développement de méthodes thermiques pour la...
TRANSCRIPT

N◦ ordre : 3936
THÈSE
présentée à
L’UNIVERSITE BORDEAUX I
École doctorale des sciences chimiques
par Cindy Hany
pour obtenir le grade de
Docteur
Spécialité : chimie-physique
Développement de méthodes thermiques
pour la caractérisation
de réactions chimiques en microfluidique
Soutenue le 3 décembre 2009
Devant la commission d’examen formée de :
Mr Laurent Servant Président du JuryMr Claude De Bellefon RapporteurMr Denis Maillet RapporteurMr Jean-Christophe Batsale ExaminateurMr Christophe Gourdon ExaminateurMr Bertrand Pavageau Examinateur
Invités :
Mr Christophe Pradère
Mr Jean Toutain

2

Remerciements
Mes premiers remerciements s’adressent à Matthieu Joanicot, ancien directeur du LoF,et à Patrick Maestro, actuel directeur du LoF, pour m’avoir accordé leur confiance sur ceprojet de thèse.
Je remercie chaleureusement Jean-Christophe Batsale, pour avoir été directeur de ceprojet et pour ses conseils avisés.
Je remercie vivement Denis Maillet et Claude De Bellefon, qui m’ont fait l’honneur dejuger ce travail, ainsi que Laurent Servant pour avoir accepté de présider mon jury de thèse.Merci également aux examinateurs du jury pour l’intérêt porté sur ce travail : BertrandPavageau pour sa vision industrielle et Christophe Gourdon pour son expertise en Géniedes Procédés .
Je suis tout particulièrement reconnaissante envers Christophe Pradère et Jean Toutainqui ont encadré ma thèse et m’ont permis de décourvrir et prendre goût au monde de lamicro-thermique. Un grand merci à tous les deux pour votre disponibilité, les nombreuseset fructueuses discussions, votre pédagogie ainsi que votre bonne humeur et votre confiance.Christophe, je te remercie pour tes rapides et nombreuses relectures du manuscrit, ainsique pour ton humour accompagnée de blagues toujours très fines ! ! ! Quant à toi, Jean,merci à ton âme de programmateur, sans qui la caméra IR ne serait pas aussi performante.
Je remercie bien amicalement Flavie Sarrazin qui a encadré, à la suite de Bertrand, lecôté industriel de cette thèse. Merci, pour tes conseils avisés, ta vision objective, ton aideet ton soutien durant toutes les aventures qui ont rythmé ces trois années.
Je remercie par ailleurs Laurent Prat et Karine Loubière, du LGC, pour leur collabo-ration et pour nous avoir permis d’étudier une application explosive.
Je tiens aussi à remercier l’atelier du CRPP, en particulier, Phillipe pour ses nombreusesréalisations de puces en serpentin.
Je remercie Pascal Panizza pour m’avoir permis d’effectuer mon stage de Master ausein du LoF, ce qui m’a ouvert les portes vers cette thèse.
J’ai effectué ma thèse entre le LoF et le TREFLE, en effectuant la plus grande partiede mon temps au LoF. Ainsi, ces trois années m’ont permis d’effectuer ma thèse dans uneambiance de travail épanouissante et très enrichissante. Je remercie toutes les personnes que
3

4
j’ai rencontrées. Tout d’abord au LoF, je remercie Philippe pour ses quelques étrennes quim’ont vraiment dépannées, Roman, Bernard et Matthieu mes collègues de bureau, Fatineet Aurélie pour leur talent en LabView, Chloé pour ses réconforts chocolatés pendant larédaction, Thomas pour ses talents de chanteur en millifluidique, Céline pour son aideadministrative et ses conseils, la petite morue, Inês, pour son amitié, Martine pour saconstante bonne humeur. Je remercie également toutes les personnes passées (Wilfried,Soca, Juan, Masa, Julie ...) et présentes au LoF (Pierre, J-B, Jacques, Simon, Fanny,Bruno ...) pour nos discussions en tout genre. Bien entendu, je remercie tous les actuelset futurs thésards, Aurore, Elise, Christophe, Julien, Oriane, Lingguo, Nicolas, Vincent,Laure, Julie, Amandine, Marta. Je remercie sincèrement les stagiaires : Hélène Lebrun etMélanie Wynn, sans lesquels une grande partie de ce travail n’aurait pas été réalisé.
Au cours de ma thèse, j’ai effectué de nombreuses visites au TREFLE et je tiens àremercier toutes les personnes du TREFLE pour leurs discussions et constante bonnehumeur : Lilian qui a débuté sa thèse en même temps que moi, Christophe R. pour sonaide sur l’estimation de Péclet, Yannick, Jean-Luc, Elena, Carolina et tous les autres.
Enfin, je remercie mon ami manouche, pour son aide, sa patience et surtout sa présence,ma petite Sandrinette pour avoir égaillé mes soirées et mes nuits, et toute ma famille, enparticulier ma maman, Roxane et Thomas, pour m’avoir toujours soutenue dans mes choix.

Table des matières
1 La thermique appliquée à la microfluidique : éléments bibliographiques 171.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 181.2 L’outil microfluidique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
1.2.1 Fabrication des microcanaux : photolithographie et technologie PDMS 181.2.2 Microréacteurs monophasiques . . . . . . . . . . . . . . . . . . . . . 20
1.2.2.1 Écoulement laminaire . . . . . . . . . . . . . . . . . . . . 201.2.2.2 Interdiffusion de deux réactifs . . . . . . . . . . . . . . . . 21
1.2.3 Microréacteurs diphasiques . . . . . . . . . . . . . . . . . . . . . . . 241.2.3.1 Formation d’un train de gouttes . . . . . . . . . . . . . . . 241.2.3.2 Mélange au sein des gouttes . . . . . . . . . . . . . . . . . 25
1.3 Champs d’applications de la microfluidique . . . . . . . . . . . . . . . . . . 271.4 De la calorimétrie classique à la microcalorimétrie . . . . . . . . . . . . . . 28
1.4.1 Microcalorimètre différentiel . . . . . . . . . . . . . . . . . . . . . . 301.4.2 Microcalorimètres intégrés . . . . . . . . . . . . . . . . . . . . . . . 311.4.3 Application des marqueurs fluorescents à la microfluidique . . . . . 321.4.4 Application des caméras Infrarouge à la microfluidique . . . . . . . 32
1.5 Conclusion et justification du travail de thèse . . . . . . . . . . . . . . . . 34
2 Développement d’un microcalorimètre différentiel par mesure macrosco-pique 372.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 382.2 Caractéristiques du dispositif . . . . . . . . . . . . . . . . . . . . . . . . . 39
2.2.1 Enceinte et système de régulation . . . . . . . . . . . . . . . . . . . 402.2.2 Pousse-seringues . . . . . . . . . . . . . . . . . . . . . . . . . . . . 412.2.3 Capteur de flux . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 412.2.4 Acquisition et étalonnage . . . . . . . . . . . . . . . . . . . . . . . . 42
2.3 Procédure expérimentale et logiciel associé . . . . . . . . . . . . . . . . . . 422.3.1 Démarche générale . . . . . . . . . . . . . . . . . . . . . . . . . . . 422.3.2 Etalonnage de l’appareil . . . . . . . . . . . . . . . . . . . . . . . . 432.3.3 Acquisition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 432.3.4 Estimation de l’enthalpie . . . . . . . . . . . . . . . . . . . . . . . . 44
2.4 Modélisation thermique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 452.4.1 Modèle thermique . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
5

6 Table des matières
2.4.2 Solution analytique . . . . . . . . . . . . . . . . . . . . . . . . . . . 472.4.3 Influence de la nature du substrat . . . . . . . . . . . . . . . . . . . 472.4.4 Répartition du flux de chaleur . . . . . . . . . . . . . . . . . . . . . 48
2.5 Étalonnage du dispositif . . . . . . . . . . . . . . . . . . . . . . . . . . . . 492.5.1 Définition du coefficient d’étalonnage . . . . . . . . . . . . . . . . . 492.5.2 Conception d’une puce étalon . . . . . . . . . . . . . . . . . . . . . 502.5.3 Principe de la mesure . . . . . . . . . . . . . . . . . . . . . . . . . . 512.5.4 Résultats . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
2.6 Détermination de l’incertitude sur la mesure de l’enthalpie . . . . . . . . . 532.6.1 Erreur sur la tension mesurée . . . . . . . . . . . . . . . . . . . . . 542.6.2 Précision sur le coefficient d’étalonnage . . . . . . . . . . . . . . . . 552.6.3 Stabilité des pousse-seringues . . . . . . . . . . . . . . . . . . . . . 562.6.4 Précision sur l’enthalpie . . . . . . . . . . . . . . . . . . . . . . . . 60
2.7 Validation du microcalorimètre . . . . . . . . . . . . . . . . . . . . . . . . 612.7.1 Choix de la réaction modèle . . . . . . . . . . . . . . . . . . . . . . 612.7.2 Conception de la puce microfluidique . . . . . . . . . . . . . . . . . 612.7.3 Principe de la mesure . . . . . . . . . . . . . . . . . . . . . . . . . . 622.7.4 Résultats lors de l’écoulement co-courant . . . . . . . . . . . . . . . 632.7.5 Résultats lors de l’écoulement gouttes . . . . . . . . . . . . . . . . . 64
2.7.5.1 Influence de l’huile lors d’un écoulement diphasique . . . . 652.7.5.2 Détermination de l’enthalpie de réaction . . . . . . . . . . 66
2.8 Applications et caractérisations de réactions chimiques . . . . . . . . . . . 682.8.1 Étude de la réaction d’estérification . . . . . . . . . . . . . . . . . . 68
2.8.1.1 Mesure de l’enthalpie de mélange . . . . . . . . . . . . . . 692.8.1.2 Mesure en écoulement co-courant . . . . . . . . . . . . . . 692.8.1.3 Mesure en écoulement gouttes . . . . . . . . . . . . . . . . 702.8.1.4 Mesure de l’enthalpie de réaction . . . . . . . . . . . . . . 71
2.8.2 Mesure de l’enthalpie molaire d’excès . . . . . . . . . . . . . . . . . 722.8.3 Dosage calorimétrique . . . . . . . . . . . . . . . . . . . . . . . . . 742.8.4 Estimation du coefficient de diffusion . . . . . . . . . . . . . . . . . 76
2.8.4.1 Conception d’un microréacteur pour la cinétique . . . . . 762.8.4.2 Résultats sur la cinétique de mélange . . . . . . . . . . . . 77
2.9 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
3 Approche locale pour l’estimation de cartographie de propriétés thermo-cinétiques par thermographie InfraRouge en puce microfluidique 813.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 823.2 Caractéristiques du dispositif . . . . . . . . . . . . . . . . . . . . . . . . . 83
3.2.1 Caméra IR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 843.2.2 Element Peltier . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 843.2.3 Puce microfluidique . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
3.3 Procédure expérimentale . . . . . . . . . . . . . . . . . . . . . . . . . . . . 853.3.1 Démarche générale . . . . . . . . . . . . . . . . . . . . . . . . . . . 85

Table des matières 7
3.3.2 Calibration du nombre de Péclet . . . . . . . . . . . . . . . . . . . 863.3.3 Acquisition et estimation . . . . . . . . . . . . . . . . . . . . . . . . 86
3.4 Présentation du système thermique . . . . . . . . . . . . . . . . . . . . . . 863.5 Méthode d’identification par approche nodale . . . . . . . . . . . . . . . . 88
3.5.1 Influence des pertes thermiques locales . . . . . . . . . . . . . . . . 883.5.2 Estimation du champ de Péclet . . . . . . . . . . . . . . . . . . . . 893.5.3 Estimation de champ de terme source . . . . . . . . . . . . . . . . . 90
3.6 Validation numérique en écoulement co-courant . . . . . . . . . . . . . . . 903.6.1 Méthode du flux local . . . . . . . . . . . . . . . . . . . . . . . . . 90
3.6.1.1 Estimation de cartographie de Péclet . . . . . . . . . . . . 903.6.1.2 Estimation des cartographies de Flux . . . . . . . . . . . . 94
3.6.2 Méthode à gradient imposé . . . . . . . . . . . . . . . . . . . . . . 953.6.2.1 Estimation de cartographie de Péclet . . . . . . . . . . . . 953.6.2.2 Estimation des cartographies de Flux . . . . . . . . . . . . 97
3.7 Validation expérimentale de mesure de Péclet en co-courant . . . . . . . . 993.7.1 Par la méthode d’un flux de chaleur local . . . . . . . . . . . . . . . 99
3.7.1.1 Démarche expérimentale . . . . . . . . . . . . . . . . . . . 993.7.1.2 Estimation du nombre de Péclet . . . . . . . . . . . . . . 1003.7.1.3 Estimation du terme source . . . . . . . . . . . . . . . . . 100
3.7.2 Par la méthode du gradient de température . . . . . . . . . . . . . 1013.7.2.1 Démarche expérimentale . . . . . . . . . . . . . . . . . . . 1013.7.2.2 Estimation du nombre de Péclet . . . . . . . . . . . . . . 102
3.8 Application à la mesure de la cinétique d’une réaction chimique . . . . . . 1043.9 Proposition d’une méthode nodale pour l’écoulement gouttes . . . . . . . . 106
3.9.1 Procédure expérimentale . . . . . . . . . . . . . . . . . . . . . . . . 1073.9.2 Démarche de la méthode . . . . . . . . . . . . . . . . . . . . . . . . 1073.9.3 Estimation de la vitesse moyenne de la goutte . . . . . . . . . . . . 1083.9.4 Méthode inverse proposée . . . . . . . . . . . . . . . . . . . . . . . 110
3.9.4.1 Estimation des cartographies du nombre de Péclet selon x 1103.9.4.2 Estimation des cartographie du nombre de Péclet selon y . 111
3.10 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113
4 Calorimètre millifluidique isopéribolique par thermographie InfraRouge1154.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1164.2 Caractéristiques du dispositif . . . . . . . . . . . . . . . . . . . . . . . . . 117
4.2.1 Système de régulation . . . . . . . . . . . . . . . . . . . . . . . . . 1174.2.2 Caméra InfraRouge . . . . . . . . . . . . . . . . . . . . . . . . . . . 118
4.3 Description du milliréacteur chimique . . . . . . . . . . . . . . . . . . . . . 1194.3.1 Description générale . . . . . . . . . . . . . . . . . . . . . . . . . . 1194.3.2 Types d’écoulements . . . . . . . . . . . . . . . . . . . . . . . . . . 121
4.4 Procédure expérimentale . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1214.4.1 Démarche générale . . . . . . . . . . . . . . . . . . . . . . . . . . . 1214.4.2 Étalonnage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122

8 Table des matières
4.4.3 Acquisition et traitement . . . . . . . . . . . . . . . . . . . . . . . . 1224.5 Modélisation thermique du milliréacteur . . . . . . . . . . . . . . . . . . . 124
4.5.1 Approche locale . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1244.5.2 Approche globale . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
4.6 Calibration du système millifluidique . . . . . . . . . . . . . . . . . . . . . 1284.6.1 Mesures des pertes thermiques . . . . . . . . . . . . . . . . . . . . . 1284.6.2 Estimation de la longueur caractéristique de thermalisation . . . . . 1294.6.3 Validation des hypothèses du modèle . . . . . . . . . . . . . . . . . 131
4.7 Validation expérimentale de l’appareil . . . . . . . . . . . . . . . . . . . . . 1334.7.1 Estimation du terme source . . . . . . . . . . . . . . . . . . . . . . 1334.7.2 Expériences réalisées . . . . . . . . . . . . . . . . . . . . . . . . . . 1334.7.3 Résultats obtenus . . . . . . . . . . . . . . . . . . . . . . . . . . . . 135
4.8 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 138
A Microcalorimètre différentiel par mesure macroscopique 145

Liste des symboles
a diffusivité thermique (m2.s−1)
C concentration en réactif limitant (mol.L−1)
Co concentration initiale (mol.L−1)
Cp capacité calorifique (J.kg−1K−1)
Ca le nombre capillaire
dh diamètre hydraulique (m)
e épaisseur (m)
G facteur de forme (m−1)
h coefficient de convection (W.m−2.K−1)
K coefficient d’étalonnage (W.K−1)
k constante de vitesse (L.mol−1.s−1 )
n nombre de mesure (adimensionnel)
P puissance injectée (W)
Q débit molaire en réactif limitant (L.s−1)
Q débit volumique (m3.s−1)
Re nombre de Reynolds
S surface (m2)
T température (◦C)
t temps (s)
Tc température de consigne (◦C ou K)
U vitesse (m.s−1)
u incertitude (%)
U tension mesurée (V)
9

10 Table des matières
v vitesse (m.s−1)
X taux de conversion
x abscisse (m)
y ordonnée (m)
Ydiff largeur de diffusion (m)
Lettres grecques
α coefficient d’étalonnage (W.V−1)
β coefficient Seebeck (V.K−1)
∆H enthalpie de réaction (J.mol−1)
λ conductivité thermique (W.m−1.K−1)
µ viscosité dynamique (Pa.s)
Φ flux de chaleur global (W)
φ flux thermique par unité de longueur (W.m−1)
ρ masse volumique (kg.m−3)
σ écart type
Sigles
ATG Analyse thermogravimétrique
DSC Differential Scanning Calorimetry
In-Sb Indium Antimoine
IR InfraRouge
MWIR Medium Wavelenght InfraRed
PEEK Polyétheréthercétone
PFA Perfluoroalkoxy
PID Proportionel Intégral Dérivé
PIV Particle Image Velocimetry
Indices
a ambiant
c consigne
conc concentration

Table des matières 11
e entrée dans la puce
exp expérimental
i milieu
m mélange
p plaque en aluminium
t tube en PFA
th théorique

12 Table des matières

Introduction
L’industrie chimique possède un rôle capital dans l’économie française et mondiale.Elle est présente dans de nombreux domaines : pharmacie, agroalimentaire, matériaux,cosmétiques, pétrochimie... Des accidents chimiques ont été provoqués par des réactionsdont la mise en oeuvre n’avait pas toujours été contrôlée : emballement thermique ouréaction secondaire non maîtrisée. Rappelons nous les catastrophes survenues à Bhopal(1984) ou Toulouse (2001) dont les conséquences dramatiques relancent la polémique surla sécurité et l’environnement.
La connaissance de la cinétique et des données thermodynamiques des réactions chi-miques est donc capitale pour le développement et la sécurité des procédés. Tout procédéchimique ou physique est associé à des phénomènes thermiques couramment caractériséspar des méthodes de calorimétrie permettant de rassembler des données thermodynamiqueset cinétiques. Dans le cas d’une réaction chimique, l’évolution du flux de chaleur est re-productible, directement mesurable et proportionnel au taux d’avancement de la réaction,les mesures calorimétriques conduisent à des données de base de cinétiques et de thermo-dynamiques. Ce type de mesure fournit non seulement des informations sur la réactionchimique, mais aussi les paramètres de dimensionnement de réacteurs pour la sécurité. Lacaractérisation de réactions chimiques par la mesure du flux de chaleur est très utilisée eta conduit au développement de nombreux calorimètres aux caractéristiques et domainesd’applications variables [1, 2]. Le calorimètre RC (Reaction Calorimeter) est un dispositifproche des réacteurs industriels avec un volume réactionnel de l’ordre du litre, une agi-tation mécanique et un système de chauffage et de refroidissement par double enveloppe.Cet appareil permet d’estimer des données telles que l’enthalpie de réaction, la capacitécalorifique des produits, etc. La DSC (Differential Scanning Calorimetry) est une techniquede microcalorimétrie dans laquelle un échantillon de quelques milligrammes est soumis àun balayage de températures par compensation de puissance thermique conduisant à desdonnées thermodynamiques. Cependant, tous ces calorimètres ne sont pas simultanémentadaptés à l’utilisation de microvolumes, à une haute sensibilité thermique, à un bon contrôledu mélange des réactifs et à des conditions isothermes.
Une des voies pour bénéficier d’un système de petite taille permettant des acquisitionsrapides de données est la microfluidique. Les réacteurs chimiques microfluidiques font par-tie des outils MEMS (Micro - Electro - Mechanical - Systems) très prometteurs pour la
13

14
caractérisation continue de réactions chimiques à petite échelle. Le principal avantage decette technologie est la petite dimension des canaux permettant entre autre le contrôle dumélange des réactifs et l’établissement d’un régime laminaire à bas nombre de Reynolds(< 100). L’écoulement diphasique ou écoulement gouttes permet la génération d’un trainde gouttes monodisperses. Ces écoulements offrent un bon contrôle du mélange des réactifspar l’absence de dispersion axiale et une meilleure gestion de l’exothermie des réactionsgrâce au transfert de chaleur dans la phase porteuse. D’autre part, ce type de microréac-teurs présentent plusieurs avantages pour le contrôle et le suivi de réactions chimiques,notamment d’isoler les produits, de transporter et mélanger efficacement les réactifs ; lemélange est environ 10 fois plus rapide que lors d’un écoulement co-courant. L’utilisationd’une faible quantité de produits est intéressante d’un point de vue économique et envi-ronnemental. En outre, cela permet de tester facilement et rapidement un grand nombrede configurations de réactions en variant les paramètres tels que la quantité de réactifs parune simple modification du débit, la température de consigne, ou encore le type de cata-lyseur [3]. De plus, le grand rapport surface sur volume augmente le transfert de chaleurdes réactions exothermiques grâce aux coefficients d’échanges entre les fluides en présence.Aussi de nouvelles conditions opératoires peuvent être testées.
Ces microsystèmes permettent d’obtenir de nouveaux produits, notamment pour lapolymérisation, l’encapsulation par émulsion double ou encore pour les opérations de cris-tallisation.
Ils constituent également de nouveaux appareils de recherche qui servent à l’acquisitionintensive de données. De nombreuses applications concernent le domaine de la physico-chimie. Un microrhéomètre a été développé par Guillot et al. : il permet de déterminerla rhéologie d’un fluide en écoulement co-courant [4]. Leng et al. ont mis au point unmicroévaporateur pour l’exploration de diagrammes de phase et des cinétiques de chan-gement d’état [5]. Ils ont également recensé les dispositifs microfluidiques adaptés à lacristallisation [6]. Les microsystèmes sont aussi utilisés pour l’étude de cinétique de réac-tions chimiques en ligne par des méthodes spectroscopiques. Plusieurs analyses consistentégalement à utiliser : un spectromètre Raman pour sonder le profil de concentration dansle microcanal [7, 8], ou encore un spectrophotomètre UV-visible [9]. D’autres études seconcentrent sur l’analyse thermique par le développement de thermopiles intégrées au mi-croréacteurs pour la détermination d’enthalpie de réaction et de cinétiques [10, 11]. Desméthodes de mesures de températures par thermographie ont aussi été explorées par Moll-mann et al. se limitant malheureusement à des résultats purement qualitatifs [12]. Dansce cadre, le récent développement de la technologie MEMS offre la possibilité d’explorerde nouvelles approches dans le domaine de la thermique pour la mesure d’enthalpie deréactions chimiques.
La mesure de température à cette échelle représente cependant la difficulté dominantealors qu’il s’agit du paramètre fondamental. À l’heure actuelle, seules quelques étudestraitent de ce problème. Une première méthode consiste à employer localement des micro-capteurs de température par la conception d’un microcalorimètre à puce microfluidiqueintégrée [10]. Une deuxième est basée sur l’utilisation de marqueurs fluorescents pour la

15
mesure de température à l’intérieur du fluide [13]. Une autre technique consiste à mesurersans contact le profil d’émission infrarouge d’une surface et mène à des analyses qualita-tives [12]. Les propriétés radiatives de surface étant peu connues, il est difficile d’obtenir unprofil de température absolue. Une étude récente montre qu’une interprétation quantitativeest possible par l’utilisation d’un modèle de transfert thermique simplifié [14].
L’objectif principal de ce travail exploratoire dans le domaine de la thermique en mi-crofluidique est de développer de nouvelles méthodes afin de déterminer l’enthalpie et lacinétique de réactions chimiques au sein de dispositifs microfluidiques. Ce travail permet-tra de fournir les premières mesures de champs de température aux microéchelles ainsi quel’estimation du flux de chaleur et de l’enthalpie de réaction. Afin d’obtenir ces donnéesexpérimentales, les recherches ont conduit :
– au développement de dispositifs et de méthodes de mesures permettant la caractéri-sation de réactions chimiques au sein de dispositifs microfluidiques,
– à l’élaboration de méthodes originales d’identification des propriétés thermodyna-miques couplées à une estimation des paramètres via des méthodes inverses,
– à la validation des dispositifs par la caractérisation d’une réaction modèle,– à la détermination de la précision d’un des dispositifs.
Une première méthode consiste à mesurer par contact le flux de chaleur global produitpar une réaction chimique au sein d’une puce microfluidique classique (microcalorimètreisotherme à flux continu). Une deuxième classe de méthodes consiste à mesurer le profilde température sans contact grâce à une caméra infrarouge. Dans un premier temps unepuce microfluidique est utilisée en tant que réacteur chimique. Les phénomènes de diffu-sion thermique autour du canal étant prépondérants, cela entraîne une modélisation pluscomplexe des effets dus au transfert de chaleur. C’est pourquoi, nous avons mis au pointune autre puce, composée d’un tube en PFA (Perfluoroalkoxy) et d’une plaque en alumi-nium. Contrairement aux puces microfluidiques classiques, l’utilisation d’un tube en PFAet d’une plaque en aluminium offre des conditions isopériboliques, où la température autourdu milieu réactionnel est constante, permettant ainsi de simplifier le modèle thermique enlimitant la diffusion.
Ce mémoire se compose de quatre parties distinctes :
Le premier chapitre est consacré à une revue bibliographique centrée sur les différentsoutils microfluidiques disponibles ainsi qu’au contexte scientifique qui a justifié les prin-cipaux objectifs de ces travaux. Tout d’abord, nous présentons la microfluidique et sesconfigurations d’écoulement : monophasique et diphasique. Ensuite, nous présentons lesdivers domaines d’application de ces microréacteurs. Puis, nous détaillons les outils baséssur une analyse thermique.
Le deuxième chapitre concerne la mesure globale d’enthalpie de réaction grâce à une mé-thode par contact à l’aide d’un fluxmètre de type Peltier. La première partie est consacrée

16
aux descriptions du dispositif et de la démarche expérimentale associée. Nous présentonsensuite la modélisation suivie de la conception du microréacteur. Puis, l’étape d’étalonnagedu dispositif est décrite et réalisée à différentes températures de consigne. La quatrièmepartie est dédiée à l’estimation de l’incertitude de mesure grâce à des expériences de répéta-bilité. Ensuite, est présentée une étape de validation de l’appareil selon deux configurationsd’écoulements : co-courant et gouttes. Enfin, la dernière partie recense les nombreuses pos-sibilités d’utilisation du microcalorimètre développé.
Le troisième chapitre traite de la mesure locale du flux de chaleur dégagé le long d’unmicrocanal lors d’une réaction chimique. Après une description de l’appareil de mesure, ladémarche expérimentale est présentée. Celle-ci concerne la mise au point de deux méthodesde calibration conduisant à l’estimation d’une cartographie du nombre de Péclet. Puis, sontdécrits le modèle thermique ainsi que la méthode d’identification des différents paramètrespar approche nodale. Ensuite, les méthodes de calibration sont validées lors de l’écoulementco-courant à l’aide de simulations aux volumes finis. À la suite de cela, une validationexpérimentale sur la mesure du nombre de Péclet puis sur le terme source dû à la réactionchimique est entreprise. Finalement, nous proposons une première approche par méthodenodale pour la caractérisation de l’écoulement en gouttes lors d’une réaction acide-base.
Le quatrième chapitre est consacré au développement d’un montage millifluidique iso-péribolique. Ce chapitre débute par une présentation du dispositif et de la démarche expé-rimentale associée et se poursuit par la mise au point d’un nouvelle méthode d’étalonnage.Enfin, l’appareil est validé par la caractérisation d’une réaction chimique modèle.

Chapitre 1
La thermique appliquée à la
microfluidique : éléments
bibliographiques
Résumé
Les microréacteurs fluidiques ont connu un essort important ces dernières années. Ilssont désormais principalement utilisés en tant qu’outil d’analyse pour l’étude phy-sique des écoulements, la mise en forme de matériaux ou encore la caractérisationde réactions chimiques. Si de nombreuses méthodes d’analyses ont émergé dans ledomaine de la mécanique des fluides et de l’analyse physicochimique, peu de travauxsont réalisés dans le domaine de la thermique. Ici, les quelques travaux couplant lathermique et la microfluidique seront analysés dans le but de justifier la nécessitéd’apporter, dans le cadre de cette thèse, de nouvelles méthodes expérimentales etthéoriques au domaine de la mesure de cinétiques chimique en microfluidique.
17

18 Chapitre 1. La thermique appliquée à la microfluidique : éléments bibliographiques
1.1 Introduction
L’objectif de ce chapitre est d’exposer les principales technologies utilisées dans le do-maine de la microfluidique. La première partie présente une méthode courante de fabri-cation des microréacteurs ainsi que les principales caractéristiques des écoulements. Puis,diverses applications de l’outil microfluidique sont présentées dans le domaine de la ca-ractérisation de réactions chimiques. Une étude bibliographique portant sur les méthodesthermiques existantes pour la caractérisation de réaction chimiques en microfluidique estréalisée. Il apparaît que ce domaine d’étude est encore peu exploré. Ainsi, nous démontronstout l’intérêt de développer de nouvelles méthodes d’analyses couplant le savoir faire duthermicien avec celui du chimiste.
1.2 L’outil microfluidique
Il existe de nombreuses techniques de fabrication des microréacteurs [15]. Dans le cas deréactions catalysées par un solide, les parois des microcanaux peuvent être réalisées par gra-vure directe dans la substance catalytique, ou, par souci d’économie, recouverts de cataly-seur actif [16]. Pour certaines applications utilisant des solvants agressifs, des matériaux trèsrésistants doivent être utilisés, par exemple : verre, silicium, PEEK (polyether-etherketone),PMMA (polyméthyl-méthacrylate), prépolymère thiolène [17], colle UV-sensible [18]. Parailleurs des puces en Kapton (polyimide) transparentes et résistantes aux rayons X ont étédéveloppées pour la détection de structures dans les microcanaux [19].
Les matériaux principalement utilisés demeurent le verre, le silicium et le polydymé-thylsiloxane (PDMS). Ces trois techniques nécessitent au préalable la reproduction de lagéométrie des microcanaux soit pour marquer le substrat à graver (technologie verre ousilicium), soit pour réaliser un moule (technologie PDMS). Cette étape est réalisée parphotolithographie. Au cours de cette thèse, toutes les puces microfluidiques sont conçuesà partir d’un moule fabriqué selon la technologie PDMS que nous détaillons ci-dessous.
1.2.1 Fabrication des microcanaux : photolithographie et techno-logie PDMS
La fabrication de puces en verre-PDMS ou silicium-PDMS s’opère en deux étapes :la réalisation du moule par photolithographie et celle de la matrice contenant les canauxpar la technologie PDMS. Les différentes étapes de microfabrication sont présentées enfigure 1.1 et détaillées par la suite.
La photolithographie douce permet de transférer les motifs d’un masque dans une résinedéposée sur un substrat de silicium. La hauteur des canaux est déterminée par la viscosité dela résine utilisée ainsi que la vitesse de rotation lors de l’étalage de la résine par spincoating.Les temps de cuisson et d’exposition dépendent de l’épaisseur de la couche de résine. Nousutilisons des résines SU8 (résine photosensible positive, société Microchem). Le substratde silicium est préalablement déshydraté à 200 ◦C pendant 30 minutes (figure 1.1 (a)). La

1.2. L’outil microfluidique 19
(a)
(b)
(c)
(d)
(e)
(f)
(g)
Fig. 1.1 – Représentation schématique des différentes étapes de la photolithographie et dumoulage de puces en PDMS : (a) déshydratation du substrat de silicium, (b) spincoatingde la résine, (c) exposition UV, (d) révélation, (e) moulage du PDMS, (f) découpage, (g)collage
résine est ensuite déposée sur ce substrat par spincoating (figure 1.1 (b)). Afin d’évaporerle solvant de la résine, une cuisson à 95 ◦C est effectuée. La résine est alors exposée à unrayonnement UV parfaitement rectiligne à travers un masque sur lequel est reproduit ennégatif la géométrie des canaux (figure 1.1 (c)). Le substrat est alors recuit à 95 ◦C pourdurcir la résine réticulée. Enfin, la résine non réticulée est dissoute via un bain révélateurde PGMEA (Propylene Glycol Methyl Ether Acetate) (figure 1.1 (d)).
Dés lors, les microcanaux peuvent être facilement réalisés par moulage dans le PDMSqui est un silicone (polyorganosyloxanes). Il s’agit de polymères comportant à la fois desliaisons Si-O et Si-C. La chaîne polysiloxane forme une colonne vertébrale extrêmementflexible, mobile et ouverte, supportant une substitution symétrique de groupements mé-thyles. Ces groupements forment un arrangement régulier apolaire qui confère au PDMSun caractère hydrophobe. Sa faible énergie de surface diminue les phénomènes d’adhésionsmoléculaires et cellulaires. Il offre un contact conforme avec le substrat, un démoulage facileet une déformation facilement contrôlable.
Un mélange de silicone et de durcisseur, en général en proportions respectives 10/1,est déposé sur le moule en silicium. Il est ensuite réticulé dans une étuve à 65 ◦C pen-dant 45 minutes (figure 1.1 (e)), puis découpé et démoulé (figure 1.1 (f)). La plaque dePDMS microstructurée est alors percée avec un emporte-pièce aux extrémités des canaux

20 Chapitre 1. La thermique appliquée à la microfluidique : éléments bibliographiques
afin de permettre l’insertion future des connectiques pour l’entrée et la sortie des fluides.Les canaux ainsi obtenus sont fermés par une plaque de verre ou de silicium (figure 1.1(g)). L’assemblage se fait par simple mise en contact après oxydation de surface dans unechambre plasma ou un ozoneur. Cette opération est réalisée au plasma ozone, pendant 20minutes pour le verre ou le silicium et 1 minute pour le PDMS. Une fois collé, le dispositifest placé plusieurs heures à 65 ◦C en étuve (figure 1.2).
Fig. 1.2 – Photographie d’une puce microfluidique composée d’un substrat de verre et dePDMS
La technologie PDMS est peu onéreuse et facile à mettre en œuvre. Le PDMS offrede multiples possibilités d’intégration de composants qui permettent de contrôler les écou-lements. Par exemple, Ismagilov et al. [20] utilisent une structure tridimensionnelle decanaux croisés en PDMS et dirigent l’écoulement des fluides par pressions externes desti-nées à changer les facteurs de forme des canaux. Kim et al. [21] proposent une micropompeen PDMS actionnée par effets thermopneumatiques et pouvant être insérée dans les mi-cropuces. Aussi, il est possible de recouvrir la lame de verre servant à fermer les canaux demotifs de l’alliage indium/tin/oxyde (ITO). Connectés à un générateur de tension, ceux-ciservent d’électrodes et permettent de générer un champ électrique au sein des fluides quicirculent dans les canaux avec lesquels ils sont en contact [22].
Deux configurations simples de microréacteurs sont désignées dans la suite par : micro-réacteur monophasique et microréacteur diphasique.
1.2.2 Microréacteurs monophasiques
1.2.2.1 Écoulement laminaire
L’écoulement d’un fluide dans une conduite est classiquement caractérisé par le nombrede Reynolds qui compare les effets inertiels et visqueux :

1.2. L’outil microfluidique 21
Re =ρvdh
µ(1.1)
v est la vitesse moyenne de l’écoulement (m.s−1), µ la viscosité du fluide (Pa.s) et ρ samasse volumique (kg.m−3), dh est le diamètre hydraulique de la canalisation défini à partirde sa section S et de son périmètre mouillé Π : dh = (4S)
Π. Un microcanal est communément
un canal dont le diamètre hydraulique est inférieur au millimètre. À ces échelles, le nombrede Reynolds est généralement largement inférieur à 1. L’hydrodynamique est donc dominéepar les effets visqueux. Le fluide est en écoulement laminaire. Dans les conduites rectilignes(de section circulaire ou rectangulaire), les vitesses sont partout parallèles aux parois etl’écoulement s’organise selon un profil de vitesse de type «Poiseuille» avec une valeur nulleaux parois et maximale au centre. Dans le cas d’une section rectangulaire à fort rapportd’aspect (de type fente : largeur ≫ hauteur), la distribution des vitesses dans un microcanalest illustrée sur la figure 1.3 et donnée par :
v(y) = vmax
(1 −
(yw2
)2)
(1.2)
y correspond à la position de la particule de fluide dans la largeur w du microcanal. Lesparois du microcanal sont situées en y = ± w/2. L’écoulement est invariant suivant lahauteur du microcanal. vmax est la vitesse maximale au centre du tube proportionnelle augradient de pression P :
vmax =w2
8µ
dP
dx(1.3)
vmax
y = w/2
y = 0
y = -w/2
x
y
vmax
y = w/2
y = 0
y = -w/2
x
y
Fig. 1.3 – Profil des vitesses dans un écoulement laminaire
1.2.2.2 Interdiffusion de deux réactifs
Parmi les possibilités d’utilisation de microcanaux en tant que réacteurs, des étudesont été présentées dans des systèmes dits en « coflow » où les deux réactifs sont injectés defaçon continue dans un même canal selon un écoulement co-courant (figure 1.4). Commel’écoulement est laminaire, il n’y a pas de convection radiale pour disperser les fluides sur lalargeur du canal. Le mélange des espèces en présence s’effectue par diffusion. La réaction a

22 Chapitre 1. La thermique appliquée à la microfluidique : éléments bibliographiques
réactif A
réactif B
100 µm
x
y
réactif A
réactif B
100 µm
x
y
>
>w
Fig. 1.4 – Écoulement co-courant réactif, le produit formé à l’interface est incolore, w étantla largeur du canal [23]
donc lieu à l’interface des deux courants, dans un cône diffusif de largeur croissante. La zoned’interdiffusion a été observée et modélisée [24, 20]. Le dispositif peut permettre d’estimerrapidement les coefficients de diffusion entre deux fluides. Considérons par exemple le casoù aucune réaction n’intervient entre les espèces, ou le cas d’une réaction extrêmementrapide devant la diffusion. En se plaçant dans un microcanal de grand facteur de forme(largeur ≫ hauteur), dans des conditions telles que l’écoulement est supposé uniformeselon x, que la dispersion axiale est négligeable et que la convection selon x domine lephénomène de diffusion selon x, le profil de concentration c(x, y) d’un soluté qui diffusedans le microcanal est solution de l’équation de diffusion :
v∂xC(x, y) = D∂2yC(x, y) (1.4)
v étant la vitesse moyenne de l’écoulement, D le coefficient de diffusion de l’espèceconsidérée. Lorsque la concentration initiale C0 du soluté est répartie telle que : C(x =0, y) = C0δ(y), le profil de concentration est donné par :
C(y) =C0√
4πDTexp
(− y2
4Dt
)(1.5)
où t = xv.
Cette équation correspond à une gaussienne dont la variance σ dépend du temps :σ(t) =
√2Dt (figure 1.5). σ est proche de la demi-largeur à mi-hauteur et correspond à
la demi-largeur ydiff du cône de diffusion. Avec cette convention, il est possible d’écrire :
(ydiff (x))2 = 2Dt = 2Dx
v(1.6)
L est la longueur de canal parcourue par les fluides en écoulement co-courant à la vitessemoyenne v pendant le temps t. Le temps de mélange total est alors défini par la relationsuivante :
t =w2
8D(1.7)
Avec ydiff = w2

1.2. L’outil microfluidique 23
Considérons maintenant le cas où des réactifs A et B sont injectés au même débit enécoulement co-courant dans un microcanal en forme de Y ou de T (figure 1.4 (a)). S’ilspossèdent les mêmes densités et viscosités et que leur coefficient d’interdiffusion D nedépend pas des concentrations, la concentration normalisée d’une espèce, par exemple A,varie dans la largeur y et la longueur x du canal comme :
CA(x, y)
CA0
=1
2
(1 + erf
(y
2√
D xv
))(1.8)
y (µm)y (µm)
CA(y)/CA0
-w/2 w/2 -w/2 w/2
CB(y)/CB0
y (µm)y (µm) y (µm)y (µm)
CA(y)/CA0
-w/2 w/2 -w/2 w/2
CB(y)/CB0
(a)
y (µm)
0-w/2 w/2
σ²(t) = 2DtC(Y
,t)/
C0
(b)
Fig. 1.5 – Profils normalisés des concentrations dans la largeur du microcanal : (a) profilà l’instant t d’un produit fabriqué dans le cône de diffusion (équation 1-6) ; (b) profils dedeux espèces diffusives dans un canal de largeur 500 µm à deux instants différents [25]
La fonction erf étant l’erreur définie par : erf(z) =∫ z
0exp(−Ψ2)dΨ où Ψ est une va-
riable. Cette solution est obtenue dans les mêmes conditions que celle de l’équation (1.4)(microcanal de grand facteur de forme et dispersion axiale négligeable). Elle fournit l’évo-lution des concentrations illustrée sur la figure 1.4 (b). Cette équation peut être modifiéedans le cas de fluides de viscosités différentes [26].
En conclusion, l’utilisation d’un écoulement co-courant de deux espèces est donc utilepour déterminer des coefficients de diffusion (équation (1.7)). Dans le cas d’utilisation d’es-pèces réactionnelles, cette configuration permet également l’étude de la cinétique initialeen repérant le premier point d’apparition du produit de la réaction à l’interface. Mais celanécessite une technique d’analyse en ligne de très haute résolution spatiale. Par ailleurs, lesécoulements co-courant ont été utilisés pour identifier des cinétiques très rapides de l’ordrede quelques millisecondes [27, 28]. Dans ce type de système, les phénomènes sont limitéspar la diffusion et une complexité inhérente peut survenir lors du couplage entre diffusionet réaction chimique.

