bases moléculaires des surdités héréditaires chez les patients marocains
TRANSCRIPT

Bases moléculaires des surdités héréditaires chez les patients
marocains
Institut Pasteur Tunis
Le 20 Octobre 2016

Projets de Recherches
• Bases moléculaire des Surdités héréditaires chez la population Marocaine
• Mediterranean Autism Project • Étude moléculaire du Diabète de type 2 et des syndromes
métaboliques • The impact of micro-RNAs and inflammatory pathways on stem
cell fate and the regenerative process in human inherited muscle disease
• Genodermatoses • Rôle du chromosome Y dans la détermination sexuelle et dans
l'infertilité masculine.
e du chromosome Y dans la détermination sexuelle et dans l'infertilité masculine.

3 3
Déficit neurosensoriel le plus fréquent
1/1000 enfant, 2.7/1000 âge < 5 ans , 3.5/1000 durant l’adolescence
Conséquences :
• Acquisition normale du langage
• Apprentissage scolaire,
• Intégration sociale et professionnelle
Surdité héréditaire
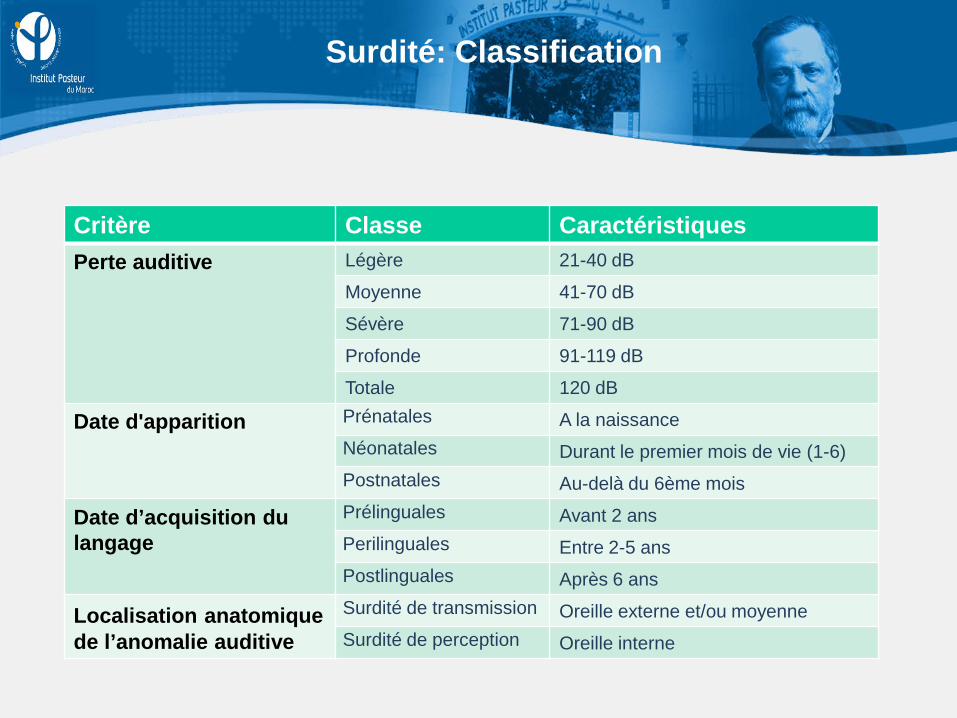
Surdité: Classification
Critère Classe Caractéristiques Perte auditive Légère 21-40 dB
Moyenne 41-70 dB
Sévère 71-90 dB
Profonde 91-119 dB
Totale 120 dB
Date d'apparition Prénatales A la naissance Néonatales Durant le premier mois de vie (1-6) Postnatales Au-delà du 6ème mois
Date d’acquisition du langage
Prélinguales Avant 2 ans Perilinguales Entre 2-5 ans Postlinguales Après 6 ans
Localisation anatomique de l’anomalie auditive
Surdité de transmission Oreille externe et/ou moyenne Surdité de perception Oreille interne

