doccours chimie organique : réactions orgaii...
TRANSCRIPT

DocCours Chimie Organique : réactions orgaII - 1/12
Figure 1 : Quelques facteurs contrôlant une réaction
Contrôle de chargeL’espèce A est pauvre en e- (électrophile), B est riche en e- (nucléophile). A et B s’attirent et vont réagir pourformer un nouveau composé : A+ + B- A-B
Contrôle orbitalaire (2ème année)Conditions pour que la réaction se fasse :les orbitales moléculaires des composés A et B doivent être :a) en phase, b) proches en énergie, et c) de symétries "compatibles"
Contrôle stériqueLe réactif A aura plus de mal à rejoindrele site + encombré (à gauche) que le site + de droite.On parle d’encombrement stérique.
En première année, les réactions seront guidées par le contrôle de charge, et dans une moindre mesure, parl’encombrement stérique.On prendra donc grand soin d’étudier la répartition des charges des composés avant d’écrire les réactions.
B CA réaction possibleentre A et C,
pas entre A et B
OA "s" OA "pz" OA "pz"+
-
+
-
+
gêne ou encombrementstérique
Me
MeMe
+ A-
approchedifficile
H
HH
+approche
facile
réaction possibleentre A et C
Figure 2 : Classification des réactifs : Acides et Bases - Nucléophiles et Electrophiles
Acide (Définitions au sens de Bronsted)Toute molécule pouvant libérer un H+ : AH → A- + H+ (A- est la base conjuguée)Exemples :
Base (Définitions au sens de Bronsted)Toute molécule pouvant capturer un H+ : B + H+ → BH+ (BH+ est l’acide conjugué)Exemples :
NucléophileToute espèce qui a une affinité pour les "noyaux", c'est-à-dire les centres chargés positivement.Concrètement : toute molécule possédant au moins un doublet riche en électron (DNL ou nuage ).Un nucléophile n’est pas forcément chargé < 0.Exemples :
ElectrophileToute espèce ayant une affinité pour les "électrons" c'est-à-dire les centres chargés négativement.Concrètement : toute molécule ayant une charge > 0, une lacune électronique, ou un atome déficitaire enélectrons. Un électrophile n’est pas forcément chargé > 0.Exemples :
R O
O
HR
OH
SH H
H H
OH H N
H
HHH
H2O H2S ROH RCOOH NH4+
ROS
HO
H R O
O
NH
HN
H
HH
HO- HS- RO- RCOO- NH3 NH2-
H2O ROH
RO
HO
H H
HS- RCOO-
SH R O
O
NH
HH
NH3HO-
OH
H2S
SH H
carbanion
RO
RO-
CHH
H
alcène
X = halogène
H CHH
H
proton
CHH
H
carboradical
X
carbocation

DocCours Chimie Organique : réactions orgaII - 2/12
Figure 3 : Différence entre basicité et nucléophilieLe concept nucléophile/électrophile est un concept de cinétique, et est donc lié aux vitesses de réaction.Le concept acide/base un concept de thermodynamique, et est donc lié aux équilibres de réactions.
Exemple : OH- et SH- ont une structure similaire, et sont tous deux basiques et nucléophiles.Concept acide/base (aspect thermodyamique)pKa (H2O/OH-) = 14 ; pKa (H2S/SH-) = 7Donc l’acide H2S est plus fort que l’acide H2Oet la base SH- est plus faible que la base OH-
OH- pourra plus facilement que SH-, arracher un H+ à un même site acide.
Concept nucléophile/électrophile (aspect cinétique)Pourtant, le doublet non liant sur S est plus gros que celui de O,car les électrons de ce doublet ont un nombre quantique n = 3(couche de valence), contre n = 2 pour O.Ils sont donc plus éloignés du noyau et plus disponible pour établirune liaison avec un autre atome.Par conséquent SH- est meilleur nucléophile que OH-
SH- réagira plus vite que OH- sur un même site électrophile.
HO-
OH
SH
HS-
Figure 4 : Conventions d’écriture des mouvements électroniquesRappel : Les mouvements électroniques sont décrits par des flèches précises :
flèche à deux pointes (ou dents) flèche à une « pointe »= mouvement de deux électrons = mouvement d’un électron
Ces flèches représentent le mouvement des électrons :elles partent donc des zones riches en électrons (doublet non liant, doublet p d’une double liaison, …)et vont vers des zones plus pauvres en électrons (atome portant une charge +, radical libre, …)Exemples : cassure d'une liaison
OHH
+-
La flèche part du milieu de la liaison qui se casseet va vers l'atome O.Un nouveau doublet non liant est créé
OH + H
OH + H OHH
La flèche part du doublet non liant qui va réagir,(et non de la charge formelle - sur O),et va vers l'atome H (et non sa charge formelle +)Une nouvelle liaison est créée.
formation d'une liaison à partir d'un doublet non liant
formation d'une liaison à partir d'un doublet d'une double liaison
La flèche part du milieu du doublet qui va réagir,et va vers l'atome H (et non sa charge formelle +)Une nouvelle liaison est créée, invisible ennotation topologique, entre un des carbones de C=Cet H.Cela donne deux carbocations possibles, selon queH se fixe sur le C de gauche (cas B) ou de droite (cas A).Bien noter que la charge formelle + est sur le C qui n'apas reçu le H.