24 Chapitre 1. La thermique appliquée à la microfluidique : éléments bibliographiques
1.2.3 Microréacteurs diphasiques
Les écoulements diphasiques en général permettent un meilleur contrôle de l’exothermiedes réactions et une intensification des transferts aux interfaces entre fluides. Par exemple,Dumman et al. [29] ont effectué la nitration très exothermique d’un composé aromatiquesimple dans un écoulement gaz/liquide et ont pu améliorer le taux de conversion de laréaction ainsi que sa sélectivité grâce à la maîtrise de la surface d’échange.
La première application pour le contrôle et le suivi de réaction chimique est présentéepar Song et al. [30]. Les avantages à réaliser la réaction chimique dans des gouttes sontmultiples car les produits sont isolés, transportés et mélangés efficacement par les effetsdiffusifs et convectifs qui ont lieu au sein de la goutte. Il est possible d’étudier précisémentla cinétique des réactions grâce à l’équivalence espace temps. En effet, mesurer le temps deréaction revient à mesurer la position de la goutte dont la vitesse est constante et connue.En fait, chaque goutte se comporte comme un réacteur batch transporté à vitesse constantevg par le courant porteur. Lorsque la position des gouttes correspondant à l’instant initial dela réaction est prise comme origine de l’axe des abscisses, la longueur x du canal parcouruesuivant cet axe est proportionnelle au temps de réaction : t = x/vg.
1.2.3.1 Formation d’un train de gouttes
L’écoulement de fluides immiscibles dans les microcanaux est contrôlé par les effets demouillage [31]. Lorsque deux fluides immiscibles sont injectés en écoulement co-courantdans une jonction en Y, deux régimes peuvent être obtenus en fonction des débits de cha-cune des phases : gouttes monodisperses ou écoulement co-courant. La transition est dueaux différences de mouillage et au confinement des fluides. L’écoulement parallèle est fa-vorisé dans le cas de canaux à section rectangulaire (de hauteur inférieure à la largeur)et de phase continue très visqueuse [32]. Dans les canaux en PDMS qui est un matériauhydrophobe, des gouttes aqueuses sont naturellement arrachées à l’intersection d’un cou-rant d’huile [33]. Deux géométries principales de l’intersection permettent de produire unarrachage contrôlé : jonction en T et jonction en croix. Nous détaillons uniquement la gé-nération de trains de gouttes par une jonction en croix (figure 1.6), technique utilisée aucours de cette thèse.
Dans une intersection en croix, deux courants de phase continue organique viennentpincer celui de la phase aqueuse et les gouttes se déplacent parallèle à l’arrivée de la phaseaqueuse (figure 1.6). Cette technique est aussi appelée «flow-focussing». Une restrictionplacée à l’endroit où se forment les gouttes peut permettre de générer une grande variétéde gouttes monodisperses ou polydisperses en modifiant les débits des phases [34].
Pour une géométrie de canal, c’est le rapport des débits qui fixe la taille des goutteset l’intervalle inter-gouttes, tandis que le débit total fixe la vitesse d’avancée. La longueurdes gouttes Lg et celle de la période du train de gouttes Lpriode peut être reliée au rapportdes débits par un coefficient A dépendant de la géométrie de l’intersection [7] :
Lg
Lpriode − Lg
= AQd
Qpc
(1.9)

1.2. L’outil microfluidique 25
phase continue
100 µm
réactif B
réactif A
phase continue
phase continue
100 µm
Fig. 1.6 – Génération de gouttes multi-constituants par «flow-focussing » [23]
De manière générale, la vitesse des gouttes est proportionnelle au débit total. Si le rap-port des débits est conservé tout en modifiant le débit total, le train de goutte conserve sespropriétés géométriques Lg et Lpriode, mais avec une vitesse différente. Par cette méthode,il est possible de changer le temps de passage des volumes réactionnels dans un canal degéométrie fixée sans changer ni le volume réactionnel ni les aires d’échange [7]. À débits im-posés, la vitesse des gouttes peut ensuite être modifiée en soutirant par exemple une partiede la phase porteuse [35]. Cela permet de modifier les temps de séjour, ou de concentrerles gouttes pour des soucis d’analyse.
1.2.3.2 Mélange au sein des gouttes
Les gouttes véhiculées au sein de microcanaux subissent le phénomène de convectionforcée qui engendre des boucles internes de recirculation. L’hydrodynamique des trainsde gouttes dans les microcanaux a été caractérisée en 3D par des expériences de micro-PIV (Particle Image Velocimetry) et par des simulations numériques [36]. Le phénomènede recirculation est mis en évidence lorsque les champs de vitesse sont tracés dans leréférentiel de la goutte. Il apparaît que les formes des lignes de courant dépendent desconditions opératoires, et notamment du nombre capillaire Ca = µv/σ (figure 1.7). Ainsi,lorsque Ca est faible, la goutte est peu déformée, l’épaisseur de film de phase continue esttrès faible et la recirculation envahit tout le demi-lobe de la goutte. Lorsque Ca augmente,la goutte est plus déformée par le profil de vitesse et des zones stagnantes apparaissentnotamment dans le nez et à l’arrière de la goutte [8]. Par ailleurs, l’efficacité des bouclesde recirculation sur le mélange (caractérisée ici par le rapport entre le temps mis pourqu’une de recirculation effectue un tour et celui mis pour que la goutte parcoure une foissa longueur) diminue lorsque Ca augmente.
Aussi, le mélange dans les gouttes résulte d’un couplage convection-diffusion avantageuxpar rapport à l’écoulement co-courant. Le tableau 1.2 récapitule les ordres de grandeurs

26 Chapitre 1. La thermique appliquée à la microfluidique : éléments bibliographiques
µm300
Fig. 1.7 – Lignes de courant dans les gouttes et la phase continue obtenues par simulationdirecte 2D [23]. Les gouttes ont le même volume mais des nombres capillaires différents :(haut) Ca = 0,157 ; (bas) Ca = 0,022.
obtenus en fonction de l’échelle du microréacteur. Les temps de mélange en gouttes sontissus de la thèse de F. Sarrazin [23] et ceux donnés pour l’écoulement co-courant sontcalculés par la formule (1.7). Globalement la configuration goutte diminue le temps demélange d’un facteur N dépendant de la dimension du canal.
Échelle du microréacteur (µm) 50 500 1000Temps de mélange en co-courant (s) 0,325 31 125
Temps de mélange en gouttes (s) 0,060 1,5 10Rapport de temps 5,4 20,7 12,5
Tab. 1.1 – Comparaison des temps de mélange en écoulement co-courant et en écoulementgouttes
Il est possible d’accélérer encore plus le mélange dans les gouttes en jouant sur l’orien-tation des réactifs au sein de la goutte ou sur la géométrie du canal. Compte tenu de laforme des boucles de recirculation, il apparaît clairement qu’une configuration initiale desréactifs telle qu’ils occupent chacun la moitié avant ou arrière de la goutte favoriseraitle mélange. Le mélange obtenu dans cette configuration a été modélisé par Handique etBurns [37] dans un canal de grand facteur de forme (largeur ≪ hauteur). Expérimentale-ment, Sarrazin et al. [38] utilisent un microcoalesceur pour forcer l’orientation des fluidesdans la direction souhaitée. Par ailleurs, le temps de mélange dans les microgouttes peutêtre réduit en générant de l’advection chaotique. Celle-ci peut être obtenue lorsque lesgouttes circulent dans des canaux coudés. À chaque rotation au passage d’un angle, lesgouttes sont déformées et les fluides internes réorientés si bien que des couches de fluidesde plus en plus fines se retrouvent empilées au sein de la goutte (figure 1.8). Song et al. [39]montrent que le phénomène suit la transformation dite « du boulanger » qui permet deprédire le temps de mélange interne aux microgouttes. Cette loi peut être approchée expéri-

1.3. Champs d’applications de la microfluidique 27
mentalement lorsque des angles coudés à 45◦ sont dessinés le long du canal [38]. Muradogluet Stone [40] utilisent des simulations en canaux coudés pour faire varier les paramètresopératoires. Comme en canal droit, l’efficacité du mélange augmente clairement lorsque lenombre capillaire diminue et la déformabilité de la goutte semble avoir un gros impact surles recirculations internes ; le mélange est meilleur lorsque le diamètre de la goutte approchede la taille du canal ou lorsque le rapport de viscosité entre la phase dispersée et la phasecontinue diminue.
Fig. 1.8 – Advection chaotique générée dans des gouttes transportées dans un canalcoudé [23]
1.3 Champs d’applications de la microfluidique
Les progrès techniques en microfabrication et en microcapteurs ont permis d’étudier lesécoulements aux micro-échelles.
La physique est maintenant mieux connue et décrite par de nombreux articles. Stone etal. [41] fournissent une revue des écoulements en microcanaux, en étudiant particulièrementles effets électrocinétiques, le mélange, la dispersion et les écoulements diphasiques [42, 43].Par ailleurs, Squires et Quake [44] étudient les comportements des fluides à la micro-échelleen s’appuyant principalement sur les nombres adimensionnels. L’écoulement confiné defluides complexes a permis d’établir des lois de comportement rhéologique adaptées auxmicrocanaux [45].
Dans une revue des outils disponibles pour les études en biologie, médecine ou chimie,Atencia et Beebe [46] se concentrent sur le contrôle des fluides et des interfaces en mi-crofluidique où les effets diffusifs ainsi que les forces visqueuses et capillaires sont souventprépondérants.
Enfin, l’estimation de cinétique de réactions chimiques est réalisée au sein de dispo-sitifs microfluidiques par des méthodes spectroscopique. DeMello [47] propose une revue

28 Chapitre 1. La thermique appliquée à la microfluidique : éléments bibliographiques
des géométries et écoulements caractéristiques qui peuvent être utilisés pour le contrôle etla détection de réactions. Au cours de plusieurs études, le profil de concentration dans lemicrocanal est mesuré par spectroscopie Raman [8, 7]. Engl et al. utilisent la spectroscopieUV-visible pour déterminer la cinétique d’une réaction [9]. La conception de puce en Kap-ton permet la mesure de concentration par détection rayon X [19]. Une autre applicationproposé par Laval et al. est de suivre la cinétique de cristalisation au sein de gouttes parmicroscopie et d’en déduire le diagramme de phase [48].
Toutes ces techniques de caractérisation sont basées sur l’estimation de profil de concen-tration par des méthodes optiques externes pour l’estimation de cinétique.
Un autre domaine est la caractérisation thermique de réactions au sein ces mi-crosystèmes. Différentes techniques sont étudiées et mènent au développement de micro-calorimètres, certaines mesurent la température du milieu ou de l’enveloppe thermostatée,d’autres s’intéressent au flux de chaleur.
1.4 De la calorimétrie classique à la microcalorimétrie
La calorimétrie est une technique largement utilisée pour déterminer les propriétés ther-modynamiques de composés chimiques ou de matériaux ainsi que pour la caractérisationde réaction chimique [1]. De nombreux calorimètres ont été développés et peuvent êtreclassifiés selon quatre types de conditions thermiques :
– isotherme : consiste à maintenir le milieu réactionel à une température constante,ainsi l’influence de la température sur le taux de conversion peut être mise en évi-dence,
– adiabatique : signifie que les échanges entre le système et le milieu extérieur sont nulsce qui permet de suivre la température au cours du temps,
– isopéribolique : ce mode implique que la température de l’environnement extérieurest constante et la température du milieu réactionnel varie,
– dynamique : consiste à appliquer une variation de température contrôlée au systèmeet permet d’obtenir une vision globale de l’activité thermique de l’échantillon.
Les calorimètres ayant de nombreuses caractéristiques, plusieurs classifications ont étéréalisées. Notamment Regenass propose de classer les calorimètres selon deux catégories :les calorimètres à accumulation de chaleur et les calorimètres à flux de chaleur [2]. Uneautre méthode de classification proposée par Zogg, divise les calorimètre en fonction duprincipe de mesure du flux et du type de contrôle thermique entre le système et le milieuenvironnant [1]. Cette classification mène à quatre catégories :
– calorimètre à flux de chaleur où la température du réacteur est contrôlé par le fluidecaloporteur. Le flux échangé est déterminé par la différence de température entre cefluide et le réacteur.
– calorimètre à bilan de chaleur, où là encore la température du fluide calorporteur im-

1.4. De la calorimétrie classique à la microcalorimétrie 29
pose la température au réacteur, par contre le flux échangé est estimé par la différencede température du fluide à l’entrée et à la sortie de l’enveloppe.
– calorimètre par compensation de puissance où le température du réacteur est imposépar la variation de puissance d’un chauffage compensatif inseré directement dansle réacteur. Le flux échangé est directement visible par la puissance consommée duchauffage.
– calorimètre à Peltier, qui permet d’imposer la température en variant la puissanceaux bornes des éléments Peltier qui offre la possibilité de chauffer ou refroidir. Cettepuissance sert à évaluer le flux échangée.
La mise en oeuvre de réactions chimiques très exothermiques dans un calorimètre peuts’avérer difficile à maîtriser et conduire à un phénomène d’emballement thermique présen-tant un réel danger pour l’homme et l’environnement. La manipulation d’une faible quantitéde matière apparait comme une solution idéale à tous points de vue, tant au niveau éco-nomique, environnemental que de la sécurité. Dans ce cadre, différents microcalorimètresexistent déjà, par exemple le calorimètre Calvet qui est un des premier calorimètre à utiliserdes volumes relativement faibles.
Le principe de fonctionnement de ce calorimètre est d’imposer une température uni-forme au sein de l’échantillon ou une rampe de température relativement lente due àl’inertie du système. La société Setaram commercialise le calorimètre Calvet C80 présentésur la figure 1.9.
Chambre référence Chambre réaction
> >
Enceinte d'isolation >
Elément chauffant >
Thermocouples >
(b)(a)
Fig. 1.9 – Schéma et photographie du calorimètre Calvet C80 de Setaram [49]
Il est composé de deux cellules identiques placées au sein d’une masse thermostatée :une cellule de référence et une cellule réaction. Un assemblage de thermocouples placésautour des cellules permet la mesure précise du flux thermique échangé entre la celluleet la masse. Le volume des cellules est de 10 cm3 et différents types de cellules existentselon leur utilisation par exemple pour l’étude de réaction liquide/liquide ou de réaction

30 Chapitre 1. La thermique appliquée à la microfluidique : éléments bibliographiques
gaz/liquide. Une cellule spécifique permet l’introduction en continu dans un réacteur semi-fermé mais la configuration d’écoulements gouttes n’est pas possible.
Cet appareil offre une sensibilité de 30 mV/W, une gamme de température variant del’ambiante à 300 ◦C avec une précision de 0,01 ◦C et la possibilité d’appliquer des pressionsjusqu’à 100 bars. Cependant le volume réactionnel reste important, de l’ordre de 10 mL,l’étude en écoulement continu est impossible dans la configuration gouttes et la mesure duflux est une information globale. Afin de palier à ces inconvénients, diverses investigationsont méné au développement de microcalorimètres.
1.4.1 Microcalorimètre différentiel
Monk et Wasdö ont mis au point un microcalorimètre continu différentiel [50]. Il estcomposé de deux cellules (figure 1.10) placés au sein d’une enceinte immergée dans un baind’eau thermostatés. Une celllule est utilisée en tant que référence et l’autre pour effectuer laréaction chimique. Le microréacteur est composé de deux plaques en argent collées par unerésine époxy. Les thermopiles sont disposées de part et d’autre du microréacteur, et assurentune mesure quasi total et global du flux de chaleur. À l’état stationnaire, les expériencesd’étalonnages indiquent une précision de 0,1 % pour une flux supérieur à 100 µcal.s−1.
EnregistrementAmplificateur
EntréeSortie
Unités calorimétriques Echangeur de chaleur
Bloc de métal
Mousse polystyrène
Cylindre en acier
inoxydable
Bain d'eau
thermostaté
Fig. 1.10 – Schéma du calorimètre développé par Monk et Wasdö
Wasdö et Markova développe un double microcalorimètre différentiel [51]. Il s’agit de

1.4. De la calorimétrie classique à la microcalorimétrie 31
deux microcalorimètres différentiels placés dans une même enceinte dont les dimensionssont semblables à un simple. L’avantage de ce dispositif est de pouvoir réaliser deux mesuresen même temps dans la même enceinte.
1.4.2 Microcalorimètres intégrés
Une méthode largement utilisée dans le développement de calorimétres microfluidiquesest l’intégration de thermopiles au sein même des microsystèmes par dépôt de couche mincepermettant la conception de résistances dépendantes en température. Un microcalorimètrecontinu a été conçu par Köhler et Zieren. [10]. Ce dispositif est composé d’un substratde silicium contenant trois thermopiles recouverte d’une membrane et d’un substrat deverre dans lequel les microcanaux sont gravés 1.11. La température peut être mesurée sur
Verre Silicium
Canal
Résistance chauffante
Thermopiles
Verre
Canal
Silicium
Membrane
Fig. 1.11 – Schéma de la puce microfluidique intégrée développée par Kölher et Zieren [10]
trois zones distinctes correspondants au mélange, à la réaction et à la fin de la réaction.Ce dispositif a permis de déterminer l’enthalpie de réaction de la neutralisation entrel’hydroxide de sodium et l’acide sulfurique.
Différentes puces microfluidiques intégrées adaptées à la caractérisation de procédésbiochimiques tels que les réactions enzymatiques ou la croissance de cellules ont été déve-loppées [52, 53, 54, 55, 56]. Lerchner et al. [52, 53] ont mis au point un dispositif où lesmicrocanaux sont moulés dans du PMMA et comporte quatre thermopiles déposées surune membrane. Le montage offre la possibilité de thermaliser les réactifs avant l’entréedans le microréacteur et de stabiliser la température du dispositif avec une précision de100 µK. Cet appareil permet l’étude de réactions enzymatiques.

32 Chapitre 1. La thermique appliquée à la microfluidique : éléments bibliographiques
(a) (b)
Fig. 1.12 – (a) Photographie de microcalorimètre avec les quatre thermopiles indépen-dantes ; (b) photographie du microcalorimètre avec microcanal et la masse de cuivre [52]
Une autre approche consiste à combiner ce type de microsystèmes intégrés avec uncalorimètre commercial, ce qui a permis la détermination de cinétiques rapides de réactionsexothermiques [57, 11] dans des conditions isothermes.
La conception de ce type de dispositif intégré n’est pas aisée, elle comporte plusieursétapes de dépôt de métal par couches minces pour la réalisation de thermopiles. De plus,la puce étant souvent constituée de silicium et de verre, les transferts thermiques ne sontpas optimisés.
1.4.3 Application des marqueurs fluorescents à la microfluidique
La mesure locale de température à l’intérieur d’un canal microfludique, consiste à injec-ter un marqueur dont l’intensité de fluorescence varie avec la température. Des rechercheseffectuées par Ross et al. sur la rhodamine B ont montré une résolution en températurede l’ordre du degré Kelvin [13] et une résolution spatiale de l’ordre du micron [58, 59].Toutefois, l’addition de rhodamine ou d’autres marqueurs peut être considérée comme uneméthode trop intrusive pour beaucoup d’applications réactives.
1.4.4 Application des caméras Infrarouge à la microfluidique
Une caméra IR est un outil non intrusif permettant la mesure locale de température. Sonprincipe de fonctionnement est de détecter les rayonnements IR dont la longueur d’ondedépend de la température du corps. La relation entre le flux du rayonnement émis et latempérature est définie par la relation suivante :
Φr = εSσB(T 4 − T 4a ) (1.10)
Φr le flux de rayonnement émis par un corps (W), σB étant la constante de Stefan-Boltzmann (5.6703.10−8 W.m−2.K−4), ε l’emissivité, Se la surface d’échange du corps (m2)
1.4. De la calorimétrie classique à la microcalorimétrie 33
et T la température du corps (K), Ta la température ambiante (K). Ainsi, une mesurequantitative de la température par caméra IR est envisageable si les propriétés du corpssont connues.
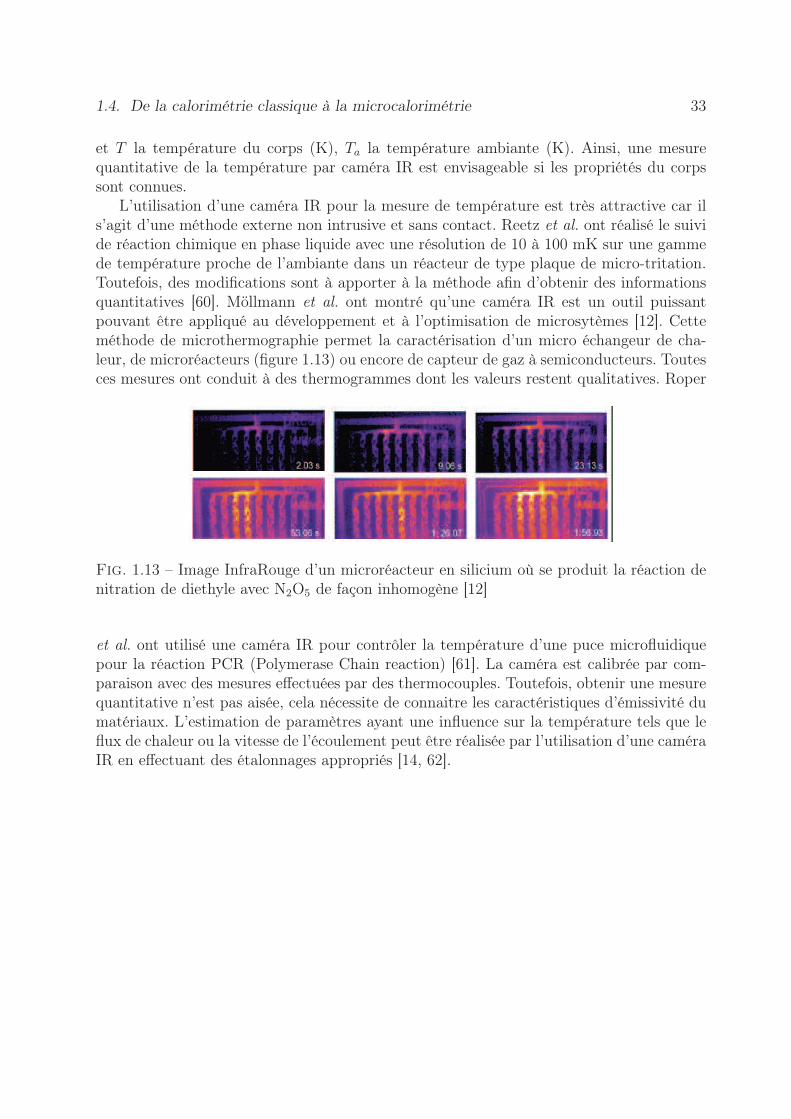
L’utilisation d’une caméra IR pour la mesure de température est très attractive car ils’agit d’une méthode externe non intrusive et sans contact. Reetz et al. ont réalisé le suivide réaction chimique en phase liquide avec une résolution de 10 à 100 mK sur une gammede température proche de l’ambiante dans un réacteur de type plaque de micro-tritation.Toutefois, des modifications sont à apporter à la méthode afin d’obtenir des informationsquantitatives [60]. Möllmann et al. ont montré qu’une caméra IR est un outil puissantpouvant être appliqué au développement et à l’optimisation de microsytèmes [12]. Cetteméthode de microthermographie permet la caractérisation d’un micro échangeur de cha-leur, de microréacteurs (figure 1.13) ou encore de capteur de gaz à semiconducteurs. Toutesces mesures ont conduit à des thermogrammes dont les valeurs restent qualitatives. Roper
Fig. 1.13 – Image InfraRouge d’un microréacteur en silicium où se produit la réaction denitration de diethyle avec N2O5 de façon inhomogène [12]
et al. ont utilisé une caméra IR pour contrôler la température d’une puce microfluidiquepour la réaction PCR (Polymerase Chain reaction) [61]. La caméra est calibrée par com-paraison avec des mesures effectuées par des thermocouples. Toutefois, obtenir une mesurequantitative n’est pas aisée, cela nécessite de connaitre les caractéristiques d’émissivité dumatériaux. L’estimation de paramètres ayant une influence sur la température tels que leflux de chaleur ou la vitesse de l’écoulement peut être réalisée par l’utilisation d’une caméraIR en effectuant des étalonnages appropriés [14, 62].

34 Chapitre 1. La thermique appliquée à la microfluidique : éléments bibliographiques
1.5 Conclusion et justification du travail de thèse
Depuis une vingtaine d’années, la microfluidique est un axe de recherche très prometteurpour le développement d’outils d’analyse sur puce. Dans ce chapitre, il apparaît clairementque les microréacteurs semblent bien adaptés à l’acquisition rapide de données cinétiquede base. En effet, le grand rapport surface sur volume favorise les transferts de matièreet de chaleur. De plus, la petite dimension du dispositif permet un temps de réponserelativement court. Les faibles quantités de produits mises en jeu sont pertinentes tantau niveau économique, environnemental ou de la sécurité dans le cas de réactions trèsexothermiques.
Si de nombreux dipositifs basés sur la spectrocopie Raman et UV-visible existent, peude systèmes s’intéressent à la mesure de température. En effet, dans les méthodescourantes, c’est essentiellement la concentration qui est obtenue.
Nous avons présenté plusieurs calorimètres microfluidiques utilisant différentes mé-thodes de mesures de températures ou de flux de chaleur. La conception de tels dispositifsnécessite la prise en compte de paramètres clés. Par exemple, le choix d’un capteur adaptéà la mesure de faible température ou de faible flux de chaleur au sein de microréacteursdont les volumes sont de l’ordre du µL. D’autre part, le transfert de chaleur doit être par-faitement maîtrisé afin de pouvoir estimer précisément les pertes et d’en déduire le flux dechaleur réel. Un autre critère important est la mise en place d’une étape d’étalonnage afind’obtenir une information quantitative.
La comparaison des performances des microcalorimètres existants montre que les sy-tèmes de puces microfluidique intégrées offrent une grande sensibilité mais nécessite unefabrication complexe.
Calorimètre Calvet Circuits intégrés Caméra IRVolume 10 mL nL µL
Sensibilité 1 µW 200 nW 1 mW
Tab. 1.2 – Comparaison des performences de différentes méthodes calorimétriques
L’analyse de ces différentes techniques existantes en calorimètrie de réactions chimiqueset en microfluidique fait ressortir deux points essentiels :
– Malgré le côté prometteur du domaine des microréacteurs, la mesure de températureou de flux par contact reste difficile et nécessite des montages rendus complexes parla contraintes des microéchelles et les perturbations associées au transfert de chaleur.
– Les techniques de cartographie de températures de surface par thermographie IRprésentent l’avantage de fournir sans contact une grande quantité de données. Cepen-dant, il reste à mettre au point des méthodes de calibration, de traitement thermiqueet à réfléchir à la conception des systèmes en vue d’une mesure thermographiquepertinente.

1.5. Conclusion et justification du travail de thèse 35
Une des voies d’exploration, proposée par ce travail de thèse, est de dévelop-per d’une part, un capteur solide par contact, simple et robuste pouvant servirde référence à une mesure thermographique et d’autre part, des méthodes lo-cales d’estimation de flux de chaleur dégagés au sein des microcanaux par lamesure de champ de température par thermographie IR.
Ainsi, dans ce mémoire, les différents dispositifs développés seront présentés selon ledéroulement suivant. Dans le chapitre 2 la conception et à la validation d’un microcalori-mètre global par contact sera décrit. Puis, nous verrons diverses applications offertes parce système, par exemple l’estimation d’enthapie de mélange ou des mesures de dosage.Le chapitre 3 et 4 sont dédiés au développement d’une mesure locale de température parthermograhie IR. Dans un premier temps, une approche microfluidique est entreprise, celanécessite la conception d’un microréacteur adapté, la mise en place d’une démarche d’éta-lonnage et d’estimation du flux. L’écoulement co-courant ainsi que l’écoulement gouttessont investigués. Le principale inconvénient est la démarche relativement complexe pourobtenir des valeurs quantitatives. Une approche millifluidique isopéribolique apparaissantplus adaptée et flexible, un nouveau dispositif est donc développé.

36 Chapitre 1. La thermique appliquée à la microfluidique : éléments bibliographiques

Chapitre 2
Développement d’un microcalorimètre
différentiel par mesure macroscopique
Résumé
Ce chapitre est consacré à la présentation d’un microcalorimètre différentiel pour lacaractérisation de réactions chimiques. Le principe original de cet appareil est l’uti-lisation d’un fluxmètre, placé sous le réacteur microfluidique, qui permet la mesuretransitoire globale du flux total dégagé par le microréacteur. Le microréacteur est com-posé d’un substrat en silicium (bon conduteur thermique) et de PDMS (bon isolantthermique). Un modèle thermique associé à ce microréacteur permet de déterminerson comportement thermique et de conclure que la réaction est effectuée dans desconditions isothermes. De plus, ce microréacteur permet d’étudier des écoulements detype : co-courant ou gouttes. Du fait de la petite dimension des microcanaux, le vo-lume réactionnel est relativement faible (quelques µL). De plus, l’important rapportsurface/volume favorise les échanges thermiques. L’écoulement au sein de tels dispo-sitifs est contrôlé par l’injection de fluides à débits volumiques imposé. Un simpleétalonnage par effet Joule permet de convertir le signal mesuré en flux. La validité del’appareil est évaluée par la détermination de l’enthalpie de la réaction entre l’acidechlorhydrique et l’hydroxyde de sodium. Le champ d’application de ce mircocalori-mètre est vaste : estimation d’enthalpie de réaction et de mélange (réaction d’estéri-fication et mélange eau-éthanol), dosage calorimétrique, cinétique de réaction et demélange.
37

38 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
2.1 Introduction
Une des méthodes les plus utilisées en chimie pour la mesure d’enthalpie est la calori-métrie. Si de nombreux dispositifs existent (DSC, ATG ...), ils ne permettent pas simulta-nément la maîtrise du mélange des réactifs, le bon contrôle des conditions thermiques etl’utilisation en continu de faibles quantités de produits. En effet, la calorimétrie classiqueutilise des volumes relativement importants, entraînant un flux de chaleur et une variationde température non négligeables pouvant être un réel danger du point de vue de la sécurité.D’autre part, les mesures sont effectuées en transitoire, les fluides n’étant pas sous écoule-ment. C’est pourquoi des microcalorimètres performants permettant l’utilisation de faiblesquantités de réactifs ont été développés. Toutefois, la mise en œuvre de tels dispositifs estrelativement complexe et parfois très coûteuse.
Le dispositif que nous présentons au cours de ce chapitre a été développé dans lebut de caractériser des réactions chimiques très exothermiques ou à cinétique rapide àmoindre coût. Comme nous l’avons vu dans le chapitre 1.2, la microfluidique consiste àconfiner les écoulements permettant, entre autre, l’utilisation de très faibles quantités deproduits (volume réactionnel de l’ordre de quelques µL), la maîtrise des écoulements etdu transfert thermique. Cet appareil consiste à utiliser une puce microfluidique commeréacteur chimique. Le flux de chaleur engendré par la réaction est ensuite mesuré grâce àdes éléments Peltier. Le coût de revient d’un tel appareil s’élève à quelques milliers d’euros.
Aussi, des réactions chimiques, très exothermiques voire explosives, peuvent être étu-diées à l’aide de ce calorimètre microfluidique. De par les faibles volumes mis en jeu, lesrisques chimiques liés à la dangerosité des réactions sont limités.
Ce chapitre s’articule en quatre parties. La première concerne la description du disposi-tif. La deuxième est consacrée à la modélisation thermique du microréacteur afin d’en opti-miser sa conception. Puis, nous présentons l’étalonnage de l’appareil. Enfin, nous validonsl’utilisation de ce microcalorimètre par la détermination de l’enthalpie de transformationschimiques modèles.

2.2. Caractéristiques du dispositif 39
2.2 Caractéristiques du dispositif
Le microcalorimètre que nous avons développé est décrit par le schéma correspondantà la figure 2.1 et par la photographie 2.2.
4
>
>
5
5a
5b
1 1r
3
2
6
>
2
3>
Fig. 2.1 – Schéma du microcalorimètre ; 1 : compartiment de réaction, 1r : compartimentde référence, 2 : microréacteur (puce microfluiduique), 3 : capteur de flux (élément Peltier),4 : bloc de laiton, 5 : plaque thermostatée, 5a : entrée du fluide caloporteur, 5b : sortie dufluide caloporteur, 6 : mousse isolante.
Il s’agit d’un calorimètre différentiel composé de deux chambres distinctes, une premièredans laquelle se produit la réaction chimique et l’autre servant de référence thermique.L’approche différentielle de cet appareil permet une mesure où les dérives potentiellesde l’appareil liées aux perturbations extérieures sont limitées. Chacune des chambres estconstituée d’une enceinte en laiton isotherme d’une épaisseur de 2,5 cm qui leur procure unegrande stabilité thermique. Dans le but de caractériser des réactions chimiques à différentestempératures, un système de régulation, placé sous la plaque de laiton, permet d’imposerune température de consigne (Tc) pouvant varier de 5 à 90 ◦C. L’enceinte en laiton et laplaque de régulation sont entourées de mousse dont le caractère isolant limite les échangesthermiques avec le milieu extérieur. Dans chacune des chambres, le réacteur chimiquerepose sur un fluxmètre et le bon contact entre ces éléments est assuré par de la graissethermique. Le réacteur utilisé est une puce microfluidique composée d’une matrice enPDMS (PolyDiMethylSiloxane) dans laquelle sont gravés les microcanaux. Cet ensemble estcollé au substrat via le procédé d’activation UV-ozone décrit précédemment (partie 1.2.1).
Les microréacteurs, ainsi constitués, sont placés dans une enceinte isolée thermique-ment. La configuration de l’ensemble, comme on le verra par la suite, permet un fonction-nement dans des conditions quasi isothermes.

40 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
Microcalorimètre
Pousse seringue
Voltmètre
Fig. 2.2 – Photographie de l’ensemble du dispositif comprenant le microclorimètre, lepousse-seringue et le voltmètre.
2.2.1 Enceinte et système de régulation
L’ensemble de l’enceinte du microcalorimètre (figure 2.3) est réalisée en laiton. L’épais-seur des plaques étant de 2,5 cm, une grande stabilité thermique est garantie. En effet, lelaiton est un alliage dont les propriétés capacitives (masse volumique, capacité thermique)procurent au système une forte inertie thermique.
Fluide
thermostaté
>
>
Laiton
Mousse isolante
Fluxmètre
Puce
microfluidique
>
>
Fig. 2.3 – Photographie du microclorimètre avec la chambre réaction ouverte. Nous pou-vons distinguer le fluxmètre, la puce microfluidique ainsi que les connexions fluidiques etélectriques.
Un système de régulation de type Lauda E200, fonctionnant par circulation d’un fluidethermostaté, est placé sous cette enceinte. Il permet d’imposer la température à laquelle lesréactions chimiques vont se produire. Le fluide caloporteur étant de l’eau, la température deconsigne du calorimètre (Tc) peut varier de 5 à 90 ◦C. Toutefois, d’autres types de fluides(huile ou eau glycolée) sont utilisables afin d’élargir la gamme de température. Compte
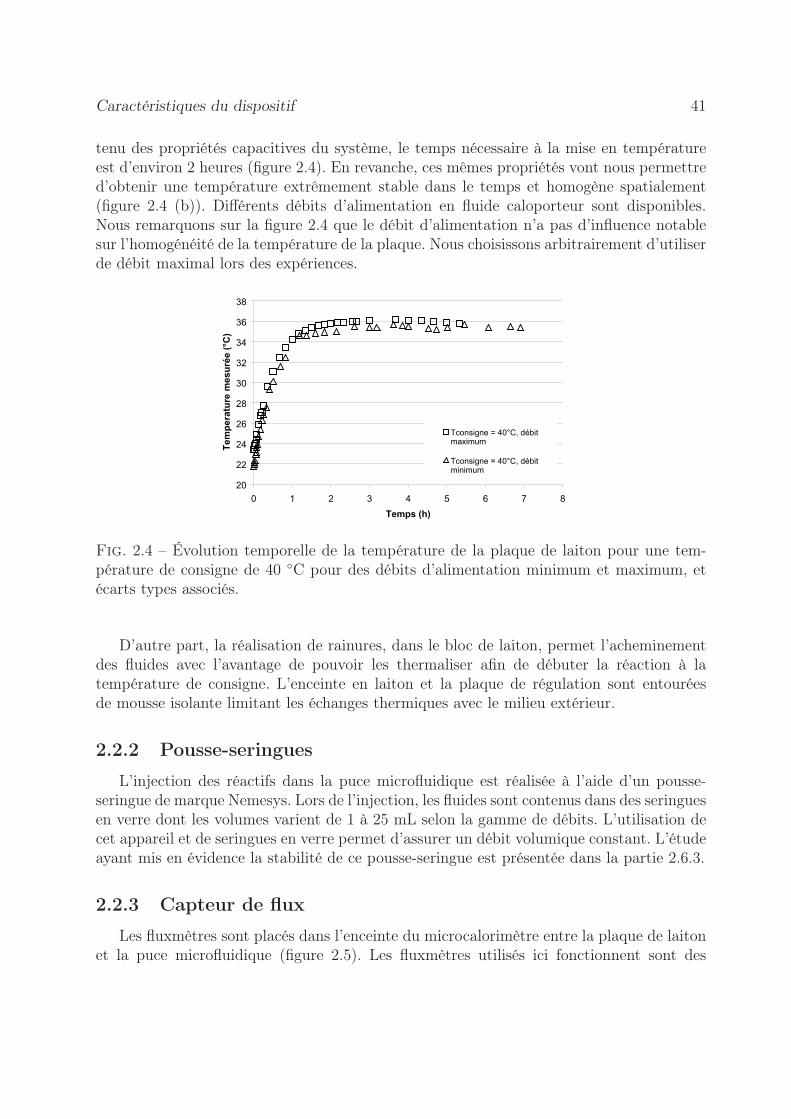
Caractéristiques du dispositif 41
tenu des propriétés capacitives du système, le temps nécessaire à la mise en températureest d’environ 2 heures (figure 2.4). En revanche, ces mêmes propriétés vont nous permettred’obtenir une température extrêmement stable dans le temps et homogène spatialement(figure 2.4 (b)). Différents débits d’alimentation en fluide caloporteur sont disponibles.Nous remarquons sur la figure 2.4 que le débit d’alimentation n’a pas d’influence notablesur l’homogénéité de la température de la plaque. Nous choisissons arbitrairement d’utiliserde débit maximal lors des expériences.
20
22
24
26
28
30
32
34
36
38
0 1 2 3 4 5 6 7 8
Temps (h)
Te
mp
era
ture
me
su
rée
(°C
)
Tconsigne = 40°C, débitmaximum
Tconsigne = 40°C, débitminimum
Fig. 2.4 – Évolution temporelle de la température de la plaque de laiton pour une tem-pérature de consigne de 40 ◦C pour des débits d’alimentation minimum et maximum, etécarts types associés.
D’autre part, la réalisation de rainures, dans le bloc de laiton, permet l’acheminementdes fluides avec l’avantage de pouvoir les thermaliser afin de débuter la réaction à latempérature de consigne. L’enceinte en laiton et la plaque de régulation sont entouréesde mousse isolante limitant les échanges thermiques avec le milieu extérieur.
2.2.2 Pousse-seringues
L’injection des réactifs dans la puce microfluidique est réalisée à l’aide d’un pousse-seringue de marque Nemesys. Lors de l’injection, les fluides sont contenus dans des seringuesen verre dont les volumes varient de 1 à 25 mL selon la gamme de débits. L’utilisation decet appareil et de seringues en verre permet d’assurer un débit volumique constant. L’étudeayant mis en évidence la stabilité de ce pousse-seringue est présentée dans la partie 2.6.3.
2.2.3 Capteur de flux
Les fluxmètres sont placés dans l’enceinte du microcalorimètre entre la plaque de laitonet la puce microfluidique (figure 2.5). Les fluxmètres utilisés ici fonctionnent sont des

42 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
éléments dit « Peltier » fonctionnant en mode passif grâce à l’effet Seebeck. Lorsqu’une desfaces du fluxmètre subit une variation de température, l’effet thermoélectrique va générerune tension proportionelle à l’écart de température entre les deux faces du fluxmètre. Leséléments Peltier utilisés proviennent du fournisseur Melcor et sont du type CP0.8-127-06L.Ils mesurent 25 mm de côté et 3,4 mm d’épaisseur, un flux maximum de 18 W peut y êtreappliqué et leur sensibilité est de 400 µV.◦C−1. Le détecteur de flux est composé de quatreéléments Peltier reliés en série offrant ainsi une surface de mesure de 50 mm × 50 mm.
Laiton
PDMS
Peltier
Microcanal
Silicium >>
>
Fig. 2.5 – Schéma de l’intérieur d’une chambre du microcalorimètre contenant le fluxmètre et lemicroréacteur.
2.2.4 Acquisition et étalonnage
Un multimètre électronique de type Agilent 34970 A est utilisé pour la mesure encontinu de la tension aux bornes des éléments Peltier. L’acquisition des données est réaliséeà une fréquence de 9 Hz. La mesure absolue correspond à la différence entre la tensionmesurée aux bornes de l’élément Peltier de la chambre réaction et celle provenant de lachambre de référence. Cette variation de tension est directement proportionnelle au fluxde chaleur dégagé ou consommé par la réaction chimique, ce coefficient de proportionalitésera estimé par un étalonnage (section 2.5). La phase d’étalonnage consiste à utiliser unerésistance chauffante recouverte d’une forte épaisseur d’isolant, ce qui permet de mesurerla tension aux bornes du Peltier en connaissant parfaitement le flux électrique injecté. Unealimentation du type Agilent E3631A est utilisée pour générer une tension aux bornesd’une résistance électrique.
2.3 Procédure expérimentale et logiciel associé
2.3.1 Démarche générale
Le déroulement d’une expérience est relativement simple. Tout d’abord, un test destabilité thermique est réalisé. Il consiste à vérifier que la soustraction des signaux aux

Procédure expérimentale et logiciel associé 43
bornes des fluxmètres est constante. Si cela est confirmé, les expériences peuvent débu-ter car le microcalorimètre est en équilibre thermique ; sinon, il est nécessaire d’attendrel’établissement de cet équilibre thermique. L’expérience se déroule en trois étapes :
– 1 stabilité thermique : la soustraction du signal référence à celui de la chambre deréaction doit être constant,
– 2 injection et début de la réaction : après environ une minute d’enregistrement,les réactifs contenus dans des seringues en verre de 5 mL sont injectés grâce aupousse-seringue ; nous constatons alors une augmentation du signal atteignant unétat stationnaire correspondant à l’équilibre thermique de la puce,
– 3 arrêt de l’injection après 20 min : les débits de réactifs sont stoppés et le signaldiminue jusqu’à sa valeur initiale.
Le signal brut correspond à une tension en Volt qui est multipliée par le coefficient d’étalon-nage (tableau 2.3) pour obtenir l’évolution temporelle du flux de chaleur en watt. Ensuite,le flux global dégagé par la réaction est calculé et en traçant le débit molaire de réactifs enfonction du flux mesuré, l’enthalpie de la réaction est estimée.
2.3.2 Etalonnage de l’appareil
L’étape d’étalonnage est primordiale pour ce type de métrologie. En effet, même si lescoefficients d’étalonnage sont donnés par les fabriquants de fluxmètre, une méthode propreassociée au dispositif est préférable. Cela permet une calibration plus générale de la fonctionde transfert du système global : puce microfluidique, élément Peltier, système de régulation,échange thermique. Cette étape consiste à générer, par effet Joule, plusieurs puissancesconnues dans la chambre réaction et de mesurer la variation de tension correspondante àchaque puissance. Ces données vont permettre de déterminer le coefficient d’étalonnage αen W.V−1 qui sera ulitisé pour estimer le flux de chaleur à partir de la tension mesurée.Ce coefficient devra être évalué à chaque température de consigne car les propriétés deséléments Peltier varient avec la température. Cette étape est décrite en détail dans lapartie 2.5.
2.3.3 Acquisition
L’acquisition des données est automatisée par un programme réalisé avec le logicielLabviewTM (figure 2.6). Grâce à ce dernier, toutes les étapes expérimentales : test destabilité, étalonnage et réaction chimique, sont automatisées ainsi que la détermination del’enthalpie. En effet, le système gère la mise en température du microcalorimètre, l’injectiondes réactifs, la mesure transitoire des flux et finalement l’estimation de l’enthlapie. Il permetaussi le tracé en temps réel du flux engendré par la réaction chimique. La durée moyennepour l’obtention de l’enthalpie est estimée à 2 heures pour une réaction très rapide et à 6heures pour une cinétique de réaction de l’ordre de l’heure.

44 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
(a)
(b)
Fig. 2.6 – Interface de commande automatisée du microcalorimètre ; (a) interface de configura-tion : entrée des différents paramètres ; (b) interface de commande (départ et arrêt) et visualisationdes mesures.
2.3.4 Estimation de l’enthalpie
A l’issue de l’expérience, le flux total de chaleur dégagée (W) par la réaction chimique estdéterminé par la différence entre la valeur du flux à l’état stationnaire et celle à l’état initial.Ce flux est positif dans le cas d’un phénomène exothermique et négatif si la transformationchimique est endothermique. Le flux de chaleur Φ(W) dépend de la variation d’enthalpiedûe à la réaction, du débit molaire et du taux de conversion. Il est égal à :
Φ = ∆HCQX (2.1)
∆H l’enthalpie de la réaction (J.mol−1), C et Q étant respectivement la concentration(mol.L−1) et le débit volumique (L.s−1) en réactif limitant et X le taux de conversion.Ainsi, en se plaçant dans des conditions où la réaction est totale, ie, X = 1, l’évolution duflux dégagé en fonction du débit molaire permet d’estimer l’enthalpie de réaction.
Lors des expériences, les débits d’injection des réactifs sont constants et l’état sta-tionnaire est rapidement atteint. Le flux de chaleur est réparti uniformément sur toute lasurface du capteur (Peltier) (section 2.4). Ainsi, le comportement du système peut êtrereprésenté très fidèlement par un modèle à une dimension dans lequel seuls les transfertsselon l’épaisseur sont pris en compte comme en atteste la réponse type échelon (figure 2.11).Pour garantir ce comportement, une phase de conception et de dimensionnement est in-dispensable.

2.4. Modélisation thermique 45
2.4 Modélisation thermique
De par la conception du dispositif, le fluxmètre est placé sous la puce microfluidique.Ainsi, pour optimiser le transfert de chaleur entre le microréacteur et le capteur, il est pri-mordial de bien dimensionner la puce microfluidique. Comme expliqué dans la partie 1.2.1,la puce microfluidique est généralement composée d’une matrice en PDMS et d’un substraten verre ou en silicium. Pour concevoir au mieux la puce, nous allons identifier la naturedu substrat qui favorise au maximum le transfert de chaleur entre le milieu réactionnelsitué dans le microcanal et la surface de l’élément Peltier. Compte tenu du fait que lecapteur soit un fluxmètre qui intégre le flux sur toute la surface du microréacteur, un mo-dèle transitoire à une dimension suffit à représenter l’évolution du transfert de chaleur lorsd’une sollicitation de type réaction chimique. En revanche, la diffusion thermique et lesélévations de température qui vont influencer la réaction chimique dépendent de la naturedu substrat. Le choix de ce dernier se révèle alors important.
2.4.1 Modèle thermique
Le système puce et élément Peltier, représenté en figure 2.7, est composé de trois milieuxhomogènes de propriétés thermiques différentes. Le milieu 1 correspond au détecteur deflux (élément Peltier) d’épaisseur e1, le milieu 2 au substrat d’épaisseur e2 et le milieu 3 à lamatrice de PDMS d’épaisseur e3. L’utilisation d’un système de régulation en températurede type Lauda va permettre d’évacuer les flux de chaleur générés par la réaction. Cecipermet de faire l’hypothèse que la température inférieure du fluxmètre est constante aucours du temps. Cette température est fixée à 0 pour cette étude et les pertes par convectionentre le PDMS et l’air sont prises en compte. La conductivité thermique du silicium (λ= 148 W.m−1.K−1) étant très supérieure à celle du PDMS (λ = 148 W.m−1.K−1), le fluxdissipé dans le canal peut être assimilé à un flux surfacique sur le substrat. De plus, le canalest supposé de très petites dimensions par rapport à e1, e2, et e3, ainsi ec/ePDMS << 1.
Flux nul :
symétrie
Flux imposé (W)
5 cm
PDMS
Substrat Si (e1 + e2 )
Fluxmètre (e1)
Pertes par convection
Température de consigne, Tc = 0°C
y
x
e3
e2
e1
b L/20
e1 + e2 + e3
Fig. 2.7 – Schéma du réacteur microfluidique et du fluxmètre. L’abscisse en x = 0 correspondau centre du canal.

46 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
Par ailleurs, nous considérons le champ de température moyenné selon z. Ainsi, le bilanthermique appliqué à ce système pour les trois milieux (i = 1, 2 ou 3) s’écrit de la façonsuivante :
∂2Ti
∂x2+
∂2Ti
∂y2=
1
ai
∂Ti
∂t, ai =
λi
ρiCp,i
(2.2)
i étant l’indice représentant le milieu considéré, T la température (K), x l’abscisse(m), y l’ordonnée (m), a la diffusivité thermique (m2.s−1), t le temps (s), λ la conducti-vité thermique (W.m−1.K−1), ρ la masse volumique (kg.m−3) et Cp la capacité calorifique(J.kg−1.K−1).
Les propriétés de ces trois milieux sont répértoriées dans le tableau 2.1.
Nature Largeur (m) e (m) λ(W.m−1.K−1) ρCp (MJ.kg−1.m−3)Silicium 0,05 500.10−6 148 1,6Verre 0,05 500.10−6 1,3 1,8PDMS 0,05 0,01 0,1 2Peltier 0,05 3.10−3 1 1
Tab. 2.1 – Propriétés thermiques des différents milieux nécessaires à la résolution du modèlethermique.
En tenant compte des conditions limites du problème pour chacun des milieux, onobtient les systèmes d’équations 2.3, 2.4 et 2.5.
Pour le milieu 1 (Peltier) on a :
∂2T1
∂x2 + ∂2T1
∂y2 = 1a1
∂T1
∂t
t = 0 → T1(t) = 0
x = 0 → ∂T1
∂x= 0
x = L → ∂T1
∂x= 0
y = 0 → T1(x, 0) = 0
y = e1 → −λ1∂T1
∂y= −λ2
∂T1
∂yet T1 = T2
(2.3)

Modélisation thermique 47
Pour le milieu 2 (substrat) on a :
∂2T2
∂x2 + ∂2T2
∂y2 = 1a2
∂T2
∂t
t = 0 → T2(t) = 0
x = 0 → ∂T2
∂t= 0
x = L → −λ2∂T2
∂x= 0
y = e1 + e2 → −λ2∂T2
∂x+ φ(x, t) = −λ3
∂T3
∂xet T2 = T3
(2.4)
Et pour le milieu 3 (PDMS), on obtient :
∂2T3
∂x2 + ∂2T3
∂y2 = 1a3
∂T3
∂t
t = 0 → T3(t) = 0
x = 0 → ∂T3
∂x= 0
x = L → ∂T3
∂x= 0
y = e1 + e2 + e3 → −λ3∂T3
∂y= hT3
(2.5)
h étant le coefficient global de transfert thermique par convection (W.m−2.K−1) et Φ leterme source dégagé dans le canal, moyenné selon z (W.m−2) : Φ (x,t) = Q0 si x < b, sinonΦ =0
2.4.2 Solution analytique
Pour résoudre ce système, nous utilisons la méthode des quadripoles qui permet detransformer le système différentiel en un problème purement algébrique. La solution sedéduit par transformés inverses de la résolution d’un système linéaire [63]. Cette méthodepermet de résoudre les problèmes de diffusion thermique par une représentation simple,sous forme matricielle, de systèmes linéaires. La résolution de ce système est présentée enannexe A.
À partir de ce modèle, une étude de dimensionnement du microréacteur est réalisée.
2.4.3 Influence de la nature du substrat
Une puce microfluidique est généralement constituée d’un substrat de verre ou de sili-cium. Grâce au modèle décrit précédemment, la température du substrat est calculée pourun flux dégagé de 1 W au centre du canal, c’est à dire en x = 0. Nous obtenons les courbesreprésentées sur la figure 2.8.
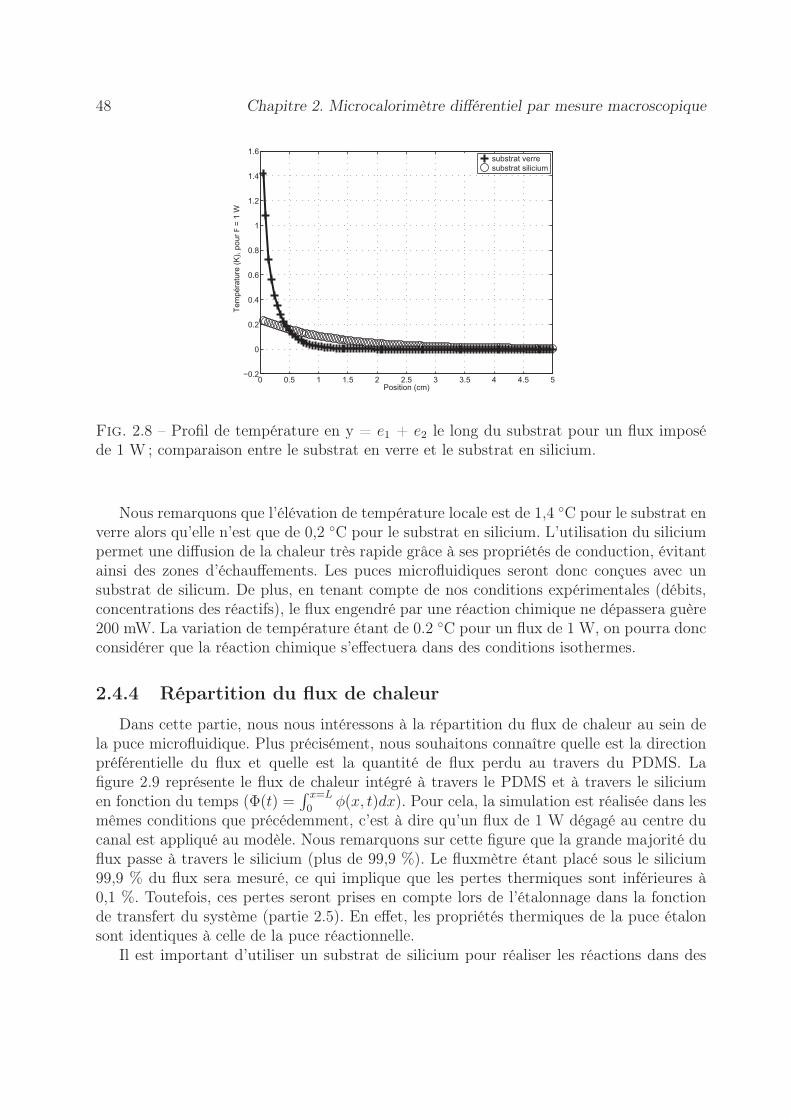
48 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 5−0.2
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
Position (cm)
Température (K), pour F = 1 W
substrat verre
substrat silicium
Fig. 2.8 – Profil de température en y = e1 + e2 le long du substrat pour un flux imposéde 1 W ; comparaison entre le substrat en verre et le substrat en silicium.
Nous remarquons que l’élévation de température locale est de 1,4 ◦C pour le substrat enverre alors qu’elle n’est que de 0,2 ◦C pour le substrat en silicium. L’utilisation du siliciumpermet une diffusion de la chaleur très rapide grâce à ses propriétés de conduction, évitantainsi des zones d’échauffements. Les puces microfluidiques seront donc conçues avec unsubstrat de silicum. De plus, en tenant compte de nos conditions expérimentales (débits,concentrations des réactifs), le flux engendré par une réaction chimique ne dépassera guère200 mW. La variation de température étant de 0.2 ◦C pour un flux de 1 W, on pourra doncconsidérer que la réaction chimique s’effectuera dans des conditions isothermes.
2.4.4 Répartition du flux de chaleur
Dans cette partie, nous nous intéressons à la répartition du flux de chaleur au sein dela puce microfluidique. Plus précisément, nous souhaitons connaître quelle est la directionpréférentielle du flux et quelle est la quantité de flux perdu au travers du PDMS. Lafigure 2.9 représente le flux de chaleur intégré à travers le PDMS et à travers le siliciumen fonction du temps (Φ(t) =
∫ x=L
0φ(x, t)dx). Pour cela, la simulation est réalisée dans les
mêmes conditions que précédemment, c’est à dire qu’un flux de 1 W dégagé au centre ducanal est appliqué au modèle. Nous remarquons sur cette figure que la grande majorité duflux passe à travers le silicium (plus de 99,9 %). Le fluxmètre étant placé sous le silicium99,9 % du flux sera mesuré, ce qui implique que les pertes thermiques sont inférieures à0,1 %. Toutefois, ces pertes seront prises en compte lors de l’étalonnage dans la fonctionde transfert du système (partie 2.5). En effet, les propriétés thermiques de la puce étalonsont identiques à celle de la puce réactionnelle.
Il est important d’utiliser un substrat de silicium pour réaliser les réactions dans des

2.5. Étalonnage du dispositif 49
Fig. 2.9 – (a) Evolution du flux de chaleur au cours du temps dans le substrat de silicium etdans le PDMS, (b) Porportion de flux de chaleur à travers le silicium (reçu par le flumètre)et le PDMS.
conditions isothermes et favoriser le transfert de chaleur vers le fluxmètre. Ainsi, un modèleà une dimension suffit à modéliser le système.
2.5 Étalonnage du dispositif
2.5.1 Définition du coefficient d’étalonnage
Le microcalorimètre permet de mesurer, au cours du temps, l’évolution de la tension auxbornes des éléments Peltier (capteur de flux). Cette variation de tension est directementproportionelle au flux dégagé par la réaction chimique selon le modèle suivant :
Φ =(λG)
βU = αU, α =
(λG)
β(2.6)
Φ étant le flux de chaleur (W), λ la conductivité thermique (W.m−1.K−1), β le coefficientSeebeck (V.K−1), G le facteur de forme (m), U la tension mesurée (V) et α le coefficientd’étalonnage (W.V−1).
Nous remarquons que ce coefficient d’étalonnage α dépend entre autre des propriétésthermiques du Peltier et donc de la température. La valeur des paramètres λ, G, et β quidéfinissent α est donnée par le fournisseur en fonction de la température (tableau 2.2).Grâce à ces données, un coefficient d’étalonnage αfournisseur peut être calculé à plusieurstempératures. Les résultats sont représentés en figure 2.13. On remarque que α diminuejusqu’à une température de 60 ◦C pour ensuite augmenter.

