bioinformatique - fil
TRANSCRIPT

BioinformatiqueJ.-S.Varre
http://www.lifl.fr/˜varre/enseignement
Bioinformatique – p.1/??

Contenu
� les banques de données
� la comparaison de séquences
� l’alignement multiple
� quelques applications
� Introduction à la bioinformatique, O’REILLY
� Introduction to bioinformatics, Prentice Hall
� Cours d’Hélène Touzet, www.lifl.fr/˜touzet
Bioinformatique – p.2/??

Les banques de données nucléiques
International Nucleotide Sequence Database Collaboration
� trois partenaires
EMBLEurope
GenBankEtats-Unis
DDJBJapon
� contributions : chercheurs et programmes de séquençage
� mêmes données, mêmes formats
� taxonomie commune
� mises à jour quotidiennes
� 32 549 400 entrées, 37 893 844 733 bases (15 février 2004)
� +10 millions d’entrées en 1 an
Bioinformatique – p.3/??

Croissance de GenBank
Bioinformatique – p.4/??

Qu’y trouve-t-on ?
� ADN génomique
� des ARNm
� des ADNc : séquence artificelle issue de la transcription inverse d’unADNc
� des EST (Expressed Sequence Tags) : séquences nucléiquescorrespondant aux extrémités d’un ADNc obtenu à partir d’un ARNmdans un type cellulaire et des conditions données
Bioinformatique – p.5/??

Qu’y trouve-t-on ?
� des génomes1978 : séquence du phage phiX174 (premier génome à ADN,5386 bp)
� virus : plusieurs centaines� bactéries :
1995 : Haemophilus Influenzae1996 : Bacillus Subtilis1996 : Escherichia Coli
aujourd’hui : environ 100 génomes
� eucaryotes :
1990 : programme international Génome Humain1996 : levure (premier eucaryote)1998 : Caenorhabditis Elegans ( pluri-cellulaire)2000 : Arabidopsis Thaliana (premier génome de plante)2000 : brouillon du génome humain
aujourd’hui : souris, drosophile, rat, zebra fish, maïsBioinformatique – p.6/??

Une entrée : les informations générales
LOCUS HSA000073 17484 bp DNA linear PRI 09-DEC-2002
DEFINITION TPA: Homo sapiens lymphocyte-specific proteintyrosine kinase (lck) gene baseline reference(distal and proximal promoters, exon 1, 1’-12,3’ UTR).
ACCESSION BN000073
Version BN000073.1 GI:28317392
KEYWORDS Third Party Annotation; TPA; LCK gene;protein tyrosine kinase.
SOURCE Homo sapiens (human)
ORGANISM Homo sapiensEukaryota; Metazoa; Chordata; Craniata;Vertebrata; Euteleostomi; Mammalia; Eutheria;Primates; Catarrhini; Hominidae; Homo.
Bioinformatique – p.7/??

Une entrée : les références bibliographiques
REFERENCE 1
AUTHORS Nervi,S., Nicodeme,S., Gartioux,C., Atlan,C.,Lathrop,M., Reviron,D., Naquet,P., Matsuda,F.,Imbert,J. and Vialettes,B.
TITLE No association between lck gene polymorphismsand protein level in type 1 diabetes
JOURNAL Diabetes 51 (11), 3326-3330 (2002)
MEDLINE 22289034
...
Medline : base de données bibliographique
� 4500 revues depuis 1960: santé, biologie,biotechnologies
� accessible via Pubmed
Bioinformatique – p.8/??

Une entrée : annotation de la séquenceFEATURES Location/Qualifierssource 1..17484
/organism="Homo sapiens"/db_xref="taxon:9606"/note="baseline reference generated fromthe working draft sequence AL121991.33"
promoter <3319..4011/note="type II distal promoter"
promoter <4533..5218/note="type I proximal promoter"
gene 5428..16817/gene="LCK"
CDS join(5428..5532,5835..5916,6091..6181,6419..6517, 6667..6770,7012..7161,7435..7587,7705..7884,10772..10848, 10942..11095,11180..11311,16615..16817)/gene="LCK"/function="immune response"/codon_start=1/product="protein tyrosine kinase"/protein_id="CAD55807.1"/db_xref="GI:28317393"/translation="MGCGCSSHPEDDWMENIDVCENCHYPIVP...EELYQLMRLCWKERPEDRPTFDYLRSVLEDFFTATEGQYQPQP"
exon 5428..5532/gene="LCK"/number=1
intron 5533..5834/gene="LCK"/number=1
...polyA_signal 17240..17245polyA_site 17261
/evidence=experimentalBioinformatique – p.9/??

Une entrée : la séquence elle-même
BASE COUNT 4014 a 4351 c 4808 g 4211 t 100 others
ORIGIN1 ccctggggct actgaaattc caggcagtgg gtgaagagga cgaggaggat gaggaggggg
61 agagcctgga ctctgtgaag gcactgacag ccaagctgca gctgcagac t cggcggccct121 catatctgga gtggacagcc caggtccaga gccaggcctg gcgcaggg cc caagccaaac
. . .
4021 cccaggctcc ctagggatgc agcagccctt tgtggctggg gagagaa gat cctcgctcaa4081 ggtcagnnnn nnnnnnnnnn nnnnnnnnnn nnnnnnnnnn nnnnnnn nnn nnnnnnnnnn4141 nnnnnnnnnn nnnnnnnnnn nnnnnnnnnn nnnnnnnnnn nnnnnng ggt gtgtgcatga4201 atgtgtgtgt gggtacgtgt gtgagagtgg gtgcctgtgt gtggggg gtg agtgtgtgtg4261 gtgggggggc acttgtggag ggtgagtgta tgtgtttact gagtgtg agt gtgggtgcct4321 gtgtgtggga gggtgagtct gtgtgtgagt gtgtggggga gtacctg tga ggggtgagtg
. . .
5401 gccccctctt ccattccctc agggaccatg ggctgtggct gcagctc aca cccggaagat5461 gactggatgg aaaacatcga tgtgtgtgag aactgccatt atcccat agt cccactggat5521 ggcaagggca cggtaagagg cgagacaggg gccttggtga gggagtt ggg tagagaatgc5581 aacccaggag aaagaaatga ccagcactac aggcccttga aagaata gag tggccctctc
. . .
17281 ccactctttg tgggtgggca gtgggggtta agaaaatggt aattag gtca ccctgagttg17341 gggtgaaaga tgggatgagt ggatgtctgg aggctctgca gacccc ttca aatgggacag17401 tgctcctcac ccctccccaa aggattcagg gtgactccta cctgga atcc cttagggaat17461 gggtgcgtca aaggaccttc ctcc
Bioinformatique – p.10/??

