ème journée française de la fibrose pulmonaire … · ... qu’il s’agisse de cellules souches...
TRANSCRIPT

3ème Journée française de la fibrose pulmonaire idiopathique
19 septembre 2014
Comité scientifique : Pr Vincent Cottin, CHU Louis Pradel – Lyon, Pr Bruno Crestani, CHU Bichat –
Paris, Pr Philippe Delaval, CHU Pontchaillou – Rennes, Pr Dominique Valeyre, Hôpital Avicenne –
Bobigny, Pr Benoït Wallaert, CHRU Calmette - Lille
Publication réalisée à l’issue de la 3ème Journée Française de la Fibrose Pulmonaire Idiopathique qui
a eu lieu le 19 septembre 2014 à Paris avec le soutien institutionnel d’InterMune.
La 3ème Journée Française de la Fibrose Pulmonaire Idiopathique a fait le point sur l’actualité
particulièrement riche de l’année 2013-2014, notamment la publication de trois études dans le
New England Journal of Medicine faisant la preuve de l’efficacité de deux médicaments dans
la Fibrose Pulmonaire Idiopathique, et d’une reclassification des pneumopathies interstitielles
idiopathiques.
REVUE DE L’ACTUALITÉ
L’origine du fibroblaste
D’après une communication du Pr Bruno Crestani, Paris
Les fibroblastes qui s’accumulent dans le foyer de fibrose se distinguent des fibroblastes
normaux par des capacités de migration augmentées d’un facteur 3 lorsqu’ils sont placés sur
une matrice extracellulaire (1), et par plus de 350 de leurs gènes différentiellement exprimés
(2).
Quelle est la source des fibroblastes ?
La question de la source de ces fibroblastes est régulièrement discutée depuis une quinzaine
d’années, et deux nouveaux acteurs ont été récemment identifiés.
Les principales sources à ce jour sont : l’amplification des cellules mésenchymateuses
naturellement présentes dans le poumon sain ; la transition épithélio-mésenchymateuse
(d’autres transitions ayant été décrites, par exemple de cellules mésothéliales ou
endothéliales) ; le recrutement de précurseurs circulants, qu’il s’agisse de cellules souches
mésenchymateuses circulantes ou de fibrocytes qui prennent leur origine dans la moelle. Le
rôle respectif de ces différentes sources reste très discuté.
Nous évoquerons plus particulièrement deux types de cellules, le lipofibroblaste et le péricyte.
Le lipofibroblaste établit des relations intimes avec la cellule épithéliale alvéolaire passant par
la libération de facteurs intervenant dans l’homéostasie de la cellule épithéliale. Il est
vraisemblable qu’une altération de la communication intercellulaire se produit au cours de la
fibrose (3). Le péricyte est une cellule mésenchymateuse très spécialisée partageant sa
membrane basale avec les cellules endothéliales. Différents marqueurs ont été identifiés
(PDGFRβ/Platelet-derived growth factor receptor-beta, NG2/neuron-glial antigen2,

CD146/cluster of differentiation 146, Angiopoiétine 1, etc.). Un modèle de fibrose induite par
la bléomycine chez la souris a permis de montrer que des péricytes sont une source
importante de myofibroblastes responsables de la fibrose (4). Il a par ailleurs été montré que
des péricytes pouvaient contribuer au développement de la fibrose pulmonaire chez l’homme
(5).
Comment le phénotype fibrosant des fibroblastes est-il maintenu ?
Il a récemment été montré que le fibroblaste interagit avec la matrice extracellulaire sur
laquelle il repose et que, dans la fibrose, cette matrice extracellulaire est rigide par rapport à
celle du poumon normal. Cette rigidité conditionne le phénotype du fibroblaste et celui-ci est
réversible ; ainsi des fibroblastes de FPI mis en culture sur une matrice saine, de rigidité
physiologique, redeviennent normaux (6). Dans une étude publiée en 2014, l’analyse de
l’expression génique de fibroblastes de poumon humain normal mis en culture sur des
sections de poumon décellularisé, normal ou fibreux, a montré que le facteur déterminant leur
phénotype n’était pas la provenance du fibroblaste (d’un poumon sain ou fibreux), mais la
matrice sur laquelle ils étaient mis en culture (7). Le phénomène était modulé in vivo par
l’inhibition des enzymes assurant la rigidité et la cohérence de la matrice : enzymes du cross
linking du collagène (lysyl oxydase-like 2) (8), transglutaminase 2 (9, 10). Par ailleurs, in vitro
et in vivo, l’inhibition de la voie Rho kinase, activée lors de l’interaction entre les fibroblastes
et la matrice, permet d’inhiber les propriétés contractiles des fibroblastes (6) et de protéger
contre la fibrose expérimentale (11).
Le cercle vicieux de la fibrose peut donc vraisemblablement être brisé en jouant sur ces deux
voies.
Nouvelle classification des PID
D’après une communication du Pr Hilario Nunes, Bobigny
La révision 2013 de la classification ATS-ERS des pneumopathies interstitielles diffuses
(PID) conforte les grandes innovations de 2002 dans le cadre des PII, notamment l’approche
multidisciplinaire et dynamique, et renforce la possibilité d’un diagnostic sans recours
obligatoire à la biopsie pulmonaire chirurgicale. Elle définit trois grands cadres au sein des
pneumopathies interstitielles idiopathiques (PII) : les PII majeures, les PII rares et les PII
inclassables. Les PII majeures se subdivisent en trois formes : les formes chroniques
fibrosantes, les formes aiguës ou subaiguës fibrosantes, et les formes liées au tabac (cf. figure
1) (12).
Figure 1 : Classification ATS-ERS des PID (12)

BR-PID : bronchiolite respiratoire avec pneumopathie interstitielle diffuse ; DIP : pneumopathie
interstitielle desquamative ; FPI : fibrose pulmonaire idiopathique ; LAM :
lymphangioléiomyomatose ; LPA : lipoprotéinose alvéolaire ; PCE : pneumopathie chronique à
éosinophiles ; PIA : pneumopathie interstitielle aiguë ; PID : pneumopathie interstitielle diffuse ; PII :
pneumopathie interstitielle idiopathique ; PINS : pneumopathie interstitielle non spécifique ; POC :
pneumopathie organisée cryptogénique.
Pneumopathies interstitielles idiopathiques majeures
Le caractère sporadique ou familial des PII majeures est à présent bien souligné avec
l’identification de différentes mutations au niveau des gènes des télomérases (TERT et
TERC), de la protéine C ou de la protéine A2 du surfactant et, plus récemment, avec la mise
en évidence d’un polymorphisme du récepteur de MUC5B, présent chez un tiers des patients
dans les formes familiale et sporadique (13, 14). La maladie a une transmission autosomique
dominante, avec une pénétrance variable et un phénomène d’anticipation. Elle s’intègre dans
le cadre du syndrome des télomères courts ou téloméropathies (15).
Ces patients ont des aspects histologiques et en TDM variés au sein de la même famille et
restent inclassables dans 60 % et 55 % des cas respectivement (16-20). Ils ont fréquemment
une exposition environnementale, ou au tabac, qui doit être prise en compte lors d’un conseil
génétique (20).
Un point majeur de la nouvelle classification ATS-ERS est la distinction de la pneumopathie
interstitielle non spécifique (PINS), à présent considérée comme une entité clinique réelle et
non plus provisoire, bien séparée de la fibrose pulmonaire idiopathique par un pronostic bien
meilleur et une réponse thérapeutique plus fréquente (21).
Les PINS peuvent être idiopathiques, mais il faut savoir rechercher des causes potentielles,
notamment les connectivites différenciées ou indifférenciées, les pneumopathies
d’hypersensibilité, les syndromes de détresse respiratoire aiguë et les causes médicamenteuses
(22, 23). Le diagnostic reste histologique et prend en compte différents éléments cliniques et
paracliniques qui permettront de tenter de distinguer la PINS de la FPI, mais aucun ne permet
un diagnostic différentiel avec certitude en dehors de la biopsie pulmonaire chirurgicale (12,

24, 25). L’aspect scanographique est très variable. Les deux éléments discriminants les plus
importantes sont le « rayon de miel » pour la FPI et le respect sous-pleural pour la PINS.
La mise à jour de la classification ATS-ERS ne comporte pas de grandes nouveautés
concernant la pneumopathie organisée cryptogénique et la pneumopathie interstitielle aiguë.
Dans les formes liées au tabac, il existe un continuum entre la bronchiolite respiratoire avec
PID et la pneumopathie interstitielle desquamative. Il est maintenant admis qu’un tableau
clinico-radiologique évocateur de pneumopathie interstitielle desquamative avec, en
particulier, une alvéolite macrophagique majeure, la présence de macrophages pigmentés,
parfois des sidérophages et des polynucléaires éosinophiles, permet souvent de ne pas réaliser
de biopsie pulmonaire chirurgicale.
Pneumopathies interstitielles idiopathiques rares
La fibroélastose pleuroparenchymateuse est une entité décrite pour la première fois en 1992,
puis dans une série américaine (26). La terminologie proposée a été « fibrose biapicale », puis
« fibroélastose pleuroparenchymateuse ». Cette entité est le plus souvent idiopathique
sporadique ou familiale, mais peut aussi survenir dans certains contextes comme le rejet
chronique post-transplantation pulmonaire, la greffe de moelle, la radiothérapie, la
chimiothérapie (alkylants), et des expositions environnementales variées. C’est une entité qui
survient sans prédominance d’âge, plutôt aux alentours de 40 ans, sans lien avec le sexe ou
avec le tabac. Les patients ont des signes fonctionnels non spécifiques, ils ont souvent un
thorax plat caractéristique (27), qui a tendance à s’aggraver en cours d’évolution.
L’hippocratisme digital et les râles crépitants sont rares. Le diagnostic est histologique, mais
la biopsie pulmonaire chirurgicale est risquée chez ces patients, avec un risque de bullage
persistant. Cette maladie est grave avec des complications à type de pneumothorax, parfois
des co-infections aspergillaires. La médiane de survie est de 11 ans (26-29).
Pneumopathies interstitielles idiopathiques inclassables
Leur prévalence est estimée entre 10 et 30 %. La terminologie d’ « inclassable » a été
débattue parce qu’il s’agit d’un groupe très hétérogène ainsi que l’a bien montré une étude
rétrospective monocentrique américaine qui a étudié des cas de patients suivis pour une PID
entre 2000 et 2011. Dans 10 % des cas, la PID était considérée comme inclassable ou
inclassée pour les raisons suivantes : 71 %, biopsie pulmonaire chirurgicale non réalisée,
quelle qu’en soit la raison (refus du patient, contre-indication du fait de comorbidités ou de la
sévérité de la maladie, etc…) ; 19 %, données cliniques, radiologiques et histologiques
contradictoires (30).
La classification clinique pragmatique, selon le comportement évolutif de la maladie, se base
sur l’évolution observée de la maladie, un objectif thérapeutique et une stratégie de suivi.
Plusieurs situations sont définies : maladie réversible avec atteinte limitée, maladie réversible
avec risque de progression, maladie stable mais persistante, maladie progressive irréversible
avec potentiel de stabilisation, maladie progressive irréversible malgré le traitement.
Conclusion
Cette nouvelle classification conforte très largement la classification de 2002. La biopsie
pulmonaire chirurgicale est de moins en moins indispensable, notamment dans les PII liées au
tabac, et dans la FPI lorsque l’aspect est typique. La classification individualise clairement la
PINS comme une entité à part entière. Elle intègre le caractère familial de certaines PII. Elle
individualise cette nouvelle entité qu’est la fibroélastose pleuroparenchymateuse. Elle tente de
définir des pneumopathies interstitielles inclassables avec un rôle de la classification
pragmatique comportementale qui doit être mieux évalué par des études prospectives. Enfin,