50 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
T (◦C) λ (W.cm−1.K−1) G (cm) β (V.K−1) α (W.V−1)-50 1,87.10−2 0,042 1,70.10−4 4,62-25 1,77.10−2 0,042 1,84.10−4 4,040 1,61.10−2 0,042 1,94.10−4 3,4825 1,51.10−2 0,042 2,02.10−4 3,1450 1.53.10−2 0,042 2,07.10−4 3,1075 1.55.10−2 0,042 2.10.10−4 3,1100 1,58.10−2 0,042 2,00.10−4 3,32125 1,63.10−2 0,042 1,96.10−4 3,49150 1,73.10−2 0,042 1,90.10−4 3,82175 1,88.10−2 0,042 1,85.10−4 4,27200 2.09.10−2 0,042 1,79.10−4 4,90
Tab. 2.2 – Valeurs du coefficient d’étalonnage αfournisseur d’après les données du construc-teur à différentes températures.
2.5.2 Conception d’une puce étalon
Comme cela est expliqué dans le paragraphe 2.3.2, il est nécessaire de déterminer le fluxde chaleur expérimental, et donc d’estimer α par un étalonnage du dispositif. Pour cela, unepuce étalon a été conçue à l’image de la puce réacteur. Cette puce étalon, représentée enfigure 2.10, est composée d’un substrat de silicium sur lequel une résistance électrique en orest déposée. L’ensemble est recouvert de PDMS, qui est un bon isolant thermique. Ainsi,les propriétés thermiques de cette puce sont les mêmes que celles du microréacteur. Larésistance électrique va permettre de reproduire, par effet Joule, le dégagement de chaleurengendré par une réaction chimique. Cela va conduire à un étalonnage complet du systèmepar la détermination du coefficient d’étalonnage α.
PDMS
Substrat silicium
Résistance éléctrique
(a) (b)
5 cm
Fig. 2.10 – (a) Schéma de la puce étalon ; (b) Photographie de la puce étalon.

Étalonnage du dispositif 51
2.5.3 Principe de la mesure
La mesure du coefficient d’étalonnage consiste à injecter différentes puissances élec-triques au travers de la résistance chauffante. En parallèle, la variation de tension auxbornes des éléments Peltier est enregistrée. Pour cela, la puce étalon est placée dansla chambre réaction du microcalorimètre et une puce microfluidique est placée dans lachambre de référence. La puissance est injectée à la résistance électrique grâce à l’utilisa-tion d’une alimentation Agilent E3631A. D’autre part, le microcalorimètre est maintenu àune température de 25 ◦C via le régulateur.
La figure 2.11 décrit l’évolution de la tension aux bornes des éléments Peltier pour unepuissance injectée de 500 mW. Cette tension correspond à la soustraction entre la tensionde la chambre réaction et celle de la chambre de référence où aucune réaction ne se produit.Cette soustraction permet de s’affranchir de toutes dérives. Dès l’injection de la puissanceaux bornes de la résistance, nous observons que la tension mesurée aux bornes des élémentsPeltier augmente jusqu’à atteindre un état stable. Celui-ci est maintenu jusqu’à l’arrêt del’injection de puissance, à cet instant la tension chute jusqu’à retrouver son état initial.Cette expérience est réalisée pour différentes valeurs de puissance allant de 100 µW à 1 W.Ici, l’hypothèse de température imposée est bien vérifiée.
0
20
40
60
80
100
120
140
0 500 1000 1500 2000 2500 3000
0
100
200
300
400
500
tension mesurée
puissance injectée
Te
nsio
n m
esu
rée
(m
V)
Pu
issa
nce
inje
cté
e (m
W)
Temps (s)
Fig. 2.11 – Évolution temporelle de la tension mesurée lors de l’étalonnage du microcalo-rimètre à T = 25 ◦C.
2.5.4 Résultats
À chaque puissance injectée (P ) correspond une variation de tension (U). D’aprèsl’équation (2.6), la puissance injectée est directement proportionelle à la tension mesu-rée via le coefficient d’étalonnage. Ainsi, nous traçons la puissance injectée en fonction
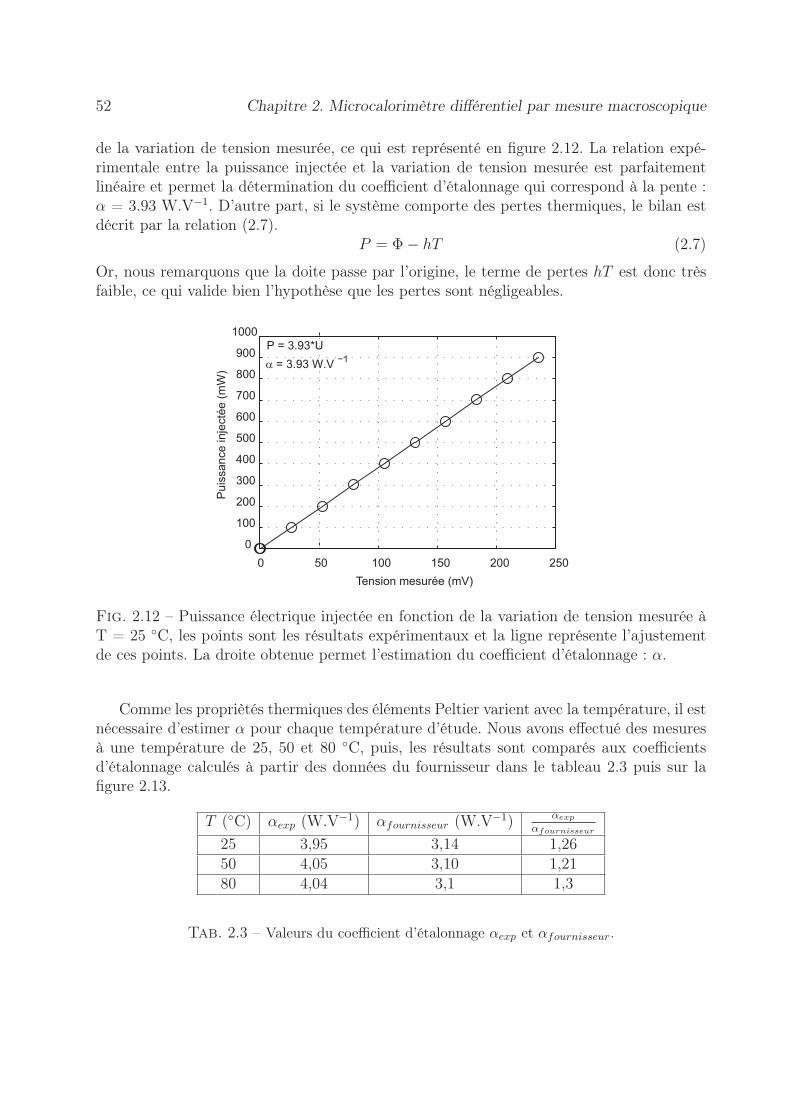
52 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
de la variation de tension mesurée, ce qui est représenté en figure 2.12. La relation expé-rimentale entre la puissance injectée et la variation de tension mesurée est parfaitementlinéaire et permet la détermination du coefficient d’étalonnage qui correspond à la pente :α = 3.93 W.V−1. D’autre part, si le système comporte des pertes thermiques, le bilan estdécrit par la relation (2.7).
P = Φ − hT (2.7)
Or, nous remarquons que la doite passe par l’origine, le terme de pertes hT est donc trèsfaible, ce qui valide bien l’hypothèse que les pertes sont négligeables.
0 50 100 150 200 250
0
100
200
300
400
500
600
700
800
900
1000 P = 3.93*U
α = 3.93 W.V−1
Puis
sance
in
jecté
e (
mW
)
Tension mesurée (mV)
Fig. 2.12 – Puissance électrique injectée en fonction de la variation de tension mesurée àT = 25 ◦C, les points sont les résultats expérimentaux et la ligne représente l’ajustementde ces points. La droite obtenue permet l’estimation du coefficient d’étalonnage : α.
Comme les propriètés thermiques des éléments Peltier varient avec la température, il estnécessaire d’estimer α pour chaque température d’étude. Nous avons effectué des mesuresà une température de 25, 50 et 80 ◦C, puis, les résultats sont comparés aux coefficientsd’étalonnage calculés à partir des données du fournisseur dans le tableau 2.3 puis sur lafigure 2.13.
T (◦C) αexp (W.V−1) αfournisseur (W.V−1) αexp
αfournisseur
25 3,95 3,14 1,2650 4,05 3,10 1,2180 4,04 3,1 1,3
Tab. 2.3 – Valeurs du coefficient d’étalonnage αexp et αfournisseur.
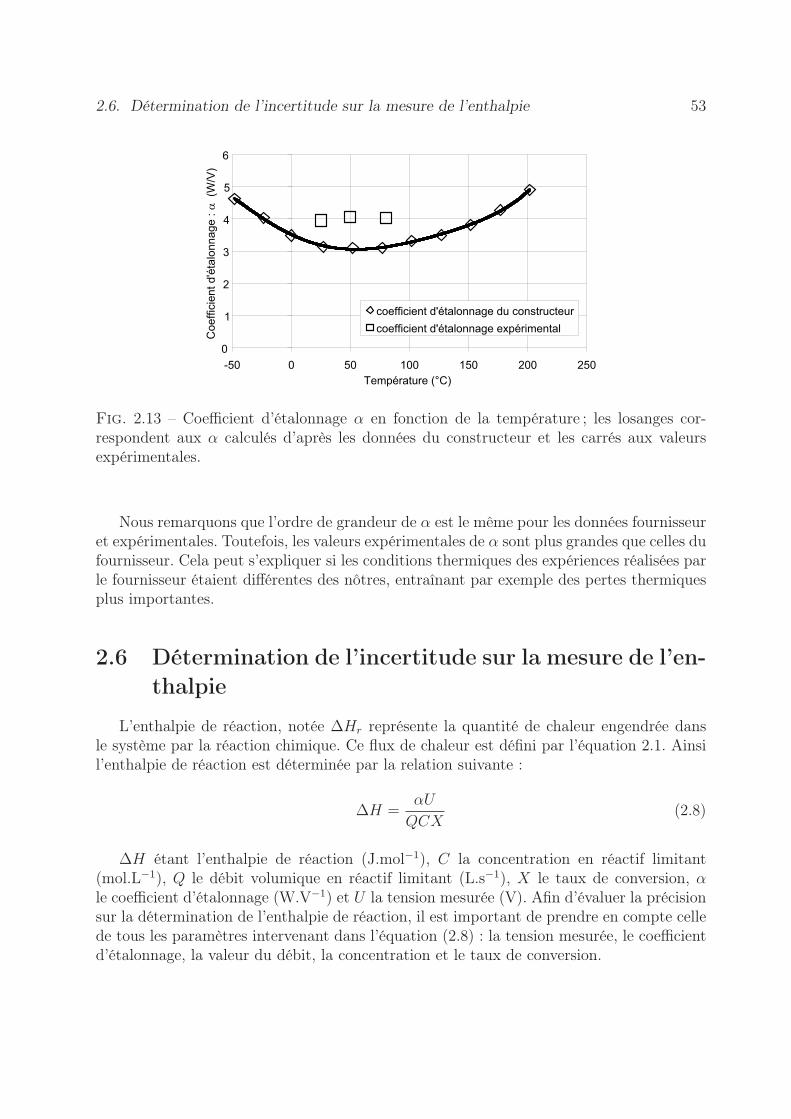
2.6. Détermination de l’incertitude sur la mesure de l’enthalpie 53
0
1
2
3
4
5
6
-50 0 50 100 150 200 250
Température (°C)
Co
eff
icie
nt d
'éta
lon
na
ge
: α
(W
/V)
coefficient d'étalonnage du constructeur
coefficient d'étalonnage expérimental
Fig. 2.13 – Coefficient d’étalonnage α en fonction de la température ; les losanges cor-respondent aux α calculés d’après les données du constructeur et les carrés aux valeursexpérimentales.
Nous remarquons que l’ordre de grandeur de α est le même pour les données fournisseuret expérimentales. Toutefois, les valeurs expérimentales de α sont plus grandes que celles dufournisseur. Cela peut s’expliquer si les conditions thermiques des expériences réalisées parle fournisseur étaient différentes des nôtres, entraînant par exemple des pertes thermiquesplus importantes.
2.6 Détermination de l’incertitude sur la mesure de l’en-thalpie
L’enthalpie de réaction, notée ∆Hr représente la quantité de chaleur engendrée dansle système par la réaction chimique. Ce flux de chaleur est défini par l’équation 2.1. Ainsil’enthalpie de réaction est déterminée par la relation suivante :
∆H =αU
QCX(2.8)
∆H étant l’enthalpie de réaction (J.mol−1), C la concentration en réactif limitant(mol.L−1), Q le débit volumique en réactif limitant (L.s−1), X le taux de conversion, αle coefficient d’étalonnage (W.V−1) et U la tension mesurée (V). Afin d’évaluer la précisionsur la détermination de l’enthalpie de réaction, il est important de prendre en compte cellede tous les paramètres intervenant dans l’équation (2.8) : la tension mesurée, le coefficientd’étalonnage, la valeur du débit, la concentration et le taux de conversion.
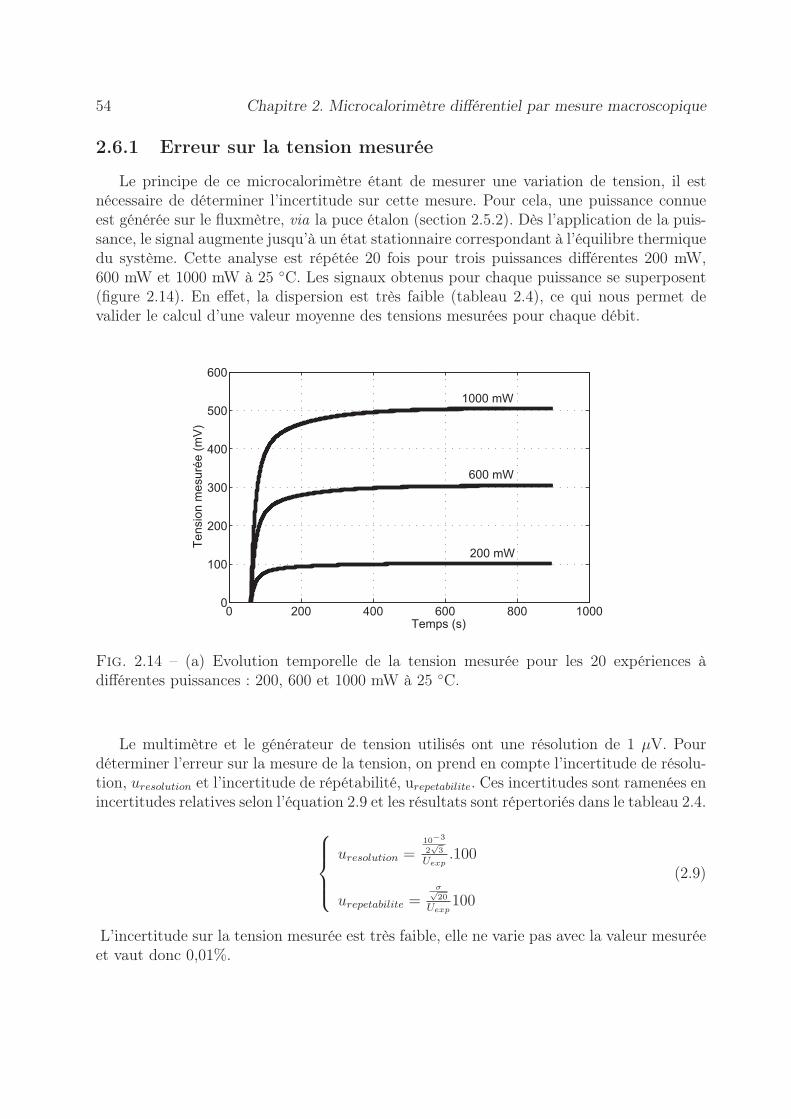
54 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
2.6.1 Erreur sur la tension mesurée
Le principe de ce microcalorimètre étant de mesurer une variation de tension, il estnécessaire de déterminer l’incertitude sur cette mesure. Pour cela, une puissance connueest générée sur le fluxmètre, via la puce étalon (section 2.5.2). Dès l’application de la puis-sance, le signal augmente jusqu’à un état stationnaire correspondant à l’équilibre thermiquedu système. Cette analyse est répétée 20 fois pour trois puissances différentes 200 mW,600 mW et 1000 mW à 25 ◦C. Les signaux obtenus pour chaque puissance se superposent(figure 2.14). En effet, la dispersion est très faible (tableau 2.4), ce qui nous permet devalider le calcul d’une valeur moyenne des tensions mesurées pour chaque débit.
0 200 400 600 800 10000
100
200
300
400
500
600
Temps (s)
Te
nsio
n m
esu
rée
(m
V)
1000 mW
600 mW
200 mW
Fig. 2.14 – (a) Evolution temporelle de la tension mesurée pour les 20 expériences àdifférentes puissances : 200, 600 et 1000 mW à 25 ◦C.
Le multimètre et le générateur de tension utilisés ont une résolution de 1 µV. Pourdéterminer l’erreur sur la mesure de la tension, on prend en compte l’incertitude de résolu-tion, uresolution et l’incertitude de répétabilité, urepetabilite. Ces incertitudes sont ramenées enincertitudes relatives selon l’équation 2.9 et les résultats sont répertoriés dans le tableau 2.4.
uresolution =10
−3
2√
3
Uexp.100
urepetabilite =σ
√
20
Uexp100
(2.9)
L’incertitude sur la tension mesurée est très faible, elle ne varie pas avec la valeur mesuréeet vaut donc 0,01%.
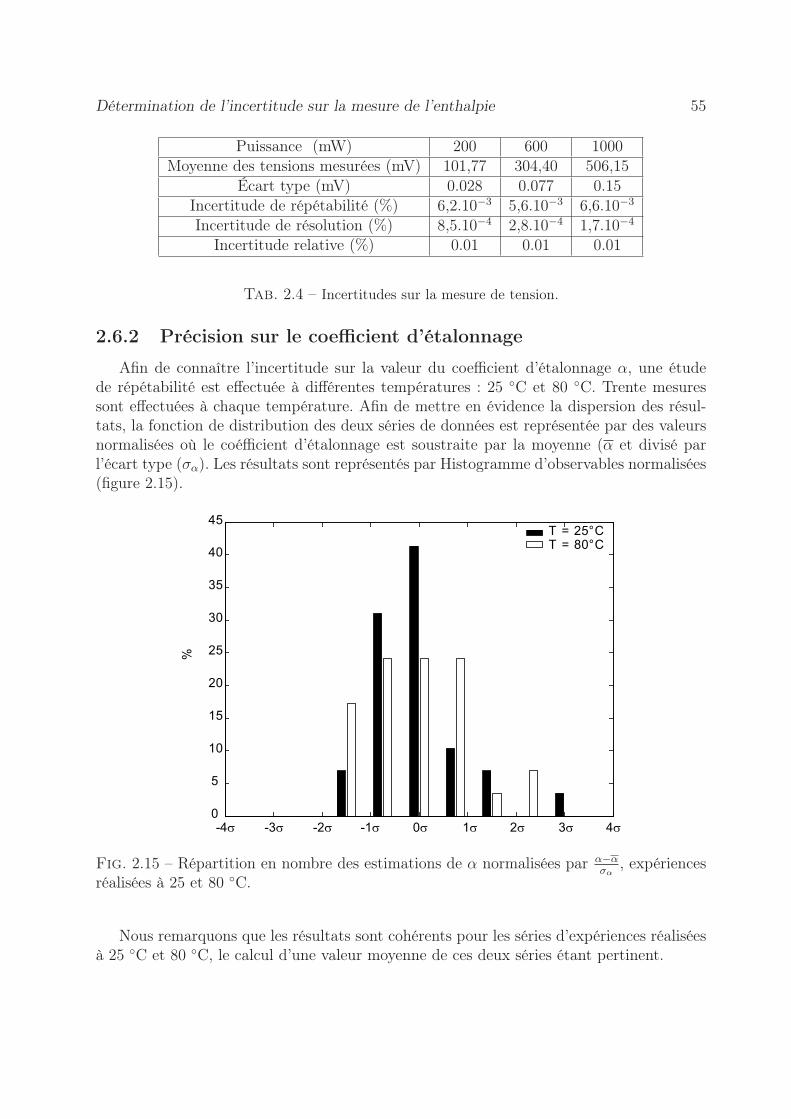
Détermination de l’incertitude sur la mesure de l’enthalpie 55
Puissance (mW) 200 600 1000Moyenne des tensions mesurées (mV) 101,77 304,40 506,15
Écart type (mV) 0.028 0.077 0.15Incertitude de répétabilité (%) 6,2.10−3 5,6.10−3 6,6.10−3
Incertitude de résolution (%) 8,5.10−4 2,8.10−4 1,7.10−4
Incertitude relative (%) 0.01 0.01 0.01
Tab. 2.4 – Incertitudes sur la mesure de tension.
2.6.2 Précision sur le coefficient d’étalonnage
Afin de connaître l’incertitude sur la valeur du coefficient d’étalonnage α, une étudede répétabilité est effectuée à différentes températures : 25 ◦C et 80 ◦C. Trente mesuressont effectuées à chaque température. Afin de mettre en évidence la dispersion des résul-tats, la fonction de distribution des deux séries de données est représentée par des valeursnormalisées où le coéfficient d’étalonnage est soustraite par la moyenne (α et divisé parl’écart type (σα). Les résultats sont représentés par Histogramme d’observables normalisées(figure 2.15).
-4σ -3σ -2σ -1σ 0σ 1σ 2σ 3σ 4σ0
5
10
15
20
25
30
35
40
45
%
T = 25°CT = 80°C
Fig. 2.15 – Répartition en nombre des estimations de α normalisées par α−ασα
, expériencesréalisées à 25 et 80 ◦C.
Nous remarquons que les résultats sont cohérents pour les séries d’expériences réaliséesà 25 ◦C et 80 ◦C, le calcul d’une valeur moyenne de ces deux séries étant pertinent.

56 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
L’écart type expérimental obtenu permet de remonter à l’incertitude de répétabilitédéfinie grâce à l’équation (2.10) :
urepetabilite =σ√n
, (2.10)
où n est le nombre de mesures effectuées. L’incertitude relative sur la valeur expérimentaledu coefficient de conversion α est donnée par l’équation (2.11).
u(%) = urepetabilite(%) =urepetabilite(W.V −1)
α(2.11)
Les résultats sont répertoriés dans le tableau 2.5. Les incertitudes relatives sont trèsacceptables 0.01 % à 25 ◦C.
Température (◦C) 25 80Moyenne de α (W.V−1) 3,95 4,04
Ecart type (W.V−1) 1,44.10−3 4,99.10−3
Incertitude de répétabilité (W.V−1) 1,6.10−2 1,76.10−3
Incertitude relative (%) 6,65.10−3 ≃ 0,01 4,35.10−2
Tab. 2.5 – Incertitudes sur la mesure du coefficient d’étalonnage.
2.6.3 Stabilité des pousse-seringues
Trois types de pousse-seringues sont à notre disposition : Bioseb, Harvard et Nemesys.Afin de déterminer le pousse-seringue le plus performant, nous nous proposons de lescomparer en évaluant la stabilité des débits puis l’écart du débit expérimental par rapportà la consigne.
Afin de tester la stabilité, nous utilisons une méthode thermique avec comme outille microcalorimètre lui-même qui consiste à mesurer le flux de chaleur dégagé par uneréaction acido-basique à différents débits variant de 500 à 2000 µL.h−1, et ce pour chaquetype de pousse-seringue. Les expériences sont réalisées à l’identique pour chaque pousse-seringue, les résultats sont représentés par les figures 2.16, 2.17 et 2.18. Nous remarquonsclairement que la valeur du plateau augmente avec celle du débit pour les trois types depousse-seringues. En effet, lorsque le débit augmente, la quantité de réactifs introduiteest plus importante et le flux de chaleur proportionellement plus grand. La stabilité desplateaux varie selon la pompe utilisée. Le pousse-seringue Nemesys permet d’obtenir desplateaux très stables, ce qui implique que la réaction se fait à un débit molaire constant. Enrevanche, des fluctuations périodiques sont observées pour les expériences réalisées avec lesappareils Bioseb et Harvard. La période de ces fluctuations a été évaluée et est comparableau pas de vis du pousse-seringue, ce qui prouve que les fluctuations sont dépendantes dutype de pousse-seringue.
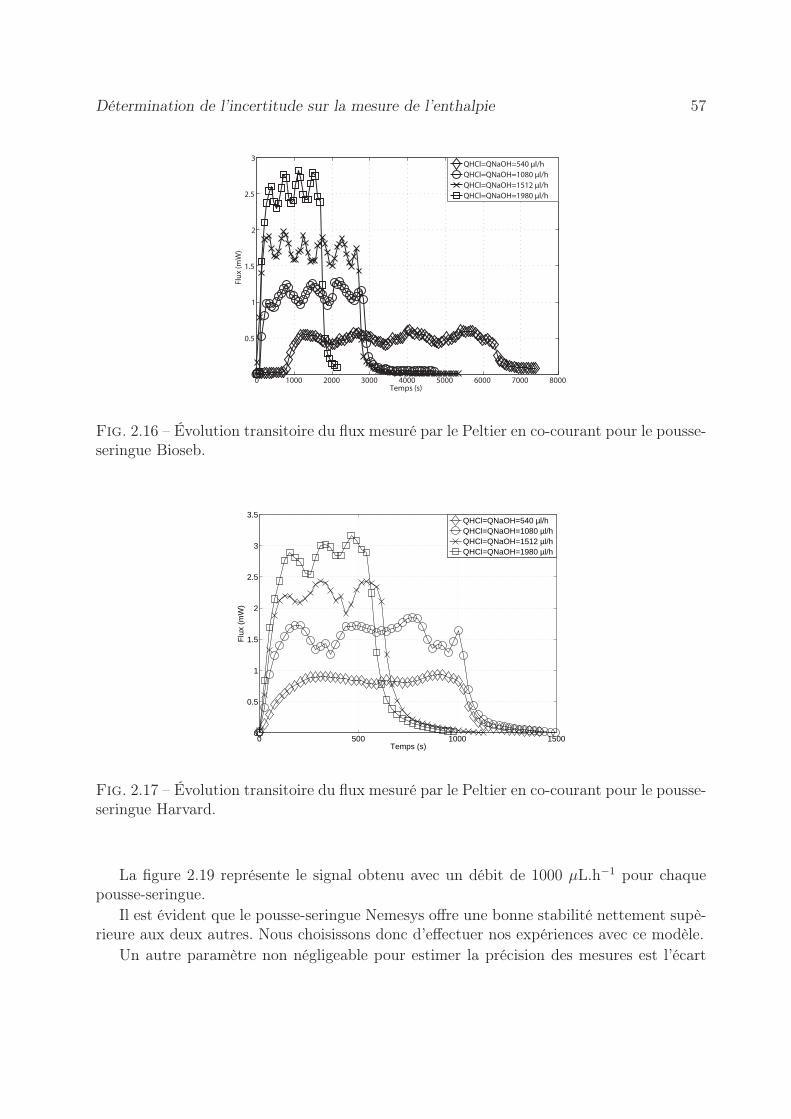
Détermination de l’incertitude sur la mesure de l’enthalpie 57
0 1000 2000 3000 4000 5000 6000 7000 80000
0.5
1
1.5
2
2.5
3
Temps (s)
Flu
x (m
W)
QHCl=QNaOH=540 µl/h
QHCl=QNaOH=1080 µl/h
QHCl=QNaOH=1512 µl/h
QHCl=QNaOH=1980 µl/h
Fig. 2.16 – Évolution transitoire du flux mesuré par le Peltier en co-courant pour le pousse-seringue Bioseb.
0 500 1000 15000
0.5
1
1.5
2
2.5
3
3.5
Temps (s)
Flu
x (m
W)
QHCl=QNaOH=540 µl/hQHCl=QNaOH=1080 µl/hQHCl=QNaOH=1512 µl/hQHCl=QNaOH=1980 µl/h
Fig. 2.17 – Évolution transitoire du flux mesuré par le Peltier en co-courant pour le pousse-seringue Harvard.
La figure 2.19 représente le signal obtenu avec un débit de 1000 µL.h−1 pour chaquepousse-seringue.
Il est évident que le pousse-seringue Nemesys offre une bonne stabilité nettement supè-rieure aux deux autres. Nous choisissons donc d’effectuer nos expériences avec ce modèle.
Un autre paramètre non négligeable pour estimer la précision des mesures est l’écart
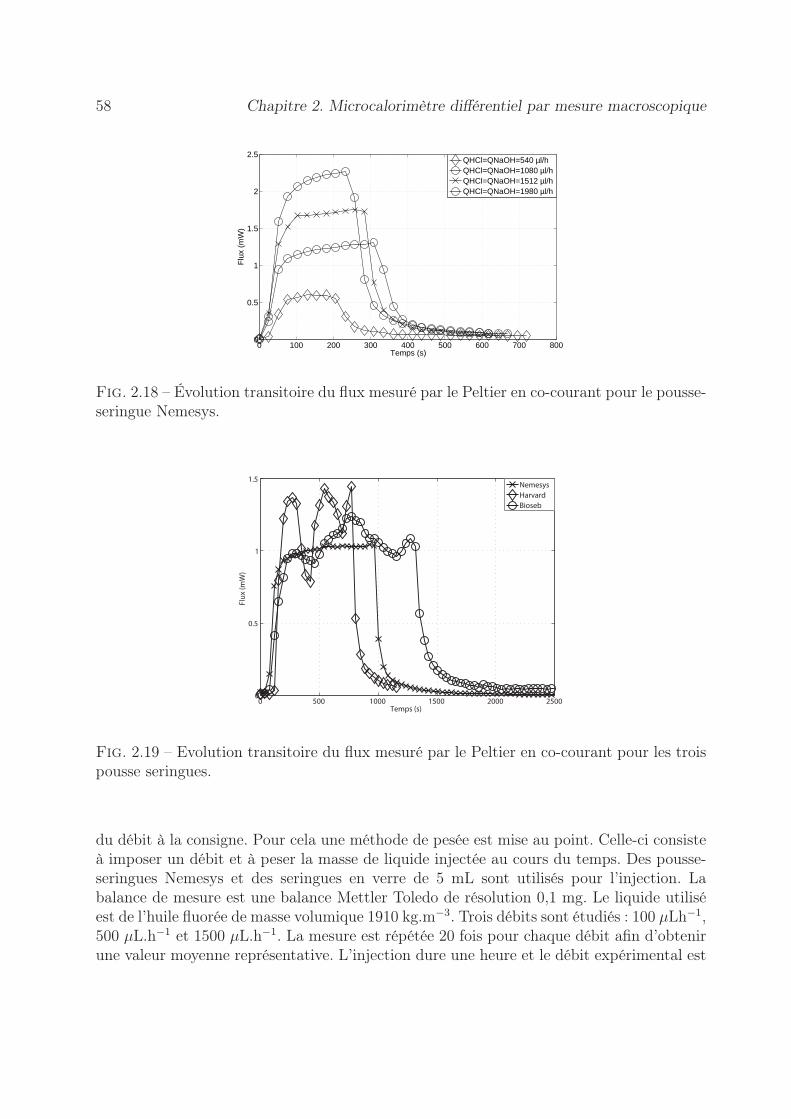
58 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
0 100 200 300 400 500 600 700 8000
0.5
1
1.5
2
2.5
Temps (s)
Flu
x (m
W)
QHCl=QNaOH=540 µl/hQHCl=QNaOH=1080 µl/hQHCl=QNaOH=1512 µl/hQHCl=QNaOH=1980 µl/h
Fig. 2.18 – Évolution transitoire du flux mesuré par le Peltier en co-courant pour le pousse-seringue Nemesys.
0 500 1000 1500 2000 25000
0.5
1
1.5
Temps (s)
Flu
x (m
W)
Nemesys
Harvard
Bioseb
Fig. 2.19 – Evolution transitoire du flux mesuré par le Peltier en co-courant pour les troispousse seringues.
du débit à la consigne. Pour cela une méthode de pesée est mise au point. Celle-ci consisteà imposer un débit et à peser la masse de liquide injectée au cours du temps. Des pousse-seringues Nemesys et des seringues en verre de 5 mL sont utilisés pour l’injection. Labalance de mesure est une balance Mettler Toledo de résolution 0,1 mg. Le liquide utiliséest de l’huile fluorée de masse volumique 1910 kg.m−3. Trois débits sont étudiés : 100 µLh−1,500 µL.h−1 et 1500 µL.h−1. La mesure est répétée 20 fois pour chaque débit afin d’obtenirune valeur moyenne représentative. L’injection dure une heure et le débit expérimental est

Détermination de l’incertitude sur la mesure de l’enthalpie 59
obtenu en traçant la masse pesée en fonction du temps. Les résultats obtenus pour chaquedébit sont représentés sur forme d’histogramme (figure 2.20).
−4σ −3σ −2σ −1σ 0σ 1σ 2σ 3σ 4σ0
10
20
30
40
50
60
%
Q = 100 µL.h−1
Q = 500 µL.h−1
Q = 1500 µL.h−1
Fig. 2.20 – Répartition en nombre des estimations du débits normalisées par Q−Q
σQ, expé-
riences réalisées à 100 µLh−1, 500 µL.h−1 et 1500 µL.h−1.
Nous remarquons que pour les trois débits testés, une faible dispersion des données estobservée ; une valeur moyenne peut alors être calculée.
Il est nécessaire d’avoir une incertitude globale pour toute la gamme de débits utilisés.L’incertitude maximale obtenue pour les trois débits étudiés est donc retenue. Deux sourcesd’erreurs sont prises en compte dans le calcul et sont ramenées à des incertitudes relatives.Il s’agit de l’incertitude sur la consigne et de l’incertitude de répétabilité (équation 2.12).
uc = Qc−Qexp
Qc.100
urepetabilite =σ
√
20
Qexp.100,
(2.12)
où u l’incertitude relative (%), Qc le débit de consigne, Qexp le débit expérimental moyenet σ l’écart type. Ainsi, l’incertitude relative totale u sur nos débits est la somme entre l’in-certitude de consigne et l’incertitude de répétabilité . D’après le tableau 2.6, l’incertitudemaximale est obtenue pour le débit de 100 µL.h−1. Puisque les débits utilisés ne descendentpas en dessous de 100 µL.h−1 lors de l’utilisation d’une seringue en verre de 5 mL, l’incer-titude relative sur le débit injecté n’excédera pas 3,23 %. Le volume de la seringue est un

60 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
Débit théorique : (Qc) (µL.h−1) 100 500 1500Débit expérimental moyen Qexp (µL.h−1) 97.47 488.32 1469.00
Ecart à la consigne Qc−Qexp
Qc(%) 2.53 2.34 2.07
Ecart type expérimental : σ (µL.h−1) 3.08 13.25 24.01Incertitude de répétabilité (%) 0,707 0,607 0,366
Incertitude relative u (%) 3,23 2,94 2,43
Tab. 2.6 – Incertitude sur le débit.
paramètre non négligeable pour le calcul de l’incertitude car il permet de définir la vitessedu moteur du pousse-seringue. Dans le cas où le débit vaut 100 µL.h−1 et le volume de laseringue vaut 5 mL, la vitesse du moteur est de 3,33.10−7 m.s−1 (équation 2.13).
Vmoteur =Qc
Sseringue
= 3, 33.10−7m.s−1, (2.13)
où Vmoteur est la vitesse du moteur (m.s−1), Qc est le débit imposé (m3.s−1) et Sseringue
représente la section interne de la seringue (m2). Pour injecter les fluides à des débitsinférieurs à 100 µL.h−1, il est nécessaire d’utiliser une seringue de volume inférieur à 5 mLpermettant une vitesse de moteur supérieure ou égale à 3.33.10−7 m.s−1. Ainsi, l’incertituderelative u définie pour un débit de 100 µL.h−1 peut être considérée.
Ce pousse-seringue permet donc d’injecter des fluides avec une précision d’environ 3 %,ce qui est raisonnable. De plus, il est très pratique d’utilisation, modulable, automatisé etoffre un gain de place appréciable surtout pour des manipulations microfluidiques.
2.6.4 Précision sur l’enthalpie
D’après l’équation (2.8), l’enthalpie de réaction dépend du coefficient d’étalonnage, dela tension mesurée, du débit, de la concentration et du taux de conversion. Pour évaluerl’enthalpie de réaction, nous nous plaçons dans les conditions où la réaction chimique esttotale au sein de la puce microfluidique c’est à dire X=1.
De plus, les produits utilisés sont des solutions commerciales dont l’incertitude surla concentration est de 2.10−4. En tenant compte des erreurs dues à la préparation dela solution, l’incertitude sur la concentration uconc est égale à 1,4.10−4 mol.L−1, ce quicorrespond à une incertitude relative de 7.10−2 % pour une concentration de 0,2 mol.L−1.
Ainsi, l’incertitude relative sur l’enthalpie (u∆H) vaut la somme des incertitudes rela-tives des différents paramètres (équation (2.14)).
u∆Hr= uα + uU + uQ + uconc (2.14)
Le microcalorimètre permet de déterminer l’enthalpie de réaction avec une précision de3,35 % à 25 ◦C et 80 ◦C.

2.7. Validation du microcalorimètre 61
2.7 Validation du microcalorimètre
2.7.1 Choix de la réaction modèle
Afin de valider le dispositif sur la mesure de l’enthalpie de réaction, nous avons choiside caractériser une réaction chimique exothermique, à cinétique rapide, dont l’enthalpiede réaction est bien connue dans la littérature. Une réaction acido-basique entre un acidefort et une base forte a été retenue (plus particulièrement la réaction entre l’acide chlor-hydrique (HCl) et l’hydroxide de sodium (NaOH)). Il s’agit d’une réaction de protonationoù l’acide chlorhydrique libère un proton (H+) et l’hydroxide de sodium en fixe un (fi-gure 2.21). La cinétique de cette réaction est très rapide et la constante de vitesse vaut−→k = 1,4.1011 L.mol−1.s−1 [?]. D’après les données de la littérature [?], cette réaction estexothermique et l’enthalpie de réaction est de 55,8 kJ.mol−1 à une température de 20 ◦C.
H2OH+ OH-
NaClH2OHCl NaOH
HCl H+ Cl-
NaOH OH-Na+
Na+ Cl- NaCl
Fig. 2.21 – Équation bilan de la réaction entre l’acide chlorhydrique et l’hydroxide desodium.
Les expériences sont réalisées avec des concentrations initiales en acide (HCl) et base(NaOH) égales à 0,2 mol.L−1. Ces solutions sont réalisées à partir de solutions commer-ciales prédosées Titrisol de Merck. L’enthalpie de la réaction à température ambiante estdéterminée via le microcalorimètre selon les deux types d’écoulements offerts par la micro-fluidique : écoulement co-courant et écoulement gouttes.
Un phénomène exothermique est défini par une variation d’enthalpie positive et unphénomène endothermique par une variation d’enthalpie négative. Cette convention nonhabituelle a été choisie car le dispositif expérimental indique une augmentation du flux dechaleur lors d’un phénomène exothermique. En effet, le repère (fluxmètre) est extérieur aumilieu réactionnel.
2.7.2 Conception de la puce microfluidique
Afin de caractériser une vaste gamme de réactions chimiques, nous avons conçu unepuce microfluidique permettant un long temps de séjour ainsi que l’étude de réactions

62 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
chimiques sous les deux types d’écoulement décrits dans le chapitre 1. Comme indiqué audébut de ce chapitre, la puce est constituée d’un substrat de silicium, de 5 cm de diamètre,recouvert d’1 cm de PDMS. La forme du canal et le système d’injection sont représentéssur la figure 2.22.
Entrée
huile
Entrées réactifs
Zone de
mélange >
>
(a) (b)
Fig. 2.22 – (a) Photographie de la puce microfluidique utilisée pour étudier la réactionacide-base dans le microcalorimètre ; dimensions : canaux carrés de 250 µm de côté et de32 cm de long ; (b) schéma de la zone d’injection des fluides.
Le canal réaction de la puce suit une forme de serpentin ce qui permet de maximisersa longueur (32 cm). Les canaux sont carrés et mesurent 250 µm ou 500 µm de côté selonle temps de séjour désiré, ce qui correspond respectivement à un volume réactionnel de20 µL ou de 80 µL. Les débits utilisés varient de 500 à 3000 µL.h−1, engendrant ainsides temps de résidence de 20 s à 10 min. Le système d’injection permet l’introduction detrois flux d’entrée et comprend une intersection en croix ; ainsi les deux types d’écoulementpeuvent être générés. Le co-écoulement est réalisé par l’injection des réactifs seuls et lesgouttes multi-constituants sont formées par ajout d’une phase continue selon la méthodede « flow-focusing». Les produits et la phase continue sont introduits par des tubes de800 µm de diamètre externe et de 500 µm de diamètre interne. Cette puce est placéedans la chambre réaction du microcalorimètre et adhère au fluxmètre grâce à la graissethermique qui favorise également les échanges de chaleur.
2.7.3 Principe de la mesure
Le déroulement des expériences est décrit dans la partie section 2.3.1.Les conditions expérimentales choisies pour cette série d’expériences permettront l’ob-
tention d’un taux de conversion égal à 1. En effet, les temps de séjour, liés à la gammede débits choisie, garantissent une réaction totale. Ainsi d’après l’équation (2.1), le fluxdépend uniquement de la variation d’enthalpie de réaction et du débit molaire. Des expé-

Validation du microcalorimètre 63
riences sont réalisées pour plusieurs débits permettant de tracer le flux total en fonctiondu débit molaire afin d’estimer l’enthalpie de la réaction.
2.7.4 Résultats lors de l’écoulement co-courant
L’écoulement co-courant consiste à injecter les deux réactifs parallèlement. Comme dé-crit dans la section 1.2.2, l’écoulement dans un microcanal est laminaire et le mélange desréactifs, lors d’un écoulement co-courant, se fait par diffusion des espèces. D’après l’équa-tion (1.7), la longueur de diffusion ou longueur de mélange dépend de la vitesse des fluides,de la largeur du canal et du coefficient de diffusion de l’espèce. Lors de la réaction acidebase, l’espèce diffusante est l’ion H+ et son coefficient de diffusion vaut 9.10−9 m.s−1 [?].Nous déterminons la longueur de diffusion pour une gamme de débit allant de 500 µL.h−1
à 3000 µL.h−1. Les résultats obtenus donnent une longueur de diffusion de 1,93 mm à11,57 mm. La longueur de la puce étant de 32 cm, le mélange sera largement terminé aumilieu du microréacteur. D’autre part, cette réaction acide base étant une réaction très ra-pide, la réaction sera totale au sein du microréacteur. Les solutions d’acide chlorhydriqueet d’hydroxide de sodium utilisées ont la même concentration (C = 0,2 mol.L−1) et laréaction s’opère mole à mole. Comme on souhaite réaliser la réaction chimique dans desconditions stoechiométriques, le débit d’HCl sera égal au débit de NaOH. Les résultatsobtenus pour les différents débits sont représentés par la figure 2.23 qui décrit l’évolutiondu flux mesuré au cours du temps.
0 500 1000 1500 2000 25000
0.5
1
1.5
2
2.5
3
3.5
4
4.5
5
Temps (s)
Flu
x (
mW
)
QHCl=QNaOH=250 µl/h
QHCl=QNaOH=500 µl/h
QHCl=QNaOH=750 µl/h
QHCl=QNaOH=1000 µl/h
QHCl=QNaOH=1250 µl/h
QHCl=QNaOH=1500 µl/h
Fig. 2.23 – Évolution temporelle du flux de chaleur dégagé par la réaction acido-basique,lors d’un écoulement co-courant pour différents débits de réactifs à T = 25 ◦C.
Nous obtenons la même évolution temporelle de courbe pour chaque débit, à savoir audépart un signal constant puis un transitoire jusqu’à l’état stationnaire. Lorsque le régime
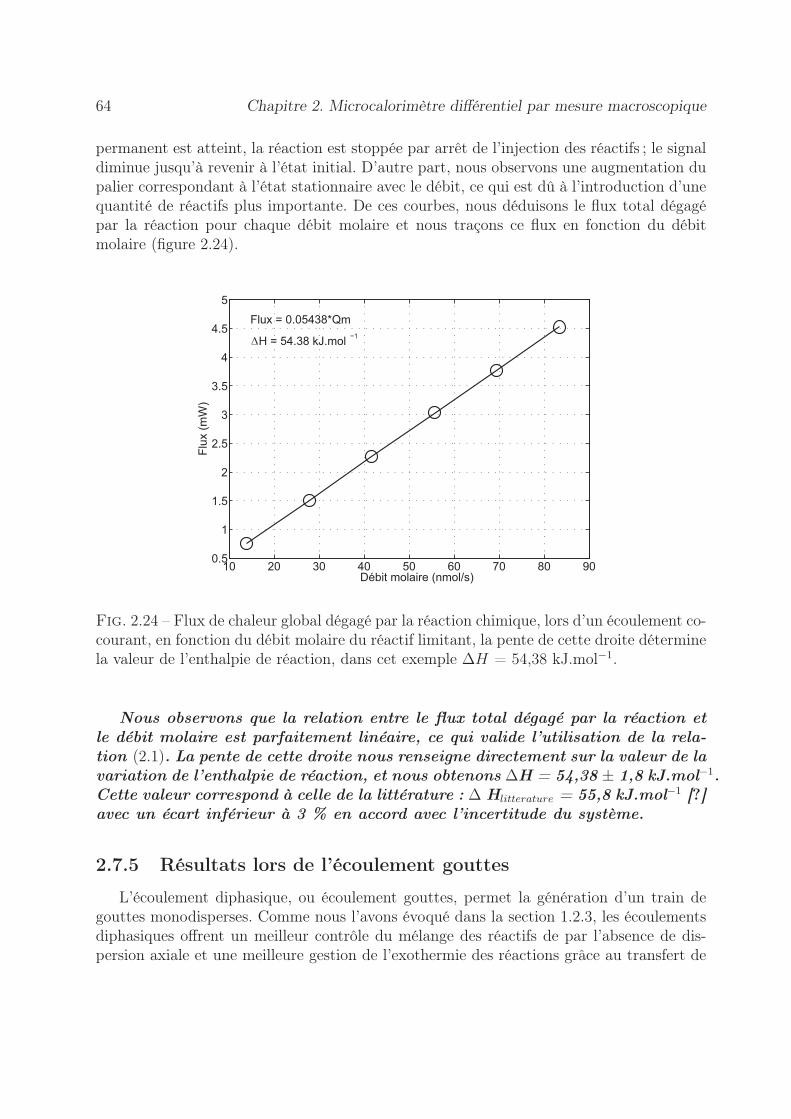
64 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
permanent est atteint, la réaction est stoppée par arrêt de l’injection des réactifs ; le signaldiminue jusqu’à revenir à l’état initial. D’autre part, nous observons une augmentation dupalier correspondant à l’état stationnaire avec le débit, ce qui est dû à l’introduction d’unequantité de réactifs plus importante. De ces courbes, nous déduisons le flux total dégagépar la réaction pour chaque débit molaire et nous traçons ce flux en fonction du débitmolaire (figure 2.24).
10 20 30 40 50 60 70 80 900.5
1
1.5
2
2.5
3
3.5
4
4.5
5
Débit molaire (nmol/s)
Flu
x (
mW
)
Flux = 0.05438*Qm
∆−1
H = 54.38 kJ.mol
Fig. 2.24 – Flux de chaleur global dégagé par la réaction chimique, lors d’un écoulement co-courant, en fonction du débit molaire du réactif limitant, la pente de cette droite déterminela valeur de l’enthalpie de réaction, dans cet exemple ∆H = 54,38 kJ.mol−1.
Nous observons que la relation entre le flux total dégagé par la réaction etle débit molaire est parfaitement linéaire, ce qui valide l’utilisation de la rela-tion (2.1). La pente de cette droite nous renseigne directement sur la valeur de lavariation de l’enthalpie de réaction, et nous obtenons ∆H = 54,38 ± 1,8 kJ.mol−1.Cette valeur correspond à celle de la littérature : ∆ Hlitterature = 55,8 kJ.mol−1 [?]avec un écart inférieur à 3 % en accord avec l’incertitude du système.
2.7.5 Résultats lors de l’écoulement gouttes
L’écoulement diphasique, ou écoulement gouttes, permet la génération d’un train degouttes monodisperses. Comme nous l’avons évoqué dans la section 1.2.3, les écoulementsdiphasiques offrent un meilleur contrôle du mélange des réactifs de par l’absence de dis-persion axiale et une meilleure gestion de l’exothermie des réactions grâce au transfert de

