métabolisme d'antidépresseurs dans différentes …bib_r_7392.p001/ref.pdf · dont le but a...
TRANSCRIPT

Métabolisme d'antidépresseurs dans différentes espèces: aspects méthodologiques et pharmacogénétiques, perspectives cliniques
Thèse de doctorat
présentée à la
Faculté de Biologie et de Médecine de l’Université de Lausanne
par
Erik Paus
Pharmacien diplômé de la Confédération Helvétique
Jury
Prof. Luc Tappy, Président du jury Prof. Pierre Baumann, Directeur de thèse
Dr. Isabelle Seif, Experte Prof. Jean-Pierre Hornung, Expert
Dr. Bertrand Rochat, Expert
Lausanne 2007

- 2 -

- 3 -
A mes parents, dont le but a toujours été le bonheur de leurs enfants.

- 4 -
Métabolisme d'antidépresseurs dans différentes espèces: aspects méthodologiques et pharmacogénétiques, perspectives cliniques
Erik Paus Unité de Biochimie et Psychopharmacologie Clinique, Centre de neurosciences Psychiatrique, Département de Psychiatrie Adulte, Faculté de Biologie et de Médecine, Université de Lausanne Lors de la prise d’un médicament, celui-ci va passer par différentes étapes que sont l’absorption, la distribution, le métabolisme et enfin l’élimination. Ces quatre étapes sont regroupées sous le nom de pharmacocinétique. A noter que ces quatre paramètres sont dynamiques et en constante évolution. Durant cette thèse, nous avons investigué différents aspects de la pharmacocinétique, tout d’abord par une revue de la littérature sur la glycoprotéine-P (Pgp). Récemment découverte, cette protéine de membrane est située aux endroits stratégiques de l’organisme comme la barrière hémato-encéphalée, le placenta ou les intestins où elle influencera l’entrée de différentes substances, en particulier les médicaments. La Pgp serait impliquée dans les phénomènes de résistances aux agents thérapeutiques en oncologie. La Pgp influence donc l’absorption des médicaments, et son impact en clinique, en termes d’efficacité de traitement et de toxicité prend chaque jour plus d’importance. Ensuite nous avons mis au point une méthode d’analyse quantitative d’un antidépresseur d’une nouvelle génération : la mirtazapine (Remeron®). La nouveauté réside dans la façon dont la mirtazapine interagit avec les neurotransmetteurs impliqués dans la dépression que sont la sérotonine et la noradrénaline. Cette méthode utilise la chromatographie liquide pour séparer la mirtazapine de ses principaux métabolites dans le sang. La spectrométrie de masse est utilisée pour les détecter et les quantifier. Les métabolites sont des substances issues de réactions chimiques entre la substance mère, la mirtazapine, et généralement des enzymes hépatiques, dans le but de rendre cette substance plus soluble en vue de son élimination. Cette méthode permet de quantifier la mirtazapine et ses métabolites dans le sang de patients traités et de déterminer la variation des taux plasmatiques chez ces patients. Puis nous avons étudié le métabolisme d’un autre antidépresseur, le citalopram, qui a un métabolisme complexe. Le citalopram est un racémate, c’est-à-dire qu’il existe sous forme de deux entités chimiques (R-(-) et S-(+) citalopram) qui ont le même nombre d’éléments mais arrangés différemment dans l’espace. La voie métabolique cérébrale du citalopram est sous le contrôle d’une enzyme, la monoamine oxydase (MAO), conduisant à une forme acide du citalopram (l’acide propionique du citalopram). La MAO existe sous deux formes : MAO-A et MAO-B. Nous avons utilisé des souris déficientes d’un gène, celui de la MAO-A, pour mieux en comprendre le métabolisme en les comparant à des souris sauvages (sans déficience de ce gène). Nous avons utilisé le citalopram et deux de ses métabolites (le déméthylcitalopram et le didéméthylcitalopram) comme substrats pour tester la formation in vitro de l’acide propionique du citalopram. Nos résultats montrent que la MAO-A favorise la formation de l’entité R-(-) et présente une plus grande affinité pour le citalopram, tandis que la MAO-B métabolise préférentiellement l’entité S-(+) et a une plus grande affinité pour les deux métabolites déméthylés. De plus, la déficience en MAO-A est partiellement compensée par la MAO-B chez les souris déficientes du gène de la MAO-A.
Résumé pour large public

- 5 -
Enfin, nous avons étudié une deuxième voie métabolique du citalopram qui s’est avérée toxique chez le chien Beagle. Celle-ci est catalysée par une autre famille d’enzymes, les cytochromes P-450, et mène aux métabolites déméthylés et didéméthylés du citalopram. Nous avons utilisé des tissus hépatiques de chiens Beagle. Plusieurs cytochromes P-450 sont impliqués dans le métabolisme du citalopram menant à sa forme déméthylée, ceci tant chez l’homme que chez le chien. Par contre, dans le métabolisme de la forme déméthylée menant à la forme didéméthylée, un seul cytochrome P-450 serait impliqué chez l’Homme, tandis qu’ils seraient plusieurs chez le chien. L’activité enzymatique produisant la forme didéméthylée est beaucoup plus importante chez le chien comparé à l’homme. Cette observation soutien l’hypothèse que des taux élevés de la forme didéméthylée participent à la toxicité spécifique du citalopram chez le chien. Nous pouvons conclure que plusieurs famille d’enzymes sont impliquées tant au niveau cérébral qu’hépatique dans la métabolisation de médicaments psychotropes. Sachant que les enzymes peuvent être stimulées ou inhibées, il importe de pouvoir suivre au plus près les taux plasmatiques des différents psychotropes et de leurs métabolites.

- 6 -
Métabolisme d'antidépresseurs dans différentes espèces: aspects méthodologiques et pharmacogénétiques, perspectives cliniques
Erik Paus Unité de Biochimie et Psychopharmacologie Clinique, Centre de neurosciences Psychiatrique, Département de Psychiatrie Adulte, Faculté de Biologie et de Médecine, Université de Lausanne La plupart des médicaments subissent une transformation enzymatique dans l’organisme. Les substances issues de cette métabolisation ne sont pas toujours dotées d’une activité pharmacologique. Il s’est avéré par conséquent indispensable de suivre les taux plasmatiques d’une substance et de ses métabolites et d’établir ou non l’existence d’une relation avec l’effet clinique observé. Ce concept nommé « therapeutic drug monitoring » (TDM) est particulièrement utile en psychiatrie ou un manque de compliance des patients est fréquemment observé. Les médicaments psychotropes ont un métabolisme principalement hépatique (cytochromes P-450) et parfois cérébral (monoamines oxydases), comme pour le citalopram par exemple. Une méthode stéréosélective de chromatographie liquide couplée à la spectrométrie de masse a été développée pour analyser les énantiomères R-(-) et S-(+) d’un antidépresseur agissant sur les récepteurs noradrénergiques et sérotoninergiques, la mirtazapine et de ses métabolites déméthylmirtazapine et 8-hydroxymirtazapine. Les données préliminaires obtenues dans les plasmas dosés suggèrent que les concentrations de R-(-)-mirtazapine sont plus élevées que celles de S-(+)-mirtazapine, à l’exception des patients qui auraient comme co-médication des inhibiteurs du CYP2D6, telle que la fluoxétine ou la thioridazine. Il y a une enantiosélectivité du métabolisme de la mirtazapine. En particulier pour la 8-hydroxymirtazapine qui est glucuroconjuguée et pour laquelle le ratio S/R varie considérablement. Cette méthode analytique présente l’avantage d’être utilisable pour le dosage stéréosélectif de la mirtazapine et de ses métabolites dans le plasma de patients ayant d’autres substances en co-médication. La glycoprotéine P fonctionne comme une pompe transmembranaire transportant les xénobiotiques depuis le milieu intracellulaire vers le milieu extracellulaire. Son induction et son inhibition, bien que moins étudiées que pour les cytochromes P-450, ont des implications cliniques importantes en termes d’efficacité de traitement et de toxicité. Cette glycoprotéine P a fait l’objet d’une recherche bibliographique. Nous avons étudié le métabolisme du citalopram, un antidépresseur de la classe des inhibiteurs spécifiques de la recapture de la sérotonine chez la souris et chez le chien. Cette substance subit un métabolisme complexe. La voie de métabolisation conduisant à la formation de l’acide propionique du citalopram, catalysée par les monoamines oxydases, a été étudiée in vitro dans les mitochondries cérébrales chez la souris déficiente du gène de la MAO-A (Tg8). La monoamine oxydase A catalyse la formation de l’énantiomère R-(-) et présente une plus grande affinité pour les amines tertiaires, tandis que la monoamine oxydase B favorise la formation de la forme S-(+) et a une affinité plus marquée pour les amines secondaires et primaires. L’étude du citalopram chez la souris Tg8 adulte a montré que la monoamine oxydase B compense la déficience de la monoamine oxydase A chez ces souris génétiquement modifiées.
Résumé

- 7 -
Une autre voie de métabolisation du citalopram conduisant à la formation de didéméthylcitalopram, catalysée par les cytochromes P-450, a été étudiée in vitro dans des microsomes hépatiques de chiens Beagle. Nos études ont montré que les cinétiques de N-déméthylation du citalopram sont biphasiques chez le chien. Les orthologues canins impliqués dans la première N-déméthylation semblent être identiques aux cytochromes P-450 humains. Par contre, dans la deuxième N-déméthylation, un seul cytochrome P-450 semble être impliqué chez l’homme (CYP2D6), tandis qu’on retrouve jusqu’à cinq orthologues chez le chien. Le CYP2D15, orthologue canin du CYP2D6, est majoritairement impliqué. De plus, l’activité enzymatique, reflétée par les clairances intrinsèques, dans la première N-déméthylation est jusqu’à 45 fois plus élevée chez le chien comparé à l’homme. Ces différentes observations soutiennent l’hypothèse que des taux élevés de didéméthylcitalopram sont responsables de la toxicité du citalopram chez le chien. Nous pouvons conclure que plusieurs famille d’enzymes sont impliquées tant au niveau cérébral qu’hépatique dans la métabolisation de médicaments psychotropes. Sachant que les enzymes peuvent être induits ou inhibés, il importe de pouvoir suivre au plus près les taux plasmatiques des différents psychotropes et de leurs métabolites.

- 8 -
Antidepressants metabolism in different species: methodological and pharmacogenetics aspects, clinical perspectives
Erik Paus Unité de Biochimie et Psychopharmacologie Clinique, Centre de neurosciences Psychiatrique, Département de Psychiatrie Adulte, Faculté de Biologie et de Médecine, Université de Lausanne Most of the drugs are metabolized in the organism. Substances issued from this metabolic activity do not always show a pharmacological activity. Therefore, it is necessary to monitor plasmatic levels of drugs and their metabolites, and establish the relationship with the clinical effect. This concept named therapeutic drug monitoring is very useful in psychiatry where lack of compliance is commonly observed. Antidepressants are mainly metabolized in the liver (cytochrome P-450) and sometimes in the brain (monoamine oxidase) like the citalopram, for exemple. A LC-MS method was developed, which allows the simultaneous analysis of R-(-) and S-(+) enantiomers of mirtazapine, an antidepressant acting specifically on noradrenergic and serotonergic receptors, and its metabolites demethylmirtazapine and 8-hydroxymirtazapine in plasma of mirtazapine treated patients. Preliminary data obtained suggested that R-(-) mirtazapine concentrations were higher than those of S-(+) mirtazapine, except in patients comedicated with CYP2D6 inhibitors such as fluoxetine or thioridazine. There is an enantioselectivity in the metabolism of mirtazapine. In particular for the 8-hydroxymirtazapine, which is glucuroconjugated and S/R ratio varies considerably. Therefore this method seems to be suitable for the stereoselective assay of mirtazapine and its metabolites in plasma of patients comedicated with mirtazapine and other drugs for routine and research purposes. P-glycoprotein is working as an efflux transporter of xenobiotics from intracellular to extracellular environment. Its induction or inhibition, although less studied than cytochrome P-450, has huge clinical implications in terms of treatment efficacy and toxicity. An extensive literature search on P-glycoprotein was performed as part of this thesis. The study of citalopram metabolism, an antidepressant belonging to the class of selective serotonin reuptake inhibitors. This substance undergoes a complex metabolism. First metabolization route leading to citalopram propionic acid, catalyzed by monoamine oxidase was studied in vitro in mice brain mitochondria. Monoamine oxidase A catalyzed the formation of R-(-) enantiomer and showed greater affinity for tertiary amines, whereas monoamine oxidase B triggered the formation of S-(+) enantiomer and demonstrated higher affinity for primary and secondary amines. Citalopram evaluation in adult Tg8 mice showed that monoamine oxidase B compensated monoamine oxidase A deficiency in those genetically transformed mice. The second metabolization route of citalopram leading to didemethylcitalopram and catalyzed by cytochrome P-450 was studied in vitro in Beagle dog’s livers. Our results showed that citalopram N-demethylation kinetics are biphasic in dogs. Canine orthologs involved in the first N-demethylation seemed to be identical to human cytochromes P-450. However, in the second N-demethylation only one cytochrome P-450 seemed to be involved in human (CYP2D6), whereas up to five canine orthologs were found in dogs. CYP2D15 canine ortholog of CYP2D6 was mainly involved. In addition, enzymatic activity reflected by intrinsic clearance in the first N-demethylation was up to 45 fold higher in dogs compared to humans. Those observations support the assumption that elevated rates of didemethylcitalopram are responsible for citalopram toxicity in dogs. We can conclude that several enzymes groups are involved in the brain, as well as in the liver, in antidepressant metabolization. Knowing that enzymes may be induced or inhibited, it makes sense to closely monitor plasmatic levels of antidepressants and their metabolites.
Summary

- 9 -
ABRÉVIATIONS 11
INTRODUCTION 12
MÉTABOLISME DES MÉDICAMENTS 12
Situation et place des médicaments psychiatriques dans la thérapeutique psychiatrique 12
Cinétique (Absorption, Distribution, Métabolisme, Élimination) 12
Enzymes impliqués dans le métabolisme 12
Polymorphisme génétique de certains enzymes 12
UDP-glucuronosyltransferase (UGT1A1), syndromes de Gilbert et de Crigler-Najjar 13
Localisation des enzymes cérébraux 13
Formation de métabolites cérébraux (toxiques ou non) 14
Rôle des métabolites dans la cinétique des psychotropes 14
Nécessité du suivi des taux plasmatiques des médicaments 14
ANTIDÉPRESSEURS 15
Mécanismes d’action des antidépresseurs 15
Médicaments racémiques 16
Mirtazapine et ses métabolites 17
Citalopram et ses métabolites 18
Toxicité du citalopram chez le chien 19
MONOAMINE OXYDASE (MAO) 20
Biochimie de la MAO. 20
Génétique de la MAO : absence de MAO-A chez une famille hollandaise 21
Intérêt de la recherche pour des modèles animaux n’exprimant pas le gène de la MAO-A ou de la MAO-B 21
ALDÉHYDE OXYDASE 22
Biochimie de l’aldéhyde oxydase 22
CYTOCHROMES P-450 22
Biochimie des cytochromes P-450 22
Polymorphisme des cytochromes P-450, notamment du CYP2D6 impliqué dans le métabolisme des psychotropes 23
Métabolisme : différences entre cytochromes P-450 humains et cytochromes canins (orthologues) 23
SITUATION ET BUTS DE LA THÈSE 25
Parcours et problèmes rencontrés lors de l’étude du métabolisme du citalopram 25
Méthodes analytiques 26
Développement d’une méthode stéréosélective pour l’analyse de la mirtazapine et de ses métabolites 26
Apport d’un travail supplémentaire dans la distribution des médicaments 26
MÉTABOLISME DU CITALOPRAM PAR LA MAO CHEZ LA SOURIS (WILD-TYPE ET KNOCK-OUT MAO-A) 27
RÉSUMÉ 27
MATÉRIEL & MÉTHODES 27
Substances et produits chimiques 27
Animaux 27
Préparation des tissus 28
Incubation 28
Extraction 28
Conditions analytiques 29
Statistiques 29
RÉSULTATS 29
DISCUSSION 34
MÉTABOLISME DU CITALOPRAM PAR LES CYTOCHROMES P-450 CHEZ LE CHIEN 37
RÉSUMÉ 37
MATÉRIEL & MÉTHODES 37
Substances et produits chimique 37
Tissus et incubation 37
Extraction 38
Conditions analytiques 38
Statistiques 38
RÉSULTATS 39
N-déméthylation du citalopram et inhibition de la formation du déméthylcitalopram 39
N-déméthylation du déméthylcitalopram et inhibition de la formation du didéméthylcitalopram 42
DISCUSSION 44
A. N-déméthylation du citalopram en déméthylcitalopram 44

- 10 -
B. N-déméthylation du déméthylcitalopram en didéméthylcitalopram 46
C. Hypothèses sur la toxicité du citalopram chez le chien 47
MISE AU POINT D’UNE MÉTHODE STÉRÉOSÉLECTIVE PAR LC-MS POUR L’ANALYSE DE LA MIRTAZAPINE ET SES APPLICATIONS 48
RÉSUMÉ 48
MATÉRIEL & MÉTHODES 48
Substances et produits chimiques 48
Extraction 48
Conditions analytiques 49
RÉSULTATS & DISCUSSION 49
Données statistiques de la méthode 50
Dosage d’échantillons plasmatiques de patients sous mirtazapine 52
GLYCOPROTÉINE-P (PGP) ET GÈNE (MDR1) : IMPLICATIONS DANS LA DISTRIBUTION DES MÉDICAMENTS 57
INTRODUCTION 57
ÉTAT ACTUEL DES CONNAISSANCES 57
Expression, fonctions et localisation de la glycoprotéine-P 57
Substrats 58
Importance de la glycoprotéine-P dans la distribution des médicaments 60
Polymorphisme du gène MDR1 60
Polymorphisme du gène MDR1 et maladie 60
PERSPECTIVES 61
CONCLUSIONS 62
RÉFÉRENCES 64

- 11 -
Abréviations 5-HT 5-hydroxytryptamine ou sérotonine ADP adénosine diphosphate ANOVA analyse de variance AO aldéhyde oxydase ATP adénosine triphosphate CIT citalopram CIT-PROP acide propionique du citalopram CYP cytochromes P-450 DCIT déméthylcitalopram DDCIT didéméthylcitalopram DMSO diméthylsulfoxyde EM métaboliseurs exhaustifs (extensive metabolizers) Fluo fluorescence CLHP (hplc) chromatographie liquide à haute pression/performance
(high pressure/performance liquid chromatography) IM métaboliseurs intermédiaires (intermediate metabolizers) IMAO inhibiteur de la monoamine oxydase ISRS (ssri) inhibiteur sélectif de la recapture de la sérotonine
(selective serotonin reuptake inhibitor) Km constante de Michaelis-Menten. Est égale à la concentration du substrat lorsque
V = ½Vmax. Le Km caractérise l’affinité d’un enzyme pour un substrat donné. LC-MS chromatographie liquide couplée à la spectrométrie de masse LOD limite de détection LOQ limite de quantification MAO monoamine oxydase MDR1 gène de résistance multiple aux principes actifs (multiple drug resistance) MPP+ ion méthyl-phényl-tétrahydro-pyridinium MPPP 1-méthyl-4phénl-4propionoxy-pipéridine MPTP 1-méthyl-4-phényl-1,2,3,6 tétrahydropyridine m/z rapport masse sur charge utilisé en spectrométrie de masse NADP+ nicotinamine adénine dinucléotide phosphate (oxydée) NADPH nicotinamine adénine dinucléotide phosphate (réduite) NaSSA antidépresseur sérotoninergique spécifique et noradrénergique PBMC cellules mononucléaire du sang périphérique
(peripheral blood mononuclear cell) Pi phosphate inorganique P-gp, Pgp glycoprotéine-P PM métaboliseurs lents (poor metabolizers) SNC système nerveux central SNP polymorphisme d’un seul nucléotide TCA antidépresseurs tricycliques TDM suivi des taux (généralement plasmatiques) des médicaments (therapeutic drug
monitoring) V taux de production initial (vélocité) Vmax taux de production maximum VIH (hiv) virus de l’immunodéficience humaine (human immuno-deficiency virus)

- 12 -
Introduction
Métabolisme des médicaments
Situation et place des médicaments psychiatriques dans la thérapeutique psychiatrique Les médicaments utilisés aujourd’hui en psychiatrie (psychotropes) sont classés en plusieurs familles. Parmi celles-ci, figurent entre autres les anxiolytiques, les stabilisateurs de l’humeur, les neuroleptiques et les antidépresseurs. La dépression est aujourd’hui l’un des troubles affectifs les plus courants (Angst, 1992; Kessler et al., 2003; Rapaport, 2001). La sévérité de ces symptômes peut varier d’une baisse de l’humeur (saisonnière ou non) à une dépression comprenant psychoses et hallucinations. Au niveau mondial, la dépression est la première cause d’incapacité et de mort prématurée. De plus, 37% des patients atteints de dépression songeront à mettre fin à leurs jours durant leur maladie et 17% passeront à l’acte (Vuoriletho et al., 2006). Pour avoir un effet, les psychotropes doivent atteindre leur cible dans le corps humain : le cerveau. Pour ce faire, plusieurs étapes sont nécessaires décrites par la cinétique.
Cinétique (Absorption, Distribution, Métabolisme, Élimination) La pharmacocinétique décrit les différentes étapes (absorption, distribution, métabolisation et élimination) que traversent tous les médicaments. Une fois absorbée, la substance active va rejoindre la circulation sanguine, où elle existe sous deux formes : une forme liée aux protéines plasmatiques et une forme libre. Seule la forme libre pourra passer entre les différentes membranes pour atteindre et se lier à son récepteur et exercer ses effets. Le passage entre ces « compartiments » dépend de la perméabilité des barrières épithéliales. Une barrière très importante, la barrière hémato-encéphalique est formée par les cellules endothéliales des capillaires cérébraux. L’altération de l’absorption et de la distribution peuvent avoir des conséquences non négligeables sur le devenir du médicament et ses effets. Ce sont principalement le métabolisme et les troubles de celui-ci qui sont le plus souvent incriminés lors de la perturbation des paramètres cinétiques.
Enzymes impliqués dans le métabolisme Si le métabolisme est l'ensemble des transformations moléculaires et des transferts d'énergie qui se déroulent dans toutes cellules, l’organe principal responsable des réactions métaboliques est le foie. Il est le site principal du métabolisme de toutes les substances endogènes et exogènes, même si d’autres organes peuvent avoir une activité métabolique (p.ex. le système digestif, les poumons, les reins, ainsi que le cerveau). Il existe principalement deux types de réactions :
Les réactions de phase I peuvent engendrer autant de métabolites actifs qu’inactifs et sont principalement des réactions d’oxydoréduction et d’hydrolyse.
Les réactions de phase II qui sont principalement des réactions de glucuro- et de sulfo-conjugaison donnent des métabolites beaucoup plus polaires et nettement moins réactifs, ce qui facilite d’autant leur élimination. Parmi les systèmes enzymatiques impliqués, les cytochromes P-450 (CYP) sont une superfamille d’enzymes, dont on connait aujourd’hui plus de 300 isozymes, importants dans la biosynthèse et la dégradation de composés endogènes comme les stéroïdes, les lipides et les vitamines. Ils métabolisent de nombreuses substances présentes dans l’environnement, dans notre alimentation et dans les médicaments (Nebert & Russell, 2002) (CF page 12).
Polymorphisme génétique de certains enzymes La réponse à un traitement médicamenteux varie d’un patient à l’autre. Après ingestion d’une dose identique d’une substance donnée, certains patients subiront des effets secondaires significatifs, tandis que d’autres patients n’auront pas encore atteint le seuil thérapeutique. Cette différence de réponse clinique est liée aux taux sanguin du médicament. Celle-ci est principalement influencée par deux composantes : la première est génétique, sous forme d’une mutation héritée (trait de caractère génétique) qui sera présente durant toute la vie. La deuxième est environnementale, influencée par des facteurs

- 13 -
exogènes (nourriture, médicaments, etc.) qui changent perpétuellement. Si, par une mutation, on observe un deuxième phénotype avec une fréquence d’au moins 1% dans une population, on parle alors d’un polymorphisme génétique (Meyer & Zanger, 1997). Les porteurs homozygotes de 2 gènes du CYP2D6 codant pour les enzymes fonctionnels sont classés comme EM (extensive metabolizers) ou métaboliseurs rapides, tout comme les porteurs d’une duplication allèlique et d’un allèle déficient. Les porteurs hétérozygotes d’un seul allèle actif sont classés comme IM (intermediate metabolizers) ou métaboliseurs intermédiaires et les porteurs homozygotes de deux allèles déficients sont classés comme métaboliseurs lents (PM poor metabolizers) ou (Kirchheiner et al., 2004). Certaines de ces différences peuvent être mises en relation avec les taux d’enzymes exprimés, en particulier pour la famille des cytochromes P-450, dont une dizaine d’isoformes sont responsables du métabolisme oxydatif de la plupart des substances médicamenteuses. Le génotypage et le phénotypage sont deux techniques utilisées pour connaître l’existence ou non d’un polymorphisme. Le génotypage recherche la séquence génétique des allèles du gène exprimant l’enzyme incriminée. Ce résultat est définitif et n’évoluera pas au cours du temps. Il n’est pas non plus influencé par des facteurs environnementaux, tels que la nourriture ou des médicaments. Le phénotypage va tester la réponse de l’enzyme à des substances tests spécifiques. Cela donne une information sur la capacité métabolique de l’enzyme concerné. Si l’enzyme est inductible, cette information peut évoluer avec le temps, car elle est dépendante à la fois de facteurs environnementaux et génétiques (Andersson et al., 2005).
UDP-glucuronosyltransferase (UGT1A1), syndromes de Gilbert et de Crigler-Najjar
La bilirubine, qui est insoluble dans l’eau, est un produit de dégradation de l’hème. La glucuronidation hépatique de la bilirubine insoluble est catalysée par l’isoenzyme 1A1 de l’UDP-glucuronosyltransférase (UGT1A1), qui est indispensable pour une excrétion efficace de la bilirubine (Sampietro & Iolascon, 1999). Les mutations du gène UGT1A1 sont responsables des syndromes de Gilbert et de Crigler-Najjar. La classification de ces syndromes sont basées sur les taux de bilirubine (Hirschfield & Alexander, 2006). Un taux normal de bilirubine est généralement inférieur à 17 µmol/l. Le syndrome de Crigler-Najjar de type I est caractérisé par une absence quasi complète de l’activité enzymatique de l’UGT1A1. Les taux de bilirubine plasmatique sont entre 340 et 685 µmol/l voir plus hauts. Dans le type II, l’activité enzymatique est sévèrement réduite avec des taux de bilirubine compris entre 100 et 340 µmol/l. L’activité enzymatique peut être induite par le phénobarbital dans le type II, mais pas dans le type I. Enfin, une hyperbilirubinémie modérée, inférieur à 50 µmol/l est souvent associée avec le syndrome de Gilbert et reflète une réduction de l’activité enzymatique de l’UGT1A1 d’environ 30% (Sampietro & Iolascon, 1999).
Localisation des enzymes cérébraux S’il est entendu que le foie représente le principal organe quant à la métabolisation des différentes substances endogènes ou exogènes (xénobiotiques) (Wilkinson, 2005), de nombreux enzymes de phase I et de phase II sont également présents dans d’autres organes y compris le cerveau. Ce sont les cytochromes P-450, les monoamines oxydases A et B et les glucuronyltransférases, qui ont été identifiés dans le système nerveux central. S’il a été démontré que les cytochromes P-450 cérébraux sont en faible quantité, et que la présence de certaines isoformes est douteuse. Les cytochromes P-450 cérébraux sont inductibles et les inducteurs endogène et exogènes agissent différemment dans l’expression des cytochromes P-450 tant au niveau hépatique que cérébral. Certains cytochromes P-450 sont exprimés de façon régulière dans le foie, alors qu’il sont inductible dans le cerveau (Miksys & Tyndale, 2004). De plus, les cytochromes P-450 cérébraux ne sont pas distribués de manière uniforme dans les différentes régions du cerveau. Ils sont surtout localisés dans certains neurones et éléments de la glie ainsi que dans les cellules endothéliales de la barrière hémato-encéphalée. On estime la concentration cérébrale des cytochromes P-450 à environ 5% comparée à celle mesurée dans le foie (Ghersi-Egea et al., 1993; Ghersi-Egea et al., 1994; Ravindranath et al., 1989). Récemment, une étude a montré la possible implication du CYP2E1 cérébral chez la souris dans le métabolisme toxique dans la maladie de Parkinson et la toxicité du MPTP (Cf. infra) (Viaggi et al., 2006). D’autres études seront nécessaires pour savoir si ces résultats sont transposables à l’Homme.

- 14 -
La distribution cérébrale des deux formes de la monoamine oxydase (MAO) montre que la MAO-A se retrouve principalement dans les neurones noradrénergiques tandis que la MAO-B se retrouve dans les neurones sérotoninergiques, les neurones histaminergiques ainsi que dans les astrocytes (Shih et al., 1999). Quelle que soit leur localisation ou leur concentration, tous ces enzymes vont former des métabolites. Il peut arriver que certains de ces métabolites soient à l’origine d’une neurotoxicité avérée, comme par exemple le MPP+ (Cf. infra).
Formation de métabolites cérébraux (toxiques ou non) Il existe peu d’études sur le métabolisme cérébral comparé au métabolisme hépatique. Néanmoins, une substance chimique, le 1-méthyl-4-phényl-1,2,3,6 tétrahydropyridine (MPTP) a été intensivement étudiée à cause de la capacité de son métabolite, le méthyl-phényl-tétrahydro-pyridinium ion (MPP+) à induire des anomalies neurologiques identiques à celles observées chez un patient atteint par la maladie de Parkinson. Ces anomalies ont été rapportées en 1982 suite à l’injection accidentelle par des patients toxicomanes d’une « héroïne de synthèse ». Ceux-ci avaient mis au point un analogue de synthèse de la mépéridine1, le MPPP (1-méthyl-4phénl-4propionoxy-pipéridine). Un des métabolites alors formé est le MPTP (Langston et al., 1983). Celui-ci est désaminé dans les cellules gliales par la MAO-B en MPP+. Ce métabolite est alors recapté par les terminaisons nerveuses dopaminergiques. Une fois dans le neurone dopaminergique, le MPP+ va agir de la même manière que le 6-hydroxydopamine en générant des peroxydes d’hydrogène et des radicaux libres. Ceux-ci vont interférer avec la respiration mitochondriale et aboutir à la destruction de ces neurones (Ebadi et al., 2002).
Rôle des métabolites dans la cinétique des psychotropes La plupart des médicaments ont des métabolites. Peu d’entres eux ont, par contre, une activité pharmacologique. Prenons l’exemple des inhibiteurs spécifiques de la recapture de la sérotonine (ISRS) : le citalopram, la fluoxétine, la fluvoxamine, la paroxétine et la sertraline. Parmi ceux-ci, le citalopram et la fluoxétine produisent des métabolites dont l’activité est cliniquement significative. La norfluoxétine est un métabolite ayant une importance en clinique par son potentiel d’inhibition du CYP2D6 et du CYP3A4. Elle a également une demi-vie de plus d’une semaine, ce qui augmente les risques d’interactions pharmacocinétiques si, un traitement avec un médicament substrat de l’un de ces deux cytochromes P-450 est initié (Baumann & Rochat, 1995). Les cytochromes P-450 sont sujets à des polymorphismes et de nombreux médicaments sont des substrats de ceux-ci, en particulier du CYP2D6 pour les psychotropes (Wilkinson, 2005). La possibilité de pouvoir suivre les taux plasmatiques d’une substance et de ses métabolites prend dès lors toute son importance. Ce concept se nomme le suivi des taux plasmatiques d’un médicament (en anglais TDM therapeutic drug monitoring).
Nécessité du suivi des taux plasmatiques des médicaments Le TDM a été introduit suite à la démonstration d’une relation entre le taux plasmatique d’une substance et/ou d’un de ses métabolites, et l’effet clinique observé. Par exemple, une telle relation directe entre les taux plasmatiques et la réponse clinique a été étudiée pour la nortriptyline (Nortrilen®), un antidépresseur tricyclique (Asberg et al., 1971). Par contre, si l’on reprend le cas des inhibiteurs sélectifs de la recapture de la sérotonine, aucun de ceux-ci n’a une relation clairement définie entre le taux plasmatique et l’efficacité clinique (Baumann, 1996). Le TDM garde néanmoins toute sa pertinence dans le cadre d’un manque de compliance de la part du patient (en particulier en psychiatrie) ou en cas de métabolisme altéré (patients âgés, métaboliseurs lents, insuffisants rénaux ou hépatiques, etc.) (Baumann et al., 2004; Baumann et al., 2005).
1 La mépéridine (médicament hors commerce en Suisse, Démérol® au Canada) est un opiacé de faible puissance.

