branche-technical report 1
TRANSCRIPT

Branche Emilie Mai 2009
Rapport technique
Master 2 : Génétique fonctionnelle et pathologie cellulaire
(46 700 caractères)
Implication du virus de l’hépatite C dans
les métabolismes lipidique et
glucidique.
Inserm U871
Physiopathologie moléculaire et nouveaux traitements des hépatites virales
151 cours Albert Thomas - 69424 LYON cedex 03
Responsable : Dr Bartosch

2
Abréviations
ACC : acétyl-CoA carboxylase AG : acides gras ApoB : apolipoprotéine B bHLH/ZIP : famille des basics helix loophelix- leucine zipper ChoRE : l’élément de réponse aux carbohydrates ChREBP : protéine liant l’élément de réponse aux carbohydrates Domaine PTB : domaine liant les tyrosines phosphorylées Domaine SH2 : domaine Src homology 2 E2 : protéine d’enveloppe du virus de l’hépatite C FAS : synthase des acides gras GK : glucokinase GSK-3 : Glycogène synthase kinase 3 G6P : glucose-6-phosphate G6PDH : glucose-6-phosphate déshydrogénase IRS : substrat du récepteur à l’insuline JNK : C-Jun N-terminal kinase L-PK : L-pyruvate kinase LXR : récepteur X du foie mTOR : mammalian target of rapamycin MTP : protéine de transfère des triglycérides microsomales PDE : phosphodiestérase PDK1 : phosphatidylinositol-dependent kinase 1 PIP2 : phosphatidylinositol biphosphate PIP3 : phosphatidylinositol triphosphate PI-3K : phosphatidylinositol 3 kinase PKB : protein kinase B PKC : protéine kinase C PTEN : homologue de la phosphatase et de la tensine deleté du chromosome 10 P85 : sous-unité régulatrice de la phosphatidylinositol-3-kinase p110 : sous-unité catalytique de la phosphatidylinositol-3-kinase RI : récepteur à l’insuline SHIP-2 : inositol 5’-phosphatase contenant un domaine SH2 SRE : élément de réponse aux carbohydrates SREBP-1c : protéine liant l’élément de réponse aux stérols VHC : virus de l’hépatite C VLVD : lipoprotéine de très faible densité TG : triglycérides

3
Résumé
L’infection par le virus de l’hépatite C (VHC) est une cause majeure de maladies
chroniques du foie avec une estimation de 170 millions de personnes infectées à
travers le monde. La gravité des maladies du foie associées au virus est très variable
allant d’une inflammation minime à la cirrhose et le carcinome hépato-cellulaire. Il
s’agit du seul virus connu pour perturber les métabolismes lipidique et glucidique
entrainant le développement de stéatose, de diabète de type 2 et/ou d’une résistance
à insuline. Les mécanismes par lesquels le VHC altère ces deux voies métaboliques
sont encore peu connus. Dans cette étude nous avons analysé la régulation de
l’expression de certaines protéines impliquées dans la voie de signalisation de
l’insuline ainsi que dans celle du glucose dans un contexte d’infection ou non par le
VHC. Pour les protéines impliquées dans la signalisation insuline, nous avons
montré que le récepteur à l’insuline ainsi que son substrat étaient sous-régulés lors
de l’infection, et que des protéines régulant négativement cette voie, comme
l’homologue de la phosphatase et de la tensine deleté du chromosome 10 (PTEN),
l’inositol 5’-phosphatase contenant un domaine SH2 (SHIP2) et la C-Jun N-terminal
kinase (JNK) étaient induites par l’infection. Nous avons montré qu’en plus de la
signalisation de l’insuline, celle du glucose était également modifiée par l’infection
par le VHC, en effet, le principal facteur de transcription sensible au glucose, la
protéine liant l’élément de réponse aux carbohydrates (ChREBP) est sur-régulé.
L’ensemble de ces résultats montre que le VHC ciblerai et modulerai l’expression et
l’activité de plusieurs protéines cellulaires afin d’induire une résistance à l’insuline
favorable à la réplication virale. Ces résultats ont été obtenus dans un système
d’infection par le VHC in vitro, il serait important de les valider dans des études
cliniques.

4
Introduction
L’agent étiologique de la majorité des cas d’hépatite non-A et non-B a été identifié
comme le virus de l’hépatite C (VHC) en 1989. En accord avec son génome, il a été
premièrement classé dans le genre des Flavivirus de la famille des Flaviviridae.
Cependant, ce virus a plusieurs particularités qui le distinguent des autres flavivirus,
comme par exemple son nombre limité d’hôtes sensibles (Pringle et al., 1999), il a de
ce fait été classé dans le genre des Hepacivirus. En effet, l’infection par le VHC est
restreinte à l’homme et au chimpanzé, alors que les Flavivirus peuvent infecter une
gamme importante d’hôtes allant des insectes aux Mammifères (Rice et al., 1996). De
plus, le VHC est limité à certains tissus : il se réplique essentiellement dans le foie
(Hoofnagle, 2002), mais des expériences cliniques et expérimentales indiquent qu’il
peut s’échapper du foie et se répliquer dans d’autres cellules, en particulier celles du
système immunitaire (Radkowski et al., 2002 ; Blackard et al., 2006 ; Pham et al.,
2006). Toutefois, ce virus est principalement hépatotropique et est une cause majeure
de maladie chronique du foie avec une estimation de 170 millions de personnes
infectées à travers le monde (Liang et al., 2000). La gravité des maladies du foie
associées au VHC est très variable allant d’une inflammation minime à une cirrhose
et le carcinome hépati-cellulaire (CHC) (Lerat et al., 2002). Le taux de progression de
l’hépatite C chronique est aussi assez variable et dépendant de différents co-facteurs
incluant l’âge, le sexe, la consommation d’alcool, le surpoids et les co-infections (Steff
LB, 2002).
Il s’agit du seul virus connu pour perturber les métabolismes lipidique et glucidique
entrainant le développement d’une stéatose (correspondant à une accumulation de
triglycérides dans les hépatocytes), d’un diabète de type 2 ou d’une résistance à
l’insuline chez l’hôte (Alaei et al., 2008). Il modulerait le métabolisme lipidique afin
de pouvoir se répliquer efficacement (Burlon et al., 2009). En fait, l’assemblage du
VHC semble intimement lié à l’activité hépatique lipidique et plus particulièrement
aux lipoprotéines de très faible densité (VLDL) (Miyanari et al., 2007). Différentes
études ont observé une association entre le VHC et les lipoprotéines in vitro et in vivo
(Burlone et al., 2009 ; Andre et al. 2002). En effet, il a été démontré que des VHC
associés aux lipoprotéines de faible densité présentent une augmentation spécifique
de l’infectiosité. De plus, certains pensent que l’association des particules virales avec
les lipoprotéines pourrait masquer des épitopes neutralisants et ainsi faciliter
l’échappement du virus au système immunitaire (Maillard et al., 2006). Le lien entre
le cycle viral du VHC et le métabolisme hépatique lipidique peut donc avoir des
effets significatifs sur le fitness viral. Le foie synthétise des acides gras (AG), stockés
sous forme de gouttelettes lipidiques avant d’être transférées dans des VLDL

