, l'oxygène restant est dosé, la différence entre
le taux d'oxygène de départ et celui du 5ème jour donnant la consommation
d'oxygène en 5 jours à la température normalisée de 20°C.

Cette méthode par dilution dans les eaux très chargées en matière
organique présente plusieurs inconvénients 1 plusieurs sous échantillons à
traiter, reproductibilité faible.
Si la D.E.O. devait être prise en compte dans le R.N.O.C.I, il
serait préférable de choisir la D.E.O. manométrique. Celle-ci consiste à
introduire une quanüté connue d'échantillon, sans dilution, dans un flacon.
L'échantillon est agité pendant 2 heures, flacon ouvert. Le liquide se
sature en oxygène. Au bout de ces 2 heures, le flacon est bouché et mis en
relation avec un manomètre à mercure. L'oxygène est consommé et la pression
baisse (le C02 produit est absorbé sur du KOR placé dans une nacelle dispo
sée dans le flacon au-dessus du liquide agi té,). Au bout de 5 jours, la lec
ture du manomètre permet de calculer directement la D.E.O.
Cette méthode est simple, précise et ne met pas en jeu des manipu
lations compliquées. Le laboratoire de la D.E.I. va posséder prochainement
un appareil à 10 postes qui permettra de pouvoir réaliser cette analyse.
Un point reste à définir: c'est celui de la température d'incuba
tion. Elle est classiquement de 20°C en Europe. L'adapter davantage aux
conditions locales (soit 30°C par exemple) permettrait de se rapprocher
davantage des phénomènes réels de biodégradation dans les milieux.
3.13 - Auto-consommation d'oxygène en 48 heures
Cette méthode n'est pas normalisée par L'AFNOR. Elle est citée
dans le Deutsche Einhei tsverfahren zur W'asser sous le numéro H5~ C'est
en quelque sorte une D.E.O. simplifiée.
L'échantillon est introduit dans 3 flacons à oxygène type AFNOR
120 ml. L'oxygène est dosé immédiatement dans le premier flacon (méthode
WINCKLER AFNOR NF T 90 - 106 (Référence V). Les 2 autres flacons sont
mis à incuber 48 heures. L'oxygène est dosé après ce laps de temps •
•soit T1 la teneur en 02 en mg/l dans le premier flacon (t = 0 heur
.soient T? et T3 . les teneurs en 02flacons lt = 48 heures).
en mg!l dans les 2 autres
L'auto-consommation en mg/l en 02 en 48 heures est donnée par:T2 + T3
A.C. 02 / 48 h = T1 - 2

~ V,16 -
Cette méthode ne nécessite que 3 flacons par échantillon, 48heures
d'incubation. Elle permet de se faire une idée de la consommation d'oxygène
dans le milieu. Il reste à déterminer la température d'incubation qui, comme
pour la D.B.O., pourrait ~tre celle du milieu naturel échantillonné.
3.14 - Essai de putrescibilité
Cette analyse simple permet de mettre en évidence l'état réducteur
du milieu et sa capacité à rendre putride les eaux. Elle n'est appropriée
qu'aux eaux très chargées en matière organique, donc plutôt dans les lagunes
qu'en mer (Référence V, NF, T 90 - 104).
Ils existent pour certains à l'état de trace dans les milieux natu
rels non pollués.
Ils peuvent être dosés dans l'eau, les sédiments et la matière
vivante. Lorsqu'il existe des particularités ~s à chacun de ces supports,
elles sont signalées dans les protocoles qui suivent.
4.1 - Détere;ents anioniques
Référence II, 1ère partie, Chapitre 1.
4.2 - Hydrocarbures dans l'eau de mer et les sédiments marins
Choix d'une technique, voir Annexe V.1 (M.MARCHAND).
- Technique par spectrophotométrie infra-rouge
Voir Annexe V.2 (.r.C.ROUSSEL).
- Technique par spectrofluorescence ultraviolette.
Un protocole d'analyse est en cours de rédaction au C.O.B. (France).
4.3 - Organochlorés
Cette classe de composés englobe simultanément les pesticides
chlorés (DDT, lindane, heptachlore, aldrine,dieldrine, et les résidus

- V,17 -
industriels:polychlorobiphényles (PCB).Voir Annexe V.3 (M.MARCHAND).
4.4 - MicrOpolluants minéraux: Ag, Cd, Cu, Zn, Co, Cr, Fe, Mn, Ni, Pb, Hg.
4.4.1 - Traces métalliques dans l'eau:
- Méthode par absorption atomique avec flamme après extraction
- Méthode ~'polarographie impulsionnelle à redissolution anodique
Référence II, 2ème partie, Chapitre 1.
4.4.2 - Mercure dans l'eau et la matière vivante
Référence II, 2ème partie, Chapitre 2•. "h j." -
4.4.3 - Traces métalliques dans les sédiments:
- Analyse de la fraction mobilisable
- Analyse totale de l'échantillon
- Mercure.
Référence II, 2ème partie, Chapitre 3.
Remarque: L'Annexe V.4 (J .L.MARTIN) expose le protocole de minéralisation du sédiment qui a été utilisé dans le cadre del'exercice d'intercalibration de l'opération test (Cf.rubrique VII ).
4.5 - Autres
- Plastifiants: inutile et abandonné dans le R.N.O. français.
- Phénols: utile seulement si pétrochimie complexe.
5 - BIOLOGIE
Les paramètres liés à la biomasse ou à l'activité des organismes
vivants sont, comme les paramètres hydrologiques et hydrochimiques des
indicateurs de la qualité du milieu aquatique.
Leur valeur excessive ou insuffisante (par rapport à des normes
à définir pour chaque milieu) est souvent le signe d'un apport polluant.

- V,18 -
Par exemple, un excès de sels nutritifs d'origine industrielle ou domesti
que entraîne une prolifération algale manifestée par des concentrations en
chlorophylle"a" supérieures à ilIa normale"; un excès de matière organique
entraîne une prolifération "anormale" ete bactéries hétél~otl'Ol'hes.
Certains par~nètres biologiques intègrent les effets de pollutions
diverses. D'autres sont plus spécifiques; ils sont susceptibles d'indiquer
la nature, l'importance et l'origine du polluant. Par exemple, les salmo
nelles sont indicateurs d'une pollution fécale; la présence de Salmonella
typhi indique le rejet d'un porteur (sain ou malade) du vecteur de la
fièvre typhoïde; la présence du m~me germe en quantité est manifeste d'un
foyer épidémique sur le proche bassin versant.
~
5.1 Chlorophylle lia" et phéopigments
La chlorophylle "a est un pigment commun à tous les végétaux.
En pleine eau, elle est spécifique des microalgues (phytoplancton) ou des
macrophytes issues des berges et du fond, ou du bassin versant. On consi
dère qu'elle est proportionnelle à la biomasse végétale. Les méthodes pro
posées pemettent de distinguer la. chlorophylle "a" de ses produits de
dégra.dation, les phéopigments.
Deux méthodes sont proposées:
Méthode par spectrophotométrieRéférence IV, Chapitre 15.
Méthode par fluorimétrieRéférence IV, C~apitre 16.
L'une et l'autre sont envisageables à Abidjan, compte tenu du
matériel existant (Cf. rubrique II).
La signification des deux méthodes est différente. La première
permet une évaluation des concentrations de chlorophylle et de phéopig
ments; la seconde une propriété des chlorophylles et phéopigments: la fluo
rescence qui n'est qu'approximativement proportionnelle à leurs concentra
tions.

- V,19 -
La première méthode est précise et son interprétation (biomasse)
aisée; la seconde est moins précise, moins facilement interprétable et
nécessi te des étalonnages fréquents. Par contre elle est très sensible,
ce qui pennet de travailler sur des volumes réduits: de l'ordre du litre
en mer, du décilitre en lagune. Elle est également plus rapide à mettre
en oeuvre.
De ce fait nous 1& conseillons pour le R.N.O., où il n'est utile
que de détecter les grandes variations de biomasse végétale.
5.2 - Zooplancton
A l'image des autres paramètres et en particulier de la chloro
phylle, la bi~;-~~~~zoopl~nctoniquequi mesure ~ indice de la richesse
du milieu aquatique, peut ~tre considérée comme un indicateur de pollution
si sa valeur se situe hors des limites habituellement rencontrée~.
La méthode décrite ci-dessous expose un protocole standard de
mesure de la biomasse.
Prélèvement du zooplancton et mesure de la biomasse
Les échantillons sont prélevés par tractions verticales (fond
surface) de filets de 40cm de diamètre d' ouve:1'ture et de 60 rm de vide
de maille. Deux ou trois traits sont effectués pour chaque échantillonnage
et le produit des p~ches est ensuite mélangé puis fractionné en deux par
ties égales. Une moitié de l'échantillon est fixée au formol à 5% et sert
à effectuer les détenninations et les comptages de chaque groupe faunisti
que. L'autre moitié est recueillie par aspiration sur filtre (oF/e p 47 )
prépesé (de poids P1), rincée au fonniate d'ammonium isotonique (pour
éliminer le sel sans altérer les organismes), puis desséchée à l'étuve
(60 0 e) pendant au moins 24 heures. Le filtre est ensuite pesé (P 2), calciné
au four (550°C) pendant 1h30, puis pesé à nouveau. (P3).
Le poids sec (P.S.) ..est donné par P.S. = P2 - Ph et le poids orga
nique (P.O.) du poids sec sans cendre (p.s.s.e.) par P.O. = P3 - P2.
5·3 - Surveillance de l'ichtyofaune
Tout comme les maillons inférieurs de la cha1ne alimentaire (phyto-

- V,20 -
plancton, zooplancton••• ) les paramètres ichtyologiques devraient égalemen
~tre soumis à une surveillance dans le cadre d'un R.N.O. Cette remarque a
été formulée par un chercheur du Centre de Recherches Océanographiques
d'Abidjan, qui propose outre le suivi physio-pathologique des espèces (en
particulier le dosage des substances toxiques au sein des différents tissu
et/ou organes des poissons), une surveillance écologique systématique de
l'ichtyofaune (peuplements, fécondité, croissance ••• ) notamment dans les
lagunes. Le contenu de cette proposition est reporté en annexe V.5 (J.J.
ALBARET) •
5.4 - Bactéries et virus
Les bactéries et les virus sont des composants normaux des milieux
naturels. Nous no'US intéressons dans leca-ril:r€k1Gl'un réseaudesuTVceislla.nce
certaines espèces ou certains groupes dont la présence et/ou l'abondance
est indicatrice d'un état de pollution, en particulier fécale, et est
susceptible de fournir des indications sur l'origine et l'importance de
cette pollution.
Les germes tests les plus couramment utilisés sont les coliformes
fécaux, les streptocoques fécaux et accessoirement Clostridium perfringens
De nombreuses méthodes sont disponibles et développées dans la
référence VII. Le choix s'est porté sur les suivantes:
5.4.1 - Numération des coliformes totaux et d'Escherichia Coli en milieuliquide
Cette analyse permet le dénombrement des coliformes totaux et des
coliformes spécialement fécaux comme E.Coli. Ce dernier germe est un germe
test de contamination fécale récente. Il est en effet aSsez fragile et dis
paraît rapidement.
La méthode en milieu liquide a été choisie pour son universalité
d'emploi, en particulier pour les milieux turbides et fortement chargés en
matières en suspension. C'est une méthode normalisée: Béférence V, Norme
AFNOR T 90 - 413.
Remarque sur le paragraphe 6.3.1 de cette norme: On peut utiliser égalemen- - - - - - - - - - - - - - .- - - -
le bouillon au pourpre de bxomocrésol, qui a la m§me composition