- 15 -
Antidépresseurs
Mécanismes d’action des antidépresseurs L’étiologie biochimique de la dépression est inconnue. Une théorie a tenté de l’expliquer en se basant sur l’observation de l’effet des antidépresseurs qui augmentent la concentration synaptique de noradrénaline et/ou de sérotonine (Figure 1). La réserpine, un alcaloïde contenu dans une plante d’origine indienne (Rauwolfia serpentina L.) a été utilisée comme antihypertenseur. Suite à son utilisation, il a été observé que cette substance diminuait le taux de catécholamines et de sérotonine et pouvait être la cause de symptômes dépressifs (Schildkraut, 1965). Le postulat de cette théorie, la théorie des monoamines, explique que la dépression serait due à une déficience de l’une ou l’autre des trois amines biogènes que sont la sérotonine, la noradrénaline et la dopamine. Cette théorie ne parvient cependant pas à expliquer pourquoi les antidépresseurs agissent sur le système des monoamines seulement après un certain délai. Il a été montré que l’augmentation de la neurotransmission semble être le résultat d’une désensibilisation (down-regulation) de certains récepteurs aux monoamines (Charney et al., 1981). Aujourd’hui, la dépression est perçue comme un dysfonctionnement neuronal avec une altération de la neurotransmission des neurones du tronc cérébral. La compensation de ce déséquilibre de neurotransmission expliquerait le délai d’action des antidépresseurs (Harro & Oreland, 1996). Tous les antidépresseurs agissent en augmentant la quantité des amines neurotransmettrices (sérotonine, noradrénaline et dopamine) dans l’espace synaptique. Cette action se fait soit par blocage de la monoamine oxydase, soit par l’inhibition de la recapture des neurotransmetteurs dans la synapse présynaptique, soit par la modulation des récepteurs post-synaptiques. La première génération d’antidépresseurs est constituée par les antidépresseurs tricycliques qui agissent en modulant autant les neurotransmetteurs sérotoninergiques que noradrénergiques. Le premier tricyclique, l’imipramine, fut introduit en 1957. Ils sont connus pour avoir de nombreux effets secondaires dus à leurs effets antagonistes sur d’autres systèmes (muscariniques, adrénergique α1 ou histaminique H1). Dans les années 1980, les inhibiteurs sélectifs de la recapture de la sérotonine ont été introduits sur le marché. Cette classe se compose aujourd’hui de 5 substances (le citalopram, la fluvoxamine, la fluoxétine, la sertraline et la paroxétine) (Hansen et al., 2005). Elle se distingue surtout des tricycliques par un profil d’effets secondaires nettement moins néfaste (Hirschfeld, 1999). De nouvelles molécules agissent de manière différente comparé aux tricycliques et aux inhibiteurs sélectifs de la recapture de la sérotonine (Kent, 2000). La venlafaxine, introduit en Suisse en 1997, inhibe la recapture de la sérotonine et à plus hautes doses, celle de la noradrénaline (Harvey et al., 2000). Son profil d’effets indésirables est proche de celui des inhibiteurs spécifiques de la recapture de la sérotonine. Par contre, la venlafaxine se lie et inactive le canal sodique des myocytes cardiaques, ce qui peut se manifester par une tachycardie sinusale lors de surdosage (Khalifa et al., 1999). La réboxétine est le chef de file de la classe des inhibiteurs sélectifs du recaptage de la noradrénaline. Contrairement aux autres familles d’antidépresseurs, elle ne bloque pas les récepteurs α-adrénergiques, muscariniques ou histaminergiques, ce qui se traduit par un profil d’effets indésirables différents (Wong et al., 2000). Ceux-ci s’apparentent aux effets anticholinergiques, non pas par inhibition des récepteurs muscariniques, mais à cause d’un déséquilibre de la balance des systèmes sympathiques/parasympathiques (Stahl et al., 2002). Enfin la mirtazapine diffère des inhibiteurs sélectifs de la recapture de la noradrénaline par un blocage des récepteurs sérotoninergiques 5-HT2A et 5-HT2C, ainsi que les récepteurs α2-adrénergiques. (Cf. infra). Les systèmes noradrénergiques, sérotoninergiques et dopaminergiques sont intimement liés et interagissent de concert. Il est virtuellement impossible d’agir uniquement sur la neurotransmission d’un seul système sans affecter les deux autres. Aujourd’hui, on tend à considérer ces trois systèmes comme un ensemble plutôt que séparément, d’où le développement de « stratégies catécholaminergiques » pour traiter la dépression (Tremblay & Blier, 2006).

- 16 -
Figure 1 : Amines biogènes potentiellement impliquées comme neuromédiateurs dans la dépression et les effets des psychotropes sur celles-ci. Schématisation du mode d’action de deux principes actifs : le citalopram et la mirtazapine.
Tryptophane
5-OH-Tryptophane
5-Hydroxytryptamine (5-HT, sérotonine)
tryptophane hydroxylase
Vésicule de sérotonine
Récepteurs 5-HT1A
Tyrosine
L-Dihydroxyphénylalanine(L-DOPA)
Dopamine
tyrosine hydroxylase
dopamine décarboxylase
Vésicule de dopamine
Récepteurs dopaminergiques
Autorécepteurs
D3
Vésicule denoradrénaline
Récepteurs noradrénergiques (α et β)
5-HIAA
Inhibition de la recapture
de la 5-HT (tricycliques;
ISRS, p.ex. citalopram)
Inhibition de
la monoamine
oxydase
MAO
Inhibition de la
monoamine
oxydase
Produits déaminés
Inhibition des
récepteurs
à la dopamine
(neuroleptiques)
Transporteur
dopamine
Inhibition de la
recapture
de la dopamine
(amphétamines,
cocaïne)
Au
toré
cep
teu
rs
αα αα2-a
dré
ner
giq
ues
Hétérorécepteurs
αααα2-adrénergiques
Récepteurs 5-HT2
Récepteurs 5-HT3
MAO
Récepteurs
αααα1-adrénergiques
Inhibition de la
recapture
de la
noradrénaline
(tricycliques)
Blocage des
hétérorécepteurs
αααα2-adrénergiques,
par la mirtazapineBlocage récepteurs
5-HT2 et 5-HT3 sérotoninergiques
et des autorécepteurs αααα2-adrénergiques
par la mirtazapine
Présynaptique
PostsynaptiqueStimulation de la transmission sérotoninergique
(suite au blocage des hétérorécepteurs αααα2)
par la mirtazapine
Stimulation de la transmission noradrénergique
(suite au blocage autorécepteur αααα2) par la
mirtazapine
Interférence
dans les
vésicules de
stockage
(réserpine)
Interférence
dans les
vésicules de
stockage
(réserpine)
inhibition du récepteur (antagoniste)
stimulation du récepteur (agoniste)
5-Hydroxytryptamine, sérotonine
noradrénaline
dopamine
Légende
Dessin modifié et adapté (Kandel et al., 2000; Timmer et al., 2000)
Médicaments racémiques Les stéréoisomères sont deux composés ayant le même nombre d’atomes connectés entre eux de la même manière, mais avec un arrangement spatial différent. Les énantiomères sont des stéréoisomères dont l’image de l’un donne celle de l’autre dans un miroir. Leurs images ne sont par contre pas superposables. Un mélange racémique, ou racémate, est un mélange équimolaire des deux énantiomères (50/50). Chaque énantiomère a des propriétés pharmacologiques qui lui sont propres. Par exemple, la (+)-carvone a une odeur de cumin, tandis que la (-)- carvone a un parfum mentholé. Un certain nombre d’antidépresseurs possédant un centre chiral (se dit d’une entité moléculaire qui n’est pas superposable à son image dans son miroir) ont été introduits sur le marché comme racémate, p.ex. le citalopram (Séropram®). Lundbeck, la société danoise qui fabrique le citalopram, a récemment mis sur le marché la forme pure de l’énantiomère S-(+)- : l’escitalopram (Cipralex®).

- 17 -
Mirtazapine et ses métabolites La mirtazapine (Remeron®) a été introduite comme mélange racémique (50/50) et il se forme des métabolites chiraux. Ils font partie d’une nouvelle famille d’antidépresseurs, les antidépresseurs sérotoninergiques spécifiques et noradrénergiques (NaSSA) (Sitsen & Moors, 1994). La mirtazapine agit en bloquant les auto- et hétérorécepteurs α2 adrénergiques. On distingue les autorécepteurs des hétérorécepteurs selon que l'agoniste endogène de ces récepteurs présynaptiques soit le neurotransmetteur libéré par le neurone lui-même (autorécepteur) ou par un autre neurone (hétérorécepteur). En bloquant les récepteurs α2, la mirtazapine diminue le rétrocontrôle négatif de la libération de noradrénaline, qui augmente sa concentration dans la fente synaptique. La mirtazapine bloque également les récepteurs sérotoninergiques 5-HT2 (5-HT2A et 5-HT2C) et 5-HT3. Le blocage des récepteurs 5-HT2A et 5-HT2C expliquerait son effet anxiolytique, tandis que le blocage des récepteurs 5-HT3 diminuerait la fréquence des nausées. Dès lors, la neurotransmission sérotoninergique se fait principalement par les récepteurs sérotoninergiques 5-HT1A (Figure 1). Ce double blocage diminue en conséquence le nombre des effets secondaires (Montgomery, 1995). La mirtazapine a une grande affinité pour les récepteurs histaminergiques H1 et peu d’affinité pour les récepteurs muscariniques et dopaminergiques, ce qui explique un effet sédatif de la mirtazapine qui peut être désiré (ou non) par le thérapeute (Davis & Wilde, 1996; Delbressine & Vos, 1997). De plus, la mirtazapine a une structure chimique tétracyclique. Chimiquement, elle est l’analogue de la miansérine (Tolvon®) avec un azote en position 6. La mirtazapine et la miansérine ont des effets similaires comme antagonistes des récepteurs α2 adrénergiques, tandis que la miansérine est 10 fois plus puissante pour inhiber les récepteurs adrénergiques α1. Cette sélectivité pour les récepteurs adrénergiques α2 montre l’importance de ceux-ci dans le profil pharmacologique de la mirtazapine (Kelder et al., 1997). La mirtazapine est vendue sous forme de racémate. Ses énantiomères R-(-)- et S-(+)- diffèrent dans leurs propriétés pharmacologiques et pharmacocinétiques (Timmer et al., 2000). La S-(+)-mirtazapine est 10 fois plus puissante que la R-(-)-mirtazapine dans l’antagonisme des récepteurs adrénergiques α2 (de Boer et al., 1995). La mirtazapine est principalement métabolisée en N-déméthylmirtazapine par N-déméthylation, en 8-hydroxymirtazapine par oxydation, ainsi que par glucuroconjugaison de l’amine tertiaire. Les voies métaboliques de la mirtazapine sont principalement sous contrôle des cytochromes P-450 CYP2D6, CYP3A4 et CYP1A2. Le CYP2D6 participe surtout à la 8-hydroxylation de l’énantiomère S-(+)-, tout comme le CYP1A2. Le CYP3A4 contribue à la N-déméthylation et la N-oxydation de l’énantiomère R-(-)- (Figure 2) (Delbressine et al., 1998). A noter que la forme S-(+) de la 8-hydroxymirtazapine est fortement glucuroconjuguée, ce qui n’est pas le cas pour la forme R-(-) (Paus et al., 2004). Enfin, une récente étude montre le possible transport de la mirtazapine dans le liquide céphalo-rachidien par la Pgp (Baumann et al., 2006).
Figure 2 : Voies métaboliques de la mirtazapine chez l’homme. (1) 8-hydroxylation suivi par la conjugaison; (2) N+ -glucuronidation; (3) N-oxydation; (4) déméthylation suivie par la sulfoconjugaison.

- 18 -
Citalopram et ses métabolites Le citalopram (CIT) (Seropram®), (+/-)-1-(3-dimethylaminopropyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile, est une amine tertiaire qui est souvent utilisée dans le traitement de la dépression. Le citalopram appartient à la classe des inhibiteurs spécifiques de la recapture de la sérotonine (ISRS). Le citalopram et ses métabolites sont des racémates, composés d’un énantiomère R-(-)- et d’un énantiomère S-(+)-. Pharmacologiquement, l’énantiomère S-(+)- du citalopram est responsable de l’activité inhibitrice sur la recapture de la sérotonine (Hyttel et al., 1992). Il augmente la neurotransmission sérotoninergique en bloquant spécifiquement la recapture de la sérotonine, tout en ayant un minimum d’effets sur la recapture de la noradrénaline et de la dopamine (Figure 1) (Hyttel, 1994). Chez l’homme, les concentrations plasmatiques sont proportionnelles à la dose administrée. Les taux thérapeutiques sont atteints en une à deux semaines. Après une prise quotidienne de 20 à 60 mg de citalopram, les taux plasmatiques moyens mesurés varient de 130 +/- 70 nmol/l pour 20mg à 400 +/- 200 nmol/l pour 60mg (Baumann & Larsen, 1995). Contrairement aux antidépresseurs tricycliques (TCA), les inhibiteurs spécifiques de la recapture de la sérotonine sont considérés comme n’ayant pas de toxicité cardiaque. Cependant, le risque de troubles du rythme cardiaque a été décrit lors d’une surdose volontaire de 400 mg de citalopram avec un allongement du QT sur l’électrocardiogramme (Catalano et al., 2001). De plus, des cas d’overdose fatale suite à l’association du citalopram à un antidépresseur inhibiteur de la MAO-A (moclobémide – Aurorix®) ont été signalés (Dams et al., 2001; Neuvonen et al., 1993). Le risque majeur de ce genre d’intoxication est le syndrome sérotoninergique. C’est un effet secondaire rare, mais craint, car pouvant rapidement engager le pronostique vital. Il est caractérisé notamment par une agitation extrême, (hyperréflexie, myoclonus et tremor), une dysphorie et un manque de coordination. Il serait principalement dû à une accumulation excessive de sérotonine dans la fente synaptique (Boyer & Shannon, 2005). Ce syndrome a également été décrit suite à l’association de moclobémide avec de la fluoxétine (Fluctine®) (Benazzi, 1996). Le métabolisme du citalopram est complexe comme le montre la Figure 3. Le citalopram est N-déméthylé en déméthylcitalopram partiellement par les CYP2C19 et CYP3A4 (Gram et al., 1993; Kobayashi et al., 1997; Olesen & Linnet, 1999; Rochat et al., 1997a; Sindrup et al., 1993; Von Moltke et al., 1999) et le déméthylcitalopram est à nouveau N-déméthylé en didéméthylcitalopram par le CYP2D6 (Sindrup et al., 1993). Une étude a montré qu’il existe une relation entre le métabolisme du citalopram et le polymorphisme oxydatif de la spartéine et de la méphénytoïne (Sindrup et al., 1993). Les observations faites laissent penser que la N-déméthylation du déméthylcitalopram est catalysée en partie par le CYP2D6. Ceci est corroboré par le fait qu’une comédication avec de la lévomépromazine, un inhibiteur du CYP2D6, n’a pas augmenté les concentrations sériques du citalopram, mais a augmenté les taux de déméthylcitalopram, suggérant fortement le métabolisme de celui-ci par le CYP2D6 (Gram et al., 1993).

- 19 -
Figure 3 : Voie métabolique du citalopram, documentée et hypothétique, chez l’homme (Dalgaard & Larsen, 1999; Rochat et al., 1998). Les métabolites hypothétiques tels que l’aldéhyde propionique du citalopram et l’alcool propionique du citalopram n’ont pas encore été identifiés. MAO, monoamine oxydase ; CYP, cytochromes ; UGT, .UDP-glucuronosyltransferase.
O
CH
2
C
H2
CH
2
NCH
3
CH3
C
F
N
O
CH
2
C
H2
CH
2
NCH
3
C
F
NH
O
CH
2
C
H2
CH
2
N
C
F
NH
H
O
CH
2
C
H2
C
C
F
N
H
OO
CH
2
C
H2
C
C
F
N
OH
O
O
CH
2
C
H2
CH
2
N
C
F
N CH3
CH3
O
OH
OH
OH
COO-
O
CH
2
C
H2
CH
2
N
C
F
N
O
OH
OH
OH
COO-
H
O
CH
2
C
H2
C
C
F
N
OH
O
O
OH
OH
OH
COO-
O
CH
2
C
H2
C
C
F
N
OH
H
H*
* *
*
*
Citalopram
MAO- AMAO- B
Al d é h y d e Ox y d a s e
CYP2 D6
CYP3 A4CYP2 C1 9CYP2 D6
*
N-déméthylcitalopram
N-didéméthylcitalopram
Acide propionique du citalopram
MAO- AMAO- B
MAO- AMAO- B
Aldéhyde propionique du citalopram
+
Citalopram-N-glucuronide
*
Didéméthylcitalopram-N-glucuronide
*
+
Acide propionique du citalopram-N-glucuronide
UGT
UGT
UGT
*
Alcool propionique du citalopram
Ré d u c t i o n p a r ?
Dans une autre voie métabolique, le citalopram, le déméthylcitalopram et le didéméthylcitalopram sont désaminés par les MAO conduisant à la formation de l’aldéhyde propionique du citalopram. Celui-ci est à son tour oxydé par l’aldéhyde oxydase en acide propionique du citalopram. L’aldéhyde peut être réduit en alcool, mais ni l’aldéhyde propionique du citalopram, ni l’alcool propionique du citalopram n’ont pu être identifiés. Deux raisons sont possibles pour expliquer cela. Les aldéhydes sont métabolisés en acides carboxyliques, car leurs constantes d’affinité (Km) sont généralement plus basses que celles des alcools. L’oxydation des aldéhydes en acides carboxyliques est difficilement réversible, contrairement à la formation des alcools (Watanabe et al., 1990). L’aldéhyde propionique du citalopram est oxydé en acide propionique du citalopram par l’aldéhyde oxydase. Un inhibiteur puissant de l’aldéhyde oxydase, la ménadione, inhibe fortement la production de l’acide propionique du citalopram (Rochat et al., 1998).
Toxicité du citalopram chez le chien
Au cours de l’évaluation préclinique et clinique sur la sécurité d’emploi du citalopram, la variabilité de la pharmacocinétique entre les espèces a été observée. Des études sur la sécurité d’emploi furent réalisées chez le rat, la souris, le babouin et le chien Beagle. Il y eu une grande inquiétude à la suite de cette étude qui montra que des doses de citalopram per os et intraveineuses, considérées comme sub-létales, aboutissaient à de sévères convulsions voir la mort du chien (Boeck & Overø, 1982). Les résultats obtenus après l’administration per os de citalopram étaient cohérents entre la clinique et les taux plasmatiques de citalopram. Les crises convulsives apparaissaient à des doses de 20-25 mg/kg ce qui correspond à des taux plasmatiques de 7000-9000 nM. Une tachycardie sinusale était également observée. Lorsque le citalopram était donné par voie intraveineuse, les crises convulsives apparaissaient à des taux plasmatiques de 7000-8000 nM. Les taux plasmatiques auxquels les chiens ont été exposés dépassaient les taux plasmatiques humains par un facteur 20 (Tableau 1) (Boeck & Overø, 1982). Une autre étude évalua l’effet de doses répétées de citalopram. Les chiens recevaient une dose de 8mg/kg par jour. Au cours de l’étude, 5 des 9 chiens décédèrent subitement. Les raisons de la mort furent attribuées à une prolongation de l’intervalle QT suivi d’une arythmie fatale due à des taux plasmatiques

- 20 -
très élevés de didéméthylcitalopram (1000 nM). Ce métabolite est considéré comme cardiotoxique combiné à des taux élevés de citalopram. Suite à cette étude, le développement clinique du citalopram a été retardé jusqu’à la confirmation que les taux plasmatiques de didéméthylcitalopram sont généralement en-dessous de la limite de détection, environ 50nM chez l’Homme (Overø & Højelse, 1994). Chez l’homme, l’effet clinique est principalement lié au citalopram, car les métabolites déméthylés et didéméthylés ont moins d’activité pharmacologique, sont présents à de faibles concentrations et passent moins facilement la barrière hémato-encéphalée (Hyttel, 1982). Le Tableau 1 compare les concentrations plasmatiques maximales (Cmax) entre l’Homme et le chien après administration orale de citalopram. Les doses de citalopram racémique étaient de 40 mg chez l’Homme (correspondant à 0.7 ± 0.1 mg/kg chez 10 patients pesant entre 51 et 74 kilos) et de 10 mg chez le chien (correspondant à 19.7 ± 0.2 mg/kg chez 3 chiens pesant entre 8 et 12 kilos).
Tableau 1 : Comparaison des Cmax [nM] entre l’Homme et le chien après administration orale de citalorpam.
Cmax [nM] R-(-)-CIT S-(+)-CIT CIT R-(-)-DCIT S-(+)-DCIT DCIT R-(-)-DDCIT S-(+)-DDCIT DDCIT
Homme 1
228.4 ± 63.6 154.2 ± 64.5 382.6 62.7 ± 17.5 46.3 ± 9.3 109 13.4 ± 3.2 6.4 ± 1.4 19.8
Chien 2, 3
5000-9000 2000 350
1 according to Sidhu, J. et al. Chirality 1997;9:686-692
2 according to Boeck, V. et al. Acta Pharmacol et Toxicol 1982;50:169-174
3 according to Overø, KF. et al. Prog Neuropsychopharmacol Biol Psychiatry 1982;6:297-309
Monoamine oxydase (MAO) La fonction principale des monoamines oxydases est de dégrader les neurotransmetteurs monoaminergiques que sont la sérotonine, la noradrénaline et la dopamine. Les monoamines oxydases sont une famille d’enzymes présentes principalement sur la membrane externe des mitochondries. Néanmoins, une activité des monoamines oxydases a été observée dans des fractions microsomales (Grothusen et al., 1996). La découverte que ces enzymes participaient au métabolisme de l’antidépresseur citalopram, d’abord dans des microsomes de foies humains, puis dans des mitochondries cérébrales chez l’homme et le rat, suscita un vif intérêt de la recherche (Kosel et al., 2002; Rochat et al., 1998).
Biochimie de la MAO. La monoamine oxydase catalyse la déamination des amines (endogènes ou exogènes) dans le cerveau et les tissus périphériques pour arriver à l’aldéhyde et à un dégagement d’H2O2 selon l’équation suivante :
CH
2
NRR1
R2CH
RO
2 R CHO H2O
2 NR1
R2HN
R1
R2+ +
H2O+
La monoamine oxydase existe sous deux formes, la MAO-A et la MAO-B, qui ont chacune une affinité différente en fonction du substrat considéré. La MAO-A a une plus grande affinité pour la sérotonine, la noradrénaline, la dopamine et son inhibiteur la clorgyline. La MAO-B a une plus grande affinité pour la phenyléthylamine, la benzylamine et son inhibiteur le L-déprényl (Tableau 2) (Shih et al., 1999). La dopamine est principalement oxydée par la MAO-A chez l’homme. Chez la souris la dopamine est uniquement métabolisée par la MAO-A aux concentrations basales et par la MAO-A et la MAO-B à hautes concentrations. Ceci à la différence du rat, où la dopamine est toujours métabolisée par la MAO-A, sans que les concentrations n’aient d’influence (Fornai et al., 1999).

- 21 -
Tableau 2 : Substrats et inhibiteurs des monoamines oxidase A et B
MAO-A MAO-B
Substrats Sérotonine (5-HT), noradrénaline Phenyléthylamine (PEA), benzylamine
Substrats non spécifiques
Dopamine, tyramine
Inhibiteurs
Clorgiline : (IC50 [µM] : 3.7 ± 0.4 si substrat = 5-HT) (IC50 [µM] : 4.0 ± 0.4 si substrat = PEA)
moclobémide
L-deprenyl (sélégiline) : (IC50 [µM] : 1.5 ± 0.1 si substrat = 5-HT) (IC50 [µM] : 2.7 ± 0.3 si substrat = PEA)
Inhibiteurs non spécifiques
Pargyline, tranylcypromine, isocarboxazide
IC50 [µM] : chez l’Homme et selon (Shih et al., 1998). Celles-ci n’ont pas été déterminées chez la souris
Ces deux enzymes sont présents dans presque tous les tissus des mammifères, que ce soit dans le système nerveux central ou d’autres organes, en particulier le foie. Dans certains tissus, seule une des deux formes de l’enzyme est exprimée. Le placenta humain exprime principalement la MAO-A, tandis que les plaquettes sanguines et les lymphocytes expriment uniquement la MAO-B (Kosel et al., 2001). Chez l’homme, la concentration des MAO est la plus haute dans le foie et la plus basse dans la rate. La MAO-A est plus importante dans les poumons et la muqueuse duodénale, tandis que la MAO-B est plus importante dans le cœur. Au contraire du rat, où la MAO-A est très importante dans le cœur et la MAO-B quasiment inexistante (Saura et al., 1992).
Génétique de la MAO : absence de MAO-A chez une famille hollandaise Ces enzymes sont codés par des gènes distincts situés sur le chromosome X dans la région Xp11.3-Xp11.23. Des variations alléliques de ces gènes pourraient être à l’origine de la variation d’activité de la MAO-B. Des délétions de la région Xp11.3, comprenant le gène NDP (Norrie disease) et pouvant inclure les gènes de la MAO-A et/ou de la MAO-B ont été retrouvées chez certains patients atteints de la maladie de Norrie. Cette affection récessive rare est liée au chromosome X. Elle est caractérisée par une dysplasie vitréorétinienne très précoce, associée à un retard mental et à une surdité d’évolution progressive et de gravité variable (Collins et al., 1992; Lenders et al., 1996). Une micro délétion sur l’exon 8 du gène de la MAO-A cause non seulement la perte de l’activité enzymatique de la MAO-A, mais est également associée à un retard mental et à un comportement impulsif voir agressif (Brunner et al., 1993a). Cette micro délétion a été décrite au sein d’une fratrie hollandaise (Brunner et al., 1993b).
Intérêt de la recherche pour des modèles animaux n’exprimant pas le gène de la MAO-A ou de la MAO-B
Le développement des modèles animaux n’exprimant pas le gène de la MAO-A ou de la MAO-B, sont des outils qui permirent l’étude du rôle de la monoamine oxydase dans les pathologies neurodégénératives ou dans l’agressivité. Une lignée de souris n’exprimant pas le gène de la MAO-A a été produite. Celle-ci avait des taux élevés de sérotonine, de noradrénaline et de dopamine, ainsi qu’un comportement très agressif, de manière identique à ce qui a été décrit chez l’homme (Cases et al., 1995). Une autre lignée de souris n’exprimant pas le gène de la MAO-B présente des taux élevés de phenyléthylamine et une grande sensibilité au stress (Grimsby et al., 1997). Elle est par contre résistante aux effets neurotoxiques du MPTP, celui-ci étant métabolisé par la MAO-B pour former le métabolite toxique (Maret et al., 1990; Shih et al., 1999).

- 22 -
Plus récemment, une lignée de souris n’exprimant ni le gène de la MAO-A ni celui de la MAO-B a été décrite (Holschneider et al., 2001). Sachant que les monoamines oxydases métabolisent le citalopram, ces modèles animaux sont utiles pour tester de quelle façon les souris déficientes du gène de la MAO-A compensent cette déficience et métabolisent le citalopram et ses métabolites.
Aldéhyde oxydase
Biochimie de l’aldéhyde oxydase L’aldéhyde oxydase (AO) est un enzyme cytoplasmique présent dans le foie ainsi que d’autres tissus. L’aldéhyde oxydase n’est pas seulement impliqué dans l’oxydation des aldéhydes en acides carboxyliques, mais également dans l’oxydation des composés hétérocycliques contenant des azotes. L’aldéhyde oxydase est impliqué dans le métabolisme de plusieurs substances comme la ziprasidone (antipsychotique) ou la zonisamide (antiépileptique), ainsi que le métabolisme de l’intermédiaire aldéhydique du citalopram. Enfin, la ménadione (IC50 0.20 ± 0.04 µM) est responsable de l’inhibition de l’aldéhyde oxydase dans différentes espèces (Obach et al., 2004). Cet enzyme est aussi impliqué dans l’hépatotoxicité de l’éthanol chez l’Homme et d’autres mammifères en oxydant l’acétaldéhyde, métabolite toxique, en acide acétique (Mendel & Bittner, 2006).
Cytochromes P-450
Biochimie des cytochromes P-450 Les cytochromes P-450 (CYP) sont une superfamille de monooxygénases impliquées dans le métabolisme de nombreux xénobiotiques ainsi que des composés endogènes comme des acides gras et des neurotransmetteurs. Ce sont principalement des enzymes hépatiques. Leur présence a également été détectée dans d’autres organes, notamment le cerveau (Voirol et al., 2000). Il y a de plus en plus d’évidences que les cytochromes P-450 soient impliqués localement, notamment au niveau cérébral, dans le métabolisme des médicaments. L’exemple du CYP2D6 est particulièrement intéressant, car de nombreux psychotropes sont substrats de celui-ci et il est exprimé de manière polymorphique chez l’homme (Dahl, 2002). Les cytochromes P-450 sont classés par homologie de structure protéique. Deux cytochromes P-450 ayant une structure protéique identique jusqu’à 40% font partie d’une même famille caractérisée par un chiffre, par exemple CYP2. Si cette similitude se situe entre 40 et 55%, ils appartiennent à la même sous-famille qui est définie par une lettre, par exemple CYP2D. Au-delà de 55% de similitude de la structure protéique, il s’agit d’un même isoenzyme que l’on distingue par un chiffre, par exemple CYP2D6 (Nebert & Russell, 2002). La réaction catalysée par les cytochromes P-450 est une monooxydation, où un atome d’oxygène est incorporé au substrat via une réductase, la NADPH, essentielle au fonctionnement des cytochromes P-450 selon la réaction suivante :
RH O2 NADPH H
+ROH NADP+ + + + H2O + +
Plus de 57 gènes expriment les différents cytochromes P-450 identifiés chez l’homme. Parmi ceux-ci, les plus importants sont les CYP1A2, CYP3A4, CYP2C9, CYP2C19, CYP2D6, CYP2B6 et CYP2E1 (Guengerich, 1997; Lewis, 2000; Rendic & Di Carlo, 1997). Ils sont impliqués dans plus de 80% du métabolisme oxydatif des xénobiotiques. Les cytochromes P-450 sont généralement spécifiques pour un xénobiotique donné ou l’un de ses deux énantiomères. Il y a souvent un recouvrement de l’activité entre les différents cytochromes P-450 (Wilkinson, 2005). Par exemple, trois cytochromes P-450 (CYP2D6, CYP3A4 et CYP1A2) sont impliqués dans le métabolisme de la mirtazapine (Delbressine et al., 1998).

