avancÉes médico- scientifi ques neuro- musculaires · chat hfmd et mutants, souris mdx, mdx(cv),...
TRANSCRIPT

savoir &comprendre
fiche
technique
AVANCÉES
médico-scientifi ques
neuro-musculaires
octobre 2012 | 5e édition
© A
FM /
Lau
rent
Aud
inet
L’élan donné à la génétique par l’Association
Française contre les Myopathies et la mise au point
des cartes du génome par Généthon ont permis aux
équipes du monde entier de localiser et d’identifier
plus facilement et plus rapidement les gènes de
nombreuses maladies génétiques dont ceux des
maladies neuromusculaires.
Ces résultats font régulièrement évoluer la
classification des maladies neuromusculaires en y
intégrant de nouveaux critères moléculaires.
Le diagnostic et le conseil génétique s‘en trouvent
ainsi améliorés, tant au niveau de leur précision
que de leur fiabilité. Ceci ouvre également la voie
à de nouvelles pistes thérapeutiques et contribue
à une meilleure définition du suivi médical et du
projet de vie des personnes concernées par ces
maladies.
Cette Fiche Technique présente dans un grand
tableau, en fonction des avancées de la recherche,
le(s) gène(s) et les protéines en cause, ainsi que
les modèles animaux et les éventuels essais
thérapeutiques pour chaque maladie.
Un index des maladies et des protéines figure à la fin
du document.

SAVOIR & COMPRENDRE FICHE TECHNIQUE
2 | Octobre 2012
Un éventail de pathologies de plus en plus large
C’est en 1868 que Guillaume Duchenne décrit pour la première fois une "paralysie pseudo-hypertrophique" ou "myopathie atrophique progressive" touchant exclusivement de jeunes garçons. En 1884, l’Allemand Erb décrit une autre forme d’atteinte musculaire caractérisée par une atteinte des ceintures scapulaire (épaules) et pelvienne (bassin), touchant aussi bien des garçons que des fi lles. C’est lui qui introduit le terme de "dystrophie musculaire". De nombreux auteurs suivront et viendront enrichir l’éventail des maladies neuromuscu-laires à partir de l’âge de début de la maladie, selon la localisation de la faiblesse muscu-laire et son évolution, et selon le mode de transmission. Il faudra attendre près de 100 ans et la découverte de beaucoup d'autres myopathies pour commencer à percer leurs mécanismes physiopathologiques.
De nombreux gènes identifi és
Depuis le début des années 1980, les tech-niques de biologie moléculaire ont révo-lutionné la connaissance des maladies neuromusculaires et permettent d’assigner une maladie à un gène en particulier. Ce concept "un gène, une maladie" est toutefois régulièrement remis en question. Pour une maladie donnée, par exemple, deux ou plusieurs gènes différents sont parfois mis en cause : les myopathies congénitales à némaline sont ainsi en rapport avec six gènes différents codant respectivement les tropomyosines 2 et 3, la troponine T1, la nébuline, l'alpha-actine et la cofi line-2. À l'inverse, les modifi cations de certains gènes entraînent parfois plusieurs tableaux cliniques différents. Les lamines A/C nucléaires sont ainsi responsables de la dystrophie musculaire d'Emery-Dreifuss autosomique dominante, de la dystrophie musculaire des ceintures LGMD1B, de la maladie de Charcot-Marie-Tooth (CMT2B1, CMT spinale), de la dystrophie muscu-laire congénitale L-CMD, d'une forme de cardiomyopathie dilatée, d'une forme de lipodystrophie ainsi que des syndromes
de vieillissement prématuré (progeria, dys-plasie mandibuloacrale, syndrome de Werner atypique, dermopathie restrictive). Des modifi cations de la sélénoprotéine de type 1 sont quant à elles retrouvées dans les myopathies congénitales à multiminicores, dans la dystrophie musculaire congénitale avec raideur de la colonne et dans une des formes de myopathie myofi brillaire.
Des mutations mieux identifiées et un diagnostic plus précis
L'identifi cation des gènes et la connaissance de plus en plus précise des mutations à l'ori-gine des différentes pathologies permettent aujourd'hui de mieux cibler les outils diagnos-tiques. Au cours des dernières années, des réseaux nationaux et internationaux regrou-pant des spécialistes des différentes maladies neuromusculaires se sont mis en place. Ils visent à mettre en commun les connaissances issues de la génétique et du suivi clinique dans le but d'élaborer des projets thérapeu-tiques et d'améliorer le diagnostic, le suivi et la prise en charge (réseaux des maladies à canaux ioniques, syndromes myasthéniques, dystrophies musculaires congénitales, mala-dies mitochondriales, CMT…).
Des mécanismes physiopathologiques mieux connus
En aval de la génétique, les mécanismes des maladies se précisent. Par exemple, l'ajout d'un sucre (glucose, mannose, ou groupe-ment sialique) sur certaines molécules de la membrane de la fi bre musculaire (dystrogly-cane, sarcoglycane) est essentiel à leur fonc-tionnement ; son absence se retrouve dans plusieurs formes de dystrophies musculaires congénitales. Plusieurs gènes codant des protéines chargées d'effectuer cette trans-formation sont impliqués (POMT1, POMT2, POMGnT1, FKRP). La découverte des méca-nismes en cause dans la dystrophie myoto-nique de Steinert, impliquant l'accumulation d'ARNs messagers dans le noyau permet éga-lement d'envisager des traitements adaptés. Enfi n, on comprend mieux les mécanismes de la dégénérescence musculaire; le rôle des cellules satellites présentes à la périphérie
Sommaire De la génétique aux thérapeutiquesLes avancées de la biologie moléculaire permettent d'affiner et de faire évoluer la classification des maladies neuromusculaires, d'optimiser les outils diagnostiques, et d'identifier de nouvelles pistes thérapeutiques.
De la génétique aux thérapeutiques ................ 2
Dystrophinopathies ............................................ 4
Dystrophies musculaires des ceintures ............ 4 - Autosomiques récessives (LGMD2) ........... 4 - Autosomiques dominantes (LGMD1) ........ 6
Autres dystrophies musculaires progressives......................................................... 6
Dystrophies musculaires congénitales ............. 8
Myopathies distales .......................................... 12
Myopathies myofi brillaires .............................. 14
Autres myopathies avec surcharge de fi laments ou inclusions ................................ 14
Myosinopathies ................................................. 16
Myopathies congénitales ................................. 16
Syndromes myotoniques ................................. 20
Canalopathies musculaires .............................. 20
Myopathies métaboliques ............................... 22
Maladies infl ammatoires du muscle ............... 24
Fibrodysplasie ossifi ante progressive ............ 24
Myasthénie ........................................................ 24
Syndromes myasthéniques congénitaux pré-synaptiques .......................... 24
Syndromes myasthéniques congénitaux synaptiques ................................. 26
Syndromes myasthéniques congénitaux post-synaptiques ........................ 26
Amyotrophies spinales proximales ................. 28
Amyotrophies spinales distales ....................... 28
Amyotrophies bulbo-spinales ......................... 30
Maladies de Charcot-Marie-Toothdémyélinisantes autosomiques dominantes ........................................................ 30
Maladies de Charcot-Marie-Toothdémyélinisantes autosomiques récessives ........................................................... 32
Maladies de Charcot-Marie-Toothaxonales ............................................................. 32
Maladies de Charcot-Marie-Toothintermédiaires ................................................... 36
Maladies de Charcot-Marie-Tooth(formes liées à l'X) ............................................ 36
Syndromes scapulo-péroniers ......................... 36
Index général .................................................... 39
Index des protéines .......................................... 42
Index des gènes ................................................ 43
Octobre 2012 | 3
Avancées médico-scientifiques neuromusculaires
des fi bres musculaires adultes et des facteurs
intervenant dans la mort cellulaire (apoptose)
sont très étudiés.
Des stratégies thérapeutiques variées et porteuses d’espoir
Pour une maladie donnée, la localisation du
gène responsable, son identifi cation, l'étude
de la protéine en cause et des voies molé-
culaires en jeu, ainsi que la mise au point
de modèles animaux constituent les étapes
préalables au développement des thérapeu-
tiques à visée curative (thérapies génique, cel-
lulaire ou pharmacologique).
La connaissance des maladies neuromus-
culaires s’appuie aussi sur les progrès de
recherche réalisés pour des maladies géné-
tiques rares non neuromusculaires. Il s’agit
alors d’établir des preuves de concept qui vont
servir de support à de nouvelles voies d’inves-
tigations dans la recherche de traitements des
maladies neuromusculaires.
Dans la dystrophie musculaire de Duchenne
(DMD), la chirurgie du gène fait ses preuves
en matière d'essais cliniques. L'injection sous-
cutanée du PRO051/GSK2402968 favorisant
le saut de l'exon 51 du gène de la dystro-
phine a donné des résultats positifs au test de
6 minutes de marche. Des essais se terminent
pour confi rmer ces résultats à plus long terme.
Un essai a aussi débuté avec des malades
ayant perdu la marche, avec pour objectif
d’améliorer la fonction des membres supé-
rieurs et la respiration.
.../... p. 38
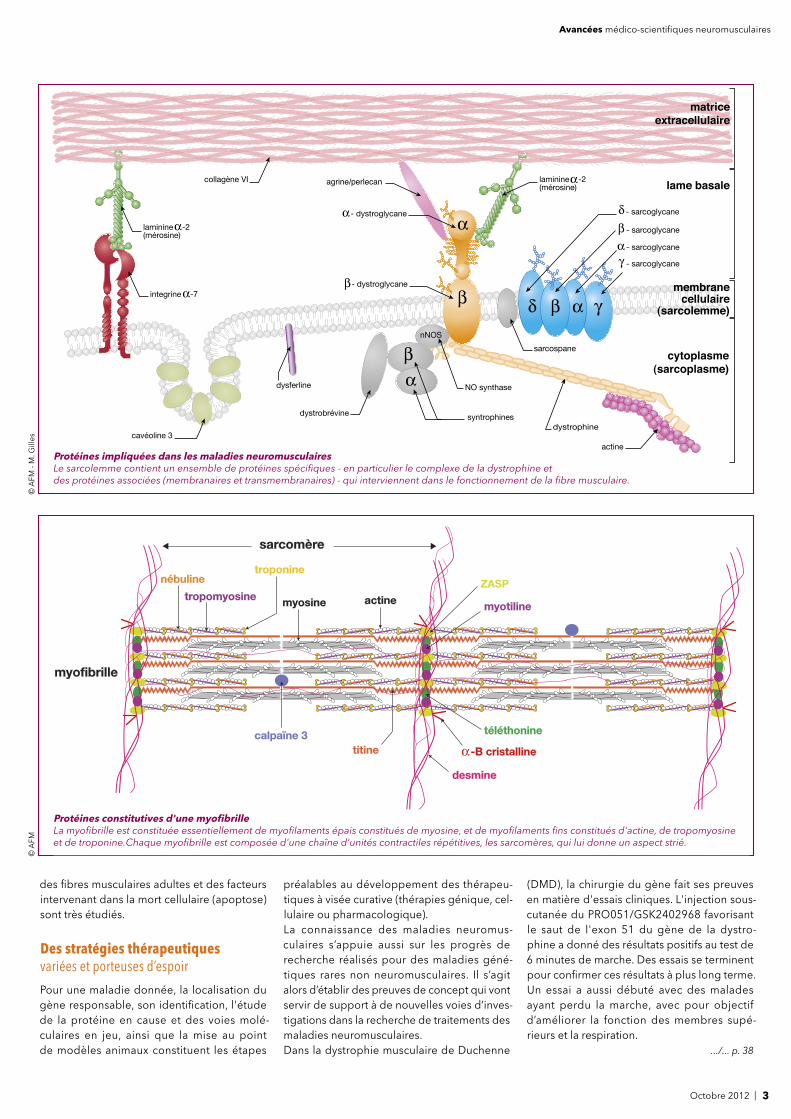
nNOS
dystrophine
- sarcoglycane- dystroglycane
- dystroglycane
syntrophines
dysferline
dystrobrévine
NO synthase
sarcospane
actine
laminine -2(mérosine)
cavéoline 3
collagène VI
integrine -7
laminine -2(mérosine)
matrice
extracellulaire
lame basale
membrane cellulaire
(sarcolemme)
agrine/perlecan
cytoplasme
(sarcoplasme)
- sarcoglycane
- sarcoglycane
- sarcoglycane
Protéines impliquées dans les maladies neuromusculairesLe sarcolemme contient un ensemble de protéines spécifi ques - en particulier le complexe de la dystrophine et des protéines associées (membranaires et transmembranaires) - qui interviennent dans le fonctionnement de la fi bre musculaire.
© A
FM
- M
. Gill
es
© A
FM
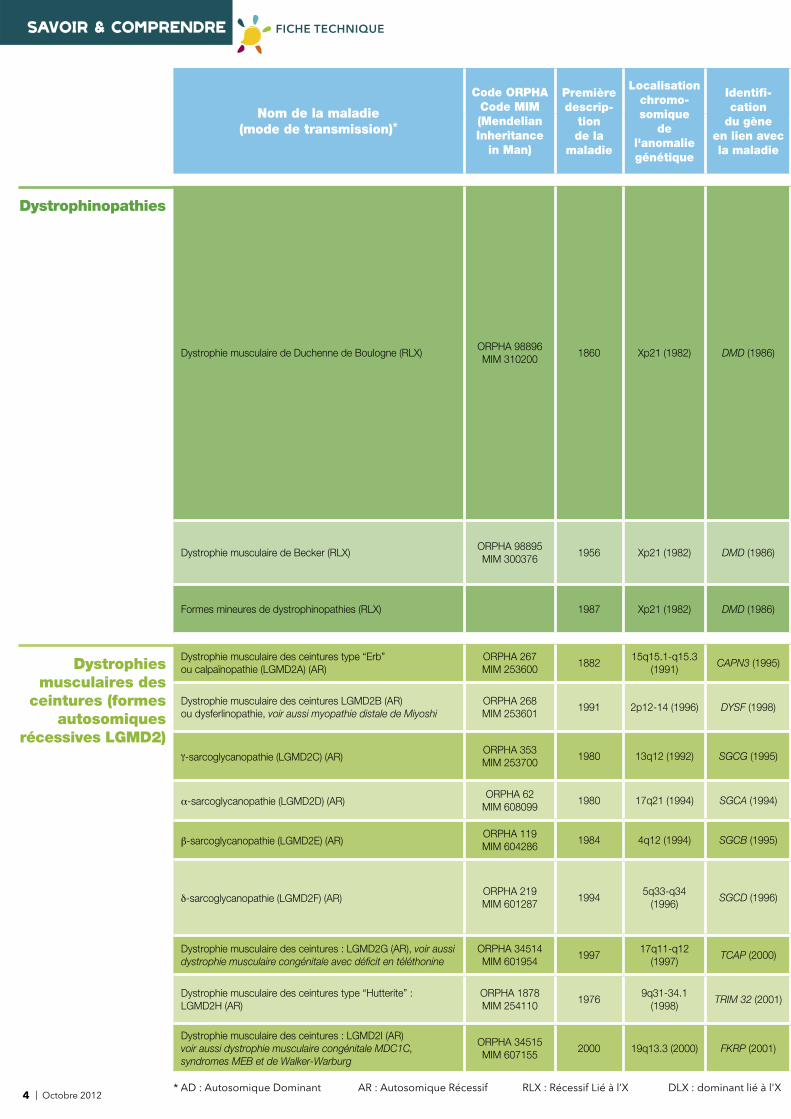
sarcomère
nébuline
tropomyosine
troponine
myosine actinemyotiline
téléthonine
-B cristalline
desmine
titine
calpaïne 3
myofibrille
Protéines constitutives d'une myofi brilleLa myofi brille est constituée essentiellement de myofi laments épais constitués de myosine, et de myofi laments fi ns constitués d'actine, de tropomyosine et de troponine.Chaque myofi brille est composée d'une chaîne d'unités contractiles répétitives, les sarcomères, qui lui donne un aspect strié.
SAVOIR & COMPRENDRE FICHE TECHNIQUE
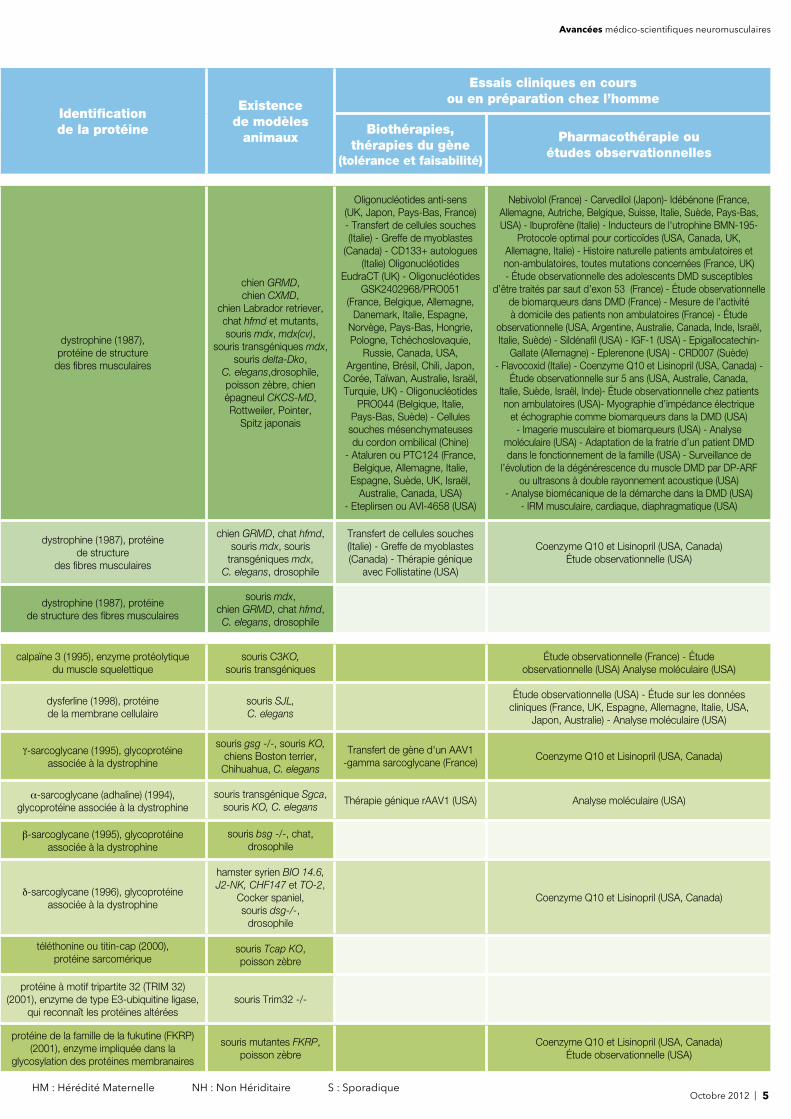
4 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Dystrophinopathies
Dystrophie musculaire de Duchenne de Boulogne (RLX) ORPHA 98896MIM 310200
1860 Xp21 (1982) DMD (1986)
Dystrophie musculaire de Becker (RLX)ORPHA 98895MIM 300376
1956 Xp21 (1982) DMD (1986)
Formes mineures de dystrophinopathies (RLX) 1987 Xp21 (1982) DMD (1986)
Dystrophies musculaires des
ceintures (formes autosomiques
récessives LGMD2)
Dystrophie musculaire des ceintures type “Erb” ou calpaïnopathie (LGMD2A) (AR)
ORPHA 267MIM 253600
188215q15.1-q15.3
(1991)CAPN3 (1995)
Dystrophie musculaire des ceintures LGMD2B (AR) ou dysferlinopathie, voir aussi myopathie distale de Miyoshi
ORPHA 268MIM 253601
1991 2p12-14 (1996) DYSF (1998)
γ-sarcoglycanopathie (LGMD2C) (AR) ORPHA 353MIM 253700
1980 13q12 (1992) SGCG (1995)
α-sarcoglycanopathie (LGMD2D) (AR)ORPHA 62
MIM 6080991980 17q21 (1994) SGCA (1994)
β-sarcoglycanopathie (LGMD2E) (AR)ORPHA 119MIM 604286
1984 4q12 (1994) SGCB (1995)
δ-sarcoglycanopathie (LGMD2F) (AR)ORPHA 219MIM 601287
19945q33-q34
(1996)SGCD (1996)
Dystrophie musculaire des ceintures : LGMD2G (AR), voir aussi dystrophie musculaire congénitale avec défi cit en téléthonine
ORPHA 34514MIM 601954
199717q11-q12
(1997)TCAP (2000)
Dystrophie musculaire des ceintures type “Hutterite” : LGMD2H (AR)
ORPHA 1878MIM 254110
19769q31-34.1
(1998)TRIM 32 (2001)
Dystrophie musculaire des ceintures : LGMD2I (AR) voir aussi dystrophie musculaire congénitale MDC1C,syndromes MEB et de Walker-Warburg
ORPHA 34515MIM 607155
2000 19q13.3 (2000) FKRP (2001)
Octobre 2012 | 5 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
dystrophine (1987), protéine de structure
des fi bres musculaires
chien GRMD, chien CXMD,
chien Labrador retriever, chat hfmd et mutants,souris mdx, mdx(cv),
souris transgéniques mdx, souris delta-Dko,
C. elegans,drosophile,poisson zèbre, chien épagneul CKCS-MD, Rottweiler, Pointer,
Spitz japonais
Oligonucléotides anti-sens (UK, Japon, Pays-Bas, France) - Transfert de cellules souches (Italie) - Greffe de myoblastes
(Canada) - CD133+ autologues (Italie) Oligonucléotides
EudraCT (UK) - Oligonucléotides GSK2402968/PRO051
(France, Belgique, Allemagne, Danemark, Italie, Espagne,
Norvège, Pays-Bas, Hongrie, Pologne, Tchéchoslovaquie,
Russie, Canada, USA, Argentine, Brésil, Chili, Japon,
Corée, Taïwan, Australie, Israël, Turquie, UK) - Oligonucléotides
PRO044 (Belgique, Italie, Pays-Bas, Suède) - Cellules souches mésenchymateuses du cordon ombilical (Chine)
- Ataluren ou PTC124 (France, Belgique, Allemagne, Italie, Espagne, Suède, UK, Israël,
Australie, Canada, USA)- Eteplirsen ou AVI-4658 (USA)
Nebivolol (France) - Carvedilol (Japon)- Idébénone (France, Allemagne, Autriche, Belgique, Suisse, Italie, Suède, Pays-Bas, USA) - Ibuprofène (Italie) - Inducteurs de l'utrophine BMN-195-
Protocole optimal pour corticoïdes (USA, Canada, UK, Allemagne, Italie) - Histoire naturelle patients ambulatoires et non-ambulatoires, toutes mutations concernées (France, UK) - Étude observationnelle des adolescents DMD susceptibles
d’être traités par saut d’exon 53 (France) - Étude observationnelle de biomarqueurs dans DMD (France) - Mesure de l'activité à domicile des patients non ambulatoires (France) - Étude
observationnelle (USA, Argentine, Australie, Canada, Inde, Israël, Italie, Suède) - Sildénafi l (USA) - IGF-1 (USA) - Epigallocatechin-
Gallate (Allemagne) - Eplerenone (USA) - CRD007 (Suède) - Flavocoxid (Italie) - Coenzyme Q10 et Lisinopril (USA, Canada) -
Étude observationnelle sur 5 ans (USA, Australie, Canada, Italie, Suède, Israël, Inde)- Étude observationnelle chez patients non ambulatoires (USA)- Myographie d’impédance électrique
et échographie comme biomarqueurs dans la DMD (USA) - Imagerie musculaire et biomarqueurs (USA) - Analyse
moléculaire (USA) - Adaptation de la fratrie d’un patient DMD dans le fonctionnement de la famille (USA) - Surveillance de
l’évolution de la dégénérescence du muscle DMD par DP-ARF ou ultrasons à double rayonnement acoustique (USA)
- Analyse biomécanique de la démarche dans la DMD (USA) - IRM musculaire, cardiaque, diaphragmatique (USA)
dystrophine (1987), protéine de structure
des fi bres musculaires
chien GRMD, chat hfmd, souris mdx, souris
transgéniques mdx,C. elegans, drosophile
Transfert de cellules souches (Italie) - Greffe de myoblastes (Canada) - Thérapie génique
avec Follistatine (USA)
Coenzyme Q10 et Lisinopril (USA, Canada)Étude observationnelle (USA)
dystrophine (1987), protéine de structure des fi bres musculaires
souris mdx, chien GRMD, chat hfmd, C. elegans, drosophile
calpaïne 3 (1995), enzyme protéolytique du muscle squelettique
souris C3KO,souris transgéniques
Étude observationnelle (France) - Étude observationnelle (USA) Analyse moléculaire (USA)
dysferline (1998), protéine de la membrane cellulaire
souris SJL,C. elegans
Étude observationnelle (USA) - Étude sur les données cliniques (France, UK, Espagne, Allemagne, Italie, USA,
Japon, Australie) - Analyse moléculaire (USA)
γ-sarcoglycane (1995), glycoprotéine associée à la dystrophine
souris gsg -/-, souris KO,chiens Boston terrier,
Chihuahua, C. elegans
Transfert de gène d'un AAV1-gamma sarcoglycane (France)
Coenzyme Q10 et Lisinopril (USA, Canada)
α-sarcoglycane (adhaline) (1994), glycoprotéine associée à la dystrophine
souris transgénique Sgca, souris KO, C. elegans
Thérapie génique rAAV1 (USA) Analyse moléculaire (USA)
β-sarcoglycane (1995), glycoprotéine associée à la dystrophine
souris bsg -/-, chat,drosophile
δ-sarcoglycane (1996), glycoprotéine associée à la dystrophine
hamster syrien BIO 14.6, J2-NK, CHF147 et TO-2,
Cocker spaniel, souris dsg-/-,
drosophile
Coenzyme Q10 et Lisinopril (USA, Canada)
téléthonine ou titin-cap (2000), protéine sarcomérique
souris Tcap KO, poisson zèbre
protéine à motif tripartite 32 (TRIM 32) (2001), enzyme de type E3-ubiquitine ligase,
qui reconnaît les protéines altéréessouris Trim32 -/-
protéine de la famille de la fukutine (FKRP)(2001), enzyme impliquée dans la
glycosylation des protéines membranaires
souris mutantes FKRP, poisson zèbre
Coenzyme Q10 et Lisinopril (USA, Canada)Étude observationnelle (USA)
SAVOIR & COMPRENDRE FICHE TECHNIQUE
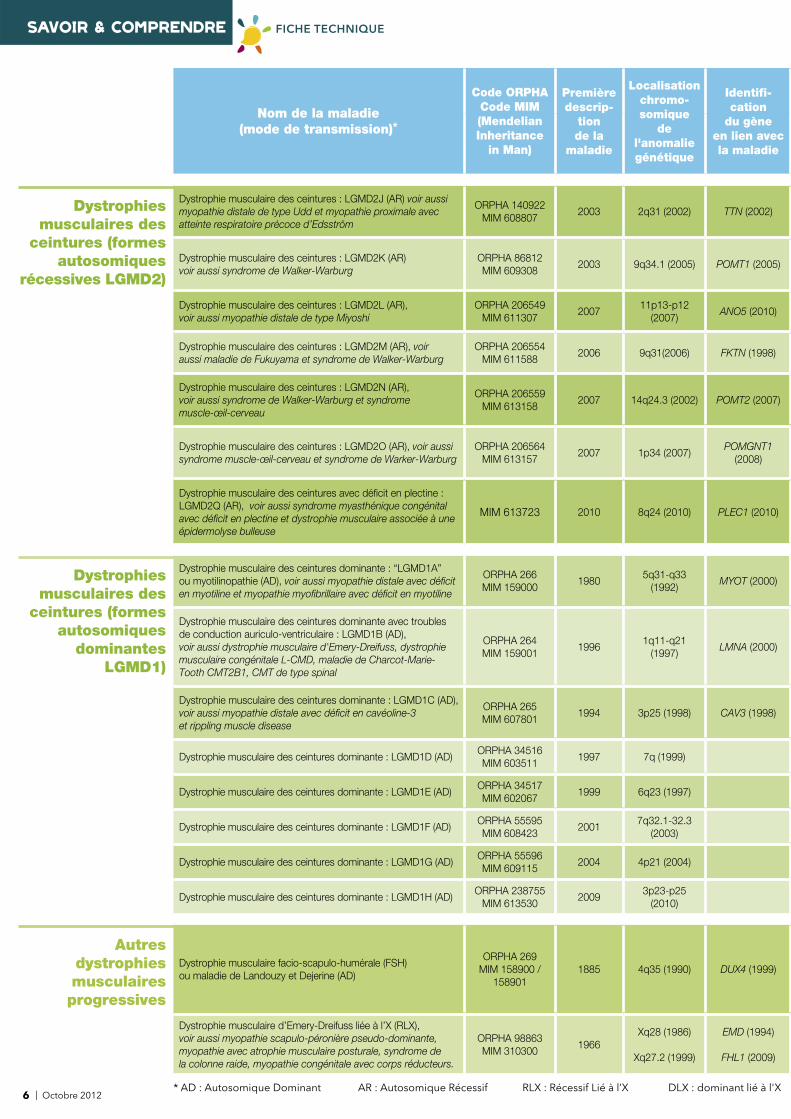
6 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Dystrophies musculaires des
ceintures (formes autosomiques
récessives LGMD2)
Dystrophie musculaire des ceintures : LGMD2J (AR) voir aussi myopathie distale de type Udd et myopathie proximale avec atteinte respiratoire précoce d’Edsström
ORPHA 140922MIM 608807
2003 2q31 (2002) TTN (2002)
Dystrophie musculaire des ceintures : LGMD2K (AR) voir aussi syndrome de Walker-Warburg
ORPHA 86812MIM 609308
2003 9q34.1 (2005) POMT1 (2005)
Dystrophie musculaire des ceintures : LGMD2L (AR),voir aussi myopathie distale de type Miyoshi
ORPHA 206549 MIM 611307
200711p13-p12
(2007)ANO5 (2010)
Dystrophie musculaire des ceintures : LGMD2M (AR), voir aussi maladie de Fukuyama et syndrome de Walker-Warburg
ORPHA 206554 MIM 611588
2006 9q31(2006) FKTN (1998)
Dystrophie musculaire des ceintures : LGMD2N (AR), voir aussi syndrome de Walker-Warburg et syndromemuscle-œil-cerveau
ORPHA 206559 MIM 613158
2007 14q24.3 (2002) POMT2 (2007)
Dystrophie musculaire des ceintures : LGMD2O (AR), voir aussi syndrome muscle-œil-cerveau et syndrome de Warker-Warburg
ORPHA 206564MIM 613157
2007 1p34 (2007)POMGNT1
(2008)
Dystrophie musculaire des ceintures avec défi cit en plectine : LGMD2Q (AR), voir aussi syndrome myasthénique congénital avec défi cit en plectine et dystrophie musculaire associée à une épidermolyse bulleuse
MIM 613723 2010 8q24 (2010) PLEC1 (2010)
Dystrophies musculaires des
ceintures (formes autosomiques
dominantes LGMD1)
Dystrophie musculaire des ceintures dominante : “LGMD1A” ou myotilinopathie (AD), voir aussi myopathie distale avec défi cit en myotiline et myopathie myofi brillaire avec défi cit en myotiline
ORPHA 266 MIM 159000
19805q31-q33
(1992)MYOT (2000)
Dystrophie musculaire des ceintures dominante avec troubles de conduction auriculo-ventriculaire : LGMD1B (AD), voir aussi dystrophie musculaire d'Emery-Dreifuss, dystrophie musculaire congénitale L-CMD, maladie de Charcot-Marie-Tooth CMT2B1, CMT de type spinal
ORPHA 264MIM 159001
19961q11-q21
(1997)LMNA (2000)
Dystrophie musculaire des ceintures dominante : LGMD1C (AD), voir aussi myopathie distale avec défi cit en cavéoline-3 et rippling muscle disease
ORPHA 265 MIM 607801
1994 3p25 (1998) CAV3 (1998)
Dystrophie musculaire des ceintures dominante : LGMD1D (AD)ORPHA 34516 MIM 603511
1997 7q (1999)
Dystrophie musculaire des ceintures dominante : LGMD1E (AD)ORPHA 34517 MIM 602067
1999 6q23 (1997)
Dystrophie musculaire des ceintures dominante : LGMD1F (AD)ORPHA 55595 MIM 608423
20017q32.1-32.3
(2003)
Dystrophie musculaire des ceintures dominante : LGMD1G (AD)ORPHA 55596 MIM 609115
2004 4p21 (2004)
Dystrophie musculaire des ceintures dominante : LGMD1H (AD)ORPHA 238755
MIM 6135302009
3p23-p25 (2010)
Autres dystrophies musculaires progressives
Dystrophie musculaire facio-scapulo-humérale (FSH) ou maladie de Landouzy et Dejerine (AD)
ORPHA 269 MIM 158900 /
1589011885 4q35 (1990) DUX4 (1999)
Dystrophie musculaire d’Emery-Dreifuss liée à l’X (RLX),voir aussi myopathie scapulo-péronière pseudo-dominante, myopathie avec atrophie musculaire posturale, syndrome dela colonne raide, myopathie congénitale avec corps réducteurs.