- V,21 -
que le bouillon lactosé, mais qui contient en plus un indicateur
de pH, le pourpre de bromocrésol, qui vire du violet au jaune à
pH 5,2 - 6,8.
5.4.2 - Numération des streptocoques et des streptocoques fécaux en milieuliquide
Cette analyse permet le dénombrement des sreptocoques totaux et des
streptocoques fécaux. Ces derniers germes sont des germes-tests de contami
nation fécale relativement ancienne. En effet, les streptocoques sont plus
résistants que les, coliformes.
La méthode par dilution en milieu liquide a été choisie pour son
universalité d'emploi, en particulier dans les milieux turbides et. chargés
en matières en suspension.
Voir annexe V.6.
5.4.3 - Ana~se bactériologique des coquillages
Les coquillages peuvent @tre un bon indioateur de pollution. En
effet, pour respirer, ils font transiter un volume important d'eau qui tra
verse leur organisme. Ils peuvent donc concentrer fortement la pollution
d'origine microbienne.
Seuls les coliformes totaux, E.Coli, les streptocoques et les strep
tocoques fécaux seront recherchés. La recherche des bactéries pathogènes
(vibrions, salmonelles, shigelles) ne se fait pas de manière routinière.
Cependant, une recherche spécifique peut @tre faite de ces germes dans des
cas bien précis (au droit d'un émissaire, en cas d'épidémie, d'endémie ••• ).
La bactériologie des coquillai$s demande une préparation, c'est-à
dire un broyage des mollusques et une dilution dans un liquide stérile. La
méthode décrite ci-dessous est celle employée par l'I.S.T.P.M. de Nantes
(Référence VII, pages 6, 8 et 9).
5.4.4 - Clostridium perfringens
Clostridium perfringens est un germe anaérobie de grande longévité
dans la nature. Répertorié dans l'échantillon en l'absence de coliformes et

- V,22 -
de streptoco~ue8 fécaux, il apporte la preuve d'une pollution ancienne.
La référence VIII propose plusieurs méthodes de dénombrement.
5.4.5 - Germes pathogènes
Dans le cas d'une contamination du milieu naturel par les coliformes
et les streptoco~ues fécaux, d'une endémie et d'une épidémie, il est suggéré
de rechercher les agents pathogènes particuliers:
- les salmonelles, en particulier S.typhi et S. paratyphi, agents
des fièvres typhordes et paratyphordes;
- le vibrion du choléra;
- le Vibrio parahaemolyticus vecteur d'une entérite;
- Shigella à l'origine des ~senteries bacillaires
...Toutes les techni~ues disponibles sont développées dans la référence
VIII et le choix de celles ~ui sont le plus adaptées à une surveillance
sanitaire de routine est fait dans la référence IX.
6 - MINERALOGIE
Il s'agit là de déterminer les espèces minéralogi~ues argileuses
(montmorillonite, kaolinite, chlorite) ou non argileuses (~uartz, calcite,
dolomite ••• ), présentes dans les matières en suspension et dans les sédiments
par analyse aux r83"0ns X. Il est évident ~ue la. ,granulométrie et la nature
du sédiment tient un rôle fondamental dans l'interprétation des résultats
d'analyse des micropolluants minéraux étant donné la méthodologie utilisée
(atta~ue sélective de surface des grains). Cela étant, l'expérience des
artisans du R.N.O. français nous enseigne ~ue cette classe de paramètres
n'est pas indispensable dans un premier temps étant donné l'information
recueillie bien modeste en face de l'effort financier exigé.
7 - HYDROCARBURES ET MACRODECHETS SUR LE LITTOR.lL
Les hydrocarbures et macrodéchets flottants sont finalement rejetés
sur le littoral (plages océani~ues et berges basses des lagunes) par le jeu

- V, 23 -
des vents et des courants. Leur quantité intègre la pollution du milieu
aquatique de la période précédente.
La méthodologie suivante est adaptée de celle suggérée par
M.BODENNEC du C.O.B. (France) qui a effectué une campagne d'évaluation
des hydrocarbures et macrodéchets sur les plages ivoiriennes pour le compte
de la Compagnie ESSO.
Prospection d'ensemble
Al' aide d'un film aérien de l'ememble du littoral, déterminer
(estimation visuelle) des zones selon leur degré de pollution en 4 ou 5
classes. Evaluer le linéaire correspondant à chaque classe.
N.B.-·""J.ai~;P'awe:pectionaérienne n'est pas.applieable aux lagunes où les---- berges sont souvent recouvertes de végétaux et où la pollution
par hydrocarbures n'est importante ~utaocidentellement.Pourlaprospection on fera une estimation visuelle à partir d'une embarcation longeant les berges.
A l'intérieur de chaque classe sélectionner au hasard une dizaine
de bandes de 1m de largeur parallèlement à la ligne des eaux et de profon
deur égale à la zone de balancement maximale des marées.
- Sur le terrain
• A l'intérieur de chaque bande, compter, peser (ou estimer le poids)
de tous les macrodéchets classés par cat'gorie: bois travaillé et non
"tft,va.illé, verre, caoutchouo, plastiques, méta.ux, papiers, v@tements,
oadavres, bouteilles, médicaments ••• Noter le maximum de détails qui per
mettront éventuellement de retrouver l'origine du polluant; par exemple
noter les indications portées sur les emball&ge~
• Sur l'ensemble de la bande ramasser les boulettes de goudron, les
peser; ramasser le sable souillé sur 1cm d'épaisseur environ, mélanger et
prélever un éohantillon.
- Au laboratoire
• Peser l' éohantillon de sable souillé, extrair~ les hydrocarbures
au chloroforme, évaporer, peser à nouveau; la différenoe de poids évalue

- V,24 -
le contenu des sables en hydrocarbures •
• Une analyse plus fine des hydrocarbures pourra éventuellement être
réalisée sur les boulettes de goudron et le sable souillé par les
méthodes précèdernrnent recommandées •
• Evaluer à partir des résultats des comptages, pesées et analyses
des bandes échantillonnées et de la prospection aérienne, la pollution par
macrodéchets et hydrocarbures du littoral ou de portions de littoral.
Un protocole analytique de l'analyse chimique d'un sable pollué est
indiqué ~ur la figure suivante. Des précisions sur la méthodologie pourront
être obtenues aùprès de G. BODENNEC au CÔi1'aê':B'rwl-~t":lFraiic~):c:
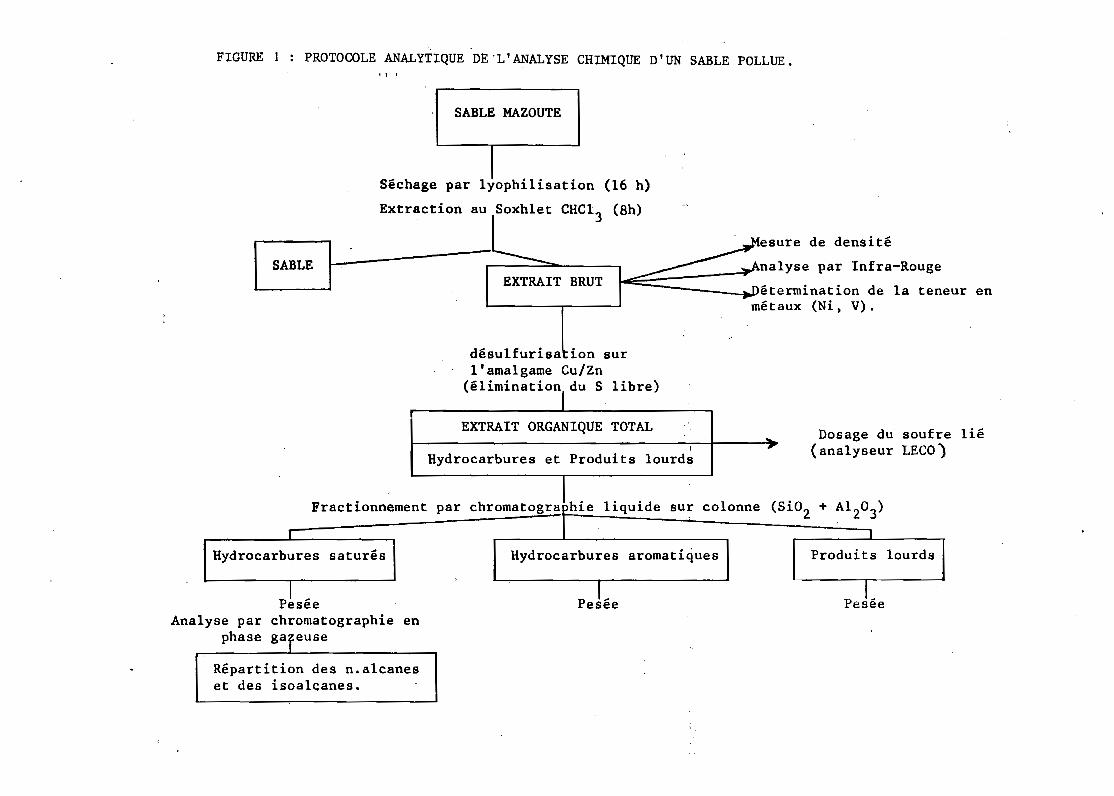
FIGURE 1 PROTOCOLE ANALYTIQUE DE 'L'ANALYSE CHIMIQUE D'UN SABLE POLLUE .• l ,
SABLE MAZOUTE
S~ch.ge par 1loPhi1isation (16 hl
Extraction au Soxh1et CHCl3
(Sh)
1 SABLE r' - .esure de dens i té
~ _na lyse par Infra-Rouge
~ :;étermination de la teneur enmétaux (Ni, V).
désu1furisation surl'amalgame Cu/Zn
(élimination. du S libre)
EXTRAIT ORGANIQUE TOTAL Dosage du soufr~
\,. (analyseur LECO ~Hydrocarbures et Produits lourds
Fractionnement par chromatogra )hie liquide sur colonne (Si02 + Al 203)
r l
Hydrocarbures saturés Hydrocarbures aromatiques Produits lourds
e lié
PèséeAnalyse par chromatographie en
phase gafeuse
Répartition des n.alcaneset des isoalcanes.
Pesée Pesee

