chapitre 7 structures covalentes des protéines 1.détermination de la structure primaire des...
TRANSCRIPT

Chapitre 7 Structures covalentes des protéines
1.Détermination de la structure primaire des protéines
A. Coupure des ponts disulfure et séquençage d'Edman
B. Réactions d'hydrolyse spécifiques de liaisons peptidiques
C. Détermination de la séquenceD. Séquençage de protéines par
spectrométrie de masse

2. Evolution chimique des protéinesA. L'anémie falciforme: influence de la sélection naturelleB. Variations entre espèces de protéines homologuesC. Evolution par duplication de gènes
3. Introduction à la bioinformatiqueA. Bases de données sur les séquencesB. Alignements de séquences et construction d'arbres phylogénétiques

4. Synthèse chimique de polypeptidesA. Stratégie générale de synthèseB. Couplage des acides aminés

Fonctions des protéines
Fonctions :- catalyse (enzymes)- transport (hémoglobine, albumine,
transporteurs membranaires)- structure (spectrine) - travail mécanique (actine et myosine)- régulation de la transcription- hormones, récepteurs (insuline,
récepteur de l’insuline)- immunoglobulines (IgG, IgM)
La fonction d'une protéine ne peut être comprise que par sa structure
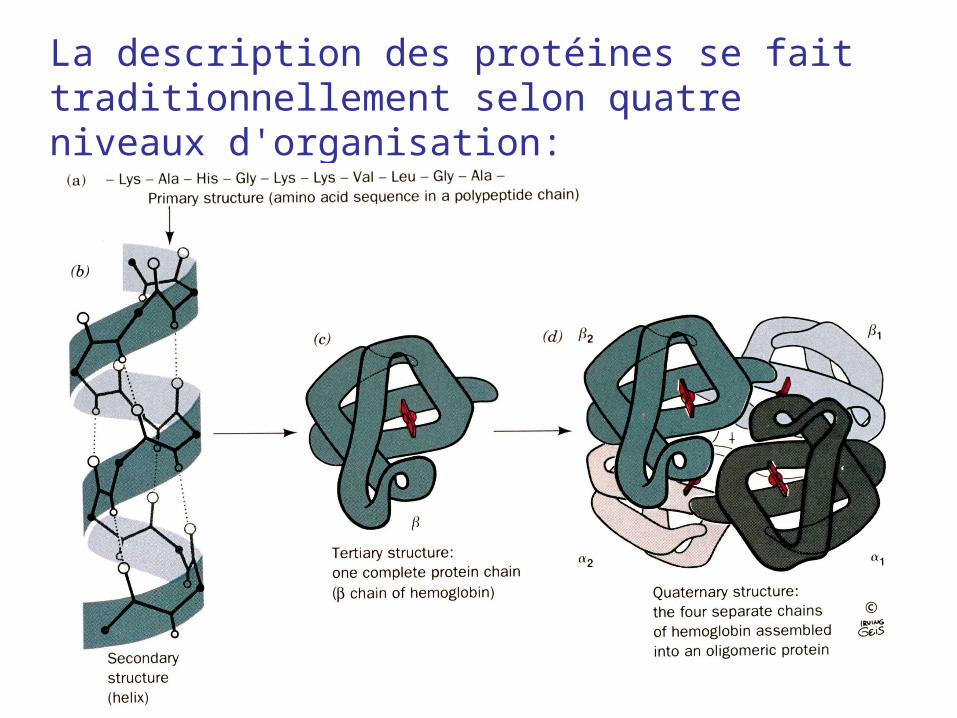
La description des protéines se fait traditionnellement selon quatre niveaux d'organisation:

1 DETERMINATION DE LA STRUCTURE PRIMAIRE DES PROTEINES
1. Séquençage de la protéine elle-même
- clivage d’une protéine en peptidesprotéasesréactifs chimiques
- méthode d’Edmancycles de réaction permettant
l’enlèvement de l’acide aminé N-terminal
- spectrométrie de masse
2. Séquençage de l’ADN (ADNc) codant la protéine

Intérêts de la détermination de la séquence en acidesaminés d'une protéine
1. La séquence d’une protéine est son identité:
-indispensable pour comprendre son mécanisme d'action au niveau moléculaire et essentielle pour la détermination de la structure tridimensionnelle
2. Permet d’identifier le gène
3. Comparaisons de séquences:
-identification des résidus les plus conservés (les plus importants pour la fonction)
-étude de l’évolution
-applications cliniques car beaucoup de maladies héréditaires sont dues à des mutations qui modifient la nature d'un acide aminé dans une protéine