5
contenant l’apolipoprotéine B (ApoB) qui vont être sécrétées dans le sang (Huang H
et al., 2007). Ce processus est régulé à différents niveaux par les voies de signalisation
du glucose et de l’insuline (Gibbons GF et al., 2002). Il a été montré que le VHC
pouvait interférer directement avec la voie de l’insuline, induire une résistance à
l’insuline menant à l’augmentation de la lipogenèse et de la synthèse des VLDL
favorisant ainsi la production du VHC (Alaei M et al., 2008 ; Kasai D et al., 2009).
Cette résistance à l’insuline peut également entrainer une stéatose ou un diabète de
type 2 (Mason et al., 1999).
On parle de résistance à l’insuline quand la concentration d’insuline nécessaire pour
avoir une réponse métabolique est plus importante que la normale, ou
alternativement quand la concentration normale d’insuline n’est plus suffisante pour
avoir une réponse métabolique normale (Bugianesi et al., 2005). La signalisation de
l’insuline est une voie très complexe impliquée dans de nombreux processus tels que
la lipolyse, la lipogenèse, la glycogenèse ou encore la protéogenèse (Figure 1). Cette
voie débute par la liaison de l’insuline sur son récepteur, un hétérotétramère
constitué de deux sous-unités α liant le ligand et de deux sous-unités β à activité
tyrosine kinase. La liaison de l’insuline sur les sous-unités α active l’une des sous-
unité β engendrant une cascade d’autophosphorylation menant à l’activation
complète du récepteur. Un des sites phosphorylés est reconnu par le domaine liant
les tyrosines phosphorylées (PTB) des substrats du récepteur à l’insuline (IRS) dont
IRS-1, IRS-2, IRS-3, IRS-4, Gab1 et Shc (Taha et al., 1999). Pendant cette interaction les
substrats du récepteur à l’insuline sont phosphorylés sur différentes tyrosines,
reconnues par des protéines possédant un domaine Src homology 2 (SH2) incluant le
complexe Grb-2-SOS, SHP2, Nck et la sous-unité régulatrice p85 de la
phosphatidylinositol-3-kinase (PI-3K). La liaison de Grb-2-SOS aux IRS ou à Shc
initie la voie des MAP kinases qui entraine l’expression de gènes cibles impliqués
dans la prolifération cellulaire. La liaison de la PI-3K à IRS active la sous-unité
catalytique, p110, de la PI-3K qui va pouvoir phosphoryler le phosphatidylinositol
biphosphate (PIP2) en phosphatidylinositol triphosphate (PIP3) (Canteley et al.,
2002). Le PIP3 va activer la kinase-1 dépendante du phosphoinositol (PDK1) qui va
alors phosphoryler AKT et mTOR. L’activation d’AKT va permettre d’activer, par
l’intermédiaire de différentes protéines, la synthèse de lipides et de glycogène et
d’inhiber la lipolyse et l’apoptose. L’activation de mTOR va stimuler la synthèse
protéique. Plusieurs protéines exercent un contrôle négatif sur différentes étapes de
cette signalisation, c’est le cas de la C-Jun N-terminal kinase (JNK) qui phosphoryle
IRS sur une sérine à proximité du domaine PTB afin de l’inactiver (Lee et al., 2003) ou
encore des deux lipides phosphatases PTEN (homologue de la phosphatase et de la
tensine déleté du chromosome 10) et SHIP-2 (l’inositol 5’-phosphatase contenant un
domaine SH2 )qui déphosphorylent le PIP3 en PIP2 (Clement S et al., 2001).
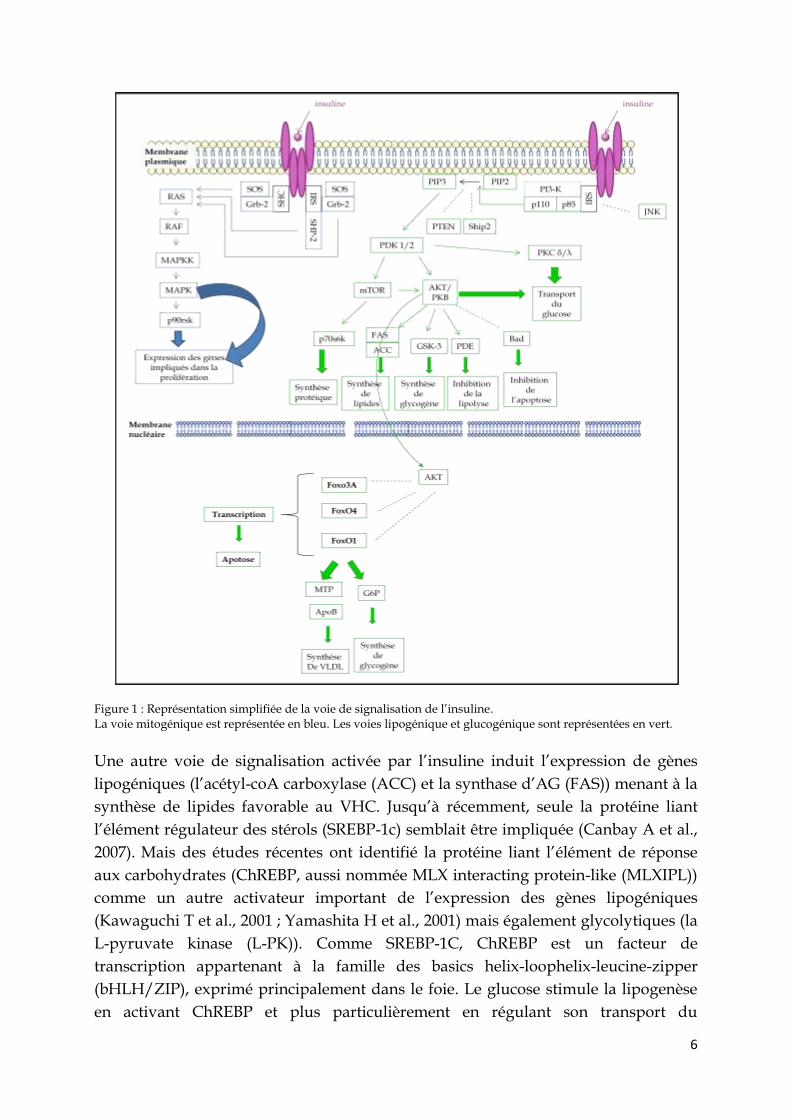
6
Figure 1 : Représentation simplifiée de la voie de signalisation de l’insuline. La voie mitogénique est représentée en bleu. Les voies lipogénique et glucogénique sont représentées en vert.
Une autre voie de signalisation activée par l’insuline induit l’expression de gènes
lipogéniques (l’acétyl-coA carboxylase (ACC) et la synthase d’AG (FAS)) menant à la
synthèse de lipides favorable au VHC. Jusqu’à récemment, seule la protéine liant
l’élément régulateur des stérols (SREBP-1c) semblait être impliquée (Canbay A et al.,
2007). Mais des études récentes ont identifié la protéine liant l’élément de réponse
aux carbohydrates (ChREBP, aussi nommée MLX interacting protein-like (MLXIPL))
comme un autre activateur important de l’expression des gènes lipogéniques
(Kawaguchi T et al., 2001 ; Yamashita H et al., 2001) mais également glycolytiques (la
L-pyruvate kinase (L-PK)). Comme SREBP-1C, ChREBP est un facteur de
transcription appartenant à la famille des basics helix-loophelix-leucine-zipper
(bHLH/ZIP), exprimé principalement dans le foie. Le glucose stimule la lipogenèse
en activant ChREBP et plus particulièrement en régulant son transport du

7
cytoplasme au noyau (Kawaguchi T et al., 2001). L’insuline, quant à elle, est capable
d’activer le récepteur X du foie (LXR) qui est un facteur de transcription permettant
l’expression des gènes ChREBP et SREBP-1C. ChREBP agirait en synergie avec
SREBP-1c en se liant à l’élément de réponse aux carbohydrates (ChoRE) situé dans
les régions promotrices des gènes lipogéniques (ACC et FAS). En revanche, il serait
seul pour induire l’expression du gène L-PK impliqué dans la glycolyse. Or il a été
montré que la lipogenèse n’était pas seulement régulée par les voies insuline/SREBP-
1C mais aussi par la signalisation du glucose (Dentin et al., 2005), par conséquent le
VHC, en régulant cette voie, pourrait augmenter la lipogenèse et donc la quantité de
lipides disponibles pour la formation de VLDL favorable à la production virale.
Figure 2 : Signalisation simplifiée du glucose et de l’insuline. La voie de signalisation du glucose est représentée en bleu et celle de l’insuline en rose. Les gènes lipogéniques peuvent être activés par deux voies distinctes, une dépendante du facteur de transcription ChREBP qui est activée par la fixation de l’insuline sur son récepteur ou par l’entrée du glucose dans la cellule et l’autre dépendante de SREBP-1C activée seulement par l’insuline
Étant donné que le VHC est impliqué dans le développement de la résistance à
l’insuline, Nous avons étudié la sur- ou sous-régulation de certaines protéines
impliquées dans les voies de signalisation de l’insuline et du glucose dans un
contexte d’infection par le VHC ou non. Nous avons sommes intéressés à la
régulation du récepteur à l’insuline (RI) et de IRS-1 ainsi qu’à celle de PTEN, SHIP2
et JNK qui sont des régulateurs négatifs de la voie de l’insuline. Concernant la
signalisation du glucose nous avons observé la régulation de la protéine ChREBP.
Nous avons utilisé pour ces études la seule lignée cellulaire capable de répliquer
efficacement le VHC, les cellules Huh 7.5, ainsi qu’une lignée cellulaire beaucoup

8
moins permissive, HepG2 CD81. Afin d’étendre ces études à d’autres lignées
cellulaires du foie ou à des tissus primaires, nous avons en parallèle développé un
génome infectieux du VHC exprimant des marqueurs de sélection nous permettant
d’obtenir une lignée cellulaire compétente pour la réplication du VHC.