- 23 -
Polymorphisme des cytochromes P-450, notamment du CYP2D6 impliqué dans le métabolisme des psychotropes
Un certain nombre de xénobiotiques peuvent augmenter l’expression des cytochromes P-450 (inducteurs) ou au contraire en diminuer l’expression (inhibiteurs) (Tableau 3). Ces composés peuvent induire des variations de « clearance », c’est-à-dire influencer le métabolisme et l’élimination du médicament. Ce qui va diminuer ou augmenter les effets de ceux-ci, aboutissant parfois à des interactions médicamenteuses. Les xénobiotiques métabolisés presque exclusivement par un seul cytochrome sont particulièrement sensibles à ces phénomènes (Tanaka, 1998).
Tableau 3 : Inducteurs et inhibiteurs des principaux cytochromes P-450 humains.
Cytochromes P-450 Inducteurs Inhibiteurs
CYP1A2 Fumée de cigarette Furafylline
CYP2C9 Carbamazépine, phénobarbital sulfaphénazole
CYP2C19 Rifampicine Oméprazole
CYP2D6 non identifié Quinidine
CYP3A4 Rifampicine Kétoconazole
CYP2E1 Éthanol, isoniazide 4-méthyl-pyrazole Ajouté à l’induction ou l’inhibition par des xénobiotiques, certains cytochromes P-450 hépatiques (CYP2D6, CYP2C19, CYP1A2 et CYP3A5) sont également associés à un polymorphisme génétique (Bolonna et al., 2004). Un des polymorphismes les mieux caractérisés est celui du CYP2D6. Entre 5 et 10% des caucasiens et ~1% des asiatiques métabolisent les substrats du CYP2D6 de manière fortement diminuée (PM ou métaboliseurs lents). A l’inverse entre 1 et 8% des caucasiens possèdent une ou plusieurs copies du gène et métabolisent les substrats du CYP2D6 de manière augmentée (UM ou métaboliseurs ultrarapides) (Bertilsson et al., 2002). Il a été démontré que les concentrations cérébrales de cytochromes P-450 représentent environ 5% de celles mesurées dans le foie (Ghersi-Egea et al., 1993). Cependant, le cerveau n’est pas un organe homogène et les taux de cytochromes P-450 dans certains neurones spécifiques pourraient être plus élevés que dans les hépatocytes. Les cytochromes P-450 cérébraux constituent un moyen de régulation locale de l’activité enzymatique, ce qui influencerait le métabolisme. Les cytochromes P-450 suivant ont été détectés dans le cerveau : CYP1A2, CYP2C9, CYP2D6 et CYP3A4 (Gervasini et al., 2004). Ces quatre cytochromes P-450 sont tous impliqués dans le métabolisme des psychotropes. Une modulation locale de l’activité de ces cytochromes P-450 pourrait être une des causes majeures de variabilité interindividuelle dans le métabolisme cérébral et influencer la réponse thérapeutique. Il y a cependant des différences entre les espèces et la signification physiologique et chimique de la présence de ces formes de cytochromes P-450 dans le cerveau humain qui restent à démontrer (Voirol et al., 2000).
Métabolisme : différences entre cytochromes P-450 humains et cytochromes canins (orthologues)
La comparaison des gènes des cytochromes P-450 des génomes humains et murins montre que 80% des gènes ont une relation orthologue 1 : 1. Cependant, certaines sous-familles de cytochromes P-450 ne sont pas identiques avec, par exemple, la perte de 4 sous-familles chez l’homme comparé aux rongeurs (CYP2G, CYP2T, CYP2AB et CYP2AC). Ces sous-familles sont uniquement présentes comme pseudogènes chez l’homme. Il se pourrait que les protéines exprimées par ces gènes chez les rongeurs ne soient plus nécessaires chez l’homme (Nelson et al., 2004). L’activité catalytique des cytochromes P-450 entre les espèces n’est pas toujours bien définie comme, par exemple, entre le CYP2D6 humain et son orthologue canin, le CYP2D15. Le Tableau 4 résume les principales sous-familles entre les différentes espèces et leurs inhibiteurs.

- 24 -
Tableau 4 : Comparaison des cytochromes P-450 entre les différentes espèces
Espèces Enzymes∗∗∗∗ Inhibiteurs Remarques Humain CYP2D6
a
Quinidine 1
1 µM de quinidine inhibe jusqu’à 80% de l’activité du CYP2D6 chez l’homme, tandis que 100 µM de quinidine inhibe jusqu’à 60% du CYP2D15 chez le chien Beagle 3, tout en n’étant pas un inhibiteur spécifique (cf. CYP1A2, ci-dessous).
Orthologue canin CYP2D15
Orthologue murin CYPd9
Orthologue rat CYP2D1 Quinine 5
La quinidine est un faible inhibiteur du CYP2D1 tandis que son stéréoisomer, la quinine, en est un puissant inhibiteur 5.
Humain CYP2C19 a
Oméprazole 1 Orthologue canin CYP2C21 / 41
b
Orthologue murin CYP2c39 Orthologue rat CYP2C11
Humain CYP2C9 a
Sulfaphénazole 1, 5
100 µM de sulfaphénazole inhibe jusqu’à 80% de l’activité du CYP2C9 chez l’homme. Le sulfaphénazole n’est pas utilisable pour inhiber le CYP2C9 chez les chiens Beagle. Son inhibition est trop faible pour être mesurée 3.
Orthologue canin CYP2C21 b
Orthologue murin CYP2c39
Orthologue rat CYP2C11 Sulfaphénazole 2, 5
Le sulfaphénazole exerce une inhibition spécifique du CYP2C9 dans les microsomes hépatiques humains, mais n’a pas d’activité inhibitrice dans les microsomes de rat 5.
Humain CYP3A4 Kétoconazole
1, 4, 5
Le kétoconazole est un puissant inhibiteur du CYP3A4, tant chez l’homme que chez le rat 5.
Orthologue canin CYP3A12 Orthologue murin CYP3a11
Orthologue rat CYP3A1 Kétoconazole 5 Le CYP3A1 n’est pas un bon modèle, car de nombreux substrats du CYP3A4 humain ne sont pas métabolisés par le CYP3A1 du rat 6.
Humain CYP1A2
Furafylline 1, 3, 5
1 µM de furafylline inhibe l’activité du CYP1A2 jusqu’à 90% chez les chiens Beagle et jusqu’à 80% chez l’homme 3.
Orthologue canin CYP1A2
Orthologue murin CYP1a2
Orthologue rat CYP1A2 Furafylline 5
L’inhibition par la furafylline est puissante et spécifique dans les microsomes hépatiques humains, mais elle inhibe également le CYP2C9 et le CYP1A2 dans les microsomes de rats 5.
Humain CYP2E1
4-méthyl-pyrazole 1
Orthologue canin CYP2E1 c
Orthologue murin CYP2e1 Orthologue rat CYP2E1
Polymorphismes connus : a (Daly, 2003), b (Blaisdell et al., 1998), c (Lankford et al., 2000) Inhibiteurs : 1 (Newton et al., 1995), 2 (Kobayashi et al., 2003), 3 (Chauret et al., 1997), 4 (Kuroha et al., 2004) 5 (Eagling et al., 1998), 6 (Zuber et al., 2002)
∗ Orthologues et orthographe des enzymes selon l’espèce considérée

- 25 -
Situation et buts de la thèse Il a récemment été démontré qu’au niveau cérébral, contrairement au niveau hépatique, ce sont les monoamines oxydases qui métabolisent préférentiellement le citalopram, le déméthylcitalopram et le didéméthylcitalopram (Kosel et al., 2002). En effet, l’activité des monoamines oxydases par rapport à ces substrats est beaucoup plus élevée que celles des cytochromes P-450. L’un de nos objectifs est de démontrer de quelle manière les monoamines oxydases sont impliquées dans le métabolisme cérébral du citalopram sur un modèle animal dans lequel l’un des deux gènes codant pour la monoamine oxydase est déficient. La MAO-A métabolise principalement les amines biogènes que sont la sérotonine, la noradrénaline et la dopamine. Or leurs taux sont beaucoup plus élevés chez la souris privée du gène de la MAO-A comparée à la souris sauvage (Cases et al., 1995). Il a été montré que la déficience du gène de la MAO-A est également associée à des troubles profonds du comportement (Brunner et al., 1993a). Ces troubles du comportement se retrouvent chez la souris déficiente du gène de la MAO-A (Cases et al., 1995). Sachant que le citalopram est métabolisé par les monoamines oxydases (Kosel et al., 2002; Rochat et al., 1998), il serait intéressant de savoir comment s’effectue son métabolisme en l’absence de la MAO-A et dans quelle mesure ces résultats sont transposables à l’Homme. Lors du développement du citalopram comme antidépresseur, une toxicité de celui-ci a été observée chez le chien Beagle, ceci était dû à des taux de citalopram et de didéméthylcitalopram très élevés (Boeck & Overø, 1982; Overø, 1982; Overø & Højelse, 1994). Le but de nos travaux est de démontrer de quelle manière les cytochromes P-450 pourraient être impliqués dans ce métabolisme toxique en testant les différents orthologues canins de ceux-ci. Pour ces études, des méthodes existantes devaient d’abord être adaptées (Kosel et al., 1998; Rochat et al., 1995a; Rochat et al., 1995b) pour l’analyse du métabolite acide propionique du citalopram dans les cerveaux de la souris sauvage et génétiquement modifiée. De nombreuses méthodes analytiques ont été développées et publiées par notre laboratoire. La mise sur le marché de la mirtazapine (Remeron®), a rendu nécessaire la mise au point d’une méthode analytique pour le dosage stéréosélectif de la mirtazapine et de ses métabolites. Une méthode de dosage par LC-MS fut mise au point durant cette thèse
Parcours et problèmes rencontrés lors de l’étude du métabolisme du citalopram La déamination du citalopram, du déméthylcitalopram ainsi que du didéméthylcitalopram par les monoamines oxydases conduisent à un métabolite, l’aldéhyde propionique du citalopram, qui est lui-même oxydé en acide propionique du citalopram. Le citalopram aldéhyde n’a jamais pu être quantifié dans nos échantillons, à cause de sa très grande instabilité et de la présence dans les échantillons biologiques d’enzymes métabolisant la forme aldéhyde (Rochat et al., 1998). De plus, l’aldéhyde propionique du citalopram n’existe pas sous une forme chimique utilisable comme standard. Le métabolisme stéréosélectif du citalopram et de ses métabolites déméthylés en acide propionique du citalopram acide a récemment été décrit dans le foie, le cerveau et le sang humain, ainsi que dans le foie et le cerveau de rat, mais pas chez la souris (Kosel et al., 2001; Kosel et al., 2002; Rochat et al., 1997a; Rochat et al., 1997b; Rochat et al., 1998). Nous avons en premier lieu vérifié si ce métabolisme se retrouvait dans le foie et le cerveau de la souris sauvage. Puis, nous avons étudié de quelle manière la souris Tg8, privée du gène de la MAO-A, métabolise le citalopram et éventuellement compense cette déficience. La déamination du diltiazem a été décrite comme étant sous le contrôle des cytochromes P-450 dans les microsomes hépatiques du rat (Nakamura et al., 1990). Nous avons également cherché à savoir si les cytochromes P-450 avaient un rôle dans la déamination du citalopram chez la souris, en étudiant celle-ci chez la souris déficiente du gène de la MAO-A. L’utilisation thérapeutique du citalopram est relativement sûre. Sa toxicité, seule ou en combinaison avec d’autres substances, est bien établie (Dams et al., 2001; Engebretsen et al., 2003; Öström et al., 1996). Cependant, la toxicité du didéméthylcitalopram chez le chien reste partiellement étudiée et expliquée. La deuxième N-déméthylation du citalopram chez l’homme est sous le contrôle du CYP2D6, dont l’orthologue canin est le CYP2D15 (Chauret et al., 1997; Roussel et al., 1998). De plus, les cytochromes P-450 canins, comme leurs homologues humains, sont également sujets à des polymorphismes (Shou et

- 26 -
al., 2003). Nous avons investigué l’implication des cytochromes P-450 canins dans la cinétique de ces deux déméthylations, ainsi que l’importance de la voie de déamination par les monoamines oxydases du citalopram chez le chien.
Méthodes analytiques Différentes méthodes analytiques ont été utilisées au cours de ce travail. La méthode stéréosélective de dosage du citalopram et de ses métabolites déméthylés a été initialement développée dans notre laboratoire. (Rochat et al., 1995b). Elle faisait appel à une colonne Cyclobond β-acétylée. Suite à un problème de manufacture, la qualité des séparations fournies par cette colonne ne donnait plus satisfaction et la méthode a dû être redéveloppée, cette fois-ci avec une colonne Chirobiotic V (Kosel et al., 1998). Les tests préliminaires à basses concentrations montraient une excellente séparation. Malheureusement, celle-ci devenait inopérante dès l’augmentation des concentrations en vue de tester la cinétique du citalopram chez le chien. Suite à la publication de Carlsson et al., sur le dosage du citalopram et de ses métabolites déméthylés, le laboratoire se dota d’une colonne Cyclobond I 2000 β-acétylée. (Carlsson & Norlander, 2001). La phase mobile fut optimisée au sein du laboratoire. La méthode stéréosélective utilisée pour doser l’acide propionique du citalopram a été également développée dans notre laboratoire (Rochat et al., 1995b).
Développement d’une méthode stéréosélective pour l’analyse de la mirtazapine et de ses métabolites
Notre laboratoire a une grande expérience dans le suivi des taux plasmatiques des médicaments psychotropes. Peu de temps avant le début de cette thèse, la firme Organon a mis sur le marché la mirtazapine (Réméron®), un antidépresseur agissant, non pas en inhibant la recapture de la sérotonine, mais en bloquant stéréosélectivement les récepteurs présynaptiques α2. Le besoin s’est fait sentir de pouvoir suivre l’évolution des taux plasmatiques de cette substance chez les patients, en particulier par des études sur la relation entre les taux plasmatiques du médicament et son effet clinique, en tenant compte du statut pharmacogénétique du patient. Pour ce faire, nous avons mis au point une méthode de dosage stéréosélectif de la mirtazapine, de la déméthylmirtazapine et de la 8-hydroxymirtazapine, par chromatographie liquide couplée à la spectrométrie de masse (LC-MS) (Paus et al., 2004).
Apport d’un travail supplémentaire dans la distribution des médicaments Un travail bibliographique additionnel a été réalisé durant cette thèse sur une protéine de membrane, la glycoprotéine-P (Pgp). Cette protéine est exprimée dans de nombreux tissus et participerait à la régulation du passage de médicaments à travers certaines membranes épithéliales. De plus, un certain nombre de polymorphismes ont été décrits pour la PGp. Enfin, les cytochromes P-450, en particulier le CYP3A4, ont de nombreux substrats communs avec la PGp. Une revue de littérature détaille les connaissances actuelles et les futurs défis posés par cette protéine (Marzolini et al., 2004).

- 27 -
Métabolisme du citalopram par la MAO chez la souris (wild-type et knock-out MAO-A)
Résumé Les incubations in vitro ont été réalisées à partir de fractions microsomales et mitochondriales de cerveaux de souris. Le citalopram, le déméthylcitalopram et le didéméthylcitalopram ont été utilisés comme substrats. Les amines secondaires et primaires que sont le déméthyl- et le didéméthylcitalopram se sont avérées être de meilleurs substrats pour les monoamines oxydases que le citalopram, l’amine tertiaire et ses énantiomères R-(-)- et S-(+)-. Des observations identiques in vitro ont été faites dans les microsomes hépatiques humains, dans des mitochondries cérébrales humaines, ainsi que des mitochondries de cerveau de rats (Kosel et al., 2002; Rochat et al., 1997a). La MAO-A va préférentiellement catalyser la formation de l’énantiomère R-(-) de l’acide propionique du citalopram avec une affinité supérieure pour les amines tertiaires. Au contraire, la MAO-B favorise la formation de l’énantiomère S-(+) de l’acide propionique du citalopram avec une affinité supérieure pour les amines secondaires et primaires. La stéréosélectivité des monoamines oxydases a déjà été démontrée dans les cerveaux humains et de rats ainsi que dans le sang humain (Kosel et al., 2002; Kosel et al., 2001; Rochat et al., 1997a). La MAO-A métabolise préférentiellement la forme R-(-)- du citalopram tandis que la MAO-B métabolise la forme S-(+)-. Les tests d’inhibitions spécifiques de la MAO-A et de la MAO-B confirment ces résultats. Enfin, les cytochromes P-450 ne sont pas impliqués dans cette voie métabolique de déamination menant à l’acide propionique du citalopram. Ces résultats montrent que la MAO-B compense la déficience de la MAO-A dans les mitochondries cérébrales des souris Tg8.
Matériel & méthodes
Substances et produits chimiques Les substances suivantes ont été offertes : les énantiomères du citalopram (S-(+)- et R-(-)-citalopram), les mélanges racémiques du citalopram, du déméthylcitalopram, du didéméthylcitalopram et de l’acide propionique du citalopram (Lundbeck AS, Copenhague, Danemark), le S-flurbiprofen (Boots Limited, Nottingham, Grande-Bretagne) et la maprotiline (Novartis, Bâle, Suisse). L’iodométhane (Fluka, Buchs, Suisse) a été utilisé comme agent de dérivation pour les acides carboxyliques. Le L-déprényl (sélégiline) a quant à lui été acheté chez Research Biochemicals Inc. (Natick, MA, USA). La triéthylamine (N° lot 90340) a été achetée chez Fluka (Buchs, Suisse) et l’acide citrique (N°lot A016250801) chez Across Organics (Belgique). Les autres substances et produits chimiques ont été achetés dans le commerce et étaient de la plus pure qualité disponible.
Animaux Les souris sauvages (C3H/HeJ) et les souris déficientes du gène de la MAO-A (Tg8) ont été hébergées et nourries dans l’animalerie de l’Institut de Biologie Cellulaire et de Morphologie (IBCM, Lausanne) avec de l’eau et de la nourriture « ad libitum », selon des cycles jour/nuit de 12 heures. Les expériences comportaient un tiers de femelles et deux tiers de mâles pour avoir assez de tissus. Les animaux vivaient en groupe jusqu’à leur mort, sauf les femelles gestantes qui étaient mises en cage seules, puis gardées avec les nouveau-nés. Les mâles et les femelles étaient dans des cages séparées. Toutes les expériences impliquant des tissus animaux ont été menées dans le plus strict respect des directives gouvernementales suisses concernant la recherche scientifique. Les souris utilisées étaient âgées de 12 semaines. La genèse et les aspects pharmacologiques des souris Tg8 ont déjà été décrits (Cases et al., 1995; Cases et al., 1998; Evrard et al., 2002).

- 28 -
Préparation des tissus Les cerveaux et foies de souris ont été homogénéisés par sonification (ultrasonic processor – Brablock Scientific, model 75038). Ce traitement n’a aucun effet sur les propriétés métaboliques des monoamines oxydases (Fowler et al., 1980). De 2 à 4 foies et 8 à 10 cerveaux de souris furent nécessaires pour la préparation des fractions cellulaires. Les tissus ont été lavés avec deux volumes de tampon A (0.32 M sucrose, 50 mM KH2PO4, 1 mM EDTA et 0.1 mM dithiothreitol, pH 7.4). Les cerveaux et les foies ont été coupés en petites fractions de 200 à 300 mg chacune, qui ont été homogénéisés durant 5 secondes à 4°C jusqu’à ce que les tissus soient entièrement dissouts.
Incubation Les préparations de mitochondries et de microsomes cérébraux et hépatiques ont été réalisées en utilisant une procédure initialement décrite par Tata et al. (Tata, 1972) et adaptée par Kosel et al. (Kosel et al., 2002). Quelques modifications ont été apportées aux procédures d’incubation. Brièvement, les préparations mitochondriales et microsomales ont été décongelées à 4°C durant 1 heure. Elles étaient ensuite pré-incubées, sans substrat, pendant 15 minutes à 37°C, puis les incubations étaient initiées par l’ajout du substrat et stoppées après 1 heure par l’addition de 100 µl d’HCl 1 M. Toutes les réactions ont été réalisées dans des tubes Eppendorf en plastique pour éviter une adsorption sur le verre. Le volume final était de 600 µl à pH 7.4 contenant 300 µg de protéines et 0.5 mM de substrats (S-(+)- et R-(-)-citalopram, et citalopram, déméthylcitalopram et le didéméthylcitalopram racémiques). Dans les études d’inhibition sélective de la MAO-A ou de la MAO-B, les échantillons ont été préincubés, sans substrat, pendant 15 minutes à 37°C avec la clorgyline ou le L-déprényl, afin d’obtenir une inhibition sélective de la MAO-A et de la MAO-B, respectivement (Donnelly & Murphy, 1977; Dostert et al., 1989). La MAO-A est inhibée par des concentrations nanomolaires de clorgyline, tandis que des concentrations micromolaires sont nécessaires pour une inhibition par le L-déprényl. L’inverse est vrai lorsque le L-déprényl est utilisé pour inhiber l’activité de la MAO-B (Strolin Benedetti, 2001). Les solutions de travail contenaient de la clorgyline à 0.02 µM ou du L-déprényl à 0.5 µM dissouts dans un mélange eau millipore / DMSO [98 : 2 (v/v)]. Il a été démontré qu’une concentration finale de 5 % de DMSO dans les incubats était sans effet sur l’activité catalytique de la monoamine oxydase (Thull & Testa, 1994). Lors de précédentes expériences dans notre laboratoire, il a été observé que le déméthylcitalopram et le didéméthylcitalopram (amines secondaires et primaires, respectivement) sont de meilleurs substrats pour la monoamine oxydase comparé au citalopram (amine tertiaire). Ceci tant chez la souris sauvage (C3H) que la knock-out MAO-A (Tg8). Cela a été montré dans le sang humain (Kosel et al., 2001) et dans les microsomes de foie humains (Rochat et al., 1998). C’est la raison pour laquelle nous avons mené la plupart de nos expériences avec le déméthylcitalopram comme substrat. La production de citalopram propionique dans les mitochondries cérébrales était linéaire durant au moins 6 heures après incubation de déméthylcitalopram. De même pour les microsomes cérébraux. La concentration finale de déméthylcitalopram était de 0.5 mM, sauf pour la cinétique où celle-ci variait entre 25, 50, 100, 250, 500, 1000 et 2500 µM.
Extraction L’acide propionique du citalopram fut extrait des incubats, en utilisant une méthode précédemment décrite par Rochat (Rochat et al., 1995b), modifiée de la façon suivante : 1 ml de KH2PO4 0.6 % (pH=3), 50 ng de S-flurbiprofen (utilisé comme standard interne) et 6 ml de n-heptane-isoamyl alcool [98.5 : 1.5 (v/v)] furent ajoutés aux incubats. Le mélange a été agité pendant 20 minutes et centrifugé à 3300 g durant 8 min à 8°C. La phase organique a été reprise et transférée dans un autre tube contenant 1.5 ml de NaOH 0.1 N. Le mélange a été agité pendant 20 minutes et centrifugé à 3300 g durant 8 min à 8°C. La phase organique fut aspirée et jetée, tandis que la phase aqueuse fut reprise et transvasée dans des tubes coniques auxquels ont été ajoutés 200 µl d’HCl 1 M et 800 µl d’acétate d’éthyle. Le mélange fut agité pendant 20 minutes et centrifugé à 2300 g durant 8 min à 8°C. La phase organique a été reprise et évaporée à sec sous flux d’azote à 45°C dans le but d’éviter toute contamination par des restes de la phase aqueuse. L’extrait sec fut re-suspendu dans 600 µl d’acétate d’éthyle. Les échantillons furent alors dérivés

- 29 -
par addition de 50 µl d’iodo-méthane en présence de 10 à 20 µg de K2CO3 durant 1 h. à 64°C. Après évaporation à sec à température ambiante sous flux d’azote, les échantillons ont été reconstitués dans 150 µl de phase mobile (n-hexane - isopropanol [50 : 50 (v/v)], qualité fluorimétrique). Finalement, des échantillons de 100 µl furent injectés dans un chromatographe HPLC avec détection fluorimétrique.
Conditions analytiques Les conditions analytiques utilisées pour la quantification des énantiomères R-(-)- et S-(+)- de l’acide propionique du citalopram sont celles précédemment décrites par Rochat (Rochat et al., 1995b), quelques modifications ayant été cependant apportées. Un chromatographe liquide Agilent - HP Série 1100 équipé d’une pompe binaire, et d’un injecteur automatique, avec une colonne analytique chirale (Chirobiotic OD, diamètre des particules 10 µm, 250 x 4.6mm de Daicel) était couplée à un détecteur fluorimétrique. La détection fluorimétrique était réalisée avec une excitation à 236 nm et une émission à 305 nm. Le flux de la phase mobile (n-hexane - isopropanol [50 : 50 (v/v)], qualité fluorimétrique) était de 0.6 ml/min. Les limites de quantification étaient de 150 pg/ml et 400 pg/ml pour les formes R-(-) et S-(+) de l’acide propionique du citalopram, respectivement.
Statistiques Les analyses statistiques ont été effectuées avec le logiciel Statistica 5.0, StatSoft Inc., Tulsa, OK, USA. Les analyses de variances (ANOVA) avec le post test de Bonferroni ont été faites avec le logiciel GraphPad Prism version 4.00 pour windows, GraphPad Software, San Diego, CA, USA. Le test de U de Mann-Whitney (p < 0.05) a été utilisé pour comparer entre les deux lignées de souris (C3H, Tg8) les différentes productions de l’acide propionique du citalopram obtenues suite aux incubations de citalopram, déméthylcitalopram et didéméthylcitalopram Les données cinétiques (Km et Vmax) ont été analysées par les moyennes du “non-linear least square curve fitting program” avec le logiciel GraphPad Prism version 4.00 pour windows, GraphPad Software, San Diego, CA, USA.
Résultats La formation de l’acide propionique du citalopram tend à augmenter avec la diminution du nombre de groupement méthyle dans le substrat, tant chez les souris sauvages (C3H) que celles déficientes du gène de la MAO-A (Tg8) (Tableau 5). Lorsque le citalopram est utilisé comme substrat, la formation des métabolites est significativement plus élevée chez les souris C3H comparé aux souris Tg8 (m vs p), l’inverse est observé lorsque le didéméthylcitalopram est utilisé comme substrat (o vs r). Il n’y a pas de différences significatives lorsque le déméthylcitalopram est utilisé comme substrat (n vs q). Il existe une stéréosélectivité dans la formation des métabolites, mais elle ne dépend que partiellement du substrat ou de la lignée comme le montre l’évolution du ratio S/R de l’acide propionique du citalopram (Tableau 5). Chez les souris C3H, la stéréosélectivité en faveur de la forme S-(+) de l’acide propionique du citalopram augmente avec le niveau de déméthylation du citalopram en didéméthylcitalopram. La réciproque ne se vérifie pas chez les souris Tg8. Dans les deux lignées murines, il n’y a pas de déamination stéréosélective du citalopram qui soit significative (a vs g ; d vs j). Dans la déamination du déméthylcitalopram, l’énantiomère S-(+) de l’acide propionique du citalopram est préférentiellement formé chez les Tg8 (e vs k) tandis que pour la déamination du didéméthylcitalopram, la forme S-(+) de l’acide propionique du citalopram est préférentiellement formée autant chez les C3H que chez les Tg8 (c vs i ; f vs l) (Tableau 5). Nos résultats montrent que la production des formes R-(-) et S-(+) de l’acide propionique du citalopram après incubation de citalopram était 5 fois (a vs d) et deux fois (g vs j) supérieures, respectivement, chez les souris C3H comparées aux souris Tg8. Après incubation de déméthylcitalopram (h vs k) et de didéméthylcitalopram (i vs l), la formation de l’énantiomère S-(+) de l’acide propionique du citalopram est inférieure chez les souris C3H comparées aux souris Tg8, tandis que les mêmes comparaisons concernant l’énantiomère R-(-)- ne montrent pas de différences significatives (b vs e ; c vs f).

- 30 -
Tableau 5 : Production de l’acide propionique du citalopram après incubation de 0.5 mM de citalopram, déméthylcitalopram ou didéméthylcitalopram dans des mitochondries cérébrales de souris sauvages (C3H) et de souris déficientes du gène de la MAO-A (Tg8).
R-(-)- S-(+)-
0.41 ± 0.16a
0.26 ± 0.06g
0.67 ± 0.14m
0.6 ± 0.05s
0.35 ± 0.08b
0.56 ± 0.08h
0.91 ± 0.13n
1.6 ± 0.18t
0.30 ± 0.08c
0.72 ± 0.13i
1.02 ± 0.25o
2.4 ± 0.33u
0.08 ± 0.03d
0.11 ± 0.04j
0.19 ± 0.03p
1.4 ± 0.34v
0.23 ± 0.05e
0.72 ± 0.07k
0.95 ± 0.26q
3.1 ± 0.24w
0.41 ± 0.04f
1.05 ± 0.10l
1.46 ± 0.35r
2.6 ± 0.47x
R- vs S+CIT-PROPC3H vs Tg8
p<0.01: a vs d; h vs k; i vs l; m vs p; o vs r; t vs w p<0.01: c vs i; e vs k; f vs l
CIT
DCIT
DDCIT
Tg8
DCIT
Comparaisons statistiques
¹ moyenne ± SD en [pmol/min/mg de protéines] (n=6)
p<0.05: g vs j
Ratio S/R d'acide
propionique du citalopram ¹
Production racémique (R+S)
d'acide propionique du
citalopram ¹
Substrat [0.5 mM]
CIT
Production d'acide propionique du citalopram ¹
Lignées
C3H DDCIT
Les résultats ci-dessus suggèrent que le déméthylcitalopram et le didéméthylcitalopram sont de meilleurs substrats de la monoamine oxydase que le citalopram, mais comme le didéméthylcitalopram est cliniquement un métabolite mineur, seule la cinétique du déméthylcitalopram a été étudiée plus en détails. Le Km et le Vmax de la déamination du déméthylcitalopram ont été calculés dans des mitochondries cérébrales des souris C3H et Tg8, à partir des graphiques d’Eadie-Hofstee (Tableau 6, Figures 4 et 5). En se référant à la formation de l’énantiomère S-(+) de l’acide propionique du citalopram, le déméthylcitalopram a une plus grande affinité (Km) pour la monoamine oxydase dans les mitochondries cérébrales de souris C3H que de souris Tg8, tandis que la production (Vmax) est similaire dans les deux lignées murines (Tableau 6). La situation s’inverse si l’on regarde le Km de la forme R-(-) de l’acide propionique du citalopram. Sa production est très faible dans les deux lignées, ce qui le rend susceptible d’être le distomère (Tableau 6). La clearance intrinsèque (Clint) était cinq fois et une fois et demie plus grande pour les énantiomères R-(-)- et S-(+)-, respectivement, chez les souris C3H comparées aux souris Tg8.

