
Les Manifestations Rhumatologiques des Principales Maladies
Systémiques
DR Hosni BEN FREDJ

INTRODUCTION
maladies auto-immunes inflammatoires chroniques
nombreux organes
maladies de système ou systémiques
liaisons anatomiques similaires: nécrose fibrinoïde du tissu conjonctif
auto-immunité: auto-anticorps: un rôle pathogénique

Les connectivites les plus fréquentes:
Le lupus érythémateux disséminé ( LED)
Le syndrome de Gougerot Sjogren (SS)
Polymyosite et dérmatopolymyosite (PM et DPM)
La Sclérodermie
connectivites mixtes ou syndrome de chevauchement

I - LE LUPUS ERYTHEMATEUX DISSEMINE
maladie auto-immune, non spécifique d'organe
un grand polymorphisme clinique
étiologie multifactorielle (facteurs génétiques et environnementaux)
évolue par poussées successives entrecoupées de rémissions
auto anticorps : AAN

9 femmes pour 1 homme.
âge de début :10 et 40 ans
incidence : 0,2 à 10/100000 habitants
prédispositions génétiques:HLA DR2 DR3
poussée de L.E.D :une exposition solaire, une infection, un stress, une prise médicamenteuse.

A- MANIFESTATIONS RHUMATOLOGIQUES
Révélatrice : 50% des cas8-9 fois/10 : au cours de l'évolution
1- Aspects cliniques :
Arthralgies:
vivestrès cortico sensible et résistent aux AINSs'accompagnent souvent de myalgies

Arthrites :
polyarthrite bilatérale et symétrique: 80% au moment du diagnostic:
aiguë volontiers fluxionnairesubaiguë avec raideur matinale et parfois nodules sous cutanés transitoires rappelant des nodules rhumatoïdes.

Elles sont rarement chroniques, réalisant 3 aspects principaux :
Soit une synovite non destructrice non déformante.Soit une atteinte déformante type main ou pied de Jaccoud ; sans destruction radiologique.Soit plus rarement déformante et destructrice.
genoux, chevilles, poignets et les doigts (MCP et IPP)
le rachis est toujours épargné

Atteintes musculaires :
Les myalgies sont fréquentes, en règle proximale. Exceptionnellement: déficit clinique, une élévation des enzymes musculaires avec des signes électriques et une nécrose histologique.
Une myopathie cortisonique peut s’observer après quelque temps d’évolution.

Ténosynovites et ruptures tendineuses:
La ténosynovite des fléchisseurs :syndrome du canal carpien.
Les ruptures tendineuses :tendon rotulien, le tendon quadricipital ou le tendon d'Achille.
Plusieurs facteurs de risque : corticothérapie prolongée - HPT secondaire à une insuffisance rénale

Ostéonecroses épihysaires aseptiques :
4 a 10% : multiples et asymptomatiques.
extrémité supérieure du fémur, condyles fémoraux, plateaux tibiaux, tête humérale.
corticothérapie et la présence d’anticorps antiphospholipides.

Arthrites septiques :
cause de mortalité chez un lupique
Facteurs favorisant :
articulation fragilisée par une synovite ou une nécrosefortes doses de corticoïdes
siège habituel : genougermes pyogènes G (+) ou G (-) , salmonelle ou BK