9
Matériel et méthode
Culture cellulaire:
La lignée Huh 7.5 est un clone dérivé de Huh 7 qui réplique fortement le VHC (Blight
KJ et al., 2002).Les cellules sont maintenes en culture à 37°C et 5% de CO2, elles sont
cultivées en DMEM (Invitrogen) supplémenté avec 10% de SVF, 2,4% de
pénicilline/streptomycine (GIBCO), glutamax (GIBCO) et 1,2% de sodium pyruvate
(GIBCO). Les cellules d’hépatocarinome humain, Hep G2, sont cultivées dans les
même conditions et le même milieu.
Production de VHCcc:
Le plasmide pJFH1, codant pour l’intégralité du génome du VHC et contenant trois
mutations adaptatives qui augmentent l’infectiosité, deux dans le protéine core,
F172C et P173S et une dans la glycoprotéine d’enveloppe E2, N534K (Delgrange et
al., 2007). Afin de réaliser une transcription in vitro, l’ADN est linéarisé avec XbaI, ce
site est situé en 3’ après la queue polyA, purifié avec le kit Qiagen, puis transcrit en
ARN avec le kit Megascript®T7 (Ambion). L’ARN est purifié par une méthode de
phénol-chloroforme acide (Ambion) et électroporé dans les cellules Huh7.5 comme
décrit précédemment (Kato et al., 2003). Pour l’électroporation la quantité d’ARN est
ajustée à 10µg, les cellules sont traitées avec de la trypsine, lavées dans du Opti-MEM
puis sont resuspendues à 2.107 cellules/ml dans du PBS froid. Les 10µg d’ARN sont
ensuite mis en contact avec 200µl de suspension cellulaire dans une cuvette
d’électroporation 0,2 cm (Bio-Rad). Les cellules sont ensuite électroporées à 140V
avec le système d’électroporation Gene Pulser Xcell (Bio-Rad). Les cellules
électroporées sont transférées dans des boites de 10cm de diamètre contenant 12 ml
de milieu DMEM complet. Après 7-15 jours, en fonction de la croissance cellulaire, le
milieu contenant les virus est récolté, filtré et ajouté sur les cellules cibles pour
l’infection.
Infection par les VHCcc:
Les cellules Huh 7.5 ou HepG2 CD81 sont ensemencées 24 h avant l’inoculation. Pour
l’infection, le milieu est remplacé par le surnageant viral dilué et les cellules sont
incubées pendant 4h à 37°C. Les surnageants sont enlevés et les cellules infectées
restent dans du milieu DMEM complet durant 72h avant d’être analysées par
immunofluorescence en utilisant un anticorps dirigé contre la protéine core du VHC
(Abcam) comme décrit précédemment (Wakita et al., 2005).

10
Immunorévélation:
L’infection des cellules Huh 7.5 et HepG2 CD81 est réalisée avec une MOI
(multiplicity of infection) supérieure à 1. Après 0, 24, 48, 72 ou 120h d’infection, les
cellules sont récoltées par un traitement à la versene puis lysées dans une solution de
lyse (0.5% triton X-100, 50mM Tris-Cl pH 7.5, 150 mM NaCl, 2 mM EDTA (inhibiteur
de protéases) (Roche)). Les lysats sont ensuite clarifiés par centrifugation à 14000 rpm
pendant 10 min à 4°C. Puis la concentration est déterminée par méthode de Bradford
(Sigma). Les lysats cellulaires sont tout d’abord préparés avec du tampon de charge
dénaturant 5X (250mM Tris pH 6,8, 10% SDS, 50% glycérol, 0,02%bleu de
bromophénol, 10% de β-mercaptoethanol). Enfin, les échantillons sont chauffés à
95°C pendant 1min puis analysés par SDS-PAGE (à 4% pour le gel de concentration
et 12% pour le gel de séparation). Les protéines sont ensuite transférées sur une
membrane de nitrocellulose (Hybond ECL, Amersham Biosciences). La membrane
est saturée durant 1h dans du TBST (25mM Tris-HCL (pH 7,5), 150mM NaCl, 0,1%
Tween 20) supplémenté de 4% lait écrémé. La membrane est incubée dans l’anticorps
primaire puis dans l’anticorps secondaire couplé à la péroxydase, tous deux dilués
dans du TBST-lait 4%. Les dilutions des anticorps ont été mises au point. Différents
anticorps primaires sont utilisés : un anticorps AP33 (Arvin Patel) anti-glycoprotéine
E2 ; un anti-récepteur à l’insuline, un anti-IRS-1, un anti PTEN, un anti-SHIP2 et un
anti-P-JNK (Santa Cruz) ; un anti-ChREBP (Thermo scientific). Les anticorps
secondaire anti-chèvre et anti-lapin proviennent de chez Dako et l’anti-souris de chez
Sigma. La membrane est ensuite révélée avec le kit de révélation ECL Supersignal
West dura (Pierce). L’actine cellulaire est détectée en utilisant un anticorps spécifique
(Sigma) afin de comparer la quantité de protéine chargée dans chaque puits.
Clonage :
Nous utilisons un plasmide approprié à la transcription de JFH1. Dans le cadre de
ouvert lecture de JFH1, le Dr Durantel (Inserm U871) avait déjà inséré les sites de
restriction Mlu et BglII permettant l’insertion de séquence ectopique en phase avec le
gène NS5A (Moradpour D et al., 2004). Nous avons inséré grâce au site MluI et BglII
un fragment amplifié par PCR en utilisant la Taq polymérase de chez NEB comme
recommandé par le fabricant. Les produits PCR codant pour les gènes de résistances
à l’hygromycine ou la blasticidine grâce aux amorces utilisées ont un site MluI au
début de la séquence codante et un site BamHI (compatible avec BglII) en C-terminal
(Hygromycine anti-sens: 5’CTAGggatccATGACCGAGTACAAGCCCAC 3’,
Hygromycine sens : 5’ GCG GacgcgtGGCACCGGGCTTGCGGG 3’, Blasticidine anti-
sens : 5’ GAACggatccATGG CCAAGCCTTTGTCTCA 3’, Blasticidine sens : 5’
GAAGacgcgtGCCCTCCCACACAT AACCA 3’). Les codons stop des gènes de
sélection sont enlevés afin de permettre la traduction en phase avec le cadre ouvert
de lecture du génome du VHC. Les produits PCR sont purifiés avec le kit de