ORPHA 98863MIM 310300
1966Xq28 (1986)
Xq27.2 (1999)
EMD (1994)
FHL1 (2009)
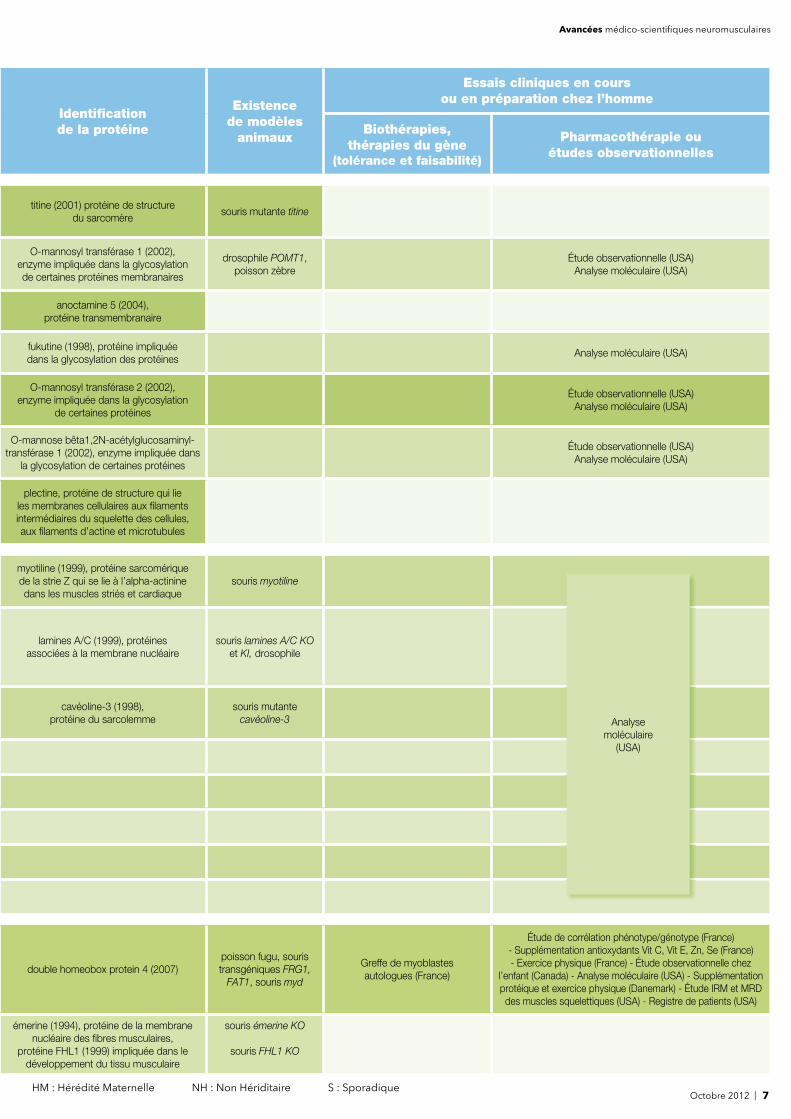
Octobre 2012 | 7 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
titine (2001) protéine de structure du sarcomère
souris mutante titine
O-mannosyl transférase 1 (2002), enzyme impliquée dans la glycosylation de certaines protéines membranaires
drosophile POMT1, poisson zèbre
Étude observationnelle (USA)Analyse moléculaire (USA)
anoctamine 5 (2004), protéine transmembranaire
fukutine (1998), protéine impliquée dans la glycosylation des protéines
Analyse moléculaire (USA)
O-mannosyl transférase 2 (2002), enzyme impliquée dans la glycosylation
de certaines protéines
Étude observationnelle (USA)Analyse moléculaire (USA)
O-mannose bêta1,2N-acétylglucosaminyl-transférase 1 (2002), enzyme impliquée dans
la glycosylation de certaines protéines
Étude observationnelle (USA)Analyse moléculaire (USA)
plectine, protéine de structure qui lie les membranes cellulaires aux fi laments intermédiaires du squelette des cellules, aux fi laments d’actine et microtubules
myotiline (1999), protéine sarcomérique de la strie Z qui se lie à l’alpha-actinine dans les muscles striés et cardiaque
souris myotiline
lamines A/C (1999), protéines associées à la membrane nucléaire
souris lamines A/C KO et KI, drosophile
cavéoline-3 (1998), protéine du sarcolemme
souris mutante cavéoline-3
double homeobox protein 4 (2007)poisson fugu, souristransgéniques FRG1,
FAT1, souris myd
Greffe de myoblastes autologues (France)
Étude de corrélation phénotype/génotype (France)- Supplémentation antioxydants Vit C, Vit E, Zn, Se (France) - Exercice physique (France) - Étude observationnelle chez
l’enfant (Canada) - Analyse moléculaire (USA) - Supplémentation protéique et exercice physique (Danemark) - Étude IRM et MRD des muscles squelettiques (USA) - Registre de patients (USA)
émerine (1994), protéine de la membrane nucléaire des fi bres musculaires,
protéine FHL1 (1999) impliquée dans le développement du tissu musculaire
souris émerine KO
souris FHL1 KO
Analyse moléculaire
(USA)
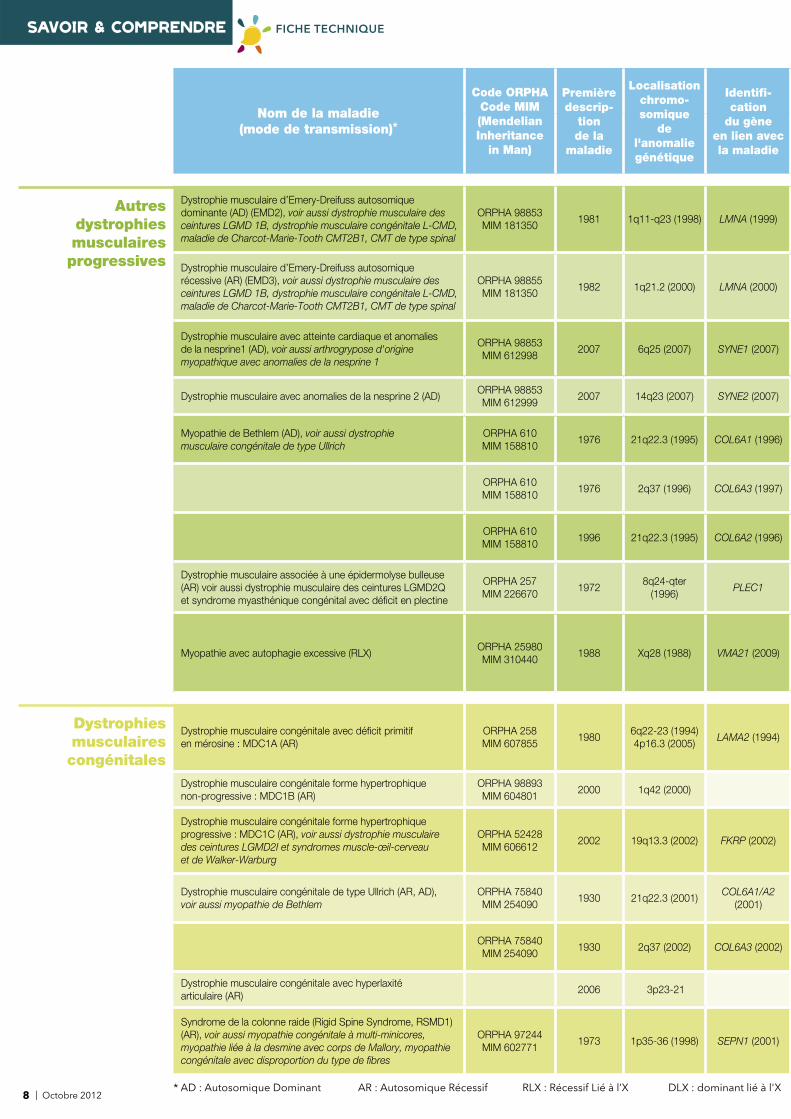
SAVOIR & COMPRENDRE FICHE TECHNIQUE
8 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Autres dystrophies musculaires progressives
Dystrophie musculaire d’Emery-Dreifuss autosomique dominante (AD) (EMD2), voir aussi dystrophie musculaire des ceintures LGMD 1B, dystrophie musculaire congénitale L-CMD, maladie de Charcot-Marie-Tooth CMT2B1, CMT de type spinal
ORPHA 98853MIM 181350
1981 1q11-q23 (1998) LMNA (1999)
Dystrophie musculaire d’Emery-Dreifuss autosomique récessive (AR) (EMD3), voir aussi dystrophie musculaire des ceintures LGMD 1B, dystrophie musculaire congénitale L-CMD, maladie de Charcot-Marie-Tooth CMT2B1, CMT de type spinal
ORPHA 98855MIM 181350
1982 1q21.2 (2000) LMNA (2000)
Dystrophie musculaire avec atteinte cardiaque et anomalies de la nesprine1 (AD), voir aussi arthrogrypose d'origine myopathique avec anomalies de la nesprine 1
ORPHA 98853MIM 612998
2007 6q25 (2007) SYNE1 (2007)
Dystrophie musculaire avec anomalies de la nesprine 2 (AD) ORPHA 98853MIM 612999
2007 14q23 (2007) SYNE2 (2007)
Myopathie de Bethlem (AD), voir aussi dystrophie musculaire congénitale de type Ullrich
ORPHA 610MIM 158810
1976 21q22.3 (1995) COL6A1 (1996)
ORPHA 610MIM 158810
1976 2q37 (1996) COL6A3 (1997)
ORPHA 610MIM 158810
1996 21q22.3 (1995) COL6A2 (1996)
Dystrophie musculaire associée à une épidermolyse bulleuse (AR) voir aussi dystrophie musculaire des ceintures LGMD2Q et syndrome myasthénique congénital avec défi cit en plectine
ORPHA 257MIM 226670
19728q24-qter
(1996)PLEC1
Myopathie avec autophagie excessive (RLX)ORPHA 25980MIM 310440
1988 Xq28 (1988) VMA21 (2009)
Dystrophies musculaires congénitales
Dystrophie musculaire congénitale avec défi cit primitif en mérosine : MDC1A (AR)
ORPHA 258 MIM 607855
19806q22-23 (1994) 4p16.3 (2005)
LAMA2 (1994)
Dystrophie musculaire congénitale forme hypertrophique non-progressive : MDC1B (AR)
ORPHA 98893 MIM 604801
2000 1q42 (2000)
Dystrophie musculaire congénitale forme hypertrophique progressive : MDC1C (AR), voir aussi dystrophie musculaire des ceintures LGMD2I et syndromes muscle-œil-cerveau et de Walker-Warburg
ORPHA 52428 MIM 606612
2002 19q13.3 (2002) FKRP (2002)
Dystrophie musculaire congénitale de type Ullrich (AR, AD), voir aussi myopathie de Bethlem
ORPHA 75840 MIM 254090
1930 21q22.3 (2001) COL6A1/A2
(2001)
ORPHA 75840MIM 254090
1930 2q37 (2002) COL6A3 (2002)
Dystrophie musculaire congénitale avec hyperlaxité articulaire (AR)
2006 3p23-21
Syndrome de la colonne raide (Rigid Spine Syndrome, RSMD1) (AR), voir aussi myopathie congénitale à multi-minicores, myopathie liée à la desmine avec corps de Mallory, myopathie congénitale avec disproportion du type de fi bres
ORPHA 97244MIM 602771
1973 1p35-36 (1998) SEPN1 (2001)
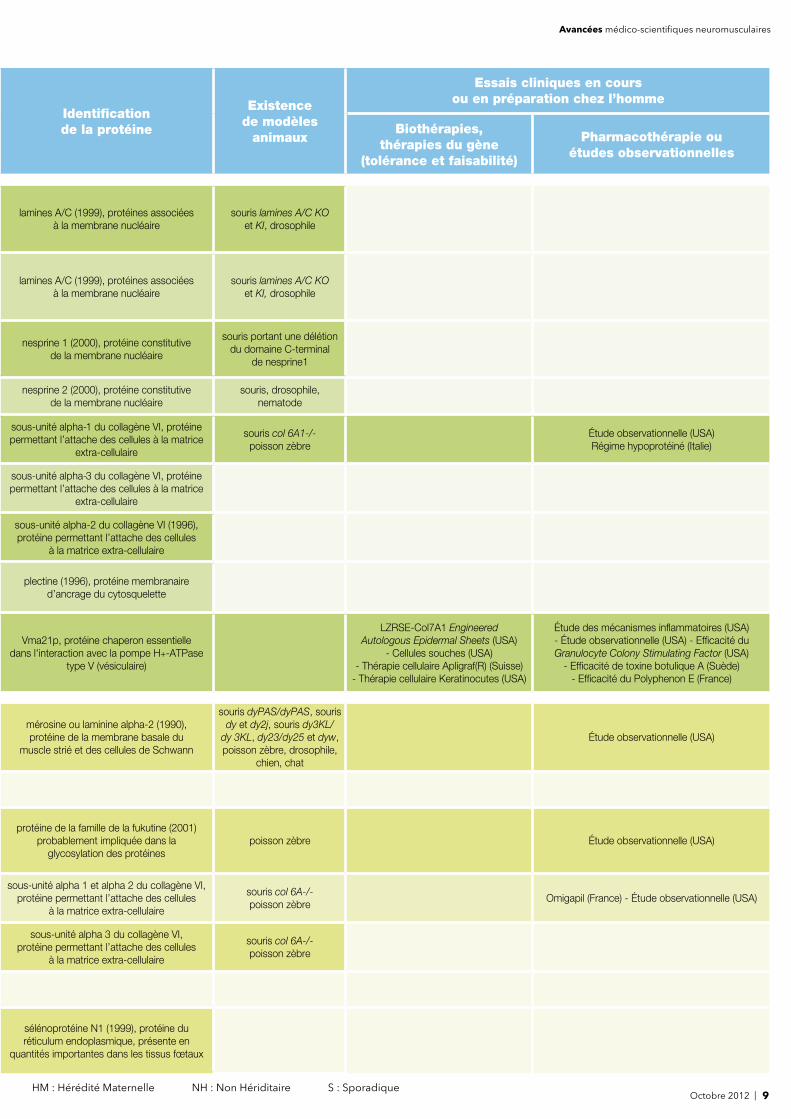
Octobre 2012 | 9 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
lamines A/C (1999), protéines associées à la membrane nucléaire
souris lamines A/C KO et KI, drosophile
lamines A/C (1999), protéines associées à la membrane nucléaire
souris lamines A/C KOet KI, drosophile
nesprine 1 (2000), protéine constitutive de la membrane nucléaire
souris portant une délétiondu domaine C-terminal
de nesprine1
nesprine 2 (2000), protéine constitutive de la membrane nucléaire
souris, drosophile, nematode
sous-unité alpha-1 du collagène VI, protéine permettant l’attache des cellules à la matrice
extra-cellulaire
souris col 6A1-/-poisson zèbre
Étude observationnelle (USA)Régime hypoprotéiné (Italie)
sous-unité alpha-3 du collagène VI, protéine permettant l’attache des cellules à la matrice
extra-cellulaire
sous-unité alpha-2 du collagène VI (1996), protéine permettant l’attache des cellules
à la matrice extra-cellulaire
plectine (1996), protéine membranaire d’ancrage du cytosquelette
Vma21p, protéine chaperon essentielle dans l'interaction avec la pompe H+-ATPase
type V (vésiculaire)
LZRSE-Col7A1 Engineered Autologous Epidermal Sheets (USA)
- Cellules souches (USA) - Thérapie cellulaire Apligraf(R) (Suisse)
- Thérapie cellulaire Keratinocutes (USA)
Étude des mécanismes infl ammatoires (USA)- Étude observationnelle (USA) - Effi cacité du Granulocyte Colony Stimulating Factor (USA)
- Effi cacité de toxine botulique A (Suède)- Effi cacité du Polyphenon E (France)
mérosine ou laminine alpha-2 (1990), protéine de la membrane basale du
muscle strié et des cellules de Schwann
souris dyPAS/dyPAS, sourisdy et dy2j, souris dy3KL/
dy 3KL, dy23/dy25 et dyw,poisson zèbre, drosophile,
chien, chat
Étude observationnelle (USA)
protéine de la famille de la fukutine (2001) probablement impliquée dans la
glycosylation des protéinespoisson zèbre Étude observationnelle (USA)
sous-unité alpha 1 et alpha 2 du collagène VI, protéine permettant l’attache des cellules
à la matrice extra-cellulaire
souris col 6A-/-poisson zèbre
Omigapil (France) - Étude observationnelle (USA)
sous-unité alpha 3 du collagène VI, protéine permettant l’attache des cellules
à la matrice extra-cellulaire
souris col 6A-/-poisson zèbre
sélénoprotéine N1 (1999), protéine du réticulum endoplasmique, présente en
quantités importantes dans les tissus fœtaux
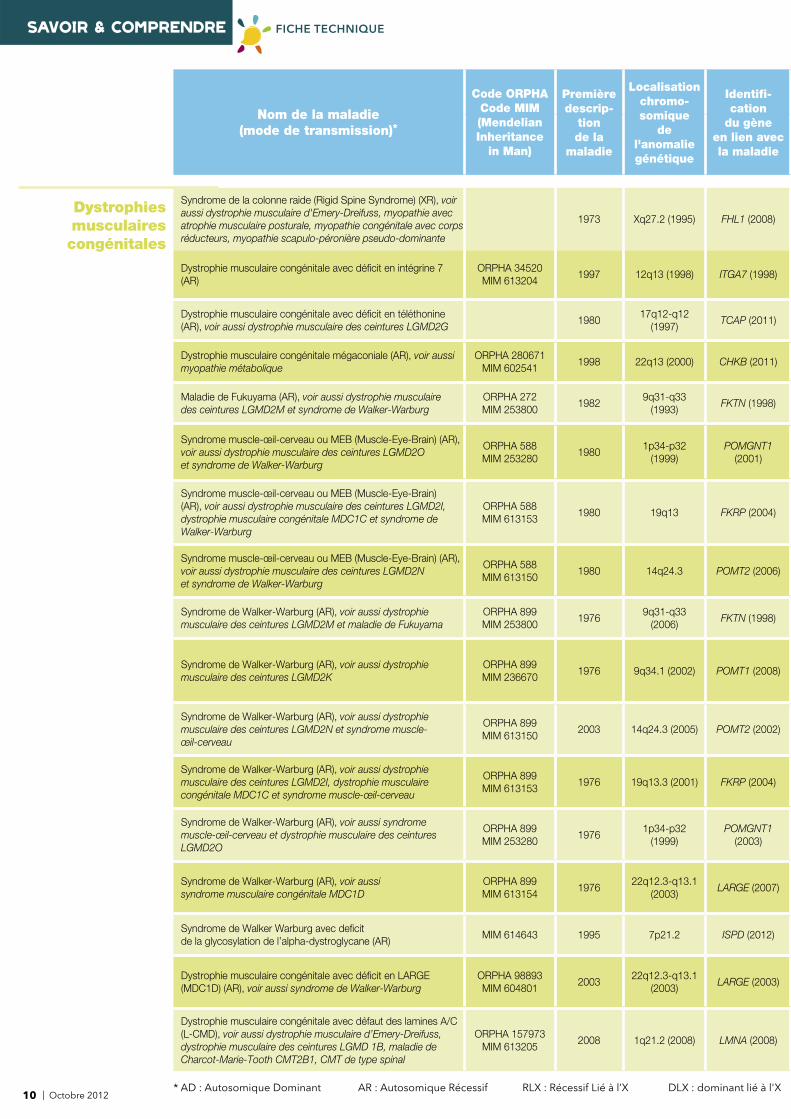
SAVOIR & COMPRENDRE FICHE TECHNIQUE
10 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Dystrophies musculaires congénitales
Syndrome de la colonne raide (Rigid Spine Syndrome) (XR), voir aussi dystrophie musculaire d'Emery-Dreifuss, myopathie avec atrophie musculaire posturale, myopathie congénitale avec corps réducteurs, myopathie scapulo-péronière pseudo-dominante
1973 Xq27.2 (1995) FHL1 (2008)
Dystrophie musculaire congénitale avec défi cit en intégrine 7 (AR)
ORPHA 34520MIM 613204
1997 12q13 (1998) ITGA7 (1998)
Dystrophie musculaire congénitale avec défi cit en téléthonine (AR), voir aussi dystrophie musculaire des ceintures LGMD2G
198017q12-q12
(1997)TCAP (2011)
Dystrophie musculaire congénitale mégaconiale (AR), voir aussi myopathie métabolique
ORPHA 280671 MIM 602541
1998 22q13 (2000) CHKB (2011)
Maladie de Fukuyama (AR), voir aussi dystrophie musculaire des ceintures LGMD2M et syndrome de Walker-Warburg
ORPHA 272 MIM 253800
19829q31-q33
(1993)FKTN (1998)
Syndrome muscle-œil-cerveau ou MEB (Muscle-Eye-Brain) (AR), voir aussi dystrophie musculaire des ceintures LGMD2O et syndrome de Walker-Warburg
ORPHA 588MIM 253280
19801p34-p32
(1999)POMGNT1
(2001)
Syndrome muscle-œil-cerveau ou MEB (Muscle-Eye-Brain) (AR), voir aussi dystrophie musculaire des ceintures LGMD2I, dystrophie musculaire congénitale MDC1C et syndrome de Walker-Warburg
ORPHA 588MIM 613153
1980 19q13 FKRP (2004)
Syndrome muscle-œil-cerveau ou MEB (Muscle-Eye-Brain) (AR), voir aussi dystrophie musculaire des ceintures LGMD2N et syndrome de Walker-Warburg
ORPHA 588MIM 613150
1980 14q24.3 POMT2 (2006)
Syndrome de Walker-Warburg (AR), voir aussi dystrophie musculaire des ceintures LGMD2M et maladie de Fukuyama
ORPHA 899MIM 253800
19769q31-q33
(2006) FKTN (1998)
Syndrome de Walker-Warburg (AR), voir aussi dystrophie musculaire des ceintures LGMD2K
ORPHA 899MIM 236670
1976 9q34.1 (2002) POMT1 (2008)
Syndrome de Walker-Warburg (AR), voir aussi dystrophie musculaire des ceintures LGMD2N et syndrome muscle-œil-cerveau
ORPHA 899MIM 613150
2003 14q24.3 (2005) POMT2 (2002)
Syndrome de Walker-Warburg (AR), voir aussi dystrophie musculaire des ceintures LGMD2I, dystrophie musculaire congénitale MDC1C et syndrome muscle-œil-cerveau
ORPHA 899MIM 613153
1976 19q13.3 (2001) FKRP (2004)
Syndrome de Walker-Warburg (AR), voir aussi syndrome muscle-œil-cerveau et dystrophie musculaire des ceintures LGMD2O
ORPHA 899MIM 253280
19761p34-p32
(1999)POMGNT1
(2003)
Syndrome de Walker-Warburg (AR), voir aussi syndrome musculaire congénitale MDC1D
ORPHA 899MIM 613154
197622q12.3-q13.1
(2003)LARGE (2007)
Syndrome de Walker Warburg avec defi cit de la glycosylation de l’alpha-dystroglycane (AR)
MIM 614643 1995 7p21.2 ISPD (2012)
Dystrophie musculaire congénitale avec défi cit en LARGE (MDC1D) (AR), voir aussi syndrome de Walker-Warburg
ORPHA 98893MIM 604801
200322q12.3-q13.1
(2003)LARGE (2003)
Dystrophie musculaire congénitale avec défaut des lamines A/C (L-CMD), voir aussi dystrophie musculaire d’Emery-Dreifuss, dystrophie musculaire des ceintures LGMD 1B, maladie de Charcot-Marie-Tooth CMT2B1, CMT de type spinal
ORPHA 157973MIM 613205
2008 1q21.2 (2008) LMNA (2008)
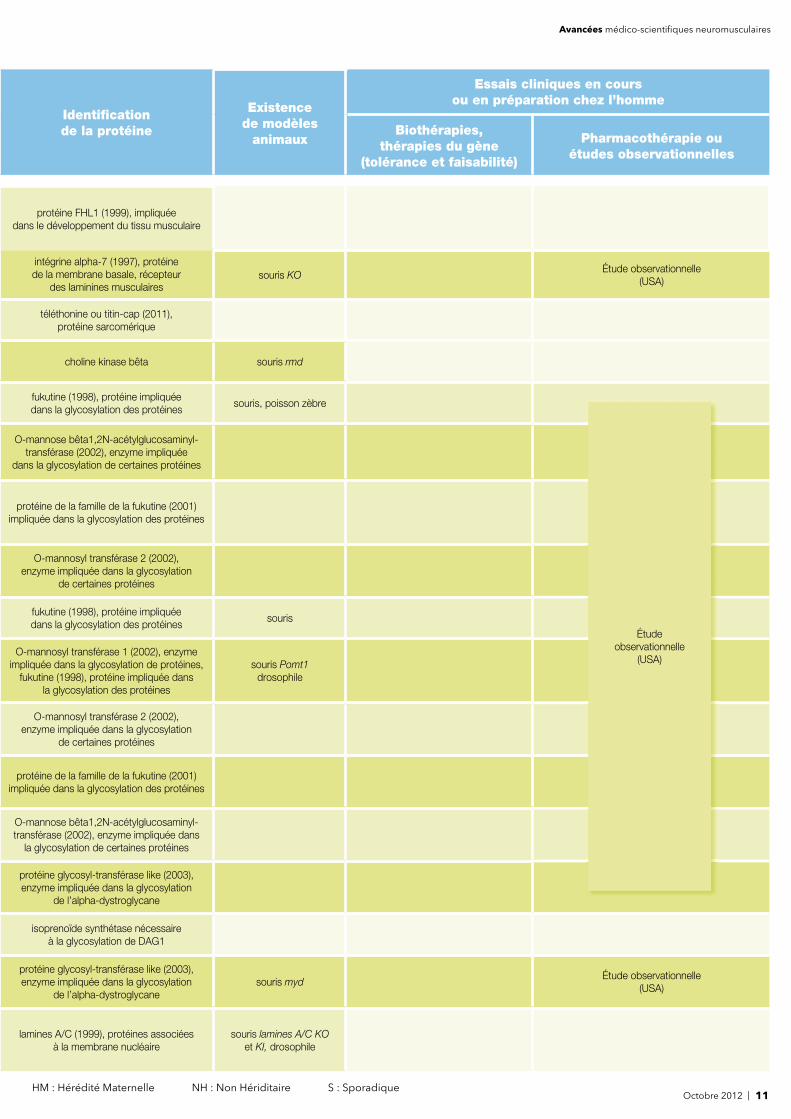
Octobre 2012 | 11 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
protéine FHL1 (1999), impliquée dans le développement du tissu musculaire
intégrine alpha-7 (1997), protéine de la membrane basale, récepteur