2-Examens complémentaires:
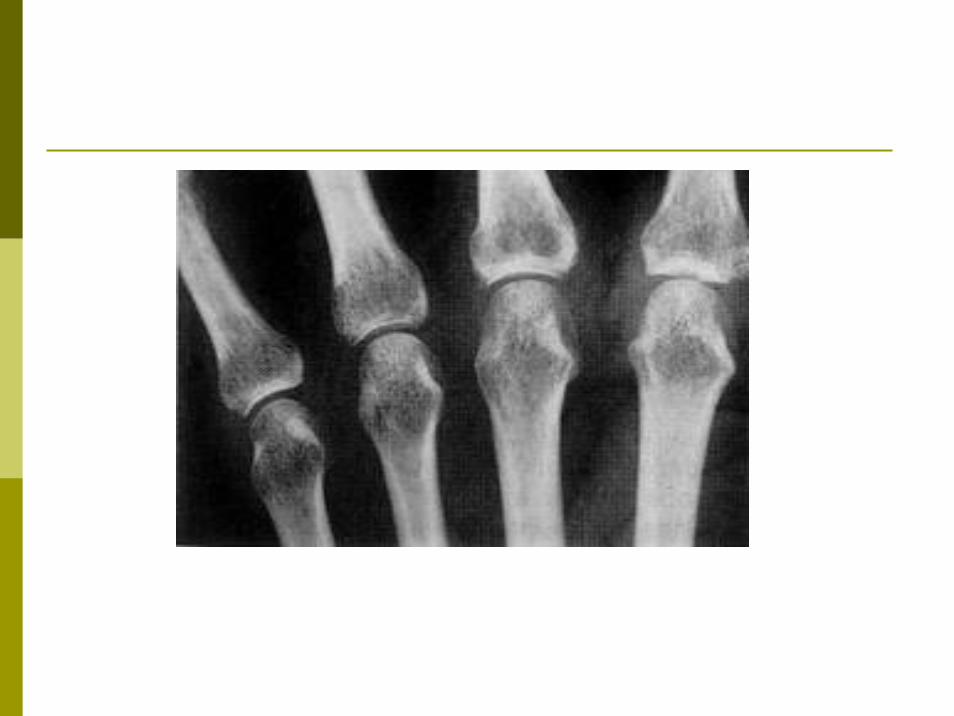
Les Rx standard:Même si déformations normales une déminéralisation épiphysaire et un gonflement des parties molles.
Le liquide articulaire :Est de type inflammatoire, peu cellulaire 2000 à
5000 éléments/mm3.L’histologie de la synoviale:Synovite inflammatoire subaiguë, non spécifique

B- MANIFESTATIONS EXTRA-ARTICULAIRES
1- Signes cutanés:Érythème facial, maculo-papuleux, siègent aux ailes du nez et les pommettes.Lupus discoïde.Photosensibilité aux zones découvertes.
2- manifestations rénales :Glomérulonéphrite : Protéinurie, hématurie,
insuffisance rénale.3- manifestations cardiaques:Pericardite, endocardite de Libman Sacks(végétations
aseptiques).

4- manifestation pleuro-pulmonaires :Pleurésie sérofibrineuse.Pneumonie lupique.Fibrose pulmonaire Interstitielle.HTAP.
5- manifestions neurologiques:Constituent un facteur pronostic.Comitialité, syndromes locaux, syndrome extra pyramidal.Manifestation neurologiques périphériques: multi ou polynévrite.
6- autres :Signes généraux : fièvre, fatigue et amaigrissement.Vasculaires: Raynaud – HTA.Hématologiques (ganglionnaires, splénomégalie).Digestive (pancréatite, hépatite).

C- DIAGONSTIC :
Le diagnostic de LED est porté sur l'association d'atteintes cliniques et d'Auto Anticorps.
Des critères diagnostic ont été proposés (ACR) : la présence de 4 critères : diagnostic (+)

Érythème facial ( ailes du nez et les pommettes)

