génomique structurale et fonctionnelle chez les animaux de...
TRANSCRIPT

10/10/2008
Génomique structurale et fonctionnelle chez les animaux de rente
Laurence FLORIGénétique Animale et Biologie Intégrative
INRA, Jouy-en-Josas

10/10/2008
Plan de l’exposéIntroduction et définitions
La génomique structurale1. La cartographie des génomes animaux2. L’amélioration génétique
- Principes
- Exemples de détections de QTLs
- Sélection assistée par marqueurs (SAM)
La génomique fonctionnelle1. Transcriptome et protéome2. Etude du transcriptome
- Techniques bas et haut-débit
- Vers le séquençage du transcriptome
- Exemples d’étude du transcriptome
La génomique génétique
Conclusion

10/10/2008
Introduction et définitions

10/10/2008
Les animaux domestiquesUne ressource pour comprendre les bases moléculaires
de la variation phénotypique
• L’élevage des animaux domestiques a abouti à une grande diversité intra-spécifique (races)
• Comprendre la base génétique de la diversité phénotypique inter et intra-spécifique est un des enjeux majeurs de la biologie moderne.

10/10/2008
Les animaux de rente
� Mammifères:Bos taurus Ovis aries Capra hircus
Sus scrofa Equus caballus Orytolagus cuniculus
� Volailles:
Gallus gallus Anas platyrhyncos
� Poissons:
Onchoryncus mykiss
Une vingtaine d’espèces domestiquées il y a plus de 8000 ans (pour la plupart).

10/10/2008
Une vision formelle de l’élevage…
• L’élevage des animaux de rente est basée sur «
maîtrise de la génération animale » (Aristote)
• Les trois éléments de la génération animale (Vissac, 2002)
� Contexte : pratiques d’élevage (nutrition)
� Contenant : techniques de reproduction (I.A.,OPU/FIV)
� Contenu : la génétique (Amélioration génétique)

10/10/2008
Quelques exemples de caractères d’intérêt zootechnique …
• Porc– Composition de la carcasse– Pathologie tumorale (Mélanome)
• Poulet– Résistance aux maladies– Modèle d’épilepsie
• Cheval– Diverses pathologies
• Espèces aquacoles– Caractéristique de la chair et
des carcasses– Résistance aux maladies virales
et bactériennes
• Mouton– Tremblante– Résistance aux salmonelles
• Lapin– Caractère Rex (pelage)
• Chèvre– Débit de traite
• Bovin– Caractères de production– Caractères fonctionnels
• Fertilité des vaches laitières• Résistance aux mammites

10/10/2008
L’amélioration génétique basée sur la sélection sur indice
• L’Amélioration Génétique s’appuie sur un choix rationalisé des
candidats à la reproduction
• Très efficace durant les dernières décennies
– Explique de 1/2 à 2/3 de l’amélioration des performances en production laitière (x4 depuis 1950)
• Les méthodes classiques sont basées sur :
– Collecte d’informations de performances individuelles
– Compilation des observations avec des informations généalogiques
– Généalogie VS Performance ⇒⇒⇒⇒ « Indice de sélection » pour chaque candidat à la reproduction
– Diffusion du progrès génétique (I.A.)

10/10/2008
Amélioration génétique et génomique
• Quelques limites de la sélection classique sur indice
– Certains caractères sont difficiles à mesurer (résistance aux maladies)
– Certains caractères ont une héritabilité faible (fertilité)
– Certains caractères sont antagonistes (QL & fert)
– Coût de l’évaluation des candidats à la reproduction (40k€/taureau d’IA)
• Apport de la dissection moléculaire des caractères d’intérêt
– Sélection directe des individus sur la base de leur génotype
– Compréhension du déterminisme moléculaire et des mécanismes physiologiques

10/10/2008
Atouts/Limites des animaux de rente
• Les Atouts :– Très bon suivis et beaucoup d’informations sur les populations exploitées
(pour les besoins de la sélection) :
• Information généalogique (Livres généalogiques)
• Information phénotypique
• Facilités pour l’obtention d’échantillons
– Possibilité de réaliser des accouplements dirigés : croisement entre 2 races (variabilité génétique !)
• Les Limites : des dispositifs expérimentaux optimaux impossibles (ou difficiles) à réaliser
– Il n’existe pas de lignées pures (=! souris, rat)
– Temps de génération souvent importants

10/10/2008
Le programme de l’INRA

10/10/2008
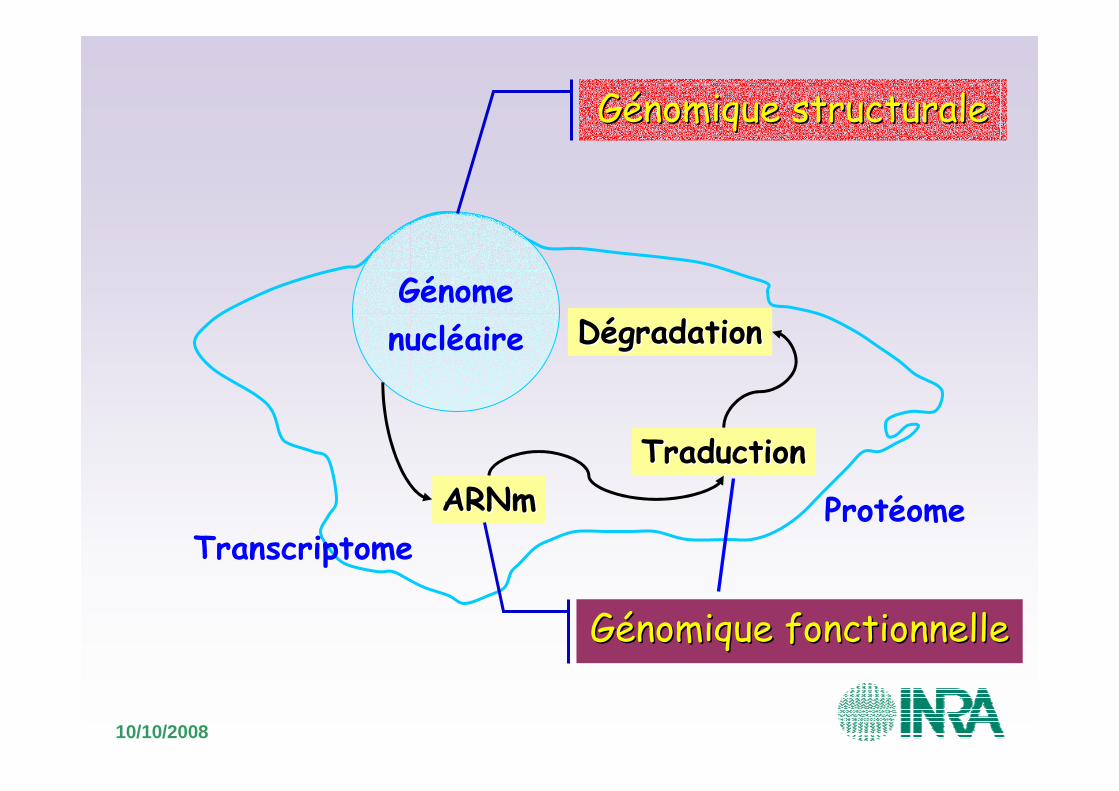
Génomenucléaire
Génomique structuraleGénomique structurale
ARNmARNmTraductionTraduction
DégradationDégradation
Génomique fonctionnelleGénomique fonctionnelle
ProtéomeTranscriptome

10/10/2008
La génomique structurale

10/10/2008
La génomique structurale
1. La cartographie des génomes animaux
2. Application: l’amélioration génétique des animaux

10/10/2008
La génomique structurale• C’est d’abord … avoir les outils pour baliser et étudier la
structure du génome– Marqueurs génétiques (microsatellites et SNP)
• Balises du génome et traceurs de l’information génétique
– Cartes génomiques
• « Charpente » et « articulation » du génome
– Collections représentatives du génome
(Banques de grands fragments d’ADN)
• BAC (Bacterial Artificial Chromosome)
• Intérêts– Connaissance fondamentale
– Amélioration génétique

10/10/2008
Cartographie génomique
• Cartographie génétique
– Loci polymorphes et familles de référence
• Cartographie physique chromosomique
– Hybrides somatiques
– Hybridation In Situ
– Hybrides d’irradiation
• Cartographie physique moléculaire
– Contig du génome
– Séquence complète de l ’ADN

10/10/2008
Cartographie génomique
• Cartographie génétique
– Loci polymorphes et familles de référence
• Cartographie physique chromosomique
– Hybrides somatiques
– Hybridation In Situ
– Hybrides d’irradiation
• Cartographie physique moléculaire
– Contig du génome
– Séquence complète de l ’ADN

10/10/2008
Cartographie génétique• Objectif: mesurer la distance entre 2 marqueurs par recombinaison méiotique et déterminer l’ordre des marqueurs le long du chromosome
• Principe: – Etude de la co-ségrégation des marqueurs au cours de la méiose– Estimation de leur ordre sur le chromosome– Calculer la distance génétique:
d=F(R) d en cM (centimorgan), R=taux de recombinaison
La mesure de la distance génétique est la fraction de recombinaison
(cM)
• Nécessite le développement de marqueurs génétiques et la collecte de familles
• Deux approches générales
– cartographie par marqueur-caractère : localiser des gènes d ’intérêt– cartographie par marqueur-marqueur : construire une charpente de cartes grâce à des marqueurs

10/10/2008
Les marqueurs moléculaires� Intérêt en cartographie :
� suivi de la transmission de segments chromosomiques au sein du pedigree
� Les 4 qualités nécessaires des marqueurs � Polymorphisme : suivi des évènements de recombinaisons au fil des générations
� Homogénéité dans la répartition génomique
� Neutralité (pas de biais de ségrégation)
� Tous les génotypes peuvent être identifiables (homozygotes et hétérozygotes)
� Types de marqueurs :
� VNTR: Mini et microsatellites
� Mutations ponctuelles: SNP (Single Nucleotide Polymorphism)
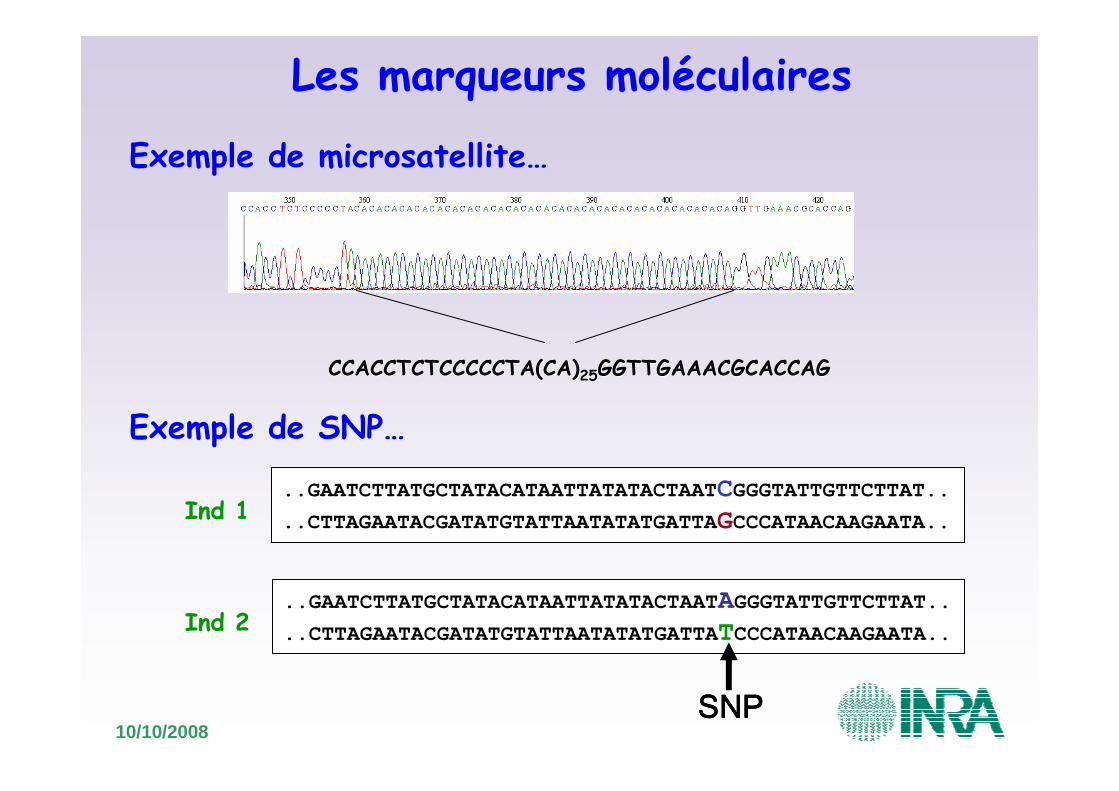
10/10/2008
CCACCTCTCCCCCTA(CA)CCACCTCTCCCCCTA(CA)2525GGTTGAAACGCACCAGGGTTGAAACGCACCAG
Exemple de microsatellite…
..GAATCTTATGCTATACATAATTATATACTAATCGGGTATTGTTCTTAT..
..CTTAGAATACGATATGTATTAATATATGATTAGCCCATAACAAGAATA..
..GAATCTTATGCTATACATAATTATATACTAATAGGGTATTGTTCTTAT..
..CTTAGAATACGATATGTATTAATATATGATTATCCCATAACAAGAATA..
Ind 1
SNPSNPSNPSNP
Ind 2
Exemple de SNP…
Les marqueurs moléculaires

10/10/2008
Cartographie génétique
• Programmes développés par l’INRA
Programmes internationaux
PIGMAP
BOVMAP
SALMAP

10/10/2008
Cartographie génomique • Cartographie génétique
– Loci polymorphes et familles de référence
• Cartographie physique chromosomique
– Hybridation In Situ
– Hybrides d’irradiation
– Cartographie comparée
• Cartographie physique moléculaire
– Contig du génome
– Séquence complète de l ’ADN
BTA7
BTA20
BTA10
BTA7
BTA20
BTA10
FISH

10/10/2008
• Objectifs
– Tirer profit des programmes génomes, en particulier humain et murin
– La séquence du génome humain est disponible depuis avril 2003 (+ souris et le rat)
– Construire des passerelles entre les génomes humain, murin et les génomes de nos espèces domestiques permettant l’extrapolation de données
• Développement de cartes comparées haute densité
– Facilité pour la cartographie de gènes : développement de cartes comparées
– Ancrage des cartes génétiques sur la carte comparée
Cartographie génomiqueCartographie comparée
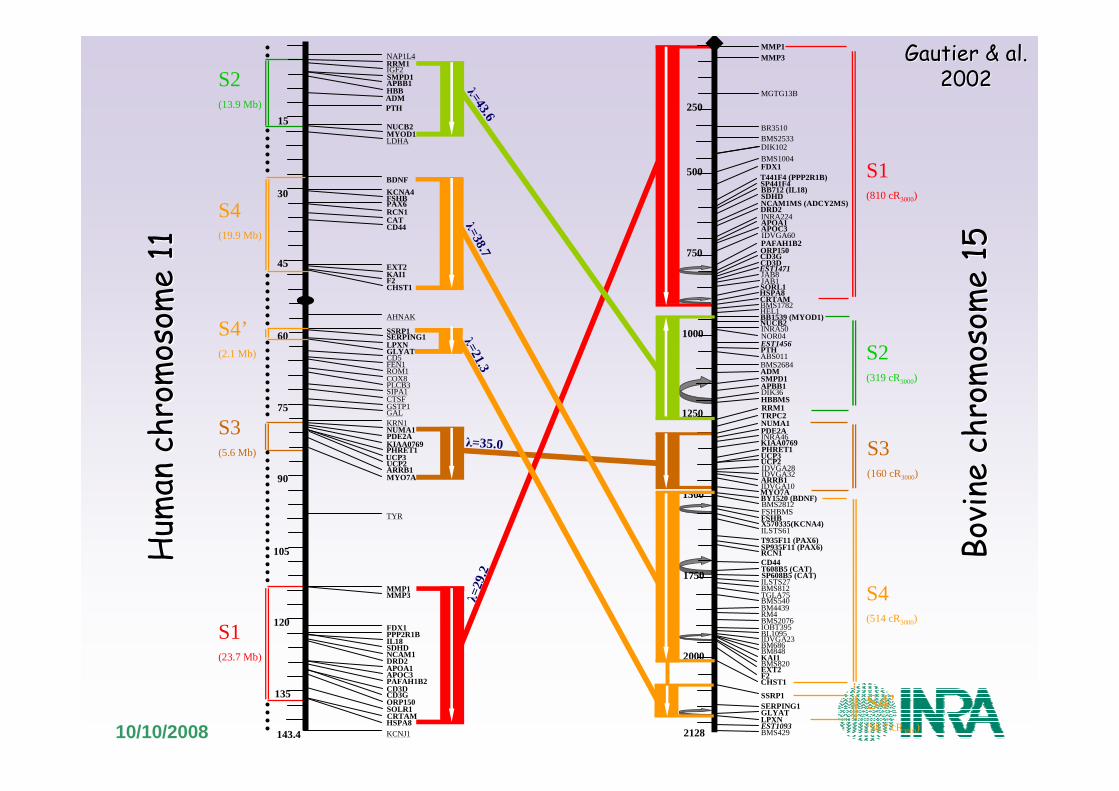
10/10/2008
T441F4 (PPP2R1B)
NCAM1MS (ADCY2MS)
BB712 (IL18)
T935F11 (PAX6)SP935F11 (PAX6)
X570335(KCNA4)
T608B5 (CAT)SP608B5 (CAT)
BB1539 (MYOD1)
MMP1MMP3
MGTG13B
BR3510BMS2533DIK102
BMS1004FDX1
SP441F4
SDHD
DRD2INRA224
APOC3APOA1
PAFAH1B2
JAB1JAB8EST1471CD3D
ORP150CD3G
HBBMSDIK36
HSPA8
NUCB2
IDVGA60
NOR04INRA50
HEL1BMS1782
EST1456
ABS011BMS2684ADM
PHRET1
TRPC2NUMA1PDE2AINRA46KIAA0769
UCP3UCP2
IDVGA10ARRB1IDVGA32IDVGA28
RCN1
ILSTS61
FSHBMSFSHB
BMS2812
CD44
ILSTS27BMS812TGLA75BMS540BM4439RM4BMS2076IOBT395BL1095IDVGA23BM686BM848KAI1BMS820EXT2F2CHST1
GLYAT
SSRP1SERPING1
LPXNEST1093BMS429
APBB1SMPD1
PTH
SORL1
CRTAM
RRM1
MYO7ABY1520 (BDNF)
NAP1L4RRM1IGF2
HBBADM
LDHA
NUCB2
KCNA4FSHBPAX6RCN1CATCD44
EXT2
CHST1
KAI1F2
AHNAK
SSRP1SERPING1LPXNGLYATCD5FEN1ROM1COX8PLCB3SIPA1CTSFGSTP1GALKRN1NUMA1PDE2AKIAA0769
UCP3UCP2ARRB1
TYR
MMP1MMP3
FDX1PPP2R1BIL18SDHDNCAM1DRD2APOA1APOC3PAFAH1B2CD3DCD3GORP150
HSPA8KCNJ1
SMPD1APBB1
PTH
MYOD1
BDNF
PHRET1
MYO7A
CRTAMSOLR1
60
75
90
105
120
135
143.4
15
30
45
S2(319 cR3000)
S1(810 cR3000)
S3(160 cR3000)
S1(23.7 Mb)
S2(13.9 Mb)
S3(5.6 Mb)
S4(19.9 Mb)
S4’(2.1 Mb)
250
500
1000
2000
2128
1250
750
1750
1500
S4’(98.7 cR3000)
S4(514 cR3000)
λ=43.6
λ=38.7λ=21.3
λ=35.0
λ=2
9.2
Human
Humanchromosome 11
chromosome 11
Bovine chromosome 15
Bovine chromosome 15
Gautier & al.Gautier & al.20022002