'""' ~- '''','). .'
8.1 Annexe V.l
CHOIX D'UNE TECHNIQUE DE MESURE DES HYDROCARBURES
,,;,'>','0,'0,", •. .' Que ce.sp~,~ pour l'e~u, l~ sédiment ou la matière vivante, l'analysE
proc~de en première étape par une extraction par un solvant organique, suivie
d'une phase de purification de l'extrait organique. Deux techniques de mesure
sont disponibles pour apprécier ce que l'on appelle des "hydrocarbures totaux"
- la spectrophotométrie infra rouge (I.R.)
- la spectrofluorescence ultraviolette (SFUV).
La première technique de mesure (IR) répond à la vibration de la
liaison C-H. Les hydrocarbures extraits sont donc dissous avant la mesure dans
un solvant ne possédant pas ce type de liaison ; on utilise le tétrachlorure
de carbone (CC14). La seconde technique (SFUV) mesure la fluorescence .des hydro
carbures aromatiques; l'extrait est dissous dans un solvant non fluorescent,
par exemple l'hexane.
Quelle technique choisir ?
La technique IR est la meilleure approche pour l'estimation des
hydrocarbures totaux, elle possède en outre une gamme de réponse linéaire grande
par rapport à la technique SFUV. Elle souffre toutefois d'une faible limite de
détectabilité. ce qui constitue un inconvénient très réel pour les mesures dans
l'eau.
On admet un seuil de 2 ~ag/l d' hydrocarbures totaux dans l'eau
comme critère de pollution. La mesure IR ne permet pas de descendre en dessous
de 20 ~g/l, tandis que la mesure par SFUV permet de déceler des teneurs inférieurE
~ 1 ~g/l. La technique par SFUV est également recommandée pour les dosages dans
la matière vivante~

v.z
En effet, la phase de purification de l'extrait ne permet
généralement pas d'éliminer tous les lipides co-extraits avec les hydrocarbures
Ces substances, par la présence de liaison C-H, interférent si l'on utilise la
mesure IR. Les inconvénients de la technique IR indiqués pour l'eau (seuil de
détectabilité) et pour la matière vivante (lipides co-extraits) n'existent pas
pour le sédiment. Ce Bera par conséquent la méthode recommandée pour ce type
d'analyse. Le tableau ci-dessous récapitule ces quelques remarques.
D'un point de vue strictement analytique, la mesure des hydro
carbures totaux (extraction, purification, mesure IR ou SFUV) ne présente pas
de difficultés majeures. Un soin doit être apporté à la propreté de la verrerie
utilisée et des réactifs. Les solvants doivent être très purs. Un soin plus par
ticulier doit être pris pour éviter toute contamination accidentelle si l'on
analyse des échantillons d'eau, compte tenu des niveaux très faibles rencontrés.
Technigues de mesures Avantages Inconvénients
i Spectrophotométrie IR.
- Répond à la liaisonC-H
- Meilleure approche pourla mesure des hydrocarbures totaux.
- Gamme linéaire de réponse : importante.
1
Limitefaible
(pour
de détectabilité
l'eau 20 ~g/l)
Spectrofluorescence UV.
- Sensible aux hydrocarbures aromatiques.(représentatifs d'unepollution pétrolièredans l'environnement)
- Méthode très'··sensible(pour l'eau 1 rg/l)
Gamme de réponse linéairefaible (effet quenching)
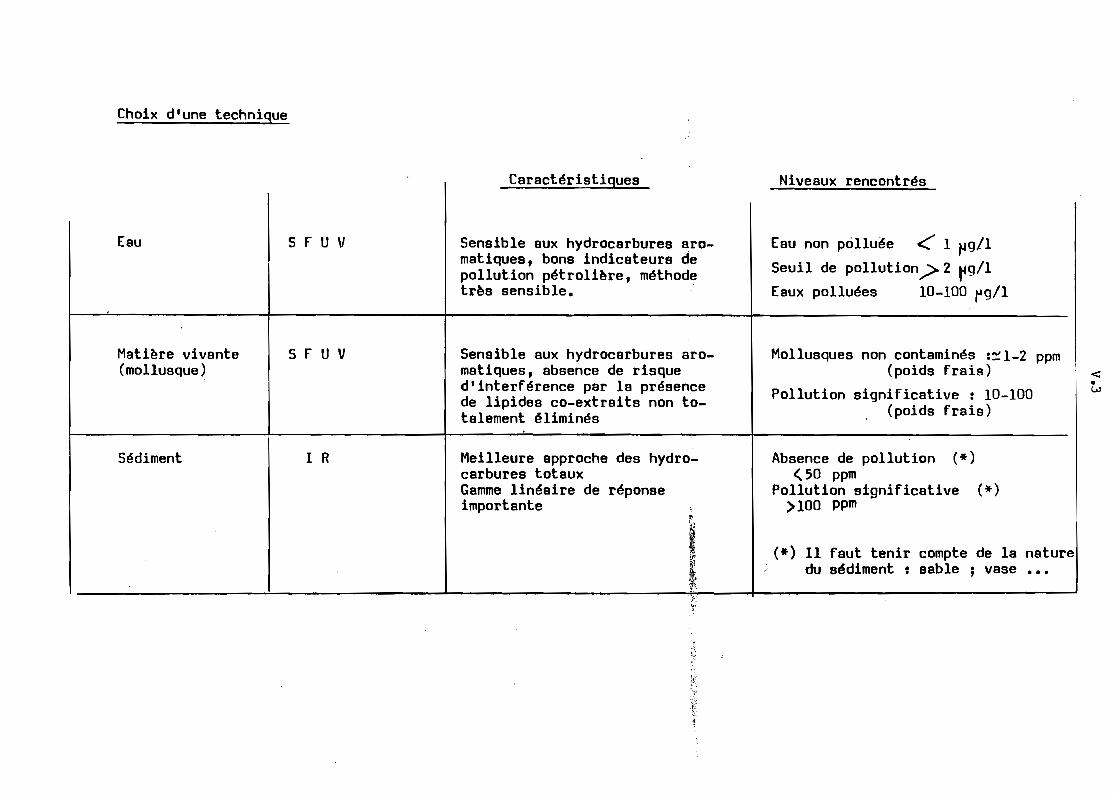
Choix d'une technique
Caractéristiques Niveaux rencontrés
Eau
Matière vivante(mollusque)
Sédiment
S r u V
S r u V
1 R
Senaible aux hydrocarburea aromatiques, bona indicateurs depollution pétrolière, méthodetrès sensible.
Sensible aux hydrocarbures aromatiques, absence de risqued'interférence par la présencede lipides co-extraits non totalement éliminés
Meilleure approche des hydrocarbures totauxGamme linéaire de réponseimportante
f:;
i~..~~":li.'
!;"c<
,'-,,.;,-~.
~,I.
:,~
~;
Eau non polluée ~ 1 ~g/l
Seuil de pollution.> 2 ~g/l
Eaux polluées 10-100 rg/l
Mollusques non contaminés :~1-2 ppm(poids frais)
Pollution significative : 10-100(poids frais)
Absence de pollution (*)<50 ppm
Pollution significative (*)>100 ppm
(*) Il faut tenir compte de la naturedu sédiment: sable; vase •••
<:•w

8.2. Annexe V,2
DOSAGE DES HYDROCARBURES DANS L'EAU DE MERET LES SEDIMENTS ~~RrNS
(J.C. ROUSSEL)
Cette méthode en ce qui concerne l'eau est ~ortement inspirée de la
norme AFNOR 90 203. elle en diffère p.ssentiellement par le protocole utilisé
en chromatographie liquide. Pour les sédiments l'étape concernant l'extrac
tion est également modifiée.
1. PRINCIPE
Las hydrocarbures sont extraits du substrat à étudier par le tétrachlo
rure de carbone. l'extrait est percolé sur un adsorbant afin de retenir les
composés polaires. l'éluat analysé par spectrométrie infrarouge. La méthode
s'applique à tous les hydrocarbures extractibles au tétrachlorure de carbone
et susceptibles d'absorber le rayonnement infrarouge dans la région comprise
antre 3000 et 2800 cm- 1 correspondant aux différentes vibrations de valence
C-H des groupes: -CH]. -CH2- et -~-H.
La limite de détection est de 0.1 mg/l pour un spectromètre ordin~ire.
Si l'appareil possède une expansion d'échelle en ordonnée üt à condition d'opé
rer avec un solvant extr~m8ment pur cett8 limite peut Gtre abdissée à 0.020 mg/l.

8.2 - v.3
2. PRELEVEMENT
Il doit se faire dans des flacons de verre, rigoureusement propres
(nett6yage au détergent puis è l'acide sulfochromique et plusieurs rinçages
à l'eau distillée ~).
L'idéal serait d'utiliser des bouchons de verre rodé sans graisse mais
ce moyen est peu commode dans le cas de transport iJrolongé. On peut utiliser
soit le bouchon de liège classique entourô de h~LJilJ8S d'eJ1uminium rincées au
tétrachlorure de carbone, soit le bouchon vissé avec disque d'étanchéité en
téflon. tout autre polymère. tout caoutchouc est à proscrire.
Afin d'éviter au maximum l'oxydation on ne laisse au haut du flacon
qu'une garde très faible.
,,",:'i'i'~'." Dan,;; le,casde,l'eau afin de diITl;inuer l'activité microbi.enne; l'échantil-
lon est acidifié après le prélèvement. (Le porter à pH inférieur è 3 en ajou
tant. par exemple, 0,1 ml d'HCl 12 N à 1 litre d'échantillonJ.Cette précaution
n'~st à prendre que si l'extraction ne se fait pas immédiatement.
Les sédiments seront congelés ou au moins acheminés dans une enceinte
réfrigérée (glaCière portative par exemple).
3. APPAREILLAGE
- Balance de précision.
- Ampoules à décanter de deux litres.
Broyeur avec bol de 1 litre sans matériau organique (élastomère, polymère.
etc ••• à proscrire).
- Soxhlets.
- Papier filtre (1).
- Colonne de chromatographie (décrite plus bas).
- Spectromètre infrarouge dispersif.
- Deux paires de cellules infrarouge (une de 1 cm, l'autre de 5 cm), en quartz (2).
- Eprouvettes graduees.
:t Eau distit Zée et non eau perrmltée.
(1) l)urieux nO 111 "vert" sans grœisse.
(2) La qualité infrasif de chez HeUma convient.

8.2 - V.4
Toute la vorrerie sera soigneusement lclVée (dC:tcf'f';c;nt. dcido sulfochro
mique) et abondamment rincée à l'eau distillée et au tétrachlorure de carbone.
4. REACTIFS
Acide chlorhydrique qualité analytique.
- Sulfate de sodium anhydre pour analyse.
_ Tétrachlorure de carbone pour spect,oscopie (3). Cette qualité est indispen
sable pour atteindre le seuil de 0,1 mgll ; au-dessus la qualité "pour ana-
lyse W suffit (4).
- Agent adsorbant silice adsorbante modifiée par la présence de magnésie
(MgO: 15,5 % - S102 : 84 % - Na S2~~< 1
0,025 et 0,250 mm (100-60 mesh) (5).
5. EXTRACTION
5.1 Eau de mer
%) de granulométrie comprise entre
Dans une ampoule à décanter verser M grammes d'eau à analyser (il est
recommandé de travailler sur 1 kg d'eau).
Porter à pH acide si l'échantillon n'a pas été acidifié auparavant
(pour l'extraction, le pH 5 est suffisant).
Prendre un volume V (~50 ml pour 1 litre d'eau) de tétrachlorure deo
carbone dont la pureté aura été vérifiée par spectrométrie infrarouge : il ne
doit pas présenter de signal à 2925 cm- l (voir figure 1). S'en servir pour laver
le flacon de prélèvement et les ajouter ensuite au contenu de l'ampoule à dé
canter. Agiter, soit manuellement, soit par agitateur mécanique pendant 1S min.
Laisser reposer 10 min. et soutirer l'extrait (qui constitue la phase
inférieure).
Filtrer cet excès sur du sulfate de sodium anhydre déposé sur un filtre
sans graisse préalablement rincé au tétrachlorure de carbone.
(3) Le produit 2209 vendu par Merk convient.
(4) Le produit 2222 vendu par Merk convient.
(5) Le produit "FZorisiZ" verulu par 8crZabo convient.
Le produit "Florigi l " vendu paI' Touzart et Matignon convient.