11
purification Qiagen puis digérés avec MluI et BamHI (NEB) et ligués dans le
plasmide JFH1 modifié et ouvert par MluI et BglII. Les ligations sont ensuite
transformées dans des bactéries compétentes Top10 (Invitrogen). Les mini et maxi
préparations sont vérifiées par séquençage des inserts.
RT-PCR
Les cellules infectées et non infectées sont récoltées après trypsination et l’ARN total
est préparé (RNeasy; QIAGEN) et quantifié. La synthèse d’ADNc est réalisée avec le
kit SuperScript III Reverse Transcriptase d’Invitrogen en mélangeant 1µg d’ARN
avec 200 ng d’oligo dT et 1 µl de dNTP 10mM. Le mélange est incubé à 65°C pour 1
min et dans la glace 1min également. Puis 4 µl de mélange de réaction First strand, 1
µl de dithiothreitol 0,1 M, 1 µl de RNAse Out et 1 µl de transcriptase inverse sont
ajoutés, le mélange est incubé à 50°C durant 60 min dans un volume final de 20 µl.
Afin d’éliminer l’ARN matrice, la RNAse H est ajoutée, suivie d’une incubation de 20
min à 37°C. Des dilutions au 10ème en séries sont effectuées avec l’ADNc obtenu et 1µl
de chaque est utilisé pour une réaction PCR, réalisé avec une Taq polymérase (NEB)
et des amorces spécifiques des gènes (tableau 1) dans les conditions standards.
Gène Amorces spécifiques
SREBP-1c sens 5’ ACACAGCGGTTTTGAACGACATC 3’
SREBP-1c anti-sens 5’ ACGGACGGGTACATCTTTACAG 3’
CHREB sens 5’ CAGCGTCGAGGTTTCACACT 3’
CHREB anti-sens 5’ CTGCTGTTTCCACGGTTGAG 3’
β-actine sens 5’ TTGTAACCAACTGGGACGATATGG 3’
β-actine anti-sens 5’ CGACCAGAGGCATACAGGGACAAC 3’ Tableau 1: Les différentes amorces utilisées pour la PCR.

12
Résultats
Régulation des protéines impliquées dans la voie métabolique
lipidique dans un contexte d’infection par le VHC.
Pour essayer de comprendre comment une infection par le VHC peut entrainer une
résistance à l’insuline, nous avons étudié la régulation de l’expression de certaines
protéines impliquées dans la signalisation de l’insuline. Pour cela, nous avons infecté
des cellules Huh 7.5 avec le VHC et suivi l’expression de ces protéines par
immunorévélation.
Toutes les cellules sont infectées par VHCcc avec une MOI supérieure à 1. En général
les cellules sont récoltées pour l’analyse avant infection (0h) puis à 24 et 72 h après
l’infection. Dans quelques expériences, du fait d’une croissance lente des cellules
infectées, nous avons changé le protocole pour maximiser la quantité de lysat
cellulaire et nous avons récolté les cellules à 0, 48 et 120 heures. En effet, l’infection
par le VHC avec une forte MOI a été décrite pour ralentir la croissance. Une
immunorévélation avec un anticorps anti-actine a été réalisé afin de contrôler la
quantité de lysat cellulaire déposée dans chaque puits (Figure 3). Ce control a été
réalisé dans toutes les expériences suivantes.
Figure 3 : Les cellules Huh 7.5 sont infectées par les VHC pendant différents temps, 0, 24 et 72 h puis sont
récoltées et lysées. Afin de voir si la quantité de protéine déposée dans chaque puits est identique, les lysats sont
analysés par immunomarquage avec un anticorps anti-actine.
En plus d’avoir vérifié que la quantité chargée de lysats cellulaire était égale dans
chaque puits, nous nous sommes assuré que les cellules avaient bien été infectées par
les VHCcc en réalisant une immunorévélation avec un anticorps AP33 dirigé contre
la protéine d’enveloppe E2 du VHC. Comme montré dans la figure 4, l’expression de
la protéine E2 augmente au cours du temps d’infection. Par conséquent nous
pouvons affirmer que les cellules Huh 7.5 sont bien infectées par le VHC. Comme
pour l’actine nous avons réalisé ce control dans toutes les expériences.

13
Figure 4 : Vérification de l’infection des cellules Huh 7.5 par les VHC. Les cellules infectées et non infectées sont
récoltées à 0h, 48h et 120 h après l’infection par le VHC. Les lysats cellulaires sont fractionnés par SDS-PAGE et
immunomarqués avec un anticorps anti-E2.
Tout d’abord nous avons étudié l’expression de RI (récepteur à l’insuline) et d’IRS-1
(substrats liant le récepteur à l’insuline), car ces protéines sont toutes deux sous-
régulées lors de la résistance à l’insuline (Okabayashi et al., 1989 ; Carvalho E et al.,
1999 ; Renström F et al., 2007). Nous observons que l’expression du récepteur à
l’insuline est diminuée après 24 h d’infection, puis semble rester stable (Figure 5A).
L’expression de IRS-1 n’est pas affectée à 24 h mais à 72 h post-infection où elle est
sous régulée de façon importante (Figure 5B); cette protéine pourrait même subir une
dégradation par le protéasome comme cela déjà été décrit ( Zlande et al., 2002).
Figure 5 : Analyse de l’expression du récepteur à l’insuline et de IRS-1 dans un contexte d’infection par le VHC.
Les cellules Huh 7.5 infectées et non infectées sont récoltées à 0 h, 48 h et 72 h après l’infection par le VHC. Les
lysats cellulaires sont fractionnés par SDS-PAGE et immunomarqués avec un anticorps polyclonal anti-récepteur
à l’insuline (A) ou anti-IRS-1 (B).
Puis nous avons étudié l’expression des différentes protéines régulant négativement
la voie de signalisation de l’insuline, PTEN, SHIP2 et JNK. Les deux lipides
phosphatases, PTEN et SHIP2 déphosphorylent le PIP3 en PIP2 empêchant ainsi la
phosphorylation de AKT. JNK et plus précisément JNK phosphorylée (P-JNK) qui est
la forme active, va phosphoryler IRS-1 sur une sérine à proximité du domaine PTB
afin de l’inactiver. En effet, IRS-1 activé est phosphorylé sur des tyrosines.
Comme montré en figure 6, ces protéines sont régulées différemment au cours du
temps mais au final elles sont toutes sur régulées. Dans les premières 24 heures après
l’infection l’expression des protéines PTEN et JNK n’est pas modifiée, mais elle est
induite après 72h (Figure 6A, C). L’expression de SHIP2, quant à elle, augmente
progressivement au cours de l’infection (Figure 6B).

14
Figure 6 : Analyse de l’expression de protéines régulatrices impliquées dans la signalisation de l’insuline dans un
contexte ou non d’infection par le VHC. Les cellules Huh 7.5 infectées et non infectées sont récoltées à 0 h, 24 h et
72 h après l’infection par le VHC. Les protéines cellulaires sont séparées par SDS-PAGE et immunomarquées avec
un anticorps anti-PTEN (A) ou un polyclonal anti-SHIP2 (B) ou un monoclonal anti-P-JNK (C).
Expression de la protéine ChREBP impliquée dans le métabolisme
glucidique.
Le lien entre l’assemblage du VHC et la production de VLDL est en accord avec les
observations faites montrant que le VHC favorise l’insulino-résistance (Alaei et al.,
2008). Il est également clair que le VHC induit une sur-régulation de la lipogenèse
(Alaei et al., 2008), probablement en fournissant une quantité de lipides nécessaire
pour augmenter le taux de synthèse de VLDL. En effet, le VHC induit l’activation de
l’un des facteur clé de la signalisation de l’insuline, le facteur de transcription SREBP-
1 (Oem et al., 2008). Cependant la lipogenèse n’est pas régulée que par la
signalisation de l’insuline, la signalisation du glucose jouant également un rôle
important (Dentin et al., 2005). Il est suggéré que le VHC sur-régulerait cette voie afin
d’augmenter la lipogenèse et donc la synthèse de VLDL favorable au VHC. Nous
avons voulu étudier le rôle de la signalisation du glucose dans la régulation
lipidique, pour cela nous avons observé les effets de l’infection sur l’expression du
principal facteur de transcription sensible au glucose, ChREBP.
Les résultats sont encore préliminaires et devront être approfondis, mais il semblerait
que l’expression de cette protéine soit induite lors de l’infection par le VHC. Dans la
lignée cellulaire Huh 7.5, la quantité de protéine ChREBP est augmentée dès 48 h
après l’infection (Figure 7A). Cette augmentation est également observée dans la
lignée hépatocytaire HepG2 CD81 mais à 72h après l’infection (Figure 7B). Ce retard
est surement due au fait que cette lignée est moins permissive à la réplication du
virus. En effet, la réplication de HCV dans les cellules HepG2 CD81 contrairement à
ce qui était observé dans les cellules Huh 7.5, était indétectable par western blot avec
l’anticorps AP33 mais nous avons détecté un signal par RT-PCR en utilisant des
amorces du brin positif du génome du VHC (données non présentées).