10/10/2008
Cartographie génomique
• Cartographie génétique
– Loci polymorphes et familles de référence
• Cartographie physique chromosomique
– Hybrides somatiques
– Hybridation In Situ
– Hybrides d’irradiation
• Cartographie physique moléculaire
– Contig du génome
– Séquence complète de l ’ADN

10/10/2008
Construction de banques génomiques� Principes généraux
� Digestion du génome en fragments de petite taille
� Clonage des fragments (intégration dans un génome bactérien hôte): Banques de BACs
� Intérêts
� Facilité de manipulation car obtention d’ADN en grande quantité de la région clonée
� Préalable à la cartographie physique d’une région : reconstituer le puzzle en recouvrant la région avec des clones chevauchants =
CONTIG
� Essentiel pour les études génétiques : recherche de polymorphismes, séquençage,

10/10/2008
ADN purifié Fragmentation
1
Copies incomplètes partant d'un
point fixe
Sens de la copie -->
Fragment d'ADN à séquencer (matrice)2a
Sen
s de
la m
igra
tion
élec
trop
horé
tique
3
Détection du signal de fluorescence à la sortie
du séquenceur
Séquence reconstituée
4
Le séquençage des génomesWhole genome shotgun
Méthode de Sanger
Clonage des fragments d’ADN (5, 50 et 150 kb)
2b
D’après Dujon

10/10/2008
5 assemblage
contig
contig 1 contig 2 contig 3 0
0.2
0.4
0.6
0.8
0 2 4 6 8 10 12Nombre de séquences (c = NL/G)
Nom
bre
de c
ontig
s(G
/L)
3X: exploratoire
6X: ébauche
12X: qualité"finale"
Type de séquence Caractéristiques Utilisation
Exploratoire Très nombreux contigs, petite taille Variations polymorphiques, biodiversitéEbauche (draft) Nombreux contigs, taille variable Premières analyses globalesFinale Peu de contigs, grands Analyse génomique fonctionnelle
Le séquençage des génomes (suite)
D’après Dujon

10/10/2008
6 Finition (supercontigs)
Ossature de supercontigs (scaffolds)
8 Annotation: ensemble de procédures informatiques qui:1- prédisent (± efficacement) les limites des gènes, des éléments de contrôle et de tout autre élément du génome2- suggèrent les fonctions des gènes à partir des comparaisons avec ce qui est déjà connu
7 Finition (remplissage des trous et zones de basse qualitévérification des assemblages, examen des séquences répétées, … )
Séquence finie, complète et de haute qualité
Le séquençage des génomes (fin)
D’après Dujon

10/10/2008
Le séquençage complet du génome bovin
• Consortium international pour la construction d’une carte physique du génome bovin
– Production de profils de restriction
• 340 000 BAC (Genome Sequencing Center, Canada)
• 100 000 BAC (INRA, LGbC)
– Séquençage d’extrémités de BAC (Nb de séquences)
• 312 000 BES (USA, Brésil, N. Zélande, Australie)
• 53 560 BES (INRA-Génoscope)
• Séquençage du génome bovin financé (50 M $) par le NIH, l’USDA, le Texas, le Canada, les Pays Bas
• Whole Genome Shot Gun Sequencing
– Couverture 7X sur un individu
– Couverture 1X à partir de plusieurs individus de différentes races : production de
marqueurs
• Actuellement (09/2007) un assemblage 7X est disponible
• La séquence du génome représente l’aboutissement de tous les efforts de cartographie

10/10/2008
Le séquençage complet du génome porcin
Consortium pour le séquençage du porcEtats-Unis, Chine, Japon, Corée, Royaume-Uni, France (INRA), Danemark, Allemagne
•Séquençage 2-3 x de BAC (MTP)
•Séquencage « whole genome shotgun » de 3-5 x
(1 x déjà fait Chine - Danemark)

10/10/2008
CompletedBCM-HGSC
Draft Assembly(7x)
MammalBos taurusCow
In progressBCM-HGSC
Draft AssemblyMammalOvis ariesSheep
In ProcessSangerDraft Assembly(BAC to BAC)
MammalSus scrofaPig
CompletedBI/MIT Draft Assembly
(7x)MammalEquus caballasHorse
CompletedWUGSC Draft AssemblyMammalFelis catusCat
CompletedBI/MIT Low Coverage WGS
(~2X)Mammal
Oryctolaguscuniculus
Rabbit
CompletedBI/MIT Draft AssemblyMammalCanis familiarisDog
CompletedWUGSC Draft Assembly
(6,6x)Non-Mammalian
VertebrateGallus gallusChicken
Genomes animaux domestiques séquencés (2008)
D’après A.Eggen
- L'ensemble des données est dans le domaine public- ”Genome Browsers” online:
ENSEMBL: http://www.ensembl.orgUCSC: http://genome.ucsc.edu/

10/10/2008
La séquence du génome : Intérêts
• Pour les efforts d’identification de gènes d’intérêt– Un nombre quasi illimités de marqueurs (microsatellites, SNP…)
– Des passerelles immédiates avec les autres génomes séquencés
– Identification des séquences codantes, des séquences régulatrices
• De manière plus générale
– Implication pour la compréhension de la dynamique de l’évolutionchez les mammifères (ex.: mise en évidence de régions conservées)

10/10/2008
2001
2008
Le séquençage haut débit
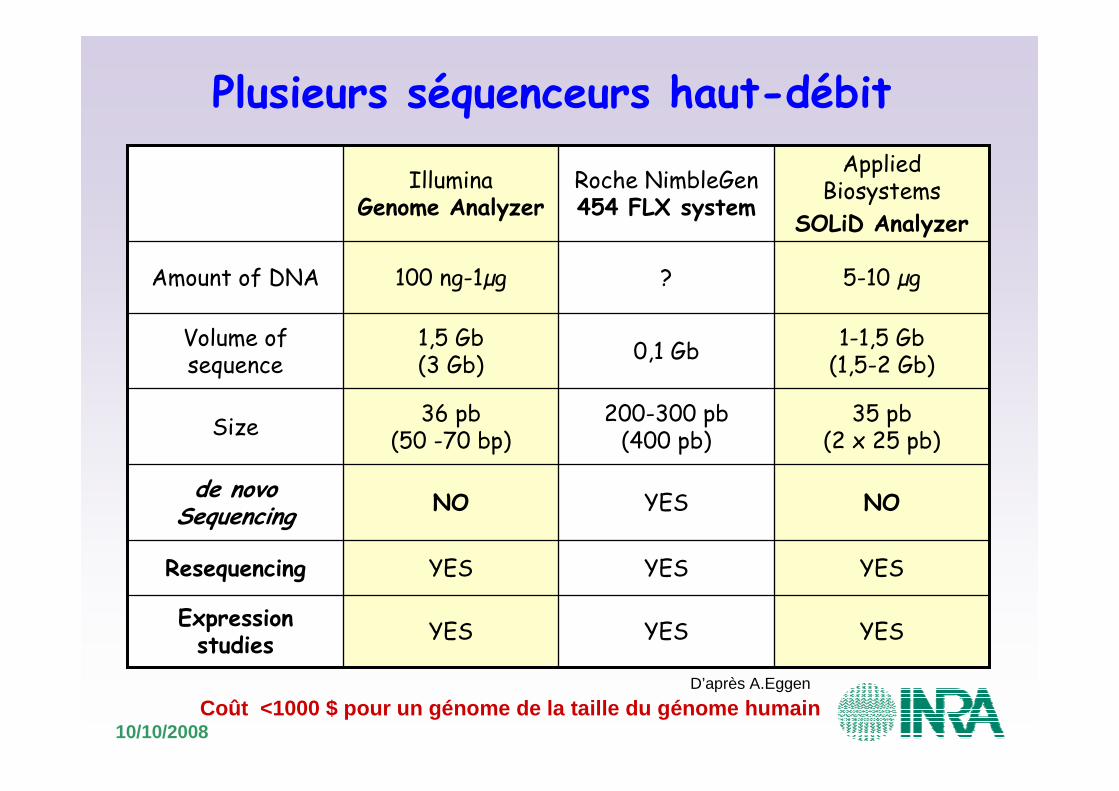
10/10/2008
YESYESYESExpression studies
YESYESYESResequencing
NOYESNOde novo Sequencing
35 pb(2 x 25 pb)
200-300 pb(400 pb)
36 pb(50 -70 bp)
Size
1-1,5 Gb(1,5-2 Gb)
0,1 Gb1,5 Gb(3 Gb)
Volume of sequence
5-10 µg?100 ng-1µgAmount of DNA
AppliedBiosystems
SOLiD Analyzer
Roche NimbleGen454 FLX system
IlluminaGenome Analyzer
D’après A.Eggen
Plusieurs séquenceurs haut-débit
Coût <1000 $ pour un génome de la taille du génome humain

10/10/2008
La détection de SNPs:valeur ajoutée du séquençage du génome
• Séquence référence (couverture 6-12x)
• Plusieurs animaux appartenant à des races différentes
• Alignement des séquences pour identifier plusieurs millers de
SNPs
• Validation des SNPs sur un nombre restreint d‘animaux de races
différentes
• Construction d‘une puce de SNPs pan-génomique (International Consortium)

10/10/2008
Digestion “in silico”
In silico estimates
135
92
70
Van Tassel et al., 2008Nature Methods 5, 247-252

10/10/2008
L’identification des SNPs
CCACGTAATCGGCACCTTAGGATCTGCaaacct….cactggATGAAGGCTC AATTGGCCATCATCGG
CCACGTAATCGGCACCTTAGGATCTGC
CCACGTAATCGGCACCTTAGGATCTGCaaacct….cactggATGAAGGCTC AATTGGCCATCATCGG
CCACGTAATC CGCACCTTAGGATCTGCaaacct….cactggATGAAGGCTCAATTGGCCATC ATCGG
CCACGTAATCGGCACCTTAGGATCTGC
CCACGTAATC CGCACCTTAGGATCTGCaaacct….cactggATGA GGGCTCAATTGGCCATCATCGG
CCACGTAATCGGCACCTTAGGATCTGCaaacct….cactggATGAAGGCTC AATTGGCCATCATCGG
CCACGTAATC CGCACCTTAGGATCTGCaaacct….cactggATGAAGGCTCAATTGGCCATC ATCGG
CCACGTAATCGGCACCTTAGGATCTGCaaacct….cactggATGAAGGCTC AATTGGCCATCATCGG
CCACGTAATCGGCACCTTAGGATCTGCaaacct….cactggATGA GGGCTCAATTGGCCATCATCGG
CCACGTAATCGGCACCTTAGGATCTGCaaacct….cactggATGAAGGCTC AATTGGCCATCATCGG
CCACGTAATC CGCACCTTAGGATCTGCaaacct….cactggATGAAGGCTCAATTGGCCATC ATCGG
GGCCACGTAATCGGCACCTTAGGATCTGCaaacct….cactggATGAAGGC TCAATTGGCCATCATCGGCCReference sequence
Holstein
Angus
BEEF
HaeIII HaeIII

10/10/2008
L’identification des SNPs bovins
• Analyse de 49 millions de lectures
– 5 022 143 tags uniques
• 62 042 SNPs putatifs identifiés avec une haute stringence
• 1 SNP / 1780 bp
• 25 125 SNPs validés et intégrés à la puce bovine: «Bovine
SNP50 genotyping BeadChip» (ILLUMINA) contenant plus
de 54 000 SNPs
Van Tassel et al., 2008Nature Methods 5, 247-252

10/10/2008
La génomique structurale
1. La cartographie des génomes animaux
2. Application: l’amélioration génétique des animaux

10/10/2008
L’amélioration génétique des animaux
Etude de caractères ayant un contrôle génétique simpledéterminisme mendélien
Phénotype qualitatifMonogénique
Etude de caractères ayant un contrôle génétique complexePhénotype qualitatifPhénotype quantitatif
=>Cartographie de QTLs (Quantitative Trait Locus)
- Gènes majeurs- Autres
=> stratégie: clonage positionnel
Sélection assistée par marqueurs (SAM)

10/10/2008
Détection de gènes majeurs
• Porcs gène RN
• Bovins gène sans corne, anomalie Mulefoot
• Ovins OAR : gène Boroola
• Caprins CHI : gène PIS
• Lapins OCU : gène Rex

10/10/2008
Phénotype
Du caractèreà la fonction
ClonageClonagepositionnel positionnel
Gènes/Allèles
• Génomiquecomparative
• Carte physique• Séquençage
des génomes
Stratégies d’identification de gènes d’intérêt

10/10/2008
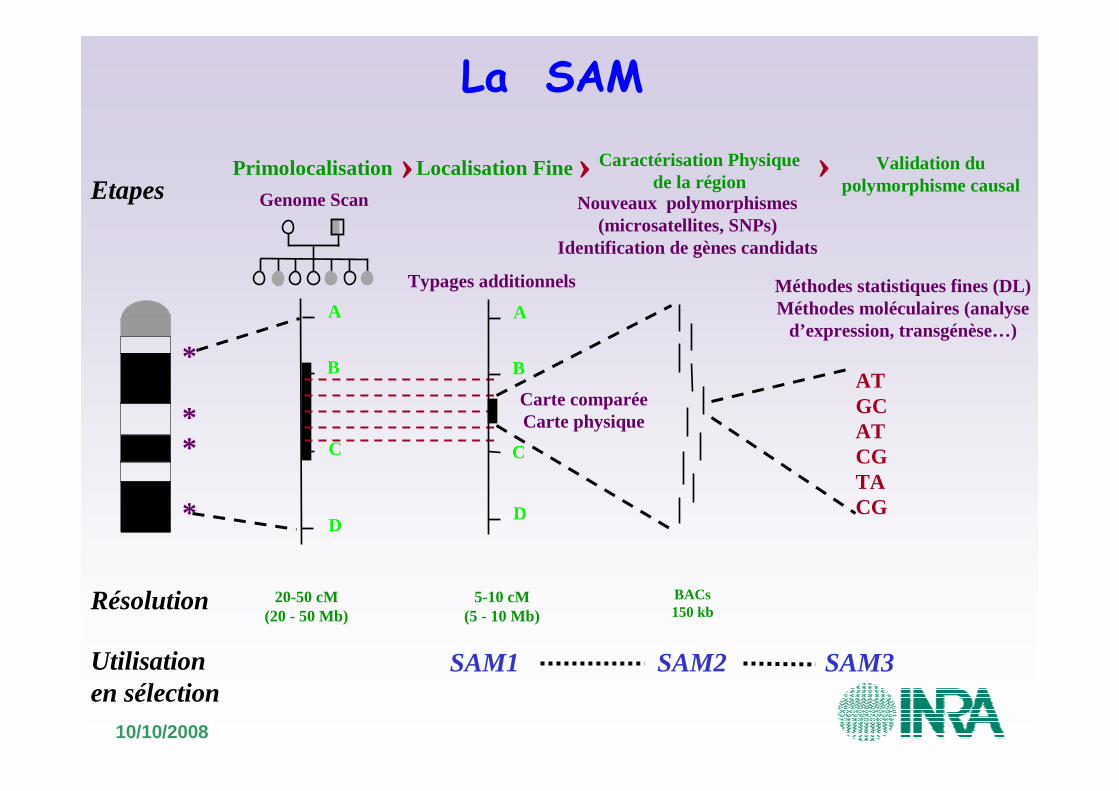
Etapes
Résolution
ATGCATCGTACG
Validation du polymorphisme causal
Méthodes statistiques fines (DL)Méthodes moléculaires (analyse d’expression, transgénèse…)
›Nouveaux polymorphismes
(microsatellites, SNPs)Identification de gènes candidats
Caractérisation Physiquede la région
BACs150 kb
Carte comparéeCarte physique
›
Conditions: Disposer de marqueurs génétiques (Microsatellites, SNP)Disposer de familles informatives
Le clonage positionnel
20-50 cM(20 - 50 Mb)
PrimolocalisationGenome Scan
B
C
**
A
*
* D
Analyses de liaison
Localisation Fine
B
A
D
C
5-10 cM(5 - 10 Mb)
Typages additionnels
›
Analyses d’association

10/10/2008
Analyses de liaison Analyses d’association
Rappelsanalyses de liaison et analyses d’association
PopulationsCas/contrôles
FamillesIntervalles: 10-20cMCaractères: Qualitatifs
QuantitatifContrôle génétique: Simple
Complexe
Familles
Intervalles: 2-5cMCaractères: Qualitatifs
QuantitatifsContrôle génétique: Simple
Complexe
Caractères: QualitatifsSimpleComplexe
Courtin et al, Infect Genet Evol. 2008 8(3):229-38.