8.2 - V.5
S.2 S~dimeYl.t8
On prélève aux environs de 300 grammes do sédiment qu'on sèche à
l'étuve è 60 0• Si le sédiment contient beaucoup d'eau on fait précéder le
chauffage d'une centrifu~ation, on recueille l'edu et on l'analyse comme il
est indiqué au paragraphe 5.1.
On place le sédiment sec dans le broyeur pour le réduire en poudre
fine et l'homogénéiser.
On mélange M grammes (environ 100 g) de la poudre obtenue èvec 20 g
de sulfate de sodium anhydre, le tout est placé dans un soxhlet dont la car
touche a été soigneusement nettoyée par une opération· à blanc avec du tétra
chlorure de carbone.
Le sédiment est extrait au soxhlet pendant huit heures par le tétrachlo~
l'ure de carbone, _le volume de tétrachlorure étant fixé par celuitle:':sol<"hi'et.
A la fin de l'opération, mesurer le voluille V de l'extrait receuillio
et le filtrer sur sulfate de sodium anhydre pour éli~iner l'eau éventuellement
entralnée.
6. CHROMATOGRAPHIE LIQUIDE
Elle est destinée à se débarrasser de composés polaires extraits en
même temps que les hydrocarbures.
On prépare une colonne de verre de 25 cm de longueur et 0.5 cm de dia
mètre intérieur munie d'un réservoir d'une cinquantaine de millitres à l'ex
trémité supérieure et d'un robinet téflon (toute graisse est à proscrire) à
l'extrémité inférieure. Cette colonne est remplie de florisil (de 1,5 è 2 g).
Laver le florisil en faisant percoler sur la ccilonne 20 ml au moins de
tétrachlorure de carbone, écarter le CC1 4 recueilli.
Verser dans le réservoir surmontant la colonne 20 ml de l'extrait, le
laisser entièrement pénétrer le florisil. recueillir le liquide élué da~s une
éprouvette graduée, puis rajouter en tête de colonne 4 ml de CC14 pur. (Ce
-volume correspond à peu près à deux fois le volume mort de la colonne) et
recueillir dans la même éprouvette. Mesurer le volume collecté soit Vl ml.
7. MESURE
Elle se fait par spectrométrie infrarouge différentielle.

8.2 - V.6
Dans le faisceau échantillon. placer une cellule de quortz remplie de
l'extrait percolé sur florisil.
Dans le faisceau de référence. placer une cellule identique mais remplie
de tétrachlorure pur également percolé sur une deuxième colonne de florisil
préalablement rincée.
Enregistrer le spectre. mesurer l'absorbance à 2G25 cm- 1 (3.42 ~) et dé
terminer la concentration d'hydrocarbures dans l'extrait par comparaison avec
une courbe d'étalonnage.
Les spectromètres infrarouge donnent 8ft g~lléral la transmission. l'absor
bance A est déterminée (voir figurs 2) par la méthode des tangentes.
Mesurer 10
et It
sur le spectre.
loA = log --
It
8. ETALONNAGE
Dans la mesure où le polluant est connu et disponible. préparer plusieurs
J~eutLo~de concentrations connues dE ce polluant dans le tétrachlorure de car~
bone. (Exprimer les concentrations en mg d'hydrocarbures par litre de CCl~).
Déterminer comme ci-dessus les densités optiques à 2925 cm- 1 (dans les
mêmes cellules que pour l'extrait) et tracer la courbe donnant l'absorbance
en fonction de la concentration. (Choisir des dilutions entre 10 et 200 mg/l).
Si le polluant est inconnu. prendre pour référence le mélange API (en %
en poids)
benzène 25
1sooctane 37,5
hexadécane 37.5
que l'on diluera comme précédemment dans le CC1 4 •
9. RESULTATS
L'absorbance A de l'extrait correspond sur la co~rbe d'étalonnage è une
concentration de C mg/litre (hydrocarbures dans le CC1 4 ).
La concentration d'hydrocarbures dans l'échantillon d'oau est

w =:
8.2 - V.7
C VIV o20 M en mg/kg
Avec 1"1
Vo
poids de 1'6chantillon de substrat (en g)
volume de CCl4 ajouté à l'eau ou recueilli après extractionsoxhlet (ml)
volume de l'extrait après passage sur florisil (en ml)
concentration lue sur la courbe {en li,g/litr8J.
10. COMMENTAIRES
Cette méthode. telle qu'elle est décrite. en utilisant des cellules de
1 cm et un appareil infrarouge de routine. permet d'atteindre sans problème des
teneurs en hydrocarbures de 10 mg par litre de tétrachlorure. soit de l'ordre~'f·ot,,. .' '{i ..... · _'::-..... ;i:;~·-__,.,,:
de 0.5 mg pa~ kilogramm~ ~'eau "(se1onle rapport eau/tétrachlorure).
En utilisant des cellules de 5 cm. du tétrachlorure de grande pureté et
un spectromère de haut de gamme (muni d'une 8xtension'du signal en ordonnée)
cette limite peut être repoussée à 0.02 mg/kg.
Pour de telles concentrations les subst~n~n~ n~turnlles présentes dans
la milieu (lipides ou autres) peuvent fiJU ssnr consirJérablemrmt l'analyse et 11
est recommandé de vérifier l'absence de produits polaires dans l'extrait. Cette
recherche qualitative peut être effectuée en laissant évaporer goutte à goutte
l'extrait sur une plaque de chlorure de sodium et enregistrer le spectre
complet de cette plaque. On ne doit pas observer de signaux entre 1800 et
1650 cm-l. Si dans ce spectre la bande carbonyle vers 1730 cm- 1 est d'intensité
comparable à celle des CH2 le dosage n'a aucun sens. La cause en est une mau
vaise séparation des composés polaires qui peut se produire. Le sédiment (c'est
rarement le cas pour l'eaU) est très chargé en ester et peu en hydrocarbures.
Il convient alors de recommencer la chromatographie en tenant compte dans le
calcul des dilutions éventuelles.

,.BIBLIOGRAPHIE
- Nonne AFNOR T90 203. Effluents aqueux des raffineries de pétrole. Dosage
des hydrocarbures totaux (Tour Europe Cédex 92DAD Paris La Défense).
- Norme AFNOR T90 114. Dosage des hydrocarbures totaux (méthode par spectro
métrie infrarouge).
Conversion for the prevention of marine pollution from land based sources.
Third meeting of the working group on oi1 pollution. London 14-16 Fev. 1979.
Annex IV Method of samping and anhydre. Di1 Industry International Explo
ration and production. Forum 37 Duke Street St James's London SW1 y GDH.

8.3. Annexe V,3
DQSAGE DES COMPOSES ORGANOCHLORES, PESTICIDES ET POLYCHLOROBIPHENYLES
DANS L'EAU DE MER, LES SEDIMENTS ET LES ORGANISMES MARINS
(M. MARCHAND)
1. INTRODUCTION
Les hydroc~rbures halogénés à haut poids molécul.aire les plus souventidentifiés dans l'environnement marin correspondent aux résidus de polychlorobiphényles (PCB) et d'insecticides chlorés: lindane, heptachlore, aldrine,dieldrine, DDT et ses deux principaux métabolites (DDD, DDE). Les polychlorobiphényles sont des composés aromatiques qui tels qu'ils sont identifiés dansl'environnement contiennent en moyenne le plus souvent 50 à 60 % de chlore parmolécule.
La détection de ces différents composés par séparation chromatographique sur colonnes remplies se heurte à de sérieuses difficultés du fait desinterférences entre les pics chromatographiques de plusieurs insecticideschlorés (aldrine, dieldrine, DDT, ODE, DDD) et ceux des résidus de PCB. L'analyse quantitative nécessite alors des traitements chimiques appropriés afin desurmonter les faiblesses inhérentes àla technique de chromatographie en phasegazeuse à faible résolution (colonnes remplies). Les principaux traitementschimiques utilisés et décrits dans le précédent manuel RNO (1977) sont:
- la séparation des insecticides chlorés des résidus de PCB par chromatographie d'adsorption (florisil, silica gel) par élution sélective avec des solvants de polarité croissante,
- des tests chimiques destructifs tels que l'attaque acide (destruction de la dieldrine) et la saponification (destruction des isomèresde l'HCH, transformation du DDT et du 000 en leurs dérivés éthyléniques correspondants, respectivement DDE et DDMU).
L'utilisation des colonnes capillaires en verre ou en silice fonduepour identifier les organochlorés par chromatographie en phase gazeuse, améliore considérablement le pouvoir de résolution par rapport aux colonnes remplies. Cette technique à haute résolution permet d'une part de séparer lesinsecticides chlorés des résidus de pes, d'autre part de mieux différencierles différents homologues et isomères des PCB.
Des exemples dp séparation chromatographique sur colonnes remplieset capillaires sont illustrés sur les figures 1 à 4 (Caprais et Marchand 1978).

". ,- .. ~: .
8.3 - V.4
Nous ne pouvons que conseiller à l'analyste de s'engager, s'il ne liadéjà fait, vers la technique de chromatographie en phase gazeuse à haute résolution. L'état actuel des procédés de préparation des colonnes capillaires deverre est décrit par 8erthou (1981). .
Les gammes de concentrations habituelles d'hydrocarbures halogénésrencontrées dans l'environnement marin varient selon la nature des échantillonsanalysés
eau de mer < 1 100 ng/l (ppt),
sédiment (poids sec) < 1 1. 000 ng/g (ppb),
moules (poids sec) 0,01 - 10 ~g/g (ppm) .
2. METHODE
L'analyse des résidus d'hydrocarbures ha1ogénés, quelque soit le typed'échantillon, comprend quatre étapes successives:
- une p~ase d'extraction des substances recherchées par un solvantorganique, suivie alUne pré-concentration de l'extrait organique,
- une phase de purification pour séparer ou détruire les composésorganiques co-extraits qui interféreraient durant l'analyse chromatographique,
- l' analys~. chromatographigue en phase gazeuse proprement dite,
des tests de confirmation.
3. PRECAUTIONS PARTICULIERES - ELIMINATION DES ARTEFACTS
Les facteurs limitant la sensibilité et l'exactitude des analysesrésiduelles sont liés essentièllement aux problèmes de contamination des réactifschimiques et de l'équipement. Tous les matériaux et produits chimiques utiliséspour l'analyse résiduelle doivent être suspectés en ce qui concerne leur propreté.Les matières plastiques (à proscrire) ainsi que nombre de produits manufacturéscontiennent des substances interférentes qui produisent des pics au cours del'analyse chromatographique. Plusieurs hydrocarbures halogénés sont fortementadsorbés sur le verre, ainsi la verrerie peut se trouver contaminée par lescomposés résiduels qui se trouvent sous forme de vapeurs dans le laboratoire.Il est essentiel que tous les solvants et matériaux employés pour l'analysesoient préalablement lavés et testés.
3.1. Verreri e
La verrerie est lavée dans de l'eau chaude contenant un détergentpuissant, puis rincée successivement à l'eau, l'acétone et l 'hexane et enfinséchée au four pendant une nuit à 300°C (à l'exception de la verrerie volumétri que).