Validation du microcalorimètre 65
chaleur dans la phase porteuse. De plus nous avons vu que le mélange est environ dix foisplus rapide que lors d’un écoulement co-courant.
Comme dans le cas des expériences en écoulement co-courant, nous réalisons la réactionchimique avec les mêmes solutions de réactifs, dans les conditions stoechiométriques et àtempérature ambiante. Les réactifs étant des solutions acqueuses, nous utiliserons, commephase continue, la phase organique suivante : huile silicone de viscosité µhuile = 20 mPa.s.
Afin de déterminer l’enthalpie de réaction lors d’un écoulement gouttes, il est nécessairede vérifier préalablement que la présence de l’huile dans notre système d’étude ne perturbepas le signal mesuré par les éléments Peltier.
2.7.5.1 Influence de l’huile lors d’un écoulement diphasique
Cette étape est très importante car nous allons mettre en évidence les perturbationsengendrées par le débit d’huile sur notre système de mesure. Pour cela, nous réalisonsplusieurs expériences où les débits de réactifs sont constants, seul le débit d’huile varie. Laquantité de réactifs étant la même pour chaque expérience, nous pouvons vérifier si le fluxglobal dégagé par la réaction est identique à chaque manipulation.
Les résultats sont représentés par la figure 2.25 où l’évolution du flux de chaleur dégagépar la réaction est représentée en fonction du temps. Les valeurs du flux ont été obtenuesen multipliant le signal de tension mesuré par le coefficient d’étalonnage.
0 1000 2000 3000 4000 5000−0.2
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
Temps (s)
Flu
x (
mW
)
Qhuile = 800 µl/h
Qhuile = 1000 µl/h
Qhuile = 1200 µl/h
Qhuile = 1400 µl/h
Qhuile = 1600 µl/h
Qhuile = 1800 µl/h
Fig. 2.25 – Flux de chaleur dégagé par la réaction acido basique lors d’un écoulementgouttes pour des débits de réactifs constants : QNaOH = QHCl = 500 µL.h−1, seul le débitd’huile varie de 800 à 1800 µL.h−1.
Nous remarquons sur la figure 2.25 que les courbes sont similaires à celles obtenueslors de l’écoulement co-courant. Le signal est stable au départ puis il augmente dès quel’injection des réactifs, atteint un état stable et diminue. D’autre part, ces courbes, corres-pondant à des débits de réactifs identiques et à des débits d’huile différents, se superposent.Le flux dégagé par la réaction est donc pratiquement identique pour chaque expérience et

66 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
l’huile n’ayant aucune influence sur la mesure, des expériences en écoulement diphasiquesont alors envisageables.
2.7.5.2 Détermination de l’enthalpie de réaction
Plusieurs débits sont expérimentés et répertoriés dans le tableau 2.7 tout en conservantle rapport de débits :
∑Qreactifs
Qhuileégal à 1. Les trains de gouttes ainsi produits, sont similaires
de par la taille des gouttes et la distance intergoutte ; seule la vitesse varie. Le débit totalmaximun est de 3000 µL.h−1 et est identique au débit maximum utilisé lors des expériencesen co-courant (tableau 2.7). Comme nous l’avons vu dans la section 1.2.3, le mélange ausein d’une goutte est plus rapide que lors d’un écoulement co-courant. Ainsi, nous avonsla certitude que le mélange sera complet.
QHCl = QNaOH (µL.h−1) 100 200 300 400 500 750QHuile (µL.h−1) 200 400 600 800 1000 1500
Tab. 2.7 – Débits d’acide chlorhydrique, d’hydroxide de sodium et d’huile silicone utiliséspour la détermination de l’enthalpie de réaction lors d’un écoulement gouttes.
Les expériences se déroulent de la façon décrite dans la section 2.7.3. Les résultatsobtenus sont représentés par la figure 2.26 qui représente l’évolution temporelle du fluxpour les différents débits étudiés. Nous remarquons clairement que le flux dégagé par laréaction acido-basique augmente avec le débit de réactifs.
Afin de déterminer l’enthalpie de réaction, le flux total dégagé par la réac-tion est représenté en fonction du débit molaire par la figure 2.27. La courbeobtenue est une droite dont la pente est égale à l’enthalpie de réaction. Aprèsrégression linéaire, la valeur de ∆H = 55,74 ± 1,84 kJ.mol−1 est obtenue.Cette valeur est en très bon accord avec celle fournie dans la littérature :∆Hlitterature = 55,8 kJ.mol−1. Un écart d’environ 0,1 % est observé. D’autre partcette valeur est proche de la valeur estimée lors de l’écoulement co-courant.

Validation du microcalorimètre 67
0 500 1000 1500 2000 25000
0.5
1
1.5
2
2.5
Temps (s)
Flu
x (
mW
)
QHCl=QNaOH=100 µl/h
QHCl=QNaOH=200 µl/h
QHCl=QNaOH=300 µl/h
QHCl=QNaOH=400 µl/h
QHCl=QNaOH=500 µl/h
QHCl=QNaOH=750 µl/h
Fig. 2.26 – Évolution temporelle du flux de chaleur dégagé par la réaction acide basiquelors d’un écoulement gouttes pour des débits différents.
5 10 15 20 25 30 35 40 450
0.5
1
1.5
2
2.5
Débit molaire (nmol/s)
Flu
x (m
W)
Flux = 0.05574*Qm
-1∆H = 55.74 kJ.mol
(b)
Fig. 2.27 – Flux de chaleur global dégagé par la réaction chimique, lors d’un écoulementgouttes, en fonction du débit molaire du réactif limitant, la pente de cette droite déterminela valeur de l’enthalpie de réaction, dans cet exemple ∆H = 55,74 kJ.mol−1.

68 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
2.8 Applications et caractérisations de réactions chimiques
L’objectif de ce chapitre est de présenter les nombreuses applications envisageablesà l’aide du microcalorimètre développé dans cette étude. En effet, cet appareil dont lafonction principale est l’estimation de l’enthalpie de réaction peut également être utilisépour
– déterminer l’enthalpie de mélange,– déterminer l’enthalpie de réaction,– réaliser des expériences de titration,– estimer les coefficients de diffusion massique.Tout d’abord, une réaction d’estérification est caractérisée par la détermination de
l’enthalpie de mélange et de réaction. Ensuite, des expériences de titration sont réalisées. Latroisième application présentée est la mesure de temps de diffusion lorsque la réaction entrel’acide chlorhydrique et l’hydroxide de sodium se produit. Enfin, le mélange eau-éthanolest caractérisé par la mesure de l’enthalpie molaire d’excès pour différentes compositionsmolaires.
2.8.1 Étude de la réaction d’estérification
La réaction d’estérification de l’anhydride propionique par le 2-butanol est largementcitée dans les publications relatives à la sécurité des procédés physico-chimiques. Cetteréaction est utilisée dans le cadre de la validation d’études théoriques concernant parexemple la détermination rapide de données cinétiques [64]. Elle possède un certain nombred’intérêts qui en font un cas d’étude modèle : la réaction s’effectue en milieu homogène,elle est fortement exothermique [65] et le modèle cinétique est connu [64, 66].
Cette réaction est décrite par l’équation suivante :
CH3
O
CH3
O
O H3C
CH3
OH
H3CO CH3
CH3
O
H3C
OH
O
anhydride propionique 2-buthanol propionate de butyle acide propionique
Fig. 2.28 – Equation de réaction entre l’ahydrire propionique et le butanol.
Cette réaction est une réaction exothermique, les données bibliographiques disponiblesproposent une valeur de l’enthalpie de réaction : ∆Hr = 62,5 kJ.mol−1 [66]. D’autre part,un effet endothermique lors du mélange entre l’anhydride propionique et le 2-butanol a étémis en évidence, son enthalpie est de ∆Hm = -4,2 kJ.mol−1 [66]. Cependant, la cinétiquede réaction est très lente : il faut 20 heures à 70 ◦C pour que la réaction soit totale.Heureusement, cette réaction peut être catalysée par l’ajout d’acide sulfurique. Cela permetde diminuer considérablement le temps de réaction qui, à 30 ◦C, avec une fraction molaire

Applications et caractérisations de réactions chimiques 69
de 3 % en acide sulfurique par rapport au 2-butanol, passe à 2 h 30 [64]. Avec de telsrésultats, il devient possible, compte tenu des temps de résidence de ces systèmes fluidiquesde mesurer cette enthalpie de mélange. Pour cela, il faut pouvoir différencier l’enthalpieendothermique, liée au mélange, de l’enthalpie exothermique liée à la réaction. En effet, depar la méthode de mesure, l’enthalpie mesurée sera globale et donc composée de ces deuxcontributions. Nous souhaitons donc distinguer expérimentalement les deux enthalpies enréalisant deux manipulations dont les temps de résidence sont différents. De plus, la réactionne sera pas catalysée lors de la mesure de l’enthalpie de mélange.
2.8.1.1 Mesure de l’enthalpie de mélange
Afin de déterminer l’enthalpie de mélange, la réaction ne doit pas se produire dans lemicroréacteur. C’est pourquoi toutes les expériences sont réalisées sans catalyseur à 30 ◦Cavec un temps de séjour maximum des réactifs de 360 s, ce qui est bien inférieur au tempsde réaction, et au minimum de 125 s, ce qui est supérieur au temps de mélange estiméà 30 s (considérant D = 10−9 m.s−1). De plus, ils sont introduits dans les proportionsstoechiométriques en tenant compte des effets de dilution. Toutes les expériences sontréalisées selon les conditions suivantes :
– puce microfluidique en serpentin de section carrée de 500 µm x 500 µm et de longueur32 cm,
– Canh = 7,76 mol.L−1,– C2bu = 10,93 mol.L−1,– huile fluorée (pour les écoulements en gouttes),– seringues de 5 mL pour l’injection des réactifs et de 10 mL pour l’injection de l’huile.
La conception de la puce permet des écoulements co-courant et en gouttes. La réactiond’estérification est caractérisée via le microcalorimètre pour ces deux types d’écoulements.
2.8.1.2 Mesure en écoulement co-courant
Une série de cinq expériences est réalisée pour différents débits (tableau 2.8). La mé-thode de mesure précédemment décrite (section 2.7.3) est appliquée.
Qanh(µL.h−1) 467,68 701,68 935,58 1169,47 1403,37Q2bu(µL.h−1) 332,21 498,32 664,42 830,53 996,63
Tab. 2.8 – Débits utilisés pour la mesure de l’enthalpie de mélange de la réaction d’estérificationlors d’un écoulement co-courant.
La figure 2.29 (a) représente le flux de chaleur en fonction du temps pour les diffé-rents débits expérimentés. Nous remarquons que le flux mesuré est négatif, ce qui traduitl’endothermicité du mélange. De plus, la valeur du plateau (régime permanent) augmentelorsque les débits injectés augmentent, ce qui est normal car la quantité de réactifs aug-mente. D’après la figure 2.29 (b), l’enthalpie de mélange est de - 3,98 kJ.mol−1. La valeur de

70 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
0 500 1000 1500 2000 2500−14
−12
−10
−8
−6
−4
−2
0
Temps (s)
Flu
x (
mW
)
Qanh = 467.68 µl/h
Qanh = 701.68 µl/h
Qanh = 935.58 µl/h
Qanh = 1169.47 µl/h
Qanh = 1403.37 µl/h
Débit molaire croissant
(a)1000 1500 2000 2500 3000 3500
−14
−12
−10
−8
−6
−4
−2
Débit molaire (mmol/s)
Flu
x (
mW
)
Flux = − 0.00398*Qm
(b)
∆−1
H = − 3.98 kJ.mol
Fig. 2.29 – (a)Évolution temporelle du flux mesuré pour différents débits de réactifs enco-courant ; (b) Flux global en fontion du débit molaire.
l’enthalpie étant connue à 3,3 % près, la valeur expérimentale vaut - 3,96 ± 0,13 kJ.mol−1.Il s’agit bien d’un phénomène endothermique, et la valeur expérimentale concorde avec unécart de 5,7 % à la littérature (∆Hm = - 4,2 kJ.mol−1).
2.8.1.3 Mesure en écoulement gouttes
Au cours de cette série d’expériences, les conditions sont telles que la somme des dé-bits des réactifs est égale au débit d’huile et le rapport Qreactifs/Qhuile est constant. Celapermet d’obtenir des microréacteurs (gouttes) de tailles identiques à chaque expérience ;seul le temps de résidence varie. Les différents débits étudiés au cours de ces manipulationssont indiqués dans le tableau 4.8. Contrairement à la réaction acido-basique étudiée pré-cédemment, les gouttes sont réalisées à l’aide d’une huile fluorée inerte aux réactifs et auxproduits mis en jeu. L’injection des réactifs est stabilisée pendant 20 minutes.
Qhuile(µL.h−1) 400 600 800 1200 1600Qanh(µL.h−1) 233,89 350,84 467,68 701,68 935,58Q2bu(µL.h−1) 166,11 249,16 332,21 498,32 664,42
Tab. 2.9 – Débits utilisés pour la mesure de l’enthalpie de mélange de la réaction d’estérificationlors d’un écoulement gouttes.
D’après les résultats expérimentaux (figure 2.30), l’enthalpie de mélange obtenue lorsd’un écoulement gouttes est de - 4,24 ± 0,14 kJ.mol−1. Le mélange est endothermique,la valeur expérimentale concorde avec la littérature avec un écart de 0,24 % (∆Hm = -4,2 kJ.mol−1).

Applications et caractérisations de réactions chimiques 71
0 500 1000 1500 2000 2500−10
−9
−8
−7
−6
−5
−4
−3
−2
−1
0
Temps (s)
Flux (mW)
Qanh = 233.89 µl/h
Qanh = 350.84 µl/h
Qanh = 467.68 µl/h
Qanh = 701.68 µl/h
Qanh = 935.58 µl/h
(a)
Débit molaire croissant
400 600 800 1000 1200 1400 1600 1800 2000 2200−10
−9
−8
−7
−6
−5
−4
−3
−2
−1
Débit molaire (mmol/s)
Flux (mW)
Flux = − 0.00424*Qm
(b)
DH = − 4.24 kJ.mol−1
Fig. 2.30 – (a)Évolution temporelle du flux mesuré pour différents débits de réactifs engouttes ; (b) Flux global en fonction du débit molaire.
2.8.1.4 Mesure de l’enthalpie de réaction
Pour déterminer l’enthalpie de réaction, il est nécessaire que la réaction soit totale ausein de la puce. Pour cela, l’analyse est effectuée à 30 ◦C en présence de catalyseur. L’acidesulfurique est dilué dans le butanol à 3 % (fraction molaire par rapport au 2-butanol).Dans ces conditions, le temps de résidence dans la puce doit être supérieur à 2,5 heurespour être sûr que la réaction soit totale. De ce fait, compte tenu de la section de 0,25 µm2
et de la longueur de 32 cm du microcanal, le débit total maximal utilisé est de 55 µL.h−1
(tableau 2.10). Les seringues de 500 µL sont utilisées pour l’injection des réactifs afin quela vitesse du moteur corresponde à celle définie dans le chapitre 2.6.3. Les réactifs sontintroduits dans les proportions stoechiométriques.
Temps de résidence (min) 141 157 176 201 235Qanh (µL.h−1) 29,24 26,31 23,39 20,47 17,54Q2bu (µL.h−1) 20,76 18,69 16,61 14,53 12,46Qtotal (µL.h−1) 50 45 40 35 30
Tab. 2.10 – Débits utilisés pour la mesure de l’enthalpie de réaction de la réaction d’esté-rification lors d’un écoulement co-courant.
D’après la figure 2.31, l’enthalpie globale peut être estimée via une régression linéaire.Ainsi l’enthalpie obtenue est de 54,8 ± 1,8 kJ.mol−1 (section 2.6.4), l’enthalpie de ré-action est estimée à 58,76 ± 1,94 kJ.mol−1. D’après [66], l’enthalpie de réaction vaut62,5 ± 1 kJ.mol−1. La valeur expérimentale concorde donc avec les données de la litté-rature. L’utilisation de ces produits et de l’acide sulfurique a détérioré le microréacteur.Ceci est dû au long temps de résidence et à l’agressivité de ces produits chimiques envers lePDMS. A l’avenir, l’étude de ce type de réaction devra être réalisée avec une puce résistante
72 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
35 40 45 50 55 60 651.4
1.6
1.8
2
2.2
2.4
2.6
2.8
3
3.2
Débit molaire (nmol/s)
Flu
x (
mW
)
Flux = 0.0548*Qm − 0.4903
∆ H = 54.8 kJ.mol−1
Fig. 2.31 – Flux dégagé par la réaction d’estérification en fonction du débit molaire lorsd’un écoulement co-flow.
aux attaques corrosives.
2.8.2 Mesure de l’enthalpie molaire d’excès
L’enthalpie molaire d’excès est une grandeur thermodynamique difficile à mesurer. Parexemple, en calorimètrie classique, il faut préalablement déterminer les grandeurs molairespartielles.
Le mélange eau-éthanol est recommandé comme modèle pour la mesure de l’enthalpiemolaire d’excès. En effet, les produits purs sont disponibles et non toxiques, et l’enthalpiedue au mélange est peu sensible aux impuretés [67, 68]. La caractérisation de ce mélangea fait l’objet de plusieurs études. Un calorimètre isotherme permettant d’appliquer despressions de 0 à 20 MPa et une mise en température jusqu’à 200 ◦C a permis de caractériserle mélange eau-éthanol à différentes températures et pressions [67, 69]. Une autre étudeutilise un calorimètre fermé dont le volume varie de 50 à 110 cm3 [70]. Le but de cette partieest d’illustrer la facilité de la mesure. Nous avons donc choisi d’investiguer le mélange eau-éthanol dont l’enthalpie molaire d’excès dépend de la fraction volumique en éthanol àtempérature et pression ambiante. Les données sont disponibles par OTT et al. [67].
Nous déterminons l’enthalpie molaire d’excès du mélange eau-éthanol à une tempéra-ture de 25 ◦C imposée par le système de régulation et à pression atmosphérique lors del’écoulement co-courant et gouttes au sein d’un microréacteur de 20 µL de volume. La frac-tion molaire en éthanol varie de 0 à 1 par pas de 0,1. Ainsi pour chaque fraction molaire,
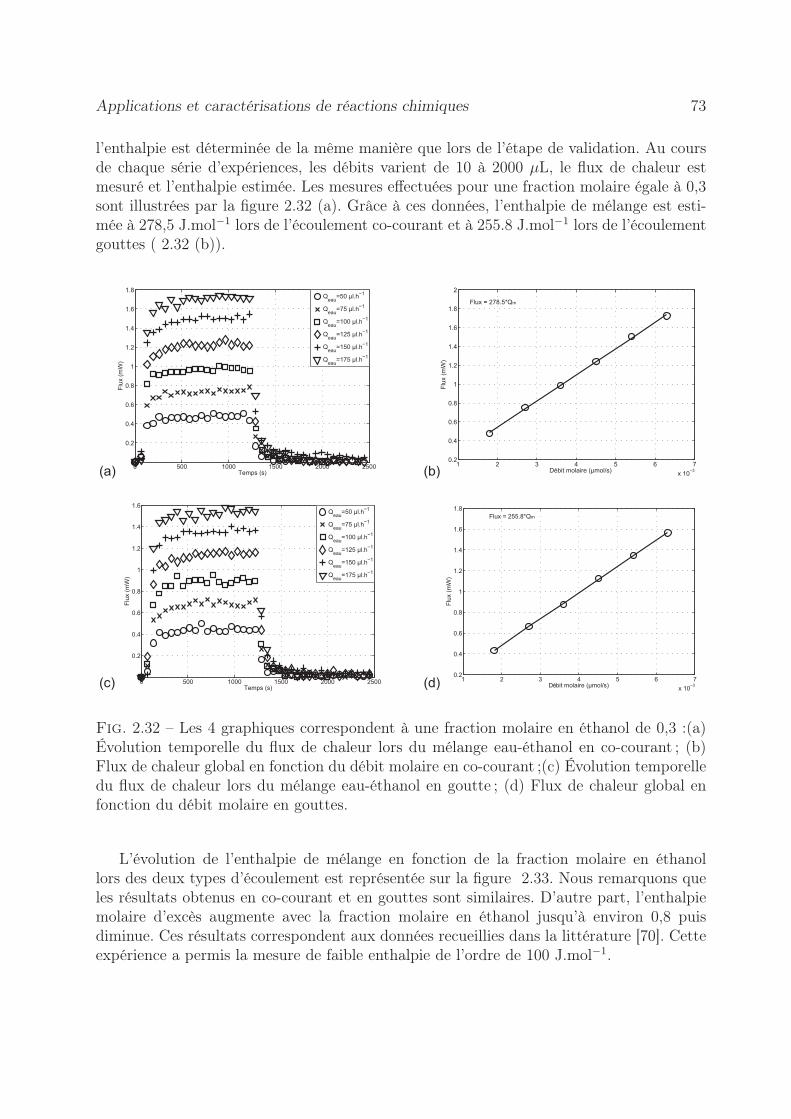
Applications et caractérisations de réactions chimiques 73
l’enthalpie est déterminée de la même manière que lors de l’étape de validation. Au coursde chaque série d’expériences, les débits varient de 10 à 2000 µL, le flux de chaleur estmesuré et l’enthalpie estimée. Les mesures effectuées pour une fraction molaire égale à 0,3sont illustrées par la figure 2.32 (a). Grâce à ces données, l’enthalpie de mélange est esti-mée à 278,5 J.mol−1 lors de l’écoulement co-courant et à 255.8 J.mol−1 lors de l’écoulementgouttes ( 2.32 (b)).
0 500 1000 1500 2000 25000
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
Temps (s)
Flu
x (
mW
)
Qeau
=50 µl.h−1
Qeau
=75 µl.h−1
Qeau
=100 µl.h−1
Qeau
=125 µl.h−1
Qeau
=150 µl.h−1
Qeau
=175 µl.h−1
1 2 3 4 5 6 7
x 10−3
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
Débit molaire (µmol/s)
Flu
x (
mW
)
Flux = 278.5*Qm
(a) (b)
1 2 3 4 5 6 7
x 10−3
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
Débit molaire (µmol/s)
Flu
x (
mW
)
Flux = 255.8*Qm
(d)0 500 1000 1500 2000 25000
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
Temps (s)
Flu
x (
mW
)
Qeau
=50 µl.h−1
Qeau
=75 µl.h−1
Qeau
=100 µl.h−1
Qeau
=125 µl.h−1
Qeau
=150 µl.h−1
Qeau
=175 µl.h−1
(c)
Fig. 2.32 – Les 4 graphiques correspondent à une fraction molaire en éthanol de 0,3 :(a)Évolution temporelle du flux de chaleur lors du mélange eau-éthanol en co-courant ; (b)Flux de chaleur global en fonction du débit molaire en co-courant ;(c) Évolution temporelledu flux de chaleur lors du mélange eau-éthanol en goutte ; (d) Flux de chaleur global enfonction du débit molaire en gouttes.
L’évolution de l’enthalpie de mélange en fonction de la fraction molaire en éthanollors des deux types d’écoulement est représentée sur la figure 2.33. Nous remarquons queles résultats obtenus en co-courant et en gouttes sont similaires. D’autre part, l’enthalpiemolaire d’excès augmente avec la fraction molaire en éthanol jusqu’à environ 0,8 puisdiminue. Ces résultats correspondent aux données recueillies dans la littérature [70]. Cetteexpérience a permis la mesure de faible enthalpie de l’ordre de 100 J.mol−1.
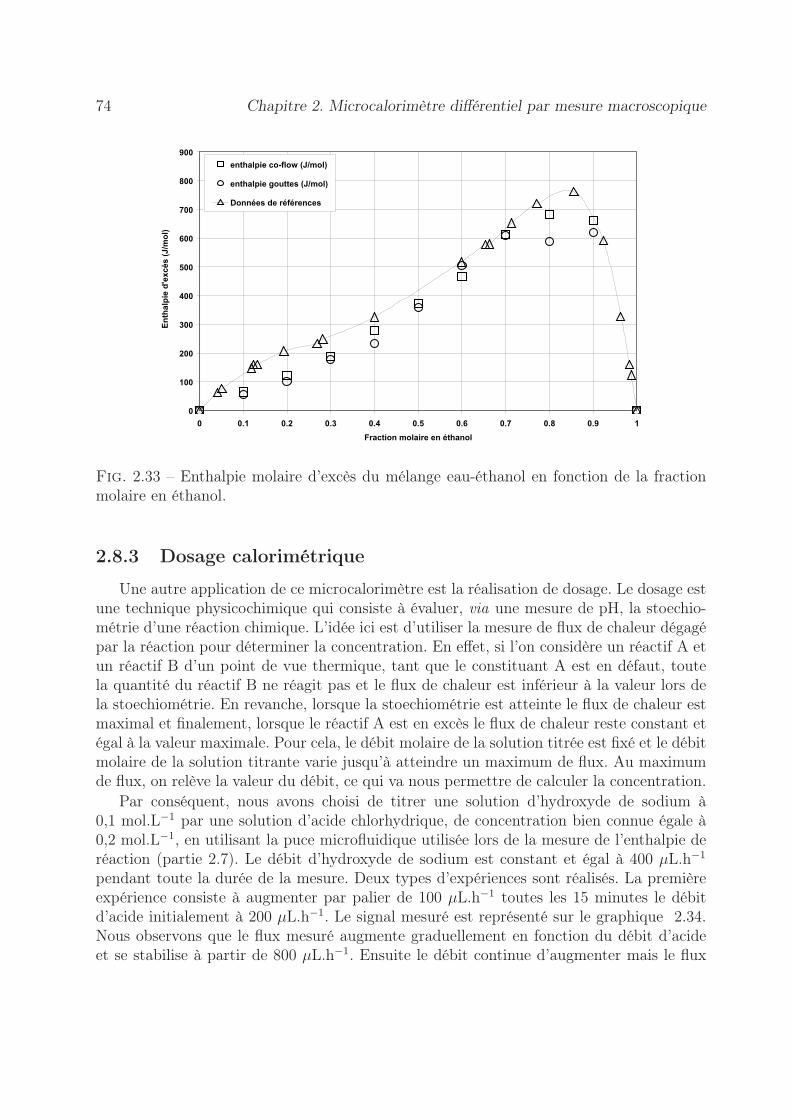
74 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
0
100
200
300
400
500
600
700
800
900
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Fraction molaire en éthanol
En
tha
lpie
d'e
xc
ès
(J
/mo
l)enthalpie co-flow (J/mol)
enthalpie gouttes (J/mol)
Données de références
Fig. 2.33 – Enthalpie molaire d’excès du mélange eau-éthanol en fonction de la fractionmolaire en éthanol.
2.8.3 Dosage calorimétrique
Une autre application de ce microcalorimètre est la réalisation de dosage. Le dosage estune technique physicochimique qui consiste à évaluer, via une mesure de pH, la stoechio-métrie d’une réaction chimique. L’idée ici est d’utiliser la mesure de flux de chaleur dégagépar la réaction pour déterminer la concentration. En effet, si l’on considère un réactif A etun réactif B d’un point de vue thermique, tant que le constituant A est en défaut, toutela quantité du réactif B ne réagit pas et le flux de chaleur est inférieur à la valeur lors dela stoechiométrie. En revanche, lorsque la stoechiométrie est atteinte le flux de chaleur estmaximal et finalement, lorsque le réactif A est en excès le flux de chaleur reste constant etégal à la valeur maximale. Pour cela, le débit molaire de la solution titrée est fixé et le débitmolaire de la solution titrante varie jusqu’à atteindre un maximum de flux. Au maximumde flux, on relève la valeur du débit, ce qui va nous permettre de calculer la concentration.
Par conséquent, nous avons choisi de titrer une solution d’hydroxyde de sodium à0,1 mol.L−1 par une solution d’acide chlorhydrique, de concentration bien connue égale à0,2 mol.L−1, en utilisant la puce microfluidique utilisée lors de la mesure de l’enthalpie deréaction (partie 2.7). Le débit d’hydroxyde de sodium est constant et égal à 400 µL.h−1
pendant toute la durée de la mesure. Deux types d’expériences sont réalisés. La premièreexpérience consiste à augmenter par palier de 100 µL.h−1 toutes les 15 minutes le débitd’acide initialement à 200 µL.h−1. Le signal mesuré est représenté sur le graphique 2.34.Nous observons que le flux mesuré augmente graduellement en fonction du débit d’acideet se stabilise à partir de 800 µL.h−1. Ensuite le débit continue d’augmenter mais le flux
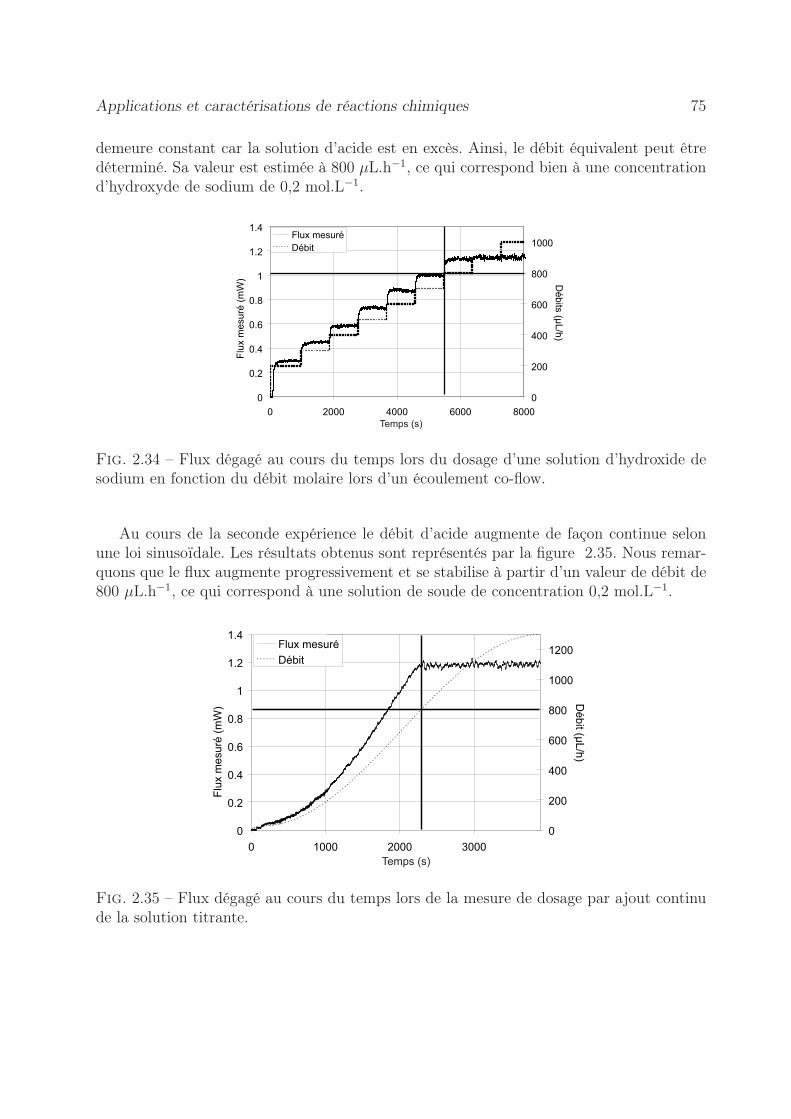
Applications et caractérisations de réactions chimiques 75
demeure constant car la solution d’acide est en excès. Ainsi, le débit équivalent peut êtredéterminé. Sa valeur est estimée à 800 µL.h−1, ce qui correspond bien à une concentrationd’hydroxyde de sodium de 0,2 mol.L−1.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 2000 4000 6000 8000
Flux m
esu
ré (
mW
)
0
200
400
600
800
1000
Débits
(µL/h
)Flux mesuré
Débit
Temps (s)
Fig. 2.34 – Flux dégagé au cours du temps lors du dosage d’une solution d’hydroxide desodium en fonction du débit molaire lors d’un écoulement co-flow.
Au cours de la seconde expérience le débit d’acide augmente de façon continue selonune loi sinusoïdale. Les résultats obtenus sont représentés par la figure 2.35. Nous remar-quons que le flux augmente progressivement et se stabilise à partir d’un valeur de débit de800 µL.h−1, ce qui correspond à une solution de soude de concentration 0,2 mol.L−1.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 1000 2000 3000
Flu
x m
esuré
(m
W)
0
200
400
600
800
1000
1200
Débit (µ
L/h
)
Flux mesuré
Débit
Temps (s)
Fig. 2.35 – Flux dégagé au cours du temps lors de la mesure de dosage par ajout continude la solution titrante.

76 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
2.8.4 Estimation du coefficient de diffusion
Les écoulements microfluidiques étant laminaires (nombre de Reynolds < à 1) le mé-lange des espèces s’opère par diffusion lors de l’écoulement co-courant. Au cours du cha-pitre 1.2.2, nous avons vu que le temps de mélange dépend du coefficient de diffusion del’espèce et de la géomètrie du microcanal : ydiff =
√2Dt. La détermination de la ci-
nétique de mélange ou de réaction nécessite des mesures de flux où le taux de conversionest inférieur à 1. Durant cette expérience, nous allons étudier le mélange lors de la réac-tion entre l’acide chlorhydrique et l’hydroxyde de sodium. Le temps de mélange de cetteréaction chimique étant très supérieur au temps de réaction, celle-ci peut être considéréecomme instantannée et la cinétique est limitée par la diffusion. Nous avons montré, aucours de la partie 2.7, que le taux de conversion vaut 1 dans la puce microfluidique enforme de serpentin. En effet, le temps de mélange entre l’acide chlorhydrique et l’hydroxidede sodium est de 0,86 s dans ce microréacteur (DH+=9,3.10−9 m2.s−1 [?]). Compte tenudu volume du microréacteur (20 µL), pour que le taux de conversion soit inférieur à 1 ausein du microréacteur, il faudrait injecter des débits de l’ordre de 80 mL.h−1, ce qui n’estpas raisonnable car le dispositif risquerait de ne pas résister aux gradients de pressionsengendrés. Par conséquent, une nouvelle puce de volume égal à 16 nL est réalisée.
2.8.4.1 Conception d’un microréacteur pour la cinétique
Nous optons pour une géométrie simple en forme de T permettant uniquement un écou-lement de type co-courant. Le microcanal mesure 100 µm de large et 80 µm de haut. Dansces conditions, le temps de mélange, entre l’acide chlorhydrique et l’hydroxide de sodium,est de 0,14 s. Afin d’utiliser une gamme de débits raisonnable, la longueur du microréacteurest prise égale à 2 mm pour que la réaction soit totale à un débit de 400 µL.h−1. La pucemicrofluidique,représentée par le schéma 2.36, est réalisée à partir d’un substrat de siliciumet de PDMS.
><
><
2 mm
100 µm
Injection Acide
<Injection Base
<
épaisseur du canal : 80 µm
Fig. 2.36 – Schéma de la puce microfluidique conçue pour la mesure de cinétique.

Applications et caractérisations de réactions chimiques 77
2.8.4.2 Résultats sur la cinétique de mélange
Les débits injectés d’acide et de base sont égaux et varient de 75 µL.h−1 à 1000 µL.h−1.Les expériences sont réalisées à différentes températures 5 et 25 ◦C. Le flux de chaleurrelatif est défini par le rapport entre le flux de chaleur mesuré et le flux correspond à untaux de conversion de 1. Il est représenté sur la figure 2.37 pour chaque débit.
0 200 400 600 800 1000 1200 1400 1600 1800−0.2
0
0.2
0.4
0.6
0.8
1
1.2
Temps (s)
Flux (mW)
QHCl=QNaOH=100 µl/h
QHCl=QNaOH=150 µl/h
QHCl=QNaOH=200 µl/h
QHCl=QNaOH=400 µl/h
QHCl=QNaOH=600 µl/h
QHCl=QNaOH=800 µl/h
QHCl=QNaOH=1000 µl/h
QHCl=QNaOH=300 µl/h
Fig. 2.37 – Évolution temporelle du flux relatif dégagé par la réaction acide base lors d’unécoulement co-courant à une température de 5 ◦C.
Nous observons sur cette figure que le flux de chaleur relatif vaut 1 pour les débitstotaux inférieurs à 400 µL.h−1, ce qui signifie que la réaction est totale pour ces temps derésidence.
D’autre part, l’enthalpie de réaction égale à 54,38 kJ.mol−1 a été déterminée dansla partie 2.7.4 en écoulement co-courant. En utilisant ce résultat et la relation 2.1, letaux de conversion peut être représenté en fonction du débit molaire sur la figure 2.38.Nous remarquons que le taux de conversion augmente avec le temps de résidence dans lemicroréacteur. De plus, un taux de conversion de 95 % atteint pour un temps de 0,15 scorrespond à un coefficient de diffusion de 8,3.10−9 m2.s−1. Ce résultat est en accord avecla littérature [?] avec un écart de 11 %.
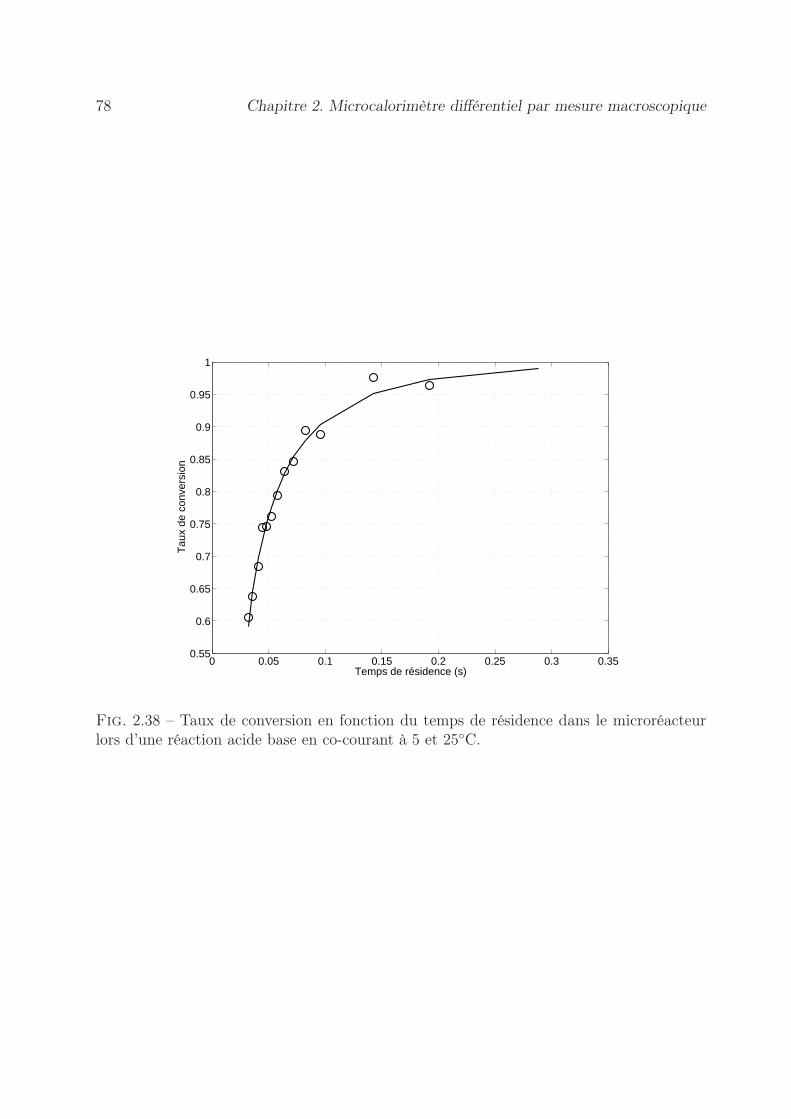
78 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
0 0.05 0.1 0.15 0.2 0.25 0.3 0.350.55
0.6
0.65
0.7
0.75
0.8
0.85
0.9
0.95
1
Temps de résidence (s)
Tau
x de
con
vers
ion
Fig. 2.38 – Taux de conversion en fonction du temps de résidence dans le microréacteurlors d’une réaction acide base en co-courant à 5 et 25◦C.

2.9. Conclusion 79
2.9 Conclusion
Un microcalorimètre isotherme continu a été développé et validé par la caractérisationd’une réaction acide base en écoulement co-courant et gouttes. Ce dispositif mesure leflux de chaleur global généré par une réaction chimique se produisant au sein d’une pucemicrofluidique. L’utilisation de la technologie microfluidique est un atout important pourcet appareil car cela permet la mesure en écoulement continu, la mise en jeu de faiblesvolumes réactionnels, le contrôle de l’écoulement et du transfert thermique. Ainsi, desréactions très exothermiques peuvent être étudiées en toute sécurité.
Le modèle thermique développé permet de dimensionner la puce afin de prévoir lecomportement thermique du réacteur. Une méthode d’étalonnage a également été mise aupoint. Il a été montré que le coefficient α = 3,95 W.V−1 obtenu à 25 ◦C permet une mesureabsolue du flux de chaleur dégagé par la réaction. D’autre part, une étude exhaustive sur ladétermination de la précision de la mesure de l’enthalpie a été réalisée et montre que cettevaleur est estimée avec une précision de 3,35 %. La caractérisation d’une réaction acide basevalide cet appareil. En effet, l’enthalpie de réaction déterminée à∆H = 54,38 ± 1,8 kJ.mol−1
lors d’un écoulement co-courant et à ∆H = 55,74 ± 1,84 kJ.mol−1 lors d’un écoulementgouttes, est en trés bon accord avec les données de la littérature (∆ Hlitterature = 55,8 kJ.mol−1
[?]).
Dans une dermière partie, les nombreuses possibilités offertes par ce dispositif ont étéillustrées :
– la mesure de l’enthalpie réaction par l’étude d’une réaction d’estérification entrel’anhydride propionique et le 2-butanol, ainsi que de la réaction acide base (HCl etNaOH),
– l’enthalpie d’excès du mélange entre l’eau et l’éthanol a été mesurée pour différentescompositions,
– la détermination de la concentration par dosage calorimétrique,– dynamique de mélange : le coefficient de diffusion des ions H+ a été évalué par l’étude
du temps de mélange de la réaction acide base.Au cours de ces mesures le taux de conversion est inférieur à 1, ce qui met en évidence ladynamique de mélange. Dans le cas où le temps de réaction serait supérieur au temps demélange et le temps de mélange très court, la cinétique de réaction pourrait être évaluée.
Finalement, l’ensemble de ce travail permet de décrire les performances du microcalo-rimètre :
– 1 : utilisation de faibles volumes réactionnels de l’ordre du nL au µL,– 2 : large gamme de temps de résidence : de la seconde à l’heure,– 3 : large gamme de débit : de l’ordre de 20 µL.h−1 à 1 mL.h−1,– 4 : large gamme de température : de 5 à 90 ◦C,– 5 : grande sensibilité de la mesure : 10 µW,– 6 : large gamme d’enthalpie mesurable : de 100 J.mol−1 au MJ.mol−1.

80 Chapitre 2. Microcalorimètre différentiel par mesure macroscopique
Cette méthode de mesure globale par contact ne remplace pas une étude plus appro-fondie relative à la cinétique de réaction et à des mesures locales de température ou de fluxdans diverses conditions (co-courant, gouttes, canal coudés ... ). Elle peut même être asso-ciée, comme mesure de référence, à des mesures thermographiques qui vont être étudiéespar la suite.

Chapitre 3
Approche locale pour l’estimation de
cartographie de propriétés
thermocinétiques par thermographie
InfraRouge en puce microfluidique
Résumé
Dans ce chapitre, des méthodes exploratoires de mesures de propriétés thermochi-miques en microfluidique par thermographie IR vont être présentées. Ces méthodesconsistent d’une part à développer une expérimentation de type microscopie ther-mique pour la mesure de champ de température dans ces systèmes hétérogènes depetites tailles. Selon la configuration de l’écoulement, co-courant ou gouttes, deuxtypes d’outils ont été développés. D’autre part, à partir de cette grande quantité demesures (≈ 80000 température en une image) nous développons une méthodologiepour l’analyse thermique. Ce type de systèmes hétérogènes avec des termes de trans-port, de réaction chimique et de diffusion nécessitent une modélisation adaptée et uneestimation précise des différents paramètres tels que : le nombre de Péclet (pour letransport), le nombre de Fourier (pour la diffusion) et le terme source (relatif à laréaction chimique). Pour cela, des méthodes modèles basées sur des estimations parapproche locale à l’aide de méthodes inverses à un paramètre seront présentées. Lechoix de telles méthodes est rendu possible par le développement de méthodes de ca-libration particulières qui nous permettent d’isoler par la physique un seul paramètredu système global complexe à traiter. Finalement, ce type d’approche va permettre lamise au point de méthodes de caractérisation de réactions chimiques par l’estimationde cartographies de propriétés thermophysiques et thermochimiques de réactions.
81

82 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
3.1 Introduction
Au cours du chapitre précédent, un microcalorimètre isotherme a été développé. Cetappareil très performant, d’une grande sensibilité et aux multiples fonctionnalités permetune mesure globale et quantitative du flux de chaleur dégagé par la réaction chimique réa-lisée au sein d’un microréacteur fluidique. L’idée de cette nouvelle approche est de pouvoirmesurer des températures locales en utilisant des systèmes de mesures tels que les camérasIR. Dans un premier temps, nous présenterons les différents dispositifs développés pour cetype d’expériences. Ensuite, nous détaillerons les différentes méthodes de calibration ex-périmentales qui vont permettre une estimation quantitative de propriétés à partir d’unemesure qualitative de champ de température. Pour cela un certain nombre de méthodesd’estmations seront nécessaires pour caractériser au mieux ces systèmes microfluidiqueshétérogènes.
Tout d’abord, nous présentons le dispositif, ses caractéristiques et la procédure expéri-mentale à suivre lors des essais. La partie suivante concerne la modélisation thermique dumicroréacteur.
La première partie est dédiée à la mesure de cinétique chimique au sein de microcannauxlors d’un écoulement co-courant. À partir de cette configuration, une méthode de mesuredes champs du nombre de Péclet sera détaillée. Puis, grâce à la connaissance de cettegrandeur, les cinétiques pourront être estimées. Cette méthode sera validée sur une réactionstandard. Enfin, nos premiers travaux sur la mesure de propriétés en écoulement gouttessera décrite. Nous proposons une première méthode originale de calibration du système avecune estimation locale de propriétés thermiques au sein d’une gouttes. Enfin, les perspectivesainsi que les difficultés seront discutées.

