cationic lanthanide complexes of neutral tripodal n,o ligands: enthalpy versus entropy-driven podate...
TRANSCRIPT

Cationic lanthanide complexes of neutral tripodal N,O ligands:enthalpy versus entropy-driven podate formation in water†
Florence Bravard, Caroline Rosset and Pascale Delangle*Laboratoire de Reconnaissance Ionique, Service de Chimie Inorganique et Biologique,Département de Recherche Fondamentale sur la Matière Condensée, CEA-Grenoble,38054 Grenoble, Cedex 09, France. E-mail: [email protected]; Fax: 33 4 38 78 50 90;Tel: 33 4 38 78 98 22
Received 9th March 2004, Accepted 5th April 2004First published as an Advance Article on the web 11th May 2004
The cationic lanthanide complexes of two neutral tripodal N,O ligands, tpa (tris[(2-pyridyl)methyl]amine) and tpaam(tris[6-((2-N,N-diethylcarbamoyl)pyridyl)methyl]amine) are studied in water. The analysis of the proton lanthanideinduced NMR shifts indicate that there is no abrupt structural change in the middle of the rare-earth series.Unexpectedly, the formation constant values of the lanthanide podates of tpaam and tpa in D2O at 298 K are similar,suggesting that the addition of the three amide groups to the ligand tpa does not lead to any increase in stability ofthe lanthanide complexes of tpaam in respect to tpa, even though the amide groups are coordinated to the metal inaqueous solution. The measurement of the enthalpy and entropy changes of the complexation reactions showsthat the two similar ligands tpa and tpaam have different driving forces for lanthanide complexation. Indeed, theformation of tpa podates benefits from an exothermic enthalpy change associated with a small entropy change,whereas the complexation reaction with tpaam is clearly entropy-driven though opposed by a positive enthalpychange. The hydration states of the europium complexes were measured by luminescence and show the coordinationof 4–5 water ligands in [Eu(tpa)]3 whereas there are only 2 in [Eu(tpaam)]3. Therefore the heptadentate ligandtpaam releases the translational entropy of more water molecules than does the tetradentate ligand tpa.
IntroductionIn recent years, there has been a resurgence of interest inthe coordination chemistry of lanthanide complexes.1–5 Thesestudies have been motivated by the numerous applications ofthese complexes in medicine, catalysis and material science. Inparticular, the development of lanthanide complexes stablewith respect to ligand dissociation in physiological conditionsis of interest for the design of lanthanide complexes active incatalyzing RNA cleavage in vivo.6,7 Cationic lanthanide com-plexes are especially effective catalysts in vivo because the Lewisacid character of the cation is greater than in neutral or anioniccomplexes.8 The design of neutral ligands that strongly bindlanthanide cations represents a real challenge and very few ofsuch complexes have been described, so far.6,8,9
The use of amide coordinating groups seems promisingwith the aim of designing effective neutral receptors forlanthanide() complexation. Several tetramide ligands derivedfrom 1,4,7,10-tetraazacyclododecane have been shown to givekinetically stable lanthanide complexes with respect to liganddissociation in water.8,10–13 Furthermore, the magnitude of thecontribution of the amide functional group to the stabilityof Gd() complexes in dtpa bisamide derivatives has beenestimated to be 3.38 log units per amide group.14
We have recently studied the complexation of lanthanide()ions by the neutral tripodal heptadentate ligand tpaam (tris[6-((2-N,N-diethylcarbamoyl)pyridyl)methyl]amine), in order toinvestigate if the addition of coordinating amide groups to thetetradentate ligand tpa (tris[(2-pyridyl)methyl]amine) wouldyield stable tripositive Ln() complexes in water.15 In the solid
† Electronic supplementary information (ESI) available: Plots of δ parai,Ln/
⟨SZ⟩Ln vs. DLn/⟨SZ⟩Ln for proton H3 in [Ln(tpaam)]3 podates in D2O at298 K (Fig. S1). Plots of δ para
i,Ln/⟨SZ⟩Ln vs. δ parak,Ln/⟨SZ⟩Ln for the H3–H5 and
H7–H5 pairs in [Ln(tpaam)]3 podates in D2O at 298 K (Fig. S2). Van’tHoff plots for europium complexation by tpa and tpaam in water (ionicstrength 1 mol L1 KCl) (Fig. S3). See http://www.rsc.org/suppdata/dt/b4/b403647f/
state, the tpaam ligand coordinates lanthanide() ions stronglywith the three amide oxygens, with Ln–O bond distances similarto those found for the corresponding triscarboxylate ligandtpaa 16 (H3tpaa = α,α,α-nitrilotri(6-methyl-2-pyridine-carboxylic acid)). Furthermore, the X-ray structures and theNMR properties of the complexes in methanol point to a weakerLn–Napical interaction in the tpaam compound than in the tpacomplexes. In water, tpaam podates partially dissociate as dothose of tpa. Unexpectedly, the formation constant values ofthe europium complexes of tpaam and tpa in D2O at 298 K aresimilar suggesting that the addition of the three amide groupsto the ligand tpa does not lead to any increase in stability of thelanthanide complexes of tpaam compared to those of tpa.
We decided to investigate further the thermodynamics ofthe complexation reaction in order to achieve a better under-standing of the parameters resulting in a similar stability of tpaand tpaam complexes toward ligand dissociation in water. Here,we report detailed studies of trivalent lanthanide complexes ofthe two neutral tripods tpa and tpaam in water. The enthalpyand entropy changes of the complexation reaction reveal thatthe driving forces for lanthanide complexation are very differ-ent for the two ligands. Indeed, the complexation reaction withtpa benefits from an exothermic enthalpy change and shows asmall entropy change, whereas the same reaction with tpaamis clearly entropy-driven though opposed by an endothermicenthalpy.
Scheme 1DO
I:1
0.1
03
9/ b
40
36
47
f
2012 D a l t o n T r a n s . , 2 0 0 4 , 2 0 1 2 – 2 0 1 8 T h i s j o u r n a l i s © T h e R o y a l S o c i e t y o f C h e m i s t r y 2 0 0 4
Publ
ishe
d on
11
May
200
4. D
ownl
oade
d by
Nip
issi
ng U
nive
rsity
on
18/1
0/20
14 0
1:40
:02.