Structure primaire de l'insuline bovine. Remarquez les ponts disulfure intra- et intercaténaires
La première détermination de la séquence complète en acides aminés d'une protéine - l'insuline de boeuf par Fred Sanger en 1953L'éucidation de la structure primaire a nécessité plus que 10 ans de travail et environ 100g de protéine!
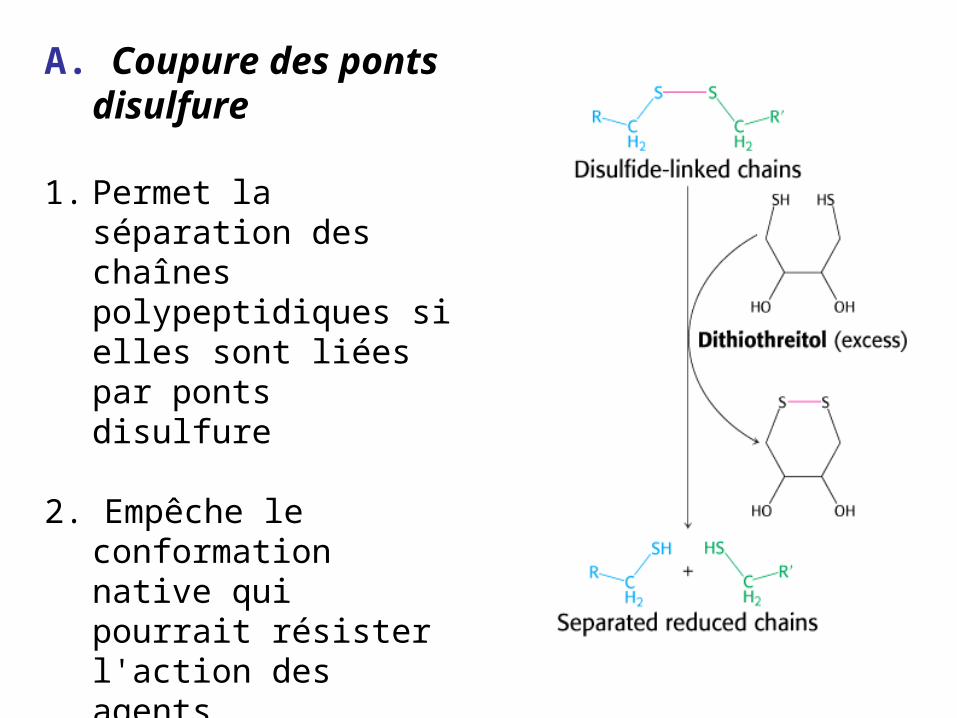
A. Coupure des ponts disulfure
1. Permet la séparation des chaînes polypeptidiques si elles sont liées par ponts disulfure
2. Empêche le conformation native qui pourrait résister l'action des agents protéolytiques
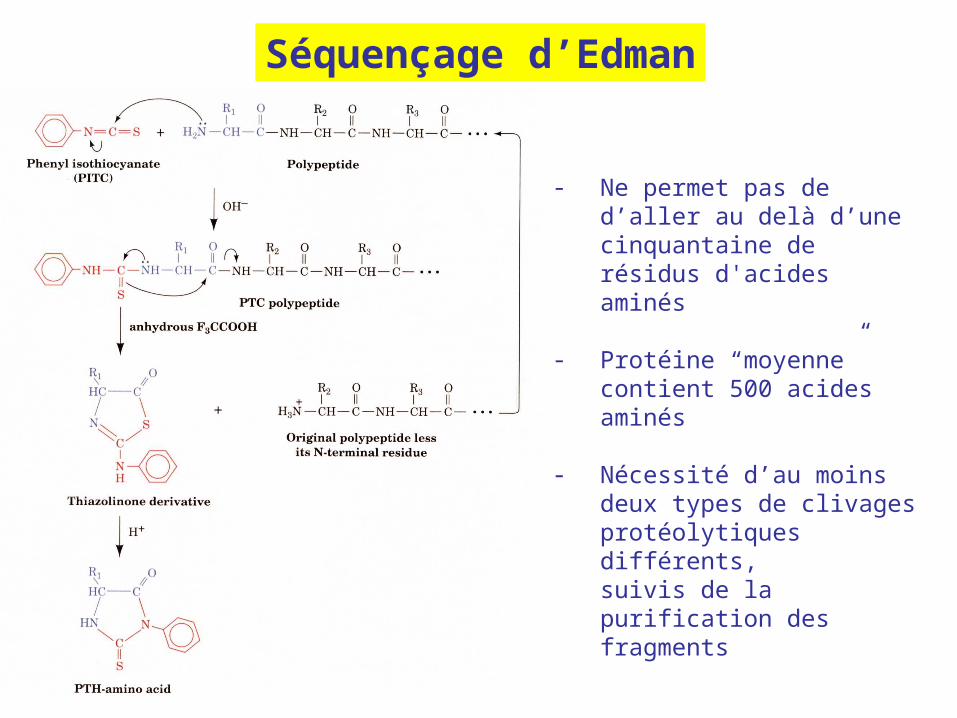
Séquençage d’Edman
- Ne permet pas de d’aller au delà d’une cinquantaine de résidus d'acides aminés
- Protéine “moyenne” contient 500 acides aminés
- Nécessité d’au moins deux types de clivages protéolytiques différents, suivis de la purification des fragments
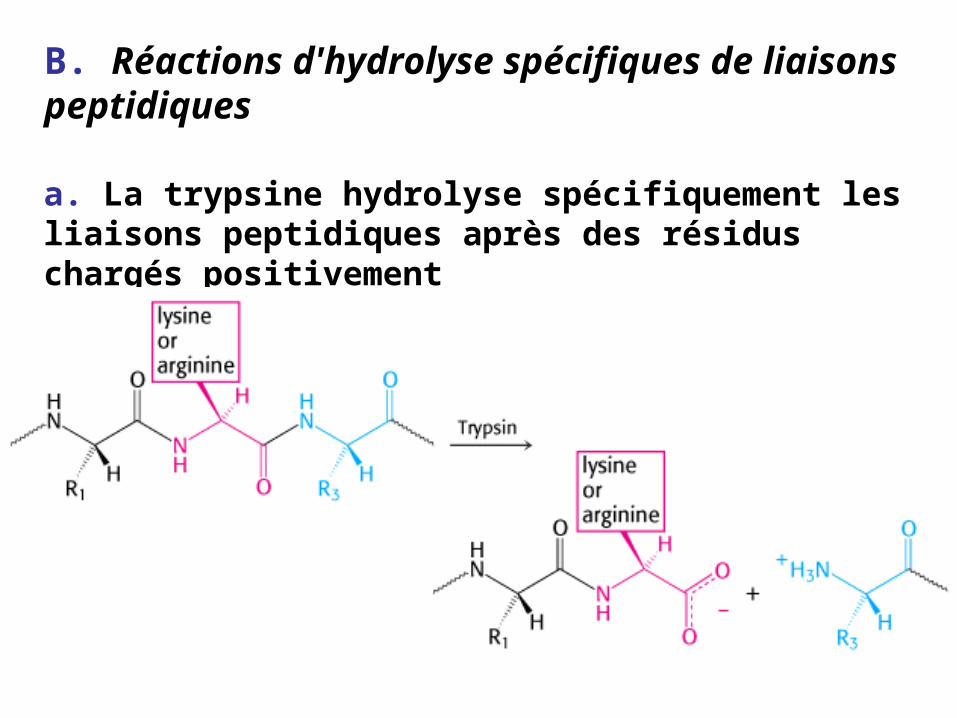
B. Réactions d'hydrolyse spécifiques de liaisons peptidiques
a. La trypsine hydrolyse spécifiquement les liaisons peptidiques après des résidus chargés positivement
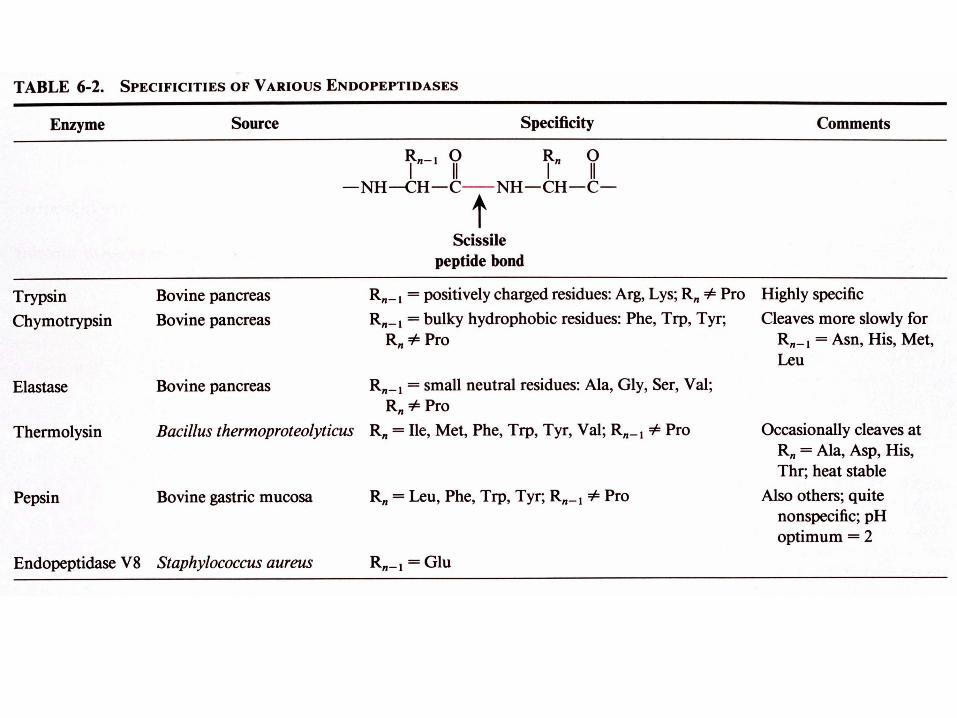
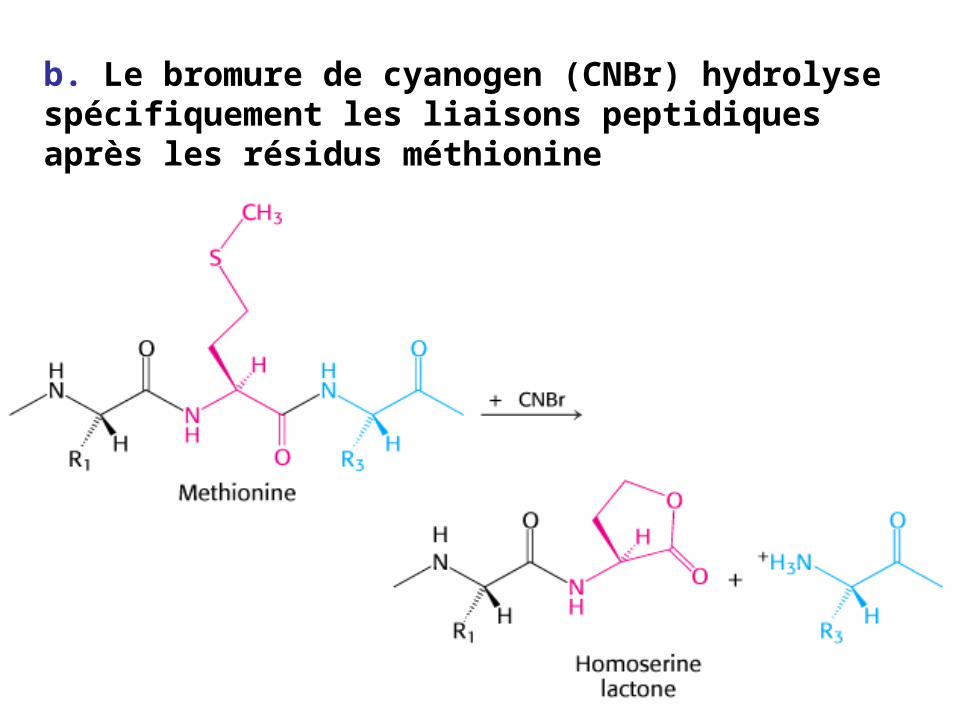
b. Le bromure de cyanogen (CNBr) hydrolyse spécifiquement les liaisons peptidiques après les résidus méthionine

