chimie gÉnÉralegensdelalune.free.fr/mp/elements de cours/c1-chimie generale/c1-4... · mp –...
TRANSCRIPT

MP – Cours de physique
Jean Le Hir, 03/10/2007 Page 1 sur 23
CHIMIE GÉNÉRALE
Chapitre 4
Cinétique chimique
La cinétique consiste en l’étude de l’avancement, en fonction du temps, d’une réaction chimique lorsque celle-ci est thermodynamiquement possible. L’étude de la cinétique chimique est enseignée en première année. Cette étude ne sera pas reprise en deuxième année, si ce n’est à l’occasion de travaux pratiques et c’est alors l’occasion de procéder à une bonne révision de ce chapitre.
4.1. Système fermé en réaction chimique
Description d’un système fermé
Hypothèse d’homogénéité
Un système est dit fermé s’il n’échange pas de matière avec l’extérieur. Nous n’étudierons l’évolution d’un système chimique fermé que dans le cas où les différentes phases (phase gazeuse, phase liquide, phases solides) qui constituent ce système réactionnel sont homogènes, ce qui signifie que tous les paramètres intensifs ont même valeur en chaque point. En particulier, la pression, la température, et l’ensemble des paramètres de constitution de chaque phase seront toujours considérés comme homogènes, y compris bien sûr lorsque l’on envisagera leur évolution au cours du temps.
Description d’une phase liquide homogène
La composition d’une phase liquide homogène est définie dès lors que l’on se donne l’ensemble des concentrations molaires ic des constituants.
Dans le cas particulier des solutions aqueuses diluées, la concentration du solvant 2H O sera toujours
considérée comme invariante.
Description d’une phase gazeuse homogène
La description d’une phase gazeuse homogène peut se faire également par la donnée des concentrations des différents constituants du mélange. La concentration molaire totale d’un mélange gazeux parfait est donnée par l’équation d’état des gaz parfaits :
n pc
V RT= =
La pression totale p étant définie, les fractions molaires ix du mélange gazeux définissent entièrement sa
constitution.

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 2 sur 23
ii
j
nx
n=∑
soit 1ix =∑
On appelle pression partielle d’un constituant d’un mélange gazeux, la pression qu’aurait ce constituant s’il était seul dans le même volume à la même température. La loi de Dalton établit que dans un mélange gazeux idéal la pression partielle ip de chaque constituant est proportionnelle à sa fraction molaire ix
dans le mélange gazeux :
i ip x p= soit ip p=∑
Bilan de matière d’une réaction chimique : notion d ’avancement ξξξξ
Coefficients stœchiométriques
Selon les normes internationales en vigueur, nous écrirons l’équation bilan d’une réaction chimique tantôt sous la forme réactifs produits= , tantôt sous la forme :
1
CP 0R
k kk
produits réactifs v=
− = =∑
R est le nombre de réactants (produits + réactifs). Les produits sont affectés de coefficients stœchiométriques positifs et les réactifs de coefficients stœchiométriques négatifs. Par exemple, nous écrirons la réaction de synthèse de l’ammoniac de l’une des façons suivantes :
2 2 3N 3H 2 NH+ = ou 3 2 22 NH N 3H 0− − =
Les trois réactants sont gazeux. L’ammoniac 3NH , seul produit de la réaction est affecté du coefficient
stœchiométrique 3NH 2ν = + , tandis que les réactifs diazote 2N et dihydrogène 2H sont affectés des
coefficients stœchiométriques 2N 1ν = − et
2H 3ν = − .
Définition de l’avancement ξξξξ (lire : « ksi »)
Les produits de réaction apparaissent proportionnellement aux coefficients stœchiométriques. Nous définissons l’avancement algébrique ξ de la réaction, à partir d’une situation initiale où les quantités de matière seront repérées par un indice inférieur 0, par le rapport :
1 1,0 2 2,0 ,0
1 2
R R
R
n n n n n n− − −ξ = = = =
ν ν ν�
Du fait de la conservation de la matière, dans le cas où les différents protagonistes de la réaction n’interviennent que dans cette réaction unique, ce rapport, homogène à une quantité de matière, est nécessairement le même pour tous les réactifs et tous les produits.
Les quantités de matière ,0k k kn n= + ν ξ doivent être positives et cela définit des valeurs extrêmes
possibles pour l’avancement : min maxξ∈ξ ⋅⋅ ξ .
La valeur maximale maxξ correspond à la disparition totale d’un premier réactif : celui-ci sera alors
qualifié de réactif limitant. Si l’on est initialement en présence de produits, minξ peut être négatif : nous
pouvons fort bien imaginer un avancement négatif.
Réactions simultanées
Dans le cas où se produisent simultanément plusieurs réactions chimiques, il convient de définir autant d’avancements que de réactions indépendantes.

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 3 sur 23
Exemple : considérons les deux réactions suivantes : 2 22CO O 2CO= + et 22CO C CO= +
Partant de conditions initiales quelconques, les quantités de matière des espèces 2CO et CO concernées
par les deux réactions simultanées seront fonctions des deux avancements de réaction. Il est alors indispensable de faire un bilan de matière unique :
2 2
2 2
2
2
2 2
CO ,0 O ,0 CO,0
1 2 CO ,0 1 2 O ,0 1 CO,0 1 2
2
CO,0 C,0 CO ,0
1 2 CO,0 1 2 C,0 2 CO ,0 1 2
2CO O 2CO
initialement :
après avancement , : 2 2 2
2CO C CO
initialement :
après avancement , : 2 2 2
n n n
n n n
n n n
n n n
= +
ξ ξ − ξ + ξ + ξ + ξ − ξ
= +
ξ ξ + ξ − ξ + ξ − ξ + ξ
4.2. Vitesses en cinétique chimique
Définition des vitesses de réaction
Vitesse de réaction associée à une équation stœchiométrique
Considérons la réaction dont le bilan est représenté par l’équation stœchiométrique :
1
A 0R
k kk
v=
=∑
où les kv représentent les coefficients stœchiométriques algébriques des réactants (négatifs pour les
réactifs et positifs pour les produits). Pour un système fermé, la variation de l’avancement ξ de la réaction est liée à la variation de la quantité de matière de chaque constituant, consécutive à la réaction, soit k kdn v d= ξ .
On appelle vitesse de réaction v associée à cette équation stœchiométrique, la dérivée par rapport au temps de l’avancement volumique :
vd
dt V
ξ =
Vitesse de formation d’un réactant
Si le volume du réacteur est constant et si le système est homogène, par agitation par exemple, la vitesse globale s’exprime en fonction des dérivées, par rapport au temps, des concentrations molaires :
1 1 1 1v k k
k k
dn dcd d
dt V V dt V dt dt
ξ ξ = = = = ν ν
La dérivée kdc
dt est la vitesse algébrique de formation du réactant A k .
Au fur et à mesure de l’avancement de la réaction, les produits sont formés : la vitesse de formation des produits est positive. Dans le même temps les réactifs disparaissent et plutôt que de parler d’une vitesse de formation négative les concernant, on préfère alors bien souvent parler de vitesse de disparition.
( )
( )
f
d
Vitesse de formation d'un produit : v A v
Vitesse de disparition d'un réactif : v A v
kk k
kk k
dc
dtdc
dt
= + = ν = − = ν

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 4 sur 23
Exemple : considérons la réaction de synthèse de l’ammoniac. 2 2 3N 3H 2NH+ =
À volume et à température constants, la vitesse de la réaction s’écrit : 3 2 2NH N H1 1v
2 3
dc dc dc
dt dt dt= + = − = −
tandis que la vitesse de formation de l’ammoniac et les vitesses de disparition du diazote et du dihydrogène, prennent la forme :
( ) 3NHf 3v NH 2v
dc
dt= + = ( ) 2H
d 2v H 3vdc
dt= − = et ( ) 2N
d 2v N vdc
dt= − =
Attention ! Si les coefficients stœchiométriques sont multipliés par une constante K quelconque, la vitesse de réaction est également multipliée par K. Par exemple, nous aurions pu écrire la réaction de synthèse de l’ammoniac sous la forme :
2 2 3
1 3N H NH
2 2+ =
La vitesse de réaction est alors divisée par deux. Toutefois, les vitesses de formation des produits et de disparition des réactifs sont indépendantes de toute écriture stœchiométrique particulière.
Remarque : lorsque les réactifs ou les produits sont à l’état gazeux, il peut être plus efficace de faire intervenir les pressions partielles, proportionnelles aux concentrations :
ii i
n RTp c RT
V= =
Facteurs de la cinétique
La première nécessité pour qu’une réaction puisse avoir lieu est à la présence de réactifs et l’on ne s’étonnera pas, a priori, que la vitesse de réaction soit d’autant plus importante que les réactifs sont en forte concentration. Si l’on imagine que tout mécanisme réactionnel suppose le choc d’une molécule de réaction avec un autre protagoniste (qui peut être une autre molécule de réactif, ou une molécule spectatrice, ou une paroi…), on comprend bien qualitativement que l’influence de la concentration sur la fréquence de ces chocs. On comprend bien aussi que lorsqu’un réactif devient très rare, la vitesse de réaction diminue : « et le combat cessa, faute de combattants ».
Nous savons que l’énergie cinétique microscopique disponible au moment du choc est proportionnelle à la température absolue. Toujours en imaginant les chocs responsables de l’avancement de réaction, on comprend assez facilement ce fait d’expérience quasi général : une augmentation de la température favorise les vitesses de réaction.
La cinétique chimique est avant tout une affaire d’expérience. Ainsi a-t-on appris à augmenter la vitesse de réactions chimique par addition de substances qui interviennent dans le mécanisme réactionnel pour en améliore l’efficacité, sans être consommées par la réaction : ces « catalyseurs » n’apparaissent pas dans le bilan de matière. En général, les vitesses de réaction sont alors d’autant plus grande que le catalyseur est abondant dans le milieu réactionnel.