3.2. Caractéristiques du dispositif 83
3.2 Caractéristiques du dispositif
Le dispositif expérimental (figure 3.1 et 3.2) permet la mesure de température d’uneréaction chimique se produisant au sein d’un microréacteur continu à l’aide d’une caméraInfraRouge.
Qentrée Qsortie
+
+
-
-
grad T
caméra IR
lame de verre
PDMS élément Peltier
substrat de silicium
masse
XY
Z
Fig. 3.1 – Représentation schématique du microcalorimètre Infrarouge
La conception de ce microréacteur est différente de celle utilisée dans le microcalorimètredu chapitre précédent de part la nature du substrat. Les réactifs sont injectés à des débitsvariant de 250 à 1000 µL.h−1 via un pousse seringue Nemesys décrit dans la partie 2.2.2.Chaque élément de ce dispositif est décrit par la suite.
Caméra IR
Puce microfluidique
Substrat silicium
Eléments Peltier
Pousse seringues
Bloc laiton
<
<<
<
<
<
Alimentation en réactifs<<
Fig. 3.2 – Photographie du dispositif expérimental

84 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
3.2.1 Caméra IR
Le principe d’une caméra IR est de mesurer le flux de rayonnements IR dont la longueurd’onde dépend de la température du corps. La caméra utilisée dans ce dispositif est unecaméra Infrarouge Cedip Jade MWIR J550 (Medium Wavelenght InfraRed) équipée d’unobjectif MWIR F/2 de 25 mm. Compte tenu de la distance entre la caméra et la surface dela puce, la résolution spatiale apparente est d’environ 200 µm. Cette caméra est sensibleaux rayonnements sur la gamme de longueur d’onde allant de 2 à 5.6 µm et permet unefréquence d’acquisition de 1 kHz. Le système de détection de la caméra est composé deplusieurs rang de détecteurs In-Sb (Antimoine d’indium) ce qui représente une matrice de240 x 320 pixels soit 76800 points de mesures.
3.2.2 Element Peltier
Les éléments Peltier sont placés de part et d’autre de la puce microfluidique (figure 3.1)pour imposer, par pompage de chaleur, un gradient de température contrôlé. Le fonctionne-ment des éléments Peltier est basé sur l’effet inverse de l’effet Seebeck utilisé précédemment.Les éléments Peltier sont placés face chauffante d’un côté de la puce et face refroidie del’autre côté de la puce. Une de leur face est en contact avec une masse en laiton, matériaucapacitif, afin de travailler dans des conditions de température imposée. Une alimentationAgilent E3631A est utilisée pour appliquer le gradient de température.
3.2.3 Puce microfluidique
Le microréacteur est constitué de PDMS et d’un substrat de verre. La fabrication com-porte deux étapes essentielles : la réalisation du moule positif représentant les microcanauxet le collage par ozone entre le PDMS gravé et le substrat (partie 1.2.1). L’observation desmicrocanaux du côté PDMS n’est pas possible car une épaisseur d’environ 1 cm est néces-saire pour une bonne isolation thermique. Ainsi, le substrat de la puce sera orienté vers lacaméra IR. L’utilisation d’un substrat de silicium n’est pas envisageable car la propriétéde conduction de ce matériau ne permet pas la visualisation de variation de température.Nous avons choisi de concevoir les microréacteurs avec un substrat de verre, matériaucouramment utilisé pour la réalisation de puces. Toutefois, le verre étant transparent auxrayons IR dans la gamme de détection de la caméra (longueur d’onde de 2 à 5,6 µm), sasurface est peinte en noir (figure 3.3).
Le substrat utilisé est un verre Corning 7740 et mesure 5 cm de diamètre et 170 µmd’épaisseur. La géométrie du microcanal (figure 3.3) est relativement simple puisqu’il s’agitd’un canal droit. Les deux types d’écoulement co-courant et gouttes sont envisageables.Trois microcanaux de largeur différentes (100, 250 et 500 µm) sont réalisés au sein de lamême puce microfluidique. Les dimensions caractéristiques du réacteur sont les suivantes :500 µm de large, 100 µm de haut et 4 cm de long, ce qui correspond à un volume de 2 µL.

3.3. Procédure expérimentale 85
0
10
20
30
40
50
60
70
80
90
100
1 2 3 4 5 6
Longueur d'onde (µm)
Tra
nsm
issio
n (
%)
zone de détection de la caméra
Fig. 3.3 – Courbe de transmission des rayons de longueurs d’onde variant de 1 à 6 µm àtravers un substrat de verre (pyrex 7740) d’une épaisseur de 500 µm
Substrat de verre>
PDMS
Entrée fluide < Sortie fluide
Entrée phase
porteuse
<
<
>>
Fig. 3.4 – Photographie du microréacteur à substrat de verre utilisé dans le cadre dumicroréacteur InfraRouge
3.3 Procédure expérimentale
3.3.1 Démarche générale
Le principe de la mesure consiste à introduire les réactifs au sein du microréacteur àl’aide du pousse-seringue. Lorsque les produits seront mis en contact, la réaction chimiquedémarre et génère un flux de chaleur. Dès lors, un film est enregistré au moyen de lacaméra IR à une fréquence de 25 Hz. Le champ de température est moyenné sur 1000ou 10000 images pour diminuer l’influence du bruit de mesure. Ainsi, une seule imagepeut être utilisée pour définir le profil de température de tout le microréacteur à l’étatstationnaire. Dans cette configuration, la réaction chimique peut être assimilée à un termesource de transport réparti spatialement dans le canal. Il est donc important de développer

86 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
une démarche suceptible de déterminer présicémment le terme de transport seul. Pour cela,une méthode expérimentale d’étalonnage est réalisée au préalable.
3.3.2 Calibration du nombre de Péclet
L’étape d’étalonnage de l’écoulement est indispensable pour effectuer une mesure quan-titative de cinétiques chimiques. Le principe est d’imposer un flux de chaleur calibré surla puce pour obtenir des champs de température parallèles entre eux. Puis un écoulementde fluide sans réaction chimique est généré, ce qui provoque une déformation des champsde température. De ces champs de température le nombre de Péclet peut être estimé. Cesinformations permettront ensuite d’estimer le terme source d’une réaction chimique. Nousproposons une méthode consistant à superposer un régime de gradient de température sta-tionnaire purement conductif à un régime de transport convectif local. La différence de cesdeux régimes revient à créer artificiellement un terme source calibré dans le canal. Deuxméthodes différentes sont mises en place : la première utilise un terme source local pareffet Joule tandis que la seconde repose sur l’imposition d’un gradient de température parl’utilisation d’éléments Peltier.
3.3.3 Acquisition et estimation
L’acquisition des images IR ou des films IR se fait par l’intermédiaire du logicielALTAIRTM développé par le fournisseur de caméra. Ce logiciel est assez complet, il per-met l’acquisition d’image et de film mais aussi le traitement de ces données. En effet, ilest possible de soustraire ou sommer des images, de réaliser des moyennes de températuressur des surfaces de formes diverses, ou encore de représenter l’évolution temporelle de latempérature en un point donné. Toutefois, dans un souci de liberté et pour maîtriser toutesles opérations de traitement, nous avons développé des programmes dans l’environnementMathlabTM permettant une mesure du profil de vitesse dans le canal par l’estimation dunombre de Péclet local. Lors de l’étape d’étalonnage, le nombre de Péclet local est estimé.Cela permet par la suite de déterminer le terme source d’une réaction chimique le longdu microcanal par l’utilisation d’une méthode inverse. Toutefois cette valeur étant quali-tative, il faudra faire une comparaison avec les résultats obtenus par le microcalorimètredu chapitre 2 pour obtenir une information quantitative.
3.4 Présentation du système thermique
Les transferts thermiques au sein du microréacteur sont des phénomènes tridimension-nels que nous ne pouvons résoudre sans établir certaines hypothèses. L’écoulement étantréalisé de manière continue, cela permet de se placer dans le cas de régime thermique sta-tionnaire. D’autre part, nous précisons que la mesure correspond à une température desurface. Le microréacteur est composé d’un substrat de verre et d’une matrice en PDMScomme l’illustre la figure 3.5.

Présentation du système thermique 87
Substrat de verre
PDMS
Microcanal
<
<
<
<
>
>Qentrée
QsortieYX
Z
>
>
1 cm> >170 µm
Fig. 3.5 – Schéma du microréacteur en 3D
Dès lors, une première comparaison entre la résistance thermique de ces deux milieuxva permettre de déterminer la répartition de flux de chaleur :
RPDMS = ePDMS
λPDMS= 1.10−1m2.K.W−1
Rverre = everre
λverre= 1, 42.10−4m2.K.W−1
(3.1)
Nous constatons que la résistance thermique du verre est nettement plus faible que celledu PDMS. De plus, les dimensions du microcanal étant petites, l’essentiel des échanges ther-miques se fait au travers de la plaque de verre. Nous considérons l’équilibre thermique localentre la plaque et le fluide dans le canal, ainsi nous faisons l’hypothèse que la distributionde chaleur a lieu uniquement dans deux dimensions selon les axes x et y.
Nous supposons que le champ de température observé par la caméra correspond à lasurface extérieure de la plaque. L’hypothèse principale consiste à supposer que les iso-thermes sont perpendiculaires au grand axe de la plaque (selon l’axe z) ou que le transfertdiffusif se fait uniquement dans le plan de la plaque (figure 3.6).
Silicium
Substrat verre
Microcanal
Laiton
Peltier
PDMS
Fig. 3.6 – Schéma des isothermes établies dans le microréacteur.
De plus, nous émettons l’hypothèse que la température du fluide dans le canal collé à laplaque est à l’équilibre avec la plaque. Ainsi, le champ de température observé peut obéirau modèle d’ailette suivant :

88 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
∂2T
∂x2+
∂2T
∂y2+ Φ(x, y) − H(T (x, y) − T∞) =
v(x, y)
a
∂T
∂x(3.2)
avec
Φ = φsource
λS
H = hp
λS
v(x,y)a
= ρCpv(x,y)
λ
(3.3)
avec v(x,y)a
∂T∂x
le terme convectif apparent, ∂2T∂x2 + ∂2T
∂y2 le terme diffusif, H les pertes thermiques(m−2) et Φ(x, y) le terme source thermique dans le canal (K.m−2), S la surface d’échange(m2), p le périmètre (m), v la vitesse dans le canal (m.s−1).
Lors des expériences, nous obtenons des cartographies de températures via la caméra IR,ce qui nous incite à utiliser une approche locale de type nodale. Ainsi, une approximationen différences finies de l’équation 3.2 pour un maillage de taille ∆x (taille apparente d’unpixel carré), donne, au noeud de coordonnées (i, j) :
(Ti+1,j + Ti−1,j + Ti,j+1 + Ti,j−1 − 4Ti,j)1
∆x2+ Hi,j(Ti,j−T∞) + Φi,j =
vi,j
a(Ti+1,j − Ti,j)
1
∆x(3.4)
En faisant apparaître un nombre de Péclet local l’équation s’écrit :
(Ti+1,j + Ti−1,j + Ti,j+1 + Ti,j−1 − 4Ti,j) + Hi,j∆x2(Ti,j−T∞) + Φi,j = Pei,j(Ti+1,j − Ti,j)(3.5)
où Pei,j =vi,j∆x
a
Nous remarquons trois paramètres inconnus : le terme de pertes thermiques, le terme detransport et le terme source. Ces paramètres doivent être estimés pour résoudre le systèmecomplet.
3.5 Méthode d’identification par approche nodale
3.5.1 Influence des pertes thermiques locales
Nous comparons la contribution des différents termes sur la température en prenant unpas spatial ∆x de 200 µm, ce qui correspond à la taille d’un pixel de la caméra, une vitessemoyenne de 1 mm.s−1 et les propriétés thermiques de l’eau. Ainsi le nombre de Péclet etle terme d’échange réduit sont estimés :
avec
Pe = v∆xa
= ∆xρCpv
λ= 1
H∆x2 = ∆x2 hp
λS= 3.10−3 (3.6)

Méthode d’identification par approche nodale 89
Il apparaît clairement que le terme de pertes locales égal à 3.10−3 est largement né-gligeable devant les termes liées au transport : Pe = 2.103 et à la diffusion (ici 4 pour lecalcul du Laplacien). Avec cette hypothèse l’équation aux différences locales finies peut seréécrire :
(Ti+1, j + Ti−1, j + Ti, j+1 + Ti, j−1 − 4Ti, j) + Φi, j = Pei, j(Ti+1, j − Ti, j) (3.7)
ou encore :△(Ti,j) + φi,j = Pei,jδ(Ti,j) (3.8)
avec :
δ(Ti,j) = Ti+1,j − Ti,j
△(Ti,j) = Ti+1,j + Ti−1,j + Ti,j+1 + Ti,j−1 − 4Ti,j
(3.9)
et :
Pei,j =vi,j ∆X
a(3.10)
vi,j étant la vitesse locale apparente (m.s−1) et a la diffusivité thermique (m2.s−1). En effet,le nombre de Péclet compare les phénomènes inertiels aux phénomènes diffusifs.
3.5.2 Estimation du champ de Péclet
D’après l’équation bilan (3.8), deux termes restent à déterminer : le terme source et leterme de transport. Pour estimer le terme de transport nous proposons une méthode decalibration où seule la thermique agit. Pour cela, un flux électrique calibré est imposé ausystème sous aucun écoulement. Dans ces conditions, l’équation se ramène à :
△(T 0i,j) + φi,j = 0 (3.11)
Ensuite, un écoulement sans réaction chimique est superposé à ce champ initial. Celaconduit au système suivant :
△(Ti,j) + φi,j = Pei,jδ(Ti,j) (3.12)
À partir de ce système de deux équations à deux inconnues, le nombre de Péclet peutêtre obtenu selon l’expression (3.13).
△(Ti,j − T 0i,j) = Pei,jδ(Ti,j) (3.13)
Sachant que la température est l’observable de notre méthode expérimentale, le problèmese ramène à un système d’équation à une inconnue. Ce système peut tout à fait être résolupar une inversion à un paramètre lorsque le Laplacien et la dérivée sont différents de 0 etpeu bruité.
Pei,j =
∑∆(Ti,j − T 0
i,j)δTi,j∑δT 2
i,j
(3.14)

90 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
Ainsi, pour éviter la résolution d’un système mal conditionné au sens des moindrescarrés, nous préconisons une méthode consistant à analyser uniquement les zones de l’imageoù le Laplacien et la dérivée sont corrélés telles que le coefficient de corrélation (calculé surune zone rectangulaire réduite à 4 pixels) vérifie :
ρi,j =
∑+2k=−2(△(Ti,j+k − T 0
i,j+k)(δ(Ti,j+k)))√∑+2k=−2(△(Ti,j+k − T 0
i,j+k))2√∑
k(δ(Ti,j+k))2
≈ 1 (3.15)
Dans ces conditions, l’estimation classique par la méthode des moindres carrés liné-raires (3.14) va être réalisée uniquement dans les zones ou la relation linéaire entre lelaplacien et la dévivée est vérifiée. Cela revient à estimer le nombre de Péclet selon laformule :
Pei,j = ρi,j
√∑+2k=−2(△(Ti,j − T 0
i,j)2)
√∑+2k=−2(δ(Ti,j − T 0
i,j))2
(3.16)
3.5.3 Estimation de champ de terme source
Considérant le domaine de température Ti,j − T 0i,j et l’équation (3.13), la relation de-
vient :△(Ti,j − T 0
i,j) = Pei,jδ(Ti,j − T 0i,j) + φ⋆
i,j (3.17)
Où le terme source φ⋆ vaut Pei,jδ(T0i,j) ce qui correspond au terme source produit. Le
nombre de Péclet ayant été déterminé prédédemment, le terme source peut être évalué.Le nombre de Péclet ainsi que le terme source sont estimés expérimentalement ou par
simulation (nombre de Péclet uniquement). Plusieurs expériences sont nécessaires : il fautconsidérer les champs de températures sans écoulement de fluides puis avec différentesvitesses.
3.6 Validation numérique en écoulement co-courant
Deux méthodes d’étalonnages sont envisageables. La première consiste à appliquer lo-calement un terme source local calibré à l’aide d’une résistance chauffante [14], la secondeimpose un gradient de température sur l’ensemble de la puce microfluidique. Dans le but demettre en évidence, les perfomance de ces deux méthodes des simulations ont été effectuéespar des volumes finis puis traitées à l’aide de MatlabTM dans le but d’étudier l’influencedu terme source sur l’estimation du nombre de Péclet dans le canal.
3.6.1 Méthode du flux local
3.6.1.1 Estimation de cartographie de Péclet
Ici, l’idée est de simuler le cas traité par Pradère et al. [14]. Selon la méthode que nousavons décrite, il est important d’introduire un terme source bien calibré dans le système
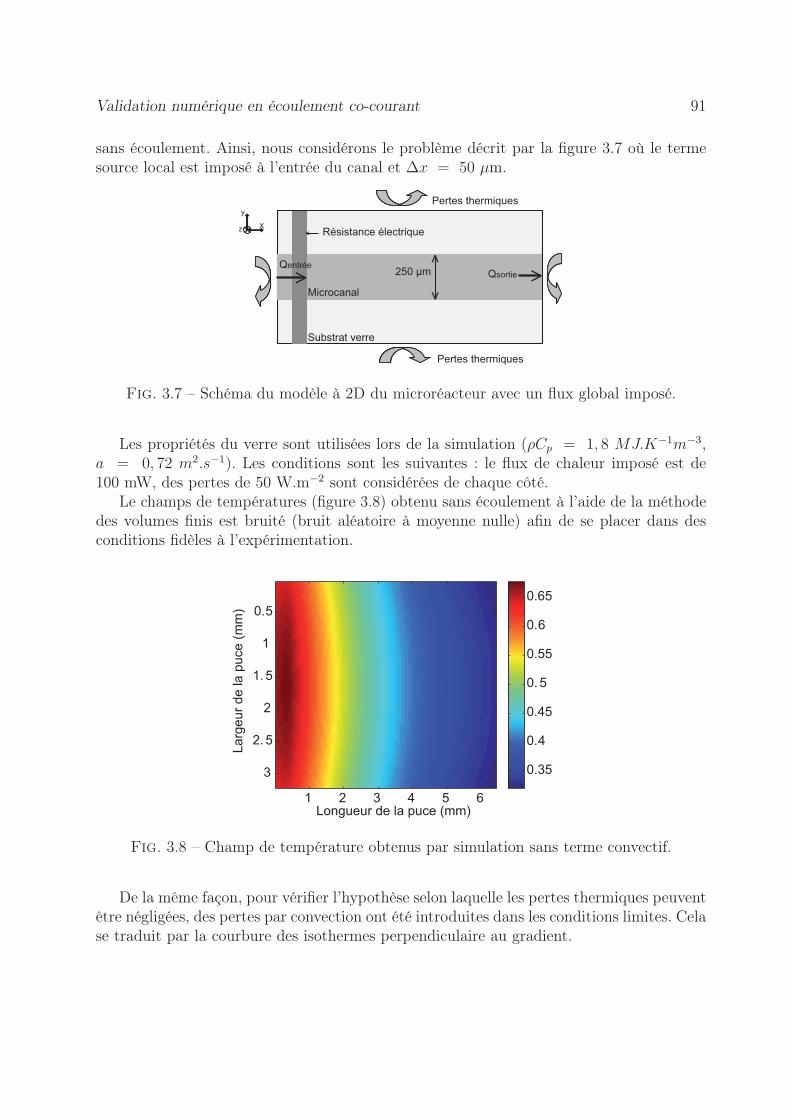
Validation numérique en écoulement co-courant 91
sans écoulement. Ainsi, nous considérons le problème décrit par la figure 3.7 où le termesource local est imposé à l’entrée du canal et ∆x = 50 µm.
< <QentréeQsortie
Microcanal
Substrat verre
Résistance électrique<Xz
y
Pertes thermiques
Pertes thermiques<
<
250 µm
Fig. 3.7 – Schéma du modèle à 2D du microréacteur avec un flux global imposé.
Les propriétés du verre sont utilisées lors de la simulation (ρCp = 1, 8 MJ.K−1m−3,a = 0, 72 m2.s−1). Les conditions sont les suivantes : le flux de chaleur imposé est de100 mW, des pertes de 50 W.m−2 sont considérées de chaque côté.
Le champs de températures (figure 3.8) obtenu sans écoulement à l’aide de la méthodedes volumes finis est bruité (bruit aléatoire à moyenne nulle) afin de se placer dans desconditions fidèles à l’expérimentation.
Longueur de la puce (mm)
La
rge
ur
de
la
pu
ce
(m
m)
1 2 3 4 5 6
0.5
1
1. 5
2
2. 5
3 0.35
0.4
0.45
0. 5
0.55
0.6
0.65
Fig. 3.8 – Champ de température obtenus par simulation sans terme convectif.
De la même façon, pour vérifier l’hypothèse selon laquelle les pertes thermiques peuventêtre négligées, des pertes par convection ont été introduites dans les conditions limites. Celase traduit par la courbure des isothermes perpendiculaire au gradient.
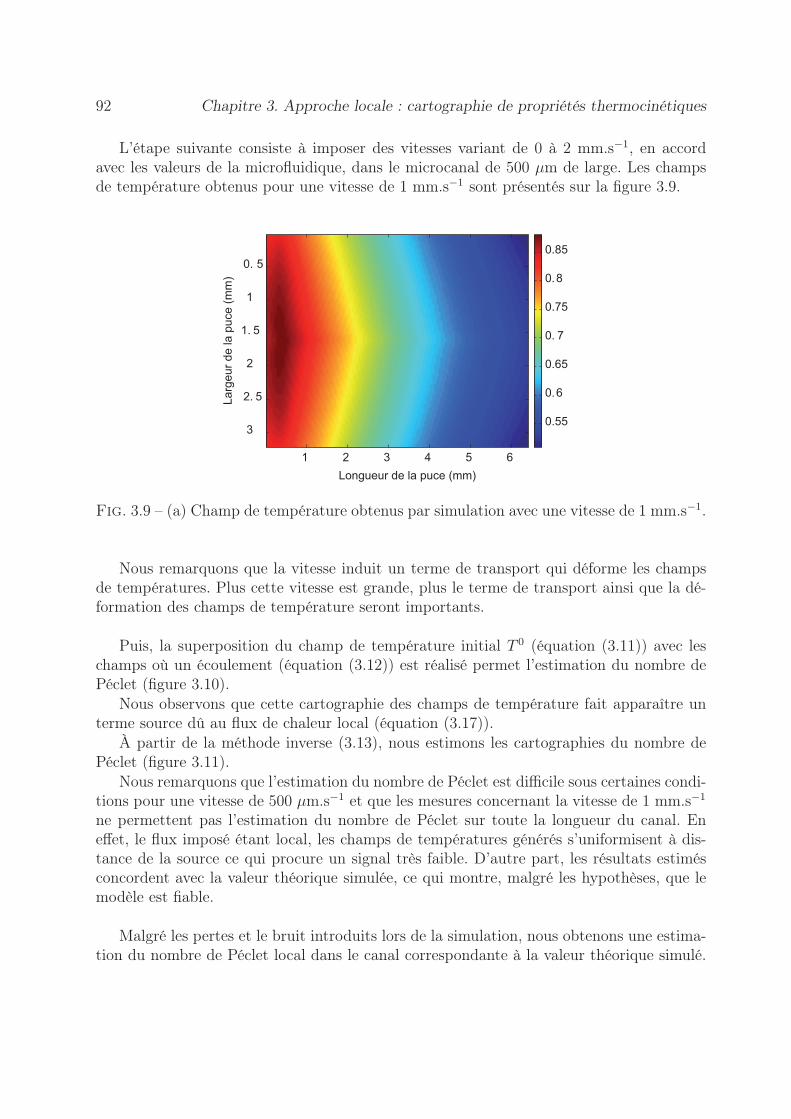
92 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
L’étape suivante consiste à imposer des vitesses variant de 0 à 2 mm.s−1, en accordavec les valeurs de la microfluidique, dans le microcanal de 500 µm de large. Les champsde température obtenus pour une vitesse de 1 mm.s−1 sont présentés sur la figure 3.9.
Longueur de la puce (mm)
Larg
eur
de la p
uce (
mm
)
1 2 3 4 5 6
0. 5
1
1. 5
2
2. 5
30.55
0.6
0.65
0. 7
0.75
0.8
0.85
Fig. 3.9 – (a) Champ de température obtenus par simulation avec une vitesse de 1 mm.s−1.
Nous remarquons que la vitesse induit un terme de transport qui déforme les champsde températures. Plus cette vitesse est grande, plus le terme de transport ainsi que la dé-formation des champs de température seront importants.
Puis, la superposition du champ de température initial T 0 (équation (3.11)) avec leschamps où un écoulement (équation (3.12)) est réalisé permet l’estimation du nombre dePéclet (figure 3.10).
Nous observons que cette cartographie des champs de température fait apparaître unterme source dû au flux de chaleur local (équation (3.17)).
À partir de la méthode inverse (3.13), nous estimons les cartographies du nombre dePéclet (figure 3.11).
Nous remarquons que l’estimation du nombre de Péclet est difficile sous certaines condi-tions pour une vitesse de 500 µm.s−1 et que les mesures concernant la vitesse de 1 mm.s−1
ne permettent pas l’estimation du nombre de Péclet sur toute la longueur du canal. Eneffet, le flux imposé étant local, les champs de températures générés s’uniformisent à dis-tance de la source ce qui procure un signal très faible. D’autre part, les résultats estimésconcordent avec la valeur théorique simulée, ce qui montre, malgré les hypothèses, que lemodèle est fiable.
Malgré les pertes et le bruit introduits lors de la simulation, nous obtenons une estima-tion du nombre de Péclet local dans le canal correspondante à la valeur théorique simulé.
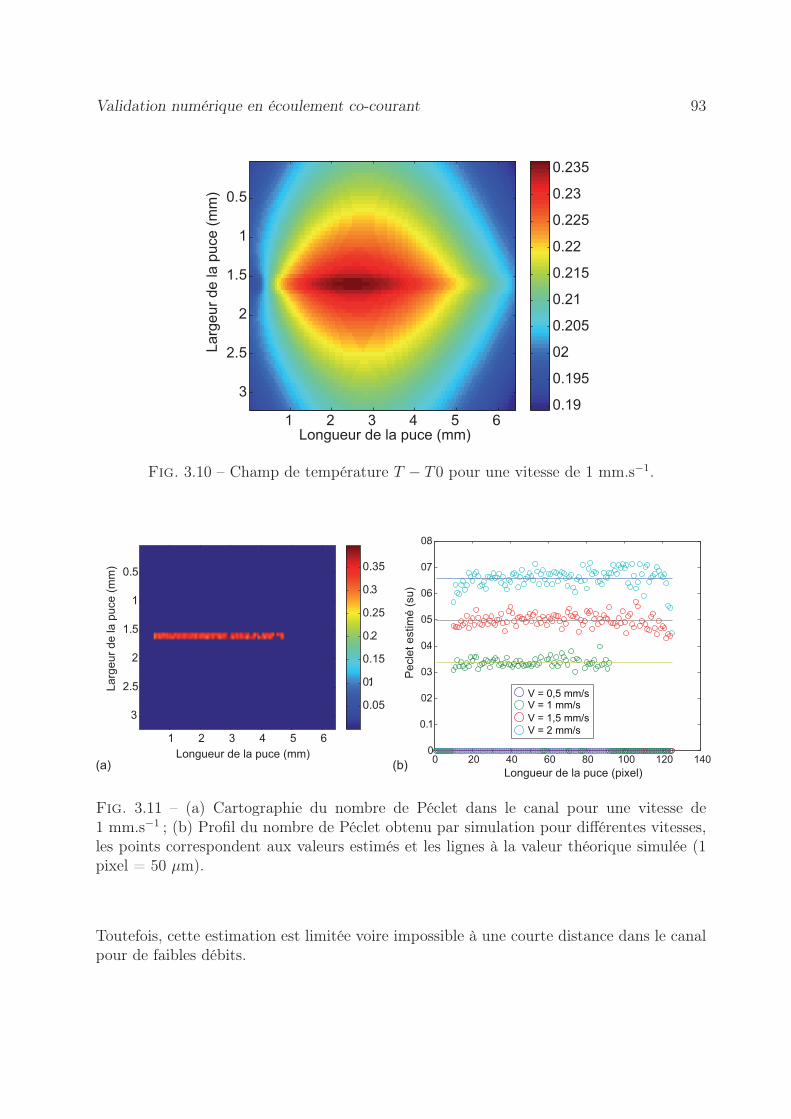
Validation numérique en écoulement co-courant 93
Longueur de la puce (mm)
La
rge
ur
de
la
pu
ce
(m
m)
1 2 3 4 5 6
0.5
1
1.5
2
2.5
30.19
0.195
0.2
0.205
0.21
0.215
0.22
0.225
0.23
0.235
Fig. 3.10 – Champ de température T − T0 pour une vitesse de 1 mm.s−1.
0 20 40 60 80 100 120 1400
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
Longueur de la puce (pixel)
Pe
cle
t e
stim
é (
su
)
V = 0,5 mm/sV = 1 mm/s
V = 1,5 mm/s
V = 2 mm/s
Longueur de la puce (mm)
La
rge
ur
de
la
pu
ce
(m
m)
1 2 3 4 5 6
0.5
1
1.5
2
2.5
30.05
0.1
0.15
0.2
0.25
0.3
0.35
(a) (b)
Fig. 3.11 – (a) Cartographie du nombre de Péclet dans le canal pour une vitesse de1 mm.s−1 ; (b) Profil du nombre de Péclet obtenu par simulation pour différentes vitesses,les points correspondent aux valeurs estimés et les lignes à la valeur théorique simulée (1pixel = 50 µm).
Toutefois, cette estimation est limitée voire impossible à une courte distance dans le canalpour de faibles débits.
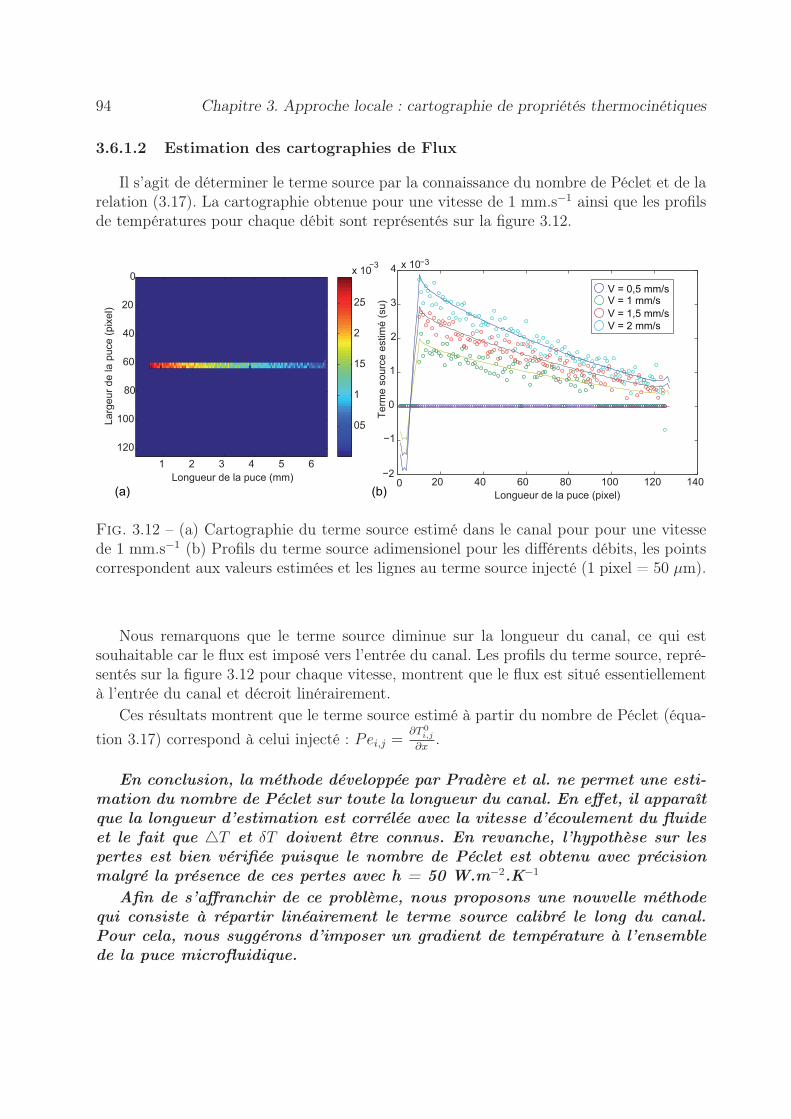
94 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
3.6.1.2 Estimation des cartographies de Flux
Il s’agit de déterminer le terme source par la connaissance du nombre de Péclet et de larelation (3.17). La cartographie obtenue pour une vitesse de 1 mm.s−1 ainsi que les profilsde températures pour chaque débit sont représentés sur la figure 3.12.
Longueur de la puce (mm)
Larg
eur
de la p
uce (
pix
el)
1 2 3 4 5 6
0
20
40
60
80
100
120
0.5
1
1.5
2
2.5
x 10−3
(a) (b)0 20 40 60 80 100 120 140
−2
−1
0
1
2
3
4 x 10−3
Longueur de la puce (pixel)
Term
e s
ourc
e e
stim
é (
su)
V = 0,5 mm/sV = 1 mm/s
V = 1,5 mm/s
V = 2 mm/s
Fig. 3.12 – (a) Cartographie du terme source estimé dans le canal pour pour une vitessede 1 mm.s−1 (b) Profils du terme source adimensionel pour les différents débits, les pointscorrespondent aux valeurs estimées et les lignes au terme source injecté (1 pixel = 50 µm).
Nous remarquons que le terme source diminue sur la longueur du canal, ce qui estsouhaitable car le flux est imposé vers l’entrée du canal. Les profils du terme source, repré-sentés sur la figure 3.12 pour chaque vitesse, montrent que le flux est situé essentiellementà l’entrée du canal et décroit linérairement.
Ces résultats montrent que le terme source estimé à partir du nombre de Péclet (équa-
tion 3.17) correspond à celui injecté : Pei,j =∂T 0
i,j
∂x.
En conclusion, la méthode développée par Pradère et al. ne permet une esti-mation du nombre de Péclet sur toute la longueur du canal. En effet, il apparaîtque la longueur d’estimation est corrélée avec la vitesse d’écoulement du fluideet le fait que △T et δT doivent être connus. En revanche, l’hypothèse sur lespertes est bien vérifiée puisque le nombre de Péclet est obtenu avec précisionmalgré la présence de ces pertes avec h = 50 W.m−2.K−1
Afin de s’affranchir de ce problème, nous proposons une nouvelle méthodequi consiste à répartir linéairement le terme source calibré le long du canal.Pour cela, nous suggérons d’imposer un gradient de température à l’ensemblede la puce microfluidique.

Validation numérique en écoulement co-courant 95
3.6.2 Méthode à gradient imposé
3.6.2.1 Estimation de cartographie de Péclet
Le système utilisé pour la modélisation est représenté par la figure 3.13.
< <Qentrée Qsortie
Microcanal
Substrat verregrad T
<
<
250 µm
Pertes par convection
Pertes par convection
f1
f1
f2
f2
Xz
y
Fig. 3.13 – Schéma du modèle à 2D du microréacteur avec un gradient imposé.
Le gradient de température est appliqué en imposant un flux positif du côté de l’entréedans le microcanal et un flux négatif en sortie du canal (figure 3.14). Lors des expériencesle gradient est imposé via des éléments Peltier (partie 3.2.2).
Longueur de la puce (mm)
Larg
eur
de la p
uce (
mm
)
1 2 3 4 5 6
0.5
1
1.5
2
2.5
3−0.3
−0.2
−0.1
0
0.1
0.2
0.3
Fig. 3.14 – Champs de températures obtenus par simulation sans terme convectif.
Nous remarquons que sans terme convectif, les variations du gradient de températuresont linéaires, parallèles entre elles et perpendiculaires au canal. De plus, l’influence despertes est encore moins importante que dans le cas précédent.
La deuxième étapes consiste à imposer des vitesses variant de 0,5 à 2 mm.s−1 par pasde 0,5 dans le but d’estimer le Pe. Le domaine de température obtenu pour une vitesse de1 mm.s−1 est représenté sur le figure 3.15.