8.3 - V.5
3.2. Solvants organiques
Les solvants organiques de qualité RP ne sont pas directement utilisables pour l'analyse résiduelle des hydrocarbures halogénés ; ces solvantsdoivent être redistillés dans un équipement en verre avant utilisation. Il estpréférable d'employer des solvants de qualité Inan09rade" ou "pes tipur", actuellement disponibles sur le marché (SOS, MERCK, MALLINKROOT, BURDICK &JACKSON, ... ).
Chaque lot de solvant doit être testé. Après injection de 5 ~l d'unefraction de solvant pré-concentré 100 fois, il ne devra pas être observé de picchromatographique (en dehors de celui du solvant) ou tout au moins, il ne devrapas être observé de pic dépassant 10 %de l'échelle de l'enregistreur dans lesconditions habituelles de sensibilité de l'appareil.
3.3. Matériaux divers
Les matériaux non combustibles, tels le sulfate de sodium anhydre. lalaine de ver.re, les feuilles d'aluminium sont passés au four à 450°C durant une.nui;t",,,-,ite&7;;broyeurs sont lavés et ri ncéspa r'}es· sol vants d' extracti on . Lespapiers filtre, sources de contamination, les adsorbants chromatographiques(florisil, alumine, silica gel) sont extraits par un solvant (hexane) avantutilisation et séchés au four à 450°C.
4. ECHANTILLONNAGE ET CONSERVATION
Il est absolument essentiel que la verrerie et les récipients utiliséspour conserver les échanti 11 ons soi ent dépourvus de toutes substances qui puissentproduire des pics d'interférence au cours de l'analyse par chromatographie enphase gazeuse (cf. § 3). Les plastiques sont à oroscrire car nombre d'entre euxcontiennent des PCB ou autres substances que Jes solvants peuvent extraire.
Le matériel solide (biologique ou sédimentaire) peut être conservédans des feuilles d1aluminium ·et congelé à - 20°C avant analyse. Il conviendrade vérifier préalablement que les feuilles d'aluminium ne contiennent pas desubstances contaminantes.
Les échantillons d'eau doivent être transferrés dans des récipientsen verre. Le délai entre le prélèvement d'eau et la prise d'essai pour l'analysedoit être le plus court possible pour éviter l'adsorption sur les 'parois. Sauftempérature excessive dans un véhicule de transport, le prélèvement peut êtretransporté à température ordinaire. La conservation doit se faire au réfrigérateur à 4°C. Pour éviter toute action bactérienne, certains auteurs recommandentd'ajouter quelques gouttes de chloroforme.
5. EAU DE ~~ER
La principale difficulté dans la recherche des hydrocarbures halogénésdans l'eau de mer est liée aux faibles concentrations résiduelles trouvées dansce milieu (de 1 'ordre du n9/l à quelques dizaines de n9/1). Le choix de la limite de détection détermine la quantité d'eau à extraire, l'importance de laconcentration de 1 'extrait organique avant l'analyse chromatographique ainsique le niveau de pureté des réactifs, solvants et verrerie. On conviendra dansle protocole exposé de considérer l'échantillon d'eau brute. La méthode manuelle

8.3- V.6
d'extraction directe au moyen d'ampoules à décanter est la plus simple à mettreen oeuvre et permet compte tenu de certaines précautions opératoires de rechercher les résidus organochlorés au niveau du ng/l.
5.1. Matériel et réactifs
Flacons en verre pyrex de deux litres ou bouteilles à vis avecjoints téflon~
- Ampoules à décanter de deux litres~ robinets téflon~
- Pipettes Pasteur,
- Tubes de centrifugation en verre~ gradués, de 15 ml ~
Solvants
- F1 ori sil,
hexane, éther éthylique~ benzène~
5.2. Mode opératoire
L'échantillon d'eau de mer (1 à 4 litres) est conservé dans un récipient en verre à 4°C. L'idéal est d'utiliser un flacon de volume égal à celuide 1'échantillon ana1ysé~ soit deux litres~ de manière à pouvoir utiliser latotalité du contenu et laver ensuite le flacon vide avec le solvant d'extractionpour récupérer la fraction résiduelle adsorbée sur les parois.
L'échantillon d'eau est introduit dans une ampoule â dêcanter de deuxlitres. Une seconde ampoule permettra de récupérer l'eau partiellement extraite.Vextraction est réalisée manuellement ou mécaniquement par trois fois 80 mld'un mélange hexane / éther éthylique (85 : 15, v/v). Les extraits successifssont recueillis et concentrés, avant de procéder à la phase de purification.
Il convient au cours de l 'opération de concentration de prendre certaines précautions pour éviter d'éventuelles pertes résiduelles par distillation.Les recommandations sont les suivantes :
- L'extrait organique doit être séché sur du sulfate de sodium anhydre.
- La température durant l'évaporation, par exemple dans un évaporateurrotatif, ne doit pas excéder 50°C. Certains auteurs recommandentl'utilisation de l'évaporateur du type KUDERNA DANISH.
- L'évaporation à sec n'est pas conseillée.
- Toute condensation d'eau qui contaminerait l'échantillon durantl'évaporation doit être évitée.
Pratiquement l'extrait est séché sur du sulfate de sodium anhydre,concentré à 5 ml à l'évaporateur rotatif, puis à 1 ml sous jet d'air ou d'azotepurifiés.

8.3 - V. 7
La purification de l'extrait concentré est réalisée sur du florisilactivé (450°C, durant une nuit), introduit dans une pipette Pasteur sur unehauteur de 5 cm. Après fixation de l'extrait au sommet de la colonne, lesrésidus organochlorés sont élués pat' 8 ml de benzène. L'éluat est recueillidans un tube de centrifugation et concentré à nouveau sous jet d'air ou d'azoteà 0,5 ml. L'extrait organique purifié et concentré est prêt pour l'analysechromatographique.
6. SEDIMENT
6.1. Matériel et réactifs
- Bocaux en verre, tamis inox,
- Appareil Soxhlet, agitateur magnétique, appareil à ultra-sons, êtuve,
- Florisil, mercure, laine de verre, erlenmeyer, colonne, petitstubes gradués,
',",,,,""""'id~"-<_ Solvants : qual Hé pes t i pur (5 bS'-'otJMERCK) .
6.2. Prélèvement et conditionnement
Les prélèvements sont effectués à l'aide d'une benne SHYPECK (fond> 15 m) ou d'une benne ECKMAN ou WILCO (fond < 15 m). Pour les prélèvementscôtiers, on utilise directement une cuillère en écrémant la surface.
Les échantillons sont stockés dans des bocaux en verre à large col(type bocaux à conserves), préalablement lavés à l'acétone et l'hexane ; unefeuille d'aluminium lavée à l'hexane est placée entre le col du bocal et lecouvercle. Les échantillons sont congelés à - 20°C dans les meilleurs délaispossibles après le prélèvement.
6.3. Mode oprratoire
6.3.1. ~~~~~9~ (élimination de l'eau interstitielle)
Deux possibilités sont utilisées:
- un séchage à l'étuve (60°C) pendant 48 h,
- une lyophilisation pendant 24 h.
Le séchage à l'étuve peut être recommandé, le risque de contaminationétant beaucoup moins grand que par lyophilisation (retour d'huile a. la pompe, , .. ).
6.3.2. Extraction
Différentes possibilités.
On peut adopter comme méthode de référence celle décrite dans le manuelRNO: extraction du sédiment sec (ou non) par de l'acétonitrile pendant 16 h (ouplus), dilution de la phase acétonitrile par cinq volumes d'eau et extraction de

8.3 - V.8
la phase aqueuse par de l 'hexane. Cette méthode donne un rendement d'extractionsupérieur à une extraction directe dans le Soxhlet par de 1'hexane~ Il y anéanmoins un certain nombre de désavnntages particulièrement dans l'utilisationd'une cartouche d'extraction qui peut être une source de contamination nonnégligeable.
Nous avons donc développé un procédé d'extraction quelque peu différent : 30 à 40 g de sédiment sec broyé et tamisé (tamis en inox de 1 mm, pouréviter une trop grande hétérogénéité de l'échantillon) sont introduits dans unerlenmeyer de 250 ml. On y ajoute 100 ml d'hexane pestipur et un barreau magnétique. L'extraction des pesticides s'effectue à l'aide d'une sonde à ultra-sonsplongeant dans 1'hexane et sous agitation magnétique pendant 20 minutes. Cetteopération est répétée une deuxième fois. Les deux extraits sont centrifugés etconcentrés à l'évaporateur rotatif puis sous jet d'air purifié à 1 ml. La purification s'effectue sur du florisil désactivé avec 5 %d/eau (colonne de0i = 6 mm, L = la cm). On élue avec 6 à 8 ml d'hexane, puis on concentre à 2 ml.A cet extrait final on ajoute 0,5 à 1 ml de mercure pour précipiter les composéssoufrés co-extraits. L'extrait purifié est prêt pour l'analyse chromatographique.
7. MATIERE VIVANTE
7.1. Matériel et réactifs
- Lyophilisateur, étuve, homogénéiseur (VIRTIS), centrifugeuse, évaporateur rotatif,
- Papier aluminium, tubes de centrifugation,
- Hexane, acétone (qualité pestipur) , acide sulfurique, alumine.
7.2. Prélèvement et conditionnement
Le matériel biologique est conservé dans des feuilles d'aluminium etcongelé à - 20°C avant analyse.
Dans le cas de mollusques, les parties molles des échantillons sontséparées de la coquille et soumises à une déshydratation par lyophilisationpendant 48 heures. Si l'on ne possède pas de lyophilisateur, l'échantillon peutêtre séché dans une étuve à 60°C ou par un mélange de sulfate de sodium anhydre.L'échantillon est ensuite broyé et homogénéisé dans un mortier.
7.3. Mode opératoire
L'extraction est réalisée sur environ la g de poids sec, dans unhomogénéiseur (type VIRTIS) par 50 ml d'hexane pendant 15 minutes. L'opérationest renouvelée une seconde fois. Les deux extraits organiques sont recueilliset centrifugés à 2 000 - 3 000 tours/minute pour se débarrasser du matérielparticulaire. La phase organique est ensuite concentrée à environ 5 ml àl'aide d'un évaporateur rotatif.

_ •. ,.__."""'.'""-~_..-~--~".,~~.,,•._'- -~----"'''-----~''-"~._~"",~"""-,--,,--,,-,,,,----~~,,"~'-"-'""->'''''''"'''-,
8.3 - V.9
La phase de purification consiste à éliminer les lipides et autresmatières organiques co-extraites de l'échantillon. La méthode employée est uneméthode destructive qui consiste à précipiter les graisses par addition d'acidesulfurique concentré (1 à 2 ml). Après agitation~ les deux phases sont séparéespar centrifugation à 3 000 tours/minute rendant la minutes.
L'extrait d'hexane purifié est prêt pour l'analyse chromatographique.Cette méthode est rapide mais n'est applicable uniquement que pour la recherchedes résidus de l 'HCH~ du DDT et de ses métabolites~ et Qes pea ; par contre, cetraitement acide détruit la dieldrine et l'endrine.
Si l'on souhaite une recherche des composés de la dieldrine et del'endrine, une purification par chromatographie d'adsorption sur alumine selonla méthode décrite par Holden et Marsden (1969) est conseillée. Une fractionaliquote de l'extrait or9anique ne contenant pas plus de 200 mg de lipides estévaporée à 1 ml dans un tube qradué sous courant d'air purifié. L'aluminebasique est activée à 800°C pendant 4 heures~ refroidie dans un dessicateuret partiellement désactivée par addition de 5 %d'eau distillée après agitationpendant 30 mn. L'adsorbant est conservé dans un récioient en verre fermé.
Placer 2 g d'alumine ainsi préparée dans une colonne chromatographiquede 0,6 cm de diamètre. Transférer .à·,;,l~i'«~od,'ft:me pipette'i,graduée 1 mldèl"léxtfaitconcentré au sommet de la colonne. Eluer par de l 'hexane jusqu'à ce que 20 mld'éluat soit recueilli au bas de la colonne. Si la purification n'est pas satisfaisante, l'opération peut être répétée sur une deuxième colonne d'alumine aprèsconcentration de l 'éluat à 1 ml.
8. ANALYSE CHROMATOGRAPHIQUE
L'identification des hydrocarbures halogénés est réalisée par chromatographie en phase gazeuse à haute résolution. Le chromatographe est équipé dluninjecteur "split-splitless" ou d'un injecteur solide type de ROS et dlun détecteur à capture d'électrons (Ni 53 ). La colonne capillaire de verre (30 m x 0,4 mm)est imprégnée dlun film silicone non polaire (SE-30). Les conditions opératoiressont les suivantes :
- Températures :
* injecteur 240°C,* détecteur 3000e~
* fourtempérature initialetempérature finaleprogrammation
- Gaz vecteur :
Hmoc~
240°C~
2°C/mn.
* débit colonne: 2 ml/mn~
* débit purge (injecteur R0S) : 50 ml/mn.
Le pouvoir de résolution de l'analyse chromatographique est donné parle nombre de plateaux théoriques évalué environ à 50 000 alors qu'il n'est quede 3 000 sur colonnes remplies.