+ H
mélange non équimolaire
A B
Figure 5 : Les deux modes de rupture d’une liaisonCassure hétérolytique :
Cassure homolytique :
+Acassure hétérolytique
(apparition de charge) ionsA B B C
HHH
ClC
HH
H
carbocation
+ ClC
HHH
Li+ LiC
HHH
carbanion ion lithium
+cassure homolytique
(pas d'apparition de charge)A B radicaux libres
A BC
HHH
Cl
carboradical
+ ClCHH
H
atome Cl

DocCours Chimie Organique : réactions orgaII - 3/12
Figure 6 : Classification des réactions (de 1ère année)
Substitution :
Symbole de la réaction : il dépend du réactif A approchant le substrat BCSN : substitution nucléophile si A est un nucléophileSE : substitution électrophile si A est un électrophile
Addition :
Symbole de la réactionAN : addition nucléophile ; si le 1er atome s’approchant de C=D est un nucléophile (il faut voir le mécanismepour le savoir)AE : addition électrophile ; si le 1er atome s’approchant de C=D est un électrophile
A + B-C A-B + Csubstitution de C par A
sur l'atome B
A-B + C=D C-Daddition
de AB sur C=D
A B
Figure 7 : Sélectivité d’une réaction
RégiosélectivitéUne réaction est dite régiosélective si elle privilégiela formation d’un isomère de constitution B parrapport à un autre isomère C.
Exemple :
StéréosélectivitéUne réaction est dite stéréosélective si elle privilégie laformation d’un stéréoisomère B par rapport à un autre B’(voir exemple plus bas)
StéréospécificitéUne réaction est dite stéréospécifique si
elle est stéréosélective et qu’un stéréoisomère A’ de A donne B et B’
dans les proportions inverses.Si B et B’ sont énantiomères, la réaction estdite énantiospécifique.Si B et B’ sont diastéréoisomères, la réaction estdite diastéréospécifique.
Exemple :
B et C sont desisomères
de constitution
B et B’ sont desstéréoisomères
A et A’ sont desstéréoisomères
B et B’ sont desstéréoisomères
Cl+ Br-
solvant
peu polaire
Br Br
+ + Cl-
composé Sminoritaire : 10 %
composé Scomposé R
majoritaire: 90 %
Cl+ Br-
Br Br
+ + Cl-
composé Smajoritaire 90 %
composé Rcomposé R
minoritaire: 10 %
réaction stéréosélective
réactionstéréospécifique
ici énantiospécifiquecar les deux composés
obtenus (R et S) sont énantiomères
solvant
peu polaire
+ HCl
Cl Cl
+
minoritaire : 18%
solvant polaire
majoritaire: 82%
+ HCl
Cl Cl
+solvant apolaire
majoritaire : 76% minoritaire: 24%

DocCours Chimie Organique : réactions orgaII - 4/12
Figure 8 : Réactivité des alcènes
Structure électronique de la liaison C=C(cas de l’éthène)Les deux carbones sont AX3E0
La géométrie est donc triangulaire planeavec des angles de base 120°.La double liaison occupe une place plus importante, donc l’angle HCH se resserre.
Il y a 5 liaisons à symétrie axiale notées et une liaison à symétrie plane notée ,perpendiculaire au plan de la molécule,et obtenue par interaction des deux OA2pz des carbones (les signes ne sont pasdes charges, mais ceux des OA)
C’est cette liaison qui empêche la libre rotationde C=C, d’où l’isomérie Z/E.
