explorations complémentaires en hématologie cellulaire · globules blancs 64 g/l ... Étalement...
TRANSCRIPT

Explorations complémentaires en
Hématologie Cellulaire
L3 Médecine
4 Octobre 2012
Dr Marina Lafage-Pochitaloff
MCU-PH en Hématologie
Laboratoire de Cytogénétique Onco-Hématologique
Département de Génétique (Pr Lévy)
CHU Timone Enfants Marseille
Aix-Marseille Université
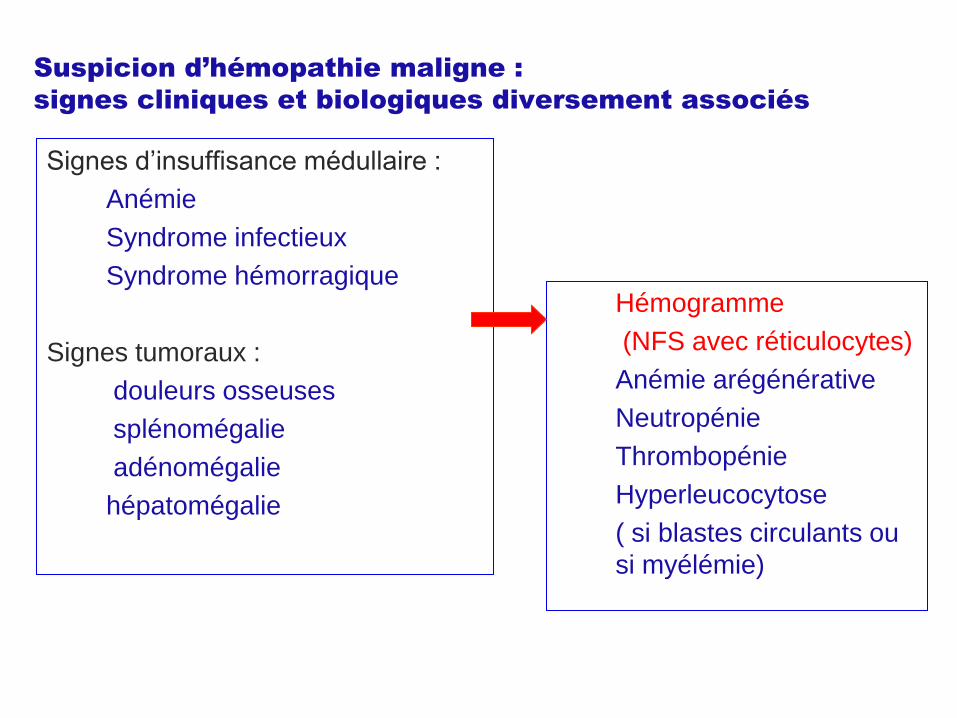
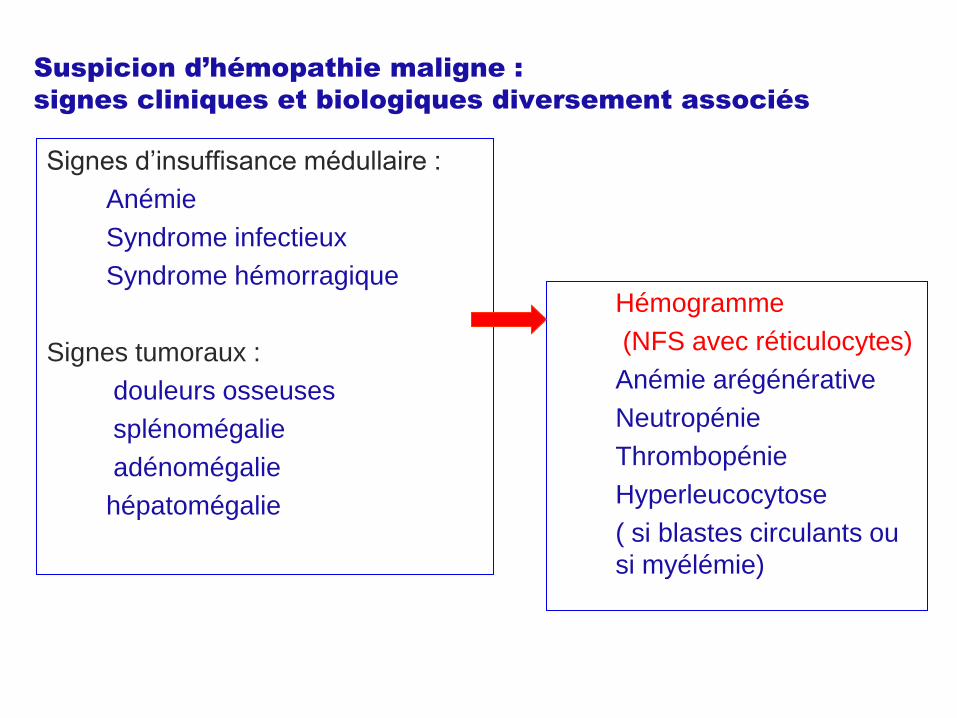
Suspicion d’hémopathie maligne :
signes cliniques et biologiques diversement associés
Signes d’insuffisance médullaire :
Anémie
Syndrome infectieux
Syndrome hémorragique
Signes tumoraux :
douleurs osseuses
splénomégalie
adénomégalie
hépatomégalie
Hémogramme
(NFS avec réticulocytes)
Anémie arégénérative
Neutropénie
Thrombopénie
Hyperleucocytose
( si blastes circulants ou
si myélémie)
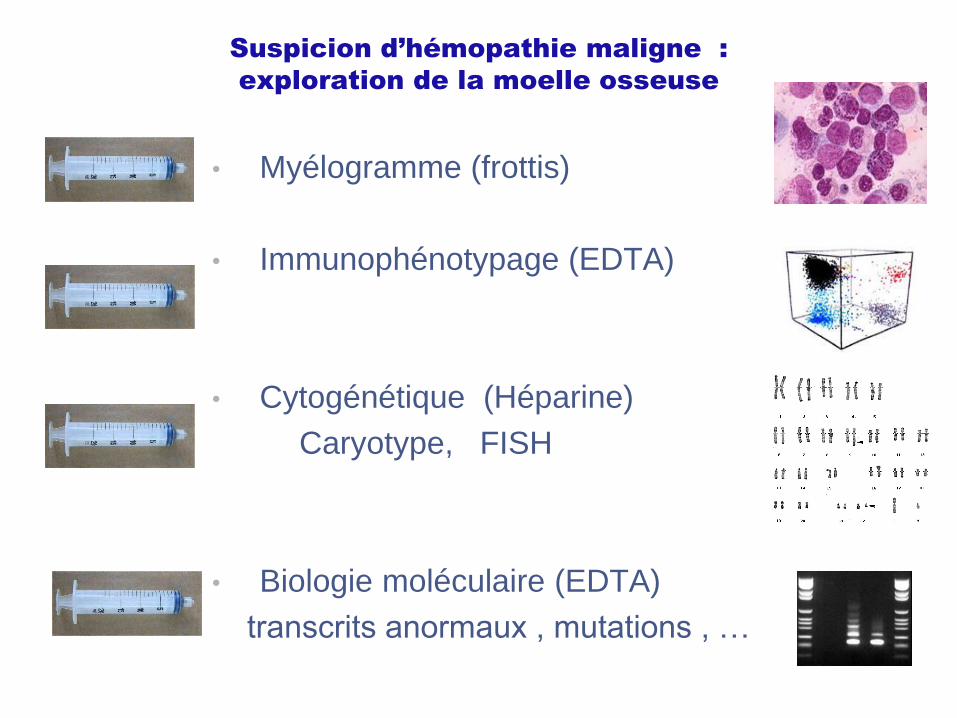
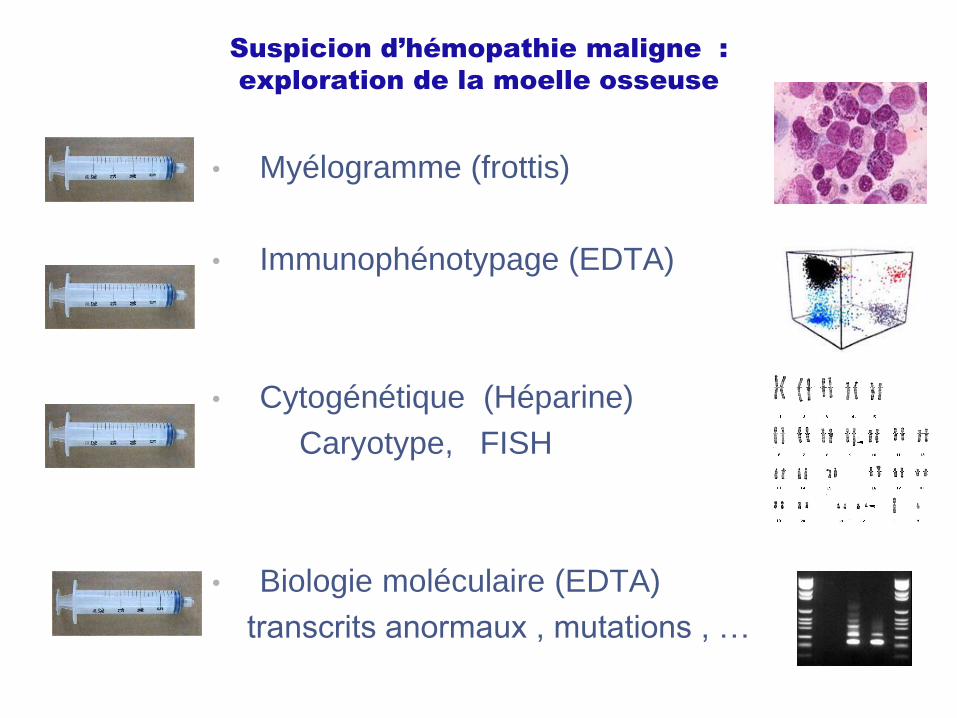
Suspicion d’hémopathie maligne :
exploration de la moelle osseuse
• Myélogramme (frottis)
• Immunophénotypage (EDTA)
• Cytogénétique (Héparine)
Caryotype, FISH
• Biologie moléculaire (EDTA)
transcrits anormaux , mutations , …

Explorations complémentaires dans le cadre
d’une hémopathie maligne
Exemple de deux hémopathies malignes :
Leucémie myéloide chronique (LMC)
Leucémie aigüe lymphoblastique (LAL)

Modèle de la Leucémie Myéloïde Chronique (LMC) (1)
Hémopathie maligne
Pathologie de la souche hématopoiétique
Syndrome myéloprolifératif chronique (SMP)
ou néoplasies myéloprolifératives de l’OMS
Evolution naturelle en 3 phases :
Phase chronique
Phase d’accélération
Phase aiguë : transformation en leucémie aiguë
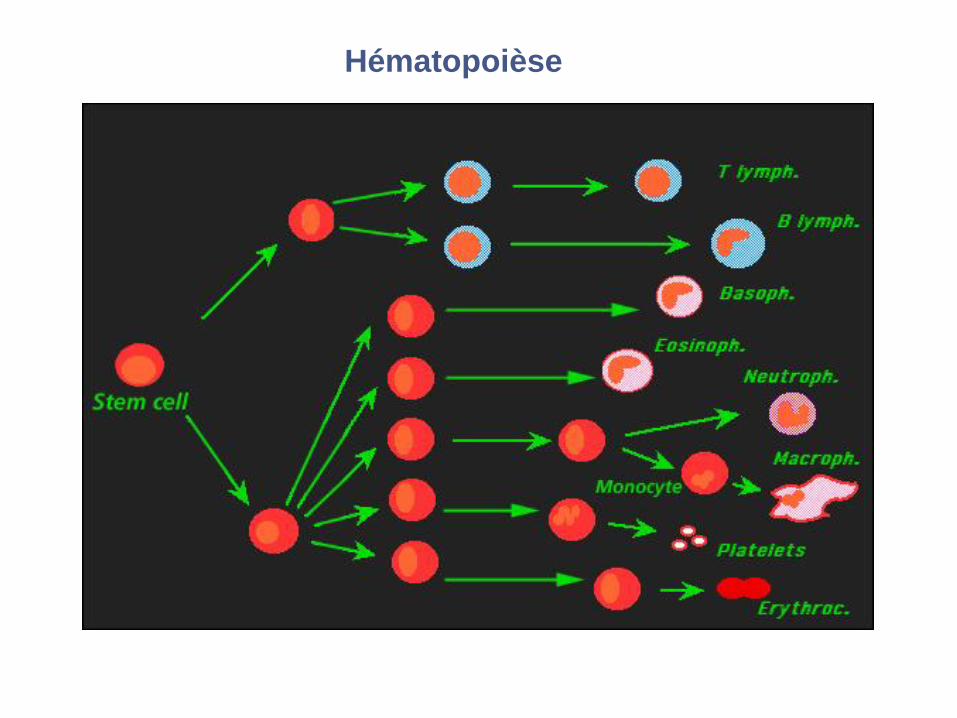
Hématopoièse
CMP = CFU-GEMM ; GMP = CFU-GM ; EMP = CFU-EMK

Modèle de la Leucémie Myéloïde Chronique (LMC) (2)
Présence du chromosome Philadelphie (Ph)
Présence du transcrit BCR-ABL
Traitement ciblé anti-tyrosine kinase (ATK)

Leucémie Myéloïde Chronique (LMC)
Epidémiologie :
Incidence = 10 nouveaux cas/ an / million d’habitants
Prévalence : en augmentation car diminution du taux
de mortalité (progrès thérapeutiques : traitement ciblé
anti-tyrosine kinase)
Facteurs risque : benzène, radiations
Age moyen au diagnostic : 54 ans
Très rare chez l’enfant, exceptionnel avant 5 ans
Sex ratio : 1,4 homme pour 1 femme

Leucémie Myéloïde Chronique (LMC) : clinique
Circonstances de découverte :
Asthénie
Hémogramme systématique +++
Examen clinique :
Le plus souvent : normal
Parfois :
signes tumoraux : splénomégalie
signes liés à l’hyperleucocytose : thromboses
veineuses ou artérielles
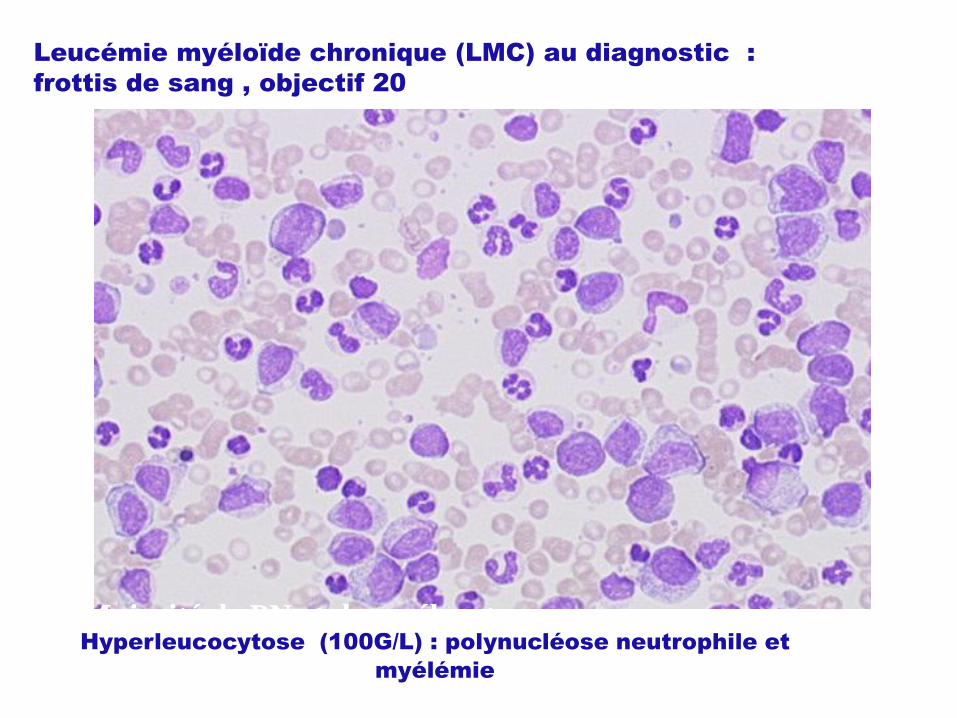
Leucémie myéloïde chronique (LMC) au diagnostic :
frottis de sang , objectif 20
Majorité de PN et de myélocytesHyperleucocytose (100G/L) : polynucléose neutrophile et
myélémie

LMC AU DIAGNOSTIC / : HEMOGRAMME
Globules rouges 5,5 T/L
Hémoglobine 15,5 g/dL
Teneur Corpusculaire Moyenne en Hémoglobine 30 pg
Volume Globulaire Moyen 90 microns cube
Globules blancs 64 G/L
Polynucléaires neutrophiles 58 % soit 37G/L
Polynucléaires éosinophiles 2 % soit 1,3 G/L
Polynucléaires basophiles 4 % soit 2,6 G/L
Monocytes 2 % soit 1,3 G/L
Lymphocytes 6 % soit 3,8 G/L
Myéloblastes 1 % soit 0,6 G/L
Promyélocytes 3 % soit 1,9 G/L
Myélocytes 18 % soit 11,5 G/L
Métamyélocytes 6 % soit 3,8 G/L
Plaquettes : 510 G/L
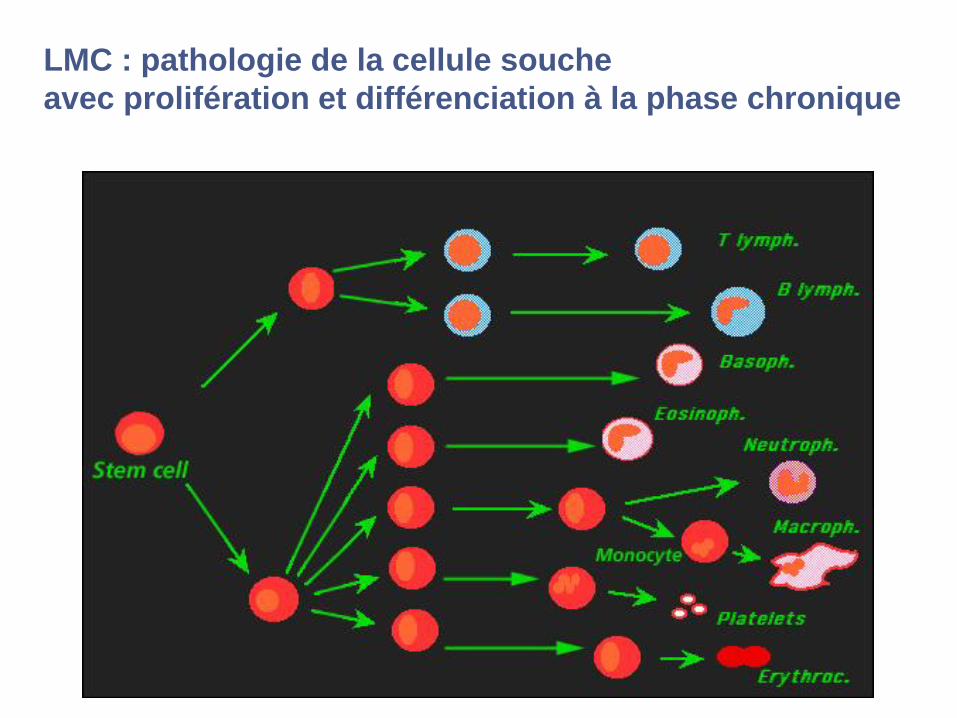
LMC : pathologie de la cellule souche
avec prolifération et différenciation à la phase chronique
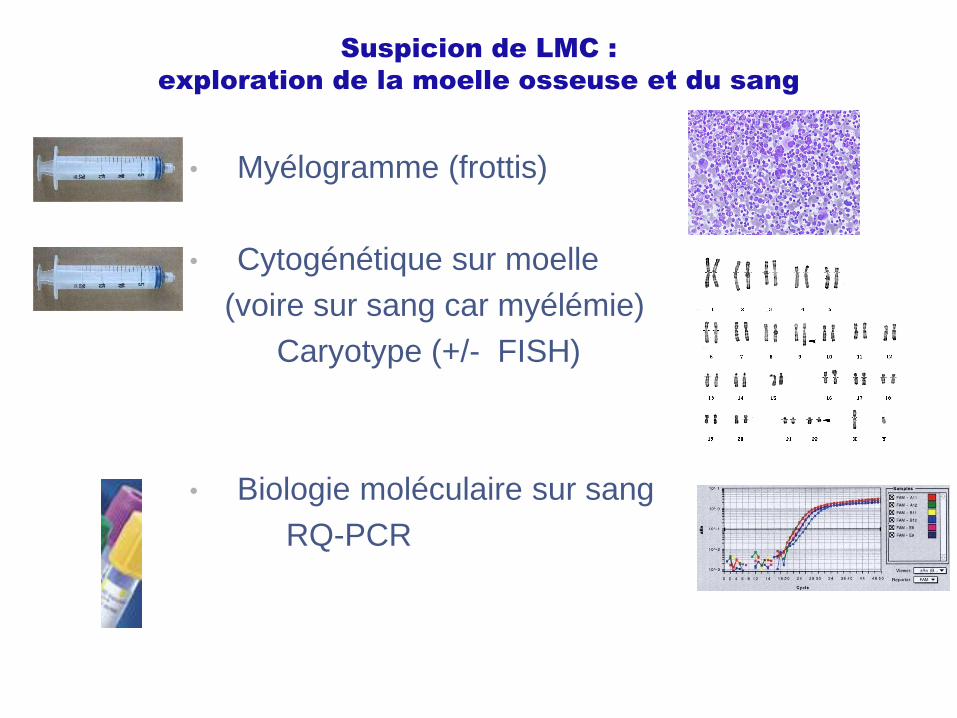
Suspicion de LMC :
exploration de la moelle osseuse et du sang
• Myélogramme (frottis)
• Cytogénétique sur moelle
(voire sur sang car myélémie)
Caryotype (+/- FISH)
• Biologie moléculaire sur sang
RQ-PCR
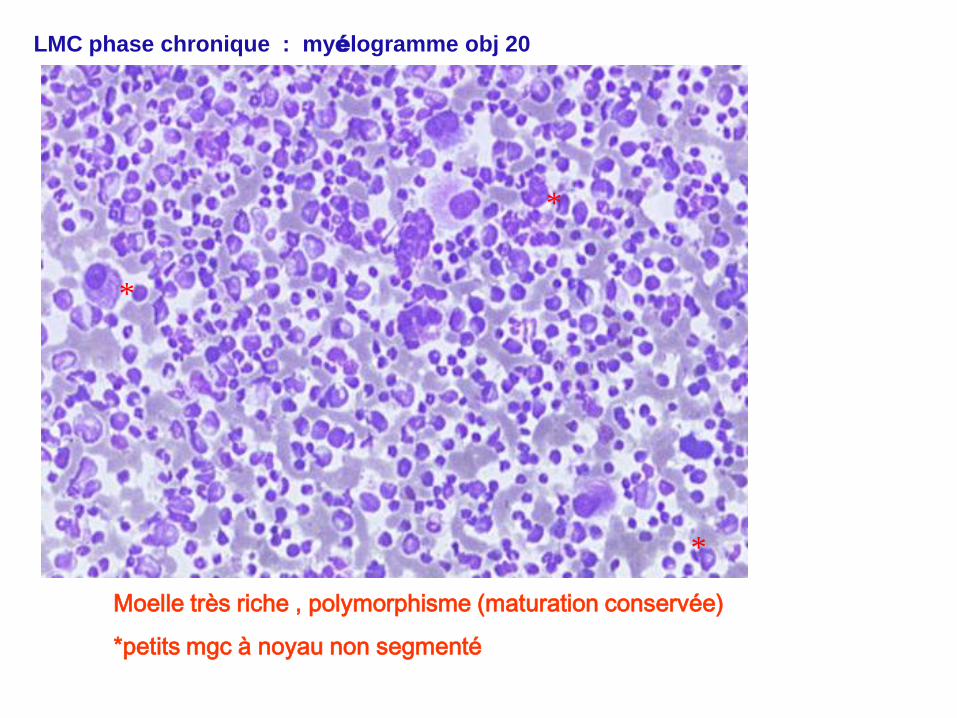
LMC phase chronique : myélogramme obj 20
Moelle très riche , polymorphisme (maturation conservée)
*petits mgc à noyau non segmenté
*
*
*

Myélogramme:
importance du % de myéloblastes
Phase chronique : myeloblastes <10%
Phase accélérée : 10%<ou = myeloblastes <20%
Phase acutisée en LAM : myeloblastes >ou =20% (TA)

LMC phase chronique : myélogramme
Richesse : très riche, en nappe
Mégacaryocytes : nombreux et de petite
taille
Lignée granuleuse : 88 %
Myéloblastes : 2 %
Promyélocytes : 3 %
Myélocytes neutrophiles : 20 %
Myélocytes éosinophiles : 3 %
Métamyélocytes neutrophiles : 27 %
Métamyélocytes éosinophiles : 3 %
Polynucléaires neutrophiles : 26 %
Polynucléaires éosinophiles : 4 %
Polynucléaires basophiles : 2 %
Lignée érythroblastique : 10%
Proerythroblastes : 0 %
Erythroblastes basophiles : 2 %
Erythroblastes polychromatophiles : 4 %
Erythroblastes acidophiles : 4 %
Monocytes: 1 %
Lymphocytes: 1%
Hyperplasie medullaire globale à prédominance granuleuse évocatrice d’un
syndrome myéloprolifératif .
Dystrophie mégacaryocytaire typique de LMC
Conclusion : LMC en phase chronique

1960
• Description du chromosome Philadelphie (Ph1 ou Ph)
•1ère anomalie chromosomique acquise caractéristique d’une tumeur
maligne
Philadelphie , 1960 : caryotype sur sang de LMC
« A minute chromosome in human chronic granulocytic leukemia »
Nowell and Hungerford, Science, 1960

Prélèvement (stérile) :
Moelle osseuse , Sang (si cellules
anormales)
Sur héparine (sans conservateur
toxique)
Transport (cellules vivantes)
rapide
température ambiante
Mise en culture
Hotte stérile
Flacon de culture
milieu de culture avec
ATB
+/- mitogène (facteurs de
croissance, …)
Caryotype hématologique : technique (1)
d’après N Nadal, CHU St Etienne

arrêt de la culture en métaphase :
synchronisation des cultures (blocage en phases S puis déblocage)
colchicine (poison du fuseau mitotique)
culture cellulaire
étuve à CO2 à 37°C
durée : 1 à 3 jours
Caryotype hématologique : technique (2)
d’après N Nadal, CHU St Etienne
Étalement des préparations
chromosomiques
Vérification au microscope à
contraste de phase : présence de
cellules en métaphase (mitoses)
Transvasement des flacons de culture pour :
Centrifugations
Choc hypotonique
Fixations
Caryotype hématologique : technique (3)
Observation des métaphases
Saisie des métaphases
puis classement des chromosomes (caryotype)
Traitement chimique des lames :
dénaturation des protéines
chromosomiques
Trypsine : bandes G
Chaleur+acide : bandes R
Coloration Giemsa
Caryotype hématologique : technique (4)
d’après N Nadal, CHU St Etienne
bandes G bandes R
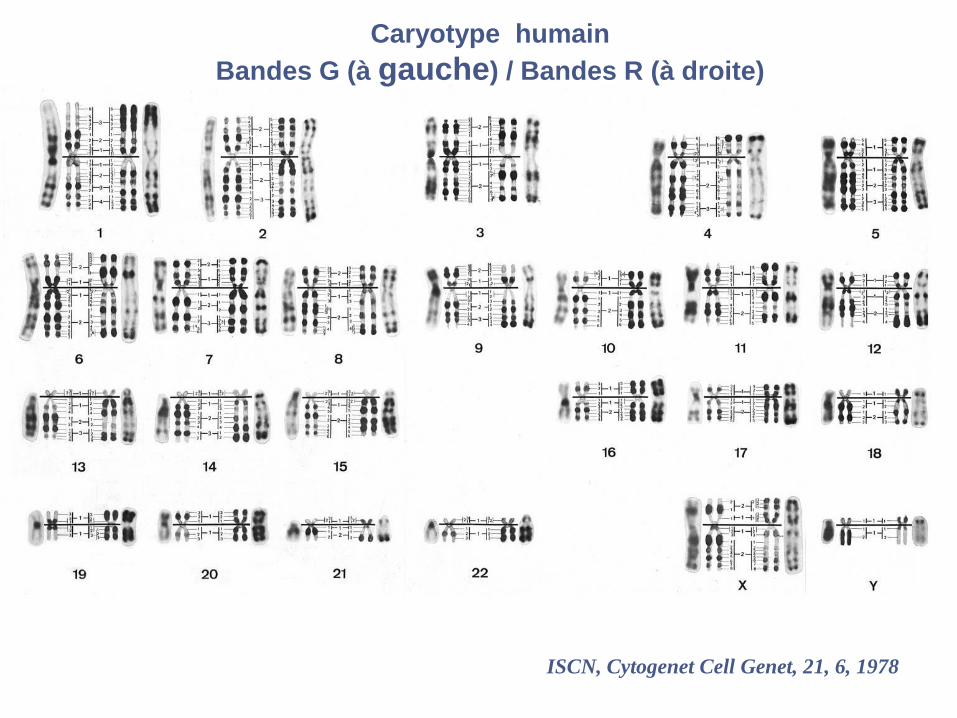
ISCN, Cytogenet Cell Genet, 21, 6, 1978
Caryotype humain
Bandes G (à gauche) / Bandes R (à droite)