Maladies monogéniques hétérogènes
Présence de manifestations cliniques avec:
• Atteintes d'autres organes,
• Degré et âge d'apparition du déficit auditif,
• Mode de transmission. Syndromique (30%) et non syndromique (70%).
Familiale et sporadique.
Liste de tous les gènes de surdité mises à jour sur le site «hereditary hearing
loss home page” (http://dnalab-www.uia.ac.be/dnalab/hhh/)
Surdité héréditaire
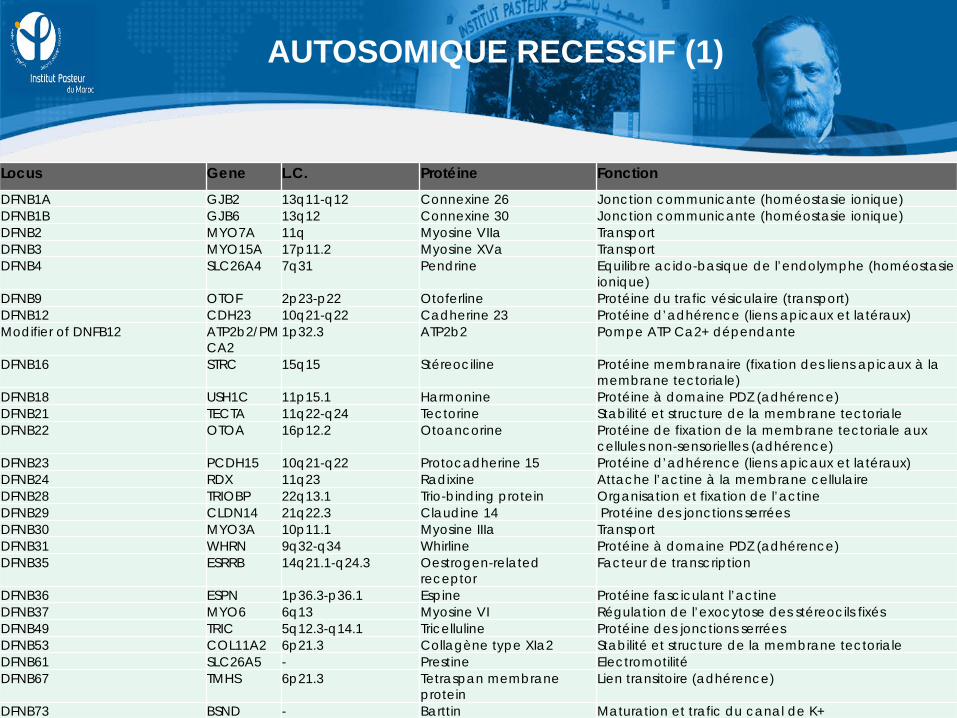
Locus Gene L.C. Protéine Fonction
DFNB1A GJB2 13q11-q12 Connexine 26 Jonction communicante (homéostasie ionique) DFNB1B GJB6 13q12 Connexine 30 Jonction communicante (homéostasie ionique) DFNB2 MYO7A 11q Myosine VIIa Transport DFNB3 MYO15A 17p11.2 Myosine XVa Transport DFNB4 SLC26A4 7q31 Pendrine Equilibre acido-basique de l’endolymphe (homéostasie
ionique) DFNB9 OTOF 2p23-p22 Otoferline Protéine du trafic vésiculaire (transport) DFNB12 CDH23 10q21-q22 Cadherine 23 Protéine d’adhérence (liens apicaux et latéraux) Modifier of DNFB12 ATP2b2/PM
CA2 1p32.3 ATP2b2 Pompe ATP Ca2+ dépendante
DFNB16 STRC 15q15 Stéreociline Protéine membranaire (fixation des liens apicaux à la membrane tectoriale)
DFNB18 USH1C 11p15.1 Harmonine Protéine à domaine PDZ (adhérence) DFNB21 TECTA 11q22-q24 Tectorine Stabilité et structure de la membrane tectoriale DFNB22 OTOA 16p12.2 Otoancorine Protéine de fixation de la membrane tectoriale aux
cellules non-sensorielles (adhérence) DFNB23 PCDH15 10q21-q22 Protocadherine 15 Protéine d’adhérence (liens apicaux et latéraux) DFNB24 RDX 11q23 Radixine Attache l’actine à la membrane cellulaire DFNB28 TRIOBP 22q13.1 Trio-binding protein Organisation et fixation de l’actine DFNB29 CLDN14 21q22.3 Claudine 14 Protéine des jonctions serrées DFNB30 MYO3A 10p11.1 Myosine IIIa Transport DFNB31 WHRN 9q32-q34 Whirline Protéine à domaine PDZ (adhérence) DFNB35 ESRRB 14q21.1-q24.3 Oestrogen-related
receptor Facteur de transcription
DFNB36 ESPN 1p36.3-p36.1 Espine Protéine fasciculant l’actine DFNB37 MYO6 6q13 Myosine VI Régulation de l’exocytose des stéreocils fixés DFNB49 TRIC 5q12.3-q14.1 Tricelluline Protéine des jonctions serrées DFNB53 COL11A2 6p21.3 Collagène type XIa2 Stabilité et structure de la membrane tectoriale DFNB61 SLC26A5 - Prestine Electromotilité DFNB67 TMHS 6p21.3 Tetraspan membrane
protein Lien transitoire (adhérence)
DFNB73 BSND - Barttin Maturation et trafic du canal de K+
AUTOSOMIQUE RECESSIF (1)

