
N° d’ordre 2008-ISAL-0127 Année 2008
THESE
présentée devant L’Institut National des Sciences Appliquées de Lyon
pour obtenir
le grade de docteur
Ecole doctorale : Matériaux de Lyon Spécialité : Matériaux polymères et composites
par
Nizar MNIF Ingénieur Matériaux
Elaboration et caractérisation de mélanges complexes à base de polypropylène en vue de son écoconception et de son recyclage dans les
véhicules hors d’usage
Soutenue le 18 décembre 2008 devant la Commission d’examen
Jury
CASSAGNAU Philippe Professeur Président DELOBEL René Professeur Rapporteur El GHARBI Rachid Professeur Rapporteur VERNEY Vincent Chargé de Recherche CNRS Examinateur GUILLET Jacques Professeur Examinateur MASSARDIER NAGEOTTE Valérie Maître de conférences Directeur de thèse ELLEUCH Boubaker Professeur Directeur de thèse KOSSENTINI KALLEL Tasnim Maître Assistante co-Directeur de thèse
1

2

3

4

Avant Propos
Ce travail de thèse a été réalisé en collaboration entre le laboratoire des matériaux
macromoléculaires / ingénierie des matériaux polymères (LMM/IMP) de l’INSA de Lyon et
le Laboratoire Eau-Energie-Environnement (L3E) de l’école nationale d’ingénieurs de Sfax –
Tunisie.
Je tiens tout d’abord à remercier le Pr. Jean François GERARD, directeur de LMM/IMP et le
Pr Hamed BEN DHIA, directeur de L3E pour m’avoir accueilli dans leurs laboratoires.
Je remercie la Région Rhône Alpes (Direction des Relations Internationales) pour avoir
financé ce projet de recherche.
Je souhaite remercier tout particulièrement mes directeurs de thèse, Mme Valérie
MASSARDIER, Mme Tasnim KALLEL et Pr Boubaker ELLEUCH, pour leur encadrement
m’apportant des suggestions appropriés, des pistes de recherche hautement judicieuses, ainsi
qu’une autonomie maîtrisée entre nos réunions de travail.
J’adresse également mes remerciements aux Professeurs René DELOBEL, Rachid
ELGHARBI et Jacques GUILLET et à Mr Vincent VERNEY, pour avoir examiné mon
travail de thèse.
Mes remerciements vont également au Pr. Philippe CASSAGNAU, pour avoir accepté de
présider mon jury de thèse.
Pour m’avoir permis de réaliser cette étude dans de meilleures conditions, j’adresse également
mes sincères remerciements à Pierre ALCOUFFE, pour avoir réalisé les expériences MEB et
à Hervé PERIER-CAMBY pour toutes se petites qui m’ont simplifié la tâche.
J’adresse finalement mes remerciements à l’ensemble des membres du LMM/IMP et du L3E :
mes collègues thésards, les autres étudiants et tous les permanents scientifiques et
administratifs, qui m’ont apporté leur contribution à l’accomplissement de ce travail.
5

6

A mes parents
A mon frère et mes sœurs
A ma bien aimée
7

8

Sommaire
Partie Bibliographique
Chapitre 1 : Valorisation de polymères................................................................................ 25 Cas particulier de l’industrie automobile ............................................................................ 25 1-1. Introduction : .................................................................................................................. 25 1-2. La réglementation sur l’automobile et l’environnement : .......................................... 26
1-2-1. Les véhicules en réparation : ..................................................................................... 26 1-2-2. Les véhicules en fin de vie : ...................................................................................... 26 1-2-3. Cycle du recyclage et de la valorisation des déchets automobiles : .......................... 27
1-3. Les polymères utilisés dans l’industrie automobile : ................................................... 28 1-4. Description des différents acteurs de la filière de recyclage des VHU : .................... 30
1-4-1. Les démolisseurs : ..................................................................................................... 30 1-4-2. Les broyeurs : ............................................................................................................ 30 1-4-3. Les recycleurs : .......................................................................................................... 31
Chapitre 2 : Les mélanges de polymères.............................................................................. 35 2-1. Introduction : .................................................................................................................. 35 2-2. Mélanges de polymères - Rappel théorique : ............................................................... 35
2-2-1. Etat de miscibilité : .................................................................................................... 35 2-2-2. Morphologies des mélanges de polymères :.............................................................. 36 2-2-3. Compatibilisation d’un mélange de polymères : ....................................................... 38
2-2-3-1. Objectifs de la compatibilisation :...................................................................... 38 2-2-3-2. Les différentes méthodes de compatibilisation : ................................................ 38 2-2-3-3. Utilisation d’un copolymère préformé : ............................................................. 39 2-2-3-4. Compatibilisation réactive in-situ : .................................................................... 42 2-2-3-5.- Comparaison des deux méthodes de compatibilisation : .................................. 43
2-3. Les mélanges PP/Elastomères utilisés dans les accessoires d’automobiles.................. 44 2-3-1. Les divers systèmes de polypropylène renforcé « chocs » :...................................... 44 2-3-2. Les mélanges mécaniques binaires (PP + EPR) : ...................................................... 45
2-3-2-1. Morphologie d’un élastomère introduit dans une matrice PP : .......................... 45 2-3-2-2. Morphologie du PP en présence d’un élastomère : ............................................ 48
2-3-3. Les mélanges ternaires (PP + élastomère + PE) : ...................................................... 49 2-3-3-1. Morphologies des systèmes ternaires :............................................................... 49
3-3-3. Les polypropylène-chocs de synthèse : ..................................................................... 51 Chapitre 3 : Les mélanges polyoléfines/CaCO3................................................................... 55 3-1. Introduction : .................................................................................................................. 55 3-2. Les renforts conventionnels : ........................................................................................ 55 3.3. Les nanocomposites et leurs spécificités : ..................................................................... 57
3.3.1. Les problèmes d'élaboration :..................................................................................... 57 3.3.1.1. Forme des particules et agrégation :.................................................................... 58 3.3.1.2. Nature des agents de couplage : .......................................................................... 59
9

3.3.2 Une zone perturbée : ................................................................................................... 60 3.3.3 Renforts et comportement mécanique :....................................................................... 61 3.3.4. Nanoparticules et propriétés thermiques : .................................................................. 63 3.3.5. Variation des propriétés rhéologiques en présence de nano-charges:........................ 64
3.4. Les interactions charges-charges et charges-polymères :............................................ 64 3.4.1. Interactions charges-charges : .................................................................................... 64 3.4.2. Interactions charges-polymères :................................................................................ 66
3.4.2.1. Extraction : .......................................................................................................... 66 3.4.2.2. Spectrométrie mécanique dynamique : ............................................................... 67
3.5. Conclusion : ..................................................................................................................... 69 Références ............................................................................................................................... 70
Partie Expérimentale Chapitre I :.............................................................................................................................. 81 Matériaux et techniques expérimentales.............................................................................. 81 I-1. Matériaux :....................................................................................................................... 81
I-1-1. Matrice Polypropylène renforcée « chocs » :............................................................. 81 I-1-2. Les compatibilisants :................................................................................................. 84 I-1-3. Les nanocharges CaCO3:............................................................................................ 87 I.1.4. Les polluants: .............................................................................................................. 88
I.1.4.1. Le polyamide (PA) et l’acrylonitrile butadiène styrène (ABS) : ......................... 88 I-1-4-2. Le polyuréthane (PU) :........................................................................................ 89
I-2. Techniques expérimentales: ........................................................................................... 91 I-2-1. Mise en œuvre des mélanges:..................................................................................... 91
I-2-1-1. Profil de vis et de température : .......................................................................... 91 I-2-1-2. Vitesse de rotation et de remplissage :................................................................ 93 I-2-1-3. Composition des mélanges : ............................................................................... 94
I-2-2. Techniques de caractérisation : .................................................................................. 94 I-2-2-1. Microscopie électronique à balayage :................................................................ 94 I-2-2-2. Comportement viscoélastique à l’état solide : .................................................... 95
PEHD................................................................................................................................... 98 I-2-2-3. Etude des propriétés thermiques par DSC : ........................................................ 98 I-2-2-3. Caractérisation mécanique en traction : .............................................................. 99 I-2-2-4. Essais chocs : ...................................................................................................... 99
Chapitre II : .......................................................................................................................... 105 Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/‛polluants’................................................................................................ 105 II-1. Introduction : ............................................................................................................... 105 II-2. Influence des compatibilisants sur les propriétés des mélanges (PP/EPR) et (PP/EPDM/talc) .................................................................................................................... 106
II-2-1. Les observations morphologiques........................................................................... 106 II-2-1-1. Morphologie des matrices (PP/EPR) et (PP/EPDM/talc)................................ 106 II-2-1-2. Morphologie des mélanges compatibilisés...................................................... 108
II-2-2. Influence des compatibilisants sur les propriétés mécaniques des mélanges : ....... 111
10

II-2-3. Influence des compatibilisants sur les propriétés thermiques et viscoélastiques à l’état solide des mélanges :................................................................................................. 113
II-2-3-1. Propriétés thermiques : .................................................................................... 113 II-2-3-1. Propriétés rhéologiques à l’état solide :........................................................... 114
II-3. Compatibilisation des mélanges pollués par l’ABS et le PA:................................... 116 II-3-1. Observations morphologiques des mélanges : ........................................................ 117 II-3-2. Caractérisation mécanique des mélanges pollués: .................................................. 120 II-3-3. Propriétés thermiques des mélanges pollués et pollués/compatibilisés:................ 123 II-3-4. Propriétés viscoélastiques à l’état solide des mélanges pollués et pollués/compatibilisés: ....................................................................................................... 126
II-4. Conclusions: ................................................................................................................. 128 Chapitre III :......................................................................................................................... 135 Elaboration et compatibilisation des mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints.............................................................................................. 135 III-1. Introduction :.............................................................................................................. 135
III-2-3- Nomenclature et composition des mélanges étudiés :........................................... 136 III-3- Résultats expérimentaux : ......................................................................................... 137
III-3-1- Propriétés thermiques :.......................................................................................... 137 III-3-2- morphologies des mélanges étudiés : .................................................................... 139 III-3-3- Propriétés mécaniques :......................................................................................... 143 III-3-4- Propriétés viscoélastiques à l’état solide :............................................................. 146
III-4. Conclusions : ............................................................................................................... 147 Chapitre IV :......................................................................................................................... 153 Caractérisation de mélanges (PP/EPR)/nano-CaCO3....................................................... 153 Influence de compatibilisants.............................................................................................. 153 IV-1. Introduction : .............................................................................................................. 153 IV-2. Etude des propriétés thermiques : ............................................................................ 153
IV-2-1. Les mélanges non compatibilisés : ........................................................................ 154 IV-2-2. Les mélanges compatibilisés : ............................................................................... 157
IV-3. Etude des propriétés viscoélastiques à l’état solide :............................................... 159 IV-3-1. Cas des mélanges non compatibilisés : ................................................................. 159 IV-3-2. Cas des mélanges compatibilisés : ........................................................................ 162
IV-4. Etude des propriétés morphologiques : .................................................................... 165 IV-4-1. Cas des mélanges non compatibilisés : ................................................................. 166 III-4-2. Etude de la morphologie des mélanges (PP/EPR)/nano-CaCO3 compatibilisés: .. 168
IV-5. Caractérisations mécaniques des mélanges renforcés : .......................................... 174 IV-5-1. Cas des mélanges renforcés nano-CaCO3 et non compatibilisés : ........................ 174 IV-4-2. Influence de compatibilisants sur les mélanges renforcés par des nano-CaCO3:.. 177
IV-6. Conclusion:.................................................................................................................. 180 Listes des figures................................................................................................................... 187 LISTE DES TABLEAUX .................................................................................................... 191
11

12

Liste des abréviations
A
ABS Acrylonitrile-butadiène-styrène
C
CaCO3 Carbonate de calcium
CPE Polyéthylène chloré
D
DCPD Dicyclopentadiène
E
EOC Copolymère d’éthylène-octène
ENB Ethylidène norborène
EPDM Ethylène propylène diène monomère
EPM Ethylène propylène monomère
EPR Ethylène propylène rubber
EPR-g-MAH Ethylène propylène rubber greffé anhydride maléique
EVA Ethylène vinyle acétate
EA Ester acrylique
G
GMA Ester de méthacrylate de glycidyle
H
HD Haxadiène
M
13

MAH Anhydride maléique
P
PA Polyamide
PABu Polyacrylate de butyle
PE Polyéthylène
PEHD Polyéthylène haute densité
PEBD Polyéthylène basse densité
PEP Copolymère bloc d’éthylène et de propylène
PMMA Polyméthacrylate de méthyle
PP Polypropylène
PP-g-2- HEMA Polypropylène 2- hydroxyéthyle méthacrylate
PP-HBP Polypropylène hyperbranchés
PS Polystyrène
PU Polyuréthane
PVAc Polyvinyle acétate
PVC Polychlorure de vinyle
T
TiO2 Oxyde de titane
TPE-g-MAH Elastomère thermoplastique greffé anhydride maléique
S
S Socal 322V (nano-CaCO3)
SEBS Poly(styrène-éthylène-butène-styrène)
SiO2 Oxyde de silicium
V
14

VHU Véhicules hors d’usage
W
W Winnofil SPM (nano-CaCO3)
15

Introduction générale
16

Introduction générale
Introduction générale
La préservation de l’environnement est devenue l’un des enjeux majeur du XXIe
siècle. La prise de conscience des limites de note planète et des impacts de l’activité des
hommes sur celle-ci depuis la fin du siècle dernier a mis en exergue la nécessité de protéger
notre cadre de vie. L’évolution de l’humanité, qui se traduit par l’amélioration de notre qualité
de vie, est liée aux évolutions technologiques et industrielles. Mais ces progrès ne sont pas
sans conséquences sur l’environnement, que ce soit en termes d’utilisation de ressources qui
ne sont pas illimitées, ou en terme de rejets, qui par leur nature ou leur quantité, ne peuvent
plus être assimilés par les cycles naturels assurant l’équilibre de notre planète.
Le développement de l’industrie s’est particulièrement accéléré au cours de la seconde
moitié du XXe siècle, cette évolution s’accentuant avec l’extension d’un mode de vie très
consommateur de ressources naturelles et producteur de nombreux déchets. Progressivement,
face à cette situation, l’industrie et la société civile ont pris conscience des risques de cette
croissance effrénée pour l’environnement. Pour réduire ces risques, des mesures ont été mises
en place, que ce soit de manière volontaire ou réglementaire, pour faire évoluer les pratiques
de conception, de fabrication, d’utilisation des produits et de gestion des déchets. L’industrie
automobile ne fait pas exception à la règle. Depuis plus d’une vingtaine d’années, des
règlementations tendent à diminuer les pollutions directes et indirectes des véhicules, qu’il
s’agisse des rejets atmosphériques dus aux moteurs à combustion, des nuisances sonores ou
de déchets issus des usines de production. Parmi ces nouvelles contraintes, l’une d’elles est
liée à la gestion de la fin de vie des véhicules et à la limitation des déchets qu’ils génèrent.
Les technologies de tri doivent répondre à des contraintes technologiques (utilisation de
formulations, pièces assemblées…) et économiques sévères qui ne permettent pas de
récupérer des polymères purs. Face à ce constat, l’élaboration de mélanges de polymères
compatibilisés pour la fabrication de pièces automobiles peut présenter une solution attractive,
d’un point de vue technologique et économique dans une perspective de recyclage de ces
matériaux. Comme on l’a dit, contrairement à d’autres stratégies de recyclage, cette voie ne
nécessite pas la séparation complète des différents constituants des déchets plastiques d’où
une simplification de l’étape de tri. On peut aussi envisager de simplifier ou de supprimer
l’étape de lavage et ne pas s’affranchir de la présence de polluants (huiles…) contenus dans
les matériaux à recycler sous forme de mélanges.
17

Introduction générale
Les travaux de recherche présentés dans ce mémoire comportent deux principales
parties :
Une partie Bibliographique qui comportera trois chapitres :
o 1er chapitre : consacré à la présentation de différentes voies de valorisation des
polymères et des mélanges de polymères particulièrement ceux utilisés dans
l’industrie automobiles.
o 2ème chapitre : où seront présentés les aspects thermodynamiques et les
propriétés de mélanges de polymères non miscibles en étudiant l’effet de
l’ajout de compatibilisants.
o 3ème chapitre : étude des propriétés des mélanges polymères/charges, effet des
interactions charges-charges et charges-polymères ainsi que de la nature des
interfaces.
Partie Expérimentale divisée en quatre chapitres :
o 1er chapitre « Matériaux et techniques expérimentales » : dans ce chapitre, nous
avons présenté tous les constituants des mélanges, à savoir les matrices
PP/ERR et PP/EPDM/talc, les polluants (acrylonitrile butadiène styrène,
polyamide et polyuréthane), les compatibilisants (copolymère à bloc
d’éthylène-b-polypropylène et un copolymère (éthylène propylène-g-anhydride
maléique) et copolymère tribloc (styrène-b-éthylène-b-butylène)) et les nano-
CaCO3. Ainsi que les conditions de mise en œuvre et les différentes techniques
de caractérisation et les conditions de chaque essai.
o 2ème chapitre « Influence de compatibilisants sur les propriétés des mélanges
(PP/élastomères)/polluants » : ce chapitre est consacré à la compatibilisation de
matrices PP/EPR et PP/EPDM/talc et de mélanges pollués en vue d’un
recyclage de déchets sous forme de mélanges. En effet contrairement à
d’autres méthodes, celle-ci ne nécessite pas la séparation complète des
différents composants des déchets plastiques d’où une simplification de l’étape
de tri. On peut aussi envisager de simplifier ou de supprimer l’étape de lavage
et ne pas s’affranchir de la présence de polluants contenus dans les matériaux à
18

Introduction générale
recycler sous forme de mélanges. Les résultats obtenus seront interprétés en
termes d’amélioration de l’interface et des propriétés mécaniques.
o 3ème chapitre « Compatibilisation de mélanges (PP/EPR)/PU en vue du
recyclage de boucliers peints » : l’objectif de cette partie est de traiter, par
ajout de compatibilisants, des déchets de pare-chocs peints dont les particules
de peinture agissent sous la forme d’impuretés non compatibles avec les
constituants des mélanges et provoquent une chute de la résilience.
o 4ème chapitre « Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence
de compatibilisants »: cette partie concerne la dispersion de deux
nanoparticules de CaCO3 dans les matrices PP/EPR et PP/EPDM/talc ainsi que
l’influence de compatibilisants sur l’interface polymères/charges et l’ensemble
des propriétés.
Ces travaux ont été réalisés dans le cadre d’une cotutelle de thèse entre le laboratoire
des matériaux macromoléculaires (LMM) de l’institut national des sciences appliquées de
Lyon (France) sous la direction de Madame. Valérie Massardier Nageotte, Maître de
conférences, et le laboratoire eau-énergie-environnement (L3E) de l’école nationale
d’ingénieurs de Sfax (Tunisie) sous la direction du Professeur Boubaker Elleuch et de
Madame. Tasnim Kossentini Kallel, Maître-assistant. Le financement (MIRA) a été accordé
par la région Rhône Alpes (Direction des Relations Internationales) durant les séjours en
France.
19

20

Partie Bibliographique
21

Chapitre 1 : Valorisation de polymères. Cas particulier de l’industrie automobile
22

Chapitre 1 : Valorisation de polymères. Cas particulier de l’industrie automobile
Chapitre 1 Valorisation de polymères
Cas particulier de l’industrie automobile
23

Chapitre 1 : Valorisation de polymères. Cas particulier de l’industrie automobile
24

Chapitre 1 : Valorisation de polymères. Cas particulier de l’industrie automobile
Chapitre 1 : Valorisation de polymères
Cas particulier de l’industrie automobile
1-1. Introduction :
Le développement de l’industrie s’est particulièrement accéléré au cours de la seconde
moitié du XXe siècle, cette évolution s’accentuant avec l’extension du mode de vie occidental
au reste de notre planète. Progressivement, face à cette situation, l’industrie et la société civile
ont pris conscience des risques de cette croissance effrénée pour l’environnement. Pour
réduire ces risques, des mesures ont été mises en place, que ce soit de manière volontaire ou
règlementaire, pour faire évoluer les pratiques de conception, de fabrication, d’utilisation des
produits et de gestion des déchets.
L’industrie automobile ne fait pas exception à la règle et depuis une vingtaine
d’années, des réglementations européennes tendent à diminuer les pollutions directes et
indirectes des véhicules. Il s’agit des rejets atmosphériques dus aux moteurs à combustion,
des nuisances sonores et des déchets issus des usines de production ou générés par des
véhicules en fin de vie.
Dans ce cadre, le recyclage de ces déchets sous forme de matières premières à
réintroduire dans le cycle industriel joue un rôle très important dans des industries comme
l’automobile. Le recyclage automobile est une activité pluridisciplinaire et complexe. La
chaîne de recyclage commence par la collecte et le tri qui est une étape complexe. Alors qu’en
Tunisie, le démontage des véhicules est largement pratiqué; en France, on a surtout développé
la filière Broyage + Tri (flottation…). Néanmoins, les contraintes économiques ne permettent
pas d’affiner le tri et si on veut avoir un fort taux de récupération des matériaux, on ne peut
pas exiger une pureté trop forte. C’est ainsi que les PP (polypropylène) automobiles à
recycler vont être « pollués » par des huiles moteurs, de l’éthylène glycol (antigel) du PE
(polyéthylène), de l’ABS (Acrylonitrile-butadiène-styrène), du PVC (polychlorure de vinyle),
des élastomères, des peintures polyuréthanes… La présence de ces impuretés n’est pas
rédhibitoire si on arrive à formuler les matériaux à recycler en compatibilisant les mélanges
issus du tri.
25

Chapitre 1 : Valorisation de polymères. Cas particulier de l’industrie automobile
1-2. La réglementation sur l’automobile et l’environnement :
Cette partie a pour objectif de développer les aspects réglementaires de la mise en
place des normes « management de la qualité et de l’environnement » dans les entreprises de
réparation et de démontage de carrosseries automobiles et de donner des exemples de
traitement des différents déchets issus de la réparation et du démontage.
L’un des enjeux du développement durable est la réduction des stocks de déchets polluants
qui ne peuvent pas être détruits.
1-2-1. Les véhicules en réparation :
Les professionnels de la réparation automobile ont la responsabilité d’organiser
l’élimination de leurs déchets. En Europe, les déchets issus de l’activité de réparation et de
maintenance sont interdits de mise en décharge depuis le 1er juillet 2002. Seuls les déchets
ultimes sont mis en décharge.
Les professionnels de la réparation doivent trier et stocker les déchets dans des installations
agréées. Les déchets sont ensuite remis à leurs propres frais à des entreprises d’élimination
agréés reconnues par les Pouvoirs publics, comme c’est le cas pour la récupération des huiles
usagées, des batteries, et des pneus.
1-2-2. Les véhicules en fin de vie :
En Europe, Un décret du 1er août 2003 relatif à la construction et à la destruction des
véhicules hors d’usage (V.H.U.) oblige la collecte de tous les véhicules hors d’usage mis sur
le marché après le 1er juillet 2002. A partir du 1er janvier 2007, cette collecte s’est étendue
aux véhicules mis sur le marché avant cette date. Les constructeurs ont pour devoir la collecte,
le retraitement et la destruction des VHU et doivent mettre en place une filière adaptée. Cette
opération est sans frais pour le possesseur du VHU, elle est effectuée par des démolisseurs
agréés qui dépolluent les véhicules avant traitement [1].
26

Chapitre 1 : Valorisation de polymères. Cas particulier de l’industrie automobile
1-2-3. Cycle du recyclage et de la valorisation des déchets automobiles : Le cycle du recyclage et de la valorisation des déchets issus du domaine automobile
contient plusieurs étapes (figure 1.1).Selon la Directive Européenne, les déchets issus des
véhicules hors usage (V.H.U) doivent être réutilisés en totalité et ceci quelque soit le type de
matériaux de base utilisé [1].
Le schéma de traitement tel que le prévoit la Directive se compose de quatre étapes :
1) Après collecte, le VHU passe par une étape obligatoire de dépollution. Cette phase
consiste à extraire tous les éléments dangereux ou polluants pour l’environnement et
pour les filières de traitement. Il s’agit essentiellement de la vidange des fluides, de
l’extraction de la batterie, de la neutralisation des éléments pyrotechniques et de
l’extraction des composants dangereux.
2) La deuxième étape est le désassemblage. Durant cette étape, la plupart des éléments
ayant une valeur marchande significative sont démontés. Il s’agit principalement de
pièces mécaniques pouvant être rénovées ou réutilisés, et donc revendus en pièces
d’occasion. D’autres pièces peuvent être démontées en vue de leur recycler, c’est le
cas des grandes pièces plastiques (comme le pare-chocs ou la planche de bord), des
mousses de sièges ou encore des vitrages.
3) La troisième étape est le broyage de la carcasse et le tri des métaux. Les métaux sont
ensuite recyclés par les acièristes (métaux ferreux) et par les affineurs (métaux non
ferreux).
4) Enfin, la dernière étape est le traitement des résidus non métalliques issus de l’étape de
broyage et de tri. Ils peuvent alors être recyclés ou servir de combustible de
substitution pour valorisation énergétique. Heureusement, jusqu’à présent, la
valorisation des polymères issus de l’automobile a surtout trouvé des débouchés pour
les pièces cachées (passages de roues…) avec cahier des charges peu contraignant.
Actuellement, il n’est pas envisageable de les utiliser pour de pièces d’aspect
(carrosseries peintes)…
27

Chapitre 1 : Valorisation de polymères. Cas particulier de l’industrie automobile
Figure 1. 1: Cycle du recyclage et de la valorisation des automobiles [1]
1-3. Les polymères utilisés dans l’industrie automobile :
Depuis plusieurs années, on assiste à une augmentation importante de la
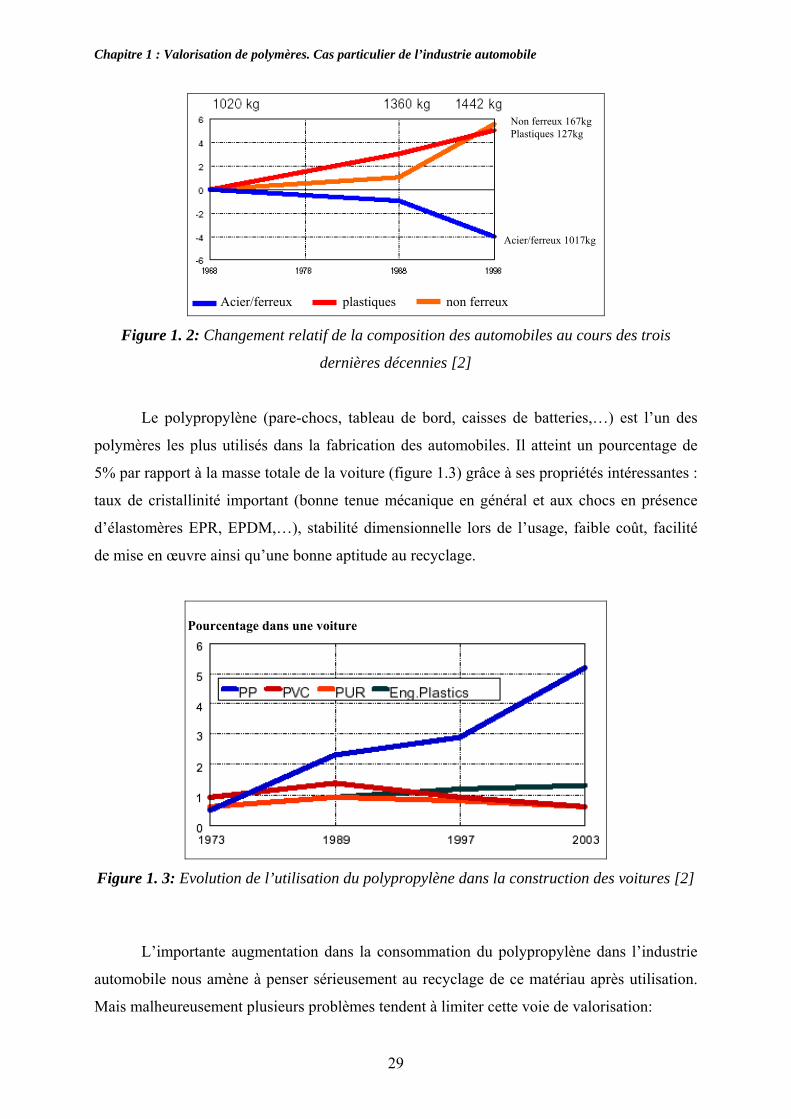
consommation des matières plastiques dans l’industrie automobile. La figure 1.2 illustre cette
variation au cours des trois décennies (1968-1998) [2]. On remarque qu’il y a une
augmentation continue de la consommation en matières plastiques.
28
Chapitre 1 : Valorisation de polymères. Cas particulier de l’industrie automobile
Non ferreux 167kg Plastiques 127kg
Acier/ferreux 1017kg
Acier/ferreux plastiques non ferreux
Figure 1. 2: Changement relatif de la composition des automobiles au cours des trois
dernières décennies [2]
Le polypropylène (pare-chocs, tableau de bord, caisses de batteries,…) est l’un des
polymères les plus utilisés dans la fabrication des automobiles. Il atteint un pourcentage de
5% par rapport à la masse totale de la voiture (figure 1.3) grâce à ses propriétés intéressantes :
taux de cristallinité important (bonne tenue mécanique en général et aux chocs en présence
d’élastomères EPR, EPDM,…), stabilité dimensionnelle lors de l’usage, faible coût, facilité
de mise en œuvre ainsi qu’une bonne aptitude au recyclage.
Pourcentage dans une voiture
Figure 1. 3: Evolution de l’utilisation du polypropylène dans la construction des voitures [2]
L’importante augmentation dans la consommation du polypropylène dans l’industrie
automobile nous amène à penser sérieusement au recyclage de ce matériau après utilisation.
Mais malheureusement plusieurs problèmes tendent à limiter cette voie de valorisation:
29

Chapitre 1 : Valorisation de polymères. Cas particulier de l’industrie automobile
- la majorité des polypropylènes utilisés dans les automobiles se présentent sous
forme de mélanges de polymères (PE, ABS, PVC,…), d’élastomères (EPR,
EPDM,…) et de charges (CaCO3, talc, noir de carbone,…),
- les différents composants d’une voiture sont revêtus par un ou plusieurs types de
peintures (polyuréthannes, acryliques,…) qui pourraient abaisser les propriétés du
matériau lors du recyclage.
1-4. Description des différents acteurs de la filière de recyclage des VHU :
Les différents acteurs intervenant dans le traitement d’un VHU sont les démolisseurs,
qui extraient les éléments ayant une forte valeur ajoutée ; les broyeurs, qui récupèrent les
pièces et les matériaux pour en faire de nouvelles matières premières ; et enfin les installations
permettant de valoriser énergétiquement les résidus combustibles issus de l’ensemble de la
filière de traitement.
1-4-1. Les démolisseurs :
Les démolisseurs sont essentiellement de petites structures travaillant de manière
artisanale. Actuellement, ils récupèrent la majeure partie des VHU. Ces derniers sont issus des
concessions, des assurances, des particuliers ou des fourrières. L’activité principale du
démolisseur est la revente de véhicules et de pièces d’occasion. En tant que premier maillon
de la chaîne de traitement, il doit assurer l’opération obligatoire de dépollution du véhicule. Il
peut également faire du démontage en vue du recyclage de certaines pièces.
1-4-2. Les broyeurs :
Si le démolisseur est dans la plupart des cas le point d’entrée de la filière de traitement
des VHU, le broyeur en est la clef de voûte. Contrairement aux démolisseurs, les broyeurs
sont des unités industrielles. Le métier du broyeur est la revente des métaux de récupération.
Les broyeurs traitent les métaux issus de la ferraille de démolition des bâtiments, des gros
produits électroménagers et des véhicules en fin de vie.
30

Chapitre 1 : Valorisation de polymères. Cas particulier de l’industrie automobile
1-4-3. Les recycleurs :
Si les broyeurs et les démolisseurs sont les acteurs principaux de la filière de
valorisation des VHU, les recycleurs en sont un maillon indispensable. Ils sont en effet les
garants de la viabilité et de la pérennité de l’ensemble de la filière de valorisation. Ils
transforment les déchets et les matériaux triés par les démolisseurs en matières premières
exploitables par l’industrie.
En suivant le schéma de traitement des VHU (figure 1.1), on distingue différents types
de recycleurs :
Pour l’étape de dépollution : recyclage des fluides, des batteries, des pneus,…
Pour l’étape de démontage : vitrages, pièces plastiques (PP, ABS, PA,….),…
Pour l’étape du broyage et du tri : recyclage des métaux,….
31

Chapitre 2 : Les mélanges de polymères
32

Chapitre 2 : Les mélanges de polymères
Chapitre 2 Les mélanges de polymères
33

Chapitre 2 : Les mélanges de polymères
34

Chapitre 2 : Les mélanges de polymères
Chapitre 2 : Les mélanges de polymères
2-1. Introduction :
L'intérêt porté depuis ces vingt dernières années aux mélanges de polymères provient
du fait qu'ils constituent un moyen peu onéreux de réaliser de nouveaux matériaux offrant des
propriétés intermédiaires et parfois même supérieures à celles de leurs constituants, en partant
de monomères et de polymères facilement accessibles. Une autre justification réside dans la
nécessité de plus en plus pressante de recycler les déchets industriels et ménagers, recyclage
qui est d'autant plus délicat que les polymères sont le plus souvent triés de façon imparfaite à
cause de contraintes économiques. En effet, plus la matière triée sera pure, plus la quantité
récupérée sera faible.
2-2. Mélanges de polymères - Rappel théorique :
La principale difficulté rencontrée dans la préparation des mélanges de polymères aux
propriétés intéressantes, est la non miscibilité quasi-générale des polymères de structure
chimique différente. Il est donc essentiel de cerner le phénomène de démixtion.
D'un point de vue fondamental, les théories les mieux établies concernent les mélanges
composés de deux polymères. Le but poursuivi est d'établir leurs diagrammes de phases [3-9].
2-2-1. Etat de miscibilité :
Les espèces chimiques en général, et les espèces polymères en particulier, sont le plus
souvent non miscibles "spontanément". D'après le second principe de la thermodynamique,
l'état de miscibilité de tout mélange est gouverné par l'enthalpie libre de mélange ∆Gm qui
s'exprime par :
(1)
Où ∆Hm et ∆Sm sont respectivement l’enthalpie et l’entropie du mélange. La condition
nécessaire, mais non suffisante, pour qu'un mélange soit miscible est que ∆Gm soit négative. A
l'inverse, le mélange se sépare en deux phases dès lors que ∆Gm est positive.
35

Chapitre 2 : Les mélanges de polymères
L'approche thermodynamique la plus largement utilisée pour exprimer ∆Gm est la
théorie de réseau de Flory-Huggins (1) selon laquelle l'enthalpie libre ∆Gm par site, dans le cas
d'un mélange binaire de constituants isomoléculaires, est donnée par :
(2)
Avec R : constante des gaz parfaits
T : température absolue
1Φ et 2Φ : fractions volumiques des constituants 1 et 2
Z1 et Z2 : définis à partir des volumes molaires V1 et V2 des constituants 1 et 2
et du volume de référence V
12χ : Paramètre d’interaction binaire
Toute étude de la miscibilité d'un mélange de polymères va finalement permettre de
classer le système considéré dans l'une des trois catégories suivantes [10] :
• les polymères sont non miscibles, quelles que soient leurs proportions relatives et la
température considérée,
• les polymères sont miscibles en toutes proportions sur tout le domaine de température
où ils sont stables,
• les polymères sont partiellement miscibles : selon les conditions (température,
composition), l’état thermodynamiquement stable correspond soit à l'existence d'une
phase unique homogène, soit à l'existence de deux phases distinctes, dont chacune est
enrichie en l'un des deux constituants.
2-2-2. Morphologies des mélanges de polymères :
L'optimisation des possibilités d'application des mélanges de polymères passe
nécessairement par le contrôle de leur morphologie finale. Les principales morphologies
rapportées à l'échelle macroscopique dans la littérature sont finalement assez bien résumées
36

Chapitre 2 : Les mélanges de polymères
sur les illustrations de la Figure 2-1. A cette échelle, la morphologie globale d'un mélange de
deux polymères 1 et 2 pourrait se limiter à l'une des trois possibilités suivantes :
• une morphologie particulaire où le polymère 1 est dispersé au sein du polymère 2,
• une morphologie bicontinue où les phases 1 et 2 sont interconnectées,
• une morphologie particulaire, mais correspondant cette fois à la dispersion du
polymère 2 dans le polymère 1.
(b) (b) (a) (c)
Figure 2. 1: Morphologie des mélanges (a) particulaire de 1 dans 2, (b) bicontinue et (c) particulaire de 2 dans 1
Bien évidemment, le passage à une échelle d'observation inférieure révèle que la
réalité est beaucoup plus complexe : notamment, l'existence de sous-structures au sein de la
phase dispersée est fréquemment citée [11].
Dans le cas particulier des copolymères à blocs qui peuvent, à l'extrême, être
considérés comme des mélanges de polymères. A leur sujet, la littérature [12] recense un
grand nombre de morphologies (dites ordonnées) selon la composition des copolymères.
Prenons l'exemple d'un copolymère diblocs poly(A-b-B) contenant majoritairement le
composant A. Trois morphologies peuvent être envisagées (dans chacun des cas, le
composant A constitue la phase continue ou matrice) [12] :
• la morphologie du copolymère correspond tout d'abord à une dispersion de particules
sphériques de B dans une matrice A selon un arrangement correspondant à celui de la
maille cristallographique d'un réseau cubique centré.
• Puis, au fur et à mesure que le pourcentage molaire du monomère B augmente,
l'arrangement s'effectue en cylindres de B disposés en réseau hexagonal dans la
matrice A.
• Enfin, pour un copolymère où la dissymétrie de composition est quasiment nulle, une
morphologie de type bicontinu est obtenue.
37

Chapitre 2 : Les mélanges de polymères
Au-delà d'un pourcentage molaire du monomère B de 50%, le composant B devient
majoritaire et forme désormais la phase continue. Le composant A va alors s'organiser tour à
tour suivant les trois morphologies décrites précédemment au fur et à mesure que la
composition du copolymère va diminuer. La dernière possibilité envisagée est celle d'un
copolymère de composition équimolaire en A et B : la morphologie observée est alors de type
lamellaire.
2-2-3. Compatibilisation d’un mélange de polymères : Les mélanges miscibles ne constituent qu’une minorité des cas rencontrés. Leur intérêt
principal est qu’ils permettent d’étendre la fenêtre d’utilisation des homopolymères qui les
composent. Les mélanges de polymères non miscibles sont beaucoup plus fréquents ; ils se
caractérisent par une structure multiphasique. Ces mélanges sont généralement incompatibles,
du fait de l’absence d’interactions favorables entre les chaînes de polymères constitutifs [12].
Les méthodes d’amélioration de l’adhésion entre les phases, impliquent l’ajout d’un
tiers-corps qui assure le contrôle et la stabilisation de la morphologie par réactions ou
interactions entre fonctions ou sites actifs sur chacune des phases en présence.
2-2-3-1. Objectifs de la compatibilisation :
Les trois effets principaux recherchés lors de la compatibilisation d’un mélange sont :
1. Diminution de la tension interfaciale pour faciliter la dispersion ;
2. Stabilisation de la morphologie afin d’éviter l’évolution de celle-ci au cours d’étapes
de transformation et de mise en œuvre du matériau ;
3. Augmentation de l’adhésion entre phases à l’état solide afin de favoriser notamment le
transfert de contrainte entre les phases et donc améliorer les propriétés mécaniques du
mélange.
En fonction de la méthode de compatibilisation choisie et du type de compatibilisant
utilisé, chacun des trois objectifs précédents peut être plus ou moins bien atteint.
2-2-3-2. Les différentes méthodes de compatibilisation : Les deux stratégies les plus fréquemment utilisées dans la compatibilisation des
mélanges de polymères non miscibles sont :
38

Chapitre 2 : Les mélanges de polymères
1. L’ajout d’un copolymère préformé, de nature et de structures adaptées, susceptible
d’interagir avec chacune des phases en présence ;
2. La formation in-situ d’un copolymère par réaction chimique à l’interface entre les
phases au cours de la préparation du mélange.
2-2-3-3. Utilisation d’un copolymère préformé : i) Principe : Cette voie de compatibilisation est similaire à l’utilisation des surfactants traditionnels
pour la préparation et la stabilisation des émulsions liquide/liquide (figure 2.1). Pour jouer son
rôle, le copolymère doit venir se positionner à l’interface entre les deux phases afin d’interagir
avec les constituants du mélange. Dans cette voie de compatibilisation, un phénomène
important doit être pris en compte : celui de la diffusion du copolymère à l’interface
liquide/liquide ; en effet, la mobilité de ce dernier, dans le milieu fondu, est beaucoup plus
réduite [12].
Le choix du copolymère comme compatibilisant est basé sur la miscibilité de ses
segments, avec au moins un des composants du mélange. Les copolymères sont considérés
comme agents interfaciaux vrais, puisqu’ils tendent à se concentrer et à agir à l’interface
comme émulsifiants [13] (figure 2.2).
Figure 2. 2: Analogie entre émulsifiant traditionnel et copolymère à bloc, en tant qu’espèces actives en surface d’un mélange
(a) (b) (c)
Bloc A Bloc B
Eau
Huile
ii)- Influence de la nature du copolymère :
Pour les mélanges à compatibiliser, le choix du compatibilisant est basé sur la
miscibilité des séquences de ce dernier avec les composants du mélange. De même, la faible
39
Chapitre 2 : Les mélanges de polymères
masse molaire des chaînes de cet agent compatibilisant va faciliter leur diffusion dans le
milieu fondu (viscosité élevée) et va privilégier leur accessibilité et leur concentration dans les
zones d’interaction c'est-à-dire l’interface entre les deux phases du mélange [13]. La solubilité
des séquences du compatibilisant dans les homopolymères, est un facteur clef [14-17] ; elle
contrôle la morphologie du mélange, sa stabilité ainsi que l’adhésion à l’interface.
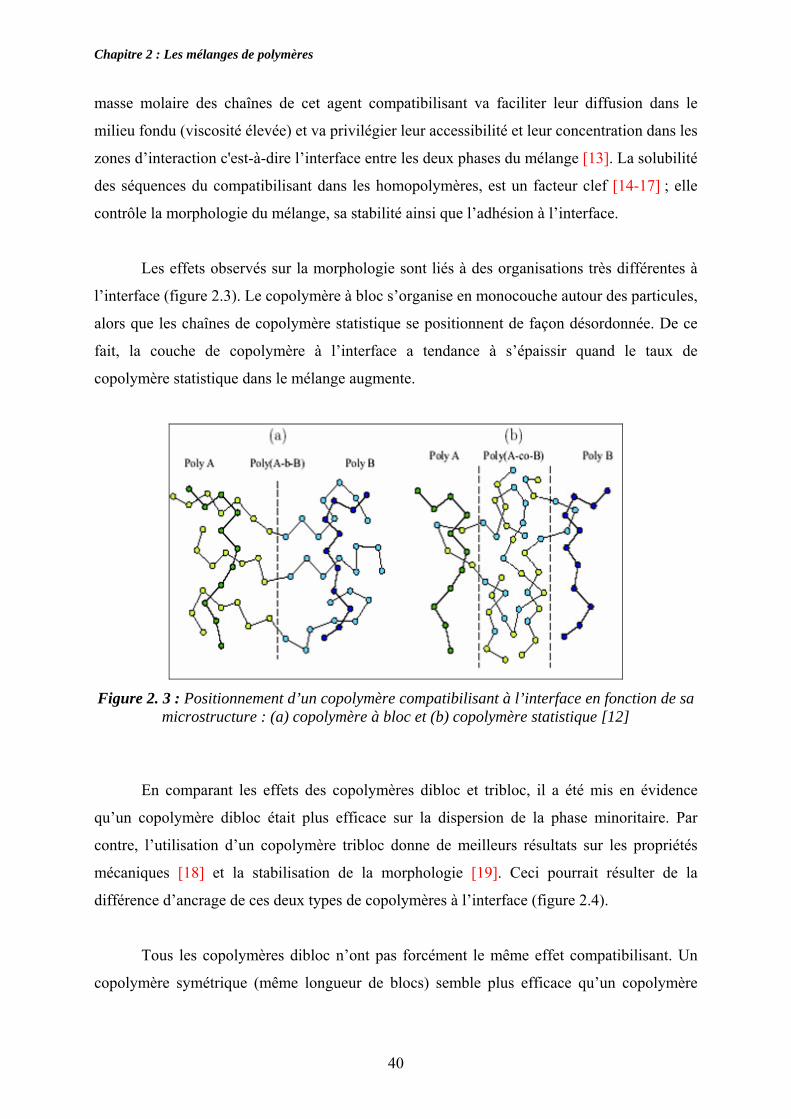
Les effets observés sur la morphologie sont liés à des organisations très différentes à
l’interface (figure 2.3). Le copolymère à bloc s’organise en monocouche autour des particules,
alors que les chaînes de copolymère statistique se positionnent de façon désordonnée. De ce
fait, la couche de copolymère à l’interface a tendance à s’épaissir quand le taux de
copolymère statistique dans le mélange augmente.
Figure 2. 3 : Positionnement d’un copolymère compatibilisant à l’interface en fonction de sa
microstructure : (a) copolymère à bloc et (b) copolymère statistique [12]
En comparant les effets des copolymères dibloc et tribloc, il a été mis en évidence
qu’un copolymère dibloc était plus efficace sur la dispersion de la phase minoritaire. Par
contre, l’utilisation d’un copolymère tribloc donne de meilleurs résultats sur les propriétés
mécaniques [18] et la stabilisation de la morphologie [19]. Ceci pourrait résulter de la
différence d’ancrage de ces deux types de copolymères à l’interface (figure 2.4).
Tous les copolymères dibloc n’ont pas forcément le même effet compatibilisant. Un
copolymère symétrique (même longueur de blocs) semble plus efficace qu’un copolymère
40

Chapitre 2 : Les mélanges de polymères
dissymétrique [20]. L’utilisation d’un copolymère dibloc partiellement alterné au centre a, par
contre, le même effet émulsifiant qu’un copolymère dibloc pur.
(a) (b) Bloc A Bloc B PolyA PolyB
Figure 2. 4: Positionnement d’un copolymère dibloc (a) ou tribloc (b) sur le polymère
iii)- Effet du taux de copolymère :
Théoriquement, quelques pourcents de copolymère dibloc symétrique suffisent pour
compatibiliser un mélange de polymères immiscibles [21].
Expérimentalement, la taille des particules de phase dispersée a tendance à diminuer
quand le taux de copolymère ajouté dans le milieu augmente, puis se stabilise ou augmente en
fonction de la nature du copolymère [20, 22]. Cependant, quelques pourcents en poids de
copolymère dibloc symétrique sont suffisants pour diminuer fortement la taille moyenne des
domaines de phase dispersée [23, 24]. Ceci a également été montré théoriquement [25]. Cette
quantité n’est pas forcément suffisante pour stabiliser le mélange. Il a été estimé que 5% à
20% de la surface des particules doivent être recouverts pour limiter la coalescence [23-25].
Parfois, un taux de copolymère plus important est nécessaire. Les écarts observés entre
la théorie et l’expérience résultent de deux phénomènes superposés :
- Les chaînes de copolymère doivent diffuser vers l’interface. La cinétique de diffusion
dépend de la viscosité du milieu mais aussi de la longueur des chaînes du copolymère.
- Il est possible que des micelles de copolymère se forment dans l’une des phases.
41