Lupus discoïde
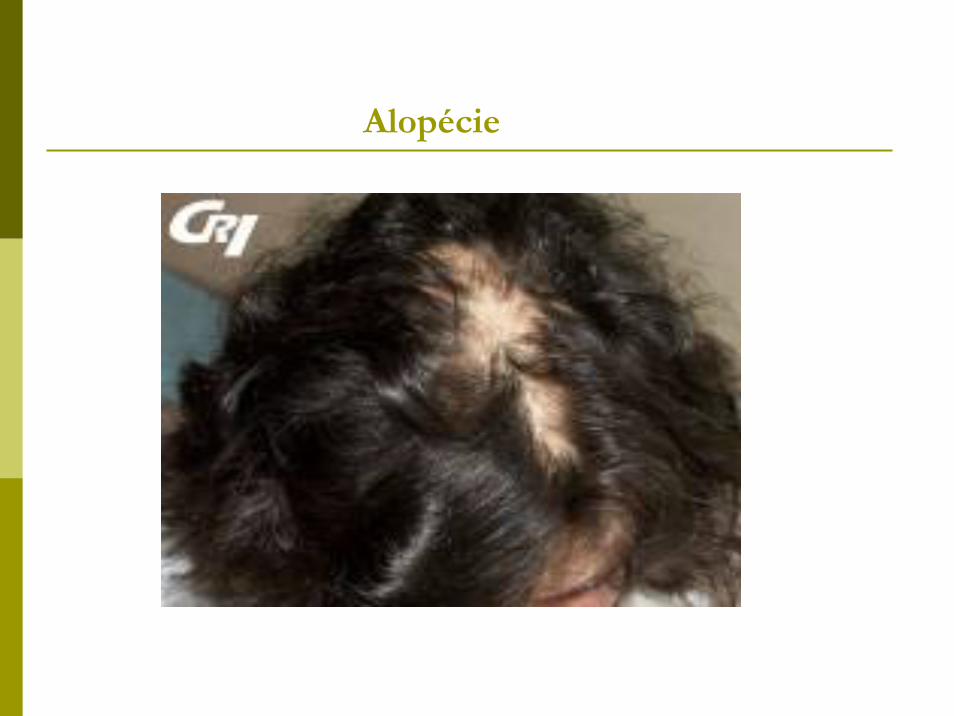
Alopécie



II - LA SCLERODERMIE SYSTEMIQUE
affection généralisée, primitivement microcirculatoire
accumulation de collagène
la peau, le TD, les poumons, et les reins PC.
maladie rare : femmes entre 20 et 50 ans
sur le plan immunologique : anticorps anticentroméres et anti scl70.

A- MANIFESTATIONS RHUMATOLOGIQUES
1 - Atteinte articulaire (50 %) :
Polyarthralgie, plus rarement polyarthrite.
L’atteinte est souvent asymétrique
Les petites et les grosses articulations : les doigts, les poignets, les genoux, les chevilles
Les signes objectifs sont habituellement mineurs, mais l'aspect peut être très inflammatoire, rouge, chaud, et sensible.

Les épanchements intra articulaires sont rares et peu abondants.
Les Rx standard:
un épaississement des parties molles périarticulairesun pincement articulaireune déminéralisation osseuse parfois une résorption de la styloïde cubitale

2- Les ténosynovites
3- Atteinte osseuse :
L’ostéonécrose de la tête du fémur
Résorption des houppes phalangiennes (acro-ostéolyse) : les phalanges peuvent disparaître rarement
la résorption osseuse peut toucher: l'extrémitéinférieure du radius, du cubitus ou la clavicule, voire même de l'angle du maxillaire inférieur, les côtes et le rachis.

4- Atteinte musculaire :
Myalgies modérés, déficit moteur minime et faible élévation des enzymes musculaires.
Dans certain cas une véritable polymyosite peut
être associée.