Stabilité de la liaison On note que E(C=C) > E(C-C)Mais : E(C=C) < 2 E(C-C)La liaison double C=C, constituée d’une liaison etd’une liaison , n’est pas aussi forte que 2 liaisons simples (liaison ).L'approximation E(C=C) = E() + E() E() + E(C-C) < 2 E(C-C) conduit à : E() < E()La liaison est plus fragile que la liaison
La grande réactivité des alcènes est donc principalement contenue dans leur liaison , car : elle est fragile
elle s’étale loin dans l’espace elle est très polarisable (déformable) elle est riche en électrons disponibles (comme un dnl)
Réactivité des alcènes(réactions de 1ère année)
les OA 2pz
interagissentet forment la
liaison
C CH
H
H
HC
H
H
CH
H
117°121,5°
+ +
- -
+
-
C-H C-C C=C
Longueur de liaison (pm) 110 154 134
Energie de liaison (kJ mol-1) 414 348 615
les alcènes sont très réactifs (par rapport auxalcanes) et se comportent comme des nucléophiles
les alcènes subissent des additions (ouverture de C=C par cassure de la liaison )
Cassure des liaisons ET sousl’action d’un oxydant comme l’ozone O3
Réactiond’ozonolyse
CH
H
CH
H
E
(électrophile = H+, X∙, …)
Cassure de la liaison (fragile et nucléophile)
à l’approche d’un électrophile ERéactions d’addition
électrophile AE
(partie la plus importante)
Figure 9 : Stabilité des alcènesLes alcènes n'ont pas tous la même stabilité ni la mêmeréactivité. Une méthode pour étudier la stabilité des alcènesest de mesurer leur énergie d’hydrogénation H° (énergielibérée au cours de l’addition d’une mole de H2).Les trois alcènes du graphique conduisent tous au mêmealcane (le butane), niveau de référence dans le graphe.Plus H° est grande, moins l’alcène de départ est stable.
On retiendra que : la stabilité d’un alcène augmente avec le nombre de groupes portés par la double liaison
(les groupes apportent des électrons par effet +I à la liaison C=C insaturée) les alcènes sont également stabilisés par les effets +M (doubles liaisons conjuguées) un alcène cis est moins stable qu’un alcène trans (encombrement stérique)
1 cal = 4,18 JEp
A : doubles liaisons conjuguées
B moins stableque A

DocCours Chimie Organique : réactions orgaII - 5/12
Figure 10 : Additions électrophiles (AE) sur la double liaison C=C - Généralités
Réaction générale :
Problème de régiosélectivité ?Si l’alcène de départ et le composé AB sont tous les deux dissymétriques, on peut obtenir un mélange de deuxproduits, isomères l’un de l’autre, selon que A se fixe sur le carbone de droite ou de gauche.Cette réaction peut donc être ou non régiosélective.C’est l’expérience et l’étude du mécanisme qui dira si elle l’est vraiment.
Problème de stéréosélectivité ?S’il y a apparition d’un carbone asymétrique, la réaction peut produire plusieurs stéréoisomères.Elle peut donc être ou non stéréosélective (ou stéréospécifique)Là encore c’est l’expérience et l’étude du mécanisme qui dira si elle l’est vraiment.
C C + A B+
C C
AB
C C
BA
+
mélange
r
Figure 11 : Additions électrophiles ioniques – Hydrohalogénation - Bilan
Il s’agit de l’addition ionique de HX sur C=C (X=halogène : F, Cl, Br ,I).On obtient un mélange de dérivés halogénés.
Bilan :
Mode opératoire : la réaction se fait à température élevée (170 °C) en phase gazeuse pour les premiers alcènes (peu réactifs) et à froid pour les alcènes plus lourds, HX gazeux barbotant dans l'alcène pur et
liquide, ou bien l'alcène et HX étant dissouts dans un solvant polaire.Il y a dégagement de chaleur (réaction exothermique).
Régiosélectivité expérimentale : Règle de Markovnikov (1869)Lors de l’addition électrophile ionique d’un composé H+─A- sur un alcène dissymétrique, A se fixe sur lecarbone le plus substitué.
Stéréosélectivité expérimentale : aucune
C CR
+ H X+
C C
XH
R
C C
HX
R
+
mélange
Figure 12 : Additions électrophiles ioniques – Hydrohalogénation - Mécanisme
Mécanisme en solvant polaireMécanisme par stades en deux étapes.
1ère étape : étape lente (la plus difficile)La liaison (nucléophile), capture le proton H+
(électrophile) de l’acide H-X. On obtient deux carbocations.
2ème étape :Les 2 carbocations sont attaqués par le nucléophile X⊖
On obtient deux composés A et B régioisomères
(voir la suite pour la détermination du composé majoritaire)
C C
H
R
+ X C C
H
R
Xcomposé A
+ X C C
X
R
H
C C
H
R
composé B
+ C C
H
R
C C
H
R
+H X+C C
R

DocCours Chimie Organique : réactions orgaII - 6/12
Figure 13 : Additions électrophiles ioniques – Hydrohalogénation – Mécanisme -Sélectivité
Absence de stéréosélectivitéLa 2ème étape du mécanisme est l’attaque du nucléophile sur le carbocation.Celui-ci est localement plan (autour du C+), et l’attaque est équiprobable de chaque côté du plan.On obtient deux composés A et A’ en même quantité.Si A et A' sont deux énantiomères, on obtient le mélange racémique.L’addition de HX est non stéréosélective.