Chapitre 2 : Les mélanges de polymères
iv)- Influence de la masse molaire du copolymère :
La concentration de copolymère à bloc symétrique nécessaire pour atteindre la taille
minimale de phase dispersée diminue lorsque la masse molaire globale du copolymère
augmente [22]. Par contre, cette taille minimale semble être indépendante de la longueur du
copolymère. A taux égal de compatibilisant dans le milieu, la morphologie du mélange est
d’autant plus stable que la masse molaire du copolymère est élevée. Ces constatations peuvent
s’expliquer par le meilleur ancrage des longues branches dans chacune des phases par
enchevêtrement. Les blocs doivent donc être d’une masse molaire supérieure ou égale à la
masse molaire critique d’enchevêtrement, pour que le copolymère soit efficace pour la
compatibilisation.
En revanche, plus la masse molaire du copolymère est élevée plus la concentration à
partir de laquelle des micelles peuvent se former diminue. De plus, plus les copolymères sont
de masses molaires élevées, plus leur cinétique de diffusion vers l’interface est lente. Il faut
donc choisir au mieux le copolymère, si cela est possible, de sorte à prendre en compte ces
deux facteurs [21].
2-2-3-4. Compatibilisation réactive in-situ :
i)- Principe :
La compatibilisation réactive consiste à générer in situ un copolymère bloc ou greffé
par réaction chimique de composés fonctionnalisés à l’interface entre les phases. Les effets
sur la tension interfaciale et la taille de la phase dispersée sont identiques à ceux observés en
compatibilisation non réactive.
ii)- Architecture des compatibilisants formés :
Il est possible de former des compatibilisants d’architecture très variée. La structure du
copolymère compatibilisant qui se forme dépend du type de réaction mise en jeu et de la
position des fonctions réactives sur les macromolécules impliquées dans la compatibilisation.
42

Chapitre 2 : Les mélanges de polymères
Les principales réactions utilisées pour la compatibilisation réactive des mélanges de
polymères sont présentées sur la figure 2.5 [26]. Les deux caractéristiques principales de ces
réactions sont les suivantes :
- Les groupements fonctionnels impliqués sont hautement réactifs et stables dans les
conditions de mise en œuvre ;
- La réaction est rapide, faiblement exothermique et irréversible.
Le rôle du procédé de mise en œuvre (généralement en extrudeuse bi-vis) dans la
compatibilisation réactive in-situ des mélanges de polymères non miscibles est de tout
premier ordre [27, 28].
Figure 2. 5: Exemples de réactions chimiques classiquement utilisées pour la
compatibilisation in-situ des mélanges de polymères immiscibles [26]
2-2-3-5.- Comparaison des deux méthodes de compatibilisation :
Les avantages et les inconvénients de deux méthodes de compatibilisation les plus
rencontrées sont représentés dans le tableau 2.1 [29, 30].
43

Chapitre 2 : Les mélanges de polymères
Tableau 2. 1 : Comparaison de la compatibilisation par ajout et formation in-situ du
copolymère compatibilisant
Ajout d’un copolymère préformé Compatibilisation réactive
Avantages- Contrôle du taux ajouté dans le
milieu - Maîtrise de la structure du
copolymère
- Formation du copolymère à l’interface
- Nombreuses structures possibles - Une seule étape de mise en
oeuvre
Inconvénients- Diffusion du copolymère vers
l’interface - Formation de micelles de
copolymère - Préparation du copolymère
- Diffusion des espèces réactives vers l’interface
- Taux de conversion de la réaction faible
- Présence d’un excès de réactifs
Pour ces deux méthodes de compatibilisation, les paramètres associés à la phase
d’élaboration (cisaillement, température, temps…) sont essentiels ; ceci afin d’obtenir une
bonne dispersion ainsi qu’une compatibilisation efficace.
2-3. Les mélanges PP/Elastomères utilisés dans les accessoires
d’automobiles
Les mélanges de polypropylène (PP) et d’élastomères [EPR (éthylène propylène
rubber) ou EPDM (éthylène propylène diène monomer)…]: composants principalement
destinés à améliorer les propriétés aux chocs) sont très utilisés dans plusieurs domaines en
particulier la fabrication des accessoires d’automobiles (exemples : les pare-chocs, les
tableaux de bords,…).
2-3-1. Les divers systèmes de polypropylène renforcé « chocs » :
Historiquement, le pare-choc tout plastique est apparu en 1975-76 sur la Fiat 127 ;
chez RENAULT, les pièces extérieures des R4, R5 et R11 étaient des copolymères dits « in
situ », alors que le PP mêlé à l’EPDM équipait la Visa de CITRÖEN ou les modèles de
FIAT ; enfin, les planches de bord et les volants étaient faits par compoundage PP + (EPDM
ou EPR) tandis qu’en réalité, ils sont formés généralement de copolymères.
Signalons par ailleurs que le terme de polypropylène « copolymère choc » est souvent
abusivement employé pour désigner tout propylène renforcé choc, qu’il s’agisse d’un
véritable copolymère à blocs (par exemple des copolymères d’éthylène et de propylène) ou
44

Chapitre 2 : Les mélanges de polymères
d’un simple mélange mécanique (exemples : PP/EPR ou PP/EPDM,..), ou encore d’un
système PP-choc de synthèse (obtenu directement au réacteur par synthèse d’une phase
élastomère dans une matrice PP).
Pour obtenir de bonnes propriétés mécaniques à partir d’un mélange de composants
chimiquement incompatibles, tels que les systèmes PP/Elastomères thermoplastiques (EPM,
EPDM,…), il est nécessaire de créer une émulsion stable de nodules d’élastomères noyés dans
la matrice PP semi-cristalline et rigide. Cette condition indispensable implique la présence du
composant minoritaire en quantité suffisante dans le mélange, ainsi que la formation
d’interactions physiques et/ou de liaisons chimiques assurant la continuité à l’interface entre
les deux phases. De plus, une bonne homogénéité et une granulométrie contrôlée et
appropriée seront recherchées.
2-3-2. Les mélanges mécaniques binaires (PP + EPR) :
Les mélanges de polypropylène et d’élastomères EPR (copolymère d’éthylène et de
propylène) sont parmi les premiers systèmes qui ont fait leur apparition sur le marché. Ils ont
été depuis longtemps abusivement appelés « copolymères », alors qu’ils sont en fait des
mélanges de polymères incompatibles et ayant une morphologie bi-phasique.
Comme signalé auparavant (paragraphe 2.2.2), la morphologie des mélanges de
polymères non miscibles, influence considérablement les propriétés finales du matériau. Il
serait donc intéressant d’étudier de près cette morphologie et de suivre ses évolutions
possibles en fonction des caractéristiques moléculaires des composants du mélange et des
paramètres de mise en œuvre.
2-3-2-1. Morphologie d’un élastomère introduit dans une matrice PP :
La morphologie du mélange composé d’un élastomère introduit mécaniquement dans
une matrice PP est la résultante de plusieurs facteurs. Parmi les plus influents, on trouve le
comportement à l’écoulement lors de la transformation à l’état fondu, les propriétés
interfaciales, les proportions volumiques dans le mélange de chaque composant et les
conditions de mise en œuvre.
45

Chapitre 2 : Les mélanges de polymères
i)- Influence de la composition et des propriétés des composants :
Danesi et al [31] estiment que la phase dispersée est constituée par le composant
quantitativement minoritaire. Selon que sa viscosité limite newtonienne à l’état fondu η0 est
inférieure ou supérieure à celle de la matrice, la dispersion sera respectivement fine et
homogène ou bien grossière.
Parallèlement, D’Orazio et al [32-34] ainsi que d’autres [35] observent que la matrice
d’un système binaire PP+EPR est formée par le composant de faible viscosité η0.
Tous s’accordent à dire que la taille et la distribution de taille des nodules de la phase
dispersée varient comme pour la viscosité relative µ, en fonction du rapport des viscosités
newtoniennes des deux entités à l’état fondu :
µ = matrice
dispersée
0
0
ηη
D’autres auteurs [36] montrent que le diamètre moyen des nodules et leur dispersion
augmentent quand la viscosité limite newtonienne de l’EPR (phase minoritaire) dans le
mélange PP/EPR augmente.
Pukanszky et al. [37] notent des différences morphologiques pour les mélanges
PP/EPDM selon que l’élastomère est statistique ou séquencé. Dans le premier cas, le système
est formé de nodules de petits diamètres (entre 0,5 et 1µm) alors que pour les copolymères
séquencés, les nodules sont plus gros (diamètres entre 8 et 10 µm). Par contre, la stabilité dans
le temps de ces derniers est plus forte que les petits nodules des copolymères statistiques qui
coalescent en quelques mois.
En ce qui concerne les nodules d’EPR, D’Orazio et al [32-34] montrent qu’une
augmentation jusqu’à 43% de leur teneur en propylène conduit à une augmentation de la taille
des nodules de la phase élastomère mais que des proportions supérieures entraînent une
réduction de leurs dimensions.
Martuscelli et al. [38] notent, en observant un film microscopique optique, que les
phases élastomères de forme nodulaire, sont rejetées à la surface du film pendant la
46

Chapitre 2 : Les mélanges de polymères
cristallisation, et qu’ils s’orientent le long des lignes de flux induites par la cristallisation de
l’iPP.
Fortenly [39] et Yang et al. [40] montrent que la taille des particules varie comme le
pourcentage d’EPR présent dans le mélange, ce qui ne prévoit pas la théorie générale de Wu
qui estime la taille des nodules seulement fonction des conditions de malaxage et de la
viscosité relative.
Fortenly [39], estiment que ce rayon est indépendant de la proportion de la phase
dispersée. Enfin, Jancar [41] aboutit à ce résultat : dans un mélange PP/EPR statistique, le PP
demeure la phase continue du système même lorsqu’il devient le composant minoritaire.
D’Orazio [42] relève le même phénomène d’inversion de phase dans le cas d’un mélange
PP/EPR vulcanisé. Pour des proportions d’élastomère de 40 à 60% et un rapport µ proche de
1, Danesi et al. [31] observent la coexistence de deux phases continues.
D’Orazio et al. [32-34] notent que la distribution de la taille des particules ainsi que la
valeur de la viscosité limite newtonienne de la phase dispersée augmentent avec la masse
molaire et l’indice de polymolécularité de l’EPR.
Yang et al. [40] mesurent une taille d’environ 0,2 à 0,5µm pour des nodules d’EPDM
présents à raison de 5 à 20% dans du PP.
ii)- Influence des conditions de mise en œuvre :
Dans la majorité des cas, la morphologie d’une pièce injectée à partir d’un mélange
PP/élastomère est évolutive depuis l’intérieur vers l’extérieur de la pièce ; elle est dite de type
cœur-peau (core-shell). En effet, elle comprend à l’intérieur un cœur constitué de nodules
sphériques de phase dispersée (EPR) noyés dans la matrice PP et à l’extérieur une peau
formée d’une couche de PP d’épaisseur comprise entre 15 et 20µm ; entre les deux, on
distingue une zone intermédiaire de structure proche de celle du cœur. La morphologie de
l’ensemble est caractérisée par une concentration en nodules d’élastomère décroissante depuis
le cœur vers l’extérieur et par une déformation elliptique de ces particules décroissante au fur
et à mesure que l’on se rapproche du cœur.
47

Chapitre 2 : Les mélanges de polymères
En s’intéressant à la morphologie des mélanges PP/EPDM, Pukanszky et al [37] ont
constaté que l’évolution de la taille des nodules de la phase dispersée EPDM lors du cycle
thermomécanique de malaxage et de plastification en extrudeuse était principalement liée aux
paramètres de mise en œuvre (vitesse, température…). De ce fait, ils ont pu noter que la taille
des nodules évolue en permanence lors du processus de mélangeage.
2-3-2-2. Morphologie du PP en présence d’un élastomère :
i)- Influence de la composition et des propriétés des composants :
Certaines influences de l’EPR sur la morphologie du mélange final PP/EPR sont
largement admises, notamment son effet nucléant sur le PP favorisant ainsi la formation des
cristallites. Beaucoup d’auteurs [41-48] observent en effet un accroissement du nombre de
sphérolites, couplé à une diminution de leur taille quand le taux d’EPR s’élève. Pour
exemples chiffrés, le diamètre des sphérolites passe de 100µm à 40µm lorsqu’on introduit
10% d’EPR dans du PP homopolymère. Jang et al [45] constatent qu’en introduisant 15%
d’EPDM, la taille des sphérolites est divisée par deux et passe de 50µm à 20µm. Ces
observations visuelles sont couplées à des mesures micro-calorimétriques mettant en évidence
une diminution de la température de fusion et une augmentation de la température de
cristallisation du système.
D’Orazio [32-34] pour un EPR ainsi que Pukanszky [37] pour un EPDM, ont
remarqué une évolution croissante de l’épaisseur de la couche amorphe inter-lamellaire de PP
accompagnée d’une diminution de l’épaisseur des lamelles cristallines de PP quand le taux
d’élastomère augmente. Ils ont alors conclu que la présence d’élastomère inhibe la croissance
cristalline du PP.
ii)- Influence des conditions de mise en œuvre :
Comme le mettent en évidence Cunha et al [49], une température de moule assez
élevée ainsi qu’une forte vitesse d’injection favorisent la croissance des sphérolites de PP et
diminuent l’effet de peau. Ils ajoutent qu’une forte vitesse d’injection va dans le sens d’une
augmentation de la proportion de phase hexagonale β des sphérolites de PP.
48
Chapitre 2 : Les mélanges de polymères
2-3-3. Les mélanges ternaires (PP + élastomère + PE) :
Les mélanges ternaires « PP-chocs » sont issus d’un mélange mécanique, comme les
systèmes binaires précédemment étudiés. Un des intérêts de ces mélanges ternaires
PP/PEHD/EPR est qu’ils possèdent à la fois une bonne résistance au choc à froid (grâce à
l’élastomère et au PEHD) et un module de rigidité correct (grâce au PEHD).
Même si d’autres configurations sont possibles [50], nous nous restreindrons dans cette étude
aux cas où le PP est le composant majoritaire.
2-3-3-1. Morphologies des systèmes ternaires :
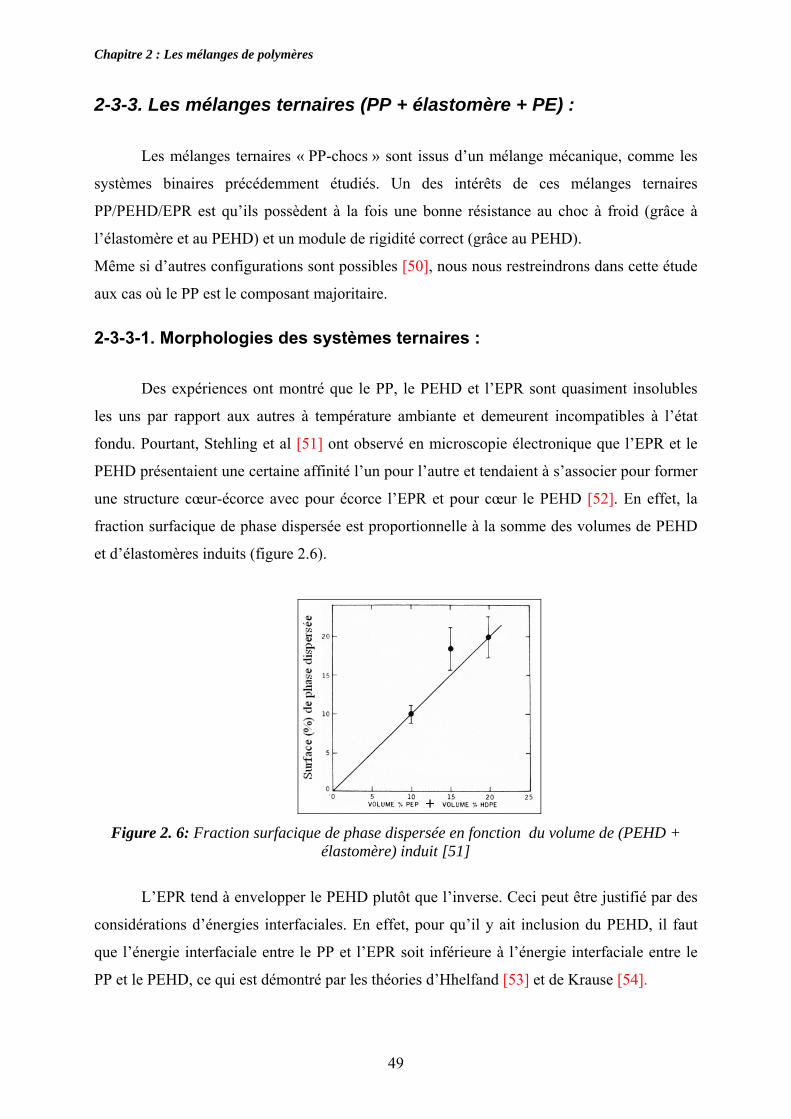
Des expériences ont montré que le PP, le PEHD et l’EPR sont quasiment insolubles
les uns par rapport aux autres à température ambiante et demeurent incompatibles à l’état
fondu. Pourtant, Stehling et al [51] ont observé en microscopie électronique que l’EPR et le
PEHD présentaient une certaine affinité l’un pour l’autre et tendaient à s’associer pour former
une structure cœur-écorce avec pour écorce l’EPR et pour cœur le PEHD [52]. En effet, la
fraction surfacique de phase dispersée est proportionnelle à la somme des volumes de PEHD
et d’élastomères induits (figure 2.6).
Figure 2. 6: Fraction surfacique de phase dispersée en fonction du volume de (PEHD +
élastomère) induit [51]
L’EPR tend à envelopper le PEHD plutôt que l’inverse. Ceci peut être justifié par des
considérations d’énergies interfaciales. En effet, pour qu’il y ait inclusion du PEHD, il faut
que l’énergie interfaciale entre le PP et l’EPR soit inférieure à l’énergie interfaciale entre le
PP et le PEHD, ce qui est démontré par les théories d’Hhelfand [53] et de Krause [54].
49

Chapitre 2 : Les mélanges de polymères
Ainsi l’EPR ajouté à un système PP/PEHD se comporte non seulement comme une
charge de renfort pour choc, mais joue également un rôle important d’agent compatibilisant
en se positionnant à l’interface du PP et du PEHD [55, 56]. D’Orazio et al [32] font
l’hypothèse d’une solubilité partielle mais mutuelle des molécules d’EPR avec celles de
PEHD ou de PP qui présentent des masses molaires et des degrés de cristallinité assez faibles.
i)- Influence des proportions et des propriétés des composants :
Le PEHD n’est que partiellement enveloppé par l’EPDM si ce dernier ne se trouve pas
en quantité suffisante dans le système, auquel cas le PEHD se retrouve lié à la matrice par
certains points d’attache. A teneur en phase dispersée (PEHD+EPDM) constante et égale à
20%, une augmentation de la teneur des nodules en EPDM entraîne un accroissement du
nombre et la taille des sphérolites PP [55].
ii)-Influence des conditions de mise en œuvre :
Une morphologie cœur-écorce se développe également quand on prépare des
prémélanges PP-EPR et PEHD-PP avant de réaliser le mélange ternaire correspondant [51]
(figure 2.7). Dans le cas où le prémélange est composé de PEHD-EPR, celui-ci présente une
morphologie de réseaux interpénétrés. Dés ajout du PP, les particules les plus grosses
participeront à la sauvegarde de ce réseau tandis que les plus petites adopteront une structure
cœur-écorce.
Figure 2. 7: Illustration schématique de la morphologie des particules composites PEP-
PEHD dans les mélanges ternaires à matrice PP : structure cœur écorce [51]
Cependant, cette solution présente l’inconvénient de nécessiter une étape onéreuse de
malaxage [57]. C’est pourquoi les producteurs de PP ont cherché à mettre au point des
50

Chapitre 2 : Les mélanges de polymères
copolymères dits séquencés PP-PE, le but étant d’incorporer des séquences de PE linéaires
qui apportent, contrairement au PP, une bonne résistance au choc à froid grâce à sa transition
vitreuse basse. Ces matériaux seront présentés dans la partie consacrée aux PP-chocs de
synthèse.
2-3-3. Les polypropylène-chocs de synthèse :
Il s’agit de matériaux polymérisés par une méthode assez récente dite « in situ » qui
permet d’obtenir divers types de matériaux :
des copolymères séquencés PP-PE (compatibles monophasés),
des copolymères statistiques Ethylène-Propylène (compatibles monophasés),
des systèmes compatibles polyphasés comprenant une phase élastomère EPR, la
matrice est le polypropylène homopolymère et des copolymères PP-EPR et PP-EPR-
PP où l’EPR possède une certaine cristallinité.
Dans le paragraphe suivant, nous évoquerons le cas des PP-chocs de synthèse
compatibles monophasés et des PP-chocs de synthèse compatibles polyphasés.
i)- Les copolymères de synthèse monophasés :
Ce sont les seuls véritables copolymères au sens chimique du terme.
Nous distinguons quatre sortes de copolymères de synthèse monophasés :
La première classe est celle des copolymères statistiques : Elle comprend en premier
lieu des caoutchoucs de composition 50% éthylène donnant des EPR ou des EPDM
qui, introduits en tant qu’élastomères dans du PP homopolymère, forment des
mélanges mécaniques [58].
Une deuxième catégorie comprend les copolymères dibloc ou tribloc composés de
polypropylène et d’EPR synthétisés par polymérisation séquencé avec le δ-TiCl3-
Et2AlCl dans l’hexane [58].
La troisième catégorie est celle des copolymères blocs constituée des matériaux
majoritairement éthyléniques (5% de propylène). Leurs propriétés ressemblent à celles
de l’éthylène-1-butène.
51

Chapitre 2 : Les mélanges de polymères
Les plus performants vis-à-vis de la résistance au choc sont ceux qui contiennent 5 à
15% d’éthylène, avec une structure de type bloc ; on les appelle parfois les
pollyallomers.
ii)- Les matériaux polyphasés :
Ils sont considérés de type blocs, mais la présence simultanée d’éthylène et de
propylène produit une phase caoutchoutique relativement importante. Comme pour les
premiers « copolymères blocs » PP-PEHD, le lien chimique entre chaque phase n’a pas été
prouvé, mais certains pensent qu’ils contiennent une certaine fraction de chaînes PP-EPR ou
PP-EPR-PP. Cette particularité leur confère des propriétés typiques dues à l’effet
compatibilisant des blocs vis-à-vis de la matrice et de l’élastomère.
52

Chapitre 3 : Les mélanges polyoléfines/CaCO3
Chapitre 3 Les mélanges polyoléfines/CaCO3
53

Chapitre 3 : Les mélanges polyoléfines/CaCO3
54

Chapitre 3 : Les mélanges polyoléfines/CaCO3
Chapitre 3 : Les mélanges polyoléfines/CaCO3
3-1. Introduction :
Les propriétés macroscopiques des matériaux composites dépendent de la nature des
constituants, de leur fraction volumique, de leur arrangement spatial et de la qualité de
l'adhésion entre constituants. Des modifications supplémentaires de comportement peuvent
également apparaître quand on s'intéresse aux particules renforçantes de taille nanométrique.
En effet, dans le cas des nanocomposites, les distances interparticulaires deviennent de l'ordre
de grandeur du rayon de giration des macromolécules (quelques nm). De plus, les valeurs
importantes du rapport surface/volume des charges induisent des augmentations de l'intensité
des interactions entre charge et polymère. Certains auteurs [59] ont montré qu'il existe au
voisinage des charges une zone où la mobilité des chaînes macromoléculaires est réduite,
généralement désignée par interphase. La formation, puis le développement de cette zone vont
être liés à la compétition des interactions entre charges et polymère-charge, éventuellement
favorisée par la présence d'agent de couplage ou de recouvrement.
3-2. Les renforts conventionnels :
Les thermoplastiques sont devenus depuis une cinquantaine d’années des matériaux
incontournables, présents dans notre quotidien, à la fois dans des applications à usage courant
ou à usage technique. Dans les polymères dits de commodité, nous pouvons citer les
polyoléfines, essentiellement le polyéthylène et le polypropylène, de faible coût, facilement
transformables et présentant un compromis de propriétés mécaniques, barrière et de surface
intéressant. Toutefois, leur usage est limité lorsque des propriétés thermomécaniques alliant
une rigidité importante avec une tenue en température sont requises. Dans ce cas, on fait appel
à un polymère plus technique, mais plus coûteux. Une autre solution consiste à incorporer
dans ces matrices de grande diffusion des renforts qui auront pour but de palier le déficit de
propriétés, sans toutefois provoquer une augmentation significative du coût des produits, nous
parlons alors de composite thermoplastique. Dans ce cas, le mode de renforcement est
spécifique à la propriété désirée.
55

Chapitre 3 : Les mélanges polyoléfines/CaCO3
Afin d’augmenter la rigidité, l’incorporation de renforts tel le talc [60], le carbonate de
calcium [61] ou le noir de carbone est la solution la plus courante. Une autre voie est
l’utilisation de renforts manufacturés, tels les fibres de verre [62] et de carbone. Cette liste est
non exhaustive et d’autres renforts sont utilisés [63]. Le gain de rigidité produit par
l’incorporation d’une deuxième phase rigide de taille micronique ou supérieure est
contrebalancé par l’apparition de concentrations de contraintes aux interfaces qui provoquent
une diminution de la résistance à la propagation de fissures dans le matériau. Afin de profiter
pleinement de l’effet renforçant des charges de fort module, il convient d’augmenter les
interactions entre le renfort et la matrice. Cela passe soit par un traitement de surface du
renfort, tel l’ensimage des fibres de verre, soit par l’ajout d’un tiers agent, appelé agent
compatibilisant, qui va favoriser le couplage fibres/matrice. Un autre défaut de ce mode de
renforcement est l’augmentation de la densité du composite, et lorsque le gain demandé en
propriétés est élevé, le taux de renforcement peut atteindre 50% en poids.
A l’opposé, l’incorporation d’un polymère élastomère (de température de transition
vitreuse inférieure à la température d’utilisation) va permettre d’améliorer le comportement au
choc. La dispersion, par voie fondue, de 5 à 20 % d’un élastomère, le plus souvent du
polybutadiène, permet l’augmentation des propriétés choc du polystyrène [64]. Là encore, les
faibles interactions interfaciales (adhésion interfaciales) peuvent limiter les performances
mécaniques des mélanges. Afin d’augmenter les interactions aux interfaces ou dans
l’interphase, des interactions peuvent être créées en utilisant par exemple des copolymères
partiellement miscibles dans chaque phase. Avec ce mode de renforcement, les propriétés
choc sont certes améliorées, mais en contrepartie, la rigidité du matériau diminue fortement
par rapport à la matrice initiale, ce qui limite le champ d’application de cette méthode.
Une autre propriété recherchée dans de nombreuses applications techniques est la
tenue au feu et à haute température des matériaux. Il est en effet essentiel d’utiliser des
matériaux capables d’empêcher ou de ralentir la propagation de flammes dans des
applications bâtiments ou transport en cas d’incendie. Des agents ignifugeants sont donc
utilisés, contenant le plus couramment des molécules halogénées. Ces dernières consomment
en effet beaucoup d’énergie lors de leur volatilisation. Mais depuis quelques années, les
contraintes environnementales ont favorisé le développement de systèmes ignifugeants sans-
halogène. L’usage de charges inorganiques est également une voie de remplacement. Le mode
d’action est alors différent : les charges présentes dans le matériau forment lors de la
56

Chapitre 3 : Les mélanges polyoléfines/CaCO3
combustion du matériau une enveloppe inorganique qui est une barrière à l’oxygène,
carburant nécessaire à la propagation des flammes. Afin d’obtenir un système ignifugé
efficace nécessaire à la formation de cette couche protectrice, le taux de charge doit être
important (environ 40% en masse). Le matériau obtenu présente alors un module mécanique
supérieur, au détriment des propriétés de tenue au choc et de la densité.
Généralement, il faut trouver des compromis de renforcement ou réaliser des
structures complexes, composées de matériaux différents. Il existe bien sur des contre–
exemples, le domaine des mélanges de polymères et des composites thermoplastiques étant
l’objet de recherches très innovantes.
3.3. Les nanocomposites et leurs spécificités : Lors de l'ajout de particules de renfort dans une matrice polymère, de nombreux
paramètres tels que la longueur et le poids moléculaire des chaînes, la taille et la distribution
des renforts, ou encore la nature des surfaces de contact et des interactions jouent un rôle sur
le comportement local (à l'interface) et global du matériau composite. Pour des inclusions de
taille macroscopique, la matrice apparaît comme une phase homogène et continue. L'étendue
des perturbations locales, dues par exemple à la nature des agents liants, est négligeable
devant les autres longueurs caractéristiques, elles n'ont ainsi pas d'influence significative sur
le comportement global. Au contraire, pour des inclusions de taille nanométrique, de
nouveaux paramètres sont à prendre en considération. La longueur des chaînes, la mobilité
locale de ces chaînes au voisinage de la nanoparticule vont alors devenir des éléments dont la
prise en compte s'avère nécessaire à la compréhension des phénomènes. De plus, pour une
fraction volumique de renforts donnée, plus la taille des particules décroît, plus elles sont
nombreuses et proches. Par conséquent, lorsque la taille des particules atteint l'échelle du
nanomètre, il peut s'avérer essentiel de considérer les interactions entre particules en plus des
interactions particules/matrice [65].
3.3.1. Les problèmes d'élaboration :
D'un point de vue expérimental et à l’échelle nanométrique, les scientifiques comme
les industriels ont du mal à réaliser des échantillons bien dispersés, avec des nanoparticules de
même taille, en conservant les mêmes procédés d'élaboration. La confrontation avec la
modélisation nécessite donc souvent la formulation d'hypothèses simplificatrices pour la mise
57

Chapitre 3 : Les mélanges polyoléfines/CaCO3
au point des modèles. Cependant, compte tenu des nombreuses avancées dans le domaine des
procédés d'élaboration, ces différents effets devraient pouvoir être découplés les uns des
autres.
3.3.1.1. Forme des particules et agrégation :
Pour les très petites tailles de particules, la forme et l'agrégation de ces particules
peuvent avoir beaucoup d'influence sur le comportement global du composite. Dans ce
domaine, on peut citer les travaux de Reynaud et al. [66] sur des polyamides (PA6) renforcés
de particules de silice nanométriques (d'un diamètre de l'ordre de 25 nm). Le PA6 est un
polymère semi-cristallin ; néanmoins, contrairement aux résultats observés par Petrovic et al.
[67] sur du polyuréthane, la présence des nanoparticules ne semble pas avoir d'influence sur la
microstructure de la phase cristalline. En revanche, même si aucun effet de taille des
particules n'est observé, l'ajout des particules de renfort se traduit par une augmentation du
module d’Young du composite, augmentation d'autant plus marquée que la fraction
volumique d'inclusions croît. Cette étude montre aussi l'importance de la forme des particules
sur le comportement mécanique du composite. Une forme complexe du renfort permet
d'assurer une meilleure cohésion entre la particule et la matrice et ainsi accroît la rigidité du
composite par rapport à des particules sphériques.
De même, les phénomènes de renfort sont plus marqués et les couplages mécaniques
plus efficaces s'il y a présence d'agrégats au lieu de particules bien dispersées. La percolation
peut posséder un effet bénéfique sur le renforcement ; pour un mauvais état de dispersion, la
limite d'élasticité est plus élevée que pour des particules parfaitement dispersées. Il est
important de noter que la taille des particules joue un rôle sur la dispersion : plus elles sont
petites et plus les phénomènes d'agrégation sont fréquents, d'où l'importance des effets
couplés (effet de taille, de percolation, etc) à l'échelle nanométrique [65].
L'existence d'agrégation est aussi mise en évidence dans les travaux de Steenbrink et
al. [68] sur un styrène acrylonitrile chargé de particules de type cœur/écorce de 100 à 600 nm
de diamètre. Le cœur est composé de polyacrylate de butyle (PABu) et l'écorce de
polyméthacrylate de méthyle (PMMA). Pour les plus petites particules, environ 100 à 150 nm
de diamètre, on observe des problèmes d'agrégation, ce qui n'est pas le cas pour des particules
plus grosses. Des essais de traction ont montré que le module décroît lorsque le taux de
58

Chapitre 3 : Les mélanges polyoléfines/CaCO3
renforts augmente (ce qui peut s'expliquer par le fait que les particules sont plus souples que
la matrice), mais n'ont pas pu révéler un effet de taille des particules sur le module de Young
ou la limite d'élasticité.
Les travaux de Thompson et al. [69] sur des films minces de polyamide renforcés par
des billes d'oxydes métalliques de 11 à 44 nm de rayon montrent aussi la dispersion des
oxydes peut jouer un rôle sur le comportement global du film. Pour les faibles taux de
renforts, il n'existe pas de percolation et les films nanostructurés montrent un comportement
fragile, plus fragile que celui attendu classiquement pour des films minces polymères. Des
essais de traction révèlent que l'ajout de renforts nanométriques rend les films plus raides que
le matériau de base dans la plupart des cas. En revanche, pour des taux de renforts plus élevés,
l'augmentation de l'allongement à la rupture laisse penser que la dispersion des oxydes
métalliques n'est pas complète.
3.3.1.2. Nature des agents de couplage :
En ce qui concerne l'influence du liant, on peut par exemple citer les travaux
d’Albérola et al. [70] sur les effets d'interface sur des élastomères renforcés de nanoparticules
de silice. En comparant des matériaux avec différents liants, elles ont montré que tout
changement de la réactivité de la surface de silice peut induire des modifications de la
mobilité des chaînes de polymère à plus ou moins longue distance de la surface. La nature du
liant modifie donc les interactions entre la particule et la matrice, entraînant une perturbation
locale de la structure et du comportement de la matrice. Dans certains cas, il est possible
d'observer la formation d'une troisième phase rigide, placée en interphase entre l'inclusion et
la matrice, au cours du procédé d'élaboration. Les travaux de Bergeret et Albérola [71] sur un
copolymère styrène - acide méthacrylique chargé de billes de verre micrométriques (5µm et
20µm) montrent que l'influence du traitement de surface du renfort observée sur la transition
vitreuse dépend notamment de la compatibilité chimique entre l'agent de couplage et la
matrice. La diminution de la mobilité due à l'ajout des particules de renfort résulte
principalement de la formation de liens physico-chimiques entre les chaînes de polymère et le
renfort.
59

Chapitre 3 : Les mélanges polyoléfines/CaCO3
3.3.2 Une zone perturbée :
De nombreuses études expérimentales mettent en avant l'existence d'une zone
spécifique entourant les nanoparticules. Les causes de cette existence peuvent être la nature
des liants utilisés lors de l'élaboration (comme précédemment évoqué dans les travaux
d’Albérola et al. [70, 71]) ou encore la présence des renforts. L'ajout de nanoparticules de
renfort affecte aussi la microstructure de la matrice. Le matériau étudié par Petrovic et al. [67]
consiste en une matrice polymère en polyuréthane (PU) renforcée par des nanoparticules de
silice de 12 nm de rayon. L'observation par microscopie à force atomique de la structure de ce
composite a révélé une distribution assez homogène des particules. Le PU étudié est un
polymère semi-cristallin, donc déjà biphasé au départ. Il possède d'un côté des sphérolites
avec une structure ordonnée et de l'autre des chaînes libres reliant ces phases cristallines. Leur
étude montre que l'ajout de nanoparticules lors de l'élaboration joue un rôle sur la
microstructure des sphérolites, effet d'autant plus grand que la fraction massique de renfort
croît. Cette modification de la microstructure, observée par microscopie, est surtout visible au
voisinage des inclusions.
Pour étudier à l'échelle du nanomètre, on peut avoir recours à la Résonance
Magnétique Nucléaire (RMN) du solide. Cette méthode repose sur l'étude de la relaxation des
moments magnétiques des atomes. En particulier, si on se focalise sur les atomes de carbone,
il est possible de rendre compte de la mobilité des chaînes carbonées dans les matériaux
hybrides inorganique/organique (par exemple composites de type inclusions de silice/matrice
polymère) ou les matériaux polymères.
Au cours de leurs travaux sur des nanocomposites particulaires, Berriot et al. [72] ont
mis en évidence l'existence d'une interphase de matrice perturbée entourant les particules de
renfort. La perturbation se traduit par une diminution locale de la mobilité des chaînes de
polymère, c'est à dire une rigidification locale de la matrice. Les matériaux étudiés sont
constitués d'inclusions sphériques de silice, de 60 nm de rayon, noyées dans une matrice
élastomère. La fraction volumique des inclusions est de 7 à 18 % suivant les échantillons
testés. Au cours de cette étude, il a aussi été montré que l'épaisseur de l'interphase décroît
lorsque la température augmente. Une étude complémentaire de Berriot et al. [73] a permis de
mettre en évidence l'influence de la dispersion des particules de renfort et de la nature du liant
sur le comportement de cette interphase en termes de mobilité moléculaire.
60

Chapitre 3 : Les mélanges polyoléfines/CaCO3
3.3.3 Renforts et comportement mécanique :
La confrontation entre différents nanocomposites à matrice polymère montre que pour
des sollicitations identiques, les propriétés macroscopiques du matériau hétérogène reflètent
l'action combinée :
- des interfaces entre les renforts et la matrice ;
- des propriétés des différentes phases ;
- de l'effet de taille des renforts ;
- de la morphologie et de la microstructure.
Les différentes matrices étudiées sont du polyéthylène (PE), du polypropylène (PP) et
du polyacrylate (PA), renforcées par des nanoparticules métalliques de 10nm de diamètre
environ. La matrice ne joue pas seulement un rôle d'environnement, mais elle influence le
comportement global du composite par la nature de ses interactions avec les particules de
renfort. Les matériaux obtenus ont montré de meilleures propriétés mécaniques et une plus
haute stabilité thermique que le polymère initial.
Dans leurs travaux, Siegel [74] et Schadler [75], et leurs collaborateurs, se sont
intéressés au cas des polymères renforcés par des particules sphériques. Ash et al. [76] ont pu
mettre en évidence une influence de la présence des nanoparticules de renfort sur la transition
vitreuse. Le matériau étudié est du polyméthacrylate de méthyle (PMMA) chargé de
particules d'alumine d'un diamètre moyen de 39 nm. Au delà d'une fraction volumique seuil
de renforts (dans le cas étudié de 1,7 %), une chute importante de la température de transition
vitreuse Tg est observée. Cette chute s'explique par des phénomènes de confinement de la
matrice, dont la mobilité des chaînes est perturbée, entre les renforts. En revanche, au dessous
de ce seuil aucun effet de ce type n'est observé. Une étude antérieure de Siegel et al. [74] sur
les mêmes matériaux avait déjà mis en évidence un comportement en traction plus ductile et
une augmentation importante de la déformation à rupture pour le matériau chargé par rapport
à la matrice pure. Siegel et al. [74,75], ont mené une étude comparative sur des systèmes
époxy chargés de particules de TiO2 micrométriques, nanométriques et la matrice pure. Des
essais de microdureté, de traction et de rayure ont été réalisés. Le nanocomposite se révèle
plus rigide que le composite classique (avec des renforts micrométriques), lui même plus
rigide que la matrice pure.
61

Chapitre 3 : Les mélanges polyoléfines/CaCO3
L'étude de microdureté des polyacrylates renforcés d'oxyde de titane (TiO2) réalisée
par Perrin, Nguyen et Vernet [77], montre que la dureté du matériau hybride croît
linéairement avec le taux de renfort, et ce jusqu'au seuil de percolation (de l'ordre de 11 % en
fraction volumique). En ce qui concerne le comportement viscoélastique du nanocomposite, la
vitesse de fluage décroît lorsque la fraction volumique de renforts augmente.
Pour enrichir la compréhension du comportement viscoélastique de ces matériaux, les
phénomènes de viscosité étant très présents dès que l'on parle de polymères, on peut noter les
travaux réalisés par Chabert au cours de sa thèse [78, 79]. L'étude d'une matrice de
polyacrylate de butyle (PABu) renforcée par des particules de polystyrène (PS)
submicroniques (100 nm) a mis en avant un effet de la fraction volumique de renforts et de
l'interaction particules/particules sur le comportement viscoélastique du nanocomposite. Par
rapport au comportement des deux phases pures (PS et PABu), le composite montre un creux
dans le plateau caoutchoutique, creux d'autant plus marqué que le taux de renforts augmente.
La confrontation avec une approche d'homogénéisation a permis d'expliquer le phénomène
par la prise en compte d'une interphase, supposée vitreuse dans l'étude, dont l'épaisseur
décroît lorsque la température augmente. Les modèles ont permis de mettre en avant
l'importance de la caractérisation mécanique de cette interphase et des interactions entre,
d'une part les inclusions et la matrice et, d'autre part, les inclusions entre elles. L'étude
expérimentale a aussi permis de mettre en évidence l'existence d'un seuil de percolation au-
delà de 20 % de fraction volumique de renforts. Au-dessus de ce seuil de percolation, le
module de cisaillement élastique G0 augmente de façon significative.
Une étude complémentaire de Chabert et al. [80] souligne l'influence particulière de la
nature des interactions particules/particules sur le comportement global des nanocomposites.
Lorsque les particules deviennent de très petite taille et très nombreuses, la distance inter-
particulaire devient de l'ordre de grandeur du rayon de giration des chaînes de polymère. Les
interactions entre les particules perturbent donc davantage le comportement de la matrice.
Par l'intermédiaire d'une confrontation entre expérience et modélisation numérique,
Colombini et Maurer [81] se sont intéressés au comportement viscoélastique de polymères
chargés en introduisant une troisième phase entre le renfort et la matrice. Cette interphase est
constituée d'un matériau différent, elle n'est pas due au seul fait de la présence des renforts.
L'étude des évolutions de tan δ et G” (représentant respectivement le facteur d'amortissement
62

Chapitre 3 : Les mélanges polyoléfines/CaCO3
et le module de dissipation ou de perte) en fonction de la température, a mis en évidence une
grande influence du comportement de cette troisième phase sur les propriétés viscoélastiques.
Au cours de cette étude, les résultats expérimentaux et les prédictions de l'approche
numérique ont montré un bon accord entre eux.
3.3.4. Nanoparticules et propriétés thermiques :
Le PP est un polymère semi-cristallin qui peut contenir différents types de cristaux
sphérolitiques de type-α, de type-β, de type-γ et de type-δ.
Le cristal sphérolitique de type-α monoclinique est le plus stable, et certains types de cristaux
sphérolitique peuvent être transformés en cristaux de type-α sous certaines conditions. Les
phases cristallines les plus probables qui apparaissent pendant le refroidissement du
polypropylène sont des cristaux de Type-α et de Type-β . Comparé aux cristaux sphérolitique
de type-β, le cristal de type-α peut subir une contrainte de flexion plus élevée, il a une densité
plus élevée, de plus grandes rigidité et dureté. Le cristal hexagonal de Type-β apparaît avec
une probabilité inférieure au cristal de type-α, celui-ci possède une bonne résistance à l'impact
et une résistance à la traction plus élevée. Il est aussi connu, que la formation de la phase
cristalline des polymères semi-cristallins est très sensible à beaucoup de facteurs, tels que
l'histoire thermique, les caractéristiques moléculaires, les impuretés etc... On a montré que les
particules nanométriques de nano charges CaCO3 sont des agents de nucléation très efficaces
et augmenteraient la quantité de cristaux de type-β dans les nano-composites, bien que la
cristallinité globale de ces composés soit à peine affectée [82-83].
Il est aussi montré que des taux autour de 4-6% en volume de particules
nanométriques de CaCO3, favorisent la formation de cristaux de type-β, augmentant ainsi la
résistance au choc du PP. A un taux de nano charge CaCO3 de 4-6 % en volume, la résistance
mécanique du PP peut être augmentée de façon optimale. Aux alentours d’un taux de nano
charges de CaCO3 de 4% en volume, l'énergie d'activation et la température maximale du pic
de fusion de cristaux de type-α montrent respectivement des valeurs minimum et maximum ;
Ces phénomènes peuvent être expliqués par la croissance préférentielle des cristaux de type-β
dans le PP à un taux de nano charge de CaCO3 de 4 à 6% en volume. A mesure que la fraction
de nanoparticules de CaCO3 augmente, le nombre de nucléons de cristaux de type-β
63

Chapitre 3 : Les mélanges polyoléfines/CaCO3
augmente, et la taille des cristaux de type-β diminue, ce qui peut causer une diminution de la
résistance mécanique des mélanges [82-83].
La plupart des auteurs [84-87] estiment que les nanocharges de CaCO3 ne modifient
pas la structure des cristallites ; cependant, les particules non traitées interfèrent sur le
processus de cristallisation de la phase polypropylène en jouant le rôle d’agents de nucléation.
Les particules de CaCO3 traitées en surface n'agissent pas en tant qu'agent de nucléation dans
le polypropylène puisque la cristallinité n'est pas augmentée.
3.3.5. Variation des propriétés rhéologiques en présence de nano-charges:
Le comportement rhéologique des polymères chargés dépend de nombreux
paramètres, comme par exemple la nature des charges (taille, forme), la concentration, les
interactions entre les charges et entre le polymère et la charge. Ces paramètres provoquent
non seulement une augmentation de la viscosité, mais aussi des phénomènes particuliers
comme, par exemple, l’existence d’un seuil d’écoulement, un comportement thixotrope,
rhéofluidifiant ou rhéoépaississant. Dans le cas des nanocomposites, on observe en général
toutes les caractéristiques rhéologiques des polymères chargés classiques. Cependant, du fait
que la taille des charges est extrêmement petite, la surface développée avec la matrice
polymère est donc très grande. Des phénomènes particuliers peuvent être observés dès les très
faibles concentrations en charges.
3.4. Les interactions charges-charges et charges-polymères :
3.4.1. Interactions charges-charges :
Les propriétés et la structure des silices dépendent directement de leur voie de
synthèse. Les différentes formes rencontrées sont les suivantes (Figure 3.1) :
- les particules élémentaires, dont la taille est comprise entre 10 et 40 nm,
- les agrégats, ou amas insécables, constitués de particules élémentaires,
64