des laminines musculairessouris KO
Étude observationnelle (USA)
téléthonine ou titin-cap (2011), protéine sarcomérique
choline kinase bêta souris rmd
fukutine (1998), protéine impliquée dans la glycosylation des protéines
souris, poisson zèbre
O-mannose bêta1,2N-acétylglucosaminyl-transférase (2002), enzyme impliquée
dans la glycosylation de certaines protéines
Étude observationnelle (2006-2020 / USA)
protéine de la famille de la fukutine (2001) impliquée dans la glycosylation des protéines
Étude observationnelle (2006-2020 / USA)
O-mannosyl transférase 2 (2002), enzyme impliquée dans la glycosylation
de certaines protéines
Étude observationnelle (2006-2020 / USA)
fukutine (1998), protéine impliquée dans la glycosylation des protéines
sourisÉtude observationnelle
(2006-2020 / USA)
O-mannosyl transférase 1 (2002), enzyme impliquée dans la glycosylation de protéines,
fukutine (1998), protéine impliquée dans la glycosylation des protéines
souris Pomt1drosophile
Étude observationnelle (2006-2020 / USA)
O-mannosyl transférase 2 (2002), enzyme impliquée dans la glycosylation
de certaines protéines
Étude observationnelle (2006-2020 / USA)
protéine de la famille de la fukutine (2001) impliquée dans la glycosylation des protéines
Étude observationnelle (2006-2020 / USA)
O-mannose bêta1,2N-acétylglucosaminyl-transférase (2002), enzyme impliquée dans
la glycosylation de certaines protéines
Étude observationnelle (2006-2020 / USA)
protéine glycosyl-transférase like (2003), enzyme impliquée dans la glycosylation
de l’alpha-dystroglycane
isoprenoïde synthétase nécessaire à la glycosylation de DAG1
protéine glycosyl-transférase like (2003), enzyme impliquée dans la glycosylation
de l’alpha-dystroglycanesouris myd
Étude observationnelle (USA)
lamines A/C (1999), protéines associées à la membrane nucléaire
souris lamines A/C KOet KI, drosophile
Étude observationnelle (2006-2020 / USA)
Étude observationnelle (2006-2020 / USA)
Étude observationnelle (2006-2020 / USA)
Étude observationnelle (2006-2020 / USA)
Étude observationnelle (2006-2020 / USA)
Étude observationnelle (2006-2020 / USA)
Étude observationnelle (2006-2020 / USA)
Étude observationnelle (2006-2020 / USA)
Étude observationnelle
(USA)
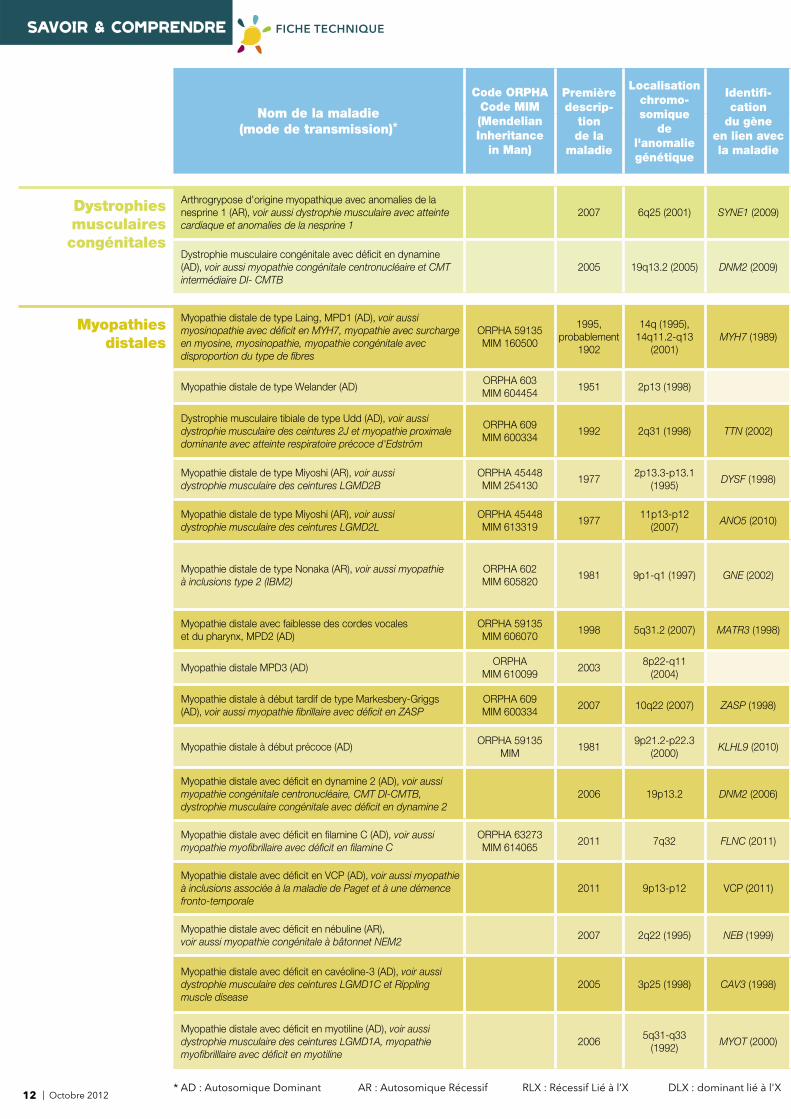
SAVOIR & COMPRENDRE FICHE TECHNIQUE
12 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Dystrophies musculaires congénitales
Arthrogrypose d'origine myopathique avec anomalies de la nesprine 1 (AR), voir aussi dystrophie musculaire avec atteinte cardiaque et anomalies de la nesprine 1
2007 6q25 (2001) SYNE1 (2009)
Dystrophie musculaire congénitale avec défi cit en dynamine (AD), voir aussi myopathie congénitale centronucléaire et CMT intermédiaire DI- CMTB
2005 19q13.2 (2005) DNM2 (2009)
Myopathies distales
Myopathie distale de type Laing, MPD1 (AD), voir aussi myosinopathie avec défi cit en MYH7, myopathie avec surcharge en myosine, myosinopathie, myopathie congénitale avec disproportion du type de fi bres
ORPHA 59135MIM 160500
1995, probablement
1902
14q (1995), 14q11.2-q13
(2001)MYH7 (1989)
Myopathie distale de type Welander (AD)ORPHA 603MIM 604454
1951 2p13 (1998)
Dystrophie musculaire tibiale de type Udd (AD), voir aussi dystrophie musculaire des ceintures 2J et myopathie proximale dominante avec atteinte respiratoire précoce d'Edström
ORPHA 609MIM 600334
1992 2q31 (1998) TTN (2002)
Myopathie distale de type Miyoshi (AR), voir aussi dystrophie musculaire des ceintures LGMD2B
ORPHA 45448MIM 254130
19772p13.3-p13.1
(1995)DYSF (1998)
Myopathie distale de type Miyoshi (AR), voir aussi dystrophie musculaire des ceintures LGMD2L
ORPHA 45448MIM 613319
197711p13-p12
(2007)ANO5 (2010)
Myopathie distale de type Nonaka (AR), voir aussi myopathie à inclusions type 2 (IBM2)
ORPHA 602MIM 605820
1981 9p1-q1 (1997) GNE (2002)
Myopathie distale avec faiblesse des cordes vocales et du pharynx, MPD2 (AD)
ORPHA 59135MIM 606070
1998 5q31.2 (2007) MATR3 (1998)
Myopathie distale MPD3 (AD)ORPHA
MIM 6100992003
8p22-q11 (2004)
Myopathie distale à début tardif de type Markesbery-Griggs (AD), voir aussi myopathie fi brillaire avec défi cit en ZASP
ORPHA 609MIM 600334
2007 10q22 (2007) ZASP (1998)
Myopathie distale à début précoce (AD)ORPHA 59135
MIM1981
9p21.2-p22.3 (2000)
KLHL9 (2010)
Myopathie distale avec défi cit en dynamine 2 (AD), voir aussi myopathie congénitale centronucléaire, CMT DI-CMTB, dystrophie musculaire congénitale avec défi cit en dynamine 2
2006 19p13.2 DNM2 (2006)
Myopathie distale avec défi cit en fi lamine C (AD), voir aussi myopathie myofi brillaire avec défi cit en fi lamine C
ORPHA 63273MIM 614065
2011 7q32 FLNC (2011)
Myopathie distale avec défi cit en VCP (AD), voir aussi myopathie à inclusions associée à la maladie de Paget et à une démence fronto-temporale
2011 9p13-p12 VCP (2011)
Myopathie distale avec défi cit en nébuline (AR), voir aussi myopathie congénitale à bâtonnet NEM2
2007 2q22 (1995) NEB (1999)
Myopathie distale avec défi cit en cavéoline-3 (AD), voir aussi dystrophie musculaire des ceintures LGMD1C et Rippling muscle disease
2005 3p25 (1998) CAV3 (1998)
Myopathie distale avec défi cit en myotiline (AD), voir aussi dystrophie musculaire des ceintures LGMD1A, myopathie myofi brilllaire avec défi cit en myotiline
20065q31-q33
(1992)MYOT (2000)
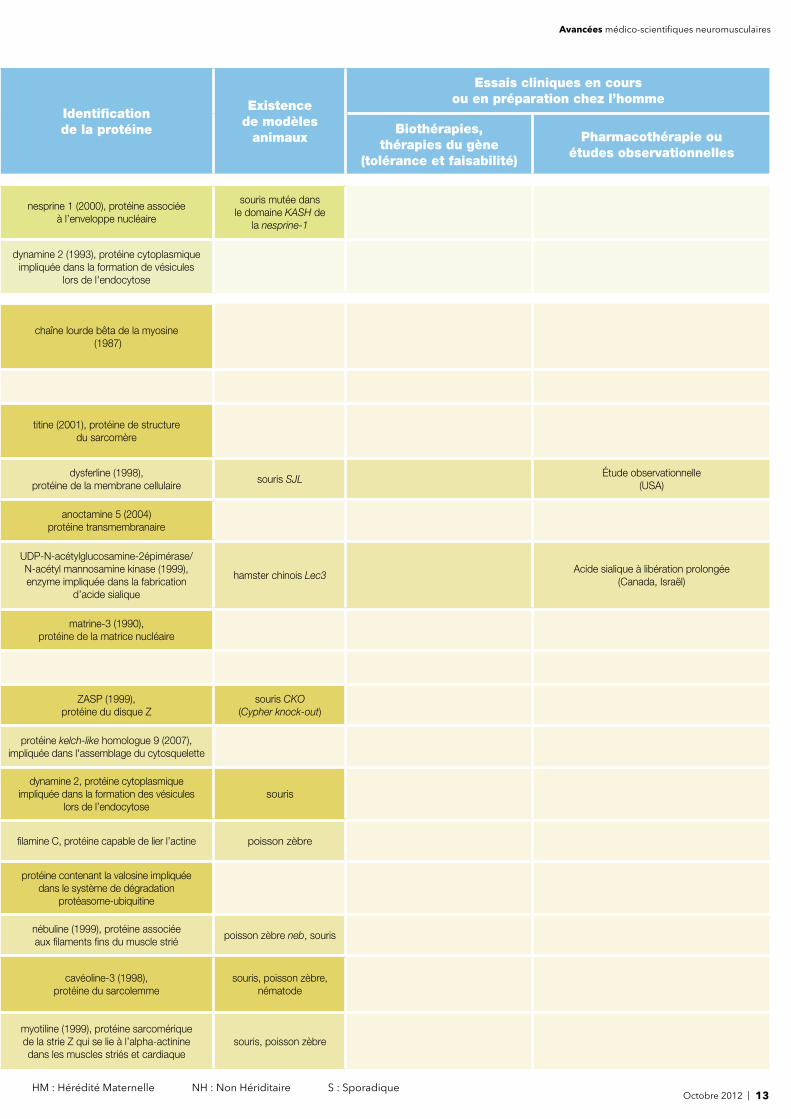
Octobre 2012 | 13 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
nesprine 1 (2000), protéine associée à l’enveloppe nucléaire
souris mutée dans le domaine KASH de
la nesprine-1
dynamine 2 (1993), protéine cytoplasmique impliquée dans la formation de vésicules
lors de l'endocytose
chaîne lourde bêta de la myosine (1987)
titine (2001), protéine de structure du sarcomère
dysferline (1998), protéine de la membrane cellulaire
souris SJLÉtude observationnelle
(USA)
anoctamine 5 (2004) protéine transmembranaire
UDP-N-acétylglucosamine-2épimérase/N-acétyl mannosamine kinase (1999), enzyme impliquée dans la fabrication
d’acide sialique
hamster chinois Lec3Acide sialique à libération prolongée
(Canada, Israël)
matrine-3 (1990), protéine de la matrice nucléaire
ZASP (1999), protéine du disque Z
souris CKO(Cypher knock-out)
protéine kelch-like homologue 9 (2007), impliquée dans l'assemblage du cytosquelette
dynamine 2, protéine cytoplasmique impliquée dans la formation des vésicules
lors de l’endocytosesouris
fi lamine C, protéine capable de lier l’actine poisson zèbre
protéine contenant la valosine impliquée dans le système de dégradation
protéasome-ubiquitine
nébuline (1999), protéine associée aux fi laments fi ns du muscle strié
poisson zèbre neb, souris
cavéoline-3 (1998), protéine du sarcolemme
souris, poisson zèbre, nématode
myotiline (1999), protéine sarcomériquede la strie Z qui se lie à l’alpha-actinine dans les muscles striés et cardiaque
souris, poisson zèbre
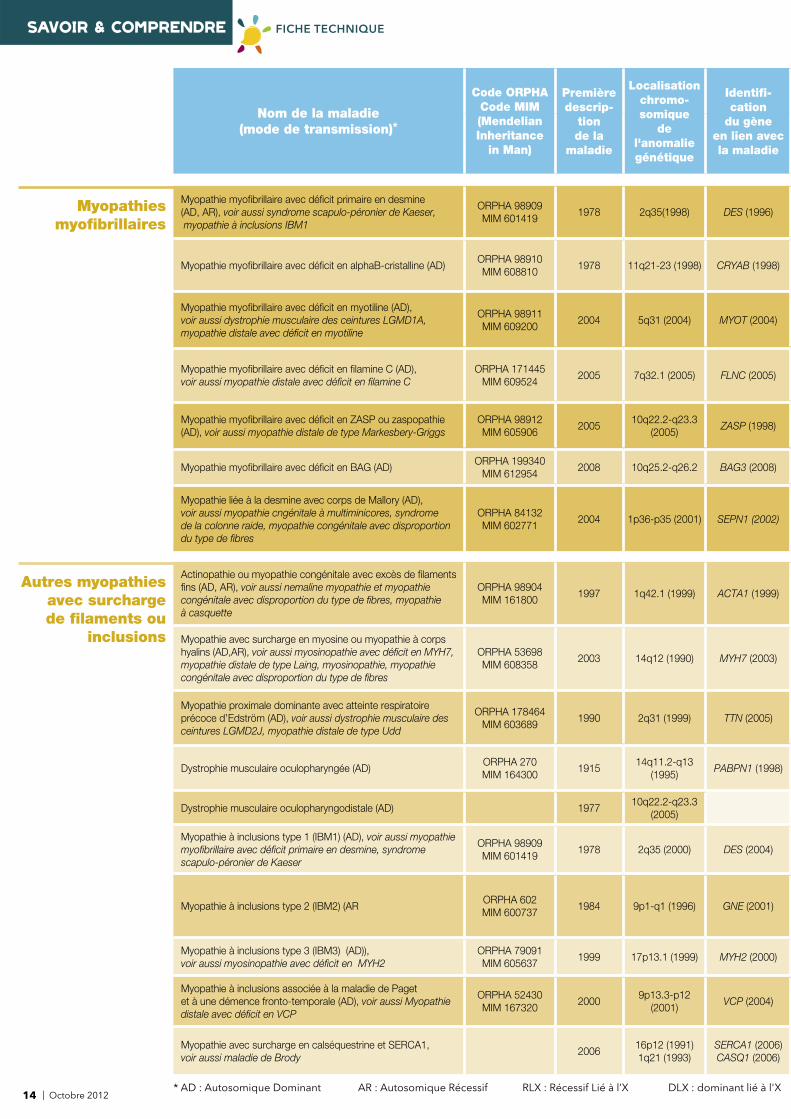
SAVOIR & COMPRENDRE FICHE TECHNIQUE
14 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Myopathies myofi brillaires
Myopathie myofi brillaire avec défi cit primaire en desmine (AD, AR), voir aussi syndrome scapulo-péronier de Kaeser, myopathie à inclusions IBM1
ORPHA 98909MIM 601419
1978 2q35(1998) DES (1996)
Myopathie myofi brillaire avec défi cit en alphaB-cristalline (AD)ORPHA 98910MIM 608810
1978 11q21-23 (1998) CRYAB (1998)
Myopathie myofi brillaire avec défi cit en myotiline (AD), voir aussi dystrophie musculaire des ceintures LGMD1A, myopathie distale avec défi cit en myotiline
ORPHA 98911MIM 609200
2004 5q31 (2004) MYOT (2004)
Myopathie myofi brillaire avec défi cit en fi lamine C (AD), voir aussi myopathie distale avec défi cit en fi lamine C
ORPHA 171445MIM 609524
2005 7q32.1 (2005) FLNC (2005)
Myopathie myofi brillaire avec défi cit en ZASP ou zaspopathie (AD), voir aussi myopathie distale de type Markesbery-Griggs
ORPHA 98912MIM 605906
200510q22.2-q23.3
(2005)ZASP (1998)
Myopathie myofi brillaire avec défi cit en BAG (AD)ORPHA 199340
MIM 6129542008 10q25.2-q26.2 BAG3 (2008)
Myopathie liée à la desmine avec corps de Mallory (AD), voir aussi myopathie cngénitale à multiminicores, syndrome de la colonne raide, myopathie congénitale avec disproportion du type de fi bres
ORPHA 84132MIM 602771
2004 1p36-p35 (2001) SEPN1 (2002)
Autres myopathies avec surcharge de fi laments ou
inclusions
Actinopathie ou myopathie congénitale avec excès de fi laments fi ns (AD, AR), voir aussi nemaline myopathie et myopathie congénitale avec disproportion du type de fi bres, myopathie à casquette
ORPHA 98904MIM 161800
1997 1q42.1 (1999) ACTA1 (1999)
Myopathie avec surcharge en myosine ou myopathie à corps hyalins (AD,AR), voir aussi myosinopathie avec défi cit en MYH7, myopathie distale de type Laing, myosinopathie, myopathie congénitale avec disproportion du type de fi bres
ORPHA 53698MIM 608358
2003 14q12 (1990) MYH7 (2003)
Myopathie proximale dominante avec atteinte respiratoire précoce d’Edström (AD), voir aussi dystrophie musculaire des ceintures LGMD2J, myopathie distale de type Udd
ORPHA 178464MIM 603689
1990 2q31 (1999) TTN (2005)
Dystrophie musculaire oculopharyngée (AD)ORPHA 270MIM 164300
191514q11.2-q13
(1995) PABPN1 (1998)
Dystrophie musculaire oculopharyngodistale (AD) 197710q22.2-q23.3
(2005)
Myopathie à inclusions type 1 (IBM1) (AD), voir aussi myopathie myofi brillaire avec défi cit primaire en desmine, syndrome scapulo-péronier de Kaeser
ORPHA 98909MIM 601419
1978 2q35 (2000) DES (2004)
Myopathie à inclusions type 2 (IBM2) (ARORPHA 602MIM 600737
1984 9p1-q1 (1996) GNE (2001)
Myopathie à inclusions type 3 (IBM3) (AD)), voir aussi myosinopathie avec défi cit en MYH2
ORPHA 79091MIM 605637
1999 17p13.1 (1999) MYH2 (2000)
Myopathie à inclusions associée à la maladie de Paget et à une démence fronto-temporale (AD), voir aussi Myopathie distale avec défi cit en VCP
ORPHA 52430MIM 167320
20009p13.3-p12
(2001)VCP (2004)
Myopathie avec surcharge en calséquestrine et SERCA1, voir aussi maladie de Brody
200616p12 (1991) 1q21 (1993)
SERCA1 (2006)CASQ1 (2006)
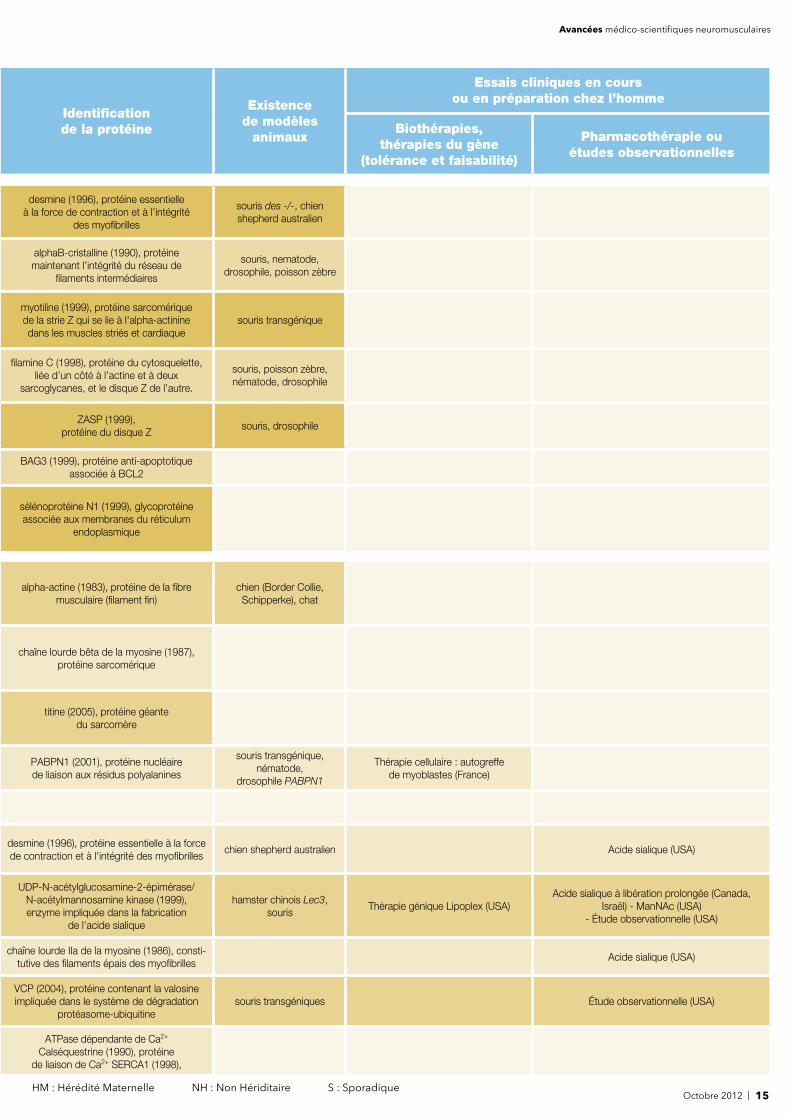
Octobre 2012 | 15 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
desmine (1996), protéine essentielle à la force de contraction et à l’intégrité
des myofi brilles
souris des -/-, chien shepherd australien
alphaB-cristalline (1990), protéine maintenant l’intégrité du réseau de
fi laments intermédiaires
souris, nematode, drosophile, poisson zèbre
myotiline (1999), protéine sarcomérique de la strie Z qui se lie à l'alpha-actinine dans les muscles striés et cardiaque
souris transgénique
fi lamine C (1998), protéine du cytosquelette, liée d’un côté à l’actine et à deux
sarcoglycanes, et le disque Z de l’autre.