il importe de distinguer les termes d’ « inclassable » et de« non classée », qui ne représentent
pas les mêmes circonstances.
Prise en charge de la fibrose pulmonaire idiopathique
D’après une communication du Dr Grégoire Prévot, Toulouse
Plus de 430 articles portant sur la prise en charge de la FPI ont été publiés depuis le début de
l’année. Ils soulignaient notamment les difficultés rencontrées dans la mise en pratique des
recommandations diagnostiques de FPI.
Diagnostic : affirmer le diagnostic de FPI
Le travail de G Raghu, publié dans le Lancet Respiratory Medicine, visait à évaluer la valeur
prédictive d’un diagnostic anatomopathologique de pneumopathie interstitielle commune
(PIC) d’aspect « possible » en TDM (PIC certaine ou probable). Dans cette perspective, il a
analysé des patients sélectionnés pour participer à un protocole thérapeutique dans la FPI
(étude ARTEMIS-IPF). Ces patients devaient être suspects de FPI avec moins de 5 % de
rayon de miel en TDM. Mille quatre-vingt-sept patients ont été sélectionnés dont 315 ont
bénéficié d’une biopsie pulmonaire chirurgicale (31).
Le diagnostic a été confirmé par la biopsie chirurgicale chez 108 sur 111 patients avec PIC
« certaine » en TDM (1 PIC « possible », 2 « non PIC), soit une valeur prédictive positive de
97,3 %. Soixante-dix-neuf sur 84 patients avec PIC « possible » en TDM ont eu un diagnostic
anatomopathologique concordant (65 PIC « certaine », 14 PIC « probable »), soit une valeur
prédictive positive de 94 %. Enfin, les investigateurs ont retrouvé un aspect de PIC chez 98
patients (76 PIC « certaine » et 22 PIC « probable ») parmi les 120 patients dont l’aspect en
TDM était incompatible avec une PIC. L’importance de cette discordance tiendrait à ce que
les patients avaient été adressés pour un protocole s’intéressant à la FPI.
Dans cette situation, est-il utile de proposer une biopsie pulmonaire chirurgicale chez un
patient avec une PIC « possible » en TDM ?
Une réponse négative sera en accord avec Fell et al. selon lesquels, dans un contexte de
suspicion de FPI survenant après 70 ans, un aspect TDM de PIC a une bonne valeur
prédictive de PIC histologique. Pourtant, en l’état actuel des choses, et malgré les résultats du
travail de G Raghu, il reste nécessaire de procéder à des biopsies pulmonaires chirurgicales
pour les PIC « possibles » en TDM (31). En effet, l’étude de G Raghu présente un biais de
sélection majeur puisqu’elle incluait des patients adressés avec un diagnostic de FPI en vue de
l’inclusion dans un essai thérapeutique. Les situations de non concordance entre PIC
« possible » en TDM et à la biopsie pulmonaire chirurgicale étaient en conséquence très sous-
estimées (32).
Diagnostic : affirmer le caractère idiopathique
Une équipe de Barcelone a étudié 46 patients chez qui avait été porté le diagnostic de FPI
selon les critères des recommandations de 2011. L’utilisation de questionnaires spécifiques de
la pneumopathie d’hypersensibilité et de tests de provocation a permis de montrer la présence
d’éléments en faveur d’une pneumopathie d’hypersensibilité chez 20 des 46 patients (33).
Malgré ses limites, cette étude pose la question de la fiabilité des critères de 2011 et souligne
que porter le diagnostic de FPI suppose une enquête exhaustive de la part du clinicien.
Les recommandations préconisent la recherche systématique d’une connectivite avec la
recherche des anticorps et des facteurs antinucléaires et anti-CCP (anti-cyclic citrullinated
peptide). Une étude de la Mayo Clinic a montré que la détection d’anticorps était positive

chez plus d’un tiers des patients avec FPI et preuve anatomopathologique de PIC. Cette
observation est cohérente avec les travaux de Lee et al. qui ont démontré que 20 % des
patients atteints d’une maladie pulmonaire idiopathique présentaient des anticorps (34). En
conséquence, la présence d’anticorps ne suffit pas à éliminer le diagnostic de FPI et il importe
d’aller plus loin dans la recherche de la connectivite.
Dans une étude portant sur 111 patients atteints de FPI, 10 ont développé une maladie
systémique (polyarthrite rhumatoïde : 4, sclérodermie : 2, polyangéite microscopique : 4)
(35). En conséquence, les patients chez qui a été posé avec certitude le diagnostic de FPI
doivent être l’objet d’un suivi attentif en raison du risque de maladie systémique encouru (30,
36).
Prise en charge et thérapeutiques
Valeyre et al. ont étudié la tolérance de la pirfénidone au long cours chez 789 patients exposés
pendant 2,6 ans en moyenne, plus de 37 % ayant été exposés à cet agent pendant plus de 3
ans, et plus de 50 % à pleine dose. Un arrêt du traitement n’a été enregistré que dans 35 % des
cas, le plus souvent en raison d’une aggravation de la FPI et non pour effet indésirable. Les
effets indésirables (EI) survenaient principalement durant les six premiers mois. Globalement,
le traitement est apparu bien toléré s’il y avait un accompagnement des patients, en particulier
durant les six premiers mois (37).
Les bénéfices de la transplantation pulmonaire ont été étudiés chez huit patients porteurs
d’une téloméropathie. Un décès s’est produit huit mois après la transplantation et les sept
autres patients étaient en vie à 1,9 ans. Les immunosuppresseurs ont dû être adaptés chez tous
les patients en raison d’EI hématologiques. Quatre patients sur huit ont du être dialysés en
raison d’une toxicité rénale aux anticalcineurines. Une colite ischémique sous mycophénolate
mofétil et des hépatites fulminantes sous azathioprine ont également été observées (38). La
transplantation pulmonaire est donc possible chez les patients avec téloméropathie, mais ses
bénéfices restent incertains et les patients doivent en être informés.
Évaluation du pronostic
L’un des principaux éléments pronostiques dans la FPI est le MRSS (Mortality Risk Scoring
System). Il lui a été ajouté un test de marche de 6 minutes (TM6) en vue de l’améliorer. Une
étude de du Bois et al. a montré que la distance parcourue au TM6 est un facteur pronostique
indépendant et la prise en compte de la distance au TM6 au diagnostic et de l’évolution à 24
semaines corrige 26 % des évènements prédits par le MRSS seul (39).
Enfin, la lysyl oxidase-like [(LOXL)-2] est un biomarqueur qui semble très prometteur. La
LOXL-2 est produite par les fibroblastes et favorise la fibrogénèse. Elle est la cible d’un
traitement en cours d’étude et elle est détectable dans le sérum (40)
Conclusion
La revue des publications récentes montre que l’application des recommandations reste
essentielle. Les rares exceptions doivent relever de décisions multidisciplinaires en centres
spécialisés. Le caractère idiopathique n’est pas toujours facile à affirmer. Il importe de se
donner les moyens de raccourcir le délai de diagnostic. L’évaluation du pronostic est
primordiale, en particulier pour les patients éligibles à la transplantation. Les scores
continuent de se développer et on ne peut que souhaiter qu’ils soient appliqués. Enfin, il y a
une place future pour les biomarqueurs.

Est-ce vraiment du rayon de miel ?
D’après une communication des Dr Marie-Pierre Debray et Claire Danel,
Paris
Un aspect scannographique en rayon de miel est l’un des quatre critères diagnostiques qui
définissent la pneumopathie interstitielle commune (PIC) « certaine » et c’est le seul qui
distingue cette dernière de la PIC « possible ». Sa présence associée à celle des autres critères
(Tableau 1) dispense de la biopsie pulmonaire chirurgicale. À l’inverse, la présence de rayon
de miel n’est pas obligatoire pour le diagnostic anatomopathologique de la PIC « certaine » et
« probable » (Tableau 2) (41).
Tableau 1. Critères diagnostiques en scanner de la pneumopathie interstitielle
commune, selon les recommandations ATS/ERS 2011 (41).
Tableau 2. Critères diagnostiques en anatomie pathologique de la pneumopathie
interstitielle commune selon les recommandations ATS/ERS 2011 (41).