C. Détermination de la séquence
L'HPLC en phase inverse permet la séparation des fragments protéolytiques avant leur séquençage par la méthode d'Edman. La séquence du polypeptide original est obtenue en comparant les séquences en acides aminés d'une série de fragments peptidiques avec celles d'une deuxième série dont les sites d'hydrolyse recouvrent ceux de la première série:
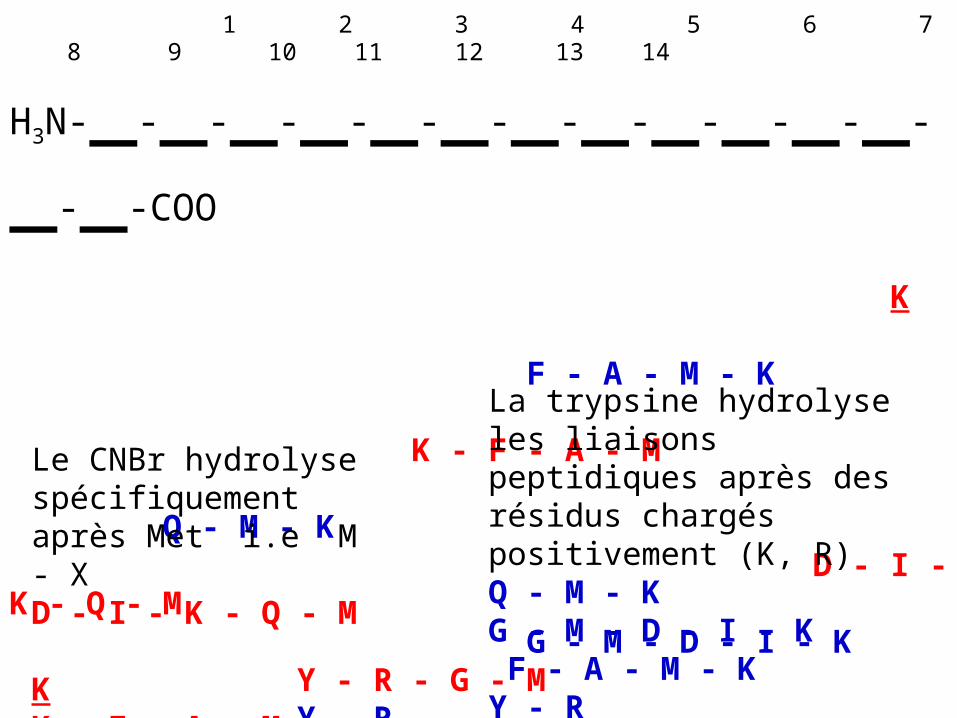
1 2 3 4 5 6 7 8 9 10 11 12 13 14
H3N-_-_-_-_-_-_-_-_-_-_-_-_-_-_-COO K F - A - M - K K - F - A - M Q - M - K D - I - K - Q - M G - M - D - I - K Y - R - G - M Y - R
Le CNBr hydrolyse spécifiquement après Met i.e M - XD - I - K - Q - M KK - F - A - MY - R - G - M
La trypsine hydrolyse les liaisons peptidiques après des résidus chargés positivement (K, R)Q - M - KG - M - D - I - K F - A - M - KY - R
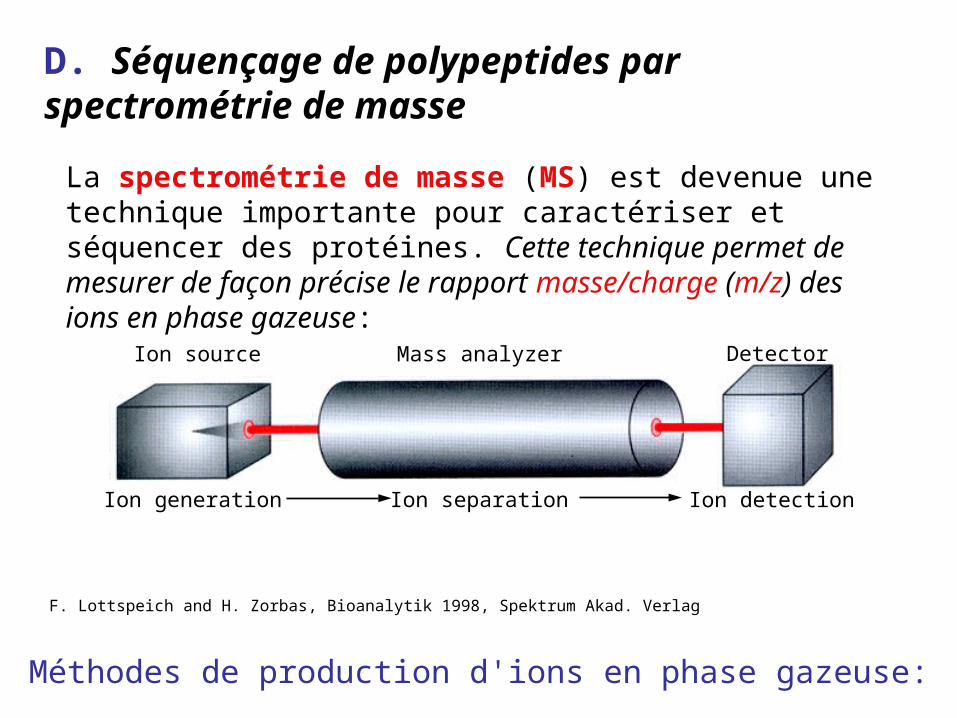
D. Séquençage de polypeptides par spectrométrie de masse
Ion source Mass analyzer Detector
Ion generation Ion separation Ion detection
F. Lottspeich and H. Zorbas, Bioanalytik 1998, Spektrum Akad. Verlag
Méthodes de production d'ions en phase gazeuse:
La spectrométrie de masse (MS) est devenue une technique importante pour caractériser et séquencer des protéines. Cette technique permet de mesurer de façon précise le rapport masse/charge (m/z) des ions en phase gazeuse:
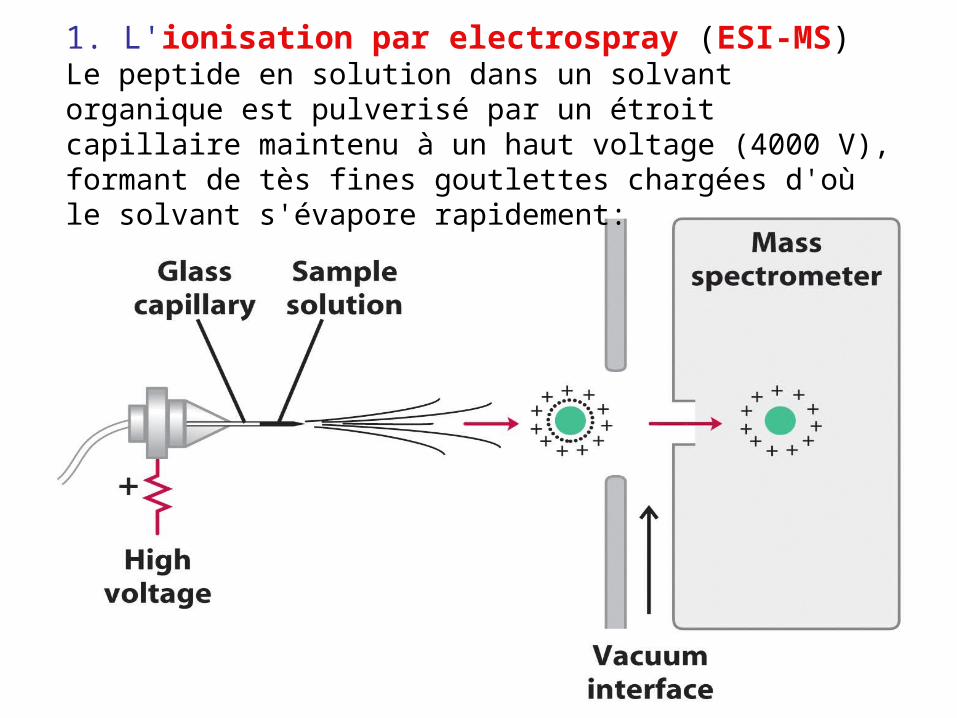
1. L'ionisation par electrospray (ESI-MS)Le peptide en solution dans un solvant organique est pulverisé par un étroit capillaire maintenu à un haut voltage (4000 V), formant de tès fines goutlettes chargées d'où le solvant s'évapore rapidement:

2. La désorption/ionisation au laser assistée par une matrice (''Matrix-Assisted Laser Desorption/Ionisation''): MALDI
Le peptide est enrobé dans une matrice cristalline et irradié par de courtes (ns) et intenses impulsions d'un rayon laser d'une longueur d'onde telle qu'elle est absorbée par la matrice. L'énergie absorbée par la matrice éjecte de sa surface les peptides intacts chargés:

• m/z: rapport masse/charge
VALEACVQAR Masse = 1059,25 Da
VALEACVQAR
H+
Masse = 1060,25 Dam/z = 1060,25/1 = 1060,25
VALEACVQAR
H+
H+ Masse = 1061,25 Dam/z = 1061,25/2 = 530,63
masse [M + nH+] = (m/z x n) - n
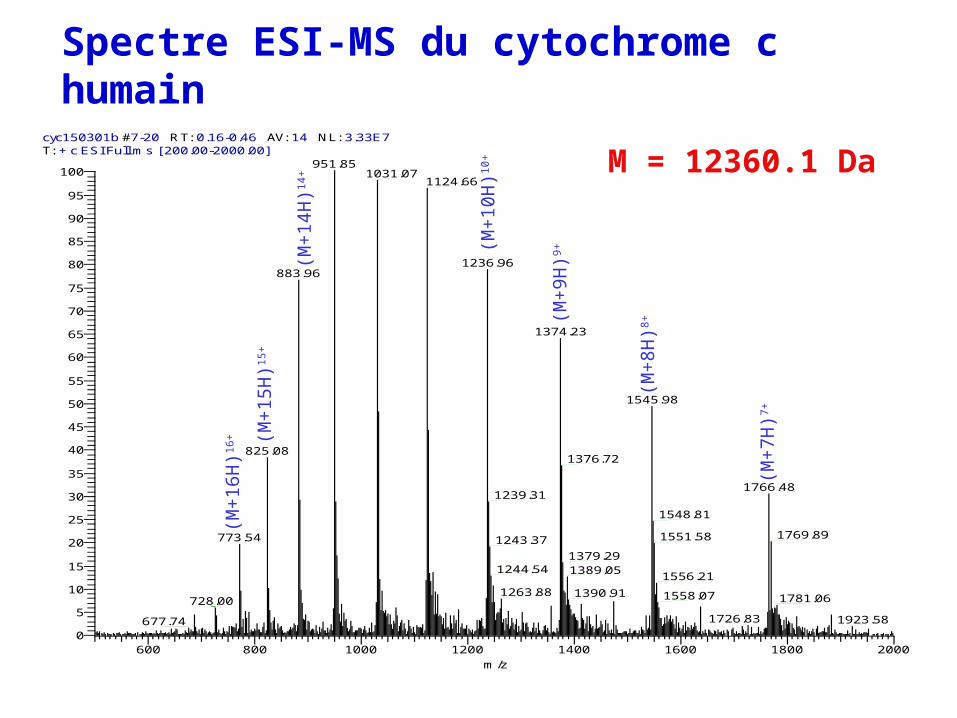
Spectre ESI-MS du cytochrome c humain
M = 12360.1 Dacyc150301b #7-20 RT: 0.16-0.46 AV: 14 NL: 3.33E7T: + c ESI Full ms [ 200.00-2000.00]
600 800 1000 1200 1400 1600 1800 2000
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Relative Abundance
951.851031.07
1124.66
1236.96883.96
1374.23
1545.98
825.081376.72
1766.481239.31
1548.81
1769.891551.58773.54 1243.37
1379.291244.54 1389.05
1556.21
1263.88 1390.91 1558.07 1781.06728.00
1726.83 1923.58677.74
(M+
10
H)1
0+
(M+
9H
)9+
(M+
8H
)8+
(M+
14
H)1
4+
(M+
15
H)1
5+
(M+
7H
)7+
(M+
16
H)1
6+
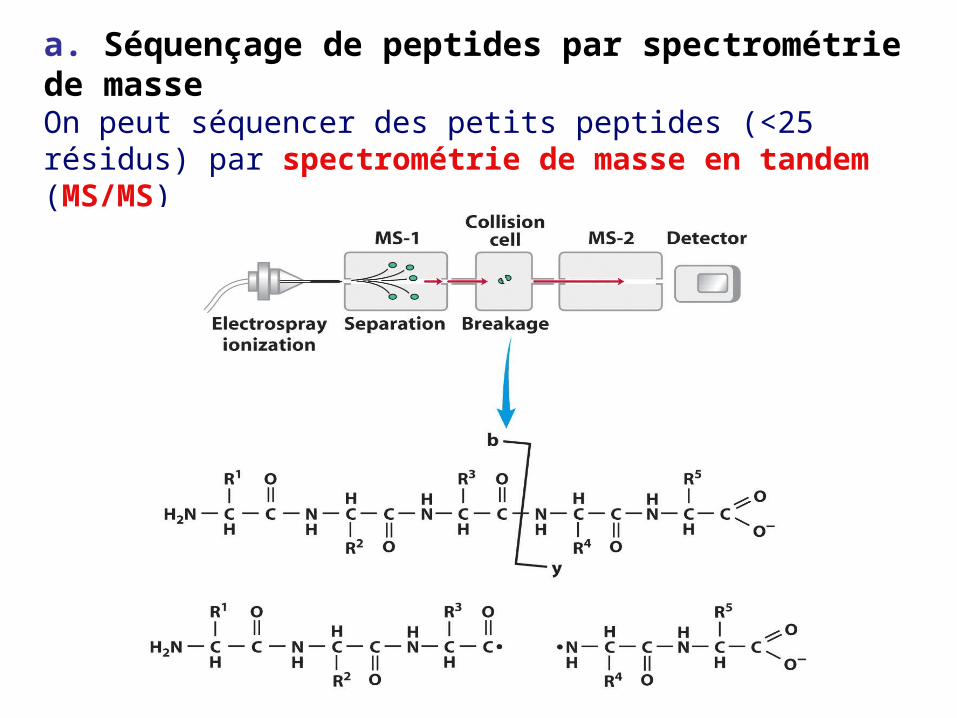
a. Séquençage de peptides par spectrométrie de masseOn peut séquencer des petits peptides (<25 résidus) par spectrométrie de masse en tandem (MS/MS)