1973 : Janet Rowley identifie l’anomalie Ph comme une translocation
t(9;22)(q34;q11)

CARYOTYPE
Indication : LMC au diagnostic
Caryotype sur : Moelle
Conditions de culture : 48h avec synchronisation
Marquage chromosomique: RHG
Nombre de mitoses photographiées et analysées : 20
Résultats :
Toutes les mitoses sont Philadelphie (Ph) positives sous forme de la
translocation classique, t(9;22)(q34;q11).
Il n’a pas été décelé d’anomalie clonale surajoutée.
Formule chromosomique :
46, XY, t(9;22)(q34;q11) [20]
Conclusion : LMC Ph positive
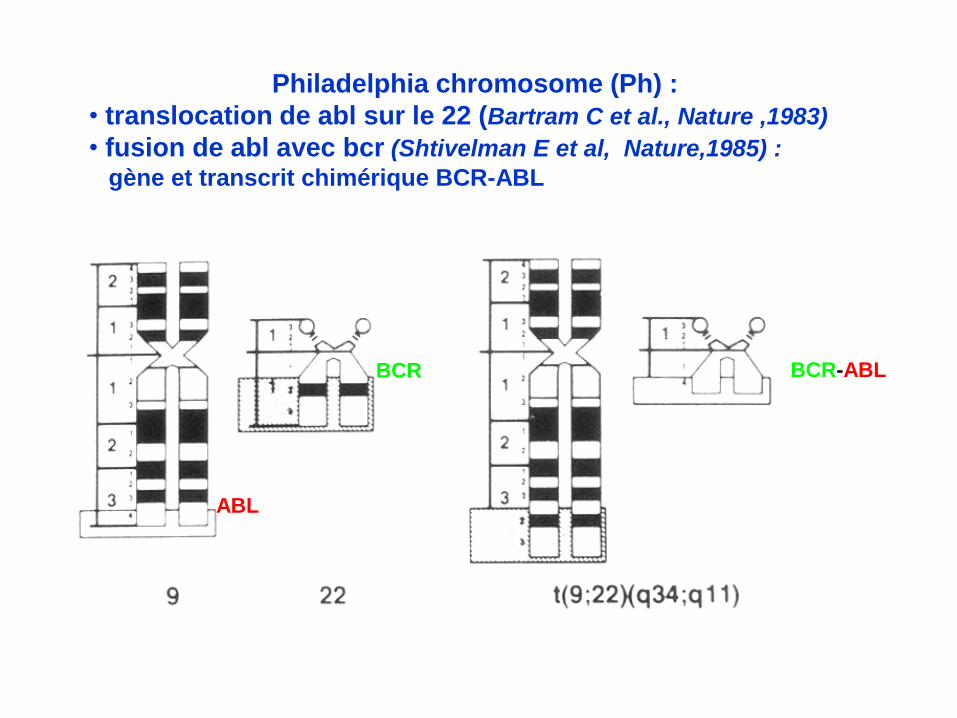
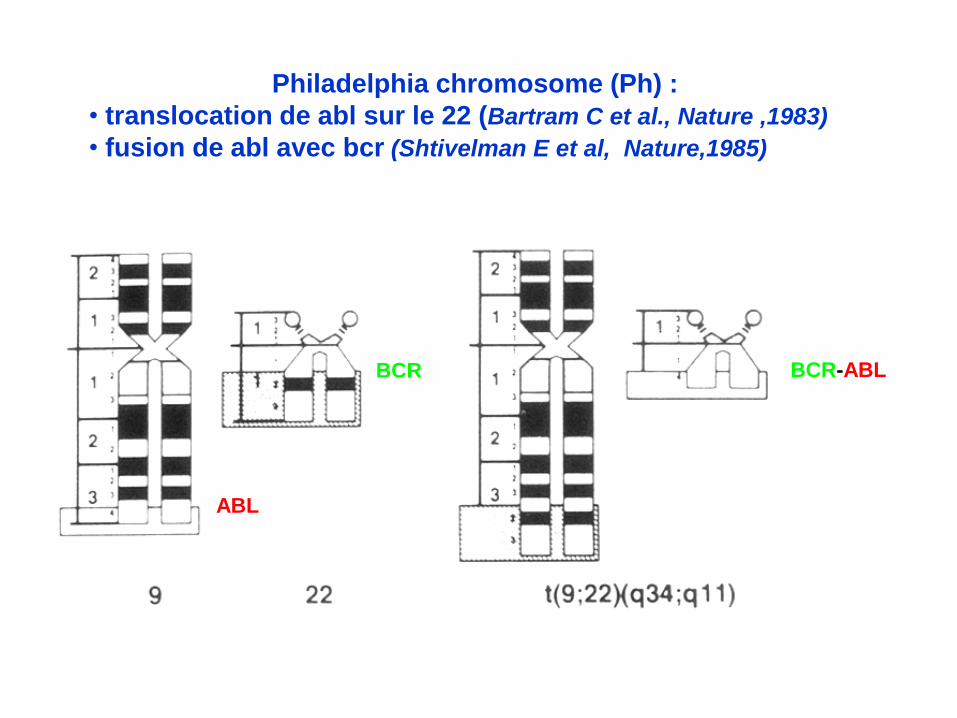
ABL
BCR BCR-ABL
Philadelphia chromosome (Ph) :
• translocation de abl sur le 22 (Bartram C et al., Nature ,1983)
• fusion de abl avec bcr (Shtivelman E et al, Nature,1985) :
gène et transcrit chimérique BCR-ABL
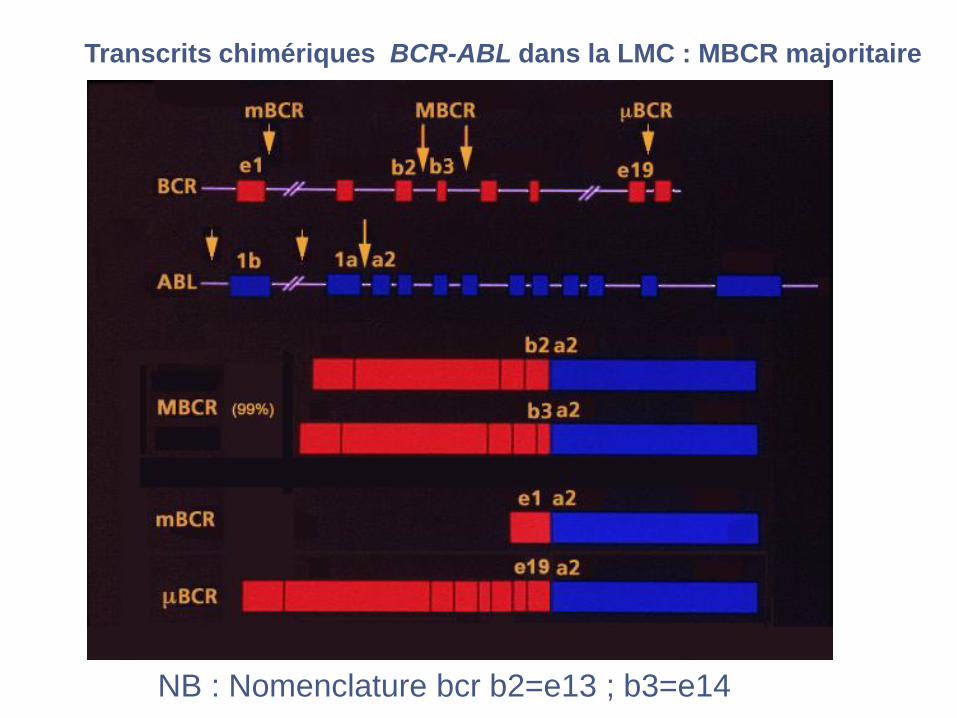
Transcrits chimériques BCR-ABL dans la LMC : MBCR majoritaire
NB : Nomenclature bcr b2=e13 ; b3=e14
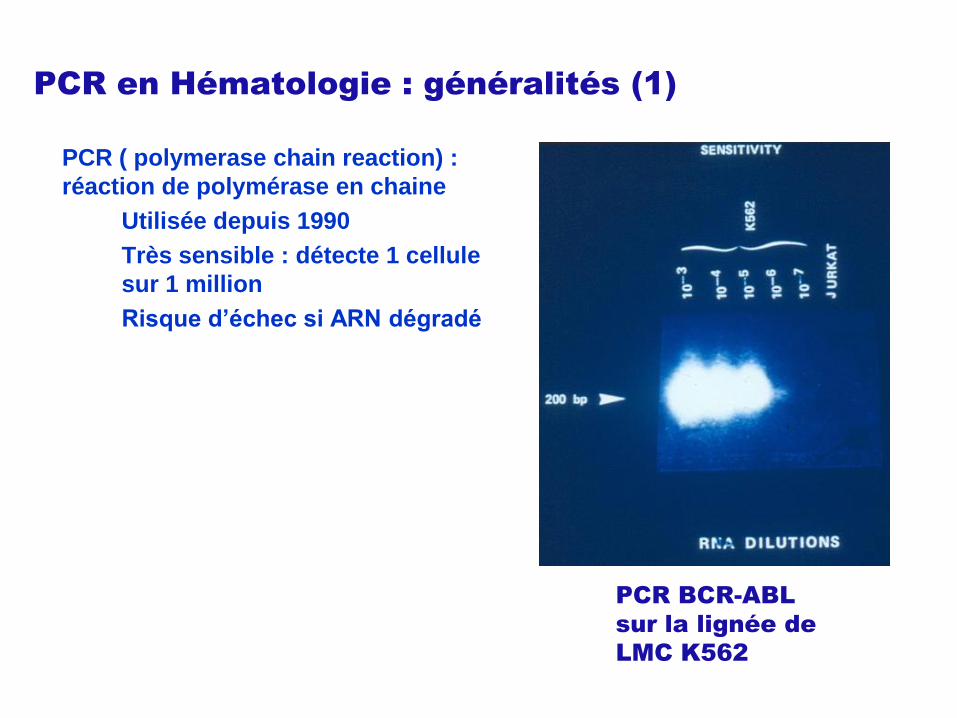
PCR en Hématologie : généralités (1)
PCR ( polymerase chain reaction) :
réaction de polymérase en chaine
Utilisée depuis 1990
Très sensible : détecte 1 cellule
sur 1 million
Risque d’échec si ARN dégradé
PCR BCR-ABL
sur la lignée de
LMC K562

Prélèvements pour PCR
Sang le plus souvent ( 1 à 2 tubes EDTA)
Moelle osseuse
Ganglion (lymphome)
Extraction de l’ADN et/ou de l’ARN à partir des cellules nucléées
1 ml de sang normal contient :
10 millions de cellules nucléées ( NFS : nbre de GB = 10G/L)
soit:
30 à 50 µg d’ADN : PCR génomique
et
1 à 10 µg ARN : RT-PCR (analyse des transcrits)
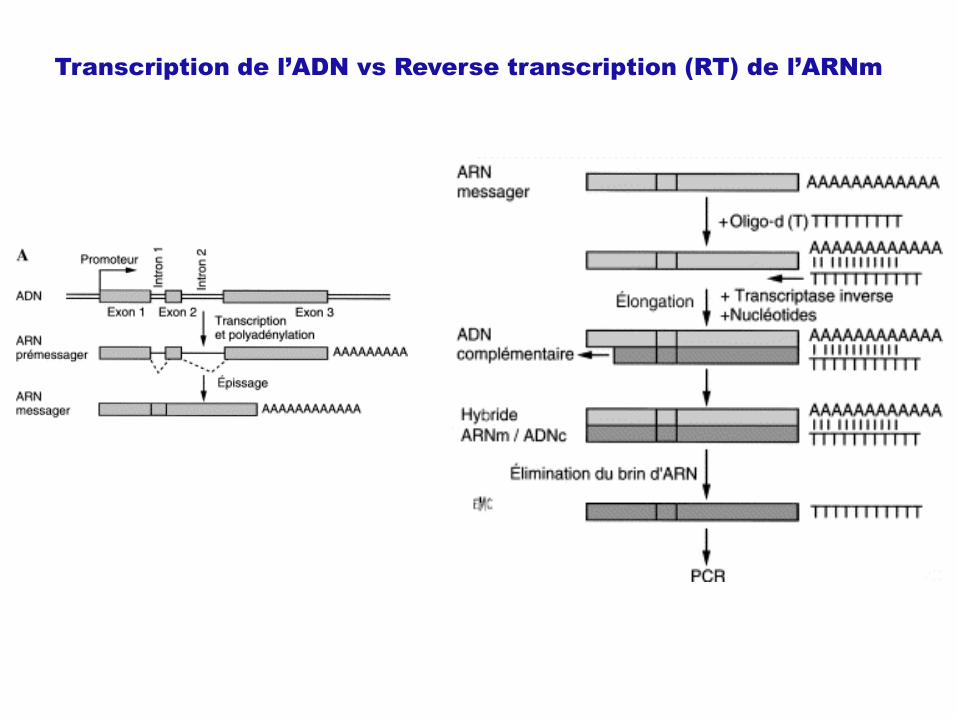
Transcription de l’ADN vs Reverse transcription (RT) de l’ARNm
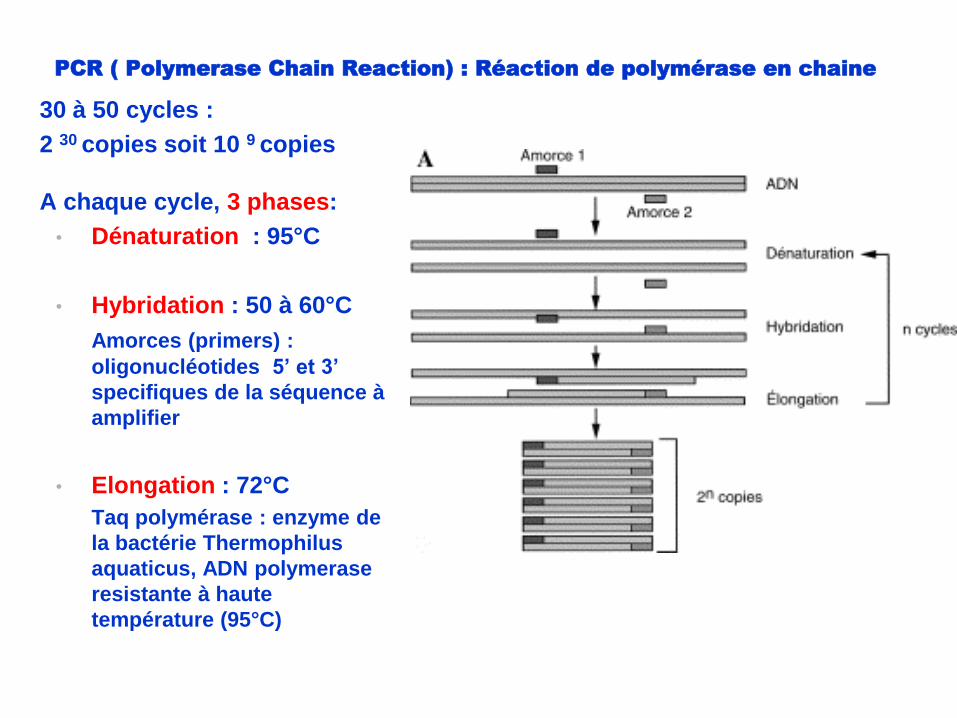
PCR ( Polymerase Chain Reaction) : Réaction de polymérase en chaine
30 à 50 cycles :
2 30 copies soit 10 9 copies
A chaque cycle, 3 phases:
• Dénaturation : 95°C
• Hybridation : 50 à 60°C
Amorces (primers) :
oligonucléotides 5’ et 3’
specifiques de la séquence à
amplifier
• Elongation : 72°C
Taq polymérase : enzyme de
la bactérie Thermophilus
aquaticus, ADN polymerase
resistante à haute
température (95°C)
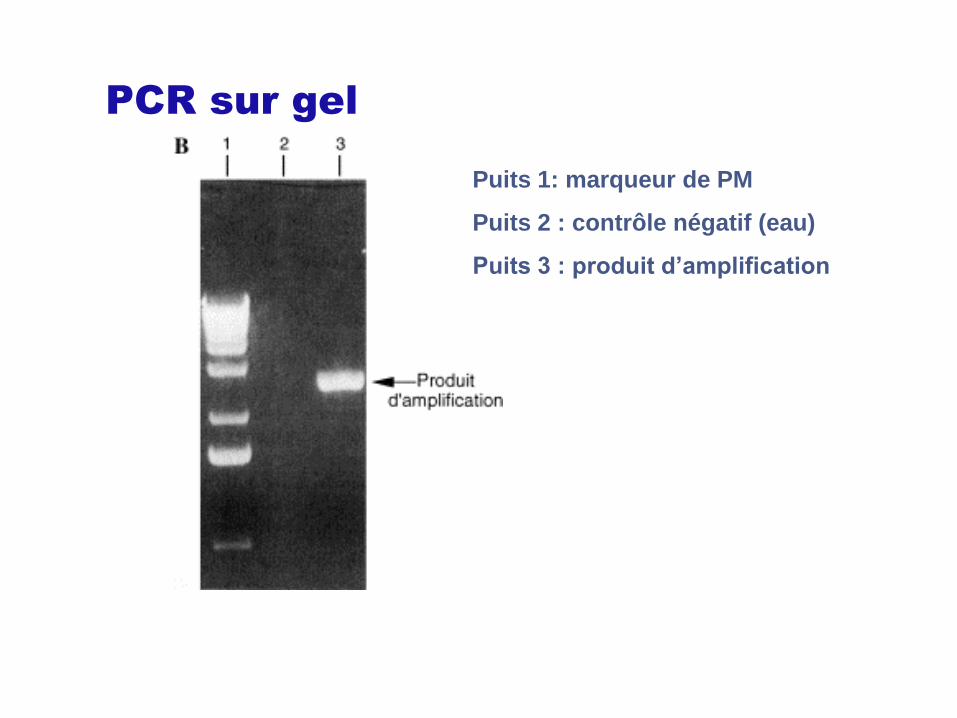
PCR sur gel
Puits 1: marqueur de PM
Puits 2 : contrôle négatif (eau)
Puits 3 : produit d’amplification

RQ-PCR en Hématologie :
RQ-PCR (real-time quantitative PCR):
Utilisée depuis 2000 en Hématologie
Très sensible : détecte 1 cellule sur 1million
Permet de quantifier et donc d’apprécier le taux
de maladie résiduelle après traitement (MRD)
Risque d’échec si ARN dégradé
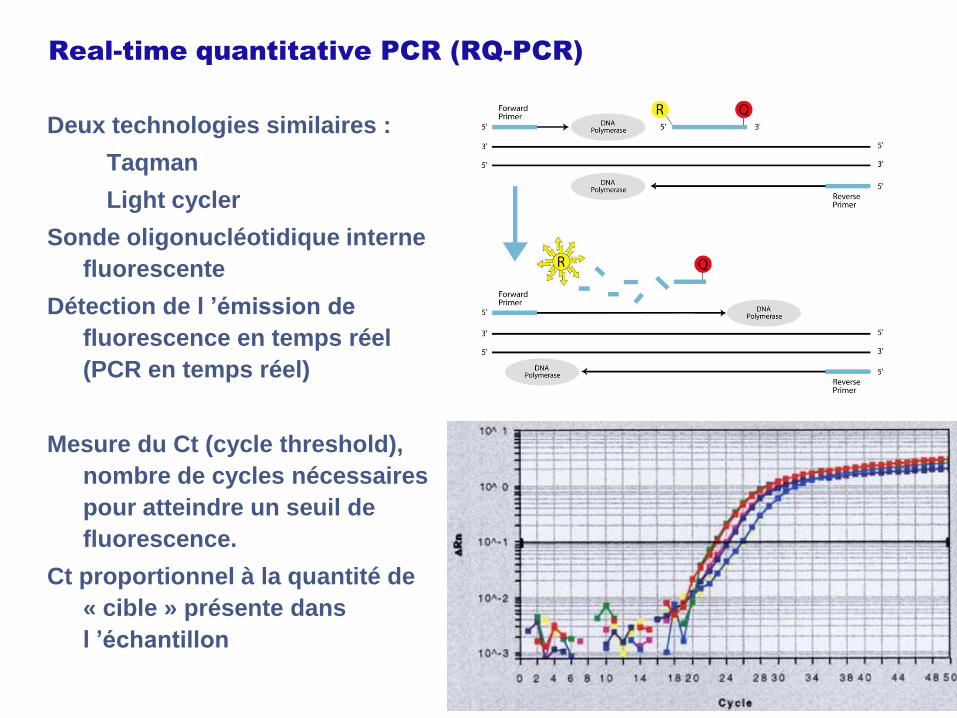
Real-time quantitative PCR (RQ-PCR)
Deux technologies similaires :
Taqman
Light cycler
Sonde oligonucléotidique interne
fluorescente
Détection de l ’émission de
fluorescence en temps réel
(PCR en temps réel)
Mesure du Ct (cycle threshold),
nombre de cycles nécessaires
pour atteindre un seuil de
fluorescence.
Ct proportionnel à la quantité de
« cible » présente dans
l ’échantillon
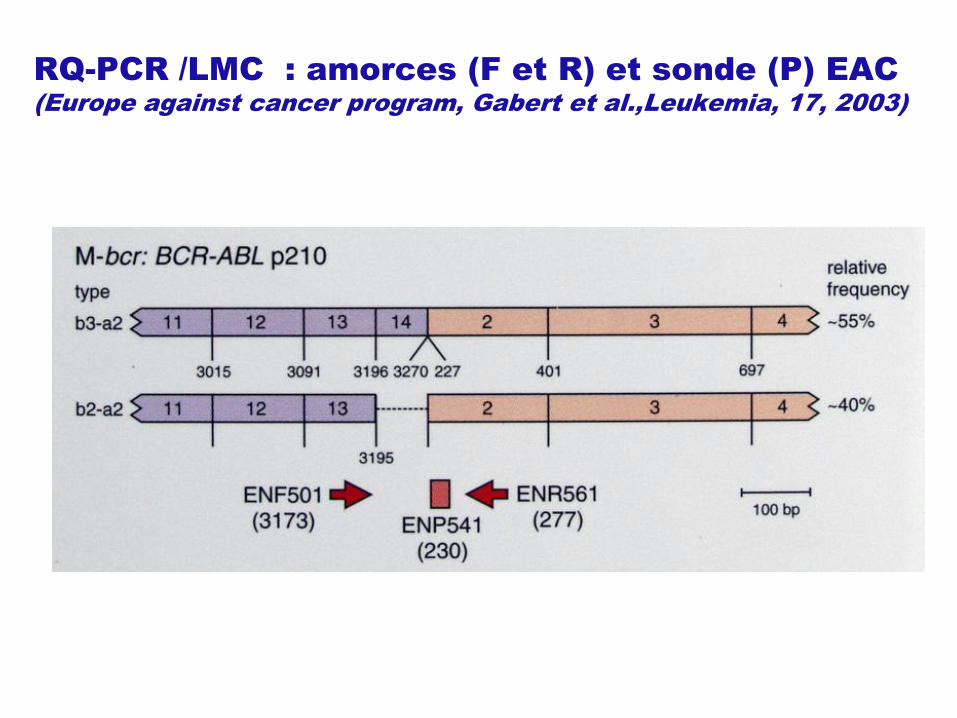
RQ-PCR /LMC : amorces (F et R) et sonde (P) EAC
(Europe against cancer program, Gabert et al.,Leukemia, 17, 2003)
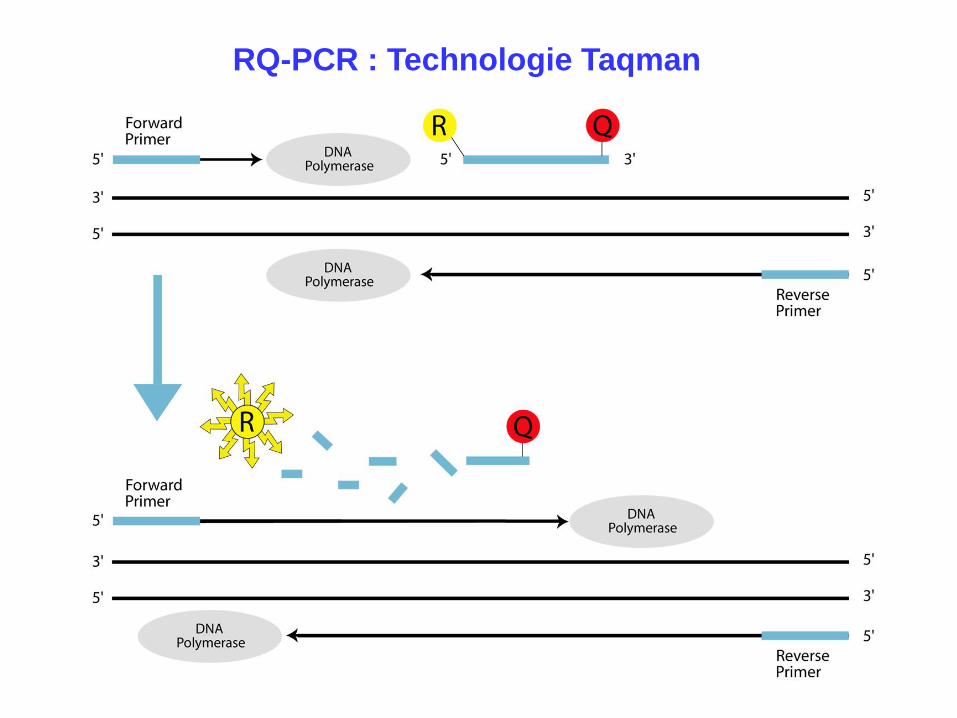
RQ-PCR : Technologie Taqman
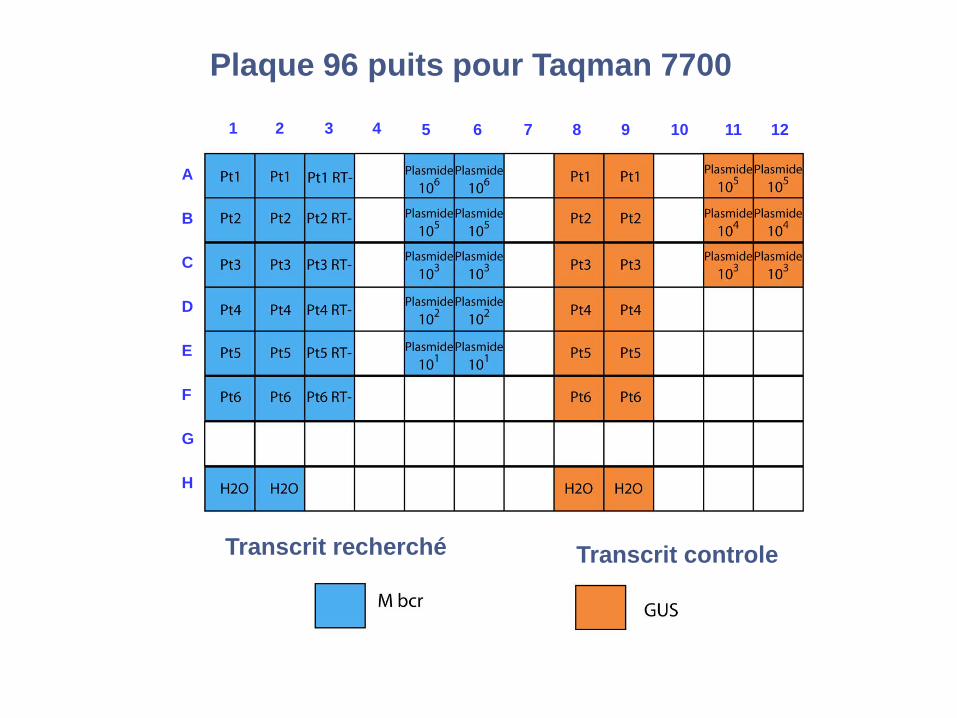
1 2 3 4 5 6 7 8 9 10 11 12
A
B
C
D
E
F
G
H
RQ-PCR Taqman : plaque 96 puitsPlaque 96 puits pour Taqman 7700
Transcrit controleTranscrit recherché
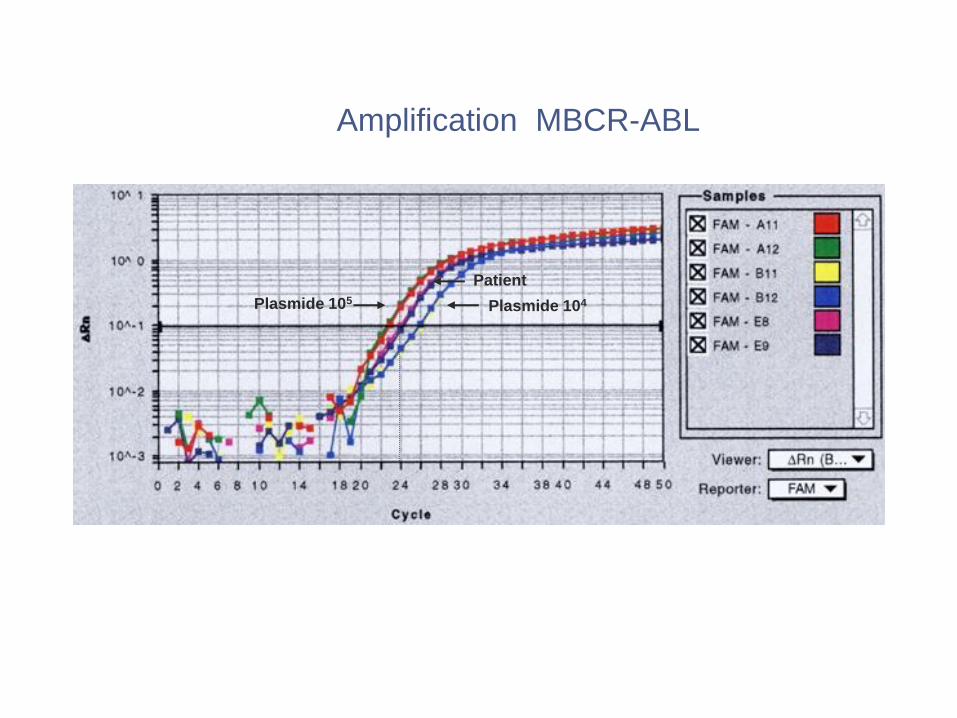
Amplification MBCR-ABL
Plasmide 105 Plasmide 104
Patient
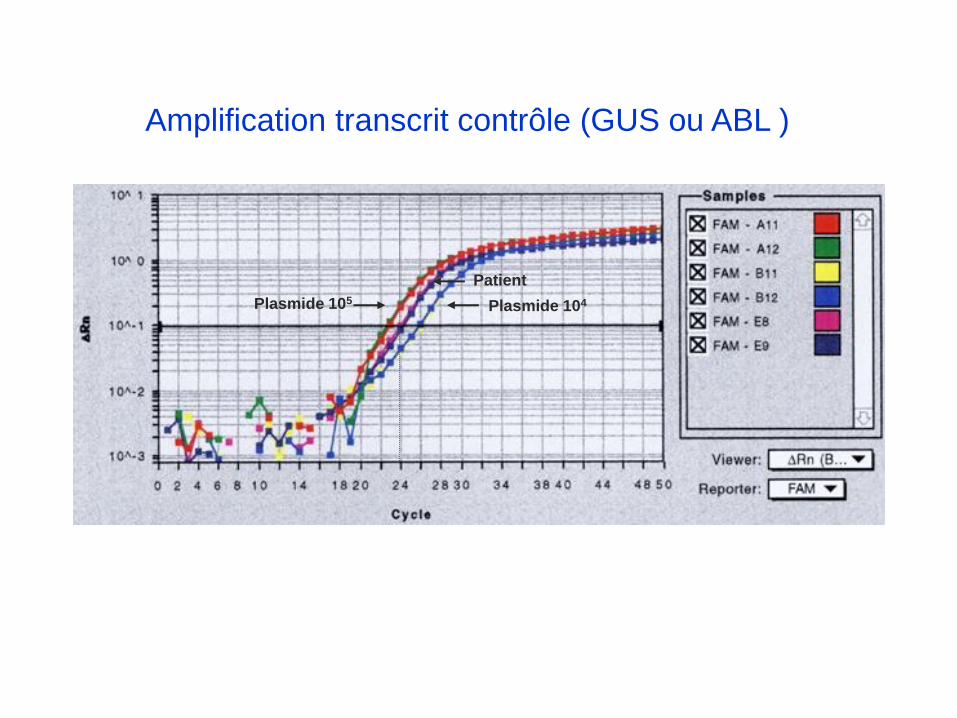
Amplification transcrit contrôle (GUS ou ABL )
Plasmide 105 Plasmide 104
Patient