Chapitre 3 : Les mélanges polyoléfines/CaCO3
- les agglomérats, de taille importante, correspondant à des associations d'agrégats,
pouvant être fractionnés lors du malaxage.
Figure 3. 1: Les différentes échelles de taille de la silice [88]
Ce sont généralement des objets fractals [89], c'est-à-dire que l'évolution des
propriétés P, comme la surface spécifique ou la masse volumique apparente, obéit à une loi de
puissance de la taille L de l'objet :
P ∝ Lm
où m est sa dimension fractale.
La figure 3.2 schématise la différence entre un objet fractal que constitue par exemple
un agrégat de silice et un objet homogène quelle que soit l'échelle d'analyse.
Figure 3. 2: (a) Matériau homogène à toutes les échelles, (b) Objet fractal [88]
Pour un pH basique, les particules sont dispersées de manière homogène dans la
matrice. Pour un pH acide, les particules de silice se sont agrégées et semblent former un
réseau percolant de charges. Les auteurs attribuent ce phénomène à l'agrégation du latex qui a
lieu à pH acide. Si la silice ne s'agrège pas, elle vient occuper les espaces vides laissés par les
particules de latex pendant le séchage.
65

Chapitre 3 : Les mélanges polyoléfines/CaCO3
3.4.2. Interactions charges-polymères :
Les interactions polymère-charge peuvent être de nature physique (liaisons hydrogène,
Van der Waals) ou chimique (liaison covalente) en fonction de la nature des constituants et
des additifs introduits dans le réacteur. Dans le cas des élastomères renforcés par de la silice
précipitée présentant certains agents de couplage, il a été montré qu'une couche de polymère
lié, ou immobilisé, se développait à la surface des charges. Les propriétés finales du
composite dépendront de celles de ses constituants (nature de la matrice, forme et dimensions
du renfort) mais également de cette interphase.
Selon le type d'interactions entre polymère et charge, on peut obtenir du polymère :
- chimiquement lié, ou chimisorbé : le polymère n'est alors pas extractible par un bon
solvant.
- physisorbé : bien que présentant des interactions importantes avec les charges, ce
polymère reste soluble.
3.4.2.1. Extraction :
La technique la plus simple permettant de mettre en évidence le polymère lié est la
technique d'extraction [77, 67]. Elle consiste à immerger les échantillons (non vulcanisés dans
le cas des élastomères) dans un bon solvant afin d'éliminer tout le polymère extractible. Le
taux de polymère lié est ensuite déterminé par thermogravimétrie sur les résidus séchés.
La figure 3.3 montre l'influence du taux de silice sur la quantité de polymère lié [77].
Si on considère le taux de polymère lié dans la matrice, on voit que celui-ci augmente
nettement avec le taux de silice, de 10 à 40% environ pour des taux de charges de 8,8 à 26,4
%vol de silice. Cependant, il reste constant pour des silices ayant des chimies de surface
différentes, à surface spécifique constante (environ 150 m²/g). L'auteur remarque également
qu'il n'y a pas de polymère lié quand un agent de recouvrement est utilisé autour de la silice,
et qu'il augmente fortement quand les échantillons contiennent un agent de couplage.
66

Chapitre 3 : Les mélanges polyoléfines/CaCO3
Figure 3. 3 : Taux de polymère lié dans des composites SBR-silice pour des fractions
volumiques de charges de 8,8 ; 16,2 ; 21,9 et 26,4 % [77]
Ono et al. [90] ont observé par microscopie électronique à transmission des
nanocomposites à matrice élastomère polyisoprène renforcée par de la silice pyrogènée et
précipitée après extraction du polymère libre dans le toluène. Ils ont constaté que dans le cas
de la silice précipitée, les particules de renfort formaient des agglomérats, emprisonnant une
grande partie de polymère lié. Dans le cas de la silice pyrogénée, les charges sont sous forme
d'agrégats entourés de polymère lié. Ils observent alors que la quantité de polymère lié est
associée au degré d'agrégation des particules de silice, et donc du nombre de fonctions
silanols à la surface des charges. Le nombre de silanols étant plus important à la surface de la
silice précipitée, son degré d'agglomération est plus élevé, ainsi que le taux de polymère lié.
3.4.2.2. Spectrométrie mécanique dynamique :
Tsagaropoulos et Eisenberg [59] ont étudié différents polymères (PVAc, PMMA…)
renforcés par des nanoparticules de silice de 7 nm de diamètre. Deux relaxations ont été mises
en évidence par spectrométrie mécanique dynamique :
- la première est associée à la relaxation principale de la matrice. Sa position en
température ne varie pas alors que l'aire du pic diminue lorsque le taux de renfort croît.
- la deuxième, plus large, est située à plus haute température. Elle est attribuée à la
présence de polymère immobilisé dans ces matériaux. Sa largeur diminue également
avec le taux de charge.
67

Chapitre 3 : Les mélanges polyoléfines/CaCO3
L'origine de cette deuxième relaxation est représentée sur la figure 3.4 :
- pour de faibles fractions volumiques, les charges sont entourées d'une couche de
polymère liée et sont bien dispersées au sein de la matrice (Figure 3.4-a),
- pour une fraction volumique de charges critique, ces zones immobilisées se
superposent, piégeant une partie du polymère, à l'origine, d'après ces auteurs, de la
seconde relaxation (Figure 3.4-b),
- quand VF augmente, les régions de mobilité réduite ont tendance à se rapprocher, puis
à se lier (Figure 3.4-c et d). Il y a alors dans le matériau une zone immobilisée,
expliquant, d'après les auteurs, la diminution de l'amplitude de la deuxième relaxation
avec VF.
Figure 3. 4 : Origine de la deuxième relaxation proposée par TSAGAROPOULOS et al. [59]
Afin de déterminer l'épaisseur de polymère lié, Boluk et Schreiber [91] utilisent les
résultats de DMA : ils ont évalué les variations du maximum du pic de tanδ associé à la
transition vitreuse du polymère en fonction du taux de charges et proposent la loi empirique
suivante :
tan δc = tan δp (1-BVF)
avec B = (1 + ∆R/R)3
Où tanδc et tanδp sont les maxima de tanδ respectivement du composite et du polymère pur
R est le rayon des charges et ∆R l'épaisseur de polymère immobilisé autour d'elles.
En utilisant ce modèle sur des composites à matrice polyéthylène chloré (CPE)
renforcée par des nanoparticules de TiO2, les auteurs obtiennent des épaisseurs de polymère
lié allant de 1 à 20 nm. Ce modèle a été également utilisé par Ou [92] sur des systèmes
68

Chapitre 3 : Les mélanges polyoléfines/CaCO3
SBR/SiO2, et Mukhopadhyay [93] sur des composites EVA/SiO2. Ainsi, ces auteurs ont
déterminé des épaisseurs de polymère immobilisé respectivement de l'ordre de 40 et 27 Å.
3.5. Conclusion :
Les travaux que nous avons rapportés ont permis de mettre en évidence l'influence de
l'introduction de nanoparticules dans des matrices polymère sur les propriétés macroscopiques
de composites.
Des variations du comportement mécanique dynamique des nanocomposites ont été
mises en évidence en fonction de la quantité de charges ou de la modification de la chimie de
surface de ces renforts. La plupart des auteurs expliquent ces phénomènes par la présence
d'une épaisseur de polymère dont la mobilité est modifiée à la surface des charges.
Cependant, nous pouvons nous demander si la morphologie, c'est-à-dire l'état de
dispersion des nanoparticules dans la matrice polymère, ne peut pas également jouer un rôle
sur le comportement mécanique global des nanocomposites. En effet, différents travaux ont
montré que la diminution de la taille des renforts favorisait l'agrégation, l'agglomération, voire
la percolation des charges. Par exemple, Pu et al. [94, 95] ont montré que le module d'Young
et la contrainte à la rupture augmentaient considérablement avec l'agrégation de la silice dans
une matrice PMMA, par rapport à de la silice distribuée aléatoirement ou régulièrement.
Les contributions respectives des effets de couplage mécanique et des effets d'interface
devraient ainsi être séparées afin de mieux comprendre les mécanismes impliqués dans le
renforcement des nanocomposites à matrice thermoplastique.
69

Chapitre 3 : Les mélanges polyoléfines/CaCO3
Références [1] Le recyclage et la valorisation des déchets en réparation Carrosserie Peinture 01-12-05,
Jean-Pierre GAVALDA, I.U.F.M. de Créteil, 1993.
[2] Schober W. The trend in automotive PP-compounds calls for submicron functional
minerals, Intertech Conference Berlin “Functional effect filers for reinforcing polymer
systems”, 13-15 September 2000.
[3] Utracki L. Polymer Alloys and Blends. Thermodynamics and Rheology: Part 1 Munich
Vienne New York: Hanser, 1989
[4] Wippler C., Nouvelle Approche Théorique de Matériaux d'Avenir: Les Alliages de
Polymères, Actua1.Chimi,1985, vol1, p 33.
[5] Paul D.R., Barlow J.W. Adv. Chem. Ser, 1979, vol 176, p 315
[6] Paul D.R., Barlow J.W. Macromol.Sci.- Rev.Macromol.Chem.C18, vol 1, p109-169
[7] Leibler L., "Thermodynamique des Mélanges Polymère" Initiation à la Chimie et à la
Physico-Chimie Macromoléculaire, 1986, vol 6, p l.
[8] Feldman D. and Aisbuch I.V. "Etude Scientifique et Technique": Alliages
Macromoléculaires, Plast. Mod. Elast, 1976, p. 58
[9] Paul D., et Bucknall C., Introduction in Polymer Blends: Formulation.Edited by D.R. Paul
and C.B. Bucknall New York: Wiley-Interscience, 2000, vol. l, p.1-14
[10] Jaziri M. Etude du greffage à l’état fondu de l’anhydride maléique sur l’ABS Influence
du composé obtenu sur l’élaboration et les propriétés des mélanges PC/ABS. Thèse Université
Jean Monnet - Saint Etienne, 1992, p. 5-6.
[11] Meynie L. Evolution et contrôle de la morphologie d’un mélange
thermoplastique/thermodurcissable polymérisé sous cisaillement. Thèse INSA de Lyon, 2003.
[12] Colbeaux A. Compatibilisation de mélanges polypropylène/polyéthylène par extrusion
réactive. Thèse INSA de Lyon, 2001.
[13] Utracki L. Commercial Polymer Blends London: Chapman & Hall, 1998, p 658.
[14] Tucker P.S. and Paul D.R. Simple model for enthalpic effects in homopolymer/block
copolymer blends. Macromolecules, 1988, vol 21, p 2801-2807.
[15] Hong K.M. and Noolandi J. Theory of phase equilibriums in systems containing block
copolymers; Macromolecules, 1983, vol 16, p 1083.
[16] Meier D., Polym. Prepr, 1977, vol. 18, p. 340.
[17] Whitmore K. and Noolandi J. Theory of phase equilibria in block copolymer-
homopolymer blends. Macromolecules, 1985, vol 18, p 2486.
70

Chapitre 3 : Les mélanges polyoléfines/CaCO3
[18] Guo H., Packirisamy S and Many R. Compatibilizing effects of block copolymers in Low
density polyethylene/polystyrene blends. Polymer, 1998, vol 39, p 2495-2505.
[19] Yang L., Smith T. and Bigio D. Melt blending of linear low density polyethylene and
polystyrene in an Haake internal mixer. I. Compatibilization and Morphology development.
Journal of applied polymer science, 1995, vol 58, p 117-127.
[20] Cigana P., Favis B. and Jérôme R. Diblock copolymers as emulsifying agents in polymer
blends: Inflence of molecular weight, Architecture, and chemical composition. Journal of
applied polymer science: Part B: Polymer physics, 1996, vol 34, p 1691-1700.
[21] Wei G., Sue H. and Chu J. Toughening and strenghthening of polypropylene using the
rigid_rigid polymer toughening concept Part I. Morphology and mechanical properties
investigations. Polymer, 2000, vol 41, p 2947-2960.
[22] Hellmann G. and Dietz M. Random ad block copolymers as compatibilizers, Directly
compared. Die Makromoleculare Chemie, Macromoleculare Symposium, 2000, vol 170, p1-8.
[23] Mocosko C., Guégan P. and Khandpur A. Compatibilizers for melt blending: Premade
block copolymers. Macromolecules, 1996, vol 29, p 5590-5598.
[24] Fayt R., Jérôme R.and Teyssié P. Molecular design of multicomponent polymer systems,
13. Control of the morphology of polyethylene/polystyrene blends block copolymers.
Macromoleculare Chemistry, 1986, vol 187, p 837-852.
[25] Tang T. and Huang B. Interfacial behavior of compatibilizers in polymers blends.
Polymer, 1994, vol 35, no 2, p 281-285.
[26] Xanthos M. and Dagli S. Compatibilization of polymer blends by reactive processing.
Polymer engineering and science, 1991, vol 31, no 13, p 929-935.
[27] Hu G., Sun Y. and Lambla M. Devolatilization : A critical sequential operation for in
situ compatibilization of immiscible polymer blends by one-step reactive extrusion. Polymer
engineering and science, 1996, vol 36, p 676-684.
[28] Ang G. Hu Y.S., Lambla M. and Kotlar H. In situ compatibilization for polypropylene
and poly(butylene terephtalate) polymer blends one-step reactive extrusion. Polymer, 1996,
vol 37, no 18, p 4119-4127.
[29] Milner S. and Xi H. How copolymers promote mixing in immiscible hopolymers.
Journal rheology, 1996, vol 40, no 4, p 663-687.
[30] Saleem M. and Baker W. In situ reactive compatibilization in polymer blends: Effects of
functional group concentrations. J of applied polymer science. 1990, vol 39, p 655-678.
[31] Danesi S. and Porter R. Blends of isotactic polypropylene and ethylene-propylene rubber
rheology, morphology and mechanics. Polymer, 1978, vol 19, p 448-457.
71

Chapitre 3 : Les mélanges polyoléfines/CaCO3
[32] D’orazio L., Mancarella C., Martuscelli E. and Sticotti G. Melt rheology, structure and
impact properties of injection-moulded samples of isotactic polypropylene/ethylene propylene
copolymer (iPP/EPR) blends: influence of molecular structure of EPR copolymer. Polymer,
1993, vol 34, p 3671-3681.
[33] D’orazio L., Mancarella C., Martuscelli E. and Polato F., Polypropylene/ethylene co-
propylene blends: influence of molecular structure and composition of EPR on melt rheology,
morphology and impact properties of injection-moulded samples. Polymer, 1991, vol 32, p
1186-1194.
[34] D’orazio L., Mancarella C., Martuscelli E., Sticotti G. Polypropylene/ethylene-co-
propylene blends: influence of molecular structure of EPR and composition on phase structure
of isothermally crustallized samples. J Materials Science, 1991, vol 26, p 4033-4047.
[35] Karger-Kocsis J. and Csikai I. Skin-core morphology and failure of injection-moulded
specimens of impact-modified polypropylene blends. Polymer engineering and science, 1987,
vol 27, p 241-253.
[36] Initiation à la chimie et la physico-chimie, 6- Mélanges de polymères, Groupe Français
d’Etude et d’Application des polymères.
[37] Pukanszky B., Tudos F., Kallo A. and Bodor G. Multiple morphology in
polypropylene/ethylene-propylene-diene terpolymer blends. Polymer, 1989, vol 30, p 1399-
1406.
[38] Martuscelli E., Silvestre C. and Abate G. Mophology, cristallization and melting
behaviour of films of isotactic polypropylene blended with ethylene-propylene copolymers
and poluisobutyle, Polymer, 1982, vol 23, p 229-237.
[39] Fortenlyi., Michalkova D., Koplikova E., Kovar J. The effect of conditions mixing of
polypropylene/ethylene-propylene elastomer blends on the morphological structure and
impact strength. Die Angewandte makromolekulare Chemie, 1990, vol 179, p 185-201.
[40] Yang D., Mancarella C., Martuscelli E. and Sticotti G. Melt rheology, phase structure
and impact properties of injection-moulded samples of isotactic propylene/ethylene propylene
copolymer (iPP/EPR) blends: influence of molecular structure of EPR copolymer. Polymer,
1993, vol 34, p 3671-3681.
[41] Jancar J., Dianselmo A., Dibenedetto A. and Kucera J. Failure mechanics in elastomers -
toughened polypropylene, Polymer, 1993, vol 34, p 1684–1694.
[42] D’orazio L., Mancarella C., Martuscelli E., Sticotti G. and Ghisellini R. Thermoplastic
elastomers from iPP/EPR blends: cristallization and phase structure development. Journal of
applied polymer science, 1994, vol 53, p 387-404.
72

Chapitre 3 : Les mélanges polyoléfines/CaCO3
[43] Graco R., Mancarella E., Martuselli E., Ragosta G. and Jinghua Y. Polyolefins blends : 2.
Effect of EPR composition on structure, morphology and mechanical properties of iPP/EPR
alloys. Polymer, 1987, vol 28, p 1929-1936.
[44] Martuscelli E., Silvestre C., Abate G. Mophology, cristallization and melting behaviour
of films of isotactic polypropylene blended with ethylene-propylene copolymers and
poluisobutyle. Polymer, 1982, vol 23, p 229-237.
[45] Jang B., Uhlmann D. and Vander-Sande J. Rubber-toughening in polypropylene. Journal
of applied polymer science, 1985, vol 30, p 2485-2504.
[46] Copolla F., Grecor R., Martuscelli E., Kammar H. and Kummerlowe C. Mechanical
properties and morphology of isoactic polypropylene/ethylene-propylene copolymer blends.
Polymer, 1987, vol 28, p 47-56.
[47] Jang B., Uhlmann D. and Vander-Sande J. Crystalline morphology of polypropylene and
rubber-modified polypropylene. Journal of applied polymer science, 1984, vol 29, p 4377-
4393.
[48] Harvey M. and Ketley A. The infrared identification of short-chain branches in
polyolefins. Journal of applied polymer science, 1961, vol 5, p 247-250.
[49] Cunha A., Pouzada A. and Crawford R. A study of the impact behaviour f injection
moulded polypropylene using two different modes of testing. Plastics, rubber and composites
processing and applications, 1992, vol 18, p 79-90.
[50] D’orazio L., Greco R., Mancarella C., Martuscelli E., Ragosta G. and Silvestre C. Effect
of the addition of ethylene-propylene random copolymers on the properties high density
polyethylene, isotactic polypropylene blends: Part 1 – Morphology and impact behaviour
molded samples. Polymer engineering and science, 1982, vol 22, p 536-544.
[51] Stehling F., Huff T., Speed C. and Wissler G. Structure and properties of rubber modified
polypropylene impact blends. Journal of applied polymer science, 1981, vol 29, p 2693-2711.
[52] Sharma Y., Patel R. and Bharaj I. The effect of elastomers of the cristallinity
polypropylene. Thermochemica Acta, 1982, vol 54, p 229-232.
[53] Halfand E., Tagami Y. Theory of the interface between immiscible polymers. Journal of
polymer science, Polymer Letters, 1971, vol 9, p 741-746.
[54] Krause S. Polymer compatibility. Journal of macromolecular science review
mocromolecular ghemist, 1972, C7(2), p 251-314.
[55] Yang D., Zhang B., Yang Y., Fang Z., Sun G. and Fenz Z. Morphology and properties of
blends of polypropylene with ethylene-propylene rubber. Polymer engineering and science,
1984, vol 24, p 612-617.
73

Chapitre 3 : Les mélanges polyoléfines/CaCO3
[56] Bing L., Chung T. C. Synthesis of Maleic Anhydride Grafted Polyethylene and
Polypropylene, with Controlled Molecular Structures. Journal of Polymer Science: Part A:
Polymer Chemistry, 2000, Vol 38, p 1337–1343.
[57] Cahier de GFP. Initiation à la chimie et à la physico-chimie macromoléculaires-Quelques
grades de polymers industriels. 1982, p 4.
[58] Wang L., Huang B. Structure and properties of sequentially polymerized propylene
[ethylene-co-propylene]-b-propylene copolymer. Journal of polymer science, Part B, Polymer
physics, 1991, vol 29, p 1447-1456.
[59] Tsagaropoulos G., Eisenberg A. Dynamic Mechanical Study of the factors affecting the
two glas transition behavior of filled polymers. Similarities and differences with random
ionomers. Macromolecules, 1995, vol 28, p 6067-6077
[60] Qiu W., Mai K. and Zeng H. Effect of silane -grafted polypropylene on the mechanical
properties and crystallization behavior of Talc/polypropylene composites. Journal of Applied
Polymer Science, 2000, vol 77, p 2974-2977.
[61] Kucera J. and Kolarik J. Mechanical properties of polyethylene and polypropylene filled
with calcium carbonate. In: Polymer Composites; Sedlacek, B., Ed. Berlin-New York: Walter
de Gruyter, 1986, p 545-552.
[62] Rodriguez-Perez M.A., Velasco J.I., Arencon D. and De-Saja J. A. Dynamic Mechanical
Behavior of PP/PET Blends Filled with Glass Beads. In: Eurofillers 03, Alicante (Spain),
2003,
[63] Sheldon R.P. Composite polymeric materials. London: Applied Science Publisher, 1982.
[64] Matsuo M. Observation of crazes in ABS-polymer and high-impact polystyrene under
the electron microscpoe. Polymer, 1966, vol 7, p 421-425.
[65] Marcadon V. Effets de taille et d'interphase sur le comportement mécanique de
nanocomposites particulaires », 2005, p 12-16.
[66] Reynaud E., Jouen T., Gauthier C., Vigier G., and Varlet J. Nano fillers in polymeric
matrix : a study on silica reinforced PA6. Polymer, 2001, vol 42, p 8759-8768.
[67] Petrovic Z. S., Cho Y. J., Javni I., Magonov S., Yerina N., Schaefer D. W., Ilavsky J.,
and Waddon A. Effect of silica nanoparticles on morphology of segmented polyurethanes.
Polymer, 2004, 45, p 4285-4295.
[68] Steenbrink A. C., Litvinov V. M. and Gaymans R J. Toughening of san with acrylic core-
shell rubber particles : particle size effect or cross-link density ? Polymer, 1998, vol 39, p
4817-4825.
74

Chapitre 3 : Les mélanges polyoléfines/CaCO3
[69] Thompson C. M., Herring H M., Gates T. S. and Connell J. W. Preparation and
characterization of metal oxide / polyimide nanocomposites. Composites Science and
Technology, 2003, vol 63 p 1591-1598.
[70] Albérola N. D., Benzarti K., Bas C. and Bomal Y. Interface e_ects in elastomers
reinforced by modi_ed precipitated silica. Polymer Composites, 2001, vol 22, 312-325.
[71] Bergeret A. and Albérola N. D. A study of the interphase in styrene-mathacrylic acid
copolymer / glass bead composites. Polymer, 1996, vol 37, p 2759-2765.
[72] Berriot J., Lequeux F., Monnerie L., Montes H., Long D. and Sotta P. Filler elastomer
interaction in model filled rubbers, 1H NMR study. Journal of Non-Crystalline Solids, 2002,
p 719-724.
[73] Berriot J., Martin F., Montes H., Monnerie L. and Sotta P. Reinforcement of model filled
elastomers ; characterization of the cross-linking density at the filler-elastomer interface by
1H NMR measurements. Polymer, 2003, vol 44, p 1437-1447.
[74] Siegel R. W., Chang S. K., Ash B. J., Stone J., Ajayan P. M., Doremus R. W. and
Schadler L. S. Mechanical behavior of polymer and ceramic matrix nanocomposites. Scripta
Mater, 2001, vol 44, p 2061-2064.
[75] Ng C. B., Schadler L. S. and Siegel R. W. Synthesis and mechanical properties of tio2
epoxy nanocomposites. Nanostructured Materials, 1999, vol 12, p 507-510.
[76] Ash B. J., Schadler L. S. and Siegel R. W. Glass transition behavior of alumina /
polymethylmethacrylate nanocomposites. Materials Letters, 2002, vol 55, p 83-87.
[77] Perrin F. X., Nguyen V. and Vernet J. L. Mechanical properties of polyacrylictitania
hybrids - microhardness studies. Polymer, 2002, vol 43, p 6159-6167.
[78] Chabert E. Propriétés mécaniques de nanocomposites à matrice polymère : approche
expérimentale et modélisation. thèse de l’ INSA de Lyon, 2002.
[79] Chabert E., Dendievel R., Gauthier C. and Cavaillé J.Y. Prediction of the elastic response
of polymer based nanocomposites: a mien field approach and a discrete simulation.
Composites Science and Technology, 2004, vol 64, p 309-316.
[80] Chabert E., Bornert M., Bourgeat-Lami E., Cavaillé J.Y., Dendievel R., Gauthier C.,
Putaux J. L. and Zaoui A. Filler-filler interactions and viscoelastic behavior of polymer
nanocomposites. Materials Science and Engineering 2004, vol 381, p 320-330.
[81] Colombini D. and Maurer F. H. J. Origin of additional mechanical transitions in
multicomponent polymeric materials. Macromolecules, 2002, vol 35, p 5891-5902.
[82] Chan C.M., Wu J.S., Li J.X. and Chueng Y.K. Polypropylene/calcium carbonate
nanocomposites. Polymer, 2002, vol 43, p 2981-2992.
75

Chapitre 3 : Les mélanges polyoléfines/CaCO3
[83] Ren X.C., Bai L.Y., Wang G.H. and Zhang B.L., China Plast, 2002, vol 14, p 22.
[84] Bartosiewicz L. and Kelly C.G. Microstructural analyses of homo and copolymer
extrusions verified with complimentary techniques. Advances in polymer technoly, 1987, vol
7, p 21-28.
[85] Menczel J. and Varga J. Influence of nucleating agent on crystallization of
polypropylene. Journal of thermal analysis, 1983, vol 28, p 161-165.
[86] Fujiyama M. and Wakino T. Role of filler particles on nucleation of polypropylene, J.
Appl. Polym. Sci, 1991, vol 42, p 2739–2742.
[87] Khareh A., Mitra A. and Radhakrishnan S. Effect of CaCO3 on the crystallization
behavior of PP. J. Mater. Sci, 1996, vol 31, p 5691-5702.
[88] Marceau S. Architecture multiéchelle et propriétés mécaniques de nanocomposites. 2003,
p17-19.
[89] Legrand A.P., Hommel H., Tuel A., Vidal A., Balard H., Papirer E., Levitz P.,
Czernichowski M., Erre R., Van Damme H., Barres O., Burneau A. and Grillet Y., Hydroxyls
of silica powders. Advances in Colloid and Interface Science, 1990, vol 33, p 91-330
[90] Ono S., Kiuchi Y., Sawanobori J. and Ito M. Structure development in silica-filled rubber
composites. Polymer International, 1999, vol 48, p 1035-1041
[91] Boluk M. Y., Schreiber H. P. Interfacial interactions and the properties of filled
polymers. I: Dynamic-mechanical responses. Polymer composites, 1986, vol 7, no 5, p 295-
301.
[92] Ou Y.C., Yu Z.Z., Vidal A. and Donnet J.B. Effects of alkylation of silicas on interfacial
interaction and molecular motions between silicas and rubber. Journal of Applied Polymer
Science, 1996, vol 59, p 1321-1328
[93] Mukhopadhyay K., Tripathy D.K. and De S.K. Dynamic mechanical properties of silica-
filled ethylene vinyl acetate rubber. Journal of Applied Polymer Science, 1993, vol 48, p
1089-1103
[94] Pu Z., Mark J.E., Jethmalani J.M. and Ford W.T. Mechanical properties of a poly(methyl
acrylate) nanocomposite containing regularly-arranged silica particles. Polymer Bulletin,
1996, p 545-551
[95] Pu Z., Mark J.E., Jethmalani J.M. and Ford W.T. Effects of dispersion and aggregation of
silica in the reinforcement of poly(methyl acrylate) elastomers. Chemistry of Materials, 1997,
vol 9, p 2442- 2447.
76

Partie Expérimentale
77

Chapitre I : Matériaux et techniques expérimentales
78

Chapitre I : Matériaux et techniques expérimentales
Chapitre I
Matériaux et techniques expérimentales
79

Chapitre I : Matériaux et techniques expérimentales
80

Chapitre I : Matériaux et techniques expérimentales
Chapitre I :
Matériaux et techniques expérimentales
Dans cette partie, nous allons tout d’abord discuter du choix des matrices utilisées et
en donner leurs propriétés. Ensuite, nous décrirons la structure, les spécificités et les
propriétés des compatibilisants et des nanocharges utilisés ainsi que leurs choix d’utilisation.
Puis, nous allons décrire le procédé de mise en œuvre des matériaux, l’extrusion bivis,
en expliquant tout d’abord les spécificités de cet outil. Nous porterons une grande attention à
la description des techniques de caractérisation et des conditions d’analyses.
I-1. Matériaux :
I-1-1. Matrice Polypropylène renforcée « chocs » :
Les matériaux PP7510 (PP/EPDM/talc) et PP108MF97 (PP/EPR) fournis par SABIC®
sont des polypropylènes renforcés « chocs » :
Le premier matériau est un mélange tri-phasique référencé sous la
dénomination de PP7510 que l’on utilise pour la fabrication des pare-chocs
arrière. Il se compose de 20% de nodules d’élastomère EPDM (ethylene-
propylene-diène monomère) de diamètre inférieur à 1µm, de 12% de feuillets
de talc de longueur moyenne 8µm, le tout noyé dans une matrice
polypropylène (PP). Pour conférer le caractère élastomère au polymère,
l’EPDM est obtenu en introduisant un motif insaturé dans la chaîne éthylène-
propylène par terpolymérisation avec un diène. De nombreux monomères
biinsaturés ont été utilisés, mais, commercialement, seuls trois d’entre eux ont
une importance :
Hexadiène 1-4 dicyclopentadiène éthylidène-5 norborène
(HD) (DCPD) (ENB)
81

Chapitre I : Matériaux et techniques expérimentales
Le choix de ces diènes est évidement lié à trois conditions essentielles :
• une seule double liaison doit être copolymérisable, pour éviter le gel ;
• aucune des doubles liaisons ne doit inactiver le site catalytique par
complexation ;
• le diène doit être suffisamment réactif pour être introduit dans la chaîne.
Le principal est l’ENB, se polymérisant exclusivement par la double liaison du cycle :
(EP) (EP)
Pour les pare-chocs avant, le matériau référencé PP108MF97, est un
copolymère choc PP/EPR (ethylene-propylene-rubber). La fraction massique
en phase élastomère est dans ce cas de 22%. Elle est formée de 50% d’éthylène
et de 50% de propylène. Le mélange est obtenu direct-réacteur par extrusion de
l’éthylène et du propylène.
Pour plus de commodité, les particules élastomères dispersées dans les matrices seront
désignées sous le terme générique de ‘nodules d’EPR et d’EPDM’. Cependant, elles
possèdent généralement une structure composite. En effet, elles sont pour la plupart
constituées d’une écorce amorphe en EPR ou EPDM et d’un cœur de PE semi-cristallin. La
géométrie des nodules est globalement sphérique, même si certains sont regroupés en
agglomérats (Figure I.1). Des particules de tailles inférieures à la moyenne, constituées d’EPR
ou d’EPDM pur, sont également observables.
82

Chapitre I : Matériaux et techniques expérimentales
Figure I. 1: Cliché MET des matrices (PP/EPR) et (PP/EPDM/talc)
Pour pouvoir interpréter l’effet de chaque constituant de mélange, nous avons utilisé
une matrice polypropylène sans élastomères commercialisés par ISPLEN® de référence PP
050 G1E.
Les caractéristiques données sur les fiches techniques des trois produits sont
regroupées sur le tableau I.1 : 2µm 5µm
Tableau I. 1: Les caractéristiques des matrices PP, PP108MF97 et PP7510
Propriétés Unités PP PP/EPDM/talc
PP7510 PP/EPR
PP108MF97 Méthodes
de test MFR à 230°C et 2,16
kg g/10 min 5,8 17 10 ISO 1133
Densité Kg/m3 905 960 905 ISO 1183 Contrainte maximale
Contrainte à la rupture Déformation à la
rupture
MPa MPa
%
35 - 8
18 15 100
19 18 500
ISO527
Module de flexion MPa 1500 1300 950 ASTM D 790
Résistance aux chocs entaillée à 23°C à 0°C
à –20°C
KJ/m2 -
4,4 - -
Non rompu Non rompu
10
Non rompu Non rompu
10
ISO 180/4A
Dureté Shore D - 68 60 62 ISO 868 Température VICAT
(10N) °C - 130 145 ISO 306/A
83

Chapitre I : Matériaux et techniques expérimentales
I-1-2. Les compatibilisants :
L’interface dans les mélanges de polymères non miscibles est d’une importance
capitale. Les interactions physiques et chimiques, aux limites des phases, contrôlent
l’ensemble des performances des mélanges. D’une interaction forte résulte une bonne
adhésion et un transfert de contrainte efficace de la phase continue à la phase dispersée. Bien
que l’adhésion interfaciale contrôle essentiellement les propriétés mécaniques, telles que la
résistance aux chocs et à la déformation, les caractéristiques rhéologiques, les conditions de
transformation et la résistance chimique sont aussi affectées par le mouillage et les
phénomènes d’absorption associés à l’adhésion.
Les méthodes d’amélioration de l’adhésion entre phases non miscibles, impliquent
l’ajout d’un tiers corps qui assure le contrôle de la morphologie et la stabilité des systèmes bi-
phasiques. Ces produits ont différentes appellations :
- Agents de couplage,
- Promoteurs d’adhésion,
- Compatibilisants,
- Agents interfaciaux.
Les agents de compatibilisation que nous avons utilisés ont été choisis en fonction des
interactions qu’ils sont susceptibles d’engendrer avec les constituants des mélanges PP/EPR et
PP/EPDM/talc que nous comptons élaborer par la suite. Ce sont des copolymères
commercialisés sous les références suivantes :
- L’Exxelor VA 1801, commercialisé par Exxon Mobil®, est chimiquement un EPR-g-
MAH (élastomère EPR greffé anhydride maléique). Il contient 69.6%wt d’éthylène,
29,8%wt de propylène et 0,6%wt d’anhydride maléique.
84
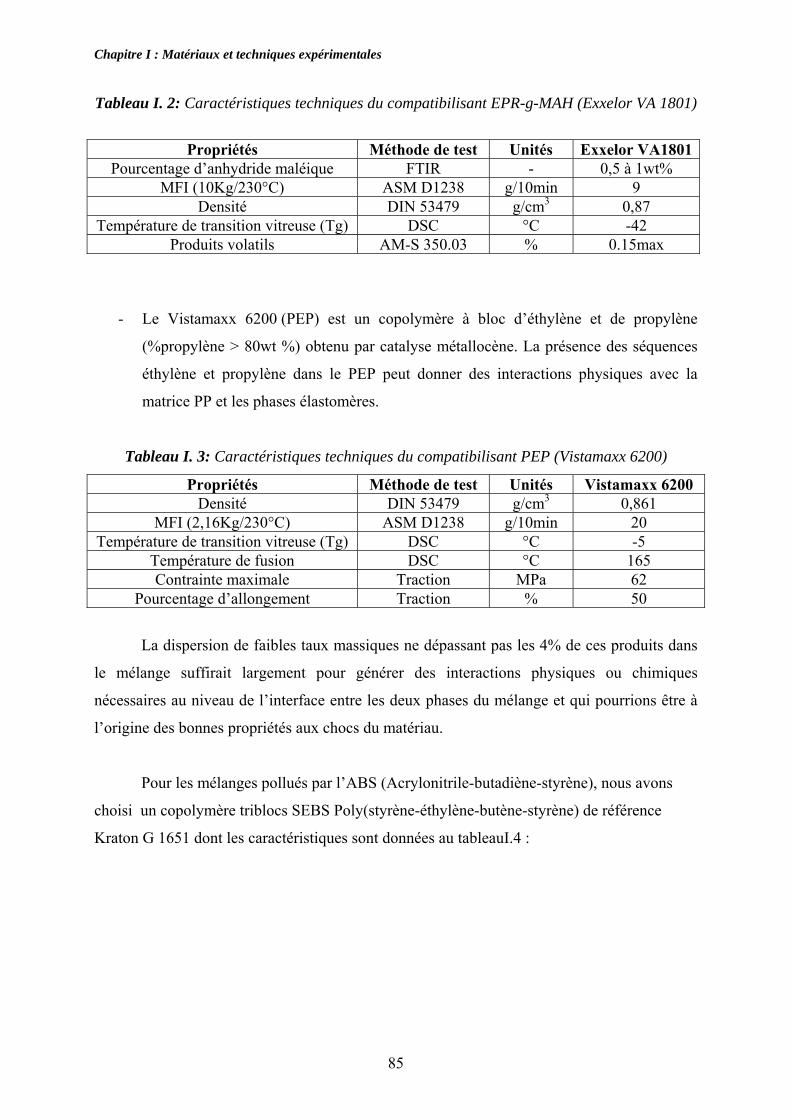
Chapitre I : Matériaux et techniques expérimentales
Tableau I. 2: Caractéristiques techniques du compatibilisant EPR-g-MAH (Exxelor VA 1801)
Propriétés Méthode de test Unités Exxelor VA1801
Pourcentage d’anhydride maléique FTIR - 0,5 à 1wt% MFI (10Kg/230°C) ASM D1238 g/10min 9
Densité DIN 53479 g/cm3 0,87 Température de transition vitreuse (Tg) DSC °C -42
Produits volatils AM-S 350.03 % 0.15max
- Le Vistamaxx 6200 (PEP) est un copolymère à bloc d’éthylène et de propylène
(%propylène > 80wt %) obtenu par catalyse métallocène. La présence des séquences
éthylène et propylène dans le PEP peut donner des interactions physiques avec la
matrice PP et les phases élastomères.
Tableau I. 3: Caractéristiques techniques du compatibilisant PEP (Vistamaxx 6200)
Propriétés Méthode de test Unités Vistamaxx 6200 Densité DIN 53479 g/cm3 0,861
MFI (2,16Kg/230°C) ASM D1238 g/10min 20 Température de transition vitreuse (Tg) DSC °C -5
Température de fusion DSC °C 165 Contrainte maximale Traction MPa 62
Pourcentage d’allongement Traction % 50
La dispersion de faibles taux massiques ne dépassant pas les 4% de ces produits dans
le mélange suffirait largement pour générer des interactions physiques ou chimiques
nécessaires au niveau de l’interface entre les deux phases du mélange et qui pourrions être à
l’origine des bonnes propriétés aux chocs du matériau.
Pour les mélanges pollués par l’ABS (Acrylonitrile-butadiène-styrène), nous avons
choisi un copolymère triblocs SEBS Poly(styrène-éthylène-butène-styrène) de référence
Kraton G 1651 dont les caractéristiques sont données au tableauI.4 :
85

Chapitre I : Matériaux et techniques expérimentales
Tableau I. 4: Les caractéristiques du Kraton G 1651
Caractéristiques Valeurs
Bloc final Polystyrène
Blocs centraux Poly(éthylène/butylène)
T° de mise en œuvre 190-260°C
Viscosité (200°C) 9.103 PI
Dans le cas des mélanges contenant du polyuréthane (chapitre IV), nous avons utilisé
deux terpolymères commercialisés par Arkema® : le Lotader AX3210 et le Lotader AX 8900.
Le Lotader AX3210 est un terpolymère d’éthylène (E), d’ester acrylique (EA) et
d’Anhydride Maléique (MAH). Quelques caractéristiques sont données au tableau I.5.
La formule chimique du Lotader AX3210 est la suivante:
Tableau I. 5: Les caractéristiques du Lotader AX3210
Caractéristiques Valeurs Unité Méthode de test
Indice de fluidité à chaud (190°C/2,16 kg)
5 g/10 min ASTM D 1238 / ISO 1133
Pourcentage de copolymère 9 % IR
Point de fusion 107 °C DSC
Module d’élasticité en flexion 120 MPa ASTM D 790 / ISO 178
Déformation au seuil d’écoulement
600 % ASTM D 638/ISOR527
86

Chapitre I : Matériaux et techniques expérimentales
Le Lotader AX8900 est un terpolymère d’éthylène (E), d’ester acrylique (AE) et
d’ester de méthacrylate de glycidyle (GMA) obtenu par un processus de polymérisation à
haute pression. La formule chimique du produit est la suivante :
.
Quelques caractéristiques du terpolymères (E-EA-GMA) sont représentées au tableau
I.6 :
Tableau I. 6: Les caractéristiques du Lotader AX8900
Caractéristiques Valeurs Unité Méthode de test
Indice de fluidité à chaud 6 g/10 min ASTM D 1238 / ISO
Pourcentage ester acrylique 24 % Méthode ATOFINA
Pourcentage de GMA 8 % Méthode ATOFINA
Point de fusion 60 °C DSC
Allongement à la rupture 1100 % ASTM D 638 Type IV
I-1-3. Les nanocharges CaCO3:
Les morphologies des nano-CaCO3 généralement utilisés dans les polymères sont
représentées sur la figure I.2. Les nano-CaCO3 peuvent être soit des calcites, soit des
aragonites ; Les nano-CaCO3 utilisés dans cette étude sont des calcites de morphologie
représentée sur la figure I.2.a.
87

Chapitre I : Matériaux et techniques expérimentales
Calcite Aragonite
(a)
(b)
(c)
Figure I. 2: Morphologie de la gamme de nano-CaCO3 proposée par SOLVAY®
Les caractéristiques techniques des nano-CaCO3 commercialisés par Solvay® sont
données dans le tableau I.7.
Tableau I. 7: Caractéristiques des nano-CaCO3
Ø des particules Traitement de surface Surface spécifique
Socal 322V 30-50nm Acide stéarique (3.5wt%)
Ratio d’enrobage = 1,1 mg/m²
25 m2/g
Winnofil-SPM 100nm Acide stéarique (2.5wt%)
Ratio d’enrobage = 1,5 mg/m²
20m2/g
I.1.4. Les polluants:
I.1.4.1. Le polyamide (PA) et l’acrylonitrile butadiène styrène (ABS) :
Les deux premiers polluants utilisés sont :
L’acrylonitrile butadiène styrène (ABS) fourni par Lustran Polymer® (Lustran ABS
2710) ayant les propriétés présentés sur le tableau I.8 :
88
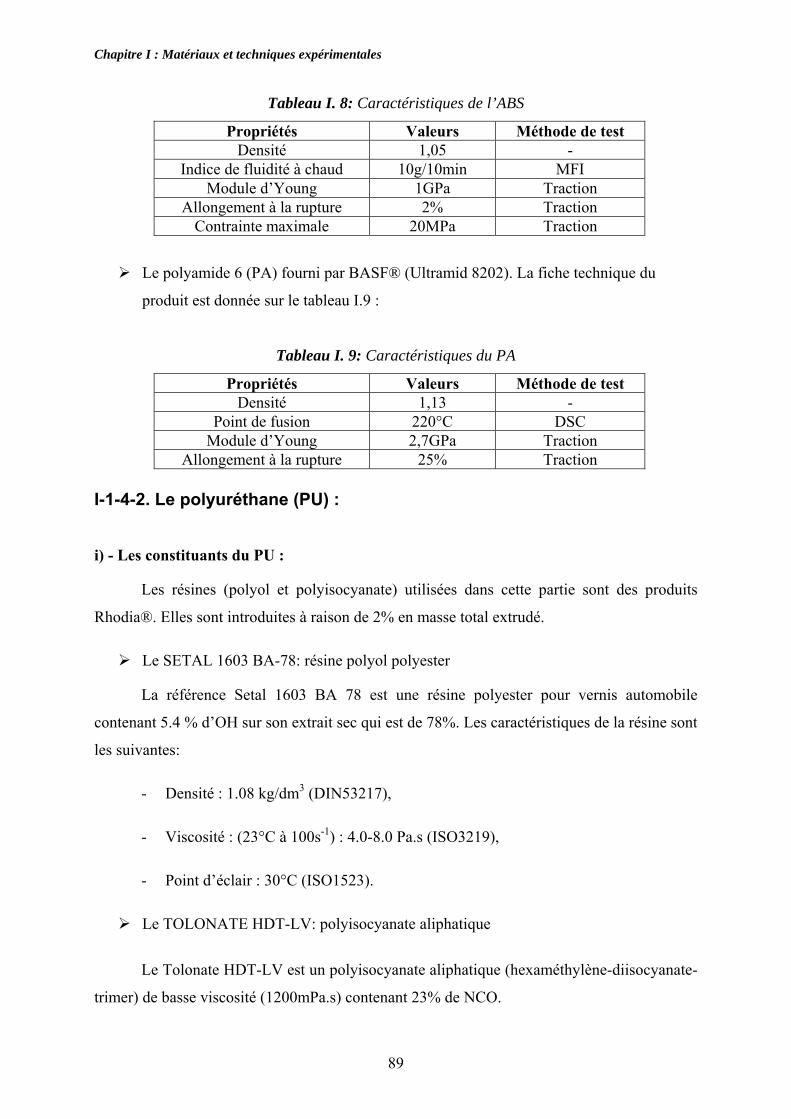
Chapitre I : Matériaux et techniques expérimentales
Tableau I. 8: Caractéristiques de l’ABS
Propriétés Valeurs Méthode de test Densité 1,05 -
Indice de fluidité à chaud 10g/10min MFI Module d’Young 1GPa Traction
Allongement à la rupture 2% Traction Contrainte maximale 20MPa Traction
Le polyamide 6 (PA) fourni par BASF® (Ultramid 8202). La fiche technique du
produit est donnée sur le tableau I.9 :
Tableau I. 9: Caractéristiques du PA
Propriétés Valeurs Méthode de test Densité 1,13 -
Point de fusion 220°C DSC Module d’Young 2,7GPa Traction
Allongement à la rupture 25% Traction I-1-4-2. Le polyuréthane (PU) : i) - Les constituants du PU :
Les résines (polyol et polyisocyanate) utilisées dans cette partie sont des produits
Rhodia®. Elles sont introduites à raison de 2% en masse total extrudé.
Le SETAL 1603 BA-78: résine polyol polyester
La référence Setal 1603 BA 78 est une résine polyester pour vernis automobile
contenant 5.4 % d’OH sur son extrait sec qui est de 78%. Les caractéristiques de la résine sont
les suivantes:
- Densité : 1.08 kg/dm3 (DIN53217),
- Viscosité : (23°C à 100s-1) : 4.0-8.0 Pa.s (ISO3219),
- Point d’éclair : 30°C (ISO1523).
Le TOLONATE HDT-LV: polyisocyanate aliphatique
Le Tolonate HDT-LV est un polyisocyanate aliphatique (hexaméthylène-diisocyanate-
trimer) de basse viscosité (1200mPa.s) contenant 23% de NCO.
89

