
UNIVERSITÉ DU QUÉBEC À MONTRÉAL
ANALYSE DE MOTIFS PROTÉIQUES PAR MÉTHODE HYBRIDE RÉSEAU DE
NEURONES ARTIFICIELS ET MODÈLE DE MARKOV CACHÉ
THÈSE
PRÉSENTÉE
COMME EXIGENCE PARTIELLE
DU DOCTORAT EN INFORMATIQUE COGNITIVE
PAR
GUYLAINE POISSON
NOVEMBRE 2004

REMERCIEMENTS
Je voudrais remercier tout particulièrement ma directrice de recherche Dr Anne Bergeron, professeur
au département d’informatique de l’UQAM, pour sa confiance en moi et en notre projet. Son support
moral et scientifique a été indispensable tout au long de cette thèse. Le succès d’un doctorat dépend
énormément de la complicité étudiante-directrice et je dois avouer que j’ai eu la chance de travailler
avec une personne extrêmement compétente mais surtout une personne formidable.
Je tiens aussi à remercier personnellement Dr Cedric Chauve, professeur adjoint au département
d’informatique de l’UQAM, pour sa collaboration précieuse dans le projet. Il est justifié de dire que
sans ses idées, cette thèse ne serait pas ce qu’elle est présentement.
Je tiens à remercier également mon codirecteur de recherche Dr Fathey Sarhan, professeur au
département de sciences biologiques de l’UQAM, ainsi que Dr Ahmed Faik, associé de recherche au
département de sciences biologiques de l’UQAM, pour leur support en biologie. Leurs commentaires
et suggestions ont été une source d’informations essentielles pour cette recherche.
Je voudrais également remercier Dr Mounir Boukadoum, professeur au département d’informatique de
l’UQAM, pour m’avoir initiée aux réseaux de neurones; Dr Pierre Poirier, professeur au département
de philosophie de l’UQAM, qui a eu des commentaires eclairants sur les aspects cognitifs de
mon projet; et Dr Mathieu Blanchette, Assistant Professor de la School of Computer Sciences de
l’université McGill, d’avoir bien voulu faire partie de mon jury de thèse. Je ne voudrais pas oublier
Johanne Gélinas, assistante à la direction du Doctorat en informatique cognitive de l’UQAM, et Dr
Ghyslain Levesque, directeur du Doctorat en Informatique Cognitive, pour leur aide plus que
précieuse.
Finalement, je veux remercier grandement mon fils William Langlois-Poisson, mon conjoint Pascal
Senez ainsi que mes parents, Paul et Paulette Poisson. Je les remercie d’être ce qu’ils sont c’est-à-dire
des gens plus qu’extraordinaires. Une thèse demande beaucoup de sacrifices et ils sont ceux qui en ont
payé le prix. Merci pour l’encouragement continuel et pour votre amour inconditionnel.

TABLE DES MATIÈRES REMERCIEMENTS............................................................................................................................ II LISTE DES FIGURES........................................................................................................................VI LISTE DES TABLEAUX ...................................................................................................................IX LISTE DES ABRÉVIATIONS...........................................................................................................XI RÉSUMÉ...........................................................................................................................................XIII INTRODUCTION ................................................................................................................................. 1 CHAPITRE I BASES BIOLOGIQUES....................................................................................................................... 6
1.1 LES SÉQUENCES..................................................................................................................... 6 1.1.1 ADN et ARN : polymères d’acides nucléiques................................................................. 8
1.1.1.1 L’ADN .......................................................................................................................................... 9 1.1.1.2 L’ARN......................................................................................................................................... 12
1.1.2 Les protéines ...................................................................................................................... 13 1.2 DE L’ADN À LA PROTÉINE........................................................................................................... 17
1.2.1 La transcription................................................................................................................... 17 1.2.2 La traduction....................................................................................................................... 19
CHAPITRE II MODIFICATIONS POST-TRADUCTIONNELLES ET ANCRE GPI......................................... 23
2.1 MODIFICATION POST-TRADUCTIONNELLE.................................................................................... 23 2.2 L’ANCRE GLYCOSYLPHOSPHATIDYLINOSITOL (GPI).................................................................... 24
2.2.1 Biosynthèse ......................................................................................................................... 26 2.2.2 Structure d’une protéine à ancre GPI................................................................................. 28
CHAPITRE III CLASSIFICATION PAR ALIGNEMENT DE SÉQUENCES ....................................................... 31
3.1 LA CLASSIFICATION............................................................................................................. 31 3.2 LA CLASSIFICATION PAR SIMILARITÉ DE SÉQUENCES .................................................................. 35
3.2.1 Alignement de deux séquences ............................................................................................ 36 3.2.2 Classification de protéines à ancre GPI et BLAST ............................................................. 45 3.2.3 Classification de protéines à ancre GPI et alignements multiples...................................... 48
CHAPITRE IV CLASSIFICATION PAR APPRENTISSAGE MACHINE............................................................. 51
4.1 INTRODUCTION ............................................................................................................................ 51 4.2 APPRENTISSAGE NEURONAL ........................................................................................................ 52
4.2.1 Historique ........................................................................................................................... 54 4.2.2 Le modèle biologique .......................................................................................................... 55

iv
4.2.3 Le modèle mathématique..................................................................................................... 57 4.2.4 Structure des connexions..................................................................................................... 59 4.2.5 L’apprentissage................................................................................................................... 61
4.2.5.1 Le modèle du perceptron ............................................................................................................. 62 4.2.5.2 Le modèle du perceptron multicouche ........................................................................................ 64
4.2.6 L’encodage des données ..................................................................................................... 68 4.2.7 Validation............................................................................................................................ 70
4.3 LES MODÈLES DE MARKOV CACHÉS............................................................................................. 71 4.3.1.1 HMM et grammaire..................................................................................................................... 79
4.4 CONCLUSION ............................................................................................................................... 85 CHAPITRE V CLASSIFICATION/PRÉDICTION D’ANCRE GPI PAR RÉSEAU DE NEURONES ARTIFICIELS ..................................................................................................................................... 86
5.1 UN MODÈLE DE RÉSEAU DE NEURONES ARTIFICIELS CLASSIFICATEUR D’ANCRE GPI................... 87 5.2 LE MODÈLE .................................................................................................................................. 87 5.3 RÉSULTATS DES TESTS ET DISCUSSION....................................................................................... 101 5.4 CONCLUSION............................................................................................................................. 104
CHAPITRE VI CLASSIFICATION/PRÉDICTION D’ANCRE GPI PAR MODÈLE DE MARKOV CACHÉ 105
6.1 UN MODÈLE DE MARKOV CACHÉ CLASSIFICATEUR D’ANCRE GPI ............................................. 105 6.2 LE MODÈLE ................................................................................................................................ 106 6.3 RÉSULTATS DES TESTS ET DISCUSSION....................................................................................... 114 6.4 UNE GRAMMAIRE GPI................................................................................................................ 117
6.4.1 La structure du signal ....................................................................................................... 117 6.5 CONCLUSION ............................................................................................................................. 119
CHAPITRE VII CLASSIFICATION/PRÉDICTION D’ANCRE GPI PAR MÉTHODE HYBRIDE RÉSEAU DE NEURONES ARTIFICIELS ET MODÈLE DE MAKKOV CACHÉ.......................................... 121
7.1 MÉTHODE DE CLASSIFICATION/PRÉDICTION HYBRIDE................................................................ 121 7.2 LA MÉTHODE ............................................................................................................................. 123 7.3 RÉSULTATS DES TESTS............................................................................................................... 125
DISCUSSION..................................................................................................................................... 130 CONCLUSION .................................................................................................................................. 134 APPENDICE A SÉQUENCES GPI DE SWISS-PROT............................................................................................. 136 APPENDICE B ALIGNEMENT MULTIPLE ........................................................................................................... 147
SORTIE CLUSTALW ......................................................................................................................... 147 APPENDICE C SÉQUENCES D’ENTRAÎNEMENT DU RÉSEAU DE NEURONES ARTIFICIELS............... 157

v
SÉQUENCES D’ENTRAÎNEMENT ........................................................................................................ 157 APPENDICE D ANALYSE ROC DU RÉSEAU DE NEURONES ARTIFICIELS................................................ 159
ANALYSE ROC................................................................................................................................ 159 APPENDICE E SÉQUENCES D’ENTRAÎNEMENT DU MODÈLE DE MARKOV CACHÉ ............................ 163
SEQUENCES D’ENTRAINEMENT ........................................................................................................ 163 APPENDICE F MODÈLE DE MARKOV CACHÉ DE INITIAL........................................................................... 167
MODELE DE MARKOV CACHE .......................................................................................................... 167 APPENDICE G ANALYSE ROC DU MODÈLE DE MARKOV CACHÉ ............................................................. 177
ANALYSE ROC................................................................................................................................ 177 APPENDICE H GRAMMAIRE STOCHASTIQUE GPI.......................................................................................... 181
PROBABILITÉ D’ÉMISSION................................................................................................................ 181 RÈGLES DE PRODUCTION STOCHASTIQUE......................................................................................... 182
APPENDICE I ANALYSE ROC MÉTHODE HYBRIDE....................................................................................... 184
ANALYSE ROC................................................................................................................................ 184 APPENDICE J PRÉDICTIONS ................................................................................................................................. 192
ARABIDOPSIS THALIANA.................................................................................................................. 192 ORYZA SATIVA ................................................................................................................................ 197
BIBLIOGRAPHIE ............................................................................................................................ 201

LISTE DES FIGURES
Figure Page
1. 1 Alphabets ou codes IUPAC-IUBMB des classes de polymères ADN, ARN et protéine ..................................................................................................................7
1. 2 Un acide nucléique en détail .................................................................................9 1. 3 Double hélice d’ADN..........................................................................................11 1. 4 Séquence d’ADN.................................................................................................11 1. 5 Structure de l’ARN..............................................................................................12 1. 6 Structure d’un acide aminé avec les groupements aminé et carboxyle. ..............14 1. 7 Structure de la protéine........................................................................................15 1. 8 Domaines protéiques. ..........................................................................................16 1. 9 Processus de transcription d’un ADN en ARN messager. ..................................18 1. 10 Traduction d’un ARN messager en protéine.. ...................................................20 2. 1 Glycosylation.......................................................................................................25 2. 2 Différentes associations protéine/membrane.....................................................25 2. 3 Biosynthèse d’une protéine à ancre GPI. ............................................................27 2. 4 Structure de la protéine à ancre GPI....................................................................29 3. 1 Classification .......................................................................................................33 3. 2 Alignement entre trois séquences d’ancre GPI différentes. ...............................34 3. 3 Alignement entre quatre différentes séquences d’ancre GPI ..............................34

vii
3. 4 Diversité du signal GPI ........................................................................................35 3. 5 Alignement entre les mots « voiture » et « toiture ». ..........................................36 3. 6 Alignement entre les mots « voiture » et « véhicule » ........................................37 3. 7 Alignement local et global. .................................................................................39 3. 8 Trois alignements possibles entre deux séquences, avec des scores différents...40 3. 9 Matrice BLOSUM62...........................................................................................41 3. 10 Alignement BLOCK...........................................................................................42 3. 11 Alignement avec espace .....................................................................................43 3. 12 Calcul d'un alignement optimal entre les séquences TCGCA et TCCA. ...........44 3. 13 Sortie du programme BLAST effectuée avec l’algorithme blastp. ...................47 3. 14 Alignement multiple..........................................................................................49 4. 1 Représentation distribuée ....................................................................................53 4. 2 Neurone biologique .............................................................................................57 4. 3 Neurone formel....................................................................................................58 4. 4 Fonctions d’activation .........................................................................................59 4. 5 Structure de connexions de réseaux de neurones artificiels. ...............................60 4. 6 Architecture du perceptron. .................................................................................63 4. 7 Architecture du perceptron multicouche. ............................................................65 4. 8 Minimum local et vrai minimum.........................................................................66 4. 9 Modèle de Markov observable ............................................................................72 4. 10 Modèle de Markov caché ..................................................................................73

viii
4. 11 Un Modèle de Markov caché. ............................................................................75 4. 12 Modèle de Markov caché avec émissions et transitions équiprobables. ............78 4. 13 Modèle de Markov caché après apprentissage. ..................................................79 4. 14 Premiers niveaux d’un arbre de dérivation........................................................82 4. 15 Grammaire dérivée d’un HMM.........................................................................84 5. 1 Séquences de protéines à ancre GPI de différentes longueurs. ............................89 5. 2 Vecteur d’entrée du réseau de neurones avant encodage. ...................................90 5. 3 Exemple de vecteur d’entrée. ..............................................................................93 5. 4 Sélection du jeu d’entrainement ..........................................................................94 5. 5 Architecture du perceptron multicouche construit pour la classification de
protéines à ancre GPI. .........................................................................................97 5. 6 Progression de l’erreur lors de la validation......................................................100 5. 7 Courbe ROC pour le test de validation du réseau de neurones. ........................101 6. 1 Structure de séquences GPI avec annotation du site d’ancrage en rouge..........108 6. 2 Modèle de Markov caché représentant le signal GPI. .......................................109 6. 3 Courbe ROC pour le test de validation du HMM .............................................113 6. 4 Les trois meilleurs chemins de la séquence PRIO_HUMAN ...........................116 7. 1 Méthode d’analyse hybride. ..............................................................................123 7. 2 Résultats de la méthode hybride........................................................................127

LISTE DES TABLEAUX Tableau Page
1. 1 Code Génétique ....................................................................................................19 2. 1 Nature des acides aminés dans la zone d’ancrage ...............................................30 4. 1 Tests de validation................................................................................................71 4. 2 Les 15 chemins différents susceptibles de générer ABAA ..................................76 5. 1 Échelles utilisées pour la conversion numérique des acides aminés...................91 5. 2 Tests de sélection du nombre de neurones de la couche cachée .........................97 5. 3 Tests d’ajustement des paramètres de l’apprentissage RPROP ..........................98 5. 4 Résultats du test de validation du réseau de neurones artificiels. .....................102 5. 5 Résultats des tests supplémentaires. ..................................................................102 5. 6 Résultats du test de prédiction à grande échelle................................................103 6. 1 Pourcentage d’occupation des acides aminés pour la zone d’ancrage. .............110 6. 2 Pourcentage d’occupation des acides aminés pour la zone intermédiaire et la
queue hydrophobe. ............................................................................................111 6. 3 Résultats du test de validation du HMM. ..........................................................115 6. 4 Résultats des jeux de test supplémentaires. .......................................................116 6. 5 Acides aminés pour le site d’ancrage selon notre méthode hybride .................118 7. 1 Annotation hybride. ...........................................................................................124 7. 2 Proportion des prédictions du réseau de neurones selon l’annotation hybride. 125

x
7. 3 Comparaison de la précision du système hybride selon la catégorie probable et du réseau de neurones artificiels seul. ...............................................................126
7. 4 Classification du protéome de Arabidopsis thaliana et Oryza sativa. ..............128 7. 5 Étude comparative de la capacité de prédiction de la méthode hybride vs les
différents prédicteurs de big-...........................................................................132

LISTE DES ABRÉVIATIONS A Adénine
a alanine
ADN Acide désoxyribonucléique
ARN Acide ribonucléique
ARNm Acide ribonucléique messager
ARNr Acide ribonucléique de transfert
ARNsn Acide ribonucléique nucléaire
ARNt Acide ribonucléique de transfert
BLAST Basic Local Alignment Search Tool
BLOSUM Blocks Substitution Matrices
C Cytosine
c cystéine
CFG Context Free Grammar (Grammaire hors contexte)
CLUSTALW Cluster Alignement
COOH Groupement carboxyle
CSG Context Sensitive Grammar (Grammaire sensible au contexte)
d acide aspartique
e acide glutamique
f phénylalanine
G Guanine
g glycine
Ghz Gigahertz
GPI Glycosylphosphatidylinositol
h histidine
HMM Hidden Markov model
i isoleucine
IA Intelligence Artificielle

xii
IUPAC International Union of Pure and Applied Chemistry
IUBMB International Union of Biochemistry and Molecular Biology
k lysine
l leucine
m méthionine
n asparagine
NCBI National Center for Biotechnology Information
NH2 Groupement aminé
p proline
PAM Point Accepted Mutation
q glutamine
r arginine
RG Regular Grammar (grammaire régulière)
ROC Receiver Operating Characteristic
RPROP Resilient Propagation
s sérine
SOM Self Organizing Maps
T Thymine
t thréonine
U Uracile
UnresG Urestricted Grammar (Grammaire sans restriction)
v valine
w tryptophane
XOR Ou exclusif
y tyrosine

RÉSUMÉ
Une ancre glycosylphosphatidylinositol (ancre GPI) est une structure d’ancrage membranaire complexe mais commune chez les protéines eucaryotes extracellulaires. Cette structure a été très bien conservée durant l’évolution de la cellule de la levure jusqu'à celle des mammifères. La fonction précise de ce type d’attachement n’est pas bien définie, mais cette conservation élevée dans l’évolution des cellules eucaryotes laisse facilement présumer un rôle fonctionnel important. Toutefois, quelques caractéristiques sont connues. Par exemple, les ancres GPI sont souvent définies comme des cibles ou des signaux positionnés à la surface des cellules. Les banques de séquences protéiques, telles que Swiss-Prot, proposent peu de séquences ayant cette modification, car leur présence n’est pas connue depuis longtemps et peu d’outils permettent l’annotation automatique des nouvelles séquences. Les différents projets de séquençage de génomes amènent une profusion de nouvelles séquences qu’il faut annoter. De plus, la prédiction de modifications post-traductionnelles des protéines fait partie intégrante d'une étude approfondie permettant la compréhension des fonctions biologiques. Elle se révèle être une étape importante, non seulement pour l’annotation des protéomes, mais aussi pour l’étude des systèmes biologiques à grande échelle. Des outils qui pourront aider à l’annotation des signaux dans les séquences sont donc une nécessité, surtout pour des structures récemment découvertes, comme les ancres GPI. Cette thèse développe une méthode d’analyse qui se base sur l’utilisation d’un réseau de neurones artificiels et d’un modèle de Markov caché (HMM). Le réseau de neurones artificiels sélectionne les séquences protéiques ayant un signal GPI potentiel et le HMM structure le signal. La combinaison des deux techniques d’apprentissage machine révèle un pouvoir prédictif intéressant, car elle exploite les propriétés physicochimiques de la molécule ainsi que la nature séquentielle de sa représentation. La méthode hybride permet de prédire 93% des séquences protéiques annotées comme protéines à ancre GPI, dans la base de données Swiss-Prot. Une caractéristique importante de la méthode d’analyse hybride que nous proposons est qu’elle cible uniquement la partie C-terminale de la protéine. Cette particularité la rend moins sensible aux erreurs si répandues dans les bases de données de séquences. De plus, cette méthode n’est pas spécifique à un seul groupe taxonomique. Elle peut être utilisée pour prédire la présence de protéines à ancre GPI chez tous les eucaryotes (plantes, animaux, champignons, protozoaires, etc.). L’utilisation d’un HMM pour structurer le signal nous permet de définir la grammaire sous-jacente au signal. Cette grammaire peut, par la suite, servir à proposer une structure du signal représentative des séquences connues à ce jour. Finalement, une technique d’annotation selon une échelle de qualité permet de combiner une très grande sensibilité ainsi qu’une annotation informative de chaque prédiction de la méthode hybride.

INTRODUCTION
En 1866, un moine tchèque du nom de Gregor Mendel établissait les premières lois de
l’hérédité grâce à son étude sur l’hybridation des plantes. De ces travaux est née la génétique
classique. Cette découverte ouvrait la porte à l’étude du transfert de l’information dans le
matériel vivant.
L’importance des gènes n’est plus un secret pour personne. De nos jours, des termes tels que
ADN (acide désoxyribonucléique) et protéine ne sont plus des termes techniques connus
seulement des experts. Les séquences biologiques ont maintenant une place capitale dans la
recherche sur le vivant. Ces séquences sont représentées par une suite de lettres provenant
d’un alphabet de 4 lettres, pour les acides nucléiques de l’ADN, et de 20 lettres, pour les
acides aminés des protéines. Depuis 1955, lors de la publication de la première séquence
protéique, l’insuline bovine (Sanger, Thompson et Kitai, 1955), le nombre de séquences
protéiques et nucléiques rendues publiques ne fait qu’augmenter. La première base de
données de séquences biologiques (Dayhoff et al., 1965) et les premiers algorithmes
d’analyse de ces données ont donc vu le jour quelques années plus tard, donnant, par la même
occasion, naissance à un nouveau domaine de recherche : la bioinformatique. Depuis
plusieurs années, des milliers de projets scientifiques, liés de près ou de loin à la découverte
d’un moine tchèque ayant vécu il y a plus de 150 ans se concentrent sur l’analyse de ces
séquences et des mystères qu’elles renferment.
Le nombre de séquences biologiques présentes dans les bases de données publiques
augmente de façon exponentielle. Par exemple, en 1982, la base de données de séquences
d’acides nucléiques du NCBI (National Center for Biotechnology Information) contenait 606
séquences. À peine 10 ans plus tard, elle contenait près de 79 000 séquences et, finalement, la
version 142 (15 juin 2004) contenait plus de 35 000 000 de séquences totalisant près de 40

2
milliards de nucléotides. La nécessité de trouver des moyens d’entreposer et, surtout,
d’analyser toute cette information fut vite un sujet de discussion et de recherche.
À l’aube du troisième millénaire, les problèmes auxquels font face les biologistes et les
informaticiens ne sont plus liés au séquençage mais plutôt à l’annotation des séquences déjà
produites. L’annotation consiste à identifier la structure et la fonction des molécules codées
par les séquences. Traditionnellement, l’annotation se fait grâce à des expériences
systématiques en laboratoire. Toutefois, avec le nombre grandissant de séquences
disponibles, il devient primordial d’automatiser, au moins partiellement, le processus
d’annotation.
Le sujet de cette thèse est le développement d’une technique d’annotation basée sur des
algorithmes de classification. Notre approche est résolument expérimentale. En effet, les
objets avec lesquels nous travaillons sont des suites de lettres qui, théoriquement, encodent
des fonctions biologiques. Un algorithme de classification ou de prédiction pourra, dans le
meilleur des cas, aider le travail des biologistes dans la détermination exacte de la fonction
d’une molécule. C’est ce critère d’utilité biologique qui, ultimement, sera le critère
fondamental pour évaluer nos algorithmes.
La complexité des informations contenues dans les séquences biologiques est un problème en
soi : certaines caractéristiques sont facilement identifiables, comme par exemple
l’hydrophobicité générale, alors que d’autres sont bien cachées, comme la présence de courts
segments ayant une fonction précise, mais ayant subi de nombreuses mutations au cours de
l’évolution. Identifier et classifier correctement ces caractéristiques requièrent souvent des
approches nouvelles, qui viennent appuyer les outils d’analyse standard, tel l’alignement de
séquences. Dans cette thèse, nous nous sommes penchés particulièrement sur le problème de
la prédiction d’une modification post-traductionnelle particulière : l’ancrage
glycosylphosphatidylinositol, auquel nous référerons par la forme abrégée ancre GPI dans la
suite du texte.

3
La structure des protéines provenant de la traduction des gènes n'est pas suffisante pour
indiquer toute la complexité de leurs fonctions. Des modifications post-traductionnelles
peuvent amener, par exemple, des changements d’activités, de localisation cellulaire et
d’interaction avec d’autres protéines (Seo et Lee, 2004). Les modifications post-
traductionnelles, comme l’ancre GPI, ont une grande importance dans le processus de
compréhension des fonctions biologiques. Toutefois, leur étude souffre d'un manque de
méthodes valables permettant l’étude à grande échelle (Mann et Jensen, 2003). La prédiction
de modification post-traductionnelle des protéines fait partie intégrante d'une étude
approfondie permettant la compréhension des fonctions biologiques. Elle se révèle être une
étape importante, non seulement pour l’annotation de protéomes, mais aussi pour l’étude des
systèmes biologiques à grande échelle.
Contribution originale
La contribution originale de ce projet est la conception d’un outil hybride de prédiction d’un
signal protéique, la modification post-traductionnelle de l’ancre GPI, important pour
l’annotation des protéomes et pour l’étude de la fonction des protéines. L’outil est hybride car
il fait appel à la fois aux techniques de réseau de neurones artificiels et à celles des modèles
de Markov cachés. L’utilisation de la méthode d’apprentissage neuronale permet une bonne
fouille préliminaire des données, tandis que l’exploitation de la nature régulière du langage
des séquences biologiques sert à structurer les prédictions et à annoter qualitativement chaque
prédiction. Cette méthode hybride donne donc un outil plus complet que ceux déjà existants
et ouvre les portes à d’autres applications en analyse de signaux protéiques. Nous proposons
enfin une grammaire formelle du signal GPI, ainsi qu’une réévaluation de nos connaissances
sur sa structure. Ces travaux ont fait l’objet de deux présentations dans des rencontres
scientifiques internationales (Poisson et al., 2003; Poisson, Bergeron et Chauve, 2004) d’un
séminaire sur invitation à l’Université de Hawaii à Manoa, au département « Information and
Computer Science ».

4
Structure de la thèse
Cette thèse s’organise en sept chapitres. Le premier chapitre propose les bases biologiques
nécessaires à la compréhension de la méthode d’analyse développée. Nous débutons par une
introduction générale aux séquences biologiques, en partant de l’ADN, jusqu’à la
construction d’une protéine. Par la suite, le chapitre 2 se concentre sur un phénomène bien
précis, celui des modifications post-traductionnelles. Dans ce chapitre, nous verrons la
mécanique derrière une modification post-traductionnelle en particulier : l’ancre GPI. La
nature même de cette modification se voit codée au sein des séquences protéiques. Nous
verrons l’importance de pouvoir classer, ou plutôt prédire, la présence de cette modification
dans les séquences biologiques.
La construction d’un outil d’analyse de cette modification post-traductionnelle demande
l’utilisation de techniques de classification de données. Le chapitre 3 présente le problème de
classification et discute des approches couramment utilisées pour l’analyse de séquences
biologiques, basées sur l’alignement de séquences. Le chapitre 4 présente deux techniques
provenant du domaine de l’apprentissage machine (réseaux de neurones artificiels et modèles
de Markov caché) qui permettront de combler les lacunes des techniques d’alignement dans
le problème qui nous intéresse.
Finalement, les 3 derniers chapitres contiennent la contribution scientifique originale de cette
thèse. Le chapitre 5 présente un modèle de réseau de neurones artificiels efficace pour
effectuer un bon nettoyage des données. Le chapitre 6 aborde la construction d’un modèle de
Markov caché représentant la structure du signal GPI ainsi que la grammaire régulière
stochastique qui en découle. Le chapitre 7 décrit la construction de la méthode hybride ainsi
que l’annotation qualitative basée sur la structuration du signal GPI par le modèle de Markov
caché.
En conclusion, nous verrons que l’utilisation de la méthode d’analyse hybride permet
d’obtenir un taux de prédiction du signal GPI plus que satisfaisant. De plus, nous
démontrerons qu’une grammaire du signal GPI proposant la structure du signal et une

5
annotation qualitative des prédictions augmente grandement la pertinence d’utilisation d’une
méthode hybride dans l’analyse de séquences biologiques.

CHAPITRE I
BASES BIOLOGIQUES Ce chapitre présente les généralités relatives aux acides nucléiques et aux protéines. Nous donnons les bases biologiques nécessaires à la compréhension du problème de la classification des séquences biologiques et, plus particulièrement, de celui lié à la classification des protéines à ancre GPI.
1.1 Les séquences
Il y a plus d’un siècle, Mendel comprit que le gène était une entité distincte. Ceci mit en
évidence un fait maintenant bien accepté : l’information nécessaire à la construction d’un
nouvel organisme se transmet d’une génération à l’autre (Lewin, 1999). Trois grandes classes
de molécules sont impliquées dans ce processus d'entreposage, de conversion et de
transmission d'information :
1. l'acide désoxyribonucléique (ADN), 2. l'acide ribonucléique (ARN) et 3. les protéines.
Ces trois types de molécules constituent les bases génétiques de la machinerie d'encodage et
de transmission de l'information cellulaire.

7
Ces molécules sont des polymères, c'est-à-dire qu'elles sont formées de suites de petites
molécules liées séquentiellement. L'ordre dans lequel ces molécules apparaissent dans un
polymère déterminera les structures, les localisations cellulaires et les fonctions biologiques.
Les molécules qui forment l'ADN et l'ARN sont appelées nucléotides. Celles qui forment les
protéines sont appelées acides aminés. Comme le nombre de ces molécules est relativement
faible, 5 pour les nucléotides et 20 pour les acides aminés, il est possible de représenter
chacune d'entre elles par une lettre de l'alphabet. Les conventions pour l'assignation de ces
lettres sont résumées dans la figure 1.1. On aura, par exemple,
1. une séquence d'ADN : "ACCCGTAGTAAA" ; 2. une séquence d'ARN : "GUACGUUUCAG" ; 3. une protéine : "aillickgrsillwwwy1".
ADN A AdénineC CytosineG Guanine T Thymine
A Adénine C Cytosine G Guanine U Urcile
ARN
PROTÉINE a alanine c cystéined acide aspartiquee acide glutamiquef phénylalanineg glycineh histidine i isoleucinek lysinel leucine
ALPHABET IUPAC-IUBMB COMPLÉMENT
R A ou G PurineY C ou T PyrimidineW A ou T Weak (faible) liaison hydrogèneS C ou G Strong (forte) liaison hydrogèneM A ou C Groupe amino même position K G ou T Groupe ketone même positionB C ou G ou T Tous sauf AD A ou G ou T Tous sauf CH A ou C ou T Tous sauf GV A ou C ou G Tous sauf T ou UN A ou C ou G ou T/U Tous
b acide aspartique ou asparaginex inconnuz acide glutamique ou glutamine. fin
m méthioninen asparaginep proline q glutamine r arginine s sérinet thréoninev valinew tryptophane y tyrosine
ALPHABET IUPAC-IUBMB COMMUN
Figure 1. 1 Alphabets ou codes IUPAC-IUBMB des classes de polymères ADN, ARN et protéine (IUPAC-IUB 1993).
1 Dans ce texte les acides aminés sont notés avec des caractéres minuscules. Toutefois certains logiciels utilisés pour faire les figures, utilisent des caractères majuscules.

8
Les protéines sont les molécules les plus diversifiées des trois. Ceci a biaisé notre
compréhension du mécanisme de la transmission de l’information pendant très longtemps.
On croyait que seules les protéines pouvaient produire une telle diversité. Cette croyance a
pris fin lorsqu’on découvrit que le matériel génétique (acides nucléiques) était le support de
l’information génétique. Les travaux de Griffith (1928) sur le virus responsable de la
pneumonie ont montré qu’une substance chimique était passée de virus morts à des virus
vivants. Cette substance fut isolée, en 1944, par Oswald Avery, Maclyn McCarty et Colin
MacLeod et se révéla être de l’ADN et non une protéine, comme on l’avait cru. Mais, ce
n’est qu’en 1952 qu’Alfred Hershey et Martha Chase démontrèrent, par marquage radioactif,
que l’ADN était le support de l’information génétique.
Dans les sections 1.1.1 et 1.1.2, nous allons décrire plus en détails ces différentes molécules.
La section 1.2 porte une attention particulière sur le processus de traduction des protéines car
nous nous intéressons à la classification/prédiction de ce type de molécules biologiques.
1.1.1 ADN et ARN : polymères d’acides nucléiques
Un acide nucléique est composé de petites molécules appelées nucléotides. Deux types
d’acides nucléiques existent : l’ADN, qui entrepose l’information génétique d’un individu et
l’ARN qui sert le plus souvent de vecteur de l’information. Un nucléotide est constitué d’une
base azotée de type purine (adénine A ou guanine G) ou de type pyrimidine (cytosine C ou
thymine/uracile T/U), d’un sucre déoxyribose (ADN) ou ribose (ARN) et d’un groupe
phosphate. La figure 1.2 montre la structure primaire d’un acide nucléique. Cette structure est
formée par un lien phosphodiester entre le phosphate en position 5’ d’un nucléotide et le
sucre en position 3’ du nucléotide suivant. Une séquence d’acide nucléique se lit en sens
5’ → 3’.

9
A
C
T
G
C
base azotée
sucre
groupe phosphate
5'
3'
nucléotide
sens
de
lect
ure
Figure 1. 2 Un acide nucléique en détail. L’encadré isole un nucléotide. Le sens de lecture est de l’extrémité 5’ de l’acide nucléique vers l’extrémité 3’.
1.1.1.1 L’ADN
Lorsque qu’une base azotée est attachée à un sucre, on la nomme un nucléoside. L’adénine
devient une adénosine, la guanine devient une guanosine, la cytosine devient une cytidine et
la thymine devient une thymidine. Une fois ces nucléosides attachés à un groupe phosphate,
on les nomme des nucléotides. Ces nucléotides forment des acides nucléiques de type ADN.
Les bases azotées formant une chaîne de nucléotides ont des affinités avec, ou une attirance
vers, un autre membre du groupe. Cette affinité les incitent à former des liens hydrogènes
entre elles, lorsque mises en contact. L’adénine A est ainsi dite complémentaire à la thymine
T et la cytosine C est complémentaire à la guanine G.

10
L’ADN typique d’un organisme consiste en une molécule formée de deux chaînes de
nucléotides entrelacées entre elles (fig. 1.3). Des liens hydrogènes les unissent, des liens forts
(trois ponts hydrogènes) entre les nucléotides C et G et des liens plus faibles (deux ponts
hydrogènes) entre A et T. Watson et Crick (1953) ont construit un modèle de la structure de
l’ADN en se basant sur trois notions importantes : 1) Des données de diffraction des rayons
X de Rosalind Franklin montrèrent une forme de double hélice. 2) La densité de l’ADN
suggérait vers la présence de deux chaînes. 3) L’affinité des bases A-T et C-G fut démontrée
par un pourcentage de G et C identique ainsi qu’un pourcentage de A et T identique dans
l’ADN (Lewin, 1999). Ces notions ont vite amené la proposition d’une structure de l’ADN en
double hélice composée de deux chaînes de nucléotides complémentaires.
L’ADN peut être représenté sous trois formes. La molécule d’ADN peut former des liens
phosphodiesters entre le sucre d’un nucléotide et le groupement phosphate du nucléotide
suivant. Cette forme d’attachement donne la structure primaire de la molécule (représentée
sous forme de chaîne de lettres figure 1.3a). Par la suite, des liens hydrogènes peuvent se
former entre les deux chaînes de la molécule, établissant ainsi la structure secondaire (fig.
1.3b). Finalement, des angles particuliers entre les liaisons reliant les nucléotides donnent la
forme en spirale caractéristique à l’ADN, sa structure tertiaire (fig. 1.3c).
La structure de l’ADN n’est pas sa caractéristique la plus importante. Ce qui importe le plus
est la séquence d’acides aminés qu’elle encode. L’ADN est une séquence de nucléotides
représentée sous forme simple ou double (fig.1.4). Comme les deux séquences sont
complémentaires, il suffit de connaître une chaîne pour pouvoir en déduire la seconde. Pour
le traitement informatique de ces séquences, la chaîne simple est normalement utilisée et sa
lecture se fait de gauche à droite, de la position 5’ vers la position 3’ (fig. 1.4).
11
A
C
T
G
C
5'
3'
G
A
C
G
3'
5'
T
ACCTGTGGAC
5'
5'
3'
3'
STRUCTURE PRIMAIRE
STRUCTURE SECONDAIRE
STRUCTURE TERTIAIREa
b
c
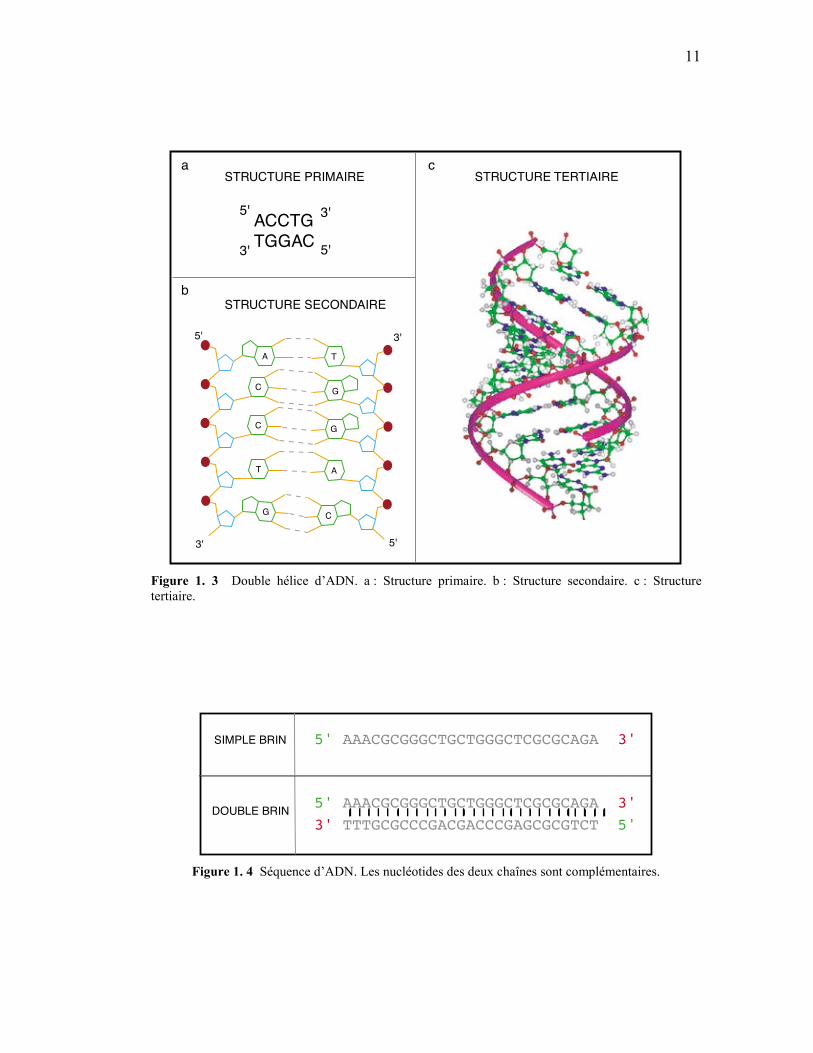
Figure 1. 3 Double hélice d’ADN. a : Structure primaire. b : Structure secondaire. c : Structure tertiaire.
5' AAACGCGGGCTGCTGGGCTCGCGCAGA 3'
5' AAACGCGGGCTGCTGGGCTCGCGCAGA 3'3' TTTGCGCCCGACGACCCGAGCGCGTCT 5'
SIMPLE BRIN
DOUBLE BRIN
Figure 1. 4 Séquence d’ADN. Les nucléotides des deux chaînes sont complémentaires.

12
1.1.1.2 L’ARN
L’ARN est, comme l’ADN, une chaîne de nucléotides. Toutefois, dans le cas de l’ARN, la
base thymine T est remplacée par l’uracile U. Le nucléoside composé de la base uracile et
d’un sucre se nomme uridine. Contrairement à L’ADN, l’ARN se retrouve sous la forme d’un
simple brin. Cette particularité fait que l’ARN forme des liens hydrogènes avec ses propres
bases, formant ainsi des structures secondaires et tertiaires très variées (fig. 1.5b et 1.5c). La
longueur de la chaîne de l’ARN est considérablement plus petite que celle de l’ADN (des
milliers au lieu de millions de nucléotides).
Figure 1. 5 Structure de l’ARN. a : Structure primaire. b : Structure secondaire. c : Structure tertiaire de l’ARNm du prion (Barrette et al., 2002).
Il existe principalement trois types d’ARN :
1. L’ARNm (messager) et pré-ARN: Il contient l’information provenant des gènes et servant à produire une protéine.

13
2. L’ARNt (transfert) : Son rôle est complémentaire à l’ARNm. Il existe environ 20 groupes d’ARNt, un pour chaque acide aminé. Sa forme de trèfle très caractéristique est composée de quatre bras. Un de ces bras, l’anticodon, est composé d’une séquence des trois bases complémentaires nécessaires à la production d’un acide aminé particulier, par exemple, le triplet UCA, code pour l’acide aminé sérine.
3. L’ARNr (ribosomal) : Accompagné de différentes protéines, l’ARNr compose les
ribosomes. Sa fonction n’est pas, comme dans le cas de l’ARNm et de l’ARNt, de nature à produire une protéine. Son rôle est plutôt structurel : il sert de charpente aux ribosomes.
Il existe aussi d’autres ARN, tels que les ARNsn (petits ARN nucléaires) ou les ARNsno
(situés dans le nucléole et impliqué dans la maturation des ribosomes).
1.1.2 Les protéines
À la différence de l’ADN et de l’ARN, la protéine est une chaîne simple brin composé
d’acides aminés (fig. 1.1). Un acide aminé se caractérise par un groupement carboxyle
(COOH), un groupement aminé (NH2) et une chaîne latérale (fig. 1.6). La chaîne latérale
différencie les acides aminés les uns des autres. La figure 1.6 montre deux exemples de
chaînes latérales, celle de la tyrosine et celle de la méthionine.
Comme pour l’ADN et l’ARN, on retrouve plusieurs niveaux de structure. La condensation
du groupement carboxyle de la tyrosine de la figure 1.6 et du groupement aminé de la
méthionine donne une liaison peptidique N-H, représentée en pourpre. Ces liaisons donnent
la structure primaire de la protéine.

14
H 2 N HC C
O
OHCH2
OH
H 2 N HC C
O
OHCH2
CHCH3 S
Tyrosine Méthionine
groupement latéralgroupement latéral
groupement aminé groupement aminégroupement carboxylegroupement carboxyle
H 2 N HC C
O
CH2
OH
HC
CO
CH2
CHCH3 S
N H
lien peptidique
Liaison peptidique entre acides aminés
Figure 1. 6 Structure d’un acide aminé avec les groupements aminé et carboxyle.
Comme pour l’ADN et l’ARN, la représentation de la structure primaire se fait sous forme
d’une chaîne de lettres que l’on peut lire de gauche à droite, c’est-à-dire de la partie NH2
(appelée N-terminale) vers la partie COOH (appelée C-terminale) (fig. 1.7a). L’agencement
de la chaîne polypeptidique dans l’espace, selon les interactions existantes (liaisons
hydrogènes) entre les acides aminés, donne la structure secondaire (fig. 1.7b). Dans la
structure secondaire, on retrouve des éléments structuraux simples comme les hélices alpha
et les feuillets béta. L’organisation des éléments de structure secondaire entre eux donne la
structure tertiaire (fig. 1.7c). Finalement, certaines chaînes polypeptidiques ont la capacité de
s’associer entre elles sous forme de dimères (deux chaînes), quadrimères etc. Cet assemblage
donne la structure quaternaire (fig. 1.7d).

15
STRUCTURE TERTIAIRE
STRUCTURE SECONDAIRE
a bSTRUCTURE PRIMAIRE
feuiller beta
hélice alpha
polypeptide
c
STRUCTURE QUATERNAIREd
polypeptide 1 polypeptide 2
polypeptide 3 polypeptide 4
feuillet betahelice alpha
feuillet beta
helice alpha
helice alpha
glggymlgsamsrpmihfgndwedryyrenmyyrypnqvyyrpvdqysnqnnfvhdcvnjtikqhtvttttkgenftetdvkmmervveqmcvtqyqkesqayy
N-terminal
C-terminal
Figure 1. 7 Structure de la protéine. a : Structure primaire de la protéine 1AG2 du prion de la souris. b : Structure secondaire de la protéine 1AG2 du prion de la souris. c : Structure tertiaire de la protéine 1AG2 du prion de la souris. d : Structure quaternaire de protéine deoxyhemoglobin2.
La capacité des protéines de se replier en conformation tridimensionnelle est très importante
au point de vue fonctionnel. Différentes conformations donnent accès à différentes portions
de la chaîne, ce qui donnera la fonction de la molécule. Il existe différents types
d’interactions qui influencent la structure des protéines. Notons particulièrement les fortes
liaisons covalentes telles que les ponts disulfures et les plus faibles liaisons non covalentes
telles que les liaisons hydrogènes, les liaisons ioniques, les interactions hydrophobes et les
forces de Van der Waals (Lewin, 1999).
Dans un exemple d’interaction, les groupements latéraux des acides aminés peuvent, par
exemple, s’avérer très hydrophobes. En présence d’eau, ces parties hydrophobes auront
tendance à se regrouper pour échapper à l’eau. Ces groupements se retrouvent donc souvent
2 Source des images : Protein DataBank www.rcsb.org.

16
au cœur de la structure de la protéine, loin de l’eau. Ces interactions vont fournir beaucoup
d’indices sur la fonction de la protéine.
Comme les protéines sont essentielles à la plupart des processus biologiques, il n’est pas
étonnant de constater qu’il existe plusieurs familles de protéines. Les protéines peuvent être
des enzymes, des transporteurs, des hormones, des régulateurs, des éléments de structure etc.
Une protéine peut aussi être organisée en domaines. Un domaine représente une région de la
protéine qui a une fonction et une structure relativement indépendante. La longueur d’un
domaine varie de 30 à 300 acides aminés (Lewin, 1999). Les domaines protéiques sont une
notion importante car ils constituent des éléments fonctionnels utiles pour la classification
des séquences. Par exemple, la figure 1.8 montre la séquence DAF humaine, une protéine
connue comme étant un facteur d’accélération de la dégradation et qui se compose d’au
moins trois domaines membranaires répertoriés selon la base de données de domaines
protéiques ProDom (version 2004.1, juin 2004). Des séquences comportant des domaines
similaires ont de fortes chances d’avoir une fonction similaire. Un domaine
transmembranaire, un domaine hydrophobe ou un domaine de la fibronectine sont des
exemples de domaines protéiques.
100 200 300 400 5000
domaine facteur sushi
domaine recepteursushi
précurseur sushi
DAF_HUMAIN
Figure 1. 8 Domaines protéiques. Domaines présents dans la protéine à ancre GPI DAF humaine. Swiss-Prot, numéro d’accession: P08174.

17
1.2 De l’ADN à la protéine
Tel que mentionné plus haut, des gènes se retrouvent le long d’une molécule d’ADN. Ces
gènes contiennent les plans nécessaires à la formation de protéines. Le processus de base
permettant le passage du gène à la protéine se décrit schématiquement comme suit :
ADN ARN ProtéineTranscription Traduction
Les sections suivantes vont traiter du processus de transcription ainsi que de la traduction de
l’ARN en protéine. Des modifications peuvent aussi se produire après la traduction. Un
exemple de telle modification sera traité dans le chapitre 2.
1.2.1 La transcription
Lors de la transcription, une chaîne d’ARN est synthétisée à partir d’un des brins de l’ADN :
le brin matrice. Le résultat de cette copie sera complémentaire à cette matrice. Le processus
de transcription est résumé dans la figure 1.9. Une région spécifique, le promoteur, sert de
point de départ à cette transcription. Une enzyme, l’ARN polymérase, se fixe sur le
promoteur et commence à synthétiser l’ARN. À ce moment, les bases thymines sont
remplacées par des uraciles. Cette synthèse se poursuit jusqu’à ce qu’un terminateur (cercle
rouge de la figure 1.9a) soit rencontré le long de la séquence d’ADN. Les gènes des
eucaryotes3 sont composés de segments de régions codantes, les exons, et non codantes, les
introns, intercalés les uns avec les autres (fig. 1.9b). Dans le transcrit primaire, ou préARN,
les introns et les exons sont présents. On note aussi la présence de la coiffe en position 5’ et
une queue polyA (fig. 1.9c). La coiffe empêche la dégradation de l’ARN en augmentant
l’affinité de l’ARN pour des enzymes de traduction, tandis que la queue polyA protège
l’extrémité 3’ de l’ARN. Lors de la maturation de l’ARN messager, le processus d’épissage
coupe les introns pour ne laisser qu’un ARN contenant les portions codantes du gène appelé
3 Les cellules eucaryotes sont caractérisées par la présence d’un noyau contenant le matériel génétique (ADN).

18
un ARN messager mature (fig. 1.9d). Cet ARN messager sera le support qui contient le code
nécessaire à la production d’une protéine précise (fig. 1.9e). Ce processus semble impliquer
un rapport :
un gène → un ARN messager → une protéine.
Toutefois ce rapport n’est pas exact, puisque des processus comme l’épissage alternatif
permettent de produire différents ARN messagers avec un seul et même gène.
exon intron
ADNbrin codant
brin matrice
5'
5'3'
3'
5' 3'préARN
préARNpromoteur terminateur
AAAAAAAAAA
coiffe queue poly A
épissage AAAAAAAAAA
5'
5' 3'
3'
AAAAAAAAAA5' 3'ARNm
préARN
ARN polimérase
Figure 1. 9 Processus de transcription d’un ADN en ARN messager.

19
1.2.2 La traduction
Le processus de transcription s’effectue, chez les eucaryotes, à l’intérieur du noyau de la
cellule. Une fois l’ARN messager à maturité, il traverse la membrane du noyau pour se
retrouver dans le cytoplasme, où s’effectue la synthèse des protéines, appelée traduction. Les
principaux acteurs dans la traduction sont l’ARN messager, les ribosomes, composés de 2
sous-unités, et les ARN de transfert. Un ARN messager est composé d’une séquence de
lettres provenant de l’alphabet des ARN (A, C, G et U). Cette séquence est traduite sous
forme de protéine en utilisant un code : le code génétique. La clé de ce code est la lecture
sous forme de triplets ou codons. Il existe 43 codons ou 64 triplets possibles. De ces 64
codons, 61 sont des acides aminés et 3 entraînent l’arrêt de la traduction (Lewin, 1999)
(tableau 1.1). Comme il n’existe que 20 acides aminés différents, plusieurs codons
représentent le même acide aminé. Par exemple, les codons GUA, GUC, GUG et GUU sont
toutes des combinaisons qui codent l’acide aminé valine.
Tableau 1. 1 Code Génétique
UUU phénylalanine fUUC phénylalanine fUUA leucine lUUG leucine l
UUCU sérine sUCC sérine sUCA sérine sUCG sérine s
UAU tyrosine yUAC tyrosine yUAA stopUAG stop
UGU cystéine cUGC cystéine cUGA stopUGG tryptophane w
U C A G
CUU leucine lCUC leucine lCUA leucine lCUG leucine l
CCCU proline pCCC proline pCCA proline pCCG proline p
CAU histidine hCAC histidine hCAA glutamine qCAG glutamine q
CGU arginine rCGC arginine rCGA arginine rCGG arginine r
AUU isoleucine iAUC isoleucine iAUA isoleucine iAUG méthionine m
AACU thréonine tACC thréonine tACA thréonine tACG thréonine t
AAU asparagine nAAC asparagine nAAA lysine kAAG lysine k
AGU sérine sAGC sérine sAGA arginine rAGG arginine r
GUU valine vGUC valine vGUA valine vGUG valine v
GGCU alanine aGCC alanine aGCA alanine aGCG alanine a
GAU acide aspartique dGAC acide aspartique dGAA acide glutanique eGAG acide glutanique e
GGU glycine gGGC glycine gGGA glycine gGGG glycine g
20
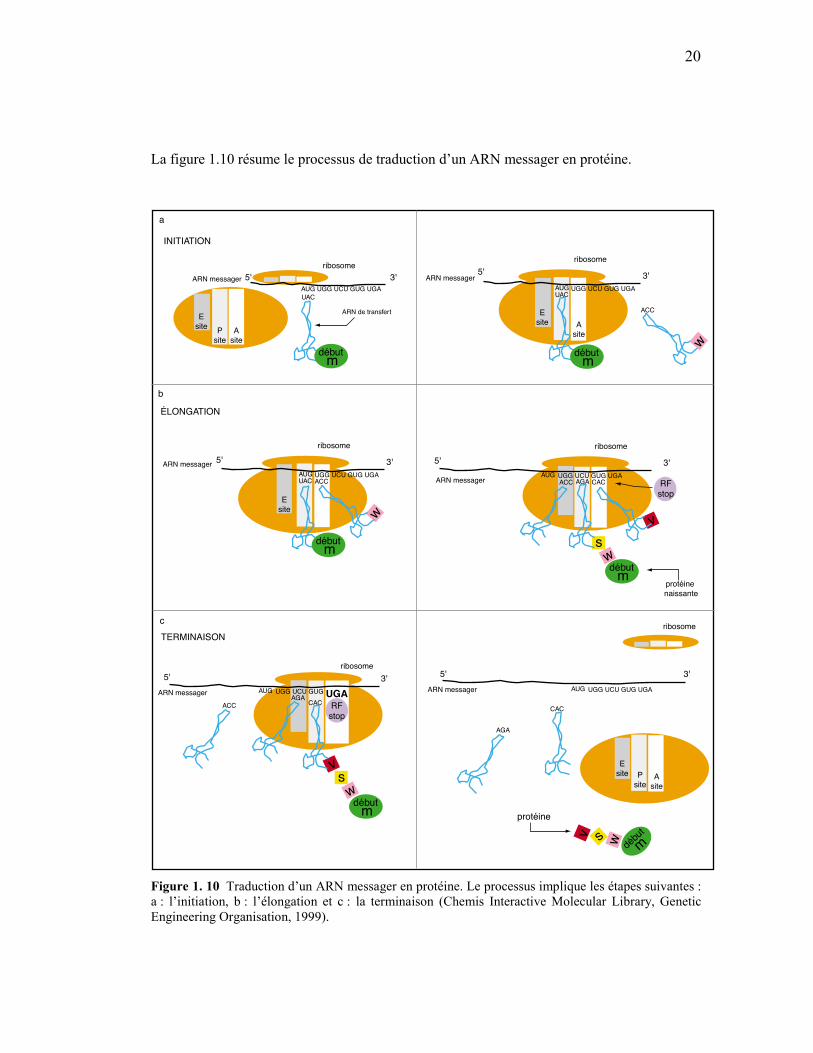
La figure 1.10 résume le processus de traduction d’un ARN messager en protéine.
CAC
a
INITIATION
ÉLONGATION
b
cTERMINAISON
5' 3'ARN messager
ARN de transfert
AUG UGG UCU GUG UGA
ribosome
UAC
mdébut
Asite
Psite
Esite
5' 3'ARN messager
ribosome
AUGUAC
UGG UCU GUG UGA
Asite
mdébut
Esite
5' 3'ARN messager
ribosome
5' 3'ARN messager
ribosome
AUG UGG UCU GUG UGA
wsv
mdébu
t
AGA
protéine
5' 3'
ARN messager
ribosome
AUG
ws
v
mdébut
RFstop
protéine naissante
5' 3'ARN messager
ribosome
ws
v
ACC
mdébut
AUG
CACUGG UCU GUG
AGA UGARF
stop
ACC
w
mdébut
AUG UGG UCU GUG UGA
w
UAC ACC
Esite
AGA CACUGG UCU GUG UGA ACC
Esite P
siteA
site
Figure 1. 10 Traduction d’un ARN messager en protéine. Le processus implique les étapes suivantes : a : l’initiation, b : l’élongation et c : la terminaison (Chemis Interactive Molecular Library, Genetic Engineering Organisation, 1999).

21
Après son passage vers le cytoplasme, l’ARN messager entre en contact avec le ribosome
pour l’initiation de la synthèse (fig. 1.10a). Le ribosome comporte deux sites pour fixer
l’ARN de transfert. Le site « A » est le site accepteur nécessaire à la fixation de l’ARN de
transfert qui vient d’arriver. Le site « P » est occupé par un ARN de transfert portant la
chaîne polypeptidique naissante. Le ribosome comprend aussi le site « E » qui sert à évacuer
les ARN de transfert une fois traités (fig.1.10a).
L’initiation signifie la mise en place des éléments nécessaires à la traduction. C’est à cette
étape que l’ARN messager entre en contact avec le ribosome et le premier ARN de transfert
(l’ARN de transfert correspondant à la méthionine m, dans la fig. 1.10a). Par la suite, l’étape
d’élongation permet la polymérisation, ou l’ajout des acides aminés correspondant à l’ARN
messager (fig. 1.10b). Finalement, la rencontre du site « A » du ribosome avec un codon stop
provoque l’arrêt de l’élongation (fig. 1.10b). Le codon stop n’est pas reconnu par un ARN de
transfert. C’est plutôt une protéine, dite facteur de relâchement, qui reconnaît ce signal
d’arrêt. Après fixation de la protéine facteur de relâchement sur le codon stop, la liaison entre
la protéine et le dernier ARN de transfert est clivée, libérant la protéine. Ce processus
s’accompagne aussi de la dissociation des sous-unités du ribosome (fig. 1.10c).
Certaines protéines devront par la suite subir une translocation, c'est-à-dire qu’elles seront
transportées vers un endroit différent de celui où la traduction a débuté. Il existe deux type de
translocation : la translocation post-traductionnelle et la translocation co-traductionnelle.
1. Translocation post-traductionnelle : Les protéines subissant la translocation post-traductionnelle sont des protéines associées aux mitochondries, aux noyaux et aux chloroplastes. Ces protéines sont synthétisées par des ribosomes libres.
2. Translocation co-traductionnelle : D’autres protéines subissent la translocation durant la traduction. Ces protéines ont un signal en position N-terminale qui dirige la suite de la traduction dans le réticulum endoplasmique. Des ribosomes associés à la membrane du réticulum endoplasmique sont utilisés lors de la synthèse de ces protéines (Lewin, 1999).

22
La traduction n’est pas toujours l’étape finale en vue de l’obtention de la protéine active.
Certaines modifications peuvent se dérouler après cette traduction. Nous parlons alors de
modifications post-traductionnelles. Dans le chapitre suivant nous discuterons d’une
modification post-traductionnelle particulière impliquant la glycosylation.

CHAPITRE II
MODIFICATIONS POST-TRADUCTIONNELLES ET ANCRE GPI Ce chapitre décrit en détails la structure et les propriétés connues d’une modification post-traductionnelle : l’ancre GPI. Cette description est d’une importance fondamentale pour le développement de logiciels de classification.
2.1 Modification post-traductionnelle
Les quelques 30 000 gènes qui composent un organisme tel que l’humain ne sont rien en
comparaison avec les 100 000 ou 200 000 protéines qu’ils encodent (Wright et Semmes,
2003). Toutefois, ces nombres sont modestes si l’on considère le nombre des différentes
modifications et interactions que ces protéines peuvent subir. On calcule que plus d’un
million de protéines différentes pourraient agir dans un organisme tel que l’humain (Wright
et Semmes, 2003).
Un type de modification rencontré est la modification post-traductionnelle. Une grande
variété de ces modifications existe. Notons la phosphorylation, l’acétylation et la
glycolysation. La phosphorylation est une modification très importante car elle joue un rôle
important dans la régulation de la fonction de plusieurs protéines. La phosphorylation est une
façon rapide de réguler une protéine. Plusieurs enzymes sont activées ou désactivées par
l’addition d’un groupe phosphate (PO4) à la protéine. La phosphorilation de l’acide aminé
sérine est la plus commune.