RQ-PCR et LMC
Calibrateur : plasmide avec un insert Mbcr
Expression des résultats :
Rapport nombre de copies BCR-ABL / nombre
de copies du gène de contrôle : nombre de
copies normalisé (NCN)
Suivi :
NCN suivi/ NCN diagnostic
cinétique au cours de l ’évolution ++

RQ-PCR
Indication : LMC au diagnostic
Prélèvement : Sang
Résultats :
Recherche du transcrit MBCR-ABL positive.
Qualité de l’ARN : très bonne (Ct ABL: 24)
Nombre de copies du transcrit MBCR-ABL pour 104 copies du transcrit contrôle (ABL) :
7564
Valeur de référence (médiane) au diagnostic des LMC en phase chronique : 8709 (100% qui
servira de reference pour le suivi de la maladie residuelle )

Chromosome Philadelphie (Ph) et LMC au diagnostic (2)
Ph absent dans 10% des cas de LMC au diagnostic :
5% des cas : BCR-ABL positif en PCR
5% des cas : BCR-ABL négatif en PCR
Compléter le caryotype et la PCR
par FISH BCR-ABL (sur le prelevement du caryotype)

Caryotype : analyse globale du génome
• 1 caryotype hématologique : 500 bandes
• 1 bande = 5 à 10.106 pb = 5 à 10.000 kb
• Rappel :
• 1 gène : 100 à 1000 kb
• génome : 3.109 pb = 3.106 kb
Caryotype
= Cytogénétique conventionnelle
ABL
BCR BCR-ABL
Philadelphia chromosome (Ph) :
• translocation de abl sur le 22 (Bartram C et al., Nature ,1983)
• fusion de abl avec bcr (Shtivelman E et al, Nature,1985)
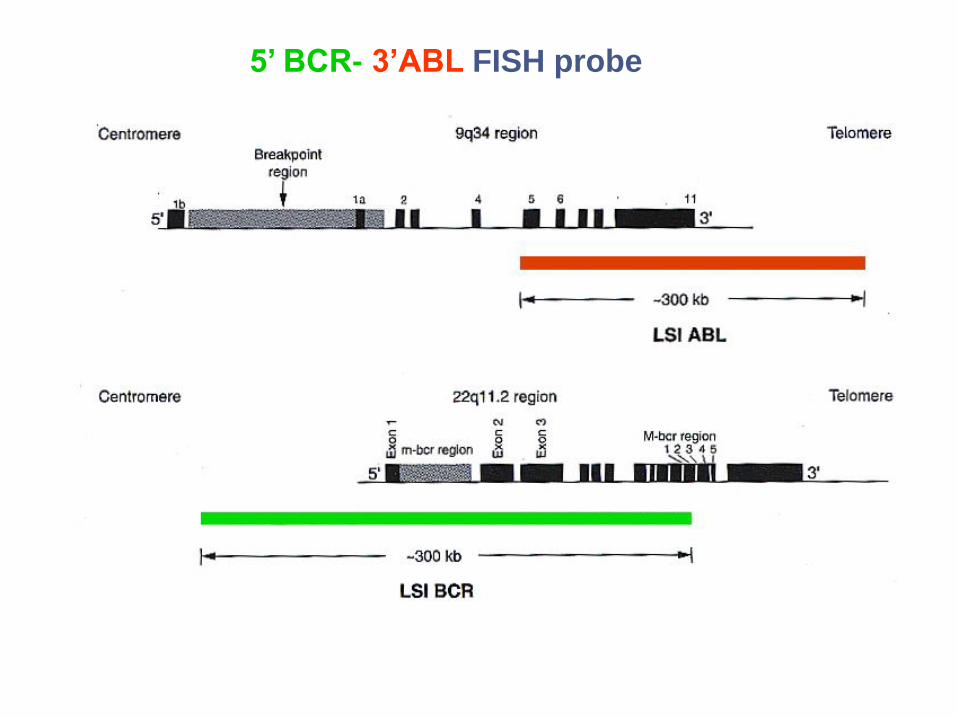
5’ BCR- 3’ABL FISH probe
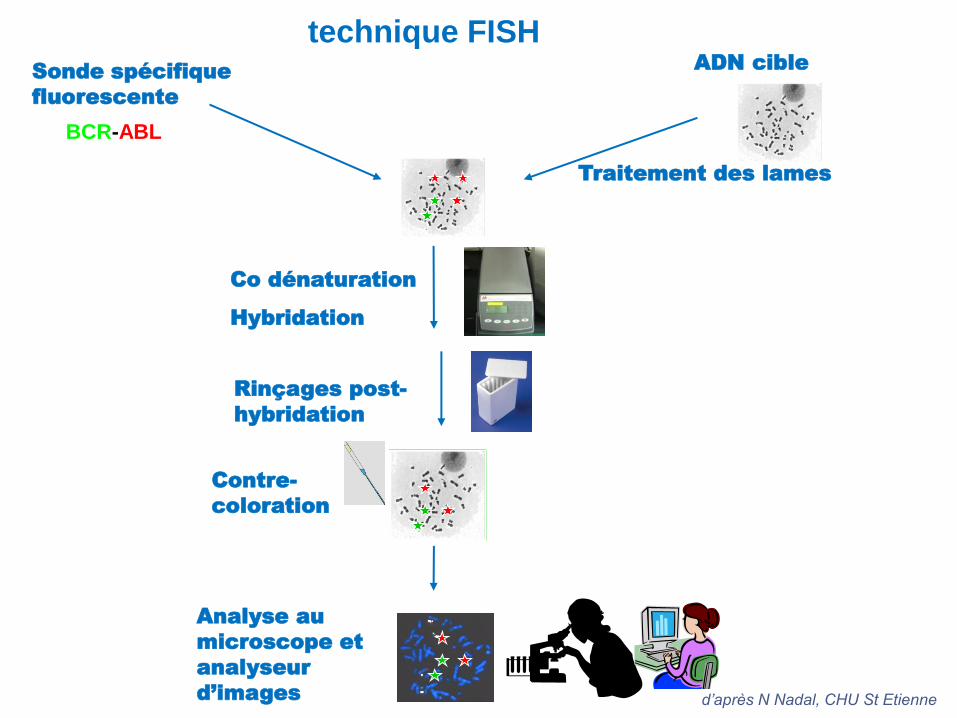
technique FISH
Traitement des lames
Sonde spécifique
fluorescente
Co dénaturation
Hybridation
Rinçages post-
hybridation
Contre-
coloration
ADN cible
BCR-ABL
Analyse au
microscope et
analyseur
d’images d’après N Nadal, CHU St Etienne
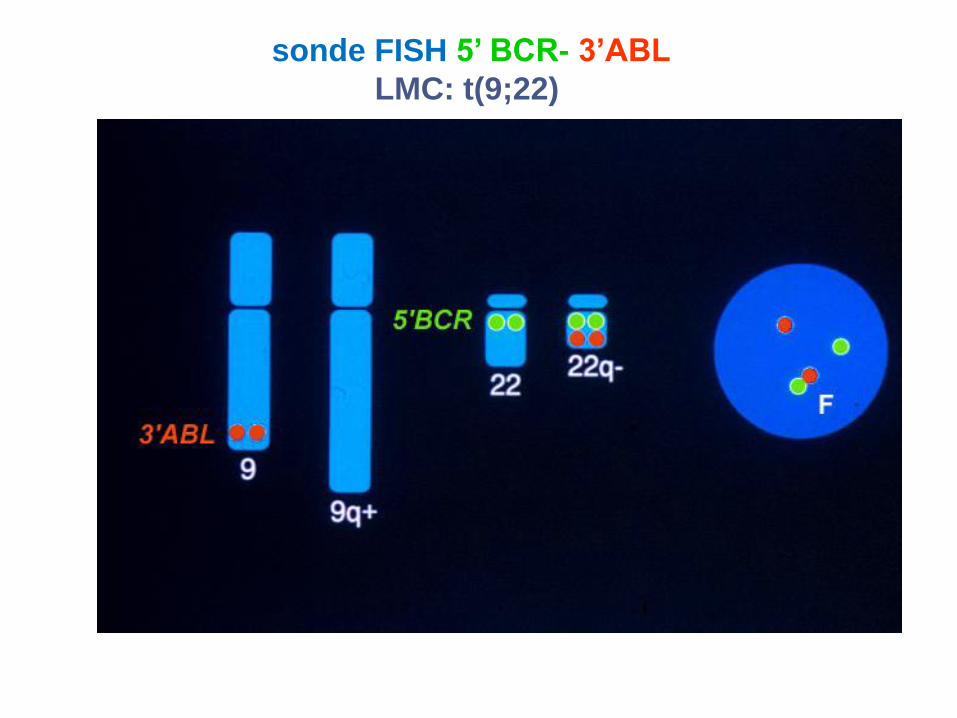
sonde FISH 5’ BCR- 3’ABL
LMC: t(9;22)
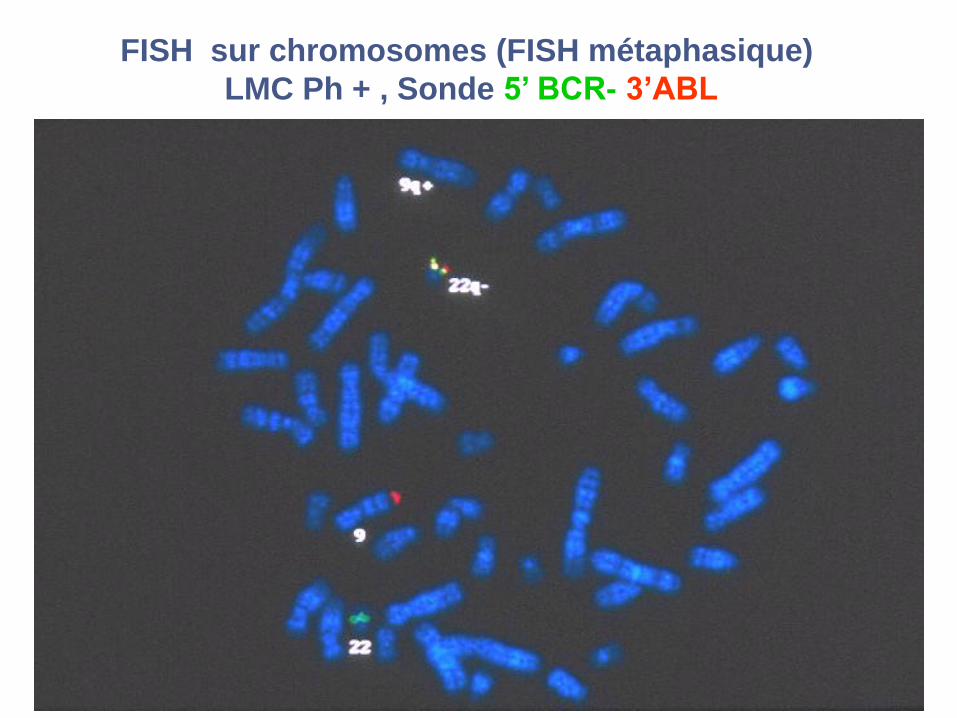
FISH sur chromosomes (FISH métaphasique)
LMC Ph + , Sonde 5’ BCR- 3’ABL
Patient LMCDonneur sain
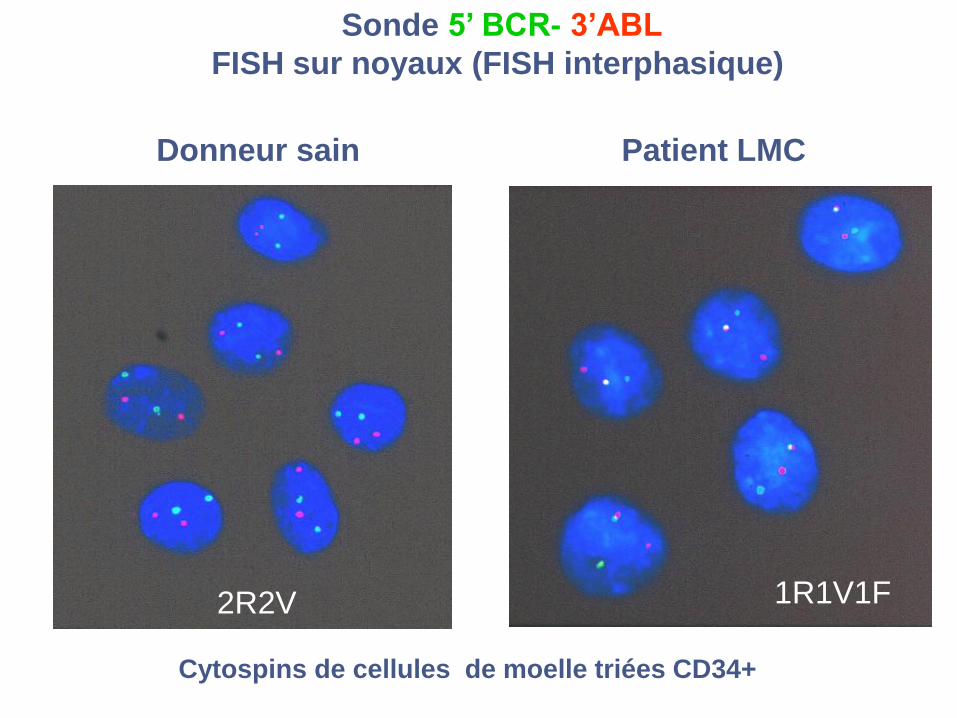
Sonde 5’ BCR- 3’ABL
FISH sur noyaux (FISH interphasique)
2R2V 1R1V1F
Cytospins de cellules de moelle triées CD34+
9
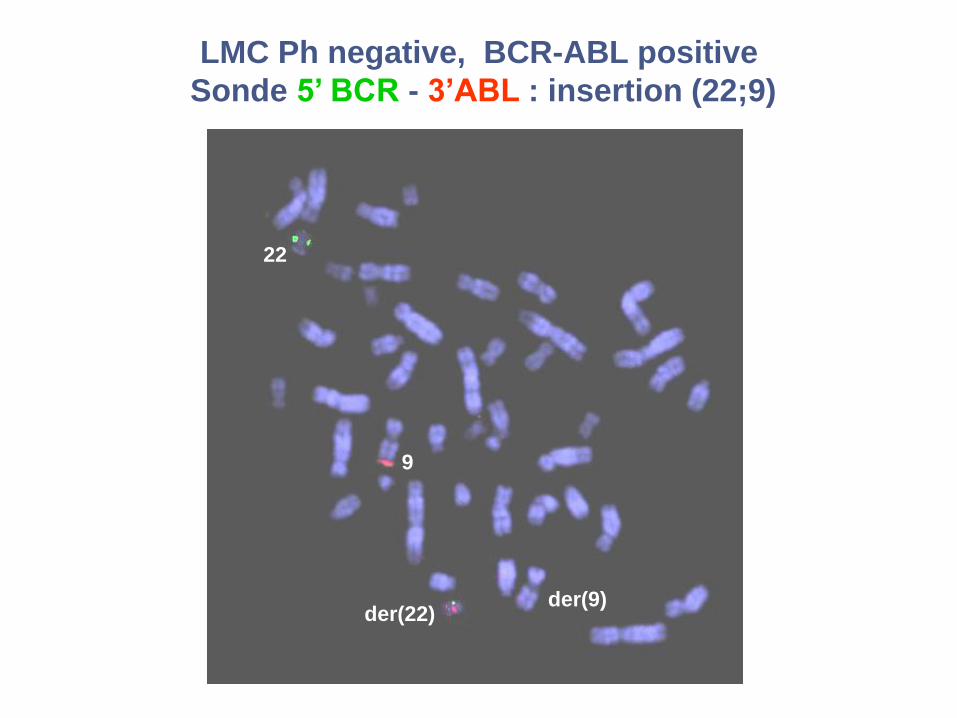
LMC Ph negative, BCR-ABL positive
Sonde 5’ BCR - 3’ABL : insertion (22;9)
22
der(9)der(22)
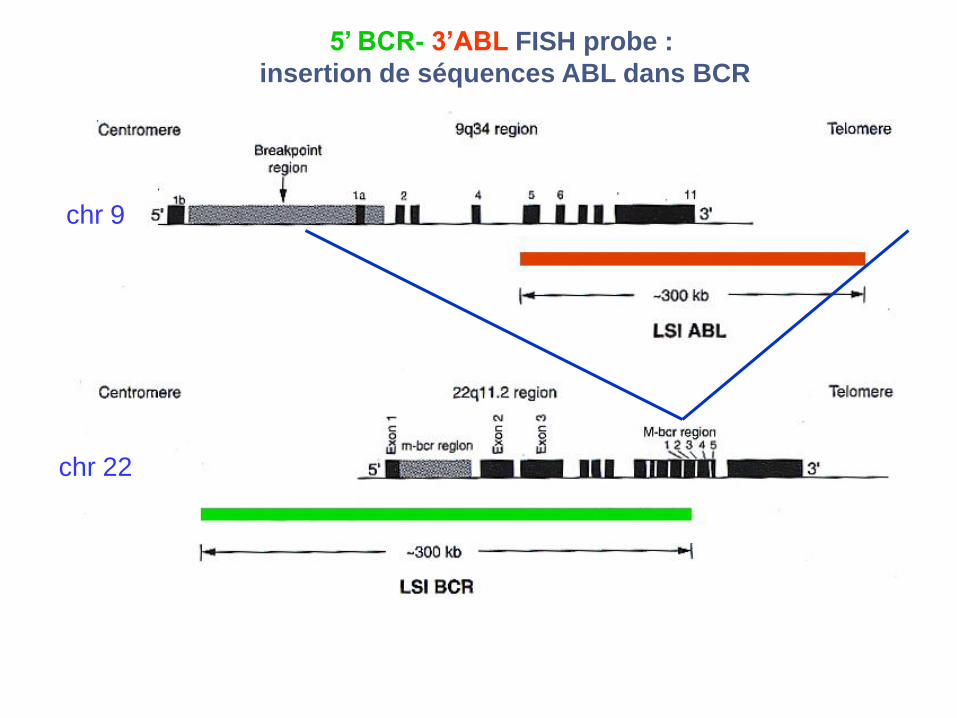
5’ BCR- 3’ABL FISH probe :
insertion de séquences ABL dans BCR
chr 9
chr 22

LMC au diagnostic : si absence de Ph au caryotype
(10% des cas)
Le plus souvent : BCR-ABL positif (en PCR et en FISH)
Ph masqué ou cryptique dû à une insertion submicroscopique
( visible uniquement en FISH ) :
insertion de 9 dans le 22
ou
insertion de 22 dans le 9
pour créer un gène chimérique BCR-ABL
répond au traitement ciblé par Imatinib
Plus rarement : BCR-ABL négatif (en PCR et en FISH)
LMC atypique, forme frontière SMP/SMD
ne répond pas au traitement ciblé par Imatinib

Syndromes myéloprolifératifs chroniques (SMP)
ou Néoplasies myéloprolifératives
Pathologies de la cellule souche hématopoiétique
Prolifération et différenciation de la lignée myéloide
Plusieurs entités (OMS) :
SMP de type LMC (Ph et/ou BCR-ABL positive)
SMP non LMC (Ph et BCR-ABL négatifs)
TE
MFP
PV
SMP /SMD (Ph et BCR-ABL négatifs)
LMMC
LMC atypiques

Classification and diagnosis of myeloproliferative neoplasms : The 2008 World Health Organization criteria and
point care diagnostic algorythms. Tefferi A and Vardiman JW, Leukemia 2008;22:14-22
Role oncogénique de BCR-ABL
Daley GQ et al,
Science,1990
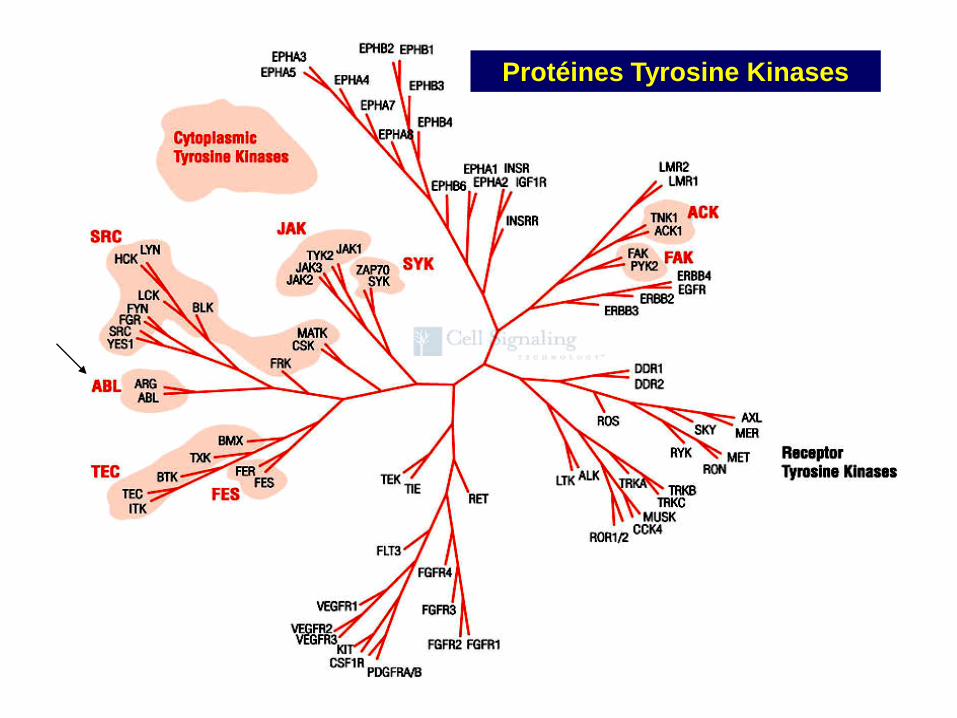
Protéines Tyrosine Kinases
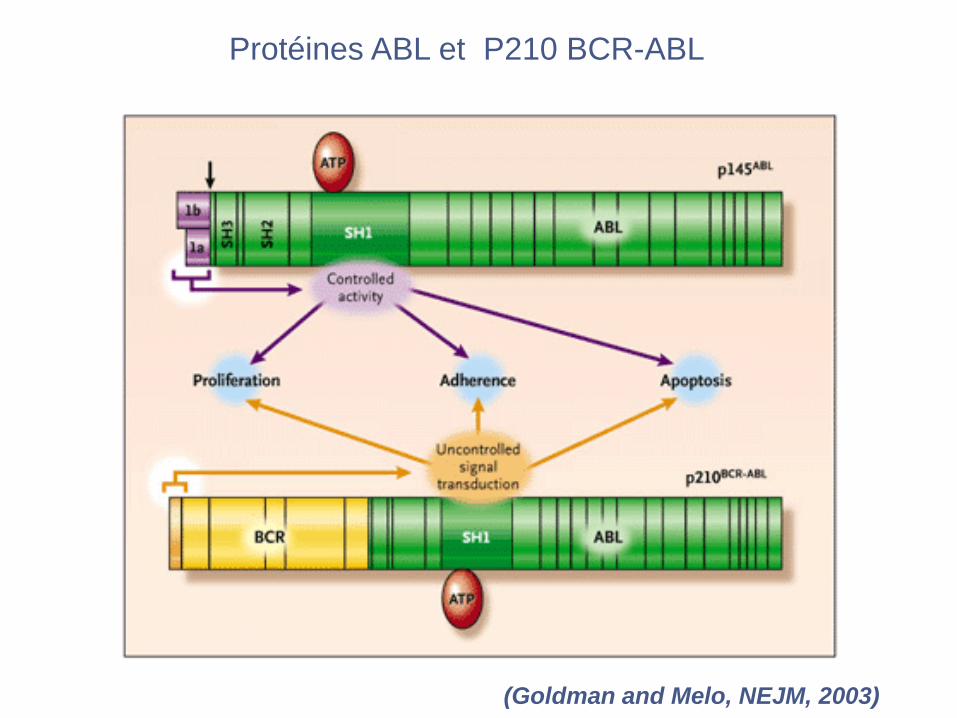
Protéines ABL et P210 BCR-ABL
(Goldman and Melo, NEJM, 2003)