- 31 -
Tableau 6 : Formation des énantiomères R-(-) et S-(+) de l’acide propionique du citalopram après incubations de concentrations croissantes (25 - 2500 µM) de déméthylcitalopram racémate dans des mitochondries cérébrales.
Lignées Produits Km a Vmax
a Clint b
R-(-)-CIT-PROP 85.4 ± 24.2 29.4 ± 2.1 0.3
S-(+)-CIT-PROP 42.6 ± 7.5 39.3 ± 1.8 0.9
R-(-)-CIT-PROP 286 ± 95.1 17.3 ± 1.4 0.06
S-(+)-CIT-PROP 70.4 ± 22.7 42.3 ± 4.1 0.6
b Clint (µl/min/mg de protéines) = Vmax/Km
C3H
Tg8
a Km (µM), Vmax (pmol/min/mg de protéines), moyenne ± SD (n=3)
La représentation graphique de la formation des formes R-(-) et S-(+) de l’acide propionique du citalopram après incubation de déméthylcitalopram montre une plus grande affinité de l’énantiomère S-(+)- pour la monoamine oxydase, aussi bien chez les souris C3H (Figure 4) que chez les souris Tg8 (Figure 5). Dans le cas où le graphique d’Eadie-Hofstee donne une représentation linéaire, une seule enzyme est impliquée. Dans ce processus de déamination, ceci semble être le cas pour chacun des énantiomères.
Figure 4 : Représentation d’Eadie-Hofstee : Formation des énantiomères R-(-) et S-(+) de l’acide propionique du citalopram après incubations de concentrations croissantes (25 to 2500 µM) de déméthylcitalopram racémate dans des mitochondries cérébrales de souris C3H.
y = -64.726x + 27.596
R2 = 0.8563
y = -44.561x + 39.641
R2 = 0.9512
0
5
10
15
20
25
30
35
40
0.00 0.10 0.20 0.30 0.40 0.50 0.60V/S
Fo
rmat
ion
de
CIT
-PR
OP
(p
mo
l/h/m
g p
rot.
)
R-(-)-CITPROP
S-(+)-CITPROP
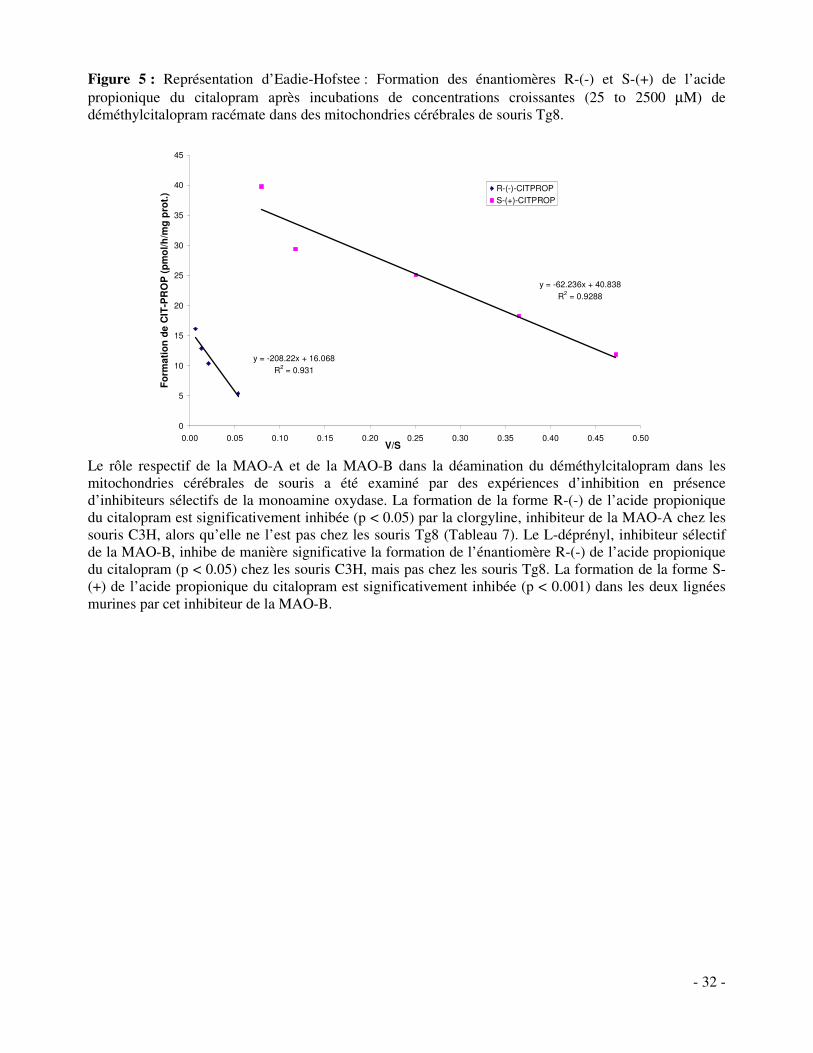
- 32 -
Figure 5 : Représentation d’Eadie-Hofstee : Formation des énantiomères R-(-) et S-(+) de l’acide propionique du citalopram après incubations de concentrations croissantes (25 to 2500 µM) de déméthylcitalopram racémate dans des mitochondries cérébrales de souris Tg8.
y = -208.22x + 16.068
R2 = 0.931
y = -62.236x + 40.838
R2 = 0.9288
0
5
10
15
20
25
30
35
40
45
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35 0.40 0.45 0.50V/S
Fo
rmat
ion
de
CIT
-PR
OP
(p
mo
l/h/m
g p
rot.
) R-(-)-CITPROP
S-(+)-CITPROP
Le rôle respectif de la MAO-A et de la MAO-B dans la déamination du déméthylcitalopram dans les mitochondries cérébrales de souris a été examiné par des expériences d’inhibition en présence d’inhibiteurs sélectifs de la monoamine oxydase. La formation de la forme R-(-) de l’acide propionique du citalopram est significativement inhibée (p < 0.05) par la clorgyline, inhibiteur de la MAO-A chez les souris C3H, alors qu’elle ne l’est pas chez les souris Tg8 (Tableau 7). Le L-déprényl, inhibiteur sélectif de la MAO-B, inhibe de manière significative la formation de l’énantiomère R-(-) de l’acide propionique du citalopram (p < 0.05) chez les souris C3H, mais pas chez les souris Tg8. La formation de la forme S-(+) de l’acide propionique du citalopram est significativement inhibée (p < 0.001) dans les deux lignées murines par cet inhibiteur de la MAO-B.

- 33 -
Tableau 7 : Contribution relative de la MAO-A et de la MAO-B dans le métabolisme de 0.5 mM de déméthylcitalopram dans les mitochondries cérébrales de souris.
Lignée Inhibiteurs R-(-)- S-(+)-
CTRL100 ± 19
[0.53 ± 0.1]
100 ± 14
[0.69 ± 0.1]
Clorgyline 81 ± 10 * 83 ± 6
CTRL100 ± 19
[0.36 ± 0.07]
100 ± 16
[0.81 ± 0.13]
L-deprenyl 75 ± 14 * 32 ± 9 ***
CTRL100 ± 23
[0.30 ± 0.07]
100 ± 9
[0.55 ± 0.05]
Clorgyline 117 ± 50 124 ± 29
CTRL100 ± 12
[0.17 ± 0.02]
100 ± 19
[0.62 ± 0.12]
L-deprenyl 124 ± 71 16 ± 10 ***
¹ Les résultats sont exprimés en pourcents de la valeur mesurée sans inhibiteurs (n=6).
Les valeurs correspondantes à 100% sont en [pmol/min/mg de protéines]
* = p<0.05
** = p<0.01
*** = p<0.001
L'activité de la MAO a été mesurée par la production stéréosélective de l'acide propionique
du citalopram dans des incubations en présence 0.02 µM de clorgyline (concentration
finale), l'inhibiteur spécifique de la MAO-A, ainsi qu'en présence de 0.5 µM de L-déprenyl
(concentration finale), l'inhibiteur spécifique de la MAO-B.
C3H
Tg8
Production d'acide propionique du citalopram ¹
Finalement, l’addition de NADP sur la formation de l’acide propionique du citalopram a été étudiée dans les microsomes cérébraux de souris C3H, afin d’évaluer la contribution des cytochromes P-450 dans la déamination du déméthylcitalopram. Le Tableau 8 montre que ce système enzymatique ne semble pas impliqué, car l’addition de ce cofacteur essentiel n’a pas influencé la production des métabolites.

- 34 -
Tableau 8 : Contribution des cytochromes P-450 dans le métabolisme de 0.5 mM de déméthylcitalopram dans les microsomes cérébraux de souris C3H.
R-(-)- S-(+)-
CTRL100 ± 32
[0.41 ± 0.13]
100 ± 13
[0.33 ± 0.05]
NADP 95 ± 39 97 ± 27
Les valeurs correspondantes à 100% sont en [pmol/min/mg de protéines]
* = p<0.05
** = p<0.01
*** = p<0.001
Production d'acide
propionique du citalopram ¹
L'activité des cytochromes a été mesurée par la production stéréosélective
d'acide propionique du citalopram dans des incubations en l'absence
(CTRL) et en présence de NADP (concentration finale: 330 µM).
¹ Les résultats sont exprimés en pourcents de la valeur mesurée sans
inhibiteurs (n=6).
Discussion Historiquement, la découverte chez une fratrie néerlandaise d’une déficience de l’enzyme de la MAO-A a montré l’influence de cet enzyme sur le comportement et le développement mental (Brunner et al., 1993a). Par la suite, une équipe a mis au point une souris génétiquement modifiée n’exprimant pas le gène de la MAO-A, afin de tester et comprendre les conséquences de cette déficience sur le comportement de l’animal (Cases et al., 1995). Cette même équipe a également constaté qu’en plus des troubles de comportement, ces souris présentaient un métabolisme des amines biogènes, en particulier la sérotonine, profondément modifié (Cases et al., 1995; Cases et al., 1998). Un peu plus tard, le rôle des monoamines oxydases cérébrales dans le métabolisme stéréosélectif du citalopram a été démontré chez l’homme et chez le rat par notre laboratoire (Kosel et al., 2002; Rochat et al., 1995b). La déamination du citalopram n’ayant jamais été étudiée chez la souris, notre laboratoire a saisi cette opportunité pour examiner cette voie métabolique du citalopram chez des souris Tg8, déficientes du gène de la MAO-A. Les amines secondaires et primaires que sont le déméthyl- et le didéméthylcitalopram se sont, en général, montrées être de meilleurs substrats pour les monoamines oxydases que le citalopram, l’amine tertiaire et ses énantiomères R-(-) et S-(+), sauf dans le cas du R-(-)-citalopram chez les souris C3H (Tableau 5). En effet, les résultats du Tableau 5 montrent qu’il y a un métabolisme stéréosélectif du citalopram et de ses métabolites déméthylés par les monoamines oxydases, ainsi qu’une affinité en fonction du substrat. La MAO-A va catalyser la formation des formes R-(-) et S-(+) de l’acide propionique du citalopram avec une affinité supérieure pour les amines tertiaires (la production de l’acide propionique du citalopram varie de 0.41 ± 0.16 chez les souris C3H versus 0.08 ± 0.03 [pmol/min/mg protéines] chez les souris Tg8 après incubation de citalopram). Au contraire, la MAO-B va favoriser la formation de l’énantiomère S-(+) de l’acide propionique du citalopram avec une affinité supérieure pour les amines secondaires et primaires. Les ratios S/R augmentent du citalopram au didéméthylcitalopram pour les deux lignées murines. A noter que chez les souris Tg8, le ratio du déméthylcitalopram est supérieur à celui du didéméthylcitalopram. Ceci peut s’expliquer par le fait que le déméthylcitalopram est le substrat pour lequel la monoamine oxydase a le plus d’affinité (Rochat et al., 1998). Une production plus importante de l’énantiomère S-(+) par la MAO-B a déjà été observée in vitro dans les microsomes hépatiques humains, dans des mitochondries cérébrales humaines, ainsi que dans des mitochondries de cerveaux de rats (Kosel et al., 2002; Rochat et al., 1998). La stéréosélectivité des monoamines oxydases a déjà été démontrée dans les

- 35 -
foies humains où les productions des énantiomères R-(-) et S-(+) de l’acide propionique du citalopram sont inhibées par la clorgyline (inhibiteur de la MAO-A) et par la sélégiline (inhibiteur de la MAO-B), respectivement (Rochat et al., 1998). Des observations identiques ont été faites dans les cerveaux humains et de rats (Kosel et al., 2002). De plus, une stéréosélectivité en fonction du substrat a également déjà été observée dans les cerveaux humains et de rats. Lors de l’utilisation du citalopram comme substrat, la forme R-(-) de l’acide propionique du citalopram est principalement produite, tandis que le déméthyl et le didéméthylcitalopram produisent préférentiellement l’énantiomère S-(+) de l’acide propionique du citalopram (Kosel et al., 2002). Malgré le fait que la production de l’acide propionique du citalopram ne reflète pas quantitativement l’activité des monoamines oxydases, puisque l’oxydation de l’aldéhyde intermédiaire ne peut être mesurée directement, les Km et Vmax apparents ont été mesurés dans les mitochondries cérébrales des deux lignées murines (Tableau 6, Figures 4 et 5). L’affinité (Km) pour l’énantiomère S-(+)- est similaire dans les deux lignées murines. Par contre, les monoamines oxydases ont trois fois moins d’affinité pour l’énantiomère R-(-)- chez les souris Tg8 comparé aux souris C3H (Tableau 6). Ce qui va de paire avec l’absence de MAO-A chez les Tg8. Il en va de même pour la production (Vmax) : si celle-ci est similaire pour l’énantiomère S-(+)-, elle est deux fois moindre pour l’énantiomère R-(-)- chez les souris Tg8 (Tableau 6). Les tests d’inhibitions spécifiques de la MAO-A et de la MAO-B montrent que la biotransformation de l’énantiomère R-(-)- du déméthylcitalopram est significativement inhibée par l’inhibiteur spécifique de la MAO-A, la clorgyline, chez les souris sauvages (C3H), mais nullement chez les souris déficientes du gène de la MAO-A (Tg8). A noter que les inhibitions des deux énantiomères R-(-) et S-(+) sont très proches (81 ± 10 et 83 ± 6 [pmol/min/mg de protéines], respectivement). Il est possible qu’en augmentant le nombre d’expériences, l’inhibition de la biotransformation de l’énantiomère S-(+) de déméthylcitalopram devienne à son tour significative (Tableau 7). L’inhibiteur spécifique de la MAO-B, le L-déprényl, inhibe de façon significative la déamination de l’énantiomère S-(+) dans les deux lignées de souris (Tableau 7). La spécificité des monoamines oxydases pour un substrat dépend de leur concentration et de leur affinité pour celui-ci, mais également de la concentration de l’enzyme, puisque l’inhibition de la MAO-B a aussi un effet inhibiteur sur la forme R-(-)- chez les souris C3H, même si cet énantiomère est principalement métabolisé par la MAO-A (Tableau 7) La spécificité des MAOs est fragile : La MAO-A et la MAO-B ont des séquences très proches, à un tel point que l’utilisation de sondes nucléiques pour étudier l’expression de ces deux gènes ne fait pas l’unanimité et est sujet à controverse. En effet, la substitution d’un seul acide aminé peut inverser la spécificité des MAOs vis-à-vis des substrats endogènes ou des inhibiteurs (Geha et al., 2001; Tsugeno & Ito, 1997). Il est important de préciser que la MAO-A est inhibée par des concentrations nanomolaires de clorgyline (concentration utilisée 0.02 µM), tandis que la MAO-B nécessite des concentrations micromolaires de L-déprényl (concentration utilisée 0.5 µM). L’inverse est vrai lorsque le L-déprényl est utilisé pour inhiber l’activité de la MAO-A (Strolin Benedetti, 2001). Il a déjà été démontré que des cytochromes P-450 ont la faculté de déaminer des aldéhydes en acides carboxyliques (Watanabe et al., 1990). Nous avons vérifié si ceci s’applique également au citalopram. Pour cela, nous avons ajouté du NADP, cofacteur essentiel des cytochromes P-450, à l’incubation de microsomes cérébraux (et non de mitochondries, pour recruter le maximum de cytochrome P-450). Les résultats présentés dans le Tableau 8 montrent que cette famille d’enzymes ne semble pas impliquée dans la voie métabolique menant à l’acide propionique du citalopram. Les cytochromes P-450 ont une fonction de N-déméthylation dans le métabolisme du citalopram, mais pas de déamination (Figure 3). Dans le cerveau de rongeurs, l’activité de la MAO-A est à peu près identique à celle de l’adulte dès la première semaine de vie, tandis que l’activité de la MAO-B est faible à la naissance et se développe durant les premiers mois de la vie en partie dû à la gliogénèse (Cases et al., 1995). A l’âge de 90 jours, l’activité de la MAO-A sur la sérotonine chez les souris déficientes du gène (Tg8) est 35 fois inférieure par rapport aux souris sauvages (C3H) (Cases et al., 1995). L’activité de la MAO-A dans la production d’acide propionique du citalopram suite à l’incubation de citalopram, n’est plus que de 5 fois inférieure chez les souris déficientes (Tg8) comparée aux souris sauvages (C3H) (Tableau 5). Il est intéressant de

- 36 -
noter que toutes les amines biogènes testées (sérotonine, dopamine et noradrénaline) sont des amines primaires. Or, le citalopram est une amine tertiaire. L’énantiomère R-(-) de l’acide propionique du citalopram suite à l’incubation de didéméthylcitalopram (amine primaire) montre une production quasi identique (Tableau 5). Cela s’explique, d’une part, parce que le didéméthylcitalopram est un meilleur substrat pour la MAO-B et, d’autre part, parce que les tests ont été effectués à l’âge adulte où les cellules gliales riches en MAO-B sont en place, contrairement aux nouveau-nés. Il a également a été observé dans la déamination des catécholamines que les deux isoenzymes (MAO-A et MAO-B) ne parviennent pas à se compenser de manière équivalente au cas où l’un des deux enzymes est déficient (Lenders et al., 1996). L’absence de MAO-A engendre des taux de sérotonine neuf fois supérieurs chez les souris déficientes du gène de la MAO-A (Tg8) comparé aux souris sauvages durant la première semaine de vie (Cases et al., 1995). A leur tour, ces taux élevés de sérotonine sont responsables de la désorganisation de l’architecture neuroanatomique du cortex somatosensoriel, en particulier des fibres thalamocorticales (Rebsam et al., 2002; Salichon et al., 2001) D’autres études sont nécessaires, tout d’abord avec ces mêmes souris déficientes du gène de la MAO-A (Tg8) mais âgées au maximum d’une semaine, afin de s’assurer d’être dans la période la plus sensible, ensuite avec les souris déficientes du gène de la MAO-B (Grimsby et al., 1997). Pour faciliter les comparaisons, les souris seraient testées aux mêmes âges (première semaine de vie et âge adulte). Enfin, les mêmes tests seraient reproduits avec les souris déficientes des deux gènes MAO-A et MAO-B (Chen et al., 2004). On pourrait ainsi comparer l’importance relative des deux isoenzymes dans la déamination du citalopram et ses métabolites déméthylés. Finalement, la caractérisation de certains métabolites encore hypothétiques, car jamais identifiés (alcool propionique du citalopram, aldéhyde propionique du citalopram), permettrait de tester de manière exhaustive l’implication des différents enzymes menant à l’acide propionique du citalopram.

- 37 -
Métabolisme du citalopram par les cytochromes P-450 chez le chien
Résumé Une étude a été menée pour mieux comprendre les causes de la toxicité du citalopram chez les chiens. Nous avons étudié les deux étapes de N-déméthylation menant au didéméthylcitalopram dont les taux se sont révélés très élevés et toxiques chez les chiens. Les résultats initiaux de cette étude ont fait l’objet d’un travail de diplôme, qui ont été repris et augmentés durant cette thèse 2. Les deux N-déméthylations ont des cinétiques biphasiques, où deux voir plusieurs enzymes sont impliqués. Le CYP2D15, orthologue canin du CYP2D6, semble être l’enzyme à haute affinité/faible capacité dans les deux réactions menant au didéméthylcitalopram. Nous avons constaté que l’activité enzymatique chez les chiens est beaucoup plus élevée que chez l’homme (clearance intrinsèque élevée). De plus, les chiens ont jusqu’à quatre cytochromes P-450 (CYP2D15, CYP3A12, CYP2C9 et CYP2E1) impliqués dans la deuxième N-déméthylation menant au didéméthylcitalopram. Ceci contrairement à l’homme, où un seul cytochrome est impliqué (le CYP2D6). La connaissance des cytochromes P-450 canins reste limitée, quand bien même les chiens sont couramment utilisés dans les études de pharmacocinétique et lors du développement de nouvelles molécules. Neuf cytochromes P-450 canins ont été identifiés par des techniques de clonage et de séquençage. Cinq d’entre eux sont connus pour présenter au moins un polymorphisme génétique (Kamimura, 2006). Des différences concernant les réactions enzymatiques chez les chiens pourraient exister dans cette étude puisque basée sur les cytochromes P-450 humains. Enfin, la principale forme des cytochromes P-450 dans les microsomes de chiens Beagles serait la sous-famille des CYP2D. Ils représenteraient 20% du total des cytochromes P-450 canins, ce qui serait unique comparé aux autres espèces. Si c’est bien cet enzyme qui a une haute affinité/faible capacité pour les deux N-déméthylation, il est probable que les taux plasmatiques des métabolites du citalopram augmentent rapidement.
Matériel & méthodes
Substances et produits chimique Les substances suivantes ont été offertes : les énantiomères du citalopram (S-(+)- et R-(-)-citalopram), les mélanges racémiques du citalopram, du déméthylcitalopram, du didéméthylcitalopram et de l’acide propionique du citalopram (Lundbeck AS, Copenhague, Danemark), le S-flurbiprofen (Boots Limited, Nottingham, Grande-Bretagne). Le S-alprenolol ainsi que les inhibiteurs spécifiques des CYP3A4/CYP3A12, CYP2D6/CYP2D15, CYP2C19, CYP2E1, CYP2C9, CYP1A2, respectivement le kétoconazole, la quinidine, l’oméprazole, le 4-methyl pyrazol, le sulphaphénazole et la furafylline, ont été achetés chez Sigma (Bâle, Suisse). Le L-déprényl (sélégiline) a quant à lui été acheté chez Research Biochemicals Inc. (Natick, MA, USA). Les autres substances et produits chimiques ont été trouvés dans le commerce et étaient de la plus pure qualité disponible.
Tissus et incubation Les foies de chiens Beagles ont été aimablement offerts par Lundbeck (Copenhague, Danemark). Les incubations des microsomes hépatiques canins ont été réalisées selon les conditions adaptées d’une procédure déjà décrite (Rochat et al., 1997a). Elles ont été pré-incubées, sans substrat, pendant 15 minutes à 37°C, puis initiées par l’ajout du substrat et stoppées après 1 heure par l’addition de 750 µl de NaOH 0.25 M. Les incubations microsomales avaient un volume final de 250 µl à pH 7.4 contenant 100 µg de protéines et des concentrations croissantes de
2 Travail de diplôme de Cédric Maître, Metabolism of Citalopram using Beagle Dog Liver Microsomes, Ecole de Pharmacie, Faculté des Sciences, Juin 2004.

- 38 -
substrats (0.5, 1, 5, 10, 50, 100, 200, 500, 750 et 1000 µM) pour les cinétiques enzymatiques ou à deux concentrations fixes de substrats (10 et 100 µM) pour les tests d’inhibitions enzymatiques. Les substrats utilisés étaient les S-(+)- et R-(-)-citalopram, le citalopram, le déméthylcitalopram et le didéméthyl citalopram racémiques. Dans les études d’inhibitions sélectives des cytochromes P-450, tous les échantillons ont préincubé avec 20 µl d’une solution de NADP 0.33 mM, du kétoconazole, de la quinidine, de l’oméprazole, de la furafylline, du 4- méthyl – pyrazole ou du sulfaphénazole pendant 15 minutes à 37°C (Moody et al., 1999). Les échantillons contrôles ne contenaient que les 20 µl d’une solution de NADP 0.33 mM. Les solutions de travail étaient composées de 5 µM de kétoconazole, de 1 mM d’oméprazole, de 2 mM de 4-méthyl-pyrazole dans du tampon phosphate 0.1 M à pH 7.4, de 0.2 mM de quinidine dans un mélange tampon phosphate à 0.1 M pH 7.4 / DMSO [90 : 10 (v/v)] et de 0.5 mM sulfaphénazole, 0.5 mM de furafylline dans un mélange tampon phosphate à 0.1 M pH 7.4 / DMSO [98 : 2 (v/v)]. Une solution pour l’inhibition non sélective des cytochromes P-450 par 0.5 mM de proadifen a été préparée dans du tampon phosphate 0.1 M à pH 7.4. Les conditions utilisées pour l’étude de l’inhibition sélective de la MAO-A ou de la MAO-B étaient identiques à celles utilisées pour les incubations de mitochondries cérébrales de souris.
Extraction Les conditions d’extraction utilisées ont été celles développées initialement dans notre laboratoire (Kosel et al., 1998; Rochat et al., 1997a). Quelques modifications furent nécessaires : 500 ng de S-alprenolol (utilisé comme standard interne), 50 µl de NaOH 1M et 1.5 ml de n-heptane-isoamyl alcool [98.5 : 1.5 (v/v)] ont été ajoutés aux incubats. Le mélange a été agité pendant 15 minutes et centrifugé à 3300 g durant 8 min à 8°C. La phase organique a été récupérée et transvasée dans des flacons injecteurs à fonds plats et évaporés à sec sous flux d’azote à 40°C. L’extrait sec a été re-suspendu dans 150 µl de phase mobile (méthanol – 0.005 mM d’acide citrique / TEA à pH 6.3 [60 : 40 (v/v)]) et transvasé dans des flacons injecteurs. Cette méthode a permis l’extraction du citalopram, du déméthylcitalopram et du didéméthylcitalopram. L’acide propionique du citalopram, ainsi que le citalopram N-oxyde n’étaient pas extraits par cette méthode. Des échantillons de 100 µl ont été injectés dans le système HPLC, sauf lorsque les concentrations de citalopram et déméthylcitalopram dépassaient les 100 µM. Pour ces échantillons plus concentrés, seuls 10 µl étaient injectés afin de permettre la quantification des autres métabolites déméthylés.
Conditions analytiques Les conditions analytiques utilisées pour la quantification des énantiomères R-(-)- et S-(+)- du citalopram, du déméthylcitalopram et du didéméthylcitalopram étaient celles décrites dans la littérature (Kosel et al., 1998; Rochat et al., 1997a) et modifiées comme suit : Un chromatographe liquide Agilent - HP Série 1100 équipé d’une pompe binaire, et d’un injecteur automatique, avec une colonne analytique chirale (Cyclobond I 2000, β-cyclodextrines acétylées, Cat n° 20124/Séries n° 17514, diamètre des particules 10 µm, 250 x 4.6mm de ASTEC Whippany, NJ 07981 USA) était couplée à un détecteur fluorimétrique. La détection fluorimétrique a été réalisée avec une excitation à 236 nm et une émission à 305 nm. Le flux était de 0.6 ml/min. La phase mobile initiale (Carlsson & Norlander, 2001) a été légèrement modifiée : méthanol – 0.005 mM d’acide citrique / TEA à pH 6.3 [60 : 40 (v/v)].
Statistiques Les analyses statistiques ont été effectuées avec le logiciel Statistica 5.0, StatSoft Inc., Tulsa, OK, USA. Les analyses de variances (ANOVA) avec le post test de Bonferroni ont été faites avec le logiciel GraphPad Prism version 4.00 pour windows, GraphPad Software, San Diego, CA, USA. Une analyse de variance de type two-way ANOVA suivie du post test de Bonferroni (p < 0.05) a été utilisée pour comparer les résultats obtenus lors des études d’inhibition des cytochromes P-450 chez les chiens. Les données cinétiques (Km et Vmax) ont été analysées par les moyennes du “non-linear least square curve fitting program” du logiciel GraphPad Prism version 4.00 pour windows, GraphPad Software, San Diego, CA, USA.

- 39 -
Résultats
N-déméthylation du citalopram et inhibition de la formation du déméthylcitalopram Les quantités de R-(-)- ou S-(+)-déméthylcitalopram ainsi que le racémate formé en fonction de la quantité de citalopram incubée sont représentées dans la Figure 6. Les valeurs obtenues pour le déméthylcitalopram racémique ont été ajustées au moyen de l’équation d’une cinétique monophasique, v = (Vmax [S]) / (Km + [S]). La N-déméthylation du citalopram en R-(-)- ou S-(+)-déméthylcitalopram n’est pas stéréosélective, car la quantité produite de chacun des énantiomères est relativement proche. Les valeurs obtenues ne sont pas distribuées de manière homogène autour de la courbe d’ajustement. Ceci suggère la possible implication de plusieurs enzymes dans la première N-déméthylation du citalopram (Figure 6).
Figure 6 : Formation de R-(-) et S-(+)-déméthylcitalopram, ainsi que de déméthylcitalopram racémique après incubations de concentrations croissantes (0.5 to 1000 µM) de citalopram racémate dans des microsomes hépatiques de chiens.
0
50
100
150
200
250
300
350
400
0 100 200 300 400 500 600 700 800 900 1000
CIT racémate [µM]
Fo
rmat
ion
de
DC
IT (
pm
ol/m
in/m
g p
rot)
S-(+)-DCIT
R-(-)-DCIT
DCIT
Courbe monophasique "ideale" rapportée au racémate
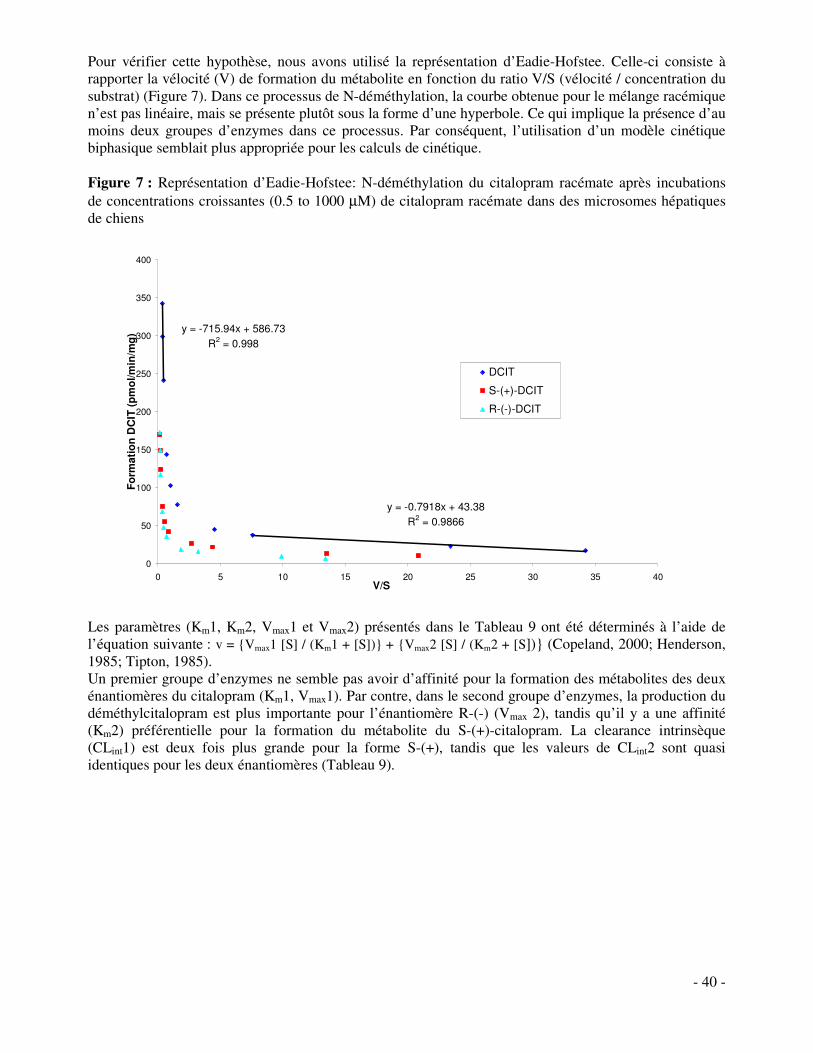
- 40 -
Pour vérifier cette hypothèse, nous avons utilisé la représentation d’Eadie-Hofstee. Celle-ci consiste à rapporter la vélocité (V) de formation du métabolite en fonction du ratio V/S (vélocité / concentration du substrat) (Figure 7). Dans ce processus de N-déméthylation, la courbe obtenue pour le mélange racémique n’est pas linéaire, mais se présente plutôt sous la forme d’une hyperbole. Ce qui implique la présence d’au moins deux groupes d’enzymes dans ce processus. Par conséquent, l’utilisation d’un modèle cinétique biphasique semblait plus appropriée pour les calculs de cinétique.
Figure 7 : Représentation d’Eadie-Hofstee: N-déméthylation du citalopram racémate après incubations de concentrations croissantes (0.5 to 1000 µM) de citalopram racémate dans des microsomes hépatiques de chiens
y = -0.7918x + 43.38
R2 = 0.9866
y = -715.94x + 586.73
R2 = 0.998
0
50
100
150
200
250
300
350
400
0 5 10 15 20 25 30 35 40V/S
Fo
rmat
ion
DC
IT (
pm
ol/m
in/m
g)
DCIT
S-(+)-DCIT
R-(-)-DCIT
Les paramètres (Km1, Km2, Vmax1 et Vmax2) présentés dans le Tableau 9 ont été déterminés à l’aide de l’équation suivante : v = {Vmax1 [S] / (Km1 + [S])} + {Vmax2 [S] / (Km2 + [S])} (Copeland, 2000; Henderson, 1985; Tipton, 1985). Un premier groupe d’enzymes ne semble pas avoir d’affinité pour la formation des métabolites des deux énantiomères du citalopram (Km1, Vmax1). Par contre, dans le second groupe d’enzymes, la production du déméthylcitalopram est plus importante pour l’énantiomère R-(-) (Vmax 2), tandis qu’il y a une affinité (Km2) préférentielle pour la formation du métabolite du S-(+)-citalopram. La clearance intrinsèque (CLint1) est deux fois plus grande pour la forme S-(+), tandis que les valeurs de CLint2 sont quasi identiques pour les deux énantiomères (Tableau 9).