24
Les deux modifications post-traductionnelle les plus communes chez les eucaryotes se
produisent en position N-terminale de la protéine. Ces deux évènements sont dits co-
traductionnels car ils se déroulent après la traduction du N-terminal mais avant la terminaison
du processus. Ce sont le clivage de la méthionine de départ de la protéine naissante et
l’acétylation en N-terminale (Polevoda et Sherman, 2000).
Les modifications en position C-terminale sont moins diversifiées. Une catégorie intéressante
de modification C-terminale implique une glycosylation : l’addition d’une ancre
glycosylphosphatidylinositol (GPI). Les sections suivantes présentent les notions de base
pour la compréhension du processus de modification post-traductionnelle. La section 2.2
décrit l’addition d’une ancre GPI, et la structure particulière des protéines à ancre GPI, en
donnant les principaux éléments qui permettront de les reconnaître ou de les prédire.
2.2 L’ancre glycosylphosphatidylinositol (GPI)
La glycosylation est une modification post-traductionnelle des plus communes (Nalivaeva et
Turner, 2001) et des plus complexe (Spiro, 2002). Ce processus implique l’ajout d’un radical
glycosyl à la molécule. La figure 2.1 montre l’ajout d’un oligosaccharide complexe lié à une
asparagine n. On retrouve principalement trois catégories de glycosylation : Les N-
glycosylations, les O-glycosylations et l’attachement d’un glycolipide (GPI) à la partie C-
terminale d’une protéine. La glycosylation a une grande importance car elle peut affecter la
stabilité, la conformation et la solubilité de la protéine (Nalivaeva et Turner, 2001).
Une ancre GPI est un type d’attachement membranaire assez récemment découvert. Sa
présence dans les cellules eucaryotes fut rendue évidente dans les années 80, grâce aux
travaux de quelques chercheurs : Futerman et al., 1985 ; Roberts et Rosenberry, 1985 ; Tse et
al., 1985 ; Ferguson, Homans et Cross, 1985 ; Ferguson et al., 1988.
Les protéines ont diverses façons de s’attacher à la membrane. Dans la figure 2.2, trois
différents types d’attachement sont illustrés : l’attachement par ancre GPI, l’attachement
transmembranaire et l’attachement par ancre lipidique. L’attachement par ancre GPI est

25
extracellulaire, dans le cas des attachements transmembranaires, les protéines sont intégrées à
la membrane et, enfin, les ancres lipidiques attachent la protéine à la membrane du côté
intracellulaire.
nglucose mannoseN-acetylglucosamine
Figure 2. 1 Glycosylation. Ajout d’un oligosaccharide complexe lié à une asparagine n.
membranedouble couche lipidique
transmembranaire
ancre GPI
ancre lipidique
espace intracellulaire
espace extracellulaire
Figure 2. 2 Différentes associations protéine/membrane. L’ancre GPI, l’attachement trans-membranaire et l’ancre lipidique.
Parmi les protéines à ancre GPI on retrouve des enzymes, des molécules d’adhésion, des
récepteurs, des antigènes d’activation, etc (Chatterjee et Mayor, 2001 ; Hooper, 2001).
Toutefois, la seule caractéristique commune reliant ces protéines est la présence de cette
ancre (Chatterjee et Mayor, 2001). La fonction exacte de cet attachement n’est pas bien
connue (Ikezawa, 2002). Néanmoins, sa conservation parmi une grande variété taxonomique
(levures, protozoaires, plantes, vertébrés et même des archéobactéries) suggère une

26
fonctionnalité importante (Low, 1999). Quelques suggestions sur la fonction de l’ancre GPI
ont été proposées. Comme ces protéines sont attachées à la membrane plasmique, elles sont
probablement une alternative à l’attachement transmembranaire. On pense aussi qu’elles
pourraient jouer un rôle dans le triage (sorting) intracellulaire, c’est-à-dire que l’ancrage
serait un signal indiquant à la cellule la position extracellulaire de la protéine (Nosjean,
Briolay et Roux, 1997), et dans la signalisation transmembranaire (Chatterjee et Mayor,
2001). Il semblerait que l’ancre GPI présent chez la protéine prion serait en cause lors de
conformation déficiente de la protéine (Lehmann et Harris, 1995). Elles seraient aussi de
bonnes cibles pharmaceutiques. Par exemple le protozoaire Plasmodium, responsable de la
malaria, se compose de plusieurs protéines ayant une ancre GPI. Certaines de ces protéines
sont directement impliquées dans l’apparition des symptômes sévères tel que les fortes
fièvres. Ces protéines sont, de la sorte, de bonnes cibles pour l’élaboration de drogues anti-
malaria (Gowda et Davidson, 1999). Les protéines à ancre GPI ont finalement une
particularité très intéressante, à savoir qu’elles sont uniquement extracellulaires. Cette
modification post-traductionnelle donne un gros avantage, lors de l’annotation de nouvelles
séquences, en précisant leur localisation cellulaire.
2.2.1 Biosynthèse
La figure 2.3 résume la biosynthèse d’une protéine à ancre GPI. Une protéine destinée à
recevoir une ancre GPI sera dirigée vers le réticulum endoplasmique lors de sa traduction,
grâce à un signal en position N-terminale. Après avoir traversé la membrane du réticulum
endoplasmique, le signal peptide sera clivé par une peptidase. Par la suite, le signal en
position C-terminale s’attachera à l’intérieur du réticulum endoplasmique pour attendre
l’addition du GPI. Ce processus d’addition implique, entre autres, un complexe transamidase
et l’ancre GPI. Ce signal est nécessaire pour l’interaction avec le complexe transamidase
(Eisenhaber B. et al., 2003).

27
espace extracellulaire
réticulum endoplasmique
golgi
espace intracellulaire
ribosome
signal N-terminal
peptidase transamidase
ancre GPI
protéine à ancrage GPI
vésicule de transfert
vésicule sécrétrice
attachement à la membrane
ARNm
Figure 2. 3 Biosynthèse d’une protéine à ancre GPI.
Cette ancre GPI a préalablement été construite du côté cytoplasmique du réticulum
endoplasmique et est transportée à l’intérieur du réticulum endoplasmique (McConville et
Menon, 2000). Par la suite, le signal C-terminal sera clivé par la transamidase pour permettre

28
l’attachement de l’ancre GPI à la nouvelle queue C-terminale (Ikezawa, 2002). Une fois cette
addition terminée, la protéine voyage par la voie de sécrétion, en passant par le golgi (où elle
subira une maturation de l’ancre), pour être finalement transportée vers son site d’ancrage.
2.2.2 Structure d’une protéine à ancre GPI
La protéine à ancre GPI est caractérisée par deux signaux de séquences (fig. 2.4a).
Premièrement, on retrouve un signal pour la translocation dans le réticulum endoplasmique
en position N-terminale. Le deuxième signal permet la reconnaissance du complexe
transamidase lors du processus d’addition de l’ancre GPI. Le signal en position N-terminale
sera clivé lors du passage dans le réticulum endoplasmique. Pour ce qui est du signal
d’addition de l’ancre GPI, il peut être divisé en quatre éléments importants (fig. 2.4b, 2.4c)
( w représente le site d’ancrage) (Eisenhaber, Bork et Eisenhaber, 1998) :
1. Une région de liaison polaire et flexible d’environ 11 acides aminés ( 11−w … 1−w ) sans structure secondaire intrinsèque ;
2. Une région de résidus de faible poids moléculaire comprenant le site
d’ancrage w : ( w … 2+w ) ;
3. Une région intermédiaire ( 3+w … 9+w ) comportant des résidus modérément polaires ;
4. Une queue commençant avec le résidu 9+w ou 10+w , jusqu'à la fin et ayant
une hydrophobicité suffisante.
L’importance de ces éléments a été démontrée dans plusieurs études. Par exemple, la nature
de l’acide aminé au site w d’ancrage est importante. Des petits acides aminés comme la
sérine, l’asparagine, l’alanine, la glycine et la cystéine sont plus efficaces que les autres
résidus lorsque retrouvés à la position du site d’attachement, probablement dû au fait que
parce que la poche de fixation de la transamidase a une largeur spécifique pour accommoder
ces petits résidus, faisant d’eux un meilleur substrat (Eisenhaber, Bork et Eisenhaber, 1998 ;
Moran et Caras, 1994; Micanovic et al., 1990).

29
} }signal en N-terminale signal en C-terminale
protéine à ancrage GPI
cliva
ge
cliva
ge
a
b
} } }zone intermédiaire queue hydrophobe
signal en C-terminale
clivage
petits aa
w-11.....w-1 w w+1w+2}polaire
w+3................w+9w+9...................................
> DAF_HUMAN 23 SGT amidated serine.ATRSTPVSRTTKHFHETTPNKGSGTTSGTTRLLSGHTCFTLTGLLGTLVTMGLLT
w...w+2 queue hydrophobezone intermédiaire
c
Figure 2. 4 Structure de la protéine à ancre GPI. a : Signaux présents dans la protéine à ancre GPI. b : Structure du signal en C-terminale. c : Structure du peptide signal dans la séquence DAF_HUMAN. Le site d’ancrage est représenté en rouge et la queue hydrophobe en bleue.
Le rôle de la queue hydrophobe est la rétention de la protéine sur la membrane jusqu’à ce que
la modification GPI s’effectue. La longueur minimale de cette zone hydrophobe dépend de la
nature des acides aminés qui la composent. Ikezawa (2002) rapporte que pour la protéine de
foie de bovin 5-nucleotidase, une longueur de 13 résidus hydrophobes semble suffisante.
La région intermédiaire a aussi quelques particularités. Des études suggèrent une zone
hydrophile de 9 à 12 acides aminés (Moran et al., 1991 et Coyen, Crisci et Lublin, 1993).
Toutefois, beaucoup de protéines s’avèrent avoir une région intermédiaire de moins de 9
résidus (Furukawa, Tsukamoto et Ikezawa, 1997). Selon Ikezawa (2002), cette région
intermédiaire, conjointement avec les résidus 1+w et 2+w , jouerait un rôle dans
l’interaction entre le résidu d’attachement et le complexe transamidase GPI. La zone du site
30
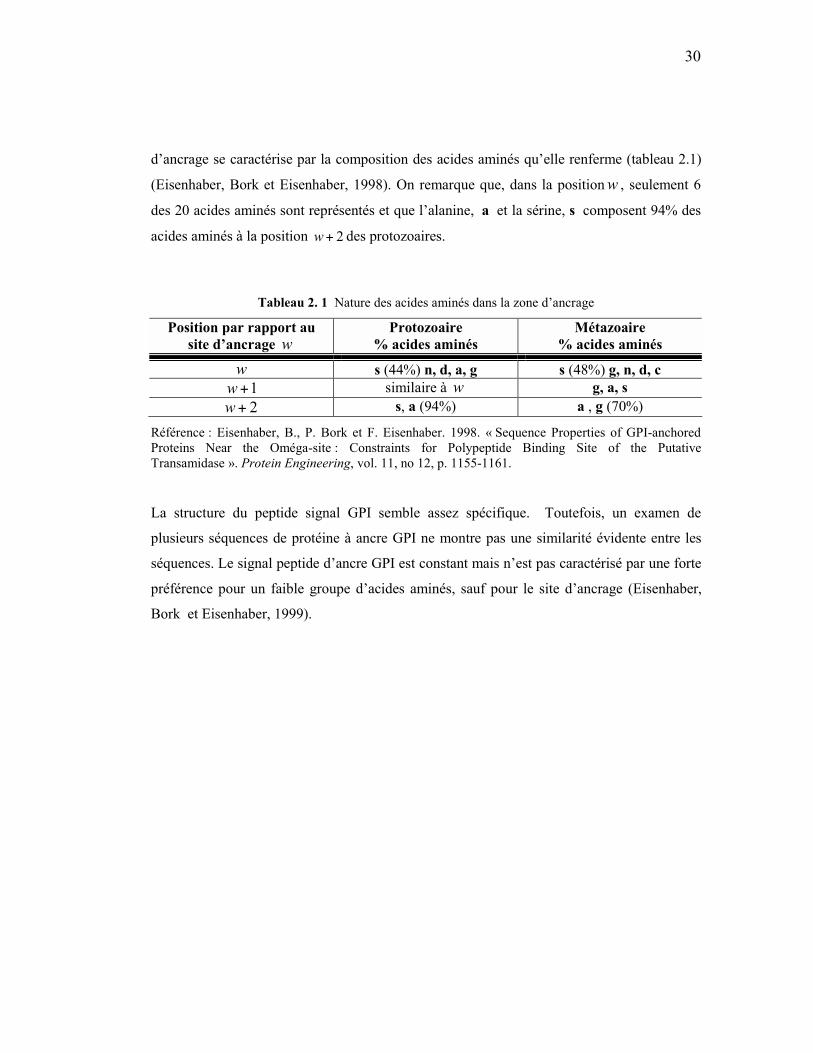
d’ancrage se caractérise par la composition des acides aminés qu’elle renferme (tableau 2.1)
(Eisenhaber, Bork et Eisenhaber, 1998). On remarque que, dans la position w , seulement 6
des 20 acides aminés sont représentés et que l’alanine, a et la sérine, s composent 94% des
acides aminés à la position 2+w des protozoaires.
Tableau 2. 1 Nature des acides aminés dans la zone d’ancrage
Position par rapport au site d’ancrage w
Protozoaire % acides aminés
Métazoaire % acides aminés
w s (44%) n, d, a, g s (48%) g, n, d, c 1+w similaire à w g, a, s 2+w s, a (94%) a , g (70%)
Référence : Eisenhaber, B., P. Bork et F. Eisenhaber. 1998. « Sequence Properties of GPI-anchored Proteins Near the Oméga-site : Constraints for Polypeptide Binding Site of the Putative Transamidase ». Protein Engineering, vol. 11, no 12, p. 1155-1161.
La structure du peptide signal GPI semble assez spécifique. Toutefois, un examen de
plusieurs séquences de protéine à ancre GPI ne montre pas une similarité évidente entre les
séquences. Le signal peptide d’ancre GPI est constant mais n’est pas caractérisé par une forte
préférence pour un faible groupe d’acides aminés, sauf pour le site d’ancrage (Eisenhaber,
Bork et Eisenhaber, 1999).

CHAPITRE III
CLASSIFICATION PAR ALIGNEMENT DE SÉQUENCES Dans ce chapitre, on introduit d’abord le problème général de la classification, puis on discute d’une technique de classification couramment utilisée en analyse de séquences biologiques : l’analyse de séquences par similarité d’alignement de paires et alignement multiple. On y verra que ces techniques traditionnelles d’analyse de séquences s’avèrent inefficaces dans le cas de recherche de certains motifs fonctionnels et de certaines caractéristiques particulières retrouvées chez des protéines ayant des fonctions différentes telles que les protéines à ancre GPI.
3.1 La classification
Classer des objets, des idées ou des concepts est une tâche que nous effectuons à tout moment
de la journée, sans même nous en rendre compte. Nos vies seraient difficiles si nous n’avions
pas cette capacité. Pour souligner l’importance de la classification, il suffit de rappeler qu’elle
est à la base de la plupart des activités intellectuelles nous caractérisant (Estes, 1994).
Lorsque nous classifions un nouvel objet, nous effectuons une généralisation d’après des
observations. Ces observations nous permettent d’extraire des caractéristiques communes à
une catégorie d’objets et, ainsi, de structurer la classe représentée. Cette capacité de
généralisation, nous l’avons dès l’enfance. Lorsque notre cerveau est mis en présence d’un
groupe d’objets divers, il tente premièrement de faire ressortir des caractéristiques communes
à ce groupe de données nouvelles. Par exemple, un individu, lorsque mis devant un groupe de
fleurs de différentes couleurs, tailles et parfums, cherchera à faire ressortir des

32
caractéristiques communes au groupe, telles que la présence de feuilles ou de pétales. Un tel
processus de généralisation est souvent imparfait, ce qui est vrai autant pour l’activité
humaine que pour la classification automatisée. La classe « fleur », par exemple, contient des
exemples et des contre-exemples qui remettent en question les clichés conventionnels. C’est
le cas de la monotrope uniflore, plante à fleur de la famille des bleuets, qui peut être
facilement confondue avec un champignon vu l’absence de chlorophylle, ou le cas des
bougainvilliers qui s’ornent de bractées colorées ayant l’apparence des pétales de fleurs.
Il existe plusieurs types de classification selon les domaines scientifiques. En taxonomie, par
exemple, on divise la classification en deux catégories : la classification cladistique et la
classification classique. En mode cladistique, les organismes sont classés selon l’état d’un
caractère (primitif ou évolué), sous forme d’arbre phylogénétique. En classification classique,
le degré de divergence entre les lignées sera utilisé pour faire la classification. En
mathématiques, par contre, on aura une catégorisation différente de la classification. Selon
Gordon (1981), on parle de méthode par partitions, de méthode hiérarchique, de méthode de
groupement ou de méthode géométrique (fig. 3.1). Dans la méthode par partitions, un objet
appartient à un seul groupe (fig. 3.1a). Dans le cas de la méthode hiérarchique, on rencontre
différents niveaux d’appartenance pour un même objet (fig. 3.1b). Pour le groupement, les
groupes peuvent se chevaucher permettant à un objet d’appartenir à plus d’un groupe (fig.
3.1c). Finalement, dans la méthode géométrique, la représentation d’un objet est
multifonctionnelle. Dans ce cas, des objets similaires au sein d’un groupe seront plus près
l’un de l’autre. Cette méthode incorpore plus d’information quant à la similarité entre les
objets. Par exemple, en ajoutant une dimension, on peu utiliser des particularités comme la
grosseur des points pour rendre la classification plus informative (fig. 3.1d).

33
Méthode par partitionsa
b
c
Méthode hiérarchique d
Méthode par groupement
Méthode géométrique
Figure 3. 1 Classification (Gordon, 1981).
Malgré les différentes appellations, descriptions et catégorisations de la classification, le but
demeure assez constant dans tous les domaines : la simplification des données (Gordon,
1981; James, 1985; Schalkoff, 1992). Dans la simplification, on recherche des motifs ou des
caractéristiques particulières qui agissent comme un filtre épurant les données en les
catégorisant, rendant ainsi la recherche et l’organisation des données plus facile.
Classification, catégorisation, groupement ou « clustering » sont tous des termes utilisés pour
parler de la même action. Certains auteurs, tels que Gordon (1981), parlent du « clustering »
comme d’un type de classification, tandis que d’autres, tels que James (1985), insistent sur le
fait que le « clustering » n’est pas de la classification, puisque la classification implique
l’affectation d’un objet à une classe et non la formation d’une classe.
Dans le problème qui nous intéresse, nous devons nous pencher sur les deux problèmes :
décider si oui ou non une séquence de protéine appartient à la classe « ancre GPI », tout en
tentant de mieux comprendre la structure de cette classe. L’exemple suivant permet de mieux
saisir la complexité de la tâche : les trois séquences de la figure 3.2, NTRI_MOUSE,
OPCM_BOVIN et LAMP_HUMAN, sont toutes annotées comme séquences « ancre GPI ».
Les colonnes en gris ombré indiquent leurs caractéristiques communes, dans ce cas des
acides aminés conservés qui ressortent clairement.

34
NTRI_MOUSE -ygnytcvasnklghtnasimlfgpgavsevnngtsrragciwllpllvlhlllkfOPCM_BOVIN -ygnytcvatnklgitnasitlygpgavidgvnsasralaclwlsgtlfahffikfLAMP_HUMAN hygnytcvaanklgvtnaslvlfrpgsvrgi-ngsislavplwllaasllcllskc
Figure 3. 2 Alignement entre trois séquences d’ancre GPI différentes (partie C-terminale) : NTRI_MOUSE, OPCM_BOVIN et LAMP_HUMAN.
Dans la section 3.2 nous verrons quelles sont les techniques classiques pour identifier de
telles caractéristiques communes dans des séquences biologiques. Hélas, la classe « ancre
GPI » échappe à une classification aussi simple. En ajoutant une quatrième séquence annotée
« ancre GPI » aux trois de la figure 3.2 on bouleverse complètement les notions qui se
dégageaient de l’observation des trois premiers exemples (voir fig. 3.3).
NTRI_MOUSE -ygnytcvasnklghtnasimlfgpgavsevnngtsrragciwllpllvlhlllkfOPCM_BOVIN -ygnytcvatnklgitnasitlygpgavidgvnsasralaclwlsgtlfahffikfLAMP_HUMAN hygnytcvaanklgvtnaslvlfrpgsvrgi-ngsislavplwllaasllcllskcPRIO_HUMAN -tetdvkmmervveqmcitqyeresqayyqrgssmvlfssppvillisfliflivg
Figure 3. 3 Alignement entre quatre différentes séquences d’ancre GPI (partie C-terminale): NTRI_MOUSE, OPCM_BOVIN, LAMP_HUMAN et PRIO_HUMAN.
Une analyse plus précise du signal GPI confirme également que la structure connue du signal
n’est pas constante. Comme le montre la figure 3.4, la structure du signal GPI de trois
protéines diffère grandement : une protéine humaine (DAF_HUMAN), une protéine de singe
de nuit (PRIO_AOTTR) et une protéine de raie électrique (5NTD_DISOM). Ces trois
exemples montrent bien la diversité du signal et surtout la flexibilité de la structure du signal.
La protéine 5NTD_DISOM ne montre aucune zone intermédiaire (la zone hydrophobe, en
bleu, chevauche la zone du site d’ancrage, en rouge). Cette séquence serait rejetée, si on
respectait la structure connue des GPI. La séquence PRIO_AOTTR a, elle aussi, une zone
intermédiaire trop petite. La protéine DAF_HUMAN, elle, respecte assez bien la structure
connue du signal, avec une zone intermédiaire hydrophile de 12 acides aminés et une queue
hydrophobe de 17 acides aminés. À la vue de ces trois exemples, il est évident que la
classification des protéines à ancre GPI basé sur l’analyse de leur séquence sera difficile.

35
> 5NTD_DISOM 30 SAT amidated serine
TDISVVSSYIKQMKVVYPAVEGRILFVENSATLPIINLKIGLSLFAFLTWFLHCS
> PRIO_AOTTR 38 SSM amidated serine
TKGENFTETDVKIMERVVEQMCITQYEKESQAYYQRGSSMVLFSSPPVILLISFL
> DAF_HUMAN 23 SGT amidated serine.
ATRSTPVSRTTKHFHETTPNKGSGTTSGTTRLLSGHTCFTLTGLLGTLVTMGLLT
Figure 3. 4 Diversité du signal GPI. Les caractères en bleu représentent la queue hydrophobe. Les caractères en rouge sont la zone d’ancrage.
Le reste de ce chapitre est consacré à la description de techniques de classification basées sur
la détection de motifs communs aux séquences biologiques : l’alignements de séquences.
3.2 La classification par similarité de séquences
En analyse de séquences biologiques, la tâche la plus souvent effectuée est sans contredit la
recherche de similarité entre deux ou plusieurs séquences (Tompa, 2000; May, 2001). Cette
similarité se calcule en comparant les caractères composant deux séquences. L’analyse de
similarité de séquences nous permet de tirer avantage des nombreuses bases de données de
séquences, grâce à des algorithmes permettant de trouver efficacement toutes les séquences
similaires à une séquence donnée. La recherche dans les bases de données de séquences est
probablement l’expérience biologique la plus rapide, la plus puissante et la plus économique
(Krawetz et Womble, 2003).
Lors du séquençage d’un génome, on détermine la séquence d’acides nucléiques des gènes
composant un organisme. La première analyse effectuée sera d’identifier le gène séquencé et
de proposer une hypothèse quant à sa fonction (Tompa, 2000). Le moyen le plus utilisé pour
connaître la fonction d’un gène est la recherche de motifs par similarité. La nature est un
bricoleur, non un inventeur (Jacob, 1977) : les séquences biologiques ne sont pas des
créations, elles sont plutôt des adaptations de séquences ayant déjà existé (Durbin et al.,
1998). La comparaison de séquences inconnues avec des séquences ayant une fonction

36
connue nous permet donc d’inférer une fonction. Un outil important est la présence de motifs
dans les séquences. Un motif conservé au cours de l’évolution est, sans aucun doute,
important au point de vue fonctionnel. Il peut nous donner un indice sur la fonction de cette
séquence. Il peut aussi nous informer sur la relation entretenue entre deux séquences au cours
de l’évolution (Brejova et al., 2000).
3.2.1 Alignement de deux séquences
Lorsque l’on calcule la similarité entre deux séquences, on cherche à voir si deux séquences
se ressemblent suffisamment pour permettre d’inférer une homologie de séquences, donc une
évolution commune, et non simplement à identifier un fort pourcentage de caractères
communs (Baxevanis et Ouellette, 2001). Puisque la représentation des molécules se fait avec
un alphabet, le calcul de la similarité entre deux séquences biologiques peut être ramené à un
calcul de la similarité entre deux séquences de lettres. Un alignement entre deux séquences de
lettres consiste à superposer les deux séquences de manière à mettre en évidence leurs
éventuelles lettres communes aux mêmes positions. La figure 3.5 présente un exemple
d’alignement entre les mots « VOITURE » et « TOITURE ». Dans cette figure, nous avons
indiqué en gris ombré les 6 lettres communes aux deux mots.
V O I T U R E
O I T U R E
T O I T U R E
Figure 3. 5 Alignement entre les mots « voiture » et « toiture ».
On peut voir, dans cet exemple, que les deux séquences de lettres s’alignent presque
parfaitement. Si on regarde la similarité entre les caractères qui les composent, seulement la
première lettre diffère. Toutefois, la similarité dans l’orthographe de ces mots ne permet pas
d’inférer une similarité dans leur signification en langue française. Si on prend, par exemple,
un alignement entre les séquences de lettres « VOITURE » et « VÉHICULE », quelle serait
la valeur de similarité entre ces séquences (fig. 3.6) ? Notons que lorsque les mots sont de

37
longueurs différentes ou même identiques, on utilise le signe «-» pour fragmenter les mots de
manière à augmenter le nombre de caractères communs.
V O I T U R E
V I U E
V É H I C U L E
Figure 3. 6 Alignement entre les mots « voiture » et « véhicule ». Alignement où le signe « » représente un espace ajouté dans une séquence pour faciliter l’alignement des autres caractères.
Dans cet exemple, on retrouve seulement 4 lettres communes. Pourtant, si on s’attarde au
sens, les mots « VOITURE » et « VÉHICULE » ont un sens beaucoup plus proche que
« VOITURE » et « TOITURE ». Le calcul de la similarité par alignement de séquences de
lettres est, dans ces exemples, plausible du point de vue de l’orthographe mais n’a pas
d’intérêt du point de vue de la signification de ces mots.
Dans le cas des séquences biologiques, le calcul de similarité entre deux séquences par
l’alignement de leurs caractères est beaucoup plus justifiable car la structure primaire, la
séquence de lettre, d’une séquence biologique détermine généralement les structures
secondaire et tertiaire qui, elles, déterminent la fonction. Ainsi, une forte similarité dans
l’orthographe de ces séquences implique une similarité de fonction. Cependant, il ne faut pas
penser que l’analyse de séquences biologiques se limite à l’inférence de la structure, donc de
la fonction d’une séquence biologique, selon sa structure primaire et qu’ainsi, une recherche
de similarité entre séquences est la solution à tous les problèmes de classement de séquences
biologiques. Il existe des cas où une faible similarité entre les lettres composant une
séquence biologique ne reflète pas des structures différentes. Par exemple, des protéines
ayant un ancêtre commun peuvent avoir une structure tridimensionnelle similaire, malgré une
structure primaire différente (Lewin, 1999). Au cours de l’évolution, les structures primaires
des séquences peuvent avoir subi des substitutions entre acides aminés ayant des propriétés
communes (Brown, 2000). Dans le cas des protéines, par exemple, les 20 acides aminés qui
composent l’alphabet permettant la représentation textuelle des séquences protéiques peuvent
avoir une structure ou des propriétés physico-chimiques similaires. Cette similarité permet

38
une substitution entre deux acides aminés sans modification de la structure tertiaire, ni de la
fonction de la séquence. Par exemple, la valine et l’isoleucine sont deux acides aminés de
petite taille et hydrophobes. Le remplacement d’une valine par une isoleucine dans une
séquence n’aura pas le même effet que le remplacement d’une valine par une glycine qui,
elle, est beaucoup moins hydrophobe.
De plus, une séquence de protéine peut se replier sous diverses formes et cette structure
dépend d’interactions complexes (Alm et al., 2002). Comme la forme de la molécule
détermine sa fonction, il devient difficile de prédire la fonction d’une protéine uniquement
par l’analyse de sa séquence de lettres. Pour certains types d’analyses de séquences, telles
que pour la prédiction de structure ou pour la recherche de certains signaux ou motifs
complexes, l’alignement de séquences n’est pas toujours la solution. Néanmoins, la
comparaison de séquences par calcul de similarité est utile dans plusieurs cas, par exemple,
pour une analyse en vue de la classification d’une séquence inconnue, en la comparant aux
séquences connues présentes dans une base de données.
Lorsque l’on effectue un alignement de séquences, différentes considérations entrent en jeu :
1. On peut vouloir faire un alignement local ou global.
2. Il faut aussi choisir un système de pointage qui permettra de quantifier nos alignements.
3. Un algorithme fera, par la suite, la recherche de l’alignement optimal.
1. Global ou Local ?
Les premières méthodes d’alignement développées cherchaient à reconnaître la similarité
globale entre deux séquences (Baxevanis et Ouellette, 2001) (fig. 3.7a). Les alignements
globaux, dans le cas des protéines, sont utiles lorsque la protéine est composée d’un seul
domaine ou pour la construction d’arbres d’évolution (Brown, 2000). Cependant, les
protéines sont rarement composées d’une seule région conservée. Elles sont composées le
plus souvent de plusieurs domaines, de façon modulaire (Doolittle et Bork 1993). Il faut donc
permettre, entre les domaines, la présence de zones intermédiaires pouvant être composées

39
d’acides aminés sans aucune similarité entre eux. Un alignement local des séquences répond
alors à cette exigence (fig. 3.7b).
a
b
global
local
séquence 1
séquence 2
séquence 1
séquence 2
Figure 3. 7 Alignement local et global.
2. Un système de pointage
Lorsqu’on aligne deux séquences, il peut exister plusieurs façons de faire (fig. 3.8). Il faut
donc utiliser un système de pointage pour évaluer l’alignement optimal. Un exemple de
système de pointage très simple est de donner un score de +2 pour une lettre commune dans
une colonne et de -1 pour une substitution ou un espace. Le score total d’un alignement sera
alors donné par la somme des scores accordés à chaque colonne de l’alignement. Le fait de
considérer des espaces (insertion ou délétion d’un caractère) à l’intérieur de la séquence a une
signification biologique. Lorsque l’on compare deux séquences, on cherche des indices à
l’effet qu’elles ont un ancêtre commun, donc une similarité possible. Lorsqu’une séquence
protéique, par exemple, se modifie au cours de l’évolution, on verra des substitutions, des
insertions et des délétions apparaître (Durbin et al., 1998).

40
1) a c - g - l l i l y - - p
a c g l i y p
a c c g g l - i - y o o p
+2+2-1+2-1+2-1+2-1+2-1-1+2
score : 8
2) a c g l - l i l y p - -
a c l i y
a c c g g l i - y o o p
+2+2-1-1-1+2+2-1+2-1-1-1
score : 3
3) a c - g l l i l y - p
a c g l i p
a c c g g l i y o o p
+2+2-1+2-1+2+2-1-1-1+2
score : 7
Figure 3. 8 Trois alignements possibles entre deux séquences, avec des scores différents.
Lorsque l’on compare des protéines, il est possible d’augmenter la justesse des alignements
en pondérant de manières différentes les substitutions entre les acides aminés. En effet,
certaines substitutions sont beaucoup plus fréquemment observées que d’autres, et ce fait a
été utilisé pour la construction de matrices de substitution. La figure 3.9 montre un exemple
d’une telle matrice, la matrice BLOSUM62. Chaque entrée de cette matrice contient le score
associé à la substitution de l’acide aminé de la ligne correspondante, par l’acide aminé de la
colonne correspondante.
Par exemple, le score de la substitution de v par g est de -3, alors que celui de la substitution
de v par i est de +1. Cette manière de différencier les substitutions reflète, entre autres, la
similarité qui existe entre les acides aminés v et i. Notons aussi que les valeurs de la
diagonale ne sont pas tout égales : la conservation, par exemple, de l’acide aminé w est

41
récompensée par un score de +11, alors que la conservation de l’acide aminé i ne donne un
gain que de +4.
Comme on peut s’y attendre, la construction de ce type de matrice repose sur l’analyse d’un
grand nombre de séquences. Plusieurs matrices différentes ont été proposées dans les
dernières décennies, la première étant due à Dayhoff et al.. (1978). La matrice de la figure 3.9
fait partie d’une famille de matrices, les matrices BLOSUM, proposé par Henikoff et
Henikoff (1992). Ces matrices ont été calculées à partir de statistiques portant sur 2000
alignements de segments de séquences contenus dans la base de données BLOCKS (Henikoff
et al., 2002)
cstpagndeqhrkmilvfyw
cstpagndeqhrkmilvfyw
9-1 4-1 1 5-3 -1 -1 7 0 1 0 -1 4-3 0 -2 -2 0 6-3 1 0 -2 -2 0 6-3 0 -1 -1 -2 -1 1 6-4 0 -1 -1 -1 -2 0 2 5-3 0 -1 -1 -1 -2 0 0 2 5-3 -1 -2 -2 -2 -2 1 -1 0 0 8-3 -1 -1 -2 -1 -2 0 -2 0 1 0 5-3 0 -1 -1 -1 -2 0 -1 1 1 -1 2 5-1 -1 -1 -2 -1 -3 -2 -3 -2 0 -2 -1 -1 5-1 -2 -1 -3 -1 -4 -3 -3 -3 -3 -3 -3 -3 1 4-1 -2 -1 -3 -1 -4 -3 -4 -3 -2 -3 -2 -2 2 2 4-1 -2 0 -2 0 -3 -3 -3 -2 -2 -3 -3 -2 1 3 1 4-2 -2 -2 -4 -2 -3 -3 -3 -3 -3 -1 -3 -3 0 0 0 -1 6-2 -2 -2 -3 -2 -3 -2 -3 -2 -1 2 -2 -2 -1 -1 -1 -1 3 7-2 -3 -2 -4 -3 -2 -4 -4 -3 -2 -2 -3 -3 -1 -3 -2 -3 1 2 11
c s t p a g n d e q h r k m i l v f y w
c s t p a g n d e q h r k m i l v f y w Figure 3. 9 Matrice BLOSUM62
La figure 3.10 donne un exemple d’un alignement BLOCK. Les segments de séquences
choisis pour construire de tels alignements sont, en général, des segments très conservés au
cours de l’évolution, et ayant un fort pourcentage de similarité. La présence fréquente dans
une même colonne, de deux acides aminés différents est une indication de leur
interchangeabilité.

42
Les matrices BLOSUM30, BLOSUM62 et BLOSUM90 sont les principales utilisées dans la
comparaison de séquences protéiques. La valeur affectée au terme BLOSUM est le
pourcentage d’identité minimum des alignements retenus dans la base de données BLOCK
pour l’estimation des scores de substitution. La matrice BLOSUM62, obtenue à l’aide
d’alignements contenant un minimum de 62% de similarité, est la plus couramment utilisée.
Une matrice BLOSUM90 sera utilisée si l’on veut comparer des séquences fortement
similaires, et une matrice BLOSUM30, pour des séquences faiblement similaires (Wishart,
2003).
D C5METTRFRASE; BLOCK
Q59606 ( 17) KILSLFSGCGGLDLGFH 12Q59797 ( 17) KILSLFSGCGGLYLGFH 28MTBF_BACSU|P17044 ( 102) TFIDLFAGIGGIRLGFE 12MTS2_SHISO|P34879 ( 73) RMIDLFAGIGGTRLGFH 18O08431 ( 73) RMIDLFAGIGGTRLGFH 18P77950 ( 73) RMIDLFAGIGGTRLGFH 18O30868 ( 5) KTIDLFAGIGGIRLGFE 11MTB1_HERAU|P25262 ( 5) RFIDLFAGIGGFRLGLE 11MTE1_HERAU|P25266 ( 5) RFIDLFAGIGGFRLGLE 11MTC2_HERAU|P25264 ( 5) RFIDLFAGIGGFRLGLE 11MTSA_LACLC|P34877 ( 80) KMIDLFAGIGGTRLGFH 18Q59958 ( 2) RFIDLFSGIGGFRLGME 23MTBA_BACAR|P19888 ( 4) KFVDLFAGIGGIRIGFE 14MTC1_HERAU|P25263 ( 3) KFIDLFAGIGGMRLGFE 11MTB1_BREEP|P10283 ( 2) KVLSLFSGCGGMDLGLE 12MTF1_FUSNU|P34906 ( 2) KLLSLFSGAGGLDLGFE 12P94630 ( 6) TIVSTFSGCGGLDLGLQ 27(bloc partiel)
Figure 3. 10 Alignement BLOCK. Dans cet alignement on peut remarquer que dans la première colonne (en rouge) on retrouve fréquemment les acides aminés k et r, et beaucoup plus rarement t. Le score de substitution de k pour r sera plus élevé que celui de k pour t, par exemple.
En plus de l’utilisation des matrices de substitution, le calcul du score d’un alignement
dépend également du nombre et de la taille des espaces. Comme on peut le voir sur la figure
3.11, certains espaces ne couvrent qu’une position, alors que d’autres s’étendent sur plusieurs
positions consécutives. La pénalité associée à un espace est composée, la plupart du temps,
par la somme de deux valeurs :
kvop +=

43
Où o est une constante pour « l’ouverture » d’un espace, v une constante pour « l’extension »
d’un espace et k est le nombre de position consécutive de l’espace. Cette approche permet de
pénaliser moins lourdement les espaces longs, puisqu’ils peuvent résulter d’une seule
mutation, insertion ou délétion, affectant plusieurs acides aminés.
arcciyrrypllvllikmmaccqlllll
a-cciyrryplli---------qlllll
gap extension de gaps
a cciyrrypll* qlllll
séquence 1
séquence 2
Figure 3. 11 Alignement avec espace.Dans cet alignement, les espaces sont indiquées par des traits "-". On remarque deux espaces, l'un de longueur 1, et le second de longueur 9. Les substitutions sont notées par des étoiles "*", et les acides aminés conservés sont marqués en gris ombré. 3. Algorithmes d’alignement.
Trouver un alignement de score optimal n'est pas une tâche simple. En effet, le nombre
d'alignements différents entre deux séquences est exponentiel en fonction de la taille des
séquences, donc il est exclu de faire une recherche exhaustive parmi tous les alignements
possibles, compte tenu que les tailles des séquences biologiques sont de l'ordre de plusieurs
centaines de caractères. Notons aussi qu'il peut exister plusieurs alignements de score optimal
pour deux séquences données.
En 1970, Needleman et Wunsch ont proposé un premier algorithme efficace pour
l'alignement global de deux séquences. Cet algorithme est basé sur la notion de
programmation dynamique qui permet de calculer tous les alignements de score optimal entre
deux séquences, ainsi que la valeur du score optimal. La programmation dynamique permet
d'obtenir les alignements optimaux de deux séquences à partir des alignements optimaux des
préfixes de ces séquences. Si les deux séquences sont de taille m et n respectivement,
l'algorithme de Needleman-Wunsch a une complexité de ( )mnO . En 1981, Smith et
Waterman ont proposé une variante de l'algorithme de Needleman-Wunsch qui permet

44
l'alignement local de séquences. Cet algorithme est également de complexité quadratique en
fonction de la longueur des séquences. Le lecteur intéressé peut trouver une présentation
détaillée de ces deux algorithmes dans Gusfiled (1997), ou dans Setubal et Meidanis (1997).
La figure 3.12 illustre le calcul d'un alignement de score optimal entre les séquences
TCGCA et TCCA, lorsque la pénalité associée à une substitution est de -1, celle associée à
un espace est de -2, et le score associé à un nucléotide conservé est de +1.
T
T C G C A
C
C
A
1
1
2
3
4
2 3 4 5
0
0
0 -2 -4 -8 -10
-2
-4
-6
-8
-6
s[i]
t[i]
1 -1 -3 -5 -7
-1 2 0 -2 -4
-3 0 1 1 -1
-5 -2 -1 0 2 Figure 3. 12 Calcul d'un alignement optimal entre les séquences TCGCA et TCCA.
Dans la figure 3.12, le calcul est effectué au moyen d'une matrice dont l'entrée ( )jia ,
contient le score optimal de l'alignement du préfixe de longueur i de la séquence TCCA, et
du préfixe de longueur j de la séquence TCGCA. L’entrée ( )jia , est calculée grâce à la
formule:
[ ][ ][ ][ ]!
"
!#
$
−−
+−−
−−
=
2,1),(1,1
21,max,
jiajipjia
jiajia ),( jip = 1, si [ ] [ ]jtis = et -1, sinon.
On relie ensuite l'entrée ( )jia , avec un pointeur vers l'entrée (ou les entrées) qui a produit la
valeur maximale dans la formule précédente.

45
Le score d'un alignement optimal est obtenu dans l'entrée en bas à droite de la matrice, et un
alignement optimal particulier peut être obtenu en « remontant » les pointeurs: un pointeur
diagonal signifiant une identité ou une substitution, et un pointeur horizontal ou vertical,
l'insertion d'un espace.
Dans l'exemple illustré à la figure 3.12, l'alignement obtenu est: T C G C A
T C – C A
dont le score est de 2.
Étant donné l'importance du problème d'alignement dans les applications biologiques, de
nombreuses améliorations ont été apportées a ces algorithmes pour tenter d'en réduire la
complexité temporelle et spatiale. Tout un domaine de recherche s'est développé autour de ce
problème, et même des livres entiers y sont consacrés; voir par exemple Navarro et Raffinot
(2002).
L'algorithme approximatif le plus couramment utilisé est sans contredit BLAST (Basic Local
Alignment Search Tool) (Atlschul et al., 1997). Cet algorithme permet d'obtenir
simultanément tous les alignements significatifs d'une séquence comparée à un ensemble de
séquences.
3.2.2 Classification de protéines à ancre GPI et BLAST
Pour identifier des protéines à ancre GPI, une première approche est d'identifier des
protéines ayant une forte similarité avec une protéine connue ayant une ancre GPI.
Typiquement, on propose à BLAST une séquence requête, ainsi qu'une base de donnée dans
laquelle rechercher des séquences similaires.

46
Par exemple, la séquence requête 5NTD_HUMAN de 574 acides aminés suivante :
>sw|P21589|5NTD_HUMAN 5’-nucleotidase precursor (EC 3.1.3.5) (Ecto-5’-nucleotidase) (5’-NT) (CD73 antigen).MCPRAARAPATLLLALGAVLWPAAGAWELTILHTNDVHSRLEQTSEDSSKCVNASRCMGGVARLFTKVQQIRRAEPNVLLLDAGDQYQGTIWFTVYKGAEVAHFMNALRYDAMALGNHEFDNGVEGLIEPLLKEAKFPILSANIKAKGPLASQISGLYLPYKVLPVGDEVVGIVGYTSKETPFLSNPGTNLVFEDEITALQPEVDKLKTLNVNKIIALGHSGFEMDKLIAQKVRGVDVVVGGHSNTFLYTGNPPSKEVPAGKYPFIVTSDDGRKVPVVQAYAFGKYLGYLKIEFDERGNVISSHGNPILLNSSIPEDPSIKADINKWRIKLDNYSTQELGKTIVYLDGSSQSCRFRECNMGNLICDAMINNNLRHTDEMFWNHVSMCILNGGGIRSPIDERNNGTITWENLAAVLPFGGTFDLVQLKGSTLKKAFEHSVHRYGQSTGEFLQVGGIHVVYDLSRKPGDRVVKLDVLCTKCRVPSYDPLKMDEVYKVILPNFLANGGDGFQMIKDELLRHDSGDQDINVVSTYISKMKVIYPAVEGRIKFSTGSHCHGSFSLIFLSLWAVIFVLYQ
a été soumise au programme BLAST du NCBI, et la base de donnée identifiée est l'ensemble
de toutes les séquences protéiques disponibles au NCBI. Notons ici qu'en date du
12 septembre 2004, le nombre de séquences disponibles atteignait l'impressionnant total de
1 144 638 séquences de protéines.
En sortie, BLAST propose un ensemble de séquences ayant une similarité statistiquement
significative avec la séquence requête. Lorsque BLAST propose un alignement avec un score
de similarité S, il donne aussi le nombre d'alignements que l'on pourrait obtenir par hasard
avec un score d'au moins S. Ce nombre est appelé e-value . Plus la e-value est petite, plus
l'alignement est significatif, une e-value supérieure à 1 signifie qu'au moins un alignement de
score S ou plus aurait pu être trouvé par hasard dans la base de donnée.
La figure 3.13 donne les 15 premières séquences trouvées par BLAST avec la séquence
requête 5NTD_HUMAN. Par exemple, la seconde séquence trouvée (NP_776554, en vert)
obtient un score de 1018 et une e-value de 0.0, c'est à dire la meilleure e-value possible.
L'alignement proposé de notre requête avec la séquence NP_776554 est un alignement local
qui s'étend de la position 28 à la position 574 de notre requête. Dans cas de NP_776554, il
s'agit bien d'une protéine à ancre GPI. Il s’agit de la protéine 5NTD_BOVIN.

47
Par contre, la neuvième séquence trouvée (NP104218.1, en rouge) obtient un score de 343 et
une e-value de 3e-93. Cette valeur correspond à 9310/3 et n'est pas loin de zero, ce qui
constitue une excellente e-value. BLAST propose un alignement de la position 29 à la
position 546 entre la séquence requête et NP104218.1. Malheureusement, cette séquence n'est
pas une protéine à ancre GPI car elle provient de la bactérie Mesorhizobium loti, un
procaryote, et les ancres GPI ne se retrouve que chez les eucaryotes et quelques
archéobactéries. Il est intéressant de noter que d’autres séquences ayant une ancre GPI, telles
que la protéine prion, n’apparaissent pas dans les séquences similaires. On voit donc que les
alignements, même s'ils permettent d'identifier certaines protéines ancre GPI, ne peuvent
servir à l'identification à grande échelle de protéines à ancre GPI.
e-valuecandidatssélectionnésdans la base de données
détail del'alignement
de la séquence requête avec un candidat
gi|4505467|ref|NP_002517.1| 5’ nucleotidase, ecto; Purine 5... 1110 0.0 gi|27806507|ref|NP_776554.1| 5’-nucleotidase, ecto (CD73) [... 1018 0.0 gi|11024643|ref|NP_067587.1| 5 nucleotidase; 5 nucleotidase... 1003 0.0 gi|6754900|ref|NP_035981.1| 5’ nucleotidase, ecto; ecto-5’-... 996 0.0 gi|41055552|ref|NP_957226.1| 5’ nucleotidase ecto; zgc:6378... 744 0.0 gi|24654424|ref|NP_725681.1| CG30104-PB [Drosophila melanog... 397 e-109 gi|19922444|ref|NP_611217.1| CG4827-PA [Drosophila melanoga... 386 e-106 gi|28573524|ref|NP_725682.2| CG30103-PA [Drosophila melanog... 366 e-100 gi|13472651|ref|NP_104218.1| 5’-nucleotidase (EC 3.1.3.5) [... 343 3e-93 gi|15966555|ref|NP_386908.1| PROBABLE 5’-NUCLEOTIDASE PRECU... 334 2e-90 gi|46199266|ref|YP_004933.1| 5’-nucleotidase [Thermus therm... 328 7e-89 gi|17937475|ref|NP_534264.1| 5’-nucleotidase [Agrobacterium... 326 5e-88
>gi|27806507|ref|NP_776554.1| 5’-nucleotidase, ecto (CD73) [Bos taurus] Length = 574
Score = 1018 bits (2631), Expect = 0.0Identities = 493/547 (90%), Positives = 527/547 (96%)
Query: 28 ELTILHTNDVHSRLEQTSEDSSKCVNASRCMGGVARLFTKVQQIRRAEPNVLLLDAGDQY 87 ELTILHTNDVHSRLEQTSEDSSKCVNASRC+GGVARL TKV QIRRAEP+VLLLDAGDQYSbjct: 28 ELTILHTNDVHSRLEQTSEDSSKCVNASRCVGGVARLATKVHQIRRAEPHVLLLDAGDQY 87...Query: 568 VIFVLYQ 574 VI +LYQSbjct: 568 VIIILYQ 574
>gi|13472651|ref|NP_104218.1| 5’-nucleotidase (EC 3.1.3.5) [Mesorhizobium loti MAFF303099] Length = 706
Score = 343 bits (880), Expect = 3e-93Identities = 202/526 (38%), Positives = 300/526 (57%), Gaps = 29/526 (5%)
Query: 29 LTILHTNDVHSRLEQTSEDSSKCV----NASRCMGGVARLFTKVQQIRRA--EPNVLLLD 82 L ILH ND HSR+E ++ S C C+GG RL T + Q R+ NVLLL+Sbjct: 28 LNILHFNDWHSRIEGNNKYESTCSADEETKGECIGGAGRLITAIAQERKKLEGQNVLLLN 87...Query: 502 ANGGDGFQMIKDELLR-HDSGDQDINVVSTYISKMKVIYPAVEGRI 546 GGDG+++ + +D G VV+ Y+ + P ++GRISbjct: 487 RQGGDGYKVFAERAKNAYDYGPGLEQVVADYLGAHRPYTPKLDGRI 532
Figure 3. 13 Sortie du programme BLAST effectuée avec l’algorithme blastp.

48
3.2.3 Classification de protéines à ancre GPI et alignements multiples
Théoriquement, il est possible de définir la notion d'alignement simultané de plusieurs
séquences, appelés alignements multiples, et de leur associer un score. Un alignement
multiple permet de mettre en évidence des régions ayant une importance fonctionnelle
(Baxevanis et Ouellette 2001), ainsi que de construire des arbres phylogénétiques.
Le problème de trouver un alignement multiple de score optimal n'est toutefois pas, encore,
résolu de manière efficace. Par exemple, l'alignement de huit séquences de longueur
moyenne, c'est à dire quelques centaines de caractères dans les applications biologiques, est
un problème pratiquement irréalisable avec les algorithmes disponibles (Thompson, Higgins
et Gibson, 1994). On se tourne donc systématiquement vers des heuristiques, qui ne
garantiront pas de trouver un alignement optimal. CLUSTALW (Higgins et Sharp, 1988 ;
Thompson, Higgins et Gibson, 1994) est l'un des algorithmes les plus utilisé. Il est basé sur
les constructions suivantes:
1. On aligne d'abord, de manière optimale, toutes les paires possibles de séquences à l'aide d'un algorithme conventionnel d'alignement. Les scores obtenus permettent de construire une matrice de distance entre les paires de séquences.
2. À partir de la matrice de distances, on construit un arbre qui permettra de « guider » l'alignement multiple. Cet arbre est calculé par des méthodes de type « neigbour-joining » (Saitou et Nei, 1987), qui regroupent progressivement les séquences qui sont les plus rapprochées.
3. Les séquences sont ensuite alignées progressivement selon l'ordre d'embranchement de l'arbre guide.
La figure 3.14 montre le processus d'alignement de 8 séquences de protéines à ancre GPI. Le
résultat de cet alignement (fig. 3.14 b) montre, une fois de plus, qu'il est bien difficile de
construire et structurer une classe pour les protéines à ancre GPI à partir de leur similarité.
Le groupe de six séquences de type 5NTD est bien structuré, comme le montre l'arbre
phylogénétique associé à l'alignement (fig. 3.14c). D'autre part, autant dans l'alignement

49
multiple que dans l'arbre, les deux séquences PRIO BOVIN et PPBT RAT se détachent du
groupe formé par les séquences 5NTD.
>5NTD_BOOMI VMKYMNSTSPITTALDGRVTFLKTNQASDACLNLASPFLVLLVLVVFYHL>5NTD_BOVIN INVVSGYISKMKVLYPAVEGRIQFSAGSHCCGSFSLIFLSVLAVIIILYQ>5NTD_DISOM VSSYIKQMKVVYPAVEGRILFVENSATLPIINLKIGLSLFAFLTWFLHCS>5NTD_HUMAN INVVSTYISKMKVIYPAVEGRIKFSTGSHCHGSFSLIFLSLWAVIFVLYQ>5NTD_MOUSE ISVVSEYISKMKVVYPAVEGRIKFSAASHYQGSFPLVILSFWAMILILYQ>5NTD_RAT ISVVSEYISKMKVIYPAVEGRIKFSAASHYQGSFPLIILSFWAVILVLYQ>Prio_BOVIN KMMERVVEQMCITQYQRESQAYYQRGASVILFSSPPVILLISFLIFLIVG>PPBT_RAT HEQNYIPHVMAYASCIGANLDHCAWASSASSPSPGALLLPLALFPLRTLF
CLUSTAL W (1.83) multiple sequence alignmentscore 1521
5NTD_BOVIN INVVSGYISKMKVLYPAVEG---RIQFSAGSHCCG-SFSLIF-LSVLAVIIILYQ--5NTD_HUMAN INVVSTYISKMKVIYPAVEG---RIKFSTGSHCHG-SFSLIF-LSLWAVIFVLYQ--5NTD_MOUSE ISVVSEYISKMKVVYPAVEG---RIKFSAASHYQG-SFPLVI-LSFWAMILILYQ--5NTD_RAT ISVVSEYISKMKVIYPAVEG---RIKFSAASHYQG-SFPLII-LSFWAVILVLYQ--5NTD_DISOM ---VSSYIKQMKVVYPAVEG---RILFVENSATLP-IINLKIGLSLFAFLTWFLHCS5NTD_BOOMI ---VMKYMNSTSPITTALDG---RVTFLKTNQASDACLNLASPFLVLLVLVVFYHL-PRIO_BOVIN -KMMERVVEQMCITQYQRES---QAYYQRGASVIL--FSSPPVILLISFLIFLIVG-PPBT_RAT --HEQNYIPHVMAYASCIGANLDHCAWASSASSPS-PGALLLPLALFPLRTLF----
PRIO BOVIN
PPBT RAT
5NTD BOOMI
5NTD DISOM
5NTD BOVIN
5NTD HUMAN
5NTD MOUSE
5NTD RAT
séquences de protéines à ancrage GPIa
alignement multiple b
arbre phylogénétiquec
Figure 3. 14 Alignement multiple. Alignement de 8 séquences de protéines à ancre GPI à l’aide du logiciel CLUSTALW.

50
L’alignement multiple permet, entre autres, la découverte de motifs communs à un groupe de
séquences ayant la même fonction. Mais, la valeur biologique de l’alignement proposé n’est
pas toujours justifiée. Il faut toujours examiner un alignement et faire des ajustements selon
la biologie sous-jacente. On ne peut pas assumer directement la présence d’un motif dans une
séquence et inférer une fonction. Car si notre alignement est incorrect, notre inférence l’est
aussi. Un alignement de séquences est sensible à l’ordre de traitement des séquences et à des
différences de longueur. Il faut donc le considérer davantage comme une information de
départ qui doit être raffinée manuellement (Thomson, 2003). De plus, dans le cas où les
séquences à aligner ont une similarité locale présente mais faible et une faible homologie
(comme les protéines à ancre GPI), l’alignement de séquences multiple ne pourra aider à
classifier les protéines appartenant à ce groupe.

CHAPITRE IV
CLASSIFICATION PAR APPRENTISSAGE MACHINE Ce chapitre introduit deux techniques d’apprentissage machine pertinents pour une recherche de motifs fonctionnels de protéines : le réseau de neurones artificiels et le modèle de Markov caché.
4.1 Introduction
Lorsque deux séquences de protéines ont une grande similarité au niveau de leur structure
primaire (un pourcentage élevé de correspondance dans la composition des lettres), il y a une
forte probabilité que ces deux protéines aient la même fonction. Un simple alignement de ces
séquences sert alors à les classifier. Toutefois deux protéines peuvent être très différentes au
niveau de la structure primaire, tout en ayant une structure tertiaire similaire (Baldi et
Brunak, 2001, Gan et al., 2001). Dans ce cas un alignement des séquences ne peut aider à les
classer. L’alignement de séquences comporte donc des lacunes pour certaines classifications.
Pour ces cas, une alternative existe : la classification de séquences biologiques par
apprentissage machine. Les outils d’apprentissage machine sont intéressants car ils peuvent
cibler des motifs cachés ou bruités qui échappent aux algorithmes d’alignement par
similarité.