View Article Online / Journal Homepage / Table of Contents for this issue
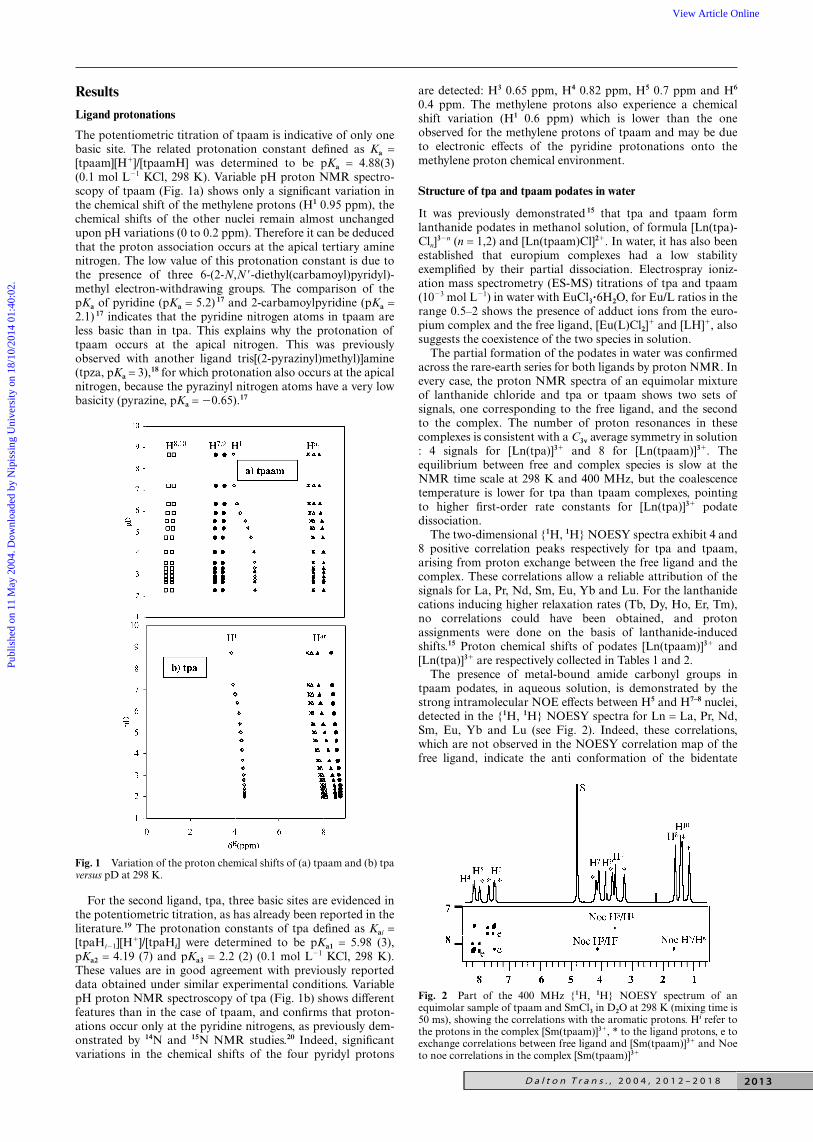
Results
Ligand protonations
The potentiometric titration of tpaam is indicative of only onebasic site. The related protonation constant defined as Ka =[tpaam][H]/[tpaamH] was determined to be pKa = 4.88(3)(0.1 mol L1 KCl, 298 K). Variable pH proton NMR spectro-scopy of tpaam (Fig. 1a) shows only a significant variation inthe chemical shift of the methylene protons (H1 0.95 ppm), thechemical shifts of the other nuclei remain almost unchangedupon pH variations (0 to 0.2 ppm). Therefore it can be deducedthat the proton association occurs at the apical tertiary aminenitrogen. The low value of this protonation constant is due tothe presence of three 6-(2-N,N-diethyl(carbamoyl)pyridyl)-methyl electron-withdrawing groups. The comparison of thepKa of pyridine (pKa = 5.2) 17 and 2-carbamoylpyridine (pKa =2.1) 17 indicates that the pyridine nitrogen atoms in tpaam areless basic than in tpa. This explains why the protonation oftpaam occurs at the apical nitrogen. This was previouslyobserved with another ligand tris[(2-pyrazinyl)methyl)]amine(tpza, pKa = 3),18 for which protonation also occurs at the apicalnitrogen, because the pyrazinyl nitrogen atoms have a very lowbasicity (pyrazine, pKa = 0.65).17
For the second ligand, tpa, three basic sites are evidenced inthe potentiometric titration, as has already been reported in theliterature.19 The protonation constants of tpa defined as Kai =[tpaHi1][H
]/[tpaHi] were determined to be pKa1 = 5.98 (3),pKa2 = 4.19 (7) and pKa3 = 2.2 (2) (0.1 mol L1 KCl, 298 K).These values are in good agreement with previously reporteddata obtained under similar experimental conditions. VariablepH proton NMR spectroscopy of tpa (Fig. 1b) shows differentfeatures than in the case of tpaam, and confirms that proton-ations occur only at the pyridine nitrogens, as previously dem-onstrated by 14N and 15N NMR studies.20 Indeed, significantvariations in the chemical shifts of the four pyridyl protons
Fig. 1 Variation of the proton chemical shifts of (a) tpaam and (b) tpaversus pD at 298 K.
are detected: H3 0.65 ppm, H4 0.82 ppm, H5 0.7 ppm and H6
0.4 ppm. The methylene protons also experience a chemicalshift variation (H1 0.6 ppm) which is lower than the oneobserved for the methylene protons of tpaam and may be dueto electronic effects of the pyridine protonations onto themethylene proton chemical environment.
Structure of tpa and tpaam podates in water
It was previously demonstrated 15 that tpa and tpaam formlanthanide podates in methanol solution, of formula [Ln(tpa)-Cln]
3n (n = 1,2) and [Ln(tpaam)Cl]2. In water, it has also beenestablished that europium complexes had a low stabilityexemplified by their partial dissociation. Electrospray ioniz-ation mass spectrometry (ES-MS) titrations of tpa and tpaam(103 mol L1) in water with EuCl36H2O, for Eu/L ratios in therange 0.5–2 shows the presence of adduct ions from the euro-pium complex and the free ligand, [Eu(L)Cl2]
and [LH], alsosuggests the coexistence of the two species in solution.