Locus Gene L.C. Protéine Fonction DFNB91 GJB3 6p25 Connexine 31 Jonction communicante (homéostasie ionique) DFNB93 CABP2 11q13.2 Calcium-binding protein 2 Homéostasie ionique DFNB94 NARS2 11q14.1 Asparaginyl-tRNA synthetase
2 Transport
DFNB97 MET 7q31.2 MET protooncogene Récepteur de surface cellulaire pour le facteur de croissance des hépatocytes (adhérence)
DFNB98 TSPEAR 21q22.3 Thrombospondin-type laminin g domain and ear repeats
Perméabilité cellulaire (transport)
DFNB99 TMEM132E 17q12 Transmembrane protein 132e Récepteur extracellulaire DFNB101 GRXCR2 5q32 Glutaredoxin, cysteine-rich 2 Organisation des stéreocils (adhérence) DFNB102 EPS8 12p12.3 Epidermal growth factor
receptor pathway substrate 8 Régulation de l’activité GEF
DFNB103 CLIC5 6p21.1 Chloride intracellular channel 5
Homéostasie ionique
- FAM65B 6p22.3 Family with sequence similarity 65 member b
Cytosquelette
AUTOSOMIQUE RECESSIF (2)

• Les mutations dans le gène GJB2 sont la première cause de surdité non syndromique chez les patients marocains
Le Gène GJB2 (Gap junction protein de type beta-2)

Le gène GJB2 (Gap junction protein de type beta-2) est le premier gène identifié qui est impliqué dans une surdité de transmission autosomique récessive non syndromique.
localisé au début du bras long du chromosome 13 en position q11.12
Responsable de de 34,59% des cas de surdités chez les patients marocains
Le Gène GJB2 (Gap junction protein de type beta-2)

Ce gène code pour une protéine membranaire appelée connexine 26 formée d’une séquence de 226 acides aminés.
Cette protéine appartient à une large famille de plus de 21 connexines codées chacune par un gène distinct.
Les Connexines jouent un rôle important dans de nombreuses fonctions biologiques telles que la différentiation et la prolifération cellulaire, le développement tissulaire, la transmission électrique et l’inflammation.
Les principales connexines dont l’altération est souvent associée à des problèmes de surdité sont la Cx26 (GJB2) dans la majorité des cas et à moindre degré la Cx30 (GJB6), la Cx31 (GJB3), la Cx32 (GJB1) et la Cx43 (GJA1).
Les mutations au niveau de ces gènes affectent le bon fonctionnement de l’épithélium sensoriel de l’oreille interne en perturbant le recyclage du K+ ce qui bloquerait le mécanisme de dépolarisation des cellules sensorielles induisant la perte auditive.
Le Gène GJB2 (Gap junction protein de type beta-2)
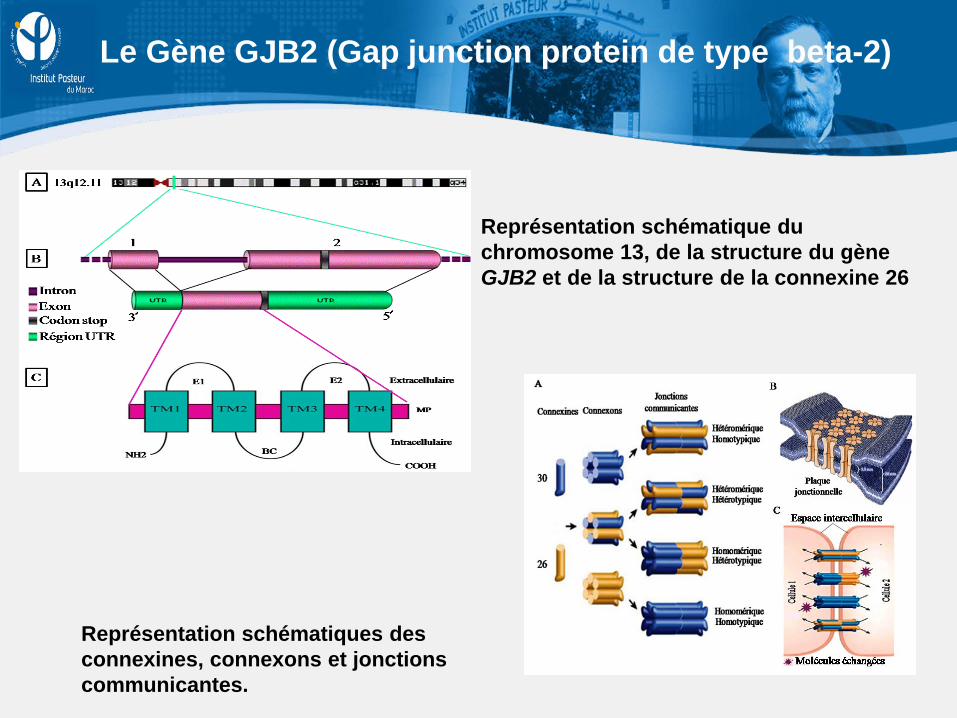
Représentation schématiques des connexines, connexons et jonctions communicantes.
Représentation schématique du chromosome 13, de la structure du gène GJB2 et de la structure de la connexine 26
Le Gène GJB2 (Gap junction protein de type beta-2)