Pré-requis techniques
Utiliser une technique scannographique appropriée
Se prononcer sur l’existence ou non d’un rayon de miel exige que le scanner soit effectué
avec une technique appropriée. On peut effectuer le scanner en mode incrémental, en réalisant
des coupes en haute résolution espacées de 10 ou 20 mm ou, de plus en plus, préférer une
acquisition volumique car cette dernière permet des reformatages dans d’autres plans
(coronal, sagittal) ainsi que divers post traitements, notamment de type minIP. L’examen doit
privilégier la résolution spatiale et donc des coupes millimétriques ou infra-millimétriques.
Les scanners thoraciques sont habituellement reconstruits en deux types de filtres, l’un
privilégiant la résolution spatiale et l’autre la résolution en densité. Les coupes reconstruites
avec le filtre spatial sont imagées en fenêtrage pulmonaire, et ce sont celles-ci qui doivent être
analysées pour la recherche du rayon de miel. Les coupes reconstruites avec le filtre de
densité, imagées en fenêtrage médiastinal, peuvent être utilisées pour les post traitements de
type minIP.
L’examen doit être effectué en bonne inspiration. Il est nécessaire de disposer de coupes en
procubitus lorsqu’existe un aspect en verre dépoli localisé ou prédominant en zones déclives.
La taille de l’échantillon pour l’examen anatomopathologique
La taille de la biopsie fournie par le chirurgien doit avoir au moins 2 cm de profondeur et
éviter des bandes parallèles à la plèvre, les territoires trop fibreux et les pointes qui sont
souvent le siège de remaniements inflammatoires non spécifiques. Le nombre de
prélèvements doit être au moins d’un fragment par lobe prélevé dans des zones qui auront été,
idéalement, pré-établies avec les radiologues et les cliniciens. Le pathologiste doit être
informé de la localisation précise de chaque biopsie.
Définition
Le terme de poumon en rayon de miel apparaît au cours du 19ème siècle. Ce terme recouvre
alors plusieurs entités débordant les pneumopathies interstitielles chroniques, et incluant en
outre les bronchectasies, les maladies kystiques pulmonaires, l’emphysème bulleux. Ce n’est
qu’avec l’avènement du scanner haute résolution qu’il sera limité à la notion de poumon en
phase terminale (« end-stage » lung), spécifique de fibrose pulmonaire.
Définition scannographique
La définition de la Fleischner Society, révisée en 2008, précise que le rayon de miel
correspond à un « cluster » ou regroupement d’espaces aériens (c’est-à-dire de densité
aérique, apparaissant noirs comme l’air intratrachéal). Les diamètres sont typiquement
similaires, le plus souvent de 3 à 10 mm. Il y a souvent, au moins dans les phases évoluées,
quelques formations aériques un peu plus grosses que les autres. Les parois sont bien définies.
Sa situation est habituellement sous-pleurale (Figure 2) (42).
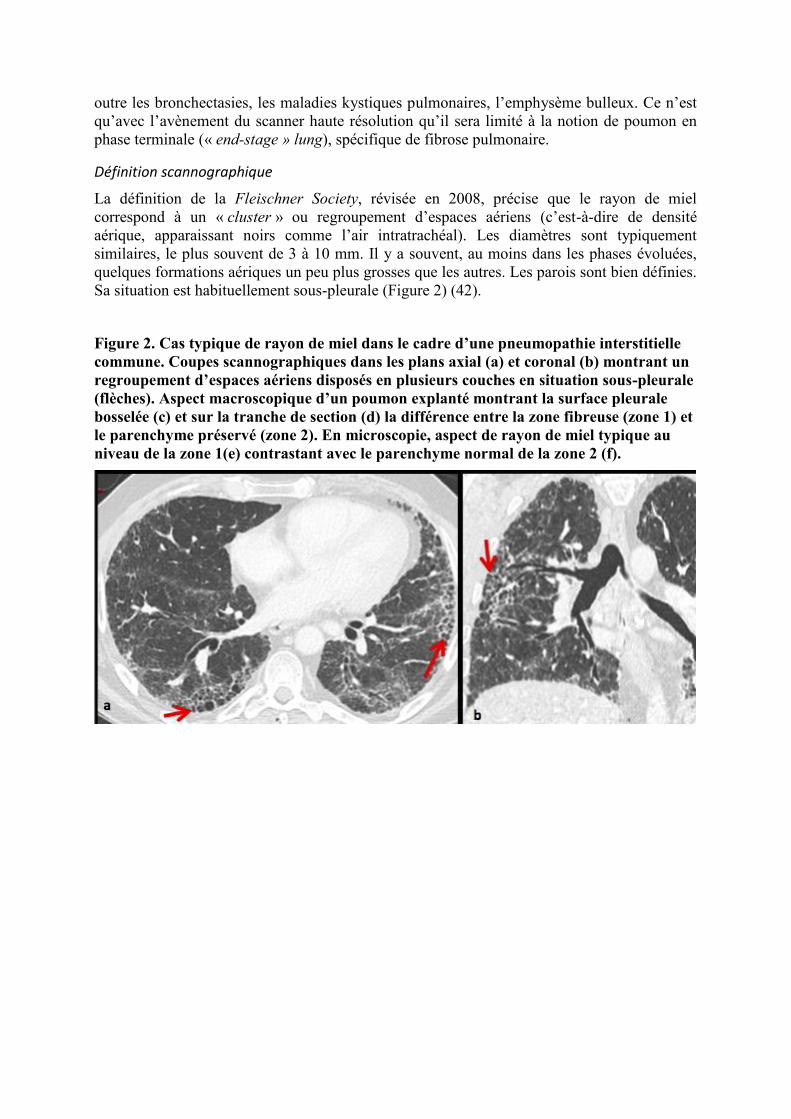
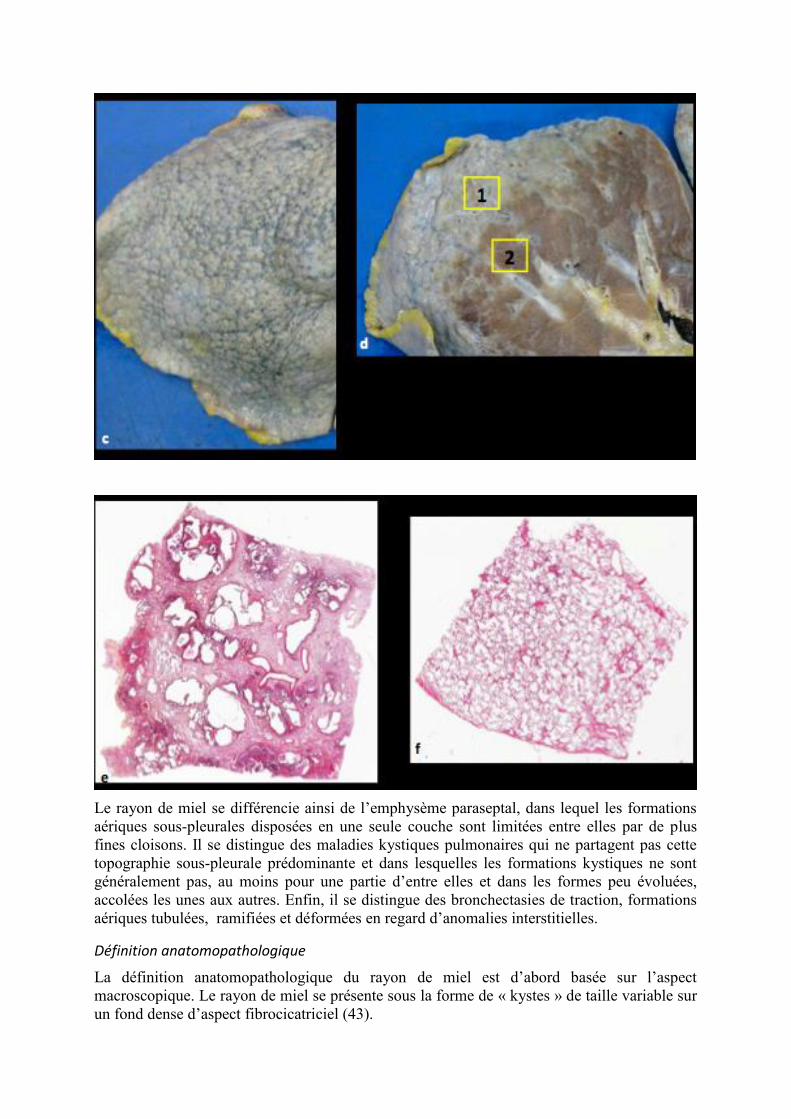
Figure 2. Cas typique de rayon de miel dans le cadre d’une pneumopathie interstitielle
commune. Coupes scannographiques dans les plans axial (a) et coronal (b) montrant un
regroupement d’espaces aériens disposés en plusieurs couches en situation sous-pleurale
(flèches). Aspect macroscopique d’un poumon explanté montrant la surface pleurale
bosselée (c) et sur la tranche de section (d) la différence entre la zone fibreuse (zone 1) et
le parenchyme préservé (zone 2). En microscopie, aspect de rayon de miel typique au
niveau de la zone 1(e) contrastant avec le parenchyme normal de la zone 2 (f).
Le rayon de miel se différencie ainsi de l’emphysème paraseptal, dans lequel les formations
aériques sous-pleurales disposées en une seule couche sont limitées entre elles par de plus
fines cloisons. Il se distingue des maladies kystiques pulmonaires qui ne partagent pas cette
topographie sous-pleurale prédominante et dans lesquelles les formations kystiques ne sont
généralement pas, au moins pour une partie d’entre elles et dans les formes peu évoluées,
accolées les unes aux autres. Enfin, il se distingue des bronchectasies de traction, formations
aériques tubulées, ramifiées et déformées en regard d’anomalies interstitielles.
Définition anatomopathologique
La définition anatomopathologique du rayon de miel est d’abord basée sur l’aspect
macroscopique. Le rayon de miel se présente sous la forme de « kystes » de taille variable sur
un fond dense d’aspect fibrocicatriciel (43).

Au plan microscopique, le rayon de miel est caractérisé par des lacunes de tailles variables,
les plus petites, millimétriques, peu ou non visibles au scanner. Ces lacunes ont un contenu
mucoïde, elles sont toujours bordées d’un épithélium, soit de type bronchiolaire métaplasique,
soit alvéolaire hypertrophique. Elles siègent au sein d’un tissu fibreux dense (Figure 2) (43).
Physiopathologie
Plusieurs hypothèses ont été émises. Selon la première d’entre elles, les aspects de rayon de
miel seraient liés à une destruction du parenchyme distal avec distensions des espaces
alvéolaires, fibrose et obstruction ou distension bronchiolaire. Ces espaces aériens néoformés
étant tapissés par deux types de revêtement épithélial, des pneumocytes ou des cellules
bronchiolaires, ciliées et mucosécrétantes (44).
Selon la deuxième hypothèse, il pourrait s’agir de la conséquence d’une bronchiolisation des
espaces aériens périphériques par une cellule souche activée au cours d’un processus de
réparation aberrant qui ferait l’acquisition d’un profil migratoire et irait se localiser et se
différencier au niveau du site de réparation (45, 46).
Difficultés en scanner
La définition des lésions en rayon de miel vues au scanner présente un certain nombre de
difficultés, notamment en raison des imprécisions de la définition de la Fleischner Society,
qui ne mentionne pas quel est le nombre d’espaces aériens permettant de parler de clusters, et
reste peu précise quant à la topographie préférentielle des lésions. Enfin, si la majorité des
radiologues considère qu’une seule couche d’espaces aériens suffit pour établir le rayon de
miel en cas d’image typique, certains en restreignent la définition aux formes multicouches.
Ce point n’est pas non plus précisé dans la définition de la Fleischner Society.
Un travail de Watadani publié en 2013 a évalué l’accord inter-observateurs pour le diagnostic
de rayon de miel en scanner, entre pneumologues experts et radiologues, experts et non
experts, de différents pays. Cet accord n’était que modéré, avec des kappa de l’ordre de 0,5,
sans différence significative entre les différents groupes de lecteurs. Les divergences portaient
principalement sur des cas d’association du rayon de miel avec des bronchectasies de traction
ou avec des lésions d’emphysème et sur la présence de kystes de grande taille (Figure 3). Il
faut toutefois souligner que les lecteurs se prononçaient sur une seule image par cas, qu’il n’y
avait pas de corrélation avec l’analyse microscopique et que les cas proposés étaient en
grande partie jugés difficiles. Il est ainsi possible que dans d’autres conditions, plus proches
de la pratique usuelle, l’accord inter-observateurs puisse être meilleur (47).
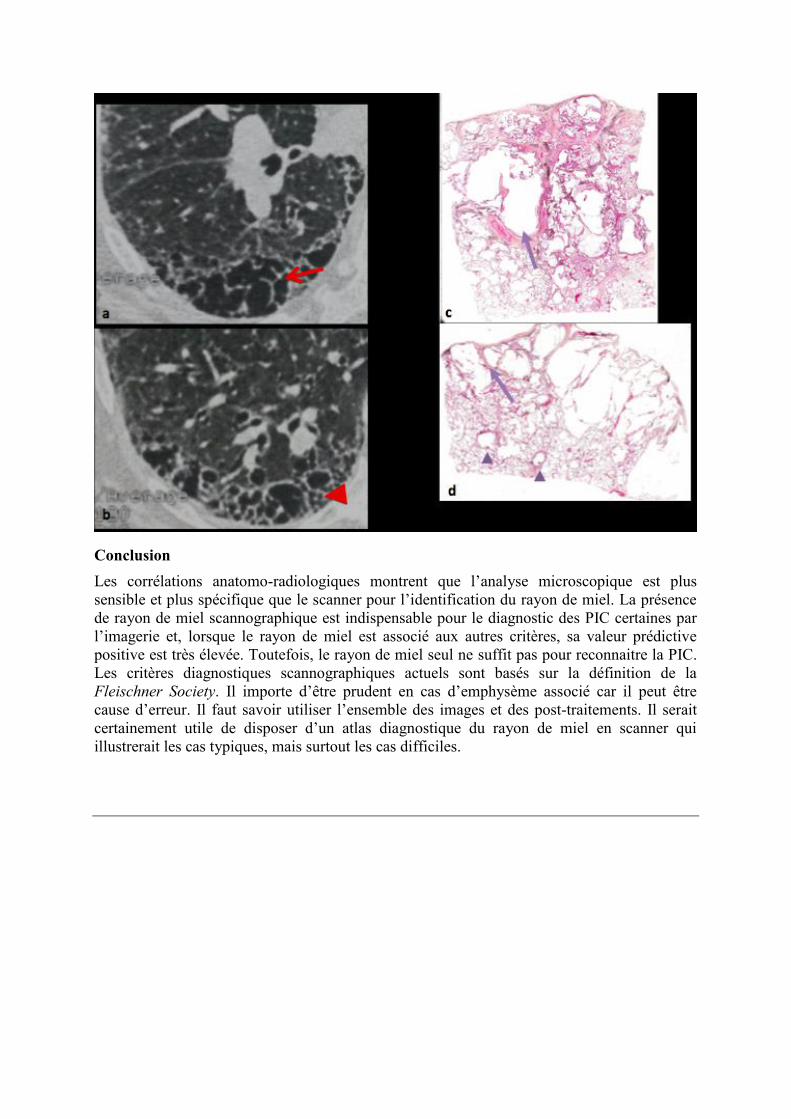
Figure 3. Diagnostic différentiel difficile en scanner (a, b). La présence de formations
aériques de grande taille avec paroi visible, bien que pouvant faire évoquer du rayon de
miel ne permet pas un diagnostic formel et peut correspondre à un emphysème remanié
(flèche), associé ici à des bronchectasies de traction (tête de flèche). Aspect
correspondant en microscopie (c, d) : emphysème à parois fibreuses (flèches) et
bronchiolectasies (têtes de flèche).
Conclusion
Les corrélations anatomo-radiologiques montrent que l’analyse microscopique est plus
sensible et plus spécifique que le scanner pour l’identification du rayon de miel. La présence
de rayon de miel scannographique est indispensable pour le diagnostic des PIC certaines par
l’imagerie et, lorsque le rayon de miel est associé aux autres critères, sa valeur prédictive
positive est très élevée. Toutefois, le rayon de miel seul ne suffit pas pour reconnaitre la PIC.
Les critères diagnostiques scannographiques actuels sont basés sur la définition de la
Fleischner Society. Il importe d’être prudent en cas d’emphysème associé car il peut être
cause d’erreur. Il faut savoir utiliser l’ensemble des images et des post-traitements. Il serait
certainement utile de disposer d’un atlas diagnostique du rayon de miel en scanner qui
illustrerait les cas typiques, mais surtout les cas difficiles.