52
4.2 Apprentissage neuronal
Un réseau de neurones artificiels est un formalisme de représentation de la connaissance.
Cette connaissance réfère, en général, à de l’information emmagasinée ou à des modèles
utilisés dans le but d’effectuer des tâches d’interprétation et de prédiction pour répondre au
monde extérieur de façon appropriée (Haykin, 1999).
Les réseaux de neurones artificiels sont inspirés des réseaux de neurones biologiques. Au
cours de l’évolution, la nature a sûrement optimisé ses modèles de traitement de
l’information. Il est donc logique de mimer les structures biologiques du raisonnement pour
obtenir des machines « intelligentes » (Morris, 1988). Les réseaux de neurones biologiques
sont fondamentalement des structures parallèles, distribuées. L’information y est
emmagasinée, traitée et communiquée de façon globale, c’est-à-dire qu’elle ne se retrouve
pas dans des sites spécifiques mais bien à la grandeur du réseau. La connaissance est
majoritairement emmagasinée dans les connexions entre les neurones, plutôt que dans les
neurones eux-mêmes (Leon, Gâlea et Zbancioc, 2002). Le concept de connaissance distribuée
y est facilement incorporé. Un réseau de neurones artificiels est ainsi caractérisé par des
interconnexions entre des unités de traitement simples (neurones) agissant en parallèle.
Chaque connexion a un poids qui lui est affecté et qui indique l’influence réciproque entre les
deux neurones. La modification de ces poids permet l’adaptation et l’apprentissage (Renders,
1995).
Pour bien illustrer ce concept, voyons comment implanter la reconnaissance du mot « DOG »
de façon distribuée (Magoulas, 2001). Dans l’IA classique, l’information indiquant comment
prononcer le mot « DOG » est emmagasinée dans un endroit précis, différent de celui où est
emmagasiné le mot « CAT ». Un lexique indiquera la position du mot ainsi que les règles à
suivre pour le prononcer et prononcer de nouveaux mots. Dans une représentation distribuée
du même problème, on aura plutôt une architecture de réseaux de neurones composée de
plusieurs couches d’unités de neurones : une couche de neurones d’entrée qui pourraient être
représentées par les lettres composant le mot « DOG », une couche cachée pour le traitement

53
intermédiaire et une couche de sortie (fig. 4.1). Ces couches sont interconnectées par des
liens auxquels sont rattachés des poids.
D
O
G
DOG
CAT+
-
Figure 4. 1 Représentation distribuée. Représentation du mot «DOG» dans un réseau de neurones artificiels. La couleur indique l’importance des connexions et l’activation des neurones dans le processus de traitement de l’information.
Dans un réseau de neurones artificiels, les lettres du mot « DOG » présentés à l’entrée
exciteront les neurones et les connexions du réseau entier (fig. 4.1). L’apprentissage se fera
en modifiant les poids rattachés à ces connexions. La connaissance du mot « DOG » sera
ainsi distribuée dans plusieurs connexions et neurones du réseau. La somme de tous ces
évènements, et non seulement un évènement ou une cascade d’évènements précis et
dépendants, est la source de la reconnaissance du mot « DOG » (Magoulas, 2001). La perte
d’une connexion n’est aucunement fatale, ce qui implique une plasticité, une capacité de
généralisation, et une tolérance au bruit remarquables.
L’idée de construire un « cerveau artificiel », c’est-à-dire une machine permettant d’imiter le
fonctionnement du cerveau humain, a connu de nombreuses incarnations, ainsi que quelques
déboires, au cours du vingtième siècle. Nous poursuivrons cette section par un bref historique
de la modélisation du cerveau, suivi d’une description de la forme et des interactions des
neurones biologiques, pour enfin donner une description détaillée de quelques algorithmes à
la base de la classification par réseau de neurones artificiels pertinents pour notre recherche.

54
4.2.1 Historique
-1943 : J. McCulloch et W. Pitts (1943) modélisent un neurone biologique en un modèle
mathématique simple. Ils démontrent que le modèle mathématique (neurone formel) peut
effectuer des opérations logiques et arithmétiques. Le modèle de neurone artificiel proposé
par McCulluch et Pitts lança la recherche sur les réseaux de neurones artificiels.
-1949 : D. Hebb (1949) propose une règle d’apprentissage basée sur des études du
comportement animal. Quand le neurone A est suffisamment excité, conjointement avec le
neurone B, le lien qui les unit se voit renforcé. Les réseaux de neurones tel que les réseaux de
Hpofield utilisent cette règle. Ces modèles sont plus proches de la réalité du cerveau.
-1957 : F. Rosenblatt (Rosenblatt 1957 ; 1958) développe le modèle du perceptron, au
laboratoire aéronautique de Cornell, dans une tentative de comprendre les processus cognitifs
humains tels que la mémoire et l’apprentissage. Le perceptron est constitué de 2 couches de
traitement. La couche d’entrée et la couche de sortie. Cette sortie ne peut avoir que des
valeurs binaires 1 ou 0 par exemple. Le perceptron ne peut résoudre que des problèmes
linéairement séparables.
-1960 : F. Rosenblatt construit le Mark I Perceptron, la première machine pouvant
« apprendre » à reconnaître des motifs optiques. Sa tâche consistait à reconnaître différentes
lettres.
-1960 : B. Windrow (Windrow et Hoff 1960) propose le modèle de Adaline (adaptive linear
neuron), un système adaptatif de classification de motifs. Le modèle de l’Adaline est
similaire au perceptron dans son architecture, mais l’apprentissage est plus proche de la
retropropagation de l’erreur (Touzet, 1992).
-1969 : M. Minsky et S. Papert publient Perceptrons, un ouvrage où ils démontrent les
limites théoriques du perceptron. Ils démontrent l’incapacité du perceptron à traiter des

55
problèmes non linéaires tels que la fonction XOR (disjonction exclusive). Ils proposent
l’hypothèse que cette limitation vaut pour les perceptrons multiples. C’est alors qu’a débuté
la période noire des réseaux de neurones artificiels, laquelle se termina en 1982. La recherche
continua, mais à moins grande échelle.
-1982 : J. J. Hopfield, un célèbre physicien, relance l’intérêt pour les réseaux de neurones
artificiels avec sa proposition d’un réseau de neurones entièrement connectés utilisant la règle
d’apprentissage de Hebb.
-1983 : La machine de Boltzmann fait son apparition et dépasse les limites du perceptron.
Son architecture inclut des neurones complètement interconnectés comme le réseau de
Hopfield. Toutefois dans la machine de Boltzmann on retrouve des neurones cachés.
L’apprentissage est inspiré de la règle de Hebb et utilise des règles d’adaptation probabilistes.
-1986 : Rumelhart et d’autres chercheurs (Rumelhart et McClelland, 1986 ; Rumelhart,
Hinton et Williams, 1986 ; LeCun 1985 ; Parker 1985) proposèrent le perceptron multicouche
et son algorithme de rétropropagation de l’erreur. Le perceptron multicouche introduit
l’utilisation de couches cachées. Avec la rétropropagation et la présence d’une fonction
d’activation sigmoïde pouvant, contrairement à la fonction de seuil, prendre des valeurs dans
un intervalle [0,1], on peut maintenant réaliser une fonction non linéaire.
4.2.2 Le modèle biologique
Le cerveau est l’inspiration biologique des réseaux de neurones artificiels. Les travaux d’un
histologiste espagnol, Santiago Ramón y Cajal (1911), sont à la source de la recherche
portant sur les neurones. Nous savons maintenant que le cerveau a une architecture
complexe : il est composé de plusieurs milliards de neurones et de trillions de connexions
entre eux (Haykin, 1999; Shepherd et Koch, 1990). Ces neurones communiquent entre eux
par des signaux électriques.

56
Le neurone biologique est composé (fig. 4.2) :
1. D’un corps cellulaire (soma) : Le soma contient le noyau de la cellule neuronale. Il est le centre où s’effectue la synthèse des constituants nécessaires à la structure et aux fonctions du neurone.
2. De dendrites : Les dendrites sont des prolongements du corps cellulaire qui acheminent l’information à l’intérieur du soma.
3. D’un axone : Une fois l’information traitée dans le corps cellulaire, elle
est entraînée le long de l’axone.
4. Des ramifications terminales de l’axone : Ces ramifications sont responsables de la transmission des signaux vers les autres neurones.
Les neurones sont connectés entre eux via des synapses qui sont les zones de contacts entre
les neurones, qui transmettent l’influx nerveux.
Il existe deux types de synapses : les synapses électriques et les synapses chimiques. Pour les
synapses chimiques, l’activité est transmise par une substance chimique : le
neurotransmetteur. Les synapses électriques, elles, transmettent directement le signal
électrique d’un neurone à l’autre. Un neurone présynaptique est un neurone conduisant
l’information vers une synapse, tandis qu’un neurone qui conduit le signal à partir d’une
synapse est un neurone postsynaptique (Vander et al., 1989). Ces synapses sont soit
excitatrices, soit inhibitrices. Une cellule neuronale a une différence de potentiel entre sa
membrane interne et sa membrane externe. Au repos, cette différence est de -60mV
(milivolt), environ. Si la différence de potentiel de la membrane s’approche d’un certain
seuil, la synapse est excitatrice. Si la différence de potentiel s’éloigne du seuil, elle est
inhibitrice. L’information passe par l’axone et un potentiel d’action (une brève inversion de
la polarité du potentiel de la membrane) sera créé. Ce potentiel d’action passe par une
synapse pour influencer l’activité d’un autre neurone qui à son tour produit un potentiel
d’action en sommant tous les signaux reçus des autres neurones. (Vander et al., 1999). Cette
notion de sommation est importante, car le modèle artificiel se basera sur cette sommation
pour effectuer le traitement à l’intérieur du neurone artificiel.

57
soma
axonedendrites
synapse
ramifications
neurone postsynaptique
neurone présynaptique
neurone présynaptique
Neurone
Figure 4. 2 Neurone biologique. Connexions entre deux neurones présynaptiques (vert) et un neurone postsynaptique (rouge).
4.2.3 Le modèle mathématique
Le modèle du neurone mathématique, ou neurone formel, proposé par McCulloch et Pitts
(1943), se veut un modèle simplifié du neurone biologique. La figure 4.3 montre un neurone
formel. Comme pour le neurone biologique, on retrouve l’équivalent des dendrites, d’un
axone et d’un corps cellulaire où le traitement de l’information s’effectue.

58
...
...
......
w1j
wij
wnj
1
ai
n
j
neurone j
θ j
y
a
a
a
Figure 4. 3 Neurone formel.
Le symbole ja représente la valeur d’activation, ou de sortie, du neurone .j La connexion du
neurone i au neurone j a un poids désigné par ijw S’il n’y a pas de connexion entre deux
neurones, on pose 0=ijw . Le seuil est noté jθ . Le calcul de ja dépend de l’entrée nette,
jnet , vers le neurone :j
....2211 njnjjj wawawanet +++=
Les deux façons les plus classiques (fig. 4.4) de calculer ja sont l’utilisation d’une fonction de
seuil :
( ) ,
si 1
si0
!"
!#$
≥
<=
jj
jjj net
netnetSeuil
θ
θ
ou une fonction sigmoïde :
( )jnetj e
netSig+
=1
1 ,
La sortie sera donc ( )jj netSeuila = ou ( )jj netSiga = .

59
1
0
1
0
a bseuil sigmoïde
Figure 4. 4 Fonctions d’activation. a : Fonction se seuil. b : Fonction sigmoïde.
Un neurone avec une fonction d’activation de seuil n’est pas très flexible, car sa sortie est
binaire. D’autre part, la fonction sigmoïde peut prendre toutes les valeurs réelles dans
l’intervalle [0,1]. Elle offre donc beaucoup de souplesse.
Le traitement de l’information semble simple lorsque décrit dans un seul neurone. Toutefois,
comme nous le verrons dans les prochaines sections, il n’est pas évident de décrire la
propagation des calculs lorsque les neurones sont en réseaux c’est-à-dire quand plusieurs
neurones sont connectés entre-eux.
4.2.4 Structure des connexions
Il existe deux types de structures de connexions. Dans les structures récurrentes (fig 4.5), on
admet des cycles dans l’architecture du réseau, alors que dans les structures non-récurrentes
(fig 4.5b), de tels cycles sont interdits. Nous allons nous intéresser à ces derniers, qui sont
pertinents pour notre recherche, et qui sont les plus couramment utilisés en biologie
moléculaire (Wu et McLarty, 2000; Baldi et Brunak, 2001).

60
a récurrent b non récurrent
Figure 4. 5 Structure de connexions de réseaux de neurones artificiels.
Les neurones non-récurrents s’organisent naturellement en couches successives. On distingue
trois types :
1. La couche d’entrée, formée par les neurones qui n’ont pas de connexions entrantes. Un neurone j sera un neurone d’entrée si 0=ijw pour chaque
valeur de .i Dans ce cas ja est donné.
2. La couche de sortie, formée par les neurones qui n’ont pas de connexions sortantes. Un neurone j sera un neurone de sortie si 0=jiw pour chaque
valeur de .i Dans ce cas ja est calculé, et constitue la « réponse » du réseau.
3. Les couches intermédiaires ou cachées, formés de neurones qui ont des
connexions entrantes et sortantes.
Lorsque les valeurs des neurones d’entrée sont connues, ainsi que les poids de toutes les
connexions, il est possible de calculer successivement toutes les valeurs ja des neurones du
réseau en utilisant les équations de la section précédente.
Une étape cruciale dans la construction d’un réseau de neurones sera la détermination des
poids des connexions, c’est l’étape d’apprentissage qui fait l’objet de la section suivante.

61
4.2.5 L’apprentissage
Avant de pouvoir servir de classificateur, un réseau de neurones doit être entraîné.
L’apprentissage, dans un réseau de neurones artificiels, est la modification des poids des
connexions. On peut diviser les réseaux de neurones selon le type d’apprentissage : les
réseaux à apprentissage supervisé et à apprentissage non supervisé. Nous nous intéresserons
principalement à l’apprentissage supervisé car il est le type d’apprentissage pertinent pour
notre recherche. Dans un tel type d’apprentissage, on présente au réseau un ensemble
d’exemples :
( ) ( ) ( ). , ,..., , , , 2211 kk DEDEDE
Où chaque iE est un ensemble de valeurs données aux neurones d’entrée, et chaque
iD correspondant représente les valeurs désirées pour les neurones de sortie. Dans un
problème de classification en deux classes (GPI ou non-GPI) comme celui qui nous intéresse,
les sorties désirées seront des valeurs binaires.
Lorsqu’un exemple iE est présenté au réseau, ce dernier calcule une « réponse » .iS L’erreur
faite par le réseau sur cet exemple est donnée par:
( )2ii SD − .
Cette quantité doit idéalement être nulle dans le cas d’un réseau parfait. En pratique, on se
contentera de ramener l’erreur sous un seuil acceptable. Le processus de minimisation de
l’erreur peut se faire un exemple à la fois, on parle alors d’apprentissage en ligne, ou encore
en soumettant tous les exemples au réseau et en calculant l’erreur globale :
( ) .1
2!=
−=k
iii SDE

62
On parle alors d’apprentissage en lots. L’apprentissage en ligne a plusieurs avantages dont la
possibilité de traiter de grandes quantités de données, d’effectuer un apprentissage plus
rapide, lorsque les données sont redondantes, et de permettre l’entrée de nouvelles données
lors de l’apprentissage (Orr et Cummins, 1999). L’apprentissage en lots est moins rapide
mais, on obtient une meilleure représentation de l’erreur globale E.
La principale technique de minimisation de l’erreur est appelée descente de gradient. Il s’agit
d’augmenter ou de diminuer chaque poids ijw d’une quantité proportionnelle à la dérivé (taux
de changement) de l’erreur par rapport à .ijw Dans les sections suivantes, nous allons
présenter plus en détails certains algorithmes utilisés dans les réseaux de neurones de types
perceptron et perceptron multicouche.
4.2.5.1 Le modèle du perceptron
Le perceptron est un réseau de neurones artificiels ayant deux couches de neurones : une
couche d’entrée et une couche de sortie (fig. 4.6). La fonction d’activation des neurones de la
couche de sortie est la fonction Seuil. Cette fonction, comme spécifié plus haut, est une
fonction ne permettant qu’une sortie binaire (0,1). Si une tâche est résoluble par un
perceptron, l’apprentissage supervisé du perceptron convergera nécessairement vers cette
solution dans un nombre fini d’itérations (Wu et McLarty, 2000).
Ce type de réseau peut être utilisé pour classifier des données en deux classes. En analyse de
séquences biologiques, le perceptron est utilisé pour classifier des sites d’attachement au
ribosome de séquences d’ARN messagers (Stormo et al., 1982). On retrouve aussi des
exemples où le perceptron a été utilisé pour la recherche de site de clivage dans les séquences
protéiques (Schneider, Rohlk et Wrede, 1993).

63
1
2
3
n
w1
w2
w3
wn
Θ
a
a
a
a
a
Figure 4. 6 Architecture du perceptron.
Grâce à son architecture simple les paramètres associés au perceptron peuvent être décrits par
n neurones d’entrée, de valeurs naa ..., ,1 , un neurone de sortie dont la valeur sera noté a ,
une valeur de seuil ,θ et les poids des connexions nwww ..., , , 21 (fig 4.7).
Au cours de l’apprentissage, les quantités ,θ et nwww ..., , , 21 vont être modifiées. On note
la variable qui indique l’étape de calcul par la lettre ,t et les valeurs correspondantes
par ( )tθ et ( ) ( ). , ... , 1 twtw n
L’apprentissage du perceptron selon l’algorithme de Windrow-Hoff (Widrow et Hoff,
1960) se déroule ainsi:
0. 0←t
1. On initialise les paramètres ( )tθ et ( ) ( ) , ... , 1 twtw n au hasard.
2. On présente un exemple au réseau ( )nxxE ... 1= dont la sortie désirée est .d

64
3. On calcule la sortie : ( ) ( )!!"
#$$%
&'==
n
itixtiwa
1 , Seuil θ
4. Si da = on passe à l’étape 5.
Si da < où da > , on modifie les poids avec l’équation ( ) [ ] ii xadtw −=+ η1 .
5. 1+← tt et on recommence à l’étape 2 avec le prochain exemple.
Le paramètre η est un nombre inférieur à 1. Il contrôle la « vitesse d’apprentissage » et est
déterminé de manière heuristique.
4.2.5.2 Le modèle du perceptron multicouche
Le perceptron multicouche est une architecture très utilisée dans le domaine de la
bioinformatique. L’architecture du perceptron se compose d’au moins trois couches de
neurones. Chaque neurone de la première couche est relié à la suivante par une connectivité
totale et ce, jusqu’à la couche de sortie (fig. 4.7). La première couche est celle d’entrée. La
dernière est la couche de sortie, tandis que les couches intermédiaires sont des couches
cachées. Typiquement, tous les neurones d’une couche sont connectés à la couche suivante et
le flot d’information passe de la couche d’entrée vers la couche de sortie. La fonction
d’activation des neurones des couches cachées et des neurones de la couche de sortie est la
fonction sigmoïde. Cette différence par rapport au perceptron, est essentielle à l’entraînement
du perceptron multicouche et permet le traitement de problèmes non linéaires.

65
entréecachée
sortie
Figure 4. 7 Architecture du perceptron multicouche.
Le nombre de neurones dans la couche d’entrée dépend de l’encodage des données. Le
nombre de neurones de la couche de sortie dépend du nombre de classes nécessaire pour la
classification des données. Il n’y a pas de convention pour décider du nombre de neurones
des couches cachées et du nombre de couches cachées. Toutefois, un réseau ayant une seule
couche cachée peut représenter la plupart des transformations entrée-sortie, s’il a un nombre
approprié de neurones et une fonction sigmoïde (Hornik, Stinchcombe et White, 1989). Le
fonctionnement du perceptron multicouche est simple : chaque neurone de la couche d’entrée
génère un signal qui sera envoyé vers les neurones de la (ou des) couche(s) cachée(s). Ces
derniers neurones génèrent un signal qui sera reçu par les neurones de la couche de sortie qui,
eux, engendrent le résultat de la classification. Contrairement au perceptron, il n’est pas
garanti que le perceptron multicouche convergera vers une solution, ce qui peut produire des
entraînements difficiles et longs dans certains cas (Wu et McLarty, 2000).
En analyse de séquences biologiques, le perceptron multicouche est une architecture très
utilisée. Comme on est en présence d’exemples en grande quantité et que la tâche à accomplir
est souvent une classification selon des classes connues, cette popularité n’est pas étonnante.
Plusieurs travaux se basent sur son utilisation. Notons, entre autres, les travaux de Quian et
Sejnowski (1988) qui ont étudié la prédiction de la structure secondaire de protéines à l’aide
d’un algorithme de rétropropagation. D’autres travaux effectués dans le domaine de l’analyse

66
de séquences protéiques sont ceux de Nakata (1995) sur l’utilisation du perceptron
multicouche et de la rétropropagation pour prédire les sites d’attachements à l’ADN de
certaines protéines. Plus récemment, Ahmad, Gromiha et Sarai (2004) ont utilisé le
perceptron multicouche pour prédire des protéines se liant à l’ADN.
L’apprentissage d’un réseau multicouche est nécessairement plus complexe que celui d’un
perceptron. Comme les neurones d’une couche cachée n’ont pas de sortie désirée, l’erreur
correspondante à ces neurones ne peut être calculée directement. Elle est plutôt calculée grâce
aux erreurs de la couche suivante. Comme l’erreur de la couche de sortie est calculable, on
peut calculer successivement l’erreur des couches antérieures, cette technique s’appelle la
rétropropagation de l’erreur (Rumelhart et McClelland, 1986; Rumelhart, Hinton et
Williams, 1986).
Dans le cas des perceptron multicouche, il n’y a pas de garantie de convergence de la
procédure de descente de gradient. L’apprentissage peut donc se retrouver piégé dans un
minimum local (fig. 4.8), ce qui peut produire des entraînements longs et difficiles.
minimum local
vrai minimum
erreur
Figure 4. 8 Minimum local et vrai minimum. En cherchant à minimiser l’erreur par descente de gradient on peut se retrouver dans un minimum local.

67
Nous allons décrire ici l’algorithme RPROP pour « Resilient Backpropagation ». La
particularité du RPROP est que seulement le signe de la dérivée est pris en compte pour
permettre l’ajustement des poids. Cet algorithme permet une convergence plus rapide,
comparativement à la rétropropagation classique. (Riedmiller et Braun, 1992; 1993). Son
fonctionnement peut se définir ainsi : il commence par une petite valeur d’ajustement et,
ensuite, il augmente cette valeur si le gradient présent a la même direction (signe) que le
gradient précédent. Toutefois, si la direction est opposée, il diminue la valeur. Cette mise à
jour est ajoutée au poids, si le gradient est positif, et soustraite du poids, s’il est négatif. Une
autre caractéristique du RPROP est que l’apprentissage est fait en lots.
L’apprentissage du perceptron multicouche selon l’algorithme RPROP (Riedmiller et Braun,
1992; 1993) se déroule ainsi :
1. Les poids ijw sont initialisés de manière aléatoire.
2. L’ensemble des exemples est soumis au système.
3. La valeur de l’erreur est calculée et les paramètres ijw sont modifiés :
( )( ) ( )( ) ( )!"
!#$
Δ+
Δ−=+
sinon.
positive.est àrapport par erreur l' de dérivée la si
1
ttwwttw
twijij
ijijijij
où ijΔ est la valeur d’incrément des poids.
4. Les incréments ijΔ sont recalculés.
( )( )( )!"
!#$
Δ
Δ=Δ +
sinon.
s.précédente itérations 2 des coursau signe de changé pas an' dérivé la si
2
11
tctc
tij
ijij
En pratique on doit avoir 12 10 cc <<< .

68
Finalement lors de l’apprentissage il est important de s’assurer d’une capacité de
généralisation la plus élevée possible. Une technique utilisée pour améliorer la généralisation
est d’ajouter à la fonction d’erreur une pénalité aux poids trop élevés.
La fonction d’erreur :
( )!=
−=k
iii sDE
1
2
devient donc,
( ) α101
2 !! +−==
ijk
iii
wsDE .
Où α représente une constante correspondant au taux de déclin. En ajustant la valeur de α ,
on améliore grandement le pouvoir de généralisation ( Krogh, 1992).
4.2.6 L’encodage des données
Pour tout type d’architecture de réseau de neurones, un élément important pour avoir un
classificateur optimal est l’encodage des données. Comme les séquences biologiques sont
représentées sous forme de chaîne de lettres, elles doivent être encodées en vecteurs pour être
présentées aux réseaux de neurones. Un encodage idéal devrait extraire le maximum
d’informations possibles et respecter la consigne que des séquences similaires ont des
vecteurs similaires (Wu et McLarty, 2000). Nous discuterons ici de deux formalismes
d’encodage couramment utilisés en analyse de séquences biologiques : l’encodage direct et
indirect.
L’encodage indirect offre une vue globale de la séquence. Il offre l’avantage de pouvoir
inclure des séquences de différentes longueurs, mais le prix à payer est celui de la perte de
l’information du positionnement des résidus.

69
La méthode de hachage N-gram est un bon exemple. Ce type d’encodage calcule la
fréquence des facteurs de N résidus dans une séquence (Wu, Whitson et McLarty, 1992).
Par exemple considérons la séquence d’acide aminés : « lllvtpfenlllvtpgdenami ». Si on
décide d’utiliser l’hydrophobicité comme propriété, on peut diviser les 20 acides aminés
selon trois groupes :
1. Hydrophile {d e n q r k} = A ; 2. Hydrophobe { a m i l v f w} = B ; 3. Intermédiaire {c s t p g h y} = C.
La séquence « lllvtpfenlllvtpgdenami » devient BBBBCCBAABBBBCCCAAABBB. Pour
N=2, les N-grams de cette séquence sont les facteurs de longueur 2 présents dans la séquence.
BB BB BB BC CC CB BA AA AB BB BB BB BC CC CC CA AA AA AB BB BB
Le nombre de facteur différents de longueur 2 avec l’alphabet {A, B, C} est 9. On peut donc
calculer la fréquence d’apparition de chacune de ces 9 paires possibles :
AA = 3 AB = 2
AC = 0 BB = 8
BA = 1 BC = 2
CC = 3 CA = 1
CB = 1,
et on obtient un vecteur de taille 9, indépendant de la longueur de la séquence:
(3, 2, 0, 8, 1, 2, 3, 1, 1).
L’encodage direct, par contre, implique la conversion des acides aminés d’une séquence
protéique sous forme d’un vecteur. Cette méthode offre l’avantage de préserver l’information

70
positionnelle présente dans la séquence. Toutefois, elle impose l’usage de fenêtres de
longueur fixe.
Les quatre bases d’acide nucléique {A, C, G, T} peuvent, par exemple, être codées par des
nombres : A = 1, C = 2, G = 3, T = 4. Par exemple, la séquence « ACCGGCTGGT »
correspondrait au vecteur d’entrée (1, 2, 2, 3, 3, 2, 4, 3, 3, 4).
Une autre façon de représenter directement les caractères d’une séquence est d’affecter des
valeurs de propriétés physico-chimiques, telle que l’hydrophobicité, à chaque acide aminé.
Dans le cas de notre modèle nous avons privilégié l’encodage direct pour préserver
l’information positionnelle.
Une étape finale de l’encodage des données est leur normalisation. Pour éviter que des
valeurs trop extrêmes dominent et limitent l’influence de valeurs moindres, il est important
de normaliser les données d’entrées d’un réseau de neurones. La normalisation des vecteurs
d’entrées donne normalement des valeurs entre [0,1] ou [-1,1].
4.2.7 Validation
La validation du modèle s’effectue en utilisant un ensemble de données test. Une méthode
d'évaluation de la performance de généralisation d’un classificateur consiste à effectuer une
mesure de qualité des prédictions. Pour un classificateur à deux classes (0 et 1), la
performance peut être évaluée en calculant les paramètres suivants : (Wu et McLarty, 2000)
(tableau 4.1) la sensibilité est la proportion de tous les vrais positifs correctement identifiés.
La spécificité est la proportion de tous les vrais négatifs correctement identifiés. La valeur de
prédiction positive est, quant à elle, la proportion qu’un vrai positif soit effectivement un vrai
positif. Pour la valeur de prédiction négative elle est la probabilité qu’un vrai négatif soit en
fait un vrai négatif. La précision du modèle est la probabilité de prédictions correctes au total.
Et finalement, le coefficient de corrélation nous donne une idée de la qualité du prédicteur.

71
Une valeur de 1 correspond à un prédicteur parfait tandis qu’une valeur de -1 correspond à un
prédicteur qui à toujours tort (Wu et McLarty, 2000).
Un autre test d'évaluation de l'efficacité de plus en plus utilisé en apprentissage machine est
l'analyse de la courbe ROC (Receiver Operating Characteristic) (Maloof, 2002). Ce type
d'analyse permet de connaître le taux de vrais positifs, ainsi que le taux de faux positifs, à
différents seuils de confiance. Pour connaître la performance des prédictions, une
approximation de l'aire sous la courbe est calculée en utilisant la règle du trapèze. Plus l'aire
sous la courbe tend vers 1, plus la performance du modèle est élevée.
Tableau 4. 1 Tests de validation
TN = Vrai négatif, TP = Vrai positif, FP = Faux positif, FN = Faux négatif Précision
du testEnsembleTNTP +
Sensibilité FNTP
TP+
Spécificité FPTN
TN+
Valeur de prédiction positive FPTP
TP+
Valeur de prédiction négativeFNTN
TN+
Coefficient de corrélation ( )( ) ( ) ( ) ( )TPFNFNTNTNFPFPTP
FNFPTNTP+×+×+×+
×−×
4.3 Les modèles de Markov cachés
Les modèles de Markov cachés sont des automates probabilistes à état finis. Un automate
probabiliste est une structure composée d'états, de transitions et d'un ensemble de probabilités
associées aux états et aux transitions. Pour bien décrire le modèle de Markov caché, il faut
tout d’abord décrire un modèle de Markov plus simple, le modèle de Markov observable.
Les modèles de Markov observables se basent sur l’hypothèse de Markov : « Le futur ne
dépend que du présent et non du passé ». Un modèle de Markov observable est un graphe
d’états dotés de transitions probabilistes et dans lequel à chaque état est associé un

72
évènement. Un chemin dans un modèle de Markov observable, à partir d’un état donné,
consiste à se déplacer d’état en état dans le graphe selon les probabilités de transitions. À un
chemin correspond donc une unique suite d’états (les états visités par ce chemin) ou de
manière équivalente, une suite d’évènements, la suite des évènements observés. La figure 4.9
représente un exemple d’un modèle de Markov observable pour un modèle de prédiction
météorologique. Ici, la question est : « En se basant sur la température d’aujourd’hui, quelle
sera la température de demain ? ». Comme on peut voir dans l’exemple, si la journée est
ensoleillée, nous avons une probabilité de transition vers une autre journée ensoleillée de 0.8,
avec seulement une probabilité de 0.1 d’avoir une journée de pluie. Dans un modèle de
Markov observable, comme chaque évènement observable est associé à un unique état du
modèle, on en déduit de manière non ambiguë un chemin dans le modèle. Dans ce cas, nous
avons une séquence d’états (soleil, pluie ou brume), car à chaque état correspond un seul
évènement observable. Cependant, pour des raisons de modélisation, il est parfois nécessaire
d’associer un même évènement à plusieurs états et vice-versa. On utilise alors les modèles de
Markov cachés.
étatbrume
étatsoleil
étatpluie
0.80.1
0.10.1
0.7
0.2 0.3
0.5
0.2
Figure 4. 9 Modèle de Markov observable. Les états sont : soleil, brume et pluie.
Un modèle de Markov caché est un modèle de Markov où les états ne sont pas des
évènements observables, mais sont munis de probabilités d’émissions des évènements
observables : chaque évènement a une probabilité, possiblement nulle, d’être émis par chaque

73
état. On se retrouve donc dans un processus stochastique double, puisque nous avons la
probabilité de transition entre les états et la probabilité d’émission d’évènements provenant
de ces états. La figure 4.10 montre un modèle de prévision météorologique doublement
stochastique. Toutefois, dans un tel modèle, à une séquence d’évènements observés donnée,
on peut possiblement associer plusieurs chemins ayant pu produire cette séquence, du fait de
la possibilité pour un évènement d’être émis par plusieurs états différents. C’est pourquoi on
parle de modèles de Markov cachés : le chemin (suite d’états) ayant produit une séquence
d’évènements observés est caché à l’observateur, seuls la séquence d’évènements est
disponible.
étatvariable
étatbon
étatmauvais
soleil 0.70brume 0.20pluie 0.10
soleil 0.20brume 0.60pluie 0.20
soleil 0.10brume 0.20pluie 0.70
Figure 4. 10 Modèle de Markov caché. Les états sont : bon, variable et mauvais. Les évènements soleil, brume et pluie sont émis par les états.
Un point important avec les modèles de Markov réside dans la possibilité d’utiliser ces outils
pour modéliser des familles de séquences (linguistiques, biologiques, …). En effet, si l’on
considère un alphabet comme un ensemble d’évènements possibles, un mot sur cet alphabet
n’est rien d’autre qu’une séquence d’évènements. On peut alors considérer que les paramètres
d’un modèle de Markov caché (nombre d’états, probabilités de transitions et probabilités
d’émissions) peuvent être calculés de sorte à capturer les propriétés statistiques d’une famille

74
donnée de séquences. Lorsqu’on a un modèle de Markov caché correspondant à un ensemble
de séquences, on distingue alors trois principaux problèmes d’intérêts.
Le problème d’évaluation : Étant donnés les paramètres d’un HMM particulier et une
séquence d’évènements observés, quelle est la probabilité que cette séquence ait été générée
par ce modèle. La probabilité qu’une séquence s provienne d’un HMM )(wM , notée
( )wsP | , où w représente les paramètres du HMM, se calcule en considérant tous les
chemins pouvant produire cette séquence. Il est nécessaire de considérer tous les chemins
car, comme nous l’avons déjà mentionné, plusieurs chemins peuvent produire une même
séquence d’évènements. À chaque chemin, on associe naturellement la probabilité que ce
chemin ait produit la séquence d’évènements considérée, en multipliant les probabilités de
transitions et d’émissions utilisées par chemin. ( )wsP | est alors la somme, sur tous les
chemins possibles, de la probabilité associée à chaque chemin. Toutefois, il est inconcevable
d’énumérer tous les chemins possibles, car leur nombre croît exponentiellement (Baldi et
Brunak, 2001). On peut cependant passer outre ce problème combinatoire en utilisant la
technique de la programmation dynamique, avec l’algorithme « Forward ». Cet algorithme
calcule un tableau [ ]jtf , bidimensionnel, dans lequel la case [ ]jtf , contient la somme des
probabilités associés aux chemins se terminant en l’état j et produisant la séquence des t
premiers évènements de la séquence s . Ce tableau peut être calculé efficacement par
programmation dynamique (Baldi et Brunak, 2001). ( )wsP | est alors obtenue en sommant
toutes les cases de la dernière colonne du tableau , c’est-à-dire les cases [ ]jtf , pour tout j et
t=n, où n est la taille de s .
Le but de l’algorithme d’évaluation d’une séquence s vis-à-vis d’un modèle de Markov
caché donné représentant une famille particulière de séquence F est de décider, en se basant
sur ( )wsP | , si la séquence s possède des caractéristiques similaires aux séquences de la
famille F et devrait être classifiée comme appartenant à cette famille. Il faut donc pouvoir
interpréter ( )wsP | dans ce but. Or de par son principe multiplicatif le long d’un chemin,
l’algorithme Forward induit une corrélation entre la longueur de la séquence s et ( )wsP | .

75
Pour remédier à ce défaut on utilisera plutôt le log-odd de ( )wsP | , une transformation
mathématique qui a un effet normalisateur par rapport à la longueur de la séquence (Baldi et
Brunak, 2001). Pour classifier s , on compare alors ce score log-odd à un seuil (Barrett,
Hughey et Karplus, 1997) déterminé de manière théorique ou expérimentale dépendant de la
connaissance de la famille F de séquences (Francke et Weynans, 2002). Le score log-odd est
le logarithme du score de la séquence divisé par la probabilité d’un modèle nul. Le modèle
nul est un modèle qui considère une séquence comme une chaîne de caractères aléatoires.
Pour mieux comprendre la difficulté du problème d’évaluation, considérons le HMM illustré
à la figure 4.11.
0 1
2
3
4p(A)= 1
2p(B)= 1
2
p(A)= 34
p(B)= 14
p(A)= 14
p(B)= 34
13
13
13
13
13
13
13
13
13
1
Figure 4. 11 Un Modèle de Markov caché.
Dans cette figure, les transitions sortant d’un état sont toutes équiprobables, et la probabilité
d’émission de la lettre A ou B est celle indiquée à coté de l’état. Par exemple, la séquence
ABAA peut être générée en parcourant la suite d’états 0, 1, 3, 3, 2, et 4. La probabilité de
générer cette suite en parcourant ce chemin est donnée par :

76
12
13
14
34
1 * *******13
13
13
14
p(A) p(B) p(A)p(A)vers état 1
vers état 4
vers état 3
vers état 3
vers état 2
= 32 * 37 4
Cette probabilité est faible, mais il existe 14 autres chemins possibles qui sont susceptibles de
générer la même séquence. L’énumération de ces chemins, donnée dans le tableau 4.2,
illustre bien l’exponentialité du nombre des chemins en fonction du nombre d’états.
Tableau 4. 2 Les 15 chemins différents susceptibles de générer ABAA
Chemins 0 1 1 1 2 4 0 1 1 1 3 4 0 1 1 2 2 4 0 1 1 2 3 4 0 1 1 3 2 4 0 1 1 3 3 4 0 1 2 2 2 4 0 1 2 2 3 4 0 1 2 3 2 4 0 1 2 3 3 4 0 1 3 2 2 4 0 1 3 2 3 4 0 1 3 3 2 4 0 1 3 3 3 4
La probabilité de générer la séquence ABAA est obtenue en additionnant les probabilités
d’obtenir ABAA pour chacun des 15 chemins possibles. C’est ce que l’algorithme
« Forward » permet de faire efficacement.
Le problème de décodage : Étant donné les paramètres d’un HMM particulier et une
séquence d’évènements, quel est le chemin dans ce modèle plus susceptible de générer cette
séquence? Ce problème est résolu par l’algorithme de Viterbi (Forney, 1973). L’algorithme
de Viterbi est une variante de l’algorithme Forward qui considère chaque chemin possible.
Cependant, au lieu de prendre la somme, il prend le chemin donnant le score maximum et
donne la séquence d’états parcourus. Il s’agit d’une technique classique dans le cadre de la
programmation dynamique.

77
Pour illustrer le problème de décodage reprenons le HMM de la figure 4.11. Étant donné la
séquence ABAA, on se pose la question d’identifier le chemin le plus probable parmi les 15
chemins de la table 4.2.
Dans cet exemple très simple, il est assez facile de se convaincre que le chemin donné par la
suite 0, 1, 2, 3, 3 et 4 est le plus probable. En effet, l’état 2 a une forte probabilité d’émettre
un B, alors que l’état 3 a une forte probabilité d’émettre un A. L’algorithme de Viterbi
permet de résoudre ce problème efficacement
Le problème d’apprentissage : Étant donnés les paramètres d’un HMM particulier et une
séquence d’évènements, quel ajustement devons-nous faire aux probabilités d’émissions
d’évènements et de transitions d’états pour que le modèle corresponde le plus possible à la
dite séquence d’évènements ? Ce problème est résolu par l’algorithme Baum-Welch. L’idée
derrière l’apprentissage Baum-Welch est d’estimer de façon itérative les paramètres d’un
modèle, en tentant de maximiser la vraisemblance du modèle selon les séquences
d’évènements ou observations.
Le problème d’apprentissage est beaucoup plus complexe que les deux précédents. En effet,
il s’agit ici de modifier les probabilités d’émission et de transition de manière à « favoriser »
certaines séquences. Considérons, par exemple, le HMM de la figure 4.12 où toutes les
émissions et transitions sont équiprobables.

78
0 1
2
3
4p(A)= 1
2p(B)= 1
2
p(A)= 12
p(B)= 12
p(A)= 12
p(B)= 12
13
13
13
13
13
13
13
13
13
1
Figure 4. 12 Modèle de Markov caché avec émissions et transitions équiprobables.
Ce HMM génère toutes les séquences possibles d’une longueur donnée ( 3≥ ), avec la même
probabilité. Supposons que nous voulions détecter des séquences répondant aux critères
suivants :
a. Le début de la séquence importe peu.
b. Au milieu, on retrouve une forte proportion de B.
c. À la fin, on retrouve une forte proportion de A.
Si les « fortes » proportions des exigences b. et c. sont connues d’avance, il est facile de
construire un HMM ayant les bonnes propriétés. Si, au contraire, les caractéristiques des
séquences recherchées nous sont inconnues à priori, mais qu’on dispose d’un ensemble
d’exemples, on peut alors, d’une manière itérative, modifier les probabilités d’émission et de
transition pour augmenter la possibilité de générer les séquences de notre ensemble. Un
ensemble d’exemples tel que :
ABABBBABBBAAAAAA AABBBBABAAABAAA AABAABBBABBBBBABAAAAAAA etc.

79
pourrait donner, suite à l’apprentissage, un HMM tel que celui de la figure 4.13 qui, à partir
de l’état 1 où les probabilités d’émissions sont équiprobables, mène, avec une forte
probabilité, à l’état 2 où la probabilité d’émission d’un B est forte, puis à l’état 3 où celle
d’émission d’un A est forte.
0 1
2
3
4p(A)= 1
2p(B)= 1
2
p(A)= 56
p(B)= 16
p(A)= 16
p(B)= 56
110
710
210
110
410
410
110
510
510
1
Figure 4. 13 Modèle de Markov caché après apprentissage.
Le problème d’apprentissage est difficile, au sens informatique du terme, et n’est
actuellement attaqué que par des heuristiques qui donnent des solutions approximatives.
4.3.1.1 HMM et grammaire
En 1953, la communauté scientifique assistait à la naissance d’une nouvelle ère, celle de
l’élucidation de la structure de l’ADN par Watson et Crick. Cette découverte ouvrait la porte
à l’étude du transfert de l’information dans le matériel vivant. De leurs travaux sur la
structure de l’ADN naissait la fameuse double hélice et la biologie moléculaire connaissait
son apothéose.

80
À la même époque, un autre évènement bouleversait un domaine complètement différent. Par
ses travaux, Noam Chomsky (Chomsky, 1957) révolutionnait le domaine de la linguistique. Il
a proposé une représentation formelle des règles syntaxiques du langage. Il a, pour ainsi dire,
décodé la « structure profonde » sous-tendant les langages. Ses observations sur la variété
infinie des langages ont donné naissance à la grammaire générative. Née dans le domaine de
la linguistique, la grammaire générative fut, par la suite, intégrée dans la théorie du calcul, et
notamment l’informatique théorique et la sémantique des langages de programmation. Ces
deux groupes de recherches ont donc, à la même époque, permis une poussée énorme dans
leurs domaines respectifs (Searls, 1993; 2001).
Mais, qu’ont donc en commun la linguistique et la biologie moléculaire ? Dès le début, les
séquences biologiques composant le génome ont suscité plusieurs comparaisons avec la
linguistique. En effet, l’utilisation d’un alphabet précis et la présence d’une représentation
textuelle des séquences biologiques ouvrent grandes les portes à une métaphore linguistique.
Le langage des séquences biologiques se prête donc bien à l’utilisation des méthodes
d’analyse du langage naturel.
Les grammaires
Comme le définit bien Searls, dans son article de 1993, « Formally, a language is simply a
set of strings of characters drawn from some alphabet… », le langage est, d’un point de vue
formel, un simple groupement de chaînes de symboles appartenant à un alphabet. Les travaux
de Chomsky ont eu pour effet de fournir des méthodes formelles de définition de langages
ou, plus simplement, de donner une théorie du modelage des chaînes de symboles présentes
dans un langage. Le but de la représentation formelle des langages est l’économie
d’expression, c’est-à-dire, la levée de l’obligation d’énumérer exhaustivement toutes les
chaînes possibles dans un langage. La puissance octroyée par cette représentation
« économique » est bien réelle. Un autre avantage est de pouvoir généraliser l’information
structurelle d’un système linguistique (Searls et Dong, 1993). Si on transpose cette définition
du langage à l’ADN, l’alphabet sera composé de quatre symboles que sont les nucléotides. La
molécule d’ADN sera ainsi représentée par une chaîne de symboles. Toutes les différentes

81
compositions de chaînes formeront un langage. Il n’est donc pas faux de dire que l’ADN est
en fait le langage du « livre de la vie » (Searls, 2002).
La hiérarchie de Chomsky
Chomsky a spécifié quatre types de grammaires basées sur les restrictions des règles de
production. Plus on monte dans la hiérarchie, plus on a la possibilité de proposer des règles
générales. Pour bien comprendre les niveaux hiérarchiques de Chomsky, il faut d’abord
définir quelques principes de notation. Il y a deux types de symboles : les variables abstraites
non-terminales (représentées par des lettres majuscules) et les symboles concrets terminaux
(représentées par des lettres minuscules). Les règles de production du langage seront notées
A a, où la partie de gauche contient au moins un symbole non-terminal qui sera
transformé, dans la partie de droite, en une chaîne terminale (composée uniquement de
symboles terminaux) ou non-terminale (comportant des symboles non-terminaux).
Une grammaire génère les chaînes composant son langage en prenant un symbole de départ
(A) et en le réécrivant. Cette réécriture se fait en recherchant, itérativement, une règle ayant
un côté gauche correspondant à un symbole non-terminal de la chaîne courante et en y
substituant le côté droit de la règle, tout ceci, jusqu’à ce que la chaîne ne contienne que des
symboles terminaux.
Pour commencer, voici un exemple de grammaire régulière. La grammaire G = (N, E, P, S)
S = Symbole initial : {A}
E = Symboles terminaux : {a, b}
N = Symboles non terminaux : {A, B}
P = Règles : {Aa, AaB, Bb, BbB}
À partir de cette grammaire, on peut dériver toutes les chaînes qui composeront le langage en
partant du symbole non terminal A.

82
Appliquons la première règle. On obtient la suite de mots [a]. On ne peut aller plus loin.
Appliquons la seconde règle. On obtient la suite [aB].
Appliquons la troisième règle à [aB]. On obtient la suite [ab]. On ne peut aller plus loin.
Appliquons la quatrième règle à [aB]. On obtient la suite [abB].
Appliquons la quatrième règle à [abB]. On obtient [abbB].
Appliquons la troisième règle à [abbB]. On obtient [abbb]. On a une suite de constantes. On
ne peut aller plus loin. On peut représenter cet exemple de grammaire grâce à un arbre de
dérivation (fig. 4.14).
A
A a A aB
B b B bB
B bB
a aB
ab abB
abbB
Figure 4. 14 Premiers niveaux d’un arbre de dérivation. Arbre relié à la grammaire décrite ci-dessus (Habrias, 2002).
Les 4 types de grammaires introduits par Chomsky :
1. La grammaire régulière (RG) : Seulement les règles de production de type Aa où A aB sont permises. Le membre gauche de toute règle contient exactement, et uniquement, un symbole non-terminal, et le membre droit en contient au plus un, sans restriction sur le nombre de symboles terminaux. Les chaînes ne peuvent donc grandir que dans une seule direction. Exemple : Voir ci-dessus.
2. La grammaire hors contexte (CFG) : Toute règle de production de type A est acceptée où représente n’importe quelle chaîne terminale ou non-terminale, excluant la chaîne vide. Exemple : avec les règles P = {A aAa, AbAb, Aaa, Abb}, une des dérivations obtenues est AaAaaaAaaaabAbaaaabaabaa.

83
3. La grammaire sensible au contexte (CSG) : Les CSG répondent au problème des copies en autorisant plus d’un symbole du côté gauche de la règle. Le côté droit de la règle est au moins aussi long que le côté gauche. Il y aura, par exemple, présence de règles de réorganisation de symboles non terminaux et de génération de symboles terminaux. Aucun algorithme fonctionnant en temps polynomial n’existe pour décider si une chaîne donnée peut être obtenue par une CSG donnée: ce problème est NP-complet. Les CSG sont donc, en pratique, non considérées.
4. La grammaire sans restriction (UnresG) : Dans une UnresG, n’importe quel symbole peut se retrouver des deux côtés de la règle. C’est la grammaire la plus générale. Aucun algorithme ne peut garantir qu’une chaîne est une dérivation valable de la grammaire dans un temps fini.
L’analogie entre la linguistique et la biologie moléculaire est plus qu’une simple métaphore.
La grande similitude entre le langage humain et celui de la cellule offre de grandes
possibilités comme, par exemple, l’utilisation des méthodes d’analyse linguistique afin de
mieux comprendre et décomposer le langage cellulaire. Plusieurs techniques
bioinformatiques puisent leurs racines de la linguistique, même si leur développement a été
indépendant. L’approche « mathématique » de la linguistique a permis une avancée
importante dans le développement de la théorie du langage formel qui sera, par la suite, un
des piliers de la recherche de motifs et de structures dans les séquences biologiques.
La plus importante utilisation des grammaires de Chomsky, en bioinformatique, est la
recherche de motifs dans les séquences biologiques via les grammaires régulières simples et
stochastiques (Betel et Hogues, 2002 ; Xuan, McCombie et Zhang, 2002 ; Baldi et Chauvin
1994; Nielsen et Krogh, 1998; DiFrancesco, Garnier et Munson, 1997; Sonnhammer, Eddy et
Durbin, 1997). Les grammaires régulières sont aussi très utilisées pour la recherche de motifs
dans les bases de données protéiques et nucléiques (Gattiker, Gasteiger et Bairoch, 2002) et
pour la prédiction de gènes dans les séquences génomiques (Burge et Karlin, 1997; Kulp et
al., 1996). La plupart des algorithmes de recherche sont donc des modèles se situant au
niveau de base de la hiérarchie de Chomsky (Durbin et al., 1998).

84
Grammaire régulière stochastique
Une grammaire stochastique est essentiellement une grammaire dans laquelle on associe une
probabilité à chaque règle de production. Dans un HMM, on retrouve des états cachés, des
matrices de transitions, des matrices d’émissions et des probabilités. Lorsque l’on veut
comparer un HMM à une grammaire, les états cachés deviennent les symboles non
terminaux, les matrices de transitions deviennent les règles de production, les matrices
d’émissions deviennent les symboles terminaux et, finalement, les probabilités restent les
probabilités. La figure 4.15 montre un exemple de grammaire stochastique dérivée d’un
modèle de HMM.
a = 0.1c = 0.9
a = 0.8c = 0.2
1 1 11 2
émissions:
état S: départétat 1: a|cétat 2: a|cétat F: fin
règles de production:
S 11 a2|c22 aF|cF
S F
règles de production stochastiques:
P(S 1) = 1P(1 a2) = 1 * 0.1 = 0.1P(1 c2) = 1 * 0.9 = 0.9P(2 aF) = 1 * 0.8 = 0.8P(2 cF) = 1 * 0.2 = 0.2
La séquence " ca " aurait une probabilité de 0.9 * 0.8 = 0.72La séquence " cc " aurait une probabilité de 0.9 * 0.2 = 0.18La séquence " ac " aurait une probabilité de 0.1 * 0.2 = 0.02La séquence " aa " aurait une probabilité de 0.1 * 0.8 = 0.08
Appartenance des séquences au HMM
4 combinaisons de séquences possiblesac aacc ca
HMM
Figure 4. 15 Grammaire dérivée d’un HMM.
Dans cet exemple, nous n’avons que deux états possibles (états 1 et 2). Les états S et F sont
les états de départ et de fin. Ils ne génèrent aucune émission de caractères. Ce HMM est
linéaire, c’est-à-dire qu’un seul chemin est possible (S 1 2 F). Ceci est dû au fait

85
que les probabilités de transition entre les états sont de 1. Chaque état émet l’un des deux
caractères, a ou c. Une probabilité est affectée à l’émission de ces caractères. Les règles de
production de cette grammaire sont donc plutôt simples. On passe de l’état initial S vers l’état
1 avec une probabilité de 1. Ensuite, on émet un c ou un a et on passe à l’état 2 avec une
probabilité de 1. On émet un a ou un c dans l’état 2 et, finalement, on passe à la sortie avec
une probabilité de 100%, une fois de plus. Les séquences générées par ce HMM sont donc
« aa », « ac », « ca » et « cc ». La séquence la plus probable est « ca » avec une probabilité
de 0.72.
4.4 Conclusion
En résumé, plusieurs choix s’offrent à nous lors d’une tâche de classification de séquences
biologiques. La nature de nos séquences, le type de classification (clustering, prédiction etc.)
ainsi que la teneur de nos connaissances, déterminent la méthode à utiliser. Il devient donc
important de bien analyser nos données avant de choisir une méthode et d’évaluer la teneur
de nos connaissances permettant une utilisation optimale de cette méthode de classification.

CHAPITRE V
CLASSIFICATION/PRÉDICTION D’ANCRE GPI PAR RÉSEAU DE NEURONES ARTIFICIELS
Les trois chapitres suivants proposent la description de la méthode de classification/prédiction hybride d’ancre GPI chez les séquences protéiques. Nous proposons une approche en trois volets. Le premier volet implique la conception d’un réseau de neurones artificiels pour le nettoyage des données. Le deuxième volet se concentre sur la structuration du signal grâce au HMM et finalement le dernier volet propose l’hybridation des deux modèles. Lorsque l’on parle de séquence protéique, la nature des acides aminés qui la compose est très importante. On constate que des propriétés physiques, chimiques et ioniques existent et influencent grandement la fonction des protéines. Des propriétés physico-chimiques intéressantes pour la classification sont les interactions de la molécule avec son environnement Par exemple l’hydropathie des acides aminés composant une protéine influence la structure que celle-ci adoptera dans un environnement. Cette structure influencera à son tour la fonction de la protéine. Il devient évident de tirer avantage de ces caractéristiques lors d’une tâche de classification. L’hydropathie et le poids moléculaire ayant été identifié expérimentalement comme des caractéristiques importantes pour le signal GPI, nous avons choisi de les utiliser dans la première étape de notre modèle. L’utilisation des réseaux de neurones artificiels et de leur grande capacité de traitement des données brouillées est proposée dans ce présent chapitre comme premier volet de classification. Toutefois l’ordre des acides aminés dans une protéine est également très important pour la biologie. L’agencement de certains acides aminés forme des signaux précis. Dans le signal GPI, la structure grammaticale est tout aussi importante que les propriétés physico-chimiques. Le HMM est une approche bien connue dans le traitement de données séquentielles tel que les séquences biologiques. L’utilisation d’un HMM pour structurer le signal GPI est présentée dans un deuxième volet (Chapitre VI). Finalement nous verrons que l’identification du signal GPI doit faire appel è la fois aux propriétés physico-chimiques et à la structure du signal. Le volet final présenté au Chapitre VII propose une approche hybridant l’utilisation de ces caractères particuliers des séquences protéiques.

87
5.1 Un modèle de réseau de neurones artificiels classificateur d’ancre GPI
Lorsqu’on a un motif complexe que l’on veut utiliser pour la classification ou l’annotation,
on se retrouve devant une quantité très importante de données et d’informations. Une
première étape, dans cette classification, est de bien résoudre le problème de nettoyage de nos
données. Les bases de données contenant les séquences biologiques, ainsi que les séquences
elles-mêmes, sont souvent incomplètes ou bruitées. Même si ces séquences sont déterminées
expérimentalement, avec une grande précision, elles subissent plusieurs manipulations avant
d’être accessibles pour fins d’analyse. Le taux d’erreur devient donc beaucoup plus important
que l’erreur initiale retrouvée normalement durant le processus expérimental (Brunak,
Engelbrecht et Knudsen, 1990). Les réseaux de neurones sont de bons « nettoyeurs » de bases
de données. En biologie moléculaire, leur utilisation a déjà servi à démontrer leur efficacité
en détectant, par exemple, de mauvaises assignations de sites d’épissage dans les ARN
messagers (Brunak, Engelbrecht et Knudsen, 1990). Les réseaux de neurones artificiels sont
un choix de modèle intéressant pour ce problème, vu leur forte capacité de généralisation et
leur performance face à des problèmes de classification impliquant une grande quantité
d’exemples ayant de l’information « cachée » ou ayant des motifs irréguliers.
Dans notre projet, le modèle de réseau de neurones artificiels est construit pour effectuer une
tâche préliminaire de classification. Un réseau de neurones artificiels permet de réduire le
nombre de séquences devant subir une classification plus pointue, donc plus demandante en
temps de calcul.
5.2 Le modèle
L’architecture de notre modèle est le perceptron multicouche présenté au chapitre 4. Le choix
de cette architecture est basé sur l’efficacité déjà prouvée du perceptron multicouche dans des
problèmes de classification et de prédiction de motifs de séquences protéiques. Un exemple
récent est donné par les travaux de Martelli, Fariselli et Casadio (2004) qui utilisent un

88
réseau de neurones artificiels pour prédire la probabilité que certains résidus cystéine puissent
être impliqués dans des ponts disulfure, pour le processus de repliement des protéines.
Nous avons utilisé un simulateur de réseaux de neurones, JavaNNS (Zell, 2002), pour
modéliser notre réseau de neurones. Pour cette architecture, l’algorithme d’apprentissage et
ses paramètres ainsi que le nombre de neurones de la couche d’entrée (vecteur d’entrée), le
nombre de couches cachées et le nombre de neurones qui les composent, ainsi que le nombre
de neurones de la couche de sortie, doivent être déterminés.
Le vecteur d’entrée
La structure connue des protéines à ancre GPI est l’instrument de base nécessaire à la
conception d’un classificateur. Comme il a été spécifié au chapitre 2, une séquence de
protéines à ancre GPI contient deux signaux: un signal en position N-terminale pour la
translocation dans le réticulum endoplasmique et un signal en position C-terminale pour
l’attachement GPI. La découverte de séquences GPI n’ayant pas de signal en N-terminale,
tels que la P137 (Ellis et Lazio, 1995), ainsi que la capacité de biosynthèse de la protéine sans
ce signal (Howell et al., 1994) nous ont incité à ne pas considérer le signal en N-terminale
dans la conception de notre classificateur.
La première étape de conception du modèle est la prise de décision quant à la zone à cibler.
Notre méthode ne cible donc que la partie C-terminale de la protéine, en vue de reconnaître le
signal d’attachement GPI. Pour la construction du modèle de réseau de neurones nous avons
porté notre attention sur deux zones importantes du signal GPI : une zone composée de trois
acides aminés de faible poids moléculaire, et une zone fortement hydrophobe.
La seconde étape implique la décision de la longueur du vecteur d’entrée et à son type
d’encodage. Une première analyse des séquences de protéines GPI présentes dans la base de
données Swiss-Prot montre que les séquences GPI peuvent avoir des longueurs très variables
(fig. 5.1).