8.3 - V.10
L'identification des substances recherchées est réalisée par rapportà l 1 in ject ion de solutions étalons d'insecticides chlorés et de PCB (les deuxtypes de profils DP-S et DP-6 sont les plus fréquemment rencontrés dans l'environnement). L'analyse quantitative des insecticides chlorés identifiés esteffectuée en considérant la hauteur de leurs pics chromatographiques caractéristiques. Pour ce qui concerne les PCB, les hauteurs des cinq principaux picschromatograohiques seront additionnées (profil DP-S : pics 5 + 8 + 13 + 14 + 19profil DP-6 : pics 9 + 10 + 12 + 20 + 21 -figures 3 et 4-).
9. METHODE DE CALCUL. LIMITES DE DETECTION
Soit :
- v : volume de l'extrait final purifié (ml),- v' : volume injecté (~l),
- X : quantité détectée dans le volume injecté (pg).
La quantité résiduelle présente dans le volume de l'extrait finalpurifié est, donc de
vX - (ng)
Vi
La concentration résiduelle dans l'échantillon analysé sera
"
Echantillon Quantité Concentrationanalysée rés i duelle
v 1Eau de mer V (litres) X- - (ng/l)
Vi V
v 1Sédiment (*) P (grammes) X- - (ng/9)
v' P
v 1Matière vivante (*) P (grammes) -- (n9/9)
Vi P
(*) : échantillon sec.
Les limites de détection pour le lindane, le DDT et les PCB sontdonnées à titre indicatif sur la base des quantités minimales détectables parchromatographie (lindane : 10 pg ; DDT: 20 pg ; PCB : 100 pg) et des condi~
tians opératoires d'analyse.
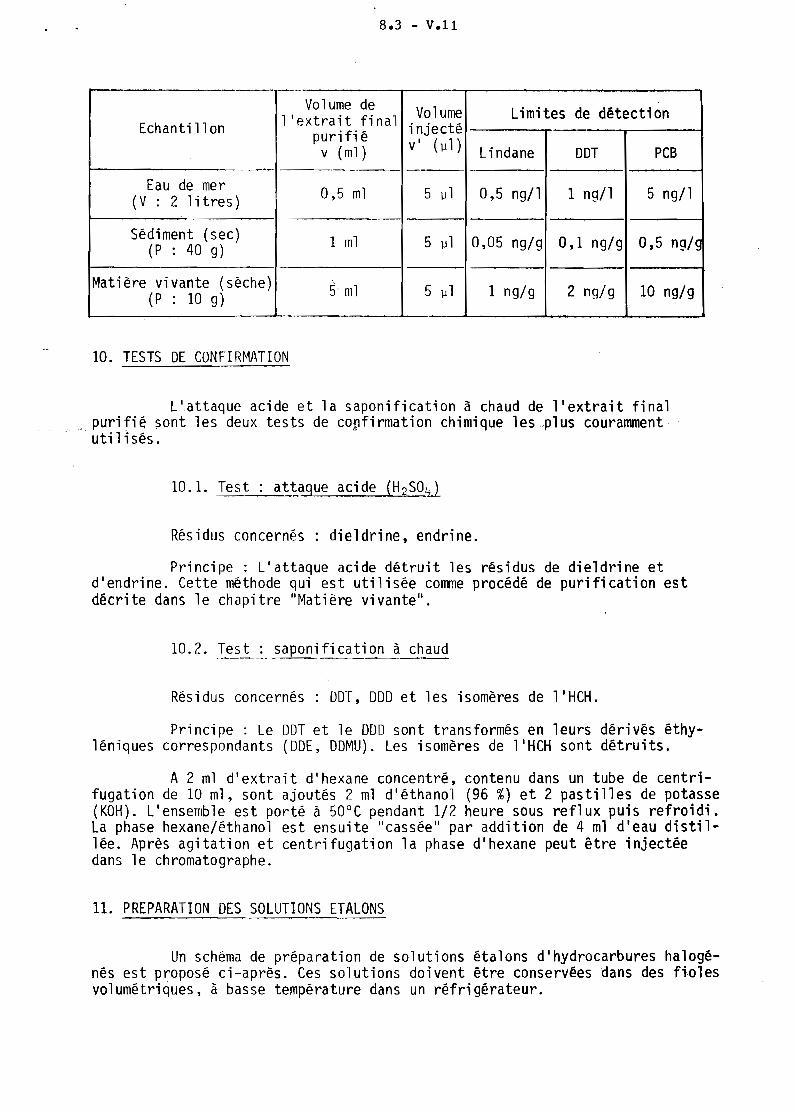
8.3 - V.ll
r---. r-----
Volume de Volume Limites de détectionEchantillon l'extrait final injectépurifié v' (111 )
V (ml) Lindane DDT PCB
Eau de mer 0,5 ml 5 1J1 0,5 ng/1 1 ng/1 5 ng/1(V : 2 litres)
Sédiment (sec) 1 ml 5 111 0,05 ng/g 0,1 ng/9 0,5 ng/g(P : 40 g)
Matière vivante (sèche) \
(P : la g) 5 ml 5 IJ 1 1 ng/g 2 ng/g 10 ng/9
10. TESTS DE CONFIRMATION
L'attaque acide et la saponification a chaud de l'extrait final_purifié sont les deux tests de copfirmation chil11ique les plus courammentutilisés.
10.1. Test: attaque acide (H 2S04 )
Résidus concernés: dieldrine, endrine.
Principe: L'attaque acide détruit les résidus de dieldrineetd'endrine. Cette méthode qui est utilisée comme procédé de purification estdécrite dans le chapitre "Matière vivante".
10.2. Jest : saponification a chaud
Résidus concernés: DDT, 000 et les isomères de l'HCH.
Principe: Le DDT et le 000 sont transformés en leurs dérivés éthyléniques correspondants (ODE, DDMU). Les isomères de l'HCH sont détruits.
A 2 ml d'extrait d'hexane concentré, contenu dans un tube de centrifugation de 10 ml, sont ajoutés 2 ml d'éthanol (96 %) et 2 pastilles de potasse(KOH). L'ensemble est porté à 50°C pendant 1/?. heure sous reflux puis refroidi.La phase hexane/éthanol est ensuite "cassée" par addition de 4 ml d'eau distillée. Après agitation et centrifugation la phase d'hexane peut être injectéedans le chromatographe.
Il. PREPARATION DES SOLUTIONS ETALONS
Un schéma de préparation de solutions étalons d'hydrocarbures halogênés est proposé ci-après. Ces solutions doivent être conservées dans des fiolesvolumétriques, à basse température dans un réfrigérateur.
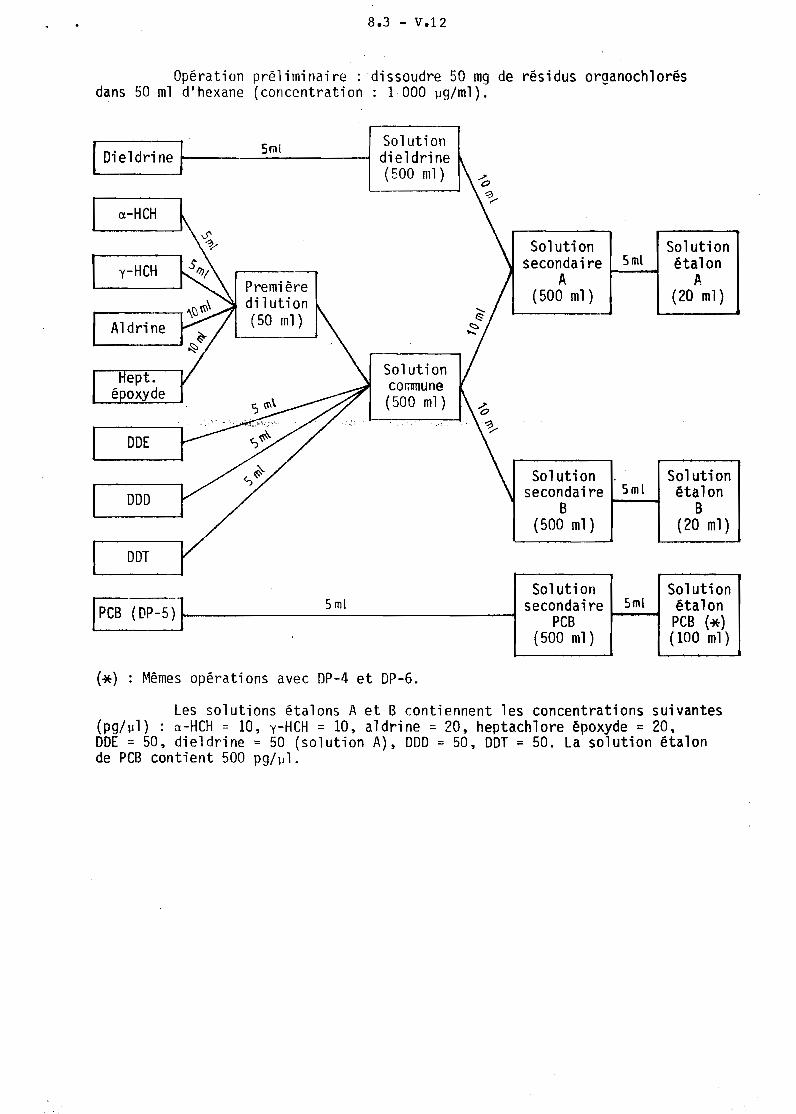
8.3 - V.12
Opération préliminaire: dissoudre 50 mg de résidus organochlorésdans 50 ml d'hexane (concentration: 1 000 ~g/ml).
Solutionétalon
A(20 ml)
Solutionsecondaire 5ml
A(500 ml)
Solutioncommune(500 ml)
Premi èredilution(50 ml)
DDE
a-HCH
y-HCH
Al dri ne
SolutionD;eldr;ne ~ sm_l ~ dieldr;ne
(500 ml)
Solution SolutionDDD secondaire Sml êtalon
B B(500 ml) (20 ml)
DDT
Solution SolutionPCB (DP-5) 5ml secondaire Sml étalon
PCB PCB (*)(500 ml) (100 ml)
(*) : Mêmes opérations avec DP-4 et DP-6.
Les solutions étalons A et B contiennent les concentrations suivantes(pg/~l) : a-HCH = 10, y-HCH = 10, aldrine = 20. heptachlore époxyde = 20.DDE = 50. dieldrine = 50 (solution A). DDD = 50, DDT = 50. La solution étalonde PCB contient 500 pg/~l.
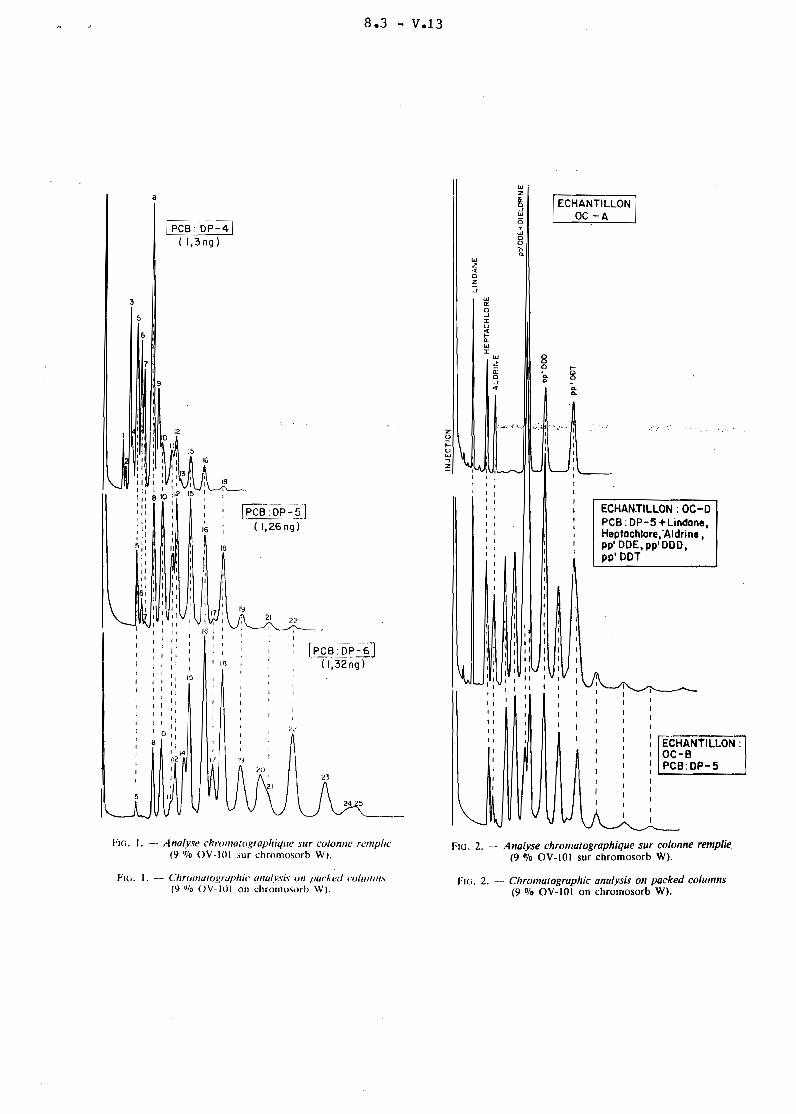
8.3 - V.13
[pcs:DP~( 1,3 ng)
wzg;.JWëi
woo-0.0.
1ECHANTILLON 1
OC -A
wz..oZ
.J
ECHANTILLON:OC-BPCB:OP-5
;""-'~--..J
ECHAN.TILLON : OC-Dpee: DP-5 +lindone ,Heptochlore,'Aldrine,pp' ODE, pp' 000,pp' DOT
1
: \ A
'v~ ~ _1 l , "'-~"'I------
1oo0.C>.
'---' :'--1
111
11,,,
8o
1"
v \J VJ ~_,J- _
, 1
" 1" ,
11 1
'" 1 \ 1l , ,v1
l ,
1
,:\-...0-.....
a:o.J<1
lUIro.JJ:U
~Q.WJ:
WZ
1:1,1:1,
't ::, ,'-, ,, ,
, ,"\,
"'1
z21-
~ \lA.:, ,l ,
1 1, ,1
23
1PCB. DP-=-G]( 1,32ng)
IpCB.DP~( 1,26ngl
lB1'-,,,,11,la
17 IY
16
iL~_: : 21 22
16: (. 1 11 1 1 1 1
l ', ,1 IrJ
12
1
'IIl
"1,1,/
l'
""l'
::,1 f 15
"l,/,
"'1
"'1'i, 14112
1
1,Il
e
6
1
7',:9,1
1 1111
'1
"'I;1""Il,I"Il'1"l",l''l': le5 ::l'l'
""Il61
1,
3
FIG. 1. - Analyse chromafOgraphiquc sur colonne remplie(9 % aV-lOI sur chromosorb \V).
FIG. 2. - Analyse chroma/Ographique sur colonne remplie(9070 aV-lOI sur chromosorb W).
FIG. l, - Chrolllu(ogruphic i/nolysis on l'od.ed ('o/tll/I!/s
(9 % UV-lOI on chrolllusllrb W).FI(;. 2. - Chromatographie analysis on paeked eolumns
(9 % aV-loI on chromosorb W).
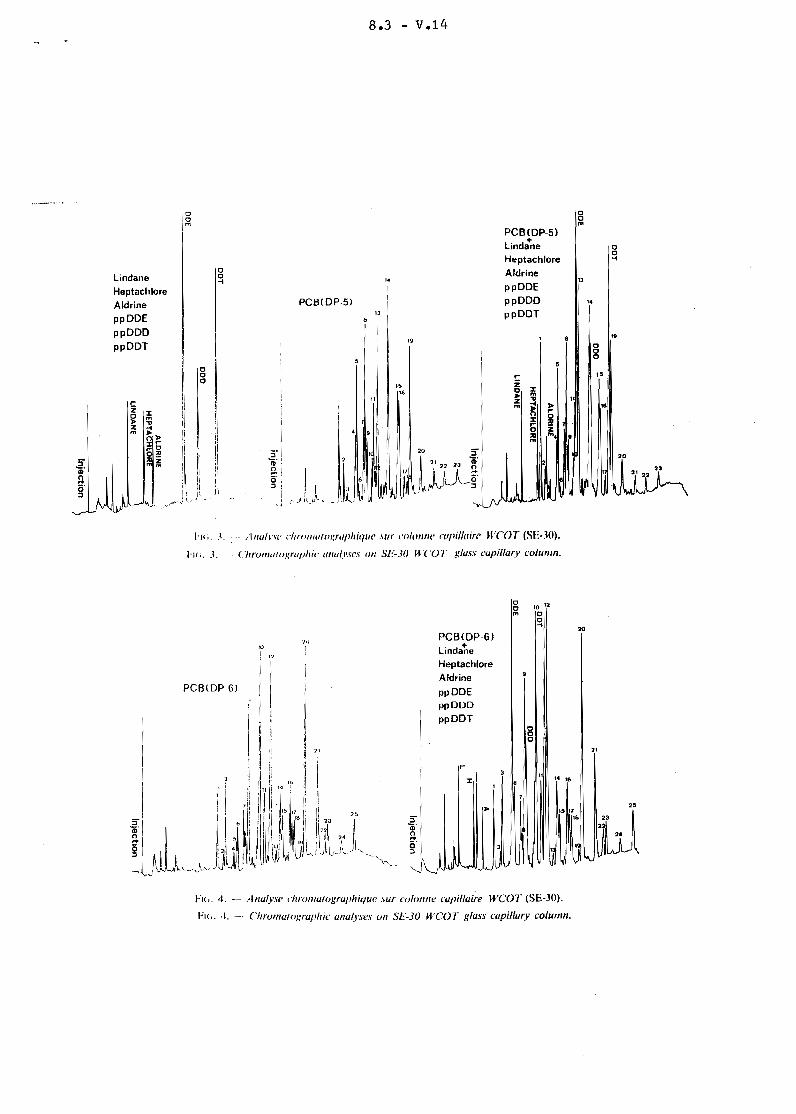
8.3 -V.14
Lindane
Heptach loreAldrineppDDE
ppDDD
ppDDT
PCBCDP-S)lJ
ï1 "
PCBCDP-S)Lindi'+neHeptachloreAldrine
ppDDEppDDDppDDT
a
8'"
lJ
1
ooo
,.
1
~w~LùJJ.D
,.Hl
r-i :r0,. mZ " 11
'" ~,.r-0 0:z: il!r-
a zil! "'
".."
5.dl
'n...ëï:)
l'Il;. 3. :- ..-llIull·\(' chrllllllllllgr0l'll/l/lIl' .'111' ,'{j/ll/IIII' l'II//il/aire WC()T (5E-.10).
"1(;. 3. ('hrulll,llllgrafJhic ([fI"lyses 011 SF-30 W( '01' glus.,' cupillary coluflln.
oom
10 12
o~
PCB\DP 6)
10
j U
PCBCDP6)Lindate
Heptachlore
Aldrine
ppDDEIJpDDDppDDT
Ir-xl, 3
~J DJ
go
" 14 18
21
Ft". 4. -- IInulyse chroIllU/iJgruphiqlli' sur ('(Jlolllle cupi/lai~e WCGT (SE.30).
Fil,. ·k - ChrulflullIgruphic aflulyses 011 S/::;·30 WCOT glass capi/lary column.