RÉSULTATS DES ESSAIS THÉRAPEUTIQUES PUBLIES EN
2014
Étude Panther
D’après une communication du Pr Benoît Wallaert, Lille
Après des résultats semblant prometteurs, l’intérêt de NAC dans la FPI n’a pas été confirmé
par l’étude Panther, mais ses résultats sont néanmoins suffisamment intéressants pour être
présentés.
La N-acétyl-cystéine
La N-acétyl-cystéine (NAC) aboutit par réductions successives à la formation du glutathion
qui compte parmi les plus efficaces des antioxydants. Un travail publié en 1990 avait
démontré qu’in vitro, l’adjonction de glutathion dans les milieux de culture inhibait la
prolifération des fibroblastes pulmonaires (48). Il existe donc des bases rationnelles soutenant
l’utilisation de la NAC et d’antioxydants comme le glutathion dans le traitement de la
prolifération des fibroblastes.
Un autre travail, mené peu après chez 17 patients atteints de FPI non-fumeurs et 14 sujets
témoins a montré que les liquides de lavage broncho-alvéolaire et les films liquidiens de
l'épithélium des patients FPI contenaient de moindres quantités de glutathion que ceux des
témoins. Un traitement par 1,8 g de NAC pendant 5 jours permettait de corriger ce déficit
(49).
Une étude randomisée multicentrique en double-aveugle, contrôlée contre placebo, a été
menée en vue d’évaluer l'efficacité sur un an d'une dose orale élevée de NAC (600 mg x 3/j)
ajoutée au traitement standard par prednisone plus azathioprine dans le traitement de la FPI.
Les critères principaux de jugement étaient l’évolution de la capacité vitale forcée (CVF) et
de la capacité de transfert du monoxyde de carbone (DLCO). Les résultats ont été
encourageants, l’adjonction de NAC paraissant préserver plus efficacement la CVF et la
DLCO. Il a cependant été montré par la suite que la méthodologie n’était pas parfaite et qu’il
manquait un vrai groupe placebo (50).
Étude Panther
Panther est une étude en double-aveugle, randomisée, contrôlée contre placebo, incluant des
patients avec FPI avec altération de la fonction pulmonaire de légère à modérée. Ils ont été
répartis en trois groupes pour recevoir : 1) une association de prednisone, d’azathioprine et de
NAC (bras triple thérapie), 2) une monothérapie par NAC seule, 3) un placebo. Le critère
principal de jugement était l’évolution de la CVF pendant un an (51).
Une analyse intermédiaire a révélé une augmentation du taux de décès chez les patients du
groupe triple thérapie (8 vs 1, p = 0,01) et d'hospitalisation (23 vs 7, P <0,001) alors qu’aucun
signe de bénéfice physiologique ou clinique en faveur de la triple thérapie n’apparaissait. Le
Conseil de surveillance a donc préconisé l’arrêt du groupe association (51).
L’étude réduite aux deux groupes NAC et placebo s’est poursuivie sans autres changements.
À un an, il n’y avait pas de différence entre les deux groupes pour la CVF, la mortalité et les
exacerbations aiguës (51).

Conclusion
Il n’y a pas lieu, aujourd’hui, de donner de la N-acétyl-cystéine en monothérapie et il n’y a
pas d’indication pour une trithérapie comportant de la NAC dans la FPI.
Il serait cependant dommage de « jeter l’enfant avec l’eau du bain ». Il reste une place en
recherche pour une évaluation de l’association de la NAC à d’autres molécules anti-
fibrotiques. Certains utilisent la NAC à haute dose dans les exacerbations des FPI. Enfin, la
N-acétyl-cystéine et surtout la trithérapie restent des options possibles dans le traitement des
PINS fibrosantes.
Nintédanib et étude INPULSIS
D’après une communication du Pr Vincent Cottin, Lyon
Le nintédanib (BIBF 1120), ou 6-méthoxycarbonyl-indolinone, est une molécule administrée
par voie orale, développée comme agent anticancéreux. C’est un inhibiteur de tyrosines
kinases, avec plusieurs cibles moléculaires, ses développeurs visant une molécule exerçant
une activité à la fois anticancéreuse et anti-angiogénique qui ne soit pas limitée par des voies
de signalisation redondantes. Des essais de phase III sont en cours pour évaluer cet agent dans
le traitement du cancer bronchique non à petites cellules et dans le cancer de l’ovaire.
Pertinence du mécanisme d’action du nintédanib
Le nintédanib cible une douzaine de récepteurs regroupés en trois grands groupes : récepteurs
du facteur de croissance vasculaire endothélial (VEGF-R), récepteurs du facteur de croissance
des fibroblastes (FGF-R), et récepteurs du facteur de croissance plaquettaire (PDGF-R). Ces
trois types de récepteurs sont impliqués à la fois dans l’angiogénèse, la prolifération des
fibroblastes, et la chémoattraction, soit différents phénomènes impliqués dans la
cancérogenèse, mais également dans la fibrogénèse (52, 53).
Le facteur initial de la fibrogénèse serait la lésion de l’épithélium alvéolaire qui déclenche des
interactions entre les cellules épithéliales et les cellules du mésenchyme pour, in fine, aboutir
à l’accumulation de fibroblastes. Les inhibiteurs de tyrosine kinase, le nintédanib notamment,
ont une action sur les étapes de progression, de recrutement et de prolifération des fibroblastes
(54).
Études de laboratoire et de phase II
Chez l’animal et in vitro, le nintedanib inhibe la transformation des fibroblastes primaires de
FPI en myofibroblastes induite par le facteur de croissance transformant bêta (TGFβ), et il a
été montré qu’il exerce un effet thérapeutique sur la fibrose et l’inflammation induites par la
bléomycine chez la souris (55).
Ces premiers résultats ont conduit à un essai de phase II dans le traitement de la FPI. Ce
travail, incluant plus de 400 patients avec une capacité vitale à plus de 50 % et une DLCO à
plus de 30 % des valeurs théoriques, utilisait la pente de déclin de la CVF à 12 mois pour
critère d’évaluation principal. À la dose de 150 mg deux fois par jour, le nintédanib était
associé à une moindre diminution de la CVF en un an par comparaison avec le placebo.
Élément intéressant, les exacerbations aiguës étaient moins fréquentes dans le groupe recevant
le nintédanib. Les diarrhées, rapportées par 55 % des patients, étaient le principal effet
indésirable du traitement (56).

Études de phase III INPULSIS-1 et INPULSIS-2
À la suite de cet essai, deux études de phase III contre placebo, INPULSIS-1 et INPULSIS-2,
ont testé pendant un an l’efficacité du nintédanib, à la posologie de 150 mg deux fois par jour,
chez 1066 patients de plus de 40 ans ayant un aspect de PIC au scanner thoracique, une
capacité vitale à plus de 50 % et une DLCO à plus de 30 % de la théorique. Le critère
d’évaluation principal était la pente de déclin de la CVF en millilitres par an ; les critères
secondaires étaient le délai de première exacerbation aiguë et le score au questionnaire
respiratoire de St Georges (SGRQ) (57).
Les deux études menées en parallèle ont abouti à des résultats similaires pour le critère
d’évaluation principal, soit une moindre diminution sur un an de la CVF chez les patients qui
recevaient le nintédanib (-114,7 ml contre -239,9 ml avec le placebo, p <0,001 dans
INPULSIS-1 ; et -113,6 ml contre -207,3 ml avec le placebo, p <0,001 dans INPULSIS-2).
Au total, la différence moyenne en un an a été de 110 ml pour la pente de déclin de la capacité
vitale par rapport au placebo (57).
La différence entre les groupes nintedanib et placebo pour le délai de première exacerbation
aiguë n’était pas significative dans INPULSIS-1. Dans INPULSIS-2, en revanche, le
nintédanib était associé à un avantage significatif par rapport au placebo (Hazard ratio : 0,38 ;
IC à 95% : 0,19-0,77 ; p = 0,005). L’évolution du score au questionnaire de St Georges a
montré une différence significative vis-à-vis du placebo dans l’une des deux études, mais pas
dans l’autre (INPULSIS-1 : -0,05 ; p = 0,9657 ; INPULSIS-2 : -2,69 ; p = 0,0197). Les
diarrhées étaient l'évènement indésirable le plus fréquent avec des taux de 61,5 % contre
18,6 % avec le placebo dans INPULSIS-1 et 63,2% et 18,3%, respectivement, dans
INPULSIS-2. Seulement 5 % des patients ont dû arrêter le traitement en raison de cet
évènement (57).
La puissance des essais était insuffisante pour évaluer la mortalité globale, mais il y avait une
tendance en faveur de sa réduction avec un risque relatif de 0,7, la différence étant non
significative dans les deux études (57).
L’effet thérapeutique était tout à fait similaire selon que la CVF à l’inclusion était inférieure
ou supérieure à 70 % de la valeur théorique. Dans les deux cas, la diminution de la CVF a été
moindre avec le nintédanib qu’avec le placebo (d’après la communication de Luca Richeldi,
ERS 2014 Munich). L’effet thérapeutique a également été tout à fait similaire en présence ou
non d’un emphysème associé. Il faut toutefois souligner que les patients inclus n’avaient que
peu ou pas d’emphysème (d’après la communication de V. Cottin, ICLAF meeting, 2014).
Les données de la tolérance ont été similaires à ce qui avait été observé dans l’étude de phase
II avec une prédominance des événements indésirables de nature digestive, de diarrhées
notamment, ainsi que de nausées et vomissements. Ces événements digestifs étaient rarement
sévères et conduisaient rarement à l’arrêt du traitement (57).
Conclusion
Le critère principal d'évaluation a été atteint dans les deux essais INPULSIS : le nintédanib a
considérablement réduit le taux annuel de diminution de la CVF par rapport au placebo. Les
résultats du critère principal étaient confirmés par les analyses de sensibilité.
Il y avait une différence statistiquement significative en faveur du nintédanib pour le délai de
première exacerbation aiguë et l’évolution du score total au questionnaire respiratoire de St
Georges dans INPULSIS-2, mais pas dans INPULSIS-1. Il y avait une différence
statistiquement significative en faveur du nintédanib pour le délai de première exacerbation
aiguë confirmée par un comité d’adjudication dans l’analyse groupée des deux études.