89
>C59A_MOUSEmraqrglillllllavfcstavsltcyhcfqpvvsscnmnstcspdqdsclyavagmqvyqrcwkqsdchgeiimdqleetklkfrccqfnlcnksdgslgktpllgtsvlvailnlcflshl>CADD_CHICKmqhktqltlsfllsqvlllacaedlectpgfqqkvfyieqpfeftedqpilnlvfddckgnnklnfevsnpdfkvehdgslvalknvseagralfvharsehaedmaeilivgadekhdalkeifkiegnlgiprqkrailatpilipenqrppfprsvgkvirsegtegakfrlsgkgvdqdpkgifrineisgdvsvtrpldreaianyelevevtdlsgkiidgpvrldisvidqndnrpmfkegpyvghvmegsptgttvmrmtafdaddpstdnallrynilkqtptkpspnmfyidpekgdivtvvspvlldretmetpkyelvieakdmgghdvgltgtatatiliddkndhppeftkkefqatvkegvtgvivnltvgdrddpatgawravytiingnpgqsfeihtnpqtnegmlsvvkpldyeisafhtllikvenedplipdiaygpsstatvqitvedvnegpvfhpnpmtvtkqenipigsivltvnatdpdtlqhqtirysvykdpaswleinptngtvattavldresphvqdnkytalflaidsgnppatgtgtlhitledvndnvpslyptlakvcddakdlrvvvlgasdkdlhpntdpfkfelskqsgpeklwrinklnnthaqvvllqnlkkanynipisvtdsgkppltnntelklqvcsckksrmdcsasdalhismtlillslfslfcl
Figure 5. 1 Séquences de protéines à ancre GPI de différentes longueurs.
Dans cet exemple, la protéine C59A_MOUSE ne contient que 123 acides aminés, tandis que
la séquence CADD_CHICK en contient plus de 700. Cette différence de longueur est due à la
fonction de la protéine, qui peut être très différente, et, aussi, à la présence de séquences
incomplètes dans les bases de données. Cette longueur hautement variable, et la position du
signal GPI en position C-terminale, ont influencé notre décision de ne cibler qu’une portion
fixe de la partie C-terminale de la protéine. De cette façon, nous n’avons pas été limité par
des protéines fragmentaires ayant la partie N-terminale tronquée.
La détermination de la longueur fixe conservée pour notre modèle est basée sur une analyse
de la longueur maximale du signal retrouvé dans les séquences de protéines à ancre GPI de
Swiss-Prot (Appendice A). Cette analyse montre que la longueur maximale de ce signal est
de 45 acides aminés. Pour laisser une certaine flexibilité à toute nouvelle protéine GPI, nous
avons sélectionné les 50 derniers acides aminés des protéines comme étant vecteurs d’entrée
de notre modèle de réseau de neurones. La figure 5.2 montre des exemples des sections de
50 acides aminés sélectionnés pour le vecteur d’entrée. Cette partie des séquences sera, par la
suite, soumise à un encodage.

90
> 5NTD_RAT isvvseyiskmkviypavegrikfsaashyqgsfpliilsfwavilvlyq> ACES_TORCA lrvqmcvfwnqflpkllnatacdgelsssgtssskgiifyvlfsilylif> ACES_TORMA rvqmcvfwnqflpkllnatacdgelsssgtssskgiifyvlfsilylify> AMPM_HELVI tsttaapttvtqptitepstptlpeltdsamtsfaslfiislgailhlil
Figure 5. 2 Vecteur d’entrée du réseau de neurones avant encodage.
L’encodage des données
La longueur fixe de nos séquences permet un encodage direct. L’encodage direct a également
été choisi pour sa capacité de préserver l’information positionnelle de la séquence. La
présence de trois zones distinctes ayant des acides aminés spécifiques est une information
particulière que l’encodage direct pourra conserver.
Le choix d’encodage requiert aussi une transformation numérique des acides aminés. Pour
avoir un prédicteur performant, il faut mettre le plus d’informations possibles dans notre
encodage. Deux caractéristiques importantes ressortent de toute analyse des séquences de
protéines à ancre GPI : l’hydrophobicité élevée de la queue terminale et le site d’ancrage
composé d’acides aminés de faible poids moléculaire. Nous avons donc sélectionné ces deux
propriétés comme base de transformation. Chaque acide aminé sera ainsi représenté par deux
valeurs numériques : son hydropathie et son poids moléculaire. Le tableau 5.1 montre les
valeurs attribuées à chaque acide aminé.
Il existe différentes échelles d’hydrophobicité (Eisenberg et al., 1982 ; Kyte et Doolitle,
1982; Engelman, Steitz et Goldman, 1986 ; White et Wimley, 1999). Nous avons choisi
l’échelle de Kyte et Doolitle pour sa grande popularité en analyse de séquences protéiques
(Tableau 5.1).

91
Tableau 5. 1 Échelles utilisées pour la conversion numérique des acides aminés.
Acide aminé échelle d’hydrophathie Kyte et Doolitle
poids moléculaire
a 1.8 89.09 d -3.5 133.10 e -3.5 147.13 g -0.4 75.07 f 2.8 165.19 l 3.8 131.18 s -0.8 105.09 y -1.3 181.19 c 2.5 121.16 w -0.9 204.23 p -1.6 115.13 h -3.2 155.16 q -3.5 146.15 r -4.5 174.20 i 4.5 131.18 m 1.9 149.21 t -0.7 119.12 n -3.5 132.12 k -3.9 146.19 v 4.2 117.15
b (d ou n) -3.5 132.61 z (e ou q) -3.5 146.64
x -0.84 136.90
En raison de la présence, dans les bases de données publiques, de trois caractères
supplémentaires x, b et z, nous les avons ajoutés à la liste des substitutions. Le caractère
x signifie que le résidu n’a pas été identifié. Dans ce cas nous attribuons les valeurs
moyennes d’hydropathie et de poids moléculaire à l’acide aminé x (dernière ligne du tableau
5.1). Le caractère b signifie que le résidu peut être une asparagine ou un acide aspartique.
Pour l’acide aminé b nous avons calculé la moyenne des valeurs d’hydropathie et de poids
moléculaire des deux acides aminé possible c’est-à-dire d et n. Le caractère z signifie que le
résidu peut être un acide aminé e ou g. Comme dans le cas de l’acide aminé b nous avons pris
la moyenne des valeurs correspondant aux deux acides aminés possibles (e et g). Quoique ces
caractères ne soient présents que dans les cas de séquençages moins précis ou incomplets,
nous avons voulu les représenter dans l’encodage pour éviter un rejet des séquences, lors de
la transformation numérique des données. L’affectation de deux valeurs à chaque acide aminé
nous donne un vecteur d’entrée de 100 neurones.

92
Finalement la dernière étape implique la normalisation des vecteurs d’entrés. Nous avons
appliqué une simple normalisation min-max.
( )( )
( ) min'min'max'*minmax
min' +−−
−=
vv
Où 'v représente la valeur normalisée de la donnée v , min' et max' représentent la valeur
minimale et maximale de l’intervalle désiré, et min et max sont la valeur minimale et
maximale de nos données réelles ( [75.07, 204.23] pour l’hydropathie et [-4.5, 4.5] pour le
poids moléculaire).
Nous avons testé le modèle de réseau de neurones avec des données normalisées et des
données brutes. L’apprentissage ainsi que la capacité de généralisation ont été évalués. La
courbe de progression de l’erreur des deux modèles est similaire dans les deux cas et les
résultats des tests de validation du modèle et des tests supplémentaires donnent une même
valeur de sensibilité et de spécificité. Une analyse plus précise des séquences sélectionnées
par les deux modèles comme étant GPI et non-GPI dans le test de validation du modèle,
montre que les mêmes séquences sont sélectionnées avec des valeurs très proches. Comme la
normalisation n’apporte pas d’amélioration nous avons décidé d’utiliser les données brutes
dans le modèle.
La figure 5.3 montre un exemple de vecteur d’entrée avant normalisation. Dans cet exemple,
on peut voir que la méthionine m, est remplacée par la valeur correspondante à son
hydrophobicité en bleu (1.9) et à son poids moléculaire en vert (149.21). Nous avons donc la
structure de notre vecteur d’entrée de 100 neurones.

93
SØquence> 5NTD_BOOMIvmkymnstspittaldgrvtflktnqasdaclnlaspflvllvlvvfyhl
Vecteur correspondant> 5NTD_BOOMI4.2 117.15 1.9 149.21 -3.9 146.19 -1.3 181.19 1.9 149.21-3.5 132.12 -0.8 105.09 -0.7 119.12 -0.8 105.09 -1.6 115.134.5 131.18 -0.7 119.12 -0.7 119.12 1.8 89.09 3.8 131.19-3.5 133.10 -0.4 75.07 -4.5 174.20 4.2 117.15 -0.7 119.122.8 165.19 3.8 131.19 -3.9 146.19 -0.7 119.12 -3.5 132.12-3.5 146.15 1.8 89.09 -0.8 105.09 -3.5 133.10 1.8 89.092.5 121.16 3.8 131.19 -3.5 132.12 3.8 131.19 1.8 89.09-0.8 105.09 -1.6 115.13 2.8 165.19 3.8 131.19 4.2 117.153.8 131.19 3.8 131.19 4.2 117.15 3.8 131.19 4.2 117.154.2 117.15 2.8 165.19 -1.3 181.19 -3.2 155.16 3.8 131.19
Figure 5. 3 Exemple de vecteur d’entrée. Vecteur avec les valeurs d’hydropathie et de poids moléculaire correspondant à chaque acide aminé.
Les jeux de données d’entraînement et de test
La conception d’un réseau de neurones artificiels requiert la sélection d’un ensemble de
séquences devant servir à l’apprentissage. Comme pour toute technique d’apprentissage
machine nous devons rassembler le plus d’exemples possibles de ces séquences. Plus la
diversité de ces séquences est grande, plus nos modèles auront une bonne capacité de
généralisation.
En date de janvier 2004, la base de donnée Swiss-Prot contenait au moins 468 séquences
protéiques ayant une annotation claire d’ancre GPI (voir Appendice A). Ces séquences
proviennent de quatre grands groupes taxonomiques d’eucaryotes. Ces groupes sont les
métazoaires, soit des animaux pluricellulaires, les plantes, les protozoaires, à savoir des
eucaryotes unicellulaires et les champignons.
Comme séquences d’entraînement et de test pour le réseau de neurones artificiels, nous avons
sélectionné les séquences GPI clairement annotées dans la base de donnée Swiss-Prot.
Plusieurs jeux de données ont été construits à partir de ces séquences:

94
Le jeu de données d’entraînement : Un alignement de séquences des 50 derniers résidus des
séquences GPI effectué à l’aide du logiciel CLUSTALW (voir Appendice B), nous a aidé à
cibler des séquences non redondantes pour éliminer le risque de biais envers un type de
séquence trop abondamment représentées (fig 5.4a). Nous avons retenu 79 des 468
séquences de protéines annotées comme ayant un ancre GPI, de la base de données Swiss-
Prot (voir Appendice C). Quelques ajustements ont été apportés au jeu d’entraînement
lorsqu’une catégorie de séquences particulière n’était pas reconnue. Par exemple, les
protéines PARA_TRYBB, PARB_TRYBB et PARC_TRYBB sont très similaires (fig. 5.4b).
Lors d’une première sélection, un seul représentant avait été choisi : PARA_TRYBB.
Comme le modèle de réseau de neurones avait de la difficulté à reconnaître les autres
membres, nous avons ajouté PARB_TRYBB au jeu d’entraînement (fig 5.4b). Cet ajout a
permis d’augmenter la capacité de prédiction du réseau pour ce type de protéine. Pour que le
modèle puisse faire une bonne discrimination, nous avons aussi construit un jeu de 79
séquences ayant une très faible probabilité d’être à ancre GPI (c’est le cas des protéines
cytoplasmiques, nucléaires, transmembranaires ou des séquences aléatoires). La combinaison
de ces deux jeux de données compose notre jeu de données d’entraînement.
prio_mansp kgenftetdvkmmervveqmcitqyekes---qayyq-----rgss-mvlfssppvillisfli------------prio_atepa -------tetdvkmmervveqmcitqyeres----qayyq-----rgss-mvlfssppvillisfliflivg-------prio_cebap -------tetdvkmmervveqmcitqyeres----qayyq-----rgss-mvlfssppvillisfliflivg-------prio_gorgo -------tetdvkmmervveqmcitqyeres----qayyq-----rgss-mvlfssppvillisfliflivg-------prio_human -------tetdvkmmervveqmcitqyeres----qayyq-----rgss-mvlfssppvillisfliflivg-------prio_pantr -------tetdvkmmervveqmcitqyeres----qayyq-----rgss-mvlfssppvillisfliflivg-------prio_ponpy -------tetdvkmmervveqmcitqyeres----qayyq-----rgss-mvlfssppvillisfliflivg-------
parb_trybb epepepepepepepepep---epepepe----pepe-----pepgaatlksvalpfaiaavglvaaf----parc_trybb epepepepepepepepep---epepepe----pepe-----pepgaatlksvalpfaiaaaalvaaf----para_trybb -----tgpeetgpeetgpe-etgp---eetgpee---tepe-----pepgaatlksvalpfavaaaalvaaf----
a
b
Figure 5. 4 Sélection du jeu d’entrainement. a : La séquence en rouge a été sélectionnée parmi un groupe de séquences très similaires. b : Les séquences en rouges ont été sélectionné malgrès leur similarité pour augmenter la capacité de reconnaître ce type particulier de signal GPI.

95
Le jeu de données de validation de l’entraînement du réseau de neurones : La généralisation
est un des plus grands avantages des réseaux de neurones. Un réseau à rétropropagation
extrapole un résultat avec des entrées qu'il n'a jamais vues. Le réseau doit voir un certain
nombre de fois les exemples proposés avant de pouvoir extrapoler un nouveau résultat.
Toutefois, il y a certains problèmes reliés à un trop grand nombre de cycles d’apprentissage.
Un problème commun est la mémorisation des entrées. Alors, comment empêcher le
surentraînement du réseau? Une méthode simple et efficace est l’utilisation d’un groupe de
données de validation. Après chaque cycle d’apprentissage effectué sur le groupe
d’entraînement, le groupe de validation est présenté au réseau et l’erreur de sortie est
calculée. L’apprentissage est arrêté lorsque l’erreur du groupe de validation est à son
minimum. Si l’apprentissage continue, l’erreur commencera à augmenter et le potentiel de
généralisation du modèle de réseau diminuera. Le réseau aura, dès lors, commencé à
mémoriser. Notre jeu de données de validation comporte 5 séquences GPI et 5 séquences
non-GPI ne se retrouvant pas dans le jeu d’entraînement.
Le jeu de données pour la validation du modèle : Pour la validation du modèle, nous avons
utilisé un jeu de données comprenant 134 séquences GPI. Nous avons effectué une sélection
comportant peu de séquences redondantes et aucune séquence du jeu d’entraînement. Nous
avons aussi sélectionné 134 séquences ayant une faible probabilité d’être à ancre GPI, pour
servir de discriminants.
Les jeux de test supplémentaires : Les bases de données comportent un grand nombre de
séquences. Une validation avec seulement 268 séquences n’est pas très précise. Nous avons
donc construit des jeux de données comportant les 468 séquences GPI de Swiss-Prot. Pour les
jeux de test supplémentaires non-GPI, nous avons sélectionné dans la base de données Swiss-
Prot :
1. Des séquences ayant une très faible probabilité d’être à ancre GPI. : Comme les protéines à ancre GPI sont exclusivement extracellulaires, les protéines cytoplasmiques et nucléaires ont une faible probabilité d’être à ancre GPI (111 séquences).

96
2. Des séquences ayant une structure ou des propriétés physico-chimiques proches des protéines à ancre GPI et étant de potentiel faux positif: L’hydrophobicité des protéines GPI est grande; des protéines ayant aussi cette particularité sont de bonnes candidates pour tester la capacité de discrimination de notre classificateur (182 séquences transmembranaires et 83 séquences de protéines de transport).
3. Des séquences générées aléatoirement : Ces séquences vont mesurer la
probabilité de classifier une séquence dans la classe GPI par simple effet du hasard (2445 séquences).
Ces tests nous donnerons une meilleure précision quant à l’évaluation de la capacité de
généralisation du modèle.
Architecture
Le seul paramètre des neurones de la couche d’entrée est leur nombre, car aucun traitement
n’y est effectué. La couche de sortie est composée d’un seul neurone, car la tâche de
classification est binaire : GPI ou non-GPI. Ce neurone a une fonction d’activation sigmoïde.
Cette fonction s’avère un choix excellent dans les cas de réseau à sortie binaire (0/1), selon
Jordan (1995). Comme les valeurs de sorties sont des nombres réels dans l’intervalle [0,1] il
faut décider d’un seuil d’acceptation de la classification. Si le seuil se retrouve plus près de 1,
notre classification sera moins sensible et plus spécifique à la classe. Si le seuil est plus près
de 0.5, la classification sera beaucoup plus sensible ou permissive ce qui entraîne une plus
forte chance d’accepter des données n’ayant pas toutes les caractéristiques recherchées.
Une seule couche cachée compose notre modèle. Le nombre de neurones composant la
couche caché a été déterminé par essai/erreur. Nous avons construit six architectures
différentes composées de 100 neurones d’entrée, d’un neurone de sortie et d’un nombre varié
de neurones dans la couche cachée (tableau 5.2). Le test de précision comportant 134
séquences GPI et 134 séquences non-GPI a été présenté à chaque modèle. Toutefois, les
résultats démontrent que le nombre de neurones de cette couche ne change pas grandement
les résultats des tests.

97
Tableau 5. 2 Tests de sélection du nombre de neurones de la couche cachée. x est la valeur du neurone de sortie
Nombre de
neurones
GPI prédits 90.0>x
non-GPI prédits 90.0<x
25 90.2% 96.2% 50 89.5% 96.2% 100 89.5% 96.2% 150 89.5% 97.0% 200 89.5% 97.0% 250 89.5% 97.0%
La structure de la couche cachée sélectionnée est celle composée de 150 neurones. Cette
structure a été préférée à celle composée de seulement 25 neurones, car plusieurs essais des
deux modèles montraient une plus grande stabilité pour le modèle ayant une couche cachée
de 150 neurones. Comme pour le neurone de la couche de sortie, la fonction d’activation de
ces neurones est une sigmoïde. L’architecture finale est donc une couche d’entrée de 100
neurones, une couche cachée de 150 neurones et une couche de sortie de 1 neurone (fig. 5.5).
entrée 100cachée 150
sortie 1
a
f
k
89.09
165.19
0.616
0.283
Figure 5. 5 Architecture du perceptron multicouche construit pour la classification de protéines à ancre GPI.

98
L’apprentissage
L’apprentissage du modèle est de type RPROP (Resilient Back Propagation). Le processus
d’apprentissage consiste à ajuster graduellement le poids des connexions, en vue d’atteindre
un score optimal pour les séquences d’entraînement ayant une ancre GPI. De nombreux
paramètres sont nécessaires pour l’apprentissage du modèle. Pour déterminer les paramètres
optimaux, des tests furent effectués en utilisant JavaNNS (tableau 5.3).
Tableau 5. 3 Tests d’ajustement des paramètres de l’apprentissage RPROP. Le symbol ijΔ représente la valeur de mise à jour des poids des connexions, maxΔ représente la valeur de mise à jour maximale,α représente la valeur de déclin des poids et x est la valeur du neurone de sortie.
ijΔ / maxΔ /α GPI
prédits 90.0>x
non-GPI prédits
90.0<x 0.1 / 50 / 5.0 89.5% 97.0% 1.0/ 50 / 5.0 89.5% 97.0% 2.0 / 50 / 5.0 88.8% 97.0%
1.0 / 10 / 5.0 88.8% 97.0% 1.0 / 50 / 5.0 89.5% 97.0%
1.0 / 100 / 5.0 88.8% 97.7%
1.0 / 50 / 0.1 66.4% 99.2% 1.0 / 50 / 1.0 84.3% 98.5% 1.0 / 50 / 5.0 89.5% 97.0%
1.0 / 50 / 10.0 89.5% 97.0% 1.0/50/50.0 100.0% 0.0%
Les paramètres de l’algorithme d’apprentissage RPROP :
1. ijΔ est la valeur d’incrément du poids ijw . Cette mise à jour sera négative ou positive selon le sens de la dérivée de l’erreur. La valeur initiale de ce paramètre n’influence pas vraiment l’apprentissage, car il s’adapte durant le processus d’apprentissage.
2. maxΔ est la valeur maximale atteignable par les valeurs de mise à jour.
Cette limite est importante pour empêcher les valeurs de mise à jour d’atteindre de trop fortes valeurs.

99
3. α est la constante correspondant au taux de déclin de la pénalité ajoutée à l’erreur. Elle correspond à l’influence de la taille des poids sur la fonction d’erreur.
Pour nous assurer que les valeurs initiales de ijΔ n’influençait pas l’apprentissage, nous
avons effectué trois tests avec des valeurs de 0.1, 1.0 et 2.0. Comme prévu, une modification
de cette valeur n’est pas critique. Nous l’avons donc mise à une valeur intermédiaire de 1.0.
La valeur maximale atteignable pour la mise à jour maxΔ n’influence pas beaucoup la
généralisation du modèle. Nous l’avons donc mise à la valeur de défaut dans javaNNS, c’est-
à-dire à 50.
Pour la valeur deα , des tests de 0.1, 1.0, 5.0, 10.0 et 50 ont été effectués. Le résultat de ces
tests démontre qu’une valeur de α près de 0 diminue, tel que prévu, le pouvoir de
généralisation du modèle en diminuant l’effet de pénalisation des poids trop grands. Le
tableau 5.3 montre que, pour une valeur α de 0.1, seulement 66.4% séquences GPI ont été
reconnues, comparativement à 89.5% pour une valeur plus élevée, telle que 5.0 ou 10.0. Nous
l’avons donc mise à 5.0. Il est intéressant de noter que pour des valeurs extrème, tel
que 50=α , l’influence de cette pénalisation des poids devient néfaste au modèle.
Les paramètres optimaux pour notre modèle sont donc :
1. Algorithme d’apprentissage : RPROP : = 1.0. max = 50.0. = 5.0
2. La fonction d’initialisation des poids est de type aléatoire, dans l’intervalle de [–1, 1] (Randomize-Weight, dans javaNNS)
3. La fonction de mise à jour de l’activation des neurones est déterminée selon un ordre
topologique, c’est-à-dire qu’elle suit la topologie du réseau (la première couche de neurones traitée est la couche d’entrée, la seconde est celle cachée, tandis que la dernière est la couche de sortie).

100
Pour empêcher la mémorisation qui diminue la capacité de généralisation nous avons utilisé
un jeu de données de validation. Grâce au graphe d’erreur nous avons pu estimer le nombre
de tour maximum permettant une capacité de généralisation optimale. Comme on peut le voir
dans la figure 5.6, après 300 cycles d’entraînement l’erreur obtenue sur les séquences de
validation augmente ce qui indique un début de mémorisation.
80
75
70
65
60
55
50
54.5
4
3.5
0
0.005
0.01
0.015
0.02
0.025
0.03
0.035
0.04
0.045
0.05
0.055
25050 200100 150 400300 350 450 500
entraînementvalidation
erreu
r2
nombre cycle d'entraînement
Figure 5. 6 Progression de l’erreur lors de la validation. La courbe rouge représente l’erreur obtenue pour le groupe de séquences de validation. La courbe verte représente l’erreur obtenue dans le groupe d’apprentissage.
Le seuil d’acceptation d’une prédiction
L’Appendice D montre une analyse ROC effectuée sur les résultats du test de validation du
réseau. Une analyse ROC permet de choisir le seuil d’acceptation d’un classement/prédiction
ou, plus spécifiquement, le meilleur ratio spécificité (capacité de discrimination de séquences
non-GPI) vs sensibilité (capacité de reconnaître le signal GPI). Comme une augmentation de
la sensibilité va toujours de pair avec une diminution de la spécificité, il est important de bien
choisir le ratio qui maximisera le pouvoir prédictif de notre modèle. Dans notre cas, un seuil

101
de 0.90 a été jugé optimal. Dans la recherche de nouvelles protéines à ancre GPI, toute
protéine qui aura une valeur de sortie du réseau égale ou supérieure à 0.90 sera prise en
considération comme étant potentiellement une protéine à ancre GPI. Dans la figure 5.7, on
peut voir un graphique représentant l’analyse ROC. L’aire sous la courbe permet de qualifier
la capacité de prédiction du modèle. Dans le cas de notre réseau, l’aire est de 0.958, ce qui est
satisfaisant.
Figure 5. 7 Courbe ROC pour le test de validation du réseau de neurones.
5.3 Résultats des tests et discussion
Un test de validation a été effectué avec le jeu de données test de 134 séquences GPI et de
134 séquences non-GPI (tableau 5.4). Ce test montre que, pour un seuil de 0.90, le modèle a
une précision de 0.93 et un bon coefficient de corrélation (0.875), ce qui implique que les
prédictions de ce modèle sont précises. Ces résultats, en plus des résultats du calcul de l’aire
sous la courbe ROC, sont de bons indices que notre modèle a une précision acceptable et une
capacité de généraliser intéressante. Toutefois, comme spécifié en section 5.2.1,
paragraphe « jeux de données », ce test de validation est peu représentatif de la diversité des
0.0 0.2 0.4 0.6 0.8 1.0
1 - spécificité
0.0
0.2
0.4
0.6
0.8
1.0
Sens
bilit
é
Courbe ROC

102
séquences présentes dans les bases de données publiques. Nous avons donc effectué des tests
supplémentaires (tableau 5.5). Ces tests montrent que le modèle a une spécificité moyenne de
0.9525, ce qui signifie que 4.75% des prédictions sont de faux positifs.
Tableau 5. 4 Résultats du test de validation du réseau de neurones artificiels.
Test de validation Précision
TotalTNTP +
= 0.93
Sensibilité
FNTPTP+
= 0.90
Spécificité
FPTNTN+
= 0.97
Valeur de prédiction positive FPTP
TP+
= 0.97
Valeur de prédiction négative FNTN
TN+
= 0.90
Coefficient de corrélation ( )( ) ( ) ( ) ( )TPFNFNTNTNFPFPTP
FNFPTNTP+×+×+×+
×−× = 0.87
Tableau 5. 5 Résultats des tests supplémentaires.
Test de spécificité : Séquences non-GPI
Cytoplasmique et nucléaire 0.98
Transmembrane 0.94
Transport 0.93
Aléatoire 0.96
Test de sensibilité : Séquences GPI
Séquences GPI Swiss-Prot 0.93
Pour la sensibilité, ou la capacité de reconnaître de vraies protéines à ancre GPI, le modèle
offre un pouvoir prédictif de 0.925, ce qui signifie que seulement 7.5% des séquences GPI

103
présentes dans Swiss-Prot ne peuvent être détectées par le modèle. Il faut noter que, vu le
faible nombre de séquences de protéines à ancre GPI présentes dans les bases de données,
nous avons utilisé toutes les séquences GPI de Swiss-Prot, sans éliminer les redondances
possibles. Il faut donc considérer la sensibilité comme étant celle du modèle de réseau de
neurones pour détecter les séquences GPI de la base de données Swiss-Prot et non comme
une sensibilité réelle. Cette sensibilité réelle se situe possiblement plus proche du 0.90 obtenu
dans notre test de validation effectué avec les séquences GPI non redondantes. Il est
important de noter que, de ces 7.5% de séquences GPI rejetées, 7% avaient une queue
C-terminale tronquée, d’où un signal GPI incomplet. Cette particularité amène un biais dans
la prédiction. De plus, 23% de ces 7.5% des séquences GPI rejetées proviennent toutes de la
même protéine, LIPL, une lipoprotéine lipase. Le modèle attribue à ces lipoprotéines lipases
un score très faible (en moyenne 0.00003). Cette observation nous porte à penser que ces
séquences sont possiblement incorrectement annotées comme séquences à ancre GPI.
Analyse à grande échelle
Nous avons effectué une analyse à grande échelle des protéines présentement séquencées
pour deux génomes, Arabidopsis thaliana (28 860 séquences) (The Arabidopsis Genome
Initiative, 2000) et Oryza sativa (28 519 séquences) (GRAMENE, 2004). Le tableau 5.6
donne les résultats obtenus pour ces deux bases de données.
Tableau 5. 6 Résultats du test de prédiction à grande échelle.
Arabidopsis thaliana Chromosome 1 à 5
Nombre total de séquences 28 860
Séquences potentiellement GPI 1 779
Pourcentage du protéome 6.2%
Oryza sativa
Nombre total de séquences 28 519
Séquences potentiellement GPI 1 575
Pourcentage du protéome 5.5%

104
Comme le montre le tableau 5.6, le pourcentage du protéome de ces deux plantes occupé par
des protéines à ancre GPI est de 6.2% et de 5.5%. Comme le réseau de neurones a un taux de
prédiction fausse d’environ 4.75%, on peut en déduire qu’une grande partie de ces
prédictions sont des faux positifs. Toutefois, notre analyse offre un bon moyen de nettoyer les
bases de données en réduisant le nombre de séquences à investiguer de près de 95%, ce qui
diminue considérablement l’espace de recherche pour une analyse en laboratoire.
Temps de traitement
La vitesse de traitement des réseaux de neurones présente un grand avantage pour la fouille
de grandes quantités de séquences biologiques. Notre modèle a effectivement bien performé
du point de vue temps de calcul. Par exemple, pour le traitement de 15 733 séquences
protéiques, il a fallu seulement 8 secondes à un Pentium 4 cadencé à 2.8 Ghz de mémoire.
5.4 Conclusion
Le modèle de réseau de neurones que nous avons construit offre une très bonne capacité de
classification/prédiction des séquences à ancre GPI, avec plus de 90% de bonnes
classifications et avec un faible temps de traitement. Toutefois, le taux de faux positifs reste
un peu élevé, avec 4.75% de fausse prédiction. Le test effectué sur des séquences aléatoires
montre effectivement 4% de prédiction au hasard. L’analyse à grande échelle de deux
protéomes montre entre 5% et 6% de séquences potentiellement GPI. Ce taux est très élevé
reflétant encore le pourcentage de faux positif trop élevé. De plus, une faiblesse du modèle
est qu’il ne donne aucune indication sur la structure du signal.
Il devient donc intéressant de se tourner vers les modèles de Markov cachés et leur capacité
d’analyse structurée des séquences. La section 4.3 détaille un modèle de Markov caché
construit pour la tâche de classification/prédiction du signal GPI.

CHAPITRE VI
CLASSIFICATION/PRÉDICTION D’ANCRE GPI PAR MODÈLE DE MARKOV CACHÉ
Ce chapitre présente le deuxième volet de la méthode hybride. On y propose un modèle de HMM pour faire la classification/prédiction du signal GPI, chez les séquences protéiques. La conception du modèle de HMM a été faite en collaboration avec Cedric Chauve et Anne Bergeron. L’implantation du modèle a été faite par Cedric Chauve. Cette section montre la puissance du HMM pour structurer le signal GPI, mais montre une moins bonne capacité de reconnaissance du signal. Cette section propose également une grammaire associée au signal.
6.1 Un modèle de Markov caché classificateur d’ancre GPI
Le signal GPI peut être vu tel une suite de mots caractéristiques dans une séquence de lettres.
Cette définition permet de dire qu’une structure linguistique existe. Cette « nature »
linguistique suggère l’utilisation des grammaires, telles que les grammaires régulières
stochastiques (HMM). Les HMM permettent l’extraction des connaissances que les
séquences contiennent sous forme de motifs ou signaux. Grâce à un « langage cellulaire »,
ces mots nous renseignent sur le rôle joué par ces séquences dans le fonctionnement de la
cellule. La définition d’une grammaire représentant le signal pourra sûrement aider à
l’établissement d’un langage cellulaire global plus précis qui, dans l’avenir, servira à mieux
comprendre le fonctionnement de tout être vivant.
La construction d’un modèle de Markov caché représentant le signal GPI offre la possibilité
de structurer ce signal. La section suivante décrit le modèle de Markov rattaché au signal GPI
ainsi que la grammaire régulière stochastique qui en découle. Le modèle est basé sur la

106
connaissance existante de la structure du signal. Cette structuration est remise en cause car,
dans l’étude de la grammaire GPI, on verra, qu’après entraînement, le modèle propose une
plus grande flexibilité dans la structure de son signal.
6.2 Le modèle
Les jeux de données
Plusieurs jeux de données ont été construits à partir des séquences de protéines de la base de
données Swiss-Prot :
Le jeu de données d’entraînement : Le groupe de séquences d’entraînement de notre modèle
de HMM est composé de 87 séquences GPI de la base de données Swiss-Prot, sélectionnées
pour la qualité de leur annotation (Appendice E). Une attention particulière a été portée à
l’annotation du site d’ancrage, car le HMM doit pouvoir prédire correctement cet
emplacement. Comme pour le réseau de neurones artificiels nous n’avons pas sélectionné des
séquences ayant une forte similarité entre elles pour empêcher un biais en leur faveur.
Le jeu de données pour la validation du modèle : Le jeu de séquences construit pour la
validation du modèle est différent de celui du réseau de neurones, car les séquences
sélectionnées pour l’entraînement sont différentes et la présence du site d’encrage doit
apparaître dans l’annotation. Ce jeu de test est composé de 66 séquences GPI (ayant un site
d’ancrage annoté) ne se retrouvant pas dans le jeu d’entraînement et non redondantes, et de
66 non-GPI.
Les jeux de test supplémentaires : Pour les mêmes raisons que pour le réseau de neurones
artificiels, nous avons utilisé des jeux de tests supplémentaires pour augmenter la qualité de
la validation du modèle. Les jeux de test supplmentaires construits pour le réseau de neurones
artificiels ont été utilisés (voir sect. 5.2.1)

107
Architecture
Pour la construction du modèle initial, nous avons utilisé une analyse des 210 séquences GPI
ayant un site d’ancrage annoté dans la base de données Swiss-Prot (fig. 6.1) ainsi que la
connaissance déjà documentée sur la structure du signal. Pour chaque séquence GPI de la
base de données Swiss-Prot, le signal GPI a été pris en note et utilisé dans la construction du
modèle. L’utilisation de cette connaissance permet l’initialisation de l’apprentissage du
modèle près de l’optimal. De la sorte, l’apprentissage se voit plus rapide et une meilleure
solution est envisageable (Baldi et Brunak, 2000).
Notre méthodologie se base sur des travaux similaires effectués en analyse de séquences
biologiques (Nielsen et Krogh, 1998). Il est à noter que la structure de départ du HMM avant
apprentissage n'est pas aléatoire, elle représente une approximation des connaissances
actuelles de la structure des séquences à ancre GPI. En d'autres termes, cette structure de
HMM peut être vue comme un résultat d'apprentissage effectué à partir d’un HMM neutre
ayant un très grand jeu de données composé d’ancres GPI aléatoires, dans un modèle
combinatoire correspondant à nos connaissances actuelles des ancres GPI. Ceci permet
d'expliquer, et justifier une phase d'apprentissage n'utilisant qu'un nombre très réduit de
séquences par rapport à la taille du modèle. Cette phase d'apprentissage doit être vue comme
une phase de raffinement du modèle d’ancres GPI aléatoires (le modèle avant apprentissage).
Cet apprentissage permettra la prise en compte des caractéristiques des séquences ayant
échappées à la modélisation combinatoire des séquences d’ancres GPI ayant servi de base à
la construction du modèle initial. Il est important de noter que le processus stochastique
générant les séquences biologiques n’est pas connu, on ne peut donc faire de justification
théorique de la méthodologie utilisée.

108
> 5NTD_BOOMI 25 nqa vmkymnstspittaldgrvtflktnqasdaclnlaspflvllvlvvfyhl
> CD24_RAT 27 ggg nqtsvapfpgnqnisaspnpsnattrgggsslqstagllalslsllhlyc
> CONT_CHICK 24 sga geyvvevrahseggdgevaqikisgatagvptlllglvlpalgvlaysgf
> NTRI_HUMAN 27 ngtcvasnklghtnasimlfgpgavsevsngtsrragcvwllpllvlhlllkf
> VSAC_TRYBB 27 dss ttdkckdktkdeckspnckwegetckdssilvtkkfalslvsaafasllf
Figure 6. 1 Structure de séquences GPI avec annotation du site d’ancrage en rouge.
Comme spécifié dans la section 2.2.2, la structure du signal GPI documentée dans la
littérature se résume ainsi ( w représente la position du site d’ancrage) (Eisenhaber, Bork et
Eisenhaber, 1998) :
1. Une région de liaison polaire et flexible d’environ 11 acides aminés ( 11−w … 1−w ) n’ayant pas de structuration secondaire intrinsèque.
2. Une région de résidus de faible poids moléculaire comprenant le site d’ancrage w : ( w … 2+w ).
3. Une région intermédiaire ( 3+w … 9+w ) comportant des résidus
modérément polaires.
4. Une queue commençant avec le résidu 9+w ou 10+w jusqu'à la fin et ayant une hydrophobicité suffisante.
Selon la littérature, le signal débuterait en position 11−w . Toutefois, notre modèle débute à la
position du site d’ancrage w car une trop grande variabilité survenant dans la région
précédente diminue la capacité de classification (fig. 6.2).

109
Début
Fin
afpghlllin....k
1 23
zone intermédiaire
queue hydrophobe
4 13i 1211 14 1516
17j41 40 39 38 37 36 35
site d'ancrage
Figure 6. 2 Modèle de Markov caché représentant le signal GPI.
Le modèle de Markov caché se résume ainsi :
1. Les trois premiers états correspondent aux petits résidus débutant par le site d’ancrage w , 1+w , 2+w .
2. Par la suite, une transition vers les 10 prochains états est possible (4 à 13). Cette section correspond au début de la partie intermédiaire. Chaque état situé dans cette partie a une probabilité de transition vers les autres états. L’état 13 représente le début d’une zone linéaire de transition composée de 3 états (14 à 16). Cette zone ferme la partie intermédiaire du signal et peut ainsi être composée de 4 à 13 acides aminés.
3. Finalement, à partir de l’état 16, des transitions sont possibles pour 20 états (17 à 36) représentant le début de la partie hydrophobe du signal. Chacun de ces 20 états a une probabilité de transition vers les autres états. L’état 36 correspond au début d’une autre zone linéaire de 5 états (37 à 41) composant la fin de la zone hydrophobe. Cette zone hydrophobe peut donc être composée de 6 à 35 acides aminés.
4. L’état final est un état particulier qui n’émet pas de signal d’acide aminé. Cet état
émet le caractère de sortie « $ » avec une probabilité de 100%.
Chaque séquence a un caractère « $ » ajouté à la fin. Pour pouvoir terminer, chaque séquence
doit passer par l’état final qui émet ce caractère de sortie. De cette façon, la longueur de la
séquence est prise en compte dans le processus d’affectation du score, ce qui permet de ne
pas biaiser les résultats en faveur des séquences courtes.

110
Les probabilités d’émission de caractères
Les probabilités d’émission des caractères pour chaque état ont été calculées à partir de
l’analyse effectuée sur les séquences de protéines GPI. Le tableau 6.1 montre les probabilités
d’émission de chaque acide aminé pour les trois premiers états.
Tableau 6. 1 Pourcentage d’occupation des acides aminés pour la zone d’ancrage. w =représente le site d’ancrage.
Acide aminé w 1+w 2+w a 0.0523 0.2618 0.3855 c 0.0285 0.0047 0.0047 d 0.0618 0.0237 0.0047 e 3.96637e-05 0.0142 0.0047 f 3.96637e-05 0.0095 0.0095 g 0.1626 0.2903 0.2284 h 3.96637e-05 0.0190 0.0095 i 3.96637e-05 0.0190 0.0190 k 3.96637e-05 0.0047 0.0047 l 3.96637e-05 0.0047 0.0428 m 3.96637e-05 0.0047 0.0095 n 0.2141 0.0047 0.0001 p 3.96637e-05 0.0285 0.0047 q 3.96637e-05 0.0142 0.0142 r 3.96637e-05 0.0237 0.0237 s 0.4666 0.2191 0.1523 t 3.96637e-05 0.0285 0.0713 v 3.96637e-05 0.0237 0.0095 w 3.96637e-05 0.0002 0.0001 y 3.96637e-05 0.0002 0.0001 $ 0 0 0
Une fois ces probabilités calculées, on remarque que, pour certains états, certains acides
aminés ont une probabilité d’émission nulle, ce qui peut être trop discriminant. Ceci n’est pas
étonnant étant donné la possibilité de retrouver peu de séquences dans les groupes
d’entraînements de problèmes biologiques. On remédie à ce problème en utilisant des
pseudo-counts (Baldi et Brunak 2001) dont le principe est le suivant: chaque probabilité
d’émission nulle, pour un acide aminé X en un état Y, est remplacée par une probabilité très
faible calculée en effectuant une moyenne des probabilités d’émission de X sur un ensemble

111
d’états fixés du HMM. Cette modification implique aussi une normalisation des probabilités
d’émission des autres acides aminés en Y, de sorte que la somme des probabilités d’émission
en Y vaut 1.
Pour la zone intermédiaire, la moyenne des probabilités d’émission est calculée sur la zone
complète (états 4 à 16). Le principe est le même pour la zone hydrophobe (voir tableau 6.2).
Par contre, pour la zone initiale, il n’était pas adapté de procéder de même du fait de la très
grande différence entre les trois états (voir tableau 6.1).
Tableau 6. 2 Pourcentage d’occupation des acides aminés pour la zone intermédiaire et la queue hydrophobe.
Acide aminé Zone intermédiaire
Queue hydrophobe
État final
a 0.1095 0.1024 0 c 0.0210 0.0195 0 d 0.0136 0.0037 0 e 0.0136 0.0056 0 f 0.0299 0.0647 0 g 0.0809 0.0436 0 h 0.0387 0.0148 0 i 0.0482 0.0606 0 k 0.0244 0.0079 0 l 0.1217 0.3252 0 m 0.0183 0.0233 0 n 0.0238 0.0056 0 p 0.0891 0.0363 0 q 0.0278 0.0145 0 r 0.0544 0.0183 0 s 0.1299 0.0891 0 t 0.0551 0.0521 0 v 0.0680 0.0790 0 w 0.0197 0.0205 0 y 0.0115 0.0123 0 $ 0 0 1

112
Pour calculer les pseudo-counts, on a donc procédé colonne par colonne, et pour chacune des
trois colonnes, on a, avant de normaliser, asigné à chacun des k acides aminés émis avec
probabilité 0 une probabilité d’émission égale à
( )wk21/1
où w est un poids déterminé axpérimentalement. Le facteur 21 vient du fait qu’il y a 21
émissions possibles, les 20 acides aminés et $.
Probabilités de transition
Les probabilités affectées aux transitions sont inversement proportionnelles au nombre de
transitions pouvant sortir de chaque état. Par exemple, l’état 1 a une seule transition possible,
soit vers l’état 2. La probabilité de cette transition sera de 1. Pour l’état 3, on a 10 transitions
possibles, soit vers les états 4 à 13. La probabilité de ces 10 transitions sera de 0.1 pour
chaque transition. Le modèle de départ avec les probabilités de transition et d’émission se
retrouve dans l’Appendice F.
L’apprentissage
Pour l’apprentissage, un ensemble de séquences d’entraînement composé de 87 séquences
annotées comme ayant une ancre GPI a été constitué. Le processus d’apprentissage a
nécessité 100 itérations de l’algorithme de Baum-Welch.
La validation
Pour valider notre modèle, nous avons effectué 500 expériences de bootstrap non
paramétrique (rééchantillonnage) sur notre ensemble de données d’apprentissage. Pour
chacune des 500 expériences, les 87 séquences du jeu d’entraînement (répétitions permises)
ont servi à construire 500 groupes d’apprentissage. Le HMM a ensuite été entraîné avec ces
nouveaux groupes de données selon la même démarche que pour le modèle initial. Chaque
« nouveau » HMM a par la suite été testé à l’aide du test de validation de 66 séquences.

113
Le seuil d’acceptation d’une prédiction
Le seuil d’acceptation d’un classement/prédiction a été effectué à l’aide d’une analyse ROC
(Appendice G). La figure 6.3 montre une courbe ROC effectuée sur les résultats du test de
validation du HMM. Pour le choix du seuil, nous avons sélectionné 4.06 comme seuil
optimal. Ce seuil nous donne une sensibilité de 0.86 et une spécificité de 0.99.
Pour l’affectation des scores, les sections d’une séquence débutant par au moins trois acides
aminés de faible poids moléculaire et ayant au minimum quinze acides aminés de longueur
ont été présentées au modèle et évaluées selon l’algorithme Forward et le principe du « log-
odd ». Le meilleur score obtenu est gardé comme le score du segment. Par la suite,
l’algorithme Viterbi a été utilisé pour trouver le chemin le plus probable et, ainsi, nous
donner la structure du signal (site d’ancrage). Pour la prédiction du site d’ancrage, les trois
meilleurs scores obtenus pour chaque séquence représentent les sites d’ancrage potentiels.
Figure 6. 3 Courbe ROC pour le test de validation du HMM
0.0 0.2 0.4 0.6 0.8 1.0
1 - Spécificité
0.0
0.2
0.6
0.8
1.0
Sens
ibili
té
Courbe ROC

114
6.3 Résultats des tests et discussion
Un test de validation a été effectué avec le jeu de données de 66 séquences GPI et de 66
séquences non-GPI (tableau 6.3).
Ce test montre que, pour un seuil de 4.06, le modèle a une précision de 0.92 et un bon
coefficient de corrélation (0.82), ce qui implique que les prédictions de ce modèle sont
précises. Le calcul de l’aire sous la courbe ROC donne une aire de 0.959, ce qui est
satisfaisant. Comme pour le réseau de neurones, ces résultats, en plus des résultats du test de
validation, sont de bons indices que notre modèle a une précision acceptable et une capacité
de généraliser intéressante.
Comme le test de validation du HMM ne comporte que 132 (66 GPI et 66 non GPI)
séquences au total, comparativement à 268 (134 GPI et 134 non GPI) séquences pour le
réseau de neurones, nous avons présenté le test de validation du réseau de neurones au HMM.
Toutefois, le résultat de ce test doit être pris avec quelques réserves, puisque certaines
séquences du jeu d’entraînement se retrouvent dans ce test. Selon ce test, la sensibilité du
modèle, à un seuil de 4.06, ne serait que de 0.77 et la spécificité serait de 0.99. Ces deux tests
de validation nous indiquent donc que la sensibilité du HMM se situe entre 0.77 et 0.86 et
que la spécificité reste près de 0.99, comparativement à 0.90 de sensibilité et à 0.97 de
spécificité pour le réseau de neurones.

115
Tableau 6. 3 Résultats du test de validation du HMM.
Test de validation Précision
du testEnsembleTNTP + = 0.92
Sensibilité
FNTPTP+
= 0.86
Spécificité
FPTNTN+
= 0.99
Valeur de prédiction positive FPTP
TP+
= 0.98
Valeur de prédiction négative FNTN
TN+
= 0.87
Coefficient de corrélation ( )( ) ( ) ( ) ( )TPFNFNTNTNFPFPTP
FNFPTNTP+×+×+×+
×−× = 0.82
À la lumière de ces résultats, il devient important de présenter nos jeux de test
supplémentaires au HMM. Le tableau 6.4 montre la sensibilité de notre modèle envers les
séquences GPI et sa spécificité envers des séquences non GPI.
Ces tests supplémentaires montrent que la sensibilité du HMM est d’environ 0.80 et la
spécificité, elle, se situe à 0.99. Si on compare ces résultats avec le réseau de neurones
artificiels, le HMM est moins sensible que le réseau de neurones, mais sa spécificité est
supérieure, avoisinant le 0.99. Comme pour le réseau de neurones artificiels, le HMM a
rejeté les protéines LIPL avec des valeurs ayant une moyenne de -18.00, ce qui est largement
en dessous du seuil d’acceptation du HMM. Nous ne pouvons que réaffirmer que, d’après nos
modèles, la protéine LIPL est probablement mal annotée comme étant une protéine à ancre
GPI.

116
Tableau 6. 4 Résultats des jeux de test supplémentaires.
Test de spécificité : Séquences non-GPI
Cytoplasmique et nucléaire 1.00
Trasnsmembrane 0.98
Transport 0.98
Aléatoire 1.00
Test de sensibilité : Séquences GPI
Séquences GPI Swiss-Prot 0.80
Temps de traitement
Les HMM sont efficaces pour la fouille de bases de données biologiques. Toutefois, dans
notre étude, le temps de traitement est beaucoup plus grand que celle du réseau de neurones
artificiels. Notre modèle a effectivement pris un temps de traitement 10 fois supérieur à celle
du réseau de neurones. Par exemple, pour le traitement de 1 575 séquences protéiques, il a
fallu 63 secondes à un Pentium 4, 2.8 Ghz.
La prédiction du site d’ancrage
Grâce à l’algorithme Viterbi, nous avons pu proposer des prédictions structurées. Pour
chaque prédiction, nous gardons les trois meilleurs chemins retracés par l’algorithme Viterbi
(fig. 6.4).
> PRIO_HUMAN12.2049 *** 27 | [ssm] vlfssppvillisfliflivg> PRIO_HUMAN11.7437 *** 26 | [gss] mvlfssppvillisfliflivg> PRIO_HUMAN 9.1819 *** 28 | [smv] lfssppvillisfliflivg
Figure 6. 4 Les trois meilleurs chemins de la séquence PRIO_HUMAN. Le site d’ancrage et les deux acides aminés suivant ( w , 1−w et 2+w ) sont représentés entre crochets. Le score obtenu pour chacun de ces chemins est représenté en gras.

117
Pour un groupe de séquences de la base de données Swiss-Prot ayant une annotation du site
d’ancrage (300 séquences), nous avons pu prédire correctement 78% des sites d’ancrages. Ce
résultat semble possiblement faible. Toutefois, 58% des sites d’ancrage manqués étaient à
moins de trois acides aminés du site annoté dans Swiss-Prot. Il faut aussi noter que beaucoup
des séquences annotées dans Swiss-Prot proviennent de prédictions. Les sites d’ancrage de
ces prédictions ne sont pas démontrés en laboratoire, donc possiblement erronés. Cette
mesure de la précision du site d’ancrage reste donc une estimation minimale de la capacité de
prédiction du HMM.
6.4 Une grammaire GPI
La construction d’un HMM représentant le signal présent chez les protéines à ancre GPI nous
offre une opportunité très intéressante : la définition d’une grammaire stochastique du signal.
Cette grammaire pourra, par la suite, servir comme source d’information sur la structure du
signal, en définissant les caractéristiques propres au signal GPI. La grammaire présentée en
Appendice H nous permet de définir une structure du signal un peu différente de la structure
proposée dans la littérature jusqu’à présent.
6.4.1 La structure du signal
La zone du site d’ancrage
Selon Eisenhaber, Bork et Eisenhaber (1998), la composition en acides aminés de la position
du site d’ancrage w serait majoritairement des s (46% en moyenne). Cette position
comporterait également les acides aminés n, d, a, g et c. Notre grammaire stochastique
représentant le signal GPI va dans le même sens. Le tableau 6.5 montre la composition en
acides aminés pour la position du site d’ancragew .

118
Tableau 6. 5 Acides aminés pour le site d’ancrage selon notre méthode hybride
acide aminé %
s 40 n 25 g 16 a 6 c 3 b 1
Pour la position 1+w , Eisenhaber la considère similaire à la position w . Dans notre cas, nous
voyons une certaine similitude mais la dominance des acides aminés s, n, d, a, g, et c n’est
pas aussi évidente. On voit que l’acide aminé g est le plus représenté avec 31% d’occurrence.
Les acides aminés s et a suivent avec respectivement 22% et 20%. Les seuls absents sont c, k,
r, w, et y. Les autres acides aminés sont faiblement représentés mais ont une probabilité
d’être émis dans certains cas.
Pour la position 2+w , Eisenhaber note une dominance de s, a et g dans la grande majorité
des cas (94% pour les protozoaires et 70% pour les métazoaires). Selon notre modèle, ces
acides aminés dominent aussi avec un total de 60% de probabilité d’émission pour les trois
acides aminés. Cependant ce plus faible pourcentage laisse la place à d’autres acides aminés
tels que l et t qui sont représentés avec une probabilité de 10%. Les seuls acides aminés
absents sont d, e, f, h, p, w et y.
Comme il a été spécifié dans le chapitre 2 section 2.2.2, la zone d’ancrage est caractérisée par
des acides aminés de faible poids moléculaire. La dominance des acides aminés de faible
poids a, s et g et l’absence ou la très faible probabilité d’occurrence des acides aminés de fort
poids moléculaire w, r, y et k prouve que le poids moléculaire est une caractéristique très
importante dans cette partie du signal GPI.
La zone hydrophile
Dans cette zone nous allons nous concentrer sur la longueur. Les chemins les plus probables
dans cette zone ont entre 7 et 12 acides aminés. La structure proposée par Eisenhaber (1998)

119
parle d’une zone de 7 acides aminés. Toutefois, on retrouve dans certains cas des chemins
ayant 6 acides aminés ou 13 acides aminés. Cette zone est principalement composée d’acides
aminés hydrophiles tel s, r, g, v et p.
La zone hydrophobe
Comme pour la région hydrophile nous avons porté notre attention sur la longueur de cette
zone et sur l’hydropathie des acides aminés qui la composent. Les chemins les plus probables
couvrent entre 19 et 25 acides aminés. Un fait intéressant est la présence d’une probabilité
faible, mais significative, de chemins n’ayant que 11 acides aminés. La nature des acides
aminés composant un chemin n’ayant que 11 acides aminés révèle la présence d’acides
aminés ayant une très forte hydropathie l et i.
Dans des travaux futurs, il serait intéressant de proposer les grammaires représentant
différents signaux protéiques. La construction d’une banque de ces grammaires pourrait
servir dans différentes analyses grammaticales telles que la recherche de motif dans les bases
de données de séquences et comme base de compréhension du langage cellulaire. L’étude des
grammaires de signaux ainsi que leurs comparaisons pourrait révéler de nouvelles
connaissances relatives aux signaux protéiques.
6.5 Conclusion
Notre modèle de HMM est plus spécifique que le modèle de réseau de neurones avec 0.99 de
spécificité, comparativement à 0.97 pour le réseau de neurones. De plus, le HMM nous offre
un grand avantage par rapport au réseau de neurones, soit celui de prédire un site d’ancrage
potentiel. Toutefois, le réseau de neurones offre une plus grande sensibilité, ou capacité de
détecter le signal GPI, que le HMM, avec un résultat de 0.90 par rapport à 0.80. Finalement
le HMM permet de définir une grammaire du signal qui nous offre beaucoup d’information
quant à la composition en acides aminés et la structure du signal GPI.

120
La grande sensibilité du réseau de neurones couplée avec une vitesse d’exécution 10 fois
supérieure au HMM suggère l’utilisation du réseau de neurones dans la tâche de nettoyage
priliminaire des données. La section suivante propose une méthode hybride utilisant le réseau
de neurones pour sélectionner les candidats potentiels à une ancre GPI et le HMM pour la
structuration du signal. Nous allons aussi introduire une annotation qualitative des résultats
de classification du réseau de neurones en utilisant une échelle de qualité basée sur les
résultats du HMM.

CHAPITRE VII
CLASSIFICATION/PRÉDICTION D’ANCRE GPI PAR MÉTHODE HYBRIDE RÉSEAU DE NEURONES ARTIFICIELS ET MODÈLE DE
MAKKOV CACHÉ Ce chapitre propose le volet final de la description de la méthode hybride Dans cette section, l’emphase est mise sur l’utilisation du HMM comme outil d’annotation des résultats de classification du réseau de neurones. Nous verrons que la méthode hybride est plus performante que les deux modèles utilisés indépendamment et que l’information obtenue est beaucoup plus complète.
7.1 Méthode de classification/prédiction hybride
Comme notre solution potentielle au problème de classification de signaux protéiques
implique deux méthodes d’apprentissage, il est intéressant de voir les différentes applications
de leur hybridation. Depuis plus de 10 ans, les hybrides réseau de neurones/HMM sont assez
communs dans la littérature scientifique. Ils sont surtout présents dans des domaines comme
la reconnaissance de l’écriture manuscrite (Bengio et al., 1995 ; Morita et al., 2003) et dans
celui de la reconnaissance de la parole (Rigoll et Willett, 1998). Cette hybridation n’est pas
étonnante, car, dans plusieurs cas, il est intéressant de pouvoir combiner le pouvoir
discriminant des réseaux de neurones avec la capacité de modéliser des séquences des HMM

122
L’hybridation est utilisée pour différentes raisons et sous différentes formes. Dans quelques
architectures hybrides, les deux modèles sont inséparables. Dans ces cas, le réseau de
neurones est utilisé pour paramétrer et moduler le HMM. Dans ces architectures,
l’apprentissage des deux modèles est unifié (Baldi et Chauvin, 1996). Un autre exemple
d’unification est l’utilisation du réseau de neurones pour l’estimation des probabilités à priori
des transitions d’états du HMM (Boulard, 1995). En bioinformatique, des travaux comme
ceux de Martelli et ses collaborateurs (Martelli et al., 2002) utilisent ce type de combinaison.
Dans d’autres architectures, les deux modèles sont entraînés séparément. Par exemple, le
réseau de neurones peut être utilisé en aval des HMM pour classifier les patrons de
probabilités d’une séquence d’évènements produits par plusieurs HMM (Cho et Kim, 1995).
Dans d’autres cas, le réseau de neurones se retrouve en amont et sert, par exemple, à calculer
les probabilités à posteriori d’appartenance à une classe. Ces cas sont des architectures
hybrides de réseaux de neurones artificiels et de HMM où le réseau de neurones est, soit en
amont, soit en aval, ou encore incorporé au HMM. Toutefois, on peut également voir
l’hybridation au point de vue de la méthode d’analyse.
L’hybridation est souvent associée à l’utilisation des réseaux de neurones artificiels ou à des
HMM pour faciliter la construction ou l’apprentissage de l’autre méthode. Cependant, cette
hybridation peut également être vue comme une combinaison des deux méthodes en tirant
avantage des forces de chacune. Le réseau de neurones et sa capacité d’avoir une vue
générale de la séquence peut servir comme prétraitement d’une séquence, tandis que le HMM
et sa capacité de structuration locale de l’information permet de raffiner la prédiction. On
pourrait ici parler plutôt de méthode hybride ou d’hybridation de méthodes, au lieu
d’architecture hybride, car l’hybridation est dans la cascade d’analyse des données, c’est-à-
dire dans l’utilisation combinée de méthodes différentes. Des travaux, tels que ceux de
Nielsen et Krogh (1998), sont un bon exemple d’utilisation combinée des HMM et des
réseaux de neurones artificiels. Ces chercheurs montrent que la classification/prédiction de
signal en partie N-terminale de protéines à l’aide d’un réseau de neurones artificiels est
raffinée par l’ajout d’un HMM permettant la discrimination entre un signal clivé et un signal
d’ancre non clivé.