RégiosélectivitéL’étape lente du mécanisme (1ère étape) conduit à 2 carbocations différents (régioisomères)Elle sera d’autant plus facile à faire que le carbocation obtenu est stable.L’addition de HX est régiosélective.
On peut d’ailleurs énoncer une version moderne plus générale de la règle de Markovnikov qu'on retiendrapar la suite.Lors de l’addition électrophile ionique d’un composé H─A sur un alcène, le composé majoritaire est issudu carbocation le plus stable.Pour conclure, il faudra étudier la stabilité des carbocations, c'est-à-dire les effets stabilisant ces carbocations,de nature peu stables.
A : 50%
C C
HR2
R1
+ X C C
H
R2
R1
X
C C
H
R2
R1
X
+
A' : 50%
Figure 14 : Stabilité des carbocations
GéométrieLe carbone central est AX3E0, (n=3) et adopte une géométrie triangulaire plane.Conséquence de cette géométrie.Les trois liaisons sont dans un planLes 2 faces sont attaquées de façon équiprobable par des nucléophiles.
Structure électronique du C+
Le carbone central est déficitaire en électrons (entourés de 6 électrons seulement, et porte une charge positive)Conséquence de cette structure :
Les carbocations sont des espèces instables et très réactives.Tout effet électronique qui diminue cette charge > 0 stabilise le carbocationTout effet électronique qui augmente cette charge déstabilise le carbocation
Facteurs influençant la stabilité des carbocations Facteurs électroniques (détail plus loin)
Effets inductifs : effets inductifs donneurs : effet +I ; effets inductifs accepteurs : effet -IEffets mésomères : effets mésomères donneurs : effet +M ; effets mésomères accepteurs : effet -M
Facteurs stériques (moins important)Une taille importante des groupes R1, R2, R3 déstabilise le carbocation
CR3R1
R2
Figure 15 : Réarrangement des carbocationsOn observe souvent que les carbocations formés se réarrangentd’eux-mêmes, pour donner des carbocations plus stables.Le réarrangement a lieu car le carbocation B obtenu est tertiaire et plus stable que le carbocation secondaire A(voir explication plus loin). Ce n’est pas de la mésomérie : il y a eu déplacement d'atomes (ici un groupeméthyl). B est un carbocation isomère de A (régioisomère)
Cas général : déplacement d'un groupe G (= H, CH3,…) voisin d'un C+
A B
G G
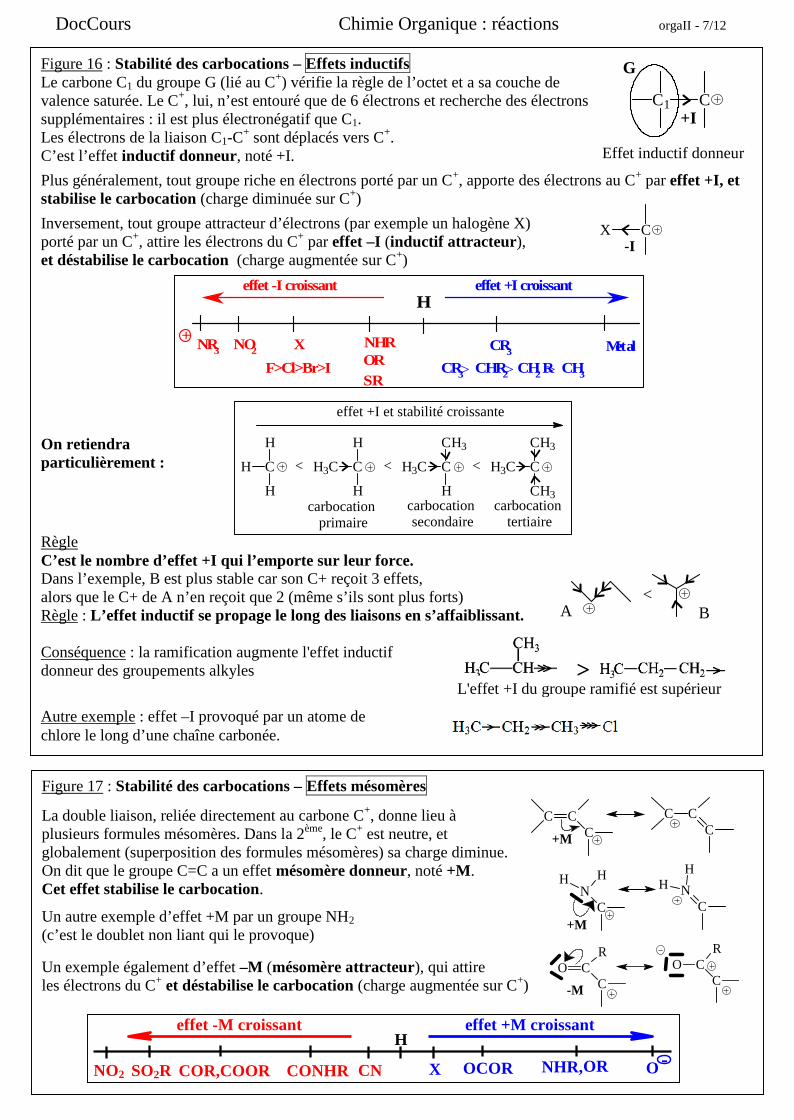
DocCours Chimie Organique : réactions orgaII - 7/12
Figure 16 : Stabilité des carbocations – Effets inductifsLe carbone C1 du groupe G (lié au C+) vérifie la règle de l’octet et a sa couche devalence saturée. Le C+, lui, n’est entouré que de 6 électrons et recherche des électronssupplémentaires : il est plus électronégatif que C1.Les électrons de la liaison C1-C
+ sont déplacés vers C+.C’est l’effet inductif donneur, noté +I.