L'événement indésirable le plus fréquent dans les groupes nintedanib était la diarrhée. La
plupart des événements étaient d'intensité légère ou modérée, et moins de 5 % des patients ont
arrêté prématurément le médicament en raison de diarrhées.
Etude ASCEND
D’après une communication du Pr Bruno Crestani, Paris
Quatre essais randomisés, contrôlés, évaluant la pirfénidone dans le traitement de la FPI chez
plus de 1100 patients au total, ont précédé l’étude Ascend : deux essais de phase III qui ont
montré un bénéfice sur la fonction ventilatoire (58, 59) et les deux études CAPACITY qui ont
eu des résultats plus contrastés, l’une étant clairement positive sur le critère principal, alors
que l’autre était négative à 72 semaines. L’analyse groupée était néanmoins positive, montrant
un bénéfice de la pirfénidone sur le déclin de la fonction respiratoire, sur la survie sans
progression, et sur la distance parcourue au test de marche (60).
À la suite de ces résultats, différents pays européens ont accordé une Autorisation de Mise sur
le Marché (AMM) à la pirfénidone dans le traitement de la FPI. Les autorités compétentes
des États-Unis désiraient cependant une troisième étude de phase III conduite dans une
population occidentale. L’étude ASCEND a été menée en vue de répondre à cette demande.
Méthodologie de l’étude ASCEND
Les critères d’inclusion de l’étude Ascend étaient stricts : symptômes présents depuis au
moins 12 mois avec un diagnostic d’ancienneté comprise entre 6 et 48 mois ; un âge compris
entre 40 et 80 ans ; un diagnostic de FPI reposant sur la combinaison d’anomalies
scannographiques et histologiques avec une lecture centralisée (adaptation selon les critères
ATS/ERS/JRS/ALAT 2011) ; au scanner, une fibrose (réticulations, rayon de miel) d’étendue
supérieure à celle de l’emphysème s’il était présent ; absence de diagnostic alternatif. La CVF
devaient être comprise entre 50 et 90 % et la DLCO entre 30 et 90 % des valeurs prédites
(avec lecture centralisée des épreuves fonctionnelles respiratoires). Le patient devait être
capable de marcher plus de 150 m au test de marche de 6 minutes. Les principaux critères
d’inéligibilité étaient un asthme ou une BPCO sévère et un rapport VEMS/CVF post-
bronchodilatateurs inférieur à 0,80 (61).
Le schéma de l’étude était très simple, avec un bras pirfénidone à la dose de 2403 mg par jour
(n = 278) et un bras placebo (n = 277), pour une durée de 52 semaines.
Le critère principal d’évaluation était la variation de CVF à la 52e semaine exprimée en
pourcentage de la valeur initiale. L’importance de l’effet a été jugé sur deux seuils
cliniquement pertinents : pourcentage de patients avec diminution d’au moins 10 % de la CVF
ou décédés ; pourcentage de patients sans diminution de la CVF (61).
Les principaux critères secondaires d’évaluation étaient la variation de la distance parcourue
au test de marche et la survie sans progression définie comme la combinaison de trois
éléments : le décès ou une diminution de la CVF d’au moins 10 % par rapport à la valeur
initiale ou une diminution d’au moins 50 mètres au TM6. Les autres critères secondaires
étaient la variation du score de dyspnée à la 52e semaine (UCSD SOBQ score), la mortalité
toutes causes et la mortalité liée à la FPI (61).
Il y a eu 20 % environ d’arrêts de traitement dans le groupe pirfénidone contre 15 % dans le
bras placebo. Les principales raisons en étaient le décès, la transplantation, ou les événements
indésirables. Ces derniers ont concerné 12 % des patients du groupe pirfénidone contre 8 %
dans le groupe placebo. Près de 95 % des patients ont pu être évalués au terme des 52
semaines de l’étude (61).

La population incluse présentait des caractéristiques proches de celles rencontrées en pratique
clinique : 68 ans d’âge moyen, sexe masculin dans plus de 75 % des cas, CVF de 68 % et
DLCO de 44 % des valeurs théoriques environ. Il y avait plus de 60 % d’anciens fumeurs
(61).
Résultats de l’étude
Efficacité
Les résultats de l’étude sont clairement positifs. Pour ce qui concerne le critère principal, dès
la 13ème semaine de suivi, le pourcentage de patients avec diminution d’au moins 10 % de la
CVF ou décédés pendant l’étude était inférieur dans le groupe pirfénidone par rapport au
placebo (p < 0,001), cette différence s’amplifiant avec le temps (61).
La proportion de patients avec diminution de la CVF de plus de 10 % était de 31,8 % à 52
semaines dans le groupe placebo contre 16,5 % dans le groupe traité (p < 0,001). De la même
façon, le pourcentage de patients qui ne diminuaient pas leur CVF pendant la durée de suivi
était deux fois plus important dans le groupe traité que dans le groupe placebo (22,7 % vs 9,7
%). Les patients ayant perdu plus de 50 mètres au test de marche ou décédés étaient
significativement moins nombreux dans le groupe traitement actif à 52 semaines (p = 0,04).
Le déclin de la CVF a été de l’ordre de 160 ml dans le groupe pirfénidone contre 280 ml dans
le groupe placebo, soit une différence hautement significative de 116 ml (p < 0,0001). Les
courbes de survie sans progression se séparaient dès le 3e mois de l’étude pour aboutir à une
différence très significative en faveur de la pirfénidone en 52e semaine (Hazard ratio : 0,57 ;
IC 95 % : 0,43 – 0,77 ; p <0,001) (61).
Les analyses de la survie sur les données groupées des études ASCEND et CAPACITY (623
patients dans le groupe pirfénidone vs 624 patients dans le groupe placebo) montrent une
diminution de 50 % des décès toutes causes avec un taux de décès toutes causes de 3,5 %
dans le groupe pirfénidone contre 6,7 % dans le groupe placebo (p = 0,01) et une diminution
de 70 % de la mortalité liée à la FPI avec un taux de décès lié à la FPI de 1,1 % dans le
groupe pirféridone et de 3,5 % dans le groupe placebo (p = 0,006). Ce bénéfice apparaissait
dès le 3e ou 4e mois de traitement (61).
Tolérance
Les évènements indésirables étaient principalement digestifs : nausées (36 % vs 13,4 %),
dyspepsie (17,6 % vs 6,1), anorexie (15,8 % vs 6,5 %). La perte de poids était retrouvée chez
12,6 % des patients contre 7,6 % dans le groupe placebo. Les rashs cutanés étaient fréquents
(28,1 % vs 8,7 %). Ces évènements indésirables conduisaient rarement à un arrêt du
traitement avec 15 % d’arrêts liés aux évènements indésirables dans le groupe pirfénidone
contre 10 % dans le groupe placebo (61).
Gestion des effets indésirables
Cette intolérance peut toutefois être gérée. Un article nous fait bénéficier de l’expérience
japonaise (la pirfénidone est utilisée au Japon depuis 2008). Il montre que la prise d’un
inhibiteur de la pompe à protons permet une meilleure tolérance digestive chez les patients
avec FPI (62). Une autre étude en « vraie vie », monocentrique, sur une petite série allemande
de 63 patients, avec un suivi médian de 11 mois, montre que les patients arrêtent le traitement
le plus souvent en raison de décès ou de progression de la maladie. Les arrêts motivés par les
EI concernaient 20 % des patients (63).

Conclusion
L’étude ASCEND confirme l’efficacité de la pirfénidone dans la FPI. Cet agent ralentit de
façon indiscutable la décroissance de la fonction respiratoire. Surtout, ASCEND montre que
la pirfénidone améliore la survie. De plus, cet essai étend le champ d’efficacité de la
pirfénidone, celle-ci ayant été donnée chez des patients dont la DLCO pouvait atteindre 30 %
de la valeur théorique alors que l’AMM réserve actuellement ce médicament aux patients
ayant une DLCO supérieure à 35 % de la valeur prédite.
Discussion
D’après une communication du Pr Jean-François Cordier, Lyon
L’histoire de la pirfénidone est relativement ancienne. La première étude expérimentale,
publiée en 1995, a évalué cette molécule dans un modèle de fibrose à la bléomycine chez le
hamster (64). Ses résultats étaient positifs, mais la pirfénidone est cependant restée ignorée.
C’est à G Raghu que l’on doit, en 1999, la première étude thérapeutique évaluant cet agent
dans le traitement de la FPI (65).
Cette étude, en ouvert, a montré un bénéfice chez les patients recevant de la pirfénidone. Elle
nous amène également à faire le constat suivant : les essais qui ont conduit à l’AMM de la
pirfénidone dans la FPI incluaient des patients atteints d’une fibrose pulmonaire associée à
une altération relativement faible de la CVF et de la DLCO. L’étude de G Raghu de 1999, en
revanche, avait porté sur 54 patients dont la CVF était de 59 % et la DLCO de 34 % des
valeurs prédites contre 68 % et 44 % respectivement dans l’étude ASCEND (61, 65).
La pirfénidone est indiquée dans une population de patients chez lesquels il a été prouvé par
des essais qu’ils peuvent être améliorés par le traitement. La question qui se pose à présent est
la suivante : les patients qui ne répondent pas aux critères de l’AMM, atteints d’une forme
plus évoluée de FPI, pourraient-ils bénéficier d’un traitement par pirfénidone ? Nous
disposons des résultats d’une étude, certes ancienne et modeste, non randomisée, qui suggère
un bénéfice chez des patients atteints d’une forme plus sévère de FPI. Nous, médecins, nous
efforçons dans notre pratique de respecter les indications des médicaments dont nous
disposons, mais ceux-ci pourraient être bénéfiques chez d’autres patients, et nous aurions
peut-être à ré-évaluer ces indications.
MISE AU POINT
État des pratiques en France dans la FPI
D’après une communication du Pr Vincent Cottin, Lyon
Une première enquête de pratique sur la FPI en France avait été coordonnée par le centre
national de référence et les centres de compétence des maladies pulmonaires rares. Elle avait
été réalisée par téléphone et internet entre le 07/12/11 et le 18/02/12 auprès de 1244
pneumologues. Près de la moitié d’entre eux (48 %), soit 20 % des pneumologues français,
avaient répondu et rempli l’enquête. Ce travail avait montré que, quel que soit leur mode
d’exercice, tous les pneumologues sont impliqués dans la prise en charge de la FPI et que
l’accès auprès de radiologues expérimentés ou experts dans cette maladie et auprès

d’anatomopathologistes spécialisés restait limité. Il relevait, en outre, d’importantes attentes
pour une aide au diagnostic, une amélioration des collaborations entre les professionnels de
santé impliqués, une formation adaptée et des recommandations spécifiques. Un certain
nombre de réponses y ont, depuis, été apportées : des mesures d’aide au diagnostic avec
l’élaboration d’une fiche de discussion multidisciplinaire ont été élaborées par les centres de
compétence ; une réflexion est actuellement en cours sur la coordination du parcours de soin ;
des démarches de formation ont été initiées ; des Recommandations pour la prise en charge de
la FPI ont été publiées en décembre 2013 (66).
Une enquête sur le vécu et les attentes des patients atteints de PID, menée dans 4 régions
pilotes, a par ailleurs révélé que ceux-ci attendent une prise en charge plus rapide, de
préférence en centre spécialisé, une annonce adaptée du diagnostic, une meilleure
coordination entre les acteurs du parcours de soins, une meilleure information sur la maladie
et un accompagnement global (67).
Deuxième enquête de pratique sur la prise en charge de la FPI
Une deuxième enquête de pratique sur la prise en charge de la FPI en France a été réalisée
entre le 16/05/2014 et le 30/08/2014. Ses objectifs étaient de connaître l’évolution des
pratiques dans la prise en charge de la FPI par rapport à la première enquête, de faire un état
des lieux du réseau de soins existant et des améliorations possibles, et de connaître l’évolution
des attentes et des besoins des pneumologues (68).
Cette deuxième enquête a été menée de façon très similaire à la première. Elle a été
coordonnée par le Centre national de référence et les centres de compétence et a été réalisée
par internet et complétée par téléphone. Elle a ciblé les pneumologues qui avaient participé à
la première enquête : 547 pneumologues ont été contactés, dont 512 (94 %) pneumologues
ayant répondu à la première enquête. Seuls les pneumologues déclarant suivre actuellement
au moins un patient avec FPI ont été interrogés, avec un taux de participation de 50 %,
similaire à celui de la première enquête.
La comparaison des résultats obtenus en 2011 et 2014 montre une tendance à l’augmentation
de la proportion de patients présentant une maladie non évoluée vus dans les centres
spécialisés, ce qui suggère un accès plus rapide ou à un stade moins avancé des patients vers
ceux-ci (68).
Le retard au diagnostic communément observé dans des maladies rares comme l’hypertension
artérielle pulmonaire ou la lymphangioléiomyomatose est également retrouvé dans la FPI. Les
patients ont vu deux médecins en moyenne avant que le diagnostic soit porté.
Cryo-biopsie sous endoscopie, pour ou contre ? Le contre
D’après une communication du Pr Jean-Michel Vergnon, Saint-Étienne
La biopsie pulmonaire suppose des prélèvements importants (atteinte disséminée), distaux
sous-pleuraux (lieu des lésions), d’excellente qualité (pas d’artefacts de prélèvements),
sécurisés (pas de saignement ni de pneumothorax), peu agressifs (patients fragiles ou âgés) et
n’aggravant pas la pathologie sous-jacente (poussées aiguës de FPI).
Biopsie chirurgicale vidéo-assistée
La biopsie chirurgicale vidéo-assistée est préconisée par les recommandations parce qu’elle
permet des prélèvements de grande taille, un abord périphérique des lésions, sans artefacts ou
très peu, le contrôle du saignement, de l’aérostase et du risque de pneumothorax par les
sutures chirurgicales. La chirurgie et la thoracoscopie donnent des résultats similaires (69,