• À ce jour, il y a eu plus de 150 mutations rapportées trouvées dans le GJB2 (The Connexin-deafness homepage: http://davinci.crg.es/deafness/)
• La mutation c.35delG est une délétion d’une des six bases guanines localisées entre les positions 30 et 35 de l’ADN codante. La première famille Marocaine portant cette mutation a été identifiée en 1998 par Lench et al.
• L’âge estimé de cette mutation chez la population Marocaine serait de 135 générations soit 2700 ans.
Mutation 35delG

Le c.35delG est la mutation la plus fréquente dans le gène GJB2 qui provoque la surdité.
Chan and Chang (2014). Laryngoscope. 124(2):E34-53.
Mutation 35delG

Cette mutation est très fréquente dans la région méditerranéenne et semble avoir surgi dans le Moyen-Orient il y a environ 500 générations (10000 ans) et se propager à travers l'Europe et autour du bassin méditerranéen (Van Laer et al., 2001 ).
Mutation 35delG
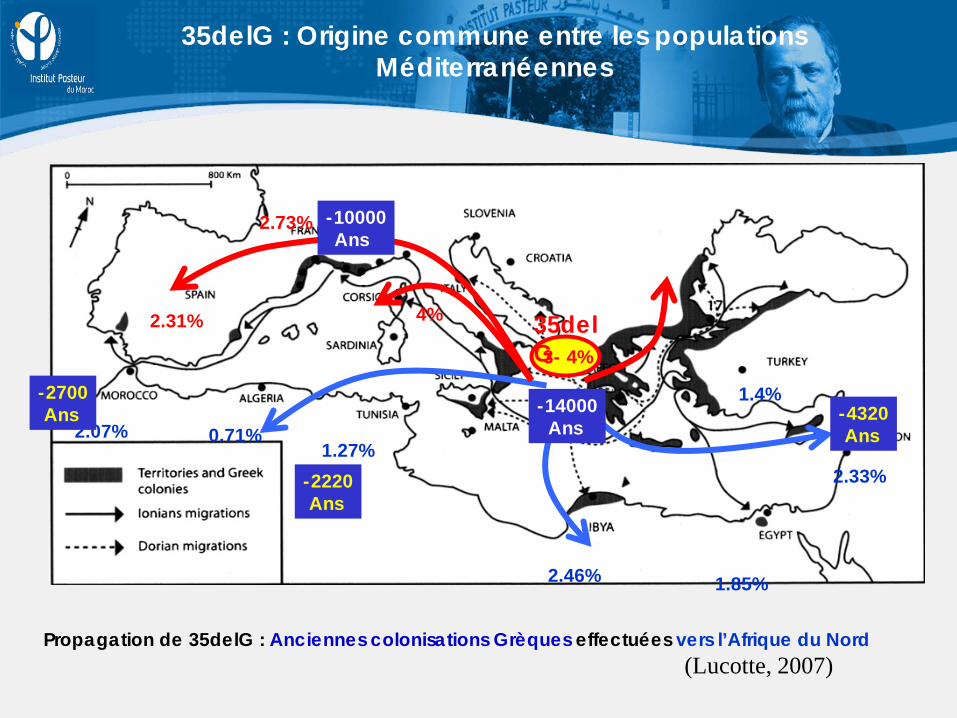
35delG : Origine commune entre les populations Méditerranéennes
0.71% 1.27%
1.4%
1.85%
2.07%
2.33%
2.31%
2.46%
2.73%
4%
3- 4%