(Goldman J and Melo JV, NEJM, 2003)

Traitement de la LMC en phase chronique :
Traitement Tolérance Efficacité
Hydréa (hydroxyurée) +++ +/-
Allogreffe +/- - (GVH) ++++ (guérison)
Interféron +/- ++
ATK Imatinib (Glivec) ++ +++
ATK 2ème génération ++ ++++
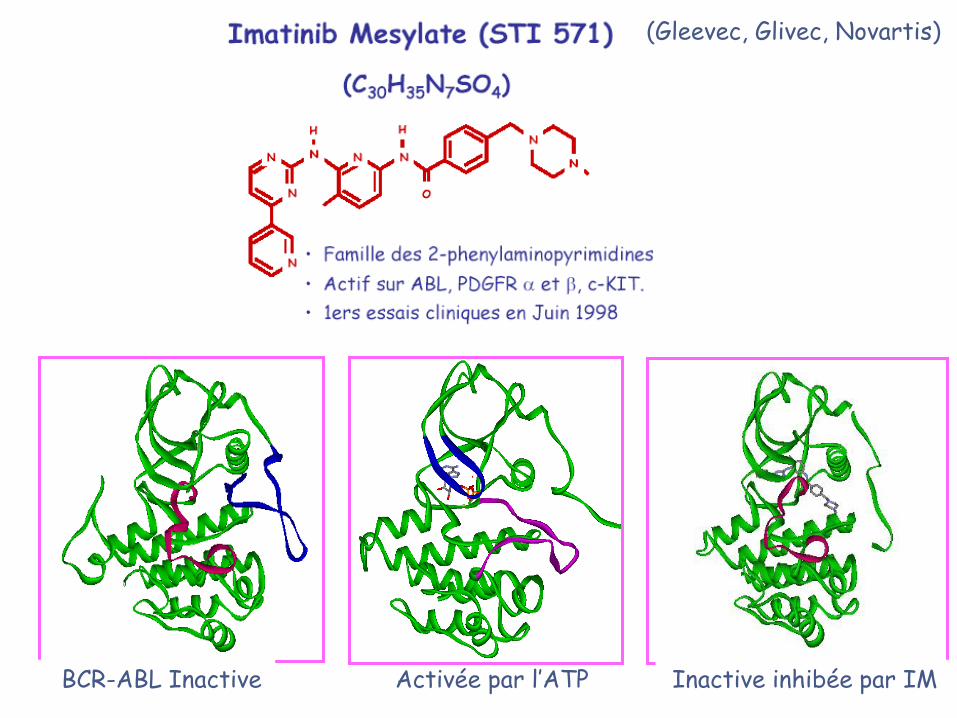
STI-
571
Inactive Active
Inactive inhibée par IMActivée par l’ATPBCR-ABL Inactive
(Gleevec, Glivec, Novartis)

Suivi des LMC par cytogénétique : critères
d ’évaluation
Kantarjian et al., NEJM, 1997 modifié en février 2002
Réponse cytogénétique majeure :
Réponse cytogénétique complète : 0% Ph
Réponse cytogénétique partielle : 1-35% Ph
Réponse cytogénétique mineure : 36-65% Ph
Réponse cytogénétique minime : 66-95% Ph
Absence de réponse cytogénétique : 96-100% Ph
NB : Caryotype médullaire avec analyse d’au moins 20
métaphases
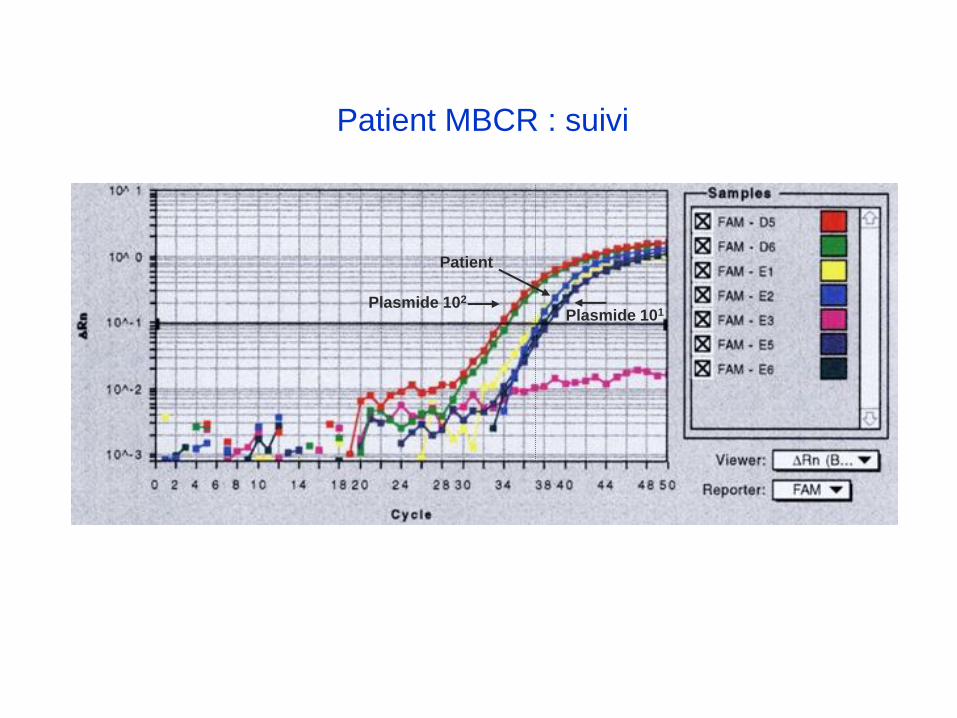
Patient MBCR : suivi
Plasmide 102
Plasmide 101
Patient

RQ-PCR MBCR-ABL
Indication : LMC Mbcr positive traitée par Glivec depuis 1 an
Prélèvement : Sang
Résultats :
Qualité de l’ARN : bonne (transcrit contrôle :ABL)
Recherche du transcrit MBCR-ABL positive.
Nombre de copies du transcrit MBCR-ABL pour 104 copies du transcrit contrôle : 4
Valeur de référence au diagnostic : 8709
Taux de maladie résiduelle (local) = 4/8709= 0.0005 =0.05%= 0.5x10-3
Facteur de conversion du laboratoire pour standardisation internationale = 0.8
Taux de maladie résiduelle (international ): TMR = 0.05% x 0.8 = 0.04%=0.4x10-3
Conclusion : Réponse moléculaire majeure (RMM car TMR <10-3 soit 0.1%)

LMC : Niveaux de maladie résiduelle
(European Leukemia Net , Baccarani et al, Blood, 2006)

Evaluation des LMC au diagnostic
Recommandations de l’ELN : Baccarani et al , JCO, dec 2009
clinique et NFS :
Age
Taille de la rate
Taux de plaquettes
Taux de blastes
Basophiles
Eosinophiles
Myelogramme :
% de myeloblastes
caryotype sur moelle :
Anomalies clonales surajoutées au Ph (CCA/Ph+)
FISH BCR-ABL :
Si Ph masqué (pour compléter le caryotype)
Si echec de caryotype
RQ-PCR sur sang

Evaluation des LMC au suivi
Recommandations de l’ELN : Baccarani et al , JCO, dec 2009
NFS : tous les 15j jusqu’à RHC : réponse hématologique complète (rate non palpable, GB<10G/L, plaq<450,pas de myelemie, basophiles < 5%)
RQ-PCR sur sang
tous les 3 mois si traitement par Glivec jusqu’à RMM (réponse moléculaire majeure)
Tous les mois dans le suivi précoce des allogreffes (6 premiers mois) , puis plus espacé (tous les ans après 2 ans post-allo)
Caryotype moelle:
1ère année à 3 mois (ELN 2010),6 mois, puis tous les 6 mois jusqu’à RCC (réponse cytogénétique complète) confirmée par un caryotype 6 mois après , puis tous les ans si pas de RQ-PCR possible
Toujours si échec de traitement (resistance 1aire ou 2aire) ou cytopénie inexpliquée
FISH BCR-ABL :
Si Ph masqué (pour compléter le caryotype)
Si echec de caryotype
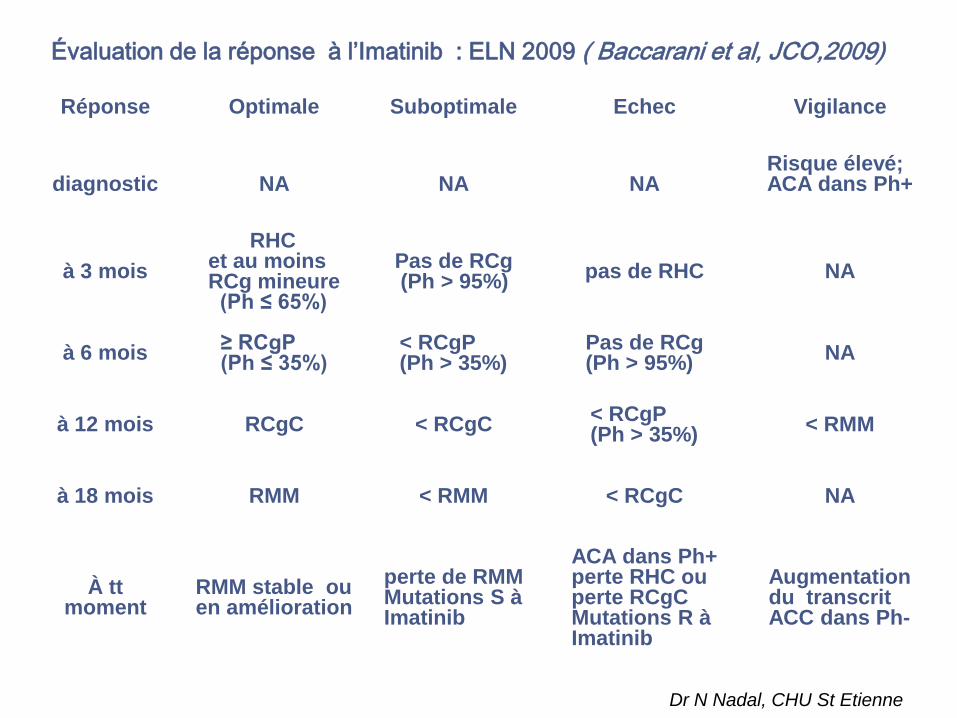
Évaluation de la réponse à l’Imatinib : ELN 2009 ( Baccarani et al, JCO,2009)
Réponse Optimale Suboptimale Echec Vigilance
diagnostic NA NA NARisque élevé;ACA dans Ph+
à 3 mois
RHCet au moins RCg mineure
(Ph ≤ 65%)
Pas de RCg(Ph > 95%)
pas de RHC NA
à 6 mois≥ RCgP (Ph ≤ 35%)
< RCgP (Ph > 35%)
Pas de RCg (Ph > 95%)
NA
à 12 mois RCgC < RCgC< RCgP (Ph > 35%)
< RMM
à 18 mois RMM < RMM < RCgC NA
À tt moment
RMM stable ou en amélioration
perte de RMMMutations S à Imatinib
ACA dans Ph+perte RHC ou perte RCgCMutations R à Imatinib
Augmentation du transcritACC dans Ph-
Dr N Nadal, CHU St Etienne

Leucémie Myéloïde Chronique (LMC)
Evolution naturelle en 3 phases :
Phase chronique
Phase d’accélération
Phase aiguë (crise blastique ou phase
blastique) :
leucémie aigue myéloïde (2/3 cas)
leucémie aigue lymphoïde (1/3 cas) : type B
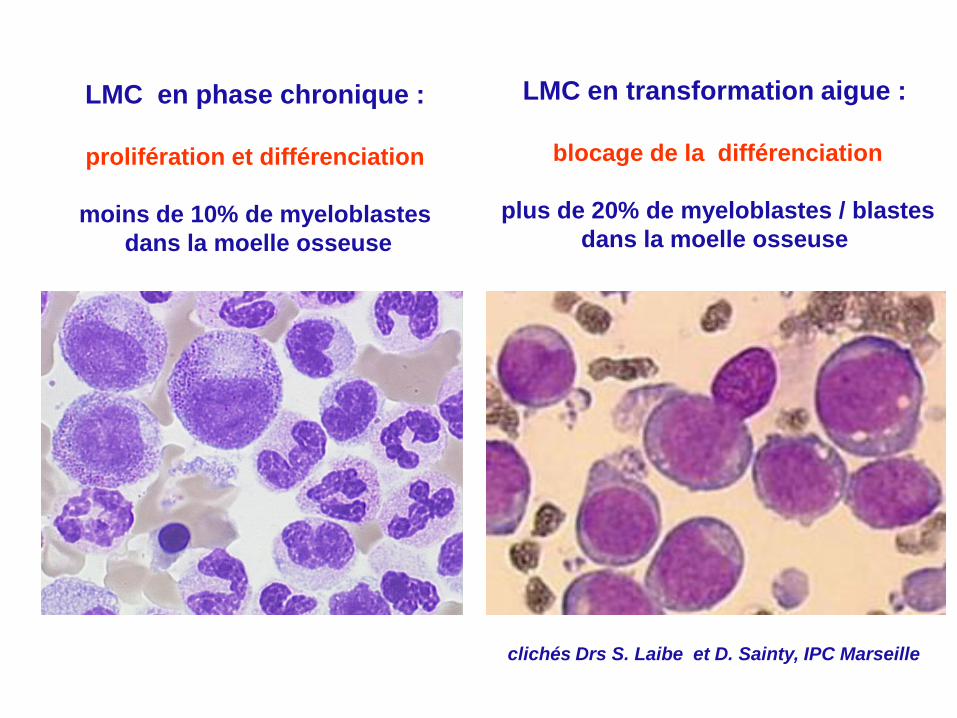
LMC en phase chronique :
prolifération et différenciation
moins de 10% de myeloblastes
dans la moelle osseuse
clichés Drs S. Laibe et D. Sainty, IPC Marseille
LMC en transformation aigue :
blocage de la différenciation
Moelle osseuse (myelogramme) :
plus de 20% de myeloblastes / blastes
dans la moelle osseuse
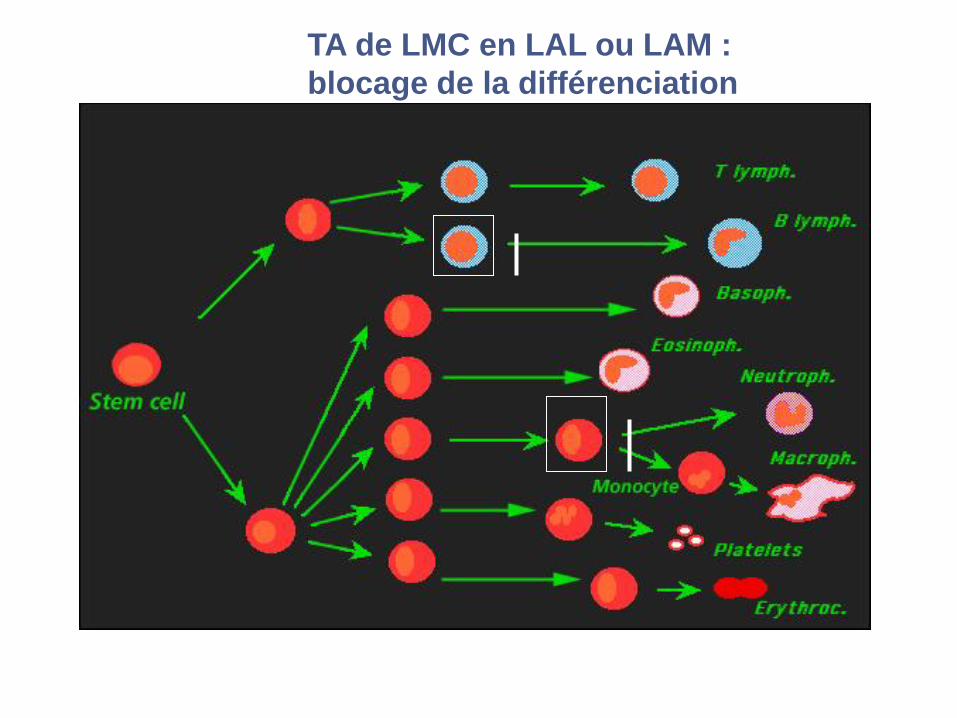
TA de LMC en LAL ou LAM :
blocage de la différenciation

Leucémie Myéloïde Chronique (LMC) et caryotype
Phase chronique :
Ph isolé
Phase d’accélération et phase d’acutisation (LA):
Ph et anomalies chromosomiques clonales
additionnelles (ACA)
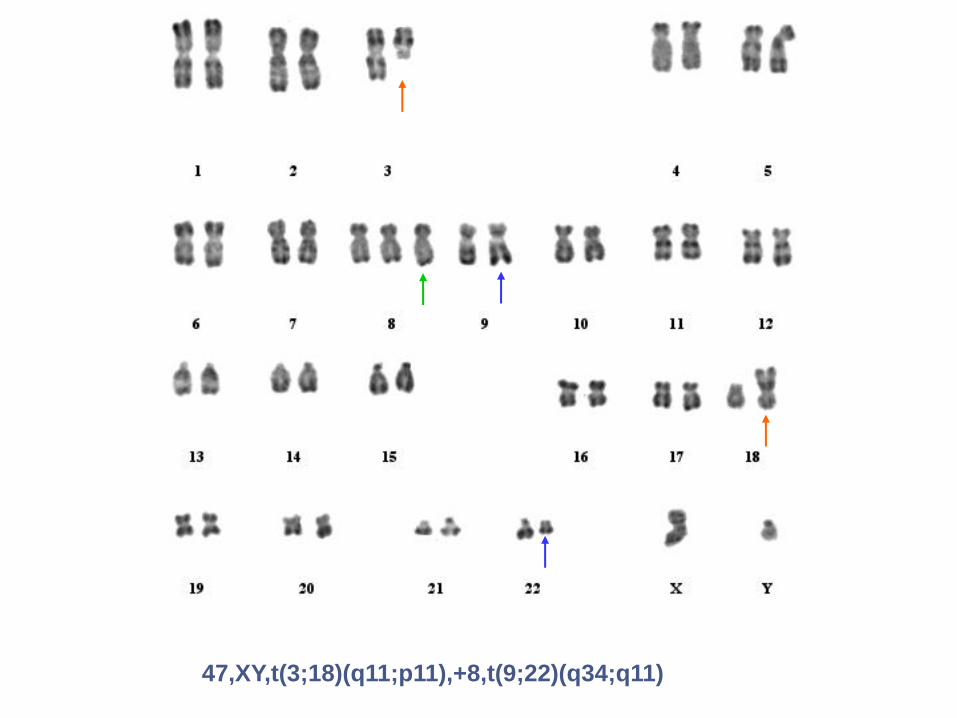
47,XY,t(3;18)(q11;p11),+8,t(9;22)(q34;q11)
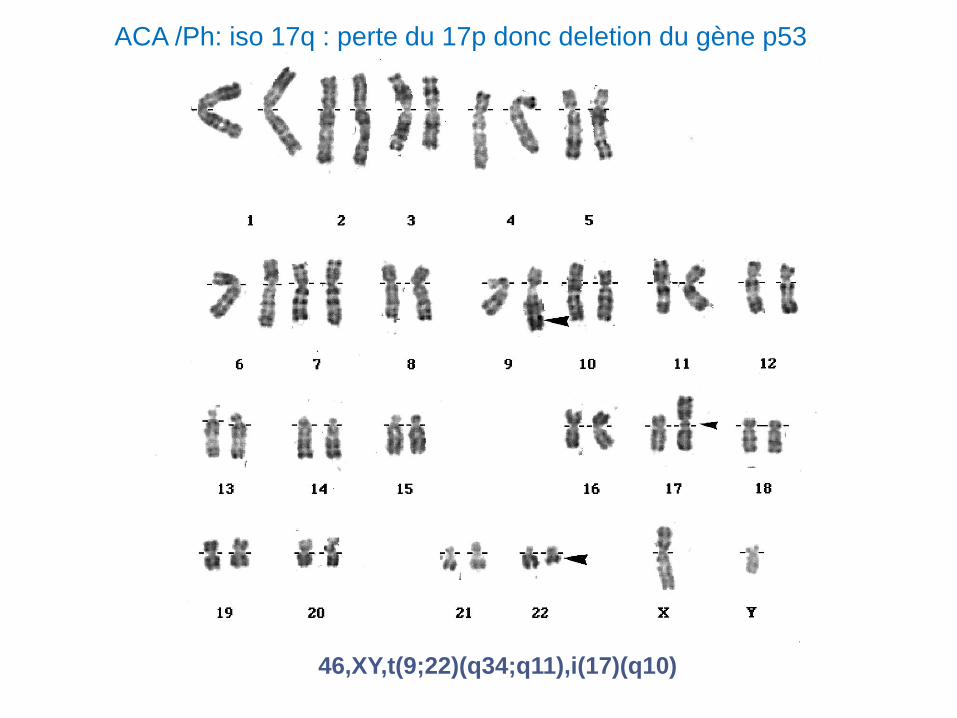
46,XY,t(9;22)(q34;q11),i(17)(q10)
ACA /Ph: iso 17q : perte du 17p donc deletion du gène p53

46,XY,t(3;21)(q26;q22),t(9;22)(q34;q11)
ACA /Ph: translocation (3;21) créant un géne chimérique AML1-EVI1

Anomalies chromosomiques clonales surajoutées au Ph
Anomalies de nombre :
trisomie 8
trisomie19
Anomalies de structure :
Isochromosome 17q : i(17)(q10) ou idic(17)(p11)
isoPh : ider(22)t(9;22)(q34;q11)
t(3;21)(q26;q22) : evi1-aml1
Anomalie de nombre et de structure :
duplication du Ph : +der(22)t(9;22)
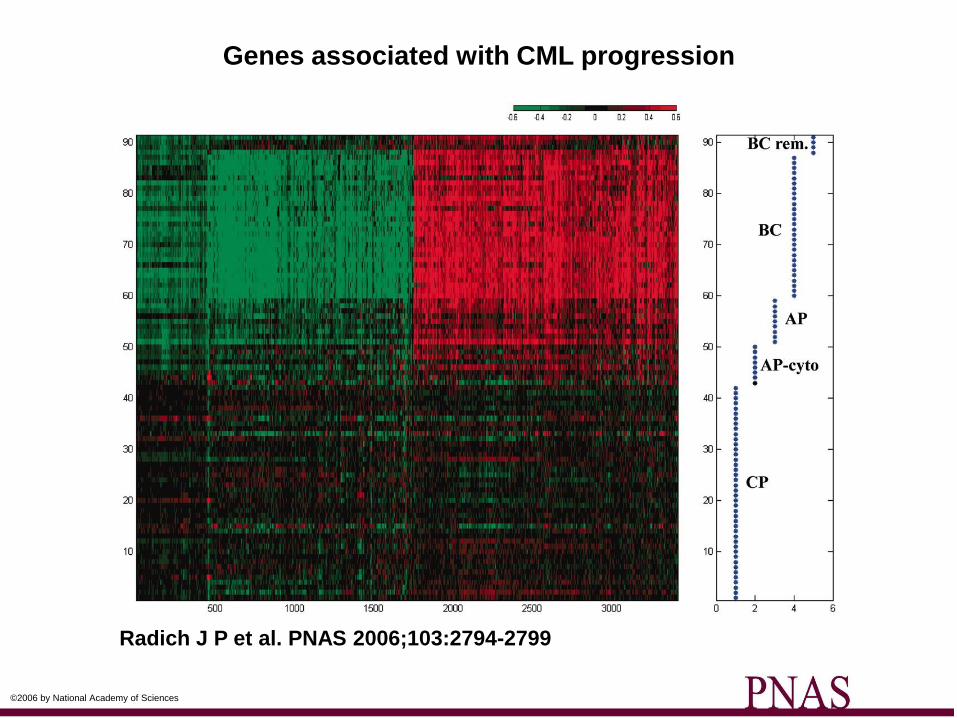
Genes associated with CML progression
Radich J P et al. PNAS 2006;103:2794-2799
©2006 by National Academy of Sciences
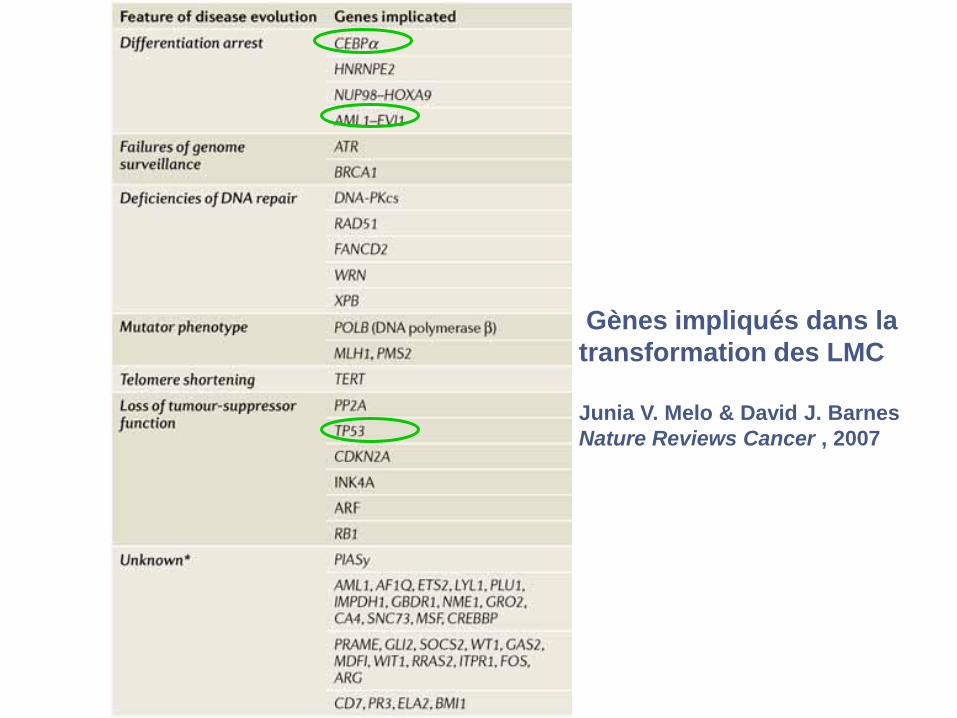
Gènes impliqués dans la
transformation des LMC
Junia V. Melo & David J. Barnes
Nature Reviews Cancer , 2007
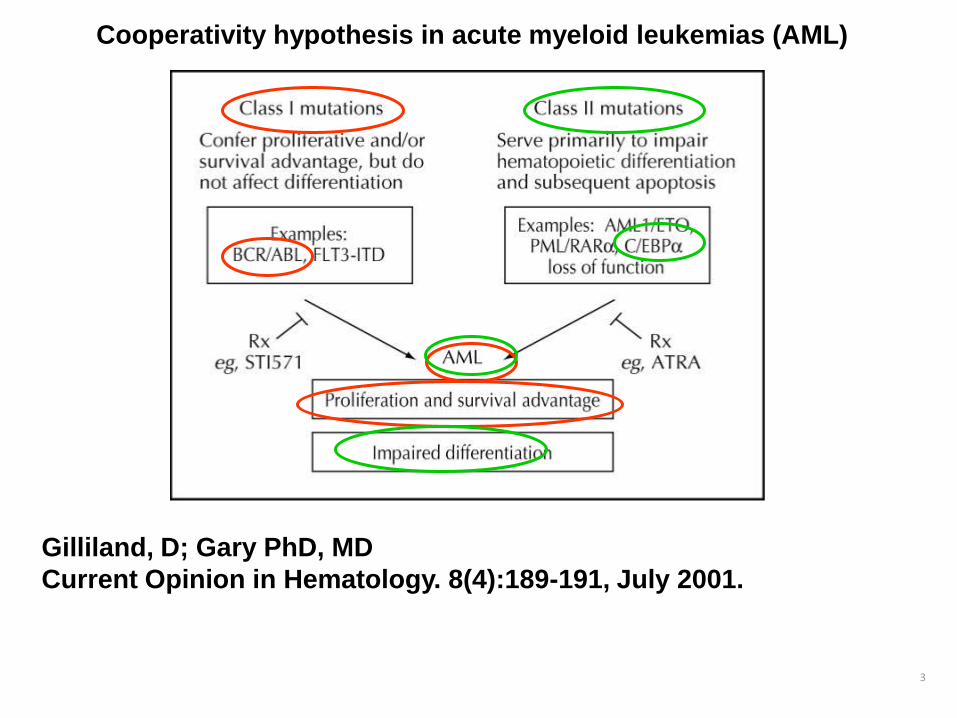
3
Gilliland, D; Gary PhD, MD
Current Opinion in Hematology. 8(4):189-191, July 2001.
Cooperativity hypothesis in acute myeloid leukemias (AML)
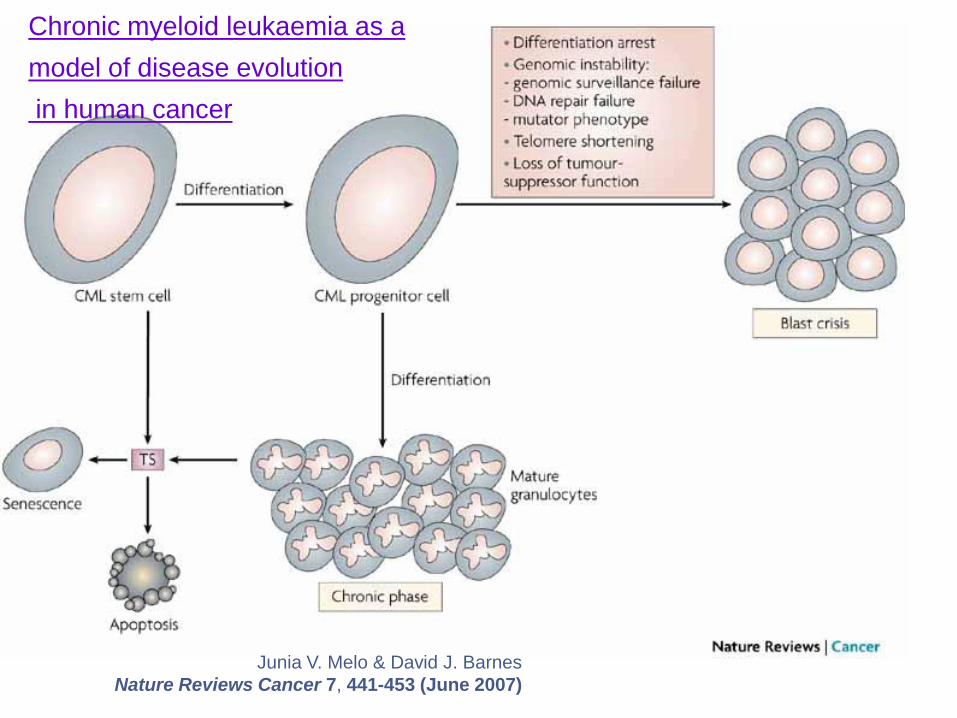
Junia V. Melo & David J. Barnes
Nature Reviews Cancer 7, 441-453 (June 2007)
Chronic myeloid leukaemia as a
model of disease evolution
in human cancer