96 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
Longueur de la puce (mm)
Larg
eur
de la p
uce (
mm
)
1 2 3 4 5 6
0.5
1
1.5
2
2.5
30.1
0.2
0.3
0.4
0.5
0.6
0.7
Fig. 3.15 – Champs de températures obtenus par simulation (v = 1 mm.s−1).
Là encore, la vitesse engendre un terme de transport qui déforme les champs de tem-pérature de façon plus ou moins importante selon sa valeur.
Puis, d’après le principe de superposition, le domaine de températures T − T0 estdéterminé (figure 3.16). Nous pouvons remarquer que le terme source est étalé sur unedistance dans le canal plus grande que lors de la méthode à flux locale.
Longueur de la puce (mm)
Larg
eur
de la p
uce (
mm
)
1 2 3 4 5 6
0. 5
1
1.5
2
2.5
3
0.37
0.38
0.39
0. 4
0.41
0.42
0.43
Fig. 3.16 – Champs de températures de domaine de température T − To.
Finalement, l’utilisation de l’équation 3.13 mènent à l’estimation de la valeur du nombrede Péclet local dans le canal pour chaque vitesse (figures 3.17).
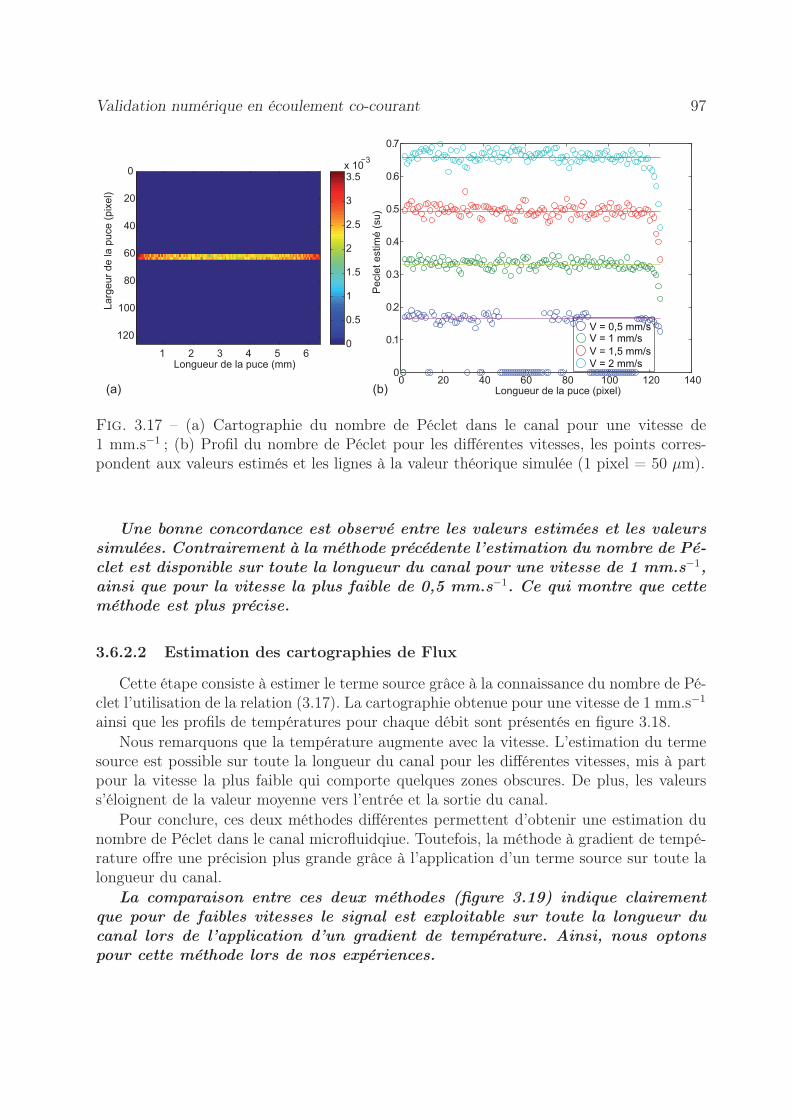
Validation numérique en écoulement co-courant 97
Longueur de la puce (mm)
Larg
eur
de la p
uce (
pix
el)
1 2 3 4 5 6
0
20
40
60
80
100
1200
0.5
1
1.5
2
2.5
3
3.5x 10
−3
0 20 40 60 80 100 120 1400
0.1
0.2
0.3
0.4
0.5
0.6
0.7
Longueur de la puce (pixel)
Pecle
t estim
é (
su)
(a) (b)
V = 0,5 mm/sV = 1 mm/s
V = 1,5 mm/s
V = 2 mm/s
Fig. 3.17 – (a) Cartographie du nombre de Péclet dans le canal pour une vitesse de1 mm.s−1 ; (b) Profil du nombre de Péclet pour les différentes vitesses, les points corres-pondent aux valeurs estimés et les lignes à la valeur théorique simulée (1 pixel = 50 µm).
Une bonne concordance est observé entre les valeurs estimées et les valeurssimulées. Contrairement à la méthode précédente l’estimation du nombre de Pé-clet est disponible sur toute la longueur du canal pour une vitesse de 1 mm.s−1,ainsi que pour la vitesse la plus faible de 0,5 mm.s−1. Ce qui montre que cetteméthode est plus précise.
3.6.2.2 Estimation des cartographies de Flux
Cette étape consiste à estimer le terme source grâce à la connaissance du nombre de Pé-clet l’utilisation de la relation (3.17). La cartographie obtenue pour une vitesse de 1 mm.s−1
ainsi que les profils de températures pour chaque débit sont présentés en figure 3.18.Nous remarquons que la température augmente avec la vitesse. L’estimation du terme
source est possible sur toute la longueur du canal pour les différentes vitesses, mis à partpour la vitesse la plus faible qui comporte quelques zones obscures. De plus, les valeurss’éloignent de la valeur moyenne vers l’entrée et la sortie du canal.
Pour conclure, ces deux méthodes différentes permettent d’obtenir une estimation dunombre de Péclet dans le canal microfluidqiue. Toutefois, la méthode à gradient de tempé-rature offre une précision plus grande grâce à l’application d’un terme source sur toute lalongueur du canal.
La comparaison entre ces deux méthodes (figure 3.19) indique clairementque pour de faibles vitesses le signal est exploitable sur toute la longueur ducanal lors de l’application d’un gradient de température. Ainsi, nous optonspour cette méthode lors de nos expériences.
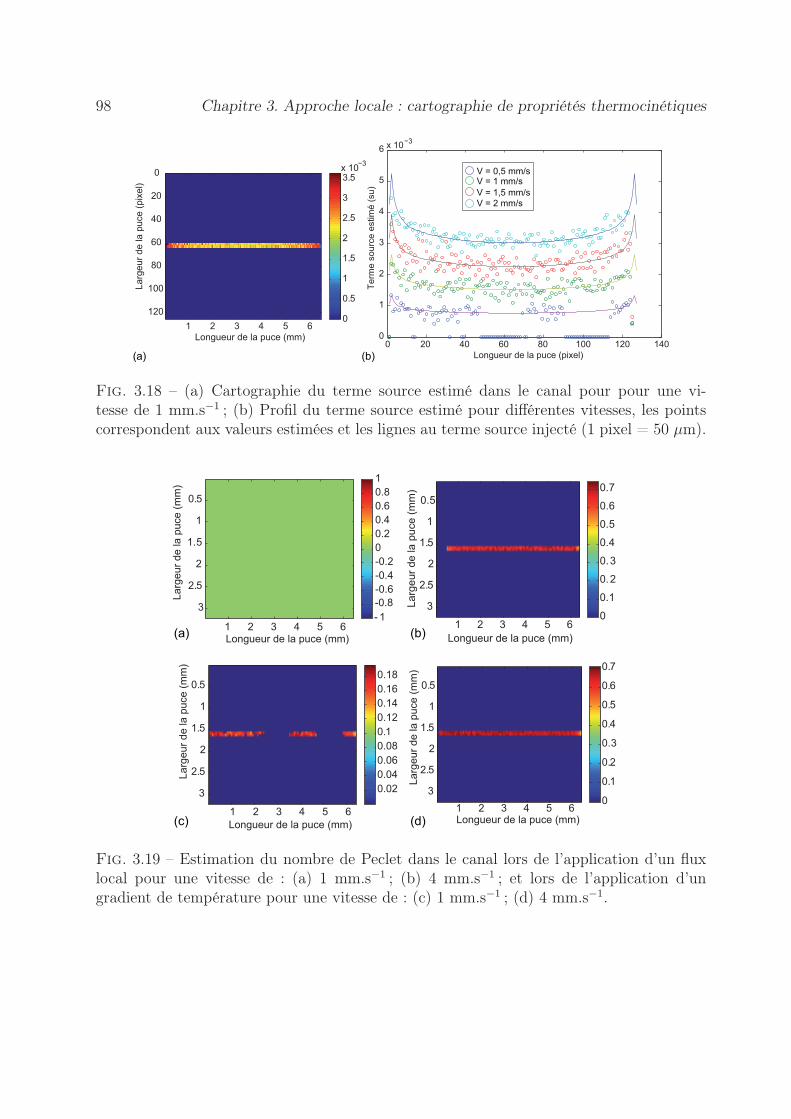
98 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
Longueur de la puce (mm)
Larg
eur
de
la
puce (
pix
el)
1 2 3 4 5 6
0
20
40
60
80
100
1200
0.5
1
1.5
2
2.5
3
3.5x 10
−3
0 20 40 60 80 100 120 1400
1
2
3
4
5
6 x 10−3
Longueur de la puce (pixel)
Term
e s
ourc
e e
stim
é (
su)
(a) (b)
V = 0,5 mm/sV = 1 mm/s
V = 1,5 mm/s
V = 2 mm/s
Fig. 3.18 – (a) Cartographie du terme source estimé dans le canal pour pour une vi-tesse de 1 mm.s−1 ; (b) Profil du terme source estimé pour différentes vitesses, les pointscorrespondent aux valeurs estimées et les lignes au terme source injecté (1 pixel = 50 µm).
Longueur de la puce (mm)
La
rge
ur
de
la
pu
ce
(m
m)
1 2 3 4 5 6
0.5
1
1.5
2
2.5
3- 1
-0.8
-0.6
-0.4
-0.2
0
0.2
0.4
0.6
0.8
1
Longueur de la puce (mm)
Larg
eur
de la p
uce (
mm
)
1 2 3 4 5 6
0.5
1
1.5
2
2.5
30
0.1
0.2
0.3
0.4
0.5
0.6
0.7
Longueur de la puce (mm)
La
rge
ur
de
la
pu
ce
(m
m)
1 2 3 4 5 6
0.5
1
1.5
2
2.5
3 0.02
0.04
0.06
0.08
0.1
0.12
0.14
0.16
0.18
Longueur de la puce (mm)
La
rge
ur
de
la
pu
ce
(m
m)
1 2 3 4 5 6
0.5
1
1.5
2
2.5
30
0.1
0.2
0.3
0.4
0.5
0.6
0.7
(a) (b)
(c) (d)
Fig. 3.19 – Estimation du nombre de Peclet dans le canal lors de l’application d’un fluxlocal pour une vitesse de : (a) 1 mm.s−1 ; (b) 4 mm.s−1 ; et lors de l’application d’ungradient de température pour une vitesse de : (c) 1 mm.s−1 ; (d) 4 mm.s−1.
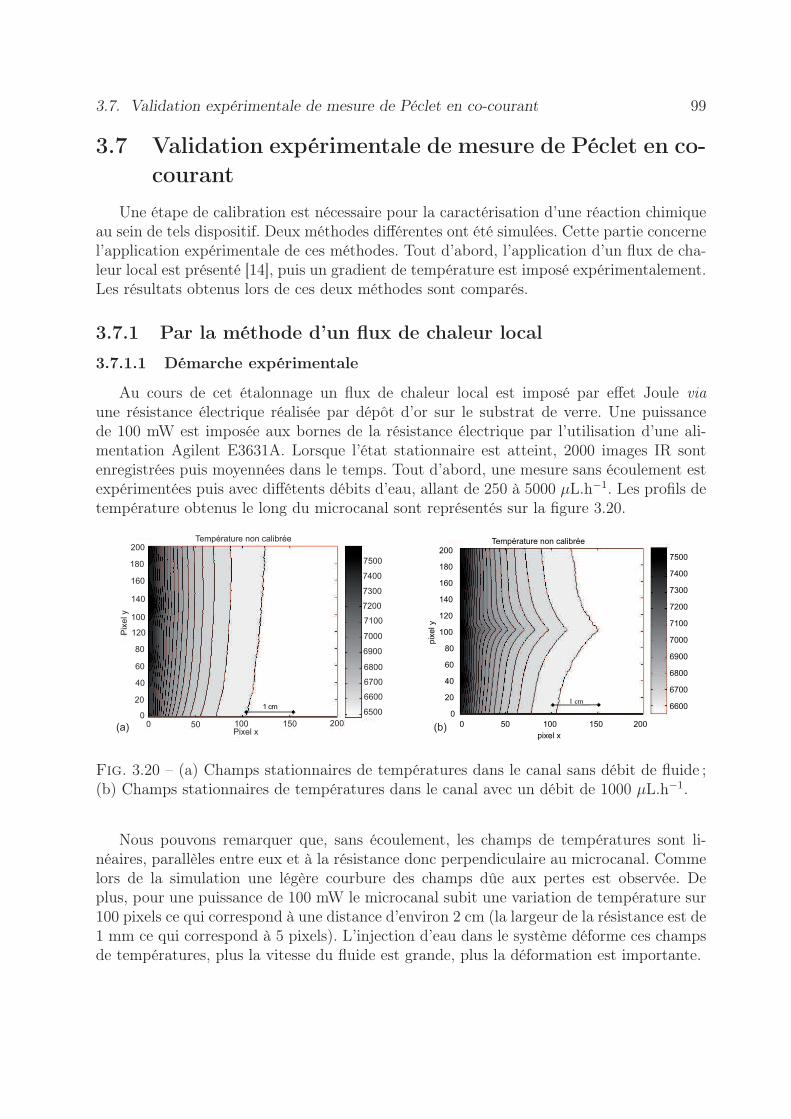
3.7. Validation expérimentale de mesure de Péclet en co-courant 99
3.7 Validation expérimentale de mesure de Péclet en co-courant
Une étape de calibration est nécessaire pour la caractérisation d’une réaction chimiqueau sein de tels dispositif. Deux méthodes différentes ont été simulées. Cette partie concernel’application expérimentale de ces méthodes. Tout d’abord, l’application d’un flux de cha-leur local est présenté [14], puis un gradient de température est imposé expérimentalement.Les résultats obtenus lors de ces deux méthodes sont comparés.
3.7.1 Par la méthode d’un flux de chaleur local
3.7.1.1 Démarche expérimentale
Au cours de cet étalonnage un flux de chaleur local est imposé par effet Joule viaune résistance électrique réalisée par dépôt d’or sur le substrat de verre. Une puissancede 100 mW est imposée aux bornes de la résistance électrique par l’utilisation d’une ali-mentation Agilent E3631A. Lorsque l’état stationnaire est atteint, 2000 images IR sontenregistrées puis moyennées dans le temps. Tout d’abord, une mesure sans écoulement estexpérimentées puis avec diffétents débits d’eau, allant de 250 à 5000 µL.h−1. Les profils detempérature obtenus le long du microcanal sont représentés sur la figure 3.20.
1 cm
200
180
160
140
100
120
80
60
40
20
00 50 100 150 200
Température non calibrée
7500
7400
7300
7200
7100
7000
6900
6800
6700
6600
6500
Pixel x
Pix
el y
1 cm
200
180
160
140
120
100
80
60
40
20
0
pix
el y
200150100500
pixel x
7500
7400
7300
7200
7100
7000
6900
6800
6700
6600
Température non calibrée
(b)(a)
Fig. 3.20 – (a) Champs stationnaires de températures dans le canal sans débit de fluide ;(b) Champs stationnaires de températures dans le canal avec un débit de 1000 µL.h−1.
Nous pouvons remarquer que, sans écoulement, les champs de températures sont li-néaires, parallèles entre eux et à la résistance donc perpendiculaire au microcanal. Commelors de la simulation une légère courbure des champs dûe aux pertes est observée. Deplus, pour une puissance de 100 mW le microcanal subit une variation de température sur100 pixels ce qui correspond à une distance d’environ 2 cm (la largeur de la résistance est de1 mm ce qui correspond à 5 pixels). L’injection d’eau dans le système déforme ces champsde températures, plus la vitesse du fluide est grande, plus la déformation est importante.
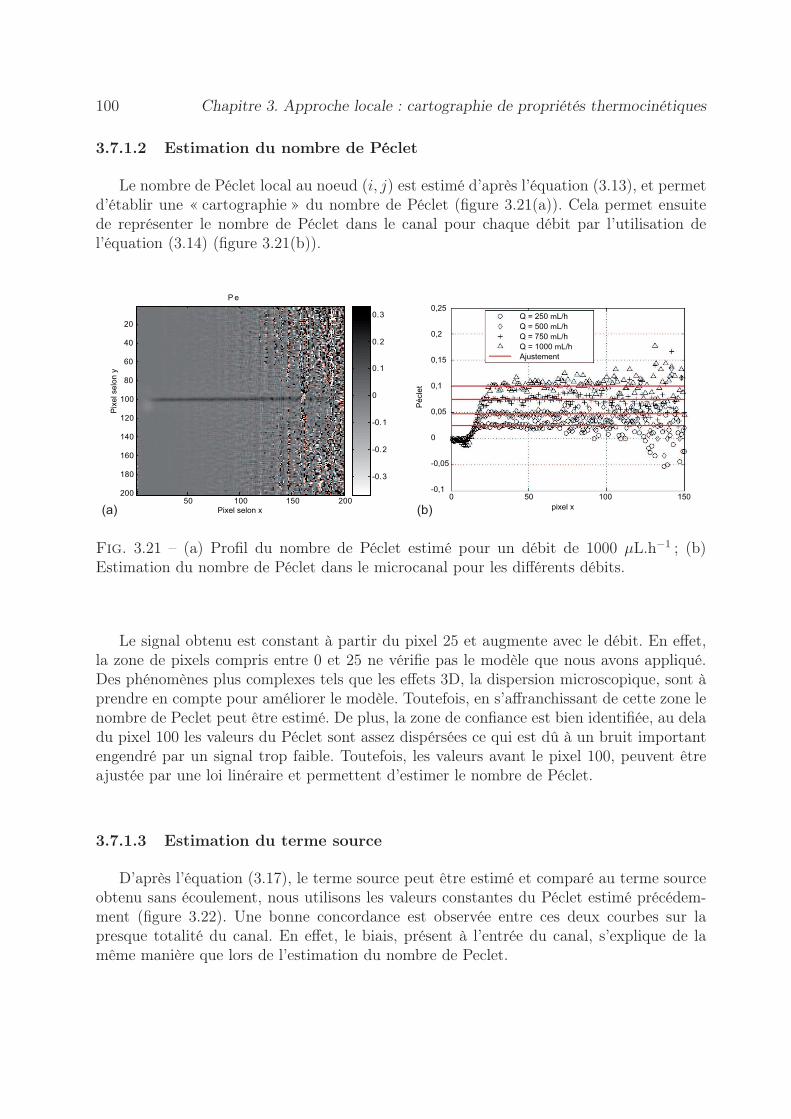
100 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
3.7.1.2 Estimation du nombre de Péclet
Le nombre de Péclet local au noeud (i, j) est estimé d’après l’équation (3.13), et permetd’établir une « cartographie » du nombre de Péclet (figure 3.21(a)). Cela permet ensuitede représenter le nombre de Péclet dans le canal pour chaque débit par l’utilisation del’équation (3.14) (figure 3.21(b)).
15010050
pixel x
0
0,25
0,2
-0,1
0,15
0,1
0,05
0
-0,05
Pé
cle
t
Q = 250 mL/h
Q = 500 mL/h
Q = 750 mL/h
Q = 1000 mL/h
Ajustement
(a) (b)Pixel selon x
Pix
el selo
n y
P e
50 100 150 200
20
40
60
80
100
120
140
160
180
200
-0.3
-0.2
-0.1
0
0.1
0.2
0.3
Fig. 3.21 – (a) Profil du nombre de Péclet estimé pour un débit de 1000 µL.h−1 ; (b)Estimation du nombre de Péclet dans le microcanal pour les différents débits.
Le signal obtenu est constant à partir du pixel 25 et augmente avec le débit. En effet,la zone de pixels compris entre 0 et 25 ne vérifie pas le modèle que nous avons appliqué.Des phénomènes plus complexes tels que les effets 3D, la dispersion microscopique, sont àprendre en compte pour améliorer le modèle. Toutefois, en s’affranchissant de cette zone lenombre de Peclet peut être estimé. De plus, la zone de confiance est bien identifiée, au deladu pixel 100 les valeurs du Péclet sont assez dispérsées ce qui est dû à un bruit importantengendré par un signal trop faible. Toutefois, les valeurs avant le pixel 100, peuvent êtreajustée par une loi linéraire et permettent d’estimer le nombre de Péclet.
3.7.1.3 Estimation du terme source
D’après l’équation (3.17), le terme source peut être estimé et comparé au terme sourceobtenu sans écoulement, nous utilisons les valeurs constantes du Péclet estimé précédem-ment (figure 3.22). Une bonne concordance est observée entre ces deux courbes sur lapresque totalité du canal. En effet, le biais, présent à l’entrée du canal, s’explique de lamême manière que lors de l’estimation du nombre de Peclet.
Validation expérimentale de mesure de Péclet en co-courant 101
Pixel selon x
Pix
el se
lon
yP e
50 100 150 200
20
40
60
80
100
120
140
160
180
200
-1
-0.5
0
0.5
1
1.5
(a) (b)0 20 40 60 80 100 120 140 160 180 200
-0.5
0
0.5
1
1.5
2
2.5
3
3.5
4
P ixels
Flu
x (
wu
)
∆(T-T0
)-P e.*d(T-T0
)
P e*dT0
Fig. 3.22 – Estimation du terme source dans le microcanal ; la ligne continu correspondantaux valeurs déterminées à partir de l’expérience sans débit et les croix aux expériences avecdébits.
3.7.2 Par la méthode du gradient de température
3.7.2.1 Démarche expérimentale
Le dispositif utilisé est représenté par la figure 3.1 où le gradient de température estappliqué par des éléments Peltier reliés en série. Une tension de 10 V est imposée auxbornes de ces éléments via une alimentation Agilent E3631A. Lorsque l’état stationnaire estatteint, 250 images IR sont enregistrées, nous avons choisi un nombre d’images relativementfaible pour tout d’abord vérifier la faisabilité de la méthode et pour ne pas surcharger laquantité d’informations. La figure 3.23 représente les champs de température établi avecun débit nul. Nous observons que ces champs sont parallèles et que l’influence des pertesest négligeable.
20 40 60 80 100 120 140 160
5
10
15
20
25
30
35 6600
6700
6800
6900
7000
7100
7200
7300
7400
7500
Longueur de la puce (pixel)
La
rge
ur
de
la
pu
ce
(p
ixe
l)
Fig. 3.23 – Champs stationnaires de températures sans débit de fluide.
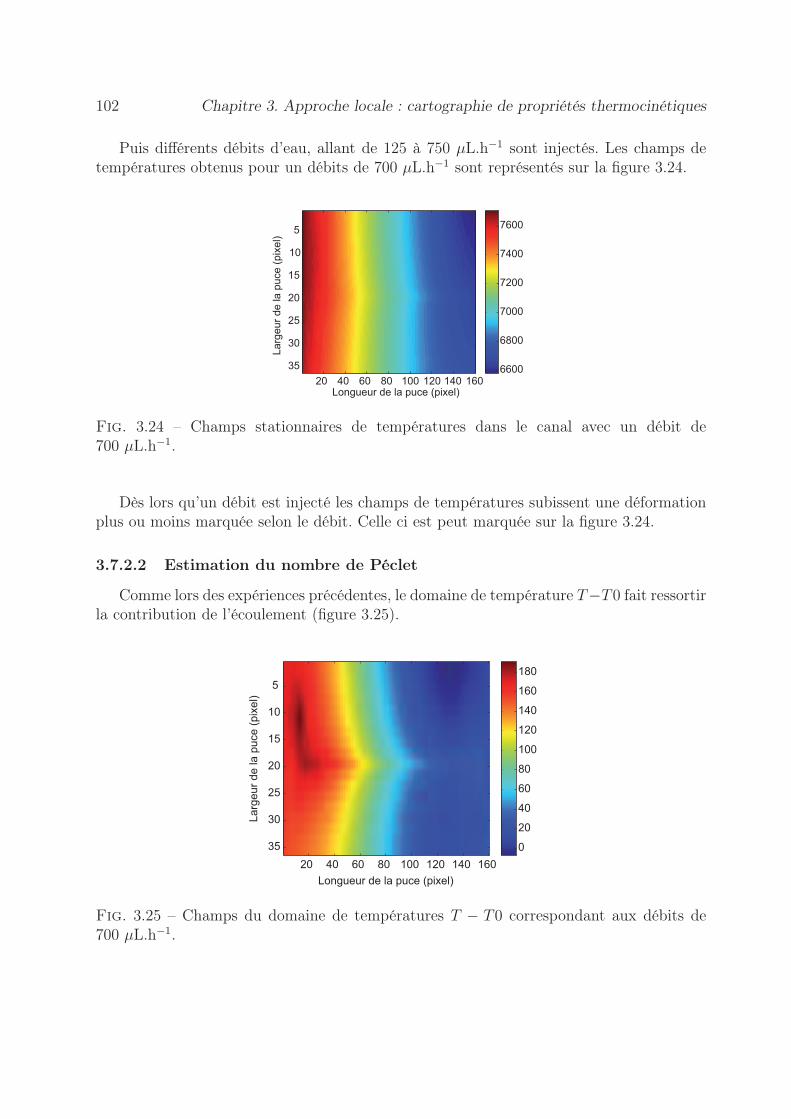
102 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
Puis différents débits d’eau, allant de 125 à 750 µL.h−1 sont injectés. Les champs detempératures obtenus pour un débits de 700 µL.h−1 sont représentés sur la figure 3.24.
20 40 60 80 100 120 140 160
5
10
15
20
25
30
35 6600
6800
7000
7200
7400
7600
Longueur de la puce (pixel)
La
rge
ur
de
la
pu
ce
(p
ixe
l)
Fig. 3.24 – Champs stationnaires de températures dans le canal avec un débit de700 µL.h−1.
Dès lors qu’un débit est injecté les champs de températures subissent une déformationplus ou moins marquée selon le débit. Celle ci est peut marquée sur la figure 3.24.
3.7.2.2 Estimation du nombre de Péclet
Comme lors des expériences précédentes, le domaine de température T−T0 fait ressortirla contribution de l’écoulement (figure 3.25).
20 40 60 80 100 120 140 160
5
10
15
20
25
30
35 0
20
40
60
80
100
120
140
160
180
Longueur de la puce (pixel)
La
rge
ur
de
la
pu
ce
(p
ixe
l)
Fig. 3.25 – Champs du domaine de températures T − T0 correspondant aux débits de700 µL.h−1.
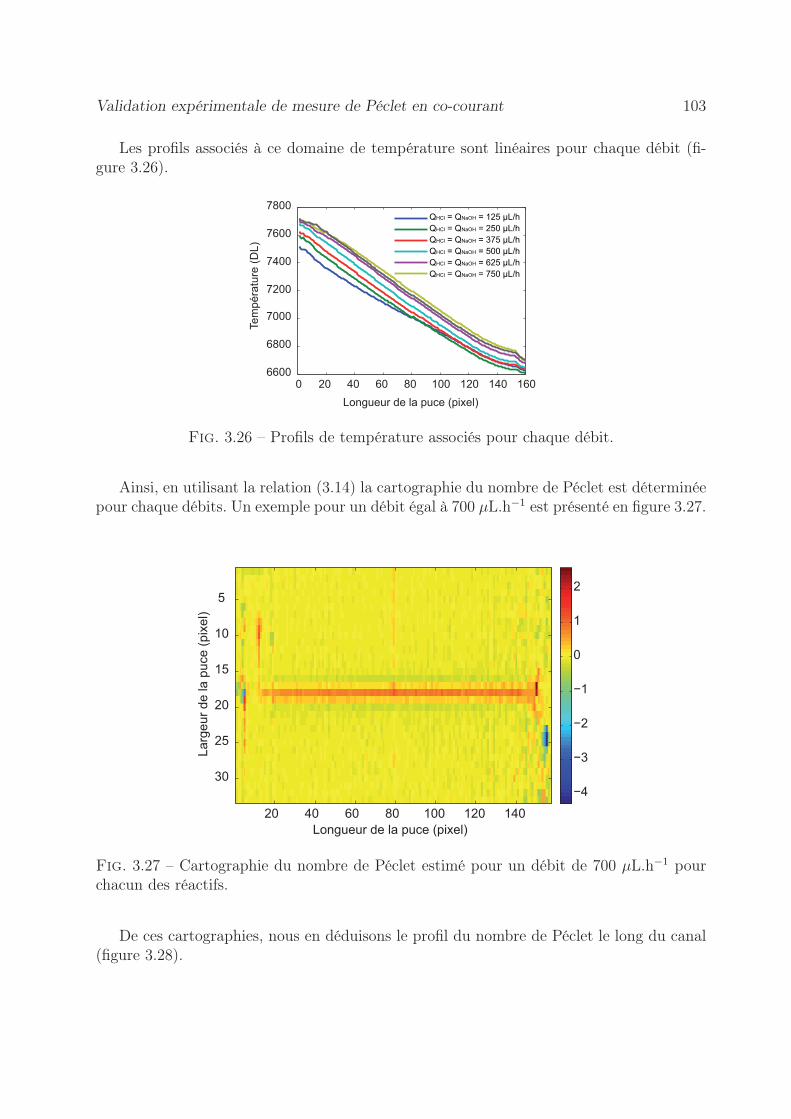
Validation expérimentale de mesure de Péclet en co-courant 103
Les profils associés à ce domaine de température sont linéaires pour chaque débit (fi-gure 3.26).
0 20 40 60 80 100 120 140 1606600
6800
7000
7200
7400
7600
7800
Longueur de la puce (pixel)
Te
mp
éra
ture
(D
L)
QHCl = QNaOH = 625 µL/h
QHCl = QNaOH = 250 µL/h
QHCl = QNaOH = 375 µL/h
QHCl = QNaOH = 500 µL/h
QHCl = QNaOH = 125 µL/h
QHCl = QNaOH = 750 µL/h
Fig. 3.26 – Profils de température associés pour chaque débit.
Ainsi, en utilisant la relation (3.14) la cartographie du nombre de Péclet est déterminéepour chaque débits. Un exemple pour un débit égal à 700 µL.h−1 est présenté en figure 3.27.
20 40 60 80 100 120 140
5
10
15
20
25
30
−4
−3
−2
−1
0
1
2
Longueur de la puce (pixel)
Larg
eur
de la p
uce (
pix
el)
Fig. 3.27 – Cartographie du nombre de Péclet estimé pour un débit de 700 µL.h−1 pourchacun des réactifs.
De ces cartographies, nous en déduisons le profil du nombre de Péclet le long du canal(figure 3.28).
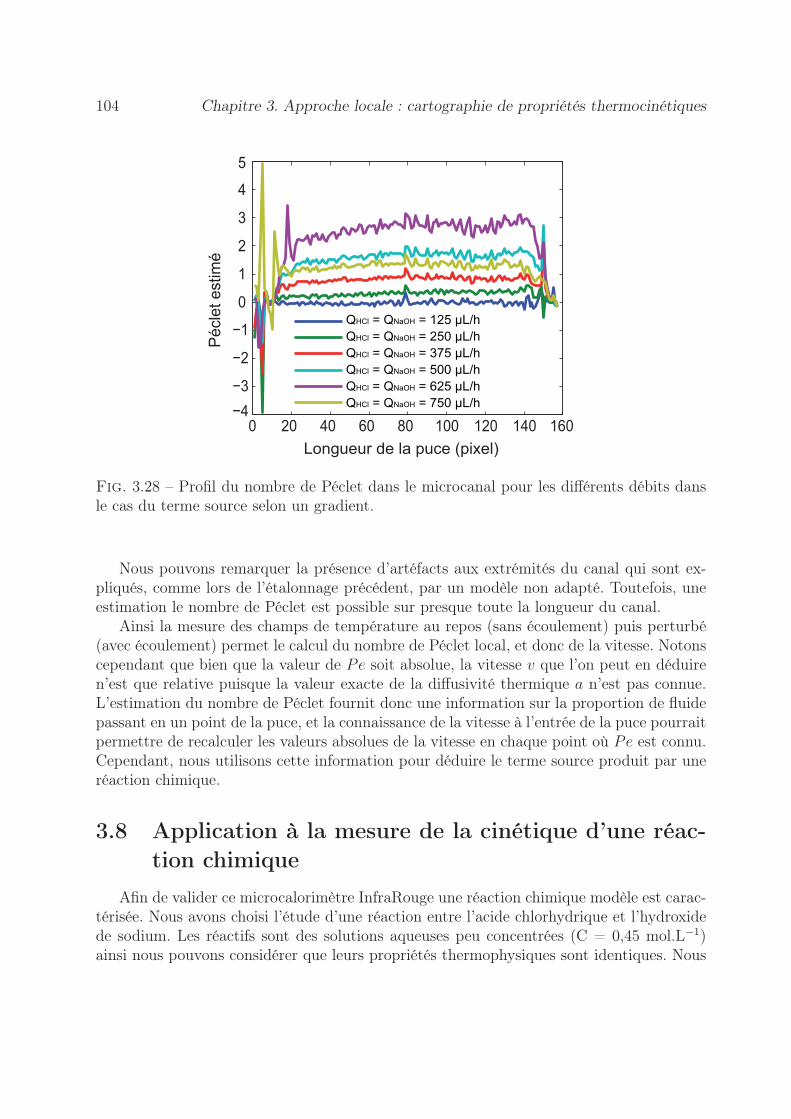
104 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
0 20 40 60 80 100 120 140 160−4
−3
−2
−1
0
1
2
3
4
5
Longueur de la puce (pixel)
Péclet estimé
QHCl = QNaOH = 625 µL/h
QHCl = QNaOH = 250 µL/h
QHCl = QNaOH = 375 µL/h
QHCl = QNaOH = 500 µL/h
QHCl = QNaOH = 125 µL/h
QHCl = QNaOH = 750 µL/h
Fig. 3.28 – Profil du nombre de Péclet dans le microcanal pour les différents débits dansle cas du terme source selon un gradient.
Nous pouvons remarquer la présence d’artéfacts aux extrémités du canal qui sont ex-pliqués, comme lors de l’étalonnage précédent, par un modèle non adapté. Toutefois, uneestimation le nombre de Péclet est possible sur presque toute la longueur du canal.
Ainsi la mesure des champs de température au repos (sans écoulement) puis perturbé(avec écoulement) permet le calcul du nombre de Péclet local, et donc de la vitesse. Notonscependant que bien que la valeur de Pe soit absolue, la vitesse v que l’on peut en déduiren’est que relative puisque la valeur exacte de la diffusivité thermique a n’est pas connue.L’estimation du nombre de Péclet fournit donc une information sur la proportion de fluidepassant en un point de la puce, et la connaissance de la vitesse à l’entrée de la puce pourraitpermettre de recalculer les valeurs absolues de la vitesse en chaque point où Pe est connu.Cependant, nous utilisons cette information pour déduire le terme source produit par uneréaction chimique.
3.8 Application à la mesure de la cinétique d’une réac-tion chimique
Afin de valider ce microcalorimètre InfraRouge une réaction chimique modèle est carac-térisée. Nous avons choisi l’étude d’une réaction entre l’acide chlorhydrique et l’hydroxidede sodium. Les réactifs sont des solutions aqueuses peu concentrées (C = 0,45 mol.L−1)ainsi nous pouvons considérer que leurs propriétés thermophysiques sont identiques. Nous

Application à la mesure de la cinétique d’une réaction chimique 105
obtenons le type d’image IR représentée par la figure 3.29 où le dégagement de chaleur dûà la réaction chimique est visible et s’étend avec le débit.
20 40 60 80 100 120 140
85
90
95
100
105
110
115 6960
6980
7000
7020
7040
7060
7080
7100
7120
7140
7160
20 40 60 80 100 120 140
85
90
95
100
105
110
115 7000
7050
7100
7150
7200
Longueur de la puce (pixel)
Larg
eur
de la p
uce (
pix
el)
Longueur de la puce (pixel)Larg
eur
de la p
uce (
pix
el)
(a) (b)
Fig. 3.29 – Profil du terme source pour les différents débits expérimentés.
Les profils de température pour chaque débit sont mesurés expérimentalement et lenombre de Péclet est estimé lors de l’étalonnage que nous venons de présenter. Ce qui nouspermet d’en déduire le terme source dû à la réaction chimique. Nous observons que le débit
0 20 40 60 80 100 120 140 160−20
−10
0
10
20
30
40
50
QHCl = QNaOH = 625 µL/h
QHCl = QNaOH = 250 µL/h
QHCl = QNaOH = 375 µL/h
QHCl = QNaOH = 500 µL/h
QHCl = QNaOH = 125 µL/h
QHCl = QNaOH = 750 µL/h
Longueur de la puce (pixel)
Te
rme
so
urc
e e
stim
é (
su
)
Fig. 3.30 – Profil du terme source pour les différents débits expérimentés.
augmente avec le flux de chaleur, ce qui s’explique par un augmentation de la quantité
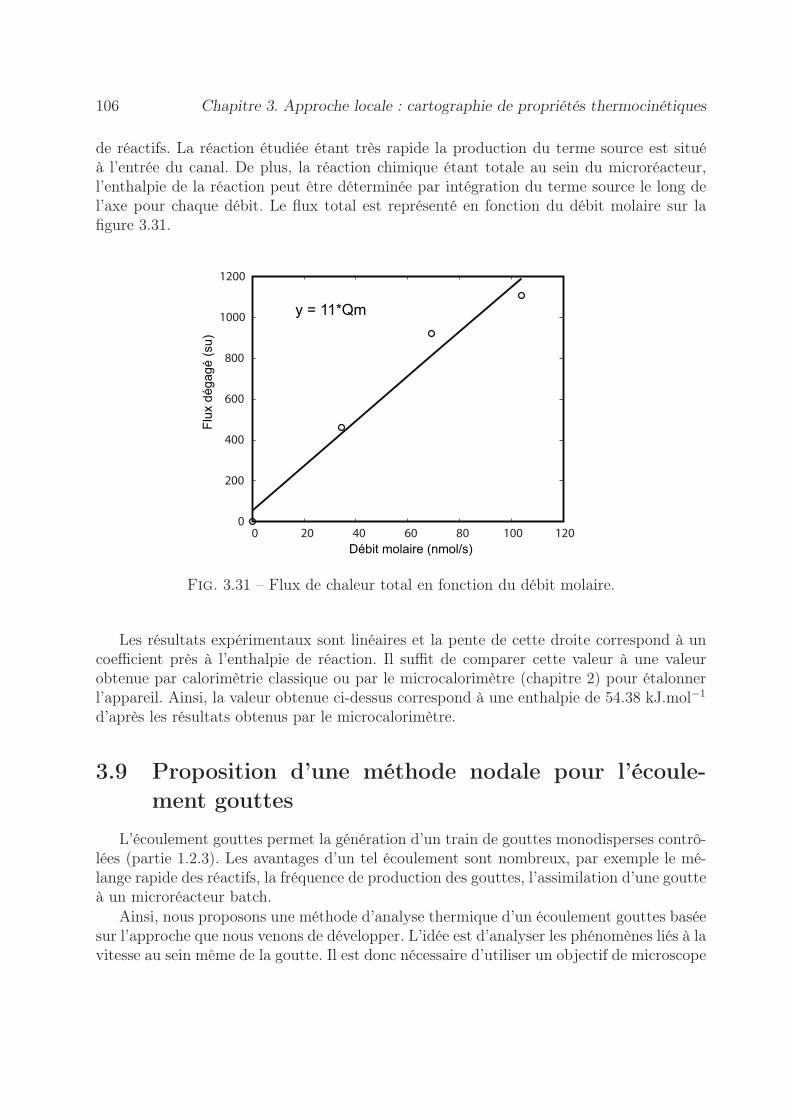
106 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
de réactifs. La réaction étudiée étant très rapide la production du terme source est situéà l’entrée du canal. De plus, la réaction chimique étant totale au sein du microréacteur,l’enthalpie de la réaction peut être déterminée par intégration du terme source le long del’axe pour chaque débit. Le flux total est représenté en fonction du débit molaire sur lafigure 3.31.
0 20 40 60 80 100 1200
200
400
600
800
1000
1200
Débit molaire (nmol/s)
Flu
x d
égagé (
su)
y = 11*Qm
Fig. 3.31 – Flux de chaleur total en fonction du débit molaire.
Les résultats expérimentaux sont linéaires et la pente de cette droite correspond à uncoefficient près à l’enthalpie de réaction. Il suffit de comparer cette valeur à une valeurobtenue par calorimètrie classique ou par le microcalorimètre (chapitre 2) pour étalonnerl’appareil. Ainsi, la valeur obtenue ci-dessus correspond à une enthalpie de 54.38 kJ.mol−1
d’après les résultats obtenus par le microcalorimètre.
3.9 Proposition d’une méthode nodale pour l’écoule-ment gouttes
L’écoulement gouttes permet la génération d’un train de gouttes monodisperses contrô-lées (partie 1.2.3). Les avantages d’un tel écoulement sont nombreux, par exemple le mé-lange rapide des réactifs, la fréquence de production des gouttes, l’assimilation d’une goutteà un microréacteur batch.
Ainsi, nous proposons une méthode d’analyse thermique d’un écoulement gouttes baséesur l’approche que nous venons de développer. L’idée est d’analyser les phénomènes liés à lavitesse au sein même de la goutte. Il est donc nécessaire d’utiliser un objectif de microscope

Proposition d’une méthode nodale pour l’écoulement gouttes 107
de résolution spatiale 25 µm. La fréquence d’acquisition est au maximum de 1 kHz pourla caméra IR.
3.9.1 Procédure expérimentale
La réaction entre l’acide chlorhydrique et l’hydroxide de sodium de concentration égaleà 0,4 mol.L−1 est réalisée lors d’un écoulement gouttes. La procédure expérimentale est lasuivante : les réactifs ainsi que la phase porteuse sont injectés par un pousse-seringue à desdébits variant de 250 à 1000 µL.h−1. Puis, un film de la scène est enregistré dès l’atteintede l’état établi, obtenu rapidement car les faibles dimensions du dispositif permettent untemps de réponse très court. Un exemple des images IR obtenues lors de l’écoulementgouttes est présentée sur la figure 3.32.
injection d’acide
injection de base
injection d'huile
Fig. 3.32 – Image IR d’une train de gouttes d’acide base.
3.9.2 Démarche de la méthode
La principale différence entre l’écoulement de type co-courant et gouttes réside dans lecaractère transitoire de ce dernier. En effet, lors d’un écoulement de type gouttes, le régimepériodique établi spatialement se superpose au régime transitoire lié au déplacement desgouttes. Dans ces conditions, il est possible d’utiliser cette périodicité pour estimer lavitesse de déplacement des gouttes. La connaissance de cette vitesse va nous permettre unchangement de repère qui va nous placer dans le référentiel local de la goutte. Ainsi, seulel’évolution transitoire du transfert thermique dans la goutte sera à traiter.
Dans ces conditions, les approches locales similaires à celles présentées dans le cas duco-courant pourront être mises en place. Dans un premier temps, la procédure de traitementd’images visant à se placer dans le repère local de la goutte est décrite, puis les méthodesd’inversions ainsi que les premiers résultats obtenus seront exposés.
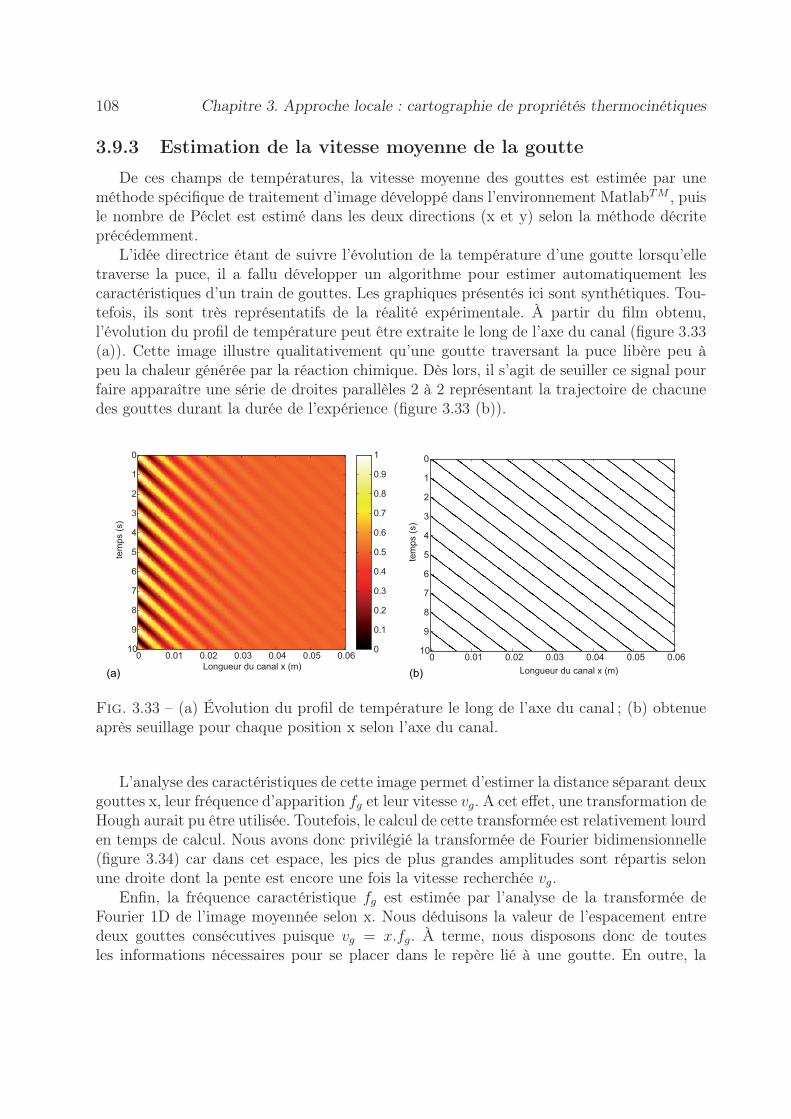
108 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
3.9.3 Estimation de la vitesse moyenne de la goutte
De ces champs de températures, la vitesse moyenne des gouttes est estimée par uneméthode spécifique de traitement d’image développé dans l’environnement MatlabTM , puisle nombre de Péclet est estimé dans les deux directions (x et y) selon la méthode décriteprécédemment.
L’idée directrice étant de suivre l’évolution de la température d’une goutte lorsqu’elletraverse la puce, il a fallu développer un algorithme pour estimer automatiquement lescaractéristiques d’un train de gouttes. Les graphiques présentés ici sont synthétiques. Tou-tefois, ils sont très représentatifs de la réalité expérimentale. À partir du film obtenu,l’évolution du profil de température peut être extraite le long de l’axe du canal (figure 3.33(a)). Cette image illustre qualitativement qu’une goutte traversant la puce libère peu àpeu la chaleur générée par la réaction chimique. Dès lors, il s’agit de seuiller ce signal pourfaire apparaître une série de droites parallèles 2 à 2 représentant la trajectoire de chacunedes gouttes durant la durée de l’expérience (figure 3.33 (b)).
Longueur du canal x (m)
tem
ps (
s)
0 0.01 0.02 0.03 0.04 0.05 0.06
0
1
2
3
4
5
6
7
8
9
10 0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
tem
ps (
s)
0 0.01 0.02 0.03 0.04 0.05 0.06
0
1
2
3
4
5
6
7
8
9
10
Longueur du canal x (m)(a) (b)
Fig. 3.33 – (a) Évolution du profil de température le long de l’axe du canal ; (b) obtenueaprès seuillage pour chaque position x selon l’axe du canal.
L’analyse des caractéristiques de cette image permet d’estimer la distance séparant deuxgouttes x, leur fréquence d’apparition fg et leur vitesse vg. A cet effet, une transformation deHough aurait pu être utilisée. Toutefois, le calcul de cette transformée est relativement lourden temps de calcul. Nous avons donc privilégié la transformée de Fourier bidimensionnelle(figure 3.34) car dans cet espace, les pics de plus grandes amplitudes sont répartis selonune droite dont la pente est encore une fois la vitesse recherchée vg.
Enfin, la fréquence caractéristique fg est estimée par l’analyse de la transformée deFourier 1D de l’image moyennée selon x. Nous déduisons la valeur de l’espacement entredeux gouttes consécutives puisque vg = x.fg. À terme, nous disposons donc de toutesles informations nécessaires pour se placer dans le repère lié à une goutte. En outre, la
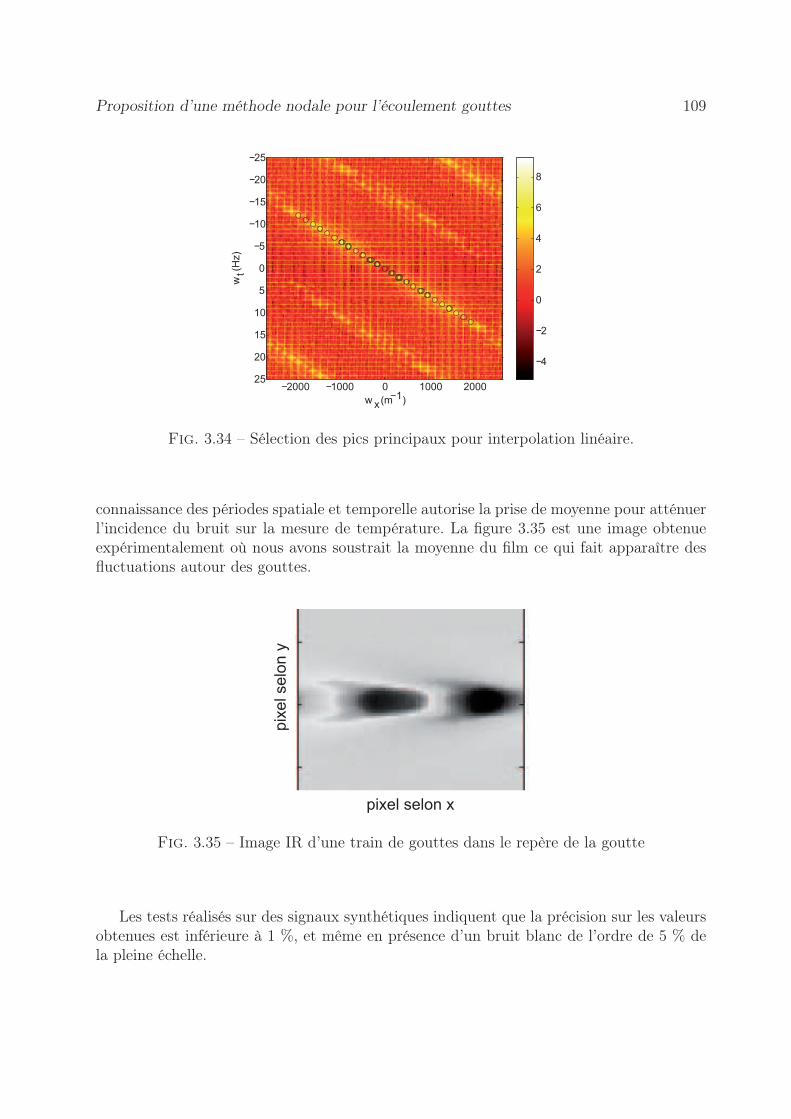
Proposition d’une méthode nodale pour l’écoulement gouttes 109
w x (m−1)
wt (
Hz)
−2000 −1000 0 1000 2000
−25
−20
−15
−10
−5
0
5
10
15
20
25
−4
−2
0
2
4
6
8
Fig. 3.34 – Sélection des pics principaux pour interpolation linéaire.
connaissance des périodes spatiale et temporelle autorise la prise de moyenne pour atténuerl’incidence du bruit sur la mesure de température. La figure 3.35 est une image obtenueexpérimentalement où nous avons soustrait la moyenne du film ce qui fait apparaître desfluctuations autour des gouttes.
pixel selon x
pix
el selo
n y
Fig. 3.35 – Image IR d’une train de gouttes dans le repère de la goutte
Les tests réalisés sur des signaux synthétiques indiquent que la précision sur les valeursobtenues est inférieure à 1 %, et même en présence d’un bruit blanc de l’ordre de 5 % dela pleine échelle.

110 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
3.9.4 Méthode inverse proposée
Le profil de température d’une goutte dans le repère local peut être suivi. Dans ce cas,le bilan thermique s’écrit de la manière suivante :
div(λ−−→gradT (x, y, t) + φ(x, y, t) = ρCp(
−→v −−→gradT (x, y, t) +
∂T (x, y, t)
∂t(3.18)
Toutes les expériences mènent à une vitesses relatives : V = −→v + reprelocal. Nousemmettons l’hypothèse que la réaction chimique est quasi instantanée, ce qui implique quela réaction chimique se produit dès le mélange des réactifs. Cela nous permet de négligerle terme source. Par l’approximation en différences finies, nous obtenons :
(T ki+1,j+T k
i−1,j+T ki,j+1+T k
i,j−1−4T ki,j) = Pexi,j(T
ki+1,j−T k
i,j)+Peyi,j(Tki+1,j−T k
i,j)+1
Foi,j
(T k+1i,j −T k
i,j)
(3.19)
avec :
Pexi,j = u∆xa
Peyi,j = v∆y
a
Foi,j = a∆t∆x2
(3.20)
Le problème d’estimation à 3 paramètres est difficile d’autant que le signal est bruité.Nous privilégions la même méthode que précédemment pour l’estimation locale du nombrede Péclet. L’estimation simultanée à trois paramètres est difficile ( [71]). Elle nécessite deconsidérer le conditionnement d’une matrice formée des covariances entre laplacien de Tet la dérivée spatiale ou temporelle. Étant donnée que les gradients dans la goutte sontpratiquement soit horizontaux soit verticaux, avec un transitoire, nous pouvons rechercheruniquement les zones des images où un seul paramètre est estimable (soit Pexi,j, soit Pexi,j,soit Foi,j). Cela revient à considérer comme en (3.15) un coefficient de corrélation entrele laplacien et une des dérivées.
3.9.4.1 Estimation des cartographies du nombre de Péclet selon x
Nous calculons le coefficient de corrélation local entre : △Ti,j et δ(Ti,j). L’estima-tion n’est effectuée que dans le cas où la corrélation est proche de 1. Ce qui mène à lacartographie du nombre de Péclet selon la direction x (figure 3.36).
Nous remarquons que la vitesse autour de la goutte, correspondant à l’huile, est néga-tive. Cela s’explique par le fait que l’estimation du nombre de Péclet est réalisés à partirdes données définies dans le repère de la goutte. La vitesse selon la direction x est quasinulle à l’avant de la goutte et très importante à l’arrière. Une zone stagnante est observéeau centre de la goutte.
Sarrazin et al. ont étudiés le mélange au sein des gouttes par la technique de microPIV.Si nous comparons les champs de vitesses au sein d’une gouttes qu’ils ont obtenu à nosrésultats, une grande similitute apparaît [23].
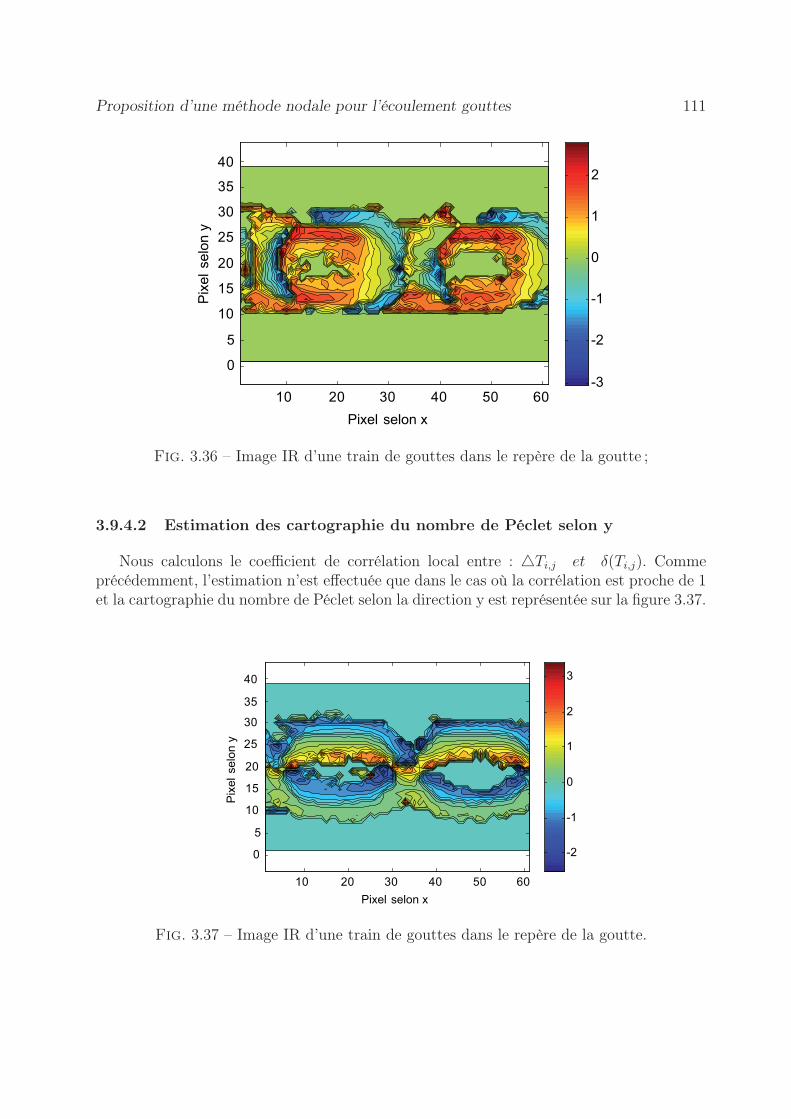
Proposition d’une méthode nodale pour l’écoulement gouttes 111
10 20 30 40 50 60
0
5
10
15
20
25
30
35
40
-3
-2
-1
0
1
2
Pix
el
se
lon
y
Pixel selon x
Fig. 3.36 – Image IR d’une train de gouttes dans le repère de la goutte ;
3.9.4.2 Estimation des cartographie du nombre de Péclet selon y
Nous calculons le coefficient de corrélation local entre : △Ti,j et δ(Ti,j). Commeprécédemment, l’estimation n’est effectuée que dans le cas où la corrélation est proche de 1et la cartographie du nombre de Péclet selon la direction y est représentée sur la figure 3.37.
Pixel selon x
Pix
el
se
lon
y
10 20 30 40 50 60
0
5
10
15
20
25
30
35
40
-2
-1
0
1
2
3
Fig. 3.37 – Image IR d’une train de gouttes dans le repère de la goutte.

112 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques
Selon la direction y, la vitesse est positive dans la partie supérieure de la goutte etnégative dans la partie inférieure. De plus, nous observons que la vitesse diminue du centrevers l’extèrieur.
Ces résultats sont très prometteurs et permettent de mettre en évidence leprofil de vitesse au sein des gouttes. Ainsi, nous pouvons en déduire la manièredont s’opère le mélange. Il est important de préciser la grande quantité d’infor-mation obtenue par ce type d’expérience. En effet, une goutte est représentatived’une expérience, nous réalisons donc n fois la même manipulation, n étant lenombre de gouttes = temps d’expérience x fg, cela permet une acquisition.