8.3 - V. 15
BIBLIOGRJI.PHIE
BERTHOU, F. 1981. Etat actuel des procédés de préparation des colonnes capillairesde verre. Spectra 2 000, .64 (9) et 65 (9), 17 pp.
CAPRAIS, J.Cl., MARCHAND M. 1981. Identification des organochlorés à haut poidsmoléculaire par chromatographie en phase gazeuse sur colonnes capillaires.Analusis, ~ (4), 140-144.
HOLDEN, A.V., K. MARSDEN. 1969. Single-stage clean-up of animal tissue extractsfor organochlorine residue analysis. J. Chromatogr., 44, 4RI-492.
RNO. 1977. Manuel des méthodes de prélèvements et d'analyses, tome ?- : Micropolluants organiques et minéraux. Ministère de la Qualité de la Vie - CNEXO, 19-52.

8.4. Annexe v,4
PROTOCOLE DE MINERALISATION DU SEDIMENTPOUR L'ANALYSE DES METAUX ADSORBES
(Fe, Mn, Cu, Zn, Cd, Pb, Co, Cr, Ni)
- 0,5 g à 1 g de fraction de sédiment < '3 ~m dans un creuset en silice ou dansun bécher:: .. pyrex" {)l.I ~"'/~...,. . , ) .- ,., .."",J,/" 1't"{i'-klllJ4't (./-2u 1110 d.I,I/" ,
- Ajouter 2 cc HCl (la qualité "pour analyse" est suffisante pour l'analyse desmétaux liés au sédiment). Si l'échantillon est de nature calcaire, ces 2 ccdoivent être ajoutés petit à petit (risques de bouillonnement et débordementde l'échantillon).
- Ajouter 4 cc HN0 3 (la qualité "pour analyse" est suffisante).
- Recouvrir le creuset d'un verre de montre.
- Chauffer.à !:: aO-90°C jusqu'à évaporation de HCl (= 1-2 heures) sur une plaquechauffante ';+., F~rmtion de vapeurs rousses.
- Porter la température à 120-130°C ; HN0 3 doit bouillonner doucement.
- Poursuivre ce chauffage jusqu'à la disparition totale ou presque totale desvapeurs rousses (2-3 heures selon la quantité de matière organique contenuepar le sédiment).
- Ajouter 3 cc HN0 3 •
- Chauffer à 120-130°C pendant!:: 1-2 heures.
- Enlever le verre de montre.
Evaporer à sec à 120-130°C (ceci peut constituer, selon la nature du sédiment,la phase la plus délicate de l'opération. En effet, au cours de l'évaporation,la solution de minéralisati'on s'épaissît, et peut former une pellicule à sasurface empêchant l'évaporation des vapeurs nitreuses sous-jacentes. Lorsquela pression de ces vapeurs augmente, des projections peuvent se produireentraînant des erreurs d'analyse par défaut. Ceci peut être évité en agitantmanuellement les creusets lors de la phase d'épaissement.
- Lorsque tout HN0 3 est évaporé, et que le creuset a refroidi: ajouter 1 cc HC1.Laisser s'imprégner le résidu de minéralisation (au besoin, aider avec unefine tige de verre).
- Ajouter 9 cc H;O distillée. Homogénéiser avec une fine tige de verre.
- Centrifuger dans des tubes coniques pendant 5-10 minutes à 3000-4000 tr/min.Selon la nature du sédiment, la filtration est possible.
- Surnageant limpide ~ Solution mère pour analyse directe ou pour dilutionsadéquates en fonction des métaux à analyser.
J.}... \of AR.'ti N'