123
Pour notre projet, nous avons opté pour l’hybridation de méthodes, et non pour une
architecture hybride. Un réseau de neurones artificiels débute la cascade d’analyse qui se
termine par un traitement effectué avec un HMM.
7.2 La méthode
Tel que vu dans le chapitre 5, notre modèle de réseau de neurones artificiels possède une
grande capacité de reconnaissance du signal GPI dans son ensemble. Il est utile pour faire un
premier tri en éliminant les séquences ayant une très faible probabilité d’être des protéines à
ancre GPI. De plus, sa grande vitesse de traitement suggère également son utilisation pour
diminuer l’espace de recherche. Par la suite, les candidats sélectionnés seront présentés au
HMM préalablement entraîné à reconnaître le signal GPI. Les scores octroyés par le HMM
serviront ensuite à qualifier la classification et à structurer le signal en identifiant la zone
d’ancrage potentielle. La figure 7.1 schématise la méthode d’analyse hybride. Comme on
peut le remarquer, l’analyse se fait en deux étapes distinctes : le nettoyage et le raffinage.
>sØquence:aacavafatyilopw
EncodageNettoyage: RØseau de
neurones artificiel
sØquences potentiellement
classe "ancre GPI"
Structure :ModŁle de Markov cachØ
>0.90
<0.90
SØquencesRejetØes
PrØdictionsstructurØes
Figure 7. 1 Méthode d’analyse hybride.

124
Le score HMM obtenu pour chaque candidat sert à qualifier la prédiction selon une échelle de
probabilité. Cette échelle s’échelonne de la classe hautement probable jusqu’à la classe
potentielle fausse positive (tableau 7.1). Les classes sont définies à l’aide d’une analyse ROC
portant sur un jeu de tests de 268 séquences (Appendice I). Ce test a été sélectionné car il
contient plusieurs exemples de séquences, ce qui nous donnera une meilleure évaluation des
classes d’annotation.
Tableau 7. 1 Annotation hybride.
Catégorie Classe Score HMM
hautement probable (classe 1)
score > 5.4
très probable (classe 2)
2.2 < score < 5.39
PROBABLE
faiblement probable (classe 3)
0.2 < score < 2.19
POTENTIELLE fausse positive potentielle (classe 4)
score < 0.19
Dans ce tableau, les classes hautement probable, très probable et faiblement probable
peuvent être regroupées sous la catégorie « probable », tandis que la classe fausse positive
potentielle peut être isolée sous la catégorie « potentielle ».
Ce type d’annotation nous permet de garder la forte sensibilité obtenue grâce au réseau de
neurones artificiels et de profiter de la structuration et de la spécificité du HMM. La
particularité de ce type d’annotation donne une grande flexibilité, laissant à l’utilisateur la
décision de privilégier la spécificité du HMM ou la sensibilité du réseau de neurones
artificiels.

125
7.3 Résultats des tests
Les résultats de la méthode hybride sont, en fait, les résultats obtenus par le réseau de
neurones artificiels sur lesquels nous effectuons une annotation. Les jeux de test
supplémentaires construits pour la validation du réseau de neurones ont été présentés au
HMM et soumis à l’annotation hybride. Le tableau 7.2 montre la proportion des prédictions
du réseau de neurones appartenant à chaque classe.
Tableau 7. 2 Proportion des prédictions du réseau de neurones selon l’annotation hybride. Résultats des tests supplémentaires.
Organisme hautement probable (classe1)
très probable (classe 2)
faiblement probable (classe 3)
fausse positive potentielle (classe 4)
GPI Métazoaire 0.83 0.10 0.03 0.04 Protozoaire 0.60 0.25 0.05 0.10 Champignon 0.67 0.19 0.07 0.07 Plante 0.77 0.13 0.05 0.05
Non-GPI Cytoplas.-nucléaire 0.00 0.00 0.00 1.00 Transmembrane 0.18 0.09 0.18 0.55 Transport 0.17 0.00 0.00 0.83 Aléatoire 0.01 0.02 0.08 0.89
Il y a donc, par exemple, 83% des prédictions métazoaires du réseau de neurones qui
appartiennent à la classe hautement probable et seulement 4% qui pourraient être considérées
comme des fausses positives potentielles. Pour les séquences non-GPI, les prédictions
sélectionnées par le réseau de neurones, dans le jeu de données de séquences cytoplasmiques
et nucléaires, se retrouvent toutes dans la classe des faux positifs. Donc, une annotation ne
considérant pas la classe fausse positive potentielle nous donnerait une spécificité de 100%
avec les séquences cytoplasmiques et nucléaires.
Le tableau 7.3 montre la précision de la méthode selon les différentes classes. Si le but est de
trouver des séquences hautement probables, seulement les prédictions de la classe supérieure

126
seront acceptées. Un seuil d’acceptation des prédictions selon une classe inférieure entraîne
une sélection de séquences GPI moins conforme à la structure de base. Cette augmentation de
la sensibilité entraîne toutefois un plus haut taux de faux positifs. Ce tableau montre la
classification des tests selon la catégorie probable comprenant les prédictions de la classe 1, 2
et 3.
Tableau 7. 3 Comparaison de la précision du système hybride selon la catégorie probable et du réseau de neurones artificiels seul.
Organisme Réseau de
neurones
Catégorie Probable
(classe 1, 2 et 3)
GPI Métazoaire 0.917 0.883 Protozoaire 0.945 0.849 Champignon 0.964 0.893 Plante 0.928 0.881
non-GPI Cytoplasmique-nucléaire
0.02 0.00
Transmembrane 0.06 0.02 Transport 0.07 0.01 Aléatoire 0.04 0.01
Le tableau 7.3 montre également qu’en éliminant la classe fausse positive potentielle (classe
4) nous perdons un peu de sensibilité. En ne conservant que les trois premières classes
(catégorie probable) on se retrouve avec une sensibilité plus basse dans tous les organismes.
Par exemple, pour les métazoaires si on garde toutes les classes (1 à 4), la sensibilité de notre
méthode hybride est identique à celle du réseau de neurone c’est-à-dire de 0.917. Si on
élimine la classe 4 on obtient une sensibilité de 0.883 chez les métazoaires. Toutefois la
spécificité de notre méthode hybride se voit avantagée par l’élimination de la classe 4. Pour
les métazoaires on passe d’une spécificité de 0.04 pour l’ensemble des classes, à une
spécificité de 0.01 pour la catégorie probable.

127
La méthode hybride offre donc une grande flexibilité. Comme le modèle de Markov caché à
été construit en se basant sur la structure typique d’un signal GPI, la classe hautement
probable rassemble les séquences ayant une structure de signal plus conventionnel. Par
contre, des séquences ayant un signal moins précis se voient classifiées dans les classes
inférieures. La présentation des résultats est très informative, laissant à l’utilisateur la
possibilité de choisir le niveau de sensibilité selon le besoin (fig. 4.13). Si, par exemple,
l’intérêt de l’étude est de trouver de nouvelles séquences à ancre GPI ayant un signal moins
conventionnel, les classes inférieures auront une forte chance de contenir ces séquences.
aNEURAL NET#7 : gb|AAC75303.1|Score: 0.0008REJECT
bNEURAL NET#267 : PSCA_HUMANScore: 0.99994ACCEPT
HYBRIDE#267 : PSCA_HUMANScore 1: 20.9146 Class 1Structure :[sga] halqpaaailallpalglllwgpgqlScore 2: 14.9032 Class 1 Structure :[aha] lqpaaailallpalglllwgpgqlScore 3: 4.9965 Class 2Structure :[nas] gahalqpaaailallpalglllwgpgql
cNEURAL NET#39 : GS28_CRIGRScore: 0.99999ACCEPT
HYBRIDE#39 : GS28_CRIGRScore 1: -1.03186 Class 4Structure :[sli] lggvigicctillllyafh
Figure 7. 2 Résultats de la méthode hybride. a : Séquence rejetée par le système. b : Séquence GPI hautement probable. c : Séquence potentiellement fausse positive.
La figure 7.2a montre un exemple de rejet de la séquence, dès la phase de nettoyage, par le
réseau de neurones artificiels. Le traitement de ces séquences se termine à cette phase. La

128
figure 7.2b montre un exemple de sortie obtenu pour une séquence ayant une forte probabilité
d’avoir un signal GPI en position C-terminale. Ici, le score du réseau de neurones artificiels
débute l’annotation. Par la suite, le score du HMM suit accompagné d’un maximum de trois
propositions de structure du signal. La partie entre crochets représente la zone du site
d’ancrage potentiel. Comme on peut voir avec l’exemple de la figure 7.2c, une prédiction
dans la classe fausse positive potentielle a normalement moins de sites d’ancrage possibles
(une seule proposition dans le cas de GS28_CRIGR) qu’une séquence de la classe hautement
probable.
Annotation à grande échelle du protéome connu de Arabidopsis et Oryza
L’analyse à grande échelle étant notre premier objectif, les séquences connues de deux
protéomes, Arabidopsis thaliana contenant 28860 séquences (The Arabidopsis Genome
Initiative, 2000) et Oryza sativa contenant 28519 séquences (GRAMENE, 2004) ont été
soumises à la méthode hybride pour annotation. Le tableau 7.4 donne les résultats obtenus
pour ces deux bases de données.
Tableau 7. 4 Classification du protéome de Arabidopsis thaliana et Oryza sativa.
Arabidopsis thaliana Chromosome 1 à 5
Nombre total de séquences 28860
Séquences potentiellement GPI sélectionnées par le réseau de neurones
1779
Annotation hybride
classe 1 2 3 4 fraction des prédictions 0.12 0.08 0.07 0.73 fraction du protéome 0.007 0.005 0.004 0.04
Oryza sativa
Nombre total de séquences 28519
Séquences potentiellement GPI 1575
Annotation hybride
classe 1 2 3 4 fraction des prédictions 0.12 0.07 0.07 0.74 fraction du protéome 0.006 0.004 0.004 0.04

129
Selon la classification choisie, la proportion occupée par les séquences GPI serait entre 0.6 %
et 1 % du protéome de Oryza sativa et entre 0.7% et 1% pour Arabidopsis thaliana (les
prédictions de la classe hautement probable pour les deux protéomes sont présentées dans
l’Appendice J)
.

DISCUSSION
Comme nous l’avons constaté dans le chapitre V, les résultats obtenus avec le modèle de
réseau de neurones artificiels sont très intéressants mais le réseau de neurones artificiels
n’offre pas une classification complète. L’utilisation des propriétés physico-chimiques des
acides aminés permet de faire un premier nettoyage des données en éliminant, entre autres,
les séquences n’ayant pas un hydrophathie compatible avec le signal GPI. La complexité du
signal demande une classification plus fine qui prend en compte d’autres critères
représentatifs du signal GPI, dont sa structure. L’introduction d’un HMM dans la méthode
d’analyse permet de prendre en compte cette structure particulière du signal GPI.
Une méthode de classification utilisant un des deux modèles (réseau de neurones artificiels
ou HMM) est envisageable, mais comme les deux systèmes s’attaquent à deux aspects
différents du signal, une hybridation des deux approches est beaucoup plus avantageuse.
Comme on peut le constater dans l’analyse des résultats de la méthode hybride, les résultats
des tests de performance sont améliorés. De plus, l’analyse à grande échelle de deux génomes
de plantes donne des résultats plus qu’intéressants. Avec une proportion entre 0.6% et 1% du
génome nous confirmons les résultats d’une étude effectuée par Eisenhaber et al. (2003) qui
estimaient la proportion du protéome de Oryza sativa occupée par les protéines à ancre GPI à
0.94% et, à 0.75% pour Arabidopsis thaliana. Nos estimations sont supérieures à l’estimation
faite par la même auteure, dans une étude antérieure sur l’analyse de la proportion occupée
par les ancres GPI dans différents protéomes (Eisenhaber, Bork et Eisenhaber 2001). Dans
cette étude, la proportion du protéome occupée par les ancres GPI était estimée à 0.5% pour
tous les eucaryotes. Cette sous-estimation peut être due au fait que le prédicteur big- de GPI
de plantes, développé par le groupe de Eisenhaber, est beaucoup plus sensible au signal GPI
(sensibilité de plus de 0.95) que les prédicteurs big- développés pour les autres eucaryotes.
Ceci nous amène à discuter de l’outil de prédiction effectuant la classification des protéines à

131
ancre GPI développé par le groupe du Dr Eisenhaber du Research Institute of Molecular
Pathology de Vienne en Autriche.
L’outil de prédiction d’ancre GPI big- , est disponible publiquement. Les bases, ainsi que la
structure de cet outil, diffèrent significativement de notre méthode hybride. À l’origine, big-
se composait de deux prédicteurs : un prédicteur spécifique aux métazoaires et un spécifique
aux protozoaires (Eisenhaber, Bork et Eisenhaber 1999). Plus récemment, le groupe de
Eisenhaber a développé un prédicteur spécifique aux plantes et un pour les champignons
(Eisenhaber et al., 2003). Notre méthode hybride, par contre, n’est pas spécifique à un
groupe taxonomique. Comme pour notre méthode hybride, la partie ciblée est la queue C-
terminale. Toutefois, dans le cas de big-, on recommande que chaque séquence soit soumise
préalablement à un outil de prédiction du signal peptide en position N-terminale. Cette
recommandation implique qu’une prédiction d’ancre GPI nécessite la présence d’un signal en
N-terminale. Ceci représente une contrainte supplémentaire qui n’est pas présente dans notre
méthode hybride.
Le principe sous-jacent à big- est une description du motif GPI basée, entre autres, sur les
propriétés physiques du signal (hydrophobicité, longueur des différentes zones composant le
signal) (Eisenhaber, Bork et Eisenhaber 2003). À partir d’une analyse des séquences GPI
connues, le logiciel big- propose une segmentation du signal en quatre zones distinctes.
Cette segmentation représente la structure du signal discutée dans le chapitre 2 (Eisenhaber et
al., 1998).
Grâce à une analyse d’un groupe de séquences d’entraînement, un score est affecté aux
séquences soumises à big- en se basant sur un alignement ainsi que sur différents calculs
effectués dans chaque zone. Cette méthode d’évaluation s’avère très spécifique au groupe
taxonomique concerné. Cette forte spécificité entraîne, toutefois, une sensibilité plus faible
pour certains groupes ayant un signal GPI moins typique tels que les protozoaires (tableau
7.5). Comme on peut le constater dans la comparaison présentée dans le tableau 7.5, la
méthode hybride est plus sensible au signal GPI que big-. On remarque une exception, dans
le cas des plantes, où big- offre une meilleure capacité de prédiction des protéines à ancre

132
GPI. Cette meilleure performance du prédicteur big- de plantes, comparativement aux autres
prédicteurs big- et à la méthode hybride, s’explique possiblement par la présence d’une
grande quantité de séquences GPI provenant de collaborateurs de Eisenhaber, Paul Dupree et
Georg Borner de l’université de Cambridge en Grande-Bretagne. Pour ce qui est de la
performance de notre méthode hybride, une plus faible sensibilité s’explique probablement
par la présence, dans la base de données Swiss-Prot, de plusieurs protéines annotées avec des
prédictions de big- (Farriol-Mathis et al., 2004). La présence de ces prédictions dans les
tests donne un avantage certain à big- dans une étude comparative.
Tableau 7. 5 Étude comparative de la capacité de prédiction de la méthode hybride vs les différents prédicteurs de big-.
Organisme Méthode hybride catégorie probable
big- prédicteur
GPI Métazoaire 0.883 0.719 Protozoaire 0.849 0.643 Champignon 0.893 0.857 Plante 0.881 0.952
non-GPI Cytoplas.-nucléaire
0.00 0.00
Transmembrane
0.02 0.00
Transport 0.01 0.00 Aléatoire 0.004 0.001
Pour le test de spécificité, on peut constater, dans le tableau 7.5, que big- est très spécifique
dans toutes les catégories, ne donnant ainsi que très peu de faux positifs. Toutefois, la
spécificité de la méthode hybride est comparable, malgré la présence d’une possibilité de
faux positifs dans la classe des protéines transmembranaires et dans celle des protéines de
transport. Cette comparaison montre donc une sensibilité supérieure de la méthode hybride
dans la plupart des groupes taxonomiques, tout en offrant une spécificité raisonnable.
Finalement notre méthode d’analyse propose une nouvelle définition de la grammaire GPI.
Cette nouvelle structuration du signal ne peut, à cette étape de la recherche, qu’être une base

133
de questionnement pour les biologistes. Il faudrait des études en laboratoire qui permettraient
de vérifier cette nouvelle structure avant de pouvoir renverser des années d’études
expérimentales ayant amené la structure du signal présentement acceptée.

CONCLUSION
L’annotation des séquences biologiques est un projet à très long terme qui va monopoliser les
efforts conjugués de biologistes, d’informaticiens et de bioinformaticiens pour plusieurs
années encore. Dans cette thèse, nous avons abordé le problème de l’annotation sous l’angle
de la classification, et nous avons montré que l’identification des signaux biologiques
nécessitait le recours à plusieurs outils de l’informatique cognitive.
L’alignement de séquences permet, dans un premier temps, d’identifier des caractéristiques
communes des séquences, comme la présence d’un groupe de nucléotides ou d’acides aminés
à une certaine position. D’autre part, certains aspects de la fonctionnalité des protéines
échappent à un traitement aussi simple, car les motifs qui leur sont associés ne dépendent pas
nécessairement de l’ordre ou de la nature des acides aminés des séquences correspondantes.
Nous avons ensuite exploré les réseaux de neurones artificiels, principalement à cause de la
vision globale des séquences que ce modèle permet d’intégrer. Notre réseau de neurones a
permis de confirmer l’importance de l’hydropathie dans la détection de protéines à ancre GPI
et même de suggérer qu’une hydropathie particulière aux GPI existe, étant donné la bonne
performance du réseau dans la tâche de classification. Malheureusement, un réseau de
neurones fonctionne souvent comme une « boîte noire », et le résultat de son apprentissage ne
fournit pas une représentation analysable des connaissances.
L’utilisation de grammaires régulières stochastiques, tels les HMM, nous a permis d’exploiter
la nature séquentielle de la structure primaire des protéines. En partant des modèles connus
du signal GPI, nous avons construit un HMM qui, après apprentissage, a raffiné nos
connaissances sur la structure de la zone d’ancrage et de la queue hydrophobe.

135
C’est toutefois l’hybridation de ces deux méthodes d’apprentissage machine qui a produit les
résultats les plus probants. En combinant la sensibilité du réseau de neurones, qui permet un
« nettoyage » des données, à la capacité du HMM de structurer le signal, nous avons construit
un classificateur à la fois spécifique et souple. Ses prédictions qualifiées de « hautement
probable » se comparent avantageusement aux outils les plus stricts, alors que ses prédictions
dans les catégories inférieures permettent d’identifier des candidats qui sortent un peu des
limites de la structure du HMM, laissant place à la découverte de protéines à ancrage GPI
ayant un signal moins spécifique.
Dans le futur il serait intéressant de tester les prédictions de notre méthode hybride de façon
expérimentale en laboratoire. La confirmation ou l’infirmation des prédictions permettrait
d’améliorer les modèles de réseau de neurones et HMM. Il serait aussi intéressant de faire
une classification de nos prédictions selon la fonction biologique des séquences sélectionnées
ayant une annotation fonctionnelle. De la sorte nous pourrions associé la présence du signal à
certaines fonctions biologiques et ainsi pouvoir aider la compréhension du rôle joué par ce
type d’attachement menbranaire. De plus l’efficacité de notre méthode hybride pour la
reconnaissance du signal GPI suggère son utilisation pour d’autres signaux protéiques.

APPENDICE A
SÉQUENCES GPI DE SWISS-PROT Cet appendice présente les 50 derniers acides aminés (signal GPI en position C-terminale) des 468 séquences de protéines à ancre GPI de la base de données Swiss-Prot version janvier 2004.
Séquences GPI > 5NTD_BOOMI 30NQA (POTENTIAL).VMKYMNSTSPITTALDGRVTFLKTNQASDACLNLASPFLVLLVLVVFYHL> 5NTD_BOVIN 30 SAG (BY SIMILARITY).INVVSGYISKMKVLYPAVEGRIQFSAGSHCCGSFSLIFLSVLAVIIILYQ> 5NTD_DISOM 30 SAT (BY SIMILARITY).VSSYIKQMKVVYPAVEGRILFVENSATLPIINLKIGLSLFAFLTWFLHCS> 5NTD_HUMAN 30 STGINVVSTYISKMKVIYPAVEGRIKFSTGSHCHGSFSLIFLSLWAVIFVLYQ> 5NTD_MOUSE 30 SAA (BY SIMILARITY).ISVVSEYISKMKVVYPAVEGRIKFSAASHYQGSFPLVILSFWAMILILYQ> 5NTD_RAT 30 SAAISVVSEYISKMKVIYPAVEGRIKFSAASHYQGSFPLIILSFWAVILVLYQ> ACES_TORCA 33 SSGLRVQMCVFWNQFLPKLLNATACDGELSSSGTSSSKGIIFYVLFSILYLIF> ACES_TORMA 32 SSGRVQMCVFWNQFLPKLLNATACDGELSSSGTSSSKGIIFYVLFSILYLIFY> AMPM_HELVI 33 DSA (POTENTIAL).TSTTAAPTTVTQPTITEPSTPTLPELTDSAMTSFASLFIISLGAILHLIL> AMPM_MANSE 33 GSG (POTENTIAL).TVAPPAETTVTPSTFPPTVAPATTPAPGSGNIAALSVVSLLVTLAINMVA> APH4_DROME 29 NGA (POTENTIAL).DDSCEDHKDGQKDRPLDKPNPKRNGATVVGASLIPILTAATAAILRGRGL> AXO1_HUMAN 27 NMA (POTENTIAL).GDGIPAEVHIVRNGGTSMMVENMAVRPAPHPGTVISHSVAMLILIGSLEL> BM86_BOOMI 32 SAA (BY SIMILARITY).KEKSEATTAATTTTKAKDKDPDPGKSSAAAVSATGLLLLLAATSVTAASL

137
> BST1_HUMAN 30 APS (POTENTIAL).LQCVDHSTHPDCALKSAAAATQRKAPSLYTEQRAGLIIPLFLVLASRTQL> BST1_MOUSE 21 SAS (POTENTIAL).LMCVDHSTHPDCIMNSASASMRRESASLHAIGDASLLISLLVALASSSQA> BST1_RAT 30 SPA (POTENTIAL).LMCVDHSTHPDCAMNSASASMWRESPALHAIGDISLIISLLVALASSSQA> BY55_HUMAN 33 SSG (BY SIMILARITY).FTETGNYTVTGLKQRQHLEFSHNEGTLSSGFLQEKVWVMLVTSLVALQAL> BY55_MOUSE 30 SSG (BY SIMILARITY).TGNHTEIRQRQRSHPDFSHINGTLSSGFLQVKAWGMLVTSLVALQALYTL> C59A_MOUSE 28 SDG (BY SIMILARITY).IMDQLEETKLKFRCCQFNLCNKSDGSLGKTPLLGTSVLVAILNLCFLSHL> C59B_MOUSE 30 NAE (POTENTIAL).AGIQSKCCQWGLCNKNLDGLEEPNNAETSSLRKTALLGTSVLVAILKFCF> CADD_CHICK 36 DAL (POTENTIAL).DSGKPPLTNNTELKLQVCSCKKSRMDCSASDALHISMTLILLSLFSLFCL> CADD_HUMAN 35 GAL (POTENTIAL).SGKPPMTNITDLRVQVCSCRNSKVDCNAAGALRFSLPSVLLLSLFSLACL> CADD_MOUSE 34 GAL (POTENTIAL).GKPPMTNITDLKVQVCSCKNSKVDCNGAGALHLSLSLLLLFSLLSLLSGL> CAH4_HUMAN 27 SGATVSMKDNVRPLQQLGQRTVIKSGAPGRPLPWALPALLGPMLACLLAGFLR> CAH4_MOUSE 27 SHA (BY SIMILARITY).KLNMKDNVRPLQPLGKRQVFKSHAPGQLLSLPLPTLLVPTLTCLVANFLQ> CD14_HUMAN 25 NSG (POTENTIAL).LDGNPFLVPGTALPHEGSMNSGVVPACARSTLSVGVSGTLVLLQGARGFA> CD14_MOUSE 25 NSG (POTENTIAL).NLSLKGNPFLDSESHSEKFNSGVVTAGAPSSQAVALSGTLALLLGDRLFV> CD14_RAT 25 NSG (POTENTIAL).SLSLTGNPFLHSESQSEAYNSGVVIATALSPGSAGLSGTLALLLGHRLFV> CD24_HUMAN 34 GGA (POTENTIAL).TGTSSNSSQSTSNSGLAPNPTNATTKAAGGALQSTASLFVVSLSLLHLYS> CD24_MOUSE 32 GGG (POTENTIAL).NQTSVAPFPGNQNISASPNPSNATTRGGGSSLQSTAGLLALSLSLLHLYC> CD24_RAT 35 SSL (POTENTIAL).NQTSVAPFSGNQSISAAPNPTNATTRSGCSSLQSTAGLLALSLSLLHLYC> CD48_HUMAN 32 SFGYTCQVSNSVSSKNGTVCLSPPCTLARSFGVEWIASWLVVTVPTILGLLLT> CD48_MOUSE 32 SSGYTCQVSNPVSSKNDTVYFTLPCDLARSSGVCWTATWLVVTTLIIHRILLT> CD48_RAT 32 SSGYTCQVSNPVSSENDTLYFIPPCTLARSSGVHWIAAWLVVTLSIIPSILLA> CD52_CANFA 33 SSL (POTENTIAL).QIQTGVLGNSTTPRMTTKKVKSATPALSSLGGGSVLLFLANTLIQLFYLS> CD52_HUMAN 30 SAS (POTENTIAL).SLLVMVQIQTGLSGQNDTSQTSSPSASSNISGGIFLFFVANAIIHLFCFS> CD52_MACFA 30 SAS (POTENTIAL).ISLLVMVQIQTGVTSQNATSQSSPSASSNLSGGGFLFFVANAIIHLFYFS> CD52_MOUSE 28 SGA (POTENTIAL).QATTAASGTNKNSTSTKKTPLKSGASSIIDAGACSFLFFANTLMCLFYLS> CD52_RAT 29 GAS (POTENTIAL).AAATTATKTTTAVRKTPGKPPKAGASSITDVGACTFLFFANTLMCLFYLS> CD59_AOTTR 29 NGG (BY SIMILARITY).LSENELKYYCCKKNLCNFNEALKNGGTTLSKKTVLLLVIPFLVAAWSLHP

138
> CD59_CALSQ 29 NGG (BY SIMILARITY).LSENELKYHCCRENLCNFNGILENGGTTLSKKTVLLLVTPFLAAAWSLHP> CD59_CERAE 29 NGG (BY SIMILARITY).LKESELQYFCCKKDLCNFNEQLENGGTSLSEKTVVLLVTLLLAAAWCLHP> CD59_HSVSA 30 NIK (POTENTIAL).QLSETQLKYHCCKKNLCNVNKGIENIKRTISDKALLLLALFLVTAWNFPL> CD59_HUMAN 29 NGGLRENELTYYCCKKDLCNFNEQLENGGTSLSEKTVLLLVTPFLAAAWSLHP> CD59_PAPSP 29 NGG (BY SIMILARITY).TLLKESELQYFCCKEDLCNEQLENGGTSLSEKTVLLLVTPLLAAAWCLHP> CD59_PIG 30 SDADFISRNLAEKKLKYNCCRKDLCNKSDATISSGKTALLVILLLVATWHFCL> CD59_RABIT 32 GTA (BY SIMILARITY).ISNRLEENSLKYNCCRKDLCNGPEDDGTALTGRTVLLVAPLLAAARNLCL> CD59_RAT 30 NGA (BY SIMILARITY).EIANVQYRCCQADLCNKSFEDKPNNGAISLLGKTALLVTSVLAAILKPCF> CD59_SAISC 29 NGG (BY SIMILARITY).LSETQLKYHCCKKNLCNVKEVLENGGTTLSKKTILLLVTPFLAAAWSRHP> CEA6_HUMAN 31 GSA (BY SIMILARITY).GSYMCQAHNSATGLNRTTVTMITVSGSAPVLSAVATVGITIGVLARVALI> CEA8_HUMAN 26 DALHTTNSATGRNRTTVRMITVSDALVQGSSPGLSARATVSIMIGVLARVALI> CEPU_CHICK 32 SGA (POTENTIAL).ASMILYEETTTALTPWKGPGAVHDGNSGAWRRGSCAWLLALPLAQLARQF> CNTR_CHICK 27 DKG (POTENTIAL).ITETTSTSTSSFMPPPTTKICDKGAGVGSGAVAVCWTAGLVLAAYGVLFI> CNTR_HUMAN 25 SGG (POTENTIAL).TSSLAPPPTTKICDPGELGSGGGPSAPFLVSVPITLALAAAAATASSLLI> CNTR_RAT 25 SGG (POTENTIAL).TSSLAPPPTTKICDPGELSSGGGPSIPFLTSVPVTLVLAAAAATANNLLI> CONN_DROME 31 AGA (POTENTIAL).SDPTELPLSRDLMDVRSNVGQDMSTAGANSLAQGMTIIVSLQVALMISRG> CONT_HUMAN 30 SGA (POTENTIAL).DGEYVVEVRAHSDGGDGVVSQVKISGAPTLSPSLLGLLLPAFGILVYLEF> CONT_MOUSE 36 SSS (POTENTIAL).DGEYVVEVRAHSDGGDGVVSQVKISGVSTLSSSLLSLLLPSLGFLVYSEF> CONT_RAT 35 SSG (POTENTIAL).GEYVVEVRAHSDGGDGVVSQVKISGVSTLSSGLLSLLLPSLGFLVFYSEF> CSA_DICDI 33 SSA (POTENTIAL).PSPTPTETATPSPTPKPTSTPEETEAPSSATTLISPLSLIVIFISFVLLI> CW12_YEAST 34 GAA (POTENTIAL).STAAPVTSTEAPKNTTSAAPTHSVTSYTGAAAKALPAAGALLAGAAALLL> CW14_YEAST 37 NVL (POTENTIAL).SAASSTVSQETVSSALPTSTAVISTFSEGSGNVLEAGKSVFIAAVAAMLI> CWP1_YEAST 33 NAG (POTENTIAL).SSPTASVISQITDGQIQAPNTVYEQTENAGAKAAVGMGAGALAVAAAYLL> CWP2_YEAST 34 NGA (POTENTIAL).EATTTAAPSSTVETVSPSSTETISQQTENGAAKAAVGMGAGALAAAAMLL> DAF1_MOUSE 27 GGD (POTENTIAL).VTKTTVRHPIRTSTDKGEPNTGGDRYIYGHTCLITLTVLHVMLSLIGYLT> DAF_CAVPO 0 ASG (IN ISOFORM 3) (POTENTIAL).KTHVYKVDSFACGASNHWLADIAKEDLRRDFSNAQNISSLLQVLGAAQTQ> DAF_HUMAN 23 SGTPVSRTTKHFHETTPNKGSGTTSGTTRLLSGHTCFTLTGLLGTLVTMGLLT> DAF_PONPY 23 SGT (BY SIMILARITY).PVSRTTKHFHETTPNKGSGTTSGTTSLLSGHKCFTLTGLLGTLVTMGLLT> DAN1_YEAST 39 NGV (POTENTIAL).KPVSSKAQSTATSVTSSASRVIDVTTNGANKFNNGVFGAAAIAGAAALLL

139
> DAN4_YEAST 40 NIF (POTENTIAL).SPIPKASSATSIAHSSASYTVSINTNGAYNFDKDNIFGTAIVAVVALLLL> EFA4_HUMAN 24 SGT (POTENTIAL).CCKERKSESAHPVGSPGESGTSGWRGGDTPSPLCLLLLLLLLILRLLRIL> FC3B_HUMAN 22 SSF (POTENTIAL).ETVNITITQGLAVSTISSFSPPGYQVSFCLVMVLLFAVDTGLYFSVKTNI> FOL1_HUMAN 32 SGARCIQMWFDPAQGNPNEEVARFYAAAMSGAGPWAAWPFLLSLALMLLWLLS> FOL1_MOUSE 32 SGA (BY SIMILARITY).RCIQMWFDPAQGNPNEEVARFYAEAMSGAGLHGTWPLLCSLSLVLLWVIS> FOL2_HUMAN 30 NAGMWFDSAQGNPNEEVARFYAAAMHVNAGEMLHGTGGLLLSLALMLQLWLLG> FOL2_MOUSE 31 SGT (POTENTIAL).IQMWFDSTQGNPNEDVVKFYASFMTSGTVPHAAVLLVPSLAPVLSLWLPG> G13A_DICDI 33 STS (POTENTIAL).PTPSTTPSTTPSTTPSSTPTQSPDDDGSTSSTLSTSFYLITLLFLIQQFI> G13B_DICDI 29 GDD (POTENTIAL).TTPSTTPSTTPSTTPSSTPTQSPGDDGSTSSTLSISFYLITLLLLTQQFI> GAS1_CAEEL 32 DSSLKNTPGVTLSPSDNSITDAPGGNDLADSSVGHGFNILSAISVYLLTVLVF> GAS1_HUMAN 13 SGG (POTENTIAL).PGSGAAASGGRGDLPYGPGRRSSGGGGRLAPRGAWTPLASILLLLLGPLF> GAS1_MOUSE 27 SGG (POTENTIAL).AAAGGRGDLPHGPGRRSSSSGSGGHWANRSAWTPFACLLLLLLLLLGSHL> GAS1_YEAST 24 NAASSSSSSSSSASSSSSSKKNAATNVKANLAQVVFTSIISLSIAAGVGFALV> GFR1_CHICK 16 SHI (POTENTIAL).GKDNTPGVSTSHISSENSFALPTSFYPSTPLILMTIALSLFLFLSSSVVL> GFR1_HUMAN 19 SHI (POTENTIAL).SNGNYEKEGLGASSHITTKSMAAPPSCGLSPLLVLVVTALSTLLSLTETS> GFR1_MOUSE 17 SHI (POTENTIAL).DYGKDGLAGASSHITTKSMAAPPSCGLSSLPVMVFTALAALLSVSLAETS> GFR1_RAT 17 SHI (POTENTIAL).DFGKDGLAGASSHITTKSMAAPPSCSLSSLPVLMLTALAALLSVSLAETS> GFR2_CHICK 35 SRH (POTENTIAL).EQSLCYSETQLTTDTMPDQKTFVDQKAAGSRHRAARILPAVPIVLLKLLL> GFR2_HUMAN 35 SRA (POTENTIAL).SKELSMCFTELTTNIIPGSNKVIKPNSGPSRARPSAALTVLSVLMLKQAL> GFR2_MOUSE 35 SCR (POTENTIAL).NSKELSMCFTELTTNISPGSKKVIKLYSGSCRARLSTALTALPLLMVTLA> GFR3_HUMAN 29 NPA (POTENTIAL).FHSQLFSQDWPHPTFAVMAHQNENPAVRPQPWVPSLFSCTLPLILLLSLW> GFR4_CHICK 27 SPA (POTENTIAL).TKVAGEERLLRGSTRLSSETSSPAAPCHQAASLLQLWLPPTLAVLSHFMM> GFR4_HUMAN 34 GRA (POTENTIAL).ASGWPPVLLDQLNPQGDPEHSLLQVSSTGRALERRSLLSILPVLALPALL> GFR4_MOUSE 32 TAG (POTENTIAL).LFTRNPCLDGAIQAFDSLQPSVLQDQTAGCCFPRVSWLYALTALALQALL> GFR4_RAT 32 NAG (POTENTIAL).PCLDGAIQAFDSSQPSVLQDQWNPYQNAGCCFLWVSSMSILTALALQALL> GP42_RAT 28 GTA (POTENTIAL).CQAENKVSRDISEPKKFPLVVSGTASMKSTTVVIWLPVSCLVGWPWLLRF> GP46_LEIAM 31 CPA (POTENTIAL).RCGARPSPCASVCVSWPRERRTECACPALFDGARLRCCALVVCAGAAPAG> GP63_LEICH 30 NAA (BY SIMILARITY).GGYITCPPYVEVCQGNVQAAKDGGNAAAGRRGPRAAATALLVAALLAVAL> GP63_LEIDO 30 NAA (BY SIMILARITY).GGYITCPPYVEVCQGNVQAAKDGGNAAAGRRGPRAAATALLVAALLAVAL

140
> GP63_LEIGU 10 DAA (BY SIMILARITY).DFEGDAADTAAMRRWRERMTALATVTAALLGIVLAAMAILVVWLLLITIP> GP63_LEIMA 30 NTAGGYITCPPYVEVCQGNVQAAKDGGNTAAGRRGPRAAATALLVAALLAVAL> GP85_TRYCR 32 ANA (POTENTIAL).IPKRGPGSQVEGGTERRHIPRIEGVRANAPVGSGLLPLLLLLGLWVFAAL> GPC1_HUMAN 27 SAA (POTENTIAL).SSRTPLTHALPGLSEQEGQKTSAASCPQPPTFLLPLLLFLALTVARPRWR> GPC1_RAT 27 SAA (POTENTIAL).SSRTPLIHALPGLSEQEGQKTSAATRPEPHYFFLLFLFTLVLAAARPRWR> GPC4_HUMAN 28 SAG (POTENTIAL).CPSEFDYNATDHAGKSANEKADSAGVRPGAQAYLLTVFCILFLVMQREWR> GPC4_MOUSE 27 SAG (POTENTIAL).PSEFEYNATDHSGKSANEKADSAGGAHAEAKPYLLAALCILFLAVQGEWR> HYA2_HUMAN 30 GAS (POTENTIAL).HFRCQCYLGWSGEQCQWDHRQAAGGASEAWAGSHLTSLLALAALAFTWTL> HYA2_MOUSE 30 NAS (POTENTIAL).HFRCQCYLGWGGEQCQRNYKGAAGNASRAWAGSHLTSLLGLVAVALTWTL> HYA2_RAT 30 DAS (POTENTIAL).HFRCHCYLGWGGEQCQWNHKRAAGDASRAWAGAHLASLLGLVAMTLTWTL> HYAP_CAVPO 18 SIS (POTENTIAL).PPITDDTSQNQDSISDITSSAPPSSHILPKDLSWCLFLLSIFSQHWKYLL> HYAP_HUMAN 36 SAT (POTENTIAL).ADGVCIDAFLKPPMETEEPQIFYNASPSTLSATMFIVSILFLIISSVASL> HYAP_MACFA 36 STT (POTENTIAL).DGVCIDASLKPPVETEGSPPIFYNTSSSTVSTTMFIVNILFLIISSVASL> HYR1_CANAL 31 NGS (POTENTIAL).IPVPHSMPSNTTDSSSSVPTIDTNENGSSIVTGGKSILFGLIVSMVVLFM> LACH_DROME 32 AGA (POTENTIAL).GEAEARVNLFETIIPVCPPACGQAYIAGAEDVSATSFALVGISARLLFAR> LACH_SCHAM 38 GDA (POTENTIAL).KAANKLGEAREEVELFETIIPVCPPACGQAYGGDAAEISTSMALILISTI> LAMP_HUMAN 32 NGS (POTENTIAL).TCVAANKLGVTNASLVLFRPGSVRGINGSISLAVPLWLLAASLLCLLSKC> LAMP_RAT 32 NGS (POTENTIAL).TCVAANKLGVTNASLVLFRPGSVRGINGSISLAVPLWLLAASLFCLLSKC> LY6A_MOUSE 33 GST (POTENTIAL).MEILGTKVNVKTSCCQEDLCNVAVPNGGSTWTMAGVLLFSLSSVLLQTLL> LY6C_MOUSE 33 GST (POTENTIAL).VPIKDPNIRERTSCCSEDLCNAAVPTAGSTWTMAGVLLFSLSSVILQTLL> LY6D_HUMAN 25 NAA (POTENTIAL).SGTSSTQCCQEDLCNEKLHNAAPTRTALAHSALSLGLALSLLAVILAPSL> LY6D_MOUSE 26 SAA (POTENTIAL).SSGSEVTQCCQTDLCNERLVSAAPGHALLSSVTLGLATSLSLLTVMALCL> LY6E_CHICK 27 SGS (POTENTIAL).GINLGIAAASVYCCDSFLCNISGSSSVKASYAVLALGILVSFVYVLRARE> LY6E_HUMAN 25 SAA (POTENTIAL).NVGVASMGISCCQSFLCNFSAADGGLRASVTLLGAGLLLSLLPALLRFGP> LY6E_MOUSE 27 AAG (POTENTIAL).LNLGVASVNSYCCQSSFCNFSAAGLGLRASIPLLGLGLLLSLLALLQLSP> LY6F_MOUSE 33 GST (POTENTIAL).MEILGTTVNVNTSCCKEDLCNAPFSTGGSTWTMTRVLLLNLGSVFLQTLL> LY6G_MOUSE 40 GVL (POTENTIAL).TEITGNAVNVKTYCCKEDLCNAAVPTGGSSWTMAGVLLFSLVSVLLQTFL> LY6H_HUMAN 30 GAG (POTENTIAL).GFINSGILKVDVDCCEKDLCNGAAGAGHSPWALAGGLLLSLGPALLWAGP> LY6H_MOUSE 26 NGA (POTENTIAL).GFINSGILKVDVDCCEKDLCNGASVAGRSPWALAGGLLLSLGPALLWAGP

141
> LY6I_MOUSE 33 GSS (POTENTIAL).KFILDPNTKMNISCCQEDLCNAAVPTGGSSWTTAGVLLFSLGSVLLQTLM> LYNX_MOUSE 32 GAG (POTENTIAL).SCFETVYDGYSKHASATSCCQYYLCNGAGFATPVTLALVPALLATFWSLL> MDP1_HUMAN 29 SGAAPEEEPIPLDQLGGSCRTHYGYSSGASSLHRHWGLLLASLAPLVLCLSLL> MDP1_MOUSE 29 SQA (BY SIMILARITY).QSPEEVPITLKELDGSCRTYYGYSQAHSIHLQTGALVASLASLLFRLHLL> MDP1_PIG 30 SAA (BY SIMILARITY).AQVPGEEPIPLGQLEASCRTNYGYSAAPSLHLPPGSLLASLVPLLLLSLP> MDP1_RABIT 29 SEA (BY SIMILARITY).QVPEEEPISLEQLGGSCRTQYGYSEAPSLHRRPGALLASLSLLLLSLGLL> MDP1_RAT 29 SRA (BY SIMILARITY).QVPEEETIPVEKLDGSCRTFYGHSRAPSIHLQIGALLASLASLVFSLHPL> MDP1_SHEEP 29 SGT (BY SIMILARITY).QAPGEEPIPLGQLEASCRTKYGYSGTPSLHLQPGSLLASLVTLLLSLCLL> MKC7_YEAST 34 NGG (POTENTIAL).ALSISKSTSSTSSTGMLSPTSSSSPRKENGGHNLNPPFFARFITAIFHHI> MM17_HUMAN 17 SGA (POTENTIAL).SEDGYEVCSCTSGASSPPGAPGPLVAATMLLLLPPLSPGALWTAAQALTL> MM17_MOUSE 35 SDA (POTENTIAL).EPLADAEDVGPGPQGRSGAQDGLAVCSCTSDAHRLALPSLLLLTPLLWGL> MM19_MOUSE 40 DSA (POTENTIAL).TNSSTGDVTPSTTDTVLGTTPSTMGSTLDIPSATDSASLSFSANVTLLGA> MM25_HUMAN 32 AAG (POTENTIAL).SGPRAPRPPKATPVSETCDCQCELNQAAGRWPAPIPLLLLPLLVGGVASR> MSA1_SARMU 31 AGS (POTENTIAL).CYLCEPDPTKKGHNDKNCAVLIAVGAGSRPTARSVFGVAAPCILALLHFT> MSLN_HUMAN 13 GGI (POTENTIAL).TLGLGLQGGIPNGYLVLDLSVQETLSGTPCLLGPGPVLTVLALLLASTLA> NAR3_HUMAN 28 SSG (BY SIMILARITY).GNINNPTPGPVPVPGPKSHPSASSGKLLLPQFGMVIILISVSAINLFVAL> NAR3_MOUSE 16 SGS (BY SIMILARITY).LILFFIKSSRSGSRSEIPSLCILWQYAPSISHGIHHFTRCFCCKLHRAIA> NAR4_HUMAN 26 ASS (POTENTIAL).NWLQLRSTGNLSTYNCQLLKASSKKCIPDPIAIASLSFLTSVIIFSKSRV> NAR4_PANTR 26 ASS (POTENTIAL).NWLQLRSTGNLSTYNCQLLKASSKKCIPDPIAIASLSFLTSVIIFSKSRV> NARA_MOUSE 26 SSL (BY SIMILARITY).KRKKSNFNCFYSGSTQAANVSSLGSRESCVSLFLVVLLGLLVQQLTLAEP> NARA_RAT 26 SSA (BY SIMILARITY).YNEIFLDSPKRKKSNYNCLYSSAGTRESCVSLFLVVLTSLLVQLLCLAEP> NARB_MOUSE 26 SIS (BY SIMILARITY).KKSNFNCFYNGSAQTVNIDFSISGSRESCVSLFLVVLLGLLVQQLTLAEP> NARB_RAT 26 SSA (BY SIMILARITY).YNEIFLDSPKRKKSNYNCLYSSAGARESCVSLFLVVLPSLLVQLLCLAEP> NTRI_HUMAN 32 NGT (POTENTIAL).CVASNKLGHTNASIMLFGPGAVSEVSNGTSRRAGCVWLLPLLVLHLLLKF> NTRI_MOUSE 32 NGT (POTENTIAL).CVASNKLGHTNASIMLFGPGAVSEVNNGTSRRAGCIWLLPLLVLHLLLKF> NTRI_RAT 32 NGT (POTENTIAL).CVASNKLGHTNASIMLFGPGAVSEVNNGTSRRAGCIWLLPLLVLHLLLKF> OPCM_BOVIN 32 NSA (POTENTIAL).YGNYTCVATNKLGITNASITLYGPGAVIDGVNSASRALACLWLSGTLFAHFFIKF> OPCM_HUMAN 32 NSA (POTENTIAL).CVATNKLGNTNASITLYGPGAVIDGVNSASRALACLWLSGTLLAHFFIKF> OPCM_RAT 32 NSA (POTENTIAL).CVATNKLGNTNASITLYGPGAVIDGVNSASRALACLWLSGTFFAHFFIKF

142
> PAG1_TRYBB 41 ADS (POTENTIAL).VVDEDSGKSFVVLGNRETVQEEKLLEEMAICGVGRADSLRRTLALLFLLF> PARA_TRYBB 33 GAATGPEETGPEETGPEETGPEETEPEPEPGAATLKSVALPFAVAAAALVAAF> PARB_TRYBB 33 GAAPEPEPEPEPEPEPEPEPEPEPEPEPEPGAATLKSVALPFAIAAVGLVAAF> PARC_TRYBB 33 GAA (BY SIMILARITY).PEPEPEPEPEPEPEPEPEPEPEPEPEPGAATLKSVALPFAIAAAALVAAF> PL13_ARATH 28 SDA (POTENTIAL).GTTRGSSSSSGDDSNVFQMIFGSDAPSRPRLTLLFSLLMISVLSLSTLLL> PONA_DICDI 30 SSS (POTENTIAL).KIPTTSYIVSCNSTPSSNSTTDSDSSSGSTVMIGLASSLLFAFATLLALF> PPB1_HUMAN 26 DAAFAACLEPYTACDLAPPAGTTDAAHPGRSVVPALLPLLAGTLLLLETATAP> PPB2_HUMAN 26 DAA (BY SIMILARITY).FAACLEPYTACDLAPPAGTTDAAHPGRSVVPALLPLLAGTLLLLETATAP> PPB3_HUMAN 26 DAA (BY SIMILARITY).FAACLEPYTACDLAPPAGTTDAAHPGRSVVPALLPLLAGTLLLLETATAP> PPBE_MOUSE 28 SAV (BY SIMILARITY).MAFAACLEPYTDCGLASPAGQSSAVSPGYMSTLLCLLAGKMLMLMAAAEP> PPBI_BOVIN 28 DAA (BY SIMILARITY).AGCVEPYTDCNLPAPTTATSIPDAAHLAASPPPLALLAGAMLLLLAPTLY> PPBI_HUMAN 30 DAA (BY SIMILARITY).HVMAFAACLEPYTACDLAPPACTTDAAHPVAASLPLLAGTLLLLGASAAP> PPBI_RAT 26 NSA (POTENTIAL).YTDCGLAPPADENRPTTPVQNSAITMNNVLLSLQLLVSMLLLVGTALVVS> PPBJ_RAT 35 NSA (POTENTIAL).RPTTPVQNSTTTTTTTTTTTTTTTTTRVQNSASSLGPATAPLAWHYWPRR> PPB_BOMMO 21 GPG (POTENTIAL).EQTHVPHRMAWAACMGPGRHVCVSAATVPTAALLSLLLAAFITLRHQCFL> PRIO_ATEGE 37 SSM (BY SIMILARITY).TETDVKMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPVILLISFLI> PRIO_ATEPA 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_CALJA 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYEKESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_CEBAP 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_CERAE 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYEKESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_CERAT 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYEKESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_CERMO 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYEKESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_CERPA 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYEKESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_CERTO 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYEKESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_COLGU 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYEKESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_CRIGR 32 SSAMMERVVEQMCVTQYQKESQAYYDGRRSSAVLFSSPPVILLISFLIFLIVG> PRIO_CRIMI 32 SSAMMERVVEQMCVTQYQKESQAYYDGRRSSAVLFSSPPVILLISFLIFLIVG> PRIO_GORGO 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_HUMAN 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG

143
> PRIO_MACFA 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYEKESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_MANSP 37 SSM (BY SIMILARITY).TETDVKMMERVVEQMCITQYEKESQAYYQRGSSMVLFSSPPVILLISFLI> PRIO_MESAU 32 SSAIMERVVEQMCTTQYQKESQAYYDGRRSSAVLFSSPPVILLISFLIFLMVG> PRIO_MOUSE 31 SSS (BY SIMILARITY).MERVVEQMCVTQYQKESQAYYDGRRSSSTVLFSSPPVILLISFLIFLIVG> PRIO_PANTR 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_PONPY 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYERESQAYYQRGSSMVLFSSPPVILLISFLIFLIVG> PRIO_PREFR 32 SSM (BY SIMILARITY).KMMERVVEQMCITQYEKESQAYYQRGSSMVFFSSPPVILLISFLIFLIVG> PRIO_RAT 32 SSA (BY SIMILARITY).MMERVVEQMCVTQYQKESQAYYDGRRSSAVLFSSPPVILLISFLIFLIVG> PRND_BOVIN 31 GAG (POTENTIAL).LYQRVLWQLIRELCSTKHCDFWLERGAGLRVTLDQPMMLCLLVFIWFIVK> PRND_HUMAN 31 GAG (POTENTIAL).LHQQVLWRLVQELCSLKHCEFWLERGAGLRVTMHQPVLLCLLALIWLMVK> PRND_MOUSE 31 GAA (POTENTIAL).LHQRVLWRLIKEICSAKHCDFWLERGAALRVAVDQPAMVCLLGFVWFIVK> PRND_SHEEP 31 GAG (POTENTIAL).LYQRVLWQLIRELCSIKHCDFWLERGAGLQVTLDQPMMLCLLVFIWFIVK> PSA_DICDI 34 GSA (POTENTIAL).VTPTVTPTVTPTPTNTPNPTPSQTSTTTGSASTVVASLSLIIFSMILSLC> PSCA_HUMAN 27 SGA (POTENTIAL).SQDYYVGKKNITCCDTDLCNASGAHALQPAAAILALLPALGLLLWGPGQL> PSCA_MOUSE 27 NGA (BY SIMILARITY).SENYYLGKKNITCCYSDLCNVNGAHTLKPPTTLGLLTVLCSLLLWGSSRL> RECK_HUMAN 26 SAG (POTENTIAL).SHVPLSALIISQVQVSSSVPSAGVRARPSCHSLLLPLSLGLALHLLWTYN> RECK_MOUSE 26 SSA (POTENTIAL).SHVHLSALIISQVQVSSSLPSSAVVGRPLFHSLLLLLSWGLTVHLLWTRP> RT4R_HUMAN 29 SGA (POTENTIAL).RTRSHCRLGQAGSGGGGTGDSEGSGALPSLTCSLTPLGLALVLWTVLGPC> RT4R_MACFA 29 SGA (POTENTIAL).RTRSHCRLGQAGSGGGGTGDSEGSGALPSLACSLAPLGLALVLWTVLGPC> RT4R_MOUSE 18 SGA (POTENTIAL).RTRSHCRLGQAGSGASGTGDAEGSGALPALACSLAPLGLALVLWTVLGPC> RT4R_RAT 29 SGA (POTENTIAL).RTRSHCRLGQAGSGSSGTGDAEGSGALPALACSLAPLGLALVLWTVLGPC> SAG1_YEAST 22 STS (POTENTIAL).SSASGSQLSGIQQNFTSTSLMISTYEGKASIFFSAELGSIIFLLLSYLLF> SM7A_HUMAN 37 AAS (POTENTIAL).EGSYFREAQHWQLLPEDGIMAEHLLGHACALAASLWLGVLPTLTLGLLVH> SM7A_MOUSE 37 AAS (POTENTIAL).EGSYLREAQHWELLPEDRALAEQLMGHARALAASFWLGVLPTLILGLLVH> SP63_STRPU 31 GSQ (POTENTIAL).RINSWDPRMNWDLSMNLDATEEPESGSQRHLPVCGVLSLVVTTLLALMLH> T10C_HUMAN 32 ASS (POTENTIAL).EETMTTSPGTPAPAAEETMTTSPGTPASSHYLSCTIVGIIVLIVLLIVFV> TEST_HUMAN 29 SGM (POTENTIAL).PNRPGVYTNISHHFEWIQKLMAQSGMSQPDPSWPLLFFPLLWALPLLGPV> TEST_MOUSE 29 NGL (POTENTIAL).PNRPGVYTNISHHYNWIQSTMIRNGLLRPDPVPLLLFLTLAWASSLLRPA> THY1_CHICK 24 CVR (BY SIMILARITY).YTGNQIKNITVIKDKLEKCVRLSLLIQNTSWLLLLLLSLPLLQAVDFVSL

144
> THY1_HUMAN 24 CEGSPPISSQNVTVLRDKLVKCEGISLLAQNTSWLLLLLLSLSLLQATDFMSL> THY1_MACMU 24 CEGSPPISSQNVTVLRDKLVKCEGISLLAQNTSWLXLLLLSLSLLQATDFMSL> THY1_MOUSE 24 CGGNPMSSNKSISVYRDKLVKCGGISLLVQNTSWMLLLLLSLSLLQALDFISL> THY1_RAT 24 CGGNPTSSNKTINVIRDKLVKCGGISLLVQNTSWLLLLLLSLSFLQATDFISL> TIP1_YEAST 33 GQR (POTENTIAL).SSKAVSSSVAPTTSSVSTSTVETASNAGQRVNAGAASFGAVVAGAAALLL> TIR1_YEAST 34 NGA (POTENTIAL).STGAKTSAISQITDGQIQATKAVSEQTENGAAKAFVGMGAGVVAAAAMLL> TR23_MOUSE 34 CSS (POTENTIAL).FPESCRPCTKCPQGIPVLQECNSTANTVCSSSVSNPRNWLFLLMLIVFCI> TREA_HUMAN 28 SGA (POTENTIAL).EGFGWDEGVVLMLLDRYGDRLTSGAKLAFLEPHCLAATLLPSLLLSLLPW> TREA_MOUSE 32 SGT (POTENTIAL).YEVQEGFGWTNGLALMLLDRYGDQLTSGTQLASLGPHCLVAALLLSLLLQ> TREA_RABIT 32 SGT (POTENTIAL).YEVQEGFGWTNGVALMLLDRYGDRLSSGTQLALLEPHCLAAALLLSFLTR> UPAR_BOVIN 25 GGA (POTENTIAL).SCCTGSGCNHPARDDQPGKGGAPKTSPAHLSFFVSLLLTARLWGATLLCT> UPAR_HUMAN 25 GAA (POTENTIAL).SCCTKSGCNHPDLDVQYRSGAAPQPGPAHLSLTITLLMTARLWGGTLLWT> UPAR_MOUSE 26 GGA (POTENTIAL).PTHLNVSVSCCHGSGCNSPTGGAPRPGPAQLSLIASLLLTLGLWGVLLWT> UPAR_RAT 26 GGA (POTENTIAL).QTHVNLSISCCNGSGCNRPTGGAPGPGPAHLILIASLLLTLRLWGIPLWT> VNL1_DROME 29 GSP (POTENTIAL).YYDNECTFGVGTEEEQLACGYRSGSPGLRILGGWLAMPLIILAIARTMSS> VNL2_DROME 44 EQL (POTENTIAL).HQVRFALRKSLEVKHLLTFGIYGNYYNNECTFGVGTAEEQLECGYKNPKI> VNL3_DROME 30 NGG (POTENTIAL).MELRQPHSQLMTFAIYGNYFDEYANGGAGRLGTLLFLLITPLIMMHLFRE> VNN1_CANFA 33 DPR (POTENTIAL).KPLSGPLLTVTLFGRIYEKDQTLKASSDPRSQVPGVMLLVIIPIVCSLSW> VNN1_HUMAN 33 GLT (POTENTIAL).KPTSGPVLTVTLFGRLYEKDWASNASSGLTAQARIIMLIVIAPIVCSLSW> VNN1_MOUSE 31 NAS (POTENTIAL).SLKPTSGPVLTIGLFGRLYGKDWASNASSDFIAHSLIIMLIVTPIIHYLC> VNN2_HUMAN 28 CGT (POTENTIAL).SGPILTVSLFGRWYTKDSLYSSCGTSNSAITYLLIFILLMIIALQNIVML> VNN3_HUMAN 24 SGA (POTENTIAL).LAPERHYEISRDGRLRSRSGAPLPVLVMALYGRVFEKDPPRLGQGSGKFQ> VNN3_MOUSE 24 GGA (POTENTIAL).LALERYYEVSRDGRLRSRGGAPLPILVMALYGRVFERDPPRLGQGPGKLQ> VSA1_TRYBB 32 DSSTPAEKCTGKKKDDCKDGCKWEAETCKDSSILLTKNFALSVVSAALVALLF> VSA8_TRYBB 32 DSSTDKCKGKLEDTCKKESNCKWEGETCKDSSILVNKQLALSVVSAAFAALLF> VSAC_TRYBB 32 DSSTTDKCKDKTKDECKSPNCKWEGETCKDSSILVTKKFALSLVSAAFASLLF> VSE2_TRYBR 38 NNS (BY SIMILARITY).RPNYRECEMRDGECNAKVAKTAEPDSKTNTTGNNSFAIKTSTLLLAVLLF> VSG2_TRYEQ 38 SNSKDGCELVEGVCKPVKQGEGENKEKTGTTNTTGSNSFVIKKAPLWLAFLLF> VSG4_TRYBR 36 DSSADKKEEKCKGKLEPECTKAPECKWEGETCKDSSILVNKQFTLSMISAAFM