Propagation de 35delG : Anciennes colonisations Grèques effectuées vers l’Afrique du Nord
-14000 Ans
-10000 Ans
-2700 Ans -4320
Ans
-2220 Ans
(Lucotte, 2007)
35delG
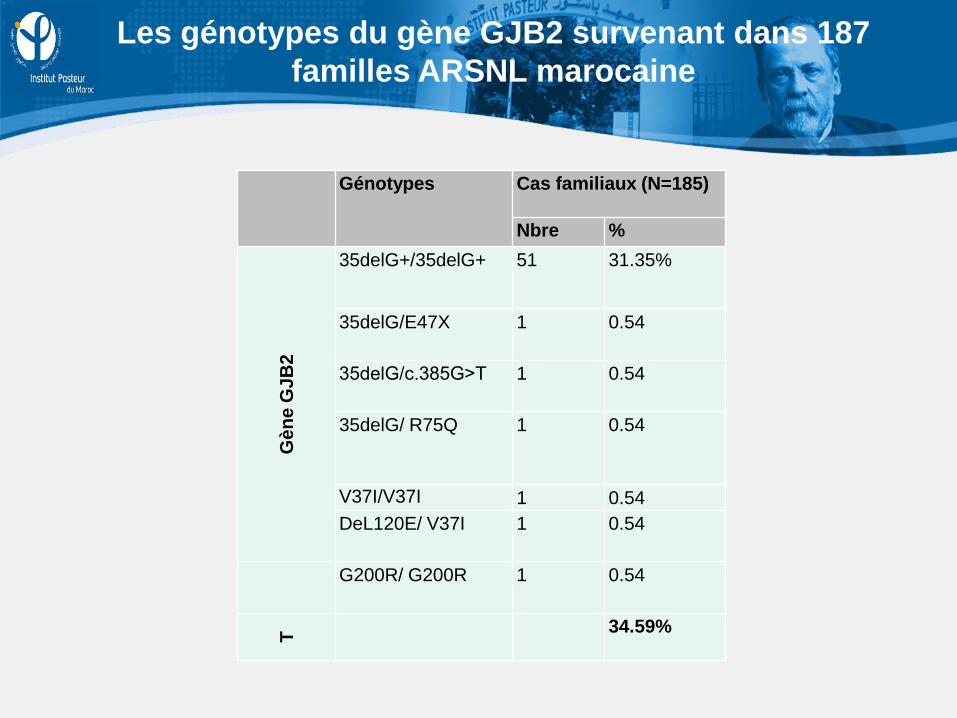
Génotypes Cas familiaux (N=185)
Nbre %
Gèn
e G
JB2
35delG+/35delG+ 51 31.35%
35delG/E47X
1 0.54
35delG/c.385G˃T
1 0.54
35delG/ R75Q
1 0.54
V37I/V37I 1 0.54 DeL120E/ V37I
1 0.54
G200R/ G200R
1 0.54
T 34.59%
Les génotypes du gène GJB2 survenant dans 187 familles ARSNL marocaine
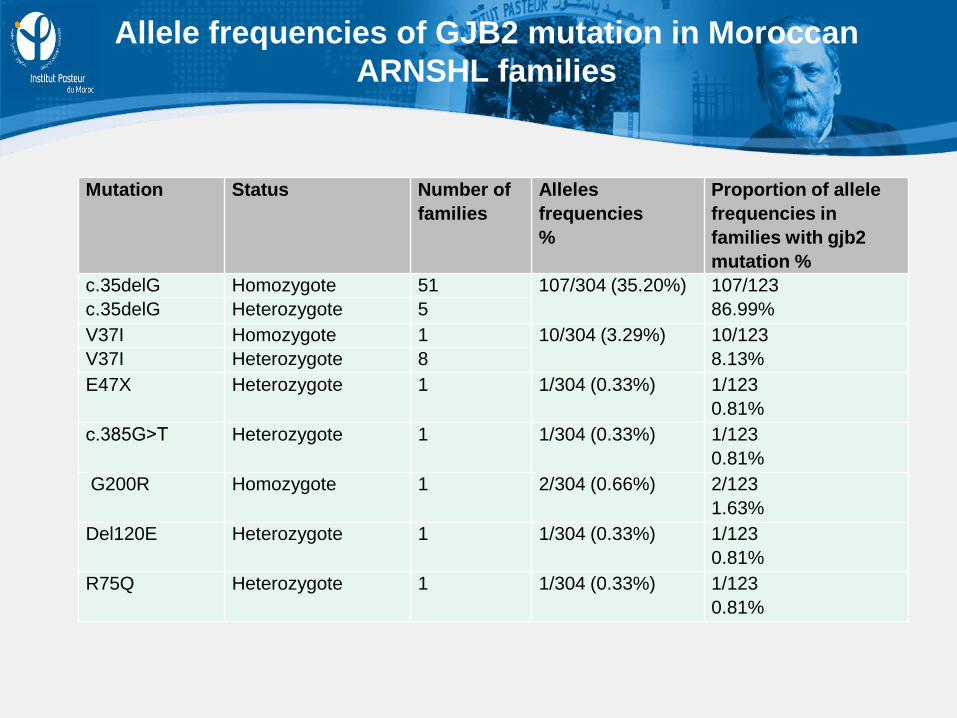
Mutation Status Number of families
Alleles frequencies %
Proportion of allele frequencies in families with gjb2 mutation %
c.35delG Homozygote 51 107/304 (35.20%) 107/123 86.99% c.35delG Heterozygote 5
V37I Homozygote 1 10/304 (3.29%) 10/123 8.13% V37I Heterozygote 8
E47X Heterozygote 1 1/304 (0.33%) 1/123 0.81%
c.385G˃T Heterozygote 1 1/304 (0.33%) 1/123 0.81%
G200R Homozygote 1 2/304 (0.66%) 2/123 1.63%
Del120E Heterozygote 1 1/304 (0.33%) 1/123 0.81%
R75Q Heterozygote 1 1/304 (0.33%) 1/123 0.81%
Allele frequencies of GJB2 mutation in Moroccan ARNSHL families