Amino Acid Residue MassesAmino Acid Residue Mass Monoisotopic AverageGlycine Gly G 57.02147 57.052Alanine Ala A 71.03712 71.079Serine Ser S 87.03203 87.078Proline Pro P 97.05277 97.117Valine Val V 99.06842 99.133Threonine Thr T 101.04768 101.105Cysteine Cys C 103.00919 103.144Isoleucine Ile I 113.08407 113.160Leucine Leu L 113.08407 113.160Asparagine Asn N 114.04293 114.104Aspartic Acid Asp D 115.02695 115.089Glutamine Gln Q 128.05858 128.131Lysine Lys K 128.09497 128.174Glutamic Acid Glu E 129.04260 129.116Methionine Met M 131.04049 131.198Histidine His H 137.05891 137.142Phenylalanine Phe F 147.06842 147.177Arginine Arg R 156.10112 156.188Tyrosine Tyr Y 163.06333 163.170Tryptophan Try W 186.07932 186.213 Carboxyamidomethyl Cysteine 160.03065 160.197Carboxymethylcysteine 161.01466 161.181

T E S T P E P T I D E+ T E S T + P E P T I D E+
b 1 T+
b 2 TE+
b 3 TES+
b 4 TEST+
b 5 TESTP+
b 6 TESTPE+
b 7 TESTPEP+
b 8 TESTPEPT+
b 9 TESTPEPTI+
b10 TESTPEPTID+
b11 TESTPEPTIDE+ - H2O
TESTPEPTIDE+ y11
ESTPEPTIDE+ y10
STPEPTIDE+ y 9
TPEPTIDE+ y 8
PEPTIDE+ y 7
EPTIDE+ y 6 PTIDE+ y 5
TIDE+ y 4 IDE+ y 3
DE+ y 2
E+ y 1
b-ions y-ions
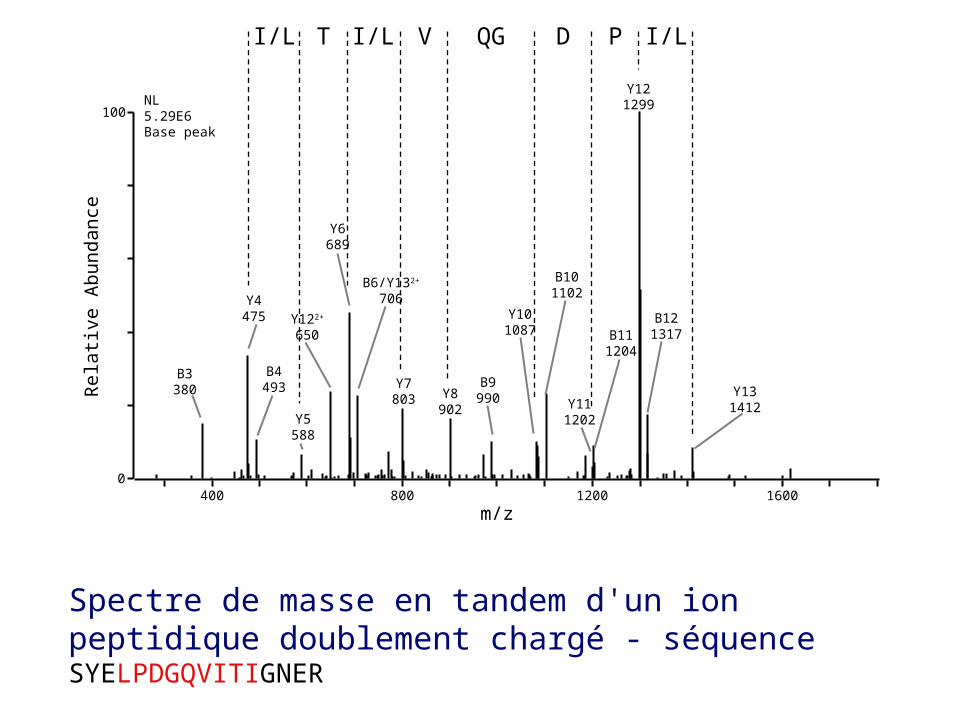
La séquence complète du peptide est ainsi elucidée. La fiabilité de la MS a encore été améliorée par la comparaison informatique du spectre de masse mesuré avec des spectres de masses prédits (''in silico'') à partir de séquences génomiques dans des bases de données
400 800 1200 1600
m/z
0
100
Rel
ativ
e A
bund
ance
Y121299
Y6689
Y4475 Y122+
650
B101102
B6/Y132+
706
Y7803
B121317
Y8902
B3380
B4493 B9
990
B111204
Y131412
Y5588
Y111202
Y101087
NL5.29E6Base peak
I/L T I/L V QG D P I/L
Spectre de masse en tandem d'un ion peptidique doublement chargé - séquence SYELPDGQVITIGNER

Avantages et inconvénients des différentes méthodes
Edman
Spectrométriede masse
Séquençage de l’ADN
Avantages
- Excellent pour 20 acides aminés standards
- OK pour longs peptides
- Rapidité relative- OK pour certaines
modificationspost-traductionnelles
Rapidité
Inconvénients
- Lenteur/lourdeur/coût- Beaucoup modifications
post-traductionnelles non
détectées
- Ambiguïté Ile/leu; Gln/Lys- Difficile avec longs
peptides
- Ignorance des modifications
post-traductionnelles- Erreurs de phase de
lecture