Chapitre I : Matériaux et techniques expérimentales
ii)- L’élaboration du PU :
Le polyuréthane a été synthétisé au laboratoire selon la formulation obtenue par
mélange des parties A et B et une cuison à 60°C :
- Partie A : 487g résine Setal 1603 (ES = 78% et %OH sur ES = 5,4%) + 62g acétate
de butyle + 18g de DBTL (dilaurate de dibutyle étain) à 1 % dans acétate de
butyle.
- Partie B : 220g Tolonate HDT LV (ES = 100% et %NCO = 23%) + 60g acétate de
butyle.
Calcul du ratio NCO/OH:
- Pour 487g de Setal, 78% d’extrait sec et 5,4% de OH sur l’ES, on obtient alors
1,2066 mol de OH,
- Pour 220g de Tolonate, 100% d’ES et 23% de NCO, on obtient donc 1,2047mol
NCO.
Le rapport: NCO/OH = 0,998
Le PU est analysé par FTIR et les résultats obtenus montrent que tous les NCO ont
réagi mais pas tous les OH.
La plaque obtenue est découpée en morceaux de taille moyenne 5x5x5mm3 et est
mélangée avec lesautres contituants de mélanges à l’état solide.
90

Chapitre I : Matériaux et techniques expérimentales
I-2. Techniques expérimentales:
I-2-1. Mise en œuvre des mélanges:
La réalisation des mélanges a été effectuée dans une extrudeuse bi-vis CLEXTRAL
BC-21 équipée de deux vis modulaires corotatives à filets interpénétrés et conjugués. Ses
caractéristiques sont les suivantes :
- Diamètre des vis : 25mm
- Entraxe : 21mm
- Longueur des vis : 900mm (L/D = 36)
I-2-1-1. Profil de vis et de température :
Le profil de vis choisi est celui de la figure I.3. Ce profil est utilisé pour l’extrusion de
mélanges Polyoléfines/CaCO3:
Matrices et compatibilisants
NanochargesCaCO3
(L = 36 D = 900mm) VIS 60° VIS 30°
Zone de Malaxage °
Figure I.
La pre
une zone de
VIS 45
3: Profils des vis et de température utilisés pour la réalisation de mélanges
mière zone de transport du polymère relativement longue (8 D), se termine par
fluidification/compression plus progressive, résultant d’un compromis entre les
91

Chapitre I : Matériaux et techniques expérimentales
profils nécessaires pour le déplacement de la matière et le malaxage des constituants du
mélange.
Une zone de malaxage assure une bonne fluidification et un prémélangeage.
L’efficacité de cette première zone de malaxage dans la dispersion a été constatée par Ess et
Hornsby [1], pour les mélanges de polypropylène avec du carbonate de calcium. Il semble que
la coexistence de polymère fondu et non fondu dans cette zone à haut cisaillement soit
responsable d’une dispersion des agglomérats du composant minoritaire.
La matière fondue subit ensuite une nouvelle séquence de décompression /
compression avec malaxage, nécessaire pour assurer une bonne dispersion. L’efficacité de ce
type de profil a été démontrée par Poitou [2], dans le cas de mélanges polyamide-élastomère.
Ce dernier a en effet constaté qu’en fonctionnement normal, l’espace entre les vis et le
fourreau n’est rempli que dans les zones qui précèdent soit une zone de malaxage à pas
inverse, soit la filière. Globalement c’est la succession de zones remplies et décomprimées qui
permet d’améliorer la dispersion.
D’après Poitou [2], du point de vue efficacité du mélangeage, il est également
préférable d’utiliser des zones de malaxages courtes ou moyennes, séparées par une portion
de vis normale plutôt qu’une zone de malaxage très longue.
La matière pénètre ensuite dans une zone de décompression où s’effectue un premier
dégazage, de manière à éliminer les composés volatils indésirables tels que : H2O vapeur,
monomères résiduels,…
A partir de la zone 5, on soumet la matière à un travail de pression, avant passage par
une nouvelle zone de malaxage suivie par une zone à pas inverse destinée à retenir plus
longtemps la matière dans la zone qui la précède. La matière pénètre ensuite dans une zone de
décompression où s’effectue un 2ème dégazage, afin de procéder à une élimination plus
poussée des produits volatils indésirables.
Enfin, le mélange est remis en pression avant la filière avec passage à travers une
dernière zone de malaxage. Cette partie de la vis (zone terminale) semble avoir un rôle
important qui est d’homogénéiser le produit sans réduire la taille des particules.
92

Chapitre I : Matériaux et techniques expérimentales
I-2-1-2. Vitesse de rotation et de remplissage :
Dans une extrudeuse bivis, le débit est essentiellement fonction du dosage de la
matière à l’alimentation et il est seulement limité par la puissance disponible pour
l’entraînement des vis. L’ajustement peut être réalisé en diminuant le remplissage moyen de
la machine par augmentation de la vitesse de rotation.
D’autre part, le temps de résidence moyen varie à peu près linéairement avec la vitesse
de rotation ; le taux de remplissage est donc proportionnel au rapport débit sur vitesse de
rotation. A vitesse constante, une augmentation du débit (donc du remplissage), induit une
plus grande efficacité du mélangeage, du fait d’un travail mécanique plus important.
On constate aussi, qu’à taux de remplissage constant, une augmentation de la vitesse
de rotation est aussi un facteur d’amélioration de la qualité de mélangeage. Malheureusement,
du fait de la limitation de puissance pour l’entraînement de la vis, les deux exigences sont
contradictoires : on ne peut augmenter simultanément à volonté vitesse de rotation et taux de
remplissage et il faut trouver un compromis.
Le malaxage des constituants a été réalisé avec une vitesse de 200 tours par minute et
un taux de remplissage (fixé par le dosage de la matière à l’alimentation) assurant un
fonctionnement de la machine à 80% environ de sa puissance maximale.
La vitesse de rotation et le dosage à l’alimentation ont été maintenus presque constants
pour tous les mélanges.
93
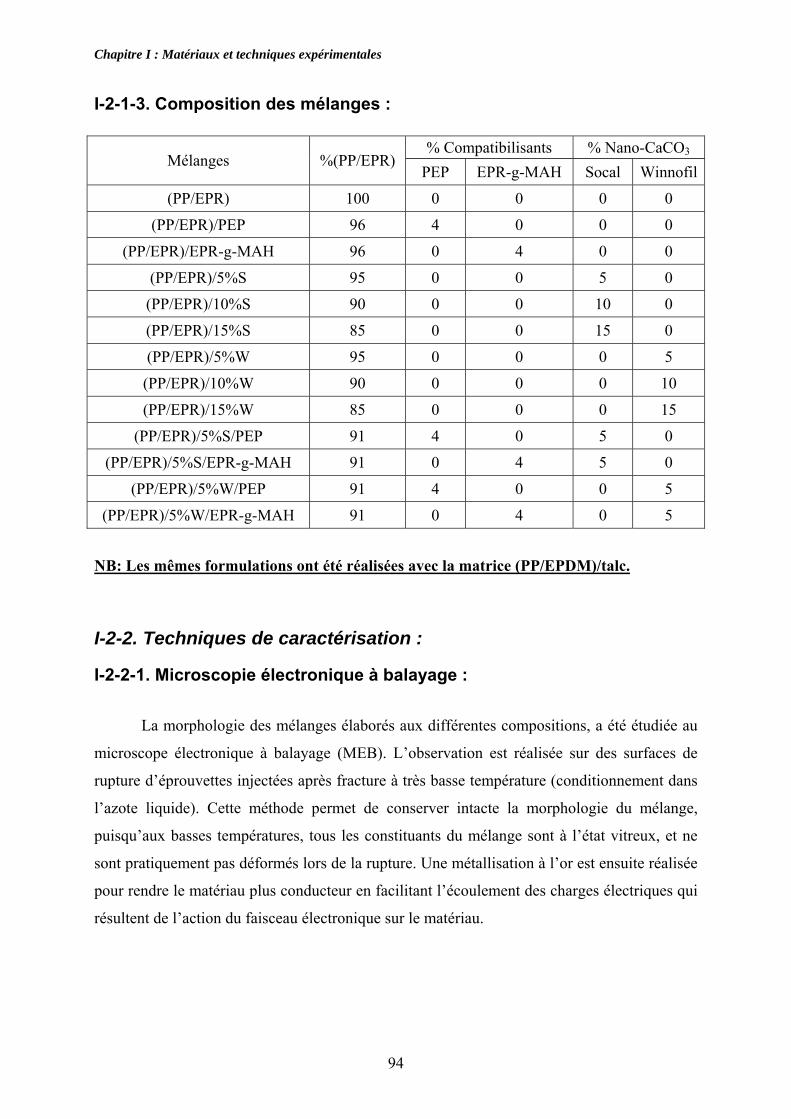
Chapitre I : Matériaux et techniques expérimentales
I-2-1-3. Composition des mélanges :
% Compatibilisants % Nano-CaCO3Mélanges %(PP/EPR)
PEP EPR-g-MAH Socal Winnofil
(PP/EPR) 100 0 0 0 0
(PP/EPR)/PEP 96 4 0 0 0
(PP/EPR)/EPR-g-MAH 96 0 4 0 0
(PP/EPR)/5%S 95 0 0 5 0
(PP/EPR)/10%S 90 0 0 10 0
(PP/EPR)/15%S 85 0 0 15 0
(PP/EPR)/5%W 95 0 0 0 5
(PP/EPR)/10%W 90 0 0 0 10
(PP/EPR)/15%W 85 0 0 0 15
(PP/EPR)/5%S/PEP 91 4 0 5 0
(PP/EPR)/5%S/EPR-g-MAH 91 0 4 5 0
(PP/EPR)/5%W/PEP 91 4 0 0 5
(PP/EPR)/5%W/EPR-g-MAH 91 0 4 0 5
NB: Les mêmes formulations ont été réalisées avec la matrice (PP/EPDM)/talc.
I-2-2. Techniques de caractérisation :
I-2-2-1. Microscopie électronique à balayage :
La morphologie des mélanges élaborés aux différentes compositions, a été étudiée au
microscope électronique à balayage (MEB). L’observation est réalisée sur des surfaces de
rupture d’éprouvettes injectées après fracture à très basse température (conditionnement dans
l’azote liquide). Cette méthode permet de conserver intacte la morphologie du mélange,
puisqu’aux basses températures, tous les constituants du mélange sont à l’état vitreux, et ne
sont pratiquement pas déformés lors de la rupture. Une métallisation à l’or est ensuite réalisée
pour rendre le matériau plus conducteur en facilitant l’écoulement des charges électriques qui
résultent de l’action du faisceau électronique sur le matériau.
94

Chapitre I : Matériaux et techniques expérimentales
I-2-2-2. Comportement viscoélastique à l’état solide :
Afin d’étudier le comportement thermomécanique des matériaux, nous avons
fréquemment recours à la spectrométrie mécanique dynamique qui s’avère relativement
sensible aux changements moléculaires du matériau. Dans le cas de nos mélanges, les
résultats obtenus par l’étude du comportement viscoélastique vont être interprétés en termes
de modification de la tension interfaciale et de la modification de l’adhésion entre les deux
phases. En effet, toutes les informations que l’on peut tirer de cette technique sont dans le cas
des mélanges, très sensibles aux variations de ces paramètres.
Nous cherchons donc, dans ce chapitre, à mettre en évidence l’influence des
compatibilisants (responsables de changements dans les propriétés morphologiques), ainsi que
leur effet sur l’adhésion. D’un autre côté, les modifications engendrées par la présence des
nano-CaCO3 sur le comportement viscoélastique des mélanges vont nous permettre de
déterminer l’influence de ces derniers sur les relaxations des différentes phases présentes dans
les mélanges.
L’étude des propriétés viscoélastiques à l’état solide a été réalisée au moyen d’un
viscoélasticimètre RHEOMETRIC RDA2. Les résultats présentés ci-après ont été obtenus en
torsion rectangulaire dynamique sur des éprouvettes parallélépipédiques dans la gamme de
température –80 à +180°C, à la vitesse de chauffage de 2K/min et à la pulsation de fréquence
de 6,28 rad/s.
a)- Rappels théoriques :
Contrairement aux mesures statiques, on applique au matériau une petite déformation
sinusoïdale (quelques pour-cent) qui aura pour expression :
ε* = ε0 . eiωt
Dans laquelle :
- ε0 est l’amplitude maximale de la déformation,
- ω est la pulsation de la fréquence.
95

Chapitre I : Matériaux et techniques expérimentales
A cette déformation périodique correspond une contrainte déphasée de la forme :
σ* = σ0 . ei(ωt+δ)
Dans laquelle :
- σ0: amplitude maximale de la contrainte,
- δ : déphasage.
Cette contrainte est déphasée d’un angle δ par rapport à la déformation du fait du
comportement partiellement visqueux du matériau.
Ainsi, le module de cisaillement est une valeur complexe :
G* = G’ + iG’’
L’angle de perte δ sera caractérisé par sa tangente donnée par :
tanδ = '''
GG = ''
'ηη
où η’ est la viscosité dynamique ou viscosité d’écoulement.
Les polymères cristallins manifestent typiquement trois relaxations indexées α, β et γ
par ordre décroissant des températures et qualifiés respectivement de sur-vitreuse, vitreuse et
sous-vitreuse. Pour les polymères amorphes, on trouve les deux types d’indexation : (β et γ)
ou (α et β), sachant qu’elles correspondent respectivement aux transitions vitreuse et sous-
vitreuse.
La transition β est associée à la transition vitreuse classique Tg des zones amorphes
des polymères semi-cristallins et correspond à la transition α (pour l’indexation α-β) des
matériaux purement amorphes ; elle est le reflet d’une relaxation intermoléculaire et la mise
en mouvement de 50 à 100 atomes de carbone dans la chaîne.
96

Chapitre I : Matériaux et techniques expérimentales
La transition γ (équivalente à la transition β pour des polymères entièrement amorphes
indexés α-β) est dite sous-vitreuse car elle apparaît pour des températures inférieures à la Tg.
Elle correspond à l’animation d’une dizaine d’atomes de carbone décrivant un mouvement de
manivelle (ou en villebrequin) autour de l’axe principal de la chaîne, conséquence d’un effet
intermoléculaire local dans des régions amorphes [3].
La transition α, située entre Tg et Tf pour les semi-cristallins, mais qui n’existe pas
pour des polymères amorphes, reste encore assez mal cernée. Elle est supposée mettre en jeu
des mouvements coopératifs intermoléculaires dans les régions cristallines [4]. Son intensité
ainsi que sa position sont fonction du taux de cristallinité et d’éventuelles modifications de la
structure des chaînes par suite d’un traitement thermique ou d’orientation ; sa position en
température est corrélable à la longueur des chaînes pendantes du cristal.
Deux types d’hypothèses sont proposés pour expliquer l’origine de cette relaxation α.
Elle pourrait résulter du cisaillement de la phase amorphe du matériau astreinte entre des
lamelles cristallines, ou bien du mouvement de rotation des chaînes autour de leur axe
longitudinal à l’intérieur du cristal.
Le PEHD est un cas particulier [4] car il peut être linéaire ou branché ou un mélange
des deux. Dans ces grades, la relaxation principale vers –110°C ne correspond pas seulement
à la transition vitreuse d’un polyéthylène linéaire mais aussi à la transition sous-vitreuse d’un
polyéthylène branché.
Enfin, il faut mentionner certains effets physico-chimiques pouvant aboutir à
l’apparition de pics inattendus supplémentaires. Ainsi, des copolymères blocs peuvent
manifester, outre les Tg de chacun de leurs composants, une troisième relaxation attribuable à
un composé intime intermédiaire ayant sa propre Tg. De même, une trompe à basse
température peut générer une morphologie métastable et ainsi aboutir à un pic unique au lieu
des deux pics classiques correspondant aux deux composants du système.
Nous rappelons dans le tableau I.10 les valeurs des diverses transitions du
polyéthylène, du polypropylène et de l’EPR que l’on rencontre dans la littérature [3 et 5-7] :
97

Chapitre I : Matériaux et techniques expérimentales
Tableau I. 10: Valeurs des diverses relaxations pour les PEHD, PP et les élastomères EPR
Matériaux Tγ (°C) Tβ (°C) Tα (°C)PEHD - -125
-100 à –110 -113
80 à 90
PP -80 -10 à +20 35 à 100 Elastomère éthylène
–propylène -120 -50 à -30 -
b)- Préparation des échantillons : Les échantillons utilisés pour l’étude de la viscoélasticité à l’état solide sont découpés
à partir de plaques obtenues par pressage à chaud (180°C), sous une pression de 30 bars et
pendant un temps de dix minutes dans un moule métallique revêtu de papier téflon. Le moule
est ensuite refroidi sous pression jusqu’à l’ambiante par circulation d’eau.
0,5 mm 1,5 mm
40 mm
Eprouvette utilisée pour les essais en RDA
I-2-2-3. Etude des propriétés thermiques par DSC :
L’analyse thermique englobe toute une série de techniques de caractérisation des
matériaux fondées sur l’étude de la variation d’une propriété physique en fonction de la
température.
Il s’agit donc essentiellement d’approches macroscopiques du comportement des
matériaux, qui font intervenir des considérations de thermodynamique des états d’équilibre et
des processus irréversibles, ainsi que de cinétique, associées aux changements d’états
(phénomènes de transition) et aux phénomènes de relaxation qui peuvent les accompagner.
Dans le cas spécifique des matériaux macromoléculaires, ou polymères, l’analyse de la
réponse thermique permet de mettre en évidence et donc de donner une interprétation
98

Chapitre I : Matériaux et techniques expérimentales
microscopique des phénomènes tels que la transition vitreuse, la fusion/cristallisation, le
vieillissement chimique ou physique, la ségrégation de phases…
Nous avons déterminé les températures de fusion des échantillons grâce à la
calorimétrie différentielle à balayage (DSC).
La température de l’échantillon est comparée en permanence à la température d’une référence.
L’échantillon et la référence sont placés dans le même four dont la température varie
linéairement avec le temps : on mesure alors ∆T (différence de température entre l’échantillon
et la référence) en fonction de la température du four. La référence est une capsule vide ; on
travaille sous balayage d’un gaz inerte (l’argon) pour éviter toute réaction du matériau à
analyser avec l’oxygène. Nous avons effectué des balayages en température allant de –100°C
à 200°C à 10°C/min. Ces mesures sont effectuées à l’aide d’un système DSC 30 de Mettler-
Toledo SA. L’appareillage est calibré avec de l’indium, du zinc et du plomb : la vérification
du calibrage se fait avec de l’indium (Tf = 156,6°C ; ∆H = 28,45 J/g). Des morceaux de
polymères sont prélevés (< 20mg de produit), et sont introduits dans une capsule en
aluminium de 40µl munie d’un couvercle percé, puis pesés avec une balance Mettler-Toledo
ayant une précision de ±1µg.
I-2-2-3. Caractérisation mécanique en traction :
Le comportement mécanique des mélanges a été réalisé à partir d’essais de traction
uniaxiale sur une machine type MTS 2/M à une vitesse de 500mm.min-1 pour les mélanges à
matrice (PP/EPR) et de 50mm.min-1 pour ceux dont la phase majoritaire est (PP/EPDM/talc)
et à une température de 22±0.5°C (éprouvettes haltères de longueur utile 25mm et de 0,5mm
d’épaisseur obtenues par injection) sur une presse à injection Battenfeld Unilog B2 ; 350 Plus
(Vitesse d’injection : 15mm/s, Maintien : 35/45 bars, Pression limite : 50 bars, Temps de
maintien : 15s).
I-2-2-4. Essais chocs :
La résistance aux chocs des différents mélanges a été étudiée grâce à des essais au
mouton-pendule charpy à une température de 22±0.5°C sur des éprouvettes injectées (figure
I.4) correspondant aux spécifications des normes (éprouvette dite « ISO1/2 »).
99

Chapitre I : Matériaux et techniques expérimentales
Les paramètres d’essais chocs sont :
- Poids = 0,917Kg,
- Longueur du pendule = 225mm,
- Vitesse = 2,93 m/s,
- Angle de chute = 160°,
- Hauteur de chute = 301,95mm.
Figure I. 4: Dimensions et forme de l'éprouvette Charpy en V
100

Chapitre I : Matériaux et techniques expérimentales
Références
[1] Ess J. W., Hornsby P. R. Twin-screw extrusion compounding of mineral filled
thermoplastics: dispersive mixing effects, Plastics and rubber processing and applications,
1987, vol 8, p147.
[2] Poitau A. Thèse de Doctorat, Ecole Nationale Supérieure des Mines de Paris, 1988.
[3] Doret C. Etude du recyclage du polypropylène issu des véhicules hors usage. Thèse
Université Jean-Monnet Saint Etienne, 1996, p 38-50.
[4] Matsuoka S. Relaxation phenomena in polymers. Hanser Publisher, 1992.
[5] Naira M. da Silva, Maria I. P. Ferreira and Maria Inês B. Tavares. Characterization of
EPDM/Atactic Polypropylene Blends, Polymer Testing, 1995, vol 14, p 329-341.
[6] McNally T. and McShane P. Rheology, phase morphology, mechanical, impact and
thermal properties of polypropylene/metallocene catalysed ethylene-1-octene copolymer
blends. Polymer, 2000, vol 43, p 3785-3793.
[7] Ana Lucia N. and Marisa C. G. Rocha. Rheological and Thermal Properties of Binary
Blends of Polypropylene and Poly(ethylene- co-1-octene). Journal of Applied Polymer
Science, 2001, Vol 79, p 1634–1639.
101

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
102

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
Chapitre II
Influence de compatibilisants sur les propriétés de mélanges
(PP/élastomères)/polluants
103

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
104

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
Chapitre II :
Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/‛polluants’
II-1. Introduction :
Ce premier chapitre est consacré à l’étude de mélanges (PP/élastomères) et
(PP/élastomères)/polluants et à l’influence de l’ajout de compatibilisants sur leurs propriétés
morphologiques, mécaniques (traction uniaxiale et choc Charpy), thermiques et
viscoélastiques à l’état solide.
Les copolymères compatibilisants présentés déjà dans le premier chapitre et utilisés dans
cette partie sont :
- un copolymère à bloc d’éthylène et de propylène appelé dans la suite PEP compatible
avec la matrice PP et ayant une affinité avec l’EPR et l’EPDM du fait de la présence
de séquences éthylène et propylène
- un élastomère EPR greffé anhydride maléique (EPR-g-MAH) utilisé pour la
compatibilisation du polluant polyamide (PA) et des nano-CaCO3.
- un copolymère triblocs Poly(styrène-éthylène-butène-styrène) (SEBS) utilisé pour la
compatibilisation des mélanges pollués ABS.
L’objectif de la deuxième partie de ce chapitre est d’étudier l’influence d’impuretés
qui pourraient se trouver en mélange avec les (PP/EPR) à cause des imperfections des
opérations de tri. Nous avons alors réalisé des mélanges avec les polluants modèles : ABS,
PA, PVC, élastomères, peintures PU, et de façon plus prospective, des nanocharges.
La mise en évidence expérimentale de la miscibilité et de la compatibilisation des
différents mélanges élaborés a été réalisée grace aux techniques de caractérisation: DSC
(analyse thermique différentielle), DMA (comportement rhéologique), MEB (microscopie
électronique à balayage), MET (microscopie électronique à transmission),…
105

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
II-2. Influence des compatibilisants sur les propriétés des mélanges (PP/EPR) et (PP/EPDM/talc)
Les morphologies possibles pour les mélanges de polymères sont très nombreuses;
leur description consiste en une définition de la distribution spatiale des constituants, de la
distribution des tailles et facteurs de forme (longueur/ épaisseur), de leur composition (des
phases) et de la nature des interfaces. Dans le cas des polymères incompatibles, le mélangeage
entraîne en général la formation d’une morphologie à deux phases, qui va gouverner les
propriétés finales du matériau [1, 2-4]. Les morphologies types souvent rencontrées dans ce
genre de mélanges, sont des structures nodulaires, fibrillaires ou co-continues. Les différentes
morphologies obtenues peuvent avoir des applications ciblées ; par exemple, les structures
nodulaires auront principalement des propriétés de résistance aux chocs exacerbées, alors que
les structures co-continues seront plutôt utilisées pour des propriétés de conduction.
II-2-1. Les observations morphologiques
II-2-1-1. Morphologie des matrices (PP/EPR) et (PP/EPDM/talc)
L’introduction de compatibilisants dans les mélanges (PP)/élastomères, dans cette
première partie de notre étude morphologique, a pour objectif :
1. La diminution de la tension interfaciale pour faciliter la dispersion (mouillage);
2. La stabilisation de la morphologie afin d'éviter l'évolution de celle-ci au cours des
étapes de transformation et de mise en œuvre du mélange;
3. L’augmentation de l'adhésion interfaciale entre phases à l'état solide notamment pour
favoriser le transfert de contrainte entre les phases et donc améliorer les propriétés
mécaniques du mélange.
Les figures II.1.(a) et (b) représentent respectivement les matrices (PP/EPR) et
(PP/EPDM/talc) ; Dans les deux cas, le faciès de rupture montre une morphologie de type
nodulaire, les phases élastomères EPR et EPDM sont dispersées dans la matrice PP sous
forme de particules sphériques (cas des nodules de petite taille) ou ellipsoïdales (cas des
nodules de grande taille). La répartition des particules d’élastomères est assez homogène dans
106

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
le mélange avec une distribution en taille des nodules respective de [0,2-0,5µm] pour la
matrice (PP/EPR) et [0,2 à 1,4µm] pour la matrice (PP/EPDM)/talc. D’autre part, on observe
sur le faciès de rupture de la figure II.1 (b) des plaquettes blanches de la charge semi-
renforçante (talc) présentes à raison de 10% en poids dans le PP ; elles sont réparties
uniformément dans le milieu. Les lamelles de talc se sont orientées dans le sens de
l’écoulement de la matière lors de la phase d’injection des éprouvettes à l’état fondu.
L’adhésion peut être évaluée qualitativement : si l’adhésion est très mauvaise, les
particules sont très distinctes de la matrice, et dans certains cas elles peuvent être délogées
lors de la rupture laissant ainsi des cavités à la place. Dans le cas contraire, les nodules restent
bien noyées dans la structure et on arrive difficilement à les distinguer de la matrice ; ces
particules ne présentent pas de vide à l’interface et peuvent même entraîner la formation
d’une interphase.
107

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
(a)
Matrice PP
Nodules EPR arrachés
Nodules EPR
Nodules EPDM
arrachés (b)
Plaquettes de talc
Matrice PP
Nodules EPDM
Figure II. 1: Morphologie des matrices (a) (PP/EPR) et (b) (PP/EPDM/talc)
Dans la suite de ce travail, nous nous intéressons surtout à la matrice (PP/EPR), les
résultats obtenus devant à priori pouvoir être extrapolés aux matériaux (PP/EPDM/talc).
II-2-1-2. Morphologie des mélanges compatibilisés
Comme nous l’avons signalé dans le paragraphe II-2-1-1, les mélanges PP/EPR (figure
II.2.(a) et (b)) présentent une morphologie nodulaire. La tension interfaciale élevée due à la
non miscibilité des constituants des mélanges est responsable des morphologies observées :
108

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
les nodules de grande taille permettent de minimiser l’énergie interfaciale (produit de la
tension interfaciale par l’aire de contact entre les phases) par réduction de la surface de
contact entre les deux matériaux ; bon nombre d’entre elles sont arrachées, laissant à la place
des cavités. Les nodules dispersés présentent des vides à l’interface, c’est l’exemple type
d’une décohésion interfaciale (mauvaise adhésion ou rupture interfaciale adhésive).
Afin de remédier à ce problème de mauvaise adhésion, nous avons ajouté des
copolymères compatibilisants à base d’éthylène et de propylène : PEP (copolymère d’éthylène
et de propylène) et EPR-g-MAH (copolymère d’éthylène et de propylène greffé anhydride
maléique). Les morphologies obtenues pour les mélanges (PP/EPR)/PEP et (PP/EPR)/EPR-g-
MAH sont présentés respectivement sur les figures II.2.(c et d) et II.2.(e et f). Les
micrographies ainsi obtenues montrent que les mélanges compatibilisés avec le copolymère
PEP présentent un faciès de rupture plus homogène avec une dispersion fine et uniforme des
particules EPR contenues dans la matrice PP. En effet, la taille des nodules de la phase
dispersée a été réduite par ajout du PEP (de [0,2-0,5µm] pour le mélange non compatibilisé à
[0,1-0,4µm] pour le mélange contenant le copolymère). D'autre part, le nombre de particules
de la phase dispersée est probablement plus important en raison de l'addition de 5% de
compatibilisant. Dans le cas des mélanges (PP/EPR)/EP-g-MAH (figure II.2.(e et f)), la
rupture est très fragile par rapport à la matrice (PP/EPR) et aux mélanges de (PP/EPR)/PEP.
Ce résultat est en accord avec l’hypothèse que les copolymères EPR-g-MAH forment une
structure core-shell (cœur-écorce) avec un noyau EPR et une écorce d’anhydride maléique
(figure II.2.(g)) [5]. La présence de cette écorce modifierait l'interface entre les phases de PP
et d’EPR et augmenterait le diamètre des particules d’environ 0,1µm initialement dans la
fourchette [0,2-0,5µm] pour le (PP/EPR) jusqu'à [0,3-0,6µm] pour les mélanges
(PP/EPR)/EPR-g-MAH.
109

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
(a) (b)
(d)(c)
(e) (f)
)
Figure II. 2: Morphologies des mélanges (a et b) (PP/Eet g) (PP/EPR)/EPR-g-MAH(x20
110
(g
PR), (c et d) (PP/EPR)/PEP and (e, f 00 and x7000)

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
II-2-2. Influence des compatibilisants sur les propriétés mécaniques des mélanges :
D’une façon générale, les propriétés mécaniques (module d’Young, allongement à la
rupture,…) des mélanges dépendent non seulement de celles des phases en présence et de
leurs fractions volumiques, mais aussi d’autres facteurs importants comme l’interaction entre
les constituants (compatibilisation et tension interfaciale), la morphologie des phases en
présence (dispersion et distribution de taille des particules dispersées), coefficient de
Poisson… puisque ces propriétés ne suivent pas une loi simple des mélanges en considérant
celles des deux constituants seuls.
Les observations morphologiques des mélanges (PP/EPR)/PEP et (PP/EPR)/EPR-g-
MAH révèlent une diminution de la taille de la phase dispersée EPR. En étudiant les mélanges
PP/Elastomères, un certain nombre d’auteurs rapportent que la tenue aux chocs à froid et plus
généralement la ductilité des systèmes renforcés sont inversement proportionnelles à la taille
des nodules de la phase dispersée [6-8] mais augmentent également avec le pourcentage
d’élastomère ajouté [9-10]. Pukanszky et al. [6] ont montré que les petits nodules
d’élastomères (<1µm) favorisent les déformations par bandes de cisaillement, tandis qu’une
morphologie à plus gros nodules privilégie le mécanisme de microcraquelure. Ces
constatations sont cohérentes avec les résultats obtenus lors de l’étude des propriétés
mécaniques (tableau II.1 et figures II.3 - II.4) où on observe qu’aux faibles déformations la
présence de compatibilisants ne modifie pas la valeur de la contrainte élastique. Par contre,
dans le domaine des grandes déformations, l’allongement et l’énergie à la rupture augmentent
considérablement, particulièrement dans le cas des mélanges (PP/EPR)/PEP.
Tableau II. 1: Propriétés mécaniques en traction uniaxiale et en choc Charpy entaillé V à
22±0.5°C
Mélanges Contrainte maximale
(MPa)
Pourcentage d’allongement
(%)
Energie à la rupture (J)
Résilience
(kJ/m2) (PP/EPR) 20,3±0,4 150±25 8,50±0,9 75±8
(PP/EPR)/PEP 20±0,4 330±30 16±0,8 90±10 (PP/EPR)/EPR-g-MAH 19,5±0,5 280±20 15,7±1,1 88±9
111
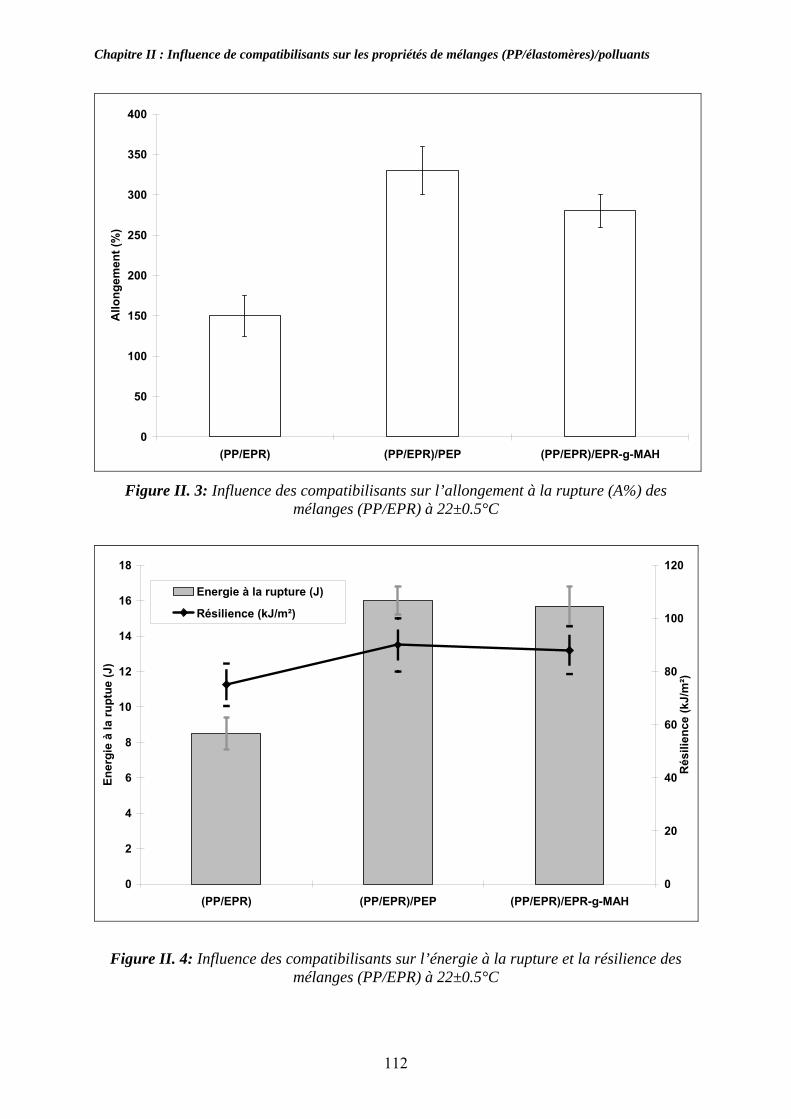
Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
0
50
100
150
200
250
300
350
400
(PP/EPR) (PP/EPR)/PEP (PP/EPR)/EPR-g-MAH
Allo
ngem
ent (
%)
Figure II. 3: Influence des compatibilisants sur l’allongement à la rupture (A%) des
mélanges (PP/EPR) à 22±0.5°C
0
2
4
6
8
10
12
14
16
18
(PP/EPR) (PP/EPR)/PEP (PP/EPR)/EPR-g-MAH
Ener
gie
à la
rupt
ue (J
)
0
20
40
60
80
100
120
Rés
ilien
ce (k
J/m
²)
Energie à la rupture (J)
Résilience (kJ/m²)
Figure II. 4: Influence des compatibilisants sur l’énergie à la rupture et la résilience des mélanges (PP/EPR) à 22±0.5°C
112

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
L’amélioration des propriétés mécaniques par l’ajout de compatibilisants
(particulièrement le copolymère PEP) résulte d’une adhésion plus importante à l’interface
entre les deux phases PP et EPR. Les résultats ainsi obtenus sont cohérents avec les
observations morphologiques (figures II.2.(a et b)). L’interface serait donc le siège
d’interactions importantes entre les deux phases du fait de la présence du compatibilisant. Ce
dernier se concentre préférentiellement à ce niveau lors de la phase d’élaboration des
mélanges à l’état fondu pour interdiffuser les blocs le constituant dans les phases
correspondantes par affinité thermodynamique. En effet, dans le cas des mélanges
(PP/EPR)/PEP une diminution de la taille de la phase dispersée EPR et une meilleure
adhésion à l’interface sont à l’origine d’une amélioration des propriétés mécaniques dans le
domaine des grandes déformations. Avec addition de PEP et d’EPR-g-MAH, la contrainte
maximale n’est pas modifiée mais le pourcentage d’allongement et l’énerge à la rupture sont
pratiquement doublés. La résilience est aussi légèrement augmentée avec ajout de PEP et
d’EPR-g-MAH.
II-2-3. Influence des compatibilisants sur les propriétés thermiques et viscoélastiques à l’état solide des mélanges :
II-2-3-1. Propriétés thermiques :
Dans le cas de nos mélanges, l’étude des propriétés thermiques par DSC de la matrice
PP/EPR (Figure II.5 et Tableau II.2) montre deux pics ; un premier pic exothermique vers
126°C qui correspond à la cristallisation du polypropylène. Un deuxième pic endothermique
(168°C) associé à la température de fusion du PP.
En présence de compatibilisants, les températures de cristallisation et de fusion du
polypropylène restent constantes. De même pour la distribution des tailles des cristallites. Par
contre, on remarque une variation de 8% de la cristallinité en présence d’EPR-g-MAH qui
joue peut être le rôle d’agent de nucléation.
Tableau II. 2 : Propriétés thermiques des mélanges
Mélanges TcPP(°C) TmPP(°C) ∆Hm(J/gPP) Cristallinité (χc) du PP(PP/EPR) 126 169 100 48
(PP/EPR)/PEP 127 169 102 49 (PP/EPR)/EPR-g-MAH 125 170 109 52
113
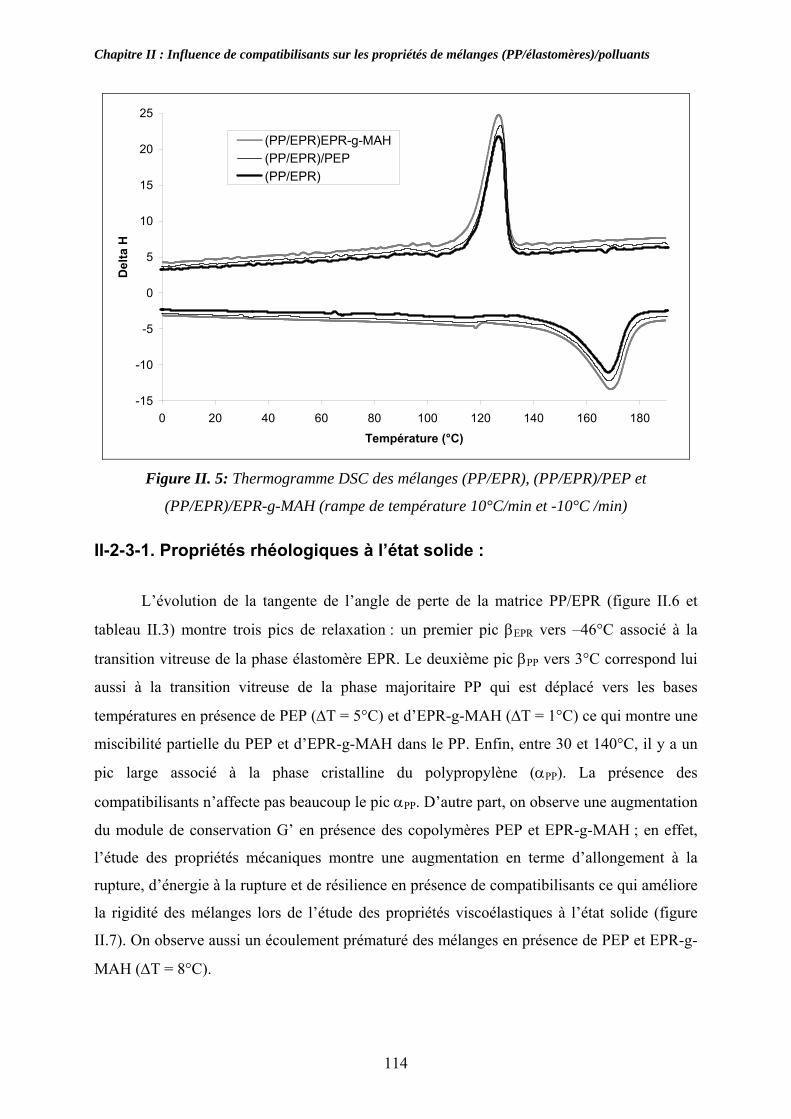
Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
-15
-10
-5
0
5
10
15
20
25
0 20 40 60 80 100 120 140 160 180
Température (°C)
Del
ta H
(PP/EPR)EPR-g-MAH(PP/EPR)/PEP(PP/EPR)
Figure II. 5: Thermogramme DSC des mélanges (PP/EPR), (PP/EPR)/PEP et
(PP/EPR)/EPR-g-MAH (rampe de température 10°C/min et -10°C /min)
II-2-3-1. Propriétés rhéologiques à l’état solide :
L’évolution de la tangente de l’angle de perte de la matrice PP/EPR (figure II.6 et
tableau II.3) montre trois pics de relaxation : un premier pic βEPR vers –46°C associé à la
transition vitreuse de la phase élastomère EPR. Le deuxième pic βPP vers 3°C correspond lui
aussi à la transition vitreuse de la phase majoritaire PP qui est déplacé vers les bases
températures en présence de PEP (∆T = 5°C) et d’EPR-g-MAH (∆T = 1°C) ce qui montre une
miscibilité partielle du PEP et d’EPR-g-MAH dans le PP. Enfin, entre 30 et 140°C, il y a un
pic large associé à la phase cristalline du polypropylène (αPP). La présence des
compatibilisants n’affecte pas beaucoup le pic αPP. D’autre part, on observe une augmentation
du module de conservation G’ en présence des copolymères PEP et EPR-g-MAH ; en effet,
l’étude des propriétés mécaniques montre une augmentation en terme d’allongement à la
rupture, d’énergie à la rupture et de résilience en présence de compatibilisants ce qui améliore
la rigidité des mélanges lors de l’étude des propriétés viscoélastiques à l’état solide (figure
II.7). On observe aussi un écoulement prématuré des mélanges en présence de PEP et EPR-g-
MAH (∆T = 8°C).
114
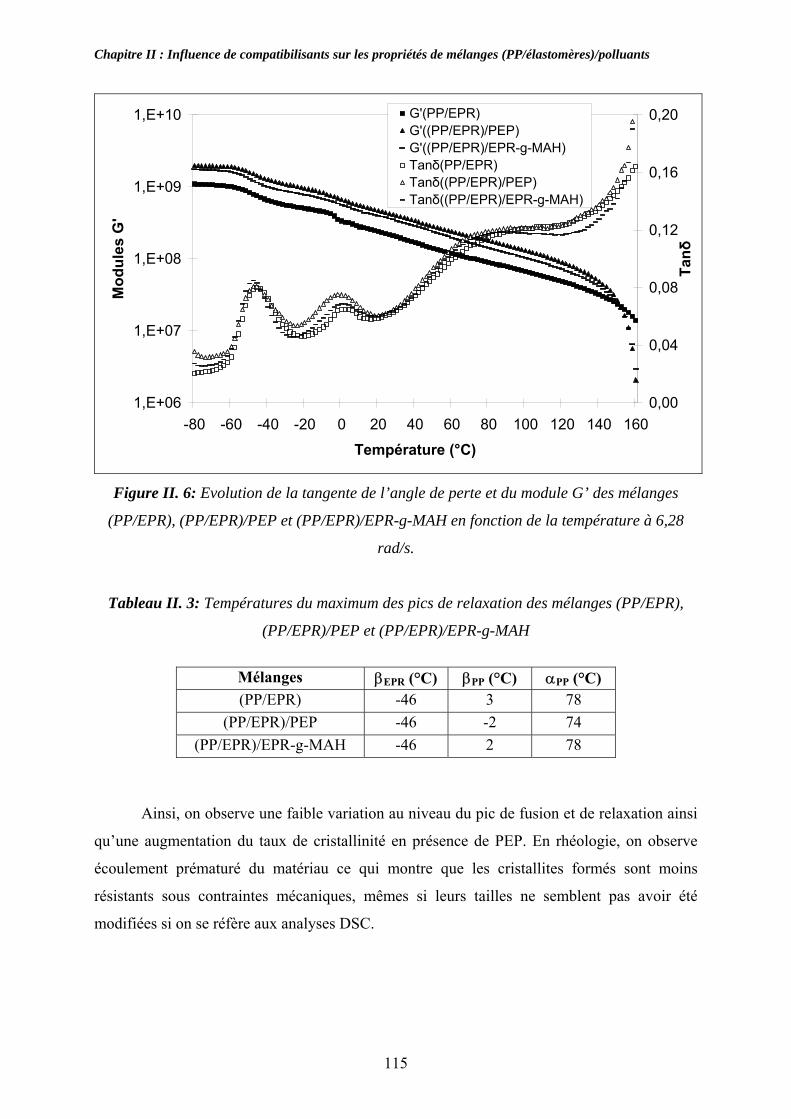
Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
1,E+06
1,E+07
1,E+08
1,E+09
1,E+10
-80 -60 -40 -20 0 20 40 60 80 100 120 140 160
Température (°C)
Mod
ules
G'
0,00
0,04
0,08
0,12
0,16
0,20
Tanδ
G'(PP/EPR)G'((PP/EPR)/PEP)G'((PP/EPR)/EPR-g-MAH)Tanδ(PP/EPR)Tanδ((PP/EPR)/PEP)Tanδ((PP/EPR)/EPR-g-MAH)
Figure II. 6: Evolution de la tangente de l’angle de perte et du module G’ des mélanges
(PP/EPR), (PP/EPR)/PEP et (PP/EPR)/EPR-g-MAH en fonction de la température à 6,28
rad/s.
Tableau II. 3: Températures du maximum des pics de relaxation des mélanges (PP/EPR),
(PP/EPR)/PEP et (PP/EPR)/EPR-g-MAH
Mélanges βEPR (°C) βPP (°C) αPP (°C) (PP/EPR) -46 3 78
(PP/EPR)/PEP -46 -2 74 (PP/EPR)/EPR-g-MAH -46 2 78
Ainsi, on observe une faible variation au niveau du pic de fusion et de relaxation ainsi
qu’une augmentation du taux de cristallinité en présence de PEP. En rhéologie, on observe
écoulement prématuré du matériau ce qui montre que les cristallites formés sont moins
résistants sous contraintes mécaniques, mêmes si leurs tailles ne semblent pas avoir été
modifiées si on se réfère aux analyses DSC.
115