- 41 -
Tableau 9 : Constantes cinétiques de la N-déméthylation du citalopram en déméthylcitalopram dans les microsomes hépatiques de chiens.
Km1 Vmax1 CLint1 Km2 Vmax2 CLint2
CIT 1.1 ± 0.3 47 ± 3 42.7 1006 ± 94 591 ± 28 0.6
S-(+)-CIT 1.0 ± 0.2 26 ± 1 26 848 ± 69 264 ± 10 0.3
R-(-)-CIT 1.4 ± 0.5 21 ± 2 15 1221 ± 161 338 ± 24 0.3
Clint (µl/min/mg de protéines) = Vmax/Km
Km (µM), Vmax (pmol/min/mg de protéines), moyenne ± SD (n=6)
Les concentrations des inhibiteurs ont été choisies selon les données de la littérature (Chauret et al., 1997; Kuroha et al., 2002; Rochat et al., 1997a) (Tableau 10). A basse concentration de citalopram racémique (10 µM), la N-déméthylation par le CYP2D15 est sensiblement inhibée par la quinidine pour les deux énantiomères R-(-)- et S-(+)- (p < 0.01 et p < 0.001, respectivement) (Tableau 10). A concentration plus élevée du substrat (100 µM), la N-déméthylation du citalopram par le CYP2D15 reste clairement inhibée par la quinidine (p < 0.001). A ceci s’ajoute l’inhibition du CYP3A12 par le kétoconazole pour les deux énantiomères (p < 0.01) (Tableau 10). L’inhibition non spécifique de la N-déméthylation des cytochromes P-450 par le proadifen est plus significative pour l’énantiomère S-(+)- (p < 0.01) que l’énantiomère R-(-)- (p < 0.05) (Tableau 10). Les résultats suggèrent aussi une inhibition de la N-déméthylation par l’oméprazole, mais elle n’est pas significative. Ceci est probablement dû à l’écart-type trop important (Tableau 10).
Tableau 10 : Inhibition de la formation de déméthylcitalopram par différents inhibiteurs des cytochromes P-450 après incubation de citalopram dans des microsomes hépatiques de chiens.
R-(-)- S-(+)-
CIT 10 µM aucun (ctrl)100 ± 25
[36 ± 9]
100 ± 11
[51 ± 6]
CYP3A12 1 µM kétoconazole 67 ± 8 75 ± 7CYP2D15 100 µM quinidine 31 ± 7 ** 27 ± 6 ***
CYP2C21 10 µM oméprazole 64 ± 22 70 ± 19
CIT 100 µM aucun (ctrl)100 ± 11
[105 ± 12]
100 ± 9
[108 ± 10]
CYP3A12 1 µM kétoconazole 75 ± 14 ** 70 ± 9 **
CYP2D15 100 µM quinidine 57 ± 28 *** 38 ± 11 ***
CYP2C21 10 µM oméprazole 84 ± 28 81 ± 20
CIT 100 µM aucun (ctrl)100 ± 4
[86 ± 3]
100 ± 3.8
[93 ± 4]
CYP1A2 50 µM furafyline 105 ± 5 110 ± 5
CYP2C9 10 µM sulfaphénazole 104 ± 2 94 ± 4CYP2E1 10 mM 4-méthyl-pyrazole 94 ± 2 94 ± 4
CYP non spécifique 100 µM proadifen [SKF-525] 82 ± 12 * 72 ± 7 **
¹ Les résultats sont exprimés en pourcents de la valeur mesurée sans inhibiteurs (n=6).
Les valeurs correspondantes à 100% sont en [pmol/min/mg de protéines]
* = p<0.05
** = p<0.01
*** = p<0.001
Formation de déméthylcitalopram 1
Enzymes Inhibiteurs

- 42 -
N-déméthylation du déméthylcitalopram et inhibition de la formation du didéméthylcitalopram
Figure 8 : Formation de R-(-)- et S-(+)-didéméthylcitalopram, ainsi que de didéméthylcitalopram racémique après incubations de concentrations croissantes (0.5 to 1000 µM) de déméthylcitalopram racémate dans des microsomes hépatiques de chiens.
0
20
40
60
80
100
120
140
160
180
200
0 100 200 300 400 500 600 700 800 900 1000
DCIT racémique [µM]
Fo
rmat
ion
DD
CIT
(p
mo
l/min
/mg
pro
t)
S-(+)-DDCIT
R-(-)-DDCIT
DDCIT
Courbe monophasique "ideale" rapportée au racémate
Les quantités de R-(-) et S-(+) didéméthylcitalopram ainsi que le racémate formé en fonction de la quantité de déméthylcitalopram incubée sont représentés dans la Figure 8. Les valeurs obtenues pour le didéméthylcitalopram ont été ajustées au moyen de l’équation d’une cinétique monophasique, v = (Vmax [S]) / (Km + [S]). Les valeurs obtenues ne sont pas distribuées de manière homogène autour de la courbe d’ajustement. Ceci suggère à nouveau la possible implication de plusieurs enzymes dans cette deuxième N-déméthylation du citalopram. A noter que dans des conditions d’incubations identiques et à concentrations maximales identiques des substrats respectifs (1000 µM), la formation de didéméthylcitalopram est deux fois moins importante que celle du déméthylcitalopram (Figure 6 et 8). Le déméthylcitalopram est préférentiellement déméthylé en S-(+)-didéméthylcitalopram à basse concentration, tandis qu’il est plutôt déméthylé en R-(-)-didéméthylcitalopram à haute concentration (Figure 8). La représentation d’Eadie-Hofstee (Figure 9) montre de nouveau une représentation hyperbolique suggérant l’implication de deux groupes d’enzymes.

- 43 -
Figure 9 : Représentation d’Eadie-Hofstee: N-déméthylation du didéméthylcitalopram racémate après incubations de concentrations croissantes (0.5 to 1000 µM) de déméthylcitalopram racémate dans des microsomes hépatiques de chiens.
y = -0.6996x + 50.592
R2 = 0.989
y = -167.48x + 214.18
R2 = 0.9954
0
20
40
60
80
100
120
140
160
180
200
0 5 10 15 20 25 30 35 40 45V/S
Fo
rmat
ion
DD
CIT
(p
mo
l/min
/mg
pro
t)
DDCIT
S-(+)-DDCIT
R-(-)-DDCIT
Les paramètres (Km1, Km2, Vmax1 et Vmax2) présentés dans la Tableau 11 ont été déterminés à l’aide de l’équation suivante : v = {Vmax1 [S] / (Km1 + [S])} + {Vmax2 [S] / (Km2 + [S])} (Copeland, 2000; Henderson, 1985; Tipton, 1985). Pour le premier groupe d’enzymes, la production de didéméthylcitalopram (Vmax1) est plus grande pour l’énantiomère S-(+), tandis que l’affinité (Km1) est plus grande pour le R-(-)-déméthylcitalopram. A l’inverse, le deuxième groupe d’enzyme montre une production (Vmax2) plus importante pour l’énantiomère R-(-) et une affinité (Km2) préférentielle pour le S-(+)-déméthylcitalopram. Les clearances intrinsèques (CLint1 et CLint2) sont une fois et demie plus grandes pour l’énantiomère S-(+)- comparé à l’énantiomère R-(-)- (Tableau11).
Tableau 11 : Constantes cinétiques de la N-déméthylation du déméthylcitalopram en didéméthylcitalopram dans les microsomes hépatiques de chiens.
Km1 Vmax1 CLint1 Km2 Vmax2 CLint2
DCIT 0.6 ± 0.2 44 ± 4 73 137 ± 18 155 ± 5 1.1
S-(+)-DCIT 0.6 ± 0.1 31 ± 3 52 66 ± 11 55 ± 3 0.8
R-(-)-DCIT 0.3 ± 0.2 11 ± 2 37 212 ± 24 105 ± 3 0.5
Clint (µl/min/mg de protéines) = Vmax/Km
Km (µM), Vmax (pmol/min/mg de protéines), moyenne ± SD (n=6)
A basse concentration de déméthylcitalopram (10 µM), la N-déméthylation par le CYP2D15 est sensiblement inhibée par la quinidine, et ceci est valable pour les deux énantiomères (p < 0.001) (Tableau 12).

- 44 -
A concentration plus élevée du même substrat (100 µM), la N-déméthylation du déméthylcitalopram par le CYP2D15 reste clairement inhibée par la quinidine pour les formes R-(-)- et S-(+)- (p < 0.001). A ceci s’ajoute également l’inhibition du CYP3A12 par le kétoconazole (p < 0.01). L’inhibition du CYP2C21 par l’oméprazole est uniquement significative pour l’énantiomère S-(+)- (p < 0.01) (Tableau 12). Le sulfaphénazole (inhibiteur spécifique du CYP2C9) et le 4-méthyl-pyrazole (inhibiteur spécifique du CYP2E1) inhibent significativement la N-déméthylation des deux énantiomères (p < 0.01). Enfin l’inhibition non spécifique par le proadifen est également significative (p < 0.001) (Tableau 12).
Tableau 12 : Inhibition de la formation de didéméthylcitalopram par différents inhibiteurs de cytochromes P-450 après incubation de déméthylcitalopram dans des microsomes hépatiques de chiens.
R-(-)- S-(+)-
DCIT 10 µM aucun (ctrl)100 ± 4
[79 ± 3]
100 ± 4
[99 ± 7]
CYP3A12 1 µM kétoconazole 96 ± 2 96 ± 3
CYP2D15 100 µM quinidine 20 ± 0.4 *** 14 ± 0.4 ***
CYP2C21 10 µM oméprazole 101 ± 1 95 ± 2
DCIT 100 µM aucun (ctrl)100 ± 3
[240 ± 6]
100 ± 3
[255 ± 8]
CYP3A12 1 µM kétoconazole 87 ± 4 ** 90 ± 4 **
CYP2D15 100 µM quinidine 38 ± 1 *** 42 ± 2 ***
CYP2C21 10 µM oméprazole 96 ± 7 92 ± 1 **
DCIT 100 µM aucun (ctrl)100 ± 1.2
[181 ± 1]
100 ± 1
[183 ± 1]
CYP1A2 50 µM furafyline 93 ± 8 95.5 ± 7
CYP2C9 10 µM sulfaphénazole 84 ± 2 ** 83 ± 1 **
CYP2E1 10 mM 4-méthyl-pyrazole 76 ± 9 ** 77 ± 8 **
CYP non specifique 100 µM proadifen [SKF-525] 69 ± 3 *** 68 ± 6 ***
¹ Les résultats sont exprimés en pourcents de la valeur mesurée sans inhibiteurs (n=6).
Les valeurs correspondantes à 100% sont en [pmol/min/mg de protéines]
* = p<0.05
** = p<0.01
*** = p<0.001
Formation of didéméthylcitalopram 1
Enzymes Inhibiteurs
Discussion Lors de l’évaluation préclinique du citalopram par la firme Lundbeck, celle-ci a rencontré un problème inattendu lors des essais chez l’animal. Les chiens Beagles ont présenté des taux plasmatiques très élevés et toxiques de didéméthylcitalopram après administration de citalopram normalement peu toxiques chez d’autres espèces, y compris chez l’Homme (Overø, 1989). Notre laboratoire a développé et validé des méthodes analytiques stéréosélectives très sensibles permettant de doser le citalopram et ses métabolites (Kosel et al., 1998; Rochat et al., 1995a; Rochat et al., 1995b). Elles ont permis d’identifier trois cytochromes P-450 impliqués dans la N-déméthylation du citalopram chez l’Homme (Rochat et al., 1997a). Nous avons utilisé ces méthodes pour identifier les cytochromes P-450 canins impliqués dans la N-déméthylation du citalopram, afin de les comparer avec les résultats obtenus chez l’Homme.
A. N-déméthylation du citalopram en déméthylcitalopram Le graphique de la formation du déméthylcitalopram après incubation de citalopram ne correspond pas à la courbe hyperbolique attendue lors d’une réaction monophasique. Ceci suggère qu’au moins deux enzymes sont impliqués (Figure 6). Ceci est confirmé par la représentation non linéaire du graphique selon Eadie-Hofstee (Figure 7). Les données analysées selon un modèle biphasique montrent qu’un des

- 45 -
enzymes a une haute affinité pour le substrat (Km1 = 1.1 µM) pour une faible capacité (Vmax1 = 47 pmol/min/mg prot.), tandis que l’autre enzyme a une très grande capacité (Vmax2 = 591 pmol/min/mg prot.) pour une faible affinité (Km2 = 1006 µM) (Tableau 9). L’enzyme présentant une haute affinité a une capacité (Vmax) 10 fois plus petite que celle de l’enzyme avec une faible affinité. A noter que, dans une étude in vitro avec des microsomes humains, il a également été rapporté que deux ou plusieurs enzymes étaient impliqués dans la N-déméthylation du citalopram chez l’Homme, ainsi qu’une capacité (Vmax) 10 fois moindre pour l’enzyme ayant une haute affinité par rapport à l’enzyme avec faible affinité (Rochat et al., 1997a). De ce point de vue, il y a une similitude entre l’Homme et le chien (Rochat et al., 1997a). Chez le chien, le groupe d’enzymes (CYP3A12 et CYP2C21 orthologues canins des CYP3A4 et CYP2C19, respectivement) ayant une faible affinité ne favorise pas la formation du S-(+)-déméthylcitalopram et ceci contrairement à ce qui a été observé chez l’Homme (Rochat et al., 1997a). En effet, la Vmax2 du R-(-)-déméthylcitalopram est plus importante que celle de la forme S-(+)- (338 versus 264 pmol/min/mg prot, respectivement). Par contre, l’enzyme à haute affinité (CYP2D15 orthologue canin du CYP2D6) a son Km en faveur du S-(+)-déméthylcitalopram et non pour la forme R-(-)-, ce qui a été observé chez l’Homme (Rochat et al., 1997a). (Km1 = 1.0 versus 1.4 µM, respectivement) (Tableau 9). Si l’élimination d’un xénobiotique est largement dépendante du métabolisme hépatique ou d’un cytochrome en particulier, la clearance intrinsèque (Vmax / Km) est donnée comme une mesure de l’activité enzymatique dans le foie (Wilkinson, 1987). Or les clearances intrinsèques pour l’enzyme à haute affinité/faible capacité sont élevées (Tableau 9).
Tableau 13 : Comparaison des paramètres cinétiques (Km et Vmax) de la N-déméthylation du citalopram dans des microsomes hépatiques chez l’Homme et le chien Beagle.
S-(+)-CIT R-(-)-CIT S-(+)-CIT R-(-)-CIT S-(+)-DCIT R-(-)-DCIT
Km1 33.8 17.3 1.0 1.4 0.7 0.4
Vmax1 1.2 1.5 1.6 1.2 1.9 0.7
CLint1 35.2 87.5 1600 857.1 2714.3 1750
Km2 202.6 184.9 855 1233 68 221
Vmax2 13.2 9.2 16 20 3.3 6.3
CLint2 65.2 49.7 18.7 16.2 48.5 28.5
Clint (µl/ h /mg de protéines) = Vmax/Km
Homme 1 Chiens Beagle
Km (µM), Vmax (nmol/ h /mg de protéines), moyenne ± SD (n=6)
1 selon Rochat, B. et al. Pharmacogenetics 1997; 7:1-10
En comparant les résultats de la première N-déméthylation à ceux obtenus par Rochat et al. (1997) chez l’Homme (Tableau 13), il ressort, d’une part une nettement plus grande affinité (Km1) 30 fois plus grande chez le chien comparée à l’Homme pour l’énantiomère S-(+) du citalopram et d’autre part une clearance intrinsèque (Clint1) 45 fois supérieure chez le chien. De plus, chez l’Homme l’affinité (Km1 et Km2) semble être en faveur de la forme R-(-), tandis que l’inverse est observé chez les chiens (Tableau 12). La deuxième N-déméthylation n’a pas été testée chez l’Homme, cependant au vu des résultats chez les chiens, on constate une affinité (Km1) encore augmentée, comparé à la première N-déméthylation, ceci tant pour la forme S-(+) que pour la forme R-(-), et une clearance intrinsèque qui a presque doublé (Tableau 13). L’activité enzymatique semble nettement plus élevée chez les chiens que chez l’Homme.
Plusieurs études sur la cinétique du citalopram chez l’Homme ont été publiées. Parmi celles-ci, une étude montre que la cinétique serait monophasique (à un seul enzyme) (Von Moltke et al., 1999), tandis que deux autres études démontrent que la cinétique serait biphasique (à deux ou plusieurs enzymes) (Olesen

- 46 -
& Linnet, 1999; Rochat et al., 1997a). Les résultats et graphiques d’Eadie-Hofstee obtenus chez les chiens sont en faveur d’une cinétique biphasique. L’inhibition de la première N-déméthylation de citalopram est sensible à la quinidine. Elle cause une diminution de production du S-(+)-déméthylcitalopram de plus de 70%. Ceci implique que le CYP2D15, l’orthologue canin du CYP2D6 est impliqué dans cette métabolisation. L’inhibition du CYP2D15 par la quinidine étant plus importante à basse concentration (10 µM de citalopram) qu’à plus haute concentration (100 µM de citalopram), fait du CYP2D15 un candidat potentiel pour être l’enzyme à haute affinité/faible capacité (Tableau 10). Les concentrations de quinidine (100µM) utilisées par Chauret et al. (1997) pour inhiber la dextromethorphane-O-déméthylase étaient 20 fois supérieures à celles utilisées par Rochat et al (1997) pour inhiber la formation de déméthylcitalopram (Chauret et al., 1997; Rochat et al., 1997a). 1 µM de quinidine inhibe jusqu’à 80% de l’activité du CYP2D6 chez l’homme, tandis que 100 µM de cette même substance n’inhibe que 60% du CYP2D15 chez le chien Beagle. Ceci est particulièrement vrai, car des différences inter espèces ont été rapportées concernant les tests d’inhibitions du CYP2D6 (Chauret et al., 1997). De plus, les concentrations de quinidine utilisées pour inhiber l’activité du CYP2D6 n’ont apparemment pas d’effet sur les autres cytochromes P-450 (Brøsen, 1990). Il est par conséquent difficile de prédire si l’inhibition du CYP2D15 se fait de manière sélective, ou si l’activité métabolique d’autres cytochromes P-450 a également été inhibée. L’inhibition spécifique du CYP3A12 par le kétoconazole (concentration finale de 1 µM) montre une diminution de la production des deux énantiomères. Enfin, on observe une diminution de production de déméthylcitalopram suite à l’inhibition du CYP2C21 par l’oméprazole, mais celle-ci n’est pas significative (Tableau 10). Le fait que le CYP2C21, orthologue canin du CYP2C19 ne soit pas diminué est plutôt surprenant, puisque le CYP2C19, avec le CYP2D6, apparaissent comme les deux enzymes principaux de la première N-déméthylation chez l’Homme (Sindrup et al., 1993). Les autres cytochromes P-450 testés ne semblent pas être impliqués.
B. N-déméthylation du déméthylcitalopram en didéméthylcitalopram Le graphique de la formation du didéméthylcitalopram après incubation de déméthylcitalopram correspond à une courbe hyperbolique attendue lors d’une réaction monophasique (Figure 8). Cependant, la représentation non linéaire du graphique selon Eadie-Hofstee suggère que deux ou plusieurs enzymes sont impliqués (Figure 9). Cette deuxième étape de N-déméthylation du citalopram semble plus stéréosélective. A basse concentration de déméthylcitalopram, l’enzyme ayant une haute affinité/faible capacité produit plus de S-(+)-didéméthylcitalopram. Aux concentrations plus élevées, c’est l’enzyme ayant une faible affinité/grande capacité qui déméthyle plus rapidement le R-(-)-déméthylcitalopram aboutissant à des plus hautes concentrations de R-(-)-didéméthylcitalopram (Tableaux 11). Un enzyme ayant une très grande affinité/faible capacité est présent dans la deuxième N-déméthylation menant à la formation de taux élevés de didéméthylcitalopram (Tableau 11). Un deuxième enzyme ayant une faible affinité/grande capacité est impliqué dans la N-déméthylation stéréosélective conduisant à la production de R-(-)-didéméthylcitalopram, ce qui a déjà été observé chez l’Homme (Olesen & Linnet, 1999). A noter que la clearance intrinsèque pour l’enzyme à haute affinité/faible capacité dans la deuxième N-déméthylation est presque deux fois plus grande que pour la première N-déméthylation (73 versus 42.7 µl/min/mg de protéines, respectivement) (Tableau 11). Il n’existe pas de résultats chez l’Homme pour la cinétique de la deuxième N-déméthylation. Cependant, en reprenant les résultats du Tableau 13, il en ressort une très nette augmentation de l’activité enzymatique chez les chiens, ainsi qu’une forte affinité (Km1) lors de la première N-déméthylation. Cette affinité augmente encore lors de la deuxième N-déméthylation (Tableau 13). Comme lors de la première N-déméthylation, la formation de didéméthylcitalopram par le CYP2D15 est inhibée par la quinidine, mais de manière encore plus importante. Ceci se voit également chez l’Homme. Nos résultats montrent l’implication d’autres cytochromes P-450 dans cette deuxième N-déméthylation,

- 47 -
tandis que chez l’Homme seul le CYP2D6 est supposé être responsable de la deuxième N-déméthylation (Tableau 11) (Sindrup et al., 1993). Outre, le CYP3A12 qui était aussi impliqué dans la première N-déméthylation, le CYP2C9 et le CYP2E1 voient leurs productions de didéméthylcitalopram significativement inhibées par le sulfaphénazole et la 4-méthyl-pyrazole, respectivement (Tableau 12). Enfin, l’inhibition du CYP2C21/41 ne serait significative qu’à haute concentration et uniquement pour la forme S-(+)-. D’autres études sont nécessaires pour mieux comprendre l’implication de ces différents cytochromes P-450 canins et en particuliers ceux présents lors de la deuxième N-déméthylation.
C. Hypothèses sur la toxicité du citalopram chez le chien Plusieurs hypothèses peuvent être émises afin d’expliquer la toxicité inattendue du citalopram chez les chiens. Il a été rapporté que la principale forme des cytochromes P-450 dans les microsomes de chiens Beagles serait la sous-famille des CYP2D. Ils représenteraient 20% du total des cytochromes P-450 canins, ce qui serait unique comparé aux autres espèces (Sakamoto et al., 1995). A titre de comparaison, chez l’Homme, les CYP2C9, CYP2C19 et CYP2D6 représenteraient le 40% du total des cytochromes P-450 humain (Ingelman-Sundberg, 2005). Si c’est bien cet enzyme qui a une haute affinité/faible capacité pour les deux N-déméthylations, il est probable que les taux plasmatiques des métabolites du citalopram augmentent rapidement. Il est donc nécessaire de faire d’autres études sur l’activité métabolique du CYP2D15. Il faudra également éclaircir à quelle concentration doit être utilisée la quinidine pour inhiber uniquement le CYP2D15. Il a été démontré que le citalopram avait une courte demi-vie (quelques heures) et une clairance élevée chez le rat, la souris, le chien et la singe. Dès lors ces espèces sont exposées à des taux réduits de citalopram à cause d’un effet de premier passage hépatique important (Overø, 1982). Ceci contrairement à l’Homme, où le citalopram a une longue demi-vie (plus de 36 heures) et chez qui l’effet de premier passage hépatique est négligeable (Overø & Højelse, 1994). Lors d’études sur la toxicité du citalopram, il a été démontré que les arythmies observées chez les chiens étaient dues à des taux élevés de citalopram et de didéméthylcitalopram (Overø & Højelse, 1994). Les résultats obtenus dans notre étude montrent qu’à haute concentration le citalopram est N-déméthylé plus lentement chez le chien que chez l’Homme (Rochat et al., 1997a). Une intense activité enzymatique règne lorsque le groupe d’enzymes ayant une haute affinité mais une faible capacité fonctionne (caractérisé par des CLint1 élevées) et ce, pour les deux N-déméthylations (Tableau 13). On pourrait imaginer que le déméthylcitalopram formé est rapidement métabolisé en didéméthylcitalopram. Dès lors, des doses répétées de citalopram pourraient conduire à des taux élevés de citalopram et des taux élevés de didéméthylcitalopram. Enfin, le fait que quatre cytochromes P-450 soient impliqués (CYP2D15, CYP3A12, CYP2C9 et CYP2E1), par rapport à un seul cytochrome (CYP2D6) chez l’homme pourrait également conduire à des taux élevés de déméthyl- et de didéméthylcitalopram. Le rôle de ces différents cytochromes P-450, tout comme celui du CYP2C21 doivent être éclaircis par d’autres études, basées non seulement sur le métabolisme de ceux-ci, mais également leur génétique (génotypes et phénotypes).

- 48 -
Mise au point d’une méthode stéréosélective par LC-MS pour l’analyse de la mirtazapine et ses applications Résultats publiés :
• Paus E, Jonzier-Perey M, Cochard N, Eap CB, Baumann P (2004) : Chirality in the new generation of antidepressants – Stereoselective analysis of enantiomers of mirtazapine, N-demethylmirtazapine and 8-hdroxymirtazapine by LC-MS. Therapeutic Drug Monitoring 26(4) :366-374.
Résumé La mirtazapine est un antidépresseur qui agit spécifiquement sur les récepteurs noradrénergiques et sérotoninergiques (de Boer, 1995). Une méthode de chromatographie liquide couplée à la spectrométrie de masse (LC-MS) a été développée, permettant l’analyse simultanée des énantiomères R-(-) et S-(+) de la mirtazapine, de la déméthylmirtazapine et de la 8-hydroxymirtazapine dans le plasma de patients sous traitement de mirtazapine (Réméron®). La méthode comprend une extraction liquide-liquide de trois étapes, suivie d’une séparation par chromatographie liquide à haute performance (HPLC) avec une colonne Chirobiotic V et une détection par spectrométrie de masse en mode électrospray. La limite de quantification (LOQ) pour tous les énantiomères est de 0.5 ng/ml. Le coefficient de variation (CV) des intra- et interday se situe entre 3.3 % et 11.7%, pour des concentrations comprises entre 5 et 50 ng/ml. La méthode permet également l’analyse quantitative de la mirtazapine et de la 8-hydroxymirtazapine glucuroconjuguée.. La mirtazapine et la 8-hydroxymirtazapine existent également sous forme glucuroconjuguée, alors que la forme conjuguée de la déméthylmirtazapine est un sulfate (Delbressine et al., 1998). La forme S-(+) de la 8-hydroxymirtazapine est principalement présente dans le plasma sous sa forme glucuronidée. Des données préliminaires suggèrent que, chez tous les patients, les concentrations de R-(-)-mirtazapine sont plus élevées que celles de la S-(+)-mirtazapine, à l’exception de ceux qui auraient comme co-médication des inhibiteurs du CYP2D6 tels que la fluoxétine et la thioridazine (Delbressine et al., 1998). De plus, la fluvoxamine semble également être un inhibiteur du métabolisme de la mirtazapine. C’est pourquoi, cette méthode est utilisable pour le dosage stéréosélectif de la mirtazapine et de ses métabolites dans le plasma de patients ayant d’autres substances comme co-médications. Ceci tant pour un dosage de routine que pour des études cliniques.
Matériel & méthodes
Substances et produits chimiques Les substances suivantes ont été offertes : mirtazapine fumarate, N-déméthylmirtazapine fumarate, 8-hydroxymirtazapine, 8-hydroxydéméthylmirtazapine et mirtazapine-N-oxyde (NV Organon, Pays-Bas), remoxipride (AstraZeneca, Suisse). La mirtazapine et la déméthylmirtazapine ont été dissoutes dans du HCl 0.1 M à une concentration de 1 mg/ml base et la 8-hydroxymirtazapine, la 8-hydroxydéméthylmirtazapine et la mirtazapine-N-oxyde, quant à elles, furent dissoutes dans du méthanol à une concentration de 1 mg/ml base, comme solutions stock conservées à -20°C. Les solutions de travail de mirtazapine, de déméthylmirtazapine, de 8-hydroxymirtazapine, de 8-hydroxydéméthylmirtazapine et de mirtazapine-N-oxyde ont été préparées quotidiennement à 10 µg/ml et 1 µg/ml à partir d’aliquotes fraichement décongelées dans de l’HCl 0.1 M. La remoxipride, utilisée comme standard interne, a été dissoute dans du méthanol à une concentration 1 mg/ml comme solution stock. La solution de travail a été préparée à partir d’aliquotes fraîchement décongelées avec une concentration finale de 2 µg/ml.
Extraction Les conditions d’extraction pour l’analyse des composés non conjugués étaient les suivantes : à 1 ml de plasma hépariné étaient ajoutés 200 ng de remoxipride, 1 ml de tampon carbonate 1 M (pH 9.4) et 6 ml de n-heptane-acétate d’éthyle (80 : 20 v/v). Le mélange a été agité pendant 15 minutes et centrifugé à

- 49 -
3300 g durant 8 min à 8°C. La phase organique a été transférée dans un autre tube contenant 1.2 ml d’HCl 0.1 M. La solution a été agitée pendant 15 minutes suivie d’une centrifugation à 3300 g durant 8 min à 8°C. La phase aqueuse a été transférée dans un autre tube contenant 1ml de tampon carbonate 1 M (pH 9.4) et 150 µl de toluène-alcool isoamylique (85 : 15 v/v). Après avoir été centrifugé et agité pendant 15 minutes, puis centrifugé à 3300 g durant 3 min. à 8°C, le solvant a été transféré dans des flacons injecteurs à fonds plats et évaporé à sec sous flux d’azote à 40°C. Les résidus ont été dissouts dans 60 µl de phase mobile et 30 µl ont été injectés dans le système LC-MS. Les conditions d’extraction pour l’analyse des composés conjugués (mirtazapine et de la 8-hydroxymirtazapine) étaient les suivantes : à 0.5 ml de plasma hépariné étaient ajoutés 200 ng de remoxipride, 300 µl de β-glucuronidase (Type HP2S Aldrich, 94500 unités/ml, ce qui correspond à ~ 30000 unités/ml de plasma), 1 ml de tampon acétate 0.2 M (pH 5.0) et 50 µl de sodium azide 4%. Les échantillons ont été ensuite incubés pendant 15 heures à 37°C. L’extraction des composés libres a été entamée après addition d’1ml de tampon glycine 1 M (pH 11.3) et 6 ml de n-heptane-acétate d’éthyle (80 : 20 v/v). Les étapes suivantes de l’extraction des composés conjugués étaient identiques à celles décrites ci-dessus pour l’extraction des composés non conjugués.
Conditions analytiques Les analyses ont été effectuées sur un chromatographe liquide Agilent - HP Série 1100 équipé d’une pompe binaire et d’un injecteur automatique. Une colonne analytique chirale (Chirobiotic V, Cat# 11024, diamètre des particules 5 µm, 250 x 4.6mm d’Astec (Whippany, NJ)) était couplée à une détection par spectrométrie de masse (Spectromètre de masse HP 1100, Agilent Technologies -Meyrin, Suisse) en mode électrospray mode positif (température des gaz à 250°C, flux de gaz desséchant (azote) à 12.0 L/min, pression de nébuliseur à 30 psig (219 kPa), fragmenté à 85 et tension capillaire (Vcap) à 3500V) et une détection par « selected ion monitoring (SIM) » (avec un rapport masse/charge (m/z) de 266.1 pour la mirtazapine, 252.1 pour la déméthylmirtazapine, 282.1 pour la 8-hydroxymirtazapine, 102.1 pour la 8-hydroxydéméthylmirtazapine, 304.1 pour la mirtazapine-N-oxyde et 371.1 pour le remoxipride). La colonne avait un flux de 0.5 ml/min, la phase mobile était constituée de 80 % de méthanol, 20 % d’eau millipore et 0.01 % d’acide acétique (v/v).
Résultats & discussion Initialement, une colonne plus courte (Chirobiotic V, Cat# 11023, 150 mm au lieu de 250mm) a été utilisée, ce qui a permis un temps d’analyse plus court, mais avec des séparations basales non satisfaisantes. La phase mobile 80 % de méthanol, 20 % d’eau (v/v) permettait une bonne séparation pour la mirtazapine et la 8-hydroxymirtazapine, mais pas pour la déméthylmirtazapine. L’addition de 0.01 % d’acide acétique a permis une bonne séparation des énantiomères des 3 composés. Dans ces conditions, la 8-hydroxydéméthylmirtazapine et la mirtazapine-N-oxyde avaient des limites de quantification inférieures à 25 ng/ml, ainsi que des temps de rétention nettement supérieurs à 17 minutes. Cette méthode n’est pas utilisable pour ces deux composés. La Figure 10 montre la séparation chirale d’un plasma blanc spiké avec 50 ng/ml de mirtazapine, déméthylmirtazapine et 8-hydroxymirtazapine. Une séparation base-base est obtenue pour chaque énantiomère avec un temps d’analyse total de 17 minutes.