145
> VSG7_TRYBR 32 DSSTECEGVKGTPPTGKAKVCGWIEGKCQDSSFLLSKQFALSVVSAAFAALLF> VSI1_TRYBB 38 SDS (BY SIMILARITY).KNECRPKKGTETTATGPGERTTPADGKANNTVSDSLLIKTSPLWLAFLLF> VSI2_TRYBB 38 NNS (BY SIMILARITY).CRTADECEMRDGECNAKVAKTAEPDSKTNTTGNNSFAIKTSTLLLAVLLF> VSI3_TRYBB 34 SFL (BY SIMILARITY).KGKTTPVCGWRKGKEGESDQDKEMCRNGSFLAKKKFALSVVSAAFTALLF> VSI4_TRYBB 32 DSS (BY SIMILARITY).TAEKCKGKGEKDCKSPDCKWEGGTCKDSSILANKQFALSVASAAFVALLF> VSI5_TRYBB 32 DSS (BY SIMILARITY).AKKCSDKKKEEECKSPNCKWDGKECKDSSILANKQFALSVASAAFVALLF> VSI6_TRYBB 32 DSSTTDKCKDKKKDDCKSPDCKWEGETCKDSSFILNKQFALSVVSAAFAALLF> VSIB_TRYBB 38 SNSKLDKEEAKRVAEQAATNQETEGKDGKTTNTTGSNSFLINKAPVLLAFLLL> VSM0_TRYBB 33 DGSGKTGDKHNCAFRKGKDGKEEPEKEKCCDGSFLVNKKFALMVYDFVSLLAF> VSM1_TRYBB 38 SNS (BY SIMILARITY).SNKCELKKDVKEKLEKESKETEGKDEKANTTGSNSFLIHKAPLLLAFLLF> VSM2_TRYBB 38 SNSNKKCTLDKEEAKKVADETAKDGKTGNTNTTGSSNSFVISKTPLWLAVLLF> VSM4_TRYBB 32 DSSTDKCKGKLEDTCKKESNCKWENNACKDSSILVTKKFALTVVSAAFVALLF> VSM5_TRYBB 32 NGS (BY SIMILARITY).PGQSAVCGFRKGKDGETDEPDKEKCRNGSFLTSKQFAFSVVSAAFMALLF> VSM5_TRYBR 33 DSS (BY SIMILARITY).TTTDKCKDKKKDDCKSPDCKWEGETCKDSSILLNKQFALMVSAAFVALLF> VSM6_TRYBB 32 DSS (BY SIMILARITY).PEKCKGKDAKTCGTTQGCKWEGETCKDSSILVTKKFALTVVSAAFVALLF> VSWA_TRYBR 38 SNS (BY SIMILARITY).DEKKRCKLSEEGKQAEKENQEGKDGKANTTGSSNSFVIKTSPLLLAVLLL> VSWB_TRYBR 38 SNS (BY SIMILARITY).GKCEAKPKAGTEAATTGPGERDAGATANTTGSSNSFVIKTSPLLFAFLLF> VSY1_TRYCO 36 SGSSSRPPSTDANTSQKGPLQRPEKSGESSHLPSGSSHGTKAIRSILHVALLM> VSY3_TRYCO 21 NSS (POTENTIAL).LGGKDMVPASEVTVPNSSNPTSRQNSVVQEPTTVSAAAITPLILPWTLLI> XPP2_HUMAN 30 AAR (BY SIMILARITY).VGPELQRRQLLEEFEWLQQHTEPLAARAPDTASWASVLVVSTLAILGWSV> XPP2_PIG 31 ARAVGPELQRRGLLEELSWLQRHTEPLSARAAPTTSLGSLMTVSALAILGWSV> YAP3_YEAST 34 NVG (POTENTIAL).TVNSSQTASFSGNLTTSTASATSTSSKRNVGDHIVPSLPLTLISLLFAFI> BCB1_ARATH 33 NAA (PROBABLE).STGGTTPPTAGGTTTPSGSSGTTTPAGNAASSLGGATFLVAFVSAVVALF> BCB2_ARATH 31 SGA (PROBABLE).SGGSPTPTTPTPGAGSTSPPPPPKASGASKGVMSYVLVGVSMVLGYGLWM> CBL1_ARATH 35 SVG (POTENTIAL).PRRIYFNGDNCVMPPPDSYPWLPNTGSHKSVGSLFAAMALLLIVFLHGNL> CBL2_ARATH 31 NAS (POTENTIAL).AFPRRIYFNGDNCVMPPPDSYPWLPNASPNIATSPFVILLITFLSVLILM> CBL4_ARATH 38 NFA (POTENTIAL).GWAFPRKVYFNGDECMLPPPDSYPFLPNSAQGNFASFSLTILLLLFISIW> CBL6_ARATH 29 SSS (POTENTIAL).LFNGDECVMPSPDDFPRLPKSAHSSSSSSAVISSVSVVFCFLLHHLLLLV> CBL7_ARATH 31 SSQ (PROBABLE).GGDGFPSKVFFNGEECSLPTILPMRSSQHRKHISVFLLALPVLALLILRA

146
> CBL8_ARATH 31 NSH (POTENTIAL).SKDGFPTKVLFNGQECSLPSVLPTSNSHRKHVSTFLLILTPFLALLFLRI> CBL9_ARATH 30 SGG (POTENTIAL).RDGFPAKVIFNGEECLLPDLLPMASGGRRNGAITVLSFITFYVAAFMVLL> CBLA_ARATH 29 SSG (POTENTIAL).DGFPTKLFFNGEECALPKHFPKKSSGHRRGISVSMSFVFATIAAFALMMD> CBLB_ARATH 23 SSG (POTENTIAL).VFFNGEECELPKYFPKKSSGMRLSGIRFLPSILLAITTFHAITDRLLTGV> COBR_ARATH 30 NGG (POTENTIAL).FPRRIYFNGDNCVMPPPDSYPFLPNGGSRSQFSFVAAVLLPLLVFFFFSA> ENL1_ARATH 33 SGS (PROBABLE).SPRHSVISPAPSPVEFEDGPALAPAPISGSVRLGGCYVVLGLVLGLCAWF> ENL2_ARATH 31 SSA (PROBABLE).MGPSGDGPSAAGDISTPAGAPGQKKSSANGMTVMSITTVLSLVLTIFLSA> ENL3_ARATH 32 GSA (PROBABLE).FFTGSSPSPAPSPALLGAPTVAPASGGSASSLTRQVGVLGFVGLLAIVLL> GLQ1_ARATH 24 STG (PROBABLE).PPLPPVSARAPTTTPGPQSTGEKSPNGQTRVALSLLLSAFATVFASLLLL> GLQ2_ARATH 26 NAQ (PROBABLE).PPLPPVTAKAPTSSPGTPSTNAQAPSGQTRITLSLLLSVFAMVLASLLLL> HIL1_ARATH 26 SSC (PROBABLE).KENSTARRNPGTSSSPSSSSSSCYKHINGFHGSLVVLFVSLSLILLGLLN> HIL2_ARATH 31 SSA (PROBABLE).CSKENTTASAGKQNPAGSAPPQPLPSSARKLCFSVFLLLSLLMMFLTLLD> PL13_ARATH 28 SDA (POTENTIAL).GTTRGSSSSSGDDSNVFQMIFGSDAPSRPRLTLLFSLLMISVLSLSTLLL> UGP1_ARATH 30 SGA (PROBABLE).DIWVVVILTTNTPEGGYSLLTTTNSGAYAFGVNGLVSSSFLFLLFCFFMF> UGP2_ARATH 25 SGA (POTENTIAL).GIWLVTVLTTNTPGGSYSNSGAFAFGVNGLVSSSLMFLHVLSHNSLSLFS> UGP3_ARATH 30 SGA (PROBABLE).SDDNWIVVVLTTSTPEGSYSPASNSGAFAFGVNGLVSSSLMFLLFCFFMF> UGP4_ARATH 29 SNG (POTENTIAL).IVVVLTTNTPEGSYSTATPTKQESNGFTFGIGLVSYLVIFMYSSFCFFLF

APPENDICE B
ALIGNEMENT MULTIPLE Cet appendice présente un alignement multiple effectué à partir des 468 séquences annoté comme ayant une ancre GPI dans la base de données Swiss-Prot, version Janvier 2004. L’alignement est effectué avec le logiciel ClustalW 1.83.
Sortie ClustalW
cd48_mouse -----------------nkstfytc-qvsnpvsskndtvyftl----pcd-------larcd48_rat -----------------nkstfytc-qvsnpvssendtlyfip----pct-------larcd48_human -----------------nysrcytc-qvsnsvsskngtvclsp----pct-------larlamp_human ------------------hygnytc-vaanklgvtnaslvlfr----pgs------vrgilamp_rat ------------------hygnytc-vaanklgvtnaslvlfr----pgs------vrgintri_mouse -------------------ygnytc-vasnklghtnasimlfg----pga------vsevntri_rat -------------------ygnytc-vasnklghtnasimlfg----pga------vsevntri_human -------------------ygnytc-vasnklghtnasimlfg----pga------vsevopcm_bovin -------------------ygnytc-vatnklgitnasitlyg----pga------vidgopcm_rat -------------------ygnytc-vatnklgntnasitlyg----pga------vidgopcm_human -------------------ygnytc-vatnklgntnasitlyg----pga------vidgcepu_chick -------------------lgntna-smilyeetttaltpwkg----pga------vhdggfr2_human ------------------lkannsk-elsmcf--telttnii-----pgsnk----vikpgfr2_mouse -----------------glkannsk-elsmcf--telttnis-----pgskk----viklgfr2_chick --------------------lnksk-eqslcysetqlttdtm-----pdqkt----fvdqcea6_human ---------------tvnnsgsymcqahnsatglnrttvtmit----vs-----------cea8_human --------------------gsyachttnsatgrnrttvrmit----vsda-----lvq-lach_drome --------------------------atn-rfgeaearvnlfe----tiip-----vcpplach_scham --------------------gkyqckaan-klgeareevelfe----tiip-----vcppc59b_mouse -----------------------srldvagiqskccqwglcn-----knldgle---epncd59_rat ---------------------ilsrleianvqyrccqadlcn-----ksfe--d---kpnc59a_mouse ----------------chgeiimdqleetklkfrccqfnlcn-----ksdg---------cd59_aottr --------------------rvsnqlsenelkyycckknlcn-----fnea------lkncd59_calsq --------------------qlsnqlsenelkyhccrenlcn-----fngi------lencd59_saisc --------------------risnqlsetqlkyhcckknlcn-----vkev------lencd59_cerae --------------------distllkeselqyfcckkdlcn-----fneq------lencd59_papsp ------------------fndistllkeselqyfcckedlc-------neq------lencd59_human --------------------dvttrlreneltyycckkdlcn-----fneq------l

148
cd59_hsvsa -------------------krisnqlsetqlkyhcckknlcn-----vnkg------iencd59_pig --------------decnfdfisrnlaekklkynccrkdlcn----------------kscd59_rabit ----------------cnfefisnrleenslkynccrkdlcn-----g---------pedly6a_mouse ----------------pniesmeilgtkvnvktsccqedlcn-----vavpng-----gsly6g_mouse ----------------ttldnteitgnavnvktycckedlcn-----aavptg-----gsly6f_mouse ----------------anlenmeilgttvnvntscckedlcn-----apfstg-----gsly6c_mouse ----------------fcpagvpikdpnirertsccsedlcn-----aavpta-----gsly6i_mouse ----------------deiekkfildpntkmnisccqedlcn-----aavptg-----gsly6h_human ----------------sdylmgfinsgilkvdvdccekdlcn-----gaagag-----hsly6h_mouse ----------------sdylmgfinsgilkvdvdccekdlcn-----gasvag-----rsly6e_human -----------------ipegvn--vgvasmgisccqsflcn-----fsaadgg---lraly6e_mouse ------------------senvnlnlgvasvnsyccqssfcn-----fsaaglg---lraly6e_chick ---------------vcpsagin--lgiaaasvyccdsflcn-----isgsss----vkapsca_human ---------------ncvddsqd--yyvgkknitccdtdlcn-----asgaha----lqppsca_mouse ---------------qceddsen--yylgkknitccysdlcn-----vngaht----lkply6d_human ------------------qgqvs----sgtsstqccqedlcnek--lhnaaptr---tally6d_mouse -----------------qqghvs----sgsevtqccqtdlcner--lvsaapgh---alltest_human --------------------vgcgrpnrpgvytnishhfewi----qklmaq-------stest_mouse --------------------igcgrpnrpgvytnishhynwi----qstmir-------nupar_bovin --------------------------thvn--vscctgsgcn----hparddqp---gkgupar_human --------------------------nhid--vscctksgcn----hpdldvqy---rsgupar_mouse --------------------vadsfpthlnvsvscchgsgcn----spt----------gupar_rat --------------------vadsfqthvnlsisccngsgcn----rpt----------gvnn1_canfa --------------------rlfsmkplsgplltvtlfgriy-----ekdqtlk---assvnn1_human --------------------rlfslkptsgpvltvtlfgrly-----ekdwasn---assvnn1_mouse ------------------dgrlvslkptsgpvltiglfgrly-----gkdwasn---assvnn3_human ---------------------lsgsqlaperhyeisrdgrlr-----srs--ga---plpvnn3_mouse ---------------------lsgsqlaleryyevsrdgrlr-----srg--ga---plpgas1_human ------------phpprpgsgaaasggrgdlpygpgrrss-------ggggr-----lapgas1_mouse -----------------pgggaaaaggrgdlphgpgrrssss-----gsggh-----wan5ntd_bovin ------------------sgdqdinvvsgyiskmkvlypaveg-----riqfs----ags5ntd_human ------------------sgdqdinvvstyiskmkviypaveg-----rikfs----tgs5ntd_mouse ------------------sgdqdisvvseyiskmkvvypaveg-----rikfs----aas5ntd_rat ------------------sgdqdisvvseyiskmkviypaveg-----rikfs----aas5ntd_disom ---------------------tdisvvssyikqmkvvypaveg-----rilfv----enscont_mouse ---------------------vpiprdgeyvvevrahsdggdgv--vsqvkis----gvscont_rat ----------------------piprdgeyvvevrahsdggdgv--vsqvkis----gvscont_human ---------------------vpiprdgeyvvevrahsdggdgv--vsqvkis----gapcadd_human -----------------imvtdsgkppmtnitdlrvqvcscrn----skvdcn----aagcadd_mouse ------------------mvtdsgkppmtnitdlkvqvcsckn----skvdcn----gagcadd_chick ----------------pisvtdsgkppltnntelklqvcsckk----srmdcs----asdthy1_human -----------------hhsghsppissqnvtvlrdklvkceg----isllaq----ntsthy1_macmu -----------------hhsghsppissqnvtvlrdklvkceg----isllaq----ntsthy1_mouse -----------------qvsganpmssnksisvyrdklvkcgg----isllvq----ntsthy1_rat -----------------rvsgqnptssnktinvirdklvkcgg----isllvq----ntsthy1_chick -----------------katndytgnqiknitvikdklekcvr----lslliq----ntsprio_atege --------------kgenftetdvkmmervveqmcitqyeres----qayyq-----rgsprio_mansp --------------kgenftetdvkmmervveqmcitqyekes----qayyq-----rgsprio_atepa -------------------tetdvkmmervveqmcitqyeres----qayyq-----rgsprio_cebap -------------------tetdvkmmervveqmcitqyeres----qayyq-----rgsprio_gorgo -------------------tetdvkmmervveqmcitqyeres----qayyq-----rgsprio_human -------------------tetdvkmmervveqmcitqyeres----qayyq-----rgsprio_pantr -------------------tetdvkmmervveqmcitqyeres----qayyq-----rgsprio_ponpy -------------------tetdvkmmervveqmcitqyeres----qayyq-----rgsprio_calja -------------------tetdvkmmervveqmcitqyekes----qayyq-----rgsprio_cerae -------------------tetdvkmmervveqmcitqyekes----qayyq-----rgsprio_cerat -------------------tetdvkmmervveqmcitqyekes----qayyq-----rgsprio_cermo -------------------tetdvkmmervveqmcitqyekes----qayyq-----rgsprio_cerpa -------------------tetdvkmmervveqmcitqyekes----qayyq-----rgsprio_certo -------------------tetdvkmmervveqmcitqyekes----qayyq-----rgsprio_colgu -------------------tetdvkmmervveqmcitqyekes----qayyq-----rgsprio_macfa -------------------tetdvkmmervveqmcitqyekes----qayyq-----rgsprio_prefr -------------------tetdvkmmervveqmcitqyekes----qayyq-----rgsprio_crigr --------------------etdvkmmervveqmcvtqyqkes----qayydg----rrsprio_crimi --------------------etdvkmmervveqmcvtqyqkes----qayydg----rrs

149
prio_rat --------------------etdvkmmervveqmcvtqyqkes----qayydg----rrsprio_mouse ---------------------tdvkmmervveqmcvtqyqkes----qayydg----rrsprio_mesau --------------------etdikimervveqmcttqyqkes----qayydg----rrsprnd_bovin --------------------kqdnklyqrvlwqlirelcstkh----cdfwle----rgaprnd_sheep --------------------kqdnklyqrvlwqlirelcsikh----cdfwle----rgaprnd_mouse --------------------kqdsklhqrvlwrlikeicsakh----cdfwle----rgaprnd_human --------------------kpdnklhqqvlwrlvqelcslkh----cefwle----rgaaces_torca -------------------kvhqrlrvqmcvfwnqflpkllna---tacdge----lsssaces_torma --------------------vhqrlrvqmcvfwnqflpkllna---tacdge----lsssbst1_mouse --------------------rpvkfl--mcvdhsthpdcimns---asasmr----resabst1_rat --------------------ppvkfl--mcvdhsthpdcamns---asasmw----respbst1_human --------------------rpvkll--qcvdhsthpdcalks---aaaatq----rkaphyap_human --------------------vdvciadgvcidaflkppmete----epqify----nasphyap_macfa ---------------------dvciadgvcidaslkppveteg---sppify----ntsscah4_human ------------------ydkeqtvsmkdnvr--plqqlgqrt---viksg-------apcah4_mouse ------------------ydedqklnmkdnvr--plqplgkrq---vfksh-------apppb1_human ---------------------ahvmafaacle--pytacdlap---pagtt----daahpppb2_human ---------------------ahvmafaacle--pytacdlap---pagtt----daahpppb3_human ---------------------ahvmafaacle--pytacdlap---pagtt----daahpppbi_human -----------------qsfvahvmafaacle--pytacdlap---pactt----daahpppbe_mouse -------------------yiahvmafaacle--pytdcglas---pagqs----savspppbi_bovin ----------------------himafagcve--pytdcnlpa---pttats-ipdaahlgfr4_mouse -----------------eafrklft-rnpcldg-aiqafdslq---psvlqd-----qtagfr4_rat ----------------------lft-rnpcldg-aiqafdssq---psvlqdqwnpyqnagfr4_human ------------------aiqafasgwppvlld-qlnpqgdpe---hsllqv-----sstcbl1_arath --------------------kgwafprriyfng---dncvmpp---pdsypw----lpntcobr_arath -------------------ekgwafprriyfng---dncvmpp---pdsypf----lpngcbl2_arath ------------------fekgwafprriyfng---dncvmpp---pdsypw----lpnacbl4_arath ----------------ftfkqgwafprkvyfng---decmlpp---pdsypf----lpnscbl6_arath ------------------------fprrilfng---decvmps---pddfpr----lpkscbl7_arath ---------------ginvpggdgfpskvffng---eecslp-----tilp-----mrsscbl8_arath ---------------gikvgskdgfptkvlfng---qecslp-----svlp-----tsnscbl9_arath ----------------invaerdgfpakvifng---eecllp-----dllp-----masgcbla_arath -----------------nipegdgfptklffng---eecalp-----khfp-----kksscblb_arath -----------------------gfpkrvffng---eecelp-----kyfp-----kkssgfr1_mouse -------------------clsdndygkdglag---asshitt----ksma----appscgfr1_rat -------------------clsdsdfgkdglag---asshitt----ksma----appscgfr1_human ----------------thlcisngnyekeglg----asshitt----ksma----appsccd52_human -----------------llltisllvmvqiqtg---lsgqndt----sqt------sspscd52_macfa ----------------fllltisllvmvqiqtg---vtsqnat----sq-------sspscd52_canfa -----------------------llvmiqiqtg---vlgnstt----prmttkkvksatpcd52_mouse ----------------tgslgqattaasgtnkn---ststkkt----plk------sgascd52_rat ----------------aattkaaattatkttta---vrktpgk----ppk------agascsa_dicdi ----------------tdt-atpsptptetatp---sptpkpt----stpe-----eteat10c_human ----------------pap-aaeetmttspgtp---apaaeet----mtts-----pgtpppbj_rat ----------------padenrpttpvqnsttt---ttttttt----tttt-----tttrbcb1_arath ----------------pg--stpstggttppta---ggtttps----gssg-----tttpg13a_dicdi ---------------eggetptpsttpsttps-----ttpsst----ptqs-----pdddg13b_dicdi ---------------tptpsttpsttpsttps-----ttpsst----ptqs-----pgddpsa_dicdi ---------------tvtptvtptvtptvtptp---tntpnpt----psqt-----stttcntr_human ----------------tttsttsslapp--ptt---kicdpge----lgsg-----ggpscntr_rat ----------------tttsttsslapp--ptt---kicdpge----lssg-----ggpsparb_trybb ---------------epepepepepepepepep---epepepe----pepe-----pepgparc_trybb ---------------epepepepepepepepep---epepepe----pepe-----pepgpara_trybb --------------tgpeetgpeetgpe-etgp---eetgpee----tepe-----pepgglq1_arath -----------------advtepplppvsarap---tttpgpq----stge-----kspnglq2_arath -----------------advtepplppvtakap---tsspgtp----stna-----qapscd14_mouse -----------------lpqvgnlslkgnpfld---seshsek----fnsg----vvtagcd14_rat -----------------lpevgslsltgnpflh---sesqsea----ynsg----vviatcd14_human --------------------vdnltldgnpflvpgtalphegs----mnsg----vvpaccw12_yeast ----------------pkngtstaapvtsteap---knttsaa----pths----vtsyttip1_yeast -----------------ssaessskavsssvap---ttssvst----stve----tasnacwp1_yeast ------------------etssassptasvisq---itdgqiq----apnt----vyeqttir1_yeast -----------------saapsstgaktsaisq---itdgqiq----atka----vseqtcwp2_yeast -----------------ttatteatttaapsst---vetvsps----stet----isqqt

150
dan1_yeast ----------------cstvtkpvsskaqstat---svtssas----rvid----vttn-dan4_yeast ----------------tskvispip-kassats---iahssas----ytvs----intn-by55_human ----------------ffsilftetgnytvtglkqrqhlefsh----negt-----lssgby55_mouse -----------------lsvlv--tgnhteirqrqrshpdfsh----ingt-----lssgvsi4_trybb ----------------agadttaekckgkg-ekdck-spdckw----eggt-----ckdsvsi5_trybb -----------------asdteakkcsdkkkeeeck-spnckw----dgke-----ckdsvsi6_trybb ----------------ggtqtttdkckdkk-kddck-spdckw----eget-----ckdsvsm5_trybr ---------------egaagtttdkckdkk-kddck-spdckw----eget-----ckdsvsa1_trybb ---------------tsrsetpaekctgkk-kddck-dg-ckw----eaet-----ckdsvsa8_trybb -----------------gteattdkckgkl-edtckkesnckw----eget-----ckdsvsm4_trybb -----------------gteattdkckgkl-edtckkesnckw----enna-----ckdsvsac_trybb ----------------tgteattdkckdkt-kdeck-spnckw----eget-----ckdsvsm6_trybb -----------------eteatpekckgkd-aktcgttqgckw----eget-----ckdsvsg4_trybr -------------agegaadkkeekckgkl-epectkapeckw----eget-----ckdsvsi3_trybb -----------------ceeenkgkttpvcgwrkgkegesdq-----dkem-----crngvsm5_trybb ------------------ekentpgqsavcgfrkgkdgetdep----dkek-----crngvsg7_trybr ------------------tiqnqtecegvkgtpptgkakvcgw----iegk-----cqdsugp1_arath -------------gigsgdiwvvvilttntpeggysllttt------nsga-----yafgugp3_arath -----------gigigsddnwivvvlttstpegsys--pas------nsga-----fafgugp2_arath -------------gidsdgiwlvtvlttntpggsys-----------nsga-----fafgugp4_arath ----------------keddwivvvlttntpegsystatptkq----esng-----ftfgnar4_human ------------------yhprgnwlqlrstgnlstyncqllk---------------asnar4_pantr ------------------yhprgnwlqlrstgnlstyncqllk---------------asvse2_trybr --------------kntgrrpnyrecemrdgecnakvaktaep------ds-------ktvsi2_trybb --------------ktqddcrtadecemrdgecnakvaktaep------ds-------ktvsg2_tryeq -----------------kedeckdgcelvegvckpvkqgegen---kektg-------ttvsm1_trybb ----------------vfntesnk-celkkdvkeklekesket---egkde-------kavsib_trybb ---------------------gkkkckldkeeakrvaeqaatnqetegkdgk------ttvsm2_trybb -----------------hndaenkkctldkeeakkvadetak----dgktg-------ntvswa_trybr ---------------dkeekdekkrckls-eegkq-aekenq----egkdg-------kavsi1_trybb -----------------eydseknecrpkkgtettatgpgertt----padg------kavswb_trybr ----------------teetigkceakpkagteaattgpgerd------aga------tacd24_mouse -----------------tqiycnqtsvapfpgnqnisaspnps----nattr-----gggcd24_rat -----------------tqiycnqtsvapfsgnqsisaapnpt----nattr-----sgccd24_human -----------------ssetttgtssnssqstsnsglapnpt----nattk-----aagfol1_human ---------------srgsgrciqmwfdpaqgnpneevarfya----aam--------sgfol1_mouse ---------------srgsgrciqmwfdpaqgnpneevarfya----eam--------sgfol2_human -------------------grciqmwfdsaqgnpneevarfya----aamhv-----nagfol2_mouse -----------------gsgrciqmwfdstqgnpnedvvkfya----sfmt-------sggp63_leich ----------------safeeggyitcppyvev-cqgnv-qaa----kdggn-----aaagp63_leido ----------------safeeggyitcppyvev-cqgnv-qaa----kdggn-----aaagp63_leima ----------------nafegggyitcppyvev-cqgnv-qaa----kdggn-----taagpc4_human ---------------------ceyqqcpsefdynatdha-gks----aneka-----dsagpc4_mouse ----------------------eyqqcpsefeynatdhs-gks----aneka-----dsahya2_mouse ------------------nylqkhfrcqcylgwggeqcqrnyk----gaagn-----asrhya2_rat ------------------sylqmhfrchcylgwggeqcqwnhk----raagd-----asrhya2_human ------------------dhlqthfrcqcylgwsgeqcqwdhr----qaagg-----asert4r_human ------------------csrknrtrshcrlgqagsggg-gtg----dsegs-----galrt4r_macfa ------------------csrknrtrshcrlgqagsggg-gtg----dsegs-----galrt4r_mouse ------------------csrknrtrshcrlgqagsgas-gtg----daegs-----galrt4r_rat ------------------csrknrtrshcrlgqagsgss-gtg----daegs-----galdaf_human -----------------atrstpvsrttkhfhettpnkgsgtt----sgttr-----llsdaf_ponpy -----------------atrstpvsrttkhfhettpnkgsgtt----sgtts-----llshil1_arath ------------------nltcskensta---rrnpgtsssps----sssss-----cykhil2_arath ----------------rcnlacskenttasagkqnpagsappq----plpss-----arkxpp2_human -----------------tirekvgpelqrrqlleefewlqqht----eplaa-----rapxpp2_pig -----------------airekvgpelqrrglleelswlqrht----eplsa-----raamdp1_human ------------------snltqape-eepipldqlggscrth----ygyss-----gasmdp1_rabit -----------------vsnqaqvpe-eepisleqlggscrtq----ygys------eapmdp1_pig ----------------qasnhaqvpg-eepiplgqleascrtn----ygys------aapmdp1_sheep -----------------asdhkqapg-eepiplgqleascrtk----ygys------gtpmdp1_mouse -----------------vssnmqspe-evpitlkeldgscrty----ygys------qahmdp1_rat -----------------vsnimqvpe-eetipvekldgscrtf----yghs------rapsm7a_human -----------------fceaqegsyfreaqhwqllpedgima----ehll------ghasm7a_mouse -----------------rceaqegsylreaqhwellpedrala----eqlm------gha

151
trea_mouse -------------ggggeyevqegfgwtnglalmlldr---yg----dqlts-----gtqtrea_rabit -------------ggggeyevqegfgwtngvalmlldr---yg----drlss-----gtqtrea_human -----------------eyevqegfgwdegvvlmlldr---yg----drlts-----gakgpc1_human -----------------srkssssrtplthalpglseqegqkt----saasc-----pqpgpc1_rat -----------------skkssssrtplihalpglseqegqkt----saatr-----pepreck_human ------------------sptlashvplsaliisqvqvsssvp----sagvr-----arpreck_mouse ------------------sptleshvhlsaliisqvqvssslp----ssavv-----grpnara_mouse ----------------sldspkrkksnfncfysgstq-aanv------sslg-----srenarb_mouse ------------------dsperkksnfncfyngsaq-tvnid----fsisg-----srenara_rat -------vrtqgyneifldspkrkksnyncly----------------ssag-----trenarb_rat -------vrtqgyneifldspkrkksnyncly----------------ssag-----arevnl1_drome ------------------------giygnyydnectfgvgtee----eqlacg----yrsvnl2_drome iseeshqvrfalrkslevkhlltfgiygnyynnectfgvgtae----eqlecg----ykncntr_chick ------------------ttevqitettststssfmpppttki----cdkg------agvvnn2_human -----------------nkngssgpiltvslfgrwytkdslys-----scg------tsnvsm0_trybb ------------------arqgtgktgdkhncafrkgkdgkee----peke------kccppbi_rat --------------------gclepytdcglappadenrpttp----vqnsa-----itmgfr1_chick ------------------nenaigkdntpgvstshissensfa----lpts------fyphyr1_canal ------------------ttvdvipvphsmpsnttdssssvpt----idtne-----ngsvnl3_drome ------------------vtrikmelrqphsqlmtfaiygnyf----deya-------ngefa4_human -------------------lqvsvcckerksesahpvgspges----gtsg------wrglynx_mouse -------------------kscvpscfetvydgyskhasatsc----cqyy-------lccw14_yeast -------------------eahsssaasstvsqetvssalpts----tavis----tfse5ntd_boomi ---------------------qdaeivmkymnstspittaldg---rvtflkt----nqagp85_trycr --------------------kdripipkrgpgsqveggterrh---iprieg-----vradaf1_mouse --------------------tqhvpvtkttvrhpirtstdkge----pntg------gdrgfr4_chick --------------------ehlqptkvageerllrgstrlss----etss------paagp42_rat -------------------gknyscqaenkvsrdisepkkfp-----lvvs-------gtsp63_strpu -----------------iygtprinswdprmnwdlsmnldate----epes-------gsyap3_yeast ----------------ggniftvnssqtasfsgnlttstasat----stss------krnpag1_trybb ------------------nkstvvvdedsgksfvvlgnretvq----eekll-----eemppb_bommo --------------------fsglyeqthvphrmawaacmgpg----rhvc-------vsgp63_leigu -------------------ikgvidfegdaadtaamrrwrerm----talat-----vtasag1_yeast ------------------liaypssasgsqlsgiqqnftstsl----mist------yegvsy1_tryco -------------------qiawessrppstdantsqkgplqr----peks------gesbcb2_arath -------------------pesppsggsptpttptpgagstsp----pppp------kasenl3_arath -------------------ghtggfftgsspspapspallgap----tvapa-----sggenl1_arath -------------------slvvisprhsvispapspvefedg----palap------apnar3_human ------------------edksqgninnptpgpvpvpgpkshp-----sas-------sgampm_helvi ----------------pststtsttaapttvtqptitepstpt----lpel-------tdpl13_arath -------------------mdvmggttrgsssssgddsnvfq-----mifgs-----dapampm_manse ----------------pseattvappaettvtpstfpptvap-----attp-------apbm86_boomi -----------------gkavckekseattaattttkakdkd-----pdpg------kssaph4_drome ------------------pakdfddscedhkdgqkdrpldkpn----pkrn------gatmm17_mouse -----------------wlvcgepladaedvgpgpqgrsgaqd-----glav-----cscmm17_human --------------------hdqsrsedgyevcsctsgasspp----gapgp----lvaamm25_human ----------------cpapssgpraprppkatpvsetcdcqc-----eln--------qconn_drome ------------------chdelsdptelplsrdlmdvrsnvg----qdms-------tagas1_yeast -------------------sgssgsssssssssasssssskkn----aatn-----vkanmkc7_yeast -----------------qtstaalsiskstsstsstgmlspts----ssspr-----kenhyap_cavpo -------------------ddddeppitddtsqnqdsisdits----sapp-------ssmsa1_sarmu ------------------sktthcylcepdptkkghndkncav-----lia------vgatr23_mouse -----------------yydpkfpescrpctkcpqgipvlqec----nstan------tvenl2_arath ----------------psgsamgpsgdgpsaagdistpagapg----qkks------sangas1_caeel -----------------llgvclkntpgvtlspsdnsitdapg-----gnd------ladpona_dicdi -------------------caanqkipttsyivscnstpssns----ttds------dssfc3b_human ------------------knvssetvnititqglavstissfs----ppgy-------qvvsy3_tryco -------------------eanislggkdmvpasevtvpnssn----ptsr------qnsaxo1_human ----------------ttgpggdgipaevhivrnggtsmmven----mavrp-----aphgfr3_human ----------------aakmrfhsqlfsqdwphptfavmahqn-----enp-------avmsln_human --------------------qddldtlglglqggipngylvld----lsvqet----lsgdaf_cavpo -------------------sdklkkthvykvdsfacgasnhwl----adia------kedmm19_mouse ------------------sqtpdtnsstgdvtpsttdtvlgtt----pstmg-----stlgp46_leiam -----------------rlgqrrcgarpspcasvcvswprerr--------------tecnar3_mouse -------------------yktvklilffikssrsgsrseips----lcilwq----yap

152
Suite de l’alignement cd48_mouse ssgvcwtatwlvvttliihrillt-------cd48_rat ssgvhwiaawlvvtlsiipsilla-------cd48_human sfgvewiaswlvvtvptilglllt-------lamp_human ngsislavp-lwllaasllcllskc------lamp_rat ngsislavp-lwllaaslfcllskc------ntri_mouse nngtsrragciwllpllvlhlllkf------ntri_rat nngtsrragciwllpllvlhlllkf------ntri_human sngtsrragcvwllpllvlhlllkf------opcm_bovin vnsasralaclwlsgtlfahffikf------opcm_rat vnsasralaclwlsgtffahffikf------opcm_human vnsasralaclwlsgtllahffikf------cepu_chick nsgawrrgscawllalplaqlarqf------gfr2_human nsgpsrarpsaaltvlsvlmlkqal------gfr2_mouse ysgscrarlstaltalpllmvtla-------gfr2_chick kaagsrhraarilpavpivllklll------cea6_human --gsapvlsavatvgitigvlarvali----cea8_human --gsspglsaratvsimigvlarvali----lach_drome acgqayiagaedvsatsfalvgisarllfarlach_scham acgqayggdaaeis-tsmalilisti-----c59b_mouse naetsslrktallgtsvlvailkfcf-----cd59_rat ngaisllgktallvtsvlaailkpcf-----c59a_mouse -----slgktpllgtsvlvailnlcflshl-cd59_aottr ggttlskktvlllvipflvaawslhp-----cd59_calsq ggttlskktvlllvtpflaaawslhp-----cd59_saisc ggttlskktilllvtpflaaawsrhp-----cd59_cerae ggtslsektvvllvtlllaaawclhp-----cd59_papsp ggtslsektvlllvtpllaaawclhp-----cd59_human ggtslsektvlllvtpflaaawslhp-----cd59_hsvsa ikrtisdk-allllalflvtawnfpl-----cd59_pig datissgk-tallvilllvatwhfcl-----cd59_rabit dgtaltgr-tvllvapllaaarnlcl-----ly6a_mouse twtmagv-llfslssvllqtll---------ly6g_mouse swtmagv-llfslvsvllqtfl---------ly6f_mouse twtmtrv-lllnlgsvflqtll---------ly6c_mouse twtmagv-llfslssvilqtll---------ly6i_mouse swttagv-llfslgsvllqtlm---------ly6h_human pwalagg-lllslgpallwagp---------ly6h_mouse pwalagg-lllslgpallwagp---------ly6e_human svtllgaglllsllpallrfgp---------ly6e_mouse sipllglglllsll-allqlsp---------ly6e_chick syavlalgilvsfv-yvlrare---------psca_human aaailal---lpalg-lllwgpgql------psca_mouse p-ttlgl---ltvlcslllwgssrl------ly6d_human ahsalslglalsllavilapsl---------ly6d_mouse ssvtlglatslsllt-vmalcl---------test_human gmsqpdpswpllffpllwalpllgpv-----test_mouse gllrpdpvplllfltlawassllrpa-----upar_bovin gapktspahlsffvsllltarlwgatllct-upar_human aapqpgpahlsltitllmtarlwggtllwt-upar_mouse gaprpgpaqlsliasllltlglwg-vllwt-upar_rat gapgpgpahliliasllltlrlwg-iplwt-vnn1_canfa dprsqvpgvmllviipivcs----lsw----vnn1_human gltaqariimliviapivcs----lsw----vnn1_mouse dfiahsliimlivt-piihy----lc-----vnn3_human vlvmalygrvfekdpprlgqgsgkfq-----vnn3_mouse ilvmalygrvferdpprlgqgpgklq-----gas1_human rgawtplas---illlllgplf---------gas1_mouse rsawtpfaclllllllllgshl---------5ntd_bovin hccgsfslif-lsvlaviiilyq--------5ntd_human hchgsfslif-lslwavifvlyq--------5ntd_mouse hyqgsfplvi-lsfwamililyq--------

153
5ntd_rat hyqgsfplii-lsfwavilvlyq--------5ntd_disom atlpiinlkiglslfafltwflhcs------cont_mouse tlsssllsll-lpslgflv-ysef-------cont_rat tlssgllsll-lpslgflvfysef-------cont_human tlspsllgll-lpafgilv-ylef-------cadd_human alrfslpsvlllslfslac-l----------cadd_mouse alhlslsllllfsllsllsgl----------cadd_chick alhismt-lillslfslfc-l----------thy1_human wllllllslsllqatdfms-l----------thy1_macmu wlxllllslsllqatdfms-l----------thy1_mouse wmlllllslsllqaldfis-l----------thy1_rat wllllllslsflqatdfis-l----------thy1_chick wllllllslpllqavdfvs-l----------prio_atege s-mvlfssppvillisfli------------prio_mansp s-mvlfssppvillisfli------------prio_atepa s-mvlfssppvillisfliflivg-------prio_cebap s-mvlfssppvillisfliflivg-------prio_gorgo s-mvlfssppvillisfliflivg-------prio_human s-mvlfssppvillisfliflivg-------prio_pantr s-mvlfssppvillisfliflivg-------prio_ponpy s-mvlfssppvillisfliflivg-------prio_calja s-mvlfssppvillisfliflivg-------prio_cerae s-mvlfssppvillisfliflivg-------prio_cerat s-mvlfssppvillisfliflivg-------prio_cermo s-mvlfssppvillisfliflivg-------prio_cerpa s-mvlfssppvillisfliflivg-------prio_certo s-mvlfssppvillisfliflivg-------prio_colgu s-mvlfssppvillisfliflivg-------prio_macfa s-mvlfssppvillisfliflivg-------prio_prefr s-mvffssppvillisfliflivg-------prio_crigr s-avlfssppvillisfliflivg-------prio_crimi s-avlfssppvillisfliflivg-------prio_rat s-avlfssppvillisfliflivg-------prio_mouse sstvlfssppvillisfliflivg-------prio_mesau s-avlfssppvillisfliflmvg-------prnd_bovin glrvtldqpmmlcllvfiwfivk--------prnd_sheep glqvtldqpmmlcllvfiwfivk--------prnd_mouse alrvavdqpamvcllgfvwfivk--------prnd_human glrvtmhqpvllcllaliwlmvk--------aces_torca g---tssskg-iifyvlfsilylif------aces_torma g---tssskg-iifyvlfsilylify-----bst1_mouse sl-haigdas-llisllvalasssqa-----bst1_rat al-haigdis-liisllvalasssqa-----bst1_human sl-yteqrag-liiplflvlasrtql-----hyap_human s---tlsatm-fivsilfliissvasl----hyap_macfa s---tvsttm-fivnilfliissvasl----cah4_human gr-plpwalpallgpmlacllagflr-----cah4_mouse gq-llslplptllvptltclvanflq-----ppb1_human grsvvpallp-llagtlllletatap-----ppb2_human grsvvpallp-llagtlllletatap-----ppb3_human grsvvpallp-llagtlllletatap-----ppbi_human ----vaaslp-llagtllllgasaap-----ppbe_mouse g--ymstllc-llagkmlmlmaaaep-----ppbi_bovin a--aspppla-llagamllllaptly-----gfr4_mouse g--ccfprvs--wlyaltalalqall-----gfr4_rat g--ccflwvs--smsiltalalqall-----gfr4_human g--ralerrs--llsilpvlalpall-----cbl1_arath gshksvgslfaamalllivflhgnl------cobr_arath gs-rsqfsfvaavllpllvffffsa------cbl2_arath spniatspfvillitflsvlilm--------cbl4_arath aq-gnfasfsltillllfisiw---------cbl6_arath ahssssssavissvsvvfcfllhhllllv--cbl7_arath qhrkhisvfllal-pvlallilra-------cbl8_arath -hrkhvstflliltpflallflri-------cbl9_arath grrngaitvlsfitfyvaafmvll-------

154
cbla_arath ghrrgisvsmsfvfatiaafalmmd------cblb_arath gmrlsgirflpsillaittfhaitdrlltgvgfr1_mouse glsslpvmvftalaallsvslaets------gfr1_rat slsslpvlmltalaallsvslaets------gfr1_human glspllvlvvtalstll--sltets------cd52_human assnisggiflffvanaiihlfcfs------cd52_macfa assnlsgggflffvanaiihlfyfs------cd52_canfa alsslgggsvllflantliqlfyls------cd52_mouse siidagacsflff-antlmclfyls------cd52_rat sitdvgactflff-antlmclfyls------csa_dicdi pssattlisplslivifisfvlli-------t10c_human asshylsctivgiivlivllivfv-------ppbj_rat vqnsasslgpataplawhywprr--------bcb1_arath agnaasslggatflvafvsavvalf------g13a_dicdi gstsstlstsfylitllfliqqfi-------g13b_dicdi gstsstlsisfylitlllltqqfi-------psa_dicdi g-sastvvaslsliifsmilslc--------cntr_human apflvsvpitlalaaaaatasslli------cntr_rat ipfltsvpvtlvlaaaaatannlli------parb_trybb aatlksvalpfaiaavglvaaf---------parc_trybb aatlksvalpfaiaaaalvaaf---------para_trybb aatlksvalpfavaaaalvaaf---------glq1_arath gqtrvalslllsafatvfasllll-------glq2_arath gqtritlslllsvfamvlasllll-------cd14_mouse apssqavals-gtlalllgdrlfv-------cd14_rat alspgsagls-gtlalllghrlfv-------cd14_human arstlsvgvs-gtlvllqgargfa-------cw12_yeast gaaakalpaa-gallagaaalll--------tip1_yeast gqrvnagaasfgavvagaaalll--------cwp1_yeast enagakaavgmgagalavaaayll-------tir1_yeast engaakafvgmgagvvaaaamll--------cwp2_yeast engaakaavgmgagalaaaamll--------dan1_yeast gankfn-ngvfgaaaiagaaalll-------dan4_yeast gaynfdkdnifgtaivavvallll-------by55_human --flqekvwvmlvtslvalqal---------by55_mouse --flqvkawgmlvtslvalqalytl------vsi4_trybb s-ilankqfalsvasaafvallf--------vsi5_trybb s-ilankqfalsvasaafvallf--------vsi6_trybb s-filnkqfalsvvsaafaallf--------vsm5_trybr s-illnkqfal-mvsaafvallf--------vsa1_trybb s-illtknfalsvvsaalvallf--------vsa8_trybb s-ilvnkqlalsvvsaafaallf--------vsm4_trybb s-ilvtkkfaltvvsaafvallf--------vsac_trybb s-ilvtkkfalslvsaafasllf--------vsm6_trybb s-ilvtkkfaltvvsaafvallf--------vsg4_trybr s-ilvnkqftlsmisaafm------------vsi3_trybb 3 s-flakkkfalsvvsaaftallf--------vsm5_trybb s-fltskqfafsvvsaafmallf--------vsg7_trybr s-fllskqfalsvvsaafaallf--------ugp1_arath vnglvsssflfllfc----ffmf--------ugp3_arath vnglvssslmfllfc----ffmf--------ugp2_arath vnglvssslmflhvlshnslslfs-------ugp4_arath i-glvsylvifmyss--fcfflf--------nar4_human skkcipdpiaiaslsfltsviifsksrv---nar4_pantr skkcipdpiaiaslsfltsviifsksrv---vse2_trybr nttgn-nsfaiktstlllavllf--------vsi2_trybb nttgn-nsfaiktstlllavllf--------vsg2_tryeq nttgs-nsfvikkaplwlafllf--------vsm1_trybb nttgs-nsflihkaplllafllf--------vsib_trybb nttgs-nsflinkapvllaflll--------vsm2_trybb nttgssnsfvisktplwlavllf--------vswa_trybr nttgssnsfviktsplllavlll--------vsi1_trybb nnt-vsdslliktsplwlafllf--------vswb_trybr nttgssnsfviktspllfafllf--------cd24_mouse sslqstagllalslsllhlyc----------

155
cd24_rat sslqstagllalslsllhlyc----------cd24_human galqstaslfvvslsllhlys----------fol1_human agpwaawpfllslalml-lwlls--------fol1_mouse aglhgtwpllcslslvl-lwvis--------fol2_human emlhgtgglllslalmlqlwllg--------fol2_mouse tvphaavllvpslapvlslwlpg--------gp63_leich grrgpraaa-tallvaallaval--------gp63_leido grrgpraaa-tallvaallaval--------gp63_leima grrgpraaa-tallvaallaval--------gpc4_human g-vrpgaqa-ylltvfcilflvmqrewr---gpc4_mouse ggahaeakp-yllaalcilflavqgewr---hya2_mouse awagshltsllglvavaltwtl---------hya2_rat awagahlasllglvamtltwtl---------hya2_human awagshltsllalaalaftwtl---------rt4r_human psltcsltp-lglalv--lwtvlgpc-----rt4r_macfa pslacslap-lglalv--lwtvlgpc-----rt4r_mouse palacslap-lglalv--lwtvlgpc-----rt4r_rat palacslap-lglalv--lwtvlgpc-----daf_human ghtcft---ltgllg-tlvtmgllt------daf_ponpy ghkcft---ltgllg-tlvtmgllt------hil1_arath hingfhgslvvlfvslslillglln------hil2_arath lc--fs---vflllsllmmfltlld------xpp2_human dta-swasvlvvstlailgwsv---------xpp2_pig ptt-slgslmtvsalailgwsv---------mdp1_human slhrhwglllaslaplvlclsll--------mdp1_rabit slhrrpgallaslsllllslgll--------mdp1_pig slhlppgsllaslvplll-lslp--------mdp1_sheep slhlqpgsllaslvtlllslcll--------mdp1_mouse sihlqtgalvaslasllfrlhll--------mdp1_rat sihlqigallaslaslvfslhpl--------sm7a_human calaas-lwlgvlptltlgllvh--------sm7a_mouse ralaas-fwlgvlptlilgllvh--------trea_mouse laslgp-hcl--vaalllslllq--------trea_rabit lallep-hcl--aaalllsfltr--------trea_human laflep-hcl--aatllpslllsllpw----gpc1_human ptfllplllflaltvarprwr----------gpc1_rat hyffllflftlvlaaarprwr----------reck_human schslllplslglalh-llwtyn--------reck_mouse lfhslllllswgltvh-llwtrp--------nara_mouse scvslflvvllgllvqqltlaep--------narb_mouse scvslflvvllgllvqqltlaep--------nara_rat scvslflvvltsllvqllclaep--------narb_rat scvslflvvlpsllvqllclaep--------vnl1_drome gspglrilggwlampliilaiartmss----vnl2_drome --------------pki--------------cntr_chick gsgavavcwtaglvlaaygvlfi--------vnn2_human saityllifillmiialqnivml--------vsm0_trybb dgsflvnkkfalmvydfvsllaf--------ppbi_rat nnvllslqllvsmlllvgtalvvs-------gfr1_chick stplilmtialslflflsssvvl--------hyr1_canal sivtggksilfglivsmvvlfm---------vnl3_drome gagrlgtllfllitplimmhlfre-------efa4_human gdtpsplcllllllllilrllril-------lynx_mouse ngagfatpvtlalvpallatfwsll------cw14_yeast gsgnvleagksvfiaavaamli---------5ntd_boomi sdaclnlaspflvllvlvvfyhl--------gp85_trycr napvgsgllplllllglwvfaal--------daf1_mouse yiyghtclitltvlhvmlsligylt------gfr4_chick pchqaasllqlwlpptlavlshfmm------gp42_rat asmksttvviwlpvsclvgwpwllrf-----sp63_strpu qrhlpvcgvlslvvttllalmlh--------yap3_yeast vgdhivpslpltlisllfafi----------pag1_trybb aicgvgradslrrtlallfllf---------ppb_bommo aatvptaallslllaafitlrhqcfl-----gp63_leigu allgivlaamailvvwlllitip--------

156
sag1_yeast kasiffsaelgsiiflllsyllf--------vsy1_tryco shlpsgsshgtkairsilhvallm-------bcb2_arath gaskgvmsyvlvgvsmvlgyglwm-------enl3_arath sassltrqvgvlgfvgllaivll--------enl1_arath isgsvrlggcyvvlglvlglcawf-------nar3_human klllpqfgmviilisvsainlfval------ampm_helvi samtsfaslfiislgailhlil---------pl13_arath srprltllfsllmisvlslstlll-------ampm_manse gsgniaalsvvsllvtlainmva--------bm86_boomi aaavsatglllllaatsvtaasl--------aph4_drome vvgaslipiltaataailrgrgl--------mm17_mouse tsdahrlalpslllltpllwgl---------mm17_human tmllllpplspgalwtaaqaltl--------mm25_human aagrwpapipllllpllvggvasr-------conn_drome ganslaqgmtiivslqvalmisrg-------gas1_yeast laqvvftsiislsiaagvgfalv--------mkc7_yeast gghnlnppffarfitaifhhi----------hyap_cavpo hilpkdlswclfllsifsqhwkyll------msa1_sarmu gsrptarsvfgvaapcilallhft-------tr23_mouse csssvsnprnwlfllmlivfci---------enl2_arath gmtvmsittvlslvltiflsa----------gas1_caeel ssvghgfnilsaisvylltvlvf--------pona_dicdi sgstvmiglassllfafatllalf-------fc3b_human sfclvmvllfavdtglyfsvktni-------vsy3_tryco vvqepttvsaaaitplilpwtlli-------axo1_human pgtvishsvamliligslel-----------gfr3_human rpqpwvpslfsctlplilllslw--------msln_human tpcllgpgpvltvlalllastla--------daf_cavpo lrrdfsnaqnissllqvlgaaqtq-------mm19_mouse dipsatdsaslsfsanvtllga---------gp46_leiam acpalfdgarlrccalvvcagaapag-----nar3_mouse sishgihhftrcfccklhraia---------

APPENDICE C
SÉQUENCES D’ENTRAÎNEMENT DU RÉSEAU DE NEURONES ARTIFICIELS
Cet appendice présente les séquences du groupe d’entraînement du réseau de neurones artificiels. Le tableau contient les 79 séquences GPI retenues comme étant représentatives du signal GPI. Il contient également 79 séquences ayant une très faible probabilité d’être à ancre GPI et qui sont utilisées comme séquences discriminantes
Séquences d’entraînement
GPI entraînement non GPI entraînement #1 : GLQ2_ARATH #2 : ENL1_ARATH #3 : CBL6_ARATH #4 : BCB1_ARATH #5 : YAP3_YEAST #6 : XPP2_HUMAN #7 : VSM0_TRYBB #8 : VSG7_TRYBR #9 : VSA1_TRYBB #10 : UPAR_BOVIN #11 : TREA_HUMAN #12 : TR23_MOUSE #13 : TIR1_YEAST #14 : THY1_CHICK #15 : TEST_HUMAN #16 : T10C_HUMAN #17 : SM7A_HUMAN #18 : SAG1_YEAST #19 : RECK_HUMAN #20 : PSCA_HUMAN #21 : PSA_DICDI #22 : PRND_HUMAN #23 : PRIO_ATEPA #24 : PPBI_BOVIN #25 : PONA_DICDI #26 : PL13_ARATH
#1 : 1-143E_HUMAN #2 : 2-7B2_HUMAN #3 : 4-TERA_HUMAN #4 : 5-PEX1_HUMAN #5 : sw:AF31_HUMAN #6 : sw:PGN_HUMAN #7 : sw:PRSX_HUMAN #8 : sw:SPAS_HUMAN #9 : sw:BAT1_HUMAN #10 : sw:ACPM_HUMAN #11 : sw:ACTA_HUMAN #12 : sw:THEA_HUMAN #13 : sw:A2A1_HUMAN #14 : sw:ILVE_HELPJ #15 : sw:BCAT_HUMAN #16 : sw:ISCS_HELPJ #17 : sw:NFS1_HUMAN #18 : sw:CSD_CHLTR #19 : sw:KYNU_HUMAN #20 : sw:BIN1_HUMAN #21 : sw:AAKG_HUMAN #22 : sw:AMY1_HUMAN #23 : sw:ANR5_HUMAN #24 : sw:ASB2_HUMAN #25 : sw:BCL3_HUMAN #26 : sw:GABB_HUMAN

158
GPI entraînement non GPI entraînement #27 : PARB_TRYBB #28 : PARA_TRYBB #29 : PAG1_TRYBB #30 : OPCM_BOVIN #31 : NTRI_HUMAN #32 : NAR3_HUMAN #33 : MSLN_HUMAN #34 : MSA1_SARMU #35 : MM25_HUMAN #36 : MM17_HUMAN #37 : MKC7_YEAST #38 : MDP1_HUMAN #39 : LYNX_MOUSE #40 : LY6I_MOUSE #41 : LY6A_MOUSE #42 : LAMP_RAT #43 : LACH_DROME #44 : HYR1_CANAL #45 : HYA2_HUMAN #46 : GPC1_RAT #47 : GP85_TRYCR #48 : GP42_RAT #49 : GFR3_HUMAN #50 : GFR2_MOUSE #51 : GAS1_CAEEL #52 : G13B_DICDI #53 : FOL1_HUMAN #54 : FC3B_HUMAN #55 : EFA4_HUMAN #56 : DAN4_YEAST #57 : DAN1_YEAST #58 : DAF1_MOUSE #59 : CW12_YEAST #60 : CSA_DICDI #61 : CONN_DROME #62 : CNTR_CHICK #63 : CEPU_CHICK #64 : CEA6_HUMAN #65 : CD59_CALSQ #66 : CD52_CANFA #67 : CD48_HUMAN #68 : CD24_HUMAN #69 : CD14_HUMAN #70 : CAH4_HUMAN #71 : CADD_HUMAN #72 : C59A_MOUSE #73 : BY55_HUMAN #74 : BST1_HUMAN #75 : BM86_BOOMI #76 : AXO1_HUMAN #77 : AMPM_MANSE #78 : ACES_TORMA #79 : 5NTD_BOVIN
#27 : sw:RN5A_MOUSE #28 : sw:P532_HUMAN #29 : sw:SYNP_HUMAN #30 : sw:V031_FOWPV #31 : sw:Y379_HUMAN #32 : sw:YB23_HUMAN #33 : sw:APL1_HUMAN #34 : sw:CG1C_HUMAN #35 : sw:CYCH_HUMAN #36 : sw:CYTB_HUMAN #37 : sw:IR01_HCMVA #38 : sw:J1L_HCMVA #39 : sw:TRS1_HCMVA #40 : sw:US04_HCMVA #41 : sw:CLPP_HUMAN #42 : sw:NBPX_HUMAN #43 : sw:COE3_HUMAN #44 : sw:DBPA_HUMAN #45 : sw:YB1_HUMAN #46 : sw:CA12_BOVIN #47 : sw:CO5A_PIG #48 : sw:HEM6_HUMAN #49 : sw:CORC_HUMAN #50 : sw:CKS2_HUMAN #51 : sw:CISY_HELPJ #52 : sw:CKS1_HUMAN #53 : sw:CIT2_HUMAN #54 : sw:CI30_HUMAN #55 : sw:DFFB_HUMAN #56 : sw:CIDA_HUMAN #57 : sw:FS27_HUMAN #58 : sw:CIT1_HUMAN #59 : sw:DCTD_HUMAN #60 : sw:RIBD_CHLTR #61 : sw:DSRA_HUMAN #62 : sw:RED1_HUMAN #63 : sw:CD5R_HUMAN #64 : sw:KVB1_HUMAN #65 : sw:CH10_HUMAN #66 : sw:EKI1_HUMAN #67 : sw:CBX2_HUMAN #68 : sw:I1BC_MOUSE #69 : sw:ICE2_HUMAN #70 : sw:CATA_HUMAN #71 : sw:CBFC_HUMAN #72 : sw:MK21_YEAST #73 : sw:MPI3_HUMAN #74 : sw:CAH2_HUMAN #75 : sw:CYNT_HELPJ #76 : sw:CAR8_HUMAN #77 : sw:TESC_HUMAN #78 : sw:A60D_DROME #79 : sw:EFER_HUMAN