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
II-3. Compatibilisation des mélanges pollués par l’ABS et le PA:
Comme nous l’avons déjà dit, le recyclage des pièces en plastique issues de véhicules
hors d'usage impose un tri par matière relativement coûteux. Il faut donc accepter de recycler
des polymères mélangés. Dans la grande majorité des cas, les polymères de natures
différentes sont immiscibles. Pour pallier l'importante perte constatée sur les propriétés
mécaniques de ces mélanges, l'utilisation d'agents compatibilisants est recommandée, car très
souvent efficace. Dans le cas présent, cependant, la faible quantité de polyamide ou d’ABS
(composants minoritaires considérés comme impuretées) introduite nécessite aussi l’ajout de
compatibilisants adéquats selon la nature et la structure chimiques des polluants. Les
présences simultanées de plusieurs polluants restent, dans ce cas, un problème important pour
la compatibilisation et le recyclage [11-14]. Le mélange se comporte plutôt comme un
polymère chargé que comme un mélange hétérogène de thermoplastiques. En conséquence,
l'introduction d'un agent compatibilisant provoque certes une émulsification et un
renforcement de la cohésion aux interfaces, mais ce dernier, qui conduisait, dans les mélanges
compatibles, à une amélioration des propriétés mécaniques globales conduit ici à une
fragilisation que l'on peut rapprocher de celles qui s'opère sur des composites tels que ceux
avec fibres de verre ensimées.
Dans le cas de mélanges PP/ABS, plusieurs types de copolymères ont été utilisés, à
savoir le PP-g-MAH [15], les copolymères d’éthylène–propylène [16] les copolymères
butadiène styrène [17], les copolymères d’éthylène et d’acétate de vinyle [18], le PP-g-acide
acrylique [19], le PP-g-2- HEMA (2- hydroxyéthyle méthacrylate) [20] et le SEBS [21]. Pour
les mélanges PP/PA, plusieurs auteurs utilisaient des copolymères fonctionnalisés anhydride
maléique pour la compatibilisation [22-23].
L’objectif de cette partie est alors d’étudier l’influence d’impuretés modèles type
acrylonitrile-butadiène-styrène (ABS) et polyamide (PA6), introduites artificiellement dans
les mélanges (PP/EPR); ceci afin d’étudier l’influence des polluants sur les propriétés de ces
mélanges. Nous avons introduit dans ces mélanges des taux de 5% de polluants (ABS et PA).
L’influence des polluants sur les propriétés finales des mélanges a été étudiée du point de vue
morphologie, propriétés viscoélastiques, des propriétés thermiques et propriétés mécaniques
en traction ou au choc.
116

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
Les compatibilisants utilisés dans cette partie et introduits à raison de 4% en masse sont :
- Le copolymère tribloc poly (styrène-ethylène-co-butylène-styrène) (SEBS) pour la
compatibilisation des mélanges contenant de l’ABS ;
- Le copolymère EPR-g-MAH décrit dans le premier chapitre utilisé pour les mélanges
(PP/EPR)/PA.
II-3-1. Observations morphologiques des mélanges :
La figure II.7.(a) et (b), représente respectivement les morphologies des mélanges
pollués ABS en absence et en présence de compatibilisant, On remarque que l’ABS est
parfaitement non miscible avec les constituants du mélange PP et EPR. Ainsi, ce polluant
modèle se présente sous forme de nodules ellipsoïdaux dispersés de taille comprise entre 2µm
et 10µm et présentant des vides à leur périphérie ; ceci traduit la tension interfaciale élevée de
entre l’ABS et les constituants du mélange. Son ajout entraîne principalement l’augmentation
du nombre de cavités vides correspondant à l’arrachement des nodules. Il est à noter que ces
vides provoquent une concentration de contraintes à leur niveau lors de l’application d’une
sollicitation et fragilisent ainsi le comportement mécanique du mélange en facilitant
l’amorçage des fissures. La présence du compatibilisant engendre une diminution de la taille
des particules ABS qui deviennent inférieures à 7µm et une amélioration de l’interface
(PP/EPR) et ABS. Ceci montre que le SEBS s’est localisé à l’interface entre la matrice et le
polluant, ce qui devrait conduire à de meilleures propriétés mécaniques.
Le deuxième polluant PA utilisé dans cette partie est aussi non miscible avec la
matrice (PP/EPR) et se présente sous forme de particules de faible adhésion avec les
constituants des mélanges (figure II.7.(c)). D’autre part, en comparant les deux faciès de
rupture des mélange (PP/EPR)/PA et (PP/EPR)/PA/EPR-g-MAH (figures II.7.(c) et II.7.(d)),
on remarque que la présence du compatibilisant EPR-g-MAH dans les mélanges pollués
(PP/EPR)/PA entraîne :
- une diminution de la taille des particules PA qui passent de [1-5µm] à [1-2µm],
- une rupture moins fragile et une meilleure continuité de la surface d’observation,
- une diminution du nombre de cavités vides qui correspondent à l’arrachement des
nodules de PA lors de la rupture.
117

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
(a) Particules ABS
Nodules EPR
Vides liés à l’arrachement
d’ABS
Matrice PP
Vides liés à l’arrachement d’EPR
(b)
Particules ABS Nodules EPR
Vides liés à l’arrachement d’EPR Matrice PP
Figure II. 7: Morphologie des mélanges (a) (PP/EPR)/ABS et (b) (PP/EPR)/ABS/SEBS
118

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
(c) Nodules EPR
Figure II. 7 (suite): Morphologie des mélanges (c) (PP/EPR)/PA et (d) (PP/EPR)/PA/EPR-g-
MAH
Les résultats ainsi obtenus viennent confirmer les travaux de plusieurs auteurs qui ont
étudié la morphologie après compatibilisation de mélanges PP/PA [24-26]. Holsti-Miettinen
et al. [24] utilisent la compatibilisation réactive en présence de SEBS-g-MAH. Jannerfeldt et
al. [25] ainsi que Byung et al. [27] comparent quant à eux l’effet du PP-g-MAH et PP-HBP
Particules PA
Matrice PP
Vides liés à l’arrachement de PA
Vides liés à l’arrachement d’EPR
(d) Nodules EPR
Vides liés à l’arrachement d’EPR
Particules PA
Vides liés à l’arrachement de PA
Matrice PP
119

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
(polypropylène hyperbranchés) sur les mélanges PP/PA. D’autres auteurs comme Yuchun Ou
et al. [28] se sont plutôt intéressés à l’ajout d’une phase élastomère fonctionnalisée (TPE-g-
MAH : élastomère thermoplastique greffé anhydride maléique) afin de pouvoir améliorer les
propriétés mécaniques aux chocs de ces mélanges : le compatibilisant TPE-g-MAH se localise
à l’interface entre les phases PP et PA en améliorant l’adhésion interfaciale (figure II.8).
Figure II. 8: Microstructure des mélanges PP/PA-6/TPE et PP/PA-6/PP-g-MAH [28]
II-3-2. Caractérisation mécanique des mélanges pollués:
L’étude des propriétés morphologiques des mélanges PP/ABS et PP/PA a montré que
les compatibilisants respectifs SEBS et EPR-g-MAH diminuent la taille de la phase dispersée
et semblent améliorer l’interface entre la matrice PP et les phases dispersées ABS et PA. Dans
cette partie, nous allons étudier le comportement mécanique des mélanges pollués/non
compatibilisés et pollués/compatibilisés. Les résultats obtenus sont présentés sur le tableau
II.4. Les figures II-9, II-10 et II-11 représentent respectivement la variation de la contrainte
maximale, de l’énergie à la rupture, l’allongement à la rupture A(%) et la résistance aux chocs
de ces mélanges.
Dans la suite de ce travail, les valeurs du module d’Young obtenues en traction ne
seront pas données. La vitesse de 500mm/min ne permet pas l’utilisation d’un extensiomètre.
Les résultats ainsi obtenus montrent que la présence des polluants ABS et PA affectent
considérablement l’ensemble des propriétés mécaniques en traction de la matrice (PP/EPR).
120

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
Cette diminution est plus importante dans le cas des mélanges (PP/EPR)/ABS et correspond à
une diminution de 60% pour l’énergie à la rupture, de 30% de la contrainte maximale et de
30% de l’allongement à la rupture A(%) ce qui confirme les oservations morphologiques où
nous avons observé une mauvaise interface entre la matrice et le polluant ABS. La présence
du compatibilisant SEBS dans les mélanges (PP/EPR)/ABS permet d’améliorer l’allongement
à la rupture A(%) et de la résilience mais sans autant pouvoir affecter l’énergie à la rupture.
Ceci confirme les observations morphologiques qui ont montré que le SEBS doit se localiser
entre les deux phases et améliorer l’interface matrice/ABS ce qui engendre une bonne
transmission des efforts entre les constituants des mélanges lors des essais mécaniques. Pour
les mélanges (PP/EPR)/PA/EPR-g-MAH, les résultats obtenus sur le tableau II.4 montrent
une augmentation de 12% de la valeur de l’allongement à la rupture A (%) et de celle de la
résilience conformément aux observations morphologiques qui ont aussi montré une bonne
adhésion à linterface (PP/EPR)/PA en présence de compatibilisant. Par contre, la valeur de
l’énergie à la rupture demeure inchangée même en présence du compatibilisant EPR-g-MAH.
Tableau II. 4: Propriétés mécaniques en traction uniaxiale et en chocs Charpy entaillé V à
22±0.5°C des mélanges pollués et pollués/compatibilisés
Mélanges Contrainte maximale
(MPa)
Allongement à la rupture
(%)
Energie à la rupture
(J)
Résilience (kJ/m2)
(PP/EPR) 20,3±0,4 150±25 8,5±0,9 75±8 (PP/EPR)/ABS 13,8±0,5 100±20 3,5±0,7 75±7
(PP/EPR)/ABS/SEBS 13,3±0,45 150±15 4,5±0,6 80±6 (PP/EPR)/PA 16±0,35 105±17 5,2±0,8 73±8
(PP/EPR)/PA/EPR-g-MAH 15,5±0,4 125±15 5,3±0,8 82±7
121
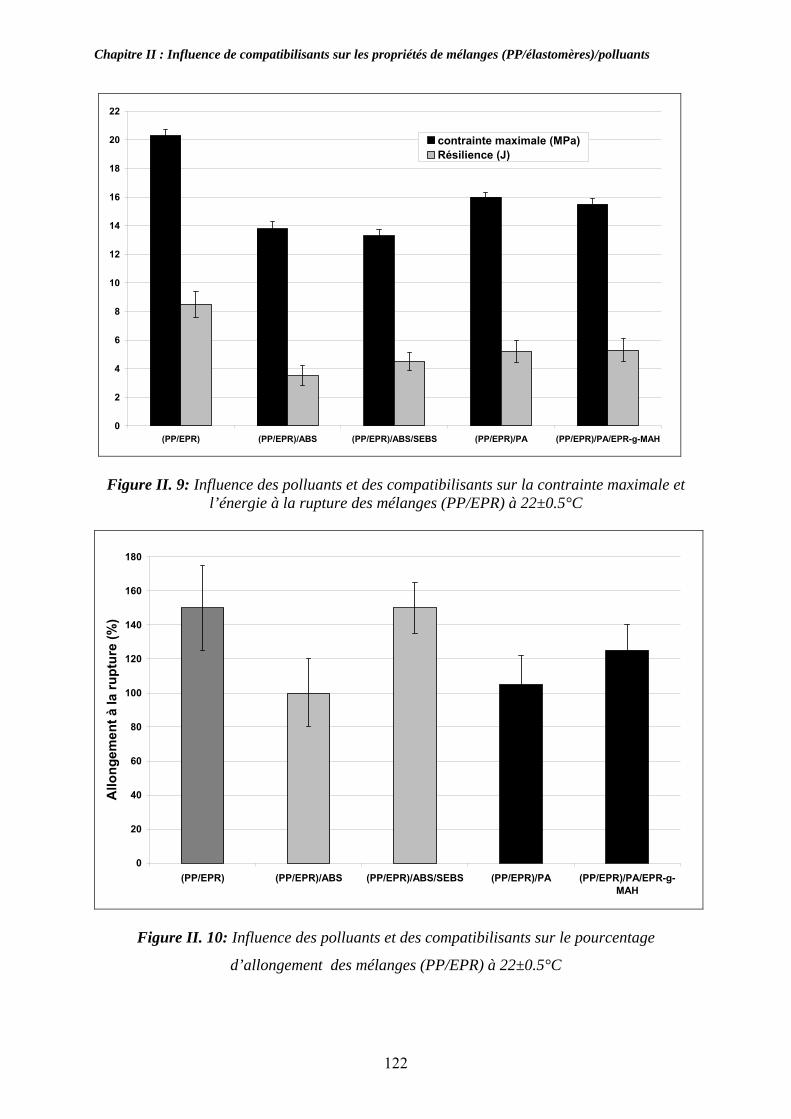
Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
0
2
4
6
8
10
12
14
16
18
20
22
(PP/EPR) (PP/EPR)/ABS (PP/EPR)/ABS/SEBS (PP/EPR)/PA (PP/EPR)/PA/EPR-g-MAH
contrainte maximale (MPa)Résilience (J)
Figure II. 9: Influence des polluants et des compatibilisants sur la contrainte maximale et l’énergie à la rupture des mélanges (PP/EPR) à 22±0.5°C
0
20
40
60
80
100
120
140
160
180
(PP/EPR) (PP/EPR)/ABS (PP/EPR)/ABS/SEBS (PP/EPR)/PA (PP/EPR)/PA/EPR-g-MAH
Allo
ngem
ent à
la ru
ptur
e (%
)
Figure II. 10: Influence des polluants et des compatibilisants sur le pourcentage
d’allongement des mélanges (PP/EPR) à 22±0.5°C
122
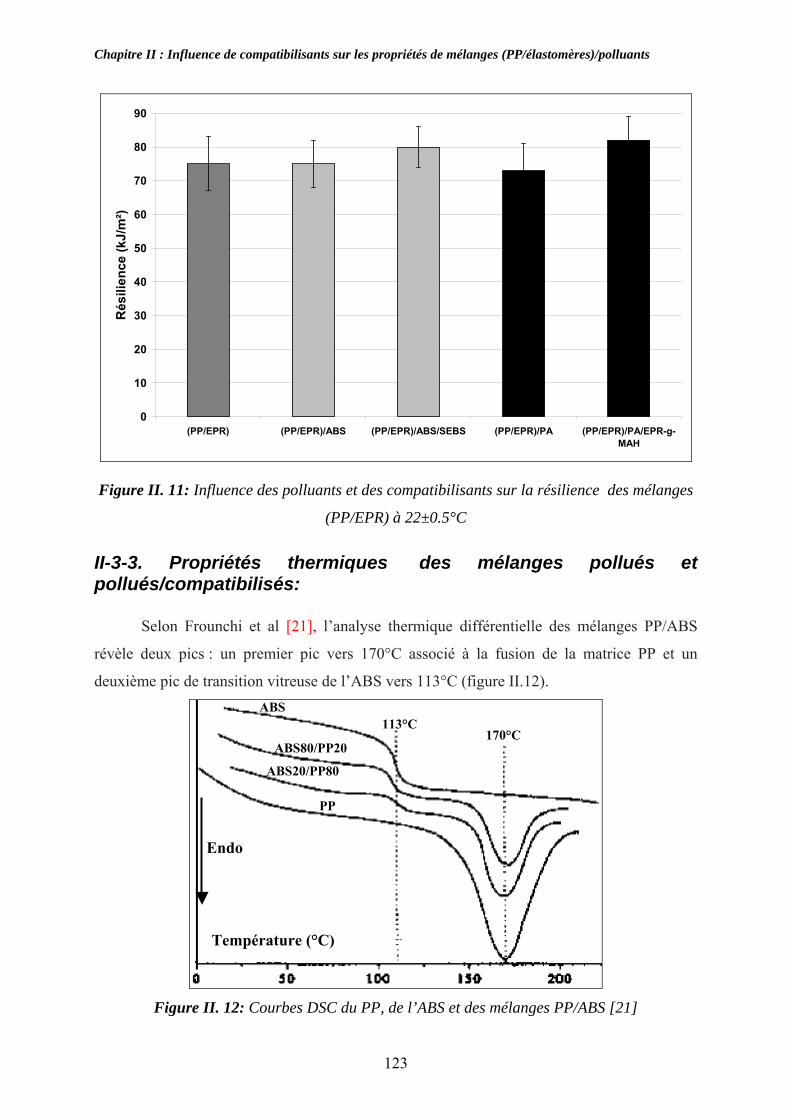
Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
0
10
20
30
40
50
60
70
80
90
(PP/EPR) (PP/EPR)/ABS (PP/EPR)/ABS/SEBS (PP/EPR)/PA (PP/EPR)/PA/EPR-g-MAH
Rés
ilien
ce (k
J/m
²)
Figure II. 11: Influence des polluants et des compatibilisants sur la résilience des mélanges
(PP/EPR) à 22±0.5°C
II-3-3. Propriétés thermiques des mélanges pollués et pollués/compatibilisés: Selon Frounchi et al [21], l’analyse thermique différentielle des mélanges PP/ABS
révèle deux pics : un premier pic vers 170°C associé à la fusion de la matrice PP et un
deuxième pic de transition vitreuse de l’ABS vers 113°C (figure II.12).
ABS 113°C
170°CABS80/PP20
ABS20/PP80
Endo
PP
Température (°C)
Figure II. 12: Courbes DSC du PP, de l’ABS et des mélanges PP/ABS [21]
123
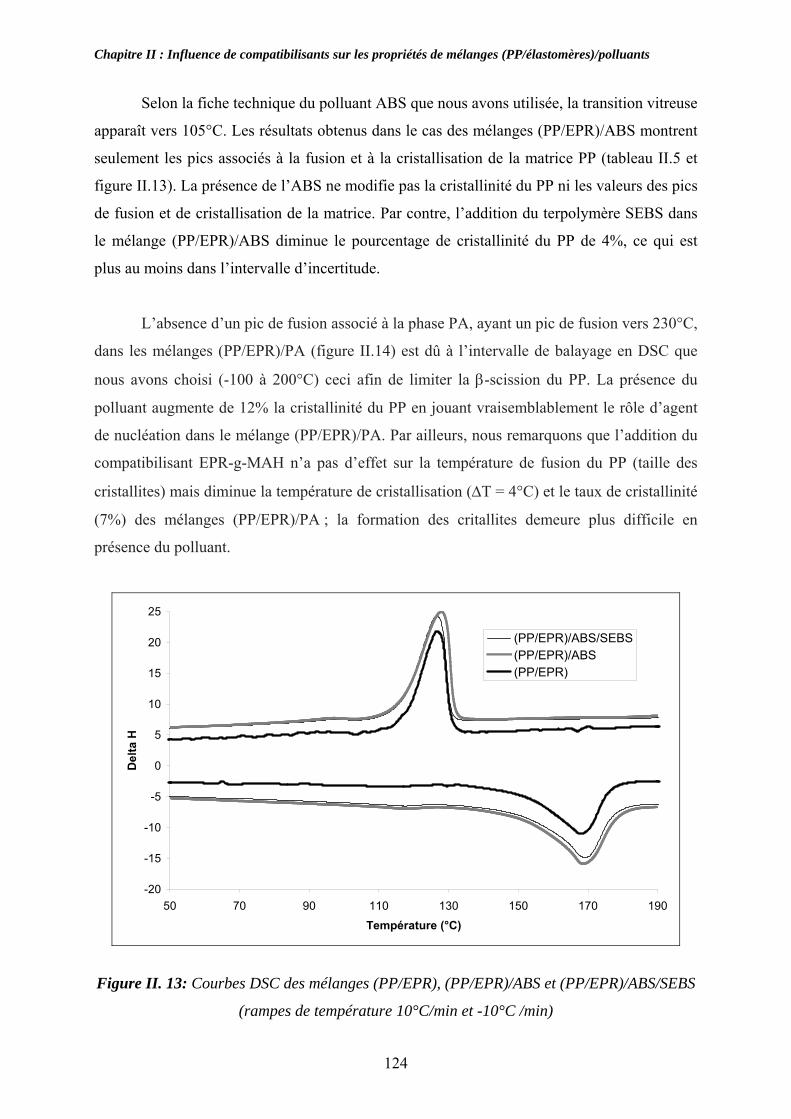
Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
Selon la fiche technique du polluant ABS que nous avons utilisée, la transition vitreuse
apparaît vers 105°C. Les résultats obtenus dans le cas des mélanges (PP/EPR)/ABS montrent
seulement les pics associés à la fusion et à la cristallisation de la matrice PP (tableau II.5 et
figure II.13). La présence de l’ABS ne modifie pas la cristallinité du PP ni les valeurs des pics
de fusion et de cristallisation de la matrice. Par contre, l’addition du terpolymère SEBS dans
le mélange (PP/EPR)/ABS diminue le pourcentage de cristallinité du PP de 4%, ce qui est
plus au moins dans l’intervalle d’incertitude.
L’absence d’un pic de fusion associé à la phase PA, ayant un pic de fusion vers 230°C,
dans les mélanges (PP/EPR)/PA (figure II.14) est dû à l’intervalle de balayage en DSC que
nous avons choisi (-100 à 200°C) ceci afin de limiter la β-scission du PP. La présence du
polluant augmente de 12% la cristallinité du PP en jouant vraisemblablement le rôle d’agent
de nucléation dans le mélange (PP/EPR)/PA. Par ailleurs, nous remarquons que l’addition du
compatibilisant EPR-g-MAH n’a pas d’effet sur la température de fusion du PP (taille des
cristallites) mais diminue la température de cristallisation (∆T = 4°C) et le taux de cristallinité
(7%) des mélanges (PP/EPR)/PA ; la formation des critallites demeure plus difficile en
présence du polluant.
-20
-15
-10
-5
0
5
10
15
20
25
50 70 90 110 130 150 170 190
Température (°C)
Del
ta H
(PP/EPR)/ABS/SEBS(PP/EPR)/ABS(PP/EPR)
Figure II. 13: Courbes DSC des mélanges (PP/EPR), (PP/EPR)/ABS et (PP/EPR)/ABS/SEBS
(rampes de température 10°C/min et -10°C /min)
124
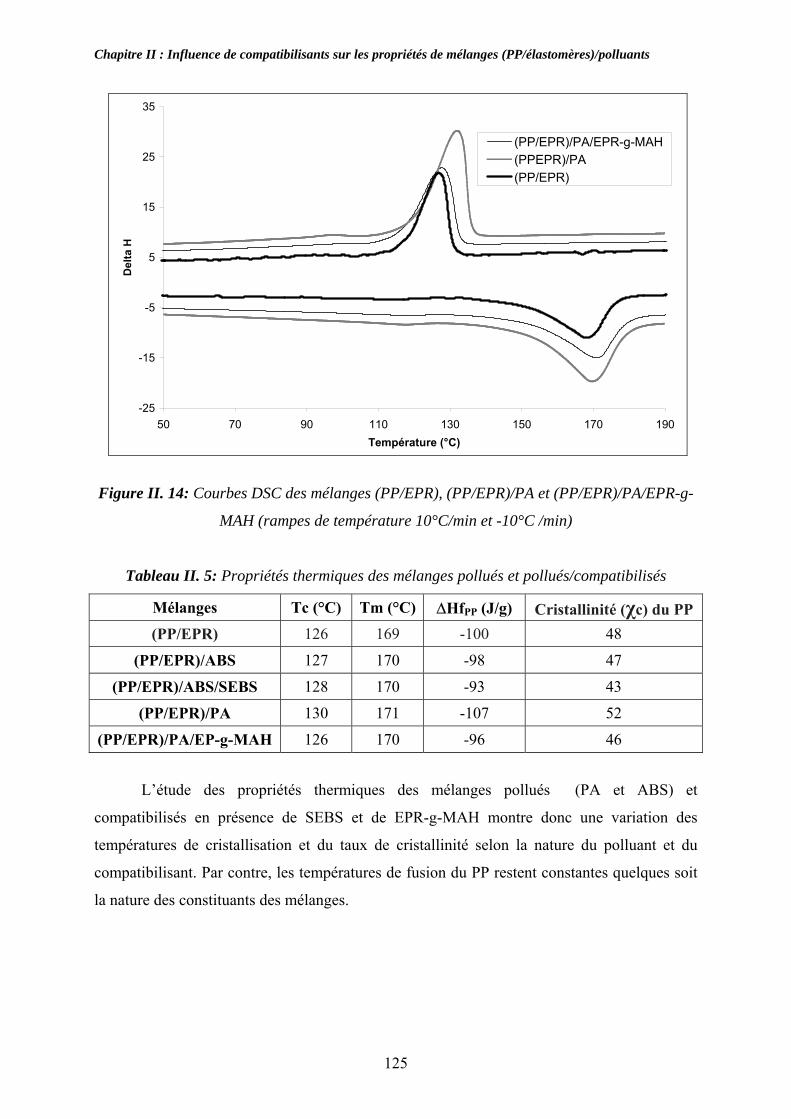
Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
-25
-15
-5
5
15
25
35
50 70 90 110 130 150 170 190Température (°C)
Del
ta H
(PP/EPR)/PA/EPR-g-MAH(PPEPR)/PA(PP/EPR)
Figure II. 14: Courbes DSC des mélanges (PP/EPR), (PP/EPR)/PA et (PP/EPR)/PA/EPR-g-
MAH (rampes de température 10°C/min et -10°C /min)
Tableau II. 5: Propriétés thermiques des mélanges pollués et pollués/compatibilisés
Mélanges Tc (°C) Tm (°C) ∆HfPP (J/g) Cristallinité (χc) du PP
(PP/EPR) 126 169 -100 48
(PP/EPR)/ABS 127 170 -98 47
(PP/EPR)/ABS/SEBS 128 170 -93 43
(PP/EPR)/PA 130 171 -107 52
(PP/EPR)/PA/EP-g-MAH 126 170 -96 46
L’étude des propriétés thermiques des mélanges pollués (PA et ABS) et
compatibilisés en présence de SEBS et de EPR-g-MAH montre donc une variation des
températures de cristallisation et du taux de cristallinité selon la nature du polluant et du
compatibilisant. Par contre, les températures de fusion du PP restent constantes quelques soit
la nature des constituants des mélanges.
125

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
II-3-4. Propriétés viscoélastiques à l’état solide des mélanges pollués et pollués/compatibilisés:
Selon Ozkoc et al. [29], l’étude des propriétés viscoélastiques à l’état solide de l’ABS
révèle deux pics de relaxations à -69,2°C et 110,3°C associés à la transition vitreuse
respective des phases polybutadiène (PB) et poly (styrène-co-acrylonitrile) (SAN) de l’ABS.
Pour les mélanges PP/PA6, en présence et en absence de TPE-g-MAH, Ou et al. [28]
observent deux pics de relaxations TgPP(14,2°C) et TgPA(80,8°C) associés respectivement à la
transition vitreuse des phases PP et PA. En présence du TPE-g-MAH, un rapprochement de
10°C environ est observé, ce qui montre que le copolymère améliore la compatibilité des deux
phases PP et PA.
Concernant le mélange (PP/EPR)/ABS, la présence de l’ABS en faible pourcentage
(5%) a donné naissance à un nouveau pic de relaxation de faible intensité dont le maximum
est situé à 115°C (tableau II.6) et qui est associé à la transition vitreuse de ce polymère. De ce
fait, l’ABS ne présente aucune miscibilité avec les autres constituants du mélange et se
présente sous forme de particules sphériques dispersées dans la matrice avec faible adhésion à
l’interface avec la matrice. Un glissement à peine significatif des pics de relaxation βPP (∆T =
2°C) et βEPR (∆T = 1°C) vers les basses températures est observé et il peut être simplement
attribué à une variation de la longueur moyenne des chaînes au cours du cycle
thermomécanique supplémentaire qui a été imposé aux mélanges pollués. Les deux pics de
relaxation αPP et βABS sont convolués, ce qui ne permet pas d’avoir accès au maximum du pic
αPP. D’autre part, une augmentation du module G’ est observée sur la figure II.15 du fait de la
présence du polluant. Pour les mélanges pollués ABS et compatibilisés SEBS, un
rapprochement des pics de relaxation de la phase PP (βPP) et ABS (βABS) (∆T = 6°C) montre
que le copolymère SEBS se place à l’interface entre les deux phases et joue le rôle d’agent de
compatibilisation, conformément aux observations morphologiques.
Concernant les mélanges (PP/EPR)/PA (tableau II.6 et figure II.16), les pics de
relaxation associés aux phases PA (βPA) et PP (αPP) sont convolués. En effet, avec un
pourcentage de 5% du polluant, l’intensité du pic de relaxation ne sera pas importante, ce qui
le rend complètement caché par celui de la phase cristalline du PP. Pour les mélanges
compatibilisés (PP/EPR)/PA/EPR-g-MAH, la présence du copolymère déplace légèrement le
126
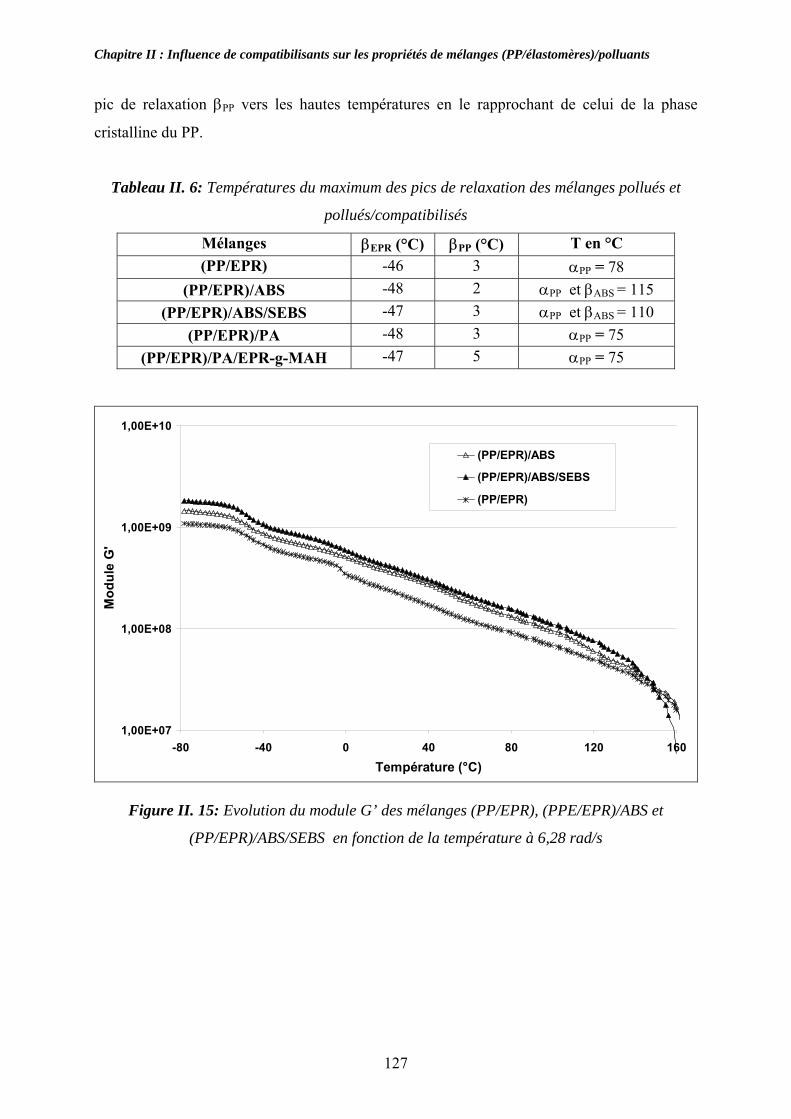
Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
pic de relaxation βPP vers les hautes températures en le rapprochant de celui de la phase
cristalline du PP.
Tableau II. 6: Températures du maximum des pics de relaxation des mélanges pollués et
pollués/compatibilisés
Mélanges βEPR (°C) βPP (°C) T en °C (PP/EPR) -46 3 αPP = 78
(PP/EPR)/ABS -48 2 αPP et βABS = 115 (PP/EPR)/ABS/SEBS -47 3 αPP et βABS = 110
(PP/EPR)/PA -48 3 αPP = 75 (PP/EPR)/PA/EPR-g-MAH -47 5 αPP = 75
1,00E+07
1,00E+08
1,00E+09
1,00E+10
-80 -40 0 40 80 120 160Température (°C)
Mod
ule
G'
(PP/EPR)/ABS
(PP/EPR)/ABS/SEBS
(PP/EPR)
Figure II. 15: Evolution du module G’ des mélanges (PP/EPR), (PPE/EPR)/ABS et
(PP/EPR)/ABS/SEBS en fonction de la température à 6,28 rad/s
127
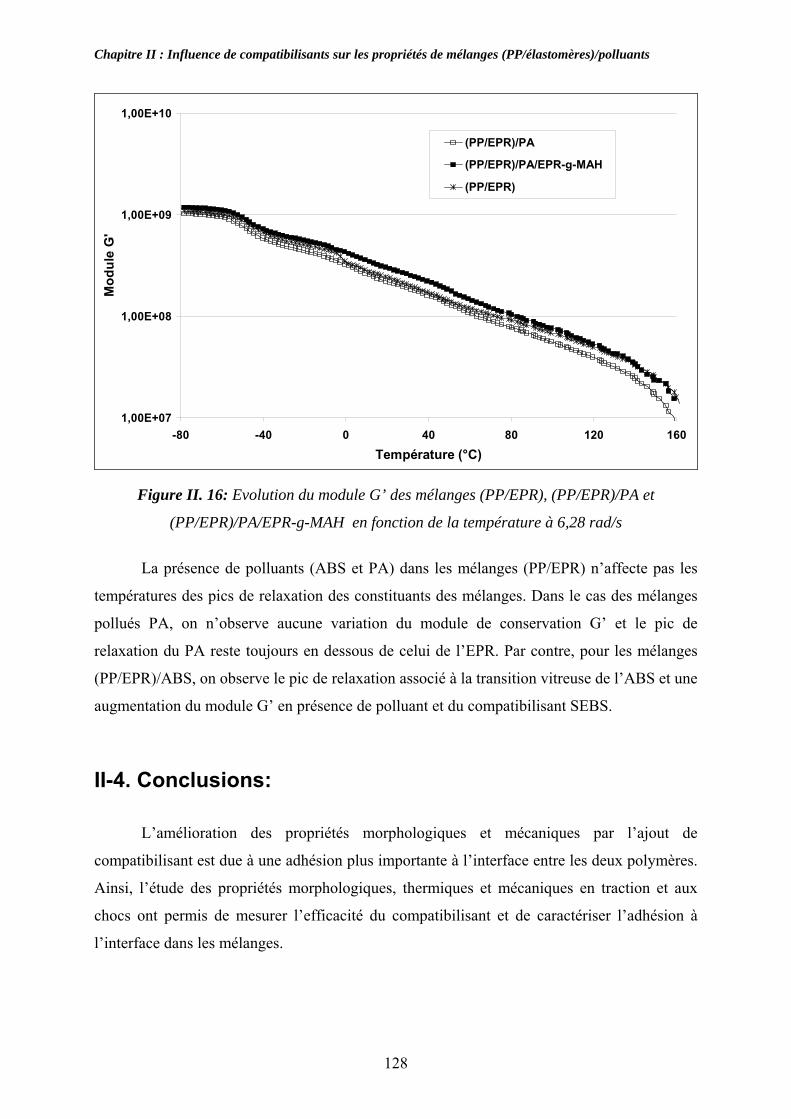
Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
1,00E+07
1,00E+08
1,00E+09
1,00E+10
-80 -40 0 40 80 120 160Température (°C)
Mod
ule
G'
(PP/EPR)/PA
(PP/EPR)/PA/EPR-g-MAH
(PP/EPR)
Figure II. 16: Evolution du module G’ des mélanges (PP/EPR), (PP/EPR)/PA et
(PP/EPR)/PA/EPR-g-MAH en fonction de la température à 6,28 rad/s
La présence de polluants (ABS et PA) dans les mélanges (PP/EPR) n’affecte pas les
températures des pics de relaxation des constituants des mélanges. Dans le cas des mélanges
pollués PA, on n’observe aucune variation du module de conservation G’ et le pic de
relaxation du PA reste toujours en dessous de celui de l’EPR. Par contre, pour les mélanges
(PP/EPR)/ABS, on observe le pic de relaxation associé à la transition vitreuse de l’ABS et une
augmentation du module G’ en présence de polluant et du compatibilisant SEBS.
II-4. Conclusions:
L’amélioration des propriétés morphologiques et mécaniques par l’ajout de
compatibilisant est due à une adhésion plus importante à l’interface entre les deux polymères.
Ainsi, l’étude des propriétés morphologiques, thermiques et mécaniques en traction et aux
chocs ont permis de mesurer l’efficacité du compatibilisant et de caractériser l’adhésion à
l’interface dans les mélanges.
128

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
Les résultats obtenus montrent une amélioration des propriétés pour les mélanges
compatibilisés avec le copolymère PEP. L’interface serait donc le siège d’interactions
importantes entre les deux phases du fait de la présence du compatibilisant qui se concentre
préférentiellement à ce niveau lors de la phase d’élaboration des mélanges à l’état fondu pour
interdiffuser les blocs le constituant dans les phases correspondantes par affinité
thermodynamique, effet particulièrement mis en évidence pour les mélanges à matrice
(PP/EPR)/PEP. Pour les mélanges (PP/EPR)/EPR-g-MAH, la présence du compatibilisant
fragilise le faciès de rupture du matériau, augmente le taux de cristallinité et les propriétés
mécaniques pour les grandes déformations mais sans modifier les pics de relaxation des
constituants des mélanges.
Pour les mélanges pollués ABS, les observations morphologiques montrent que les
constituants des mélanges ne sont pas miscibles. L’ABS se présente sous forme de particules
de taille inférieure à 10µm et possédant un pic de relaxation vers 115°C. Sa présence ne
modifie pas les propriétés thermiques des mélanges mais diminue de 30 à 60% la contrainte
maximale, l’allongement et l’énergie à la rupture. L’ajout de compatibilisant SEBS améliore
l’adhésion à l’interface, l’allongement à la rupture et diminue la taille des nodules ABS (<7
µm).
Dans le cas des mélanges (PP/ERP)/PA, le polluant se présente aussi sous forme de
particules non miscibles et de tailles dans la fourchette [1-5µm]. Sa présence augmente de 8%
le taux de cristallinité mais n’affecte pas les propriétés rhéologiques à l’état solide et diminue
légèrement les propriétés mécaniques. La présence du compatibilisant EPR-g-MAH diminue
la taille des particules PA [1-2µm] et le nombre de cavités vides qui correspondent à
l’arrachement du polluant lors de la rupture.
129

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
Références [1] Xanthos M. and Dagli S. Compatibilization of Polymer Blends by Reactive Processing,
Polymer Engineering and Science, 1991, vol 31, n° 13, p 929-935.
[2] Kunori T. and Geil P. H. Morphology-property relationships in polycarbonate-based
blends. I. Modulus, J Macromol Sci Phys, 1980, Vol 18, Issue 1, p93-134
[3] Rösch J. Modeling the mechanical properties in polypropylene/polyamide-6 blends with
core shell morphology, Polym Eng Sci, 1995, vol 35, p1917-1922.
[4] Gonzalez-Montiel A., Keskkula H., Paul D. R. Impact-modified nylon 6/polypropylene
blends: 1. Morphology-property relationships, Polymer, 1995, vol 36, p 4587.
[5] Mnif N., Massardier V., Kallel T. and Elleuch B. Study of the modification of the
properties of (PP/EPR) blends with a view to preserving natural resources when elaborating
new formulation and recycling polymers Polym Comp, 2008, DOI10.1002.
[6] Pukanszky B. and al. Particle break-up and coalescence in heterogeneous PP/EPDM
blends; effect of particle size on some mechanical properties, Plastics, Rubber and
Composites Processing and Applications, 1991, vol 15, No 1, p 31-38.
[7] Ivan Fortelný, Danuse Michálková, Jaroslava Koplíková, Edita Navrátilová, Josef Kovár.
The effect of conditions of mixing of polypropylene/ethylene-propylene elastomer blends on
their morphological structure and impact strength, Die Angewandte Makromolekulare
Chemie, 1990, vol 179, p185-201.
[8] D'orazio L., Mancarella C., Martuscelli E., Sticotti G., Polypropylene/ethylene-co-
propylene blends: influence of molecular structure of EPR and composition on phase structure
of isothermally crystallized samples, Journal of materials science, 1991, vol 26, no15, p 4033-
4047.
[9] Jancar J., Dianselmo A., Dibenetto A., Kucera J. failure mechanics in elastomer toughened
polypropylene, Polymer, 1993, vol 34, p 1684-1694.
[10] Chodhary V., Varma H. S., Varma I. K. Polyolefin blends: effect of EPDM rubber on
crystallization, morphology and mechanical properties of polypropylene/EPDM blends,
POLYMER, 1991, vol 32, no14, p 2534-2540.
[11] Mnif N., Massardier V. and Jaziri M. Influence of an ethylene-octene copolymer and of
pollutants in (PP/EPR) blends. Journal of Applied Polymer Science, Vol 104, 2007, p 3220–
3227.
130

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
[12] Jaziri M., Massardier V., Mnif N and Perier-Camby H. Rheological, Thermal, and
Morphological Properties of blends based on poly(propylene), ethylene propylene rubber and
ethylene-1-octene copolymer that could result from End of Life Vehicles (ELV). Effect of
maleic anhydride grafted poly(propylene). Polymer engineering and science, 2007, vol 47, p
1009-1015.
[13] Kallel T., Jaziri M. and Elleuch B. Étude d'une voie de valorisation des mélanges PE/PP
et PE/PS: Aspect rhéologique. Dechets Sci Tech, 2002, vol 25, p 40.
[14] Kallel T., Massardier V., Jaziri M. Gerard J.F. and Elleuch B. Effect of model pollutants
on the recycling of PE/PS plastic blends. Progress in rubber, plastics and recycling
technology, 2003, vol 19, p 61-75.
[15] Hiromasu, K. A. Jpn. Patent 06 248 154, 1995 (to Hitachi Chemical Co. Ltd.).
[16] Yamamoto, Y.; Aibe, H. Jpn. Patent 05 93 110, 1993 (to Mitsui Petrochemical Ind.).
[17] Padwa, A. R. U.S. Patent 35 082 742, 1992 (to Monsato Co.).
[18] TDK Electronics Co. Ltd. Jpn. Patents 82 53 550, 82 53 549, 1982.
[19] Alpesh C. Patel, Ragesh B. Bramhatt, Sarawade B. D., Surekha D. Morphological and
Mechanical Properties of PP/ABS Blends Compatibilized with PP- g-acrylic Acid, Journal of
Applied Polymer Science, 2001, Vol 81, p 1731–1741.
[20] Alpesh C. Patel, Ragesh B. Brahmbhatt, Surekha D. Mechanical Properties and
Morphology of PP/ABS Blends Compatibilized with PP-g-2-HEMA, 2003, Journal of
Applied Polymer Science, Vol 88, p 72–78.
[21] Frounchi M., Burford R. P., State of Compatibility in Crystalline Polypropylene/ABS
Amorphous Terpolymer Thermoplastic Blends. Effect of Styrenic Copolymers as
Compatibilisers, Iran J Polym Sci Technol,1993, vol 2, p 59.
[22] Omonov T.S., Harrats C. and Groeninckx G., Co-continuous and encapsulated three
phase morphologies in uncompatibilized and reactively compatibilized polyamide
6/polypropylene/polystyrene ternary blends using two reactive precursors, Polymer, 2005, vol
46, p12322-36.
[23] Jose S., Francis B., Thomas S. and Karger-Kocsis J. Morphology and mechanical
properties of polyamide 12/polypropyleneblends in presence and absence of reactive
compatibiliser,, Polymer, 2006, vol 47, p 3874–3888.
[24] Holsti-Miettinen R. M., Seppälä J. V., Ikkala O. T. and Reima I. T. Functionalized
elastomeric compatibilizer in PA 6/PP blends and binary interactions between compatibilizer
and polymer, Polymer Engineering and Science, 1994, Vol 34, p 395-404.
131

Chapitre II : Influence de compatibilisants sur les propriétés de mélanges (PP/élastomères)/polluants
[25] Jannerfeldt G., Boogh L. and Månson J.A. E. Influence of hyperbranched polymers on
the interfacial tension of polypropylene/polyamide-6 blends, Journal of Polymer Science Part
B: Polymer Physics, 1999, Vol 37, p 2069-2077.
[26] Duvall J., Sellitti C., Myers C., Hiltner A. and Baer E. Effect of compatibilization on the
properties of polypropylene/polyamide-66 (75/25 wt/wt) blends, 1994, Journal of Applied
Polymer Science, Vol 52, p 195-206.
[27] Byung S. Y., Joon Y. J., Moon H. S., Yong M. L. and Suck H. L. Mechanical properties
of polypropylene/polyamide 6 blends: Effect of manufacturing processes and
compatibilisation, 1997, Polymer Composites, Vol 18, p 757-764.
[28] Yuchun O., Yuguo L., Xiaoping F., Guisheng Y. Maleic Anhydride Grafted
Thermoplastic Elastomer as an Interfacial Modifier for Polypropylene/Polyamide 6 Blends,
2004, Journal of Applied Polymer Science, 2004, Vol 91, p 1806-1815.
[29] Guralp O., Goknur B. and Erdal B. Effects of Olefin-Based Compatibilizers on the
Morphology, Thermal and Mechanical Properties of ABS/Polyamide-6 Blends”, 2007,
Journal of Applied Polymer Science, Vol 104, p 926-935.
132

Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
Chapitre III Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en
vue de recyclage des boucliers peints
133

Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
134

Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
Chapitre III :
Elaboration et compatibilisation des mélanges (PP/EPR)/PU réticulés en vue de recyclage des
boucliers peints
III-1. Introduction :
Un des objectifs de ce travail était de traiter le problème des déchets de pare-chocs
peints dont les particules de peinture agissent sous la forme d’impuretés non compatibles et
provoquent une chute catastrophique de la résilience. A l’échelle industrielle, une extrusion à
température élevée dégrade préférentiellement le PU mais pourra aussi provoquer la β-
scission du PP et la diminution des propriétés mécaniques des mélanges.
Une solution consiste à filtrer les impuretés (salissures) et les particules de peinture en bout de
vis mais cela n’est pas satisfaisant.
Afin d’apporter une autre solution dégradant moins le PP, nous avons recherché une
voie de compatibilisation de ce polluant PU thermodurcissable.
Deux possibilités peuvent être explorées :
les particules de peinture restent à l’état de paillettes solides réticulées. Il faut alors
synthétiser un agent de couplage selon la nature de la peinture utilisée (polyuréthane
ou autre) comme on le ferait pour une charge minérale ou organique. La dispersion
peut se faire pendant la phase de compoundage.
la peinture est traitée par solvolyse, ce traitement pouvant se faire sur les granulés de
rebroyé ou directement dans l’extrudeuse.
Cette dernière possibilité est plus aléatoire compte-tenu du temps nécessaire pour
mener à bien le traitement par solvolyse.
135
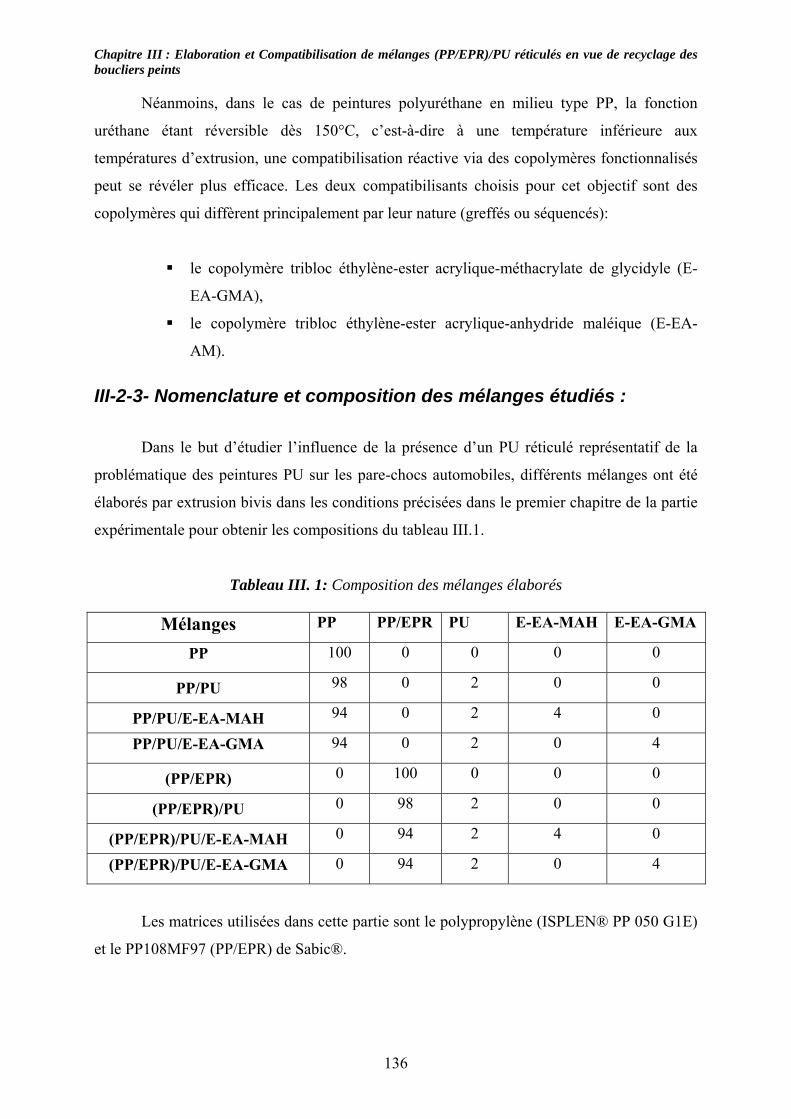
Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
Néanmoins, dans le cas de peintures polyuréthane en milieu type PP, la fonction
uréthane étant réversible dès 150°C, c’est-à-dire à une température inférieure aux
températures d’extrusion, une compatibilisation réactive via des copolymères fonctionnalisés
peut se révéler plus efficace. Les deux compatibilisants choisis pour cet objectif sont des
copolymères qui diffèrent principalement par leur nature (greffés ou séquencés):
le copolymère tribloc éthylène-ester acrylique-méthacrylate de glycidyle (E-
EA-GMA),
le copolymère tribloc éthylène-ester acrylique-anhydride maléique (E-EA-
AM).
III-2-3- Nomenclature et composition des mélanges étudiés :
Dans le but d’étudier l’influence de la présence d’un PU réticulé représentatif de la
problématique des peintures PU sur les pare-chocs automobiles, différents mélanges ont été
élaborés par extrusion bivis dans les conditions précisées dans le premier chapitre de la partie
expérimentale pour obtenir les compositions du tableau III.1.
Tableau III. 1: Composition des mélanges élaborés
Mélanges PP PP/EPR PU E-EA-MAH E-EA-GMA
PP 100 0 0 0 0
PP/PU 98 0 2 0 0
PP/PU/E-EA-MAH 94 0 2 4 0
PP/PU/E-EA-GMA 94 0 2 0 4
(PP/EPR) 0 100 0 0 0
(PP/EPR)/PU 0 98 2 0 0
(PP/EPR)/PU/E-EA-MAH 0 94 2 4 0
(PP/EPR)/PU/E-EA-GMA 0 94 2 0 4
Les matrices utilisées dans cette partie sont le polypropylène (ISPLEN® PP 050 G1E)
et le PP108MF97 (PP/EPR) de Sabic®.
136

Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
III-3- Résultats expérimentaux :
III-3-1- Propriétés thermiques :
Les figures III.1 et III.2 et le tableau III.2 présentent les résultats DSC obtenus pour les
matrices PP et (PP/EPR) et leurs mélanges en présence du PU et de compatibilisants choisis
pour cette étude.
Les courbes DSC des constituants PP et (PP/EPR) utilisé comme matrice dans les
mélanges présentent deux transitions différentes : la première transition obtenue sous forme
d’un pic endothermique à 170°C est attribuée à la fusion du polypropylène (TmPP). La
deuxième transition exothermique apparaît respectivement à 110°C et 126°C et est associée
aux températures de cristallisation du polypropylène (TcPP) dans les matrices PP et (PP/EPR).
Pour les mélanges contenant la peinture PU et les compatibilisants (E-EA-MAH et E-EA-
GMA), on remarque que les températures de fusion restent constantes. Par contre, la présence
de PU augmente la température de cristallisation et diminue le taux de cristallinité dans le cas
des mélanges à matrice (PP/EPR). Ceci montre qu’on forme plus difficilement des cristallites
en présence du PU et d’EPR (la taille des cristallites demeure inchangée : TmPP est constante).
Malheureusement, nous n’avons pas pu avoir séparément la matrice PP du (PP/EPR) pour
étudier l’effet de l’ajout du PU sur le taux de cristallinité du polypropylène. D’après les
observations MEB, étudiées dans la suite de ce chapitre, le PU se disperse dans le PP ce qui
peut être à l’origine de cette différence observée au niveau de la cristallinité du PP ; en effet,
la concentration du PU dans le PP passe de 2% pour les mélanges PP/PU à 2,5% dans les
mélanges (PP/EPR)/PU.
137
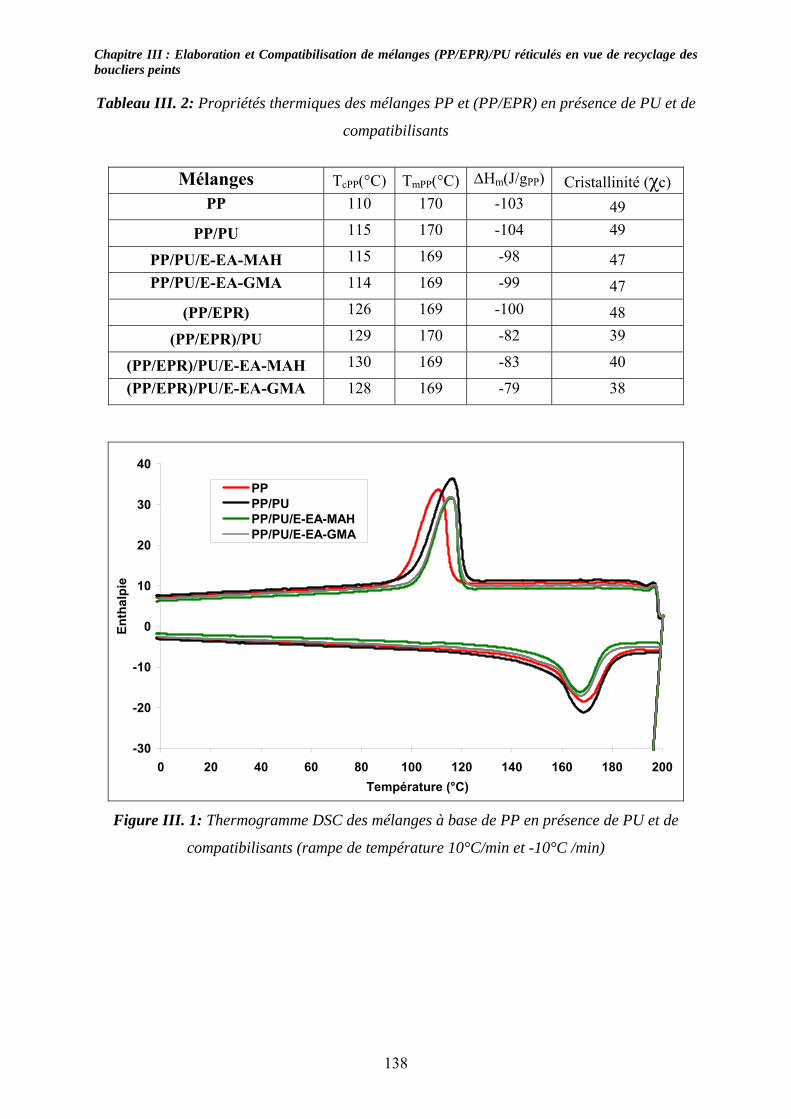
Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints Tableau III. 2: Propriétés thermiques des mélanges PP et (PP/EPR) en présence de PU et de
compatibilisants
Mélanges TcPP(°C) TmPP(°C) ∆Hm(J/gPP) Cristallinité (χc)
PP 110 170 -103 49
PP/PU 115 170 -104 49
PP/PU/E-EA-MAH 115 169 -98 47 PP/PU/E-EA-GMA 114 169 -99 47
(PP/EPR) 126 169 -100 48
(PP/EPR)/PU 129 170 -82 39
(PP/EPR)/PU/E-EA-MAH 130 169 -83 40
(PP/EPR)/PU/E-EA-GMA 128 169 -79 38
-30
-20
-10
0
10
20
30
40
0 20 40 60 80 100 120 140 160 180 200Température (°C)
Enth
alpi
e
PPPP/PUPP/PU/E-EA-MAHPP/PU/E-EA-GMA
Figure III. 1: Thermogramme DSC des mélanges à base de PP en présence de PU et de
compatibilisants (rampe de température 10°C/min et -10°C /min)
138
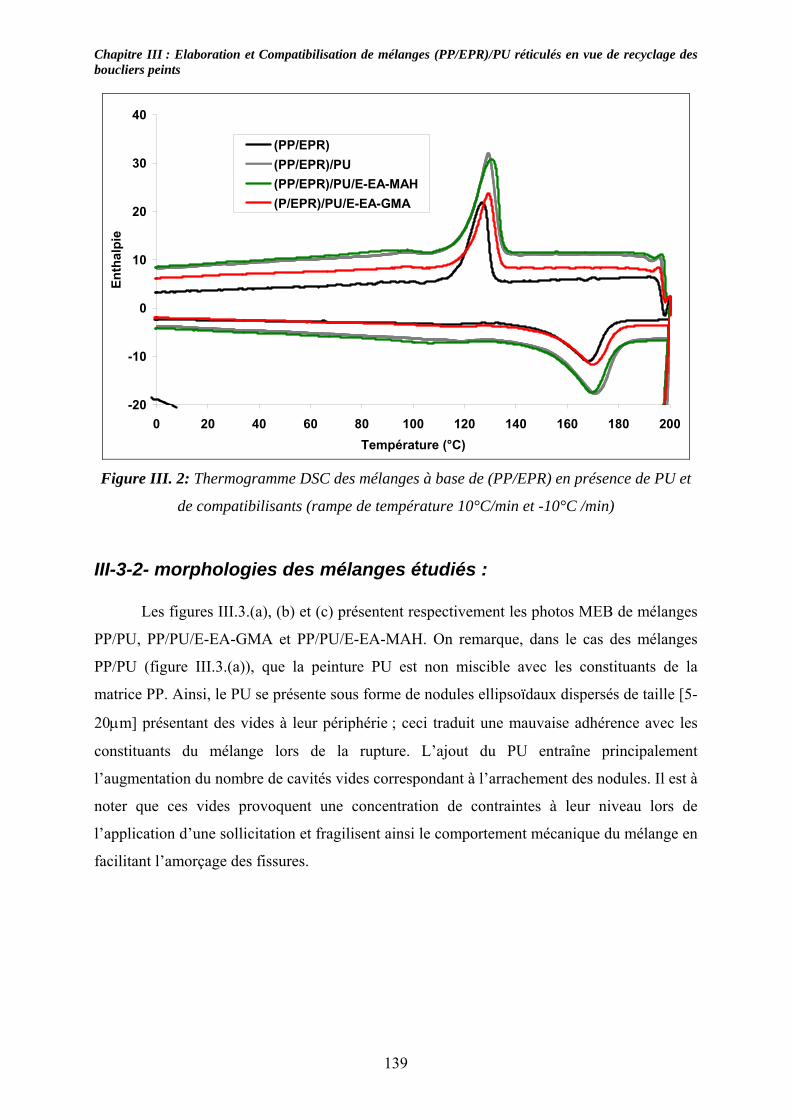
Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
-20
-10
0
10
20
30
40
0 20 40 60 80 100 120 140 160 180 200Température (°C)
Enth
alpi
e
(PP/EPR)(PP/EPR)/PU(PP/EPR)/PU/E-EA-MAH(P/EPR)/PU/E-EA-GMA
Figure III. 2: Thermogramme DSC des mélanges à base de (PP/EPR) en présence de PU et
de compatibilisants (rampe de température 10°C/min et -10°C /min)
III-3-2- morphologies des mélanges étudiés :
Les figures III.3.(a), (b) et (c) présentent respectivement les photos MEB de mélanges
PP/PU, PP/PU/E-EA-GMA et PP/PU/E-EA-MAH. On remarque, dans le cas des mélanges
PP/PU (figure III.3.(a)), que la peinture PU est non miscible avec les constituants de la
matrice PP. Ainsi, le PU se présente sous forme de nodules ellipsoïdaux dispersés de taille [5-
20µm] présentant des vides à leur périphérie ; ceci traduit une mauvaise adhérence avec les
constituants du mélange lors de la rupture. L’ajout du PU entraîne principalement
l’augmentation du nombre de cavités vides correspondant à l’arrachement des nodules. Il est à
noter que ces vides provoquent une concentration de contraintes à leur niveau lors de
l’application d’une sollicitation et fragilisent ainsi le comportement mécanique du mélange en
facilitant l’amorçage des fissures.
139
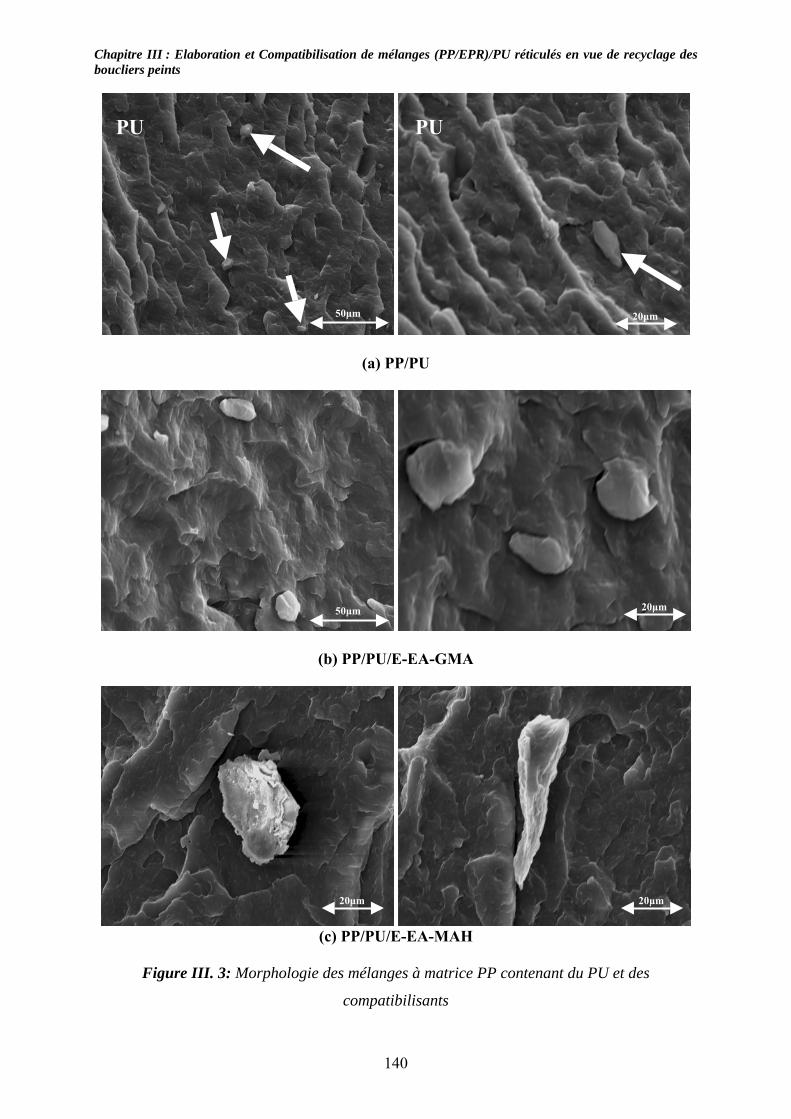
Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
PU PU
50µm 20µm
(a) PP/PU
20µm 50µm
(b) PP/PU/E-EA-GMA
20µm 20µm
(c) PP/PU/E-EA-MAH
Figure III. 3: Morphologie des mélanges à matrice PP contenant du PU et des
compatibilisants
140

Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
Dans le cas des mélanges PP/PU compatibilisés en présence des copolymères E-EA-
GMA et E-EA-MAH (figure III.3.(b) et (c)), la peinture se présente toujours sous forme de
particules non miscibles avec la matrice PP avec une mauvaise adhésion à l’interface. Les
résultats obtenus montrent qu’aucune interaction entre le PU et les compatibilisants ne s’est
produite lors de l’extrusion des mélanges.
141
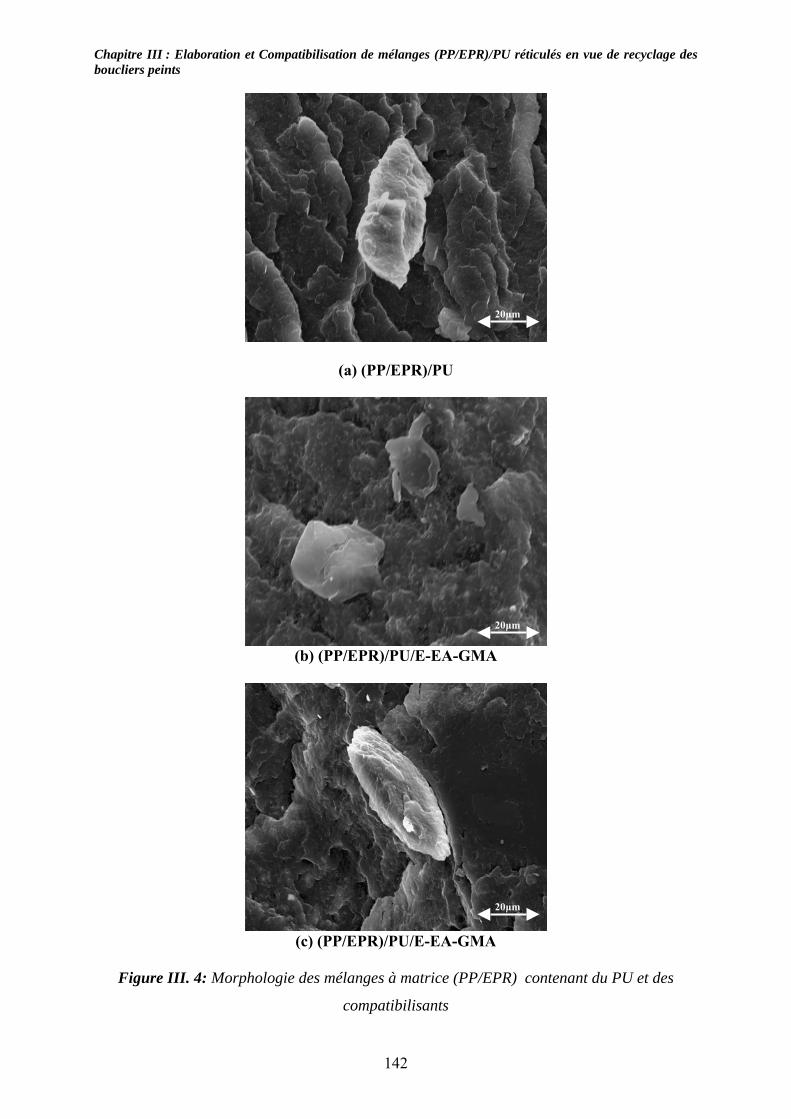
Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
20µm
(a) (PP/EPR)/PU
20µm
(b) (PP/EPR)/PU/E-EA-GMA
20µm
(c) (PP/EPR)/PU/E-EA-GMA
Figure III. 4: Morphologie des mélanges à matrice (PP/EPR) contenant du PU et des
compatibilisants
142

Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints La figure III.4 montre aussi que dans le cas des mélanges à matrice (PP/EPR)/PU, la
peinture se présente sous forme de particules non miscibles avec les différents constituants
même en présence de compatibilisants. La taille des nodules PU appartient à la fourchette [5-
40µm].
III-3-3- Propriétés mécaniques :
L’ensemble des résultats obtenus lors de l’étude mécanique en traction et en choc des
mélanges à matrice PP et (PP/EPR) en présence du PU et de compatibilisants sont donnés sur
le tableau III.3 et les figures III.5-6-7-8.
Les résultats obtenus dans le cas des mélanges à matrice PP montrent que :
- la contrainte maximale et la résilience restent pratiquement constantes en présence
de PU et de compatibilisants. Ceci est cohérent avec les propriétés thermiques qui
montrent l’absence de variation des taux de cristallinité des mélanges.
- les valeurs de l’allongement et de l’énergie à la rupture diminuent de 50%. Ceci
confirme les observations morphologiques où nous avons observé une faible
adhésion à l’interface PP/PU,
- en présence de compatibilisants E-EA-MAH et E-EA-GMA, on n’observe aucune
amélioration des propriétés mécaniques. Ces résultats viennent alors confirmer les
observations morphologiques.
Pour les mélanges (PP/EPR)/PU, on observe une diminution de 40 à 60% de la
contrainte à la rupture, de l’allongement (%) et de la résilience et de plus de 70% de l’énergie
à la rupture due à une mauvaise adhésion entre la matrice (PP/EPR) et le PU ainsi que de la
diminution du taux de cristallinité observé lors de l’étude des propriétés thermiques. En
présence de copolymères E-EA-MAH et E-EA-GMA, aucune amélioration n’est observée, ce
qui est en accord avec les observations morphologiques.
143
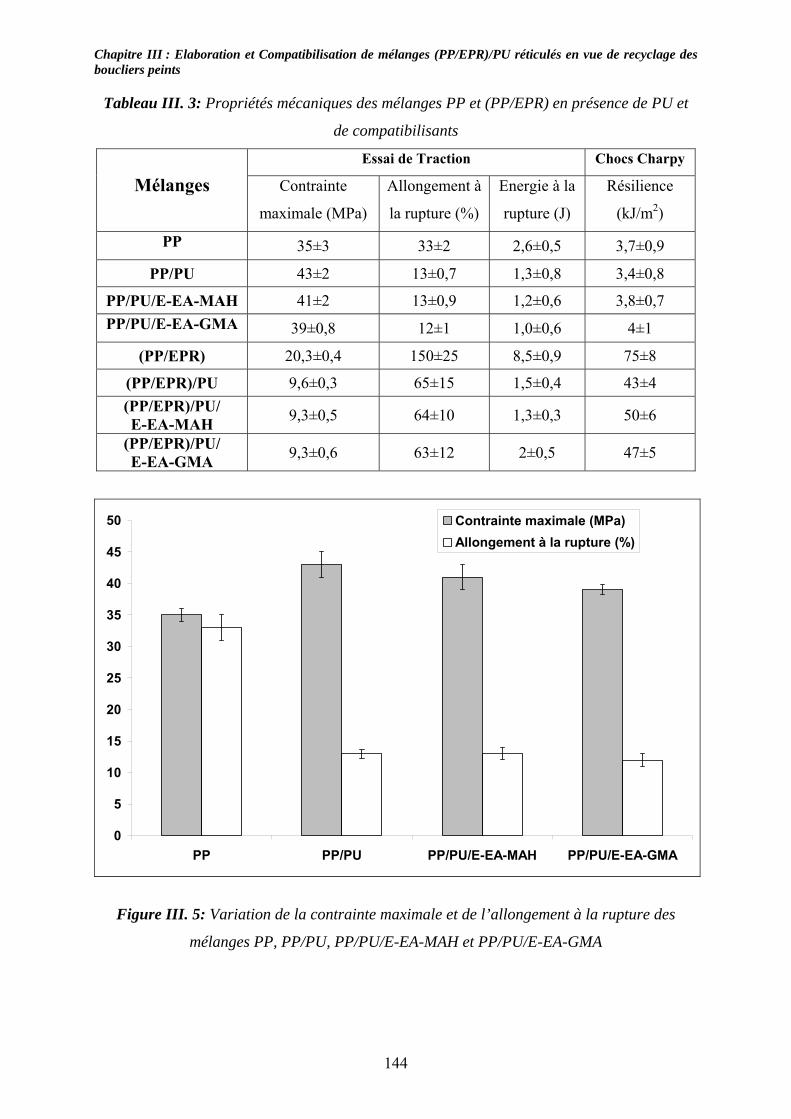
Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
Tableau III. 3: Propriétés mécaniques des mélanges PP et (PP/EPR) en présence de PU et
de compatibilisants
Essai de Traction Chocs Charpy
Mélanges Contrainte
maximale (MPa)
Allongement à
la rupture (%)
Energie à la
rupture (J)
Résilience
(kJ/m2)
PP 35±3 33±2 2,6±0,5 3,7±0,9
PP/PU 43±2 13±0,7 1,3±0,8 3,4±0,8
PP/PU/E-EA-MAH 41±2 13±0,9 1,2±0,6 3,8±0,7 PP/PU/E-EA-GMA 39±0,8 12±1 1,0±0,6 4±1
(PP/EPR) 20,3±0,4 150±25 8,5±0,9 75±8
(PP/EPR)/PU 9,6±0,3 65±15 1,5±0,4 43±4 (PP/EPR)/PU/ E-EA-MAH 9,3±0,5 64±10 1,3±0,3 50±6
(PP/EPR)/PU/ E-EA-GMA 9,3±0,6 63±12 2±0,5 47±5
0
5
10
15
20
25
30
35
40
45
50
PP PP/PU PP/PU/E-EA-MAH PP/PU/E-EA-GMA
Contrainte maximale (MPa)Allongement à la rupture (%)
Figure III. 5: Variation de la contrainte maximale et de l’allongement à la rupture des
mélanges PP, PP/PU, PP/PU/E-EA-MAH et PP/PU/E-EA-GMA
144
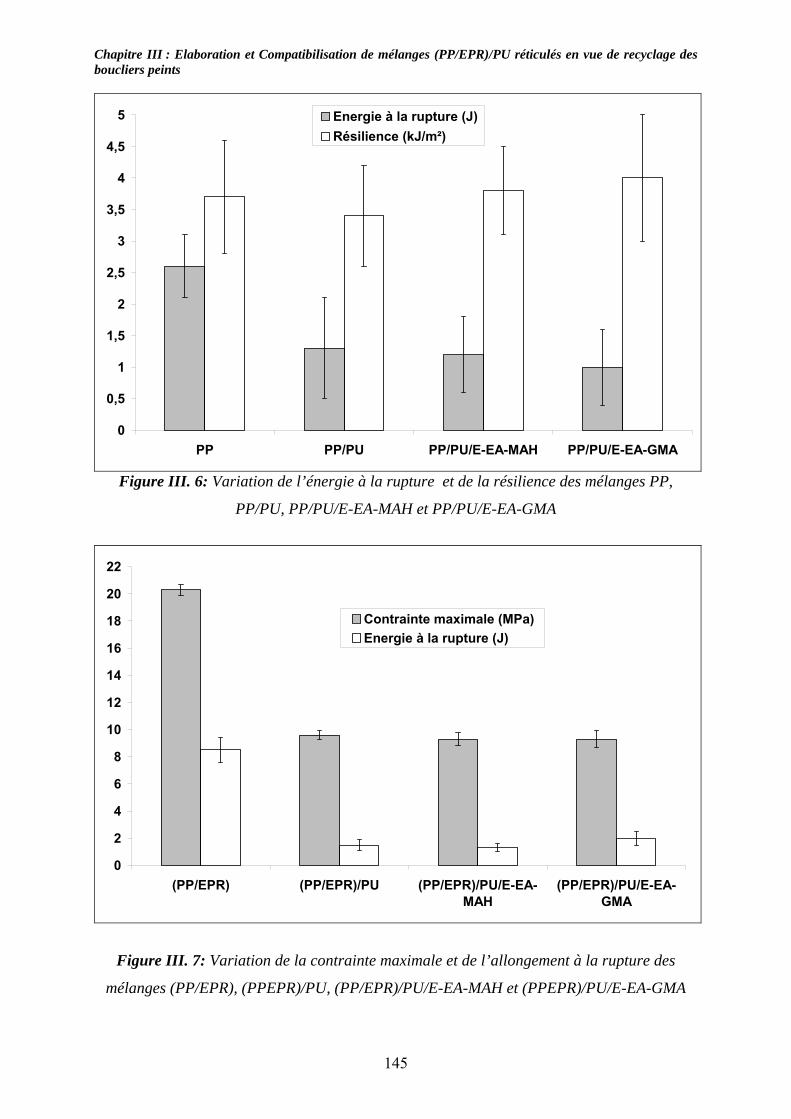
Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
0
0,5
1
1,5
2
2,5
3
3,5
4
4,5
5
PP PP/PU PP/PU/E-EA-MAH PP/PU/E-EA-GMA
Energie à la rupture (J)Résilience (kJ/m²)
Figure III. 6: Variation de l’énergie à la rupture et de la résilience des mélanges PP,
PP/PU, PP/PU/E-EA-MAH et PP/PU/E-EA-GMA
0
2
4
6
8
10
12
14
16
18
20
22
(PP/EPR) (PP/EPR)/PU (PP/EPR)/PU/E-EA-MAH
(PP/EPR)/PU/E-EA-GMA
Contrainte maximale (MPa)Energie à la rupture (J)
Figure III. 7: Variation de la contrainte maximale et de l’allongement à la rupture des
mélanges (PP/EPR), (PPEPR)/PU, (PP/EPR)/PU/E-EA-MAH et (PPEPR)/PU/E-EA-GMA
145
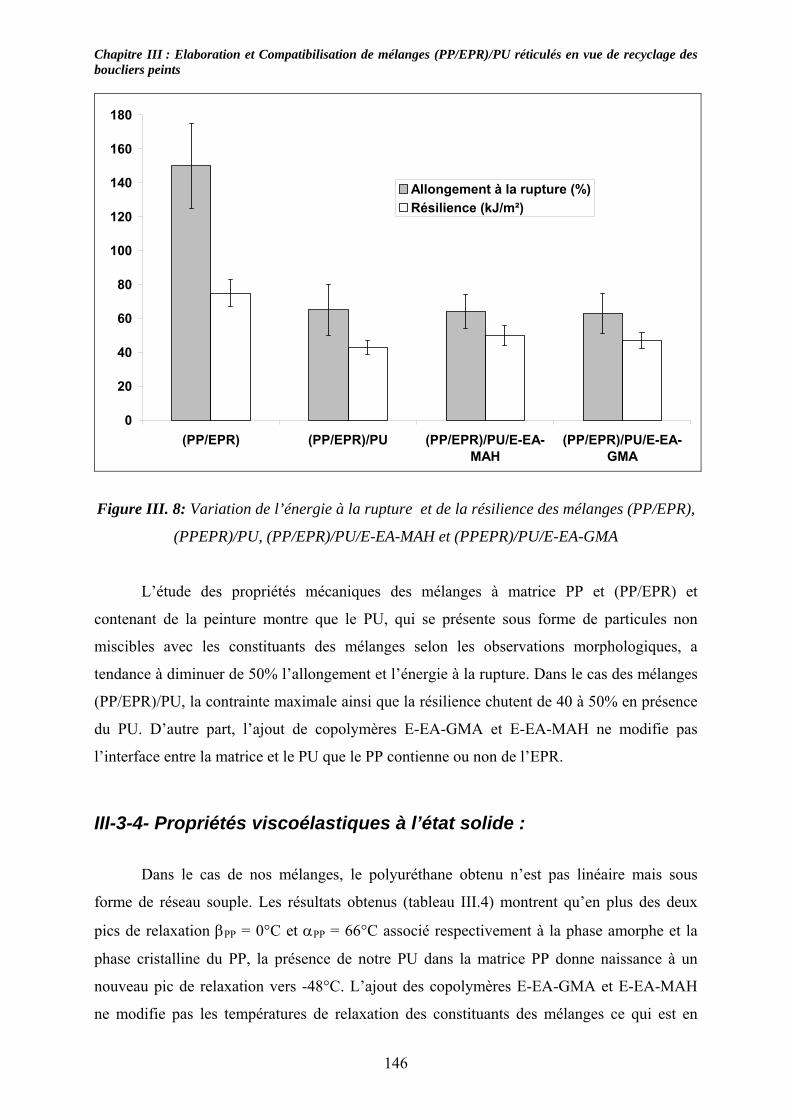
Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
0
20
40
60
80
100
120
140
160
180
(PP/EPR) (PP/EPR)/PU (PP/EPR)/PU/E-EA-MAH
(PP/EPR)/PU/E-EA-GMA
Allongement à la rupture (%)Résilience (kJ/m²)
Figure III. 8: Variation de l’énergie à la rupture et de la résilience des mélanges (PP/EPR),
(PPEPR)/PU, (PP/EPR)/PU/E-EA-MAH et (PPEPR)/PU/E-EA-GMA
L’étude des propriétés mécaniques des mélanges à matrice PP et (PP/EPR) et
contenant de la peinture montre que le PU, qui se présente sous forme de particules non
miscibles avec les constituants des mélanges selon les observations morphologiques, a
tendance à diminuer de 50% l’allongement et l’énergie à la rupture. Dans le cas des mélanges
(PP/EPR)/PU, la contrainte maximale ainsi que la résilience chutent de 40 à 50% en présence
du PU. D’autre part, l’ajout de copolymères E-EA-GMA et E-EA-MAH ne modifie pas
l’interface entre la matrice et le PU que le PP contienne ou non de l’EPR.
III-3-4- Propriétés viscoélastiques à l’état solide :
Dans le cas de nos mélanges, le polyuréthane obtenu n’est pas linéaire mais sous
forme de réseau souple. Les résultats obtenus (tableau III.4) montrent qu’en plus des deux
pics de relaxation βPP = 0°C et αPP = 66°C associé respectivement à la phase amorphe et la
phase cristalline du PP, la présence de notre PU dans la matrice PP donne naissance à un
nouveau pic de relaxation vers -48°C. L’ajout des copolymères E-EA-GMA et E-EA-MAH
ne modifie pas les températures de relaxation des constituants des mélanges ce qui est en
146
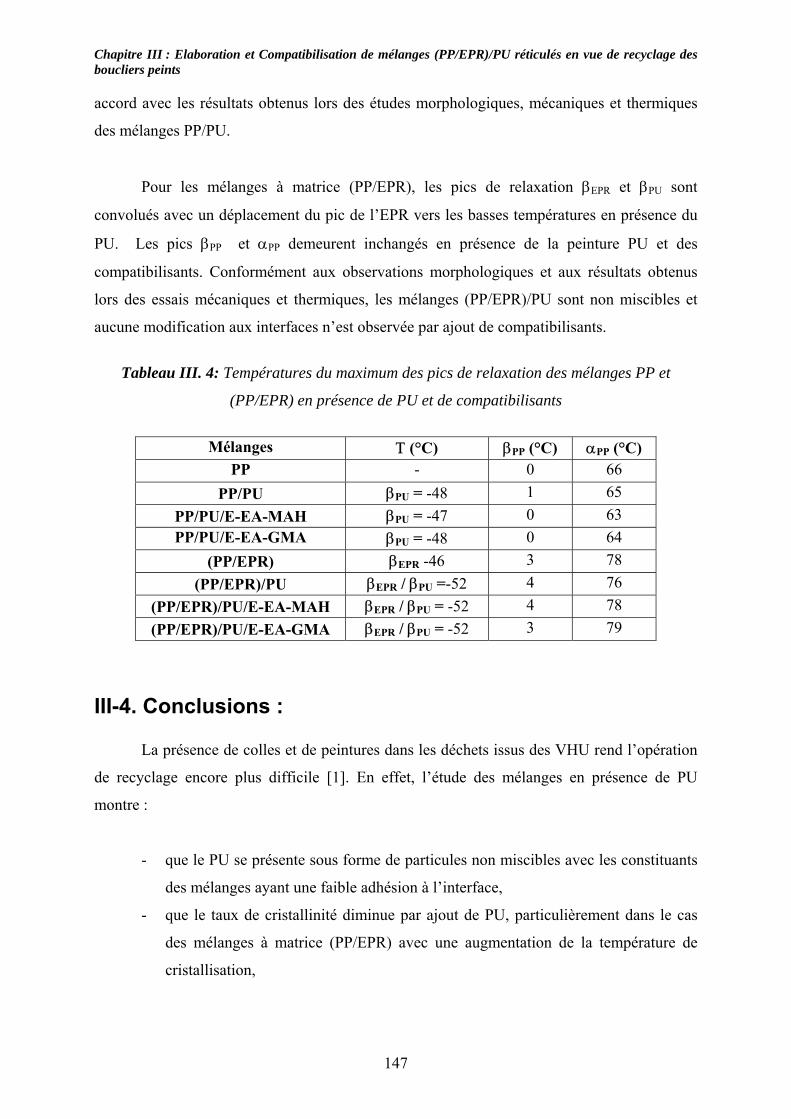
Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints accord avec les résultats obtenus lors des études morphologiques, mécaniques et thermiques
des mélanges PP/PU.
Pour les mélanges à matrice (PP/EPR), les pics de relaxation βEPR et βPU sont
convolués avec un déplacement du pic de l’EPR vers les basses températures en présence du
PU. Les pics βPP et αPP demeurent inchangés en présence de la peinture PU et des
compatibilisants. Conformément aux observations morphologiques et aux résultats obtenus
lors des essais mécaniques et thermiques, les mélanges (PP/EPR)/PU sont non miscibles et
aucune modification aux interfaces n’est observée par ajout de compatibilisants.
Tableau III. 4: Températures du maximum des pics de relaxation des mélanges PP et
(PP/EPR) en présence de PU et de compatibilisants
Mélanges Τ (°C) βPP (°C) αPP (°C)
PP - 0 66 PP/PU βPU = -48 1 65
PP/PU/E-EA-MAH βPU = -47 0 63 PP/PU/E-EA-GMA βPU = -48 0 64
(PP/EPR) βEPR -46 3 78 (PP/EPR)/PU βEPR / βPU =-52 4 76
(PP/EPR)/PU/E-EA-MAH βEPR / βPU = -52 4 78 (PP/EPR)/PU/E-EA-GMA βEPR / βPU = -52 3 79
III-4. Conclusions : La présence de colles et de peintures dans les déchets issus des VHU rend l’opération
de recyclage encore plus difficile [1]. En effet, l’étude des mélanges en présence de PU
montre :
- que le PU se présente sous forme de particules non miscibles avec les constituants
des mélanges ayant une faible adhésion à l’interface,
- que le taux de cristallinité diminue par ajout de PU, particulièrement dans le cas
des mélanges à matrice (PP/EPR) avec une augmentation de la température de
cristallisation,
147

Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
- une chute de l’allongement à la rupture et de l’énergie à la rupture due à la
mauvaise adhésion entre le PU et la matrice avec une diminution de la contrainte
maximale et de la résilience par modification du taux de cristallinité,
- l’apparition d’un nouveau pic de relaxation du PU et une faible variation des pics
de relaxation des autres constituants lors de l’étude des propriétés viscoélastiques à
l’état solide (fréquence 1Hz sur le domaine de températures -80-160°C),
- que la présence de compatibilisants E-EA-MAH et E-EA-GMA ne modifie pas les
propriétés thermiques, morphologiques, viscoélastiques à l’état solide et
mécaniques des mélanges en présence du PU.
A l’issue de cette étude, nous proposons d’autres alternatives qui pourrons être
étudiées dans la suite à savoir :
- l’utilisation d’autres compatibilisants,
- le changement des conditions de mise en œuvre en augmentant par exemple la
température d’extrusion des mélanges ou le temps de séjour pour favoriser les
réactions PU-compatibilisants. Industriellement, une extrusion à température
élevée (240°C) dégrade le PU mais aussi la matrice polypropylène.
- La modification du mode de préparation des échantillons en essayant d’étudier des
mélanges plus représentatifs obtenus par broyage de plaques revêtues de fines
couches de PU.
148

Chapitre III : Elaboration et Compatibilisation de mélanges (PP/EPR)/PU réticulés en vue de recyclage des boucliers peints
Références
[1] Gavalda J.P. Le recyclage et la valorisation des déchets en réparation Carrosserie Peinture
01-12-05, 1993, I.U.F.M. de Créteil.
149

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
150

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
Chapitre IV
Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3.
Influence de compatibilisants
151

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
152

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
Chapitre IV :
Caractérisation de mélanges (PP/EPR)/nano-CaCO3.
Influence de compatibilisants
IV-1. Introduction :
L'introduction de charges de taille nanométrique dans une matrice polymère permet
d'obtenir des matériaux aux propriétés améliorées ou nouvelles, comme les propriétés
thermiques, mécaniques, électriques, optiques, de retardateur de feu ou barrière. Dans le cas
particulier des mélanges PP/élastomères, la présence de l’EPR augmente la résilience mais
diminue le module d’Young et la contrainte maximale des mélanges. L’ajout d’un certain
pourcentage de nanoparticules peut, dans ce cas, en fonction de la qualité de l’interface et des
constituants, améliorer les propriétés des mélanges renforcés pour les faibles déformations.
Dans ce chapitre, on se propose d’étudier l’influence de l’ajout des nano-CaCO3 sur
les propriétés morphologiques, mécaniques, rhéologiques à l’état solide et thermiques des
mélanges (PP/EPR) en présence et en absence de compatibilisants. Les résultats obtenus
serviront dans la suite à :
Optimiser le pourcentage de nanocharges à introduire dans la matrice (PP/EPR),
Prédire les propriétés des mélanges renforcés selon la nature du traitement de surface
appliqué sur les nano-CaCO3,
Etudier les interactions polypropylène/élastomère/charges minérales,
Connaître l’effet de compatibilisants sur l’interface polymère/nano-CaCO3,…
IV-2. Etude des propriétés thermiques :
Le carbonate de calcium est connu comme étant un agent de nucléation. Il a
généralement pour effet d’augmenter le taux de cristallinité et la température de cristallisation
du polypropylène dans les mélanges PP/CaCO3. Par contre, pour des mélanges plus
153

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants complexes, certains auteurs [2-4] ont pu observer d’autres phénomènes liés à la composition
et aux propriétés des constituants des mélanges.
Chan et al. [1] ont étudié l’effet de l’ajout du CaCO3 non traité sur le taux de
cristallisation du PP pour des échantillons à différents pourcentages de CaCO3 (de
granulométrie de l’ordre de 44 nm) par analyse calorimétrique différentielle DSC. Les
résultats ont montré que l’ajout de CaCO3 ne modifie pas le taux de cristallinité ; par contre il
contribue à l’augmentation de la température de cristallisation de 10°C. Ils remarquent aussi
qu’il y a eu formation d’un nouveau pic révélant la formation d’une nouvelle phase du PP qui
est la phase β (structure hexagonale) et qui pourra être à l’origine d’une augmentation de la
déformation et de la contrainte à la rupture. Zebarjad et al. [2] ont aussi étudié l’influence de
l’ajout de CaCO3 non traités sur la cristallisation du PP par des analyses DSC. Les résultats
montrent une augmentation de la température de cristallisation et une diminution de
l’enthalpie de fusion des mélanges dues à la présence de l’agent de nucléation (CaCO3) dans
la matrice PP. Dans le cas des mélanges PP/EOC (ethylene-octene copolymer) contenant des
nano-CaCO3 traités par l’acide stéarique, Premphet et al. [3] montrent que la présence de
l’acide stérique diminue l’énergie libre de surface des nanocharges par rapport à ceux non
traités et la variation de la température de cristallisation devient alors moins significative (∆T
= 4°C pour les nano-CaCO3 traités acide stéarique et ∆T = 22°C pour les nano-CaCO3 non
traités). Les mêmes résultats ont été obtenus pour des mélanges PP/EPDM/CaCO3 selon les
travaux de Kolarik et al. [4]
Les propriétés thermiques de nos mélanges renforcés par des nano-CaCO3 à écorce
formée d’acide gras avec et sans addition d’agent compatibilisant, ont été analysées par
calorimétrie différentielle à balayage à 10°C/min et à -10°C/min afin d'observer l'influence de
l'addition de ces charges sur les températures de cristallisation et de fusion et sur le taux de
cristallinité du polymère.
IV-2-1. Les mélanges non compatibilisés :
Les figures IV.1 et IV.2 ou le tableau IV.1 présentent les propriétés thermiques des
mélanges renforcés nano-CaCO3 (Socal et Winnofil) et non compatibilisés. On remarque que,
contrairement à ce qui a été décrit dans la bibliographie pour les mélanges PP/CaCO3 [5-6], la
présence des nano-CaCO3 dans nos mélanges (PP/EPR) ne modifie pas les températures de
154