10/10/2008
La génomique structurale
1. La cartographie des génomes animaux
2. Application: l’amélioration génétique des animaux
a) Identification de gènes majeurs
Exemple 1: clonage positionnel de la mutation RN- chez le porc

10/10/2008
Clonage positionnel chez le porcLa mutation RN-
Clonage positionnel d'un gène influençant la qualité de la viande (rendement technologique du jambon cuit, tendreté & jutosité)
Identification d’un gène régulant la quantité de glycogène musculaire, et donc le pH post-mortem de la viande

10/10/2008
La mutation RN-
• Phénotype RN-: chez les porcs Hampshire
• Mutation dominante
• Effet:• favorable: croissance• défavorable: taux de glycogène élevé dans les muscles squelettiques
Effet négatif sur la qualité de la viande et la transformation (pH)
Pertes économiques importantes
• Objectif:

10/10/2008
Localisation de la mutation RN-
• Mutation détectée par analyse de liaison (1000 méioses informatives)
• Première analyse de liaison: localisation sur le chromosome 15 => région de 2,5Mb
• Construction d’un contig de BAC de 2,5Mb– But: identifier de nouveaux marqueurs microsatellites et de nouveaux marqueurs SNP
• Nouvelles analyses de liaison

10/10/2008
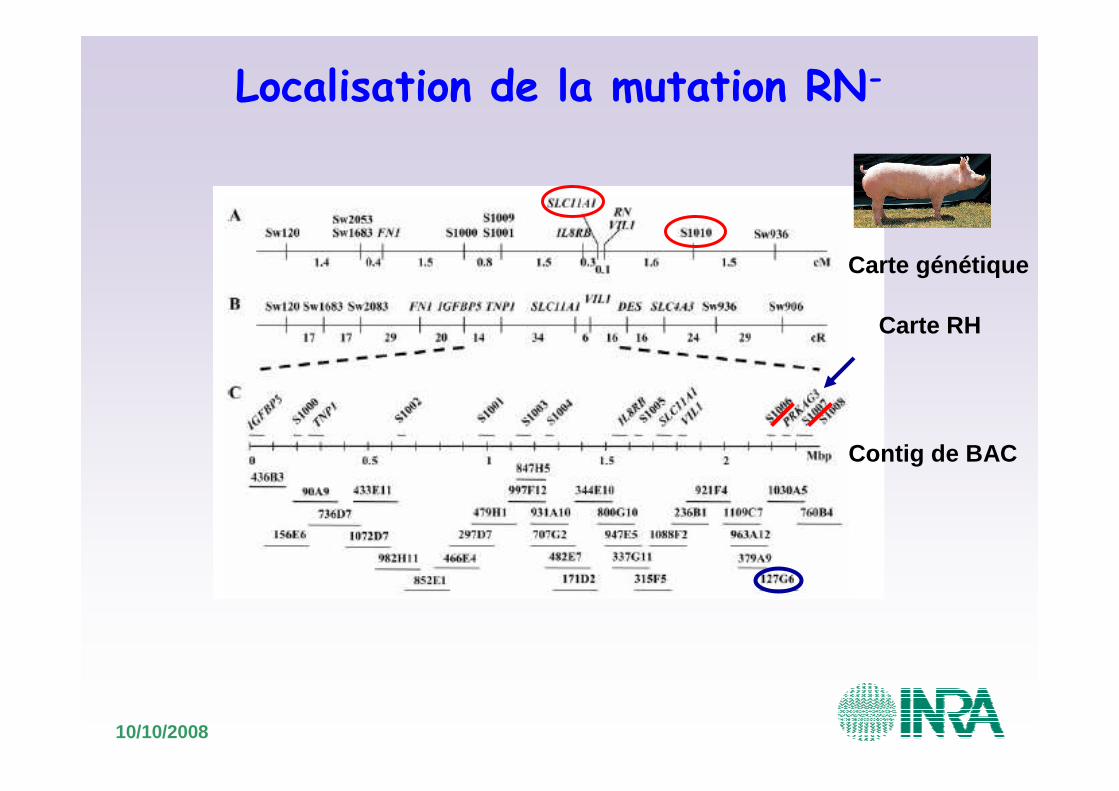
Localisation de la mutation RN-
•Nouvelles analyses de liaison: exclusion de la région proximale de SLC11A1 et de la région distale SNP S1010
Carte génétique
Carte RH
Contig de BAC
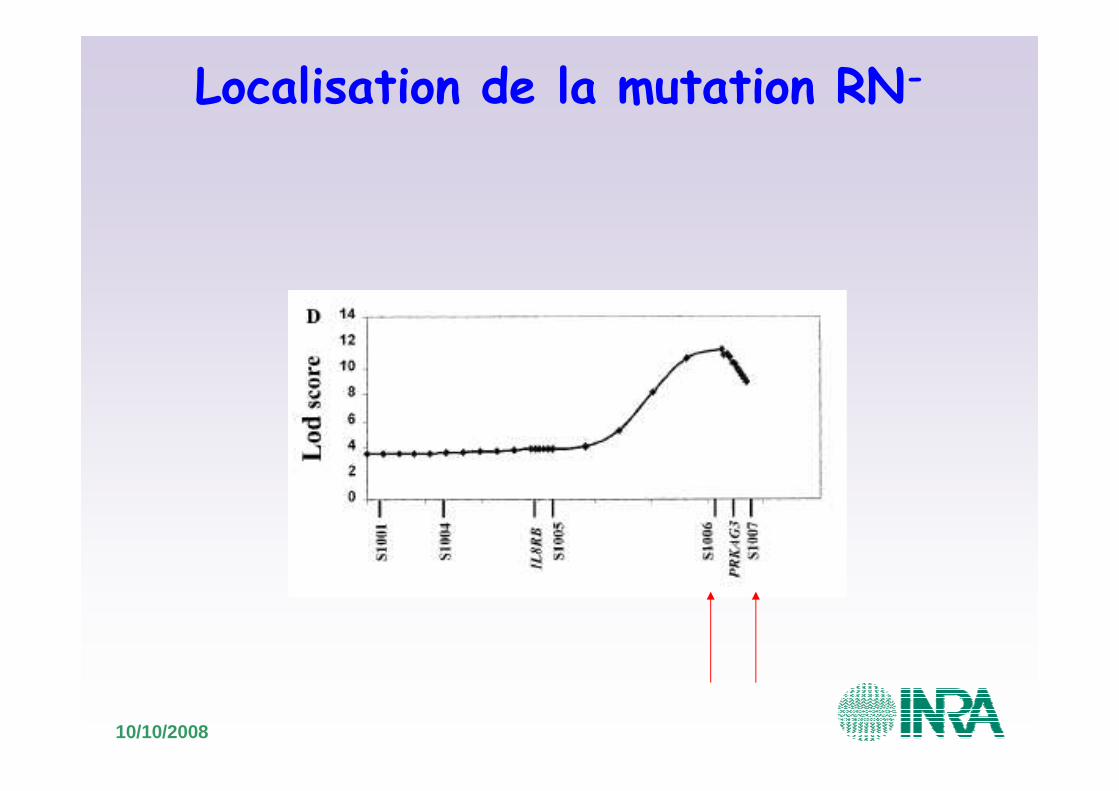
10/10/2008
Localisation de la mutation RN-

10/10/2008
• Cartographie comparée:homme (2q), souris (chr1) =>pas de gènes candidats évidents
• Analyse de déséquilibre de liaison:association complète entre les allèles des marqueurs S1006, S1007 (BAC 127G6) et RN-
Localisation de la mutation RN-
DL sur 91 porcs Hampshire
10/10/2008
Carte génétique
Carte RH
Contig de BAC
Localisation de la mutation RN-

10/10/2008
• Séquençage shotgun du BAC 127G6 (1000 individus)3 séquences codantes
-KIAA173-CYP27A1-3e gènes
• KIAA173 et CYP27A1 ne sont pas des gènes candidats
• 3e gène: similarité avec la sous-unité γ de l’AMP activated proteinkinase
• Rôle des AMPK: Rôle clef dans la régulation du métabolisme énergétique de la cellule eucaryote
Augmentation du ratio AMP/ATP => activation AMPK=>production ATP et inhibition de la consommation d’ATP=>inactivation de la glycogen synthase (Enzyme régulant la synthèse du
glycogène).
Bon candidat positionnel et fonctionnel
Localisation de la mutation RN-

10/10/2008
• Détermination de la séquence du gène par RT-PCR et amplification rapide des extrémités de cDNA
Chez le porc (RN+/RN+) et chez l’homme
=> gène différent des autres isoformes et orthologue du gène humain (BLAST)
=> PRKAG3
• Expression (Northern blot)=> uniquement dans le muscle
Localisation de la mutation RN- dans le gène PKARG3
• Comparaison de la séquence de PRKAG3 d’individus RN-/RN- et individus RN+/RN+
=> Différence de 7 nucléotides dont 4 substitutions non synonymes

10/10/2008
•Animaux: Hampshire+ autres PC (15 races)
•Phénotype: mesure du taux de glycogène des muscles squelettiques
•Génotype: mutation R200Q du gène PRKAG3
Localisation de la mutation RN- dans le gène PKARG3
Analyse d’association des mutations avec le phénotype RN-

10/10/2008
Caractérisation fonctionnelle de la mutation RN-
•Mesure de l’activité de l’AMPK dans des extraits musculaires:
l’activité est 3 fois supérieure chez les porcs RN+ que chez les porcs RN- (en présence et en absence d’AMP)
•La mutation semble inhiber l’activation de l’AMPK et la dégradation du glycogène

10/10/2008
La génomique structurale
1. La cartographie des génomes animaux
2. Application: l’amélioration génétique des animaux
a) Identification de gènes majeurs
Exemple 2: chez le moutonLes brebis callipyges
10/10/2008
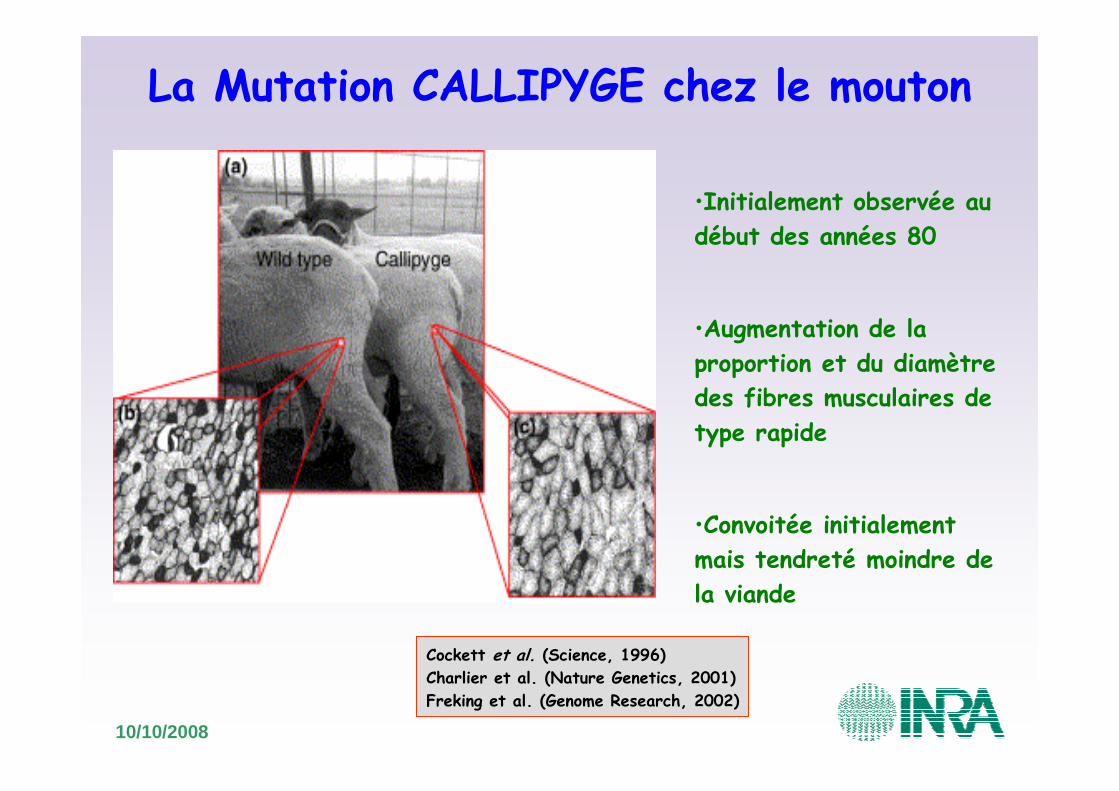
La Mutation CALLIPYGE chez le mouton
•Initialement observée au début des années 80
•Augmentation de la proportion et du diamètre des fibres musculaires de type rapide
•Convoitée initialement mais tendreté moindre de la viande
Cockett et al. (Science, 1996)Charlier et al. (Nature Genetics, 2001)Freking et al. (Genome Research, 2002)

10/10/2008
Un mode de transmission original
• Observé pour la première fois: ~15% de descendants d’un bélier
(Solid Gold) atteints
• Après croisement 50% de descendants issus d’un mâle callipyge
(CLPG) atteints
• Mais…
– 0% des descendants issus d’une femelle CLPG atteints
– 25% des descendants d’un croisement Femelle CLPG x Mâle CLPG (au lieu de 50% sous l’hypothèse de l’implication d’un gène soumis à une empreinte paternelle)

10/10/2008
La surdominance polaire…
CLPGPAT
clpgMAT
clpgPAT CLPGPAT
CLPGMATCLPGMAT
Mode de transmission non mendélien.Seuls les hétérozygotes ayant hérité la mutation de leur père expriment
le phénotype…
CLPG: allèle mutéclpg: allèle non muté
10/10/2008
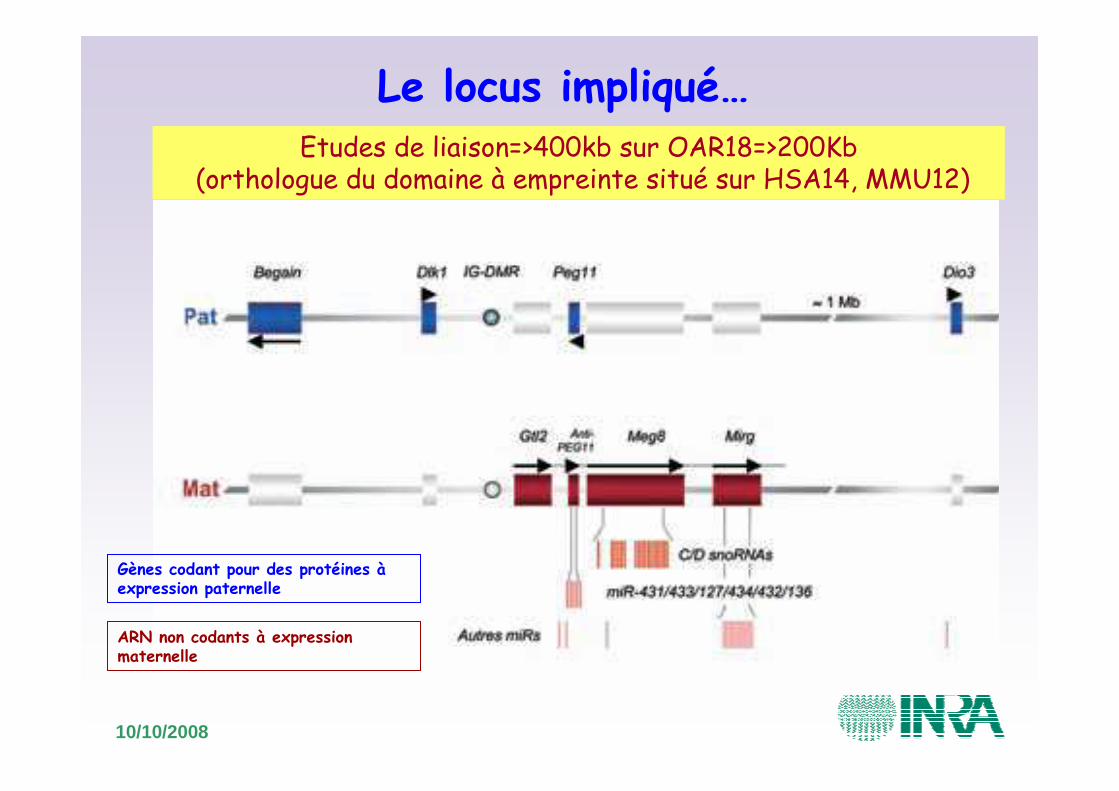
Le locus impliqué…Etudes de liaison=>400kb sur OAR18=>200Kb
(orthologue du domaine à empreinte situé sur HSA14, MMU12)
Gènes codant pour des protéines à expression paternelle
ARN non codants à expression maternelle