Plus généralement, tout groupe riche en électrons porté par un C+, apporte des électrons au C+ par effet +I, etstabilise le carbocation (charge diminuée sur C+)
Inversement, tout groupe attracteur d’électrons (par exemple un halogène X)porté par un C+, attire les électrons du C+ par effet –I (inductif attracteur),et déstabilise le carbocation (charge augmentée sur C+)
On retiendraparticulièrement :
RègleC’est le nombre d’effet +I qui l’emporte sur leur force.Dans l’exemple, B est plus stable car son C+ reçoit 3 effets,alors que le C+ de A n’en reçoit que 2 (même s’ils sont plus forts)Règle : L’effet inductif se propage le long des liaisons en s’affaiblissant.
Conséquence : la ramification augmente l'effet inductifdonneur des groupements alkyles
Autre exemple : effet –I provoqué par un atome dechlore le long d’une chaîne carbonée.
Figure 17 : Stabilité des carbocations – Effets mésomères
La double liaison, reliée directement au carbone C+, donne lieu àplusieurs formules mésomères. Dans la 2ème, le C+ est neutre, etglobalement (superposition des formules mésomères) sa charge diminue.On dit que le groupe C=C a un effet mésomère donneur, noté +M.Cet effet stabilise le carbocation.
Un autre exemple d’effet +M par un groupe NH2
(c’est le doublet non liant qui le provoque)
Un exemple également d’effet –M (mésomère attracteur), qui attireles électrons du C+ et déstabilise le carbocation (charge augmentée sur C+)
L'effet +I du groupe ramifié est supérieur
Effet inductif donneur
G
C1 C+I
-ICX
H
NHRORSR
X
F>Cl>Br>I
NO2NR3+
CR3 Metal
CR3 CHR2 CH2 R CH3> > >
effet -I croissant effet +I croissant
CH3C
H
H
C
H
H
H < CH3C
CH3
H
< CH3C
CH3
CH3
<
carbocation primaire
carbocation secondaire
carbocation tertiaire
effet +I et stabilité croissante
<BA
>
+M
C CC
C CC
+M
NC
HHN
C
HH
-M
O CC
R
O CC
R
H
NHR,ORX
effet -M croissant effet +M croissant
NO2 O -OCORSO2R COR,COOR CONHR CN

DocCours Chimie Organique : réactions orgaII - 8/12
Figure 18 : Stabilité des carbocations – compétition entre effets I et M
Certains groupes peuvent donner à la fois un effet inductif et mésomère.Parfois même dans des sens contraires.C’est le cas des halogènes qui sont +M et –I :
RègleAssez généralement, c’est l’effet mésomère quil’emporte sur l’effet inductif.
CX-I
+M
CX
<
carbocation secondairemais plus stable car effet +M de la double liaison
carbocation tertiaire(pas de mésomérie)
+M
Figure 19 : Additions électrophiles ioniques – Hydrohalogénation - Résumé
Réaction bilan
Conditions opératoiresSouvent dans un solvant polaire, qui va stabiliser le carbocation.
Mécanisme ionique en deux étapes
L’hydrohalogénation est régiosélective et non stéréosélective.
Figure 20 : Additions électrophiles ioniques – Hydratation - Bilan
Il s’agit de l’addition de H2O sur C=C . On obtient un mélange d’alcools.
Bilan :
Mode opératoire :La réaction se fait dans une solution aqueuse d'acide sulfurique H2SO4
(ou phosphorique H3PO4) diluée, en chauffant à reflux.