- 50 -
Figure 10 : Chromatogramme LC-MS en mode SIM obtenu après extraction d’un plasma blanc contenant 50 ng/ml des énantiomères R-(-)- et S-(+)- de la déméthylmirtazapine [R-(-)-DMIR & S-(+)-DMIR] (A), de la R-(-)-mirtazapine et de la S-(+)-mirtazapine [R-(-)-MIR & S-(+)-MIR] (B), des énantiomères R-(-)- et S-(+)- de la 8-hydroxymirtazapine [R-(-)-8-OH-MIR &, S-(+)-8-OH-MIR] (C), ainsi que le standard interne, la remoxipride (D).
Données statistiques de la méthode Le Tableau 14 montre les données sur la qualité de la méthode. Si le rendement d’extraction est relativement bon pour la mirtazapine et la déméthylmirtazapine, il est relativement faible pour la 8-hydroxymirtazapine. La droite de calibration est linéaire de 1 à 100 ng/ml pour chaque énantiomère et la limite de quantification (LOQ) est de 0.5 ng/ml pour chaque énantiomère. La remoxipride a été choisie comme standard interne (ISTD) suite à son retrait du marché comme antipsychotique, il y a de cela plusieurs années. Dès lors, la probabilité de la trouver comme co-médication était très faible.

- 51 -
Tableau 14 : Données statistiques de l’analyse stéréosélective de la mirtazapine et de ses métabolites dans un échantillon de plasma
R-(-) S-(+) R-(-) S-(+) R-(-) S-(+)
Variations durant la journée (n=8)Intervalle des concentrations (ng/ml) 1-100 1-100 1-100 1-100 1-100 1-100
r (moyenne de 10 expériences) 0.996 0.998 0.997 0.997 0.997 0.997
RécupérationConcentrations utilisées (ng/ml) 5 5 5 5 5 5
Récupération mésurée (%) :
moyenne ± écart-ype (CV en %)
Concentrations utilisées (ng/ml) 50 50 50 50 50 50
Récupération mésurée (%) :
moyenne ± écart-ype (CV en %)
Variations durant la journée (n=8)Concentration théorique (ng/ml) 5 5 5 5 5 5
Concentration mésurée (ng/ml) :
moyenne ± écart-ype (CV en %)
Concentration théorique (ng/ml) 25 25 25 25 25 25
Concentration mésurée (ng/ml) :
moyenne ± écart-ype (CV en %)
Concentration théorique (ng/ml) 50 50 50 50 50 50
Concentration mésurée (ng/ml) :
moyenne ± écart-ype (CV en %)
Variation entre les jours (n= 8)Concentration théorique (ng/ml) 5 5 5 5 5 5
Concentration mésurée (ng/ml) :
moyenne ± écart-ype (CV en %)
Concentration théorique (ng/ml) 25 25 25 25 25 25
Concentration mésurée (ng/ml) :
moyenne ± écart-ype (CV en %)
Concentration théorique (ng/ml) 50 50 50 50 50 50
Concentration mésurée (ng/ml) :
moyenne ± écart-ype (CV en %)
Remoxipride
5 ± 0.3 (5.7)
Mirtazapine Déméthylmirtazapine 8-Hydroxymirtazapine
4.6 ± 05 (11.6) 4.6 ± 04 (7.9) 5.1 ± 0.5 (9.1) 5.2 ± 0.4 (7.7)
75 ± 3 (4) 73 ± 3 (4) 51 ± 2 (4) 53 ± 2 (3)
27.8 ± 1.8 (6.4)
42 ± 2 (5) 40 ± 2 (4) 88 ± 2 (3)
52 ± 5 (9) 46 ± 6 (8) 41 ± 3 (8) 78 ± 2 (3)
4.9 ± 0.4 (8.2)
25.6 ± 1.8 (7) 25 ± 1.6 (6.3)
47.6 ± 2.1 (4.3) 46.5 ± 1.7 (3.6) 45.6 ± 2.2 (4.8) 47.4 ± 1.6 (3.3) 45 ± 1.7 (3.7) 45.6 ± 1.8 (4)
4.8 ± 0.5 (10.7) 5 ± 0.3 (6.3) 5 ± 0.3 (6.1)4.7 ± 0.5 (10.6) 4.7 ± 0.5 (9.9) 4.7 ± 0.4 (7.9)
27.8 ± 2.5 (9) 26.1 ± 2 (7.5) 27.8 ± 1.9 (6.8)
70 ± 4 (5) 65 ± 4 (7) 47 ± 4 (8)
25.1 ± 2.4 (9.7) 23.6 ± 1.5 (6.4) 23.8 ± 2.3 (9.7) 24.2 ± 2.5 (10.3) 24.8 ± 2.4 (9.7) 24.7 ± 2 (7.9)
47.5 ± 5.2 (11) 46.5 ± 3.4 (7.3) 47.3 ± 4.4 (9.4) 47.1 ± 4.9 (10.3) 48.4 ± 5.5 (11.4) 49.9 ± 5.8 (11.7)

- 52 -
Dosage d’échantillons plasmatiques de patients sous mirtazapine Les Figures 11 (sans hydrolyse) et 12 (avec hydrolyse) montrent les chromatogrammes de l’analyse stéréosélective de mirtazapine et de ses métabolites dans le plasma de patients traités avec 30 mg par jour de mirtazapine. La Tableau 15 présente les concentrations plasmatiques des énantiomères de la mirtazapine, de la déméthylmirtazapine et de la 8-hydroxymirtazapine chez 13 patients psychiatriques traités avec de la mirtazapine. Les concentrations mesurées pour les composés non-conjugués correspondent bien aux valeurs de la courbe de calibration, excepté pour le patient n° 1 traité avec des doses inhabituellement élevées de mirtazapine (150 mg par jour). Pour l’analyse des échantillons plasmatiques soumis à l’hydrolyse, certains échantillons ont dû être dilués de manière appropriée à cause d’une conjugaison très prononcée de la mirtazapine et de la 8-hydroxymirtazapine chez certains patients (par exemple, le patient n° 3).
Figure 11 : Chromatogramme LC-MS en mode SIM obtenu après extraction du plasma du patient 13 traité avec 30 mg/jour de mirtazapine. Les composés ne sont pas conjugués.
CF Figure 10 pour les abréviations.
Figure 12 : Chromatogramme LC-MS en mode SIM obtenu après extraction du plasma du patient 13 traité avec 30 mg/jour de mirtazapine. La Figure 12 présente la somme des composés conjugués et non conjugués.
CF Figure 10 pour les abréviations.

- 53 -
Tableau 15 : Concentrations (ng/ml) des énantiomères de la mirtazapine et de ses métabolites mesurés dans le plasma de 13 patients psychiatriques.
R-(-) S-(+) S-(+)/R-(-) R-(-) S-(+) S-(+)/R-(-) R-(-) S-(+) S-(+)/R-(-) R-(-) S-(+) S-(+)/R-(-) R-(-) S-(+) S-(+)/R-(-) R-(-) S-(+) R-(-) S-(+)
a âge en annéesb nq, non quantifié (se situe en dessous de la limite de détection)c uniquement pour les patients (n=8) traité avec 30mg/jour de mirtazaoine.
Non conjugué
5.9 313 53
Mirtazapine Déméthylmirtazapine 8-Hydroxymirtazapine
2
Total (conjugué et non conjugué) Non conjugué/Mirtazapine
totale (%)Mirtazapine 8-Hydroxymirtazapine
188 97 0.563 0.6
Co-médications
81 39 0.5 1.7 3.5
Patient n° Agea/Sex
1 46/F 55103 65 29
Non conjugué/8-
Hydroxymirtazapine totale (%)
1.1 150
Mirtazapine
(mg/jour)
venlafaxine, lithium
2 55/F 15 19 1.2 8 2 0.3 nq 1 - 24 15 0.6 1.5 1 6.6 62 121 - 10.6 80aténolol, clorazepate, flurazepam,
amoxicilline, fluoxetine
3 87/f 39 37 0.9 74 15 0.2 2 3.5 1.8 247 136 0.6 9.5 451 47 16 27 21 0.8 60
acide acétylsalicylique, nitroglycérine,
hydrochlorothiazide + amiloride,
clométhiazole
4 82/F 48 20 0.4 32 5 0.2 0.7 1.5 2 106 32 0.3 4.6 105 23 45 63 16 1.4 45
venlafaxine, tramadol, zopiclone,
paracetamol, lévothyroxine, acide
acétylsalicylique
5 75/F 34 23 0.7 35 15 0.4 0.4 2.2 5.8 99 48 0.5 4.8 104 22 34 48 7.9 2.1 45clozapine, clomethiazole, propranolol,
midazolam
6 41/M 46 26 0.6 13 2 0.1 nq 2.2 - 105 34 0.3 8.1 134 17 44 76 - 1.7 30olanzapine, flurazempam, corazepate,
fluvoxamine
7 45/F 14 18 1.3 2 nq - 0.2 0.9 4 22 22 1 2.2 8.7 3.9 64 81 10 10.1 30 fluoxétine, lithium, lorazepam
8 72/F 11 28 2.7 12 7 0.6 nq 1.4 - 53 59 1.1 4.1 91 23 20 48 - 1.5 30thioridazine, pantoprazol, pizotifène,
morphine.fulrazépam
9 42/M 24 6 0.2 11 3 0.2 nq 0.9 - 37 8 0.2 4.8 24 5 66 75 - 3.9 30 olanzapine, chlorprothixène, alprazolam
10 66/M 11 4 0.4 6 2 0.2 nq 0.9 - 42 8 0.2 3.4 50 15 26 52 - 1.9 30 clorazépate
11 37/F 8 16 2 5 nq - 0.3 1.1 4 8 14 1.7 1.9 11 59 100 118 14 9.5 30fluoxétine, olanzapine, clorazepate,
zopiclone
12 75/M 9 87 9.4 5 4 0.8 1.7 2.1 1.3 26 126 4.8 2.2 22 10 36 69 77 9.4 30
clozapine, zopiclone, omeprazole,
gabapentine, metformine, acide
acétylsalicyclique, venlafaxine, linisopril,
rofecoxib
13 84/F 34 19 0.6 26 5 0.2 0.9 1.7 1.9 . 74 29 0.4 10.1 8.8 1.4124 12 46 30losartan, clozapine, bromazepam,
esméprazole
20 ± 14 26 ± 26 2.1 ±3
63
10 ± 7 3.6 ± 2.1 0.4 ± 0.3 1,2 ± 1.60.8 ± 0.71.4 ± 0.5 2.8 ± 1.4 50 ± 26 73 ± 22 28 ± 33Moyenne ± écart-type c 4.9 ± 44.6 ± 3 58 ± 51 11.4 ± 6.446 ± 31 37 ± 39

- 54 -
Les ratios S/R, dans les résultats des composés non conjugués, montrent que les concentrations de la R-(-)-mirtazapine sont plus élevées que celles de la S-(+)-mirtazapine chez 8 des 13 patients. La forme R-(-) est considérée comme la forme pharmacologiquement active de l’antidépresseur (de Boer et al., 1995; de Boer, 1996). Dans une étude méthodologique déjà mentionnée, les concentrations de la R-(-)-mirtazapine sont plus élevées que celles de la S-(+)-mirtazapine chez tous les patients (Dodd et al., 2000). Il est important de préciser que ces patients, participant à une étude clinique, n’étaient probablement pas co-médiqués avec des inhibiteurs des cytochromes P-450. Dans la présente étude, les ratios S/R étaient inférieurs à 1 pour la déméthylmirtazapine et supérieurs à 1 pour la 8-hydroxymirtazapine chez tous les patients. Cependant, les concentrations de la R-(-)-8-hydroxymirtazapine sont très faibles et au-dessous de la limite de quantification uniquement chez quelques patients. La déméthylmirtazapine est supposée contribuer faiblement à l’effet global de la mirtazapine (Delbressine et al., 1998). Les concentrations de la R-(-)-déméthylmirtazapine étaient inférieures à celles de la R-(-)-mirtazapine chez 10 des 13 patients testés. Aucun des patients traités ne montra une concentration de S-(+)-déméthylmirtazapine supérieure à celle de la S-(+)-mirtazapine. De nombreux patients étaient co-médiqués avec différents médicaments, incluant de puissants inhibiteurs du CYP2D6 comme la fluoxétine (patients 2, 7 et 11) (Baumann, 1996) ou la thioridazine (patient 8) (von Bahr et al., 1985). D’autres étaient co-médiqués avec la fluvoxamine (inhibiteur du CYP1A2, CYP2C9 et CYP2C19) (patient 6) (Baumann, 1996) l’oméprazole (inhibiteur du CYP2C19) (patient 12) ou l’esoméprazole (patient 13) (Andersson et al., 2001; Ko et al., 1997) (Tableau 15). Il est à noter que 4 des 5 patients co-médiqués avec la fluoxétine ou la thioridazine avaient des concentrations de S-(+)-mirtazapine plus élevées que celles de la R-(-)-mirtazapine (patients 2, 7, 8 et 11). Malgré l’inhibition du CYP2D6 par la fluoxétine ou la thioridazine, la formation de S-(+)-8-hydroxymirtazapine n’était pas complètement abolie. Ces observations correspondent à celles déjà observées où la S-(+)-mirtazapine est préférentiellement hydroxylée en position 8 par le CYP2D6, alors que la R-(-)-mirtazapine est préférentiellement métabolisée par N-glucuronidation (Timmer et al., 2000). De plus, au sein du sous-groupe des patients traités par 30 mg par jour de mirtazapine, le patient n°6 (co-médiqué avec de la fluvoxamine) avait le plus haut taux plasmatique de R-(-)-mirtazapine (46 ng/ml). Des études in vitro ont montré que les CYP3A4 et CYP1A2 contribuent à la N-déméthylation de la mirtazapine (Störmer et al., 2000a) (Tableau 15). Ces interprétations doivent être considérées comme extrêmement préliminaires, car ces données ne sont pas le résultat d’une étude clinique contrôlée. De plus, aucun de ces patients n’a été génotypé pour le CYP2D6 et la compliance de certains a pu être lacunaire. C’est pourquoi ces observations stimulantes ont besoin d’être confirmées par d’autres études pharmacocinétiques cliniques plus approfondies. En effet, dans une étude comprenant des sujets phénotypés pour le CYP2D6, les paramètres pharmacocinétiques de la mirtazapine racémique n’ont pas montré de différences entre les métaboliseurs rapides (EM) et les métaboliseurs lents (PM) après l’absorption d’une dose unique (Dahl et al., 1997). Cependant, une analyse stéréosélective a montré que chez les métaboliseurs exhaustifs, la S-(+)-mirtazapine était plus rapidement éliminée que la R-(-)-mirtazapine, tandis que chez les métaboliseurs lents, aucune stéréosélectivité n’a été observée. De plus, il a été montré que l’élimination de la R-(-)-mirtazapine est indépendante du statut pharmacogénétique (Timmer et al., 2000). Enfin, il semble que le patient 11 puisse souffrir du syndrome de Gilbert puisque le ratio non conjuguée/mirtazapine totale dépasse les 100% (Tableau 15). Dans le cadre d’une étude contrôlée, ce patient sera probablement un « outlier ». Il serait intéressant de connaître les taux de bilirubine de ce patient, et de les relier à une hyperbilirubinémie modérée, caractéristique du syndrome de Gilbert, reflétant une réduction de l’activité de l’enzyme UGT1A1 d’environ 30% (Borlak et al., 2000).

- 55 -
Figure 13 : Principales voies métaboliques de la mirtazapine chez l’homme. Figure adaptée de (Delbressine et al., 1998; Paus et al., 2004).
NN
NCH
3
H
NN
NCH
3
H
O
NN
NCH
3
OH
H
NN
NH
H
R-(-)-, S-(+)-mirtazapine
R-(-)-, S-(+)-mirtazapine-N-oxyde
R-(-)-, S-(+)-8-hydroxymirtazapine
R-(-)-, S-(+)-N-déméthylmirtazapine
R-(-)-, S-(+)-N-mirtazapine-glucuronide
R-(-)-, S-(+)-8-hydroxymirtazapine-glucuronide
UGT, UGT1A1?
UGT
CYP1A2CYP3A4CYP2C9CYP2C19CYP2B6
CYP1A2CYP3A4
CYP2D6CYP1A2CYP2C9CYP3A4CYP2C19CYP2B6
En caractère gras, l’enzyme ayant la plus grande affinité (Km), ainsi que l’énantiomère préférentiellement formé. En italique, les enzymes étant accessoires pour cette voie métabolique (Störmer et al., 2000b). La Figure 12 montre le chromatogramme du plasma d’un patient traité avec la mirtazapine soumis à une hydrolyse enzymatique. La comparaison des concentrations des énantiomères de la mirtazapine et de la 8-hydroxymirtazapine non conjuguées par rapport aux concentrations totales (non-conjuguées et conjuguées) montrent que dans le plasma ces composés sont également présents sous forme glucuronidée (Tableau 15). Chez les patients traités par la mirtazapine, les produits glucuronidés de la mirtazapine et de la 8-hydroxymirtazapine ont été précédemment décrits comme étant éliminées dans les urines. Suite à une analyse chirale, il a été montré que la 8-hydroxy-glucuronide est préférentiellement formée à partir de la

- 56 -
S-(+)-8-hydroxymirtazapine, tandis que le glucuronide de l’ammonium quaternaire est principalement formé par la R-(-)-mirtazapine (Delbressine et al., 1998). Aucune donnée quantitative n’a été publiée sur les produits glucuronidés de la mirtazapine dans le plasma. Cette étude confirme le fait que la S-(+)-8-hydroxymirtazapine est hautement conjuguée dans le plasma puisque le ratio des non-conjuguées/total de la S-(+)-8-hydroxymirtazapine varie de 0.8 % à 10.1 % entre les individus (Tableau 15). Les concentrations moyennes de la R-(-)-8-hydroxymirtazapine sont extrêmement faibles, inférieures à 10 % comparé à l’autre énantiomère. Le ratio moyen S/R-mirtazapine des substances non-conjuguées était de 2.1, tandis qu’il était de 1.2 pour la totalité des substances. Ceci confirme une glucuroconjugaison préférentielle de la R-(-)-mirtazapine, comme suggéré par l’étude sur les urines (Delbressine et al., 1998), mais avec une stéréosélectivité plus faible que celle observée avec les composés hydroxylés. Toutefois, en raison de l’absence de standards pour les produits conjugués de la mirtazapine et de ses métabolites, la procédure pour cette analyse n’a pas pu être complètement validée. En conclusion, cette méthode sensible et stéréosélective permet l’analyse quantitative de la mirtazapine, de la déméthylmirtazapine et de la 8-hydroxymirtazapine dans le plasma de patients traités avec la mirtazapine. L’utilisation de la chromatographie liquide couplée à la spectrométrie de masse permet l’analyse d’échantillons plasmatiques de patients co-médiqués avec différents principes actifs, comme illustré dans cette étude. Ces résultats préliminaires mais prometteurs suggèrent la nécessité d’études supplémentaires sur le potentiel d’interaction stéréosélective des inhibiteurs des cytochromes P-450 avec la mirtazapine en clinique.

- 57 -
Glycoprotéine-P (Pgp) et gène (MDR1) : implications dans la distribution des médicaments Revue publiée :
• Marzolini C, Paus E, Buclin T, Kim RB (2004) Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clinical Pharmacology and Therapeutic 75(1):13-33.
Introduction Le gène MDR1 (Multiple Drug Resistance) code pour une protéine, la glycoprotéine-P (Pgp), qui a été étudiée de manière exhaustive dans son rôle associé à un phénotype de résistance à de multiples substances médicamenteuses dans certains cancers. Cependant, ce transporteur est normalement exprimé dans les tissus sains. Il a été démontré qu’il joue un rôle important dans la réponse de l’organisme à certaines substances. Plus récemment, une hétérogénéité génétique en termes de polymorphismes d’un seul nucléotide (SNP) de ce transporteur a particulièrement attiré l’attention. Il se pourrait que celui-ci soit l’un des déterminants dans le devenir du médicament et de son efficacité.
État actuel des connaissances
Expression, fonctions et localisation de la glycoprotéine-P
La glycoprotéine-P est un membre de la famille des « ATP-binding cassettes » et le gène ABCB1, appelé MDR1 code pour cette protéine. Chez l’homme, il existe deux formes du gène (MDR1 et MDR3) tandis que trois gènes sont présents chez le rongeur : mdr1a, mdr1b et mdr2 (Schinkel, 1997). La glycoprotéine-P encodée par les gènes humains MDR1 ou murins mdr1a/b ont une fonction de transporteur faisant sortir la substance de la cellule, tandis que celles encodées par les gènes humains MDR3 ou murins mdr2 ont une fonction de phospholipides flippases (van Helvoort et al., 1996). La glycoprotéine-P humaine est une protéine transmembranaire phosphorylée et glycosylée de 1280 acides aminés, composée de deux séquences homologues et symétriques, chacune d’entre elles comprenant six domaines transmembranaires. La glycoprotéine-P fonctionne comme une pompe transmembranaire qui transporte les xénobiotiques ayant pénétrés dans la membrane plasmatique vers le milieu extracellulaire. Elle peut aussi interagir avec les substances enchevêtrées dans la double couche de la membrane lipidique (Figure 14).
Figure 14 : Fonction de la glycoprotéine-P (Pgp) Ce modèle montre le transport de xénobiotiques à travers les membranes cellulaires ou depuis le compartiment intracellulaire.

- 58 -
L’hydrolyse de l’ATP fournit l’énergie nécessaire pour un transport actif des xénobiotiques lui permettant de fonctionner contre des gradients de concentrations (Higgins & Gottesmann, 1992). La métaphore d’un « aspirateur » est souvent utilisée pour illustrer son mécanisme d’action. La glycoprotéine-P a été initialement identifiée à cause de sa surexpression dans les cultures de cellules cancéreuses associée à une résistance croisée contre de multiples agents cytotoxiques (Juliano & Ling, 1976). Depuis, ce transporteur a été identifié comme étant exprimé dans de nombreux tissus normaux, suggérant une fonction physiologique. On trouve la glycoprotéine-P à la surface canaliculaire des hépatocytes, à la surface apicale des cellules du tube proximal des reins, dans la bordure en brosse des entérocytes intestinaux, dans les cellules souches hématopoïétiques, dans les macrophages, dans les cellules de la lignée blanche « natural killer » et dans les cellules dendritiques présentant les antigènes (Thiebaut et al., 1987). De plus, la glycoprotéine-P est exprimée dans l’épithélium du plexus choroïde dans le cerveau, (forme la barrière entre le sang et le liquide céphalorachidien), ainsi qu’à la surface luminale des cellules épithéliales des capillaires cérébraux (forme la barrière hémato-encéphalée). La glycoprotéine-P est également présente dans les autres tissus connus pour avoir une barrière entre le sang et les tissus, tels que le placenta, les ovaires et les testicules (Cordon-Cardo et al., 1989; Sugawara et al., 1988). C’est pourquoi, la localisation anatomique et la fonction de la glycoprotéine-P suggèrent que ce transporteur agit comme une barrière protectrice qui maintient les toxines hors des tissus en excrétant dans la bile, les urines et la lumière intestinale. Ceci empêche l’accumulation des toxines dans des organes critiques comme le cerveau, le fœtus, les gonades et la moelle osseuse.
Substrats La relation structure-activité des substrats de la glycoprotéine-P n’est pas encore bien définie. Le caractère lipophile de la molécule et la présence, ou non, d’atomes d’azote liés apparaissent comme des paramètres pertinents (Ecker et al., 1999). Les molécules transportées de la manière la plus efficace sont généralement des bases faibles, bien que certains composés acides puissent également être transportés. Presque tous les substrats de la glycoprotéine-P paraissent être de nature amphiphile. La Tableau 16 fournit une liste des composés qui sont substrats et/ou inhibiteurs, ou inducteurs de la glycoprotéine-P (Avendano & Menendez, 2002; Brinkmann et al., 2001; Fromm, 2002; Kim, 2002; Sakaeda et al., 2002; Seelig, 2003). La diversité des substrats transportés par la glycoprotéine-P est grande. Elle inclut de nombreux médicaments, aux profils pharmacologiques très différents, comme des anticancéreux, des antihypertenseurs, des antiallergiques, anti-infectieux, des immunosuppresseurs et des anti-inflammatoires. Il est aussi évident que de nombreuses substances substrats de la glycoprotéine-P sont également des substrats des cytochromes P-450, en particulier du CYP3A4. Ce recouvrement entre les substrats de la glycoprotéine-P et du CYP3A4 pourrait être le résultat d’une régulation coordonnée entre l’expression du gène du CYP3A4 et celui du MDR1. Leur deux gènes sont localisés sur le même chromosome et à proximité (7q22.1 et 7q21.1 pour le CYP3A4 et le MDR1, respectivement). Cependant, le recouvrement de leurs substrats n’est pas complet, puisque certains substrats du CYP3A4 (p. ex. le midazolam) ne sont pas transportés par la glycoprotéine-P (Eap et al., 2004; Kim et al., 1999). De la même manière, la digoxine, le fexofénadine et la talinolol sont des substrats de glycoprotéine-P qui n’ont pas d’interactions significatives avec les cytochromes P-450 (CYP3A4 inclus). Depuis la publication de la revue, de nouveaux substrats ont été découvert pour la Pgp. Parmi ceux-ci, la doxépine, la venlafaxine et la paroxétine sont des substrats de la Pgp, alors que la mirtazapine n’est pas un substrat (Uhr et al., 2003). L’acide valproïque n’est pas un substrat des Pgp (Baltes et al., 2007). Une autre approche est basée sur les pharmacophores pour déterminer l’activité des substrats. Chang, C. et al. a ainsi testé 7 substrats (acitretin, cholécalciférol, misoprostol, nafcillin, repaglinide, salmétérol, and telmisartan) sur lesquels aucune donnée en relation avec la Pgp n’était disponible. Ce groupe montra que ces 7 molécules sont substrats de la Pgp (Chang et al., 2006). Enfin, le millepertuis est non seulement un substrat des cytochromes P-450 (CYP3A4, CYP1A2 et CYP2C9) mais également des Pgp (Pal & Mitra, 2006). Le millepertuis étant en vente libre, cela risque de poser des problèmes face à l’automédication croissante des patients.