The partial formation of the podates in water was confirmedacross the rare-earth series for both ligands by proton NMR. Inevery case, the proton NMR spectra of an equimolar mixtureof lanthanide chloride and tpa or tpaam shows two sets ofsignals, one corresponding to the free ligand, and the secondto the complex. The number of proton resonances in thesecomplexes is consistent with a C3v average symmetry in solution: 4 signals for [Ln(tpa)]3 and 8 for [Ln(tpaam)]3. Theequilibrium between free and complex species is slow at theNMR time scale at 298 K and 400 MHz, but the coalescencetemperature is lower for tpa than tpaam complexes, pointingto higher first-order rate constants for [Ln(tpa)]3 podatedissociation.
The two-dimensional 1H, 1H NOESY spectra exhibit 4 and8 positive correlation peaks respectively for tpa and tpaam,arising from proton exchange between the free ligand and thecomplex. These correlations allow a reliable attribution of thesignals for La, Pr, Nd, Sm, Eu, Yb and Lu. For the lanthanidecations inducing higher relaxation rates (Tb, Dy, Ho, Er, Tm),no correlations could have been obtained, and protonassignments were done on the basis of lanthanide-inducedshifts.15 Proton chemical shifts of podates [Ln(tpaam)]3 and[Ln(tpa)]3 are respectively collected in Tables 1 and 2.
The presence of metal-bound amide carbonyl groups intpaam podates, in aqueous solution, is demonstrated by thestrong intramolecular NOE effects between H5 and H7–8 nuclei,detected in the 1H, 1H NOESY spectra for Ln = La, Pr, Nd,Sm, Eu, Yb and Lu (see Fig. 2). Indeed, these correlations,which are not observed in the NOESY correlation map of thefree ligand, indicate the anti conformation of the bidentate
Fig. 2 Part of the 400 MHz 1H, 1H NOESY spectrum of anequimolar sample of tpaam and SmCl3 in D2O at 298 K (mixing time is50 ms), showing the correlations with the aromatic protons. Hi refer tothe protons in the complex [Sm(tpaam)]3, * to the ligand protons, e toexchange correlations between free ligand and [Sm(tpaam)]3 and Noeto noe correlations in the complex [Sm(tpaam)]3
2013D a l t o n T r a n s . , 2 0 0 4 , 2 0 1 2 – 2 0 1 8
Publ
ishe
d on
11
May
200
4. D
ownl
oade
d by
Nip
issi
ng U
nive
rsity
on
18/1
0/20
14 0
1:40
:02.
View Article Online
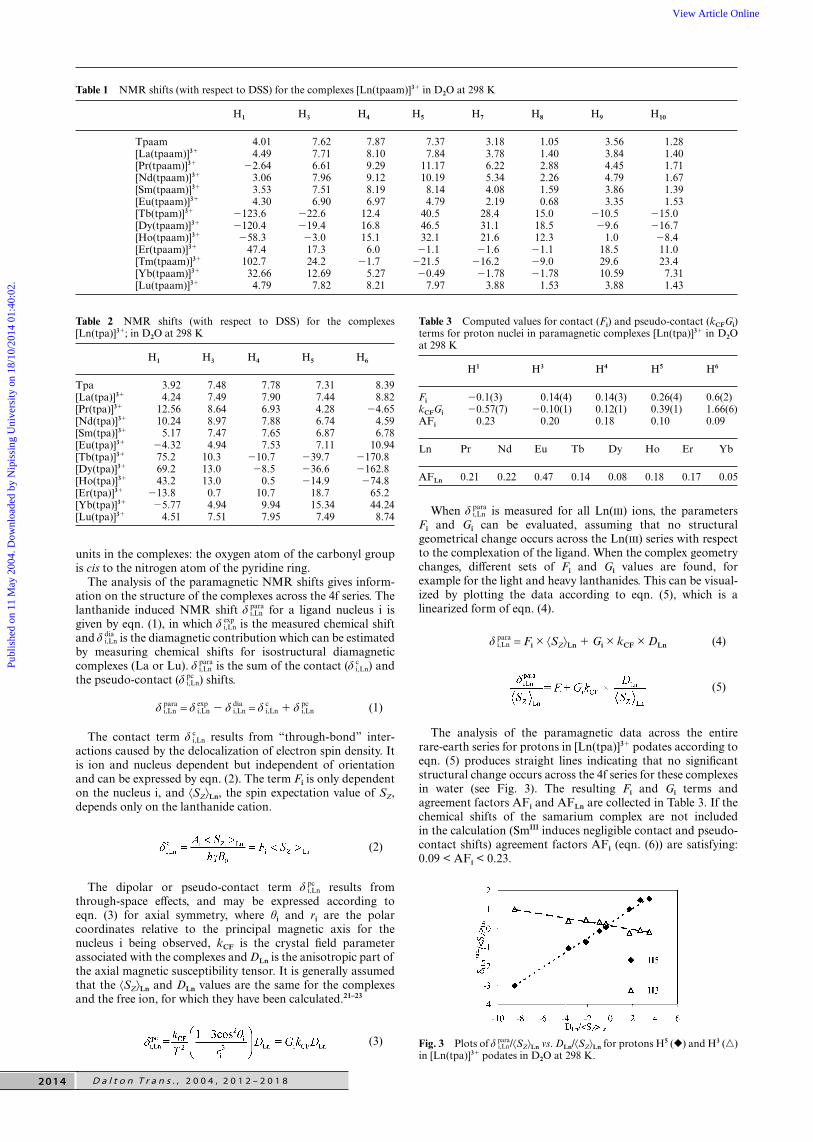
Table 1 NMR shifts (with respect to DSS) for the complexes [Ln(tpaam)]3 in D2O at 298 K
H1 H3 H4 H5 H7 H8 H9 H10
Tpaam 4.01 7.62 7.87 7.37 3.18 1.05 3.56 1.28[La(tpaam)]3 4.49 7.71 8.10 7.84 3.78 1.40 3.84 1.40[Pr(tpaam)]3 2.64 6.61 9.29 11.17 6.22 2.88 4.45 1.71[Nd(tpaam)]3 3.06 7.96 9.12 10.19 5.34 2.26 4.79 1.67[Sm(tpaam)]3 3.53 7.51 8.19 8.14 4.08 1.59 3.86 1.39[Eu(tpaam)]3 4.30 6.90 6.97 4.79 2.19 0.68 3.35 1.53[Tb(tpam)]3 123.6 22.6 12.4 40.5 28.4 15.0 10.5 15.0[Dy(tpaam)]3 120.4 19.4 16.8 46.5 31.1 18.5 9.6 16.7[Ho(tpaam)]3 58.3 3.0 15.1 32.1 21.6 12.3 1.0 8.4[Er(tpaam)]3 47.4 17.3 6.0 1.1 1.6 1.1 18.5 11.0[Tm(tpaam)]3 102.7 24.2 1.7 21.5 16.2 9.0 29.6 23.4[Yb(tpaam)]3 32.66 12.69 5.27 0.49 1.78 1.78 10.59 7.31[Lu(tpaam)]3 4.79 7.82 8.21 7.97 3.88 1.53 3.88 1.43
units in the complexes: the oxygen atom of the carbonyl groupis cis to the nitrogen atom of the pyridine ring.