souris, poisson zèbre, nématode, drosophile
ZASP (1999), protéine du disque Z
souris, drosophile
BAG3 (1999), protéine anti-apoptotiqueassociée à BCL2
sélénoprotéine N1 (1999), glycoprotéine associée aux membranes du réticulum
endoplasmique
alpha-actine (1983), protéine de la fi bre musculaire (fi lament fi n)
chien (Border Collie, Schipperke), chat
chaîne lourde bêta de la myosine (1987), protéine sarcomérique
titine (2005), protéine géante du sarcomère
PABPN1 (2001), protéine nucléaire de liaison aux résidus polyalanines
souris transgénique, nématode,
drosophile PABPN1
Thérapie cellulaire : autogreffe de myoblastes (France)
desmine (1996), protéine essentielle à la force de contraction et à l'intégrité des myofi brilles
chien shepherd australien Acide sialique (USA)
UDP-N-acétylglucosamine-2-épimérase/ N-acétylmannosamine kinase (1999), enzyme impliquée dans la fabrication
de l’acide sialique
hamster chinois Lec3, souris
Thérapie génique Lipoplex (USA)Acide sialique à libération prolongée (Canada,
Israël) - ManNAc (USA)- Étude observationnelle (USA)
chaîne lourde IIa de la myosine (1986), consti- tutive des fi laments épais des myofi brilles
Acide sialique (USA)
VCP (2004), protéine contenant la valosineimpliquée dans le système de dégradation
protéasome-ubiquitinesouris transgéniques Étude observationnelle (USA)
ATPase dépendante de Ca2+
Calséquestrine (1990), protéine de liaison de Ca2+ SERCA1 (1998),
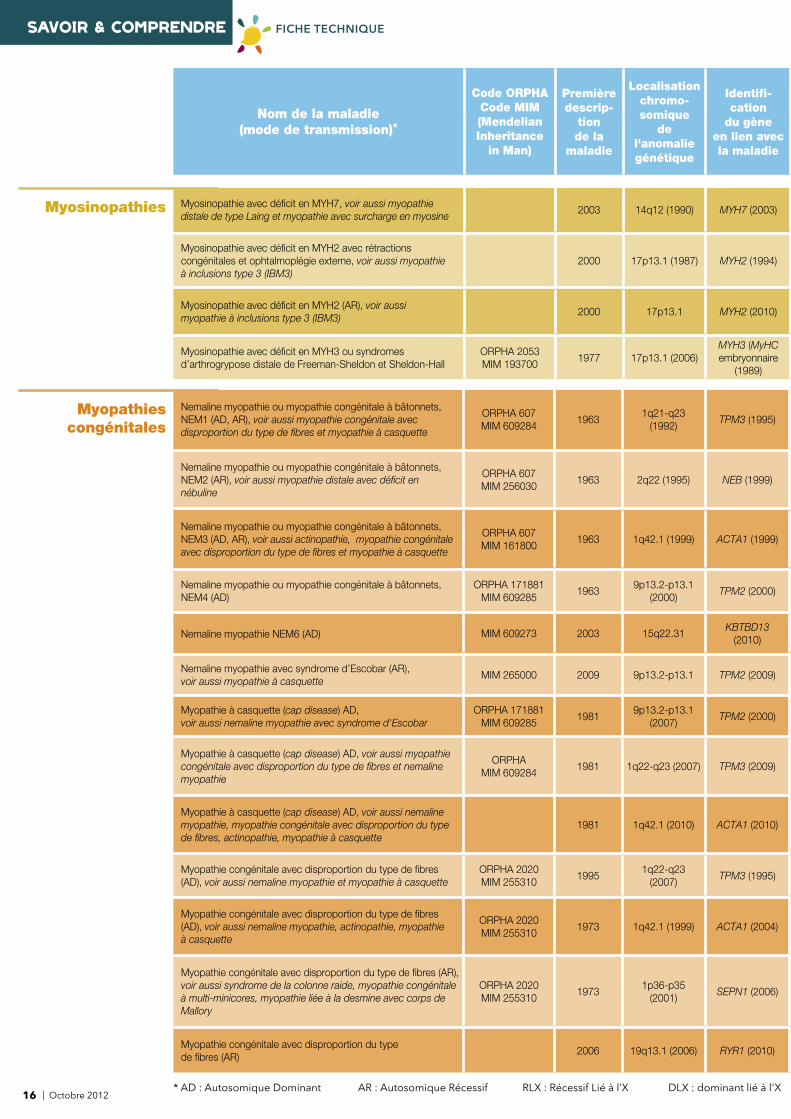
SAVOIR & COMPRENDRE FICHE TECHNIQUE
16 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Myosinopathies Myosinopathie avec défi cit en MYH7, voir aussi myopathie distale de type Laing et myopathie avec surcharge en myosine
2003 14q12 (1990) MYH7 (2003)
Myosinopathie avec défi cit en MYH2 avec rétractions congénitales et ophtalmoplégie externe, voir aussi myopathie à inclusions type 3 (IBM3)
2000 17p13.1 (1987) MYH2 (1994)
Myosinopathie avec défi cit en MYH2 (AR), voir aussi myopathie à inclusions type 3 (IBM3)
2000 17p13.1 MYH2 (2010)
Myosinopathie avec défi cit en MYH3 ou syndromes d’arthrogrypose distale de Freeman-Sheldon et Sheldon-Hall
ORPHA 2053MIM 193700
1977 17p13.1 (2006)MYH3 (MyHC embryonnaire
(1989)
Myopathies congénitales
Nemaline myopathie ou myopathie congénitale à bâtonnets, NEM1 (AD, AR), voir aussi myopathie congénitale avec disproportion du type de fi bres et myopathie à casquette
ORPHA 607MIM 609284
19631q21-q23
(1992)TPM3 (1995)
Nemaline myopathie ou myopathie congénitale à bâtonnets, NEM2 (AR), voir aussi myopathie distale avec défi cit en nébuline
ORPHA 607MIM 256030
1963 2q22 (1995) NEB (1999)
Nemaline myopathie ou myopathie congénitale à bâtonnets, NEM3 (AD, AR), voir aussi actinopathie, myopathie congénitale avec disproportion du type de fi bres et myopathie à casquette
ORPHA 607MIM 161800
1963 1q42.1 (1999) ACTA1 (1999)
Nemaline myopathie ou myopathie congénitale à bâtonnets, NEM4 (AD)
ORPHA 171881MIM 609285
19639p13.2-p13.1
(2000)TPM2 (2000)
Nemaline myopathie NEM6 (AD) MIM 609273 2003 15q22.31KBTBD13
(2010)
Nemaline myopathie avec syndrome d’Escobar (AR), voir aussi myopathie à casquette
MIM 265000 2009 9p13.2-p13.1 TPM2 (2009)
Myopathie à casquette (cap disease) AD, voir aussi nemaline myopathie avec syndrome d'Escobar
ORPHA 171881MIM 609285
19819p13.2-p13.1
(2007)TPM2 (2000)
Myopathie à casquette (cap disease) AD, voir aussi myopathie congénitale avec disproportion du type de fi bres et nemaline myopathie
ORPHAMIM 609284
1981 1q22-q23 (2007) TPM3 (2009)
Myopathie à casquette (cap disease) AD, voir aussi nemaline myopathie, myopathie congénitale avec disproportion du type de fi bres, actinopathie, myopathie à casquette
1981 1q42.1 (2010) ACTA1 (2010)
Myopathie congénitale avec disproportion du type de fi bres (AD), voir aussi nemaline myopathie et myopathie à casquette
ORPHA 2020MIM 255310
19951q22-q23
(2007)TPM3 (1995)
Myopathie congénitale avec disproportion du type de fi bres (AD), voir aussi nemaline myopathie, actinopathie, myopathie à casquette
ORPHA 2020MIM 255310
1973 1q42.1 (1999) ACTA1 (2004)
Myopathie congénitale avec disproportion du type de fi bres (AR), voir aussi syndrome de la colonne raide, myopathie congénitale à multi-minicores, myopathie liée à la desmine avec corps de Mallory
ORPHA 2020MIM 255310
19731p36-p35
(2001)SEPN1 (2006)
Myopathie congénitale avec disproportion du type de fi bres (AR)
2006 19q13.1 (2006) RYR1 (2010)
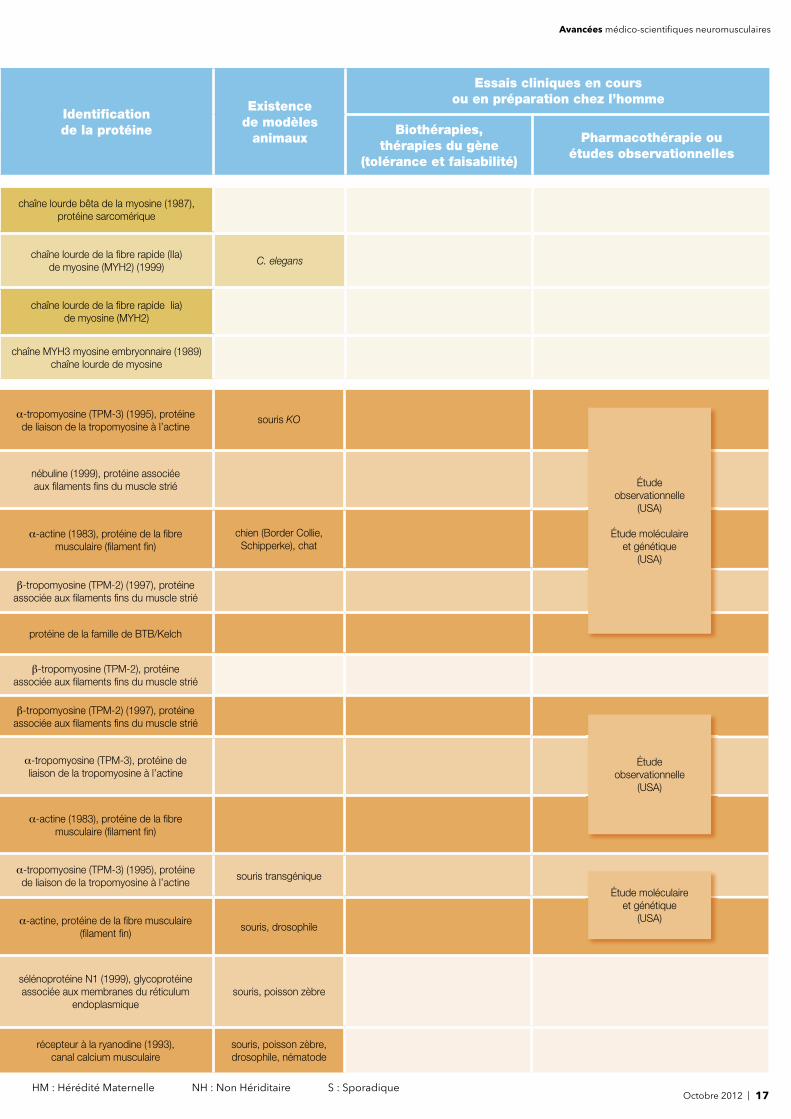
Octobre 2012 | 17 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
chaîne lourde bêta de la myosine (1987), protéine sarcomérique
chaîne lourde de la fi bre rapide (IIa) de myosine (MYH2) (1999)
C. elegans
chaîne lourde de la fi bre rapide Iia) de myosine (MYH2)
chaîne MYH3 myosine embryonnaire (1989) chaîne lourde de myosine
α-tropomyosine (TPM-3) (1995), protéine de liaison de la tropomyosine à l’actine
souris KO
nébuline (1999), protéine associée aux fi laments fi ns du muscle strié
α-actine (1983), protéine de la fi bre musculaire (fi lament fi n)
chien (Border Collie, Schipperke), chat
β-tropomyosine (TPM-2) (1997), protéine associée aux fi laments fi ns du muscle strié
protéine de la famille de BTB/Kelch
β-tropomyosine (TPM-2), protéine associée aux fi laments fi ns du muscle strié
β-tropomyosine (TPM-2) (1997), protéine associée aux fi laments fi ns du muscle strié
α-tropomyosine (TPM-3), protéine de liaison de la tropomyosine à l’actine
α-actine (1983), protéine de la fi bre musculaire (fi lament fi n)
α-tropomyosine (TPM-3) (1995), protéine de liaison de la tropomyosine à l’actine
souris transgénique
α-actine, protéine de la fi bre musculaire (fi lament fi n)
souris, drosophile
sélénoprotéine N1 (1999), glycoprotéine associée aux membranes du réticulum
endoplasmiquesouris, poisson zèbre
récepteur à la ryanodine (1993), canal calcium musculaire
souris, poisson zèbre, drosophile, nématode
Étude observationnelle
(USA)
Étude moléculaire et génétique
(USA)
Étude observationnelle
(USA)
Étude moléculaire et génétique
(USA)
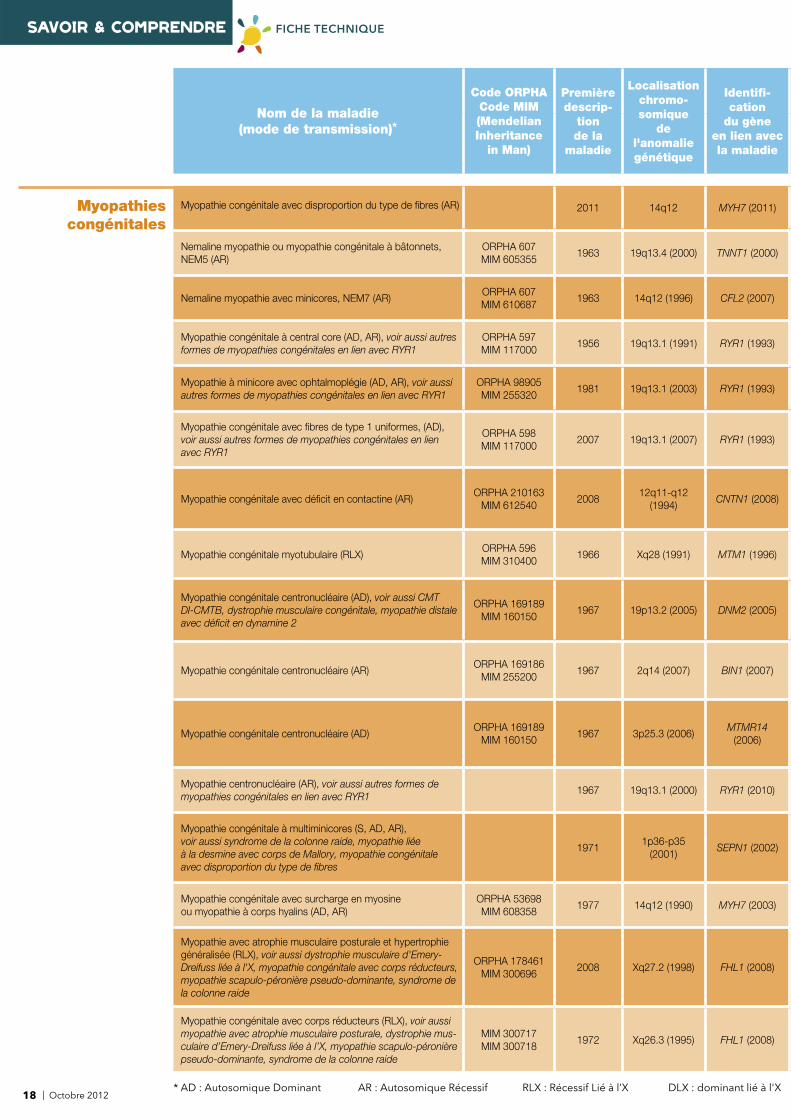
SAVOIR & COMPRENDRE FICHE TECHNIQUE
18 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Myopathies congénitales
Myopathie congénitale avec disproportion du type de fi bres (AR) 2011 14q12 MYH7 (2011)
Nemaline myopathie ou myopathie congénitale à bâtonnets, NEM5 (AR)
ORPHA 607MIM 605355
1963 19q13.4 (2000) TNNT1 (2000)
Nemaline myopathie avec minicores, NEM7 (AR)ORPHA 607MIM 610687
1963 14q12 (1996) CFL2 (2007)
Myopathie congénitale à central core (AD, AR), voir aussi autres formes de myopathies congénitales en lien avec RYR1
ORPHA 597MIM 117000
1956 19q13.1 (1991) RYR1 (1993)
Myopathie à minicore avec ophtalmoplégie (AD, AR), voir aussi autres formes de myopathies congénitales en lien avec RYR1
ORPHA 98905MIM 255320
1981 19q13.1 (2003) RYR1 (1993)
Myopathie congénitale avec fi bres de type 1 uniformes, (AD), voir aussi autres formes de myopathies congénitales en lien avec RYR1
ORPHA 598MIM 117000
2007 19q13.1 (2007) RYR1 (1993)
Myopathie congénitale avec défi cit en contactine (AR)ORPHA 210163
MIM 6125402008
12q11-q12 (1994)
CNTN1 (2008)
Myopathie congénitale myotubulaire (RLX)ORPHA 596MIM 310400
1966 Xq28 (1991) MTM1 (1996)
Myopathie congénitale centronucléaire (AD), voir aussi CMT DI-CMTB, dystrophie musculaire congénitale, myopathie distale avec défi cit en dynamine 2
ORPHA 169189MIM 160150
1967 19p13.2 (2005) DNM2 (2005)
Myopathie congénitale centronucléaire (AR)ORPHA 169186
MIM 2552001967 2q14 (2007) BIN1 (2007)
Myopathie congénitale centronucléaire (AD)ORPHA 169189
MIM 1601501967 3p25.3 (2006)
MTMR14 (2006)
Myopathie centronucléaire (AR), voir aussi autres formes de myopathies congénitales en lien avec RYR1
1967 19q13.1 (2000) RYR1 (2010)
Myopathie congénitale à multiminicores (S, AD, AR), voir aussi syndrome de la colonne raide, myopathie liée à la desmine avec corps de Mallory, myopathie congénitale avec disproportion du type de fi bres
19711p36-p35
(2001)SEPN1 (2002)
Myopathie congénitale avec surcharge en myosine ou myopathie à corps hyalins (AD, AR)
ORPHA 53698MIM 608358
1977 14q12 (1990) MYH7 (2003)
Myopathie avec atrophie musculaire posturale et hypertrophie généralisée (RLX), voir aussi dystrophie musculaire d'Emery-Dreifuss liée à l'X, myopathie congénitale avec corps réducteurs, myopathie scapulo-péronière pseudo-dominante, syndrome de la colonne raide
ORPHA 178461MIM 300696
2008 Xq27.2 (1998) FHL1 (2008)
Myopathie congénitale avec corps réducteurs (RLX), voir aussi myopathie avec atrophie musculaire posturale, dystrophie mus-culaire d’Emery-Dreifuss liée à l’X, myopathie scapulo-péronière pseudo-dominante, syndrome de la colonne raide
MIM 300717MIM 300718
1972 Xq26.3 (1995) FHL1 (2008)
Octobre 2012 | 19 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
chaîne lourde bêta de la myosine, protéine sarcomérique
troponine T (1989), protéine associée aux fi laments fi ns du muscle strié
Étude observationnelle (USA)
cofi line-2 (1996), protéine régulatrice des fi laments d'actine
récepteur à la ryanodine (1993), canal calcium musculaire
porc stressé, souris ryr1
récepteur à la ryanodine (1993), canal calcium musculaire
récepteur à la ryanodine (1993), canal calcium musculaire
contactine-1 (1994), protéine d'adhésion neuronale avec une localisation préférentielle
au niveau de la jonction neuromusculairesouris Cntn1-/-
myotubularine (1996), enzyme de la famille des tyrosine-phosphatases
labrador retriever cnm,souris myotubularine (-),chien Manchester terrier
Étude observationnelle (USA)Étude moléculaire et génétique (USA)
dynamine-2 (2005), protéine cytoplasmique impliquée dans la formation de vésicules
lors de l'endocytose
souris, chien Border Collie
amphiphysine (1992), impliquée dans l'architecture interne de la cellule
et le transport membranairenématode
protéine (n°14) de la famille des myotubularines, phosphatase exprimée
de façon prédominante dans les muscles squelettiques et le coeur
souris MTMR14 -/-
récepteur à la ryanodine (1993), canal calcium musculaire
souris, poisson zèbre, drosophile, nématode
sélénoprotéine N1 (1999), glycoprotéine associée aux membranes du réticulum
endoplasmique
souris, poisson zèbre
chaîne lourde bêta de la myosine (1987), protéine sarcomérique
protéine FHL1 (1999), impliquée dans le développement du tissu musculaire
protéine FHL1, impliquée dans le développement musculaire
souris transgénique FHL1
Étude observationnelle (USA)
SAVOIR & COMPRENDRE FICHE TECHNIQUE
20 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Syndromes myotoniques Dystrophie myotonique de Steinert (DM1) (AD)
ORPHA 273MIM 160900
191819q13.2-13.3
(1988)DMPK (1992)
Dystrophie myotonique de type 2 (DM2) : Myopathie myotonique proximale (PROMM = proximal myotonic myopathy) (AD)
ORPHA 606MIM 602668
1994 3q21 (1999) ZNF9 (2001)
Rippling muscle disease (AD) ORPHA 97238MIM 600332
1989 1q41 (1994)
Rippling muscle disease (AR), voir aussi dystrophie musculaire des ceintures LGMD 1C et myopathie distale avec défi cit en cavéoline-3
ORPHA 97238MIM 606072
1989 3p25 (2001) CAV3 (2001)
Maladie de Brody (AR), voir aussi myopathie avec surcharge en SERCA1
ORPHA 53347MIM 601003
1969 16p12 (1996) SERCA1 (1996)
Syndrome de Schwartz-Jampel de type I ou myotonie chondrodysplasique (AR)
ORPHA 800MIM 255800
19621p34-p36.1
(1995)SJS1 (2000)
Canalopathiesmusculaires
Myotonie congénitale de Thomsen (AD), voir aussi myotonie congénitale de Becker
ORPHA 614MIM 160800
1876 7q35 (1992) CLCN1 (1992)
Myotonie congénitale de Becker (AR), voir aussi myotonie congénitale de Thomsen
ORPHA 614MIM 255700
1966 7q35 (1993) CLCN1 (1993)
Paralysie périodique hyperkaliémique (adynamie épisodique de Gamstorp) (AD), voir aussi paramyotonie d’Eulenburg, myotonie avec crampes douloureuses, myotonie fl uctuante, paralysie périodique hypokaliémique, SMC avec défi cit du canal sodium
ORPHA 682MIM 170500
1956 17q13.1-q13.3
(1990)SCN4A (1990)
Paramyotonie d’Eulenburg (AD), voir aussi paralysies périodiques hyperkaliémique, hypokaliémique, myotonie avec crampes douloureuses, myotonie fl uctuante, SMC avec défi cit du canal sodium
ORPHA 684MIM 168300
1886 17q13.1-q13.3
(1991)SCN4A (1991)
Myotonie avec crampes douloureuses (AD), voir aussi paralysies périodiques hyperkaliémique, hypokaliémique, paramyotonie d’Eulenburg, myotonie fl uctuante, SMC avec défi cit du canal sodium
ORPHA 997736MIM 608390
197117q13.1-q13.3
(1994)SCN4A (1994)
Myotonie fl uctuante (AD), voir aussi paralysies périodiques hyperkaliémique, hypokaliémique, paramyotonie d’Eulenburg, myotonie avec crampes douloureuses, SMC avec défi cit du canal sodium
ORPHA 997734MIM 608390
199017q13.1-q13.3
(1994)SCN4A (1994)
Paralysie périodique hypokaliémique type I (maladie de Westphal) (AD)
ORPHA 681MIM 170400
1979 1q31-32 (1994)CACNAIS (1994)(ex CACNL1A3)
Paralysie périodique hypokaliémique type II (AD)ORPHA 681MIM 613345
197917q13.1-q13.3
(1999)SCN4A (1999)
Paralysie périodique avec dysrythmie cardiaque, sensible au potassium ou syndrome d'Andersen-Tawil (AD)
ORPHA 37553MIM 170390
1971 17q23 (2001) KCNJ2 (2001)
Paralysie périodique (AD, AR) ORPHA 206976 200111q13-q14
(2001)KCNE3 (2001)
Octobre 2012 | 21 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
expansion de triplets dans DMPK (1994), avec piégeage d'autres
ARN messagers dans le noyau
caille, souris DM20, DM55, DM300, DM320, DM1200,
DM1700, souris DMPK, souris DM15, DM1, souris
Mbn12, souris transgéniques
Somatokine (USA)Méthylphénidate (Canada)
Mexilétine (USA)Pronostic cardiaque et respiratoire (France)
Étude sur les arythmies (USA)
expansion de quadruplets dans une protéine doigt de zinc (ZNF9) (1989),
impliquée dans le piégeage intranucléaire d'ARN messagers
cavéoline-3 (1998), protéine de la membrane cellulaire
souris cavéoline-3
adénosine triphosphatase Ca++ dépendante du reticulum endoplasmique
(1995), protéine impliquée dans la production d'énergie
bœuf Chianina ATP2A1 -/-, poisson zèbre
ATP2A1/SERCA1
perlecan (1993), protéoglycane de la membrane basale dans l’enveloppe
des faisceaux musculaires
souris Hspg2 -/-,nématode
canal chlore musculaire (1992), canal ionique membranaire
souris adr, mto, chow chow, chèvre,
poisson torpille, souris ko
Mexilétine (France)Étude observationnelle (USA, Canada, UK)
canal chlore musculaire (1992), canal ionique membranaire
souris adr/adr, chienMiniature Schnauzer
Étude observationnelle (USA, Canada, UK)
sous-unité α du canal sodium musculaire (1990), canal ionique
membranaire
souris Scn4a, cheval Quarter Horse
Dichlorphénamide (UK, Italie, USA)
sous-unité α du canal sodium musculaire (1990), canal ionique
membranaire
souris Scn4a, cheval Quarter Horse
Mexilétine (France)Étude observationnelle (USA, Canada, UK)
sous-unité α du canal sodium musculaire (1990), canal ionique
membranaire
souris Scn4a, cheval Quarter Horse
sous-unité α du canal sodium musculaire (1990), canal ionique
membranaire
souris Scn4a, cheval Quarter Horse
sous-unité α du récepteur aux dihydropyridines (1994) canal ionique
souris mdg, souris NaV1.4-R669H, souris SCN4A, né-
matode, hamster, drosophileDichlorphénamide (UK, Italie, USA)
sous-unité α du canal sodium musculaire (1990) canal ionique membranaire
souris CACNA1S Dichlorphénamide (UK, Italie, USA)
sous-unité α indépendante du voltage d'un canal potassium (1994),
canal ionique membranairesouris KCNJ2 Étude observationnelle (USA, Canada, Italie, UK)
sous unité β du canal potassium (1999), canal ionique membranaire
SAVOIR & COMPRENDRE FICHE TECHNIQUE
22 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Myopathies métaboliques
Myopathies mitochondriales (HM, AD, AR) ORPHA 206966 1967
Myopathie mégaconiale, voir aussi dystrophie musculaire congénitale mégaconiale
ORPHA 280671MIM 602541
1964 22q13 (2000) CHKB (2011)
Nouvelle forme de myopathie mitochondrialeORPHA 26791MIM 231680
2007 4q32-qter (1993)
ETFDH (1993)(electron-
transferring-fl avoprotein
dehydrogenase)
Lipidose musculaire : défi cit en carnitine palmitoyl transférase de type II (CPT II) (AR)
ORPHA 157MIM 255110
1970 1p32 (1991) CPT2 (1994)
Lipidose musculaire : défi cit primaire systémique en carnitine (AR)
ORPHA 158MIM 212140
1975 5q31.1 (1998)SLC22A5
(1999)
Lipidose musculaire : défi cit en acyl CoA déshydrogénase (AR)
ORPHA 26792MIM 201470
198412q22-qter
(1988)
ACADS (= SCAD)
(1988)
Lipidose musculaire : défi cit en VLCAD (Very Long Chain AcylCoA Dehydrogenase)
ORPHA 26793MIM 201475
1998 17p.13 (1996)ACADVL
(= VLCAD) (1996)
Rhabdomyolyse et myoglobinurie récurrente (AR) ORPHA 99845MIM 605518
2008 2p25.1 LPIN (2008)
Myopathie à surcharge lipidique (AR)ORPHA 98908MIM 610717
2007 11p15.5 (2007) PNPLA2 (2007)
Glycogénose musculaire (type II) : maladie de Pompe (AR)ORPHA 365MIM 232300
193217q25.2-25.3
(1979)GAA (1990)
Glycogénose lysosomale à activité maltase acide normale (type IIb) : maladie de Danon (RLX), voir aussi dystrophie musculaire scapulo-péronière avec retard mental et cardiomyopathie léthale
ORPHA 34587MIM 300257
1981 Xq24 (1990) LAMP2 (1995)
Glycogénose musculaire (type IIIa) : maladie de Forbes, maladie de Cori (AR)
ORPHA 366MIM 232400
1989 1p21(1992) AGL (1996)
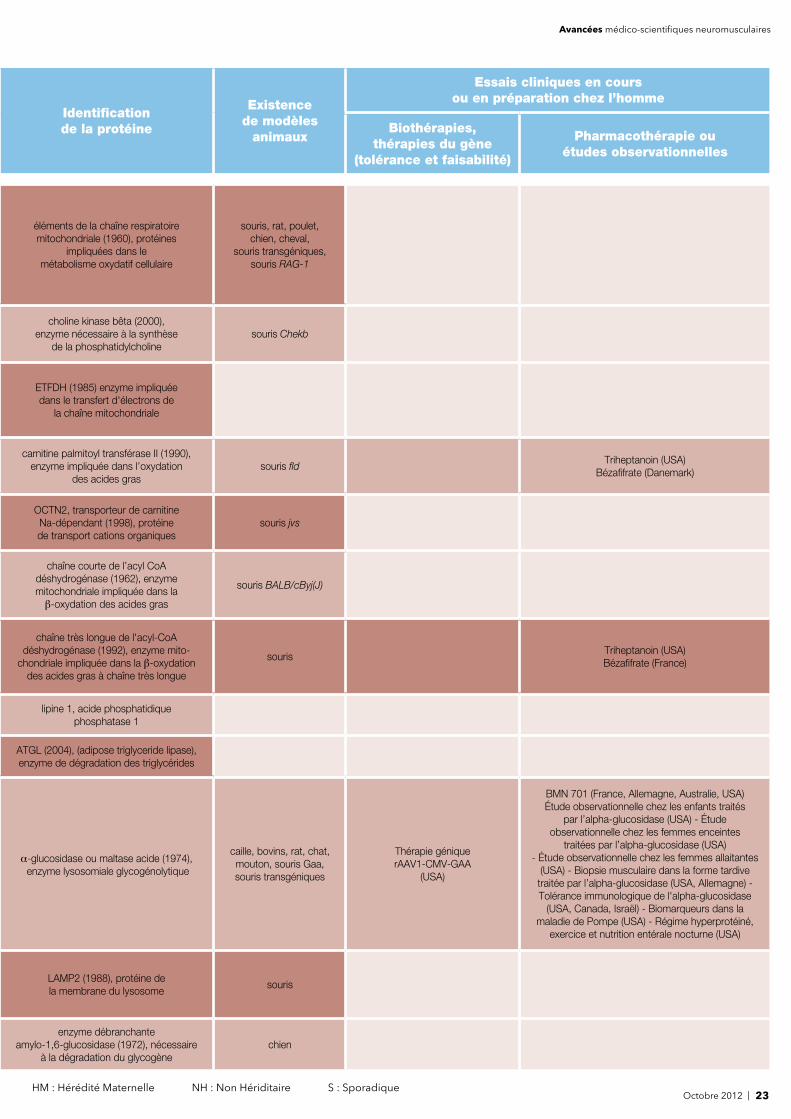
Des altérations génétiques liées à certaines de ces maladies, situées sur l'ADN mitochondrial (codant 13 sous-unités de la chaîne respiratoire qui comporte plus d'une centaine de sous-unités) ou sur l'ADN nucléaire,
ont été identifi ées dès la fi n des années 1980.