APPENDICE D
ANALYSE ROC DU RÉSEAU DE NEURONES ARTIFICIELS Cet appendice présente l’analyse ROC effectué sur le jeu de séquences du test de validation du réseau de neurones artificiels. Cette analyse a permis la détermination du seuil d’acceptation des prédictions (0.90)
Analyse ROC
Positif si ≥ Sensibilité 1 - Spécificité-1.00000 1.000 1.000
.00001 1.000 .888
.00002 .985 .769
.00003 .978 .679
.00004 .963 .642
.00005 .963 .604
.00006 .955 .567
.00006 .955 .545
.00008 .955 .530
.00009 .955 .515
.00011 .955 .500
.00012 .955 .493
.00013 .955 .485
.00014 .955 .478
.00016 .955 .463
.00018 .955 .455
.00021 .955 .448
.00023 .955 .433
.00026 .955 .425

160
Positif si ≥ Sensibilité 1 - Spécificité
.00039 .955 .403
.00043 .955 .396
.00051 .955 .388
.00056 .955 .381
.00071 .955 .373
.00087 .955 .366
.00089 .955 .358
.00096 .955 .351
.00108 .955 .343
.00119 .955 .336
.00140 .955 .328
.00159 .955 .321
.00164 .955 .313
.00173 .955 .306
.00215 .955 .299
.00254 .955 .291
.00263 .955 .284
.00269 .955 .276
.00277 .948 .276
.00303 .948 .269
.00326 .948 .261
.00347 .948 .254
.00388 .948 .246
.00452 .948 .239
.00499 .948 .231
.00509 .948 .224
.00563 .948 .216
.00670 .948 .209
.00788 .948 .201
.00912 .948 .194
.00998 .948 .187
.01046 .948 .179
.01185 .948 .172
.01312 .948 .164
.01356 .940 .164
.01386 .940 .157
.01414 .940 .149
.01533 .940 .142
.01637 .940 .134
.01694 .940 .127

161
Positif si ≥ Sensibilité 1 - Spécificité
.01694 .940 .127
.01792 .940 .119
.02127 .940 .112
.03034 .940 .104
.04442 .933 .104
.05710 .933 .097
.06247 .933 .090
.09170 .933 .082
.13811 .925 .082
.16051 .925 .075
.16901 .925 .067
.21070 .918 .067
.38235 .918 .060
.60521 .910 .060
.71156 .903 .060
.73189 .903 .052
.74027 .903 .045
.81220 .896 .045
.88600 .896 .037
.89993 .896 .030
.92121 .896 .022
.93895 .888 .022
.94674 .881 .022
.96438 .881 .015
.97781 .873 .015
.98500 .866 .015
.99180 .858 .015
.99221 .851 .015
.99317 .843 .015
.99400 .836 .015
.99515 .828 .015
.99695 .821 .015
.99786 .813 .015
.99818 .799 .015
.99857 .791 .015
.99869 .784 .015
.99873 .776 .015
.99891 .761 .015
.99909 .754 .015
.99919 .746 .015

162
Positif si ≥ Sensibilité 1 - Spécificité.99919 .746 .015.99927 .746 .007.99927 .739 .007.99932 .731 .007.99937 .724 .007.99939 .716 .007.99943 .709 .007.99953 .701 .007.99961 .679 .007.99962 .672 .007.99966 .657 .007.99971 .649 .007.99975 .642 .007.99976 .634 .007.99978 .627 .007.99980 .619 .007.99981 .612 .007.99982 .604 .007.99983 .597 .007.99984 .582 .007.99985 .575 .007.99986 .567 .007.99988 .560 .007.99988 .530 .007.99990 .500 .000.99991 .478 .000.99992 .463 .000.99993 .440 .000.99994 .410 .000.99995 .388 .000.99996 .373 .000.99997 .358 .000.99998 .299 .000.99999 .231 .000
1.00000 .067 .0002.00000 .000 .000

APPENDICE E
SÉQUENCES D’ENTRAÎNEMENT DU MODÈLE DE MARKOV CACHÉ Cet appendice présente les séquences du groupe d’entraînement du modèle de Markov caché. L’appendice contient les 87 séquences GPI, ayant une annotation de la position du site d’ancrage retenues comme étant représentatives du signal GPI.
Séquences d’entraînement
> 5NTD_HUMAN 25 STGSTGSHCHGSFSLIFLSLWAVIFVLYQ> ACES_TORMA 27 SSGSSGTSSSKGIIFYVLFSILYLIFY> AMPM_HELVI 28 DSADSAMTSFASLFIISLGAILHLIL> APH4_DROME 24 NGANGATVVGASLIPILTAATAAILRGRGL> AXO1_HUMAN 22 NMANMAVRPAPHPGTVISHSVAMLILIGSLEL> BM86_BOOMI 27 SAASAAAVSATGLLLLLAATSVTAASL> BST1_HUMAN 25 APSAPSLYTEQRAGLIIPLFLVLASRTQL> BY55_HUMAN 28 SSGSSGFLQEKVWVMLVTSLVALQAL> C59A_MOUSE 23 SDGSDGSLGKTPLLGTSVLVAILNLCFLSHL> CADD_CHICK 31 DALDALHISMTLILLSLFSLFCL> CAH4_HUMAN 22 SGASGAPGRPLPWALPALLGPMLACLLAGFLR> CD14_HUMAN 20 NSGNSGVVPACARSTLSVGVSGTLVLLQGARGFA> CD24_HUMAN 29 GGAGGALQSTASLFVVSLSLLHLYS

164
> CD48_HUMAN 27 SFGSFGVEWIASWLVVTVPTILGLLLT> CD52_HUMAN 25 SASSASSNISGGIFLFFVANAIIHLFCFS> CD59_HUMAN 24 NGGNGGTSLSEKTVLLLVTPFLAAAWSLHP> CEA6_HUMAN 26 GSAGSAPVLSAVATVGITIGVLARVALI> CEA8_HUMAN 21 DALDALVQGSSPGLSARATVSIMIGVLARVALI> CEPU_CHICK 27 SGASGAWRRGSCAWLLALPLAQLARQF> CNTR_HUMAN 20 SGGSGGGPSAPFLVSVPITLALAAAAATASSLLI> CONT_MOUSE 31 SSSSSSLLSLLLPSLGFLVYSEF> CONN_DROME 2 AGAAGANSLAQGMTIIVSLQVALMISRG> DAF_HUMAN 18 SGTSGTTSGTTRLLSGHTCFTLTGLLGTLVTMGLLT> EFA4_HUMAN 19 SGTSGTSGWRGGDTPSPLCLLLLLLLLILRLLRIL> FOL1_HUMAN 27 SGASGAGPWAAWPFLLSLALMLLWLLS> FOL2_HUMAN 25 NAGNAGEMLHGTGGLLLSLALMLQLWLLG> GAS1_CAEEL 27 DSSDSSVGHGFNILSAISVYLLTVLVF> GFR1_CHICK 11 SHISHISSENSFALPTSFYPSTPLILMTIALSLFLFLSSSVVL> GFR3_HUMAN 24 NPANPAVRPQPWVPSLFSCTLPLILLLSLW> GFR4_RAT 27 NAGNAGCCFLWVSSMSILTALALQALL> GP42_RAT 23 GTAGTASMKSTTVVIWLPVSCLVGWPWLLRF> GPC1_HUMAN 22 SAASAASCPQPPTFLLPLLLFLALTVARPRWR> HYA2_HUMAN 25 GASGASEAWAGSHLTSLLALAALAFTWTL> LACH_SCHAM 33 GDAGDAAEISTSMALILISTI> LAMP_HUMAN 27 NGSNGSISLAVPLWLLAASLLCLLSKC> LY6G_MOUSE 35 GVLGVLLFSLVSVLLQTFL> MDP1_HUMAN 24 SGASGASSLHRHWGLLLASLAPLVLCLSLL> MM17_MOUSE 30 SDASDAHRLALPSLLLLTPLLWGL> MM19_MOUSE 35 DSADSASLSFSANVTLLGA> MM25_HUMAN 27 AAGAAGRWPAPIPLLLLPLLVGGVASR> MSLN_HUMAN 8 GGIGGIPNGYLVLDLSVQETLSGTPCLLGPGPVLTVLALLLASTLA> NAR3_HUMAN 23 SSGSSGKLLLPQFGMVIILISVSAINLFVA

165
> NTRI_RAT 27 NGTNGTSRRAGCIWLLPLLVLHLLLKF> OPCM_RAT 27 NSANSASRALACLWLSGTFFAHFFIKF> PPB1_HUMAN 21 DAADAAHPGRSVVPALLPLLAGTLLLLETATAP> PPB_BOMMO 16 GPGGPGRHVCVSAATVPTAALLSLLLAAFITLRHQCFL> PPBE_MOUSE 23 SAVSAVSPGYMSTLLCLLAGKMLMLMAAAEP> PPBI_HUMAN 25 DAADAAHPVAASLPLLAGTLLLLGASAAP> PPBJ_RAT 30 NSANSASSLGPATAPLAWHYWPRR> PRIO_CRIGR 27 SSASSAVLFSSPPVILLISFLIFLIVG> PRND_BOVIN 26 GAGGAGLRVTLDQPMMLCLLVFIWFIVK> PSCA_MOUSE 22 NGANGAHTLKPPTTLGLLTVLCSLLLWGSSRL> RECK_MOUSE 21 SSASSAVVGRPLFHSLLLLLSWGLTVHLLWTRP> RT4R_HUMAN 24 SGASGALPSLTCSLTPLGLALVLWTVLGPC> SM7A_MOUSE 32 AASAASFWLGVLPTLILGLLVH> SP63_STRPU 26 GSQGSQRHLPVCGVLSLVVTTLLALML> T10C_HUMAN 27 ASSASSHYLSCTIVGIIVLIVLLIVFV> TEST_HUMAN 24 SGMSGMSQPDPSWPLLFFPLLWALPLLGPV> TEST_MOUSE 24 NGLNGLLRPDPVPLLLFLTLAWASSLLRPA> THY1_HUMAN 19 CEGCEGISLLAQNTSWLLLLLLSLSLLQATDFMSL> TR23_MOUSE 29 CSSCSSSVSNPRNWLFLLMLIVFCI> TREA_HUMAN 23 SGASGAKLAFLEPHCLAATLLPSLLLSLLPW> UPAR_MOUSE 21 GGAGGAPRPGPAQLSLIASLLLTLGLWGVLLWT> VNN1_HUMAN 28 GLTGLTAQARIIMLIVIAPIVCSLSW> VNN2_HUMAN 23 CGTCGTSNSAITYLLIFILLMIIALQNIVML> VNN3_MOUSE 19 GGAGGAPLPILVMALYGRVFERDPPRLGQGPGKLQ> XPP2_HUMAN 25 AARAARAPDTASWASVLVVSTLAILGWSV> BCB1_ARATH 28 NAANAASSLGGATFLVAFVSAVVALF> CBL1_ARATH 30 SVGSVGSLFAAMALLLIVFLHGNL> CBL2_ARATH 26 NASNASPNIATSPFVILLITFLSVLILM> CBL7_ARATH 26 SSQSSQHRKHISVFLLALPVLALLILRA
166
> CBLA_ARATH 24 SSGSSGHRRGISVSMSFVFATIAAFALMMD> COBR_ARATH 25 NGGNGGSRSQFSFVAAVLLPLLVFFFFSA> ENL1_ARATH 28 SGSSGSVRLGGCYVVLGLVLGLCAWF> GLQ1_ARATH 19 STGSTGEKSPNGQTRVALSLLLSAFATVFASLLLL> HIL1_ARATH 21 SSCSSCYKHINGFHGSLVVLFVSLSLILLGLLN> UGP1_ARATH 25 SGASGAYAFGVNGLVSSSFLFLLFCFFMF> PL13_ARATH 23 SDASDAPSRPRLTLLFSLLMISVLSLSTLLL> CW12_YEAST 29 GAAGAAAKALPAAGALLAGAAALLL> CW14_YEAST 32 NVLNVLEAGKSVFIAAVAAMLI> DAN1_YEAST 34 NGVNGVFGAAAIAGAAALLL> HYR1_CANAL 26 NGSNGSSIVTGGKSILFGLIVSMVVLFM> MKC7_YEAST 29 NGGNGGHNLNPPFFARFITAIFHHI> SAG1_YEAST 17 STSSTSLMISTYEGKASIFFSAELGSIIFLLLSYLLF> TIP1_YEAST 28 GQRGQRVNAGAASFGAVVAGAAALLL> YAP3_YEAST 29 NVGNVGDHIVPSLPLTLISLLFAFI> CD59_HSVSA 25 NIKNIKRTISDKALLLLALFLVTAWNFPL

APPENDICE F
MODÈLE DE MARKOV CACHÉ DE Initial Cet appendice présente le modèle de Markov caché conçu à partir des connaissances de la structure GPI proposée dans la littérature.
Modèle de Markov caché
Probabilité de transition entre états -------------------------------------------------------- [ (0:1) ] -------------------------------------------------------- 0:[ (1:1) ] 1:[ (2:1) ] 2:[ (3:0.1) (4:0.1) (5:0.1) (6:0.1) (7:0.1) (8:0.1) (9:0.1) (10:0.1) (11:0.1) (12:0.1) ] 3:[ (4:0.111111) (5:0.111111) (6:0.111111) (7:0.111111) (8:0.111111) (9:0.111111) (10:0.111111) (11:0.111111) (12:0.111111) ] 4:[ (5:0.125) (6:0.125) (7:0.125) (8:0.125) (9:0.125) (10:0.125) (11:0.125) (12:0.125) ] 5:[ (6:0.142857) (7:0.142857) (8:0.142857) (9:0.142857) (10:0.142857) (11:0.142857) (12:0.142857) ] 6:[ (7:0.166667) (8:0.166667) (9:0.166667) (10:0.166667) (11:0.166667) (12:0.166667) ]

168
7:[ (8:0.2) (9:0.2) (10:0.2) (11:0.2) (12:0.2) ] 8:[ (9:0.25) (10:0.25) (11:0.25) (12:0.25) ] 9:[ (10:0.333333) (11:0.333333) (12:0.333333) ] 10:[ (11:0.5) (12:0.5) ] 11:[ (12:1) ] 12:[ (13:1) ] 13:[ (14:1) ] 14:[ (15:1) ] 15:[ (16:0.05) (17:0.05) (18:0.05) (19:0.05) (20:0.05) (21:0.05) (22:0.05) (23:0.05) (24:0.05) (25:0.05) (26:0.05) (27:0.05) (28:0.05) (29:0.05) (30:0.05) (31:0.05) (32:0.05) (33:0.05) (34:0.05) (35:0.05) ] 16:[ (17:0.0526316) (18:0.0526316) (19:0.0526316) (20:0.0526316) (21:0.0526316) (22:0.0526316) (23:0.0526316) (24:0.0526316) (25:0.0526316) (26:0.0526316) (27:0.0526316) (28:0.0526316) (29:0.0526316) (30:0.0526316) (31:0.0526316) (32:0.0526316) (33:0.0526316) (34:0.0526316) (35:0.0526316) ] 17:[ (18:0.0555556) (19:0.0555556) (20:0.0555556) (21:0.0555556) (22:0.0555556) (23:0.0555556) (24:0.0555556) (25:0.0555556) (26:0.0555556) (27:0.0555556) (28:0.0555556) (29:0.0555556) (30:0.0555556) (31:0.0555556) (32:0.0555556) (33:0.0555556) (34:0.0555556) (35:0.0555556) ] 18:[ (19:0.0588235) (20:0.0588235) (21:0.0588235) (22:0.0588235) (23:0.0588235) (24:0.0588235) (25:0.0588235) (26:0.0588235) (27:0.0588235) (28:0.0588235) (29:0.0588235) (30:0.0588235) (31:0.0588235) (32:0.0588235) (33:0.0588235) (34:0.0588235) (35:0.0588235) ] 19:[ (20:0.0625) (21:0.0625) (22:0.0625) (23:0.0625) (24:0.0625) (25:0.0625) (26:0.0625) (27:0.0625) (28:0.0625) (29:0.0625) (30:0.0625) (31:0.0625) (32:0.0625) (33:0.0625) (34:0.0625) (35:0.0625) ] 20:[ (21:0.0666667) (22:0.0666667) (23:0.0666667) (24:0.0666667) (25:0.0666667) (26:0.0666667) (27:0.0666667) (28:0.0666667) (29:0.0666667) (30:0.0666667) (31:0.0666667) (32:0.0666667) (33:0.0666667) (34:0.0666667) (35:0.0666667) ] 21:[ (22:0.0714286) (23:0.0714286) (24:0.0714286) (25:0.0714286) (26:0.0714286) (27:0.0714286) (28:0.0714286) (29:0.0714286) (30:0.0714286) (31:0.0714286) (32:0.0714286) (33:0.0714286) (34:0.0714286) (35:0.0714286) ]

169
22:[ (23:0.0769231) (24:0.0769231) (25:0.0769231) (26:0.0769231) (27:0.0769231) (28:0.0769231) (29:0.0769231) (30:0.0769231) (31:0.0769231) (32:0.0769231) (33:0.0769231) (34:0.0769231) (35:0.0769231) ] 23:[ (24:0.0833333) (25:0.0833333) (26:0.0833333) (27:0.0833333) (28:0.0833333) (29:0.0833333) (30:0.0833333) (31:0.0833333) (32:0.0833333) (33:0.0833333) (34:0.0833333) (35:0.0833333) ] 24:[ (25:0.0909091) (26:0.0909091) (27:0.0909091) (28:0.0909091) (29:0.0909091) (30:0.0909091) (31:0.0909091) (32:0.0909091) (33:0.0909091) (34:0.0909091) (35:0.0909091) ] 25:[ (26:0.1) (27:0.1) (28:0.1) (29:0.1) (30:0.1) (31:0.1) (32:0.1) (33:0.1) (34:0.1) (35:0.1) ] 26:[ (27:0.111111) (28:0.111111) (29:0.111111) (30:0.111111) (31:0.111111) (32:0.111111) (33:0.111111) (34:0.111111) (35:0.111111) ] 27:[ (28:0.125) (29:0.125) (30:0.125) (31:0.125) (32:0.125) (33:0.125) (34:0.125) (35:0.125) ] 28:[ (29:0.142857) (30:0.142857) (31:0.142857) (32:0.142857) (33:0.142857) (34:0.142857) (35:0.142857) ] 29:[ (30:0.166667) (31:0.166667) (32:0.166667) (33:0.166667) (34:0.166667) (35:0.166667) ] 30:[ (31:0.2) (32:0.2) (33:0.2) (34:0.2) (35:0.2) ] 31:[ (32:0.25) (33:0.25) (34:0.25) (35:0.25) ] 32:[ (33:0.333333) (34:0.333333) (35:0.333333) ] 33:[ (34:0.5) (35:0.5) ] 34:[ (35:1) ] 35:[ (36:1) ] 36:[ (37:1) ] 37:[ (38:1) ] 38:[ (39:1) ] 39:[ (40:1) ]

170
40:[ (41:1) ] 41:[ ] Probabilité d’émission des états Les numéros correspondent aux acides aminés 0 = a 10 = m 1 = c 11 = n 2 = d 12 = p 3 = e 13 = q 4 = f 14 = r 5 = g 15 = s 6 = h 16 = t 7 = I 17 = v 8 = k 18 = w 9 = l 19 = y 20 = $ -------------------------------------------------------- 0:[ (0:0.052356) (1:0.0285578) (2:0.0618753) (3:3.96637e-05) (4:3.96637e-05) ] 0:[ (5:0.166587) (6:3.96637e-05) (7:3.96637e-05) (8:3.96637e-05) (9:3.96637e-05) ] 0:[ (10:3.96637e-05) (11:0.214184) (12:0.00475964) (13:3.96637e-05) (14:3.96637e-05) ] 0:[ (15:0.466445) (16:0.00475964) (17:3.96637e-05) (18:3.96637e-05) (19:3.96637e-05) ] 0:[ (20:0) ] 1:[ (0:0.26178) (1:0.00475964) (2:0.0237982) (3:0.0142789) (4:0.00951928) ] 1:[ (5:0.290338) (6:0.0190386) (7:0.0190386) (8:0.00475964) (9:0.00951928) ] 1:[ (10:0.00475964) (11:0.00475964) (12:0.0285578) (13:0.0142789) (14:0.0237982) ] 1:[ (15:0.214184) (16:0.0285578) (17:0.0237982) (18:0.000237982) (19:0.000237982) ] 1:[ (20:0) ] 2:[ (0:0.385531) (1:0.00475964) (2:0.00475964) (3:0.00475964) (4:0.00951928) ] 2:[ (5:0.228463) (6:0.00951928) (7:0.0190386) (8:0.00475964) (9:0.0428367) ] 2:[ (10:0.00951928) (11:0.000158655) (12:0.00475964) (13:0.0142789) (14:0.0237982) ] 2:[ (15:0.152308) (16:0.0713946) (17:0.00951928) (18:0.000158655) (19:0.000158655) ] 2:[ (20:0) ] 3:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 3:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 3:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 3:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 3:[ (20:0) ]

171
4:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 4:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 4:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 4:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 4:[ (20:0) ] 5:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 5:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 5:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 5:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 5:[ (20:0) ] 6:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 6:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 6:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 6:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 6:[ (20:0) ] 7:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 7:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 7:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 7:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 7:[ (20:0) ] 8:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 8:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 8:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 8:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 8:[ (20:0) ] 9:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 9:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 9:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 9:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 9:[ (20:0) ] 10:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 10:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 10:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 10:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 10:[ (20:0) ] 11:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 11:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ]

172
11:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 11:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 11:[ (20:0) ] 12:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 12:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 12:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 12:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 12:[ (20:0) ] 13:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 13:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 13:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 13:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 13:[ (20:0) ] 14:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 14:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 14:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 14:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 14:[ (20:0) ] 15:[ (0:0.109524) (1:0.0210884) (2:0.0136054) (3:0.0136054) (4:0.029932) ] 15:[ (5:0.0809524) (6:0.0387755) (7:0.0482993) (8:0.0244898) (9:0.121769) ] 15:[ (10:0.0183673) (11:0.0238095) (12:0.0891156) (13:0.0278912) (14:0.0544218) ] 15:[ (15:0.129932) (16:0.055102) (17:0.0680272) (18:0.0197279) (19:0.0115646) ] 15:[ (20:0) ] 16:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 16:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 16:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 16:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 16:[ (20:0) ] 17:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 17:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 17:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 17:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 17:[ (20:0) ] 18:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 18:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 18:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 18:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ]

173
18:[ (20:0) ] 19:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 19:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 19:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 19:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 19:[ (20:0) ] 20:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 20:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 20:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 20:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 20:[ (20:0) ] 21:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 21:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 21:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 21:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 21:[ (20:0) ] 22:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 22:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 22:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 22:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 22:[ (20:0) ] 23:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 23:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 23:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 23:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 23:[ (20:0) ] 24:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 24:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 24:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 24:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 24:[ (20:0) ] 25:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 25:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 25:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 25:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 25:[ (20:0) ]

174
26:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 26:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 26:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 26:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 26:[ (20:0) ] 27:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 27:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 27:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 27:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 27:[ (20:0) ] 28:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 28:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 28:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 28:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 28:[ (20:0) ] 29:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 29:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 29:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 29:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 29:[ (20:0) ] 30:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 30:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 30:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 30:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 30:[ (20:0) ] 31:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 31:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 31:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 31:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 31:[ (20:0) ] 32:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 32:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 32:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 32:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 32:[ (20:0) ] 33:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 33:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ]

175
33:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 33:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 33:[ (20:0) ] 34:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 34:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 34:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 34:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 34:[ (20:0) ] 35:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 35:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 35:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 35:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 35:[ (20:0) ] 36:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 36:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 36:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 36:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 36:[ (20:0) ] 37:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 37:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 37:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 37:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 37:[ (20:0) ] 38:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 38:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 38:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 38:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 38:[ (20:0) ] 39:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 39:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 39:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 39:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ] 39:[ (20:0) ] 40:[ (0:0.102402) (1:0.0195954) (2:0.00379267) (3:0.005689) (4:0.0647914) ] 40:[ (5:0.0436157) (6:0.0148546) (7:0.0606827) (8:0.00790139) (9:0.325221) ] 40:[ (10:0.0233881) (11:0.005689) (12:0.0363464) (13:0.0145386) (14:0.0183312) ] 40:[ (15:0.0891277) (16:0.0521492) (17:0.0790139) (18:0.0205436) (19:0.0123262) ]

176
40:[ (20:0) ] 41:[ (0:0) (1:0) (2:0) (3:0) (4:0) ] 41:[ (5:0) (6:0) (7:0) (8:0) (9:0) ] 41:[ (10:0) (11:0) (12:0) (13:0) (14:0) ] 41:[ (15:0) (16:0) (17:0) (18:0) (19:0) ] 41:[ (20:1) ]

APPENDICE G
ANALYSE ROC DU MODÈLE DE MARKOV CACHÉ Cet appendice présente l’analyse ROC effectué sur le jeu de séquences du test de validation du modèle de Markov caché. Cette analyse a permis la détermination du seuil d’acceptation des prédictions (4.06).
Analyse ROC
Positif si ≥ Sensibilité 1 - Spécificité-130.9740 1.000 1.000-100.3043 1.000 .985
-70.0919 1.000 .970-64.6060 .985 .970-55.4391 .985 .955-50.1545 .985 .939-47.4981 .985 .924-42.9586 .985 .909-39.6593 .985 .894-39.1454 .985 .879-38.3041 .985 .864-37.1962 .985 .848-35.0567 .985 .833-32.8017 .985 .818-32.2445 .985 .803-31.5099 .985 .788-30.5648 .985 .773-30.3170 .985 .758-29.2388 .985 .742
178
Positif si ≥ Sensibilité 1 - Spécificité
-28.1578 .985 .727-27.9751 .985 .712-27.0816 .985 .697-25.9900 .985 .682-25.2441 .985 .667-24.7227 .985 .652-24.5436 .985 .636-23.9543 .970 .636-22.6563 .970 .621-21.8518 .970 .606-21.6638 .970 .591-21.0570 .970 .576-20.1328 .970 .561-19.6065 .970 .545-19.5209 .970 .530-19.3858 .970 .515-19.2032 .970 .500-19.0145 .970 .485-18.7405 .970 .470-18.3186 .970 .455-17.9946 .970 .439-17.2377 .970 .424-16.3429 .970 .409-16.0575 .970 .394-15.8336 .970 .379-15.6831 .970 .364-15.6013 .970 .348-14.9256 .970 .333-14.0684 .970 .318-13.6879 .970 .303-13.3658 .970 .288-12.8991 .970 .273-12.5839 .970 .258-12.5476 .970 .242-12.5282 .955 .242-12.4082 .955 .227-12.0672 .955 .212-11.6922 .939 .212-11.4169 .939 .197-11.2216 .939 .182-11.1037 .939 .167

179
Positif si ≥ Sensibilité 1 - Spécificité
-10.7137 .939 .152-9.7289 .939 .136-8.5161 .939 .121-7.7638 .939 .106-7.2049 .924 .106-6.0013 .924 .091-4.6696 .924 .076-2.7353 .924 .061-1.2497 .909 .061-.7189 .894 .061.0006 .879 .061.4212 .879 .045
1.6873 .864 .0453.1218 .864 .0304.0603 .864 .0154.7855 .848 .0155.6475 .833 .0156.4128 .818 .0157.2031 .803 .0158.5886 .788 .0159.3235 .773 .0159.5479 .758 .0159.6811 .742 .015
10.0141 .727 .01510.9471 .712 .01511.8113 .697 .01512.1591 .682 .01512.6793 .667 .01513.6032 .652 .01514.1753 .636 .01514.2842 .621 .01514.4655 .606 .01514.6496 .591 .01514.7155 .576 .01514.7702 .561 .01514.9972 .545 .01515.3026 .530 .01515.6030 .515 .01515.9688 .515 .00016.5249 .500 .00016.9249 .485 .000

180
Positif si ≥ Sensibilité 1 - Spécificité
16.9901 .470 .00017.2923 .455 .00017.6959 .439 .00017.9814 .424 .00018.6005 .409 .00019.4418 .394 .00020.0143 .379 .00021.0939 .364 .00022.0600 .348 .00022.5085 .333 .00022.8683 .318 .00022.9592 .303 .00023.0787 .288 .00023.2409 .273 .00023.4228 .258 .00023.5270 .242 .00023.6071 .227 .00023.7527 .212 .00023.9069 .197 .00024.4451 .182 .00025.6716 .167 .00026.5989 .152 .00026.8402 .136 .00027.0458 .121 .00027.2307 .106 .00027.4733 .091 .00027.8510 .076 .00028.4029 .061 .00029.5027 .045 .00031.8686 .030 .00034.1874 .015 .00035.8590 .000 .000
-130.9740 1.000 1.000-100.3043 1.000 .985

APPENDICE H
GRAMMAIRE STOCHASTIQUE GPI Cet appendice présente la première partie de la grammaire stochastique GPI dérivé du modèle de Markov caché entraîné. Cette grammaire est décrite en énumérant les probabilitéd’émission des états ainsi que les règles de production stochastiques. La grammaire complète est accessible sur le disque annexé.
Probabilité d’émission
Chaque état a la possibilité d’émettre les caractères représentant les 20 acides aminés ainsi
que le symbole z.
État S : départ
État 1-41 : a|c|d|e|f|g|h|i|k|l|m|n|p|q|r|s|t|v|w|y|z
État F : fin
La section suivante contient les règles de production stochastique de la grammaire du signal
GPI.

182
Règles de production stochastique
P(S1)= 1.0000 P(1a2)= 1.0 0.0697 0.0697P(1c2)= 1.0 0.0341 0.0341P(1d2)= 1.0 0.0111 0.0111P(1e2)= 1.0 0.0000 0.0000P(1f2)= 1.0 0.0000 0.0000P(1g2)= 1.0 0.1627 0.1627P(1h2)= 1.0 0.0000 0.0000P(1i2)= 1.0 0.0000 0.0000P(1k2)= 1.0 0.0000 0.0000P(1l2)= 1.0 0.0000 0.0000P(1m2)= 1.0 0.0000 0.0000P(1n2)= 1.0 0.2551 0.2551P(1p2)= 1.0 0.0000 0.0000P(1q2)= 1.0 0.0000 0.0000P(1r2)= 1.0 0.0000 0.0000P(1s2)= 1.0 0.3953 0.3953P(1t2)= 1.0 0.0000 0.0000P(1v2)= 1.0 0.0000 0.0000P(1w2)= 1.0 0.0000 0.0000P(1y2)= 1.0 0.0000 0.0000P(1z2)= 1.0 0.0000 0.0000
P(2a3)= 1.0 0.2093 0.2093 P(3a4)= 0.5351 0.3720 0.1991c 1.0 0.0000 0.0000 c 0.5351 0.0116 0.0062d 1.0 0.0465 0.0465 d 0.5351 0.0000 0.0000e 1.0 0.0116 0.0116 e 0.5351 0.0000 0.0000f 1.0 0.0116 0.0116 f 0.5351 0.0000 0.0000g 1.0 0.3139 0.3139 g 0.5351 0.2441 0.1306h 1.0 0.0116 0.0116 h 0.5351 0.0000 0.0000i 1.0 0.0116 0.0116 i 0.5351 0.0116 0.0062k 1.0 0.0000 0.0000 k 0.5351 0.0116 0.0062l 1.0 0.0116 0.0116 l 0.5351 0.0511 0.0273m 1.0 0.0116 0.0116 m 0.5351 0.0116 0.0062n 1.0 0.0000 0.0000 n 0.5351 0.0000 0.0000p 1.0 0.0341 0.0341 p 0.5351 0.0000 0.0000q 1.0 0.0116 0.0116 q 0.5351 0.0232 0.0124r 1.0 0.0000 0.0000 r 0.5351 0.0232 0.0124s 1.0 0.2209 0.2209 s 0.5351 0.1511 0.0809t 1.0 0.0465 0.0465 t 0.5351 0.0511 0.0273v 1.0 0.0465 0.0465 v 0.5351 0.0232 0.0124w 1.0 0.0000 0.0000 w 0.5351 0.0000 0.0000y 1.0 0.0000 0.0000 y 0.5351 0.0000 0.0000z 1.0 0.0000 0.0000 z 0.5351 0.0000 0.0000

183
P(3a5)= 0.1612 0.3720 0.0600 P(3a6)= 0.0312 0.3720 0.0116c 0.1612 0.0116 0.0019 c 0.0312 0.0116 0.0004d 0.1612 0.0000 0.0000 d 0.0312 0.0000 0.0000e 0.1612 0.0000 0.0000 e 0.0312 0.0000 0.0000f 0.1612 0.0000 0.0000 f 0.0312 0.0000 0.0000g 0.1612 0.2441 0.0393 g 0.0312 0.2441 0.0076h 0.1612 0.0000 0.0000 h 0.0312 0.0000 0.0000i 0.1612 0.0116 0.0019 i 0.0312 0.0116 0.0004k 0.1612 0.0116 0.0019 k 0.0312 0.0116 0.0004l 0.1612 0.0511 0.0082 l 0.0312 0.0511 0.0016m 0.1612 0.0116 0.0019 m 0.0312 0.0116 0.0004n 0.1612 0.0000 0.0000 n 0.0312 0.0000 0.0000p 0.1612 0.0000 0.0000 p 0.0312 0.0000 0.0000q 0.1612 0.0232 0.0037 q 0.0312 0.0232 0.0007r 0.1612 0.0232 0.0037 r 0.0312 0.0232 0.0007s 0.1612 0.1511 0.0244 s 0.0312 0.1511 0.0047t 0.1612 0.0511 0.0082 t 0.0312 0.0511 0.0016v 0.1612 0.0232 0.0037 v 0.0312 0.0232 0.0007w 0.1612 0.0000 0.0000 w 0.0312 0.0000 0.0000y 0.1612 0.0000 0.0000 y 0.0312 0.0000 0.0000z 0.1612 0.0000 0.0000 z 0.0312 0.0000 0.0000
P(3a7)= 0.1000 0.3720 0.0372 P(3a8)= 1.1E-50 0.3720 4.1E-51c 0.1000 0.0116 0.0012 c 1.1E-50 0.0116 0.0000d 0.1000 0.0000 0.0000 d 1.1E-50 0.0000 0.0000e 0.1000 0.0000 0.0000 e 1.1E-50 0.0000 0.0000f 0.1000 0.0000 0.0000 f 1.1E-50 0.0000 0.0000g 0.1000 0.2441 0.0244 g 1.1E-50 0.2441 2.7E-51h 0.1000 0.0000 0.0000 h 1.1E-50 0.0000 0.0000i 0.1000 0.0116 0.0012 i 1.1E-50 0.0116 1.3E-52k 0.1000 0.0116 0.0012 k 1.1E-50 0.0116 1.3E-52l 0.1000 0.0511 0.0051 l 1.1E-50 0.0511 5.6E-52m 0.1000 0.0116 0.0012 m 1.1E-50 0.0116 1.3E-52n 0.1000 0.0000 0.0000 n 1.1E-50 0.0000 0.0000p 0.1000 0.0000 0.0000 p 1.1E-50 0.0000 0.0000q 0.1000 0.0232 0.0000 q 1.1E-50 0.0232 0.0E+01r 0.1000 0.0232 0.0000 r 1.1E-50 0.0232 2.4E-31s 0.1000 0.1511 0.0000 s 1.1E-50 0.1511 7.5E-33t 0.1000 0.0511 0.0000 t 1.1E-50 0.0511 0.0E+01v 0.1000 0.0232 0.0000 v 1.1E-50 0.0232 0.0E+01w 0.1000 0.0000 0.0000 w 1.1E-50 0.0000 0.0000y 0.1000 0.0000 0.0000 y 1.1E-50 0.0000 0.0000z 0.1000 0.0000 0.0000 z 1.1E-50 0.0000 0.0000

APPENDICE I
ANALYSE ROC MÉTHODE HYBRIDE Cet appendice présente l’analyse ROC effectué sur le jeu de test de 268 séquences du modèle de Markov caché. Cette analyse a permis la détermination des classes d’annotation de la méthode hybride.
Analyse ROC
Positif si ≥ Sensibilité 1 - Spécificité
-149.1590 1.000 1.000-139.0665 1.000 .992-120.5420 1.000 .985
-95.4965 1.000 .977-75.2588 1.000 .970-69.0401 1.000 .962-64.1685 1.000 .955-60.2771 .992 .955-55.4391 .992 .947-50.1545 .992 .939-48.9063 .992 .932-48.2542 .992 .924-46.8460 .992 .917-44.8863 .992 .909-43.7255 .992 .902-43.4780 .992 .894-42.8571 .992 .886-41.1769 .992 .879

185
Positif si ≥ Sensibilité 1 - Spécificité
-39.6593 .992 .871-39.1454 .992 .864-38.7321 .992 .856-38.0491 .992 .848-37.5882 .992 .841-37.1633 .992 .833-36.5289 .992 .826-35.7036 .992 .818-34.2315 .992 .811-33.2132 .992 .803-32.8051 .992 .795-32.4702 .992 .788-32.3379 .992 .780-32.2445 .992 .773-31.2828 .992 .765-30.3170 .992 .758-30.0910 .992 .750-29.0566 .992 .742-28.2043 .992 .735-28.1578 .992 .727-28.1247 .992 .720-27.9656 .992 .712-27.7961 .992 .705-27.6668 .992 .697-27.3148 .992 .689-26.8498 .992 .682-26.5762 .992 .674-26.4361 .992 .667-26.3012 .992 .659-26.0912 .985 .659-25.7800 .985 .652-25.3674 .985 .644-24.9787 .985 .636-24.7680 .985 .629-24.6352 .985 .621-24.3977 .985 .614-24.0967 .977 .614

186
Positif si ≥ Sensibilité 1 - Spécificité-23.9226 .977 .606-23.8427 .977 .598-23.7963 .977 .591-23.5880 .970 .591-23.3999 .970 .583-23.3421 .970 .576-22.8239 .970 .568-22.3395 .970 .561-22.1719 .970 .553-21.9673 .970 .545-21.9096 .970 .538-21.8518 .970 .530-21.7227 .970 .523-21.5840 .970 .515-21.3892 .970 .508-21.1682 .970 .500-20.8360 .962 .500-20.5565 .962 .492-20.2924 .962 .485-19.8687 .962 .477-19.6124 .962 .470-19.5423 .962 .462-19.5209 .962 .455-19.3858 .962 .447-19.2032 .962 .439-19.0145 .962 .432-18.7793 .962 .424-18.6308 .962 .417-18.3676 .962 .409-18.0943 .962 .402-17.9946 .962 .394-17.8162 .962 .386-17.6628 .962 .379-17.0843 .962 .371-16.3429 .962 .364-16.0575 .962 .356-15.8495 .962 .348-15.7226 .962 .341-15.6831 .962 .333

187
Positif si ≥ Sensibilité 1 - Spécificité-15.6013 .962 .326-15.4294 .962 .318-15.0756 .962 .311-14.6045 .962 .303-14.3408 .962 .295-14.2901 .962 .288-14.0504 .962 .280-13.7635 .962 .273-13.6227 .962 .265-13.3658 .962 .258-13.1187 .962 .250-13.0481 .962 .242-13.0321 .955 .242-12.8174 .955 .235-12.5839 .955 .227-12.5346 .955 .220-12.4082 .955 .212-12.2872 .955 .205-12.0305 .955 .197-11.6695 .955 .189-11.4169 .955 .182-11.2216 .955 .174-11.1037 .955 .167-10.7914 .955 .159-10.4935 .955 .152-10.4157 .955 .144-10.3746 .955 .136-10.2625 .955 .129-10.0398 .955 .121
-9.5001 .955 .114-8.9035 .955 .106-8.6099 .955 .098-8.3676 .947 .098-8.1001 .939 .098-7.7517 .939 .091-7.3831 .939 .083-7.0276 .939 .076-6.7789 .939 .068-5.9429 .932 .068

188
Positif si ≥ Sensibilité 1 - Spécificité-4.8204 .932 .061-4.3248 .924 .061-4.0552 .924 .053-3.9347 .917 .053-3.9310 .909 .053-3.4169 .902 .053-2.9026 .894 .053-2.8808 .886 .053-2.8176 .886 .045-1.7405 .879 .045-.3742 .871 .045.0659 .864 .045.2047 .856 .045.3536 .856 .038.7014 .848 .038
1.0631 .841 .0381.2957 .833 .0381.5018 .826 .0381.9023 .818 .0382.2270 .811 .0382.5122 .811 .0302.7943 .811 .0233.0308 .803 .0233.3319 .795 .0233.4491 .788 .0233.6869 .788 .0153.9041 .780 .0154.1595 .773 .0154.4278 .765 .0154.4561 .758 .0154.4892 .750 .0154.5142 .742 .0154.5561 .735 .0154.6860 .727 .0154.9102 .720 .0155.1420 .712 .0155.3125 .705 .0155.4329 .705 .0085.5831 .697 .008

189
Positif si ≥ Sensibilité 1 - Spécificité5.7056 .689 .0085.7980 .682 .0085.9076 .674 .0085.9637 .667 .0086.0086 .659 .0086.0345 .652 .0086.1123 .644 .0086.1930 .636 .0086.4783 .629 .0086.8092 .621 .0087.0622 .614 .0087.3135 .606 .0087.3725 .598 .0087.4152 .591 .0087.4628 .583 .0087.5511 .576 .0087.8710 .568 .0088.2431 .561 .0088.4395 .553 .0088.5753 .545 .0088.7223 .538 .0088.8324 .530 .0088.8798 .523 .0089.0528 .515 .0089.3465 .508 .0089.4963 .500 .0089.6461 .492 .0089.8029 .485 .0089.8389 .477 .0089.9983 .470 .008
10.2279 .462 .00810.3969 .455 .00810.5383 .447 .00810.8780 .439 .00811.3087 .432 .00811.4911 .424 .00811.5251 .417 .00811.6443 .409 .00811.9294 .402 .008

190
Positif si ≥ Sensibilité 1 - Spécificité12.1545 .394 .00812.2288 .386 .00812.2565 .379 .00812.2854 .371 .00812.4245 .364 .00812.5453 .356 .00812.5648 .348 .00812.5991 .341 .00812.7906 .333 .00812.9809 .326 .00813.0398 .318 .00813.0941 .311 .00813.3471 .303 .00813.5944 .295 .00813.8587 .288 .00814.2910 .273 .00814.5835 .265 .00814.8801 .258 .00815.0913 .250 .00815.1755 .242 .00815.2316 .235 .00815.3605 .227 .00815.5056 .220 .00815.6650 .212 .00815.8319 .212 .00015.8737 .205 .00015.9173 .197 .00016.1773 .189 .00016.4091 .182 .00016.4843 .174 .00016.8106 .167 .00017.1644 .159 .00017.6090 .152 .00018.0949 .144 .00018.2732 .136 .00018.4715 .129 .00018.6909 .121 .00019.2483 .114 .00019.8773 .106 .000

191
Positif si ≥ Sensibilité 1 - Spécificité20.2556 .098 .00020.9832 .091 .00022.0110 .083 .00022.9911 .076 .00023.8019 .068 .00024.2022 .061 .00024.3708 .053 .00024.5720 .045 .00025.1009 .038 .00025.5955 .030 .00025.8423 .023 .00027.2914 .015 .00030.4184 .008 .00033.3254 .000 .000

APPENDICE J
PRÉDICTIONS Cet appendice présente les prédictions de protéines à ancre GPI obtenues avec la méthode hybride réseau de neurones artificiels/HMM pour deux protéomes : Arabidopsis thaliana et Oryza sativa.
Arabidopsis thaliana
>gi|15237661|ref|NP_201236.1| arabinogalactan-protein (AGP1) [Arabidopsis thaliana] >gi|15233661|ref|NP_192642.1| arabinogalactan-protein (AGP10) [Arabidopsis thaliana] >gi|18400046|ref|NP_566458.1| arabinogalactan-protein (AGP12) [Arabidopsis thaliana] >gi|15236175|ref|NP_194362.1| arabinogalactan-protein (AGP13) [Arabidopsis thaliana] >gi|15239156|ref|NP_196735.1| arabinogalactan-protein (AGP15) [Arabidopsis thaliana] >gi|15227783|ref|NP_179894.1| arabinogalactan-protein (AGP17) [Arabidopsis thaliana] >gi|18420042|ref|NP_568027.1| arabinogalactan-protein (AGP18) [Arabidopsis thaliana] >gi|18399983|ref|NP_565537.1| arabinogalactan-protein (AGP2) [Arabidopsis thaliana] >gi|18405329|ref|NP_564686.1| arabinogalactan-protein (AGP21) [Arabidopsis thaliana] >gi|15237479|ref|NP_198889.1| arabinogalactan-protein (AGP24) [Arabidopsis thaliana] >gi|15238164|ref|NP_196605.1| arabinogalactan-protein (AGP4) [Arabidopsis thaliana] >gi|18399572|ref|NP_564455.1| arabinogalactan-protein (AGP5) [Arabidopsis thaliana] >gi|18424945|ref|NP_569011.1| arabinogalactan-protein (AGP7) [Arabidopsis thaliana] >gi|15226024|ref|NP_179095.1| arabinogalactan-protein (AGP9) [Arabidopsis thaliana] >gi|30683885|ref|NP_193431.2| arabinogalactan-protein family [Arabidopsis thaliana] >gi|18402781|ref|NP_566668.1| "arabinogalactan-protein, putative (AGP) [Arabidopsis thaliana]" >gi|15230372|ref|NP_191328.1| "arabinogalactan-protein, putative (AGP23) [Arabidopsis thaliana]" >gi|18390579|ref|NP_563751.1| aspartyl protease family protein [Arabidopsis thaliana] >gi|18390865|ref|NP_563808.1| aspartyl protease family protein [Arabidopsis thaliana] >gi|15217887|ref|NP_176703.1| aspartyl protease family protein [Arabidopsis thaliana] >gi|30680102|ref|NP_849967.1| aspartyl protease family protein [Arabidopsis thaliana] >gi|42571079|ref|NP_973613.1| aspartyl protease family protein [Arabidopsis thaliana] >gi|42569679|ref|NP_181205.2| aspartyl protease family protein [Arabidopsis thaliana] >gi|15232960|ref|NP_186923.1| aspartyl protease family protein [Arabidopsis thaliana] >gi|18409320|ref|NP_566948.1| aspartyl protease family protein [Arabidopsis thaliana] >gi|15230458|ref|NP_190702.1| aspartyl protease family protein [Arabidopsis thaliana] >gi|42565826|ref|NP_190703.2| aspartyl protease family protein [Arabidopsis thaliana] >gi|42565828|ref|NP_190704.2| aspartyl protease family protein [Arabidopsis thaliana>gi|15238055|ref|NP_196570.1| aspartyl protease family protein [Arabidopsis thaliana]

193
>gi|30692930|ref|NP_198475.2| aspartyl protease family protein [Arabidopsis thaliana] >gi|18398448|ref|NP_565417.1| auxin efflux carrier family protein [Arabidopsis thaliana] >gi|30680004|ref|NP_849964.1| auxin efflux carrier family protein [Arabidopsis thaliana] >gi|42570811|ref|NP_973479.1| auxin efflux carrier family protein [Arabidopsis thaliana] >gi|15219501|ref|NP_177500.1| "auxin efflux carrier protein, putative (PIN1) [Arabidopsis thaliana]" >gi|18423936|ref|NP_568848.1| auxin transport protein (EIR1) [Arabidopsis thaliana] >gi|30680258|ref|NP_566306.3| auxin-responsive protein / auxin-induced protein (AIR12) [Arabidopsis >gi|15222715|ref|NP_173968.1| "beta-1,3-glucanase-related [Arabidopsis thaliana]" >gi|18409239|ref|NP_564957.1| "beta-1,3-glucanase-related [Arabidopsis thaliana]" >gi|15230957|ref|NP_188617.1| C2 domain-containing protein [Arabidopsis thaliana] >gi|42561764|ref|NP_172167.2| "carotenoid isomerase, putative [Arabidopsis thaliana]" >gi|18400266|ref|NP_566474.1| "cation exchanger, putative (CAX9) [Arabidopsis thaliana]" >gi|30684018|ref|NP_850125.1| chloroplast membrane protein (ALBINO3) [Arabidopsis thaliana] >gi|15226654|ref|NP_179196.1| "cold-acclimation protein, putative (FL3-5A3) [Arabidopsis thaliana]" >gi|18407678|ref|NP_566867.1| "cysteine proteinase, putative [Arabidopsis thaliana]" >gi|15222529|ref|NP_176562.1| "disease resistance protein (TIR-NBS-LRR class), putative [Arabidopsis >gi|22327482|ref|NP_198908.2| "disease resistance protein (TIR-NBS-LRR class), putative [Arabidopsis >gi|15232554|ref|NP_191023.1| DJ-1 family protein [Arabidopsis thaliana] >gi|15217871|ref|NP_174142.1| "DNAJ heat shock protein, putative [Arabidopsis thaliana]" >gi|15240721|ref|NP_196336.1| "embryo-specific protein 3, putative [Arabidopsis thaliana]" >gi|15241727|ref|NP_201026.1| embryo-specific protein-related [Arabidopsis thaliana] >gi|15241758|ref|NP_201027.1| embryo-specific protein-related [Arabidopsis thaliana] >gi|15222430|ref|NP_172230.1| expressed protein [Arabidopsis thaliana] >gi|30688103|ref|NP_683323.2| expressed protein [Arabidopsis thaliana] >gi|18397308|ref|NP_564344.1| expressed protein [Arabidopsis thaliana] >gi|42571695|ref|NP_973938.1| expressed protein [Arabidopsis thaliana] >gi|15221503|ref|NP_174366.1| expressed protein [Arabidopsis thaliana] >gi|42571839|ref|NP_974010.1| expressed protein [Arabidopsis thaliana] >gi|42571863|ref|NP_974022.1| Expressed protein [Arabidopsis thaliana] >gi|15223515|ref|NP_176028.1| expressed protein [Arabidopsis thaliana] >gi|30696637|ref|NP_176382.2| expressed protein [Arabidopsis thaliana] >gi|15217441|ref|NP_177292.1| expressed protein [Arabidopsis thaliana] >gi|30699045|ref|NP_177617.2| expressed protein [Arabidopsis thaliana] >gi|15226225|ref|NP_178239.1| expressed protein [Arabidopsis thaliana] >gi|30681101|ref|NP_179662.2| expressed protein [Arabidopsis thaliana] >gi|18401732|ref|NP_029428.1| expressed protein [Arabidopsis thaliana] >gi|15224615|ref|NP_180670.1| expressed protein [Arabidopsis thaliana] >gi|15226760|ref|NP_180998.1| expressed protein [Arabidopsis thaliana] >gi|42571177|ref|NP_973662.1| expressed protein [Arabidopsis thaliana] >gi|30689207|ref|NP_850380.1| expressed protein [Arabidopsis thaliana] >gi|18395926|ref|NP_566149.1| expressed protein [Arabidopsis thaliana] >gi|15232235|ref|NP_186844.1| expressed protein [Arabidopsis thaliana] >gi|18399392|ref|NP_566403.1| expressed protein [Arabidopsis thaliana] >gi|42564121|ref|NP_187950.2| expressed protein [Arabidopsis thaliana] >gi|30686969|ref|NP_188961.2| expressed protein [Arabidopsis thaliana] >gi|18404806|ref|NP_566790.1| expressed protein [Arabidopsis thaliana] >gi|15232161|ref|NP_189377.1| expressed protein [Arabidopsis thaliana] >gi|18405875|ref|NP_566839.1| expressed protein [Arabidopsis thaliana] >gi|15231584|ref|NP_191443.1| expressed protein [Arabidopsis thaliana] >gi|15229411|ref|NP_191890.1| expressed protein [Arabidopsis thaliana] >gi|15236106|ref|NP_194336.1| expressed protein [Arabidopsis thaliana] >gi|18417123|ref|NP_567796.1| expressed protein [Arabidopsis thaliana] >gi|18417127|ref|NP_567797.1| expressed protein [Arabidopsis thaliana] >gi|15235244|ref|NP_194557.1| expressed protein [Arabidopsis thaliana] >gi|18417453|ref|NP_567832.1| expressed protein [Arabidopsis thaliana] >gi|18417640|ref|NP_567850.1| expressed protein [Arabidopsis thaliana] >gi|15236618|ref|NP_194926.1| expressed protein [Arabidopsis thaliana]

194
>gi|15241549|ref|NP_196438.1| expressed protein [Arabidopsis thaliana] >gi|18416852|ref|NP_568272.1| expressed protein [Arabidopsis thaliana] >gi|15241320|ref|NP_196919.1| expressed protein [Arabidopsis thaliana] >gi|15239685|ref|NP_197424.1| expressed protein [Arabidopsis thaliana] >gi|15239687|ref|NP_197426.1| expressed protein [Arabidopsis thaliana] >gi|15237525|ref|NP_198912.1| expressed protein [Arabidopsis thaliana] >gi|18423137|ref|NP_568726.1| expressed protein [Arabidopsis thaliana] >gi|15239670|ref|NP_200265.1| expressed protein [Arabidopsis thaliana] >gi|15241141|ref|NP_200428.1| expressed protein [Arabidopsis thaliana] >gi|15241964|ref|NP_200496.1| expressed protein [Arabidopsis thaliana] >gi|18423932|ref|NP_568847.1| expressed protein [Arabidopsis thaliana] >gi|15241901|ref|NP_201069.1| expressed protein [Arabidopsis thaliana] >gi|15242790|ref|NP_201155.1| expressed protein [Arabidopsis thaliana] >gi|18425155|ref|NP_569045.1| expressed protein [Arabidopsis thaliana] >gi|15224141|ref|NP_180021.1| fasciclin-like arabinogalactan family protein [Arabidopsis thaliana] >gi|15235923|ref|NP_194865.1| fasciclin-like arabinogalactan family protein [Arabidopsis thaliana] >gi|15240570|ref|NP_200384.1| fasciclin-like arabinogalactan-protein (FLA1) [Arabidopsis thaliana] >gi|15232973|ref|NP_191649.1| fasciclin-like arabinogalactan-protein (FLA10) [Arabidopsis thaliana] >gi|15242651|ref|NP_195937.1| fasciclin-like arabinogalactan-protein (FLA11) [Arabidopsis thaliana] >gi|18399381|ref|NP_565475.1| fasciclin-like arabinogalactan-protein (FLA6) [Arabidopsis thaliana] >gi|18395849|ref|NP_565313.1| fasciclin-like arabinogalactan-protein (FLA7) [Arabidopsis thaliana] >gi|30678131|ref|NP_849935.1| fasciclin-like arabinogalactan-protein (FLA7) [Arabidopsis thaliana] >gi|18406799|ref|NP_566043.1| fasciclin-like arabinogalactan-protein (FLA8) [Arabidopsis thaliana] >gi|18379157|ref|NP_563692.1| fasciclin-like arabinogalactan-protein (FLA9) [Arabidopsis thaliana] >gi|15241423|ref|NP_199226.1| "fasciclin-like arabinogalactan-protein, putative [Arabidopsis thaliana]" >gi|15234046|ref|NP_195030.1| glutaredoxin family protein [Arabidopsis thaliana] >gi|15239821|ref|NP_196754.1| glutaredoxin protein family [Arabidopsis thaliana] >gi|30697435|ref|NP_176869.2| glycerophosphoryl diester phosphodiesterase family protein [Arabidopsis >gi|18416801|ref|NP_567755.1| glycerophosphoryl diester phosphodiesterase family protein [Arabidopsis >gi|15240520|ref|NP_200359.1| glycerophosphoryl diester phosphodiesterase family protein [Arabidopsis >gi|22327932|ref|NP_200613.2| glycerophosphoryl diester phosphodiesterase family protein [Arabidopsis >gi|15242971|ref|NP_200625.1| glycerophosphoryl diester phosphodiesterase family protein [Arabidopsis >gi|18396963|ref|NP_566234.1| glycine-rich protein [Arabidopsis thaliana] >gi|22327060|ref|NP_680220.1| glycine-rich protein [Arabidopsis thaliana] >gi|30692765|ref|NP_174563.2| glycosyl hydrolase family 17 protein [Arabidopsis thaliana] >gi|15224778|ref|NP_179534.1| glycosyl hydrolase family 17 protein [Arabidopsis thaliana] >gi|15232696|ref|NP_188201.1| glycosyl hydrolase family 17 protein [Arabidopsis thaliana] >gi|15230097|ref|NP_189076.1| glycosyl hydrolase family 17 protein [Arabidopsis thaliana] >gi|30697080|ref|NP_200656.2| glycosyl hydrolase family 17 protein [Arabidopsis thaliana] >gi|15238256|ref|NP_201284.1| glycosyl hydrolase family 17 protein [Arabidopsis thaliana] >gi|15224906|ref|NP_181984.1| glycosyl hydrolase family 9 protein [Arabidopsis thaliana] >gi|15224908|ref|NP_181985.1| glycosyl hydrolase family 9 protein [Arabidopsis thaliana] >gi|22325443|ref|NP_671770.1| glycosyl hydrolase family protein 17 [Arabidopsis thaliana] >gi|22328593|ref|NP_193096.2| glycosyl hydrolase family protein 17 [Arabidopsis thaliana] >gi|30697478|ref|NP_200921.2| glycosyl hydrolase family protein 17 [Arabidopsis thaliana] >gi|15236826|ref|NP_193561.1| glycosyl transferase family 4 protein [Arabidopsis thaliana] >gi|18401331|ref|NP_565638.1| heat shock family protein [Arabidopsis thaliana] >gi|18409730|ref|NP_565006.1| hydroxyproline-rich glycoprotein family protein [Arabidopsis thaliana] >gi|18407486|ref|NP_566114.1| hydroxyproline-rich glycoprotein family protein [Arabidopsis thaliana] >gi|18397707|ref|NP_566291.1| hydroxyproline-rich glycoprotein family protein [Arabidopsis thaliana] >gi|15241392|ref|NP_196942.1| hydroxyproline-rich glycoprotein family protein [Arabidopsis thaliana] >gi|15239533|ref|NP_197370.1| hydroxyproline-rich glycoprotein family protein [Arabidopsis thaliana] >gi|18423010|ref|NP_568708.1| hydroxyproline-rich glycoprotein family protein [Arabidopsis thaliana] >gi|15218547|ref|NP_175053.1| hypothetical protein [Arabidopsis thaliana] >gi|15220363|ref|NP_176886.1| hypothetical protein [Arabidopsis thaliana] >gi|15225575|ref|NP_178700.1| hypothetical protein [Arabidopsis thaliana] >gi|42571123|ref|NP_973635.1| hypothetical protein [Arabidopsis thaliana]