8.5. Annexe V,5
PROPOSITION POUR LA SURVEILLANCE ËCOLOGIQUE
DE L'ICHTYOFAUNE DE LA LAGUNE EBRIË
La !~u~iillance écologique de l 'ichtyofaune d'un milieu peutêtre envisagée à trOis niveaux différents et complémentaires
Le peupl~mènt , on s'efforcera de mettre en évidence les modificationspouvant$qtvenir dans la composition, la structure et la biomasse despeuplements.
2°) Les populations, on évaluera les modific!1tjons,p.Ol./vantsurvenirdans............'--_-..,._ . .. -'." - -. .:;.,/t.~r.-i::~:·":"--L:.~~~:,-"~-:-~~-' - -. - -" .". .
la valeur des principaux paramètres biologigues caractéristiques decertaines populations (croissance, fécondité ... ).
Enfin on effectuera une surveillance physio-pathologique des individus(en particulier dosage des substances toxiques au sein des différentstissus et/ou organes de poissons appartenant aux principales espècescommercialisées) ..
Les trois points (mais en particulier les 2 premiers) nécessitent une connaissance approfondie des conditions "normales" de fonctionnement de l'écosystème.
aO) En ce qui concerne la composition et la structure des peuplementsichtyologiques, une étude synécologique actuellement en voie d'achèvement constitue une bonne base de référence. Elle montre en particulierque le milieu lagunaire Ebrié est, en ce qui concerne l'ichtyocoenose,d'une grande complexité de fonctionnement liée à une diversité et unevariabilité spatio-temporelle extrême et qu'il convient de distinguer.- très schématiquement - le secteur abidjanais sous forte influencemarine (grossièrement. de la digue de Jacqueville à l'extrémité est dePetit Bassam) du reste de la lagune.
Outre une bonne connaissance "initiale" du milieu il faut pour réaliserune bonne surveillance des peuplements. disposer d'un protocole adaptéfondé sur la réalisation de pêches expérimentales au moyen d'une technique peu sélective et d'une reproductibilité satisfaisante.

- 2 -
La senne tournante, qui a fourni l'essentiel des' données de l'étudede référence, parait bien adaptée. Cependant, son uti 1i sation s'es trévélée logistiquement fort lourde et une couverture lien routinelldel'ensemble du réseau lagunaire semble difficilement envisageable.Par contre, le choix de quelques stations représentatives semble possible. Il convient en particulier de suivre le secteur abidjanaisdont l'importance écologique et économique a déjà été soulignée(ALBARET 1980, projet UNESCO) de même que sa complexité liée àune très forte variabilité (crues, saisons marines, marées ... ).En conséquence seules pourront être appréhendées des modificationsdrastiques de la nature et/ou de la structure des peuplements liés àune péjoration grave des conditions de ce milieu soumisà de nombreusesagre~sions.
Nous disposons d'une bonne série de données relatives à la baie deCocody (suivie mensuellement, pendant 18 mois à raison de 5 stationspar moi s) qu' i 1 convient de prolonger au même rythme. De plus, 1a ba iede Biétri siège d'une pollution importante (et objet d'une attentiontoute particulière des chimistes, devrait être suivie au même rythmeainsi que le chenal central ou la région portuaire.
En ce qui concerne le reste de la lagune quelques stations environ·unedizaine pourraient être suivies bimestrie11ement.
bO) L'étude de 1lévo1ution de certains paramètres biologiques (de croissance et de reproduction notamment) peut dans URe certaine mesure,permettre d'évaluer l'impact des pollutions sur les populations ichtyalogiques. Cependant, et compte tenu de variations importantes (saisonnières et individuelles surtout), une connaissance très fine"de labiologie des espèces choisie s'avère nécessaire afin de pouvoir différencier ce qui relève de la variabilité roca1e, saisonnière ou individuelle de ce qui est (éventuellement) imputable à une dégradàtion desconditions de milieu. De plus, il est inconcevable d'isoler un quelconqueparamètre et clest en termes de stratégie que doit être posé le problème .
. ../ ...

l~'ik . -3 -
En effet/et par exemple/l'évaluation même très preclse de la féconditéunitaire individuelle ne sert de rien pour l'évaluation de la féconditéd'une population, si l'on ignore les modifications quant au nombre depontes annuelles, à la taille de première maturité ou encore au sexratio.
On aura donc soin de choisir quelques espèces dont certains aspects del'écologie et la biologie seront bien et précisément connues. A titred'exemple une étude fine de la reproduction des deux espèces de Tilapialagunaires est en cours, appréhendant les variations locales et saisonnières de fécondité,. la taille de première maturité (dans la nature eten élevage), les variations de sex-ratio, etc ...
• "t; . :..-,'C",' .c ..,~ ."
cOlle troisième volet concerne la recherche des zones d'accumulation etle dosage de substances toxiques (métaux lourds, pesticides ... ) au seindes organismes de poissons appartenant à des espèces se situant à
différents niveaux de la pyramide trophique (ou en bout de chatnestrophiques différentes).
Il semble le plus facile à réaliser pratiquement et fournir les résultatsles plus immédiatement utilisables.
En fonction de ce que l'on sait de la biologie et l'écologie (régimealimentaire, relations trophiques en particulier) on sélectionnera 4à 5 espèces sur lesquelles seront prélevés les organes (foie, gonades,muscles) destinés aux dosages.
1 Prédateur ichtyophageou Elops par exemple).
(Sphyraena ou Epinephalus
1 Malacophage (C~rysichthys ou Tylochrom~s).
1 Filtreur (Ethmalosa).
1 Mangeur benthique détritivore (Gerres).
J.J. ALBARET

Principe
Les principes généraux de cette méthode sont ceux décrits dans l'exposéde la colimétrie en milieux liquides. Cependant, alors que le tube primairecontient déjà une certaine quantité d'azide de sodium, le repiquage destubes « positifs» sur un milieu nettement plus inhibiteur (plus forte concentration en azide de sodium et présence d'éthyl violet), ne laisse se développer que les streptocoques fécaux.
Milieux de culture
- Milieu de Rothe normal.
Dans 1 litre d'eau distillée, dissoudre par chauffage doux
peptone. .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 20 gglucose '" . .. . . .. .. .. .. 5 gchlorure de sodium......... .. . . .. . .. .. . .. .. .. . . .. .. .. . 5 gphosphate dipotassique... .. ..... .. .. . .. .. ... .. . .. . .. .. .. 2,7 gphosphate monopotassique.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2,7 gazothydrate de sodium................................. 0,2 g
Ajuster le pH à 6,8-7. Répartir (l0 ml exactement) dans des tubes de 160x 16 mm.Stériliser 20 minutes à l'autoclave à 120"C.
- Milieu de Rothe double concentration.Procéder de la même manière, mais en doublant la masse des réactifs. Le pH doit êtrecompris entre 6,8 et 7. Répartir en tubes de 220x22mm (lOml) ou en tubes de330 x 33 mm (50 ml). Stériliser 20 minutes à l'autoclave à 1200c.Ces deux milieux doivent être mis à l'abri de toute évaporation; ne pas utiliser de bouchonde coton. Ils existent commercialement sous forme déshydratée ou prêts à l'emploi.
- Milieu de Litsky.
Dans 1 litre d'eau distillée, dissoudre par chauffaae doux :
peptone. •. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 20 'g
glucose , . . . . . . . . . . . . . . . . . . .. . . . . . . . . . . 5 gchlorure de sodium... . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5 gphosphate dipotassique , 2,7 gphosphate monopotassique... . . .. .. .. .. .. . .. . . 2,7 gazothydrate de sodium....... . . . . . . . . . . . . . . . . . . . . . . . . . 0, 3 gsolution d'éthyl violet. " 5 ml environ
Répartir (10 ml) dans des tubes de J60xI6mm. Stériliser à l'autoclave à 1200C.Existe commercialement sous forme déshydratée ou prêt à l'emploi. La teneur en éthylviolet doit être déterminée en préparant des milieux de teneur variable (3,5-4-4,5-55,5-6-6,5 mlfl) et en les ensemençant d'un Bacillus subtilis, d'un Streptococcus fecalisou d'un Streptococcus havis. La teneur à retenir est celle pour laquelle la culture de Bacil/usest inhibée, alors que les Streptocaccus sc développent.
Mode opératoire
Test présomptif
Ensemencer le nombre choisi de tubes de milieu de Rothe (constituantles tubes « primaires »), en utilisant soit du milieu à double concentration(pour les ensemencements de 10 ou 50 ml) soit du milieu à simple concen·tration (pour les ensemencements de 1 ml d'eau ou de dilution de l'eau).Veiller à ce qu'aucune évaporation ne se soit produite dans le milieu depuissa préparation, car celle-ci entraînerait une concentration des produitsinhibiteurs. Homogénéiser soigneusement, par agitation, le contenu destubes; s'assurer, une fois cel,le-ci terminée, que la teinte du bouillon estuniforme en haut et en bas dil tube, de façon à ce que la concentration eninhibiteur soit identique en tous points.Incuber les tubes à 37°C et l*s examiner après 24 et 48 heures. Les tubesprésentant un trouble micr~bien pendant cette période sont présuméscon.tenir un streptocoque féc\il et sont soumis au test confirmatif.
Test confirmatif
Après agitation des tubes positifs, prélever sur chacun d'eux successivement3 oses bouclées (de 3 mm de diamètre) ou quelques gouttes de pipettePasteur, et les reporter dans ties tubes de milieu de Litsky A l'éthyl violetet azide de sodium. Incuber tA 37°C pendant 24-48 heures.
L'apparition d'un trouble mitrobien confirme la présence d'un streptocoque fécal. Parfois la culture s'agglomère au fond du tube en fixant lecolorant et en formant une pastille violette de signification identique Acelle du trouble.Avec un milieu parfaitement préparé il n'y a pratiquement pas de faussesréactions. Éventuellement c0l1-trôler le diagnostic bactériologique par unsimple examen microscopique/; (direct ou après coloration de Gram) quidoit faire apparaitre la présence de Cocci à Gram positif, en courteschaînettes ou en diplocoques: et l'absence de bacilles A Gram positif,qui pourrait faire soupçonneria présence de Boeil/us, germes responsablesdes fausses réactions les plus fréquentes.
Expression des résultats. Choix des prises d'essai
Les résultats de dénombrement des streptocoques fécaux sont expriméscomme ceux d'Escherichia coli en nombre de germes par 100 ml.n est souhaitable de préciser « nombre le plus probable» avant le nombrereprésentant le résultat et d'indiquer à la suite les résultats extrêmes dansla limite de confiance de 95 % donnée par les tables.Le même type d'ensemencement utilisé pour E. coli peut être employé,pour un même type d'eau, pour les streptocoques fécaux.
00•0\•P>:;j:;j(Il~(Il
<Z ..~
0\
t'1
~~H0Z
0t'1CIl
CIlt'1 ~Z :::a
t'1~ "tlH ~l":' 0H C)t'1 0c::.ot"' ~H CIl
'8t'1H ~0t'1 0
t'1CIl
CIl~
~"tl~0C)0.0c::t'1CIl
l'%:lt'1
&;~