• Les mutations dans le gène LRTOMT sont la deuxième cause de surdité non syndromique chez les patients marocains.
Le Gène LRTOMT

Le Gène LRTOMT
o Identifié en 2007, responsable d’une surdité non syndromique autosomique récessive DFNB63.
o Protéine COMT2 contient un domaine Catéchol-O-MéthylTransférase et intervient dans la régulation du niveau de l'adrénaline, la noradrénaline et la dopamine dans le cerveau.
o LRTOMT : essentiel à la fonction vestibulaire et auditive. exprimé dans les cellules ciliées sensorielles de l'oreille interne.

Structure du gène LRTOMT (Leucine Rich transmembrane and 0-
Methyltransferase domain containing)
LRTOMT1 LRTOMT2:COMT2
Le Gène LRTOMT

Liste des mutations identifiées au niveau du gène LRTOMT
LRTOMT2 (ADN)
LRTOMT2 (Protéine)
LRTOMT1 (ADN)
LRTOMT1 (Protéine)
Exon Population Reference
c.47T>C p.Leu16Pro c.450T>C p.Thr150Thr 2 Iranienne Du et al., 2008
c.213C>G p.Tyr71X c.736C>G 3’UTR 3 Iranienne Du et al., 2008
c.358 +4A>C p.Ala29SerfsX54 c.761 4A>C p.Gly163ValfsX4 8 Turque Ahmed et al., 2008
c.242G>A p.Arg81Gln c.645G>A p.Ala215Ala 8 Tunisienne Marocaine
Ahmed et al., 2008 Charif et al., 2012
c.313T>C p.Trp105Arg c.716T>C 3’UTR 8 Tunisienne Ahmed et al., 2008
c.328G>A p.Glu110Lys c.731G>A 3’UTR 8 Pakistanais Ahmed et al., 2008
c.104delC p.Ser35SerfsX13 c.507delC p.Val169ValfsX258 8 Iranienne Vanwesemael et al.,2011
Le Gène LRTOMT

Génotype
Cas familiaux
(N=185)
Témoins
(N=120)
Nombre Pourcentage Nombre Pourcentage
242G>A+/ + 18 9,72% 0 0
242G>A+/ - 0 0 5 4.16%
Fréquence de la mutation 242G>A identifiée au niveau du gène LRTOMT
Le Gène LRTOMT
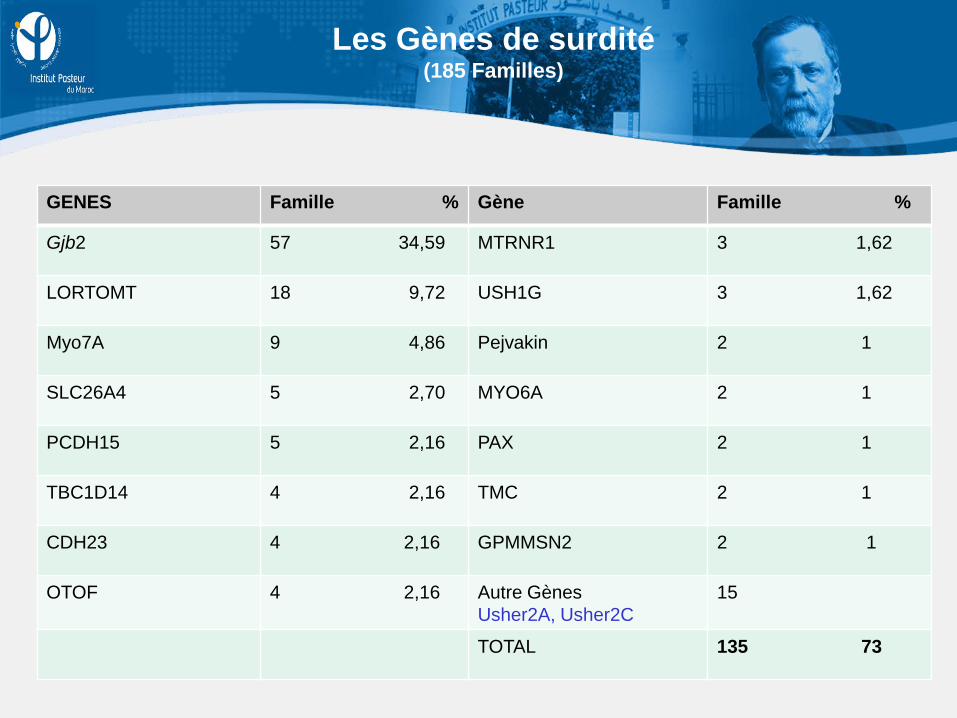
GENES Famille % Gène Famille %
Gjb2 57 34,59 MTRNR1 3 1,62
LORTOMT 18 9,72 USH1G 3 1,62
Myo7A 9 4,86 Pejvakin 2 1
SLC26A4 5 2,70 MYO6A 2 1
PCDH15 5 2,16 PAX 2 1
TBC1D14 4 2,16 TMC 2 1
CDH23 4 2,16 GPMMSN2 2 1
OTOF 4 2,16 Autre Gènes Usher2A, Usher2C
15
TOTAL 135 73
Les Gènes de surdité (185 Familles)