3.10. Conclusion 113
3.10 Conclusion
L’approche microfluidique consistant à étudier la réponse de surface d’une plaque aucontact d’un microcanal a été abordée en suivant les méthodes développés par Pradère etal. [14]. Nous avons d’abord cherché à améliorer le montage initial par la mise au point d’unsystème à pompages latéraux par éléments Peltier, puis nous avons appliqué la méthoded’estimation pour la caractérisation de gouttes.
Ainsi, ce travail a permis d’élaborer une méthode de mesure par thermographie Infra-Rouge pour la caractérisation de réactions en microfluidique. Une méthode d’estimationà un paramètre a été développée et des cas synthétiques simulés. Nous avons vu que lacalibration du dipositif est une étape très importante qui a menée à deux techniques dif-férentes. Une méthode à gradient de température où le nombre de Peclet peut être estimésur toute la longueur du canal et une méthode de flux local généré par effet Joule quis’avère moins sensible. Ce dispositif a ensuite été validé par la caractérisation d’une réac-tion acide-base lors de l’écoulement co-courant. L’évolution temporelle du terme source àété évaluée pour différents débits, puis la relation linéaire entre le flux global dégagé parla réaction et la quantité de réactif a été observée.
La seconde application que nous avons explorée est la caractérisation d’un écoulementdiphasique avec une résolution spatiale de 25 µm. Tout d’abord, il est nécessaire de détecterles gouttes pour estimer leur vitesse et de se placer au sein du référentiel lié à la goutte. Puis,des cartographies du nombre de Péclet ont été obtenues par le même principe d’estimationque lors de l’écoulement co-courant. Celles-ci font apparaître la manière dont s’opère lemélange qui est comparable aux études menées par PIV. Le nombre de Péclet étant reliéà la vitesse, celle-ci peut être déduite de ces cartographies, nous pouvons assimiler cesmesures à de la « PIV thermique ».
Les méthodes d’estimation sont ici extrêmement simplifiées par l’estimation à un pa-ramètre par recherche de zone de corrélation. Toutefois, elles présentent déjà l’avantagede fournir des cartographies et de remonter au flux de chaleur dégagé par une réactionchimique. Il est bien évident qu’à l’avenir d’autres méthodes doivent être explorées, parexemple la méthode Bayesienne tenant compte de l’erreur d’estimation du nombre de Pé-clet, ou l’estimation simultanée dans le cas des gouttes.
Cependant, des phénomènes locaux situés à l’entrée du microcanal ne correspondentpas au modèle simplifié que nous avons établi. Cela n’a pas d’influence notable sur lamesure de l’enthalpie, ce qui ne serait pas le cas lors de l’estimation de la cinétique d’uneréaction.
Afin de s’affranchir de ces phénomènes, plusieurs solutions sont envisageables, commedéfinir un modèle plus complexe ou encore limiter expérimentalement la diffusion. Cettedernière option est une des motivations du prochain chapitre.
Ce dispositif est une technique non intrusive permettant la mesure de champ de vitessequi rend possible l’investigation de dispositifs composés de matériaux opaques.

114 Chapitre 3. Approche locale : cartographie de propriétés thermocinétiques

Chapitre 4
Calorimètre millifluidique isopéribolique
par thermographie InfraRouge
Résumé
Le but de ce chapitre est de présenter un calorimètre sans contact permettant lamesure de cinétique et d’enthalpie de réactions chimiques très exothermiques. L’ori-ginalité de ce travail réside dans la conception d’une puce millifluidique aux propriétésthermiques particulières (conditions thermiques dites « isopériboliques ») et à l’utili-sation d’une caméra InfraRouge comme capteur. Plusieurs avantages sont offerts parce dispositif, comme la faible taille du canal qui engendre un faible volume réactionnel(de l’ordre du mL). D’autre part, les expériences sont réalisées en continu avec un boncontrôle du mélange des réactifs. Le système d’injection développé permet l’étude enécoulement co-courant et gouttes.Les propriétés isopériboliques du milliréacteur permettent une simplification du mo-dèle thermique. Ce modèle comporte deux coefficients de transfert estimés par deuxétalonnages, ce qui permet ensuite une estimation du flux de chaleur local (cinétique)par l’utilisation d’une méthode inverse. Un profil de température de 1200 mesures detempératures le long du milliréacteur. Ainsi, l’enthalpie de réaction et sa répartitionspatiale peuvent être déterminées. Ce dispositif de mesure est validé par la caracté-risation d’une réaction acide (HCl) - base (NaOH) lors d’un écoulement co-courant.Le mélange des espèces par diffusion est mis en évidence et l’enthalpie de réaction estdéterminée.
115

116 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
4.1 Introduction
Nous avons vu, au chapitre précédent, qu’une estimation quantitative du flux est pos-sible à partir d’une mesure qualitative d’un champ de température obtenu à l’aide d’unecaméra IR. En effet, les données thermodynamiques, comme l’enthalpie d’une réaction chi-mique se produisant en continu au sein d’un microcanal, peuvent être estimées. Selon lespropriétés thermiques du système (puce millifluidique) la modélisation thermique sera plusou moins complexe.
L’idée de cette partie est alors de concevoir une nouvelle puce où les phénomènes liés à ladiffusion spatiale (dans le plan de la caméra) sont mieux maîtrisés. Le travail présenté dansce chapitre est consacré au développement d’un calorimètre millifluidique sans contact oùla mesure de température est obtenue par une caméra IR. Le milliréacteur est composé d’untube en PFA (isolant thermique) et d’une plaque en aluminium (conducteur thermique).La combinaison de ces deux matériaux confère les propriétés isopériboliques où les pertespar diffusion sont maîtrisées, ce qui permet un bon contrôle du transfert de chaleur.
Ce chapitre débute par une présentation générale du dispositif de mesure. Puis, lemodèle thermique appliqué à la puce millifluidique est décrit, ainsi que son inversion. Latroisième partie détaille les deux étapes de l’étalonnage de l’appareil. Au cours de la dernièrepartie, nous validerons le dispositif par la mise en évidence du mélange par diffusion desespèces lors d’un écoulement co-courant, et par l’estimation de l’enthalpie de réaction d’uncas modèle.

4.2. Caractéristiques du dispositif 117
4.2 Caractéristiques du dispositif
Une photographie (figure 4.1) ainsi qu’un schéma (figure 4.2) représentent l’ensembledu dispositif de mesure. Ce dispositif est essentiellement composé d’un milliréacteur etd’une caméra InfraRouge. Le milliréacteur, présenté en détail dans la section 4.3, est placésur un système de régulation qui permet de maintenir la puce à une température constante.Les fluides sont introduits par un pousse-seringue de type Nemesys (voir partie 2.2.2). Lacaméra IR, située au dessus du milliréacteur, permet la mesure du champ de température.
Fig. 4.1 – Photographie du millicalorimètre dans son ensemble.
4.2.1 Système de régulation
La régulation de la puce millifluidique à une température constante et uniforme estréalisé par un système de thermalisation AMS07-005 de la société AMS technologie. Cedernier utilise deux éléments Peltier placés sous une plaque d’aluminium mesurant 18 cmde long sur 16 cm de largeur et 5 mm d’épaisseur (figure 4.3). Ce module est contrôlé parun régulateur PID (AMS technologie : Ali-2410) relié à une sonde Pt100 qui mesure latempérature au centre de la plaque. La température de consigne peut varier de 5 à 90 ◦Cavec une précision de 0,1 ◦C. Ce système permet la régulation d’une surface de 18 cm delong et 16 cm de large utilisée pour thermaliser le milliréacteur et préchauffer les réactifs.

118 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
régulateur thermique PIDsystème de
régulation
> >>
camera IR
pousse seringue puce isopéribolique
plaque en aluminium
tube PFAmilieu réactionnel
Fig. 4.2 – Représentation schématique du millicalorimètre.
sonde Pt 100 plaque
éléments
Peltieréchangeur
thermiqueventilateur
contrôleur de température
Fig. 4.3 – Photographie du système de régulation comprenant l’appareil de thermalisationet le régulateur de température.
4.2.2 Caméra InfraRouge
La caméra InfraRouge, Cedip Jade MWIR J550 (Medium Wavelenght InfraRed), dontnous avons fait usage au cours du chapitre précédent, est à nouveau employée pour observerl’ensemble du milliréacteur. Cette caméra est sensible au rayonnement dans le moyen In-fraRouge sur une gamme de longueur d’onde allant de 2 à 5,6 µm. Le système de détection

4.3. Description du milliréacteur chimique 119
de la caméra est composé de plusieurs rangs de détecteurs In-Sb (Antimoine d’indium)dont le nombre de pixels est de 240 x 320, ce qui correspond à 76800 points de mesures.Pour l’étude présentée ici, un objectif 25 mm MWIR est également utilisé. Compte tenude la distance entre la caméra et la surface de la puce, la résolution spatiale apparente estd’environ 200 µm.
4.3 Description du milliréacteur chimique
4.3.1 Description générale
Le milliréacteur utilisé pour réaliser les réactions chimiques est présenté sur la figure 4.4.Ce système est composé d’un tube en PFA (Perfluoroalkoxy) et d’une plaque en aluminium.Le tube en PFA est un bon isolant thermique (λt = 0,25 W.m−1.K−1) alors que l’aluminiumest un bon conducteur (λp = 237 W.m−1.K−1). Le tube en PFA mesure 3,2 mm de diamètreexterne et 1,6 mm de diamètre interne, la paroi du tube a une épaisseur égale à 800 µm, cequi engendre une résistance thermique de 3,37 m2.K.W−1. De plus, la plaque en aluminiumest maintenue à une température constante (Tp) (partie 4.2). Ainsi, la réaction chimiquea lieu dans des conditions isopériboliques, autrement dit, la température autour du milieuréactionnel est constante et contrôlée. Cette plaque en aluminium a subi un traitementde surface par anodisation afin d’éliminer toutes réflexions et d’obtenir une émissivitéuniforme.
système d’injection
>plaque en alumium
anodisée
>
>tube en PFA
> zone de préchauffage
> zone réactionnelle
Fig. 4.4 – Photographie du milliréacteur isopéribolique avec le système d’injection desréactifs.
La plaque en aluminium mesure 12 cm x 12 cm et contient une rainure en forme deserpentin où le tube en PFA est inséré (figure 4.5).
Cette forme de serpentin a été choisie afin d’optimiser le volume du réacteur. La lon-gueur du tube étant de 47 cm, le volume interne est de 950 µL. En effet, cela est important

120 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
plaque d'aluminium
tube PFA
>
>
milieu réactionel
< >12 cm
<
<8 mm
<
<
3.2 mm
<
<
3.2 mm
Fig. 4.5 – Schéma de la puce isopéribolique vue en coupe.
dans le cas d’une réaction relativement lente et permet d’ajuster le temps de résidence.Une gamme de débits de 2 à 60 mL.h−1 correspond à des temps de résidence de 1 à 30 min.
Les réactifs sont injectés séparément dans des tubes en PEEK (Polyétheréthercétone)dont le diamètre externe est de 800 µm et le diamètre interne 500 µm. Ces tubes en PEEKsont ensuite insérés parallèlement dans le tube en PFA grâce à une connectique en formede croix (figure 4.6).
Point de mélange
>
Tube en PFA
>
>Tube en PEEK
>
Connectique en croix
>
>Tube en PEEK
><
1,6 mm
Fig. 4.6 – Schéma représentant la connectique en croix et l’insertion des tubes en PEEKau sein du tube en PFA du milliréacteur isopéribolique.
Ce système d’injection astucieux offre une bonne maîtrise du point de rencontre desréactifs qui a lieu à l’issue de ces tubes. Ainsi, il suffit d’insérer les tubes contenant lesréactifs jusqu’au point de mélange désiré, ce qui permet un préchauffage des réactifs. Unautre avantage apporté par ce système d’injection est la possibilité de générer les deuxtypes d’écoulements, soit l’écoulement co-courant, soit l’écoulement gouttes.

4.4. Procédure expérimentale 121
4.3.2 Types d’écoulements
L’utilisation d’une connectique en croix permet d’introduire jusqu’à trois fluides dif-férents et ainsi de produire un écoulement co-courant ou un écoulement gouttes. Lors del’écoulement co-courant, représenté en figure 4.7, une des entrées est bouchée et les deuxautres servent à l’introduction des petits tubes contenant les réactifs.
>
Acide
>
Base
>
Zone interdiffusion
Point de mélange des réactifs>
Tube en PFA
>
>
Tubes en PEEK
>Fig. 4.7 – Schéma représentant la génération d’un écoulement co-courant au sein du mil-liréacteur isopéribolique.
Comme décrit dans le chapitre 1.2.2, le mélange des réactifs se fait par interdiffusiondes espèces. Le temps de mélange correspond au temps de diffusion sur toute la largeur ducanal. Il peut être déterminé par l’expression 1.7.
L’écoulement gouttes (figure 4.8) est produit de la même manière que l’écoulementco-courant avec en plus l’introduction d’une phase porteuse immiscible par la troisièmeentrée.
Pour les expériences présentées ici, les réactifs sont des solutions acido-basique aqueuses,et la phase porteuse est une phase organique non miscible. Plus précisémment, nous utili-sons une huile silicone de viscosité 10 mPa.s qui ne présente aucune intéraction avec nosréactifs. L’écoulement gouttes, décrit dans la partie 1.2.3, présente l’avantage de diminuerle temps de mélange par rapport à celui d’un écoulement co-courant.
4.4 Procédure expérimentale
4.4.1 Démarche générale
Avant de débuter une mesure, il est important de mettre le dispositif à la températurede consigne souhaitée. Lorsque que le système a atteint le régime permanent, la tempéra-ture est constante et uniforme à la surface de la plaque. Le principe de la mesure consiste

122 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
>
Huile silicone
>
Acide
>
Base
>
Huile silicone
>
Gouttes d'acide et de base
>>>>>>>>>>>>>
Tube en PFA
>
>
Tubes en PEEK
>
Fig. 4.8 – Schéma représentant la génération d’un écoulement gouttes au sein du milli-réacteur isopéribolique.
à introduire les réactifs dans la puce millifluidique via des pousse-seringues. Alors, la ré-action chimique débute, créant un terme source thermique dans le canal. Dès l’obtentiondu nouvel équilibre thermique, une image ou un film peuvent être enregistrés à l’aide dela caméra IR. Cet état stationnaire est défini par une stabilité des températures et dépenddu temps de résidence ainsi que du temps de réponse du système thermique qui va pilo-ter le régime transitoire. Une seule image suffit pour définir le profil de température detout le milliréacteur à l’état stationnaire. La température de la plaque est notée Tp et latempérature du tube qui sert de réacteur TR.
4.4.2 Étalonnage
Au cours de l’étape d’étalonnage, la relation entre la puissance dissipée dans le tubeet l’écart de température entre la plaque et le tube est établie. Les valeurs des coefficientsd’étalonnage dépendent des propriétés des fluides et des matériaux. Ils devront être estiméspour chaque température de consigne et pour chaque système réactionnel étudié.
4.4.3 Acquisition et traitement
L’acquisition des images IR est effectuée par l’intermédiaire du logiciel ALTAIRTM .Cependant, ses multiples fonctionnalités ne permettent pas d’exploiter directement leschamps de températures obtenus lors des expériences. Dans ce but, un outil de post-traitement dans l’environnement MatlabTM a été réalisé. Le fonctionnement de ce dernierpeut se décliner en trois étapes :
– Prétraitement : il s’agit tout d’abord de construire un maillage curviligne respectantla topologie en accord avec les dimensions des puces (plans de fabrication) permettant

4.4. Procédure expérimentale 123
de générer toutes les variables relatives à la géométrie du système (figure 4.9). Par lasuite, une bibliothèque de géométries est constituée.
– Mise à l’échelle : dans cette étape, après avoir sélectionné sa géométrie, l’utilisateurvient placer un masque directement sur le champ de température. Ce masque corres-pond à la zone d’intérêt, i.e., celle correspondant au tube. Du fait de la résolution del’image, ce placement demeure approximatif. C’est pourquoi un algorithme spécifiqueest appliqué pour positionner précisément le maillage (qui servira de grille d’interpo-lation) sur l’image. Le processus d’optimisation mis en place repose sur l’idée que leprofil de température doit être symétrique (ou presque) par rapport à l’axe du tube,compte tenu des dimensions caractéristiques de la puce, notamment du rayon externedu tube, et du rayon de courbure des virages (voir encart de la figure 4.9). À terme,les paramètres de mise à l’échelle de l’image, comprenant, entre autres, la taille despixels dans chacune des directions, sont obtenus. Une étude numérique, utilisant desimages synthétiques, a établi que cet algorithme positionne le maillage à une préci-sion de l’ordre de 0.1 µm. Pour une même série d’expériences, la détermination desparamètres de mise à l’échelle n’est à effectuer qu’une seule fois.
– Extraction : une fois le maillage curviligne correctement positionné, le profil detempérature le long de l’axe du tube est obtenu par interpolation linéaire (figure 4.10).De plus, la température moyenne de la plaque est calculée.
40 45 50 55 60
95
100
105
110
y (
mm
)
x (mm)
90 95 100 105 110 115
60
65
70
75
x (mm)
y (
mm
)
0 20 40 60 80 100 120
20
30
40
50
60
70
80
90
100
x (mm)
y (
mm
)
Fig. 4.9 – Schéma représentant le maillage de l’ensemble du canal du milliréacteur.
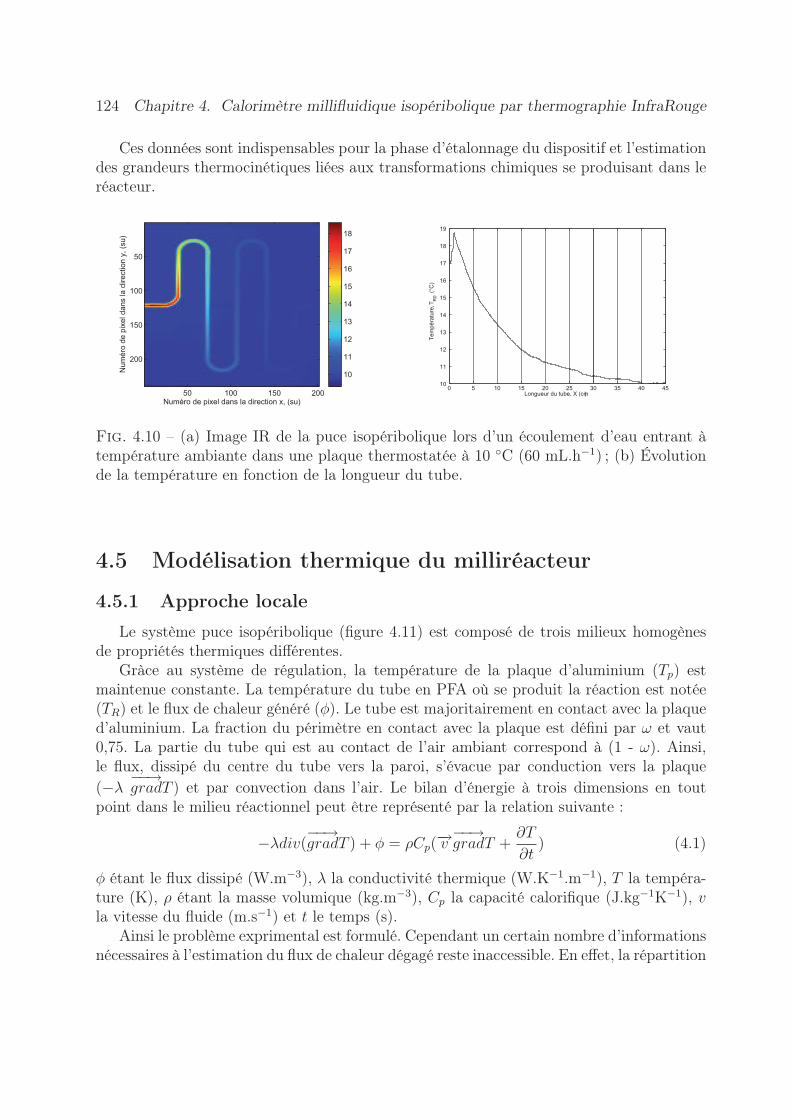
124 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
Ces données sont indispensables pour la phase d’étalonnage du dispositif et l’estimationdes grandeurs thermocinétiques liées aux transformations chimiques se produisant dans leréacteur.
Numéro de pixel dans la direction x, (su)
Nu
mé
ro d
e p
ixe
l d
an
s la
dire
ctio
n y
, (s
u)
50 100 150 200
50
100
150
200
10
11
12
13
14
15
16
17
18
0 5 10 15 20 25 30 35 40 4510
11
12
13
14
15
16
17
18
19
Longueur du tube, X (cm)
Tem
péra
ture
, T R
0 (
°C)
Fig. 4.10 – (a) Image IR de la puce isopéribolique lors d’un écoulement d’eau entrant àtempérature ambiante dans une plaque thermostatée à 10 ◦C (60 mL.h−1) ; (b) Évolutionde la température en fonction de la longueur du tube.
4.5 Modélisation thermique du milliréacteur
4.5.1 Approche locale
Le système puce isopéribolique (figure 4.11) est composé de trois milieux homogènesde propriétés thermiques différentes.
Gràce au système de régulation, la température de la plaque d’aluminium (Tp) estmaintenue constante. La température du tube en PFA où se produit la réaction est notée(TR) et le flux de chaleur généré (φ). Le tube est majoritairement en contact avec la plaqued’aluminium. La fraction du périmètre en contact avec la plaque est défini par ω et vaut0,75. La partie du tube qui est au contact de l’air ambiant correspond à (1 - ω). Ainsi,le flux, dissipé du centre du tube vers la paroi, s’évacue par conduction vers la plaque(−λ
−−→gradT ) et par convection dans l’air. Le bilan d’énergie à trois dimensions en tout
point dans le milieu réactionnel peut être représenté par la relation suivante :
−λdiv(−−→gradT ) + φ = ρCp(
−→v −−→gradT +
∂T
∂t) (4.1)
φ étant le flux dissipé (W.m−3), λ la conductivité thermique (W.K−1.m−1), T la tempéra-ture (K), ρ étant la masse volumique (kg.m−3), Cp la capacité calorifique (J.kg−1K−1), vla vitesse du fluide (m.s−1) et t le temps (s).
Ainsi le problème exprimental est formulé. Cependant un certain nombre d’informationsnécessaires à l’estimation du flux de chaleur dégagé reste inaccessible. En effet, la répartition

4.5. Modélisation thermique du milliréacteur 125
Plaque d'aluminium
Tube PFA
>
>
Echanges par
conduction
>
>
Milieu réactionel
Pertes par convection
Flux imposé>
>
>z
yx
TR
TP
>
>Entrée
du fluide
>
Sortie du fluide
Fig. 4.11 – Schéma du milliréacteur isopéribolique.
de h selon l’axe, les propriétés du fluides ou encore les champs de vitesses ne sont pasfacilement ni toujours disponibles. Une simplification du problème s’avère essentielle, nousavons donc décidé d’effectuer un bilan d’énergie sur une longueur élémentaire.
4.5.2 Approche globale
Comme nous venons de le voir, une modélisation locale est inaccessible. L’objectif étantde construire un outil de caractérisation, un modèle de représentation de l’évolution de latempérature dans le tube doit être défini. Selon les hypothèses que les propriétés physico-chimiques sont indépendantes de la composition et de la température et que le régimepermanent est établi, un bilan élémentaire sur une tranche de longueur dx du tube faitapparaître trois contributions [?] :
– un terme convectif représentant la variation d’enthalpie sur ce volume élémentaire,– un terme lié aux pertes thermiques qui résultent de deux contributions ; d’une part
celles relatives à la conduction, et d’autre part celles représentant les échanges avecl’air ambiant,
– un dernier terme intégrant les effets thermiques liés aux transformations chimiquesdans le milieu réactionnel φ (W.m−1).
Nous émettons les hypothèses suivantes sur le système :

126 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
– le milieu est homogène avec une conductivité thermique constante,– les variations de température dues à la réaction chimique sont faibles, ce qui suppose
que les propriétés des matériaux sont constantes,– la diffusion selon la direction de l’écoulement n’est pas prise en compte,– les conditions isopériboliques engendrent un contrôle du transfert de chaleur entre le
milieu réactionnel et la plaque,– les écoulements microfluidiques étant stationnaires, l’équivalence espace temps : x =
vt, peut être appliquée.Ainsi, nous en déduisons l’équation différentielle, décrivant l’évolution de la température àla surface du tube (équation (4.2)) :
(ρCp)appQdTR(x)
dx= φ(x) − K
L(TR(x) − Tp) −
Hcv
L(TR(x) − Ta) (4.2)
où Q est le débit volumique (m3.s−1), φ le flux (W.m−1), (ρCp)app la capacité calorifiquevolumique équivalente au système (tube + milieu réactionnel) (J.K−1.m−3), TR la tempéra-ture du tube, Ta la température de l’air, et L la longueur totale du tube (m). K représentele coefficient de perte par conduction et Hcv celui par convection (W.K−1).
En introduisant, Kcv, qui représente l’inverse d’une longueur caractéristique, l’équationprécédente peut s’écrire :
φ(x) =K
L
[1
Kcv
dTR(x)
dx+ (TR(x) − Tp) +
Hcv
K(TR(x) − Ta)
](4.3)
La représentation en coupe de notre système (figure 4.12) révèle que la majorité dutube est en contact avec la plaque entraînant ainsi des pertes par conduction.
plaque d'aluminium
tube PFA
>>
milieu réactionel
pertes par convection
pertes par conduction
Fig. 4.12 – Schéma en coupe du milliréacteur

4.5. Modélisation thermique du milliréacteur 127
L’autre partie du tube étant en contact avec l’air, des pertes par convection demeurent.Les coefficients de pertes thermiques peuvent être représentés par ces relations :
K = ωπdLλt
et
Hcv = (1 − ω)πdLh
Kcv = 1(ρCp)appQ
KL
(4.4)
et étant l’épaisseur du tube (m), λt la conductivité thermique du tube (W.m−1.K−1), hle coefficient global de transfert thermique par convection (W.m−2K−1), ω la fraction desurface en contact avec la plaque et d le diamètre externe du tube (m).
Afin de comparer les pertes par conduction et celles par convection, l’équation (4.3) estréécrite de la façon suivante :
φ(x) =1
L
[K
Kcv
dTR(x)
dx+ K
(1 +
Hcv
K
)TR(x) − K
(1 +
HcvTa
KTp
)Tp
](4.5)
De par la faible épaisseur du tube (et ≈ 800 µm) et sa faible conductivité thermique(λt ≈ 1 W.m−1.K−1), la résistance thermique du tube est très basse (équation 4.6) :
Rt =et
λt
≈ 10−3m2.K.W−1 (4.6)
De plus, la fraction du périmètre du tube en contact avec la plaque est nettement supérieureà celle en contact avec l’air où des pertes par convection ont lieu : ω = 0.75. Pour unetempérature de consigne de 5 ◦C et considérant que la valeur du coefficient de convectionh est proche de 10 W.m−2.K−1, nous déterminons :
Hcv
K= 1.10−4
HcvTa
KTp= 1.10−4
(4.7)
Le rapport entre HcvTa et KTp est donc très faible et bien inférieur à 1 (équation 4.7).Nous faisons l’hypothèse que les pertes par convection peuvent être négligées et l’équationbilan (4.3) peut être simplifiée et réécrite de la façon suivante :
φ(x) =K
L
[1
Kcv
dTR(x)
dx+ (TR(x) − Tp)
](4.8)
D’après ce modèle, trois paramètres, K, Kcv et φ, sont inconnus. La partie suivantedécrit les protocoles expérimentaux mis en place pour leur estimation.

128 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
4.6 Calibration du système millifluidique
4.6.1 Mesures des pertes thermiques
Cette étape d’étalonnage permet de relier le flux de chaleur dissipé à l’intérieur dutube en PFA à la température de surface mesurée par la caméra IR. Le principe est degénérer, par effet Joule, un flux de chaleur bien calibré à l’intérieur du tube, puis demesurer la température en surface. Pour cela, un fil d’étain est introduit dans le canalrempli d’eau (figure 4.13).
fil d'étain
plaque d'aluminium (Tc) tube en PFA (TR)
> >
>
Fig. 4.13 – Schéma représentant la puce isopéribolique lors de l’étalonnage des pertesthermiques
Une puissance est dissipée par effet Joule via le fil d’étain, ce qui engendre une élévationde température uniforme tout le long du tube, comme nous pouvons l’observer sur lafigure 4.13. Lorsque la température du tube ainsi que celle de la plaque sont stabilisées,une seule image infrarouge ou un film moyenné de tout le milliréacteur est enregistrée. Ainsi,la différence de température entre le tube et la plaque peut être déterminée. Nous réalisonscette expérience pour plusieurs valeurs de puissance, de 0,25 à 3 W. La température dela plaque est fixée à 10◦C. Dans cette situation particulière, le terme source par unité delongueur est uniforme et le profil de température qui en résulte est plat. Conformément àl’équation (4.8), la puissance dissipée équilibre parfaitement les pertes thermiques, ce quise traduit par la relation suivante :
P = K(TR − Tp) (4.9)
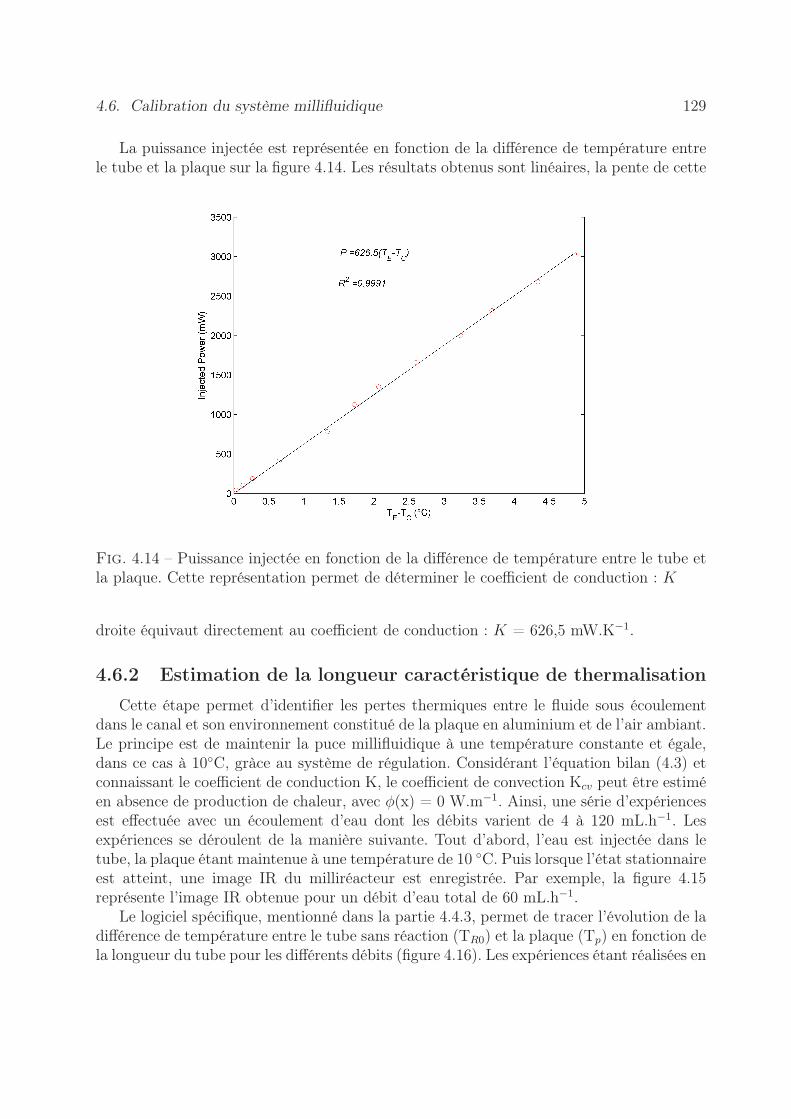
4.6. Calibration du système millifluidique 129
La puissance injectée est représentée en fonction de la différence de température entrele tube et la plaque sur la figure 4.14. Les résultats obtenus sont linéaires, la pente de cette
Fig. 4.14 – Puissance injectée en fonction de la différence de température entre le tube etla plaque. Cette représentation permet de déterminer le coefficient de conduction : K
droite équivaut directement au coefficient de conduction : K = 626,5 mW.K−1.
4.6.2 Estimation de la longueur caractéristique de thermalisation
Cette étape permet d’identifier les pertes thermiques entre le fluide sous écoulementdans le canal et son environnement constitué de la plaque en aluminium et de l’air ambiant.Le principe est de maintenir la puce millifluidique à une température constante et égale,dans ce cas à 10◦C, gràce au système de régulation. Considérant l’équation bilan (4.3) etconnaissant le coefficient de conduction K, le coefficient de convection Kcv peut être estiméen absence de production de chaleur, avec φ(x) = 0 W.m−1. Ainsi, une série d’expériencesest effectuée avec un écoulement d’eau dont les débits varient de 4 à 120 mL.h−1. Lesexpériences se déroulent de la manière suivante. Tout d’abord, l’eau est injectée dans letube, la plaque étant maintenue à une température de 10 ◦C. Puis lorsque l’état stationnaireest atteint, une image IR du milliréacteur est enregistrée. Par exemple, la figure 4.15représente l’image IR obtenue pour un débit d’eau total de 60 mL.h−1.
Le logiciel spécifique, mentionné dans la partie 4.4.3, permet de tracer l’évolution de ladifférence de température entre le tube sans réaction (TR0) et la plaque (Tp) en fonction dela longueur du tube pour les différents débits (figure 4.16). Les expériences étant réalisées en

130 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
50
100
150
200
Nu
mé
ro d
e p
ixe
l d
an
s la
dire
ctio
n y
20 40 60 80 100 120 140 160 180 200
Numéro de pixel dans la direction x
18
17
16
15
14
13
12
11
10
Fig. 4.15 – Image IR du milliréacteur isopéribolique, maintenu à une température de 10 ◦C,lors d’un écoulement d’eau à un débit de 60 mL.h−1, l’eau étant initialement à températureambiante (≈ 20 ◦C).
débit continu, au sein d’une puce isopéribolique et sans réaction chimique, l’équation (4.8)se réduit donc à :
dTR0
dx= −Kcv(TR0 − Tp), (4.10)
avec
Kcv =K
(ρCp)pappQL=
ωπd
(ρCp)pappRtQ(4.11)
La solution analytique de l’équation (4.10) peut être définie par l’expression suivante :
TR0(x) − Tp
T 0R0 − Tp
= exp(−Kcvx), (4.12)
avec T 0R0 la température du tube en x=0 (K).
La figure 4.17, permet d’estimer le coefficient de pertes thermiques par convection (Kcv)pour chaque débit. Nous pouvons remarquer que pour chaque débit, une droite est obtenueet la pente détermine la valeur de Kcv. Or log(Kcv) = a − log(Q), avec a = K
L(ρCp)app,
ainsi ces courbes donnent aussi accès à la valeur de (ρCp)app.Les résultats obtenus par ajustement des données montrent que la température à l’entrée
de la puce est constante pour les différents débits et égale à la température ambiante
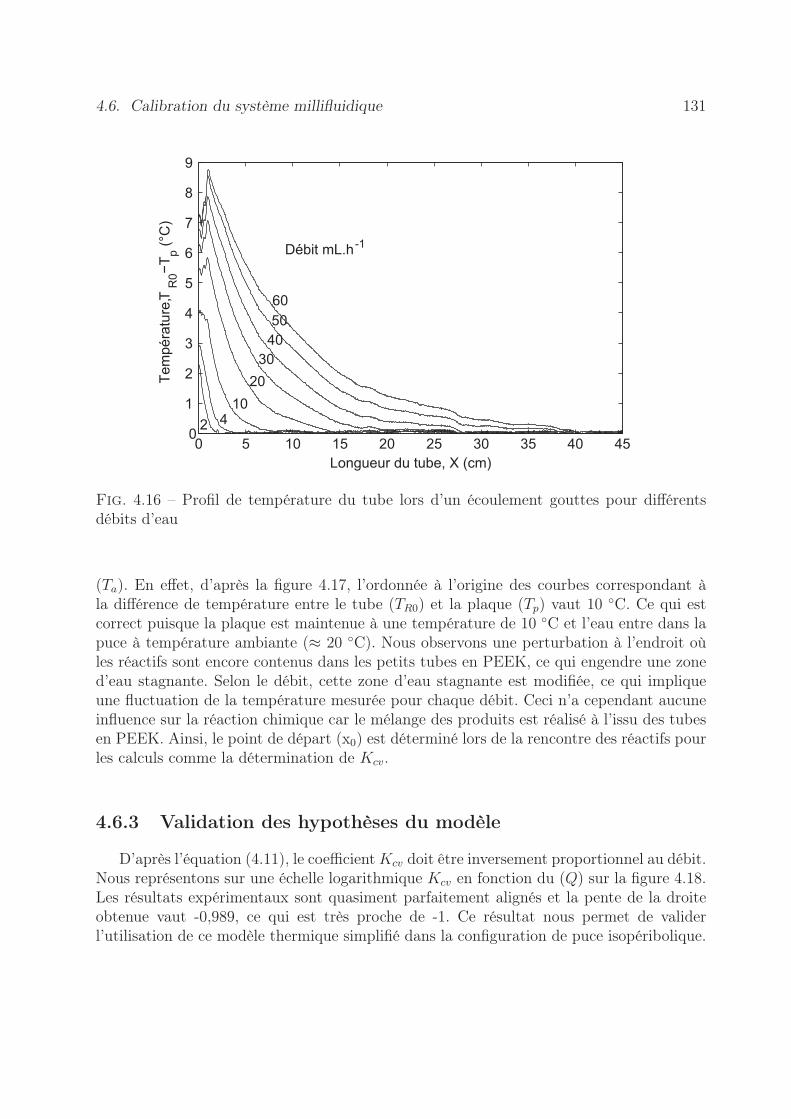
4.6. Calibration du système millifluidique 131
0 5 10 15 20 25 30 35 40 450
1
2
3
4
5
6
7
8
9
Longueur du tube, X (cm)
Tem
péra
ture
, TR
0−
Tp (
°C)
2
40
30
10
20
4
50
60
Débit mL.h -1
Fig. 4.16 – Profil de température du tube lors d’un écoulement gouttes pour différentsdébits d’eau
(Ta). En effet, d’après la figure 4.17, l’ordonnée à l’origine des courbes correspondant àla différence de température entre le tube (TR0) et la plaque (Tp) vaut 10 ◦C. Ce qui estcorrect puisque la plaque est maintenue à une température de 10 ◦C et l’eau entre dans lapuce à température ambiante (≈ 20 ◦C). Nous observons une perturbation à l’endroit oùles réactifs sont encore contenus dans les petits tubes en PEEK, ce qui engendre une zoned’eau stagnante. Selon le débit, cette zone d’eau stagnante est modifiée, ce qui impliqueune fluctuation de la température mesurée pour chaque débit. Ceci n’a cependant aucuneinfluence sur la réaction chimique car le mélange des produits est réalisé à l’issu des tubesen PEEK. Ainsi, le point de départ (x0) est déterminé lors de la rencontre des réactifs pourles calculs comme la détermination de Kcv.
4.6.3 Validation des hypothèses du modèle
D’après l’équation (4.11), le coefficient Kcv doit être inversement proportionnel au débit.Nous représentons sur une échelle logarithmique Kcv en fonction du (Q) sur la figure 4.18.Les résultats expérimentaux sont quasiment parfaitement alignés et la pente de la droiteobtenue vaut -0,989, ce qui est très proche de -1. Ce résultat nous permet de validerl’utilisation de ce modèle thermique simplifié dans la configuration de puce isopéribolique.
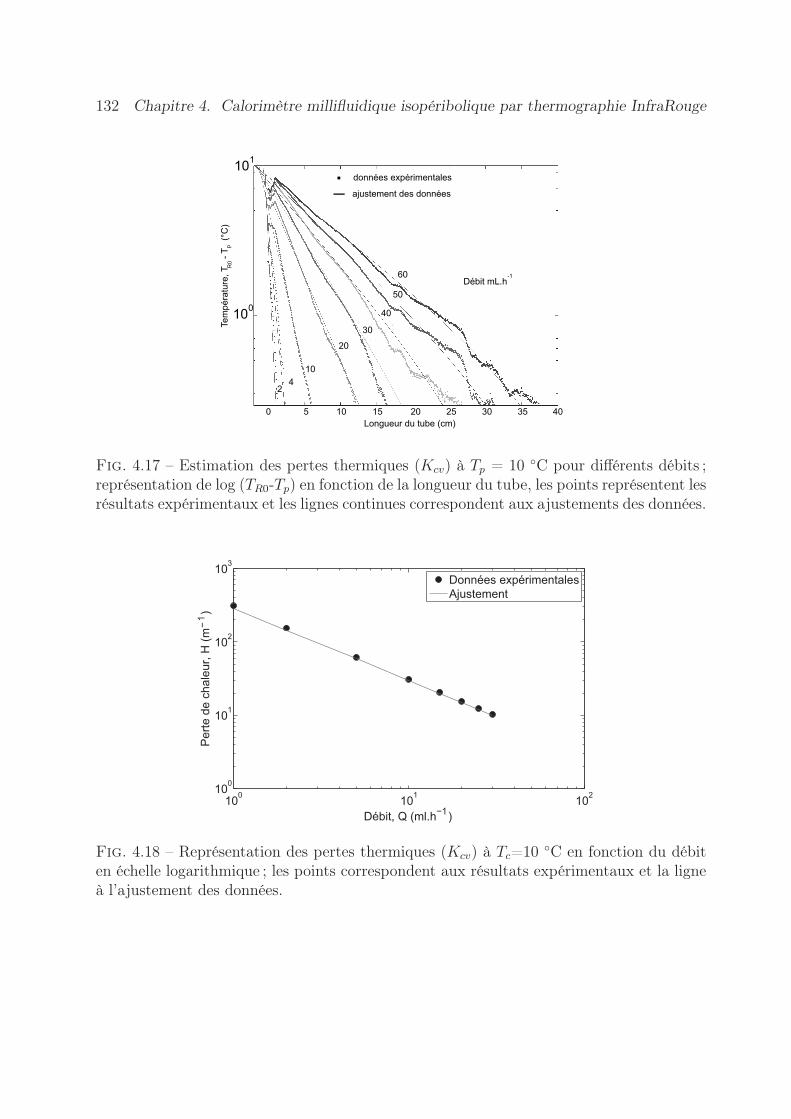
132 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
101
010
Tem
péra
ture
, T
-
T (
°C)
R0
p
Longueur du tube (cm)
0 5 10 15 20 25 30 35 40
Débit mL.h-1
données expérimentales
ajustement des données_ .
60
50
40
30
20
10
42
Fig. 4.17 – Estimation des pertes thermiques (Kcv) à Tp = 10 ◦C pour différents débits ;représentation de log (TR0-Tp) en fonction de la longueur du tube, les points représentent lesrésultats expérimentaux et les lignes continues correspondent aux ajustements des données.
100
101
102
100
101
102
103
Débit, Q (ml.h −1
)
Pe
rte
de
cha
leu
r, H
(m
−1)
Données expérimentales
Ajustement
Fig. 4.18 – Représentation des pertes thermiques (Kcv) à Tc=10 ◦C en fonction du débiten échelle logarithmique ; les points correspondent aux résultats expérimentaux et la ligneà l’ajustement des données.