10/10/2008
Expression des gènes selon le génotype de la mutation
Profil d’expression unique dans le génotype affectéPas de perturbations de l’empreinte parentale. Mutation active la transcription des gènes en cis
DLK1:- Gène à expression paternelle- Effecteur de l’hypertrophie musculaire

10/10/2008
Découverte de la mutation
184 kb
Substitution
A to G
10/10/2008
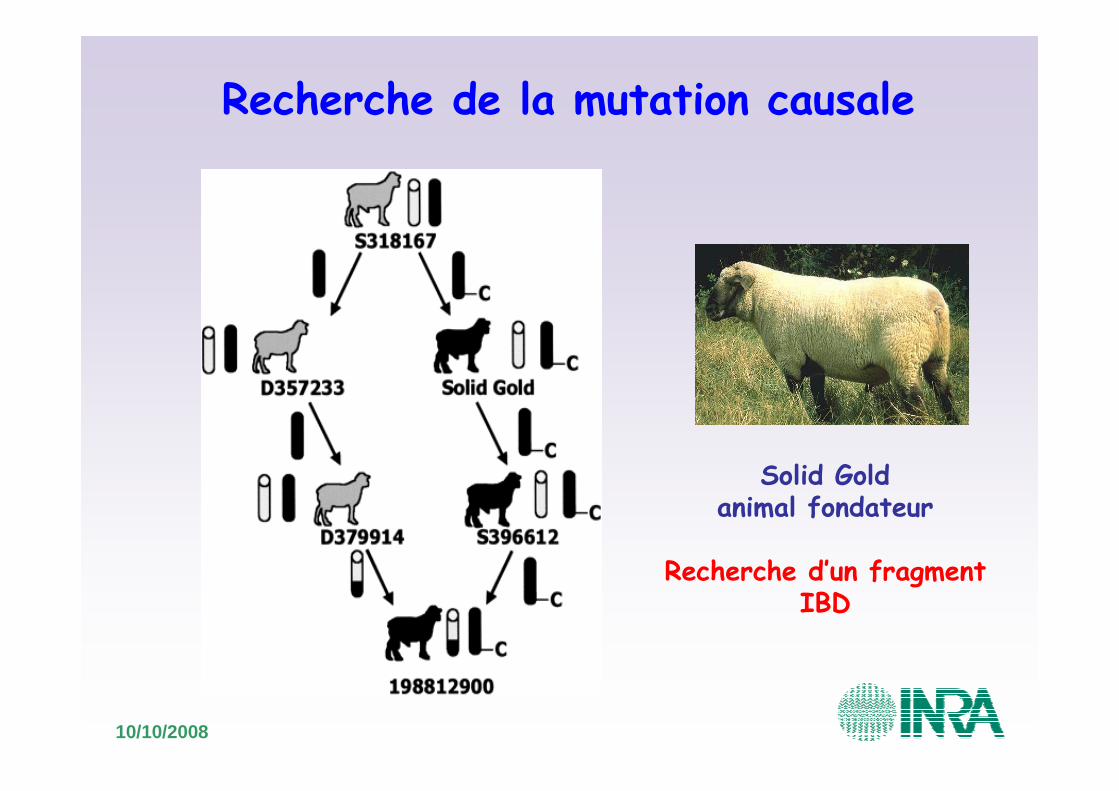
Recherche de la mutation causale
Solid Goldanimal fondateur
Recherche d’un fragmentIBD
10/10/2008
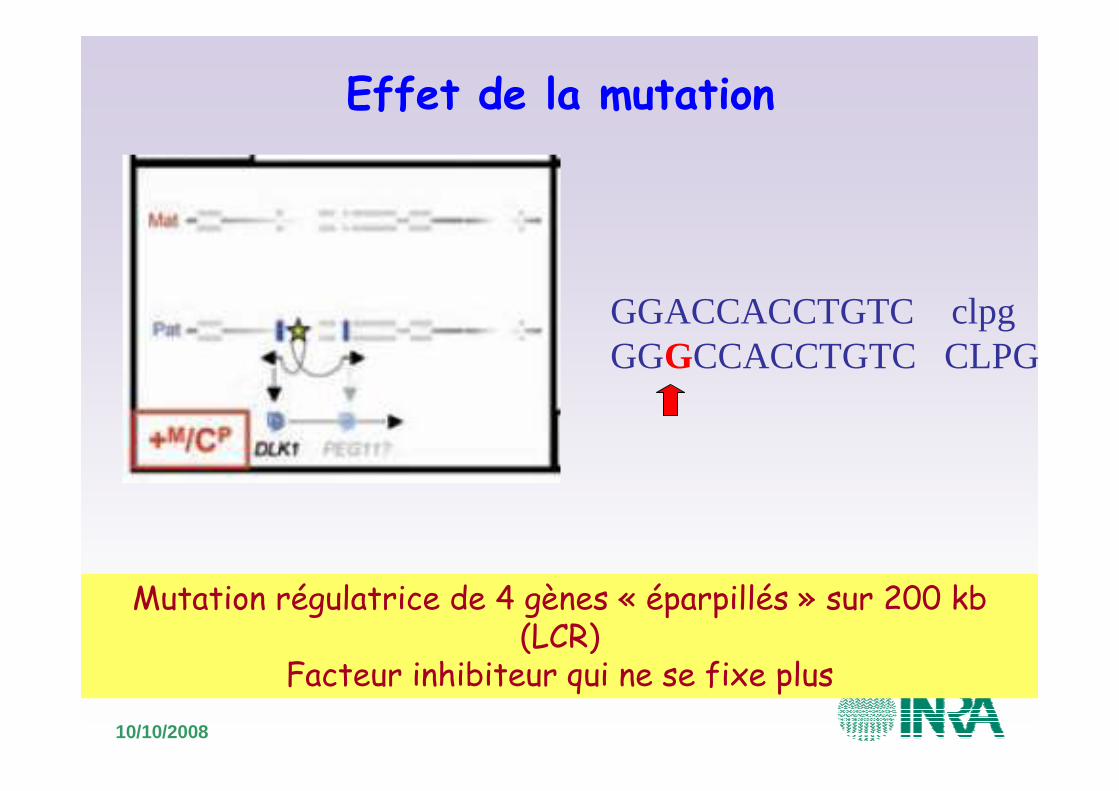
Effet de la mutation
GGACCACCTGTC clpgGGGCCACCTGTC CLPG
Mutation régulatrice de 4 gènes « éparpillés » sur 200 kb(LCR)
Facteur inhibiteur qui ne se fixe plus

10/10/2008
L’empreinte génétique : rappels
• Forme de régulation génique dépendante de l’origine parentale
=>expression monoallélique de certains gènes nucléaires
– Décrite dans plusieurs espèces de mammifères placentaires
– Soit la copie de l’allèle paternel (ex IGF2R) soit la copie de l’allèle
maternel (ex IGF2) est transcriptionnellement active
– Nb= environ 80 . Les gènes soumis à empreinte sont organisés en
grappe + ICE = élément régulateur commun
– Région contenant systématiquement des ARN non codants
– En général conservés entre espèces
• Variation des empreintes au cours du développement

10/10/2008
Effacement et établissement des empreintes génétiques au cours du développement
Exemple souris13e jour du devtembryonnaire

10/10/2008
L’empreinte et la théorie du conflit…
• Paradoxe
– L’expression mono-allélique augmente la vulnérabilité aux mutations
délétères
• La théorie du conflit (Wilkins et Haig, Nature Review Genetics, 2003)
– Intérêts génétiques parfois conflictuels entre la mère et le père
chez les espèces polygames (majorité des mammifères). Pour
augmenter la propagation de ses gènes
• Le mâle transmet à sa descendance des gènes qui vont favoriser la croissance fœtale
(au détriment de l’avenir reproductif de la mère)
• La femelle transmet à sa descendance des gènes qui vont favoriser une économie de
ressources et une augmentation du nombre de portée.
– La plupart des gènes soumis à empreinte interviennent dans des
mécanismes de transfert de ressources maternelles à la descendance(favorisés par les gènes à expression paternelle, inhibés par ceux à expression maternelle)

10/10/2008
La génomique structurale
1. La cartographie des génomes animaux
2. Application: l’amélioration génétique des animaux
a) Identification de gènes majeurs
b) Cartographie de QTLs
Exemple 1: chez les bovins

10/10/2008
La primolocalisation :Dispositif animal (exemple français chez les bovins laitiers)
20-50 cM(20 - 50 Mb)
Etapes
Résolution
Utilisation en sélection
PrimolocalisationGenome Scan
B
C**
A
*
* D
….
….
� 26 familles de demi-frères Prim’Holstein: 2138 fils
….
….
� 9 familles Normandes: 548 fils
….
….
� 6 familles Montbéliardes: 370 fils
Prim'Holstein Normande Montbéliarde

10/10/2008
Pourquoi un tel dispositif « petite-fille » ?
�Le dispositif existe déjà (coût réduit au seul génotypage)
•Les taureaux d ’IA sont les descendants d’un nombre limité de pères
•Les taureaux sont évalués en routine par testage sur descendance
�Le phénotype des fils est similaire à la moyenne des D performances (petite-filles)
•Haute précision dans l’évaluation (variance résiduelle réduite)
•Puissance de détection augmentée pour un nombre donné de génotypes
�Détection de QTL ségrégeant dans les populations exploitées

10/10/2008
Cartographie de QTLs chez les bovins laitiers
• 6 QTL sur 3 chromosomes
– Caractères de production• Quantité de matière protéique (BTA7 & BTA26)
• Quantité de matière grasse (BTA26)
– Caractères fonctionnels• Numération cellulaire (BTA15)
• Fertilité femelle (BTA7)
– Caractères morphologiques• Epaisseur du talon (BTA15)

10/10/2008
La génomique structurale
1. La cartographie des génomes animaux
2. Application: l’amélioration génétique des animaux
a) Identification de gènes majeurs
b) Cartographie de QTL
Exemple 2: chez les porcs

10/10/2008
Détection de QTLs chez le porc
• Caractères de productionCroissance
Epaisseur lard dorsal
Quantité de lipides intramusculaires
…
• Santé et résistance aux maladiesCaractères immunologiques (en cours)
Infection par le PrV
Développement et régression des mélanomes cutanés

10/10/2008
QTL “muscle et epaisseur de gras” chez le porc
Nezer et al., 1999, Nature Genetics 21 : 155-156.
Van Laere et al., 2003, Nature 425 : 832-836.
LWxPietrain (1032 F2)
Performance de croissancesupérieure
Musculature exceptionnelle peu de gras
21 phénotypes mesurant les performances de croissance, les proportions corporelles, la musculature, le dépôt de graisse, la qualité de la viande
Objectif: Localiser des QTLs responsables des différences génétiques entre les deux races

10/10/2008
QTL sur chromosome 2 soumis à l’empreinteparentale : seul l’allèle paternel a un effet
H2: QTL soumis à empreinte paternelle
HSA11p : IGF2 MYOD1
Log10(H1/H0)
Log10(H2/H0)
Log10(H3/H0)
H3: QTL soumis à empreinte maternelle
10/10/2008
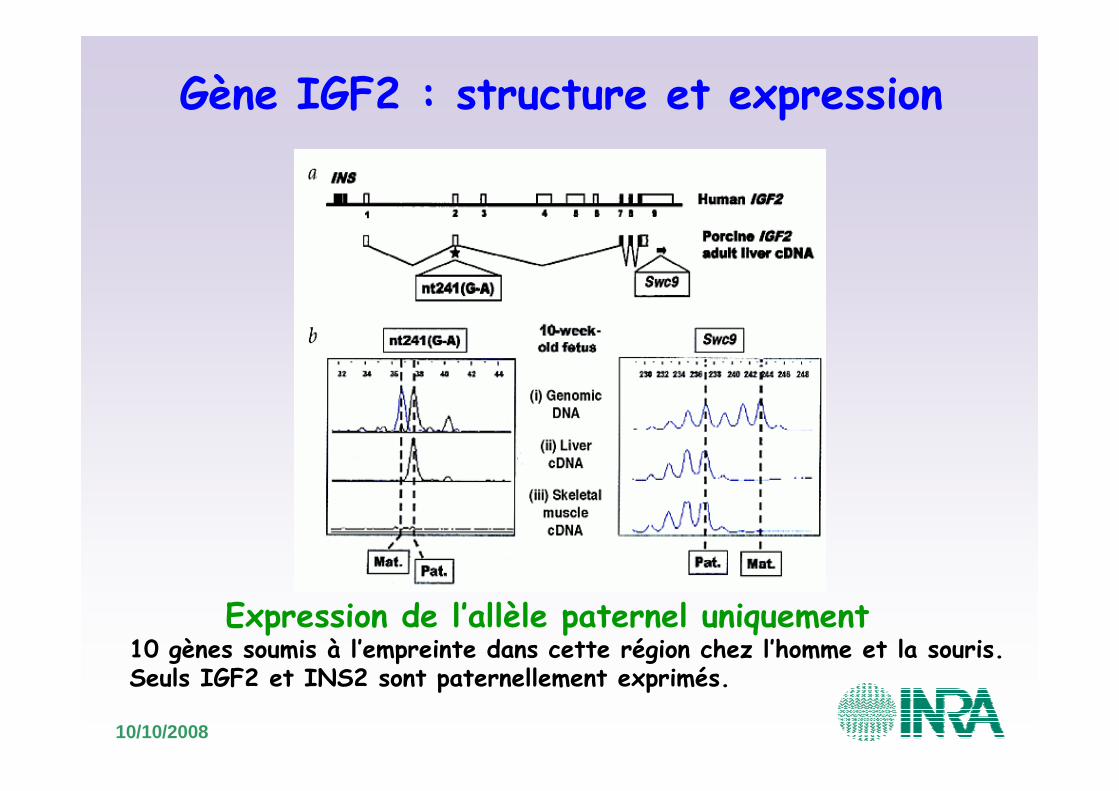
Gène IGF2 : structure et expression
Expression de l’allèle paternel uniquement10 gènes soumis à l’empreinte dans cette région chez l’homme et la souris. Seuls IGF2 et INS2 sont paternellement exprimés.

10/10/2008
Découverte de la mutation causale
Substitution dans une région non-codante (intron 3)

10/10/2008

10/10/2008
Effet de la mutation
Substitution A/G
•Empêche la fixation d’un facteur inhibiteur de l’expression d’IGF2 dans le muscle squelettique et le muscle cardiaque,et uniquementaprès la naissance.
•Ne perturbe pas l’empreinte parentale
•Augmente la masse musculaire de 3 à 4 %

10/10/2008
La génomique structurale
1. La cartographie des génomes animaux
2. Application: l’amélioration génétique des animaux
a) Identification de gènes majeurs
b) Cartographie de QTLs
Exemple 3: chez le mouton
10/10/2008
Le dogme central et les ARN non codants
ARN Non Codants :- miRNA- siRNA

10/10/2008
Les micro-ARN : quelques éléments
• Ce sont des ARNs non codants (20-23 nt)
– Décrits pour la première fois chez C.elegans : Lee et al. (Cell, 1992)
– Interviennent dans la régulation post-transcriptionnelle de gènes cibles
(plantes et animaux)
– Impliqués dans une multitude de fonctions biologiques (développement,
prolifération cellulaire, apoptose, réponse au stress, tumorigénèse)
– Hautement conservés entre espèces

10/10/2008
Biosynthèse, structure et action
Pre-miRNA
Wienholds and Plasterk, FEBS Letters, 2005
22 nt
70-80 nt
Riboonucleoproteincomplex

10/10/2008
Abondance des miRNAs
– 1-5% des gènes prédits (analyse de la séquence des génomes).• 1 000 chez l’homme• 250 chez C. elegans
– > 1/3 de l’ensemble des gènes ont des miRNA-bindingsequences.
– Cela suggère que les miRNA sont impliqués dans de nombreuses fonctions cellulaires.
– Octobre 2008 : 8619 miRNA
Base de données http://microrna.sanger.ac.uk/

10/10/2008
Juillet 2006
Exemple de cartographie de QTL chez le mouton

10/10/2008
Introduction…
• Mouton Texel
– race à viande, hypertrophie musculaire.
• Programme de recherche de QTL
– Romanov X Texel, F2, 258 petits
• 37 mesures : muscle, dépôt graisse, composition…
• 153 marqueurs pour le criblage total du génome