Le format FASTA
>gi|28317392|tpe|BN000073.1|HSA000073 TPA: Homo sapien s lymphocyte-specific proteinCCCTGGGGCTACTGAAATTCCAGGCAGTGGGTGAAGAGGACGAGGAGGATGAGGAGGGGGAGAGCCTGGACTCTGTGAAGGCACTGACAGCCAAGCTGCAGCTGCAGACTCGGCGGCCCTCATATCTGGAGTGGACAGCCCAGGTCCAGAGCCAGGCCTGGCGCAGGGCCCAAGCCAAACCTGGACCAGGGGGACCTGGGGACATCTGTGGTTTCGACTCAATGGACTCCGCCCTTGAGTGGCTCCGACGGGAGCTGGTGAGTCTGGTGGGGTGGGCAGCTTTGCCCAGGGCCTTCAGGCTGGTCCTCCTTAGGGTCCAAGACCACAGGCATGGGGAAGGGATGAGAATGGACCCTGCCTCCAGCAACCGCCCGCTCCGGTGTCCTGGGCTGAGGAGCCGGGAATGGACTTGTCCAGTGTTGTATTCAGGGAGGCTTGCTGAAGAAGGATTCCATCCAATCTACTGACCTTGTGCTCCTGTTGCTTAGGGGACAGAGCACTGGTGTTGGGGGGTGTGGGGCACCATTATAAATAGGGAAATACACAGCCTCCCCTACTCTGGGACTTTCTATGGCCTAAAGATACAGCGAAGTGCAAAAGGAGGCACTGGCCATGCTGCAGGCAGATTCTGAAGAGGTGAAGTTTCAGTAGGTGGCTCCCAACTCAACATGTGCAAATATAAAGATACCCAGGCTCCAACACAGAGATTCTGCTTAGTTGATTTGGGATGGGGCCCAGGCAGTTGGATTTGCTTAATAATGACTCCTTAGCTGGGTTTGAGAACCACCAGGTGAAGCCAGATTGGGAGATAGGGAGAGATCAAGCTGAGGTGAGAGGGGGCTGGGGAGGACACAGTCAGTGCAAAGGCCCAGCGTTGCCAGGAAGGGAGAAACGGCGCGCGGCCGAGGGCCTGTCCAGTAGGCTCACGCCCCCTCTCGGCCGCAGCGGGAGATGCAGGCGCAGGACAGGCAGCTGGCAGGGCAGCTGCTGCGGCTGCGGGCCCAGCTGCACCGACTGAAGATGGACCAAGCCTGTCACCTGCACCAGGAGCTGCTGGATGAGGCCGAGCTGGAGCTGGAGCTGGAGCCCGGGGCCGGCCTAGCCCTGGCCCCGCTGCTGC...
Bioinformatique – p.11/??
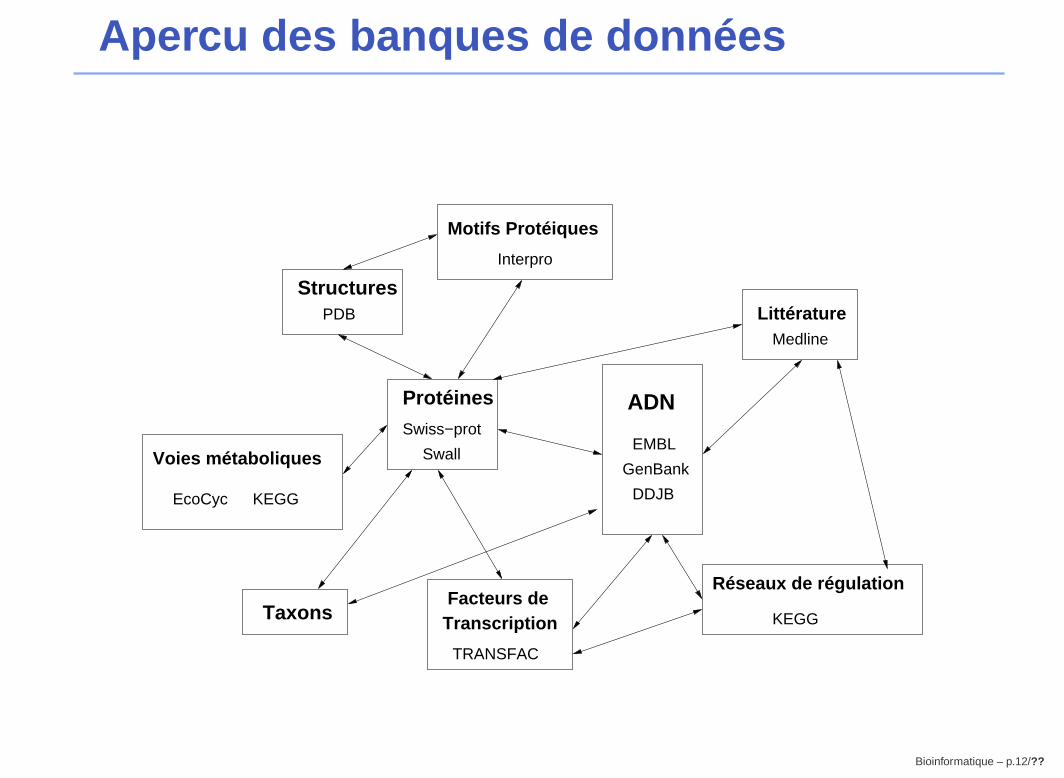
Apercu des banques de données
GenBank
ADN
EMBL
DDJB
Protéines
Swiss−prot
Swall
Facteurs de Transcription
TRANSFAC
Motifs Protéiques
Interpro
StructuresPDB Littérature
Medline
Voies métaboliques
KEGGEcoCyc
KEGG
Réseaux de régulation
Taxons
Bioinformatique – p.12/??

Pourquoi comparer des séquences ?
� l’évolution se fait par mutations successives
� homologie ⇒ similarité
� même séquence ⇒ même fonction ?
Bioinformatique – p.13/??

Exemple : l’insuline
el ephant FVNQHLCGSHLVEALYLVCGERGFFYTPKTGIVEQCCTGVCSLYQLENYCN||||||||||||||||||||||||||||| ||| |||| |||||||||||
hamster FVNQHLCGSHLVEALYLVCGERGFFYTPKSGIVDQCCTSICSLYQLENYCN
el ephant FVNQHLCGSHLVEALYLVCGERGFFYTPKTGIVEQCCTGVCSLYQLENYCN||||||||||||||||||||||||||||| ||||||| |||||||||||
baleine FVNQHLCGSHLVEALYLVCGERGFFYTPKAGIVEQCCASTCSLYQLENYCN
el ephant FVNQHLCGSHLVEALYLVCGERGFFYTPKTGIVEQCCTGVCSLYQLENYCN|| ||||||| ||||||||||||| || ||||||| |||||||||||
alligator AANQRLCGSHLVDALYLVCGERGFFYSPKGGIVEQCCHNTCSLYQLENYCN
Bioinformatique – p.14/??

Le dotplot
Bioinformatique – p.15/??

Dotplot
� un outil graphique pour la comparaison
� principe� mettre les séquences le long des axes d’une matrice� mettre un point là où il y a un match
une diagonale (une suite de points en diagonale) ⇒ une région similaire
Bioinformatique – p.16/??

Dotplot - exemple
match (identité) → �
mismatch → �
A C T C G A G C T A T C G
A � � �
C � � � �
T � � �
A � � �
T � � �
G � � �
A � � �
G � � �
A � � �
T � � �
A � � �
T � � �
G � � �
Bioinformatique – p.17/??

Dotplot
⇒ problème de bruit
Bioinformatique – p.18/??

Mise en pratique
� filtrage� les éléments ne sont pas traités un par un, mais par fenêtre (de taille
25 généralement)� seules les fenêtres avec un score suffisamment élévé sont retenues
→ élimination des similitudes courtes, nonsignificatives
� matrice de score� variation d’intensité : on mesure la similarité entre deux bases ou
deux acides aminés plus finement que par � et �
→ Prise en compte des propriétés des acidesaminés avec des matrices de pour les correspondances entre acides
aminés (PAM, BLOSUM)
Bioinformatique – p.19/??

Dotplot
Sans filtrage
Bioinformatique – p.20/??

Dotplot
Filtrage à 60%
Bioinformatique – p.20/??
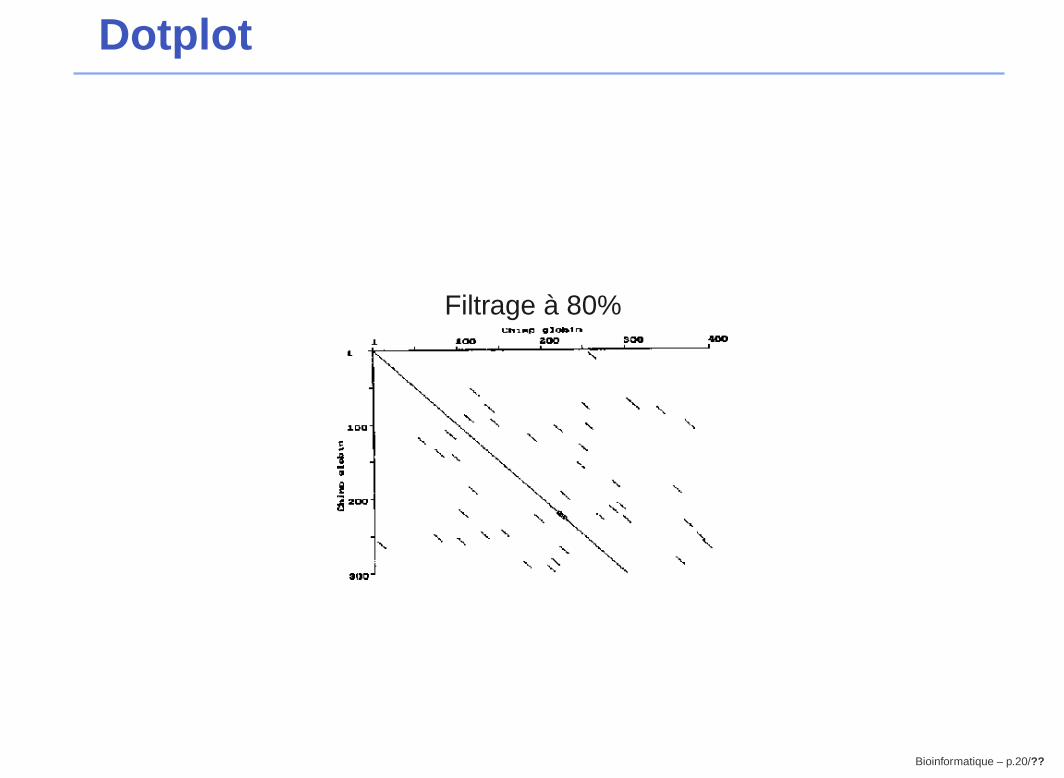
Dotplot
Filtrage à 80%
Bioinformatique – p.20/??