Régiosélectivité expérimentale : Règle de MarkovnikovL'hydratation des alcènes conduit majoritairement à l'alcool le plus substitué
Stéréosélectivité expérimentale : aucune
C CR
+ H X+
C C
XH
R
C C
HX
R
+
mélange
+ C C
H
R
C C
H
R
+H X+C C
R
maj
1)
C C
H
R
+ X C C
H
R
X
2)
AE
Étape lente : addition électrophile de H+
Formation du carbocation plan = intermédiaireréactionnel (IR) instable.Le carbocation majoritaire est le plus stable.Possibilité de réarrangement.
2ème étape (plus facile)Attaque du nucléophile X- et formation desdérivés halogénés dont un est majoritaire, celuiqui provient du carbocation le plus stable : c'estla règle de Markovnikov
+ C C
H
R
OH
C C
H
R
OH
+H OH+C C
R
mélange
chauffage à reflux

DocCours Chimie Organique : réactions orgaII - 9/12
Figure 21 : Additions électrophiles ioniques – Hydratation - Mécanisme
Mécanisme de l’hydratation (catalysée ici par H2SO4)C’est un mécanisme ionique en 4 étapes tout a fait similaire à celui de l’hydrohalogénation.L’acide sulfurique est régénéré en dernière étape. C’est un catalyseur.
maj
2)
3)
+ C C
H
R
C C
H
R
H+C CR
S
O
O
O
O
H
H
H+ +AB
AE
C C
H
R
1)= -O-SO3H= HSO4-
ion hydrogénosulfate
OHH
+ C C
H
R
OHH
S
O
O
O
O
H
AB+C C
H
R
OHH
O-SO3H + H2SO4C C
H
R
OH
4)
Etape 1 : l’acide fort H2SO4 libère facilement 1H+ pour donner sa base -OSO3H
Etape 2 (lente)Addition électrophile de H+ sur le nuage .Deux carbocations sont formés dont un estmajoritaire, le plus stable.
Etape 3 (facile)Attaque du nucléophile H2O sur le carbocation.
Etape 4 (facile)La base -OSO3H arrache le H+ porté parl’oxygène. L’alcool est formé et H2SO4 régénéré.Le bilan s’obtient par 1+2+3+4
Figure 22 : Additions électrophiles ioniques – Hydratation – Remarques et réactions secondaires
Choix de l'acide catalyseurOn ne peut pas utiliser d’acide halogéné HX (X = F, Cl, Br, I), car l’étape 1) conduit à l’anion halogénure X -,bon nucléophile. C’est cet ion qui se fixerait sur le C+ dans l’étape 3, ce qui conduit à une hydrohalogénation(1ère addition électrophile étudiée dans ce cours).Pour catalyser l’hydratation, on doit donc utiliser des acides dont la base conjuguée est peu nucléophile. C’estle cas de H2SO4 et H3PO4.
Réarrangements possiblesComme dans l’hydrohalogénation, le carbocation peut se réarranger et conduire à un mélange d'alcools.
Exemple :
Réactions secondaires au cours de l'hydratationL’étape 3 est une attaque nucléophile. Tout nucléophile du milieu est susceptible de s’ajouter sur le C+.Il y a 3 nucléophiles du milieu, plus ou moins forts :
a) L’eauCela conduit à l’étape classique 3) et plus tard à la formation d’un alcool.
b) L’ion hydrogénosulfate HSO4-
Il est peu nucléophile, mais dans le cas ou l’acide sulfuriqueest concentré, cet ion est en concentration importante et entreraen compétition avec l’eau, pour donner un hydrogénosulfated’alkyle par la réaction 3bis)
c) L’alcène lui-même est nucléophile (nuage ).Il peut s’ajouter sur C+ (par 3ter), s’il est suffisamment concentré, et donne deux nouveaux carbocations,dimères du précédent (seul le plus stable est représenté). La réaction peut se poursuivre et conduit à unepolymérisation.
OHA OHB
OHC
H2O
H2SO4+ +
3bis) C C
H
R
+ C C
H
R
OHO3S
O-SO3H
a) b) c) C CR
O-SO3HOHH
3ter) C C
H
R H
HH + C CR
H
H
H
1 2 34 12CC
H
RH
H
H
C C H
RH
H
43
majoritaire
...C C
R
H
H
H

DocCours Chimie Organique : réactions orgaII - 10/12
Figure 23 : Additions électrophiles ioniques – Halogénation - Bilan
Il s’agit de l’addition ionique de X2 sur C=C (X=halogène : F, Cl, Br ,I).On obtient un mélange de dérivés dihalogénés.