10/10/2008
Identification d’un QTL avec un effet majeur sur la musculature
– Localisé sur OAR02h2=0,05-0,25Explique 1/5 à 1/3 des différences entre les 2 races
– Après cartographie fine : région comprenant le gène Myostatine (GDF8)…
10/10/2008
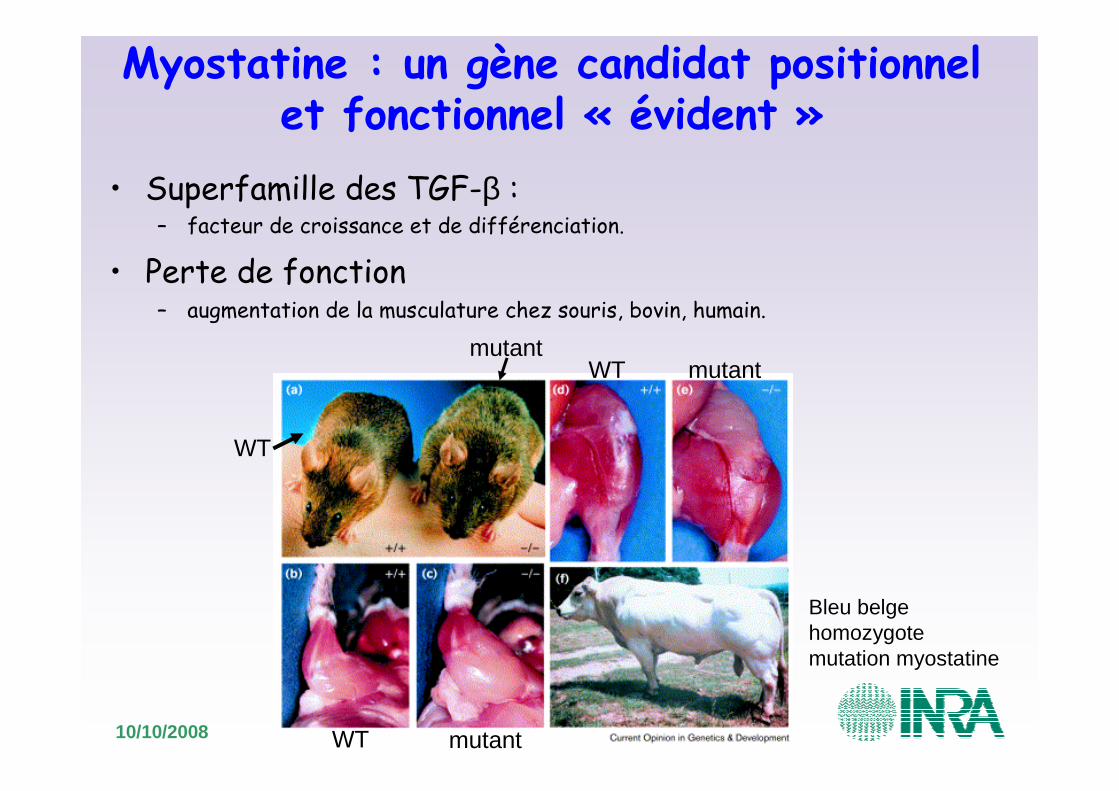
Myostatine : un gène candidat positionnel et fonctionnel « évident »
• Superfamille des TGF-β : – facteur de croissance et de différenciation.
• Perte de fonction– augmentation de la musculature chez souris, bovin, humain.
WT
mutant
WT
WT mutant
mutant
Bleu belge homozygotemutation myostatine

10/10/2008
• Séquençage région codante du gène :
– 3 Texel F0
– 7 contrôles (5 Romanov F0, 1 Dorset, 1Tarasconnais)
• Aucun polymorphisme identifié…
Mais…

10/10/2008
• Northern blot
-Transcrit de taille attendue chez Texel / contrôle
- Même intensité des bandes.
Transcrit GDF8
Ld : longissimus dorsi; St : semitendinosusTx : Texel; Rv : Romanov
Les transcrits de GDF8 (myostatine)
• ARN provenant muscle Texel/contrôleAmplification de l’ORF : RT-PCR, séquençage
ARN messager normal chez animaux Texel – niveau séquence

10/10/2008
3 Texel et 7 contrôles– 20 SNPs dans 10,2 kb– Aucun dans des éléments conservés
Génotypage de tous les SNPs– 42 Texel et 90 contrôles (11 races, 4
Texel/Romanov)– « monomorphisme virtuel des Texel»
– 18 SNPs éliminés : au moins 1 des 4 T/Rhomozygotes
– SNP g-2449C-G éliminé :
• à 2,5kb en amont site d’initiation de la transcription
• 1 Texel hétérozygote pour cet SNP et homozygotes pour tous les autres SNPs
Un seul SNP candidat : g+6723G-A
Recherche de SNPs : région non codante

10/10/2008
Étude du SNP g+6723G-A
• En 3’UTR
• Allèle A: crée un motif octamérique –cible de microARN
– décrit par Xie et al (Nature, 2005)
– miR-1 (.1 et .2), miR-206, miR122a reconnaissent cette cible
– miR-1 : très exprimé dans muscle et coeur souris– Les gènes de ces 4 miARN retrouvés chez le mouton
– Expression chez le mouton
Zhao et al., Nature, 2005

10/10/2008
Effet de la mutation
myostatine
myostatine
T : texel; W : contrôle
Immunoprécipitation : chez les Texel, la bande est 3 fois moins intense
La mutation entraîne une instabilité des transcrits
• Hypothèse : La mutation provoque un création d’un site cible de miRNA
• Test de l’hypothèse : diminution de la myostatine circulante?
• Résultats
10/10/2008
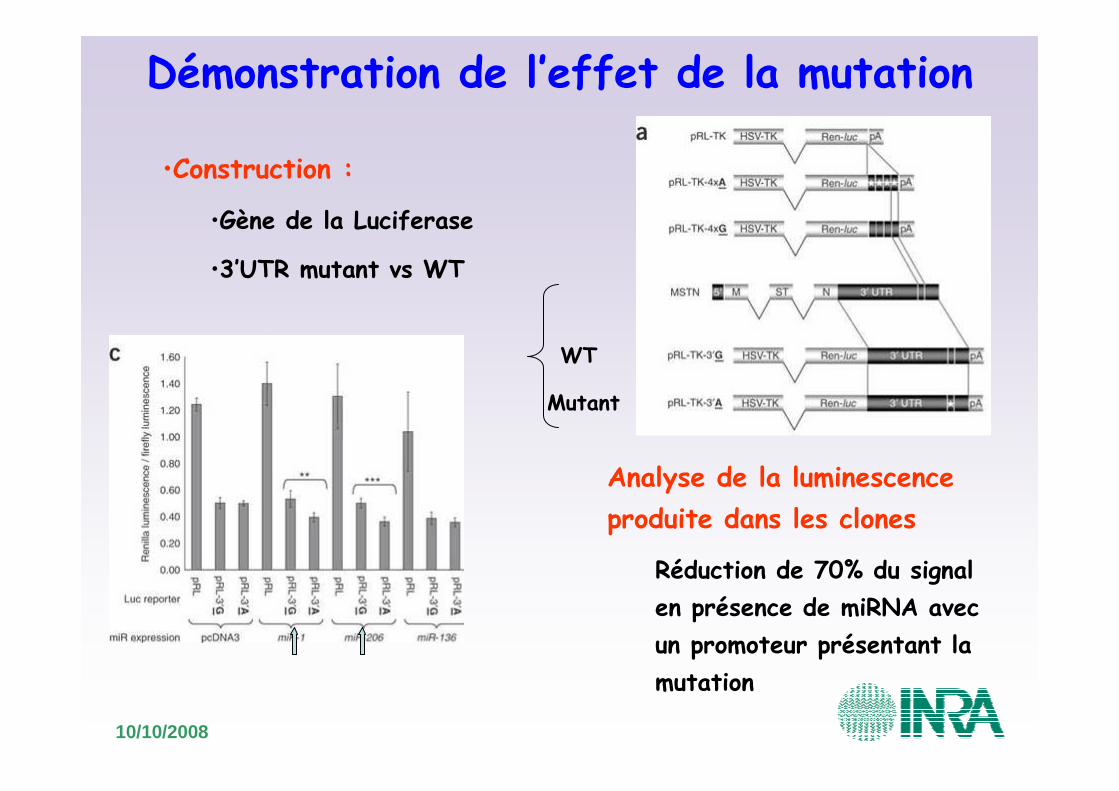
Démonstration de l’effet de la mutation
Mutant
WT
•Construction :
•Gène de la Luciferase
•3’UTR mutant vs WT
Analyse de la luminescence produite dans les clones
Réduction de 70% du signal en présence de miRNA avec un promoteur présentant la mutation

10/10/2008
Conclusions de l’étude
• Une mutation en 3’ du gène de la myostatine crée une cible pour au moins 2 miRNA exprimé dans les muscles squelettiques.
• Diminution de la quantité de Myostatine (limitant la croissance des tissus musculaire) dans le muscle =>Hypertrophie musculaire

10/10/2008
Développement d’une base de donnée Patrocles (http://www.patrocles.org)
Liaison SNPs/site cible miARN
– Chez l’homme• 73 497 SNPs dans 3’UTR de 13 621 gènes– Crée ou détruit 1 octamer décrit par Xie, Nature, 2005– 2 490 SNPs putatifs crée un site– 2 597 SNPs putatifs détruit un site– Dont 483 affectent un octamère conservé entre 4 espèces mammifères
– Chez Souris• 77 283 SNPs dans 3’UTR de 10 200 gènes– 1 183 SNPs putatifs crée un site– 1 321 SNPs putatifs détruit un site» Dont 234 conservés au cours de l’évolution

10/10/2008
Base de données de QTLs chez les animaux (http://www.animalgenome.org/QTLdb/)

10/10/2008
La génomique structurale
1. La cartographie des génomes animaux
2. Application: l’amélioration génétique des animaux
c) La sélection assistée par marqueur (SAM)

10/10/2008
Amélioration génétique
• Exemple bovins/porcs
• Très efficace durant les dernières décennies par exemple performances de croissance et efficacité alimentaire
• Méthodes classiques basées sur :– Collecte d’informations de performances individuelles (paramètres
de production)
– Compilation des observations avec des informations généalogiques
– « Index de sélection » pour identifier les meilleurs candidats à la
sélection

10/10/2008
Quelques limites …
• Efficacité diminue si le :
– Phénotype difficile à mesurer
– Caractère possède une faible héritabilité
• Se limitent aux caractères mesurables dans un grand
nombre d’animaux
• Nouveaux critères de sélection :
– Adéquation aux « produits finis »
– Qualité et acceptabilité pour les consommateurs
– Bien être animal, résistance aux maladies …
10/10/2008
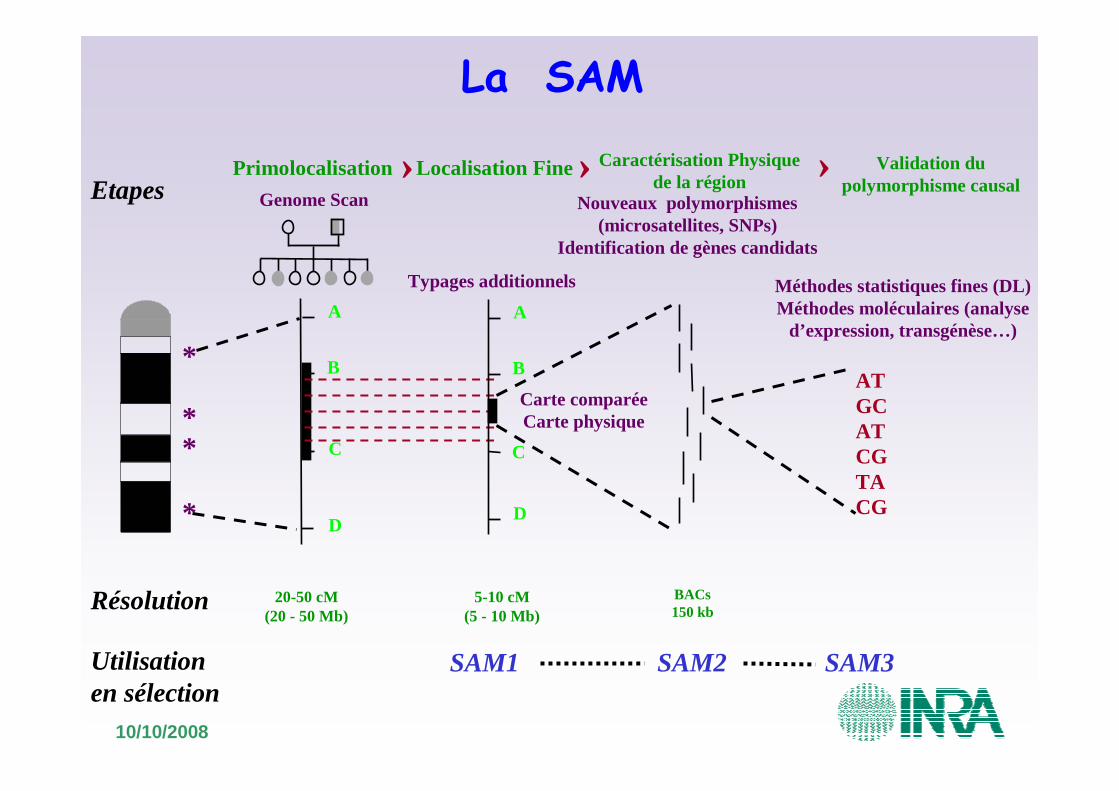
20-50 cM(20 - 50 Mb)
Primolocalisation
Genome Scan
Localisation Fine
B
A
D
C
5-10 cM(5 - 10 Mb)
Typages additionnels
›Etapes
Résolution
Utilisation en sélection
SAM1 SAM2 SAM3
B
C
**
A
*
* D
ATGCATCGTACG
Validation du polymorphisme causal
Méthodes statistiques fines (DL)Méthodes moléculaires (analyse
d’expression, transgénèse…)
›Nouveaux polymorphismes
(microsatellites, SNPs)Identification de gènes candidats
BACs150 kb
Caractérisation Physiquede la région
Carte comparéeCarte physique
›
La SAM

10/10/2008
La SAM chez les bovins

10/10/2008
1 1
22
3 3
44
55
66
77
99
88
NOM : INRA83
POS : chrm1 - 15
•Les Marqueurs : bornes du génome
La SAM chez les bovins

10/10/2008
MELKIOR LEHOUX
La SAM chez les bovins
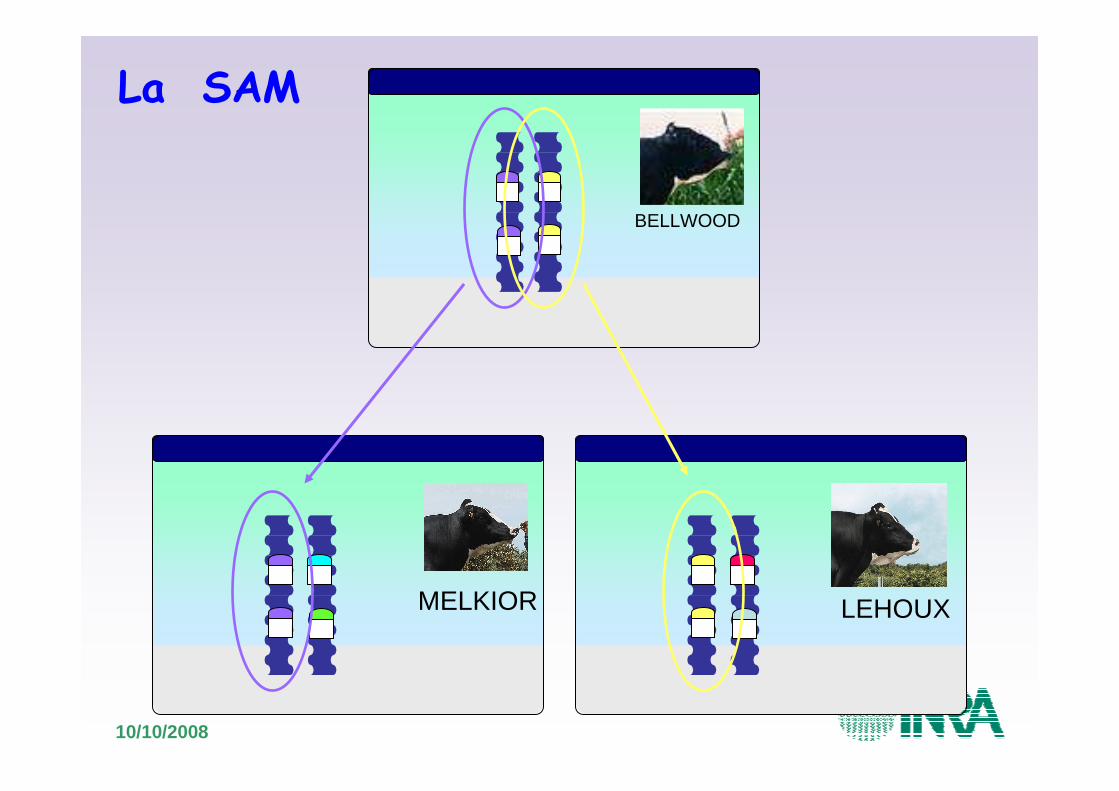
10/10/2008
BELLWOOD
MELKIOR LEHOUX
La SAM
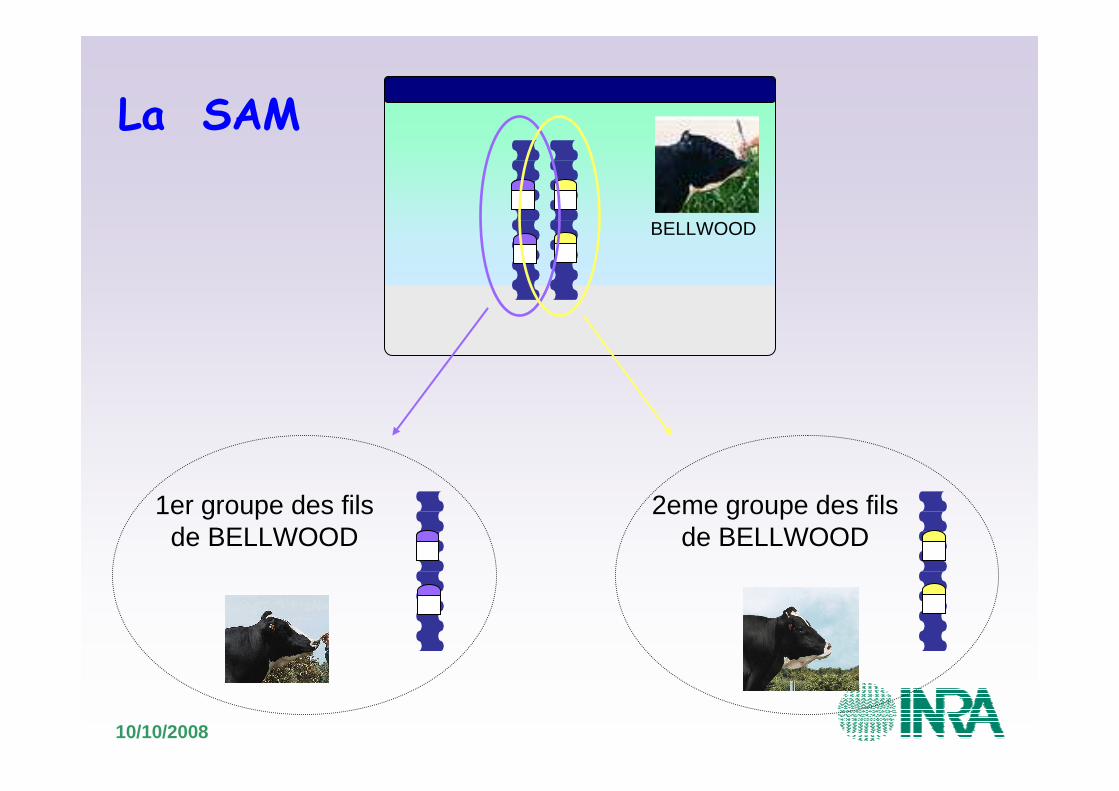
10/10/2008
BELLWOOD
1er groupe des fils de BELLWOOD
2eme groupe des fils de BELLWOOD
La SAM