- 59 -
Tableau 16 : Substrats inhibiteurs et inducteurs de la glycoprotéine-P (Situation : janvier 2004).
Substances substrat inhibiteur inducteur Substances substrat inhibiteur inducteur
anticancereux antivirauxactinomycin D ☯☯☯☯ adefovir ☯☯☯☯
adriamycin ☯☯☯☯ amprenavir ☯☯☯☯ ☯☯☯☯
cisplatin ☯☯☯☯ indinavir ☯☯☯☯ ☯☯☯☯ ☯☯☯☯
daunorubicin ☯☯☯☯ ☯☯☯☯ nelfinavir ☯☯☯☯ ☯☯☯☯ ☯☯☯☯
docetaxel ☯☯☯☯ ritonavir ☯☯☯☯ ☯☯☯☯ ☯☯☯☯
doxorubicin ☯☯☯☯ saquinavir ☯☯☯☯ ☯☯☯☯ ☯☯☯☯
etoposide ☯☯☯☯
imatinib ☯☯☯☯ antibiotiquesirinotecan ☯☯☯☯ cefazolin ☯☯☯☯
mitomycin C ☯☯☯☯ clarithromycin ☯☯☯☯
mitoxantrone ☯☯☯☯ clofazimine ☯☯☯☯ ☯☯☯☯
paclitaxel ☯☯☯☯ erythromycin ☯☯☯☯ ☯☯☯☯
tamoxifen ☯☯☯☯ ☯☯☯☯ fleroxacin ☯☯☯☯ ☯☯☯☯
teniposide ☯☯☯☯ lévofloxacin ☯☯☯☯ ☯☯☯☯
topotecan ☯☯☯☯ norfloxacine ☯☯☯☯ ☯☯☯☯
vinblastin ☯☯☯☯ ☯☯☯☯ rifampin ☯☯☯☯
vincristin ☯☯☯☯ ☯☯☯☯ sparfloxacin ☯☯☯☯ ☯☯☯☯
vindesine ☯☯☯☯ ☯☯☯☯ tetracycline ☯☯☯☯
antihypertenseurs antimalariquescarvedilol ☯☯☯☯ chloroquine ☯☯☯☯ ☯☯☯☯
celiprolol ☯☯☯☯ mefloquine ☯☯☯☯ ☯☯☯☯
diltiazem ☯☯☯☯ ☯☯☯☯
felodipine ☯☯☯☯ ☯☯☯☯ antimycotiqueslosartan ☯☯☯☯ clotrimazole ☯☯☯☯
nicardipine ☯☯☯☯ ☯☯☯☯ itraconazole ☯☯☯☯ ☯☯☯☯
nifedipine ☯☯☯☯ ☯☯☯☯ ketoconazole ☯☯☯☯
nitrendipine ☯☯☯☯ ☯☯☯☯
prazosin ☯☯☯☯ immunosuppresseurspropanolol ☯☯☯☯ ☯☯☯☯ cyclosporin ☯☯☯☯ ☯☯☯☯
reserpine ☯☯☯☯ rapamycin ☯☯☯☯ ☯☯☯☯
talinolol ☯☯☯☯ sirolimus ☯☯☯☯
tacrolimus ☯☯☯☯ ☯☯☯☯
antiarhythmiques valspodar ☯☯☯☯ ☯☯☯☯
amiodarone ☯☯☯☯ ☯☯☯☯
digoxin ☯☯☯☯ antidépresseurspropafenone ☯☯☯☯ ☯☯☯☯ amitriptyline ☯☯☯☯ ☯☯☯☯
quinidine ☯☯☯☯ ☯☯☯☯ millepertuis ☯☯☯☯
verapamil ☯☯☯☯ ☯☯☯☯
neuroleptiquesdiurétiques chloropromazine ☯☯☯☯ ☯☯☯☯
amiloride ☯☯☯☯ ☯☯☯☯ clozapine ☯☯☯☯
spironolactone ☯☯☯☯ flupenthixol ☯☯☯☯
fluphenazine ☯☯☯☯ ☯☯☯☯
glucocorticoïdes haloperidol ☯☯☯☯
aldosterone ☯☯☯☯ perphenazine ☯☯☯☯
cortisol ☯☯☯☯ phenothiazine ☯☯☯☯
dexamethasone ☯☯☯☯ ☯☯☯☯ promazine ☯☯☯☯
hydrocortisone ☯☯☯☯ ☯☯☯☯ thioridazine ☯☯☯☯
methylprednisolone ☯☯☯☯
antiépileptiquesautres phénobarbital ☯☯☯☯
atorvastatin ☯☯☯☯ ☯☯☯☯ phénytoin ☯☯☯☯
bromocriptine ☯☯☯☯
colchicine ☯☯☯☯ antiacidesdipyridamole ☯☯☯☯ ☯☯☯☯ cimétidine ☯☯☯☯
emetine ☯☯☯☯ ranitidine ☯☯☯☯
estradiol ☯☯☯☯
fexofenadine ☯☯☯☯ opiacésflunitrazepam ☯☯☯☯ fentanyl ☯☯☯☯
ivermectin ☯☯☯☯ méthadone ☯☯☯☯
loperamide ☯☯☯☯ morphine ☯☯☯☯ ☯☯☯☯
progesterone ☯☯☯☯ pentazocine ☯☯☯☯
retinoic acid ☯☯☯☯
rhodamine 123 ☯☯☯☯ antiémétiquesterfenadine ☯☯☯☯ ☯☯☯☯ domperidon ☯☯☯☯
vecuronium ☯☯☯☯ ondansetron ☯☯☯☯

- 60 -
Importance de la glycoprotéine-P dans la distribution des médicaments La co-localisation de la glycoprotéine-P et du CYP3A4 dans l’intestin grêle et le foie suggère que ce transporteur a aussi un rôle important dans l’absorption, la distribution et l’excrétion des xénobiotiques. Les modèles animaux en ont apporté l’illustration. Une lignée de souris (CF-1) naturellement déficiente du gène mdr1a montra une grande sensibilité aux effets neurotoxiques de l’ivermectine un antiparasitaire substrat de la glycoprotéine-P (Schinkel et al., 1994). L’absence de la glycoprotéine-P au niveau de la barrière hémato-encéphalée chez ces souris déficientes résultait en une accumulation d’ivermectine 80 fois supérieure à la normale entrainant une neurotoxicité (Lankas & Cartwright, 1997). Les souris CF-1 étaient également nettement plus sensibles à des malformations in utero induites par l’ivermectine (Lankas et al., 1998). Des études comparatives de souris mdr1a (-/-) et mdr1b (-/-) ont montré que le gène mdr1a (-/-) est la forme principale de la glycoprotéine-P murine exprimée dans les capillaires cérébraux, le tractus digestif et le placenta, tandis que le gène mdr1b (-/-) est exprimé uniquement dans le foie et les reins. Les deux isoformes murines semblent remplir les mêmes fonctions que le gène MDR1 codant pour la glycoprotéine-P humaine (Schinkel et al., 1997). L’induction et l’inhibition de la glycoprotéine-P, bien que moins étudiées que pour les cytochromes P-450, ont des implications cliniques importantes en termes d’efficacité de traitement et de toxicité. Un exemple d’interaction cliniquement pertinente est celle entre la digoxine et l’administration concomitante d’anti-arythmiques comme le vérapamil, la quinidine ou l’amiodarone. Dans le cas de la digoxine associée au vérapamil, les concentrations plasmatiques de digoxine augmentent de 40% probablement suite à l’inhibition intestinale, hépatique et rénale de la glycoprotéine-P (Verschraagen et al., 1999).
Polymorphisme du gène MDR1 Entre deux brins d’ADN, il peut y avoir une différence seulement dans un nucléotide. Cela peut être à la base d’un polymorphisme appelé SNP (single nucleotide polymorphism) et représente la variation de séquence la plus commune dans le génome humain. Basé sur leur localisation dans le génome, le polymorphisme d’un seul nucléotide (SNP) peut être classé comme SNP aléatoire (SNP le plus courant retrouvé dans les introns des gènes), SNP codant (retrouvé dans les exons représentant la région traduite d’un gène) et SNP non-codant (retrouvé en dehors de la région traduite d’un gène, comme le promoteur). Les SNPs codants présentent un intérêt majeur en pharmacogénétique car ils peuvent coder pour un changement d’acides aminés. Les SNPs non synonymes se réfèrent à un polymorphisme qui aura comme conséquence un changement d’acides aminés, tandis que les SNPs synonymes n’ont pas de conséquence. Le gène MDR1 humain est composé de 28 exons. Le premier polymorphisme a été identifié in vitro sur des cellules cancéreuses (Kioka et al., 1989). Au moment de la publication (2004), 29 SNPs ont été identifiés sur le gène MDR1. Un SNP sur l’exon 21 à la position 2677 peut résulter en deux changements distincts d’acides aminés qui sont Ala893Ser (G2677T) et Ala893Thr (G2677A). Cependant, un SNP synonyme sur l’exon 26 (C3435T) a été la première mutation à être associée à une expression protéique altérée, quand bien même ce SNP ne change pas la séquence des acides aminés (Ile) (Hoffmeyer et al., 2000). Dans la même étude, l’expression de la glycoprotéine-P dans le duodénum des individus avec l’allèle T homozygote (muté) était diminuée comparée à celle des individus avec l’allèle C (sauvage). D’autres ont montré que le SNP sur l’exon 21 (G2677T/A) et sur l’exon 1b (T129C) peut être associé avec une expression ou un transport altéré (Kim et al., 2001; Tanabe et al., 2001).
Polymorphisme du gène MDR1 et maladie Il a été suggéré que l’allèle 3435T sur l’exon 26 du gène MDR1 pourrait avoir un rôle protecteur pour des patients atteints de Parkinsonisme et en particulier chez ceux ayant été exposés à des pesticides (Drozdzik et al., 2003). Ce qui signifie que les pesticides sont en général des substrats de la glycoprotéine-P et que

- 61 -
l’exposition du système nerveux central à de tels agents peut conduire à des maladies neurodégénératives. Un autre exemple du rôle protecteur de l’allèle 3435T sur l’exon 26 est lié aux maladies inflammatoires des intestins comme la colite ulcérative et la maladie de Crohn (Schwab et al., 2003). Des études épidémiologiques ont montré une incidence moins élevée de la colite ulcérative chez les Africains comparés aux Caucasiens, ce qui est cohérent avec les différences ethniques dans la fréquence du polymorphisme de l’allèle 3435T sur l’exon 26 (Schaeffeler et al., 2001). Le polymorphisme MDR1 a également été étudié en relation avec la thérapeutique conte le sida. Il est connu que tous les inhibiteurs des protéases du VIH sont transportés par la glycoprotéine-P et que leur absorption intestinale, tout comme leur pénétration dans le système nerveux central, dépendent fortement de la glycoprotéine-P (Roberts et al., 2002). De plus, la pénétration d’autres barrières physiologiques comme les testicules et le placenta, est limitée par ces mêmes inhibiteurs des protéases du VIH (Choo et al., 2000). Récemment, Fellay et al. ont trouvé une relation entre l’expression de la glycoprotéine-P dans les cellules mononucléaires du sang périphérique (PBMC) de patients infectés par le virus du VIH et les lymphocytes CD4 (Fellay et al., 2002). Les patients ayant l’allèle 3435T sur l’exon 26 voyaient le nombre de cellules CD4 très nettement augmenté 6 mois après le début de la thérapie. Il est fort probable que le bénéfice associé avec l’allèle T pourrait être le résultat d’une meilleure pénétration des inhibiteurs des protéases du VIH dans les cellules CD4. En plus d’influencer la concentration intracellulaire des xénobiotiques, la surexpression de la glycoprotéine-P in vitro a montré une diminution de la susceptibilité des lymphocytes CD4 humains à l’infection par le virus de VIH. Ceci, en interférant avec l’adhésion virale à la cellule et probablement dans la libération de virus dans la circulation (Lee et al., 2000; Speck et al., 2002). Cependant, la pertinence clinique d’un tel rôle pour la glycoprotéine-P doit encore être déterminée.
Perspectives Étant donnée l’importance de la glycoprotéine-P dans le devenir du médicament et sa réponse, il n’est pas surprenant de constater un intérêt croissant pour le polymorphisme du gène MDR1. Afin de minimiser au maximum les risques d’associations douteuses entre un génotype MDR1 et un phénotype in vivo, une attention particulière sera portée aux haplotypes, aux facteurs environnementaux ainsi qu’à la taille de l’échantillon. En effet, ce genre d’étude devrait s’assurer que les données démographiques des sujets choisis ne varient pas en fonction des différents SNPs du gène MDR1. Par exemple, l’expression de la glycoprotéine-P chez une population ayant une maladie ne devrait pas être comparée avec une autre population n’ayant pas cette maladie. De plus, d’autres études concernant la spécificité des substrats, les différences inter-espèces dans l’expression et la régulation de la glycoprotéine-P sont nécessaires. Enfin, la standardisation des tests utilisés pour quantifier la glycoprotéine-P et son ARN messager est nécessaire. Ceci dans le but de mieux cerner le rôle des polymorphismes du gène MDR1 et la fonction de ces transporteurs. Depuis la parution de cette revue en 2004, plusieurs méthodes ont été publiées, afin de mesurer l’activité in vivo des cytochromes (généralement de CYP3A4) et la Pgp. Parmi celle-ci, la méthode de Kirby et al. permet de phénotyper simultanément le CYP3A4 et la Pgp, sans interaction pharmacocinétique portant à conséquence (Kirby et al., 2006). Ce type de « cocktail » existait déjà pour les cytochromes (Blakey et al., 2004; Christensen et al., 2003; Clement Jerdi et al., 2005), et permettait de doser jusqu’à 5 cytochromes P-450. Il serait souhaitable que dans un avenir proche, il existe la possibilité de phénotyper 5 cytochromes ou plus, avec la Pgp, et ceci sans interaction pharmacocinétique majeure.

- 62 -
Conclusions La première partie de cette thèse a été consacrée à l’étude du métabolisme du citalopram dans le cerveau de souris sauvages et de souris génétiquement modifiées. Nous avons montré que la formation de l’acide propionique du citalopram dépend principalement des monoamines oxydases et que les cytochromes P-450 ne seraient pas impliqués dans cette voie métabolique. Nous avons également confirmé dans le cas particulier du citalopram la stéréosélectivité des monoamines oxydases. La MAO-A va préférentiellement catalyser la formation de l’énantiomère R-(-) de l’acide propionique du citalopram. Elle a également une plus grande affinité pour les amines tertiaires. Au contraire, la MAO-B va favoriser la formation de la forme S-(+) de l’acide propionique du citalopram. Elle a plus d’affinité pour les amines secondaires et primaires. Nous avons également étudié le métabolisme du citalopram chez la souris Tg8, déficiente du gène de la MAO-A, initialement développée pour des études sur le comportement. Nos résultats suggèrent une compensation partielle de la déficience de la MAO-A dans les mitochondries cérébrales des souris Tg8 par la MAO-B murine. Les souris n’exprimant pas le gène de la MAO-A étant viables, elles se sont donc adaptées à cette déficience. Cependant, les expériences ont été réalisées sur des souris adultes, or si l’activité de la MAO-A est à peu près identique durant la vie d’un rongeur, celle de la MAO-B augmente progressivement jusqu’à l’âge adulte (Cases et al., 1995). Les taux de sérotonine étant dramatiquement plus élevés chez la jeune souris déficiente du gène de la MAO-A (Tg8), sont également responsable de la désorganisation anatomique du cortex somatosensoriel (Cases et al., 1996). Pour ces deux raisons, d’autres études sont nécessaires d’une part avec de jeunes souris déficientes du gène de la MAO-A (Tg8) pour être dans la période sensible de la déficience de la MAO-A, et d’autre part, avec des souris déficientes du gène de la MAO-B (Grimsby et al., 1997). Etant donné que le rapport MAO-A/MAO-B change avec l’âge du cerveau, il serait utile de tester des souris jeunes, âgées d’une semaine, et des souris adultes. La genèse récente de souris doubles knock-out MAO-A/B (Chen et al., 2004) permettrait de caractériser l’implication des deux isoenzymes dans le processus de déamination du citalopram et de ses formes déméthylées. Un autre aspect du métabolisme du citalopram a été étudié chez les chiens, l’objectif étant de mieux comprendre les causes possibles de la toxicité liée au didéméthylcitalopram. Nos résultats montrent que les cinétiques de N-déméthylation sont biphasiques chez les chiens. Le CYP2D15, orthologue canin du CYP2D6, semble être l’enzyme à haute affinité/faible capacité dans les deux réactions menant au didéméthylcitalopram. Cet enzyme fait partie de la sous-famille des CYP2D qui représenterait 20% du total des cytochromes P-450 canins, ce qui serait unique comparé aux autres espèces. En effet, chez l’Homme, les CYP2C9, CYP2C19 et CYP2D6 représentent ensemble 40% de la totalité des cytochromes P-450. Il est intéressant de constater que les clearances intrinsèques, reflet de l’activité enzymatique dans le foie (Wilkinson, 1987), sont beaucoup plus élevées chez le chien que chez l’Homme dans la première N-déméthylation et restent très élevées chez le chien dans la deuxième N-déméthylation. Enfin, il semblerait que dans la deuxième N-déméthylation menant au didéméthylcitalopram, quatre orthologues canins des cytochromes P-450 soient impliqués (CYP2D15, CYP3A12, CYP2C9 et CYP2E1), tandis qu’un seul cytochrome P-450 (CYP2D6) serait impliqué chez l’Homme. Ceci reste hypothétique et demande de plus amples vérifications. La connaissance exhaustive des inhibiteurs spécifiques des orthologues canins des cytochromes P-450 humains, ainsi que leurs concentrations efficaces, permettrait de tester de manière sans équivoque l’implication réelle des différents orthologues dans cette étape de N-déméthylation. Ces différentes constatations permettent d’expliquer en partie les taux très élevés de didéméthylcitalopram chez les chiens et donc la toxicité observée. Dans un troisième chapitre de cette thèse, nous avons développé une méthode pour le dosage stéréosélectif par LC-MS de la mirtazapine et de ses métabolites principaux (Paus et al., 2004) dans des échantillons plasmatiques de patients dans le cadre du « therapeutic drug monitoring » (suivi des taux plasmatiques des principes actifs) effectué au sein de notre laboratoire.

- 63 -
Les résultats d’une étude préliminaire nous ont montré que des médicaments coadministrés pouvaient également être la source de perturbations du métabolisme de la mirtazapine. Par exemple, certains patients étaient co-médiqués avec différents médicaments, incluant parfois de puissants inhibiteurs du CYP2D6 comme la fluoxétine (Baumann, 1996) ou la thioridazine (von Bahr et al., 1985). La formation de la forme 8-hydroxylée de la mirtazapine est sous le contrôle du CYP2D6 et malgré son inhibition médicamenteuse, la formation de S-(+)-8-hydroxymirtazapine n’était pas complètement abolie. Ces observations correspondent avec celles rapportées dans un article de revue où la S-(+)-mirtazapine subit préférentiellement une 8-hydroxylation sous contrôle du CYP2D6, alors que la R-(-)-mirtazapine était préférentiellement métabolisée par N-glucuronidation et que son élimination était indépendante du statut pharmacogénétique (Timmer et al., 2000). Le suivi des taux plasmatiques de la mirtazapine et de ses métabolites chez les patients offre la possibilité au thérapeute de suivre les risques liés au traitement médicamenteux. Enfin, une revue de littérature sur la glycoprotéine-P, constitue la dernière partie de cette thèse. L’importance des protéines de transport y est bien mise en évidence. Si le métabolisme est un domaine de recherches intensives, la distribution fait elle aussi l’objet de nombreuses recherches, en particulier suite aux polymorphismes décrits au sein de ces protéines de transport.

- 64 -
Références
Andersson,T., Flockhart,D.A., Goldstein,D.B., Huang,S.M., Kroetz,D.L., Milos,P.M., Ratain,M.J. & Thummel,K. (2005). Drug-metabolizing enzymes: evidence for clinical utility of pharmacogenomic tests. Clin.Pharmacol.Ther., 78: 559-581.
Andersson,T., Hassan-Alin,M., Hasselgren,G. & Rohss,K. (2001). Drug interaction studies with esomeprazole, the (S)-isomer of omeprazole. Clinical Pharmacokinetics, 40: 523-537.
Angst,J. (1992). Epidemiology of depression. Psychopharmacology, 106: S71-S74.
Asberg,M., Cronholm,B., Sjöqvist,F. & Tuck,D. (1971). Relationship between plasma level and therapeutic effect of nortriptyline. British Medical Journal, 3: 331-334.
Avendano,C. & Menendez,J. (2002). Inhibitors of multidrug resistance to antitumor agents (MDR). Curr Med Chem, 9: 159-193.
Baltes,S., Fedrowitz,M., Luna Tortós,C., Potschka,H. & Löscher,W. (2007). Valproic Acid Is Not a Substrate for P-glycoprotein or Multidrug Resistance Proteins 1 and 2 in a Number of in Vitro and in Vivo Transport Assays. J Pharmacol.Exp.Ther, 320: 331-343.
Baumann,P. (1996). Pharmacokinetic-pharmacodynamic relationship of the selective serotonin reuptake inhibitors. Clinical Pharmacokinetics, 31: 444-469.
Baumann,P., Hiemke,C., Ulrich,S., Eckermann,G., Gaertner,I., Kuss,H.J., Laux,G., Müller-Oerlinghausen,B., Rao,M.L., Riederer,P. & Zernig,G. (2004). The AGNP-TDM expert group consensus guidelines: therapeutic drug monitoring in psychiatry. Pharmacopsychiatry, 37: 243-265.
Baumann,P., Jonzier-Perey,M., Paus,E. & Nikisch,G. (2006). Mirtazapine Enantiomers in Blood and Cerebrospinal Fluid. Neuropsychobiology, 54: 179-181.
Baumann,P. & Larsen,F. (1995). The pharmacokinetics of citalopram. Reviews in Contemporary Pharmacotherapy, 6: 287-295.
Baumann,P. & Rochat,B. (1995). Comparative pharmacokinetics of selective serotonin reuptake inhibitors: a look behind the mirror. International Clinical Psychopharmacology, 10 (suppl. 1): 15-21.
Baumann,P., Ulrich,S., Eckermann,G., Gerlach,M., Kuss,H.J., Laux,G., Muller-Oerlinghausen,B., Rao,M.L., Riederer,P., Zernig,G. & Hiemke,C. (2005). The AGNP-TDM Expert Group Consensus Guidelines: focus on therapeutic monitoring of antidepressants. Dialogues.Clin.Neurosci., 7: 231-247.
Benazzi,F. (1996). Serotonin syndrome with moclobemide-fluoxetine combination. Pharmacopsychiatry, 29: 162.
Bertilsson,L., Dahl,M.L., Dalen,P. & Al Shurbaji,A. (2002). Molecular genetics of CYP2D6: clinical relevance with focus on psychotropic drugs. British Journal of Clinical Pharmacology, 53: 111-122.
Blaisdell,J., Goldstein,J.A. & Bai,S.A. (1998). Isolation of a new canine cytochrome P450 CDNA from the cytochrome P450 2C subfamily (CYP2C41) and evidence for polymorphic differences in its expression. Drug Metabolism and Disposition, 26: 278-283.

- 65 -
Blakey,G.E., Lockton,J.A., Perrett,J., Norwood,P., Russell,M., Aherne,Z. & Plume,J. (2004). Pharmacokinetic and pharmacodynamic assessment of a five-probe metabolic cocktail for CYPs 1A2, 3A4, 2C9, 2D6 and 2E1. British Journal of Clinical Pharmacology, 57: 162-169.
Boeck,V. & Overø,K.F. (1982). Studies on acute toxicity and drug levels of citalopram in the dog. Acta Pharmacologica et Toxicologica, 50: 169-174.
Bolonna,A.A., Arranz,M.J., Mancama,D. & Kerwin,R.W. (2004). Pharmacogenomics - can genetics help in the care of psychiatric patients? International Review of Psychiatry, 16: 311-319.
Borlak,J., Thum,T., Landt,O., Erb,K. & Hermann,R. (2000). Molecular diagnosis of a familial nonhemolytic hyperbilirubinemia (Gilbert's syndrome) in healthy subjects. Hepatology, 32: 792-795.
Boyer,E.W. & Shannon,M. (2005). The serotonin syndrome. N.Engl.J Med., 352: 1112-1120.
Brinkmann,U., Roots,I. & Eichelbaum,M. (2001). Pharmacogenetics of the human drug-transporter gene MDR1: impact of polymorphisms on pharmacotherapy. Drug Discov.Today, 6: 835-839.
Brøsen,K. (1990). Recent developments in hepatic drug oxidation. Implications for clinical pharmacokinetics. Clinical Pharmacokinetics, 18: 220-239.
Brunner,H.G., Nelen,M., Breakefield,X.O., Ropers,H.H. & van Oost,B.A. (1993a). Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science, 262: 578-580.
Brunner,H.G., Nelen,M.R., van Zandvoort,P., Abeling,N.G.G.M., van Gennip,A.H., Wolters,E.C., Kuiper,M.A., Ropers,H.H. & van Oost,B.A. (1993b). X-linked borderline mental retardation with prominent behavioral disturbance: phenotype, genetic localization, and evidence for disturbed monoamine metabolism. American Journal of Human Genetics, 52: 1032-1039.
Carlsson,B. & Norlander,B. (2001). Optimization and characterization of the chiral separation of citalopram and its demethylated metabolites by response-surface methodology. Chromatographia, 53: 266-272.
Cases,O., Lebrand,C., Giros,B., Vitalis,T., De Maeyer,E., Caron,M.G., Price,D.J., Gaspar,P. & Seif,I. (1998). Plasma membrane transporters of serotonin, dopamine, and norepinephrine mediate serotonin accumulation in atypical locations in the developing brain of monoamine oxidase A knock-outs. Journal of Neuroscience, 18: 6914-6927.
Cases,O., Seif,I., Grimsby,J., Gaspar,P., Chen,K., Pournin,S., Müller,U., Aguet,M., Babinet,C., Shih,J.C. & De Maeyer,E. (1995). Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science, 268: 1763-1767.
Cases,O., Vitalis,T., Seif,I., De Maeyer,E., Sotelo,C. & Gaspar,P. (1996). Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron, 16: 297-307.
Catalano,G., Catalano,M.C., Epstein,M.A. & Tsambiras,P.E. (2001). QTc interval prolongation associated with citalopram overdose: a case report and literature review. Clinical Neuropharmacology, 24: 158-162.
Chang,C., Bahadduri,P.M., Polli,J.E., Swaan,P.W. & Ekins,S. (2006). Rapid Identification of P-glycoprotein Substrates and Inhibitors. Drug Metabolism and Disposition, 34: 1976-1984.

- 66 -
Charney,D.S., Menkes,D.B. & Heninger,G.R. (1981). Receptor sensitivity and the mechanism of action of antidepressant treatment: implication for the etiology and therapy of depression. Arch Gen Psychiatry, 38: 1160-1180.
Chauret,N., Gauthier,A., Martin,J. & Nicoll-Griffith,D.A. (1997). In vitro comparison of cytochrome P450-mediated metabolic activities in human, dog, cat, and horse. Drug Metabolism and Disposition, 25: 1130-1136.
Chen,K., Holschneider,D.P., Wu,W., Rebrin,I. & Shih,J.C. (2004). A spontaneous point mutation produces monoamine oxidase A/B knock-out mice with greatly elevated monoamines and anxiety-like behavior. J Biol.Chem., 279: 39645-39652.
Choo,E.F., Leake,B., Wandel,C., Imamura,H., Wood,A.J. & Wilkinson,G.R. (2000). Pharmacological inhibition of P-glycoprotein transport enhances the distribution of HIV-1 protease inhibitors into brain and testes. Drug Metabolism and Disposition, 28: 655-660.
Christensen,M., Andersson,K., Dalen,P., Mirghani,R.A., Muirhead,G.J., Nordmark,A., Tybring,G., Wahlberg,A., Yasar,U. & Bertilsson,L. (2003). The Karolinska cocktail for phenotyping of five human cytochrome P450 enzymes. Clinical Pharmacology and Therapeutics, 73: 517-528.
Clement Jerdi,M., Daali,Y., Kondo Oestreicher,M., Cherkaoui,S. & Dayer,P. (2005). A simplified analytical method for a phenotyping cocktail of major CYP450 biotransformation routes. Journal of Pharmaceutical and Biomedical Analysis, 35: 1203-1212.
Collins,F.A., Murphy,D.L., Reiss,A.L., Sims,K.B., Lewis,J.G., Freund,L., Karoum,F., Zhu,D.P., Maumenee,I.H. & Antonarakis,S.E. (1992). Clinical, Biochemical, and Neuropsychiatric Evaluation of A Patient with A Contiguous Gene Syndrome Due to A Microdeletion Xp11.3 Including the Norrie Disease Locus and Monoamine-Oxidase (Maoa and Maob) Genes. American Journal of Medical Genetics, 42: 127-134.
Copeland,R.A. (2000). Kinetics of single-substrate enzyme reaction. Enzymes, pp. 1-396. Wiley-VCH. 396 pp.
Cordon-Cardo,C., O'Brien,J.P., Casals,D., Rittman-Grauer,L., Biedler,J.L., Melamed,M.R. & Bertino,J.R. (1989). Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc.Natl.Acad.Sci.U.S.A, 86: 695-698.
Dahl,M.L. (2002). Cytochrome P450 phenotyping/genotyping in patients receiving antipsychotics: useful aid to prescribing? Clinical Pharmacokinetics, 41: 453-470.
Dahl,M.L., Voortman,G., Alm,C., Elwin,C.-E., Delbressine,L., Vos,R., Bogaards,J.J.P. & Bertilsson,L. (1997). In vitro and in vivo studies on the disposition of mirtazapine in humans. Clinical Drug Investigation, 13: 37-46.
Dalgaard,L. & Larsen,C. (1999). Metabolism and excretion of citalopram in man: identification of O-acyl- and N-glucuronides. Xenobiotica, 29: 1033-1041.
Daly,A.K. (2003). Pharmacogenetics of the major polymorphic metabolizing enzymes. Fundamental and Clinical Pharmacology, 17: 27-41.
Dams,R., Benijts,T.H.P., Lambert,W.E., Van Bocxlaer,J.F., Van Varenbergh,D., Peteghem,C.V. & De Leenheer,A.P. (2001). A fatal case of serotonin syndrome after combined moclobemide-citalopram intoxication. Journal of Analytical Toxicology, 25: 147-151.

- 67 -
Davis,R. & Wilde,M.I. (1996). Mirtazapine - A review of its pharmacology and therapeutic potential in the management of major depression. CNS Drugs, 5: 389-402.
de Boer,T. (1995). The effects of mirtazapine on central noradrenergic and serotonergic neurotransmission. International Clinical Psychopharmacology, 10 (suppl. 4): 19-23.
de Boer,T. (1996). The pharmacologic profile of mirtazapine. Journal of Clinical Psychiatry, 57 (suppl. 4): 19-25.
de Boer,T., Ruigt,G.S.F. & Berendsen,H.H.G. (1995). The α2- selective adrenoceptor antagonist org 3770 (mirtazapine, remeron) enhances noradrenergic and serotonergic transmission. Hum.Psychopharmacol.Clin.Exp., 10: S107-S118.
Delbressine,L.P.C., Moonen,M.E.G., Kaspersen,F.M., Wagenaars,G.N., Jacobs,P.L., Timmer,C.J., Paanakker,J.E., Vanhal,H.J.M. & Voortman,G. (1998). Pharmacokinetics and biotransformation of mirtazapine in human volunteers. Clinical Drug Investigation, 15: 45-55.
Delbressine,L.P.C. & Vos,R.M.E. (1997). The clinical relevance of preclinical data: mirtazapine, a model compound. Journal of Clinical Psychopharmacology, 17: 29S-33S.
Dodd,S., Burrows,G.D. & Norman,T.R. (2000). Chiral determination of mirtazapine in human blood plasma by high-performance liquid chromatography. Journal of Chromatography B, 748: 439-443.
Donnelly,C.H. & Murphy,D.L. (1977). Substrate- and inhibitor-related characteristics of human platelet monoamine oxidase. Biochemical Pharmacology, 26: 853-858.
Dostert,P.L., Strolin Benedetti,M. & Tipton,K.F. (1989). Interactions of monoamine oxidase with substrates and inhibitors. Medicinal Research Reviews, 9: 45-89.
Drozdzik,M., Bialecka,M., Mysliwiec,K., Honczarenko,K., Stankiewicz,J. & Sych,Z. (2003). Polymorphism in the P-glycoprotein drug transporter MDR1 gene: a possible link between environmental and genetic factors in Parkinson's disease. Pharmacogenetics, 13: 259-263.
Eagling,V.A., Tjia,J.F. & Back,D.J. (1998). Differential selectivity of cytochrome P450 inhibitors against probe substrates in human and rat liver microsomes. British Journal of Clinical Pharmacology, 45: 107-114.
Eap,C.B., Buclin,T., Hustert,E., Bleiber,G., Powell Golay,K., Aubert,A.C., Baumann,P., Telenti,A. & Kerb,R. (2004). Pharmacokinetics of midazolam in CYP3A4 - and CYP3A5-genotyped subjects. European Journal of Clinical Pharmacology, 60: 231-236.
Ebadi,M., Sharma,S.G., Shavali,S. & El Refaey,H. (2002). Neuroprotective actions of Delegiline. Journal of Neuroscience Research, 67: 285-289.
Ecker,G., Huber,M., Schmid,D. & Chiba,P. (1999). The importance of a nitrogen atom in modulators of multidrug resistance. Molecular Pharmacology, 56: 791-796.
Engebretsen,K.M., Harris,C.R. & Wood,J.E. (2003). Cardiotoxicity and late onset seizures with citalopram overdose. Journal of Emergency Medicine, 25: 163-166.
Evrard,A., Malagie,I., Laporte,A.M., Boni,C., Hanoun,N., Trillat,A.C., Seif,I., De Maeyer,E., Gardier,A., Hamon,M. & Adrien,J. (2002). Altered regulation of the 5-HT system in the brain of MAO-A knock-out mice. European Journal of Neuroscience, 15: 841-851.