Caractéristiques de la Leucémie Myéloïde Chronique (LMC)
Hémopathie maligne
Pathologie de la souche hématopoiétique
Syndrome myéloprolifératif chronique (SMP)
ou néoplasies myéloprolifératives de l’OMS
Présence du chromosome Philadelphie (Ph)
Présence du transcrit BCR-ABL
Traitement ciblé anti-tyrosine kinase (ATK)

Conclusion : Historique de LMC : avancées tous les 10 ans
1960 : Nowell et Hungerford, chercheurs à Philadelphie mettent en évidence la même anomalie chromosomique dans toutes les cellules de plusieurs cas de leucémie myéloïde chronique (LMC) : le chromosome Philadelphie (Ph)
1973 : Janet Rowley identifie l’anomalie Ph comme une t(9;22)(q34;q11)
1985 : Traitement de la LMC par alpha-interféron : surveillance cytogénétique
1990 : Clonage de la t(9;22) : fusion des gènes BCR et ABL : diagnostic moléculaire et mise en évidence de l’activité oncogénique de BCR-ABL
2000 : Traitement par Imatinib (Glivec), inhibiteur de l’activité tyrosine-kinase de BCR-ABL :surveillance cytogénétique et moléculaire (PCR quantitative)
2005 : ATK de 2ème génération (Dasatinib et Nilotinib) chez les patients resistants au Glivec très actifs (sauf si mutation T315 : allogreffe proposée ).


Explorations complémentaires dans le cadre
d’une hémopathie maligne
2ème exemple d’hémopathie maligne :
Leucémie aigüe lymphoblastique (LAL)
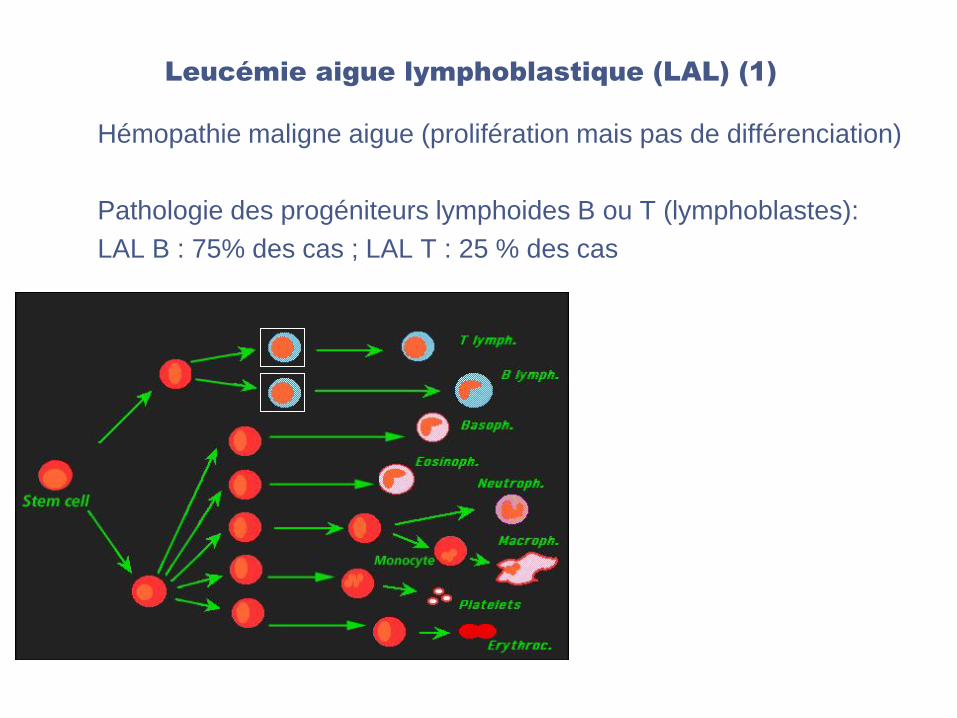
Leucémie aigue lymphoblastique (LAL) (1)
Hémopathie maligne aigue (prolifération mais pas de différenciation)
Pathologie des progéniteurs lymphoides B ou T (lymphoblastes):
LAL B : 75% des cas ; LAL T : 25 % des cas

Leucémie aigue lymphoblastique (LAL) (2)
Rare mais la plus fréquente des néoplasies malignes
de l’enfant
Urgence diagnostique et thérapeutique
Thérapeutique adaptée dépend des explorations
complémentaire

Suspicion d’hémopathie maligne aigue :
signes cliniques et biologiques
Signes d’insuffisance médullaire :
Anémie
Syndrome infectieux
Syndrome hémorragique
Signes tumoraux :
douleurs osseuses
splénomégalie
adénomégalie
hépatomégalie
Hémogramme urgent
Anémie (arégénérative)
Neutropénie
Thrombopénie
Hyperleucocytose
( si blastes circulants )
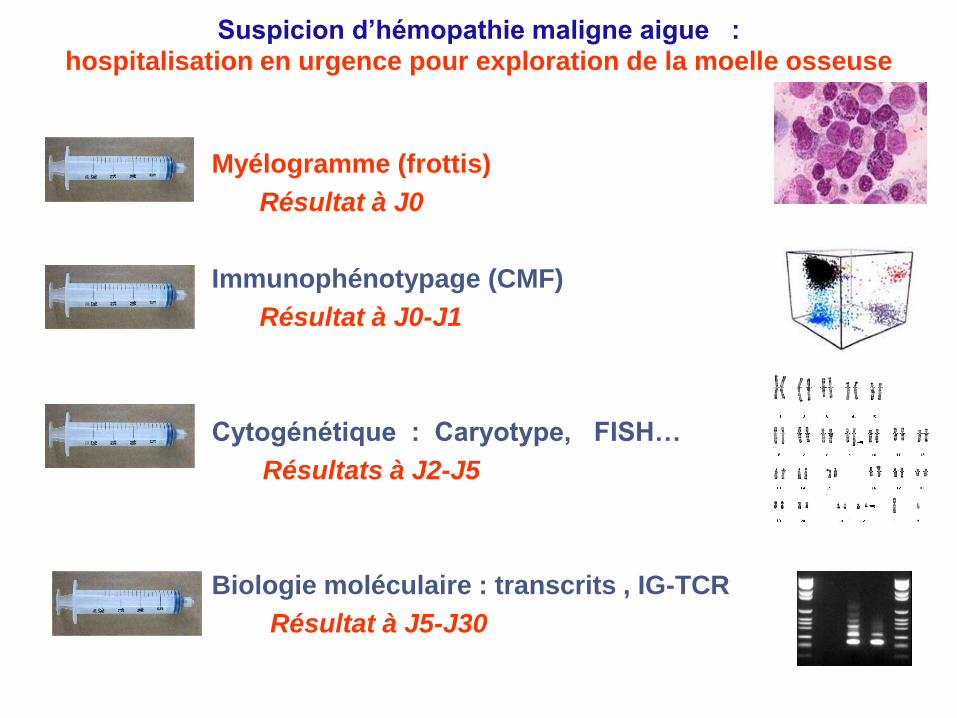
Suspicion d’hémopathie maligne aigue : hospitalisation en urgence pour exploration de la moelle osseuse
Myélogramme (frottis)
Résultat à J0
Immunophénotypage (CMF)
Résultat à J0-J1
Cytogénétique : Caryotype, FISH…
Résultats à J2-J5
Biologie moléculaire : transcrits , IG-TCR
Résultat à J5-J30

w
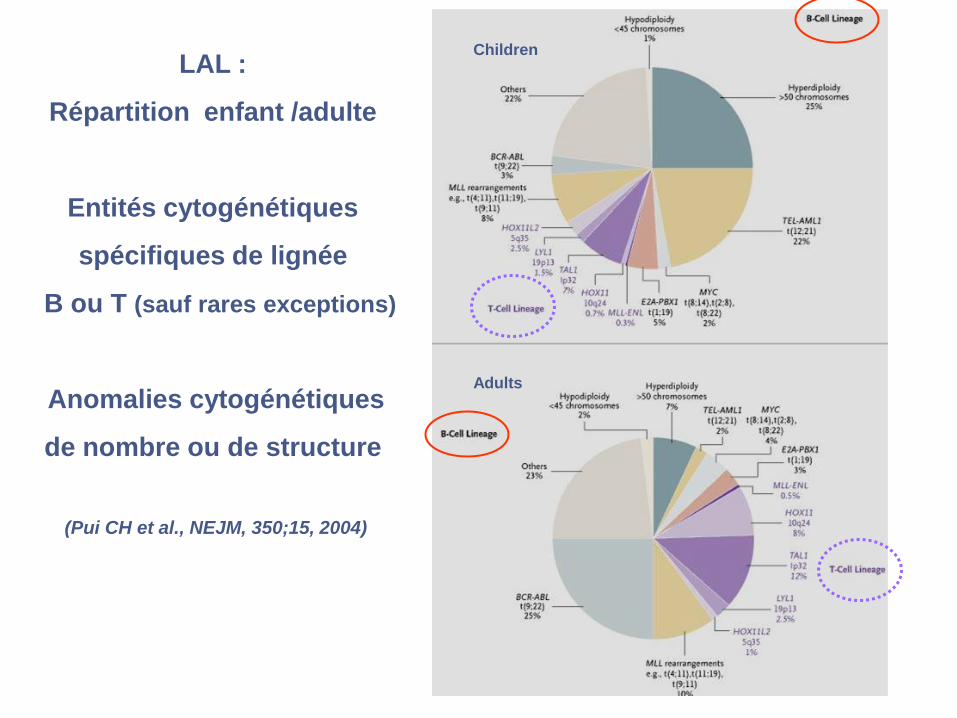
LAL :
Répartition enfant /adulte
Entités cytogénétiques
spécifiques de lignée
B ou T (sauf rares exceptions)
Anomalies cytogénétiques
de nombre ou de structure
(Pui CH et al., NEJM, 350;15, 2004)
Children
Adults

Valeur pronostique de la cytogénétique
dans les LAL de l’enfant
Survie sans événement (EFS) des LAL de l’enfant du St Jude’s Children
Hospital de 1991 à 1999 : 3 protocoles successifs, 467 enfants
Pui CH et al., NEJM, 350;15, 2004
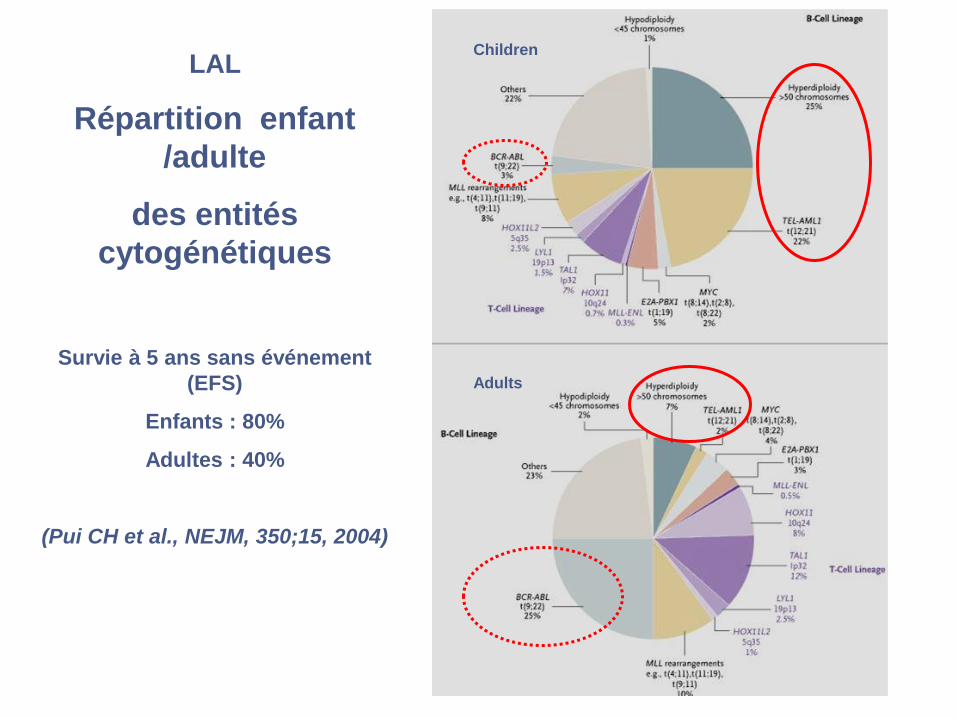
LAL
Répartition enfant
/adulte
des entités
cytogénétiques
Survie à 5 ans sans événement
(EFS)
Enfants : 80%
Adultes : 40%
(Pui CH et al., NEJM, 350;15, 2004)
Children
Adults

C. Fossat, CHU Marseille
Acheminement rapide : 2-3h
Température ambiante
Sang, moelle osseuse, liquides biologiques
Anticoagulant : EDTA, Héparine
Tissu solide : ganglion
milieu humidifié
Sous la direction des anatomo pathologistes
Typage des blastes : prélèvement pour CMF

D’après C. Fossat, CHU Marseille
Ac mono ou polyclonaux : 350 CD (cluster de différenciation)
Plusieurs clones/ CD
Un épitope antigènique/clone
Ig G1, Ig G2, Ig M : Origine murine
Lyophilisés, liquides, purs ou marqués
MARQUAGE CELLULAIRE pour CMF :
membranaire, cytoplasmique, nucléaire
D’après C. Fossat, CHU Marseille
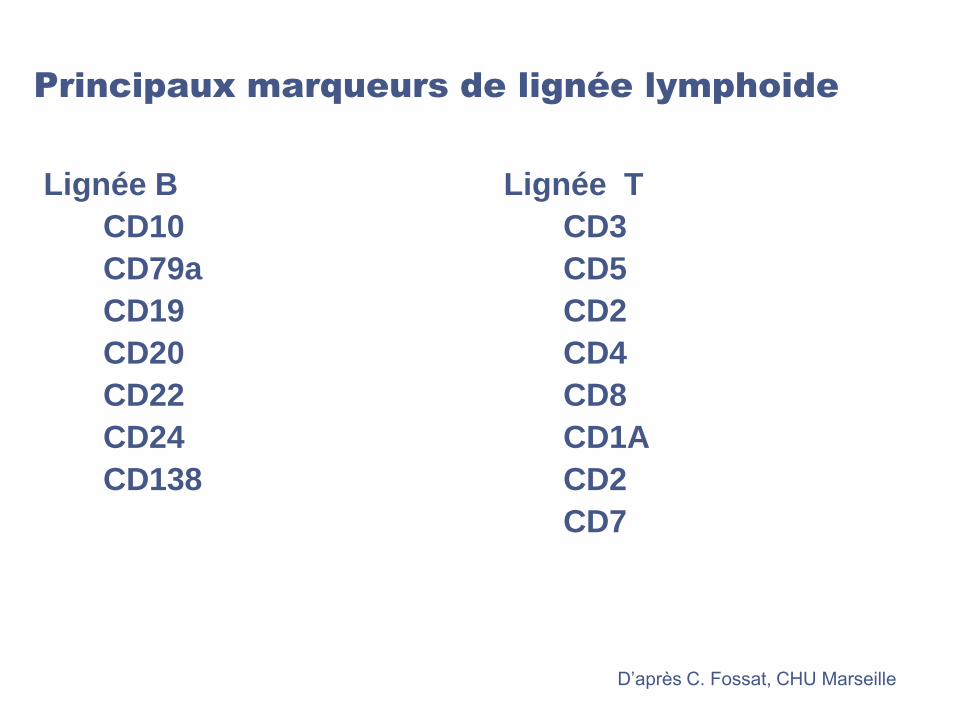
Principaux marqueurs de lignée lymphoide
Lignée T
CD3
CD5
CD2
CD4
CD8
CD1A
CD2
CD7
Lignée B
CD10
CD79a
CD19
CD20
CD22
CD24
CD138
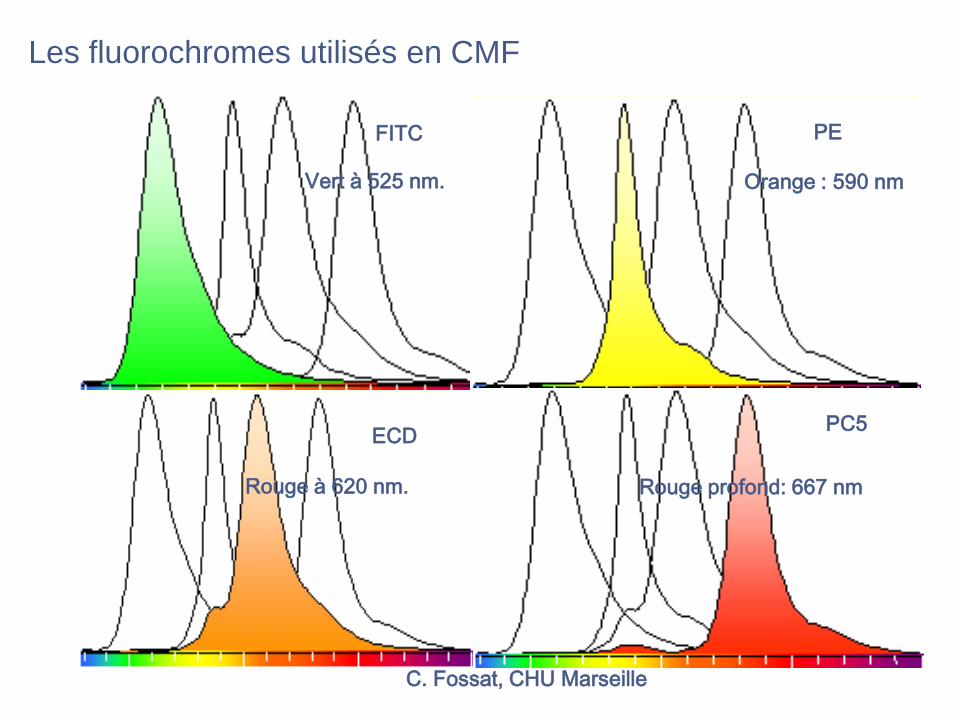
C. Fossat, CHU Marseille
FITC PE
ECDPC5
Vert à 525 nm. Orange : 590 nm
Rouge profond: 667 nm Rouge à 620 nm.
Les fluorochromes utilisés en CMF
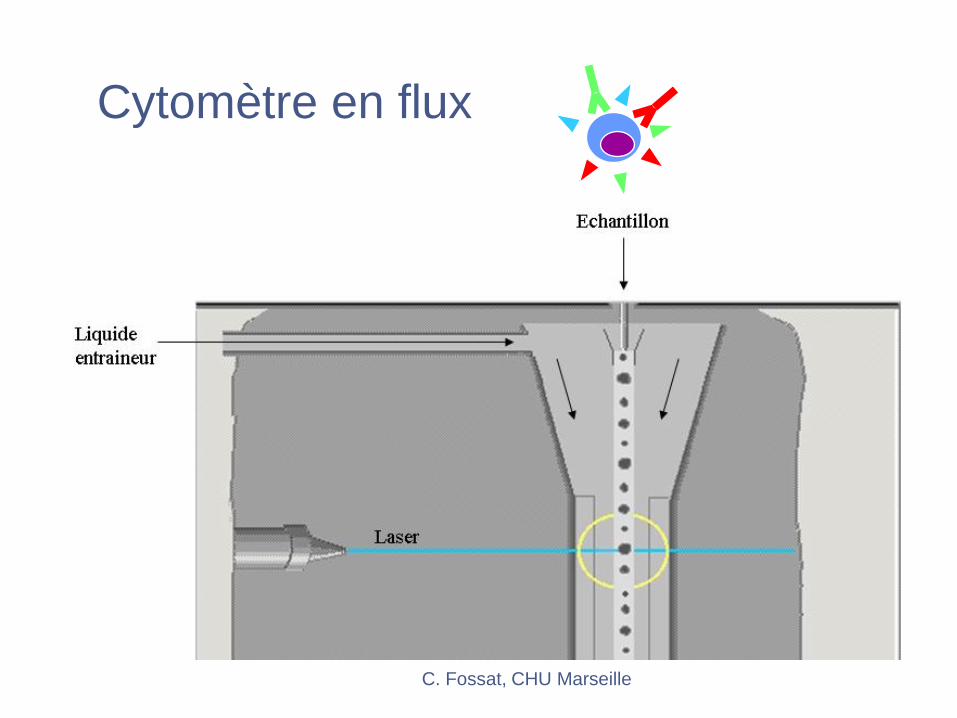
C. Fossat, CHU Marseille
Cytomètre en flux
C. Fossat, CHU Marseille
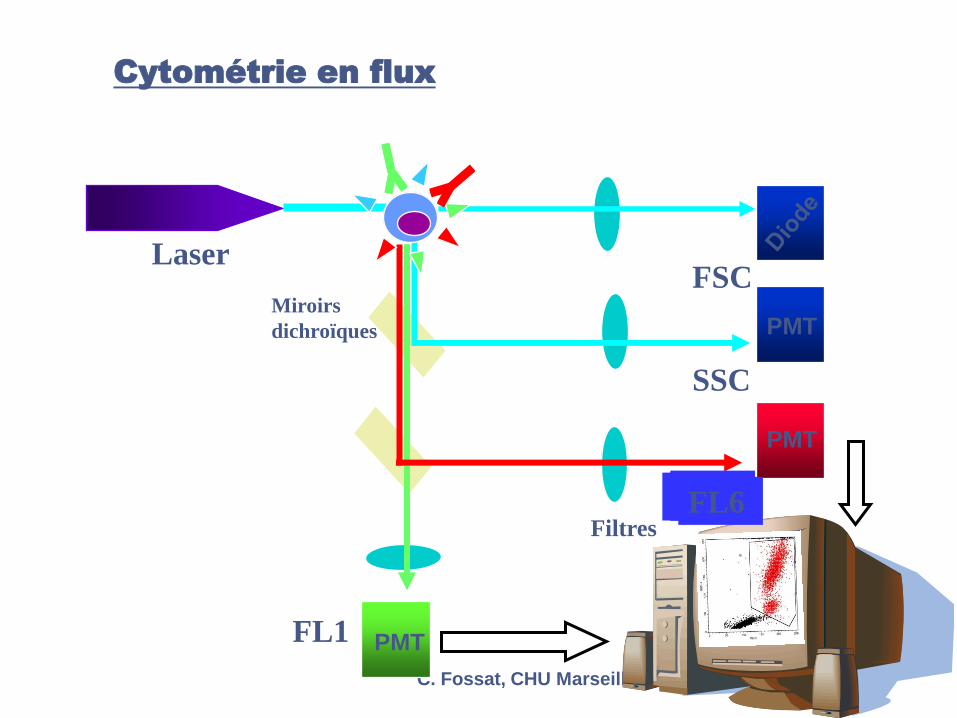
Cytométrie en flux
FL1
FL2
SSC
FSCLaser
Miroirs
dichroïques
Filtres
PMT
PMT
PMT
FL3FL4FL6FL5FL6
C. Fossat, CHU Marseille
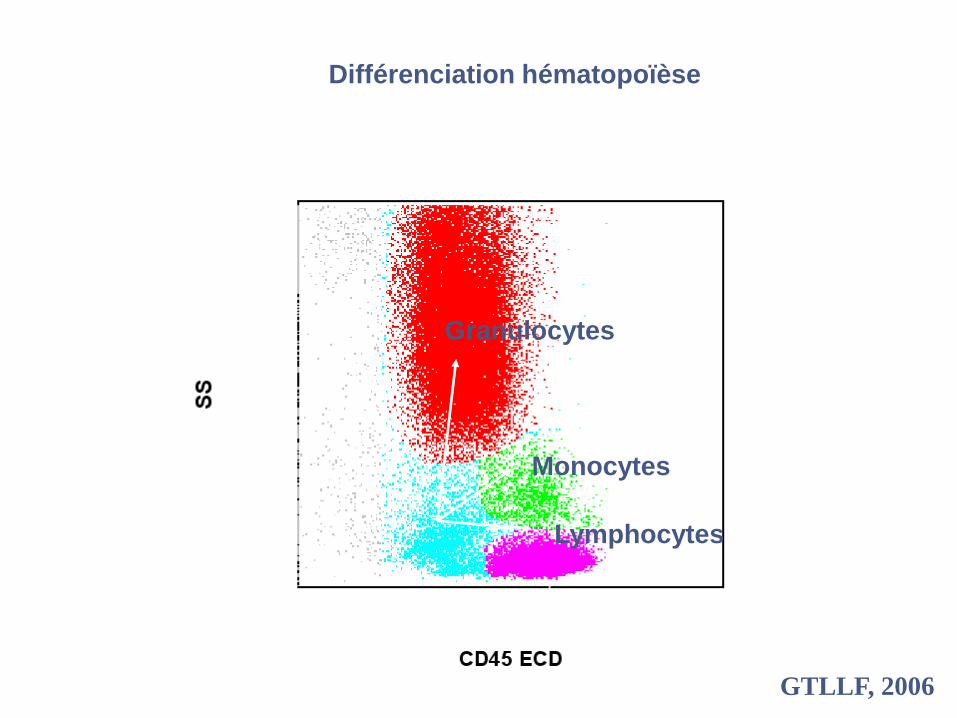
Granulocytes
Monocytes
Lymphocytes
Différenciation hématopoïèse
GTLLF, 2006
C. Fossat, CHU Marseille
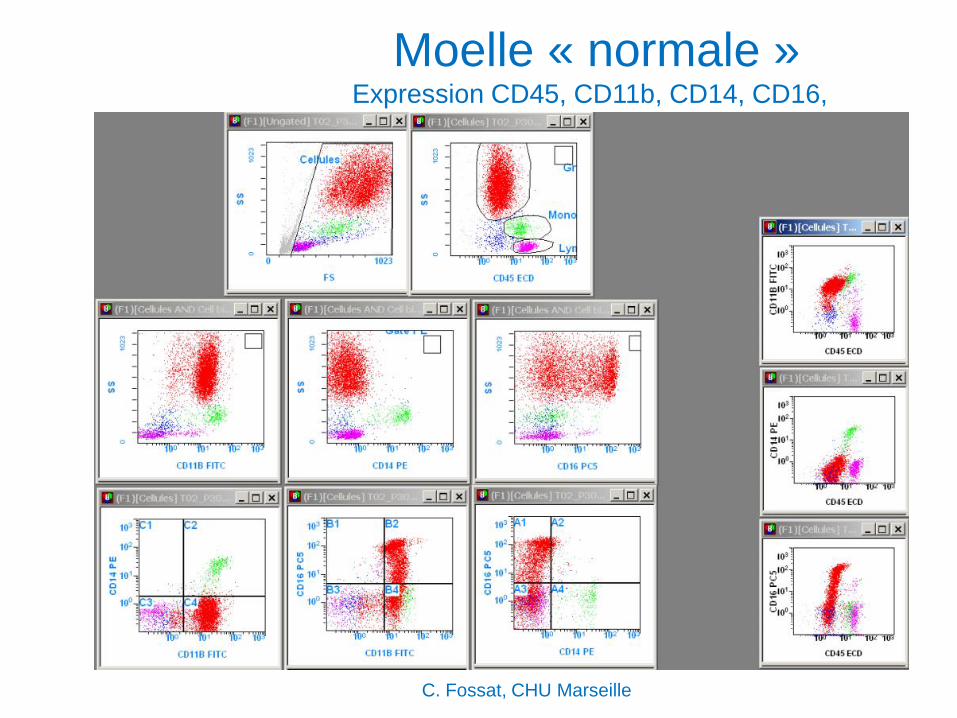
Moelle « normale » Expression CD45, CD11b, CD14, CD16,
C. Fossat, CHU Marseille
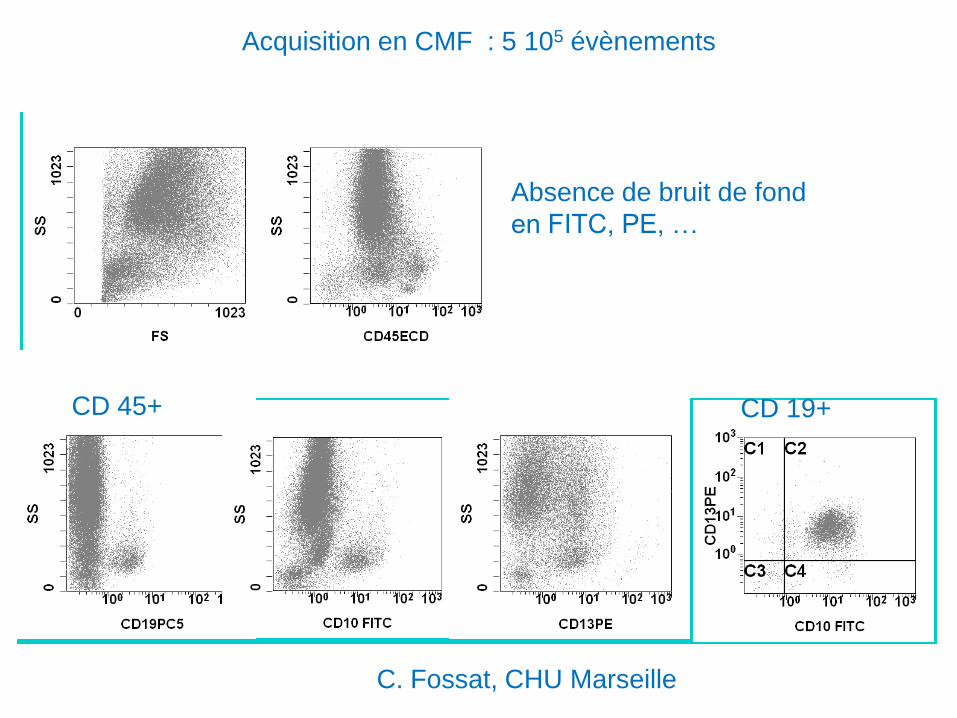
Acquisition en CMF : 5 105 évènements
Absence de bruit de fond
en FITC, PE, …
CD 45+ CD 19+