Principaux facteurs cinétiques
— Les vitesses de réaction sont initialement plus importantes quand les réactifs sont en forte concentration et décroissent jusqu’à s’annuler au fur et à mesure que les réactifs sont consommés par l’avancement de réaction.
— Les vitesses de réaction sont des fonctions croissantes de la température.
— Les vitesses de réaction peuvent être améliorées par catalyse : ajout de substances modifiant le mécanisme réactionnel sans intervenir dans le bilan de matière.

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 5 sur 23
4.3. Cinétique formelle des réactions avec ordre
Définition d’un ordre
Le plus souvent, la variation avec les concentrations des vitesses des réactions chimiques obéissent à des lois assez complexes et la compréhension de ces lois de variation passe par la connaissance des mécanismes réactionnels. Nous étudierons quelques exemples plus loin.
Pour quelques réactions, la loi de vitesse est particulièrement simple, les vitesses s’exprimant sous la forme d’un monôme des concentrations des réactifs. On dit alors que ces réactions chimiques sont des réactions avec ordre.
Loi cinétique générale des réactions avec ordre
réactifs
v ipik c==== ∏∏∏∏
Les nombres ip , ordres partiels, sont des nombres réels quelconques, positifs, négatifs ou nuls, Ils sont déterminés de façon expérimentale et ont a priori des valeurs différentes des valeurs absolues iνννν des coefficients stœchiométriques des réactifs dans la réaction.
La somme des ordres partiels correspond à l’ordre global de la réaction. Les ordres partiels sont des données expérimentales : nous étudierons quelques méthodes permettant leur détermination.
La constante k s’appelle constante de vitesse, elle a une dimension physique, et par conséquent une unité, qui dépend de l’ordre global de la réaction.
Exemple 1 : la réaction 2 2 2 22 HI H O 2 H O I+ = + a pour loi cinétique 2HI H Ov k c c= . L’ordre partiel par
rapport à HI, qui s’identifie ici à celui du peroxyde d’hydrogène 2 2H O , vaut 1. L’ordre global est donc 2.
Exemple 2 : la réaction 22 NO Cl 2 NOCl+ = a pour loi cinétique 2
2NO Clv k c c= . L’ordre global est
égal à 3. Les ordres partiels s’identifient pour cet exemple aux coefficients stœchiométriques de la réaction : c’est un cas particulier.
Exemple 3 : lorsque le produit de réaction HBr est éliminé du milieu réactionnel au fur et à mesure qu’il apparaît, la réaction 2 2H Br 2 HBr+ = a pour loi cinétique
2 2
1/ 2Br Hv k c c= . L’ordre global est égal à 3/2.
Pour cet exemple, l’ordre partiel concernant le dibrome n’est pas entier.
Attention ! La vitesse v ne peut pas toujours être représentée par une formule monôme du type précédent. Dans ce cas, on dit que la réaction n’a pas d’ordre. Dans exemple 3 ci-dessus, la synthèse du bromure d’hydrogène étudiée par Bodenstein, le produit de réaction intervient pour limiter la vitesse de réaction. Si HBr n’est pas évacué du milieu réactionnel, L’expression de la vitesse est la suivante :
2 2
2
1/ 2Br H
HBr
Br
v1
c ck
ck
c
=′+
Par contre, pour cette réaction, à l’instant initial, ( )HBr 0 0c = et le produit HBr de la réaction
n’intervient pas dans le processus réactionnel. La vitesse initiale a pour expression ( ) ( ) ( )
2 2
1/ 2Br Hv 0 0 0k c c= . La réaction est sans d’ordre, mais elle a un ordre initial.

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 6 sur 23
Étude formelle d’une loi cinétique d’ordre 1
Équation différentielle
Le cas le simple est celui d’une réaction dont la vitesse ne dépend que de la concentration d’un seul réactif par une loi cinétique d’ordre 1. On écrit préférentiellement l’équation bilan de telle sorte que le réactif intervenant dans la loi cinétique ait un coefficient stœchiométrique 1− .
Exemple : 2 5 2 2
1N O 2 NO O
2= + 2 5
2 5
N ON Ov
dck c
dt= = −
La concentration en réactif ( )c t obéit à une équation différentielle linéaire du premier ordre sans second
membre et son intégration ne pose aucun problème particulier. Écrivons cette équation sous la forme canonique :
( ) ( )0
dc t c t
dt+ =
τ en posant
1
kτ =
Intégration
La solution générale est une fonction exponentielle de constante de temps τ et nous ajustons la constante multiplicative de telle sorte que cela corresponde à la concentration initiale ( )0c en réactif.
Temps de demi-réaction
Le temps au bout duquel la moitié du réactif initial a disparu s’appelle « temps de demi-réaction ». Dans le cas d’une loi cinétique d’ordre 1, ce temps est indépendant de la concentration initiale. Cette propriété est caractéristique de l’ordre 1.
1/ 2
ln 2ln 2t
k= τ =
Remarque : les désintégrations radioactives obéissent à de telles lois cinétiques. Dans ce cas, le temps de demi-réaction s’appelle généralement « demi-vie » : c’est le temps nécessaire pour que la moitié des atomes d’un échantillon donné de matériau radioactif se désintègre. Ce temps est indépendant de toute contingence extérieure. Nous pouvons interpréter cela de la façon suivante : la probabilité de désintégration d’un atome donné ne varie pas au cours du temps. Le fait d’avoir « survécu » pendant une longue période n’augmente pas la probabilité de désintégration.
Dégénérescence de l’ordre
Pour l’exemple cité, lorsque la décomposition de ( )2 5 gN O se produit alors que le réactif gazeux est en
équilibre avec le solide ( )2 5 sN O , la pression partielle de ( )2 5 gN O reste fixe à température constante.
Nous avons alors : ( )2 5 gN O
tec C= et ( ) ( )2 25 g 5 g
1 0N O N Ov k c k c k′ ′= = = avec
( )2 5 gN Ok k c′ =
Ainsi, par dégénérescence, on observe une cinétique d’ordre 0, ce qui est rare. k′ est alors appelée constante cinétique apparente.
( ) ( )0t
c t c e−
τ=( )0c
0
0 τ t1/ 2 ln 2t = τ
( )0
2
c
CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 7 sur 23
Étude formelle d’une loi cinétique d’ordre 2 à un s eul réactif
Équation différentielle
De la même façon que précédemment, on écrit l’équation bilan de telle sorte que le réactif intervenant dans la loi cinétique ait un coefficient stœchiométrique 1− .
Exemple : 2 2
1NO NO O
2= + 2
2
NO2NOv
dck c
dt= = −
La concentration en réactif ( )c t obéit à une équation différentielle du premier ordre à variables
séparables. Écrivons cette équation sous la forme de variables séparées :
2
dck dt
c= −
Intégration
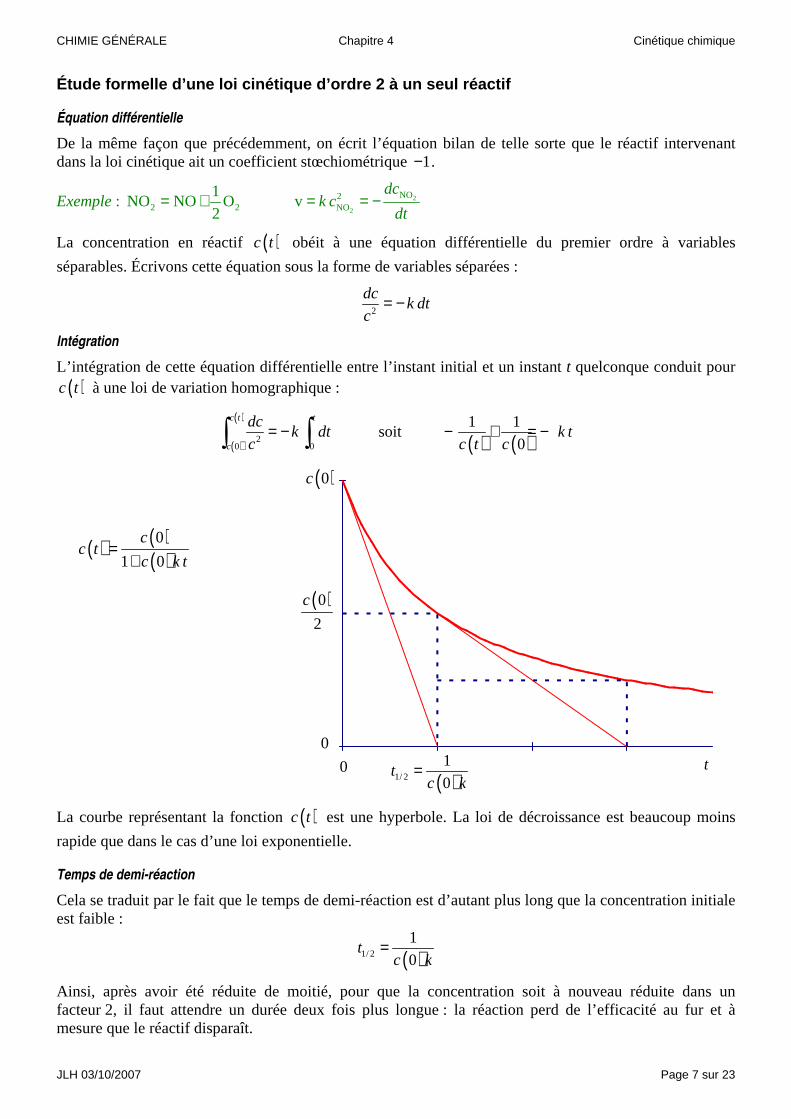
L’intégration de cette équation différentielle entre l’instant initial et un instant t quelconque conduit pour ( )c t à une loi de variation homographique :
( )
( )
20 0
c t t
c
dck dt
c= −∫ ∫ soit
( ) ( )1 1
0k t
c t c− + = −
La courbe représentant la fonction ( )c t est une hyperbole. La loi de décroissance est beaucoup moins
rapide que dans le cas d’une loi exponentielle.