B- DIAGNSOTIC
Repose sur l'existence des autres signes extra articulaires:
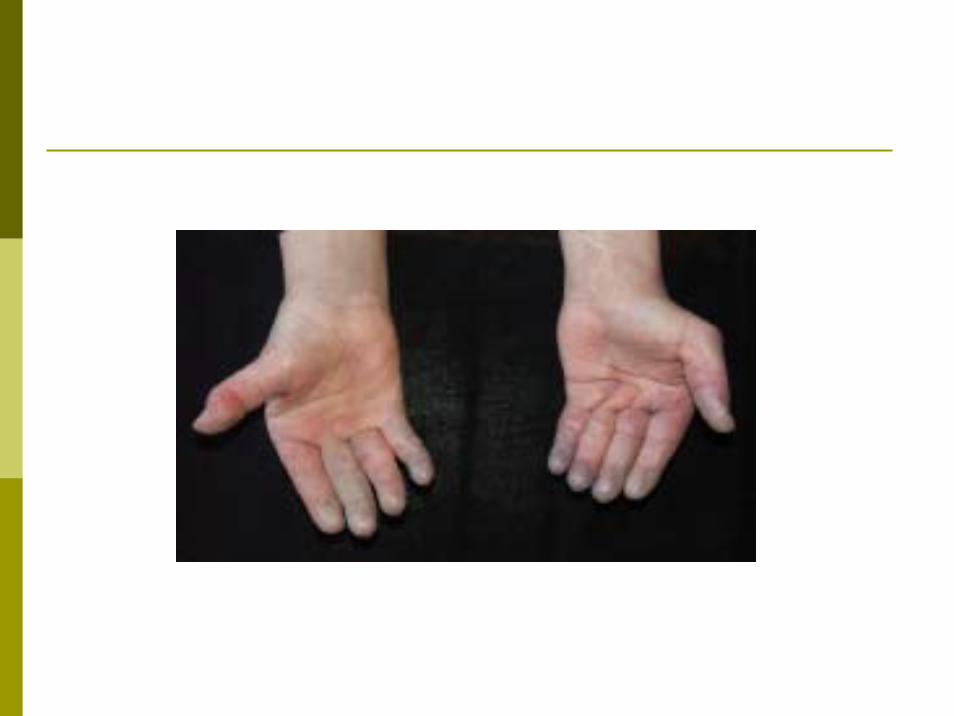
1- Atteinte cutanée:La peau est atrophique indurée, rétractée difficile à plisser.Au visage : effacement des rides et des plis, avec limitation de l'ouverture de la bouche.Au mains: sclérodactylie : la peau est difficile à plisser, doigts effilés en légère flexion. Télangiectasies à la face, au tronc et aux mains. Calcifications sous cutanées à la palpation et sur les radiographies.
2- Syndrome de Raynaud :Très fréquent souvent plus sévère et plus mutilant que le
Raynaud idiopathique.

3- Atteinte digestive :Oesophage : dysphagie, reflux, objectivés par une manométrie œsophagienne ou une fibroscopie gastrique qui montrent une dyskinésie oesophagienne et la diminution du péristaltisme du sphincter inférieur de l'œsophage.
4- Cœur :- Myocardite, troubles de la conduction et du rythme.- Péricardite.- Insuffisance ventriculaire droite.
5- Rein :Protéinurie, insuffisance rénale et HTA.
6- Poumon:Atteinte grave:fibrose pulmonaire irréversible, diminution de
la de diffusion du Co et/ou HTAP.

Faciès Sclérodermique

Résorption des houppes phalangiennes

Ostéolyse phalangiennes

Capillaroscopie: Capillaroscopie: mméégacapillairegacapillaire

III - POLYMYOSITE (PM)
ET DERMATO-POLYMYOSITE (DPM)
Groupe d'affections musculaire (muscle strié) en apparence primitive, de topographie volontiers rhizomélique, de nature inflammatoire évoluant sur un mode aigue subaiguë ou chronique.
Il peuvent intéresser la peau, le tissu sous cutané, et rarement les viscères.
Polymyosite sans atteinte cutanéDermatopolymyosite avec signes cutanés
Femmes : 40 - 60 ans

A - Atteintes articulaires (15 à 30%) :
arthralgies, rarement des arthritessouvent atteinte oligo ou polyarticulairesles poignets, genoux, épaules et doigts.
Les arthrites guérissent en quelques semainesNi déformantes, ni érosives mais parfois récidivantes.
Ces arthrites orientent parfois à tort vers le diagnostic de PR; si elles précèdent les signes musculaires, mais les critères de la PR sont absents et il n' y a pas de modifications radiologiques.

B- Diagnostic :Repose sur l'existence des autres signes:
1- Signes musculaires :Myalgies inflammatoires des ceintures (scapulaire et surtout pelvienne), faiblesse musculaire.
2- Atteinte cutanée:Erythro-œdème, photosensible et prédominant sur les zones découvertes. Erythème orbitaires en lunette (coloration liliacée prédominant sur les paupières supérieures) : signe quasipathognomique. Erythème peri-unguéal douloureux à la pression (signe de manucure). Parfois : télangiectasies et syndrome Raynaud.