Dotplot
Repérer une similarité globale
horizontalement : ADN codant pour la chaîne α de l’hémoglobine humaineverticalement : ADN codant pour la chaîne β de l’hémoglobine humaine
Bioinformatique – p.21/??

Dotplot
Repérer des similarités locales
horizontalement : séquence nucléaire du gène de l’actine de muscle de Pisaster ochraceusverticalement : cDNA de ce même gène
Bioinformatique – p.22/??

Dotplot
Repérer des similarités locales
horizontalement : séquence nucléaire du gène de l’actine de muscle de Pisaster ochraceusverticalement : cDNA de ce même gène
Bioinformatique – p.22/??

Dotplot
Repérer des répétitions
protéine ribosomale S1 de Escherichia Coli sur elle-même
Bioinformatique – p.23/??

Dotplot
Repérer des répétitions
protéine ribosomale S1 de Escherichia Coli sur elle-même
Bioinformatique – p.23/??

Dotplot
Repérer des répétitions
protéine ribosomale S1 de Escherichia Coli sur elle-même
Bioinformatique – p.23/??

Avantages et inconvénients
� Les plus:� simple� très informatif
� Les moins:� interprétation ⇒ pas de mesure objective� identification ⇒ pas de méthode de détection automatique
⇒ besoin d’une mesure quantitative de similarité
Bioinformatique – p.24/??

Matrices de score
Bioinformatique – p.25/??

Matrice BLOSUM-62
# Matrix made by matblas from blosum62.iij# * column uses minimum score# BLOSUM Clustered Scoring Matrix in 1/2 Bit Units# Blocks Database = /data/blocks_5.0/blocks.dat# Cluster Percentage: >= 62# Entropy = 0.6979, Expected = -0.5209
A R N D C Q E G H I L K M F P S T W Y V B Z X *A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 -1 0 -4R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 0 -1 -4N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 0 -1 -4D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 1 -1 -4C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 -3 -2 -4Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 3 -1 -4E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 -2 -1 -4H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 0 -1 -4I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 -3 -1 -4L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 -3 -1 -4K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 0 1 -1 -4M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -3 -1 -1 -4F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -3 -3 -1 -4P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -2 -1 -2 -4S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 0 0 -4T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -1 -1 0 -4W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4 -3 -2 -4Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -3 -2 -1 -4V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -3 -2 -1 -4B -2 -1 3 4 -3 0 1 -1 0 -3 -4 0 -3 -3 -2 0 -1 -4 -3 -3 4 1 -1 -4Z -1 0 0 1 -3 3 4 -2 0 -3 -3 1 -1 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4X 0 -1 -1 -1 -2 -1 -1 -1 -1 -1 -1 -1 -1 -1 -2 0 0 -2 -1 -1 -1 -1 -1 -4* -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 1
Bioinformatique – p.26/??

Comment obtenir une telle matrice ?
� matrices log odds ratio
Sij = logqij
pipj
� exprime le ratio entre:la probabilité que deux résidus i et j soient alignés par descendanceet la probabilité que ceux-ci soient alignés par chance
� explication :� qij = la fréquence que l’alignement de i et j soit observé dans les
séquences� pi = la fréquence d’occurrence de i
� un score est > 0 si la proba d’un match significatif est > à la probad’un match aléatoire
⇒ matrices PAM et BLOSUM
Bioinformatique – p.27/??

Matrice BLOSUM
BLocks SUbstitutions MatricesHenikoff & Henikoff, 1992
� fréquence de changements entre deux acides aminés avec conservationde structureéchantillon : BLOCKS
� BLOSUM-N : seuil de similarité, N = % de similarité
� convient bien pour la recherche de similarités locales
� la plus courante : BLOSUM-62 (matrice par défaut de BLAST)
Bioinformatique – p.28/??
Matrice BLOSUM - Matériel
Bioinformatique – p.29/??

Matrice BLOSUM - Construction
1. élimination des séquences identiques à moins de n% ⇒ BLOSUM-n
2. calcul du nombre total de paires d’aa i et j: fij
qij =fij
# total de paires
3. calcul de la matrice log odds
Sij = logMij
fi
4. un score est > 0 si la proba d’un match significatif est > à la proba d’unmatch aléatoire
Bioinformatique – p.30/??

Alignement de 2 séquences
Bioinformatique – p.31/??

Alignement
� mise en correspondance de deux séquences (ADN ou protéines)
R D I S L V - - - K N A G I| | | | | | | |R N I - L V S D A K N V G I
� 3 événements mutationnels élémentaires :
• substitution
• insertion
• délétion
indel
� score� substitution : matrice de similarité� indel : pénalité
Le score de l’alignement est la somme des scores des événementsélémentaires.
Bioinformatique – p.32/??

Alignement
� 2 séquences → plusieurs alignements possiblesR D I S L V - - - K N A G I| | | | | | | |R N I - L V S D A K N V G I
R D I - - S L V K N A - - - G I| | | | | |R N I L V S - - - D A K N V G I
R D I - - S L V K N A G I| | | | | | |R N I L V S D A K N V G I
� bon/mauvais alignement ? ⇒ score
Mismatch : Match :
DN : 1AV, LD : 0
G, N : 6R, K : 5
A, I, L, S, V : 4
Indel : −5
Bioinformatique – p.33/??

Alignement global
Needleman & Wunsch - 1970
� évaluation d’une ressemblance globale entre deux séquences
� donn ees :
� deux séquences (nucléotides ou acides aminés)� des scores de similarité et des pénalités
� probl eme :
Quel est l’alignement de score maximal ?
Bioinformatique – p.34/??
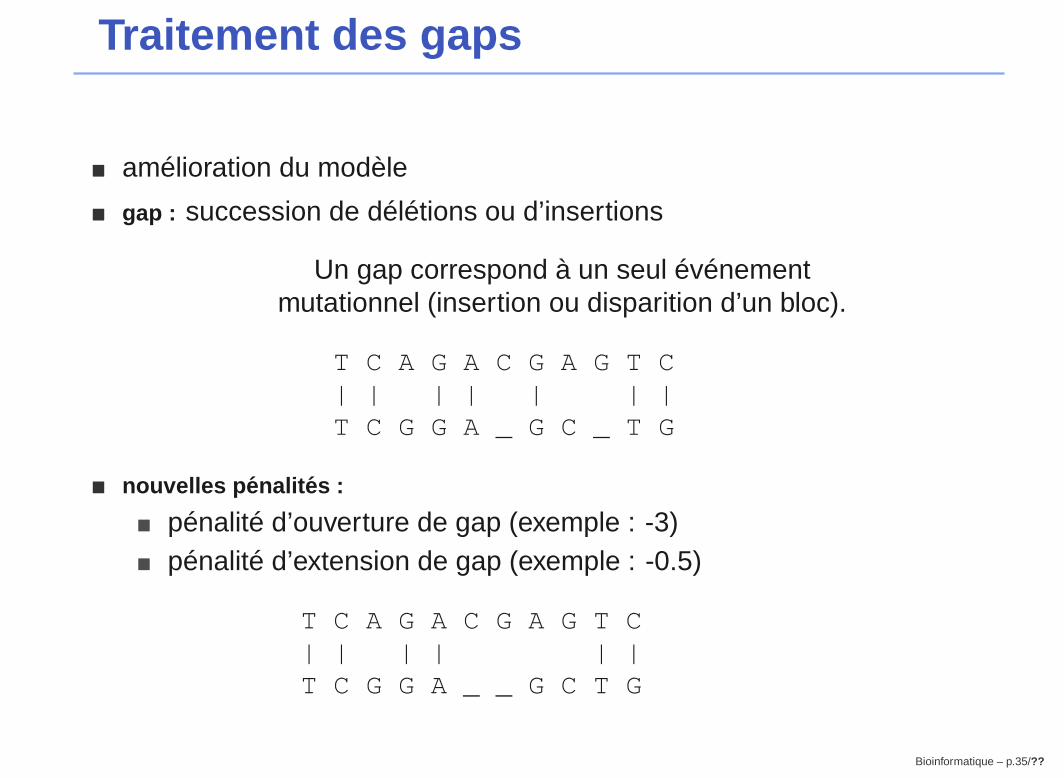
Traitement des gaps
� amélioration du modèle
� gap : succession de délétions ou d’insertions
Un gap correspond à un seul événementmutationnel (insertion ou disparition d’un bloc).
T C A G A C G A G T C| | | | | | |T C G G A _ G C _ T G
� nouvelles p enalit es :
� pénalité d’ouverture de gap (exemple : -3)� pénalité d’extension de gap (exemple : -0.5)
T C A G A C G A G T C| | | | | |T C G G A _ _ G C T G
Bioinformatique – p.35/??