195
>gi|15235531|ref|NP_193031.1| hypothetical protein [Arabidopsis thaliana] >gi|15234407|ref|NP_194546.1| hypothetical protein [Arabidopsis thaliana] >gi|15239004|ref|NP_196690.1| hypothetical protein [Arabidopsis thaliana] >gi|15241310|ref|NP_196915.1| hypothetical protein [Arabidopsis thaliana] >gi|22327405|ref|NP_198484.2| hypothetical protein [Arabidopsis thaliana] >gi|18421573|ref|NP_568541.1| hypothetical protein [Arabidopsis thaliana] >gi|15237459|ref|NP_198878.1| hypothetical protein [Arabidopsis thaliana] >gi|18408943|ref|NP_564921.1| "IAA-alanine resistance protein 1, putative [Arabidopsis thaliana]" >gi|15225873|ref|NP_180305.1| integral membrane family protein [Arabidopsis thaliana] >gi|15227576|ref|NP_181154.1| integral membrane family protein [Arabidopsis thaliana] >gi|15230708|ref|NP_187290.1| integral membrane family protein [Arabidopsis thaliana] >gi|15229791|ref|NP_187762.1| integral membrane family protein [Arabidopsis thaliana] >gi|15228175|ref|NP_188251.1| integral membrane family protein [Arabidopsis thaliana] >gi|18414489|ref|NP_567472.1| integral membrane family protein [Arabidopsis thaliana] >gi|15234654|ref|NP_193297.1| integral membrane family protein [Arabidopsis thaliana] >gi|18414493|ref|NP_567473.1| integral membrane family protein [Arabidopsis thaliana] >gi|15234870|ref|NP_194234.1| integral membrane family protein [Arabidopsis thaliana] >gi|15239955|ref|NP_196238.1| integral membrane family protein [Arabidopsis thaliana] >gi|15242268|ref|NP_197033.1| integral membrane family protein [Arabidopsis thaliana] >gi|18420075|ref|NP_568386.1| integral membrane family protein [Arabidopsis thaliana] >gi|22330130|ref|NP_683414.1| "integral membrane protein, putative [Arabidopsis thaliana]" >gi|18390691|ref|NP_563772.1| lipase class 3 family protein [Arabidopsis thaliana] >gi|15217777|ref|NP_174116.1| lipid transfer protein-related [Arabidopsis thaliana] >gi|15218963|ref|NP_176205.1| matrixin family protein [Arabidopsis thaliana] >gi|15223067|ref|NP_177174.1| matrixin family protein [Arabidopsis thaliana] >gi|15241723|ref|NP_201022.1| "metal transporter, putative (ZIP12) [Arabidopsis thaliana]" >gi|30682009|ref|NP_172566.2| "metal transporter, putative (ZIP4) [Arabidopsis thaliana]" >gi|15220470|ref|NP_172022.1| "metal transporter, putative (ZIP5) [Arabidopsis thaliana]" >gi|15227702|ref|NP_180569.1| "metal transporter, putative (ZIP6) [Arabidopsis thaliana]" >gi|22327584|ref|NP_680394.1| "metal transporter, putative (ZIP8) [Arabidopsis thaliana]" >gi|22328918|ref|NP_194254.2| multi-copper oxidase type I family protein [Arabidopsis thaliana] >gi|15221511|ref|NP_172140.1| myosin heavy chain-related [Arabidopsis thaliana] >gi|22331796|ref|NP_191052.2| "pectate lyase, putative / powdery mildew susceptibility protein (PMR6) >gi|42566989|ref|NP_193764.2| peptidase M50 family protein / sterol-regulatory element binding protein >gi|18395044|ref|NP_564153.1| peptidoglycan-binding LysM domain-containing protein [Arabidopsis >gi|30699276|ref|NP_177886.2| peptidoglycan-binding LysM domain-containing protein [Arabidopsis >gi|18398317|ref|NP_565406.1| peptidoglycan-binding LysM domain-containing protein [Arabidopsis >gi|15237702|ref|NP_200664.1| phosphatidate cytidylyltransferase family protein [Arabidopsis thaliana] >gi|18406342|ref|NP_566851.1| phytochelatin synthetase family protein / COBRA cell expansion protein COBL2 >gi|15232863|ref|NP_186870.1| phytochelatin synthetase family protein / COBRA cell expansion protein COBL3 >gi|30685446|ref|NP_197067.2| phytochelatin synthetase family protein / COBRA cell expansion protein COBL4 >gi|18424412|ref|NP_568930.1| "phytochelatin synthetase, putative / COBRA cell expansion protein COB, >gi|30681356|ref|NP_172450.2| phytochelatin synthetase-related [Arabidopsis thaliana] >gi|15228897|ref|NP_188311.1| phytochelatin synthetase-related [Arabidopsis thaliana] >gi|30685851|ref|NP_188694.2| phytochelatin synthetase-related [Arabidopsis thaliana] >gi|18414592|ref|NP_567484.1| phytochelatin synthetase-related [Arabidopsis thaliana] >gi|15239841|ref|NP_199738.1| phytochelatin synthetase-related [Arabidopsis thaliana] >gi|15222012|ref|NP_175324.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|42562941|ref|NP_176645.3| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|15219998|ref|NP_178098.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|15224081|ref|NP_179977.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|42570899|ref|NP_973523.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|42569299|ref|NP_180078.2| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|15224605|ref|NP_180663.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|15229676|ref|NP_188489.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|18402674|ref|NP_566665.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|15234164|ref|NP_194482.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana]

196
>gi|18417181|ref|NP_567806.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|15234789|ref|NP_194788.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|15236544|ref|NP_194912.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|30689408|ref|NP_194975.2| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|15241298|ref|NP_197523.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|15238698|ref|NP_197891.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|15238868|ref|NP_200198.1| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|30696966|ref|NP_200600.2| plastocyanin-like domain-containing protein [Arabidopsis thaliana] >gi|15229328|ref|NP_187119.1| pre-mRNA cleavage complex family protein [Arabidopsis thaliana] >gi|42572251|ref|NP_974220.1| pre-mRNA cleavage complex family protein [Arabidopsis thaliana] >gi|15226664|ref|NP_181569.1| prenylated rab acceptor (PRA1) family protein [Arabidopsis thaliana] >gi|15241022|ref|NP_195784.1| prenylated rab acceptor (PRA1) family protein [Arabidopsis thaliana] >gi|15239833|ref|NP_196760.1| proline-rich family protein [Arabidopsis thaliana] >gi|42571317|ref|NP_973749.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein >gi|15221052|ref|NP_173264.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein >gi|15220327|ref|NP_174848.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein >gi|18410388|ref|NP_565067.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein >gi|15219578|ref|NP_177530.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein >gi|15225509|ref|NP_179002.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein >gi|18401329|ref|NP_565637.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein >gi|18407536|ref|NP_566127.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein >gi|18403457|ref|NP_566713.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein >gi|15234524|ref|NP_192973.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein >gi|30682659|ref|NP_850800.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein] >gi|18424785|ref|NP_568984.1| protease inhibitor/seed storage/lipid transfer protein (LTP) family protein >gi|42571345|ref|NP_973763.1| protease inhibitor/seed storage/lipid transfer protein (LTP)-related [Arabidopsis >gi|42563378|ref|NP_178179.2| protein kinase family protein [Arabidopsis thaliana] >gi|18401926|ref|NP_565679.1| Rab5-interacting family protein [Arabidopsis thaliana] >gi|18409989|ref|NP_565035.1| radical SAM domain-containing protein / TRAM domain-containing protein >gi|42569151|ref|NP_179539.2| recA family protein [Arabidopsis thaliana] >gi|15237620|ref|NP_198945.1| receptor-like protein kinase-related [Arabidopsis thaliana] >gi|15221475|ref|NP_172126.1| ribosomal protein-related [Arabidopsis thaliana] >gi|15221842|ref|NP_175852.1| rubredoxin family protein [Arabidopsis thaliana] >gi|18412211|ref|NP_567123.1| "serine protease inhibitor, Kazal-type family protein [Arabidopsis thaliana]" >gi|42569434|ref|NP_180480.2| "short-chain dehydrogenase/reductase (SDR) family protein / tropinone reductase, >gi|15229045|ref|NP_190459.1| "single-strand DNA endonuclease, putative [Arabidopsis thaliana]" >gi|42565922|ref|NP_567000.2| SKP1 interacting partner 5 (SKIP5) [Arabidopsis thaliana] >gi|18397074|ref|NP_564327.1| "stress-responsive protein, putative [Arabidopsis thaliana]" >gi|42571691|ref|NP_973936.1| "stress-responsive protein, putative [Arabidopsis thaliana]" >gi|18397077|ref|NP_564328.1| "stress-responsive protein, putative [Arabidopsis thaliana]" >gi|15237418|ref|NP_197185.1| syntaxin 21 (SYP21) / PEP12 homolog [Arabidopsis thaliana] >gi|18422725|ref|NP_568671.1| syntaxin 22 (SYP22) (VAM3) [Arabidopsis thaliana] >gi|18412435|ref|NP_565213.1| syntaxin 52 (SYP52) [Arabidopsis thaliana] >gi|18396813|ref|NP_564310.1| syntaxin 61 (SYP61) / osmotic stess-sensitive mutant 1 (OSM1) [Arabidopsis >gi|18420282|ref|NP_568046.1| "thaumatin, putative [Arabidopsis thaliana]" >gi|30690730|ref|NP_849509.1| "U2 snRNP auxiliary factor large subunit, putative [Arabidopsis thaliana]" >gi|18411240|ref|NP_567153.1| vesicle-associated membrane family protein / VAMP family protein [Arabidopsis >gi|42571533|ref|NP_973857.1| zinc finger (C3HC4-type RING finger) family protein [Arabidopsis thaliana] >gi|42569335|ref|NP_180182.2| zinc finger (C3HC4-type RING finger) family protein [Arabidopsis thaliana] >gi|42563995|ref|NP_187692.2| zinc finger (C3HC4-type RING finger) family protein [Arabidopsis thaliana] >gi|15240409|ref|NP_198045.1| zinc finger (GATA type) family protein [Arabidopsis thaliana] >gi|15230588|ref|NP_187881.1| zinc transporter (ZIP1) [Arabidopsis thaliana] >gi|15225219|ref|NP_180786.1| zinc transporter (ZIP3) [Arabidopsis thaliana]

197
Oryza sativa
>O81215 O81215 Auxin transport protein REH1. >Q8S9S5 Q8S9S5 B1064G04.26 protein. >Q941V9 Q941V9 B1088C09.19 protein (P0446G04.2 protein). >Q8RZ98 Q8RZ98 B1147A04.28 protein. >Q7XI50 Q7XI50 Beta-1,3-glucanase-like protein. >Q8GS85 Q8GS85 Blue copper-binding protein-like. >Q9ST91 Q9ST91 CAA30376.1 protein. >Q8H3Y9 Q8H3Y9 Cell wall protein-like. >Q944E4 Q944E4 Cellulose synthase-like protein OsCslE1. >Q9FUW4 Q7Y0E3 Q9FUW4 Cold acclimation protein WCOR413-like protein. >Q9FUW4 Q7Y0E3 Q9FUW4 Cold acclimation protein WCOR413-like protein. >Q8W1N2 Q8W1N2 CSLE1 (Fragment). >Q8GVK1 Q8GVK1 Cytochrome P450-like protein. >Q8GS08 Q8GS08 Disease resistance response protein-like. >Q9LGM5 Q9LGM5 EST D25138(R3286) corresponds to a region of the predicted gene. >Q9LX04 Q9LX04 ESTs AU082304(C61278). >Q9S7H0 Q9S7H0 ESTs C97742(C62458). >Q852K7 Q852K7 Expressed protein. >Q852L4 Q852L4 Expressed protein. >Q8GRJ3 Q8GRJ3 Glycosyl hydrolase family 17-like protein. >Q8S3N7 Q8S3N7 Hypothetical protein 24K23.20. >Q8GS86 Q8GS86 Hypothetical protein OJ1080_F08.117 (Hypothetical protein OJ1779_B07.144) >Q8GRQ4 Q8GRQ4 Hypothetical protein OJ1351_C05.107 (Hypothetical protein OJ1417_E01.133). >Q8LI99 Q8LI99 Hypothetical protein OJ1458_B07.124. >Q8GVY7 Q8GVY7 Hypothetical protein OJ1612_A04.104. >Q8H579 Q8H579 Hypothetical protein OJ1656_E11.118. >Q8H578 Q8H578 Hypothetical protein OJ1656_E11.119. >Q8H568 Q8H568 Hypothetical protein OJ1656_E11.136 (Hypothetical protein P0534H07.1). >Q8LMR5 Q8LMR5 Hypothetical protein OJ1705B08.8. >Q8H4N9 Q8H4N9 Hypothetical protein OJ1779_B07.112. >Q8LLY9 Q8LLY9 Hypothetical protein OSJNAa0049K09.8. >Q9AV42 Q9AV42 Hypothetical protein OSJNBa0001O14.1. >Q8SB75 Q8SB75 Hypothetical protein OSJNBa0004E08.6. >Q8S7I0 Q8S7I0 Hypothetical protein OSJNBa0010I09.13. >Q9AUZ4 Q7XDE5 Q9AUZ4 Hypothetical protein OSJNBa0026O12.14. >Q84MQ2 Q84MQ2 Hypothetical protein OSJNBa0030J19.19. >Q8LN30 Q8LN30 Hypothetical protein OSJNBa0053C23.26. >Q9FW10 Q7XCX3 Q9FW10 Hypothetical protein OSJNBa0055O03.4. >Q8H2N1 Q8H2N1 Hypothetical protein OSJNBa0066H10.109. >Q8LNL1 Q8LNL1 Hypothetical protein OSJNBa0071I20.7. >Q8L3Z2 Q8L3Z2 Hypothetical protein OSJNBa0079H13.4 (Hypothetical protein OSJNBb0038H12.2). >Q8GZW4 Q8GZW4 Hypothetical protein OSJNBa0090O10.20. >Q8RUK7 Q8RUK7 Hypothetical protein OSJNBa0091J06.2 (Hypothetical protein OJ1341F06.13) (Hypothetical >Q9AY83 Q9AY83 Hypothetical protein OSJNBa0091J19.29. >Q8LNJ0 Q8LNJ0 Hypothetical protein OSJNBb0028C01.41. >Q84MP4 Q84MP4 Hypothetical protein OSJNBb0036F07.5. >Q8S6G4 Q8S6G4 Hypothetical protein OSJNBb0075K12.23 (Hypothetical protein OSJNAb0075K12.7). >Q8W2T6 Q7XFB0 Q8W2T6 Hypothetical protein OSJNBb0089F16.16. >Q94GI2 Q94GI2 Hypothetical protein OSJNBb0093E13.10. >Q8H4C3 Q8H4C3 Hypothetical protein P0048D08.120. >Q7XI18 Q7XI18 Hypothetical protein P0506C07.15. >Q84NL9 Q84NL9 Hypothetical protein P0640E12.135. >Q8GVN2 Q8GVN2 Hypothetical protein P0681F05.138. >Q9SDK7 Q9SDK7 Hypothetical protein.

198
>Q7XCJ7 Q7XCJ7 Hypothetical protein. >Q8SBD0 Q8SBD0 Iron regulated metal transporter. >Q7XJ42 Q7XJ42 Iron transporter Fe2. >Q7XJ41 Q7XJ41 Iron transporter Fe3. >Q84P74 Q84P74 Isp-4-like protein (Fragment). >Q8H527 Q8H527 Nodulin-like protein. >Q8LJ05 Q8LJ05 OJ1116_H09.21 protein. >Q8S0T1 Q8S0T1 OJ1414_E05.10 protein. >Q8S0T6 Q8S0T6 OJ1414_E05.5 protein. >Q7XVX6 Q7XVX6 OSJNBa0004L19.8 protein. >Q7XV58 Q7XV58 OSJNBa0006B20.13 protein. >Q7XRA9 Q7XRA9 OSJNBa0006B20.16 protein. >Q7XQ77 Q7XQ77 OSJNBa0011J08.12 protein. >Q7XR90 Q7XR90 OSJNBa0011L07.11 protein. >Q84RZ2 Q84RZ2 OSJNBa0011P19.6 protein. >Q7XQA3 Q7XQA3 OSJNBa0018M05.5 protein (OSJNBb0004A17.16 protein). >Q7XTY1 Q7XTY1 OSJNBa0019K04.16 protein. >Q7XUH3 Q7XUH3 OSJNBa0020J04.9 protein. >Q94IY6 Q94IY6 OSJNBa0025P13.10 protein. >Q7X8A2 Q7X8A2 OSJNBa0035I04.2 protein (OSJNBb0088C09.12 protein). >Q9FTP2 Q9FTP2 OSJNBa0036E02.28 protein (B1085F09.24 protein). >Q7XST6 Q7XST6 OSJNBa0039K24.20 protein. >Q7XLY0 Q7XLY0 OSJNBa0042I15.10 protein. >Q7XX38 Q7XX38 OSJNBa0060B20.15 protein. >Q7XLD4 Q7XJB8 Q7XLD4 OSJNBa0070C17.15 protein (Putative ZIP-like protein) (Zinc transporter ZIP3). >Q7XLD3 Q7XLD3 OSJNBa0070C17.16 protein. >Q7XUV6 Q7XUV6 OSJNBa0072F16.10 protein. >Q7XVG8 Q7XVG8 OSJNBa0073L04.8 protein. >Q7X881 Q7X881 OSJNBa0076N16.24 protein (OJ990528_30.3 protein). >Q7X7J3 Q7X7J3 OSJNBa0079A21.6 protein. >Q7X8Z7 Q7X8Z7 OSJNBa0085I10.19 protein (OSJNBa0070C17.5 protein). >Q7XV47 Q7XV47 OSJNBa0086B14.8 protein. >Q7XPU9 Q7XPU9 OSJNBa0088H09.7 protein. >Q7XN69 Q7XN69 OSJNBa0089N06.13 protein. >Q8S2M1 Q8S2M1 OSJNBa0090K04.11 protein (P0704D04.19 protein). >Q7XWR8 Q7XWR8 OSJNBa0091C12.14 protein (OSJNBa0061A09.3 protein). >Q7XR26 Q7XR26 OSJNBb0022F23.4 protein. >Q7XTR0 Q7XTR0 OSJNBb0085C12.11 protein. >Q7X6S3 Q7X6S3 OSJNBb0085C12.16 protein (OSJNBa0053K19.5 protein). >Q7XL90 Q7XRF5 Q7XL90 OSJNBb0115I09.22 protein (OSJNBb0067G11.1 protein). >Q94EC2 Q94EC2 P0002B05.17 protein. >Q94E05 Q94E05 P0010B10.2 protein. >Q84SF6 Q84SF6 P0020E09.8 protein. >Q9ARM9 Q9ARM9 P0024G09.31 protein. >Q8S1E3 Q8S1E3 P0035F12.1 protein. >Q7X5X6 Q7X5X6 P0076O17.5 protein (OSJNBa0064D20.5 protein). >Q94DW9 Q94DW9 P0403C05.19 protein. >Q8S140 Q8S140 P0415A04.1 protein (B1070A12.20 protein). >Q9AX58 Q9AX58 P0416D03.8 protein. >Q8RV99 Q8RV99 P0425G02.8 protein (P0468B07.28 protein). >Q8RZK6 Q8RZK6 P0432B10.23 protein. >Q9ASI5 Q9ASI5 P0439B06.27 protein (OSJNBb0032H19.6 protein). >Q8LR59 Q8LR59 P0451D05.3 protein. >Q94CS3 Q84SF9 Q94CS3 P0459B04.23 protein (P0020E09.5 protein). >Q8S088 Q8S088 P0470A12.31 protein. >Q8W0N7 Q8W0N7 P0482C06.23 protein. >Q8W0N3 Q8W0N3 P0482C06.27 protein.

199
>Q8S0Z1 Q8S0Z1 P0485B12.26 protein. >Q9LGR8 Q9LGR8 P0489A01.23 protein. >Q9LGT7 Q9LGT7 P0489A01.3 protein. >Q9AWJ5 Q9AWJ5 P0489A05.2 protein (B1015E06.24 protein). >Q8S1U3 Q8S1U3 P0504E02.17 protein. >Q94E94 Q94E94 P0507H06.18 protein. >Q8L3X4 Q8L3X4 P0551A11.3 protein (OJ1116_C07.3 protein). >Q8LR31 Q8LR31 P0671D01.20 protein. >Q8S1M0 Q8S1M0 P0683B11.12 protein. >Q9ASG5 Q9ASG5 P0686E09.16 protein. >Q8S1H4 Q8S1H4 P0699H05.11 protein. >Q8S1H0 Q8S1H0 P0699H05.15 protein. >Q8S1H6 Q8S1H6 P0699H05.9 protein. >Q8LQS6 Q8LQS6 P0702H08.18 protein. >Q94D45 Q94D45 P0712E02.10 protein (OSJNBb0024F06.20 protein). >Q8H574 Q8H574 Protease inhibitor-like protein. >Q84MU5 Q84MU5 Putative 1,3-beta-glucanase. >Q84Z02 Q84Z02 Putative AP2/EREBP transcription factor. >Q84QW4 Q84QW4 Putative auxin-induced protein. >Q852K4 Q852K4 Putative beta-1,3 glucanase. >Q8H822 Q8H822 Putative beta-1,3-glucanase. >Q94ED2 Q94ED2 Putative beta-glucosidase. >Q8H3F0 Q8H3F0 Putative BLE2 protein. >Q852J1 Q852J1 Putative blue copper-binding protein. >Q9AUW1 Q9AUW1 Putative blue copper-binding protein. >Q8SB26 Q8SB26 Putative blue copper-binding protein. >Q8SB30 Q8SB30 Putative chloroplast nucleoid DNA-binding protein. >Q8H3X4 Q8H3X4 Putative disease resistance response protein-related/ dirigent protein-related.PNATTI >Q8S5V0 Q8S5V0 Putative endosperm specific protein. >Q8H3S1 Q8H3S1 Putative fasciclin-like arabinogalactan-protein. >Q8GTK3 Q8GTK3 Putative glucan endo-1,3-beta-glucosidase. >Q7Y157 Q7Y157 Putative glucanase. >Q9AX79 Q9AX79 Putative H+-transporting ATPase. >Q8LH82 Q8LH82 Putative hexokinase. >Q851L1 Q851L1 Putative LRR receptor-like protein kinase. >Q94LV0 Q7XCG2 Q94LV0 Putative membrane protein. >Q94LV0 Q7XCG2 Q94LV0 Putative membrane protein. >Q94LQ4 Q7XC94 Q94LQ4 Putative metalloproteinase. >Q94LQ4 Q7XC94 Q94LQ4 Putative metalloproteinase. >Q84S07 Q84S07 Putative nodulin. >Q84Z90 Q84Z90 Putative pathogenesis-related protein. >Q8L3W2 Q8L3W2 Putative peroxidase 1. >Q8H4P7 Q8H4P7 Putative photosystem II 10 kD polypeptide. >Q84M48 Q84M48 Putative phytocyanin. >Q8L3Q4 Q8L3Q4 Putative pollen specific protein (Putative ascorbate oxidase). >Q852L3 Q852L3 Putative protease inhibitor. >Q94GN3 Q94GN3 Putative receptor-associated protein. >Q8RYT6 Q8RYT6 Putative RNA helicase, DRH1. >Q94LG3 Q94LG3 Putative selenium-binding protein-like. >Q94DL9 Q94DL9 Putative syntaxin. >Q94HS1 Q94HS1 Putative thaumatin-like protein. >Q94HS3 Q94HS3 Putative thaumatin-like protein. >Q7XGU7 Q7XGU7 Putative thaumatin-like protein. >Q7XGV0 Q7XGV0 Putative thaumatin-like protein. >Q949F3 Q949F3 Putative vesicle-associated membrane protein (VAMP). >Q8LN94 Q8LN94 Putative vesicle-associated membrane protein. >Q7Y247 Q7Y247 Putative zinc transporter OsZIP2.

200
>Q8H385 Q8H385 Putative zinc transporter protein ZIP1. >Q7XJ47 Q7XJ47 Putative zinc transporter. >Q84L19 Q84L19 Putative ZIP-like zinc transporter. >Q9LDK7 Q9LDK7 Similar to Arabidopsis thaliana mRNA for MYB-related protein. >Q9LIY5 Q9LIY5 Similar to mavicyanin. >Q94FP2 Q94FP2 Succinate dehydrogenase subunit 3. >Q949E8 Q949E8 Uclacyanin 3-like protein. >Q8L555 Q8L555 Uclacyanin 3-like protein-like protein. >Q8H0B6 Q7X8J9 Q8H0B6 UDP-galactose 4-epimerase-like protein (OSJNBa0058K23.4 protein)

BIBLIOGRAPHIE Ahmad, S., M.M. Gromiha et A. Sarai. 2004. « Analysis and Prediction of DNA-binding Proteins and their Binding Residues Based on Composition, Sequence and Structural Information ». Bioinformatics, vol. 20, no 4, p. 477-86.
Alm, E., A.V. Morozov, T. Kortemme et D. Baker. 2002. « Simple Physical Models Connect Theory and Experiment in Protein Folding Kinetics ». Journal of Molecular Biology, vol. 322, no 2, p. 463 –76.
Altschul S.F., T.L. Madden, A.A. Schaefer, J. Zhang, Z. Zhang, W. Miller et D. J. Lipman DJ. 1997. « Gapped BLAST and PSI-BLAST:a New Generation of Protein Database Search Programs ». Nucleic Acids Research, vol. 25, no 17, p. 3389 –402.
Altschul, S.F., et B. W. Erickson. 1986. « Locally Subalignments Using Nonlinear Similarity Functions ». Bulletin of Mathematical Biology, vol. 48, no 5/6, p. 633-660.
Anderson, J., A. Pellionisz et E. Rosenfeld. 1990. Neurocomputing 2 : Directions for Research, Cambridge (MA) : MIT Press.
Avery, O.T., C.M. Macleod et M. McCarty. 1944. « Studies on the Chemical Nature of the Substance Inducing Transformation of Pneumococcal Types : Induction of Transformation by a Desoxyribonucleic Acid Fraction Isolated from Pneumococcus Type III ». Journal of Experimental Medecine, vol. 79, p. 137-159.
Baldi, P. et S. Brunak. 2001. Bioinformatics : The Machine Learning Approach. Cambridge (MA) : MIT Press.
Baldi, P., et Y. Chauvin. 1996. « Hybrid modeling, HMM/NN architectures, and protein applications ». Neural Computation, vol. 8, p.1541-1565.
Baldi, P. et Y. Chauvin. 1994. « Hidden Markov Models of the G-protein-coupled receptor family ». Journal of Computational Biology, vol. 1, no 4, p. 311-335.

202
Barrett C., R. Hughey et K. Karplus 1997. « Scoring Hidden Markov Models ». Computer Applications in the Biosciences, vol. 13, no 2, p. 191-199.
Barrette, I., G. Poisson, P. Gendron et F. Major. 2001. « Pseudoknots in Prion Protein mRNAs Confirmed by Comparative Sequence Analysis and Pattern Searching ». Nucleic Acids Research, vol. 29, no 3, p. 753-758.
Baxevanis, A.D. et B.F.F. Ouellette 2001. Bioinformatics : A Practical Guide to the Analysis of Genes and Proteins. Second Edition, Wiley-Interscience. Bengio, Y., Y.LeCun, C.Nohl et C. Burges. 1995. « LeRec : a NN/HMM Hybrid for On-line Handwriting Recognition. Neural Computation, vol. 7, no 5, p. 1289-1303.
Betel, D. et C.W. Hogues. 2002. « Kangaroo – A Pattern-Matching Program for Biological Sequences ». BMC Bioinformatcs. vol. 3, no 1, p. 20.
Boeckmann, B., A. Bairoch, R. Apweiler, M-C. Blatter, A. Estreicher, E. Gasteiger, M.J. Martin, K. Michoud, C. O’Donovan, I. Phan, S. Pilbout et M. Schneider. 2003. « The SWISS-PROT Protein Knowledgebase and its Supplement TrEMBL in 2003 ». Nucleic Acids Research, vol. 31, no 1, p. 365-370.
Bork, P., C. Ouzounis, C. Sander, M. Scharf, R. Schneider et E. Sonnhammer. 1992. « Comprehensive Sequence Analysis of the 182 Predicted Open Reading Frames of Yeast Chromosone III ». Protein Science, vol. 1, no 12, p.1677-1690.
Bourlard, H. et S. Bengio. 2002. « Hidden Markov Models and Other Finite State Automata for Sequence Processing ». chap. in The Handbook of Brain Theory and Neural Networks, 2e Édition. Cambridge (MA) : The MIT Press.
Bourlard H. 1995. « REMAP: Recursive Estimation and Maximization of a Posteriori Probabilities in Connectionist Speech Recognition ». Proceedings of Eurospeech-95, Madrid. p. 1663-1666.
Brejova, B., C. DiMarco, T. Vinar, S.R. Hidalgo, C. Holguin et C. Patten. 2000. Finding Patterns in Biological Sequences. Project Report for CS798g. University of Waterloo.

203
Brown, S.M. 2000. Bioinformatics: A Biologist’s Guide to Biocomputing and the Internet. Natick (MA) : Eaton Publishing.
Brunak, S., J. Engelbrecht et S. Knudsen. 1990. « Neural Network Detecs Errors in the Assignment of pre-mRNA Splice Site ». Nucleic acids Research, vol. 18, no 16, p. 4797-4801.
Brunak, S., J. Engelbrecht et S. Knudsen. 1991. « Prediction of Human mRNA Donor and Acceptor Sites from DNA Sequences ». Journal of Moecular Biology, vol. 220, no 1, p. 49-65.
Burge, C. et S. Karlin. 1997. « Prediction of Complete Gene Structures in Human Genomic DNA ». Journal of Molecular Biology, vol. 268, no 1, p. 78-94.
Chatterjee, S. et S. Mayor. 2001. « The GPI-anchor and Protein Sorting ». Cellular and Molecular Life Sciences, vol. 58, no 14, p. 1969-1987.
Cho, S.-B. et J.H. Kim. 1995. « An HMM/MLP Architecture for Sequence Recognition ». Neural Computation, vol. 7. p. 358-369.
Chomsky, N. 1957. Syntactic Structures. The Hague : Mouton.
Comet, J-P. 1998. Programmation dynamique et alignement de séquences biologiques. Thèse de Doctorat, Université de technologie de Compiègne.
Coyen, K.E., A. Crisci et D.M. Lublin. 1993. « Construction of Synthetic Signals for Glycosyl-phosphatidylinositol Anchor Attachment. Analysis of Amino Acid Sequence Requirements for anchoring ». Journal of Biological Chemistry, vol. 268, no 9, p. 6689-6693.
Dayhoff, M.O., R.M. Schwartz et B. Orcutt. 1978. « A Model of Evolutionary Change in Proteins ». Chap. In Atlas of Protein Sequence and Structure, p.345-352. Washington : M. O. Dayhoff éd. Dayhoff, M.O., R.V. Eck, M.A. Chang et M.R. Sochard. 1965. « Atlas of Protein Sequence and Structure » vol. 1. Silver Spring (MD): National Biomedical Research Foundation.

204
DiFrancesco, V., J. Garnier et P.J. Munson. 1997. « Protein Topology Recognition from Secondary Structure Sequences Applications of the Hidden Markov Models to Alpha Class Proteins ». Journal of Molecular Biology, vol. 267, no 2, p. 446-463.
Doolittle, R. J. et P. Bork. 1993. « Evolutionarily Mobile Modules in Proteins ». Scientific American, vol. 269, no 4, p. 50-56.
Duda, R.O., P.E. Hart et D.G. Stork. 2001. Pattern Classification 2e ed. NewYork (NY) : John Wiley & Sons.
Durbin, R., S. Eddy, A. Krogh et G. Mitchison. 1998. « Biological Sequences Analysis- Probabilistic Models of Proteins and Nucleic Acids ». Cambridge (MA) : University Press.
Eddy, S.R. 1995. « Multiple Alignment Using Hidden Markov Models ». chap. in Proceedings of the Third International Conference on Intelligent System for Molecular Biology. Menlo park (CA) : IAAA press.
Eddy, S. R. 1996. « Hidden Markov Models ». Current Opinion in Structural Biology, vol. 6, p. 361-365.
Eisenberg, D., R.M. Weiss, T.C. Terwilliger et W. Wilcox. 1982. « Hydrophobic Moments and Protein Structure ». Faraday Symposia of the Chemical Society, vol. 17, p. 109-120.
Eisenhaber, B., M. Wildpaner, A.J. Schultz, G.H H. Borner, P. Dupree et F. Eisenhaber. 2003. « Glycosylphosphatidylinositol Lipid Anchoring of Plant Proteins. Sensitive Prediction from Sequence- and Genome-wide Studies for Arabidopsis and Rice ». Plant physiology, vol. 133, no 4, p. 1691-1701.
Eisenhaber, B., P. Brok et F. Eisenhaber. 2001. « Post-translational GPI Lipid Anchor Modification of Proteins in Kingdoms of Life: Analysis of Protein Sequence Data from Complete Genomes ». Protein Engeeniring, vol. 14, no 1, p. 17-25.
Eisenhaber, B., P. Brok, et F. Eisenhaber. 1999. « Prediction of Potential GPI-modification Sites in Proprotein Sequences ». Journal of Molecular Biology, vol. 292, no 3, p. 741-758.

205
Eisenhaber, B., P. Bork et F. Eisenhaber. 1998. « Sequence Properties of GPI-anchored Proteins Near the Oméga-site : Constraints for Polypeptide Binding Site of the Putative Transamidase ». Protein Engineering, vol. 11, no 12, p. 1155-1161.
Ellis J.A. et J.P. Lazio. 1995. «Identification and Caracterization of a Novel Protein (p137) which Transcytoses Bidirectionally in Caco-2 Cells ». Journal of Biological Chemistry, vol. 270, no 35, p. 20717-20723.
Engelman, D.M., T.A. Steitz et A. Goldman. 1986. « Identifying Nonpolar Transbilayer Helices in Amino Acid Sequences of Membrane Proteins. Annual Review of Biophysics and Biophysical Chemistry, vol.15, p. 321-353.
Estes, W.K. 1994. Classification and Cognition. Oxford (NY) : Oxford University Press.
Farriol-Mathis, N., J.S. Garavelli, B. Boeckmann, S. Duvaud, E. Gasteiger, A. Gateau, A-L. Veuthey et A. Bairoch. 2004. “Annotation of Post-translational Modifications in the Swiss-Prot Knowledge base”. Proteomics, vol. 4, p. 1537-1550.
Feng, D.F. et R.F. Doolitle. 1987. « Progressive Sequence Alignment As a Prerequisite to Correct Phylogenetic Trees ». Journal of Molecular Evolution, vol. 25, no 4, p. 351-360.
Ferguson, M.A., S.W. Homans, R.A. Dwek et T.W. Rademacher. 1988. « Glycosyl-phosphatidylinositol Moiety that Anchor Trypanosoma brucei Variant Surface Glycoprotein to the Membrane ». Science, vol. 239. no 4841 pt1, p. 753-759.
Ferguson, M.A., K. Haldar et G. A. Cross. 1985. « Trypanosoma brucei Variant Surface Glycoprotein has a sn-1,2-dimyristyl Glycerol Membrane Anchor at its COOH Terminus ». Journal of Biological Chemistry, vol. 260, no 8, p. 4963-4968.
Forney, G.D.Jr. 1973. « The Viterbi Algorithm ». Proceedings of the IEEE, vol. 61, no 3, p. 268-278.
Francke, S. et L. Weynans. 2002. Étude sur les Modèles de Markov cachés et les applications à la bioinformatique. Rapport de stage, École Nationale Supérieure de Techniques Avancées (ENSTA).

206
Furukawa, Y., K. Tsukamoto et H. Ikezawa. 1997. « Mutational Analysis of the C-terminal Signal Peptide of Bovine Liver 5-nucleotidase for GPI Anchoring: a Study on the Significance of the Hydrophilic Spacer Region ». Biochimica et Biophysica Acta, vol. 1328, no 2, p. 185-196.
Futerman, A.H., M.G. Low, K.E. Ackermann, W. R.Sherman, et I. Silam. 1985. « Identification of Covalently Bound Inositol in the Hydrophobic-anchoring Domain of Torpedo Acelylcholinesterase ». Biochemical and Biophysical Research Communications, vol. 129, no 1, p. 312-317.
Gan, H.H., R. A. Perlow, S. Roy, J. Ko, M. Wu, J. Huang, S. Yan, A. Nicoletta, J. Vafai, D. Sun, L. Wang, J.E. Noah, S. Pasquali et T. Schlick. « Analysis of Protein Sequence/Structure Similarity Relationships ». Biophysical Journal, vol. 83, p. 2781-2791.
Gattiker, A., E. Gasteiger et A. Bairoch. 2002. « ScanProsite: a Reference Implementation of a PROSITE Scanning Tool ». Applied Bioinformatics, vol. 1, no 2, p.107-108.
Gautheret, D., F. Major, R. Cedergren. 1990. « Pattern searching/alignment with RNA Primary and Secondary Structure: an Effective Descriptor for tRNA ». Computer Applications in the Biosciences, vol. 6, no 4, p. 325-331.
Gordon A.D. 1981. Classification Methods for the Exploratory Analysis of Multivariate Data. London (UK) : Chapman and Hall.
Gosselin, B. 2000. Traitement de l’information—Classification et Reconnaissance Statistique de Formes. Notes de cours. Faculté Polytechnique de Mons.
Gowda, D. C. et E. A. Davidson. 1999. « Protein Glycosylationin the Malari parasite ». Parasitology Today, vol. 15, no 4, p. 147-152. GRAMENE, 2004. A Comparative Mapping Resource. for Grains. http://www.gramene.org/ Griffith, F. 1928. « The Significance of Pneumococcal Types ». Journal of Hygiene, vol. 27, p. 113-159
Gusfield, D. 1997. Algorithms on Strings, Trees, and sequences. Cambridge(MA) : Cambridge University Press.

207
Habrias, H. 2002. Génie logiciel Module de spécification 2. Instituts Universitaires de technologie (IUT). Université Nantes.
Haykin, S. 1999. Neural Networks, A Comprehensive Foundation: second edition. New York (NY) : MacMillan College Publishing,
Hebb, D.O. 1949. The Organization of Behavior. New York: Wiley.
Henikoff, J.G., E.A. Greene, S. Pietrokovski et S. Henikoff, « Increased Coverage of Protein Families with the BLOCKS Database Servers », Nucleic Acids Research, vol. 28, p. 228-230. (2000).
Henikoff, S. et J.G. Henikoff. 1992. « Amino Acid Substitution Matrices from Protein Blocks ». Proceedings of the Natural Academy of Sciences of the United States of America, vol. 89, no 22, p.10915-10919.
Heyshey, A.D. et M. Chase. 1952. « Independent Functions of Viral Protein and Nucleic Acid in Growth of Bacteria ». Journal of General Physiology, vol. 36, no. 1, p. 39-56.
Higgins, D.G. et P.M. Sharp. 1988. « CLUSTAL: a Package for Performing Multiple Sequence Alignment on a Microcomputer ». Gene, vol. 73, no 1, p. 237-244.
Holmström, L., P. Koistinen, J. Laaksonen et E. Oja. 1996. Comparison of Neural and Statistical Classifiers: Theory and Practice. Rolf Nevanlinna Institute Research Reports A13, Helsinki.
Hooper, N.M. 2001. « Determination of Glycosyl-phosphatidylinositol Membrane Protein Anchorage ». Proteomics, vol. 1, no 6, p. 718-755.
Hopfield, J.J. 1982. « Neural Networks and Physical Systems with Emergent Collective Computational Abilities ». Proceedings of the Natural Academy of Sciences of the United States of America, vol. 79, no 8, p. 2554-2558.
Hornik, K., M. Stinchcombe et H. White.1989. « Multilayer Feedforward Networks are Universal Approximators ». Neural Networks, vol. 2, no 5, p. 359-366.

208
Howell, S., C. Lanctot, G. Boileau et P. Crine. 1994. «A Cleavage N-terminal Signal Peptide is not a Prerequiste for the Biosynthesis of Glycosylphosphatidylinositol-anchored Proteins ». Journal of Biological Chemistry, vol. 269, no 25, p. 16993-16996.
Ikezawa, H. 2002. « Glycosylphophatidylinositol (GPI)-anchored proteins ». Biological and Pharmaceutical Bulletin, vol. 25, no 4, p. 409-417.
IUPAC-IUB 1993, « Joint Commission on Biochemical Nomenclature (JCBN). Nomenclature and Symbolism for Amino Acids and Peptides: Corrections to Recommendations 1983 ». European Journal of Biochemistry, vol. 213, no 1, p.1-3.
Jacob, F. 1977. « Evolution and Tinkering ». Science, vol. 196, no 4295, p. 1161-1166.
James, M. Classification Algorithms. New York (NY) : John Wiley & Son.
Jordan, M.I. 1995. Why the logistic Function? A Tutorial Discussion on Probabilities and Neural Network. Computational Cognitive Science Report 9503. Cambridge (MA) : MIT press.
Kulp, D., D. Haussler, M.G. Reese et F.H. Eeckman. 1996. « A Generalized Hidden Markov Model for the Recognition of Human Genes in DNA ». Proceedings of the International Conference on Intelligent Systems Molecular Biology, vol. 4, p.134-42 Krawetz, S.A. et D.D. Womble. 2003. Introduction to Bioinformatics : A Theoretical and Practical Approach . Totowa (N-J): Humana Press.
Krogh, A. et J.A. Hertz. 1992. A simple weight decay can improve generalization. dans J.E. Moody, S.J. Hanson, and R.P. Lippmann, editors, Advances in Neural Information Processing Systems 4, p. 450—957. Kyte, J. et R.F. Doolittle. 1982. « A Simple Method for Displaying the Hydropathic Character of a Protein ». Journal of Molecular Biology, vol. 157, no 1, p. 105-132.
LeCun, Y. 1985. « A Learning Scheme for Asymmetric Threshold Networks ». Proceedings of Cognitiva 85, p.599-604. Paris, France.

209
Lehmann, S. et D.A. Harris. 1995. « A Mutant Prion Protein Displays an Aberrant Membrane Association When Expressed in Cultured Cells ». Journal of Biological Chemistry, vol. 270, no 41, p. 24589-24597
Leon, F., D. Gâlea, et M. Zbancioc 2002. « Knowledge Representation Through Interactive Networks ». Proceedings of the European Conference on Intelligent Systems, Iasi.
Lewin, B. 1999. Gènes VI 6e éd, traduction de l’anglais par Chrystelle Sanlaville. Bruxelles : DeBoeck Université.
Low, M. 1999. « GPI-anchored Biomolecules- an Overview ». chap. In GPI-anchored Membrane Proteins and Carbohydrates, p.1-14, Austin (TX) : Landes.
Magoulas, G. 2001. Neural and Genetic Computing. Brunel University, Uxbridge, United Kingdom.
Mann, M. et O. Jensen. 2003. « Proteomic Analysis of Post-translational Modifications ». Nature Biotechnology, vol. 21, no 3, p. 255-261.
Martelli P. L., P. Fariselli et R. Casadio. 2004. « Prediction of Disulfide-bonded Cysteines in Proteomes with a Hidden Neural Network ». Proteomics, vol. 4, no 6, p. 1665-1671.
Martelli, PL, P. Fariselli, L. Malaguti et R. Casadio 2002. « Prediction of the Disulfide Bonding State of Cysteines in Proteins with Hidden Neural Networks ». Protein Engeeniring, vol. 15, no 12, p. 951-953.
May, A.C.W. 2001. « Optimal Classification of Protein Sequences and Selection of Representative Sets From Multiple Alignments: Application to Homologous Families and Lessons for Structural Genomics ». Protein Engineering, vol. 14, no 4, p 209-217.
McConville, M.J. et Menon A.K. 2000. « Recent Developments in the Cell Biology and Biochemistry of Glycosylphosphatidylinositol Lipids ». Molecular and Membrane Biology, vol. 17, no 1, p. 1-16
McCulloch, W.S. et W.H. Pitts. 1943. « A logical Calculus of the Ideas Immanent in Nervous Activity ». Bulletin of Mathematical Biophysics, vol. 5, p. 115-133.

210
Micanovic, R., L.D. Gerber, J. Berger, K. Kodukula et S. Udenfriend. 1990. « Selectivity of the Cleavage/Attachment Site of Phosphatidylinositol-glycan-anchored Membrane Proteins Determined by Site-specific Mutagenesis at Asp-484 of Placental Alkaline Phosphatase ». Proceedings of the National Academy of Sciences of the United States of America, vol. 87,no 1, p. 157-161.
Minsky, M. et S. Papert. 1969. Perceptrons. Cambridge (MA) : MIT Press.
Moran, P., H. Raab, W.J. Kohr et I.W. Caras. 1991. « Glycophospholipid Membrane Anchor Attachment Molecular Analysis of the Cleavage/Attachment Site ». Journal of Biological Chemistry, vol. 266, no 2, p. 1250-1257.
Moran, P., I.W. Caras. 1994. « Requirements for Glycosylphosphatidylinositol Attachment Are Similar but Not Identical in Mammalian Cells and Parasitic Protozoa ». Journal of Cell Biology, vol. 125, no 2, p. 333-343.
Morita, M., R. Sabourin, F. Bortolozzi et C.Y. Suen. 2003. « Segmentation and Recognition of Handwritten date : an HMM/MLP Hybrid Approach ». International Journal on Document Analysis and Recognition, vol. 6, p. 248-262.
Morris, W.F. 1988. Artificial Intelligence: a Knowledge-based Approach. Boston (MA) : Boyd and Fraser éditeur.
Nakata, K. 1995. « Prediction of Zinc Finger DNA Binding Protein ». Computer Applications in the Biosciences, vol. 11, p. 125-131. Nalivaeva, N.N. et A.J. Turner. 2001. « Post-translational Modifications of Proteins : Acetylcholinesterase as a Model System ». Proteomics, vol. 1, no 6, p. 735-747.
Natt, NK., H. Kaur, G.P. Raghava. 2004. « Prediction of Transmembrane Regions of Beta-barrel Proteins Using ANN- and SVM-based Methods ». Proteins, vol. 56, no 1, p.11-8.
Navarro, G et M. Raffinot. 2002. Flexible Pattern Matching in Strings. Cambridge (MA) : Cambridge University Press.

211
Needleman , S.B., C. Wunssch. 1970. « A General Method Applicable to the Search for Similarities in the Amino Acid Sequence of Two Proteins ». Journal of Molecular Biology, vol. 48, no 3, p. 443-453.
Nielsen, H. et A. Krogh. 1998. « Prediction of Signal Peptides and Signal Anchors by a Hidden Markov Model ». Proceedings of the International Conference on Intelligent Systems Molecular Biology, vol. 6, p.122-130.
Niles, L.T. et H.F. Silverman. 1990. « Combining Hidden Markov Models and Neural Network Classifiers »., Proceedings of IEEE International Conference on Acoustics, Speech, and Signal Processing, p. 417-420.
Nosjean, O., A. Briolay et B. Roux. 1997. « Mammalian GPI Proteins : Sorting, Membrane Residence and Functions. Biochimica et Biophysica Acta, vol. 1331, p. 153-186.
Orr, G. et F. Cummins. 1999. Neural Network : Lecture Notes. Willamate University Oregon.
Parker, D.B. 1985. Learning Logic. Tech Report TR 47 Center for Computational Research in Economics and Management Science. Cambridge (MA) : MIT press.
Poisson G., A. Bergeron, C. Chauve et B. 2003. « Prediction of Post-translational GPI-Anchor Modification by Machine Learning ». Human Proteome Organisation, HUPO Molecular and Cellular Biology, Special, vol. 2, no 9, p.826. Poisson, G., A. Bergeron et C. Chauve. 2004. Artificial Neural Network and Hidden Markov Model for GPI-Anchored Protein Predictions. 12th International Conference on Intelligent System for Molecular Biology, Glascow (GB).
Polevoda, B. et F. Sherman. 2000. « N-terminal Acetylation of Eukaryotic Proteins ». Journal of Biological Chemistry, vol. 275, no 47, p. 36479-36482.
Quian, N. et T.J. Sejnowski. 1988. « Predicting the Secondary Structure of Globular Proteins Using Neural Network Models ». Journal of Molecular Biology, vol. 202, no 4, p. 865-884. Ramón y Cajal, S. 1911. Histologie du système nerveau de l’homme et des vertébrés, Paris : Maloine.

212
Renders, J-M. 1995. Algorithmes génétiques et réseaux de neurones. Paris : Hermès Science éditeur.
Riedmiller, M. et H. Braun. 1992. « RPROP A Fast Adaptive Learning Algorithm ». Proceedings of the 1992 International Symposium on Computer and Information Sciences, Antalya, Turquie. p. 279-285.
Riedmiller, M. et H. Braun. 1993. « A Direct Adaptive Method for Faster Backpropagation Learning: The RPROP Algorithm ». Proceedings of the IEEE International Conference on Neural Networks, p. 586-591.
Rigoll, G. et D. Willett. 1998. « A NN/HMM Hybrid for Continuous Speech Recognition with a Discriminant Nonlinear Feature Extraction ». Proceedings of the IEEE-ICASSP, p. 9-12.
Ripley, B.D. 1993. Statistical Aspects of Neural Networks—Networks on Chaos: Statistical and probabilities Aspects. U. Bornndorff-Nielsen, J. Jensen, and W. Kendal, eds., Chapman and Hall
Ripley, B.D. 1996. Pattern Recognition and Neural Networks. Cambridge (MA) : University Press.
Roberts, W.L. et T.L. Rosenberry. 1985. « Identification of Covalently Attached Fatty Acids in the Hydrophobic Membrane-binding Domain of Human Erythrocyte Acetylcholinesterase ». Biochemical and Biophysical Research Communications, vol. 133, no 2, p. 621-627.
Rosenblatt, F. 1957. The Perceptron: A Perceiving and Recognizing Automaton (Project PARA). Technical Report 85-460-1, Cornell Aeronautical Laboratory.
Rosenblatt, F. 1958. « The Perceptron: a Probabilistic Model for Information Storage and Organization in the Brain ». Psychological Review, vol. 65, no 6, p. 386-408.
Rumelhart, D.E., G.E. Hinton et R.J. Williams. 1986. « Learning Representations by Back-propagating Errors ». Nature, vol. 323, no 9, p. 533-536.

213
Rumelhart, D.E. et J.L. McClelland. 1986. Parallel Distributed Processing: Explorations in the Microstructure of Cognition. Cambridge (MA) : The MIT Press.
Saitou, N. et M. Nei. 1987. « The Neighbor-joining Method: a New Method for Reconstructing Phylogenetic Trees ». Molecular Biology and Evolution, vol. 4, no 4, p. 406-425.
Sanger, F., E.O.P. Thompson, et R. Kitai. 1955. « The Amine Groups of Insulin ». Biochemistry Journal, vol. 59, p. 509-514.
Sarle, W.S. 1994. « Neural Networks and Statistical Models ». Proceedings of the Nineteenth Annual SAS Users Group International Conference, Cary, NC, SAS Institute, p. 1538-1550. Schalkoff, R. 1992. Pattern Recognition: Statistical, Structural and Neural Approaches. New York (NY) : John Wiley & Sons éditeur.
Scheinder, G., S. Rohlk et P. Wrede. 1993. « Analyse of Cleavage-site Pattern in Protein Precursor Sequences with Perceptron-type Neural Network ». Biochemical and Biophysical Research Communications, vol. 194, p. 951-959.
Searls, D.B. et S. Dong. 1993. « A Syntactic Pattern Recognition System for DNA Sequences ». Proceedings of the Second International Conference on Bioinformatics, Supercomputing, and Complex Genome Analysis. p. 89-101.
Searls, D.B. 1993. « The Computational Linguistics of Biological Sequences » chap 2. in Artificial Intelligence and Molecular Biology, p. 47-120 Cambridge (MA) : AAAI Press.
Searls, D.B. 2001. « Reading the Book of Life ». Bioinformatics, vol. 17, no 7, p. 579-580.
Searls, D.B. 2002. « The Language of Genes ». Nature, vol 420, no 6912, p. 211-217.
Senior, A. 1994. « Off–Line Cursive Handwriting Recognition using Recurrent Neural Networks ». Thèse de Doctorat, University of Cambridge.

214
Seo, J. et K-J Lee. 2004. « Post-translational Modifications and Their Biological Functions: Proteomic Analysis and Systematic Approaches ». Journal of Biochemistry and Molecular Biology, vol. 37, no 1, p. 35-44. Setubal, J. et J. Meidanis. 1997. Introduction to Computational Molecular Biology Boston (MA): PWS Publishing Co.
Shepherd, G.M. et C. Koch. 1990. « Introduction to Synaptic Circuits », dans The Synaptic Organization of the Brain, p.3-31. NewYork: Oxford University Press.
Smith, T.F. et M.S. Waterman. 1981. « Identification of Common Molecular Subsequences ». Journal of Molecular Biology, vol. 47, no 1, p. 195-197.
SNNS, 1998. Stuttgart Neural Network Simulator (SNNS) version 4.2. Breitwiesenstrasse (Allemagne) : University of Stuttgart.
Sonnhammer, E.L.L., S.R. Eddy et R. Durbin. 1997. « Pfam a Comprehensive Database of Protein Domain Families Based on Seed Alignments ». Proteins, vol. 28, no 3, p. 405-420.
Spiro, R.G. 2002. « Protein Glycosylation : Nature, Distribution, Enzymatic Formation, and Disease Implication of Glycopeptide Bounds ». Glycobiology, vol. 12, no 4, p. 43R-56R.
Stormo, G.D., T.D. Schneider, L. Gold et A. Ehrenfeucht. 1982. « Use of the Perceptron Algorithm to Distinguish Translational Initiation Site in E.coli ». Nucleic Acids Research, vol. 10, no 9, p. 2997-3011.
The Arabidopsis Genome Initiative. 2000. « Analysis of the Genome Sequence of the Flowering Plant Arabidopsis thaliana ». Nature, vol. 408, no 6814, p. 796-815.
Thomson, S.M. 2003. « An Introduction to Multiple Sequence Alignment and Analysis » chap. in Introduction to Bioinformatics : A Theoretical and Practical Approach, Totowa (NJ) : Humana Press.
Thompson, J.D., D.G. Higgins et T.J. Gibson. 1994. « CLUSTALW: Improving the Sensibility of Progressive Multiple Sequence Alignment through Sequence Weighting, Positions-specific Gap Penalities and Weight Matrix Choice ». Nucleic Acids Research, vol. 22, no 22, p. 4673-4680.

215
Tompa, M. 2000. Lectures Notes on Biological Sequences Analysis. Technical report Department and Computer Science and Engineering. Seattle (WA) : University of Washington.
Touzet, C. 1992. Les réseaux de neurons artificiels : Introduction au connexionnisme. http://saturn.epm.ornl.gov/~touzetc/
Tse, A.G., A.N. Barclay, A. Watts et A.F. Williams. 1985. « A Glycophospholipid Tail at the Carboxyl Terminus of the Thy-1 Glycoprotein of Neurons and Thymocytes ». Science, vol. 230, no 4729. p. 1003-1008.
Vander A.J., J.H. Sherman, D. S. Luciano et J. R. Gontier. 1989. Physiologie Humaine 2e éd. McGraw Hill.
Waterman, M.S. et M. Eggert. 1987. « A New Algorithm for Best Subsequence Alignments with Applications to tRNA-rRNA Comparisons ». Journal of Molecular Biology, vol. 197, no 4, p. 723-728.
Watson, J. et F. Crick. 1953. « Molecular Structure of Nucleic Acids: A structure for Deoxyribose Nucleic Acid ». Nature, vol 171, p. 737. White, S.H. et W.C. Wimley. 1999. « Membrane Protein Folding and Stability: Physical Principles ». Annual Review of Biophysics and Biomolecular. Structure, vol. 28, p. 319-365. Widrow, B. et M.E. Hoff. 1960. « Adaptive Switching Circuits ». 1960 IRE WESCON Convention Record New-York, p. 96-104.
Wishart, D.S. 2003. « Sequence Similarity and Database Searching ». Introduction to Bioinformatics : A theoretical and practical approach, Totowa (NJ) : Humana Press.
Wright, G.L.jr et O. J. Semmes. 2003. « Proteomics Health and Disease ». Journal of Biomedecine and Biotechnology, vol. 4, p. 215-216.
Wu, C. G., Whitson, J. McLarty, A. Ermongkonchai et T. Chang. 1992. « Protein Classification Artificial Neural System ». Protein Science, vol. 1, p. 667-677.

216
Wu, C. et J.W. McLarty. 2000. Neural Networks and Genome Informatics Methods in Computational Biology and Biochemistry 1, NewYork (NY) : Elsevier publishing.
Xuan, Z., W.R. McCombie, et M. Q. Zhang. 2002. « GFScan: A Gene Family Search Tool at Genomic DNA Level ». Genome Research, vol. 12, no 7, p. 1142-1149.
Zell, A. 2002. JavaNNS 2002, Stuttgart Neural Networks Simulator (SNNS). Breitwiesenstrasse (Allemagne) : University of Stuttgart.http://www-ra.informatik.uni-tuebingen.de/