2 EVOLUTION CHIMIQUE DES PROTEINES
Les changements au cours de l'évolution, dus à des mutations qui se font au hasard, modifient souvent la structure primaire d'une protéine. Une mutation dans une protéine, si elle doit se propager, doit augmenter la probabilité de survie. En de rares occasions, une mutation défavorable améliore l'adaptation de son hôte à son environnement naturel:
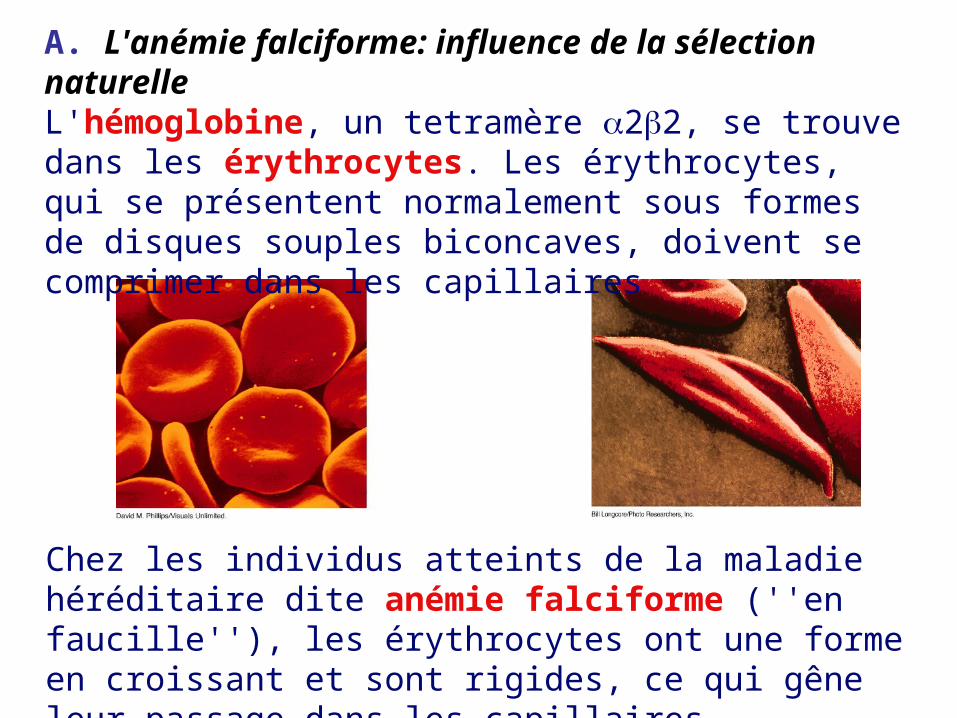
A. L'anémie falciforme: influence de la sélection naturelleL'hémoglobine, un tetramère 22, se trouve dans les érythrocytes. Les érythrocytes, qui se présentent normalement sous formes de disques souples biconcaves, doivent se comprimer dans les capillaires
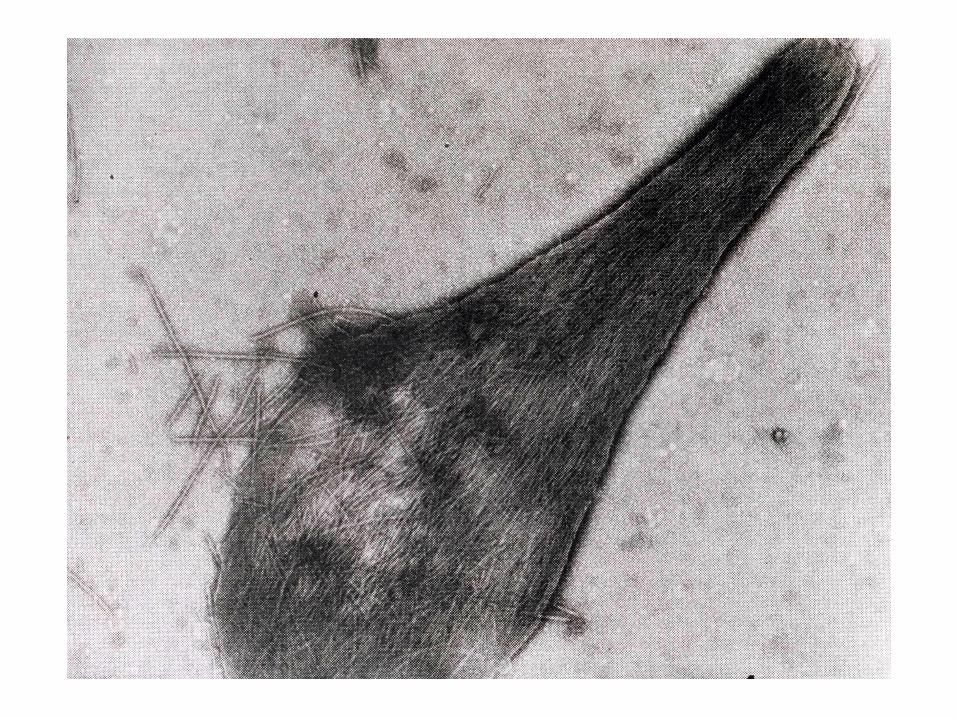
Chez les individus atteints de la maladie héréditaire dite anémie falciforme (''en faucille''), les érythrocytes ont une forme en croissant et sont rigides, ce qui gêne leur passage dans les capillaires

a. L'anémie falciforme est une maladie moléculaire
En 1945, Linus Pauling a postulé que l'anémie falciforme est due à la présence d'une hémoglobine mutante. Par des études électrophorétiques, il a montré que l'hémoglobine normale (HbA) a une charge anionique plus négative que l'hémoglobine de l'anémie falciforme (HbS):
Cette différence vient du remplacement du Glu 6 de HbA par une Val dans HbS(Glu 6 Val)
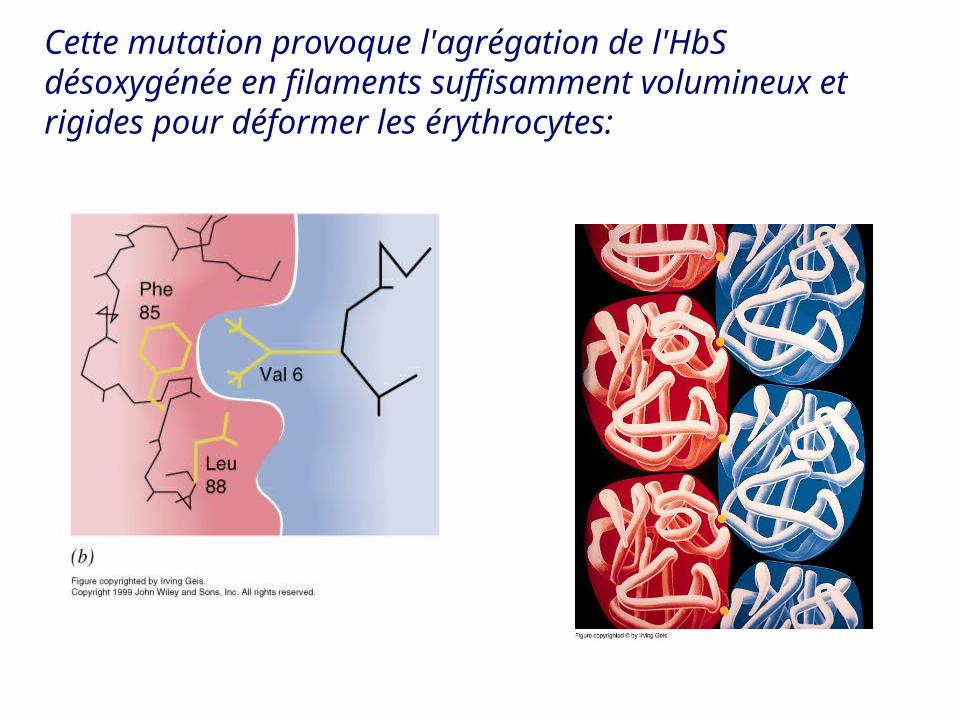
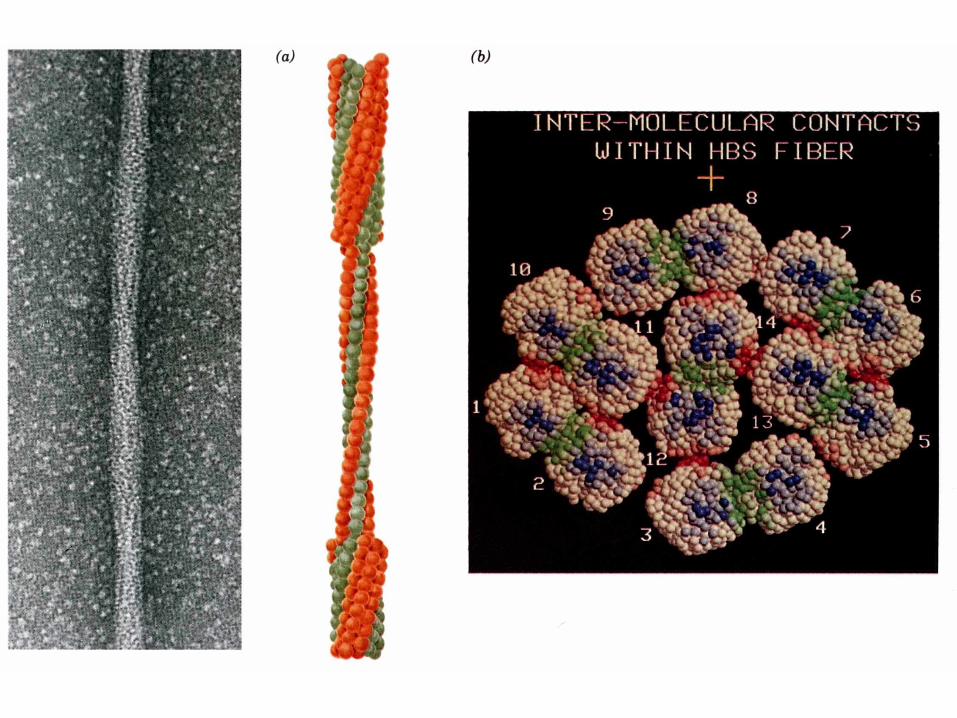
Cette mutation provoque l'agrégation de l'HbS désoxygénée en filaments suffisamment volumineux et rigides pour déformer les érythrocytes:

b. Le ''trait'' anémie falciforme confère la résistance à la malaria
Mutations du gène d’une des deux globines entraînant:-Modification de l’affinité de Hb pour O2
-Anémie falciforme- Maladie récessive- Mutations des sous-unités ß (Gluß6 en Val) de
l'HbS- Polymérisation de la désoxy-hémoglobine- Déformation des globules rouges et hémolyse- Hétérozygotes: non malades, mais porteurs du
“trait”- Fréquence très élevée dans région d’endémie de
malaria- Avantage des hétérozygotes (résistance relative à
malaria)- Au premier stade de l'infection, les érythrocytes
infectés sont retires de la circulation par la rate. Aux stades ultérieurs, la forme en faucille désorganise le parasite mécaniquement et/ou métaboliquement
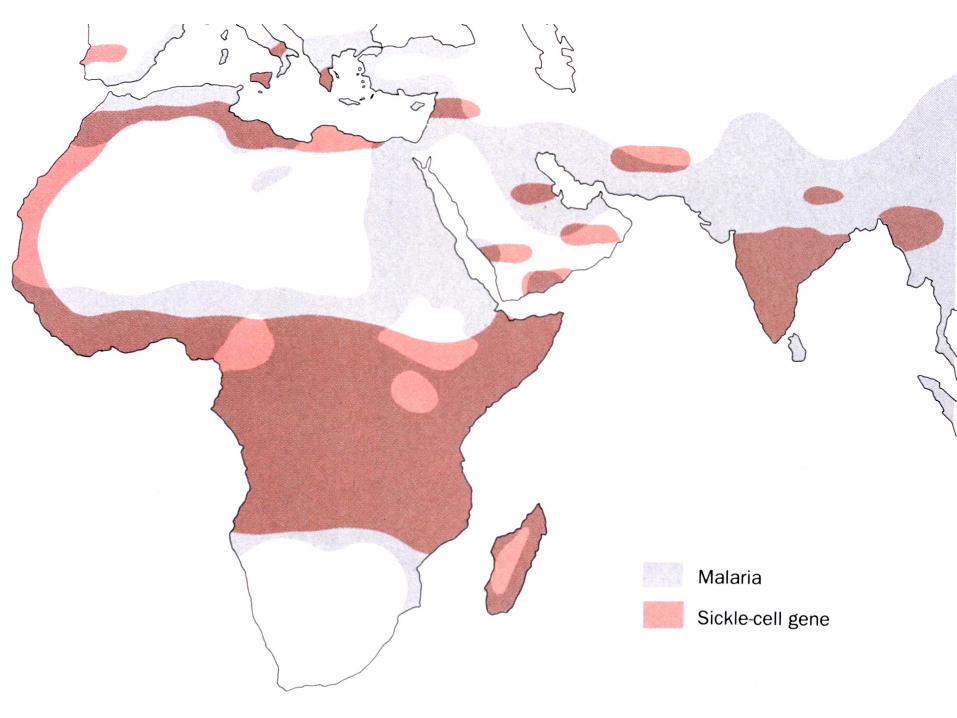

B. Variations entre espèces de protéines homologues: effets de la dérive naturelle
Les structures primaires d'une protéine donnée d'espèces voisines sont très semblables
Une protéine bien adapté à sa fonction continue néanmoins à évoluer
La dérive naturelle - modification d'une protéine par mutation aléatoire avec le temps sans affecter significativement sa fonction
La comparaison des structures primaires de protéines homologues indique quels sont des acides aminés qui sont indispensables à sa fonction, ceux qui ont moins d'importance, et ceux qui n'ont pas de rôle spécifique

Résidu invariant - acide aminé seul capable d'assurer un rôle essentiel à un endroit particulier de la séquence en acides aminés d'une série de protéines homologues - dû à ses propriétés chimiques/structurelles particulières
Substitutions conservatrices - position de la séquence en acides aminés occupée par des résidus qui ont des propriétés physico-chimiques similaires (par exemple ceux à propriétés acides: Asp et Glu)
Position hypervariable - beaucoup de résidus d'acides aminés différents peuvent être tolérés en certaines positions
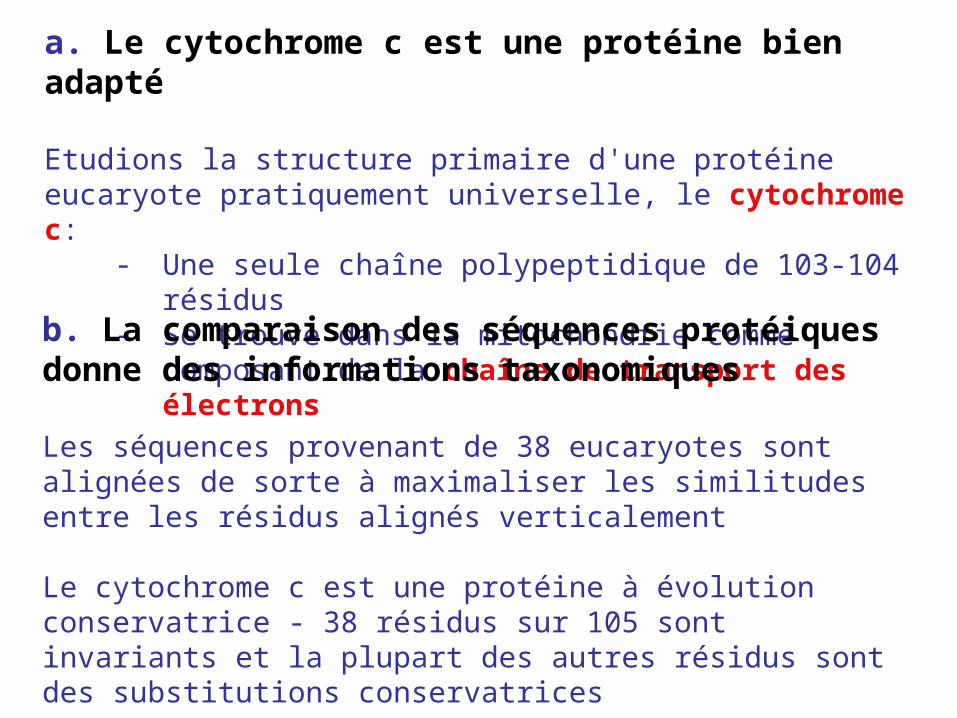
a. Le cytochrome c est une protéine bien adapté
Etudions la structure primaire d'une protéine eucaryote pratiquement universelle, le cytochrome c:
- Une seule chaîne polypeptidique de 103-104 résidus
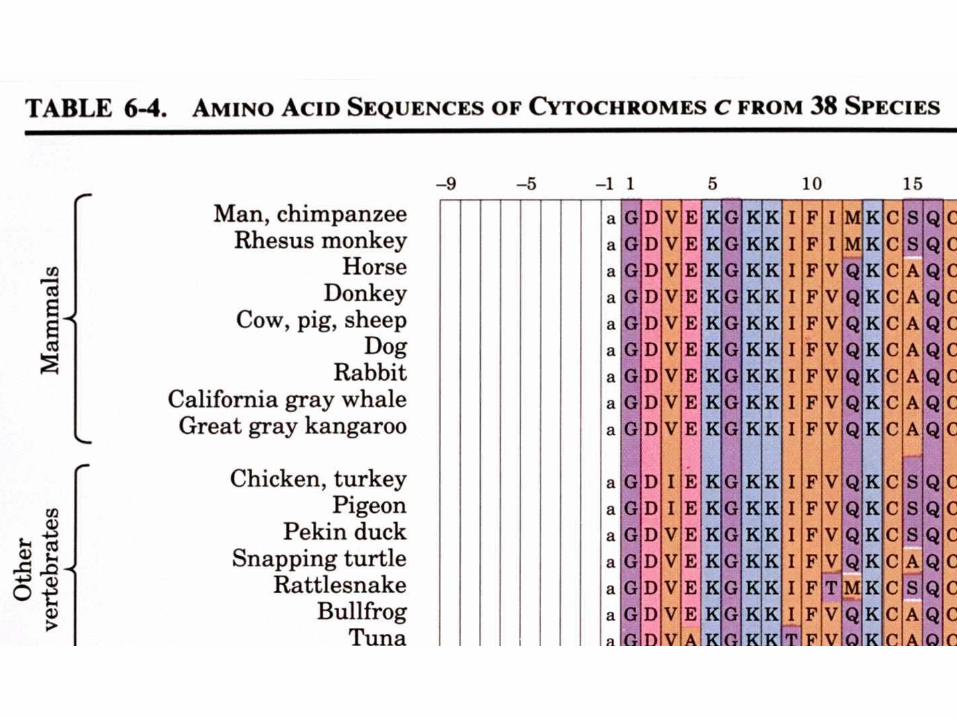
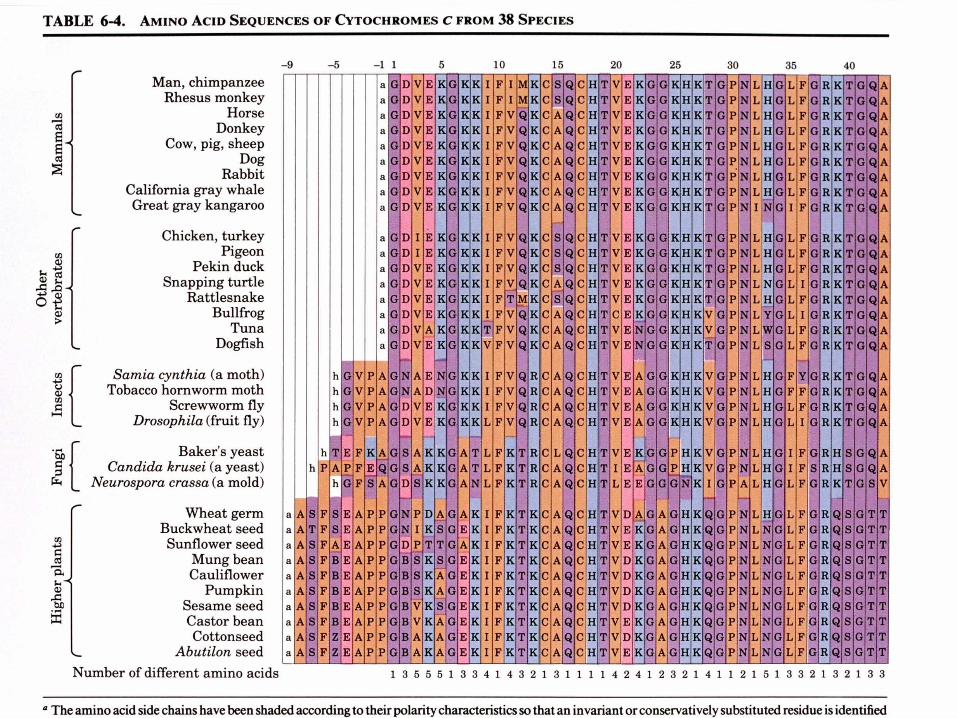
- Se trouve dans la mitochondrie comme composant de la chaîne de transport des électronsb. La comparaison des séquences protéiques
donne des informations taxonomiques
Les séquences provenant de 38 eucaryotes sont alignées de sorte à maximaliser les similitudes entre les résidus alignés verticalement
Le cytochrome c est une protéine à évolution conservatrice - 38 résidus sur 105 sont invariants et la plupart des autres résidus sont des substitutions conservatrices
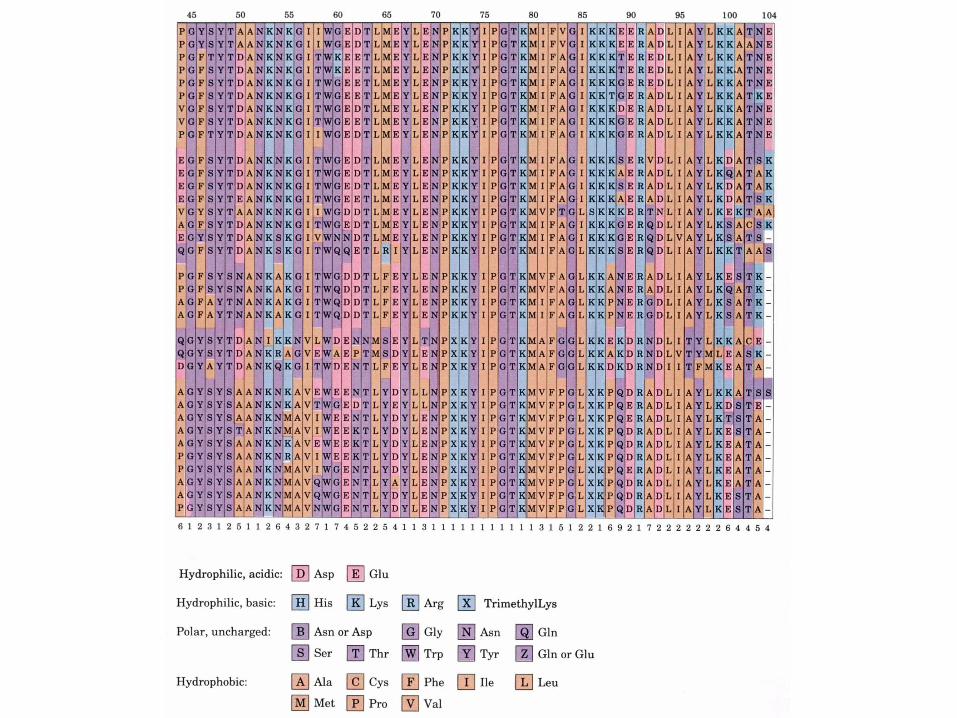