3- Atteinte Viscérale:
Fibrose pulmonaire interstitielle diffuse.Myocardite.
4- Examens complémentaires:
Syndrome inflammatoireCPK, Aldolases, LDH et Transaminases.AC spécifiques de la PM et de la DP : Anti-JO1 (anti-synthétase), Anti-ku, Anti-Mi1 et Anti-Mi2.E.M.G: tracé de type myogène.Histologie : infiltration inflammatoire lymphoplasmocytaire musculaire avec zone de nécrose et de régénération.
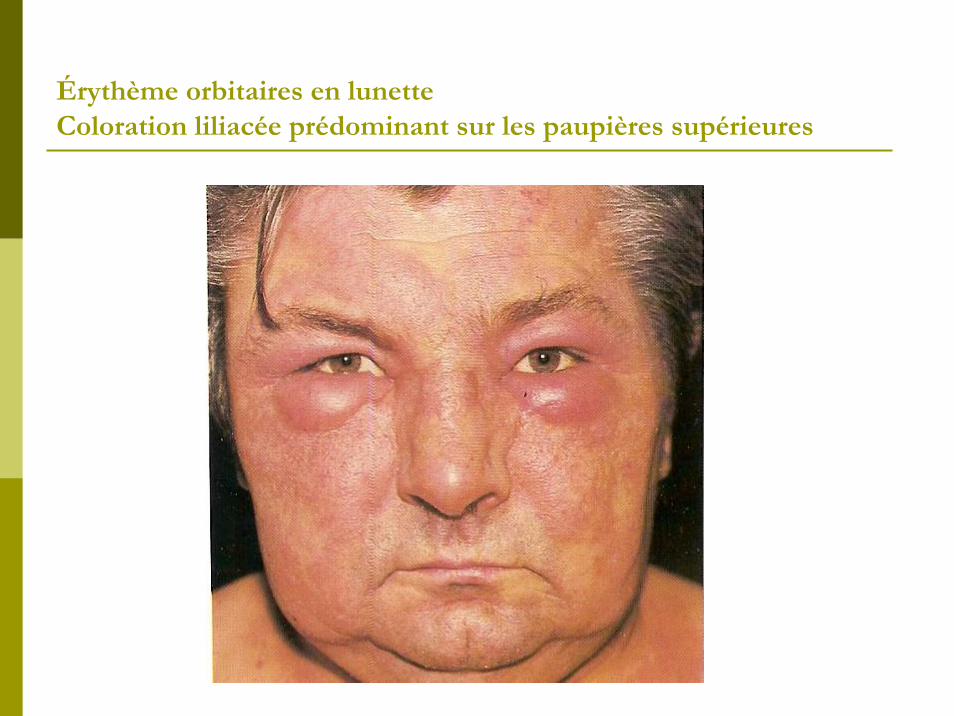
Érythème orbitaires en lunette Coloration liliacée prédominant sur les paupières supérieures


IV - SYNDROME DE GOUGEROT-SJOGREN
syndrome de SJOGREN (SS) ou syndrome sec
maladie auto-immune caractérisée par une infiltration lymphoïde des glandes exocrines de l'organisme en particulier lacrymales et salivaires.
SS primitif isolé
SS secondaire associé (PR, LED, DPM ou sclérodermie)
Fréquence : 0,1à 0,4 %: 2éme maladie auto-immune systémique
9 femmes pour un homme : autour de 50 ans.
Il existe une liaison significative avec les groupes HLA B8 et DR3.

A - Atteintes articulaires
75% des patients.
souvent polyarthralgie, parfois polyarthrite.Elle précède ou accompagne l'installation du syndrome sec.
symétrique, intermittente, plutôt que permanente, mais peut durer plusieurs mois.
Elle touche de préférence les MCP.
Une déformation de type Jaccoud est possible.