15
Figure 7 : Analyse de l’expression de la protéine ChREBP dans des cellules infectées par le VHC. Les cellules Huh
7.5 (A) et HepG2 (B) sont infectées par les VHC pendant différents temps, 0, 24 et 72 h puis sont récoltées et
lysées. Les lysats cellulaires sont fractionnés par SDS-PAGE et immunomarqués avec un anticorps polyclonal
anti-ChREBP.
Afin de voir si l’augmentation de niveau de la protéine ChREBP est due à une
augmentation de la transcription, à une réduction de la dégradation ou à autre chose,
nous avons réalisé une RT-PCR avec des amorces spécifiques de ChREBP et d’autres
gènes contrôles que sont SREBP-1c et l’actine. La transcription de SREBP-1c a été
décrite pour être sur-régulé par le VHC, et donc, sert de control positif. La
transcription du gène de l’actine est utilisée comme contrôle négatif comme elle n’est
pas modifiée par le VHC. En effet, la détection de transcrits de l’actine est identique
dans les cellules infectées (Figure 8Aa) et non infectées (Figure 8Ab). Le premier
couple d’amorces utilisé pour détecter les transcrits de SREBP-1c est peu sensible
mais nous pouvons voir que SREBP-1c est visible seulement dans les cellules
infectées par le VHC (avec l’ADNc le moins dilué), illustrant que ce gène est plus
activement transcrit dans les cellules infectées (Figure 8Ba) que dans les cellules non
infectées (Figure 8Bb). Ce résultat est confirmé par un deuxième couple d’amorces
toujours spécifique de SREBP-1c (Figure 8C). Nous observons qu’il y a une quantité
plus importante de SREBP-1c transcrit dans les cellules infectées. En effet, à la plus
forte dilution (1/100), les transcrits sont détectables uniquement dans les cellules
infectées (Figure 8Ca). Des résultats similaires sont observés pour ChREBP (Figure
8D), confirmant et appuyant les résultats obtenus par western blot. Mais cette
expérience nous apporte une information complémentaire, puisqu’elle nous indique
que la surexpression de protéine observée précédemment est due, au moins en partie
à une sur-régulation de la transcription du gène ChREBP.

16
Figure 8 : Analyse de la régulation de la quantité de d’ARNm codant ChREBP dans les cellules Huh 7.5 infectées
par le VHC. Après 72 heures d’infection les cellules sont récoltées, les ARN totaux sont extraits et préparés. Puis
l’ADNc est synthétisé, des dilutions au 10ème en séries sont effectuées et 1µl de chaque est utilisé pour les
différentes réactions PCR réalisées avec des couples d’amorces spécifiques de l’actine (A), SREBP-1C (B,C) et de
ChREBP (D). Nous détectons la différence de transcription de ces gènes dans les cellules infectées (a) et celles non
infectées (b).
Développement de nouvelles lignées capables de répliquer HCV
efficacement.
La lignée cellulaire hépatocytaire Huh 7.5 est hautement transformée, par conséquent
son utilisation du glucose est très probablement altérée. En effet les cellules
tumorales utilisent beaucoup de glucose du fait de leur fort taux de réplication. De
plus, la réduction de la formation et de la sécrétion des lipides dans cette lignée
cellulaire a déjà été décrite (Icard et al., 2009). Il semble donc nécessaire d’étudier les
effets du VHC sur le métabolisme cellulaire lipidique et glucidique dans un contexte
plus physiologique. Pour cette raison nous avons commencé à développer un
système cellulaire alternatif plus physiologique du point de vue du métabolisme
lipidique et glucidique qui serait également capable de supporter l’infection par le
VHC. Un génome adapté du VHC exprimant différents marqueurs de sélection a été
développé, afin de permettre la sélection de clones et de lignées cellulaire permissifs
à la production du VHC.
Comme pour les études sur les régulations des protéines impliquées dans les
métabolismes glucidique et lipidique, nous avons utilisé le génome JFH1 contenant
trois mutations spécifiques décrites pour augmenter l’infectiosité (Delgrange et al.,
2007). Nous avons inséré dans ce plasmide des sites de restriction en phase avec le
cadre de lecture ouverte du gène NS5A, nous permettant d’insérer des fragments
obtenus par PCR codant pour des marqueurs de sélection comme les gènes de

17
résistance à l’hygromycine ou à la blasticidine. En effet, il a été montré que l’insertion
de gènes dans la région N-terminale de NS5A ne perturbait pas la réplication virale
(Moradpour et al., 2004). Les plasmides obtenus sont ensuite vérifiés par séquençage
et testés pour leur fonctionnalité. Après une transcription in vitro, les génomes de
sélection sont électroporés dans les cellules Huh 7.5, qui sont ensuite cultivées trois
jours sans pression de sélection, puis des quantités appropriées minimales
d’antibiotiques nécessaires pour tuer les cellules non transfectées sont ajoutées dans
le milieu. L’électroporation des génomes JFH1 codant pour l’hygromycine ou la
blasticidine mène à un retard de croissance et des clones sont obtenus après 7-15
jours de culture dans le milieu sélectif. Une titration préliminaire indique que le titre
viral est de 6.102 FFU/ml. Ce titre est probablement fortement sous-estimé (plusieurs
magnitudes) comme les cultures n’ont pas été effectuées de manière appropriée due
à des raisons de logistique. En effet, les pièces de culture cellulaire du laboratoire ont
été fermées pour le déménagement de l’unité Inserm (avril-mai). Nous reprendrons
ces cultures prochainement afin de réaliser une titration plus correcte.

18
Discussion
L’infection par le VHC induit une résistance à l’insuline, un syndrome métabolique
associé à une augmentation de la lipogenèse et de la biosynthèse de VLDL pourrait
favoriser la production et le fitness du virus. Les observations faites chez l’hôte
démontrent que la résistance à l’insuline induite par le VHC a de fréquentes et
sérieuses conséquences pathologiques qui se manifestent sous forme de stress
oxydant ou de stéatose. L’interaction entre la résistance à l’insuline, le stress oxydant
et la stéatose favorise fortement l’induction de la réponse inflammatoire et conduit à
la fibrose du foie. L’infection chronique par le VHC peut donc créer un
environnement extrêmement oxydatif qui favorise l’instabilité génomique, et qui,
combiné avec les effets mitogènes directs de certaines protéines du VHC, doit
probablement contribuer au processus de transformation maligne conduisant au
cancer du foie (Caldwell et al., 2004 ; McGivern et al., 2008). En effet, l’infection
chronique par le VHC est un facteur de risque majeur pour le développement d’un
CHC (carcinome hépato-cellulaire); chaque année, 4-5% des patients infectés par le
VHC développent un CHC. Des marqueurs sérologiques de l’infection par le VHC
sont trouvés dans 27 à 80% des CHC, ce pourcentage variant en fonction de la
localisation géographique. L’infection par le VHC augmente de 17 fois le risque de
développer un CHC comparé à des patients sains (Donato et al., 2002 ; El-Serag HB
2002 ; Imazeki et al., 2003 ; Sun et al., 2003).
Comme l’infection chronique du VHC est impliquée dans cette pathologie, il
est important de comprendre le rôle de la résistance à l’insuline dans le cycle viral et
ces effets dans la lipogenèse et l’assemblage de VLDL. Il apparait que le VHC induit
une résistance à l’insuline et par conséquent une sur-régulation de la lipogenèse
(Alaei et al., 2008) probablement pour fournir la quantité de lipides nécessaire pour
augmenter le taux de synthèse de VLDL. En effet, le VHC permet l’activation de
SREBP-1c, qui est un régulateur majeur de la lipogenèse induite par l’insuline (Oem
et al., 2008). Cependant la lipogenèse n’est pas régulée uniquement par la voie
insuline/SREBP-1c, mais également par la signalisation du glucose (Dentin et al.,
2005), suggérant que le VHC peut sur-réguler cette voie de signalisation afin
d’assurer l’augmentation de la lipogenèse.
Dans cette étude nous avons étudié la régulation de l’expression de différentes
protéines impliquées dans la signalisation de l’insuline comme du glucose, dans le
but de mieux comprendre leurs impacts dans le processus d’assemblage, le cycle
viral et plus particulièrement le rôle des lipoprotéines dans ce cycle. Comme le seul
système in vitro de réplication du VHC disponible est basé sur un seul isolat viral très