Octobre 2012 | 23 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
éléments de la chaîne respiratoire mitochondriale (1960), protéines
impliquées dans le métabolisme oxydatif cellulaire
souris, rat, poulet, chien, cheval,
souris transgéniques,souris RAG-1
choline kinase bêta (2000), enzyme nécessaire à la synthèse
de la phosphatidylcholinesouris Chekb
ETFDH (1985) enzyme impliquée dans le transfert d'électrons de
la chaîne mitochondriale
carnitine palmitoyl transférase II (1990), enzyme impliquée dans l’oxydation
des acides grassouris fl d
Triheptanoin (USA)Bézafi frate (Danemark)
OCTN2, transporteur de carnitine Na-dépendant (1998), protéine de transport cations organiques
souris jvs
chaîne courte de l’acyl CoA déshydrogénase (1962), enzyme mitochondriale impliquée dans la β-oxydation des acides gras
souris BALB/cByj(J)
chaîne très longue de l'acyl-CoA déshydrogénase (1992), enzyme mito-
chondriale impliquée dans la β-oxydation des acides gras à chaîne très longue
sourisTriheptanoin (USA)Bézafi frate (France)
lipine 1, acide phosphatidique phosphatase 1
ATGL (2004), (adipose triglyceride lipase), enzyme de dégradation des triglycérides
α-glucosidase ou maltase acide (1974), enzyme lysosomiale glycogénolytique
caille, bovins, rat, chat,mouton, souris Gaa, souris transgéniques
Thérapie génique rAAV1-CMV-GAA
(USA)
BMN 701 (France, Allemagne, Australie, USA)Étude observationnelle chez les enfants traités
par l’alpha-glucosidase (USA) - Étude observationnelle chez les femmes enceintes
traitées par l’alpha-glucosidase (USA) - Étude observationnelle chez les femmes allaitantes
(USA) - Biopsie musculaire dans la forme tardive traitée par l’alpha-glucosidase (USA, Allemagne) - Tolérance immunologique de l'alpha-glucosidase
(USA, Canada, Israël) - Biomarqueurs dans la maladie de Pompe (USA) - Régime hyperprotéiné,
exercice et nutrition entérale nocturne (USA)
LAMP2 (1988), protéine de la membrane du lysosome
souris
enzyme débranchante amylo-1,6-glucosidase (1972), nécessaire
à la dégradation du glycogènechien
SAVOIR & COMPRENDRE FICHE TECHNIQUE
24 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Myopathies métaboliques
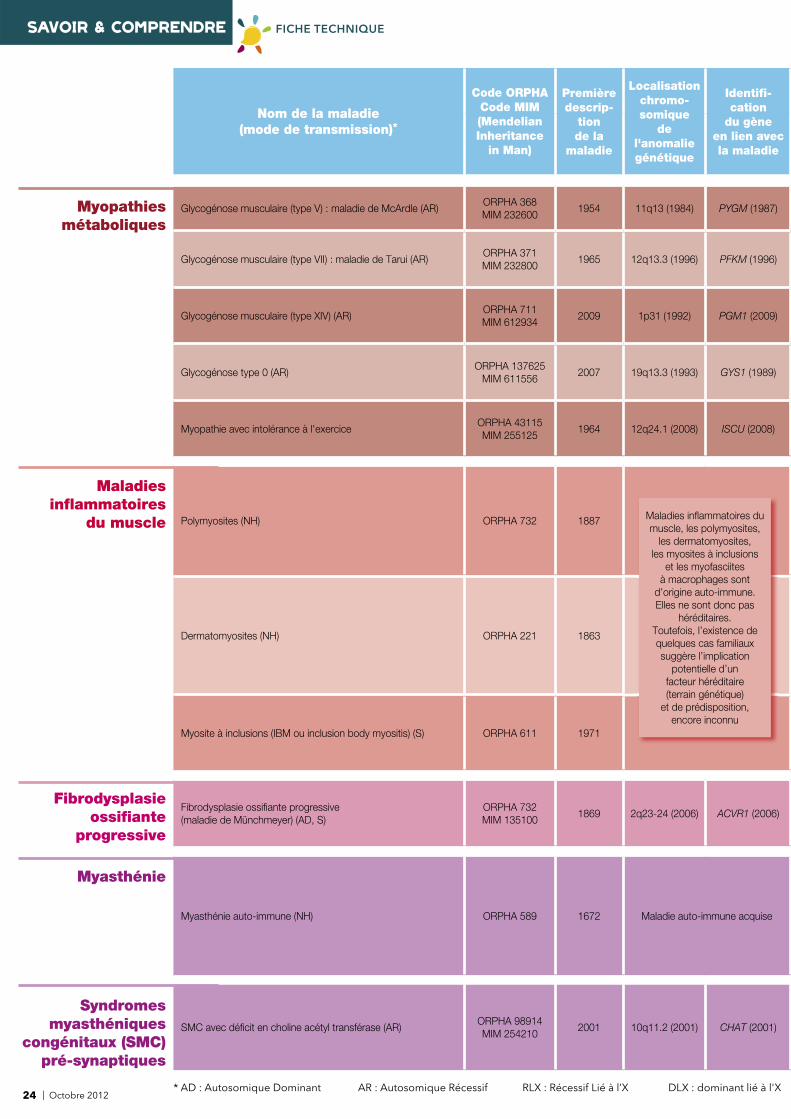
Glycogénose musculaire (type V) : maladie de McArdle (AR)ORPHA 368MIM 232600
1954 11q13 (1984) PYGM (1987)
Glycogénose musculaire (type VII) : maladie de Tarui (AR)ORPHA 371MIM 232800
1965 12q13.3 (1996) PFKM (1996)
Glycogénose musculaire (type XIV) (AR)ORPHA 711MIM 612934
2009 1p31 (1992) PGM1 (2009)
Glycogénose type 0 (AR)ORPHA 137625
MIM 6115562007 19q13.3 (1993) GYS1 (1989)
Myopathie avec intolérance à l'exerciceORPHA 43115MIM 255125
1964 12q24.1 (2008) ISCU (2008)
Maladies infl ammatoires
du muscle Polymyosites (NH) ORPHA 732 1887
Dermatomyosites (NH) ORPHA 221 1863
Myosite à inclusions (IBM ou inclusion body myositis) (S) ORPHA 611 1971
Fibrodysplasie ossifi ante
progressive
Fibrodysplasie ossifi ante progressive (maladie de Münchmeyer) (AD, S)
ORPHA 732MIM 135100
1869 2q23-24 (2006) ACVR1 (2006)
Myasthénie
Myasthénie auto-immune (NH) ORPHA 589 1672 Maladie auto-immune acquise
Syndromesmyasthéniques
congénitaux (SMC)pré-synaptiques
SMC avec défi cit en choline acétyl transférase (AR)ORPHA 98914MIM 254210
2001 10q11.2 (2001) CHAT (2001)
Maladies infl ammatoires dumuscle, les polymyosites,
les dermatomyosites, les myosites à inclusions
et les myofasciites à macrophages sont
d’origine auto-immune. Elles ne sont donc pas
héréditaires. Toutefois, l’existence de quelques cas familiaux suggère l’implication
potentielle d’un facteur héréditaire (terrain génétique)
et de prédisposition, encore inconnu

Octobre 2012 | 25 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
phosphorylase musculaire, enzyme musculaire de la glycogénolyse
mouton, bovins,souris
phosphofructokinase musculaire (1983), enzyme musculaire de la glycolyse
chiens (springer spaniel anglais, cocker américain)
souris Pfkm -/-
phosphoglucomutase 1 (1993), enzyme impliquée dans la glycolyse
et la gluconéogénèse
glycogène synthétase musculaire (1989), enzyme impliquée dans la synthèse
du glycogène
ISCU (1990) , protéine impliquée dans les processus de repliement de certains
groupements fer-soufre
rat, macaque, sourisC57BL/6, souris CIC
greffe de cellules souches hématopoïétiques
(USA)
Rituximab (France) - Infl iximab (USA)- MEDI-545 (USA) - Investigation génétique (UK)- Methotrexate + corticoïdes (Tchécoslovaquie)
- Abatacept (Suède, Tchécoslovaquie)- BAF312 (USA, UK, Suède, Tchécoslovaquie,
Hongrie, Pologne) - Étude observationnelle (USA)- Investigation de gènes (UK) - Étude par IRM (France)
Gènes de susceptibilité (UK)
chiensgreffe de cellules souches
hématopoïétiques (USA)
Infl iximab (USA) - Rituximab (France) - MEDI-545 (USA) - Etanercept (USA) - Métothrexate + corticoïdes (Tchécoslovaquie) - Investigation génétique (UK)
- Abatacept (Suède, Tchécoslovaquie) - BAF312 (USA, UK, Suède, Tchécoslovaquie, Hongrie, Pologne)
- Étude observationnelle (USA) - Gènes de susceptibilité (UK)- Étude par IRM (France) -Apremilast (USA)
- Créatine (Brésil)
souris mdm, nématode, drosophile
autogreffe de lymphocytes T régulateurs (cellules Treg) (France)- thérapie génique avec Follistatine (USA) - greffe de cellules souches
hématopoïétiques (USA)
Etanercept (USA) - Arimoclomol (USA, UK)- Rituximab (France) - Étude par IRM (France)
- Étude observationnelle (USA)- Gènes de susceptibilité (UK)
ACVR1 (2002), récepteur des protéines osseuses morphogénétiques BMP rôle dans la régulation du développement
et du modelage osseux
souris transgénique
souris EAMG, rat EAMG,cochon d'Inde EAMG,félin spontané EAMG,chien spontané Diane
Shelton WMS
Cellules souches hématopoïétiques (USA)
Rituximab (France) - Prednisone (France)- Exercice physique (USA) - Acide mycophénolique (USA,
Taïwan) - Methotrexate (USA) - Sagramostim ou GM-CSF (USA) - Étude observationnelle du rôle
du thymus (Allemagne)- Tacrolimus (Chine) - Prednisone (France) - Belimumab (USA, Canada)
- CK-2017357 (USA) - Thymectomie (USA)- Autoanticorps (USA)
choline acétyltransférase (1990), enzyme nécessaire à la synthèse de l’acétylcholine
SAVOIR & COMPRENDRE FICHE TECHNIQUE
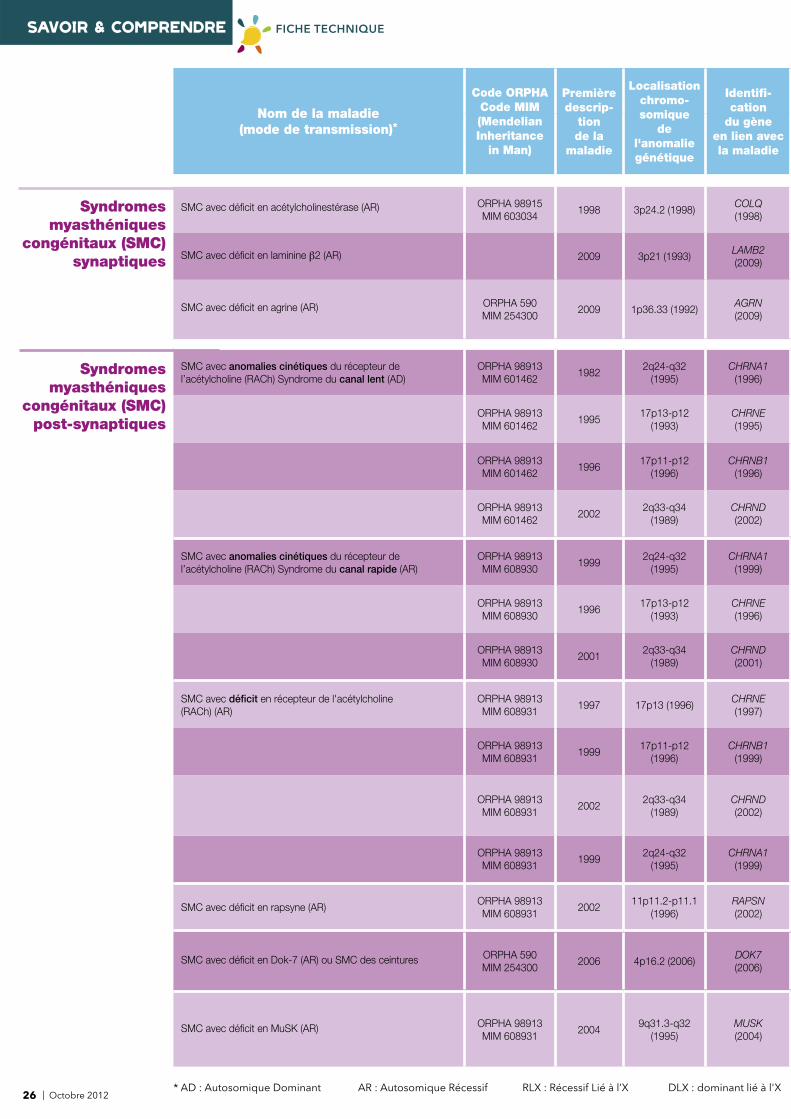
26 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Syndromesmyasthéniques
congénitaux (SMC)synaptiques
SMC avec défi cit en acétylcholinestérase (AR) ORPHA 98915MIM 603034
1998 3p24.2 (1998)COLQ (1998)
SMC avec défi cit en laminine β2 (AR) 2009 3p21 (1993)LAMB2 (2009)
SMC avec défi cit en agrine (AR) ORPHA 590MIM 254300
2009 1p36.33 (1992)AGRN (2009)
Syndromesmyasthéniques
congénitaux (SMC)post-synaptiques
SMC avec anomalies cinétiques du récepteur de l’acétylcholine (RACh) Syndrome du canal lent (AD)
ORPHA 98913MIM 601462
19822q24-q32
(1995)CHRNA1
(1996)
ORPHA 98913MIM 601462
199517p13-p12
(1993)CHRNE (1995)
ORPHA 98913MIM 601462
199617p11-p12
(1996)CHRNB1
(1996)
ORPHA 98913MIM 601462
20022q33-q34
(1989)CHRND (2002)
SMC avec anomalies cinétiques du récepteur de l’acétylcholine (RACh) Syndrome du canal rapide (AR)
ORPHA 98913MIM 608930
19992q24-q32
(1995)CHRNA1
(1999)
ORPHA 98913MIM 608930
199617p13-p12
(1993)CHRNE (1996)
ORPHA 98913MIM 608930
20012q33-q34
(1989)CHRND (2001)
SMC avec défi cit en récepteur de l'acétylcholine (RACh) (AR)
ORPHA 98913MIM 608931
1997 17p13 (1996)CHRNE (1997)
ORPHA 98913MIM 608931
199917p11-p12
(1996)CHRNB1
(1999)
ORPHA 98913MIM 608931
20022q33-q34
(1989)CHRND (2002)
ORPHA 98913MIM 608931
19992q24-q32
(1995)CHRNA1
(1999)
SMC avec défi cit en rapsyne (AR) ORPHA 98913MIM 608931
200211p11.2-p11.1
(1996)RAPSN (2002)
SMC avec défi cit en Dok-7 (AR) ou SMC des ceintures ORPHA 590MIM 254300
2006 4p16.2 (2006)DOK7 (2006)
SMC avec défi cit en MuSK (AR) ORPHA 98913MIM 608931
20049q31.3-q32
(1995)MUSK (2004)
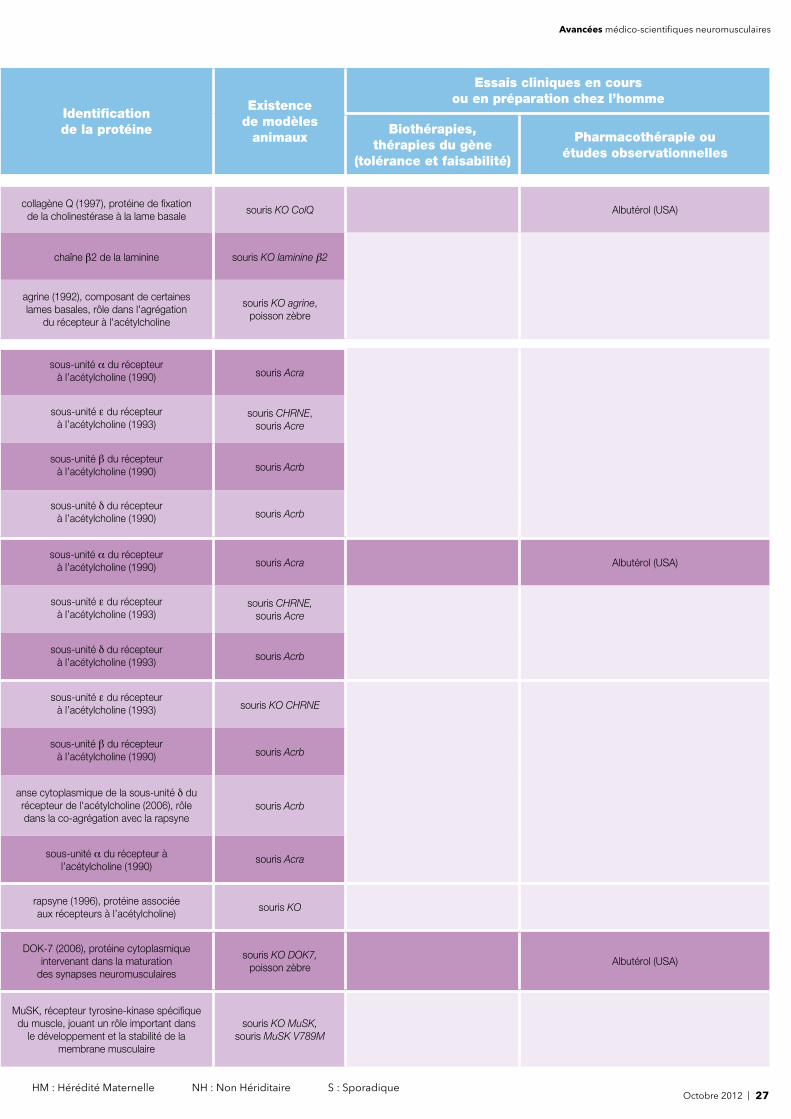
Octobre 2012 | 27 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
collagène Q (1997), protéine de fi xation de la cholinestérase à la lame basale
souris KO ColQ Albutérol (USA)
chaîne β2 de la laminine souris KO laminine β2
agrine (1992), composant de certaines lames basales, rôle dans l'agrégation
du récepteur à l'acétylcholine
souris KO agrine, poisson zèbre
sous-unité α du récepteur à l’acétylcholine (1990) souris Acra
sous-unité ε du récepteur à l’acétylcholine (1993)
souris CHRNE, souris Acre
sous-unité β du récepteur à l’acétylcholine (1990) souris Acrb
sous-unité δ du récepteur à l’acétylcholine (1990) souris Acrb
sous-unité α du récepteur à l’acétylcholine (1990) souris Acra Albutérol (USA)
sous-unité ε du récepteur à l’acétylcholine (1993)
souris CHRNE, souris Acre
sous-unité δ du récepteur à l’acétylcholine (1993) souris Acrb
sous-unité ε du récepteur à l’acétylcholine (1993) souris KO CHRNE
sous-unité β du récepteur à l’acétylcholine (1990) souris Acrb
anse cytoplasmique de la sous-unité δ durécepteur de l'acétylcholine (2006), rôle dans la co-agrégation avec la rapsyne
souris Acrb
sous-unité α du récepteur àl’acétylcholine (1990)
souris Acra
rapsyne (1996), protéine associée aux récepteurs à l’acétylcholine)
souris KO
DOK-7 (2006), protéine cytoplasmique intervenant dans la maturation
des synapses neuromusculaires
souris KO DOK7,poisson zèbre
Albutérol (USA)
MuSK, récepteur tyrosine-kinase spécifi que du muscle, jouant un rôle important dans
le développement et la stabilité de la membrane musculaire
souris KO MuSK,souris MuSK V789M
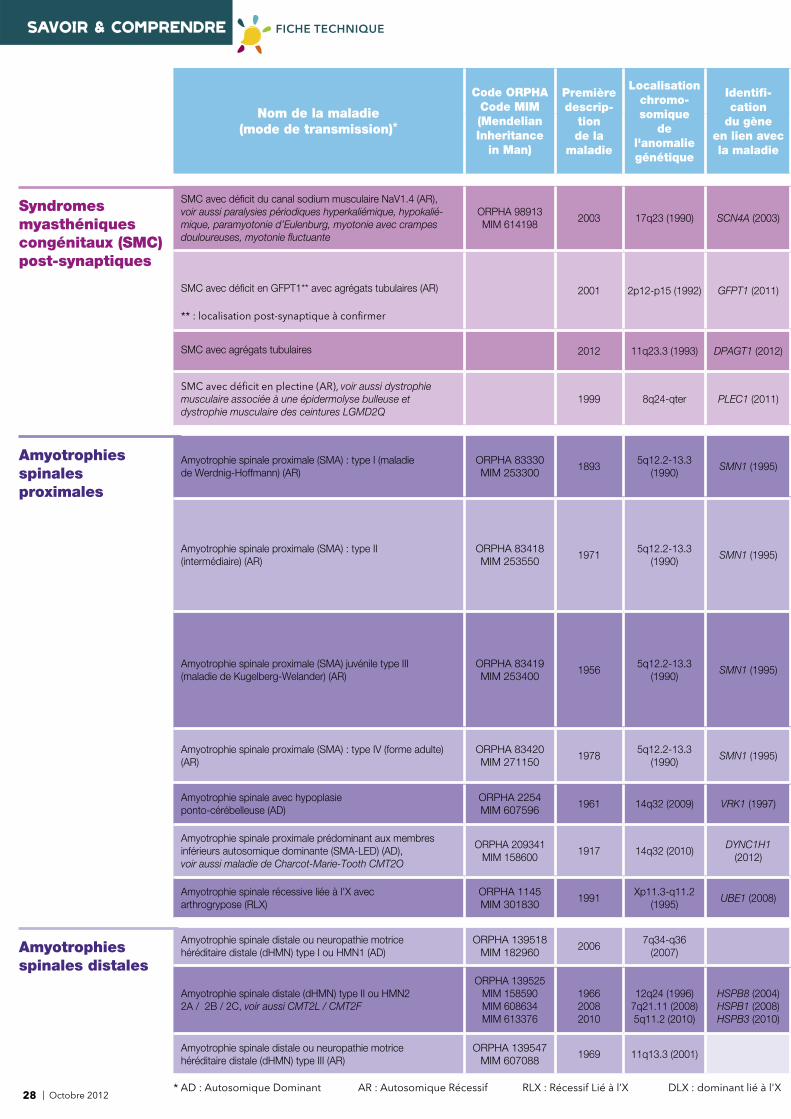
SAVOIR & COMPRENDRE FICHE TECHNIQUE
28 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Syndromesmyasthéniquescongénitaux (SMC)post-synaptiques
SMC avec défi cit du canal sodium musculaire NaV1.4 (AR), voir aussi paralysies périodiques hyperkaliémique, hypokalié-mique, paramyotonie d’Eulenburg, myotonie avec crampes douloureuses, myotonie fl uctuante
ORPHA 98913MIM 614198
2003 17q23 (1990) SCN4A (2003)
SMC avec défi cit en GFPT1** avec agrégats tubulaires (AR)
** : localisation post-synaptique à confi rmer
2001 2p12-p15 (1992) GFPT1 (2011)
SMC avec agrégats tubulaires 2012 11q23.3 (1993) DPAGT1 (2012)
SMC avec défi cit en plectine (AR), voir aussi dystrophie musculaire associée à une épidermolyse bulleuse et dystrophie musculaire des ceintures LGMD2Q
1999 8q24-qter PLEC1 (2011)
Amyotrophiesspinales proximales
Amyotrophie spinale proximale (SMA) : type I (maladie de Werdnig-Hoffmann) (AR)
ORPHA 83330MIM 253300
18935q12.2-13.3
(1990)SMN1 (1995)
Amyotrophie spinale proximale (SMA) : type II (intermédiaire) (AR)
ORPHA 83418MIM 253550
19715q12.2-13.3
(1990)SMN1 (1995)
Amyotrophie spinale proximale (SMA) juvénile type III (maladie de Kugelberg-Welander) (AR)
ORPHA 83419MIM 253400
19565q12.2-13.3
(1990)SMN1 (1995)
Amyotrophie spinale proximale (SMA) : type IV (forme adulte) (AR)
ORPHA 83420MIM 271150
19785q12.2-13.3
(1990)SMN1 (1995)
Amyotrophie spinale avec hypoplasie ponto-cérébelleuse (AD)
ORPHA 2254MIM 607596
1961 14q32 (2009) VRK1 (1997)
Amyotrophie spinale proximale prédominant aux membres inférieurs autosomique dominante (SMA-LED) (AD), voir aussi maladie de Charcot-Marie-Tooth CMT2O
ORPHA 209341MIM 158600
1917 14q32 (2010)DYNC1H1
(2012)
Amyotrophie spinale récessive liée à l'X avec arthrogrypose (RLX)
ORPHA 1145MIM 301830
1991Xp11.3-q11.2
(1995)UBE1 (2008)
Amyotrophiesspinales distales
Amyotrophie spinale distale ou neuropathie motrice héréditaire distale (dHMN) type I ou HMN1 (AD)
ORPHA 139518MIM 182960
20067q34-q36
(2007)
Amyotrophie spinale distale (dHMN) type II ou HMN22A / 2B / 2C, voir aussi CMT2L / CMT2F
ORPHA 139525MIM 158590MIM 608634MIM 613376
196620082010
12q24 (1996)7q21.11 (2008)5q11.2 (2010)
HSPB8 (2004)HSPB1 (2008)HSPB3 (2010)
Amyotrophie spinale distale ou neuropathie motrice héréditaire distale (dHMN) type III (AR)
ORPHA 139547MIM 607088
1969 11q13.3 (2001)

Octobre 2012 | 29 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
sous-unité α du canal sodium musculaire (1990), canal ionique membranaire
souris Scn4A
glutamine fructose-6-phosphate amidotransférase (2001), rôle dans la