Sensibilité aux paramètres
� match 2, mismatch -1, indel -1
A C G G C T - A T C| | | | | |A C T G - T A A T G
� match 1, mistmatch -1, indel -2
A C G G C T A T C| | | | |A C T G T A A T G
L’alignement optimal dépend de la matrice de similarité et despénalités pour les indels.
Bioinformatique – p.36/??

Alignement local
Smith & Waterman -1981
� donn ees :
� deux séquences (nucléotides ou acides aminés),� des scores de similarité.
� probl eme :
Quelles sont les régions de forte similarité entre les deux séquences ?
Bioinformatique – p.37/??

GGCTGACCACCTTGTA vs. GATCACTTCCATGGCAGTA
alignement global :1 G G C T G A C C A C C _ T T G T A - - - 16
| | | | | | | | |1 G A _ T C A C T T C C A T G G C A G T A 19
dotplot :G G C T G A C C A C C T T G T A
G ◦ ◦ � ◦
A � ◦ ◦
T ◦ ◦ ◦ ◦
C ◦ ◦ � ◦ ◦
A ◦ �
C ◦ ◦ ◦ ◦ �
T ◦ � ◦ ◦
T ◦ ◦ � ◦
C ◦ ◦ ◦
A ◦ ◦ ◦
T ◦ ◦ ◦ ◦
G ◦ ◦ ◦ ◦
G ◦ ◦ ◦ ◦
C ◦ ◦ ◦
A ◦ ◦ ◦
G ◦ ◦ ◦ �
T ◦ ◦ ◦ �
A ◦ ◦ �
similarité non révélée par l’alignement globalBioinformatique – p.38/??

Exemple
alignement local :
5 G A C C A C C T T 13| | | | | | |
1 G A T C A C _ T T 8
14 G T A 16| | |
17 G T A 19
Bioinformatique – p.39/??

Test de la robustesse du score
1. S : score de l’alignement entre U et V
2. génération de 200 (500, 1000, . . . )permutations aléatoires de V (même longueur , même composition)
3. alignements avec U
4. distribution des scores d’alignement
Où se situe S dans cette distribution ?
Bioinformatique – p.40/??

Exemple
Human alpha haemoglobin (141 aa) vs. Human myoglobin (153 aa )
VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF-DLS-----HGSAQVKGHGKKVADALTNAVAHVDDMPNALSAL:: .. : ..::::.:. ..:.:.: :.: . :.: . : .: .:. ..:...: :: .: .::.. . . .. .....:
GLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDKFKHLKSEDEMKASEDLKKHGATVLTALGGILKKKGHHEAEIKPL
SDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR------.. :: : .. ....:.:.. .:... :..:.........: :. .. ..:.:.AQSHATKHKIPVKYLEFISECIIQVLQSKHPGDFGADAQGAMNKALELFRKDMASNYKELGFQG
Chicken lysozyme (129 aa) vs. Bovine ribonuclease (124 aa)
KVFGRCELAAAMKRHGLDNYRGYSLGNWVCAAKFESNFNTQATNRNTDGSTDYGILQINSRWWCNDGRTP--GSRNLCNIPCSALLSSD: . :: ..:. .:. . . .. :.....:. :.. . ... .. .. .... :.. ... .... : .. :.. .:KETA----AAKFERQHMDSSTSAASSSNYCNQMMKSRNLTKDRCKPVNTFVHESLADVQAV--CSQKNVACKNGQTNCYQSYSTMSITD
ITASVNCAKKIVSDGDGMNAWVAWRNRCKGTDVQAWIRGCRL.. ...: .. .. .: :.:. . . ..
CRET-GSSKYPNCAYKTTQANKHIIVACEGNPYVPVHFDASV
Bioinformatique – p.41/??

PRSS sur les globin
< 20 0 0:22 0 0: one = represents 1 library sequences24 0 0:26 0 0:28 0 0:30 1 1:*32 3 3:==*34 9 7:======*==36 25 15:==============*==========38 37 25:========================*============40 29 34:============================= *42 33 42:================================= *44 51 46:============================================ =*=====46 41 47:========================================= *48 32 45:================================ *50 51 41:========================================*=== =======52 31 36:=============================== *54 24 31:======================== *56 30 26:=========================*====58 18 21:================== *60 24 17:================*=======62 19 14:=============*=====64 4 11:==== *66 10 9:========*=68 5 7:===== *70 4 5:====*72 7 4:===*===74 3 3:==*76 2 3:==*78 3 2:=*=80 1 2:=*82 0 1:*84 1 1:*86 0 1:*88 1 1:*90 0 0: unshuffled s-w score: 17792 1 0:= For 500 sequences, a score >= 177 is expected 3.096e-0 6 times94 0 0:
Bioinformatique – p.42/??

PRSS poulet/bovin
< 20 0 0:22 0 0: one = represents 1 library sequences24 0 0:26 0 0:28 0 0:30 0 1:*32 3 3:==*34 9 7:======*==36 17 15:==============*==38 24 25:========================*40 35 34:=================================*=42 44 42:=========================================*==44 57 46:============================================ =*===========46 50 47:============================================ ==*===48 44 45:============================================ *50 45 41:========================================*=== =52 25 36:========================= *54 28 31:============================ *56 20 26:==================== *58 24 21:====================*===60 17 17:================*62 10 14:========== *64 8 11:======== *66 10 9:========*=68 3 7:=== *70 6 5:====*=72 6 4:===*==74 2 3:==*76 2 3:==*78 2 2:=*80 5 2:=*===82 1 1:*84 0 1:*86 2 1:*=88 0 1:*90 0 0:92 0 0: unshuffled s-w score: 4394 1 0:= For 500 sequences, a score >= 43 is expected 267.1 time s96 0 0:
Bioinformatique – p.43/??

BLAST
Bioinformatique – p.44/??

BLAST
Basic Local Alignment Search ToolAltschul et al. - 1997
� programme pour la recherche de similarités dans les bases de données
� utilise un algorithme très rapide pour construire des alignements locauxapprochés
� séquences nucléiques et protéiques
� connecté aux principales banques de données
Bioinformatique – p.45/??

Exemple de résultat
Query= Felis catus DRD4 gene fordopamine receptor D4(276 letters)
Database: All GenBank+EMBL+DDBJ+PDB sequences1,174,453 sequences; 5,001,591,585 total letters
Score ESequences producing significant alignments: (bits) Value
gi|AB069665 Felis catus DRD4 gene f... 210 5e-52
gi|AB069662 Nyctereutes procyonoide... 157 7e-36
gi|AB069661 Canis lupus DRD4 gene f... 157 7e-36
gi|AB069666 Bos taurus DRD4 gene fo... 143 1e-31
gi|291947| Homo sapiens Dopamine D4 recep... 135 2e-29
Bioinformatique – p.46/??

Exemple de résultats
>gi|18143632|dbj|AB069662.1|AB069662 Nyctereutes proc yonoides
DRD4 gene fordopamine receptor D4. Length = 393
Score = 157 bits (79), Expect = 7e-36Identities = 94/99 (94%)Strand = Plus / Plus
Query 1 ttcttcctaccctgcccgctcatgctgctgctctactgggccacg ttcc 48|||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct 1 ttcttcctaccctgcccgctcatgctgctgctctactgggccacg ttcc 48
Query 49 ggggcctgcggcgctgggaggcggctcgccaggccaagctgcac tgccgg 99|||||||||||||||||||||| || || | ||||||||||||| |||||
Sbjct 49 ggggcctgcggcgctgggaggccgcgcgtcgggccaagctgcac ggccgg 99
Score = 107 bits (54), Expect = 5e-21Identities = 60/62 (96%)Strand = Plus / Plus
Query 215 ggaggcgcgccaagatcaccggccgggagcgcaaggccatgag ggtcct 252|||| |||||||||||||| |||||||||||||||||||||||||||||
Sbjct 332 ggagacgcgccaagatcacgggccgggagcgcaaggccatgag ggtcct 379
Query 253 tgccggtggtggtc 276||||||||||||||
Sbjct 380 tgccggtggtggtc 393
Bioinformatique – p.47/??