Classification des différents type du syndrome d’Usher
Surdité syndromique: syndrome d’Usher
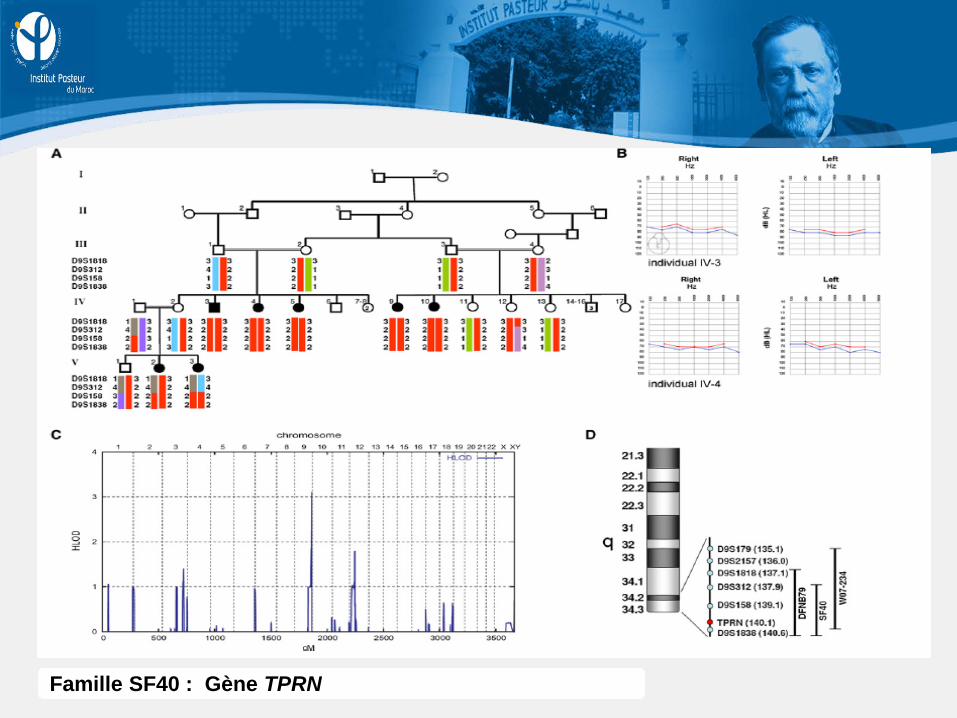
Famille SF40 : Gène TPRN
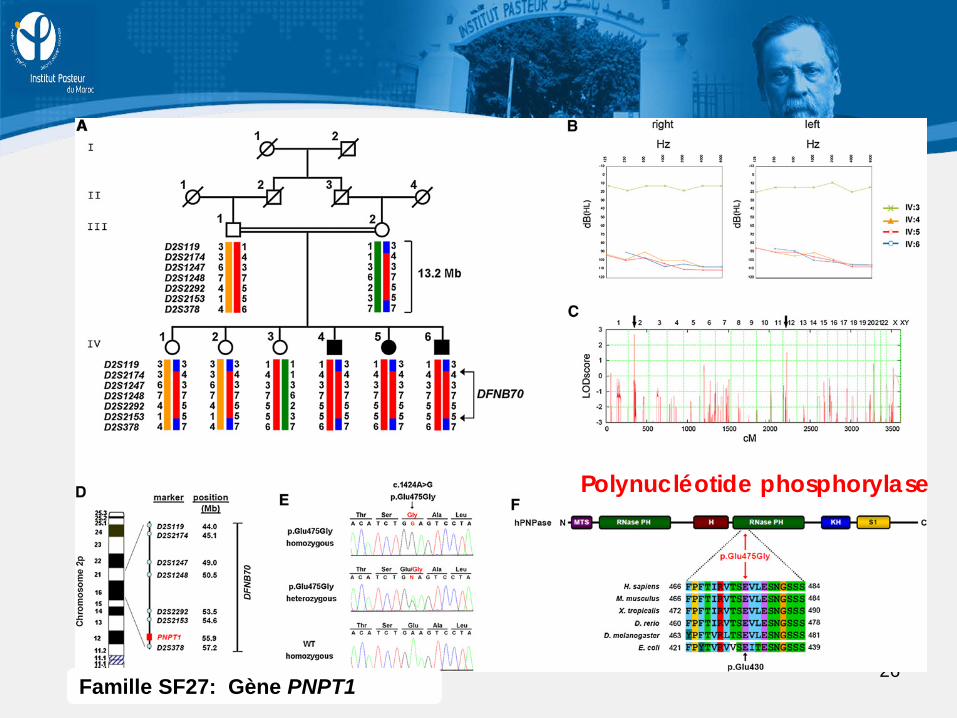
26 Famille SF27: Gène PNPT1
Polynucléotide phosphorylase