production de glucosamine-6-phosphate pour la synthèse des glycoprotéines,
glycolipides et protéoglycanes
poisson zèbre
enzyme impliquée dans la synthèsedes glycoprotéines
plectine, protéine membranaire d’ancrage du cytosquelette
SMN1 (1995), protéine de survie du motoneurone, jouant un rôle dans le
métabolisme des ARN messagers
Acide valproïque et carnitine (USA, Canada, Allemagne) Sodium phenylbutyrate (USA) - RG3039 (USA) - Histoire naturelle (USA) - ISIS-SMNRx (USA)
- Étude observationnelle (USA)
SMN1 (1995), protéine de survie du motoneurone, jouant un rôle dans le métabolisme des ARN messagers
ISIS-SMNRx (USA) - Étude observationnelle (France) - Étude observationnelle sur le port d'orthèses (Italie)
- Étude observationnelle (USA) - TRO19622 ou Olesoxime (France, Allemagne, Italie, UK, Pologne, Pays-Bas, Belgique) - (USA) - Phenylbutyrate (USA)
- Valproate et levocarnitine (Inde) - Entraînement progressif (USA) - RG3039 (USA)
- Exercice physique (USA)
SMN1 (1995), protéine de survie du motoneurone, jouant un rôle dans le métabolisme des ARN messagers
ISIS-SMNRx (USA) - Étude observationnelle (France) - Étude observationnelle sur le port d'orthèses (Italie)
- Valproate et levocarnitine (Inde) - Étude observation-nelle (USA) - TRO19622 ou Olesoxime (France,
Allemagne, Italie, Angleterre, , Pologne, Pays-Bas, Belgique)- Hydroxyurée (USA) - 4-aminopyridine (USA)- Exercice physique (USA) - Entraînement progressif
(USA)– Nombre de copies SMN (Mali)
SMN1 (1995), protéine de survie du motoneurone, jouant un rôle dans le
métabolisme des ARN messagers
Acide valproïque (USA) - Exercice physique (USA) - Nombre de copies SMN (Mali)
VRK1, kinase essentielle à la formation de l'enveloppe nucléaire
chaîne lourde de la dynéine cytoplasmique 1, participant au transport axonal
E1 (UBE1) (2007), enzyme d'activation impliquée dans la voie ubiquitine-protéasome
Goserelin (Zoladex®) (Thailande)
HSPB8, HSPB1, HSPB3,protéines de choc thermique
souris smn, naip, dt, mdf, par,
mnd, pma, spa, spd,new, wr, vache suisse,
danoise, cheval, chien, porc
sus scrofa, lapin, macaque,C. elegans,
poisson zèbre, drosophile
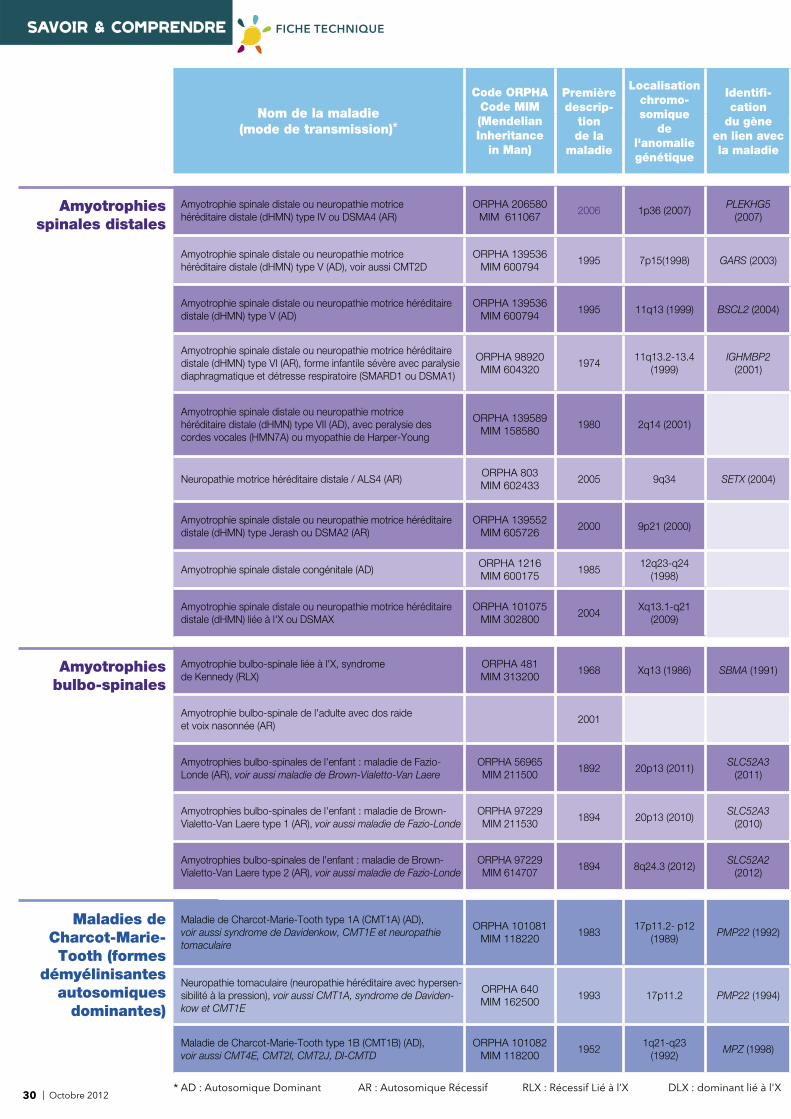
SAVOIR & COMPRENDRE FICHE TECHNIQUE
30 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Amyotrophiesspinales distales
Amyotrophie spinale distale ou neuropathie motrice héréditaire distale (dHMN) type IV ou DSMA4 (AR)
ORPHA 206580MIM 611067
2006 1p36 (2007) PLEKHG5
(2007)
Amyotrophie spinale distale ou neuropathie motrice héréditaire distale (dHMN) type V (AD), voir aussi CMT2D
ORPHA 139536MIM 600794
1995 7p15(1998) GARS (2003)
Amyotrophie spinale distale ou neuropathie motrice héréditaire distale (dHMN) type V (AD)
ORPHA 139536MIM 600794
1995 11q13 (1999) BSCL2 (2004)
Amyotrophie spinale distale ou neuropathie motrice héréditaire distale (dHMN) type VI (AR), forme infantile sévère avec paralysie diaphragmatique et détresse respiratoire (SMARD1 ou DSMA1)
ORPHA 98920MIM 604320
197411q13.2-13.4
(1999)IGHMBP2
(2001)
Amyotrophie spinale distale ou neuropathie motrice héréditaire distale (dHMN) type VII (AD), avec peralysie des cordes vocales (HMN7A) ou myopathie de Harper-Young
ORPHA 139589MIM 158580
1980 2q14 (2001)
Neuropathie motrice héréditaire distale / ALS4 (AR)ORPHA 803MIM 602433
2005 9q34 SETX (2004)
Amyotrophie spinale distale ou neuropathie motrice héréditaire distale (dHMN) type Jerash ou DSMA2 (AR)
ORPHA 139552MIM 605726
2000 9p21 (2000)
Amyotrophie spinale distale congénitale (AD)ORPHA 1216MIM 600175
198512q23-q24
(1998)
Amyotrophie spinale distale ou neuropathie motrice héréditaire distale (dHMN) liée à l'X ou DSMAX
ORPHA 101075MIM 302800
2004Xq13.1-q21
(2009)
Amyotrophiesbulbo-spinales
Amyotrophie bulbo-spinale liée à l'X, syndrome de Kennedy (RLX)
ORPHA 481MIM 313200
1968 Xq13 (1986) SBMA (1991)
Amyotrophie bulbo-spinale de l'adulte avec dos raide et voix nasonnée (AR)
2001
Amyotrophies bulbo-spinales de l'enfant : maladie de Fazio-Londe (AR), voir aussi maladie de Brown-Vialetto-Van Laere
ORPHA 56965MIM 211500
1892 20p13 (2011)SLC52A3
(2011)
Amyotrophies bulbo-spinales de l'enfant : maladie de Brown-Vialetto-Van Laere type 1 (AR), voir aussi maladie de Fazio-Londe
ORPHA 97229MIM 211530
1894 20p13 (2010)SLC52A3
(2010)
Amyotrophies bulbo-spinales de l'enfant : maladie de Brown-Vialetto-Van Laere type 2 (AR), voir aussi maladie de Fazio-Londe
ORPHA 97229MIM 614707
1894 8q24.3 (2012)SLC52A2
(2012)
Maladies de Charcot-Marie-Tooth (formes
démyélinisantes autosomiques
dominantes)
Maladie de Charcot-Marie-Tooth type 1A (CMT1A) (AD),voir aussi syndrome de Davidenkow, CMT1E et neuropathie tomaculaire
ORPHA 101081MIM 118220
198317p11.2- p12
(1989)PMP22 (1992)
Neuropathie tomaculaire (neuropathie héréditaire avec hypersen-sibilité à la pression), voir aussi CMT1A, syndrome de Daviden-kow et CMT1E
ORPHA 640 MIM 162500
1993 17p11.2 PMP22 (1994)
Maladie de Charcot-Marie-Tooth type 1B (CMT1B) (AD),voir aussi CMT4E, CMT2I, CMT2J, DI-CMTD
ORPHA 101082MIM 118200
19521q21-q23
(1992)MPZ (1998)
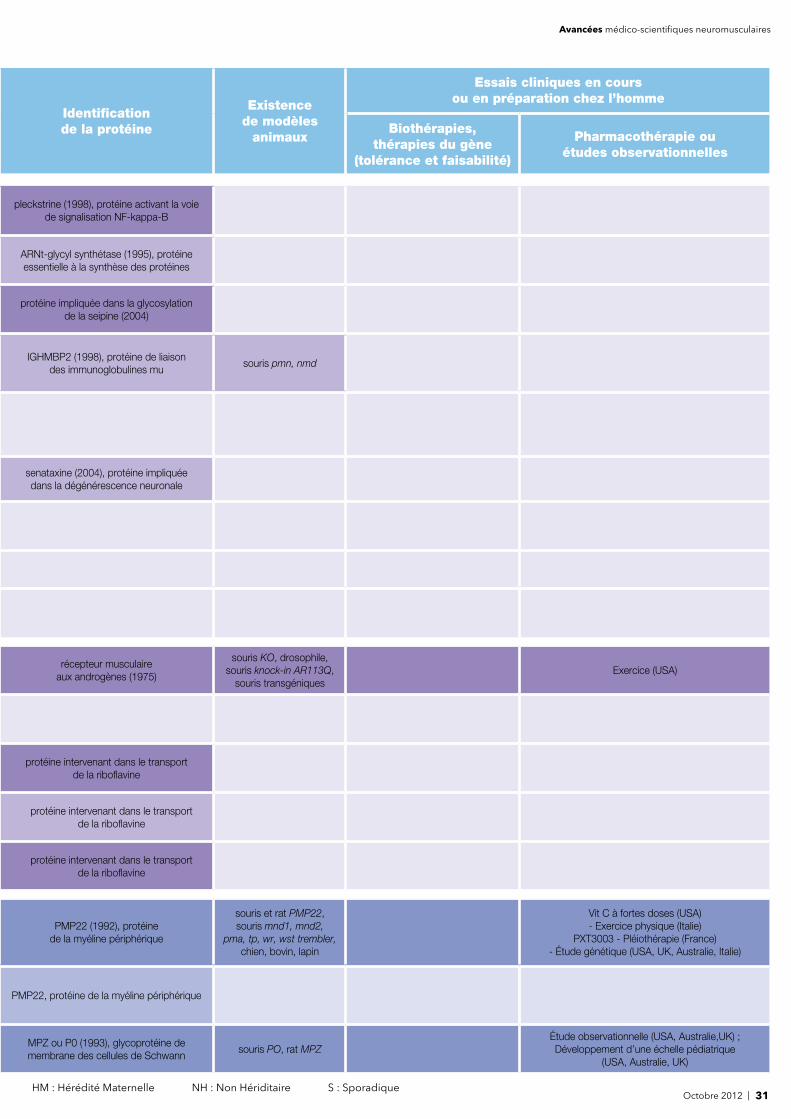
Octobre 2012 | 31 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
pleckstrine (1998), protéine activant la voie de signalisation NF-kappa-B
ARNt-glycyl synthétase (1995), protéine essentielle à la synthèse des protéines
protéine impliquée dans la glycosylation de la seipine (2004)
IGHMBP2 (1998), protéine de liaison des immunoglobulines mu
souris pmn, nmd
senataxine (2004), protéine impliquée dans la dégénérescence neuronale
récepteur musculaire aux androgènes (1975)
souris KO, drosophile,souris knock-in AR113Q,
souris transgéniquesExercice (USA)
protéine intervenant dans le transport de la ribofl avine
protéine intervenant dans le transport de la ribofl avine
protéine intervenant dans le transport de la ribofl avine
PMP22 (1992), protéine de la myéline périphérique
souris et rat PMP22,souris mnd1, mnd2,
pma, tp, wr, wst trembler, chien, bovin, lapin
Vit C à fortes doses (USA)- Exercice physique (Italie)
PXT3003 - Pléiothérapie (France)- Étude génétique (USA, UK, Australie, Italie)
PMP22, protéine de la myéline périphérique
MPZ ou P0 (1993), glycoprotéine de membrane des cellules de Schwann
souris PO, rat MPZÉtude observationnelle (USA, Australie,UK) ;Développement d’une échelle pédiatrique
(USA, Australie, UK)
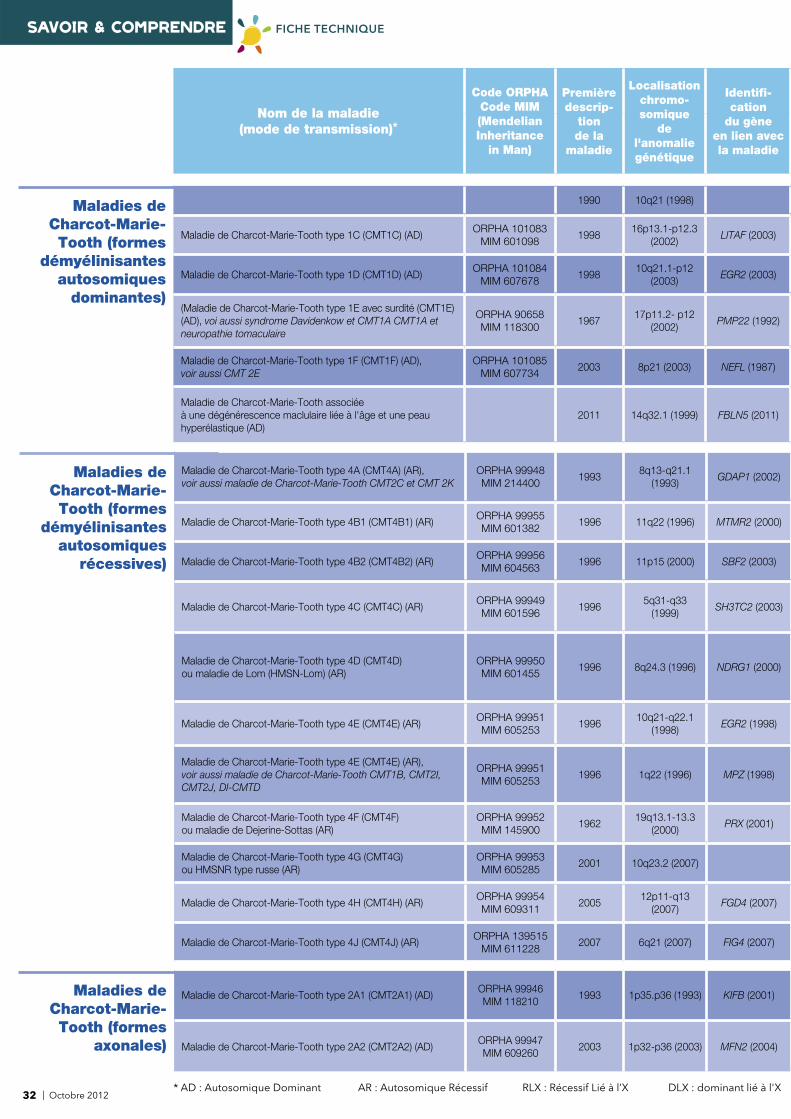
SAVOIR & COMPRENDRE FICHE TECHNIQUE
32 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Maladies de Charcot-Marie-Tooth (formes
démyélinisantes autosomiques
dominantes)
1990 10q21 (1998)
Maladie de Charcot-Marie-Tooth type 1C (CMT1C) (AD)ORPHA 101083
MIM 6010981998
16p13.1-p12.3 (2002)
LITAF (2003)
Maladie de Charcot-Marie-Tooth type 1D (CMT1D) (AD)ORPHA 101084
MIM 6076781998
10q21.1-p12 (2003)
EGR2 (2003)
(Maladie de Charcot-Marie-Tooth type 1E avec surdité (CMT1E) (AD), voi aussi syndrome Davidenkow et CMT1A CMT1A et neuropathie tomaculaire
ORPHA 90658MIM 118300
196717p11.2- p12
(2002)PMP22 (1992)
Maladie de Charcot-Marie-Tooth type 1F (CMT1F) (AD), voir aussi CMT 2E
ORPHA 101085MIM 607734
2003 8p21 (2003) NEFL (1987)
Maladie de Charcot-Marie-Tooth associée à une dégénérescence maclulaire liée à l'âge et une peau hyperélastique (AD)
2011 14q32.1 (1999) FBLN5 (2011)
Maladies de Charcot-Marie-Tooth (formes
démyélinisantes autosomiques
récessives)
Maladie de Charcot-Marie-Tooth type 4A (CMT4A) (AR), voir aussi maladie de Charcot-Marie-Tooth CMT2C et CMT 2K
ORPHA 99948MIM 214400
19938q13-q21.1
(1993)GDAP1 (2002)
Maladie de Charcot-Marie-Tooth type 4B1 (CMT4B1) (AR)ORPHA 99955MIM 601382
1996 11q22 (1996) MTMR2 (2000)
Maladie de Charcot-Marie-Tooth type 4B2 (CMT4B2) (AR)ORPHA 99956MIM 604563
1996 11p15 (2000) SBF2 (2003)
Maladie de Charcot-Marie-Tooth type 4C (CMT4C) (AR)ORPHA 99949MIM 601596
19965q31-q33
(1999)SH3TC2 (2003)
Maladie de Charcot-Marie-Tooth type 4D (CMT4D) ou maladie de Lom (HMSN-Lom) (AR)
ORPHA 99950MIM 601455
1996 8q24.3 (1996) NDRG1 (2000)
Maladie de Charcot-Marie-Tooth type 4E (CMT4E) (AR)ORPHA 99951MIM 605253
199610q21-q22.1
(1998)EGR2 (1998)
Maladie de Charcot-Marie-Tooth type 4E (CMT4E) (AR), voir aussi maladie de Charcot-Marie-Tooth CMT1B, CMT2I, CMT2J, DI-CMTD
ORPHA 99951MIM 605253
1996 1q22 (1996) MPZ (1998)
Maladie de Charcot-Marie-Tooth type 4F (CMT4F) ou maladie de Dejerine-Sottas (AR)
ORPHA 99952MIM 145900
196219q13.1-13.3
(2000)PRX (2001)
Maladie de Charcot-Marie-Tooth type 4G (CMT4G) ou HMSNR type russe (AR)
ORPHA 99953MIM 605285
2001 10q23.2 (2007)
Maladie de Charcot-Marie-Tooth type 4H (CMT4H) (AR)ORPHA 99954MIM 609311
200512p11-q13
(2007)FGD4 (2007)
Maladie de Charcot-Marie-Tooth type 4J (CMT4J) (AR)ORPHA 139515
MIM 6112282007 6q21 (2007) FIG4 (2007)
Maladies de Charcot-Marie-Tooth (formes
axonales)
Maladie de Charcot-Marie-Tooth type 2A1 (CMT2A1) (AD)ORPHA 99946MIM 118210
1993 1p35.p36 (1993) KIFB (2001)
Maladie de Charcot-Marie-Tooth type 2A2 (CMT2A2) (AD)ORPHA 99947MIM 609260
2003 1p32-p36 (2003) MFN2 (2004)
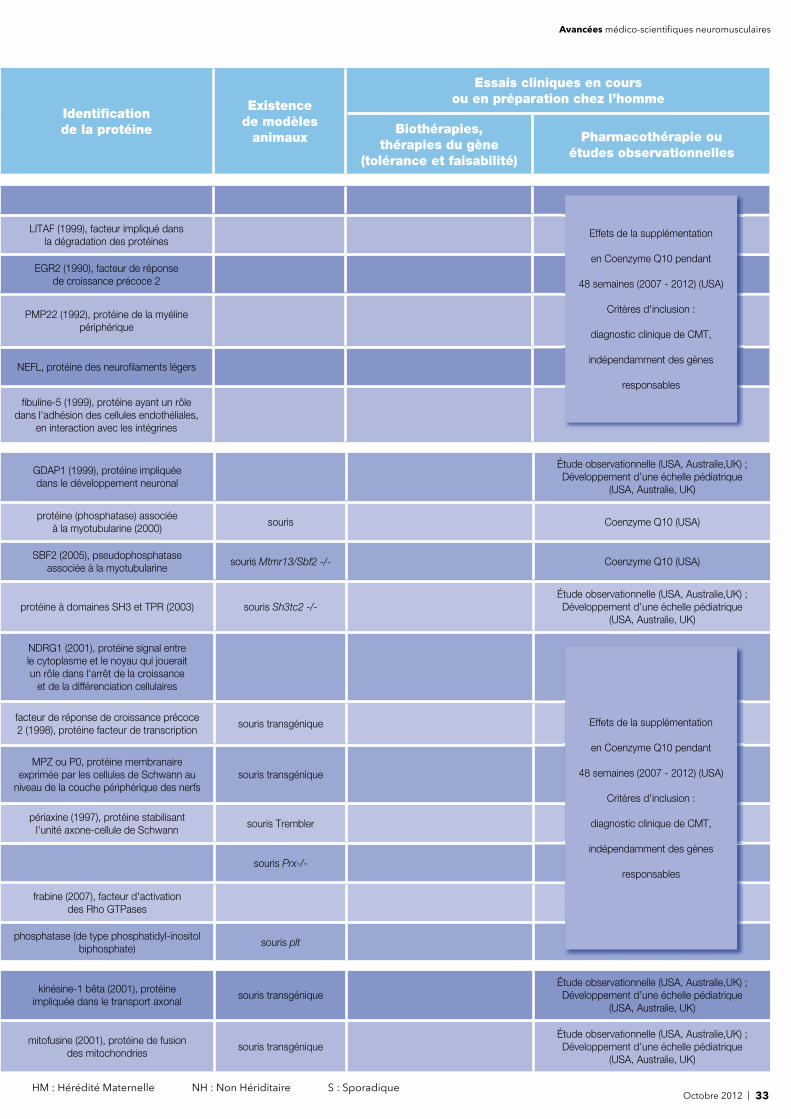
Octobre 2012 | 33 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
Coenzyme Q10 (USA)
LITAF (1999), facteur impliqué dans la dégradation des protéines
Coenzyme Q10 (USA)
EGR2 (1990), facteur de réponse de croissance précoce 2
Coenzyme Q10 (USA)
PMP22 (1992), protéine de la myéline périphérique
Coenzyme Q10 (USA)
NEFL, protéine des neurofi laments légers Coenzyme Q10 (USA)
fi buline-5 (1999), protéine ayant un rôle dans l'adhésion des cellules endothéliales,
en interaction avec les intégrinesCoenzyme Q10 (USA)
GDAP1 (1999), protéine impliquée dans le développement neuronal
Étude observationnelle (USA, Australie,UK) ;Développement d’une échelle pédiatrique
(USA, Australie, UK)
protéine (phosphatase) associée à la myotubularine (2000)
souris Coenzyme Q10 (USA)
SBF2 (2005), pseudophosphatase associée à la myotubularine
souris Mtmr13/Sbf2 -/- Coenzyme Q10 (USA)
protéine à domaines SH3 et TPR (2003) souris Sh3tc2 -/-Étude observationnelle (USA, Australie,UK) ;Développement d’une échelle pédiatrique
(USA, Australie, UK)
NDRG1 (2001), protéine signal entre le cytoplasme et le noyau qui jouerait un rôle dans l'arrêt de la croissance
et de la différenciation cellulaires
Coenzyme Q10 (USA)
facteur de réponse de croissance précoce 2 (1998), protéine facteur de transcription
souris transgénique Coenzyme Q10 (USA)
MPZ ou P0, protéine membranaire exprimée par les cellules de Schwann au
niveau de la couche périphérique des nerfssouris transgénique Coenzyme Q10 (USA)
périaxine (1997), protéine stabilisant l'unité axone-cellule de Schwann
souris Trembler Coenzyme Q10 (USA)
souris Prx-/- Coenzyme Q10 (USA)
frabine (2007), facteur d'activation des Rho GTPases
Coenzyme Q10 (USA)
phosphatase (de type phosphatidyl-inositolbiphosphate)
souris plt Coenzyme Q10 (USA)
kinésine-1 bêta (2001), protéine impliquée dans le transport axonal
souris transgéniqueÉtude observationnelle (USA, Australie,UK) ;Développement d’une échelle pédiatrique
(USA, Australie, UK)
mitofusine (2001), protéine de fusion des mitochondries
souris transgéniqueÉtude observationnelle (USA, Australie,UK) ;Développement d’une échelle pédiatrique
(USA, Australie, UK)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Effets de la supplémentation
en Coenzyme Q10 pendant
48 semaines (2007 - 2012) (USA)
Critères d'inclusion :
diagnostic clinique de CMT,
indépendamment des gènes
responsables
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Effets de la supplémentation
en Coenzyme Q10 pendant
48 semaines (2007 - 2012) (USA)
Critères d'inclusion :
diagnostic clinique de CMT,
indépendamment des gènes
responsables
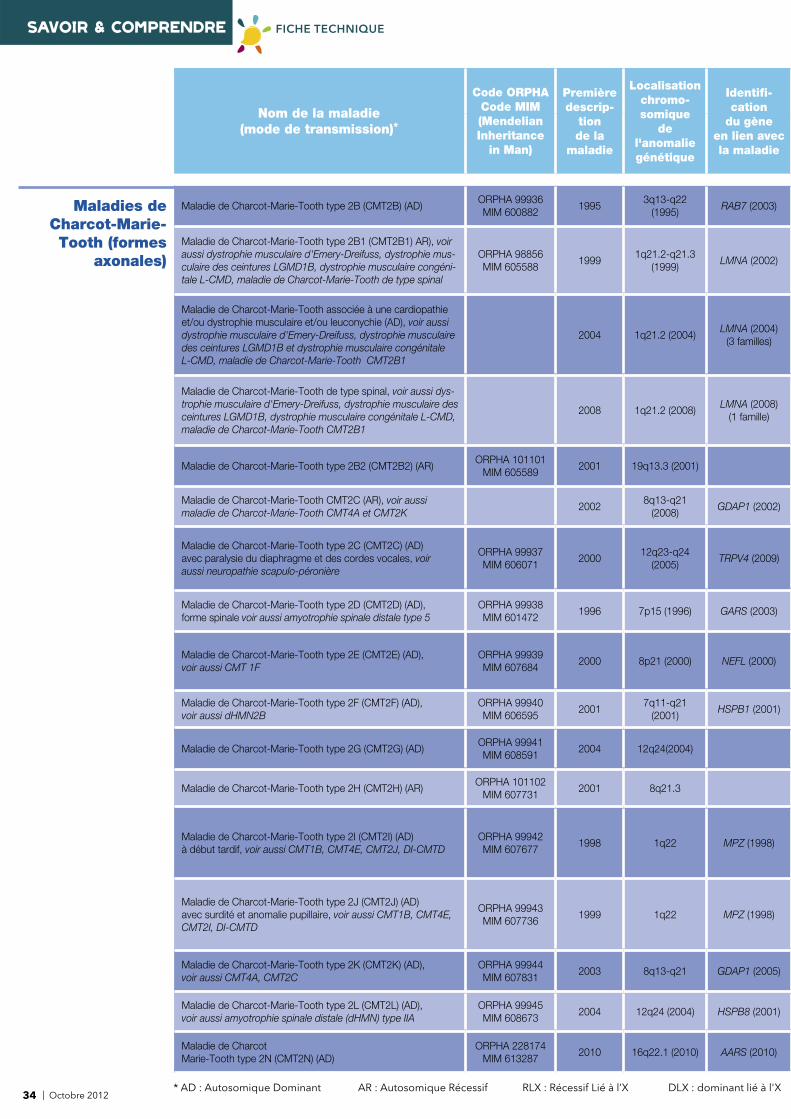
SAVOIR & COMPRENDRE FICHE TECHNIQUE
34 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Maladies de Charcot-Marie-Tooth (formes
axonales)
Maladie de Charcot-Marie-Tooth type 2B (CMT2B) (AD)ORPHA 99936MIM 600882
19953q13-q22
(1995)RAB7 (2003)