Alignement Felis Catus/ Nyctereute
1 ttcttcctaccctgcccgctcatgctgctgctctactgggccacg | 145 g gcgagc......................................
||||||||||||||||||||||||||||||||||||||||||||| | ||| || |
1 ttcttcctaccctgcccgctcatgctgctgctctactgggccacg | 181 g gc.agcccggacggcacccccggcccgccgccccccgacggcac
|
46 ttccggggcctgcggcgctgggaggcggctcgccaggccaagctg | 152 ............................................c
|||||||||||||||||||||||||| || || | |||||||||| | |
46 ttccggggcctgcggcgctgggaggccgcgcgtcgggccaagctg | 225 ccccgatgacacccccgacgccaccccctgccccccgccccccgc
|
91 cactgccgggcgcctcgtcggcccagcggccccggcccaccgccc | 153 ccccgacgccgtcgcgccccccgacgccgtcccagccgagccgcc
||| ||||| | || || | ||||||||||||||||| || ||| | ||||||||||| ||| |||||||| ||||| ||| || ||||| ||
91 cacggccggacaccgcgcagacccagcggccccggcccgccaccc | 270 ccccgacgccgccgcgccccccgccgccgaccctgcggagccccc
|
136 cccga.ggt...................................c | 19 8 gcggcaggcacccaggaggaggcgcgccaagatcaccggccggga
||||| ||| | | | ||||| ||| || | |||| |||||||||||||| ||||||||
136 cccgacggtacccccggccccccgccccccgacggcagccccgac | 31 5 gtggcagccacgcaagcggagacgcgccaagatcacgggccggga
|
| 243 gcgcaaggccatgagggtcctgccggtggtggtc
| ||||||||||||||||||||||||||||||||||
| 360 gcgcaaggccatgagggtcctgccggtggtggtc
Bioinformatique – p.48/??

Raccoon-dog
Bioinformatique – p.49/??
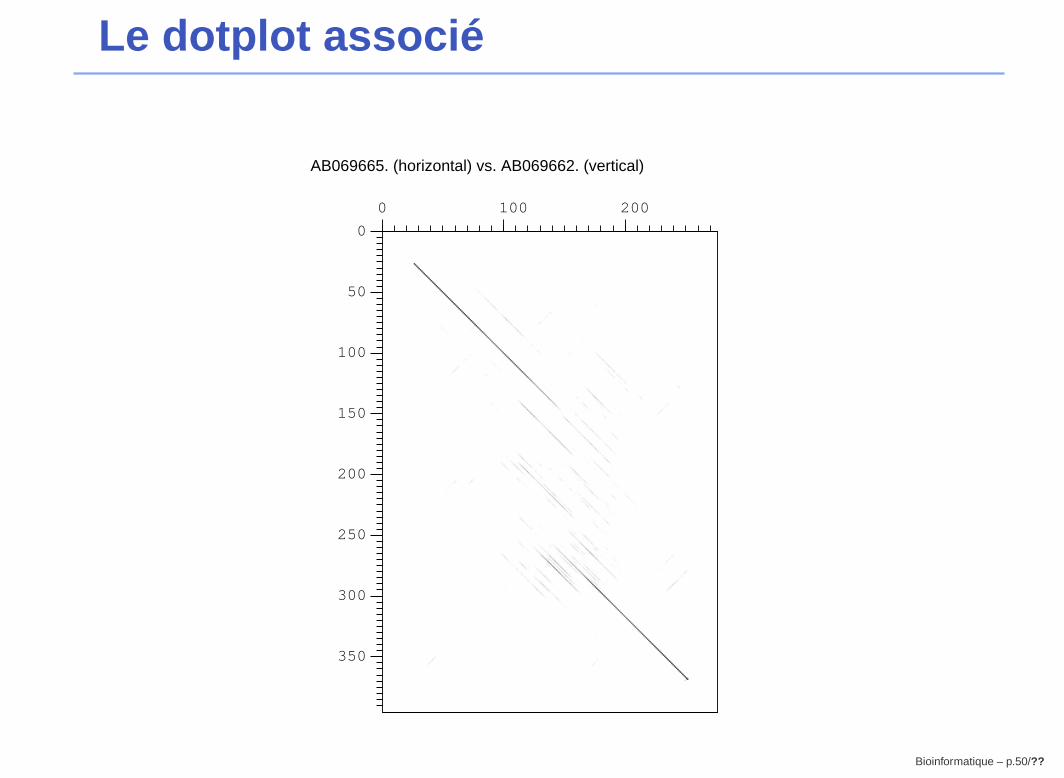
Le dotplot associé
AB069665. (horizontal) vs. AB069662. (vertical)
0 100 200
0
50
100
150
200
250
300
350
Bioinformatique – p.50/??

Alignement multiple
Bioinformatique – p.51/??

Définition de l’alignement multiple
� entrée : k séquences
* * * * * * * * * * * * ** * * * * * * * * ** * * * * * * * * * * * ** * * * * * * * * *
� sortie : un tableau contenant les k séquences, avec des indels
* * * * * * * * * - * * * ** * * - - - * * * - * * * ** * * - * * * * * * * * * ** * * - - * * - - * * * * *
Bioinformatique – p.52/??

Al. multiple vs al 2 à 2
Alignement 2 à 2
Deux séquences quelconques↓
Détecter une similarité syntaxique↓
Il y a-t-il une fonction commune ?
Alignement multiple
Famille de séquences avec la même fonction↓
À quelle conservation syntaxique cela correspond-il ?
Bioinformatique – p.53/??

Application 1 : modélisation demotifs
Bioinformatique – p.54/??