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants fusion et de cristallisation ainsi que le taux de cristallinité du PP ; en effet, les nano-CaCO3
utilisés dans notre étude comportent une « écorce » acide gras (majoritairement acide
stéarique) qui isole, au moins partiellement, le CaCO3 du PP et de l’EPR, avec des chaînes
aliphatiques. Dans ce cas, seule la fraction non enrobée des particules de nano-CaCO3 pourra
jouer le rôle d’agent de nucléation décrit dans la bibliographie [5-6].
A vu de nos résultats en DSC, il semble qu’une large fraction de la surface de nos
nano-CaCO3 soit recouverte d’acides gras ce qui ne leurs permet pas d’augmenter le taux de
cristallinité et modifier les températures de fusion et de cristallisation.
Tableau IV. 1: Propriétés thermiques des mélanges (PP/EPR) renforcés CaCO3 et non
compatibilisés
Mélanges TcPP(°C) TmPP(°C) ∆Hm(J/gPP) Cristallinité (χc) (PP/EPR) 126 169 -100 48
(PP/EPR)/5%W 127 169 -105 50 (PP/EPR)/10%W 126 167 -105 50
(PP/EPR)/15%W 124 168 -102 49 (PP/EPR)/5%S 126 170 -104 50 (PP/EPR)/10%S 126 167 -104 50 (PP/EPR)/15%S 126 168 -102 49
155
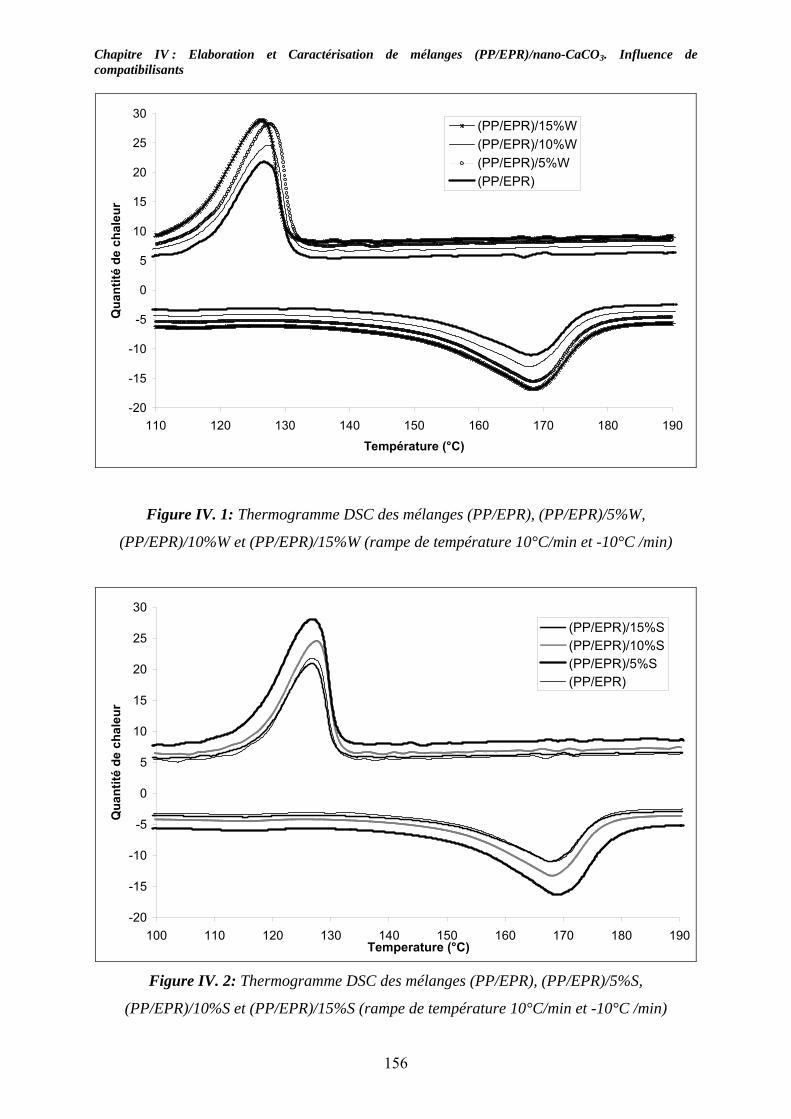
Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
-20
-15
-10
-5
0
5
10
15
20
25
30
110 120 130 140 150 160 170 180 190
Température (°C)
Qua
ntité
de
chal
eur
(PP/EPR)/15%W(PP/EPR)/10%W(PP/EPR)/5%W(PP/EPR)
Figure IV. 1: Thermogramme DSC des mélanges (PP/EPR), (PP/EPR)/5%W,
(PP/EPR)/10%W et (PP/EPR)/15%W (rampe de température 10°C/min et -10°C /min)
-20
-15
-10
-5
0
5
10
15
20
25
30
100 110 120 130 140 150 160 170 180 190Temperature (°C)
Qua
ntité
de
chal
eur
(PP/EPR)/15%S(PP/EPR)/10%S(PP/EPR)/5%S(PP/EPR)
Figure IV. 2: Thermogramme DSC des mélanges (PP/EPR), (PP/EPR)/5%S,
(PP/EPR)/10%S et (PP/EPR)/15%S (rampe de température 10°C/min et -10°C /min)
156
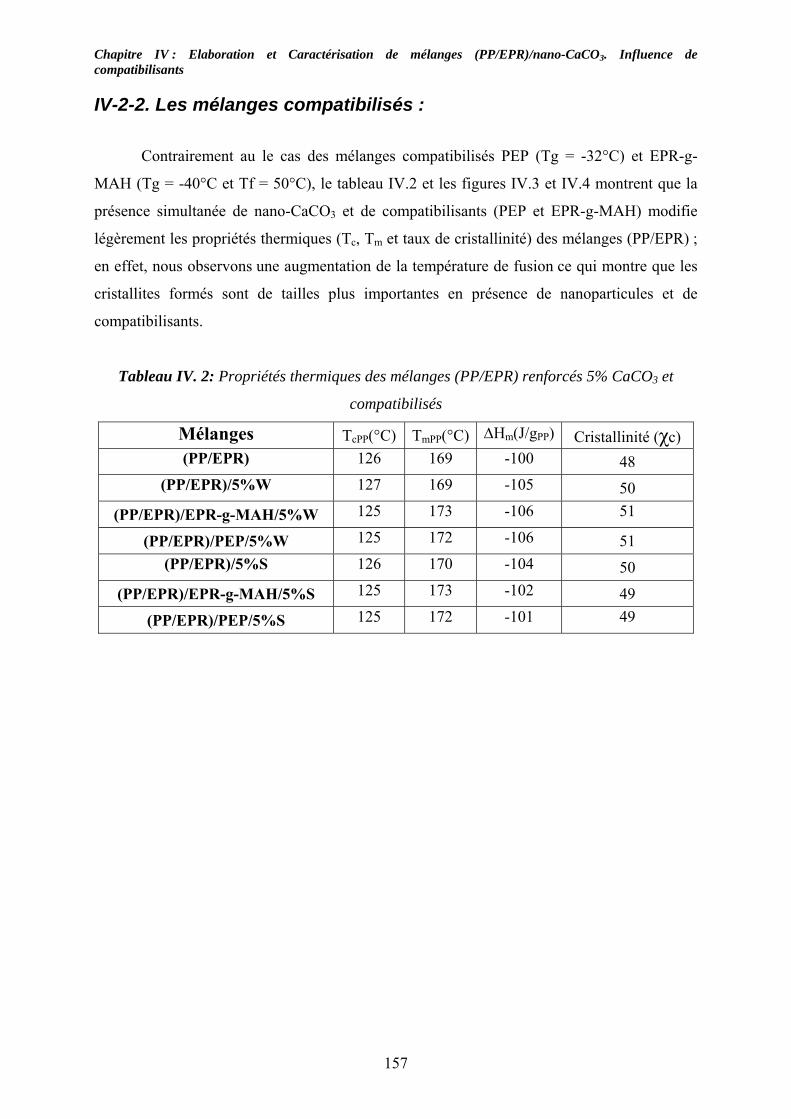
Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants IV-2-2. Les mélanges compatibilisés :
Contrairement au le cas des mélanges compatibilisés PEP (Tg = -32°C) et EPR-g-
MAH (Tg = -40°C et Tf = 50°C), le tableau IV.2 et les figures IV.3 et IV.4 montrent que la
présence simultanée de nano-CaCO3 et de compatibilisants (PEP et EPR-g-MAH) modifie
légèrement les propriétés thermiques (Tc, Tm et taux de cristallinité) des mélanges (PP/EPR) ;
en effet, nous observons une augmentation de la température de fusion ce qui montre que les
cristallites formés sont de tailles plus importantes en présence de nanoparticules et de
compatibilisants.
Tableau IV. 2: Propriétés thermiques des mélanges (PP/EPR) renforcés 5% CaCO3 et
compatibilisés
Mélanges TcPP(°C) TmPP(°C) ∆Hm(J/gPP) Cristallinité (χc) (PP/EPR) 126 169 -100 48
(PP/EPR)/5%W 127 169 -105 50
(PP/EPR)/EPR-g-MAH/5%W 125 173 -106 51
(PP/EPR)/PEP/5%W 125 172 -106 51 (PP/EPR)/5%S 126 170 -104 50
(PP/EPR)/EPR-g-MAH/5%S 125 173 -102 49
(PP/EPR)/PEP/5%S 125 172 -101 49
157
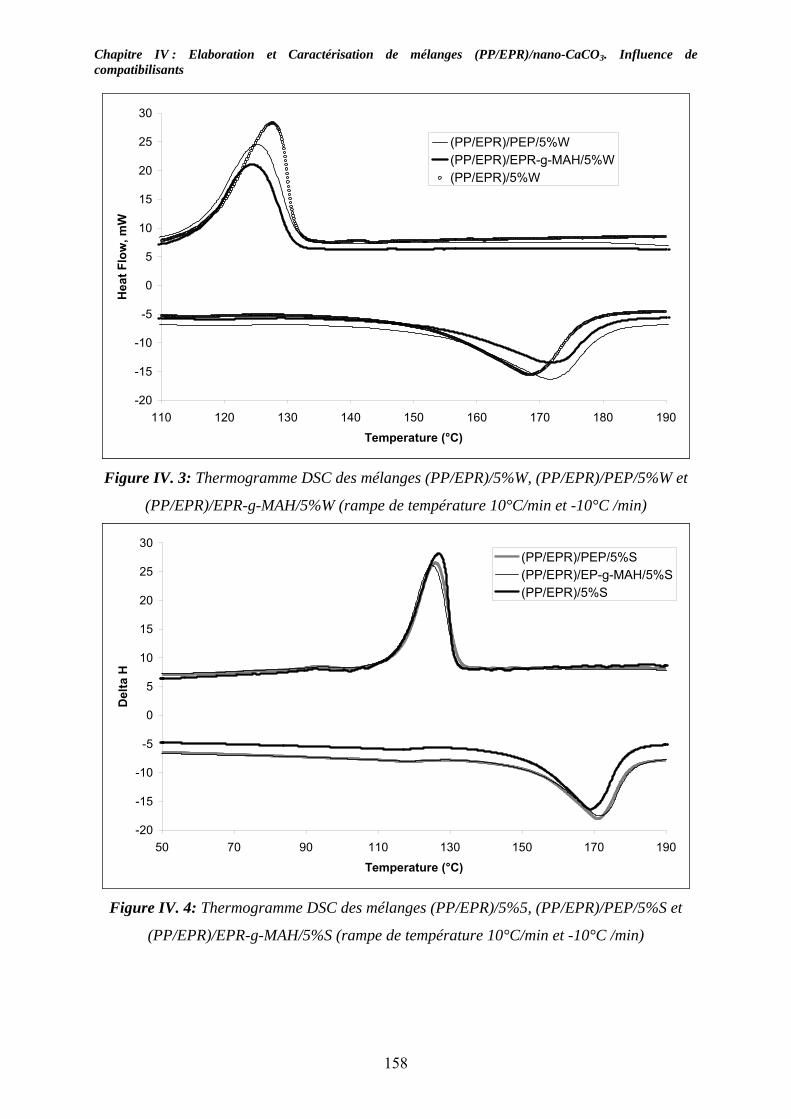
Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
-20
-15
-10
-5
0
5
10
15
20
25
30
110 120 130 140 150 160 170 180 190
Temperature (°C)
Hea
t Flo
w, m
W
(PP/EPR)/PEP/5%W(PP/EPR)/EPR-g-MAH/5%W(PP/EPR)/5%W
Figure IV. 3: Thermogramme DSC des mélanges (PP/EPR)/5%W, (PP/EPR)/PEP/5%W et
(PP/EPR)/EPR-g-MAH/5%W (rampe de température 10°C/min et -10°C /min)
-20
-15
-10
-5
0
5
10
15
20
25
30
50 70 90 110 130 150 170 190
Temperature (°C)
Del
ta H
(PP/EPR)/PEP/5%S(PP/EPR)/EP-g-MAH/5%S(PP/EPR)/5%S
Figure IV. 4: Thermogramme DSC des mélanges (PP/EPR)/5%5, (PP/EPR)/PEP/5%S et
(PP/EPR)/EPR-g-MAH/5%S (rampe de température 10°C/min et -10°C /min)
158

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
L’étude des propriétés thermiques des mélanges renforcés nano-CaCO3 comportant
une écorce ne modifie que légèrement les propriétés thermiques des mélanges. En effet, le
traitement appliqué à la surface des nanoparticules isole, au moins partiellement, le CaCO3 du
PP ce qui fait que nos nano-CaCO3 ne joue pas ou peu un rôle d’agent de nucléation ce qui
signifie que le taux de recouvrement de nos particules par les acides gras est important.
L’ajout de compatibilisants aux mélanges renforcés diminue la température de cristallisation,
augmente la taille des cristallites et modifie légèrement le taux de cristallinité (chapitre II).
IV-3. Etude des propriétés viscoélastiques à l’état solide : Lors de l’étude en spectroscopie mécanique dynamique, nous avons mentionné dans le
paragraphe 3.4.2.2 du chapitre 3 de la partie bibliographique que la présence de nanocharges
dans un polymère peut donner naissance à de nouveaux pics de relaxation différents de ceux
de la matrice. En effet, la fraction de polymères moins mobile autour des nano-CaCO3 peut
être à l’origine de nouveaux pics : la mise en mouvement des macromolécules autour des
nanocharges devient plus difficile, ce qui peut aussi engendrer un déplacement des pics de
relaxation vers les hautes températures. Dans le cas contraire, si l’adhésion à l’interface
polymères/charges minérales est faible, les nanocharges peuvent jouer le rôle de plastifiant et
la mise en mouvement des macromolécules devient plus facile, ce qui déplace les
températures des pics de relaxation vers les basses températures.
IV-3-1. Cas des mélanges non compatibilisés :
Les résultats obtenus lors de l’étude des propriétés viscoélastiques à l’état solide des
mélanges renforcés par des nano-CaCO3 (S et W) à 5%, 10% et 15% sont donnés sur le
tableau IV.3 et les figures IV.5 et IV.6.
On remarque qu’à taux de nano-CaCO3 croissant, on observe :
une augmentation de la valeur de la partie réelle du module de cisaillement G' sur
toute la gamme de température analysée,
Contrairement à ce que nous avons obtenu (décalage des maxima des pics de
relaxations vers les basses températures), Premphet at al. [3, 8] et Kolarik et al [4] ont
159
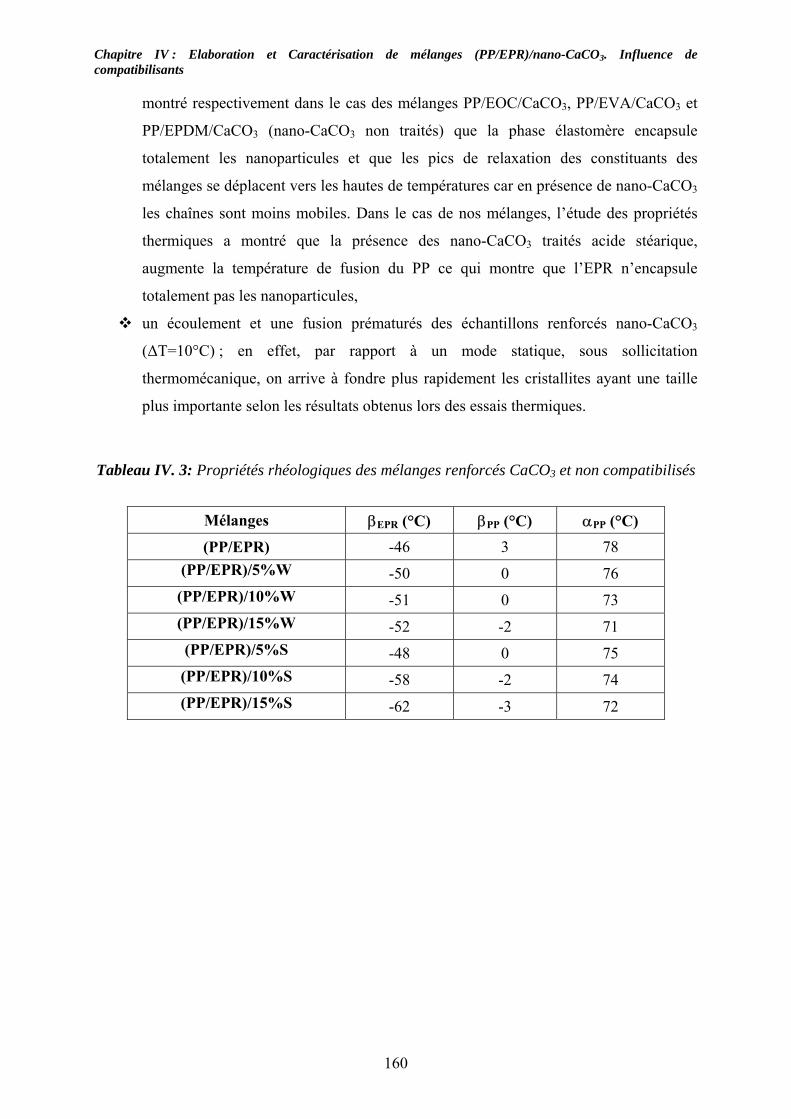
Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
montré respectivement dans le cas des mélanges PP/EOC/CaCO3, PP/EVA/CaCO3 et
PP/EPDM/CaCO3 (nano-CaCO3 non traités) que la phase élastomère encapsule
totalement les nanoparticules et que les pics de relaxation des constituants des
mélanges se déplacent vers les hautes de températures car en présence de nano-CaCO3
les chaînes sont moins mobiles. Dans le cas de nos mélanges, l’étude des propriétés
thermiques a montré que la présence des nano-CaCO3 traités acide stéarique,
augmente la température de fusion du PP ce qui montre que l’EPR n’encapsule
totalement pas les nanoparticules,
un écoulement et une fusion prématurés des échantillons renforcés nano-CaCO3
(∆T=10°C) ; en effet, par rapport à un mode statique, sous sollicitation
thermomécanique, on arrive à fondre plus rapidement les cristallites ayant une taille
plus importante selon les résultats obtenus lors des essais thermiques.
Tableau IV. 3: Propriétés rhéologiques des mélanges renforcés CaCO3 et non compatibilisés
Mélanges βEPR (°C) βPP (°C) αPP (°C) (PP/EPR) -46 3 78
(PP/EPR)/5%W -50 0 76 (PP/EPR)/10%W -51 0 73 (PP/EPR)/15%W -52 -2 71 (PP/EPR)/5%S -48 0 75 (PP/EPR)/10%S -58 -2 74 (PP/EPR)/15%S -62 -3 72
160
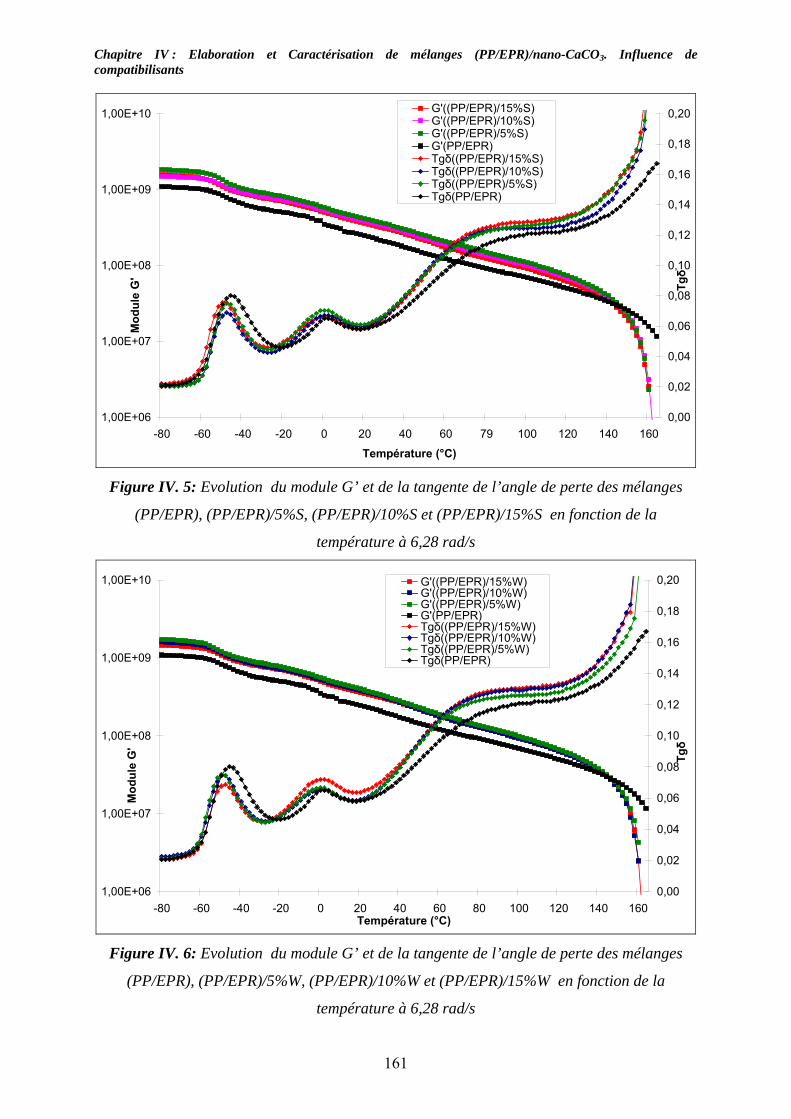
Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
1,00E+06
1,00E+07
1,00E+08
1,00E+09
1,00E+10
-80 -60 -40 -20 0 20 40 60 79 100 120 140 160
Température (°C)
Mod
ule
G'
0,00
0,02
0,04
0,06
0,08
0,10
0,12
0,14
0,16
0,18
0,20
Tgδ
G'((PP/EPR)/15%S)G'((PP/EPR)/10%S)G'((PP/EPR)/5%S)G'(PP/EPR)Tgδ((PP/EPR)/15%S)Tgδ((PP/EPR)/10%S)Tgδ((PP/EPR)/5%S)Tgδ(PP/EPR)
Figure IV. 5: Evolution du module G’ et de la tangente de l’angle de perte des mélanges
(PP/EPR), (PP/EPR)/5%S, (PP/EPR)/10%S et (PP/EPR)/15%S en fonction de la
température à 6,28 rad/s
1,00E+06
1,00E+07
1,00E+08
1,00E+09
1,00E+10
-80 -60 -40 -20 0 20 40 60 80 100 120 140 160Température (°C)
Mod
ule
G'
0,00
0,02
0,04
0,06
0,08
0,10
0,12
0,14
0,16
0,18
0,20
Tgδ
G'((PP/EPR)/15%W)G'((PP/EPR)/10%W)G'((PP/EPR)/5%W)G'(PP/EPR)Tgδ((PP/EPR)/15%W)Tgδ((PP/EPR)/10%W)Tgδ((PP/EPR)/5%W)Tgδ(PP/EPR)
Figure IV. 6: Evolution du module G’ et de la tangente de l’angle de perte des mélanges
(PP/EPR), (PP/EPR)/5%W, (PP/EPR)/10%W et (PP/EPR)/15%W en fonction de la
température à 6,28 rad/s
161

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
Dans le cas des mélanges PP/EPR, nous avons remarqué que la présence de nano-
CaCO3 décale toutes les valeurs des pics de relaxation du PP et de l’EPR vers les basses
températures (∆T = 3°C pour PP, ∆T = 4°C pour l’EPR). En effet, les nanoparticules se
dispersent dans la matrice en s’intercalant entre les macromolécules et en augmentant la
distance intermoléculaire ce qui facilite la mobilité des chaînes.
IV-3-2. Cas des mélanges compatibilisés :
Afin qu’on puisse mieux interpréter les résultats pour les mélanges (PP/EPR)
renforcés nano-CaCO3 et compatibilisés, nous avons réalisé des essais simplifiés avec une
matrice PP sans EPR en présence de PEP. Les résultats obtenus peuvent être résumé comme
suit :
- le spectre relatif au mélange PP/PEP, présente deux pics de relaxation vers -2°C
et 70°C qui correspondent respectivement à la transition vitreuse de la phase
amorphe et à la relaxation de la phase cristalline du PP et des blocs
polypropylènes du PEP. Le pic de relaxation associé au bloc PE n’a pu être
observé car il faut descendre à des températures plus basses (-120°C).
- dans le cas des mélanges PP/PEP/S, la présence des nano-CaCO3 (Socal) décale
le pic de relaxation de la phase amorphe βPP vers les hautes températures (∆T =
4°C), conformément aux résultats obtenus par Premphet et al. [3] et Hornsby et
al [8]. Contrairement aux mélanges contenant l’EPR, la présence des
nanoparticules dans le PP rend la mobilité des chaînes plus difficile, ce qui tend
encore à montrer que dans les mélanges (PP/EPR), les nano-CaCO3 vont
préférentiellement dans la phase EPR,
- les mêmes résultats sont obtenus dans le cas des mélanges PP/PEP/W.
Dans le cas des mélanges à matrice (PP/EPR), les résultats obtenus lors de l’étude des
propriétés viscoélastiques à l’état solide des mélanges renforcés par des nano-CaCO3 (W et S)
en présence de compatibilisants (PEP et EPR-g-MAH) ont donné les résultats présentés sur le
tableau IV.4 et les figures IV.7 et IV.8.
162
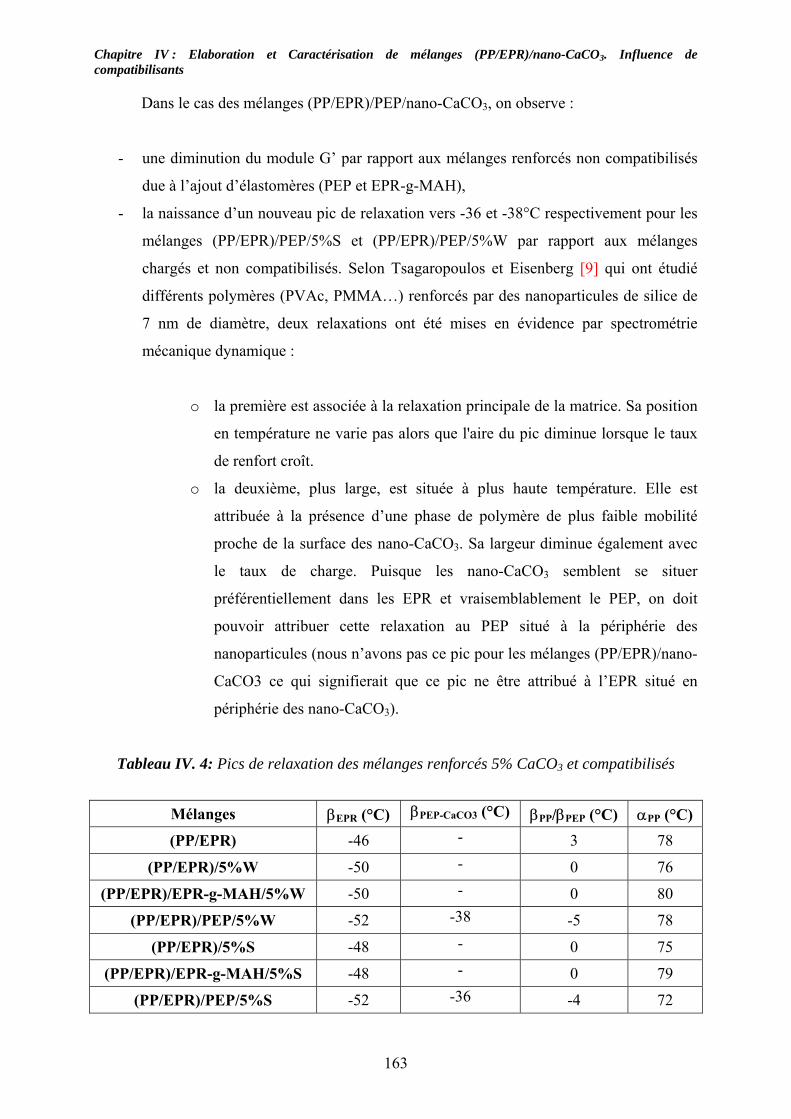
Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
Dans le cas des mélanges (PP/EPR)/PEP/nano-CaCO3, on observe :
- une diminution du module G’ par rapport aux mélanges renforcés non compatibilisés
due à l’ajout d’élastomères (PEP et EPR-g-MAH),
- la naissance d’un nouveau pic de relaxation vers -36 et -38°C respectivement pour les
mélanges (PP/EPR)/PEP/5%S et (PP/EPR)/PEP/5%W par rapport aux mélanges
chargés et non compatibilisés. Selon Tsagaropoulos et Eisenberg [9] qui ont étudié
différents polymères (PVAc, PMMA…) renforcés par des nanoparticules de silice de
7 nm de diamètre, deux relaxations ont été mises en évidence par spectrométrie
mécanique dynamique :
o la première est associée à la relaxation principale de la matrice. Sa position
en température ne varie pas alors que l'aire du pic diminue lorsque le taux
de renfort croît.
o la deuxième, plus large, est située à plus haute température. Elle est
attribuée à la présence d’une phase de polymère de plus faible mobilité
proche de la surface des nano-CaCO3. Sa largeur diminue également avec
le taux de charge. Puisque les nano-CaCO3 semblent se situer
préférentiellement dans les EPR et vraisemblablement le PEP, on doit
pouvoir attribuer cette relaxation au PEP situé à la périphérie des
nanoparticules (nous n’avons pas ce pic pour les mélanges (PP/EPR)/nano-
CaCO3 ce qui signifierait que ce pic ne être attribué à l’EPR situé en
périphérie des nano-CaCO3).
Tableau IV. 4: Pics de relaxation des mélanges renforcés 5% CaCO3 et compatibilisés
Mélanges βEPR (°C) βPEP-CaCO3 (°C) βPP/βPEP (°C) αPP (°C) (PP/EPR) -46 - 3 78
(PP/EPR)/5%W -50 - 0 76
(PP/EPR)/EPR-g-MAH/5%W -50 - 0 80
(PP/EPR)/PEP/5%W -52 -38 -5 78
(PP/EPR)/5%S -48 - 0 75
(PP/EPR)/EPR-g-MAH/5%S -48 - 0 79
(PP/EPR)/PEP/5%S -52 -36 -4 72
163
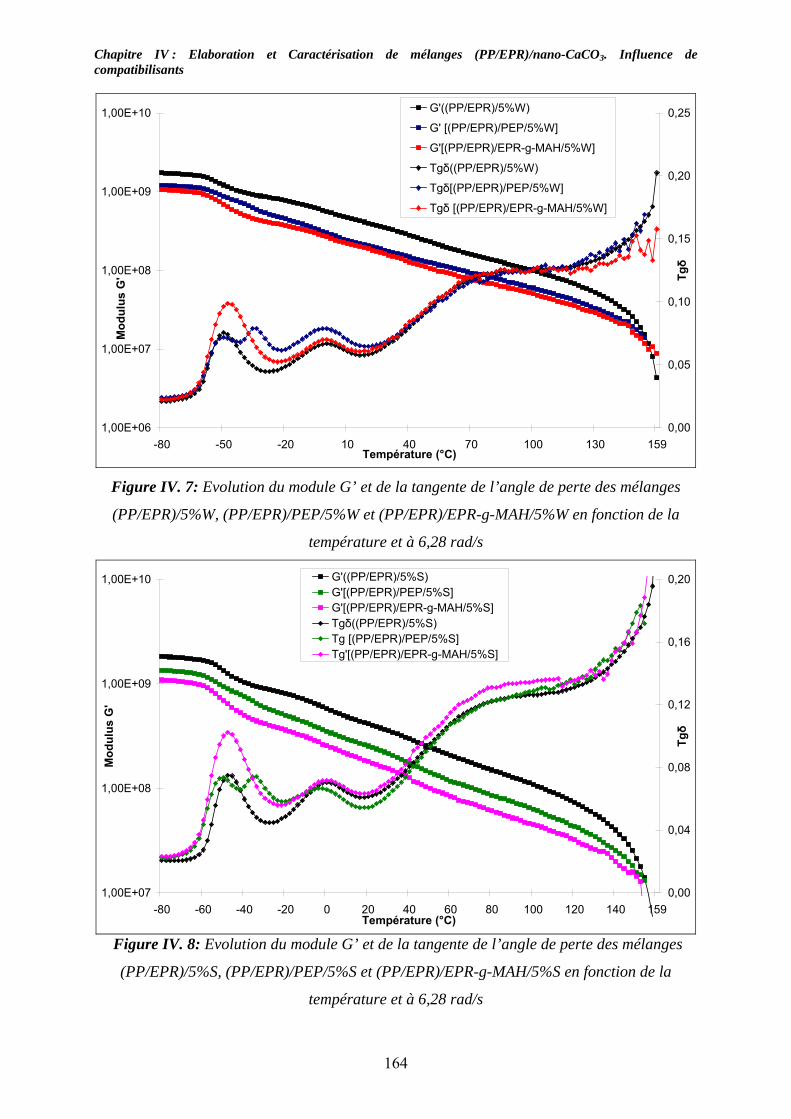
Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
1,00E+06
1,00E+07
1,00E+08
1,00E+09
1,00E+10
-80 -50 -20 10 40 70 100 130 159Température (°C)
Mod
ulus
G'
0,00
0,05
0,10
0,15
0,20
0,25
Tgδ
G'((PP/EPR)/5%W)
G' [(PP/EPR)/PEP/5%W]
G'[(PP/EPR)/EPR-g-MAH/5%W]
Tgδ((PP/EPR)/5%W)
Tgδ[(PP/EPR)/PEP/5%W]
Tgδ [(PP/EPR)/EPR-g-MAH/5%W]
Figure IV. 7: Evolution du module G’ et de la tangente de l’angle de perte des mélanges
(PP/EPR)/5%W, (PP/EPR)/PEP/5%W et (PP/EPR)/EPR-g-MAH/5%W en fonction de la
température et à 6,28 rad/s
1,00E+07
1,00E+08
1,00E+09
1,00E+10
-80 -60 -40 -20 0 20 40 60 80 100 120 140 159Température (°C)
Mod
ulus
G'
0,00
0,04
0,08
0,12
0,16
0,20
Tgδ
G'((PP/EPR)/5%S)G'[(PP/EPR)/PEP/5%S]G'[(PP/EPR)/EPR-g-MAH/5%S]Tgδ((PP/EPR)/5%S)Tg [(PP/EPR)/PEP/5%S]Tg'[(PP/EPR)/EPR-g-MAH/5%S]
Figure IV. 8: Evolution du module G’ et de la tangente de l’angle de perte des mélanges
(PP/EPR)/5%S, (PP/EPR)/PEP/5%S et (PP/EPR)/EPR-g-MAH/5%S en fonction de la
température et à 6,28 rad/s
164

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
L’étude des propriétés à l’état solide des mélanges renforcés par des nano-CaCO3 et
renforcés/compatibilisés par du PEP ou de l’EPR-g-MAH montre que la présence de nano-
CaCO3 affecte l’ensemble des pics de relaxation des constituants des mélanges en les
déplaçant vers les basses températures ; en effet, les nanoparticules traitées par l’acide
stéarique s’intercalent entre les macromolécules en facilitant le mouvement des chaînes. Le
traitement appliqué à la surface des nanocharges diminue la tension de surface et limite le
phénomène d’encapsulation observé pour les mélanges PP/EOC et PP/EPDM [3-4]. Dans le
cas des mélanges renforcés et compatibilisés par l’EPR-g-MAH, on n’observe qu’un faible
déplacement du pic de relaxation associé à la phase cristalline du PP. Pour les mélanges
(PP/EPR)/PEP/nanocharges, la présence du copolymère PEP donne naissance à un nouveau
pic de relaxation vers -38°C associé à une fraction volumique de charges critiques, selon
Tsagaropoulos et Eisenberg [9], qui réduisent la mobilité d’une partie des polymères et sont à
l'origine d’une seconde relaxation.
IV-4. Etude des propriétés morphologiques :
Lors de l’étude bibliographique, nous avons montré que la majorité des auteurs se sont
focalisés sur le contrôle des morphologies en agissant sur les interactions physico-chimiques
développées aux interfaces. Ces études mettent également en évidence le faible niveau
d’interactions créées entre les nanocharges fonctionnalisées et la matrice polypropylène
apolaire et la nécessité d’introduire un agent compatibilisant interagissant avec ces deux
composés. Généralement, cet agent est un polypropylène greffé anhydride maléique, noté
PP-g-MAH dans le cas des mélanges à matrice polypropylène. L’anhydride maléique polaire
peut interagir avec la surface des nanocharges, les chaînes du PP-g-MAH peuvent interagir
avec celles de même nature que la matrice par inter-diffusion et/ou co-cristallisation.
Deux paramètres entrent alors en compte dans le choix du compatibilisant : quelle
quantité de PP-g-MAH doit être introduite pour créer suffisamment d’interactions avec les
nanocharges et quelles doivent être les caractéristiques des chaînes macromoléculaires du
compatibilisant pour former une interphase avec la matrice ? Afin de répondre à ces
interrogations, nous avons observé, dans un premier temps, l’influence de différentes
nanocharges sur la matrice (PP/EPR) puis l’effet de l’introduction de compatibilisants sur les
165

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants propriétés des mélanges chargés sur l’état de dispersion des nanocharges à l’aide de notre
extrudeuse bivis.
IV-4-1. Cas des mélanges non compatibilisés : Sur la figure IV.9 sont représentées les morphologies des mélanges (PP/EPR) et
(PP/EPDM/talc) en présence de 5% de nano-CaCO3 (Winnofil et Socal). La dispersion des
nano-CaCO3 est comparable dans les matrices (PP/EPR) et (PP/EPDM)/talc, et, de ce fait, par
la suite, nous nous intéresserons seulement aux mélanges (PP/EPR).
Dans le cas des mélanges renforcés avec les particules CaCO3, la dispersion des
nanocharges dans la matrice (PP/EPR) est fonction de la nature du traitement appliqué à la
surface, du protocole et du procédé de mise en œuvre et du pourcentage de nano-CaCO3 [10].
Dans le cas des mélanges (PP/EPR)/5%W et (PP/EPDM/talc)/5%W (figure III.9.(c) et (d)), la
dispersion des nanocharges sur l’ensemble du faciès de rupture est bonne et la taille des
agglomérats de particules ne dépasse pas 2µm. Par contre, pour les mélanges (PP/EPR)/5%S
et (PP/EPDM/talc)/5%S (figure III.9.(a) et (b)), les agglomérats de particules sont plus gros,
de forme rectangulaire ou ellipsoïdale (jusqu’à 12x5µm²), avec un faciès de rupture plus
fragile que dans le cas des mélanges (PP/EPR)/5%W et (PP/EPDM/talc)/5%W. Ceci montre
que la présence d’acide stéarique sur la surface des particules (figure III.9.(a) et (b)) ne permet
pas une bonne dispersion des nanocharges lors de la mise en œuvre par extrusion. Selon
Zhang et al. [11], dans le cas de mélanges PP/EOC (Copolymère d’éthylène-octène)
l’adhésion entre la matrice et les particules est moins bonne pour des CaCO3 avec écorce
acide stéarique comparativement à des nano-CaCO3 non traités ; en effet, l’énergie libre de la
surface est plus importante dans le cas des nanocharges traitées ce qui minimise les
interactions charges/polymères ; en effet, avec les fonctions –COOH, on peut avoir formation
de liaisons hydrogène entre les particules d’acide stéarique en surface des nano-CaCO3. Pour
les mélanges (PP/EPR)/W et (PP/EPDM/talc)/W, un meilleur ratio d’enrobage (1,5mg/m²)
des nanoCaCO3 (Winnofil) conduit à des agrégats d’environ 1 à 2µm alors que dans le cas
du Socal 322V, des particules de petites tailles (30-50nm) et un ratio d’enrobage de 1,1 mg/m²
conduit à des agrégats de taille comprise entre 3 et 12µm .
166

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
(a) : (PP/EPR)/5%S (b) : (PP/EPDM/talc)/5%S
(c) : (PP/EPR)/5%W (d) : (PP/EPDM/talc)/5%W
Figure IV. 9: Morphologie des mélanges (a) (PP/EPR)/5%S, (b) (PP/EPDM/talc)/5%S,
(c) (PP/EPR)/5%W et (d) (PP/EPDM/talc)/5%W
167

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants III-4-2. Etude de la morphologie des mélanges (PP/EPR)/nano-CaCO3 compatibilisés: La mauvaise dispersion des nano-CaCO3 dans la matrice (PP/EPR) et la formation
d’agglomérats de grande taille est défavorable du point de vue des propriétés mécaniques. De
ce fait, dans cette partie, nous allons nous intéresser à l’étude de la morphologie de mélanges
(PP/EPR) chargés nano-CaCO3 en présence de compatibilisants. D’après les travaux menés
par Xu Wang et al. [10] sur les mélanges PP/EPDM/nano-CaCO3, la dispersion des nano-
CaCO3 (traités acide stéarique et de diamètre 40nm) dans une matrice PP/EPDM dépend du
protocole et du procédé de mise en œuvre. Trois configurations peuvent se présenter :
Si les constituants PP, EPDM et nano-CaCO3 sont mélangés dans une extrudeuse bivis
(200°C) en même temps, alors la morphologie (Figure IV.10.a) comportera une partie
de nano-CaCO3 dispersés dans le PP et une deuxième localisée dans l’EPDM : à priori
ce protocole est le plus proche de notre travail de thèse,
Une morphologie (Figure IV.10.b) nodulaire sans aucune interaction entre EPDM et
nano-CaCO3 si le PP et les nano-charges sont prémélangés et l’EPDM ajouté dans
l’extrudeuse,
Les nano-CaCO3 sont complètement noyés dans l’EDPM s’ils sont prémélangés
ensembles avant d’être extrudés avec le PP (Figure IV.10.c).
Lors de l’étude des propriétés mécaniques en traction et aux chocs, Xu Wang et al. [10]
ont montré que :
- la contrainte maximale des mélanges présentés sur les figures IV.10.(a) et IV.10.(b),
diminue par ajout de nano-CaCO3,
- la résilience des mélanges IV.10.(a) et IV.10.(b) augmente en fonction du taux de
nanoparticules avec un maximum respectivement vers 4% et 10% en masse,
- les propriétés aux chocs des mélanges IV.10.(c) s’améliorent par ajout de nano-CaCO3
pour atteidre un optimum à 22% en masse,
- le module d’Young des trois mélanges augmente pour atteindre un maximum entre 4
et 10% de nanocharges.
168

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
(a) (b) (c)
Figure III. 10: Dispersion des nano-CaCO3 et de l’EPDM dans le PP [10]
C’est alors en fonction de la morphologie souhaitée (figures IV.10.(a), IV.10.(b) et
IV.10.(c)) que l’on devra choisir le procédé et peut être les compatibilisants les plus
pertinents. En effet, dans le cas des figures IV.10.(a) et IV.10.(b), un compatibilisant qui
pourra interagir avec les nano-CaCO3 améliorera les propriétés des mélanges. Par contre, pour
une morphologie type figure IV.10.(c), une amélioration de l’interface PP/élastomères
conduira à de meilleures propriétés. Pour cela, nous allons nous intéresser à l’étude des
mélanges (PP/EPR)/nano-CaCO3 en présence de compatibilisants fonctionnalisés, EPR-g-
MAH, et non fonctionnalisés, PEP, qui seront dispersés dans les mélanges chargés pour
essayer d’améliorer respectivement les interactions à l’interface polymères/charges minérales
et polymères/élastomères.
Les figures IV.9.(a)-(b) et IV.11 présentent les morphologies des mélanges
(PP/EPR)/5%W et (PP/EPR)/5%S avec et sans compatibilisants (PEP et EPR-g-MAH). Pour
mieux observer l’adhésion aux interfaces, nous avons choisi un agrandissement plus
important que celui utilisé dans le paragraphe II.2.1 où nous avons montré que la dispersion
des nano-CaCO3 dans la matrice (PP/EPR) dépend du traitement appliqué à la surface des
nanoparticules.
La présence du copolymère EPR-g-MAH dans les mélanges (PP/EPR)/5%W et
(PP/EPR)/5%S (figures IV.11.(c) et IV.11.(d)) provoque :
- une augmentation de la taille des nodules de la phase dispersée EPR,
- l’apparition de nodules EPR contenant des nano-CaCO3,
169

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
- une fragilisation de l’ensemble du faciès de rupture comme dans le cas des mélanges
non chargés observés sur la figure II.2 du chapitre II,
- une mauvaise dispersion des nanoparticules et l’apparition d’agrégats de particules
même dans le cas des nano-CaCO3 winnofil bien dispersés dans la matrice en absence
de compatibilisants,
- une agrégation des nanoparticules CaCO3 sur le faciès de rupture comme dans le cas
des mélanges non compatibilisés. Cette mauvaise dispersion est due à de fortes
interactions particule/particule. Les particules CaCO3 possèdent une énergie de surface
libre importante et elles tendent donc à se regrouper et à former des agrégats de taille
plus ou moins importante en présence du compatibilisant EPR-g-MAH. Ces résultats
peuvent être interprétés avec l’hypothèse que l’anhydride maléique, qui est
vraisemblablement partiellement hydraté en acide maléique va former des liaisons H
avec l’acide stéarique en surface des nanocharges ce qui va entraîner la formation
d’agglomérats plus importants qu’en absence d’EPR-g-MAH.
Dans le cas des mélanges (PP/EPR)/PEP/5%W et (PP/EPR)/PEP/5%S (figures IV.11.(e)
et IV.11.(f)), la présence du copolymère PEP engendre :
- une adhésion meilleure entre les constituants de la matrice PP et EPR observée dans le
chapitre II et confirmée dans le paragraphe suivant lors de l’étude des propriétés
mécaniques,
- un faciès de rupture ductile avec une meilleure dispersion de la phase élastomère EPR,
- l’absence d’agglomérats de particules de CaCO3, particulièrement dans le cas des
nanocharges Winnofil qui possèdent un taux de recouvrement moyen en acide
stéarique plus important (1,5mg/m²) par rapport au Socal (1,1mg/m²),
- une meilleure dispersion des différents constituants des mélanges (PP, EPR et nano-
CaCO3),
- une diminution de la taille de la phase EPR.
170

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
(a)
Figure IV. 11: Morphologie des mélanges (a) (PP/EPR)/5%W, (b) (PP/EPR)/5%S
(Partie 1/3)
Matrice PP
Nodule EPR
Particules CaCO3 (W)
Cavités vides d’EPR arrachés
(b) Nodule EPR Particules CaCO3 (S)
Agglomérats de particules CaCO3 (S)
Matrice PP Cavités vides
d’EPR arrachés
171

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
(c) Matrice PP
Nodules EPR/CaCO3 (W)
Nodules EPR et /ou EPR-g-MAH
Agglomérats de particules CaCO3 (S)
(d)
Nodules EPR et/ou EPR-g-MAH
Nodules EPR/CaCO3
Matrice PP
Figure IV.11 : Morphologie des mélanges (c) (PP/EPR)/EPR-g-MAH/5%W, (d)
(PP/EPR)/EPR-g-MAH/5%S (Partie 2/3)
172

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
(e)
Matrice PP
Cavités d’EPR et/ou PEP
Particules CaCO3 (W)
Nodules EPR et/ou PEP
(f)
Cavités d’EPR et/ou PEP
Particule CaCO3 (S)
Nodules EPR et/ou PEP
Matrice PP
Figure IV.11 : Morphologie des mélanges (e) (PP/EPR)/PEP/5%W et (f)
(PP/EPR)/PEP/5%S (Partie 3/3)
173