C. Fossat, CHU Marseille
Immaturité HLA Dr, CD34, TdT, CD10
Lymphoïde B cytCD79a, cytCD22, CD19
Lymphoïde T cytCD3
Myélo-Mono MPO, cytCD13, CD13, CD33, CD117, CD14, CD4
Plaquettaire CD41, CD42, CD61
Erythroïde Glycophorine A, CD36, CD71
Nature de la prolifération
pathologique d’après le profil en CMF

C. Fossat, CHU Marseille
LAL B : profil antigénique associé à la leucémie
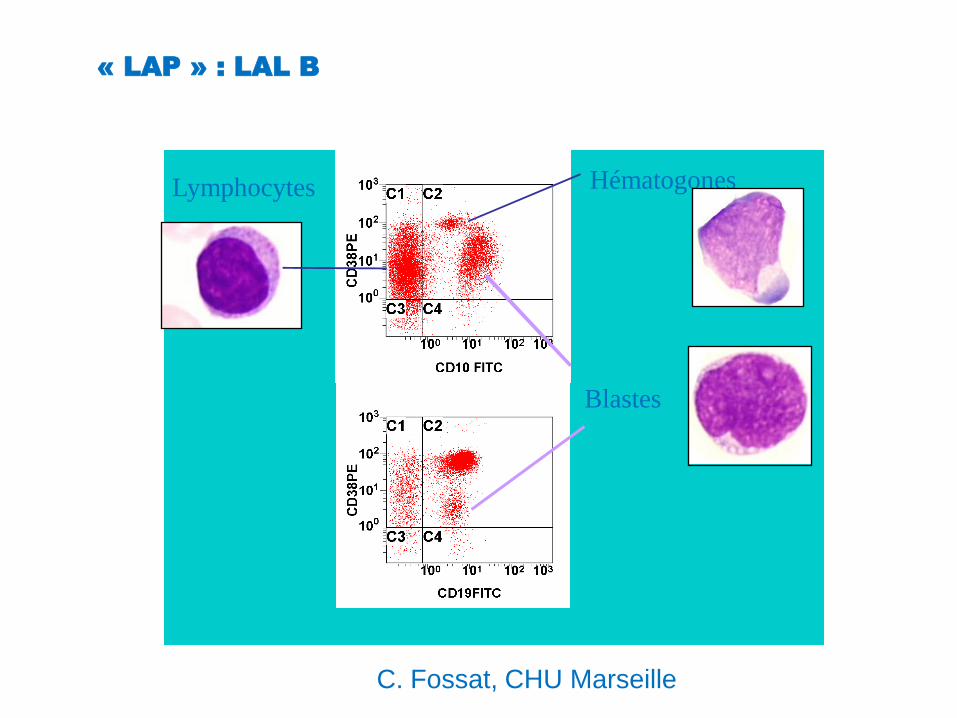
« LAP » (leukemia associated profile)
Intensité d’expression : 100%
CD45, CD10, CD 38, CD19
Asynchronisme de maturation : 50%CD10/CD22, CD34/CD22,
Infidélité de lignée : 20%
CD13, CD33, CD7, CD2,...
Evolution du clone : 20%
Plusieurs combinaisons
C. Fossat, CHU Marseille
« LAP » : LAL B
Blastes
HématogonesLymphocytes

C. Fossat, CHU Marseille
« LAP » des LAL T
Intensité d’expression : 100%
cCD3, CD99
Asynchronisme de maturation : 50%TdT/cCD3, CD1a/TcR,
Infidélité de lignée : 20%
CD13, CD33, CD15,
Evolution du clone : 10%
Plusieurs combinaisons

C. Fossat, CHU Marseille
Classification EGIL
LAL B : cCD79a, cCD22
BI ProB Tdt+, CD10-
BII B Commune Tdt+, CD10+
BIII Pre B Tdt+/-, CD10+, mc+
BIV B Mature Tdt-, CD10+/-, mc+,Igs+
C. Fossat, CHU Marseille
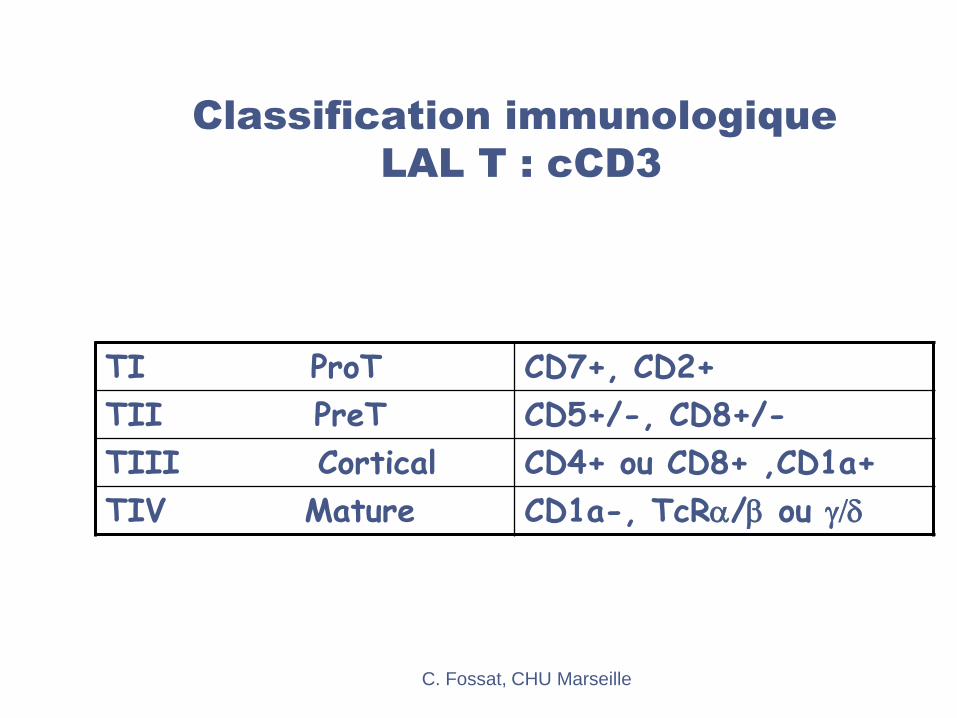
Classification immunologique
LAL T : cCD3
TI ProT CD7+, CD2+
TII PreT CD5+/-, CD8+/-
TIII Cortical CD4+ ou CD8+ ,CD1a+
TIV Mature CD1a-, TcRa/b ou g/d

Leucémie aigüe lymphoblastique (LAL) :
Entités les plus fréquentes
LAL B:
Adulte : LAL Ph
Enfant : Hyperdiploidie et t(12;21)
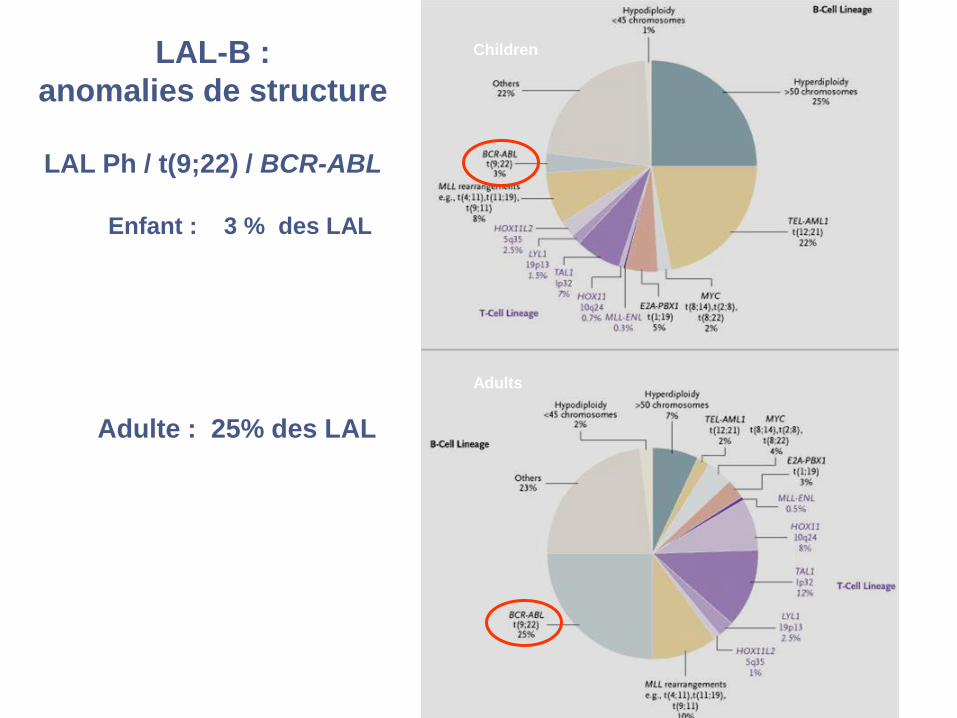
LAL-B :
anomalies de structure
LAL Ph / t(9;22) / BCR-ABL
Enfant : 3 % des LAL
Adulte : 25% des LAL
Children
Adults
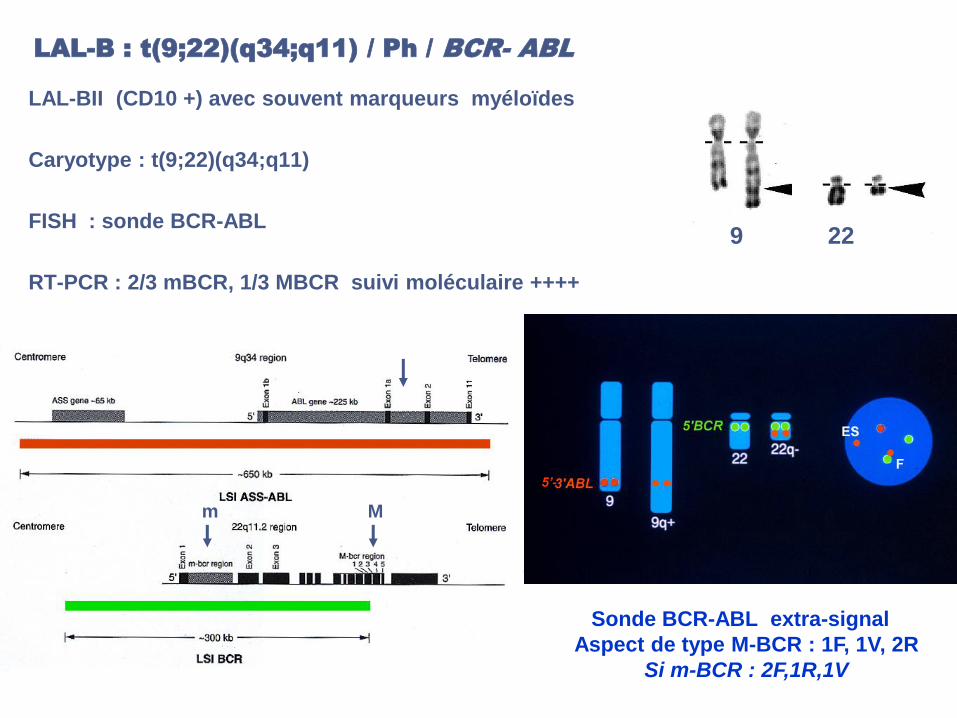
LAL-B : t(9;22)(q34;q11) / Ph / BCR- ABL
LAL-BII (CD10 +) avec souvent marqueurs myéloïdes
Caryotype : t(9;22)(q34;q11)
FISH : sonde BCR-ABL
RT-PCR : 2/3 mBCR, 1/3 MBCR suivi moléculaire ++++
9 22
5’-
Sonde BCR-ABL extra-signal :
Aspect de type M-BCR : 1F, 1V, 2R
Si m-BCR : 2F,1R,1V
m M
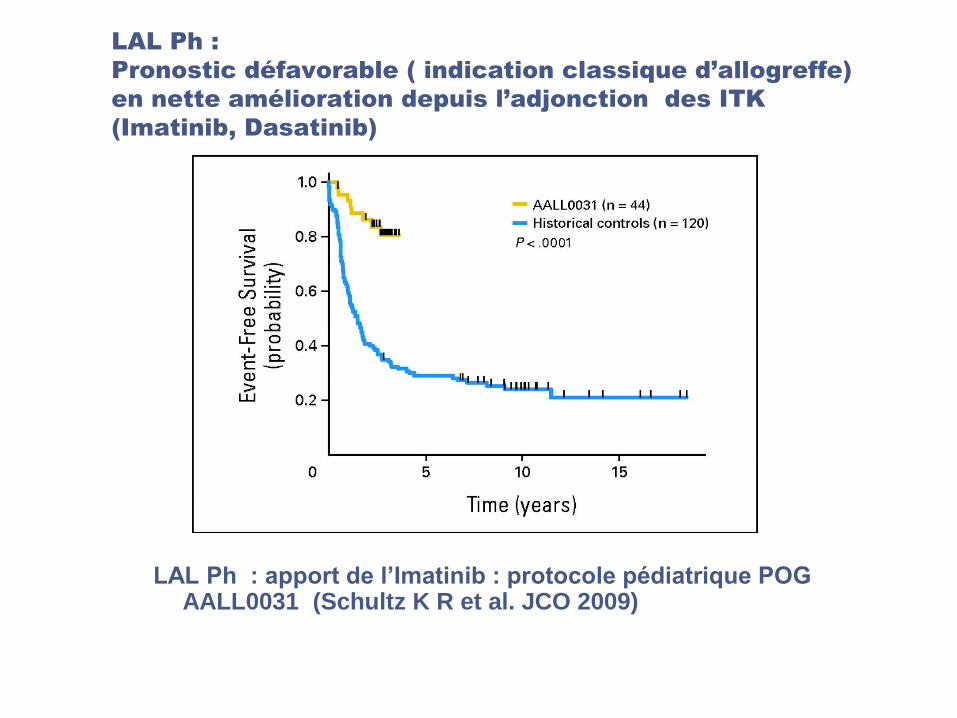
LAL Ph :
Pronostic défavorable ( indication classique d’allogreffe)
en nette amélioration depuis l’adjonction des ITK
(Imatinib, Dasatinib)
LAL Ph : apport de l’Imatinib : protocole pédiatrique POG AALL0031 (Schultz K R et al. JCO 2009)
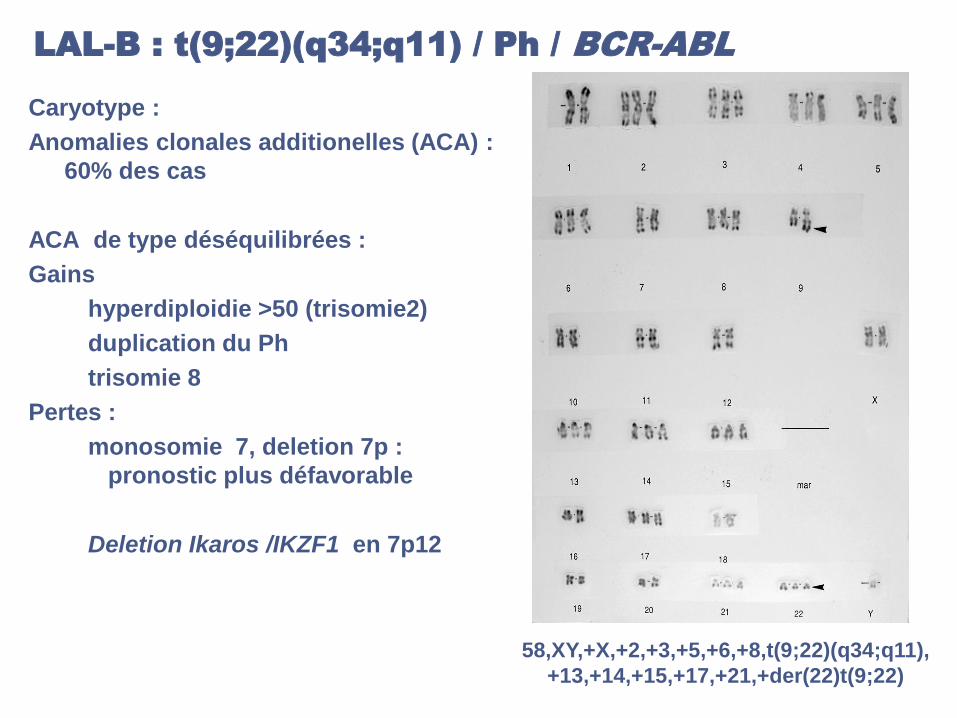
LAL-B : t(9;22)(q34;q11) / Ph / BCR-ABL
Caryotype :
Anomalies clonales additionelles (ACA) :
60% des cas
ACA de type déséquilibrées :
Gains
hyperdiploidie >50 (trisomie2)
duplication du Ph
trisomie 8
Pertes :
monosomie 7, deletion 7p :
pronostic plus défavorable
Deletion Ikaros /IKZF1 en 7p12
58,XY,+X,+2,+3,+5,+6,+8,t(9;22)(q34;q11),
+13,+14,+15,+17,+21,+der(22)t(9;22)
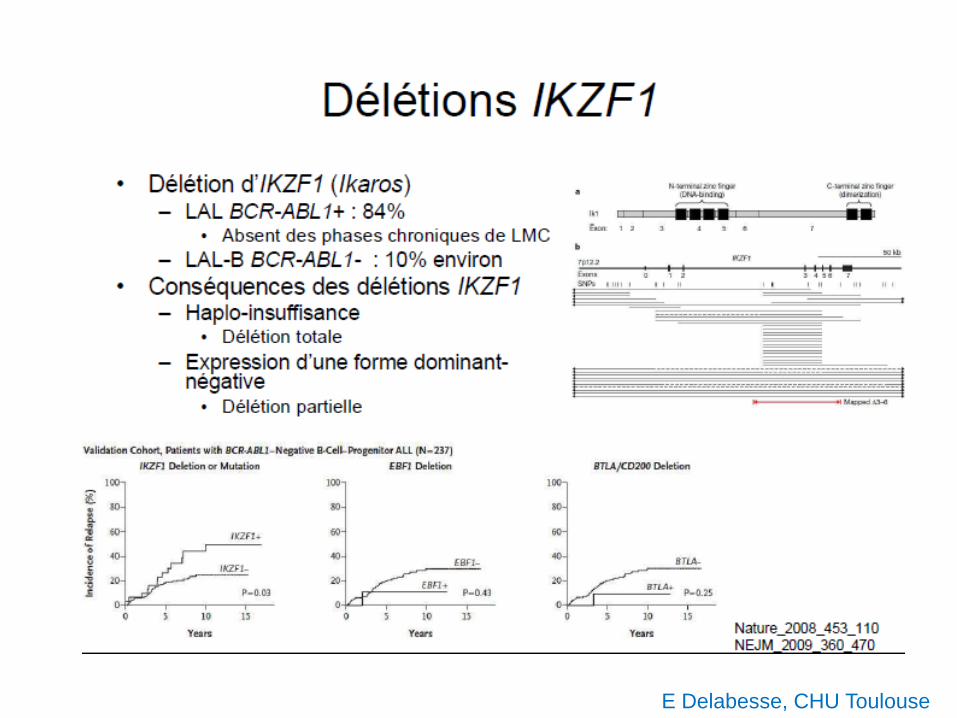
E Delabesse, CHU Toulouse

E Delabesse, CHU Toulouse

E Delabesse, CHU Toulouse

E Delabesse, CHU Toulouse
E Delabesse, CHU Toulouse
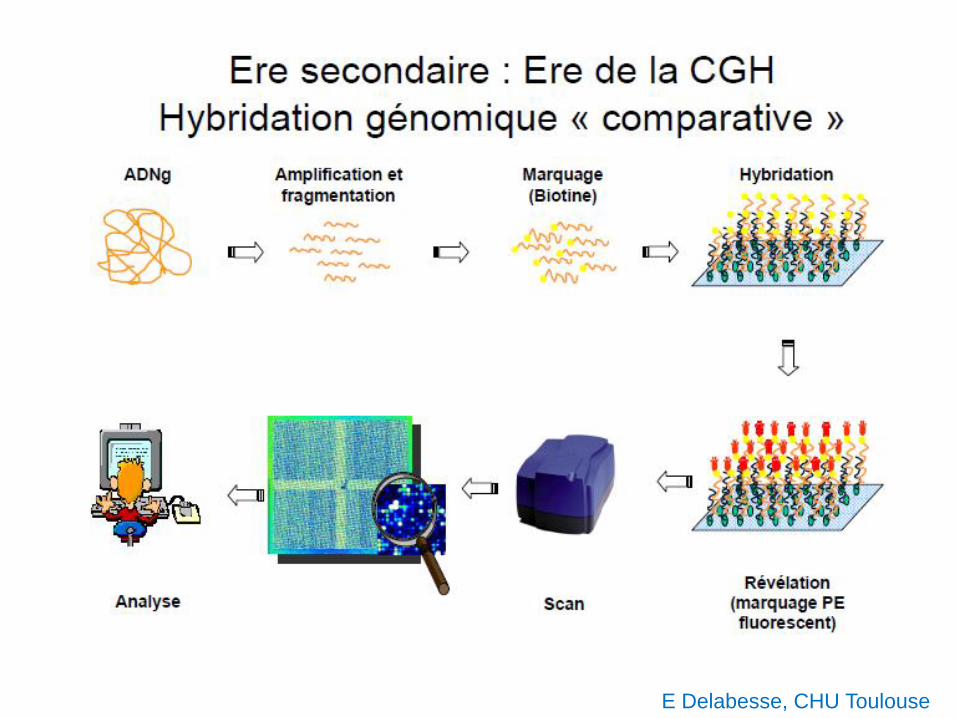
E Delabesse, CHU Toulouse