HommeSinge rhésus
LapinPoulet
PingouinSerpent à sonnettes
ThonBlé
Saccharomyces cerevisiaeCandida crusei
01 09 8 013 12 8 014 12 8 2 014 15 18 19 20 021 21 17 17 18 26 043 43 44 46 46 46 49 045 45 45 46 45 47 47 47 051 51 50 51 50 51 48 50 27 0
Hom
me
Sin
ge r
hésu
s
Lapi
n Poule
t
Thon
Blé
Sacc
haro
myce
sce
revis
iae
Pin
gouin
Serp
ent
àso
nnett
es
Candid
a c
ruse
i
Le moyen le plus facile de comparer les différences évolutives entre protéines homologues consiste à compter les différences en acides aminés entre ces protéines. L'ordre de ces différences est en accord avec la taxonomie classique
Matrice des différences en acides aminés pour 10 séquences de cytochrome c d'espèces différentes

L'analyse par ordinateur des données de la matrice des différences permet de construire un arbre phylogénétique qui indique les relations ancestrales entre les organismes qui produisent ces protéines
Chaque point de branchement de l'arbre indique l'existence probable d'un ancêtre commun à tous les organismes qui se trouvent au-dessus
Les distances évolutives relatives qui séparent deux points de branchement voisins sont exprimées en nombre de différences en acides aminés pour 100 résidus de la protéine (''Percentage of Accepted pont Mutations'' ou unités PAM). Ceci permet de mesurer quantitativement le degré de relation entre les espèces, ce que la taxonomie classique ne peut pas faire


Fibrinopeptides: peptides libérés lors de la conversion du fibrinogène en fibrine (pas de fonction propre)
Hémoglobine: protéine transporteuse d’O2, libre dans le globule rouge
Cytochrome c: transporteur d’électrons, qui doit interagir avec complexesIII et IV de la chaîne respiratoire
Histone H4: protéine servant à “l’emballage” de l’ADN,interagissant avec d’autres histones (octamère) et avec ADN
c. Les protéines évoluent à des vitesses qui leur sont propresOn peut porter en graphique les différences moyennes en unités PAM des séquences en acides aminés des deux côtés d'un point de branchement en fonction du temps, selon des données paléontologiques, depuis que les espèces correspondantes ont divergé de leur ancêtre commun
En comparant quatre protéines non apparentées:
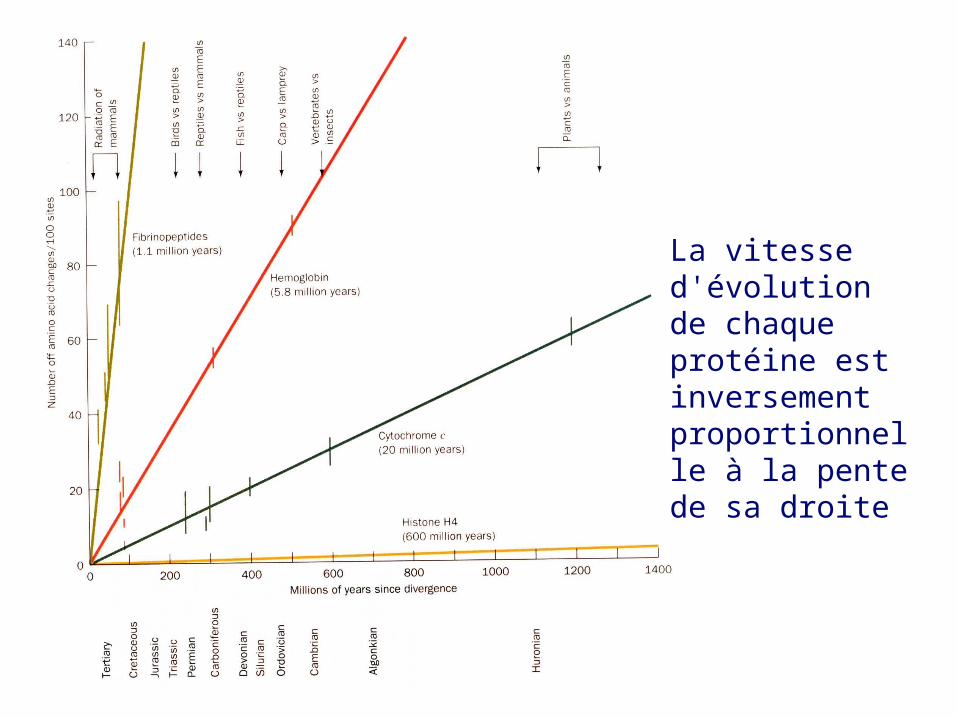
La vitesse d'évolution de chaque protéine est inversement proportionnelle à la pente de sa droite

Cytochrome c: transporteur d’électrons qui doit interagir avec des complexes de grande taille sur une grande partie de sa surface; tout changement par mutation affectera très vraisemblablement ces interactions
Histone H4: protéine servant à “l’emballage” de l’ADN,interagissant avec d’autres histones (octamère) et avec ADN. Son rôle essentiel dans le compactage de l'ADN dans la chromatine la rend tout à fait intolérante à tout changement mutationnel

C. Evolution par duplication de gènesLa plupart des protéines ont des similitudes de séquences très importantes avec d'autres protéines d'un même organisme. De telles protéines se sont formées par duplication de gène. La duplication de gène est un moyen d'évolution particulièrement efficace - un des gènes dupliqués peut évoluer vers une nouvelle fonction par sélection naturelle tandis que son homologue continue sa fonction ancestrale indispensable
Exemple - les protéines de la famille globine:• Les séquences des sous unités et de l'hémoglobine
tétramérique, 22, et de la chaîne de la myoglobine monomérique sont très semblables
• La globine ancestrale fonctionnait sans doute simplement comme une protéine de stockage d'oxygène
• La duplication du gène a permis l'évolution vers une hémoglobine monomérique avec une affinité faible pour l'oxygène pour pouvoir transférer l'oxygène à la myoglobine
• La duplication de la chaîne a donné naissance à la chaîne et la structure tétramérique qui a fortement améliorée sa capacité à transporter l'oxygène

QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

Exemple - les protéines de la famille globine:5. L'hémoglobine foetale est un tetramère a2, par duplication du gène en, avec une plus grande affinité pour l'oxygène que l'hémoglobine a2 maternelle
On trouve encore une hémoglobine monomérique chez la lamproie qui a conservé sa morphologie proche de l'anguille depuis > 400 millions d'années:

Les protéines homologues appartenant à un même organisme, et les gènes qui les codent, sont dits ''paralogues''. Les chaînes globines , , et la myoglobine humaine sont donc des paralogues
Les protéines et gènes homologues d'organismes différentes et issus de la divergence des espèces (les différents cytochrome c, par exemple) sont dits ''orthologues''

3 INTRODUCTION A LA BIOINFORMATIQUE
La profusion de séquences protéiques à partir de projets de séquençage génomiques et la disponibilité de données structurales au cours des dernières années a donné naissance à la bioinformatique - analyse de séquences et de structures tridimensionnelles par ordinateur
A. Bases de données sur les séquences
1. On recherche les séquences d'intérêt dans une banque de données via le Web, par exemple SWISS-PROT (http://expasy.org/sprot/)
2. On peut rechercher des séquences homologues en utilisant l'algorithme ''BLAST'' (Basic Local Alignment Search Tool) - http://www.expasy.org/tools/blast/