Temps de demi-réaction
Cela se traduit par le fait que le temps de demi-réaction est d’autant plus long que la concentration initiale est faible :
( )1/ 2
1
0t
c k=
Ainsi, après avoir été réduite de moitié, pour que la concentration soit à nouveau réduite dans un facteur 2, il faut attendre un durée deux fois plus longue : la réaction perd de l’efficacité au fur et à mesure que le réactif disparaît.
( )0c
0
0 t( )1/ 2
1
0t
c k=
( )0
2
c
( ) ( )( )0
1 0
cc t
c k t=
+

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 8 sur 23
Étude formelle d’une loi cinétique d’ordre global 2 , à deux réactifs
Équation différentielle
Deux espèces chimiques, que nous noterons A et B, réagissent entre elles selon une loi cinétique d’ordre partiel égal à 1 par rapport à chaque réactif. Nous écrirons l’équation bilan de telle sorte que chacun des réactifs y apparaisse avec un coefficient stœchiométrique 1− . L’équation bilan s’écrit donc :
A B produits+ =
Exemple : la réaction de saponification, dont les réactifs sont un ester et l’ion hydroxyde est de ce type.
3 2 5 2 5 3CH COOC H OH C H OH CH COO− −+ = + 3 2 5CH COOC HOH
v k c c−=
Étudions les variations au cours du temps de l’avancement volumique xV
ξ= de la réaction.
A0 B0
A0 B0
A B
'
produits
initialement c c
à l instant t c x c x
+ =
− −
L’avancement ( )x t obéit à une équation différentielle du premier ordre à variables séparables :
( )( )A0 B0
dxk c x c x
dt= − −
Écrivons cette équation sous la forme de variables séparées :
( )( )A0 B0
dxk dt
c x c x=
− −
Intégration
L’intégration se fait par décomposition de la fraction rationnelle en éléments simples :
( )( )0 0 0A0 B0 A0 B0 B0 A0
1 1x x tdx dxk dt
c x c x c x c x c c
= − = − − − − − ∫ ∫ ∫
Nous en déduisons : ( )( )
A0 B0
B0 A0 B0 A0
1ln
c c xk t
c c c c x
−=
− − soit : ( )
A0 B0
A0 B0A0 B0B0 A0
c k t c k t
c k t c k t
e ex t c c
c e c e
− −
− −
−=−
Note : la décomposition en éléments simple ne se fait de cette façon que si B0 A0c c≠ . Si, au contraire, les
concentrations initiales en réactif A et B sont égales, le problème est formellement équivalent au cas étudié précédemment d’une réaction d’ordre 2 à un seul réactif.
Temps de demi-réaction
Si l’on fait l’hypothèse que A est le réactif limitant ( )A0 B0c c< , le temps de demi-réaction est défini par la
diminution de la moitié de la concentration Ac , soit A0 A0 / 2c x x c− = = . Cette relation définit le
temps 1/ 2t :
( )A0
1/ 2B0 A0 B0
1ln 2
ct
k c c c
= − −
CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 9 sur 23
4.4. Vitesse de réaction et température : loi d’Arr henius
Les réfrigérateurs permettent de conserver les aliments plus longtemps en diminuant, par abaissement de la température, la vitesse des réactions de dégradation biochimique. En analyse chimique, on procède à des blocages cinétiques en effectuant une « trempe » du milieu réactionnel au moyen d’une chute brutale de la température.
Inversement, la vitesse des réactions croît, en général très vite, avec la température. Au voisinage de la température ordinaire, la constante de vitesse double ou triple pour une élévation de température de l’ordre de grandeur de 10°C.
Le Suédois Svante Arrhenius énonce, en 1889, une loi phénoménologique qui rend bien compte de la variation avec la température des vitesses de réaction avec ordre.
Loi d’Arrhenius
Les constantes de vitesse k des réactions chimiques avec ordre sont des fonctions généralement croissantes de la température. L’expression est donnée par la formule :
maE
RTk Ae−−−−
====
maE , énergie molaire, est l’énergie d’activation de la réaction, A est le facteur préexponentiel
d’Arrhenius, R est la constante des gaz parfaits (((( ))))1 18,31 J mol KR − −− −− −− −= ⋅ ⋅= ⋅ ⋅= ⋅ ⋅= ⋅ ⋅ et T la température
absolue.
Arrhenius introduire la notion de choc efficace : seuls réagissent les réactifs ayant acquis, par activation thermique, une énergie microscopique suffisante aE . Rapportée à l’échelle macroscopique, cette énergie,
comme nous le verrons plus loin, n’est pas sans rapport avec l’énergie d’activation maE .
L’ordre de grandeur de ces énergies d’activation varie entre 30 et 200 1kJ mol−⋅ .
Le coefficient A est encore appelé facteur de fréquence. Sa dimension est celle de la constante de vitesse k.
Graphiquement, il est possible de déterminer la valeur de l’énergie d’activation en mesurant la pente,
égale à maE
R− , de la courbe représentant ln k en fonction de
1
T.
Remarque. Citons une exception à la règle de croissance de la vitesse avec la température, la réaction d’oxydation du monoxyde d’azote : 2 22 NO O 2NO+ =
1
T croissantT
ln k
mapente E
R−

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 10 sur 23
4.5. Méthodes expérimentales
Suivi d’une concentration au cours du temps
Quelle que soit la méthode expérimentale choisie pour étudier la cinétique d’une réaction chimique, il faut d’une façon ou d’une autre mesurer l’avancement de la réaction et pour cela connaître l’évolution au cours du temps de la concentration d’un réactif ou d’un produit. De nombreuses techniques existent, en voici quelques unes.
pH-métrie
On s’intéresse particulièrement à la concentration des ions oxonium 3H O+ que l’on mesure par
potentiométrie à l’aide d’un pH-mètre.
Conductimétrie
Dans le cas d’une réaction consommant ou produisant des espèces chimiques ioniques, la conductivité de la solution varie avec l’avancement. Dans le cas d’une réaction unique et d’une solution suffisamment diluée, la conductivité de la solution est une fonction affine de l’avancement de réaction.
Rappelons que pour les solutions idéales, la conductivité électrique γ est la somme des conductivités
partielles iγ dues à chaque espèce ionique :
ii
γ = γ∑ avec i i icγ = λ
où iλ est la conductivité molaire partielle de chaque espèce ionique.
Exemple : dans le cas d’une réaction de saponification, des ions hydroxyde OH− , très conducteurs, disparaissent tandis qu’apparaissent des ions carboxylate RCOO− dont la conductivité est très inférieure.
Pour un avancement volumique x, la conductivité de la solution est une fonction de x de la forme :
( ) ( ) ( )OH RCOO0x x− −γ = γ − λ − λ
Spectrophotométrie
Cette méthode est utilisée pour évaluer la concentration en solution d’un ion qui en modifie la couleur : la lumière qui traverse une substance colorée est en partie absorbée.
L’absorption de la lumière dépend de la longueur d’onde, de l’épaisseur traversée, de la concentration en corps dissous et aussi du solvant. Pour les solutions, elle dépend légèrement de la température.
réseau
fente
source
photomultiplicateur
cuve contenantl’échantillon
0I I

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 11 sur 23
Notons 0I l’intensité lumineuse incidente sur la cuve contenant l’échantillon de solution et I l’intensité
lumineuse transmise. On appelle transmittance le rapport 0/I I et absorbance l’opposé du logarithme
décimal de la transmittance (ainsi définie, l’absorbance est positive) :
010 10
0
log logII
AI I
= − =
Selon la loi de Beer-Lambert, l’absorbance d’une solution diluée est la somme d’absorbances partielles proportionnelles à la longueur L de la cuve et à la concentration ic du soluté absorbant :
( )i i
i
A c L= ε λ∑
Le coefficient de proportionnalité ( )iε λ est l’absorbance linéique molaire ou coefficient d’extinction
pour la longueur d’onde λ sélectionnée. Ce coefficient d’extinction dépend également de la température.
Une mesure de concentration suppose que l’on réalise un étalonnage du dispositif, c’est-à-dire que l’on mesure l’absorbance pour la même longueur d’onde et pour une concentration nulle.
Mise en évidence d’un ordre simple
Méthode différentielle de Van’t Hoff
Si l’on trace le graphe de la fonction ( )c t , les pentes ( )dc t
dt peuvent être tracées et lues sur le graphe.
Traçons alors un deuxième graphe en portant en ordonnée le logarithme ( )log v de la vitesse et en
abscisse le logarithme ( )log c de la concentration. Pour une loi cinétique obéissant à un ordre p, la vitesse
est de la forme v pk c= .
Le graphe de la fonction ( ) ( )( ) ( ) ( )log v log log logf c k p c= = + est une droite dont la pente est égale à
l’ordre p de la réaction, l’ordonnée à l’origine étant égale au logarithme de la constante de vitesse.
La méthode n’est pas très précise, mais elle permet d’évaluer rapidement si une réaction a un ordre et quelle est la valeur possible de cet ordre. Une fois l’ordre présumé, la mise en œuvre d’une méthode intégrale permet de s’en assurer.
Remarque : on peut tout aussi bien utiliser des logarithmes décimaux ou des logarithmes népériens. D’autre part, si le seul but est de déterminer l’ordre de la réaction, le choix des unités n’a aucune importance. En effet, le fait de changer d’unité ne change pas la pente d’un diagramme log-log.