E Delabesse, CHU Toulouse

E Delabesse, CHU Toulouse
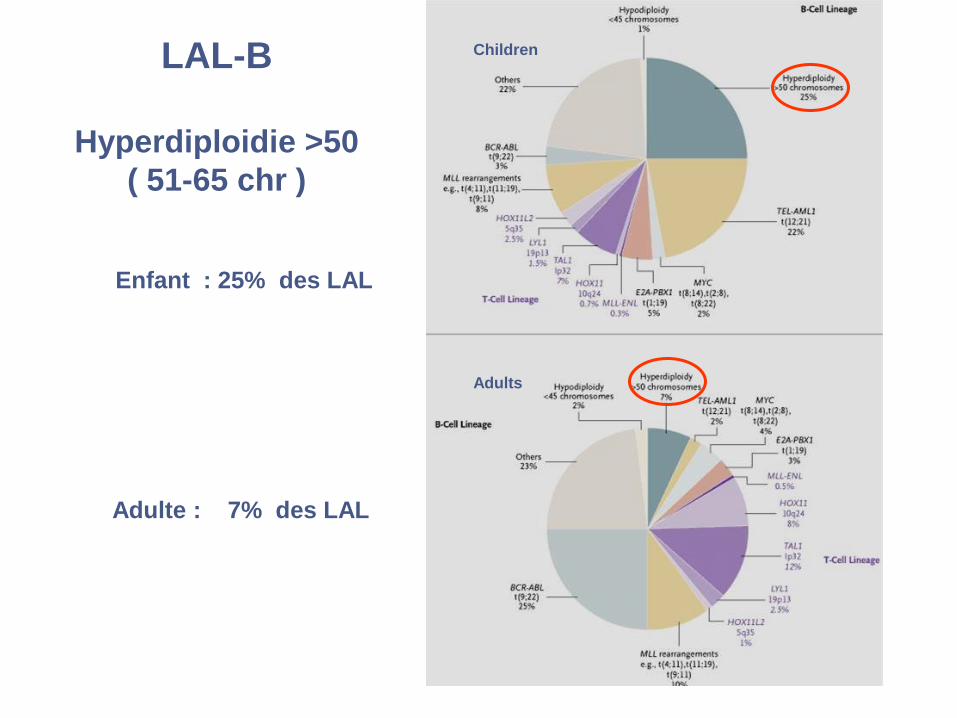
LAL-B
Hyperdiploidie >50
( 51-65 chr )
Enfant : 25% des LAL
Adulte : 7% des LAL
Children
Adults
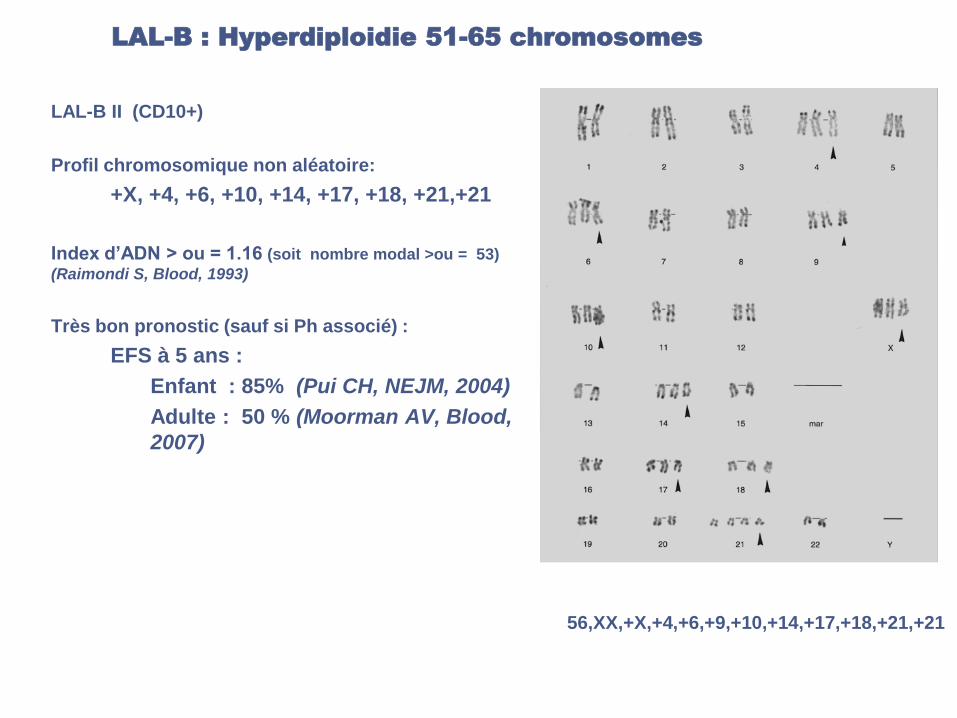
LAL-B : Hyperdiploidie 51-65 chromosomes
LAL-B II (CD10+)
Profil chromosomique non aléatoire:
+X, +4, +6, +10, +14, +17, +18, +21,+21
Index d’ADN > ou = 1.16 (soit nombre modal >ou = 53)
(Raimondi S, Blood, 1993)
Très bon pronostic (sauf si Ph associé) :
EFS à 5 ans :
Enfant : 85% (Pui CH, NEJM, 2004)
Adulte : 50 % (Moorman AV, Blood,
2007)
56,XX,+X,+4,+6,+9,+10,+14,+17,+18,+21,+21
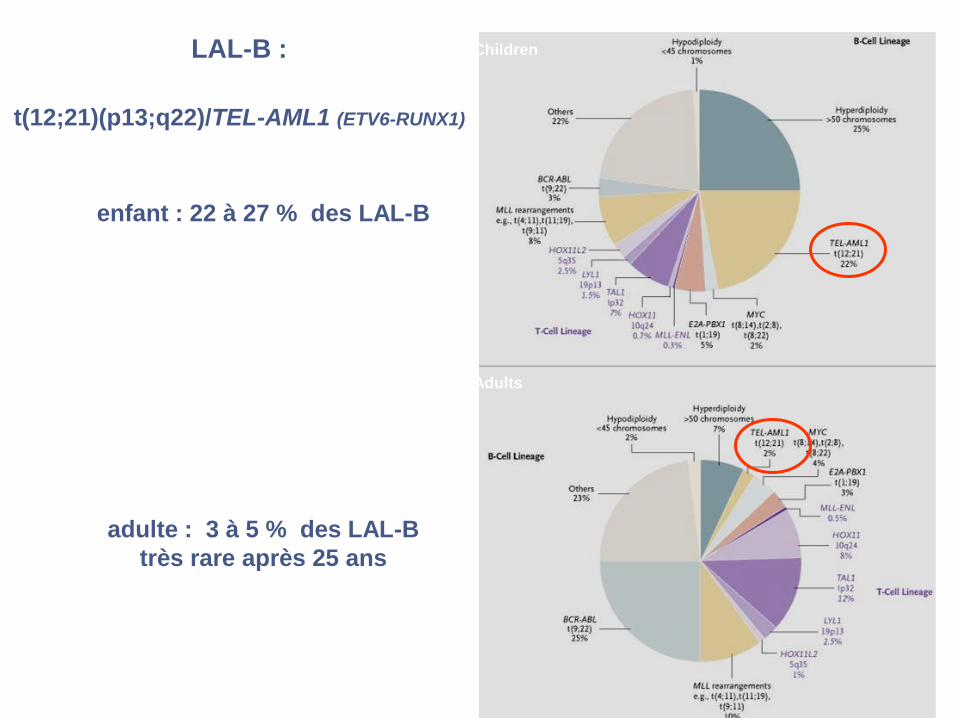
LAL-B :
t(12;21)(p13;q22)/TEL-AML1 (ETV6-RUNX1)
enfant : 22 à 27 % des LAL-B
adulte : 3 à 5 % des LAL-B
très rare après 25 ans
Children
Adults
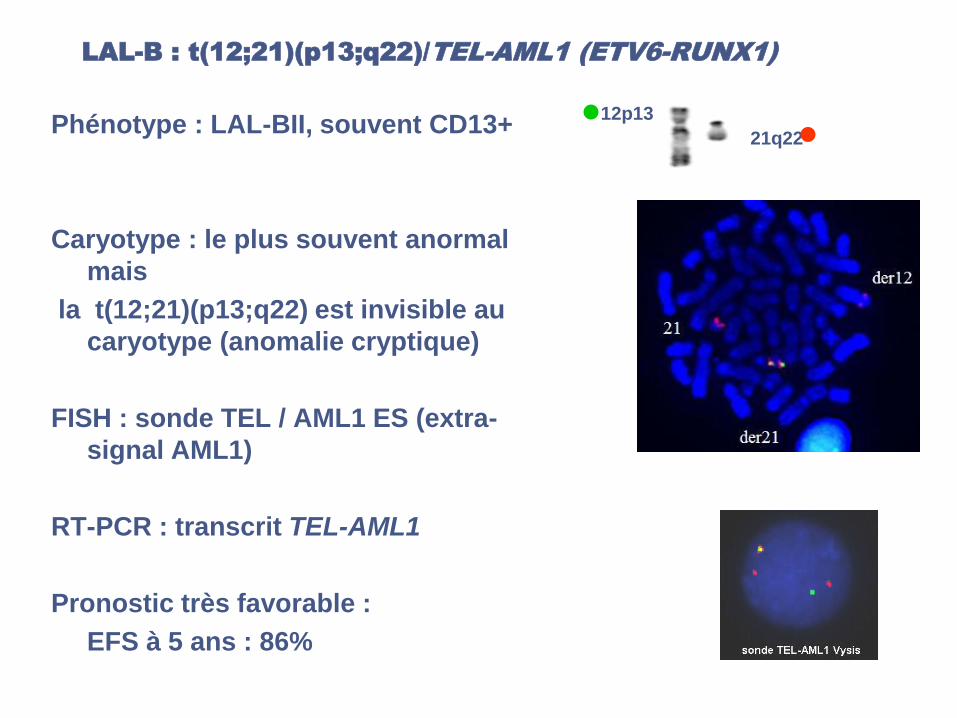
LAL-B : t(12;21)(p13;q22)/TEL-AML1 (ETV6-RUNX1)
Phénotype : LAL-BII, souvent CD13+
Caryotype : le plus souvent anormal
mais
la t(12;21)(p13;q22) est invisible au
caryotype (anomalie cryptique)
FISH : sonde TEL / AML1 ES (extra-
signal AML1)
RT-PCR : transcrit TEL-AML1
Pronostic très favorable :
EFS à 5 ans : 86%
12p13
21q22
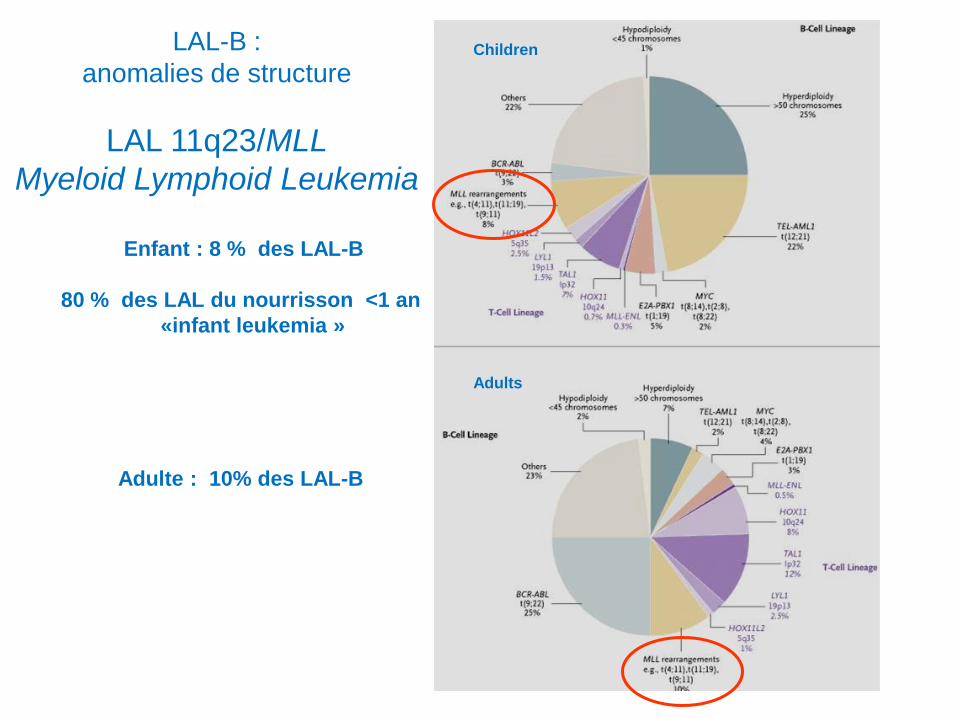
LAL-B :
anomalies de structure
LAL 11q23/MLL
Myeloid Lymphoid Leukemia
Enfant : 8 % des LAL-B
80 % des LAL du nourrisson <1 an
«infant leukemia »
Adulte : 10% des LAL-B
Children
Adults
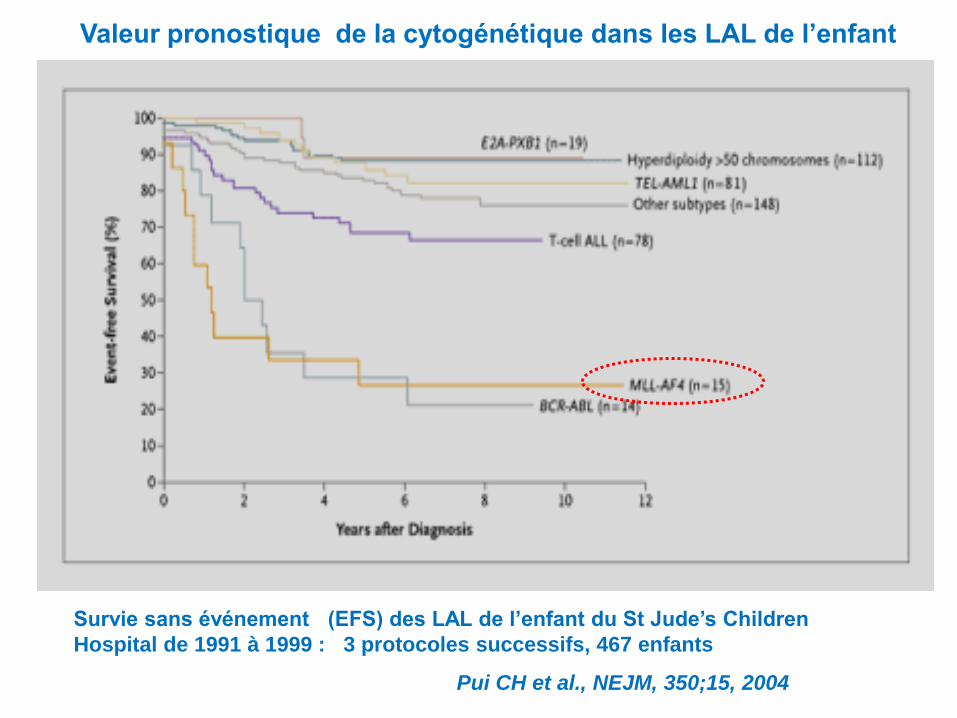
Valeur pronostique de la cytogénétique dans les LAL de l’enfant
Survie sans événement (EFS) des LAL de l’enfant du St Jude’s Children
Hospital de 1991 à 1999 : 3 protocoles successifs, 467 enfants
Pui CH et al., NEJM, 350;15, 2004
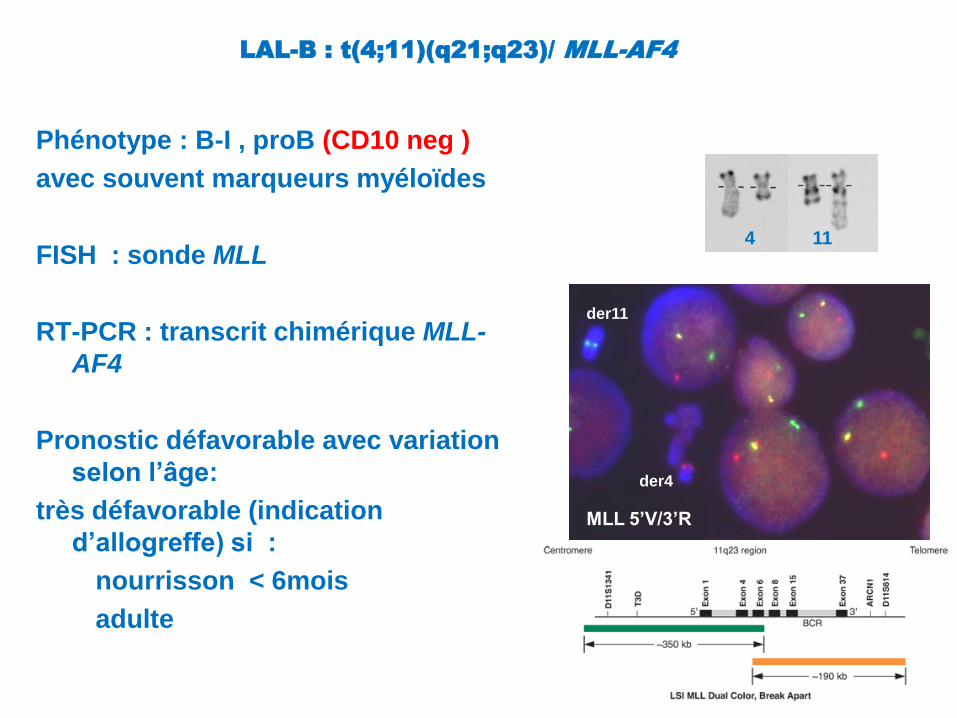
LAL-B : t(4;11)(q21;q23)/ MLL-AF4
Phénotype : B-I , proB (CD10 neg )
avec souvent marqueurs myéloïdes
FISH : sonde MLL
RT-PCR : transcrit chimérique MLL-
AF4
Pronostic défavorable avec variation
selon l’âge:
très défavorable (indication
d’allogreffe) si :
nourrisson < 6mois
adulte
4 11
MLL 5’V/3’R
der11
der4
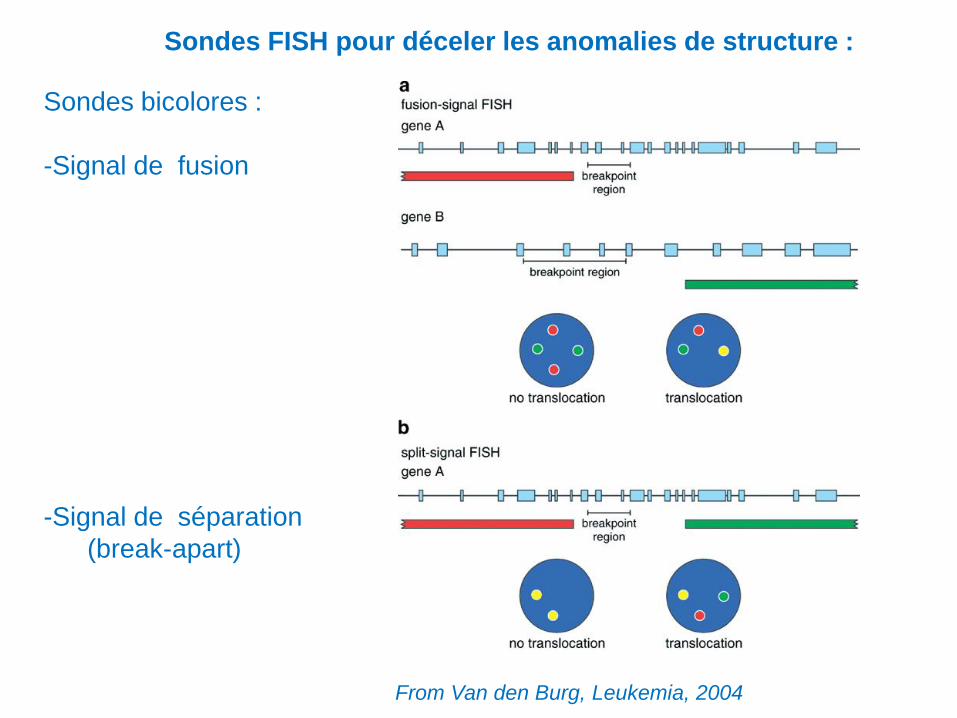
From Van den Burg, Leukemia, 2004
Sondes FISH pour déceler les anomalies de structure :
Sondes bicolores :
-Signal de fusion
-Signal de séparation
(break-apart)
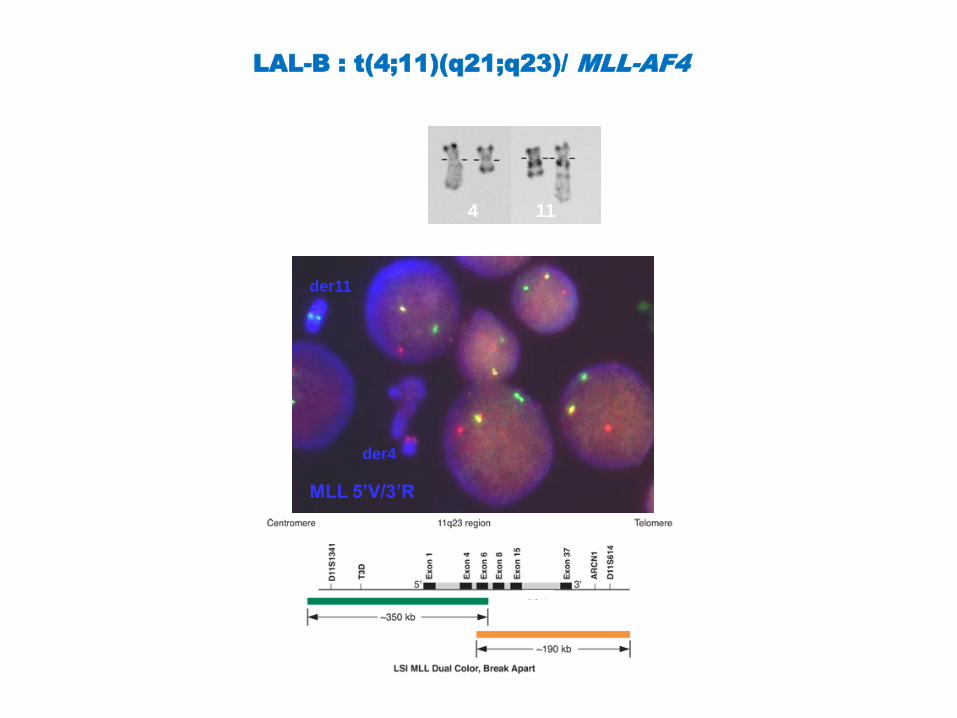
LAL-B : t(4;11)(q21;q23)/ MLL-AF4
4 11
MLL 5’V/3’R
der11
der4

Leucémies aiguës lymphoblastiques (LAL) :
Valeur pronostique de la maladie résiduelle
Maladie residuelle minime (MRD pour minimal residual disease) :
maladie détectable en dessous du seuil de la cytologie
(5% de blastes residuels )
Recherchée après chaque phase de chimiothérapie
induction soit J30 post diagnostic
1ère consolidation soit J60
2ème consolidation soit J90
MRD moléculaire : valeur pronostique défavorable si
Présence du transcrit bcr-abl dans les LAL Ph
Niveau Ig-TCR >ou = 10-2 après induction dans les LAL de
l’enfant
MRD par CMF : validée aux USA, en cours d’évaluation en Europe
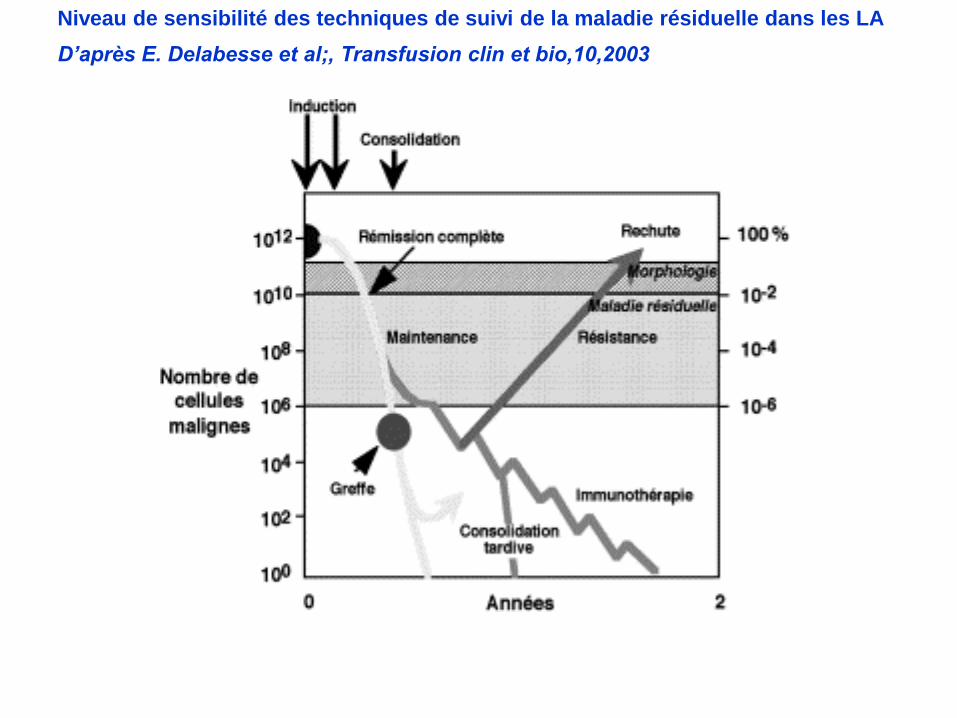
Niveau de sensibilité des techniques de suivi de la maladie résiduelle dans les LA
D’après E. Delabesse et al;, Transfusion clin et bio,10,2003

C. Fossat, CHU Marseille
Infidélité de lignée
Asynchronisme de maturation
Intensité d’expression
Profil phénotypique associé
à la leucémie : « LAP »
Apport de la CMF dans le suivi de la maladie :Etude de la maladie résiduelle