Doigt de zinc
TYY1_HUMAN/383-407 YVCPF-DGCN---KKFAQSTNLKSHILT---HYKQ8_CAEEL/78-102 YKCT---VCR---KDISSSESLRTHMFKQ-HHBASO_HUMAN/719-742 FQCD---ICK---KTFKNACSVKIHHKN--MHZG29_XENLA/62-84 FVCT---VCG---KTYKYKHGLNTHLHS---HP43_XENBO/106-130 LKCSV-PGCK---RSFRKKRALRIHVSE---HIKAR_MOUSE/488-512 FECN---MCG---YHSQDRYEFSSHITRG-EHQ92610/1043-1069 YTCG---YCTEDSPSFPRPSLLESHISL--MHTRA1_CAEEL/306-331 YKCEF-ADCE---KAFSNASDRAKHQNR--THZN10_HUMAN/383-405 YKCN---QCG---IIFSQNSPFIVHQIA---HGLI1_XENLA/283-310 FVCHW-QDCSRELRPFKAQYMLVVHMRR---HXFIN_XENLA/276-298 FRCS---ECS---RSFTHNSDLTAHMRK---HTF3A_BUFAM/72-97 CKCET-ENCN---LAFTTASNMRLHFKR--AHZG58_XENLA/174-196 FVCT---ECN---LSFAGLANLRSHQHL---HP43_XENBO/163-187 YRCSY-EDCQ---TVSPTWTALQTHLKK---HTSH_DROME/354-378 FRCV---WCK---QSFPTLEALTTHMKDS-KHZN76_HUMAN/165-189 FRCGY-KGCG---RLYTTAHHLKVHERA---HTF3A_BUFAM/219-244 YRCPR-ENCD---RTYTTKFNLKSHILT--FHSUHW_DROAN/349-373 YACK---ICG---KDFTRSYHLKRHQKYS-SCZN76_HUMAN/285-309 YTCPE-PHCG---RGFTSATNYKNHVRI---HSRYC_DROME/469-492 FKCN---YCP---RDFTNFPNWLKHTRR--RHEVI1_HUMAN/761-784 YRCK---YCD---RSFSISSNLQRHVRN--IH...
extrait de Pfam, entrée zf-C2H2
Bioinformatique – p.55/??
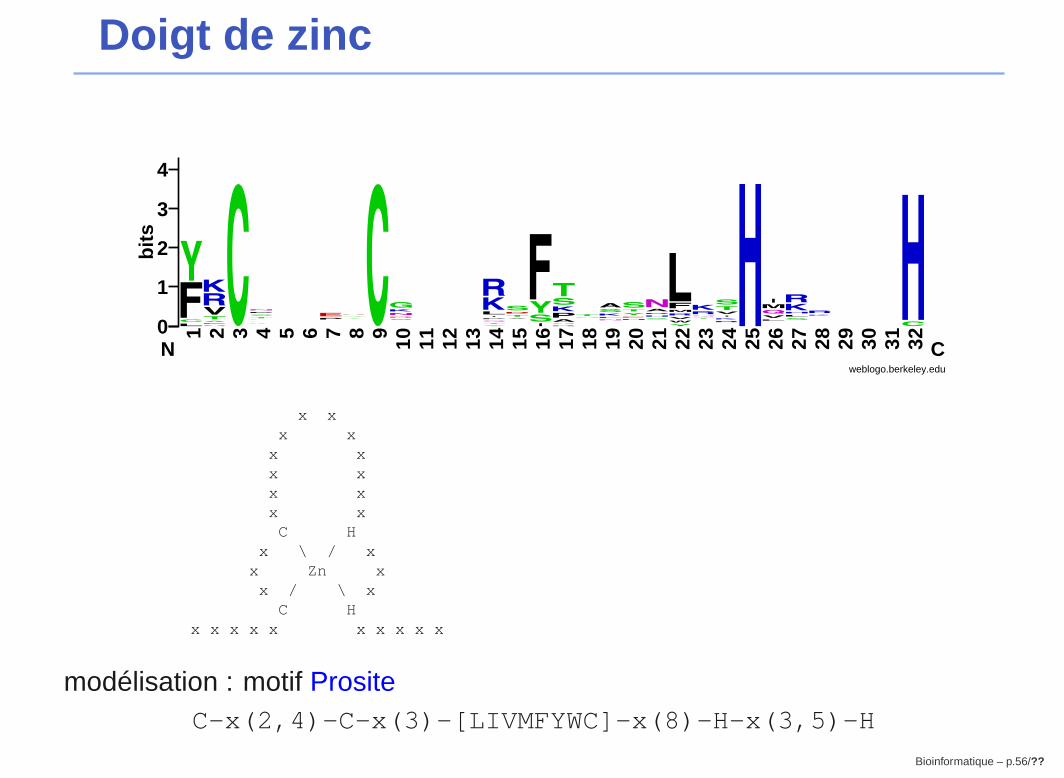
Doigt de zinc
weblogo.berkeley.edu
0
1
2
3
4bi
ts
N
1
L
C
FY
2
Q
E
A
T
VRK
3
C4
K
G
E
T
S
P
N
5 6 7P
E
8
Y
G
D
9
C10
S
D
N
K
G
11 12 13 14
Y
T
Q
P
I
L
KR
15
A
T
D
S
16
I
SYF
17
R
Q
A
PK
ST
18
N
T
19
N
L
K
S
A
20
H
E
Y
T
S
21
S
H
D
A
N
22
Y
W
V
R
M
FL
23
T
Q
R
K
24
R
K
I
V
T
S
25
H
26
L
V
Q
M
I
27
S
L
H
KR
28
K
R
29 30 31 32
C
HC
x xx x
x xx xx xx x
C Hx \ / x
x Zn xx / \ x
C Hx x x x x x x x x x
modélisation : motif PrositeC-x(2,4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3,5)-H
Bioinformatique – p.56/??

Site de fixation de la cellulose
CEL1_AGABI/292-319 CGGig---wtGgtgCvapyqCkv--iNdYysQCFAEB_PENFN/325-352 CGGig---ysGctaCaspytCqk--aNdYysQCGUN1_TRILO/435-462 CGGig---ytGcktCtsgttCqy--gNdYysQCGUN1_TRIRE/431-458 CGGig---ysGcktCtsgttCqy--sNdYysQCGUN2_TRIRE/29-56 CGGig---wsGptnCapgsaCst--lNpYyaQCGUN3_HUMIN/24-51 CGGvg---fsGstsCvsgytCvy--lNdWysQCGUN4_TRIRE/315-342 CGGsg---ysGptrCappatCst--lNpYyaQCGUN5_TRIRE/213-240 CGGag---wtGpttCqapgtCkv--qNqWysQCGUNB_FUSOX/33-60 CGGqn---wsGtpcCtsgnkCvk--lNdFysQCGUNF_FUSOX/25-52 CGGng---wtGattCasglkCek--iNdWyyQCGUNK_FUSOX/343-373 CGGsksaypnGnlaCatgskCvk--qNeYysQCGUX1_HUMGR/497-524 CGGig---ftGptqCeepyiCtk--lNdWysQCGUX1_PENJA/509-536 CGGng---wtGpttCvspytCtk--qNdWysQCGUX1_PHACH/488-515 CGGig---ytGsttCaspytChv--lNpYysQCGUX1_TRIRE/485-512 CGGig---ysGptvCasgttCqv--lNpYysQCGUX1_TRIVI/485-512 CGGig---yiGptvCasgstCqv--lNpYysQCGUX2_AGABI/478-505 CGGqg---wtGptaCqspstChv--iNdFysQCGUX2_TRIRE/34-61 CGGqn---wsGptcCasgstCvy--sNdYysQCGUX3_AGABI/28-55 CGGng---wtGpttCasgstCvk--qNdFysQCGUXC_FUSOX/486-513 CGGqn---ysGpttCkspftCkk--iNdFysQCPSBP_PORPU/30-57 CGGig---fdGvtcCseglmCmk--mGpYysQCPSBP_PORPU/73-100 CGGmn---ysGktmCspgfkCve--lNeFfsQCPSBP_PORPU/132-160 CGGem---fmGakcCkfglvCye-tsGkWqsQCPSBP_PORPU/176-205 CGGmg---ymGstmCvggykCmaiseGsMykQC
extrait de Prosite, entrée PS00562Bioinformatique – p.57/??

Site de fixation de la cellulose
weblogo.berkeley.edu
0
1
2
3
4
bits
N
1
C2
G3
G4
V
E
A
S
M
N
QI
5
M
K
NG
6 7 8 9
P
FWY
10
N
I
D
M
TS
11
G12
V
T
N
K
G
A
S
C
P
13
P
L
KT
14
V
M
A
C
T
15
C
16
E
S
Q
K
TVA
17
T
G
F
E
A
PS
18
PG
19
N
G
A
F
T
L
SY
20
V
Q
M
I
A
KT
21
C
22
T
S
M
H
K
Q
V
23
A
T
E
YVK
24 25 26
M
G
E
A
S
QIL
27
GN
28
S
Q
K
E
PD
29
M
FWY
30
Q
F
Y
31
Y
K
A
S
32
Q
33
CC
C-G-G-x(4,7)-G-x(3)-C-x(5)-C-x(3,5)-[NHG]-x-[FYWM]-x(2)-Q-C
+----------------+| +-----|---------+| | | |
xxxxxxCxxxxxxxxxxCxxxxxCxxxxxxxxxCx****************************
les 4 cystéines sont impliquées dans des liaisons di-sulfures.
Bioinformatique – p.58/??

Site de fixation du fact. de transcription SP1
ctccgcccgaaccccgcccacaccccgcccccaccccgcccccaccccgcccccgccacgccccca
A C G T
1 0 6 0 0
2 0 5 0 1
3 1 5 0 0
4 0 6 0 0
5 0 0 6 0
6 0 6 0 0
7 0 6 0 0
8 0 6 0 0
9 1 4 1 0
Bioinformatique – p.59/??

Application 2 : Structure d’ARN
Bioinformatique – p.60/??

Le principe
G A G C C C A G U U C
A G G A C U C U U C
A A U C A C C C G A U
Changement de base compensatoire:
Quand une base impliquée dans un appariement mute, la basecomplémentaire mute également, pour préserver la paire, et donc, la
structure secondaire.
Bioinformatique – p.61/??