70). Cette technique est toutefois agressive, avec un taux de mortalité liée au geste ou à une
poussée de FPI d’environ 4,3 % et une morbidité atteignant 20 % (71, 72). Le rapport risque-
bénéfice est à discuter en discussion multidisciplinaire, en particulier chez les gens âgés (73).
Biopsies transbronchiques
Les biopsies transbronchiques sont associées à une très faible mortalité (0,1 %), mais elles
s’intéressent à des affections infiltrantes à distribution péribronchique et comportant une
signature histologique typique, sur un prélèvement limité, dans un contexte caractéristique
(sarcoïdose, lymphangite carcinomateuse, pneumonie organisée cryptogénique, silicose, etc.).
Dans les FPI, les recommandations internationales sont très réservées vis-à-vis des biopsies
transbronchiques : “Transbronchial biopsy should not be used in the evaluation of IPF in the
majority of patients, but may be appropriate in a minority.” (weak) (42) en raison de
prélèvements de petite taille, d’artéfacts (crush), de prélèvements péribronchiques, sans
contrôle des saignements et du risque de pneumothorax.
Certains auteurs montrent toutefois qu’elle permettrait 30 % ou plus de diagnostic de PIC (44,
74).
La cryo-biopsie : des réticences
Les réticences vis-à-vis de la cryo-biopsie s’expliquent par différentes raisons. L’adhérence
au tissu étant très forte, la cryo-biopsie réalise un véritable arrachement pulmonaire. Ces
prélèvements sont, certes, plus gros, atteignant les structures alvéolaires distales, mais sans
contrôle a priori du saignement et du risque de pneumothorax. La méthode n’est pas
standardisée. Il y a une variabilité du temps de congélation de 3 à 10 secondes, du type de
sonde (1,9 ou 2,4 mm). Les effets secondaires décrits sont très hétérogènes (pneumothorax ou
saignement). La mise en œuvre de la technique n’est pas simple : anesthésie générale ou
sédation profonde, intubation ou bronchoscope rigide, amplificateur de brillance, sonde de
type Fogarti, courbe d’apprentissage non négligeable (40 examens).
Une étude d’une équipe de Tel Aviv, présentée lors du dernier congrès de l’ERS, a retrouvé 4
à 5 % d’hémoptysies ou de pneumothorax avec la cryo-biopsie, mais une autre étude d’une
équipe espagnole présentée à ce même congrès trouvait 50 % d’hémorragies et 14 % de
pneumothorax. La question est d’être ou ne pas être près de la plèvre. Une biopsie effectuée
loin de la plèvre, mais près des vaisseaux, entraîne des saignements ; une biopsie proche de la
plèvre permettra d’obtenir plus de tissu pathologique, mais entraînera plus de pneumothorax.
Ce risque de pneumothorax augmente encore en cas de PIC en raison de la structure
particulière du tissu et atteint 34 % dans une étude récente de l’équipe du Pr V. Poletti (75).
La cryo-biopsie est associée à la formation d’artefacts liés au froid dans 58 % des cas (75).
S’ils ne gênent pas l’observation en microscopie optique, la microscopie électronique est
confrontée à la destruction des structures intracellulaires.
Au plan de son efficacité, une comparaison récente des cryo-biopsies et des biopsies
conventionnelles dans les pneumopathies interstitielles a montré que la biopsie
conventionnelle permettait le diagnostic chez 11 patients sur 38 et la cryo-biopsie chez 20 sur
39 (76). Dans les affections fibrosantes, une autre étude relève que 6 sur 69 patients ne sont
pas diagnostiqués et que sur 47 patients avec FPI, 11 ont une UIP avec confiance limitée de
l’anatomopathologiste. Cette étude comptant également 8 patients inclassables, 19 FPI
possibles et 6 échecs, nous aboutissons à une absence de diagnostic final chez 33 malades sur
69 (75).
La frilosité à l’égard des cryo-biopsies s’explique en outre par l’absence d’étude randomisée
contre la biopsie chirurgicale. Elle présente des limites (anticoagulants, hypertension
pulmonaire).

Conclusion
La cryo-biopsie pourrait être une étape permettant d’éviter certaines biopsies chirurgicales,
mais cela nécessite encore une meilleure standardisation et une meilleure validation. La cryo-
biopsie n’est sûrement pas un geste anodin, elle nécessite compétence et précautions.
Cryobiopsie sous endoscopie, pour ou contre ? Le pour
D’après une communication du Pr Venerino Poletti, Forli, Italie
Nous avons adopté le « jumbo forceps », utilisé par les gastroentérologues, à la fin du siècle
dernier. Nous avons ainsi appris que plus de 10 spécimens pouvaient être obtenus sans
augmenter le risque de pneumothorax. La fréquence des saignements était supérieure à celle
observée avec les sondes flexibles, mais l'utilisation d’une sonde à ballonnet de Fogarty s’est
révélée très efficace pour contrôler le saignement. Le « jumbo forceps » permettait de
prélever des échantillons de tissu de grande taille (2,5 x 1,9 mm vs 1,4 x 1 mm avec les pinces
classiques), le tissu obtenu étant, de plus, dépourvu d’artefacts. Il faut souligner que dans une
minorité de cas, les échantillons prélevés comportaient du tissu provenant de la périphérie du
lobule secondaire (plèvre, cloisons interlobulaires et veines).
Principe de la cryobiopsie transbronchique
La cryo-biopsie fonctionne par effet Joule-Thomson qui veut qu'un gaz comprimé libéré à
débit élevé se dilate en générant une très basse température. L'agent de refroidissement, du
dioxyde de carbone ou de l'oxyde nitrique, est appliqué sous haute pression (45 bars) à travers
le canal central de la sonde. Le gaz se dilate en raison de la différence soudaine de pression et
entraîne une chute de la température à -80°C à la pointe de la sonde.
Le poids et le diamètre des cryobiopsies sont positivement corrélés avec la durée d'activation
et le diamètre de la sonde cryogénique (1,9 et 2,4 mm). Avec la sonde cryogénique de
2,4 mm, la taille des échantillons est nettement supérieur à celui obtenu avec des pinces à
biopsie standard pour toutes les durées d'activation de 1 à 3 secondes, et avec la cryosonde de
1,9 mm après des durées de congélation de 2 et 3 secondes. Nous avons choisi un temps de
refroidissement de 5-6 secondes et de rester très proches de la plèvre (moins d’un centimètre).
Au plan pratique, les cryobiopsies transbronchiques de tissu pulmonaire sont réalisées alors
que les patients sont sous sédation profonde et intubés avec une sonde endotrachéale armée ou
un tube rigide. Un ballonnet de Fogarty est positionné à l'entrée de la bronche segmentaire
présélectionnée. La sonde cryogénique est introduite dans la zone sélectionnée sous guidage
fluoroscopique via un bronchoscope souple. Une distance d'environ 10 à 20 mm de la paroi
thoracique et une relation perpendiculaire entre la paroi thoracique et la sonde sont
considérées comme optimales. Une fois mise en place, la sonde est refroidie pendant 3 à 6
secondes. Le tissu congelé fixé à la pointe de la sonde est ensuite retiré et le spécimen est
décongelé dans une solution saline puis fixé au formol. Le nombre de biopsies est
généralement de 3 à 6.
Les échantillons ont un diamètre de 5 à 7 mm au maximum. En raison de la plus grande taille
des échantillons, de l’absence d’artefacts, de la présence de structures périphériques du lobule
pulmonaire secondaire (plèvre, cloisons interlobulaires et veines), des aspects
histopathologiques complexes peuvent être identifiés (PIC, PINS, DIP, etc.).
Efficacité et sécurité de la cryobiopsie transbronchique
La cryobiopsie transbronchique a été utilisée avec succès chez des patients atteints de
pneumopathie interstitielle.

Dans une étude incluant 41 patients PID évalués par cryobiopsie transbronchique, la taille des
échantillons prélevés était significativement plus grande que celle des échantillons obtenus
par des pinces à biopsie flexibles (11,11 mm2 vs 5,82 mm2). Un pneumothorax a été observé
chez deux patients (4,87 %) et résolu par drainage. L'architecture tissulaire et les structures
cellulaires étaient bien conservées (77).
Dans une autre petite série de 10 patients PID, la cryobiospie transbronchique n’a été associée
à aucune complication majeure (78).
Dans une étude incluant 25 patients PID, les cryobiopsies avaient une superficie moyenne de
64,2 mm2. Un diagnostic précis a pu être porté chez 19 des patients, soit un rendement
diagnostique de 80 % (79).
Une étude prospective a porté sur 69 cas de PID (CVF > 50 % et DLCO > 35 % des valeurs
théoriques) sans diagnostic scannographique. La cryobiopsie a été associée à un cas de
saignement prolongé malgré l'utilisation préventive du ballonnet de Fogarty. Un
pneumothorax s’est produit chez 19 patients (27 %). Un patient (1,4 %) est décédé d'une
exacerbation aiguë de FPI. Des cryobiopsies appropriées ont été obtenues dans 67 cas (97 %),
leur taille moyenne était de 43,11 mm2. Des aspects histopathologiques spécifiques ont été
établis avec un niveau de confiance élevé chez 52 patients (76 %). L’accord entre les
pathologistes pour le diagnostic de PIC était très bon avec un coefficient kappa de 0,83. Les
investigateurs ont souligné la sécurité et le bon rendement diagnostic de la cryobiopsie
transbronchique dans le diagnostic des PID. Le taux de pneumothorax était relativement
élevé, peut-être en raison de prélèvements à proximité de la plèvre (1 cm ou moins de la paroi
thoracique) (80).
Le suivi par cryobiopsie transbronchique de 40 transplantés pulmonaires n’a été associé à
aucune complication majeure. Le diamètre moyen des échantillons en cryo-biopsie était de 10
mm2 contre 2 mm2 avec des pinces à biopsie classiques (p <0,05). La grande taille et la bonne
qualité des biopsies obtenues en cryobiopsie étaient associées à une augmentation
significative du pourcentage de tissu alvéolaire (65 % vs 34 %, respectivement, p <0,05), ce
qui a permis une bonne détection histologique des rejets aigus (n = 4), pneumonies (n = 3) et
lésions alvéolaires diffuses (n = 1) (81).
Une étude pilote a comparé les résultats de cryobiopsie transbronchique avec ceux de pinces à
biopsie transbronchiques standard chez 21 patients transplantés pulmonaires. Aucune
différence significative entre les deux techniques n’a été observée pour les saignements,
aucune hémorragie sévère et aucun pneumothorax périopératoire n'ayant été observés dans les
deux groupes (82).
L'efficacité et la sécurité des cryobiopsies transthoraciques ont été évaluées chez des patients
immunodéprimés avec infiltrats pulmonaires. Aucune complication majeure n'a eu lieu. La
surface moyenne de l'échantillon était de 9 mm2 (83).
La cryobiopsie transbronchique a été adoptée dans notre centre en mars 2011, et depuis lors,
176 patients PID ont bénéficié de cette procédure. Cette cohorte est constituée de 105
hommes et 71 femmes, l'âge moyen est de 57 ans (26 à 79 ans). Un aspect histopathologique
caractéristique a été reconnu dans 80 % des cas. Aucune hémorragie majeure n’a été
observée ; nous avons constaté un pneumothorax dans 23 % des cas, et une exacerbation
aiguë mortelle de FPI.
Conclusion
La cryobiopsie transbronchique pourrait être considérée comme une alternative à la biopsie
pulmonaire chirurgicale chez les patients atteints de PID. L'approche chirurgicale permet le
prélèvement de plus grands échantillons, mais elle est associée à des risques importants :