C. Fossat, CHU Marseille
MRD par CMF : exemple d’une LAL de
l’enfant
LAL B II (Mars 2002) – Georgie
Mai 2004 : pris en charge au CHU Timone, pour greffe
allogénique intrafamiliale
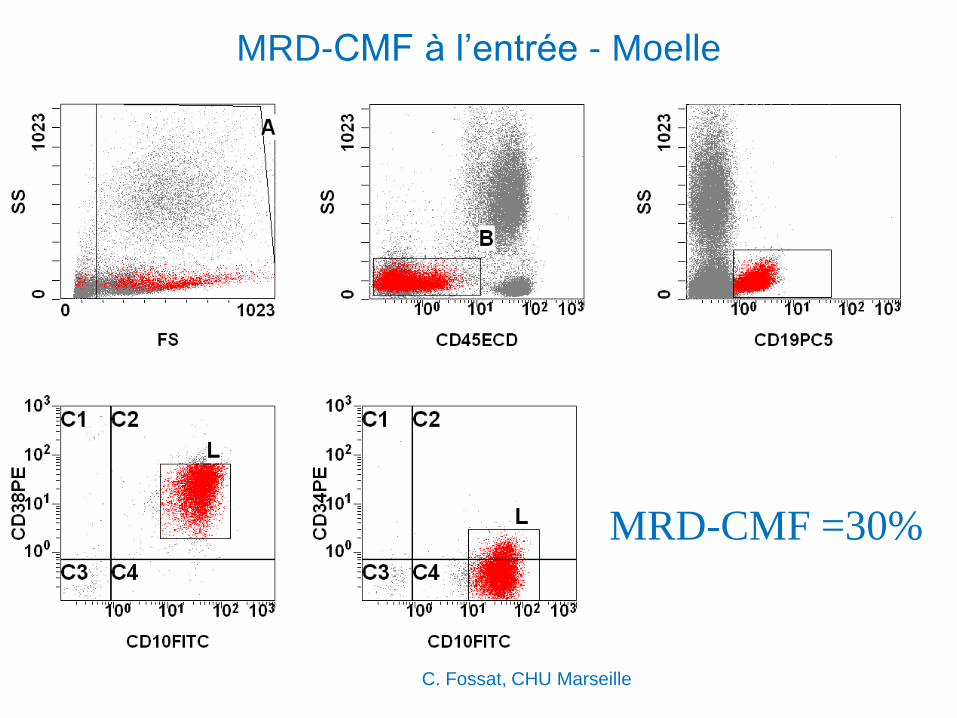
C. Fossat, CHU Marseille
MRD-CMF à l’entrée - Moelle
MRD-CMF =30%
C. Fossat, CHU Marseille
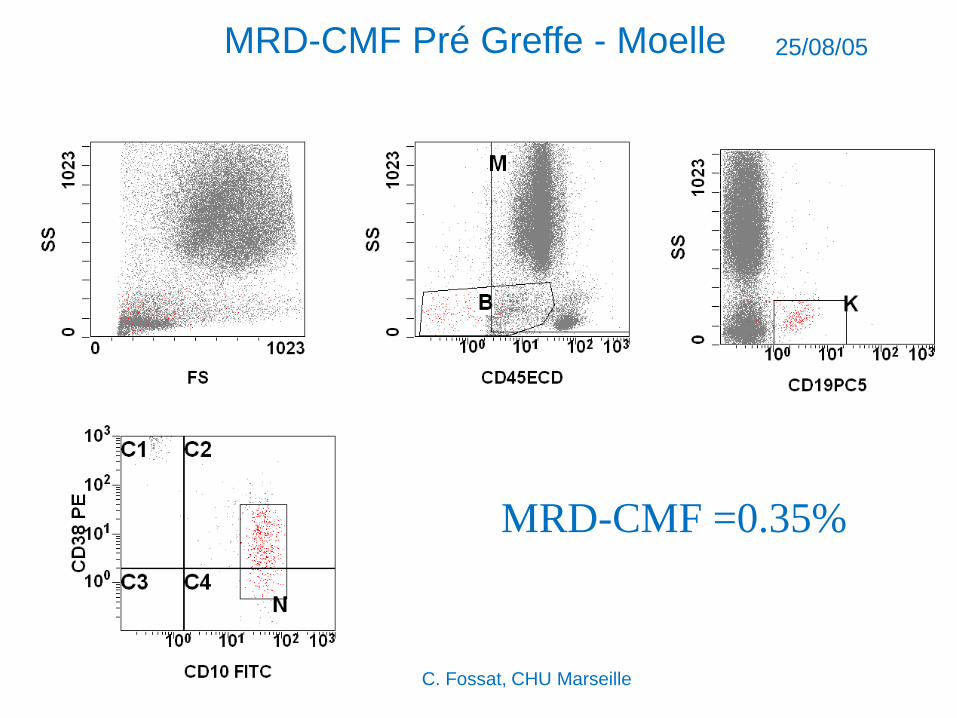
MRD-CMF =0.35%
MRD-CMF Pré Greffe - Moelle 25/08/05
C. Fossat, CHU Marseille
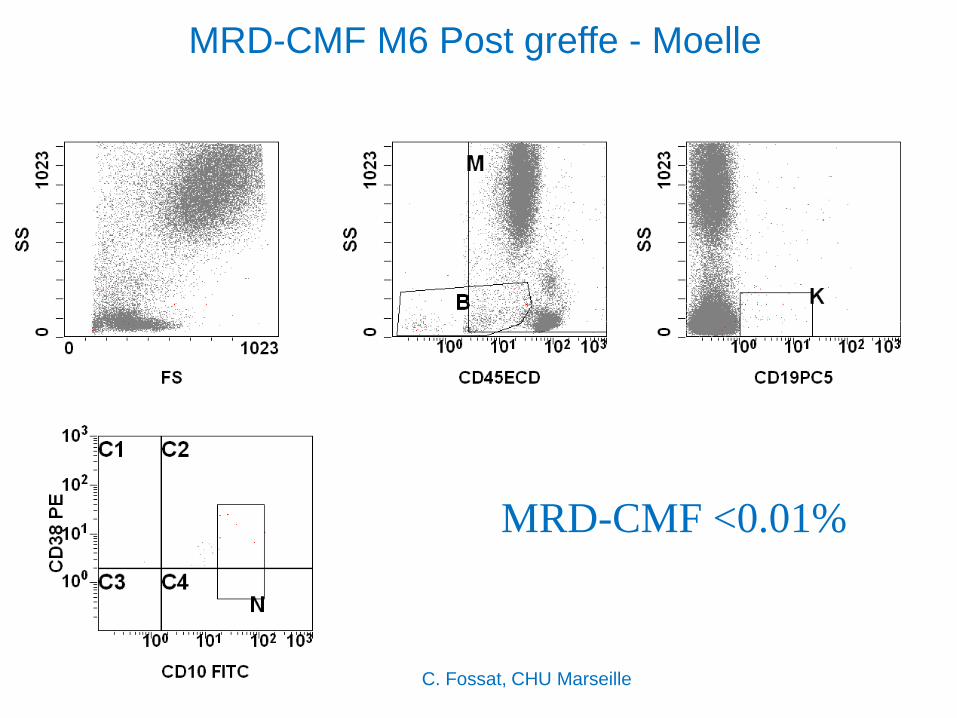
MRD-CMF M6 Post greffe - Moelle
MRD-CMF <0.01%
C. Fossat, CHU Marseille
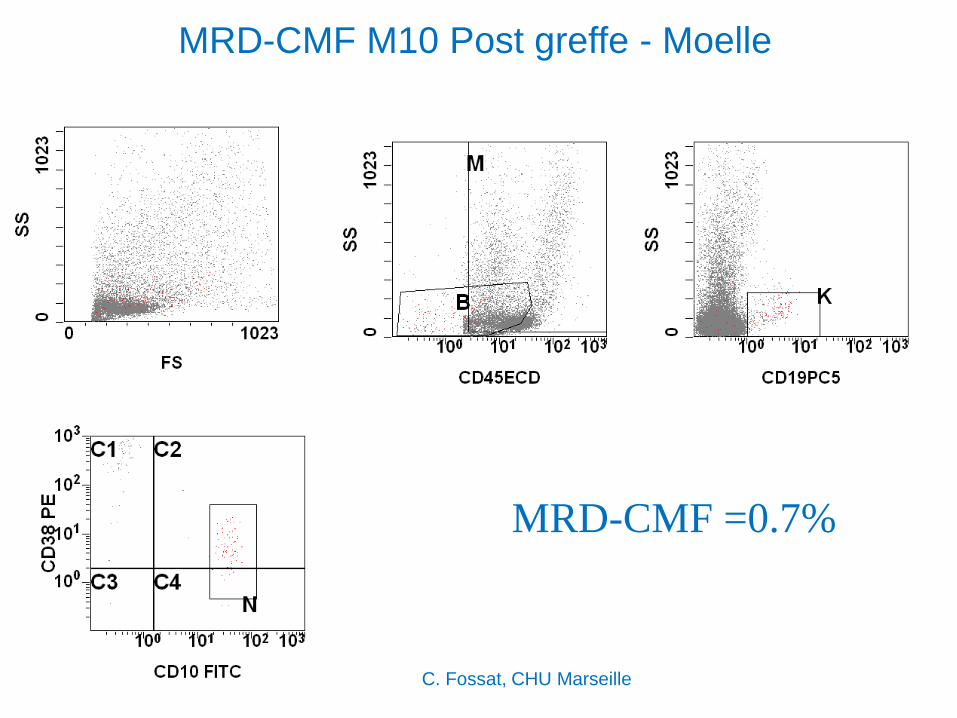
MRD-CMF M10 Post greffe - Moelle
MRD-CMF =0.7%
C. Fossat, CHU Marseille
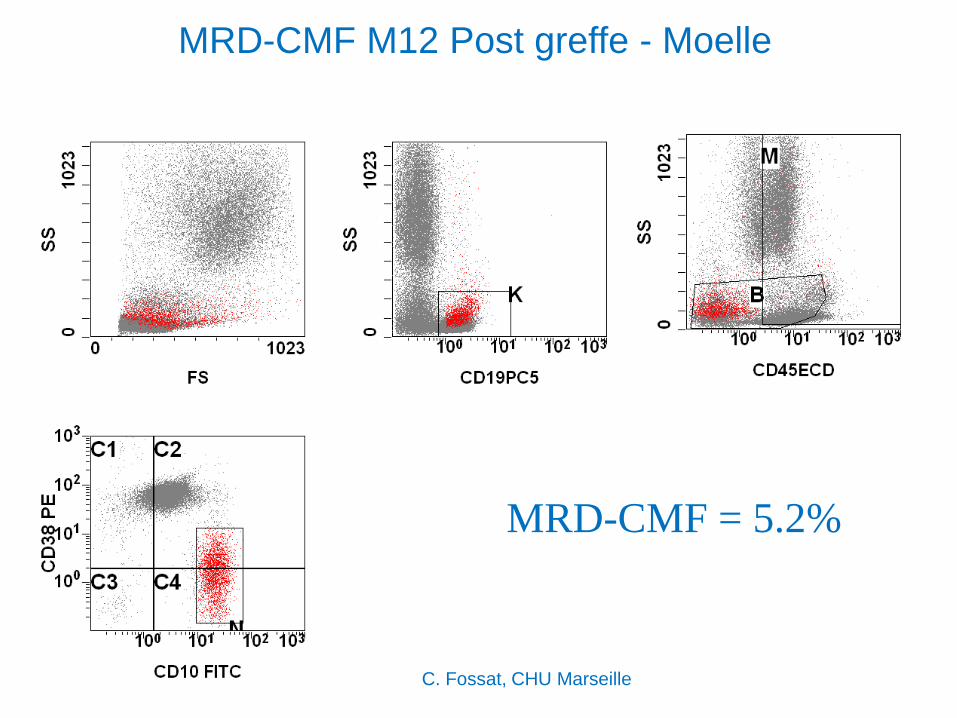
MRD-CMF M12 Post greffe - Moelle
MRD-CMF = 5.2%
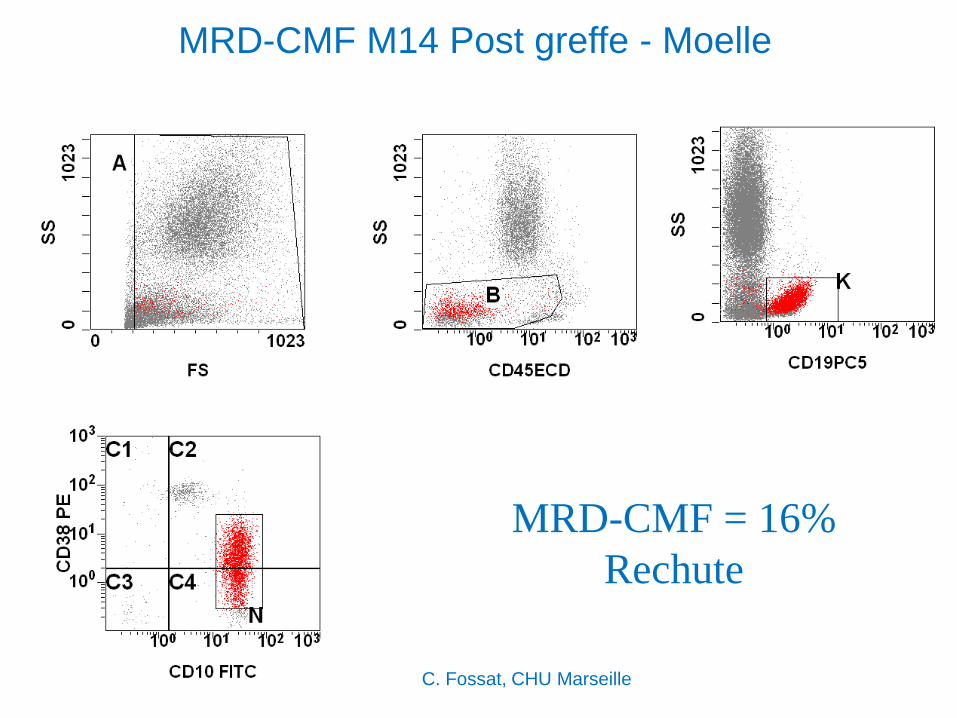
C. Fossat, CHU Marseille
MRD-CMF M14 Post greffe - Moelle
MRD-CMF = 16%
Rechute
C. Fossat, CHU Marseille
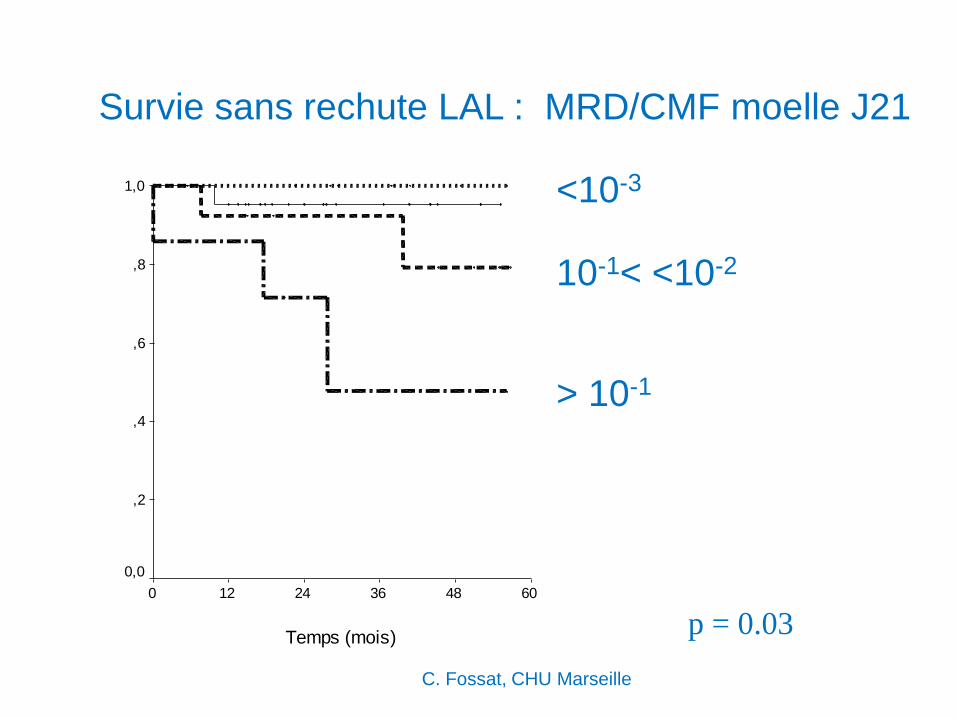
Temps (mois)
60483624120
Su
rvie
san
s R
echu
tes
1,0
,8
,6
,4
,2
0,0
p = 0.03
Survie sans rechute LAL : MRD/CMF moelle J21
> 10-1
<10-3
10-1< <10-2
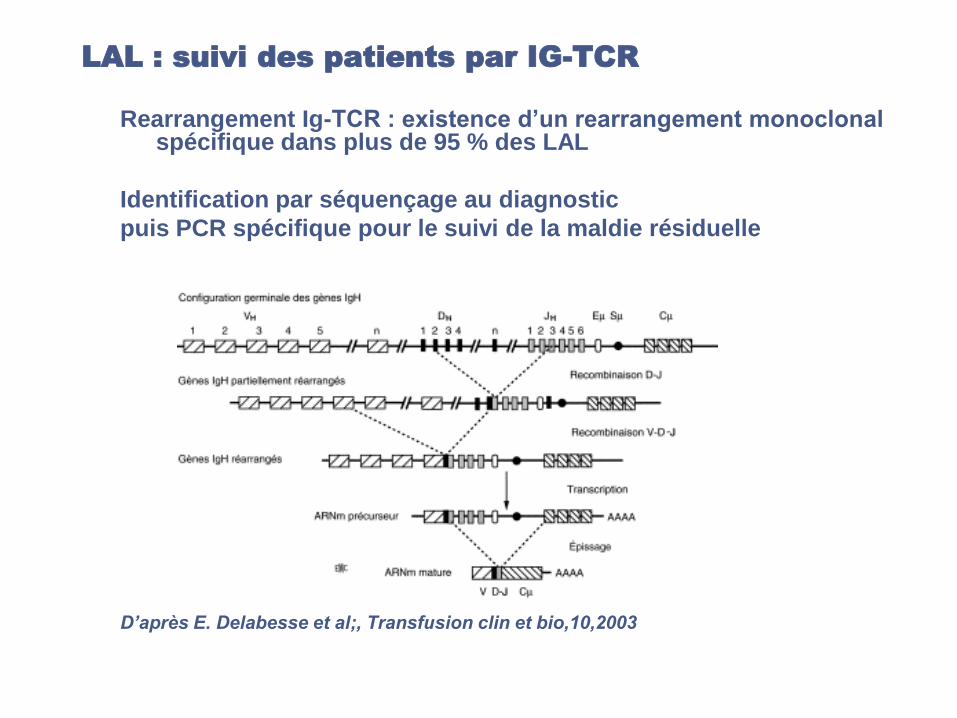
LAL : suivi des patients par IG-TCR
Rearrangement Ig-TCR : existence d’un rearrangement monoclonal spécifique dans plus de 95 % des LAL
Identification par séquençage au diagnostic
puis PCR spécifique pour le suivi de la maldie résiduelle
D’après E. Delabesse et al;, Transfusion clin et bio,10,2003
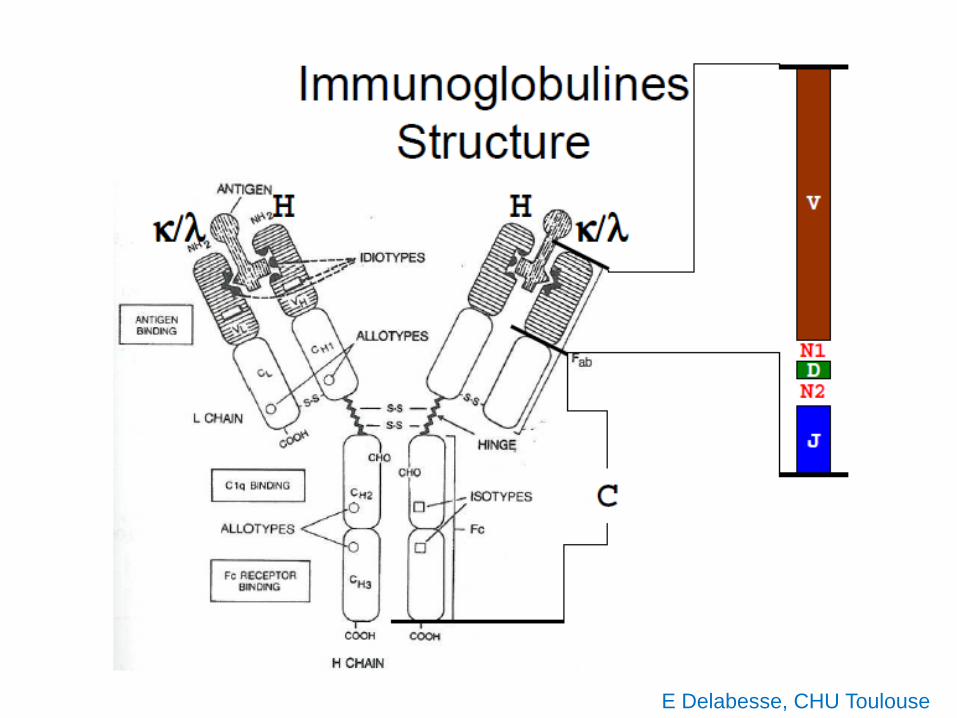
E Delabesse, CHU Toulouse
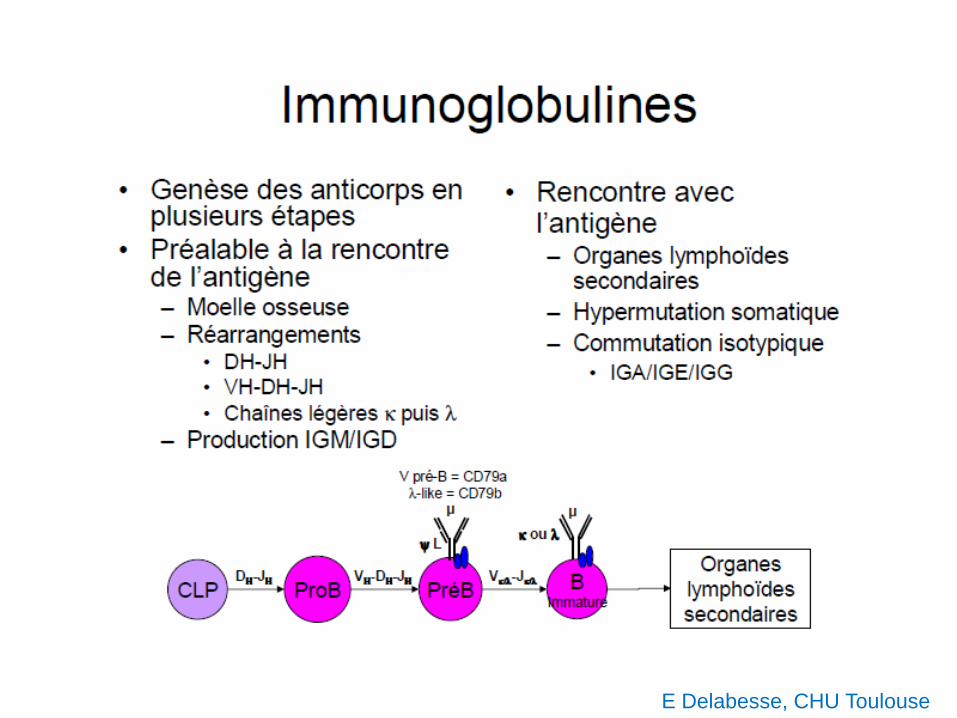
E Delabesse, CHU Toulouse
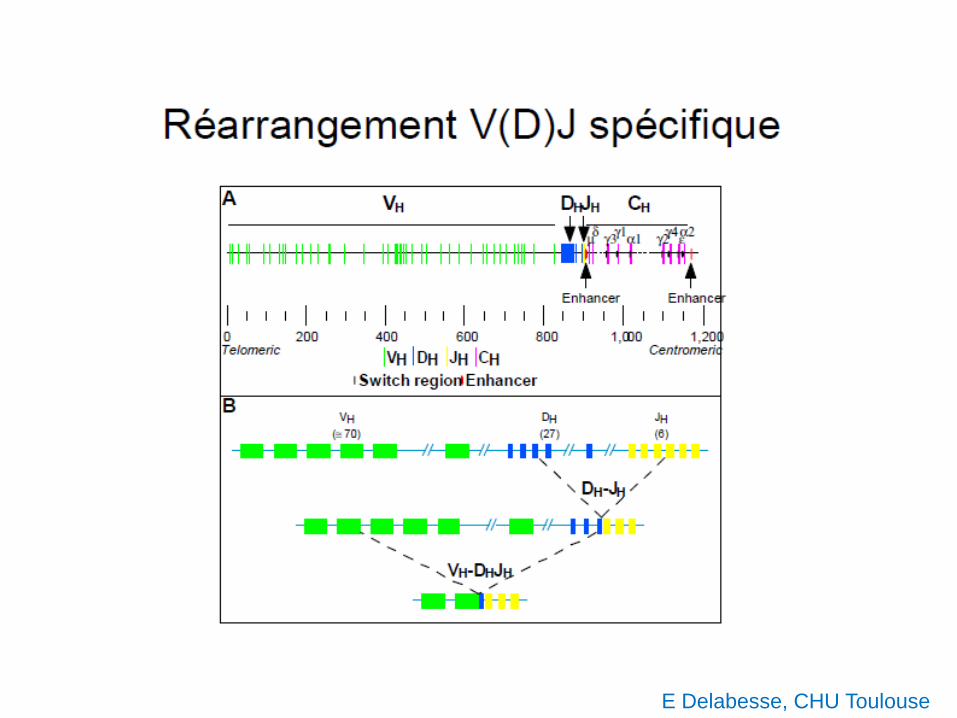
E Delabesse, CHU Toulouse

E Delabesse, CHU Toulouse
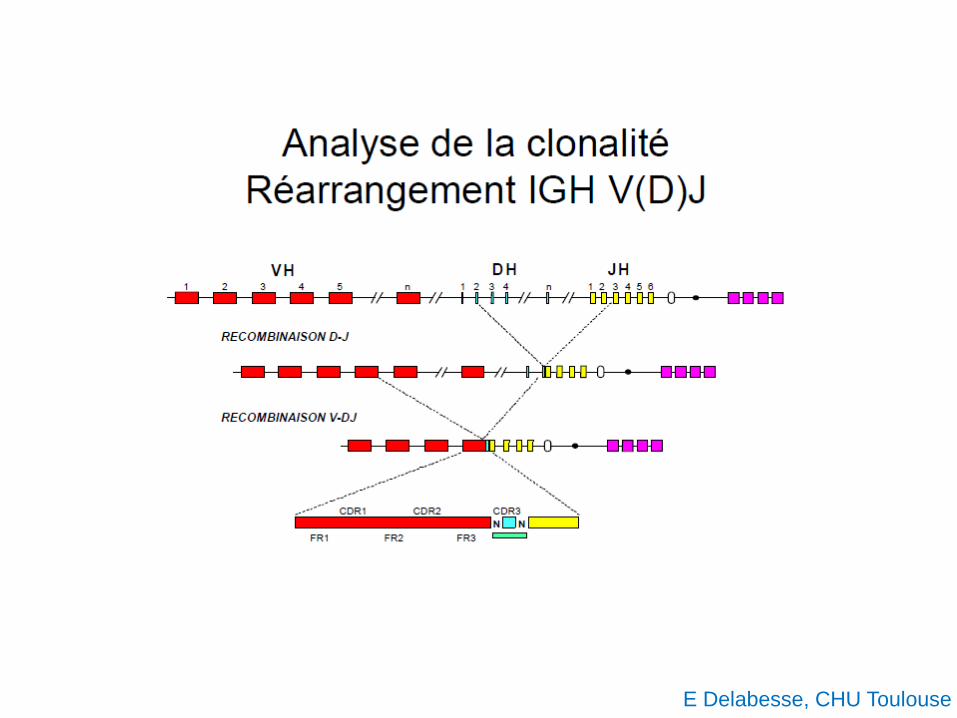
E Delabesse, CHU Toulouse
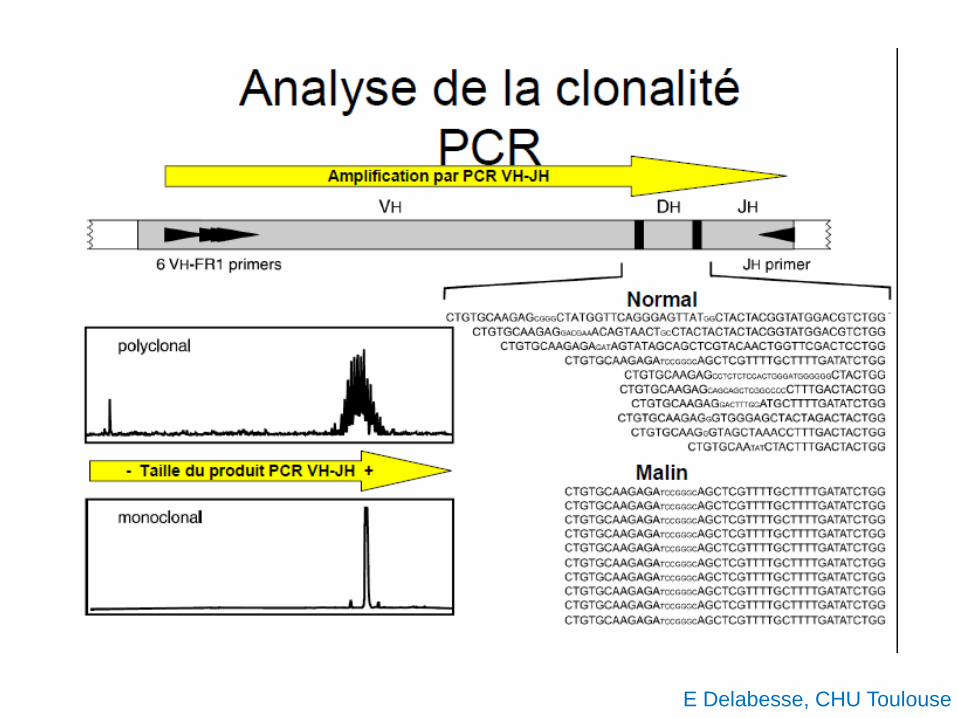
E Delabesse, CHU Toulouse

Bilan cytogénétique, immunophénotypique et moléculaire
au diagnostic (1)
Anomalies cytogénétiques et/ou moléculaires acquises présentes
dans toutes les hémopathies malignes
Valeur diagnostique des anomalies de type primaire (exemple du Ph)
La majorité de ces anomalies a une valeur pronostique indépendante :
anomalies de type primaire : TEL-AML1 versus BCR-ABL dans LAL
anomalies de type secondaire : ACA dans LMC et LAL Ph, deletion Ikaros
dans les LAL
Prise en compte dans les protocoles thérapeutiques ( décision d’
intensification , allogreffe dans les LAL, …)

Bilan cytogénétique, immunophénotypique et moléculaire
au diagnostic (2)
Marqueurs de clonalité à établir au diagnostic pour le suivi des
patients (maladie résiduelle ou MRD)
Suivi cytogénétique : LMC
Suivi moléculaire : LMC et LA (leucémies aigues)
Suivi par CMF : LA
Complémentarité des techniques :
Analyse globale du génome :
caryotype (résolution 10 000kb)
Analyses ciblées :
FISH (resolution : 50 à 100kb) ,
PCR (transcrits chimériques) ,
séquençage des gènes d’Ig et TCR dans LAL
Suspicion d’hémopathie maligne :
signes cliniques et biologiques diversement associés
Signes d’insuffisance médullaire :
Anémie
Syndrome infectieux
Syndrome hémorragique
Signes tumoraux :
douleurs osseuses
splénomégalie
adénomégalie
hépatomégalie
Hémogramme
(NFS avec réticulocytes)
Anémie arégénérative
Neutropénie
Thrombopénie
Hyperleucocytose
( si blastes circulants ou
si myélémie)
Suspicion d’hémopathie maligne :
exploration de la moelle osseuse
• Myélogramme (frottis)
• Immunophénotypage (EDTA)
• Cytogénétique (Héparine)
Caryotype, FISH
• Biologie moléculaire (EDTA)
transcrits anormaux , mutations , …