B. Alignements de séquences et construction d'arbres phylogénétiques
On fait un alignement de séquences multiples en utilisant le programme CLUSTALW (http://align.genome.jp/).
Les séquences sont envoyées en format ''FASTA''. Cet algorithme permet la construction d'un arbre phylogénétique à partir d'une matrice de
différences en acides aminés

>AAPK2_MOUSEMAEKQKHDGRVKIGHYVLGDTLGVGTFGKVKIGEHQLTGHKVAVKILNRQKIRSLDVVGKIKREIQNLKLFRHPHIIKLYQVISTPTDFFMVMEYVSGGELFDYICKHGRVEEVEARRLFQQILSAVDYCHRHMVVHRDLKPENVLLDAQMNAKIADFGLSNMMSDGEFLRTSCGSPNYAAPEVISGRLYAGPEVDIWSCGVILYALLCGTLPFDDEHVPTLFKKIRGGVFYIPDYLNRSVATLLMHMLQVDPLKRATIKDIREHEWFKQDLPSYLFPEDPSYDANVIVDEAVKEVCEKFECTESEVMNSLYSGDPQDQLAVAYHLIIDNRRIMNQASEFYLASSPPSGSFMDDSAMHIPPGLKPHPERMPPLIADSPKARCPLDALNTTKPKSLAVKKAKWHLGIRSQSKACDIMAEVYRAMKQLGFEWKVVNAYHLRVRRKNPVTGNYVKMSLQLYLVDSRSYLLDFKSIDDEVVEQRSGSSTPQRSCSAAGLHRARSSFDSSTAENHSLSGSLTGSLTGSTLSSASPRLGSHTMDFFEMCASLITALAR
>AAPK1_DANIOMATDKQKHEGRVKIGHYILGDTLGVGTFGKVKVGQHELTKHQVAVKILNRQKIRSLDVVGKIRREIQNLKLFRHPHIIKLYQVISTPTDIFMVMEYVSGGELFDYICKNGKLDEKESRRLFQQIISGVDYCHRHMVVHRDLKPENVLLDAHMNAKIADFGLSNMMSDGEFLRTSCGSPNYAAPEVISGRLYAGPEVDIWSSGVILYALLCGTLPFDDDHVPTLFKKICDGIFFTPQYLNPSVISLLKHMLQVDPMKRATIKEIREDEWFKQDLPKYLFPEDAAYSSNMIDEEALKEVCEKCECTEEEVLNCLYSRNHQDPLAVAYHLIIDNRRIMSEAKDFYLASSPPDSFLDDLPAHHSAKVHPERVPFLVAESQPRPRHTLDELNPQKSKHLGVRRAKWHLGIRSQSRPNDIMSEVCRAMKQLDYEWKVVNPYYLRVRRKNPVTGMHTKMSLQLYQVDSRTYLLDFRSIDDDMMEVKSGTATPHRSGSVGNYRTTLKNDKSEKNECEDAAKGEASAPSTPPISASKVAEGSLASSLTSSVDSTGGEILPRPGSHTIEFFEMCANLIKLLAR
>AAPK2_CAENORHMPPSGRFDRTIALAGTGHLKIGNFVIKETIGKGAFGAVKRGTHIQTGYDVAIKILNRGRMKGLGTVNKTRNEIDNLQKLTHPHITRLFRVISTPSDIFLVMELVSGGELFSYITRKGALPIRESRRYFQQIISGVSYCHNHMIVHRDLKPENLLLDANKNIKIADFGLSNYMTDGDLLSTACGSPNYAAPELISNKLYVGPEVDPWSCGVILYAMLCGTLPFDDQNVPTLFAKIKSGRYTVPYSMEKQAADLISTMLQVDPVKRADVKRIVNHSWFHIDLPYYLFPECENESSIVDIDVVQSVAEKFDVKEEDVTGALLAEDHHHFLCIAYRLEVNHKRNADESSQKAMEDFWEIGKTMKMGSTSLPVGATTKTNVGRKILEGLKKEQKKLTWNLGIRACLDPVETMKHVFLSLKSVDMEWKVLSMYHIIVRSKPTPINPDPVKVSLQLFALDKKENNKGYLLDFKGLTEDEEAVPPSRCRSRAASVSVTLAKSKSDLNGNSSKVPMSPLSPMSPISPSVNIPKVRVDDADASLKSSLNSSIYMADIENSMESLDEVSTQSSEPEAPIRSQTMEFFATCHIIMQALLAE
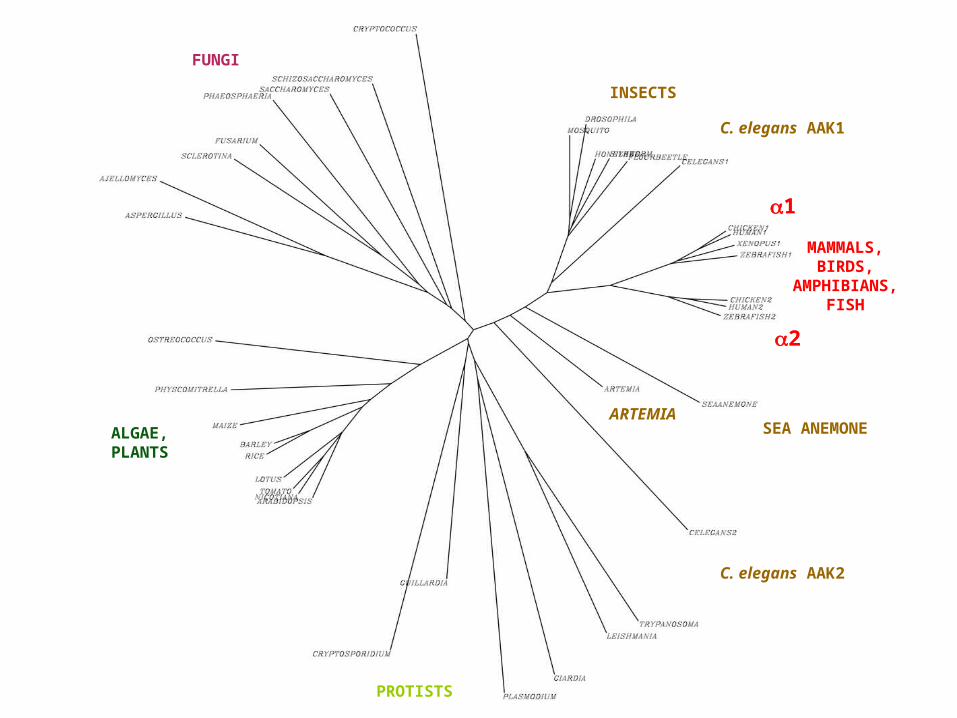
FUNGI
ALGAE,PLANTS
PROTISTS
INSECTS
SEA ANEMONE
C. elegans AAK1
MAMMALS, BIRDS,
AMPHIBIANS, FISH
1
2
C. elegans AAK2
ARTEMIA

4. Synthèse chimique de polypeptidesA. Stratégie générale de synthèse
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
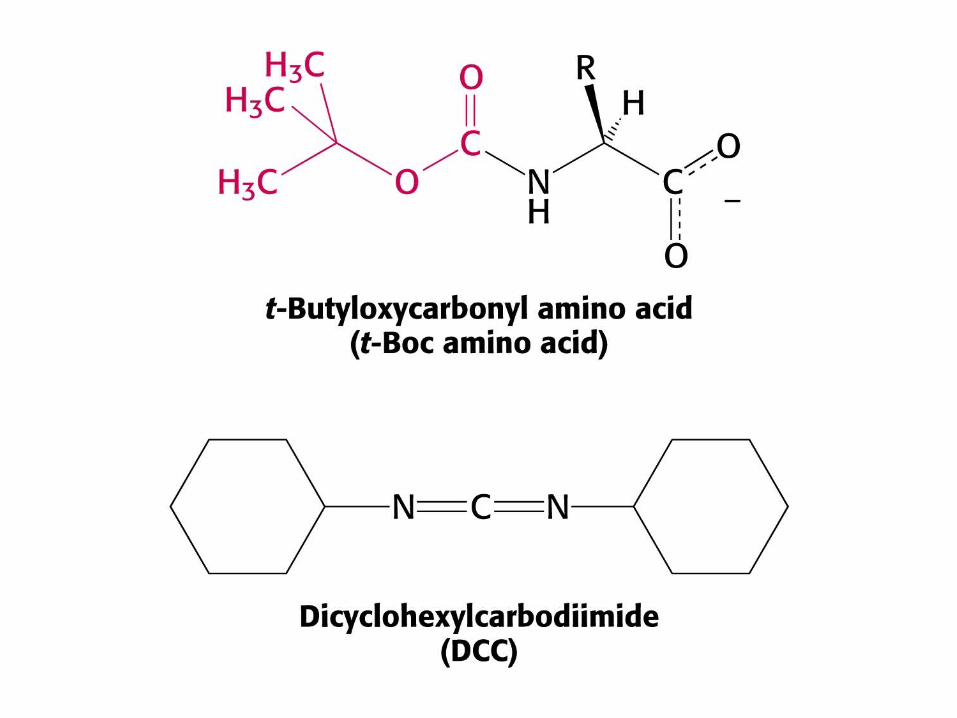
Suite des réactions pour la synthèse d'un polypeptide en phase solide - le symbole Mn
correspond au n ième résidu d'acide aminé qui doit être ajouté. Les polypeptides sont synthétisés par addition d'acides aminés à l'extrémité N-terminale. Sn est le groupement protecteur de sa chaîne latérale. Y symbolise le protecteur du groupe amine. Le couplage utilise une carbodiimide (comme le dicyclohexylcarbodiimide ou DCCD)

B. Couplage des acides aminés