mortalité dans les 90 jours de 2 à 4 % (ces chiffres sont encore plus élevés chez les sujets avec
un diagnostic définitif de FPI), fuites d'air prolongées, douleur thoracique persistante,
arythmies cardiaques et complications infectieuses. La durée moyenne du séjour à l'hôpital est
significativement plus importante chez les patients subissant une biopsie pulmonaire
chirurgicale. Enfin, la cryobiopsie transbronchique peut être effectuée en ambulatoire.
Des essais multicentriques évaluant les différents aspects techniques (refroidissement à l'aide
de monoxyde de carbone ou d'oxyde nitrique ; biopsies réalisées à moins d’un centimètre de
la paroi thoracique ou plus centrales ; nombre d'échantillons à obtenir ; comparaison entre les
différentes sondes (sondes 1,9 mm vs 2,4 mm) ; utilité des biopsies dans les différents lobes ;
impact clinique de cette méthode sur la décision diagnostique multidisciplinaire finale)
permettront de préciser les aspects techniques et l'utilité clinique de cet outil diagnostique.
Prise en charge symptomatique de la toux et de la dyspnée
D’après une communication du Pr Benoît Wallaert, Lille
Toux et FPI
La toux est un signe d’appel extrêmement fréquent dans la FPI, plus encore que dans l’asthme
(84). Ses mécanismes sont multiples et relativement mal connus. Ils pourraient mettre en jeu
des récepteurs mécaniques, une augmentation de l’hyperréactivité, un reflux gastro-
œsophagien (RGO), un syndrome d’apnée du sommeil (SAS), des neurotrophines (BDNF,
NGF, facteurs neurotrophiques…). On notera toutefois que l’intensité de la toux est beaucoup
plus importante dans la journée que durant la nuit, ce qui est contradictoire avec une
participation majeure du RGO. La concentration de NGF dans le lavage alvéolaire est élevée
chez les patients atteints de fibrose pulmonaire, mais son rôle réel reste à déterminer (85).
Les remèdes : peu d’informations car peu d’études !
Les études portant sur le traitement de la toux chez les patients avec FPI sont rares. Une étude
en cross-over, contre placebo, menée chez 20 patients FPI, a montré que le thalidomide à une
dose de 50 mg puis 100 mg par jour per os permet une diminution du score de toux. L’intérêt
du thalidomide dans la FPI reste cependant limité en raison des contraintes réglementaires
associées à sa prescription (dossier complexe, double contraception chez l’homme et chez la
femme, test de grossesse mensuel chez la femme) (86). Une étude contre placebo qui a
exploré l’efficacité d’un traitement du RGO associant l’oméprazole (40 mg x2/j) et la
ranitidine (300 mg/j) pendant 8 semaines chez 18 patients avec FPI n’a montré aucun effet sur
la toux (87). L’étude de deux dosages de pirfénidone versus placebo chez 275 patients FPI a
montré une moindre aggravation de la toux, qui reste tout de même plus importante après un
an de traitement qu’au début (88). Une évaluation du WO2013117503 et du WO2013117504
est en cours dans le traitement de la toux de la fibrose, mais les résultats ne sont encore
publiés. De petites séries montreraient que de petites doses d’interféron alpha pourraient
diminuer la toux chez certains patients.
En réalité, les essais thérapeutiques menés dans la FPI ne portent pas sur la toux, qui est certes
un symptôme gênant, mais qui ne constitue pas une priorité thérapeutique, celle-ci étant
d’arrêter l’évolution de la fibrose ou d’améliorer des critères comme la CVF, la mortalité ou
les exacerbations.

Dyspnée et FPI
La dyspnée a deux composantes, subjective (gène éprouvée par le patient, dans sa dimension
sensorielle et affective) et objective (limitation à l’effort observée par le médecin) qui doivent
être distinguées. La composante subjective peut être explorée à l’aide de questionnaires de
dyspnée (MRC, score de Sadoul, NYHA, BDI-TD, Multidimensional Dyspnea Profile,
dyspnea 12) et de questionnaires de qualité de vie.
De nombreuses études ont recherché des corrélations entre la dyspnée et différents
paramètres. Un travail de G Raghu publié en 2006 a montré que la mise en place d’un
traitement contre le RGO s’accompagne d’une diminution de la dyspnée (89). Un essai
contrôlé évaluant le sildénafil dans la FPI avancée a montré un bénéfice significatif sur la
dyspnée au Shortness of Breath Questionnaire (p = 0,006) (90).
Deux études ont évalué l’oxygénothérapie. L’une, en cross-over, menée chez 20 patients
ayant une FPI (CVF 71 %, DLCO 57 %, PaO2 73 mmHg) qui ont reçu soit un placebo (de
l’air) soit de l’oxygène, n’a montré aucune amélioration du test de marche de 6 minutes (91).
L’autre travail mené chez 54 patients dont 34 avec FPI, a montré une franche amélioration de
la distance au TM6, de la sensation de dyspnée et du temps de récupération (92).
Différentes études ont exploré l’effet de la réhabilitation pulmonaire sur la dyspnée chez des
patients ayant une FPI. L’effet est variable, de prononcé à nul, mais au-delà de la dyspnée, la
réhabilitation est une prise en charge globale qui a un effet important sur la façon dont les
gens vont vivre leur maladie (93). L’activité physique quotidienne des patients atteints de
fibrose est moindre que celle des sujets témoins. Un sujet en bonne santé fait environ 12 000
pas par jour, les patients atteints de fibrose en font 4 000, et la durée de leurs activités
physiques supérieures à 2,5 mètres est très basse (94). Une étude menée chez 46 patients
ayant une FPI, dont les résultats sont en cours de publication, montre que le niveau d’activité
physique quotidienne est le même avant et après réhabilitation. Toutefois, le niveau de
dyspnée était un peu moindre, et les troubles anxio-dépressifs étaient significativement
diminués.
Conclusion
La toux est très fréquente chez les patients ayant une FPI. Nous la traitons dans notre service
par corticothérapie à faible dose. Pour ce qui concerne la dyspnée, nous faisons
systématiquement appel à la réhabilitation respiratoire et nous instaurons fréquemment une
oxygénothérapie de déambulation, dont nous discutons avec le patient de façon à savoir s’il
en ressent un bénéfice (certains patients se sentent beaucoup mieux sous oxygène, d’autres
non). Quand la gêne associée à la dyspnée est très importante, nous faisons appel à des
opiacés. Le sildénafil, qui améliore la qualité de vie et diminue la dyspnée, peut être envisagé.
Bibliographie
1. Li Y, Jiang D, Liang J, et al. Severe lung fibrosis requires an invasive fibroblast
phenotype regulated by hyaluronan and CD44. J Exp Med 2011;208:1459-71.
2. Vuga LJ, Ben-Yehudah A, Kovkarova-Naumovski E, et al. WNT5A is a regulator of
fibroblast proliferation and resistance to apoptosis. Am J Respir Cell Mol Biol
2009;41:583-9.
3. Barkauskas CE, Cronce MJ, Rackley CR, et al. Type 2 alveolar cells are stem cells in
adult lung. J Clin Invest 2013;123:3025-36.

4. Hung C, Linn G, Chow YH, et al. Role of lung pericytes and resident fibroblasts in the
pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med 2013;188:820-30
5. Rock JR, Barkauskas CE, Cronce MJ, et al. Multiple stromal populations contribute to
pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl
Acad Sci USA 2011;108:E1475-83.
6. Marinković A, Liu F, Tschumperlin DJ. Matrices of physiologic stiffness potently
inactivate idiopathic pulmonary fibrosis fibroblasts. Am J Respir Cell Mol Biol
2013;48:422-30.
7. Parker MW, Rossi D, Peterson M, et al. Fibrotic extracellular matrix activates a
profibrotic positive feedback loop. J Clin Invest 2014;124:1622-35.
8. Barry-Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidase-
like-2 impedes the development of a pathologic microenvironment. Nat Med
2010;16:1009-17.
9. Olsen KC, Sapinoro RE, Kottmann RM, et al. Transglutaminase 2 and its role in
pulmonary fibrosis. Am J Respir Crit Care Med 2011;184:699-707.
10. Oh K, Park HB, Byoun OJ, et al. Epithelial transglutaminase 2 is needed for T cell
interleukin-17 production and subsequent pulmonary inflammation and fibrosis in
bleomycin-treated mice. J Exp Med 2011;208:1707-19.
11. Zhou Y, Huang X, Hecker L, et al. Inhibition of mechanosensitive signaling in
myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest
2013;123:1096-108. Travis WD, Costabel U, Hansell DM, et al. An official American
Thoracic Society/European Respiratory Society statement: Update of the international
multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit
Care Med 2013;188(6):733-48.
12. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic
Society/European Respiratory Society statement: Update of the international
multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit
Care Med 2013;188(6):733-48.
13. Garcia JG. Genomic investigations into acute inflammatory lung injury. Proc Am Thorac
Soc 2011;8:167-72.
14. Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and
pulmonary fibrosis. N Engl J Med 2011;364:1503-12.
15. Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by
mutations in telomerase. Proc Natl Acad Sci USA 2007;104:7552-7.
16. Steele MP, Speer MC, Loyd JE, et al. Clinical and pathologic features of familial
interstitial pneumonia. Am J Respir Crit Care Med 2005;172:1146-52.
17. Fernandez BA, Fox G, Bhatia R, et al. A Newfoundland cohort of familial and sporadic
idiopathic pulmonary fibrosis patients: clinical and genetic features. Respir Res
2012;13:64.
18. Diaz de Leon A, Cronkhite JT, Katzenstein AL, et al. Telomere lengths, pulmonary
fibrosis and telomerase (TERT) mutations. PLoS One 2010;5:e10680.
19. Lee HY, Seo JB, Steele MP, et al. High-resolution CT scan findings in familial interstitial
pneumonia do not conform to those of idiopathic interstitial pneumonia. Chest
2012;142:1577-83.
20. Leslie KO, Cool CD, Sporn TA, et al. Familial idiopathic interstitial pneumonia:
histopathology and survival in 30 patients. Arch Pathol Lab Med 2012;136:1366-76.
21. Travis WD, Matsui K, Moss J, Ferrans VJ. Idiopathic nonspecific interstitial pneumonia:
prognostic significance of cellular and fibrosing patterns: survival comparison with usual
interstitial pneumonia and desquamative interstitial pneumonia. Am J Surg Pathol
2000;24:19-33.