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
IV-5. Caractérisations mécaniques des mélanges renforcés :
La dispersion de charges (CaCO3, mica, talc,…) dans les matrices thermoplastiques est
actuellement une pratique courante dans les industries plastiques afin de réduire les coûts de
production de pièces et d’améliorer leurs propriétés. De nombreux auteurs ont montré que les
charges minérales dispersées dans une matrice PP augmentent la rigidité des polymères, mais
elles diminuent plus ou moins la ductilité en fonction de la nature de la matrice, les
constituants des mélanges et essentiellement du traitement appliqué à la surface des
nanoparticules [12-15]. Pour améliorer les interfaces entre les constituants, des
compatibilisants sont introduits dans les mélanges [18] . Maiti et al. [12], en étudiant les
mélanges PP/CaCO3, ont observé des variations significatives de l’ensemble des propriétés
mécaniques, dont l'augmentation de 25% de la résistance aux chocs avec des nano-CaCO3 non
traités.
L’étude des propriétés mécaniques de nanocomposites nous permet de compléter les
informations issues des analyses morphologiques précédentes. La structuration des
nanocharges influence le comportement mécanique du composite. Conformément aux
résultats obtenus par Boucard [17], lorsque les nanocharges sont sous forme d’amas
micrométriques, le composite présente un module de flexion élevé et une faible résistance aux
chocs. Toutefois, ces observations ne peuvent être attribuées à la structuration seule car celles-
ci dépendent également des constituants présents dans les mélanges (compatibilisants,
polluants,…). Or, ces derniers ont également un effet prépondérant sur les propriétés
mécaniques des nanocomposites.
IV-5-1. Cas des mélanges renforcés nano-CaCO3 et non compatibilisés :
Dans le cas des mélanges (PP/EPR), nous allons nous intéresser, dans cette partie, à
l’étude de l’influence du pourcentage et de la nature des nanocharges sur les propriétés
mécaniques en traction et en choc Charpy. Le tableau IV.5 regroupe l’ensemble des résultats
obtenus en étudiant les propriétés mécaniques de mélanges (PP/EPR)/nanocharges pour les
deux références Socal et Winnofil, ceci pour des pourcentages de 5, 10 et 15% de nano-
CaCO3.
174

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants Dans la suite de ce travail, la valeur des modules d’Young ne sera pas mentionnée car
les essais de traction ont été réalisés sans extensiomètre ; en effet, vue la grande vitesse de la
traverse utilisée dans notre étude (500 mm/min), la fixation de l’extensiomètre était trop
difficile. Cette vitesse a été utilisée car l’allongement à la rupture des mélanges était très
important.
Les résultats obtenus montrent que :
- la contrainte maximale reste constante quelle que soit la nature et le pourcentage des
nano-CaCO3 ajoutés. La faible variation observée est due aux erreurs expérimentales
engendrées par l’hétérogénéité au niveau des échantillons (figure IV.12),
- L’énergie à la rupture représentée par l’aire au dessous de la courbe σ = f(ε) est deux
fois plus importante en présence de nanocharges par rapport aux mélanges non
renforcés PP/EPR. Elle reste constante pour des mélanges (PP/EPR)/W contenant 5 et
15% de nano-CaCO3 et pour les mélanges (PP/EPR)/S contenant entre 5 et 10%. Une
diminution importante de presque 400% est observée pour le mélange
(PP/EPR)/15%S ; la mauvaise dispersion des nano-CaCO3 Socal et la faible adhésion
à l’interface avec la matrice (PP/EPR) observée lors de l’étude morphologique est à
l’origine de cette variation (figure IV.12),
- les valeurs de l’allongement à la rupture décroissent pour les pourcentages de 10 et
15% de nanocharges. Cette diminution est moins marquée dans le cas des mélanges
(PP/EPR)/W qui présentent une meilleure dispersion et une bonne adhésion avec la
matrice (PP/EPR) selon l’étude morphologique (figure IV.13),
- L’ajout de nanocharges fait diminuer la résilience, même pour un faible pourcentage
(5%). Cette diminution est plus marquée pour les mélanges à fort pourcentage de
nanocharges (15%), ce qui est encore une fois en bon accord avec les observations
morphologiques (figure IV.13) où on semblait voir une fragilisation du faciès de
rupture.
175

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants Tableau IV. 5: Propriétés mécaniques des mélanges renforcés CaCO3 et non compatibilisés
Essai de Traction Chocs Charpy
Mélanges Contrainte
maximale (MPa)
Allongement à
la rupture (%)
Energie à la
rupture (J)
Résilience
(kJ/m2)
(PP/EPR) 20.3±0.4 150±25 8.50±0.9 75±8 (PP/EPR)/5%W 19.4±0.35 150±35 15.9±1.2 60±12 (PP/EPR)/10%W 18.9±0.45 115±30 15.75±1.1 48±10 (PP/EPR)/15%W 18.4±0.3 85±25 15.65±1.15 41±9 (PP/EPR)/5%S 19.3±0.45 110±40 16.2±1.1 50±15 (PP/EPR)/10%S 18.9±0.35 80±20 16.2±1.2 30±11 (PP/EPR)/15%S 18.8±0.4 64±30 3.5±1.0 27±13
0
2
4
6
8
10
12
14
16
18
20
22
(PP/EPR) (PP/EPR)/5%W (PP/EPR)/10%W (PP/EPR)/15%W (PP/EPR)/5%S (PP/EPR)/10%S (PP/EPR)/15%S
Contrainte maximale (MPa)Energie à la rupture (J)
Figure IV. 12: Contrainte maximale et énergie à la rupture en fonction de la nature et du
taux de nano-CaCO3
176
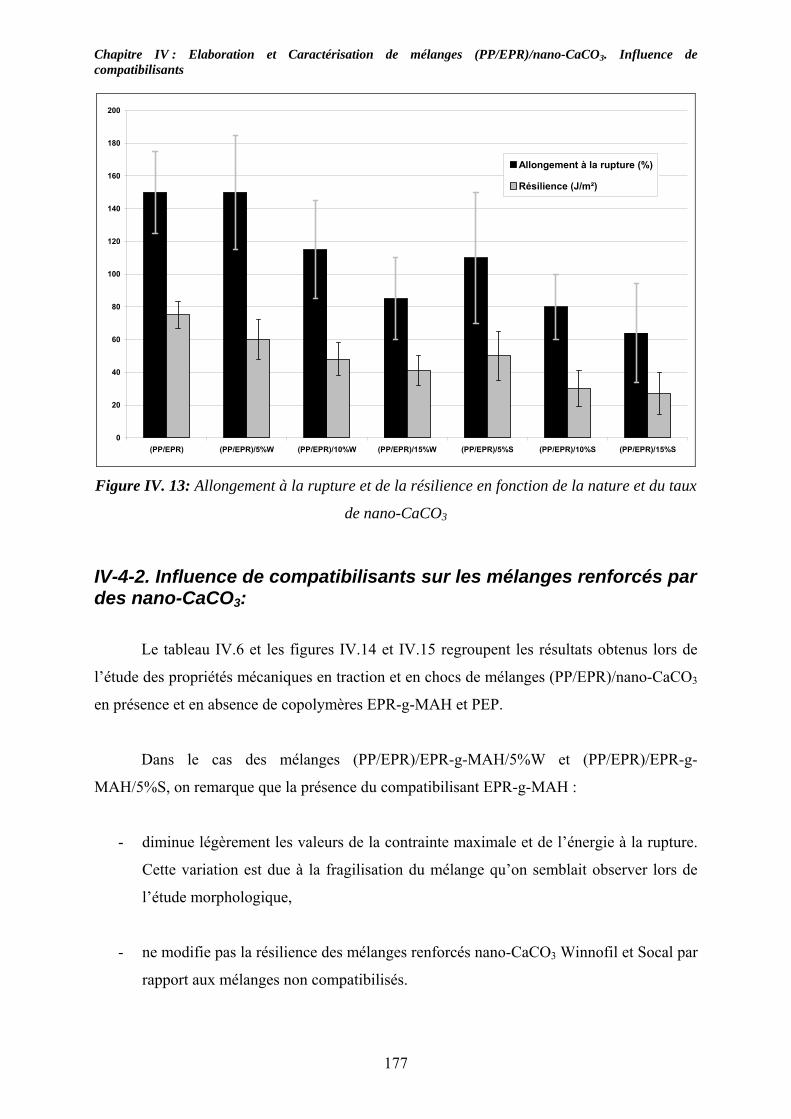
Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
0
20
40
60
80
100
120
140
160
180
200
(PP/EPR) (PP/EPR)/5%W (PP/EPR)/10%W (PP/EPR)/15%W (PP/EPR)/5%S (PP/EPR)/10%S (PP/EPR)/15%S
Allongement à la rupture (%)
Résilience (J/m²)
Figure IV. 13: Allongement à la rupture et de la résilience en fonction de la nature et du taux
de nano-CaCO3
IV-4-2. Influence de compatibilisants sur les mélanges renforcés par des nano-CaCO3:
Le tableau IV.6 et les figures IV.14 et IV.15 regroupent les résultats obtenus lors de
l’étude des propriétés mécaniques en traction et en chocs de mélanges (PP/EPR)/nano-CaCO3
en présence et en absence de copolymères EPR-g-MAH et PEP.
Dans le cas des mélanges (PP/EPR)/EPR-g-MAH/5%W et (PP/EPR)/EPR-g-
MAH/5%S, on remarque que la présence du compatibilisant EPR-g-MAH :
- diminue légèrement les valeurs de la contrainte maximale et de l’énergie à la rupture.
Cette variation est due à la fragilisation du mélange qu’on semblait observer lors de
l’étude morphologique,
- ne modifie pas la résilience des mélanges renforcés nano-CaCO3 Winnofil et Socal par
rapport aux mélanges non compatibilisés.
177
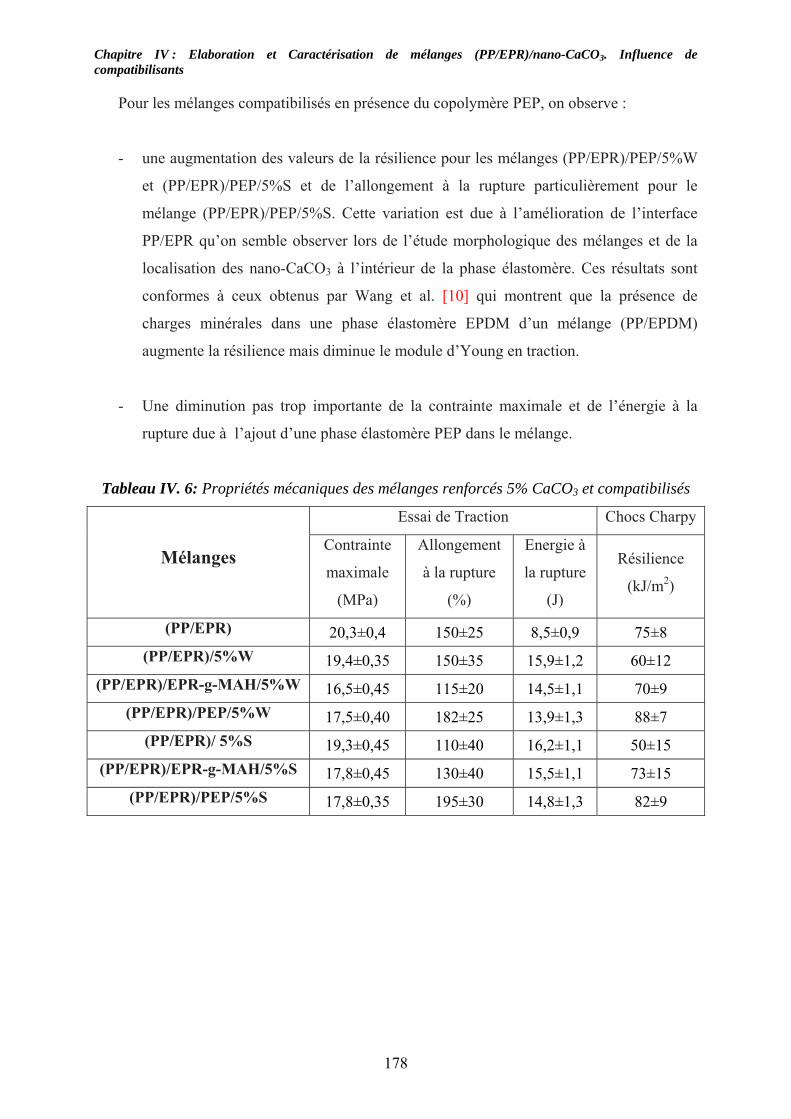
Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
Pour les mélanges compatibilisés en présence du copolymère PEP, on observe :
- une augmentation des valeurs de la résilience pour les mélanges (PP/EPR)/PEP/5%W
et (PP/EPR)/PEP/5%S et de l’allongement à la rupture particulièrement pour le
mélange (PP/EPR)/PEP/5%S. Cette variation est due à l’amélioration de l’interface
PP/EPR qu’on semble observer lors de l’étude morphologique des mélanges et de la
localisation des nano-CaCO3 à l’intérieur de la phase élastomère. Ces résultats sont
conformes à ceux obtenus par Wang et al. [10] qui montrent que la présence de
charges minérales dans une phase élastomère EPDM d’un mélange (PP/EPDM)
augmente la résilience mais diminue le module d’Young en traction.
- Une diminution pas trop importante de la contrainte maximale et de l’énergie à la
rupture due à l’ajout d’une phase élastomère PEP dans le mélange.
Tableau IV. 6: Propriétés mécaniques des mélanges renforcés 5% CaCO3 et compatibilisés
Essai de Traction Chocs Charpy
Mélanges Contrainte
maximale
(MPa)
Allongement
à la rupture
(%)
Energie à
la rupture
(J)
Résilience
(kJ/m2)
(PP/EPR) 20,3±0,4 150±25 8,5±0,9 75±8 (PP/EPR)/5%W 19,4±0,35 150±35 15,9±1,2 60±12
(PP/EPR)/EPR-g-MAH/5%W 16,5±0,45 115±20 14,5±1,1 70±9 (PP/EPR)/PEP/5%W 17,5±0,40 182±25 13,9±1,3 88±7
(PP/EPR)/ 5%S 19,3±0,45 110±40 16,2±1,1 50±15 (PP/EPR)/EPR-g-MAH/5%S 17,8±0,45 130±40 15,5±1,1 73±15
(PP/EPR)/PEP/5%S 17,8±0,35 195±30 14,8±1,3 82±9
178
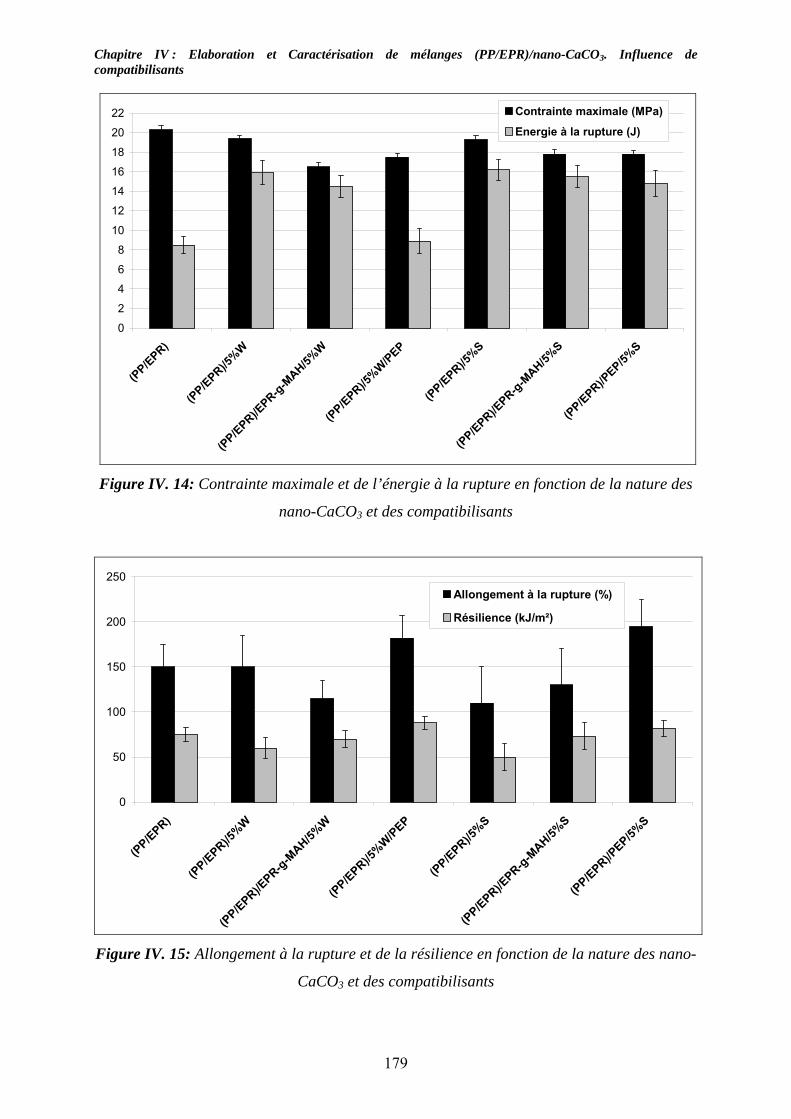
Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
0
2
4
6
8
10
12
14
16
18
20
22
(PP/EPR)
(PP/EPR)/5
%W
(PP/EPR)/E
PR-g-MAH/5%
W
(PP/EPR)/5
%W/PEP
(PP/EPR)/5
%S
(PP/EPR)/E
PR-g-MAH/5%
S
(PP/EPR)/P
EP/5%S
Contrainte maximale (MPa)
Energie à la rupture (J)
Figure IV. 14: Contrainte maximale et de l’énergie à la rupture en fonction de la nature des
nano-CaCO3 et des compatibilisants
0
50
100
150
200
250
(PP/EPR)
(PP/EPR)/5
%W
(PP/EPR)/E
PR-g-MAH/5%
W
(PP/EPR)/5
%W/PEP
(PP/EPR)/5
%S
(PP/EPR)/E
PR-g-MAH/5%
S
(PP/EPR)/P
EP/5%S
Allongement à la rupture (%)
Résilience (kJ/m²)
Figure IV. 15: Allongement à la rupture et de la résilience en fonction de la nature des nano-
CaCO3 et des compatibilisants
179

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
IV-6. Conclusion:
Dans ce chapitre, nous avons étudié l’influence de l’introduction des nano-CaCO3 et
de compatibilisants (PEP et EPR-g-MAH) sur les propriétés des mélanges (PP/EPR) utilisés
dans la fabrication de boucliers automobiles. Les résultats expérimentaux montrent que :
La dispersion des nanoparticules dans une matrice PP/EPR dépend de la nature des
nano-CaCO3 utilisés. La présence du compatiblisant PEP diminue la taille des
particules EPR et améliore l’interface entre les constituants des mélanges avec une
meilleure dispersion des nanoparticules dans la matrice. Pour le copolymère EPR-g-
MAH, on observe une fragilisation du faciès de rupture, une augmentation de la taille
des phases dispersées et une dégradation de l’interface PP/EPR ;
La température de fusion (taille des cristallites) étudiés par DSC augmentent
légèrement en présence de nanocharges et de compatibilisants contrairement aux
nanocharges non traitées étudiées par différents auteurs [2] et qui ont montré plutôt
une variation de la température de cristallisation et du taux de cristallinité;
La présence des nano-CaCO3 dans les mélanges PP/EPR augmente le module de
conservation G’, décale les pics de relaxation de la phase élastomère vers les basses
températures et provoque un écoulement prématuré des mélanges à haute température.
La présence du PEP donne naissance à un nouveau pic de relation associé aux
polymères « piégés » à l’interface des nanoparticules en présence du compatibilisant ;
Le comportement mécanique en traction et aux chocs des mélanges renforcés nano-
CaCO3 dépend de la morphologie obtenue et des taux de nanoparticules introduites. La
présence du compatibilisant PEP engendre une bonne dispersion des constituants et
une meilleure adhésion à l’interface PP/EPR. Par contre, le copolymère EPR-g-MAH
a tendance à modifier légèrement l’ensemble des propriétés mécaniques.
180

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants
Références
[1] Marisa C.G. Rocha, Antonio H.M.F.T. Silva, Fernanda M.B. Coutinho and Ana Lucia N.
Silva. Study of composites based on polypropylene and calcium carbonate by experimental
design. Polymer Testing, 2005, vol 24, p 1049–1053.
[2] Zebarjad S.M., Tahani M. and Sajjadi S.A. Influence of filler particles on deformation and
fracture mechanism of isotactic polypropylene. Journal of Materials Processing Technology,
2004, vol 155, p 1459–1464.
[3] Premphet K. and Horanont P. Influence of stearic acid treatment of filler particles on the
structure and properties of ternary-phase propylene composites. J Appl Polym Sci, 1999, Vol
74, p 3445-3454.
[4] Kolarik J., Lednicky F., Jancar J. and Pukanszky B. Phase structure of ternary composites
consisting of polypropylene/elastomer/filler. Effect of functionalized components. Polymer
commun, 1990, vol 31, p 201-204.
[5] Chan C.M., Wu J.S., Li J.X. and Chueng Y.K. Polypropylene/calcium carbonate
nanocomposites. Polymer, 2002, vol 43, p 2981-2992.
[6] Ren X.C., Bai L.Y., Wang G.H. and Zhang B.L., China Plast, 2002, vol 14, p 22.
[7] Albérola N.D., Bas C. and Mélé P. Composites particulaires: modélisation du
comportement viscoélastique assorti du concept de percolation. Comptes Rendus de
l'Académie des Sciences, 1994, t319, série II, p. 1129-1134
[8] Hornsby P.R. and Premphet K. Influence of phase microstructure of the mechanical
properties of ternary phase propylene composites. J Appl Polym Sci, 1998, vol 70, p 587-597.
[9] Tsagaropoulos G., Eisenberg A. Dynamic Mechanical Study of the factors affecting the
two glas transition behavior of filled polymers. Similarities and differences with random
ionomers. Macromolecules, 1995, vol 28, p 6067-6077
[10] Wang X., Sun J. and Huang R. Influence of the compounding route on the properties of
polypropylene/nano-CaCO3/ethylene-propylene-diene terpolymer tercomponent composites.
J Appl Polym Sci, 2006, vol 99, p 2268-2272.
[11]Zhang L., Li C. and Rui Huang R. Toughness mechanism of
polypropylene/elastomer/filler composites. Journal of Polymer Science: Part B: Polymer
Physics, 2005, vol. 43, p 1113–1123.
[12] Maiti S. and Mahapatro P. Mechanical properties of iPP/CaCO3 composites. J Appl
Polym Sci 1991, vol 42, p 3101-3110.
181

Chapitre IV : Elaboration et Caractérisation de mélanges (PP/EPR)/nano-CaCO3. Influence de compatibilisants [13] Maiti S and Lopez B. Tensile properties of polypropylene/kaolin composites. J Appl
Polym Sci, 1992, vol 44, p 353-360.
[14] Tjong S., Li R. and Chueng T. Mechanical behavior of CaCO3 particulate-filled B-
crystalline phase polypropylene composites. Polym Eng Sci, 1997, vol 37, p 166-172.
[15] Mitsuishi K., Kodama S. and Kawasaki H. Mechanical properties of polypropylene filled
with calcium carbonate. Polym Eng Sci, 1985, vol 25, p 1069-1073.
[16] Mnif N., Massardier V., Kallel T. and Elleuch B. Study of the modification of the
properties of (PP/EPR) blends with a view to preserving natural resources when elaborating
new formulation and recycling polymers Polym Comp, 2008, DOI10.1002.
[17] Boucard S. Développement de formulations polyoléfines / silicates lamellaires: contrôle
de la nanostructuration par la gestion des interactions physico-chimiques et le procédé de mise
en oeuvre dans le fondu. Thèse INSA de Lyon 2004.
182

Conclusion générale
Conclusion Générale
L’utilisation de deux copolymères PEP et EPR-g-MAH, afin d’améliorer les interfaces
(PP/EPR), ne montre une amélioration des propriétés que pour les mélanges compatibilisés
avec le copolymère PEP ; l’interface serait donc le siège d’interactions importantes entre les
deux phases (PP et élastomères) du fait de la présence du compatibilisant qui se concentre
préférentiellement à ce niveau lors de la phase d’élaboration des mélanges à l’état fondu. Pour
les mélanges (PP/EPR)/EPR-g-MAH, la présence du compatibilisant fragilise le faciès de
rupture du matériau, augmente le taux de cristallinité et les propriétés mécaniques pour les
grandes déformations mais sans modifier les pics de relaxation des constituants des mélanges.
Alors que la compatibilisation de mélanges binaires a été largement étudiée, celle de
mélanges ternaires constitue une originalité importante, les comportements de mélanges
binaires ne pouvant pas forcément s’extrapoler aux ternaires.
Dans le cas des mélanges (PP/EPR) pollués ABS et PA, qui se présentent sous forme
de nodules non miscibles avec les constituants des mélanges, l’ajout de compatibilisant SEBS
aux mélanges (PPEPR)/ABS améliore l’adhésion à l’interface, l’allongement à la rupture et
diminue la taille des nodules ABS. D’autre part, la présence du copolymère EPR-g-MAH
diminue la taille des particules PA dans les mélanges (PP/EPR)/PA et le nombre de cavités
vides qui correspondent à l’arrachement du polluant lors de la rupture.
L’étude des propriétés morphologiques en MEB des mélanges renforcés nano-CaCO3
a montré que la dispersion des nanoparticules dépend, en plus des conditions de mise en
œuvre et des constituants des mélanges, de la taille et de l’état de surface des nano-CaCO3
(taux de recouvrement par des acides gras… et surface spécifique). La présence du
compatibilisant PEP diminue la taille des particules EPR et améliore l’interface entre les
constituants des mélanges avec une meilleure dispersion des nanoparticules dans la matrice.
Pour le copolymère EPR-g-MAH, on observe une fragilisation du faciès de rupture, une
augmentation de la taille des phases dispersées et une mauvaise adhésion à l’interface
PP/élastomère. L’analyse thermique a montré, dans le cas des mélanges renforcés nano-
CaCO3 et renforcés/compatibilisés, que le taux de cristallinité et la température de fusion
(taille des cristallites) augmentent légèrement contrairement aux nanocharges non traitées.
183

Conclusion générale
L’étude des propriétés viscoélastiques à l’état solide révèle une augmentation du
module de conservation G’, un déplacement du pic de relaxation de la phase élastomère vers
les basses températures (les nanoparticules jouent le rôle de plastifiant en s’intercalant entre
les macromolécules et en facilitant leurs déplacements). La présence du PEP donne naissance
à un nouveau pic de relaxation associé aux polymères « piégés » à l’interface des
nanoparticules en présence du compatibilisant. Le comportement mécanique en traction et aux
chocs des mélanges renforcés nano-CaCO3 dépend de la morphologie obtenue et des taux de
nanoparticules introduites. La présence du compatibilisant PEP engendre une bonne
dispersion des constituants et une meilleure adhésion à l’interface PP/EPR. Par contre, le
copolymère EPR-g-MAH a tendance à modifier légèrement l’ensemble des propriétés
mécaniques.
Afin de simuler le recyclage des boucliers peints, nous avons étudié les propriétés des
mélanges contenant du polyuréthane (PU) en présence et en absence de compatibilisants. Les
résultats obtenus ont montré que le PU se présente sous forme de particules non miscibles
avec les constituants de mélanges avec une faible adhésion à l’interface, sa présence diminue
le taux de cristallinité et augmente la température de cristallisation. Nous avons observé aussi,
lors de l’étude des propriétés mécaniques (traction et chocs), une chute de l’allongement à la
rupture et de l’énergie à la rupture due à la mauvaise adhésion entre le PU et la matrice et une
diminution de la contrainte maximale et de la résilience par modification du taux de
cristallinité. L’apparition d’un nouveau pic de relaxation du PU et une faible variation des
pics de relaxation des autres constituants par étude des propriétés viscoélastiques à l’état
solide confirment alors l’hypothèse de la non miscibilité des constituants. La présence de
compatibilisants E-EA-MAH et E-EA-GMA ne modifie pas les propriétés thermiques,
morphologiques, viscoélastiques à l’état solide et mécaniques des mélanges en présence du
PU.
L’utilisation des nano-CaCO3 dans les mélanges (PP/EPR), utilisés dans la fabrication
de boucliers automobiles, a pour but d’améliorer les propriétés chocs et de remplacer une
fraction de la matrice par une charge minérale moins coûteuse et moins polluante. D’autres
nanoparticules pourront être étudiées comme suite à ce travail tel que des nano-SiO2, de la
montmorillonite…
184

Conclusion générale
Pour les mélanges contenant du PU :
• une première solution a été étudiée et pourra être approfondie dans la suite : les
particules de peinture restent à l’état de paillettes solides réticulées. Il faut alors
synthétiser un agent de couplage selon la nature de la peinture utilisée (polyuréthane
ou autre).
• une deuxième solution pourra aussi être développée : la peinture est traitée par
solvolyse pourra se faire directement dans l’extrudeuse. Cette dernière possibilité est
plus aléatoire compte-tenu du temps nécessaire pour mener à bien le traitement par
solvolyse.
185

186

Listes des figures Partie Bibliographique Chapitre 1 : FIGURE 1. 1: CYCLE DU RECYCLAGE ET DE LA VALORISATION DES AUTOMOBILES
[1]..................................................................................................................................... 28 FIGURE 1. 2: CHANGEMENT RELATIF DE LA COMPOSITION DES AUTOMOBILES AU
COURS DES TROIS DERNIERES DECENNIES [2] ...................................................... 29 FIGURE 1. 3: EVOLUTION DE L’UTILISATION DU POLYPROPYLENE DANS LA
CONSTRUCTION DES VOITURES [2] .......................................................................... 29 Chapitre 2 : FIGURE 2. 1: MORPHOLOGIE DES MELANGES (A) PARTICULAIRE DE 1 DANS 2, (B)
BICONTINUE ET (C) PARTICULAIRE DE 2 DANS 1................................................... 37 FIGURE 2. 2: ANALOGIE ENTRE EMULSIFIANT TRADITIONNEL ET COPOLYMERE A
BLOC, EN TANT QU’ESPECES ACTIVES EN SURFACE D’UN MELANGE .............. 39 FIGURE 2. 3 : POSITIONNEMENT D’UN COPOLYMERE COMPATIBILISANT A
L’INTERFACE EN FONCTION DE SA MICROSTRUCTURE : (A) COPOLYMERE A BLOC ET (B) COPOLYMERE STATISTIQUE [12]........................................................ 40
FIGURE 2. 4: POSITIONNEMENT D’UN COPOLYMERE DIBLOC (A) OU TRIBLOC (B) SUR LE POLYMERE ....................................................................................................... 41
FIGURE 2. 5: EXEMPLES DE REACTIONS CHIMIQUES CLASSIQUEMENT UTILISEES POUR LA COMPATIBILISATION IN-SITU DES MELANGES DE POLYMERES IMMISCIBLES [26] ......................................................................................................... 43
FIGURE 2. 6: FRACTION SURFACIQUE DE PHASE DISPERSEE EN FONCTION DU VOLUME DE (PEHD + ELASTOMERE) INDUIT [51] ................................................. 49
FIGURE 2. 7: ILLUSTRATION SCHEMATIQUE DE LA MORPHOLOGIE DES PARTICULES COMPOSITES PEP-PEHD DANS LES MELANGES TERNAIRES A MATRICE PP : STRUCTURE CŒUR ECORCE [51] .................................................... 50
Chapitre 3 : FIGURE 3. 1: LES DIFFERENTES ECHELLES DE TAILLE DE LA SILICE [88] ............... 65 FIGURE 3. 2: (A) MATERIAU HOMOGENE A TOUTES LES ECHELLES, (B) OBJET
FRACTAL [88] ................................................................................................................ 65 FIGURE 3. 3 : TAUX DE POLYMERE LIE DANS DES COMPOSITES SBR-SILICE POUR
DES FRACTIONS VOLUMIQUES DE CHARGES DE 8,8 ; 16,2 ; 21,9 ET 26,4 % [77].......................................................................................................................................... 67
FIGURE 3. 4 : ORIGINE DE LA DEUXIEME RELAXATION PROPOSEE PAR TSAGAROPOULOS ET AL. [59]..................................................................................... 68
187

Partie expérimentale Chapitre I : FIGURE I. 1: CLICHE MET DES MATRICES (PP/EPR) ET (PP/EPDM/TALC) ................. 83 FIGURE I. 2: MORPHOLOGIE DE LA GAMME DE NANO-CACO3 PROPOSEE PAR
SOLVAY®......................................................................................................................... 88 FIGURE I. 3: PROFILS DES VIS ET DE TEMPERATURE UTILISES POUR LA
REALISATION DE MELANGES...................................................................................... 91 FIGURE I. 4: DIMENSIONS ET FORME DE L'EPROUVETTE CHARPY EN V ................ 100 Chapitre II : FIGURE II. 1: MORPHOLOGIE DES MATRICES (A) (PP/EPR) ET (B) (PP/EPDM/TALC)
........................................................................................................................................ 108 FIGURE II. 2: MORPHOLOGIES DES MELANGES (A ET B) (PP/EPR), (C ET D)
(PP/EPR)/PEP AND (E, F ET G) (PP/EPR)/EPR-G-MAH(X2000 AND X7000) ........ 110 FIGURE II. 3: INFLUENCE DES COMPATIBILISANTS SUR L’ALLONGEMENT A LA
RUPTURE (A%) DES MELANGES (PP/EPR) A 22±0.5°C .......................................... 112 FIGURE II. 4: INFLUENCE DES COMPATIBILISANTS SUR L’ENERGIE A LA RUPTURE
ET LA RESILIENCE DES MELANGES (PP/EPR) A 22±0.5°C.................................... 112 FIGURE II. 5: THERMOGRAMME DSC DES MELANGES (PP/EPR), (PP/EPR)/PEP ET
(PP/EPR)/EPR-G-MAH (RAMPE DE TEMPERATURE 10°C/MIN ET -10°C /MIN) .. 114 FIGURE II. 6: EVOLUTION DE LA TANGENTE DE L’ANGLE DE PERTE ET DU
MODULE G’ DES MELANGES (PP/EPR), (PP/EPR)/PEP ET (PP/EPR)/EPR-G-MAH EN FONCTION DE LA TEMPERATURE A 6,28 RAD/S. ............................................. 115
FIGURE II. 7: MORPHOLOGIE DES MELANGES (A) (PP/EPR)/ABS ET (B) (PP/EPR)/ABS/SEBS...................................................................................................... 118
FIGURE II. 8: MICROSTRUCTURE DES MELANGES PP/PA-6/TPE ET PP/PA-6/PP-G-MAH [28] ....................................................................................................................... 120
FIGURE II. 9: INFLUENCE DES POLLUANTS ET DES COMPATIBILISANTS SUR LA CONTRAINTE MAXIMALE ET L’ENERGIE A LA RUPTURE DES MELANGES (PP/EPR) A 22±0.5°C.................................................................................................... 122
FIGURE II. 10: INFLUENCE DES POLLUANTS ET DES COMPATIBILISANTS SUR LE POURCENTAGE D’ALLONGEMENT DES MELANGES (PP/EPR) A 22±0.5°C ...... 122
FIGURE II. 11: INFLUENCE DES POLLUANTS ET DES COMPATIBILISANTS SUR LA RESILIENCE DES MELANGES (PP/EPR) A 22±0.5°C .............................................. 123
FIGURE II. 12: COURBES DSC DU PP, DE L’ABS ET DES MELANGES PP/ABS [21] .. 123 FIGURE II. 13: COURBES DSC DES MELANGES (PP/EPR), (PP/EPR)/ABS ET
(PP/EPR)/ABS/SEBS (RAMPES DE TEMPERATURE 10°C/MIN ET -10°C /MIN)..... 124 FIGURE II. 14: COURBES DSC DES MELANGES (PP/EPR), (PP/EPR)/PA ET
(PP/EPR)/PA/EPR-G-MAH (RAMPES DE TEMPERATURE 10°C/MIN ET -10°C /MIN)........................................................................................................................................ 125
FIGURE II. 15: EVOLUTION DU MODULE G’ DES MELANGES (PP/EPR), (PPE/EPR)/ABS ET (PP/EPR)/ABS/SEBS EN FONCTION DE LA TEMPERATURE A 6,28 RAD/S ..................................................................................................................... 127
FIGURE II. 16: EVOLUTION DU MODULE G’ DES MELANGES (PP/EPR), (PP/EPR)/PA ET (PP/EPR)/PA/EPR-G-MAH EN FONCTION DE LA TEMPERATURE A 6,28 RAD/S........................................................................................................................................ 128
188

Chapitre III : FIGURE III. 1: THERMOGRAMME DSC DES MELANGES A BASE DE PP EN PRESENCE
DE PU ET DE COMPATIBILISANTS (RAMPE DE TEMPERATURE 10°C/MIN ET -10°C /MIN)..................................................................................................................... 138
FIGURE III. 2: THERMOGRAMME DSC DES MELANGES A BASE DE (PP/EPR) EN PRESENCE DE PU ET DE COMPATIBILISANTS (RAMPE DE TEMPERATURE 10°C/MIN ET -10°C /MIN) ............................................................................................ 139
FIGURE III. 3: MORPHOLOGIE DES MELANGES A MATRICE PP CONTENANT DU PU ET DES COMPATIBILISANTS...................................................................................... 140
FIGURE III. 4: MORPHOLOGIE DES MELANGES A MATRICE (PP/EPR) CONTENANT DU PU ET DES COMPATIBILISANTS......................................................................... 142
FIGURE III. 5: VARIATION DE LA CONTRAINTE MAXIMALE ET DE L’ALLONGEMENT A LA RUPTURE DES MELANGES PP, PP/PU, PP/PU/E-EA-MAH ET PP/PU/E-EA-GMA ............................................................................................................................... 144
FIGURE III. 6: VARIATION DE L’ENERGIE A LA RUPTURE ET DE LA RESILIENCE DES MELANGES PP, PP/PU, PP/PU/E-EA-MAH ET PP/PU/E-EA-GMA ................. 145
FIGURE III. 7: VARIATION DE LA CONTRAINTE MAXIMALE ET DE L’ALLONGEMENT A LA RUPTURE DES MELANGES (PP/EPR), (PPEPR)/PU, (PP/EPR)/PU/E-EA-MAH ET (PPEPR)/PU/E-EA-GMA ......................................................................................... 145
FIGURE III. 8: VARIATION DE L’ENERGIE A LA RUPTURE ET DE LA RESILIENCE DES MELANGES (PP/EPR), (PPEPR)/PU, (PP/EPR)/PU/E-EA-MAH ET (PPEPR)/PU/E-EA-GMA............................................................................................... 146
Chapitre IV : FIGURE IV. 1: THERMOGRAMME DSC DES MELANGES (PP/EPR), (PP/EPR)/5%W,
(PP/EPR)/10%W ET (PP/EPR)/15%W (RAMPE DE TEMPERATURE 10°C/MIN ET -10°C /MIN)..................................................................................................................... 156
FIGURE IV. 2: THERMOGRAMME DSC DES MELANGES (PP/EPR), (PP/EPR)/5%S, (PP/EPR)/10%S ET (PP/EPR)/15%S (RAMPE DE TEMPERATURE 10°C/MIN ET -10°C /MIN)..................................................................................................................... 156
FIGURE IV. 3: THERMOGRAMME DSC DES MELANGES (PP/EPR)/5%W, (PP/EPR)/PEP/5%W ET (PP/EPR)/EPR-G-MAH/5%W (RAMPE DE TEMPERATURE 10°C/MIN ET -10°C /MIN) ............................................................................................ 158
FIGURE IV. 4: THERMOGRAMME DSC DES MELANGES (PP/EPR)/5%5, (PP/EPR)/PEP/5%S ET (PP/EPR)/EPR-G-MAH/5%S (RAMPE DE TEMPERATURE 10°C/MIN ET -10°C /MIN) ............................................................................................ 158
FIGURE IV. 5: EVOLUTION DU MODULE G’ ET DE LA TANGENTE DE L’ANGLE DE PERTE DES MELANGES (PP/EPR), (PP/EPR)/5%S, (PP/EPR)/10%S ET (PP/EPR)/15%S EN FONCTION DE LA TEMPERATURE A 6,28 RAD/S ................. 161
FIGURE IV. 6: EVOLUTION DU MODULE G’ ET DE LA TANGENTE DE L’ANGLE DE PERTE DES MELANGES (PP/EPR), (PP/EPR)/5%W, (PP/EPR)/10%W ET (PP/EPR)/15%W EN FONCTION DE LA TEMPERATURE A 6,28 RAD/S ................ 161
FIGURE IV. 7: EVOLUTION DU MODULE G’ ET DE LA TANGENTE DE L’ANGLE DE PERTE DES MELANGES (PP/EPR)/5%W, (PP/EPR)/PEP/5%W ET (PP/EPR)/EPR-G-MAH/5%W EN FONCTION DE LA TEMPERATURE ET A 6,28 RAD/S..................... 164
189

FIGURE IV. 8: EVOLUTION DU MODULE G’ ET DE LA TANGENTE DE L’ANGLE DE PERTE DES MELANGES (PP/EPR)/5%S, (PP/EPR)/PEP/5%S ET (PP/EPR)/EPR-G-MAH/5%S EN FONCTION DE LA TEMPERATURE ET A 6,28 RAD/S ...................... 164
FIGURE IV. 9: MORPHOLOGIE DES MELANGES (A) (PP/EPR)/5%S, (B) (PP/EPDM/TALC)/5%S, ................................................................................................ 167
FIGURE III. 10: DISPERSION DES NANO-CACO3 ET DE L’EPDM DANS LE PP [10] .. 169 FIGURE IV. 11: MORPHOLOGIE DES MELANGES (A) (PP/EPR)/5%W, (B)
(PP/EPR)/5%S ............................................................................................................... 171 FIGURE IV. 12: CONTRAINTE MAXIMALE ET ENERGIE A LA RUPTURE EN FONCTION
DE LA NATURE ET DU TAUX DE NANO-CACO3 ...................................................... 176 FIGURE IV. 13: ALLONGEMENT A LA RUPTURE ET DE LA RESILIENCE EN
FONCTION DE LA NATURE ET DU TAUX DE NANO-CACO3 ................................. 177 FIGURE IV. 14: CONTRAINTE MAXIMALE ET DE L’ENERGIE A LA RUPTURE EN
FONCTION DE LA NATURE DES NANO-CACO3 ET DES COMPATIBILISANTS.... 179 FIGURE IV. 15: ALLONGEMENT A LA RUPTURE ET DE LA RESILIENCE EN
FONCTION DE LA NATURE DES NANO-CACO3 ET DES COMPATIBILISANTS.... 179
190

LISTE DES TABLEAUX Partie Bibliographique Chapitre 2 : TABLEAU 2. 1 : COMPARAISON DE LA COMPATIBILISATION PAR AJOUT ET
FORMATION IN-SITU DU COPOLYMERE COMPATIBILISANT................................ 44 Partie Expérimentale Chapitre I : TABLEAU I. 1: LES CARACTERISTIQUES DES MATRICES PP108MF97 ET PP7510 ...... 83 TABLEAU I. 2: CARACTERISTIQUES TECHNIQUES DU COMPATIBILISANT EPR-G-
MAH (EXXELOR VA 1801) ............................................................................................. 85 TABLEAU I. 3: CARACTERISTIQUES TECHNIQUES DU COMPATIBILISANT PEP
(VISTAMAXX 6200) ......................................................................................................... 85 TABLEAU I. 4: LES CARACTERISTIQUES DU KRATON G 1651........................................ 86 TABLEAU I. 5: LES CARACTERISTIQUES DU LOTADER AX3210 .................................... 86 TABLEAU I. 6: LES CARACTERISTIQUES DU LOTADER AX8900 .................................... 87 TABLEAU I. 7: CARACTERISTIQUES DES NANO-CACO3.................................................. 88 TABLEAU I. 8: CARACTERISTIQUES DE L’ABS ................................................................. 89 TABLEAU I. 9: CARACTERISTIQUES DU PA....................................................................... 89 TABLEAU I. 10: VALEURS DES DIVERSES RELAXATIONS POUR LES PEHD, PP ET LES
ELASTOMERES EPR....................................................................................................... 98 Chapitre II : TABLEAU II. 1: PROPRIETES MECANIQUES EN TRACTION UNIAXIALE ET EN CHOC
CHARPY ENTAILLE V A 22±0.5°C .............................................................................. 111 TABLEAU II. 2 : PROPRIETES THERMIQUES DES MELANGES ..................................... 113 TABLEAU II. 3: TEMPERATURES DU MAXIMUM DES PICS DE RELAXATION DES
MELANGES (PP/EPR), (PP/EPR)/PEP ET (PP/EPR)/EPR-G-MAH........................... 115 TABLEAU II. 4: PROPRIETES MECANIQUES EN TRACTION UNIAXIALE ET EN CHOCS
CHARPY ENTAILLE V A 22±0.5°C DES MELANGES POLLUES ET POLLUES/COMPATIBILISES....................................................................................... 121
TABLEAU II. 5: PROPRIETES THERMIQUES DES MELANGES POLLUES ET POLLUES/COMPATIBILISES....................................................................................... 125
TABLEAU II. 6: TEMPERATURES DU MAXIMUM DES PICS DE RELAXATION DES MELANGES POLLUES ET POLLUES/COMPATIBILISES ......................................... 127
191

Chapitre III : TABLEAU III. 1: PROPRIETES THERMIQUES DES MELANGES (PP/EPR) RENFORCES
CACO3 ET NON COMPATIBILISES............................................................................. 155 TABLEAU III. 2: PROPRIETES THERMIQUES DES MELANGES (PP/EPR) RENFORCES
5% CACO3 ET COMPATIBILISES................................................................................ 157 TABLEAU III. 3: PROPRIETES RHEOLOGIQUES DES MELANGES RENFORCES CACO
ET NON COMPATIBILISES3
.......................................................................................... 160TABLEAU III. 4: PICS DE RELAXATION DES MELANGES RENFORCES 5% CACO3 ET
COMPATIBILISES......................................................................................................... 163 TABLEAU III. 5: PROPRIETES MECANIQUES DES MELANGES RENFORCES CACO3 ET
NON COMPATIBILISES................................................................................................ 176 TABLEAU III. 6: PROPRIETES MECANIQUES DES MELANGES RENFORCES 5% CACO
ET COMPATIBILISES3
................................................................................................... 178 Chapitre IV : TABLEAU IV. 1: COMPOSITION DES MELANGES ELABORES....................................... 136 TABLEAU IV. 2: PROPRIETES THERMIQUES DES MELANGES PP ET (PP/EPR) EN
PRESENCE DE PU ET DE COMPATIBILISANTS....................................................... 138 TABLEAU IV. 3: PROPRIETES MECANIQUES DES MELANGES PP ET (PP/EPR) EN
PRESENCE DE PU ET DE COMPATIBILISANTS....................................................... 144 TABLEAU IV. 4: TEMPERATURES DU MAXIMUM DES PICS DE RELAXATION DES
MELANGES PP ET (PP/EPR) EN PRESENCE DE PU ET DE COMPATIBILISANTS........................................................................................................................................ 147
192

ANNEXES
(Articles publiés)
193