Sur les radiographies on trouve :
Une déminéralisation épiphysaire.
Parfois un discret pincement de l'interligne,
jamais d'érosions ni de destruction,
ce qui permet d'établir la différence entre les manifestations articulaires du SS primitif et celles d'une PR associée à un SS.

B- Éléments du diagnostic:
1 - Les signes oculaires: (xérophtalmie) Douleur, rougeur et sensation de brûlure ou de sable
dans les yeux.
2 - Les signes bucco pharyngés (xérostomie) Sensation de bouche sèche avec polydypsie.
3 - les autres atteintes muqueuses:Le nez, le larynx, les bronches, les voies génitales.
4 - Les atteintes viscérales:L'atteinte rénale: néphropathie interstitielle L'atteinte pulmonaire: pneumopathie interstitielle

5 - Signes biologiques :
Hyper gamma polyclonaleFR souvent positif AAN dirigés contre des antigènes nucléaires solubles (Anti SSA et/ou Anti SSB) Ac Anti-Alpha-Fodrine
6 - Biopsie labialeInfiltration plasmocytaire des glandes salivaires.

Xérostomie

Hypertrophie parotidienne

V - PERIARTERITE NOUEUSE (PAN)
Vascularite nécrosante touchant les artères de moyen calibre
Elle se voit surtout entre 40 et 60 ans
Les 2 sexes sont également touchés
Relation avec le virus de l'hépatite B (Ag Hbs)

A - Atteintes articulaires
1- Arthralgies :Migratrices sans gonflement articulaire touchant le plus souvent les grosses articulations : genoux, chevilles, coudes et poignets.Il n'y a pas de déformation, ni destruction articulaire.
2- plus rarement tableau de polyarthrite chronique (PAN +PR) chez un sujet préalablement porteur d'une PR (PR maligne)
3- manifestations osseuses:remaniements osseux périostés surtout aux membres
inférieurs.

B - DIAGNOSTIC DE LA PAN :Se base sur les autres signes de la maladie
1 - Signes généraux2 - Signes cutanés :
Livedo racemosa (marbré) ou reticularis (en réseau).Purpura, urticaire, érythème, gangrène des extrémités.
3 - Signes Neurologiques:Périphériques (fréquents) : multinévrite sensitivomotrice distale atteignant très souvent le SPECentraux : céphalées, convulsion, hémiplégie, syndrome méningé (hémorragie) confusion et AVC.

4 - Cardio-vasculaire:Myocardite.Insuffisance cardiaque secondaire a une coronarite
5 - Atteinte rénale6 - pleuro-pulmonaire7 - Gastro-intestinale
Ulcères.Infarctus intestinal.Perforations.
8 - Signes biologiques :Syndrome inflammatoire.Ag HBS (+) dans 40 à 50 % des cas.Habituellement, pas d'AC dans les F. primitives de PAN.
9 – Histologie :Artérite segmentaire nécrosante avec granulome.

Gangrène des extrémités

VI - LES CONNECTIVITES MIXTES
Les connectivites mixtes associent des signes de différentes connectivites, en général de lupus, de sclérodermie et de dermatopolymyosite.
5 à 10 femmes pour un homme: entre 30 et 40 ans.
Le syndrome de Sharp est une entité, associant des signes modéré de 3 connectivites : LED, Sclérodermie et PM.

A - Atteinte articulaire :Arthralgies ou polyarthrite souvent fixe, symétrique et progressive ; évoquant une PR débutante:IPP, MCP, poignets, coudes, chevilles, MTP et genoux. modérée, non déformante, sauf parfois par une main de Jaccoud (20 a 30% des cas).Absence de destruction radiologique.certains patients peuvent développer un tableau de PR typique.Les ténosynovites sont rares.
B - Diagnostic :association des signes de LED, Scl et PM

Toutes ces propositions concernant le LED sont exactes, sauf une Laquelle ?
A C’est une maladie de la femme jeuneB Une polyarthrite est souvent observée aumoment du diagnosticC L’atteinte articulaire résiste aux AINSD Le rachis est épargnéE Il peut se compliquer d’une arthropathie
déformante et destructrice de type Jaccoud