Maladie de Charcot-Marie-Tooth type 2B1 (CMT2B1) AR), voir aussi dystrophie musculaire d'Emery-Dreifuss, dystrophie mus-culaire des ceintures LGMD1B, dystrophie musculaire congéni-tale L-CMD, maladie de Charcot-Marie-Tooth de type spinal
ORPHA 98856MIM 605588
19991q21.2-q21.3
(1999)LMNA (2002)
Maladie de Charcot-Marie-Tooth associée à une cardiopathie et/ou dystrophie musculaire et/ou leuconychie (AD), voir aussi dystrophie musculaire d'Emery-Dreifuss, dystrophie musculaire des ceintures LGMD1B et dystrophie musculaire congénitale L-CMD, maladie de Charcot-Marie-Tooth CMT2B1
2004 1q21.2 (2004)LMNA (2004)
(3 familles)
Maladie de Charcot-Marie-Tooth de type spinal, voir aussi dys-trophie musculaire d'Emery-Dreifuss, dystrophie musculaire des ceintures LGMD1B, dystrophie musculaire congénitale L-CMD, maladie de Charcot-Marie-Tooth CMT2B1
2008 1q21.2 (2008)LMNA (2008)
(1 famille)
Maladie de Charcot-Marie-Tooth type 2B2 (CMT2B2) (AR)ORPHA 101101
MIM 6055892001 19q13.3 (2001)
Maladie de Charcot-Marie-Tooth CMT2C (AR), voir aussi maladie de Charcot-Marie-Tooth CMT4A et CMT2K
20028q13-q21
(2008)GDAP1 (2002)
Maladie de Charcot-Marie-Tooth type 2C (CMT2C) (AD) avec paralysie du diaphragme et des cordes vocales, voir aussi neuropathie scapulo-péronière
ORPHA 99937MIM 606071
200012q23-q24
(2005)TRPV4 (2009)
Maladie de Charcot-Marie-Tooth type 2D (CMT2D) (AD), forme spinale voir aussi amyotrophie spinale distale type 5
ORPHA 99938MIM 601472
1996 7p15 (1996) GARS (2003)
Maladie de Charcot-Marie-Tooth type 2E (CMT2E) (AD), voir aussi CMT 1F
ORPHA 99939MIM 607684
2000 8p21 (2000) NEFL (2000)
Maladie de Charcot-Marie-Tooth type 2F (CMT2F) (AD), voir aussi dHMN2B
ORPHA 99940MIM 606595
20017q11-q21
(2001)HSPB1 (2001)
Maladie de Charcot-Marie-Tooth type 2G (CMT2G) (AD)ORPHA 99941MIM 608591
2004 12q24(2004)
Maladie de Charcot-Marie-Tooth type 2H (CMT2H) (AR)ORPHA 101102
MIM 6077312001 8q21.3
Maladie de Charcot-Marie-Tooth type 2I (CMT2I) (AD) à début tardif, voir aussi CMT1B, CMT4E, CMT2J, DI-CMTD
ORPHA 99942MIM 607677
1998 1q22 MPZ (1998)
Maladie de Charcot-Marie-Tooth type 2J (CMT2J) (AD) avec surdité et anomalie pupillaire, voir aussi CMT1B, CMT4E, CMT2I, DI-CMTD
ORPHA 99943MIM 607736
1999 1q22 MPZ (1998)
Maladie de Charcot-Marie-Tooth type 2K (CMT2K) (AD), voir aussi CMT4A, CMT2C
ORPHA 99944MIM 607831
2003 8q13-q21 GDAP1 (2005)
Maladie de Charcot-Marie-Tooth type 2L (CMT2L) (AD), voir aussi amyotrophie spinale distale (dHMN) type IIA
ORPHA 99945MIM 608673
2004 12q24 (2004) HSPB8 (2001)
Maladie de CharcotMarie-Tooth type 2N (CMT2N) (AD)
ORPHA 228174MIM 613287
2010 16q22.1 (2010) AARS (2010)
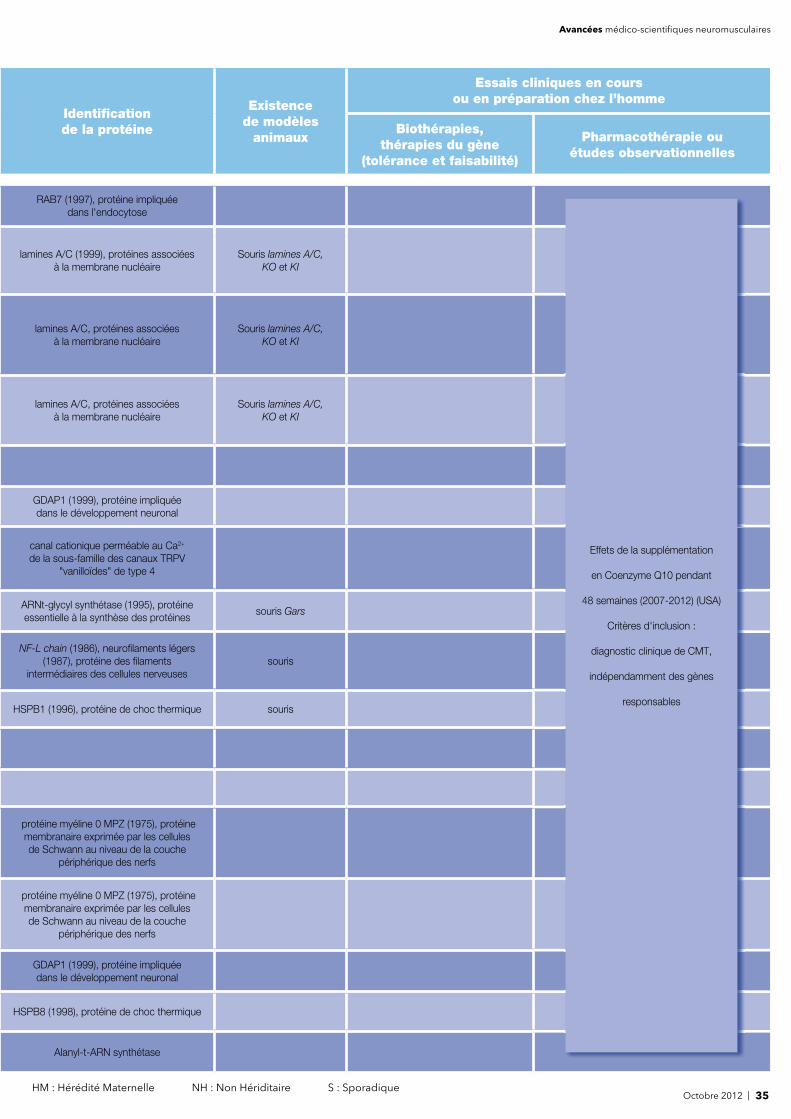
Octobre 2012 | 35 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
RAB7 (1997), protéine impliquée dans l'endocytose
Coenzyme Q10 (USA)
lamines A/C (1999), protéines associées à la membrane nucléaire
Souris lamines A/C, KO et KI
Coenzyme Q10 (USA)
lamines A/C, protéines associées à la membrane nucléaire
Souris lamines A/C, KO et KI
Coenzyme Q10 (USA)
lamines A/C, protéines associées à la membrane nucléaire
Souris lamines A/C, KO et KI
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
GDAP1 (1999), protéine impliquée dans le développement neuronal
Coenzyme Q10 (USA)
canal cationique perméable au Ca2+ de la sous-famille des canaux TRPV
"vanilloïdes" de type 4Coenzyme Q10 (USA)
ARNt-glycyl synthétase (1995), protéine essentielle à la synthèse des protéines
souris Gars Coenzyme Q10 (USA)
NF-L chain (1986), neurofi laments légers (1987), protéine des fi laments
intermédiaires des cellules nerveusessouris Coenzyme Q10 (USA)
HSPB1 (1996), protéine de choc thermique souris Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
protéine myéline 0 MPZ (1975), protéine membranaire exprimée par les cellules de Schwann au niveau de la couche
périphérique des nerfs
Coenzyme Q10 (USA)
protéine myéline 0 MPZ (1975), protéine membranaire exprimée par les cellules de Schwann au niveau de la couche
périphérique des nerfs
Coenzyme Q10 (USA)
GDAP1 (1999), protéine impliquée dans le développement neuronal
Coenzyme Q10 (USA)
HSPB8 (1998), protéine de choc thermique Coenzyme Q10 (USA)
Alanyl-t-ARN synthétase
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Effets de la supplémentation
en Coenzyme Q10 pendant
48 semaines (2007-2012) (USA)
Critères d'inclusion :
diagnostic clinique de CMT,
indépendamment des gènes
responsables
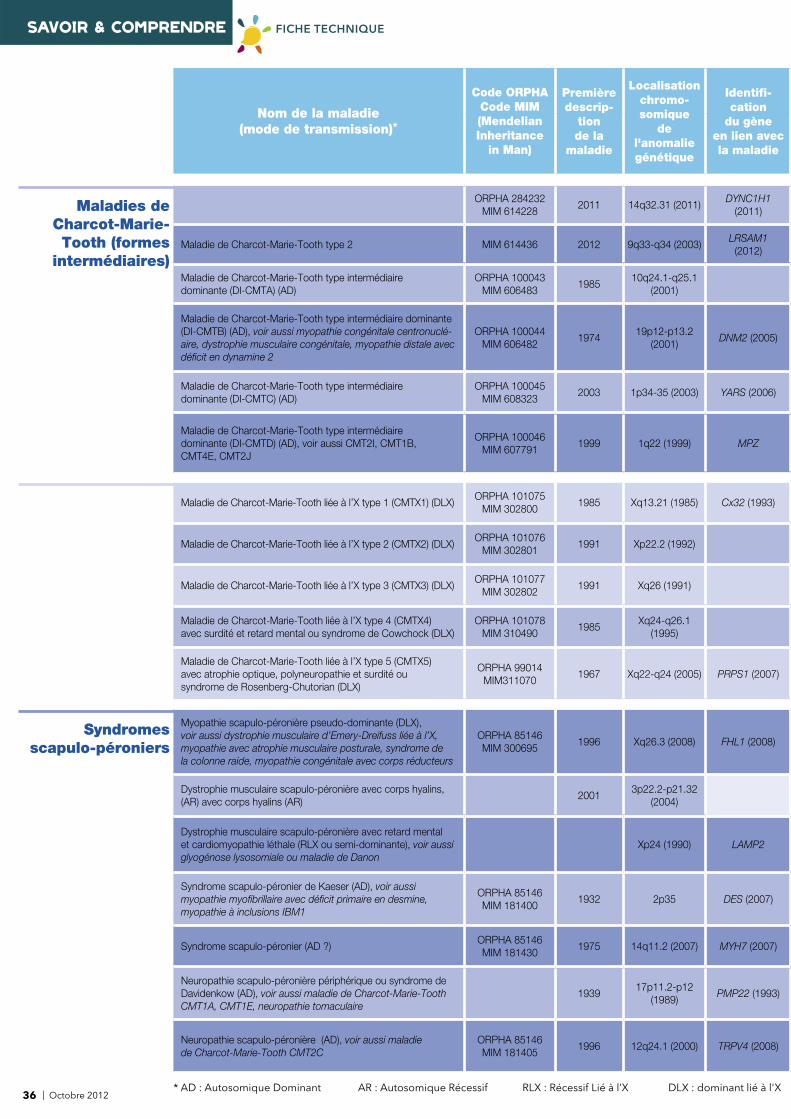
SAVOIR & COMPRENDRE FICHE TECHNIQUE
36 | Octobre 2012* AD : Autosomique Dominant AR : Autosomique Récessif RLX : Récessif Lié à l’X DLX : dominant lié à l'X
Nom de la maladie (mode de transmission)*
Code ORPHACode MIM(Mendelian Inheritance
in Man)
Première descrip-
tion de la
maladie
Localisationchromo-somique
de l'anomalie génétique
Identifi -cation
du gèneen lien avecla maladie
Maladies de Charcot-Marie-Tooth (formes
intermédiaires)
ORPHA 284232MIM 614228
2011 14q32.31 (2011)DYNC1H1
(2011)
Maladie de Charcot-Marie-Tooth type 2 MIM 614436 2012 9q33-q34 (2003)LRSAM1
(2012)
Maladie de Charcot-Marie-Tooth type intermédiaire dominante (DI-CMTA) (AD)
ORPHA 100043MIM 606483
198510q24.1-q25.1
(2001)
Maladie de Charcot-Marie-Tooth type intermédiaire dominante (DI-CMTB) (AD), voir aussi myopathie congénitale centronuclé-aire, dystrophie musculaire congénitale, myopathie distale avec défi cit en dynamine 2
ORPHA 100044MIM 606482
197419p12-p13.2
(2001)DNM2 (2005)
Maladie de Charcot-Marie-Tooth type intermédiaire dominante (DI-CMTC) (AD)
ORPHA 100045MIM 608323
2003 1p34-35 (2003) YARS (2006)
Maladie de Charcot-Marie-Tooth type intermédiaire dominante (DI-CMTD) (AD), voir aussi CMT2I, CMT1B, CMT4E, CMT2J
ORPHA 100046MIM 607791
1999 1q22 (1999) MPZ
Maladie de Charcot-Marie-Tooth liée à l’X type 1 (CMTX1) (DLX)ORPHA 101075
MIM 3028001985 Xq13.21 (1985) Cx32 (1993)
Maladie de Charcot-Marie-Tooth liée à l’X type 2 (CMTX2) (DLX)ORPHA 101076
MIM 3028011991 Xp22.2 (1992)
Maladie de Charcot-Marie-Tooth liée à l’X type 3 (CMTX3) (DLX)ORPHA 101077
MIM 3028021991 Xq26 (1991)
Maladie de Charcot-Marie-Tooth liée à l’X type 4 (CMTX4) avec surdité et retard mental ou syndrome de Cowchock (DLX)
ORPHA 101078MIM 310490
1985Xq24-q26.1
(1995)
Maladie de Charcot-Marie-Tooth liée à l’X type 5 (CMTX5) avec atrophie optique, polyneuropathie et surdité ou syndrome de Rosenberg-Chutorian (DLX)
ORPHA 99014MIM311070
1967 Xq22-q24 (2005) PRPS1 (2007)
Syndromesscapulo-péroniers
Myopathie scapulo-péronière pseudo-dominante (DLX), voir aussi dystrophie musculaire d’Emery-Dreifuss liée à l’X, myopathie avec atrophie musculaire posturale, syndrome de la colonne raide, myopathie congénitale avec corps réducteurs
ORPHA 85146MIM 300695
1996 Xq26.3 (2008) FHL1 (2008)
Dystrophie musculaire scapulo-péronière avec corps hyalins, (AR) avec corps hyalins (AR)
20013p22.2-p21.32
(2004)
Dystrophie musculaire scapulo-péronière avec retard mental et cardiomyopathie léthale (RLX ou semi-dominante), voir aussi glyogénose lysosomiale ou maladie de Danon
Xp24 (1990) LAMP2
Syndrome scapulo-péronier de Kaeser (AD), voir aussi myopathie myofi brillaire avec défi cit primaire en desmine, myopathie à inclusions IBM1
ORPHA 85146MIM 181400
1932 2p35 DES (2007)
Syndrome scapulo-péronier (AD ?)ORPHA 85146MIM 181430
1975 14q11.2 (2007) MYH7 (2007)
Neuropathie scapulo-péronière périphérique ou syndrome de Davidenkow (AD), voir aussi maladie de Charcot-Marie-Tooth CMT1A, CMT1E, neuropathie tomaculaire
193917p11.2-p12
(1989)PMP22 (1993)
Neuropathie scapulo-péronière (AD), voir aussi maladie de Charcot-Marie-Tooth CMT2C
ORPHA 85146MIM 181405
1996 12q24.1 (2000) TRPV4 (2008)
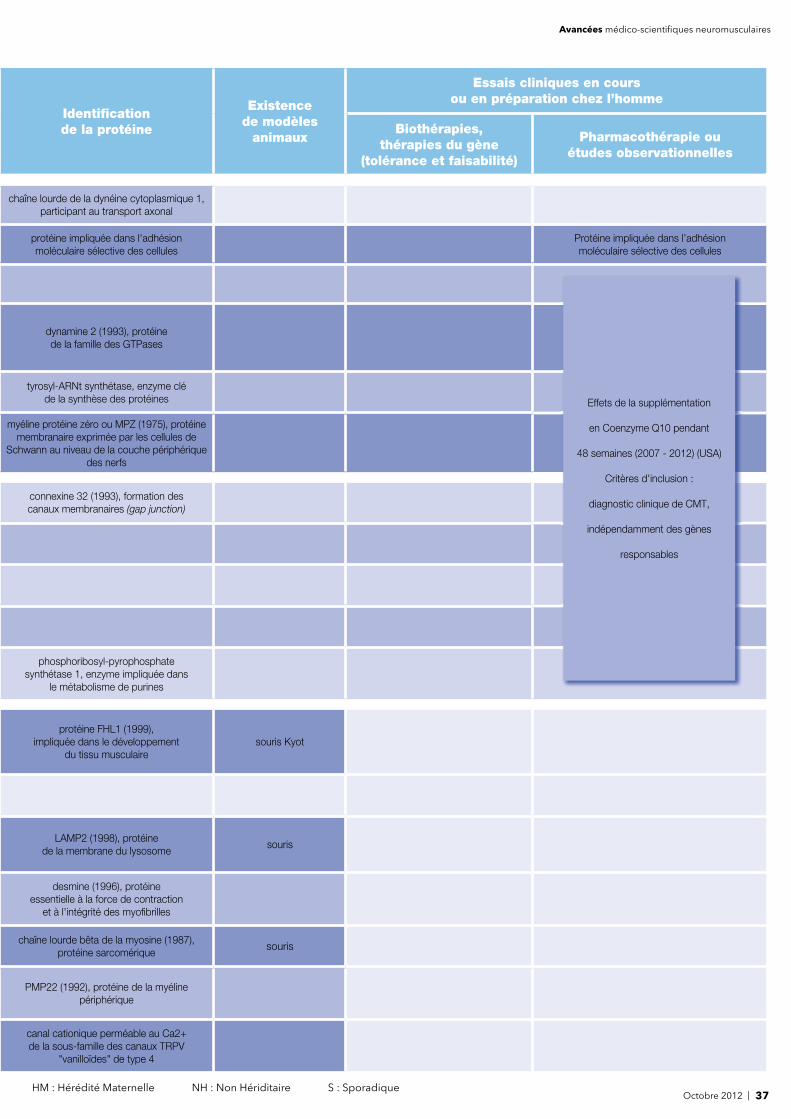
Octobre 2012 | 37 HM : Hérédité Maternelle NH : Non Hériditaire S : Sporadique
Avancées médico-scientifiques neuromusculaires
Identifi cation de la protéine
Existencede modèles
animaux
Essais cliniques en cours ou en préparation chez l’homme
Biothérapies, thérapies du gène
(tolérance et faisabilité)
Pharmacothérapie ouétudes observationnelles
chaîne lourde de la dynéine cytoplasmique 1, participant au transport axonal
protéine impliquée dans l'adhésion moléculaire sélective des cellules
Protéine impliquée dans l’adhésion moléculaire sélective des cellules
dynamine 2 (1993), protéine de la famille des GTPases
Coenzyme Q10 (USA)
tyrosyl-ARNt synthétase, enzyme clé de la synthèse des protéines
Coenzyme Q10 (USA)
myéline protéine zéro ou MPZ (1975), protéine membranaire exprimée par les cellules de
Schwann au niveau de la couche périphérique des nerfs
Coenzyme Q10 (USA)
connexine 32 (1993), formation des canaux membranaires (gap junction)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
phosphoribosyl-pyrophosphate synthétase 1, enzyme impliquée dans
le métabolisme de purinesCoenzyme Q10 (USA)
protéine FHL1 (1999), impliquée dans le développement
du tissu musculairesouris Kyot
LAMP2 (1998), protéine de la membrane du lysosome
souris
desmine (1996), protéine essentielle à la force de contraction
et à l'intégrité des myofi brilles
chaîne lourde bêta de la myosine (1987), protéine sarcomérique
souris
PMP22 (1992), protéine de la myéline périphérique
canal cationique perméable au Ca2+ de la sous-famille des canaux TRPV
"vanilloïdes" de type 4
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Coenzyme Q10 (USA)
Effets de la supplémentation
en Coenzyme Q10 pendant
48 semaines (2007 - 2012) (USA)
Critères d'inclusion :
diagnostic clinique de CMT,
indépendamment des gènes
responsables
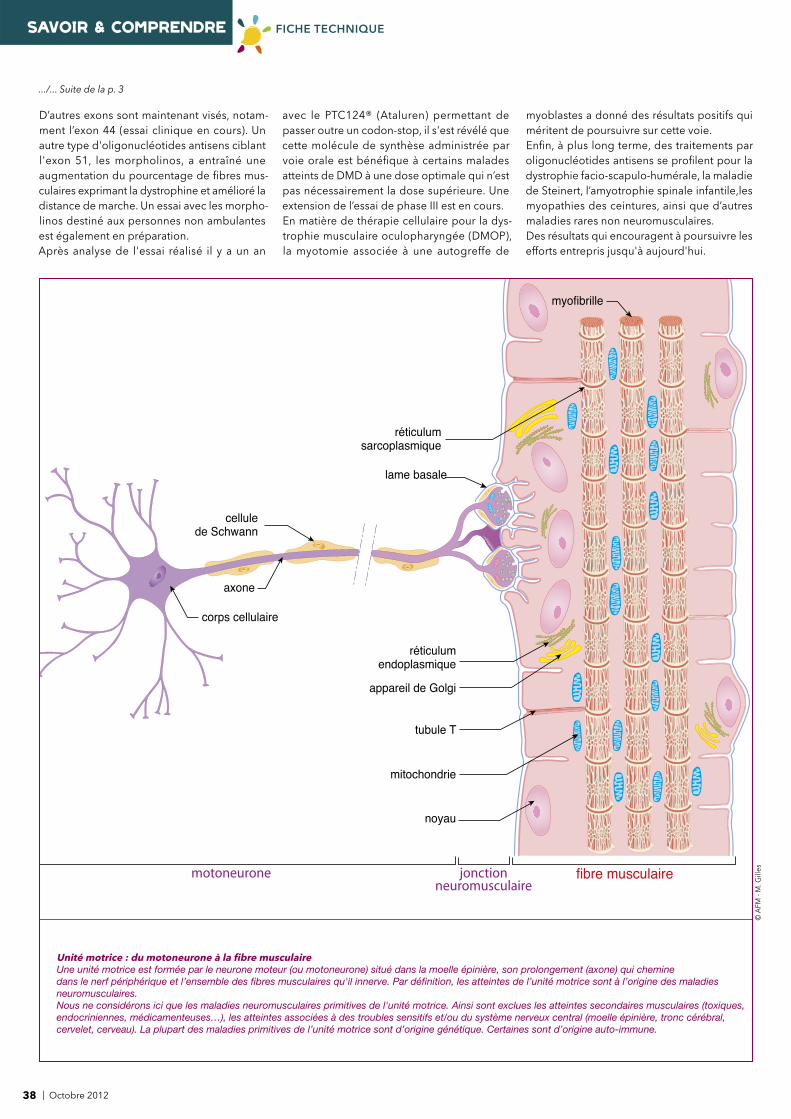
SAVOIR & COMPRENDRE FICHE TECHNIQUE
38 | Octobre 2012
.../... Suite de la p. 3
D’autres exons sont maintenant visés, notam-
ment l’exon 44 (essai clinique en cours). Un
autre type d'oligonucléotides antisens ciblant
l'exon 51, les morpholinos, a entraîné une
augmentation du pourcentage de fi bres mus-
culaires exprimant la dystrophine et amélioré la
distance de marche. Un essai avec les morpho-
linos destiné aux personnes non ambulantes
est également en préparation.
Après analyse de l'essai réalisé il y a un an
avec le PTC124® (Ataluren) permettant de
passer outre un codon-stop, il s'est révélé que
cette molécule de synthèse administrée par
voie orale est bénéfi que à certains malades
atteints de DMD à une dose optimale qui n’est
pas nécessairement la dose supérieure. Une
extension de l’essai de phase III est en cours.
En matière de thérapie cellulaire pour la dys-
trophie musculaire oculopharyngée (DMOP),
la myotomie associée à une autogreffe de
myoblastes a donné des résultats positifs qui
méritent de poursuivre sur cette voie.
Enfi n, à plus long terme, des traitements par
oligonucléotides antisens se profi lent pour la
dystrophie facio-scapulo-humérale, la maladie
de Steinert, l’amyotrophie spinale infantile,les
myopathies des ceintures, ainsi que d’autres
maladies rares non neuromusculaires.
Des résultats qui encouragent à poursuivre les
efforts entrepris jusqu'à aujourd'hui.
cellulede Schwann
réticulumendoplasmique
appareil de Golgi
lame basale
tubule T
mitochondrie
noyau
myofibrille
axone
corps cellulaire
réticulumsarcoplasmique
fibre musculairemotoneurone jonctionneuromusculaire
© A
FM
- M
. Gill
es
Unité motrice : du motoneurone à la fi bre musculaire
Une unité motrice est formée par le neurone moteur (ou motoneurone) situé dans la moelle épinière, son prolongement (axone) qui chemine
dans le nerf périphérique et l’ensemble des fi bres musculaires qu'il innerve. Par défi nition, les atteintes de l’unité motrice sont à l’origine des maladies
neuromusculaires.
Nous ne considérons ici que les maladies neuromusculaires primitives de l'unité motrice. Ainsi sont exclues les atteintes secondaires musculaires (toxiques,
endocriniennes, médicamenteuses…), les atteintes associées à des troubles sensitifs et/ou du système nerveux central (moelle épinière, tronc cérébral,
cervelet, cerveau). La plupart des maladies primitives de l’unité motrice sont d’origine génétique. Certaines sont d’origine auto-immune.