4.7. Validation expérimentale de l’appareil 133
4.7 Validation expérimentale de l’appareil
4.7.1 Estimation du terme source
Dès lors que les paramètres K et Kcv du modèle sont déterminés, l’équation (4.8)permet d’évaluer directement le terme source à partir de la mesure expérimentale. Danscette expression, la dérivée spatiale est évaluée par un schéma de différence finie. Afind’exacerber l’influence de ce terme, nous pouvons également combiner les équations (4.8)et (4.10) pour faire apparaître une température TRch, représentant la différence entre leprofil de température en présence de réaction chimique et celui où le fluide se met entempérature. L’évolution de cet écart est régit par la relation (4.13) :
φ(x) =K
L
(TRch(x) +
dTRch(x)
dx
1
Kcv
)(4.13)
L’estimation du flux le long du tube permet d’évaluer les grandeurs caractéristiques d’uneréaction. Néanmoins, comme nous l’avons vu précédemment, dans un écoulement de typeco-courant, le mélange des espèces et donc des réactifs, se traduit par un temps de mélangedurant lequel la réaction chimique peut être limitée par la diffusion. Deux cas peuventdonc se produire. Si la réaction est très rapide et le temps de séjour supérieur au tempsde mélange, la réaction est totale et de ce fait l’intégrale du terme source le long tubedonne directement accès à l’enthalpie de réaction, connaissant le débit volumique Q et laconcentration molaire initiale en réactif limitant C :
Φ =
∫ x=L
x=0
φ(x)dx = QC∆H (4.14)
Si, au contraire le temps de mélange est très faible devant le temps caractéristique de laréaction, on peut directement relier le terme source au degré d’avancement de la réaction :
X(x) =1
QC∆H
∫ u=x
u=0
φ(u)du (4.15)
Compte tenu du fait que le régime stationnaire est atteint, il y a parfaite correspondanceentre l’espace et le temps : x = vt, où v est la vitesse débitante du fluide réactionnel.Ainsi, X découle directement la cinétique chimique au sein du milliréacteur. Il dépenddirectement des grandeurs caractéristiques intrinsèques de la vitesse de réaction, à savoir lesordres partiels par rapport à chacun des réactifs, la constante pré-exponentielle et l’énergied’activation (si le modèle d’Arrhénius est applicable). Ces paramètres sont identifiablespar optimisation sur un jeu d’expériences pour lesquelles les conditions initiales varient,i.e., les concentrations en réactifs de chacune des alimentations, ou plus simplement, lesrapports des débits de réactifs.
4.7.2 Expériences réalisées
Afin de valider le millicalorimètre sur la mesure de l’enthalpie de réaction, une réac-tion dont les données thermodynamiques sont bien connues est étudiée. Comme pour la
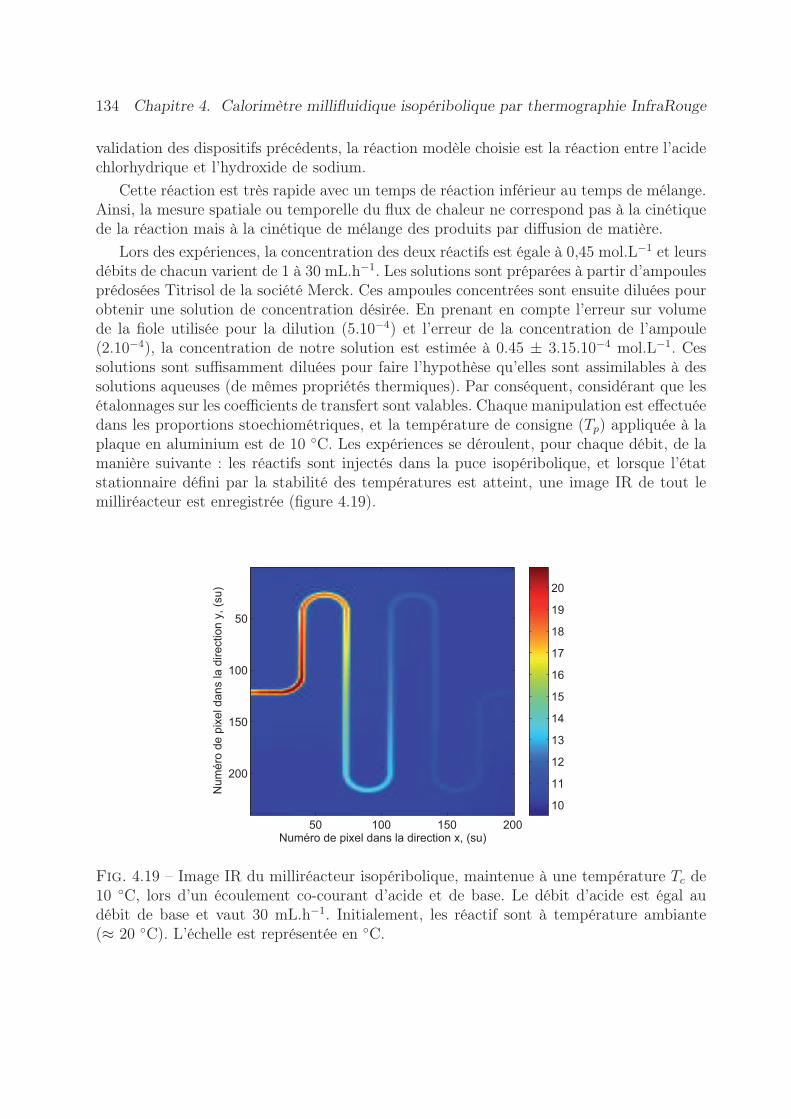
134 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
validation des dispositifs précédents, la réaction modèle choisie est la réaction entre l’acidechlorhydrique et l’hydroxide de sodium.
Cette réaction est très rapide avec un temps de réaction inférieur au temps de mélange.Ainsi, la mesure spatiale ou temporelle du flux de chaleur ne correspond pas à la cinétiquede la réaction mais à la cinétique de mélange des produits par diffusion de matière.
Lors des expériences, la concentration des deux réactifs est égale à 0,45 mol.L−1 et leursdébits de chacun varient de 1 à 30 mL.h−1. Les solutions sont préparées à partir d’ampoulesprédosées Titrisol de la société Merck. Ces ampoules concentrées sont ensuite diluées pourobtenir une solution de concentration désirée. En prenant en compte l’erreur sur volumede la fiole utilisée pour la dilution (5.10−4) et l’erreur de la concentration de l’ampoule(2.10−4), la concentration de notre solution est estimée à 0.45 ± 3.15.10−4 mol.L−1. Cessolutions sont suffisamment diluées pour faire l’hypothèse qu’elles sont assimilables à dessolutions aqueuses (de mêmes propriétés thermiques). Par conséquent, considérant que lesétalonnages sur les coefficients de transfert sont valables. Chaque manipulation est effectuéedans les proportions stoechiométriques, et la température de consigne (Tp) appliquée à laplaque en aluminium est de 10 ◦C. Les expériences se déroulent, pour chaque débit, de lamanière suivante : les réactifs sont injectés dans la puce isopéribolique, et lorsque l’étatstationnaire défini par la stabilité des températures est atteint, une image IR de tout lemilliréacteur est enregistrée (figure 4.19).
Numéro de pixel dans la direction x, (su)
Nu
mé
ro d
e p
ixe
l d
an
s la
dire
ctio
n y
, (s
u)
50 100 150 200
50
100
150
200
10
11
12
13
14
15
16
17
18
19
20
Fig. 4.19 – Image IR du milliréacteur isopéribolique, maintenue à une température Tc de10 ◦C, lors d’un écoulement co-courant d’acide et de base. Le débit d’acide est égal audébit de base et vaut 30 mL.h−1. Initialement, les réactif sont à température ambiante(≈ 20 ◦C). L’échelle est représentée en ◦C.
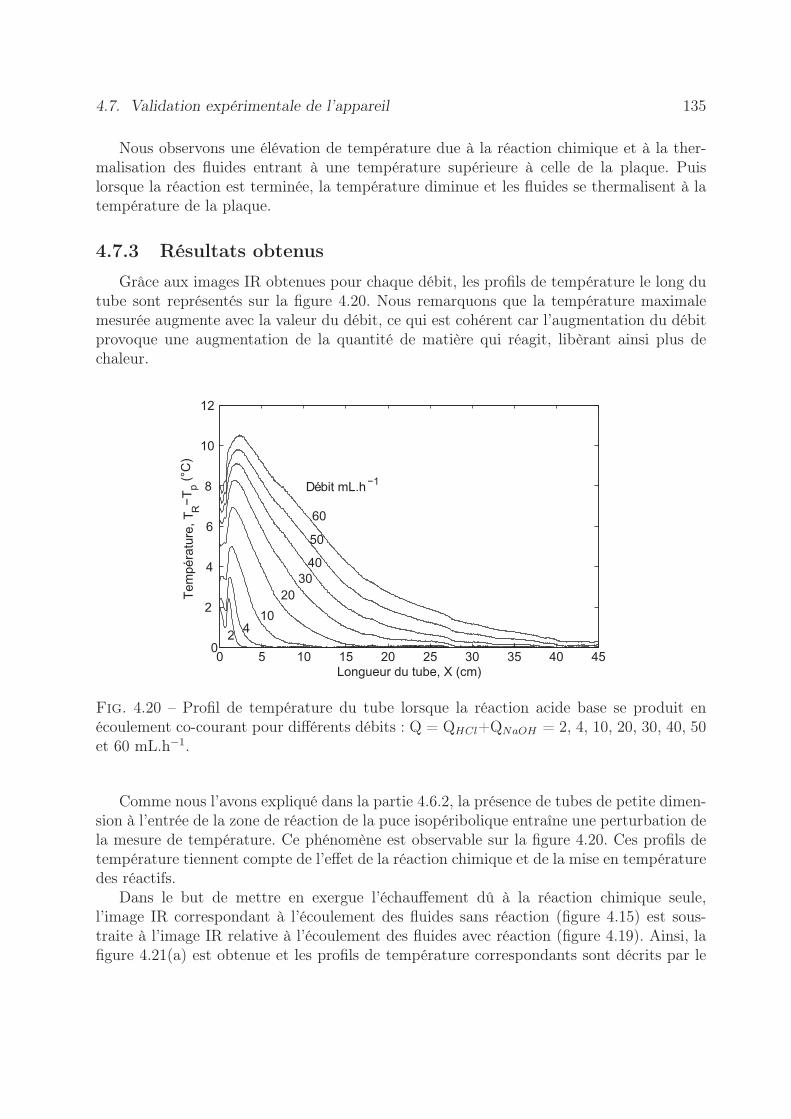
4.7. Validation expérimentale de l’appareil 135
Nous observons une élévation de température due à la réaction chimique et à la ther-malisation des fluides entrant à une température supérieure à celle de la plaque. Puislorsque la réaction est terminée, la température diminue et les fluides se thermalisent à latempérature de la plaque.
4.7.3 Résultats obtenus
Grâce aux images IR obtenues pour chaque débit, les profils de température le long dutube sont représentés sur la figure 4.20. Nous remarquons que la température maximalemesurée augmente avec la valeur du débit, ce qui est cohérent car l’augmentation du débitprovoque une augmentation de la quantité de matière qui réagit, libèrant ainsi plus dechaleur.
0 5 10 15 20 25 30 35 40 450
2
4
6
8
10
12
Longueur du tube, X (cm)
Tem
péra
ture
, T
R−
Tp (
°C)
24
10
20
30
40
50
60
Débit mL.h−1
Fig. 4.20 – Profil de température du tube lorsque la réaction acide base se produit enécoulement co-courant pour différents débits : Q = QHCl+QNaOH = 2, 4, 10, 20, 30, 40, 50et 60 mL.h−1.
Comme nous l’avons expliqué dans la partie 4.6.2, la présence de tubes de petite dimen-sion à l’entrée de la zone de réaction de la puce isopéribolique entraîne une perturbation dela mesure de température. Ce phénomène est observable sur la figure 4.20. Ces profils detempérature tiennent compte de l’effet de la réaction chimique et de la mise en températuredes réactifs.
Dans le but de mettre en exergue l’échauffement dû à la réaction chimique seule,l’image IR correspondant à l’écoulement des fluides sans réaction (figure 4.15) est sous-traite à l’image IR relative à l’écoulement des fluides avec réaction (figure 4.19). Ainsi, lafigure 4.21(a) est obtenue et les profils de température correspondants sont décrits par le
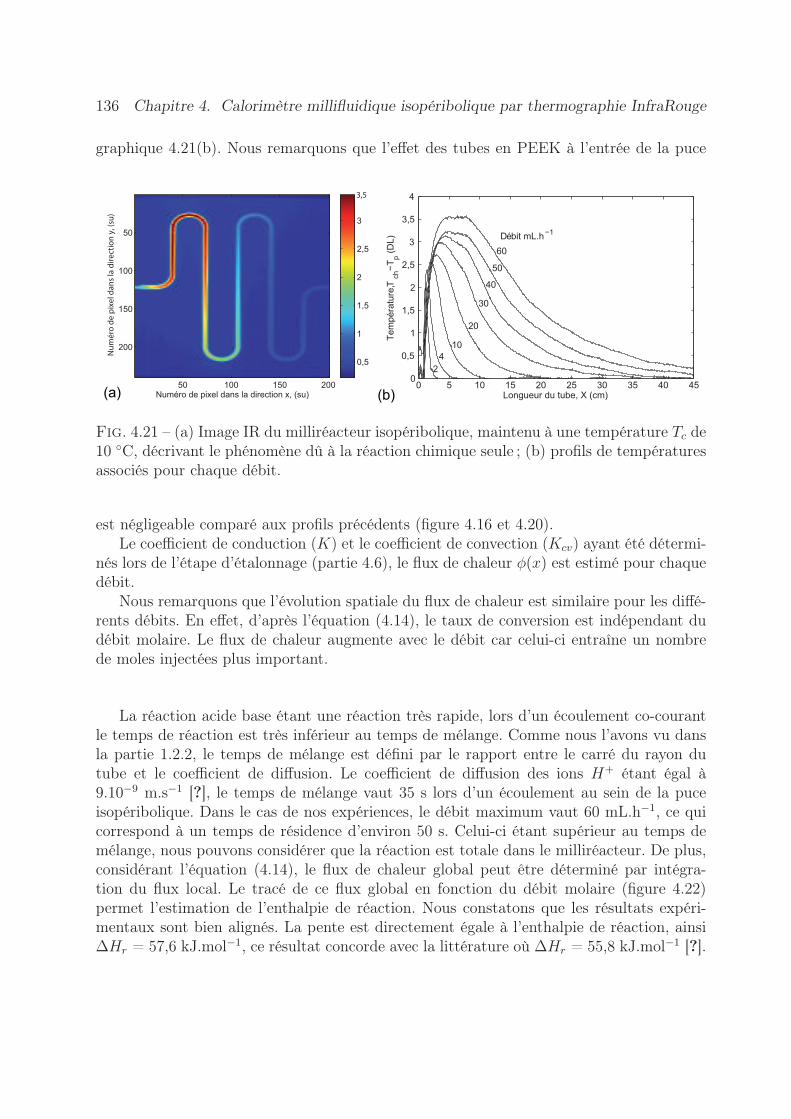
136 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
graphique 4.21(b). Nous remarquons que l’effet des tubes en PEEK à l’entrée de la puce
Numéro de pixel dans la direction x, (su)
Pix
el nu
mber
in y
direction,
(wu)
50 100 150 200
50
100
150
200
0,5
1
1,5
2
2,5
3
0 5 10 15 20 25 30 35 40 450
0,5
1
1,5
2
2,5
3
3,5
4
Longueur du tube, X (cm)T
em
péra
ture
, Tch−
Tp (
DL
)
2
4
10
20
60
Débit mL.h−1
30
50
40
(b)
Nu
mé
ro d
e p
ixe
l da
ns
la d
ire
ctio
n y
, (su
)
3,5
(a)
Fig. 4.21 – (a) Image IR du milliréacteur isopéribolique, maintenu à une température Tc de10 ◦C, décrivant le phénomène dû à la réaction chimique seule ; (b) profils de températuresassociés pour chaque débit.
est négligeable comparé aux profils précédents (figure 4.16 et 4.20).Le coefficient de conduction (K) et le coefficient de convection (Kcv) ayant été détermi-
nés lors de l’étape d’étalonnage (partie 4.6), le flux de chaleur φ(x) est estimé pour chaquedébit.
Nous remarquons que l’évolution spatiale du flux de chaleur est similaire pour les diffé-rents débits. En effet, d’après l’équation (4.14), le taux de conversion est indépendant dudébit molaire. Le flux de chaleur augmente avec le débit car celui-ci entraîne un nombrede moles injectées plus important.
La réaction acide base étant une réaction très rapide, lors d’un écoulement co-courantle temps de réaction est très inférieur au temps de mélange. Comme nous l’avons vu dansla partie 1.2.2, le temps de mélange est défini par le rapport entre le carré du rayon dutube et le coefficient de diffusion. Le coefficient de diffusion des ions H+ étant égal à9.10−9 m.s−1 [?], le temps de mélange vaut 35 s lors d’un écoulement au sein de la puceisopéribolique. Dans le cas de nos expériences, le débit maximum vaut 60 mL.h−1, ce quicorrespond à un temps de résidence d’environ 50 s. Celui-ci étant supérieur au temps demélange, nous pouvons considérer que la réaction est totale dans le milliréacteur. De plus,considérant l’équation (4.14), le flux de chaleur global peut être déterminé par intégra-tion du flux local. Le tracé de ce flux global en fonction du débit molaire (figure 4.22)permet l’estimation de l’enthalpie de réaction. Nous constatons que les résultats expéri-mentaux sont bien alignés. La pente est directement égale à l’enthalpie de réaction, ainsi∆Hr = 57,6 kJ.mol−1, ce résultat concorde avec la littérature où ∆Hr = 55,8 kJ.mol−1 [?].
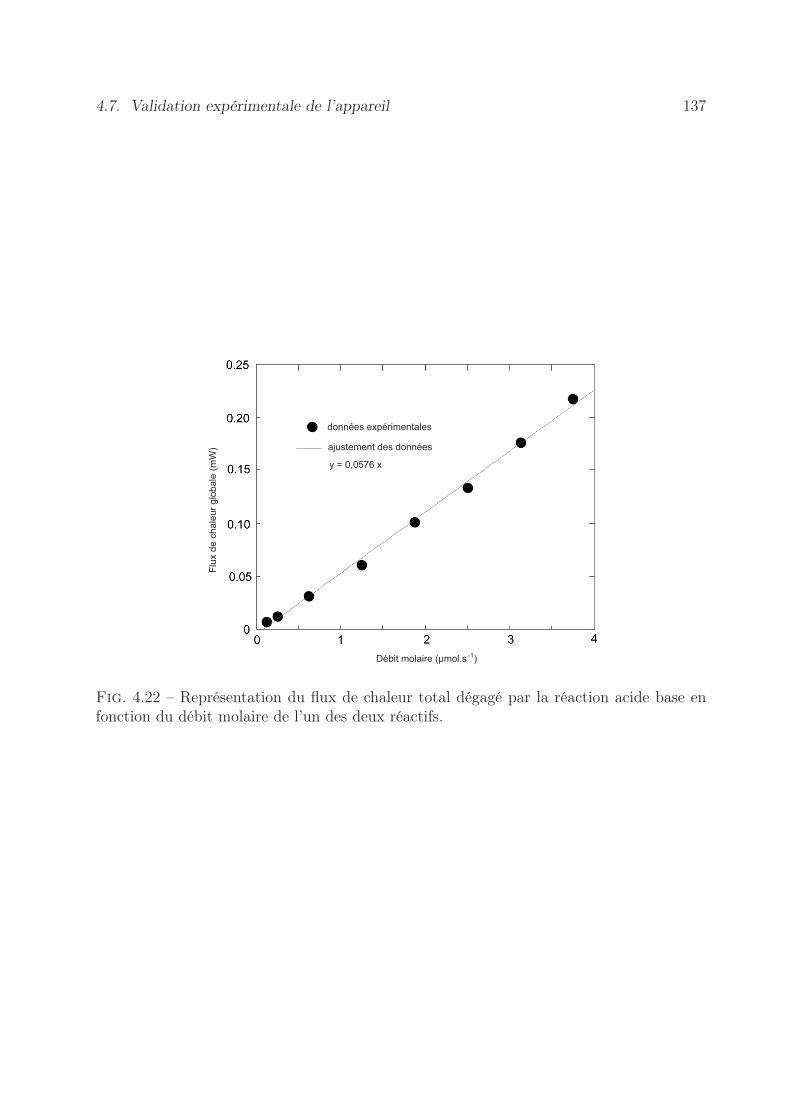
4.7. Validation expérimentale de l’appareil 137
données expérimentales
ajustement des données
y = 0,0576 x
Débit molaire (µmol.s )-1
Flu
x d
e c
ha
leu
r g
lob
ale
(m
W)
Fig. 4.22 – Représentation du flux de chaleur total dégagé par la réaction acide base enfonction du débit molaire de l’un des deux réactifs.

138 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge
4.8 Conclusion
Un millicalorimètre sans contact, composé d’une caméra IR et d’une puce de typeisopéribolique, a été développé puis validé en écoulement co-courant par la caractérisationd’une réaction chimique.
Au cours de ce chapitre, nous avons démontré comment obtenir une mesure quantita-tive de flux de chaleur à partir de températures mesurées par une caméra IR. Pour celaune procédure d’étalonnage visant à déterminer les coefficients de conduction et de convec-tion associés à un modèle thermique simplifié sont nécessaires. Ensuite, l’utilisation d’uneméthode inverse permet d’estimer la valeur du terme source le long du canal. De plus, ungrand nombre de points de mesure est obtenu le long du canal (de l’ordre de 1200 points).Cela permet, en fonction du débit, le suivi de cinétique aux temps courts. Par exemple, untemps de résidence de 1 min correspond à une mesure toutes les 0,05 s.
Un autre point important et original de ce travail est la conception simple d’une puceisopéribolique à partir d’un tube en PFA et d’une plaque en aluminium. Ce type de puceest très simple à mettre en oeuvre et possède un caractère modulable (figure 4.23). En effet,il est très facile de faire varier le volume du réacteur ou le temps de résidence en jouant parexemple sur le diamètre interne du tuyau, en modifiant les dimensions de la plaque ou leparcours du tube. Il est aussi envisageable de juxtaposer plusieurs plaques afin d’imposerdifférentes températures de consigne ou bien différents volumes, de sorte à se conformer leplus précisément possible aux procédés industriels.
Tc1=100°C
Tc2=30°C Tc3=30°C
Tc4= -15°C
(b)(a)
Fig. 4.23 – Aspect modulable de milliréacteur (a) Schéma de la juxtaposition de quatremilliréacteurs ; (b) Photographie d’un milliréacteur isopéribolique de 2 mL de volume.

4.8. Conclusion 139
Cette étude a nécessité un travail expérimental considérable qui a permis d’établirun riche retour d’expérience, ainsi que la mise en oeuvre d’une démarche expérimentalecomposée d’une étape d’étalonnage, de thermalisation et de réaction. Au cours des diversesexpériences effectuées, nous avons pu remarquer que la détermination du coefficient dethermalisation (Kcv) est précise et efficace si l’écart de température entre les fluides àl’entrée et la plaque est au minimum de 10 ◦C. Un autre point important concerne l’étapede réaction où les fluides doivent être thermalisés pour améliorer la mesure de flux dechaleur. L’ensemble de ce travail permet de décrire les performances de ce dispositif :
– 1 : utilisation de faibles volumes réactionnels de l’ordre du µL au mL,– 2 : large gamme de temps de résidence : de la minute à l’heure,– 3 : large gamme de débit : de l’ordre du mL.h−1 à 100 mL.h−1,– 4 : large gamme de température : de 5 à 90 ◦C,– 5 : bonne sensibilité de la mesure 100 mW,– 6 : large gamme d’enthalpie mesurable : du kJ.mol−1 au MJ.mol−1.
Le temps de mélange lors d’un écoulement co-courant est non négligeable au sein dece dispositif, il est d’environ 30 s lors de la diffusion des ions H+ dans l’eau. Ainsi, lacaractérisation de la cinétique d’une réaction très rapide est compromise ou nécessiteraitun couplage complexe entre le bilan d’énergie et le bilan matière. Ceci dit, nous avonspu retrouver l’enthalpie de cette réaction, validant ainsi notre dispositif qui pourra êtreutilisé pour la détermination de cinétiques de réactions plus lentes (de quelques minutes àquelques heures).

140 Chapitre 4. Calorimètre millifluidique isopéribolique par thermographie InfraRouge

Conclusions et perspectives
Au cours de cette thèse nous avons mis au point plusieurs méthodes d’analyses ther-miques appliquées à la caractérisation de réactions chimiques au sein de dipositifs micro-fluidiques. De part leurs nombreux avantages, les dispositifs microfluidiques s’avèrent trèsprometteurs pour :
– la mise en œuvre de réactions chimiques en continu sous deux types d’écoulements :co-courant et gouttes
– l’utilisation de faibles volumes réactionnels.
Après avoir présenté la microfluidique, ses configurations d’écoulement ainsi que sesprincipales applications en tant qu’outil d’analyse, nous avons focalisé notre attention surles appareils couplant microfluidique et analyse thermique. Nous avons constaté que latechnique prépondérante consiste à intégrer un ou plusieurs capteurs de température ausein de la puce. Dans le but d’étoffer ce type de techniques, nous avons proposé plusieursméthodes au cours des différents chapitres de ce mémoire.
La première a consisté en une mesure globale par un capteur de flux externe en contactavec le microréacteur. Le microcalorimètre ainsi développé permet la mesure du flux dechaleur lors des deux types d’écoulement. Le microréacteur, dont le volume vaut quelquesdizaines de µL, a été conçu en optimisant son transfert thermique par l’application d’unmodèle thermique (pertes thermiques < 0,1 %). La mesure du flux de chaleur est possiblesi un étalonnage préalable a été effectué. D’après une étude de l’incertitude de la mesure,la précision de cet appareil est de 3,4 % et sa limite de détection est estimée à environ 100mJ.mol−1. La caractérisation d’une réaction acido-basique a permis de valider ce dispositifpour la mesure de l’enthalpie lors de l’écoulement co-courant et gouttes. D’autre part,diverses fonctionnalités du dipositif ont été explorées :
– estimation de l’enthalpie de réaction,– estimation de l’entalpie molaire d’excès,– mesure de la concentration par dosage calorimètrique,– mise en évidence de la dynamique de mélange.
L’utilisation de cet appareil est assez simple et est totalement automatisée. Le temps demise en œuvre pour obtenir une enthalpie de réaction est d’environ 2 heures. En effet,notre méthode nécessite d’établir un régime permanent pour effectuer les mesures. Uneamélioration que l’on pourrait apporter serait de s’intéresser au régime transitoire qui
141

142
permettrait de diminuer considérablement le temps expérimental qui passerait à moins de30 minutes.
Une technique de mesure de température de surface par TIR a été explorée dans ledeuxième chapitre. Nous avons établi un modèle simplifié qui, par une méthode d’estimationà un paramètre, permet de déterminer une cartographie du nombre de Péclet et du termesource. Pour cela, une étape préalable de calibration étant primordiale, nous avons élaboréet testé par simulation et expérimentalement une méthode à flux local imposé et une autreà gradient de température. Cette dernière engendre un champ de température sur toute lalongueur du canal, ce qui permet une estimation du nombre de Péclet sur l’ensemble ducanal. Puis, une réaction acido-basique a été caractérisée par l’estimation temporelle duterme source pour différents débits, conduisant à l’enthalpie de réaction.
Nous avons appliqué ces méthodes d’estimation à un écoulement gouttes. La réactionacide-base étant quasi-instantanée, elle est utilisée comme production de flux afin de cali-brer le dispositif. Ainsi, des cartographies du nombre de Péclet de la goutte ont été obte-nues. Ces premiers résultats étant comparables aux données représentatives des champs devitesse à l’intérieur de la gouttes par analyse PIV sont très encourageants et prometteurs.
Enfin, nous avons développé un nouveau réacteur millifluidique isopéribolique où lesphénomènes liés à la diffusion spatiale sont mieux maîtrisés. Ainsi, une mesure quantitativede l’enthalpie de réaction est possible par thermographie IR. Une méthode de calibration,nécessitant l’estimation des pertes thermiques et du coefficient relatif à la longueur dethermalisation, a été definie puis expérimentée. Enfin, l’appareil a été validé par la me-sure d’enthalpie d’une réaction acido-basique standard dont les valeurs concordent avecla littérature. Ce dispositif présente l’avantage de contrôler la diffusion thermique et ainsid’obtenir une information quantitative. Toutefois, au vu des dimensions du milliréacteurla résolution spatiale est nettement plus faible que le technique précédente.
Nous pouvons noter qu’une partie de ce travail de recherche a permis d’établir différentesméthodes expérimentales d’étalonnage adaptées au dispositif de mesure. Tout d’abord,l’utilisation d’une résistance chauffante intégrée à la puce microfluidique a été mise aupoint pour étalonner le microcalorimètre. La relation entre la puissance injectée et la tensionmesurée a été déterminée. Puis, une méthode à flux local a été employée pour configurer lesméthodes TIR. De plus, une autre manière d’étalonnage a consisté à appliquer un gradientde température sur le microréacteur en obtenant un champ de température distinct surtoute la longueur du microcanal. De cette façon, le nombre de Péclet a pu être estimé.Dans le cadre de la caractérisation de l’écoulement gouttes par TIR, une nouvelle méthodea été investiguée. Celle-ci consiste à utiliser la réaction acido-basique comme terme source.En effet, cette réaction est quasi instantanée ce qui correspond à la réponse impulsionelleen thermique. La quatrième méthode concerne le calorimètre isopéribolique et utilise unfil chaud et une mesure de différence de température.

143
Le microcalorimètre et le millicalorimètre ont été validé et permettent des mesuresquantitatives. Leurs proriétés intrinsèques étant différentes, il est intéressant de comparerleurs performances :
Microcalorimètre Millicalorimètrevolume du réacteur nL au µL µL au mL
gamme de temps de résidence de la seconde à l’heure de la minute à l’heuregamme de débit 20 µL.h−1 à 1 mL.h−1 1 mL.h−1 à 100 mL.h−1
gamme de température de 5 à 90 ◦C de 5 à 90 ◦Csensibilité de la mesure 10 µW 100 mW
gamme d’enthalpie mesurable 100 J.mol−1 au MJ.mol−1 kJ.mol−1 au MJ.mol−1
Le microcalorimètre offre une meilleure précision et un volume réactionnel plus faible.Ainsi, les transformations chimiques à faibles enthalpies seront caractérisées par ce dernier.Toutefois, le PDMS, matrice du microréacteur, est perméable à de nombreux solvants etfaiblement inerte chimiquement, ce qui limite les applications à des réactions mettant enœuvre des produits très concentrés. Le millicalorimètre permet de palier ces problèmes parl’utilisation de tubes de natures variables selon des réactifs et produits.
Les perspectives d’applications de ces dispositifs sont nombreuses, quelques exemplessont cités par la suite.
Bien que le dispositif microfluidique associé à la caméra IR permette une approchelocale de résolution spatiale importante, cette méthode reste à optimiser. En effet un cou-plage avec le capteur global (microcalorimètre) est indispensable pour obtenir une valeurquantitative du terme source. D’autre part, une modélisation plus précise est à envisager :
– analyse du déséquilibre de la température entre la plaque et le fluide,– étude en régime transitoire,– estimations simultanées d’un grand nombre de points.En collaboration avec le laboratoire de génie des procédés de Toulouse, nous débu-
tons l’étude de la réaction très exothermique de la nitration du toluène dans le dispositifmillifluidique qui présente un faible volume réactionnel (1 mL) et des matériaux inerteschimiquement.
Des études ont été initiées pour la caractérisation de réactions chimiques d’intérêt pourRhodia, pour lesquells les propriétés thermodynamiques sont nécessaires pour le dimen-sionnement des réacteurs.
Il serait aussi très intéressant de coupler le millicalorimètre isopéribolique avec unetechnique d’analyses spectroscopiques tels que la spectroscopie Raman. Cela permettraitde cumuler les informations thermiques et de concentration en une seule étape.

144

Annexe A
Microcalorimètre différentiel par mesure
macroscopique
Modélisation thermique
Solution analytique
Nous avons vu au cours de la partie modélisation thermique (partie 2.4, 45) que le bilanthermique appliqué à ce système pour les trois milieux (i = 1, 2 ou 3) s’écrit de la façonsuivante :
∂2Ti
∂x2+
∂2Ti
∂y2=
1
ai
∂Ti
∂t, ai =
λi
ρiCp,i
(A.1)
i étant l’indice représentant le milieu considéré, T la température (K), x l’abscisse(m), y l’ordonnée (m), a la diffusivité thermique (m2.s−1), t le temps (s), λ la conducti-vité thermique (W.m−1.K−1), ρ la masse volumique (kg.m−3) et Cp la capacité calorifique(J.kg−1.K−1).
Les propriétés de ces trois milieux sont répértoriées dans le tableau A.1.
Nature Largeur (m) e (m) λ(W.m−1.K−1) ρCp (MJ.kg−1.m−3)Silicium 0,05 500.10−6 148 1,6Verre 0,05 500.10−6 1,3 1,8PDMS 0,05 0,01 0,1 2Peltier 0,05 3.10−3 1 1
Tab. A.1 – Propriétés thermiques des différents milieux nécessaires à la résolution du modèlethermique
En tenant compte des conditions limites du problème pour chacun des milieux, onobtient les systèmes d’équations A.10, A.11 et A.12.
145

146
Pour le milieu 1 (Peltier) on a :
∂2T1
∂x2 + ∂2T1
∂y2 = 1a1
∂T1
∂t
t = 0 → T1(t) = 0
x = 0 → ∂T1
∂x= 0
x = L → ∂T1
∂x= 0
y = 0 → T1(x, 0) = 0
y = e1 → −λ1∂T1
∂y= −λ2
∂T1
∂yet T1 = T2
(A.2)
Pour le milieu 2 (substrat) on a :
∂2T2
∂x2 + ∂2T2
∂y2 = 1a2
∂T2
∂t
t = 0 → T2(t) = 0
x = 0 → ∂T2
∂t= 0
x = L → −λ2∂T2
∂x= 0
y = e1 + e2 → −λ2∂T2
∂x+ φ(x, t) = −λ3
∂T3
∂xet T2 = T3
(A.3)
Et pour le milieu 3 (PDMS), on obtient :
∂2T3
∂x2 + ∂2T3
∂y2 = 1a3
∂T3
∂t
t = 0 → T3(t) = 0
x = 0 → ∂T3
∂x= 0
x = L → ∂T3
∂x= 0
y = e1 + e2 + e3 → −λ3∂T3
∂y= hT3
(A.4)
h étant le coefficient global de transfert thermique par convection (W.m−2.K−1) et φ leterme source dégagé dans le canal, moyenné selon z (W.m−2).
Pour résoudre ce système, nous utilisons la méthode des quadripoles qui permet detransformer le système différentiel en un problème purement algébrique. La solution sedéduit par transformés inverses, de la résolution d’un système linéaire [63]. Cette méthode

147
permet de résoudre les problèmes de diffusion thermique par une représentation simple,sous forme matricielle, de systèmes linéaires.
Il est possible d’appliquer à la température une transformée de Fourier (équation A.5)et de Laplace (équation A.6), ce qui donne la transformée θ représentée par l’équation A.7.
θ(y, αn) =∫ L
x=0T (x, y) cos(αnx)dx
avec αn = nπL
(A.5)
θ(p) =
∫ +∞
t=0
T (t)exp(−pt)dt (A.6)
7−→
θ(y, αn, p) =∫ +∞
t=0
∫ L
x=0T (x, y, t) cos(αnx)exp(−pt)dxdt
avec αn = nπL
(A.7)
Nous appliquons cette transformée aux milieux i et nous obtenons, pour chacun desmilieux, la formulation suivante :
d2θi(y, αn, p)dy2 − β2
nθi(y, αn, p) = 0
β2n = α2
n + p
ai
(A.8)
Ce système ( A.8) a pour solution générale l’expression suivante :
θi(y, αn, p) = Ach(βny) + Bsh(βny)
Φi(y, αn, p) = −λdθi(y, αn)dy
= −λiβi[Ash(βny) + Bch(βny)](A.9)
Les systèmes d’équations pour les différents milieux sont réécrits dans l’espace trans-formé et s’écrivent de la façon suivantes :
Milieu 1
d2θ1(y, αn, p)dy2 − β2
nθ1(y, αn, p) = 0
y = 0 → θ1(y, αn, p) = 0
y = e1 → −λ1dθ1(y, αn, p)
dy= −λ2
dθ2(y, αn, p)dy
et θ1(y, αn, p) = θ2(y, αn, p)
(A.10)

148
Milieu 2
d2θ2(y, αn, p)dy2 − β2
nθ2(y, αn, p) = 0
y = e1 → −λ1dθ1(y, αn, p)
dy= −λ2
dθ2(y, αn, p)dy
, et θ1(y, αn, p) = θ2(y, αn, p)
y = e1 + e2 → −λ2dθ2(y, αn, p)
dy+ Φ̃(αn, p) = −λ3
dθ3(y, αn, p)dy
et dθ2(y, αn, p) = θ3(y, αn, p)(A.11)
Milieu 3
d2θ3(y, αn, p)dy2 − β2
nθ3(y, αn, p) = 0
y = e1 + e2 → −λ2dθ2(y, αn, p)
dy+ Φ̃(αn, p) = −λ3
dθ3(y, αn, p)dy
et θ2(y, αn, p) = θ3(y, αn, p)
y = e1 + e2 + e3 → −λ3dθ3(y, αn, p)
dy= hθ3(y, αn, p)
(A.12)
Dans ces conditions le terme du flux s’écrit de la manière suivante dans l’espace trans-formé :
Φ̃(αn, p) =Q0sin(αnb)
αn
∫ +∞
0
H(t)e−1
pdt (A.13)
En appliquant les conditions aux limites en y=ei−1 et y=ei, on peut écrire le système sousla forme matricielle suivante :
{θi(0, βn)Φi(0, βn)
}=
(ch(βnei)
sh(βnei)λiβn
λiβnsh(βnei) ch(βnei)
){θi(ei, βn)Φi(ei, βn)
}(A.14)
Le problème à résoudre peut ensuite se réécrire sous ce formalisme :
{0Φ1(0, βn)
}=
(A1 B1
C1 D1)
)(A2 B2
C2 D2)
){θ2(e1 + e2, βn)Φ2(e1 + e2, βn)
}(A.15)
avec
{θ2(e1 + e2, βn)Φ2(e1 + e2, βn)
}=
{θ3(e1 + e2, βn)
Φ3(e1 + e2, βn) − Φ̃(βn, p)
}(A.16)
et {θ3(e3 + e2, βn)Φ3(e3 + e2, βn)
}=
(A3 B3
C3 D3
){θ3(e1 + e2 + e3, βn)hθ3(e1 + e2 + e3, βn)
}(A.17)
avec
{Ai = Di = ch(βnei)
Bi = sh(βnei)λiβn
et Ci = λiβnsh(βnei)(A.18)

149
De ces systèmes, la température du substrat (e1+e2) peut être déterminée dans l’espacetransformé :
θ2(e1 + e2, βn) =
(A1B2+B1D2
A1A2+B1C2
)
1 +(
A1B2+B1D2
A1A2+B1C2
)( C3h
+D3
A3h
+B3
)
QOsin(αnb)F (P )
αn
(A.19)
Finalement le calcul dans l’espace réel de θ2(e1 + e2, βn) est fait par une inversion deStehfest pour le domaine temporel et de Fourier pour le domaine spatial :
T2(x, e1 + e2, t) = 1L(τ(0, e1 + e2, t) + 2)
∑+∞
n=1 τ(αn, e1 + e2, t)cos(αn, xn)
avec :τ(αn, e1 + e2, t) = F−1(θ(αn, e1 + e2, p))
où :θ(0, e1 + e2, βn) =
(A1B2+B1D2A1A2+B1C2
)
1+(
A1B2+B1D2A1A2+B1C2
)( C3h
+D3
A3h
+B3
)
QObF (P )
(A.20)
À partir de ce modèle, une étude de dimensionnement du microréacteur peut êtreréalisée (chapitre 2.4).

150

Bibliographie
[1] A. Zogg, F. Stoessel, U. Fischer, and K. Hungerbuhler. Isothermal reaction calorimetryas a tool for kinetic analysis. Thermochimica Acta, 419(1-2) :1–17, 2004.
[2] W. Regenass. The development of heat flow calorimetry as a tool for process optimi-zation and process safety. Journal of Thermal Analysis, 49(3) :1661–1675, 1997.
[3] P. Tabeling. Introduction à la microfluidique. Belin ; Collection Echelles, 2003.
[4] P. Guillot et al. A rheometer on a microfluidic chip. Houille Blanche-Revue Interna-tionale de l Eau, (3) :43–49, 2006.
[5] J. Leng, B. Lonetti, P. Tabeling, M. Joanicot, and A. Ajdari. Microevaporators forkinetic exploration of phase diagrams. Physical Review Letters, 96(8), 2006.
[6] J. Leng and J. B. Salmon. Microfluidic crystallization. Lab on A Chip, 9(1) :24–34,2009.
[7] G. Cristobal et al. On-line laser raman spectroscopic probing of droplets engineeredin microfluidic devices. Lab on A Chip, 6(9) :1140–1146, 2006.
[8] F. Sarrazin, T. Bonometti, L. Prat, C. Gourdon, and J. Magnaudet. Hydrodynamicstructures of droplets engineered in rectangular micro-channels. Microfluidics andNanofluidics, 5(1) :131–137, 2008.
[9] W. Engl, M. Tachibana, A. Colin, and P. Panizza. A droplet-based high-throughputtubular platform to extract rate constants of slow chemical reactions. Chemical En-gineering Science, 63(6) :1692–1695, 2008.
[10] J. M. Köhler and Martin Zieren. Chip reactor for microfluid calorimetry. Thermochi-mica Acta, 310(1-2) :25–35, 1998.
[11] M. A. Schneider and R. Stoessel. Determination of the kinetic parameters of fast exo-thermal reactions using a novel microreactor-based calorimeter. Chemical EngineeringJournal, 115(1-2) :73–83, 2005.
[12] K. P. Mollmann, N. Lutz, and M. Vollmer. Thermography of microsystems. Inframa-tion 2004 Proceedings, ITC 104 A 2004-07-27, 2004.
[13] David Ross, Michael Gaitan, and Laurie E. Locascio. Temperature measurementin microfluidic systems using a temperature-dependent fluorescent dye. AnalyticalChemistry, 73(17) :4117–4123, 2001.
151

152 Bibliographie
[14] C. Pradere, M. Joanicot, J. C. Batsale, J. Toutain, and C. Gourdon. Processingof temperature field in chemical microreactors with infrared thermography. QIRT(Quantitative InfraRed Thermography), 3(1) :117–135, 2006.
[15] A. Gavriilidis, P. Angeli, E. Cao, K. K. Yeong, and Y. S. S. Wan. Technology andapplications of microengineered reactors. Chemical Engineering Research and Design,80(A1) :3–30, 2002.
[16] R. Wunsch, M. Fichtner, O. Gorke, K. Haas-Santo, and K. Schubert. Process ofapplying al2o3 coatings in microchannels of completely manufactured microstructuredreactors. Chemical Engineering and Technology, 25(7) :700–703, 2002.
[17] T. Wu, Y. Mei, J. T. Cabral, C. Xu, and K. L. Beers. A new synthetic methodfor controlled polymerization using a microfluidic system. Journal of the AmericanChemical Society, 126(32) :9880–9881, 2004.
[18] C. Harrison, J. Cabral, C. M. Stafford, A. Karim, and E. J. Amis. A rapid prototypingtechnique for the fabrication of solvent-resistant structures. Journal of Micromecha-nics and Microengineering, 14(1) :153–158, 2004.
[19] R. Barrett et al. X-ray microfocussing combined with microfluidics for on-chip x-rayscattering measurements. Lab on A Chip, 6(4) :494–499, 2006.
[20] R. F. Ismagilov et al. Pressure-driven laminar flow in tangential microchannels : anelastomeric microfluidic switch. Analytical Chemistry, 73(19) :4682–4687, 2001.
[21] J. H. Kim, K. H. Na, C. J. Kang, D. Jeon, and Y. S. Kim. A disposablethermopneumatic-actuated microvalve stacked with pdms layers and ito-coated glass.Microelectronic Engineering, 73-4 :864–869, 2004.
[22] D. R. Link et al. Electric control of droplets in microfluidic devices. AngewandteChemie-International Edition, 45(16) :2556–2560, 2006.
[23] F. Sarrazin. Microréacteurs diphasiques pour le développement rapide des procédés.2006.
[24] A. E. Kamholz, B. H. Weigl, B. A. Finlayson, and P. Yager. Quantitative analysis ofmolecular interaction in a microfluidic channel : The t-sensor. Analytical Chemistry,71(23) :5340–5347, 1999.
[25] F. Sarrazin, J. B. Salmon, D. Talaga, and L. Servant. Chemical reaction imagingwithin microfluidic devices using confocal raman spectroscopy : The case of water anddeuterium oxide as a model system. Analytical Chemistry, 80(5) :1689–1695, 2008.
[26] J. B. Salmon et al. In situ raman imaging of interdiffusion in a microchannel. AppliedPhysics Letters, 86(9), 2005.
[27] Charles N. Baroud, Fridolin Okkels, Laure Menetrier, and Patrick Tabeling. Reaction-diffusion dynamics : confrontation between theory and experiment in a microfluidicreactor. Phys Rev E Stat Nonlin Soft Matter Phys, 67(6 Pt 1), 2003.
[28] J. B. Salmon et al. An approach to extract rate constants from reaction - diffusiondynamics in a microchannel. Analytical Chemistry, 77(11) :3417–3424, 2005.

Bibliographie 153
[29] G. Dummann et al. The capillary-microreactor : a new reactor concept for the in-tensification of heat and mass transfer in liquid-liquid reactions. Catalysis Today,79(1-4) :433–439, 2003.
[30] H. Song, J. D. Tice, and R. F. Ismagilov. A microfluidic system for controlling reactionnetworks in time. Angewandte Chemie-International Edition, 42(7) :768–772, 2003.
[31] R. Dreyfus, P. Tabeling, and H. Willaime. Ordered and disordered patterns in two-phase flows in microchannels. Physical Review Letters, 90(14), 2003.
[32] Pierre Guillot and Annie Colin. Stability of parallel flows in a microchannel after a tjunction. Phys Rev E Stat Nonlin Soft Matter Phys, 72(6 Pt 2), 2005.
[33] T. Thorsen, R. W. Roberts, F. H. Arnold, and S. R. Quake. Dynamic pattern forma-tion in a vesicle-generating microfluidic device. Physical Review Letters, 86(18) :4163–4166, 2001.
[34] S. L. Anna, N. Bontoux, and H. A. Stone. Formation of dispersions using "flowfocusing" in microchannels. Applied Physics Letters, 82(3) :364–366, 2003.
[35] L. Prat et al. Performance evaluation of a novel concept "open plate reactor" appliedto highly exothermic reactions. Chemical Engineering and Technology, 28(9) :1028–1034, 2005.
[36] F. Sarrazin et al. Experimental and numerical study of droplets hydrodynamics inmicrochannels. Aiche Journal, 52(12) :4061–4070, 2006.
[37] K. Handique and M. A. Burns. Mathematical modeling of drop mixing in a slit-typemicrochannel. Journal of Micromechanics and Microengineering, 11(5) :548–554, 2001.
[38] F. Sarrazin et al. Mixing characterization inside microdroplets engineered on a micro-coalescer. Chemical Engineering Science, 62(4) :1042–1048, 2007.
[39] H. Song, M. R. Bringer, J. D. Tice, C. J. Gerdts, and R. F. Ismagilov. Experimentaltest of scaling of mixing by chaotic advection in droplets moving through microfluidicchannels. Applied Physics Letters, 83(22) :4664–4666, 2003.
[40] M. Muradoglu and H. A. Stone. Mixing in a drop moving through a serpentinechannel : A computational study. Physics of Fluids, 17(7), 2005.
[41] H. A. Stone, A. D. Stroock, and A. Ajdari. Engineering flows in small devices :Microfluidics toward a lab-on-a-chip. Annual Review of Fluid Mechanics, 36 :381–411,2004.
[42] S. Waelchli and P. R. von Rohr. Two-phase flow characteristics in gas-liquid micro-reactors. International Journal of Multiphase Flow, 32(7) :791–806, 2006.
[43] A. Wegmann and P. R. von Rohr. Two phase liquid-liquid flows in pipes of smalldiameters. International Journal of Multiphase Flow, 32(8) :1017–1028, 2006.
[44] T. M. Squires and S. R. Quake. Microfluidics : Fluid physics at the nanoliter scale.Reviews of Modern Physics, 77(3) :977–1026, 2005.
[45] J. Goyon, A. Colin, G. Ovarlez, A. Ajdari, and L. Bocquet. Spatial cooperativity insoft glassy flows. Nature, 454(7200) :84–87, 2008.

154 Bibliographie
[46] J. Atencia and D. J. Beebe. Controlled microfluidic interfaces. Nature, 437(7059) :648–655, 2005.
[47] A. J. deMello. Control and detection of chemical reactions in microfluidic systems.Nature, 442(7101) :394–402, 2006.
[48] P. Laval, N. Lisai, J. B. Salmon, and M. Joanicot. A microfluidic device based ondroplet storage for screening solubility diagrams. Lab on A Chip, 7(7) :829–834, 2007.
[49] Setaram. Calvet calorimeter c80. 2009.
[50] P. Monk and I. Wadso. A flow micro reaction calorimeter. Acta Chemica Scandinavica,22(6) :1842, 1968.
[51] L. Wadso and N. Markova. A double twin isothermal microcalorimeter. Thermochi-mica Acta, 360(2) :101–107, 2000.
[52] J. Lerchner et al. A new micro-fluid chip calorimeter for biochemical applications.Thermochimica Acta, 445(2) :144–150, 2006.
[53] J. Lerchner, A. Wolf, G. Wolf, and I. Fernandez. Chip calorimeters for the investigationof liquid phase reactions : Design rules. Thermochimica Acta, 446(1-2) :168–175, 2006.
[54] J. Higuera-Guisset et al. Calorimetry of microbial growth using a thermopile basedmicroreactor. Thermochimica Acta, 427(1-2) :187–191, 2005.
[55] G. Velve Casquillas et al. Thermo-resistance based micro-calorimeter for continuouschemical enthalpy measurements. Microelectronic Engineering, 85(5-6) :1367–1369,2008.
[56] Seung I. Yoon, Se C. Park, and Yong J. Kim. A micromachined microcalorimeter withsplit-flow microchannel for biochemical sensing applications. Sensors and ActuatorsB : Chemical, 134(1) :158–165, 2008.
[57] M. A. Schneider, T. Maeder, P. Ryser, and F. Stoessel. A microreactor-based systemfor the study of fast exothermic reactions in liquid phase : characterization of thesystem. Chemical Engineering Journal, 101(1-3) :241–250, 2004.
[58] S. Ebert, K. Travis, B. Lincoln, and J. Guck. Fluorescence ratio thermometry in amicrofluidic dual-beam laser trap. Optics Express, 15(23) :15493–15499, 2007.
[59] H. F. Arata et al. Temperature distribution measurement on microfabricated thermo-device for single biomolecular observation using fluorescent dye. Sensors and ActuatorsB-Chemical, 117(2) :339–345, 2006.
[60] M. T. Reetz, M. H. Becker, K. M. Kühling, and A. Holzwarth. Time-resolved ir-thermographic detection and screening of enantioselectivity in catalytic reactions.Angewandte Chemie-International Edition, 37(19) :2647–2650, 1998.
[61] M. G. Roper, C. J. Easley, L. A. Legendre, J. A. C. Humphrey, and J. P. Landers.Infrared temperature control system for a completely noncontact polymerase chainreaction in microfluidic chips. Analytical Chemistry, 79(4) :1294–1300, 2007.
[62] C. Hany, C. Pradere, J. Toutain, and J. C. Batsale. A millifluidic calorimeter withinfrared thermography for the measurement of chemical reaction enthalpy and kinetics.QIRT (Quantitative InfraRed Thermography), 5(2) :121–129, 2008.

Bibliographie 155
[63] D. Maillet, S. André, J. C. Batsale, A. Degiovanni, and C. Moyne. Thermal Quadru-poles, Solving the heat equation through integral transforms. Wiley, 2000.
[64] I. M. Galvan, J. M. Zaldivar, H. Hernandez, and E. Molga. The use of neural networksfor fitting complex kinetic data. Computers and Chemical Engineering, 20(12) :1451–1465, 1996.
[65] J. A. Feliu, B. Grau, M. A. Alos, and J. J. ias Hernandez. Match your processconstraints using dynamic simulation. Chemical Engineering Progress, 99(12) :42–48,2003.
[66] O. Ubrich, B. Srinivasan, P. Lerena, D. Bonvin, and F. Stoessel. Optimal feed profilefor a second order reaction in a semi-batch reactor under safety constraints - experi-mental study. Journal of Loss Prevention in the Process Industries, 12(6) :485–493,1999.
[67] J. B. Ott et al. Excess-enthalpies for (ethanol + water) at 298.15-k and pressures of0.4, 5, 10, and 15 mpa. Journal of Chemical Thermodynamics, 18(1) :1–12, 1986.
[68] C. J. Wormald, L. Badock, and M. J. Lloyd. Excess enthalpies for (water plus metha-nol) at t=423 k to t=523 k and pressures up to 20 mpa, a new flow mixing calorimeter.Journal of Chemical Thermodynamics, 28(6) :603–613, 1996.
[69] J. B. Ott et al. Excess-enthalpies for (ethanol + water) at 398.15, 423.15-k, 448.15-k, and 473.15-k and at pressures of 5 and 15 mpa - recommendations for choosing(ethanol + water) as an hme reference mixture. Journal of Chemical Thermodynamics,19(4) :337–348, 1987.
[70] M. J. Costigan, L. J. Hodges, K. N. Marsh, R. H. Stokes, and C. W. Tuxford. Theisothermal displacement calorimeter - design modifications for measuring exothermicenthalpies of mixing. Australian Journal of Chemistry, 33(10) :2103–2119, 1980.
[71] M. Bamford, J. C. Batsale, D. Reungoat, and O. Fudym. Simultaneous velocity anddiffusivity mapping in the case of 3-d transient heat diffusion : heat pulse thermogra-phy and ir image sequence analysis. QIRT (Quantitative InfraRed Thermography),5(1) :97–126, 2008.