- 68 -
Fellay,J., Marzolini,C., Meaden,E.R., Back,D.J., Buclin,T., Chave,J.P., Decosterd,L.A., Furrer,H., Opravil,M., Pantaleo,G., Retelska,D., Ruiz,L., Schinkel,A.H., Vernazza,P., Eap,C.B. & Telenti,A. (2002). Response to antiretroviral treatment in HIV-1-infected individuals with allelic variants of the multidrug resistance transporter 1: a pharmacogenetics study. Lancet, 359: 30-36.
Fornai,F., Chen,K., Giorgi,F.S., Gesi,M., Alessandri,M.G. & Shih,J.C. (1999). Striatal dopamine metabolism in monoamine oxidase B-deficient mice: a brain dialysis study. Journal of Neurochemistry, 73: 2434-2440.
Fowler,C.J., Callingham,B.A., Oconnor,M.D.L. & Matthews,E.K. (1980). The Effect of Sonication Upon Monoamine Oxidase-A and Oxidase-B in the Rat-Liver. Biochemical Pharmacology, 29: 1185-1188.
Fromm,M.F. (2002). The influence of MDR1 polymorphisms on P-glycoprotein expression and function in humans. Adv.Drug.Deliv.Rev., 54: 1295-1310.
Geha,R.M., Rebrin,I., Chen,K. & Shih,J.C. (2001). Substrate and inhibitor specificities for human monoamine oxidase A and B are influenced by a single amino acid. Journal of Biological Chemistry, 276: 9877-9882.
Gervasini,G., Carrillo,J.A. & Benitez,J. (2004). Potential role of cerebral cytochrome P450 in clinical pharmacokinetics - Modulation by endogenous compounds. Clinical Pharmacokinetics, 43: 693-706.
Ghersi-Egea,J.F., Leninger-Muller,B., Suleman,G., Siest,G. & Minn,A. (1994). Localization of drug-metabolizing enzyme activities to blood- brain interfaces and circumventricular organs. Journal of Neurochemistry, 62: 1089-1096.
Ghersi-Egea,J.F., Perrin,R., Leininger-Muller,B., Grassiot,M.-C., Jeandel,C., Floquet,J., Cuny,G., Siest,G. & Minn,A. (1993). Subcellular localization of cytochrome P450, and activities of several enzymes responsible for drug metabolism in the human brain. Biochemical Pharmacology, 45: 647-658.
Gram,L.F., Hansen,M.G.J., Sindrup,S.H., Brøsen,K., Poulsen,J.H., Aaes-Jørgensen,T. & Fredricson Over∅,K. (1993). Citalopram: interaction studies with levomepromazine, imipramine, and lithium. Therapeutic Drug Monitoring, 15: 18-24.
Grimsby,J., Toth,M., Chen,K., Kumazawa,T., Klaidman,L., Adams,J.D., Karoum,F., Gal,J. & Shih,J.C. (1997). Increased stress response and beta-phenylethylamine in MAOB-deficient mice. Nat.Genet., 17: 206-210.
Grothusen,A., Hardt,J., Bräutigam,L., Lang,D. & Böcker,R. (1996). A convenient method to discriminate between cytochrome P450 enzymes and flavin-containing monooxygenases in human liver microsomes. Archives of Toxicology, 71: 64-71.
Guengerich,F.P. (1997). Comparisons of catalytic selectivity of cytochrome P450 subfamily enzymes from different species. Chemico-Biological Interactions, 106: 161-182.
Hansen,R.A., Gartlehner,G., Lohr,K.N., Gaynes,B.N. & Carey,T.S. (2005). Efficacy and safety of second-generation antidepressants in the treatment of major depressive disorder. Annals of Internal Medicine, 143: 415-426.
Harro,J. & Oreland,L. (1996). Depression as a spreading neuronal adjustment disorder. European Neuropsychopharmacology, 6: 207-223.
Harvey,A.T., Rudolph,R.L. & Preskorn,S.H. (2000). Evidence of the dual mechanisms of action of venlafaxine. Arch Gen Psychiatry, 57: 503-509.

- 69 -
Henderson,P.J.F. (1985). Techniques in the life sciences, B1/II supplement Protein and enzyme biochemistry, BS114. Ireland Ltd.: Elsevier Scientific Publishers Ireland Ltd. 48 pp.
Higgins,C.F. & Gottesmann,M.M. (1992). Is the multidrug transporter a flippase? Trends.Biochem.Sci., 17: 18-21.
Hirschfeld,R.M.A. (1999). Efficacy of SSRIs and newer antidepressants in severe depression: comparison with TCAs. Journal of Clinical Psychiatry, 60: 326-335.
Hirschfield,G.M. & Alexander,G.J. (2006). Gilbert's syndrome: an overview for clinical biochemists. Annals of Clinical Biochemistry, 43: 340-343.
Hoffmeyer,S., Burk,O., von Richter,O., Arnold,H.P., Brockmöller,J., Johne,A., Cascorbi,I., Gerloff,T., Roots,I., Eichelbaum,M. & Brinkmann,U. (2000). Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proceedings of the National Academy of Sciences of the United States of America, 97: 3473-3478.
Holschneider,D.P., Chen,K., Seif,I. & Shih,J.C. (2001). Biochemical, behavioral, physiologic, and neurodevelopmental changes in mice deficient in monoamine oxidase A or B. Brain Research Bulletin, 56: 453-462.
Hyttel,J. (1982). Citalopram - pharmacological profile of a specific serotonin uptake inhibitor with antidepressant activity. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 6: 277-295.
Hyttel,J. (1994). Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs). International Clinical Psychopharmacology, 9 (suppl.1): 19-26.
Hyttel,J., Bøgesø,K.P., Perregaard,J. & Sanchez,C. (1992). The pharmacological effect of citalopram resides in the (S)-(+)- enantiomer. Journal of Neural Transmission - General Section, 88: 157-160.
Ingelman-Sundberg,M. (2005). Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics Journal, 5: 6-13.
Juliano,R.L. & Ling,V. (1976). A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim.Biophys.Acta, 455: 152-162.
Kamimura,H. (2006). Genetic polymorphism of cytochrome P450s in beagles: possible influence of CYP1A2 deficiency on toxicological evaluations. Arch Toxicol..
Kandel,E.R., Schwartz,J.H. & Jessell,T.M. (2000). Disorder of Mood: Depression, Mania and Anxiety Disorders. Principales of Neural Science, pp. 1-1321. McGraw-Hill. 1321 pp.
Kelder,J., Funke,C., de Boer,T., Delbressine,L., Leysen,D. & Nickolson,V. (1997). A comparison of the physicochemical and biological properties of mirtazapine and mianserin. Journal of Pharmacy and Pharmacology, 49: 403-411.
Kent,J.M. (2000). SNaRIs, NaSSAs, and NaRIs: new agents for the treatment of depression. Lancet, 355: 911-918.
Kessler,R.C., Berglund,P., Demler,O., Jin,R., Koretz,D., Merikangas,K.R., Rush,A.J., Walters,E.E. & Wang,P.S. (2003). The epidemiology of major depressive disorder. JAMA, 289: 3095-3105.
Khalifa,M., Daleau,P. & Turgeon,J. (1999). Mechanism of sodium channel block by venlafaxine in guinea pig ventricular myocytes. J Pharmacol.Exp.Ther, 291: 280-284.

- 70 -
Kim,R.B. (2002). Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metabolism Reviews, 34: 47-54.
Kim,R.B., Leake,B.F., Choo,E.F., Dresser,G.K., Kubba,S.V., Schwarz,U.I., Taylor,A., Xie,H.G., McKinsey,J., Zhou,S., Lan,L.B., Schuetz,J.D., Schuetz,E.G. & Wilkinson,G.R. (2001). Identification of functionally variant MDR1 alleles among european americans and african americans. Clinical Pharmacology and Therapeutics, 70: 189-199.
Kim,R.B., Wandel,C., Leake,B., Cvetkovic,M., Fromm,M.F., Dempsey,P.J., Roden,M.M., Belas,F., Chaudhary,A.K., Roden,D.M., Wood,A.J.J. & Wilkinson,G.R. (1999). Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein. Pharmaceutical Research, 16: 408-414.
Kioka,N., Tsubota,J., Kakehi,Y., Komano,T., Gottesman,M.M., Pastan,I. & Ueda,K. (1989). P-glycoprotein gene (MDR1) cDNA from human adrenal: normal P-glycoprotein carries Gly185 with an altered pattern of multidrug resistance. Biochemical and Biophysical Research Communications, 162: 224-231.
Kirby,B., Kharasch,E.D., Thumme,K.T., Narang,V.S., Hoffer,C.J. & Unadkat,J.D. (2006). Simultaneous measurement of in vivo P-glycoprotein and cytochrome P450 3A activities. J Clin Pharmacol., 46: 1313-1319.
Kirchheiner,J., Nickchen,K., Bauer,M., Wong,M.L., Licinio,J., Roots,I. & Brockmöller,J. (2004). Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol.Psychiatry, 9: 442-473.
Ko,J.W., Sukhova,N., Thacker,D., Chen,P. & Flockhart,D.A. (1997). Evaluation of omeprazole and lansoprazole as inhibitors of cytochrome P450 isoforms. Drug Metabolism and Disposition, 25: 853-862.
Kobayashi,K., Chiba,K., Yagi,T., Shimada,N., Taniguchi,T., Horie,T., Tani,M., Yamamoto,T., Ishizaki,T. & Kuroiwa,Y. (1997). Identification of cytochrome P450 isoforms involved in citalopram N-demethylation by human liver microsomes. Journal of Pharmacology and Experimental Therapeutics, 280: 927-933.
Kobayashi,K., Urashima,K., Shimada,N. & Chiba,K. (2003). Selectivities of human cytochrome P450 inhibitors toward rat P450 isoforms: study with cDNA-expressed systems of the rat. Drug Metabolism and Disposition, 31: 833-836.
Kosel,M., Amey,M., Aubert,A.C. & Baumann,P. (2001). In vitro metabolism of citalopram by monoamine oxidase B in human blood. European Neuropsychopharmacology, 11: 75-78.
Kosel,M., Eap,C.B., Amey,M. & Baumann,P. (1998). Analysis of the enantiomers of citalopram and its demethylated metabolites using chiral liquid chromatography. Journal of Chromatography B: Biomedical Applications, 719: 234-238.
Kosel,M., Gnerre,C., Voirol,P., Amey,M., Rochat,B., Bouras,C., Testa,B. & Baumann,P. (2002). In vitro biotransformation of the selective serotonin reuptake inhibitor citalopram, its enantiomers and demethylated metabolites by monoamine oxidase in rat and human brain preparations. Molecular Psychiatry, 7: 181-188.
Kuroha,M., Azumano,A., Kuze,Y., Shimoda,M. & Kokue,E. (2002). Effect of multiple dosing of ketoconazole on pharmacokinetics of midazolam, a cytochrome P-450 3A substrate in beagle dogs. Drug Metabolism and Disposition, 30: 63-68.

- 71 -
Kuroha,M., Shirai,Y. & Shimoda,M. (2004). Multiple oral dosing of ketoconazole influences pharmacokinetics of quinidine after intravenous and oral administration in beagle dogs. J Vet.Pharmacol.Ther, 27: 355-359.
Langston,J.W., Ballard,P., Tetrud,J.W. & Irwin,I. (1983). Chronic parkinsonism in humans due to a product of meperidine-analog systhesis. Science, 219: 979-980.
Lankas,G.R. & Cartwright,M.E. (1997). P-glycoprotein deficiency in a subpopulation of CF-1 mice enhances ivermectin-induced neurotoxicity. Toxicology and Applied Pharmacology, 143: 357-365.
Lankas,G.R., Wise,L.D., Cartwright,M.E., Pippert,T.R. & Umbenhauer,D.R. (1998). Placental P-glycoprotein deficiency enhances susceptibility to chemically induced birth defects in mice. Reproductive Toxicology, 12: 457-463.
Lankford,S.M., Bai,S.A. & Goldstein,J.A. (2000). Cloning of canine cytochrome P450 2E1 cDNA: identification and characterization of two variant alleles. Drug Metabolism and Disposition, 28: 981-986.
Lee,C.G., Ramachandra,M., Jeang,K.T., Martin,M.A., Pastan,I. & Gottesman,M.M. (2000). Effect of ABC transporters on HIV-1 infection: inhibition of virus production by the MDR1 transporter. FASEB.J., 14: 516-522.
Lenders,J.W.M., Eisenhofer,G., Abeling,N.G.G.M., Berger,W., Murphy,D.L., Konings,H., Bleeker Wagemakers,L.M., Kopin,I.J., Karoum,F., van Gennip,A.H. & Brunner,H.G. (1996). Specific genetic deficiencies of the A and B isoenzymes of monoamine oxidase are characterized by distinct neurochemical and clinical phenotypes. Journal of Clinical Investigation, 97: 1010-1019.
Lewis,D.F.V. (2000). On the recognition of mammalian microsomal cytochrome P450 substrates and their characteristics - Towards the prediction of human P450 substrate specificity and metabolism. Biochemical Pharmacology, 60: 293-306.
Maret,G., Testa,B., Jenner,P., Eltayar,N. & Carrupt,P.A. (1990). The MPTP story -MAO activates tetrahydropyridine derivatives to toxins causing parkinsonism. Drug Metabolism Reviews, 22: 291-332.
Marzolini,C., Paus,E., Buclin,T. & Kim,R.B. (2004). Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clinical Pharmacology and Therapeutics, 75: 13-33.
Mendel,R.R. & Bittner,F. (2006). Cell biology of molybdenum. Biochim.Biophys.Acta, 1763: 621-635.
Meyer,U.A. & Zanger,U.M. (1997). Molecular mechanisms of genetic polymorphisms of drug metabolism. Annual Review of Pharmacology and Toxicology, 37: 269-296.
Miksys,S. & Tyndale,R.F. (2004). The unique regulation of brain cytochrome P450 2 (CYP2) family enzymes by drugs and genetics. Drug Metabolism Reviews, 36: 313-333.
Montgomery,S.A. (1995). Safety of mirtazapine: a review. International Clinical Psychopharmacology, 10 (suppl. 4): 37-45.
Moody,G.C., Griffin,S.J., Mather,A.N., McGinnity,D.F. & Riley,R.J. (1999). Fully automated analysis of activities catalysed by the major human liver cytochrome P450(CYP) enzymes: assessment of human CYP inhibition potential. Xenobiotica, 29: 53-75.
Nakamura,S., Ito,Y., Fukushima,T., Sugawara,Y. & Ohashi,M. (1990). Metabolism of diltiazem. III. Oxidative deamination of diltiazem in rat liver microsomes. Journal of Pharmacobio-Dynamics, 13: 612-621.

- 72 -
Nebert,D.W. & Russell,D.W. (2002). Clinical importance of the cytochromes P450. Lancet, 360: 1155-1162.
Nelson,D.R., Zeldin,D.C., Hoffman,M.G., Maltais,L.J., Wain,H.M. & Nebert,D.W. (2004). Comparqaison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics, 14: 1-18.
Neuvonen,P.J., Pohjola-Sintonen,S., Tacke,U. & Vuori,E. (1993). Five fatal cases of serotonin syndrome after moclobemide-citalopram or moclobemide-clomipramine overdoses. Lancet, 342: 1419.
Newton,D.J., Wang,R.W. & Lu,A.Y.H. (1995). Cytochrome P450 inhibitors - Evaluation of specificities in the in vitro metabolism of therapeutic agents by human liver microsomes. Drug Metabolism and Disposition, 23: 154-158.
Obach,R.S., Huynh,P., Allen,M.C. & Beedham,C. (2004). Human liver aldehyde oxidase: inhibition by 239 drugs. Journal of Clinical Pharmacology, 44: 7-19.
Olesen,O.V. & Linnet,K. (1999). Studies on the stereoselective metabolism of citalopram by human liver microsomes and cDNA-expressed cytochrome P450 enzymes. Pharmacology, 59: 298-309.
Öström,M., Eriksson,A., Thorson,J. & Spigset,O. (1996). Fatal overdose with citalopram. Lancet, 348: 339-340.
Overø,K.F. (1982). Kinetics of citalopram in test animals: drug exposure in safety studies. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 6: 297-309.
Overø,K.F. (1989). The pharmacokinetic and safety evaluation of citalopram from preclinical and clinical data. In: Montgomery,S.A. (Ed.), Citalopram. The new antidepressant from Lundbeck research. Proceedings of a symposium 11 august 1988, pp. 22-30. Amsterdam: Excerpta Medica. 30 pp.
Overø,K.F. & Højelse,F. (1994). The unexpected preclinical finding - Elucidation of dog toxicity - Citalopram. Association of the Swedish Pharmaceutical Industry, 113-115.
Pal,D. & Mitra,A.K. (2006). MDR- and CYP3A4-mediated drug–herbal interactions. Life Sciences, 78: 2131-2145.
Paus,E., Jonzier-Perey,M., Cochard,N., Eap,C.B. & Baumann,P. (2004). Chirality in the new generation of antidepressants: Stereoselective analysis of the enantiomers of mirtazapine, N-demethylmirtazapine and 8-hydroxymirtazapine by LC-MS. Therapeutic Drug Monitoring, 26: 366-374.
Rapaport,M.H. (2001). Prevalence, recognition, and treatment of comorbid depression and anxiety. Journal of Clinical Psychiatry, 62 (Suppl 24): 6-10.
Ravindranath,V., Anandatheerthavarada,H.K. & Shankar ,S.K. (1989). Xenobiotic metabolism in human brain - presence of cytochrome P-450 and associated mono-oxygenases. Brain Research, 496: 331-335.
Rebsam,A., Seif,I. & Gaspar,P. (2002). Refinement of thalamocortical arbors and emergence of barrel domains in the primary somatosensory cortex: A study of normal and monoamine oxidase a knock-out mice. Journal of Neuroscience, 22: 8541-8552.
Rendic,S. & Di Carlo,F.J. (1997). Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metabolism Reviews, 29: 413-580.

- 73 -
Roberts,R.L., Joyce,P.R., Mulder,R.T., Begg,E.J. & Kennedy,M.A. (2002). A common P-glycoprotein polymorphism is associated with nortriptyline-induced postural hypotension in patients treated for major depression. Pharmacogenomics Journal, 2: 191-196.
Rochat,B., Amey,M. & Baumann,P. (1995a). Analysis of enantiomers of citalopram and its demethylated metabolites in plasma of depressive patients using chiral reverse-phase liquid chromatography. Therapeutic Drug Monitoring, 17: 273-279.
Rochat,B., Amey,M., Gillet,M., Meyer,U.A. & Baumann,P. (1997a). Identification of three cytochrome P450 isozymes involved in N-demethylation of citalopram enantiomers in human liver microsomes. Pharmacogenetics, 7: 1-10.
Rochat,B., Amey,M., van Gelderen,H., Testa,B. & Baumann,P. (1995b). Determination of the enantiomers of citalopram, its demethylated and propionic acid metabolites in human plasma by chiral HPLC. Chirality, 7: 389-395.
Rochat,B., Kosel,M., Baumann,P., Boss,G., Testa,B. & Gillet,M. (1997b). MAO-A and MAO-B differ in their stereoselective biotransformation of citalopram in human liver. pp. S137-S138. S138 pp.
Rochat,B., Kosel,M., Boss,G., Testa,B., Gillet,M. & Baumann,P. (1998). Stereoselective biotransformation of the selective serotonin reuptake inhibitor, citalopram, and its demethylated metabolites by monoamine oxidases in human liver. Biochemical Pharmacology, 56: 15-23.
Roussel,F., Duignan,D.B., Lawton,M.P., Obach,R.S., Strick,C.A. & Tweedie,D.J. (1998). Expression and characterization of canine cytochrome P450 2D15. Archives of Biochemistry and Biophysics, 357: 27-36.
Sakaeda,T., Nakamura,T. & Okumura,K. (2002). MDR1 genotype-related pharmacokinetics and pharmacodynamics. Biological and Pharmaceutical Bulletin, 25: 1391-1400.
Sakamoto,K., Kirita,S., Baba,T., Nakamura,Y., Yamazoe,Y., Kato,R., Takanaka,A. & Matsubara,T. (1995). A new cytochrome P450 form belonging to the CYP2D in dog liver microsomes: purification, cDNA cloning, and enzyme characterization. Archives of Biochemistry and Biophysics, 319: 372-382.
Salichon,N., Gaspar,P., Upton,A.L., Picaud,S., Hanoun,N., Hamon,M., De Maeyer,E., Murphy,D.L., Mossner,R., Lesch,K.P., Hen,R. & Seif,I. (2001). Excessive activation of serotonin (5-HT) 1B receptors disrupts the formation of sensory maps in monoamine oxidase a and 5-HT transporter knock-out mice. Journal of Neuroscience, 21: 884-896.
Sampietro,M. & Iolascon,A. (1999). Molecular pathology of Crigler-Najjar type I and II and Gilbert's syndromes. Haematologica, 84: 150-157.
Saura,J., Kettler,R., Da Prada,M. & Richards,J.G. (1992). Quantitative enzyme radioautography with 3H-Ro 41-1049 and 3H-Ro 19-6327 in vitro: localization and abundance of MAO-A and MAO-B in rat CNS, peripheral organs, and human brain. J.Neurosci., 12: 1977-1999.
Schaeffeler,E., Eichelbaum,M., Brinkmann,U., Penger,A., Asante-Poku,S., Zanger,U.M. & Schwab,M. (2001). Frequency of 3435T polymorphism of MDR1 gene in African people. Lancet, 358: 383-384.
Schildkraut,J.J. (1965). The catecholamine hypothesis of affective disorders : a review of supporting evidence. American Journal of Psychiatry.
Schinkel,A.H. (1997). The physiological function of drug-transporting P-glycoproteins. Semin.Cancer Biol., 8: 161-170.

- 74 -
Schinkel,A.H., Mayer,U., Wagenaar,E., Mol,C.A.A.M., Van Deemter,L., Smit,J.J.M., Van Der Valk,M.A., Voordouw,A.C., Spits,H., Van Tellingen,O., Zijlmans,J.-M., Fibbe,W.E. & Borst,P. (1997). Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoprotein. Proc Natl Acad Sci, 94: 4028-4033.
Schinkel,A.H., Smit,J.J., Van Tellingen,O., Beijnen,J.H., Wagenaar,E. & Van Deemter,L.e.al. (1994). Disruption of the mouse mdr1a P-Glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell, 77: 491-502.
Schwab,M., Schaeffeler,E., Marx,C., Fromm,M.F., Kaskas,B. & Metzler,J. (2003). Association between the C3435T MDR1 gene polymorphism and susceptibility for ulcerative colitis. Gastroenterology, 124: 26-33.
Seelig,A. (2003). A general pattern for substrate recognition by P-glycoprotein. European Journal of Biochemistry, 251: 252-261.
Shih,J.C., Chen,K. & Geha,R.M. (1998). Determination of regions important for monoamine oxidase (MAO) A and B substrate and inhibitor selectivities. Journal of Neural Transmission, (Suppl. 52): 1-8.
Shih,J.C., Chen,K. & Ridd,M.J. (1999). Monoamine Oxydase: From genes to behavior. Ann.Rev.Neurosci., 22: 197-217.
Shou,M., Norcross,R., Sandig,G., Lu,P., Li,Y., Lin,Y., Mei,Q., Rodrigues,A.D. & Rushmore,T.H. (2003). Substrate specificity and kinetic properties of seven heterologously expressed dog cytochromes p450. Drug Metabolism and Disposition, 31: 1161-1169.
Sindrup,S.H., Brøsen,K., Hansen,M.G.J., Aaes-Jørgensen,T., Fredricson Over∅,K. & Gram,L.F. (1993). Pharmacokinetics of citalopram in relation to the sparteine and the mephenytoin oxidation polymorphisms. Therapeutic Drug Monitoring, 15: 11-17.
Sitsen,J.M.A. & Moors,J. (1994). Mirtazapine, a novel antidepressant, in the treatment of anxiety symptoms - Results from a placebo-controlled trial. Drug Investigation, 8: 339-344.
Speck,R.R., Yu,X.F., Hildreth,J. & Flexner,C. (2002). Differential effects of p-glycoprotein and multidrug resistance protein-1 on productive human immunodeficiency virus infection. J.Infect.Dis., 186: 332-340.
Stahl,S.M., Mendels,J. & Schwartz,G.E. (2002). Effects of reboxetine on anxiety, agitation, and insomnia: results of a pooled evaluation of randomized clinical trials. Journal of Clinical Psychopharmacology, 22: 388-392.
Störmer,E., Von Moltke,L.L. & Greenblatt,D.J. (2000a). Scaling drug biotransformation data from cDNA-expressed cytochrome P-450 to human liver: a comparison of relative activity factors and human liver abundance in studies of mirtazapine metabolism. Journal of Pharmacology and Experimental Therapeutics, 295: 793-801.
Störmer,E., Von Moltke,L.L., Shader,R.I. & Greenblatt,D.J. (2000b). Metabolism of the antidepressant mirtazapine in vitro: contribution of cytochromes P-450 1A2, 2D6, and 3A4. Drug Metabolism and Disposition, 28: 1168-1175.
Strolin Benedetti,M. (2001). Biotransformation of xenobiotics by amine oxidases. Fundamental and Clinical Pharmacology, 15: 75-84.
Sugawara,I., Kataoka,I., Morishita,Y., Hamada,H., Tsuro,T., Itoyama,S. & Mari,S. (1988). Tissue distribution of P-glycoprotein encoded by a multidrug-resistant gene as revealed by a monoclonal antibody MRP16. Cancer Res., 48: 1926-1929.

- 75 -
Tanabe,M., Ieiri,I., Nagata,N., Inoue,K., Ito,S., Kanamori,Y., Takahashi,M., Kurata,Y., Kigawa,J., Higuchi,S., Terakawa,N. & Otsubo,K. (2001). Expression of P-glycoprotein in human placenta: relation to genetic polymorphism of the multidrug resistance (MDR)-1 gene. J.Phamacol.Exp.Ther, 297: 1137-1143.
Tanaka,E. (1998). Clinically important pharmacokinetic drug-drug ineractions: role of cytochrome P450 enzymes. J.Clin.Pharm.Ther., 23: 403-416.
Tata,J.R. (1972). Preparations and properties of microsomal and submicrosomal fractions from secretory and non-secretory tissues. In: Birnie,G.D. (Ed.), Subcellular components - Preparation and fractionation, pp. 185-214. London: Butterworths. 214 pp.
Thiebaut,F., Tsuruo,T., Hamada,H., Gottesman,M.M., Pastan,I. & Willingham,M.C. (1987). Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl.Acad.Sci.U.S.A, 84: 7735-7738.
Thull,U. & Testa,B. (1994). Screening of unsubstituted cyclic compounds as inhibitors of monoamine oxidases. Biochemical Pharmacology, 47: 2307-2310.
Timmer,C.J., Sitsen,J.M.A. & Delbressine,L.P. (2000). Clinical pharmacokinetics of mirtazapine. Clinical Pharmacokinetics, 38: 461-474.
Tipton,K.F. (1985). Techniques in the life sciences, B1/II supplement - Protein and enzyme biochemistry, BS113. Ireland Ltd.: Elsevier Scientific Publishers. 61 pp.
Tremblay,P. & Blier,P. (2006). Catecholaminergic strategies for the treatment of major depression. Curr Drug Targets, 7: 149-158.
Tsugeno,Y. & Ito,A. (1997). A key amino acid responsible for substrate selectivity of monoamine oxidase A and B. J Biol Chem, 272: 14033-14036.
Uhr,M., Grauer,M.T. & Holsboer,F. (2003). Differential enhancement of antidepressant penetration into the brain in mice with abcb1ab (mdr1ab) P-glycoprotein gene disruption. Biological Psychiatry, 54: 840-846.
van Helvoort,A., Smith,A.J., Sprong,H., Fritzsche,I., Schinkel,A.H., Borst,P. & van Meer,G. (1996). MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell, 87: 507-517.
Verschraagen,M., Koks,C., Schellens,J.H. & Beijnen,J.H. (1999). P-glycoprotein system as a determinant of drug interactions: the case of digoxin-verapamil. Pharmacological Research, 40: 301-306.
Viaggi,C., Pardini,C., Vaglini,F. & Corsini,G.U. (2006). Cytochrome P-450 and Parkinson's disease: protective role of neuronal CYP 2E1 from MPTP toxicity. J Neural Transm.Suppl, 70: 173-176.
Voirol,P., Jonzier-Perey,M., Porchet,F., Reymond,M.J., Janzer,R.C., Bouras,C., Strobel,H.W., Kosel,M., Eap,C.B. & Baumann,P. (2000). Cytochrome P-450 activities in human and rat brain microsomes. Brain Research, 855: 235-243.
von Bahr,C., Spina,E., Birgersson,C., Ericsson,Ö., Göransson,M., Henthorn,T. & Sjöqvist,F. (1985). Inhibition of desmethylimipramine 2-hydroxylation by drugs in human liver microsomes. Biochemical Pharmacology, 34: 2501-2505.
Von Moltke,L.L., Greenblatt,D.J., Grassi,J.M., Granda,B.W., Venkatakrishnan,K., Duan,S.X., Fogelman,S.M., Harmatz,J.S. & Shader,R.I. (1999). Citalopram and desmethylcitalopram in vitro:

- 76 -
human cytochromes mediating transformation, and cytochrome inhibitory effects. Biological Psychiatry, 46: 839-849.
Vuoriletho,M.S., Melartin,T.K. & Isometsa,E.T. (2006). Suicidal behaviour among primary-care patients with depressive disorders. Psychol Med, 36: 203-210.
Watanabe,K., Narimatsu,S., Yamamoto,I. & Yoshimura,H. (1990). Hepatic microsomal oxygenation of aldehydes to carboxylic acids. Biochemical and Biophysical Research Communications, 166: 1308-1312.
Wilkinson,G.R. (1987). Clearance approaches in pharmacology. Pharmacol.Rev., 39: 1-47.
Wilkinson,G.R. (2005). Drug metabolism and variability among patients in drug response. N.Engl.J.Med, 352: 2211-2221.
Wong,E.H.F., Sonders,M.S., Amara,S.G., Tinholt,P.M., Piercey,M.F.P., Hoffman,W.P., Hyslop,D.K., Franklin,S., Porsolt,R.D., Bonsignori,A., Carfagna,N. & McArthur,R.A. (2000). Reboxetine: a pharmacologically potent, selective, and specific norepinephrine reuptake inhibitor. Biological Psychiatry, 47: 818-829.
Zuber,R., Anzenbacherova,E. & Anzenbacher,P. (2002). Cytochromes P450 and experimental models of drug metabolism. J Cell Mol.Med, 6: 189-198.