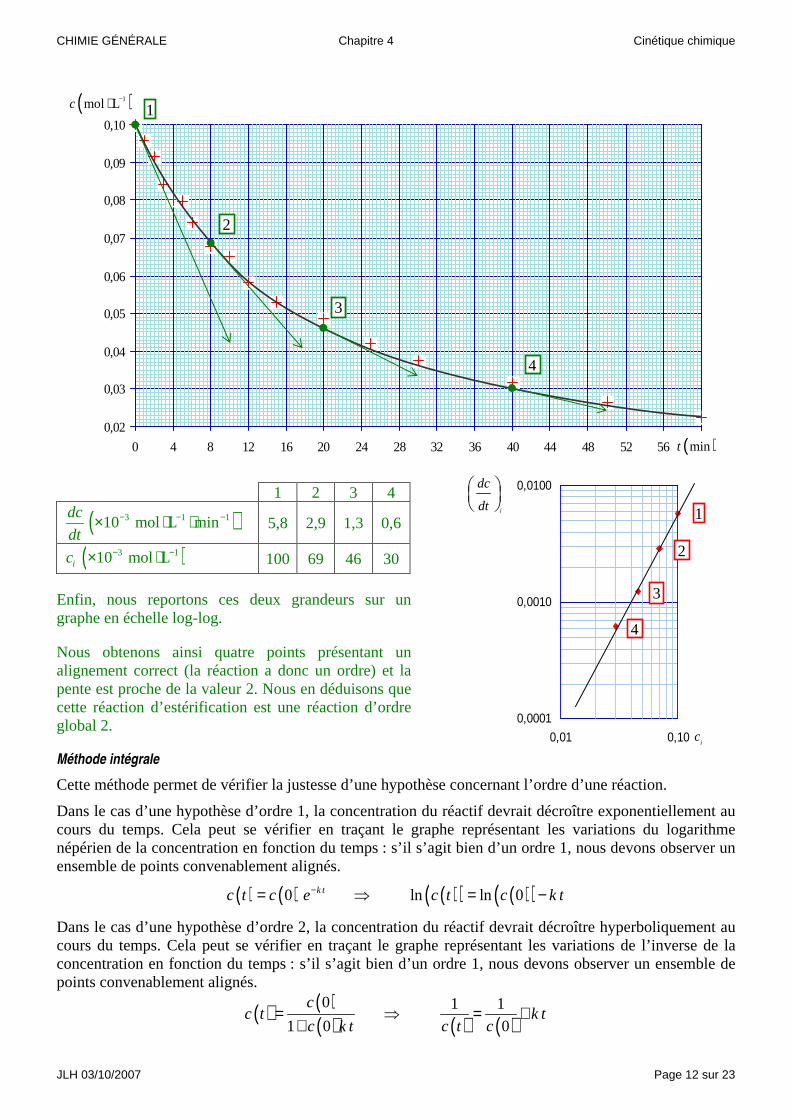
Exemple : voici un ensemble de mesure de concentration c en ions oxonium 3H O+ au cours d’une
réaction d’estérification de l’acide éthanoïque par le butan-1-ol. Les concentrations des réactifs étaient initialement égales.
Nous reportons ces points sur un graphe et traçons, du mieux possible, une courbe représentant une loi de variation ( )c t compatible avec nos mesures.
Sur cette courbe, en quatre points, nous mesurons les pentes i
dc
dt
ainsi que les concentrations ic
correspondantes.
CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 12 sur 23
1 2 3 4
( )3 1 110 mol L mindc
dt− − −× ⋅ ⋅ 5,8 2,9 1,3 0,6
( )3 110 mol Lic − −× ⋅ 100 69 46 30
Enfin, nous reportons ces deux grandeurs sur un graphe en échelle log-log.
Nous obtenons ainsi quatre points présentant un alignement correct (la réaction a donc un ordre) et la pente est proche de la valeur 2. Nous en déduisons que cette réaction d’estérification est une réaction d’ordre global 2.
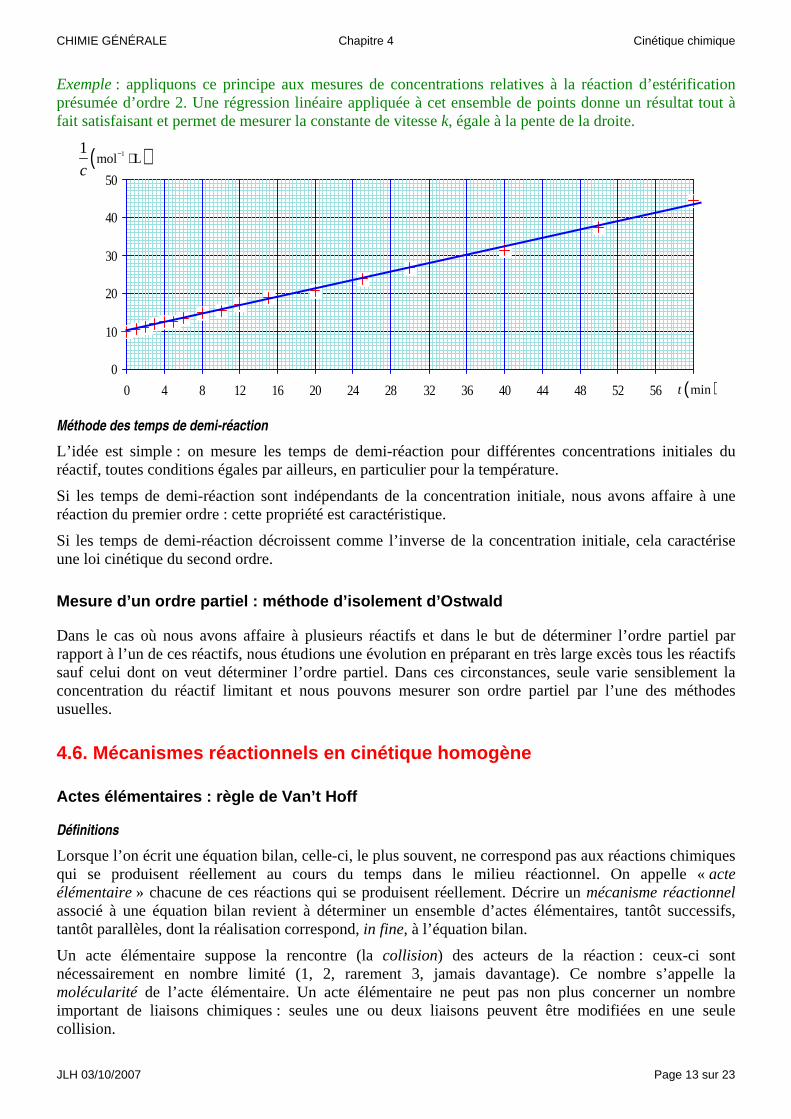
Méthode intégrale
Cette méthode permet de vérifier la justesse d’une hypothèse concernant l’ordre d’une réaction.
Dans le cas d’une hypothèse d’ordre 1, la concentration du réactif devrait décroître exponentiellement au cours du temps. Cela peut se vérifier en traçant le graphe représentant les variations du logarithme népérien de la concentration en fonction du temps : s’il s’agit bien d’un ordre 1, nous devons observer un ensemble de points convenablement alignés.
( ) ( )0 k tc t c e−= ⇒ ( )( ) ( )( )ln ln 0c t c k t= −
Dans le cas d’une hypothèse d’ordre 2, la concentration du réactif devrait décroître hyperboliquement au cours du temps. Cela peut se vérifier en traçant le graphe représentant les variations de l’inverse de la concentration en fonction du temps : s’il s’agit bien d’un ordre 1, nous devons observer un ensemble de points convenablement alignés.
( ) ( )( )0
1 0
cc t
c k t=
+ ⇒
( ) ( )1 1
0k t
c t c= +
0,02
0,03
0,04
0,05
0,06
0,07
0,08
0,09
0,10
0 4 8 12 16 20 24 28 32 36 40 44 48 52 56 60
( )1mol Lc −⋅
( )mint
1
2
3
4
0,0001
0,0010
0,0100
0,01 0,10
1
2
3
4
i
dc
dt
ic
CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 13 sur 23
Exemple : appliquons ce principe aux mesures de concentrations relatives à la réaction d’estérification présumée d’ordre 2. Une régression linéaire appliquée à cet ensemble de points donne un résultat tout à fait satisfaisant et permet de mesurer la constante de vitesse k, égale à la pente de la droite.
Méthode des temps de demi-réaction
L’idée est simple : on mesure les temps de demi-réaction pour différentes concentrations initiales du réactif, toutes conditions égales par ailleurs, en particulier pour la température.
Si les temps de demi-réaction sont indépendants de la concentration initiale, nous avons affaire à une réaction du premier ordre : cette propriété est caractéristique.
Si les temps de demi-réaction décroissent comme l’inverse de la concentration initiale, cela caractérise une loi cinétique du second ordre.
Mesure d’un ordre partiel : méthode d’isolement d’O stwald
Dans le cas où nous avons affaire à plusieurs réactifs et dans le but de déterminer l’ordre partiel par rapport à l’un de ces réactifs, nous étudions une évolution en préparant en très large excès tous les réactifs sauf celui dont on veut déterminer l’ordre partiel. Dans ces circonstances, seule varie sensiblement la concentration du réactif limitant et nous pouvons mesurer son ordre partiel par l’une des méthodes usuelles.
4.6. Mécanismes réactionnels en cinétique homogène
Actes élémentaires : règle de Van’t Hoff
Définitions
Lorsque l’on écrit une équation bilan, celle-ci, le plus souvent, ne correspond pas aux réactions chimiques qui se produisent réellement au cours du temps dans le milieu réactionnel. On appelle « acte élémentaire » chacune de ces réactions qui se produisent réellement. Décrire un mécanisme réactionnel associé à une équation bilan revient à déterminer un ensemble d’actes élémentaires, tantôt successifs, tantôt parallèles, dont la réalisation correspond, in fine, à l’équation bilan.