22. Katzenstein AL, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis. Histologic
features and clinical significance. Am J Surg Pathol 1994;18:136-47.
23. Cottin V, Donsbeck AV, Revel D, Loire R, Cordier JF. Nonspecific interstitial
pneumonia. Individualization of a clinicopathologic entity in a series of 12 patients. Am J
Respir Crit Care Med 1998;158:1286-93.
24. American Thoracic Society; European Respiratory Society. American Thoracic
Society/European Respiratory Society International Multidisciplinary Consensus
Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the
American Thoracic Society (ATS), and the European Respiratory Society (ERS) was
adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee,
June 2001. Am J Respir Crit Care Med 2002;165:277-304.
25. Travis WD, Hunninghake G, King TE Jr, et al. Idiopathic nonspecific interstitial
pneumonia: report of an American Thoracic Society project. Am J Respir Crit Care Med
2008;177:1338-47.
26. Frankel SK1, Cool CD, Lynch DA, Brown KK. Idiopathic pleuroparenchymal
fibroelastosis: description of a novel clinicopathologic entity. Chest 2004;126:2007-13.
27. Harada T et al, PMID:24881083, Eur Respir Rev.2014 Jun;23(132):263-6.
28. Reddy TL, Tominaga M, Hansell DM, et al. Pleuroparenchymal fibroelastosis: a spectrum
of histopathological and imaging phenotypes. Eur Respir J 2012;40:377-85.
29. Watanabe K. Pleuroparenchymal Fibroelastosis: Its Clinical Characteristics. Curr Respir
Med Rev 2013;9:299-237.
30. Ryerson CJ, Urbania TH, Richeldi L, et al. Prevalence and prognosis of unclassifiable
interstitial lung disease. Eur Respir J 2013;42:750-7.
31. Raghu G, Lynch D, Godwin JD, et al. Diagnosis of idiopathic pulmonary fibrosis with
high-resolution CT in patients with little or no radiological evidence of honeycombing:
secondary analysis of a randomised, controlled trial. Lancet Respir Med 2014;2:277-84.
32. Johannson KA, de Boer K, Wolters PJ, Golden JA, Lee JS, Collard HR. Diagnosis of
idiopathic pulmonary fibrosis with high-resolution CT. Lancet Respir Med 2014;2:e5.
33. Morell F, Villar A, Montero MÁ, et al. Chronic hypersensitivity pneumonitis in patients
diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet
Respir Med 2013;1:685-94.
34. Lee JS, Kim EJ, Lynch KL, et al. Prevalence and clinical significance of circulating
autoantibodies in idiopathic pulmonary fibrosis. Respir Med 2013;107:249-55.
35. Kono M, Nakamura Y, Enomoto N, et al. Usual interstitial pneumonia preceding collagen
vascular disease: a retrospective case control study of patients initially diagnosed with
idiopathic pulmonary fibrosis. PLoS One 2014;9:e94775.
36. Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N, Spagnolo P. Differential
diagnosis of usual interstitial pneumonia: when is it truly idiopathic ? Eur Respir Rev
2014;23:308-319.
37. Valeyre D, Albera C, Bradford WZ, et al. Comprehensive assessment of the long-term
safety of pirfenidone in patients with idiopathic pulmonary fibrosis. Respirology
2014;19:740-7.
38. Silhan LL, Shah PD, Chambers DC, et al. Lung transplantation in telomerase mutation
carriers with pulmonary fibrosis. Eur Respir J 2014;44:178-87.
39. du Bois RM. 6-minute walk distance as a predictor of outcome in idiopathic pulmonary
fibrosis. Eur Respir J 2014;43:1823-4.
40. Chien, Eur Respir J 2013.
41. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement:
idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management.
Am J Respir Crit Care Med 2011;183:788-824.

42. Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, Remy J. Fleischner
Society: glossary of terms for thoracic imaging. Radiology 2008;246:697-722.
43. Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual
interstitial pneumonia. Chest 2006;129:1126-31.Johkoh T, Sumikawa H, Fukuoka J, et al.
Do you really know precise radiologic-pathologic correlation of usual interstitial
pneumonia? Eur J Radiol 2014;83:20-6.
44. Johkoh T, Sumikawa H, Fukuoka J, et al. Do you really know precise radiologic-
pathologic correlation of usual interstitial pneumonia? Eur J Radiol 2014;83:20-6.
45. Plantier L, Crestani B, Wert SE, et al. Ectopic respiratory epithelial cell differentiation in
bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 2011;66:651-7.
46. Seibold MA, Smith RW, Urbanek C, et al. The idiopathic pulmonary fibrosis honeycomb
cyst contains a mucocilary pseudostratified epithelium. PLoS One 2013;8:e58658.
47. Watadani T, Sakai F, Johkoh T, et al. Interobserver variability in the CT assessment of
honeycombing in the lungs. Radiology 2013;266:936-44.
48. Cantin AM, Larivée P, Bégin RO. Extracellular glutathione suppresses human lung
fibroblast proliferation. Am J Respir Cell Mol Biol 1990;3:79-85.
49. Meyer A, Buhl R, Magnussen H. The effect of oral N-acetylcysteine on lung glutathione
levels in idiopathic pulmonary fibrosis. Eur Respir J 1994;7:431-6.
50. Demedts M, Behr J, Buhl R, et al. High-dose acetylcysteine in idiopathic pulmonary
fibrosis. N Engl J Med 2005;353:2229-42.
51. Idiopathic Pulmonary Fibrosis Clinical Research Network, Martinez FJ, de Andrade JA,
Anstrom KJ, King TE Jr, Raghu G. Randomized trial of acetylcysteine in idiopathic
pulmonary fibrosis. N Engl J Med 2014;370:2093-101.
52. Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with
sustained receptor blockade and good antitumor efficacy. Cancer Res 2008;68:4774-82.
53. Woodcock HV, Molyneaux PL, Maher TM. Reducing lung function decline in patients
with idiopathic pulmonary fibrosis: potential of nintedanib. Drug Des Devel Ther
2013;7:503-10.
54. King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011;378:1949-
61.
55. Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-
inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models
of lung fibrosis. J Pharmacol Exp Ther 2014;349:209-20.
56. Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in
idiopathic pulmonary fibrosis. N Engl J Med 2011;365:1079-87.
57. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic
pulmonary fibrosis. N Engl J Med 2014;370:2071-82.
58. Azuma A, Nukiwa T, Tsuboi E, et al. Double-blind, placebo-controlled trial of
pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med
2005:171:1040-7.
59. Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur
Respir J 2010;35:821-9.
60. Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic
pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011;377:1760-9.
61. King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in
patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2083-92.
62. Arai T, Inoue Y, Sasaki Y, et al. Predictors of the clinical effects of pirfenidone on
idiopathic pulmonary fibrosis. Respir Investig 2014;52:136-43.
63. Oltmanns U, Kahn N, Palmowski K, et al. Pirfenidone in idiopathic pulmonary fibrosis:
real-life experience from a german tertiary referral center for interstitial lung diseases.
Respiration 2014;88:199-207.

64. Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of
pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med
1995;125:779-85.
65. Raghu G, Johnson WC, Lockhart D, Mageto Y. Treatment of idiopathic pulmonary
fibrosis with a new antifibrotic agent, pirfenidone: results of a prospective, open-label
Phase II study. Am J Respir Crit Care Med 1999;159:1061-9.
66. Cottin V, Crestani B, Valeyre D, et al. Recommandations pratiques pour le diagnostic et la
prise en charge de la fibrose pulmonaire idiopathique. Élaborées par le centre national de
référence et les centres de compétence pour les maladies pulmonaires rares sous l’égide de
la Société de pneumologie de langue française. Rev Mal Respir 2013;30:879-902.
67. Cottin V, Crestani B, Bourdin A, Prévot G, Clerson P, Guérin M, Bouquillon B. Enquête
sur le vécu et les attentes des patients atteints de PID dans 4 régions pilotes. CPLF 2014.
68. Cottin V, Cadranel J, Crestani B, et al. Management of idiopathic pulmonary fibrosis in
France: a survey of 1244 pulmonologists. Respir Med 2014;108:195-202.
69. Ayed AK, Raghunathan R. Thoracoscopy versus open lung biopsy in the diagnosis of
interstitial lung disease: a randomised controlled trial. J R Coll Surg Edinb 2000;45:159-
63.
70. Miller JD, Urschel JD, Cox G, et al. A randomized, controlled trial comparing
thoracoscopy and limited thoracotomy for lung biopsy in interstitial lung disease. Ann
Thorac Surg 2000;70:1647-50.
71. Kreider ME, Hansen-Flaschen J, Ahmad NN, et al. Complications of video-assisted
thoracoscopic lung biopsy in patients with interstitial lung disease. Ann Thorac Surg
2007;83:1140-4.
72. Park JH, Kim DK, Kim DS, et al. Mortality and risk factors for surgical lung biopsy in
patients with idiopathic interstitial pneumonia. Eur J Cardiothorac Surg 2007;31:1115-9.
73. Fell CD, Martinez FJ, Liu LX, et al. Clinical predictors of a diagnosis of idiopathic
pulmonary fibrosis. Am J Respir Crit Care Med 2010;181:832-7.
74. Tomassetti S, Cavazza A, Colby TV, et al. Transbronchial biopsy is useful in predicting
UIP pattern. Respir Res 2012;13:96.
75. Casoni GL, Tomassetti S, Cavazza A, et al. Transbronchial lung cryobiopsy in the
diagnosis of fibrotic interstitial lung diseases. PLoS One 2014;9:e86716.
76. Pajares V, Puzo C, Castillo D, et al. Diagnostic yield of transbronchial cryobiopsy in
interstitial lung disease: a randomized trial. Respirology 2014;19:900-6.
77. Babiak A, Hetzel J, Krishna G, et al. Transbronchial cryobiopsy: a new tool for lung
biopsies. Respiration 2009;78:203-8.
78. Pajares V, Torrego A, Puzo C, Lerma E, Gil De Bernabé MA, Franquet T.
[Transbronchial lung biopsy using cryoprobes]. Arch Bronconeumol 2010;46:111-5.
79. Kropski JA, Pritchett JM, Mason WR, et al. Bronchoscopic cryobiopsy for the diagnosis
of diffuse parenchymal lung disease. PLoS One 2013;8:e78674.
80. Casoni GL, Tomassetti S, Cavazza A, et al. Transbronchial lung cryobiopsy in the
diagnosis of fibrotic interstitial lung diseases. PLoS One 2014;9:e86716.
81. Fruchter O, Fridel L, Rosengarten D, Raviv Y, Rosanov V, Kramer MR. Transbronchial
cryo-biopsy in lung transplantation patients: first report. Respirology 2013;18:669-73.
82. Yarmus L, Akulian J, Gilbert C, et al. Cryoprobe transbronchial lung biopsy in patients
after lung transplantation: a pilot safety study. Chest 2013;143:621-6.
83. Fruchter O, Fridel L, Rosengarten D, Rahman NA, Kramer MR. Transbronchial
cryobiopsy in immunocompromised patients with pulmonary infiltrates: a pilot study.
Lung 2013;191:619-24.
84. Key AL, Holt K, Hamilton A, Smith JA, Earis JE. Objective cough frequency in
Idiopathic Pulmonary Fibrosis. Cough 2010;6:4.

85. Harrison NK. Cough, sarcoidosis and idiopathic pulmonary fibrosis: raw nerves and bad
vibrations. Cough 2013;9:9.
86. Horton MR, Santopietro V, Mathew L, et al. Thalidomide for the treatment of cough in
idiopathic pulmonary fibrosis: a randomized trial. Ann Intern Med 2012;157:398-406.
87. Kilduff CE, Counter MJ, Thomas GA, Harrison NK, Hope-Gill BD. Effect of acid
suppression therapy on gastroesophageal reflux and cough in idiopathic pulmonary
fibrosis: an intervention study. Cough 2014;10:4.
88. Azuma A, Taguchi Y, Ogura T, et al. Exploratory analysis of a phase III trial of
pirfenidone identifies a subpopulation of patients with idiopathic pulmonary fibrosis as
benefiting from treatment. Respir Res 2011;12:143.
89. Raghu G, Yang ST, Spada C, Hayes J, Pellegrini CA. Sole treatment of acid
gastroesophageal reflux in idiopathic pulmonary fibrosis: a case series. Chest
2006;129:794-800.
90. Idiopathic Pulmonary Fibrosis Clinical Research Network, Zisman DA, Schwarz M, et al.
A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med
2010;363:620-8.
91. Nishiyama O, Miyajima H, Fukai Y, et al. Effect of ambulatory oxygen on exertional
dyspnea in IPF patients without resting hypoxemia. Respir Med 2013;107:1241-6.
92. Visca D, Montgomery A, de Lauretis A, et al. Ambulatory oxygen in interstitial lung
disease. Eur Respir J 2011;38:987-90.
93. Kenn K, Gloeckl R, Behr J. Pulmonary rehabilitation in patients with idiopathic
pulmonary fibrosis--a review. Respiration 2013;86:89-99.
94. Wallaert B, Monge E, Le Rouzic O, et al. Physical activity in daily life of patients with
fibrotic idiopathic interstitial pneumonia. Chest 2013;144:1652-8.