Méthode des covariations
Etape 1 : construction d’un alignement multiple
G A G C − C C A G U U C
− A G G A C − U C U U C
A A U C A C C C G A U −
− A G G A C − U C U U C
Etape 2 : détection des positions corréléesprésomption d’appariement
Bioinformatique – p.62/??
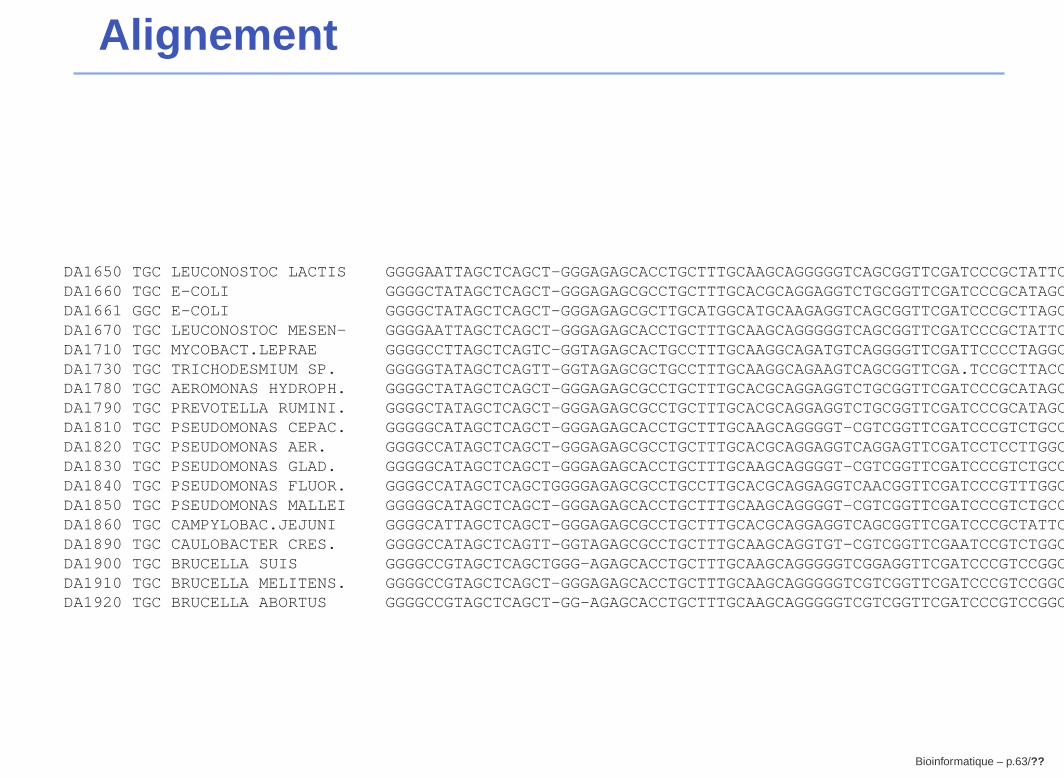
Alignement
DA1650 TGC LEUCONOSTOC LACTIS GGGGAATTAGCTCAGCT-GGGAGAGCACCTGCTTTGCAAGCAGGGGGTCAGCGGTTCGATCCCGCTATTCTCCA---DA1660 TGC E-COLI GGGGCTATAGCTCAGCT-GGGAGAGCGCCTGCTTTGCACGCAGGAGGTCTGCGGTTCGATCCCGCATAGCTCCACCADA1661 GGC E-COLI GGGGCTATAGCTCAGCT-GGGAGAGCGCTTGCATGGCATGCAAGAGGTCAGCGGTTCGATCCCGCTTAGCTCCACCADA1670 TGC LEUCONOSTOC MESEN- GGGGAATTAGCTCAGCT-GGGAGAGCACCTGCTTTGCAAGCAGGGGGTCAGCGGTTCGATCCCGCTATTCTCCA---DA1710 TGC MYCOBACT.LEPRAE GGGGCCTTAGCTCAGTC-GGTAGAGCACTGCCTTTGCAAGGCAGATGTCAGGGGTTCGATTCCCCTAGGCTCCA---DA1730 TGC TRICHODESMIUM SP. GGGGGTATAGCTCAGTT-GGTAGAGCGCTGCCTTTGCAAGGCAGAAGTCAGCGGTTCGA.TCCGCTTACCCCCA---DA1780 TGC AEROMONAS HYDROPH. GGGGCTATAGCTCAGCT-GGGAGAGCGCCTGCTTTGCACGCAGGAGGTCTGCGGTTCGATCCCGCATAGCTCCACCADA1790 TGC PREVOTELLA RUMINI. GGGGCTATAGCTCAGCT-GGGAGAGCGCCTGCTTTGCACGCAGGAGGTCTGCGGTTCGATCCCGCATAGCTCCACCADA1810 TGC PSEUDOMONAS CEPAC. GGGGGCATAGCTCAGCT-GGGAGAGCACCTGCTTTGCAAGCAGGGGT-CGTCGGTTCGATCCCGTCTGCCTCCACCADA1820 TGC PSEUDOMONAS AER. GGGGCCATAGCTCAGCT-GGGAGAGCGCCTGCTTTGCACGCAGGAGGTCAGGAGTTCGATCCTCCTTGGCTCCACCADA1830 TGC PSEUDOMONAS GLAD. GGGGGCATAGCTCAGCT-GGGAGAGCACCTGCTTTGCAAGCAGGGGT-CGTCGGTTCGATCCCGTCTGCCTCCACCADA1840 TGC PSEUDOMONAS FLUOR. GGGGCCATAGCTCAGCTGGGGAGAGCGCCTGCCTTGCACGCAGGAGGTCAACGGTTCGATCCCGTTTGGCTCCA---DA1850 TGC PSEUDOMONAS MALLEI GGGGGCATAGCTCAGCT-GGGAGAGCACCTGCTTTGCAAGCAGGGGT-CGTCGGTTCGATCCCGTCTGCCTCCACCADA1860 TGC CAMPYLOBAC.JEJUNI GGGGCATTAGCTCAGCT-GGGAGAGCGCCTGCTTTGCACGCAGGAGGTCAGCGGTTCGATCCCGCTATTCTCCACCADA1890 TGC CAULOBACTER CRES. GGGGCCATAGCTCAGTT-GGTAGAGCGCCTGCTTTGCAAGCAGGTGT-CGTCGGTTCGAATCCGTCTGGCTCCACCADA1900 TGC BRUCELLA SUIS GGGGCCGTAGCTCAGCTGGG-AGAGCACCTGCTTTGCAAGCAGGGGGTCGGAGGTTCGATCCCGTCCGGCTCCACCADA1910 TGC BRUCELLA MELITENS. GGGGCCGTAGCTCAGCT-GGGAGAGCACCTGCTTTGCAAGCAGGGGGTCGTCGGTTCGATCCCGTCCGGCTCCACCADA1920 TGC BRUCELLA ABORTUS GGGGCCGTAGCTCAGCT-GG-AGAGCACCTGCTTTGCAAGCAGGGGGTCGTCGGTTCGATCCCGTCCGGCTCCACCA
Bioinformatique – p.63/??

Structure de l’ARNt
Bioinformatique – p.64/??

Application 3 : phylogéniemoléculaire
Bioinformatique – p.65/??

Problématique
� retracer l’historique des espèces à partir des mutations observées
� données : gènes communs aux familles étudiées, pas trop divergents
� résultat : classification sous forme d’arbre phylogénétique
Bioinformatique – p.66/??

Méthodes de parcimonie
Rasoir d’Occam
"Pluralitas non estponenda sine neccesitate"
� privilégier l’arbre qui minimise le nombre de mutations
� le nombre global de mutations est obtenu en faisant la somme desmutations le long de chaque branche
Bioinformatique – p.67/??