Un acte élémentaire suppose la rencontre (la collision) des acteurs de la réaction : ceux-ci sont nécessairement en nombre limité (1, 2, rarement 3, jamais davantage). Ce nombre s’appelle la molécularité de l’acte élémentaire. Un acte élémentaire ne peut pas non plus concerner un nombre important de liaisons chimiques : seules une ou deux liaisons peuvent être modifiées en une seule collision.
0
10
20
30
40
50
0 4 8 12 16 20 24 28 32 36 40 44 48 52 56 60
( )1mol L1
c− ⋅
( )mint

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 14 sur 23
Nous écrirons donc un acte élémentaire sous la forme suivante :
1 1 2 2 1 1 2 2A A A Am m m m′ ′ ′ ′+ + → + +� �
La flèche → remplace le signe « égal » conventionnel pour une équation bilan. Les molécularités partielles im sont des nombres entiers tels que i
réactifs
m∑ a pour valeur 1, 2, rarement 3, jamais davantage.
Attention ! Les molécularités partielles ne doivent pas être confondues avec les coefficients stœchiométriques d’une équation bilan qui sont définis algébriquement et peuvent ne pas être entiers.
Règle de Van’t Hoff
Un acte élémentaire ne peut se produire qu’à l’occasion de la rencontre des protagonistes. Van’t Hoff démontre dans le cadre de ses Études de dynamique chimique (modélisation microscopique de la réaction chimique) que dans un intervalle de temps donné, le nombre de rencontres est proportionnel à la probabilité de présence dans l’espace de chacun des acteurs, c’est-à-dire à leur concentration. Il en déduit la forme de la loi de vitesse, que nous appellerons « règle de Van’t Hoff ».
Règle de Van’t Hoff
Les actes élémentaires sont des réactions avec ordre. L’ordre global est égal à la molécularité m de l’acte, définie comme le nombre d’entités microscopiques régissantes. Les ordres partiels relatifs à chaque réactif A i sont entiers et ont pour valeur 1 ou 2, valeur de la molécularité
partielle im du réactif.
Av i
i
m
réactifs
k c==== ∏∏∏∏ et {{{{ }}}}1, 2, 3iréactifs
m m= ∈= ∈= ∈= ∈∑∑∑∑
Théorie de l’état de transition : complexe activé, profil réactionnel
Existence de complexes activés
Pour qu’un acte élémentaire se produise, il ne suffit pas que les acteurs se rencontrent. Il faut aussi, pour que le choc soit efficace, que la rencontre se fasse sous un angle favorable. Il faut également que l’énergie disponible1 dans le référentiel du centre de masse des entités réagissantes soit suffisante pour franchir certaines barrières de potentiel : il doit se former un complexe activé, structure apparaissant de façon fugitive, mais passage indispensable pour que la réaction ait lieu.
Exemple : considérons l’action d’un réactif nucléophile tel que l’ion hydronium OH− sur le monochlorométhane 3CH Cl . La réaction 3 3OH CH Cl CH OH Cl− −+ → + est un acte élémentaire que
l’on qualifie de substitution nucléophile bimoléculaire : NS 2. Pour que cette réaction se produise, il ne
suffit pas qu’un ion OH− rencontre une molécule 3CH Cl , il faut aussi que l’approche du substituant
nucléophile se fasse du bon coté de la molécule de chlorométhane, à l’opposé de l’atome de chlore.
De plus, la réaction ne se produit que si l’énergie des réactifs est suffisante pour passer par un état de
transition, un complexe activé pentacoordonné que l’on peut noter 1/ 2 1/ 23HO CH Cl
≠− − � � .
1 Cette énergie, rappelons-le, est une fonction croissante de la température.

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 15 sur 23
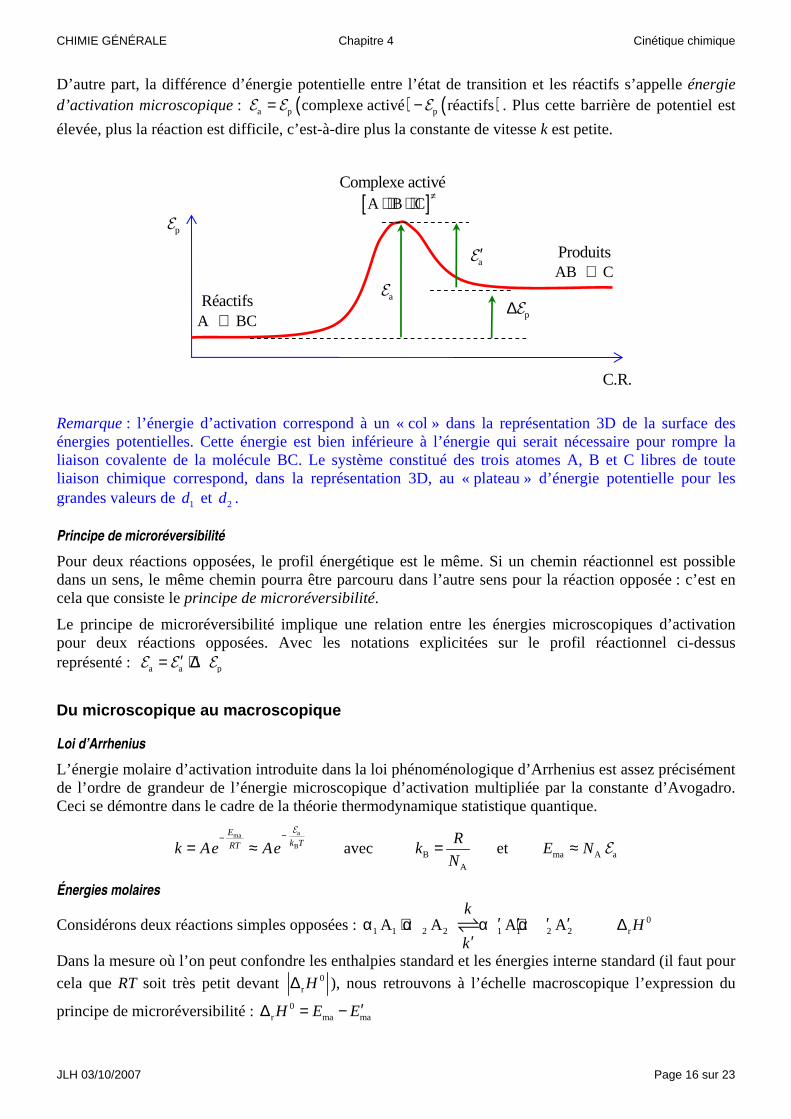
Considérons le schéma réactionnel suivant : [ ]A BC A B C AB C
réactifs produitscomplexeactivé
≠+ → ⋅⋅ ⋅⋅ → +����� ����������
Et plaçons nous dans le cas simple où A, B et C représentent trois atomes et que l’approche de l’atome A s’effectue de façon parfaitement axiale par rapport à l’orientation de la molécule BC.
Exemple : [ ]K BrH K Br H KBr H≠+ → ⋅⋅ ⋅⋅ → +
L’énergie potentielle d’un système de trois atomes alignés est une fonction ( )p 1 2,d dE à deux variables
( )1 A Bd d= − et ( )2 B Cd d= − qui peut être représentée sous la forme d’une surface dans un graphe à
trois dimensions.
Initialement l’atome A se trouve très loin de la molécule BC. La distance 1d est donc très grande et la
distance 2d correspond à la longueur de la liaison B C− , telle que l’énergie potentielle soit minimale.
Au fur et à mesure de l’approche de l’atome A, le point représentatif du système des trois atomes se déplace sur la surface des énergies potentielles au fond d’une « vallée ». Ce point décrit un chemin réactionnel et l’on peut caractériser son évolution par son abscisse curviligne sur ce chemin, que l’on appellera coordonnée réactionnelle (C.R.).
La représentation de l’évolution de l’énergie potentielle en fonction de la coordonnée réactionnelle définit le profil réactionnel de la réaction. Ce profil réactionnel met en évidence deux grandeurs énergétiques importantes.
D’une part, la différence entre l’énergie potentielle des produits et l’énergie potentielle des réactifs définit le bilan énergétique de la réaction : cette différence ( ) ( )p p pproduits réactifs∆ = −E E E multipliée par la
constante d’Avogadro, correspond à l’énergie interne de réaction :
r A pU N∆ = ∆E
[ ]≠⋅ ⋅ ⋅ ⋅Complexe activé
A B C
ValléeCol
Plateau
( )p 1 2,d dE
1d
2d
molécule BC
atome A
Réactifs
molécule AB
atome C
Produits
CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 16 sur 23
D’autre part, la différence d’énergie potentielle entre l’état de transition et les réactifs s’appelle énergie d’activation microscopique : ( ) ( )a p pcomplexe activé réactifs= −E E E . Plus cette barrière de potentiel est
élevée, plus la réaction est difficile, c’est-à-dire plus la constante de vitesse k est petite.
Remarque : l’énergie d’activation correspond à un « col » dans la représentation 3D de la surface des énergies potentielles. Cette énergie est bien inférieure à l’énergie qui serait nécessaire pour rompre la liaison covalente de la molécule BC. Le système constitué des trois atomes A, B et C libres de toute liaison chimique correspond, dans la représentation 3D, au « plateau » d’énergie potentielle pour les grandes valeurs de 1d et 2d .
Principe de microréversibilité
Pour deux réactions opposées, le profil énergétique est le même. Si un chemin réactionnel est possible dans un sens, le même chemin pourra être parcouru dans l’autre sens pour la réaction opposée : c’est en cela que consiste le principe de microréversibilité.