Mutations hétérozygote composites ont été identifiées au niveau du gène TBC1D24 chez deux familles marocaines grâce au WES

En haut toutes les mutations qui causent la surdité. En bas mutations du gène TBC1D24 en relation avec l’épilepsie, en haut toutes les mutations qui causent la surdité.
Le gène TBC1D24
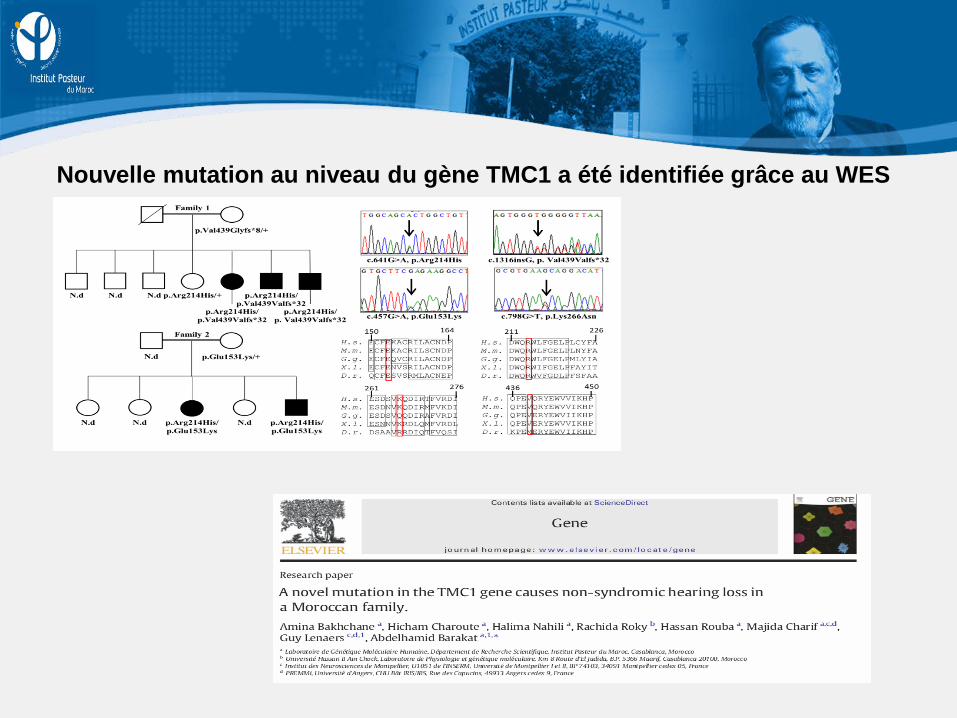
Nouvelle mutation au niveau du gène TMC1 a été identifiée grâce au WES
La base de données MGDD (http://mgdd.pasteur.ma) donne un aperçu sur le spectre des troubles génétiques trouvés chez la population marocaine.
La MFMD (http://mfmd.pasteur.ma) une base de données créé pour offrir un accès à l’ensemble des mutations fondatrice chez la population méditerranéenne.
The Mediterranean Founder Mutation Database(MFMD)
The Moroccan Genetic Disease Database (MGDD)
Laboratoire de Génétique Moléculaire Humaine

Amina bakchane, Amale Bousfiha, Sara Salim, Soukaina Elraichi, Hicham Charoute Laboratoire de Génétique moléculaire Humaine Institut Pasteur du Maroc
Dr G. Lenaers PREMML, Mitochondrial Medicine Research Center, Université d’Angers
Pr C. Petit Institut Pasteur Paris Institut de Vision Paris
Remerciement
Dr S Abdelhak Institut Pasteur Tunis

Merci pour votre attention