10/10/2008
1er groupe des fils de BELLWOOD
Moyenne = 29
2eme groupe des fils de BELLWOOD
Moyenne = 13>
Il est donc plus intéressant d’avoir reçu que .
Comparaison des performances• Groupe de fils de BELLWOOD pour la MG
La SAM chez les bovins

10/10/2008
JOCKO
Fille de JOCKO
BESNE
...
...
•Informations marqueurs
On peut avoir de
l’information pour
toutes les familles
typées
La SAM chez les bovins

10/10/2008
La SAM permet de lire la carte d’identité d’animaux sans performance (candidats).
MELKIORLEHOUX
...
BELLWOOD
Veau 1 Veau 2 Veau 3 Veau 4
JOCKO
Fille de JOCKO
BESNE
...
...
La SAM

10/10/2008
Chez les BV laitiers:
• Génotypage des jeunes taureaux et génisses laitiers pour > 40 marqueurs pour 12 régions QTLdifférentes soit 10.000 animaux typés / an.
La SAM chez les bovins
10/10/2008
20-50 cM(20 - 50 Mb)
Primolocalisation
Genome Scan
Localisation Fine
B
A
D
C
5-10 cM(5 - 10 Mb)
Typages additionnels
›Etapes
Résolution
Utilisation en sélection
SAM1 SAM2 SAM3
B
C
**
A
*
* D
ATGCATCGTACG
Validation du polymorphisme causal
Méthodes statistiques fines (DL)Méthodes moléculaires (analyse
d’expression, transgénèse…)
›Nouveaux polymorphismes
(microsatellites, SNPs)Identification de gènes candidats
BACs150 kb
Caractérisation Physiquede la région
Carte comparéeCarte physique
›
La SAM

10/10/2008
Sélection: perspectives
• SAM deuxième et troisième génération
• Introduction de paramètres immunologiques dans les schémas de sélection
– Etude en cours chez le porc pour estimer la faisabilité et déterminer les meilleurs paramètres (projet IMMOPIG)
– Suite: études QTL sur ces paramètressélection divergente

10/10/2008
La génomique fonctionnelle
1. Transcriptome et protéome
2. Etude du transcriptome
a) Méthodes bas-débitb) Méthodes haut-débitc) Le séquençage du
transcriptomed) Exemples d’études
du transcriptome à l’aide des puces à ADN

10/10/2008
La génomique fonctionnelle
1. Transcriptome et protéome
2. Etude du transcriptome
a) Méthodes bas-débitb) Méthodes haut-débitc) Le séquençage du
transcriptomed) Exemples d’études
du transcriptome à l’aide des puces à ADN
10/10/2008
Génomenucléaire
Génomique structuraleGénomique structurale
ARNmARNmTraductionTraduction
DégradationDégradation
Génomique fonctionnelleGénomique fonctionnelle
ProtéomeTranscriptome

10/10/2008
Génome, transcriptome et protéome

10/10/2008
Etude du protéome
Approches utilisées: Après extraction de protéines- Electrophorèse sur gel 2D:Séparation des protéines et recherche des protéines par analyse d’image
- Spectrométrie de masseIdentification de protéines à partir de leur empreinte peptidique massique.
Ex: MS MALDI-TOF
-Protein arrays (intéractions protéine-protéine)
-Séquençage

10/10/2008
Etude du transcriptome
• Etude d’un nombre limité de gènes
� Western blot
� PCR quantitative en temps reel (qRT-PCR)
• Techniques “haut-débit” :
• Nécessitant de connaître les sondes spécifiques de gènes: puces à ADN
globales & différentielles
ADNc ou oligonucléotides
– Approche réseau dédié (CREA exhaustif pour une région donnée, d’une fonction donnée)
– Approche pan-génomique
• Méthodes exhaustives: ne nécessitant pas de connaître les séquences ADN à étudier
– Banques d’hybridation soustractive (SSH)
– Serial Analysis for Gene Expression (SAGE)
– Le séquençage du transcriptome
10/10/2008
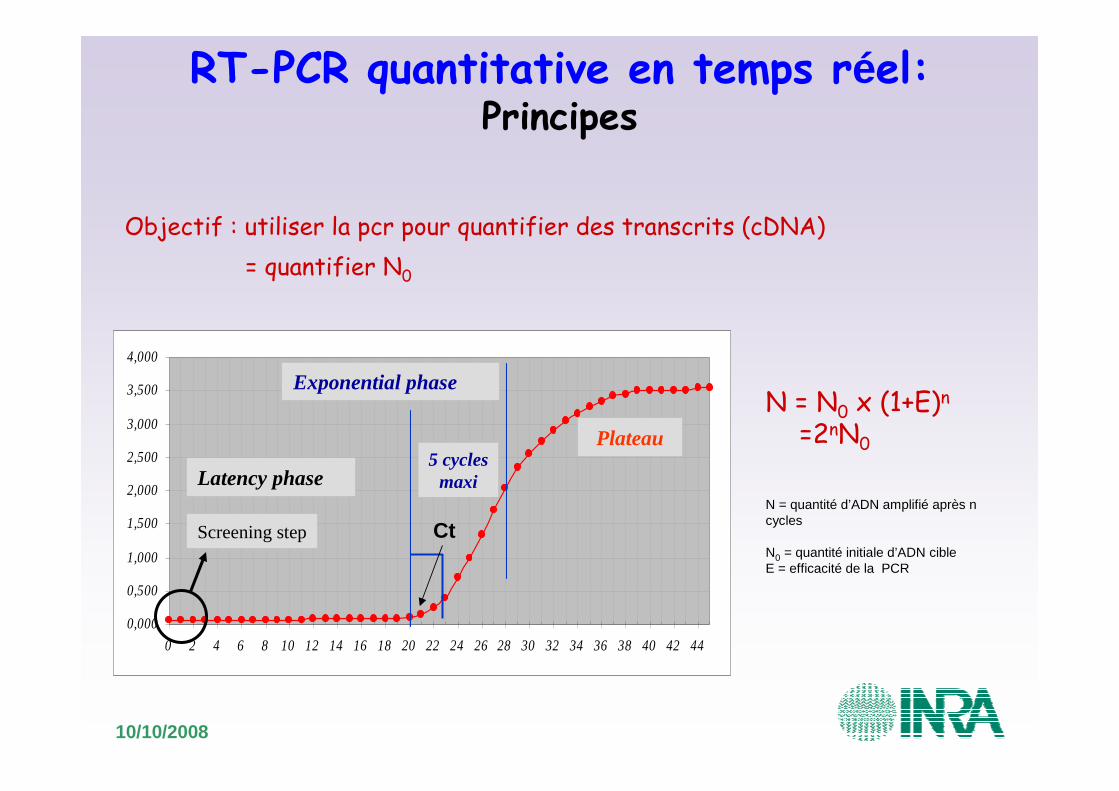
RT-PCR quantitative en temps réel:Principes
0,000
0,500
1,000
1,500
2,000
2,500
3,000
3,500
4,000
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44
Latency phase
Screening step
Exponential phase
Plateau5 cycles
maxi
Ct
Objectif : utiliser la pcr pour quantifier des transcrits (cDNA)
N = N0 x (1+E)n
=2nN0
N = quantité d’ADN amplifié après n cycles
N0 = quantité initiale d’ADN cibleE = efficacité de la PCR
= quantifier N0
10/10/2008
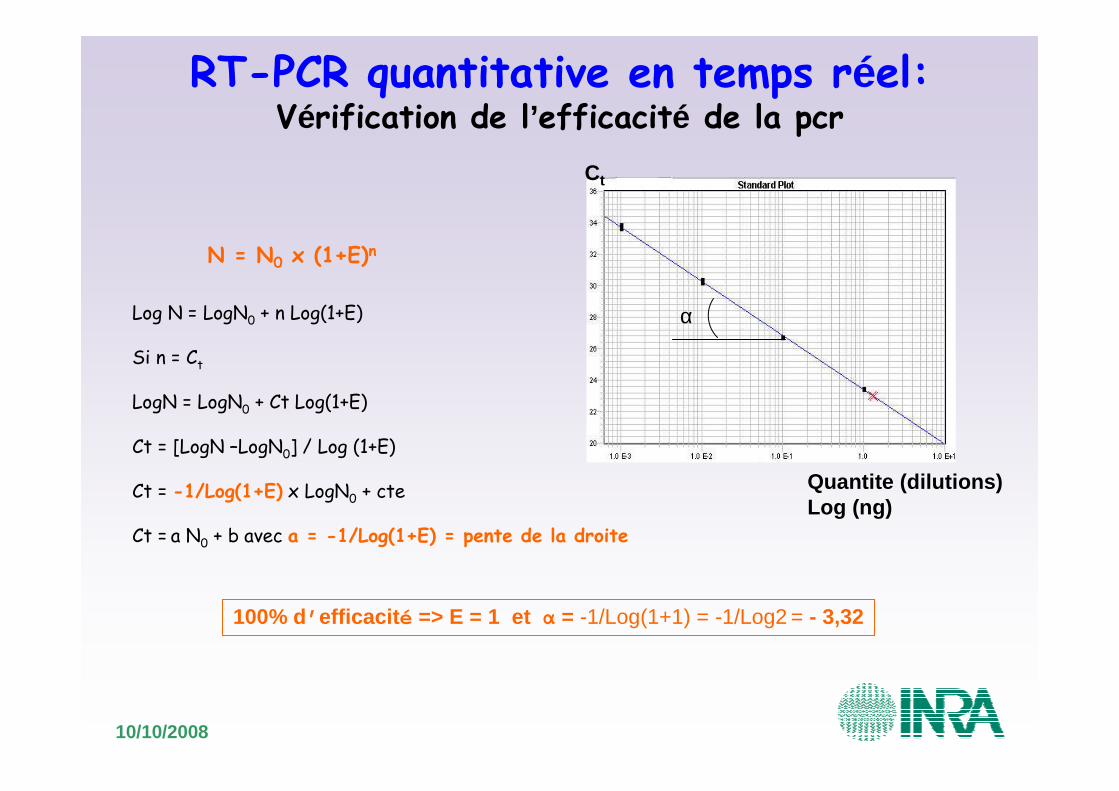
N = N0 x (1+E)n
Quantite (dilutions)Log (ng)
Ct
Log N = LogN0 + n Log(1+E)
Si n = Ct
LogN = LogN0 + Ct Log(1+E)
Ct = [LogN –LogN0] / Log (1+E)
Ct = -1/Log(1+E) x LogN0 + cte
Ct = a N0 + b avec a = -1/Log(1+E) = pente de la droite
100% d’ efficacit é => E = 1 et α = -1/Log(1+1) = -1/Log2 = - 3,32
α
RT-PCR quantitative en temps réel:Vérification de l’efficacité de la pcr

10/10/2008
1) Normalisation avec le gène de référence
∆ Ct = CTGcible – CT Gref
2) Quantification relative des données normalisées entre une condition et un calibrateur
∆∆ Ct = (CTGcible – CT Gref)cond1 – (CTGcible – CT Gref) calibrator
Formule de quantification relative 2 –∆∆∆∆∆∆∆∆Ct
RT-PCR quantitative en temps réel:Quantification relative
qRT-PCR utilisée pour confirmer les résultats obtenus à l’aide des puces à ADN

10/10/2008
La génomique fonctionnelle
1. Etude du protéome
2. Etude du transcriptome
a) Techniques bas-débit
b) Techniques haut-débit
c) Exemples d’études du transcriptome à l’aide des puces à ADN

10/10/2008
Etude du transcriptome
• Etude d’un nombre limité de gènes
� Western blot
� PCR quantitative en temps reel (qRT-PCR)
• Techniques “haut-débit” :
• Nécessitant de connaître les sondes spécifiques de gènes: puces à ADN
globales & différentielles
ADNc ou oligonucléotides
– Approche réseau dédié (CREA exhaustif pour une région donnée, d’une fonction donnée)
– Approche pan-génomique
• Méthodes exhaustives: ne nécessitant pas de connaître les séquences ADN à étudier
– Serial Analysis for Gene Expression (SAGE)
– Le séquençage du transcriptome

10/10/2008
Puces à ADN Serial Analysis of Gene ExpressionQuelques principes
Courtin et al, 2007

10/10/2008
Analyse et validation des résultats
• Importance du traitement statistique des données
• Validation des résultats au niveau des transcrits (qRT-PCR) et au
niveau protéique (immunohistochimie, Cytométrie en flux…)
• Analyse bioinformatiques
-Gene Ontology (GO):(biological process, cellular component, molecular function)
ex: DAVID, PANTHER…
-Réseaux de gènes, voie biologiques et canoniques
Ex: INGENUITY

10/10/2008
Puces à ADN et réseaux dédiés:approche région candidate
Objectif 1: Localiser finement un gène et une mutation responsable du contrôle d’un phénotype
- Tous les BAC ou plasmides ou oligonucléotides (ciblant des régions codantes et non codantes) d’une région
chromosomique candidate (QTL) sont déposés sur membrane ou lame de verre
- Hybridation d’ARN sain/ ARN malade
- Identification des BACs, plasmides ou oligonucléotides qui présentent une expression différentielle
- Identification des gènes candidats
Objectif 2: étudier l’expression d’une région chromosomique contenant des gènes d’intérêts dans plusieurs conditions
- Co-régulation des gènes
- cDNA ou oligonucléotides spécifiques de tous les gènes de la région
- Expression différentielle
Objectif 3: étude des régions codantes et non codantes d’une portion chromosomique
- Tiling array (oligonucléotides)
- Chip on chip:étude de la transcription

10/10/2008
Puces à ADN et réseaux pan-génomiques
Puce de gènes• « Maison » INRA-CRB (centre de ressources biologiques)
– Membranes (cDNA) Truite, Porcs– Lames de verre (oligos) Bovin, Porc, Poulet
• « Commerciaux »– Set d’oligos commerciaux (Qiagen, Opéron)– Puces affymetrix– Puces Agilent– Puces Nimblgen
Tiling arrays• « Maison »• « Commerciaux » en développement pour les animaux de rente
CHip on chipObjectif: détecter si une proteine donnée se fixe spécifiquement sur une séquence d’ADN
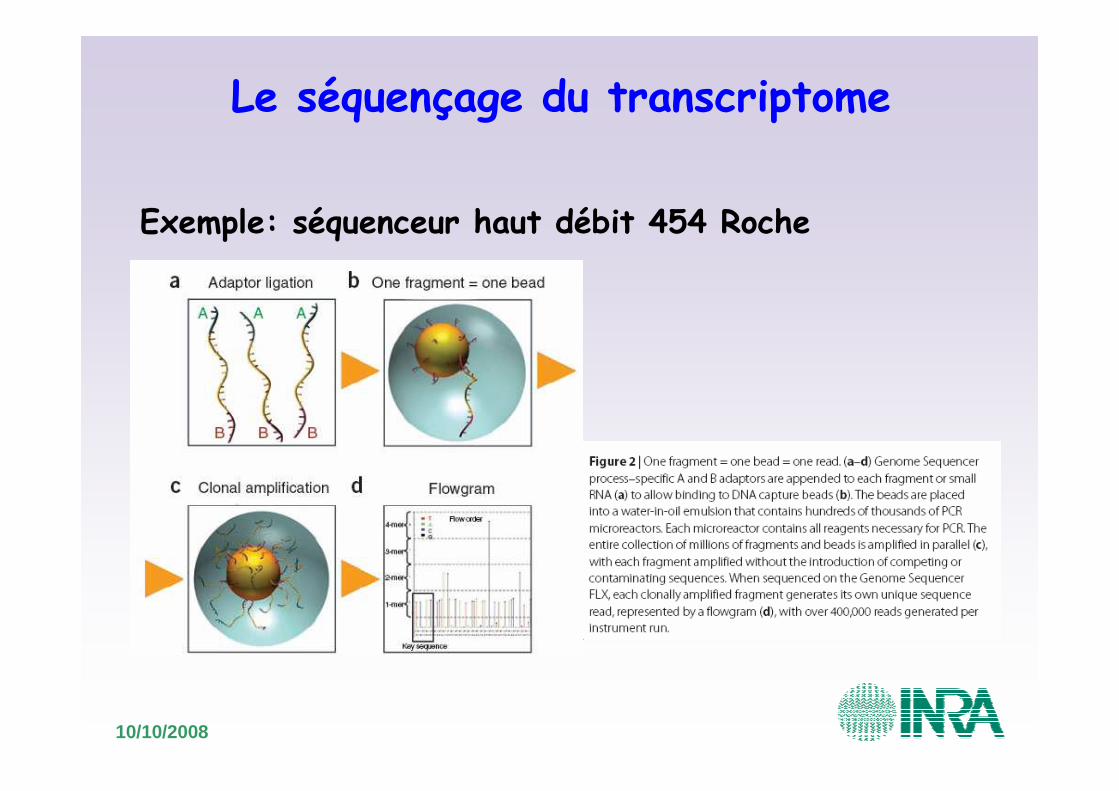
10/10/2008
Le séquençage du transcriptome
Exemple: séquenceur haut débit 454 Roche