Bilan :
Mode opératoire : la réaction se fait violemment avec F2 (destruction de l’alcène par oxydation) facilement avec Cl2 et Br2 à température ordinaire dans un solvant (di ou tri ou tétrachloroéthane). C’est la
base du test des alcènes : décoloration rapide du dibrome orange en présence d’un alcène. difficilement avec I2
Régiosélectivité expérimentale :Ne se pose pas puisqu’on ajoute un composé X2 symétrique.
Stéréosélectivité expérimentale :On remarque expérimentalement une forte stéréospécificité. Dans l’exemple, 3 stéréoisomères (et non 4 carplan de symétrie) peuvent être à priori obtenus. On n’en obtient que certains, selon l'alcène de départ (Z ou E).
C CR3
R4
R1
R2
+ X X C C R4
R3
XX
R2
R1mélange de
stéréoisomères
Br2Br Br
CH2Cl2 CH2Cl2
Br2+
mélange racémique
Br
Br
Br
Br1 2composé méso
Figure 24 : Additions électrophiles ioniques – Halogénation - Mécanisme
AE+
CH
CH
Br Br CH
CH
Br
+
ion bromoniumou ion ponté (a)
1) + Br
a
b
2)
mélange racémique
CH
CH
Br
CH
C
HBr
Br
+CH
C
H Br
Br1
2
1 2
1 = (S,S)
+ Br
2 = (R,R)
Etape 1 (étape lente)La liaison Br-Br n’est pas polarisée par nature, mais elle estpolarisable.A l’approche du nuage de l'alcène, elle se polarise : il apparaîtune charge + sur le Br le plus proche, qui, en se fixant sur lesdeux carbones à la fois conduit à un intermédiaire réactionnelappelé ion ponté, ou ion halonium (ici ion bromonium).C’est bien une attaque électrophile de la liaison C=C (par Br+ ).Note : il y a en fait 2 ions pontés, selon la face de l'alcèneattaquée (attaque a ou b), qui conduiront aux mêmes produits.
Etape 2Le nucléophile Br- attaque l’un des deux carbones (les 2attaques sont ici équiprobables), et forcément du côté opposé dupremier Br fixé : on parle d’attaque anti.On obtient les deux énantiomères 1 et 2 en même quantité, doncun mélange racémique.L'attaque de l'ion ponté (b) donne le même résultat.
Figure 25 : Additions électrophiles ioniques – Halogénation – Mécanisme - Stéréospécificité
Stéréosélectivité du mécanisme
"L’attaque anti" implique donc le schémasuivant. On notera que les composés 1 et 2sont des énantiomères et que le composé 3est achiral (plan de symétrie dans une desconformations, composé méso).
La bromation des alcènes est donc stéréospécifique à 100% (et même diastéréospécifique, car le couple 1-2et le composé 3 sont des diastéréoisomères).On retiendra que l'halogénation est une "anti addition", cela signifie que le 2ème brome se fixe sur la faceopposée à celle attaquée par le 1er brome.
produits
réactifs
50% 50% 0%
0% 0% 100%
CH
C
H Br
Br 1C
HC
HBr
Br2
C
H
CHBr
Br3

DocCours Chimie Organique : réactions orgaII - 11/12
Figure 26 : Additions électrophiles ioniques – Halogénation – Remarques
Dans certains cas, on remarque une certaine proportion de "syn addition".C’est lorsque que l’ion halonium s’ouvre, pour former un carbocation relativement stable. L’halogénure pourraalors s’ajouter sur les deux faces du C+ d’où le mélange d’addition anti et syn.
C’est le cas de la chloration : l’ion chloronium, moins stable que l’ion bromonium, évolue partiellement versles deux carbocations de la figure.
Cela se passe aussi dans le cas où le carbocation formé est particulièrement stable (carbocation typebenzylique dans l’exemple).
CH
CH
Cl 21
1 2
CH
CH
ClCl
CH
CH
Clsyn syn
anti
Figure 27 : Additions électrophiles radicalaires – Hydrohalogénation – Bilan
Il s’agit de l’addition de HX sur C=C mais ici dans des conditions favorisant l'apparition de radicaux et nond'ions. On obtient là encore un mélange de dérivés halogénés.
Bilan :
Mode opératoire : la réaction se fait à reflux dans un solvant apolaire et aprotique (dioxanne, tétrachlorure de carbone).
solvant aprotique = solvant dont la molécule ne peut pas libérer de H+
en présence d'une petite quantité d'initiateur, en général un peroxyde (composé du type Z-O-O-Z'). Cecomposé n’est pas régénéré, mais n'apparaît pas dans le bilan (composé minoritaire).