Le principe de microréversibilité implique une relation entre les énergies microscopiques d’activation pour deux réactions opposées. Avec les notations explicitées sur le profil réactionnel ci-dessus représenté : a a p′= + ∆E E E
Du microscopique au macroscopique
Loi d’Arrhenius
L’énergie molaire d’activation introduite dans la loi phénoménologique d’Arrhenius est assez précisément de l’ordre de grandeur de l’énergie microscopique d’activation multipliée par la constante d’Avogadro. Ceci se démontre dans le cadre de la théorie thermodynamique statistique quantique.
ama
B
Ek TRTk Ae Ae
−−= ≈
E
avec BA
Rk
N= et ma A aE N≈ E
Énergies molaires
Considérons deux réactions simples opposées : 1 1 2 2 1 1 2 2A A A Ak
k′ ′ ′ ′α + α α + α
′� 0
rH∆
Dans la mesure où l’on peut confondre les enthalpies standard et les énergies interne standard (il faut pour
cela que RT soit très petit devant 0rH∆ ), nous retrouvons à l’échelle macroscopique l’expression du
principe de microréversibilité : 0r ma maH E E′∆ = −
pE
aE
p∆ERéactifs
Produits
Complexe activé
C.R.
A BC+
[ ]A B C≠⋅ ⋅ ⋅ ⋅
AB C+a′E

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 17 sur 23
Constante de vitesse et constante d’équilibre
Pour tout couple de réactions opposées ayant un ordre, caractérisées par des ordres partiels ip pour la
réaction directe et ip′ pour la réaction inverse, la vitesse globale de la réaction s’écrit :
A Av i i
i i
p p
réactifs produits
k c k c ′′′= −∏ ∏
À l’équilibre, à température constante, la cinétique nous donne v 0= , soit : A
A
i
i
i
i
p
produits
p
réactifs
ck
k c
′′
=′
∏∏
Du point de vue thermodynamique, l’équilibre est caractérisé par la constante d’équilibre ( )0K T et la loi
d’action de masse en milieu homogène, s’écrit :
( ) ( ) r
AA0 00
A
ii i
i
i
i
nproduits
réactantsréactifs
cc
K T cc c
′αν ′
−∆′α
= =
∏∏ ∏
avec r i i iréactants produits réactifs
n ′∆ = ν = α − α∑ ∑ ∑
On en déduit, pour une température fixée, une relation entre grandeurs thermodynamique et cinétique, pour deux réactions d’ordre simple opposées, suivant la règle de Van’t Hoff dans les deux sens, c’est-à-dire deux réactions dont les ordres partiels dans les deux sens s’identifient aux coefficients iα , valeurs
absolues des coefficients stœchiométriques iν de l’équation bilan : i i ip = α = ν
( ) ( ) ( )r r
A0 0 0
A
i
i
i
i
p
n nproduits
p
réactifs
ck
K T c cc k
′′
−∆ −∆= =
′
∏∏
Approximation des états quasi stationnaires et Appr oximation de l’étape cinétiquement limitante
Étude générale du mécanisme A → B → C
Considérons cette suite de deux transformations A 1k
→ B puis B 2k
→ C, toutes deux monomoléculaires et, par conséquent, du premier ordre conformément à la règle de Van’t Hoff. On part du réactif A à la concentration initiale 0c pour aboutir à la transformation totale de A en C.
La vitesse de disparition de A obéit à une loi cinétique du premier ordre : A1 A
dck c
dt= −
Compte tenu de la condition initiale ( )A 00c c= , nous en déduisons : ( ) 1A 0
k tc t c e−=
Dès lors, nous pouvons écrire la loi cinétique concernant B, consommé par la deuxième réaction et produit par la première :
B1 A 2 B
dck c k c
dt= + − soit 1B
2 B 1 0k tdc
k c k c edt
−+ =
La solution générale de cette équation est la somme de la solution générale 2k tK e− de l’équation sans second membre et d’une solution particulière de l’équation complète que nous rechercherons de la même forme que le second membre, soit 1
Bpk tc Ae−= .
Déterminons la constante A par identification : 1 1 11 2 1 0
k t k t k tk Ae k Ae k c e− − −− + = 10
2 1
kA c
k k⇒ =
−
Il reste à déterminer la constante K en écrivant la condition initiale ( )B 0 0c = .
CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 18 sur 23
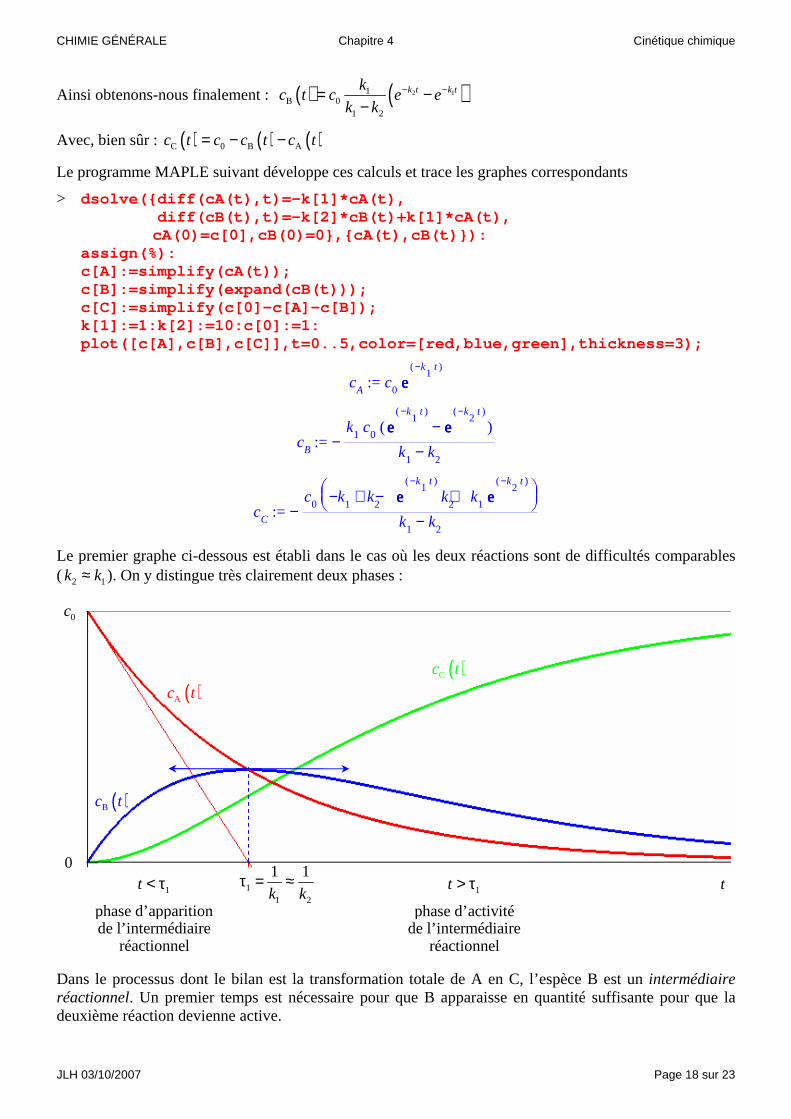
Ainsi obtenons-nous finalement : ( ) ( )2 11B 0
1 2
k t k tkc t c e e
k k− −= −
−
Avec, bien sûr : ( ) ( ) ( )C 0 B Ac t c c t c t= − −
Le programme MAPLE suivant développe ces calculs et trace les graphes correspondants
> dsolve({diff(cA(t),t)=-k[1]*cA(t), diff(cB(t),t)=-k[2]*cB(t)+k[1]*cA(t),
cA(0)=c[0],cB(0)=0},{cA(t),cB(t)}): assign(%): c[A]:=simplify(cA(t)); c[B]:=simplify(expand(cB(t))); c[C]:=simplify(c[0]-c[A]-c[B]); k[1]:=1:k[2]:=10:c[0]:=1: plot([c[A],c[B],c[C]],t=0..5,color=[red,blue,green],thickness=3);
:= cA
c0
eeee( )−k
1t
:= cB
−k
1c
0( ) − eeee
( )−k1
t
eeee( )−k
2t
− k1
k2
:= cC
−c
0
− + − + k
1k
2eeee
( )−k1
t
k2
k1
eeee( )−k
2t
− k1
k2
Le premier graphe ci-dessous est établi dans le cas où les deux réactions sont de difficultés comparables ( 2 1k k≈ ). On y distingue très clairement deux phases :
Dans le processus dont le bilan est la transformation totale de A en C, l’espèce B est un intermédiaire réactionnel. Un premier temps est nécessaire pour que B apparaisse en quantité suffisante pour que la deuxième réaction devienne active.