Exemple
1 A A G A G T G C A2 A G C C G T G C G3 A G A T A T C C A4 A G A G A T C C G
3 arbres non enracinés possiblesInconv enient : long
Bioinformatique – p.68/??
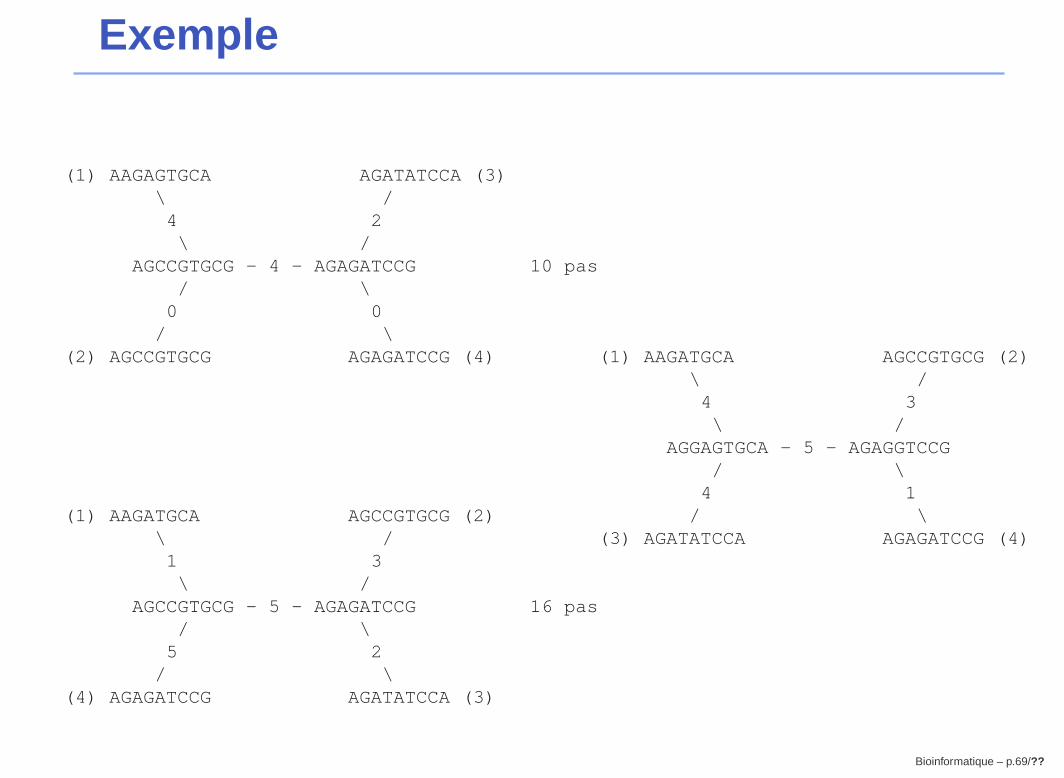
Exemple
(1) AAGAGTGCA AGATATCCA (3)\ /
4 2\ /
AGCCGTGCG - 4 - AGAGATCCG 10 pas/ \
0 0/ \
(2) AGCCGTGCG AGAGATCCG (4) (1) AAGATGCA AGCCGTGCG (2)\ /
4 3\ /
AGGAGTGCA - 5 - AGAGGTCCG 14 pas/ \
4 1(1) AAGATGCA AGCCGTGCG (2) / \
\ / (3) AGATATCCA AGAGATCCG (4)1 3
\ /AGCCGTGCG - 5 - AGAGATCCG 16 pas
/ \5 2
/ \(4) AGAGATCCG AGATATCCA (3)
Bioinformatique – p.69/??

Méthodes de distance
� point de départ : alignement multiple
� matrice de toutes les distances deux à deux
� classification hiérarchique
On construit l’arbre à partir des feuilles en regroupantprogressivement les noeuds 2 à 2 pour former des clusters .
Bioinformatique – p.70/??

Exemple : UPGMA
Unweight Pair Group Method with Arithmetic mean
� à chaque étape, on regroupe les deux clusters les plus proches
� la distance est calculée en faisant la moyenne arithmétique
Reprise de l’exemple précédentMatrice initiale Après une itération
1 2 3 4
1 0 4 5 6
2 0 5 4
3 0 2
4 0
1 2 3 + 4
1 0 4 5, 5
2 0 4, 5
3 + 4 0
Bioinformatique – p.71/??

Construction d’un alignementmultiple
Bioinformatique – p.72/??

Clustal
Thompson et al. - 1994
CLUSTAL = cluster + alignement
Etape 1 : alignements globaux 2 à 2Etape 2 : arbre phylogénétique (clusters)Etape 3 : alignement multiple obtenu par
combinaisons des alignements 2 à 2
Exemple :
s1 cgatgagtcattgtgactg
s2 cgagccattgtagctactg
s3 cgaccattgtagctacctg
s4 cgatgagtcactgtgactg
indel : -2, substitution : -1, identité : 1Bioinformatique – p.73/??

Etape 1
Calcul des scores de similarité de tous les alignements globaux 2 à 2
s1 cgatgagtcattgt-g--actg s2 cgagccattgtagcta-ctg||| | |||||| | |||| ||| |||||||||||| |||
s2 cga-g--ccattgtagctactg s3 cga-ccattgtagctacctg
s1 cgatgagtcattg-tgactg s2 cga-g--ccattgtagctactg||| | | | | | ||| ||| | || ||| | ||||
s3 cgacca-ttgtagctacctg s4 cgatgagtcactgt-g--actg
s1 cgatgagtcattgtgactg s3 cgaccattgtagctacctg|||||||||| |||||||| ||| | | | |||
s4 cgatgagtcactgtgactg s4 cgatgagtcactgtgactg
Tableau des scores d’alignement:
s1 s2 s3 s4
s1 2 0 17
s2 2 14 0
s3 0 14 −1
s4 17 0 −1
n séquences
↓
n(n − 1)/2 calculs
Bioinformatique – p.74/??
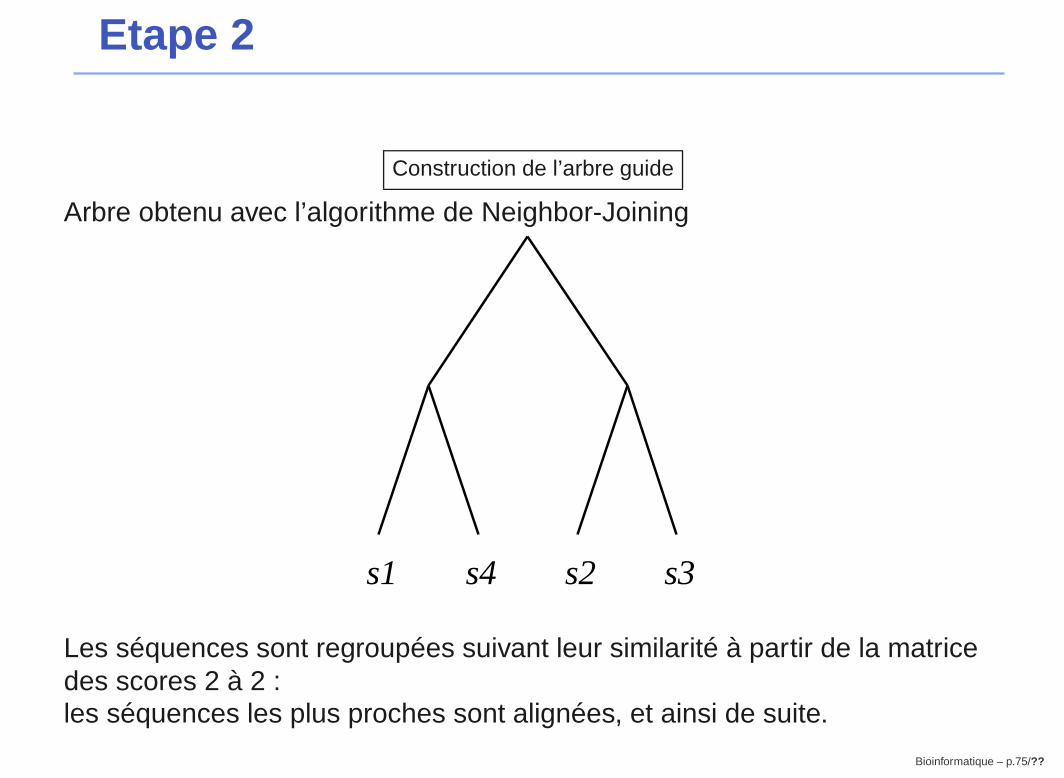
Etape 2
Construction de l’arbre guide
Arbre obtenu avec l’algorithme de Neighbor-Joining
s4s1 s2 s3
Les séquences sont regroupées suivant leur similarité à partir de la matricedes scores 2 à 2 :les séquences les plus proches sont alignées, et ainsi de suite.
Bioinformatique – p.75/??

Etape 3
Construction de l’alignement multiple final
Alignement progressif des séquences, en les incorporant dans l’ordre del’arbre guide.
s1 cgatgagtcattgtgactg|||||||||| ||||||||
s4 cgatgagtcactgtgactg
s2 cgagccattgtagcta-ctg||| |||||||||||| |||
s3 cga-ccattgtagctacctg↓s2 CGA---GCCATTGTAGCTAC-TGs3 CGA----CCATTGTAGCTACCTGs1 CGATGAGTCATTGT-G--AC-TGs4 CGATGAGTCACTGT-G--AC-TG
Once a gap, always a gap
Bioinformatique – p.76/??