10/10/2008
La génomique fonctionnelle
1. Etude du protéome
2. Etude du transcriptome
a) Techniques bas-débit
b) Techniques haut-débit
c) Exemples d’études du transcriptome à l’aide des puces à ADN

10/10/2008
2007
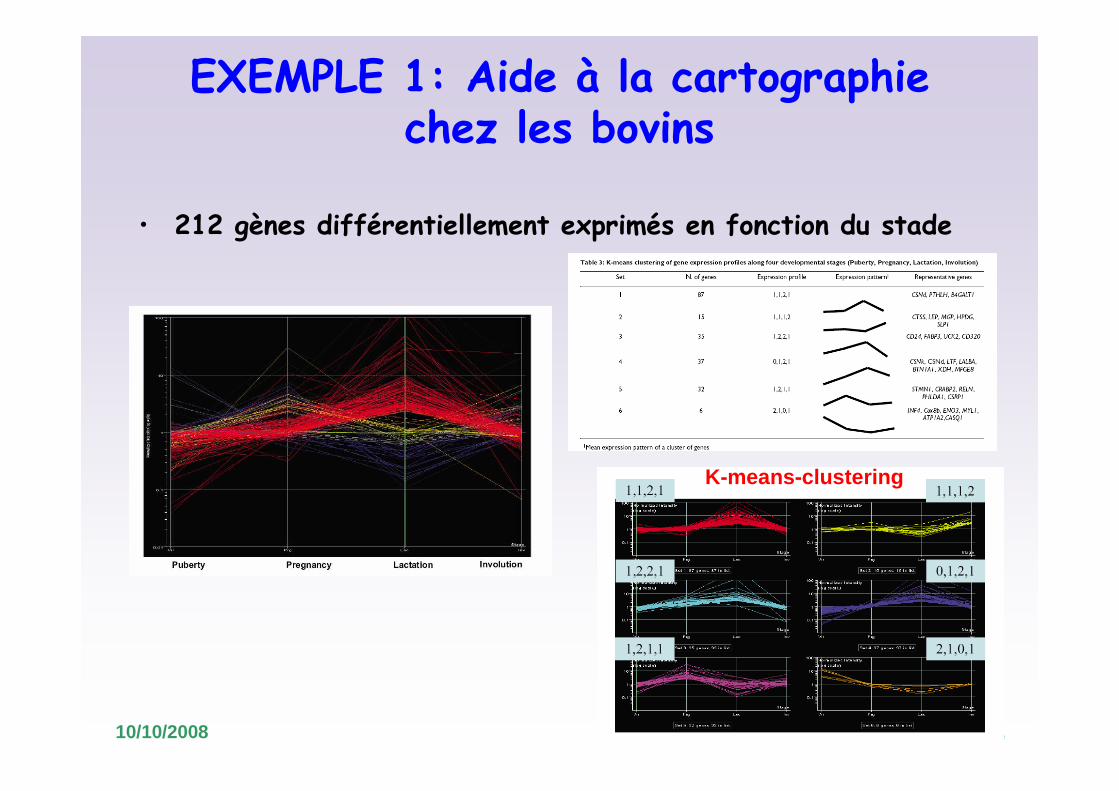
EXEMPLE 1: Aide à la cartographie chez les bovins

10/10/2008
Aide à la cartographie de QTLS
Arbilly et al, Animal Genetics, 2006

10/10/2008
• Point de départ: Etudes QTL sur les caractères de production laitière dans différentes race de bovins
Identification de plusieurs QTL contenant quelques centaines de gènes
Identification d’un QTN: mutation de DGAT1 le gène causal affectant la quantité de matière grasse du lait sur BTA14
• Objectif : étudier l’expression des gènes dans la glande mammaire de souris à différents stades (puberté, gestation, lactation, involution) et combiner ces données avec les résultats des études QTL chez les bovins pour proposer des gènes candidats à tester.
• Outil: Puce Affymetrix: 12488 sondesTissu: glande mammaire souris C57BL/6J
EXEMPLE 1: Aide à la cartographie chez les bovins

10/10/2008
EXEMPLE 1: Aide à la cartographie chez les bovins
• Analyse différentielle (ANOVA) et comparaison avec d’autres études d’expression
249 sondes en commun correspondant à 212 gènes bovins
10/10/2008
EXEMPLE 1: Aide à la cartographie chez les bovins
• 212 gènes différentiellement exprimés en fonction du stade
K-means-clustering

10/10/2008
EXEMPLE 1: Aide à la cartographie chez les bovins
• Comparaison avec les données de cartographiePbl: beaucoup de gènes ont des fonctions inconnues
DGAT1 est surexpriméUtilisation de la base de donnée cgQTL pour proposer des gènes candidats
clusters
BTA6
http://cowry.agri.huji.ac.il/QTLMAP/qtlmap.htm

10/10/2008
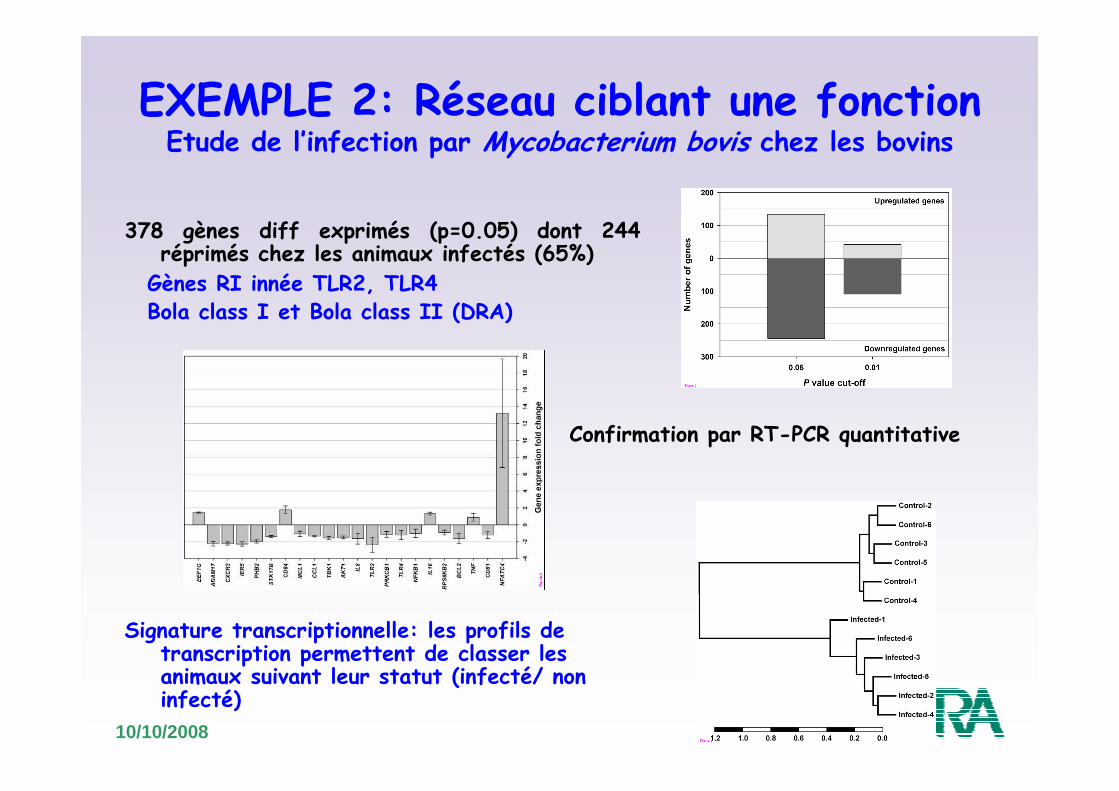
EXEMPLE 2: Réseau ciblant une fonctionEtude de l’infection par Mycobacterium bovis chez les bovins
Meade et al, 2007, BMC Genomics
• Objectif : comparaison des niveaux de transcrits de PBMC provenant de 6 bovins infectés par Mycobacterium bovis/ 6 bovins non infectés
• Outil: Puce cDNA dédiée immunospécifique: 1391 gènes (spots)
• Analyse PBMCAnimaux infectés Contrôles
10/10/2008
378 gènes diff exprimés (p=0.05) dont 244 réprimés chez les animaux infectés (65%)
Signature transcriptionnelle: les profils de transcription permettent de classer les animaux suivant leur statut (infecté/ non infecté)
Gènes RI innée TLR2, TLR4Bola class I et Bola class II (DRA)
Confirmation par RT-PCR quantitative
EXEMPLE 2: Réseau ciblant une fonctionEtude de l’infection par Mycobacterium bovis chez les bovins
10/10/2008
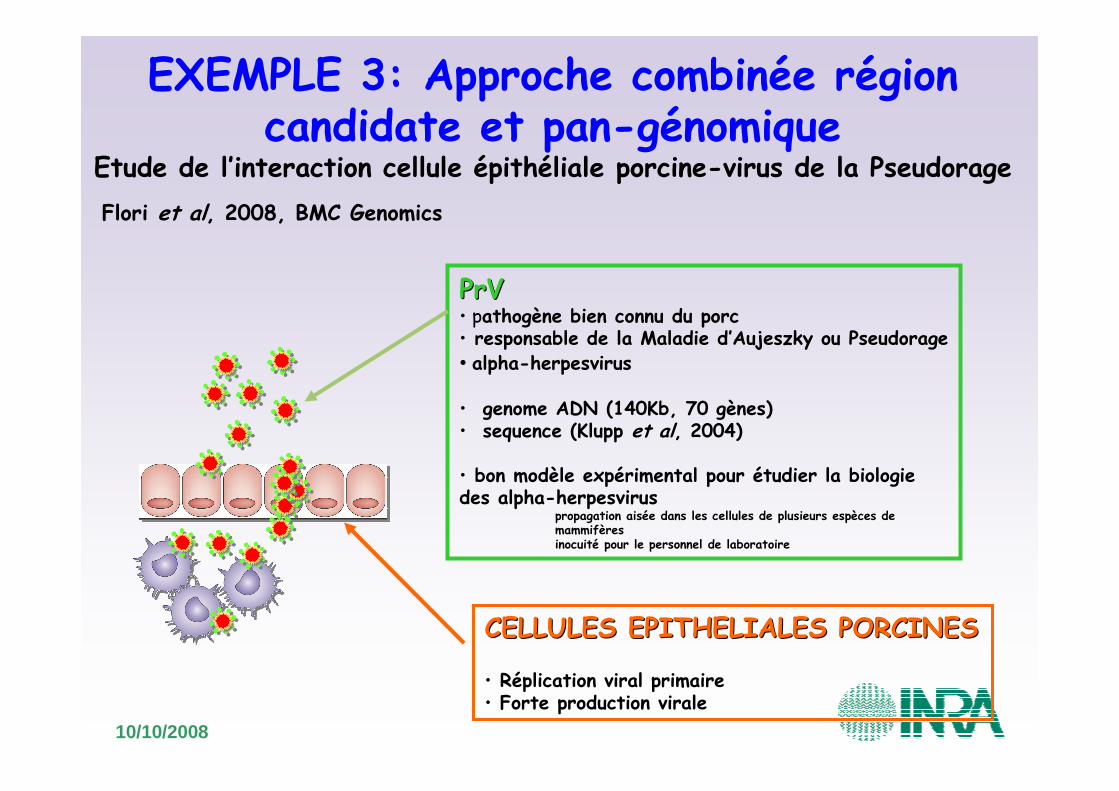
EXEMPLE 3: Approche combinée région candidate et pan-génomique
Etude de l’interaction cellule épithéliale porcine-virus de la Pseudorage
PrVPrV• pathogène bien connu du porc• responsable de la Maladie d’Aujeszky ou Pseudorage• alpha-herpesvirus
• genome ADN (140Kb, 70 gènes)• sequence (Klupp et al, 2004)
• bon modèle expérimental pour étudier la biologiedes alpha-herpesvirus
propagation aisée dans les cellules de plusieurs espèces de mammifèresinocuité pour le personnel de laboratoire
CELLULES EPITHELIALES PORCINESCELLULES EPITHELIALES PORCINES
• Réplication viral primaire• Forte production virale
Flori et al, 2008, BMC Genomics

10/10/2008
Etude des intéractions cellule hôte –pathogène: approches transcriptomiques
• Puces à ADN: outils efficaces pour analyser les interactions cellule hôte-pathogèneHossein H et al, 2006, Curr. Opin. Immunol.
• Etudes transcriptomiques:- Analyse du transcriptome de la cellule OU du pathogène
-Peu d’études simulatanées des transcriptomes de la cellule et du pathogène
P.berghei ANKA et souris (brain) Lovegrove FE et al, 2006, BMC GenomicsEBV et NK/T cells Zhang YJH et al, 2005, Br. J. Cancer
� Etudes transcriptomiques des intéraction PrV-cellule- Analyse du transcriptome cellulaire
- Etudes cinétiques
- Modèles hétérologues REF cells / rat chip Ray and Enquist. 2004. J. Virol.
Brukman and Enquist. 2006. J. Virol.
HEK-293 cells / human chip Blanchard et al, 2006. Vet. Res.

10/10/2008
Objectifs
Analyser in vitro le dialogue entre les cellules épithéliales porcines et le PrVEtablir un lien temporel direct entre l’expression des gènes viraux et cellulaires.
- Analyse simultanée des modifications du transcriptome viral et cellulaire- Etude cinétique- Système homologue (PrV/cellules porcines / puces porcines)
Suivre la mock-infection et l’infection par le PrV des cellules épithéliales porcines PK15
Détecter les gènes viraux et cellulaires différentiellement exprimés durant l’infection entre les cellules infectées (I) et mock-infected (MI) à différents temps

10/10/2008
Couverture ~ 72.5% de la région SLA(Swine Leucocyte Antigen)
70 gènes du PrV
1515 genesMicroarray dédié SLA/PrVconstruit au laboratoire (CRB-GADIE/LREG)
Microarray générique Qiagen-NRSP8 8541 gènes
⇒ADNcs et exons sous-clonés
⇒GEO GPL5622
=> oligonucleotides
Outils et méthodes
1.5 % gènes impliqués dans la réponse immunitaire
• Microarrays
• PCR quantitative en temps réél
Zhao et al, 2005, Genomics
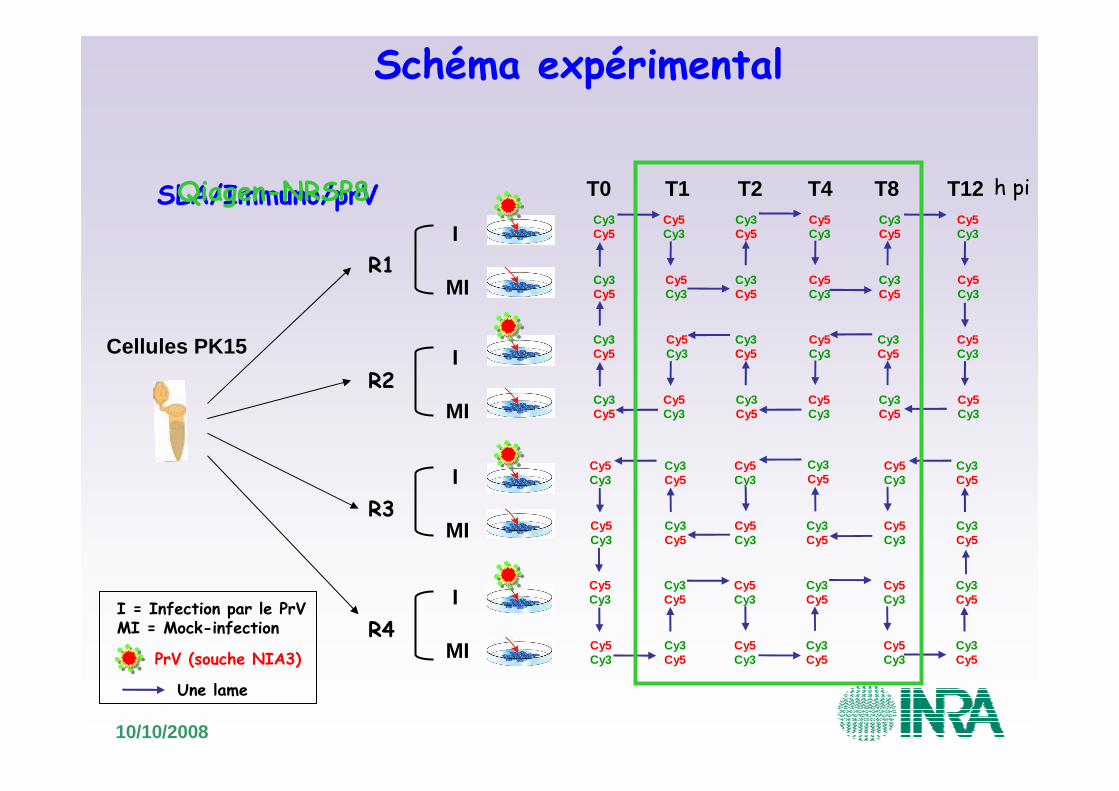
10/10/2008
Cellules PK15
I
MI
I
MI
I
MI
I
MI
Schéma expérimental
I = Infection par le PrVMI = Mock-infection
T0 T1 T2 T4 T8 T12Cy3Cy5
Cy5Cy3
Cy3Cy5
Cy3Cy5
Cy3Cy5
Cy5Cy3
Cy5Cy3
Cy5Cy3
Cy3Cy5
Cy3Cy5
Cy5Cy3
Cy5Cy3
Cy3Cy5
Cy5Cy3
Cy3Cy5
Cy3Cy5
Cy3Cy5
Cy5Cy3
Cy5Cy3
Cy5Cy3
Cy3Cy5
Cy3Cy5
Cy5Cy3
Cy5Cy3
Cy3Cy5
Cy5Cy3
Cy3Cy5
Cy3Cy5
Cy3Cy5
Cy5Cy3
Cy5Cy3
Cy5Cy3
Cy3Cy5
Cy3Cy5
Cy5Cy3
Cy5Cy3
Cy3Cy5
Cy5Cy3
Cy3Cy5
Cy3Cy5
Cy3Cy5
Cy5Cy3
Cy5Cy3
Cy5Cy3
Cy3Cy5
Cy3Cy5
Cy5Cy3
Cy5Cy3
h piSLA/Immuno/prVQiagen-NRSP8
Une lame
R1
R3
R4
R2
PrV (souche NIA3)