The analysis of the paramagnetic NMR shifts gives inform-ation on the structure of the complexes across the 4f series. Thelanthanide induced NMR shift δ para
i,Ln for a ligand nucleus i isgiven by eqn. (1), in which δ exp
i,Ln is the measured chemical shiftand δ dia
i,Ln is the diamagnetic contribution which can be estimatedby measuring chemical shifts for isostructural diamagneticcomplexes (La or Lu). δ para
i,Ln is the sum of the contact (δ ci,Ln) and
the pseudo-contact (δ pci,Ln) shifts.
The contact term δ ci,Ln results from “through-bond” inter-
actions caused by the delocalization of electron spin density. Itis ion and nucleus dependent but independent of orientationand can be expressed by eqn. (2). The term Fi is only dependenton the nucleus i, and ⟨SZ⟩Ln, the spin expectation value of SZ,depends only on the lanthanide cation.
The dipolar or pseudo-contact term δ pci,Ln results from
through-space effects, and may be expressed according toeqn. (3) for axial symmetry, where θi and ri are the polarcoordinates relative to the principal magnetic axis for thenucleus i being observed, kCF is the crystal field parameterassociated with the complexes and DLn is the anisotropic part ofthe axial magnetic susceptibility tensor. It is generally assumedthat the ⟨SZ⟩Ln and DLn values are the same for the complexesand the free ion, for which they have been calculated.21–23
δ parai,Ln = δ exp
i,Ln δ diai,Ln = δ c
i,Ln δ pci,Ln (1)
(2)
(3)
Table 2 NMR shifts (with respect to DSS) for the complexes[Ln(tpa)]3; in D2O at 298 K
H1 H3 H4 H5 H6
Tpa 3.92 7.48 7.78 7.31 8.39[La(tpa)]3 4.24 7.49 7.90 7.44 8.82[Pr(tpa)]3 12.56 8.64 6.93 4.28 4.65[Nd(tpa)]3 10.24 8.97 7.88 6.74 4.59[Sm(tpa)]3 5.17 7.47 7.65 6.87 6.78[Eu(tpa)]3 4.32 4.94 7.53 7.11 10.94[Tb(tpa)]3 75.2 10.3 10.7 39.7 170.8[Dy(tpa)]3 69.2 13.0 8.5 36.6 162.8[Ho(tpa)]3 43.2 13.0 0.5 14.9 74.8[Er(tpa)]3 13.8 0.7 10.7 18.7 65.2[Yb(tpa)]3 5.77 4.94 9.94 15.34 44.24[Lu(tpa)]3 4.51 7.51 7.95 7.49 8.74 When δ para
i,Ln is measured for all Ln() ions, the parametersFi and Gi can be evaluated, assuming that no structuralgeometrical change occurs across the Ln() series with respectto the complexation of the ligand. When the complex geometrychanges, different sets of Fi and Gi values are found, forexample for the light and heavy lanthanides. This can be visual-ized by plotting the data according to eqn. (5), which is alinearized form of eqn. (4).
The analysis of the paramagnetic data across the entirerare-earth series for protons in [Ln(tpa)]3 podates according toeqn. (5) produces straight lines indicating that no significantstructural change occurs across the 4f series for these complexesin water (see Fig. 3). The resulting Fi and Gi terms andagreement factors AFi and AFLn are collected in Table 3. If thechemical shifts of the samarium complex are not includedin the calculation (SmIII induces negligible contact and pseudo-contact shifts) agreement factors AFi (eqn. (6)) are satisfying:0.09 < AFi < 0.23.
δ parai,Ln = Fi × ⟨SZ⟩Ln Gi × kCF × DLn (4)
(5)
Fig. 3 Plots of δ parai,Ln/⟨SZ⟩Ln vs. DLn/⟨SZ⟩Ln for protons H5 () and H3 ()
in [Ln(tpa)]3 podates in D2O at 298 K.
Table 3 Computed values for contact (Fi) and pseudo-contact (kCFGi)terms for proton nuclei in paramagnetic complexes [Ln(tpa)]3 in D2Oat 298 K
H1 H3 H4 H5 H6
Fi 0.1(3) 0.14(4) 0.14(3) 0.26(4) 0.6(2)kCFGi 0.57(7) 0.10(1) 0.12(1) 0.39(1) 1.66(6)AFi 0.23 0.20 0.18 0.10 0.09
Ln Pr Nd Eu Tb Dy Ho Er Yb
AFLn 0.21 0.22 0.47 0.14 0.08 0.18 0.17 0.05
2014 D a l t o n T r a n s . , 2 0 0 4 , 2 0 1 2 – 2 0 1 8
Publ
ishe
d on
11
May
200
4. D
ownl
oade
d by
Nip
issi
ng U
nive
rsity
on
18/1
0/20
14 0
1:40
:02.