( )Ac t
( )Cc t
( )Bc t
11 2
1 1
k kτ = ≈ t
0c
0
1
phase d’apparitionde l’intermédiaire
réactionnel
t < τ 1
phase d’activitéde l’intermédiaire
réactionnel
t > τ
CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 19 sur 23
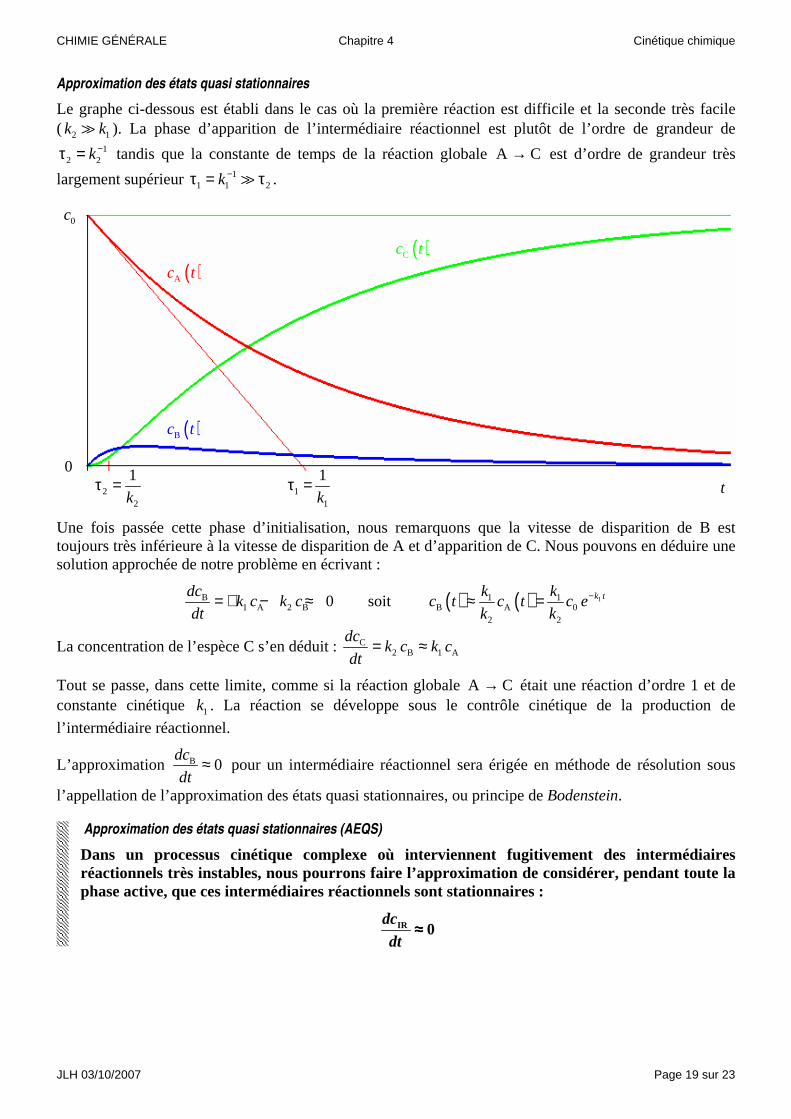
Approximation des états quasi stationnaires
Le graphe ci-dessous est établi dans le cas où la première réaction est difficile et la seconde très facile ( 2 1k k� ). La phase d’apparition de l’intermédiaire réactionnel est plutôt de l’ordre de grandeur de
12 2k−τ = tandis que la constante de temps de la réaction globale A C→ est d’ordre de grandeur très
largement supérieur 11 1 2k−τ = τ� .
Une fois passée cette phase d’initialisation, nous remarquons que la vitesse de disparition de B est toujours très inférieure à la vitesse de disparition de A et d’apparition de C. Nous pouvons en déduire une solution approchée de notre problème en écrivant :
B1 A 2 B 0
dck c k c
dt= + − ≈ soit ( ) ( ) 11 1
B A 02 2
k tk kc t c t c e
k k−≈ =
La concentration de l’espèce C s’en déduit : C2 B 1 A
dck c k c
dt= ≈
Tout se passe, dans cette limite, comme si la réaction globale A C→ était une réaction d’ordre 1 et de constante cinétique 1k . La réaction se développe sous le contrôle cinétique de la production de
l’intermédiaire réactionnel.
L’approximation B 0dc
dt≈ pour un intermédiaire réactionnel sera érigée en méthode de résolution sous
l’appellation de l’approximation des états quasi stationnaires, ou principe de Bodenstein.
Approximation des états quasi stationnaires (AEQS)
Dans un processus cinétique complexe où interviennent fugitivement des intermédiaires réactionnels très instables, nous pourrons faire l’approximation de considérer, pendant toute la phase active, que ces intermédiaires réactionnels sont stationnaires :
IR 0dc
dt≈≈≈≈
( )Ac t
( )Cc t
( )Bc t
11
1
kτ = t
0c
0
22
1
kτ =
CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 20 sur 23
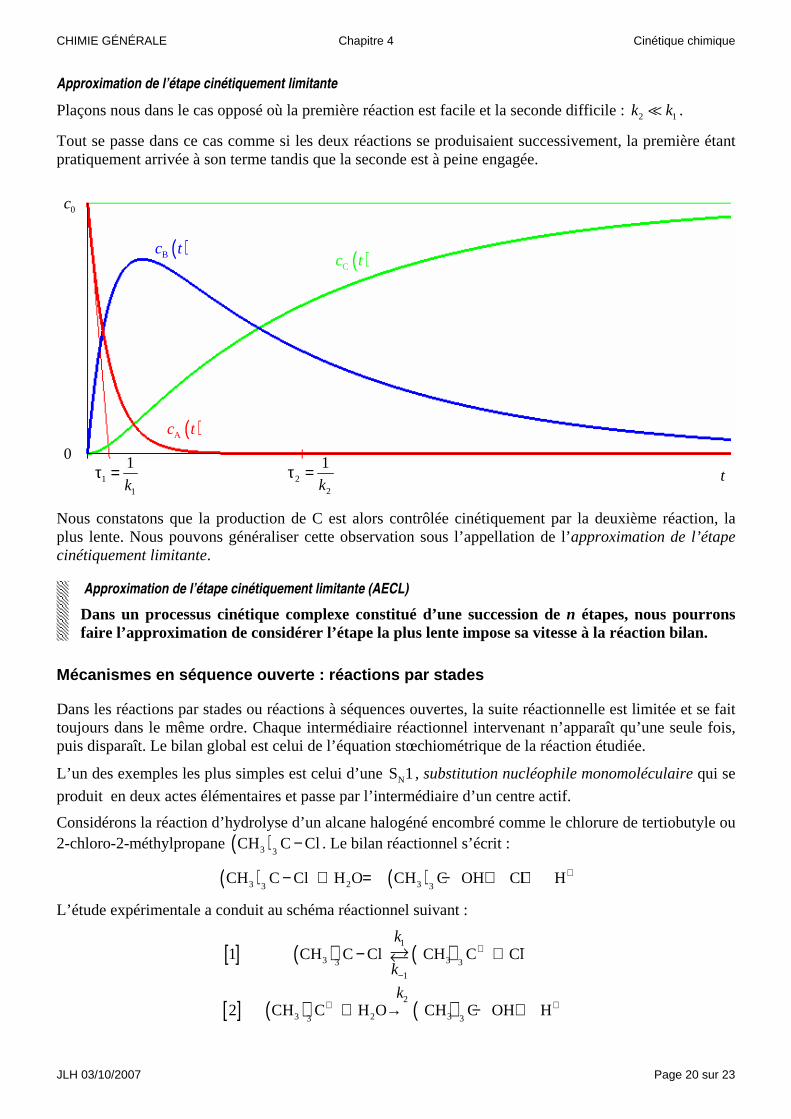
Approximation de l’étape cinétiquement limitante
Plaçons nous dans le cas opposé où la première réaction est facile et la seconde difficile : 2 1k k� .
Tout se passe dans ce cas comme si les deux réactions se produisaient successivement, la première étant pratiquement arrivée à son terme tandis que la seconde est à peine engagée.
Nous constatons que la production de C est alors contrôlée cinétiquement par la deuxième réaction, la plus lente. Nous pouvons généraliser cette observation sous l’appellation de l’approximation de l’étape cinétiquement limitante.
Approximation de l’étape cinétiquement limitante (AECL)
Dans un processus cinétique complexe constitué d’une succession de n étapes, nous pourrons faire l’approximation de considérer l’étape la plus lente impose sa vitesse à la réaction bilan.
Mécanismes en séquence ouverte : réactions par stad es
Dans les réactions par stades ou réactions à séquences ouvertes, la suite réactionnelle est limitée et se fait toujours dans le même ordre. Chaque intermédiaire réactionnel intervenant n’apparaît qu’une seule fois, puis disparaît. Le bilan global est celui de l’équation stœchiométrique de la réaction étudiée.
L’un des exemples les plus simples est celui d’une NS 1, substitution nucléophile monomoléculaire qui se
produit en deux actes élémentaires et passe par l’intermédiaire d’un centre actif.
Considérons la réaction d’hydrolyse d’un alcane halogéné encombré comme le chlorure de tertiobutyle ou 2-chloro-2-méthylpropane ( )3 3
CH C Cl− . Le bilan réactionnel s’écrit :
( ) ( )3 2 33 3CH C Cl H O CH C OH Cl H− +− + = − + +
L’étude expérimentale a conduit au schéma réactionnel suivant :
[ ] ( ) ( )
[ ] ( ) ( )
1
3 33 3
1
2
3 2 33 3
1 CH C Cl CH C Cl
2 CH C H O CH C OH H
k
k
k
+ −
−
+ +
− +
+ → − +
0c
22
1
kτ = t1
1
1
kτ =
( )Ac t
( )Cc t( )Bc t
0
CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 21 sur 23
L’expérience montre également une expression de la vitesse globale R Clv k c −= , indépendante du
nucléophile. R − symbolise le groupe tertiobutyle ( )3 3CH C− . Vérifions que cette loi cinétique est bien
compatible avec le mécanisme proposé.
La vitesse globale de la réaction est définie comme la vitesse d’apparition de l’alcool : R OHvdc
dt−=
Dans ce cas, d’après la règle de Van’t Hoff appliquée à l’étape élémentaire [ ]2 , nous avons aussi :
22 H ORv k c c+=
R+ est un intermédiaire réactionnel, il ne peut donc pas apparaître dans l’expression de la vitesse v, mais on lui applique l’approximation des états quasi stationnaires :
2
R1 R Cl 1 2 H OR Cl R
0dc
k c k c c k c cdt
+
+ − +− −= = − − soit 2
1 R ClR
1 2 H OCl
k cc
k c k c+
−
−
−
=+
Puis, en reportant cette expression dans la formule de la vitesse v, nous obtenons :
2
2
1 2 R Cl H O
1 2 H OCl
vk k c c
k c k c−
−
−
=+
Si toutes les étapes ont des vitesses dont les ordres de grandeur sont comparables, on ne retrouve pas la loi simple expérimentale. La réaction n’admet pas d’ordre. Mais si l’acte [ ]2 est très rapide par rapport à
l’acte [ ]1− , alors 22 H O 1 Cl
k c k c −−� , et l’expression de la vitesse se simplifie :
1 R Clv k c −=
L’étape [ ]1 de rupture hétérolytique du substrat est limitante. L’ordre est égal à 1, la réaction est
monomoléculaire.