Régiosélectivité expérimentale : Règle de anti-Markovnikov ou effet KharaschLors de l’addition électrophile radicalaire d’un composé H─X sur un alcène dissymétrique, X se fixe sur lecarbone le moins substitué.
Stéréosélectivité expérimentale : aucune
C CR
+ H X+
C C
XH
R
C C
HX
R
+
mélange
Figure 28 : Additions électrophiles radicalaires – Hydrohalogénation – Mécanisme
Il s'agit d'une réaction en chaîne radicalaire. (attention : flèches à une pointe, pas d’ion)Le peroxyde Z-O-O-Z possède une liaison O-O fragile, qui se casse sous l’action de la chaleur. C'estl'initiateur), qui sert à amorcer la réaction (étape d'initiation).
majoritaire
C CH
H
R
H+ C C H
H
Br
H
R
+Br
1 2
C C H
H
H
R
Br 21
C C H
H
Br
H
R
Br H + C C H
H
Br
H
R
H
Br+
maillon
1) Z O O Z Z O2chaleur
Z O + H Br Z O H + Br2)
3)
4)
5) Br + Br Br2
Initiation
propagation
propagation
propagation
rupture
L’étape 3 est l’addition de l’électrophile Br(atome de brome demandeur d’électron)
Il y a formation de deux carboradicaux, dontun est plus stable (effets inductifs donneurs)
La connaissance du mécanisme permet dedonner une version plus générale de la règlede Kharasch :Lors de l’addition électrophile radicalaired’un composé H─A sur un alcène, lecomposé majoritaire est issu ducarboradical le plus stable.

DocCours Chimie Organique : réactions orgaII - 12/12
Figure 29 : Coupure oxydante de la liaison C=C – Ozonolyse - Bilan
L’ozone O3 est un oxydant puissant, et peut être représenté par les 3 formes mésomères suivantes :
C’est la forme III qui permet d’expliquer la réaction d’ozonolyse (découverte par Harries en 1903), dont lemécanisme complet n’est pas au programme.1ère phase : l’alcène est traité à -78°C en solution dans le dichloroéthane (CH2Cl2) par l’ozone (gaz). Onobtient un ozonide stable à cette température. Les liaisons ondulées signifient que tous les types destéréochimie sont possibles.
2ème phase (hydrolyse)Il y a une légère différence selon que cette phase est réalisée dans des conditions réductrices ou oxydantes.
Hydrolyse réductriceOn ramène le milieu à température ambiante et on ajoute un réducteur : du zinc en poudre en solution dans l'd’acide éthanoïque ; ou encore le diméthylsulfure (CH3)2S).L'ozonide est coupé. Le bilan est une coupure de liaison C=C avec formation d’aldéhydes et/ou de cétones.
Hydrolyse oxydanteOn ramène le milieu à température ambiante. L’ozonide est ensuite hydrolysé en présence d’eau et deperoxyde d’hydrogène (H2O2 : oxydant)Cela conduit à des cétones, les aldéhydes éventuels sont transformés en acides carboxyliques (c'est la seuledifférence par rapport à l'hydrolyse réductrice)
C'est le bilan qu'il faut savoir écrire et non toutes ces étapes. Exemple de bilan des 2 phases :
O O O OOO OOO
I II III
O3
R1
R2
R3
R4
O3
CH2Cl2 (-78 °C) R1R2
R3R4
O OO
instable
R1R2
O
O
O
R4
R3
ozonide stable
R1R2
O
O
O
HR3
Zn (poudre)
CH3COOH / eauO
R1
R2
+ OR3
H
R1R2
O
O
O
HR3
H2O2eau
OR1
R2
+ OR3
OH
1) O3 / CH2Cl2
2) Zn /CH3COOH
OO
H
1
61
61) O3 / CH2Cl2
2) H2O2, H2O
OO
OH
1
61
6
Figure 30 : Coupure oxydante de la liaison C=C – Ozonolyse - Utilisation
Recherche de structureLa réaction est largement utilisée pour localiser la position des doubles liaisons dans les molécules organiques.
Exemple : trouver l’alcène A.
Réaction de synthèse de composés dicarbonylés
Autre agent d’oxydation : l’ion permanganate MnO4-
On fait réagir l’alcène sur une solution concentrée à chaud d’ion MnO4- (oxydant). La liaison double se coupe
et conduit à des cétones, des acides carboxyliques (dans le cas d’un H sur le carbone) et à CO2 dans le cas dedeux H.
1) O 3 / CH 2Cl2 (-78 °C)
2) Zn / CH3COOH / eauO
H
H+ Oalcène inconnu A
R1
R2
2) Zn / CH3COOH / eau
1) O3 / CH2Cl2 (-78 °C)
MnO4- concentré
à chaudO
OH
OO
OH+ CO2+