View Article Online

Table 4 Computed values for contact (Fi) and pseudo-contact (kCFGi) terms for proton nuclei in paramagnetic complexes [Ln(tpaam)]3 in D2O at298 K
H1 H3 H4 H5 H7 H8 H9 H10
Pr–Eu Fi 0.33(5) 0.16(4) 0.11(5) 0.24(6) 0.12(6) 0.05(3) 0.1(1) 0.0(3) kCFGi 0.74(2) 0.14(2) 0.08(2) 0.24(3) 0.19(3) 0.12(1) 0.03(4) 0.03(2)Tb–Yb Fi 0.2(7) 0.1(1) 0.08(9) 0.01(1) 0.1(1) 0.1(1) 0.5(2) 0.2(2) kCFGi 1.5(1) 0.27(2) 0.13(2) 0.43(5) 0.30(3) 0.17(2) 0.31(4) 0.29(3) AFi 0.14 0.15 0.24 0.17 0.18 0.18 0.21 0.23
Ln Pr Nd Eu Tb Dy Ho Er Tm Yb
AFLn 0.004 0.03 0.10 0.10 0.13 0.20 0.22 0.20 0.13
It has been shown that variations of the second-order crystalfield parameter kCF can mask structural changes occurring inthe middle of the lanthanide series.24 eqn. (7), proposed byGeraldes et al.,25 does not depend on crystal field parametersand any deviation from linearity in δi,Ln/⟨SZ⟩Ln versus δk,Ln/⟨SZ⟩Ln
plots along the lanthanide series can safely be ascribed tostructural changes.
The analysis of the paramagnetic chemical shifts, for[Ln(tpa)]3 podates, with the two nuclei crystal-field independ-ent method according to eqn. (7) gives straight lines (see Fig. 4)confirming that no structural changes occur in aqueoussolution for these complexes.
The paramagnetic data for [Ln(tpaam)]3 podates in waterare better fitted with two distinct series (Pr–Eu and Tb–Yb),according to eqn. (5) (see Fig. S1 ESI † and Table 4). Never-theless the change in Gi.kCF between Eu and Tb cannot be safelyassigned to structural changes affecting Gi since the variation ofthe crystal field parameter kCF is unknown. The use of thecrystal-field independent method according to eqn. (7), givesstraight lines for all couples of protons (see Fig. S2 ESI †) exceptthose containing H9 and H10. These two latter groups ofprotons are indeed situated in a flexible ethyl chain which issyn to the lanthanide cation, and their arrangement may thus bevery sensitive to the size of the cation. These data indicate thatno abrupt structural change exist across the lanthanide seriesfor [Ln(tpaam)]3 podates, even though the diethylcarbamoylgroup may be rather flexible.
Equilibrium constants
As the complexes are partially dissociated for concentrations of1–10 mmol L1, and the exchange is slow at the NMR time
(6)
(7)
Fig. 4 Plots of δ parai,Ln/⟨SZ⟩Ln vs. δ para
k,ln /⟨SZ⟩Ln for H3–H5 and H6–H5 pairs in[Ln(tpa)]3 podates in D2O at 298 K.
scale, 1H NMR is an appropriate method for the determinationof the equilibrium constants. The apparent equilibriumconstants (298 K, KCl 1 mol L1, pD = 6.5–7) were measuredin D2O by integration of the 1H NMR signals correspondingto the complex and the free ligand. The constants β110 definedin eqn. (8) and the apparent constant β app
110 are related byeqn. (9), where K L
a is the first protonation constant of the ligand(K L
a = [L][H]/[LH])) and K Lna is the hydrolysis constant of
the lanthanide cation (K Lna = [Ln(OH)][H]/[Ln]).26 The
calculated logβ110 values are collected in Tables 5 and 6, anddo not deviate significantly from log β app
110 in our experimentalconditions.
From these measurements, it can be deduced that for bothligands, the equilibrium constants do not follow an electrostatictrend, i.e. an increase with atomic number of the lanthanide,indeed tpa shows a slight maximum in affinity for NdIII andtpaam for TbIII. Furthermore, at 298 K tpaam podates haveglobally the same stability as tpa podates suggesting that theaddition of amide groups to the ligand tpa does not lead to anyincrease in stability of the lanthanide complexes of tpaam inrespect to tpa even though the amide groups are coordinated tothe metal in aqueous solution.
Enthalpy and entropy changes
To better understand the similarity in the values of the equi-librium constants of tpa and tpaam lanthanide podates, theenthalpy and entropy changes of the complexation reactionwere measured. The equilibrium constants were calculated fromthe 1H NMR spectra in D2O at a constant ionic strength (1 molL1 KCl) in a small temperature range (typically 5 to 25 C for[Ln(tpa)]3 and 5 to 30 C for [Ln(tpaam)]3 complexes) and thevalues of the enthalpy and entropy changes of reaction (8) werecalculated from the van’t Hoff plots (eqn. (10)). Indeed,thermodynamic data determined by NMR and the van’t Hoffequation have been demonstrated to be in good agreement withcalorimetric results for weak lanthanide() complexes inwater.27–29 This analysis was not possible for Tb, Dy and Ho,because of the very small values of the proton transverserelaxation times, giving rise to important errors in theequilibrium constant determination.
The van’t Hoff plots obtained for Eu() complexes (Fig. S3ESI †) show a first striking feature: the signs of the slopes aredifferent for the two ligands. The formation of the europiumcomplex of tpa is exothermic (∆ rH < 0), whereas that of tpaam
(8)
(9)
(10)
2015D a l t o n T r a n s . , 2 0 0 4 , 2 0 1 2 – 2 0 1 8
Publ
ishe
d on
11
May
200
4. D
ownl
oade
d by
Nip
issi
ng U
nive
rsity
on
18/1
0/20
14 0
1:40
:02.
View Article Online

is endothermic (∆ rH > 0). This indicates that these two verysimilar ligands have different driving forces for europium cationcomplexation. The thermodynamic parameters of reaction (8)are collected in Tables 5 and 6, respectively, for tpaam and tpa.The thermodynamic data indicate that lanthanide com-plexation by tpaam is driven by a positive entropy but opposedby an unfavorable (positive) enthalpy. In contrast, tpa podateformation is clearly driven by an exothermic enthalpy withsmaller entropic contributions, which are unfavorable (neg-ative) at the beginning of the series (La, Pr), and becomefavorable (positive) after Nd (see Fig. 5).