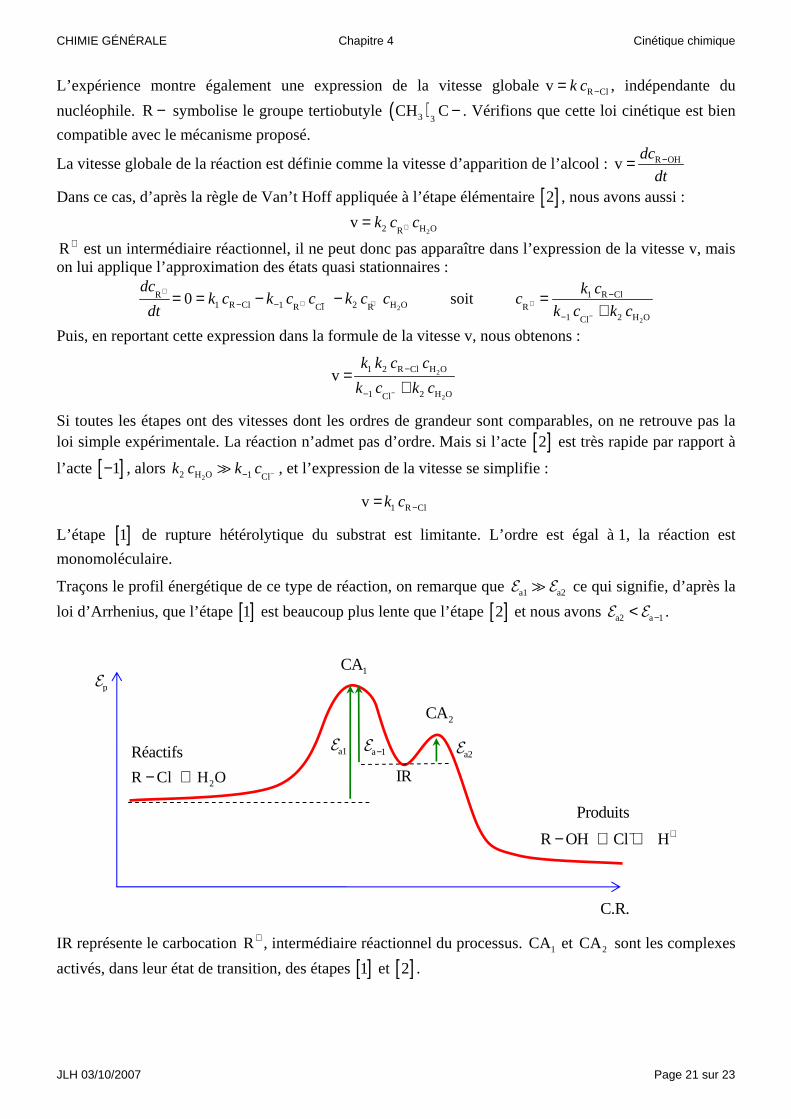
Traçons le profil énergétique de ce type de réaction, on remarque que a1 a2E E� ce qui signifie, d’après la
loi d’Arrhenius, que l’étape [ ]1 est beaucoup plus lente que l’étape [ ]2 et nous avons a2 a 1−<E E .
IR représente le carbocation R+ , intermédiaire réactionnel du processus. 1CA et 2CA sont les complexes
activés, dans leur état de transition, des étapes [ ]1 et [ ]2 .
pE
a1E
R OH Cl H− +− + +
Réactifs
Produits
1CA
C.R.
2R Cl H O− +
2CA
a 1−E a2E
IR

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 22 sur 23
Mécanismes en séquence fermée : réactions en chaîne
Dans les réactions en chaîne ou réactions à séquence fermée, un des intermédiaires réactionnels se trouve cette fois régénéré au cours d’une des étapes et peut à nouveau entrer en réaction. C’est le porteur de chaîne. On obtient alors un enchaînement d’actes élémentaires qui se reproduisent de façon cyclique, d’où leur nom.
Les réactions en chaîne droite ou linéaire
Si, à partir d’un centre actif unique, naît un seul centre actif, il s’agit d’une réaction en chaîne droite ou linéaire, pour laquelle on peut appliquer l’approximation des états quasi stationnaires aux intermédiaires réactionnels. De nombreuses réactions chimiques se produisent en chaîne droite : les additions et les substitutions radicalaires, pyrolyse d’alcanes, synthèse d’acide bromhydrique ou chlorhydrique, polymérisation radicalaire, etc.
C’est après avoir terminé l’étude de la réaction de l’hydrogène sur l’iode que Bodenstein se tourne vers celle de l’hydrogène sur le brome, vers 1903, espérant probablement rencontrer un nouvel exemple de cinétique bimoléculaire. Les résultats sont étonnamment différents, la réaction n’a pas d’ordre et Bodenstein a observé la loi de vitesse suivante :
2 2H Br 2 HBr+ = 2 2
2
1/2H Br
HBr
Br
v1
c ck
ck
c
=′+
Cette « curieuse » loi de vitesse est interprétée seulement treize ans plus tard par Christiansen, Herzfeld et Polanyi, indépendamment les uns des autres. Ils proposent le cycle de réactions en chaîne suivant :
[ ]
[ ]
[ ]
[ ]
[ ]
1
2
2
2
3
2
2
2
1
2
1 Br 2 Br
2 Br H HBr H
3 H Br HBr Br
4 HBr H Br H
5 2 Br Br
k
k
k
k
k
−
−
→
+ → +
+ → +
+ → +
→
Cette réaction, qui passe par des intermédiaires réactionnels radicalaires, est appelée substitution radicalaire.
Définissons, sur cet exemple, quelques termes caractéristiques des réactions en chaîne. On distingue trois phases : l’amorçage (souvent remplacé par un anglicisme : initiation), la propagation et la rupture ou terminaison.
— l’amorçage permet la formation du centre actif initial, encore nommé porteur de chaîne, sous forme d’un intermédiaire réactionnel radicalaire, par apport d’énergie photochimique, thermique ou par action des peroxydes. Dans l’exemple de la synthèse de HBr, la réaction est déclenchée par la dissociation thermique du dibrome, vers 200°C-300°C : c’est l’étape [ ]1 .
— la propagation est la phase de progression des réactions en chaîne. Le porteur de chaîne, en général par action sur un réactif, engendre un centre actif qui lui-même régénère le centre actif initial. La réaction peut se poursuivre par itération de ces étapes que l’on nomme maillon de chaîne. Le bilan de la réaction en chaîne s’identifie au bilan d’un maillon de chaîne.

CHIMIE GÉNÉRALE Chapitre 4 Cinétique chimique
JLH 03/10/2007 Page 23 sur 23
Dans notre exemple, les réactions [ ]2 et [ ]3 constituent le maillon de la chaîne, elles génèrent le
produit HBr de la réaction et reconstituent le porteur de chaîne Br, prêt pour un autre cycle. L’ensemble constitue la phase de propagation de la réaction.
[ ] [ ] 2 22 3 H Br 2 HBr+ + →
— la rupture ou terminaison est la phase de disparition des centres actifs, la plupart du temps par recombinaison des porteurs de chaîne, leur concentration étant plus importante. La réaction s’arrête donc. Dans le cas de la synthèse de HBr, il s’agit des étapes [ ]4 et [ ]5 .
Une réaction en chaîne est aussi caractérisée par sa longueur de chaîne . La longueur d’une chaîne est le nombre de maillons ou cycles auxquels peut donner naissance, en moyenne, un porteur de chaîne avant d’être détruit dans la phase de rupture.
Revenons à la modélisation de la loi cinétique de Bodenstein :
Afin de définir la vitesse globale de la réaction, il est intéressant, par souci de simplification, d’utiliser l’espèce qui intervient le moins souvent dans le schéma réactionnel, soit ici 2H :
2
2
H2 Br H 4 H HBrv
dck c c k c c
dt= − = −
Br et H sont des intermédiaires réactionnels. On peut leur appliquer l’approximation des états quasi stationnaires :
[ ]
[ ]
2 2 2
2 2
2Br1 Br 2 Br H 3 H Br 4 H HBr 5 Br
H2 Br H 3 H Br 4 H HBr
2 2 0
0
dck c k c c k c c k c c k c
dtdc
k c c k c c k c cdt
α = − + + − =
β = − − =
La somme [ ] [ ]α + β fournit : 2
1/ 2
1Br Br
5
kc c
k
=
Puis, à partir de [ ]β , nous obtenons : 2
2
2
1/ 2
2 H1H Br
5 3 Br 4 HBr
k ckc c
k k c k c
= +
Après report dans l’expression de la vitesse v, nous trouvons finalement : 2 2
2
1/ 2 1/2H Br1
24 HBr5
3 Br
v1
c ckk
k ckk c
=
+
En posant 1/ 2
12
5
kk k
k
=
et 4
3
kk
k′ = , cela s’écrit : 2 2
2
1/2H Br
HBr
Br
v1
c ck
ck
c
=′+
Cette expression est en accord avec l’expérience.
Les réactions en chaîne ramifiées
Si, au contraire, une étape engendre plusieurs centres actifs, c’est une réaction en chaîne ramifiée, plus difficile à modéliser. La multiplication des intermédiaires réactionnels provoque l’accélération de la réaction et peut conduire à une explosion. L’exemple le plus classique de réaction en chaîne ramifiée est le « mélange tonnant » ou synthèse de l’eau, mais c’est aussi le mécanisme de nombreuses autres oxydations.