19
particulier et une seule lignée cellulaire hépatocytaire (Huh 7.5), nous avons analysé
des modifications des signalisations de insuline et du glucose induite par le VHC
dans ce système.
Le récepteur à l’insuline qui est la première protéine jouant un rôle la
signalisation de l’insuline, est, dans notre étude, sous-régulée dans les cellules
infectées par le VHC ; ces résultats contredisent ceux observés dans l’étude de
Kawaguchi et al. Ils observent que le niveau d’expression de ce récepteur reste
inchangé après une expression de la protéine core du VHC que ce soit dans des
cellules HepG2 transfectées ou dans des souris transgénique pour cette protéine
(Kawaguchi et al., 2004). Cette différence peut-être due au fait que les systèmes
d’étude ne sont pas identiques. En effet, notre étude est réalisée dans des cellules
Huh 7.5 infectées par l’isolat JFH1 qui est de génotype 2a alors que Kawaguchi et al.
travaillent dans des cellules HepG2 transfectées par la protéine core d’un virus de
génotype 1a. Il est possible que les protéines régulées par le VHC pour induire une
résistance à l’insuline varient en fonction des génotypes. La lignée cellulaire peut
également avoir son importance, mais nous pouvons aussi penser que la diminution
de l’expression du récepteur à l’insuline est due à une autre protéine que le core du
VHC.
Nous avons également étudié la régulation de IRS-1, qui une fois
phosphorylée sur des tyrosines va pouvoir activer différentes voies dont la synthèse
lipidique. La sous-régulation hépatique de cette protéine mène à une augmentation
de la résistance à l’insuline (Araki et al., 1994). Lors de l’infection par le VHC,
l’expression de IRS-1 est diminuée dans notre modèle d’étude. Ceci est en accord
avec différentes études plus ou moins récentes qui ont montré que la protéine core
du VHC induit la dégradation de IRS-1 (Kawaguchi et al., 2004 ; Pazienza et al.,
2007). En effet, Rui L et al. ont observé que les protéines suppressives de la
signalisation des cytokines, SOCS1 et SOCS3 induisent l’ubiquitination et la
dégradation par le protéasome de IRS-1 dans l’ensemble des cellules et des tissus
animaux étudiés, contribuant ainsi à l’établissement de la résistance à l’insuline ( Rui
et al., 2002). Une étude semble contredire le fait que IRS-1 soit sous-régulée par le
VHC. Elle démontre que l’association du récepteur à l’insuline avec IRS-1 est
diminuée malgré l’augmentation de la quantité de ces deux protéines chez les
patients infectés. Il semblerait que cette faible association soit due à la diminution de
la phosphorylation des tyrosines de IRS-1 (Aytug et al., 2004). Ceci étant, les auteurs
de l’étude admettent que l’observation de l’augmentation de l’expression de ces deux
protéines est inattendue car on s’attend plutôt à la réduction de leurs expressions due
à une dégradation protéolytique excessive médiée par une signalisation exagérée via
le relargage de cytokines du fait de l’inflammation induite par le VHC (Petit et al.,
2001).

20
Le VHC sous-régule la signalisation de l’insuline également en induisant
l’expression de deux lipides phosphatases, SHIP2 et PTEN. La seule étude qui
observe l’expression de PTEN dans un contexte d’infection par le VHC est celle de
Waris et al.. Ils montrent que l’expression de PTEN dans les cellules Huh 7.5 infectées
avec le VHC est inchangée par rapport à celle des cellules non infectées, de manière
intéressante ils observent une augmentation de PTEN-phosphorylé, la forme inactive
de PTEN, dans les cellules infectées (Waris et al., 2007). Pour SHIP2 aucune étude
dans la littérature n’avait encore suggéré que l’expression de cette lipide phosphatase
était modulée par le VHC.
JNK est une protéine appartenant à la famille de MAPkinases (Mitogen-
activated protein kinases), elle est activée par les cytokines pro-inflammatoires mais
également par l’insuline. Cette protéine exerce un rétrocontrôle négatif sur voie de
l’insuline en phosphorylant IRS sur un résidu sérine proche du domaine PTB où
normalement le récepteur à l’insuline phosphoryle une tyrosine. Nos résultats
montrent que le VHC augmente l’expression de cette protéine JNK, ce qui est en
accord avec différentes études qui observaient l’implication du VHC dans le
développement de CHC. En effet, JNK ne joue pas uniquement un rôle de régulateur
négatif de la voie de l’insuline mais également un rôle activateur d’AP-1, permettant
la stimulation de la prolifération cellulaire. Différents travaux montraient que des
protéines du VHC telle que la protéine core, NS3 (dans des cellules HepG2) et NS5A
(dans des cellules de rein embryonnaire, les 293T) induisaient l’expression de JNK.
(Erhardt et al., 2002 ; Hassan et al., 2005 ; Park et al., 2003). Ces résultats suggèrent
que l’inhibition de JNK ou l’interférence dans l’association d’IRS-1 avec JNK pourrait
être une bonne cible thérapeutique pour réduire la résistance à l’insuline induite ou
non par le VHC.
L’ensemble de ces résultats montre que le VHC cible et module l’expression et
l’activité de plusieurs protéines cellulaires afin d’induire une résistance à l’insuline.
D’un coté, le fait de diminuer l’expression du récepteur à l’insuline ainsi que celle de
IRS-1 diminue la sensibilité à l’insuline puisque l’hormone se fixe moins et le signal
est peu transmis. Et d’un autre coté l’augmentation de l’expression des protéines
PTEN, SHIP2 et P-JNK régulant négativement la signalisation intracellulaire de
l’insuline, contribue également à une diminution de la sensibilité à l’insuline.
La réduction de la signalisation de l’insuline favorise la lipogenèse et donc
probablement le processus d’assemblage du VHC. En effet, SREBP-1c, un régulateur
majeur de la lipogenèse induite par l’insuline, est sur-régulée par l’infection du VHC.
Cependant la synthèse lipidique n’est pas seulement régulée par la signalisation de
l’insuline mais également par celle du glucose. Le glucose peut effectivement activer
la synthèse de lipides via le facteur de transcription ChREBP. Cette protéine peut
également être activée par l’insuline par l’intermédiaire de LXR. Similairement aux
travaux de recherche effectués sur SREBP-1c, nous trouvons que l’expression de la