Hydration state
As hydration is an important factor in lanthanide complexformation in water, we have measured the number of watermolecules present in the first coordination sphere of europiumin the podates. The solvation states of tpa and tpaam complexesof Eu() were studied by comparison of their luminescencedecays in H2O and D2O. Due to the different quenchingefficiencies of the O–H and O–D oscillators, the measurementof Eu3 phosphorescence lifetimes (τ) allows an estimation ofthe number of coordinated water molecules present in solution,qEu, using the equation of Parker and co-workers 30 (eqn. (11))which is a corrected version of the empirical equation ofHorrocks and Sudnick 31,32 accounting for closely diffusing OHoscillators. The observed lifetimes (Table 7) are in agreementwith the presence of 4.4 ± 0.5 coordinated water ligands in the[Eu(tpa)]3 complex and only 2.1 ± 0.2 in [Eu(tpaam)]3. These
Table 5 Thermodynamic properties of lanthanide complexation reac-tion with tpaam in water (KCl 1 mol L1). Logβ110 is given at 298 K
Ln logβ110 ∆H /kJ mol1 ∆S /J mol1 K1
La 1.99(4) 15.5(7) 90(3)Pr 1.99(4) 18(1) 99(2)Nd 1.92(4) 21(3) 107(3)Sm 2.29(4) 18(2) 104(7)Eu 2.34(4) 20(2) 111(5)Tb 2.37(4) Dy 2.32(4) Ho 2.21(4) Er 2.14(4) 27(2) 133(3)Tm 2.10(4) 29(2) 138(3)Yb 1.94(4) 28(2) 133(2)Lu 1.79(4) 29(1) 133(7)
Table 6 Thermodynamic properties of lanthanide complexation reac-tion with tpa in water (KCl 1 mol L1). Logβ110 is given at 298 K
Ln logβ110 ∆H /kJ mol1 ∆S /J mol1 K1
La 1.99(4) 17(1) 21(2)Pr 2.25(4) 13(1) 2(1)Nd 2.54(4) 13(2) 5(2)Sm 2.51(4) 12(2) 6(1)Eu 2.49(4) 13(2) 3(1)Tb 2.00(4) Dy 1.87(4) Ho 1.79(4) Er 1.86(4) 3(2) 23(2)Yb 2.03(4) 6(1) 18(2)Lu 2.07(4)
Table 7 Luminescence data
τH2O/ms τD2O/ms q a
[Eu(tpa)]3 0.22(2) 1.52(5) 4.4(5)[Eu(tpaam)]3 0.34(2) 1.05(5) 2.1(2)
a Water molecules of the coordination sphere calculated according toeqn. (11).
values are consistent with a total coordination number of 8–9for [Eu(tpa)]3 and 9 for [Eu(tpaam)]3. The number ofcoordinated water molecules in the tpaam podate is the sameas in its analogue bearing three acetate functions tpaa,16
confirming that the three amide groups are coordinated to thecation in water.
DiscussionThe complexation process of a hydrated ion can be decom-posed into two steps. The first one involves the desolvation ofboth the metal cation and the ligand and is highly enthalpyunfavorable since the hydration enthalpies are very large fortrivalent lanthanide cations.33 In the second step, the twodesolvated partners combine to form the complex with a favor-able enthalpy change. The overall changes reflect the sum ofthese opposed contributions and most often, the combinationenthalpy cannot compensate the dehydration enthalpy leadingto entropy stabilized lanthanide complexes. Entropy effects areassociated with freedom of motion in both the metal ion andthe ligand, and consist of translational entropy of the solutesinvolved as well as the internal forms of entropy such as free-dom of vibration and rotation.34,35 The equilibrium constantsof the lanthanide complexes of tpa and tpaam do not increasewith the hardness of the cation across the whole lanthanideseries, contrary to many studied systems.3,36–38 On the otherhand, the thermodynamic quantities of the complexationreaction, ∆ r H and ∆ r S , are globally increasing across therare-earth series, reflecting the evolution of the dehydrationcontributions.
The complexation process with tpa is dominated by theenthalpy-driven combination step; indeed the reaction is exo-thermic across the rare-earth series and has a small entropiccontribution. These small entropy values are in line with thefact that the release of the water molecules from the co-
Fig. 5 Thermodynamic parameters for (a) [Ln(tpaam)]3 and(b) [Ln(tpa)]3 podates formation in water KCl 1 mol L1 and 298 K.
qEu =1.2(1/τH2O 1/τD2O 0.25) with τH2O and τD2O in ms (11)
2016 D a l t o n T r a n s . , 2 0 0 4 , 2 0 1 2 – 2 0 1 8
Publ
ishe
d on
11
May
200
4. D
ownl
oade
d by
Nip
issi
ng U
nive
rsity
on
18/1
0/20
14 0
1:40
:02.
View Article Online

ordination sphere of the lanthanide cation nearly compensatethe decrease in internal entropy of the ligand and the negativeentropy change due to the loss of translational entropy of thereagents upon complexation. Indeed, the hydration state of[Eu(tpa)]3 indicates that only 3 or 4 water molecules are dis-placed from the cation coordination sphere in [Eu(H2O)8]
3.39
Moreover, in the case of lanthanide complexation with poly-aminocarboxylates, it has been shown that the entropy changesof the complexation reactions are correlated to the number ofcoordinated carboxylate functions,40 whereas neutral groupslike amide, induce only very small positive contributions to theentropy change.41,42 The large positive entropies obtained forlanthanide complexes with carboxylato-containing ligands areattributed to the charge neutralization occuring when thesenegatively charged ligands react with trivalent lanthanidecations 34 and also to the increase of translational entropyproduced upon desolvation of the charged receptor.43 Theneutral character of the ligand combined with the limitednumber of solvent molecules displaced during the complex-ation process may thus explain the small entropy changesmeasured for [Ln(tpa)]3 podates.
Contrary to tpa, the formation of lanthanide podates oftpaam is endothermic. Furthermore, the entropy changes aremuch more positive than for tpa (an increase of about 100 J mol1
K1 for a given cation). Indeed, the dehydration contributionsare expected to be greater for tpaam which is heptadentate asdemonstrated by the hydration state of the europium complexpointing to the displacement of 6 water ligands during the reac-tion, that produces a greater increase of translational entropyupon complexation. Another neutral nitrogen-donor ligandtpen, (N,N,N,N-tetrakis(2-pyridylmethyl)ethylenediamine) 44
which is hexadentate, gives intermediate thermodynamicparameters between tpa and tpaam for lanthanide complex-ation reaction. The entropy change is favorable (51 J mol1
K1 for La() cation), and the reaction is exothermic with anegative enthalpy change of 4.9 kJ mol1.