10/10/2008
Acquisition des données, normalisation et analyse statistique
1/ Acquisition des données
AA AA
MM
BlocsBlocs
MM
BlocsBlocs
• Soustraction de la médiane de chaque bloc
M = Log2(Cy5/Cy3)
A = 0.5*Log2(Cy5*Cy3)
MM MM• Transformation Log2• Locally weighted polynomial regression
=> Lowess (f=0.3)
2/ Normalisation
• Scan des lames Scanarray (LCE,CEA) and Virtek (PICT, INRA)
• Quantification Imagene software
• Stockage des données BASE (Sigenae)
3/ Selection des gènes différentiellement exprimés• Modèle linéaire Ycfnds = µc + ααααf + ββββn + δδδδd + Ecflje• Comparaisons Student t test - MI vs I à chaque temps => 1 pvalue pour chaque gène
• Correction pour tests multiples : False Discovery Rate (FDR) =0,05
• Hierarchical clustering : HCL et k-means (TmeV software)4/ Exploration des données
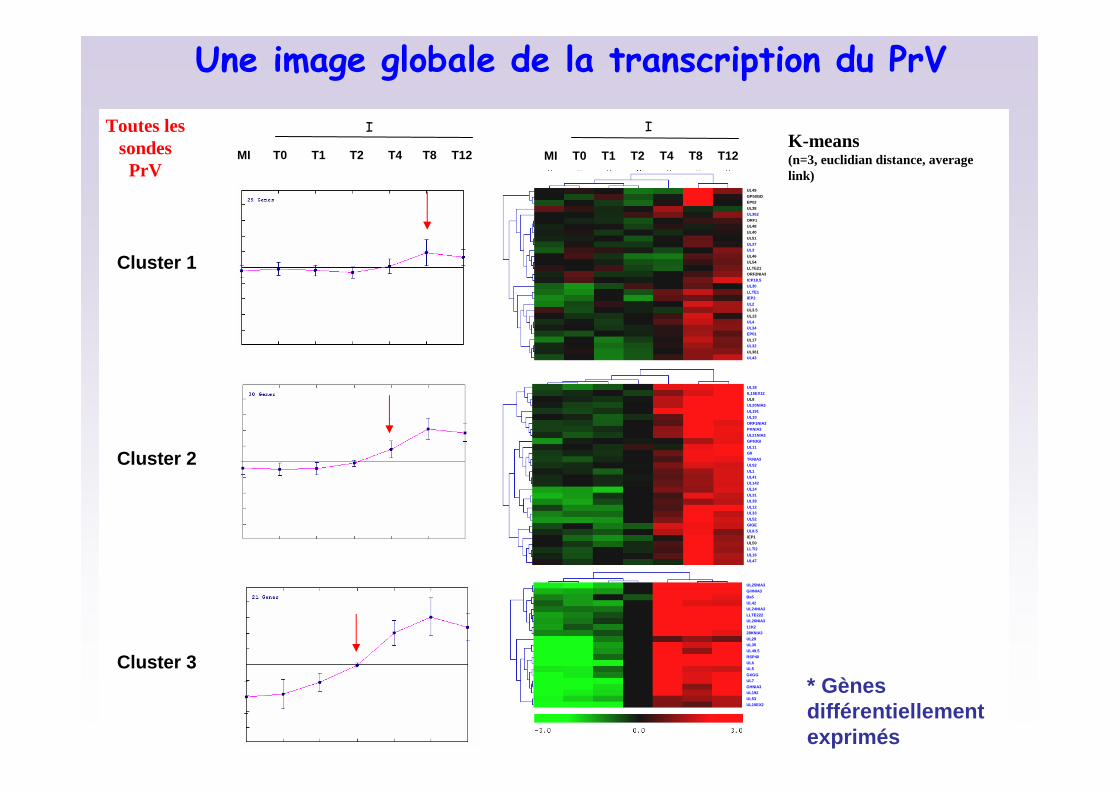
10/10/2008
* Gènes différentiellementexprimés
Cluster 1
Cluster 2
Cluster 3
T8MI T0 T1 T4 T12T2
UL49
GP50GDEP02
UL38UL362
ORF1UL48
UL40UL51
UL37UL3
UL46UL54
LLTE21ORF2NIA3
ICP18.5UL30
LLTE1IEP2
UL2UL3.5
UL13UL4
UL34EP01
UL17UL32
UL361UL43
UL18IL15EX12
UL8UL20NIA3
UL191UL10
ORF1NIA3PKNIA3
UL21NIA3GP63GI
UL11GII
TKNIA3UL92
UL1UL41
UL142UL14
UL31UL39
UL12UL33
UL52GIGE
UL8.5IEP1
UL50LLTI2
UL16UL47
UL25NIA3
GIIINIA3Ba5
UL42UL24NIA3
LLTE222UL26NIA3
11K228KNIA3
UL29UL35
UL49.5RSP40
UL6UL5
GXGGUL7
GHNIA3UL192
UL53UL15EX2
Une image globale de la transcription du PrV
T8MI T0 T1 T4 T12T2
I IK-means(n=3, euclidian distance, averagelink)
Toutes les sondes PrV
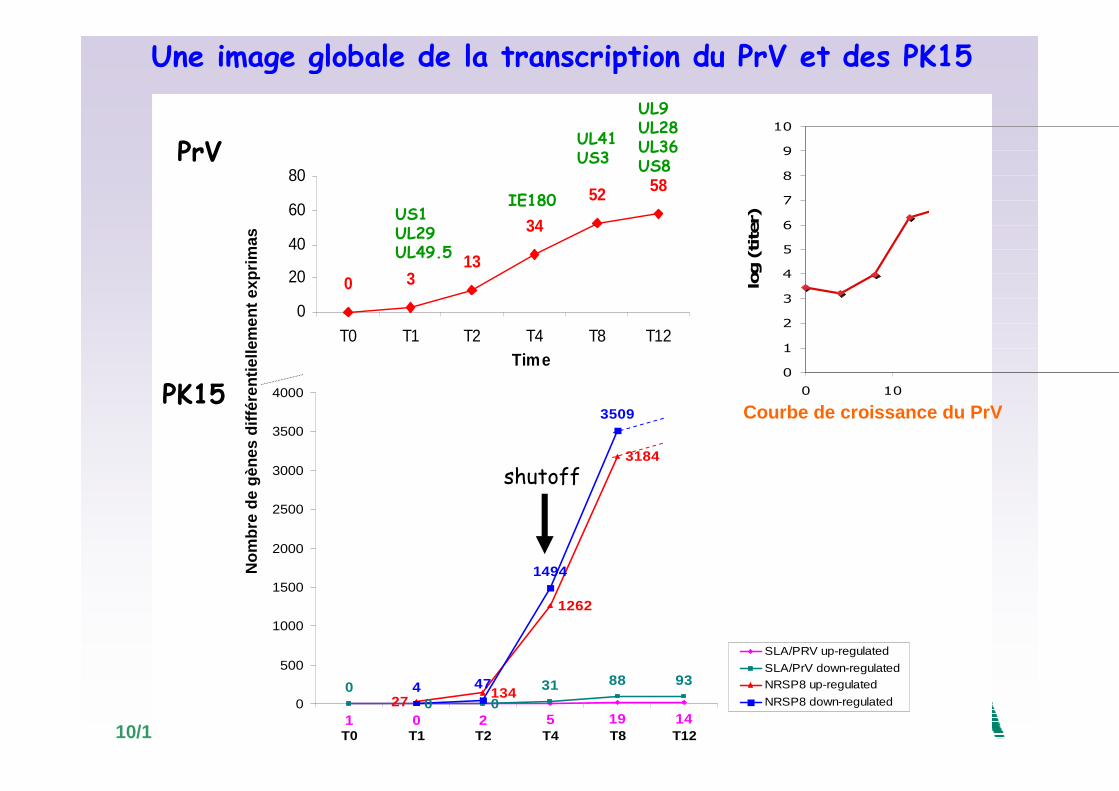
10/10/2008
0 313
52 58
34
0
20
40
60
80
T0 T1 T2 T4 T8 T12Time
PrV
US1UL29UL49.5
IE180
UL9UL28UL36US8
UL41US3
Nom
bre
de g
ènes
diff
éren
tielle
men
texp
rimas
1 0 2 5 19 140 0
1262
3184
4 47
1494
3509
0 938831134270
500
1000
1500
2000
2500
3000
3500
4000
T0 T1 T2 T4 T8 T12
SLA/PRV up-regulated
SLA/PrV down-regulatedNRSP8 up-regulated
NRSP8 down-regulated
PK150
1
2
3
4
5
6
7
8
9
10
0 10
log (titer)
Courbe de croissance du PrV
shutoff
Une image globale de la transcription du PrV et des PK15

10/10/2008
• Augmentation du nombre de gènes différentiellement exprimés au cours du temps à partir de 2h pi parallèlement au nombre de gènes viraux.
• Beaucoup de gènes cellulaires sont réprimés. Cette down-régulationeest détéctée à partir de 4h pi et augmente à 8 et 12h pi.
���� Pas de shutoff précocemais shutoff plus précoce dans les cellules porcines que dans les autres cellules (8h pi)
• Le gène viral UL41 codant pour la virion host shutoff protein (vhs) est détectée 8h pi
���� shutoff probablement induit par les protéines vhsnéosynthétisées
PrV et shutoff cellulaire

10/10/2008
L’infection par le PrV altère de multiples processus biologiques et fonctions cellulaires
Ingenuity Pathways analysisGene number/Top function
T1
T2
T4
T8
SLA-IaTAP1TAP2Calnexine
IRFsTLR8 PPIA
Gene ExpressionMolecularTransport
CellCycle
CellDeath
Cellular Movment
Cellular Assembly
andOrganization
Immune Response
DNA Replicationand Repair
Cellsignalling
Gene Expression
MolecularTransport
DrugMetabolism
Nucleic acidmetabolism
BCl-2FAIM2Caspase1Caspase 3Caspase 7NF-KB2
US3 8h piUS1 1h pi
F-actinB-actinMyosin
US3 8h pi
HIST1H2ALHISTAH4JHISTAH2BK
HDAC2HDAC10HDAC3HDAC6HDAC7AHDAC9
US3 8h pi
Up-regulatedDown-regulated
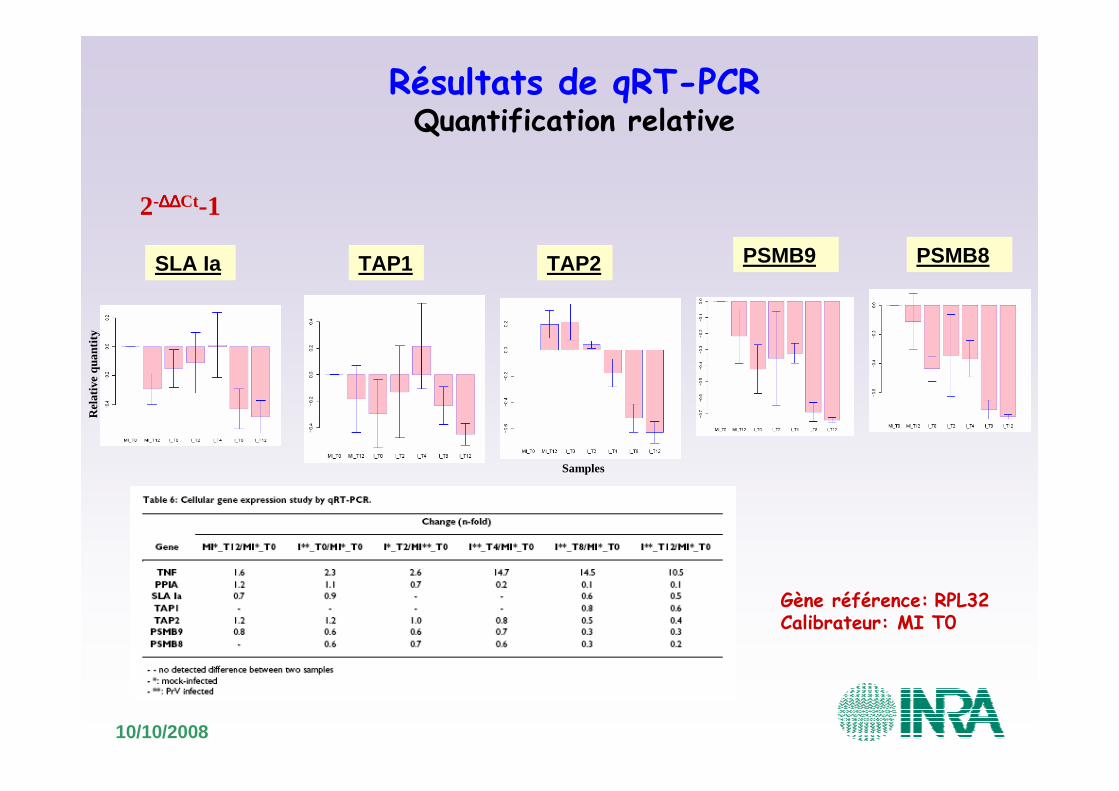
10/10/2008
Résultats de qRT-PCRQuantification relative
2-∆∆∆∆∆∆∆∆Ct-1
SLA Ia TAP2
Gène référence: RPL32Calibrateur: MI T0
Rel
ativ
e qu
antit
y
TAP1 PSMB9 PSMB8
Samples

10/10/2008
Diminution de SLA Ia à la surface des PK15 Cytométrie en flux
10/10/2008
Rupert Abele, Robert Tampe´FEBS Letters 580 (2006) 1156–1163
Stratégie du PrV pour se soustraire à la voie de présentation antigénique du CMH de classe I
Inhibition de l’activité du transporteur TAPAmbagala et al, 2000, J. Immunol.
/interaction avec la protéine gN codée par le gène UL49.5Koppers-Lalic et al, 2005, PNASUL49.5 (gN)1h pi
UL41 (vhs)8h pi
PrV
Diminution des niveaux de transcrits dans les cellules infectées
SLA-IaTAP1TAP2PSMB8 (LMP7)PSMB9 (LMP2)
PK15
Le PrV developpe differentes strategies pour échapper à la voie de présentation antigénique du CMH de classe I: -arrêter la pompe à peptides
-diminuer la transcription de molécules clefs
Diminution de l’expression des molécules du CMH Cl I à la surface des cellulesMellencamp et al, 1991, J. Virol.

10/10/2008
Conclusions et perspectives
Perspectives• Analyse des intéractions PrV-cellules porcines dans d’autres cellules cibles comme
les cellules dendritiques immatures
• Etude de l’expression des gènes viraux et cellules en utilisant des virus mutants
Conclusions• L’expression des gènes du PrV et des cellules porcines peut être analysée
conjointement avec des puces à ADN
• Chronologie de la transcription des gènes du PrV jusqu’alors jamais décrite
• Image globale de la transcription avec un lien temporel direct entre l’expressiondes gènes viraux et cellulaires
• Nouvelles données sur les stratégies virale d’échappement à la réponse immunitaire(voie de présentation antigénique par les molécules de classe I du CMH)

10/10/2008
Bilan études du transcriptome chez le porc Puces à ADN
Tuggle et al, Int.J.Biol.Sci., 2007, 3(3):132-152
• Muscle: 5 études
•Appareil reproducteur (utérus, ovaire, testis): 9 études
•Réponse immunitaire: 12 études

10/10/2008
La génomique génétique

10/10/2008
� Intégrer des génotypes et des données d’expression
– Objectif: identifier des régions du génome intervenant dans la variabilité de l’expression des gènes
– Principe: Etudier la quantité de transcrits produits pour beaucoup (tous) de gènes comme autant de caractères quantitatifs
analyse de liaison et d’association
La génomique génétique (« Genetical Genomics »)
Schadt et al, Nature, 2003, 422:297-302
� Article pionnier

10/10/2008
� Cartographie de QTL d’expression (eQTL)
2 types d’eQTL� Cis-acting eQTL: le gène codant pour le transcrit est situé dans l’intervalle de confiance de l’eQTL
� Trans-acting eQTL: l’intervalle de localisation est situé dans une région distante du gène codant pour le transcrit considéré
Cis et trans-régulation de l’abondance des transcrits
Drake et al, 2006, Mammalian Genome
La génomique génétique (« Genetical Genomics »)

10/10/2008
La génomique génétique (« Genetical Genomics »)

10/10/2008
� Proposer et classer les gènes candidats
� Définir des sous-types moléculaires à l’intérieur de la population
pour un phénotype donné
� Identifier des voies métaboliques et cellulaires, impliquées dans
l’apparition du phénotype
� Identifier des facteurs causaux impliqués dans une variation
phénotypique
La génomique génétiqueIntérêts

10/10/2008CONCLUSION

10/10/2008
• Les animaux de rente– Possibilité de croisements dirigés
– Des espèces modèles pour des mécanismes parfois originaux
– Des mutations régulatrices importantes dans le contrôle des
variations phénotypiques
• La sélection– L’information moléculaire acquise peut être valorisée (Sélection
assistée par marqueurs)
– Ajout de nouveaux paramètres immunologiques dans les critères
de sélection
– Utilisation des données d’expression

10/10/2008
Merci de votre attention…