21
protéine ChREBP est augmentée dans les cellules infectées. Ce résultat est en accord
avec de précédentes études où la protéine core du VHC pouvait induire une
accumulation de lipides dans les cellules Huh 7.5 (Abid et al., 2005 ; Jackel-Cram C
et al., 2007 ; Waris et al., 2007). Nous avons donc identifié un nouveau facteur de
transcription en plus de SREBP-1c, ChREBP, qui est induit par le VHC pour
augmenter la synthèse de lipides et l’assemblage de VLDL.
Des études complémentaires seront nécessaires pour savoir si la sur-régulation
du niveau de protéine ChREBP due à l’augmentation de sa transcription est corrélée
avec l’accentuation de la lipogenèse et avec l’assemblage des VLDL. La sur- ou sous-
expression du récepteur à l’insuline, de IRS-1, PTEN, SHIP2, JNK ou ChREBP, pourra
nous aider pour étudier le rôle exact de ces protéines dans le cycle viral et les
pathologies associées.
Même si les résultats précédents nous aident à comprendre le rôle de la
résistance à l’insuline et des métabolismes lipidique et glucidique dans le cycle viral,
il est important de rappeler qu’ils ont été obtenus dans un système in vitro assez
artificiel. La souche virale JFH1 de génotype 2a dont la sequence diffère des isolats
d’hépatite C chronique a été isolée à partir d’un patient ayant une hépatite
fulminante (Kato et al., 2001). Ce genre d’hépatite virale sévère aigüe est caractérisé
par une réplication virale importante et une intense réponse immunitaire de l’hôte
contre les cellules ou les tissus infectés. Quelques cas d’hépatites fulminantes dus au
VHC ont été rapportés après l’arrêt de chimiothérapie ou de drogues
immunosuppressives. Dans ces cas, de hauts titres viraux étaient associés et il y avait
l’apparition de sérieux problèmes hépatiques dues à une réplication viral illimitée,
du fait des conditions d’immunosuppression (Vento et al., 1996). Ceci étant, ces
hépatites restent assez atypiques et limitent notre système d’étude. De plus nous
avons effectué la plupart de notre travail dans des cellules Huh 7.5. Cette lignée
cellulaire hépatocytaire est transformée, entrainant surement des modifications dans
son utilisation de glucose. Il semblerait également que le métabolisme lipidique de
ces cellules soit assez altéré, entrainant un défaut dans l’assemblage des lipoprotéines
(Icard et al., 2009). Donc le développement de lignées cellulaires permissives à la
réplication du VHC ayant un métabolisme glucidique et lipidique physiologique est
important. Dans cette optique, nous avons mis au point un génome du VHC
sélectionnable. L’insertion des gènes de résistance dans le génome viral permettra à
terme d’isoler une lignée cellulaire capable de supporter une infection par le VHC et
permettra également de sélectionner des cellules infectées. Nous avons commencé à
tester la fonctionnalité des génomes sélectionnables dans les lignées cellulaires Huh
7.5 et HepG2 CD81. Dès que nous aurons terminé la caractérisation basique de ces
génomes, nous pourrons infecter des hépatocytes primaires humains et essayer
d’isoler des clones hautement permissifs à la réplication du virus. Pour le moment,

22
nous validons les résultats obtenus avec les cellules Huh 7.5 infectées par JFH1 pour
ensuite envisager de réaliser des études cliniques.
L’identification de l’altération des métabolismes glucidique et lipidique par le
VHC ne nous éclaire pas seulement sur la nécessité de la lipogenèse pour la
production virale mais nous aide également à mieux comprendre la pathogénèse
associée à l’infection chronique et à établir de nouvelles approches thérapeutiques ou
à revoir celles existantes (sensibilisateur à l’insuline ou médicaments diminuant le
cholestérol) pour les traitements de l’infection chronique. Il existe différents
traitement contre le diabète de type 2, comme les biguanides qui favorisent l’action
de l’insuline au niveau du foie ou les Thiazolidinediones qui améliorent la sensibilité
à l’insuline et réduisent la glycémie. Ces derniers activent le récepteur nucléaire
PPARgamma (Peroxisome Proliferator-Activated Receptor gamma) afin d’altérer
l’expression de gènes impliqués dans l’homéostasie lipidique et glucidique.
L’utilisation de ces thérapies pour le traitement de l’infection chronique du VHC
pourrait être considérée. De plus, notre étude pourra permettre d’identifier de
nouvelles cibles thérapeutiques cellulaires ciblant les métabolismes glucidique et
lipidiques aussi bien que l’infection chronique du VHC, telles que les lipides
phosphatases (PTEN et SHIP2) et JNK.
Remerciements
Tout d’abord, je remercie le Dr Durantel de m’avoir fourni la construction du
plasmide JFH1 muté contenant les sites de restriction, également Mr Jammard pour
m’avoir assisté lors de manipulations mais aussi également pour ses conseils. Bien
évidemment, je remercie ma responsable, le Dr Bartosch de m’avoir aussi bien
encadré, tant dans la pratique que dans la rédaction. Et enfin je remercie le Dr
Zoulim de m’avoir accueilli dans son équipe ainsi que toute l’unité Inserm U871 pour
sa gentillesse et sa convivialité qui a rendu mon travail agréable.

23
Références -Abid K, Pazienza V, de Gottardi A, Rubbia-Brandt L, Conne B, Pugnale P, Rossi C, Mangia A, Negro F. An in vitro model of hepatitis C virus genotype 3a-associated triglycerides accumulation. J Hepatol. 2005;42:744-51.
- Alaei M, Negro F. Hepatitis C virus and glucose and lipid metabolism. Diabetes Metab. 2008;34:692-700.
-André P, Komurian-Pradel F, Deforges S, Perret M, Berland JL, Sodoyer M, Pol S, Bréchot C, Paranhos-Baccalà G, Lotteau V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J Virol. 2002;76:6919-6928
- Araki E, Lipes MA, Patti ME, Brüning JC, Haag B 3rd, Johnson RS, Kahn CR. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372:186-190.
- Aytug S, Reich D, Sapiro LE, Bernstein D, Begum N. Impaired IRS-1/PI3-kinase signaling in patients with HCV: a mechanism for increased prevalence of type 2 diabetes. Hepatology. 2003;38:1384-1392.
- Blackard JT, Kemmer N, Sherman KE. Extrahepatic replication of HCV: insights into clinical manifestations and biological consequences. Hepatology. 2006;44:15-22 - Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001-130014.
-Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chonic liver diseases. Hepatology. 2005;42:987-1000.
- Burlone ME, Budkowska A. Hepatitis C virus cell entry: role of lipoproteins and cellular receptors. J Gen Virol. 2009; 90: 1055-1070.
- Caldwell SH, Crespo DM, Kang HS, Al-Osaimi AM. Obesity and hepatocellular carcinoma. Gastroenterology. 2004;127:97-103. - Canbay A, Bechmann L, Gerken G Lipid metabolism in the liver. Z Gastroenterol 2007;45: 35-41 - Cantley LC. The phophoinositide 3-kinase pathway. Science. 2002;296:1655-1657.
- Carvalho E, Jansson PA, Axelsen M, Eriksson JW, Huang X, Groop L, Rondinone C, Sjöström L, Smith U. Low cellular IRS 1 gene and protein expression predict insulin resistance and NIDDM. FASEB J. 1999;13:2173-2178
- Clément S, Krause U, Desmedt F, Tanti JF, Behrends J, Pesesse X, Sasaki T, Penninger J, Doherty M, Malaisse W, Dumont JE, Le Marchand-Brustel Y, Erneux C, Hue L, Schurmans S. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature. 2001; 409: 92-97
- Delgrange D, Pillez A, Castelain S, Cocquerel L, Rouillé Y, Dubuisson J, Wakita T, Duverlie G, Wychowski C. Robust production of infections viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol. 2007;88:2495-2503.