Finally, it has been demonstrated that the driving forces forlanthanide() complexation by the two neutral podands tpaand tpaam are very different. Whereas the complexationreaction with tpa benefits from an exothermic enthalpy change,the reaction with tpaam is clearly entropy-driven thoughopposed by an endothermic enthalpy. The removal of morewater molecules from the lanthanide coordination sphere andfrom the ligand produces a favorable increase in translationalentropy, but also a large positive enthalpic term, which is notcounterbalanced by the gain associated with the three additivecoordinating amide groups.
Indeed, the combination terms are difficult to comparebecause although tpaam has three supplementary bindingcarbonyl groups in comparison to tpa, the nitrogen–metalinteractions may have a lower contribution: (i) the electron-withdrawing effect of the amide groups decreases the σ-donorcharacter of the pyridine nitrogen atoms, as evidenced bythe dissociation constant values of pyridinium (pKa = 5.2) and2-carbamoyl-pyridinium (pKa = 2.1), (ii) the steric interactionsmay weaken the Ln–Napical bond.15 The contribution of 3.38 logunits per amide functional group, to the stability of thelanthanide complexes of DTPA derivatives, as evaluated byPaul-Roth and Raymond 14 is not found here, when tpaam iscompared to tpa, because of the lower contributions of thenitrogen–metal interactions.
Experimental
General information
The ligands tris[(2-pyridyl)methyl)]amine (tpa) 45 and (tris[6-((2-N,N-diethylcarbamoyl)pyridyl)methyl]amine (tpaam) 15
were prepared according to literature procedures. Lanthanidechloride salts were purchased from Aldrich, and deuterium
oxide (99.9 at.% D) from Eurisotop. The mass spectra wereacquired on a LCQ-ion trap (Finnigan–Thermoquest, San Jose,USA) equipped with an electrospray source.
Potentiometry. All titrant solutions were prepared usingwater purified by passing through a Millipore Milli-Q reverse-osmosis cartridge system (resistivity 18 MΩ cm). Carbonate-free 0.1 mol L1 KOH and 0.1 mol L1 HCl were preparedfrom Fisher Chemicals concentrates. Potentiometric titrationswere performed in 0.1 mol L1 aqueous KCl under an argonatmosphere, the temperature was controlled to ±0.1 C with acirculating water bath. The p[H] (p[H] = log[H], concen-tration in molarity) was measured in each titration with a com-bined pH glass electrode (Metrohm) filled with 3 mol L1 KCland the titrant addition was automated by use of a 751 GPDtitrino (Metrohm). The electrode was calibrated in hydrogenion concentration by titration of HCl with KOH in 0.1 mol L1
KCl.46 A plot of meter reading versus p[H] allows the deter-mination of the electrode standard potential (E ) and the slopefactor ( f ). Continuous potentiometric titrations with HCl 0.1mol L1 were conducted on 20 mL of aqueous solutionscontaining 103 mol L1 of the ligand. Back titration with0.1 mol L1 KOH was performed after each experiment tocheck whether equilibration had been achieved. 100 points weremeasured with a 2 min delay between the measurements.
Experimental data were refined using the computer programHyperquad 2000.47,48 All equilibrium constants are concen-tration quotients rather than activities. The ionic product ofwater at 25 C and 0.1 mol L1 ionic strength is pKw = 13.78.26
All values and errors (one standard deviation) reportedrepresent the average of at least three independentexperiments.
NMR spectroscopy. NMR spectra were recorded either on aUNITY or a MERCURY 400 Varian spectrometer. The methylprotons of DSS (3-(trimethylsilyl)-1-propane-sulfonic acid)were used as external reference. Samples for NMR spectroscopywere prepared by mixing appropriate volumes of stocksolutions of the ligand (∼ 0.01 mol L1) and the lanthanidechloride salt (∼ 0.01 mol L1) in deuterium oxide at a fixedionic strength (1 mol L1 KCl). Ligand concentrations weredetermined by potentiometric titration, and the metalconcentrations by EDTA titrations using xylenol orange asindicator. The pD of the samples were measured according topD = pHread 0.41.49
Quantitative proton NMR spectra were recorded with 10 srelaxation delays. The apparent equilibrium constants werecalculated from the free ligand and complex concentrations,determined by deconvolution of the 1H NMR signals givingprecise peak integral values. The use of different metal/ligandratios did not affect the value of the stability constant.
The measurement of the stability constants at no less thanten different temperatures, typically from 5 to 25 C for tpacomplexes, and from 5 to 30 C for tpaam, gave linear van’tHoff plots (ln(β110) = f(1/T )), allowing the determinationof ∆ rH /R (slope) and ∆ rS /R (origin), R being the perfectgas constant. Equilibrium constants, enthalpy and entropychanges are the result of at least two independent experimentalmeasurements.
Luminescence measurements. The lifetimes of europiumcomplexes were measured in H2O and D2O, in samples wherethe europium cation is totally complexed : [Eu] = 0.001 mol L1
and [L] = 0.05 mol L1. They were measured on a Perkin–ElmerLS50B spectrofluorimeter. Reported lifetimes, τ, are averages ofthree separate measurements calculated by monitoring theemission intensity at 616 nm after at least 20 different delaytimes covering two or more lifetimes. The gate time was 1 msand slit widths of 10 nm were employed.
2017D a l t o n T r a n s . , 2 0 0 4 , 2 0 1 2 – 2 0 1 8
Publ
ishe
d on
11
May
200
4. D
ownl
oade
d by
Nip
issi
ng U
nive
rsity
on
18/1
0/20
14 0
1:40
:02.
View Article Online

Acknowledgements
We thank Dr Christelle Gateau and Dr Marinella Mazzanti forfruitful discussions. This work was supported by the Directionde l’Energie Nucléaire at the Commissariat à l’EnergieAtomique.
References
1 V. Alexander, Chem. Rev., 1995, 95, 273.2 D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and
J. A. K. Howard, Chem. Rev., 2002, 102, 1977.3 C. Piguet and J.-C. G. Bünzli, Chem. Soc. Rev., 1999, 28, 347.4 J.-C. Bünzli and C. Piguet, Chem. Rev., 2002, 102, 1897.5 P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem.