A
B
C
D
E
F
Index général
- Actinopathie : voir Autres myopathies avec surcharge de fi laments ou inclusions ................................................................................................... 14 - ACVR1 : voir Fibrodysplasie ossifi ante progressive ....................................................................................................................................................... 24- Acyl CoA déshydrogénase (Défi cit en) : voir Myopathies métaboliques (Lipidose musculaire).............................................................................. 22- Acétylcholinestérase (Défi cit en) : voir Syndromes myasthéniques congénitaux ...................................................................................................... 26- Adhaline ou α-sarcoglycane : voir Dystrophies musculaires des ceintures autosomiques récessives (Sarcoglycanopathies)...............................5- Adynamie épisodique de Gamstorp ou Paralysie périodique hyperkaliémique : voir Canalopathies musculaires ............................................. 20- Agrine : voir Syndromes myasthéniques congénitaux synaptiques ............................................................................................................................ 26- Alpha-actine : voir Autres myopathies avec surcharge en fi laments ou inclusions, Myopathies congénitales ................................................15-17- Alpha-B cristalline : voir Myopathies myofi brillaires ...................................................................................................................................................... 14- Alpha-dystroglycane : voir Dystrophies musculaires congénitales.............................................................................................................................. 11- Alpha-sarcoglycane : voir Dystrophies musculaires des ceintures autosomiques récessives (Sarcoglycanopathies) .............................................5- Alpha-tropomyosine : voir Myopathies congénitales .................................................................................................................................................... 17- Amphiphysine : voir Myopathies congénitales .............................................................................................................................................................. 19- Andersen-Tawil (syndrome d') : voir Canalopathies musculaires ................................................................................................................................. 20- Anoctamine : voir Dystrophies musculaires des ceintures autosomiques récessives ..................................................................................................7- Arthrogrypose : voir Dystrophies musculaires congénitales ........................................................................................................................................ 12
- Bêta-sarcoglycane (Dystrophie musculaire avec défi cit en) ou β-sarcoglycanopathie : voir Dystrophies musculaires des ceintures autosomiques récessives (Sarcoglycanopathies) ...................................................................................5
- Becker (Dystrophie musculaire de) : voir Dystrophinopathies ........................................................................................................................................4- Becker (Myotonie congénitale de) : voir Canalopathies musculaires .......................................................................................................................... 18- Bethlem : voir Autres dystrophies musculaires progressives ..........................................................................................................................................8- Brody (maladie de) : voir syndromes myotoniques ....................................................................................................................................................... 20- Brown-Vialetto-Van Laere (maladie de) : voir Amyotrophies bulbo-spinales ............................................................................................................. 30
- Calpaïne : voir Dystrophies musculaires des ceintures autosomiques récessives ........................................................................................................5- Calpaïnopathie : voir Dystrophies musculaires des ceintures autosomiques récessives ............................................................................................4- Calséquestrine : voir Autres myopathies avec surcharge de fi laments ou inclusions ............................................................................................... 14- Canal lent (Syndrome du) : voir Maladies de la jonction neuromusculaire (Syndromes myasthéniques congénitaux) ....................................... 26- Canal rapide (Syndrome du) : voir Maladies de la jonction neuromusculaire (Syndromes myasthéniques congénitaux)................................... 26- Cap disease : voir Myopathies congénitales .................................................................................................................................................................. 16- Carnitine (Défi cit en) : voir Myopathies métaboliques (Lipidose musculaire) ............................................................................................................ 22- Carnitine-palmitoyl transférase de type II (Défi cit en) : voir Myopathies métaboliques (Lipidose musculaire) ..................................................... 22- Casquette (Myopathie à) : voir Myopathies congénitales ............................................................................................................................................. 16- Cavéoline-3 : voir Dystrophies musculaires des ceintures autosomiques dominantes, Myopathies distales, Syndromes myotoniques 7-13-21- Central core (Myopathie congénitale à) : voir Myopathies congénitales .................................................................................................................... 18- Centronucléaire (Myopathie congénitale) : voir Myopathies congénitales ...........................................................................................................16-18- Charcot-Marie-Tooth (CMT) ......................................................................................................................................................................................30 à 37- Chondrodysplasique (myotonie) voir Syndromes myotoniques ................................................................................................................................. 20- Chutorian (Syndrome de) : voir Maladies de Charcot-Marie-Tooth formes liées à l'X ............................................................................................... 36- Cofi line : voir Myopathies congénitales .......................................................................................................................................................................... 19- Collagène VI : voir Autres dystrophies musculaires progressives, Dystrophies musculaires congénitales ...............................................................9- Colonne raide (Syndrome de la) : voir Dystrophies musculaires congénitales .............................................................................................................8- Connexine : voir Neuropathies héréditaires sensitivo-motrices ................................................................................................................................... 37- Contactine : voir Myopathies congénitales ..................................................................................................................................................................... 18- Cori (Maladie de) ou maladie de Forbes : voir Myopathies métaboliques (Glycogénose musculaire) .................................................................. 22- Cowchock (Syndrome de) : voir Maladies de Charcot-Marie-Tooth ............................................................................................................................ 34
- Davidenkow (syndrome de) : voir Maladies de Charcot-Marie-Tooth démyélinisantes autosomiques dominantes ............................................ 30- Dejerine : voir Autres dystrophies musculaires progressives ..........................................................................................................................................6- Dermatomyosites : voir Maladies infl ammatoires du muscle ....................................................................................................................................... 24- Desmine : voir Myopathies myofi brillaires, Syndromes scapulo-péroniers ...........................................................................................................15-37- DMOP : voir Dystrophie musculaire oculopharyngée ................................................................................................................................................... 14- Duchenne de Boulogne (Dystrophie musculaire de) : voir Dystrophinopathies ..........................................................................................................4- Dynamine : voir Dystrophies musculaires congénitales, Myopathes congénitales, Neuropathies héréditaires sensitivo-motrices ........13-19-37- Dysferline : voir Dystrophies musculaires des ceintures autosomiques récessives, Myopathies distales ...........................................................5-13- Dysferlinopathie : voir Dystrophies musculaires des ceintures autosomiques récessives ..........................................................................................4- Dystrophine : voir Dystrophinopathies ...............................................................................................................................................................................5
- Emery-Dreifuss : voir Autres dystrophies musculaires progressives ...............................................................................................................................6- Emerine : voir Autres dystrophies musculaires progressives...........................................................................................................................................7- Erb (Dystrophie musculaire des ceintures de type) ou Calpaïnopathie : voir Dystrophies musculaires des ceintures autosomqiues récessives 4 - Enzyme débranchante (Défi cit en) : voir Myopathies métaboliques (Glycogénose musculaire) ............................................................................ 23- Eulenburg (Paramyotonie d’) : voir Canalopathies musculaires ................................................................................................................................... 20
- Fazio-Londe (maladie de) : voir Amyotrophies bulbo-spinales .................................................................................................................................... 30- FHL1 : voir Myopathies congénitales ............................................................................................................................................................................... 19- Filamine : voir Myopathies myofi brillaires ....................................................................................................................................................................... 12
Octobre 2012 | 39
Avancées médico-scientifiques neuromusculaires

SAVOIR & COMPRENDRE FICHE TECHNIQUE
G
H
I
J
K
L
M
- Forbes (Maladie de) ou maladie de Cori : voir Myopathies métaboliques (Glycogénose musculaire) .................................................................. 22- Frabine : voir Maladies de Charcot-Marie-Tooth autosomiques récessives ................................................................................................................ 33- FOP : voir Fibrodysplasie ossifi ante progressive ............................................................................................................................................................ 24- Freeman (syndrome de) : voir Myosinopathies .............................................................................................................................................................. 16- FSH : voir Autres dystrophies musculaires progressives (Dystrophie facio-scapulo-humérale) .................................................................................6- Fukutine : voir Dystrophies musculaires des ceintures, Dystrophies musculaires congénitales ...........................................................................7-11- Fukuyama (Dystrophie musculaire congénitale de type) : voir Dystrophies musculaires congénitales ................................................................. 10
- γ-sarcoglycane (Dystrophie musculaire avec défi cit en) ou γ-sarcoglycanopathie : voir Dystrophies musculaires des ceintures autosomiques récessives ..........................................................................................................................5
- Gamstorp (Adynamie épisodique de) ou Paralysie périodique hyperkaliémique : voir Canalopathies musculaires ........................................... 20- Griggs : voir Myopathies myofi brillaires .......................................................................................................................................................................... 12
- Hall (syndrome de) : voir Myosinopathies ....................................................................................................................................................................... 16- Harper-Young (myopathie de) : voir Amyotrophies spinales distales .......................................................................................................................... 30- Hutterite : voir Dystrophies musculaires des ceintures.....................................................................................................................................................4
- Intégrine : voir Dystrophies musculaires congénitales .................................................................................................................................................. 10
- Jampel (syndrome de) : voir Syndromes myotoniques ................................................................................................................................................. 20- Jerash : voir Amyotrophies spinales distales .................................................................................................................................................................. 30
- Kaeser (syndrome) : voir Syndromes scapulo-péroniers ............................................................................................................................................... 36- Kennedy (syndrome de) : voir Amyotrophies bulbo-spinales ...................................................................................................................................... 30- Kinésine : voir Maladies de Charcot-Marie-Tooth formes axonales ............................................................................................................................. 33- Kugelberg-Welander (Maladie de) ou Amyotrophie spinale infantile type III : voir Amyotrophies spinales proximales ..................................... 28
- Laing (Myopathie distale de) : voir Myopathies distales ............................................................................................................................................... 12- Lamines A/C : voir Dystrophies musculaires des ceintures autosomiques dominantes, Autres dystrophies musculaires
progressives, Dystrophies musculaires congénitales, Neuropathies héréditaires sensitivo-motrices ........................................................7-9-11-35- Laminine alpha2 : voir Dystrophies musculaires congénitales ........................................................................................................................................9- Laminine �2 : voir Syndromes myasthéniques congénitaux synaptiques ................................................................................................................... 27- LAMP2 : voir Myopathies métaboliques .......................................................................................................................................................................... 23- Landouzy-Dejerine (Myopathie de) : voir Autres dystrophies musculaires progressives (Dystrophie musculaire facio-scapulo-humérale) ........6- LARGE (DMC avec mutation du gène) : voir Dystrophies musculaires congénitales ................................................................................................ 11- LGMD : voir Dystrophies musculaires des ceintures .................................................................................................................................................. 4 à 7- Londe (maladie de Fazio-) : voir Amyotrophies bulbo-spinales ................................................................................................................................... 30- Lom (maladie de) : voir Maladies de CHarcot-Marie-Tooth récessive ......................................................................................................................... 32
- McArdle (Maladie de) : voir Myopathies métaboliques (Glycogénose musculaire) .................................................................................................. 24- Mallory (corps de) : voir myopathies myofi brillaires ...................................................................................................................................................... 14- Markesbery-Griggs (Myopathie de) ou Dystrophie musculaire tibiale de type Udd : voir Myopathies distales ................................................... 12- Matrine : voir Myopathies distales .................................................................................................................................................................................... 13- MEB : voir Dystrophies musculaires congénitales .......................................................................................................................................................8-10- Mégaconiale : voir Myopathies métaboliques ............................................................................................................................................................... 22- Mérosine (Dystrophie musculaire avec défi cit en) : voir Dystrophies musculaires congénitales ................................................................................8- Minicores : voir Myopathies congénitales ....................................................................................................................................................................... 18- Mitochondriales (myopathies) .......................................................................................................................................................................................... 22- Mitofusine : voir Maladies de Charcot-Marie-Tooth formes axonales ......................................................................................................................... 33- Miyoshi (Myopathie de) : voir Myopathies distales ........................................................................................................................................................ 12- Multiminicores : voir Myopathies congénitales .............................................................................................................................................................. 18- Münchmeyer : voir Fibrodysplasie ossifi ante progressive ............................................................................................................................................ 24- Muscle-œil-cerveau (syndrome de) : voir Dystrophies musculaires Congénitales .................................................................................................8-10- Myasthénie .......................................................................................................................................................................................................................... 24- Myasthénique congénital (Syndrome) .....................................................................................................................................................................24 à 29- Myopathie à central core : voir Myopathies congénitales ............................................................................................................................................ 18- Myopathie à inclusions autosomique récessive (IBM2) ou Myopathie de Nonaka : voir Myopathies distales ...................................................... 12- Myopathie à multiminicores : voir Myopathies congénitales ....................................................................................................................................... 18- Myopathie centronucléaire : voir Myopathies congénitales ......................................................................................................................................... 18- Myopathie myotubulaire : voir Myopathies congénitales ............................................................................................................................................. 18- Myopathies à bâtonnets : voir Myopathies congénitales .............................................................................................................................................. 16- Myosine : voir Myopathies distales, Autres myopathies avec surcharge de fi laments ou inclusions, Myosinopathies,
Myopathies congénitales ............................................................................................................................................. 12-14-16-19- Myosite à inclusions : voir Maladies infl ammatoires du muscle ................................................................................................................................... 24- Myosite ossifi ante : voir Fibrodysplasie ossifi ante progressive .................................................................................................................................... 24- Myotiline : voir Dystrophies musculaires des ceintures LGMD1, Myopathies distales, Myopathies myofi brillaires ..................................... 7-13-15- Myotonie chondrodystrophique ou syndrome de Schwartz-Jampel : voir Syndromes myotoniques .................................................................... 20
40 | Octobre 2012

- Myotonie de Steinert : voir Syndromes myotoniques (Dystrophie myotonique) ....................................................................................................... 20- Myotubularine : voir Myopathies congénitales .............................................................................................................................................................. 19
- Nébuline : voir Myopathies distales, Myopathies congénitales ..............................................................................................................................13-17- Nemaline myopathies ou Myopathies congénitales à bâtonnets : voir Myopathies congénitales .......................................................................... 16- Nesprine : voir Autres dystrophies musculaires progressives, Dystrophies musculaires congénitales ...............................................................9-13- Nonaka (Myopathie de) : voir Myopathies distales ........................................................................................................................................................ 12
- O-mannosyl transférase : voir Dystrophies musculaires des ceintures autosomiques récessives ..............................................................................7
- PABPN1 : voir Autres myopathies avec surcharge de fi laments ou inclusions ........................................................................................................... 15- Paramyotonie d’Eulenburg : voir Canalopathies musculaires ...................................................................................................................................... 20- Périaxine : voir Neuropathies héréditaires sensitivo-motrices autosomiques récessives ......................................................................................... 33- Perlecan : voir Syndromes myotoniques ......................................................................................................................................................................... 21- Phosphofructokinase : voir Myopathies métaboliques (Glycogénose musculaire) ................................................................................................... 25- Phosphorylase : voir Myopathies métaboliques (Glycogénose musculaire) .............................................................................................................. 25- Pleckstrine : voir Amyotrophies spinales distales ........................................................................................................................................................... 31- Plectine : voir Autres dystrophies musculaires progressives ...........................................................................................................................................9- Polymyosites : voir Maladies infl ammatoires du muscle ............................................................................................................................................... 24- Pompe (Maladie de) : voir Myopathies métaboliques (Glycogénose musculaire) .................................................................................................... 22- PROMM (ou Dystrophie myotonique de type 2) : voir Syndromes myotoniques ...................................................................................................... 20
- Rapsyne : voir Syndromes myasthéniques congénitaux post-synaptiques ................................................................................................................ 26- Récepteur de l’acétylcholine : voir Syndromes myasthéniques congénitaux post-synaptiques .............................................................................. 26- Rigid Spine Syndrome : voir Dystrophies musculaires congénitales ..............................................................................................................................8- Rippling : voir Syndromes myotoniques ......................................................................................................................................................................... 20- Rosenberg-Chutorian (Syndrome de) : voir Maladies de Charcot-Marie-Tooth liées à l'X ....................................................................................... 34- Ryanodine : voir Myopathies congénitales ................................................................................................................................................................17-19
- Sarcoglycanopathie, Sarcoglycane : voir Dystrophies musculaires des ceintures autosomiques récessives ....................................................... 4-5- Schwartz-Jampel (Syndrome de) ou Myotonie chondrodystrophique : voir Syndromes myotoniques ................................................................. 20- Sélénoprotéine : voir Dystrophies musculaires congénitales, Myopathies myofi brillaires, Myopathies congénitales .......................... 9-15-17-19- Senataxine : voir Amyotrophies spinales distales .......................................................................................................................................................... 29- SERCA1 : voir Autres myopathies avec surcharge de fi laments ou inclusions ........................................................................................................... 14- Sheldon (syndrome) voir Myosinopathies ....................................................................................................................................................................... 16- SMA (Spinal Muscular Atrophy) voir Amyotrophies spinales proximales.................................................................................................................... 28- SMN1 : voir Amyotrophies spinales proximales ............................................................................................................................................................. 29- Sottas (maladie de Dejerine-) : voir Maladies de Charcot-Marie-Tooth démyélinisantes autosomiques récessives ............................................. 32- Steinert (dystrophie myotonique de) : voir Syndromes myotoniques (Dystrophies myotoniques) ......................................................................... 20- Syndrome de la colonne raide (Rigid spine syndrome) : voir Dystrophies musculaires congénitales .......................................................................8- Syndrome de Walker-Warburg : voir Dystrophies musculaires congénitales ............................................................................................................. 10- Syndrome du canal lent : voir Syndromes myasthéniques congénitaux ..................................................................................................................... 26- Syndrome du canal rapide : voir Syndromes myasthéniques congénitaux ................................................................................................................ 26- Syndrome MEB ou Muscle-Eye-Brain syndrome : voir Dystrophies musculaires congénitales ............................................................................... 10
- Tarui (Maladie de) : voir Myopathies métaboliques (Glycogénose musculaire) ........................................................................................................ 24- Tawil (syndrome de) : voir Canalopathies musculaires .................................................................................................................................................. 20- Téléthonine : voir Dystrophies musculaires des ceintures autosomiques récessives ...................................................................................................5- Thomsen (Myotonie congénitale de) : voir Canalopathies musculaires...................................................................................................................... 20- Titine : voir Dystrophies musculaires des ceintures, Myopathies distales, Autres myopathies avec surcharge de fi laments
ou inclusions ............................................................................................................................................................................................................... 7-13-15- TRIM 32 : voir Dystrophies musculaires des ceintures autosomiques récessives .........................................................................................................5- Tropomyosine : voir Myopathies congénitales ..........................................................................................................................................................15-17- Troponine : voir Myopathies congénitales ...................................................................................................................................................................... 19
- Udd (Dystrophie musculaire tibiale de type) : voir Myopathes distales ...................................................................................................................... 12- Ullrich (Syndrome d’) : voir Dystrophies musculaires congénitales ................................................................................................................................8
- Van Laere (maladie de Brown-Vialetto) : voir Amyotrophies bulbo-spinales ............................................................................................................. 30- Vialetto ( maladie de Brown) : voir Amyotrophies bulbo-spinales ............................................................................................................................... 30
- Walker-Warburg (Syndrome de) : voir Dystrophies musculaires congénitales .......................................................................................................... 10- Welander (Myopathie de) : voir Myopathies distales .................................................................................................................................................... 12- Werdnig-Hoffmann (Maladie de) ou Amyotrophie spinale infantile type I : voir Amyotrophies spinales proximales .......................................... 28- Westphal (Maladie de) ou Paralysie périodique hypokaliémique : voir Canalopathies musculaires ...................................................................... 20
- Young (syndrome de) : voir Amyotrophies spinales distales ........................................................................................................................................ 30
N
O
P
R
S
T
U
V
W
Y
Octobre 2012 | 41
Avancées médico-scientifiques neuromusculaires

SAVOIR & COMPRENDRE FICHE TECHNIQUE
acétylcholine ......................................27
actine ............................................15, 17
ACVR1 ................................................25
acyl CoA déshydrogénase ................23
adénosine triphosphatase Ca++ .......21
agrine .................................................27
alpha-actine .................................15, 17
alphaB-cristalline ...............................15
alpha-glucosidase .............................23
alpha-tropomyosine ..........................17
amphiphysine ....................................19
amylo-1,6 glucosidase ......................23
androgènes (récepteurs aux) ...........31
anoctamine 5 .................................7, 13
ARNt-glycyl synthétase ...............31, 35
ATPase dépendante de Ca++ ............15
ATGL ...................................................23
bêta-tropomyosine ............................17
BAG3 ..................................................15
calpaïne 3 ............................................. 5
calséquestrine....................................15
canal chlore ........................................21
canal potassium .................................21
canal sodium ................................21, 29
canal TRPV “vaniloïde" ................35, 37
carnitine palmitoyl transférase II ......23
cavéoline-3 ...............................7, 13, 21
chaîne respiratoire mitochondriale .23
choline acétyl-transférase .................25
choline kinase bêta .....................11, 23
cofi line-2 .............................................19
collagène VI ......................................... 9
collagène Q .......................................27
connexine 32 .....................................37
contactine-1 .......................................19
croissance (facteur de réponse de) .33
desmine ........................................15, 37
dihydropyridines (récepteurs aux)...21
DMPK ..................................................21
double homeobox protein ................. 7
DOK-7 .................................................27
dynamine 2 ........................... 13, 19, 37
dynéine cytoplasmique 1 ...........29, 37
dysferline ........................................5, 13
dystrophine .......................................... 5
E1 ........................................................29
EGR2 ...................................................33
émerine ................................................ 7
ETFDH.................................................23
FHL1 ................................... 7, 11, 19, 37
fi buline-5 ............................................33
fi lamine C .....................................13, 15
FKRP ............................................5, 9, 11
frabine ................................................33
fukutine ...........................................7, 11
GDAP1 ..........................................33, 35
glutamine fructose-6 phosphate
amidotransférase ...............................29
glycogène synthétase .......................25
glycosyl-transférase like ....................11
HSPB1 ...........................................29, 35
HSPB3 .................................................29
HSPB8 ...........................................29, 35
IGHMBP2 ............................................31
intégrine alpha-7 ...............................11
ISCU ....................................................25
isoprenoïde synthétase ....................11
Kelch-like homologue 9 ....................13
kinésine-1 bêta ..................................33
lamines A/C ..........................7, 9, 11, 35
laminine ..............................................27
laminine alpha-2 .................................. 9
LAMP2 ..........................................23, 37
lipine 1 ................................................23
LITAF ...................................................33
maltase acide .....................................23
matrine-3 ............................................13
mérosine .............................................. 9
mitofusine ..........................................33
MPZ ........................................ 31, 33, 37
MuSK ...................................................27
MYH3 myosine ...................................17
myosine ..................... 13, 15, 17, 19, 37
myotiline ...................................7, 13, 15
myotubularine ...................................19
NDRG1 ...............................................33
NF-L chain ..........................................35
nébuline .......................................13, 17
NEFL ...................................................33
nesprine 1 ......................................9, 13
nesprine 2 ............................................ 9
OCTN2 ...............................................23
O-mannose bêta,
2N-acétylglucosaminyl-transférase 7, 11
O-mannosyl transférase 1 .............7, 11
O-mannosyl transférase 2 .............7, 11
P0 ........................................... 31, 33, 37
PABPN1 ..............................................15
périaxine.............................................33
perlecan .............................................21
pleckstrine ..........................................31
plectine .......................................7, 9, 29
phosphatase (phosphatidyl-inositol
biphosphate)......................................33
phosphatidyl-inositol biphosphate .33
phosphofructokinase ........................25
phosphoglucomutase .......................25
phosphoribosyl-pyrophosphate
synthétase 1 .......................................37
phosphorylase ...................................25
PMP22 ................................... 31, 33, 37
RAB7 ...................................................35
rapsyne ...............................................27
ribofl avine (transport de la) ..............31
ryanodine .....................................17, 19
sarcoglycane (α, β, γ, δ) ....................... 5
SBF2 ....................................................33
SH3 (protéine à domaine) ................33
seipine ................................................31
sélénoprotéine N1 ........... 9, 15, 17, 19
senataxine ..........................................31
SMN1 ..................................................29
téléthonine .....................................5, 11
titin-cap ..........................................5, 11
titine ..........................................7, 13, 15
TPR (protéine à domaine) .................33
TRIM 32................................................. 5
tropomyosine .....................................17
troponine T .........................................19
tyrosine-kinase (récepteur) ...............27
tyrosyl-ARNt synthétase ....................37
UBE1 ...................................................29
UDP-N-acétylglucosamine-2épimérase/
N-acétyl mannosamine kinase ...13, 15
valosine ..............................................13
VCP .....................................................15
Vma21p ................................................ 9
VRK1 ...................................................29
ZASP .............................................13, 15
ZNF9 ...................................................21
A
B
C
D
E
F
G
H
I
K
L
M
N
O
P
R
S
T
U
V
Z
Index des protéines
42 | Octobre 2012
AARS ............................................... 34ACADS ............................................ 22ACADVL .......................................... 22ACTA1 ....................................... 14, 16ACVR1 ............................................. 24AGL ................................................. 22AGRN .............................................. 26ANO5 .......................................... 6, 12
BAG3 ............................................... 14BIN1 ................................................ 18BSCL2 ............................................. 30
CACNAIS ........................................ 20CACNL1A3 ..................................... 20CAPN3 .............................................. 4CASQ1 ............................................ 14CAV3 ..................................... 6, 12, 20CFL2 ................................................ 18CHAT ............................................... 24CHKB ......................................... 10, 22CHRNA1 ......................................... 26CHRNB1 .......................................... 26CHRND ........................................... 26CHRNE ............................................ 26CLCN1 ............................................ 20CNTN1 ............................................ 18COL6A1 ............................................ 8COL6A2 ............................................ 8COL6A3 ............................................ 8COLQ .............................................. 26CPT2 ............................................... 22CRYAB ............................................. 14Cx32 ................................................ 36
DES ........................................... 14, 36DMD.................................................. 4DMPK .............................................. 20DNM2 .................................12, 18, 36DOK7 .............................................. 26DPAGT1 .......................................... 28DUX4................................................. 6DYNC1H1 ................................. 28, 36DYSF............................................ 4, 12
EGR2 ............................................... 32EMD .................................................. 6ETFDH ............................................. 22
FBLN5 ............................................. 32FGD4 ............................................... 32FHL1 ................................6, 10, 18, 36FIG4 ................................................ 32FLNC ......................................... 12, 14
FKRP ........................................ 4, 8, 10FKTN ........................................... 6, 10
GAA ................................................ 22GARS ......................................... 30, 34GDAP1 ...................................... 32, 34GFPT1 ............................................. 28GNE ........................................... 12, 14GYS1 ............................................... 24
HSPB1 ....................................... 28, 34HSPB3 ............................................. 28HSPB8 ....................................... 28, 34
IGHMBP2 ........................................ 30ISCU ................................................ 24ISPD ................................................ 10ITGA7 .............................................. 10
KBTBD13 ........................................ 16KCNE3............................................. 20KCNJ2 ............................................. 20KIFB ................................................. 32KLHL9 .............................................. 12
LAMA2 .............................................. 8LAMB2 ............................................ 26LAMP2 ...................................... 22, 36LARGE ............................................. 10LITAF ............................................... 32LMNA ................................ 6, 8, 10, 34LPIN1 .............................................. 22LRSAM1 .......................................... 36
MATR3 ............................................ 12MFN2 .............................................. 32MPZ ...............................30, 32, 34, 36MTM1 .............................................. 18MTMR2 ........................................... 32MTMR14 ......................................... 18MUSK .............................................. 26MYH2 ........................................ 14, 16MYH3 .............................................. 16MYH7 ..................... 12, 14, 16, 18, 36MYOT .................................... 6, 12, 14
NDRG1 ............................................ 32NEB ........................................... 12, 16NEFL.......................................... 32, 34
PABPN1 .......................................... 14PFKM ............................................... 24PGM1 .............................................. 24PLEC1 ..................................... 6, 8, 28
PLEKHG5 ........................................ 30PNPLA2 ........................................... 22PMP22 ................................30, 32, 36POMGNT1 .................................. 6, 10 POMT1 ........................................ 6, 10POMT2 ........................................ 6, 10PRPS1 .............................................. 36PRX .................................................. 32PYGM .............................................. 24
RAB7 ............................................... 34RAPSN ............................................. 26RYR1 .......................................... 16, 18
SBF2 ................................................ 32SBMA .............................................. 30SCAD .............................................. 22SCN4A ...................................... 20, 28SEPN1 .............................8, 14, 16, 18SERCA1 .................................... 14, 20SETX ................................................ 30SGCA ................................................ 4SGCB ................................................ 4SGCD ................................................ 4SGCG ................................................ 4SH3TC2 ........................................... 32SJS1 ................................................ 20SLC22A5 ......................................... 22SLC52A2 ......................................... 30SLC52A3 ......................................... 30SMN1 .............................................. 28SYNE1 ......................................... 8, 12SYNE2 ............................................... 8
TCAP ........................................... 4, 10TNNT1 ............................................. 18TPM2 ............................................... 16TPM3 ............................................... 16TRIM 32 ............................................. 4TRPV4 ........................................ 34, 36TTN ........................................ 6, 12, 14
UBE1 ............................................... 28
VCP ........................................... 12, 14VLCAD ............................................ 22VMA21 .............................................. 8VRK1 ................................................ 28
YARS ................................................ 36
ZASP ......................................... 12, 14ZNF9 ............................................... 20
A
B
C
D
E
F
G
H
I
K
L
M
N
P
R
S
T
U
V
Y
Z
Index des gènes
Octobre 2012 | 43
Avancées médico-scientifiques neuromusculaires

Association reconnue d’utilité publique
1, rue de l’Internationale - BP 59 - 91002 Évry cedexTél. : 33 (0) 1 69 47 28 28 - Fax : 33 (0) 1 60 77 12 16
Siège social : AFM - Institut de Myologie47-83, boulevard de l’Hôpital - 75651 Paris cedex 13
www.afm-telethon.fr
SAVOIR & COMPRENDRE FICHE TECHNIQUE Avancées médico-scientifiques neuromusculaires
© A
FM 1
0/20
12 (5
e éd
.) •
ISSN
: 17
69-1
850
• Re
pro
duc
tion
sans
but
lucr
atif
auto
risée
en
men
tionn
ant l
'orig
ine
• Ré
dac
tion
: TN
. Brig
nol,
J.A
. Urt
izb
erea
• V
alid
atio
n : S
. Bra
un •
Mis
e en
pag
e : a
2i g
rap
hic
• Ic
ono
gra
phi
e : M
. Gill
es •
em
ail :
myo
info
@af
m.g
enet
hon.
fr •
Imp
ress
ion
: Taa
g 0
1 69
25
40 4
0
Site Internet AFMwww.afm-telethon.frRubrique "Médiathèque" : publications médico-scientifi ques de l'AFM, rédigées et validées par une équipe de rédacteurs spécialisés (PDF téléchargeables).
www.myobase.orgBase documentaire sur les maladies neuromusculaires et le handicap moteur éditée par le service documentation de l'AFM.
www.musclegenetable.fr
Principales maladies neuromusculairesFiche Technique Savoir et Comprendre, AFM, 2012.
Myobank : une banque de tissus pour la rechercheFiche Technique Savoir et Comprendre, AFM, 2009.
Polymyosite pureFiche Technique Savoir et Comprendre, AFM, 2010.
Dermatomyosite pureFiche Technique Savoir et Comprendre, AFM, 2010.
Myosites idiopathiquesFiche Technique Savoir et Comprendre, AFM, 2010.
Myosite de chevauchementFiche Technique Savoir et Comprendre, AFM, 2010.
Myosite à inclusions sporadiqueFiche Technique Savoir et Comprendre, AFM, 2010.
Canalopathies musculairesFiche Technique Savoir et Comprendre, AFM, 2010.
Paralysies périodiquesFiche Technique Savoir et Comprendre, AFM, 2010.
Myotonies congénitalesFiche Technique Savoir et Comprendre, AFM, 2010.
Myotonies du canal sodiumFiche Technique Savoir et Comprendre, AFM, 2010.
Paramyotonie congénitaleFiche Technique Savoir et Comprendre, AFM, 2010.
EN
SAVO
IR +
Nous remercions tout particulièrement Gisèle Bonne, Daniel Hantaï, Yann Péréon pour leur aide précieuse dans la relecture du tableau concernant les laminopathies, les syndromes myasthéniques congénitaux et les maladies de Charcot-Marie-Tooth.