24
- Dentin R, Girard J, Postic C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie. 2005;87:81-86
- Donato F, Tagger A, Gelatti U, Parrinello G, Boffetta P, Albertini A, Decarli A, Trevisi P, Ribero ML, Martelli C, Porru S, Nardi G. Alcohol and hepatocellular carcinoma: the effect of lifetime intake and hepatitis virus infections in men and women. Am J Epidemiol. 2002;155: 323-331
- El-Serag HB Hepatocellular carcinoma: an epidemiologic view. J Clin Gastroenterol. 2002; 35:72-78
- Erhardt A, Hassan M, Heintges T, Häussinger D. Hepatitis C virus core protein induces cell proliferation and activates ERK, JNK, and p38 MAP kinases together with the MAP kinase phosphatase MKP-1 in a HepG2 Tet-Off cell line. Virology. 2002;292:272-284.
-Gibbons GF, Brown AM, Wiggins D, Pease R. The roles of insulin and fatty acids in the regulation of hepatic very-low-density lipoprotein assembly. J R Soc Med.2002;95:23-32.
- Hassan M, Ghozlan H, Abdel-Kader O. Activation of c-Jun NH2-terminal kinase (JNK)
signaling pathway is essential for the stimulation of hepatitis C virus (HCV) non-structural
protein 3 (NS3)-mediated cell growth. Virology. 2005;333:324-336.
- Hoofnagle JH. Course and outcome of hepatitis C. Hepatology. 2002;36:21-29.
- Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M Jr, Ye J. Hepatitis C virus production by
human hepatocytes dependent on assembly and secretion of very low-density lipoproteins.
Proc Natl Acad Sci U S A.2007;104:5848-5853.
- Icard V, Diaz O, Scholtes C, Perrin-Cocon L, Ramière C, Bartenschlager R, Penin F, Lotteau
V, André P. Secretion of hepatitis C virus envelope glycoproteins depends on assembly of
apoloprotein B positive lipoproteins. Plos. 2009;4:1-13.
- Imazeki F, Yokosuka O, Fukai K, Saisho H. Favorable prognosis of chronic hepatitis C after
interferon therapy by long-term cohort study. Hepatology. 2003;38:493-502.
-Jackel-Cram C, Babiuk LA, Liu Q. Up-regulation of fatty acid synthase promoter by
hepatitis C virus core protein: genotype-3a core has a stronger effect than genotype-1b core. J
Hepatol. 2007;46:985-1007.
- Kasai D, Adachi T, Deng L, Nagano-Fujii M, Sada K, Ikeda M, Kato N, Ide YH, Shoji I,
Hotta H. HCV replication suppresses cellular glucose uptake through down-regulation of
cell surface expression of glucose transporters. J Hepatol. 2009;50:883–894.
- Kato T, Furusaka A, Miyamoto M, Date T, Yasui K, Hiramoto J, Nagayama K, Tanaka T, Wakita T. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J Med Virol. 2001;64:334-339. - Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology. 2003; 125:1808-1817.

25
- Kawaguchi T, Takenoshita M, Kabashima T, Uyeda K. Glucose and cAMP regulate the Ltype pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc Natl Acad Sci U S A. 2001;98:13710-13715.
- Kawaguchi T, Yoshida T, Harada M, Hisamoto T, Nagao Y, Ide T, Taniguchi E, Kumemura
H, Hanada S, Maeyama M, Baba S, Koga H, Kumashiro R, Ueno T, Ogata H, Yoshimura A,
Sata M. Hepatitis C virus down-regulates insulin receptor subtrates 1 and 2 through up-
regulation of supressor of cytokine signalling 3. Am J Pathol. 2004;165:1499-1508.
- Lee YH, Giraud J, Davis RJ, White MF. c-Jun N-terminal kinase (JNK) mediates feedback
inhibition of the insulin signaling cascade. J Biol Chem. 2003;278:2896-2902.
-Lerat H, Honda M, Bears MR, Loesch K, Sun J, Yang Y, Okuda M, Gosert R, Xiao SY,
Weinman SA, Lemon SM. Steatosis and liver cancer in transgenic mice expressing the
structural and non structural proteins of hepatitis C virus. Gastroenterology. 2002;122:352-
365.
- Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. Pathogenesis, natural history, treatment,
and prevention of hepatitis C. Ann Intern Med. 2000;132:296-305.
- Maillard P, Huby T, Andréo U, Moreau M, Chapman J, Budkowska A. The interaction of
natural hepatitis C virus with human scavenger receptor SR-BI/Cla1 is mediated by ApoB-
containing lipoproteins. FASEB J. 2006;20:735-757.
- Mason AL, Lau JY, Hoang N, Qian K, Alexander GJ, Xu L, Guo L, Jacob S, Regenstein FG,
Zimmerman R, Everhart JE, Wasserfall C, Maclaren NK, Perrillo RP. Association of diabetes
mellitus ans chronic hepatitis C virus infection. Hepatology. 1999;29:328-333.
- McGivern DR, Lemon SM. Tumor suppressors, chromosomal instability, and hepatitis C
virus-associated liver cancer. Annu Rev Pathol. 2009;4:399-415.
- Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R,
Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis
C virus production. Nat Cell Biol. 2007;9:1089-1097.
- Moradpour D, Evans MJ, Gosert R, Yuan Z, Blum HE, Goff SP, Lindenbach BD, Rice CM.
Insertion of green fluorescent protein into nonstructural protein 5A allows direct
visualization of functional hepatitis C virus replication complexes. J Virol. 2004;78:7400-7409.
-Oem JK, Jackel-Cram C, Li YP, Zhou Y, Zhong J, Shimano H, Babiuk LA, Liu Q. Activation
of sterol regulatory element-binding protein 1c and fatty acid synthase transcription by
hepatitis C virus non-structural protein 2. J Gen Virol. 2008;89:1225-1230.
- Okabayashi Y, Maddux BA, McDonald AR, Logsdon CD, Williams JA, Goldfine ID.
Mechanisms of insulin-induced insulin-receptor downregulation. Decrease of receptor
biosynthesis ans mRNA levels. Diabetes. 1989;38:182-187.

26
- Park KJ, Choi SH, Choi DH, Park JM, Yie SW, Lee SY, Hwang SB. Hepatitis C virus NS5A protein modulates c-Jun N-terminal kinase through interaction with tumor necrosis factor receptor-associated factor 2. J Biol Chem. 2003;278:30711-30718. - Pazienza V, Clément S, Pugnale P, Conzelman S, Foti M, Mangia A, Negro F. The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology. 2007;45:1164-71. - Petit JM, Bour JB, Galland-Jos C, Minello A, Verges B, Guiguet M, Brun JM, Hillon P. Risk factors for diabetes mellitus and early insulin resistance in chronic hepatitis C. J Hepatol. 2001;35:279-283. - Pham TN, Michalak TI. Occult hepatits C virus persistence: identification and characteristics. MLO Med Lab Obs. 2006;38:20-22. -Pringle CR. Virus taxonomy. The universal system of virus taxonomy, updated to include
the new proposals ratified by the International Committee on Toxonomy of viruses during
1998. Arch Virol 1999;144:421-429.
- Radkowski M, Wilkinson J, Nowicki M, Adair D, Vargas H, Ingui C, Rakela J, Laskus T.
Search for hepatitis C virus negative-strand RNA sequences and analysis of viral sequences in
the central nervous system: evidence of replication. J Virol. 2002;76:600-608.
- Renström F, Burén J, Svensson M, Eriksson JW. Insulin resistance induced by high glucose
and high insulin precedes insulin receptor substrate 1 protein depletion in human
adipocytes. Metabolism. 2007;56:190-198.
-Rice CM. Flaviviridae : the viruses and their replication. In:Fields BN, Knipe DM, Howley
PM et al. Fields Virology, 3rd en, vol 1, Philadelphia: Lippincott-Raven. 1996:931-959.
- Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin
signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002;277:42394-
42398.
-Steff LB. Natural history of chronic hepatitis C. Hepatology. 2002;36:35-46.
- Sun CA, Wu DM, Lin CC, Lu SN, You SL, Wang LY, Wu MH, Chen CJ Incidence and
cofactors of hepatitis C virus-related hepatocellular carcinoma: a prospective study of 12,008
men in Taiwan. Am J Epidemiol. 2003;157:674-82.
-Taha C, Klip A. The insulin signaling pathway. J Membr Biol. 1999;169:1-12.
- Vento S, Cainelli F, Mirandola F, Cosco L, Di Perri G, Solbiati M, Ferraro T, Concia E.
Fulminant hepatitis on withdrawal of chemotherapy in carriers of hepatitis C virus. Lancet.
1996;347:92-93.

27
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, Bartenschlager R, Liang TJ. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005; 11: 791-796. - Waris G, Felmlee DJ, Negro F, Siddiqui A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J Virol. 2007;81:8122-8130. - Yamashita H, Takenoshita M, Sakurai M, Bruick RK, Henzel WJ, Shillinglaw W, Arnot D,Uyeda K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver.Proc Natl Acad Sci U S A 2001;98:9116-9121. - Zlande R, Mitchell JJ, Wu J, Sun XJ. Molecular mechanism of insulin-induced degradation
of insulin receptor substrate 1. Molecular and cellulair biology. 2002;22:1016-1026.