Rev., 1999, 99, 2293.6 J. R. Morrow, L. A. Buttrey, V. M. Shelton and K. A. Berback,
J. Am. Chem. Soc., 1992, 114, 1903.7 M. Komiyama, N. Takeda and H. Shigekama, Chem. Commun.,
1999, 1443.8 S. Amin, J. R. Morrow, C. H. Lake and M. R. Churchill,
Angew. Chem., Int. Ed. Engl., 1994, 33, 773.9 J. R. Morrow, S. Amin, C. H. Lake and M. R. Churchill,
Inorg. Chem., 1993, 32, 4566.10 D. Parker and J. A. G. Williams, J. Chem. Soc., Perkin Trans. 2, 1995,
1305.11 R. S. Dickins, J. A. K. Howard, C. W. Lehmann, J. M. Moloney,
D. Parker and R. D. Peacock, Angew. Chem., Int. Ed. Engl., 1997,36, 521.
12 S. Aime, A. Barge, J. I. Bruce, M. Botta, J. A. K. Howard,J. M. Moloney, D. Parker, A. S. de Sousa and M. Woods, J. Am.Chem. Soc., 1999, 121, 5762.
13 A. Bianchi, L. Calabi, C. Giorgi, P. Losi, P. Mariani, P. Paoli,P. Rossi, B. Valtancoli and M. Virtuani, J. Chem. Soc., DaltonTrans., 2000, 697.
14 C. Paul-Roth and K. N. Raymond, Inorg. Chem., 1995, 34, 1408.15 F. Bravard, Y. Bretonnière, R. Wietzke, C. Gateau, M. Mazzanti,
P. Delangle and J. Pécaut, Inorg. Chem., 2003, 42, 7978.16 Y. Bretonnière, M. Mazzanti, J. Pécaut, F. A. Dunand and
A. E. Merbach, Inorg. Chem., 2001, 40, 6737.17 D. D. Perrin, Dissociation Constants of Organic Bases in Aqueous
Solution, Butterworths, London, 1965.18 R. Wietzke, M. Mazzanti, J.-M. Latour, J. Pécaud, P.-Y. Cordier and
C. Madic, Inorg. Chem., 1998, 37, 6690.19 G. Anderegg and F. Wenk, Helv. Chim. Acta, 1967, 50, 2330.20 G. Anderegg, K. Popov and P. S. Pregosin, Helv. Chim. Acta, 1986,
69, 329.21 B. Bleaney, J. Magn. Reson., 1972, 8, 91.22 B. Bleaney, C. M. Dobson, B. A. Levine, R. B. Martin,
R. J. P. Williams and A. V. Xavier, J. Chem. Soc., Chem. Commun.,1972, 791.
23 R. M. Golding and M. P. Halton, Aust. J. Chem., 1972, 25,2577.
24 N. Ouali, B. Bocquet, S. Rigault, P.-Y. Morgantini, J. Weber andC. Piguet, Inorg. Chem., 2002, 41, 1436.
25 C. Platas, F. Avecilla, A. de Blas, C. F. G. C. Geraldes, T. Rodriguez-Blas, H. Adams and J. Mahia, Inorg. Chem., 1999, 38, 3190.
26 R. M. Smith, A. E. Martell and R. J. Motekaitis, NIST CriticallySelected Stability Constants of Metal Complexes Database, NISTStandard Reference Database 46, 2001.
27 Y. Israëli, C. Bonal, C. Detellier, J.-P. Morel and N. Morel-Desrosiers, Can. J. Chem., 2002, 80, 163.
28 Y. Israeli and C. Detellier, Carbohydr. Res., 1997, 297, 201.29 Z. Chen, N. Morel-Desrosiers, J.-P. Morel and C. Detellier,
Can. J. Chem., 1994, 72, 1753.30 A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker,
L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem.Soc., Perkin Trans. 2, 1999, 493.
31 W. DeW. Horrocks, Jr. and D. R. Sudnick, J. Am. Chem. Soc., 1979,101, 334.
32 W. DeW. Horrocks, Jr. and D. R. Sudnick, Acc. Chem. Res., 1981,14, 384.
33 E. N. Rizkalla and G. R. Choppin, in Handbook on the Physics andChemistry of Rare-Earths, ed. K. A. Gschneidner, Jr. and L. Eyring,Elsevier, Amsterdam, 1991, vol. 15, pp. 393–442.
34 A. E. Martell, R. D. Hancock and R. J. Motekaitis, Coord. Chem.Rev., 1994, 133, 39.
35 V. Vallet, U. Wahlgren and I. Grenthe, J. Am. Chem. Soc., 2003, 125,14941.
36 G. R. Choppin, Pure Appl. Chem., 1971, 27, 23.37 G. R. Choppin, J. Less-Common Met., 1985, 112, 193.38 J.-C. G. Bünzli, Lanthanide Probes in Life, Chemical and Earth
Sciences, ed. J.-C. G. Bünzli and G. R. Choppin, Elsevier, 1989.39 E. N. RizkallaG. R. Choppin, in Handbook on the Physics and
Chemistry of Rare-Earths, ed. K. A. Gschneidner, Jr., L. Eyring,G. R. Choppin, and G. H. Lander, Elsevier, Amsterdam, 1994,vol. 18, pp. 559–590.
40 G. R. Choppin, Thermochim. Acta, 1993, 227, 1.41 E. N. Rizkalla, G. R. Choppin and W. Cacheris, Inorg. Chem., 1993,
32, 582.42 H. Imura, G. R. Choppin, W. P. Cacheris, L. A. D. Learie, T. J. Dunn
and D. H. White, Inorg. Chim. Acta, 1997, 258, 227.43 J.-M. Senegas, G. Bernardinelli, D. Imbert, J.-C. Bünzli,
P.-Y. Morgantini, J. Weber and C. Piguet, Inorg. Chem., 2003, 42,4680.
44 M. P. Jensen, L. R. Morss, J. V. Beitz and D. D. Ensor, J. AlloysCompd., 2000, 303–304, 137.
45 G. Anderegg and F. Wenk, Helv. Chim. Acta, 1971, 51, 224.46 A. E. MartellR. J. Motekaitis, Determination and Use of Stability
Constants, VCH, New York, 1992.47 P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739.48 L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca,
Coord. Chem. Rev., 1999, 184, 311.49 P. K. Glasoe and F. A. Long, J. Phys. Chem., 1960, 64, 188.
2018 D a l t o n T r a n s . , 2 0 0 4 , 2 0 1 2 – 2 0 1 8
Publ
ishe
d on
11
May
200
4. D
ownl
oade
d by
Nip
issi
ng U
nive
rsity
on
18/1
0/20
14 0
1:40
:02.
View Article Online